Область изобретения
Изобретение относится к пиридильному неароматическому азотсодержащему гетероцикло-1-карбоксилатному производному или его фармацевтически приемлемой соли, полезным в качестве лекарственного средства, в частности, в качестве лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли, обладающего активностью ингибирования жирно-кислотной амид-гидролазы (далее указана как FAAH). Настоящее изобретение также относится к способу скрининга для определения ингибитора активности FAAH, служащего в качестве лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли; и к фармацевтической композиции для лечения частого мочеиспускания и недержания мочи, для лечения повышенной активности мочевого пузыря и/или для лечения боли, которая содержит вещество, полученное в соответствии со способом скрининга по настоящему изобретению, или содержит вещество, которое ингибирует активность жирно-кислотной амид-гидролазы.
Предпосылки изобретения
Известно, что жирно-кислотная амид-гидролаза (FAAH) гидролизует эндоканнабиноид, инкактивируя его (см. непатентные ссылочные документы 1-4). Эндоканнабиноид - это родовой термин для обозначения биологического вещества, которое действует на каннабиноидный рецептор для проявления его физиологической активности. Типичные эндоканнабиноиды представляют собой анандамид, пальмитоилэтаноламид, олеамид, 2-арахидоноилглицерин; и известно, что они гидролизуются под действием FAAH с утратой их активности. Известно, что Δ9-тетрагидроканнабинол, который считается активным ингредиентом Cannabis (марихуана), активирует каннабиноидный рецептор (см. непатентный ссылочный документ 5).
У млекопитающих на сегодняшний день известны два типа каннабиноидных рецепторов - CB1 и CB2. CB1 экспрессируется в центральной и периферической нервной системе и при активации проявляет действие на психику и аналгетическое действие. CB2 экспрессируется в иммунной системе и при активации проявляет противовоспалительное действие и аналгетическое (и противовоспалительное) действие.
С другой стороны, в модели цистита у крыс агонист каннабиноидного рецептора увеличивает емкость мочевого пузыря и порог мочеиспускания (непатентный ссылочный документ 6 и непатентный ссылочный документ 7); и побочные эффекты, такие как галлюцинации, бред, тахикардия, ортостатическая гипотензия, наблюдаемые при введении агониста каннабиноидного рецептора животным, не наблюдаются при введении им ингибитора FAAH (непатентный ссылочный документ 8). Учитывая вышесказанное, ожидают, что ингибитор FAAH будет представлять собой лекарственное средство для лечения частого мочеиспускания и недержания мочи, лекарственное средство для лечения повышенной активности мочевого пузыря и/или лекарственное средство для лечения боли.
В качестве соединений, обладающих активностью ингибирования FAAH, известны соединения, которые могут служить в качестве аналгетического средства, средства против страха, антиэпилептического средства, антидепрессанта, противорвотного, сердечно-сосудистого средства или средства против глаукомы [C1-4 алкильные или полициклические ароматические эфирные производные содержащих ароматическое кольцо или фенил-замещенный алифатический углеводород карбаминовых кислот (патентный ссылочный документ 1) и фенилциклогексилкарбамата (патентный ссылочный документ 2)]. Диоксан-2-алкилкарбаматные производные, которые представляют собой соединения, обладающие активностью ингибирования FAAH, описаны как лекарственные средства от недержания мочи, что является одним из вариантов большого количества расстройств, перечисленных в ссылочном документе (патентный ссылочный документ 3). Однако патентный ссылочный документ 3 не раскрывает результаты экспериментов, подтверждающие целительный эффект при лечении частого мочеиспускания и недержания мочи и/или лечении повышенной активности мочевого пузыря, в нем нет никакого предположения такого эффекта. 4-Аминопиридилпиперидин-1-карбоксилат, представляющий собой тип пиридильных неароматических азотсодержащих гетероцикло-1-карбоксилатов, описан как ингибитор ацетилхолинэстеразы (непатентный ссылочный документ 9); однако в этом ссылочном документе ничего не сказано о том, что соединение является лекарственным средством для лечения частого мочеиспускания и недержания мочи и/или лекарственным средством для лечения повышенной активности мочевого пузыря.
Патентный ссылочный документ 1: WO2003/065989
Патентный ссылочный документ 2: WO2004/033422
Патентный ссылочный документ 3: JP-A 2003-192659
Непатентный ссылочный документ 1: Prostaglandins Leukotrienes and Essential Fatty acids, (England), 2002, Vol. 66, pp. 143-160
Непатентный ссылочный документ 2: British Journal of Pharmacology (England), 2004, Vol. 141, pp. 253-262
Непатентный ссылочный документ 3: Nature (England), 1996, Vol. 384, pp. 83-87
Непатентный ссылочный документ 4: Biochemical Pharmacology, (USA), 2001, Vol. 62, pp. 517-526
Непатентный ссылочный документ 5: Current Medicinal Chemistry (USA), 1999, Vol. 6, pp. 635-664
Непатентный ссылочный документ 6: The Journal of Neuroscience, 2002, Vol. 22, pp. 7147-7153
Непатентный ссылочный документ 7: Pain, 1998, Vol. 76, pp. 189-199
Непатентный ссылочный документ 8: Nature Medicine, (England), 2003, Vol. 9, pp. 76-81
Непатентный ссылочный документ 9: Journal of Pharmaceutical Science, 1992, Vol. 81, pp. 380-385
Раскрытие изобретения
Задачи, решаемые настоящим изобретением
Целью настоящего изобретения является обеспечение лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли, которые не имеют или имеют значительно меньше связанных с каннабиноидом побочных эффектов и проблем зависимости. Другие цели состоят в обеспечении способа скрининга для определения вещества, обладающего активностью ингибирования FAAH, или лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли; и в обеспечении фармацевтической композиции для лечения частого мочеиспускания и недержания мочи, для лечения повышенной активности мочевого пузыря и/или для лечения боли, которая содержит вещество, полученное в соответствии со способом скрининга по настоящему изобретению, или вещество, способное ингибировать активность жирно-кислотной амид-гидролазы.
Средства для решения задач
Авторы настоящего изобретения тщательно исследовали проблему получения соединения, обладающего активностью ингибирования FAAH, и в результате были найдены новые пиридильные азотсодержащие гетероцикло-1-карбоксилатные производные.
Кроме того, авторами настоящего изобретения было впервые обнаружено, что, когда соединение, обладающее активностью ингибирования FAAH, вводят крысам, страдающим частым мочеиспусканием, индуцируемым циклофосфаамидом, тогда эффективная емкость мочевого пузыря крыс увеличивается, и, кроме того, было обнаружено, что соединение, обладающее активностью ингибирования FAAH, обладает отличным терапевтическим эффектом в модели боли у крыс, в результате был обеспечен способ скрининга лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли путем отбора ингибитора FAAH, таким образом, было создано настоящее изобретение.
Конкретно, настоящее изобретение относится к:
[1] Пиридильному неароматическому азотсодержащему гетероцикло-1-карбоксилатному производному общей формулы (I), и его фармацевтически приемлемой соли:

где:
HET1 представляет собой 5-7-членное неароматическое азотсодержащее гетерокольцо,
R1, R2 и R3 являются одинаковыми или отличными друг от друга, и каждый представляет собой
(1) H,
(2) OH,
(3) необязательно этерифицированный карбоксил,
(4) циано,
(5) низший алкил-CO-,
(6) оксо (=O),
(7) формулу [R101-(O)m1]m2-[ALK1, необязательно замещенный OH]-(O)n1-,
(m1 и n1 являются одинаковыми или отличными друг от друга, где каждый имеет значение 0 или 1),
m2 имеет значение от 1 до 5,
ALK1 представляет собой низший алкилен, низший алкенилен или низший алкинилен,
R101 представляет собой
(i) H,
(ii) Ar1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей:
(a) H2N-,
(b) галоген,
(c) циано,
(d) необязательно этерифицированный карбоксил,
(e) группу R1011aR1012aN-CO-,
(f) HET2,
(g) Ar1a, необязательно замещенный галогеном, циано, OH, низшим алкил-О- или низшим алкилом,
Ar1a представляет собой арил,
(h) низший алкил,
(j) OH,
(k) низший алкил-O-, необязательно замещенный группой Ar1a или галоген-Ar1a,
(l) HET2-CO-, необязательно замещенный галогеном, Ar1a или HETAr1a,
HET2 представляет собой азотсодержащее гетерокольцо,
HETAr1a представляет собой азотсодержащий гетероарил,
(s) HET2-CONR1011a-,
(t) H2NCONH- и
(u) необязательно этерифицированный карбоксил-ALK2a,
ALK2a представляет собой низший алкил или низший алкенил,
(iii) ALK2a, необязательно замещенный группой R1011aR1012aN или Ar1a,
R1011a и R1012a являются одинаковыми или отличными друг от друга, и каждый представляет собой
(a) H,
(b) cALK,
cALK представляет собой циклоалкил,
(c) ALK2a, необязательно замещенный галогеном, cALK, OH, низший алкил-О- или Ar1a, или
(d) Ar1a-SO2-, необязательно замещенный галогеном,
(iv) HET2, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) ALK2a, необязательно замещенный группой Ar1a или галоген-Ar1a,
(b) Ar1a,
(c) HETAr1a, необязательно замещенный низшим алкилом,
(d) Ar1a-CO- или галоген-Ar1a-CO-,
(v) cALK, необязательно замещенный ALK2a,
или
(vi) необязательно этерифицированный карбоксил,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R101-(O)m1] могут быть одинаковыми или отличными друг от друга),
(8) группу R102-ALK1-N(R103)-CO-,
(R102 представляет собой
(i) H,
(ii) cALK,
(iii) HETAr1a или
(iv) Ar1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) HO,
(b) ALK2a-O-,
(c) cALK-ALK1-O-,
(d) cALK-Ar1a-ALK1-O- и
(e) Ar1a-ALK1-O-,
R103 представляет собой
(i) H,
(ii) cALK,
(iii) ALK2a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) HET2,
(b) Ar1a и
(c) галоген-Ar1a,
(iv) HETAr1a, или
(v) Ar1a-[CO]m1, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) cALK,
(b) H2N,
(c) группу R1011aR1012aN-CO- или
(d) ALK2a),
(9) группу R104aR105aN-[CO]m1-ALK1-,
(R104a и R105a являются одинаковыми или отличными друг от друга, и каждый представляет собой группу R103),
(10) группу R106-ALK3-L1-,
(R106 представляет собой
(i) группу R101-(O)m1-,
(ii) группу R104R105N-,
(iii) группу ALK2a-CONH- или
(iv) группу Ar1a-CONH-,
ALK3 представляет собой низший алкилен, низший алкенилен или циклоалкилен,
L1- представляет собой -C(=O)- или -SO2-),
(11) ALK2a-CONH-, необязательно замещенный группой Ar1a,
(12) Ar1a, замещенный галогеном,
(13) группу [R107-(O)m1]m2-Ar2-(O)n1-,
(Ar2 представляет собой арилен,
R107 представляет собой
(i) H,
(ii) галоген,
(iii) ALK2a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) HO,
(b) cALK,
(c) HET2,
(d) Ar1a, необязательно замещенный галогеном, низшим алкилом, низший алкил-O-, группой R1011aR1012aN-[CO]p-, циано или необязательно этерифицированным карбоксилом,
(e) необязательно этерифицированный карбоксил,
(f) HET2-[CO]p-, необязательно замещенный группой R1011aR1012aN-[CO]p-, и
(g) группу R1011aR1012aN-[CO]p-,
p имеет значение 0 или 1,
(iv) группу R1011aR1012aN-[CO]p- или
(v) группу R1011aR1012aN-[CO]p-Ar1a,
при этом когда m2 имеет значение от 2 до 5, тогда группы [R107-(O)m1] могут быть одинаковыми или отличными друг от друга, и, кроме того, группа [R107-(O)m1]m2 может представлять собой метилендиокси с образованием кольца),
(14) группу [R107-(O)m1]m2-Ar2-N(R103) -CO-,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R107-(O)m1] могут быть одинаковыми или отличными друг от друга),
(15) группу [R1011aR1012aN-[CO]m1]m2-Ar2-(O)n1-,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R1011aR1012aN-[CO]m1] могут быть одинаковыми или отличными друг от друга),
(16) группу [R108]m2-Ar2-L2-,
[R108 представляет собой
(i) H,
(ii) галоген,
(iii) HO,
(iv) cALK-O-,
(v) группу R109-ALK1-(O)m1-,
(R109 представляет собой
(a) H,
(b) cALK,
(c) Ar1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(1') галоген,
(2') циано,
(3') N2O,
(4') ALK2a, необязательно замещенный галогеном,
(5') HO,
(6') ALK2a-O-, необязательно замещенный галогеном,
(7') необязательно этерифицированный карбоксил или
(8') группу R104R105N,
(d) HETAr1a или
(e) группу R104R105N-[CO]m1-),
(vi) группу R1013R1014N-,
R1013 и R1014 являются одинаковыми или отличными друг от друга, и каждый представляет собой
(i) H,
(ii) ALK2a,
(iii) cALK-ALK1- или
(iv) Ar1a-ALK1-, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(1') галоген,
(2') циано,
(3') ALK2a, необязательно замещенный галогеном,
(4') ALK2a-O-, необязательно замещенный галогеном,
(vii) HET2-(O)m1-, необязательно замещенный низшим алкилом,
L2 представляет собой -CO- или -S(O)q-,
q имеет значение 0, 1 или 2,
при этом когда m2 имеет значение от 2 до 5, тогда группы [R108] могут быть одинаковыми или отличными друг от друга],
(17) группу [R101]m2-Ar2-CONH-,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R101] могут быть одинаковыми или отличными друг от друга),
(18) группу [R111]m2-HETAr2-(O)m1-,
(R111 представляет собой
(i) H,
(ii) галоген,
(iii) оксо (=O) или
(iv) группу R103a-(O)n1-,
R103a представляет собой
(i) H,
(ii) cALK,
(iii) ALK2a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) HET2,
(b) Ar1a,
(c) cALK и
(d) галоген-Ar1a,
(iv) HETAr1a или
(v) Ar1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей (a) cALK, (b) H2N и (c) группу R1011aR1012aN-CO-,
HETAr2 представляет собой азотсодержащий гетероарилен,
при этом когда m2 имеет значение от 2 до 5, тогда группы [R111] могут быть одинаковыми или отличными друг от друга),
(19) формулу [R112]m2-HETAr2-N(R103)-CO-,
(R112 представляет собой
(i) H,
(ii) cALK,
(iii) ALK2a или
(iv) Ar1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) галоген,
(b) HO,
(c) ALK2a-O- и
(d) Ar1a-ALK1-O-,
при этом когда m2 имеет значение от 2 до 5, тогда группы [R112] могут быть одинаковыми или отличными друг от друга,
(20) формулу [R108]m2-HETAr2-L2-,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R108] могут быть одинаковыми или отличными друг от друга),
при условии, что, когда какая-либо из групп R1, R2 и R3 представляет собой группу [R111]m2-HETAr2-(O)m1-, и когда m1 имеет значение 0, тогда остальные группы R1, R2 и R3 представляют собой H;
R4, R5, R6 и R7 являются одинаковыми или отличными друг от друга, и каждый представляет собой
(1) H,
(2) галоген,
(3) необязательно этерифицированный карбоксил,
(4) HO,
(5) группу R113-ALK4-(O)m3-,
(ALK4 представляет собой низший алкилен, низший алкенилен, или низший алкинилен,
m3 имеет значение 0 или 1,
R113 представляет собой
(i) H,
(ii) HO,
(iii) низший алкил-O-, необязательно замещенный необязательно этерифицированным карбоксилом,
(iv) необязательно этерифицированный карбоксил,
(v) низший алкил-CO-O- или
(vi) группу R104bR105bN-[CO]m3- (R104b и R105b являются одинаковыми или отличными друг от друга, и каждый представляет собой группу R103),
(6) R114R115N (R114 и R115 являются одинаковыми или отличными друг от друга, и каждый представляет собой
(i) H или
(ii) ALK2b, необязательно замещенный группой R104bR105bN,
ALK2b представляет собой низший алкил или низший алкенил),
(7) группу R116-(ALK4)n2-N(R117)-CO-,
(n2 имеет значение 0 или 1,
R116 представляет собой
(i) H,
(ii) HO,
(iii) низший алкил-O-,
(iv) необязательно этерифицированный карбоксил,
(v) группу R104bR105bN-[CO]m3-,
(vi) Ar1b, необязательно замещенный (a) OH или (b) ALK2b-O-,
Ar1b представляет собой арил,
(vii) HET3, необязательно замещенный группой R104bR105bN-[CO]m3- или необязательно этерифицированным карбоксилом,
HET3 представляет собой азотсодержащее гетерокольцо,
(viii) Ar1b, необязательно замещенный группой R104R105N-[CO]m3-, или
(ix) SO3H),
R117 представляет собой ALK2b, необязательно замещенный (i) H или(ii) Ar1b),
(8) Ar1b, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей необязательно этерифицированный карбоксил и группу R1011bR1012bN-[(CO)]m3-,
R1011b и R1012b являются одинаковыми или отличными друг от друга, и каждый представляет собой
(i) H,
(ii) cALK,
(iii) ALK2b, необязательно замещенный галогеном, cALK, OH, низший алкил-O- или Ar1b, или
(iv) Ar1b-SO2-, необязательно замещенный галогеном,
(9) HET3, необязательно замещенный необязательно этерифицированным карбоксилом,
(10) HET3-CO-, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей ALK2b и группу R104bR105bN-[CO]m3-, или
(11) циано,
при условии, что 4-аминопиридин-3-ил пиперидин-1-карбоксилат исключается - это условие применяется к указанному ниже].
[2] Соединению [1], представленному общей формулой (II):
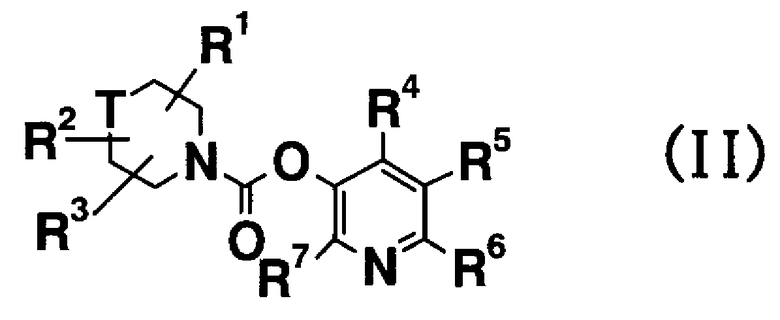
где:
R1-R7 имеют значения, определенные в [1],
T представляет собой CH2, NH, NHCH2 или O,
и также включается случай, где водород в T замещен R1-R3 - то же относится и к описанному ниже].
[3] Соединению [2], где R1-R3 являются одинаковыми или отличными друг от друга, и каждый представляет собой группу [R101-(O)m1]m2-[ALK1, необязательно замещенный OH]-(O)n1-, группу R102-ALK1-N(R103)-CO-, группу R106-ALK3-L1-, группу [R107-(O)m1]m2-Ar2-(O)n1-, группу [R107-(O)m1]m2-Ar2-N(R103)-CO- или группу [R108]m2-Ar2-L2-.
[4] Пиридильному неароматическому азотсодержащему гетероцикло-1-карбоксилатному производному общей формулы (III) и его фармацевтически приемлемой соли:
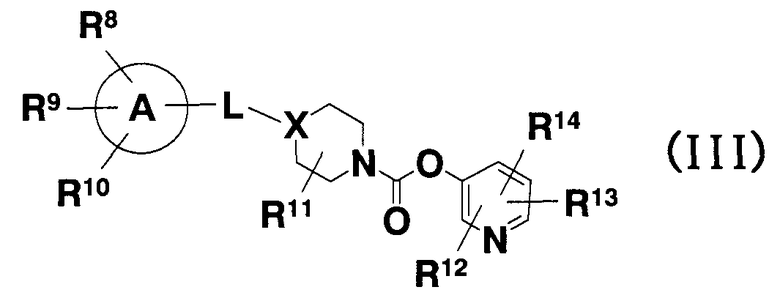
где:
кольцо A представляет собой бензольное кольцо, циклопентановое кольцо, циклогексановое кольцо, циклогептановое кольцо или 5-7-членное азотсодержащее гетерокольцо;
L представляет собой простую связь, низший алкилен, низший алкенилен, -N(R15)-C(=O)-, -C(=O)-N(R15)-, -(низший алкенилен)-C(=O)-, -O- или -C(=O)-,
R15 представляет собой H или низший алкил,
X представляет собой CH или N,
R8-R10 являются одинаковыми или отличными друг от друга, и каждый представляет собой
группу, выбранную из представленной ниже группы G,
арил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
азотсодержащий гетероарил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
R16-(низший алкилен)-O-,
R16-(низший алкилен)-N(R15)- или
R17R18N-C(=O)-,
R16 представляет собой
арил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
азотсодержащий гетероарил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G, или
3-8-членный циклоалкил,
R17 и R18 являются одинаковыми или отличными друг от друга, и каждый представляет собой H, низший алкил или 3-8-членный циклоалкил,
(кроме того, R17 и R18 могут образовывать, вместе с атомом N, связанным с ними, 3-8-членное азотсодержащее гетерокольцо),
группа G включает H, галоген, -CN, -CF3, низший алкил или -O-низший алкил,
R11 представляет собой H, низший алкил или оксо (=O),
R12-R14 являются одинаковыми или отличными друг от друга, и каждый представляет собой H, низший алкил, -C(=O)-O-(низший алкил), -CO2H или -CONH2].
[5] Соединению [4], где кольцо A представляет собой бензольное кольцо, циклогексановое кольцо, пиперидиновое кольцо или пиперазиновое кольцо.
[6] Соединению [5], где R9, R10, R11, R12 и R13 представляют собой H.
[7] Пиридильному неароматическому азотсодержащему гетероцикло-1-карбоксилатному производному общей формулы (IV) и его фармацевтически приемлемой соли:
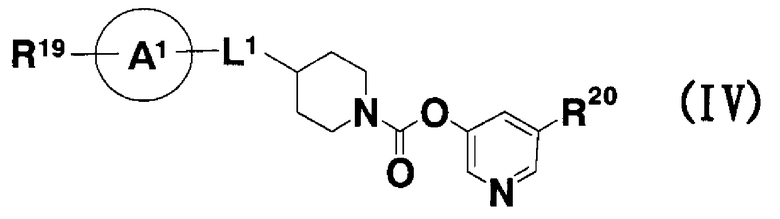
где:
кольцо A1 представляет собой бензольное кольцо, пиперидиновое кольцо или пиперазиновое кольцо;
L1 представляет собой низший алкилен, низший алкенилен, -N(R15)-C(=O)- или -O-;
R15 представляет собой H или низший алкил,
R19 представляет собой
группу, выбранную из представленной ниже группы G,
азотсодержащий гетероарил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
R16-(низший алкилен)-O- или R17R18N-C(=O)-,
R16 представляет собой
арил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
азотсодержащий гетероарил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G, или
3-8-членный циклоалкил,
R17 и R18 являются одинаковыми или отличными друг от друга, и каждый представляет собой H или низший алкил,
(кроме того, R17 и R18 могут образовывать, вместе с атомом N, связанным с ними, 5- или 6-членное азотсодержащее гетерокольцо),
группа G включает H, галоген, -CN, -CF3, низший алкил или -O-низший алкил,
R20 представляет собой H, -C(=O)-O-(низший алкил), -CO2H или -CONH2].
[8] Пиридильному неароматическому азотсодержащему гетероцикло-1-карбоксилатному производному общей формулы (V) и его фармацевтически приемлемой соли:

где:
L2 представляет собой низший алкилен, низший алкенилен или -(низший алкенилен)-C(=O)-,
R21 представляет собой H, галоген, -CN, -CF3, низший алкил или -O-низший алкил,
R22 представляет собой H, -C(=O)-O-(низший алкил), -CO2H или -CONH2].
[9] Соединению [1], выбранному из следующей группы:
пиридин-3-ил 4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилат,
5-{[(4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-ил)карбонил]окси}никотиновая кислота,
5-({[4-(2-фенилэтил)пиперидин-1-ил]карбонил}окси)-никотиновая кислота,
5-[({4-[4-(2-циклогексилэтокси)фенокси]пиперидин-1-ил}карбонил)окси]никотиновая кислота,
5-[({4-[(E)-2-фенилвинил]пиперидин-1-ил}карбонил)-окси]никотиновая кислота,
5-{[(4-[3-[1-(6-метилпиридин-2-ил)пиперидин-4-ил]пропил}пиперидин-1-ил)карбонил]окси}никотиновая кислота,
5-(аминокарбонил)пиридин-3-ил 4-{2-[3-(аминокарбонил)фенил]этил}пиперидин-1-карбоксилат,
5-(аминокарбонил)пиридин-3-ил 4-(2-{3-[(диметиламино)карбонил]фенил}этил)пиперидин-1-карбоксилат,
5-(аминокарбонил)пиридин-3-ил 4-{2-[3-(пиперидин-1-илкарбонил)фенил]этил}пиперидин-1-карбоксилат,
5-(аминокарбонил)пиридин-3-ил 4-{2-[3-(пирролидин-1-илкарбонил)фенил]этил}пиперидин-1-карбоксилат,
пиридин-3-ил 4-[(2E)-3-фенилпроп-2-еноил]пиперазин-1-карбоксилат,
пиридин-3-ил 4-(анилинокарбонил)пиперидин-1-карбоксилат,
5-(аминокарбонил)пиридин-3-ил 4-(2-фенилэтил)пиперидин-1-карбоксилат,
пиридин-3-ил 4-(2-фенилэтил)пиперазин-1-карбоксилат,
5-(метоксикарбонил)пиридин-3-ил 4-(2-фенилэтил)пиперазин-1-карбоксилат,
5-(аминокарбонил)пиридин-3-ил 4-[2-(3-фторфенил)этил]пиперидин-1-карбоксилат,
5-(аминокарбонил)пиридин-3-ил 4-[2-(3-цианофенил)этил]пиперидин-1-карбоксилат.
[10] Фармацевтической композиции, включающей соединение [1] в качестве ее активного ингредиента.
[11] Фармацевтической композиции [10], которая представляет собой ингибитор FAAH.
[12] Фармацевтической композиции [10], которая представляет собой лекарственное средство для лечения частого мочеиспускания, недержания мочи и/или повышенной активности мочевого пузыря.
[13] Фармацевтической композиции [10], которая представляет собой лекарственное средство для лечения боли.
[14] Применению соединения [1] для получения ингибитора FAAH или лекарственного средства для лечения частого мочеиспускания, недержания мочи и/или повышенной активности мочевого пузыря.
[15] Применению соединения [1] для получения ингибитора FAAH или лекарственного средства для лечения боли.
[16] Способу лечения частого мочеиспускания, недержания мочи и/или повышенной активности мочевого пузыря, включающему введение пациенту терапевтически эффективного количества соединения [1].
[17] Способу лечения боли, включающему введение пациенту терапевтически эффективного количества соединения [1].
[18] Способу скрининга для определения лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли, включающему (1) стадию контактирования испытываемого вещества с полипептидом, который содержит (a) аминокислотную последовательность, представленную SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, (b) аминокислотную последовательность, полученную из аминокислотой последовательности, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8 путем делеции, замещения и/или вставки в эту последовательность от 1 до 10 аминокислот, (c) аминокислотную последовательность, имеющую гомологию, по меньшей мере, 70% с аминокислотой последовательностью, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, или (d) аминокислотную последовательность полной аминокислотной последовательности, кодируемой полинуклеотидом, представленным SEQ ID NO:1, SEQ ID NO:3, SEQ ID NO:5 или SEQ ID NO:7, или полинуклеотидом, способным гибридизоваться с ее комплементарной последовательностью в жестких условиях, или ее частью, не содержащей, по меньшей мере, содержащего трансмембранную область амино-концевого участка этой последовательности, и который может гидролизовать субстрат, (2) стадию анализа полипептида на изменение его активности, и (3) стадию выбора вещества, способного ингибировать активность полипептида,
(где "субстрат", с которым контактирует FAAH или функциональная FAAH, может представлять собой любой эндоканнабиноид, способный гибридизоваться под действием FAAH или функциональной FAAH; и конкретно, он включает анандамид, пальмитоилэтаноламид, 2-арахидоноилглицерин и олеамид; и субстрат, меченный 3H или 14C, а также можно использовать смесь меченого субстрата и немеченного субстрата - то же относится и к указанному ниже).
[19] Способу скрининга для определения лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли, включающему (1) стадию контактирования испытываемого вещества с полипептидом, который содержит (a) аминокислотную последовательность, представленную SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, (b) аминокислотную последовательность, полученную из аминокислотной последовательности, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8 путем делеции, замещения и/или вставки в эту последовательность от 1 до 10 аминокислот, (c) аминокислотную последовательность, имеющую гомологию, по меньшей мере, 70% с аминокислотной последовательностью, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, или (d) аминокислотную последовательность полной аминокислотной последовательности, кодируемой полинуклеотидом, представленным SEQ ID NO:1, SEQ ID NO:3, SEQ ID NO:5 или SEQ ID NO:7, или полинуклеотидом, способным гибридизоваться с ее комплементарной последовательностью в жестких условиях или ее частью, не содержащей, по меньшей мере, трансмембранную область амино-концевого участка этой последовательности, и который может гидролизовать субстрат, в присутствии субстрата полипептида, (2) стадию измерения количества гидролизованного продукта, преобразованного из субстрата, и (3) стадию выбора вещества, способного ингибировать гидролиз субстрата.
[20] Способу скрининга для определения лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли, включающему (1) стадию контактирования испытываемого вещества с клеткой или тканью, экспрессирующей полипептид, который содержит (a) аминокислотную последовательность, представленную SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, (b) аминокислотную последовательность, полученную из аминокислотной последовательности, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8 путем делеции, замещения и/или вставки в эту последовательность от 1 до 10 аминокислот, (c) аминокислотную последовательность, имеющую гомологию, по меньшей мере, 70% с аминокислотной последовательностью, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, или (d) аминокислотную последовательность полной аминокислотной последовательности, кодируемой полинуклеотидом, представленным SEQ ID NO:1, SEQ ID NO:3, SEQ ID NO:5 или SEQ ID NO:7, или полинуклеотидом, способным гибридизоваться с ее комплементарной последовательностью в жестких условиях или ее частью, не содержащей, по меньшей мере, содержащего трансмембранную область амино-концевого участка этой последовательности, и который может гидролизовать субстрат, или с лизатом или гомогенатом клетки или ткани, в присутствии субстрата полипептида, (2) стадию измерения количества гидролизованного продукта, преобразованного из субстрата, и (3) стадию выбора вещества, способного ингибировать гидролиз субстрата.
[21] Способу скрининга для определения лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли, включающему (1) стадию контактирования испытываемого вещества с жирно-кислотной амид-гидролазой, (2) стадию анализа фермента на изменение его активности и (3) стадию выбора вещества, способного ингибировать активность фермента.
Результаты изобретения
Фармакологические испытания Примеров 438 - 442 подтвердили эффективность соединений по настоящему изобретению. Например, типичные соединения, представленные в Таблице 64, обладают отличным эффектом ингибирования FAAH; типичные соединения, представленные в Примере 441, являются полезными в качестве лекарственного средства для лечения частого мочеиспускания и недержания мочи, а также лекарственного средства для лечения повышенной активности мочевого пузыря; и типичные соединения, представленные в Примере 442, являются полезными в качестве лекарственного средства для лечения боли. Кроме того, соединения по настоящему изобретению являются высокостабильными в водных растворах и обладают отличными качествами как лекарственные средства.
Изобретение, описанное в патентном ссылочном документе 2, является полезным в качестве аналгетического средства, средства против страха, антиэпилептического средства, антидепрессанта, противорвотного, сердечно-сосудистого средства или средства против глаукомы; однако авторы настоящего изобретения обнаружили, что настоящее изобретение является полезным для лекарственного средства для лечения частого мочеиспускания и недержания мочи и/или лекарственного средства для лечения повышенной активности мочевого пузыря, отличного от патентного ссылочного документа 2. Кроме того, соединения по настоящему изобретению обладают отличным эффектом ингибирования FAAH и поэтому являются полезными для лекарственных средств для лечения (1) нейропсихиатрических расстройств (например, страха, депрессии, эпилепсии), (2) психических расстройств, нейродегенеративных расстройств (например, травмы головы, церебральной ишемии, деменции), (3) иммунологических и воспалительных заболеваний, (4) рвоты, (5) расстройств питания, (6) синдрома раздраженной толстой кишки, язвенного колита, (7) гипертензии, (8) глаукомы или (9) расстройств сна. Кроме того, соединения не имеют или имеют значительно меньше связанных с каннабиноидом побочных эффектов и проблем зависимости.
Кроме того, в соответствии со способом скрининга по настоящему изобретению, лекарственное средство для лечения частого мочеиспускания и недержания мочи, лекарственное средство для лечения повышенной активности мочевого пузыря и/или лекарственное средство для лечения боли, которые не имеют или имеют значительно меньше связанных с каннабиноидом побочных эффектов и проблем зависимости, могут быть выбраны на основании активности ингибирования FAAH. Вещества, полученные в соответствии со способом скрининга, и вещества, обладающие активностью ингибирования FAAH, могут образовывать фармацевтические композиции, полезные для лечения частого мочеиспускания и недержания мочи, для лечения повышенной активности мочевого пузыря и/или для лечения боли.
Лучший способ осуществления изобретения
Ниже представлено подробное описание настоящего изобретения.
Соединения по настоящему изобретению описаны подробно ниже.
Определения
Если специально не указано иное, термин "низший" в определении структурных формул в настоящем описании означает линейную или разветвленную углеродную цепь, содержащую от 1 до 6 атомов углерода.
"Низший алкил" включает, например, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, трет-пентил, гексил, изогексил; предпочтительно - метил, этил, пропил, бутил, трет-бутил.
"Низший алкенил" означает алифатическую углеводородную группу, содержащую, по меньшей мере, одну двойную связь, включая, например, винил, пропенил, аллил, изопропенил, 1,3-бутадиенил, гексенил.
"Циклоалкил" означает моно - три-циклическую алифатическую насыщенную углеводородную кольцевую группу, содержащую от 3 до 14 атомов углерода, включая, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, бициклогептил, бициклооктил, трициклододеканил, бицикло[2.2.1]гептил, бицикло[2.2.2]октил, предпочтительно - циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил.
"Арил" означает моно- три-циклическую ароматическую углеводородную кольцевую группу, содержащую от 6 до 14 атомов углерода, в которой фенил может быть конденсирован с циклоалкилом. Например, такая группа включает фенил, инденил, нафтил, антрил, фенантрил, инданил, тетрагидронафтил, предпочтительно - фенил, нафтил.
"Гетероциклический" означает 4-16-членное, моноциклическое, бициклическое или трициклическое, насыщенное или ненасыщенное кольцо, содержащее от 1 до 4 гетероатомов, выбранных из N, S и O. Гетероциклическая группа может быть поперечно-связанной или иметь спиро-структуру. Ненасыщенное кольцо включает ароматическое кольцо (гетероарил) и неароматическое кольцо. Моноциклическая группа включает азетидинил, оксетанил, пирролидинил, 1,3-диоксоланил, пиразолидинил, пиперазинил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, фурил, тиенил, пирролил, имидазолил, пиразолил, тиазолил, оксазолил, пиридил, пиразинил, пиримидинил, триазолил, тиадиазолил, пиридазинил, оксадиазолил, тетразолил; бициклическая группа включает индолил, изоиндолил, 3,4-метилендиоксифенил, 3,4-этилендиоксифенил, бензофуранил, бензотиенил, бензотиадиазолил, бензотиазолил, бензимидазолил, индолил, изоиндолил, хинолил, изохинолил, 1,2,3,4-тетрагидрохинолил, 1,2,3,4-тетрагидроизохинолил, декагидроизохинолил, хиноксалинил; трициклическая группа включает карбазолил, акридинил, фенотиазинил. Поперечно-связанная гетероциклическая группа включает хинуклидинил, 2,5-диазабицикло[2.2,1]гептил, 8-азабицикло[3.2.1]октил, 7-азабицикло[2.2.1]гептил. Имеющая спиро-структуру гетероциклическая группа включает 1,4-диокса-8-азаспиро[4,5]деканил.
"Азотсодержащий гетероарил" означает 4-10-членный, моно- или би-циклический ароматический азотсодержащий гетероарил, содержащий от 1 до 3 атомов азота в указанной выше гетероциклической группе. Такая группа включает, например, пирролил, имидазолил, тиазолил, пиразолил, триазолил, тетразолил, пиридил, пиридазинил, пиримидинил, пиразинил, индолил, изоиндолил, бензимидазолил, бензопиразолил, хинолил, изохинолил, хиноксалинил, предпочтительно - имидазолил, тиазолил, пиридил, бензимидазолил, хинолил.
"Азотсодержащая насыщенная гетероциклическая группа" означает 3-10-членную, моно- или би-циклическую азотсодержащую гетероциклоалкильную группу, содержащую от 1 до 3 атомов азота в указанной выше гетероциклической группе. Такая группа включает, например, азиридинил, азетидинил, пирролидинил, пиперидил, пиперазинил, морфолинил, гексагидроазепинил, 1,4-диазепинил, 1,4-оксазепинил, хинуклидинил, 2,5-диазабицикло[2.2.1]гептил, азабициклооктил (например, азабицикло[3.2,1]октил), диазабициклооктил, азабициклононил, азабициклодеканил, 1,4-диокса-8-азаспиро[4,5]деканил, предпочтительно - пирролидинил, пиперидил, пиперазинил, морфолинил, гексагидроазепинил, 1,4-диазепинил, 1,4-оксазепинил, хинуклидинил, 2,5-диазабицикло[2.2.1]гептил, азабицикло[3.2.1]октил.
"Азотсодержащее гетерокольцо" означает указанную выше азотсодержащую гетероарильную группу, указанную выше азотсодержащую насыщенную гетероциклическую группу или конденсированную группу азотсодержащего гетероарила и азотсодержащего гетероциклоалкила. Предпочтительно, она представляет собой пирролидинил, пиперидил, пиперазинил, морфолинил, гексагидроазепинил, азабицикло[3.2.1]октил, 1,4-диокса-8-азаспиро[4.5]деканил, имидазолил, пиридил, хинолил.
"Неароматическое азотсодержащее гетерокольцо" означает азотсодержащую насыщенную гетероциклическую группу и ненасыщенную азотсодержащую гетероциклическую группу, за исключением азотсодержащего гетероарила из указанной выше азотсодержащей гетероциклической группы. Предпочтительно, такая группа представляет собой 5-7-членную неароматическую азотсодержащую гетероциклическую группу.
"Низший алкилен", "низший алкенилен", "циклоалкилен", "арилен" и "азотсодержащий гетероарилен" представляют собой двухвалентные группы, полученные из указанных выше низшего алкила, низшего алкенила, циклоалкила, арила и азотсодержащего гетероарила путем удаления из них любого атома водорода.
"Этерифицированный карбоксил" означает низший алкил-O-CO-, арил-низший алкил-O-CO- или H2N-CO-арил-низший алкил-O-CO-.
"Галоген" означает группу галогена, конкретно включающую фтор, хлор, бром, иод, предпочтительно - фтор, хлор.
"Необязательно замещенный" означает "незамещенный" или "замещенный одинаковыми или отличными друг от друга 1-5 заместителями".
В зависимости от типа заместителей в таком соединении, соединение (I) по настоящему изобретению может содержать оптические изомеры (оптически активные изомеры, диастереомеры) или геометрические изомеры. Соответственно, соединение (I) по настоящему изобретению включает смеси или выделенные соединения таких оптических изомеров или геометрических изомеров.
Соединение (I) по настоящему изобретению может образовывать фармацевтически приемлемые соли, такие как кислотно-аддитивные соли или соли с основаниями. Например, соли включают кислотно-аддитивные соли, образованные с неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота; или органической кислотой, такой как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота, глутаминовая кислота; а также соли с неорганическим основанием, такие как соли натрия, калия, магния, кальция, алюминия; или органическим основанием, таким как метиламин, этиламин, моноэтаноламин, диэтаноламин, триэтаноламин, циклогексиламин, лизин, орнитин. Кроме того, соединение (I) или его фармацевтически приемлемая соль по настоящему изобретению могут образовывать гидраты, сольваты с этанолом или т.п. и кристаллические полиморфы.
Кроме того, соединение (I) по настоящему изобретению включает все соединения, способные метаболизоваться в условиях организма с преобразованием в соединение (I) или его фармацевтически приемлемую соль по настоящему изобретению, которые представляют собой пролекарства. Группа, образующая пролекарства соединения (I) по настоящему изобретению, включает соединения, описанные в Prog. Med., 5:2157-2161 (1985), и соединения, описанные в “PHARMACEUTICAL RESEARCH and DEVELOPMENT”, VOLUME 7 Drug Design, pp. 163-198 by Hirokawa Publishing, 1990. Конкретно, это группы, способные преобразовываться в первичный амин или вторичный амин или HO-, HO-CO- или т.п. в настоящем изобретении через гидролиз или сольволиз или в физиологических условиях. Пролекарства HO- представляют собой, например, необязательно замещенный низший алкил-CO-O-, необязательно замещенный арил-CO-O-, необязательно замещенный гетероарил-CO-O-, RO-CO-необязательно замещенный низший алкилен-CO-O- (R означает H- или низший алкил, то же относится и к указанному ниже), RO-CO-необязательно замещенный низший алкенилен-CO-O-, RO-CO-низший алкилен-O-низший алкилен-CO-O-, RO-CO-CO-O-, ROS(=O)2-необязательно замещенный низший алкенилен-CO-O-, фталидил-O-, 5-метил-1,3-диоксолен-2-он-4-ил-метилокси.
"Частое мочеиспускание", как этот термин используется в настоящем описании, означает состояние, при котором частота мочеиспускания увеличивается по сравнению с нормальными пределами. "Недержание мочи" означает непроизвольное мочеиспускание, которое представляет социальную и гигиеническую проблему.
"Повышенная активность мочевого пузыря", как этот термин используется в настоящем описании, означает синдром, диагностируемый как субъективный симптом, такой как частое мочеиспускание или позывы на мочеиспускание (Neurourology and Urodynamics, USA, 2002, Vol. 21, pp. 167-178). Патогенная причина включает, например, невропатию (например, вызванную нейрогенным мочевым пузырем, церебральным инфарктом), непроходимость нижних мочевых путей (например, доброкачественная гипертрофия предстательной железы) и старение; и в качестве патогенного механизма, общего для указанных состояний, гиперактивность капсаицин-чувствительных афферентных нейронов.
Повышенную активность мочевого пузыря можно лечить путем облегчения состояния частого мочеиспускания, недержания мочи и позывов на мочеиспускание. Это очевидно, например, из того факта, что введение антихолестеринергического средства, оксибутинин гидрохлорида (Japan Standard Product Classification Number 87259; by Aventis Pharma) пациенту, страдающему повышенной активностью мочевого пузыря, при дозе от 2 до 3 мг/введение и три раза в день может облегчить состояние частого мочеиспускания, недержания мочи и позывов на мочеиспускание, и такое введение поэтому является эффективным для лечения повышенной активности мочевого пузыря.
Наличие эффекта лечения частого мочеиспускания и недержания мочи и/или эффекта лечения повышенной активности мочевого пузыря может быть подтверждено способами, известными специалистам в данной области, или способами, являющимися модификацией таких способов. Например, в данной области техники часто используют патогенную модель, индуцируемую введением от 50 до 200 мг циклофосфаамида (CPA) крысам, морским свинкам, собакам или т.п. (Ozawa et al., The Journal of Urology, Vol. 162, pp. 2211-2216, 1999; Boucher et al., The Journal of Urology, Vol. 164, pp. 203-208, 2000). Это патогенная модель, которая сопутствует геморрагическому циститу, и поскольку капсаицин-чувствительный афферентный нейрон участвует в патогенном механизме частого мочеиспускания, можно считать, что эта модель может быть подходящей патологической моделью для различных типов повышенной активности мочевого пузыря, включая невропатический мочевой пузырь (Carlo Alberto Maggi et al., Journal of the Autonomic Nervous System, Vol. 38, pp. 201-208, 1992). Состояние частого мочеиспускания можно подтвердить уменьшением эффективной емкости мочевого пузыря. Патологической модели животного эффективную дозу фармацевтической композиции вводят перорально, внутрибрюшинно или внутривенно, один или несколько раз; и когда эффективная емкость мочевого пузыря животного увеличивается, тогда может быть подтвержден эффект фармацевтической композиции для лечения частого мочеиспускания и недержания мочи и/или для лечения повышенной активности мочевого пузыря.
"Боль", как этот термин используется в настоящем описании, представляет собой родовой термин для невропатической боли, ноцисептивной боли и воспалительной боли, из которых "невропатическая боль" означает боль, вызванную нарушением функции периферической или центральной нервной системы, и включает диабетическую невропатическую боль, боль при раке, невралгию тройничного нерва, фантомную боль, постгерпетическую боль и таламическую боль. Основной клинический симптом невропатической боли включает боль как будто сжимающую, боль типа жжения, гиперальгезию и аллодинию.
Нестероидные противовоспалительные лекарственные средства и наркотические аналгетики, такие как морфин, являются обычными аналгетиками, известными как слабо эффективные при невропатической боли. В медицинских учреждениях для облегчения боли используют антиэпилептическое средство, такое как габапентин, и средство против аритмии, такое как мексилетин, но их аналгетическая активность не является достаточной.
Наличие эффекта лечения невропатической боли может быть подтверждено способами, известными специалистам в данной области, или способами, являющимися модификацией таких способов. Например, используя крыс L5/L6 с лигированным спинным нервом, что осуществляют в соответствии с частично модифицированным способом Kim и Chung (Pain, Vol. 50, pp. 355-363, 1992), оценивают облегчающий эффект соединения, выражающийся в значительном снижении порога ответа на тактильную стимуляцию (аллодиния), и на основании этого может быть подтвержден эффект испытываемого соединения для лечения невропатической боли.
Соединение по настоящему изобретению включает соединения, эффективные для лечения частого мочеиспускания и недержания мочи, а также повышенной активности мочевого пузыря; соединения, эффективные для лечения боли, в частности, невропатической боли; и соединения, эффективные в обоих вышеуказанных случаях.
Способы получения
Соединение и его фармацевтически приемлемую соль по настоящему изобретению можно получить с применением различных известных способов получения, используя характеристики, исходя из основной структуры соединения или типа заместителей в этом соединении.
В зависимости от типа функциональной группы в соединении, часто может быть эффективным, с точки зрения технологии его получения, замещение функциональной группы подходящей защитной группой (способной легко преобразовываться в функциональную группу) на стадии его исходного вещества или промежуточного соединения. Функциональная група включает, например, аминогруппу, гидроксильную группу и карбоксильную группу; и их защитные группы представляют собой, например, группы, описанные в “Protective groups in Organic Synthesis (2nd Ed)” by Greene & Wuts. Такие группы можно подходящим образом выбрать и использовать в зависимости от реакционных условий.
В данном способе защитные группы удаляют, если это необходимо, после их введения и осуществления реакции с получением желаемого соединения.
Типичные способы получения соединений по настоящему изобретению и их промежуточных соединений описаны ниже.
(Аббревиатуры, используемые ниже в описании, означают следующее:
ДМФА: N,N-диметилформамид,
ДМСО: диметилсульфоксид,
ТГФ: тетрагидрофуран,
ТФУ: трифторуксусную кислоту,
Tol: толуол,
EtOAc: этилацетат,
DCE: 1,2-дихлорэтан,
TEA: триэтиламин)
Типичные способы получении соединений по настоящему изобретению описаны ниже, однако, настоящее изобретение не органичивается ими.
В случае, когда в определенном положении соединения по настоящему изобретению существует подобный заместитель, а не такой, как указан в формуле реакции в способе получения соединения, соединение, охватываемое настоящим изобретением, легко можно получить через модификацию заместителя.
Способ получения 1 (Образование карбамата):
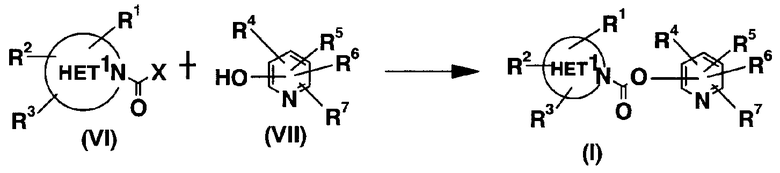
где X представляет собой удаляемую группу, выгодную для реакции, то же относится и к указанному ниже.
Эта реакция представляет собой этерификацию кетонового производного общей формулы (VI) и соответствующего для данной реакции количества гидроксипиридинового производного общей формулы (VII), в растворителе, инертном к реакции, при перемешивании, при охлаждении, или при комнатной температуре, или при нагревании. Удаляемая группа X включает, например, атом галогена, низшую алкоксигруппу, феноксигруппу, имидазолильную группу. Инертный растворитель включает, например, ДМФА, диметилацетамид, ТГФ, диоксан, диметоксиэтан, диэтоксиэтан, бензол, Tol, ксилол и смеси таких растворителей. Для промотирования реакции, предпочтительно, к реакционной смеси добавляют основание (например, натрий, гидрид натрия, метоксид натрия, этоксид натрия).
Способ получения 2 (Образование карбамата):
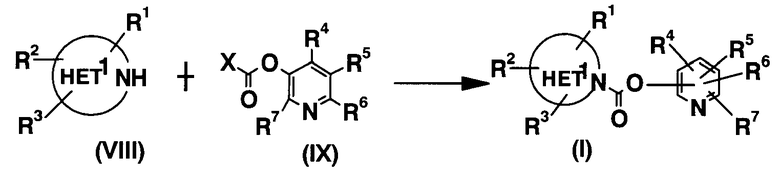
Эту реакцию осуществляют путем перемешивания азотсодержащего гетероциклического соединения общей формулы (VIII) и соответствующего для данной реакции количества пиридинового производного общей формулы (IX) в растворителе, инертном к реакции, при охлаждении, или при комнатной температуре, или при нагревании. Для промотирования реакции, предпочтительно, к реакционной смеси добавляют основание (например, натрий, гидрид натрия, метоксид натрия, этоксид натрия, TEA, пиридин).
Способ получения 3 (Гидролиз):
Соединение (I-3) по настоящему изобретению, содержащее карбоксильную группу, можно получить через гидролиз соответствующего соединения, содержащего этерифицированную карбоксильную группу, например, в соответствии с удалением защитной группы, описанным в “Protective groups in Organic Synthesis (2nd Ed)” by Greene & Wuts.
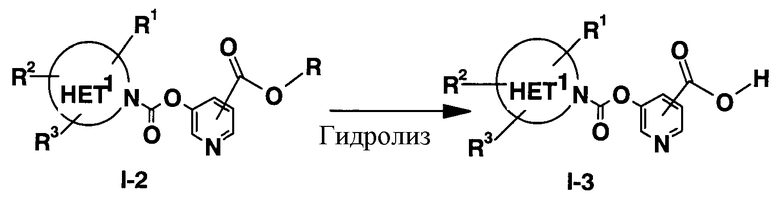
где ROCO- означает этерифицированную карбоксильную группу, то же относится и к указанному ниже.
Способ получения 4 (Амидирование):

Соединение (I-3) или соединение, где R1 представляет собой карбоновую кислоту, может взаимодействовать с амином, а соединение, где R1 представляет собой амин, может взаимодействовать с карбоновой кислотой, таким образом, могут быть получены различные амидные соединения. Когда азот-содержащее гетероциклическое соединение представляет собой пиперидин, тогда его можно подвергнуть взаимодействию с карбоновой кислотой или сульфоновой кислотой или их реакционно-способным производным с получением различных типов амидных соединений. Реакцию можно осуществлять в присутствии агента конденсации (например, дициклогексилкарбодиимида (DCC), диизопропилкарбодиимида (DIPC), 1-этил-3-(3-диметиламинопропил)карбодиимида (WSC), 1,1'-карбонилбис-1H-имидазола (CDI)) и, необязательно, кроме того в присутствии добавки (например, N-гидроксисукцинимида (HONSu) 1-гидроксибензотриазола (HOBt), диметиламинопиридина (DMAP)). Реакционно-способное производное карбоновой кислоты или сульфоновой кислоты включает галогенангидриды кислот, ангидриды кислот, активные сложные эфиры. Реакцию также можно осуществлять, например, в соответствии со способами, описанными в “Jikken Kagaku koza (Courses in Experimental Chemistry, 4th Ed)”, Vol. 22, edited by the Chemical Society of Japan, Maruzen, 1992.
Способ получения 5 (Реакция сочетания):
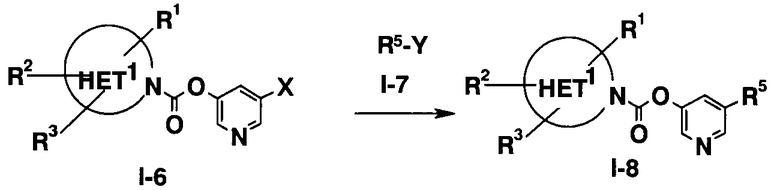
где X представляет собой галоген или -O-SO2CF3, и Y представляет собой -B(OH)2, диалкилбор, диалкоксибор или триалкилолово. X может представлять собой -B(OH)2, диалкилбор, диалкоксибор или триалкилолово, и Y может представляет собой галоген или -O-SO2CF3.
Два ароматических кольца или, в частности, комбинацию соединения (I-6) и соединения (I-7), подвергают взаимодействию, предпочтительно, в присутствии катализатора на основе переходного металла и подходящей добавки, таким образом, получая биарильное соединение (I-8). Типичные способы получения описаны в “Jikken Kagaku koza (Courses in Experimental Chemistry, 4th Ed)”, Vol. 25, Organic Synthesis VII, pp. 353-366, pp. 396-427, 1991 (Maruzen). Предпочтительный катализатор на основе переходного металла для использования в настоящем изобретении включает различные комплексы палладия, такие как тетракис(трифенилфосфин)палладий, и различные комплексы никеля, такие как дибромбис(трифенилфосфин)никель. Также предпочтительная для использования в настоящем изобретении добавка включает трифенилфосфин, карбонат натрия, цинк; и их соответственно выбирают в засисимости от способа, в котором их используют. Как правило, реакцию осуществляют в растворителе при комнатной температуре или при нагревании. Помимо описанной реакции, также предпочтительно использовать реакцию образования биарильной структуры, например, реакцию галогенированного арильного соединения с арильным реагентом Гриньяра в присутствии подходящего катализатора на основе переходного металла.
Способы получения исходных соединений
Исходные соединения, используемые для получения соединений по настоящему изобретению, могут представлять собой известные соединения или могут быть получены путем необязательной обработки известных соединений в соответствии с указанными выше способами получения или в соответствии со способами, хорошо известными специалистам в данной области (J. March, ADVANCED ORGANIC CHEMISTRY (John WILEY & SONS (1992)) (например, ацилирования, алкилирования, образования мочевины, окисления, восстановления (предпочтительно, COMPREHENSIVE ORGANIC SYNTHESIS 8 REDUCTION (Pergamon Press) (1991)), галогенирования).
Способ получения (i):
Реакция Мицунобу:
Исходное соединение (X) можно получить через реакцию Мицунобу спиртов общей формулы (XI) и (XII). Эту реакцию осуществляют путем перемешивания соединений (XI) и (XII) в присутствии эквивалентного или избыточного количества трифенилфосфина и диэтилазодикарбоксилата, в инертном растворителе, как в способе получения 1, в условиях от охлаждения до нагревания.
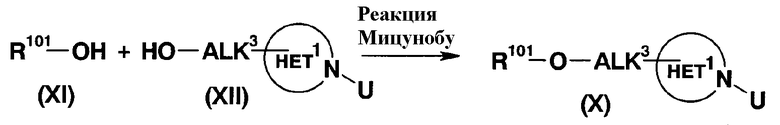
где:
U представляет собой амино-защитную группу,
ALK3 представляет собой ALK1, необязательно замещенный HO, и то же относится и к указанному ниже.
Способ получения (ii):
Реакция замещения:
Эта реакция представляет собой алкилирование. Первичный амин, вторичный амин, спирт, тиол, первичный амид или вторичный амид подвергают взаимодействию с соответствующим для данной реакции количеством соединения, содержащего удаляемую группу, в растворителе, инертном к реакции, в эквивалентом соотношении этих двух компонентов или в таком соотношении, когда какой-либо из них находится в избыточном количестве, при перемешивании, в условиях от охлаждения до нагревания. Бывают случаи, когда реакцию выгодно осуществлять в присутствии основания (например, неорганического основания, такого как карбонат калия, карбонат натрия, карбонат цезия; органического основания, такого как TEA, диизопропилэтиламин; алкоксида металла, такого как трет-бутоксид калия, трет-бутоксид натрия; гидрида натрия, гидрида лития) и добавки (иодида тетра-н-бутиламмония, иодида калия, иодида натрия) для ровного промотирования реакции. Растворитель, инертный к реакции, включает, например, дихлорметан, DCE, хлороформ, бензол, Tol, ксилол, простой эфир, ТГФ, диоксан, EtOAc, этанол, метанол, 2-пропанол, ацетонитрил, ДМФА, N,N-диметилацетамид, N-метилпирролидон, диметилимидазолидинон, ДМСО, ацетон, метилэтилкетон, воду, а также гомогенные или гетерогенные смеси таких растворителей. Растворитель можно соответствующим образом выбрать в зависимости от различных используемых реакционных условий.

