Данное изобретение относится к производным хиназолинтиазолинона, которые обладают CDK1 антипролиферативной активностью и используются в качестве противораковых агентов.
Циклинзависимые киназы (CDKs) являются серинтреонинпротеинкиназами, которые играют решающую роль в регуляции переходов между различными фазами клеточного цикла, такого как переход из находящейся в покое стадии в G1 (гэп между митозом и началом ДНК репликации из-за нового раунда клеточного деления) в S фазу (период активного синтеза ДНК) или перехода от G2 в М фазу, в которой происходит активный митоз и клеточное деление. (См., например, статьи, собранные в Science, 274: 1643-1677 (1996); и Ann. Rev. Cell Dev. Biol., 13: 261-291 (1997)). CDK комплексы формируются посредством объединения регуляторных циклиновых субъединиц (например, циклин A, B1, B2, D1, D2, D3 и Е) и каталитической киназной субъединицы (например, CDK1, CDK2, CDK4, CDK5 и CDK6). Это наименование подразумевает, что CDKs проявляют полную зависимость от циклиновых субъединиц, чтобы фосфорилировать субстраты, являющиеся их мишенями, и функция различных киназа/циклиновых пар состоит в регуляции перехода через специфические фазы клеточного цикла.
Как отмечено выше, эти протеинкиназы представляют собой класс белков (ферментов), регулирующих различные клеточные функции. Это сопровождается фосфорилированием специфических аминокислот в белковых субстратах, в результате чего происходит конформационное изменение субстратного белка. Конформационное превращение модулирует активность субстрата или его способность к взаимодействию с другими связывающими партнерами. Ферментная активность протеинкиназы связана со скоростью, с которой киназа присоединяет фосфатные группы к субстрату. Она может быть измерена, например, определением количества субстрата, которое превращается в продукт, в виде функции времени. Фосфорилирование субстрата происходит на активном участке протеинкиназы.
С точки зрения приведенных выше свойств эти киназы принимают важное участие в распространении сигнальной трансдукции фактора роста, которая приводит к клеточной пролиферации, дифференцировке и миграции. Фактор роста фибропласта (FGF) и васкулярный эндотелиальный фактор роста (VEGF) были признаны в качестве важнейших медиаторов, промотирующих опухолевый ангиогенез. VEGF активирует эндотелиальные клетки посредством передачи сигнала через два близко родственных рецептора, одним из которых является киназная вставка доменсодержащего рецептора (KDR) (See, Hennequin L. F. et. al., J. Med. Chem. 45(6): 1300 (2002)). FGF активирует эндотелиальные клетки посредством передачи сигнала через FGF рецептор (FGFR). Рост твердых опухолей зависит от образования новых кровеносных сосудов (ангиогенез). Соответственно ингибиторы рецепторов FGFR и KDR, которые препятствуют росту сигнальной трансдукции и вследствие этого ослабляют или предотвращают ангиогенез, служат агентами профилактики и лечения твердых опухолей (See, Klohs W.E. et. al., Current Opinion in Biotechnology, 10:544 (1999).
Вследствие того что CDKs, например CDK1, служат главными активаторами клеточного деления, ингибиторы CDK1 могут быть использованы в качестве антипролиферативных агентов. Эти ингибиторы могут служить для развития терапевтического вмешательства в подавление разрегурированного развития клеточного цикла.
В соответствии с изобретением было показано, что соединения формулы I
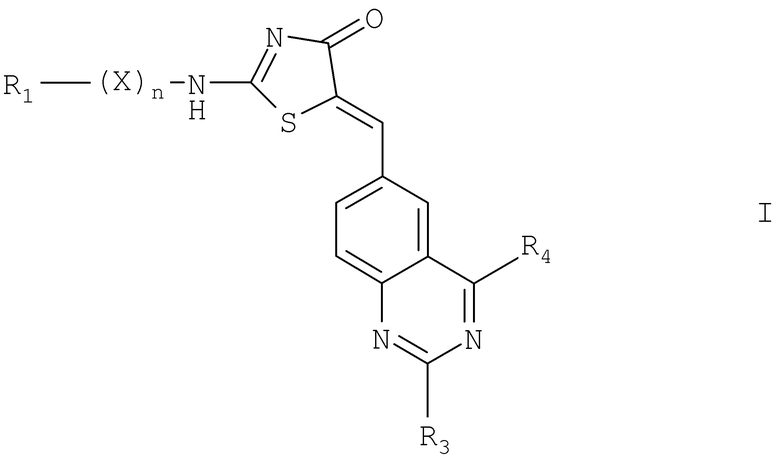
где
R1 обозначает водород, низший алкил или 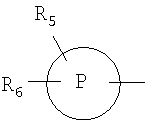 ;
;
Х выбирают из низшего алкилена, цикло(низшего алкилена), содержащего от 3 до 6 атомов углерода, и гидрокси(низшего алкилена);
 выбирают из
выбирают из
арильного кольца,
циклоалкильного кольца, содержащего от 3 до 6 атомов углерода,
4-6-членного гетероциклоалкильного кольца, содержащего от 3 до 5 атомов углерода, и от 1 до 2 гетероатомов, выбранных из группы, состоящей из кислорода, азота и серы, и
5- или 6-членного гетероароматического кольца, содержащего от 1 до 2 гетероатомов, выбранных из группы, состоящей из кислорода, серы и азота;
R5 и R6 независимо выбирают из группы, включающей водород, гидроксигруппу, гидроксигруппу(низший алкил), низший алкил, галоген, перфтор(низший алкил) и низшую алкоксигруппу;
R3 выбирают из водорода, -NH-R7 и -NH-C(O)-R8;
R4 выбирают из водорода, низшего алкила и -O(H2H2O)у-R10;
R7 обозначает водород или низший алкил;
R8 и R10 обозначают низший алкил;
n обозначает целое число от 0 до 1; и
y обозначает целое число от 0 до 3;
при условии, что когда n обозначает 0 и R1 обозначает водород или низший алкил, тогда R3/R4 не могут оба обозначать водород; или
N-оксиды соединений, где R1 содержит азот в гетероароматическом кольце, сульфоны, где R1 содержит серу в гетероциклоалкильном кольце или гетероароматическом кольце; или
их фармацевтически приемлемые соли, ингибируют активность CDKs, в частности CDK1. Эти агенты по изобретению и фармацевтические композиции, содержащие такие агенты, применяются в лечении различных болезней и болезненных состояний, связанных с неконтролируемой или нежелательной клеточной пролиферацией, такой как рак, с аутоиммунными болезнями, вирусными болезнями, грибковыми заболеваниями, нейродегенеративными расстройствами и сердечно-сосудистыми заболеваниями.
Ингибирование и/или модуляция активности CDKs, в частности CDK1, делает такие соединения формулы I и композиции, содержащие эти соединения, применимыми при лечении болезней, модулируемых киназной активностью, особенно в качестве противоопухолевых агентов при лечении раковых заболеваний, более предпочтительно твердых опухолей, таких как рак грудной железы, рак легкого, рак толстой кишки и рак простаты.
Как отмечено выше, соединения формулы I являются потенциальными анти-пролиферативными агентами и служат для опосредования и/или ингибирования активности CDKs, в частности CDK1, являясь вследствие этого противоопухолевыми агентами для лечения рака или других болезней, связанных с неконтролируемой или аномальной клеточной пролиферацией.
В одном предпочтительном варианте по настоящему изобретению предлагаются соединения формулы I,
где
R1 обозначает водород, или ;
Х выбирают из низшего алкилена, цикло(низшего алкилена), содержащего от 3 до 6 атомов углерода, и гидрокси (низшего алкилена);
выбирают из
арильного кольца,
цикло(низший алкил)кольца, содержащего от 3 до 6 атомов углерода,
4-6-членного гетероциклоалкильного кольца, содержащего от 3 до 5 атомов углерода и от 1 до 2 гетероатомов, выбранных из группы, состоящей из кислорода, азота и серы, и
5- или 6-членного гетероароматического кольца, содержащего от 1 до 2 гетероатомов, выбранных из группы, состоящей из кислорода, серы и азота;
R5 и R6 независимо выбирают из группы, включающей водород, гидроксигруппу, гидроксигруппу(низший алкил), низший алкил, галоген, перфтор(низший алкил) и низшую алкоксигруппу;
R3 выбирают из водорода, -NH-R7 и -NH-C(O)-R8;
R4 выбирают из водорода, низшего алкила и -O(CH2CH2O)y-R10;
R7 обозначает водород или низший алкил;
R8 и R10 обозначают низший алкил;
n обозначает целое число от 0 до 1; и
y обозначает целое число от 0 до 3;
при условии, что когда n=0, один из R1, R3 и R4 не является водородом; или
N-оксиды соединений, где R1 содержит азот в гетероароматическом кольце, сульфоны, где R1 содержит серу в гетероциклоалкильном кольце или гетероароматическом кольце; или
их фармацевтически приемлемые соли.
Предпочтительными соединениями формулы I являются соединения, где n=0. Эти соединения включают соединения формулы:
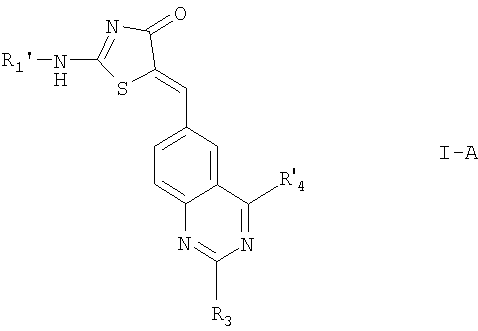
где R1' обозначает водород или низший алкил;
R4' обозначает низший алкил или -O(H2H2O)y-R10; и
R3, R10 и у определены выше; или
их фармацевтически приемлемые соли.
Соединения формулы I, где n=0, также включают соединения:
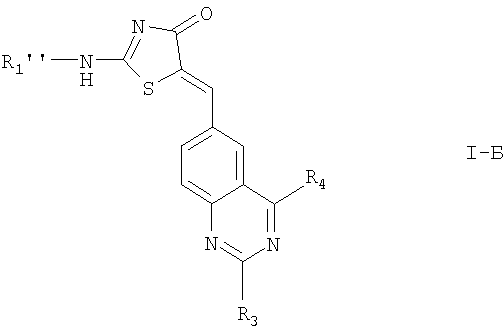
где
R1'' обозначает ;
R3, R4, R5, R6 и определены выше; или
N-оксиды соединений, где R1'' содержит азот в гетероароматическом кольце, сульфоны, где R1'' содержит серу в гетероциклическом кольце или гетероароматическом кольце; или
их фармацевтически приемлемые соли.
Когда в соединении формулы I n=1, это соединение имеет формулу:
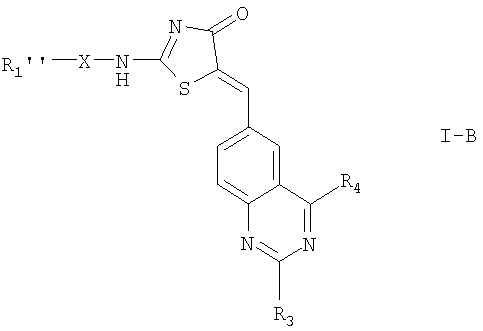
где
R1'', X, R3, R4 и определены выше; или
N-оксиды соединений, где R1'' содержит азот в гетероароматическом кольце, сульфоны, где R1'' содержит серу в гетероциклическом кольце или гетероароматическом кольце; или
их фармацевтически приемлемые соли.
