Настоящее изобретение относится к новым производным β-карболина, полезным в качестве ингибиторов фосфодиэстеразы. Кроме того, изобретение относится к синтезу производных β-карболина и промежуточных соединений, используемых при их получении. Дополнительно, настоящее изобретение относится к применению описанных производных для лечения заболеваний и болезненных состояний, относящихся к PDE (фосфодиэстераза), например эректильной дисфункции у мужчин.
Эректильную дисфункцию (ED) определяют как неспособность достижения или сохранения достаточно стойкой эрекции для удовлетворительного полового сношения. По данным оценки в настоящее время приблизительно 7-8% мужского населения страдает от определенной степени ED, что эквивалентно, по крайней мере, 20 миллионам мужчин только в Соединенных Штатах. Поскольку вероятность ED увеличивается с возрастом, предполагается, что процент данного заболевания будет расти в будущем, поскольку увеличивается средний рост населения.
Эректильная дисфункция у мужчин может быть следствием психогенного и/или органического факторов. Хотя ED является многофакторной, более вероятно, что у некоторых подгрупп мужского населения присутствуют симптомы заболевания. В частности, среди пациентов с диабетом, гипертензией (повышенным кровяным давлением), сердечными заболеваниями и рассеянным склерозом имеется особенно высокая распространенность ED. Кроме того, пациенты, принимающие некоторые классы лекарственных средств, таких как противогипертонические средства, антидепрессанты, седативные средства и транквилизаторы, более подвержены к проявлению ED.
Лечение ED включает множество фармакологических агентов, вакуумные устройства и протезы мужского полового члена. Среди фармакологических агентов в настоящее время на практике используют папаверин, фентоламин и альпростадил. Данные агенты являются эффективными только после непосредственной инъекции в пещеристое тело или внутриуретральной инъекции и связаны с побочным действием, таким как приапизм (болезненная эрекция полового члена), фиброз, болезненность полового члена и гематома в месте инъекции. Вакуумные устройства представляют собой атравматичное альтернативное лечение ED. Такие устройства приводят к эрекции путем создания отрицательного давления вокруг оси пениса, приводя к повышенному кровотоку в пещеристое тело посредством пассивного артериального расширения. Хотя данная форма терапии часто является успешной при ED органического происхождения, неудовлетворенность вызывает недостаток естественности и времени, необходимого для использования механического устройства, и трудности и дискомфорт при эякуляции. Множество полужестких или надувных протезов полового члена использовалось с определенным успехом, в частности у мужчин с диабетом. Данные устройства обычно рассматриваются в том случае, когда другие методы лечения не имели успеха и связаны с повышенным риском инфекции и ишемией.
Недавно ингибитор фосфодиэстеразы V (PDEV), силденафил (Viagra®), был одобрен Управлением по контролю за продуктами и лекарствами США (FDA) в качестве перорального эффективного лекарственного средства для лечения ED. Силденафил, 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)фенил]-1-метил-3-н-пропил-6,7-дигидро-1-н-пиразоло[4,3-d]пиримидин-7-он и ряд родственных аналогов и их применение в качестве антиангинальных (противостенокардических) агентов описаны в патентах США 5250534 и 5346901. Применение силденафила и родственных аналогов для лечения эректильной дисфункции у мужчин описано в Публикации Международной заявки РСТ № WO 94/28902, опубликованной 22 декабря 1994 года. При клинических исследованиях лекарственное средство улучшало половую функцию примерно у 70% мужчин, которые страдали от ED психогенной и органической этиологии. Однако лекарственное средство показало менее впечатляющую эффективность у пациентов, которые подверглись радикальной простатэктомии, при улучшении эрекции у 43% пациентов, которые принимали силденафил по сравнению с 15% у принимавших плацебо. Кроме того, применение силденафила связано с некоторыми нежелательными побочными действиями, включая головную боль, приток крови и нарушение цветового зрения, являющимися результатом неселективного действия на ряд тканей. Несмотря на данные недостатки, лекарственное средство рассматривается пациентами как предпочтительное по сравнению с другими способами лечения, которые включают введение лекарства непосредственно в пенис посредством инъекции, применение внешних устройств или хирургическое вмешательство.
Daugan и др. в патенте США 5859009 (ЕР 0740668 В1 и WO 95/19978) описывают синтез тетрациклических производных в качестве ингибиторов циклического гуанозин 3',5'-монофосфата (цГМФ), а именно фосфодиэстеразы, и их применение для лечения сердечно-сосудистых заболеваний. Daugan и др. в WO 97/03675 указывают на применение тетрациклических производных при лечении импотенции.
Bombrun и др. в WO 97/43287 описывают ряд производных карболина, более конкретно, 2-(замещенный алкилкарбонил)замещенные производные карболина и их применение для лечения сердечно-сосудистых заболеваний в качестве ингибиторов циклического гуанозин 3',5'-монофосфата, а именно фосфодиэстеразы.
Ellis и др. в WO 94/28902 и ЕР 0702555 В1 описывают ряд производных пиразолпиримидинона и их применение для лечения эректильной дисфункции. Campbell S.F. в WO 96/16657 описывает применение бициклических гетероциклических соединений для лечения импотенции (пиразолопиримидоны); наряду с тем, что Campbell и др. в WO 96/16644 указывают на применение селективных цГМФ PDE ингибиторов для лечения эректильной дисфункции.
Ohashi и др. в WO 97/45427 описывают тетрациклические производные пиридокарбазола, обладающие цГМФ PDE ингибирующим действием.
Fourtillan и др. в WO 96/08490 A1 описывают ряд производных карболина и их применение для лечения заболеваний, связанных с нарушениями мелатониновой активности. Ueki и др. в патенте США 5126448 описывают производные пиридина и 1,2,3,4-тетрагидропиридина, полезные в качестве психотропных лекарственных средств, обладающих успокаивающим действием. Atkinson и др. в патенте США 3328412 описывают производные 1-арил- и 1-гетероарил-2-ацил-1,2,3,4-тетрагидро-β-карболина, обладающие продолжительными анальгетическими (болеутоляющими) свойствами.
Сексуально стимулированная эрекция мужского полового члена является результатом сложного взаимодействия физиологических процессов, включающих центральную нервную систему, периферическую нервную систему и мышцы гладкой мускулатуры. В частности, высвобождение оксида азота из неадренергических, нехолинергических нервов и эндотелия активирует гуанилилциклазу и увеличивает внутриклеточные уровни цГМФ в пещеристом теле. Увеличение внутриклеточного цГМФ снижает внутриклеточные уровни кальция, приводя к трабекулярной релаксации гладкой мышцы, что, в свою очередь, приводит к расширению объема тела и сокращению субоболочечных венул, приводя к эрекции мужского полового члена.
PDEV был найден в тромбоцитах человека и сосудистой гладкой мышце, наводя на мысль о роли данного фермента в регулировании внутриклеточных концентраций цГМФ в сердечно-сосудистой ткани. Действительно, показано, что ингибиторы PDEV проводят к эндотелиально-зависимому расширению сосудов посредством потенцирования увеличения внутриклеточного цГМФ, индуцированного оксидом азота. Более того, PDEV ингибиторы селективно снижают легочное артериальное давление на животных моделях застойной сердечной недостаточности и легочной гипертензии. Следовательно, в дополнение к их полезности при ED, PDEV ингибиторы вероятно могут дать терапевтическую пользу при таких заболеваниях как сердечная недостаточность, легочная гипертензия и стенокардия.
Предполагается, что агенты, увеличивающие концентрацию цГМФ в ткани мужского полового члена либо посредством повышенного высвобождения, либо сниженного разложения цГМФ, будут представлять собой эффективные лекарственные средства при ED. Внутриклеточные уровни цГМФ регулируются ферментами, включенными в его образование и разложение, а именно гуанилатциклазами и циклическими нуклеотидфосфодиэстеразами (PDE). К настоящему времени описано, по меньшей мере, девять семейств PDE млекопитающих, пять из которых способно гидролизовать активный цГМФ в неактивный ГМФ (гуанозин монофосфат) при физиологических условиях (PDE I, II, V, VI и IX). PDE V представляет собой преобладающую изоформу в пещеристом теле человека. Следовательно, можно было бы ожидать, что ингибиторы PDEV будут увеличивать концентрацию цГМФ в пещеристом теле и усиливать продолжительность и частоту эрекции мужского полового члена.
Кроме того, известно, что селективные PDE ингибиторы могут использоваться при лечении различных заболеваний и болезненных состояний, включая эректильную дисфункцию у мужчин (ED), дисфункцию полового возбуждения у женщин, половую дисфункцию у женщин, относящуюся к кровотоку и продуцированию окиси азота в тканях вагины и клитора, преждевременные роды, дисменоррею, сердечно-сосудистые заболевания, атеросклероз, артериальные окклюзионные заболевания, тромбоз, рестеноз коронарной артерии, стенокардию, инфаркт миокарда, сердечную недостаточность, ишемические сердечные заболевания, гипертензию, легочную гипертензию, астму, перемежающуюся хромоту и диабетические осложнения.
Соответственно задача настоящего изобретения заключается в определении соединений, которые увеличивают концентрацию цГМФ в ткани мужского полового члена посредством ингибирования фосфодиэстераз, в частности PDEV. Другой задачей настоящего изобретения является определение соединений, которые могут использоваться для лечения полового расстройства, в частности эректильной дисфункции и/или импотенции у животных мужского пола и полового расстройства у животных женского пола. Еще одной задачей изобретения является установление способов лечения полового расстройства, особенно эректильной дисфункции, с использованием соединений настоящего изобретения.
Другая задача изобретения состоит в определении соединений, которые могут использоваться для лечения болезненных расстройств, опосредованных PDEV, таких как эректильная дисфункция у мужчин, женское половое расстройство, сердечно-сосудистые заболевания, атеросклероз, артериальные окклюзионные заболевания, тромбоз, рестеноз коронарной артерии, стенокардия, инфаркт миокарда, сердечная недостаточность, ишемические сердечные заболевания, гипертензия, легочная гипертензия, астма, перемежающаяся хромота или диабетические осложнения.
В данном описании авторы настоящего изобретения описывают ряд производных β-карболина, обладающих способностью ингибировать фосфодиэстеразу типа V в ферментативном анализе и увеличивать концентрацию цГМФ в ткани пещеристого тела in vitro.
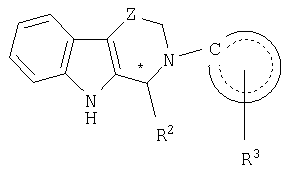
Настоящее изобретение относится к новым производным β-карболина, которые могут использоваться в качестве ингибиторов фосфодиэстеразы. Более конкретно, настоящее изобретение относится к соединениям общей формулы (I):
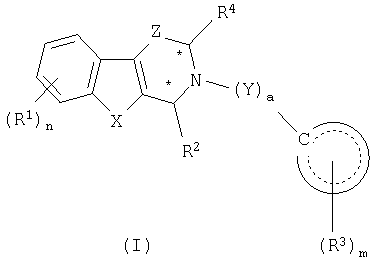
где R1 независимо выбирают из группы, состоящей из галогена, нитро, гидрокси, С1-С8алкила, С1-С8алкокси, -NH2, -NHRA, -N(RA)2, -O-RA, -C(O)NH2, -C(O)NHRA, -C(O)N(RA)2, -NC(O)-RA, -SO2NHRA, -SO2N(RA)2, фенила (необязательно замещенного 1-3 RB) и гетероарила (необязательно замещенного 1-3 RB);
где каждый RA выбирают независимо из группы, состоящей из С1-С8алкила, арила (необязательно замещенного 1-3 RB), С1-С8аралкила (необязательно замещенного 1-3 RB) и гетероарила (необязательно замещенного 1-3 RB);
где каждый RВ выбирают независимо из группы, состоящей из галогена, гидрокси, нитро, циано, С1-С8алкила, С1-С8алкокси, С1-С8алкоксикарбонила, карбоксиС1-С8алкила, С1-С8алкилсульфонила, трифторметила, трифторметокси, амино, ацетиламино, ди(С1-С8алкил)амино, ди(С1-С8алкил)аминоС1-С8алкокси, ди(С1-С8алкил)аминоацетилС1-С8алкила, ди(С1-С8алкил)аминоацетиламино, карбоксиС1-С8алкилкарбониламино, гидроксиС1-С8алкиламино, -NHRA, -N(RA)2 и гетероциклоалкилС1-С8алкокси;
n равно целому числу от 0 до 4;
Х выбирают из группы, состоящей из О, S и NRD;
где RD выбирают из группы, состоящей из водорода, гидрокси, -ORA, C1-C8алкила (где алкил необязательно замещен одним-тремя заместителями, независимо выбранными из галогена, карбокси, амино, С1-С8алкиламино, ди(С1-С8алкил)амино, С1-С8алкоксикарбонила, гетероарила или гетероциклоалкила), гетероарила и гетероарилкарбонила (где гетероарил необязательно может быть замещен фенилом или замещенным фенилом, где заместители фенила представляют собой один-три RB);
R2 выбирают из группы, состоящей из С5-С10алкила (необязательно замещенного 1-3 RC), арила (необязательно замещенного 1-3 RB), гетероарила (необязательно замещенного 1-3 RB) и гетероциклоалкила (необязательно замещенного 1-3 RB);
где каждый RC независимо выбирают из группы, состоящей из галогена, гидрокси, нитро, NH2, NHRA и N(RA)2;
Z выбирают из группы, состоящей из СН2, СНОН и С(О); при условии, что когда Z представляет СНОН или С(О), то Х представляет NH;
R4 выбирают из группы, состоящей из водорода, гидрокси, карбокси, С1-С6алкилкарбонила, С1-С6алкоксикарбонила, ди(С1-С8алкил)аминоалкоксикарбонила, ди(С1-С8алкил)аминоС1-С8алкиламинокарбонила и -CORF;
где RF выбирают из группы, состоящей из С1-С8алкила, NH2, NHRA, NRA 2, -С1-С8алкил-NH2, -С1-С8алкил-NHRA, -С1-С8алкил-NRA 2 и -NH-С1-С8алкилNRA 2;
а равно целому числу от 0 до 1;
Y выбирают из группы, состоящей из СН2, С(О), С(О)О, С(О)-NH и SO2;
 выбирают из группы, состоящей из нафтила, гетероарила и гетероциклоалкила;
выбирают из группы, состоящей из нафтила, гетероарила и гетероциклоалкила;
m равно целому числу от 0 до 2;
R3 независимо выбирают из группы, состоящей из галогена, нитро, С1-С8алкила, С1-С8алкокси, трифторметила, трифторметокси, фенила (необязательно замещенного 1-3 RB), фенилсульфонила, нафтила, С1-С8аралкила, гетероарила (необязательно замещенного 1-3 RB), NH2, NHRA и N(RA)2;
при условии, что если представляет 2-фурил или 2-тиенил, то m равно целому числу от 1 до 2;
и их фармацевтически приемлемым солям.
