Изобретение относится к области химии, в частности к катализаторам, для глубокой гидроочистки нефтяных фракций и органического сырья, и может быть использовано в нефтеперерабатывающей и нефтехимической промышленности.
В последнее время потребность в производстве моторных топлив со сверхнизким содержанием серы (менее 10 ppm) растет. В ближайшие годы самым крупнотоннажным процессом нефтепереработки остается гидроочистка, и роль ее будет возрастать в связи с ужесточением норм на содержание серы в бензине и дизельном топливе, а также с увеличением глубины переработки нефти и вовлечении нетрадиционных углеводородных ресурсов в нефтепереработку.
Абсолютное большинство катализаторов гидроочистки представляет собой нанесенные Co(Ni)Mo(W)/Al2O3 системы, включающие иногда в свой состав и другие элементы периодической системы для улучшения или модификации каталитических свойств. Синергизм каталитического действия сульфидов Co(Ni) и Mo(W) обеспечивается образованием в процессе сульфидирования катализатора фазы Co(Ni)MoS, в которой высокодисперсные кристаллиты MoS2 декорированы атомами промотора (Со или Ni). Различают как минимум два типа активных компонентов сульфидных катализаторов: фазы Co(Ni)MoS типов I и II. Фаза CoMoS типа II значительно превосходит по активности фазу типа I и характеризуется слабым взаимодействием активных компонентов с носителем и, следовательно, большей полнотой сульфидирования. В большинстве случаев фазы типа II имеют вид мультислойных упаковок MoS2, хотя это и не является их обязательным свойством.
Недостатком традиционных отечественных катализаторов СоМо/Al2O3 и NiMo(W)/Al2O3 является низкая каталитическая активность, не позволяющая обеспечивать производство моторных топлив со сверхнизким содержанием серы, обусловленная наличием на поверхности активной фазы I типа, а также низкоактивных в катализе отдельных сульфидов переходных металлов и неактивных шпинелей типа Ni(Co)Al2O4, образующихся в процессе миграции атомов промотора на высокотемпературных стадиях прокаливания.
В последние годы для создания катализаторов глубокой гидроочистки углеводородного сырья используют методы формирования активного компонента на поверхности оптимального по текстуре и свойствам носителя за счет следующих подходов:
1. Использование носителей с оптимальными текстурными характеристиками, позволяющими максимально диспергировать активный компонент и равномерно нанести его в поры, доступные для молекул-реагентов. Для этих целей синтезируют носители с развитой удельной поверхностью (не ниже 200 м2/г), объемом пор (0,5-0,9 см3/г) и узким распределением пор по размерам, при этом доля пор размером 75-350 Å должна составлять не менее 70-75% от общего объема пор (US 3840472, B01J 27/19, C10G 45/08, 8.10.74; 2003/0173256; 4818743, B01J 23/85, 04.04.89; 4879265, B01J 23/24, 07.11.89, РФ №2192923, C10G 45/08, B01J 27/188, B01J 35/10, 20.11.2002; РФ №2377067, B01J 21/04, C10G 45/04, B01J 23/58, B01J 23/75, B01J 23/755, B01J 27/18, 27.12.2009 и др.). Такое распределение пор по размерам обеспечивает не только высокую доступность серосодержащих соединений к поверхности активного компонента, но и снижает возможность закоксовывания пор.
2. Использование алюмооксидных носителей, модифицированных добавками бора и/или фосфора и/или редкоземельного металла (US 3755150, C10G 45/08, 28.08.1973; US 4392985, B01J 27/188, C10G 45/08; US 4500424, B01J 027/185, 19.02.1985; US 4818743, B01J 027/185, 4.04.1989, РФ №2192923, C10G 45/08, B01J 27/188, B01J 35/10, 20.11.2002; РФ №2197323, B01J 23/88, B01J 21/12, B01J 23/882, B01J 23/883, B01J 23/885, B01J 23/887, C10G 45/08, 27.01.2003; РФ №2306978, B01J 23/88, B01J 23/83, B01J 37/02, 27.09.2007 и др.), выполняющие функции снижения концентрации основных центров на поверхности носителя, уменьшения взаимодействия оксидного предшественника, а также повышающими механическую прочность и термостабильность. Недостаток данного способа проявляется при высоком содержании добавок, которые приводят к снижению каталитической стабильности вследствие ускоренного отложения кокса.
3. Модифицирование алюмооксидных носителей добавками оксидов титана (РФ 2155637, B01J 23/88, 10.09.2000 и др.), циркония и кремния (US 6267874, 31.07.2001; РФ №2197323, B01J 23/88, B01J 21/12, B01J 23/882, B01J 23/883, B01J 23/885, B01J 23/887, C10G 45/08, 27.01.2003 и др.), добавками аморфных алюмосиликатов и цеолитов (US 5686374, B01J 29/10, 11.11.97, РФ №2311959, B01J 37/02, B01J 23/88, B01J 29/40, B01J 29/18, B01J 32/00, C10G 45/06 и др.) для увеличения кислотных свойств носителя. Основным недостатком этого способа является дорогостоящая технология синтеза модифицированных алюмооксидных носителей, например использование в качестве предшественников алкоксидов кремния и алюминия и повышенных температур (40-90°C) для проведения стадий соосаждения и старения гелей в случае использования неорганических соединений кремния и алюминия (US 6267874, 31.07.2001).
Общим недостатком для вышеперечисленных носителей является повышенная кислотность, которая приводит к ускоренным отложениям кокса и дезактивации катализаторов. Техническим решением настоящего изобретения является присутствие в составе носителя переходного или благородного металла, который в условиях процесса гидроочистки способен активировать молекулярный водород. Активированный водород в силу своей высокой реакционной способности интенсивно участвует в целевых реакциях и процессе гидрирования предшественников кокса, тем самым увеличивая каталитическую стабильность. Таким образом, использование нового носителя, представляющего собой гамма-оксид алюминия, модифицированный кислотной и металлической добавкой, обеспечивает высокую эффективность работы катализатора.
Высокоактивные катализаторы гидроочистки углеводородного сырья получают с применением следующих подходов:
1. Использование «мягких» режимов термообработки катализатора при температурах не выше 200°C (US 2003/0173256, 18.09.2003; US 7618916 В2, 17.11.2009; РФ №2402380, B01J 23/882, B01J 23/883, B01J 21/02, B01J 21/04, B01J 37/02, C10G 45/08, B01J 38/62, 27.10.2010 и др.) и сульфидирования (US 4879265, 7.11.1989, US 2003/0173256, 18.09.2003 и др.). Недостаток этого способа заключается в том, что оксидный предшественник может не полностью сульфидироваться, а дисперсность активной фазы катализатора является низкой.