В соединениях I, I-Б и I-B, где R1 и R1'' являются заместителями, содержащими арильный фрагмент, предпочтительным арильным фрагментом является фенил. Используемые в данном описании галогены включают все четыре галогена: хлор, фтор, бром и йод.
Как использовано в описании, термин "низший алкил", самостоятельно или в комбинации, означает моновалентную, прямолинейную или разветвленную насыщенную углеводородную группу, содержащую от 1 до 6 атомов углерода, такую как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил и подобные им.
Термин "циклоалкил" означает цикло(низший алкил) заместитель, который представляет собой 3-6-членное насыщенное углеводородное кольцо. Предпочтительными циклоалкильными заместителями являются циклопропил, циклобутил, циклогексил и т.п., при этом циклопропил является особенно предпочтительным.
Термин "низшая алкоксигруппа" обозначает прямолинейную или разветвленную -O-низшую алкильную группу, образованную из низшего алкила, содержащего от 1 до 6 атомов углерода, такую как метоксигруппа, этоксигруппа, н-пропоксигруппа, изопропоксигруппа, н-бутоксигруппа, трет-бутоксигруппа и подобные им.
Термин "арил" обозначает моновалентное, моно- или бициклическое незамещенное ароматическое углеводородное кольцо, такое как фенил или нафтил, при этом фенил является предпочтительным.
Термин "гетероциклоалкил" относится к 4-6-членному моноциклическому насыщенному кольцу, содержащему от 3 до 5 атомов углерода и 1 или 2 гетероатома, выбранных из группы, включающей кислород, азот или серу. Предпочтительные гетероциклические алкильные группы включают морфолинил, тиопиранил или тетрагидропиранил.
Термин "гетероароматическое кольцо" относится к моновалентному 5 или 6-членному моноциклическому гетероароматическому кольцу, содержащему от 4 до 5 атомов углерода и 1 или 2 гетероатома, выбранных из группы, включающей кислород, азот или серу. Предпочтительные гетероароматические группы включают тиофенил, тиоазол, пиридинил, фуранил и т.п.
Термин "гидроксигруппа" или "гидроксил" означает -ОН.
Термин "гидрокси(низший алкил)" означает низшую алкильную группу, по определению выше, которая является замещенной, предпочтительно монозамещена гидроксильной группой.
Термин "низший алкенил" означает дивалентный, насыщенный, прямолинейный или разветвленный углеводородный заместитель, содержащий от 1 до 6 атомов углерода.
Термин "циклоалкилен" или "цикло(низший алкилен)" означает цикло(низший) алкиленовый заместитель, который является дивалентным, незамещенным, 3-6-членным, насыщенным углеводородным кольцом. Предпочтительными циклоалкиленовыми заместителями являются циклопропенил или циклобутенил.
Термин "низшая алканоилоксигруппа-низший алкилен" означает низший алкиленовый заместитель, замещенный, предпочтительно монозамещенный, низшей алканоилоксигруппой, при этом "низшая алканоилоксигруппа" означает группу -С(O)O-низший алкил, а термин "низший алкил" определен выше.
Термин "низшая алкоксигруппа-низший алкилен" означает низший алкиленовый заместитель, как описано выше, замещенный, предпочтительно монозамещенный, низшей алкоксигруппой, где низшая алкоксигруппа определена выше.
Термин " гидроксигруппа(низший алкилен)" означает низший алкиленовый заместитель, замещенный, предпочтительно монозамещенный, гидроксильной группой.
Термин "низшая алкоксигруппа-низший алкил" означает низший алкильный заместитель, как определено выше, замещенный, предпочтительно монозамещенный, низшей алкоксигруппой, где низшая алкоксигруппа определена выше.
Термин "перфтор(низший алкил)" означает любую низшую алкильную группу, в которой все атомы водорода замещены фтором. Предпочтительными перфтор(низший алкил) группами являются трифторметил, пентафторэтил, гептафторпропил, при этом особенно предпочтительной группой является трифторметильная группа.
Термин «фармацевтически приемлемая соль» относится к стандартным кислотно-аддитивным или основно-аддитивным солям, которые сохраняют биологическую эффективность и свойства соединений формул I, II, III, IV и V и образуются из соответствующих нетоксичных органических и неорганических кислот или органических или неорганических оснований. Примеры кислотно-аддитивных солей включают соли, полученные из неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота и азотная кислота, и соли, полученные из органических кислот, таких как п-толуолсульфокислота, салициловая кислота, метансульфоновая кислота, щавелевая кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота и подобных им. Примеры основноаддитивных солей включают соли, полученные из гидроксидов аммония, калия, натрия и четвертичного аммониевого основания, такого как, например, гидроксид тетраметиламмония. Технология химической модификации фармацевтических соединений (например, лекарств) в соли для получения соединений с улучшенной физической и химической стабильностью, гигроскопичностью, сыпучестью и растворимостью хорошо известна химикам-фармацевтам. См., например, Н. Ansel et. al., Pharmaceutical Dosage Forms and Drug Delivery Systems (6th Ed. 1995) стр.196 и 1456-1457.
В соответствии с настоящим изобретением соединения формулы I могут быть получены из соединений формулы:
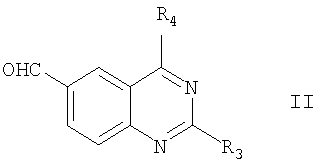
где R3 и R4 определены выше.
Соединение формулы II превращается в соединение формулы I по следующей реакционной схеме 1, где X, R1, R3, R4 и n определены выше.
Схема 1
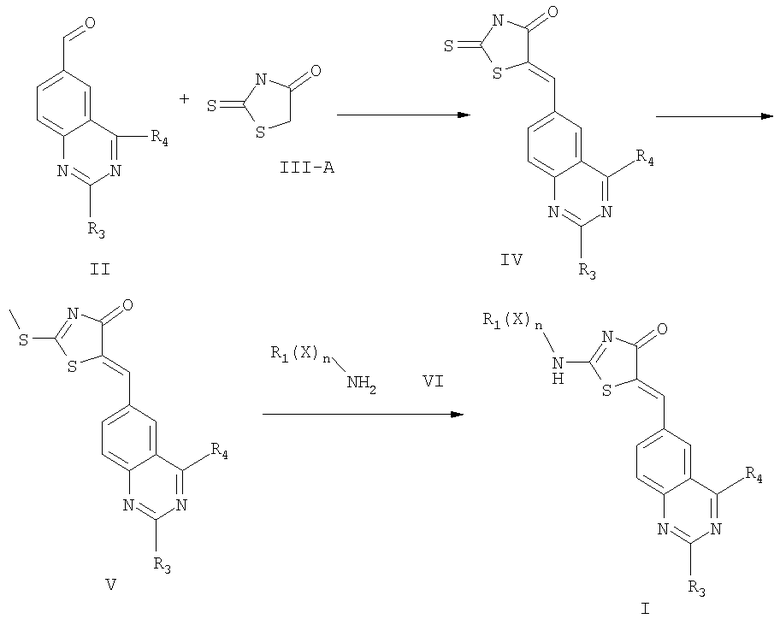
В соответствии с настоящим изобретением соединение формулы II вводят в реакцию с соединением формулы III-A [роданин (2-тио-4-тиазолин-4-он)] посредством реакции Кновенагеля с получением соединения формулы IV. При этом при проведении этой конденсации может быть использовано любое из стандартных условий реакции Кновенагеля. Обычно эту реакцию проводят при нагревании с обратным холодильником в присутствии ацетата щелочного металла и уксусной кислоты.
На следующей стадии этого синтеза образовавшийся замещенный тиазолидин формулы IV обрабатывают метилирующим агентом для метилирования тиогруппы соединения формулы IV с получением соединения формулы V. Предпочтительным метилирующим агентом является йодметан. Эта реакция проводится в органическом аминном основании, таком как диизопропилэтиламин (ДИЭА). При проведении этой реакции температура и давление не являются критическими, и эта реакция проводится при комнатной температуре и атмосферном давлении. В действительности, при проведении этой реакции может быть использовано любое из стандартных условий метилирования тиогруппы.
На следующей стадии синтеза соединение формулы V вводят в реакцию с соединением формулы VI для получения соединения формулы I. Соединение формулы VI является амином, поэтому в этой реакции может быть использовано любое стандартное условие для аминного замещения метилтиогруппы. В соответствии с одним вариантом это замещение проводят посредством реакции соединения формулы VI с соединением формулы V в присутствии стандартного растворителя, такого как ацетонитрил. Обычно эта реакция проводится в присутствии органического аминного основания, такого как диизопропилэтиламин.
С другой стороны, соединение формулы I может быть получено реакцией соединения формулы II с соединением формулы:

где R1 обозначен выше.
Реакцию соединения формулы VII с соединением формулы II для получения соединения формулы I проводят в органическом растворителе, таком как бензол или толуол, при высокой температуре от 100°С до 200°С в замкнутой системе. При использовании этого метода реакция проводится при высоких температурах и давлении. Соединение формулы VII может быть получено непосредственным замещением с использованием реакции соединения формулы
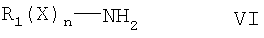
где R1, X и n определены выше,
с соединением формулы III-A. Реакция замещения проводится обычно в присутствии активатора и амина в качестве основания. Предпочтительным активатором является хлорид ртути. Эту реакцию проводят в инертном органическом растворителе. При этом может быть использован любой стандартный инертный органический растворитель, такой как ацетонитрил, метиленхлорид и т.п. При проведении этой реакции используется аминное основание, такое как диизопропилэтиламин. При проведении этой реакции температура и давление не являются критическими, и эта реакция проводится при комнатной температуре и атмосферном давлении. При проведении этой реакции может быть использован любой из стандартных методов замещения меркаптогруппы амином.
В соответствии с вариантом по настоящему изобретению соединение формулы II, где R4 обозначает -O(CH2CH2O)y-R10, a R10 и у определены выше, имеет формулу:
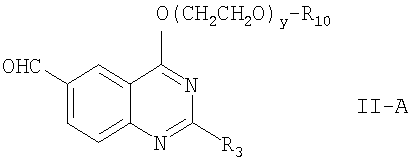
где R3, R10 и у определены выше.
Соединение формулы II-A может быть получено из соединения формулы
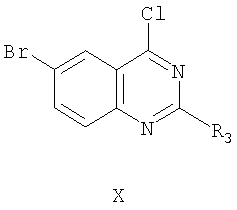
реакцией с соединением формулы,
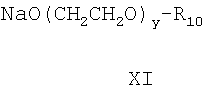
где R10 и у определены выше, с получением соединения
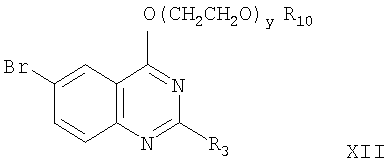
где R10, R3 и y определены выше.
Реакцию соединения формулы Х с соединением формулы XI для получения соединения формулы XII проводят по стандартной методике путем взаимодействия хлорида с алкоксидом щелочного металла с получением простого эфира. Любые стандартные условия взаимодействия хлорида с алкоксидом щелочного металла могут быть использованы для получения соединения формулы XII. На следующей стадии этой реакции для получения соединения формулы II-А используют реакцию формилирования для превращения брома в СНО заместитель фенильного кольца. Эту реакцию проводят введением соединения формулы XI в реакцию с моноксидом углерода в присутствии дифенилпропилфосфина (дфф) и основания с использованием ацетата палладия в качестве катализатора при температуре от 60 до 100°С. Давление, применяемое в этой реакции, обычно составляет от 480 до 550 кПа. Любой стандартный метод формилирования для превращения галоидной группы фенильного кольца посредством реакции с моноксидом углерода может быть применен при проведении этой конденсации.