Изобретение относится к фармацевтической композиции, включающей фармацевтически приемлемый носитель и любое из описанных выше соединений. Изобретение относится к фармацевтической композиции, полученной смешиванием любого из описанных выше соединений и фармацевтически приемлемого носителя. Изобретение относится к способу получения фармацевтической композиции, включающему смешивание любого из описанных выше соединений и фармацевтически приемлемого носителя.
Изобретение относится к способу лечения полового расстройства у нуждающегося в этом субъекта, например, эректильной дисфункции у мужчин, импотенции, женской половой дисфункции, например дисфункции полового возбуждения у женщин, половой дисфункции у женщин, относящейся к кровотоку и продуцированию окиси азота в тканях вагины и клитора, преждевременных родов и дисменорреи, включающему введение субъекту терапевтически эффективного количества любого описанного выше соединения или фармацевтической композиции.
Изобретение относится к способу увеличения концентрации цГМФ в ткани мужского полового члена посредством ингибирования фосфодиэстераз, конкретно PDEV, у нуждающегося в этом субъекта мужского пола, включающему введение субъекту любого описанного выше соединения или фармацевтической композиции.
Кроме того, изобретение относится к способу продуцирования эндотелиально-зависимого расширения сосудов у нуждающегося в этом субъекта посредством потенцирования увеличения внутриклеточного цГМФ, индуцируемого оксидом азота, включающему введение субъекту любого описанного выше соединения или фармацевтической композиции.
Кроме того, изобретение относится к способу лечения болезненного состояния, выбранного из группы, состоящей из эректильной дисфункции (ED) у мужчин, импотенции, женской половой дисфункции, дисфункции полового возбуждения у женщин, половой дисфункции у женщин, относящейся к кровотоку и продуцированию окиси азота в тканях вагины и клитора, преждевременных родов, дисменорреи, сердечно-сосудистых заболеваний, атеросклероза, артериальных окклюзионных заболеваний, тромбоза, рестеноза коронарной артерии, стенокардии, инфаркта миокарда, сердечной недостаточности, ишемических сердечных заболеваний, гипертензии, легочной гипертензии, астмы, перемежающейся хромоты и диабетических осложнений, у нуждающегося в этом субъекта, включающему введение субъекту любого описанного выше соединения или фармацевтической композиции.
Изобретение также относится к применению любого из описанных выше соединений для получения лекарственного средства для: (а) лечения полового расстройства, особенно эректильной дисфункции у мужчин, (b) лечения импотенции, (с) увеличения концентрации цГМФ в ткани мужского полового члена посредством ингибирования фосфодиэстеразы, конкретно, PDEV и/или (d) лечения заболевания, выбранного из группы, состоящей из преждевременных родов, дисменорреи, сердечно-сосудистых заболеваний, атеросклероза, артериальных окклюзионных заболеваний, тромбоза, рестеноза коронарной артерии, стенокардии, инфаркта миокарда, сердечной недостаточности, ишемических сердечных заболеваний, гипертензии, легочной гипертензии, астмы, перемежающейся хромоты и диабетических осложнений, у нуждающегося в этом субъекта.
В настоящем изобретении разработаны новые производные β-карболина, которые можно использовать для лечения полового расстройства, в частности мужской эректильной дисфункции (ED). Хотя соединения настоящего изобретения в первую очередь полезны для лечения полового расстройства у мужчин или эректильной дисфункции, их также можно использовать для лечения женского полового расстройства, например дисфункции полового возбуждения у женщин, половой дисфункции у женщин, относящейся к кровотоку и продуцированию окиси азота в тканях вагины и клитора, преждевременных родов и дисменорреи.
Более конкретно, соединения настоящего изобретения имеют формулу
где все значения радикалов являются такими, как определено выше.
Предпочтительно n равно 0. Предпочтительно m равно целому числу от 0 до 1.
В варианте осуществления настоящего изобретения Х выбирают из S или NRD, где RD выбирают из группы, состоящей из водорода, галогенС1-С6алкила, ди(С1-С4алкил)аминоС1-С6алкила, гетероарила, гетероарилС1-С4алкила, гетероциклоалкилС1-С4алкила, карбоксиС1-С4алкила, С1-С4алкоксикарбонилС1-С4алкила и гетероарилкарбонила; где гетероарил необязательно дополнительно замещен фенилом или замещенным фенилом, где заместители у фенила представляют собой один или два заместителя, независимо выбранных из RB; и где каждый RB независимо выбирают из группы, состоящей из галогена, нитро, С1-С4алкила, С1-С4алкокси, трифторметила, трифторметокси, амино и ди(С1-С4алкил)амино. Предпочтительно Х выбирают из S или NRD, где RD выбирают из группы, состоящей из водорода, ди(метил)аминоэтила, ди(метил)амино-н-пропила, ди(этил)аминоэтила, ди(этил)амино-н-бутила, N-пирролидинилэтила, N-морфолинилэтила, 2-пиридилметила, 4-пиридилметила, 5-(4-метилфенил)-2-пиримидинила, карбоксиметила, карбоксиэтила, 4-хлор-н-бутила, 2-(5-(3-трифторметилфенил)фурил)карбонила, 2-(5-(3-нитрофенил)фурил)карбонила, метоксикарбонилметила, метоксикарбонилаэтила и 2-бензоксазолила. Более предпочтительно, Х представляет NRD, где RD выбирают из группы, состоящей из водорода, ди(метил)аминоэтила, 4-пиридилметила, 2-пиридилметила, N-морфолинилэтила, карбоксиэтила, карбоксиметила, ди(этил)аминоэтила, N-пирролидинилэтила и 5-(4-метилфенил)-2-пиримидинила. Наиболее предпочтительно, Х представляет NRD, где RD выбирают из группы, состоящей из водорода, ди(метил)аминоэтила, N-морфолинилэтила, карбоксиметила и N-пирролидинилэтила.
Предпочтительно Z выбирают из группы, состоящей из СН2 и С(О); при условии, что когда Z представляет С(О), то Х представляет NH.
Предпочтительно Y выбирают из группы, состоящей из С(О), SO2 и СН2. Более предпочтительно Y выбирают из группы, состоящей из С(О) и СН2. Наиболее предпочтительно Y представляет С(О).
В варианте осуществления настоящего изобретения выбирают из группы, состоящей из нафтила и гетероарила. Предпочтительно выбирают из группы, состоящей из нафтила, 2-пиримидинила, 2-фурила, 3-фурила, 2-бензофурила, 2-тиенила, 2-бензотиенила, 2-бензотиазолила, 2-бензоксазолила, 2-бензимидазолила, 4-тиазолила, 2-тиазолила, 3-пиразолила, 4-пиразолила, 5-празолила, 3-(1,2,5-триазолила), 4-изоксазолила, 2-пиридила и 3-пиридила. Более предпочтительно выбирают из группы, состоящей из нафтила, 2-пиримидинила, 2-фурила, 2-бензофурила, 2-тиенила, 2-бензотиенила, 2-бензотиазолила, 2-бензоксазолила, 2-тиазолила, 4-тиазолила и 2-пиридила. Наиболее предпочтительно выбирают из группы, состоящей из 2-пиримидинила, 2-фурила, 2-бензофурила, 2-бензоксазолила, 2-тиазолила и 2-пиридила.
В варианте осуществления настоящего изобретения R2 выбирают из группы, состоящей из 3,4-метилендиоксифенила, 3,4-(дифтор)метилендиоксифенила, 2,3-дигидробензофурила, 2,3-дигидробензо[1,4]диоксин-6-ила, пиридила, фенила и замещенного фенила, где заместители фенила представляют собой один-два заместителя, независимо выбранных из галогена, С1-С4алкила, С1-С4алкокси, трифторметила, циано, нитро, С1-С4алкоксикарбонила, ди(С1-С4)алкил)амино или ди(С1-С4алкил)аминоС1-С4алкокси. Предпочтительно R2 выбирают из группы, состоящей из фенила, 3,4-метилендиоксифенила, 3,4-(дифтор)метилендиоксифенила, 2,3-дигидробензофурила, 2,3-дигидробензо[1,4]диоксин-6-ила, 4-пиридила, 3-пиридила, 4-цианофенила, 3-нитрофенила, 4-нитрофенила, 4-трифторметилфенила, 4-метоксифенила, 3,4-диметилфенила, 3,5-диметилфенила, 3,4-диметоксифенила, 3-трифторметил-4-хлорфенила, 3,4-дихлорфенила, 4-хлорфенила, 4-метоксикарбонилфенила, 3,4-диметоксифенила, 4-(диметиламино)фенила и 4-(N-(3-диметиламино)-н-пропокси)фенила. Более предпочтительно R2 выбирают из группы, состоящей из 3,4-метилендиоксифенила, 2,3-дигидроксибензофурила и 2,3-дигидробензо[1,4]диоксин-6-ила. Наиболее предпочтительно R2 выбирают из группы, состоящей из 3,4-метилендиоксифенила и 2,3-дигидроксибензофурила.
Предпочтительно R4 выбирают из группы, состоящей из водорода, карбокси, С1-С4алкоксикарбонила, ди(С1-С4алкил)аминоС1-С4алкоксикарбонила и ди(С1-С4алкил)аминоС1-С4алкиламинокарбонила. Более предпочтительно R4 выбирают из группы, состоящей из водорода, карбокси, диметиламиноэтоксикарбонила, диметиламиноэтиламинокарбонила и метоксикарбонила. Наиболее предпочтительно R4 представляет водород.
В варианте осуществления настоящего изобретения R3 независимо выбирают из группы, состоящей из галогена, нитро, С1-С4алкила, С1-С4алкокси, трифторметила, С1-С4аралкила, пиразинила, пиридила, замещенного галогеном пиридила, диметилзамещенного имидазолила, фенила, фенилсульфонила и замещенного фенила; где заместители в фениле представляют собой один или несколько заместителей, независимо выбранных из галогена, гидрокси, С1-С4алкила, С1-С4алкокси, трифторметила, трифторметокси, нитро, амино, ацетиламино, С1-С4алкилсульфонила, карбоксиС1-С4алкилкарбониламино, гидроксиС1-С4алкиламино, ди(С1-С4алкил)аминоС1-С4алкокси, ди(С1-С4алкил)аминоацетиламино или гетероциклоалкилС1-С4алкокси. Предпочтительно R3 независимо выбирают из группы, состоящей их хлора, брома, метила, н-пропила, трет-бутила, метокси, трифторметила, нитро, фенила, бензила, фенилсульфонила, 4-гидроксифенила, 4-хлорфенила, 4-метилфенила, 3,4-диметоксифенила, 3-трифторметилфенила, 4-трифторметилфенила, 5-трифторметилфенила, 4-метоксифенила, 2-нитрофенила, 3-нитрофенила, 4-нитрофенила, 3-аминофенила, 4-аминофенила, 2-нитро-4-хлорфенила, 2-нитро-4-метилфенила, 2-нитро-4-метилсульфонилфенила, 3-ацетиламинофенила, 4-ацетиламинофенила, 4-(3-карбокси-н-пропил)карбониламинофенила, 2-хлор-5-трифторметилфенила, 4-(4-гидрокси-н-бутил)аминофенила, 2-(диметиламино)ацетиламинофенила, 4-[2-(N-пирролидинил)этокси]фенила, 4-[2-(4-морфолинил)этокси]фенила, 4-(2-(диметиламино)этокси)фенила, 4-пиразинила, 2,3-диметил-3Н-имидазолила, 2-пиридила и 3-пиридила. Более предпочтительно R3 выбирают из группы, состоящей из брома, трет-бутила, метокси, трифторметила, нитро, фенила, 4-хлорфенила, 3,4-диметоксифенила, 3-трифторметилфенила, 4-метилфенила, 4-метоксифенила, 2-нитрофенила, 3-нитрофенила, 4-нитрофенила, 3-аминофенила, 2-нитро-4-хлорфенила, 2-нитро-4-метилфенила, 2-нитро-4-метилсульфонилфенила, 4-(3-карбокси-н-пропил)карбониламинофенила, 2-хлор-5-трифторметилфенила, 4-(4-гидрокси-н-бутил)аминофенила, 2,2-(диметиламино)ацетил аминофенила, 4-пиразинила, 2-пиридила и 2,3-диметил-3Н-имидазол-4-ила. Наиболее предпочтительно R3 выбирают из группы, состоящей из третбутила, метокси, нитро, фенила, 4-хлорфенила, 4-метилфенила, 4-метоксифенила, 3,4-диметоксифенила, 3-трифторметилфенила, 2-нитрофенила, 3-нитрофенила, 4-нитрофенила, 3-аминофенила, 2-нитро-4-метилсульфонилфенила, 2-(диметиламино)ацетиламинофенила, 2-пиридила и 2,3-диметил-3Н-имидазол-4-ила.
Для применения в медицине соли соединений по данному изобретению относятся к нетоксичным «фармацевтически приемлемым солям». Однако другие соли могут использоваться для получения соединений согласно данному изобретению или их фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли соединений включают в себя кислотно-аддитивные соли, которые, например, могут быть образованы смешиванием раствора соединения с раствором фармацевтически приемлемой кислоты, такой как хлористо-водородная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, бензойная кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Кроме того, когда соединения изобретения содержат кислотный фрагмент, их подходящие фармацевтически приемлемые соли могут включать в себя соли щелочных металлов, например соли натрия или калия; соли щелочно-земельных металлов, например соли кальция или магния, и соли, образованные подходящими органическими лигандами, например соли четвертичного аммония. Таким образом, примерами фармацевтически приемлемых солей являются следующие:
ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, эдетат калия, камзилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глютамат, гликоллиларзанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, аммониевую соль N-метилглюкамина, олеат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат.
В объем настоящего изобретения включены пролекарства соединений данного изобретения. В общем, такие пролекарства будут представлять собой функциональные производные соединений, которые легко превращаются in vivo в целевое соединение. Таким образом, в способах лечения согласно настоящему изобретению термин «введение» будет охватывать лечение различных описываемых заболеваний с использованием конкретного раскрываемого соединения или соединения, которое не раскрывается конкретно, но которое превращается в точно определенное соединения in vivo после введения пациенту. Обычные методики выбора и получения подходящих пролекарственных производных описаны, например, в "Design of Prodrugs", ed. H.Bundgaard, Elsevier, 1985.
