Изобретение относится к области очистки ароматических соединений, более конкретно к способу гидрирования бензола, смесей бензола и толуола, смесей бензола и ксилола или изомерной смеси ксилола или смесей бензола, толуола и ксилола или изомерной смеси ксилола, содержащих сернистые ароматические соединения, и к способу их десульфирования.
Сильно экзотермическая реакция гидрирования требует тщательного контроля за температурой и временем контакта, чтобы достичь полноты превращения при высокой селективности. В особенности следует подавлять образование значительных количеств метилциклопентана, которое предпочтительно протекает при повышенных температурах. Типичные спецификации циклогексана требуют, чтобы остаточное содержание бензола составляло <100 ч. на млн и метилциклопентана <200 ч. на млн. Равным образом критичным является также содержание н-парафинов (как, например, н-пентан, н-гексан). Эти нежелательные соединения также возникают предпочтительно при повышенных температурах гидрирования, и так же, как метилциклопентан, их можно отделить от желательного циклогексана только с помощью трудоемких операций по разделению (как, например, экстракция, ректификация или применение молекулярных сит, как описано в патенте Великобритании GB 1341057). Используемый катализатор также оказывает сильное влияние на масштаб образования нежелательных побочных продуктов, как метилциклопентан, н-гексан, н-пентан и т.д.
На этом фоне стоит добиваться того, чтобы проводить гидрирование при возможно более низких температурах. Это, однако, с другой стороны, ограничивается тем, что в зависимости от характера используемого катализатора гидрогенизации, только начиная с повышенных температур, достигается необходимая активность катализатора гидрирования, которая, со своей стороны, достаточна, чтобы получать рентабельный выход.
Никелевые и платиновые катализаторы, применяемые для гидрирования бензола, обнаруживают ряд недостатков. Никелевые катализаторы очень чувствительны к серосодержащим примесям в бензоле, так что приходится или использовать для гидрирования очень чистый бензол, или, как описано в патенте Великобритании GB 1104275, применять в основном реакторе платиновый катализатор, который работает при повышенном содержании серы и таким образом защищает реактор окончательного гидрирования, содержащий никелевый катализатор. Другая возможность состоит в том, чтобы катализатор гидрирования легировать рением, как описано в патенте Великобритании GB 1155539, или в катализатор гидрирования вводить ионит, как сообщается в патенте Великобритании GB 1144499. Получение таких катализаторов, однако, является трудоемким и дорогостоящим.
Платиновые катализаторы проявляют меньше недостатков, чем никелевые катализаторы, но они очень дороги.
В качестве альтернативы поэтому в литературе недавнего времени указывают на катализаторы гидрирования бензола в циклогексан, содержащие рутений.
В патенте SU 319582 описываются суспензионные рутениевые катализаторы для получения циклогексана из бензола, легированные палладием, платиной или родием. Однако, ввиду использования палладия, платины или рутения они очень дороги, и к тому же в случае суспензионных катализаторов приготовление и регенерация катализатора как трудоемки, так и дороги.
В патенте США US 3917540 для получения циклогексана из бензола описываются катализаторы, нанесенные на Al2O3. В качестве активного металла они содержат благородный металл из VIII побочной подгруппы периодической системы, а также щелочной металл, технеций или рений. Далее в патенте США US 3244644 описаны рутениевые катализаторы гидрирования, нанесенные на η-Al2O3, которые также должны подходить для гидрирования бензола. Эти катализаторы, однако, содержат по меньшей мере 5% активного металла. Кроме того, получение модификации η-Al2O3 - процесс как трудоемкий, так и дорогой.
Далее в международном патенте WO 00/63142 описывается, в частности, гидрирование незамещенных ароматических соединений с применением катализатора, который в качестве активного металла содержит по меньшей мере один металл VIII побочной подгруппы периодической системы и который нанесен на основу с макропористой структурой. В качестве активного металла особенно подходит рутений, а в качестве носителей наиболее подходящими являются окись алюминия и диоксид циркония.
Преимущество этих способов заключается в сравнительно выгодной цене рутения, применяемого в катализаторе в качестве активного металла, по сравнению с затратами, которые возникают при использовании в катализаторах гидрирования других металлов, а именно палладия, платины или родия. Однако и здесь неблагоприятным является тот факт, что эти рутениевые катализаторы чувствительны к сернистым примесям.
Из европейского патента ЕР 600406 известно, что ненасыщенные углеводороды, а именно алкены (например, этилен), которые содержат тиофен в качестве примеси, можно десульфурировать, если их обработать водородом в количестве от 0,01 до 4 об.% в присутствии медно-цинкового десульфуризатора, с атомным отношением медь/цинк, приблизительно равным 1:~0,3-10. Особенно подчеркивается, что количество водорода не должно превышать приведенное значение, так как это ведет к нежелательному гидрированию подлежащих очистке ненасыщенных углеводородов.
В основу настоящего изобретения положена исходная задача разработать такой способ гидрирования ароматических углеводородов или их смесей, содержащих сернистые ароматические соединения, в соответствующие алициклические соединения или их смеси, в особенности бензола с получением циклогексана, который сделает возможным получать алициклические соединения или их смеси с очень высокой селективностью и выходом.
В соответствии с этим настоящее изобретение касается способа превращения ароматического углеводорода, содержащего сернистые ароматические соединения, или смеси ароматических углеводородов, содержащие сернистые ароматические соединения, причем на первой стадии удаляют серосодержащие ароматические соединения, при необходимости в присутствии водорода (стадия а); это десульфирование проводят в присутствии медно-цинкового десульфуризатора с атомным отношением медь:цинк от 1:0,3 до 1:10, который можно получать способом соосаждения. На второй стадии полученный таким образом ароматический углеводород или соответственно смесь углеводородов гидрируют в присутствии нанесенного рутениевого катализатора и водорода в соответствующее алициклическое соединение, или их смесь (стадия b), причем катализатор нанесен на основу с мезопористой и/или макропористой структурой.
В предпочтительном варианте исполнения в качестве ароматического углеводорода применяют бензол, который в присутствии водорода гидрируется в циклогексан.
В другом предпочтительном варианте исполнения применяют смесь ароматических углеводородов, которую в присутствии водорода гидрируют в соответствующую смесь алициклических соединений. В качестве смеси ароматических углеводородов при этом применяют такие, которые содержат бензол и толуол, или бензол и ксилол, или смесь изомерных ксилолов, или бензол, толуол и ксилол, или смесь изомерных ксилолов. При гидрировании из бензола получается циклогексан, из толуола - метилциклогексан и из ксилолов - соответствующие диметилциклогексаны.
На стадии а) ароматический углеводород или смесь ароматических углеводородов, который содержит примесь сернистых ароматических соединений, десульфурируют. Среди серосодержащих ароматических соединений имеют в виду особенно тиофен, бензотиофен, дибензотиофен или соответствующие алкильные производные, более всего - тиофен. Наряду с этими ароматическими серосодержащими соединениями в ароматическом углеводороде или смеси ароматических углеводородов могут присутствовать также другие серосодержащие примеси, например нижеследующие неароматические сернистые соединения, а именно сероводород, меркаптаны, как метилмеркаптан, тетрагидротиофен, дисульфиды, как диметилдисульфид, COS или CS2. Кроме того, могут содержаться и другие примеси, а именно вода, алканы с 5-7 атомами углерода, например н-гептан, алкены с 5-7 атомами углерода, например пентен или гексен, причем двойная связь может находиться в любом месте углеродного скелета; циклоалканы с 5-7 атомами углерода, например метилциклопентан, этилциклопентан, диметилциклопентан, циклогексан, метилциклогексан, или циклоалкены с 5-7 атомами углерода, например циклогексен.
Ароматический углеводород, который применяют в особом варианте исполнения, как правило, имеет чистоту >98 мас.%, особенно предпочтительно >99 мас.%, преимущественно >99,5 мас.%, наиболее предпочтительно >99,9 мас.%. Если применяется смесь ароматических углеводородов, то доля ароматических углеводородов в используемой смеси составляет >98 мас.%, особенно предпочтительно >99 мас.%, преимущественно >99,5 мас.%, наиболее предпочтительно >99,9 мас.%. В обоих случаях количество серосодержащих ароматических примесей составляет до 2 мас. ч. на млн, преимущественно до 1 мас. ч. на млн. Суммарное содержание сернистых примесей может составлять до 5 мас. ч. на млн, преимущественно до 3 мас. ч. на млн, наиболее предпочтительно до 2 мас. ч. на млн, в особых случаях до 1 мас. ч. на млн. Остальные примеси могут составлять до 2 мас.%, преимущественно до 0,5 мас.%, наиболее предпочтительно до 0,1 мас.%. Вода в ароматическом углеводороде или в соответствующих смесях ароматических углеводородов может содержаться в количестве до 0,1 мас.%, преимущественно до 0,07 мас.%, наиболее предпочтительно до 0,05 мас.%.
Десульфирование проводят на медно-цинковом десульфуризаторе, при необходимости в присутствии водорода. Этот медно-цинковый десульфуризатор содержит по меньшей мере медь и цинк, причем атомное отношение медь:цинк составляет от 1:0,3 до 1:10, преимущественно от 1:0,5 до 1:3 и наиболее предпочтительно от 1:0,7 до 1:1,5. Он получается способом соосаждения, и его можно применять как в окисленной, так и в восстановленной форме.
В особом варианте исполнения медно-цинковый десульфуризатор содержит по меньшей мере медь, цинк и алюминий, причем атомное отношение медь:цинк:алюминий составляет от 1:0,3:0,05 до 1:10:2, преимущественно от 1:0,6:0,3 до 1:3:1 и наиболее предпочтительно от 1:0,7: 0,5 до 1:1,5:0,9.
Десульфуризаторы можно получать различными способами. Например, можно смешать водный раствор, который содержит соединение меди, в частности водорастворимое, например нитрат или ацетат меди, и соединение цинка, в частности водорастворимое, например нитрат или ацетат цинка, с водным раствором соединения основного характера (например, карбоната натрия, карбоната калия), с образованием осадка (способ соосаждения). Образовавшийся осадок отфильтровывают, промывают водой или сначала промывают, затем фильтруют и после этого высушивают. Затем прокаливают при температуре приблизительно от 270 до 400°С. В заключение полученный твердый материал взмучивают в воде, отфильтровывают и высушивают. Полученный таким образом медно-цинковый десульфуризатор ("окисленная форма") можно применять в этой форме при десульфировании.
В другом варианте исполнения полученный таким образом смешанный оксид можно подвергнуть восстановлению водородом. Его проводят при температурах приблизительно от 150 до 350°С, преимущественно приблизительно от 150 до 250°С, в присутствии водорода, причем водород разбавляют инертным газом, например азотом, аргоном, метаном, наиболее предпочтительно азотом, чтобы содержание водорода составляло 10 об.% или менее, преимущественно 6 об.% или менее, наиболее предпочтительно от 0,5 до 4 об.%. Полученный таким образом медно-цинковый десульфуризатор ("восстановленная форма") можно применять в этой форме при десульфировании.
Кроме того, медно-цинковый десульфуризатор может содержать также металлы, принадлежащие к VIII группе периодической системы (а именно Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt), к группе IB (например Ag, Au) или группе VIB (как, например, Cr, Mo, W). Такие реагенты можно получать, если в вышеописанном процессе получения добавлять соответствующие соли металлов.
Далее твердый материал, полученный после прокаливания или также после гидрогенизации, можно формовать или экструдировать в таблетки или другие формы, причем может оказаться полезным добавлять присадки, например связующие, как графит.
В другом варианте исполнения водный раствор, который содержит соединение меди, в частности водорастворимое, например нитрат или ацетат меди, соединение цинка, в частности водорастворимое, например нитрат или ацетат цинка, и соединение алюминия, как, например, гидроксид алюминия, нитрат алюминия, алюминат натрия, можно смешать с водным раствором соединения основного характера (например, карбоната натрия, карбоната калия), с образованием осадка (способ соосаждения). Образовавшийся осадок отфильтровывают, промывают водой или сначала промывают, затем фильтруют и после этого высушивают. Затем прокаливают при температуре приблизительно от 270 до 400°С. В заключение полученный твердый материал взмучивают в воде, отфильтровывают и высушивают. Полученный таким образом медно-цинковый десульфуризатор ("окисленная форма") можно применять в этой форме при десульфировании.
В следующем варианте исполнения полученный таким образом смешанный оксид можно подвергнуть восстановлению водородом. Его проводят при температурах приблизительно от 150 до 350°С, преимущественно приблизительно от 150 до 250°С, в присутствии водорода, причем водород разбавляют инертным газом, например азотом, аргоном, метаном, наиболее предпочтительно азотом, чтобы содержание водорода составляло 10 об.% или менее, преимущественно 6 об.% или менее, наиболее предпочтительно от 0,5 до 4 об.%. Полученный таким образом медно-цинковый десульфуризатор ("восстановленная форма") можно применять в этой форме при десульфировании.
Кроме того, медно-цинковый десульфуризатор может содержать также металлы, принадлежащие к VIII группе периодической системы (а именно Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt), к группе IB (например, Ag, Au) или группе VIB (например, Cr, Mo, W). Такие реагенты можно получать, если в вышеописанном процессе получения добавлять соответствующие соли металлов.
Далее твердый материал, полученный после прокаливания или также после гидрогенизации, можно формовать или экструдировать в таблетки или другие формы, причем может оказаться полезным добавлять присадки, например, связующие, как графит.
В следующем варианте исполнения соосаждение можно проводить с pH-контролем, если, например, так регулировать скорость подачи растворов солей, чтобы в процессе осаждения поддерживалось значение рН примерно от 7 до 7,5. Возможно также осадок, образующийся при осаждении, после промывания подвергать распылительной сушке.
Еще в одном варианте исполнения соосаждение можно проводить таким образом, чтобы компоненты оксид меди - оксид цинка осаждались из водных растворов соответствующих солей (например, нитратов или ацетатов) с помощью веществ со щелочной реакцией (например, карбонатов щелочных металлов, карбоната аммония) в присутствии коллоидно-дисперсных (в виде геля или золя) оксида алюминия, гидроксида алюминия.
Прокаливание, желательную при необходимости гидрогенизацию и формование можно производить, как описано выше.
Кроме того, можно применять имеющиеся на рынке катализаторы, как, например, катализатор R 3-12 фирмы BASF или G-132A фирмы Süd-Chemie.
В предпочтительном варианте исполнения медно-цинковый десульфуризатор применяют в восстановленной форме. Может быть выгодным смешанный оксид, полученный вышеописанным способом, подвергать восстановлению водородом, которое можно исполнять следующим образом (написание [кат] ниже означает катализатор).
1. Смешанный оксид нагревают до температуры от 100 до 140°С, наиболее предпочтительно до 120±5°С, в токе азота с объемным расходом от 200 до 400 нм3/м3 [КАТ]·ч, наиболее предпочтительно - 300±20 нм3/м3 [КАТ]·час.
2. В начале восстановления к вышеприведенному току азота добавляют 0,5±0,1 об.% водорода до тех пор, пока повышение температуры не составит 15-20°С и останется постоянным. После этого ток водорода повышают до 1,0±0,1 об.% до тех пор, пока суммарное повышение температуры не составит не более 30±5°С, и температура останется постоянной.