Когда R3 в соединении формулы Х обозначает -NHR7, это соединение имеет формулу:
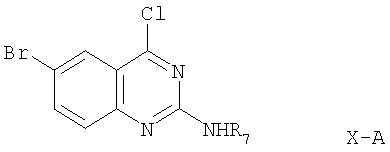
где R7 обозначен выше.
Соединение формулы Х-А может быть получено из соединения формулы:
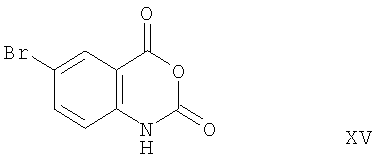
реакцией соединения формулы XV (синтез соединения формулы XV описан в примере 12б) с соединением:
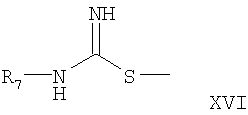
где R7 обозначен выше,
с получением соединения формулы:
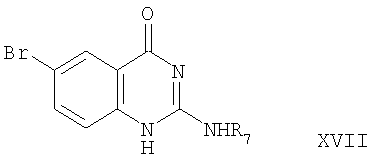
где R7 обозначен выше,
которое может быть превращено в соединение формулы Х-А.
Соединение формулы XV превращают в соединение формулы XVII реакцией соединения формулы XV с соединением формулы XVI. Эта реакция проводится добавлением соединения формулы XVI к соединению формулы XV в инертном растворителе. В качестве реакционной среды может быть использован любой инертный растворитель, такой как ацетонитрил и вода. Реакция протекает при нагревании образовавшейся смеси с обратным холодильником. Соединение формулы XVII может быть превращено в соединение формулы Х-А путем обработки стандартным хлорирующим агентом, например оксихлоридом фосфора. Эта реакция проводится обычно при нагревании с обратным холодильником в атмосфере азота.
Соединение формулы XVI коммерчески доступно или может быть получено из соответствующей тиомочевины, R7-NH-C(=S)-NH2, с алкилирующим агентом, таким как йодметан.
В соединениях формулы I, I-Б и I-B предпочтительным является такой класс соединений, где R3 и R4 оба обозначают водород, и такой класс соединений, где R3 обозначает -NHR7 и R4 обозначает водород или -O(CH2CH2O)y-R10.
В соединениях формулы I и I-Б, где n=1, заместители R1 и R1' обозначают кольца, причем предпочтительным кольцом является фенил, который может быть незамещенным или замещенным заместителями R5 и R6.
Одним из вариантов соединения формулы I-A являются соединения, где R1' обозначает водород. Среди этого класса соединений предпочтительными являются соединения, где R4' обозначает -O(CH2CH2O)y-R10.
Другим вариантом настоящего изобретения являются соединения формулы I-Б, где n=0 и обозначает фенил или гетероароматическое кольцо, содержащее от 1 до 2 гетероатомов. Предпочтительными гетероароматическими кольцами являются кольца, содержащие два гетероатома, один из которых является азотом, а другой серой, при этом тиазол является наиболее предпочтительным. Другим предпочтительным гетероароматическим кольцом является кольцо, содержащее один гетероатом, предпочтительно серу.
В соответствии с другим вариантом соединением формулы I-Б, где n=1, являются такие соединения, где Х обозначает низший алкиленовый заместитель. Этот вариант включает класс соединений, где R1 содержит незамещенное или замещенное фенильное кольцо. Другим классом соединений являются соединения, где n=1, и такие соединения, где Х является низшим алкиленовым заместителем и R1' обозначает гетероароматическое кольцо. Предпочтительными гетероароматическими кольцами являются такие, которые содержат два гетероатома, один из которых является азотом, а другой серой, при этом тиазол является наиболее предпочтительным. Другим предпочтительным гетероароматическим кольцом является кольцо, содержащее один гетероатом, предпочтительно серу, при этом особенно предпочтительным является тиофен. В этом предпочтительном классе соединений R4 предпочтительно обозначает -O(CH2CH2O)y-R10.
Другим вариантом соединения формулы I-Б, где n=1, являются соединения, где Х обозначает гидрокси(низший алкилен). Этот класс соединений включает соединения, где R1 обозначает гетероароматическое кольцо. Предпочтительными гетероароматическими кольцами являются такие, которые содержат два гетероатома, один из которых является азотом, а другой серой, при этом тиазол является наиболее предпочтительным. Другим предпочтительным гетероароматическим кольцом является кольцо, содержащее один гетероатом, предпочтительно серу, при этом особенно предпочтительным является тиофен. В этом предпочтительном классе соединений R4 предпочтительно обозначает -O(CH2CH2O)y-R10. Другим классом соединений, где n=1 и X' обозначает гидрокси(низший алкилен), являются такие соединения, где R1 обозначает незамещенный или замещенный фенил. В этом предпочтительном классе соединений R4 предпочтительно обозначает -O(CH2CH2O)y-R10.
Фармацевтические композиции по изобретению могут, альтернативно или в дополнение к соединению формулы I, включать в качестве активного ингредиента фармацевтически приемлемые лекарства, фармацевтически активные метаболиты и фармацевтически приемлемые соли таких соединений и метаболитов. Такие соединения, пролекарства, мультимеры, соли и метаболиты иногда относят к общей группе под названием "активные агенты" или "агенты."
В случае твердых агентов специалистам в данной области техники известно, что представленные в изобретении соединения и соли могут существовать в различных кристаллических или полиморфных формах, все из которых входят в объем данного изобретения и конкретные формулы.
Терапевтически эффективные количества активных агентов по изобретению могут применяться для лечения болезней, опосредованных модуляцией или регуляцией протеинкиназ CDK1. "Эффективное количество" подразумевает такое количество агента, которое существенно ингибирует пролиферацию и/или предупреждает дедифференциацию эукариотической клетки, например клетки млекопитающего, насекомого, растительной или грибной клетки, и является эффективным для указанного применения, например специфического терапевтического лечения.
Количество данного агента, соответствующего эффективному количеству, варьируется в зависимости от таких факторов, как применяемое соединение, болезненное состояние и его тяжесть, идентификация организма (например, масса тела) субъекта или реципиента, нуждающегося в лечении, но может быть, тем не менее, стандартно определено методом, известным из области техники в соответствии с обстоятельствами, связанными с заболеванием, включая, например, специфичность вводимых агентов, метод введения, болезненное состояние, требующее лечения, и субъекта или реципиента, подлежащего лечению. "Лечение" подразумевает, по крайней мере, облегчение болезненного состояния у субъекта, такого как млекопитающее (например, человек), которое вызывается, по крайней мере частично, активностью CDK1 протеинкиназы, и включает: предотвращение заболевания, встречающегося у млекопитающего, особенно, когда, как было найдено, млекопитающее имеет предрасположение к такому заболеванию, но которое еще не диагностировано в качестве имеющегося; модуляцию и/или ингибирование такого болезненного состояния; и/или облегчение болезненного состояния.
Далее, настоящее изобретение относится к способам модуляции или ингибирования активности протеинкиназы CDK1, например, в тканях млекопитающих, посредством введения агента по изобретению. Антипролиферативная активность агентов легко определяется известными методами, например, при использовании целых клеточных культур с помощью МТТ анализа. Активность агентов по изобретению в качестве модуляторов CDK1 протеинкиназной активности может быть определена любым из методов, доступных специалистам в области техники, включая анализы в условиях in vivo и/или in vitro. Примеры соответствующих анализов для измерения активности включают примеры, описанные в международной заявке WO 99/21845; Parast et al., Biochemistry, 37, 16788-16801 (1998); Connell-Crowley and Harpes, Cell Cycle: Materials and Methods (Michele Pagano, ed. Springer, Berlin, Germany) (1995); международных заявках WO 97/34876 и WO 96/14843. Эти свойства могут оцениваться, например, посредством одного или более биологических тестирующих методов, изложенных в приведенных ниже примерах.
Активные агенты по изобретению могут быть введены в фармацевтические композиции, как описано ниже. Фармацевтические композиции по изобретению включают эффективное модулирующее, регулирующее или ингибирующее количество соединения формулы I и инертный фармацевтически приемлемый носитель или разбавитель. В одном из вариантов фармацевтических композиций эффективные уровни агентов по изобретению включают количество, необходимое для осуществления терапевтической поддержки, включая антипролиферативное действие. Под "эффективными уровнями" имеются в виду уровни, при которых пролиферация ингибируется или контролируется. Эти композиции изготавливаются в форме единичных доз, соответствующих методу введения, например, парентеральному или оральному введению.
Агент по изобретению может быть введен в стандартных дозах, комбинирующих терапевтически эффективное количество агента (например, соединения формулы I) в качестве активного ингредиента с соответствующими фармацевтическими носителями или разбавителями с помощью стандартных методик. Эти методики могут включать смешивание, гранулирование и прессование или растворение ингредиентов в соответствии с изготовлением необходимой формы.
Применяемый фармацевтический носитель может представлять собой твердое вещество или жидкость. Примерами твердых носителей могут служить лактоза, сахароза, тальк, желатин, агар, пектин, камедь, стеарат магния, стеариновая кислота и им подобные. Примерами жидких носителей могут служить сироп, масло земляного ореха, оливковое масло, вода и подобные им. Аналогично носитель или разбавитель может включать материалы, способствующие задержке во времени или реализации во времени, такие как глицерилмоностеарат или глицерилдистеарат, в отдельности, или в смеси с воском, этилцеллюлозой, гидроксипропилметилцеллюлозой метилметакрилатом и им подобными.
Может использоваться целый ряд фармацевтических форм. Так, если используется твердый носитель, изготовление может осуществляться посредством таблетирования, в форме порошков или пелет, заполняемых в твердые желатиновые капсулы, в форме таблеток или леденцов. Количество твердого носителя также варьируется. Если используется жидкий носитель, изготовление осуществляется в виде сиропа, эмульсии, мягких желатиновых капсул, стерильных инъекционных растворов или суспензий в ампулах или пузырьках, или неводных жидких суспензий.
Чтобы получить стабильные водорастворимые дозированные формы, фармацевтически приемлемая соль агента по изобретению может быть растворена в водном растворе органической или неорганической кислоты. Если растворимая солевая форма является неприемлемой, агент может быть растворен в соответствующем сорастворителе или комбинациях растворителей.
Если будет признано, что существующие дозированные формы агентов, применяемых в композициях по настоящему изобретению, должны изменяться по отношению к данному комплексу, подлежащему применению, формируется особая композиция, выбирается особое место введения, реципиент и болезнь, подлежащая лечению. Оптимальные дозы для данного набора условий могут быть установлены специалистами в данной области техники с использованием стандартных тестов определения дозы на основе экспериментальных данных для агента.
Композиции по изобретению могут быть получены методами, в основном известными в приготовлении фармацевтических композиций, с использованием стандартных технологий, таких как смешивание, растворение, гранулирование, дражирование, растирания в порошок, эмульгирование, инкапсулирование, включение в полимер или лиофилизация. Фармацевтические композиции могут быть сформированы с использованием одного или более физиологически приемлемых носителей, выбираемых из наполнителей и вспомогательных веществ, которые облегчают функционирование активных соединений в фармацевтических препаратах.