Когда соединения согласно данному изобретению имеют по крайней мере один хиральный центр, они, соответственно, могут существовать в виде энантиомеров. Когда соединения имеют два или более хиральных центров, они дополнительно могут существовать в виде диастреомеров. Следует понимать, что все такие изомеры и их смеси включены в объем настоящего изобретения. Кроме того, некоторые кристаллические формы соединений могут существовать в виде полиморфов, и подразумевается, что они, как таковые, включены в настоящее изобретение. Дополнительно, некоторые соединения могут образовывать сольваты с водой (то есть гидраты) или обычными органическими растворителями, и подразумевается, что такие сольваты включены в объем данного изобретения.
Как использовано в данном описании, если не указано другого, «галоген» будет означать хлор, бром, фтор и иод.
Термин «алкил», когда он использован сам по себе или как часть замещающей группы, будет означать линейные или разветвленные алканы, имеющие один-десять атомов углерода, или любое число в данном диапазоне. Например, алкильные радикалы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 3-(2-метил)бутил, 2-пентил, 2-метилбутил, неопентил, н-гексил и 2-метилпентил.
Термин «алкокси» будет обозначать кислородный простой эфирный радикал описанной выше линейной или разветвленной алкильной группы. Например, алкоксирадикалы включают метокси, этокси, н-пропокси, н-бутокси, втор-бутокси, трет-бутокси и тому подобное.
Термин «арил» относится к ароматическим группам, таким как фенил, нафтил и тому подобное.
Термин «аралкил» означает алкильную группу, замещенную арильной группой. Например, бензил, фенетил и тому подобное.
Термин «гетероарил», как он использован в данном описании, представляет собой стабильную пяти или шестичленную моноциклическую ароматическую кольцевую систему, содержащую один-три гетероатома, независимо выбранных из N, O или S; и любую девяти или десятичленную бициклическую ароматическую кольцевую систему, содержащую атомы углерода и один-четыре гетероатома, независимо выбранных из N, O или S. Гетероарильная группа может быть присоединена через любой гетероатом или атом углерода, что приводит к созданию стабильной структуры. Примеры гетероарильной группы включают, но не ограничиваются ими, пиридил, пиримидинил, тиенил, фурил, имидазолил, изоксазолил, оксазолил, пиразолил, пиразинил, пирролил, тиазолил, тиадиазолил, триазолил, бензимидазолил, бензофуранил, бензотиенил, бензизоксазолил, бензоксазолил, индазолил, индолил, бензотиазолил, бензотиадиазолил, бензотриазолил, хинолинил или изохинолинил. Особенно предпочтительные гетероарильные группы включают пиридил, пиразолил, фурил, тиазолил, тиенил, имидазолил, изоксазолил, пиразинил, пиримидинил, триазолил, бензофурил, бензотиенил, бензимидазолил, бензотиазолил и бензоксазолил.
Термин «циклоалкил», как он использован в данном описании, представляет собой стабильную трех-восьмичленную моноциклическую кольцевую структуру, состоящую из насыщенных атомов углерода. Подходящие примеры включают циклопропил, циклобутил, циклопентил, циклгексил, циклогептил и циклооктил.
Термин «гетероциклоалкил» представляет собой стабильную насыщенную или частично ненасыщенную трех-восьмичленную моноциклическую кольцевую структуру, содержащую атомы углерода и один-четыре, предпочтительно один-два, гетероатома, независимо выбранных из N, O или S; или любую стабильную насыщенную, частично ненасыщенную или частично ароматическую девяти-десятичленную бициклическую кольцевую систему, содержащую атомы углерода и один-четыре гетероатома, независимо выбранных из N, O или S. Гетероциклоалкил может быть присоединен через любой гетероатом или атом углерода, что приводит к созданию стабильной структуры. Подходящие примеры гетероциклоалкильных групп включают пирролидинил, пиразолидинил, пиперидинил, пиперазинил, морфолинил, дитианил, тритианил, диоксоланил, диоксанил, тиоморфолинил, 3,4-метилендиоксифенил, 2,3-дигидробензофурил, 2,3-дигидробензо[1,4]диоксин-6-ил, 2,3-дигидрофуро[2,3-b]пиридил, 1,2-(метилендиокси)циклогексан и тому подобное. Особенно предпочтительные гетероциклоалкильные группы включают пирролидинил, морфолинил, 3,4-метилендиоксифенил, 2,3-дигидробензофурил и 2,3-дигидробензо[1,4]диоксин-6-ил.
Как использовано в данном описании, условное обозначение «*» будет означать присутствие стереогенного центра.
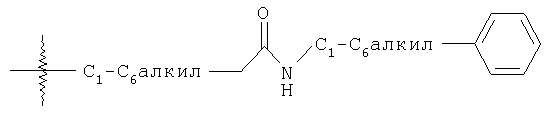
В соответствии со стандартной номенклатурой, использованной в данном описании, терминальная часть обозначенной боковой цепи охарактеризована первой, с последующим описанием смежных функциональных групп впередистоящих по месту присоединения. Таким образом, например, заместитель фенилС1-С6алкиламинокарбонилС1-С6алкил относится к группе формулы
Подразумевается, что определение любого заместителя или переменной в конкретном месторасположении молекулы не зависит от его определений где-либо еще в данной молекуле. Следует понимать, что заместители и образцы замещения в соединениях данного изобретения могут быть выбраны средним специалистом в данной области для создания соединений, которые являются химически стабильными, и которые легко могут быть синтезированы способами, известными в данной области, а также способами, изложенными в данном описании. Кроме того, подразумевается, что когда n или m>1, соответствующие заместители R1 или R3 могут быть одинаковыми или различными.
Термин «половая дисфункция», как он использован в данном описании, включает в себя мужскую половую дисфункцию, мужскую эректильную дисфункцию, импотенцию, женскую половую дисфункцию, дисфункцию полового возбуждения у женщин, половую дисфункцию у женщин, относящуюся к кровотоку и продуцированию окиси азота в ткани вагины и клитора.
Термин «субъект», как он использован в данном описании, относится к животному, предпочтительно к млекопитающему, наиболее предпочтительно к человеку, который является объектом лечения, наблюдения или эксперимента.
Термин «терапевтически эффективное количество», как он использован в данном описании, означает такое количество активного соединения или фармацевтического агента, которое вызывает биологическую или лекарственную ответную реакцию в системе тканей животного или человека, которая является искомой для исследователя, ветеринара, лечащего врача или другого клинического врача, что включает ослабление симптомов подвергаемого лечению заболевания или болезненного состояния.
Как использовано в данном описании, термин «композиция» предназначен для охвата продукта, включающего определенные ингредиенты в определенных количествах, а также любого продукта, который является результатом, прямо или косвенно, комбинации определенных ингредиентов в определенных количествах.
Следующие сокращения использованы в данном описании, в частности, в реакциях и примерах:
Соединения формулы (I) могут быть получены способами, охарактеризованными ниже более подробно.
Соединения формулы (I), где (Y)a представляет С(О), могут быть получены в соответствии со способом, показанным в общих чертах на схеме 1.
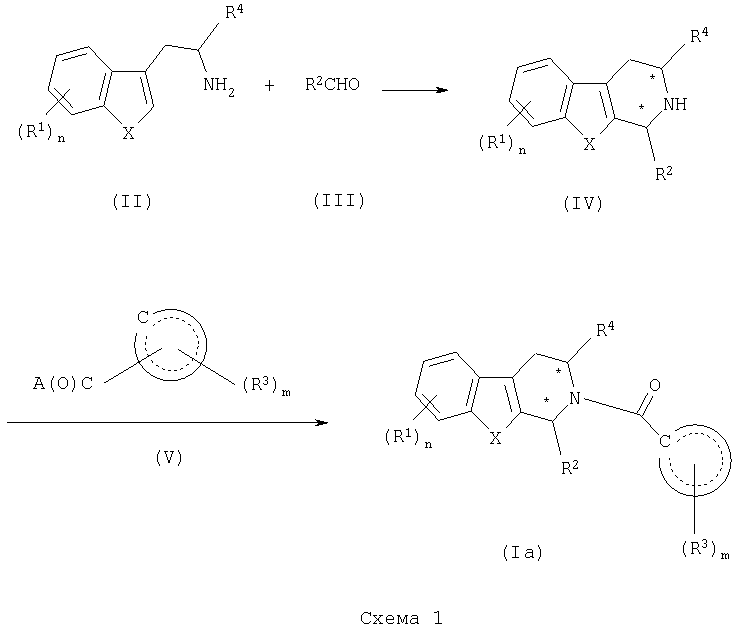
Более конкретно, для получения трициклического соединения формулы (IV) соединение формулы (II), где Х представляет О, S или NH, представляющее собой известное соединение или соединение, получаемое известными способами, подвергают взаимодействию с подходящим образом замещенным альдегидом формулы (III) в органическом растворителе, таком как ДХМ, ТГФ, толуол и тому подобное, в присутствии кислотного катализатора, такого как NFA, тозиловая кислота и тому подобное.
Для получения соответствующего соединения формулы (Ia) соединение формулы (IV) подвергают взаимодействию с подходящим образом замещенным соединением формулы (V), где А представляет галоген, в присутствии основания, такого как триэтиламин (TEA), диизопропилэтиламин (DIPEA), карбонат натрия и тому подобное, в органическом растворителе, таком как дихлорметан (ДХМ), N,N'-диметилформамид (ДМФ), тетрагидрофуран (ТГФ) и тому подобное; или с подходящим образом замещенным соединением формулы (V), где А представляет гидрокси, в присутствии конденсирующего агента, такого как DCC, DIC, PyBop, PyBrop и тому подобное, в органическом растворителе, таком как дихлорметан (ДХМ), N,N'-диметилформамид (ДМФ), тетрагидрофуран (ТГФ) и тому подобное.
Альтернативно, соединения формулы (I), где Х представляет О, S или NH, и (Y)a представляет С(О), могут быть получены в соответствии со способом, показанным в общих чертах на схеме 2.
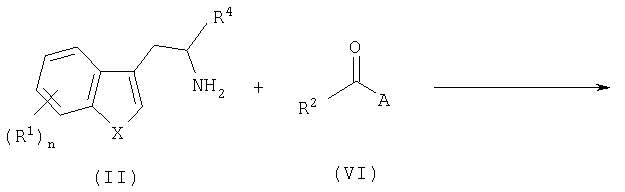
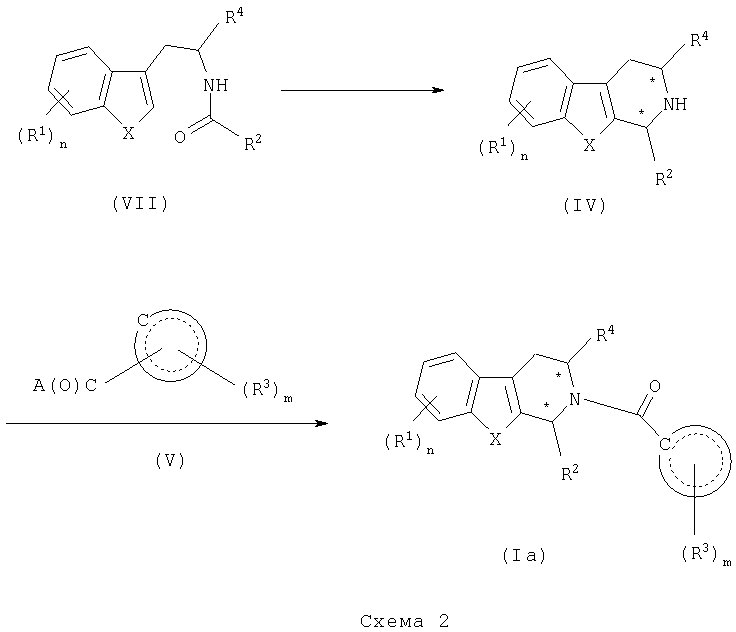
Более конкретно, соединение формулы (II), где Х представляет О, S или NH, подвергают взаимодействию с замещенным подходящим образом соединением формулы (VI), где А представляет галоген или гидрокси, в органическом растворителе, таком как ДХМ, ТГФ, ДМФ и тому подобное, получая соответствующее соединение формулы (VII).
Соединение формулы (VII) подвергают циклизации, обрабатывая POCl3 в органическом растворителе, таком как толуол, бензол и тому подобное, с последующим восстановлением NaBH4, в органическом растворителе, таком как этанол, изопропанол и тому подобное, получая соответствующее соединение формулы (IV).
Соединение формулы (IV) затем подвергают взаимодействию с замещенным подходящим образом соединением формулы (V), получая соединение формулы (Ia), как показано в общем виде на схеме 1.
Соединения формулы (I), где Х представляет О, S или NH, и (Y)a представляет SO2, могут быть получены способом, как показано в общем виде на схеме 3.

Соответственно, подходящим образом замещенное соединение формулы (IV) подвергают взаимодействию с подходящим образом замещенным соединением формулы (VIII), где А представляет галоген или гидрокси, являющимся известным соединением или соединением, получаемым известными способами, в органическом растворителе, таком как ДХМ, хлороформ, ТГФ и тому подобное, с образованием соединения формулы (Ib).
Соединения формулы (I), где Х представляет О, S или NH, и (Y)a представляет СН2, могут быть получены способом, как показано в общем виде на схеме 4.

Соответственно, подходящим образом замещенное соединение формулы (Ia) обрабатывают восстанавливающим агентом, таким как LAH, диборон и другие, предпочтительно LAH, в органическом растворителе, таком как метанол, ТГФ, диэтиловый эфир и тому подобное, предпочтительно при температуре в диапазоне (-20)-40°С, получая соответствующее соединение формулы (Ic).
Соединения формулы (I), где (Y)a представляет СН2, и Х представляет NH, альтернативно, могут быть получены в соответствии со способом, представленным в общем виде на схеме 5.
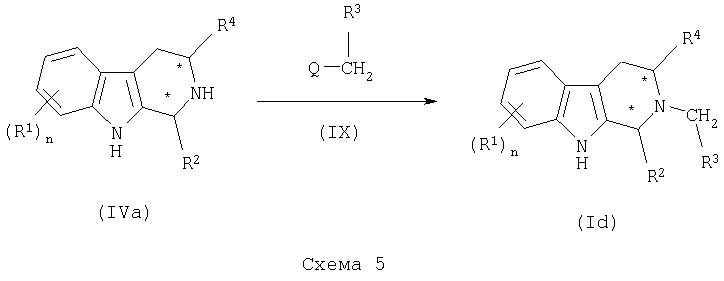
Соответственно, соединение формулы (IVa) подвергают взаимодействию с замещенным подходящим образом соединением формулы (IX), где Q представляет галоген, О-тозилат и О-мезолат, в органическом растворителе, таком как ДХМ, ТГФ и тому подобное, получая соответствующее соединение формулы (Id).