3. После этого ток водорода повышают до 2,0±0,2 об.%, причем температура катализатора не должна превышать 230°С, преимущественно 225°С.
4. Теперь ток водорода повышают до 4,0±0,4 об.% и одновременно повышают температуру азота до 200±10°С, причем и здесь температура катализатора не должна превышать 230°С, преимущественно 225°С.
5. Теперь ток водорода повышают до 6,0±0,6 об.% и одновременно поддерживают температуру катализатора 220±10°С.
6. В заключение реакционную смесь в токе азота с объемным расходом от 200 до 400 нм3/м3 [КАТ]·ч, наиболее предпочтительно - 300±20 нм3/м3 [КАТ]·ч, охлаждают до температуры ниже 50°С, причем скорость охлаждения не должна превышать 50±5 К/ч.
Полученный таким образом медно-цинковый десульфуризатор находится в "восстановленной форме" и может применяться таким образом. Его можно, однако, также хранить до момента использования в атмосфере инертного газа. Далее, возможно также сохранять медно-цинковый десульфуризатор в инертном растворителе. Иногда бывает выгодно хранить медно-цинковый десульфуризатор в окисленной форме и проводить активирование "в нужный момент". В связи с этим может быть также выгодным перед активированием проводить стадию высушивания. При этом прокаленный медно-цинковый десульфуризатор, находящийся в окисленной форме, нагревают в токе азота с объемным расходом от 200 до 400 нм3/м3 [КАТ]·ч, наиболее предпочтительно - 300±20 нм3/м3 [КАТ]·ч, до температуры от 180 до 220°С, наиболее предпочтительно до 200±10°С, причем скорость нагревания не должна превышать 50 К/ч. Как только удалена вода, можно охлаждать (массу) до температуры от 100 до 140°С, наиболее предпочтительно до 120±5°С, причем скорость охлаждения не должна превышать 50 К/ч, и проводить активирование, как описано выше.
В наиболее предпочтительном варианте исполнения применяют медно-цинковый десульфуризатор, который содержит от 35 до 45 мас.%, преимущественно от 38 до 41 мас.% оксида меди, от 35 до 45 мас.%, преимущественно от 38 до 41 мас.% оксида цинка и от 10 до 30 мас.%, преимущественно от 18 до 24 мас.% оксида алюминия, а также при необходимости другие оксиды металлов.
В чрезвычайно предпочтительном варианте исполнения применяют медно-цинковый десульфуризатор, который содержит от 38 до 41 мас.% оксида меди, от 38 до 41 мас.% оксида цинка и от 18 до 24 мас.% оксида алюминия.
Эти медно-цинковые десульфуризаторы получают из соответствующих прокаленных смешанных оксидов вышеприведенным способом. В одном варианте исполнения десульфирование ароматического углеводорода или смеси ароматических углеводородов, преимущественно бензола, проводят на медно-цинковом десульфуризаторе в окисленной форме, без участия водорода.
В другом варианте исполнения десульфирование ароматического углеводорода или смеси ароматических углеводородов, преимущественно бензола, проводят на медно-цинковом десульфуризаторе в окисленной форме, в присутствии водорода.
В следующем варианте исполнения десульфирование ароматического углеводорода или смеси ароматических углеводородов, преимущественно бензола, проводят на медно-цинковом десульфуризаторе в восстановленной форме, без участия водорода.
В следующем варианте исполнения десульфирование ароматического углеводорода или смеси ароматических углеводородов, преимущественно бензола, проводят на медно-цинковом десульфуризаторе в восстановленной форме, в присутствии водорода.
Обычно десульфирование проводят при температурах от 40 до 200°С, особенно предпочтительно от 50 до 180°С, наиболее предпочтительно от 60 до 160°С, преимущественно - от 70 до 120°С, и давлении от 1 до 40 бар, особенно предпочтительно от 1 до 32 бар, преимущественно от 1,5 до 5 бар, наиболее предпочтительно - от 2,0 до 4,5 бар. Десульфирование можно проводить в присутствии инертного газа, например азота, аргона или метана. Как правило, однако, десульфирование проводят без добавления инертного газа.
Обычно - по собственному усмотрению - при этом применяют водород с чистотой ≥99,8 об.%, наиболее предпочтительно ≥99,9 об.%, преимущественно - ≥99,95 об.%. Эта же степень чистоты актуальна для водорода, который применяют при проведении, при необходимости, активирования катализаторов.
Обычно массовое соотношение ароматического углеводорода, или смеси ароматических углеводородов, и водорода составляет от 40000:1 до 1000:1, особенно предпочтительно от 38000:1 до 5000:1, наиболее предпочтительно от 37000:1 до 15000:1, преимущественно от 36000:1 до 25000:1, в особом случае от 35000:1 до 30000:1.
Как правило, часовая объемная скорость жидкости (LHSV, "Liquid Hourly Space Velocity") составляет от 0,5 до 10 кг ароматического углеводорода на объемную долю катализатора в час (кг/(м3 [КАТ]·ч)), наиболее предпочтительно от 1 до 8 кг/(м3 [КАТ]·ч), преимущественно от 2 до 6 кг/(м3 [КАТ]·ч).
Десульфурированный таким образом ароматический углеводород, или смесь ароматических углеводородов, преимущественно бензол, теперь содержит ароматические сернистые соединения в количествах не более 0,07 мг/кг, преимущественно не более 0,05 мг/кг, а общее содержание серы составляет в сумме ≤0,20 мг/кг, преимущественно ≤0,15 мг/кг, наиболее предпочтительно ≤0,10 мг/кг.
Вышеописанные десульфуризаторы предоставляют также возможность уменьшить содержание или полностью удалить из ароматического углеводорода или смеси ароматических углеводородов хлор, мышьяк и/или фосфор, то есть соответствующие хлор-, мышьяк- и/или фосфорсодержащие соединения.
Десульфирование ароматического углеводорода или смеси ароматических углеводородов можно производить в одном или нескольких реакторах, соединенных параллельно или последовательно. Эти реакторы обычно работают в жидкофазном режиме, причем газ и жидкость вводят прямотоком или противотоком, преимущественно - прямотоком. Существует, однако, возможность использования реакторов в оросительном режиме, причем газ и жидкость вводят прямотоком или противотоком, преимущественно - противотоком.
В случае необходимости десульфуризатор можно также вновь удалить из реактора. Если десульфуризатор находится в восстановленной форме, то может быть выгодным перед удалением подвергнуть его окислению. В качестве окислителя применяют кислород или смесь кислорода с одним или несколькими инертными газами, например воздухом. Окисление производится по известному в технике обычному способу. Можно, например, проводить окисление следующим образом.
1. Десульфуризатор сначала промывают током азота с объемным расходом от 200 до 400 нм3/м3 [КАТ]·ч, наиболее предпочтительно - 300±20 нм3/м3 [КАТ]·ч.
2. В начале окисления к вышеприведенному току азота добавляют ток воздуха, с объемным расходом от 5 до 10 нм3/м3 [KAT]·ч, наиболее предпочтительно - 7±1 нм3/м3 [КАТ]·ч, причем температура повышается приблизительно на 50°С. После этого ток воздуха увеличивают в течение промежутка времени от 0,5 час до 2,0 ч, преимущественно в течение 1±0,2 ч, до объемного расхода от 10 до 18 нм3/м3 [КАТ]·ч, наиболее предпочтительно - до 14±1 нм3/м3 [КАТ]·ч, и выдерживают в этих условиях от 6 до 10 ч, преимущественно 8±0.5 ч.
3. После этого поток воздуха увеличивают в течение промежутка времени от 0,5 ч до 2,0 ч, преимущественно в течение 1±0,2 ч, до объемного расхода от 20 до 35 нм3/м3 [КАТ]·ч, наиболее предпочтительно - до 28±2 нм3/м3 [КАТ]·ч, причем температура десульфуризатора не должна превышать 230°С, преимущественно 225°С, и поддерживается в течение промежутка времени от 3 до 5 ч, преимущественно 4±0,5 ч.
4. Затем поток воздуха увеличивают до объемного расхода от 120 до 180 нм3/м3 [КАТ]·ч, преимущественно - до 150±10 нм3/м3 [КАТ]·ч, и одновременно поток азота уменьшают также до объемного расхода от 120 до 180 нм3/м3 [КАТ]·ч, преимущественно - до 150±10 нм3/м3 [КАТ]·ч, причем температура десульфуризатора не должна превышать 230°С, преимущественно 225°С. Этот режим сохраняется до тех пор, пока не упадет температура и не уравняется содержание кислорода в отходящем и поступающем газах.
5. В заключение, при понижении расхода азота до нуля поток воздуха увеличивают до объемного расхода от 200 до 400 нм3/м3 [КАТ]·ч, наиболее предпочтительно - до 300±20 нм3/м3 [КАТ]·ч. Этот режим сохраняют около 1 ч, пока не завершится окисление.
Полученный таким образом медно-цинковый десульфуризатор теперь можно извлекать.
Теперь на стадии b) десульфурированный ароматический углеводород или смесь ароматических углеводородов гидрируют в соответствующее алициклическое соединение или смесь алициклических соединений в присутствии нанесенного рутениевого катализатора, причем катализатор наносят на пористую основу, имеющую мезо- и/или макропоры.
Принципиально можно применять любые носители с макропорами, т.е. как носители, имеющие только макропоры, так и те, которые наряду с макропорами содержат также мезопоры и/или микропоры. Понятия "макропоры", "мезопоры" и "микропоры" в рамках настоящего изобретения применяются таким образом, как они определены в Pure Appl. Chem. 46, 71 (1976), а именно как поры, диаметр которых превышает 50 нм (макропоры), или составляет от 2 до 50 нм (мезопоры), или <2 нм (микропоры).
Наиболее подходящими носителями являются соответствующие активированный уголь, карбид кремния, окись алюминия, оксид кремния, окись титана, двуокись циркония или также их смеси. Предпочтительно применяют соответствующие окись алюминия, диоксид циркония или оксид кремния, наиболее предпочтительно - γ-окись алюминия или оксид кремния.
- В особо предпочтительном варианте исполнения применяется рутениевый катализатор, нанесенный на γ-окись алюминия.
В общем содержание рутения составляет от 0,01 до 30 мас.%, преимущественно от 0,01 до 5 мас.%, наиболее предпочтительно от 0,1 до 1,5 мас.% соответственно, в пересчете на общую массу катализатора.
В предпочтительном варианте исполнения применяют нанесенный рутениевый катализатор, причем средний диаметр пор носителя составляет по меньшей мере 50 нм, ВЕТ-поверхность - не более 30 м2/г, а количество рутения - от 0,01 до 30 мас.%, в пересчете на общую массу катализатора. Наиболее предпочтительны нанесенные рутениевые катализаторы, причем средний диаметр пор носителя составляет от 100 нм до 200 мкм, а ВЕТ-поверхность - не более 15 м2/г.
В другом предпочтительном варианте исполнения применяют нанесенный рутениевый катализатор, причем количество рутения составляет от 0,01 до 30 мас.%, в пересчете на общую массу катализатора, от 10 до 50% объема пор носителя образуют макропоры с диаметром от 50 нм до 10000 нм и от 50 до 90% объема пор носителя образуют мезопоры с диаметром от 2 до 50 нм и причем сумма долей объемов пор составляет 100% (Определение среднего диаметра пор и распределения пор по размерам проводят способом ртутной порометрии в соответствии с ДИН 66133).
Нанесенные рутениевые катализаторы получают нанесением рутения на основу. Это может происходить, как правило, с помощью пропитки носителя водными растворами солей рутения или путем опрыскивания носителя соответствующим раствором соли рутения. Подходящими солями рутения являются нитраты, нитрозилнитраты, галогениды, карбонаты, карбоксилаты, ацетилацетонаты, комплексные хлориды и нитриты или аминные комплексы, наиболее благоприятными являются нитрат и нитрозилнитрат.
Носитель, пропитанный раствором соли рутения или покрытый слоем этой соли, в заключение, как правило, сушат при температурах от 100 до 150°С и по выбору прокаливают при температурах от 200 до 600°С, преимущественно - от 350 до 450°С.
Полученный таким образом прокаленный нанесенный рутениевый катализатор теперь активируют обработкой газовым потоком, содержащим свободный водород, при температурах от 30 до 600°С, преимущественно - от 150 до 450°С. Как правило, газовый поток содержит от 50 до 100 об.% водорода и до 50 об.% азота.
Обычно раствор соли рутения наносят на основу в таком количестве, чтобы содержание рутения составляло от 0,01 до 30 мас.%, преимущественно от 0,01 до 5 мас.%, особенно предпочтительно от 0,01 до 1 мас.% и наиболее предпочтительно от 0,05 до 1 мас.% соответственно в пересчете на общую массу катализатора.
В особом варианте исполнения применяют такие носители, которые имеют макропористую структуру и обнаруживают средний диаметр пор не менее 50 нм, преимущественно не менее 100 нм, наиболее предпочтительно - не менее 500 нм, и поверхность которых, соответственно BET, составляет не более 30 м2/г, преимущественно не более 15 м2/г, особенно предпочтительно не более 5 м2/г и наиболее предпочтительно - от 0,5 до 3 м2/г. Средний диаметр пор этого носителя преимущественно составляет от 100 нм до 200 мкм, преимущественно от 500 нм до 50 мкм (Поверхность носителя определяли по способу BET с помощью адсорбции N2, наиболее предпочтительно - в соответствии с ДИН 66131).
Металлическая поверхность полученного таким образом нанесенного рутениевого катализатора составляет от 0,01 до 10 м2/г, предпочтительно от 0,05 до 5 м2/г и наиболее предпочтительно от 0,05 до 3 м2/г (Поверхность металла определяют способом хемосорбции, описанным J.Lemaire et al. in "Characterisation of Heterogeneous Catalysts", Hrsg. Francis Delanney, Marcel Dekker, New York, 1984, S.310-324).
Отношение металлической поверхности к поверхности носителя катализатора составляет при этом не более 0,05, наиболее предпочтительно не более 0,005.
Преимущественно распределение пор носителя (по размерам) может быть приблизительно бимодальным. Преимущественно подобное бимодальное распределение пор по диаметру имеет максимумы около 600 нм и около 20 мкм.
В следующем предпочтительном варианте исполнения применяют носители, которые содержат макропоры и мезопоры. Они в особенности (в частности) показывают такую структуру распределения пор, что от 5 до 50%, преимущественно от 10 до 45%, особенно предпочтительно от 10 до 30% и наиболее предпочтительно - от 15 до 25% общего объема пор образуют макропоры, с диаметром пор от 50 нм до 10000 нм, а от 50 до 95%, преимущественно от 55 до 90%, особенно предпочтительно от 70 до 90% и наиболее предпочтительно - от 75 до 85% общего объема пор образуют мезопоры, с диаметром пор от 2 до 50 нм. Сумма долей объемов пор составляет при этом 100%.
Суммарный объем пор использованного при этом носителя составляет от 0,05 до 1,5 см3/г, преимущественно от 0,1 до 1,2 см3/г и наиболее предпочтительно - от 0,3 до 1,0 см3/г.
Средний диаметр пор использованного при этом носителя составляет от 5 до 20 нм, преимущественно от 8 до 15 нм и наиболее предпочтительно от 9 до 12 нм (Определение среднего диаметра пор проводят способом ртутной порометрии в соответствии с ДИН 66133).