Для орального применения соединения могут быть быстро сформированы посредством комбинации соединений с фармацевтически приемлемыми носителями, известными из уровня техники. Такие носители способствуют формированию соединений по изобретению в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и им подобных, для орального применения пациентом, подлежащим лечению. Фармацевтические препараты для орального применения могут быть получены с использованием твердых наполнителей в смеси с активным ингредиентом (агентом), необязательно с растиранием образующейся смеси, и обработкой смеси гранул после добавления соответствующих вспомогательных веществ, если желательно, для получения таблеток или драже.
Изобретение иллюстрируется далее следующими примерами, которые не ограничивают объем настоящего изобретения. В названных примерах температуры приведены в градусах Цельсия (°С), если не оговорено особо.
Примеры
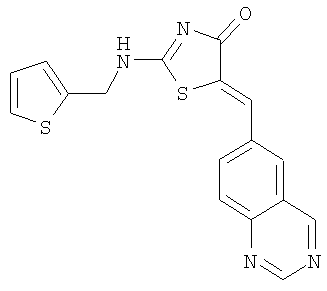
Пример 1: 5-[1-Хиназолин-6-илмет-(Z)-илиден]-2-[(тиофен-2-илметил)амино]тиазол-4-он
а) Получение 6-хиназолинкарбоксальдегида
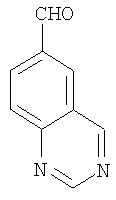
Смесь 6-метилхиназолина (5,0 г, 34,7 ммолей) и диоксида селена (7,7 г, 69,4 ммолей) нагревают при 160°С в течение 12 ч. После охлаждения до комнатной температуры при перемешивании добавляют метанол. Затем удаляют твердое вещество фильтрованием и фильтрат концентрируют. Ускоренная хроматография (Merck силикагель 60, 230-400 мэш, 0%-30% этилацетата в гексане в течение 30 мин) дает 6-хиназолинкарбоксальдегид (выход 2,4 г, 43,7%) в виде белого твердого вещества: ЖХ-МС m/e 159 (МН+).
б) Получение 5-хиназолин-6-илметилен-2-тиоксотиазолидидин-4-она
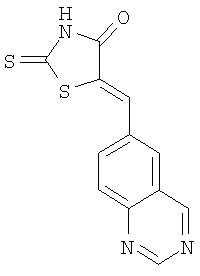
Суспензию 6-хиназолинкарбоксальдегида (пример 1а, 1,5 г, 9,5 ммолей), роданина (1,26 г, 9,5 ммолей) и ацетата натрия (3,11 г, 38 ммолей) в уксусной кислоте (10 мл) перемешивают при 130°С в течение 12 ч. После охлаждения до комнатной температуры добавляют воду (40 мл). Твердое вещество отделяют фильтрованием, промывают водой и высушивают, получая 5-хиназолин-6-илметилен-2-тиоксотиазолидин-4-он (выход 2,6 г, 100%) в виде твердого вещества. ЖХ-МС m/е 274 (МН+).
в) Получение 2-метилсульфанил-5 -хиназолин-6-илметилентиазол-4-она
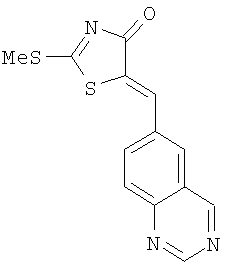
Суспензию 5-хиназолин-6-илметилен-2-тиоксотиазолидин-4-она (пример 16, 2,6 г, 9,52 ммолей), йодметана (1,2 мл, 19,0 ммолей) и диизопропилэтиламина (ДИЭА) (2,4 мл, 14,3 ммолей) в безводном этаноле (100 мл) перемешивают при комнатной температуре в течение 12 ч. После добавления воды (200 мл) твердое вещество отделяют фильтрованием, промывают водой и высушивают, получая 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-он (выход 2,5 г, 92%) в виде черного твердого вещества. ЖХ-МС m/е 288 (МН+).
г) Получение 5-[1-хиназолин-6-илмет-(Z)-илиден]-2-[(тиофен-2-илметил)амино]тиазол-4-она
Суспензию 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она (пример 1в, 58 мг, 0,2 ммоля), тиофенметиламина (45,3 мг, 0,4 ммоля) и диизопропилэтиламина (ДИЭА) (70 мкл, 0,4 ммоля) в ацетонитриле (1 мл) нагревают до 145°С посредством микроволнового облучения в течение 20 мин. После охлаждения до комнатной температуры твердое вещество отделяют фильтрованием, промывают небольшим количеством ацетонитрила и высушивают. Ускоренная хроматография (Merck силикагель 60, 230-400 мэш, 0%-5% метанола в метиленхлориде в течение 30 мин) дает 5-[1-хиназолин-6-илмет-(Z)-илиден]-2-[(тиофен-2-илметил)амино]тиазол-4-он в виде слегка желтого твердого вещества: ЖХ-МС m/е 353 (МН+).
Пример 2: 5-[1-Хиназолин-6-илмет-(Z)-илиден]-2-(тиазол-2-иламино)тиазол-4-он
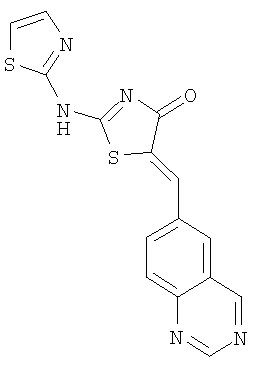
Используют методику, аналогичную описанной в примере 1 г, и исходя из 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она, тиазол-2-иламина и ДИЭА получают 5-[1-хиназолин-6-илмет-(Z)-илиден]-2-(тиазол-2-иламино)тиазол-4-он: ЖХ-МС m/е 340 (МН+).
Пример 3: 2-[2-(3-Фторфенил)этиламино]-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он
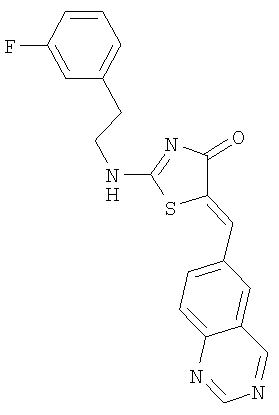
Используют методику, аналогичную описанной в примере 1 г, и исходя из 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она, 2-(3-фторфенил)этиламина и ДИЭА, получают 2-[2-(3-фторфенил)этиламино]-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он: ЖХ-МС m/е 379 (МН+).
Пример 4: 2-(2-Этоксифениламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он
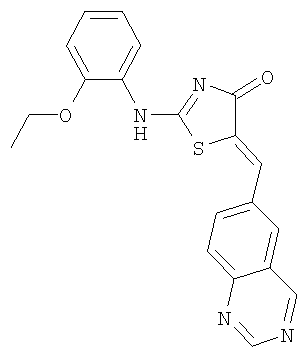
Используют методику, аналогичную описанной в примере 1 г, и исходя из 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она, 2-этоксианилина и ДИЭА, получают 2-(2-этоксифениламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он: ЖХ-МС m/е 377 (МН+).
Пример 5: 2-(4-Фтор-2-метоксифениламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он
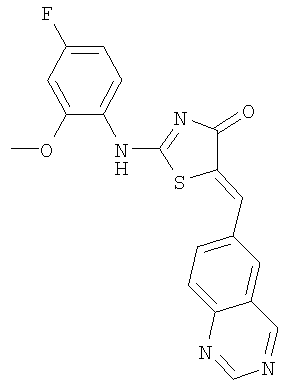
Используют методику, аналогичную описанной в примере 1 г, и исходя из 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она, 4-фтор-2-метоксианилина и ДИЭА, получают 2-(4-фтор-2-метоксифениламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он: ЖХ-МС m/е 381 (МН+).
Пример 6: 2-(3-Фторфениламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он
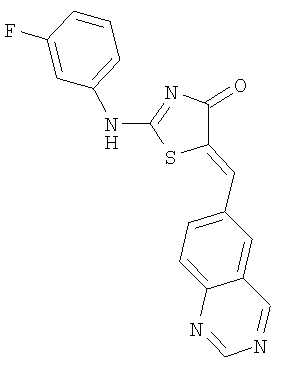
Используют методику, аналогичную описанной в примере 1 г, и исходя из 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она, 3-фторанилина и ДИЭА, получают 2-(3-фторфениламино)-5-[1 -хиназолин-6-илмет-(Z)-илиден]тиазол-4-он: ЖХ-МС m/е 351 (МН+).
Пример 7: 2-((R)-1-Гидроксиметил-2-фенилэтиламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он
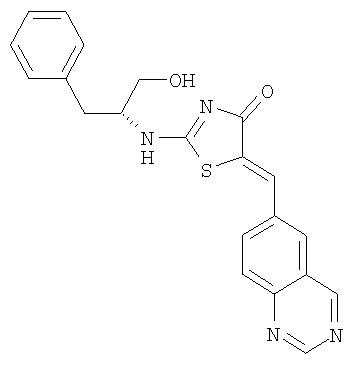
Используют методику, аналогичную описанной в примере 1 г, и исходя из 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она, 2-(R)-1-гидроксиметил-2-фенилэтиламина и ДИЭА, получают 2-((R)-1-гидроксиметил-2-фенилэтиламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он: ЖХ-МС m/е 391 (МН+).
Пример 8: 2-(3-Фторбензиламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он
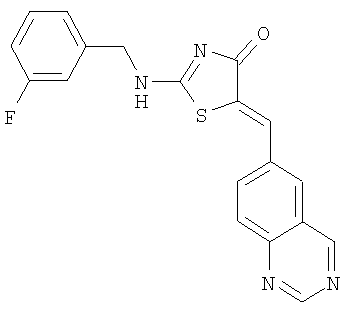
Используют методику, аналогичную описанной в примере 1 г, и исходя из 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она, 3-фторбензиламина и ДИЭА, получают 2-(3-фторбензилмино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он: ЖХ-МС m/е 379 (МН+).
Пример 9: 2-(2,4-Диметоксифениламино)-5-[1-хиназолин-6-илмет-(Z)-илиден1 тиазол-4-он
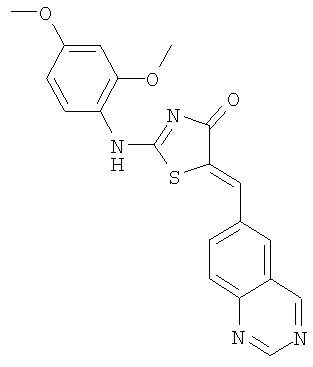
Используют методику, аналогичную описанной в примере 1 г, и исходя из 2-метилсульфанил-5-хиназолин-6-илметилентиазол-4-она, 2,4-диметоксифениламина и ДИЭА, получают 2-(2,4-диметоксифениламино)-5-[1-хиназолин-6-илмет-(Z)-илиден]тиазол-4-он: ЖХ-МС m/е 393 (МН+).
