Данное изобретение предлагает производные тиазолинонзамещенных хинолинов с замещением хинолинового кольца по 2-му положению, которые проявляют CDK1 (циклинзависимая киназа) антипролиферативную активность и применимы как противораковые агенты.
Циклинзависимые киназы CDK представляют собой серин-треонин протеиновые киназы, которые играют ключевую роль в регуляции переходов между различными фазами клеточного цикла, таких как продвижение от стадии покоя в G1 (промежуток между митозом и началом ДНК репликации в новом цикле деления клетки) к S (период активного синтеза ДНК) или переход от G2 к фазе М, при которой происходит активный митоз и деление клетки (см., например, статьи, собранные в Science, 274, 1996, с.1643-1677; Ann.Rev.Cell Dev. Biol., 13, 1997, с.261-291). CDK комплексы формируются путем ассоциации регуляторной циклиновой субъединицы (например, циклин А, В1, В2, D1, D2, D3 и Е) и каталитической субъединицы киназы (например, CDK1, CDK2, CDK4, CDK5 и CDK6). Как предполагает их название, CDK киназы проявляют абсолютную зависимость от субъединицы циклина для фосфорилирования их субстратных мишеней и различную функцию киназ/циклиновых пар для регулирования прохождения через специфические фазы клеточного цикла.
Из вышесказанного следует, что указанные протеинкиназы являются классом белков (ферментов), регулирующих различные клеточные функции. Это достигается за счет фосфорилирования определенных аминокислот в белковых субстратах, приводящего к конформационному изменению в белковом субстрате. Конформационное изменение модулирует активность субстрата или его способность взаимодействовать с другими связывающими партнерами. Ферментная активность протеинкиназы означает скорость, с которой киназа присоединяет фосфатные группы к субстрату. Она может быть измерена, например, путем определения количества субстрата, превращаемого в продукт, как функцию времени. Фосфорилирование субстрата происходит в активном центре протеинкиназы.
Благодаря описанным выше свойствам, эти киназы играют важную роль в прохождении сигнальной трансдукции фактора роста, что ведет к клеточной пролиферации, дифференциации и миграции. Фактор роста фибробластов (ФРФ) и фактор роста эндотелия сосудов (ФРЭС) определены как важные медиаторы ангиогенеза, вызванного опухолью. ФРЭС активирует эндотелиальные клетки, проводя сигнал через два высокоаффинных рецептора, один из которых является рецептором с доменом, содержащим киназу в качестве вставки (КДР). (См. Hennequin L.F. и др., J. Med. Chem. 46, N6, 2002, с.1300). ФРФ активирует эндотелиальные клетки, проводя сигнал через ФРФ-рецептор (ФРФР). Твердые опухоли нуждаются в формировании новых кровеносных сосудов (ангиогенез) для роста. Соответственно, ингибиторы рецепторов ФРФР и КДР, препятствующие трансдукции сигнала роста и таким образом замедляющие или предотвращающие ангиогенез, применимы как агенты для предотвращения образования или лечения твердых опухолей.
Поскольку CDK киназы, такие как CDK1, являются основными активаторами клеточного деления, ингибиторы CDK1 могут применяться как антипролиферативные агенты. Эти ингибиторы могут применяться для терапевтического воздействия, приводящего к подавлению нерегулируемого развития клеточного цикла.
Согласно предлагаемому изобретению было показано, что соединение формулы:
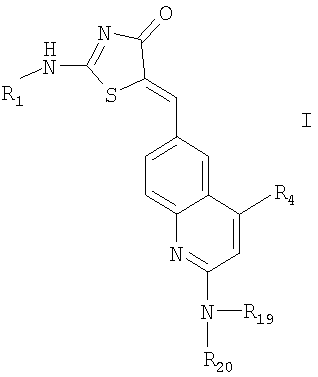
в котором
R1 означает водород, низший алкил, арилоксизамещенный низший алкил, -С(O)2[CH2CH2O]р-R9, -[CH2CH2O]V-R8, или R2-(X)n-;
Х означает низший алкилен, гидроксизамещенный низший алкилен, циклозамещенный низший алкилен, арилзамещенный низший алкилен, карбоксизамещенный низший алкилен, амидозамещенный низший алкилен, моно- или дигалоидзамещенный низший алкилен, аминозамещенный низший алкилен, моно- или ди-низший алкиламинозамещенный низший алкилен или имидозамещенный низший алкилен;
R2 означает
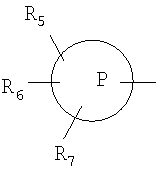
 означает арильное кольцо; циклоалкил, содержащий 3-6 атомов углерода; 4-6-членное гетероциклоалкильное кольцо, содержащее 3-5 атомов углерода и 1-2 гетероатома, выбранных из группы, состоящей из кислорода, азота и серы; или 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из кислорода, серы и азота;
означает арильное кольцо; циклоалкил, содержащий 3-6 атомов углерода; 4-6-членное гетероциклоалкильное кольцо, содержащее 3-5 атомов углерода и 1-2 гетероатома, выбранных из группы, состоящей из кислорода, азота и серы; или 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из кислорода, серы и азота;
R5, R6 и R7 независимо выбраны из группы, состоящей из гидрокси, низшего алкилсульфона, гидроксизамещенного низшего алкила, водорода, низшего алкила, галоида, перфторзамещенного низшего алкила, низшей алкокси, амино, моно- или ди-низшей алкиламино, или когда два из заместителей R5, R6 и R7 присоединены к смежным атомам углерода кольца  , эти два заместителя могут вместе со смежными присоединенными атомами углерода образовывать арильное кольцо; 3-6-членный циклоалкил; 4-6-членное гетероциклоалкильное кольцо; 4-6-членное гетероароматическое кольцо; указанное гетероциклоалкильное кольцо и указанное гетероароматическое кольцо, содержащие 1-2 гетероатома, выбранных из группы, состоящей из кислорода, азота или серы;
, эти два заместителя могут вместе со смежными присоединенными атомами углерода образовывать арильное кольцо; 3-6-членный циклоалкил; 4-6-членное гетероциклоалкильное кольцо; 4-6-членное гетероароматическое кольцо; указанное гетероциклоалкильное кольцо и указанное гетероароматическое кольцо, содержащие 1-2 гетероатома, выбранных из группы, состоящей из кислорода, азота или серы;
R4 означает водород, -(O)k(CH2CH2O)у-R10,
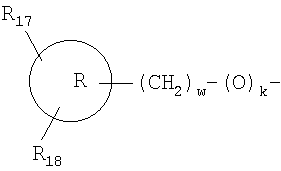
или -O-(СН2)nR14
R19 означает водород;
R20 означает водород, низший алкил или -С(O)-R11;
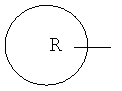 означает арильное кольцо; циклоалкил, содержащий 3-6 атомов углерода; 4-6-членное гетероциклическое алкильное кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из кислорода, серы и азота; или 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из кислорода, серы и азота;
означает арильное кольцо; циклоалкил, содержащий 3-6 атомов углерода; 4-6-членное гетероциклическое алкильное кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из кислорода, серы и азота; или 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из кислорода, серы и азота;
R8 и R9 независимо означают водород или низший алкил;
R10 и R11 означают низший алкил;
R14 означает перфторзамещенный низший алкил;
R17 и R18 независимо означают водород, низший алкил или -(СН2)z-C(O)OR11;
n и k означают целые числа от 0 до 1;
w, у и z означают целые числа от 0 до 3;
р означает целое число от 0 до 6; и
v и m означают целые числа от 1 до 6;
или N-окиси соединений, где R2 содержит азот в гетероароматическом кольце, сульфоны, где R2 содержит серу в гетероциклоалкильном кольце или гетероароматическом кольце;
или их фармацевтически приемлемые соли, которые ингибируют активность киназ CDK, особенно CDK1.
Эти агенты изобретения и фармацевтические композиции, содержащие такие агенты, применимы при лечении различных заболеваний или болезненных состояний, связанных с неконтролируемой или нежелательной клеточной пролиферацией, таких как рак, аутоиммунные заболевания, вирусные заболевания, грибковые заболевания, нейродегенеративные заболевания и кардиоваскулярные заболевания.
Ингибирование и/или модулирование активности киназ CDK, в частности, CDK1, соединениями этой формулы и композициями, содержащими эти соединения, делает их полезными при лечении заболеваний, опосредованных активностью киназы, в частности, в качестве противоопухолевых агентов при лечении различных видов рака.
Как здесь указано, соединения формулы I являются потенциальными антипролиферативными агентами и потому применимы для опосредования и/или ингибирования активности киназ CDK, в частности CDK1, представляя, таким образом, противоопухолевые агенты для лечения рака или других заболеваний, связанных с неконтролируемой или анормальной клеточной пролиферацией.
Одними из предпочтительных соединений формулы I являются соединения, соответствующие формуле
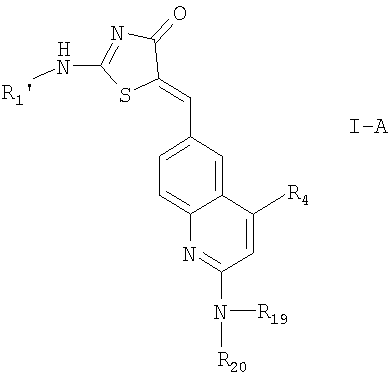
в которых R1' означает водород или низший алкил или низший алкоксиалкил, а R4, R19 и R20 имеют значения, указанные выше, или
их фармацевтически приемлемые соли и соединения формулы
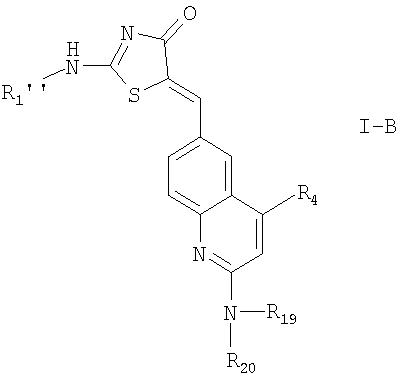
в которых R1'' означает R2'-(X')n-;
n, R4, R19 и R20 имеют значения, указанные выше, и
Х означает низший алкилен, гидроксизамещенный низший алкилен, циклозамещенный низший алкилен или моно- или дигалоидзамещенный низший алкилен;
R2' означает
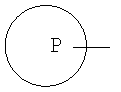 означает арильное кольцо, циклоалкильное кольцо, содержащее 3-6 атомов углерода, 4-6-членное гетероциклоалкильное кольцо, содержащее 3-5 атомов углерода и 1-2 гетероатома, выбранных из группы, состоящей из кислорода, азота или серы, и 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из кислорода, серы или азота,
означает арильное кольцо, циклоалкильное кольцо, содержащее 3-6 атомов углерода, 4-6-членное гетероциклоалкильное кольцо, содержащее 3-5 атомов углерода и 1-2 гетероатома, выбранных из группы, состоящей из кислорода, азота или серы, и 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из кислорода, серы или азота,
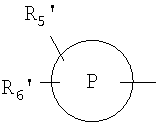
R5' и R6' независимо выбраны из группы, состоящей из гидрокси, низшего алкилсульфона, гидроксизамещенного низшего алкила, водорода, низшего алкила, галоида, перфторированного низшего алкила, низшей алкокси, амино, моно- или ди замещенной низшим алкилом аминогруппы;
или N-окиси соединений, в которых R2' содержит азот в гетероароматическом кольце, сульфоны, в которых R2' содержит серу в гетероциклоалкильном кольце или в гетероароматическом кольце;
или их фармацевтически приемлемые соли.
В соединениях I и I-B, где R1, R1'', R2 и Х содержат арильный радикал, предпочтительным арилом является фенил. Как используется здесь, понятие галоид включает все четыре галоида, таких как хлор, фтор, бром и йод.
Как используется в спецификации, термин «низший алкил», один или в комбинации, означает моновалентную прямоцепную или разветвленную насыщенную углеводородную группу, содержащую один-шесть атомов углерода, такую как метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил и тому подобные.