Поверхность использованного при этом носителя составляет от 50 до 500 м2/г, преимущественно от 200 до 350 м2/г и наиболее предпочтительно - от 200 до 300 м2/г (Поверхность носителя определяли по способу BET с помощью адсорбции N2, наиболее предпочтительно - в соответствии с ДИН 66131).
- В другом варианте исполнения можно использовать катализатор, поверхностный слой которого в качестве активного металла содержит рутений, индивидуально или совместно по меньшей мере еще с одним металлом из побочных групп IB, VIIB или VIII периодической системы элементов (Ch. Ab. Service-версия), нанесенный на подложку на основе двуокиси кремния.
Этот поверхностный катализатор отличается тем, что количество активного металла составляет <1 мас.%, предпочтительно от 0,1 до 0,5 мас.%, особенно предпочтительно от 0,25 до 0,35 мас.%, в пересчете на общую массу катализатора, причем по меньшей мере 60 мас.%, предпочтительно 80 мас.% активного металла, в пересчете на общее количество активного металла, находится в поверхностном слое зерен катализатора, с глубиной пропитки до 200 мкм. Вышеприведенные данные получены с помощью SEM (сканирующей электронной микроскопии), ЕРМА (электронного микроанализа образца) - EDXS (энергорассеивающего рентгеновского анализа) и представляют усредненное значение. Дополнительные сведения по вышеуказанным способам измерения и техническим приемам приведены, например, в "Spectroscopy in Catalysis", J.V.Niemantsverdriet, VCH, 1995.
Поверхностный катализатор отличается тем, что преобладающее количество активного металла находится в поверхностном слое зерен, до глубины пропитки 200 мкм, следовательно, близко к поверхности катализатора. Внутри (зерна) катализатора (в толще), напротив, активный металл не присутствует вовсе или находится только в очень малых количествах.
Предпочтительным является такой поверхностный катализатор, у которого не удается обнаружить активный металл в толще (внутри зерна) катализатора, т.е. активный металл находится только в самом наружном слое (зерен катализатора), например в зоне с глубиной пропитки от 100 до 200 мкм.
В следующем особенно предпочтительном варианте исполнения поверхностный катализатор отличается тем, что по данным (FEG)-TEM (способ просвечивающей электронной микроскопии с автоэмиссионной пушкой) с помощью способа EDXS (энергорассеивающего рентгеновского анализа) удается обнаружить частицы активного металла только в самом наружном слое, с глубиной пропитки 200 мкм, предпочтительно 100 мкм, особенно предпочтительно - 50 мкм. Частицы менее 1 нм невозможно обнаружить.
Рутений можно применять в качестве активного металла, индивидуально или совместно по меньшей мере еще с одним металлом из побочных групп IB, VIIB или VIII периодической системы элементов (Сh.Аb. Service-версия). Наряду с рутением другими подходящими активными металлами являются, например, платина, родий, палладий, иридий, кобальт или никель, или смесь двух или более из них. Среди металлов побочных групп IB и/или VIIB, также пригодных для применения, подходящими являются, например, медь и/или рений. Предпочтительно в качестве активного металла в поверхностном катализаторе применяют рутений индивидуально или совместно с платиной или иридием; в высшей степени предпочтительно в качестве активного металла применяют один рутений.
Поверхностный катализатор проявляет вышеупомянутую очень высокую активность при малой загрузке активного металла, которая составляет <1 мас.%, в пересчете на общую массу катализатора. Предпочтительно количество активного металла в адсорбционном катализаторе составляет от 0,1 до 0,5 мас.%, особенно предпочтительно от 0,25 до 0,35 мас.%. Было найдено, что глубина пропитки носителя активным металлом зависит от загрузки катализатора активным металлом. Уже при загрузке катализатора 1 мас.% или более, например 1,5 мас.% (активного металла) в толще (внутри зерна) катализатора, т.е. на глубине проникновения от 300 до 1000 мкм находится существенное количество активного металла, который влияет на активность катализатора гидрирования, особенно при большой продолжительности гидрирования, особенно для быстрых реакций, когда в толще (внутри зерна) катализатора может проявиться недостаток водорода.
В варианте исполнения с поверхностным катализатором по меньшей мере 60 мас.% активного металла, в пересчете на общее количество активного металла, находится в поверхностном слое зерен катализатора, с глубиной пропитки до 200 мкм. Предпочтительно в поверхностном катализаторе по меньшей мере 80 мас.% активного металла, в пересчете на общее количество активного металла, находится в оболочке катализатора, с глубиной пропитки до 200 мкм. В высшей степени предпочтительным является такой поверхностный катализатор, у которого не удается обнаружить активный металл в толще (внутри зерна) катализатора, т.е. активный металл находится только в самой оболочке, например в зоне с глубиной проникновения от 100 до 200 мкм. В другом предпочтительном варианте исполнения 60 мас.%, предпочтительно 80 мас.% активного металла, в пересчете на общее количество активного металла, находится в оболочке катализатора, с глубиной проникновения до 150 мкм. Вышеприведенные данные получены с помощью SEM (сканирующей электронной микроскопии), ЕРМА (электронного микроанализа образца) - EDXS (энергорассеивающего рентгеновского анализа) и представляют усредненное значение. Для определения глубины проникновения частиц активного металла несколько гранул катализатора (например, 3, 4 или 5) срезают поперек оси прутка (если катализатор существует в форме прутков). Затем с помощью строчной развертки регистрируют профили отношения концентраций активный металл/Si. На каждой измерительной линии отмеряли несколько, например 15-20, точек измерения на равных расстояниях; размер зоны ("пятна") измерения составляет около 10 мкм·10 мкм. После интеграции количества активного металла по всей глубине можно определить распространенность активного металла в данной зоне.
В высшей степени предпочтительно количество активного металла, в пересчете на отношение концентраций активного металла к Si, на поверхности поверхностного катализатора, составляет от 2 до 25%, предпочтительно от 2 до 10%, особенно предпочтительно от 4 до 6%, определенное с помощью способов SEM - ЕРМА - EDXS. Анализ поверхности происходит с помощью анализа способом площадей областей (зон) размером 800 мкм × 2000 мкм, с информационной глубиной около 2 мкм. Элементный состав определяют в мас.% (приведенных к 100%). Среднее отношение концентраций (активный металл/Si) усредняют по 10 областям измерения.
Под названием "поверхность" поверхностного катализатора по содержанию настоящего описания следует понимать наружную оболочку катализатора до глубины пропитки около 2 мкм. Эта глубина пропитки соответствует информационной глубине при упомянутом выше анализе поверхности.
В высшей степени предпочтительным является поверхностный катализатор, в котором количество активного металла, в пересчете на массовое отношение активного металла к Si (мас./мас. в %), составляет на поверхности адсорбционного катализатора от 4 до 6%, на глубине пропитки 50 мкм - от 1,5 до 3% и на глубине пропитки от 50 до 150 мкм - от 0,5 до 2%, определенное с помощью способов SEM EPMA (EDXS). Приведенные величины представляют усредненные значения.
Далее анализ с помощью способов (FEG)-TEM показывает, что размер частиц активного металла предпочтительно уменьшается с увеличением глубины пропитки.
Активный металл в поверхностном катализаторе предпочтительно находится частично или полностью в кристаллическом состоянии. В предпочтительных случаях в поверхностном слое (зерен) адсорбционного катализатора с помощью способов SAD (микродифракции, дифракции электронов от выбранного участка) или XRD (дифракции рентгеновских лучей, рентгенографии) удается обнаружить микрокристаллический активный металл.
Поверхностный катализатор может дополнительно содержать ионы щелочно-земельных металлов (М2+), следовательно, М=Be, Mg, Ca, Sr и/или Ва, наиболее предпочтительно Mg и/или Ca, в высшей степени предпочтительно Mg. Содержание ионов щелочно-земельных металлов (М2+) в катализаторе составляет предпочтительно от 0,01 до 1 мас.%, наиболее благоприятно от 0,05 до 0,5 мас.%, в высшей степени благоприятно от 0,1 до 0,25 мас.% соответственно, в пересчете на массу носителя - двуокиси кремния.
Основным компонентом катализатора является носитель на основе двуокиси кремния, в общем случае аморфной. Под определением "аморфный" в связи с этим подразумевают, что доля кристаллической фазы двуокиси кремния составляет меньше 10 мас.% носителя. Носители, используемые для приготовления катализаторов, могут, правда, обнаруживать надмолекулярные структуры, которые образуются упорядоченной системой пор носителя.
В качестве носителей в основном рассматриваются аморфные образцы двуокиси кремния, которые по меньшей мере на 90 мас.% состоят из двуокиси кремния, причем оставшиеся 10 мас.%, преимущественно не более 5 мас.% носителя, могут представлять собой другой окисный материал, например MgO, CaO, TiO2, ZrO2, Fe2O3, и/или окислы щелочных металлов.
В предпочтительном варианте исполнения изобретения носитель не содержит галогенов, наиболее благоприятно не содержит хлора, т.е. содержание галогена в носителе составляет менее 500 мас.ч. на млн, например от 0 до 400 мас.ч. на млн. Таким образом, предпочтителен адсорбционный катализатор, который в пересчете на общую массу катализатора, содержит менее 0,05 мас.% галогенида (определенного способом ионообменной хроматографии).
Предпочтительными являются носители, которые имеют удельную поверхность от 30 до 700 м2/г, предпочтительно от 30 до 450 м2/г (ВЕТ-поверхность, в соответствии с ДИН 66131).
Подходящие аморфные носители на основе двуокиси кремния технически легкодоступны и являются продажными реактивами (см., например, O.W.Flörke, "Silica" в Ullmann's Encyclopedia of Industrial Chemistry 6th Edition on CD-ROM). Они могут быть как естественного происхождения, так и получены искусственно. Примерами аморфных носителей на основе двуокиси кремния являются силикагели, кизельгур, пирогенные кремниевые кислоты и осажденные кремниевые кислоты. В предпочтительном варианте исполнения изобретения катализаторы содержат силикагели в качестве носителей.
В зависимости от формы исполнения изобретения носитель может отличаться по внешнему виду. При использовании поверхностного катализатора в неподвижном слое обычно применяют формованные изделия из носителя, которые можно получать экструдированием, горячим прессованием или таблетированием и которые могут иметь форму шариков, таблеток, цилиндров, прутков, колец, полых цилиндров, звездочек и т.п. Размеры этих формовок обычно колеблются от 0,5 мм до 25 мм. Часто применяют катализатор в виде прутков диаметром от 1,0 до 5 мм и длиной от 2 до 25 мм. С прутками меньших размеров, вообще говоря, достигается более высокая активность; однако они не проявляют достаточной механической устойчивости в процессе гидрирования. Поэтому в высшей степени предпочтительно применяют прутки диаметром от 1,5 до 3 мм.
Получение поверхностного катализатора предпочтительно происходит таким образом, что сначала раствором ацетата рутения (III), индивидуально или совместно с раствором по меньшей мере еще одной соли из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов (Ch.Ab. Service-версия), пропитывают носитель, одно- или многократно; полученный твердый материал высушивают и после этого восстанавливают, причем раствор по меньшей мере еще одной соли из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов можно наносить на одной или нескольких стадиях пропитки совместно с раствором ацетата рутения (III), или на одной или нескольких стадиях пропитки отдельно от раствора ацетата рутения (III). Отдельные стадии процесса ниже описаны более подробно.
Получение поверхностного катализатора, включающее стадии:
1) одно- или многократная пропитка носителя, содержащего двуокись кремния, раствором ацетата рутения (III), индивидуально или совместно с раствором по меньшей мере еще одной соли из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов (Ch.Ab. Service-версия);
2) последующее высушивание;
3) последующее восстановление;
причем раствор по меньшей мере еще одной соли из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов можно наносить на одной или нескольких стадиях пропитки совместно с раствором ацетата рутения (III) или на одной или нескольких стадиях пропитки отдельно от раствора ацетата рутения (III).
На вышеназванной стадии 1) происходит одно- или многократная пропитка носителя, содержащего двуокись кремния, раствором ацетата рутения (III), индивидуально или совместно с раствором по меньшей мере еще одной соли из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов (Ch.Ab. Service-версия). Так как количество активного металла в поверхностном катализаторе очень мало, в предпочтительном варианте исполнения производится однократная пропитка. Ацетат рутения (III) или соли металлов побочных групп IB, VIIB или VIII периодической системы элементов представляют собой предшественники активного металла. При применении раствора ацетата рутения (III) в качестве предшественника можно наиболее благоприятно получать поверхностные катализаторы, которые в том числе отличаются тем, что основная часть активного металла, предпочтительно одного рутения, помещается в поверхностном катализаторе до глубины пропитки 200 мкм. В толще (внутри зерен) катализатора не обнаруживается совсем или только немного активного металла.
Подходящими растворителями для приготовления раствора ацетата рутения (III) или раствора по меньшей мере еще одной соли из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов являются вода или также смеси на основе воды, или растворитель, содержащий до 50 об.% одного или нескольких смешивающихся с водой органических растворителей, например смеси с алканолами с 1-4 атомами углерода, как метанол, этанол, н-пропанол или изопропанол. Можно также применять водную уксусную кислоту или ледяную уксусную кислоту. Все смеси следует выбирать таким образом, чтобы существовал раствор или фаза. Предпочтительными растворителями являются уксусная кислота, вода или их смеси. Особенно предпочтительным растворителем является смесь воды и уксусной кислоты, так как ацетат рутения (III) обычно имеется в виде раствора в уксусной или ледяной уксусной кислоте. Можно, однако, использовать ацетат рутения (III) в виде твердого вещества (для приготовления раствора). Можно также получать катализатор без применения воды.
Раствор по меньшей мере еще одной соли из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов можно наносить в одну или несколько стадий пропитки совместно с раствором ацетата рутения (III), или в одну или несколько стадий пропитки отдельно от раствора ацетата рутения (III).
Это означает, что пропитку можно производить раствором, в котором имеется ацетат рутения (III), а также по меньшей мере еще одна соль из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов. Пропитка этим раствором может происходить одно- или многократно. Возможно, однако, также, что сначала (носитель) пропитывают раствором ацетата рутения (III), a после этого, на отдельной стадии пропитки - раствором, который содержит по меньшей мере еще одну соль из числа металлов побочных групп IB, VIIB или VIII периодической системы элементов. Последовательность пропитки может быть также и обратной. Также возможно, что одну из двух стадий пропитки или обе стадии повторяют одно- или многократно в любой последовательности. После каждой пропитки обычно высушивают материал.
Подходящими солями других металлов побочных групп IB, VIIB или VIII периодической системы элементов, которые можно применять на стадии пропитки, являются, например, нитраты, ацетонаты и ацетаты, причем предпочтительными являются ацетаты.
Особенно предпочтительно пропитка производится индивидуальным раствором ацетата рутения (III) в одну стадию.
