Изобретение относится к новым производным 4-аминотиазола, которые ингибируют циклин-зависимые киназы. Указанные соединения и их фармацевтически приемлемые соли и эфиры обладают антипролиферативной активностью и используются для лечения или подавления развития рака, прежде всего солидных опухолей. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим такие соединения, и к способам лечения или подавления развития рака, наиболее предпочтительно лечения и подавления роста опухолей молочной железы, легких, ободочной кишки и предстательной железы. Наконец, настоящее изобретение относится к новым промежуточным соединениям, которые используются при получении описанных в тексте заявки новых производных 4-аминотиазола.
Неконтролируемая пролиферация клеток является маркером рака. Опухолевые клетки обычно содержат какую-либо форму повреждения генов, которые непосредственно или опосредованно регулируют цикл клеточного деления.
Развитие клеток в различных фазах клеточного цикла регулируется серией мультиферментных комплексов, состоящих из регуляторного белка циклина и киназы. Такие киназы называются циклин-зависимыми киназами (Cdk). Киназы Cdk экспресссируются в ходе клеточного цикла, в то время как уровень циклинов изменяется в зависимости от стадии клеточного цикла.
Четыре главных фазы клеточного цикла называются G1, S, G2 и M. Ключевыми ферментами регуляции клеточного цикла являются циклин D/Cdk4, циклин D/Cdk6, циклин E/Cdk2, циклин A/Cdk2 и циклин B/Cdk1 (известный также, как Cdc2/циклин В). Циклин D/Cdk4, циклин D/Cdk6 и циклин E/Cdk2 контролируют прохождение G1-фазы и переход из G1- в S-фазу за счет фосфорилирования фосфопротеина ретинобластомы pRb. Циклин A/Cdk2 регулирует прохождение S-фазы, а циклин B/Cdk1 контролирует точку контроля G2 и регулирует вступление в М фазу (митоз).
Развитие клеточного цикла регулируется киназами Cdk1 (cdc2) и Cdk2 сразу после G1, когда клетки переходят к цитокинезу. Следовательно, можно предположить, что ингибирование лекарственными средствами указанных киназ Cdk не только блокирует клеточную пролиферацию, но и инициирует гибель клеток за счет апоптоза. После перехода клеток через точку контроля G1 и вступления в S фазу они становятся независимыми от стимуляции ростовым фактором для развития клеточного цикла.
После завершения репликации ДНК клетки переходят в G2 фазу клеточного цикла для подготовки к М фазе и цитокинезу. Установлено, что киназа Cdk1 регулирует прохождение клетками через две последние фазы клеточного цикла в ассоциации с циклинами А и В. Для полной активации Cdk1 необходимо связывание обоих циклинов и специфическое фосфорилирование (Morgan D.O., De Bondt H.L., Curr. Opin. Cell. Biol., 6, 239-246 (1994)). После активации комплексы Cdk1/циклин подготавливают клетку к делению в течение М фазы.
Переход из фазы G1 в фазу S, указанный выше, регулируется комплексом Cdk4 с циклином D и Cdk2 с циклином Е. Указанные комплексы фосфорилируют опухолевый супрессорный белок ретинобластомы (pRb), высвобождая фактор транскрипции E2F и способствуя экспресии генов, необходимых в S фазе (Nevins J.R., Science, 258, 424-429 (1992), Lavia P., BioEssays, 21, 221-230 (1999)). Блокирование активности комплексов Cdk4/циклин D и Cdk2/циклин Е останавливает клеточный цикл в фазе G1. Например, белки семейства INK4, включая белок p16INK4a, который блокирует киназную активность комплекса Cdk4/циклин D, вызывает остановку в фазе G1 (Sherr C.J., Science, 274, 1672-1677 (1996)). Специфичность блока подробно описана в литературе (см. статью Vidal A., Gene, 247, 1-15 (2000)).
Современные данные свидетельствуют о том, что комплекс Cdk4 с циклином D3 также играет определенную роль в развитии клеточного цикла при прохождении фазы G2. Ингибирование этого комплекса белком р16 или с использованием доминантно негативной Cdk4 приводит к остановке в фазе G2 клеток, которые не экспрессируют pRb (Gabrielli B.G. и др., J. Biol. Chem., 274, 13961-13969 (1999)).
Установлено, что многочисленные дефекты в пути pRb наблюдаются при различных видах рака. Например, сверхэкспрессия Cdk4 наблюдается в случаях наследственной меланомы (Webster K.R., Exp. Opin. Invest. Drugs, 7, 865-887 (1998)), циклин D сверхэкспрессирован во многих видах рака человека (Sherr C.J., Science, 274, 1672-1677 (1996)), белок р16 мутирован или отсутствует во многих опухолях (Webster K.R., Exp. Opin. Invest. Drugs, 7, 865-887 (1998)), a функция pRb утрачена из-за мутаций или делеции при многих типах рака человека (Weinberg R.A., Cell, 81, 323-330 (1995)). Установлено, что дефекты в этом пути оказывают влияние на прогноз. Например, потеря белка р16 коррелирует с отрицательным прогнозом при немелкоклеточной карциноме легких (NSCLC) и злокачественной меланоме (Tsihlias J. и др., Annu. Rev. Med., 50, 401-423 (1999)). Дефекты циклина D1 и/или pRb на уровне гена и/или экспрессии присутствуют в более 90% случаев немелкоклеточного рака легких, свидетельствуя о том, что циклин D1 и/или pRb представляют собой важную стадию в процессе онкогенеза легких (Marchetti А. и др., Int. J. Cancer, 75, 573-582 (1998)). В 49 из 50 случаев карциномы поджелудочной железы (98%) путь pRb/pl6 подавляется исключительно за счет инактивации гена белка р16 и связанного с ним циклина D (Schutte M. и др., Cancer Res., 57, 3126-3134 (1998)). Взаимосвязь экспрессии pRb и циклин/циклин-зависимых киназ в ряде тканей описана в статье Teicher В.A., Cancer Chemother. Pharmacol., 46, 293-304 (2000).
В связи с участием пути Cdk4/циклин D/pRb в процессее онкогенеза в организме человека за счет регуляции перехода клеточного цикла из фазы G1 в фазу S, и благодаря эффективному лечебному действию при модулировании этого пути, существует необходимость в разработке агентов, которые ингибируют или стимулируют отдельные стадии этого пути. Например, установлено воздействие на раковые клетки антител, антисмысловых олигонуклеотидов и сверхэкспрессии или при добавлении белков, принимающих участие в этом пути. См., например, Lukas J. и др., Nature, 79, 573-582 (1995), Nevins J.R., Science, 258, 424-429 (1992), Lim I.K. и др., Molecular Carcinogenesis, 23, 25-35 (1998), Tam S.W. и др., Oncogene, 9, 2663-2674 (1994), Driscoll В. и др., Am.J. Physiol., 273 (Lung Cell. Mol. Physiol.), L941-L949 (1997) и Sang J. и др., Chin. Sci. Bull., 44, 541-544 (1999)).
Роль киназ cdk в регуляции клеточной пролиферации является таким образом вполне доказанной. Например, как указано выше, существует множество данных, подтверждающих возможность применения соединений, ингибирующих мишени в путях Cdk4, Cdk2 и Cdk1, в качестве антипролиферативных терапевтических агентов. Таким образом, ингибиторы клеточной пролиферации действуют в качестве обратимых цитостатических агентов, которые применяются при лечении патологических состояний, ассоциированных с аномальным ростом клеток, таких, как различные типы рака и другие нарушения пролиферации клеток, включающие, например, воспалительный процесс (например, доброкачественную гиперплазию предстательной железы, семейный аденоматоз, полипоз, нейрофиброматоз, атеросклероз, фиброз легких, артрит, псориаз, воспалительное заболевание кишечника, инфекции в связи с отторжением трансплантата), вирусные инфекции (включая, без ограничения перечисленным, вирус герпеса, поксвирус, вирус Эпштейна-Барра), аутоиммунное заболевание (например, обыкновенная волчанка, ревматоидный артрит, псориаз, воспалительное заболевание кишечника), нейродегенеративные нарушения (включая, без ограничения перечисленным, болезнь Альцгеймера) и нейродегенеративные заболевания (например, болезнь Паркинсона, боковой амиотрофический склероз, пигментный ретинит, мышечная атрофия и церебральная дегенерация).
В качестве ингибиторов киназ Cdk идентифицировано несколько отдельных классов низкомолекулярных соединений: оломоуцин и другие аналоги пурина, флавопиридол, стауроспорин, UCN-01 и другие индолокарбазолы, 9-гидроксиэллиптицин, индирубин, пауллоны, диарилмочевины, хиназолины, индопиразолы, [2, 3-d]пиридопиримидины, фаскаплизин, аминотиазолы, диаминотиазолы, пара-теридиноны и пиразолы (Carlson и др., Cancer Res., 56, 2973-2978 (1996), De Azevedo и др., Eur. J. Biochem., 243, 518-526 (1997), Bridges A.J., Exp. Opin. Ther. Patents., 5, 1245-1257 (1995), Reinhold и др., J. Biol. Chem., 278, 3803-3807 (1998), Kakeya H. и др., Cancer Res., 58, 704-710 (1998), Harper J.W., Cancer Surveys, 29, 91-107 (1997), Harrington E.A. и др., Proc. Natl. Acad. Sci. USA, 95, 11945-11950 (1998), Meijer L. и др., Eur. J. Biochem., 267, 1-13 (2000), Garrett M.D. и др., Current Opin. Genetics Develop., 9, 104-111 (1999), Mgbonyebi О.Р. и др., Cancer Res., 59, 1903-1910 (1999), Hoessel и др., Nature Cell Biology, 1, 60-67 (1999), Zaherevitz и др., Cancer Res., 59, 2566-2569 (1999), Honma Т. и др., 221st National ACS Meeting., Medi 136 (2001). Sielecki T.M. и др., Bioorg. Med. Chem. Lett., 11, 1157-1160 (2001), Nugiel D.A. и др., J. Med. Chem., 44, 1334-1336 (2001), Fry D.W. и др., J. Biol. Chem., 276, 16617-15523 (2001), Soni R. и др., Biochem. Biophys. Res. Commun., 275, 877 (2000), Ryu C-K. и др., Bioorg. Med. Chem. Lett., 10, 461 (2000), Jeong H-W. и др., Bioorg. Med. Chem. Lett., 10, 1819 (2000), Toogood и др., J. Med. Chem., 43, 4606-4616 (2000), Chong W., Fischer, Curr. Opin. in Drug Discov. and Develop., 4, 623-634 (2001), WO 0009921845, Toogood. P., WO 0119825, Toogood P., WO 0138315, Reich S.H., WO 0179198, Webster K. US 6262096.
Обзор соединений, ингибирующих путь Cdk4/циклин D, приводится в литературе, см. Harris W. и Wilkinson S., Emerging Drugs, 5, 287-297 (2000), Dumas J., Exp. Opin. Ther. Patents., 11, 405-429 (2001), Sielecki Т. и др., J. Med. Chem., 43, 1-18 (2000), WO 99/21845, 6569878 B1, 2003/0220326 A1 и WO 2003011843 A1, причем в каждом указанном документе описаны 4-аминотиазолы общей формулы
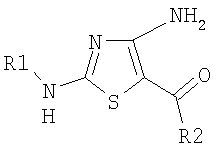 .
.
Установлено, что указанные соединения являются ингибиторами циклин-зависимых киназ.
Настоящее изобретение относится к новым производным 4-аминотиазола формулы
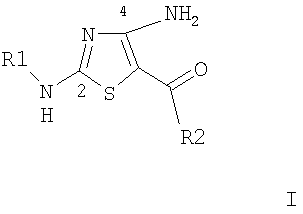 ,
,
где
R1 выбирают из группы, включающей
(a) (низш.)алкил, замещенный арилом,
(b)
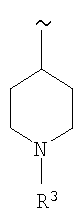 , и
, и
(с)
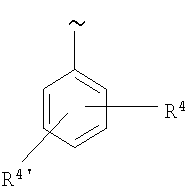
R2 выбирают включающей арил, гетероарил, циклоалкил и гетероцикл, причем каждый из них замещен 1-4 заместителями, назависимо выбранными из группы, включающей (а) (низш.)алкил, (b) галоген, (с) OR5, (d) NH2 и (е) NO2,
R3 выбирают из группы, включающей (а) H, (b) (низш.)алкил, (с) CO2R6,
(d) C(O)R6, (e) SO2R6 и (f) SO2NR5R6,
R4 и R4' каждый независимо выбирают из группы, включающей (а) Н, (b) (низш.)алкил, необязательно замещенный группами оксо, CO2R6, OR6 и/или NH2, (с) S(O)nR7, (d) OR8, (e) NR5R6 и (f) CO2R6,
R5 и R6 каждый независимо выбирают из группы, включающей (а) Н, (b) N, (c) (низш.)алкил, (d) (низш.)алкил, замещенный группами оксо, CO2R9, OR9, и/или NR10R11, (e) арил, необязательно замещенный галогеном, (f) гетероарил, (g) N-арил, где арильная группа необязательно замещена одним или более галогенами, и (h) арил, замещенный галогеном или CF3,
R7 означает (низш.)алкил или арил,
R8 выбирают из группы, включающей (а) Н, (b) (низш.)алкил и (с) (низш.)алкил, замещенный NR5R6,
R9 выбирают из группы, включающей Н и (низш.)алкил,
R10 и R11 каждый независимо выбирают из группы, включающей Н и (низш.)алкил, а
n равно 0, 1 или 2,
или к их фармацевтически приемлемым солям или сложным эфирам.