где:
Q представляет собой O, S или NH,
Z представляет собой удаляемую группу (например, Cl, Br, I или OMs).
Способ получения (iii):
Этот способ получения включает взаимодействие альдегида или кетона общей формулы (XVI) с реагентом Виттига (Wittig) или с реагентом Хорнера-Эммонса общей формулы (XVII) с получением, таким образом, соединения (XVIII).
Эту реакцию осуществляют в присутствии эквивалентного или избыточного количества основания (например, органического основания, такого как TEA, диизопропилэтиламин; неорганического основания, такого как карбонат калия, карбонат натрия, карбонат цезия), путем перемешивания соединения (XVI) и соединения (XVII) в указанном выше инертном растворителе, в эквивалентном соотношении этих двух компонентов или в таком соотношении, когда какой-либо из них находится в избыточном количестве, в условиях от охлаждения до нагревания. Бывают случаи, когда выгодно добавление в систему добавки (например, иодида тетра-н-бутиламмония, иодида калия) для ровного промотирования реакции.
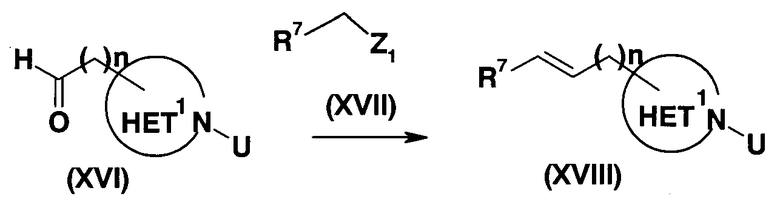
Z1 представляет собой группу, используемую в реагенте Виттига или в реагенте Хорнера-Эммонса (например, соль фосфония или диэфир фосфорной кислоты),
n имеет значение 0 или 1.
[1] Способ скрининга по настоящему изобретению:
Жирно-кислотная амид-гидролаза (далее может быть указана как FAAH) включает ферменты, обладающие активностью гидролизации анандамида, пальмитоилэтаноламида, олеамида и/или 2-арахидоноилглицерина, и поскольку они идентифицированы как вещества одного молекулярного вида, их можно выделить из любых видов, например, млекопитающих, таких как человек (GenBank Accession Number NM_001441), мышь (GenBank Accession Number NM__010173), крыса (GenBank Accession Number NM__024132), свинья (GenBank Accession Number AB027132), кролик, овца, курица, собака, кошка, хомяк, белка, медведь, олень, обезьяна. Кроме того, она не ограничена природным полипептидом и может включать искусственно полученные мутанты.
Относительно (a) полипептида, который содержит аминокислотную последовательность полной аминокислотной последовательности, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, или часть такой аминокислотной последовательности, не содержащую, по меньшей мере, содержащего трансмембранную область амино-концевого участка этой последовательности, и который может гидролизовать анандамид, пальмитоилэтаноламид, олеамид и/или 2-арахидоноилглицерин;
(b) полипептида, который содержит аминокислотную последовательность полной аминокислотной последовательности, выделенной из аминокислотной последовательности, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, путем делеции, замещения и/или вставки от 1 до 10, предпочтительно от 1 до 7, более предпочтительно от 1 до 5 содержащихся в ней аминокислот, или часть такой аминокислотной последовательности, не содержащую, по меньшей мере, содержащего трансмембранную область амино-концевого участка этой последовательности, и который может гидролизовать анандамид, пальмитоилэтаноламид, олеамид и/или 2-арахидоноил глицерин;
(c) полипептида, который содержит аминокислотную последовательность, имеющую гомологию, по меньшей мере, 70%, предпочтительно, по меньшей мере, 80%, более предпочтительно, по меньшей мере, 90%, наиболее предпочтительно, по меньшей мере, 95% с аминокислотной последовательностью, представленной SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6 или SEQ ID NO:8, и который может гидролизовать анандамид, пальмитоилэтаноламид, олеамид и/или 2-арахидоноил глицерин;
(d) полипептида, который содержит аминокислотную последовательность полной аминокислотной последовательности, кодируемой полинуклеотидом, представленным SEQ ID NO:1, SEQ ID NO:3, SEQ ID NO:5 или SEQ ID NO:7, или полинуклеотидом, способным гибридизоваться с ее комплементарной последовательностью в жестких условиях или ее частью, не содержащей, по меньшей мере, содержащего трансмембранную область амино-концевого участка этой последовательности, и который может гидролизовать анандамид, пальмитоилэтаноламид, олеамид и/или 2-арахидоноилглицерин;
указанный выше (a)-(d) имеет родовое название "функциональная FAAH".
Указанный выше "содержащий трансмембранную область амино-концевой участок", как указано в настоящем описании, означает амино-концевой участок, который включает внеклеточную область по амино-концу и трансмембранную область, находящуюся в клеточной мембране, с образованием сэндвической конструкции между внеклеточной областью и внутриклеточной областью. Существование и местоположение трансмембранной области может быть предсказано на основании аминокислотной последовательности белка с использованием программы предопределения мембранной структуры белка, TMpred, PSORT, SOSUI. Конкретно, "содержащий трансмембранную область амино-концевой участок" представляет собой, например, участок от 1-й до 30-й в SEQ ID NO:2 и участок от 1-й до 29-й в SEQ ID NO:6. Известно, что полипептид, представленный аминокислотами от 30-й до 579-й в SEQ ID NO:6, с исключением участка от 1-й до 29-й в SEQ ID NO:6, также обладает такой же ферментативной активностью, как и полипептид, у которого не исключен этот участок (Matthew et al., Biochemistry, Vol. 37, pp. 15177-15178, 1998).
"Гомология", как указано в настоящем описании, означает идентичность величин, полученных путем использования параметров, подготовленных в недостаточном количестве через поиск при помощи программы Clustal V (Higgins & Sharp, Gene, Vol. 73, pp. 237-244, 1998; Thompson et al., Nucleic Acid Res., Vol. 22, pp. 4673-7680, 1994). Эти параметры следующие:
Попарно выстроенные параметры,
K tuple 1
Gap Penalty 3
Window 5
Diagonals Saved 5.
Указанные выше "жесткие условия" для гибридизации, как указано в настоящем описании, означают условия, не вызывающие какого-либо неспецифического связывания. Конкретно, например, гибридизацию осуществляют в растворе, включающем 50% формамида, 5×SSC (0,75 M NaCl, 0,075 M цитрата натрия, pH 7), 5Ч раствор Denhardt (0,1% Ficoll 400, 0,1% поливинилпирролидона, 0,1% BSA), ДНК модифицированной спермы лосося (50 г/мл), 0,1% SDS и 10% сульфата декстрана, в температурных условиях от 37 до 42°C, в течение от около 12 до 18 часов, и затем, необязательно, после предварительной промывки осуществляют промывку промывочным раствором (0,2×SSC, 0,1% SDS) в температурных условиях от 50 до 60°C.
Указанный выше "гидролиз анандамида, пальмитоилэтаноламида, олеамида и/или 2-арахидоноилглицерина", как указано в настоящем описании, конкретно означает, что в соответствии со способом, описанным в Примерах 1-4, анандамид (N-арахидоноилэтаноламин) разлагается на арахидоновую кислоту и этаноламин; пальмитоилэтаноламид (N-пальмитоил этаноламин) - на пальмитиновую кислоту и этаноламин; олеамид (цис-9,10-октадеценамид) - на олеиновую кислоту и аммиак, а 2-арахидоноилглицерин - на арахидоновую кислоту и глицерин, в результате гидролиза в буфере, имеющем pH от 7 до 9, при температуре от 4°C до 37°C, в течение времени от 30 минут до 90 минут.
Способ скрининга по настоящему изобретению включает способ скрининга для определения лекарственного средства для лечения частого мочеиспускания и недержания мочи, лекарственного средства для лечения повышенной активности мочевого пузыря и/или лекарственного средства для лечения боли, включающий (1) стадию контактирования испытываемого вещества с FAAH или функциональной FAAH, (2) стадию анализа этого вещеаства на активность FAAH или функциональной FAAH и (3) стадию отбора вещества, которое ингибирует активность FAAH или функциональной FAAH.
(1) Стадия контактирования испытываемого вещества с FAAH или функциональной FAAH:
Для контактирования испытываемого вещества с FAAH или функциональной FAAH испытываемое вещество можно добавлять к любому из следующих:
a) клетке или ткани, экспрессирующей FAAH или функциональную FAAH,
b) трансформанту, трансформированному вектором экспрессии, содержащим полинуклеотид, который кодирует FAAH или функциональную FAAH,
c) лизату или гомогенату a) или b),
d) очищенному продукту FAAH или функциональной FAAH, очищенному из c) и инкубированному в течение заданного периода времени; или
e) тканевому гомогенату или крови испытываемого животного, которому вводили испытываемое вещество.
a) Клетка или ткань, экспрессирующая FAAH или функциональную FAAH:
Конкретно, клетка, экспрессирующая FAAH или функциональную FAAH, включает нейроны, глиальные клетки, эпителиальные клетки, эндотелиальные клетки, лимфоциты, макрофаги, тромбоциты, тучные клетки, моноциты, дендритные клетки, гепатоциты, клетки почек, энтероциты, панкреатические клетки, клетки мочи, плацентальные клетки, клетки мочевого пузыря, клетки предстательной железы, кератинизирующие клетки и мышечные клетки. При условии, что они экспрессируют FAAH или функциональную FAAH, эти клетки могут быть выделены из любых видов; и, например, в данном изобретении используют клетки, выделенные у млекопитающих, таких как человек, мышь, крыса, свинья, кролик, овца, курица, собака, кошка, хомяк, белка, медведь, олень, обезьяна.
Для используемых клеток установлены клеточные линии; и также можно использовать клетки, отшелушенные или выделенные из тканей животных. Установленные клеточные линии, используемые в настоящем изобретении, включают клетки 5673 клеточной линии рака эпителия мочевого пузыря человека, клетки PC-3 клеточной линии рака предстательной железы человека, клетки RBL-2H3 базофильной клеточной линии лейкоза крысы, клетки N18TG2 клеточной линии нейробластомы крысы, клетки C6 клеточной линии глиомы крысы, клетки J774 клеточной линии макрофагов крысы, клетки PC-12 выделенной из мозгового вещества надпочечников клеточной линии феохромоцитомы крысы, клетки U937 клеточной линии моноцитов человека, клетки MFC-7 клеточной линии рака молочной железы человека, клетки EFM-19 клеточной линии рака молочной железы человека, клетки CaCo-2 клеточной линии рака толстой кишки человека (эти клеточные линии доступны от American Type Culture Collection (ATCC)), клетки HaCaT клеточной линии эпидермальных кератиноцитов человека и клетки CHP100 клеточной линии нейробластомы человека. Предпочтительными являются клетки 5673 клеточной линии рака эпителия мочевого пузыря человека и клетки RBL-2H3 базофильной клеточной линии лейкоза крысы.
Ткань, экспрессирующая FAAH или функциональную FAAH, конкретно включает головной мозг, мочевой пузырь, предстательную железу, почку, печень, яички, мышцы, сосуды, поджелудочную железу, пищеварительный тракт, легкое, матку, плаценту, кожу, лимфоцит, тромбоцит, макрофаг, моноцит, тучную клетку и предстательную железу. Предпочтительно, используют головной мозг, печень и моноцит. При условии, что они экспрессируют FAAH или функциональную FAAH, эти ткани могут быть выделены из любых видов. Например, можно использовать ткани, выделенные у млекопитающих, таких как человек, мышь, крыса, свинья, кролик, овца, курица, собака, кошка, хомяк, белка, медведь, олень, обезьяна.
Для определения, экспрессирует или нет клетка или ткань FAAH или функциональную FAAH, клеточный или тканевый экстракт можно использовать и анализировать при помощи анализа вестерн-блоттинга с использованием антитела, способного к детекции представляющего интерес полипептида, или путем ПЦР (полимеразной цепной реакции) с использованием праймеров, способных к специфической детекции полинуклеотида, который кодирует представляющий интерес полипептид. Кроме того, лизат или гомогенат клетки или ткани подвергают взаимодействию с субстратом, таким как анандамид, пальмитоилэтаноламид, олеамид и/или 2-арахидоноилглицерин, в буфере, имеющем pH от 7 до 9, при температуре 4°C до 37°C, в течение времени от 30 минут до 90 минут, после чего в системе определяют, гидролизован или нет субстрат для предназначенного определения.
b) Трансформант, трансформированный при помощи вектора экспрессии, содержащего полинуклеотид, который кодирует FAAH или функциональную FAAH:
Полинуклеотид, который кодирует FAAH или функциональную FAAH, может быть выделен из библиотеки кДНК путем скрининга при помощи ПЦР или гибридизации с использованием праймеров и зонда, сконструированных и синтезированных на основании информации об известных аминокислотных последовательностях и последовательностях оснований.
Фрагмент, который содержит выделенный полинуклеотид, вставляют в подходящий вектор экспрессии, и его можно трансфицировать в клетку хозяина эукариота или прокариота; и в клетке хозяина полипептид, кодируемый трансфицированным полинуклеотидом, может, таким образом, экспрессироваться. Вектор экспрессии может быть любым известным вектором, выбранным в засисимости от клетки хозяина, для которой также можно использовать векторную плазмиду, подходящим образом выбранную в засисимости от клетки хозяина и содержащую вставленную в нее подходящую промоторную и связанную с экспрессией фенотипа последовательность. Также можно использовать вектор экспрессии со специфической последовательностью, вставленной в него таким образом, чтобы полипептид, кодируемый вставленным полинуклеотидом, мог экспрессироваться как слитый с глутатион-S-трансферазой (GST) или с меткой, такой как Flag или His. В случае, когда одна клетка одновременно трансформируется некоторыми другими типами полинуклеотидов, тогда используемый один вектор экспрессии может быть сконструирован таким образом, чтобы он включал такие разные типы полинуклеотидов, или такие полинуклеотиды могут присутствовать отдельно в разных векторах экспрессии. Альтернативно, можно получить клетку с хромосомной ДНК с такого типа строением, и ее можно использовать.
Вектор экспрессии с желаемым полинуклеотидом, вставленным в него, может быть введен в клетку хозяина в соответствии со способом DEAE-декстран (Luthman et al., Nucleic Acids Res., Vol. 11, pp. 1295-1308, 1983), способом со-осаждения фосфата кальция-ДНК (Graham et al., Virology, Vol. 52, pp. 456-457, 1973), способом с использованием коммерчески доступного реагента для трансфекции, Lipofectamine 2000 (от Invitrogen) или FeGENE 6 (от Roche Molecular Biochemicals) или способом электропорации (Neumann et al., EMBO J., Vol. 1, pp. 841-845, 1982) для желаемой трансформации. В случае, когда в качестве клетки хозяина используют E. coli, компетентную клетку E. coli получают в присутствии одновременно CaCl2, MgCl2 или RbCl, в соответствии со способом Hanahan (Hanahan et al., Mol. Biol. Vol. 166, pp. 557-580, 1983), и вектор экспрессии с вставленным в него желаемым полинуклеотидом вводят в нее для трансформации клетки.
c) Лизат или гомогенат a) или b):
Клеточный гомогенат можно получить путем промывки клетки несколько раз при помощи буфера с последующей гомогенизацией с использованием гомогенизатора Potter-Elvehjem или т.п., с получением, таким образом, однородного раствора. Тканевый гомогенат можно получить добавлением буфера, охлажденного льдом, к ткани в 5-10-кратном объеме в расчете на массу ткани, с последующей гомогенизацией с использованием гомогенизатора Potter-Elvehjem во льду с получением, таким образом, однородного раствора, а затем подвергая его дальнейшей ультразвуковой гомогенизации в течение нескольких секунд. Буфер может представлять собой буфер Tris (50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA) или буфер Hepes (1 мМ EDTA, 100 мМ NaCl, 12,5 мМ Hepes, pH 8,0). Например, в данном случае применимы способы испытаний Примера 265 и Примера 266. Лизат E. coli, трансформированной вектором экспрессии, который содержит полинуклеотид, кодирующий FAAH или функциональную FAAH, можно получить путем сбора клеток E. coli при помощи центрифугирования, а затем растворения их в буфере для лизиса (например, 20 мМ Трис-HCl (pH 8,0), 500 мМ NaCl, 10% глицерина, 0,2 мМ EDTA, 0,5 мМ DTT, 10 мМ имидазола, 1% н-октил-β-D-глюкопиранозид).
d) Очищенный продукт FAAH или функциональной FAAH, очищенный из c):
Очищенный продукт FAAH или функциональной FAAH можно получить из a) клетки или ткани, экспрессирующей FAAH или функциональную FAAH, или b) лизата или гомогената трансформанта, трансформированного вектором экспрессии, который содержит полинуклеотид, кодирующий FAAH или функциональную FAAH, в соответствии с обычным способом очистки, таким как аффинная хроматография, электрохроматография, гель-фильтрационная хроматография, ионообменная хроматография или хроматография разделения.
Конкретно, очистка представляет собой следующее: Клетку или ткань, экспрессирующую FAAH или функциональную FAAH, гомогенизируют в растворителе, содержащем сахарозу, а затем подвергают центрифугированию и сверхвысокоскоростному центрифугированию с получением фракции микросом, затем ее растворяют в растворителе, содержащем Triton-X, и снова центрифугируют для удаления осадка и полученный белок-лизат обрабатывают в системе жидкостной экспресс-хроматографии белков (FPLC) (от Pharmacia) (Ueda et al., J. Biol. Chem., Vol. 270, pp. 23813-23827, 1995).
Альтернативно, E. coli, трансформированную таким образом, чтобы она экспрессировала слитую с His меткой FAAH или функциональную FAAH, растворяют в буфере для лизиса, затем обрабатывают ультразвуком и центрифугируют (например, при 10000 × g в течение 20 минут) и полученную надосадочную жидкость смешивают со смолой, предварительно уравновешенной с буфером для лизиса и имеющей высокое сродство с His меткой, при низкой температуре, в течение, по меньшей мере, 12 часов. Затем смолу промывают, и слитую с His меткой FAAH или функциональную FAAH высвобождают из смолы с получением ее очищенного продукта.
Для контактирования испытываемого вещества с указанной выше клеткой или тканью или клеточным или тканевым лизатом или гомогенатом, полученным указанным выше способом, или очищенным продуктом FAAH или функциональной FAAH можно применить способ инкубации в течение заданного периода времени с добавлением к ним, или без добавления, испытываемого вещества. Конкретно, испытываемое вещество растворяют в растворе, выбранном соответствующим образом в засисимости от растворимости в нем этого вещества, таком как дистиллированная вода или диметилсульфоксид (ДМСО), и добавляют к указанной выше клетке или ткани или клеточному или тканевому лизату или гомогенату или очищенному продукту FAAH или функциональной FAAH с получением концентраций от 0,003 нМ до 10 мкМ. Клеточный или тканевый образец инкубируют в CO2 инкубаторе при температуре 37°C в течение от 30 до 60 минут; а остальные вещества при температуре от 4°C до 37°C в течение от 30 до 90 минут, достигая, таким образом, желаемый контакт с испытываемым веществом.
e) Тканевый гомогенат или кровь испытываемого животного, которому вводили испытываемое вещество:
Когда испытываемое вещество вводят испытываемому животному, тогда испытываемое вещество может контактировать с FAAH или функциональной FAAH, существующей в ткани или крови испытываемого животного. Испытываемое животное включает, например, млекопитающих, таких как мышь, крыса, собака. Испытываемое вещество можно вводить испытываемому животному следующим образом: Испытываемое вещество суспендируют или растворяют в носителе, который обычно используют в соответствии со свойствами испытываемого вещества, таком как физиологический водный раствор, раствор диметилформамида или 10% раствор метилцеллюлозы, и его можно вводить испытываемому животному перорально, подкожно, внутрибрюшинно или внутривенно. После введения ткань извлекают и эту ткань гомогенизируют в соответствии со способом, описанным выше в c), таким образом, получая тканевый гомогенат. Конкретно, например, от 1 до 3 мг/кг испытываемого вещества перорально вводят 9-недельным крысам и извлеченные у них через 30 минут после этого головной мозг, печень или моноциты гомогенизируют с получением тканевого гомогената. Альтернативно, от 0,3 до 3 мг/кг испытываемого вещества внутривенно вводят 13-18-месячным собакам и извлеченные у них через 30 минут после этого головной мозг, печень или моноциты гомогенизируют с получением тканевого гомогената. Более конкретно, например, тканевый гомогенат может быть получен в соответствии со способом, описанным в Примере 267. Кровь можно брать из сердца или нисходящей аорты испытываемого животного, которому вводили испытываемое вещество.
(2) Стадия анализа изменения активности FAAH или функциональной FAAH:
Для анализа изменения активности FAAH или функциональной FAAH можно использовать способ определения изменения ферментативной активности FAAH или функциональной FAAH, основанный на наличии или отсутствии контакта с испытываемым веществом. Ферментативную активность FAAH или функциональной FAAH можно определить путем контактирования FAAH или функциональной FAAH с субстратом в течение заданного периода времени и измерения количества разложившегося продукта субстрата. Альтернативно, это также можно определить путем измерения количества эндоканнабиноида, который является эндогенным субстратом для FAAH, содержащейся в ткани или крови испытываемого животного.
Для анализа зависимого от испытываемого вещества изменения ферментативной активности FAAH или функциональной FAAH субстрат подвергают контакту с FAAH или функциональной FAAH в течение заданного периода времени в присутствии или в отсутствие испытываемого вещества и получают отношение количества разложившегося продукта субстрата в присутствии испытываемого вещества к количеству разложившегося продукта субстрата в отсутствие испытываемого вещества для предполагаемого анализа.
Альтернативно, FAAH или функциональную FAAH, которую предварительно подвергали контактированию с испытываемым веществом, и FAAH или функциональную FAAH, которая не контактировала с испытываемым веществом, по отдельности подвергают контакту с субстратом в течение заданного периода времени и получают отношение количества разложившегося продукта субстрата в результате действия FAAH или функциональной FAAH, которую предварительно подвергали контактированию с испытываемым веществом, к количеству разложившегося продукта субстрата в результате действия FAAH или функциональной FAAH, которая не контактировала с испытываемым веществом, посредством чего можно определить зависимое от испытываемого вещества изменение ферментативной активности.
Кроме того, зависимое от испытываемого вещества изменение ферментативной активности также можно определить путем измерения количества эндоканнабиноида в ткани или крови испытываемого животного до и после введения испытываемого вещества испытываемому животному с последующим получением отношения количества эндоканнабиноида после введения испытываемого вещества к количеству эндоканнабиноида до введения испытываемого вещества; или путем измерения количества эндоканнабиноида в ткани или крови испытываемого животного, которому вводили или которому не вводили испытываемое вещество с последующим получением отношения количества эндоканнабиноида в ткани или крови испытываемого животного, которому вводили испытываемое вещество, к количеству эндоканнабиноида в ткани или крови испытываемого животного, которому не вводили испытываемое вещество, посредством чего можно определить зависимое от испытываемого вещества изменение ферментативной активности.
FAAH и функциональную FAAH можно подвергнуть контактированию с субстратом в условиях, указанных ниже, в соответствии с состоянием FAAH или функциональной FAAH.
Для контактирования FAAH или функциональной FAAH, экспрессируемой в клетке или ткани a) или b) в (1) выше, с субстратом можно использовать способ добавления субстрата к культивируемым клеткам или ткани в буфере, имеющем pH от 7 до 9, и взаимодействия их в CO2 инкубаторе при температуре 37°C или комнатной температуре, предпочтительно, в течение от 30 до 60 минут. Реакцию можно остановить путем перенесения клеток или ткани на лед для их быстрого охлаждения, после чего ингибитор FAAH можно подвергнуть контактированию с ними при его достаточной концентрации; или путем добавления 1:1 (по объему) раствора хлороформа и метанола. Клетки или ткань лизируют или гомогенизируют в соответствии со способом, описанным в (1)c) выше, таким образом, получая их лизат или гомогенат.
Для контактирования FAAH или функциональной FAAH в лизате или гомогенате клетки или ткани в c) или e) из (1) выше с субстратом можно использовать способ добавления субстрата к лизату или гомогенату, который был разбавлен буфером, имеющим pH от 7 до 9, таким образом, чтобы концентрация белка, предпочтительно, составляла от 10 до 100 мкг/мл, и взаимодействия их в температурных условиях от 4°C до 37°C. Время реакции можно подходящим образом определить в засисимости от условий, таких как количество добавленного фермента, количество добавленного субстрата и температуры реакции. Например, когда осуществляют взаимодействие при комнатной температуре, время реакции может быть от 30 до 90 минут.
Для контактирования очищенной FAAH или функциональной FAAH в (1)d) выше с субстратом можно использовать способ добавления субстрата к лизату или гомогенату, который был разбавлен буфером, имеющим pH от 7 до 9, и взаимодействия их в температурных условиях от 4°C до 37°C. Время реакции можно соответствующим образом определить в засисимости от условий, таких как количество добавленного фермента, количество добавленного субстрата и температуры реакции. Например, когда осуществляют взаимодействие при комнатной температуре, время реакции может быть от 30 до 90 минут.
Для измерения количества разложившегося продукта субстрата непрореагировавший субстрат и разложившийся продукт в ферментном реакционном растворе отделяют друг от друга, и можно измерять количество разложившегося продукта. Для отделения непрореагировавшего субстрата от разложившегося продукта можно использовать водорастворимость разложившегося продукта, этаноламин. Например, 1:1 (по объему) раствор хлороформа и метанола добавляют к реакционному раствору фермента в количестве в 2 раза больше, чем количество реакционного раствора, с последующим перемешиванием, а затем центрифугированием, в результате чего содержащий разложившийся продукт верхний слой, водно/этанольный слой можно отделить от непрореагировавшего субстрата, содержащегося в нижнем слое - хлороформном слое. Альтернативно, систему можно смешать с жидким агентом, представляющим собой сцинтилляционную жидкость, который не абсорбирует воду, при этом жирорастворимый непрореагировавший радиоактивный субстрат может быть помещен в такую жидкость, и разложившийся продукт можно, таким образом, отделить от непрореагировавшего субстрата. Еще в одном альтернативном способе непрореагировавший субстрат можно отделить от разложившегося продукта при помощи тонкослойной хроматографии или высоко-эффективной жидкостной хроматографии.
В случае, когда используют 3H- или 14C-меченный субстрат или смесь меченого субстрата и немеченного субстрата, количество разложившегося продукта или количество непрореагировавшего субстрата можно измерить при помощи жидкостного сцинтилляционного счетчика, или его можно получить в виде рентгеновского латентного снимка на фотографической пластине и измерить при помощи считывающего устройства для фотографических пластин.
В случае, когда используют немеченный субстрат, поглощение в системе при 205 нМ можно отслеживать методом высоко-эффективной жидкостной хроматографии и, таким образом, можно измерить количество разложившегося продукта или количество непрореагировавшего субстрата (Lang et al., Anal. Biochem., Vol. 238, pp. 40-45, 1996).
Когда измерено количество непрореагировавшего субстрата, тогда количество непрореагировавшего субстрата можно вычесть из количества субстрата, добавленного до реакции, и, таким образом, можно получить количество разложившегося продукта. Альтернативно, количество разложившегося продукта субстрата, измеренное в буфере, не содержащем FAAH или функциональную FAAH, в качестве контроля, можно вычесть из количества разложившегося продукта субстрата с FAAH или функциональной FAAH, таким образом, можно получить чистое количество разложившегося продукта субстрата с FAAH или функциональной FAAH.
Количество эндоканнабиноида в тканевом гомогенате можно измерить, например, путем гомогенизации образца ткани с 2:1:1 (по объему) раствором хлороформа, метанола и 50 мМ Tris (pH 8,0), а затем измерения количества эндоканнабиноида, содержащегося в органическом слое (хлороформный слой) при помощи жидкостной хроматографии/масс-спектрометрии с использованием изотопа (Cravatt et al., Proc., Natl. Acad. Sci. USA, Vol. 98, pp. 9371-9376, 2001).
Количество эндоканнабиноида в крови можно измерить, например, следующим образом: Плазму отделяют от образца крови и белок плазмы удаляют при помощи центрифугирования вместе с таким же количеством добавленного ацетона (-20°C). Ацетон выпаривают при помощи струи азота, прилагаемой к системе, добавляют 1:2 (по объему) раствор метанола и хлороформа и измеряют количество эндоканнабиноида, содержащегося в органическом слое (хлороформный слой) при помощи жидкостной хроматографии/масс-спектрометрии с использованием изотопа (Giuffraida et al., Eur. J. Pharmacol., Vol. 408, pp. 161-168, 2000).
(3) Стадия выбора вещества, которое ингибирует активность FAAH или функциональной FAAH:
Вещество, которое ингибирует активность FAAH или функциональной FAAH, можно выбрать следующим образом: Испытываемое вещество подвергают контактированию с FAAH или функциональной FAAH, результат сравнивают со случаем, когда они не контактировали с испытываемым веществом, и можно выбрать вещество, которое снижает количество разложившегося продукта субстрата.
Конкретно, испытываемое вещество подвергают контактированию с FAAH или функциональной FAAH и результат сравнивают со случаем, когда они не контактировали с испытываемым веществом. При этом, вещество, в присутствии которого количество разложившегося продукта фермента снижается, предпочтительно, до 1/2 или меньше, можно скринировать на лекарственное средство для лечения частого мочеиспускания и недержания мочи, лекарственное средство для лечения повышенной активности мочевого пузыря и/или лекарственное средство для лечения боли.
Альтернативно, испытываемое вещество в различных концентрациях подвергают контактированию с FAAH или функциональной FAAH; и на основании количества разложившегося продукта субстрата, не контактировавшего с испытываемым веществом, взятого за 100%, получают относительную величину (%) разложившегося продукта субстрата, контактировавшего с испытываемым веществом, используемым в различных концентрациях; или на основании количества разложившегося продукта субстрата, не контактировавшего с испытываемым веществом, взятого за 100%, и на основании количества разложившегося продукта субстрата в случае, когда известный ингибитор FAAH, имеющий достаточную концентрацию, контактирует с FAAH или функциональной FAAH в течение достаточного периода времени, взятого за 0%, получают относительную величину (%) разложившегося продукта субстрата, контактировавшего с испытываемым веществом, используемым в различных концентрациях. С использованием кривой ингибирования на графике, где относительная величина (%) разложившегося продукта субстрата представлена по вертикальной оси, а концентрация испытываемого вещества по горизонатльной оси, рассчитывают концентрацию испытываемого вещества, которая дает относительную величину, 50%, разложившегося продукта субстрата (значение ИК50); и вещество, у которого значение ИК50 составляет, предпочтительно, не более 1 мкМ, более предпочтительно, не более 100 нМ, скринируют на лекарственное средство для лечения частого мочеиспускания и недержания мочи, лекарственное средство для лечения повышенной активности мочевого пузыря и/или лекарственное средство для лечения боли. Например, см. испытания в Примере 438 - Примере 440.
Еще в одном альтернативном варианте испытываемое вещество вводят испытываемому животному и количество эндоканнабиноида в ткани или крови животного сравнивают до и после введения испытываемого вещества; и вещество, которое увеличивает количество, предпочтительно, до 1,5 раз, может быть выбрано в качестве вещества, которое ингибирует активность FAAH или функциональной FAAH, а именно, это вещество можно скринировать на лекарственное средство для лечения частого мочеиспускания и недержания мочи, лекарственное средство для лечения повышенной активности мочевого пузыря и/или лекарственное средство для лечения боли.
[2] Испытываемое вещество:
Специальным образом не определенное, испытываемое вещество для использования в способе скрининга по настоящему изобретению включает, например, коммерчески доступные продукты (включая пептиды), различные известные соединения, зарегистрированные в Chemical File (включая пептиды), группы соединений, полученных в соответствии с методами комбинаторной химии (Terrett et al., J. Steele. Tetrahedron, Vol. 51, pp. 8135-8173, 1995), культуральные супернатанты, выделенные из микроорганизмов, полученные из растений или морских организмов природные компоненты, экстракты тканей животных, а также соединения (включая пептиды), полученные путем химической или биологической модификации соединений (включая пептиды), выбранных в соответствии со способом скрининга по настоящему изобретению.
[3] Фармацевтическая композиция для лечения частого мочеиспускания и недержания мочи, для лечения повышенной активности мочевого пузыря и/или для лечения боли:
В качестве активного ингредиента фармацевтической композиции по настоящему изобретению используют вещество, которое ингибирует активность FAAH или функциональной FAAH, где ингибирующее вещество может быть выбрано, например, в соответствии со способом скрининга по настоящему изобретению.
Фармацевтическая композиция по настоящему изобретению не ограничивается фармацевтической композицией, которая содержит, в качестве ее активного ингредиента, вещество, полученное в соответствии со способом скрининга по настоящему изобретению, но может включать любую и каждую фармацевтическую композицию для лечения частого мочеиспускания и недержания мочи, для лечения повышенной активности мочевого пузыря и/или для лечения боли, которая содержит, в качестве ее активного ингредиента, вещество для ингибирования активности FAAH или функциональной FAAH; и предпочтительно, она представляет собой фармацевтическую композицию для лечения частого мочеиспускания и недержания мочи, для лечения повышенной активности мочевого пузыря и/или для лечения боли.
Эффект лечения частого мочеиспускания и недержания мочи, эффект лечения повышенной активности мочевого пузыря и/или эффект лечения боли может быть подтвержден, как указано выше.
Композиция, содержащая, в качестве ее активного ингредиента, вещество, которое ингибирует активность FAAH или функциональной FAAH, например, ДНК, белок (включая антитело или фрагмент антитела), пептид или любое другое соединение, может быть получена в виде фармацевтической композиции с использованием фармацевтически приемлемого носителя, эксципиента и/или любой другой добавки, обычно используемой для получения фармацевтических композиций, в зависимости от типа их активного ингредиента.
Введение композиции можно осуществить, например, путем перорального введения в форме таблеток, пилюль, капсул, гранул, тонкодисперсных гранул, порошков или пероральных жидкостей; или парентерального введения путем инъекций, таких как внутривенная, внутримышечная или внутрисуставная инъекции, суппозиториев, чрескожных препаратов или препаратов для введения в слизистую оболочку. В частности, для пептидов, которые перевариваются в желудке, парентеральное введение, такое как внутривенная инъекция, является предпочтительным.
Твердые композиции для перорального введения могут включать смесь, по меньшей мере, одного или нескольких активных ингредиентов и, по меньшей мере, одного инертного разбавителя, например, лактозы, маннита, глюкозы, микрокристаллической целлюлозы, гидроксипропилцеллюлозы, крахмала, поливинилпирролидона или алюминометасиликата магния. Помимо инертных разбавителей, твердые композиции могут содержать другие добавки, что является обычным, например, смазочные вещества, разрыхлители, стабилизаторы, солюбилизирующие вещества или вещества, способствующие солюбилизации. Таблетки и пилюли, необязательно, могут иметь сахарное покрытие или могут иметь гастро- или энтеросолюбильное покрытие.