2. Увеличение количества вносимого металла (например, до 20-29 мас.% MoO3), оптимизация соотношения Co(Ni)/Mo(W) и условий сульфидирования катализатора для увеличения количества фазы II типа (US 4879265, B01J 027/19, 7.07.1989; US 2003/0173256; US 2006/00545536, 16.03.2006 и др.). Основным недостатком этого способа синтеза является трудность приготовления концентрированных пропиточных растворов, стадии нанесения активных компонентов, а также снижение их дисперсности.
3. Введение в состав активного компонента добавок, обладающих гидрирующей способностью, например Ni, W, или металлов платиновой группы (Pt, Pd, Rh) для увеличения гидрирующей способности катализатора (US 6267874, B01J 21/12, 31.07.2001). Основным недостатком этого способа является дорогостоящая технология синтеза модифицированных алюмооксидных носителей, обусловленная совместным использованием модифицированных кремнийсодержащими добавками носителей, так чтобы концентрация бренстедовских центров составляла не менее 50 мкмоль/г, и металлов платиновой группы.
4. Использование в качестве предшественников биметаллических соединений, среди них гетерополисоединений (РФ №2103065 B01J 37/02, B01J 23/882, B01J 23/883, B01J 27/188, 27.01.1998; US 2004/0132614 А1, 08.07.2004; US 007687430 В2, 30.03.2010; РФ №2385764 B01J 23/882, B01J 37/02, 10.04.2010; РФ №2386476 B01J 23/88, B01J 23/882, B01J 27/199, B01J 37/02, 20.04.2010; РФ №2387475 B01J 21/04, B01J 27/19, B01J 27/24, C10G 45/08, 27.04.2010; РФ №2414963 B01J 21/04, B01J 21/08, B01J 23/30, B01J 23/14, B01J 37/02, B01J 23/755, B01J 23/28, B01J 23/20, B01J 27/14, C10G 45/08, 27.03.2011 и др.). Основным недостатком этого способа является трудность контролирования состава гетерополисоединений на поверхности носителя, вызванная оксид-оксидным взаимодействием предшественник - носитель, особенно, на термических стадиях приготовления.
5. Использование органических добавок, снижающих образование окристаллизованных фаз типа CO3O4 и CoAl2O4 и оксид-оксидное взаимодействие предшественника с носителем (US 4879265, 7.11.1989; US 2003/0173256, 18.09.2003; US 6540908 B1, 01.04.2003; US 6923904 B1, 02.08.2005; US 7235173 B2, 26.06.2007; US 7605107 B2, 20.10.2009; US 7618916 B2, 17.11.2009; РФ №2402380, B01J 23/882, B01J 23/883, B01J 21/02, B01J 21/04, B01J 37/02, C10G 45/08, B01J 38/62, 27.10.2010 и др.). Недостатком данных катализаторов является взаимодействие органических добавок с Mo(W) предшественником, снижающим скорость его сульфидирования, а также использование растворов, содержащих контр-ионы (катионы аммония, нитрат анионы и др.), способные снижать дисперсность оксидного предшественника, а также препятствовать взаимодействию Mo(W) и Co(Ni) частиц.
Общим недостатком для вышеперечисленных катализаторов и способов их приготовления является то, что с их использованием не удается достичь ультранизкого остаточного содержания серы, низкого содержания азотсодержащих соединений и ароматических углеводородов в получаемых продуктах, а также быстрая дезактивация катализаторов, вызванная отложениями кокса, агломерацией (спеканием) частиц активной фазы и образованием отдельных сульфидов в процессе эксплуатации катализаторов, особенно при повышенных температурах.
Наиболее близким к предлагаемому решению является способ приготовления катализатора для глубокой гидроочистки углеводородного сырья, в который на стадии пропитки вносят комплексное кислородсодержащее соединение молибдена и кобальта и/или никеля. В качестве носителя катализатор содержит оксид алюминия или оксид алюминия с добавкой оксида кремния или цеолита. Способ получения катализатора включает растворение оксида молибдена и карбоната кобальта в смеси комплексообразующих органических кислот (лимонной, молочной, малоновой, уксусной, муравьиной) [RU 2314154, B01J 23/882, B01J 23/883, 10.01.2008].
Недостатком данного способа приготовления катализатора является то, что молибденсодержащие анионы в зависимости от pH раствора могут иметь различную степень полимеризации молибдена, образуя изополианионы. Использование в качестве комплексообразователя органических кислот приводит к тому, что степень полимеризации молибдена в растворе является неопределенной и зависит от многих факторов. В результате невозможно создание оксидного предшественника активной фазы сульфидных молибденсодержащих катализаторов с определенной, заранее заданной степенью полимеризации молибдена. Каталитические свойства катализатора-прототипа не позволяют получать продукты с ультранизким содержанием серы, низким содержанием ароматических углеводородов.
Техническим результатом настоящего изобретения является создание нового состава и способа приготовления носителя и катализатора глубокой гидроочистки углеводородного сырья. Технический результат достигается за счет введения биметаллического комплексного соединения металлов VIII и VIB групп Периодической системы, на модифицированный носитель, который состоит из композиции гамма-оксида алюминия, оксидов неметалла: фосфора и/или бора, оксидов переходного металла: никеля и/или кобальта, или одного из оксидов благородного металла: платины, родия, рутения, при следующем содержании, мас.%: оксиды неметалла 0,5-1,5, оксиды переходного металла 0,5-5,0 или оксиды благородного металла 0,3-0,8, оксид алюминия 99,2-91,5%; катализатор имеет удельную поверхность 180-350 м2/г, объем пор 0,3-0,6 см3/г, средний диаметр пор 8,5-13,0 нм.
Биметаллическое комплексное соединение содержит как минимум один из следующих гетерополианионов [CO2Mo10O38H4]6-, [Co(OH)6Mo6O18]4-, [Ni(OH)6Mo6O18]4-, [Ni2Mo10O38H4]6-, [P2Mo5O23]6-, [SiMo12O40]4-, [PMo12O40]3-, при этом содержание в прокаленном при 550°С катализаторе MoO3 составляет 14,0-23,0% мас., СоО и/или NiO - 4,0-6,5% мас. Катализатор имеет форму цилиндров или трехлистников.