Пропитка носителя может происходить различными способами и в известной мере определяется видом носителя. Можно, например, опрыскивать или спринцевать носитель раствором-предшественником или суспендировать носитель в этом растворе. Например, можно суспендировать носитель в водном растворе предшественника активного металла и отфильтровать от водной фазы по прошествии определенного времени. По количеству поглощенной жидкости и концентрации активного металла в растворе можно затем простым способом оценить содержание активного металла в катализаторе. Пропитка носителя может, например, также происходить таким образом, что носитель обрабатывают определенным количеством раствора-предшественника активного металла, соответствующим максимальному количеству жидкости, которое может поглотить носитель. Для этого, например, носитель опрыскивают нужным количеством жидкости. Походящей аппаратурой для этого являются аппараты, обычно применяемые для смешивания жидкостей с твердыми веществами (см. Vauck/MÜller, Grundoperationen chemischer Verfahrenstechnick, 10. Aufgabe, Deutscher Verlag für Grundstoffindustrie, 1994, S. 405 ff.), например, качающиеся сушилки, барабаны для замачивания (пропитки), барабанные смесители, лопаточные смесители и т.п. Монолитные носители обычно опрыскивают водными растворами предшественника активного металла.
Для пропитки преимущественно применяют растворы с низким содержанием галогена, особенно (в частности) с низким содержанием хлора, т.е. они не содержат совсем или содержат менее 500 мас. ч. на млн, наиболее предпочтительно менее 100 мас. ч. на млн галогена, например, от 0 до <80 мас. ч. на млн галогена, в пересчете на суммарную массу раствора.
Концентрация предшественника активного металла в растворах естественно определяется количеством наносимого предшественника активного металла и поглотительной емкостью носителя по отношению к раствору и составляет <20 мас.%, предпочтительно от 0,01 до 6 мас.%, особенно предпочтительно от 0,1 до 1,1 мас.%, в пересчете на общую массу использованного раствора.
На стадии 2) проводят высушивание. Оно может происходить в соответствии с обычными способами высушивания твердых веществ при соблюдении приведенных ниже температурных пределов. Для качества, т.е. активности катализатора, важным является соблюдение верхнего предела температур сушки. Несоблюдение приведенных ниже температур сушки ведет к заметной потере активности. Прокаливание при повышенных температурах, например выше 300°С или даже 400°С, как предлагается в современной технике, не только излишне, но и оказывает вредное влияние на активность катализатора. Для достижения достаточной скорости высушивания сушку предпочтительно проводят при повышенной температуре, предпочтительно при температуре ≤180°С, особенно предпочтительно ≤160°С, и по меньшей мере не ниже 40°С, наиболее предпочтительно не ниже 70°С, в особых случаях не ниже 100°С и в высшей степени предпочтительно - при температурах от 110°С до 150°С.
Высушивание твердого материала, пропитанного предшественником активного металла, обычно происходит при нормальном давлении, причем для стимулирования высушивания можно применять также разрежение. Часто для содействия высушиванию над высушиваемым материалом или через него пропускают поток газа, например воздуха или азота.
Продолжительность высушивания, естественно, зависит от требуемой степени осушки и температуры и предпочтительно составляет от 1 ч до 30 ч, преимущественно от 2 до 10 ч.
Предпочтительно высушивание обрабатываемого носителя проводят таким образом, чтобы содержание воды или летучих компонентов растворителя перед последующим восстановлением составляло менее 5 мас.%, наиболее предпочтительно не более 2 мас.%, в пересчете на суммарную массу твердого материала. Приведенные массовые доли при этом относятся к потерям массы твердого материала, определенным при температуре 160°С, давлении 1 бар и продолжительности (сушки) 10 мин. Таким образом активность используемого катализатора может быть еще увеличена.
На стадии 3) твердый материал, полученный после высушивания, переводят в каталитически активную форму. Это происходит благодаря восстановлению твердого материала при температурах в общем от 150°С до 450°С, предпочтительно от 250°С до 350°С известными способами.
Для этого твердый материал, полученный после высушивания, при указанных выше температурах вводят в контакт с водородом или смесью водорода с инертным газом. Абсолютное давление водорода имеет второстепенное значение для результата восстановления и может, например, варьироваться от 0,2 бар до 1,5 бар. Часто гидрирование материала катализатора происходит при нормальном давлении водорода в токе водорода. Преимущественно восстановление происходит при движении твердого материала, например при проведении восстановления во вращающейся трубчатой печи или вращающейся шаровидной печи. Таким способом активность используемого катализатора может быть еще увеличена. Используемый водород преимущественно не содержит каталитических ядов, как СО- и S-содержащие соединения, например H2S, COS и другие.
Восстановление можно производить также с помощью органических восстановителей, как гидразин, формальдегид, формиаты или ацетаты.
По окончании восстановления можно для большего удобства в обращении известным способом перевести катализатор в пассивное состояние, если, например, провести краткосрочную обработку катализатора газом, содержащим кислород, например воздухом, преимущественно, однако, - инертной газовой смесью, содержащей от 1 до 10 об.% кислорода. Здесь можно применять также СО2 или смеси СО2/О2.
Активный катализатор можно также хранить в инертном органическом растворителе, например этиленгликоле.
Для получения поверхностного катализатора в другой форме проведения можно предшественник активного металла катализатора, например, полученный по вышеописанному или согласно описанию в международном патенте WO-A2-02/100538 (BASF AG), пропитать раствором одной или нескольких солей щелочно-земельного металла (II).
Предпочтительными солями щелочно-земельных металлов (II) являются соответствующие нитраты, особенно нитрат магния и нитрат кальция.
Предпочтительным растворителем для солей щелочно-земельных металлов (II) на этой стадии пропитки является вода. Концентрация соли щелочно-земельного металла (II) в растворе составляет от 0,01 до 1 моль/л.
Например, вмонтированный в трубу катализатор - активный металл/SiO2, вводят в контакт с потоком водного раствора соли щелочно-земельного металла (II). Катализатор, подлежащий пропитке, можно также обрабатывать стационарно - действием находящегося над ним слоя раствора соли щелочно-земельного металла.
Предпочтительно таким образом осуществляется насыщение катализатора - активный металл/SiO2, особенно его поверхности, ионом или ионами щелочно-земельных металлов.
Избыточную соль щелочно-земельного металла и незафиксированные на поверхности ионы щелочно-земельного металла смывают с катализатора (промывка водой, промывка катализатора).
Для облегчения манипулирования, например введения в трубку реактора, катализатор после пропитки можно высушить. Для этого сушку можно, например, проводить в печи при температурах <200°С, например, от 50 до 190°С, особенно предпочтительно при температурах <140°С, например, от 60 до 130°С.
Этот процесс пропитки можно проводить ex situ или in situ: первое означает - перед введением катализатора в реактор, второе означает - в реакторе (после введения катализатора).
В варианте способа пропитка катализатора ионами щелочно-земельных металлов также может происходить in situ, благодаря тому, что к раствору гидрируемого ароматического субстрата (исходное вещество) прибавляют ионы щелочно-земельных металлов, например, в виде растворов солей щелочно-земельных металлов. Для этого, например, сначала растворяют соответствующее количество соли в воде и затем добавляют к субстрату, растворенному в органическом растворителе.
В соответствии с вариантом можно в процессе гидрирования катализатор применять в комбинации с гидрируемым субстратом, в составе которого находится раствор, содержащий ионы щелочно-земельных металлов. При этом содержание ионов щелочно-земельных металлов в гидрируемом субстрате составляет в общем от 1 до 100 мас. ч. на млн, наиболее благоприятно от 2 до 10 мас. ч. на млн.
По условиям получения активный металл в катализаторах находится в металлическом состоянии.
Благодаря применению при получении адсорбционного катализатора предшественника активного металла и растворителя с низким содержанием галогенов, особенно хлора, содержание галогенидов, особенно хлоридов, в адсорбционных катализаторах в результате находится ниже 0,05 мас.% (от 0 до значения <500 мас. ч. на млн, например, от 0 до 400 мас. ч. на млн), в пересчете на общую массу катализатора.
Содержание хлора определяют, например, описанным ниже способом ионообменной хроматографии.
В выбранном варианте предпочтительно, чтобы процентное отношение структур Q2 и Q3 - Q2/Q3, определенное способом 29Si-ЯMP твердого тела, было меньше 25, предпочтительно меньше 20, особенно предпочтительно меньше 15, например, от 0 до 14 или от 0,1 до 13. Это означает, что степень конденсации двуокиси кремния в используемом носителе особенно высока.
С помощью способа 29Si-ЯМР твердого тела производится идентификация Qn - структур (n=2, 3, 4) и определение процентных отношений.
Qn=Si(OSi)n(OH)4-n при n=1, 2, 3 или 4.
Обнаруживают сигнал Qn для n=4 при -110,8 ч. на млн; n=3 при -100,5 ч. на млн и n=2 при - 90,7 ч. на млн (эталон: тетраметилсилан) (Q0 и Q1 не идентифицировали). Анализ проводят в условиях "магического угла вращения" (МУВ) при комнатной температуре (20°С) (МУВ 5500 Гц) с круговой поляризацией (КП 5 ms) и с применением развязки дипольного взаимодействия с протонами 1Н. Из-за частичного перекрывания сигналов интенсивности оценивают по анализу формы линий. Анализ формы линий проводили с помощью стандартного пакета программ фирмы Galactic Industrie, причем рассчитывали линию, выровненную по способу наименьших квадратов.
Преимущественно носитель содержит не более 1 мас.%, и особенно не более 0,5 мас.%, и наиболее предпочтительно <500 мас. ч. на млн оксида алюминия, в расчетах - Al2O3.
Поскольку на конденсацию двуокиси кремния могут повлиять также алюминий и железо, то суммарная концентрация Al(III) и Fe(II и/или III) предпочтительно составляет меньше 300 мас. ч. на млн, особенно предпочтительно - меньше 200 мас. ч. на млн, например, от 0 до 180 мас. ч. на млн.
Доля оксидов щелочных металлов предпочтительно образуется в результате получения носителя и может составлять до 2 мас.%. Часто она составляет менее 1 мас.%. Подходящими являются также носители, не содержащие оксидов щелочных металлов (от 0 до <0,1 мас.%). Доля MgO, CaO, TiO2 или ZrO2 в носителе может составлять до 10 мас.% и преимущественно не превышает 5 мас.%. Подходящими, однако, являются также носители, которые не содержат этих оксидов металлов в заметных количествах (от 0 до <0,1 мас.%).
Поскольку Al(III) и Fe(II и/или III) в структуре двуокиси кремния могут создавать кислотные центры, предпочтительно, чтобы в носителе осуществлялась компенсация зарядов, предпочтительно катионами щелочно-земельных металлов (М2+, М=Be, Mg, Ca, Sr, Ba). Это означает, что массовое отношение М(II) к Al(III) + Fe(II и/или III) больше 0,5, предпочтительно >1, особенно предпочтительно больше 3 (Римские цифры в скобках при символе элемента означают степень окисления элемента).
Гидрирование десульфурированного ароматического углеводорода или смеси десульфурированных ароматических углеводородов, преимущественно бензола, на описанном выше нанесенном рутениевом катализаторе в алициклическое соединение или соответствующую смесь алициклических соединений, преимущественно в циклогексан, в присутствии водорода можно проводить в жидкой фазе или газовой фазе. Предпочтительно процесс гидрирования проводят в жидкой фазе - как правило, при температуре от 50 до 250°С, преимущественно от 60 до 200°С, наиболее предпочтительно - от 70 до 170°С. Применяемые при этом давления составляют от 1 до 200 бар, преимущественно от 10 до 50 бар, наиболее предпочтительно от 19 до 40 бар и в специальном случае - от 25 до 35 бар.
Обычно при гидрировании применяют водород с чистотой ≥99,8 об.%, наиболее предпочтительно ≥99,9 об.%, преимущественно - ≥99,95 об.%.
Особенно предпочтительно при этом происходит полное гидрирование ароматического углеводорода или смеси ароматических углеводородов, причем под полным гидрированием следует понимать степень превращения гидрируемого соединения в общем >98%, предпочтительно >99%, особенно предпочтительно >99,5%, в высшей степени предпочтительно >99,9%, наиболее предпочтительно >99,99% и в специальном случае >99,995%.
Обычно массовое соотношение ароматического углеводорода или смеси ароматических углеводородов и водорода составляет от 8:1 до 5:1, преимущественно от 7,7:1 до 5,5:1, наиболее предпочтительно от 7,6:1 до 6:1 и в специальном случае - от 7,5:1 до 6,5:1.
Гидрирование десульфурированного ароматического углеводорода или смеси десульфурированных ароматических углеводородов можно производить в одном или нескольких реакторах, соединенных параллельно или последовательно. Эти реакторы обычно работают в оросительном режиме. При этом газ и жидкость вводят прямотоком или противотоком, преимущественно - прямотоком. Существует, однако, возможность использования последовательно соединенных реакторов в жидкофазном режиме.
Как правило, часовая объемная скорость жидкости (LHSV, "Liquid Hourly Space Velocity") составляет от 0,1 до 10 кг ароматического углеводорода на объемную долю катализатора в час (кг/(м3 [КАТ]·ч)), преимущественно от 0,3 до 1,5 кг/(м3 [КАТ]·ч). Плотность орошения обычно составляет от 20 до 100 м3 ароматического углеводорода на обтекаемую площадь сечения слоя катализатора в час (м3/м2·ч), преимущественно от 60 до 80 м3/м2·ч.
Может быть выгодным достигать в первом реакторе степени превращения ароматического углеводорода от 95 до 99,5%, а в последующем - степени превращения >99,9%, наиболее предпочтительно >99,99%, преимущественно >99,995%. В подобном случае, как правило, отношение объемов загрузки основного реактора и последующего реактора составляет от 20:1 до 3:1, наиболее предпочтительно - от 15:1 до 5:1.
В следующем варианте исполнения основной реактор можно использовать в режиме циркуляции. Рециркуляционное отношение (отношение подачи в кг/ч к возвратному потоку в кг/ч) составляет обычно от 1:5 до 1:100, преимущественно от 1:10 до 1:50, предпочтительно - от 1:15 до 1:35. При этом возможно также частично или полностью отводить теплоту, выделяющуюся в реакции, если пропускать возвратный поток через теплообменник.
В другом варианте исполнения дополнительный реактор можно также встроить в основной реактор.
Время от времени может потребоваться регенерировать катализатор гидрирования из-за уменьшения его активности. Это происходит в соответствии с известными в технике способами, общепринятыми для катализаторов на основе благородных металлов, как рутениевые катализаторы. Здесь следует назвать, например, обработку катализатора кислородом, как описано в патенте Бельгии BE 882279, обработку разбавленными минеральными кислотами, не содержащими галогена, как описано в патенте США US 4072628, или обработку перекисью водорода, например, в виде водных растворов с концентрацией от 0,1 до 35 мас.%, или обработку другими окислителями, преимущественно в виде растворов, не содержащих галогенов. Обычно после реактивации и перед повторным использованием катализатор промывают растворителем, например водой.
Полученный в соответствии с данным способом продукт реакции, следовательно, алициклическое соединение или смесь соответствующих алициклических соединений, можно на стадии с) подвергать дальнейшей очистке.
