Настоящее изобретение относится к новым антитромботическим двойным ингибиторам, включающим метку в виде биотина или производного биотина, способу их получения, фармацевтическим композициям, содержащим указанные соединения в качестве активных ингредиентов, а также к применению указанных соединений для получения лекарственных средств.
Недавний прогресс в поиске антагонистов GpIIb/IIIa, имеющих предсказываемое антитромботическое действие, предпочтительно с более длинным периодом полувыведения (для достижения согласующихся уровней ингибирования агрегации тромбоцитов), привел в результате к новым антитромботическим двойным ингибиторам, имеющим смешанный фармакологический профиль. Эти новые соединения ингибируют две ключевые мишени как в системе свертывания крови (фактор Ха), так и в пути агрегации тромбоцитов (GpIIb/IIIa) (описанные в ЕР 1574516).
В качестве предупредительной меры в области антикоагулянтной и антиатеротромботической терапии существует потребность в антидоте, способном эффективно нейтрализовать или минимизировать активность используемого антикоагулянтного или антиатеротромботического лекарственного средства. Такая потребность существует, поскольку хорошо известно, что у пациента, подвергаемого лечению, по какой-либо случайной причине может начаться кровотечение. Кроме того, это необходимо при хирургическом вмешательстве для пациента, подвергаемого антиатеротромботическому или антикоагулянтному лечению. Кроме того, во время некоторых хирургических операций антикоагулянты могут использоваться в высокой дозе так, чтобы предотвратить свертывание крови, и в конце операции необходимо их нейтрализовать. Кроме того, клинически эффективных антидотов все еще нет в наличии в антитромбоцитарной терапии, когда используют ингибиторы GpIIb/IIIa. Следовательно, является полезным иметь в распоряжении антиатеротромботические и/или антикоагулянтные агенты, которые можно нейтрализовать для того, чтобы прекратить в любое время антиатеротромботическую и/или антикоагулянтную активность.
В заявке US 2004/0024197 (WO 02/24754) описывается, что в экстренном случае антикоагулянтную активность некоторых полисахаридов можно частично уменьшить, используя авидин, если эти полисахариды содержат по крайней мере ковалентную связь с биотином или производным биотина. О. Roger и др. в Carbohydrate Polymers 50 (2002) 273-278 обсуждают дериватизацию углеводов с помощью восстановительного аминирования, в том числе агентов биотинилирования. Это обсуждение относится к неизбирательной модификации полидисперсных встречающихся в природе полисахаридов.
Настоящее изобретение относится к новым нейтрализуемым двойным ингибиторам, получаемым из двойных ингибиторов, описанных в ЕР 1574516. Обнаружено, что определенную "метку" в виде биотина, являющуюся группой  , на которую в этом документе также приводится ссылка в виде "ВТ" (происходящую из гексагидро-2-оксо-1Н-тиено[3,4-d]имидазол-4-пентановой кислоты, предпочтительно D(+)-изомера), или его аналога можно присоединить или ввести в структуру соединений, описанных в ЕР 1574516, что приводит в результате к получению нейтрализуемых двойных ингибиторов.
, на которую в этом документе также приводится ссылка в виде "ВТ" (происходящую из гексагидро-2-оксо-1Н-тиено[3,4-d]имидазол-4-пентановой кислоты, предпочтительно D(+)-изомера), или его аналога можно присоединить или ввести в структуру соединений, описанных в ЕР 1574516, что приводит в результате к получению нейтрализуемых двойных ингибиторов.
Таким образом, настоящее изобретение относится к соединениям формулы (I)
олигосахарид-спейсер-антагонист GpIIb/IIIa (I),
где
олигосахарид представляет собой отрицательно заряженный олигосахаридный остаток, включающий от четырех до двадцати пяти моносахаридных единиц, при этом заряд компенсируется положительно заряженными противоинонами, и происходящий из олигосахарида, который сам по себе обладает (опосредуемой АТ-III) анти-Ха активностью;
спейсер представляет собой связь или по существу фармакологически неактивный связывающий остаток;
антагонист GpIIb/IIIa представляет собой остаток, имитирующий RGD- и/или K(QA)GD-фрагмент фибриногена, включающий карбоксилатную составляющую и основную составляющую, находящиеся в пределах остатка на расстоянии 10-20 Å друг от друга;
и их фармацевтически приемлемой соли, или пролекарству, или сольвату, где соединение формулы I, кроме того, включает по крайней мере одну ковалентную связь с меткой в виде биотина или его аналога.
Соединения настоящего изобретения являются эффективными антитромботическими агентами благодаря как опосредуемому АТ-III ингибированию фактора Ха свертывания крови, так и ингибированию агрегации тромбоцитов путем противодействия связыванию фибриногена со своим рецептором. Они применимы для лечения и (возможно) профилактики тромботических заболеваний. Такие заболевания включают ряд тромботических или протромботических состояний, при которых активируется система свертывания крови, которые включают, но без ограничения, тромбоз глубоких вен, эмболию сосудов легких, тромбофлебит, окклюзию артерий тромбом или эмболом, реокклюзию артерий во время или после пластической операции на сосудах, рестеноз после повреждения артерий или инвазивных кардиологических процедур, послеоперационный тромбоз или эмболия вен, удар и инфаркт миокарда.
Метка в виде биотина (или его аналога) в соединениях настоящего изобретения быстро узнается специфическим антидотом, являющимся авидином (The Merck Index, Twelfth edition, 1996, M. N. 920, pages 151-152) или стрептавидином, двумя тетрамерными белками с соответствующими массами, равными приблизительно 66000 и 60000 Да, которые обладают очень высоким сродством к биотину, и связывается с ним. Таким образом, в экстренной ситуации действие двойного ингибитора настоящего изобретения можно быстро нейтрализовать, используя авидин или стрептавидин, например, путем инъекции фармацевтического раствора, содержащего авидин или стрептавидин. Также можно использовать аналоги авидина и стрептавидина, обладающие высоким сродством к биотину. Получаемый в результате неактивный комплекс антидот-ингибитор удаляется из кровотока.
В сравнительных исследованиях с использованием соответствующих не биотилированных соединений обнаружено, что введение биотиновой метки в двойные ингибиторы не блокирует их ингибиторную в отношении агрегации тромбоцитов активность или их опосредуемую антитромбином III (АТ-III) анти-Ха активность. Кроме того, введение авидина или стрептавидина приводит к быстрой и вплоть до количественной нейтрализации антитромботической активности соединений формулы I.
Аналоги биотина, которые можно использовать в качестве метки в соответствии с настоящим изобретением, можно выбрать, но без ограничения, из аналогов биотина, продемонстрированных в каталоге Piersce, 1999-2000, страницы 62-81, например 6-биотинамидогексаноат, 6-(6-биотинамидогексанамидо)гексаноат и 2- биотинамидоэтантиол и т.п. В таких аналогах биотиновая метка ВТ, определенная ранее, является характерной частью структуры. Другими аналогами являются, например, аналоги биотина, в которых алкилирована биотинамидная связь (где алкил представляет собой (1-4(алкил), предпочтительно метил), которая является устойчивой к расщеплению биотинидазой, или другие аналоги биотина, включающие, например, гидроксиметилен, карбоксилат или ацетат альфа в биотинамидной связи.
Предпочтительные аналоги биотина имеют формулу -(NH-CO)n-(CH2)p-X-BT, где n представляет собой число 0 или 1, p представляет собой число 4 или 5, X=NH, N(1-4)алкил, -NH-CH(CH2OH)-CH2-C(O)-NH-, -NH-CH(CH3)-CH2-C(O)-NH-, -NH-CH(COOH)-CH2-C(O)-NH-, -NH-CH(CH2COOH)-CH2-C(O)-NH-, а ВТ представляет собой метку, определенную ранее.
Для соединений настоящего изобретения подходит любой отрицательно заряженный олигосахаридный остаток, состоящий из от четырех до двадцати пяти моносахаридных единиц. Подходящими соединениями настоящего изобретения являются соединения, в которых олигосахарид представляет собой сульфатированный олигосахаридный остаток. Предпочтительно олигосахаридный остаток происходит из олигосахарида, который сам по себе обладает (опосредуемой АТ-III) анти-Ха активностью, такого как олигосахариды, раскрытые в ЕР 0454220, ЕР 0529715, WO 97/47659, WO 98/03554 и WO 99/36443. Предпочтительными, кроме того, являются олигосахаридные остатки, имеющие от четырех до шестнадцати моносахаридных единиц. Самым предпочтительным олигосахаридом является сульфатированный пентасахаридный остаток. Предпочтительные пентасахаридные остатки имеют структуру А
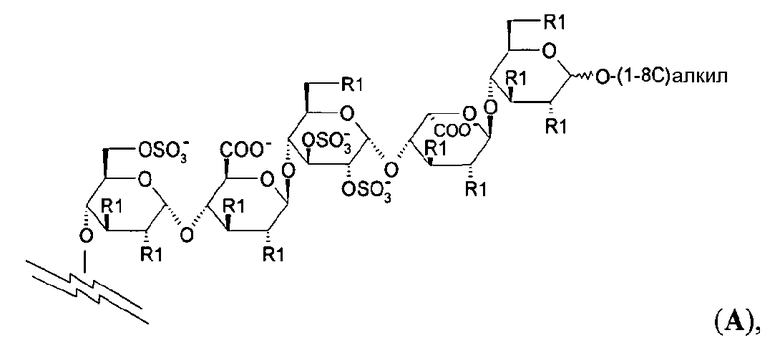
где R1 представляет собой независимо метку в виде биотина или его аналога, OSO3 - или (1-8С)алкокси.
Особенно предпочтительные пентасахариды имеют структуру В
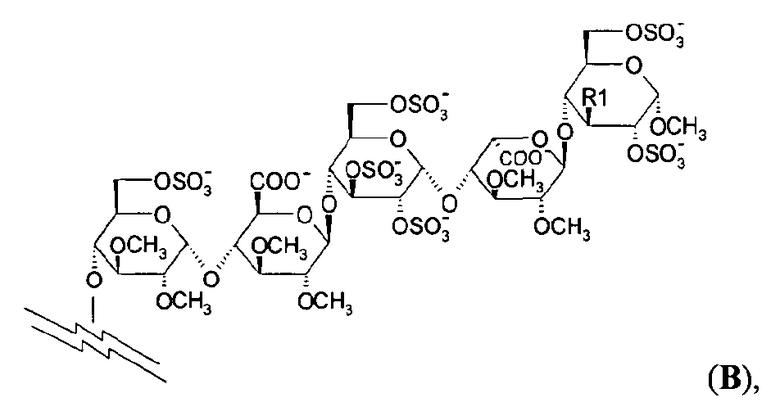
где R1 представляет собой ОСН3 или OSO3 -. В самых предпочтительных пентасахаридах структуры В R1 представляет собой OSO3 -.
Спейсер представляет собой связь или по существу фармакологически неактивный, гибкий, связывающий остаток. Используемый здесь термин "по существу фармакологически неактивный" означает, что спейсер не содержит атомов или групп, которые проявляют фармакологическую активность саму по себе в дозах, при которых соединения настоящего изобретения являются терапевтически эффективными. Таким образом, в дозах, в которых соединения настоящего изобретения используются в качестве антитромботических агентов, природа спейсера не приводит к демонстрируемым фармакологическим побочным эффектам. В предпочтительных вариантах осуществления настоящего изобретения спейсер представляет собой по существу фармакологически неактивный связывающий остаток, предпочтительно имеющий 1-50 атомов, подсчитываемых вдоль "остова" спейсера, не включая кислород олигосахаридного остатка. Спейсер может включать (до некоторой степени) жесткие элементы, такие как кольцевые структуры и ненасыщенные связи. Спейсер соединений настоящего изобретения является предпочтительно гибким. Квалифицированный в данной области специалист может легко сконструировать подходящие спейсеры. Наиболее предпочтительной длиной спейсера является 10-35 атомов, в частности 10-28. По причинам синтеза более длинные спейсеры считаются менее подходящими, однако более длинные спейсеры могут, тем не менее, использоваться в соединениях настоящего изобретения. В высокой степени подходящие спейсеры включают по крайней мере один -(СН2СН2О)-элемент. Более предпочтительные спейсеры включают более, предпочтительно шесть -(СН2СН2О)-элементов.
Сайт присоединения спейсера к остатку антагониста GpIIb/IIIa может быть выбран по существу произвольно при условии, что не отменяется активность антагониста GpIIb/IIIa. Таким образом, должны оставаться не затронутыми обычно присутствующие карбоксилатная составляющая (необязательно этерифицированная) и основная составляющая.
В предпочтительных соединениях в соответствии с настоящим изобретением остаток антагониста GpIIb/IIIa выбирают из остатков, происходящих из Ro 435054, SC 54701 (ксемилофибана), RWJ 50042, субрафибана (Ro 443888), ламифибана (Ro 449883),GPI 562, FK 633, тирофибана (MK 383), орбофибана (SC 57101), эптифибатида (С68 22), роксифибана (XV 459), эларофибана (RWJ 53308), SR 121787, лефрадафибана (BIBU 52), лотрафибана (SB 214857), гантофибана (YM 028), T-250, EF 5077, ZD 2486, TAK 029, TP 9201, L 703014, SR 121566 (активной формы SR 121787) и UR-3216. Производные указанных остатков также включают химически модифицированные остатки, в которых сохраняется часть, включающая (необязательно этерифицированную) карбоксилатную составляющую и основную составляющую (или защищенную основную составляющую). В предпочтительном варианте осуществления изобретения выбирают SR 121566 (активную форму SR 121787). Самыми предпочтительными являются соединения, в которых остаток антагонста GpIIb/IIIa происходит из тирофибана.
