Настоящее изобретение относится к очистке углеводородов, содержащих сернистые соединения. Более конкретно оно относится к процессу обессеривания жидких углеводородов, содержащих сераорганические соединения, путем экстракции труднокрекингируемых сераорганических (ТКС) соединений в регенерируемую ионную жидкость, модифицированную солями переходных металлов, включая кобальт, никель, железо, медь, в том числе различные комплексы кобальта (II).
По причинам экономического и экологического характера имеется все возрастающая потребность в жидких углеводородных смесях, содержащих очень низкие концентрации серы. Например, в топливе для автомобилей уровень содержания серы не должен превышать 30 ppm.
В настоящее время для промышленной очистки жидких углеводородов нефти наиболее часто применяется гидроочистка. Этот метод, как известно, обеспечивает практически полное удаление меркаптанов, сульфидов и дисульфидов из жидких углеводородов (см. «Технология переработки нефти и газа», ч.3, «Химия», Москва, 1966). Недостатком этого способа является высокое остаточное содержание тиофенов и высокая стоимость процесса.
Другие известные способы очистки жидкого углеводородного сырья включают окисление сераорганических соединений путем контактирования сырья с водным раствором серной кислоты, содержащим ионы металла, выбранного из группы, включающей марганец, ванадий, хром, кобальт, церий или их смесь, и предварительно обработанный электролизом в условиях окисления, повышающих степень окисления указанных ионов. Полученную в результате контактирования смесь разделяют с получением очищенного жидкого углеводородного сырья и отработанного рабочего раствора, содержащего восстановленные ионы металла, с последующей регенерацией электролизом в условиях окисления ионов металла в степени окисления, превышающей их минимальную степень окисления и возвращением в процесс (см. патент РФ №2101320). Недостатками этого способа являются невозможность его применения для очистки газообразных углеводородов и высокие концентрации серной кислоты. Эти недостатки были устранены путем окисления сернистых соединений при контактировании углеводородного сырья с рабочим реагентом, содержащим ионы переходных металлов в степени окисления, превышающей их минимальную степень окисления, разделение полученной смеси с получением очищенного углеводородного сырья и отработанного рабочего реагента, который дополнительно содержит ионы по меньшей мере одного и/или сам металл, выбранный из группы непереходных металлов, включающих медь, свинец, олово, золото, нанесенные на поверхность сорбента. Наличие ионов металлов или самих металлов увеличивает окислительную способность рабочего реагента и обеспечивает дополнительное комплексообразование с сернистыми соединениями, находящимися в очищаемом углеводородном сырье. Соли металлов, нанесенные на поверхность сорбента, позволяют упростить процесс очистки углеводородного сырья, поскольку разделение смеси, полученной в результате контактирования, может быть осуществлено простым фильтрованием (см. патент RU №2166530). К недостаткам этого метода можно отнести использование металлов (медь, свинец, олово, золото) в качестве окислителей, и вообще, выбранные металлы и их ионы являются экологически опасными (медь, свинец, олово) или дорогостоящими (золото).
Улучшение качества углеводородного сырья за счет более полного удаления сераорганических веществ достигается обработкой очищаемых углеводородов водным раствором хлорида одновалентного катиона в турбулентном режиме под воздействием электрического поля с градиентом напряженности не менее 0,01 В/см2. В качестве водного раствора хлорида одновалентного катиона используют водные растворы соединений NaCl, KCl, LiCl, NH4Cl или их смеси в соотношении целевой продукт: водный раствор хлорида одновалентного катиона 1÷1 - 1÷50. Этим способом можно очистить парафиновые, олефиновые и ароматические углеводороды, например, бензол, этилбензол, изопропилбензол и другие углеводороды в жидком состоянии (см. патент RU №2026335). Недостатком этого метода окисления сераорганических соединений в условиях генерации активного хлора является возможность образования различных супертоксичных хлорорганических ароматических соединений, в том числе диоксинов, особенно на углеродных электродах (см. Савельева Т.В. «Деструкция ароматических соединений в условиях электрохимической генерации активного хлора». Дисс. канд. хим. наук, М. МГУ, 2001; Г.А.Богдановский, Т.В.Савельева, Т.С.Сабурова, Электрохимия, т.37, в.8, с.1008, 2001).