Жидкая композиция для перорального введения включает, например, эмульсии, растворы, суспензии, сиропы и эликсиры и могут содержать обычные инертные разбавители, например, дистиллированную воду или этанол. Помимо инертных разбавителей, жидкая композиция также может содержать, например, смачивающие вещества, суспендирующие вещества, подсластители, ароматизаторы или антисептики.
Инъекции для парентерального введения включают асептические водные или неводные растворы, суспензии или эмульсии. Водорастворимые растворы или суспензии могут содержать, например, дистиллированную воду для инъекций или физиологический раствор, в качестве разбавителя. Разбавители для не растворимых в воде растворов или суспензий включают, например, пропиленгликоль, полиэтиленгликоль, растительное масло (например, оливковое масло), спирты (например, этанол) или Polysorbate 80. Такие композиции могут, кроме того, содержать смачивающие вещества, эмульгаторы, диспергирующие вещества, стабилизаторы, солюбилизирующие вещества или вещества, способствующие солюбилизации, или антисептики. Такие композиции можно стерилизовать, например, фильтрованием через бактериальный фильтр или путем добавления к ним гермицида или путем облучения. Если желательно, могут быть получены твердые композиции, не содержащие патогенных микроорганизмов, перед применением их растворяли в не содержащей патогенных микроорганизмов воде или любой другой не содержащей патогенных микроорганизмов среде для инъекций.
Дозу композиции соответственно определяют в засисимости от уровня активности активного ингредиента или, конкретно, вещества, полученного в соответствии со способом скрининга по настоящему изобретению, и от симптома, возраста, пола субъекта, которому ее вводят.
Например, при пероральном введении доза, как правило, может составлять от около 0,1 до 100 мг/день, предпочтительно от 0,1 до 50 мг/день для взрослого (масса тела 60 кг). При парентеральном введении доза инъекции может составлять от 0,01 до 50 мг/день, предпочтительно от 0,01 до 10 мг/день.
ПРИМЕРЫ
Настоящее изобретение описывается более подробно со ссылкой на следующие Примеры. Соединения по настоящему изобретению не должны ограничиваться соединениями, описанными в представленных ниже Примерах. Способы получения исходных соединений представлены в Ссылочных Примерах. Некоторые соединения по настоящему изобретению также могут быть исходными соединениями для других; и для удобства способы их получения могут быть представлены в настоящей заявке как Ссылочные Примеры. Химические структурные формулы и физико-химические свойства соединений, полученнных в Ссылочных Примерах, представлены в Таблицах 1-15. Химические структурные формулы соединений, полученных в Примерах, представлены в Таблицах 16-34; и их физико-химические свойства представлены в Таблицах 35-63. Структуры других соединений по настоящему изобретению представлены в Таблицах 65-73. Эти соединения могут быть легко получены в соответствии с указанными выше способами получения или способами, описанными в представленных ниже Ссылочных Примерах и Примерах, или в соответствии со способами, являющимися очевидными для специалистов в данной области, или в соответствии с модификациями таких способов.
Когда используют коммерчески доступные наборы, можно ссылаться на письменные инструкции, прилагаемые к ним.
Используемые в настоящем описании аббревиатуры имеют следующие значения:
Rex: Ссылочный Пример
Ex: Пример
Str: структурная формула
DAT: физико-химические свойства
1H-ЯМР δ(м.д.), растворитель: спектр ядерного магнитного резонанса
В физико-химических данных соединений Примеров:
ДМСО: ДМСО-d6
MS m/z: данные масс-спектрометрии
Com: соединение
NC: циано
Ph: фенил
Me: метил
diMe: диметил
Et: этил
Pr: пропил
iPr: изопропил
Bu: бутил
tBu: трет-бутил
iBu: изобутил
Pen: пентил
Hex: гексил
Hep: гептил
Oct: октил
cPr: циклопропил
cPen: циклопентил
cHex: циклогексил
cHep: циклогептил
cOct: циклооктил
Ac: ацетил
Cl: хлор
diCl: дихлор
CN: циано
F: фтор
diF: дифтор
FPh фторфенил
NCPh: цианофенил
diFPh: дифторфенил
O2N: нитро
MeO: метокси
diMeO: диметокси
Br: бром
diBr: дибром
BrPh: бромфенил
F3C: трифторметил
AcO: ацетокси
MeOCO или COOMe: метоксикарбонил
tBuOCO или COOtBu: трет-бутоксикарбонил
HO: гидрокси
HOPh: гидроксифенил
H2N: амино
PhCONH: бензоиламино
EtCONH: этилкарбониламино
Me2N: диметиламино
Et2N: диэтиламино
BIP2: 2-бифенил
BIP3: 3-бифенил
BIP4: 4-бифенил
BIP5: 5-бифенил
BIP6: 6-бифенил
Thiop2: тиофен-2-ил
Thiop3: тиофен-3-ил
Thiop4: тиофен-4-ил
Thiop5: тиофен-5-ил
PYRR1: пирролидин-1-ил
PYRR2: пирролидин-2-ил
PYRR3: пирролидин-3-ил
PYRR4: пирролидин-4-ил
PYRR5: пирролидин-5-ил
Py2: пиридин-2-ил
Py3: пиридин-3-ил
Py4: пиридин-4-ил
Py5: пиридин-5-ил
IM1: имидазол-1-ил
IM2: имидазол-2-ил
IM3: имидазол-3-ил
IM4: имидазол-4-ил
BenzIM1: бензимидазол-1-ил
BenzIM2: бензимидазол-2-ил
BenzIM3: бензимидазол-3-ил
BenzIM4: бензимидазол-4-ил
BenzIM5: бензимидазол-5-ил
BenzIM6: бензимидазол-6-ил
Pyrazi1: пиразин-1-ил
Pyrazi2: пиразин-2-ил
Pyrazi3: пиразин-3-ил
Pyrazi4: пиразин-4-ил
Pyrazi5: пиразин-5-ил
Pyrazi6: пиразин-6-ил
PIPE1: пиперидин-1-ил
PIPE2: пиперидин-2-ил
PIPE3: пиперидин-3-ил
PIPE4: пиперидин-4-ил
PIPE5: пиперидин-5-ил
PIPE6: пиперидин-6-ил
PIPERA: пиперазин
PIPERA1: пиперазин-1-ил
PIPERA2: пиперазин-2-ил
PIPERA3: пиперазин-3-ил
PIPERA4: пиперазин-4-ил
PIPERA5: пиперазин-5-ил
Pyrazo1: пиразол-1-ил
Pyrazo2: пиразол-2-ил
Pyrazo3: пиразол-3-ил
Pyrazo4: пиразол-4-ил
Pyrazo5: пиразол-5-ил
Mo: морфолин
Mo2: морфолин-2-ил
Mo3: морфолин-3-ил
Mo4: морфолин-4-ил
Mo5: морфолин-5-ил
Azep: азепин
Azep1: азепин-1-ил
Azep2: азепин-2-ил
Azep3: азепин-3-ил
Azep4: азепин-4-ил
Thiaz2: тиазол-2-ил
Thiaz3: тиазол-3-ил
Thiaz4: тиазол-4-ил
Thiaz5: тиазол-5-ил
QUI1: хинолин-1-ил
QUI2: хинолин-2-ил
QUI3: хинолин-3-ил
QUI4: хинолин-4-ил
QUI5: хинолин-5-ил
QUI6: хинолин-6-ил
QUI7: хинолин-7-ил
QUI8: хинолин-8-ил
ISOQUI2: изохинолин-2-ил
ISOQUI3: изохинолин-3-ил
ISOQUI4: изохинолин-4-ил
ISOQUI5: изохинолин-5-ил
ISOQUI6: изохинолин-6-ил
ISOQUI7: изохинолин-7-ил
ISOQUI8: изохинолин-8-ил
NAPH1: нафталин-1-ил
NAPH2: нафталин-2-ил
NAPH3: нафталин-3-ил
NAPH4: нафталин-4-ил
NAPH5: нафталин-5-ил
TEA: триэтиламин
Sal: дополнительная соль
HCl: гидрохлорид
oxal: оксалат
fum: фумарат
p-tol: g-толуолсульфонат
Ссылочный Пример 1:
Раствор в ТГФ (10 мл), содержащий фенол (471 мг) и диэтилазодикарбоксилат (2,83 г, 40% Tol раствор), добавляли по каплям к раствору в ТГФ (15 мл), содержащему трет-бутил 4-(гидроксиметил)пиперидин-1-карбоксилат (1,57 г) и трифенилфосфин (1,70 г), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. К реакционному раствору добавляли воду (40 мл) с последующей экстракцией при помощи EtOAc. Органический слой промывали водным раствором 1 M гидроксида натрия и насыщенным солевым раствором, в указанном порядке, и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=4:1 (об./об.)) с получением бесцветного масла (1,14 г). Полученное соединение растворяли в EtOAc, добавляли раствор 4 M хлористый водород/EtOAc (9,6 мл) с последующим перемешиванием при комнатной температуре в течение 5 часов с получением гидрохлорида 4-(феноксиметил)пиперидина (680 мг) в виде бесцветного порошка.
Таким же способом, как в Ссылочном Примере 1, получали соединения Ссылочных Примеров 2 до 27.
Ссылочный Пример 28:
Воду (10 мл), карбонат натрия (4,76 г) и тетракистрифенилфосфинпалладий (866 мг) добавляли в указанном порядке к раствору в диметоксиэтане (50 мл), содержащему 3-бромбензамид (3,0 г) и (3-гидроксифенил)борную кислоту (2,27 г), с последующим перемешиванием при температуре 60°C в течение 24 часов. Реакционный раствор охлаждали, разбавляли EtOAc и органический слой промывали водой и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: EtOAc) с получением бледно-желтого порошка (2,74 г). С использованием полученного соединения и такого же способа, как в Ссылочном Примере 1, получали соединение Ссылочного примера 28.
Ссылочный Пример 29:
Раствор в ТГФ (80 мл), содержащий 4-(бензилокси)фенол (8,0 г) и диэтилазодикарбоксилат (26 мл, 40% Tol раствор), добавляли по каплям к раствору в ТГФ (80 мл), содержащему трет-бутил 4-гидроксипиперидин-1-карбоксилат (12 г) и трифенилфосфин (16 г), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. К реакционному раствору добавляли воду (40 мл) с последующей экстракцией при помощи EtOAc. Органический слой промывали водным раствором 1 M гидроксида натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=8:1 (об./об.)) с получением бесцветного масла (12,4 г).
10% Палладий на углероде (каталитическое количество) добавляли к этанольному (100 мл) раствору, содержащему полученное соединение (5,18 г), с последующим перемешиванием в атмосфере газообразного водорода при комнатной температуре при нормальном давлении в течение 16 часов. Катализатор удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении с получением бледно-коричневого твердого вещества (4,0 г).
1-(Бромметил)-3-фторбензол (2,5 мл) и карбонат калия (2,8 г) добавляли к раствору в ацетонитриле (100 мл), содержащему полученное соединение (4,0 г) с последующим нагреванием при температуре 80°C в течение 22 часов. Твердые вещества удаляли фильтрованием, полученный фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=8:1 (об./об.)) с получением бесцветного твердого вещества (5,15 г).
Полученное соединение (5,15 г) растворяли в EtOAc (20 мл), добавляли раствор 4М хлористый водород/EtOAc (20 мл) с последующим перемешиванием при комнатной температуре в течение 5 часов. Затем растворитель выпаривали при пониженном давлении. Остаток растворяли в воде, нейтрализовали водным раствором 1 M гидроксида натрия и образовавшееся твердое вещество сушили с получением 4-{4-[(3-фторбензил)окси]фенокси}пиперидина (3,70 г).
Таким же способом, как в Ссылочном Примере 29, получали соединения Ссылочных Примеров 30-36.
Ссылочный Пример 37:
Диэтилазодикарбоксилат (11 мл, 40% Tol раствор) добавляли по каплям к раствору в ТГФ (30 мл), содержащему трет-бутил 4-гидроксипиперидин-1-карбоксилат (4,6 г), трифенилфосфин (6,1 г) и 6-хлор-2-пиридинол (2,0 г) при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. К реакционному раствору добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали водным раствором 1М гидроксида натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=10:1 (об./об.)) с получением трет-бутил 4-[(6-хлор-2-пиридинил)окси]-1-пиперидинкарбоксилата (3,8 г).
(3-Фторфенил)метанол (220 мг) и трет-бутоксид калия (200 мг) добавляли к раствору в ДМФА (5 мл), содержащему трет-бутил 4-[(6-хлор-2-пиридинил)окси]-1-пиперидинкарбоксилат (500 мг), с последующим нагреванием при температуре 100°C в течение 30 минут. Затем к смеси добавляли (3-фторфенил)метанол (220 мг) и трет-бутоксид калия (200 мг) с последующим нагреванием при температуре 110°C в течение 30 минут. К реакционному раствору добавляли воду, а затем экстрагировали при помощи EtOAc. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=10:1 (об./об.)) с получением белого твердого вещества (420 мг).
Полученное соединение (400 мг) растворяли в EtOAc (5 мл), добавляли раствор 4М хлористый водород/EtOAc (3 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида 2-[(3-фторбензил)окси]-6-(4-пиперидинокси)пиридина (310 мг).
Таким же способом, как в Ссылочном Примере 37, получали соединение Ссылочного примера 38.
Ссылочный Пример 39:
Воду (4 мл), карбонат натрия (610 мг) и тетракистрифенилфосфинпалладий (110 мг) добавляли в указанном порядке к раствору в Tol (10 мл), содержащему трет-бутил 4-[(6-хлор-2-пиридинил)окси]-1-пиперидинкарбоксилат (500 мг) и [3-(аминокарбонил)фенил]борную кислоту (320 мг), с последующим нагреванием в течение ночи при температуре 100°C. Реакционный раствор охлаждали и разбавляли EtOAc. Органический слой промывали водным раствором безводного гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:2 (об./об.)) с получением бледно-желтого порошка (590 мг).
Полученное соединение (590 мг) растворяли в EtOAc (5 мл) и добавляли раствор 4М хлористый водород/EtOAc (5 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида 3-[6-(4-пиперидинилокси)-2-пиридинил]бензамида (440 мг).
Ссылочный Пример 40:
TEA (4,6 мл) и метансульфонилхлорид (2,0 мл) добавляли по каплям к раствору в метиленхлориде (80 мл), содержащему трет-бутил 4-(2-гидроксиэтил)пиперидин-1-карбоксилат (5,0 г), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 3 часов. К реакционному раствору добавляли водный раствор гидрокарбоната натрия и метанол с последующим перемешиванием при комнатной температуре в течение 30 минут. Смесь экстрагировали хлороформом и органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=10:1 (об./об.)) с получением бесцветного твердого вещества (6,1 г).
Гидрид натрия (541 мг, 60% в масле) добавляли к раствору в ДМФА (80 мл), содержащему полученное соединение (2,0 г) и фенилпропанол (1,3 г), при температуре 0°C с последующим нагреванием при температуре 100°C в течение 20 часов. Реакционный раствор охлаждали, добавляли воду с последующей экстракцией при помощи EtOAc. Смесь промывали водным раствором 1 M хлористоводородной кислоты, насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=20:1 (об./об.)) с получением желтого масла (1,96 г).
Полученное соединение (1,96 г) растворяли в EtOAc (5 мл) и добавляли раствор 4М хлористый водород/EtOAc (10 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов. Образовавшееся твердое вещество собирали фильтрованием и сушили с получением гидрохлорида 4-[2-(3-фенилпропокси)этил]пиперидин (1,55 г).
Ссылочный Пример 41:
TEA (2,30 мл) и метансульфонилхлорид (1,22 мл) добавляли по каплям к раствору в ТГФ (40 мл), содержащему трет-бутил 4-гидроксипиперидин-1-карбоксилат (3,02 г), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли EtOAc (50 мл) и воду (50 мл). Органический слой промывали водным 5% раствором лимонной кислоты, насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении с получением бледно-оранжевого масла. Полученное масло растворяли в DMA (25 мл) и карбонат цезия (5,38 г) и к смеси добавляли 4-сульфанилфенол (1,89 г) с последующим нагреванием при температуре 50°C в течение 2 часов. Реакционный раствор охлаждали, добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали водным раствором 1 M хлористоводородной кислоты и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=4:1 (об./об.)) с получением трет-бутил 4-[(4-гидроксифенил)сульфанил]пиперидин-1-карбоксилата (3,40 г) в виде бесцветного порошка.
1-(Бромметил)-3-фторбензол (0,436 мл) и карбонат калия (670 мг) добавляли к раствору в ацетонитриле (15 мл), содержащему трет-бутил 4-[(4-гидроксифенил)сульфанил]пиперидин-1-карбоксилат (1,00 г), с последующим нагреванием при температуре 80°C в течение 2 часов. Реакционный раствор охлаждали, добавляли к нему насыщенный солевой раствор с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом натрия, растворитель выпаривали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=8:1 (об./об.)) с получением трет-бутил 4-({4-[(3-фторбензил)окси]фенил}сульфанил)пиперидин-1-карбоксилата (1,50 г) в виде бесцветного порошка.
Трет-бутил 4-({4-[(3-фторбензил)окси]фенил}-сульфанил)пиперидин-1-карбоксилат (501 мг) растворяли в EtOAc (5 мл) и добавляли раствор 4М хлористый водород/EtOAc (3 мл) с последующим перемешиванием при комнатной температуре в течение 3 часов. Затем растворитель выпаривали при пониженном давлении. Остаток растворяли в воде, нейтрализовали водным раствором 1М гидроксида натрия с последующей экстракцией хлороформом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении с получением 4-({4-[(3-фторбензил)окси]фенил}сульфанил)пиперидина (328 мг).
Таким же способом, как в Ссылочном Примере 41, получали соединение Ссылочного Примера 42.
Ссылочный Пример 43:
mCPBA (1,64 г) добавляли к раствору в хлороформе (20 мл), содержащему трет-бутил 4-({4-[(3-фторбензил)окси]фенил}сульфанил)пиперидин-1-карбоксилат (1,50 г), полученный в способе Ссылочного примера 41, при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 17 часов. Твердое вещество удаляли фильтрованием и к фильтрату добавляли 10% водный раствор сульфата натрия с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=2:1 (об./об.)) с получением бесцветного порошка (1,58 г). Полученный порошок (1,56 г) растворяли в EtOAc (10 мл), добавляли раствор 4М хлористый водород/EtOAc (8 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов. Затем твердое вещество собирали фильтрованием и промывали EtOAc с получением гидрохлорида 4-({4-[(3-фторбензил)окси]фенил}сульфонил)пиперидина (1,13 г) в виде бесцветного порошка.
Таким же способом, как в Ссылочном Примере 43, получали соединения Ссылочных Примеров 44-46.
Ссылочный Пример 47:
Раствор в ТГФ (5 мл) трет-бутил 4-[(4-гидроксифенил)сульфанил]пиперидин-1-карбоксилата (495 мг), полученного в способе Ссылочного Примера 41, и диэтилазодикарбоксилат (1,04 г, 40% Tol раствор) добавляли по каплям к раствору в ТГФ (5 мл), содержащему циклогексилметанол и трифенилфосфин (629 мг), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. К реакционному раствору добавляли воду (40 мл) с последующей экстракцией при помощи EtOAc. Органический слой промывали водным раствором 1М гидроксида натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=9:1 (об./об.)) с получением трет-бутил 4-{[4-(циклогексилметокси)фенил]сульфонил}пиперидин-1-карбоксилата (744 мг) в виде бледно-желтого масла.
Полученный трет-бутил 4-{[4-(циклогексилметокси)фенил]сульфонил}пиперидин-1-карбоксилат (635 мг) растворяли в EtOAc (7 мл) и добавляли раствор 4М хлористый водород/EtOAc (3,6 мл) с последующим перемешиванием при комнатной температуре в течение 6 часов. Твердое вещество собирали фильтрованием и промывали EtOAc с получением гидрохлорида 4-{[4-(циклогексилметокси)фенил]-сульфонил}пиперидина (485 мг) в виде бесцветного порошка.
Таким же способом, как в Ссылочном Примере 47, получали соединение Ссылочного Примера 48.
Ссылочный Пример 49:
Гидрид натрия (355 мг, 60% в масле) и бензил бромид (1,0 мл) добавляли к раствору в ТГФ (40 мл), содержащему трет-бутил 4-гидроксипиперидин-1-карбоксилат (1,5 г), с последующим нагреванием при температуре 60°C в течение 13 часов. Реакционный раствор охлаждали, добавляли воду с последующей экстракцией при помощи EtOAc. Смесь промывали водным раствором 1 M хлористоводородной кислоты, насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=10:1 (об./об.)) с получением бесцветного масла (1,91 г).
Полученное соединение (1,8 г) растворяли в EtOAc (5 мл) и добавляли раствор 4М хлористый водород/EtOAc (15 мл) с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционный раствор разбавляли простым изопропиловым эфиром и образовавшееся твердое вещество собирали фильтрованием и сушили с получением гидрохлорида 4-(бензилокси)пиперидина (1,32 г).
Таким же способом, как в Ссылочном Примере 49, получали соединения Ссылочных Примеров 50-53.
Ссылочный Пример 54:
Диэтилазодикарбоксилат (2,6 мл, 40% Tol раствор) добавляли по каплям к раствору в ТГФ (10 мл), содержащему (3-фторфенил)метанол (730 мг), трифенилфосфин (1,5 г) и 6-хлор-3-пиридинол (500 мг), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. Реакционный раствор разбавляли при помощи EtOAc. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=8:1 (об./об.)) с получением белого твердого вещества (810 мг).
Трет-бутил 4-гидроксипиперидин-1-карбоксилат (1,0 г) и трет-бутоксид калия (570 мг) добавляли к раствору в ДМФА (10 мл), содержащему полученное белое твердое вещество (800 мг), с последующим нагреванием при температуре 130°C в течение 1 часа. Затем к смеси добавляли трет-бутоксид калия (400 мг), а затем продолжали нагревание при температуре 130°C в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры, разбавляли при помощи EtOAc, промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=7:1 (об./об.)) с получением белого твердого вещества (350 мг).
Полученное соединение (345 мг) растворяли в EtOAc (3 мл) и добавляли раствор 4М хлористый водород/EtOAc (2 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида 6-[(3-фторбензил)окси]-2-(4-пиперидинокси)пиридина (260 мг).
Ссылочный Пример 55:
[1-(Трет-бутоксикарбонил)пиперидин-4-ил]уксусную кислоту (0,60 г) растворяли в диметилформамиде (12 мл) и к смеси добавляли 1-[3-(диметиламино)пропил]-3-этилкарбодиимид гидрохлорид (0,89 г), 1-гидроксибензотриазол (0,50 г) и бензиламин (0,40 г) с последующим перемешиванием при комнатной температуре в течение 15 часов. К реакционному раствору добавляли воду и перемешивали в течение 1 часа. Затем к смеси добавляли раствор гидрокарбоната натрия с последующей экстракцией при помощи EtOAc. Органический слой промывали раствором 0,5 M хлористоводородной кислоты и насыщенным солевым раствором, в указанном порядке. Органический слой сушили над безводным сульфатом магния, растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:2 (об./об.)) с получением бесцветного порошка (0,69 г).
Полученное соединение (0,69 г) растворяли в EtOAc (10 мл) и добавляли раствор 4М хлористый водород/EtOAc (2,2 мл) с последующим перемешиванием при комнатной температуре в течение 20 часов. Реакционный раствор концентрировали до сухого твердого вещества с получением гидрохлорида N-бензил-2-пиперидин-4-илацетамида (0,62 г).
Ссылочный Пример 56:
Фосфорную кислоту (7 мл) и дифосфорпентоксид (14 г) нагревали при температуре 150°C в течение 30 минут, к смеси добавляли N-метилбензол-1,2-диамин (1,3 г) и гидрохлорид 4-пиперидин-4-илбутановой кислоты (1,5 г) с последующим нагреванием при температуре 120°C в течение 3 часов. Реакционный раствор выливали в воду, нейтрализовали водным раствором гидроксида натрия и затем экстрагировали хлороформом. Органический слой сушили над безводным сульфатом магния, растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол:водный аммиак=10:1:0,1 (об./об./об.)) с получением 1-метил-2-(3-пиперидин-4-илпропил)-1H-бензимидазола (1,61 г).
Ссылочный Пример 57 и Ссылочный Пример 58:
трет-Бутоксид калия (1,72 г) добавляли к раствору в ТГФ (30 мл), содержащему [4-(метоксикарбонил)бензил](трифенил)фосфонийбромид (7,51 г), при температуре 0°C с последующим перемешиванием в течение 1 часа. К реакционному раствору добавляли по каплям раствор в ТГФ (20 мл), содержащий трет-бутил 4-формилпиперидин-1-карбоксилат (Beilstein Registry No. 7704210, 2,96 г), при температуре 0°C с последующим перемешиванием в течение 14 часов. К реакционному раствору добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=9:1 (об./об.)) с получением желтого масла (3,77 г).
Полученное соединение (3,75 г) растворяли в метаноле (20 мл) и ТГФ (10 мл) и добавляли водный раствор 1 M гидроксида натрия (16,3 мл) с последующим перемешиванием при температуре 50°C в течение 4 часов. Реакционный раствор охлаждали и растворитель выпаривали при пониженном давлении. Смесь подкисляли добавлением раствора 1 M хлористоводородной кислоты и осажденное твердое вещество собирали фильтрованием и промывали водой с получением бледно-коричневого порошка (2,82 г).
Хлорид аммония (2,26 г), гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (3,24 г), 1-гидроксибензотриазол (1,14 г) и TEA (5,88 мл) добавляли к раствору, содержащему полученное соединение (2,80 г), в ДМФА (30 мл) с последующим перемешиванием при комнатной температуре в течение 32 часов. К реакционному раствору добавляли воду и осажденное твердое вещество собирали фильтрованием и промывали водой с получением бледно-коричневого порошка (2,61 г).
Полученное соединение (2,58 г) растворяли в EtOAc (15 мл) и добавляли раствор 4М хлористый водород/EtOAc (15 мл) с последующим перемешиванием при комнатной температуре в течение 8 часов. Образовавшееся твердое вещество собирали фильтрованием, промывали EtOAc и сушили с получением гидрохлорида 4-[(E)-2-пиперидин-4-илвинил]бензамида (1,92 г) (Ссылочный Пример 57).
10% Палладий на углероде (каталитическое количество) добавляли к раствору, содержащему гидрохлорид 4-[(E)-2-пиперидин-4-илвинил]бензамида (800 мг), в метаноле (15 мл)/воде (5 мл) с последующим перемешиванием в атмосфере газообразного водорода при комнатной температуре при нормальном давлении в течение 4 часов. Катализатор удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении. Полученное твердое вещество перекристаллизовывали из этанола/ацетонитрила с получением гидрохлорида 4-(2-пиперидин-4-илэтил)бензамида (451 мг) (Ссылочный Пример 58).
Ссылочный Пример 59:
Триацетоксиборогидрид натрия (2,2 г) добавляли к раствору в дихлорметане (30 мл), содержащему трет-бутил 4-(4-аминофенокси)-1-пиперидинкарбоксилат (2,0 г, Beilstein Registry No. 9262581), циклогексанкарбальдегид (770 мг) и уксусную кислоту (1,25 г), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционному раствору добавляли насыщеннй водный раствор гидрокарбоната натрия с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и полученное твердое вещество перекристаллизовывали из EtOAc/гексана с получением бледно-коричневых кристаллов (2,0 г).
Триацетоксиборогидрид натрия (1,1 г) добавляли к раствору в дихлорметане (20 мл), содержащему полученное кристаллическое вещество (970 мг), водный 37% раствор формальдегида (0,94 мл) и уксусную кислоту (0,75 г), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционному раствору добавляли насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и полученное масло растворяли в EtOAc (15 мл). Добавляли раствор 4М хлористый водород/EtOAc (5 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида N-(циклогексилметил)-N-метил-4-(4-пиперидинилокси)анилина (820 мг).
Ссылочный Пример 60:
В атмосфере потока аргона трис(дибензилиденацетон)дипалладий (95 мг) добавляли к раствору в Tol (10 мл), содержащему бензил 3-иодфениловый простой эфир (1,1 г), трет-бутил 1-пиперазинкарбоксилат (640 мг), трет-бутоксид натрия (500 мг) и 2-бифенилил(дициклогексил)фосфин (70 мг), с последующим нагреванием при температуре 80°C в течение 1 часа. Реакционный раствор охлаждали, разбавляли при помощи EtOAc и органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=5:1 (об./об.)) с получением коричневого твердого вещества (950 мг).
Полученное твердое вещество (940 мг) растворяли в EtOAc (5 мл) и добавляли раствор 4М хлористый водород/EtOAc (5 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением дигидрохлорида 1-[3-(бензилокси)фенил]пиперазина (840 мг).
Ссылочный Пример 61:
Диэтилазодикарбоксилат (4,8 мл, 40% Tol раствор) добавляли по каплям к раствору в ТГФ (60 мл), содержащему 4-(бензилокси)-2-хлорфенол (1,7 г, Beilstein Registry No. 6582932), трифенилфосфин (2,8 г) и трет-бутил 4-гидроксипиперидин-1-карбоксилат (2,1 г) при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. Реакционный раствор разбавляли при помощи EtOAc. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=5:1 (об./об.)) с получением белого твердого вещества (2,3 г).
Полученное соединение (1,0 г) растворяли в EtOAc (10 мл) и добавляли раствор 4М хлористый водород/EtOAc (10 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида 4-[4-(бензилокси)-2-хлорфенокси]пиперидина (690 мг).
Ссылочный Пример 62:
Тионилхлорид (10 мл) добавляли по каплям к раствору 4-гидроксибензолсульфоната натрия (1,00 г) в ДМФА (5 мл) с последующим нагреванием при температуре 65°C в течение 3 часов. Реакционный раствор охлаждали и добавляли Tol (10 мл). Растворитель выпаривали при пониженном давлении, добавляли воду с последующей экстракцией хлороформом. Органический слой промывали водным насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением бесцветного твердого вещества (587 мг).
При температуре 0°C, раствор полученного на предыдущей стадии соединения (579 мг) в ацетонитриле (10 мл) добавляли к раствору в ацетонитриле (10 мл), содержащему 1-трет-бутоксикарбонилпиперидин (672 мг) и пиридин (0,58 мл), с последующим перемешиванием при комнатной температуре в течение 2 часов. Растворитель выпаривали при пониженном давлении, добавляли Tol (10 мл) и подвергали азеотропной прегонке. Затем добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением бесцветного твердого вещества (0,41 г).
Карбонат калия (248 мг) добавляли к раствору в ацетонитриле (20 мл), содержащему полученное соединение (0,41 г) и 1-(бромметил)-3-фторбензол (340 мг), с последующим нагреванием при температуре 80°C в течение 3 часов. Твердое вещество удаляли фильтрованием, полученный фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=5:1 (об./об.)) с получением бесцветного твердого вещества (469 мг).
Полученное соединение (460 мг) растворяли в смешанном растворе EtOAc (5 мл) и ТГФ (5 мл) и добавляли раствор 4 M хлористый водород/EtOAc (20 мл) с последующим перемешиванием при температуре 70°C в течение 3 часов. Затем растворитель выпаривали при пониженном давлении. Остаток растворяли в воде, нейтрализовали водным раствором 1М гидроксида натрия и образовавшееся твердое вещество сушили с получением 4-{4-[(3-фторбензил)окси]бензолсульфонил}пиперазина (304 мг).
Ссылочный Пример 63
Диэтилазодикарбоксилат (3,3 мл, 40% Tol раствор) добавляли по каплям к раствору в ТГФ (30 мл), содержащему 4-(бензилокси)-3-хлорфенол (1,2 г, Beilstein Registry No. 5527577), трифенилфосфин (1,9 г) и трет-бутил 4-гидроксипиперидин-1-карбоксилат (1,5 г), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. Реакционный раствор разбавляли при помощи EtOAc и органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=5:1 (об./об.)) с получением белого твердого вещества (1,7 г).
Полученное соединение (1,6 г) растворяли в EtOAc (20 мл) и добавляли раствор 4М хлористый водород/EtOAc (15 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида 4-[4-(бензилокси)-3-хлорфенокси]пиперидина (1,3 г).
Ссылочный Пример 64:
3-Фторбензолсульфонилхлорид (3,2 г) добавляли к раствору в пиридине (30 мл), содержащему трет-бутил 4-(4-аминофенокси)-1-пиперидинкарбоксилат (4,0 г, Beilstein Registry No. 9262581), при температуре 0°C с последующим перемешиванием в течение ночи при комнатной температуре. Растворитель выпаривали при пониженном давлении и разбавляли хлороформом. Органический слой промывали водным 10% раствором лимонной кислоты, водой и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=60:1 (об./об.)) с получением белого твердого вещества (5,3 г).
Карбонат калия (280 мг) и метилиодид (0,28 мл) добавляли к раствору в ацетонитриле (10 мл), содержащему полученное соединение (700 мг), с последующим перемешиванием при температуре 50°C в течение 3 часов. Реакционный раствор разбавляли при помощи EtOAc, органический слой промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=3:1 (об./об.)) с получением бесцветного масла (700 мг).
Полученное масло (700 мг) растворяли в EtOAc (10 мл) и добавляли раствор 4М хлористый водород/EtOAc (5 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида 3-фтор-N-метил-N-[4-(4-пиперидинилокси)фенил]бензолсульфонамида (480 мг).
Ссылочный Пример 65:
Гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (630 мг) и 1-гидроксибензотриазол (440 мг) добавляли к раствору в ДМФА (10 мл), содержащему 1-[(бензилокси)карбонил]-4-(трет-бутоксикарбонил)-2-пиперидинкарбоновую кислоту (1,0 г), с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем добавляли концентрированный водный раствор аммиака (2 мл) с последующим перемешиванием при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду и осажденное твердое вещество собирали фильтрованием, промывали водой и сушили при пониженном давлении с получением бесцветного твердого вещества (870 мг).
Полученное твердое вещество (860 мг) растворяли в EtOAc (10 мл) и добавляли раствор 4М хлористый водород/EtOAc (5 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида бензил 2-(аминокарбонил)-1-пиперазинкарбоксилата (700 мг).
Ссылочный Пример 66:
Пиридин (1,62 мл) и 4-нитрофенилхлоридкарбонат (2,22 г) добавляли к раствору в ацетонитриле (20 мл), содержащему метил 4-(гидроксиметил)бензоат при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционному раствору добавляли 5% водный раствор лимонной кислоты, а затем экстрагировали при помощи EtOAc. Органический слой промывали насыщенным водным раствором гидрокарбоната и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=4:1 (об./об.)) с получением бледно-коричневого порошка (2,39 г).
Трет-бутил пиперидин-1-карбоксилат (1,47 г) добавляли к раствору в ацетонитриле (30 мл), содержащему полученное соединение (2,37 г), с последующим перемешиванием при комнатной температуре в течение 8 часов. Реакционный раствор разбавляли при помощи EtOAc и промывали водным раствором 0,5 M гидроксида натрия. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=2:1 (об./об.)) с получением бесцветного твердого вещества (3,32 г).
К раствору в ТГФ (30 мл), содержащему полученное соединение (3,30 г), добавляли метанол (0,34 мл) и водный раствор 1 M гидроксида натрия (8,52 мл) с последующим перемешиванием при комнатной температуре в течение 26 часов. Растворитель выпаривали при пониженном давлении, к остатку добавляли водный раствор 1 M хлористоводородной кислоты с последующей экстракцией хлороформом. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Полученный остаток перекристаллизовывали из гексана/EtOAc с получением бесцветного порошка (2,37 г).
Хлорид аммония (321 мг), гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (767 мг), 1-гидроксибензотриазол (270 мг) и TEA (0,83 мл) добавляли к раствору в ДМФА (10 мл), содержащему полученное соединение (729 мг), с последующим перемешиванием при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду и осажденное твердое вещество собирали фильтрованием и промывали водой с получением бледно-коричневого порошка (722 мг).
Полученное соединение (700 мг) растворяли в EtOAc (6 мл), добавляли раствор 4М хлористый водород/EtOAc (4,8 мл) с последующим перемешиванием при комнатной температуре в течение 3 часов. Образовавшееся твердое вещество собирали фильтрованием, промывали EtOAc и сушили с получением гидрохлорида 4-(аминокарбонил)бензил пиперидин-1-карбоксилата (541 мг).
Ссылочный Пример 67:
Раствор в ТГФ (5 мл), содержащий метил 4-гидроксибензоат (460 мг) и диэтилазодикарбоксилат (0,71 мл), добавляли по каплям к раствору в ТГФ (5 мл), содержащему циклогексилметанол (510 мг) и трифенилфосфин (1,18 г), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. К реакционному раствору добавляли водный раствор 1 M гидроксида натрия (40 мл) с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=4:1 (об./об.)) с получением бесцветного твердого вещества (930 мг).
Водный раствор 1 M гидроксида натрия (4,4 мл) добавляли к раствору в метаноле (5 мл)/ТГФ (3 мл), содержащему полученное соединение (920 мг), с последующим перемешиванием при температуре 50°C в течение 6 часов. Смесь охлаждали до комнатной температуры и добавляли EtOAc (40 мл) и воду (30 мл) с последующим перемешиванием. Органический слой экстрагировали водным раствором 1М гидроксида натрия. Водные слои объединяли и доводили до pH 1 при помощи концентрированной хлористоводородной кислоты. Затем водный слой экстрагировали хлороформом и затем сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток перекристаллизовывали из гексана/EtOAc с получением 4-(циклогексилметокси)бензойной кислоты (600 мг).
Гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (359 мг) и 1-гидроксибензотриазол (254 мг) добавляли к раствору в ДМФА (10 мл), содержащему полученное соединение (370 мг), и добавляли трет-бутил 1-пиперазинкарбоксилат (350 мг) с последующим перемешиванием при комнатной температуре в течение 12 часов. К реакционному раствору добавляли воду и осажденное твердое вещество собирали фильтрованием, промывали водой и сушили при пониженном давлении с получением бесцветного твердого вещества (610 мг).
Полученное соединение (600 мг) растворяли в EtOAc (6 мл) и добавляли раствор 4М хлористый водород/EtOAc (4 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденное твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида 1-[4-(циклогексилметокси)бензоил]пиперазина (580 мг).
Таким же способом, как в Ссылочном Примере 67, получали соединения Ссылочных Примеров 68-72.
Ссылочный Пример 73:
При -70°C раствор 1,59 M нормального-бутиллития/ТГФ (14,6 мл) добавляли к раствору 2 M диметиламина/ТГФ (11,6 мл) с последующим перемешиванием в течение 10 минут. Смесь нагревали до 0°C и добавляли 3-хлор-5-гидроксипиридин (1,00 г) с последующим перемешиванием в течение ночи при комнатной температуре. Добавляли этанол (15 мл) и растворитель выпаривали при пониженном давлении. К остатку добавляли воду с последующей экстракцией хлороформом. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=10:1 (об./об.)) с получением 3-диметиламино-5-гидроксипиридиан (176 мг).
Ссылочный Пример 74:
Трис-дибензилиденацетонпалладий (21 мг), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (124 мг) и трет-бутоксид натрия (160 мг) добавляли, в указанном порядке, к раствору в Tol (10 мл), содержащему 3-бензилокси-5-бромпиридин (400 мг) и морфолин (158 мг), с последующим нагреванием при температуре 85°C в течение 4 часов. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматограффией на силикагеле (элюент: хлороформ:метанол=20:1 (об./об.)) с получением бесцветного масла (372 мг).
10% Палладий на углероде (каталитическое количество) добавляли к этанольному раствору (20 мл), содержащему полученное соединение (370 мг), и в атмосфере газообразного водорода смесь перемешивали при комнатной температуре и при нормальном давлении в течение 1,5 часов. Катализатор удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении с получением 5-гидрокси-3-морфолинилпиридина (248 мг).
Таким же способом, как в Ссылочном Примере 74, получали соединения Ссылочных Примеров 75 и 76.
Ссылочный Пример 77:
Метоксид натрия (393 мг) добавляли к метанольному раствору (20 мл), содержащему 5-(бензолсульфонилокси)-2-(бромметил)пиридин (Beilstein Registry No. 7430370, 800 мг) с последующим перемешиванием при комнатной температуре в течение 4 часов. К реакционному раствору добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: EtOAc) с получением 6-(метоксиметил)пиридин-3-ола (200 мг).
Ссылочный Пример 78:
TEA (0,21 мл) и ди-трет-бутил дикарбонат (463 мг) добавляли в указанном порядке к раствору в ТГФ (10 мл) 3-бензилокси-5-аминопиридина (250 мг) с последующим нагреванием при температуре 60°C в течение 3 часов. Растворитель выпаривали при пониженном давлении, добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:1 (об./об.)) с получением бесцветного твердого вещества (153 мг).
10% Палладий на углероде (каталитическое количество) добавляли к этанольному раствору (20 мл), содержащему полученное соединение (240 мг), и в атмосфере газообразного водорода смесь перемешивали при комнатной температуре при нормальном давлении в течение 1,5 часов. Катализатор удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении с получением трет-бутил (5-гидроксипиридин-3-ил)карбамата (167 мг).
Ссылочный Пример 79:
При температуре 0°C раствор гидрида натрия (60% масляная смесь, 139 мг) в ТГФ (10 мл) добавляли к раствору в ТГФ (10 мл) метилдиэтилфосфоноацетата (732 мг) с последующим перемешиванием в течение 15 минут. Затем добавляли 5-(бензилокси)никотинальдегид (495 мг) с последующим перемешиванием при комнатной температуре в течение 4 часов. К реакционному раствору добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением бесцветного твердого вещества (680 мг).
10% Палладий на углероде (каталитическое количество) добавляли к этанольному раствору (20 мл), содержащему полученное соединение (330 мг), и в атмосфере газообразного водорода смесь перемешивали при комнатной температуре при нормальном давлении в течение 2 часов. Катализатор удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении с получением метил 3-(5-гидроксипиридин-3-ил)пропаноата (150 мг).
Ссылочный Пример 80:
При температуре -78°C раствор в ТГФ (30 мл) метил 5-(бензилокси)никотината (3,52 г) добавляли к раствору в ТГФ (100 мл) литийалюминийгидрида (1,49 г) с последующим перемешиванием в течение 15 минут и затем перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор охлаждали до 0°C и затем добавляли воду (1,49 мл), 15% водный раствор гидроксида натрия (1,49 мл) и воду (4,47 мл), в указанном порядке. Твердое вещество удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=10:1 (об./об.)) с получением бесцветного твердого вещества (1,41 г).
Трет-бутил бромацетат (609 мг), бисульфат тетрабутиламмония (35 мг) и 50% водный раствор гидроксида натрия (2 мл) добавляли, в указанном порядке, к бензольному раствору (20 мл), содержащему полученное соединение (450 мг), с последующим перемешиванием в течение ночи при комнатной температуре. Смесь нейтрализовали водным раствором 1 M хлористоводородной кислоты с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=6:4 (об./об.)) с получением бесцветного масла (576 мг).
10% палладий на углероде (каталитическое количество) добавляли к этанольному раствору (20 мл), содержащему полученное соединение (570 мг), и в атмосфере газообразного водорода смесь перемешивали при комнатной температуре при нормальном давлении в течение 1 часа. Катализатор удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=15:1 (об./об.)) с получением трет-бутил [(5-гидроксипиридин-3-ил)метокси]ацетата (400 мг).
Ссылочный Пример 81:
Пентаметилбензол (826 мг) добавляли к раствору в ТФУ (10 мл), содержащему метил (2E)-3-[5-(бензилокси)пиридин-3-ил]акрилат (300 мг), с последующим перемешиванием в течение ночи при температуре 60°C. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=10:1 (об./об.)) с получением трет-бутил (5-гидроксипиридин-3-ил)ацетата (180 мг).
Ссылочный Пример 82:
Диизопропилэтиламин (2,05 мл) и метоксиметилхлорид (0,89 мл) добавляли в указанном порядке к раствору в ТГФ (60 мл) метил 3-гидроксиникотината (1,50 г) и затем перемешивали в течение ночи при комнатной температуре. Растворитель выпаривали при пониженном давлении, добавляли воду, а затем экстрагировали хлороформом. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением бесцветного масла (2,01 г).
При температуре -78°C раствор в ТГФ (20 мл) полученного соединения (1,98 г) добавляли к раствору в ТГФ (50 мл) литийалюминийгидрида (838 мг) с последующим перемешиванием в течение 30 минут и затем перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор охлаждали до 0°C и добавляли воду (0,84 мл), 15% водный раствор гидроксида натрия (0,84 мл) и воду (2,52 мл), в указанном порядке. Твердое вещество удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: EtOAc) с получением бесцветного масла (838 г).
К раствору в пиридине (10 мл), содержащему полученное соединение (828 мг), добавляли уксусный ангидрид (1,39 мл) с последующим перемешиванием при комнатной температуре в течение 1,5 часов. Растворитель выпаривали при пониженном давлении, добавляли Tol (10 мл) и подвергали азеотропной прегонке с получением бесцветного масла (1,01 г).
Раствор 4 M хлористый водород/диоксан (3,58 мл) добавляли к раствору полученного соединения (1,01 г) в диоксане (10 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. Растворитель выпаривали при пониженном давлении с получением гидрохлорида (5-гидроксипиридин-3-ил)метилацетата (973 мг).
Ссылочный Пример 95:
Трифенилфосфин (2,8 г) добавляли к раствору в Tol (50 мл) 3-цианобензилбромида (2,0 г) с последующим перемешиванием при температуре 80°C в течение 5 часов. Смесь охлаждали до комнатной температуры и осажденное твердое вещество собирали фильтрованием и промывали Tol. Смесь сушили при пониженном давлении с получением (3-цианобензил)(трифенил)фосфонийбромида (3,4 г).
При охлаждении льдом, гидрид натрия (60% в масле, 141 мг) добавляли к раствору в ДМФА (20 мл) (3-цианобензил)(трифенил)фосфонийбромида (1,6 г) и трет-бутил 4-формил-1-пиперидинкарбоксилата (0,75 г) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную жидкость разбавляли при помощи EtOAc, промывали водой и сушили над безводным сульфатом магния. Растворитель выпаривали и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=6:1 (об./об.)) с получением масла. 10% Палладий на углероде (100 мг) добавляли к раствору полученного масла в EtOAc (30 мл) с последующим перемешиванием в атмосфере потока водорода в течение 2 часов. Катализатор удаляли через целит и растворитель концентрировали с получением масла. Полученное масло растворяли в EtOAc (10 мл) и к смеси добавляли раствор 4 M хлористый водород/EtOAc (5 мл), затем перемешивали при комнатной температуре в течение 6 часов, а затем концентрировали. Полученное твердое вещество промывали простым эфиром и сушили при пониженном давлении с получением гидрохлорида 3-[2-(4-пиперидинил)этил]бензонитрила (506 мг).
Таким же способом, как в Ссылочном Примере 95, получали соединения Ссылочных Примеров 96-101.
Ссылочный Пример 102:
Трифенилфосфин (85,8 г) добавляли к раствору в Tol (400 мл) метил 3-бромметилбензоата (50,0 г) с последующим перемешиванием при температуре 80°C в течение 10 часов. После охлаждения смеси до комнатной температуры, осажденные кристаллы собирали фильтрованием и промывали Tol. Смесь сушили при пониженном давлении с получением (3-метоксикарбонилбензил)(трифенил)фосфонийбромида (107,6 г).
При охлаждении льдом, трет-бутоксид калия (22,5 г) добавляли к раствору в ДМФА (250 мл) (3-метоксикарбонилбензил)(трифенил)фосфонийбромида (84,6 г) с последующим перемешиванием при комнатной температуре в течение 30 минут. Затем к смеси при охлаждении льдом добавляли раствор трет-бутил 4-формил-1-пиперидинкарбоксилат (30,6 г) в ДМФА (50 мл) и затем перемешивали в течение ночи при комнатной температуре. К реакционной жидкости добавляли уксусную кислоту (11,5 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем смесь разбавляли при помощи EtOAc, промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=7:1 (об./об.)). Остаток растворяли в EtOAc, к смеси добавляли активированный уголь с последующим перемешиванием при комнатной температуре в течение 2 часов. Активированный уголь удаляли через целит и растворитель выпаривали при пониженном давлении с получением бесцветного масла.
10% Палладий на углероде (4,58 г) добавляли к раствору полученного масла в EtOAc (400 мл) с последующим перемешиванием в атмосфере потока водорода в течение 2 часов. Катализатор удаляли через целит и растворитель концентрировали с получением трет-бутил 4-{2-[3-(метоксикарбонил)фенил]этил}-1-пиперидина (45,4 г).
Таким же способом, как в Ссылочном Примере 102, получали соединение Ссылочного Примера 103.
Ссылочный Пример 104:
Водный раствор 1 M гидроксида натрия (196 мл) добавляли к раствору трет-бутил 4-{2-[3-(метоксикарбонил)фенил]этил}-1-пиперидина (45,4 г) в ТГФ (200 мл)/метаноле (50 мл) с последующим перемешиванием при температуре 60°C в течение 2 часов. Органический растворитель выпаривали при пониженном давлении и при охлаждении льдом к остатку добавляли 0,5 M раствор хлористоводородной кислоты (400 мл). Реакционную жидкость разбавляли при помощи EtOAc, промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель выпаривали с получением 3-{2-[1-(трет-бутоксикарбонил)-4-пиперидинил]этил}бензойной кислоты (43,5 г).
Таким же способом, как в Ссылочном Примере 104, получали соединение Ссылочного Примера 105.
Ссылочный Пример 106:
3-{2-[1-(Трет-бутоксикарбонил)-4-пиперидинил]этил}-бензойную кислоту (17,8 г) растворяли в ДМФА (200 мл) и к смеси добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (15,4 г) и 1-гидроксибензотриазол (10,8 мг) с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционному раствору добавляли аммонийхлорид (8,57 г) и TEA (22,3 мл) с последующим перемешиванием в течение ночи при комнатной температуре. К реакционному раствору добавляли насыщенный водный раствор гидрокарбоната натрия, осажденные кристаллы собирали фильтрованием и сушили с получением трет-бутил 4-{2-[3-(аминокарбонил)фенил]этил}-1-пиперидина (10,8 г).
Таким же способом, как в Ссылочном Примере 106, получали соединения Ссылочных Примеров 107-118.
Ссылочный Пример 119:
Трет-бутил 4-[2-(4-{[(2-гидроксиэтил)амино]карбонил}-фенил)этил]пиперидин-1-карбоксилат (280 мг), тетрабромида углерод (247 мг) и 2,6-лутидин (103 мг) растворяли в дихлорметане (5,6 мл) и при охлаждении льдом добавляли трифенилфосфин (195 мг) с последующим перемешиванием при комнатной температуре в течение 3 часов. Растворитель выпаривали и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=3:7 (об./об.)) с получением трет-бутил 4-{2-[4-(1-азиридинилкарбонил)фенил]этил}-1-пиперидинкарбоксилата (136 мг) в виде бесцветного масла.
Ссылочный Пример 120:
Трет-бутил 4-{2-[3-(аминокарбонил)фенил]этил}-1-пиперидин (13,8 г) растворяли в EtOAc (200 мл) и добавляли раствор 4 M хлористый водород/EtOAc (130 мл) с последующим перемешиванием при комнатной температуре в течение 4 часов, а затем концентрировали. К полученному остатку добавляли ацетонитрил с последующим нагреванием и осажденные кристаллы собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении с получением гидрохлорида 3-[2-(4-пиперидинил)этил]бензамида (11,2 г).
Таким же способом, как в Ссылочном Примере 120, получали соединения Ссылочных Примеров 121-139.
Ссылочный Пример 140:
В атмосфере потока аргона карбонат натрия (0,43 г) и тетракис(трифенилфосфин)палладий (80 мг) добавляли к раствору в Tol (6 мл)/воде (2 мл) трет-бутил 4-[2-(3-бромфенил)этил]-1-пиперидинкарбоксилата (0,50 г) и фенилбороновой кислоты (0,20 г) с последующим нагреванием при перемешивании при температуре 100°C в течение 7 часов. Смесь охлаждали до комнатной температуры, разбавляли при помощи EtOAc и промывали насыщенным водным раствором гидрокарбоната натрия. Смесь сушили над безводным сульфатом магния, затем растворитель выпаривали и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=10:1 (об./об.)) с получением трет-бутил 4-[2-(3-бифенил)этил]-1-пиперидинкарбоксилата (0,41 г).
Раствор 4 M хлористый водород/EtOAc (1,5 мл) добавляли к раствору трет-бутил 4-[2-(3-бифенил)этил]-1-пиперидинкарбоксилата (0,41 г) в EtOAc (4 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Осажденные кристаллы собирали фильтрованием, промывали EtOAc/гексаном и сушили при пониженном давлении с получением гидрохлорида 4-[2-(3-бифенил)этил]пиперидина (0,31 г).
Таким же способом, как в Ссылочном Примере 140, получали соединения Ссылочных Примеров 141 и 142.
Ссылочный Пример 143:
При охлаждении льдом ди-трет-бутилдикарбонат (2,6 г) добавляли к раствору 4,4'-(1,3-пропан-диил)дипиперидина (5,0 г) в дихлорметане (50 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную жидкость разбавляли хлороформом, промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол:водный концентрированный раствор аммиака=4:1:0,1 (об./об.)) с получением трет-бутил 4-[3-(4-пиперидинил)пропил]-1-пиперидинкарбоксилата (2,2 г).
В атмосфере аргона трет-бутоксид натрия, (0,52 г), трис(дибензилиденацетон)дипалладий (100 мг) и 2-(дициклогексилфосфино)бифенил (76 мг) добавляли к раствору в Tol (22 мл) 2-хлор-6-метилпиридина (0,56 г) и трет-бутил 4-[3-(4-пиперидинил)пропил]-1-пиперидинкарбоксилата (1,1 г) с последующим нагреванием при перемешивании при температуре 100°C в течение 1 часа. Смесь охлаждали до комнатной температуры, разбавляли при помощи EtOAc и промывали водным насыщенным раствором гидрокарбоната натрия. Смесь сушили над безводным сульфатом магния, растворитель выпаривали и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=10:1 (об./об.)) с получением трет-бутил 4-{3-[1-(6-метил-2-пиридинил)-4-пиперидил]пропил}-1-пиперидиндикарбоксилата (1,3 г).
Раствор 4 M хлористый водород/EtOAc (10 мл) добавляли к раствору трет-бутил 4-{3-[1-(6-метил-2-пиридинил)-4-пиперидинил]пропил}-1-пиперидиндикарбоксилата (1,3 г) в EtOAc (25 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную жидкость концентрировали, затем добавляли 2-пропанол/диэтиловый простой эфир с последующим перемешиванием. Осажденное твердое вещество собирали фильтрованием и сушили при пониженном давлении с получением дигидрохлорида 2-метил-6-{4-[3-(4-пиперидинил)пропил]-1-пиперидил}пиридина (1,1 г).
Таким же способом, как в Ссылочном Примере 143, получали соединения Ссылочных Примеров 144 и 145.
Ссылочный Пример 146:
Метансульфонилхлорид (2,7 мл) добавляли по каплям к раствору трет-бутил 4-(3-гидроксипропил)пиперидин-1-карбоксилата (8,00 г) и TEA (4,8 мл) в метиленхлориде (200 мл) при температуре 0°C с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную жидкость промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, затем сушили над безводным сульфатом магния, и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: EtOAc:гексан=1:3 (об./об.)) с получением трет-бутил 4-{3-[(метилсульфонил)окси]пропил}пиперидин-1-карбоксилата (10,1 г).
Суспензию трет-бутил 4-{3-[(метилсульфонил)окси]пропил}пиперидин-1-карбоксилат (1,00 г), дигидрохлорида 1-пиперазин-1-ил-изохинолина (980 мг), карбоната цезия (1,02 г) и иодида натрия (467 мг) в DMI (20 мл) перемешивали при температуре 140°C в течение 1 часа. К реакционной жидкости добавляли EtOAc, промывали водой и насыщенным водным раствором гидрокарбоната натрия, в указанном порядке, затем сушили над безводным сульфатом магния и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:1 (об./об.)) с получением трет-бутил 4-[3-(4-изохинолин-1-илпиперазин-1-ил)пропил]пиперидин-1-карбоксилата (1,07 г) в виде бледно-желтого масла.
Раствор 4 M хлористый водород/EtOAc раствор (5,0 мл) добавляли по каплям к раствору трет-бутил 4-[3-(4-изохинолин-1-илпиперазин-1-ил)пропил]пиперидин-1-карбоксилата (1,44 г) в EtOAc (15 мл) с последующим перемешиванием в течение ночи. Растворитель выпаривали, твердое вещество промывали EtOAc и собирали фильтрованием с получением дигидрохлорида 1-[4-(3-пиперидин-4-илпропил)пиперазин-1-ил]изохинолина (1,32 г) в виде белого твердого вещества.
Таким же способом, как в Ссылочном Примере 146, получали соединение Ссылочного Примера 154.
Ссылочный Пример 147:
4-Нитрофенилхлорформиат (7,0 г) добавляли к раствору раствору метил 5-гидроксиникотината (5,3 г) и диизопропилэтиламина (6,1 мл) в дихлорметане (100 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционную жидкость промывали водой и сушили над безводным сульфатом магния. Растворитель выпаривали и полученное твердое вещество промывали EtOAc/гексаном и сушили при пониженном давлении с получением метил 5-{[(4-нитрофенокси)карбонил]окси}никотината (8,4 г).
Таким же способом, как в Ссылочном Примере 147, получали соединение Ссылочного Примера 148.
Ссылочный Пример 151:
Раствор в ДМФА (15 мл) 3-{2-[1-(трет-бутоксикарбонил)-4-пиперидинил]этил}бензойной кислоты (1,25 г), гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (863 мг) и 1-гидроксибензотриазола (608 мг) перемешивали при комнатной температуре в течение 1 часа и затем добавляли раствор гидробромида 2-бромэтиламина (2,30 г) в TEA (1,6 мл) с последующим перемешиванием в течение ночи. К реакционной жидкости добавляли насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией при помощи EtOAc, затем промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и растворитель выпаривали с получением неочищенного продукта трет-бутил 4-[2-(3-{[(2-бромэтил)амино]карбонил}фенил)этил]пиперидин-1-карбоксилата.
Раствор 4 M хлористый водород/EtOAc (5 мл) добавляли к раствору неочищенного трет-бутил 4-[2-(3-{[(2-бромэтил)амино]карбонил}фенил)этил]пиперидин-1-карбоксилата в EtOAc (15 мл) при комнатной температуре с последующим перемешиванием в течение ночи. Растворитель выпаривали при пониженном давлении с получением гидрохлорида N-(2-бромэтил)-3-(2-пиперидин-4-илэтил)бензамида (1,27 г) в виде белого твердого вещества.
TEA (0,90 мл) добавляли по каплям к суспензии N-(2-бромэтил)-3-(2-пиперидин-4-илэтил)бензамид гидрохлорида (1,20 г) и метил 5-{[(4-нитрофенокси)карбонил]окси}никотината (1,02 г) в ацетонитриле (30 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Растворитель реакционной смеси выпаривали при пониженном давлении, затем к смеси добавляли насыщенный водный раствор гидрокарбоната натрия, экстрагировали при помощи EtOAc и сушили над безводным сульфатом магния. Смесь фильтровали, растворитель выпаривали и остаток очищали два раза колоночной хроматограффией на силикагеле (основной диоксид кремния с элюентом: гексан:EtOAc=1:2 (об./об.), затем нейтральный диоксид кремния с элюентом: хлороформ:метанол=19:1 (об./об.)) с получением метил 5-[{(4-[2-(3-{[(2-бромэтил)амино]карбонил}фенил)этил]пиперидин-1-ил}карбонил)окси]никотината (762 мг) в виде белого порошка.
Суспензию метил 5-[{(4-[2-(3-{[(2-бромэтил)амино]карбонил}фенил)этил]пиперидин-1-ил}карбонил)окси]никотината (750 мг), карбоната калия (300 мг) и иодида калия (361 мг) в ДМФА (10 мл) перемешивали при температуре 80°C в течение 1 часа. Реакционную жидкость оставляли для охлаждения, затем к смеси добавляли EtOAc, промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, в указанном порядке, сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=20:1 (об./об.)) с получением метил 5-{[(4-{2-[3-(азиридин-1-илкарбонил)фенил]этил}пиперидин-1-ил)карбонил]окси}никотината (630 мг) в виде бесцветного масла.
Ссылочный Пример 152:
При охлаждении льдом дифенилфосфорилазид (540 мг) добавляли к раствору 3-{2-[1-(трет-бутоксикарбонил)-4-пиперидил]этил]бензойной кислоты (600 мг) и TEA (0,3 мл) в Tol (10 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционному раствору добавляли EtOAc, промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением бесцветного масла (630 мг). Раствор полученного масла (400 мл) в Tol (10 мл) перемешивали при температуре 110°C в течение 1 часа. Смесь охлаждали до комнатной температуры и добавляли водный 30% раствор аммиака (0,2 мл) с последующим перемешиванием при комнатной температуре в течение 15 часов. К реакционному раствору добавляли EtOAc, затем промывали 1 н. водным раствором хлористоводородной кислоты и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=95:5 (об./об.)) с получением трет-бутил 4-(2-{3-[(аминокарбонил)амино]фенил}этил)-1-пиперидинкарбоксилата (227 мг).
Раствор 4 M хлористый водород/EtOAc (4 мл) добавляли к раствору трет-бутил 4-(2-{3-[(аминокарбонил)амино]фенил}этил)-1-пиперидинкарбоксилата (227 мг) в EtOAc (9 мл) с последующим перемешиванием при комнатной температуре в течение 3 часов. Растворитель выпаривали при пониженном давлении с получением гидрохлорида 1-{3-[2-(4-пиперидил)этил]фенил}мочевины (185 мг).
Метил 5-{[(4-нитрофенокси)карбонил]окси}никотинат (228 мг) добавляли к раствору в ацетонитриле (5 мл) гидрохлорида 1-{3-[2-(4-пиперидинил)этил]фенил}мочевины (185 мг) и TEA (0,2 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную жидкость разбавляли при помощи EtOAc, промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом магния. Растворитель выпаривали и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=10:1 (об./об.)) с получением метил 5-({[4-(2-{3-[(аминокарбонил)амино]фенил}-этил)-1-пиперидил]карбонил}окси)никотината (183 мг).
Таким же способом, как в Ссылочном Примере 152, получали соединение Ссылочного Примера 153.
Ссылочный Пример 155:
Трет-бутил 4-этинилпиперидин-1-карбоксилат (12,5 г) и иодбензол (12,8 г) растворяли в смеси растворителей ТГФ:TEA=1:1 (об./об.) (125 мл), затем при комнатной температуре к смеси добавляли иодид меди (455 мг) и комплекс палладий-тетракистрифенилфосфин (1,38 г), в указанном порядке, с последующим перемешиванием в течение ночи при комнатной температуре. Растворитель выпаривали, к смеси добавляли EtOAc и промывали водным раствором 1 M хлористоводородной кислоты, водой и насыщенным солевым раствором, в указанном порядке. Смесь сушили над сульфатом магния и растворитель выпаривали с получением светло-коричневого масла. Смесь очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=19:1 (об./об.)) с получением трет-бутил 4-(фенилэтинил)пиперидин-1-карбоксилата (15,5 г) в виде светло-коричневого масла.
Раствор 4 M хлористый водород/EtOAc (70 мл) добавляли к трет-бутил 4-(фенилэтинил)пиперидин-1-карбоксилату (7,0 г) с последующим перемешиванием при комнатной температуре в течение 30 минут. Растворитель выпаривали с получением гидрохлорида 4-(фенилэтинил)пиперидина (5,4 г) в виде белого порошка.
Пример 1:
3-Гидроксипиридин (400 мг), TEA (1,17 мл) и DMAP (каталитическое количество) добавляли в указанном порядке к раствору в ТГФ (10 мл), содержащему пиперидин-1-карбонилхлорид (745 мг), и затем нагревали при температуре 60°C в течение 5 часов. Реакционный раствор охлаждали, затем добавляли воду (3 мл) и экстрагировали при помощи EtOAc. Экстракт промывали водой и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:1 (об./об.)) с получением бесцветного масла. Полученное масло растворяли в этаноле и к смеси добавляли этанольный раствор щавелевой кислоты (378 мг) с получением бесцветного порошка. Смесь перекристаллизовывали из гексана/этанола с получением (пиридин-3-ил) пиперидин-1-карбоксилатоксалата (761 мг).
Пример 2:
Раствор в метиленхлориде (20 мл), содержащий 3-гидроксипиридин (568 мг) и пиридин (724 мкл), добавляли по каплям к раствору в метиленхлориде (25 мл), содержащему трифосген (590 мг), с последующим перемешиванием при комнатной температуре в течение 1 часа. Растворитель выпаривали при пониженном давлении, остаток растворяли в пиридине (30 мл), затем к смеси добавляли соединение (1,2 г), полученное в Ссылочном Примере 22, с последующим нагреванием при температуре 70°C в течение 4 часов. Реакционный раствор концентрировали при пониженном давлении, затем добавляли хлороформ и водный раствор гидрокарбоната натрия и органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:2 (об./об.)) с получением бесцветного порошка. Смесь перекристаллизовывали из гексана/EtOAc с получением (пиридин-3-ил) 4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилата (861 мг).
Таким же способом, как в Примере 2, получали соединения Примеров 3-118, 389-391, 416 и 417 и Ссылочных Примеров 83-93.
Пример 119:
Раствор в метиленхлориде (20 мл), содержащий 3-гидроксипиридин (1,43 г) и пиридин (1,46 мл), добавляли по каплям к раствору в метиленхлориде (30 мл), содержащему трифосген (1,48 г), с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли по каплям раствор в метиленхлориде (5 мл), содержащий трет-бутил 1-пиперазинкарбоксилат (2,0 г) и пиридин (0,97 мл), затем к смеси добавляли пиридин (20 мл) с последующим нагреванием при температуре 70°C в течение 4 часов. Реакционный раствор концентрировали при пониженном давлении, разбавляли при помощи EtOAc и органический слой промывали насыщенным водным раствором гидрокарбоната натрия, а затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на основном силикагеле (элюент: гексан:EtOAc=4:1 (об./об.)) с получением бесцветного твердого вещества (3,0 г).
Полученное соединение (3,0 г) растворяли в EtOAc (20 мл)/2-пропаноле (10 мл), затем к смеси добавляли раствор 4 M хлористый водород/EtOAc (10 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении и полученное твердое вещество промывали EtOAc и сушили при пониженном давлении с получением дигидрохлорида 3-пиридил 1-пиперидинкарбоксилата (2,66 г).
Гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (150 мг), 1-гидроксибензотриазол (110 мг) и диизопропилэтиламин (0,23 мл) добавляли к раствору в ДМФА (5 мл), содержащему полученное соединение (190 мг) и 4-(циклооктилметокси)бензойную кислоту (176 мг), полученную из циклооктилметанола в соответствии со Ссылочным Примером 70, с последующим перемешиванием в течение ночи при комнатной температуре. Реакционный раствор разбавляли при помощи EtOAc, органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток перекристаллизовывали из EtOAc/гексана с получением 3-пиридил 4-[4-(циклооктилметокси)бензоил]-1-пиперазинкарбоксилата (240 мг).
Таким же способом, как в Примере 119, получали соединения Примеров 120-136.
Пример 137:
трет-Бутоксид калия (810 мг) добавляли к раствору в ДМФА (10 мл), содержащему 6-хлорникотинонитрил (1,0 г) и 3-хлорбензиловый спирт (1,0 г), с последующим перемешиванием в течение ночи при комнатной температуре. К реакционному раствору добавляли воду и осажденное твердое вещество собирали фильтрованием, промывали водой и гексаном, в указанном порядке, и сушили при пониженном давлении с получением коричневого твердого вещества (1,3 г).
К этанольному раствору (10 мл), содержащему полученное соединение (1,3 г), добавляли водный раствор 5 M гидроксида натрия (10 мл) с последующим перемешиванием при температуре 100°C в течение 4 часов. После охлаждения смеси до комнатной температуры добавляли 1 н. раствор хлористоводородной кислоты (56 мл) и осажденное твердое вещество собирали фильтрованием, промывали водой и сушили при пониженном давлении с получением бесцветного твердого вещества (0,82 г).
К раствору в ДМФА (5 мл), содержащему полученное соединение (176 мг) и дигидрохлорид 3-пиридил 1-пиперазинкарбоксилата (166 мг), добавляли гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (150 мг), 1-гидроксибензотриазол (110 мг) и диизопропилэтиламин (0,23 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционный раствор разбавляли при помощи EtOAc, органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на основном силикагеле (элюент: гексан:EtOAc=1:2 (об./об.)) с получением бесцветного масла (140 мг).
К раствору в 2-пропаноле, содержащему полученное соединение (140 мг), добавляли щавелевую кислоту (35 мг) с последующим перемешиванием в течение 30 минут. Осажденное твердое вещество собирали фильтрованием, промывали 2-пропанолом/гексаном и сушили при пониженном давлении с получением 3-пиридил 4-({6-[(3-хлорбензил)окси]-3-пиридил}карбонил)-1-пиперазинкарбоксилат 0,5-оксалата (120 мг).
Таким же способом, как в Примере 137, получали соединение Примера 138.
Пример 139:
Карбонат калия (1,04 г) и бром EtOAc (0,610 мл) добавляли к раствору в ацетонитриле (15 мл), содержащему 4-гидроксибензамид (686 мг), с последующим нагреванием при температуре 80°C в течение 2 часов. Реакционный раствор охлаждали, добавляли воду (45 мл) и осажденное твердое вещество собирали фильтрованием, промывали водой и сушили с получением этил [4-(аминокарбонил)фенокси]ацетата (893 мг) в виде бледно-коричневого порошка.
Полученное соединение (870 мг) растворяли в ТГФ (10 мл) и к смеси добавляли этанол (0,274 мл) и водный раствор 1 M гидроксида натрия (4,68 мл) с последующим перемешиванием при комнатной температуре в течение 4 часов. Реакционный раствор концентрировали при пониженном давлении, подкисляли водным раствором 1 M хлористоводородной кислоты и осажденное твердое вещество собирали фильтрованием и сушили с получением бледно-коричневого порошка [4-(аминокарбонил)фенокси]уксусной кислоты (714 мг).
TEA (0,251 мл), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (259 мг), 1-гидроксибензотриазол (122 мг) и полученную выше [4-(аминокарбонил)фенокси]уксусную кислоту (184 мг) добавляли к раствору в ДМФА (5 мл), содержащему дигидрохлорид 3-пиридил 1-пиперидинкарбоксилата (252 мг), полученный в способе Примера 121, с последующим перемешиванием при комнатной температуре в течение 5 часов. К реакционному раствору добавляли насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=95:5 (об./об.)) и полученное твердое вещество перекристаллизовывали из EtOAc/ацетонитрила с получением пиридин-3-ил 4-{[4-(аминокарбонил)фенокси]ацетил}пиперидин-1-карбоксилата (274 мг).
Таким же способом, как в Примере 139, получали соединения Примеров 140 и 141.
Пример 142:
TEA (0,23 мл) и бензолсульфонилхлорид (0,075 мл) добавляли к раствору в дихлорметане (5 мл), содержащему дигидрохлорид 3-пиридил 1-пиперазинкарбоксилата (150 мг), с последующим перемешиванием в течение ночи при комнатной температуре. Реакционный раствор разбавляли хлороформом, органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ) и полученное твердое вещество перекристаллизовывали из 2-пропанола с получением 3-пиридил 4-(фенилсульфонил)-1-пиперазинкарбоксилата (130 мг).
Таким же способом, как в Примере 142, получали соединение Примера 143.
Пример 144:
К раствору в пиридине (3 мл), содержащему дигидрохлорид 3-пиридил 1-пиперазинкарбоксилата (150 мг), добавляли бензилхлорформиат (91 мг) с последующим перемешиванием при комнатной температуре в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, разбавляли при помощи EtOAc и органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, остаток разбавляли 2-пропанолом (3 мл) и добавляли гидрат толуолсульфоновой кислоты (100 мг) с последующим перемешиванием. Осажденные кристаллы собирали фильтрованием и перекристаллизовывали из 2-пропанола с получением тозилата бензил 3-пиридил 1,4-пиперазиндикарбоксилата (98 мг).
Таким же способом, как в Примере 144, получали соединения Примеров 145 и 146.
Пример 147:
10% Палладий на углероде (каталитическое количество) добавляли к раствору в ТГФ (20 мл)/2-пропаноле (20 мл), содержащему 3-пиридил 4-[(4-бензилокси)бензоил]-1-пиперазинкарбоксилат (1,3 г), и в атмосфере газообразного водорода смесь перемешивали при комнатной температуре при нормальном давлении в течение 12 часов. Катализатор удаляли фильтрованием, фильтрат концентрировали при пониженном давлении и полученное твердое вещество перекристаллизовывали из EtOAc/гексана с получением 3-пиридил 4-(4-гидроксибензоил)-1-пиперазинкарбоксилата (950 мг).
Раствор В ТГФ (5 мл), содержащий 3-пиридил 4-(4-гидроксибензоил)-1-пиперазинкарбоксилат (300 мг) и диэтилазодикарбоксилат (0,62 мл, 40% Tol раствор), добавляли по каплям к раствору в ТГФ (5 мл), содержащему 3-хлорбензиловый спирт (200 мг) и трифенилфосфин (360 мг), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 3 дней. Реакционный раствор разбавляли хлороформом, промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=95:5 (об./об.)) и полученное твердое вещество перекристаллизовывали из 2-пропанола с получением 3-пиридил 4-{4-[(3-хлорбензоил)окси]бензил}-1-пиперазинкарбоксилата (260 мг).
Таким же способом, как в Примере 147, получали соединения Примеров 148-166.
Пример 167:
Карбонат калия (270 мг) добавляли к раствору в ацетонитриле (10 мл), содержащему 3-пиридил 4-(4-гидроксибензоил)-1-пиперазинкарбоксилат (530 мг) и метил 3-(бромметил)бензоат (450 мг), с последующим перемешиванием при температуре 80°C в течение 1 часа. К реакционному раствору добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали водой и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:4 (об./об.)) с получением бесцветного твердого вещества (470 мг).
Полученное твердое вещество (100 мг) перекристаллизовывали из EtOAc с получением 3-пиридил 4-(4-{[3-(метоксикарбонил)бензил]окси}бензоил)-1-пиперазинкарбоксилата (88 мг).
Пример 168:
4-Этил 1-пиридин-3-ил пиперидин-1,4-дикарбоксилат (0,732 г) растворяли в ТГФ (15 мл) и этаноле (8,0 мл) и при охлаждении льдом к смеси добавляли по каплям водный раствор 1 M гидроксида натрия (3,9 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и нейтрализовали при помощи 1 M хлористоводородной кислоты (0,5 мл). Реакционную жидкость концентрировали при пониженном давлении, к остатку добавляли метанол и осажденную соль удаляли фильтрованием с отсосом. Фильтрат концентрировали с получением 1-[(пиридин-3-илокси)карбонил]пиперидин-4-карбоновой кислоты (0,727 г) в виде бесцветного твердого вещества.
Полученное соединение (0,60 г) растворяли в диметилформамиде (10 мл) и к смеси добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,93 г), 1-гидроксибензотриазол (0,51 г) и циклогексанметиламин (0,43 г) с последующим перемешиванием при комнатной температуре в течение 15 часов. К реакционному раствору добавляли воду, а затем снова перемешивали в течение 1 часа. Затем к смеси добавляли раствор гидрокарбоната натрия с последующей экстракцией при помощи EtOAc. Органический слой промывали раствором 0,5 M хлористоводородной кислоты и насыщенным солевым раствором, в указанном порядке. Органический слой сушили над безводным сульфатом магния, растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:4 (об./об.)) с получением бесцветного порошка (0,69 г). Смесь перекристаллизовывали из этанола и гексана с получением (пиридин-3-ил) 4-{[(циклогексилметил)амино]карбонил}пиперидин-1-карбоксилата (261 мг).
Таким же способом, как в Примере 168, получали соединения Примеров 169-192, 383-388 и Ссылочного Примера 94.
Пример 193:
3-Пиридинилхлоридкарбонат (330 мг) добавляли к раствору в пиридине (10 мл), содержащему 1-бензил 2-метил-1,2-пиперазиндикарбоксилат (660 мг, Beilstein Registry No. 4236331), с последующим перемешиванием при температуре 80°C в течение 7 часов. Реакционный раствор концентрировали при пониженном давлении, разбавляли хлороформом и органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на основном силикагеле (элюент: гексан:EtOAc=1:1 (об./об.)) с получением бесцветного масла (700 мг).
К раствору в ТГФ (5 мл), содержащему полученное соединение (430 мг), добавляли водный раствор 1 M гидроксида натрия (1,2 мл) с последующим перемешиванием при температуре 50°C в течение 3 часов. К смеси добавляли водный раствор 1 M гидроксида натрия (0,8 мл) и продолжали нагревание при температуре 50°C в течение 1 часа, затем охлаждали до комнатной температуры и добавляли 1 н. раствор хлористоводородной кислоты (2 мл). Реакционный раствор экстрагировали при помощи EtOAc, органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и осажденное твердое вещество промывали EtOAc/гексаном и сушили при пониженном давлении с получением 1-[(бензилокси)карбонил]-4-[(3-пиридилокси)карбонил]-2-пиперадинкарбоновой кислоты (140 мг).
Таким же способом, как в Примере 193, получали соединения Примеров 194 и 195.
Пример 196:
Пиридин-3-ил 4-({[2-(метиламино)фенил]амино}карбонил)пиперидин-1-карбоксилат (0,41 г) растворяли в уксусной кислоте (10 мл) с последующим нагреванием при температуре кипения с обратным холодильником в течение 2 часов. Растворитель выпаривали и остаток перекристаллизовывали из метанола и диэтилового эфира с получением (пиридин-3-ил) 4-(1-метил-1H-бензимидазол-2-ил)пиперидин-1-карбоксилата (307 мг).
Пример 197:
Пиридин-3-ил 4-[(трет-бутоксикарбонил)амино]пиперидин-1-карбоксилат (0,249 г) растворяли в ТГФ (5,0 мл) и при охлаждении льдом к смеси добавляли раствор 4 M хлористый водород/EtOAc (2,10 мл) с последующим перемешиванием при комнатной температуре в течение 24 часов. Реакционный раствор концентрировали досуха с получением дигидрохлорида пиридин-3-ил 4-аминопиперидин-1-карбоксилата (0,280 г).
Полученное соединение (0,28 г) растворяли в диметилформамиде (10 мл) и к смеси добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,28 г), 1-гидроксибензотриазол (0,16 г), TEA (0,54 мл) и 6-фенилгексановую кислоту (0,18 г) с последующим перемешиванием при комнатной температуре в течение 15 часов. К реакционному раствору добавляли воду и продолжали перемешивание в течение 1 часа. Затем к смеси добавляли раствор гидрокарбоната натрия с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: EtOAc) с получением бесцветного порошка. Смесь перекристаллизовывали из метанола и диэтилового эфира с получением (пиридин-3-ил) 4-[(6-фенилгексаноил)амино]пиперидин-1-карбоксилата (108 мг).
Пример 198:
10% Палладий на углероде (каталитическое количество) добавляли к раствору в ТГФ (75 мл)/2-пропаноле (75 мл), содержащему 3-пиридил 4-[3-(бензилокси)фенокси]-1-пиперидинкарбоксилат (4,0 г), и в атмосфере газообразного водорода смесь перемешивали при комнатной температуре при нормальном давлении в течение 24 часов. Катализатор удаляли фильтрованием, фильтрат концентрировали при пониженном давлении и полученное твердое вещество промывали EtOAc/гексаном и сушили при пониженном давлении с получением 3-пиридил 4-(3-гидроксифенокси)-1-пиперидинкарбоксилата (2,2 г).
Пример 199:
10% Палладий на углероде (каталитическое количество) добавляли к раствору в ТГФ (75 мл)/2-пропаноле (75 мл), содержащему 3-пиридил 4-[4-(бензилокси)фенокси]-1-пиперидинкарбоксилат (3,7 г), и в атмосфере газообразного водорода смесь перемешивали при комнатной температуре при нормальном давлении в течение 24 часов. Катализатор удаляли фильтрованием, фильтрат концентрировали при пониженном давлении и полученное твердое вещество промывали EtOAc/гексаном и сушили при пониженном давлении с получением 3-пиридил 4-(4-гидроксифенокси)-1-пиперидинкарбоксилата (2,4 г).
Пример 200:
Диэтилазодикарбоксилат (0,35 мл, 40% Tol раствор) добавляли по каплям к раствору в ТГФ (5 мл), содержащему 3-пиридил 4-(3-гидроксифенокси)-1-пиперидинкарбоксилат (160 мг), циклогексилметанол (87 мг) и трифенилфосфин (200 м), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. Реакционный раствор разбавляли хлороформом, промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:1 (об./об.)). Полученное масло растворяли в EtOAc (5 мл), добавляли раствор 4 M хлористый водород/EtOAc (1 мл) с последующим перемешиванием при комнатной температуре. Растворитель выпаривали при пониженном давлении и осажденное твердое вещество промывали EtOAc/2-пропанолом и сушили при пониженном давлении с получением 3-пиридил гидрохлорида 4-[3-(циклогексилметокси)фенокси]-1-пиперидинкарбоксилата (94 мг).
Таким же способом, как в Примере 200, получали соединения Примеров 201 до 205.
Пример 206:
Диэтилазодикарбоксилат (0,35 мл, 40% Tol раствор) добавляли по каплям к раствору в ТГФ (5 мл), содержащему 3-пиридил 4-(4-гидроксифенокси)-1-пиперидинкарбоксилат (160 мг), 3-хлорбензиловый спирт (110 мг) и трифенилфосфин (200 м), при температуре 0°C с последующим перемешиванием при комнатной температуре в течение 24 часов. Реакционный раствор разбавляли хлороформом, промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:3 (об./об.)). Полученное масло растворяли в EtOAc (5 мл) и добавляли раствор 4 M хлористый водород/EtOAc (1 мл) с последующим перемешиванием при комнатной температуре. Растворитель выпаривали при пониженном давлении и осажденное твердое вещество перекристаллизовывали из EtOAc/2-пропанола с получением гидрохлорида 3-пиридил 4-{4-[(3-хлорбензил)окси]фенокси}-1-пиперидинкарбоксилата (45 мг).
Таким же способом, как в Примере 206, получали соединения Примеров 207-212.
Пример 213:
10% Палладий на углероде (каталитическое количество) добавляли к этанольному раствору (100 мл), содержащему метил 5-[({4-[4-(бензилокси)фенокси]пиперидин-1-ил}карбонил)окси]никотинат, и в атмосфере газообразного водорода смесь перемешивали в течение ночи при комнатной температуре при нормальном давлении. Катализатор удаляли фильтрованием, полученный фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=15:1 (об./об.)) с получением бесцветного масла (1,08 г).
К раствору в ТГФ (20 мл), содержащему полученное соединение (450 мг) и 3-циклогексил-1-пропанол (315 мг), добавляли 2,2 M диэтилазодикарбоксилат (1,01 мл) и трифенилфосфин (581 мг) с последующим нагреванием при температуре 50°C в течение 22 часов. К реакционному раствору добавляли воду и затем экстрагировали хлороформом. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=2:1 (об./об.)) с получением метил 5-[({4-[4[(3-циклогексилпропокси)фенокси]пиперидин-1-ил}карбонил)окси]-никотината (242 мг).
Таким же способом, как в Примере 213, получали соединения Примеров 214-216.
Пример 217:
10% Палладий на углероде (каталитическое количество) добавляли к раствору в ТГФ (10 мл), содержащему 5-[({4-[4-(бензилокси)фенокси]пиперидин-1-ил}карбонил)окси]никотиновую кислоту (200 мг), и в атмосфере газообразного водорода смесь перемешивали при комнатной температуре при нормальном давлении в течение 3 часов. Катализатор удаляли фильтрованием и полученный фильтрат концентрировали при пониженном давлении с получением оксалата 5-[({4-[4-(гидрокси)фенокси]пиперидин-1-ил}карбонил)окси]никотиновой кислоты (55 мг).
Пример 218:
Соединение (4,0 г) Примера 29, полученное таким же способом, как в Примере 2, растворяли в ТГФ (30 мл) и метаноле (15 мл) и к смеси добавляли по каплям при охлаждении льдом водный раствор 1 M гидроксида натрия (12 мл). Смесь перемешивали при комнатной температуре в течение 30 минут, а затем при охлаждении льдом, смесь нейтрализовали раствором 1 M хлористоводородной кислоты (12 мл). Бесцветное осажденное твердое вещество собирали фильтрованием с получением 5-{[(4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-ил)карбонил]окси}-никотиновой кислоты (3,52 г).
Таким же способом, как в Примере 218, получали соединения Примеров 219-224 и Примеров 226-243.
Пример 225:
Раствор в метиленхлориде (30 мл), содержащий метил 5-гидроксиникотинат (2,20 г) и пиридин (4 мл), добавляли по каплям к раствору в метиленхлориде (50 мл), содержащему трифосген (1,56 г), с последующим перемешиванием при комнатной температуре в течение 1 часа. Растворитель выпаривали при пониженном давлении, остаток растворяли в пиридине (50 мл) и к смеси добавляли гидрохлорид 4-(2-фенилэтил)пиперидина (2,70 г) с последующим нагреванием в течение ночи при температуре 80°C. Реакционный раствор концентрировали при пониженном давлении, затем добавляли EtOAc и водный раствор гидрокарбоната натрия. Органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:1 (об./об.)) с получением бесцветного порошка. Смесь перекристаллизовывали из гексана/EtOAc с получением метил 5-({[4-(2-фенилэтил)пиперидин-1-ил]карбонил}окси)никотината (3,95 г).
Метил 5-({[4-(2-фенилэтил)пиперидин-1-ил]карбонил}-окси)никотинат (3,95 г) растворяли в ТГФ (32 мл) и метаноле (16 мл) и при охлаждении льдом к смеси добавляли по каплям водный раствор 1 M гидроксида натрия (16 мл). Смесь перемешивали при комнатной температуре в течение 30 минут и при охлаждении льдом смесь нейтрализовали раствором 1 M хлористоводородной кислоты (16 мл). Бесцветное осажденное твердое вещество собирали фильтрованием и перекристаллизовывали из метанола/воды с получением 5-({[4-(2-фенилэтил)пиперидин-1-ил]карбонил}окси)никотиновой кислоты (3,70 г).
Пример 244:
Соединение Примера 219, 5-{[(4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-ил)карбонил]окси}никотиновую кислоту (0,50 г), растворяли в ДМФА (8,0 мл) и к смеси добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,38 г), 1-гидроксибензотриазол (0,22 г) и трет-бутиловый эфир глицина (0,21 г) с последующим перемешиванием при комнатной температуре в течение 15 часов. К реакционному раствору добавляли воду с последующим перемешиванием в течение 1 часа. Затем к смеси добавляли раствор гидрокарбоната натрия с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:1 (об./об.)) с получением бесцветного масла (0,444 г).
Полученное соединение (0,444 г) растворяли в метиленхлориде (5,0 мл) и при охлаждении льдом добавляли ТФУ (1,15 мл). Смесь перемешивали при указанной температуре в течение 24 часов и затем реакционную жидкость концентрировали с получением желтого твердого вещества. Смесь перекристаллизовывали из этанола и диэтилового эфира с получением {[(5-{[(4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-ил)карбонил]окси}пиридин-3-ил)карбонил]амино}уксусной кислоты (348 мг).
В соответствии со способом амидирования, как описано в Примере 244, получали соединения Примеров 245-257.
Пример 258:
Воду (4 мл), карбонат натрия (337 мг) и тетракистрифенилфосфин палладий (115 мг) добавляли, в указанном порядке, к раствору в диметоксиэтане (12 мл), содержащему соединение (400 мг) Примера 54 и [3-(аминокарбонил)фенил]бороновую кислоту (176 мг), с последующим нагреванием при температуре 80°C в течение 5 часов. Реакционный раствор охлаждали и разбавляли при помощи EtOAc. Органический слой промывали водой и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:5 (об./об.)) с получением 5-[3-(аминокарбонил)фенил]пиридин-3-ил-4-бензилпиперидин-1-карбоксилата (205 мг).
Таким же способом, как в Примере 258, получали соединения Примеров 259, 265, 266 и 399.
Пример 260:
Раствор 4 M хлористый водород/диоксан (1,8 мл) добавляли к раствору в ТГФ (10 мл), содержащему 5-[(трет-бутоксикарбонил)амино]пиридин-3-ил 4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилат (174 мг), с последующим перемешиванием при температуре 60°C в течение 4 часов. Растворитель выпаривали при пониженном давлении с получением гидрохлорида 5-аминопиперидин-3-ил 4-{4-[(3-фторбензил)окси]фенокси}пиридин-1-карбоксилата (74 мг).
Пример 261:
К раствору в ТГФ (10 мл), содержащему 5-[4-(этоксикарбонил)пиперидин-1-ил]пиридин-3-ил оксалат 4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилата (240 мг) добавляли водный раствор 1 M гидроксида натрия (3,24 мл) с последующим перемешиванием при температуре 60°C в течение 5 часов. К реакционному раствору добавляли раствор 1 M хлористоводородной кислоты (3,24 мл) и растворитель выпаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=10:1 (об./об.)). Полученное масло растворяли в этаноле/воде, затем к смеси добавляли щавелевую кислоту (24 мг) для кристаллизации с получением оксалата 1-(5-{[(4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-ил)карбонил]окси}пиридин-3-ил)пиперидин-4-карбоновой кислоты (93 мг).
Пример 262:
К раствору в метиленхлориде (10 мл), содержащему 5-[(2-трет-бутокси-2-оксоэтокси)метил]пиридин-3-ил 4-{4-[(3-(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилат (333 мг), добавляли ТФУ (1,0 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Растворитель выпаривали при пониженном давлении с получением [(5-{[(4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-ил)карбонил]окси}пиридин-3-ил}метокси]уксусной кислоты (232 мг).
Пример 263:
К раствору в ТГФ (20 мл), содержащему 5-[(ацетокси)метил]пиридин-3-ил оксалат 4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилата (1,10 г) добавляли водный раствор 1 M гидроксида натрия (7,65 мл) с последующим перемешиванием при температуре 65°C в течение 3 часов. Реакционную жидкость нейтрализовали раствором 1 M хлористоводородной кислоты с последующей экстракцией хлороформом и сушкой над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=12:1 (об./об.)) с получением 5-(гидроксиметил)пиперидин-3-ил 4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилата (770 мг).
Пример 264:
К раствору в ТГФ (5 мл), содержащему 5-[(1E)-3-метокси-3-оксопроп-1-ен-1-ил]пиридин-3-ил 4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилат (158 мг), добавляли водный раствор 1 M гидроксида натрия (1,11 мл) с последующим перемешиванием при температуре 60°C в течение 3 часов. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=10:1 (об./об.)) с получением (2E)-3-(5-{[(4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-ил)карбонил]окси}пиридин-3-ил)акриловой кислоты (88 мг).
Пример 267:
(a) Метил 5-{[(4-нитрофенокси)карбонил]окси}никотинат (723 мг) добавляли к раствору гидрохлорида 3-[2-(4-пиперидил)этил]бензонитрила(475 мг) и TEA (0,58 мл) в ацетонитриле (10 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную жидкость разбавляли при помощи EtOAc, затем промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали, полученный остаток подвергали колоночной хроматографией на основном силикагеле (элюент: гексан:EtOAc=1:1 (об./об.)) и удаляли побочный продукт - нитрофенол. Затем смесь очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=3:2 (об./об.)) с получением метил 5-[({4-[2-(3-цианофенил)этил]-1-пиперидил}карбонил)окси]никотината (284 мг).
(b) К раствору метил 5-[({4-[2-(3-цианофенил)этил]-1-пиперидил}карбонил)окси]никотината (272 мг) в ТГФ (5 мл)/воде (4 мл) добавляли водный раствор 1 M гидроксида натрия (0,69 мл) с последующим перемешиванием в течение ночи при комнатной температуре. К реакционной жидкости добавляли раствор 1 M хлористоводородной кислоты (0,69 мл) и осажденные кристаллы собирали фильтрованием. Кристаллы промывали горячим раствором метанол/вода и сушили с получением 5-[({4-[2-(3-цианофенил)этил]-1-пиперидил}карбонил)окси]никотиновой кислоты (240 мг).
Таким же способом, как на стадии (a) Примера 267, получали соединения Ссылочных Примеров 149-150 и Примеров 268-272, 392, 396, 400, 402, 413, 419, 421 и 422.
В соответствии с таким же способом, который содержит стадию (b) после стадии (a), как в Примере 267, получали соединения Примеров 273-317, 393-395, 401, 403, 405, 406, 414 и 418.
Пример 318:
Гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (62 мг), 1-гидроксибензотриазол (43 мг), хлорид аммония (43 мг) и TEA (0,038 мл) добавляли к раствору 5-[({4-[2-(3-цианофенил)этил]-1-пиперидил}карбонил)окси]никотиновой кислоты (102 мг) в ДМФА (3,0 мл) с последующим перемешиванием в течение ночи при комнатной температуре. К реакционной жидкости добавляли насыщенный водный раствор гидрокарбоната натрия и осажденные кристаллы собирали фильтрованием и сушили. Полученное кристаллическое вещество перекристаллизовывали из EtOAc/гексана с получением 5-(аминокарбонил)-3-пиридил 4-[2-(3-цианофенил)этил]-1-пиперидинкарбоксилата (81 мг).
Таким же способом получали соединения Примеров 319-382, 397, 398, 404, 408-412, 415, 420 и 423.
Пример 407:
При охлаждении льдом трет-бутоксид калия (2,73 г) добавляли к раствору гидрохлорида трифенил (пиридин-4-илметил)фосфонийхлорида (4,75 г) и трет-бутил 4-формилпиперидин-1-карбоксилата (1,91 г) в ДМФА (50 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную жидкость разбавляли при помощи EtOAc, промывали водой и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом магния. Растворитель выпаривали и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=1:2 (об./об.)) с получением белого твердого вещества (2,05 г).
Полученное твердое вещество (2,04 г) растворяли в EtOAc (30 мл) и к смеси добавляли 10% палладий на углероде (200 мг) с последующим перемешиванием в присутствии водорода при комнатной температуре в течение 3 часов. Катализатор удаляли фильтрованием, растворитель концентрировали и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:этил ацетат=1:1 (об./об.)) с получением трет-бутил 4-[(E)-2-пиридин-4-илвинил]пиперидин-1-карбоксилата (1,70 г) в виде белого твердого вещества.
Раствор 4М хлористый водород/EtOAc (0,88 мл) и оксид платины (100 мг) добавляли к этанольному (25 мл) раствору трет-бутил 4-[(E)-2-пиридин-4-илвинил]пиперидин-1-карбоксилата (1,02 г) с последующим перемешиванием в присутствии водорода (3,5 атм) в течение 24 часов. Смесь продували аргоном, разбавляли метанолом, фильтровали через целит и концентрировали при пониженном давлении. Осажденное твердое вещество промывали EtOAc/гексаном и сушили при пониженном давлении с получением гидрохлорида трет-бутил 4-(2-пиперидин-4-илэтил)пиперидин-1-карбоксилата (850 мг) в виде белого твердого вещества.
2-(Дициклогексилфосфино)бифенил (71 мг) и (1E,4E)-1,5-дифенил-1,4-пентадиен-3-он-палладий (93 мг) добавляли к раствору в толуоле (10 мл) гидрохлорида трет-бутил 4-(2-пиперидин-4-илэтил)пиперидин-1-карбоксилата (1,13 г), 2-хлор-6-метилпиридина (431 мг) и трет-бутоксида натрия (487 мг) с последующим перемешиванием при температуре 120°C в течение 1 часа. Реакционную жидкость оставляли для охлаждения, затем к смеси добавляли насыщенный водный раствор карбоната натрия с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Затем растворитель выпаривали и остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:EtOAc=10:1 (об./об.)) с получением трет-бутил 4-{2-[1-(6-метилпиридин-2-ил)пиперидин-4-ил]этил}пиперидин-1-карбоксилата (660 мг) в виде красного масла.
К раствору трет-бутил 4-{2-[1-(6-метилпиридин-2-ил)пиперидин-4-ил]этил}пиперидин-1-карбоксилата (650 мг)в EtOAc (10 мл) добавляли раствор 4М хлористый водород/EtOAc (2 мл) с последующим перемешиванием при комнатной температуре в течение 2 дней. Реакционную жидкость концентрировали с получением дигидрохлорида 2-метил-6-[4-(2-пиперидин-4-илэтил)пиперидин-1-ил]пиридина (644 мг) в виде желтого аморфного вещества.
Метил 5-{[(4-нитрофенокси)карбонил]окси}никотинат (505 мг) добавляли к раствору в ацетонитриле (10 мл) дигидрохлорида 2-метил-6-[4-(2-пиперидин-4-илэтил)пиперидин-1-ил]пиридина (520 мг) и TEA (0,50 мл) с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционную жидкость разбавляли при помощи EtOAc, промывали насыщенным водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель выпаривали и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=98:2 (об./об.)) с получением метил 5-{[(4-{2-[1-(6-метилпиридин-2-ил)пиперидин-4-ил]этил}пиперидин-1-ил}карбонил]окси}никотината (424 мг).
К раствору метил 5-{[(4-{2-[1-(6-метилпиридин-2-ил)пиперидин-4-ил]этил}пиперидин-1-ил)карбонил]окси}никотината (208 мг) в ТГФ (5 мл) добавляли водный раствор 1 M гидроксида натрия (0,45 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную жидкость концентрировали с получением 5-{[(4-{2-[1-(6-метилпиридин-2-ил)пиперидин-4-ил]этил}пиперидин-1-ил)карбонил]окси}никотината натрия (158 мг).
Гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (103 мг), 1-гидроксибензотриазол (90 мг) и хлорид аммония (119 мг) добавляли к раствору в ДМФА (10 мл) 5-{[(4-{2-[1-(6-метилпиридин-2-ил)пиперидин-4-ил]этил}пиперидин-1-ил)карбонил]окси}никотината натрия (210 мг) с последующим перемешиванием в течение ночи при комнатной температуре.
Реакционную жидкость разбавляли при помощи EtOAc, промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и полученный остаток перекристаллизовывали из EtOAc/гексана с получением 5-(аминокарбонил)пиридин-3-ил 4-{2-[1-(6-метилпиридин-2-ил)пиперидин-4-ил]этил}пиперидин-1-карбоксилата (150 мг).
Пример 438:
Скрининг для определения вещества, обладающего активностью ингибирования FAAH с использованием гомогената головного мозга крысы:
(1) Приготовление гомогената головного мозга крысы:
Голову мужской особи 10-недельной крысы линии SD (Japan SLC) отсекали и головной мозг извлекали и взвешивали. Добавляли охлажденный льдом буфер (50 мМ Трис-HCl (pH 7,4), 0,32 M сахарозы) в пятикратном объеме в расчете на массу животного и смесь гомогенизировали с использованием гомогенизатора во льду с получением однородной суспензии. Смесь центрифугировали (1500×g, 4°C, 15 минут) и супернатант снова центрифугировали (15000×g, 4°C, 20 минут) с получением осадка. Затем, используя ультразвуковой генератор (UR-20P, Tommy Seiko), смесь обрабатывали ультразвуком (power dial 4) в течение 5 секунд. Концентрацию белка в полученном гомогенате измеряли в соответствии со способом окрашивания-связывания (protein assay CBB solution, Nacalai Tesque). Используя буфер (50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA, 0,1 мг/мл BSA, 100 мМ NaCl), суспензию головного мозга крысы разбавляли так, чтобы концентрация белка была 60 мкг/мл, с получением, таким образом, раствора фермента.
(2) Скрининг для определения вещества, обладающего активностью ингибирования FAAH:
Получали раствор субстрата, включающий 2 мкКи/мл радиомеченного анандамида (Анандамид [этаноламин 1-3H] (American Radiolabelled Chemical)), 8 мкМ анандамида (Funakoshi), 50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA, 0,1 мг/мл BSA и 100 мМ NaCl. Получали растворы ипытываемых веществ, растворяли их в ДМСО с получением концентраций от 1 нМ до 100 мкМ. 50 мкл раствора субстрата и 1 мкм раствора испытываемого вещества добавляли к 50 мкл раствора фермента и оставляли на 1 час. В качестве контроля использовали ДМСО вместо раствора испытываемого вещества. К раствору добавляли 200 мкл 1:1 (по объему) раствор хлороформ/метанол, а затем перемешивали с завихрением. Смесь центрифугировали (15000 об/мин, 2 минуты), в результате чего разложившийся продукт этаноламин (этаноламин 1-3H) отделялся в верхнем слое (водно/метанольный слой), а непрореагировавший радиомеченный анандамид (Анандамид [этаноламин 1-3H]) был в нижнем слое (хлороформный слой). 30 мкл верхнего слоя переносили в 96-луночный стойкий к органическим растворителям белый микропланшет (PicoPlate-96; Perkin Elmer), добавляли 150 мкл Microscint-20 (Perkin Elmer) и смесь измеряли при помощи сцинтилляционного счетчика для микропланшетов (TopCountTM; Beckman). По сравнению с контролем, вещество, дающее пониженное значение, выбирали в качестве вещества, ингибирующего активность FAAH.
(3) Измерение значения ИК50 для вещества, ингибирующего активность FAAH:
Испытываемое соединение растворяли в ДМСО с получением различных концентраций от 1 нМ до 100 мкМ для получения растворов испытываемого вещества. В соответствии со способом, указанным выше, соединение анализировали на его влияние на активность FAAH. В качестве контроля использовали ДМСО. Измеренное значение для случая, когда буфер (50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA, 0,1 мг/мл BSA, 100 мМ NaCl) подвергали взаимодействию вместо раствора фермента, вычитали из каждого измеренного значения. На основании измеренного значения контроля, 100%, получали значение ИК50 для испытываемого вещества. Например, значение ИК50 для соединений Примеров 2, 151, 225, 228, 273, 324, 325 и 359 составило 0,14 нМ, 27 нМ, 0,37 нМ, 0,19 нМ, 0,65 нМ, 0,54 нМ, 2,5 нМ и 1,3 нМ, соответственно.
Представленные выше результаты подтверждают, что, когда испытываемое вещество контактирует с гомогенатом ткани, которая экспрессирует FAAH или функциональную FAAH, и когда можно измерить зависимое от испытываемого вещества изменение активности FAAH, тогда его можно скринировать на вещество, ингибирующее активность FAAH, или, иными словами, на лекарственное средство для лечения частого мочеиспускания и недержания мочи, лекарственное средство для лечения повышенной активности мочевого пузыря и/или лекарственное средство для лечения боли.
Пример 439:
Скрининг для определения вещества, обладающего активностью ингибирования FAAH с использованием раковой клетки эпителия мочевого пузыря человека:
(1) Скрининг для определения вещества, обладающего активностью ингибирования FAAH:
Клетки 5678 раковой клетки эпителия мочевого пузыря человека (HTB-9; ATCC) высевали в 48-луночный культуральный планшет в количестве 1 × 105 клеток/лунка с использованием среды RPMI1640 (Invitrogen), содержащей 10% фетальную бычью сыворотку (HyClone). После инкубирования при температуре 37°C в течение, по меньшей мере, 12 часов, клетки промывали 400 мкл/лунка буфером (сбалансированный солевой раствор Хэнка, 20 мМ Hepes-NaOH (pH 7,4)). Испытываемое вещество, растворенное в ДМСО, добавляли к раствору субстрата (указанный выше буфер, содержащий 3 мкКи/мл радиомеченного анандамида (Анандамид [этаноламин 1-3H]) и 10 мкМ анандамида) таким образом, чтобы получить концентрацию от 0,003 нМ до 30 нМ. В качестве контроля добавляли только ДМСО. К указанным выше клеткам добавляли 100 мкл/лунка раствора субстрата и инкубировали в CO2 инкубаторе при температуре 37°C в течение 30 минут. Затем культуральный планшет с клетками переносили на лед и раствор субстрата удаляли отсосом; и 75 (мкл/лунка цитолитического раствора (указанный выше буфер, содержащий 0,5% Triton X-100, и добавляли 10 мкМ соединения, обладающего активностью ингибирования FAAH, 3'-карбамоилбифенил-3-ил циклогексилкарбамата (URB597; Cayman chemical; Kathuria et al., Nature Med., Vol. 9, pp. 76-81, 2003)), с последующим перемешиванием. Полученный клеточный лизат из каждой лунки отдельно переносили в 1,5-мл пробирку для образца, в которую добавляли 150 мкл раствора 1:1 (по объему) хлороформ/метанол, и затем перемешивали с завихрением. Смесь центрифугировали (15000 об/мин, 2 минут), в результате чего разложившийся продукт, этаноламин (этаноламин 1-3H) отделялся в верхний слой (водно/метанольный слой), а непрореагировавший радиомеченный анандамид был в нижнем слое (хлороформный слой). 25 мкл верхнего слоя переносили в 96-луночный стойкий к органическим растворителям белый микропланшет (PicoPlate-96; Perkin Elmer), добавляли 150 мкл Microscint-20 (Perkin Elmer) и смесь измеряли при помощи сцинтилляционного счетчика для микропланшетов (TopCountTM; Beckman). По сравнению с контролем, вещество, дающее пониженное значение, выбирали в качестве вещества, ингибирующего активность FAAH.
(2) Измерение значения ИК50 для вещества, ингибирующего активность FAAH:
Испытываемое соединение, растворенное в ДМСО с получением концентрации 10 мМ, растворяли в растворе субстрата так, чтобы получить различные концентрации от 0,003 нМ до 30 мкМ. В соответствии со способом, указанным выше, соединение анализировали на его влияние на активность FAAH. В качестве отрицательного контроля использовали ДМСО. В качестве положительного контроля к раствору субстрата добавляли URB597 с получением концентрации 10 мкМ. На основании измеренного значения положительного контроля, 0%, и измеренного значения отрицательного контроля, 100%, получали значение ИК50 для испытываемого вещества. Результаты испытания представлены в Таблице 64.
Представленные выше результаты подтверждают отличную ингибиторную активность типичных соединений по настоящему изобретению в отношении FAAH. Кроме того, данные показывают, что, когда испытываемое вещество контактирует с клеткой, которая экспрессирует FAAH или функциональную FAAH, и когда можно измерить зависимое от испытываемого вещества изменение активности FAAH, тогда его можно скринировать на вещество, ингибирующее активность FAAH, или, иными словами, на лекарственное средство для лечения частого мочеиспускания и недержания мочи, лекарственное средство для лечения повышенной активности мочевого пузыря и/или лекарственное средство для лечения боли.
Пример 440:
Скрининг для определения вещества, обладающего активностью ингибирования FAAH, с использованием гомогената ткани крысы, которой вводили испытываемое вещество:
(1) Введение крысе и приготовление гомогената ткани:
Испытываемое вещество, суспендированное в 0,5% растворе метилцеллюлозы (MC), перорально вводили двум 9-недельным самцам крыс Wistar (Japan SLC) при дозе от 1 до 3 мг/кг. В качестве контроля двум другим крысам вводили 0,5% раствор MC. Через 30 минут у каждой крысы брали кровь под эфирной анестезией через аорту. При этом голову каждой крысы отсекали и извлекали головной мозг.
3 мл собранной крови разбавляли таким же количеством водного физиологического раствора и осторожно помещали на 3 мл гемоцит-отделяющего агента (Nycoplep; AXIS-SHIELD) в пробирку для центрифугирования. Смесь центрифугировали (400 x g, 20 минут) для сбора слоя моноцитов. Полученные моноциты промывали два раза физиологическим раствором, замораживали и хранили при температуре -20°C до их использования для измерений.
К собранному мозгу крыс добавляли буфер (50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA) в пятикратном объеме в расчете на массу животного и смесь гомогенизировали с использованием гомогенизатора во льду с получением однородной суспензии. Затем, используя ультразвуковой генератор (UR-20P, (power dial 4) Tommy Seiko), смесь обрабатывали ультразвуком в течение 5 секунд. К указанным замороженным моноцитам добавляли 100 мкл охлажденного льдом буфера (50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA) и, используя ультразвуковой генератор (UR-20P, (power dial 4) Tommy Seiko), смесь обрабатывали ультразвуком в течение 5 секунд. Концентрацию белка в каждом гомогенате головного мозга и моноцитов измеряли в соответствии со способом окрашивания-связывания (protein assay CBB solution, Nacalai Tesque). Используя буфер (50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA, 0,1 мг/мл BSA, 100 мМ NaCl), гомогенаты головного мозга и моноцитов разбавляли так, чтобы концентрация белка была 80 мкг/мл и 400 мкг/мл, с получением, таким образом, ферментных растворов.
(2) Измерение активности FAAH:
50 мкл ферментного раствора подвергали взаимодействию с 50 мкл раствора субстрата (2 мкКи/мл радиомеченного анандамида (Анандамид [этаноламин 1-3H] (American Radiolabeled Chemical)), добавляли 8 мкМ анандамида (Funakoshi), 50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA) при комнатной температуре в течение 1 часа. К смеси добавляли 200 мкл раствора 1:1 (по объему) хлороформа и метанола и затем перемешивали с завихрением. Смесь центрифугировали (12000 х g, 2 минут), в результате чего разложившийся продукт этаноламина (этаноламин 1-3H) отделялся в верхний слой (водно/метанольный слой), а непрореагировавший радиомеченный анандамид (Анандамид [этаноламин 1-3H]) был в нижнем слое (хлороформный слой). 25 мкл верхнего слоя переносили в 96-луночный стойкий к органическим растворителям белый микропланшет (PicoPlate-96; Perkin Elmer), добавляли 150 мкл Microscinti-20 (Perkin Elmer) и смесь измеряли при помощи сцинтилляционного счетчика для микропланшетов (TopCountTM; Beckman).
На основании активности FAAH в контроле, не содержащем испытываемое вещество гомогенате головного мозга или моноцитов крысы, 100%, и на основании активности FAAH в не содержащем тканевый гомогенат буфере (50 мМ Трис-HCl (pH 8,0), 1 мМ EDTA, 0,1 мг/мл BSA, 100 мМ NaCl), 0%, получали относительное значение (%) активности FAAH в тканевом гомогенате крысы, которой вводили испытываемое вещество. Вещество, которое снижало относительную величину активности FAAH, было выбрано в качестве вещества, ингибирующего активность FAAH.
Представленные выше результаты подтверждают, что, когда испытываемое вещество вводят испытываемому животому и когда можно измерить зависимое от испытываемого вещества изменение активности FAAH в гомогенате ткани животного, тогда его можно скринировать на вещество, ингибирующее активность FAAH, или, иными словами, на лекарственное средство для лечения частого мочеиспускания и недержания мочи, лекарственное средство для лечения повышенной активности мочевого пузыря и/или лекарственное средство для лечения боли.
Пример 441:
Эффект соединения на циклофосфамид (CPA)-индуцируемое частое мочеиспускание у крыс:
Соединения испытывали на их действие по облегчению раздражения мочевого пузыря, используя патологические модели. Известно, что системное введение циклофосфамида (CPA) преобразовывает соединение в его метаболит, акролеин, и, когда он присутствует в моче, он повреждает слизистую мочевого пузыря. У крыс введение CPA вызывает боль в мочевом пузыре или частое мочеиспускание, сопровождаемое геморрагическим циститом, и поэтому, используя таких крыс, можно оценить активность лекарственного средства в отношении этих симптомов. В данном эксперименте использовали 9-недельных самок крыс Wistar (Charles River). Крысам внутрибрюшинно вводили CPA (100 мг/кг) и через 2 дня крыс испытывали. Крысам вводили перорально (п.о.) испытываемое соединение; и через 15 минут перорально вводили принудительным образом дистиллированную воду (30 мл/кг). Крыс помещали в метаболическую клетку и постоянно измеряли количество мочи в течение 1 часа. Общее количество мочи делили на общее количество мочеиспусканий и, таким образом, рассчитывали эффективную емкость мочевого пузыря. В результате, в группе, которой вводили растворитель, 0,5% раствор метилцеллюлозы (MC), эффективная емкость мочевого пузыря уменьшалась, и крысы демонстрировали частое мочеиспускание. При пероральном введении эффективная доза соединений Примеров 2, 218 и 261 составила 3 мг/кг; а эффективная доза соединений Примеров 225, 228, 273, 313, 324, 325 и 359 составила 1 мг/кг. Эти соединения увеличивали уменьшенную эффективную емкость мочевого пузыря и облегчали состояние частого мочеиспускания.
Пример 442:
Эффект соединений против аллодинии у крыс с лигированным спинным нервом L5/L6 (модель невропатической боли):
5-6-недельных самцов крыс SD подвергали операции по лигированию левосторонних L5 и L6 спинных нервов с использованием шелковых нитей. Для оценки аналгетического эффекта испытываемого вещества использовали испытание иглой von Frey. Вкратце, заднюю лапу животного кололи иглой, в результате минимальную силу иглы, вызывающую отдергивание лапы, принимали за пороговое значение ответа (log gram) на механическую стимуляцию. В предварительном испытании было подтверждено, что порог ответа оперированной лапы животного заметно снижался в течение 7-14 дней после операции (при аллодинии), и эффект у испытываемого соединения против аллодинии оценивали в любой день от 7 до 14 после операции. В день перед испытанием измеряли пороговое значение ответа перед введением испытываемого соединения. Испытываемых животных делили на группы таким образом, чтобы средняя разница и отклонение порогового значения до введения испытываемого соединения в группах были небольшими. В оценочном испытании испытываемых соединений измеряли пороговое значение ответа после введения испытываемых соединений. Испытываемое соединение перорально вводили за 60 минут до измерения порогового значения ответа. На основании порогового значения ответа оперированных и неоперированных лап в группе, которой вводили растворитель, 0% и 100%, соответственно, рассчитывали активность испытываемого соединения на его эффект против аллодинии. В результате, при пероральном введении при дозе 10 мг/кг соединения Примера 126 это соединение показало активность против аллодинии 74%.
(M+H)+