Предпочтительные соединения в соответствии с настоящим изобретением включают одну ковалентную связь с меткой в виде биотина или его аналога.
Метка в виде биотина (или его аналога) может присутствовать во всех частях соединения формулы I. Таким образом, вариантами осуществления настоящего изобретения являются соединения, в которых (а) олигосахаридный остаток соединения формулы I включает ковалентную связь с меткой в виде биотина или его аналога, (b) спейсер соединения формулы I включает ковалентную связь с меткой в виде биотина или его аналога и (с) остаток антагониста GpIIb/IIIa в соединении формулы I включает ковалентную связь с меткой в виде биотина или его аналога.
Предпочтительными являются соединения, включающие одну ковалентную связь с аналогом биотина, причем олигосахаридный остаток соединения формулы I включает одну ковалентную связь с аналогом биотина формулы -(NH-CO)n-(CH2)p-X-BT, где n представляет собой число 0 или 1 (в этих соединениях предпочтительно n=1), p представляет собой число 4 или 5 (в этих соединениях предпочтительно р=5), X и ВТ имеют значения, определенные ранее.
Другие предпочтительные соединения настоящего изобретения включают одну ковалентную связь с аналогом биотина, причем спейсер соединения формулы I включает одну ковалентную связь с аналогом биотина формулы -(CH2)4-X-BT, где Х и ВТ имеют значения, определенные ранее.
Типичные примеры биотинилированных двойных ингибиторов настоящего изобретения имеют структуры
 и
и
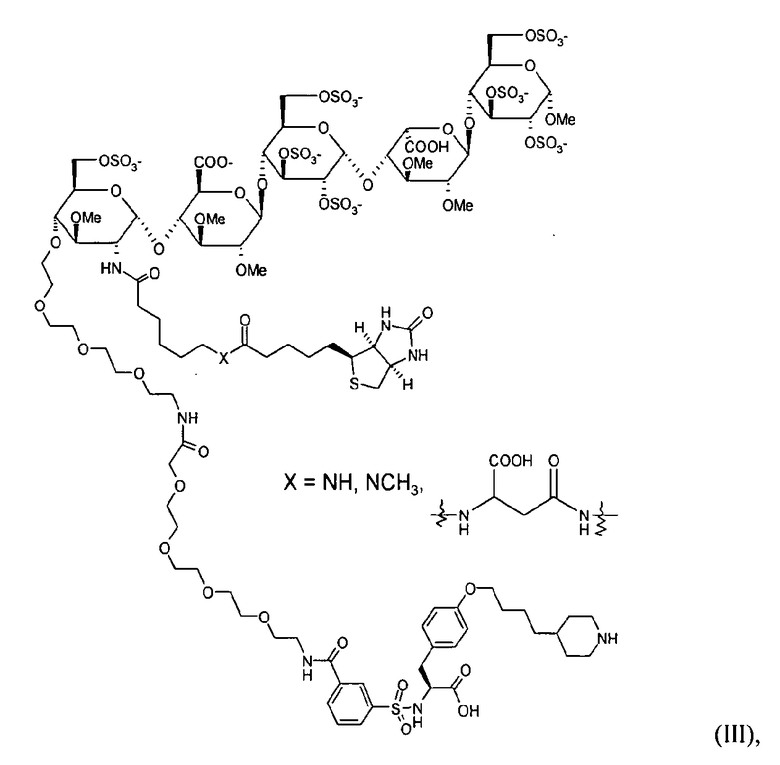
но также являются соединениями формулы I, в которых спейсер присоединен к олигосахариду в другом положении, и/или соединениями, в которых метка в виде биотина (аналога) присутствует в других положениях молекулы. Соединения формулы II являются предпочтительными примерами настоящего изобретения.
Положительно заряженные противоионы означают H+, Na+, K+, Ca+ и т.п. Предпочтительно соединения формулы I находятся в форме их натриевой соли.
Термин "основная составляющая" означает любую хорошо известную основную составляющую, такую как амин, амидин, гуанидин, пиперидин и т.п.
Под фразой "на расстоянии 10-20 Å друг от друга" подразумевается пространственная ориентация двух групп относительно другой, не только измеряемая вдоль связей. В распоряжении квалифицированного в данной области специалиста имеются хорошо известные методы моделирования для определения расстояния (см., например, J. Med. Chem. 1994, 37, 2537-2551).
Термин (1-8С)алкил означает разветвленную или не разветвленную алкильную группу, имеющую 1-8 атомов углерода, например метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, гексил и октил. Предпочтительными алкильными группами являются метил и этил.
В термине (1-8С)алкокси алкильный остаток имеет значение, определенное ранее.
Термин "пролекарство" означает соединение, которое метаболизируется в организме в активное соединение, например соединение, в котором основная составляющая (такая, как амино- или бензамидино-группа) в остатке антагониста GpIIb/IIIa в соединении формулы I защищена, например, гидроксигруппой, (1-6С)алкоксильной или (1-6)алкоксикарбонильной группой. Примерами пролекарств также являются соединения формулы I, в которых карбоксилатная группа в остатке антагониста GpIIb/IIIa является этерифицированной.
Сольваты в соответствии с настоящим изобретением включают гидраты.
Соединения настоящего изобретения можно приготовить необязательно путем модифицирования ранее описанных антагонистов GpIIb/IIIa, которые происходят, например, из тирофибана, SR 121566, Ro 435054, RWJ 50042 или SC 54701 (фармакологически активной формы ксемилофибана), ламифибана или их аналогов, с использованием аминокислот, пептидомиметиков или дополнительных функциональных групп (например, -СООН, -NH2, -SH, -OH, -N3, концевого алкина и т.п.), используя методы, как правило, известные в данной области техники. Пример синтеза такого модифицированного RGD-аналога описывается в публикации Bioorganic Chemistry 29, 357-379 (2001), в которой соединение предлагается в качестве возможного вектора для целенаправленной доставки лекарственного средства. В соответствии с настоящим изобретением необязательно модифицированную часть антагониста GpIIb/IIIa (а) связывают непосредственно с олигосахаридом, или (b) связывают с остатком олигосахарид-спейсер, или (с) связывают со спейсером, который впоследствии связывают с остатком олигосахарид-спейсер (например, с помощью методов, известных из заявок WO 99/65934; WO 01/42262). Для этой цели можно использовать любой подходящий олигосахарид, например олигосахариды, известные из литературы (например, из ЕР 0454220 и ЕР 0529715, не ограничиваясь этими источниками), или коммерчески доступные олигосахариды. Олигосахариды можно фосфорилировать в соответствующий момент времени, используя методы, известные в данной области техники, например, методы, описанные Buijsman R. et al. (Bioorg. Med. Chem. Lett. 1999, 9, 2013-2018). Связывание спейсера с олигосахаридом можно, например, выполнить, используя методы, описанные в ЕР 0649854 или ЕР 04005343.1.
Что касается метода, с помощью которого биотиновую метку присоединяют к соединениям формулы I, в литературе по химии предлагаются несколько возможностей, которые можно использовать и при которых можно использовать различные наборы защитных групп, хорошо известных квалифицированному в данной области техники специалисту. Биотиновую метку, включающую реакционноспособную группу, например, типа активированного сложного эфира, малеимида, иодацетила или первичного амина, предпочтительно подвергают реакции с функциональной аминогруппой, или тиольной функциональной группой, или функциональной группой карбоновых кислот, или альдегидной функциональной группой, при этом реакция происходит в соответствии с условиями, описанными в литературе (ср. Savage et al. Avidin-Biotin Chemistry: A Handbook, Pierce Chemical Company, 1992).
Биотиновую метку можно, например, связать непосредственно с (отрицательно заряженным) олигосахаридным остатком, или через функциональную необязательно N-(1-4С)алкилированную аминогруппу остатка олигосахарид-спейсер, или через необязательно N-(1-4С)алкилированный аминокислотный остаток с функциональной необязательно N-(1-4С)алкилированной аминогруппой олигосахаридного остатка соединения формулы I.
В другом аспекте настоящего изобретения биотиновую метку можно, например, связать непосредственно с остатком антагониста GpIIb/IIIa, или через функциональную необязательно N-(1-4С)алкилированную аминогруппу связывающего остатка, или через необязательно N-(1-4С)алкилированный аминокислотный остаток с функциональной необязательно N-(1-4С)алкилированной аминогруппой остатка антагониста GpIIb/IIIa соединения формулы I.
В еще одном аспекте настоящего изобретения биотиновую метку можно, например, ввести поэтапно с помощью, во-первых, связывания непосредственно с остатком антагониста GpIIb/IIIa, или через функциональную необязательно N-(1-4С)алкилированную аминогруппу части спейсера формулы I, или через необязательно N-(1-4С)алкилированный аминокислотный остаток с функциональной необязательно N-(1-4С)алкилированной аминогруппой остатка антагониста GpIIb/IIIa в соединении формулы I и, во-вторых, связывания непосредственно с олигосахаридом, или через функциональную необязательно N-(1-4С)алкилированную аминогруппу части спейсера формулы I, или через необязательно N-(1-4С)алкилированный аминокислотный остаток с функциональной необязательно N-(1-4С)алкилированной аминогруппой (отрицательно заряженного) олигосахарида соединения формулы I, или наоборот.
В другом аспекте настоящего изобретения необязательно N-алкилированные аминокислотные остатки или α-N-замещенные (бета-)аминокислотные аналоги можно ввести с помощью пептидного связывания, используя известные в данной области техники методы. Азидогруппа является подходящей латентной функциональной аминогруппой, которую можно использовать в предшественниках соединения формулы I для последующего введения биотиновой метки.
Также другими примерами известных антагонистов GpIIb/IIIa могут служить в качестве (основы для) части антагониста GpIIb/IIIa в соединениях настоящего изобретения (но без ограничения этими примерами) соединения Ro 43 8857, Ro 48 3657, BIBL 12, FK 633, GR 144053, EMD 76 334, SR 121566, SB 208651, SC 54684, SC 52012, DMP 754, FR 158999, GR 200976, XV 788, MK 383 (тирофибан), RWJ 53308, ZD 2486, L 709780, RGD 891, T 250, C 6822, BIBU 104, SB 214857, SC 57101, G 7453, TAK 029, XV 454, XV 459, L 734 217, DMP 802, SR 121787, TP 9201, DMP 757, SC 52012, RPR 109891, YM 68128, ME 3229, ME 3230, CT 50352, MK 852, S 1197, DMP 728, SC 57345, L 738 167, GR 233548, Ro 438857, TA 993, YM 337, BIBW 194, BIBU 129, BIBW 98, тетрафибрицин, L 703 014, BIBU 251, GR 91669, RG 13965, G 7446, PS 028, XR 300, NSL 9403, L 756568, S 1762, L 746 223, L 767685, NSL 95301, G 4120, SB 207043, GR 83895, P246, L 739 758, XR 299, SV 873, RWJ 50228, XQ 870, EF 5154, AR 0510, G 7570, G 7442, G 7464, RWJ 52656, TAK 024, MS 180, MS 28168, XU 063, XU 065, L 734115, SM 20302, TS 943, NSL 96184, UR 12947, XU 057, L 750034, UR 3216, UR 2922, CP 4632, AR 0598, SC 79992, SC 4992, RGD 039, ME 3277, T 250, SC 57099B, SKF 106760, SKF 107260, RWJ 52654, PSA 0613, CGH 400, NSL 95317, XT 111, RWJ 27755, L 736622, SC 46749, SM 20302, YM 570029, CY 311176 и антагонисты GpIIb/IIIa, описанные в ЕР 0529858, WO 96/20172, EP 0496378, EP 0530505, Bioorg. & Med. Chem. 3, 539 (1995), WO 93/08174, J. Am. Chem. Soc. 115, 8861 (1993), J. Med. Chem. 43, 3453 (2000), Bioorg. Med. Chem. 3, 337 (1995), US 5239113, US 5344957, US 5973003, US 5703125, WO 96/37464, WO 93/07867, US 5378712, EP 445796, US 5273982, US 5770575, WO 01/602813, EP 656348, US 5726185, EP 505868, EP 560730, US 5561112, EP 513675, US 5574016, WO 94/09030, EP 478363, US 5292756, US 5206373, WO 93/16994, US 5312923, EP 743302, US 5658929, US 5880136, US 5814643, US 6040317, Expert Opin. Ther. Patents 13(8), 1173-1188 (2003) и Curr. Pharm. Design 14, 1567-1609 (2004).
Кроме того, в настоящее изобретение включены соединения, включающие вновь сконструированные остатки антагонистов GpIIb/IIIa, имитирующие RGD- и/или K(QA)GD-фрагмент фибриногена, обычно включающие карбоксилатную составляющую (необязательно этерифицированную) и основную составляющую, находящиеся в пределах остатка на расстоянии 10-20 Å друг от друга.