Термин «циклоалкил» означает циклический низший алкил, который образует 3-6-членное моновалентное незамещенное насыщенное углеводородное кольцо. Предпочтительными циклоалкильными заместителями являются циклопропил, циклобутил, циклогексил и т.д.
Термин «низшая алкокси группа» означает прямоцепную или разветвленную алкоксигруппу, сформированную из низшего алкила, содержащего один-шесть углеродных атомов, такую как метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси и тому подобные.
Термин «арил» означает моновалентное моно- или бициклическое незамещенное ароматическое углеводородное кольцо, такое как фенильное или нафтильное, причем фенильное является предпочтительным.
Термин «гетероциклоалкил» означает 4-6-членное моноциклическое насыщенное кольцо, содержащее 3-4 атома углерода и один или два гетероатома, выбранных из группы, состоящей из кислорода, азота или серы. Предпочтительные гетероциклические алкильные группы включают морфолинил, тетрагидротиопиранил или тетрагидропиранил.
Термин «гетероароматическое кольцо» относится к моновалентному 5- или 6-членному моноциклическому ароматическому кольцу, содержащему 4-5 атомов углерода и 1-2 гетероатома, выбранных из группы, состоящей из кислорода, азота или серы. Предпочтительными гетероароматическими группами являются тиофенил, тиазол, пиридинил, фуранил и т.д.
Термин «циклический низший алкилен» означает бивалентную циклоалкильную группу, как она определена выше.
Термин «низший алкилен» означает дивалентный насыщенный прямоцепной или разветвленный углеводородный заместитель, содержащий один-шесть атомов углерода.
Термин «арилзамещенный низший алкилен» означает низшую алкиленовую группу, как она определена выше, замещенную, предпочтительно однозамещенную арильной группой, как она определена выше. Примерами являются бензил, 1-фенилэтил или 2-фенилэтил.
Термин «карбоксизамещенный низший алкилен» относится к низшему алкиленовому заместителю с карбоксирадикалом.
Термин «гидроксизамещенный низший алкилен» относится к низшей алкиленовой группе, замещенной, предпочтительно однозамещенной, гидроксигруппой, а там, где описывается амидозамещенный низший алкилен, это подразумевает низшую алкиленовую группу, как она определена здесь выше, замещенную амидным заместителем.
Термин «моно- или дигалоидзамещенный низший алкилен» относится к низшему алкиленовому заместителю, как он описан выше, монозамещенному или дизамещенному по одному или двум атомам углеродной цепи низшего алкилена, где указанным заместителем является галоид.
Термин «галоид» означает фтор, хлор, бром или йод.
Термин «моно- или дизамещенный низшим алкиламино-заместителем низший алкилен» относится к низшему алкилену, как он описан выше, который моно- или дизамещен по одному или двум атомам углеродной цепи низшего алкилена моно- или ди- низшей алкиламино-группой.
Термин «аминозамещенный низший алкилен» описывает низший алкиленовый заместитель, как он определен выше, замещенный, предпочтительно монозамещенный, аминогруппой.
Термин «амидозамещенный низший алкилен» относится к низшему алкиленовому заместителю, как он определен выше, замещенному по одному положению амидогруппой.
Термин «имидозамещенный низший алкилен» относится к низшему алкиленовому заместителю, как он определен выше, замещенному по одному положению имидогруппой.
Термин «арилокси» относится к арилоксизаместителю, где арил определен выше. Предпочтительной арильной группой является фенил и предпочтительной арилоксигруппой является феноксигруппа.
Термин «перфторированный низший алкил» означает любую низшую алкильную группу, в которой все атомы водорода в низшей алкильной группе замещены или заменены фтором. К предпочтительным перфторированным низшим алкилам относятся такие группы, как перфторметил, пентафторэтил, гептафторпропил и т.д., причем трифторметильная группа наиболее предпочтительна.
Термин «фармацевтически приемлемые соли» относится к обычным кислотно-аддитивным солям или основно-аддитивным солям, которые сохраняют биологическую эффективность и свойства соединений формул I, II, III, IV и V и которые сформированы из подходящих нетоксичных органических или неорганических кислот или органических или неорганических оснований. Примеры кислотно-аддитивных солей включают производные неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота и азотная кислота, и производные органических кислот, таких как п-толуенсульфоновая кислота, салициловая кислота, метансульфоновая кислота, щавелевая кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота и тому подобные. Примеры основно-аддитивных солей включают производные гидроксидов аммония, калия, натрия и четвертичного аммония, такого, например, как тетраметиламмоний гидроксид. Химическое превращение фармацевтического соединения (т.е. лекарства) в соль хорошо известно специалистам в области фармацевтической химии и проводится с целью получения улучшенной физической и химической стабильности, гигроскопичности, текучести и растворимости соединений. См., например, Н. Ansel и др., Pharmaceutical Dosage Forms and Drug Delivery Systems (6-е изд., 1995), ее. 196 и 1456-1457.
Согласно предлагаемому изобретению соединения формулы I могут быть получены в соответствии со схемой реакции 1, в которой, если только специально не оговорено другое, R1, R4, R19 и R20 имеют значения, приведенные здесь выше.
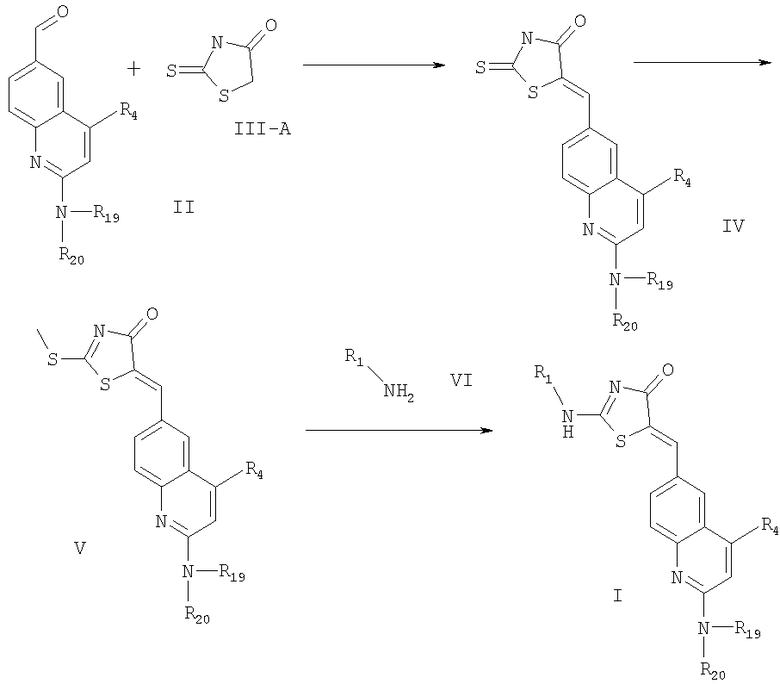
Схема 1
В соответствии с предлагаемым изобретением соединение формулы II вводят в реакцию Кновенагеля с соединением формулы III-A (роданин (2-тиоксотиазолин-4-он)) с получением соединения формулы IV. Любые условия, традиционно применяемые в реакции Кновенагеля, могут быть применены для проведения этой конденсации. Обычно эту реакцию проводят при температуре кипения в присутствии ацетата щелочного металла и уксусной кислоты.
На следующей стадии синтеза полученный замещенный тиазолидин формулы IV обрабатывают метилирующим агентом для метилирования тиогруппы в соединении формулы IV с получением соединения формулы V. Предпочтительным метилирующим агентом является йодистый метил. Реакцию проводят в среде органического амина, такого как диизопропилэтиламин (ДИЭА). Температура реакции и давление несущественны при проведении данной реакции, и она может быть осуществлена при комнатной температуре и атмосферном давлении. Фактически, любые условия, используемые обычно для метилирования тиогруппы, могут быть применены.
На следующей стадии синтеза соединение формулы V вводят в реакцию с соединением формулы VI, получая соединение формулы I. Соединение формулы VI является амином, и при проведении этой реакции могут быть применены любые способы, традиционно используемые для замены тиогруппы на аминогруппу. В соответствии с одним из разделов это замещение проводят реакцией соединения формулы VI с соединением формулы V в присутствии традиционного растворителя, такого как ацетонитрил. В общем случае эту реакцию проводят в присутствии аминного основания, такого как диизопропилэтиламин.
С другой стороны, соединения формулы I могут быть получены реакцией соединения формулы II с соединением формулы:
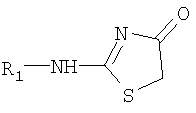
в котором R1 имеет значение, приведенное выше.
Для получения соединения формулы I реакцию соединения формулы VII с соединением формулы II проводят в высококипящем органическом растворителе, таком как бензол или толуол, при высоких температурах от 150°С до 250°С в закрытой системе. Таким образом, эту реакцию проводят при высоких температурах и давлении. Эта реакция особенно выгодна тогда, когда желательно получить соединения формулы I, в которых группа R содержит галоиды в цепи Х или в кольце Р. Соединение формулы VII можно прямо получить путем непосредственного замещения по реакции соединения формулы VI 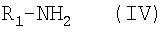 , где R1 имеет значение, приведенное выше, с соединением формулы III-A. Реакцию замещения обычно проводят в присутствии активатора тиенильной группы в тиенильном соединении формулы IX и в присутствии аминного основания. Одним из предпочтительных активаторов является двухлористая ртуть. Реакцию проводят в инертном органическом растворителе. Может быть применен любой обычный инертный органический растворитель, такой как ацетонитрил, хлористый метилен и т.д. с использованием такого аминного основания, как диизопропилэтиламин. Температура и давление не являются существенными для данной реакции, и она может быть проведена при комнатной температуре и атмосферном давлении. Для проведения данной реакции может быть использован любой традиционный метод замещения тиенильной группы амином.
, где R1 имеет значение, приведенное выше, с соединением формулы III-A. Реакцию замещения обычно проводят в присутствии активатора тиенильной группы в тиенильном соединении формулы IX и в присутствии аминного основания. Одним из предпочтительных активаторов является двухлористая ртуть. Реакцию проводят в инертном органическом растворителе. Может быть применен любой обычный инертный органический растворитель, такой как ацетонитрил, хлористый метилен и т.д. с использованием такого аминного основания, как диизопропилэтиламин. Температура и давление не являются существенными для данной реакции, и она может быть проведена при комнатной температуре и атмосферном давлении. Для проведения данной реакции может быть использован любой традиционный метод замещения тиенильной группы амином.
Соединения формулы VI, в которых R1 означает Х и Х означает гидроксизамещенный низший алкилен, могут быть получены из соответствующих аминокислот или эфиров аминокислот восстановлением борогидридом щелочного металла. С другой стороны, эти гидроксизамещенные низшие алкиленовые соединения могут быть получены из соответствующих эфиров цианокарбоновых кислот восстановлением алюмогидридом лития. Восстановление превращает цианогруппу в аминогруппу и эфирную группу в гидроксильную группу. Это восстановление должно иметь место перед реакцией соединения формулы VI с соединением формулы V.
С другой стороны, соединения формулы VI, в которых R1 означает R2X и Х означает карбоксизамещенный низший алкилен, амидозамещенный низший алкилен или имидозамещенный низший алкилен, могут быть прямо превращены в соединения формулы I реакцией соответствующего соединения формулы VI с соединением формулы V или с соединением формулы III-A, как описано выше.
Там, где кольца  или
или  означают N-окись в азотсодержащем цикле, который образует кольца или , эти N-окиси могут быть образованы из четвертичного кольцевого атома азота окислением. Может быть применен любой традиционный метод окисления четвертичного кольцевого атома азота в N-окись. Предпочтительным окислительным агентом является мета-хлорнадбензойная кислота (МХНБК).