Указанные соединения ингибируют циклин-зависимые киназы. Указанные соединения и их фармацевтически приемлемые соли и сложные эфиры обладают антипролиферативной активностью и используются при лечении или подавлении развития рака, прежде всего солидных опухолей.
Настоящее изобретение относится также к фармацевтическим композициям, включающим одно или более соединений по изобретению или их фармацевтически приемлемую соль или эфир и фармацевтически приемлемый носитель или эксципиент.
Кроме того, настоящее изобретение относится к применению соединений формулы I для получения лекарственных средств, предназначенных для лечения рака, предпочтительно солидных опухолей и более предпочтительно рака молочной железы, легких, ободочной кишки и предстательной железы.
Кроме того, настоящее изобретение относится к способу лечения или подавления развития рака, прежде всего для лечения или подавления развития солидных опухолей, наиболее предпочтительно для лечения или подавления развития рака молочной железы, легких, ободочной кишки и предстательной железы, причем указанный способ включает введение пациенту, который нуждается в таком лечении, терапевтически эффективного количества соединений формулы I или его фармацевтически приемлемой соли или эфира.
Наконец, настоящее изобретение относится к новым промежуточным соединениям, которые используются для получения соединений формулы I.
Термины, используемые в описании заявки, имеют следующие значения.
«Арил» означает одновалентный моноциклический или бициклический ароматический карбоциклический углеводородный радикал, предпочтительно 6-10-членную ароматическую циклическую систему. Предпочтительные арильные группы включают, без ограничения перечисленным, фенил, нафтил, толил и ксилил.
«Циклоалкил» означает неароматический, частично или полностью насыщенный одновалентный циклический углеводородный радикал, содержащий от 3 до 8 атомов. Примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил и циклогексил.
«Эффективное количество» означает количество (материала), которое эффективно предотвращает, ослабляет или подавляет симптомы заболевания или продлевает срок жизни субъекта, нуждающегося в лечении.
«Галоген» означает фтор, хлор, бром или иод, предпочтительно фтор или хлор.
«Гетероатом» означает атом, выбранный из N, О и S.
«Гетероарил» означает ароматическую гетероциклическую циклическую систему, содержащую до двух циклов. Предпочтительные гетероарильные группы включают, без ограничения перечисленным, тиенил, фурил, индолил, пирролил, пиридинил, пиридин, пиразинил, оксазолил, тиаксолил, хинолинил, пиримидинил, имидазол, бензофуран и тетразолил.
«Гетероциклил» означает насыщенный или частично ненасыщенный неароматический циклический радикал, содержащий в цикле 3-8 атомов, из которых 1-3 атома являются гетероатомами, выбранными из атомов азота, кислорода, S(O)n (где n равно целому числу от 0 до 2), или из их комбинаций, а остальные атомы в цикле являются атомами углерода. Примерами предпочтительных гетероциклилов являются пиперидин, пиперазин, пирролидин, морфолин, индолин, тетрагидропиранил, тиоморфолино, пентаметиленсульфид и пентаметиленсульфон.
«KI» является мерой термодинамического связывания лиганд/ингибитор (т.е. соединения по изобретению) с белком-мишенью. Ki определяют, как описано ниже в примере 18.
Термин «(низш.)алкил», используемый отдельно или в составе другого термина, например, (низш.)алкилгетероциклил, означает насыщенный алифатический углеводород с прямой или разветвленной цепью, содержащий от 1 до включительно 6, предпочтительно от 1 до 4 атомов углерода. Типичные (низш.)алкильные группы включают метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил, гексил и т.п.
«Оксо» означает группу =O.
«Фармацевтически приемлемый сложный эфир» означает этерифицированное соединение формулы I, содержащее карбоксильную группу, причем сложные эфиры сохраняют биологическую активность и свойства соединений формулы I и расщепляются in vivo (в организме) с образованием соответствующей активной карбоновой кислоты. Примерами сложноэфирных групп, которые расщепляются (в данном случае гидролизуются) in vivo с образованием соответствующих карбоновых кислот (R40C(=O)ОН), являются (низш.)алкиловые эфиры, замещенные группой NR41R42, где R41 и R42 означают (низш.)алкил, или где NR41R42 вместе образуют моноциклический алифатический гетероцикл, такой, как пирролидин, пиперидин, морфолин, N-метилпиперазин и т.п.; ацилоксиалкиловые эфиры формулы R40C(=O)OCHR43OC(=O)R44, где R43 означает водород или метил, а R44 означает (низш.)алкил или циклоалкил; эфиры угольной кислоты (карбонаты) формулы R40C(=O)OCHR43OC(=O)OR45, где R43 означает водород или метил, а R45 означает (низш.)алкил или циклоалкил; или аминокарбонилметиловые эфиры формулы R40C(=O)OCH2C(=O)NR41R42, где R41 и R42 означают водород или (низш.)алкил, или где NR41R42 вместе образуют моноциклический алифатический гетероцикл, такой, как пирролидин, пиперидин, морфолин, N-метилпиперазин и т.п. В описании заявки R40 имеет значения, аналогичные значениям R2, R3, R4 и R4'.
Примерами (низш.)алкиловых эфиров являются метиловый, этиловый и н-пропиловый эфиры и т.п. Примерами (низш.)алкиловых эфиров, замещенных группой NR41R42, являются диэтиламиноэтиловый, 2-(4-морфолинил)этиловый и 2-(4-метилпиперазин-1-ил)этиловый эфиры и т.п. Примеры ацилоксиалкиловых эфиров являются пивалоилоксиметиловый, 1-ацетоксиэтиловый и ацетоксиметиловый эфиры. Примерами эфиров угольной кислоты являются 1-(этоксикарбонилокси)этиловый и 1-(циклогексилоксикарбонилокси)этиловый эфиры. Примерами аминокарбонилметиловых эфиров являются N,N-диметилкарбамоилметиловый и карбамоилметиловый эфиры.
Дополнительная информация относительно примеров и применения эфиров для доставки фармацевтических соединений содержится в монографии Design of Prodrugs, Bundgaard H ed., Elsevier (1985). См. также Н.Ansel и др., Pharmaceutical Dosage Forms and Drug Delivery Systems, 6th Ed., 108-109 (1995), Krogsgaard-Larsen и др., Textbook of Drug Design and Development, 2d Ed., 152-191 (1996).
«Фармацевтически приемлемая соль» означает обычные кислотно-аддитивные соли или основно-аддитивные соли, которые сохраняют биологическую активность и свойства соединений формулы I и образуются из пригодных нетоксических органических или неорганических кислот или органических или неорганических оснований. Примеры кислотно-аддитивынх солей включают соли неорганических кислот, таких, как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота и азотная кислота, и соли органических кислот, таких, как пара-толуолсульфоновая кислота, салициловая кислота, метансульфоновая кислота, щавелевая кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота и т.п. Примеры основно-аддитивных солей включают соли аммония, калия, натрия и гидроксидов четвертичного аммонийного основания, таких например, как гидроксид тетраметиламмония. Химическая модификация фармацевтического соединения (т.е. лекарственного средства) с образованием соли известна фармакологам и используется для повышения физической и химической стабильности, улучшения гигроскопичности, текучести и растворимости соединений. См., например, Н.Ansel и др., Pharmaceutical Dosage Forms and Drug Delivery Systems, 6th Ed., 196, 1456-1457 (1995).
«Фармацевтически приемлемый», такой, как фармацевтически приемлемый носитель, эксципиент и т.п., означает фармакологически приемлемый материал и в основном нетоксичный для субъекта, которому вводится указанное соединение.
«Замещенный», такой, как замещенный алкил, означает, что заместитель находится в одном или более положений, если не указано иное, а заместители в каждом положении независимо выбирают из указанных значений.
«Терапевтически эффективное количество» означает количество по меньшей мере одного соединения формулы I или его фармацевтически приемлемой соли или эфира, которое эффективно ингибирует пролиферацию и/или предотвращает дифференциацию опухолевых клеток человека, включая клеточные линии опухолей человека.
В одном предпочтительном варианте осуществления настоящего изобретения предлагаются соединения формулы I, где
R1 выбирают из группы, включающей (низш.)алкил, замещенный арилом,
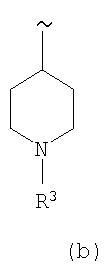 и
и 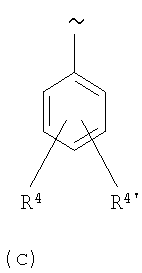 ,
,
R2 выбирают из группы, включающей арил, гетероарил, циклоалкил и гетероциклил, каждый из которых содержит 1-4 заместителя, независимо выбранных из группы, включающей (низш.)алкил, галоген, OR5, NH2 и NO2,
R3 выбирают из группы, включающей CO2R6 и SO2R6,
R4 и R4' каждый независимо выбирают из группы, включающей Н, (низш.)алкил, необязательно замещенный группами оксо, CO2R6, OR6 и/или NH2; S(O)nR7, OR8, NR5R6 и CO2R6,
R5 и R6 каждый независимо выбирают из группы, включающей Н, N, (низш.)алкил, замещенный группами оксо, CO2R9, OR9 и NR10R11; арил, необязательно замещенный галогеном, гетероарил, N-арил, где арил необязательно замещен (h) одним или более галогенами, и арил, замещенный галогеном или CF3,
R7 означает (низш.)алкил или арил,
R8 выбирают из группы, включающей Н, (низш.)алкил и (низш.)алкил, замещенный группой NR5R6,
R9 выбирают из группы, включающей Н и (низш.)алкил,
R10 и R11 каждый независимо выбирают из группы, включающей Н и (низш.)алкил, а
n равно 0, 1 или 2,
или их фармацевтически приемлемые соли или сложные эфиры.
В предпочтительном варианте соединений формулы I R2 означает фенил, предпочтительно фенил, замещенный галогеном, наиболее предпочтительно F, и OR5, где R5 означает (низш.)алкил. В наиболее предпочтительном варианте R2 означает фенил, замещенный одним иди двумя F и одной группой OR5, где R5 означает (низш.) алкил, предпочтительно метил.
В другом предпочтительном варианте предлагаются соединения формулы I, в которых R2 имеет значения, указанные выше, а R1 означает
где
R3 выбирают из группы, включающей Н, CO2R6, C(O)R6, SO2R6 и SO2NR5R6,
R5 и R6 каждый независимо выбирают из группы, включающей Н и (низш.)алкил,
или их фармацевтически приемлемые соли или эфиры.
Более предпочтительны соединения, указанные выше, в которых R2 означает фенил, содержащий один, два или три заместителя, независимо выбранные из группы, включающей галоген или -O- (низш.) алкил.
В еще одном варианте настоящего изобретения предлагаются соединения формулы I, где
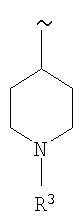
R1 означает
 ;
;
R2 означает 3-фторфенил, необязательно замещенный одним или двумя заместителями, выбранными из группы, включающей -F и -O-СН3,
R3 выбирают из группы, включающей Н, CO2R6, C(O)R6, SO2R6 и SO2NR5R6,
R5 и R6 каждый независимо выбирают из группы, включающей Н и (низш.)алкил,
или их фармацевтически приемлемые соли или эфиры.
Примеры соединений формулы I, указанной выше, включают, без ограничения перечисленным,
трет-бутиловый эфир 4-[4-амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты,
трет-бутиловый эфир 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты,
трет-бутиловый эфир 4-[4-амино-5-(2,3-дифтор-6-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты,
[4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанон,
[4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанон,
[4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон,
1-[4-[4-амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-ил]этанон,
[4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанон,
1-[4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-ил]этанон,
[4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанон,
диметиламид 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-сульфоновой кислоты,
[4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон и
[4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,6-дифторфенил)метанон.
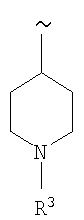
В еще одном варианте изобретения R1 означает
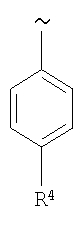
где R4 означает S(O)nR7, где R7 означает (низш.)алкил, предпочтительно метил.
Наиболее предпочтительно R4 означает -S(O)2СН3.
Примеры соединений формулы I, указанной выше, включают
[4-амино-2-(4-метансульфонилфениламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон.
Заявленные соединения формулы I, указанной выше, могут существовать в виде таутомеров или структурных изомеров. Подразумевается, что изобретение включает любые таутомеры или структурные изомеры указанных соединений или смеси указанных изомеров, и не ограничивается любой одной таутомерной или изомерной формой, представленной в вышеуказанной формуле.
В другом варианте изобретения примеры соединений по изобретению включают, без ограничения перечисленным, следующие соединения (см.таблицу).