Пептидное связывание, процедурную стадию в описанном выше способе получения соединений настоящего изобретения, можно выполнить с использованием обычно известных в данной области техники методов связывания - или конденсации - пептидных фрагментов, таких как азидный метод, метод смешанных ангидридов, метод активированных сложных эфиров, карбодиимидный метод или, предпочтительно под влиянием солей аммония/урония, таких как TBTU, особенно с добавлением каталитических и супрессирующих рацемизацию соединений, подобных N-гидроксисукцинимиду, N-гидроксибензотриазолу и 7-аза-N-гидроксибензотриазолу. Обзоры приводятся в The Peptides, Analysis, Synthesis, Biology, Vol. 3, E. Gross and J. Meienhofer, eds. (Academic Press, New York, 1981) и в Peptides: Chemistry and Biology, N. Sewald and H.-D. Jakubke (Wiley-VCH, Weinheim, 2002).
Функциональные аминогруппы, присутствующие в соединениях, можно защитить во время процедуры синтеза с помощью N-защитной группы, которая означает группу, обычно используемую в химии пептидов для защиты α-аминогрупп, подобную трет-бутилоксикарбонильной (Вос) группе, бензилоксикарбонильной (Z) группе, 9-флуоренилметилоксикарбонильной (Fmoc) группе или фталоильной (Phth) группе, или можно ввести путем демаскировки азидной составляющей. Обзоры аминозащитных групп и способов их удаления приводятся в упоминаемых выше The Peptides, Analysis, Synthesis, Biology, Vol. 3 и в Peptides: Chemistry and Biology.
Функциональные группы амидинов, если присутствуют, можно оставить незащищенными на стадии связывания или их можно защитить, используя карбамат, такой как аллилоксикарбонил или бензилоксикарбонил. Функциональную группу амидина предпочтительно вводят в мягких условиях, используя составляющую 1,2,4-оксадиазолин-5-она в качестве предшественника.
Группы карбоновых кислот можно защитить с помощью группы, обычно используемой в химии пептидов для защиты группы α-карбоновых кислот, такой как трет-бутиловый эфир. Группы карбоновых кислот модифицированных антагонистов GpIIb/IIIa предпочтительно защищать в виде бензиловых эфиров. Удаление защитных групп может происходить различными путями в зависимости от природы этих защитных групп. Обычно снятие защиты происходит в кислотных условиях и в присутствии акцепторов или в условиях восстановления, таких как каталитическое гидрирование.
Условием, необходимым для конъюгации антагониста GpIIb/IIIa с олигосахаридом, является присутствие ортогонально реакционноспособной закрепляющей группы, такой как карбоксилатная группа, которую можно связать непосредственно с олигосахаридным остатком или с производным олигосахарид-спейсер или через спейсер с производным олигосахарид-спейсер. Для того чтобы сделать возможной такую конъюгацию, во многих случаях требуется дополнительная модификация антагониста GpIIb/IIIa.
Конструкцию из разделяемых спейсерами строительных блоков для синтеза соединений можно получить различными путями, используя известные в данной области техники методы, или линейным способом с помощью поэтапного введения аминокислот, их производных или пептидомиметиков, или конвергентным способом с помощью связывания блоков в виде промежуточных конструкций.
Соединения настоящего изобретения, которые могут находиться в форме свободного основания, можно выделить из реакционной смеси в форме фармацевтически приемлемой соли. Фармацевтически приемлемые соли можно также получить с помощью обработки свободного основания формулы (I) органической или неорганической кислотой, такой как хлористоводородная, бромистоводородная, иодистоводородная, серная, фосфорная, уксусная, пропионовая, гликолевая, малеиновая, малоновая, метансульфокислота, фумаровая, янтарная, винная, лимонная, бензойная, аскорбиновая и т.п.
Соединения настоящего изобретения или их промежуточные продукты могут обладать хиральными атомами углерода и могут, следовательно, быть получены в виде чистого энантиомера, или в виде смеси энантиомеров, или в виде смеси, содержащей диастереомеры. В данной области техники хорошо известны методы получения чистых энантиомеров, например кристаллизация солей, полученных из оптически активных кислот и рацемических смесей, или хроматография с использованием хиральных колонок. Для диастереомеров можно использовать колонки для прямо-фазовой или обращенно-фазовой хроматографии.
Соединения настоящего изобретения можно назначать энтерально или парентерально. Точная доза и схема введения этих соединений и их композиций обязательно зависит от потребностей индивидуального субъекта, которому лекарственное средство назначается, степени заболевания или потребности и оценки практикующего врача. Как правило, при парентеральном введении требуются более низкие дозы, чем при других способах введения, которые в большей мере зависят от абсорбции. Однако для людей ежедневные дозы составляют предпочтительно 0,0001-10 мг на кг веса тела, более предпочтительно 0,001-1 мг на кг веса тела.
Лекарственное средство, изготовленное с использованием соединений настоящего изобретения, можно также использовать в качестве адъювантов при (экстренной) антитромботической терапии. В таком случае лекарственное средство вводят с другими соединениями, применимыми для лечения таких болезненных состояний, такими как аспирин, клопидогрел или статины.
Смешанные с фармацевтически подходящими вспомогательными веществами, например, описанными в стандартном ссылочном документе, Gennaro et al., Remington'a Pharmaceutical Science, (18th ed., Mack Publishing Company, 1990, см. особенно Part 8: Pharmaceutical Preparations and Their Manufacture) соединения можно подвергнуть прессованию в твердые единицы доз, такие как пилюли, таблетки, или подвергнуть обработке в капсулы или суппозитории. При помощи фармацевтически приемлемых жидкостей соединения можно также применять в форме раствора, суспензии, эмульсии и т.п. для использования в виде препарата для инъекции.
Для получения единиц дозы, например таблеток, предусматривается использование традиционных добавок, таких как наполнители, красящие вещества, полимерные связующие вещества и т.п. Как правило, можно использовать любую фармацевтически приемлемую добавку, которая не мешает функции активных компонентов. Подходящие носители, с которыми можно вводить соединения, включают лактозу, крахмал, производные целлюлозы и т.п. или их смеси, используемые в подходящих количествах.
Краткое описание чертежей
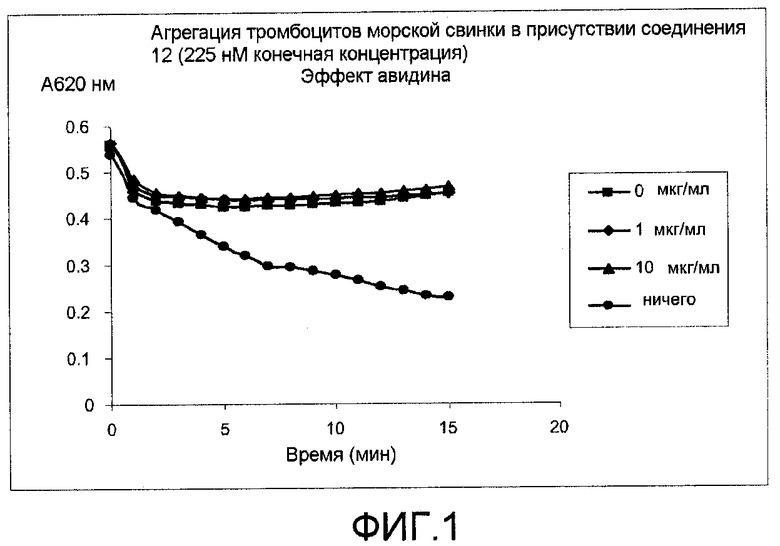
Фиг.1: Эффект авидина на ингибирование агрегации тромбоцитов с помощью соединения 12. Предварительная инкубация соединения и авидина.
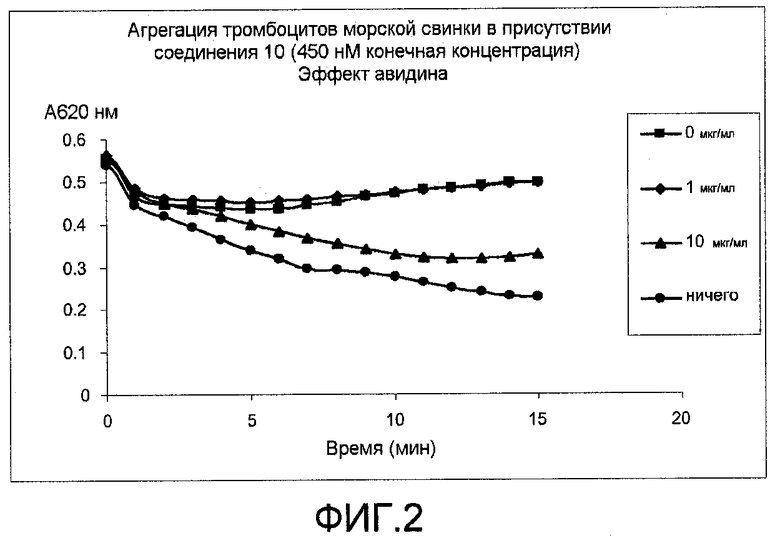
Фиг.2: Эффект авидина на ингибирование агрегации тромбоцитов с помощью соединения 10. Предварительная инкубация соединения и авидина.
Фиг.3: Эффект авидина на ингибирование агрегации тромбоцитов морских свинок с помощью соединения 12 (добавление авидина через 9 минут после ADP-индуцированной агрегации тромбоцитов).
Фиг.4: Эффект авидина на ингибирование агрегации тромбоцитов с помощью соединения 10 (добавление авидина через 9 минут после ADP-индуцированной агрегации тромбоцитов).
Фиг.5: Эффект авидина на ингибирование агрегации тромбоцитов человека с помощью соединения 17 (добавление авидина через 9 минут после ADP-индуцированной агрегации тромбоцитов).
Фиг.6: Демонстрирует усредненные данные после подкожного введения 500 нмоль/кг соединения.
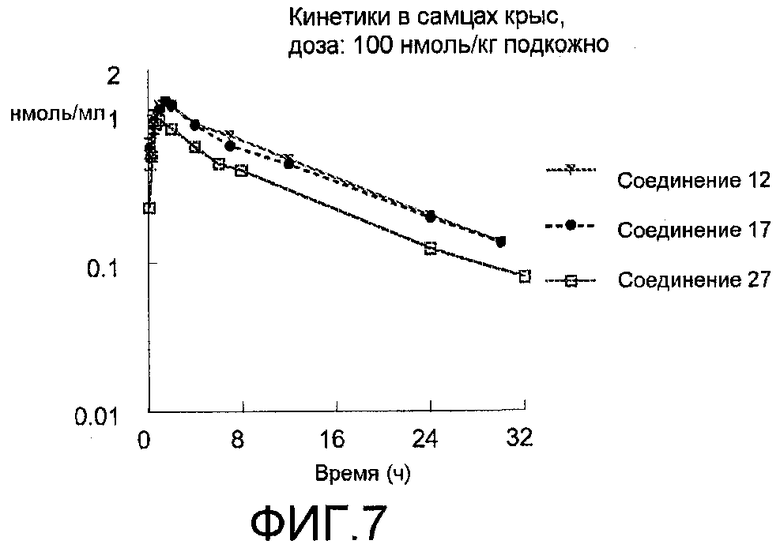
Фиг.7: Усредненные данные после подкожного введения 100 нмоль/кг соединения.
Фиг.8: Демонстрирует усредненные данные после подкожного введения 100 нмоль/кг соединения. В момент времени, равный 1 ч, внутривенно вводили авидин (10 мг/кг) тем крысам, которых обрабатывали соединением 10 или 12. Фармакокинетическое поведение соединения 27 (пентасахаридной составляющей) в отсутствие авидина изображено для сравнения.
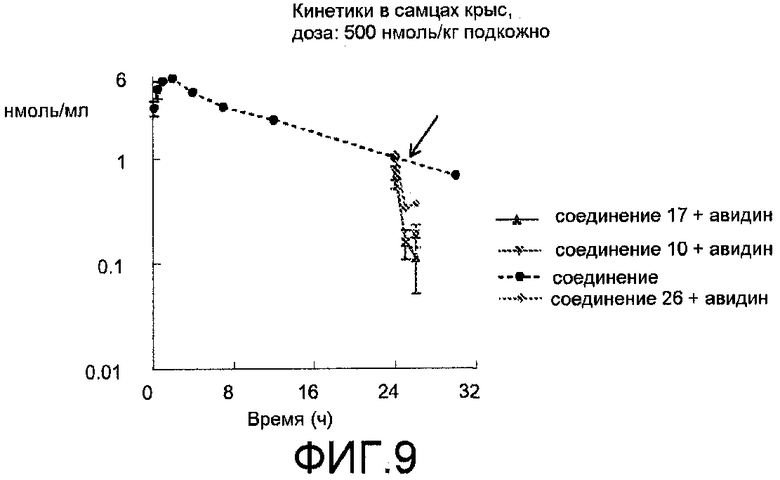
Фиг.9: Усредненные данные после подкожного введения 500 нмоль/кг соединения 10, 17 или 26. В момент времени, равный 24 ч, внутривенно вводили авидин (10 мг/кг), после чего образцы крови брали в моменты времени, равные 25 и 26 ч. Для сравнения величины 100 нмоль/кг подкожно соединения 17 нормализовали в отношении дозы 500 нмоль/кг.
Настоящее изобретение далее иллюстрируется следующими примерами.