Задача снижения содержания сераорганических соединений в конечных продуктах нефтепереработки может быть решена за счет повышения степени извлечения сернистых соединений из углеводородного сырья их электрохимическим окислением с использованием ионных жидкостей (см. патент US 6274026 B1). Метод основан на электрохимическом окислении в ионной жидкости серосодержащих соединений с последующим удалением продуктов окисления центрифугированием и (или) отгонкой углеводородной фракции. Способ применим к присутствующим в нефти меркаптанам и немеркаптановым серосодержащим соединениям, присутствующим в нефти в виде тиофенов и бензотиофенов и способным при электрохимическом окислении образовывать димеры или олигомеры, осаждающиеся на аноде или образующие в ионной жидкости нерастворимый осадок. В качестве ионных жидкостей используют 1-бутил-3-метилимидазолий гексафторфосфат, 1-этил-3-метилимидазолий тетрахлоралюминат, 1-бутилпиридиний нитрат, 1-бутил-3-метилимидазолий тетрафторборат и их смеси. Окисление проводят в электрохимическом ректоре при потенциале 1,0-2,5 В по хлорсеребряному электроду сравнения при плотности тока 1-10 мА/см2 на платиновых, никелевых, графитовых электродах в интервале температур 0-100°С. Недостатком этого способа является ограничение по применению его только в случае серосодержащих соединений, способных к окислительной электрополимеризации, а такие соединения как, например, дибензотиофен не удаляются.
Наиболее близким к предлагаемому способу по совокупности существенных признаков является способ экстракции сераорганических соединений из углеводородов ионными жидкостями (см. патент US 7001504 В2). Метод основан на удалении серосодержащих органических соединений экстракцией из жидкой или газообразной углеводородной фазы в ионную жидкость с последующим разделением образующейся двухфазной системы декантацией или центрифугированием и регенерацией ионной жидкости. В качестве ионных жидкостей используют 1-бутил-3-метилимидазолий гексафторфосфат, 1-этил-3-метилимидазолий тетрахлоралюминат, 1-бутилпиридиний нитрат, 1-бутил-3-метилимидазолий тетрафторборат и их смеси. Оптимизация процесса экстракции проводится посредством подбора времени контакта (5-60 мин), температуры (30-50°С), давления (1-50 атм), парциальным окислением серосодержащих соединений до сульфоксидов или сульфонов до или в процессе экстракции. Окисление проводится биологическим или химическим способом с использованием в качестве катализаторов окисления солей или оксидов металлов, выбранных из группы платина, палладий, ванадий, никель. Способ удаления серосодержащих соединений из ионной жидкости выбирается из известных методик: нагревание ионной жидкости для испарения серосодержащих соединений, экстракция серосодержащих соединений из ионной жидкости другими растворителями, пропусканием через ионную жидкость инертных газов, испарением серосодержащих соединений под вакуумом, окислением серосодержащих соединений до сернистого газа, экстракцией сверхкритическим СО2 или комбинацией этих методов. Содержание серосодержащих соединений в углеводородной фазе после контакта с ионной жидкостью без стадии окисления снижается на 10-15%. Для снижения содержания серосодержащих соединений в углеводородной фазе на 50-80% требуется многократное проведение стадии экстракции. Недостатком этого метода является необходимость проведения многократной экстракции для снижения содержания серосодержащих соединений в углеводородной фазе на 50-80% с постоянной регенерацией ионной жидкости после одной стадии экстракции для восстановления абсорбционной емкости.
Целью настоящего изобретения является улучшение качества углеводородного сырья за счет более полного удаления серосодержащих органических веществ.
Предлагаемый способ снижения содержания серосодержащих соединений в углеводородном сырье основан на более высокой растворимости серосодержащих соединений в ионной жидкости по сравнению с углеводородами. Процесс экстракции включает в себя смешивание углеводородной фазы с ионной жидкостью с последующим отделением углеводородной фазы и регенерацией ионной жидкости. Предлагаемый способ может быть реализован в относительно мягких условиях с использованием стандартных операций, что приводит к значительному снижению капитальных затрат.
Предлагаемый способ применим для удаления серосодержащих органических соединений, в том числе и труднокрекингируемых (ТКС - тиофенов, бензо- и дибензотиофенов), из любых типов углеводородного сырья без ограничений.
Ионные жидкости (ИЖ) стали альтернативой классическим растворителям благодаря своим уникальным свойствам. Они негорючи, имеют пренебрежимо малое давление паров, термически устойчивы, нетоксичны, проводят электрический ток. Дополнительным преимуществом использования ионных жидкостей для очистки углеводородного сырья является экстракция азотсодержащих соединений. Кроме перечисленного, ионные жидкости удовлетворяют всем основным принципам «зеленой химии» (см. Л.М.Кустов, И.П.Белецкая, Рос. хим. ж., 2004, т.48, №6, с.3).