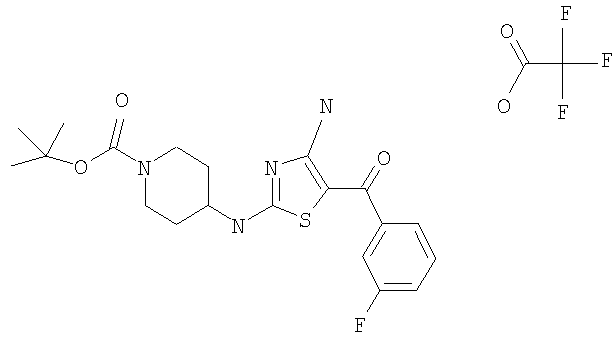
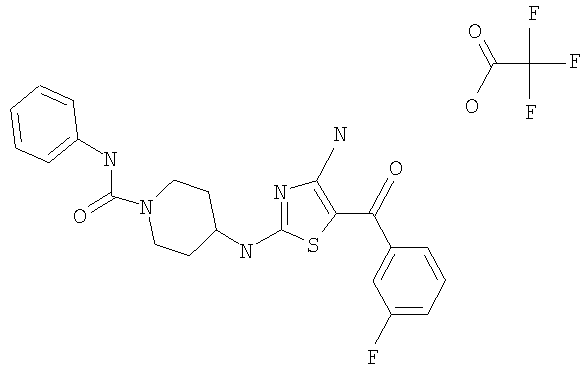
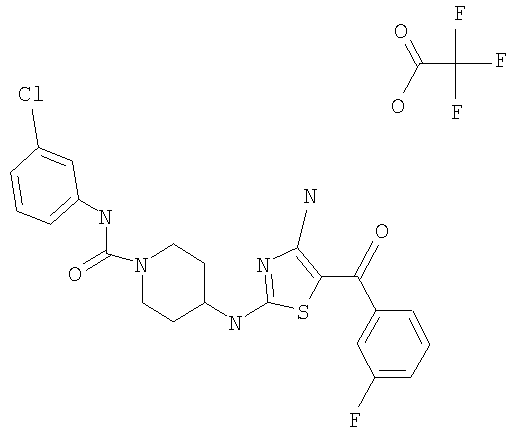
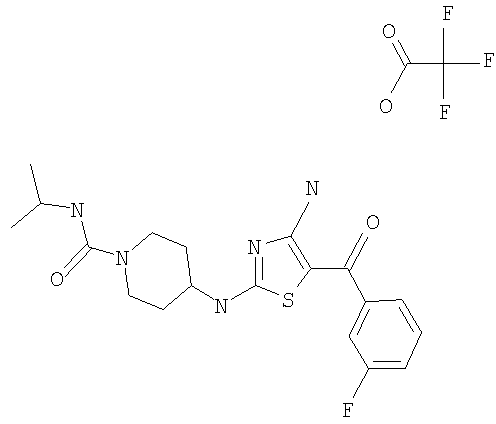
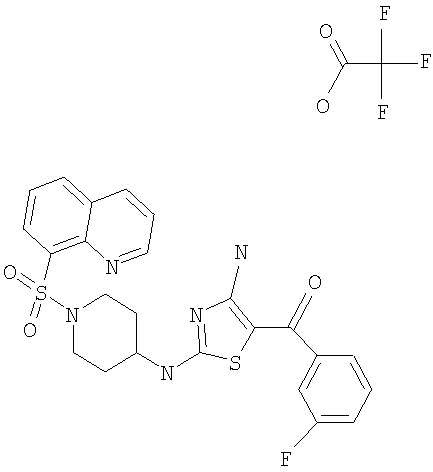
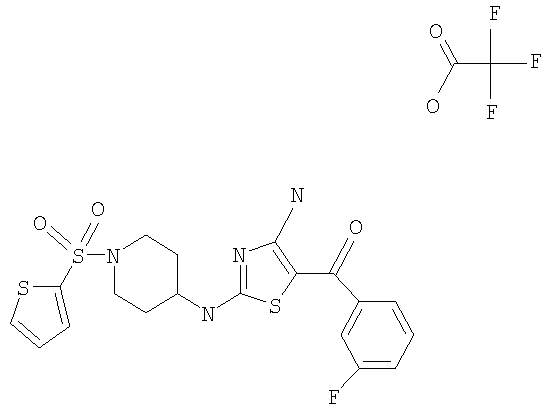
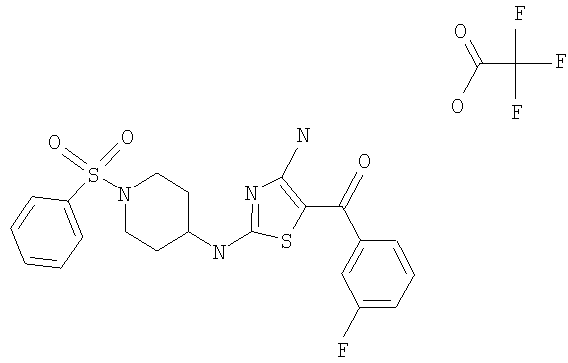
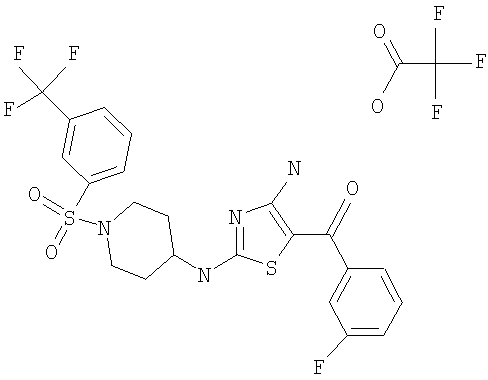
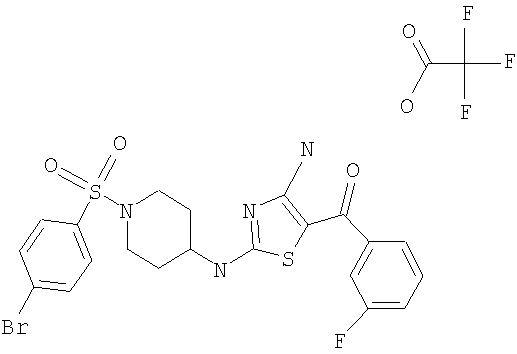
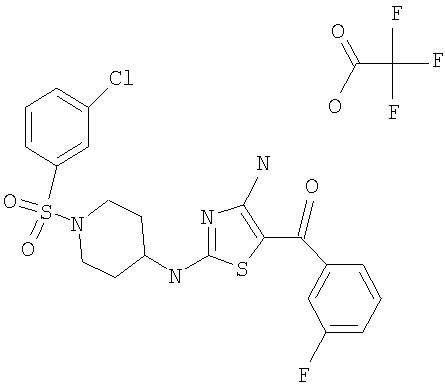
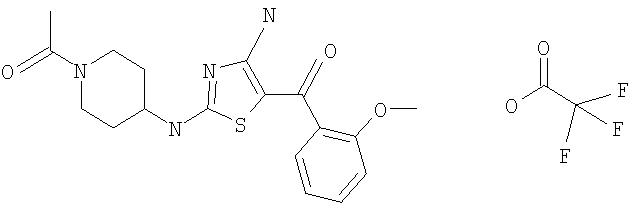
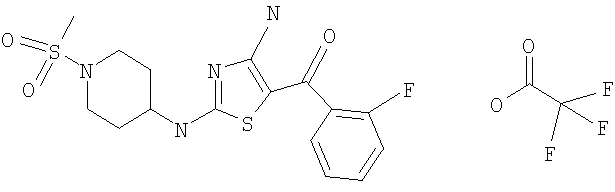
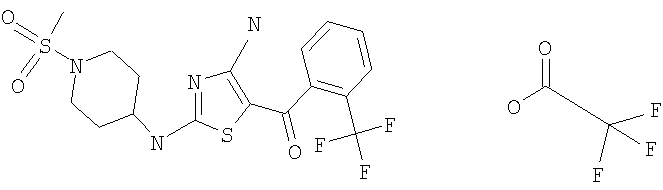
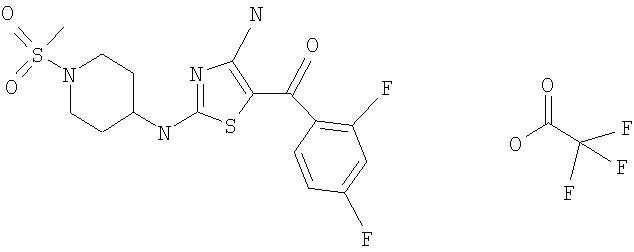
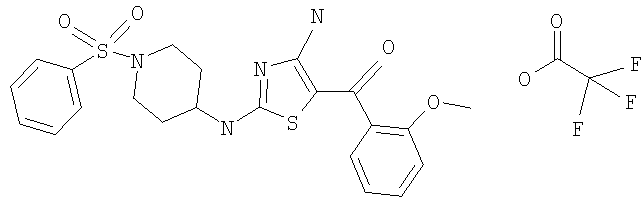
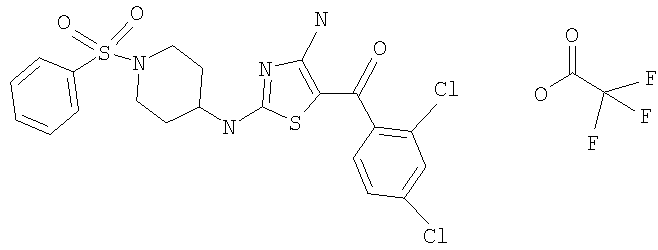
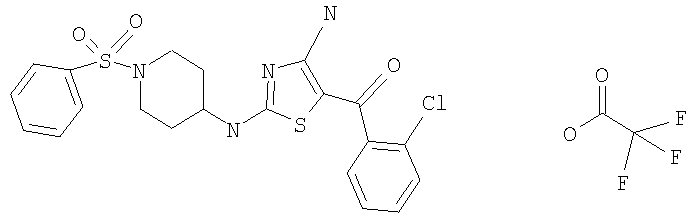
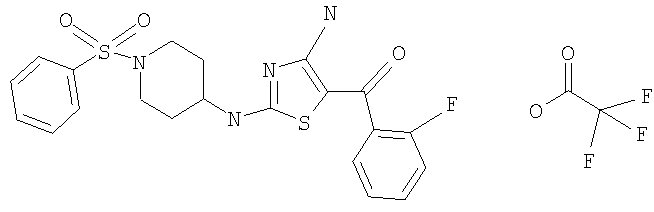
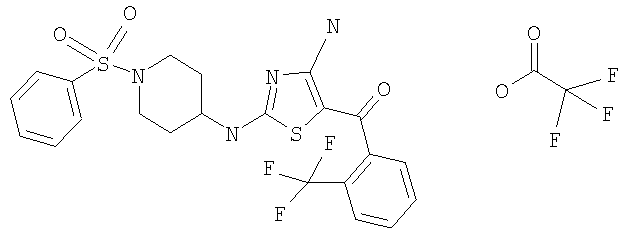
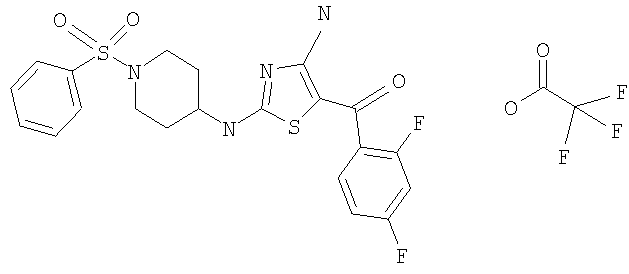
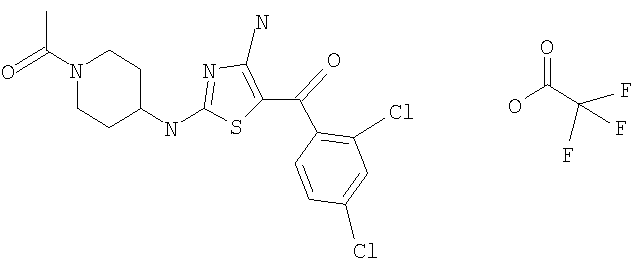
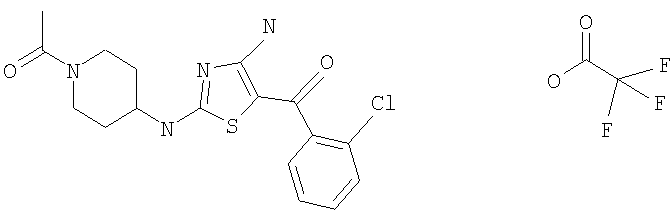
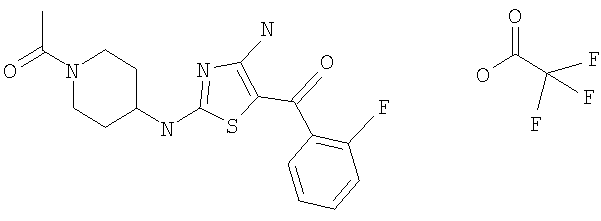
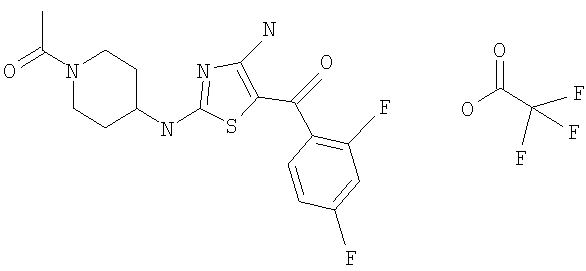
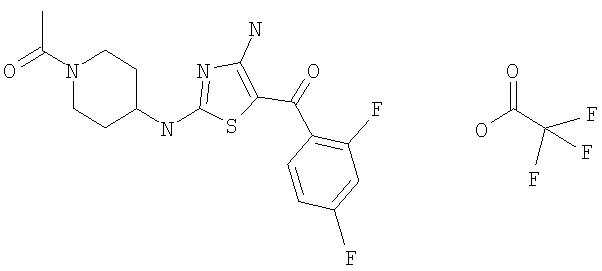
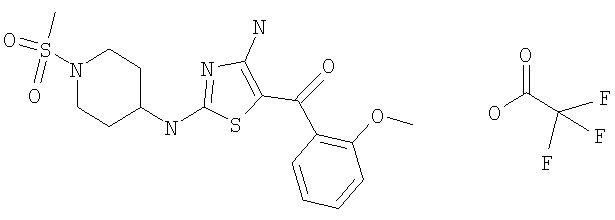
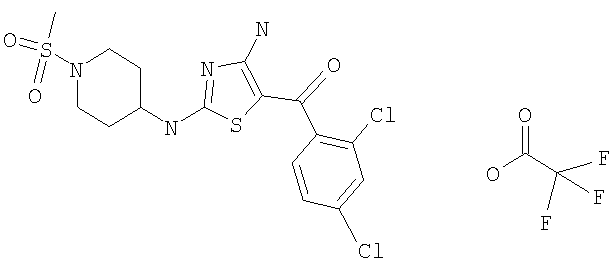
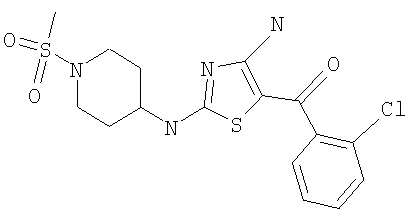
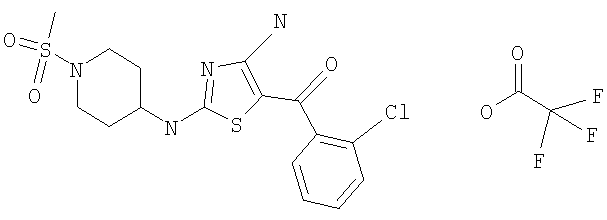
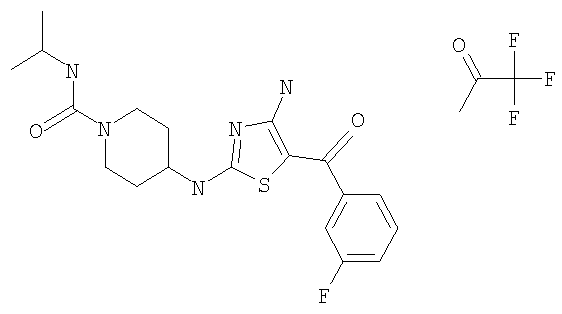
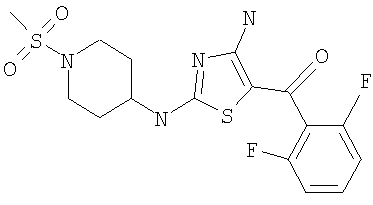
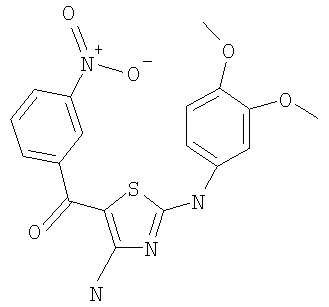
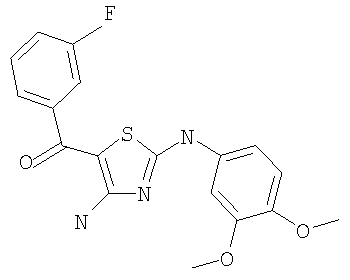
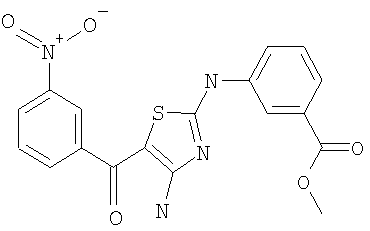
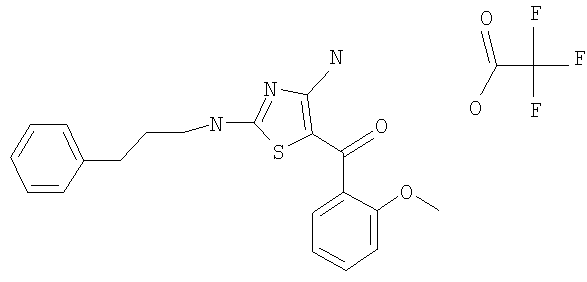
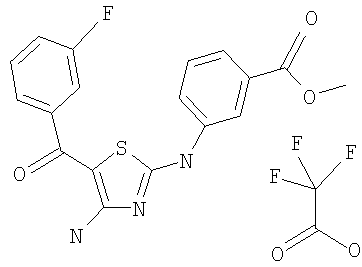
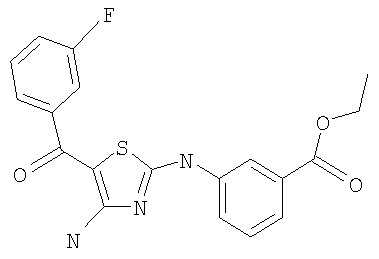
Соединения по настоящему изобретению получают любым известным способом. Пригодные методы синтеза указанных соединений приводятся в примерах. В общем случае соединения формулы I можно получить по одной из указанных ниже схем.
Замыкание цикла
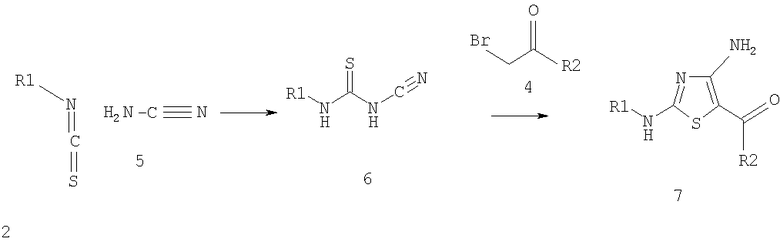
Соединения по изобретению получают алкилироанием и циклизацией ряда производных тиомочевины, как показано на схеме 3, с использованием известных реакций. Производные мочевины, которые используются для этих целей, включают производные нитроамидинотиомочевины (Binu R. и др., Org. Prep. Proced Int., 1998, 30, 93-96 (1998)), 1-[(арилтиокарбамоил)амино]-3,5-диметилпиразолы (Jenardanan G.C. и др., Synth. Commun., 27, 3457-3462 (1997)) и N-(аминоиминометил)-N'-фенилтиомочевину (Rajasekharan K.N. и др., Synthesis, 353-355 (1986)).
Другим производным тиомочевины, которое используются для получения соединений по изобретению с использованием алкилирования и циклизации, является N-цианотиомочевина (Gewald K. и др., J. Prakt. Chem., 97-104 (1967). Например, как показано ниже на схеме 3, N-цианотиомочевину формулы 6 вводят в реакцию с галогенметилкетоном, таким, как бромметилкетон формулы 4, при температуре от приблизительно комнатной температуры до приблизительно 65°С с образованием соединения формулы 7. Исходные материалы формулы (2) и (4), которые не являются коммерческими препаратами, указаны в конкретных примерах.
Схема 1
Схема 2

Схема 3
В другом варианте соединения по изобретению получают также по реакции иммобилизованного на смоле метилового эфира аминотиооксометилзамещенной карбамидотиокислоты формулы 9 с бромметилкетоном формулы 4, как показано ниже на схеме 4.
Схема 4
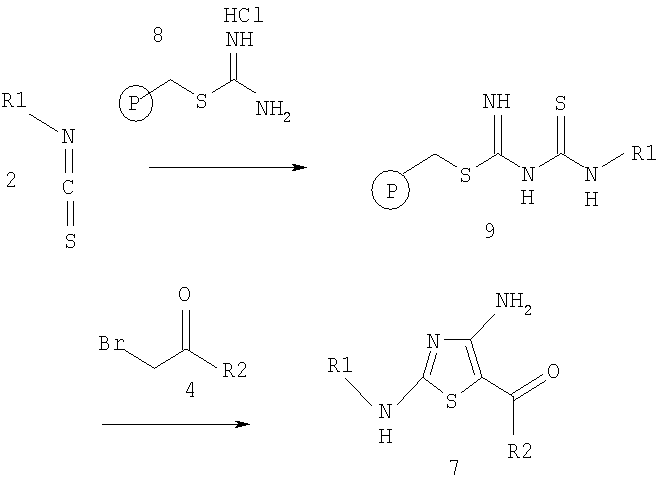
Иммобилизованное на смоле производное тиомочевины формулы 9 получают по любой методике, известной специалисту в области органической химии. Например, такое соединение можно получить по реакции иммобилизованной на смоле соли тиоурония формулы 8 с изотиоцианатом формулы 2 в присутствии основания, такого, как третичный амин (например, триэтиламин или диизопропилэтиламин) в инертном растворителе, таком, как полярный апротонный растворитель (например, N,N-диметилформамид). Обычно реакцию проводят приблизительно при комнатной температуре. Затем из иммобилизованного на смоле производного тиомочевины формулы 9 получают соединение формулы 7, например, при взаимодействии с галогенметилкетоном (например, бромметилкетоном формулы 4) в пригодном инертном растворителе, таком, как полярный апротонный растворитель (например, N,N-диметилформамид) при приблизительно комнатной температуре.
Разделение смеси стереоизомеров на оптически чистые стереоизомеры (если соединение формулы I является хиральным соединением)
Необязательное разделение изомеров формулы I проводят известными способами, такими, например, как хиральная жидкостная хроматография высокого давления (известная, как ЖХВР). Методы разделения изомеров известны и подробно описаны в специальной литературе (см., например, Jacques J. и др., Enantiomers, Racemates, and Resolutions», John Wiley and Sons, NY (1981). Методы разделения хиральной ЖХВР также известны и подробно описаны в литературе (см. например, Pirkle W.H. и Finn J., «Separation of Enantiomers by Liquid Chromatographic Methods» в монографии «Asymmetric Synthesis», t.1, c.87-124, Morrison J.D., Ed., Academic Press, Inc., NY (1983)).