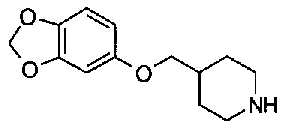

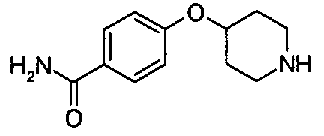
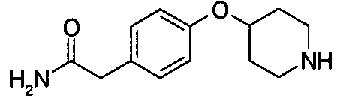
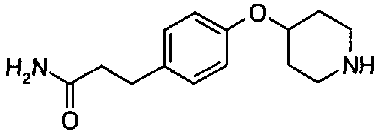
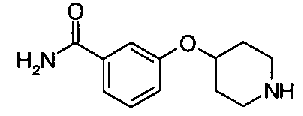
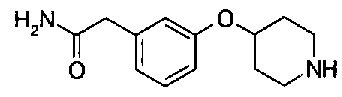
(M+H)+

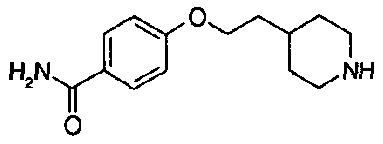
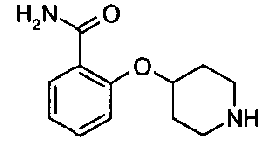
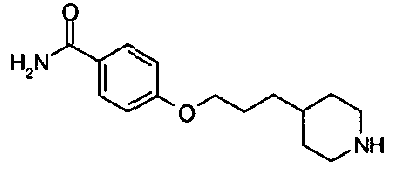
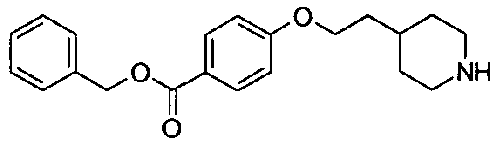
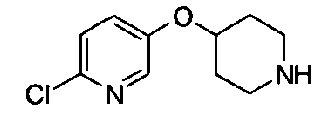

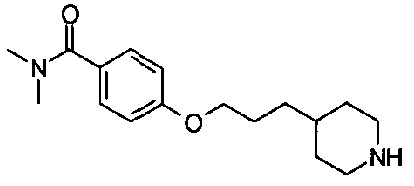
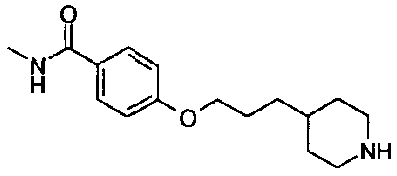

(M+H)+



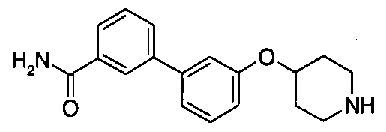


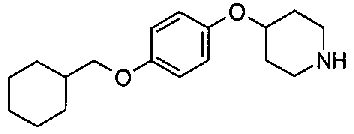

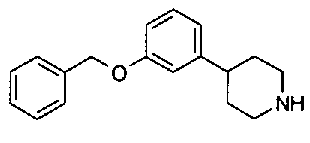
(M+H)+
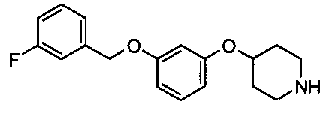
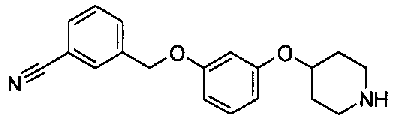
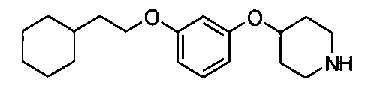
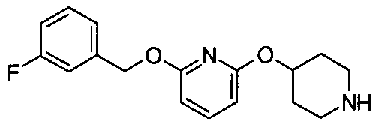
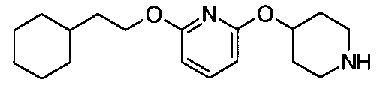
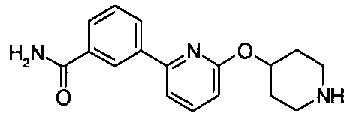

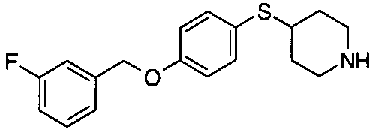
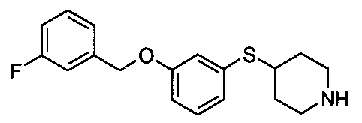

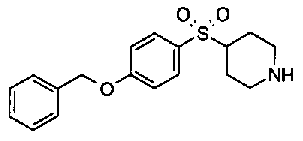
(M+H)+
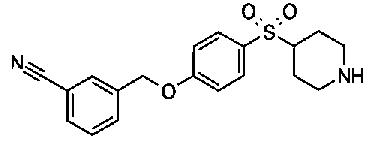
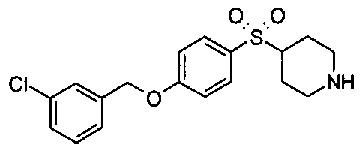

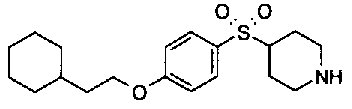
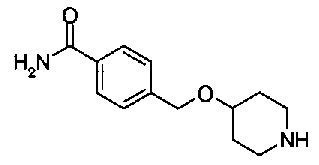
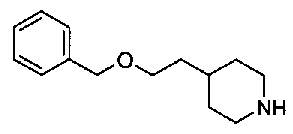
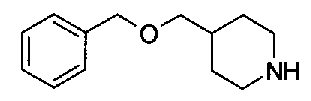

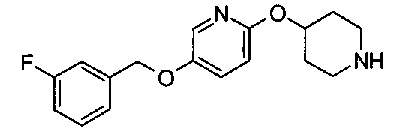
(M+H)+
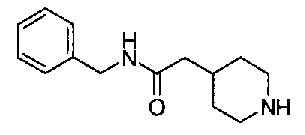
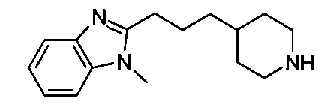
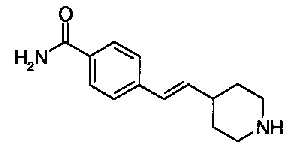
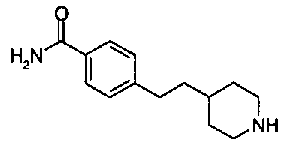
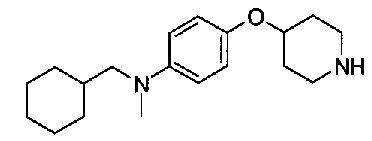
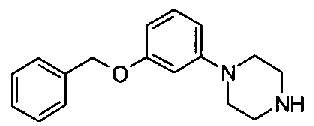

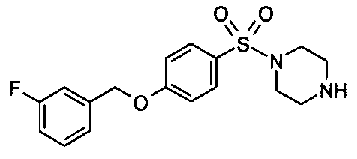
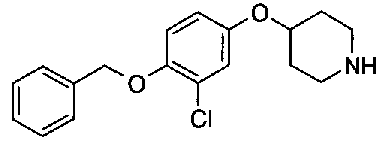
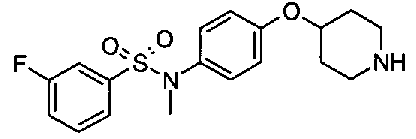
(M+H)+


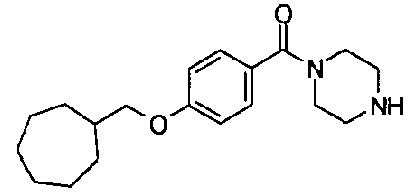
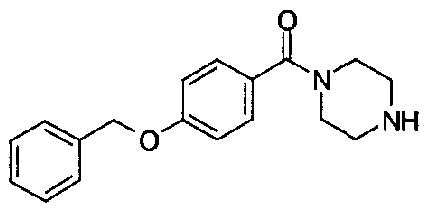
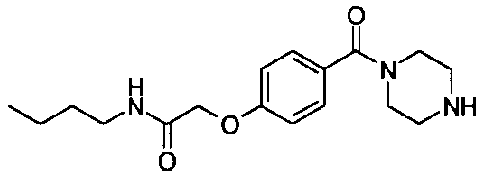

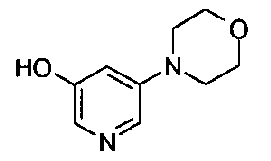
(M+H)+

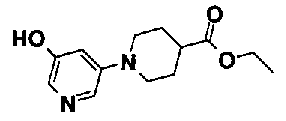
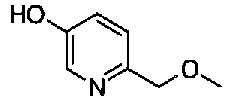
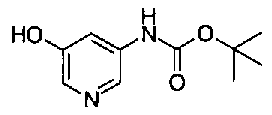

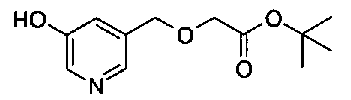
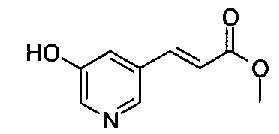
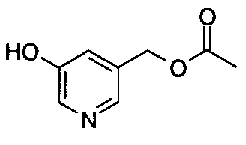
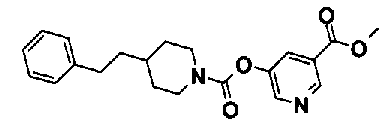

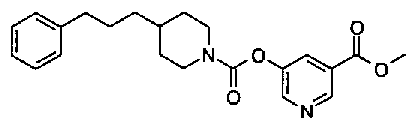
(M+H)+
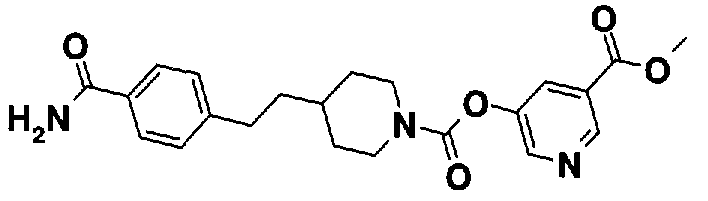
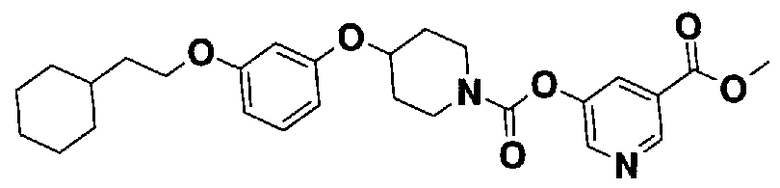

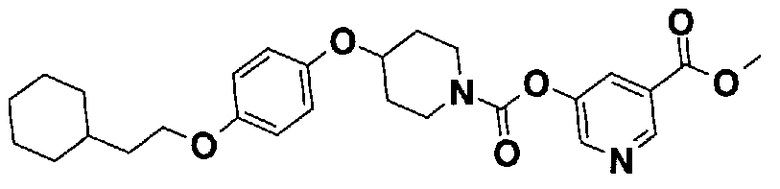

(M+H)+ или
(М-Н)-или (М)+
FAB или
ESI или
EI
FAB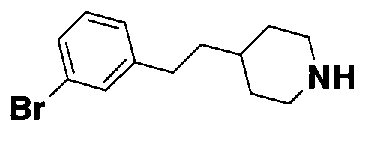
FAB
FAB
FAB
FAB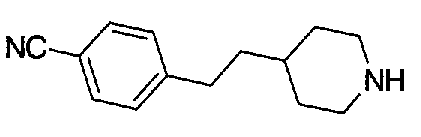
FAB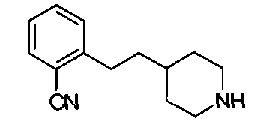
FAB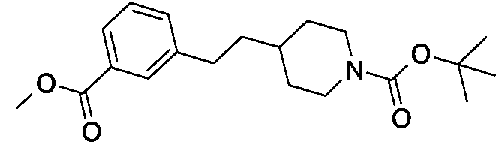
FAB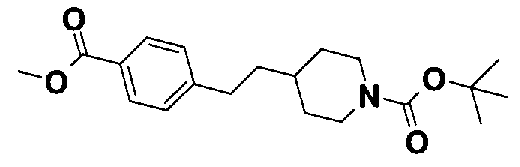
ESI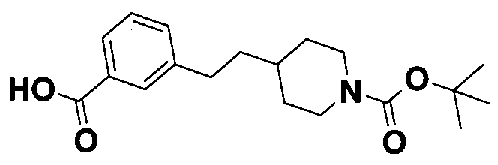
ESI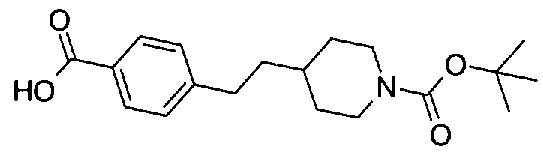
ESI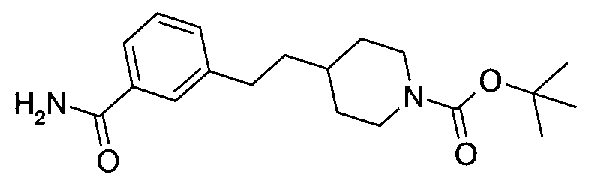
ESI
(M+H)+ или
(М-Н)-или (М)+
FAB или
ESI или
EI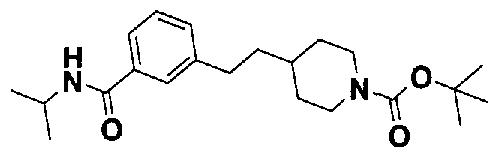
ESI
ESI
API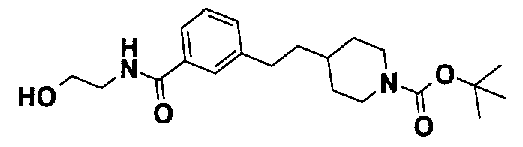
API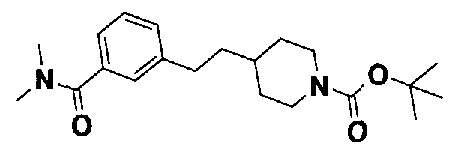
ESI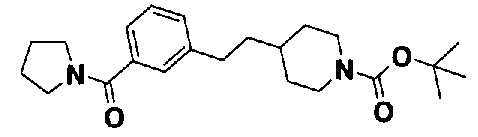
FAB
FAB
ESI
ESI
ESI
ESI
FAB
(M+H)+ или
(М-Н)-или (М)+
FAB или
ESI или
EI
ESI
FAB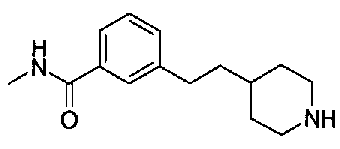
FAB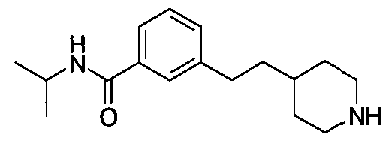
ESI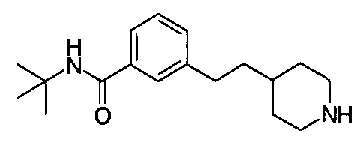
ESI
FAB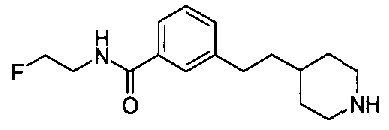
ESI
ESI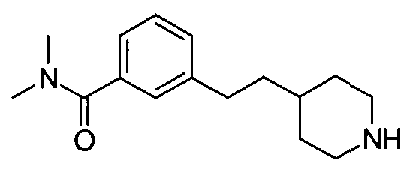
FAB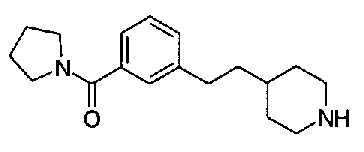
ESI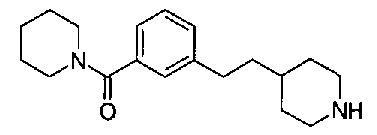
ESI
ESI
(M+H)+ или
(М-Н)-или (М)+
FAB или
ESI или
EI
ESI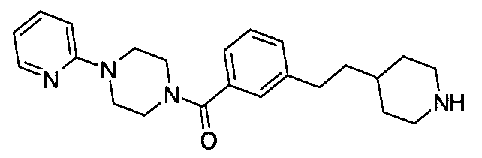
ESI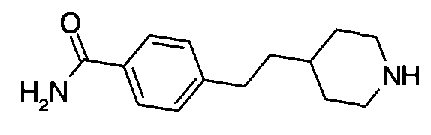
ESI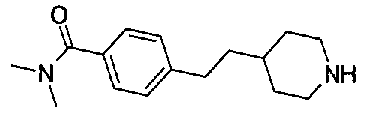
ESI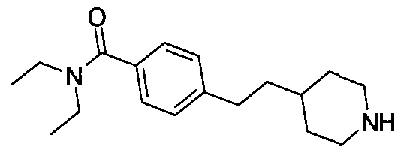
ESI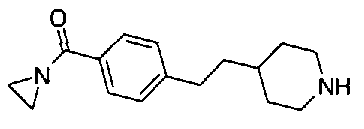
ESI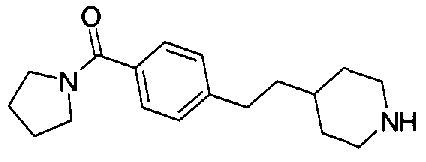
ESI
ESI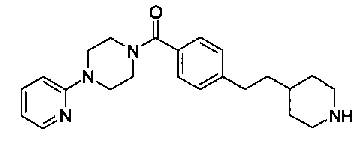
ESI
FAB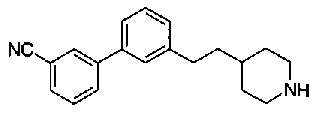
FAB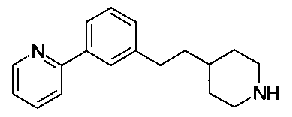
FAB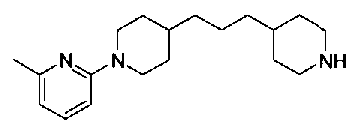
FAB
(M+H)+ или
(М-Н)-или (М)+
FAB или
ESI или
EI
ESI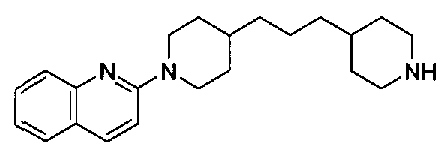
FAB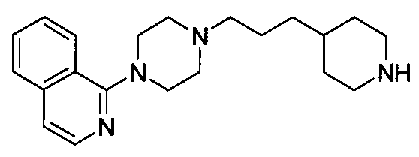
ESI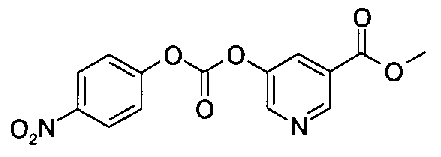
ESI
ESI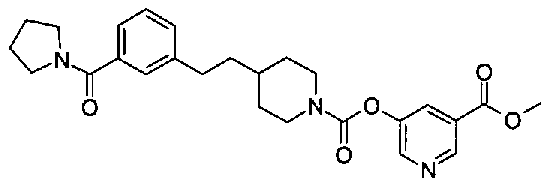
FAB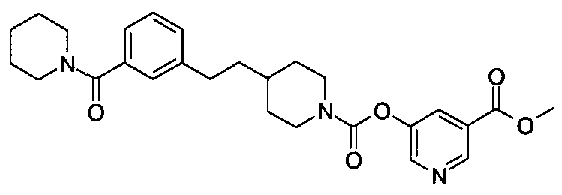
ESI
ESI
ESI
FAB
(M+H)+ или
(М-Н)-или (М)+
FAB или
ESI или
EI
ESI
ESI
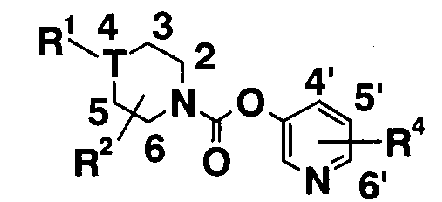