В случае, если в качестве исходного использован ароматический углеводород и получено соответствующее алициклическое соединение, то можно полученный продукт реакции подвергнуть ректификации, чтобы при необходимости отделить образовавшиеся побочные продукты, например низкокипящие по сравнению с соответствующим алициклическим соединением, как н-гексан и н-пентан, или также высококипящие. Если, например, в качестве исходного использовали бензол, то полученный циклогексан может содержать в качестве примесей, например, н-гексан и н-пентан, которые можно отделить как низкокипящие. В качестве более высококипящих можно при необходимости рассмотреть метилциклогексан, который также можно отделить перегонкой. При ректификации чистый циклогексан получают через боковой отвод колонны, в то время как легкокипящие компоненты отводят из головки колонны, а высококипящие компоненты - из куба колонны. Альтернативно очистка продукта может происходить также в колонне с диафрагмой (разделительной перегородкой), причем здесь чистый циклогексан отводят на высоте диафрагмы.
Если в качестве исходного используют смесь ароматических углеводородов, то отдельные компоненты образующейся смеси алициклических соединений разделяют перегонкой и при необходимости перегонкой отделяют другие примеси.
Выделяющуюся при экзотермической реакции гидрирования теплоту можно при необходимости в случае соответствующего выбора режима давления при перегонке использовать для того, чтобы поддерживать работу испарителя дистилляционной колонны. Для этого можно горячий выгружаемый продукт реакции вводить непосредственно в испаритель колонны или при необходимости нагревать вторичный носитель (например, генерирование пара) и вводить его в испаритель колонны.
Отдельные стадии процесса, как и процесс в целом, можно проводить в непрерывном, полунепрерывном или периодическом режиме.
С помощью способа согласно изобретению можно, таким образом, получать продукты гидрирования, которые не содержат гидрируемого исходного вещества совсем или содержат очень малые остаточные концентрации.
Кроме того, настоящее изобретение касается способа десульфирования ароматического углеводорода, который содержит сернистые ароматические соединения, при необходимости в присутствии водорода, как описано выше на стадии а).
Стадия регенерации
В процессе гидрирования, в котором применяют полученные выше катализаторы, после определенного периода работы можно наблюдать дезактивацию катализатора. Такой дезактивированный катализатор можно вернуть в состояние первоначальной активности путем промывания. Активность может вновь составлять до >90%, преимущественно >95%, более предпочтительно >98%, наиболее предпочтительно >99%, в высшей степени предпочтительно >99,5% от первоначальной величины. Дезактивация объясняется действием следов или остатков воды, адсорбированной на катализаторе. Поразительным образом это можно устранить с помощью промывания инертным газом. Таким образом, способ регенерации согласно изобретению можно охарактеризовать как сушку катализатора или удаление из него воды.
"Промывание" означает, что катализатор вводят в контакт с инертным газом. Обычно для этого с помощью подходящего известного в технике конструктивного решения инертный газ пропускают над катализатором.
Промывание инертным газом проводят при температуре приблизительно от 10 до 350°С, предпочтительно приблизительно от 50 до 250°С, особенно предпочтительно приблизительно от 70 до 180°С, в высшей степени предпочтительно - приблизительно от 80 до 130°С.
Давление при промывании составляет от 0,5 до 5 бар, преимущественно от 0,8 до 2 бар, наиболее предпочтительно - от 0,9 до 1,5 бар.
Согласно изобретению обработку катализатора проводят преимущественно инертным газом. Предпочтительные инертные газы включают азот, углекислый газ, гелий, аргон, неон или их смеси. В высшей степени предпочтительным является азот.
Согласно особому варианту исполнения изобретения процесс регенерации согласно изобретению проводят в том же реакторе, в котором происходило гидрирование, не извлекая катализатор из него. В особенно удобном варианте промывание катализатора согласно настоящему изобретению проводят в реакторе при значениях температуры и давления, которые соответствуют или близки параметрам реакции гидрирования, благодаря чему в результате получается только очень короткое временное прекращение процесса реакции.
Согласно настоящему изобретению промывание инертным газом проводят с объемным расходом от 20 до 200 нл/ч, предпочтительно с объемным расходом от 50 до 200 нл/ч на литр катализатора.
Промывание инертным газом предпочтительно проводят на протяжении от 10 до 50 ч, особенно предпочтительно - от 10 до 20 ч. Например, расчетное время высушивания слоя катализатора на промышленной установке производства циклогексана с предполагаемой влажностью от 2 до 5 мас.% составляет приблизительно от 18 до 30 ч. Промывание в способе согласно изобретению проводят как в нисходящем (down flow), так и в восходящем потоке (up flow).
Следующим предметом настоящего изобретения является объединенный способ гидрирования ароматического углеводорода в присутствии рутениевого катализатора, включающий стадию регенерации катализатора. При этом сначала ароматический углеводород или смесь ароматических углеводородов, в каждом конкретном случае содержащих сернистые ароматические соединения, в соответствии с способом согласно изобретению на стадии а) десульфурируют, а затем на стадии b) гидрируют. После этого на стадии регенерации, как описано выше, катализатор гидрирования регенерируют с помощью инертного газа до достижения полной или частичной первоначальной активности.
Ароматическим углеводородом преимущественно является бензол, в предпочтительном варианте исполнения углеводород представляет собой смесь бензола и толуола, или смесь бензола и ксилола или изомерной смеси ксилолов, или смесь бензола, толуола и ксилола или изомерной смеси ксилолов.
Способ согласно изобретению пригоден, кроме того, для высушивания катализаторов, которые во время различных операций, как техническое обслуживание или хранение, впитали воду.
Приведенные ниже примеры должны прояснять изобретение.
Примеры десульфирования ароматических углеводородов или смеси ароматических углеводородов (стадия а)
Опыты проводили в трубчатом реакторе непрерывного действия с внутренней термопарой (⌀ 6 мм), с сопроводительным обогревом (матрац с обогревом) и устройством для дозировки жидкости.
В качестве десульфуризатора применяли катализатор R 3-12 фирмы BASF AG в виде таблеток 5×3 мм - далее именуемый катализатором А.
Высушивание десульфуризатора происходило согласно вышеописанному. Для этого десульфуризатор нагревали в токе азота с объемным расходом 300±20 нм3/м3 [КАТ]·ч до температуры 200±10°С, причем скорость нагревания не превышала 50 К/ч. Как только была удалена вода, смесь охлаждали до температуры 120±5°С, причем скорость охлаждения не превышала 50 К/ч. Процедуру высушивания производили в оросительном режиме (направление потока сверху вниз).
В некоторых случаях десульфуризатор применяли в его восстановленной форме. При этом, как описано выше, десульфуризатор с помощью водорода переводили из его окисленной формы в восстановленную. Для этого высушенный десульфуризатор (в окисленной форме) нагревали до температуры 120±5°С в токе азота с объемным расходом 300±20 нм3/м3 [КАТ]·ч. Затем к указанному току азота добавляли 0,5±0,1 об.% водорода до тех пор, пока повышение температуры не составило от 15 до 20°С и она осталась постоянной. После этого ток водорода повышали до 1,0±0,1 об.% до тех пор, пока суммарное повышение температуры оказалось не более 30±5°С и температура вновь осталась постоянной. Тогда ток водорода повышали до 2,0±0,2 об.%, причем температура катализатора не превышала 225°С. Ток водорода повышали теперь до 4,0±0,4 об.% и одновременно повышали температуру азота до 200±10°С, причем и здесь температура катализатора не превышала 225°С. Дальнейшее повышение тока водорода до 6,0±0,6 об.% приводило к повышению температуры катализатора до 220±10°С, которую и поддерживали.
По прошествии 1 часа (смесь) охлаждали в токе азота с объемным расходом 300±20 нм3/м3 [КАТ]·ч до температуры ниже 50°С, причем скорость охлаждения не превышала 50±5 К/ч. Процедуру восстановления производили в оросительном режиме (направление потока сверху вниз).
В качестве исходного вещества применяли бензол с чистотой >99,95%.
Анализ используемого бензола и продуктов реакции производили способом газовой хроматографии с указанием площадей под пиками в % (Прибор: HP 5890-2, с пробоотборником; диапазон (?): 4; колонка: 30 м DB1; толщина пленки: 1 мкм; внутренний диаметр колонки: 0,25 мм; объем пробы: 5 мкл; газ-носитель: гелий; скорость потока 100 мл/мин; температура инжектора: 200°С; детектор: FID; температура детектора: 250°С; программа установки температурного режима: 6 мин при 40°С, 10°С/мин до 200°С за 8 мин, общая продолжительность 30 мин).
Анализ на общее содержание серы в используемом бензоле, а также в продуктах реакции производили способом ионообменной хроматографии после сжигания по Уикболду. Для этого от 4 до 6 г образца смешивают с ацетоном (Merck Suprasolv Artikel-Nr. 1.0012.1000) в отношении 1:1 и затем сжигают в кислородно-водородном пламени в агрегате сгорания Уикболда ("сжигание по Уикболду"). Конденсат продуктов сгорания собирают в щелочной приемник, который содержит 40 ммоль КОН (Merck Suprapure, Artikel-Nr. 1.050.020.500) (водный раствор). Образовавшийся из серы сульфат, собранный в приемнике, определяют способом ионообменной хроматографии.
(Система ионообменной хроматографии: модульная система, Fa. Metrohm; предколонка: DIONEX АО 12, 4 мм; разделительная колонка: DIONEX AS 12, 4 мм; элюент: 2,7 ммоль Na2CO3 (Merck Suprapure, Artikel-Nr. 1.063.950.500) и 0,28 ммоль NaHCO3 (Riedel de Haen, p.A. Artikel-Nr. 31437); скорость потока 1 мл/мин; детектирование: проводимость после химического подавления; устройство для подавления: например, MSM, Fa. Metrohm).
Пример а1
100 мл катализатора А, который сушили в соответствии с вышеописанной процедурой сушки, в окисленной форме вводили в описанный выше трубчатый реактор (⌀ 25 мм × 40 см), причем катализатор укладывали в инертную сыпучую массу из кольцевой насадки V4A, которая находится выше и ниже засыпки собственно катализатора. Высота слоя массы собственно катализатора составляла около 22 см. Опыт проводили в жидкофазном режиме при давлении 20 бар, причем во время опыта пропускали азот со скоростью 30 нл/ч, прямотоком с потоком жидкости.
Приведенные в таблице 1 данные ясно показывают, что с помощью катализатора А - в окисленной форме - можно проводить десульфирование используемого бензола.
Пример а2
100 мл катализатора А, который сушили в соответствии с вышеописанной процедурой сушки и восстанавливали в соответствии с вышеописанной процедурой активирования, в восстановленной форме вводили в описанный выше трубчатый реактор (⌀ 25 мм × 80 см), причем катализатор укладывали в инертную сыпучую массу из кольцевой насадки V4A, которая находится выше и ниже засыпки собственно катализатора. Высота слоя массы собственно катализатора составляла около 22 см. Опыт проводили в жидкофазном режиме при давлении 20 бар, причем во время опыта пропускали смесь азота и водорода, прямотоком с потоком жидкости.
Данные, приведенные в таблице 2, ясно показывают, что с помощью катализатора А - в восстановленной форме - можно проводить десульфирование используемого бензола.
Пример а3
100 мл катализатора А, который сушили в соответствии с вышеописанной процедурой сушки и восстанавливали в соответствии с вышеописанной процедурой активирования, в восстановленной форме вводили в описанный выше трубчатый реактор (⌀ 25 мм × 40 см), причем катализатор укладывали в инертную сыпучую массу из кольцевой насадки V4A, которая находится выше и ниже засыпки собственно катализатора. Высота слоя массы собственно катализатора составляла около 22 см. Опыт проводили в жидкофазном режиме при давлении 20 бар, причем во время опыта пропускали водород со скоростью 2 нл/ч, прямотоком с потоком жидкости.
Данные, приведенные в таблице 3, ясно показывают, что с помощью катализатора А - в восстановленной форме - десульфирование используемого бензола можно проводить также при длительной эксплуатации (в непрерывном режиме). Кроме того, эти данные ясно показывают, что восстанавливаются в циклогексан лишь ничтожно малые количества бензола.
По окончании этого длительного опыта использованный катализатор извлекали и анализировали. Для этого катализатор медленно окисляли при температуре около 25-30°С смесью воздуха с азотом или чистым воздухом. Окисленный катализатор извлекали в виде десяти отдельных фракций приблизительно равного объема, в каждом отдельном случае отбирали пробу и подвергали ее элементному анализу. Результаты анализа приведены в Таблице 4. Пробы пронумерованы по направлению потока (жидкофазный режим, фракция 1 внизу, фракция 10 вверху).
Как и следовало ожидать, фракция катализатора на входе в реактор (фракция 1) обнаруживает самую высокую концентрацию серы, в то время как в последней фракции (фракция 10) концентрация наименьшая.
Пример а4
50 мл катализатора А, который сушили в соответствии с вышеописанной процедурой сушки и восстанавливали в соответствии с вышеописанной процедурой активирования, в восстановленной форме вводили в описанный выше трубчатый реактор (⌀ 25 мм × 40 см), причем катализатор укладывали в инертную сыпучую массу из кольцевой насадки V4A, которая находится выше и ниже засыпки собственно катализатора. Высота слоя массы собственно катализатора составляла около 11 см. Опыт проводили в жидкофазном режиме при давлении 3 бар, причем во время опыта пропускали водород со скоростью 2 нл/ч, прямотоком с потоком жидкости.
Данные, приведенные в таблице 5, ясно показывают, что десульфирование можно проводить при давлении 3 бар, температуре 80° и нагрузках на катализатор >5 кгбензол/лкатализатор·ч.
Пример а5
В трубчатый реактор непрерывного действия (⌀ 46 мм × 3500 мм) вводили 3700 мл катализатора А, причем катализатор укладывали в инертную сыпучую массу, которая находится выше и ниже засыпки собственно катализатора (800 соответственно 500 мл). Введенный катализатор А затем сушили и восстанавливали в соответствии с процедурой, представленной в таблице 6.
В заключение проводили десульфирование при давлении от 3 до 32 бар в режиме орошения напуском.