Превращение соединения формулы I, содержащего группу азотистого основания, в фармацевтически приемлемую кислотно-аддитивную соль
Необязательное превращение соединения формулы I, содержащего группу азотистого основания, в фармацевтически приемлемую кислотно-аддитивную соль проводят известными способами. Например, соединение обрабатывают неорганической кислотой, такой, например, как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, или соответствующей органической кислотой, такой, как уксусная кислота, лимонная кислота, винная кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота или т.п.
Превращение соединения формулы I, содержащего карбоксильную группу, в фармацевтически приемлемую соль щелочного металла
Необязательное превращение соединения формулы I, содержащего карбоксильную группу, в фармацевтически приемлемую соль щелочного металла проводят известными способами. Например, соединение обрабатывают неорганическим основанием, таким, как гидроксид лития, гидроксид натрия, гидроксид калия и т.п.
Превращение соединения формулы I, содержащего карбоксильную группу, в фармацевтически приемлемый сложный эфир
Необязательное превращение соединения формулы I, содержащего карбоксильную группу, в фармацевтически приемлемый сложный эфир проводят известными способами. Условия получения сложного эфира зависят от устойчивости других функциональных групп в условиях реакции этерификации. Если другие группы достаточно устойчивы в кислотной среде, этерификацию обычно проводят при нагревании раствора соединения в спирте в присутствии минеральной кислоты (например, серной кислоты). Другие способы получения сложных эфиров, которые обычно используют в том случае, если соединение неустойчиво в кислотной среде, включают взаимодействие соединения со спиртом в присутствии конденсирующего агента и необязательно в присутствии дополнительных агентов, катализирующих реакцию. Специалисту в области органической химии известно множество таких конденсирующих агентов. В качестве примеров можно привести дициклогексилкарбодиимид и трифенилфосфин/диэтилазодикарбоксилат. Если в качестве конденсирующего агента используется дициклогексилкарбодиимид, реакцию обычно проводят при обработке кислоты карбодиимидом в присутствии спирта и необязательно в присутствии каталитического количества (0-10 мол.%) N,N-диметиламинопиридина в инертном расторителе, таком, как галогенированный углеводород (например, дихлорметан) при температуре от приблизительно 0°С до приблизительно комнатной температуры, предпочтительно при приблизительно комнатной температуре. Если в качестве конденсирующего агента используется трифенилфосфин/диэтилазодикарбоксилат, реакцию обычно проводят при обработке кислоты трифенилфосфином и диэтилазодикарбоксилатом в присутствии спирта в инертном растворителе, таком, как простой эфир (например, тетрагидрофуран) или ароматический углеводород (например, бензол) при температуре от приблизительно 0°С до приблизительно комнатной температуры, предпочтительно при приблизительно 0°С.
Композиции/составы
В другом варианте настоящее изобретение включает фармацевтические композиции, содержащие по меньшей мере одно соединение формулы I или его фармацевтически приемлемую соль или эфир и фармацевтически приемлемый эксципиент и/или носитель.
Указанные фармацевтические композиции можно вводить пероральным способом, например, в форме таблеток, таблеток с покрытием, драже, твердых или мягких желатиновых капсул, растворов, эмульсий или суспензий. Композиции можно также вводить ректальным способом, например, в форме суппозиториев, или парентеральным способом, например, в форме инъекционных растворов.
Фармацевтические композиции по настоящему изобретению, включающие соединения формулы I и/или их фармацевтически приемлемые соли или эфиры, получают известным в данной области техники способом, например, с использованием процессов смешивания, инкапсулирования, растворения, гранулирования, эмульгирования, включения в матрицу, дражирования или лиофилизации. Указанные фармацевтические препараты можно перерабатывать в смеси с терапевтически инертными неорганическими или органическими носителями. При получении таблеток, таблеток с покрытием, драже и твердых желатиновых капсул в качестве таких носителей используются лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли. Пригодные носители для мягких желатиновых капсул включают растительные масла, воски и жиры. В зависимости от природы активного соединения в случае мягких желатиновых капсул обычно не требуется никаких носителей. Пригодными носителями при получении растворов и сиропов являются вода, полиолы, сахароза, инвертированный сахар и глюкоза. Пригодными носителями для инъекций являются вода, спирты, полиолы, глицерин, растительные масла, фосфолипиды и ПАВ. Пригодными носителями для суппозиториев являются природные или отвержденные масла, воски, жиры и полужидкие полиолы.