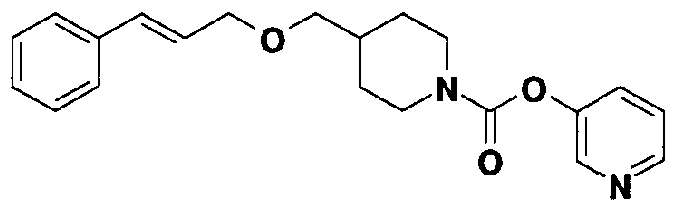
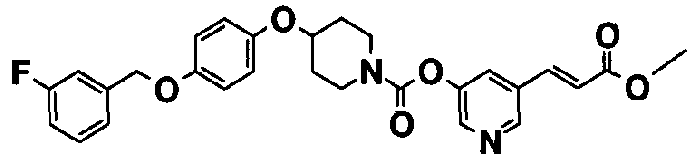

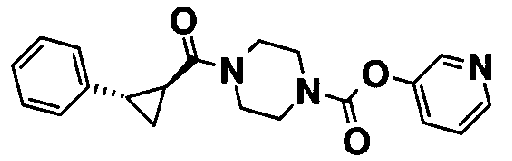
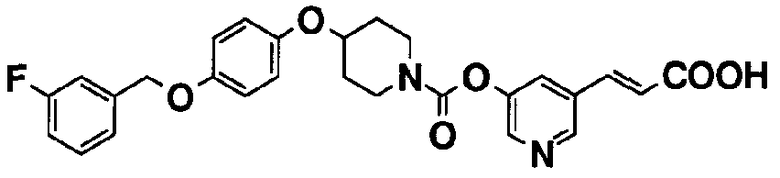
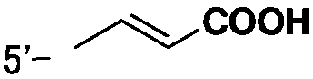
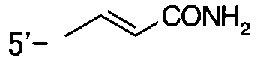
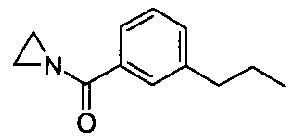



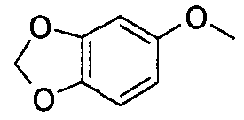
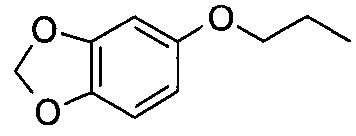
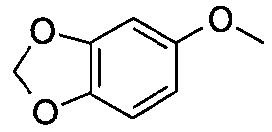
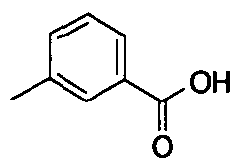
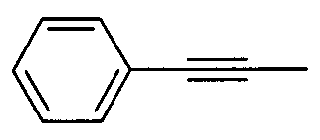


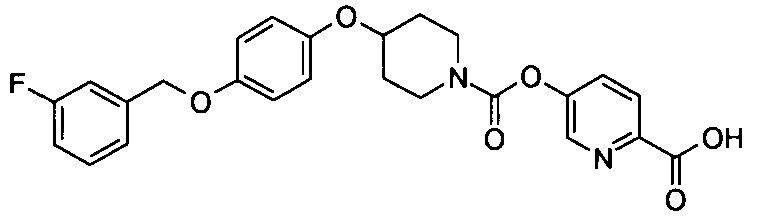
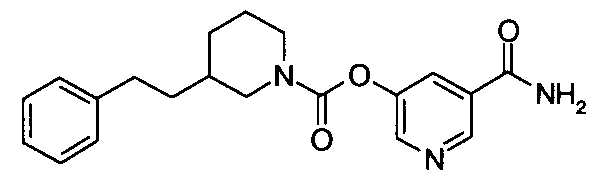
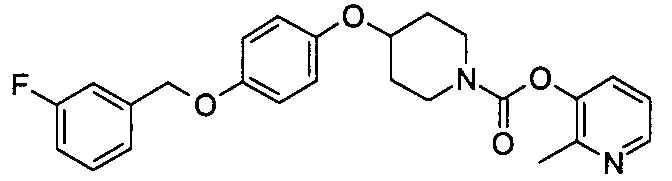

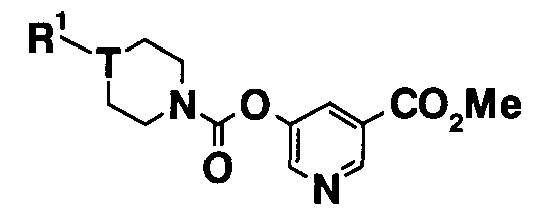

1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
1H-ЯМР δ (м.д.), растворитель: MS m/z
439(M+H)+FAB
ДМСО:455(M+H)+FAB
437(M+H)+FAB
431(M+H)+FAB
1H-ЯМР δ (м.д.), растворитель: MS m/z
465(M+H)+FAB
ДМСО:327(M+H)+FAB
422(M+H)+FAB
495(M+H)+FAB
1H-ЯМР δ (м.д.), растворитель: MS m/z
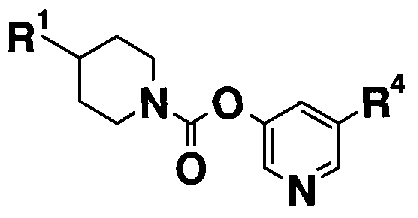
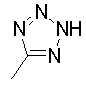
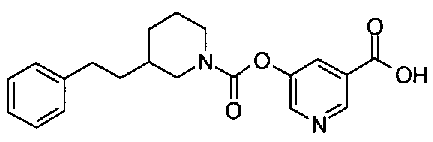
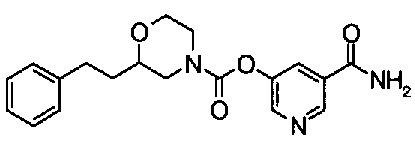

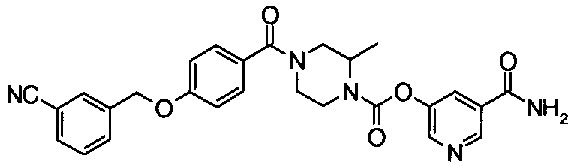
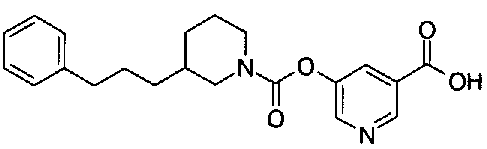
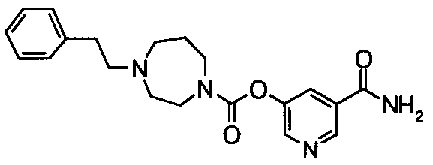
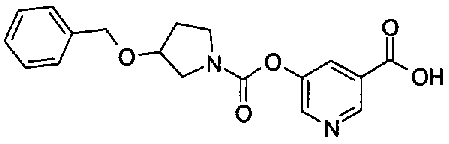
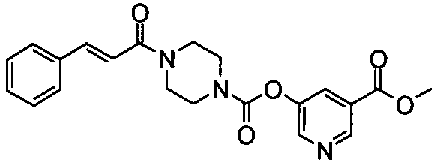

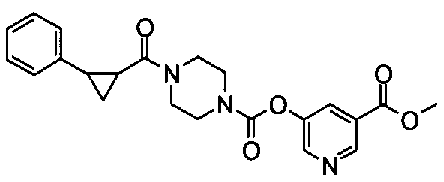

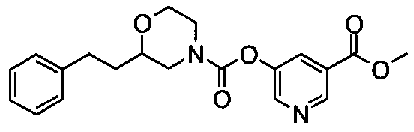
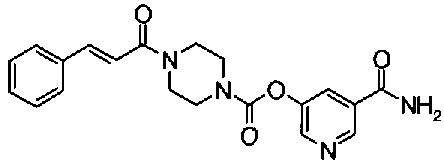



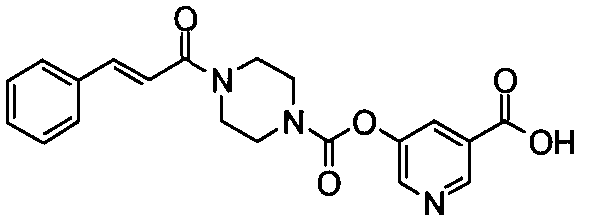
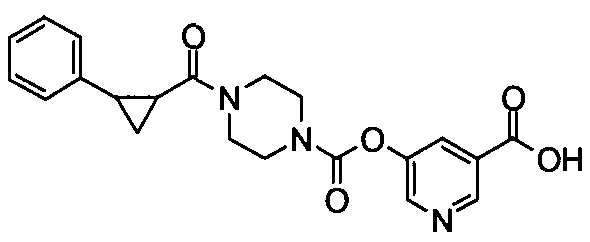
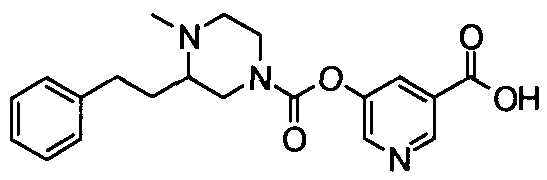

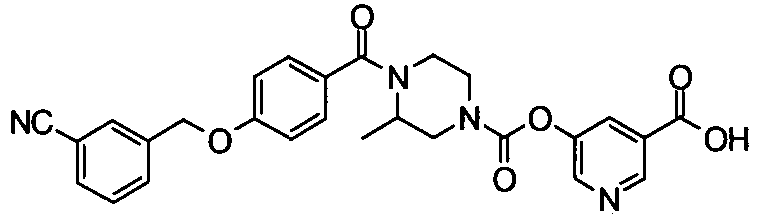
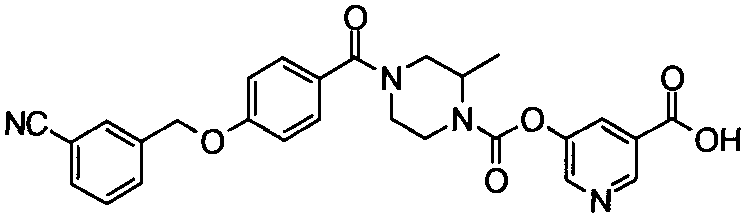
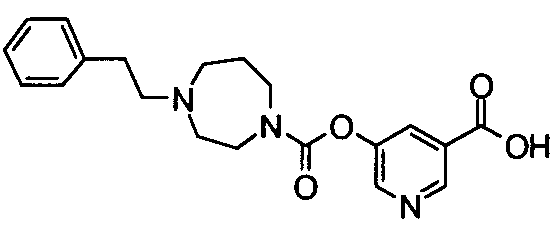
Промышленная применимость
Соединения по настоящему изобретению обладают отличной активностью ингибирования FAAH и являются полезными для лечения связанных с FAAH расстройств, в частности, частого мочеиспускания и недержания мочи, повышенной активности мочевого пузыря и/или боли.
Отдельный перечень последовательностей
Автор изобретения указан в графе, имеющей номер <223> SEQ ID NO:1 в прилагаемом ниже перечне последовательностей.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИДИЛЬНОЕ НЕАРОМАТИЧЕСКОЕ АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛО-1-КАРБОКСИЛАТНОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2408581C2 |
| КОНДЕНСИРОВАННЫЕ БИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛЫ, ОБЛАДАЮЩИЕ DGAT ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ | 2003 |
|
RU2342388C2 |
| ПРОИЗВОДНЫЕ АРИЛМЕТОКСИ ИЗОИНДОЛИНА И КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ИХ, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2567753C2 |
| ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ | 2003 |
|
RU2333203C2 |
| АЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2493154C2 |
| ИНГИБИТОРЫ С-FMS КИНАЗЫ | 2007 |
|
RU2475483C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 7-(ГЕТЕРО)АРИЛ-4,5,6,7-ТЕТРАГИДРО[1,2,3]ТРИАЗОЛО[1,5-A]ПИРИДИНА | 2013 |
|
RU2563254C2 |
| БЕНЗИЛАМИНОВЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2660421C2 |
| КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2389731C2 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2014 |
|
RU2673819C2 |





































Изобретение относится к соединениям с общими формулами I, II, IV и V 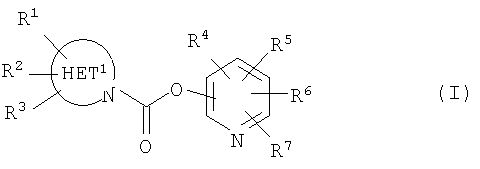 ,
, 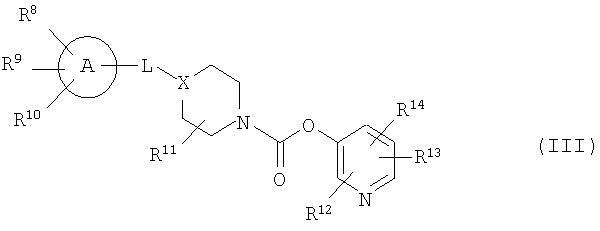 ,
, 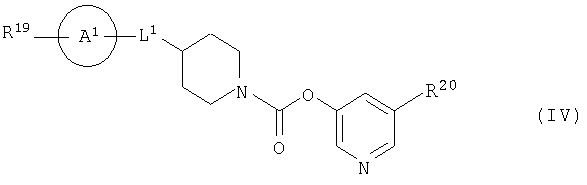 ,
, 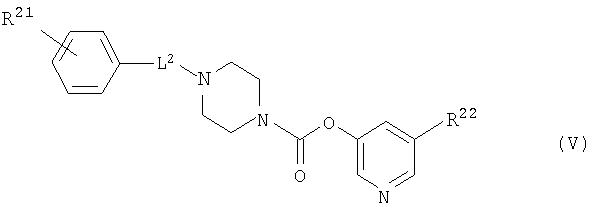 , значения радикалов, такие, как представлено в формуле изобретения. Также представленное изобретение относится к фармацевтической композиции на основе вышеописанных соединений, к их применению, а также к способу лечения частого мочеиспускания, недержания мочи и повышенной активности мочевого пузыря, кроме того, к способу лечения боли. Технический результат: получены и описаны новые соединения, которые могут быть полезны для лечения заболеваний, связанных с жирнокислотной амид-гидролазой (FAAH), в частности, для лечения частого мочеиспускания и недержания мочи, повышенной активности мочевого пузыря и/или боли. 9 н. и 7 з.п.ф-лы, 73 табл.
, значения радикалов, такие, как представлено в формуле изобретения. Также представленное изобретение относится к фармацевтической композиции на основе вышеописанных соединений, к их применению, а также к способу лечения частого мочеиспускания, недержания мочи и повышенной активности мочевого пузыря, кроме того, к способу лечения боли. Технический результат: получены и описаны новые соединения, которые могут быть полезны для лечения заболеваний, связанных с жирнокислотной амид-гидролазой (FAAH), в частности, для лечения частого мочеиспускания и недержания мочи, повышенной активности мочевого пузыря и/или боли. 9 н. и 7 з.п.ф-лы, 73 табл.
1. Пиридильное неароматическое азотсодержащее гетероцикло-1-карбоксилатное производное общей формулы (I) или его фармацевтически приемлемая соль:

где НЕТ1 представляет собой 5-7-членное неароматическое азотсодержащее гетерокольцо,
R1, R2 и R3 являются одинаковыми или отличными друг от друга, и каждый представляет собой
(1) Н,
(2) ОН,
(3) НО-СО-, низший алкил-O-СО-, арил-низший алкил-O-СО- или H2N-CO-арил-низший алкил-O-СО-,
(4) циано,
(5) низший алкил-СО-,
(6) оксо (=O),
(7) формулу [R101-(O)ml]m2-[ALK1, необязательно замещенный ОН]-(O)n1-,
(m1 и n1 являются одинаковыми или отличными друг от друга, где каждый имеет значение 0 или 1),
m2 имеет значение от 1 до 5,
ALK1 представляет собой низший алкилен, низший алкенилен или низший алкинилен,
R101 представляет собой
(i) Н,
(ii) Аr1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей:
(a) H2N-,
(b) галоген,
(c) циано,
(d) необязательно этерифицированный карбоксил,
(e) группу R1011aR1012aN-CO-,
(f) НЕТ2,
(g) Ar1a, необязательно замещенный галогеном, циано, ОН, низшим алкил-O- или низшим алкилом,
Аr1a представляет собой арил,
(h) низший алкил,
(j) ОН,
(k) низший алкил-O-, необязательно замещенный группой Аr1a или
галоген-Аr1a,
(l) HET2-CO-, необязательно замещенный галогеном, Аr1a или HETAr1a,
НЕТ2 представляет собой азотсодержащее гетерокольцо,
НЕТАr1a представляет собой азотсодержащий гетероарил,
(s) HET2-CONR1011a-,
(t) H2NCONH- и
(u) необязательно этерифицированный карбоксил-АLК2a,
ALK2a представляет собой низший алкил или низший алкенил,
(iii) ALK2a, необязательно замещенный группой R1011aR1012aN или Аr1a, R1011a и R1012a являются одинаковыми или отличными друг от друга, и каждый представляет собой
(a) Н,
(b) cALK,
cALK представляет собой циклоалкил,
(c) ALK2a, необязательно замещенный галогеном, cALK, ОН, низший алкил-0- или Аr1a или
(d) Arla-SO2-, необязательно замещенный галогеном,
(iv) НЕТ2, необязательно замещенный, по меньшей мере, одним
заместителем, выбранным из группы, включающей
(a) ALK2a, необязательно замещенный группой Аr1a или галоген-Аr1a,
(b) Аr1a,
(c) НЕТАr1a, необязательно замещенный низшим алкилом,
(d) Аr1a-СО- или галоген-Аr1a-СО-,
(v) cALK, необязательно замещенный ALK2a,
или
(vi) необязательно этерифицированный карбоксил,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R101-(O)m1] могут быть одинаковыми или отличными друг от друга),
(8) группу R102-ALK1-N(R103)-CO-,
(R102 представляет собой
(i) H,
(ii) cALK,
(iii) HETAr1a или
(iv) Ar1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) НО,
(b) ALK2a-O-,
(c) cALR-ALK1-О-,
(d) cALK-Ar1a-ALK1-O-, и
(e) Ar1a-ALK1-O-,
R103 представляет собой
(i)H,
(ii) cALK,
(iii) ALK2a, необязательно замещенный, по меньшей мере, одним
заместителем, выбранным из группы, включающей
(a) НЕТ2,
(b) Аr1a и
(c) галоген-Аr1a,
(iv) НЕТАr1a или
(v) Ar1a-[CO]m1, необязательно замещенный, по меньшей мере, одним
заместителем, выбранным из группы, включающей
(a) cALK,
(b) H2N,
(c) группу R1011aR1012aN-CO- или
(d) ALK2a),
(9) группу R104aR105aN-[CO]m1-ALK1-,
(R104a и R105a являются одинаковыми или отличными друг от друга, и каждый представляет собой группу R103),
(10) группу R106-ALK3-L1-,
(R106 представляет собой
(i) группу R101-(O)m1-,
(ii) группу R104aR105aN-,
(iii) группу ALK2a-CONH- или
(iv) группу Ar1a-CONH-,
ALK3 представляет собой низший алкилен, низший алкенилен или циклоалкилен,
L1 - представляет собой -С(=O)- или -SO2-),
(11) ALK2a-CONH-, необязательно замещенный группой Аr1a,
(12) Аr1a, замещенный галогеном,
(13) группу [R107(O)m1]m2-Ar2-(O)n1-,
(Аr2 представляет собой арилен,
R107 представляет собой
(i) Н,
(ii) галоген,
(iii) ALK2a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) НО,
(b) cALK,
(c) НЕТ2,
(d) Ar1a, необязательно замещенный галогеном, низшим алкилом, низшим алкил-O-, группой Rl011aRl012aN-[CO]p-, циано или необязательно этерифицированным карбоксилом,
(e) необязательно этерифицированный карбоксил,
(f) НЕТ2-[СО]p-, необязательно замещенный группой R1011aR1012aN-[CO]p-, и
(g) группу R1011aR1012aN-[CO]p-,
p имеет значение 0 или 1,
(iv) группу R1011aR1012aN-[CO]p- или
(v) группу R1011aR1012aN-[CO]p-Ar1a,
при этом когда m2 имеет значение от 2 до 5, тогда группы [R107-(O)m1] могут быть одинаковыми или отличными друг от друга, и, кроме того, группа [R107-(O)m1]m2 может представлять собой метилендиокси с образованием кольца),
(14) группу [R107-(O)m1]m2-Ar2-N(R103)-CO-,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R107-(O)m1] могут быть одинаковыми или отличными друг от друга),
(15) группу R1011aR1012a-N-[СО]m1]m2-Аr2(О)n1-,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R1011aR1012a-[CO]m1] могут быть одинаковыми или отличными друг от друга),
(16) группу [R108]m2-Ar2-L2-,
[R108 представляет собой
(i)H,
(ii) галоген,
(iii) НО,
(iv) cALK-O-,
(v) группу R109-ALK1-(O)m1-,
(R109 представляет собой
(a) Н,
(b) cALK,
(c) Аr1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(1') галоген,
(2') циано,
(3') NO2,
(4') ALK2a, необязательно замещенный галогеном,
(5') НО,
(6') ALK2a-O-, необязательно замещенный галогеном,
(7') необязательно этерифицированный карбоксил или
(8') группу R104aR105aN,
(d) HETAr1a или
(e) группу R104aR105aN-[CO]m1-),
(vi) группу R1013R1014N-,
R1013 и R1014 являются одинаковыми или отличными друг от друга, и каждый представляет собой
(i) H,
(ii) ALK2a,
(iii) cALK-ALK1- или
(iv) Ar1a-ALK1-, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(1') галоген,
(2') циано,
(3') ALK2a, необязательно замещенный галогеном,
(4') ALK2a-O-, необязательно замещенный галогеном,
(vii) HET2-(O)m1-, необязательно замещенный низшим алкилом,
L2 представляет собой -СО- или -S(O)q-,
q имеет значение 0, 1 или 2,
при этом когда m2 имеет значение от 2 до 5, тогда группы [R108] могут быть одинаковыми или отличными друг от друга],
(17) группу [R101]m2-Ar2-CONH-,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R101] могут быть одинаковыми или отличными друг от друга),
(18) группу [R111]m2-НЕТАr2-(О)m1-,
(R111 представляет собой
(i) H,
(и) галоген,
(iii) оксо (=O) или
(iv) группу R103a-(O)n1-,
R103a представляет собой
(i) Н,
(ii) cALK,
(iii) ALK2a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) НЕТ2,
(b) Аr1a,
(c) cALK и
(d) галоген-Аr1a,
(iv) НЕТАr1a или
(v) Аr1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей (a) cALK, (b) H2N и (с) группу R1011aR1012aN-CO-,
НЕТАr2 представляет собой азотсодержащий гетероарилен,
при этом когда m2 имеет значение от 2 до 5, тогда группы [R111] могут быть одинаковыми или отличными друг от друга),
(19) формулу [R112]m2-HETAr2-N(R103)-CO-,
(R112 представляет собой
(i) H,
(ii) cALK,
(iii) ALK2a или
(iv) Ar1a, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из группы, включающей
(a) галоген,
(b) НО,
(c) ALK2a-O- и
(d) Ar1a-ALK1-O-,
при этом когда m2 имеет значение от 2 до 5, тогда группы [R112] могут быть одинаковыми или отличными друг от друга,
(20) формулу [R108]m2-HETAr2-L2-,
(при этом когда m2 имеет значение от 2 до 5, тогда группы [R108] могут быть одинаковыми или отличными друг от друга),
при условии, что, когда какая-либо из групп R1, R2 и R3 представляет собой группу [Rlll]m2-HETAr2-(O)m1-, и когда m1 имеет значение 0, тогда остальные группы
R1, R2 и R3 представляют собой Н;
R4, R5, R6 и R7 представляют собой Н;
при условии, что пиридин-3-ил 4-метилпиперазин-1-карбоксилат и пиридин-2-ил 2-оксопиридин-1(2Н)-карбоксилат исключаются.
2. Соединение по п.1, представленное общей формулой (II), или его фармацевтически приемлемая соль:

где R1-R7 имеют значения, определенные в п.1,
Т представляет собой СН2, NH, NHCH2 или О,
и также включается случай, где водород в Т замещен R1-R3.
3. Соединение по п.2, где R1-R3 являются одинаковыми или отличными друг от друга, и каждый представляет собой H, группу [R101-(O)m1]m2-[ALK1, необязательно замещенный ОН]-(O)n1-, группу R102-ALK1-N(R103)-СО-, группу R106-ALK3-L1-, группу [R107-(O)m1]m2-Ar2-(O)n1-, группу [R107-(O)m1]m2-Ar2-N(R103)-CO- или группу [R108]m2-Ar2L2-, или его фармацевтически приемлемая соль.
4. Пиридильное неароматическое азотсодержащее гетероцикло-1-карбоксилатное производное общей формулы (III) или его фармацевтически приемлемая соль:
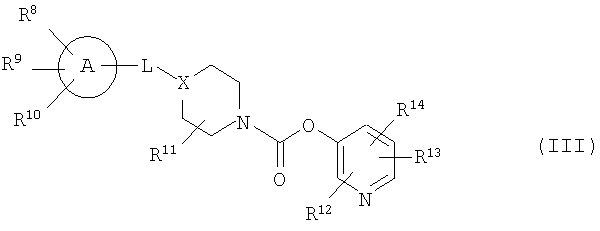
где кольцо А представляет собой бензольное кольцо, циклопентановое кольцо, циклогексановое кольцо, циклогептановое кольцо или 5-7-членное азотсодержащее гетерокольцо;
L представляет собой простую связь, низший алкилен, низший алкенилен, -N(R15)-C(=O)-, -C(=O)-N(R15)-, -(низший алкенилен)-С(=О)-, -О- или -С(=O)-,
R15 представляет собой Н или низший алкил,
Х представляет собой СН или N,
R8-R10 являются одинаковыми или отличными друг от друга, и каждый представляет собой
группу, выбранную из представленной ниже группы G,
арил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
азотсодержащий гетероарил, необязательно замещенный одинаковыми
или отличными друг от друга группами, выбранными из представленной ниже группы G,
R16-(низший алкилен)-O-,
R16-(низший алкилен)-N(R15)- или
R17R18N-C(=O)-,
R16 представляет собой
арил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
азотсодержащий гетероарил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G, или
3-8-членный циклоалкил,
R17 и R18 являются одинаковыми или отличными друг от друга, и каждый представляет собой Н, низший алкил или 3-8-членный циклоалкил,
(кроме того, R17 и R18 могут образовывать, вместе с атомом N, связанным с ними, 3-8-членное азотсодержащее гетерокольцо),
группа G включает Н, галоген, -CN, -СF3, низший алкил или -O-низший алкил,
R11 представляет собой Н, низший алкил или оксо (=O),
R12-R14 представляют собой Н.
5. Соединение по п.4, где кольцо А представляет собой бензольное кольцо, циклогексановое кольцо, пиперидиновое кольцо или пиперазиновое кольцо, или его фармацевтически приемлемая соль.
6. Соединение по п.5, где R9, R10 и R11 представляют собой Н, или его фармацевтически приемлемая соль.
7. Пиридильное неароматическое азотсодержащее гетероцикло-1-карбоксилатное производное общей формулы (IV) или его фармацевтически приемлемая соль:
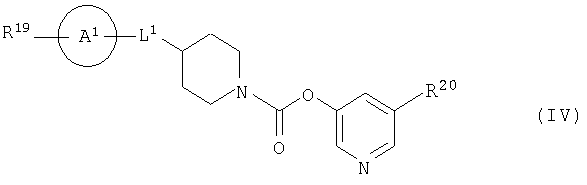
где кольцо А1 представляет собой бензольное кольцо, пиперидиновое кольцо или пиперазиновое кольцо;
L1 представляет собой низший алкилен, низший алкенилен, -N(R15)-C(=O)-или -O-;
R15 представляет собой Н или низший алкил,
R19 представляет собой
группу, выбранную из представленной ниже группы G,
азотсодержащий гетероарил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
R16-(низший алкилен)-O- или R17R18N-C(=O)-,
R16 представляет собой
арил, необязательно замещенный одинаковыми или отличными друг от друга группами, выбранными из представленной ниже группы G,
азотсодержащий гетероарил, необязательно замещенный одинаковыми
или отличными друг от друга группами, выбранными из представленной ниже группы G, или
3-8-членный циклоалкил,
R17 и R18 являются одинаковыми или отличными друг от друга, и каждый представляет собой Н или низший алкил,
(кроме того, R17 и R18 могут образовывать, вместе с атомом N, связанным с ними, 5- или 6-членное азотсодержащее гетерокольцо),
группа G включает Н, галоген, -CN, -СF3, низший алкил или -O-низший алкил,
R20 представляет собой Н.
8. Пиридильное неароматическое азотсодержащее гетероцикло-1-карбоксилатное производное общей формулы (V) или его фармацевтически приемлемая соль:
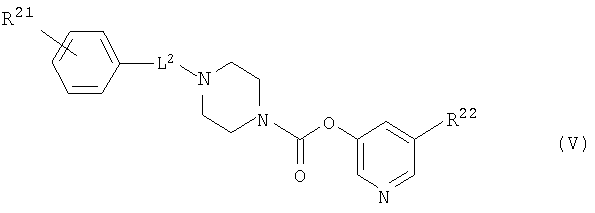
где L2 представляет собой низший алкилен, низший алкенилен или -(низший алкенилен)-С(=O)-,
R21 представляет собой Н, галоген, -CN, -СF3, низший алкил или -O- низший алкил,
R22 представляет собой Н.
9. Соединение по п.1, выбранное из следующей группы:
пиридин-3-ил4-{4-[(3-фторбензил)окси]фенокси}пиперидин-1-карбоксилат,
пиридин-3-ил 4-[(2Е)-3-фенилпроп-2-еноил]пиперазин-1-карбоксилат,
пиридин-3-ил 4-(анилинокарбонил)пиперидин-1-карбоксилат,
пиридин-3-ил 4-(2-фенилэтил)пиперазин-1-карбоксилат,
или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, обладающая FAAH ингибирующей активностью, содержащая соединение по любому из пп.1-9 или его фармацевтически приемлемую соль в качестве ее активного ингредиента.
11. Фармацевтическая композиция по п.10, которая представляет собой лекарственное средство для лечения частого мочеиспускания, недержания мочи и/или повышенной активности мочевого пузыря.
12. Фармацевтическая композиция по п.10, которая представляет собой лекарственное средство для лечения боли.
13. Применение соединения по любому из пп.1-9 или его фармацевтически приемлемой соли для получения ингибитора FAAH или лекарственного средства для лечения частого мочеиспускания, недержания мочи и/или повышенной активности мочевого пузыря.
14. Применение соединения по любому из пп.1-9 или его фармацевтически приемлемой соли для получения ингибитора FAAH или лекарственного средства для лечения боли.
15. Способ лечения частого мочеиспускания, недержания мочи и/или повышенной активности мочевого пузыря, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-9 или его фармацевтически приемлемой соли.
16. Способ лечения боли, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп. 1-9 или его фармацевтически приемлемой соли.
| Накопитель короткомерных лесоматериалов | 1974 |
|
SU477903A1 |
| J | |||
| Med | |||
| Chem | |||
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| WO 00/59510, 12.10.2000 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| J | |||
| Pharm | |||
| Sci, vol.81, no.4, 1992, p.380-385 | |||
| Chemische berichte, vol.118, no.2, 1985, p.468-482 | |||
| RU 98105684, 10.02.2000. | |||