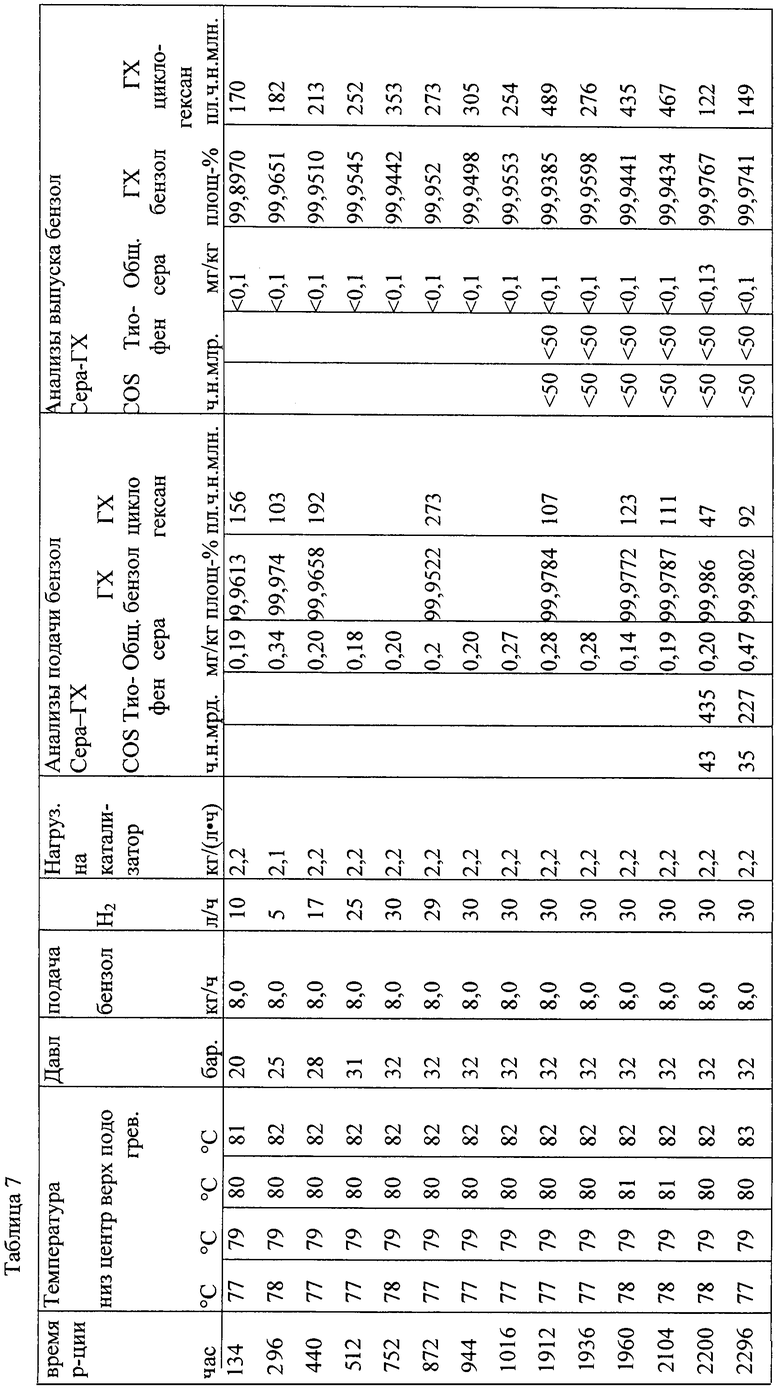
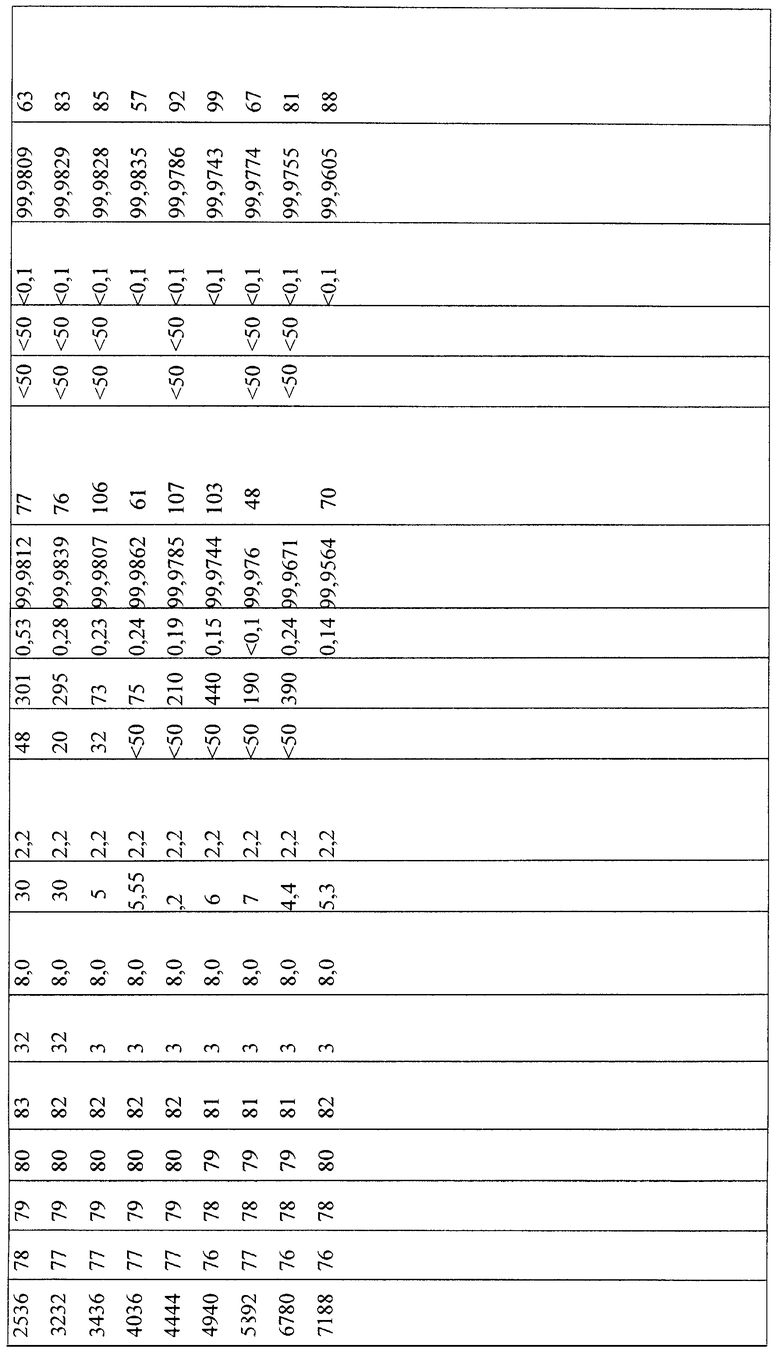
Результаты таблицы 7 ясно показывают, что содержание сернистых ароматических соединений можно понизить до значения ниже 70 ч. на млрд част. (частей на миллиард частей).
Примеры гидрирования ароматического углеводорода или смеси ароматических углеводородов (стадия b).
Общее описание способа 1 (AVB 1)
Опыт проводили в реакторе непрерывного действия (⌀ 12 мм × 1050 мм), с двойной рубашкой, с тремя масляными нагревательными контурами, равномерно распределенными по длине реактора. Реактор эксплуатировали в непрерывном оросительном режиме с циркуляцией жидкости с регулируемым расходом (высокопроизводительный насос). Далее опытная установка была оснащена отделителем жидкой фазы для разделения газа и жидкости, со стабилизацией уровня, устройством для регулирования отходящих газов, внешним теплообменником и пробоотборником. Подачу водорода производили регулируемым давлением (в бар), избыточный водород измеряли регулированием расхода (в нл/ч), подачу исходного бензола производили с помощью насоса высокой производительности. Выгрузку продуктов реакции производили через вентиль, регулировкой уровня. Температуру измеряли с помощью термоэлемента в начале (на входе) и в конце (на выходе) реактора или в массе катализатора. Общее содержание серы в исходном бензоле составляло <0,1 мг/кг (по данным ионообменной хроматографии). В качестве катализатора использовали Ru/Al2O3 мезо-/макропористой структуры, с содержанием рутения 0,47 мас.% Ru (катализатор В), или Ru/SiO2 мезопористой структуры с содержанием рутения 0,32 мас.% Ru (катализатор С). Их получали согласно приведенному описанию. Например, катализатор С получают следующим образом.
50 кг носителя SiO2 (D 11-10 (BASF); 3-миллиметровые (Nr. 04/19668), водопоглощение 0,95 мл/г, поверхность BET 135 м2/г) помещают в барабан для пропитки и насыщают при водопоглощении 96-98 мас.%. Водный пропиточный раствор содержит 0,176 кг Ru в виде его ацетата фирмы Unicore, 4,34 мас.% Ru, (партия 0255). Пропитанный катализатор сушат в печи в статических условиях при температуре 145°С до значения остаточной влажности около 1%. Восстановление проводят в токе водорода (около 75% Н2 в N2, причем N2 используют как продувочный газ; 1,5 нм3/ч Н2 - 0,5 нм3/ч N2) при подвижной массе катализатора, при температуре 300°С и продолжительности 90 мин (1-2 ч). Пассивирование проводят в токе разбавленного воздуха (воздух в N2). Подачу воздуха регулируют таким образом, чтобы температура катализатора держалась ниже 30-35°. Готовый катализатор содержит 0,31-0,32 мас.% Ru.
Ниже этот катализатор описан более подробно.
N2-сорбция: BET 130-131 м2/г (ДИН 66131)
Средний диаметр пор 26-27 нм (ДИН 66134)
Объем пор: 0,84-0,89 мл/г
Hg-порометрия (ДИН 66133): BET 119-122 м2/г
Средний диаметр пор 4V/A 28-29 нм
Объем пор: 0,86-0,87 мл/г
Восстановленный катализатор С содержит, по меньшей мере частично, кристаллический рутений в самой наружной зоне (поверхность прутка). Рутений существует в носителе в виде отдельных частичек размером 1-10 нм (>5 нм иногда): в большинстве случаев 1-5 нм. Размер частиц убывает в направлении снаружи внутрь.
Частицы рутения наблюдаются до глубины 30-50 мкм от поверхности прутка. В этой оболочке рутений присутствует, по меньшей мере частично, в кристаллической форме. Основная часть рутения находится, следовательно, также в этой оболочке (>90% в пределах первых 50 мкм).
Общее описание опыта 2 (AVB 2)
В обогреваемый лабораторный автоклав объемом 1,2 л (внутренний диаметр 90 мм, высота сосуда: 200 мм, изготовлен из спецстали), с 4-лопастной барботажной мешалкой; с перегородками - разделителями потока, и внутренним нагнетательным стояком для отбора проб или для наполнения и освобождения автоклава, - вводят соответствующее используемое количество (по объему или по массе) катализатора в „катализаторной ячейке" (из спецстали).
Для испытания давлением автоклав герметически закрывают и подают 50 бар азота. В заключение снимают давление, с помощью вакуумного насоса в автоклаве создают разрежение, затем соединение с насосом перекрывают, и в автоклав через нагнетательный стояк всасывается исходное вещество или раствор исходного вещества.
Для удаления остаточных количеств кислорода в автоклав поочередно подают и сбрасывают давление - дважды по 10-15 бар азота и дважды по 10-15 бар водорода.
Включают мешалку, устанавливают скорость перемешивания 1000 об/мин и нагревают раствор до температуры реакции. Необходимая температура достигается не позднее чем через 15 мин. Водород до соответствующего требуемого давления нагнетают в пределах 5 мин. Рассчитывают расход водорода и постоянно поддерживают соответствующее необходимое значение давления.
Через равные промежутки времени через нагнетательный стояк отбирают предварительные пробы (для промывания стояка) и пробы реакционной смеси для наблюдения за ходом реакции.
По прошествии соответствующего времени отключают нагревание, охлаждают автоклав до 25°С, медленно снимают избыточное давление и с помощью небольшого избыточного давления выгружают реакционную смесь через стояк. После этого с помощью вакуумного насоса в автоклаве создают разрежение, затем соединение с насосом перекрывают, и в автоклав через нагнетательный стояк всасывается новое исходное вещество или раствор исходного вещества.
Эта процедура позволяет использовать катализатор многократно. Используемый водород имел чистоту не менее 99,9-99,99 об.% (в пересчете на сухой газ). Примесями являются окись углерода (не более 10 об.ч. на млн), азот (не более 100 об.ч. на млн), аргон (не более 100 об.ч. на млн) и вода (не более 400 об.ч. на млн).
Используемые реакционные смеси анализировали способом газовой хроматографии с указанием ГХ-площадей в % (Прибор: HP 5890-2, с пробоотборником; степень: 4; колонка: 30 м DB1; толщина пленки: 1 мкм; внутренний диаметр колонки: 0,25 мм; объем пробы: 5 мкл; газ-носитель: гелий; скорость потока 100 мл/мин; температура инжектора: 200°С; детектор: FID; температура детектора: 250°С; программа установки температурного режима: 6 мин при 40°С, 10°С/мин до 200°С за 8 мин, общая продолжительность 30 мин).
Анализ на общее содержание серы в используемом бензоле, а также в продуктах реакции производили способом ионообменной хроматографии после сжигания по Уикболду. Для этого от 4 до 6 г образца смешивают с ацетоном (Merck Suprasolv Artikel-Nr. 1.0012.1000) в отношении 1:1 и затем сжигают в кислородно-водородном пламени в агрегате сгорания Уикболда ("сжигание по Укболду"). Конденсат продуктов сгорания собирают в щелочной приемник, который содержит 40 ммоль КОН (Merck Suprapure, Artikel-Nr. 1.050.020.500) (водный раствор). Образовавшийся из серы сульфат, собранный в приемнике, определяют способом ионообменной хроматографии.
(Система ионообменной хроматографии: модульная система, Fa. Metrohm; предколонка: DIONEX AG 12, 4 мм; разделительная колонка: DIONEX AS 12, 4 мм; элюент: 2,7 ммоль Na2CO3 (Merck Suprapure, Artikel-Nr. 1.063.950.500 и 0,28 ммоль NaHCO3 (Riedel de Haen, p.A. Artikel-Nr. 31437); скорость потока 1 мл/мин; детектирование: проводимость после химического подавления; устройство для подавления: например, MSM, Fa. Metrohm).
Пример b1 (по AVB 1)
104 мл (63,9 г) катализатора В использовали для непрерывного гидрирования при давлении водорода 32 бар, отходящем газе 1-5 нл/ч, при температуре на входе в реактор 88-100°С и отношении подача/циркуляция 1:30. Результаты сведены в таблицу 8.
Пример b2 (по AVB 1)
104 мл (63,9 г) катализатора В использовали для непрерывного гидрирования при давлении водорода 19 бар, отходящем газе 1-5 нл/ч, при температуре на входе в реактор 88-100°С и отношении подача/циркуляция 1:30. Результаты сведены в таблицу 9.
Пример b3 (по AVB 1)
104 мл (45,0 г) катализатора С использовали для непрерывного гидрирования при давлении водорода 32 бар, отходящем газе 1-5 нл/ч, при температуре на входе в реактор 88-100°С и отношении подача/циркуляция 1:30. Результаты сведены в таблицу 10.