Фармацевтические композиции могут также включать консерванты, солюбилизирующие агенты, стабилизирующие агенты, смачивающие агенты, эмульгирующие агенты, подсластители, красители, ароматизаторы, соли для регуляции осмотического давления, буферные вещества, обволакивающие агенты и антиоксиданты. Кроме того, они могут содержать другие терапевтически ценные соединения, включая дополнительные активные ингредиенты, отличающиеся от соединений формулы I.
Дозировки
Как указано выше, соединения по настоящему изобретению, включая соединения формулы I, используются для лечения или подавления нарушений клеточной пролиферации, включая химиопрофилактику рака. Химиопрофилактика означает ингибирование развития инвазивного рака за счет подавления индукции мутагенного процесса или подавление развития предраковых клеток, которые уже подвергались воздействию факторов, направленных на подавление рецидивирующей опухоли. Указанные соединения и составы, содержащие указанные соединения, прежде всего пригодны для лечения или подавления роста солидных опухолей, таких например, как опухоли молочной железы, ободочной кишки, легких и предстательной железы.
Терапевтически эффективное количество соединения по настоящему изобретению означает количество соединения, которое при введении субъекту эффективно предотвращает развитие, подавляет симптомы заболевания или снижает их интенсивность или продлевает жизнь субъекта, нуждающегося в лечении. Терапевтически эффективное количество определяется лечащим врачом.
Терапевтически эффективное количество или доза соединения по настоящему изобретению изменяется в широком интервале и определяется известным способом. Такая доза должна соответствовать индивидуальным требованиям в каждом конкретном случае в зависимости от типа введенного соединения (соединений), способа введения, состояния, подлежащего лечению, а также от состояния пациента. В общем случае, при пероральном или парентеральном введении взрослому человеку массой приблизительно 70 кг, пригодной является суточная доза от приблизительно 10 мг до приблизительно 10000 мг, предпочтительно от приблизительно 200 мг до приблизительно 1000 мг, хотя верхний предел можно превысить в зависимости от показаний. Суточную дозу можно вводить в виде разовой дозы или раздельными дозами, или при парентеральном введении дозу можно вводить непрерывным вливанием.
Комбинации
Соединение по настоящему изобретению можно использовать в комбинации (при одновременном или последовательном введении) с известными противораковыми методами лечения, такими, как лучевая терапия, или с цитостатическими или цитотоксическими агентами, такими, например, как, без ограничения перечисленным, ДНК-взаимодействующими агентами, такими, как цисплатин или доксорубицин, ингибиторами топоизомеразы II, такими, как этопозид, ингибиторами топоизомеразы I, такими, как СРТ-11 или топотекан, агентами, взаимодействующими с микротрубочками, такими, как паклитаксель, доцетаксель, или эпотилоны, гормональными агентами, такими, как тамоксифен, ингибиторами тимидилатсинтаз, такими, как 5-фторурацил, и антиметаболитами, такими, как метотрексат. Соединения формулы I можно также использовать в комбинации с модуляторами трансактивации р53.
Если лекарственное средство получают в форме фиксированной дозы, вышеуказанные комбинации включают соединения по настоящему изобретению в интервале доз, указанных выше, и другой фармацевтически активный агент, или лечение проводят с учетом указанного интервала доз. Например, недавно установлено, что ингибитор cdk1 оломуцин оказывает синергическое действие с известными цитотоксическими агентами, вызывая апоптоз (J. Cell Sci., 108, 2897-2904 (1995)). Соединения формулы I можно также вводить последовательно с известными противоопухолевыми или цитотоксическими агентами, если одновременное введение или введение комбинации невозможно. В настоящем изобретении схема введения не ограничивается: соединения формулы I можно вводить до или после введения известного противоопухолевого или цитотоксического агента. Например, цитотоксическая активность ингибитора cdk флавопиридола зависит от последующего введения противоопухолевых агентов (Cancer Research, 57, 3375 (1997).
Примеры
Следующие примеры иллюстрируют предпочтительные методы синтеза и применения соединений и композиций по настоящему изобретению. Указанные примеры и препараты не ограничивают объем изобретения. Подразумевается, что в пределах сущности и объема изобретения, определяемыми формулой изобретения, возможны различные его варианты и модификации.
Пример 1
трет-Бутиловый эфир 4-изотиоцианатопиперидин-1-карбоновой кислоты
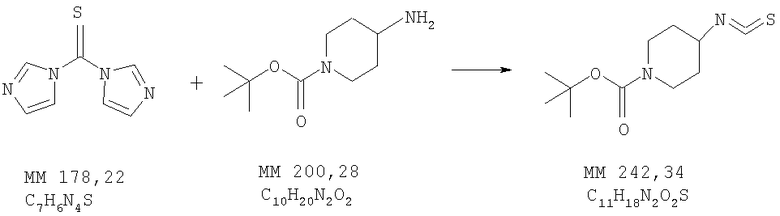
трет-Бутиловый эфир 4-аминопиперидин-1-карбоновой кислоты (5,0 г, 25 ммолей, фирма Astatech, Inc) растворяли в диметилформамиде (120 мл), охлаждали до -15°С и в раствор медленно добавляли тиокарбонилдиимидазол (4,8 г, 27 ммолей, фирма Aldrich) в диметилформамиде (100 мл), охлажденный до -10°С. Смесь перемешивали при комнатной температуре в течение 14 ч, растворитель удаляли в вакууме, остаток растворяли в хлористом метилене (200 мл) и дважды промывали водой. Затем растворитель удаляли и полученный остаток растирали в гексане и фильтровали. В полученный раствор добавляли норит, фильтровали через слой целита и удаляли растворитель, при этом получали трет-бутиловый эфир 4-изотиоцианатопиперидин-1-карбоновой кислоты (5,7 г, выход 94%) в виде масла. МС-НР (EI(+)): 242 (MM).
Пример 2
2-Бром-1-(3-фторфенил)этанон
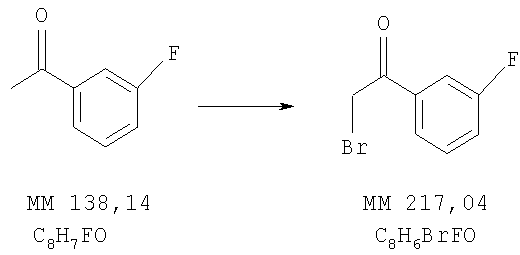
В раствор 1-(3-фтофенил)этанона (14,0 г, 100 ммолей, фирма Aldrich) в диоксане (250 мл) при 12-15°С в течение 30 мин добавляли по каплям раствор брома (17 г, 105 ммолей) в диоксане (120 мл). Полученную смесь перемешивали в течение 15 мин и удаляли основное количество растворителя. Полученный остаток переносили в гексан (200 мл), дважды промывали водой, сушили над сульфатом магния и удаляли растворитель, при этом получали 2-бром-1-(3-фторфенил)этанон.
1Н-ЯМР (300 МГц, CDCl3,): δ 3,71 (s, 2H, СН2), 7,30-7,77 (m, 4H, ароматич.).
Пример 3
трет-Бутиловый эфир 4-[4-амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты
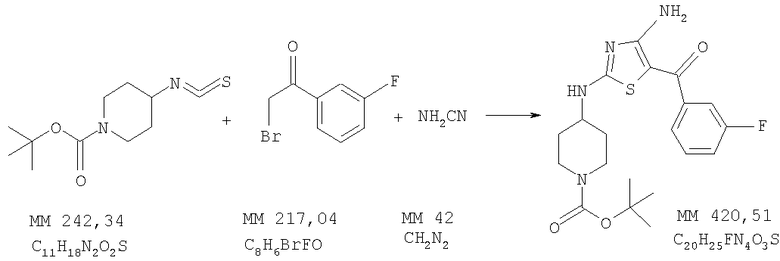
В смесь трет-бутилового эфира 4-изотиоцианатопиперидин-1-карбоновой кислоты (2,4 г, 10 ммолей, пример 1), цианоамида (0,42 г, 10 ммолей, фирма Aldrich) в трет-бутиловом спирте (5 мл) и ацетонитриле (35 мл) при температуре 20°С в течение 15 мин добавляли раствор трет-бутоксида калия (1,0 М раствор в ТГФ, 10 мл, 10 ммолей, фирма Aldrich). Затем в смесь добавляли 2-бром-1-(3-фторфенил)этанон (2,0 г, 9,2 ммоля, пример 2), полученную суспензию перемешивали в течение 2 ч, растворитель удаляли и твердый остаток очищали методом хроматографии на колонке с силикагелем (элюент: этилацетат/гексан, от 60% до 70%), при этом получали трет-бутиловый эфир 4-[4-амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты (2,4 г, выход 57%).
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,36 (m, 2H, NCH2), 1,41 (s, 9H, 3СН3), 1,88 (m, 2H, NCH2), 2,88 (ушир., 2H, NCH2), 3,6-4,0 (m, 1H, CH), 3,87 (m, 2H, NCH2), 7,31 (t, 1H, ароматич.), 7,38 (d, 1H, ароматич.), 7,48 (t, 1H, ароматич.), 7,51 (q, 1H, ароматич.), 8,01 (ушир., 1H, NH), 8,48 (ушир,, 1H, NH), 8,70 (ушир., 1H, NH).
Пример 4
трет-Бутиловый эфир 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты
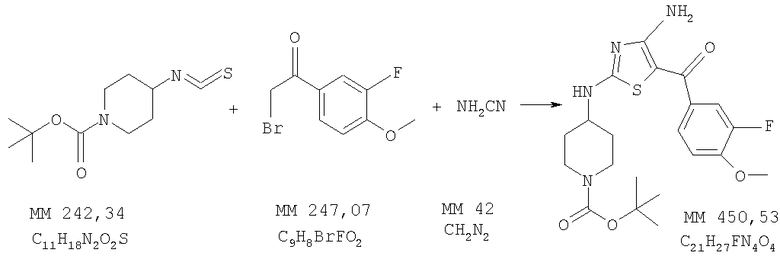
трет-Бутиловый эфир 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты получали бромированием 1-(3-фтор-4-метоксифенил)этанона (фирма Aldrich) аналогично тому, как описано в примере 2, и последующей циклизацией полученного продукта, как описано в примере 3. МС-ВР: рассч. 451,1810, найд. 451,1813 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,37 (m, 2H, СН2), 1,41 (s, 9H, 3СН3), 1,80 (m, 2Н, СН2), 2,78 (ушир., 2H, NCH2), 3,32 (m, 2H, NCH2), 3,6-4,0 (ушир., 1Н, СН), 3,85 (m, 3H, ОСН3), 7,22 (t, 1H, ароматич.), 7,45 (d, 1H, ароматич.), 7,48 (m, 1H, ароматич.), 7,94 (ушир., 1H, NH), 8,45 (ушир., 1H, NH), 8,65 (ушир., 1H, NH).
Пример 5
трет-Бутиловый эфир 4-[4-амино-5-(2,3-дифтор-6-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты
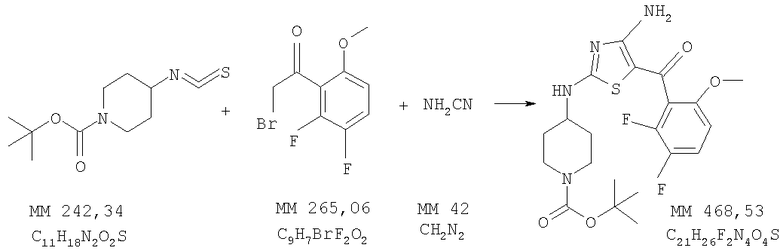
трет-Бутиловый эфир 4-[4-амино-5-(2,3-дифтор-6-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты получали бромированием 1-(2,3-дифтор-6-метоксифенил)этанона (фирма Matrix) аналогично тому, как описано в примере 2, и последующей циклизацией полученного продукта, как описано в примере 3. МС-НР (ES(+/-)): 468 (MM).
1H-ЯМР (300 МГц, ДМСО-d6): δ 1,48 (m, 2H, СН2), 1,92 (m, 2H, СН2), 2,55 (s, 9H, 3СН3), 2,75 (m, 2H, CH2), 3,39 (m, 2H, СН2), 3,6-4,0 (ушир., 1H, СН), 3,81 (s, 3H, ОСН3), 6,92 (m, 1H, ароматич.), 7,50 (m, 1H, ароматич.), 7,6 (2× ушир., 1H, NH), 7,8 (ушир., 1H, NH), 7,99 (m, 1H, ароматич.), 9,0 (ушир., 1H, NH).
Пример 6
[4-Амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанон
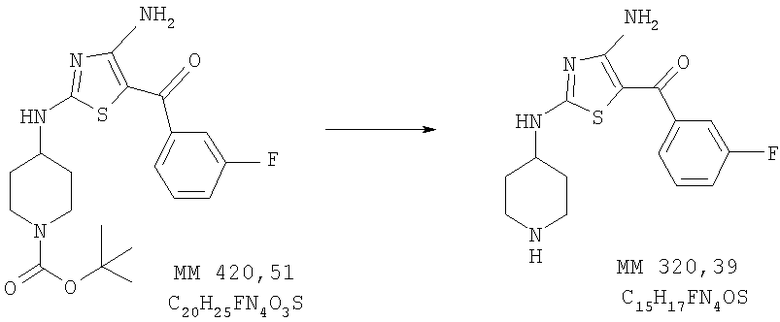
трет-Бутиловый эфир 4-[4-амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты (0,82 г, 1,95 ммоля, пример 3) растворяли в смеси трифторуксусной кислоты (16 мл) и хлористого метилена (30 мл), выдерживали в течение 1 ч, растворитель удаляли, остаток растворяли в хлористом метилене (300 мл), промывали 10% раствором Na2CO3 (50 мл), сушили над Na2SO4 и концентрировали, при этом получали твердое вещество. Продукт растирали в этиловом эфире и фильтровали, при этом получали [4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанон (450 мг, выход 72%). МС-НР (ES(+/-)): 320 (MM).