означают N-окись в азотсодержащем цикле, который образует кольца или , эти N-окиси могут быть образованы из четвертичного кольцевого атома азота окислением. Может быть применен любой традиционный метод окисления четвертичного кольцевого атома азота в N-окись. Предпочтительным окислительным агентом является мета-хлорнадбензойная кислота (МХНБК).
Соединение формулы I, в котором R1 означает водород, может быть получено реакцией Кновенагеля соединения формулы II с соединением формулы VII с получением соединения формулы IV. Данная конденсация может быть проведена в любых условиях, обычных для реакции Кновенагеля. Обычно эту реакцию проводят при температуре кипения в присутствии ацетата щелочного металла и уксусной кислоты. При реакции Кновенагеля соединения формулы VII с соединением формулы II, в котором R20 означает -С(=O)-R11, R20 образует амид. Этот амид гидролизуют с получением амина во 2-м положении соединения формулы I, в котором R20 означает водород. Основание, используемое в реакции Кновенагеля, гидролизует амидную группу до соответствующей аминной.
В соответствии с предлагаемым изобретением, соединение формулы II, в котором R4 означает водород, т.е. соединение формулы
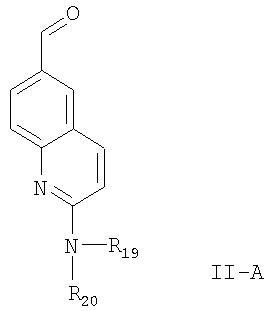
где R19 и R20 имеют значения, указанные выше,
может быть получено из соединения формулы
по реакции, приведенной ниже на схеме 2, где R19 и R20 имеют значения, указанные выше.
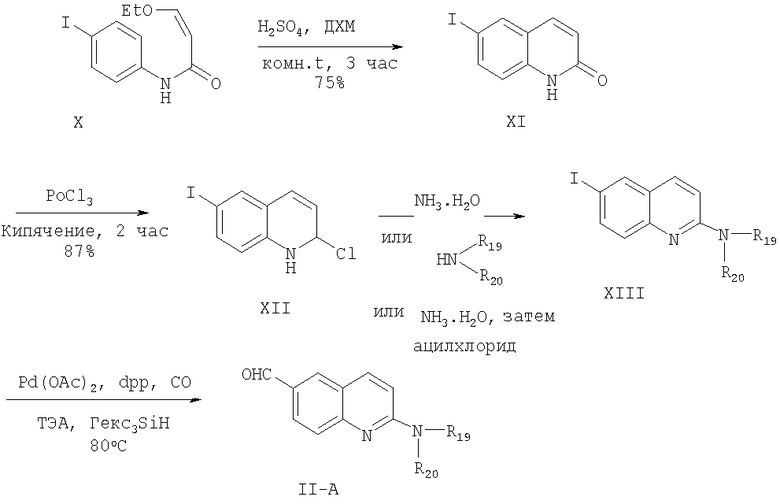
Схема 2
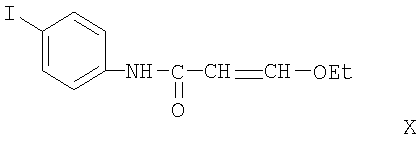
Соединение формулы X, которое может быть получено из 4-йоданилина с 3-этоксиакролоилхлоридом, циклизуют в соединение формулы XI действием серной кислоты. Обычно эту реакцию проводят в инертном растворителе, таком как дихлорметан. Циклизация осуществляется по альфа, бета-ненасыщенной двойной связи в эфирной группе соединения формулы Х при использовании серной кислоты При проведении данной циклизации температура и давление несущественны, и реакция циклизации может быть проведена при комнатной температуре и атмосферном давлении.
Циклизованное соединение формулы XI, содержащее оксозаместитель, может быть переведено в хлорированное соединение формулы XII обработкой хлорирующим агентом, таким как хлорокись фосфора. Обычно применение такого высококипящего жидкого хлорирующего агента, как хлорокись фосфора, предпочтительно. При кипячении реакционной смеси происходит превращение оксогруппы в хлор с высокими выходами. Любые условия, обычно используемые для превращения оксогруппы в хлор, могут применяться при проведении данной реакции. На следующей стадии этой реакции соединение формулы XII вводят в реакцию с гидроксидом аммония с получением соединения формулы XIII. Реакцию с гидроксидом аммония проводят под давлением при температуре 100-200°С, предпочтительно при температуре 150°С в течение 1-4 ч. Если это желательно, вторичный амин с заместителями R19 и R20 может быть получен реакцией соединения формулы XII с замещенным амином или любыми обычными методами превращения первичного амина во вторичный амин с низшим алкилом. 2-Амидозамещенное соединение формулы II-A с заместителем -C(=P)R11 может быть получено реакцией соединения с первичной аминной группой с ацилхлоридом. На следующей стадии этого синтеза соединение формулы XIII превращали в соединение формулы II-A, используя реакцию формилирования для превращения йодида в заместитель СНО в фенильном кольце. Реакцию проводят взаимодействием соединения формулы XIII с монооксидом углерода в присутствии дифенилпропилфосфина (dpp) и основания, используя ацетат палладия в качестве катализатора. При проведении реакции монооксид углерода добавляют к реакционной смеси под давлением при температуре 60-100°С. Используют, как правило, давление 70-80 psi. Для проведения этого превращения может быть применен любой обычный способ превращения галоида в альдегидную группу в фенильном кольце с использованием реакции с монооксидом углерода.
Синтез 2,4-дизамещенного соединения формулы I, интермедиат формулы
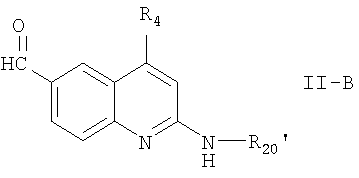
где R20' означает -C(=O)-R11, a R4 и R11 имеют значения, приведенные выше,
может быть осуществлен в соответствии с приведенной далее схемой реакции 3, где R20' и R4 имеют значения, указанные выше.
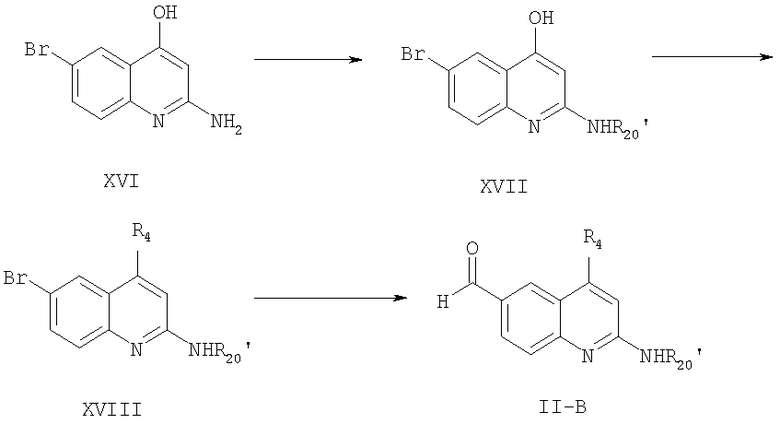
Схема 3
Соединение формулы XVII может быть получено из соединения формулы XVI (получение соединения XVI описано в примере 2) реакцией соединения формулы XVI с реакционно-способным производным карбоновой кислоты формулы R11-C(=O)-OH, где R11 имеет значение, указанное выше.
Любые обычные способы превращения аминов в амиды реакцией с активными производными карбоновой кислоты, такими как галоидангидриды или ангидриды, могут применяться для проведения данной реакции. Соединение формулы XVII, содержащее гидроксигруппу, может быть превращено в соединение формулы XVIII реакцией гидроксигруппы соединения формулы XVII с галоидом заместителя R4, если желательно ввести этот заместитель в положение 4 соединения формулы I. Эту реакцию проводят, вводя соответствующий галогенид во взаимодействие с соединением формулы XVII при температуре кипения инертного органического растворителя, используемого в качестве среды. В проведении этой реакции может быть применен любой обычный метод взаимодействия гидроксигруппы с галогенидом. На последней стадии данного синтеза соединение формулы XVIII превращают в соединение формулы II-B, применяя реакцию формилирования для превращения йодгруппы в заместитель СНО в фенильном кольце. Эту реакцию можно проводить, как описано здесь ранее, взаимодействием соединения формулы XVIII с монооксидом углерода в присутствии основания и с использованием тетракис(трифенилфосфин)палладиевого катализатора при температуре 60-140°С. При проведении реакции монооксид углерода добавляют к реакционной среде под давлением. Обычно используют давление 40-80 psi. Любой обычный метод формилирования галоидной группы в альдегидную в фенильном кольце с использованием реакции с монооксидом углерода может быть применен для превращения соединения формулы XVIII в соединение формулы II-B.
Среди соединений формулы I и тех ее частей, которые включают соединения формулы I-A и формулы I-B, предпочтительными являются те соединения, в которых арильные группы во всех арильных заместителях означают предпочтительно фенил.
Предпочтительными среди представителей класса соединений формулы I-A являются те соединения формулы I-A, в которых R1 означает водород. Особенно предпочтительными представителями этого класса соединений являются те соединения, в которых R4 означает -(O)k(CH2CH2O)у-R10.
В этом случае соединения, в которых R20 означает -С(=O)-R11, особенно предпочтительны.
Предпочтительным представителем соединений формулы I-B является класс тех соединений, в которых n означает 0 и R2' означает циклоалкильное кольцо низшего алкила, особенно циклопропил. Среди этого класса соединений предпочтительными являются те соединения, в которых R4 означает -(O)k(CH2CH2O)у-R10.
В этом классе соединения, в которых R20 означает -С(=O)-R11, особенно предпочтительны.
Другая часть предлагаемого изобретения представляет те соединения формулы I-B, в которых n означает 1 и Х означает низший алкилен, гидроксизамещенный низший алкилен, циклический низший алкилен или моно- или дигалоидзамещенный низший алкилен. В этом случае R4 предпочтительно означает -(O)k(CH2CH2O)у-R10.
В этой предпочтительной части особенно предпочтительными являются соединения, в которых R20 означает -С(=O)-R11.
Фармацевтические композиции предлагаемого изобретения могут, альтернативно или в дополнение к соединению формулы I, включать в качестве активного ингредиента фармацевтически приемлемые пролекарства, фармацевтически активные метаболиты и фармацевтически приемлемые соли таких соединений и метаболитов. Такие соединения, пролекарства, мультимеры, соли и метаболиты иногда обозначаются здесь как «активные агенты» или «агенты».
В случае твердых агентов специалистам понятно, что предлагаемые соединения и соли могут существовать в различных кристаллических или полиморфных формах, которые все предполагаются в тексте предлагаемого изобретения и в форме изобретения.
Терапевтически эффективные количества активных агентов предлагаемого изобретения могут применяться для лечения заболеваний, опосредованных модуляцией или регуляцией протеиновых киназ CDK1. «Эффективное количество» предполагает количество агента, которое существенно ингибирует пролиферацию и/или препятствует де-дифференциации эукариотической клетки, например клеток людей, насекомых, растений, грибов, и которое эффективно для указанного применения, например, для специального терапевтического лечения.
Количество данного агента, соответствующего такому количеству, будет варьироваться в зависимости от некоторых факторов, таких как конкретное соединение, болезненное состояние и его острота, идентичность (например, вес) субъекта или «хозяина», нуждающегося в лечении, но может быть определено обычным способом, известным специалистам, в соответствии с конкретными обстоятельствами данного случая, включая, например, конкретный назначаемый агент, тип введения, состояние, подвергающееся лечению, и субъект или «хозяин», которого лечат. «Лечение» подразумевает, по крайней мере, уменьшение степени заболевания у субъекта, такого как млекопитающее (например, человек), вызванного, по крайней мере частично, активностью CDK1 протеинкиназы; предотвращение болезненного состояния у млекопитающего, в частности, в тех случаях, когда млекопитающее предрасположено к болезненному состоянию, но оно еще не диагностировано как наличествующее; изменение или ингибирование болезненного состояния и/или облегчение болезненного состояния.