ПРИМЕРЫ
Используемые сокращения
ADP=аденозин-5'-дифосфат
Aq.=водный
AT III=антитромбин III
Bn=бензил
Boc=трет-бутилоксикарбонил
DCM=дихлорметан
DiPEA=N,N-диизопропилэтиламил
DMF=N,N-диметилформамид
Fmoc=9-флуоренилметилкарбамат
NMM=N-метилморфолин
Me=метил
sat.=насыщенный
PRP=богатая в отношении тромбоцитов плазма
PPP=бедная в отношении тромбоцитов плазма
RT=комнатная температура
TBTU=2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония тетрафторборат
TFA=трифторуксусная кислота
THF=тетрагидрофуран
TRAP=пептид-агонист рецептора тромбина
Z=бензилоксикарбонил
ПРИМЕР 1
трет-Бутил-15-N-(9-флуоренилметилоксикарбонил)-15-аза-3,6,9,12-тетраокса-пентадеканоат (2)
трет-Бутил-15-амино-3,6,9,12-тетраокса-пентадеканоат (1) (0,5 г, 1,45 ммоль), который получали, как описано в ЕР 1574516, растворяли в THF (7,5 мл) и H2O (5 мл). Добавляли 4 н. раствор NaOH до достижения рН приблизительно 9. Порциями добавляли N-9-флуоренилметилкарбоната сукцинимид (FmocOSu, 0,54 г, 1,6 ммоль, 1,1 эквивалента). Через 10 мин добавляли дополнительно 4 N раствор NaOH до достижения рН приблизительно 9. Через 3 часа реакционную смесь подкисляли с помощью 1 н. раствора HCl до рН 6-7. Добавляли H2O в реакционную смесь, которую затем экстрагировали трижды EtOAc. Органическую фазу промывали соляным раствором и обезвоживали над MgSO4. После фильтрации растворитель удаляли при пониженном давлении (50 мбар, 50ºС). Неочищенное масло очищали с помощью хроматографии с использованием колонки с силикагелем (DCM/MeOH, 1/0→95/5, объем/объем) с получением соединения 2 в виде желтоватого масла (0,61 г, 79%). Rf 0,64 (DCM/MeOH, 95/5 объем/объем).
ПРИМЕР 2
15-N-(9-Флуоренилметилоксикарбонил)-15-аза-3,6,9,12-тетраокса-пентадеканоат (3)
Соединение 2 растворяли в DCM (3,5 мл) и добавляли TFA (3,5 мл) в атмосфере азота. После перемешивания в течение 1,5 часов реакционную смесь концентрировали при пониженном давлении. Затем избыток TFA удалили путем повторного концентрирования в толуоле. Добавляли DCM/Et2O (100 мл, 1/2, объем/объем) и раствор промывали 1 н. HCl. Водный слой экстрагировали DCM/Et2O (100 мл, 1/2, объем/объем). Объединенные органические слои промывали солевым раствором и обезвоживали над MgSO4. После фильтрации растворитель удаляли при атмосферном давлении (50ºС). Неочищенное масло очищали с помощью хроматографии с использованием колонки с силикагелем (DCM/MeOH/AcOH, 99/0/1→89/10/15, объем/объем/объем) с получением соединения 3. Остающийся AcOH удаляли растворением продукта в виде неочищенного масла в DCM/Et2O (1/2, объем/объем) и промыванием H2O (3×) и соленым раствором с последующим обезвоживанием над MgSO4. После фильтрации растворитель удаляли при атмосферном давлении (50ºС) с получением соединения 3 в виде желтоватого масла (0,37 г, 67%). Rf 0,32 (DCM/MeOH/AcOH, 89/10/1, объем/объем/объем).
ПРИМЕР 3
трет-Бутил-15-N-(9-флуоренилметилоксикарбонил)-15-аза-3,6,9,12-тетраокса-пентадеканоил-ε-N-(бензилоксикарбонил)-L-лизин (4)
Соединение 3 (0,37 мг, 0,77 ммоль) растворяли в DCM (18 мл). Впоследствии добавляли DiPEA (0,4 мкл, 2,31 ммоль, 3 эквивалента) и TBTU (0,25 г, 0,77 ммоль) в атмосфере N2 и раствору давали возможность перемешаться в течение 10 мин. Затем добавляли ε-(Z)-L-Lys-OtBu. HCl (0,29 г, 0,77 ммоль) и смесь перемешивали в течение дополнительных 1,5 часов. Реакционную смесь разбавляли DCM и промывали H2O, 1 н. HCl, насыщенным раствором NaHCO3 и солевым раствором. Органическую фазу обезвоживали (MgSO4) и концентрировали при атмосферном давлении. Очистку проводили с помощью хроматографии с использованием колонки с силикагелем (DCM/MeOH, 1/0→9/1, объем/объем) с получением соединения 4 в виде желтоватого масла (0,51 г, 83%). Rf 0,85 (DCM/MeOH, 9/1, объем/объем). ESI-MS: 792,6 [M+H]+, 814,6 [M+Na]+, 736,4 [M-tBu+H]+
ПРИМЕР 4
трет-Бутил-15-N-трет-бутилоксикарбонил-15-аза-3,6,9,12-тетраокса-пентадеканоил-ε-N-(бензилоксикарбонил)-L-лизин (5)
Соединение 4 (0,26 г, 0,32 ммоль) растворяли в THF (5 мл). Добавляли Et2NH (1 мл) и раствору давали возможность перемешаться в течение 24 часов. Избыток Et2NH и растворителя удаляли при пониженном давлении (50ºС). Толуол добавляли и удаляли при пониженном давлении (50ºС, 65 мбар) с получением N-незащищенного продукта (0,21 г, 0,32 ммоль). Rf 0,23 (DCM/MeOH, 9/1, объем/объем). ESI-MS: 570,4 [M+H]+, 514,4 [M-tBu+H]+.
Неочищенный продукт растворяли в DCM (3 мл). Добавляли Et3N (0,11 мл) с последующим добавлением ди-трет-бутилдикарбоната (73 мг, 0,34 ммоль, 1,1 эквивалента) в атмосфере N2. После перемешивания в течение 5 ч смесь добавляли к холодному (5ºС) раствору 1 н. HCl и экстрагировали EtOAc. Органический слой промывали солевым раствором и обезвоживали (MgSO4). После фильтрации растворители удаляли при пониженном давлении (180 мбар, 50ºС). Очистку проводили с помощью хроматографии с использованием колонки с силикагелем (DCM/MeOH, 1/0→95/5, объем/объем) с получением бесцветного масла (0,17 г, 82%). Rf 0,5 (DCM/MeOH, 9/1, объем/объем). ESI-MS: 670,6 [M+H]+, 692,4 [M+Na]+, 570,4 [M-Boc+H]+, 514,1 [M-Boc-tBu+H]+.
ПРИМЕР 5
15-Аза-3,6,9,12-тетраокса-пентадеканоил-ε-[D-(+)-биотинил]-L-лизин (6)
Соединение 5 (0,23 г, 0,34 ммоль) растворяли в ETOH (5 мл) и Н2О (1,2 мл). После продувания раствора азотом в течение 5 минут добавляли Pd/C 10% (0,11 г). Через раствор пропускали водород в течение 4 ч. Азот продували через раствор в течение 10 минут с удалением какого-либо водорода. Смесь фильтровали через декалит и концентрировали при пониженном давлении (170 мбар, 50ºС) с получением N-L-лизин-незащищенного промежуточного продукта в виде бесцветного масла (0,15 г, 81%). Rf 0,02 (DCM/EtOAc, 9/1, объем/объем).
D-(+)-биотин (75 мг, 0,31 ммоль) суспендировали в DCM (7 мл). Впоследствии добавляли DiIPEA (0,11 мл, 0,62 ммоль, 2 эквивалента) и TBTU (0,10 г, 0,31 ммоль) в атмосфере N2 и раствору давали возможность перемешаться в течение 1 ч. К реакционной смеси добавляли раствор описанного выше ε-N-L-лизин-незащищенного промежуточного продукта в DCM (3 мл). Смеси давали возможность перемешаться в течение 16 ч. Добавляли Н2О и экстрагировали DCM (3×). Органический слой обезвоживали (MgSO4), фильтровали и концентрировали при пониженном давлении (850 мбар, 50ºС). Очистку проводили с помощью хроматографии с использованием колонки с силикагелем (элюция: DCM/MeOH, 1/0→9/1, объем/объем) с получением масла (0,13 г, 60%). Rf 0,48 (DCM/MeOH, 9/1, объем/объем). ESI-MS: 762,6 [M+H]+, 784,6 [M+Na]+, 662,4 [M-Boc+H]+, 606,4 [M-Boc-tBu+H]+. Масло растворяли в сухом 4 н. растворе HCl в диоксане (4 мл) и перемешивали. Через час появлялось нерастворимое масло, после чего удаляли растворитель при пониженном давлении (100 мбар, 50ºС) с получением соединения 6 с количественным выходом. ESI-MS: 606,4 [M+H]+, 628,4 [M+Na]+.
ПРИМЕР 6
Бензил-N-<3-{[ε-(D-(+)-биотинил)-L-лизин-15-аза-3,6,9,12-тетраокса-пентадеканоил]-карбонил}-бензолсульфонил>-4-О-{4-бензилоксикарбонил-4-пиперидинил)-бутил}-L-тирозин (8)
Соединение 7 (0,10 г, 0,16 ммоль), которое готовили, как описано в ЕР 1574516, растворяли в DMF (4 мл). Впоследствии добавляли DiPEA (56 мкл, 0,32 ммоль, 2 эквивалента) и TBTU (51 мг, 0,16 ммоль) в атмосфере N2 и раствору давали возможность перемешаться в течение 45 мин. Соединение 6 (0,17 ммоль) растворяли в DMF (1 мл) и к реакционной смеси добавляли DiPEA (28 мкл, 0,16 ммоль, 1 эквивалент). Смеси давали возможность перемешаться в течение 16 ч. Реакционную смесь разбавляли DCM и промывали 1 н. раствором HCl. Органическую фазу промывали Н2О (3×), обезвоживали (MgSO4), фильтровали и концентрировали при пониженном давлении (850 мбар, 50ºС). Очистку проводили с помощью хроматографии с использованием колонки с силикагелем (элюция: DCM/MeOH/AcOH, 99/0/1→79/20/1, объем/объем) с получением соединения 8 (56 мг, 27%). Rf 0,15 (DCM/MeOH, 9/1, объем/объем). ESI-MS: 1316,6 [M+H]+, 1338,8 [M+Na]+, 1182,6 [M-Z+H]+, 1204,6 [M-Z+Na]+, 1314,8 [M-H]+.
ПРИМЕР 7
Общая методика получения соединений 10, 12, 17 и 26
Производное карбоновой кислоты (33 мкмоль) (т.е. соединение 8, 11, 16 или 25) сушили с помощью повторного концентрирования в сухом DMF (2×2 мл), растворяли в DMF (1 мл) и перемешивали в присутствии TBTU (11 мг, 33 мкмоль) и DiPEA (5,7 мкл, 33 мкмоль) в атмосфере N2. Через 1 час добавляли пентасахарид 9 (31 мкмоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре и анализировали с помощью ионообменной (Mono-Q) и обращенно-фазовой (Luna C18) хроматографии. Реакционную смесь концентрировали (<50ºС, 15 мм рт.ст.). С (неочищенного) продукта (10 мг/мл в H2O/t-BuOH, 1/1, объем/объем) снимали защиту с помощью гидрирования (Н2) с использованием 10% Pd/C (добавляли равное по весу количество относительно неочищенного продукта). Через 16 ч раствор дегазировали, фильтровали через фильтр 0,45 мкМ HPLC и концентрировали при пониженном давлении (<50ºС, 15 мм рт.ст.). Конъюгат очищали с помощью ионообменной хроматографии (Q-sepharose, буфер: Н2О→2 M NaCl) с последующими обессоливанием на колонке Sephadex G25 (Н2О) и лиофилизацией.