1Н-ЯМР (300 МГц, ДМСО-d6,): δ 1,38 (m, 2H, СН2), 1,88 (m, 2H, СН2), 2,95 (m, 2H, CH2), 3,32 (ушир., 2H, CH2), 3,6-4,0 (ушир., 1Н, CH), 3,78 (s, 3H, ОСН3), 7,31 (t, 1Н, ароматич.), 7,38 (d, 1Н, ароматич.), 7,48 (t, 1Н, ароматич.), 7,51 (q, 1H, ароматич.), 8,01 (ушир., 1Н, NH), 8,48 (ушир., 1Н, NH), 8,70 (ушир., 1Н, NH).
Пример 7
[4-Амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанон
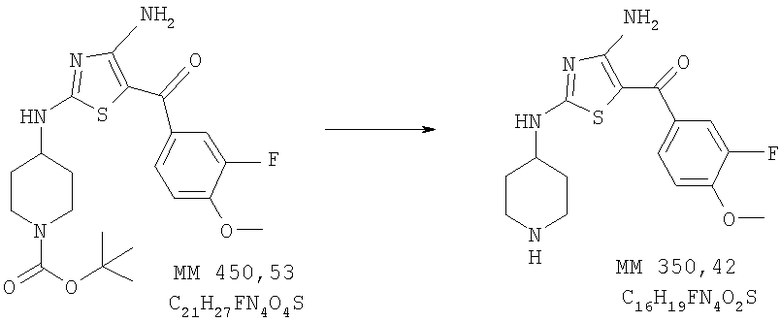
[4-Амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанон (220 мг, выход 90%) получали удалением защитных групп в составе трет-бутилового эфира 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты (0,30 г, 0,66 ммоля, пример 4) аналогично тому, как описано в примере 6. МС-НР (ES(+/-)): 350 (MM).
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,36 (m, 2H, СН2), 1,75 (m, 2H, СН2), 2,88 (ушир., 2H, СН2), 3,32 (m, 2H, CH2), 3,2-3,4 (ушир., 1Н, NCH), 3,78 (s, 3Н, ОСН3), 7,23 (t, 1Н, ароматич.), 7,48 (t, 1Н, ароматич.), 7,62 (t, 1Н, ароматич.), 8,01 (ушир., 1Н, NH), 8,48 (ушир., 1Н, NH), 8,70 (ушир., 1Н, NH).
Пример 8
[4-Амино-2-(пиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон
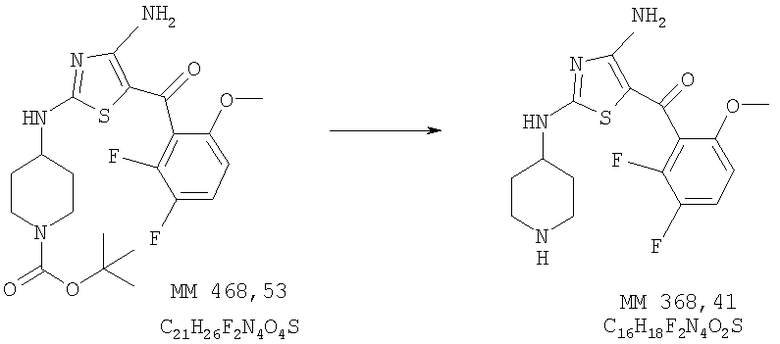
[4-Амино-2-(пиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон (123 мг, выход 52%) получали удалением защитных групп в составе трет-бутилового эфира 4-[4-амино-5-(2,3-дифтор-6-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты (0,30 г, 0,64 ммоля, пример 5) аналогично тому, как описано в примере 6. МС-НР (ES(+/-)): 368 (MM).
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,38 (m, 2H, CH2), 1,78 (m, 2H, СН2), 2,91 (ушир., 2Н, СН2), 3,35 (m, 3Н, СН и NCH2), 3,75 (s, 3Н, ОСН3), 6,95 (m, 1H, ароматич.), 7,43 (m, 1Н, ароматич.), 8,01 (ушир., 1Н, NH), 8,48 (ушир., 1Н, NH), 8,70 (ушир., 1H, NH).
Пример 9
1-[4-[4-Амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-ил]этанон
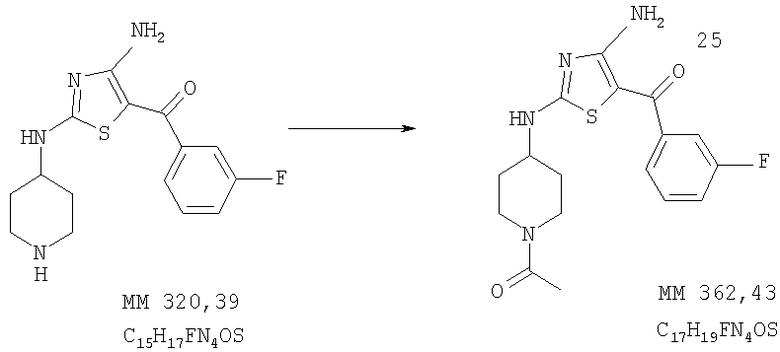
[4-Амино-2-(пиперидин-4-иламино)тиазол-5-ил]-(3-фторфенил)метанон (0,10 г, 0,31 ммоля, пример 6) растворяли в смеси тетрагидрофурана (20 мл), хлороформа (6 мл) и пиридина, охлаждали до -10°С. Затем добавляли ацетилхлорид (0,032 г, 43 ммоля), перемешивали при комнатной температуре в течение 0,5 ч, разбавляли охлажденным хлористым метиленом (100 мл), дважды промывали 10% раствором Na2CO3 (вод.), сушили над Na2SO4, удаляли растворитель и остаток отделяли от смеси тетрагидрофурана и гексана, при этом получали 1-[4-[4-амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-ил]этанон (35 мг, выход 30%). МС-ВР: рассч. 363,1286, найд. 363,1288 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,28 (m, 1Н, СН), 1,41 (m, 1H, CH), 1,92 (m, 2H, CH2), 2,75 (t, 1H, NCH), 3,14 (t, H, NCH), 3,32 (s, 3Н, СОСН3), 3,6-4,0 (ушир., 1H, CH), 3,77 (d, 1H, NCH), 4,22 (d, 1H, NCH), 7,31 (t, 1H, ароматич.), 7,38 (d, 1H, ароматич.), 7,48 (t, 1H, ароматич.), 7,51 (q, 1H, ароматич.), 8,01 (ушир., 1H, NH), 8,48 (ушир., 1H, NH), 8,72 (ушир., 1H, NH).
Пример 10
[4-Амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанон
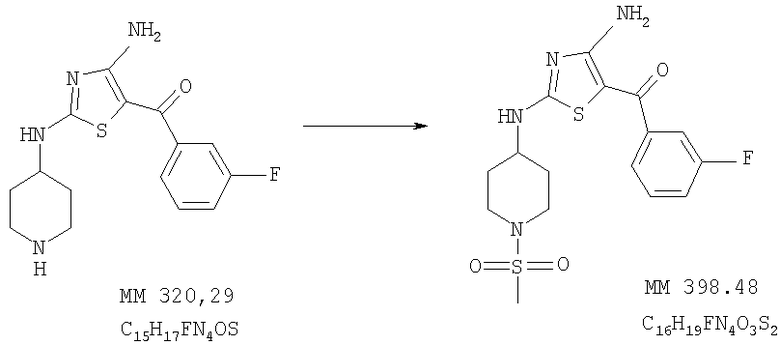
[4-Амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанон (35 мг, выход 40%) получали при взаимодействии [4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанона (0,09 г, 0,28 ммоля, пример 6) и метансульфонилхлорида аналогично тому, как описано в примере 9. МС-ВР: рассч. 399,0956, найд. 399,0960 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,54 (m, 2H, СН2), 2,03 (m, 2H, CH2), 2,88 (m, 5Н, NCH2 и SCH3), 3,6-4,0 (ушир., 1H, СН), 3,51 (d, 2H, NCH2), 7,32 (t, 1H, ароматич.), 7,38 (d, 1H, ароматич.), 7,48 (t, 1H, ароматич.), 7,52 (q, 1H, ароматич.), 7,99 (ушир., 1H, NH), 8,48 (ушир., 1H, NH), 8,77 (ушир., 1H, NH).
Пример 11
1-[4-[4-Амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-ил]этанон
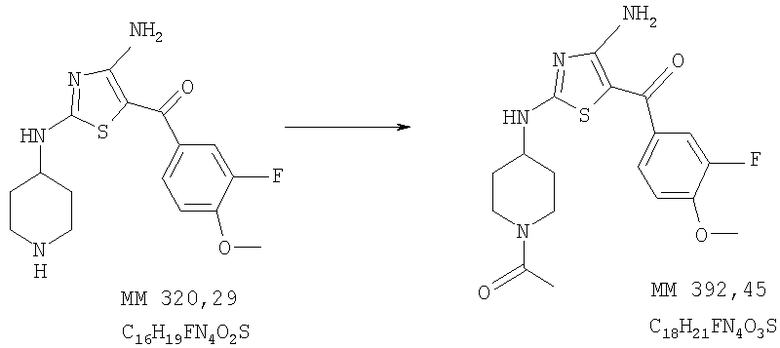
1-[4-[4-Амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-ил]этанон (100 мг, выход 77%) получали при взаимодействии [4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанона (0,125 г, 0,36 ммоля, пример 7) и ацетилхлорида аналогично тому, как описано в примере 9. МС-ВР: рассч. 393,1391, найд. 393,1396 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,38 (dm, 2H, СН2), 1,92 (m, 2H, СН2), 2,02 (s, 3Н, СОСН3), 2,76 (t, 1H, NCH), 3,26 (t, 1H, NCH), 3,6-4,0 (ушир., 1H, CH), 3,77 (m, 1H, NCH), 3,90 (s, 3Н, ОСН3), 4,22 (m, 1H, NCH), 7,23 (t, 1H, ароматич.), 7,47 (t, 1H, ароматич.), 7,50 (t, 1H, ароматич.), 7,9-8,5 (ушир., 2H, 2NH), 8,69 (ушир., 1H, NH).
Пример 12
[4-Амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанон
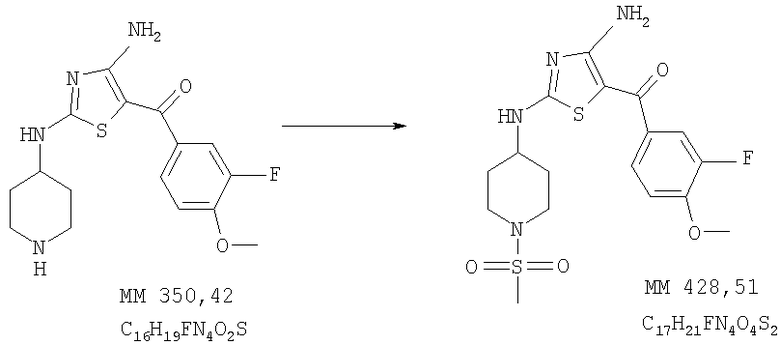
[4-Амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанон (30 мг, выход 23%) получали при взаимодействии [4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанона (0,11 г, 0,31 ммоля, пример 7) и метансульфонилхлорида аналогично тому, как описано в примере 9. МС-ВР (EI): рассч. 428,0988, найд. 428,0982 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,46 (m, 2H, СН2), 2,01 (m, 2H, СН2), 2,90 (m, 5Н, СН2 и SCH3), 3,55 (m, 2HCH2), 3,6-4,0 (ушир., 1Н, СН), 3,91 (s, 3H, ОСН3), 7,24 (t, 1Н, ароматич.), 7,47 (t, 1Н, ароматич.), 7,59 (t, 1Н, ароматич.), 7,9-8,5 (ушир., 2H, 2NH), 8,71 (ушир., 1Н, NH).
Пример 13
Диметиламид 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-сульфоновой кислоты
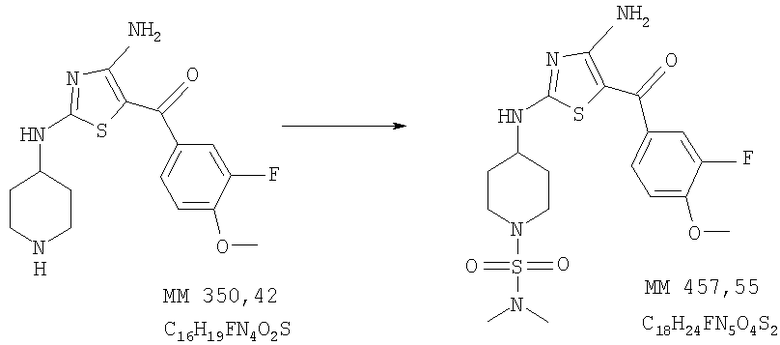
Диметиламид 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-сульфоновой кислоты (30 мг, выход 22%) получали при взаимодействии [4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанона (0,08 г, 0,022 ммоля, пример 7) и диметилсульфамоилхлорида аналогично тому, как описано в примере 9, за исключением того, что в качестве катализатора использовали диизопропилэтиламин, а продукт очищали на короткой колонке с силикагелем (для предварительной очистки продукта использовали этилацетат). МС-ВР: рассч. 458,1327, найд. 458,1331 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6,): δ 1,51 (m, 2H, CH2), 1,97 (m, 2H, CH2), 2,77 (s, 6H, 2×NCH2), 2,99 (m, 2H, CH2), 3,52 (m, 2H, СН2), 3,6-4,0 (ушир., 1Н, СН), 3,89 (s, 3H, ОСН3), 7,9-8,5 (ушир., 2H, 2NH), 8,71 (ушир., 1Н, NH).