Предлагаемое изобретение, кроме того, направлено на разработку методов модуляции или ингибирования активности протеинкиназы CDK1, например, в ткани млекопитающего, путем введения ему агента изобретения. Антипролиферативная активность агентов легко измеряется различными известными методами, например, используя культуры цельных клеток в МТТ (МТТ -время прохождения) анализе. Активность агентов предлагаемого изобретения как модуляторов активности протеинкиназы CDK1 может быть измерена любыми методами, доступными специалистам, включая анализы in vivo и/или in vitro. Примеры подходящих анализов для измерений активности включают описанные в International Publication № WO 99/21845; Parast и др., Biochemistry, 37, 1998, с.16788-16801; Connell-Crowley и Harpes, Cell Cycle: Materials and Methods (Michele Pagano, ред., Springer, Берлин, Германия), 1995: International Publication № WO 97/34876 и International Publication № WO 96/14843. Эти свойства можно оценить, например, используя одну или более биологических процедур, приведенных в нижеизложенных примерах.
Активные агенты предлагаемого изобретения могут быть скомпонованы в фармацевтические композиции, описанные ниже. Фармацевтические композиции предлагаемого изобретения включают эффективно модулирующее, регулирующее или ингибирующее количество соединения формулы I и инертный фармацевтически приемлемый носитель или разбавитель. В одном разделе фармацевтических композиций эффективные уровни агентов изобретения подбирают так, чтобы эффективные уровни обеспечивали терапевтический эффект, заключающийся в антипролиферативной способности. Термин «эффективные уровни» означает такие уровни, при которых пролиферация ингибируется или ограничивается. Эти композиции готовят в форме единичной дозы, подходящей для способа введения, например, парентерального или орального введения.
Агент изобретения может вводиться в традиционной дозовой форме, полученной комбинацией терапевтически эффективного количества агента (например, соединения формулы I) в качестве активного ингредиента с подходящими фармацевтическими носителями или разбавителями по обычным процедурам. Эти процедуры могут включать смешивание, гранулирование и спрессовывание или растворение ингредиентов, что более подходит для желаемого препарата.
Применяемый фармацевтический носитель может быть как твердым, так и жидким. Примерами твердых носителей являются лактоза, сукроза, тальк, желатина, агар, пектин, гуммиарабик, стеарат магния, стеариновая кислота и тому подобное. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло, вода и тому подобное. Носитель или разбавитель может также включать материал продленного или постепенного высвобождения, известный специалистам, такой как моностеарат глицерина или дистеарат глицерина, один или с воском, этилцеллюлоза, гидроксипропилметилцеллюлоза, метилметакрилат и тому подобное.
Различные фармацевтические формы могут быть использованы. Так, если применяется твердый носитель, препарат может быть таблетирован, помещен в твердую желатиновую капсулу в форме порошка или таблетки или в быть в форме таблетки или лепешки. Количество твердого носителя может варьироваться. Если используется жидкий носитель, препарат может быть в виде сиропа, эмульсии, мягкой желатиновой капсулы, стерильного раствора для инъекции или суспензии в ампуле или флаконе или неводной жидкой суспензии.
Для получения стабильной водорастворимой дозовой формы фармацевтически приемлемая соль заявляемого в изобретении агента может быть растворена в водном растворе органической или неорганической кислоты. Если растворимая солевая форма недоступна, агент может быть растворен в подходящем со-растворителе или комбинациях со-растворителей.
Следует понимать, что актуальные дозы агентов, используемых в композициях предлагаемого изобретения, будут различаться согласно использованию индивидуального комплекса, применяемой индивидуальной композиции, способу введения и месту, организму "хозяина" и заболеванию, требующему лечения. Оптимальные дозы для данного набора условий могут быть установлены специалистами в этой области с использованием обычных тестов, применяемых для определения доз на основании экспериментальных данных для агента.
Композиции предлагаемого изобретения могут быть получены известными способами получения фармацевтических композиций, например, используя обычную технику приготовления, такую как смешивание, растворение, гранулирование, изготовление драже, растирание, эмульгирование, энкапсулирование, связывание и лиофилизацию. Фармацевтические композиции могут быть включены в лекарственную форму традиционным способом с использованием одного или более физиологически приемлемых носителей, которые могут быть отобраны из наполнителей и дополнительных веществ, облегчающих процесс перевода активных соединений в лекарственные препараты, применимые фармацевтически.
Для орального введения соединения могут быть легко превращены в лекарственную форму комбинацией соединений с фармацевтически приемлемыми носителями, известными специалистам. Такие носители способны формировать соединения предлагаемого изобретения в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, густых смесей, суспензий и тому подобное, для орального проглатывания больным, подвергающимся лечению. Фармацевтические препараты для орального употребления могут быть получены с использованием твердого наполнителя в смеси с активным ингредиентом (агентом), необязательным размельчением полученной смеси и получением смеси гранул после добавления подходящих дополнительных материалов, если требуется, для получения таблеток или внутреннего содержимого драже.
Предлагаемое изобретение иллюстрируется далее следующими сопровождающими рабочими примерами, которые ни в коей мере не ограничивают объем изобретения.
Примеры
Пример 1
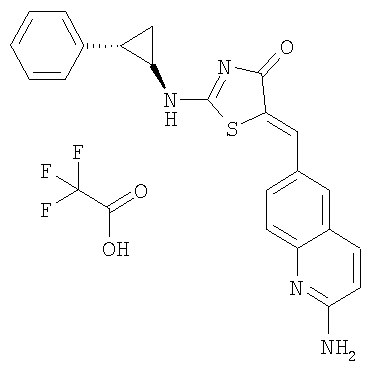
5-[1-(2-Аминохинолин-6-ил)мет-(Z)-илиден]-2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-он; соединение с трифторуксусной кислотой.
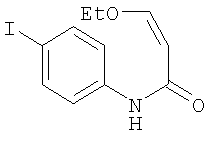
Получение 3-этокси-N-(4-йодфенил)акриламида
К раствору оксалилхлорида (40 г, 0,555 моль) медленно добавляли при 0°С этилвиниловый эфир (105,6 г, 0,84 моль) и смесь перемешивали в течение 2 ч при 0°С и 12 ч при комнатной температуре. После удаления части растворителя на роторном испарителе черную смесь кипятили при 120°С в течение 30 мин. После удаления растворителя на роторном испарителе и затем в вакууме масляного насоса был получен в виде черной жидкости 3-этоксиакрилоилхлорид (69,9 г), который прямо использовали на следующей стадии реакции без последующей очистки.
К смеси 4-йоданилина (14 г, 64 ммоль) и пиридина (10,5 мл, 128 ммоль) в метиленхлориде (85 мл) добавляли 3-этоксиакрилоилхлорид (10 г, 75 ммоль). После перемешивания в течение 5 ч дополнительно добавляли 3-этоксиакрилоилхлорид (5 г, 38 ммоль) и пиридин (10,5 мл, 64 ммоль). После перемешивания в течение 2 дней реакционную смесь промывали водой (3×100 мл), сушили над MgSO4 и концентрировали, получая 3-этокси-N-(4-йодфенил)акриламид в виде черного масла (11,32 г, 56%). ЖХ-МС m/е 318 (МН+).
Получение 6-йодо-1Н-хинолин-2-она
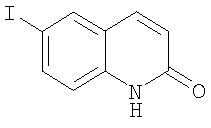
Серную кислоту медленно добавляли к 3-этокси-N-(4-йодфенил)акриламиду (11,3 г, 36 ммоль) при перемешивании. После перемешивания в течение 3 ч реакционную смесь медленно выливали на лед (~300 г). Осадок собирали фильтрованием, промывали водой и сушили. После флэш-хроматографии (Merck силикагель 60, 230-400 меш, 0%-15% метанола в метиленхлориде в течение 40 мин) выделяли 6-йодо-1Н-хинолин-2-он (7,23 г, 75%) в виде черного твердого вещества. ЖХ-МС m/е 272 (МН+).
Получение 2-хлор-6-йодхинолина
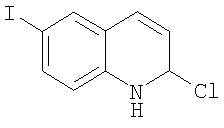
Смесь 6-йодо-1Н-хинолин-2-она (6,23 г, 23 ммоль) и хлорокиси фосфора (25 мл) нагревали при кипении в атмосфере N2 в течение 2 ч. После охлаждения растворитель удаляли на роторном испарителе и затем в вакууме масляного насоса. Затем медленно добавляли насыщенный раствор бикарбоната натрия (100 мл). Твердое вещество собирали фильтрованием, промывали насыщенным раствором бикарбоната натрия, водой, сушили и получали 2-хлор-6-йодхинолин (5,78 г, 87%) в виде черного твердого вещества. ЖХ-МС m/е 290 (МН+).
Получение 6-йодохинолин-2-иламина
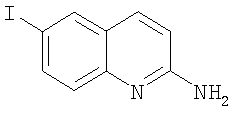
Суспензию 2-хлор-6-йодхинолина (1 г, 3,46 ммоль) в гидроокиси аммония (28%, 20 мл) нагревали при 140°С в течение 3 дней в вакуумированной пробирке. После охлаждения растворитель удаляли на роторном испарителе. Твердое вещество собирали фильтрованием, промывали водой, сушили и получали 6-йодхинолин-2-иламин (0,78 г, 84%) в виде черного твердого вещества. ЖХ-МС m/е 271 (МН+).
Получение 2-аминохинолин-6-карбальдегида
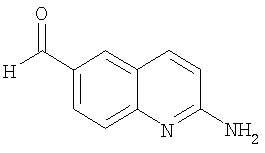
Смесь 6-йодхинолин-2-иламина (200 мг, 0,74 ммоль), триэтиламина (0,26 мл, 1,85 ммоль), дифенилпропилфосфина (dpp, 17 мкл, 0,074 ммоль) и ацетата палладия (II) (17 мг, 0,074 ммоль) в сухом N,N-диметилформамиде (4 мл) перемешивали в атмосфере окиси углерода в вакуумированной пробирке при 75 psi при комнатной температуре в течение 10 мин. После добавления тригексилсилана (0,53 мл, 1,5 ммоль) смесь перемешивали в атмосфере окиси углерода при 75 psi в течение 4 ч при 80°С. Затем реакционной смеси давали охладиться до 25°С, после чего экстрагировали метиленхлоридом (2×50 мл). Объединенные органические слои последовательно промывали водой (3×50 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После флэш-хроматографии (Merck силикагель 60, 70-230 меш, этилацетат) получали 2-аминохинолин-6-карбальдегид (30 мг, 24%) в виде твердого вещества.
Получение 2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-она.
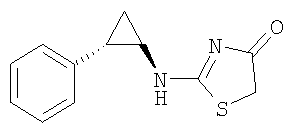
К суспензии хлоргидрата (1R,2S)-2-фенилциклопропиламина (0,85 г, 5 ммоль) и роданина (2-тиоксотиазолин-4-она) (0,68 г, 5 ммоль) в ацетонитриле (20 мл) добавляли при комнатной температуре N,N-диизопропилэтиламин (ДИЭА) (2,61 мл, 15 ммоль) Затем этот раствор охлаждали до 0°С и к нему в две порции за период в 10 мин добавляли дихлорид ртути (1,35 г, 5 ммоль). После добавления суспензии давали нагреться до комнатной температуры и перемешивали в течение 2 дней. Полученные твердые черные вещества были профильтрованы через пластинку целлита и промыты этилацетатом (500 мл). Из фильтрата удаляли растворитель в вакууме и твердый остаток разбавляли водой (100 мл) и этилацетатом (100 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические экстракты промывали солевым раствором и высушивали над безводным сульфатом магния. После фильтрования осушающего агента и удаления растворителя в вакууме получали сырой остаток, который очищали, используя колоночную хроматографию на Biotage силикагеле, получая 0,474 г (выход 42%) 2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-она в виде белого аморфного твердого вещества: EI-HRMC (масс-спектрометрия высокого разрешения с электронной ионизацией) m/e вычисленное для C12H12N2OS (M+) 232,0670, найденное 232,0665.