Соединения формулы (I), где Х представляет О, S или NH, и (Y)a представляет (Y)0 (т.е. где а равно 0, так что Y отсутствует), могут быть получены в соответствии со способом, показанным в общем виде на схеме 6.
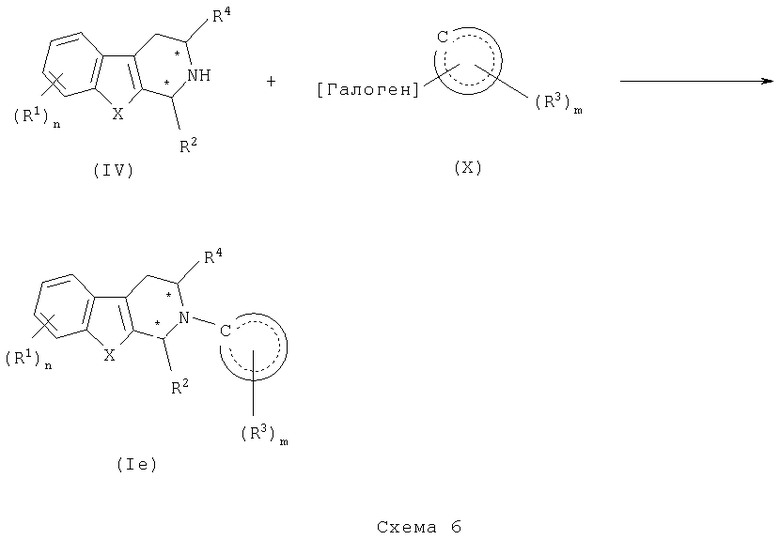
Более конкретно, соединение формулы (IV), представляющее собой известное соединение или соединение, получаемое известными способами, подвергают взаимодействию с замещенным подходящим образом галогенидом формулы (Х), представляющим собой известное соединение или соединение, получаемое известными способами, в органическом растворителе, таком как толуол, ДМФ, 1-метил-2-пирролидинон и тому подобное, предпочтительно при температуре в диапазоне примерно от 80 до 250°С, получая соответствующее соединение формулы (Ie).
Соединения формулы (I), где Х представляет NRD, может быть получено в соответствии со способом, показанным в общем виде на схеме 7.
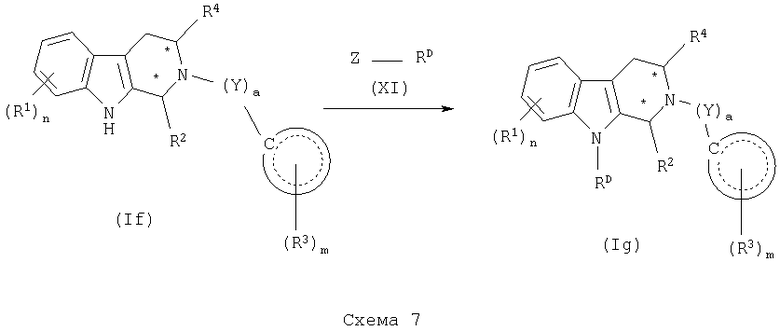
Соответственно, соединение формулы (If) подвергают взаимодействию с соединением формулы (XI), где Z представляет галоген, гидрокси, О-тозилат или О-мезолат, и основанием, таким как гидрид натрия, трет-бутоксид калия и тому подобное, в растворителе, таком как ДМФ, 1-метил-2-пирролидинон и тому подобное, получая соответствующее соединение формулы (Ig).
Соединения формулы (I), где Z представляет СН-ОН или С(О), могут быть получены в соответствии со способом, показанным в общем виде на схеме 8.
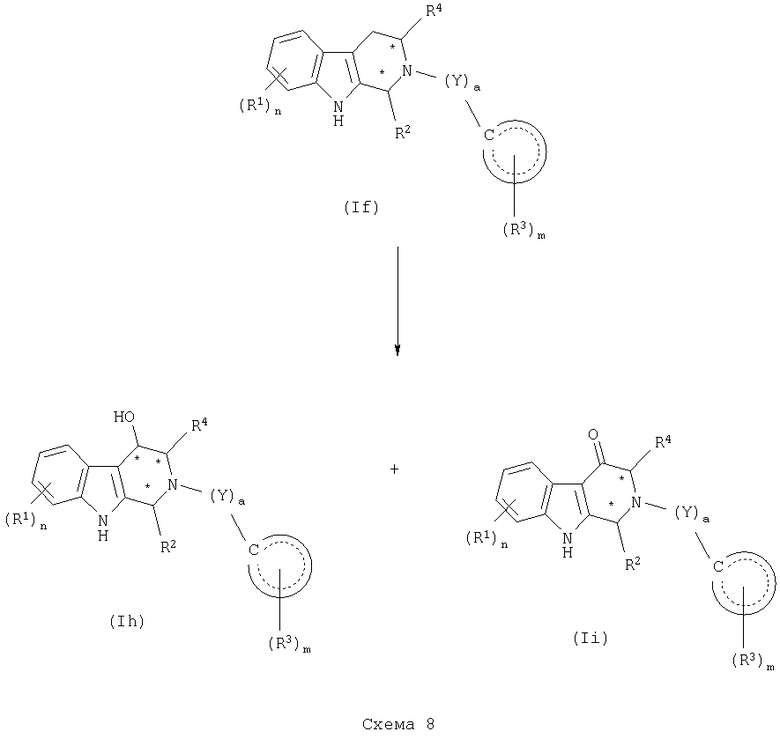
Более конкретно, соединение формулы (If) обрабатывают окислителем, таким как DDQ, хлоранил и тому подобное, в растворителе, таком как ТГФ, метанол, вода и тому подобное, предпочтительно при температуре в диапазоне примерно от -78 до примерно 30°С, получая смесь соответствующих соединений формулы (Ih) и (Ii). Предпочтительно соединения формул (Ih) и (Ii) разделяют известными способами, такими как перекристаллизация, колоночная хроматография и тому подобное.
Соединения формулы (I), где представляет 2-тиазолил, могут быть получены в соответствии со способом, показанным в общем виде на схеме 9.
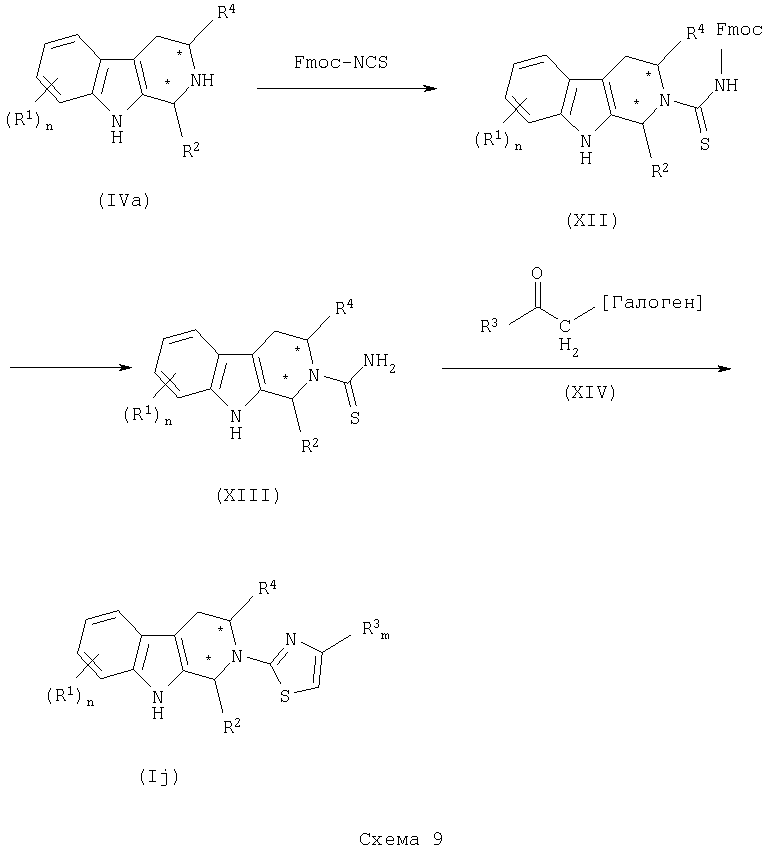
Соответственно, замещенное подходящим образом соединение формулы (IVa) подвергают взаимодействию с Fmoc-NCS в органическом растворителе, таком как ДХМ, ДМФ, ТГФ и тому подобное, предпочтительно при комнатной температуре, получая соответствующее соединение формулы (XII).
Соединение (XII) подвергают взаимодействию с 20%-ным пиперидином в спирте, таком как метанол, этанол и тому подобное, получая соответствующий амин формулы (XIII).
Амин формулы (XIII) подвергают взаимодействию с замещенным подходящим образом α-галогенметилкетоном формулы (XIV) в присутствии органического растворителя или смеси, такой как ДМФ, этанол:диоксан и тому подобное, в присутствии основания, такого как TEA, DIPEA и тому подобное, предпочтительно при температуре примерно 70°С, получая соответствующее соединение формулы (Ij).
Соединения формулы (I), где (Y)a представляет С(О)О, могут быть получены в соответствии со способом, показанным в общем виде на схеме 10.
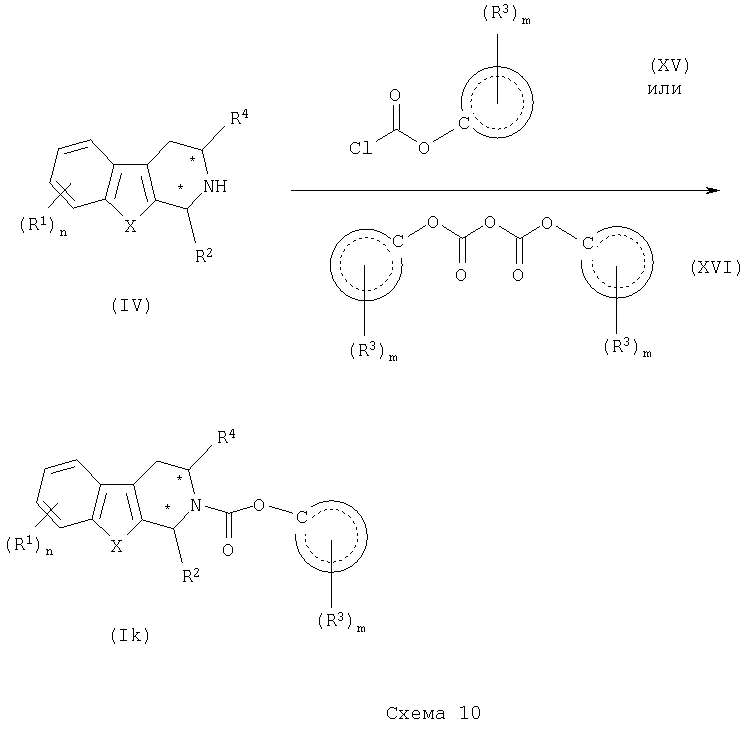
Более конкретно, соединение формулы (IV) подвергают взаимодействию с замещенным подходящим образом хлорформиатом формулы (XV) или ангидридом формулы (XVI) в органическом растворителе, таком как ДХМ, ДМФ, ТГФ и тому подобное, получая соответствующее соединение формулы (Ik).
Соединения формулы (I), где (Y)a представляет С(О)-NH, могут быть получены в соответствии со способом, показанным в общем виде на схеме 11.
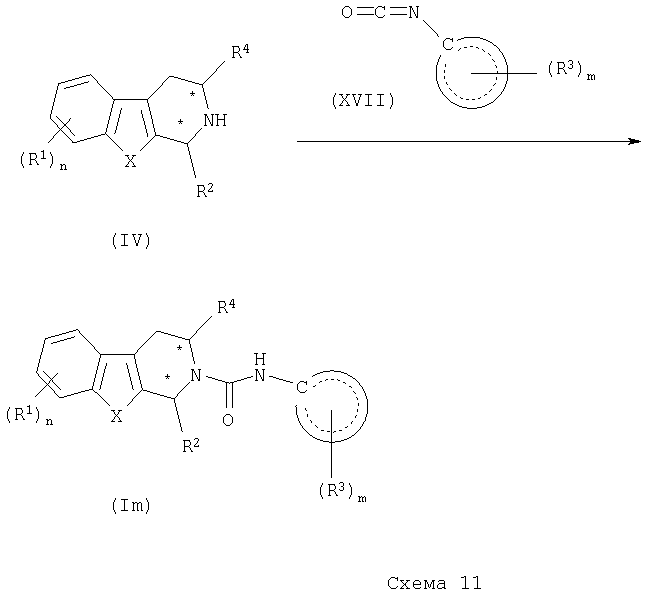
Соответственно, соединение формулы (IV) подвергают взаимодействию с замещенным подходящим образом соединением формулы (XVII) в органическом растворителе, таком как ДХМ, ДМФ, ТГФ и тому подобное, получая соответствующее соединение формулы (Im).
В том случае, когда способы получения соединений согласно изобретению приводят к смеси стереоизомеров, данные изомеры могут быть разделены обычными способами, такими как препаративная хроматография. Соединения могут быть получены в рацемической форме, или могут быть получены индивидуальные энантиомеры либо с использованием энантиоселективного синтеза или разделением. Например, соединения могут быть разделены на составляющие их энантиомеры стандартными методами, такими как образование диастереоизомерных пар посредством солеобразования с оптически активной кислотой, такой как (-)-ди-п-толуоил-D-винная кислота и/или (+)-ди-п-толуоил-L-винная кислота, с последующей дробной кристаллизацией и регенерированием свободного основания. Соединения также могут быть разделены путем образования диастереомерных сложных эфиров или амидов с последующим хроматографическим разделением и удалением хиральной вспомогательной добавки. Альтернативно соединения могут быть разделены с использованием хиральной ВЭЖХ колонки.
В процессе любого из способов получения соединений настоящего изобретения может оказаться необходимо и/или желательно защитить чувствительные или реакционноспособные группы в любой из рассматриваемых молекул. Это может быть осуществлено с использованием обычных защитных групп, таких, например, как описанные в Protective Groups in Organic Chemistry, ed. J.F.W.McOmie, Plenum Press, 1973; и Т.W.Greene & P.G.М.Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991. Защитные группы могут быть удалены на подходящей последующей стадии с использованием способов, известных специалистам в данной области.
Полезность соединений для лечения половой дисфункции может быть определена в соответствии с методиками, описанными в данном описании в примерах 10, 11 и 12.
Следовательно, настоящее изобретение относится к способу лечения половой дисфункции у нуждающегося в этом субъекта, включающему введение ему любого из определенных в данном описании соединений в количестве, эффективном для лечения полового расстройства. Соединение можно вводить пациенту с использованием любого обычного пути введения, включая, но, не ограничиваясь ими, внутривенный, пероральный, подкожный, внутримышечный, внутрикожный и парентеральный. Количество соединения, которое является эффективным для лечения полового расстройства, составляет между 0,1 мг на 1 кг и 2 мг на 1 кг массы тела субъекта.