Алюмооксидный носитель катализаторов глубокой гидроочистки углеводородного сырья состоит из композиции гамма-оксида алюминия, оксидов неметалла: фосфора и/или бора, а также содержит на поверхности оксид переходного металла: никеля и/или кобальта, или один из оксидов благородного металла: платины, родия, рутения, при следующем содержании, мас.%: оксиды неметалла 0,5-1,5, оксиды переходного металла 0,5-5,0 или оксиды благородного металла 0,3-0,8, оксид алюминия 99,2-91,5%; имеет удельную поверхность 200-370 м2/г, объем пор 0,5-1,0 см3/г, средний диаметр пор 8,5-13,5 нм.
Способ получения носителя для катализатора глубокой гидроочистки углеводородного сырья заключается в том, что гамма-оксид алюминия пропитывают соединениями бора и/или фосфора, переходного металла (никеля и/или кобальта) или благородного металла (одного из ряда платины, родия, рутения), с последующей сушкой при 80-120°C (5 ч) и прокаливанием при 300°C (2 ч). В качестве соединений бора используют как минимум одно, выбранное из ряда: ортоборная кислота H3BO3, пироборная кислота H2B4O7, метаборат аммония NH4BO2, борат аммония (NH4)3BO3, тетраборат аммония (NH4)2B4O7, в качестве соединений фосфора - ортофосфорная кислота Н3РО4, метафосфорная кислота HPO3, пирофосфорная кислота H4P2O7, гидрофосфат аммония (NH4)2HPO4, метафосфат аммония NH4PO3, пирофосфат аммония (NH4)4P2O7. В качестве соединений переходных металлов используют как минимум одно, выбранное из ряда: гексагидрат нитрата кобальта Co(NO3)2·6H2O, тетрагидрат ацетата кобальта Со(СН3СОО)2·4H2O, тетрагидрат сульфата кобальта CoSO4·7H2O, тетрагидрат хлорида кобальта CoCl2·4H2O, дигидрат цитрата кобальта Со3(C6H5O7)2·2H2O, гексагидрат нитрата никеля Ni(NO3)2·6H2O, тетрагидрат ацетата никеля Ni(CH3COO)2·4H2O, тетрагидрат сульфата никеля NiSO4·7H2O, тетрагидрат хлорида никеля NiCl2·4H2O, дигидрат цитрата никеля Ni(C6H5O7)2·14H2O. В качестве соединений благородных металлов используют как минимум одно, выбранное из ряда платинохлористоводородная кислота H2PtCl6, тетрахлорид гексаамминплатины [Pt(NH3)6]Cl4, дихлородиамминоплатина(II), [PtCl2(NH3)2], тригидрат хлорида родия RhCl3·3H2O, дигидрат нитрата родия Rh(NO3)3·2H2O, трихлорид гексаамминродия [Rh(NH3)6]Cl3, пентагидрат хлорида рутения RuCl4·5H2O, гексагидрат нитрата рутения Ru(NO3)3·6H2O, трихлорид гексаамминрутения [Ru(NH3)6]Cl3.
Способ приготовления катализатора глубокой гидроочистки углеводородного сырья пропиткой носителя по влагоемкости или с избытком раствором предшественников активного компонента заключается в том, что носитель однократно пропитывают водным раствором, имеющим pH 1,5-3,0, содержащим как минимум один из гетерополианионов ряда [CO2Mo10O38H4]6-, [Co(OH)6Mo6O18]4-, [Ni(OH)6Mo6O18]4-, [Ni2Mo10O38H4]6-, [P2Mo5O23]6-, [SiMo12O40]4-, [PMo12O40]3-, в качестве соединения кобальта используется одно из ряда гидроксид кобальта Со(ОН)2·nH2O, кобальт углекислый CoCO3, кобальт углекислый основной 2CoCO3·3Со(ОН)2·nH2O, в качестве соединения никеля используется одно из ряда гидроксид никеля Ni(OH)2·nH2O, никель углекислый NiCO3, никель углекислый основной NiCO3·nNi(OH)2·mH2O, в качестве стабилизатора пропиточного раствора используют карбоновую кислоту, содержащую, по меньшей мере, одну карбоксильную группу и 1-20 углеродных атомов. Гетерополианион [P2Mo5O23]6- или [PMo12O40]3- может формироваться путем последовательного растворения оксида молибдена MoO3 или молибденовой кислоты H2MoO4 в 85%-ой фосфорной кислоте H3PO4 и добавлении 30%-го раствора H2O2, при температуре 50-90°С и pH 1,0-1,5 в соотношениях, соответствующих стехиометрии в гетерополианионе. Один из гетерополианионов [CO2Mo10O38H4]6-, [Co(OH)6Mo6O18]4-, [Ni(OH)6Mo6O18]4-, [Ni2Mo10O38H4]6- может формироваться путем последовательного растворения оксида молибдена MoO3 или молибденовой кислоты H2MoO4 в 30%-ном растворе пероксида водорода при температуре 50-90°С, с последующим добавлением соли Со или Ni в соотношениях, соответствующих стехиометрии в гетерополианионе. Для приготовления катализатора в качестве стабилизатора используется лимонная кислота. Для приготовления катализатора используют либо пропитку носителя по влагоемкости, либо из избытка раствора. Пропитка гранул носителя проводится после создания вакуума в сосуде, содержащем носитель. Пропитка гранул носителя после создания вакуума проводится пропиточным раствором при температурах 20-50°C. После пропитки катализатор сушат при температуре 120-180°C в потоке воздуха или азота.
Носитель, а следовательно, и катализатор, преимущественно, выполнены в виде экструдатов цилиндров или трехлистников, диаметром 1,0-1,5 мм и длиной 3-5 мм. Использование экструдатов в форме «трехлистника» также позволяет увеличить эффективность гидрообессеривания нефтяных фракций.
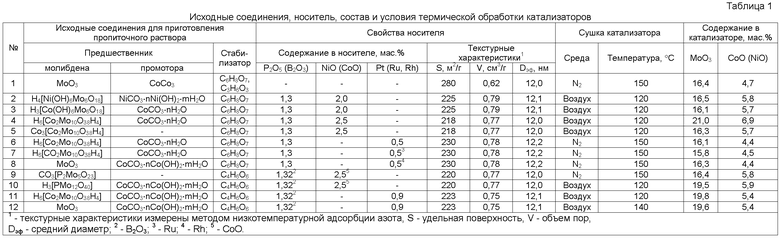
Исходные соединения для приготовления совместного пропиточного раствора, состав и текстурные характеристики носителя, а также условия сушки катализаторов приведены в табл.1.