Получение 5-[1-(2-аминохинолин-6-ил)мет-(Z)-илиден]-2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-она, соединение с трифторуксусной кислотой
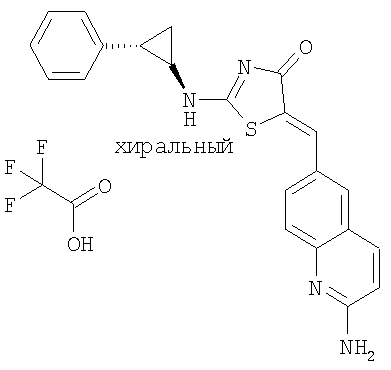
К суспензии 2-аминохинолин-6-карбальдегида (30 мг, 0,174 ммоль) и 2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-она (26 мг, 0,11 ммоль) в толуоле (1 мл) добавляли бензойную кислоту (3 мг, 0,011 ммоль) и пиперидин (3 мкл, 0,011 ммоль). Смесь нагревали до 150°С в микроволновой печи в течение 20 мин. После охлаждения до комнатной температуры отфильтровывали твердое вещество, промывали толуолом и сушили. Сырой продукт очищали ВЭЖХ (сорбент для обращеннофазной ВЭЖХ С 18, 10%-90% ацетонитрила в воде в течение 10 мин), получая 5-[1-(2-аминохинолин-6-ил)мет-(Z)-илиден]-2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-он, соединение с трифторуксусной кислотой (36 мг, 84%) в виде твердого желтого вещества. ЖХ-МС m/e 271 (МН+).
Пример 2
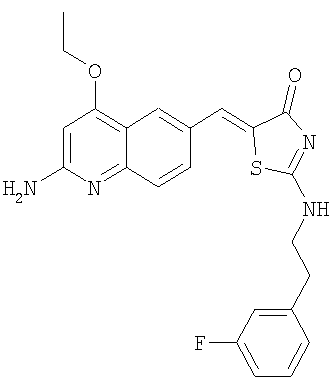
5-(2-Амино-4-этоксихинолин-6-илметилен)-2-[2-(3-фторфенил)этиламино]тиазол-4-он
Получение 6-бром-1Н-бензо[d][1,3]оксазин-2,4-диона
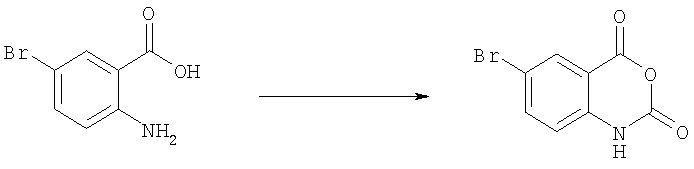
Раствор 2-амино-5-бромбензойной кислоты (280 г, 1,3 моль) в ацетонитриле (1,3 л) нагревали до 50-55°С. Добавляли по каплям одновременно пиридин (206 г, 2,61 моль) и раствор трифосгена (128,8 г, 0,43 моль) в дихлорметане (720 мл). После завершения добавления смесь перемешивали при 50-55°С еще 2 ч. Удаляли растворитель при пониженном давлении и добавляли воду. Осадок отделяли фильтрованием, промывали последовательно водой и охлажденным дихлорметаном и затем сушили в вакууме, получая желаемый продукт 6-бром-1Н-бензо[о][1,3]оксазин-2,4-дион (304 г, 98%). Это вещество использовали на следующей стадии без дополнительной очистки.
Получение 2-амино-6-бромхинолин-4-ола.
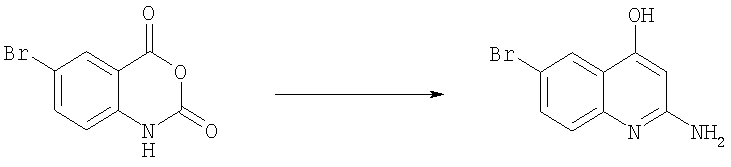
Раствор 6-бром-1Н-бензо[d][1,3]оксазин-2,4-диона (90 г, 0,373 моль) в N,N-диметилформамиде (ДМФ) (400 мл) добавляли к раствору малонитрила (37 г, 0,41 моль) и триэтиламина (41,4 г, 0,41 моль) в ДМФ (150 мл) при 50-60°С. Реакционную смесь выдерживали при 50-60°С в течение 30 мин. Выливали реакционную смесь в ледяную 0,2н. HCl (400 мл), образовавшийся осадок отфильтровывали и сушили в вакууме. Затем его растворяли в 8н. КОН (2 л) и раствор кипятили в течение 40 ч. После охлаждения до комнатной температуры смесь нейтрализовали HCl и полученный осадок отфильтровывали и высушивали на воздухе, получая 2-амино-6-бромхинолин-4-ол в виде бледно-желтого твердого вещества (95 г, 97%). Это вещество использовали на следующей стадии без дополнительной очистки.
Получение N-(6-бром-4-гидроксихинолин-2-ил)ацетамида
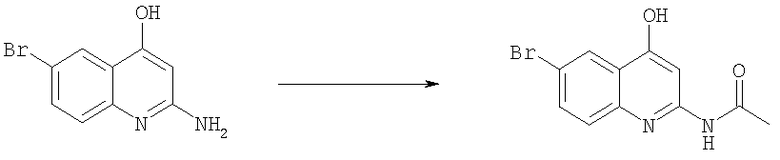
Смесь 2-амино-6-бромхинолин-4-ола (29,4 г, 0,12 моль), уксусного ангидрида (36,7 г, 0,36 моль) и серной кислоты (20 мл) в ледяной уксусной кислоте (300 мл) кипятили в течение 30 мин. Смеси давали охладиться до комнатной температуры, затем выливали в воду. Осадок отфильтровывали и высушивали, получая продукт N-(6-бром-4-гидроксихинолин-2-ил)ацетамид в виде коричневого твердого вещества (32 г, 93%). Этот продукт использовали на следующей стадии без дополнительной очистки.
Получение N-(6-бром-4-этоксихинолин-2-ил)ацетамида
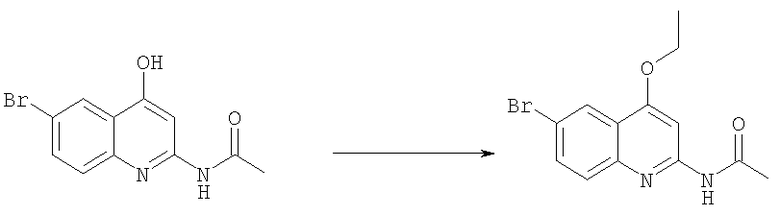
Смесь N-(6-бром-4-гидроксихинолин-2-ил)ацетамида (32 г, 0,114 мол), йодистого этила (26,77 г, 0,171 моль) и карбоната калия (130 г, 0,912 моль) в ацетонитриле (250 мл) кипятили в течение 2 ч, затем растворитель удаляли, и остаток растирали с водой. Осадок выделяли фильтрованием и высушивали, получая N-(6-бром-4-этоксихинолин-2-ил)ацетамид в виде бледно-желтого твердого вещества (28 г, 80%). Этот продукт использовали на следующей стадии без дополнительной очистки.
Получение N-(4-этокси-6-формилхинолин-2-ил)ацетамида
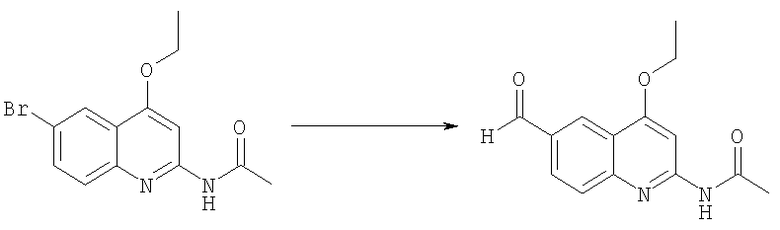
Смесь тетракис(трифенилфосфин)палладия (Pd(PPh3)4) и формиата натрия (5 г, 48 ммоль) в ацетонитриле (30 мл) насыщали азотом. Раствор N-(6-бром-4-этоксихинолин-2-ил)ацетамида (2,5 г, 8,12 ммоль) в ДМСО (30 мл) добавляли через резиновую перегородку. Сосуд выдерживали в атмосфере окиси углерода (50 psi) и нагревали до 120°С в течение 30 мин, затем смесь охлаждали до комнатной температуры, ацетонитрил удаляли при пониженном давлении, добавляли воду и полученный осадок отфильтровывали, высушивали, получая желаемый альдегид N-(4-этокси-6-формилхинолин-2-ил)ацетамид в виде бледно-желтого твердого вещества (0,8 г, 40%).
Получение 5-(2-амино-4-этоксихинолин-6-илметилен)-2-[2-(3-фторфенил)этиламино]тиазол-4-она
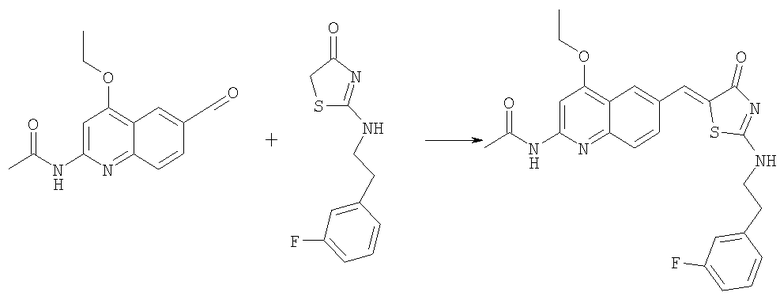
Раствор N-(4-этокси-6-формилхинолин-2-ил)ацетамида (пример 2е, 50 мг, 0,19 ммоль) в уксусной кислоте (1,5 мл) обрабатывали 2-[2-(3-фторфенил)этиламино]тиазол-4-оном (69 мг, 0,29 ммоль) и ацетатом натрия (63 мг, 0,77 ммоль) в микроволновом синтезаторе при 180°С в течение 60 мин, добавляли воду (0,5 мл) и реакционную смесь выдерживали в микроволновом синтезаторе при 140°С в течение 15 мин. Затем смесь гасили 1н. NaOH, осадок отфильтровывали с отсасыванием и последовательно промывали водой, эфиром и дихлорметаном. Сырой осадок затем растворяли в ДМФ и концентрировали до сухого состояния. Этот продукт растирали с горячим диоксаном и фильтровали. Маточную жидкость концентрировали при уменьшенном давлении, получая 5-(2-амино-4-этоксихинолин-6-илметилен)-2-[2-(3-фторфенил)этиламино]тиазол-4-он в виде порошка (23 мг, 28%). ЖХ-МС m/e 437 (МН+).
Пример 3
2-Амино-5-(2-амино-4-этоксихинолин-6-илметилен)тиазол-4-он
Раствор N-(4-этокси-6-формилхинолин-2-ил)ацетамида (пример 2е, 50 мг, 0,19 ммоль) в уксусной кислоте (1,5 мл) обрабатывали псевдотиогидантоином (34 мг, 0,29 ммоль) и ацетатом натрия (63 мг, 0,77 ммоль) в микроволновом синтезаторе при 180°С в течение 45 мин. Смесь разделяли между 1н. NaOH и дихлорметаном. Водный слой, содержащий целевой продукт, концентрировали до сухого состояния, и сырой остаток очищали обращеннофазной ВЭЖХ, получая продукт в виде соли с ТФУ (5 мг, 8%). ЖХ-МС m/e 315 (МН+).