ПРИМЕР 8
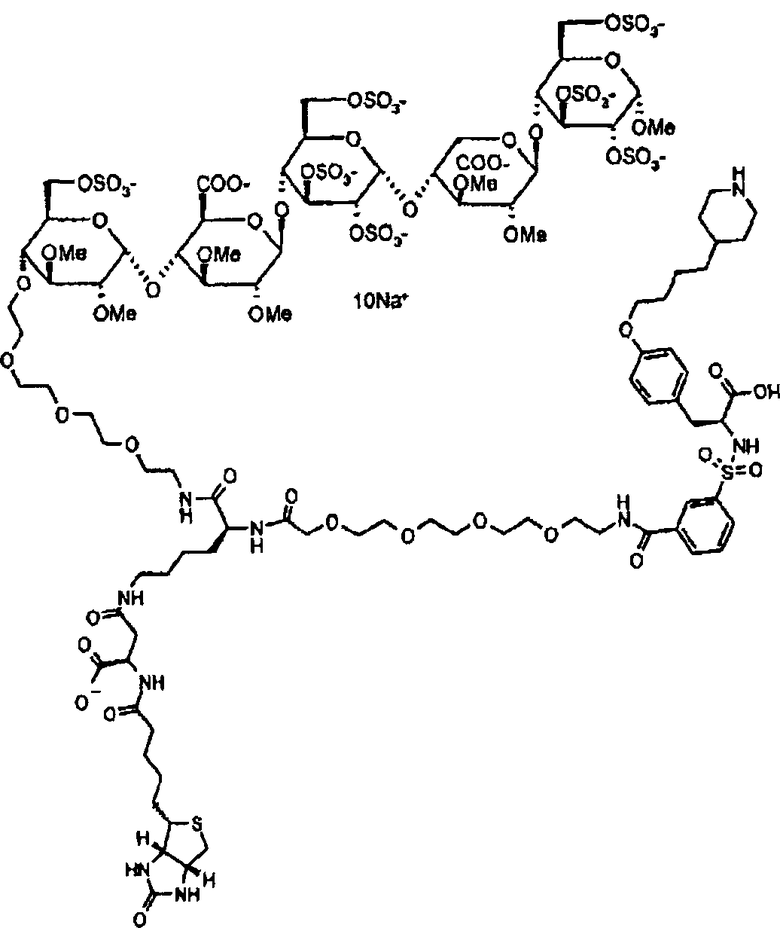
Нонакиснатриевая соль метил-О-2,3-ди-О-метил-4-O<<<12-N-<<N ε -(D-(+)-биотинил)-N-<3-{[15-N-(15-аза-1-кето-3,6,9,12-тетраокса-пентадецил)]-карбонил}-бензолсульфонил>-4-О-{4-(4-пиперидинил)-бутил}-L-тирозил>-лизил>>-12-аза-3,6,9-триокса-додецил>>>6-О-сульфо-альфа-D-глюкопиранозил-(1->4)-О-2,3-ди-О-метил-бета-D-глюкопирануронозил-(1->4)-О-2,3,6-три-О-сульфо-альфа-D-глюкопиранозил-(1->4)-О-2,3-ди-О-метил-альфа-L-идопирануронозил-(1->4)-О-2,3,6-три-О-сульфо-альфа-D-глюкопиранозида (10)
Конъюгацию карбоновой кислоты 8 (37,8 мг, 28,7 мкмоль) с пентасахаридом 9 (51,7 мг, 27,4 мкмоль) [который можно получить связыванием дериватизированного моносахарида 5, описанного в WO 01/42262, с тетрасахаридом 48, описанным в US 2004/0024197, используя методы, сходные с теми, которые описаны в настоящей заявке, в том числе снятие защиты и сульфатирование] с последующими очисткой и снятием защиты проводили в соответствии с общей методикой. Конъюгат 10 получали в виде белого твердого вещества с выходом 13,6 мг (16%, 2 стадии).
1H-ЯМР (D2O, 600 МГц, HH-COSY): δ 7,85 (д, 1Н), 7,78 (д, 1Н), 7,63 (д, 1Н), 7,42 (т, 1Н), 6,85 (д, 1Н), 6,50 (д, 1Н), 5,39 (д, 1Н), 5,33 (д, 1Н), 5,13 (ушир.с, 1Н), 5,08 (д, 1Н), 4,58 (м, 1Н), 4,58 (д, 1Н), 4,49 (м, 1Н), 4,48 (м, 1Н), 4,33 (м, 1Н), 4,29 (м, 1Н), 4,28 (дд, 1Н), 4,22 (дд, 1Н), 4,21 (м, 2H), 4,20 (м, 1Н), 4,17 (м, 2H), 4,09 (м, 1Н), 4,07 (м, 1Н), 4,05 (д, 1Н), 4,02 (м, 1Н), 3,96 (с, 2H), 3,89 (м, 1Н), 3,86 (м, 1Н), 3,85 (м, 2H), 3,79 (м, 2H), 3,73 (м, 1Н), 3,69 (м, 1Н), 3,65 (м, 1Н), 3,61-3,51 (м, 26H), 3,60-3,38 (8 × с, 34H), 3,48 (м, 2H), 3,42 (м, 1Н), 3,38 (м, 1H), 3,32 (м, 1Н), 3,19 (м, 3H), 2,92-2,89 (м, 1Н), 2,67 (д, 1Н), 2,48 (м, 1Н), 2,13 (т, 2H), 1,91 (д, 1Н), 1,70 (м, 2H), 1,65-1,37 (м, 5H).
ESI-MS: как обнаружено: m/z 1381,3 [M+H]2-, 1392,3 [M+Na]2-, 1402,8 [M+2Na]2-, 1413,8 [M+3Na]2-, 920,2 [M-3H]3-, 927,5 [M-3H+Na]3-, 934,5 [M-3H+2Na]3-, 690,2 [M-4H]4-, 694,7 [M-4H+Na]4-.
ПРИМЕР 9
Нонакиснатриевая соль метил-О-2,3-ди-О-метил-4-O<<12-N-<3-{[15-N-(15-аза-1-кето-3,6,9,12-тетраокса-пентадецил)]-карбонил}-бензолсульфонил>-4-О-{4-(4-пиперидинил)-бутил}-L-тирозил>-12-аза-3,6,9-триокса-додецил>>6-О-сульфо-альфа-D-глюкопиранозил-(1->4)-О-2,3-ди-О-метил-бета-D-глюкопирануронозил-(1->4)-О-2,3,6-три-О-сульфо-альфа-D-глюкопиранозил-(1->4)-О-2,3-ди-О-метил-альфа-L-идопирануронозил-(1->4)-О-2,3,6-три-О-сульфо-альфа-D-глюкопиранозида (12)
Соединение 12 получали связыванием соединения 9 (101 мг, 53,9 мкмоль) с соединением 11 (60 мг, 56,6 мкмоль), которое получали, как описано в ЕР 1574516, в соответствии с общей методикой. Выход 72 мг (52%).
1H-ЯМР (D2O, 600 МГц, HH-COSY): δ 7,84 (м, 1Н), 7,75 (м, 1Н), 7,62 (м, 1Н), 7,42 (т, 1Н), 6,83 (д, 2H), 6,48 (д, 2H), 5,38 (д, 1Н), 5,33 (м, 1Н), 5,08 (д, 1H), 5,02 (ушир.с, 1Н), 4,58 (д, 1Н), 4,46 (ушир.с, 2H), 4,28 (м, 2H), 4,17 (м, 1Н), 4,22 (м, 1Н), 4,20 (м, 2H), 4,11 (м, 2H), 4,04 (д, 1Н), 4,03 (м, 1Н), 4,02(м, 1Н), 3,88 (м, 2H), 3,84 (м, 2H), 3,82 (м, 1H), 3,79 (м, 1Н), 3,72 (1Н, м), 3,66 (м, 2H), 3,62-3,33 (м, 54H), 3,20 (dд, 1Н), 3,18 (м, 1Н), 2,92 (м, 2H), 1,92 (м, 2H), 1,70 (м, 2H), 1,58 (м, 1Н), 1,44 (м, 2H), 1,33 (м, 4H).
ESI-MS: m/z 1225,2 [M+5H+2Na]2-, 823,8 [M+3Na+3H]3-, 816,5 [M+2Na+4H]3-, 809,1 [M+Na+5H]3-.
ПРИМЕР 10
Альфа-бензиловый эфир [D-(+)-биотинил]-L-аспарагиновой кислоты (14)
К суспензии D-биотина (2,0 г, 8,19 ммоль) в DMF (52 мл) добавляли пентафторфенол (1,6 г, 15,6 ммоль), а затем DCC (2,5 г, 12,3 ммоль). Реакционной смеси давали возможность перемешиваться в атмосфере азота в течение ночи при комнатной температуре. Реакционная смесь оставалась в прежнем состоянии в виде суспензии, и ее отфильтровывали и концентрировали. Остаток переносили в Et2O и перемешивали в течение нескольких минут, после чего суспензию фильтровали и сушили под вакуумом с получением белого твердого вещества (2,58 г). ESI-MS: 411 [M+H]+. Твердое вещество растворяли в DMF (90 мл) и добавляли Et3N (1,24 мл, 8,82 ммоль, 1,4 эквивалента). Порциями добавляли H-Asp-OBn в виде твердого вещества. После приблизительно 15 мин реакционная смесь становилась прозрачной, и ей предоставляли возможность перемешиваться в течение дополнительных 2 ч. Реакционную смесь концентрировали при пониженном давлении и добавляли воду, а затем MeOH (3:1). Образовавшееся твердое вещество отфильтровывали, промывали Et2O и сушили под вакуумом с получением соединения 14 в виде белого твердого вещества (2,92 г, 77%). ESI-MS: 450 [M+H]+.
ПРИМЕР 11
N-15-аза-3,6,9,12-тетраокса-пентадеканоил-ε<N-[D-(+)-биотинил]-L-аспартил-альфа-бензиловый эфир>-L-лизин (15)
Соединение 13 (3,60 г, 6,72 ммоль, 1,07 эквивалента), которое получали, как описано выше для синтеза соединения 6, и соединение 14 (2,92 г, 6,30 ммоль) растворяли в DMF (80 мл). Добавляли DiPEA (2,2 мл, 12,6 ммоль, 2 эквивалента) в атмосфере азота, а затем TBTU (2,53 г, 7,88 ммоль, 1,25 эквивалента). Реакционной смеси давали возможность перемешаться в течение ночи, после чего растворитель удалили при пониженном давлении. Очистку проводили с использованием HPLC (ACN/H2O) с получением желтоватого масла (2,31 г). ESI-MS: 967 [M+H]+. Снятие защиты проводили в DCM/TFA (1:1, 60 мл) при перемешивании в течение 2 ч при комнатной температуре. Добавляли DCM (150 мл) и растворители удаляли с помощью нагревания (45ºС, атмосферное давление). Этот процесс повторяли 3× с последующей повторной концентрацией в толуоле (3×). Соединение 15 собирали в виде желтоватого твердого вещества (3,29 г, 30%). ESI-MS: 811 [M+H]+.
ПРИМЕР 12
ε-N-(D-(+)-биотинил-бета-L-аспартил)-N-<3-{[15-N-(15-аза-1-кето-3,6,9,12-тетраокса-пентадецил)]-карбонил}-бензолсульфонил>-4-О-{4-(4-пиперидинил)-бутил}-L-тирозил>-L-лизин (16)
К раствору соединения 7 (1,41 г, 1,94 ммоль) в DMF (50 мл) добавляли DiPEA (1,0 мл, 5,8 ммоль, 3 эквивалента), в атмосфере азота, а затем TBTU (685 мг, 2,1 ммоль, 1,1 эквивалента) и смеси давали возможность перемешаться в течение 1 ч. Затем добавляли раствор соединения 15 в DMF (20 мл) и перемешивали в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении и неочищенный продукт очищали с использованием HPLC (ACN/H2O/TFA) с получением желтоватого твердого вещества (783 мг, 26%). ESI-MS: 1522 [M+H]+.
ПРИМЕР 13
Декакиснатриевая соль метил-2,3-ди-О-метил-4-O<<<12-N-<<ε-N-(D-(+)-биотинил-бета-L-аспартил)-N-<3-{[15-N-(15-аза-1-кето-3,6,9,12-тетраокса-пентадецил)]-карбонил}-бензолсульфонил>-4-О-{4-(4-пиперидинил)-бутил}-L-тирозил>-L-лизил>>-12-аза-3,6,9-триокса-додецил>>>6-О-сульфо-альфа-D-глюкопиранозил-(1->4)-О-2,3-ди-О-метил-бета-D-глюкопирануронозил-(1->4)-О-2,3,6-три-О-сульфо-альфа-D-глюкопиранозил-(1->4)-О-2,3-ди-О-метил-альфа-L-идопирануронозил-(1->4)-2,3,6-три-О-сульфо-альфа-D-глюкопиранозида (17)
Синтез соединения 17 в своей основе проводили в соответствии с общей методикой. Таким образом, к раствору соединения 16 (768 мг, 0,505 ммоль) в DMF (50 мл) добавляли DiPEA (106 мкл, 0,61 ммоль, 1,2 эквивалента), а затем TBTU (178 мг, 0,56 ммоль, 1,1 эквивалента) в атмосфере азота. После перемешивания раствора в течение 1 ч добавляли соединение 9 (906 мг, 0,48 ммоль) и перемешивание продолжали в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и очищали на Q-sepharose (H2O→2 M NaCl). Обессоливание проводили на Sephadex-G25 с получением прозрачного масла (1,5 г). Масло растворяли в H2O (37 мл) и t-BuOH (37 мл) и добавляли 10% Pd/C (670 мг) в атмосфере азота. Через раствор пропускали водород в течение 16 ч. После удаления катализатора Pd/C с помощью фильтрации соединение снова очищали на Q-sepharose и обессоливали на Sephadex-G25 с получением соединения 17 с выходом 465 мг (34%).
1H-ЯМР (D2O, 600 МГц, HH-COSY): δ 7,94 (м, 1Н), 7,86 (т, 1Н), 7,71 (м, 1Н), 7,52 (т, 1Н), 6,93 (д, 1Н), 6,58 (д, 1Н), 5,45 (д, 1Н), 5,42 (д, 1Н), 5,18 (ушир.с, 1Н), 5,15 (д, 1Н), 4,56 (м, 1H), 4,33-4,41 (м, 2H), 4,30-4,21 (м, 4H), 4,13-4,05 (м, 4H), 3,89-3,96 (м, 5H), 3,87-3,82 (м, 3H), 3,81-3,70 (м, 7H), 3,69-3,60 (м, 39H), 3,59-3,48 (м, 13H), 3,47-3,35 (м, 8H), 3,30-3,24 (м, 3H), 3,11 (т, 2H), 3,06-2,93 (м, 4H), 2,79-2,73 (м, 2H), 2,60-2,52 (м, 2H), 2,26 (т, 2H), 2,01-1,96 (м, 2H), 1,81-1,74 (м, 3H), 1,71-1,25 (м, 20H).