Настоящее изобретение также относится к фармацевтической композиции, включающей одно или более соединений по данному изобретению в сочетании с фармацевтически приемлемым носителем. Предпочтительно данные композиции существуют в виде единичных препаративных лекарственных форм, таких как таблетки, пилюли, капсулы, порошки, гранулы, стерильные парентеральные растворы или суспензии, дозированные аэрозоли или жидкие спреи, капли, ампулы, автоинъектирующие устройства или суппозитории; для перорального, парентерального, внутриназального, сублингвального или ректального введения или для введения посредством ингаляции или инсуфляции. Альтернативно композиция может быть представлена в форме, подходящей для введения один раз в неделю или один раз в месяц; например, нерастворимая соль активного соединения, такая как деканоатная соль, может быть приспособлена для разработки депо-препарата для внутримышечной инъекции. Для получения твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим носителем, например с обычными ингредиентами для таблетирования, такими как кукурузный крахмал, лактоза, сахароза, сорбит, тальк, стеариновая кислота, стеарат магния, дикальцийфосфат или камеди, и другими фармацевтическими разбавителями, например с водой, для получения твердой таблеточной композиции, содержащей гомогенную смесь соединения настоящего изобретения или его фармацевтически приемлемой соли. При упоминании данных предварительных композиций как гомогенных подразумевается, что активный ингредиент равномерно распределен в композиции таким образом, что композиция легко может быть разделена на равно эффективные препаративные лекарственные формы, такие как таблетки, пилюли и капсулы. Такую твердую предварительную композицию впоследствии разделяют на единичные препаративные лекарственные формы описанного выше типа, содержащие от 1 до 1000 мг активного ингредиента по настоящему изобретению. Таблетки или пилюли новой композиции могут быть покрыты оболочкой или компаундированы другим образом для создания препаративной лекарственной формы, дающей преимущество пролонгированного действия. Например, таблетка или пилюля может включать в себя внутреннюю дозу и внешнюю дозу компонентов, где последний из них представляет собой оболочку для первого. Два компонента могут быть разделены энтеральным (растворимым в кишечнике) слоем, который служит для подавления разложения в желудке и позволяет внутреннему компоненту проходить неповрежденным в двенадцатиперстную кишку или замедленно высвобождаться. Для таких энтеральных слоев или покрытий можно использовать множество материалов, такие материалы включают ряд полимерных кислот с такими веществами как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы для перорального введения или введения путем инъекций, в которые могут быть введены новые композиции настоящего изобретения, включают жидкие растворы, ароматизированные подходящим образом сиропы, водные или масляные суспензии и ароматизированные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические средства переноса. Подходящие диспергирующие или суспендирующие агенты для водных суспензий включают синтетические и природные камеди, такие как трагакант, гуммиарабик, альгинат, декстран, натрийкарбоксиметилцеллюлоза, метилцеллюлоза, поливинилпирролидон или желатин.
Способ лечения половых расстройств, в частности эректильной дисфункции у мужчин (ED), описанный в настоящем изобретении, также может быть осуществлен с использованием фармацевтической композиции, включающей любое из описанных здесь соединений и фармацевтически приемлемый носитель. Фармацевтическая композиция может содержать от 1 мг до 1000 мг, предпочтительно до 10 до 500 мг соединения и может быть получена в виде любой формы, подходящей для выбранного способа введения. Носители включают необходимые и инертные фармацевтические эксципиенты, включая, но, не ограничиваясь ими, связующие вещества, суспендирующие агенты, смазочные агенты, вкусовые добавки, подсластители, консерванты, красители и агенты покрывающих оболочек. Композиции, подходящие для перорального введения, включают твердые формы, такие как пилюли, таблетки, каплеты, капсулы (каждая из которых включает рецептуры немедленного высвобождения, рассчитанного по времени высвобождения и продолжительного непрерывного высвобождения), гранулы и порошки, и жидкие формы, такие как растворы, сиропы, эликсиры, эмульсии и суспензии. Формы, используемые для парентерального введения, включают стерильные растворы, эмульсии и суспензии.
Преимущественно, соединения настоящего изобретения можно вводить в виде единичной дневной лекарственной дозы, или суммарную дневную дозировку можно вводить в виде разделенных доз два, три или четыре раза в день. Кроме того, соединения настоящего изобретения можно вводить в виде внутриназальной формы посредством местного применения с помощью подходящих внутриназальных носителей или посредством чрескожных пластырей для кожи, хорошо известных среднему специалисту в данной области. При введении в виде чрескожной системы доставки введение лекарственного средства на протяжении режима дозирования, конечно, скорее будет непрерывным, чем периодическим.
Например, для перорального введения в виде таблетки или капсулы активный компонент лекарственного средства можно объединять с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное. Более того, при желании или при необходимости в смесь могут быть включены подходящие связующие вещества, смазочные вещества, дезинтегрирующие агенты и красители. Подходящие связующие вещества включают, без ограничения, крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические камеди, такие как гуммиарабик, трагакант или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Дезинтеграторы включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное.
Жидкие формы могут включать подходящим образом ароматизированные суспендирующие или диспергирующие агенты, такие как синтетические и природные камеди, например, трагакант, гуммиарабик, метилцеллюлозу. Для парентерального введения требуются стерильные суспензии и растворы. Когда желательно внутривенное введение, используют изотонические препараты, которые обычно содержат подходящие консерванты.
Соединение настоящего изобретения также можно вводить в виде липосомных систем доставки, таких как небольшие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть получены из множества фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Соединения настоящего изобретения также могут доставляться с использованием моноклональных антител в качестве индивидуальных носителей, к которым присоединены молекулы соединения. Соединения настоящего изобретения также могут быть связаны с растворимыми полимерами в качестве носителей лекарственного средства, способных достигать цели. Такие полимеры могут включать поливинилпирролидон, сополимеры пирана, полигидроксипропилметакриламинфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Кроме того, соединения настоящего изобретения могут быть связаны с классом биоразрушаемых полимеров, используемых для достижения контролируемого высвобождения лекарственного средства, например, полиактиковой кислотой, полиэпсилонкапролактоном, полигидроксиянтарной кислотой, сложными полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и сшитыми или амфипатическими блок-сополимерами гидрогелей.
В каждом случае, когда требуется лечение полового расстройства, в частности эректильной дисфункции (ED) у мужчин, соединения данного изобретения можно вводить в виде любой из представленных выше композиций и в соответствии с режимами дозировки, определенными в данной области.
Дневная дозировка продуктов может варьироваться в широком диапазоне от 1 до 1000 мг в день для взрослого человека. Для перорального введения композиции предпочтительно получают в виде таблеток, содержащих 5,0, 10,0, 15,0, 25,0, 50,0, 100, 250 и 500 миллиграммов активного ингредиента для симптоматического регулирования дозировки для пациента, подвергаемого лечению. Эффективное количество лекарственного средства обычно обеспечивается при уровне дозировки примерно от 0,1 мг/кг до примерно 20 мг/кг массы тела в сутки. Предпочтительно диапазон составляет от примерно 0,2 мг/кг до примерно 10 мг/кг массы тела в сутки и особенно предпочтительно от примерно 0,5 мг/кг до примерно 10 мг/кг массы тела в сутки. Соединения можно вводить при режиме дозировки 1-4 раза в сутки.
Оптимальные дозировки для введения легко могут быть определены специалистами в данной области, и они будут изменяться в зависимости от конкретного используемого соединения, способа введения, интенсивности препарата, способа введения и развития заболевания. Дополнительно к необходимости регулировать дозировку приводят факторы, связанные с конкретным подвергаемым лечению пациентом, включая возраст пациента, масса, режим питания и время введения.
Для того чтобы помочь пониманию изобретения представлены следующие примеры, которые не предназначены и не истолковываются как ограничивающие каким-либо образом изобретение, изложенное в следующей ниже формуле изобретения.
Если не указано другого, спектры 1Н ЯМР регистрировали на приборе Bruker AC-300.
Пример 1
1-(3,4-Метилендиоксифенил)-2-[5-(3-трифторметилфенил)фуроил]-2,3,4,9-тетрагидро-1Н-β-карболин (№58)
К суспензии 5-(3-трифторметилфенил)фуранкарбоновой кислоты (256 мг, 1 ммоль) в ДХМ (20 мл, безводный) добавляли оксалилхлорид (165 мг, 1,3 ммоль) с последующим добавлением двух капель ДМФ. Смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли раствор 1-(3,4-метилендиоксифенил)-2,3,4,9-тетрагидро-1Н-β-карболина (292 мг, 1 ммоль) (полученного в соответствии со способом, описанным в WO 97/43287, промежуточное соединение 7, стр.24) и триэтиламина (0,4 мл) в ДХМ (10 мл, безводный), и смесь перемешивали при комнатной температуре в течение 16 часов, промывали последовательно водным NaHCO3, насыщенным раствором соли (2х), 1н. HCl и насыщенным раствором соли (2х) и сушили MgSO4. После упаривания растворителя получали белое твердое вещество.
Т.пл.: 126-129°С
Масс-спектр (m/z): 531 (MH+);
1H-ЯМР (CDCl3) δ: 2,96 (д, J=8 Гц, 1H), 3,24 (м, 1H), 3,56 (м, 1H), 4,60 (д, J=8 Гц, 1H), 5,90 (с, 2H), 6,70 (д, J=8 Гц, 1H), 6,83-6,99 (м, 4H), 7,13-7,34 (м, 4H), 7,55 (м, 3H), 7,87 (д, J=7 Гц, 1H), 7,95 (с, 1H), 8,23 (с, 1H).
Пример 2
9-[2-(Пирролидин-1-ил)этил]-1-(3,4-метилендиоксифенил)-2-[5-(3-трифторметилфенил)фуроил]-2,3,4-тригидро-1Н-β-карболин (№75)
К раствору 1-(3,4-метилендиоксифенил)-2-[5-(3-трифторметилфенил)фуроил]-2,3,4,9-тетрагидро-1Н-β-карболина (полученного, как в примере 1) (600 мг, 1,14 ммоль) в ДМФ (15 мл, безводный) добавляли при комнатной температуре гидрид натрия (60%, 105 мг, 2,6 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли гидрохлорид N-хлорэтилпирролидина (214 мг, 1,26 ммоль) и 15-краунэфир-5 (1 капля). Смесь перемешивали при комнатной температуре в течение 16 часов, гасили NH4Cl, экстрагировали этилацетатом и сушили MgSO4. После упаривания растворителя остаток очищали колоночной хроматографией (силикагель, этилацетат:гексаны=3:1), получая белое твердое вещество.
Масс-спектр (m/z): 628 (MH+);
1H-ЯМР (CDCl3) δ: 1,26 (м, 4H), 2,64 (м, 4H), 2,89 (м, 2H), 3,05 (д, J=8 Гц, 1H), 3,28 (т, J=8 Гц, 1H), 3,59 (т, J=8 Гц, 1H), 3,96 (м, 1H), 4,16 (м, 1H), 4,58 (д, J=8 Гц, 1H), 5,96 (с, 2H), 6,75 (д, J=8 Гц, 1H), 6,84 (м, 2H), 7,02 (д, J=8 Гц, 1H), 7,15-7,29 (м, 4H), 7,43 (с, 1H), 7,59 (м, 3H), 7,89 (д, J=7 Гц, 1H), 7,96 (с, 1H).
Для получения продукта для биологического испытания соответствующую соль метансульфоновой кислоты получали добавлением 1,0 эквивалента метансульфоновой кислоты к раствору указанного соединения в ДХМ.
Т.пл.: 122-124°С
Пример 3
1-(3,4-Метилендиоксифенил)-2-[5-(3,4-диметоксифенил)пиримидин-2-ил]-2,3,4,9-тетрагидро-1Н-β-карболин (№7)
Раствор 1-(3,4-метилендиоксифенил)-2,3,4,9-тетрагидро-1Н-β-карболина (3,73 г, 12,8 ммоль) (полученного в соответствии со способом, описанным в WO 97/43287, промежуточное соединение 7, стр.24) и 2-хлор-5-(3,4-диметоксифенил)пиримидина (1,6 г, 6,4 ммоль) в ДМФ (50 мл, безводный) нагревали при 120°С при перемешивании в течение 16 часов. Реакционную смесь гасили NH4Cl и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором соли (2х) и сушили MgSO4. Колоночная хроматография (силикагель, этилацетат:гексаны=2:3) давала белое твердое вещество.
Т.пл.: 173-175°С
Масс-спектр (m/z): 507 (MH+);
1H-ЯМР (CDCl3) δ: 2,91 (д, J=9 ГЦ, 1H), 3,02 (тд, J=9,1 Гц, 1H), 3,39 (тд, J=9,1 Гц, 1H), 3,92 (с, 3H), 3,94 (с, 3H), 5,02 (д, J=9,1 Гц, 1H), 5,92 (с, 2H), 6,72 (д, J=8 Гц, 1H), 6,87-7,03 (м, 4H), 7,11-7,17 (м, 3H), 7,31 (д, J=8 Гц, 1H), 7,56 (д, J=8 Гц, 1H), 7,80 (с, 1H), 8,56 (с, 2H).
Пример 4
1-(3,4-Метилендиоксифенил)-2-[5-(3,4-диметоксифенил)пиримидин-2-ил]-9-диметиламиноэтил-2,3,4-тригидро-1Н-β-карболин (№5)
В соответствии с методикой, описанной в примере 1, подвергали взаимодействию 1-(3,4-метилендиоксифенил)-2-[5-(3,4-диметоксифенил)пиримидин-2-ил]-2,3,4,9-тетрагидро-1Н-β-карболин (полученный, как описано в примере 3) (1,0 г, 1,97 ммоль), гидрохлорид 2-хлор-N,N-диметилэтиламина (0,342 г, 2,37 ммоль), гидрид натрия (60%, 0,190 г, 4,74 ммоль) и 15-краунэфир-5, получая продукт в виде слегка желтоватого твердого вещества (после колоночной хроматографии на силикагеле, этилацетат).