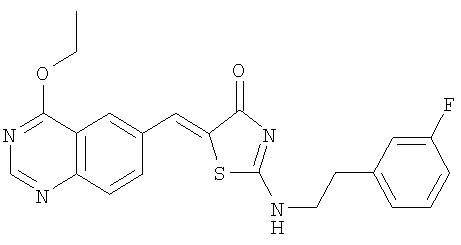
Пример 10: 5-[1-(4-Этоксихиназолин-6-ил)мет-(Z)-илиден]-2-[2-(3-фторфенил)этиламино]тиазол-4-он
а) Получение 6-бром-4-этоксихиназолина
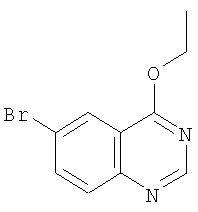
Суспензию 6-бром-4-хлорхиназолина (4,87 г, 20 ммолей), этоксида натрия (95%, 14,32 г, 200 ммолей) в безводном этиловом спирте (150 мл) перемешивают при комнатной температуре в течение 4 ч. После окончания реакции растворитель выпаривают, добавляют ледяную воду, а затем добавлением 3-нормального водного раствора HCl доводят рН до 9, при этом выпадает осадок. Твердое вещество отделяют и трижды промывают водой, а затем высушивают. Ускоренная хроматография (Merck силикагель 60, 230-400 мэш, 10%-50% этилацетата в гексане в течение 30 мин) дает 6-бром-4-этоксихиназолин (выход 2,13 г, 55%) в виде слегка желтого твердого вещества. ЖХ-МС m/е 254 (MH+).
б) Получение 4-этоксихиназолин-6-карбальдегида
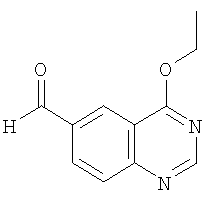
В смесь 6-бром-4-этоксихиназолина (пример 10а, 506 мг, 2 ммоля,), дифенилпропилфосфина (45,6 мг, 0,2 ммоля), ацетата палладия (44,8 мг, 0,2 ммоля) и триэтиламина (505 мг, 5 ммолей) в безводном N,N-диметилформамиде (ДМФ) (25 мл) вводят моноксид углерода под давлением 516 кПа. После перемешивания реакционной смеси при комнатной температуре в течение 15 мин моноксид углерода удаляют и добавляют тригексилсилан (1140 мг, 4 ммолей). В образовавшуюся реакционную смесь вводят моноксид углерода под давлением 516 кПа и нагревают при 80 С в течение 18 ч. После охлаждения реакции до комнатной температуры добавляют дихлорметан. Образовавшийся раствор трижды экстрагируют водой. Органический слой отделяют и концентрируют, получая желтое твердое вещество. Ускоренная хроматография (Merck силикагель 60, 230-400 мэш, 10%-40% этилацетата в гексане в течение 40 мин) дает 4-этоксихиназолин-6-карбальдегид (выход 256 мг, 66%) в виде слегка желтого твердого вещества. ЖХ-МС m/е 203 (МН+).
в) Получение 2-[(3-фторфенил)этиламино]тиазол-4-она.
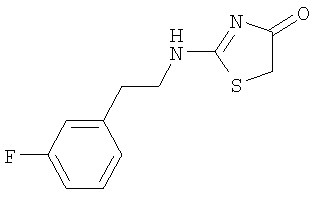
К раствору (3-фторфенил)этиламина (3,06 г, 22 ммоля) и роданина (2,66 г, 20 ммолей) в ацетонитриле (70 мл) добавляют ДИЭА (7,66 мл, 44 ммоля) при комнатной температуре. Затем раствор охлаждают до 0°С и двумя порциями добавляют хлорид ртути (5,97 г, 22 ммоля). После добавления смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 3 дней. Образовавшееся черное твердое вещество фильтруют через слой целита и промывают дихлорметаном (500 мл) и метанолом (250 мл). Растворители удаляют в вакууме, а сырой остаток растворяют в горячем этилацетате (25 мл) и хранят в холодильнике в течение ночи. Затем твердое вещество отделяют фильтрованием и промывают этилацетатом. После высушивания на воздухе выделяют с выходом 3,65 г (76,6%) 2-[(3-фторфенил)этиламино]тиазол-4-он в виде белого аморфного твердого вещества:
ВР-ЭС (+) m/е вычислено для C11H11FN2OS (М+Н)+ 239,0649, найдено 239,0647.
г) Получение 5-[1-(4-этоксихиназолин-6-ил)мет-(Z)-илиден]-2-[2-(3-фторфенил)этиламино]тиазол-4-он
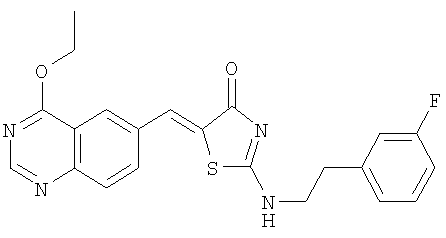
Смесь 4-этоксихиназолин-6-карбальдегида (пример 106, 20 мг, 0,1 ммоля,), 2-[2-(3-фторфенил)этиламино]тиазол-4-она (пример 10в, 24 мг, 0,1 ммоля) и пиперидина (10 мкл, 0,1 ммоля) в безводном этиловом спирте (1 мл) подвергают микроволновому облучению при 160°С в течение 25 мин. После охлаждения реакции до комнатной температуры твердое вещество отделяют фильтрованием, затем промывают МеОН и высушивают, получая 5-[1-(4-этоксихиназолин-6-ил)мет-(Z)-илиден]-2-[2-(3-фторфенил)этиламино]тиазол-4-он (выход 12 мг, 30%) в виде слегка желтого твердого вещества. ЖХ-МС m/е 423 (МН+).
Пример 11: 2-амино-5-[1-(4-этоксихиназолин-6-ил)мет-(Z)-илиден]тиазол-4-он
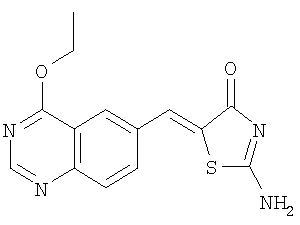
Суспензию 4-этоксихиназолин-6-карбальдегида (пример 10б) (1 экв.) псевдотиогидантоина (1 экв.) и ацетата натрия (4 экв.) в уксусной кислоте при перемешивании нагревают с обратным холодильником в течение 12 ч. После охлаждения до комнатной температуры добавляют воду. Твердое вещество отделяют фильтрованием, промывают водой и высушивают, получая 2-амино-5-[1-(4-этоксихиназолин-6-ил)мет-(Z)-илиден]тиазол-4-он в виде слегка желтого твердого вещества. ЖХ-МС m/е 256 (МН+).
Пример 12: 5-[1-(4-Этокси-2-метиламинохиназолин-6-ил)мет-(Z)-илиден]-2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-он
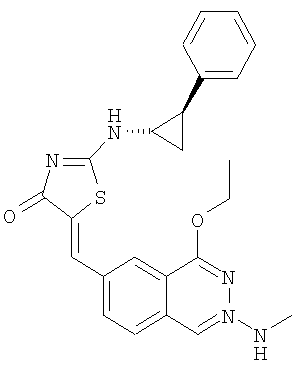
а) Получение 2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-она
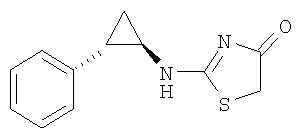
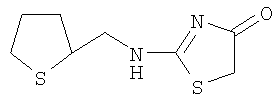
Используют методику, аналогичную описанной в примере 10в, и исходя из (1R,2S)-2-фенилциклопропиламина, роданина, хлорида ртути и ДИЭА, получают 2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-он. ЖХ-МС m/е 232 (МН+).
б) Получение 6-бром-1Н-бензо[d][1,3]оксазин-2,4-диона
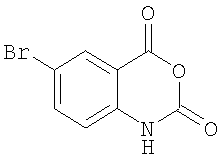
К суспензии 2-амино-5-бромбензойной кислоты (270 г, 1,25 молей) в ацетонитриле (1250 мл) одновременно добавляют раствор трифосгена (123,8 г, 416,7 ммолей) в дихлорметане (ДХМ) (500 мл) и пиридин (197,5 г, 2,5 мол) при 55°С.Образовавшуюся смесь перемешивают далее в течение 3 ч, а затем охлаждают до комнатной температуры. Осадок отделяют фильтрованием, промывают ацетонитрилом и высушивают, получая 6-бром-1Н-бензо[d][1,3]оксазин-2,4-дион в виде светлого порошка (выход 280 г, 93%). +Н ЯМР (ДМСО-d6): δ 11,83 (s, 1Н), 7,98 (s, 1H), 7,89-7,87 (d, 1H,), 7,09-7,07 (d, 1H).
б) Получение 6-бром-2-метиламино-1Н-хиназолин-4-она
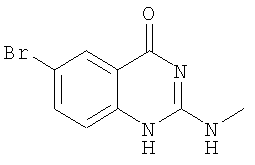
К раствору ацетонитрила (360 мл) и воды (90 мл) прибавляют 6-бром-1H-бензо[d][1,3]оксазин-2,4-дион (24,2 г, 0,1 моля), 1,2-диметилизотиомочевину (23,2 г, 0,1 моля), а затем безводный карбонат натрия (11,7 г, 0,11 моля) и нагревают смесь с обратным холодильником в течение 3 ч. После охлаждения осадок отделяют фильтрованием и промывают водой, получая 6-бром-2-метиламино-1H-хиназолин-4-он (выход 16,5 г, 65 ммолей, 65%) в виде слегка желтого порошка. 1Н ЯМР (ДМСО-d6): δ 11,20 (b, 1H), 7,91-7,90 (d, 1H), 7,64-7,63 (d, 1H), 7,20-7,18 (d, 1H), 6,30 (b, 1H), 2.82-2,81 (d, 3H).
в) Получение (6-бром-4-хлорхиназолин-2-ил)метиламина
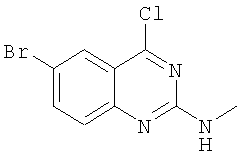
К раствору оксихлорида форфора (50 мл) добавляют порошкообразный 6-бром-2-метиламино-1Н-хиназолин-4-он (10,0 г, 39,4 ммолей), а затем диметилфениламин (8 мл) и нагревают смесь с обратным холодильником в течение получаса в атмосфере N2. После охлаждения смесь переносят в лед и подщелачивают 2-молярным раствором NaOH. Осадок отделяют фильтрованием и очищают с помощью колоночной хроматографии, получая (6-бром-4-хлорхиназолин-2-ил)метиламин (выход 5 г, 18,3 ммолей, 46,6%) в виде желтого твердого вещества. 1Н ЯМР (ДМСО-d6): δ 8,03 (s, 1Н), 7,86-7,84 (m, 2H), 7,47-7,45 (d, 1Н), 2,86-2.84 (d, 3H).
г) Получение (6-бром-4-этоксихиназолин-2-ил)метиламина
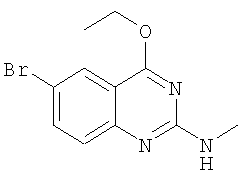
К раствору этоксида натрия (8,11 г, 119,3 ммолей) в абсолютном спирте (150 мл) добавляют одной порцией порошкообразный (6-бром-4-хлорхиназолин-2-ил)метиламин (13 г, 47,7 ммолей) и смесь перемешивают в течение 3 ч при комнатной температуре в атмосфере N2. Избыток спирта удаляют в вакууме, а остаток очищают с помощью колоночной хроматографии, получая (6-бром-4-этоксихиназолин-2-ил)метиламин (выход 4 г, 14,2 ммолей, 29,8%) в виде слегка желтого твердого вещества. 1H ЯМР (ДМСО-d6): δ 7,90-7,89 (d, 1Н), 7,70-7,67 (dd, 1Н), 7,33-7,31 (m, 1Н), 7,21-7,20 (dd, 1Н), 4,45-4,44 (q, 2H), 2,83-2,82 (d, 3H), 1,40-1,37 (t,3H).