Пример 4
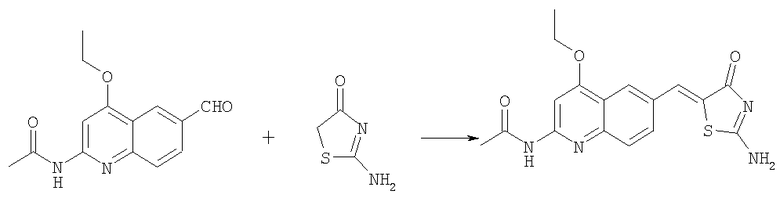
N-(4-Этокси-6-{4-оксо-2-[(тетрагидропиран-4-илметиламино)-4Н-тиазол-5-илиденметил}хинолин-2-ил]ацетамид
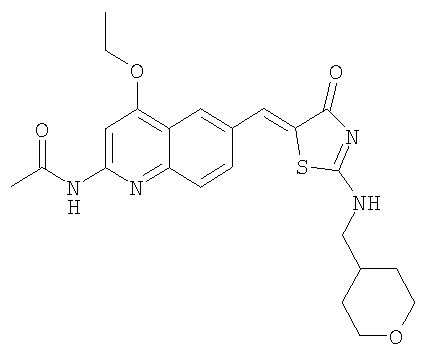
Получение ацетата с-(тетрагидропиран-4-ил)метиламмония
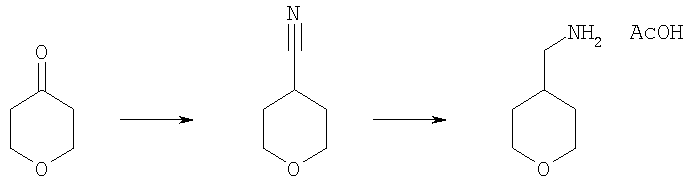
Охлажденный на ледяной бане раствор тетрагидро-4Н-пиран-4-она (7,5 г, 75 ммоль) и тозилметилизоцианида (16,05 г, 82,4 ммоль) в ДМЭ (125 мл) обрабатывали суспензией трет-бутоксида калия (16,8 г, 150 ммоль) в трет-бутиловом спирте (250 мл). Реакционную смесь перемешивали 3,5 ч при комнатной температуре и затем разбавляли эфиром (250 мл). Смесь последовательно промывали водой и солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали быстрой перегонкой с коротким холодильником, получая нитрил в виде бесцветного масла (2,98 г). Это вещество растворяли в 1М боран/тетрагидрофуран (ТГФ) (134 мл, 134 ммоль) и перемешивали в течение ночи. Избыток борана «гасили» метанолом (комнатная температура, 1 ч) и затем смесь концентрировали до сухого остатка. Остаток растворяли в 4н. HCl/диоксан, перемешивали 1 ч при комнатной температуре и концентрировали при пониженном давлении. Твердое вещество растирали с эфиром и отфильтровывали отсасыванием. Суспензию этого вещества (1,81 г, 11,9 ммоль) в ТГФ (30 мл) обрабатывали 1н. NaOH (11,9 мл, 11,9 ммоль) в течение получаса при комнатной температуре. Удаляли отгонкой ТГФ, водный раствор насыщали NaCl и экстрагировали дихлорметаном. Органический слой высушивали над сульфатом натрия и концентрировали при пониженном давлении. Остаток обрабатывали уксусной кислотой (0,68 мл, 11,9 ммоль), получая после высушивания в вакуумном шкафу ацетат с-(тетрагидропиран-4-ил)метиламмония (1,71 г).
Получение N-(4-этокси-6-{4-оксо-2-[(тетрагидропиран-4-илметиламино)-4Н-тиазол-5-илиденметил}хинолин-2-ил]ацетамида
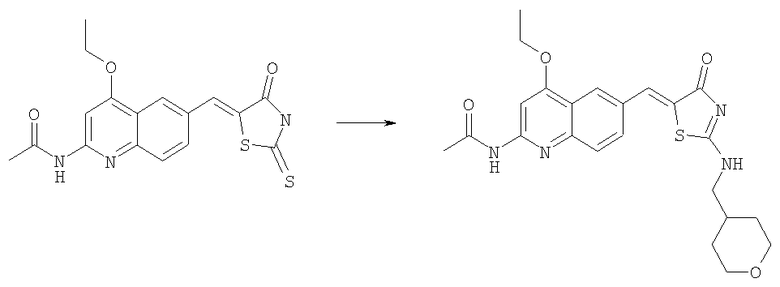
Суспензию N-[4-этокси-6-(4-оксо-2-тиоксотиазолидин-5-илиденметил)хинолин-2-ил]ацетамида (пример 6а, 50 мг, 0,12 ммоль) в ацетонитриле (2 мл) вводили в реакцию с диизопропилэтиламином (0,20 мл, 1,2 ммоль) и йодистым метилом (0,15 мл, 2,3 ммоль) при комнатной температуре в течение 45 мин. Смесь концентрировали до сухого остатка, остаток суспендировали в ацетонитриле (2 мл). Последовательно добавляли при комнатной температуре диизопропилэтиламин (0,20 мл, 1,2 ммоль) и ацетат с-(тетрагидропиран-4-ил)метиламмония (100 мг, 0,58 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Остаток собирали фильтрованием с отсасыванием, адсорбировали на SiO2 и очищали на колонке с силикагелем в смеси метанол-этилацетат с градиентом 0-10%, получая твердый продукт (29 мг, 56%). ЖХ-МС m/e 455 (МН+).
Пример 5
N-[6-(2-Циклопропиламино-4-оксо-4Н-тиазол-5-илиденметил)-4-этоксихинолин-2-ил]ацетамид
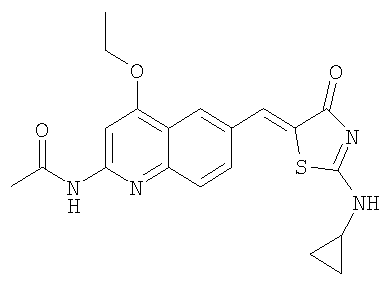
Получение 2-циклопропиламинотиазол-4-она
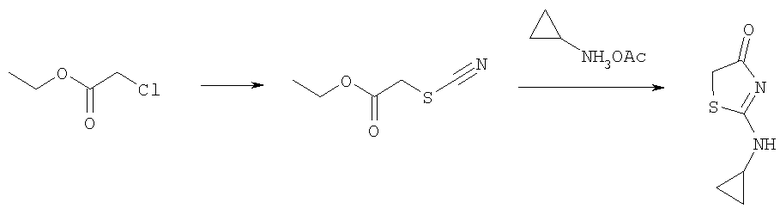
Раствор этилхлорацетата (20 г, 0,16 моль) и тиоцианата калия (12,7 г, 0,13 моль) в этаноле (100 мл) кипятили в течение 3 ч. Затем раствор фильтровали и концентрировали при пониженном давлении, получая 18,7 г этилового эфира тиоцианатуксусной кислоты (97%). Смесь ацетата циклопропиламмония (0,50 г, 4,27 ммоль) и этилового эфира тиоцианатуксусной кислоты (0,62 г, 4,27 ммоль) нагревали до 90°С в течение 3 ч и оставляли на ночь при комнатной температуре. Затем реакционную смесь распределяли между 6н. HCl и дихлорметаном. Слои разделяли. Водный слой подщелачивали добавлением 6н. гидроокиси аммония и затем концентрировали до сухого состояния. Сырой продукт растирали с дихлорметаном и осадок отфильтровывали. Фильтрат сушили над сульфатом натрия и концентрировали до сухого состояния. Сырой продукт очищали, пропуская через колонку с силикагелем с 100% этилацетатом, получая 2-циклопропиламинотиазол-4-он (244 мг, 37%).
Получение N-[6-(2-циклопропиламино-4-оксо-4Н-тиазол-5-илиденметил)-4-этоксихинолин-2-ил] ацетамида
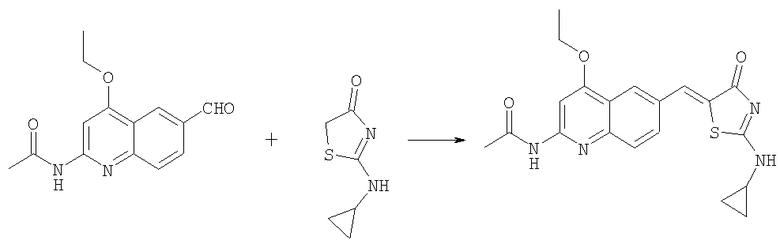
Раствор N-(4-этокси-6-формилхинолин-2-ил)ацетамида (100 мг, 0,39 ммоль) в уксусной кислоте (2 мл) обрабатывали 2-циклопропиламинотиазол-4-оном (61 мг, 0,39 ммоль) и ацетатом натрия (127 мг, 1,55 ммоль) в микроволновом синтезаторе при 180°С в течение 90 мин. Осадок собирали фильтрованием с отсасыванием, последовательно промывали водой и эфиром, получая после высушивания в вакуумном сушильном шкафу продукт N-[6-(2-циклопропиламино-4-оксо-4Н-тиазол-5-илиденметил)-4-этоксихинолин-2-ил]ацетамид в виде твердого вещества (32 мг, 21%). ЖХ-МС m/e 397 (МН+).
Пример 6
N-(4-Этокси-6-{4-оксо-2-[2-тетрагидропиран-4-ил)этиламино]-4Н-тиазол-5-илиденметил}хинолин-2-ил)ацетамид:
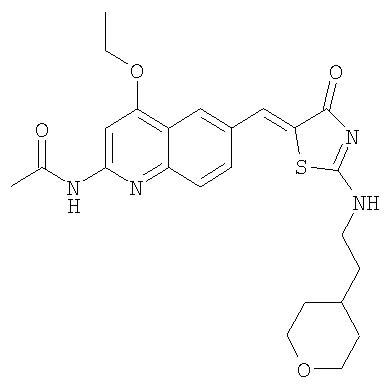
Получение N-[4-этокси-6-(4-оксо-2-тиоксотиазолидин-5-илиденметил)хинолин-2-ил]ацетамида
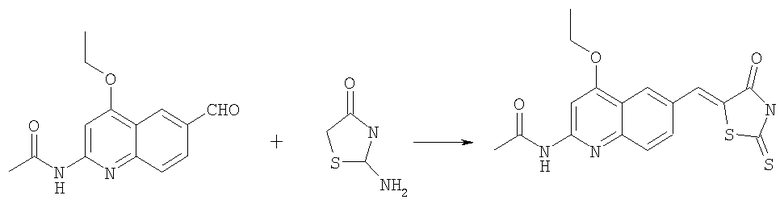
Раствор N-(4-этокси-6-формилхинолин-2-ил)ацетамида (400 мг, 1,55 ммоль) в уксусной кислоте (6 мл) обрабатывали роданином (320 мг, 0,29 ммоль) и ацетатом натрия (530 мг, 6,5 ммоль) в микроволновом синтезаторе при 160°С в течение 25 мин. Осадок собирали фильтрованием с отсасыванием, промывали уксусной кислотой, водой и эфиром, затем высушивали в вакуумном шкафу, получая промежуточный N-[4-этокси-6-(4-оксо-2-тиоксотиазолидин-5-илиденметил)хинолин-2-ил]ацетамид в виде твердого коричневого вещества (396 мг, 68%).