Масс-спектр (m/z): 578 (MH+);
1H-ЯМР (CDCl3) δ: 2,21 (с, 6H), 2,22 (м, 1H), 2,61 (м, 1H), 2,89 (дд, J=13,4 Гц, 1H), 3,03 (тд, J=13,4 Гц, 1H), 3,35 (тд, J=13,4 Гц, 1H), 3,91 (м, 1H), 3,92 (с, 3H), 3,95 (с, 3H), 4,06 (м, 1H), 4,96 (дд, J=13,4 Гц, 1H), 5,93 (с, 2H), 6,72 (д, J=8 Гц, 1H), 6,83 (с, 1H), 6,85-6,98 (м, 4H), 7,12 (д, J=8 Гц, 1H), 7,21 (д, J=8 Гц, 1H), 7,31 (д, J=8 Гц, 1H), 7,34 (с, 1H), 7,58 (д, J=8 Гц, 1H), 8,56 (с, 1H).
Пример 5
1-(3,4-Метилендиоксифенил)-2-[5-(4-метоксифенил)пиримидин-2-ил]-4-оксо-2,3,4,9-тетрагидро-1Н-β-карболин (№157) и
1-(3,4-Метилендиоксифенил)-2-[5-(4-метоксифенил)пиримидин-2-ил]-4-гидрокси-2,3,4,9-тетрагидро-1Н-β-карболин (№158)
К смеси DDQ (113,5 мг, 0,5 ммоль) и 1-(3,4-метилендиоксифенил)-2-[5-(3,4-диметоксифенил)пиримидин-2-ил]-2,3,4,9-тетрагидро-1Н-β-карболин (полученного, как описано в примере 3) (51 мг, 0,1 ммоль) при -78°С добавляли смешанный растворитель ТГФ:вода (9:1). Смесь перемешивали при 0°С и оставляли нагреваться до комнатной температуры в течение 15 часов. Колоночная хроматография (силикагель, гексаны:этилацетат=1:1) давала оксо- и гидроксипроизводные, соответственно, в виде белых твердых веществ.
№157
Масс-спектр (m/z): 521 (МН+), 519 (М-1);
1H-ЯМР (CDCl3) δ: 3,90 (д, J=18 Гц, 1H), 3,89 (с, 3H), 3,91 (с, 3H), 5,43 (д, J=18 Гц, 1H), 5,84 (с, 2H), 6,62 (д, J=8 Гц, 1H), 6,71 (д, J=8 Гц, 1H), 6,88-7,00 (м, 4H), 7,29-7,43 (м, 3H), 7,53 (с, 1H), 8,25 (м, 1H), 8,51 (с, 2H), 9,55 (с, 1H).
№158
Масс-спектр (m/z): 523 (МН+), 521 (MH+);
1H-ЯМР (CDCl3) δ: 3,30 (т, J=6 Гц, 1H), 3,69 (д, J=6 Гц, 1H), 3,92 (с, 3H), 3,94 (с, 3H), 5,97 (с, 2H), 6,11 (с, 1H), 6,71 (д, J=8 Гц, 1H), 6,93-7,05 (м, 4H), 7,18 (д, J=8 Гц, 1H), 7,23 (д, J=8 Гц, 1H), 7,40 (т, J=6 Гц, 1H), 7,49 (д, J=8 Гц, 1H), 7,82 (д, J=8 Гц, 1H), 8,43 (с, 2H), 9,15 (с, 1H).
Пример 6
1-(3,4-Метилендиоксифенил)-2-[4-(4-метоксифенил)тиазол-2-ил]-2,3,4,9-тетрагидро-1Н-β-карболин (№169)
А: 1-(3,4-Метилендиоксифенил)-2-[3-(флуоренилметилоксикарбонил)тиокарбамоил]-2,3,4,9-тетрагидро-1Н-β-карболин
Смесь 1-(3,4-метилендиоксифенил)-2,3,4,9-тетрагидро-1Н-β-карболина (2,66 г, 9,08 ммоль) (полученного в соответствии со способом, описанным в WO 97/43287, промежуточное соединение 7, стр.24) и Fmoc-изотиоцианата (2,82 г, 10,14 ммоль) растворяли в сухом дихлорметане (50 мл). Смесь перемешивали в течение 16 часов при температуре окружающей среды, а затем концентрировали в вакууме. Очистка флэш-хроматографией (0-10% метанол в дихлорметане) давала защищенную тиомочевину в виде бледно-желтого твердого вещества.
Масс-спектр (m/z): 574 (MH+);
1H-ЯМР (CDCl3) δ: 2,86 (дд, J=12,9, 5,1 Гц, 1H), 3,09 (дт, J=17,1, 6,9 Гц, 1H), 3,56 (дт, J=12,9, 5,1 Гц, 1H), 4,19 (т, J=6,9 Гц, 1H), 4,43-4,53 (м, 2H), 5,91 (с, 2H), 6,70 (д, J=8 Гц, 1H), 6,90 (ушир.д, J=7,6 Гц, 1H), 6,97 (ушир.с, 1H), 7,11-7,78 (ряд м, 17H).
В: 1-(3,4-Метилендиоксифенил)-2-(тиокарбамоил)-2,3,4,9-тетрагидро-1Н-β-карболин
Раствор защищенной тиомочевины из части А (4,78 г, 8,33 ммоль) в 20% (об./об.) пиперидине в метаноле нагревали при кипячении с обратным холодильником в течение 5 часов. Смесь концентрировали в вакууме, получая неочищенный остаток, который очищали флэш-хроматографией (SiO2, 0-10% метанол в дихлорметане), получая желтое твердое вещество.
Масс-спектр (m/z): 352 (MH+);
1H-ЯМР (CDCl3) δ: 2,69-2,87 (ряд м, 2H), 3,10-3,19 (м, 1H), 4,24 (ушир.с, 1H), 6,00 (д, J=3,3 Гц, 2H), 6,72 (д, J=8,0 Гц, 1H), 6,87 (д, J=8,0 Гц, 1H), 7,00-7,11 (ряд м, 3H), 7,30 (д, J=8,0 Гц, 1H), 7,46 (д, J=7,7 Гц, 1H), 7,74 (ушир.с, 3H), 11,06 (с, 1H).
C. 1-(3,4-Метилендиоксифенил)-2-[4-(4-метоксифенил)тиазол-2-ил]-2,3,4,9-тетрагидро-1Н-β-карболин (№169)
К раствору тиомочевины из части В (223 мг, 0,63 ммоль) в смеси 1:1 диоксан:этанол (5 мл) добавляли 4-метоксифенил-2'-бромацетофенон (175 мг, 0,76 ммоль) и триэтиламин (0,40 мл). Смесь нагревали при 70°С в течение 3 часов, охлаждали до комнатной температуры и концентрировали на роторном испарителе. Остаток очищали флэш-хроматографией (SiO2, 0-10% метанол в дихлорметане), получая бесцветное твердое вещество.
Масс-спектр (m/z): 482 (MH+);
1H-ЯМР (CDCl3) δ: 2,86-2-3,07 (ряд м, 2H), 3,61-3,71 (м, 1H), 3,78 (с, 3H), 3,91-4,02 (м, 1H), 5,99 (д, J=3,3 Гц, 2H), 6,58 (с, 1H), 6,80-7,11 (ряд м, 8H), 7,31 (д, J=7,8 Гц, 1H), 7,48 (д, J=7,6 Гц, 1H), 7,82 (д, J=8,7 Гц, 2H), 10,93 (с, 1H).
Пример 7
1-(3,4-Метилендиоксифенил)-2-[4-фенилтиазол-2-ил]-2,3,4,9-тетрагидро-1Н-β-карболин (№170)
А: 1-(3,4-Метилендиоксифенил)-2-[3-(флуоренилметилоксикарбонил)тиокарбамоил]-2,3,4,9-тетрагидро-1Н-β-карболин
Смесь 1-(3,4-метилендиоксифенил)-2,3,4,9-тетрагидро-1Н-β-карболина (2,66 г, 9,08 ммоль) (полученного в соответствии со способом, описанным в WO 97/43287, промежуточное соединение 7, стр.24) и Fmoc-изотиоцианата (2,82 г, 10,14 ммоль) растворяли в сухом дихлорметане (50 мл). Смесь перемешивали в течение 16 часов при температуре окружающей среды, а затем концентрировали в вакууме. Очистка флэш-хроматографией (0-10% метанол в дихлорметане) давала защищенную тиомочевину в виде бледно-желтого твердого вещества.
Масс-спектр (m/z): 574 (MH+);
1H-ЯМР (CDCl3) δ: 2,86 (дд, J=12,9, 5,1 Гц, 1H), 3,09 (дт, J=17,1, 6,9 Гц, 1H), 3,56 (дт, J=12,9, 5,1 Гц, 1H), 4,19 (т, J=6,9 Гц, 1H), 4,43-4,53 (м, 2H), 5,91 (с, 2H), 6,70 (д, J=8 Гц, 1H), 6,90 (ушир.д, J=7,6 Гц, 1H), 6,97 (ушир.с, 1H), 7,11-7,78 (ряд м, 17H).
В: 1-(3,4-Метилендиоксифенил)-2-(тиокарбамоил)-2,3,4,9-тетрагидро-1Н-β-карболин
Раствор защищенной тиомочевины из части А (4,78 г, 8,33 ммоль) в 20% (об./об.) пиперидине в метаноле нагревали при кипячении с обратным холодильником в течение 5 часов. Смесь концентрировали в вакууме, получая неочищенный остаток, который очищали флэш-хроматографией (SiO2, 0-10% метанол в дихлорметане), получая желтое твердое вещество.
Масс-спектр (m/z): 352 (MH+);
1H-ЯМР (CDCl3) δ: 2,69-2,87 (ряд м, 2H),3,10-3,19 (м, 1H), 4,24 (ушир.с, 1H), 6,00 (д, J=3,3 Гц, 2H), 6,72 (д, J=8,0 Гц, 1H), 6,87 (д, J=8,0 Гц, 1H), 7,00-7,11 (ряд м, 3H), 7,30 (д, J=8,0 Гц, 1H), 7,46 (д, J=7,7 Гц, 1H), 7,74 (ушир.с, 3H), 11,06 (с, 1H).
С. 1-(3,4-Метилендиоксифенил)-2-[4-фенилтиазол-2-ил]-2,3,4,9-тетрагидро-1Н-β-карболин (№170)
К раствору тиомочевины из части В (227 мг, 0,65 ммоль) добавляли β-бромацетофенон (159 мг, 0,80 ммоль) и триэтиламин (0,40 мл). Данную смесь нагревали при 70°С в течение 3 часов, охлаждали до комнатной температуры и концентрировали на роторном испарителе. Остаток очищали флэш-хроматографией (SiO2, 0-10% метанол в дихлорметане), получая бледно-желтое твердое вещество.
Масс-спектр (m/z): 452 (MH+);
1H-ЯМР (CDCl3) δ: 2,87-2-3,06 (ряд м, 2H), 3,63-3,73 (м, 1H), 3,93-3,99 (м, 1H), 5,99 (д, J=3,3 Гц, 2H), 6,59 (с, 1H), 6,81-7,11 (ряд м, 5H), 7,25-7,69 (ряд м, 6H), 7,89 (д, J=7,4 Гц, 2H), 10,95 (с, 1H).
Пример 8
1-(2,3-Дигидробензофуран-5-ил)-2-[5-(2,3-диметил-3Н-имидазол-4-ил)пиримидин-2-ил]-2,3,4,9-тетрагидро-1Н-β-карболин (№190)
2-(5-Бром-2-пиримидинил)-1-(2,3-дигидро-5-бензофуранил)-2,3,4,9-тетрагидро-1Н-β-карболин (0,45 г, 1,00 ммоль), 1,2-диметил-1Н-имидазол (0,18 г, 1,87 ммоль), Pd(OAc)2 (12 мг, 0,05 ммоль), PPh3 (26 мг, 0,1 ммоль) и К2СО3 (0,28 г, 2 ммоль) перемешивали в 3,5 мл ДМФ при 140°С в течение 14 часов. Смесь выливали в водный 10%-ный раствор NaOH (50 мл). Полученный раствор экстрагировали СН2Cl2 (3×50 мл) и сушили над Na2SO4. Очистка препаративной ТСХ давала указанный в заголовке продукт в виде желтого порошка.
1H-ЯМР 300 МГц (CDCl3) δ: 2,21 (с, 3H), 2,35 (с, 3H), 2,90 (м, 2H), 3,10 (т, 2H, J=8,8 Гц), 3,35 (м, 1H), 4,52 (т, 2H, J=8,8 Гц), 4,91 (м, 1H), 6,68-7,61 (м, 10H).
Масс-спектр (m/z): 463 (MH+), 461 (MH-).
Пример 9
2-[2,3']Бипиридинил-6'-ил-1-(2,3-дигидробензофуран-5-ил)-2,3,4,9-тетрагидро-1Н-β-карболин (№191)
А: 2-(5-Бромпиридин-2-ил)-1-(2,3-дигидробензофуран-5-ил)-2,3,4,9-тетрагидро-1Н-β-карболин
1-(2,3-Дигидро-5-бензофуранил)-2,3,4,9-тетрагидро-1Н-β-карболин (11,6 г, 40 ммоль), 2,5-дибромпиридин (10,42 г, 44 ммоль), Pd2dba3 (1,465 г, 1,6 ммоль), dppp (1,32 г, 3,2 ммоль) и NaOt-Bu (5,38 г, 56 ммоль) перемешивали в 60 мл ДМФ при 80°С в течение 3 дней. Реакционную смесь фильтровали через слой целита с использованием CH2Cl2. Затем реакционную смесь концентрировали, неочищенную смесь загружали на колонку Foxy (110 г силикагеля) и элюировали смесью этилацетат/гексан (3:7). Продукт кристаллизовался в пробирках. Продукт концентрировали, а затем перекристаллизовывали из ТГФ, получая продукт в виде желтых кристаллов.
1H-ЯМР 400 МГц (ТГФ-d8) δ: 0,91 (м, 1H), 1,15 (м, 1H), 1,25 (т, 2H, J=9,5 Гц), 1,60 (м, 1H), 2,31 (м, 1H), 2,60 (т, 2H, J=9,5 Гц), 4,75 (д, 1H, J=7,6 Гц), 5,02 (д, 1H, J=7,6 Гц), 5,10-5,28 (м, 4H), 5,380 (м, 2H), 5,58 (м, 1H), 5,72 (м, 1H), 6,28 (с, 1H), 8,12 (с, 1H).
Масс-спектр (m/z): 446, 448 (MH+), 444, 446 (MH-).
В: 2-[2,3']Бипиридинил-6'-ил-1-(2,3-дигидробензофуран-5-ил)-2,3,4,9-тетрагидро-1Н-β-карболин
Продукт с указанной выше стадии А (0,4 г, 0,896 ммоль), 2-трибутилстаннанилпиридин (0,8 г, 2,17 ммоль) и Pd(PPh3)4 (0,12 г, 0,104 ммоль) перемешивали в 1,4-диоксане (5 мл) при 88°С в течение 24 часов. Реакционную смесь фильтровали через слой целита с использованием CH2Cl2, а затем концентрировали до небольшого объема. Препаративная ТСХ (3:7 этилацетат/гексан; затем 5% CH3OH/CH2Cl2) давала продукт в виде желтого твердого вещества.