Критериями выбора ионной жидкости для экстракции являются температура плавления (Тпл ниже 50°С) и растворимость углеводородов в ИЖ (менее 1%). Для этой цели могут быть использованы ионные жидкости, содержащие следующие типы катионов и анионов.
Катионы: алкилимидазолий, алкилпиридиний, полиалкиламмоний, алкилпиперидиний и др.
Анионы: тетрафторборат, гексафторфосфат, трифлат, бис(трифлат)имид и др.
Наиболее подходящими для выбранной цели являются ИЖ на основе замещенного катиона имидазолия с анионом тетрафторбората. На экстракционную способность имидазолиевой ионной жидкости также оказывает влияние длина углеводородной цепи заместителя. В предлагаемом способе использованы ионные жидкости 1-метил-3-бутилимидазолий тетрафторборат (MBImBF4) и 1-метил-3-октилимидазолий тетрафторборат (MOImBF4). Увеличение экстракционной емкости может быть достигнуто также добавлением катионов различных металлов, что обеспечивает дополнительное комплексообразование с сернистыми соединениями, находящимися в очищаемом углеводородном сырье.
Поставленная задача достигается экстракцией серосодержащих соединений из углеводородных смесей в ионную жидкость в процессе их смешивания. Увеличение экстракционной емкости ионной жидкости достигается использованием ионной жидкости с катионом имидазолия, замещенным в третьем положении на бутильный или октильный радикал, и добавлением одного и более ионов металла, выбранных из группы, включающей кобальт, никель, железо, медь, в том числе различные комплексы кобальта (II): двухводный ацетилацетонат, комлекс с ацетоуксусным эфиром, безводный ацетилацетонат, пивалат, 4,5-дикарбоксифталоцианиновый и N,N-бис(салицилиден)этилендиаминовый комплексы.
Заявляемый способ осуществляется следующим образом.
Модельную смесь бензол - октан (объемное соотношение 1:5) (I) или гептан (II) с концентрацией бензотиофена (БТ) или дибензотиофена (ДБТ) 1500 ppm (для I) или 500 ppm (для II) объемом 1 мл смешивают с 1 мл рабочего реагента - ионной жидкостью. Экстракцию проводят при перемешивании в течение 15-60 минут при температуре 25°С. После перемешивания смесь разделяется на две фазы: ионная жидкость находится снизу, углеводородная фаза сверху. Углеводородную фазу отделяют и анализируют на остаточное содержание серосодержащих соединений. Анализ остаточного содержания БТ и ДБТ в гептане проводят, например, на спектрофотометре в УФ-диапазоне по градуировочному графику при длине волны λ=322 нм для БТ и при длине волны λ=327 нм для ДБТ. Анализ остаточного содержания БТ и ДБТ в модельной смеси (I) определяют с помощью газового хроматографа, например, с использованием колонки 3 м × 3 мм, содержащей 5% SE-30 на хромосорбе. Рабочий реагент готовят путем растворения расчетного количества нитратов металлов, выбранных из группы, включающей кобальт, никель, железо, медь, или различных комплексов кобальта (II): двухводного ацетилацетоната (А), комлекса с ацетоуксусным эфиром (В), безводного ацетилацетоната (С), пивалата (D), 4,5-дикарбоксифталоцианинового (Е) и N,N-бис(салицилиден)этилендиаминового комплексов (F). Концентрация добавок составляет С=10-3 моль/л. Регенерацию ионной жидкости проводят электрохимическим окислением или восстановлением серосодержащих соединений в трехэлектродном реакторе с неразделенным катодным и анодным пространством. Электрохимическое воздействие проводят в электрохимическом ректоре при потенциале -1,0÷+2,5 В по квазиобратимому платиновому электроду сравнения при плотности тока 1-10 мА/см2 на платиновых или графитовых электродах в интервале температур 25-50°С. Продукты электрохимического окисления осаждаются на электроде в твердом виде и легко удаляются с поверхности. Продукты электрохимического восстановления скапливаются на дне реактора и могут быть отделены от ионной жидкости с помощью стандартных процедур. Коэффициент распределения D рассчитывают по формуле
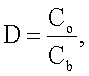
где Со и Cb - концентрации вещества в ионной жидкости и в углеводородной фазе соответственно.
Степень извлечения R определяют по формуле
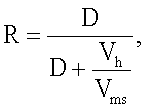
где Vh и Vms - объемы углеводородной фазы и ионной жидкости соответственно.