Получение N-(4-этокси-6-{4-оксо-2-[2-тетрагидропиран-4-ил)этиламино]-4Н-тиазол-5-илиденметил} хинолин-2-ил)ацетамида
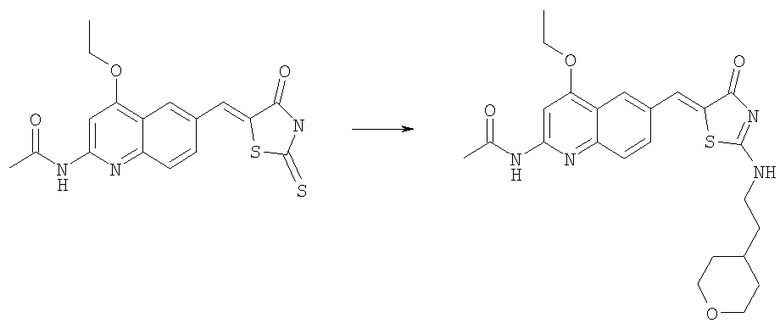
Суспезию N-[4-этокси-6-(4-оксо-2-тиоксотиазолидин-5-илиденметил)хинолин-2-ил]ацетамида (пример 6а, 50 мг, 0,11 ммоль) в ацетонитриле (1,5 мл) обрабатывали в течение 30 мин при комнатной температуре диизопропилэтиламином (0,200 мл, 1,15 ммоль) и йодистым метилом (0,15 мл, 2,3 ммоль), затем смесь концентрировали до сухого состояния и остаток суспендировали в ацетонитриле (1,5 мл). К нему последовательно добавляли при комнатной температуре диизопропилэтиламин (0,20 мл, 1,15 ммоль) и 4-(2-аминоэтил)тетрагидропиран (0,075 мл, 0,58 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Осадок собирали фильтрованием с отсасыванием и промывали ацетонитрилом. Затем его адсорбировали на SiO2 и очищали колоночной хроматографией на силикагеле при градиенте метанол/этилацетат 0-10%, получая продукт N-(4-этокси-6-{4-оксо-2-[2-тетрагидропиран-4-ил)этиламино]-4Н-тиазол-5-илиденметил}хинолин-2-ил)ацетамид в виде бледно-желтого твердого вещества (22 мг, 50%). ЖХ-МС m/e 469 (MH+).
Пример 7
N-(6-{2-Амино-4-оксо-4Н-тиазол-5-илиденметил}-4-этоксихинолин-2-ил)ацетамид
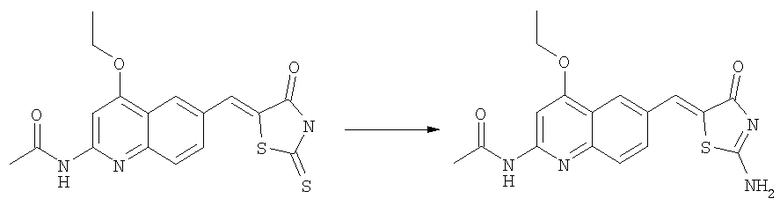
Суспензию N-[4-этокси-6-(4-оксо-2-тиоксотиазолидин-5-илиденметил)хинолин-2-ил]ацетамида (пример 6а, 80 мг, 0,21 ммоль) в ацетонитриле (3 мл) вводили в реакцию с диизопропилэтиламином (0,40 мл, 2,2 ммоль) и йодистым метилом (0,30 мл, 4,6 ммоль) в течение 30 мин при комнатной температуре. Смесь концентрировали до сухого состояния и остаток суспендировали в ДМФ (0,5 мл). Добавляли при комнатной температуре раствор аммиака в метаноле (7н., 5 мл, 35 ммоль) и смесь перемешивали при комнатной температуре в течение 24 ч. Затем смесь концентрировали до сухого состояния и твердый остаток растирали с водой. Осадок собирали фильтрованием с отсасыванием, последовательно промывали водой и эфиром и затем высушивали на воздухе, получая продукт в виде светлокоричневого твердого вещества (50 мг, 63%). ЖХ-МС m/е 357 (МН+).
Пример 8
N-[6-(2-Циклопропилметиламино-4-оксо-4Н-тиазол-5-илиденметил)-4-этоксихинолин-2-ил]ацетамид
Получение 2-циклопропилметиламинотиазол-4-она
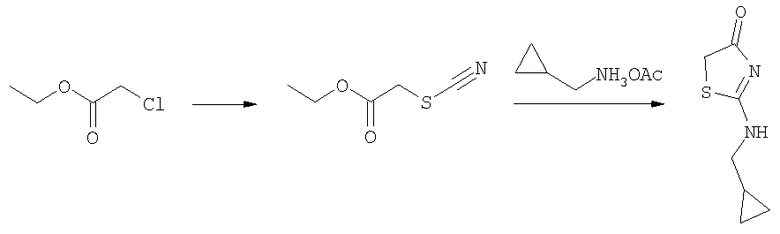
Смесь ацетата циклопропилметиламмония (0,45 г, 3,44 ммоль) и этилового эфира тиоцианатуксусной кислоты (0,5 г, 3,44 ммоль) нагревали 2 ч при 90°С. Реакционную смесь разделяли между 6н. HCl и дихлорметаном. Слои разделяли. Водный слой подщелачивали 6н. гидроксидом аммония и затем экстрагировали дихлорметаном. Органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении. Водный слой концентрировали до сухого остатка, который растирали с ДМФ. Раствор в ДМФ фильтровали и концентрировали до сухого состояния. Остатки объединяли и очищали на колонке с силикагелем со 100% этилацетатом, получая 2-циклопропилметиламинотиазол-4-он (275 мг, 47%).
Получение N-[6-(2-циклопропилметиламино-4-оксо-4Н-тиазол-5-илиденметил)-4-этоксихинолин-2-ил]ацетамида
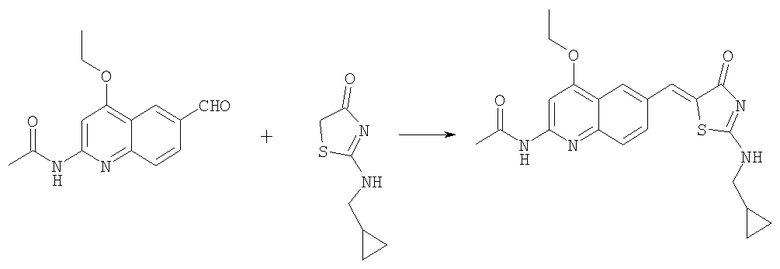
Раствор N-(4-этокси-6-формилхинолин-2-ил)ацетамида (100 мг, 0,39 ммоль) в уксусной кислоте (2 мл) обрабатывали 2-циклопропилметиламинотиазол-4-оном (66 мг, 0,39 ммоль) и ацетатом натрия (127 мг, 1,55 ммоль) в микроволновом синтезаторе при 180°С в течение 2 ч. Реакционную смесь затем распределяли между 1н. NaOH и смесью этилацетат/дихлорметан (1:1). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали до сухого состояния. Сырой остаток очищали колоночной хроматографией на силикагеле при градиенте метанол/этилацетат 0-7%, получая продукт N-[6-(2-циклопропилметиламино-4-оксо-4Н-тиазол-5-илиденметил)-4-этоксихинолин-2-ил]ацетамид в виде твердого вещества (10 мг, 7%). ЖХ-МС m/e 411(MH+).
Пример 9
N-(6-{2-[([1,4]Диоксин-2-илметил)амино]-4-оксо-4Н-тиазол-5-илиденметил}-4-этоксихинолин-2-ил)ацетамид
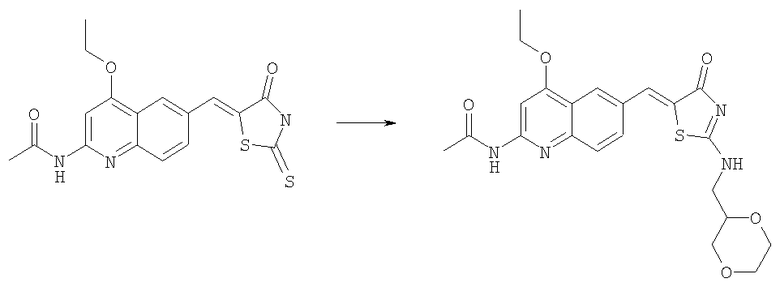
Суспензию N-[4-этокси-6-(4-оксо-2-тиоксотиазолидин-5-илиденметил)хинолин-2-ил]ацетамида (пример 6а, 50 мг, 0,12 ммоль) в ацетонитриле (1,5 мл) вводили в реакцию с диизопропилэтиламином (0,20 мл, 1,2 ммоль) и йодистым метилом (0,15 мл, 2,3 ммоль) при комнатной температуре в течение 30 мин. Смесь концентрировали до сухого состояния, и остаток суспендировали в ацетонитриле (1,5 мл). Последовательно при комнатной температуре добавляли диизопропилэтиламин (0,20 мл, 1,2 ммоль) и с-[1,4]диоксин-2-илметиламин (68 мг, 0,58 ммоль) и смесь оставляли перемешиваться при комнатной температуре в течение ночи. Осадок отбирали фильтрованием с отсасыванием, адсорбировали на SiO2 и очищали на колонке с силикагелем со 100% этилацетатом, получая продукт N-(6-{2-[([1,4]диоксин-2-илметил)амино]-4-оксо-4Н-тиазол-5-илиденметил}-4-этоксихинолин-2-ил)ацетамид в виде твердого вещества (34 мг, 63%). ЖХ-МС m/e 457 (МН+).
Пример 10
Фармакологические анализы
Фармакологические свойства соединений предлагаемого изобретения могут быть подтверждены рядом фармакологических опытов. Ниже приведены примеры фармакологических опытов, которые были проведены с соединениями предлагаемого изобретения и их солями. Соединения предлагаемого изобретения проявляют CDKl/циклин В активность со значениями Ki меньшими 5,0 мкМ. Это означает, что все эти соединения активны в ингибировании CDK-1/циклин В.
Для определения ингибиторной CDK1 активности были осуществлены анализы или FlashPlateTM (NENTM -Life Science Products) или HTRF (метод гомогенной разрешенной по времени флуоресценции). Оба типа анализов на киназы проводили с использованием рекомбинантного человеческого CDK1/циклин В комплекса. GST (глутатион-3-трансфераза)-циклин В и CDK1 кДНК клоны в векторах бакуловирусов были предоставлены д-р W. Harper из Baylor College of Medicine, Хьюстон, ТХ. Белки были коэкспрессированы в High FiveTM клетки насекомых, и комплекс очищали на глутатионовой смоле Sepharose (Pharmacia, Piscataway, NJ), как описано ранее (Harper J.W. и др., Cell, 75, 1993, сс.805-816). бх-Гистидин меченную укороченную форму белка ретинобластомы (Рб) (аминокислота 386-928) использовали как субстрат в анализе CDKl/циклин В (экспрессионный пластид был предоставлен Dr. Veronica Sullivan, Department of Molecular Virology, Roche Research Center, Welwyn Garden City, Англия). Рб белок является природным субстратом для фосфорилирования киназой CDK1 (см. Herwig и Strauss Eur. J. Biochem., 246, 1997, с.581-60 и приведенные там ссылки). Экспрессию 62Kd белка контролировал индуцированный IPTG (изопропил-бета-D-тиогалактопиранозид) промотер в штамме Ml 5 E.coli. Клетки лизировали ультразвуком и проводили очистку через связывание лизатов при рН 8,0 на Ni-хелатированной агарозной колонке, предварительно обработанной 1 мМ имидазолом. Смолу затем несколько раз промывали буфером, постепенно понижающим рН до 6,0 и элюировали 500 мМ имидазолом. Элюированный белок диализовали против 20 мМ ГЕПЕС с рН 7,5, 30% глицерина, 200 мМ NaCl и 1 мМ ДТТ (дитиотреитол, реактив Клеланда). В запасных очищенных сливах белка Рб определяли концентрацию белка, доводили до аликвоты и хранили при -70°С.
При проведении FlashPlate анализа киназы, 96-луночную плашку FlashPlate покрывали Рб белком с концентрацией 10 мкг/мл, используя 100 мкл на ячейку. Плашки инкубировали при 4°С в течение ночи или 3 ч на шейкере при комнатной температуре. Для контроля неспецифического фосфорилирования один ряд ячеек покрывали защитным буфером 100 мкл/ячейка (20 мМ ГЕПЕС, 0,2 М NaCl). Плашки затем дважды промывали промывочным буфером (0,01% Твин 20 в фосфатно-буферном солевом растворе). Тестируемые соединения (тест-соединения) добавляли в ячейки при 5х конечной концентрации. Реакции инициировали немедленным добавлением 40 мкл реакционной смеси (25 мМ ГЕПЕС, 20 мМ MgCl2, 0,002% Твин 20, 2 мМ ДТТ, 1 мкМ АТФ, 4 нМ 33Р-АТФ) и достаточного количества фермента, так, чтобы сигналы превышали нулевой уровень, по крайней мере, в десять раз. Плашки инкубировали при комнатной температуре на шейкере в течение 30 мин. Плашки промывали четыре раза промывочным буфером, закрывали и снимали показания TopCount сцинциляционного счетчика (Packard Instrument Co., Downers Grove, IL). Процент ингибирования фосфорилирования Рб белка, который является мерой ингибирования CDK активности, определяли по следующей формуле:
100 × (1-тест-соединение-неспециф.)/(общее-неспециф.)