1H-ЯМР (CDCl3) δ: 2,82 (м, 1H), 3,10 (м, 3H), 3,58 (м, 1H), 4,31 (м, 1H), 4,53 (т, 2H, J=9,5 Гц), 6,71 (д, 1H, J=7,6 Гц), 6,85 (д, 1H, J=7,6 Гц).
Масс-спектр (m/z): 445 (MH+), 443 (MH-).
В соответствии с описанными выше методиками были получены соединения, перечисленные в таблицах 1-6.
диоксифенил
аминоэтил
диоксифенил
диоксифенил
аминоэтил
диоксифенил
аминоэтил
диоксифенил
фенил)
аминоэтил
диоксифенил
фенил)
диоксифенил
фенил)
диоксифенил
сульфонил)фенил
диоксифенил
фенил)
диоксифенил
диоксифенил
фенил)
диоксифенил
диоксифенил
диоксифенил
диоксифенил
сульфонил
фенил)
диоксифенил
диоксифенил
диоксифенил
диоксифенил
фенил)
диоксифенил
диоксифенил
фенил)
диоксифенил
диоксифенил
диоксифенил
фенил)
диоксифенил
фенил)
диоксифенил
фенил)
фенил)
диоксифенил
фенил)
амино)
этокси)
фенил)
диоксифенил
[1,4]диоксин-6-ил
[1,4]диоксин-6-ил
фенил)
диоксифенил
диокси
фенил
метилфенил)
диокси
фенил
метилфенил)
аминоэтил
диокси
фенил
фурил
диокси
фенил
фурил
диокси
фенил
фурил
диоксифенил
фурил
аминоэтил
диоксифенил
тиенил
диокси
фенил
тиенил
фенил)-5-фурил
карбонил
метилен
диокси)фенил
метил
фенил)-5-фурил
карбонил
метилен
диокси)
фенил
метилфенил)
метилен
диокси)
фенил
метилен
диокси)
фенил
метилфенил)
карбонил
этил
диокси
фенил
метилфенил)
карбонил
метил
диокси
фенил
карбонил
метил
диокси
фенил
метилфенил)
метил
диокси
фенил
метилфенил)
диокси
фенил
метилфенил)
этил
диокси
фенил
метилфенил)
этил
диокси
фенил
метилфенил)
метил
диокси
фенил
метил
диокси
фенил
метилфенил)
аминоэтил
диокси
фенил
метилфенил)
аминобутил
диоксифенил
метилфенил)
диокси
фенил
метилфенил)
аминоэтил
диокси
фенил
аминоэтил
диокси
фенил
аминоэтил
диоксифенил
аминоэтил
диоксифенил
аминоэтил
диоксифенил
аминоэтил
диоксифенил
аминоэтил
диоксифенил
аминоэтил
диоксифенил
фенил)
амино
пропил
диоксифенил
фенил)
метил
диоксифенил
фенил)
динилэтил
диоксифенил
фенил)
диоксифенил
фенил)
диоксифенил
фенил)
диоксифенил
диоксифенил
диоксифенил
аминофенил)
диоксифенил
фенил)
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
диоксифенил
фенил
метил-4-хлорфенил
фенил
фенил
фенил
карбонил фенил
фенил
фенил
фенил
фенил
фенил
фенил
фенил
фенил
фенил
фенил
фенил
фенил
фенил
карбонил
фенил
карбонил
фенил
карбонил
Пример 10
Исследование in vitro
Анализ циклической нуклеотидфосфодиэстеразы (PDE)
Выделение PDEV
PDEV выделяли из тканей кролика и человека в соответствии с протоколом, описанным Boolell et al. (Boolell М., Allen М.J., Ballard С.A., Geo-Attee С., Muirhead G.J., Naylor A.М., Osterloh I.H., and Gingell C.) в International Journal of Impotence Research 1996, 8, 47-52, с незначительными модификациями.
Кратко, ткани кролика или человека гомогенизировали в охлаждаемом льдом буферном растворе, содержащем 20 мМ HEPES (рН 7,2), 0,25 М сахарозы, 1 мМ EDTA и 1 мМ фенилметилсульфонилфторида (PMSF). Гомогенизат центрифугировали при 100000g в течение 60 минут при 4°С. Супернатант фильтровали через 0,2 мкМ фильтр и загружали в Pharmacia Mono Q анионообменную колонку (объем слоя 1 мл), которую уравновешивали 20 мМ HEPES, 1 мМ EDTA и 0,5 мМ PMSF. После вымывания несвязанных белков ферменты элюировали с использованием линейного градиента 100-600 мМ NaCl в том же буфере (от 30 до 50 мл в сумме, в зависимости от ткани). Ферменты мышц скелета, пещеристого тела, сетчатки, сердца и тромбоцитов элюировали 35, 40, 45, 50 и 50 мл соответственно. Колонку обрабатывали при скорости потока 1 мл/мин и собирали фракции объемом 1 мл. Фракции, содержащие PDE различной активности, собирали по отдельности и использовали в последующих исследованиях.
Измерение ингибирования PDEV
PDE анализ проводили так, как описано Thompson и Appleman в Biochemistry 1971, 10, 311-316, с незначительными модификациями, как указано ниже.
Анализы были приспособлены к 96-луночному планшету. Фермент анализировали в 5 мМ MgCl2, 15 мМ Tris-HCl (pH 7,4), 0,5 мг/мл бычьего сывороточного альбумина, 1 мМ цГМФ или цАМФ, 0,1 мкКи [3H]-цГМФ или [3H]-цАМФ и 2-10 мкл элюента для колонки. Суммарный объем для анализа составлял 100 мкл. Реакционную смесь инкубировали при 30°С в течение 30 минут. Реакцию останавливали посредством кипячения в течение 1 минуты и затем охлаждали на льду. Полученные [3H] 5'-мононуклеотиды дополнительно преобразовывали в незаряженные [3H]-нуклеозиды добавлением 25 мкл 1 мг/мл змеиного яда (Ophiophagus Hannah) и инкубированием при 30°С в течение 10 минут. Реакцию останавливали добавлением 1 мл Bio-Rad AG1-X2 суспензии смолы (1:3). Все заряженные нуклеотиды связывались смолой и только незаряженные [3H]-нуклеозиды оставались в супернатанте после центрифугирования. Отбирали аликвоту в 200 мкл и обсчитывали с использованием жидкостной сцинтилляции. PDE активность выражали как часть моля гидролизованного циклического нуклеотида/мин/мл ферментативного препарата.
Исследования ингибирования проводили в буфере для анализа при 10%-ной конечной концентрации ДМСО. В данных условиях гидролиз продукта увеличивался со временем и ростом концентрации фермента в линейной зависимости.
Пример 11
In Vitro определение Ki ингибиторов фосфодиэстеразы
Анализ адаптировали для 96-луночного планшета. Фосфодиэстеразу анализировали в присутствии 5 мМ MgCl2, 15 мМ Tris-HCl (pH 7,4), 0,5 мг/мл бычьего сывороточного альбумина, 30 нМ [3H]-цГМФ и исследуемого соединения в различных концентрациях. Количество используемого фермента для каждой реакции было таким, что менее 15% первоначального субстрата конвертировалось за время анализа. Для всех измерений исследуемое соединение растворяли и разбавляли в 100% ДМСО (2% ДМСО в анализе). Общий анализируемый объем составлял 100 мкл. Реакционную смесь инкубировали при 30°С в течение 90 минут. Реакцию останавливали кипячением в течение 1 минуты, а затем немедленно охлаждали, перенося на ледяную баню. В каждую лунку затем добавляли 25 мкл 1 мг/мл змеиного яда (Ophiophagus Hannah) и реакционную смесь инкубировали при 30°С в течение 10 минут. Реакцию останавливали добавлением 1 мл суспензии смолы Bio-Rad AG1-X2 (1:3). Отбирали аликвоту в 200 мкл и обсчитывали с использованием жидкостной сцинтилляции.
% Ингибирования максимального превращения субстрата (ферментом в отсутствие ингибитора) рассчитывали для каждой концентрации исследуемого соединения. Для определения IC50 строили график % ингибирования относительно log концентрации исследуемого соединения с использованием GraphPad Prism нелинейного регрессионного анализа (сигмоидальная кривая доза-ответ). В условиях, когда концентрация субстрата ≪Кm фермента (Кm - концентрация субстрата, при которой достигается половина максимальной скорости фермента), Кi эквивалентен значению IC50.
Данные масс-спектров и PDEV ингибирующей активности для иллюстративных соединений по настоящему изобретению описаны в таблицах 6 и 7. Данные ингибирования представлены либо как IC50(мкМ), как процент ингибирования при данной концентрации тестируемого соединения, либо как значение Ki.
Пример 12
Исследование in vivo
В соответствии с методикой, описанной Carter et al., (Carter et al., Ballard С.A. and Naylar A.М.) в Journal of Urology 1998, 160, 242-246, была исследована эффективность in vivo соединений настоящего изобретения.
Пример 13
В качестве конкретного варианта осуществления пероральной композиции 100 мг соединения примера 7 вводили в состав лекарственного средства вместе с достаточно мелко измельченной лактозой, получая общее количество 580-590 мг для заполнения твердых желатиновых капсул 0 размера.
Хотя представленное выше описание вместе с примерами, приведенными в целях иллюстрации, указывает принципы настоящего изобретения, следует понимать, что практическое осуществление изобретения охватывает все обычные варианты, адаптированные и/или модифицированные, проявляющиеся в объеме следующей формулы изобретения и ее эквивалентах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО ПИРРОЛОПИРИДИНОНА, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2001 |
|
RU2267490C2 |
| БЕТА-КАРБОЛИНОВЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБЫ СВЯЗЫВАНИЯ, ДОСТИЖЕНИЯ АГОНИСТИЧЕСКОГО/АНТАГОНИСТИЧЕСКОГО ЭФФЕКТА | 1999 |
|
RU2233841C2 |
| ИСПОЛЬЗОВАНИЕ ИНГИБИТОРОВ СGМР-ФОСФОДИЭСТЕРАЗЫ ДЛЯ ЛЕЧЕНИЯ ИМПОТЕНЦИИ | 1996 |
|
RU2181288C2 |
| ПРОИЗВОДНЫЕ β-КАРБОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2210571C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1- И 1,1-ДИЗАМЕЩЕННЫХ-4-ФЕНИЛ-2,3,4,9-ТЕТРАГИДРО-1H-БЕТТА-КАРБОЛИНОВ | 2004 |
|
RU2332418C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНОХИНОЛИНА | 1994 |
|
RU2168511C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2259202C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОЛОЖИТЕЛЬНЫХ МОДУЛЯТОРОВ МЕТАБОТРОПНОГО ГЛУТАМАТНОГО РЕЦЕПТОРА 2 (РЕЦЕПТОРА MGLU2) | 2008 |
|
RU2479577C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ | 2001 |
|
RU2286343C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2278863C2 |
Настоящее изобретение относится к новым производным β-карболина общей формулы (I), обладающим свойствами ингибитора действия фосфодиэстеразы V(PDEV). Соединения могут найти применение при лечении заболеваний и болезненных состояний, относящихся к PDE, например, эректильной дисфункции у мужчин. В общей формуле (I)

R1 означает водород; n равен 0; Х выбирают из группы, состоящей из О, S и NRD; R2 выбирают из группы: фенила (необязательно замещенного 1-3 RB), 6-членного азотсодержащего гетероарила и 5-6-членного гетероциклоалкила, содержащего 1-2-атома кислорода, и конденсированного с бензольным кольцом (необязательно замещенного 1-3 RB); R4 выбирают из группы, состоящей из водорода, карбокси, С1-С6алкилкарбонила, ди(С1-С8алкил)аминоалкоксикарбонила, ди(С1-С8алкил)аминоС1-С8алкиламинокарбонила; а равно целому числу от 0 до 1; Y выбирают из группы, состоящей из СН2, С(О); Z выбирают из группы, состоящей из СН2, СНОН и С(О); при условии, что когда Z представляет СНОН или С(О), то Х представляет NH; выбирают из группы, состоящей из нафтила, 5-6-членного гетероарила, содержащего 1-3 гетероатома, выбираемых из азота, кислорода и/или серы, возможно конденсированного с бензольным кольцом; m равно целому числу от 0 до 2; R3 независимо выбирают из группы, состоящей из галогена, нитро, С1-С8алкила, С1-С8алкокси, трифторметила, фенила (необязательно замещенного 1-3 RB), фенилсульфонила, нафтила, С1-С8аралкила, 5-6-членного гетероарила, содержащего 1-3 атома азота в цикле (необязательно замещенного 1-3 RB). Изобретение также относится к фармацевтической композиции, способу ее получения и способам ингибирования действия фосфодиэстеразы V(PDEV), а также увеличения концентрации цГМФ. 7 н. и 7 з.п. ф-лы, 7 табл.
выбирают из группы, состоящей из нафтила, 5-6-членного гетероарила, содержащего 1-3 гетероатома, выбираемых из азота, кислорода и/или серы, возможно конденсированного с бензольным кольцом; m равно целому числу от 0 до 2; R3 независимо выбирают из группы, состоящей из галогена, нитро, С1-С8алкила, С1-С8алкокси, трифторметила, фенила (необязательно замещенного 1-3 RB), фенилсульфонила, нафтила, С1-С8аралкила, 5-6-членного гетероарила, содержащего 1-3 атома азота в цикле (необязательно замещенного 1-3 RB). Изобретение также относится к фармацевтической композиции, способу ее получения и способам ингибирования действия фосфодиэстеразы V(PDEV), а также увеличения концентрации цГМФ. 7 н. и 7 з.п. ф-лы, 7 табл.