Примеры осуществления способа.
Пример 1. Способ осуществлен в лабораторных условиях. В реакторе смешивают модельную смесь (I) с БТ и ионную жидкость MBImBF4, перемешивают с помощью магнитной мешалки в течение 30 мин при температуре 25°С. Остаточное содержание БТ в модельной смеси составляет 1000 ppm.
Пример 2. Способ осуществлен, как описано в примере 1. В качестве рабочей жидкости использован MBImBF4 с добавкой рассчитанного количества нитрата меди (I). Остаточное содержание БТ в модельной смеси составляет 900 ppm.
Пример 3. Способ осуществлен, как описано в примере 1. В качестве рабочей жидкости использован MBImBF4 с добавкой рассчитанного количества нитрата кобальта (II). Остаточное содержание БТ в модельной смеси составляет 750 ppm.
Пример 4. Способ осуществлен, как описано в примере 1. В качестве рабочей жидкости использован MBImBF4 с добавкой рассчитанного количества нитрата никеля (II). Остаточное содержание БТ в модельной смеси составляет 850 ppm.
Пример 5. Способ осуществлен, как описано в примере 1. В качестве рабочей жидкости использован MBImBF4 с добавкой рассчитанного количества нитрата железа (II). Остаточное содержание БТ в модельной смеси составляет 850 ppm.
Пример 6. Способ осуществлен в лабораторных условиях. В реакторе смешивают гептан с БТ и ионную жидкость MBImBF4, перемешивают с помощью магнитной мешалки в течение 30 мин при температуре 25°С. Результаты приведены в таблице.
Пример 7. Способ осуществлен в лабораторных условиях, в реакторе смешивают гептан с БТ и ионную жидкость MOImBF4, перемешивают с помощью магнитной мешалки в течение 30 мин при температуре 25°С. Результаты приведены в таблице.
Пример 8. Способ осуществлен, как описано в примере 7. В качестве рабочей жидкости использован MOImBF4 с добавкой рассчитанного количества 4,5-дикарбоксифталоцианинового комплекса.
Результаты анализа приведены в таблице.
Примеры 9-10. Способ осуществлен в лабораторных условиях. В реакторе смешивают гептан с ДБТ и ионную жидкость MBImBF4 (9) или MOImBF4 (10), перемешивают с помощью магнитной мешалки в течение 30 мин при температуре 25°С. Результаты приведены в таблице.
Примеры 11-12. Способ осуществлен, как описано в примере 10. В качестве рабочей жидкости использован MOImBF4 с добавками рассчитанного количества различных комплексов кобальта (II): 4,5-дикарбоксифталоцианинового и N,N-бис(салицилиден)этилендиаминового комплексов. Результаты анализа приведены в таблице.
Представленные в таблице данные показывают, что использование ионных жидкостей обеспечивает достаточно высокое снижение концентрации серосодержащих соединений в исходном углеводородном сырье. Увеличение экстракционной емкости ионной жидкости по отношению к БТ так и ДБТ достигается использованием ионной жидкости с катионом имидазолия, замещенным на алкильный радикал, особенно в случае октильного радикала. Добавление к ионной жидкости ионов металла, выбранных из группы, включающей кобальт, никель, железо, медь, в том числе различные комплексы кобальта (II): 4,5-дикарбоксифталоцианиновый и N,N-бис(салицилиден)этилендиаминовый комплексы также увеличивает коэффициент распределения и степень извлечения серосодержащих соединений. Наиболее существенное улучшение качества очистки показали добавки 4,5-дикарбоксифталоцианинового и N,N-бис(салицилиден)этилендиаминового комплексов кобальта (II).