где «тест-соединение» относится к средним значениям импульсов в мин тестируемого соединения в двойном повторении, «неспециф.» относится к средним значениям импульсов в мин в отсутствие CDKl/циклина В и «общее» относится к средним значениям импульсов в мин в отсутствие соединения. Величина IC50 означает концентрацию тестируемого соединения, которая уменьшает на 50% вызываемую протеинкиназой инкорпорацию радиоактивной метки в описанных условиях теста. Величина константы ингибитора Ki вычисляется по следующему уравнению: Ki=IC50/(1+[S]/Km), где [S] означает концентрацию АТФ и Km означает константу Михаэлиса.
Анализ киназы методом гомогенной разрешенной по времени флуоресценции (HTRF) проводили в 96-луночных полипропиленовых плашках (BD Biosciences, Bedford, MA). Тестируемые соединения сначала растворяли в ДМСО, а затем разбавляли буфером 1 для анализа киназы (25 мМ ГЕПЕС, рН 7,0, 8 мМ MgCl2, 1,5 мМ ДТТ и 162 мкМ АТФ) с концентрацией ДМСО 15%. CDKl/циклин В фермент разбавляли буфером 2 для анализа киназы (25 мМ ГЕПЕС, рН 7,0, 8 мМ MgCl2, 0,003% Твин 20, 0,045% БСА, 1,5 мМ ДТТ и 0,338 мкМ Рб белок). Для инициирования киназной реакции 20 мкл раствора соединения смешивали с 40 мкл раствора CDK1/циклин В в аналитических плашках с конечной концентрацией CDK1/циклин В и Рб 0,1 мкг/мл и 0,113 мкМ соответственно и инкубировали 30 мин при 37°С. Добавляли 15 мкл антифосфоРб антитела (Ser 780) (Cell Signaling Technology, Beverly, MA) с разбавлением антитела 1:7692. Продолжали инкубировать при 37°С в течение 25 мин, после чего в ячейки добавляли меченое LANCE Eu-W1024 анти-LgG кролика и анти-His антитело, конъюгированные с реагентом для флуоресценции SureLightAllophucocyanin (20 нМ PerkinElmer, Wellesley, MA). Инкубацию продолжали еще 40 мин при 37°С. В конце инкубации 35 мкл реакционной смеси помещали в свежие черные полистирольные платы с 384 лунками (Corning incorporated. Corning, NY) и считывали на флуоресцентном планшет-ридере показания при длине волны возбуждения 340 нм и длине волны излучения 665/615 нм.
Значения Ki, показывающие активность CDK1/циклин В, которые относятся к соединениям предлагаемого изобретения, находятся в пределах от примерно 0,001 мкМ до примерно 5,000 мкМ. Специфические данные для некоторых примеров следующие:
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИНАЗОЛИНИЛМЕТИЛЕНТИАЗОЛИНОНЫ В КАЧЕСТВЕ CDK-1 ИНГИБИТОРОВ | 2005 |
|
RU2405782C2 |
| НОВЫЕ АЗАИНДОЛТИАЗОЛИНОНЫ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2005 |
|
RU2391342C2 |
| 4-МОНОЗАМЕЩЕННЫЕ ТИАЗОЛИНОНХИНОЛИНЫ | 2005 |
|
RU2397983C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,4-ДИАМИНОТИАЗОЛ-5-ОНА | 2005 |
|
RU2395501C2 |
| 1, 5-НАФТИРИДИНАЗОЛИДИНОНЫ, ОБЛАДАЮЩИЕ CDK1 АНТИПРОЛИФЕРАТИВНОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2405781C2 |
| ПРОИЗВОДНЫЕ ЦЕФАЛОСПОРИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1994 |
|
RU2130939C1 |
| СОЕДИНЕНИЯ | 2018 |
|
RU2774952C2 |
| ЗАМЕЩЕННЫЕ ПИРИДАЗИН-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ СОЕДИНЕНИЙ, ИНГИБИРУЮЩИХ КИНАЗЫ | 2009 |
|
RU2526618C2 |
| ПИРАЗОЛПИРИМИДИНЫ | 2005 |
|
RU2412186C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
Настоящее изобретение относится к соединениям формулы I и их фармацевтически приемлемым солям. Соединения настоящего изобретения обладают ингибирующей активностью в отношении CDK1 киназы. В формуле I
R1 означает водород или R2-(X)n-; X означает низший алкилен или зациклизованный низший алкилен; R2 означает ; где означает фенил; циклоалкил, содержащий 3-6 атомов углерода; 4-6-членное гетероциклоалкильное кольцо, содержащее 3-5 атомов углерода и 1-2 атома кислорода; R5, R6 и R7 независимо выбраны из группы, включающей водород или галоид; R4 означает водород или -(O)k(CH2CH2O)y-R10; R19 означает водород; R20 означает водород или -C(O)-R11; R10 и R11 означают низший алкил; n и k означают целое число от 0 до 1; у означает целое число от 0 до 3. Изобретение также относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении CDK1 киназы, содержащей одно или более соединений изобретения. 2 н. и 13 з.п. ф-лы.
1. Соединение формулы
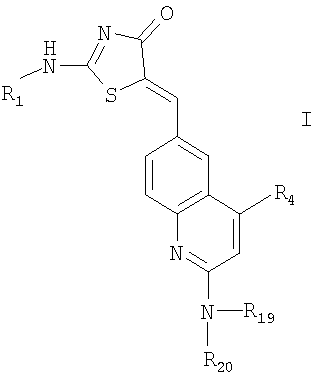
где R1 означает водород или R2-(X)n-;
X означает низший алкилен или зациклизованный низший алкилен;
R2 означает
;
где означает фенил; циклоалкил, содержащий 3-6 атомов углерода; 4-6-членное гетероциклоалкильное кольцо, содержащее 3-5 атомов углерода и 1-2 атома кислорода;
R5, R6 и R7 независимо выбраны из группы, включающей водород или галоид;
R4 означает водород или -(O)k(CH2CH2O)y-R10;
R19 означает водород;
R20 означает водород или -C(O)-R11;
R10 и R11 означают низший алкил;
n и k означают целое число от 0 до 1;
у означает целое число от 0 до 3;
или их фармацевтически приемлемые соли.
2. Соединение по п.1 формулы I-A
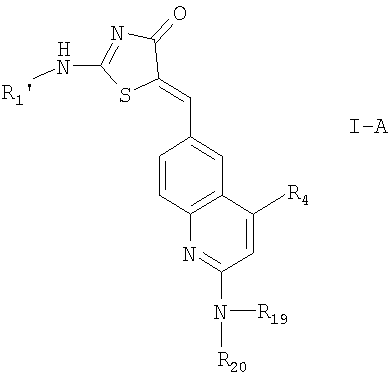
где R1' означает водород; и
R4, R19 и R20 имеют значения, указанные в п.1;
или их фармацевтически приемлемые соли.
3. Соединение по п.2, где
R1' означает водород;
R4 означает -(O)k(CH2CH2O)y-R10; и
R10, k и у имеют значения, указанные в п.1.
4. Соединение по п.3, где указанное соединение выбрано из группы, включающей
N-(6-{2-амино-4-оксо-4Н-тиазол-5-илиденметил}-4-этоксихинолин-2-ил)ацетамид, и
2-амино-5-(2-амино-4-этоксихинолин-6-илметилен)тиазол-4-он.
5. Соединение по п.1 формулы I-B
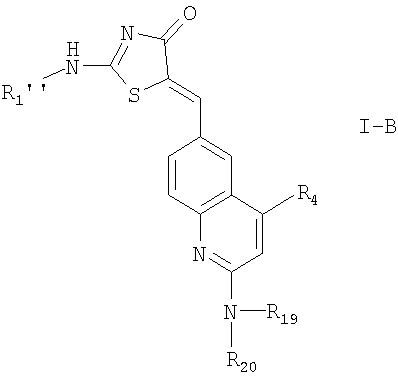
где R1'' означает R2'-(X')n-;
n, R4, R19 и R20 имеют значения, указанные в п.1, и
X' означает низший алкилен или зациклизованный низший алкилен;
R2' означает
где означает фенил; циклоалкильное кольцо, содержащее 3-6 атомов углерода; 4-6-членное гетероциклоалкильное кольцо, содержащее 3-5 атомов углерода и 1-2 атома кислорода;
R5' и R6' независимо выбраны из группы, включающей водород или галоид;
или их фармацевтически приемлемые соли.
6. Соединение по п.5, где
X' означает низший алкилен;
R2' означает фенил, необязательно замещенный галоидом;
R4 означает -O-СН2-СН3;
n равно 1.
7. Соединение по п.6, где указанное соединение представляет собой 5-(2-амино-4-этоксихинолин-6-илметилен)-2-[2-(3-фторфенил)этиламино]тиазол-4-он.
8. Соединение по п.5, где
X' означает низший алкилен;
R2' означает гетероциклическое кольцо, содержащее 3-5 атомов углерода и 1-2 атома кислорода;
R4 означает -O-СН2-СН3;
n равно 1.
9. Соединение по п.8, где указанное соединение выбрано из группы, включающей
N-(4-этокси-6-{4-оксо-2-[2-(тетрагидропиран-4-ил)этиламино]-4Н-тиазол-5-илиденметил}хинолин-2-ил)ацетамид;
N-(4-этокси-6-{4-оксо-2-[(тетрагидропиран-4-илметиламино)-4Н-тиазол-5-илиденметил}хинолин-2-ил]ацетамид; и
N-(6-{2-[([1,4]диоксин-2-илметил)амино]-4-оксо-4Н-тиазол-5-илиденметил}-4-этоксихинолин-2-ил]ацетамид.
10. Соединение по п.5, где
X' означает низший алкилен;
R2' означает циклопропил;
R4 означает водород или -O-СН2-СН3;
n равно 0 или 1.
11. Соединение по п.10, где указанное соединение выбрано из группы, включающей
N-[6-(2-циклопропилметиламино-4-оксо-4Н-тиазол-5-илиденметил)-4-этоксихинолин-2-ил]ацетамид;
N-[6-(2-циклопропиламино-4-оксо-4Н-тиазол-5-илиденметил)-4-этоксихинолин-2-ил]ацетамид; и
5-[1-(2-аминохинолин-6-ил)мет-(Z)-илиден]-2-((1R,2S)-2-фенилциклопропиламино)тиазол-4-он.
12. Соединение формулы I по п.1, обладающее ингибирующей активностью в отношении CDK1 киназы.
13. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении CDK1 киназы, включающая одно или более соединений формулы I по п.1 вместе с фармацевтически приемлемыми вспомогательными веществами.
14. Соединение формулы I по п.1 для получения лекарственного средства, обладающего ингибирующей активностью в отношении CDK1 киназы.
15. Соединение формулы I по п.1 для получения лекарственного средства, предназначенного для лечения рака, особенно твердых опухолей.
| ПРОИЗВОДНЫЕ ХИНОЛИНА | 1995 |
|
RU2137770C1 |
| EP 1215208 A2, 19.06.2002 | |||
| WO 2004007491 A1, 22.01.2004 | |||
| WO 2004006916 A1, 22.01.2004 | |||
| WO 2004047760 A2, 10.06.2004. | |||