д) Получение 4-этокси-2-метиламинохиназолин-6-карбальдегида
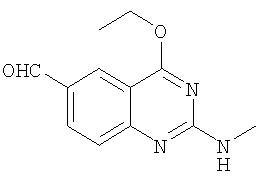
К раствору ацетонитрила (40 мл) и ДМСО (40 мл) добавляют тетракис(трифенилфосфин)палладий(0) (1,65 г, 1,43 ммолей), безводный формиат натрия (5,82 г, 85,6 ммолей), а затем порошкообразный (6-бром-4-этоксихиназолин-2-ил)метиламин (4 г, 14,3 ммолей) и образовавшуюся смесь нагревают до 85°С, в атмосфере моногидроксида углерода под давлением 344 кПа. После перемешивания в течение 48 ч охлажденную смесь переносят в воду и экстрагируют ДХМ (трижды по 200 мл). Объединенные органические фазы промывают рассолом, высушивают над Na2SO4, выпаривают и получают коричневое твердое вещество, которое очищают с помощью колоночной хроматографии, получая 4-этокси-2-метиламино-хиназолин-6-карбальдегид (выход 1 г, 4,3 ммолей, 30%) в виде светлого твердого вещества. 1Н ЯМР (ДМСО-d6): δ 9,95 (s, 1Н), 8,38 (s, 1H), 8,00-7,97 (d, 1H), 7,59-7,56 (m, 1H), 7,46-7,44 (d, 1H), 4,53-4,48 (q, 2H), 2,89-2,87 (d, 3H), 1,44-1,40 (t, 3H).
e) Получение 5-[1-(4-этокси-2-метиламинохиназолин-6-ил)мет-(Z)-илиден]-2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-она
К суспензии 2-(транс)фенилциклопропиламинотиазол-4-она (пример 12а, 38,0 мг, 0,16 ммоля) и 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 45,5 мг, 0,20 ммоля) в 2 мл толуола в микроволновой трубке добавляют бензойную кислоту (2,0 мг, 0,016 ммоля) и пиперидин (1,5 мг, 0,02 ммоля). Реакционную смесь нагревают до 150°С посредством микроволнового облучения в течение 1 ч. Затем реакционную смесь охлаждают до комнатной температуры и разбавляют толуолом. Твердое вещество отделяют фильтрованием и промывают толуолом, H2Cl2 и эфиром, получая в результате 5-[1-(4-этокси-2-метиламинохиназолин-6-ил)мет-(Z)-илиден]-2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-он в виде коричневого твердого вещества с выходом 52 мг (72,9%), МС: m/е 446 (МН+).
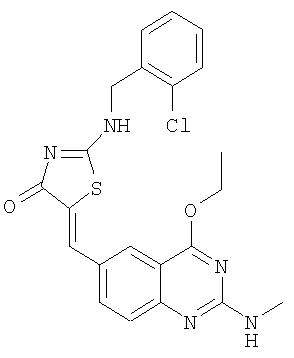
Пример 13: 2-(2-Хлорбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он
а) Получение 2-(2-хлорбензиламино)тиазол-4-она
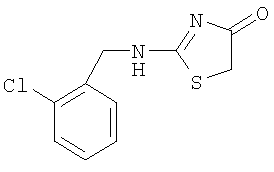
Используют методику, аналогичную описанной в примере 10в, и исходя из 2-хлорбензиламина, роданина, хлорида ртути и ДИЭА, получают 2-(2-хлорбензиламино)тиазол-4-он.
ЖХ-МС m/е 241 (МН+).
б) Получение 2-(2-хлорбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-она
К суспензии 2-(2-хлорбензиламино)тиазол-4-она (пример 13а, 38,5. мг, 0,16 ммоля), и 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 45,5 мг, 0,20 ммоля) в 2 мл толуола в микроволновой трубке добавляют бензойную кислоту (2,0 мг, 0,016 ммоля) и пиперидин (1,5 мг, 0,02 ммоля). Реакционную смесь нагревают до 150°С посредством микроволнового облучения в течение 45 мин. Затем реакционную смесь охлаждают до комнатной температуры и твердое вещество отфильтровывают, промывают толуолом, метиловым спиртом и эфиром и получают 2-(2-хлорбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он в виде коричневого твердого вещества с выходом 43,2 мг (59,5%), МС: m/е 454 (МН+).
Пример 14: 2-амино-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он
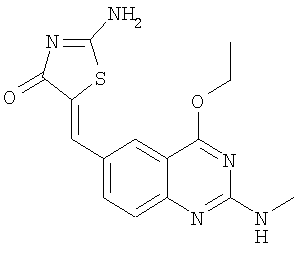
К суспензии псевдотиогидантоина (40,6 мг, 0,35 ммоля), 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 81,0 мг, 0,35 ммоля) и NaOAc (82,0 мг, 1,0 ммоль) в 2,5 мл ксилола добавляют уксусную кислоту (78,6 мг, 1,3 ммоля). Реакционную смесь нагревают с обратным холодильником в течение ночи. Затем реакционную смесь охлаждают до комнатной температуры и твердое вещество отфильтровывают, промывают MeCN и получают сырой продукт, который тритурируют с эфиром, получая 2-амино-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он в виде коричневого твердого вещества с выходом 4,5 мг (3,9%). МС: m/е 330 (МН+).
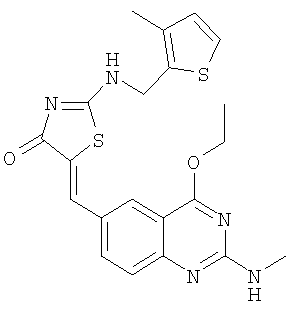
Пример 15: 5-(4-Этокси-2-метиламинохиназолин-6-илметилен)-2-[(3-метилтиофен-2-илметил)амино]тиазол-4-он
а) Получение 2-[(3-метилтиофен-2-илметил)амино]тиазол-4-она.
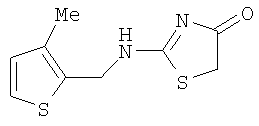
К раствору 3-метилтиофен-2-илметиламина (700 мг, 5,5 ммолей) и роданина (732 мг, 5,5 ммолей) в ацетонитриле (30 мл) добавляют диизопропилэтиламин (ДИЭА) (1,91 мл, 11 ммолей) при комнатной температуре. Затем раствор охлаждают до 0°С и одной порцией добавляют хлорид ртути (1,52 г, 5.6 ммолей). После добавления суспензию оставляют нагреваться до комнатной температуры и перемешивают в течение 3 дней. Образовавшееся черное твердое вещество отфильтровывают через слой целита и промывают ацетонитрилом (200 мл) и этилацетатом (250 мл). Фильтраты удаляют в вакууме, а сырой остаток растворяют в дихлорметане (150 мл) и промывают водой и солевым раствором. После высушивания над сульфатом магния фильтрат удаляют в вакууме, а осадок растворяют в дихлорметане (10 мл) и разбавляют гексаном (10 мл). После хранения в течение ночи в холодильнике твердое вещество отделяют фильтрованием и промывают дихлорметаном. Затем высушивают на воздухе и выделяют с выходом 390 мг (31,5%) 2-[(3-метилтиофен-2-илметил)амино]тиазол-4-он в виде слегка желтого аморфного твердого вещества:
ЭИ-ВРМС m/е вычислено для C9H10N2OS2 (M+) 226,0235, найдено 226,0232.
б) Получение 5-(4-этокси-2-метиламинохиназолин-6-илметилен)-2-[(3-метилтиофен-2-илметил)амино]тиазол-4-она
К суспензии 2-[(3-метилтиофен-2-илметил)амино]тиазол-4-она (36,2. мг, 0,16 ммоля) и 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 45,5 мг, 0,20 ммоля) в 2 мл толуола в микроволновой трубке добавляют бензойную кислоту (2,0 мг, 0,016 ммоля) и пиперидин (1,5 мг, 0,02 ммоля). Реакционную смесь нагревают до 150°С посредством микроволнового облучения в течение 45 мин. Затем реакционную смесь охлаждают до комнатной температуры и твердое вещество отфильтровывают, промывают толуолом, метиловым спиртом и эфиром, получая 5-(4-этокси-2-метиламинохиназолин-6-илметилен)-2-[(3-метилтиофен-2-илметил)амино]тиазол-4-он в виде коричневого твердого вещества с выходом 56,5 мг (80,4%), МС: m/е 440 (МН+).
Пример 16: 2-(3-Хлор-4-фторбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он
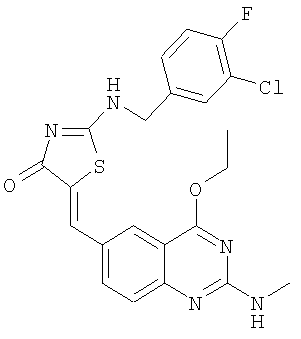
а) Получение 2-(3-хлор-4-фторбензиламино)тиазол-4-она
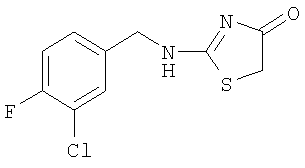
Используют методику, аналогичную описанной в примере 15 а, и исходя из 3-хлор-4-фторбензиламина, роданина, хлорида ртути и ДИЭА, получают 2-(3-хлор-4-фторбензиламино)тиазол-4-он. ЖХ-МС m/е 259 (МН+).
б) Получение 2-(3-хлор-4-фторбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-она
К суспензии 2-(3-хлор-4-фторбензиламинотиазол-4-она (пример 16а, 41,4. мг, 0,16 ммоля) и 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 45,5 мг, 0,20 ммоля) в 2 мл толуола в микроволновой трубке добавляют бензойную кислоту (2,0 мг, 0,016 ммоля) и пиперидин (1,5 мг, 0,02 ммоля). Реакционную смесь нагревают до 150°С посредством микроволнового облучения в течение 30 мин. Затем реакционную смесь охлаждают до комнатной температуры и твердое вещество отфильтровывают, промывают толуолом и получают сырой продукт с выходом 58,8 мг (77,9%), который растворяют в 0,5 мл горячего ДМФ и разбавляют водой при комнатной температуре. Осадок отделяют и промывают водой, ацетоном и эфиром, высушивают над Na2SO4 и концентрируют, получая 2-(3-хлор-4-фторбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он в виде слегка желтого твердого вещества с выходом 43,6 мг (57,7%), МС: m/е 472 (МН+).
Пример 17: 2-(2-Хлор-4-фторбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он
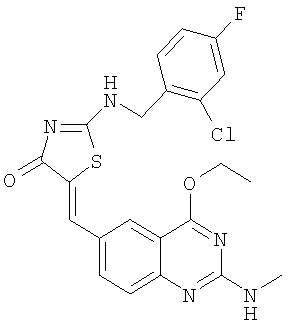
а) Получение 2-(2-хлор-4-фторбензиламино)тиазол-4-она
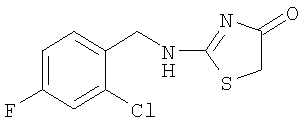
Используют методику, аналогичную описанной в примере 15а, и исходя из 2-хлор-4-фторбензиламина, роданина, хлорида ртути и ДИЭА, получают 2-(2-хлор-4-фторбензиламино)тиазол-4-он. ЖХ-МС m/е 259 (МН+).