ESI-MS: m/z 1481,9 [M+4Na-4H]2-, 1471,0 [M+3Na-3H]2-, 980,7 [M+3Na-3H]3-, [M+Na+5H]3-.
ПРИМЕР 14
ε-N-Метил-ε-N-трифторацетил-L-лизин (19)
Соединение 18 (1,26 г, 3,2 ммоль) растворяли в DMF (15 мл) и добавляли K2CO3 (2,2 г, 16 ммоль, 5 эквивалентов), а затем метилиодид (1,6 мл, 25,6 ммоль, 8 эквивалентов). Раствор нагревали до 100ºС и перемешивали в течение 24 ч в герметичном сосуде. Реакционную смесь охлаждали и разбавляли EtOAc. Получаемое в результате твердое вещество отфильтровывали, промывали соляным раствором и сушили на MgSO4. Неочищенный продукт очищали с помощью хроматографии с использованием колонки с силикагелем (DCM/MeOH, 95/5) с получением масла (1,18 г), которое растворяли в смеси DCM (5 мл) и TFA (5 мл) и перемешивали в течение 1 ч. Затем растворители удаляли при пониженном давлении и неочищенный продукт концентрировали в толуоле (3х) с получением соединения 19 (1,57 г, 87%). ESI-MS: m/z 271,2 [M+H]+.
ПРИМЕР 15
Метил-15-азидо-3,6,9,12-тетраокса-пентадеканоил-ε-N-метил-ε-N-трифторацетил-L-лизин (21)
Соединения 19 (1,57 г, 2,16 ммоль) и 20 (0,60 г, 2,16 ммоль), которое готовили снятием защиты с соответствующего производного трет-бутилового эфира, описанного в ЕР 1574516, связывали, как описано для синтеза соединения 4, с получением соединения 21 с выходом 86% (980 мг, 1,85 ммоль). ESI-MS: m/z 530,2 [M+H]+, 552,2 [M+Na]+.
ПРИМЕР 16
15-Азидо-3,6,9,12-тетраокса-пентадеканоил-ε-N-метил-L-лизин (22)
Соединение 21 (0,98 г, 1,85 ммоль) растворяли в TNF (6 мл), добавляли 1 н. LiOH (6 мл) и получаемый в результате раствор перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь нейтрализовали добавлением 1 н. HCl и впоследствии концентрировали при пониженном давлении с получением неочищенного соединения 22 со снятой защитой, которое использовали без дальнейшей очистки в следующей реакции. ESI-MS: m/z 420,2 [M+H]+.
ПРИМЕР 17
15-Азидо-3,6,9,12-тетраокса-пентадеканоил-ε-N-[D-(+)-биотинил]-ε-N-метил-L-лизин (23)
Соединения 22 (1,85 ммоль, неочищенное) связывали с D-(+)-биотином (0,54 г, 2,22 ммоль, 1,2 эквивалента), как описано выше для получения соединения 6, с получением биотинилированного производного лизина 23, которое использовали без очистки в следующей реакции. ESI-MS: m/z 646,4 [M+H]+, 668,6 [M+Na]+.
ПРИМЕР 18
15-Аза-3,6,9,12-тетраокса-пентадеканоил-ε-N-[D-(+)-биотинил]-ε-N-метил-L-лизин (24)
Соединение 23 (1,85 ммоль, неочищенное) растворяли в t-BuOH (50 мл) и H2O (50 мл). Добавляли 10% Pd/C (750 мг) в атмосфере азота. Через раствор пропускали водород в течение приблизительно 4 ч. Pd/C удаляли с помощью фильтрации через декалит и раствор промывали EtOH. Растворители удаляли при пониженном давлении (50 мбар, 50ºС) с получением неочищенного соединения 24 в виде масла (>100%, оставшийся растворитель). ESI-MS: m/z 620,4 [M+H]+.
ПРИМЕР 19
Бензил-N-<3-{[-ε-(D-(+)-биотинил-ε-N-метил)-L-лизин-15-аза-3,6,9,12-тетраокса-пентадеканоил]-карбонил}-бензолсульфонил>-4-О-{4-(N-бензилоксикарбонил-4-пиперидинил)-бутил}-L-тирозин (25)
Связывание соединения 24 (0,58 г, 0,93 ммоль, неочищенного) с соединением 7 (0,50 г, 0,68 ммоль) проводили, как описано выше для синтеза соединения 8. Очистка с помощью хроматографии с использованием колонки с силикагелем и препаративной HPLC привела к получению ε-N-метилированного производного лизина 25 с выходом 77 мг (8,5% на протяжении последних четырех стадий, начиная с соединения 21). ESI-MS: m/z 1330,6 [M+H]+.
ПРИМЕР 20
Нонакиснатриевая соль метил-О-2,3-ди-О-метил-4-O<<<12-N-<<N ε -(D-(+)-биотинил-ε-N-метил)-N-<3-{[15-N-(15-аза-1-кето-3,6,9,12-тетраокса-пентадецил)]-карбонил}-бензолсульфонил>-4-О-{4-(4-пиперидинил)-бутил}-L-тирозил>-лизил>>-12-аза-3,6,9-триокса-додецил>>>6-О-сульфо-альфа-D-глюкопиранозил-(1->4)-О-2,3-ди-О-метил-бета-D-глюкопирануронозил-(1->4)-О-2,3,6-три-О-сульфо-альфа-D-глюкопиранозил-(1->4)-О-2,3-ди-О-метил-альфа-L-идопирануронозил-(1->4)-2,3,6-три-О-сульфо-альфа-D-глюкопиранозида (26)
Соединение 25 (77 мг, 58 мкмоль) связывали с пентасахаридным производным 9 (0,10 г, 55 мкмоль), как описано в общей методике. За очисткой и обессоливанием неочищенного продукта, как описано в общей методике, следовала лиофилизация с получением целевого конъюгата 26 с выходом 50 мг (27%). ESI-MS: m/z 2762,5 [M+H]+.
Схема 1а

Схема 1b
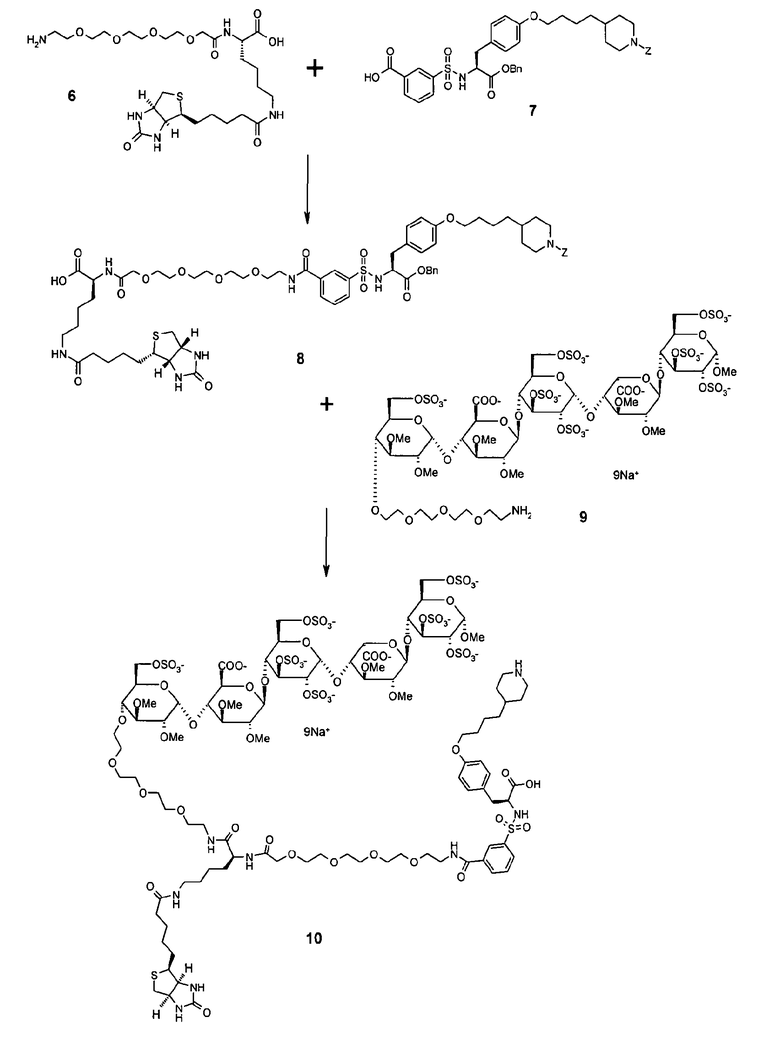
Схема 2

Схема 3а
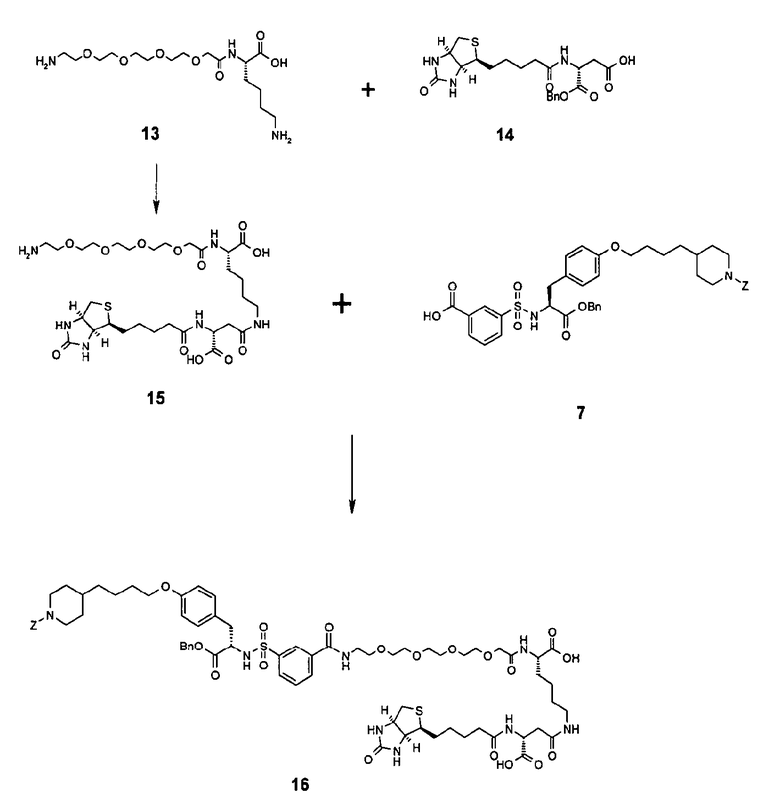
Схема 3b

Схема 4a
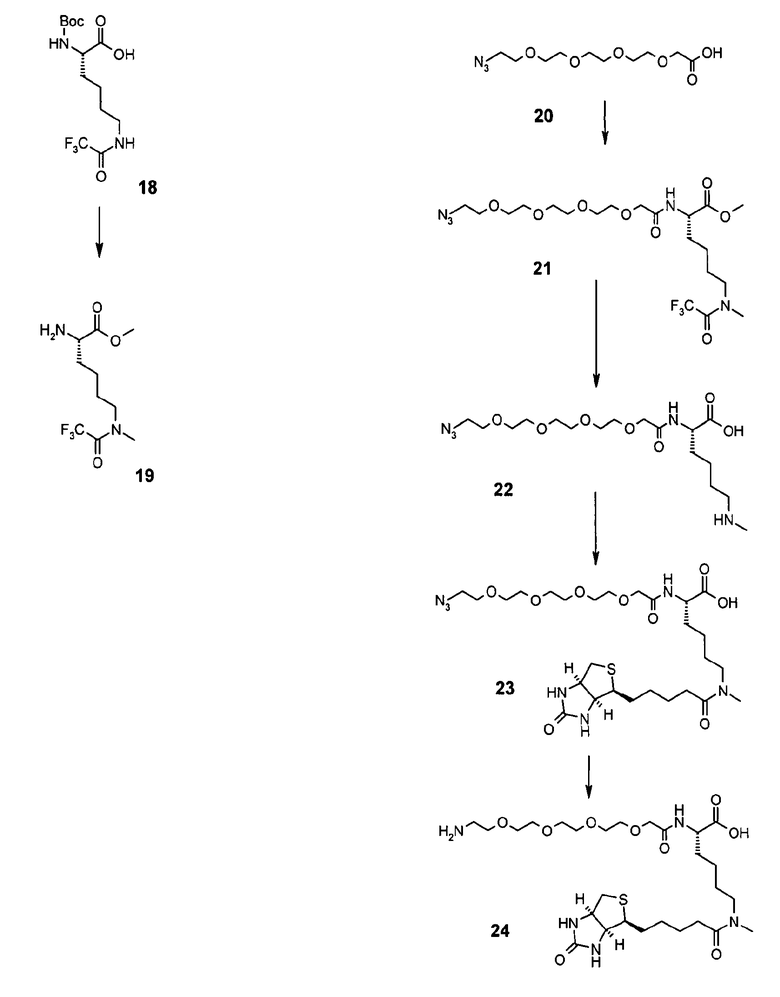
Схема 4b

Эталонные соединения

ПРИМЕР 21
Фармакология
1.1. In vitro тест на ингибирование агрегации тромбоцитов морских свинок, индуцированной ADP
Добавление аденозиндифосфата (ADP) в богатую в отношении тромбоцитов плазму (PRP) человека или морской свинки in vitro индуцирует агрегацию тромбоцитов. Эту агрегацию можно оценить путем измерения изменения оптической плотности (OD) PRP. Использовали следующий in vitro тест для оценки проверяемого соединения в отношении ингибирования ADP-индуцированной агрегации тромбоцитов морских свинок.