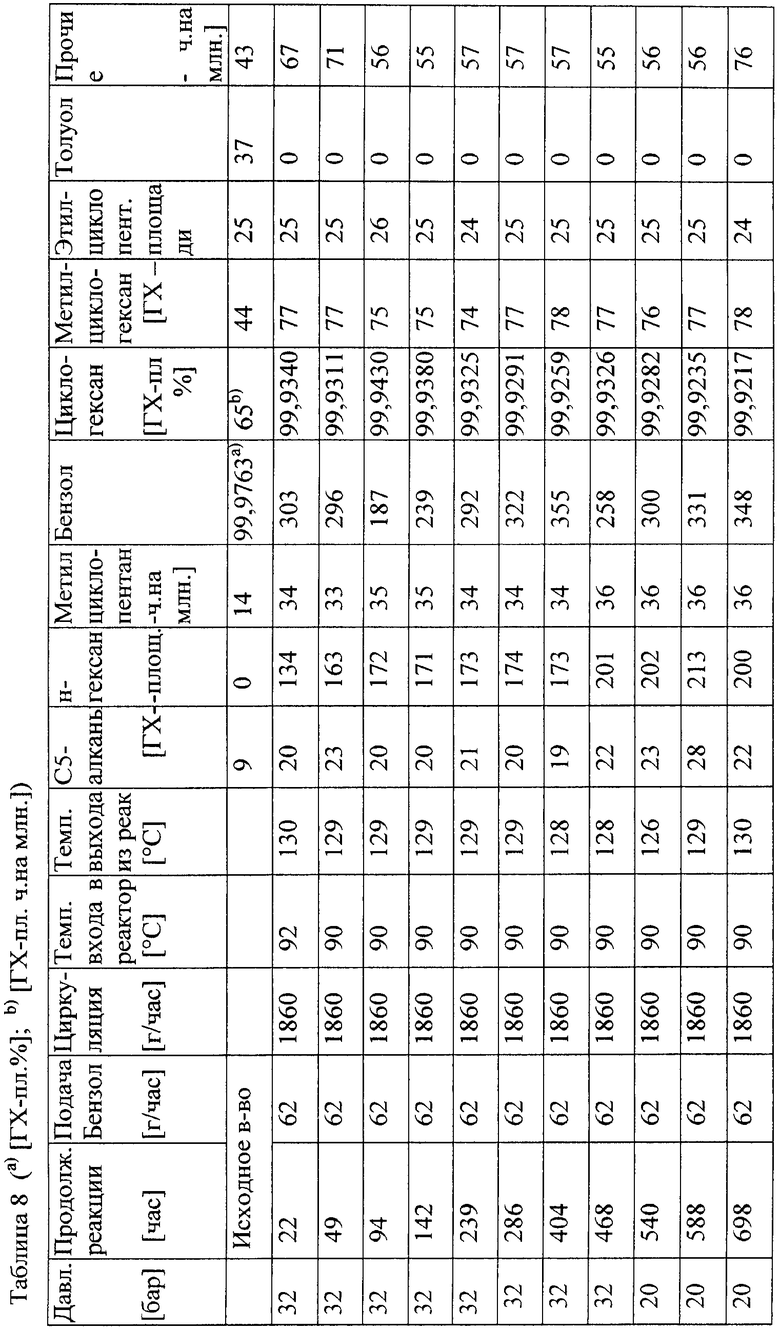
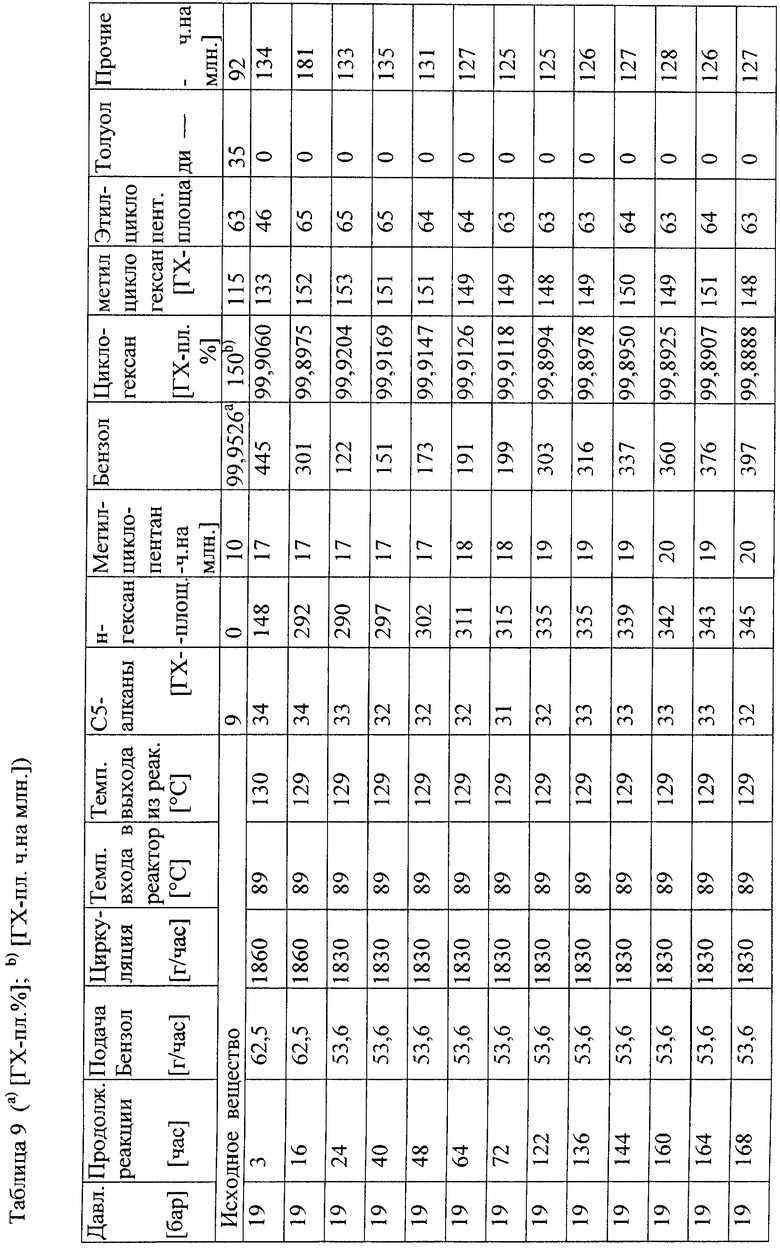
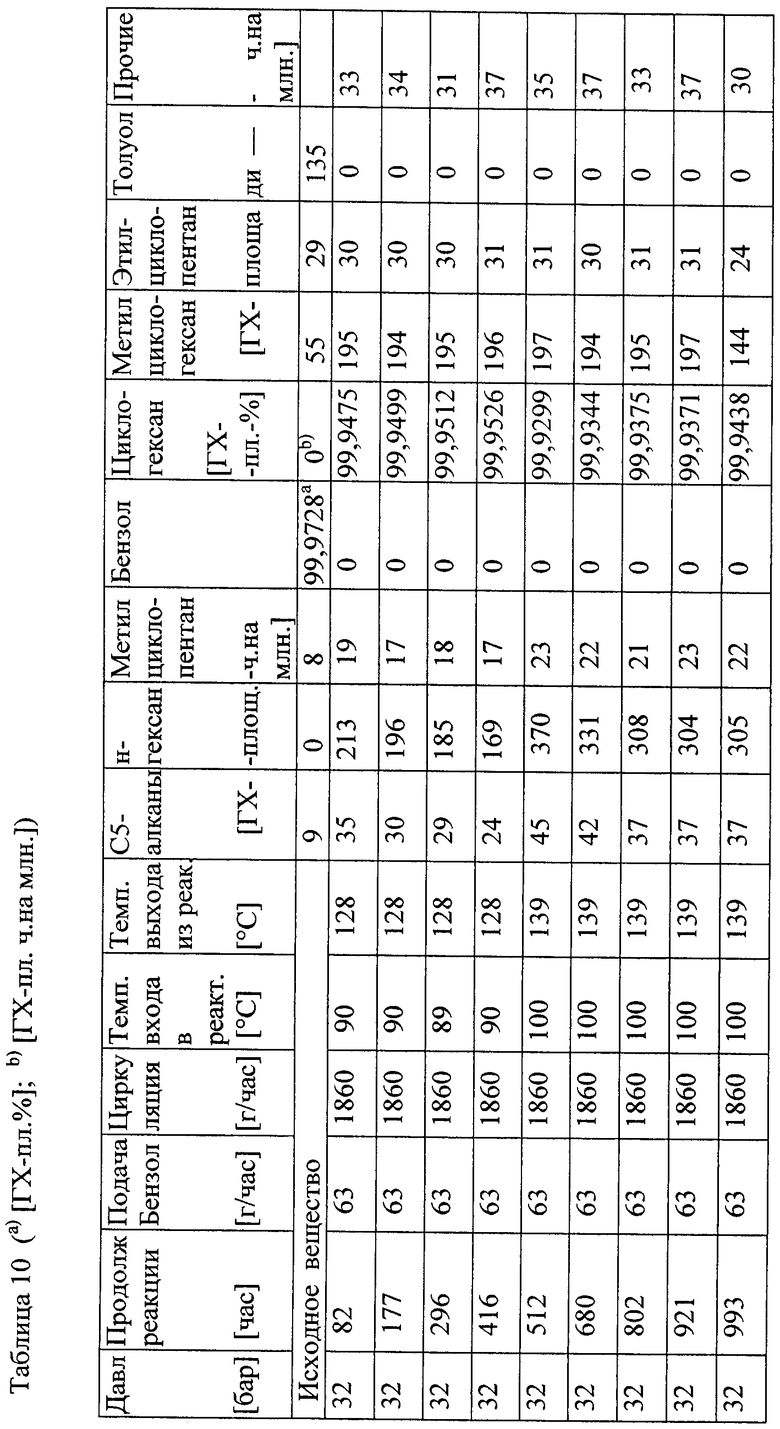
Данные, сведенные в таблицы 8-10, ясно показывают, что можно получать циклогексан с отличной селективностью.
Пример b4
Установка для гидрирования состоит из складской емкости для десульфурированного бензола, приемного резервуара, дозировочного насоса для бензола, основного реактора (⌀ 45×2000 мм) с отделителем жидкой фазы для разделения газа и жидкости и регулирующим устройством для стабилизации уровня; системой циркуляции жидкости с насосом и теплообменником для отвода выделяющейся теплоты реакции; из дополнительного реактора (⌀ 22 мм, L 1500 мм) с отделителем для разделения газа и жидкости и регулировкой для стабилизации уровня, а также складской емкостью для продуктов гидрирования. Основной и дополнительный реакторы были снабжены внутренним термоэлементом (⌀ 6 мм в основном реакторе, ⌀ 3 мм в дополнительном реакторе). Оба реактора работали в оросительном режиме. Жидкость и газ подавали прямотоком.
В основной реактор загружали 2700 мл (1870 г), в дополнительный - 340 мл (229 г) катализатора В. Для изоляции и сопутствующего подогрева основной реактор был снабжен электрическими нагревательными плитами. Пространство выше и ниже слоя катализатора заполняли инертной сыпучей массой (колечки проволочной сетки из спецстали).
Исходным веществом был десульфурированный бензол, полученный аналогично описанному в примерах а4 или а5, с общим содержанием серы <0,1 мг/кг.
Бензол и циклогексан анализировали способом газовой хроматографии с указанием ГХ-площадей в % или ГХ-пл. в ч. на млн; анализы проводили без внутреннего стандарта (Прибор: HP 5890-2, с пробоотборником; диапазон (?): 4; колонка: 30 м DB1; толщина пленки: 1 мкм; внутренний диаметр колонки: 0,25 мм; объем пробы: 5 мкл; газ-носитель: гелий; скорость потока 100 мл/мин; температура инжектора: 200°С; детектор: FID; температура детектора: 250°С; программа установки температурного режима: 6 мин при 40°С, 10°С/мин до 200°С за 8 мин, общая продолжительность 30 мин).
Анализ на общее содержание серы в используемом бензоле, а также в продуктах реакции производили способом ионообменной хроматографии после сжигания по Уикболду. Для этого от 4 до 6 г образца смешивают с ацетоном (Merck Suprasolv Artikel-Nr. 1.0012.1000) в отношении 1:1 и затем сжигают в кислородно-водородном пламени в агрегате сгорания Уикболда ("сжигание по Уикболду"). Конденсат продуктов сгорания собирают в щелочной приемник, который содержит 40 ммоль КОН (Merck Suprapure, Artikel-Nr. 1.050.020.500) (водный раствор). Образовавшийся из серы сульфат, собранный в приемнике, определяют способом ионообменной хроматографии.
(Система ионообменной хроматографии: модульная система, Fa. Metrohm; предколонка: DIONEX AG 12, 4 мм; разделительная колонка: DIONEX AS 12, 4 мм; элюент: 2,7 ммоль Na2CO3 (Merck Suprapure, Artikel-Nr. 1.063.950.500 и 0,28 ммоль NaHCO3 (Riedel de Haen, p.A. Artikel-Nr. 31437); скорость потока 1 мл/мин; детектирование: проводимость после химического подавления; устройство для подавления: например, MSM, Fa. Metrohm).
Частично содержание серы определяли способом газовой хроматографии (пределы обнаружения - по 50 ppb для COS и тиофена (разделительная колонка: СР SIL88 (100% цианопропилполисилоксан), длина: 50 м; толщина пленки: 0,2 мкм; внутренний диаметр: 0,25 мм; газ-носитель: гелий; давление подкачки: 1,5 бар; дробная доза: на колонке (мл/мин); продувание мембраны: 5 мл/мин; температура печи: 60°С; время предварительного нагрева: 10 мин; скорость 1: 5°С/мин; температура печи 1:200°С; время дополнительного нагрева 1:10 мин; скорость 2: -; температура печи 2: -; время дополнительного нагрева 2: -; температура инжектора: на колонке (°С); температура детектора: 220°С; ввод в хроматограф: ГХ-автоматический пробоотборник; объем пробы: 1,0 мкл; тип детектора: PFPD (пламенный фотометр; ГХ-способ: массовые проценты с внешней калибровкой; специфика: ввод НА колонку и особый пламефотометрический детектор).
Сначала установка работала при давлении 20 бар, после 860 часов работы давление на установке повысили до 32 бар. В смежном дополнительном реакторе гидрировали до полного превращения; среди продуктов реакции бензол практически не обнаруживался.
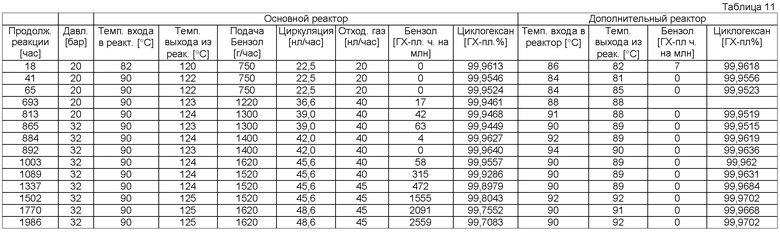
Данные результаты показывают, что способ согласно изобретению делает возможным осуществлять полное превращение бензола и получать циклогексан высокой степени чистоты.
Пример b5
Опыт проводили в той же установке и при тех же условиях, которые описаны в примере b4). Правда, в основной реактор загружали 2700 мл (1218 г) катализатора С, а в дополнительный - 340 мл (153 г) катализатора С. Кроме того, установка с самого начала работала при давлении 32 бар.
Полученные здесь результаты также показывают, что бензол практически не обнаруживается среди продуктов реакции.
Далее по прошествии 5347 час работы в подачу исходного вещества добавили 4,3 мас.% толуола. В продуктах реакции обнаруживали соответствующие количества метилциклогексана, но толуол отсутствовал.
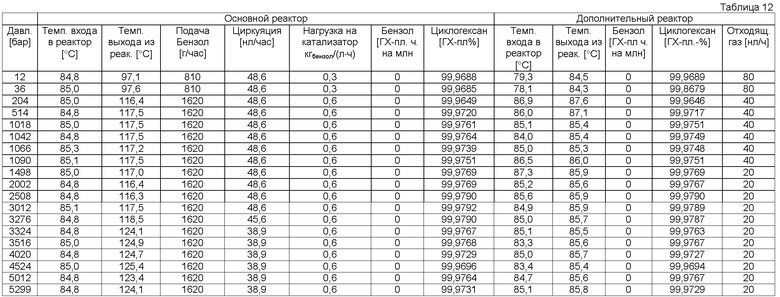
Пример b6 (по AVB 2)
750 мл 5%-ного раствора бензола в циклогексане в присутствии 9,0 г (около 22 мл) катализатора С гидрировали водородом при температуре 100°С и давлении 20 бар.