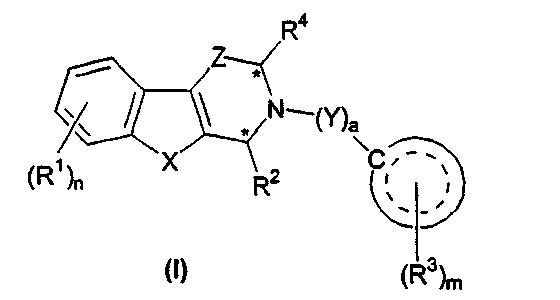
где
R1 означает водород;
n равен 0;
Х выбирают из группы, состоящей из О, S и NRD;
R2 выбирают из группы фенила (необязательно замещенного 1-3 RB), 6-членного азотсодержащего гетероарила и 5-6-членного гетероциклоалкила, содержащего 1-2-атома кислорода, и конденсированного с бензольным кольцом (необязательно замещенного 1-3 RB);
R4 выбирают из группы, состоящей из водорода, карбокси, С1-С6алкилкарбонила,ди(С1-С8алкил)аминоалкоксикарбонила, ди(С1-С8алкил)аминоС1-С8алкиламинокарбонила;
а равно целому числу от 0 до 1;
Y выбирают из группы, состоящей из СН2, С(О);
Z выбирают из группы, состоящей из СН2, СНОН и С(О); при условии, что когда Z представляет СНОН или С(О), то Х представляет NH;
 выбирают из группы, состоящей из нафтила, 5-6-членного гетероарила,содержащего 1-3 гетероатома, выбираемых из азота, кислорода и/или серы, возможно конденсированного с бензольным кольцом;
выбирают из группы, состоящей из нафтила, 5-6-членного гетероарила,содержащего 1-3 гетероатома, выбираемых из азота, кислорода и/или серы, возможно конденсированного с бензольным кольцом;
m равно целому числу от 0 до 2;
R3 независимо выбирают из группы, состоящей из галогена, нитро, С1-С8алкила, С1-С8алкокси, трифторметила, фенила (необязательно замещенного 1-3 RB), фенилсульфонила, нафтила, С1-С8аралкила, 5-6-членного гетероарила,содержащего 1-3 атома азота в цикле (необязательно замещенного 1-3 RB);
где каждый RВ выбирают независимо из группы, состоящей из галогена, гидрокси, нитро, циано, С1-С8алкила, С1-С8алкокси, С1-С8алкоксикарбонила, карбоксиС1-С8алкила, С1-С8алкилсульфонила, трифторметила, амино, ацетиламино, ди(С1-С8алкил)амино, ди(С1-С8алкил)аминоС1-С8алкокси, ди(С1-С8алкил)аминоацетилС1-С8алкила, ди(С1-С8алкил)аминоацетиламино, карбоксиС1-С8алкилкарбониламино, гидроксиС1-С8алкиламино;
где RD выбирают из группы, состоящей из водорода, ди(С1-С4алкил)аминоC1-C6алкила,5-6-членного гетероарила, 5-6-членного гетероарилC1-C4алкила, 5-6-членного гетероциклоалкилC1-C4алкила, (причем, в каждом из трех последних значений гетероарил или гетероциклоалкил содержит 1-2-гетероатома, выбираемых из азота и/или кислорода, и может быть конденсированным с бензольным кольцом), карбоксиC1-C4алкила, С1-С8алкоксикарбонилC1-C4алкила, и 5-6-членного кислородсодержащего гетероарилкарбонила (где гетероарил необязательно может быть замещен фенилом или замещенным фенилом, где заместители фенила представляют собой один-три RB);
где каждый RВ выбирают независимо из группы, состоящей из галогена, гидрокси, нитро, циано, С1-С8алкила, С1-С8алкокси, С1-С8алкоксикарбонила, карбоксиС1-С8алкила, С1-С8алкилсульфонила, трифторметила, амино, ацетиламино, ди(С1-С8алкил)амино, ди(С1-С8алкил)аминоС1-С8алкокси, ди(С1-С8алкил)аминоацетилС1-С8алкила, ди(С1-С8алкил)аминоацетиламино, карбоксиС1-С8алкилкарбониламино, гидроксиС1-С8алкиламино;
при условии, что если представляет 2-фурил или 2-тиенил, то m равно целому числу от 1 до 2;
и его фармацевтически приемлемые соли.
Х выбирают из S или NRD, где RD выбирают из группы, состоящей из водорода, ди(С1-С4алкил)аминоC1-C6алкила,5-6-членного гетероарила, 5-6-членного гетероарилC1-C4алкила, 5-6-членного гетероциклоалкилC1-C4алкила, (причем, в каждом из трех последних значений гетероарил или гетероциклоалкил содержит 1-2-гетероатома, выбираемых из азота и/или кислорода, и может быть конденсированным с бензольным кольцом), карбоксиC1-C4алкила, С1-С8алкоксикарбонилC1-C4алкила, и 5-6-членного кислородсодержащего гетероарилкарбонила, где гетероарил необязательно может быть замещен фенилом или замещенным фенилом, где заместители фенила представляют собой один-три RB;
где каждый RB выбирают независимо из группы, состоящей из галогена, нитро, С1-С4алкила, С1-С4алкокси, трифторметила, амино и ди(С1-С4алкил)амино;
R2 выбирают из группы, состоящей из 3,4-метилендиоксифенила, 3,4-(дифтор)метилендиоксифенила, 2,3-дигидробензофурила, 2,3-дигидробензо[1,4]диоксин-6-ила, пиридила, фенила и замещенного фенила, где фенильные заместители представляют собой один-два заместителя, независимо выбранных из галогена, С1-С4алкила, С1-С4алкокси, трифторметила, циано, нитро, С1-С4алкоксикарбонила, ди(С1-С4алкил)амино или ди(С1-С4алкил)аминоС1-С4алкокси;
R4 выбирают из группы, состоящей из водорода, карбокси, С1-С4алкоксикарбонила, ди(С1-С4алкил)аминоС1-С4алкоксикарбонила и ди(С1-С4алкил)аминоС1-С4алкиламинокарбонила;
Y выбирают из группы, состоящей из СН2,С(О);
выбирают из группы, состоящей из нафтила и 5-6-членного гетероарила, содержащего 1-3 гетероатома, выбираемых из азота, кислорода и/или серы, возможно конденсированного с бензольным кольцом;
R3 независимо выбирают из группы, состоящей из галогена, нитро, С1-С4алкила, С1-С4алкокси, трифторметила, С1-С4аралкила, пиразинила, пиридила, замещенного галогеном пиридила, диметилзамещенного имидазолила, фенила, фенилсульфонила и замещенного фенила; где заместители в фениле представляют собой один или более заместителей, независимо выбранных из галогена, гидрокси, С1-С4алкила, С1-С4алкокси, трифторметила, трифторметокси, нитро, амино, ацетиламино,
С1-С4алкилсульфонила, карбоксиС1-С4алкилкарбониламино, гидроксиС1-С4алкиламино, ди(С1-С4алкил)аминоС1-С4алкокси, ди(С1-С4алкил)аминоацетиламино или гетероциклоалкилС1-С4алкокси;
при условии, что когда представляет 2-фурил или 2-тиенил, тогда m равно целому числу 1 или 2;
и его фармацевтически приемлемые соли.
Х выбирают из S или NRD, где RD выбирают из группы, состоящей из водорода, ди(метил)аминоэтила, ди(метил)амино-н-пропила, ди(этил)аминоэтила, ди(этил)амино-н-бутила, N-пирролидинилэтила, N-морфолинилэтила, 2-пиридилметила, 4-пиридилметила, 5-(4-метилфенил)-2-пиримидинила, карбоксиметила, карбоксиэтила, 2-(5-(3-трифторметилфенил)фурил)карбонила, 2-(5-(3-нитрофенил)фурил)карбонила,метоксикарбонилметила, метоксикарбонилэтила и 2-бензоксазолила;
R2 выбирают из группы, состоящей из фенила, 3,4-метилендиоксифенила, 3,4-(дифтор)метилендиоксифенила, 2,3-дигидробензофурила, 2,3-дигидробензо[1,4]диоксин-6-ила, 4-пиридила, 3-пиридила, 4-цианофенила, 3-нитрофенила, 4-нитрофенила, 4-трифторметилфенила, 4-метоксифенила, 3,4-диметилфенила, 3,5-диметилфенила, 3,4-диметоксифенила, 3-трифторметил-4-хлорфенила, 3,4-дихлорфенила, 4-хлорфенила, 4-метоксикарбонилфенила, 3,4-диметоксифенила, 4-(диметиламино)фенила и 4-(N-(3-диметиламино)-н-пропокси)фенила;
R4 выбирают из группы, состоящей из водорода, карбокси, диметиламиноэтоксикарбонила, диметиламиноэтиламинокарбонила и метоксикарбонила;
выбирают из группы, состоящей из нафтила, 2-пиримидинила, 2-фурила, 3-фурила, 2-бензофурила, 2-тиенила, 2-бензотиенила, 2-бензотиазолила, 2-бензоксазолила, 2-бензимидазолила, 4-тиазолила, 2-тиазолила, 3-пиразолила, 4-пиразолила, 5-пиразолила, 3-(1,2,5-триазолила), 4-изоксазолила, 2-пиридила и 3-пиридила;
R3 независимо выбирают из группы, состоящей их хлора, брома, метила, н-пропила, трет-бутила, метокси, трифторметила, нитро, фенила, бензила, фенилсульфонила, 4-гидроксифенила, 4-хлорфенила, 4-метилфенила, 3,4-диметоксифенила, 3-трифторметилфенила, 4-трифторметилфенила, 5-трифторметилфенила, 4-метоксифенила, 2-нитрофенила, 3-нитрофенила, 4-нитрофенила, 3-аминофенила, 4-аминофенила, 2-нитро-4-хлорфенила, 2-нитро-4-метилфенила, 2-нитро-4-метилсульфонилфенила, 3-ацетиламинофенила, 4-ацетиламинофенила, 4-(3-карбокси-н-пропил)карбониламинофенила, 2-хлор-5-трифторметилфенила, 4-(4-гидрокси-н-бутил)аминофенила, 2-(диметиламино)ацетиламинофенила,4-[2-N-пирролидинил)этокси]фенила, 4-[2-(4-морфолинил)этокси]фенила, 4-(2-диметиламино)этокси)фенила, 4-пиразинила, 2,3-диметил-3Н-имидазолила, 2-пиридила и 3-пиридила;
при условии, что когда представляет 2-фурил или 2-тиенил, тогда m равно целому числу 1 или 2;
и его фармацевтически приемлемые соли.
Х представляет NRD, где RD выбирают из группы, состоящей из водорода, ди(метил)аминоэтила, 4-пиридилметила, 2-пиридилметила, N-морфолинилэтила, карбоксиэтила, карбоксиметила, ди(этил)аминоэтила, N-пирролидинилэтила и 5-(4-метилфенил)-2-пиримидинила;
R2 выбирают из группы, состоящей из 3,4-метилендиоксифенила, 2,3-дигидроксибензофурила и 2,3-дигидробензо[1,4]диоксин-6-ила;
R4 представляет водород;
Y выбирают из группы, состоящей из С(О) и СН2;
выбирают из группы, состоящей из нафтила, 2-пиримидинила, 2-фурила, 2-бензофурила, 2-тиенила, 2-бензотиенила, 2-бензотиазолила, 2-бензоксазолила, 2-тиазолила, 4-тиазолила и 2-пиридила;
m равно целому числу от 0 до 1;
R3 выбирают из группы, состоящей из брома, трет-бутила, метокси, трифторметила, нитро, фенила, 4-хлорфенила, 3,4-диметоксифенила, 3-трифторметилфенила, 4-метилфенила, 4-метоксифенила, 2-нитрофенила, 3-нитрофенила, 4-нитрофенила, 3-аминофенила, 2-нитро-4-хлорфенила, 2-нитро-4-метилфенила, 2-нитро-4-метилсульфонилфенила,4-(3-карбокси-н-пропил)карбониламинофенила, 2-хлор-5-трифторметилфенила, 4-(4-гидрокси-н-бутил)аминофенила, 2,2-(диметиламино)ацетиламинофенила, 4-пиразинила, 2-пиридила и 2,3-диметил-3Н-имидазол-4-ила;
при условии, что когда представляет 2-фурил или 2-тиенил, тогда m равно 1;
и его фармацевтически приемлемые соли.
Х представляет NRD, где RD выбирают из группы, состоящей из водорода, ди(метил)аминоэтила, N-морфолинилэтила, карбоксиметила и N-пирролидинилэтила;
R2 выбирают из группы, состоящей из 3,4-метилендиоксифенила и 2,3-дигидроксибензофурила;
Z выбирают из группы, состоящей из СН2 и С(О); при условии, что когда Z представляет С(О), тогда Х представляет NH;
Y представляет С(О);
выбирают из группы, состоящей из 2-пиримидинила, 2-фурила, 2-бензофурила, 2-бензоксазолила, 2-тиазолила и 2-пиридила;
R3 выбирают из группы, состоящей из трет-бутила, метокси, нитро, фенила, 4-хлорфенила, 4-метилфенила, 4-метоксифенила, 3,4-диметоксифенила, 3-трифторметилфенила, 2-нитрофенила, 3-нитрофенила, 4-нитрофенила, 3-аминофенила, 2-нитро-4-метилсульфонилфенила, 2-(диметиламино)ацетиламинофенила, 2-пиридила и 2,3-диметил-3Н-имидазол-4-ила;
при условии, что когда представляет 2-фурил, тогда m равно 1;
и его фармацевтически приемлемые соли.
1-(3,4-метилендиоксифенил)-2-[(5-фенил-2-фурил)карбонил]-2,3,4,9-тетрагидро-1Н-β-карболин;
1-(3,4-метилендиоксифенил)-2-[5-(2-пиридил)-2-пиримидинил]-9-ди(метил)аминоэтил-2,3,4,9-тетрагидро-1Н-β-карболин;
1-(3,4-метилендиоксифенил)-2-[5-(3,4-диметоксифенил)-2-пиримидинил]-1,2,3,9-тетрагидро-4-оксо-4Н-β-карболин;
1-(3,4-метилендиоксифенил)-2-[5-(4-метилфенил)-2-пиримидинил]-1,2,3,9-тетрагидро-4-оксо-4Н-β-карболин;
1-(3,4-метилендиоксифенил)-2-[5-(4-метоксифенил)-2-пиримидинил]-1,2,3,4-тетрагидро-4-оксо-4Н-β-карболин;
1-(3,4-метилендиоксифенил)-2-[4-(4-метоксифенил)-2-тиазолил]-2,3,4,9-тетрагидро-1Н-β-карболин;
1-(3,4-метилендиоксифенил)-2-(4-фенил-2-тиазолил)-2,3,4,9-тетрагидро-1Н-β-карболин;
2-[2,3']бипиридинил-6'-ил-1-(2,3-дигидробензофуран-5-ил)-2,3,4,9-тетрагидро-1Н-β-карболин;
1-(2,3-дигидробензофуран-5-ил)-2-[5-(2,3-диметил-3Н-имидазол-4-ил)-2,3,4,9-тетрагидро-1Н-β-карболин; и их фармацевтически приемлемые соли.
| С.А.ПОГОСЯН и др.Химико-фармацевтический журнал | |||
| - М.: Медицина, 1987 | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| Шеститрубный элемент пароперегревателя для котлов с жаровыми и прогарными трубами | 1918 |
|
SU678A1 |