Пример 14
[4-Амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон
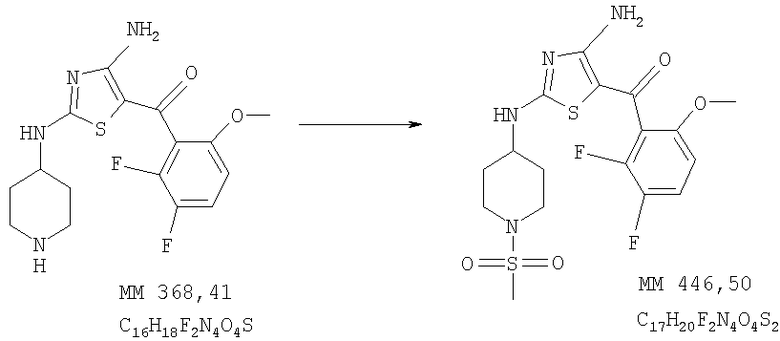
[4-Амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон (50 мг, выход 90%) получали при взаимодействии [4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанона (0,05 г, 0,14 ммоля, пример 8) и метансульфонилхлорида аналогично тому, как описано в примере 9, за исключением того, что в качестве катализатора использовали диизопропилэтиламин. ЖХ/МС-НР: 446 (MM, M+H).
1Н-ЯМР (300 МГц, ДМСО-d6): δ 1,57 (m, 2H, СН2), 1,92 (m, 2H, СН2), 2,85 (m, 5Н, SCH3 и NCH2), 3,53 (m, 2H, NCH2), 3,6-4,0 (ушир., 1Н, СН), 3,88 (s, 3Н, ОСН3), 6,72 (m, 1Н, ароматич.), 7,42 (m, 1Н, ароматич.), 7,6 (2× ушир., 1Н, NH), 7,8 (ушир., 1Н, NH), 7,99 (m, 1Н, ароматич.), 9,0 (ушир., 1Н, NH).
Ki CDK1 составляет 0,001 мкМ, Ki CDK2 составляет 0,001 мкМ, Ki CDK4 составляет 0,002 мкМ.
Пример 15
Изотиоцианато-4-метансульфонилбензол
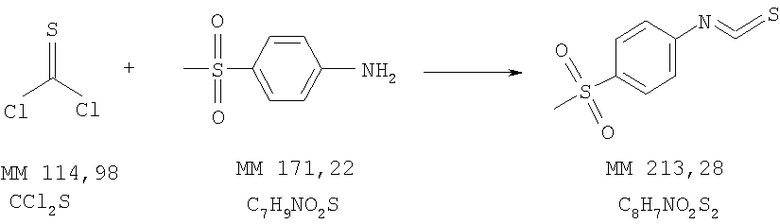
4-Метансульфонилфениламин (2,4 г, 14 ммолей) растворяли в смеси воды (30 мл) и соляной кислоты (9 мл, 37%), в полученный раствор при комнатной температуре добавляли по каплям тиофосген (1,5 г, 13,2 ммоля) и тщательно перемешивали в течение 1 ч. Затем полученную суспензию фильтровали, остаток на фильтре промывали водой и сушили над P2O5, при этом получали 1-изотиоцианато-4-метансульфонилбензол (2,4 г, выход 85%).
1Н-ЯМР (300 МГц, COCl3): δ 3,09 (s, 3H, СН3), 7,41 (d, 2H, ароматич.), 7,98 (d, 2Н, ароматич.).
Пример 16
[4-Амино-2-(4-метансульфонилфениламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон
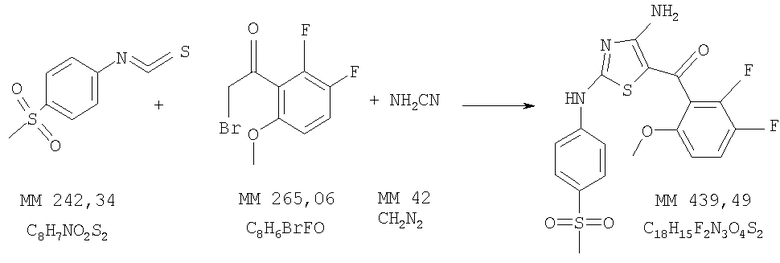
[4-Амино-2-(4-метансульфонилфениламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон (15 мг, выход 12%) получали в две стадии: сначала бромировали 1-(2,3-дифтор-6-метоксифенил)этанон (фирма Matrix) аналогично тому, как описано в примере 2, затем получали диаминотиазол аналогично тому, как описано в примере 3. Полученное твердое вещество очищали хроматографией на колонке с силикагелем (элюент: 2,5% метанол/хлористый метилен), очищенный продукт растирали в эфире и отделяли фильтрованием. МС-ВР: рассч. 440,0545, найд. 440,0545 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6,): δ 3,18 (s, 3H, SCH3), 3,68 (s, 3H. ОСН3), 6,95 (m, 1H, ароматич.), 7,50 (m, 1H, ароматич.), 7,74 (dd, 4H, ароматич.), 8,2 (ушир., 1Н, NH), 11,21 (s, 1H, NH).
Пример 17
[4-Амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,6-дифторфенил)метанон
2-Метилизотиоуреидопроизводное смолы (34350-95)
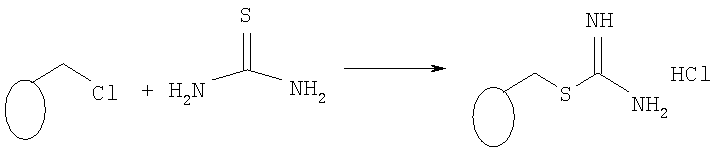
В смесь этанола и 1,4-диоксана (500 мл, 1:4) добавляли смолу Меррифилда (50 г, емкость 4,3 ммоля Cl/г, фирма Fluka) и тиомочевину (5 экв., 82 г, 1,8 моля, фирма Aldrich) и полученную смесь встряхивали при температуре 85°С в течение 5 сут. Затем смесь фильтровали, полученную смолу трижды промывали диметилформамидом (3×50 мл), изопропанолом (3×50 мл), дихлорметаном (3×50 мл) и этиловым эфиром (3×50 мл) и продукт сушили в эксикаторе в течение 3 сут, при этом получали 77,5 г 2-метилизотиоуреидопроизводного смолы грязно-белого цвета (емкость 3 ммоля/г по данным микроанализа).
Смола, модифицированная трет-бутиловым эфиром 4-[3-(2-метилизотиоуреидо)тиоуреидопиперидин-1-карбоновой кислоты
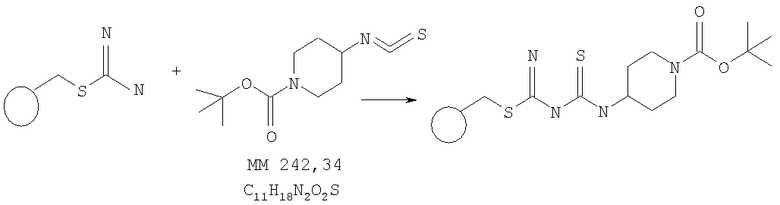
В колбу объемом 100 мл помещали смолу, содержащую метилизотиоуреидопроизводное смолы (2,9 г, пример 17а), раствор трет-бутилового эфира 4-изотиоцианатопиперидин-1-карбоновой кислоты в диметилформамиде (пример 1) и диизопропилэтиламин (4 экв.) и полученную смесь встряхивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали и продукт трижды промывали диметилформамидом (3×30 мл), изопропанолом (3×30 мл), дихлорметаном (3×30 мл) и диэтиловым эфиром (3×30 мл) и сушили в эксикаторе в течение 3 сут, при этом получали продукт грязно-белого цвета (3,93 г, емкость 1,8 ммоля/г).
трет-Бутиловый эфир 4-[4-амино-5-(2,6-дифторбензоил)тиазол-2-иламино-пиперидин-1-карбоновой кислоты
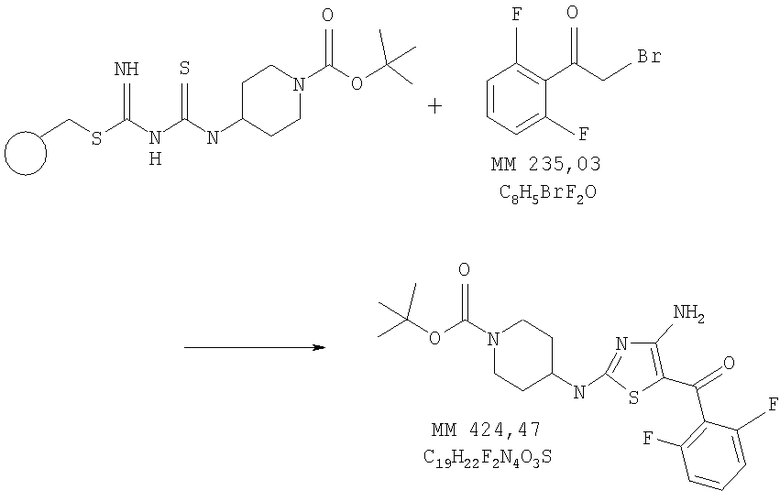
В колбу помещали смесь диметилформамида (безв., 10 мл), смолу, модифицированную трет-бутиловым эфиром 4-[3-(2-метилизотиоуреидо)тиоуреидо]пиперидин-1-карбоновой кислоты (0,60 г, 1,08 ммоля), 2-бром-1-(2,6-дифторфенил)этанона (0,616 г, 2,6 ммоля, 2 экв., пример 2), диизопропилэтиламин, иммобилизованный на полимере (PS-DIEA, фирма Aldrich, 823 мг, 3 экв.) и встряхивали в течение ночи. Затем в реакционную смесь добавляли смолу-акцептор, PS-трисамин (0,612 г, 2,5 экв., фирма Argonaut), интенсивно встряхивали в течение ночи и фильтровали через короткий картридж с силикагелем. Полученную смолу промывали дихлорметаном (3×10 мл) и промывной раствор объединяли с исходным фильтратом. Объединенный раствор концентрировали и очищали на колонке с силикагелем, при этом получали трет-бутиловый эфир 4-[4-амино-5-(2,6-дифторбензоил)тиазол-2-иламинопиперидин-1-карбоновой кислоты (0,276 г, выход 62%) в виде твердого вещества грязно-белого цвета.
[4-Амино-2-(пиперидин-4-иламино)тиазол-5-ил](2,6-дифторфенил)метанон
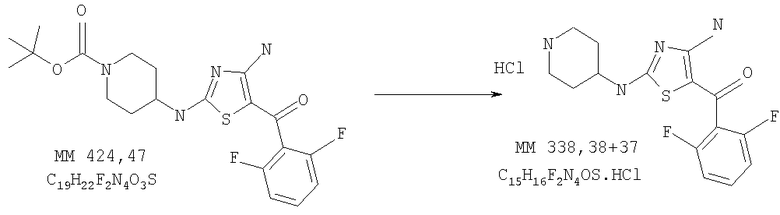
В колбу, содержащую трет-бутиловый эфир 4-[4-амино-5-(2,6-дифторбензоилтиазол-2-иламино]пиперидин-1-карбоновой кислоты (0,276 г, 0,63 ммоля), добавляли 4н. раствор HCl (2 мл) в 1,4-диоксане (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем смесь концентрировали и остаток растирали в этиловом эфире, при этом получали неочищенный [4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](2,6-дифторфенил)метанон (0,262 г), который использовали на следующей стадии без дополнительной очистки.
[4-Амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,6-дифторфенилметанон
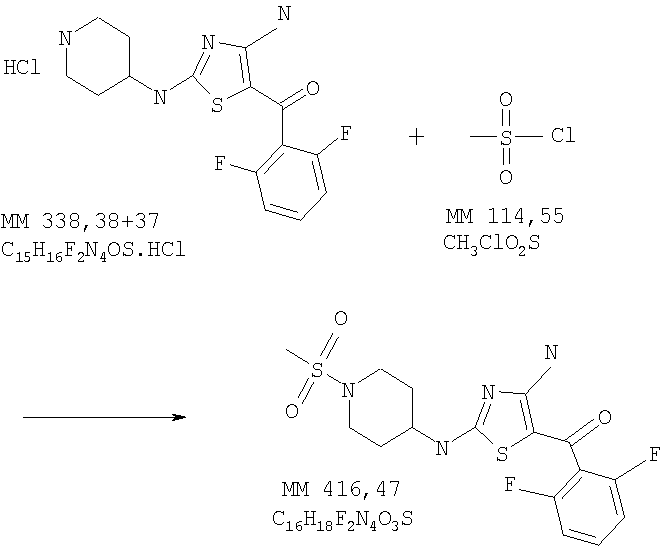
В колбу помещали [4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](2,6-дифторфенил)метанон (0,25 г, неочищенный продукт, полученный на предыдущей стадии), дихлорметан (безв., 1,5 мл) и метансульфонилхлорид (3 экв., 155 мкл), в полученную смесь добавляли диэтилизопропиламин (6 экв., 697 мкл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь концентрировали и продукт очищали на колонке с силикагелем, при этом получали [4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,6-дифторфенил)метанон (0,38 г) в виде твердого вещества светло-коричневого цвета (суммарный выход 15%). МС-ВР: рассч. 417,0861, найд. 417,0865 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6,): δ 1,45-1,57 (m, 2H, СН2), 1,95-2,05 (m, 2H, СН2), 2,82-2,90 (m, 5Н, СН3, СН2), 3,45-3,55 (m, 2H, СН2), 3,6-4,0 (ушир., 1Н, СН), 7,14-7,20 (m, 2H, ароматич.), 7,45-7,55 (m, 1Н, ароматич.), 8,15-8,25 (ушир., 1Н, NH2), 8,7-8,9 (ушир., 1Н, NH).