Катализатор в каждом из пяти последовательных опытов использовали повторно. Пробы отбирали по прошествии 10, 20, 30, 40, 60, 90, 120 и 180 минут реакции.
В таблице 13 приведены данные понижения содержания бензола во времени. Использованы средние значения результатов пяти опытов, а также максимальные отклонения вверх и вниз от среднего значения результатов соответствующих проб. Определение содержания бензола производили способом ГХ-анализа по ГХ-площадям в %.
Пример b7 (по AVB 2)
750 мл 5%-ного раствора бензола в циклогексане в присутствии 9,0 г (около 22 мл) катализатора С гидрировали водородом при температуре 100°С и давлении 32 бар.
Катализатор в каждом из пяти последовательных опытов использовали повторно. Пробы отбирали по прошествии 10, 20, 30, 40, 60, 90, 120 и 180 минут реакции.
В таблице 14 приведены данные понижения содержания бензола во времени. Использованы средние значения результатов пяти опытов, а также максимальные отклонения вверх и вниз от среднего значения результатов соответствующих проб. Определение содержания бензола производили способом ГХ-анализа по ГХ-площадям в %.
Пример с
Регенерация катализатора гидрирования
Пример получения рутениевого катализатора
Мезо-/макропористый алюмооксидный носитель в виде 3-5 мм сфер с суммарным объемом пор 0,44 см3/г, причем 0,09 см3/г (20% суммарного объема) составляют поры с диаметром от 50 нм до 10000 нм, и 0,35 см3/г (80% суммарного объема) составляют поры с диаметром от 2 нм до 50 нм (средний диаметр пор - около 11 нм и поверхность - 286 м2/г), пропитывали водным раствором нитрата рутения (III). При этом объем раствора, поглощенного во время пропитки, приблизительно соответствовал объему пор использованного носителя. В заключение носитель, пропитанный раствором нитрата рутения (III), сушили при 120°С и активировали водородом (восстанавливали) при 200°С в токе водорода.
Полученный таким образом катализатор содержал 0,5 мас.% рутения, в пересчете на массу катализатора. Поверхность рутения составляла 0,72 м2/г. Отношение поверхности рутения к поверхности носителя составляло 0,0027.
Пример 1. Исследования сорбции
С помощью измерений поглощения водяного пара на катализаторе, полученном согласно описанному выше (0,5% Ru/γ-Al2O3), определяли сродство катализатора к воде.
Оказалось, что уже при относительно небольшом давлении пара (30%) катализатор поглощает 5% воды. Если в реакторе или в исходных веществах вода также присутствует только в следовых количествах, катализатор может сорбировать эту воду.
Пример 2. Испытание на долговечность при гидрировании бензола
В установке для получения циклогексана в присутствии катализатора рутений/оксид алюминия с содержанием 0,5% Ru на носителе γ-Al2O3 в процессе производства наблюдается постоянное снижение активности катализатора и возрастающее содержание бензола. Дальнейшее наблюдение за реакцией показало, что в течение испытания катализатора на долговечность при гидрировании бензола остаточное содержание бензола на выходе из основного реактора за время работы около 3400 ч возрастает по величине от нескольких сотен ч.на млн. до нескольких тысяч ч. на млн. Расчет показал, что при подаче 16620 кг/ч бензола с содержанием воды от 30 до 50 ч. на млн в установку вводится 0,8 кг воды в час. Сюда попадают еще дополнительные 3,5 кг/ч воды, которые приносит газообразный водород.
В случае выключения установки после 3394 часов работы установка работала с остаточным содержанием бензола 0,2% при нагрузке на катализатор 0,6 гбензол/млкат·ч. Во время отключения в установку нагнетали азот при температуре от 70 до 100°С, а затем снимали давление. После повторного запуска установка выпускает реакционную смесь с остаточным содержанием бензола от 0,01% до 0,4% при нагрузке на катализатор 0,6 гбензол/млкат·ч.
Наблюдаемый в этом случае эффект высушивания катализатора был вновь подтвержден данными после 7288 часов работы. При нагрузке на катализатор 0,9 гбензол/млкат·ч остаточное содержание бензола на выходе из установки составляло 0,2% и даже повышалось до 0,56%. После выключения установки катализатор сушили током азота 100 нл/ч при температуре 110°С в течение 34 ч. После повторного запуска установки с нагрузкой на катализатор 0,6 гбензол/млкат·ч остаточное содержание бензола составляло от 0,03% до 0,07%, что можно объяснить заметным повышением активности катализатора благодаря высушиванию.
В обоих случаях высушивание катализатора приводило к значительно более высокой активности катализатора, которая была близка по величине или равна первоначальной активности катализатора.
Пример 3. Изучение влияния воды на гидрирование бензола
Для моделирования влияния воды на гидрирование бензола в присутствии рутениевого катализатора проводили серию опытов в автоклаве до и после насыщения катализатора водой, а также после высушивания катализатора. В лабораторный автоклав загружали 5%-ный раствор бензола в циклогексане вместе с рутениевым катализатором, смесь нагревали до температуры 100°С и следили за ходом реакции при давлении водорода 32 бар с помощью регулярного отбора проб. В заключение пробы анализировали способом газовой хроматографии.
Были проведены 23 опыта гидрирования, затем катализатор вводили в контакт с водой. После этого были проведены следующие 13 опытов гидрирования. Катализатор показывал заметно пониженную, но почти постоянную активность. После высушивания катализатора в реакционной трубке в токе азота при 100°С были проведены следующие 5 опытов; катализатор показывал активность гидрирования, подобную той, что была до насыщения водой.
Опыты подтверждают, что активность используемых катализаторов рутений/оксид алюминия после контакта с водой значительно понижается, однако катализатор можно вновь реактивировать с помощью высушивания в токе азота и почти полностью восстановить первоначальную активность.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗОЛА, ТОЛУОЛА И КСИЛОЛОВ | 1990 |
|
RU2026852C1 |
| СПОСОБ ГИДРИРОВАНИЯ АРОМАТИЧЕСКОГО СОЕДИНЕНИЯ ПОСРЕДСТВОМ РЕАКЦИОННОЙ ДИСТИЛЛЯЦИИ | 2001 |
|
RU2277079C2 |
| СЕЛЕКТИВНАЯ ИЗОМЕРИЗАЦИЯ КСИЛОЛОВ И КОНВЕРСИЯ ЭТИЛБЕНЗОЛА | 2000 |
|
RU2233260C2 |
| Способ приготовления катализатора для гидроочистки бензол-толуол-ксилольной фракции пироконденсатов | 1990 |
|
SU1734818A1 |
| Способ получения бензола и ксилолов и катализатор для получения бензола и ксилолов | 1990 |
|
SU1796604A1 |
| РУТЕНИЕВЫЕ КАТАЛИЗАТОРЫ | 2002 |
|
RU2322293C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА НА ОСНОВЕ МОЛИБДЕНА И ПЛАТИНЫ ДЛЯ СИНТЕЗА БЕНЗОЛА ПУТЕМ ТРАНСАЛКИЛИРОВАНИЯ | 2017 |
|
RU2757851C2 |
| НАНОСТРУКТУРИРОВАННЫЙ КАТАЛИЗАТОР ГИДРИРОВАНИЯ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ С6-С8 | 2019 |
|
RU2696957C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОМЕРОВ КСИЛОЛА (ВАРИАНТЫ) | 2006 |
|
RU2484078C2 |
| СПОСОБ ДИСПРОПОРЦИОНИРОВАНИЯ ТОЛУОЛА | 1993 |
|
RU2131862C1 |
Изобретение относится к вариантам способа гидрирования бензола, смесей бензола и толуола, смесей бензола и ксилола или изомерной смеси ксилола или смесей бензола, толуола и ксилола или изомерной смеси ксилола, содержащих сернистые ароматические соединения, в одном из которых на первой стадии, при необходимости в присутствии водорода, содержание сернистых ароматических соединений снижают в присутствии десульфуризатора, содержащего медь и цинк в атомном отношении от 1:0,3 до 1:10 (стадия а), и на второй стадии бензол, смеси бензола и толуола, смеси бензола и ксилола или изомерной смеси ксилола или смеси бензола, толуола и ксилола или изомерной смеси ксилола гидрируют в присутствии нанесенного на носитель рутениевого катализатора, содержащего от 0,01 до 30 мас.% рутения, в пересчете на общую массу катализатора, в присутствии водорода (стадия b). Также изобретение относится к способу десульфуризации, использующему тот же самый десульфуризатор. Применение настоящего способа позволяет получать алициклические соединения или их смеси с высокой селективностью и выходом. 3 н. и 21 з.п. ф-лы, 14 табл.
1. Способ гидрирования бензола, смесей бензола и толуола, смесей бензола и ксилола, или изомерной смеси ксилола, или смесей бензола, толуола и ксилола, или изомерной смеси ксилола, содержащих сернистые ароматические соединения, в котором на первой стадии, при необходимости в присутствии водорода, содержание сернистых ароматических соединений снижают в присутствии десульфуризатора, содержащего медь и цинк в атомном отношении от 1:0,3 до 1:10 (стадия а), и на второй стадии бензол, смеси бензола и толуола, смеси бензола и ксилола, или изомерной смеси ксилола, или смеси бензола, толуола и ксилола, или изомерной смеси ксилола гидрируют в присутствии нанесенного на носитель рутениевого катализатора, содержащего от 0,01 до 30 мас.% рутения, в пересчете на общую массу катализатора, в присутствии водорода (стадия b).
2. Способ по п.1, в котором на стадии а) содержание сернистых ароматических соединений понижают до значения ≤70 ч. на млрд. ч., а общее содержание серы до суммарного значения ≤200 ч. на млрд. ч. при температуре от 40 до 200°С и давлении от 1 до 40 бар.
3. Способ по п.1, в котором на стадии а) используют десульфуризатор, содержащий медь и цинк в атомном отношении от 1:0,5 до 1:3.
4. Способ по п.1, в котором десульфуризатор содержит от 35 до 45 мас.% оксида меди, от 35 до 45 мас.% оксида цинка, от 10 до 30 мас.% оксида алюминия.
5. Способ по п.1 или 2, в котором стадию а) проводят в отсутствии водорода.
6. Способ по п.1 или 2, в котором стадию а) проводят в присутствии водорода.
7. Способ по п.1 или 2, в котором десульфуризатор на стадии а) используют в окисленной форме.
8. Способ по п.1 или 2, в котором десульфуризатор на стадии а) используют в восстановленной форме.
9. Способ по п.1 или 2, в котором стадию а) проводят при температуре от 50 до 180°С и давлении от 1 до 32 бар.
10. Способ по п.1 или 2, в котором содержание рутения в нанесенном на носитель рутениевом катализаторе, используемом на стадии b), составляет от 0,01 до 5 мас.%, в пересчете на общую массу катализатора.
11. Способ по п.1 или 2, в котором нанесенный на носитель рутениевый катализатор, используемый на стадии b), в качестве носителя содержит оксид алюминия.
12. Способ по п.1 или 2, в котором нанесенный на носитель рутениевый катализатор, используемый на стадии b), в качестве носителя содержит оксид кремния.
13. Способ по п.12, в котором нанесенный на носитель рутениевый катализатор представляет собой поверхностный катализатор, в котором по меньшей мере 60 мас.% активного металла находится в оболочке катализатора, с глубиной проникновения до 200 мкм.
14. Способ по п.1 или 2, в котором стадию b) проводят при температуре от 50 до 250°, предпочтительно от 60 до 200°С, особенно предпочтительно от 70 до 170°С, и давлении от 1 до 200 бар, особенно от 10 до 50 бар, предпочтительно от 19 до 40 бар, наиболее предпочтительно от 25 до 35 бар.
15. Способ по п.1 или 2, в котором гидрированию подвергают бензол.
16. Способ по п.1, в котором стадию а) проводят при давлении от 2 до 4,5 бар, температуре от 50 до 180°С, в присутствии десульфуризатора, содержащего от 35 до 45 мас.% оксида меди, от 35 до 45 мас.% оксида цинка, от 10 до 30 мас.% оксида алюминия, используемого в восстановленной форме, а стадию b) проводят при давлении от 19 до 40 бар, температуре от 70 до 170°С, в присутствии нанесенного на оксид алюминия рутениевого катализатора, содержание рутения в котором составляет от 0,01 до 30 мас.%, в пересчете на общую массу катализатора.
17. Способ по п.1, в котором стадию а) проводят при давлении от 2 до 4,5 бар, температуре от 50 до 180°С, в присутствии десульфуризатора, содержащего от 35 до 45 мас.% оксида меди, от 35 до 45 мас.% оксида цинка, от 10 до 30 мас.% оксида алюминия, используемого в восстановленной форме, а стадию b) проводят при давлении от 19 до 40 бар, температуре от 70 до 170°С, в присутствии нанесенного на оксид кремния рутениевого катализатора, содержание рутения в котором составляет от 0,01 до 30 мас.%, в пересчете на общую массу катализатора.
18. Способ по п.16 или 17, в котором стадию а) проводят в присутствии водорода.
19. Способ гидрирования бензола, смесей бензола и толуола, смесей бензола и ксилола, или изомерной смеси ксилола, или смесей бензола, толуола и ксилола, или изомерной смеси ксилола, содержащих сернистые ароматические соединения, который кроме указанных в пп.1-18 стадий а) и b) и их параметров включает стадию с), на которой полученный продукт реакции подвергают дальнейшей очистке.
20. Способ по п.19, в котором дальнейшую очистку на стадии с) осуществляют путем дистилляции.
21. Объединенный способ гидрирования бензола, смесей бензола и толуола, смесей бензола и ксилола или изомерной смеси ксилола или смесей бензола, толуола и ксилола или изомерной смеси ксилола, содержащих сернистые ароматические соединения, который кроме указанных в пп.1-20 стадий а) и b) и их параметров включает стадию регенерации, включающую продувку катализатора инертным газом при температуре от 10 до 350°С и давлении от 0,5 до 5 бар в течение от 10 до 50 ч.
22. Способ десульфирования бензола, смесей бензола и толуола, смесей бензола и ксилола, или изомерной смеси ксилола, или смесей бензола, толуола и ксилола, или изомерной смеси ксилола, содержащих сернистые ароматические соединения, включающий обработку десульфуризатором, содержащим от 35 до 45 мас.% оксида меди, от 35 до 45 мас.% оксида цинка, от 10 до 30 мас.% оксида алюминия, при атомном отношении меди и цинка, равном от 1:0,3 до 1:10, при необходимости в присутствии водорода, причем содержание сернистых ароматических соединений понижается до значения ≤70 ч. на млрд. ч., а общее содержание серы до суммарного значения ≤200 ч. на млрд. ч.
23. Способ по п.22, причем десульфирование проводят в присутствии водорода.
24. Способ по п.22, причем десульфирование проводят в отсутствие водорода.
| Тепловой расходомер | 1983 |
|
SU1157356A1 |
| Способ управления кровлей при разработке пластовых месторождений | 1981 |
|
SU1104275A1 |
| US 2003113598 А1, 19.06.2003 | |||
| 0 |
|
SU403658A1 | |