Катализаторы испытывали в виде частиц размером 0,25-0,50 мм, приготовленных путем измельчения и рассеивания исходных гранул прокаленного катализатора.
Каталитическую активность в реакции гидродесульфуризации (ГДС) дибензотиофена (ДБТ) определяли на лабораторной проточной установке с микрореактором. В реактор загружали катализатор в количестве 0,3000±0,0005 г (фракция 0,25-0,50 мм), разбавленный SiC до общего объема 1 см3. Реакцию ГДС изучали при следующих условиях: температура 300°C, давление водорода 3.0 МПа, соотношение водород/сырье 600 нл/л, объемная скорость подачи сырья 50,0 ч-1. В качестве сырья использовали модельную смесь ДБТ (1500 ppm серы) в гептане с добавлением внутреннего стандарта - н-гексадекана (1.0 мас.%). Каждый час проводили отбор катализата для анализа его состава на газовом хроматографе Кристалл-5000. Разделение продуктов реакций осуществляли на кварцевой капиллярной колонке с привитой фазой OV-101.
Активность катализаторов в ГДС ДБТ оценивали по константе скорости ГДС, рассчитанной по уравнению 1-го порядка
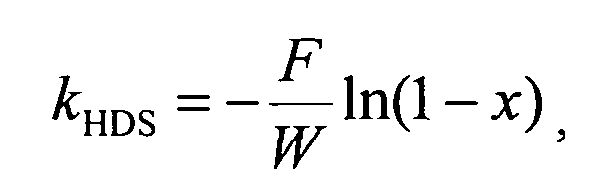
где F - расход ДБТ, моль/ч;
W - масса катализатора, г;
х - конверсия ДБТ, %.
Основными продуктами ГДС ДБТ были бифенил (БФ), образующийся по маршруту прямого обессеривания (DS) ДБТ, циклогексилбензил (ЦГБ) и дициклогексил (ДЦГ) - по маршруту гидрирования (HYD) ДБТ. Следы продуктов неполного гидрирования ДБТ (тетрагидродибензотиофена (ТГДБТ)) наблюдали в продуктах реакций всех катализаторов. Относительную HYD/DS селективность рассчитывали с использованием следующего уравнения:
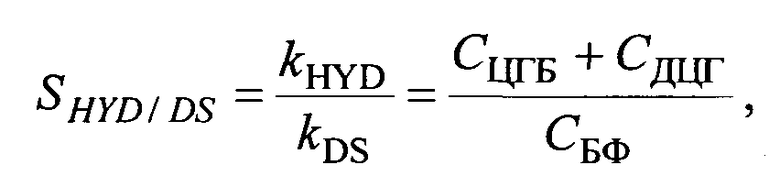
где СЦГБ, СДЦГ, СБФ - содержание циклогексилбензола, дициклогексила и бифенила в продуктах реакции, соответственно (мас.%).
Результаты каталитических испытаний представлены в табл.2.
Катализаторы испытывали также в процессе гидроочистки дизельных фракций. В трубчатый реактор загружали 15 см3 катализатора, разбавленного SiC до общего объема 30 см3. Сульфидирование проводили смесью диметилдисульфида и керосиновой фракции при 240°С в течение 10 ч и при 340°С в течение 8 ч. Сырье для проведения данных тестовых испытаний представляло собой смесь прямогонной дизельной фракции (далее ПДФ) и легкого газойля каталитического крекинга (далее ЛГКК) в массовом соотношении 90/10% мас. и имело следующие характеристики: ρ4 20=0,848; nD 20=1,479; содержание серы 1,14% мас. (11400 ppm); температура начала кипения 180°С; температура выкипания 96% объема 360°С. Условия испытания: давление водорода 4,0 МПа, кратность циркуляции водорода 500 нл/л сырья, объемная скорость подачи сырья 1,6-2,0 ч-1, температура в реакторе 340-350°С.
Стабильность работы катализаторов оценивали в жестких условиях по ускоренной степени дезактивации: для этого процесс гидроочистки проводили с использованием смесевого сырья, состоящего из 70% прямогонной дизельной фракции, 16% легкого газойля каталитического крекинга и 14% легкого газойля замедленного коксования, при следующих условиях: температура реактора 370°C, давление 3,5 МПа, объемная скорость подачи сырья 2,0 ч-1, кратность циркуляции водород/сырье 350 нл/л, продолжительность 50 ч.
Стабильность работы катализатора оценивали по степени ускоренной дезактивации и рассчитывали по формуле:
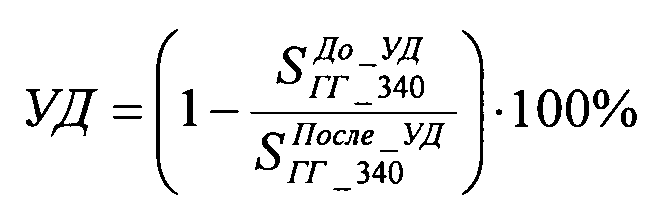
где 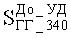
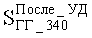
Гидрогенизаты отделяли от водорода в сепараторах высокого и низкого давления, затем подвергали обработке 10%-ным раствором NaOH в течение 15 мин, отмывали дистиллированной водой до нейтральной реакции промывных вод, высушивали в течение суток над прокаленным CaCl2. Содержание серы определяли с помощью рентгенофлюоресцентных энерго- и волнодисперсионных анализаторов. Содержание отдельных групп ароматических углеводородов определяли методом квазинормально-фазовой ВЭЖХ в изотермическом режиме.
Результаты испытаний катализаторов представлены в табл.2.
Сущность изобретения иллюстрируется следующими примерами.
Пример 1
Состав катализатора и способ его приготовления согласно известному техническому решению - прототипу.
Для приготовления пропиточного раствора к 5,89 г кобальта углекислого CoCO3 добавляют 42 см3 дистиллированной воды и нагревают до 80°C. Затем при перемешивании добавляют порциями 6,93 г моногидрата лимонной кислоты C6H8O7·H2O, при этом наблюдают интенсивное выделение CO2. Дождавшись полного растворения осадка, добавляют 12,14 г оксида молибдена MoO3 и 4 см3 80%-ной молочной кислоты C3H6O3. Продолжают перемешивать раствор при 80°С до полного растворения. После растворения объем раствора составляет 50 мл, pH равен 2,5. В качестве носителя используют экструдаты из оксида алюминия, диаметром 1,3-1,4 мм и длиной 4-5,2 мм, имеющие удельную поверхность 280 м2/г, объем пор 0,62 см3/г и диаметр пор 11,9 нм, определенные из изотерм адсорбции азота. На долю пор размером 70-130 Å приходится 65% от общего объема пор. К 30 г прокаленных экструдатов добавляют 27 см3 пропиточного раствора и выдерживают 15 мин. Пропитанные экструдаты сушат при комнатной температуре в потоке азота и подвергают термообработке в потоке азота при 150°С в течение 10 ч. После прокаливания на воздухе в течение 2 ч при 550°С катализатор содержит 16,4% мас. МоО3, 4,7% мас. NiO (табл.1).
Пример 2
Для приготовления модифицированного оксида алюминия используют гранулы гамма-оксида алюминия с удельной поверхностью 230 м2/г, объемом пор 0,81 см3/г и диаметром пор 12,2 нм. 2,50 г гидрофосфата аммония (NH4)2HPO4 растворяют в 85 см3 дистиллированной воды при 20-50°C, далее полученный раствор приливают к 100 г гамма-оксида алюминия. Полученный фосфорсодержащий носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 2 ч. 7,95 г нитрата никеля Ni(NO3)2·6H2O растворяют в 85 см3 воды, а далее приливают к фосфорсодержащему носителю. Полученный модифицированный носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 4 ч. После прокаливания гранулы модифицированного носителя имеют удельную поверхность 225 м2/г, объем пор 0,79 см3/г и средний диаметр пор 12,1 нм.
Для приготовления пропиточного раствора 26,90 г гексамолибдоникелевой гетерополикислоты H4[Ni(OH)6Mo6O18] и 7,45 г гидроксокарбоната никеля NiCO3·nNi(ОН)2·mH2O растворяют в 65 см3 воды при 40-60°C и перемешивании. После окончания выделения CO2 в полученный раствор добавляют 11,7 г моногидрата лимонной кислоты C6H8O7·H2O и доводят объем водой до 85 см3. pH пропиточного раствора равен 2.0-3.0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат на воздухе при комнатной температуре, а далее при 120°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 16,5% мас. MoO3, 5,8% мас. NiO (табл.1).
Пример 3
Модифицированный носитель готовят по примеру 2.
Для приготовления пропиточного раствора 26,06 г гексамолибдокобальтовой гетерополикислоты H4[Co(OH)6Mo6O18] и 6,31 г карбоната кобальта CoCO3·nH2O растворяют в 65 см3 воды при 40-60°C и перемешивании. После окончания выделения CO2 в полученный раствор добавляют 11,3 г моногидрата лимонной кислоты С6Н8О7·Н2О и доводят объем водой до 85 см3. pH пропиточного раствора равен 2,0-3,0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат на воздухе при комнатной температуре, а далее при 120°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 16,1% мас. MoO3, 5,7% мас. СоО и NiO (табл.1).
Пример 4
Для приготовления модифицированного оксида алюминия используют гранулы гамма-оксида алюминия с удельной поверхностью 230 м2/г, объемом пор 0,81 см3/г и диаметром пор 12,2 нм. 2,50 г гидрофосфата аммония (NH4)2HPO4 растворяют в 85 см3 дистиллированной воды при 20-50°C, далее полученный раствор приливают к 100 г гамма-оксида алюминия. Полученный фосфорсодержащий носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 2 ч. 10,00 г нитрата никеля Ni(NO3)2·6H2O растворяют в 85 см3 воды, а далее приливают к фосфорсодержащему носителю. Полученный модифицированный носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 4 ч. После прокаливания гранулы модифицированного носителя имеют удельную поверхность 218 м2/г, объем пор 0,77 см3/г и средний диаметр пор 12,0 нм.
Для приготовления пропиточного раствора 36,30 г декамолибдодикобальтовой гетерополикислоты H6[CO2Mo10O38H4] и 8,38 г карбоната кобальта CoCO3·nH2O растворяют в 65 см3 воды при 40-60°C и перемешивании. После окончания выделения CO2 в полученный раствор добавляют 15,0 г моногидрата лимонной кислоты C6H8O7·H2O и доводят объем водой до 85 см3. pH пропиточного раствора равен 2,0-3,0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат на воздухе при комнатной температуре, а далее при 120°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 21,0% мас. MoO3, 6,9% мас. СоО и NiO (табл.1).
Пример 5
Модифицированный носитель готовят по примеру 4.
Для приготовления пропиточного раствора 28,67 г кобальтовой соли декамолибдодикобальтовой гетерополикислоты Со3[Со2Мо10О38Н4] и 10,1 г моногидрата лимонной кислоты C6H8O7·H2O растворяют в 75 см3 воды при 40-60°C и перемешивании. pH пропиточного раствора равен 2.0-3.0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат на воздухе при комнатной температуре, а далее при 120°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 16,3% мас. MoO3, 5,7% мас. СоО и NiO (табл.1).
Пример 6
Для приготовления модифицированного оксида алюминия используют гранулы гамма-оксида алюминия с удельной поверхностью 230 м2/г, объемом пор 0,81 см3/г и диаметром пор 12,2 нм. 2,50 г гидрофосфата аммония (NH4)2HPO4 растворяют в 85 см3 дистиллированной воды при 20-50°C, далее полученный раствор приливают к 100 г гамма-оксида алюминия. Полученный фосфорсодержащий носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 2 ч.
Фосфорсодержащий носитель пропитывают по влагоемкости 85 см3 раствором платинохлористоводородной кислоты H2PtCl6 с концентрацией платины 5,9 мг/см3. Полученный модифицированный носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 4 ч. После прокаливания гранулы модифицированного носителя имеют удельную поверхность 230 м2/г, объем пор 0,78 см3/г и средний диаметр пор 12,2 нм.
Для приготовления пропиточного раствора 25,76 г декамолибдодикобальтовой гетерополикислоты Н6[Со2Мо10О38Н4] и 6,17 г карбоната кобальта CoCO3·nH2O растворяют в 65 см3 воды при 40-60°C и перемешивании. После окончания выделения CO2 в полученный раствор добавляют 11,1 г моногидрата лимонной кислоты C6H8O7·H2O и доводят объем водой до 85 см3. pH пропиточного раствора равен 2,0-3,0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат в атмосфере азота при комнатной температуре, а далее при 150°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 16,1% мас. МоО3, 4,4% мас. СоО (табл.1).
Пример 7
101 г фосфорсодержащего носителя, полученного согласно примеру 6, пропитывают по влагоемкости 85 см3 раствором нитрата рутения Ru(NO3)3 с концентрацией рутения 5,9 мг/см3. Полученный модифицированный носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 4 ч. После прокаливания гранулы модифицированного носителя имеют удельную поверхность 230 м2/г, объем пор 0,78 см3/г и средний диаметр пор 12,2 нм.
Для приготовления пропиточного раствора 25,21 г декамолибдодикобальтовой гетерополикислоты H6[CO2Mo10O38H4] и 6,27 г карбоната кобальта CoCO3·nH2O растворяют в 65 см3 воды при 40-60°C и перемешивании. После окончания выделения CO2 в полученный раствор добавляют 11,2 г моногидрата лимонной кислоты C6H8O7·H2O и доводят объем водой до 85 см3. рН пропиточного раствора равен 2,0-3,0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат в атмосфере азота при комнатной температуре, а далее при 150°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 15,8% мас. МоО3, 4,5% мас. СоО (табл.1).
Пример 8
101 г фосфорсодержащего носителя, полученного согласно примеру 6, пропитывают по влагоемкости 85 см3 раствором хлорида родия RhCl3 с концентрацией родия 5,9 мг/см3. Полученный модифицированный носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 4 ч. После прокаливания гранулы модифицированного носителя имеют удельную поверхность 230 м2/г, объем пор 0,78 см3/г и средний диаметр пор 12,2 нм.
Для приготовления пропиточного раствора 20,2 г оксида молибдена растворяют в 70 см3 воды при добавлении 3,8 г 85%-ного раствора ортофосфорной кислоты и температуре 60-90°C. Далее в полученный раствор добавляют 7,01 г гидрокарбоната кобальта CoCO3·nCo(ОН)2·mH2O и 11,0 г моногидрата лимонной кислоты C6H8O7·H2O. После окончания выделения CO2 объем полученного раствора доводят водой до 85 см3. pH пропиточного раствора равен 2.0-3.0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат в атмосфере азота при комнатной температуре, а далее при 150°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 16,3% мас. MoO3, 4,4% мас. СоО (табл.1).
Пример 9
Для приготовления модифицированного оксида алюминия используют гранулы гамма-оксида алюминия с удельной поверхностью 230 м2/г, объемом пор 0,81 см3/г и диаметром пор 12,2 нм. 3,80 г метабората аммония метаборат аммония NH4BO2·2H2O растворяют в 85 см3 дистиллированной воды при 20-50°C, далее полученный раствор приливают к 100 г гамма-оксида алюминия. Полученный борсодержащий носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 2 ч. 10,00 г нитрата кобальта Co(NO3)2·6H2O растворяют в 85 см3 воды, а далее приливают к борсодержащему носителю. Полученный модифицированный носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 4 ч. После прокаливания гранулы модифицированного носителя имеют удельную поверхность 220 м2/г, объем пор 0,77 см3/г и средний диаметр пор 12,0 нм.
Для приготовления пропиточного раствора 33,67 г кобальтовой соли пентамолибдодифосфорной кислоты Со3[P2Mo5O23] растворяют в 70 см3 воды при добавлении 9,3 г винной кислоты С4Н6О6. Далее объем полученного раствора доводят водой до 85 см3. pH пропиточного раствора равен 1,5-2,5.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат в атмосфере азота при комнатной температуре, а далее при 150°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 16,4% мас. МоО3, 5,8% мас. СоО (табл.1).
Пример 10
Модифицированный носитель готовят по примеру 9.
Для приготовления пропиточного раствора 34,41 г додекамолибдофосфорной кислоты Н3[РМо12О40] растворяют в 60 см3 воды при добавлении 7,30 г гидрокарбоната кобальта CoCO3·nCo(ОН)2·mH2O и 10,2 г винной кислоты C4H6O6 при 40-70°C. Далее объем полученного раствора доводят водой до 85 см3. pH пропиточного раствора равен 1,5-2,5.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат в воздушной атмосфере при комнатной температуре, а далее при 120°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 19,5% мас. МоО3, 5,9% мас. СоО (табл.1).
Пример 11
101 г борсодержащего носителя, полученного согласно примеру 9, пропитывают по влагоемкости 85 см3 раствором платинохлористоводородной кислоты H2PtCl6 с концентрацией платины 10,6 мг/см3. Полученный модифицированный носитель сушат при 60, 80 и 120°C по 2 ч на воздухе, а далее прокаливают со скоростью 1°C/мин до 500°C и выдерживают в течение 4 ч. После прокаливания гранулы модифицированного носителя имеют удельную поверхность 223 м2/г, объем пор 0,75 см3/г и средний диаметр пор 12,1 нм.
Для приготовления пропиточного раствора 33,53 г декамолибдодикобальтовой гетерополикислоты Н6[Со2Мо10О38Н4] и 8,80 г гидрокарбоната кобальта СоСо3·nCo(ОН)2·mH2O растворяют в 70 см3 воды при 40-60°C и перемешивании. После окончания выделения CO2 в полученный раствор добавляют 12,3 г винной кислоты C4H6O6 и доводят объем водой до 85 см3. pH пропиточного раствора равен 2,0-3,0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат в воздушной атмосфере при комнатной температуре, а далее при 120°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 19,8% мас. MoO3, 5,4% мас. СоО (табл.1).
Пример 12
Модифицированный носитель готовят по примеру 11.
Для приготовления пропиточного раствора 25,6 г MoO3 растворяют в 77 г 30%-ного раствора H2O2 при интенсивном перемешивании и температуре 70-90°C. Далее в полученный раствор добавляют 8,77 г гидрокарбоната кобальта CoCO3·nCo(ОН)2·mH2O. После окончания выделения CO2 в полученный раствор добавляют 12,3 г винной кислоты C4H6O6 и доводят объем водой до 85 см3. рН пропиточного раствора равен 2,0-3,0.
Модифицированный носитель массой 100 г выдерживают в вакууме 30 мин, затем заливают пропиточным раствором, имеющим температуру 40°C. Носитель выдерживают в пропиточном растворе в течение 15 мин. Полученный катализатор сушат в воздушной атмосфере при комнатной температуре, а далее при 140°C в течение 8 ч.
После прокаливания на воздухе в течение 2 ч при 550°C катализатор содержит 19,6% мас. MoO3, 5,4% мас. СоО (табл.1).
Катализаторы заявляемого состава и способа приготовления имеют высокую каталитическую активность и обеспечивают получение продуктов с низким содержанием серы, менее 50 и 10 ppm (табл.2). Использование в качестве модификатора носителя переходного или благородного металла обеспечивает более высокую каталитическую стабильность.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И ПРОЦЕСС СЕЛЕКТИВНОГО ГИДРООБЕССЕРИВАНИЯ ОЛЕФИНСОДЕРЖАЩЕГО УГЛЕВОДОРОДНОГО СЫРЬЯ | 2013 |
|
RU2557248C2 |
| КАТАЛИЗАТОР ГИДРООБЕССЕРИВАНИЯ, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И ПРОЦЕСС ГЛУБОКОЙ ГИДРООЧИСТКИ УГЛЕВОДОРОДНОГО СЫРЬЯ | 2014 |
|
RU2573561C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРОВ ДЛЯ ГЛУБОКОЙ ГИДРООЧИСТКИ НЕФТЯНЫХ ФРАКЦИЙ | 2008 |
|
RU2385764C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРОВ И КАТАЛИЗАТОР ДЛЯ ГЛУБОКОЙ ГИДРООЧИСТКИ НЕФТЯНЫХ ФРАКЦИЙ | 2012 |
|
RU2486010C1 |
| КАТАЛИЗАТОР ГЛУБОКОЙ ГИДРООЧИСТКИ НЕФТЯНЫХ ФРАКЦИЙ И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2015 |
|
RU2631424C2 |
| КАТАЛИЗАТОР, СПОСОБ ПРИГОТОВЛЕНИЯ НОСИТЕЛЯ, СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА И СПОСОБ ГИДРООЧИСТКИ УГЛЕВОДОРОДНОГО СЫРЬЯ | 2015 |
|
RU2639159C2 |
| СОСТАВ И СПОСОБ СИНТЕЗА КАТАЛИЗАТОРА ГИДРОДЕОКСИГЕНАЦИИ КИСЛОРОДСОДЕРЖАЩЕГО УГЛЕВОДОРОДНОГО СЫРЬЯ | 2012 |
|
RU2492922C1 |
| Способ получения катализатора гидроочистки дизельных фракций, катализатор гидроочистки дизельных фракций и способ его применения | 2022 |
|
RU2800668C1 |
| КАТАЛИЗАТОР ГИДРООЧИСТКИ ТЯЖЕЛЫХ НЕФТЯНЫХ ФРАКЦИЙ И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2008 |
|
RU2414963C2 |
| КАТАЛИЗАТОР ГИДРООЧИСТКИ МАСЛЯНЫХ ФРАКЦИЙ И РАФИНАТОВ СЕЛЕКТИВНОЙ ОЧИСТКИ И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2012 |
|
RU2497585C2 |
Изобретение относится к катализатору глубокой гидроочистки углеводородного сырья, состоящему из одно или несколько биметаллических комплексных соединений металлов VIII и VIB групп, нанесенных на модифицированный носитель определенного состава. Катализатор имеет удельную поверхность 180-350 м2/г, объем пор 0,3-0,6 см3/г, средний диаметр пор 8,5-13,0 нм. Изобретение также относится к носителю катализатора, который состоит из композиции гамма-оксида алюминия, оксидов неметалла: фосфора и/или бора, оксидов переходного металла: никеля и/или кобальта, или одного из оксидов благородного металла: платины, родия, рутения, при следующем содержании, мас.%: оксиды неметалла 0,5-1,3, оксиды переходного металла 0,5-5,0 или оксиды благородного металла 0,3-0,8, оксид алюминия 99,0-93,5%; имеет удельную поверхность 200-370 м2/г, объем пор 0,5-1,0 см3/г, средний диаметр пор 8,5-13,5. Изобретение имеет отношение к способу получения этого носителя, который осуществляют путем пропитывания гамма-оксида алюминия соединениями бора и/или фосфора, переходного металла (никеля и/или кобальта) или благородного металла (одного из ряда платины, родия, рутения), с последующей сушкой при 80-120°C (5 ч) и прокаливанием при 300°C (2 ч). Далее, способ получения катализатора включает однократную пропитку носителя водным раствором, имеющим рН 1,5-3,0, содержащим как минимум один из гетерополианионов ряда [CO2Mo10O38H4]6-, [Co(OH)6Mo6O18]4-, [Ni(OH)6Mo6O18]4-, [Ni2Mo10O38H4]6-, [P2Mo5O23]6-, [SiMo12O40]4-, [PMo12O40]3-. В качестве соединения кобальта используется одно из ряда гидроксид кобальта, кобальт углекислый CoCO3, кобальт углекислый основной. В качестве соединения никеля используется одно из ряда гидроксид никеля, никель углекислый NiCO3, никель углекислый основной. В качестве стабилизатора пропиточного раствора используют карбоновую кислоту, содержащую, по меньшей мере, одну карбоксильную группу и 1-20 углеродных атомов. Технический результат настоящего изобретения - увеличение каталитической стабильности катализатора, высокая эффективность работы катализатора. 4 н. и 11 з.п. ф-лы, 2 табл., 12 пр.
1. Катализатор глубокой гидроочистки углеводородного сырья, состоящий из соединений металлов VIII и VIB групп, нанесенных на алюмооксидный носитель, отличающийся тем, что содержит одно или несколько биметаллических комплексных соединений металлов VIII и VIB групп, нанесенных на модифицированный носитель, который состоит из композиции гамма-оксида алюминия, оксидов неметалла: фосфора и/или бора, оксидов переходного металла: никеля и/или кобальта, или одного из оксидов благородного металла: платины, родия, рутения, при следующем содержании, мас.%: оксиды неметалла 0,5-1,3, оксиды переходного металла 0,5-5,0 или оксиды благородного металла 0,3-0,8, оксид алюминия 93,5-99,0; катализатор имеет удельную поверхность 180-350 м2/г, объем пор 0,3-0,6 см3/г, средний диаметр пор 8,5-13,0 нм.
2. Катализатор по п.1, отличающийся тем, что в прокаленном при 550°С катализаторе MoO3 составляет 14,0-23,0 мас.%, CoO и/или NiO - 4,0-6,5 мас.%.
3. Катализатор по п.1, отличающийся тем, что имеет форму цилиндров или трехлистников.
4. Носитель катализаторов глубокой гидроочистки углеводородного сырья, который состоит из композиции гамма-оксида алюминия, оксидов неметалла: фосфора и/или бора, отличающийся тем, что содержит на поверхности оксид переходного металла: никеля и/или кобальта, или один из оксидов благородного металла: платины, родия, рутения, при следующем содержании, мас.%: оксиды неметалла 0,5-1,3, оксиды переходного металла 0,5-5,0 или оксиды благородного металла 0,3-0,8, оксид алюминия 99,0-93,5; имеет удельную поверхность 200-370 м2/г, объем пор 0,5-1,0 см3/г, средний диаметр пор 8,5-13,5 нм.
5. Способ получения носителя для катализатора глубокой гидроочистки углеводородного сырья по п.4, отличающийся тем, что гамма-оксид алюминия пропитывают соединениями бора и/или фосфора, переходного металла (никеля и/или кобальта) или благородного металла (одного из ряда платины, родия, рутения), с последующей сушкой при 80-120°C (5 ч) и прокаливанием при 300°C (2 ч).
6. Способ получения носителя по п.5, отличающийся тем, что в качестве соединений бора используют как минимум одно, выбранное из ряда ортоборная кислота H3BO3, пироборная кислота H2B4O7, метаборат аммония NH4BO2, борат аммония (NH4)3BO3, тетраборат аммония (NH4)2B4O7, в качестве соединений фосфора - ортофосфорная кислота H3PO4, метафосфорная кислота HPO3, пирофосфорная кислота Н4Р2О7, гидрофосфат аммония (NH4)2HPO4, метафосфат аммония NH4PO3, пирофосфат аммония (NH4)4P2O7.
7. Способ получения носителя по п.5, отличающийся тем, что в качестве соединений переходных металлов используют как минимум одно, выбранное из ряда гексагидрат нитрата кобальта Co(NO3)2·6H2O, тетрагидрат ацетата кобальта Со(СН3СОО)2·4H2O, тетрагидрат сульфата кобальта CoSO4·7H2O, тетрагидрат хлорида кобальта CoCl2·4H2O, дигидрат цитрата кобальта Со3(C6H5O7)2·2H2O, гексагидрат нитрата никеля Ni(NO3)2·6H2O, тетрагидрат ацетата никеля Ni(CH3COO)2·4H2O, тетрагидрат сульфата никеля NiSO4·7H2O, тетрагидрат хлорида никеля NiCl2·4H2O, дигидрат цитрата никеля Ni(C6H5O7)2·14H2O.
8. Способ получения носителя по п.5, отличающийся тем, что в качестве соединений благородных металлов используют как минимум одно, выбранное из ряда платинохлористоводородная кислота H2PtCl6, тетрахлорид гексаамминплатины [Pt(NH3)6]Cl4, дихлородиамминоплатина(II), [PtCl2(NH3)2], тригидрат хлорида родия RhCl3·3H2O, дигидрат нитрата родия Rh(NO3)3·2H2O, трихлорид гексаамминродия [Rh(NH3)6]Cl3, пентагидрат хлорида рутения RuCl4·5H2O, гексагидрат нитрата рутения Ru(NO3)3·6H2O, трихлорид гексаамминрутения [Ru(NH3)6]Cl3.
9. Способ приготовления катализатора глубокой гидроочистки углеводородного сырья по п.1 пропиткой носителя по влагоемкости или с избытком раствором предшественников активного компонента, отличающийся тем, что носитель однократно пропитывают водным раствором, имеющим рН 1,5-3,0, содержащим как минимум один из гетерополианионов ряда [CO2Mo10O38H4]6-, [Co(OH)6Mo6O18]4-, [Ni(OH)6Mo6O18]4-, [Ni2Mo10O38H4]6-, [P2Mo5O23]6-, [SiMo12O40]4-, [PMo12O40]3-, в качестве соединения кобальта используется одно из ряда гидроксид кобальта, кобальт углекислый CoCO3, кобальт углекислый основной, в качестве соединения никеля используется одно из ряда гидроксид никеля, никель углекислый NiCO3, никель углекислый основной, в качестве стабилизатора пропиточного раствора используют карбоновую кислоту, содержащую, по меньшей мере, одну карбоксильную группу и 1-20 углеродных атомов.
10. Способ приготовления катализатора по п.9, отличающийся тем, что гетерополианион [P2Mo5O23]6- или [PMo12O40]3- формируется путем последовательного растворения оксида молибдена MoO3 или молибденовой кислоты H2MoO4 в 85%-й фосфорной кислоте H3PO4 и добавлении 30%-ного раствора H2O2, при температуре 50-90°С и pH 1,0-1,5 в соотношениях, соответствующих стехиометрии в гетерополианионе.
11. Способ приготовления катализатора по п.9, отличающийся тем, что один из гетерополианионов [CO2Mo10O38H4]6-, [Со(ОН)6Mo6O18]4-, [Ni(OH)6Mo6O18]4-, [Ni2Mo10O38H4]6- формируется путем последовательного растворения оксида молибдена MoO3 или молибденовой кислоты H2MoO4 в 30%-ном растворе пероксида водорода при температуре 50-90°C, с последующим добавлением соли Со или Ni в соотношениях, соответствующих стехиометрии в гетерополианионе.
12. Способ приготовления катализатора по любому из пп.9, 10 или 11, отличающийся тем, что для приготовления катализатора в качестве стабилизатора используется лимонная кислота.
13. Способ приготовления катализатора по любому из пп.9, 10 или 11, отличающийся тем, что пропитка гранул носителя проводится после создания вакуума в сосуде, содержащем носитель.
14. Способ приготовления катализатора по любому из пп.9, 10 или 11, отличающийся тем, что пропитка гранул носителя после создания вакуума проводится пропиточным раствором при температурах 20-50°С.
15. Способ приготовления катализатора по любому из пп.9, 10 или 11, отличающийся тем, что после пропитки катализатор сушат при температуре 120-180°С в потоке воздуха или азота.
| КАТАЛИЗАТОР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРОЦЕСС ГИДРООБЕССЕРИВАНИЯ ДИЗЕЛЬНЫХ ФРАКЦИЙ | 2006 |
|
RU2314154C1 |
| US 20060054536 А1, 16.03.2006 | |||
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ДЛЯ ГИДРООЧИСТКИ НЕФТЯНОГО СЫРЬЯ | 1996 |
|
RU2103065C1 |
| US 4818743 A1, 04.04.1989 | |||
| КАТАЛИЗАТОР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРОЦЕСС ГИДРООБЕССЕРИВАНИЯ ДИЗЕЛЬНЫХ ФРАКЦИЙ | 2006 |
|
RU2314154C1 |