Ki в отношении CDK1 составляет 0,003 мкМ, в отношении CDK2 0,005 мкМ, в отношении CDK4 0,006 мкМ.
Фармакологические свойства соединений по настоящему изобретению оценивают с использованием ряда методов фармакологического анализа. Фармакологические анализы, приведенные ниже, проводят с использованием соединений по изобретению и их солей. Соединения по изобретению проявляют активность в отношении киназ, например, Ki в отношении cdk4 составляет менее 3 мкМ, предпочтительно менее 0,5 мкМ, Ki в отношении cdk2 составляет менее 8 мкМ, предпочтительно менее 0,5 мкМ, и Ki в отношении cdk1 составляет менее 10 мкМ, предпочтительно менее 0,5 мкМ.
Пример 18
Анализ киназной активности
Определение Ki
Эксперимент проводили с использованием рекомбинантных комплексов циклин/B-CDK1 человека, циклин/Е-CDK2 человека или циклин/D-CDK4. Клоны кДНК GST-циклина Е (GST-cycE), CDK2, GST-циклина В (GST-cycB), CDK1, GST-CDK4 и циклина D1 (cycD1) в бакуловирусных векторах были получены д-ром Харпером (Dr. W.Harper, Baylor College of Medicine, Houston, TX). Белки совместно экспрессировали в клетках насекомых High Five™ и комплекс очищали на глутатион-сефарозе (фирма Pharmacia, Piscataway, NJ), как описано ранее (Harper J.W. и др., Cell, 75, 805-816 (1993)). При анализе cycD1-CDK4, cycB-CDK1 и the cycE-CDK2 в качестве субстрата использовали меченую гексагистидином укороченную форму белка ретинобластомы (Rb) (фрагмент 386-928) (экспрессионная плазмида предоставлена д-ром Veronica Sullivan, Department of Molecular Virology, Roche Research Centre, Welwyn Garden City, United Kingdom). Белок Rb является природным субстратом фосфорилирования киназами CDK4, CDK2 и CDK1 (см. Herwig и Strauss, Eur. J. Biochem., 246, 581-601 (1997) и ссылки, приведенные в описании заявки).
Экспрессию белка 62 кДа проводили под контролем IPTG-индуцируемого промотора в клетках штамма Ml5 Е. coli. Клетки лизировали при обработке ультразвуком, лизат при pH 8,0 наносили на колонку с Ni-агарозой, предварительно промытой 1 мМ раствором имидазола. Затем сорбент несколько раз промывали буферными растворами, постепенно снижая величину pH до 6,0, и белок элюировали 500 мМ раствором имидазола. Элюат диализовали против буферного раствора следующего состава: 20 мМ раствор HEPES, pH 7,5, 30% глицерин, 200 мМ NaCl и 1 мМ DTT. В образцах очищенного гибридного белка Rb определяли концентрацию белка, разделяли на аликвотные части и хранили при -70°С.
Анализы проводили методом HTRF с использованием гибридных белков, указанных выше, CDK1, CDK2 и CDK4. Реакции проводили в 96-луночных планшетах, а флуоресценцию определяли в 384-луночных планшетах. Анализ выполняли при концентрациях, в три раза превышающих соответствующие Km для АТФ.
При анализе CDK4 исходные растворы анализируемых соединений разбавляли втрое (3×) до конечной концентрации буферным раствором следующего состава: 25 мМ Hepes, pH 7,0, 6,25 мМ MgCl2, 1,5 мМ ДТТ, 135 мкМ АТФ. Концентрация ДМСО составляла не более 4,76%. В лунки 96-луночного планшета помещали по 20 мкл раствора анализируемых соединений. Киназную реакцию инициировали добавлением по 40 мкл в лунку реакционной смеси следующего состава: 0,185 мкМ Rb, 2,25 мкг/мл CDK4 в 25 мМ HEPES, pH 7,0, 6,25 мМ MgCl2, 0,003% твин 20, 0,3 мг/мл БСА, 1,5 мМ ДТТ. Контрольные лунки не содержали CDK4. Планшеты инкубировали при 37°С в течение 30 мин при встряхивании. Киназную реакцию останавливали добавлением 15 мкл в лунку 1,6 мкМ раствора антител антифосфо-Rb (Ser 780) (фирма Cell Signaling Inc.) в 25 мМ Hepes, pH 7,0, 24 мМ EDTA, 0,2 мг/мл БСА. Инкубацию проводили при 37°С в течение 30 мин, а затем в лунки добавляли (по 15 мкл в лунку, 3 нМ) антикроличий IgG, меченный реактивом LANCE Eu-W1024, и 60 нМ конъюгат аллофикоцианин/анти-His6 (фирма PerkinElmer, Life Sciences) в 25 мМ Hepes, pH 7,0, 0,5 мг/мл БСА. Инкубацию продолжали при 37°С в течение еще 1 ч. После завершения инкубации 35 мкл реакционной смеси при двойном повторе из каждой лунки переносили в 384-луночные черные планшеты и определяли интенсивность флуоресценции на планшет-ридере ViewLux или Victor V (фирма PerkinElmer Life Sciences) при волне возбуждения 340 нм и волне испускания 615 и 665 нм.
Величины IC50 (концентрацию анализируемых соединений, при которой флуоресценция, наблюдаемая в контрольных лунках, снижается на 50%) рассчитывали при сравнении с фоном при 665 нм, нормализованным по европию (615 нм). Для конкурентных ингибиторов АТФ константу ингибирования Ki рассчитывали по следующему уравнению Ki=IC50/(1+[S]/Km), где [S] означает концентрацию субстрата (АТФ), a Km означает константу Михаэлиса-Ментен для АТФ.
Анализы с использованием киназ CDK1 и CDK2 проводили по аналогичной методике с небольшими изменениями концентраций реагентов и белков.
Буферные растворы, в которых растворяли анализируемые соединения и ферменты, содержали 10 мМ MgCl2.
При анализе CDK1 и CDK2 концентрация АТФ составляла 162 мкМ и 90 мкМ, соответственно. CDK1 использовали при концентрации (в растворе реагента) 0,15 нг/мкл, а CDK2 при концентрации 0,06 нг/мкл. Концентрации реагентов в растворе для детектирования составляла 3-12 нМ Eu-Ab и 60-90 нМ АРС-антиНis6, соотношение сигнала к фону составляло по меньшей мере 10:1.
Пример 19
Методика получения
Смешивают компоненты 1, 2 и 3 в соответствующем смесителе в течение 15 мин.
Гранулируют смесь, полученную на стадии 1, в присутствии 20% раствора повидона K30 (компонент 4).
Высушивают гранулят, полученный на стадии 2 при 50°С.
Пропускают гранулят, полученный на стадии 3 через соответствующую мельницу.
К размолотому грануляту, полученному на стадии 4, добавляют компонент 5 и смесь перемешивают в течение 3 мин.
Прессуют гранулят, полученный на стадии 5, на соответствующем прессе.
Пример 20
Методика получения
Смешивают компоненты 1, 2 и 3 в соответствующем смесителе в течение 15 мин.
Добавляют компоненты 4 и 5 и перемешивают в течение 3 мин.
Смесью заполняют соответствующие капсулы.
Пример 21
Методика получения
Растворяют компонент 1 в компоненте 2.
К компоненту 6 добавляют компоненты 3, 4 и 5 и смесь перемешивают до образования дисперсии, а потом гомогенизируют.
К смеси, полученной на стадии 2, добавляют раствор, полученный на стадии 1, гомогенизируют до образования прозрачного раствора.
Раствор стерилизуют фильтрованием через фильтр с диаметром пор 0,2 мкм и заполняют флаконы.
Пример 22
Получение инъекционного раствора/эмульсии
Растворяют компонент 1 в компоненте 2.
К компоненту 6 добавляют компоненты 3, 4 и 5 и смесь перемешивают до образования дисперсии, а потом гомогенизируют.
К смеси, полученной на стадии 2, добавляют раствор, полученный на стадии 1, гомогенизируют до образования прозрачного раствора.
Раствор стерилизуют фильтрованием через фильтр с диаметром пор 0,2 мкм и заполняют флаконы.
В то время как настоящее изобретение иллюстрируется конкретными и предпочтительными вариантами его осуществления, для специалиста в данной области представляется очевидным, что благодаря экспериментам и использованию настоящего изобретения на практике возможны его различные изменения и модификации. Таким образом, подразумевается, что настоящее изобретение не ограничивается описанием заявки, а определяется прилагаемыми пунктами формулы изобретения и их эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРАЗОЛПИРИМИДИНЫ | 2005 |
|
RU2412186C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,6-ДИАМИНОПИРИДИН-3-ОНА | 2005 |
|
RU2385866C2 |
| ДИАМИНОТИАЗОЛЫ, ОБЛАДАЮЩИЕ СВОЙСТВАМИ ИНГИБИТОРА ЦИКЛИН-ЗАВИСИМОЙ КИНАЗЫ 4 | 2003 |
|
RU2311414C2 |
| СОЕДИНЕНИЯ | 2008 |
|
RU2461559C2 |
| ХИНАЗОЛИНИЛМЕТИЛЕНТИАЗОЛИНОНЫ В КАЧЕСТВЕ CDK-1 ИНГИБИТОРОВ | 2005 |
|
RU2405782C2 |
| НОВЫЕ АЗАИНДОЛТИАЗОЛИНОНЫ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2005 |
|
RU2391342C2 |
| Производные бензимидазола, способы их получения и применения | 2017 |
|
RU2779534C2 |
| 1, 5-НАФТИРИДИНАЗОЛИДИНОНЫ, ОБЛАДАЮЩИЕ CDK1 АНТИПРОЛИФЕРАТИВНОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2405781C2 |
| АРИЛКАРБОНИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ | 2003 |
|
RU2340605C2 |
| ИНГИБИТОРЫ JAK | 2010 |
|
RU2538204C2 |
Изобретение относится к соединениям формулы I и их фармацевтически приемлемым солям и сложным эфирам. Соединения настоящего изобретения обладают ингибирующей активностью в отношении циклин-зависимых киназ. В формуле I
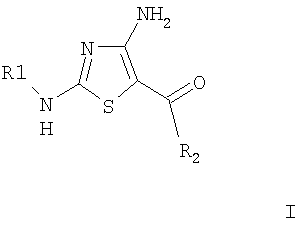
R1 означает , R3 выбирают из группы, состоящей из Н, CO2R6, C(O)R6, SO2R6 и SO2NR5R6, R5 и R6 каждый независимо выбирают из группы, включающей Н и (низш.)алкил, R2 означает фенил, содержащий один, два или три заместителя, независимо выбранных из группы, включающей галоген или -O-(низш.)алкил. Изобретение также относится к фармацевтической композиции, включающей в качестве активного ингредиента эффективное количество соединения формулы I. 2 н. и 4 з.п. ф-лы, 1 табл.
1. Соединение формулы
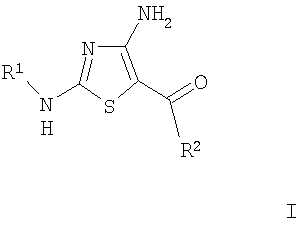
где R1 означает
R3 выбирают из группы, состоящей из Н, CO2R6, C(O)R6, SO2R6 и SO2NR5R6,
R5 и R6 каждый независимо выбирают из группы, включающей Н и (низш.)алкил,
R2 означает фенил, содержащий один, два или три заместителя, независимо выбранных из группы, включающей галоген или -O-(низш.)алкил, и
его фармацевтически приемлемые соли или сложные эфиры.
2. Соединение по п.1, где R2 означает 3-фторфенил, необязательно замещенный одним или двумя заместителями, выбранными из группы, включающей -F и -O-СН3.
3. Соединение по п.1, выбранное из группы, включающей
трет-бутиловый эфир 4-[4-амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты,
трет-бутиловый эфир 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты,
трет-бутиловый эфир 4-[4-амино-5-(2,3-дифтор-6-метоксибензоил)тиазол-2-иламино]пиперидин-1-карбоновой кислоты,
[4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанон,
[4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанон,
[4-амино-2-(пиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон и
1-[4-[4-амино-5-(3-фторбензоил)тиазол-2-иламино]пиперидин-1-ил]этанон.
4. Соединение по п.1, выбранное из группы, включающей
[4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](3-фторфенил)метанон,
1-[4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-ил]этанон,
[4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](3-фтор-4-метоксифенил)метанон,
диметиламид 4-[4-амино-5-(3-фтор-4-метоксибензоил)тиазол-2-иламино]пиперидин-1-сульфоновой кислоты,
[4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,3-дифтор-6-метоксифенил)метанон и
[4-амино-2-(1-метансульфонилпиперидин-4-иламино)тиазол-5-ил](2,6-дифторфенил)метанон.
5. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении циклин-зависимых киназ, включающая в качестве активного ингредиента эффективное количество соединения по п.1 и фармацевтически приемлемый носитель или эксципиент.
6. Фармацевтическая композиция по п.5, пригодная для парентерального введения.
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННЫХ 4-ФЕНИЛТИАЗОЛОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2124515C1 |
| Способ изготовления бумаги для электрохимической записи | 1944 |
|
SU66810A1 |
| WO 03011843 A1, 13.02.2003 | |||
| 0 |
|
SU156567A1 | |