Материалы
Богатая в отношении тромбоцитов плазма (PRP): Свободно текущую кровь берут у здорового добровольца или морской свинки и помещают в 0,1 объема цитрата натрия. 2Н2О, 3,8% в дистиллированной Н2О (вес/объем). Конечная концентрация цитрата натрия составляет 0,38%. Кровь с цитратом натрия центрифугируют при 1600 Н/кг (160g, т.е. 900 оборотов/мин) в Hettich Rotanta/AP при комнатной температуре. Через 15 минут центрифугирование прекращают с выключенным тормозом и отбирают супернатант (PRP). Свежий раствор ADP (аналитической степени чистоты), 50 мкМ в 0,9% NaCl в MQ-воде, используют немедленно.
В этом исследовании тирофибан (AGGRASTAT® (MSD)), приобретенный в виде 0,25 мг/мл концентрата для внутривенной инфузии, ингибирует агрегацию тромбоцитов человека, индуцированную 5 мкМ ADP, на 50% при концентрации 30-60 нМ (IC50).
Оборудование
1. Sysmex-счетчик форменных элементов крови модель КХ-21.
2. Считывающее устройство MF Labsystems iEMS с фильтром 620 нм, орбитальной установкой для покачивания при 1000 оборотов/мин и постоянной температурой 37ºС. Поглощение измеряют с использованием программы Labsystems iEMS.
3. Система для сбора крови 600 мл с иголкой, артикль Р4203 (NPBI).
4. 96-луночные микропланшеты с плоским дном (Greiner Labortechnik).
Методика проведения
Тромбоциты в супернатанте (PRP) подсчитывают, используя Sysmex-счетчик форменных элементов крови, и супернатант разбавляют РРР (бедной в отношении тромбоцитов плазмой) с получением PRP, содержащей приблизительно 400000±50000 тромбоцитов/мл. Тромбоциты следует стабилизировать при комнатной температуре в течение по крайней мере 20 мин, но не дольше 3 ч.
150 мкл PRP переносят пипеткой в лунку микропланшета. Добавляют 30 мкл проверяемого соединения в диапазоне концентраций (7 концентраций на соединение) или носителя и микропланшет помещают в считывающее устройство MF Labsystems iEMS при 37ºС. Оптическую плотность (OD620) измеряют при 620 нм. После покачивания в течение 2 минут в считывающем устройстве (1000 оборотов/мин) OD измеряют во второй раз. Это делают для подтверждения стабильности тромбоцитов (отсутствия спонтанной агрегации тромбоцитов). Затем добавляют 20 мкл 50 мкМ раствора ADP и OD кинетически измеряют каждую минуту в течение 14 минут при 620 нм. Между двумя измерениями планшет покачивают в течение 40 секунд при 1000 оборотов/мин. Каждое проверяемое соединение исследуют в по крайней мере 2 экспериментах, используя PRP от различных добровольцев.
Оценка ответа
Среднеарифметическую OD при каждой концентрации соединения (в том числе носителя) рассчитывают для моментов времени (t), равных 0 и 10 мин. Процент ингибирования при каждой концентрации рассчитывают, используя формулу:

IC50 проверяемого соединения - это концентрация, при которой ADP-индуцированная агрегация тромбоцитов уменьшается на 50%. Для этого строят график зависимости величин процента ингибирования от концентрации соединения и IC50 рассчитывают, используя Graphpad Prism 3.0 (с различным спадом).
1.2. In vitro тест на ингибирование агрегации тромбоцитов человека, индуцированной TRAP
Добавление пептида-агониста рецептора тромбина (TRAP) к отмытым тромбоцитам (WPL) человека или морской свинки in vitro индуцирует агрегацию тромбоцитов. Эту агрегацию можно определить путем измерения оптической плотности WPL. Используют описанный здесь in vitro тест для анализа активности проверяемого соединения в отношении ингибирования TRAP-индуцированной агрегации тромбоцитов человека. Считывающее устройство для микропланшетов используют для измерения активности нескольких соединений одновременно.
Материалы
∗Состав буфера Watson:
рН доводят до 7,4 с помощью NaOH (1 моль/л).
∗Состав TNP-буфера:
рН раствора доводят при 37ºС до 7,4 с помощью HCl (10 моль/л).
∗Раствор PGI2:
Матричный раствор простагландина I2 1 мг/мл в КОН (1 моль/л) хранят при -20ºС. Непосредственно перед использованием готовят раствор 5 мкг/мл в охлажденном на льду NaCl (9 г/л).
∗Богатая в отношении тромбоцитов плазма (PRP):
Свободно текущую кровь берут у здорового добровольца или морской свинки и помещают в 0,1 объема 3,8% цитрата натрия. 2Н2О в MQ-воде (вес/объем). Конечная концентрация цитрата натрия составляет 0,38%. Кровь с цитратом натрия центрифугируют при 1600 Н/кг (160g) в центрифуге Hettich Rotanta/AP при комнатной температуре. Через 15 минут центрифугирование прекращают с выключенным тормозом. Супернатант (=PRP) отбирают и разбавляют бедной в отношении тромбоцитов плазмой с получением суспензии, содержащей приблизительно 400000 тромбоцитов/мл.
∗Бедная в отношении тромбоцитов плазма (PPP):
Кровь с цитратом натрия центрифугируют при приблизительно 20000 Н/кг в течение 10 минут при комнатной температуре и РРР сифонируют.
∗Отмытые тромбоциты (WPI):
Аликвоту 1 мкл раствора PGI2 добавляют в 1 мл PRP и после этого центрифугируют при 20000 Н/кг в течение 10 минут при комнатной температуре. Плазму сифонируют и к тромбоцитам в осадке добавляют буфер Watson, содержащий 5 нг/мл PGI2, и тромбоциты ресуспендируют в первоначальном объеме с помощью осторожного перемешивания пластиковой палочкой-мешалкой. Суспензию тромбоцитов снова центрифугируют при 20000 Н/кг. Тромбоциты ресуспендируют в буфере Watson для того, чтобы получить суспензию, содержащую приблизительно 400000 тромбоцитов/мл.
∗Раствор TRAP:
TRAP растворяют в Н2О с получением раствора, содержащего 50 мкмоль/л. Свежий раствор должен готовиться ежедневно. Для всех водных растворов используют ультрачистую Н2О (Milli-Q-качества).
∗Фибриноген человека (Kordia/ERL, артикль: FIB 2 порошок): 0,5 г фибриногена в виде порошка растворяют в 50 мл MQ-воды под вакуумом. Этот матричный раствор хранят в аликвотах 100 мкл при -20ºС. Непосредственно перед использованием готовят раствор 0,5 мг/мл в солевом растворе.
В этом исследовании тирофибан (AGGRASTAT® (MSD)), приобретенный в виде 0,25 мг/мл концентрата для внутривенной инфузии, ингибирует агрегацию тромбоцитов человека, индуцированную 5 мкМ TRAP, на 50% при конечной концентрации 30-60 нМ (IC50).
Методика проведения
Концентрацию WPL определяют в Sysmex-счетчике форменных элементов крови и суспензию разбавляют буфером Watson для получения концентрации приблизительно 400000 тромбоцитов/мл. Перед использованием WPL дают возможность стабилизировать при комнатной температуре в течение по крайней мере 20 мин, но не дольше 3-4 часов.
150 мкл WPL переносят пипеткой в лунку микропланшета. Добавляют 15 мкл раствора проверяемого соединения или носителя и 15 мкл раствора фибриногена и микропланшет помещают в считывающее устройство для микропланшета при 37ºС. Затем измеряют оптическую плотность (OD) при 405 нм и после покачивания в течение 2 минут в считывающем устройстве OD405 измеряют снова для подтверждения стабильности тромбоцитов (отсутствия спонтанной агрегации тромбоцитов). Добавляют 20 мкл 50 мкМ раствора TRAP и OD405 кинетически измеряют каждую минуту в течение 14 минут при 405 нм. Между двумя измерениями планшет покачивают в течение 40 секунд при 1000 оборотов/мин. Для определения IC50 проверяемого соединения каждое соединение исследуют в по крайней мере 2 экспериментах, используя WPL от различных добровольцев.
Оценка ответа
Среднеарифметическую OD при каждой концентрации соединения (в том числе носителя) рассчитывают для моментов времени (t), равных 0 и 10 мин. Процент ингибирования при каждой концентрации рассчитывают, используя Microsoft Excel, по формуле:
Строят график зависимости процента ингибирования от концентраций соединения. IC50 рассчитывают, используя Graphpad Prism 3.0 (с различным спадом). IC50 проверяемого соединения - это концентрация, при которой TRAP-индуцированная агрегация тромбоцитов уменьшается на 50%.
1.3. In vitro тест для определения анти-фактор Ха активности в плазме человека
Анти-фактор Ха активность проверяемых соединений в плазме человека измеряли амидолитически с использованием S2222 (Chromogenix, Chromogenics Ltd, Molndal, Швеция), используя способ, описанный Teien и Lie (Teien AN, Lie M. Evaluation of an amidolytic heparin assay method increased sensitivity by adding purified antithrombin III. Thromb. Res. 1977, 10: 399-410). Антифактор Ха активность выражают в Е/мкмоль после сравнения амидолитической активности с калибровочной кривой стандартного гепарина.
Краткое изложение in vitro антитромботических активностей
2.1. In vitro нейтрализация антитромботической активности
А. In vitro нейтрализация ингибирования агрегации тромбоцитов
(предварительная инкубация соединения с авидином)
Как проведено в описанном выше протоколе для ADP-индуцированной агрегации тромбоцитов. Агрегацию проводили в присутствии 225 нМ соединения 12 или 450 нМ соединения 10 в концентрациях, при которых достигается максимальное ингибирование агрегации тромбоцитов морских свинок. Перед индуцированием агрегации тромбоцитов морских свинок с помощью добавления ADP соединение и авидин (из белого яйца, Sigma) в различных концентрациях предварительно инкубировали в течение 2 минут при комнатной температуре (фиг.1 и 2).
А. In vitro нейтрализация ингибирования агрегации тромбоцитов
(после задержанного добавления авидина)
Как проведено в описанном выше протоколе для ADP-индуцированной агрегации тромбоцитов. Агрегацию проводили в присутствии 225 нМ соединения 12 или 450 нМ соединения 10 в концентрациях, при которых достигается максимальное ингибирование агрегации тромбоцитов морских свинок или человека. Через 7 минут определение агрегации тромбоцитов прерывали и добавляли авидин (из белого яйца, Sigma) в различных концентрациях в момент времени t, равный 9 мин. В пределах 1 минуты возобновляли определение агрегации тромбоцитов (фиг.3 и 4).
На фиг.5 демонстрируется эффект авидина на ингибирование с помощью соединения 17 агрегации тромбоцитов человека (добавление авидина через 7 минут после для ADP-индуцированной агрегации тромбоцитов).
Вывод: введение авидина в пробу агрегации тромбоцитов морских свинок, содержащую соединение 10, приводит к немедленному восстановлению агрегации тромбоцитов (=нейтрализации антитромботической, анти-GpIIb/IIIa активности), в то время как ингибиторная активность не биотинилированного эквивалентного антитромботического соединения 12 не могла быть восставлена. Соединение авидина с соединением 17 приводит к полному восстановлению агрегации тромбоцитов человека.
3.1 Фармакокинетики
Фармакокинетические свойства соединений 10, 12, 17 и 27 исследовали на самцах крыс Wistar весом 300-400 г. Крыс подвергали анестезии ингаляцией смеси О2/N2/изофлуран, после чего правую яремную вену канюлировали. На следующий день крысам вводили подкожно дозы 100 или 500 нмоль/кг. После подкожного введения брали образцы крови с различными интервалами времени. Затем кровь центрифугировали, после чего плазму сифонировали и хранили при -20ºС до использования. Концентрацию проверяемого соединения измеряли амидолитически с использованием S2222 (Chromogenix, Chromogenics Ltd, Molndal, Швеция) путем определения анти-Ха активности на основе способа Teien и Lie в полученных образцах плазмы в сравнении с калибровочной кривой, построенной с использованием матричного раствора самого проверяемого соединения (Teien AN, Lie M. Evaluation of an amidolytic heparin assay method increased sensitivity by adding purified antithrombin III. Thromb. Res. 1977, 10: 399-410). Концентрацию в образцах выражали в нмоль/мл и кинетические параметры рассчитывали с использованием модели без дробления WinNonlin (фиг.6 и 7).
Фармакокинетические параметры после подкожного введения крысе соединения 10 или 12 (500 нмоль/кг). Эксперимент проводили с n=3/обработку
Фармакокинетические параметры после подкожного введения крысе соединения 12, 17 или 27 (100 нмоль/кг). Эксперимент проводили с n=3/обработку
(двойной эталонный ингибитор)
(эталонный пентасахарид)
Делается вывод, что в пределах вариабельности эксперимента соединения 10, 12, 17 и 27 демонстрируют одинаковое фармакокинетические поведение в крысах.