Таким образом, использование предложенного способа очистки от серосодержащих соединений обеспечивает достижение поставленной цели - улучшение качества углеводородного сырья.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ ДИЗЕЛЬНОГО ТОПЛИВА ОТ СЕРОСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 2018 |
|
RU2673539C1 |
| СПОСОБ ОЧИСТКИ ЖИДКИХ МОТОРНЫХ ТОПЛИВ ОТ СЕРОСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 2013 |
|
RU2541315C1 |
| СПОСОБ ОЧИСТКИ УГЛЕВОДОРОДНЫХ СМЕСЕЙ ОТ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2011 |
|
RU2460760C1 |
| СПОСОБ ОЧИСТКИ УГЛЕВОДОРОДНОГО СЫРЬЯ ОТ СЕРАОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 1998 |
|
RU2125080C1 |
| СПОСОБ ГЛУБОКОЙ ОКИСЛИТЕЛЬНО-АДСОРБЦИОННОЙ ДЕСУЛЬФУРИЗАЦИИ ЖИДКИХ УГЛЕВОДОРОДНЫХ ТОПЛИВ И СОРБЕНТЫ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2012 |
|
RU2482162C1 |
| СПОСОБ ОЧИСТКИ ЖИДКОГО УГЛЕВОДОРОДНОГО СЫРЬЯ ОТ СЕРООРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 1996 |
|
RU2101320C1 |
| КАТАЛИЗАТОР ГИДРОПЕРЕРАБОТКИ НЕФТЯНЫХ ФРАКЦИЙ (ВАРИАНТЫ) | 2016 |
|
RU2640210C1 |
| КАТАЛИЗАТОРЫ СУЛЬФООКИСЛЕНИЯ И СПОСОБЫ И СИСТЕМЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2472841C2 |
| СОСТАВ И СПОСОБ ПРИГОТОВЛЕНИЯ НОСИТЕЛЯ И КАТАЛИЗАТОРА ГЛУБОКОЙ ГИДРООЧИСТКИ УГЛЕВОДОРОДНОГО СЫРЬЯ | 2012 |
|
RU2569682C2 |
| Катализатор и способ осуществления реакции Фишера-Тропша с его использованием | 2015 |
|
RU2614420C1 |
Предлагаемый способ относится к способу очистки жидких углеводородных смесей от труднокрекингируемых сераорганических (ТКС) соединений. Изобретение касается способа очистки углеводородных смесей от серосодержащих гетероциклических соединений, включающего экстракцию серосодержащих соединений из органической фазы в ионные жидкости, отделение углеводородной фракции от ионной жидкости, электрохимическую регенерацию ионной жидкости. В качестве ионной жидкости используют: 1-метил-3-бутилимидазолий тетрафторборат (MBImBF4), 1-метил-3-октилимидазолий тетрафторборат (MOImBF4), содержащую растворенные соединения нитратов переходных металлов, выбранных из группы, включающей кобальт, никель, железо, медь, а также комплексы кобальта (II): 4,5-дикарбоксифталоцианиновый, N,N-бис(салицилиден)этилендиаминовый. Технический результат - улучшение качества углеводородного сырья за счет более полного удаления серосодержащих органических веществ. 2 з.п. ф-лы, 1 табл.
1. Способ очистки углеводородных смесей от серосодержащих гетероциклических соединений, включающий экстракцию серосодержащих соединений из органической фазы в ионные жидкости, отделение углеводородной фракции от ионной жидкости, электрохимическую регенерацию ионной жидкости, отличающийся тем, что в качестве ионной жидкости используют 1-метил-3-бутилимидазолий тетрафторборат (MBImBF4), 1-метил-3-октилимидазолий тетрафторборат (MOImBF4), содержащую растворенные соединения нитратов переходных металлов, выбранных из группы, включающей кобальт, никель, железо, медь, а также комплексы кобальта (II): 4,5-дикарбоксифталоцианиновый, N,N-бис(салицилиден)этилендиаминовый.
2. Способ по п.1, отличающийся тем, что регенерацию ионной жидкости проводят электрохимическим окислением или восстановлением серосодержащих соединений в трехэлектродном реакторе с неразделенным катодным и анодным пространством.
3. Способ по п.2, отличающийся тем, что электрохимическое воздействие проводят в электрохимическом ректоре при потенциале -1,0÷+2,5 В по квазиобратимому платиновому электроду сравнения при плотности тока 1-10 мА/см2 на платиновых или графитовых электродах в интервале температур 25-50°С.
| US 20030085156 A1, 08.05.2003 | |||
| СПОСОБ ЗАБОРА КРОВИ ИЗ РЕТРООРБИТАЛЬНОГО ВЕНОЗНОГО СПЛЕТЕНИЯ У МЕЛКИХ ЛАБОРАТОРНЫХ ЖИВОТНЫХ | 2024 |
|
RU2840917C1 |
| СПОСОБ ОЧИСТКИ УГЛЕВОДОРОДНОГО СЫРЬЯ ОТ СЕРНИСТЫХ СОЕДИНЕНИЙ | 2000 |
|
RU2166530C1 |
| СПОСОБ ОЧИСТКИ ЖИДКОГО УГЛЕВОДОРОДНОГО СЫРЬЯ ОТ СЕРООРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 1996 |
|
RU2101320C1 |