б) Получение 2-(2-хлор-4-фторбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-она
К суспензии 2-(2-хлор-4-фторбензиламино)тиазол-4-она (пример 17а, 41,4. мг, 0,16 ммоля) и 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 45,5 мг, 0,20 ммоля) в 2 мл толуола в микроволновой трубке добавляют бензойную кислоту (2,0 мг, 0,016 ммоля) и пиперидин (1,5 мг, 0,02 ммоля). Реакционную смесь нагревают до 150°С посредством микроволнового облучения в течение 30 мин. Затем реакционную смесь охлаждают до комнатной температуры и твердое вещество отфильтровывают, промывают толуолом, метиловым спиртом и эфиром, получая 2-(2-хлор-4-фторбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он в виде слегка коричневого твердого вещества с выходом 46,6 мг (61,7%), МС: m/е 472 (МН+).
Пример 18: 2-(2-Хлор-6-метилбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он
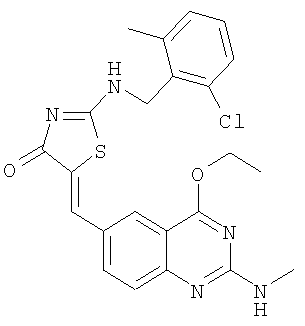
а) Получение 2-(2-хлор-6-метилбензиламино)тиазол-4-она
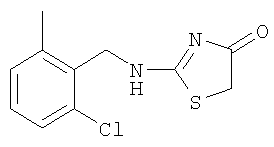
Используют методику, аналогичную описанной в примере 15а, и исходя из 2-хлор-6-метилбензиламина, роданина, хлорида ртути и ДИЭА, получают 2-(2-хлор-6-метилбензиламино)тиазол-4-он. ЖХ-МС m/е 259 (МН+).
б) Получение 2-(2-хлор-6-метилбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-она
К суспензии 2-(2-хлор-6-метилбензиламино)тиазол-4-она (пример 18а, 40,8 мг, 0,16 ммоля) и 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 45,5 мг, 0,20 ммоля) в 2 мл толуола в микроволновой трубке добавляют бензойную кислоту (2,0 мг, 0,016 ммоля) и пиперидин (1,5 мг, 0,02 ммоля). Реакционную смесь нагревают до 150°С посредством микроволнового облучения в течение 30 мин. Затем реакционную смесь охлаждают до комнатной температуры и твердое вещество отфильтровывают, промывают толуолом, метиловым спиртом и эфиром, получая 2-(2-хлор-6-метилбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он в виде слегка коричневого твердого вещества с выходом 45,9 мг (61,3%), МС: m/е 468 (МН+).
Пример 19: 5-(4-Этокси-2-метиламинохиназолин-6-илметилен)-2-[(тиофен-2-илметил)амино]тиазол-4-он
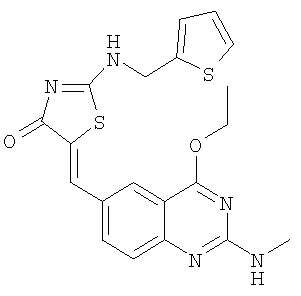
а) Получение 2-(тиофен-2-илметил)амино)тиазол-4-она
Используют методику, аналогичную описанной в примере 15а, и исходя из тиофен-2-илметиламина, роданина, хлорида ртути и ДИЭА, получают 2-(тиофен-2-илметиламино)тиазол-4-он. ЖХ-МС m/е 259 (МН+).
б) Получение 5-(4-этокси-2-метиламинохиназолин-6-илметилен)-2-[(тиофен-2-илметил)амино]тиазол-4-она
К суспензии 2-[(тиофен-2-илметил)амино]тиазол-4-она (34,0 мг, 0,16 ммоля) и 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 45,5 мг, 0,20 ммоля) в 2 мл толуола в микроволновой трубке добавляют бензойную кислоту (2,0 мг, 0,016 ммоля) и пиперидин (1,5 мг, 0,02 ммоля). Реакционную смесь нагревают до 150°С посредством микроволнового облучения в течение 30 мин. Затем реакционную смесь охлаждают до комнатной температуры и твердое вещество отфильтровывают, промывают толуолом, метиловым спиром и эфиром, получая 5-(4-этокси-2-метиламинохиназолин-6-илметилен)-2-[(тиофен-2-илметил)амино]тиазол-4-он в виде коричневого твердого вещества с выходом 48,6 мг (69,1%). МС: m/е 426 (МН+).
Пример 20: 5-(4-Этокси-2-метиламинохиназолин-6-илметилен)-2-[2-(3-фторфенил)этиламино]тиазол-4-он
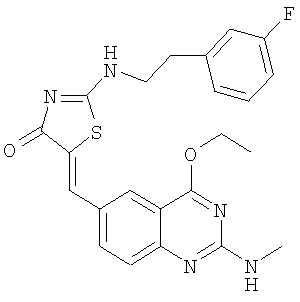
К суспензии 2-[2-(3-фторфенил)этиламино]тиазол-4-она (пример 10в, 38,1 мг, 0,16 ммоля) и 4-этокси-2-метиламинохиназолин-6-карбальдегида (пример 12е, 45,5 мг, 0,20 ммоля) в 2 мл толуола в микроволновой трубке добавляют бензойную кислоту (2,0 мг, 0,016 ммоля) и пиперидин (1,5 мг, 0,02 ммоля). Реакционную смесь нагревают до 150°С посредством микроволнового облучения в течение 30 мин. Затем реакционную смесь охлаждают до комнатной температуры и твердое вещество отфильтровывают, промывают толуолом, метиловым спиртом и эфиром, получая 5-(4-этокси-2-метиламинохиназолин-6-илметилен)-2-[2-(3-фторфенил)этиламино]тиазол-4-он в виде слегка коричневого твердого вещества с выходом 52,3 мг (72,4%). МС: m/е 452 (МН+).
Пример 21
Фармакологические свойства соединений по настоящему изобретению могут быть подтверждены рядом фармакологических анализов. Примеры фармакологических анализов, которые следует применять, были проведены с соединениями по настоящему изобретению и их солями. Соединения по изобретению проявляют CDK1/циклин В активность с Ki величинами менее 5,0 мкМ, что демонстрирует активность этих соединений при ингибировании CDK1/циклин В.
Для определения ингибирования Cdk1 активности анализы киназ проводят с использованием FlashPlate™ анализа (NEN™-Life Science Products) или HTRF анализа. Оба типа анализа киназ осуществляют, используя рекомбинантный человеческий Cdk1/циклин В-комплекс. GST-циклин В (GST-cycB) и Cdk1 кДНК клоны в бакуловирусных векторах предоставлены Dr. W. Harper из Baylor College of Medicine, Houston, TX. Белки соэкспрессируют в High Five™ клетках насекомых и комплекс очищают с использованием глутатионовой Sepharose смолы (Pharmacia, Piscataway, NJ), как ранее описано (J.W.Harper et al Cell, 1993, 75, 805-816). 6х-Гистидинмеченые укороченные формы белка ретинобластомы (Rb) (аминокислота 386-928) используются в качестве субстрата для Cdk1/циклин В анализа (экспрессионная плазмида предоставлена Dr. Veronica Sillivan, Departament of Molecular Virology, Roshe Research Centre, Welwyn Garden City, United Kingdom). Rb белок является натуральным субстратом для фосфорилирования с помощью Cdk1 (см. Herwig and Strauss, Eur. J. Biochem., vol.246 (1997), pp.581-601 и приведенные там ссылки). Экспрессию 62Kd белка контролируют IPTG индуцируемым промотором в Ml 5 линии E.coli. Клетки лизируют ультразвуком и проводят очистку лизатов при рН 8,0 через наполненную Ni-хелированной агарозой колонку, предварительно обработанную 1 мМ имидазола. Смолу затем несколько раз промывают буфером с постепенно уменьшающимися значениями рН до 6,0 и элюируют 500 мл имидазола. Элюированный белок подвергается диализу с использование 20 мМ HEPES рН 7,5, 30% глицерина, 200 мМ NaCl и 1 мМ DTT. В очищенном Rb слитом белке количественно оценивают концентрацию белка, разделяют его на аликвоты и хранят при -70°С.
Для FlashPlate™ анализа киназ 96-ячеистые Flash планшеты покрывают Rb белком при 10 мкг/мл, используя 100 мкл на ячейку. Планшеты инкубируют при 4°С в течение ночи или в течение 3 ч при комнатной температуре на вибраторе. Чтобы контролировать неспецифическое фосфорилирование, один ряд ячеек покрывают 100 мкл/ячейку буфером для покрытия (20 мМ HEPES, 0,2 М NaCl). Планшеты затем дважды отмывают буфером для отмывания (0,01% Tween 20 в фосфат-буферированном рассоле). Соединения, подлежащие тестированию («тестируемые соединения»), добавляют в ячейки с пятикратной конечной концентрацией. Реакции инициируют немедленным добавлением 40 мкл реакционной смеси (25 мМ HEPES, 20 мМ MgCl2, 0,002% Tween 20, 2 мМ DTT, 1 мкМ АТФ, 4 нМ 33P-АТР), а добавление достаточного количества фермента дает импульсы, по крайней мере, десятикратно превышающие фоновые значения. Планшеты инкубируют при комнатной температуре на вибраторе в течение 30 мин, а затем четыре раза отмывают буфером для отмывания, запаивают и считывают на TopCount сцинтилляционном счетчике (Packard Instrument Co., Downers Grove, IL). Процент ингибирования Rb фосфорилирования, являющийся мерой ингибирования Cdk активности, определяют согласно следующей формуле:

где «тестируемое соединение» относится к среднему числу импульсов в минуту тестируемых дублетов, «неспецифическое» относится к среднему числу импульсов в минуту, когда Cdk1-/циклин В и другие не были добавлены, и «общее» относится к среднему числу импульсов в минуту, когда никакое соединение не было добавлено. Величина IC50 обозначает концентрацию тестируемого соединения, которая на 50% уменьшает индуцируемое протеинкиназой внедрение радиометок при описанных условиях тестирования. Величина константы ингибирования Ki вычисляется следующим образом: Ki=IC50/(1+[S]/Km), где [S] обозначает концентрацию АТФ и Km обозначает константу Михаелиса.
Гомогенный разрешенный во времени флюоресцентный киназный анализ (HTRF) проводится в 96-ячеистых полипропиленовых планшетах (BD Biosciences, Bedford, MA). Тестируемые соединения сначала растворяют в ДМСО, а затем разбавляют буфером 1 для анализа киназ (25 мМ HEPES, рН 7,0, 8 мМ MgCl2, 1,5 мМ DTT, и 162 мкМ АТФ) с ДМСО с концентрацией 15%. CDK1/циклин В разбавляют буфером 2 для анализа киназ (25 мМ HEPES, рН 7,0, 8 мМ MgCl2, 0,003% Tween 20, 0.045% ВСА, 1,5 мМ DTT и 0,675 мМ Rb белка). Для инициирования киназной реакции 20 мкл раствора соединения смешивают с 40 мкл CDK1/циклин В раствора в планшетах для анализа с конечными концентрациями CDK1/циклин В и Rb, составляющими 0,1 мкг/мл и 0,225 мкМ соответственно, и инкубируют при 37°С в течение 30 мин. Добавляют 15 мл анти-фосфо-Rb (Ser 780) антитела (Cell Signaling Technology, Beverly, MA,) с разбавлением антитела 1:7692. Инкубирование продолжают при 37°С в течение 25 мин, после чего добавляют в ячейки LANCE Eu-W1024 меченый антикроличий IgG (I nM, PerkinElmer, Wellesley, MA) и анти-His антитело, конъюгированное с SureLight-Allophucocyanin (20 нМ, PerkinElmer, Wellesley, MA). Инкубирование продолжают при 37°С дополнительно в течение 40 мин. После завершения инкубирования 35 мкл реакционной смеси переносят в 384-ячеистый черный полистирольный планшет (Corning Incorporated, Corning, NY) и считывают с помощью флюоресцентного считывающего планшета при длине волны возбуждения 340 нм и эмиссионной длиной волны 665/615 нм.
Ki величины, отражающие CDK1/циклин В активность, относящиеся к соединениям, составляющим сущность изобретения, варьируются приблизительно от 0,001 мкМ до 5,000 мкМ. Конкретные данные для некоторых примеров составляют:
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ АЗАИНДОЛТИАЗОЛИНОНЫ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2005 |
|
RU2391342C2 |
| ТИАЗОЛИНОН-2-ЗАМЕЩЕННЫЕ ХИНОЛИНЫ | 2005 |
|
RU2395509C2 |
| 1, 5-НАФТИРИДИНАЗОЛИДИНОНЫ, ОБЛАДАЮЩИЕ CDK1 АНТИПРОЛИФЕРАТИВНОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2405781C2 |
| 4-МОНОЗАМЕЩЕННЫЕ ТИАЗОЛИНОНХИНОЛИНЫ | 2005 |
|
RU2397983C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,4-ДИАМИНОТИАЗОЛ-5-ОНА | 2005 |
|
RU2395501C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА | 2002 |
|
RU2302244C2 |
| ПРОИЗВОДНЫЕ 6-АМИНОХИНАЗОЛИНА ИЛИ 3-ЦИАНОХИНОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРНЫХ ТИРОЗИНКИНАЗ EGFR ИЛИ HER-2 | 2010 |
|
RU2536102C2 |
| СОЕДИНЕНИЯ АМИНОХИНАЗОЛИНОВ | 2005 |
|
RU2382034C2 |
| ИМИДАЗО[1,5-а] ПИРИМИДО[5,4-d][1] БЕНЗАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА GABA A | 2002 |
|
RU2287531C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕКСАФТОРИЗОПРОПАНОЛА | 2005 |
|
RU2389718C2 |
Настоящее изобретение относится к соединению формулы I и его фармацевтически приемлемым солям. Соединения настоящего изобретения обладают ингибирующей активностью в отношении CDK1 киназы и могут быть полезны при лечении рака, в частности рака грудной железы, рака легкого, рака толстой кишки и рака простаты. В формуле I
R1 обозначает водород или
;
выбран из фенила и 5-членного гетероароматического кольца, содержащего от 1 до 2 гетероатомов, выбранных из группы, состоящей из серы и азота; Х выбран из низшего алкилена, зациклизованного низшего алкилена, содержащего от 3 до 6 атомов углерода, и гидрокси(низшего алкилена); R5 и R6 независимо выбраны из группы, включающей водород, низший алкил, галоген и низшую алкоксигруппу; R3 выбран из водорода и -NH-R7; R4 выбран из водорода и -O(CH2CH2O)y-R10; R7 обозначает низший алкил; R10 обозначает низший алкил; n обозначает целое число от 0 до 1; и y равно 0 при условии, что когда n равно 0 и R1 обозначает водород, тогда R3/R4 не могут оба обозначать водород. Изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения изобретения. 2 н. и 43 з.п. ф-лы.
1. Соединение формулы
где
R1 обозначает водород или
X выбран из низшего алкилена, зациклизованного низшего алкилена, содержащего от 3 до 6 атомов углерода, и гидрокси(низшего алкилена);
выбран из фенила, и
5-членного гетероароматического кольца, содержащего от 1 до 2 гетероатомов, выбранных из группы, состоящей из серы и азота;
R5 и R6 независимо выбраны из группы, включающей водород, низший алкил, галоген и низшую алкоксигруппу;
R3 выбран из водорода и -NH-R7;
R4 выбран из водорода и -O(CH2CH2O)y-R10;
R7 обозначает низший алкил;
R10 обозначает низший алкил;
n обозначает целое число от 0 до 1;
y равно 0;
при условии, что когда n равно 0 и R1 обозначает водород, тогда R3/R4 не могут оба обозначать водород;
или его фармацевтически приемлемые соли.
2. Соединение по п.1 формулы I-A
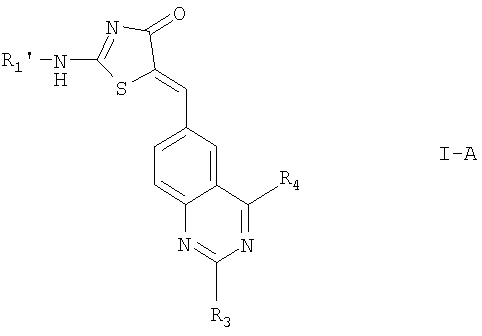
где R1' обозначает водород;
R4' обозначает -O(CH2CH2O)y-R10;
R3, R10 и y определены в п.1;
или его фармацевтически приемлемые соли.
3. Соединение по п.2, где R1' обозначает водород.
4. Соединение по п.3, где указанным соединением является 2-амино-5-[1-(4-этоксихиназолин-6-ил)метилиден]тиазол-4-он.
5. Соединение по п.2, где R3 обозначает -NHR7 и R7 определен в п.1.
6. Соединение по п.5, где указанным соединением является 2-амино-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он.
7. Соединение по п.1 формулы I-Б
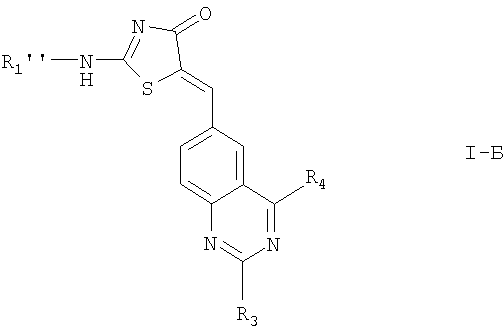
где R1'' обозначает
;
R3, R4, R5, R6 и определены в п.1;
или его фармацевтически приемлемые соли.
8. Соединение по п.7, где
R1'' обозначает и обозначает фенил, а
R5 и R6 определены в п.1.
9. Соединение по п.8, где указанным соединением является 2-(2,4-диметоксифениламино)-5-[1-хиназолин-6-илметилиден]тиазол-4-он.
10. Соединение по п.8, где указанным соединением является 2-(2-этоксифениламино)-5-[1-хиназолин-6-илметилиден]тиазол-4-он.
11. Соединение по п.8, где указанным соединением является 2-(4-фтор-2-метоксифениламино)-5-[1-хиназолин-6-илметилиден]тиазол-4-он.
12. Соединение по п.7, где обозначает гетероароматическое кольцо, содержащее от 1 до 2 гетероатомов.
13. Соединение по п.12, где указанное гетероароматическое кольцо содержит два гетероатома, один из которых является серой, а второй азотом.
14. Соединение по п.13, где указанным соединением является 5-[1-хиназолин-6-илметилиден]-2-(тиазол-2-иламино)тиазол-4-он.
15. Соединение по п.1 формулы
где
R1'' имеет такое же значение как R1 в п.1;
Х, R3, R4 и определены в п.1;
или его фармацевтически приемлемые соли.
16. Соединение по п.15, где Х обозначает низший алкилен.
17. Соединение по п.16, где R3 обозначает водород, R4 обозначает водород или -O(CH2CH2O)y-R10, R10 и y определены в п.1.
18. Соединение по п.17, где
R1'' обозначает и обозначает фенил;
R5 и R6 определены в п.1.
19. Соединение по п.18, где указанным соединением является 2-[2-(3-фторфенил)этиламино]-5-[1-хиназолин-6-илметилиден]тиазол-4-он.
20. Соединение по п.18, где указанным соединением является 2-(3-фторбензиламино)-5-[1-хиназолин-6-илметилиден]тиазол-4-он.
21. Соединение по п.18, где указанным соединением является 5-[1-(4-этоксихиназолин-6-ил)метилиден]-2-[2-(3-фторфенил)этиламино]тиазол-4-он.
22. Соединение по п.16, где
R1'' обозначает и фенил;
R3 обозначает -NHR7,
R4 обозначает водород или -O(CH2CH2O)y-R10,
R5, R6, R7, R10 и y определены в п.1.
23. Соединение по п.22, где указанным соединением является 2-(2-хлорбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он.
24. Соединение по п.22, где указанным соединением является 2-(3-хлор-4-фторбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он.
25. Соединение по п.22, где указанным соединением является 2-(2-хлор-4-фторбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он.
26. Соединение по п.22, где указанным соединением является 2-(2-хлор-6-метилбензиламино)-5-(4-этокси-2-метиламинохиназолин-6-илметилен)тиазол-4-он.
27. Соединение по п.22, где указанным соединением является 5-(4-этокси-2-метиламинохиназолин-6-илметилен)-2-[2-(3-фторфенил)этиламино]тиазол-4-он.
28. Соединение по п.16, где
R1'' обозначает и обозначает гетероароматическое кольцо, содержащее от 1 до 2 гетероатомов, и R5 и R6 имеют значения, приведенные в п.1.
29. Соединение по п.28, где указанное гетероароматическое кольцо содержит два гетероатома, один из которых является серой, а второй азотом.
30. Соединение по п.29, где указанное гетероароматическое кольцо является тиазольным кольцом.
31. Соединение по п.28, где указанное гетероароматическое кольцо содержит один гетероатом, который является атомом серы.
32. Соединение по п.31, где R3 и R4 обозначают водород.
33. Соединение по п.32, где указанным соединением является 5-[1-хиназолин-6-илметилиден]-2-[(тиофен-2-илметил)амино]тиазол-4-он.
34. Соединение по п.31, где R3 обозначает -NHR4, R4 и R7 определены в п.1.
35. Соединение по п.34, где указанным соединением является 5-(4-этокси-2-метиламинохиназолин-6-илметилен)-2-[(3-метилтиофен-2-илметил)амино]тиазол-4-он.
36. Соединение по п.34, где указанным соединением является 5-(4-этокси-2-метиламинохиназолин-6-илметилен)-2-[(тиофен-2-илметил)амино]тиазол-4-он.
37. Соединение по п.15, где Х обозначает зациклизованный низший алкилен.
38. Соединение по п.37, где
R1'' обозначает и обозначает фенил; и
R5 и R6 определены в п.1.
39. Соединение по п.38, где указанным соединением является 5-[1-(4-этокси-2-метиламинохиназолин-6-ил)мет-(Z)-илиден]-2-(2-фенилциклопропиламино)тиазол-4-он.
40. Соединение по п.15, где Х обозначает гидрокси(низший алкилен).
41. Соединение по п.40, где
R1'' обозначает и обозначает фенильное кольцо;
R5 и R6 определены в п.1.
42. Соединение по п.41, где указанным соединением является 2-(1-гидроксиметил-2-фенилэтиламино)-5-[1-хиназолин-6-илметилиден]тиазол-4-он.
43. Соединения формулы I по п.1 для лечения рака, особенно твердых опухолей, наиболее предпочтительно рака легкого, рака грудной железы, рака толстой кишки и рака простаты.
44. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении CDK1 киназы, включающая терапевтически эффективное количество по крайней мере одного соединения формулы I по п.1 вместе с фармацевтически приемлемыми вспомогательными веществами.
45. Фармацевтическая композиция по п.44, предназначенная для лечения рака, особенно твердых опухолей, наиболее предпочтительно рака легкого, рака грудной железы, рака толстой кишки и рака простаты.
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2151770C1 |
| WO 2004007491 A1, 22.01.2004 | |||
| WO 2004006916 A1, 22.01.2004. | |||