3.2 Фармакокинетики - эксперимент по нейтрализации
Крысам подкожно вводили соединение 10, 12 или 27 в дозе 100 нмоль/кг. Через 1 ч брали образец крови и внутривенно вводили 10 мг/кг авидина (из белого яйца, Sigma) крысам, обработанным соединением 10 или 12. Впоследствии брали образцы крови через 0,5-1-3-6 и 23 часов. Кровь обрабатывали, как описано в фармакокинетическом эксперименте, и концентрацию образцов определяли путем измерения (остаточной) анти-Ха активности (фиг.8).
Фармакокинетические параметры после подкожного введения 100 нмоль/кг соединения 10 или 12 и авидина (10 мг/кг) в момент времени t, равный 1 ч. Эксперимент проводили с n=3/обработку
Делается вывод, что после подкожного введения соединения 10 (100 нмоль/кг) антитромботическую активность, определяемую путем измерения (оставшейся) анти-Ха активности, можно нейтрализовать с помощью введения внутривенно 10 мг/кг авидина. Отражением нейтрализации соединения 10 с помощью авидина является сильно уменьшенное общее Т1/2 выведения, сильно уменьшенная общая площадь под кривой концентрации в плазме, вычерченной во времени, экстраполированном в бесконечное время, и сильно увеличенный клиренс по сравнению с соединением 12. Кроме того, на фармакокинетическое поведение не биотинилированного эквивалентного соединения 12 не влияет добавление авидина (также при сравнении с эталонным пентасахаридом 27, который демонстрирует сходный профиль). Последнее из двух вышеуказанных наблюдений подтверждает, что нейтрализация связана с присутствием биотиновой метки и что присутствие такой метки не влияет на фармакокинетическое поведение двойного ингибитора.
В отдельном эксперименте соединение 10, 17 или 26 вводили в дозе 500 нмоль/кг подкожно, после чего брали кровь спустя 24 часа. Затем внутривенно вводили 10 мг/кг авидина и кровь брали через 25 и 26 часов (фиг.9).
Уровни в плазме после подкожного введения 500 нмоль/кг соединения 10, 17 и 26 в момент времени t, равный 24 часам, и через 2 часа после введения авидина (10 мг/кг) в момент времени t, равный 24 ч. Эксперимент проводили с n=3/обработку, величины приведены в виде среднеарифметического ± SEM
Делается вывод, что в пределах 2 часов после введения авидина (10 мг/кг) концентрация в плазме соединений 10, 17 и 26 уменьшается на 75, 83 и 66% соответственно. Эксперимент проводили спустя 24 часа после подкожного введения биотинилированных соединений, что показывает, что линкер между биотиновой составляющей и антитромботическим компонентом является стабильным in vivo.
4. In vivo нейтрализация антитромботической активности
Внутрисосудистая инфузия суспензии коллагена индуцирует агрегацию тромбоцитов и вызывает транзиторную тромбоцитопению у крыс. Этот тест используют для оценки влияния проверяемого соединения на тяжесть тромбоцитопении, индуцированной коллагеном, у крыс.
В первом эксперименте самцам морских свинок подкожно вводили соединение 10 в дозе 75 нмоль/кг или носитель за 4 часа до инфузии коллагена. Во втором эксперименте соединение 12 и 17 подкожно вводили в дозе 100 нмоль/кг в один и тот же момент времени. Самцов морских свинок подвергали анестезии с помощью внутримышечного введения кетамина + седамуна (90+10 мг/кг соответственно). Через 15 минут одну из общих сонных артерий вскрывали и осуществляли канюлирование с помощью канюли PE 50 (Clay Adams). Два образца крови по 0,5 мл собирали в пластиковые флаконы, содержащие 25 мкл 0,2 М раствора Na2EDTA. Канюлю затем соединяли со шприцом, содержащим 0,25 мг/мл суспензии коллагена (Hormon Chemie, Munich, Западная Германия, разведенной изотоническим буфером, рН 2,8). Эту суспензию вводили через инфузию 225 мкл в течение 30 секунд. Затем шприц отсоединяли и брали 2 образца крови 0,5 мл через 85 и 95 секунд после начала инфузии коллагена. Затем животных умерщвляли с использованием эвтазата, после чего определяли число тромбоцитов на образец с использованием Sysmex-счетчика форменных элементов крови модели КХ-21. В том случае, когда использовали авидин, внутривенно вводили 10 мг/кг авидина в момент времени, равный 4 ч после подкожного введения соединения 10, 12 или 17, после чего осуществляли инфузию коллагена в пределах 5 минут.
После подсчета числа тромбоцитов в отобранных образцах крови уменьшение числа тромбоцитов рассчитывают путем деления среднеарифметического числа тромбоцитов в образцах крови, отобранных в t=85 и 95 секунд, на среднеарифметическую величину в образцах крови, отобранных в t=0. Тест проводили с n=4.
Число тромбоцитов после введения соединения 10 (75 ммоль/кг) с или без авидина в in vivo модели (морской свинки), основанной на индуцированной коллагеном агрегации тромбоцитов
Число тромбоцитов после введения соединения 12 или 17 (100 ммоль/кг, подкожно) с или без авидина в in vivo модели (морской свинки), основанной на индуцированной коллагеном агрегации тромбоцитов
После введения дозы 75 нмоль/кг подкожно соединения 10 или 100 нмоль/кг подкожно соединения 12 или 17 соединения ингибировали индуцированную коллагеном агрегацию тромбоцитов более чем на 66% через 4 часа после введения. Введение 10 мг/кг авидина как раз перед инфузией коллагена вызывало немедленную и количественную нейтрализацию ингибиторной в отношении агрегации тромбоцитов активности соединений 10 и 17, но не соединения 12, соединения, у которого отсутствует биотиновая составляющая. Эти результаты показывают, что нейтрализация анти-GpIIb/IIIa-опосредуемых антитромбоцитарных активностей соединений 10 и 17 опосредуется только биотиновой меткой и основана на специфическом связывании с авидином. Кроме того, сравнение относительного уменьшения числа тромбоцитов под действием соединений 10, 12 и 17 показывает, что биотиновая метка не мешала существенной антитромботической активности двойных ингибиторов.
Дополнительная фармакология
Зависимое от дозы кровотечение, индуцированное у морских свинок после введения соединения 17, было немедленно остановлено введением авидина (внутривенно 10 мг/кг).
| название | год | авторы | номер документа |
|---|---|---|---|
| ДВОЙНЫЕ АНТИТРОМБОТИЧЕСКИЕ ИНГИБИТОРЫ, СОДЕРЖАЩИЕ ОСТАТОК БИОТИНА | 2005 |
|
RU2403259C2 |
| КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ИЛИ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ | 1997 |
|
RU2194038C2 |
| ТРИЦИКЛИЧЕСКИЕ АНТАГОНИСТЫ ТРОМБИНОВОГО РЕЦЕПТОРА | 2003 |
|
RU2329264C9 |
| ПРОИЗВОДНЫЕ НИПЕКОТИНОВОЙ КИСЛОТЫ КАК ВЕЩЕСТВА, ПРЕПЯТСТВУЮЩИЕ ТРОМБООБРАЗОВАНИЮ | 1995 |
|
RU2135470C1 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ПРОИЗВОДНЫЕ ТИЕНОИНДОЛА ДЛЯ ЛЕЧЕНИЯ РАКОВОГО ЗАБОЛЕВАНИЯ | 2013 |
|
RU2632199C2 |
| Аминоалкилдезоксипроизводное целлюлозы, способ его получения и средство, обладающее антитромбоцитарной активностью | 2018 |
|
RU2694342C1 |
| ИЗОКСАЗОЛИНЫ И ИЗОКСАЗОЛЫ, СПОСОБ ПОДАВЛЕНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОДАВЛЯЮЩАЯ АГРЕГАЦИЮ ТРОМБОЦИТОВ | 1994 |
|
RU2149871C1 |
| НИТРООКСИПРОИЗВОДНЫЕ ФЛУВАСТАТИНА, ПРАВАСТАТИНА, ЦЕРИВАСТАТИНА, АТОРВАСТАТИНА И РОЗУВАСТАТИНА В КАЧЕСТВЕ ХОЛЕСТЕРИНСНИЖАЮЩИХ АГЕНТОВ С УЛУЧШЕННОЙ ПРОТИВОВОСПАЛИТЕЛЬНОЙ, АНТИТРОМБОТИЧЕСКОЙ И АНТИТРОМБОЦИТАРНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2362770C2 |
| КОНЪЮГАТЫ ПОЛИПЕПТИДА И ОЛИГОСАХАРИДА | 2006 |
|
RU2443713C2 |
| НОВЫЕ КОНЪЮГАТЫ АНАЛОГОВ СС-1065 И БИФУНКЦИОНАЛЬНЫЕ ЛИНКЕРЫ | 2011 |
|
RU2578719C9 |





Описывается новое соединение общей формулы (I):
олигосахарид-спейсер-антагонист GpIIb/IIIa (1),
где олигосахарид представляет собой отрицательно заряженный пентасахаридный остаток формулы (В)
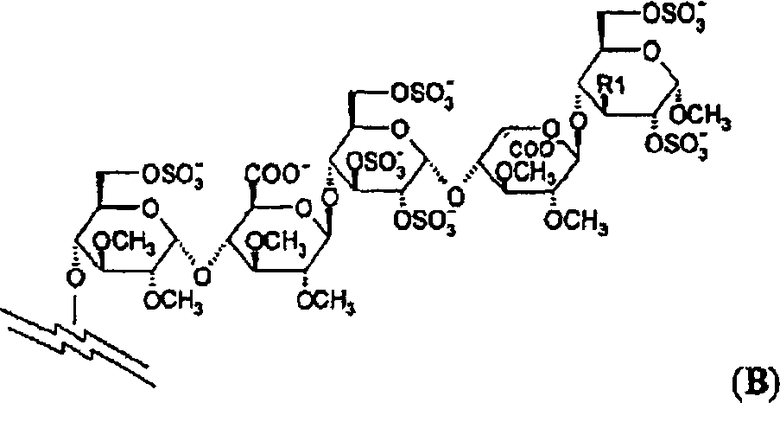 ,
,
где R1 означает ОСН3 или OSO3 -, при этом заряд компенсируется положительно заряженными противоионами; спейсер представляет собой по существу фармакологически неактивный связывающий остаток, имеющий длину 10-35 атомов; антагонист GpIIb/IIIa представляет собой остаток тирофибана (МК 383), при этом спейсер включает ковалентную связь с аналогом биотина формулы -(СН2)4-Х-ВТ, где X=NH, N(1-4С)алкил, -NH-СН(CH2OH)-СН2-С(O)-NH-, -NH-CH(CH3)-CH2-C(O)-NH-, -NH-CH(COOH)-CH2-C(O)-NH- или -NH-CH(CH2COOH)-CH2-C(O)-NH-, а ВТ представляет собой метку  ; или его фармацевтически приемлемая соль. Соединения обладают антитромботической активностью и могут использоваться для лечения или профилактики тромботических заболеваний. 3 н. и 5 з.п. ф-лы, 7 табл., 9 ил.
; или его фармацевтически приемлемая соль. Соединения обладают антитромботической активностью и могут использоваться для лечения или профилактики тромботических заболеваний. 3 н. и 5 з.п. ф-лы, 7 табл., 9 ил.
1. Антитромботическое соединение формулы (I)
олигосахарид-спейсер-антагонист GpIIb/IIIa (I),
где олигосахарид представляет собой отрицательно заряженный пентасахаридный остаток формулы В
,
где R1 представляет собой ОСН3 или OSO3 -, при этом заряд компенсируется положительно заряженными противоинонами,
спейсер представляет собой, по существу, фармакологически неактивный связывающий остаток, имеющий длину 10-35 атомов;
антагонист GpIIb/IIIa представляет собой остаток тирофибана (МК 383);
где спейсер соединения формулы I включает ковалентную связь с аналогом биотина формулы -(СН2)4-Х-ВТ, где X=NH, N(1-4С)алкил, -NH-CH(CH2OH)-CH2-C(O)-NH-, -NH-CH(CH3)-CH2-C(O)-NH-, -NH-CH(COOH)-CH2-C(O)-NH- или -NH-CH(CH2COOH)-CH2-C(O)-NH-, a
ВТ представляет собой метку ;
и его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором спейсер включает по меньшей мере один - (CH2CH2O)-элемент.
3. Соединение по п.2, являющееся

4. Соединение по п.3 в форме его натриевой соли.
5. Соединение по п.4, являющееся
6. Соединение по любому из пп.1-5 для применения в антитромботической терапии.
7. Фармацевтическая композиция, обладающая антитромботической активностью, включающая эффективное количество соединения по любому из пп.1-5 и фармацевтически подходящие вспомогательные вещества.
8. Применение соединения по любому из пп.1-5 для получения лекарственного средства для лечения или профилактики тромбоза или других связанных с тромбином заболеваний.
| Устройство для перегрузки грузов | 1988 |
|
SU1574516A1 |
| Ветряный двигатель | 1926 |
|
SU5133A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| АНТИТРОМБОТИЧЕСКИЕ СОЕДИНЕНИЯ | 1999 |
|
RU2225869C2 |
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |

