ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение относится к фармацевтическим составам для контролируемого высвобождения активных соединений. Эти составы существуют в виде полимерных микрочастиц, гелей in-situ или твердых имплантатов. Они основаны на биоразлагаемых полимерах и особенно полезны для контролируемой доставки в ткани лечебных протеинов или пептидов. Кроме того, изобретение относится к полимерным микрочастицам, включенным в упомянутые составы и к методам изготовления таких частиц. В дополнительных аспектах изобретение относится к фармацевтическим наборам, которые включают упомянутые составы, и к использованию таких наборов.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Парентеральные лекарственные формы, обладающие способностью медленно выделять лекарство, были созданы в ответ на потребность в улучшении терапевтического использования лекарственных веществ, которые из-за их физико-химических свойств нельзя вводить оральным путем и которые имеют относительно короткий период полувыведения, что заставляет делать частые инъекции. Частые инъекции неудобны для больных, а если их делают врачи или медицинские сестры, то это обходится довольно дорого. Ощущения дискомфорта и боли могут привести к нарушению дисциплины больных в выполнении врачебного назначения и поставить под угрозу успех лечения.
Число лекарственных веществ, которые нельзя вводить в организм удобным оральным способом, неизбежно возрастает, главным образом, вследствие новейших достижений биотехнологических исследований в области фармакологии, приведших к появлению множества мощных пептидных и белковых лекарств. Возможно, за исключением некоторых мелких пептидов, эти соединения довольно нестабильны в желудочно-кишечной среде, а также, что еще более важно, слишком велики и гидрофильны как молекулы, чтобы всасываться через слизистую оболочку кишечника в достаточном количестве. Для некоторых из веществ с такими свойствами разрабатываются инъекционные или имплантируемые рецептуры с контролируемым высвобождением, позволяющие уменьшить частоту введения доз, то есть снижающие дискомфорт для больных, что приводит к повышению дисциплины и увеличению вероятности конечного успеха терапии.
Парентеральные лекарственные формы с контролируемым высвобождением обычно выпускаются в виде макроскопических твердых одиночных или множественных имплантатов (например, полимерных стержней и вафельных пластинок), взвесей микрочастиц, а в последние годы также и гелей, включая гели, образующиеся in-situ. Твердые имплантаты с загруженными лекарствами выпускаются в виде неразлагающихся полимерных, керамических или металлических устройств, которые необходимо удалять хирургическим путем после определенного периода воздействия лекарства, или в виде биоразлагаемых полимерных форм, которые не требуют удаления из организма. Примером не разрушающегося имплантата служит устройство Viadur® компании Байер, которое высвобождает пептидное лекарство лейпролид в течение одного года. Примером биологически разрушающегося имплантата служит Zoladex® компании AstraZeneca, представляющий собой полимерный стержень, способный высвобождать пептидное лекарство госелерин в течение одного или трех месяцев, соответственно.
Вскоре после появления на рынке первых биоразлагаемых имплантатов стали выпускаться микрочастицы с контролируемым высвобождением, например, депонированные лекарственные формы Lupron® от компании Takeda, которые высвобождают лейпролид в течение одного, трех и четырех месяцев, соответственно. Для того чтобы ввести такие микрочастицы через инъекцию, их надо суспендировать в водном носителе. Однако по соображениям стабильности депонированные микрочастицы нельзя длительно хранить в виде водной суспензии, а надо восстанавливать из сухого порошка непосредственно перед применением.
Различные конструкции микрочастиц с загрузкой лекарств и методы их приготовления описаны в публикации E. Mathiowitz et al., Microencapsulation (Микрокапсулирование), в книге: Encyclopedia of Controlled Drug Delivery (Энциклопедия контролируемой доставки лекарств в организм) (под редакцией E. Mathiowitz), Vol. 2, p. 493-546, John Wiley & Sons (1999), которая упоминается здесь в качестве ссылки.
Для осуществления инъекций систем доставки лекарств через особо тонкие иглы, создающие повышенный комфорт для больного, ученые, занимающиеся проблемой доставки лекарств, в последние годы начали разрабатывать инъекционные гели, способные образовывать подкожные или внутримышечные депо. В одной из концепций создаются лекарственные формы в виде гелей, обладающие высокой способностью к разжижению при сдвиге и тиксотропностью. Прикладывая к этим гелям усилие сдвига перед введением в организм, можно существенно снизить их вязкость, что позволяет выполнить инъекцию относительно небольшой иглой, причем после введения геля его прочность медленно восстанавливается. В соответствии с другой концепцией создаются жидкие лекарственные композиции, которые после введения в организм образуют гели в ответ на изменения окружающей среды, в частности, pH, температуры и ионной силы. Согласно третьему подходу в организм посредством инъекции вводят жидкие полимерные лекарства, содержащие безводный растворитель. После введения растворитель диффундирует, уходя из места инъекции, что приводит к преципитации полимерных частиц или к образованию геля.
Подробное обсуждение биологически разлагающихся инъекционных гелей представлено в публикации A. Hatefi et al., Journal of Controlled Release 80 (2002), 9-28, которая упоминается здесь для ссылки.
Терапевтическая полезность разных полимерных носителей для контролируемого высвобождения лекарственных веществ, особенно полимеров и сополимеров молочной кислоты и гликолевой кислоты, была продемонстрирована для нескольких активных соединений, в частности лейпролида, гозерелина, бусерелина и трипторелина, - все они представляют собой пептиды с очень большим терапевтическим индексом, т.е. с весьма малой токсичностью даже в дозах, намного превышающих терапевтически эффективные концентрации. В отличие от этого, для хуже переносимых активных соединений, например эритропоэтинов и интерферонов, когда необходима точно контролируемая доставка, позволяющая достичь лечебного эффекта без неприемлемых побочных эффектов, лекарственные формы с контролируемым высвобождением разрабатывались с гораздо меньшим успехом. Основная трудность заключается в том, что биоразлагаемые полимерные носители, которые успешно использовались раньше, очевидно, неспособны обеспечить профиль высвобождения нулевого или близкого к нулевому порядка. Вместо этого при введении в организм они создают весьма нежелательное начальное взрывное высвобождение. Более того, аутокаталитический распад полимеров и сополимеров молочной кислоты и гликолевой кислоты также может приводить к эффектам демпинга дозы на более поздних стадиях высвобождения лекарства. С другой стороны, другие новые полимеры, которые обсуждались как улучшенные носители для контролируемого высвобождения лекарственных соединений, не имеют профиля безопасности, характерного для поли(лактидов) и поли(гликолидов).
Таким образом, существует потребность в новых полимерных системах доставки с доказанной биологической совместимостью, которые в то же время способны лучше контролировать высвобождение относительно токсичных терапевтических соединений, чем прежние носители.
Поэтому цель изобретения состоит в том, чтобы удовлетворить потребность в новых лекарственных составах с контролируемым высвобождением, содержащих один или более полимерных носителей с очень хорошей биологической совместимостью, а также относительно токсичное лекарственное соединение, которое вводят в организм неоральным способом, например, протеин.
Еще одна цель изобретения - обеспечить пользователей микрочастицами, имплантатами и гелевыми композициями, содержащими активное соединение, которое высвобождается с контролируемой скоростью. Кроме того, цель изобретения - обеспечить пользователей наборами, которые содержат такие лекарственные композиции, и рекомендациями по их фармацевтическому применению. Другие цели и возможности применения данного изобретения станут очевидными на основе последующего описания и пунктов формулы изобретения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение предусматривает фармацевтическую композицию для контролируемого высвобождения интерферонов. Точнее говоря, лекарственная композиция, предлагаемая изобретением, включает биоразлагаемый полимер и активное соединение, выбираемое из группы интерферонов. Биоразлагаемый полимер это блок-сополимер, состоящий из поли(этиленгликоль)терефталата (ПЭГТ) и поли(бутилентерефталата) (ПБТ). Предпочтительное активное соединение это интерферон, выбираемый из семейства альфа-интерферонов.
В другом варианте композиция, предлагаемая изобретением, создана таким образом, что содержит микрочастицы с блок-сополимером и, по меньшей мере, с некоторой частью интерферона, включенного в состав лекарства. Такая композиция особенно полезна как парентеральная лекарственная форма с контролируемым высвобождением, которую можно вводить в организм посредством внутримышечных или подкожных инъекций.
В другом варианте изобретение предусматривает фармацевтический набор, состоящий из первого и второго герметичных отделений, причем первое отделение содержит упомянутую лекарственную композицию на основе микрочастиц, главным образом, в сухом виде, а второе отделение содержит жидкий водный носитель для восстановления композиции в виде инъекционной суспензии микрочастиц.
В другом варианте лекарственная композиция, предлагаемая изобретением, имеет форму твердого имплантата.
Другие варианты реализации изобретения включают методы изготовления фармацевтической композиции и способы ее фармацевтического использования.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 наглядно представляет высвобождение интерферона из частиц сополимера in vitro и у хомяков.
Фиг.2 наглядно представляет высвобождение интерферона из частиц сополимера in vitro и у обезьян.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В процессе открытия, приведшего к данному изобретению, было установлено, что многие из полимеров, которые предполагались как агенты для контролируемого высвобождения активных соединений, в частности полимеры молочной и/или гликолевой кислоты, не очень хорошо подходят для доставки относительно токсичных активных соединений, в частности интерферонов. В особенности необходимо отметить то, что характер высвобождения выглядит плохо контролируемым, особенно, если полимеры сформированы в виде микрочастиц или гелей. Например, при использовании традиционных полимерных носителей, по-видимому, трудно избежать так называемого взрывного высвобождения, то есть быстрого выброса значительной части включенного в лекарственный состав активного соединения вскоре после введения. В зависимости от терапевтического индекса соответствующего активного соединения такое взрывное высвобождение может вызвать у больных довольно выраженные токсические эффекты.
В отличие от этого, неожиданно было обнаружено, что блок-сополимеры ПЭГТ и ПБТ способны инкорпорировать и высвобождать интерфероны (вышеупомянутые соединения) контролируемым образом лучше, при этом с незначительным или практически отсутствующим взрывным эффектом, что будет обсуждаться далее в этом описании.
Следовательно, данное изобретение предлагает фармацевтическую композицию для контролируемого высвобождения, включающую биоразлагаемый полимер и активное соединение, выбираемое из группы интерферонов, причем биоразлагаемый полимер представляет собой блок-сополимер, состоящий из поли(этиленгликоль)терефталата (ПЭГТ) и поли(бутилентерефталата) (ПБТ).
Изобретателями также было обнаружено, что описанные выше блок-полимеры могут образовывать удивительно удобную матрицу для инкорпорирования интерферонов и их последующего контролируемого высвобождения. Особенно важно, что они могут инкорпорировать интерфероны в большом количестве без потери их биологической активности.
Другой довод в пользу особой пригодности указанных сополимеров заключается в том, что они способны контролировать высвобождение инкорпорированных интерферонов в широком диапазоне профилей, что можно считать весьма желательным в зависимости от конкретного варианта терапевтического применения. Полимерный носитель может быть создан в разных формах, например, в виде микрочастиц, пленок, гелей и твердых имплантатов, а также в широком диапазоне молекулярного веса и степеней гидрофильности, что наряду с геометрической структурой конкретной формы или созданного на ее основе лекарственного состава позволяет достичь разной продолжительности высвобождения интерферона и разных по типу профилей его высвобождения.
Фармацевтическая композиция определяется как состав, который типично используется в лечебных или диагностических целях или для поддержания хорошего состояния здоровья и профилактики заболеваний. Если одни фармацевтические композиции созданы и составлены для немедленного высвобождения инкорпорированных активных соединений, то другие обладают свойством контролируемого высвобождения активного вещества, что придает лекарству пролонгированное действие и длительную эффективность. Для описания разных типов контролируемого высвобождения было предложено несколько терминов. В этом документе термин контролируемое высвобождение включает любое высвобождение активного соединения, в частности, задержанное высвобождение, пролонгированное высвобождение, константное высвобождение или высвобождение нулевого порядка, растянутое высвобождение, непрерывное высвобождение, медленное высвобождение, двухфазное высвобождение и т.д.
Композиция содержит в своем составе биоразлагаемый полимер. В соответствии с терминологией IUPAC (Международного союза теоретической и прикладной химии) полимер определяется как вещество, состоящее из макромолекул. В свою очередь, макромолекула это молекула с высокой относительной молекулярной массой, строение которой, главным образом, представляет собой множественное повторение однотипных структурных единиц. Однако в общеязыковом смысле различие между полимером и макромолекулами, из которых он состоит, проводится не всегда. Это также справедливо и для данного текста, в котором признаки, строго говоря, присущие полимеру, могут приписываться макромолекулам.
Подверженность биологическому распаду можно определить как способность вещества разрушаться химически в физиологических условиях, в физиологической окружающей среде или под воздействием ферментов. В контексте изобретения предпочтительно, чтобы полимер, подверженный биологическому распаду (биоразлагаемый полимер), разрушался в физиологической окружающей среде, то есть в физиологических жидкостях при температуре тела (даже при отсутствии ферментов), в том смысле, что существенный распад полимера должен происходить в течение часов, дней, недель, месяцев или лет. В распаде полимера могут участвовать разные химические механизмы, включая гидролиз и/или окисление. Во избежание неверных толкований надо отметить, что подверженность биологическому распаду вовсе не означает, что полимер должен распадаться на соответствующие мономерные единицы. Важно понимать, что процесс распада ведет к возникновению растворимых видов молекул, которые могут выводиться из организма в процессах почечной или печеночной экскреции. В данном изобретении полимер является носителем активного соединения и контролирует его высвобождение.
Кроме того, биоразлагаемый полимер, выбирается из группы блок-сополимеров, которые состоят из поли(этиленгликоль)терефталата (ПЭГТ) и поли(бутилен)-терефталата (ПБТ). Сополимер определяется как полимер, полученный из мономеров более чем одного вида. В блок-сополимере (или блок-полимере) составные макромолекулы содержат смежные блоки, структурно отличающиеся друг от друга, иначе говоря, смежные блоки содержат структурные единицы, полученные из разных видов мономера или из одного и того же вида мономера, но с разным составом или разной последовательностью распределения структурных единиц. Блок можно определить как часть макромолекулы, содержащую множество структурных единиц, которые обладают, по меньшей мере, одним признаком, отсутствующим в соседних частях.
Многие блок-сополимеры, содержащие ПЭГТ и ПБТ, были описаны в прототипах, например, J. M. Bezemer et al. (J. Control Release 1999, 62 (3), 393-405; J. Biomed. Mater. Res. 2000, 52 (1), 8-17; J. Control Release 2000, 66 (2-3), 307-320; J. Control Release 2000, 67 (2-3), 249-260; J. Control Release 2000, 67 (2-3), 233-248; J. Control Release 2000, 64 (1-3), 179-192), R. Dijkhuizen-Radersma et al. (Biomate-rials 2002, 23 (24), 4719-4729; J. Biomed. Mater. Res. 2004, 71A (1), 118; Biomate-rials 2002, 23 (6), 1527-1536; Pharm. Res. 2004, 21 (3), 484-491; Int. J. Pharm. 2002, 248 (1-2), 229-237; Eur. J. Pharm. Biopharm. 2002, 54 (1), 89-93) и J. Sohier et al. (J. Control Release 2003, 87 (1-3), 57-68; Eur. J. Pharm. Biopharm. 2003, 55 (2), 221-228), а также в патентных заявках WO 93/21858, EP 0 830 859 A2 и EP 1 090 928 Al, причем все эти документы включены в данное описание во всей полноте.
Можно понимать, что эти сополимеры состоят из повторяющихся блоков гидрофильного поли(этиленгликоля) (ПЭГ) и гидрофобного поли(бутилентерефталата) (ПБТ). Эти сложные полиэфиры, состоящие из простых полиэфиров, в типичном случае получают поликонденсацией ПЭГ, бутандиола и диметил-терефталата. Альтернативное понимание заключается в том, что они состоят из повторяющихся блоков поли(этиленгликоль)терефталата (ПЭГТ) и ПБТ. Такие сополимеры обычно обладают свойствами термопластичных эластомеров. В водной среде они образуют гидрогели или подобные им полимерные сети, в которых цепочки полимера имеют не химические, а физические перекрестные связи. Полагают, что перекрестные связи являются результатом ассоциации "твердых" сегментов ПБТ в кристаллические домены, тогда как аморфные части, состоящие из "мягких" сегментов ПЭГ и отчасти ПБТ, обусловливают разбухание в воде. В отличие от химических перекрестных связей эти физические перекрестные связи носят обратимый характер и могут разрываться при повышении температуры или в соответствующих растворителях.
В соответствии с изобретением активное соединение выбирается из группы интерферонов. Интерфероны представляют собой семейство протеинов природного происхождения, извлекаемых из клеток человека и вовлеченных в разные функции иммунной системы, в частности в борьбу с вирусными инфекциями. Некоторые интерфероны были разработаны в виде фармацевтических продуктов и в настоящее время выпускаются как продукты генной инженерии для использования при лечении лейкозов, гепатита, рассеянного склероза и других тяжелых болезней.
В отличие от некоторых других активных пептидов и протеинов, успешно введенных в лекарственные продукты с контролируемым высвобождением, интерфероны имеют относительно малый терапевтический индекс. Иначе говоря, они проявляют существенную токсичность на уровне, превышающем терапевтически эффективную концентрацию. Поэтому для достижения лечебного эффекта без неприемлемых побочных эффектов необходимо обеспечить их точно контролируемую доставку в организм.
Один из основных классов интерферонов - это альфа-интерфероны (альфа-ИФН или α-ИФН). Альфа-интерфероны включают множество природных и модифицированных протеинов со сходными молекулярным весом и функциональностью (см. D. J. A. Crommelin et al., Pharmaceutical Biotechnology, Harwood Academic Publishers (1997), 219-222). Одним из главных источников происхождения этих протеинов у человека являются лейкоциты. Известны, по меньшей мере, 23 разных природных подтипа и несколько модифицированных вариантов альфа-ИФН, а некоторые из них выпускаются в виде фармацевтических продуктов. Например, разработана и выпускается в промышленных масштабах смесь нескольких подтипов природных альфа-ИФН, извлеченных из инфицированных лейкоцитов человека. В настоящее время самыми важными членами группы альфа-ИФН считаются рекомбинантные варианты альфа-ИФН-2a и альфа-ИФН-2b. Еще один рекомбинантный альфа-ИФН, используемый в терапевтической практике, это ИФН-альфакон-1.
Основная функция этих интерферонов заключается в повышающей регуляции иммунной системы, в частности, в стимуляции иммунных клеток, способных распознавать и прямым или косвенным образом разрушать раковые клетки и вирусы. К терапевтическим показаниям для использования альфа-интерферонов относятся (хронический) гепатит B, (хронический) гепатит C, лейкоз с волосковыми клетками, (хронический) миелолейкоз, множественная миелома, фолликулярная лимфома, карциноидная опухоль, злокачественная меланома, бородавки гениталий, карцинома мочевого пузыря, карцинома шейки матки, почечно-клеточный рак (гипернефрома), папилломатоз гортани, грибовидный микоз (фунгоидная гранулема, болезнь Алибера), остроконечная кондилома, ТОРС (атипичная пневмония) и (связанная со СПИДом) саркома Капоши. Фактически, в настоящее время в соответствии с данным изобретением наиболее предпочтительно, чтобы активное соединение был выбрано из группы альфа-интерферонов.
Альфа-интерфероны природного происхождения имеют молекулярную массу от 19 до 26 кДа и состоят из белков с длиной цепочки 156-166 аминокислот и 172 аминокислоты. Все альфа-ИФН обладают общим регионом консервативной последовательности между позициями аминокислот 115-151, тогда как аминоконцевые последовательности варьируются. Многие подтипы альфа-ИФН отличаются по своим последовательностям только в одной или двух позициях. Природные варианты также включают протеины, усеченные на 10 аминокислот на карбоксиконцевом участке.
Другой большой класс интерферонов - это бета-интерфероны (бета-ИФН), а самыми важными представителями этого класса в терапии являются бета-ИФН-1a и бета-ИФН-1b. Эти интерфероны используются, например, при лечении определенных форм рассеянного склероза, особенно, рецидивирующих форм рассеянного склероза, замедляя процесс накопления физической инвалидности и снижая частоту клинических обострений. Больные с рассеянным склерозом, у которых была продемонстрирована эффективность этих средств, включают лиц, перенесших первый клинический эпизод болезни и проявляющих на МРТ (магнитно-резонансных томограммах) характерные признаки рассеянного склероза.
Еще один класс интерферонов, имеющих терапевтическое значение, это гамма-интерфероны (гамма-ИФН). Эти интерфероны обладают противовирусной, антипролиферативной и иммуномодулирующей активностью. Один представитель гамма-интерферонов, гамма-ИФН-1b, в настоящее время выпускается на рынок для лечения тяжелых инфекций при хронических гранулематозах.
В последние годы были открыты и описаны некоторые дополнительные классы интерферонов, включая эпсилон-ИФН, каппа-ИФН и лямбда-ИФН (см. Kontsek et al., Acta Virol. 2003;47(4):201-15).
Согласно данному изобретению особенно хорошими свойствами обладает фармацевтическая композиция, в которой интерферон выбирается из группы альфа-интерферонов, предпочтительно, из группы, включающей альфа-ИФН, альфа-ИФН-2a, альфа-ИФН-2b, ИФН-альфакон-1, пегилированный альфа-ИФН-2a, пегилированный альфа-ИФН-2b, усеченный альфа-ИФН-2a, усеченный альфа-ИФН-2b, гибридные белки из альфа-ИФН и альбумина и их функциональные производные. В этом контексте альфа-интерферон также может представлять собой смесь разных вариантов альфа-интерферона, в частности, смесь природных альфа-интерферонов, которые трудно или не нужно разделять и очищать. Интерферон можно экстрагировать из живых организмов, изолированных клеток или клеточных культур. Для получения нужного интерферона клетки и/или организмы, из которых этот интерферон будет получен, могут быть модифицированы, например, инфекцией.
В соответствии с данным изобретением особенно хорошими свойствами обладает композиция, в которой интерферон представляет собой рекомбинантный продукт, полученный из генетически сконструированных клеток или организмов, причем эти клетки или организмы предпочтительно выбираются из клеток или организмов млекопитающих, насекомых, бактерий, дрожжей, грибов и высших растений.
Один из самых пригодных интерферонов для реализации преимуществ данного изобретения это усеченный вариант альфа-ИФН-2b или, по выбору, смесь, состоящая из более чем одного усеченного варианта альфа-ИФН-2b. Например, при помощи современных методов генной инженерии можно создать молекулы с аминокислотной последовательностью альфа-ИФН-2b, в которой удалены последние 5-10 аминокислот на N-конце. В другом варианте реализации изобретения предпочтительны такие разновидности альфа-ИФН-2b, которые укорочены на 7-8 N-концевых аминокислот.
Предпочтительно, чтобы композиция, предлагаемая данным изобретением, проявляла высвобождение активного соединения, по меньшей мере, на протяжении 7-дневного периода. Более предпочтительно, чтобы высвобождение интерферона происходило, по меньшей мере, 10 дней или, по меньшей мере, 14 дней. В других вариантах реализации изобретения высвобождение происходит на протяжении, по меньшей мере, 3 недель, 4 недель, 6 недель и 2 месяцев, соответственно. В настоящее время наиболее предпочтительным считается высвобождение на протяжении периода времени от 10 дней до 1 месяца. Разновидность выбираемого полимера и дополнительные специальные характеристики, необходимые для достижения необходимой длительности высвобождения, по меньшей мере, частично зависят от выбранной конструкции лекарственной формы, что будет более подробно описано ниже.
Изобретение также связано с фармацевтической композицией для контролируемого высвобождения, которая содержит биоразлагаемый полимер, а также одно или более активных соединений, выбираемых из группы интерферонов, причем, по меньшей мере, 80% вес. активного соединения в расчете на общий вес активного соединения высвобождается в мономерной (не агрегированной) форме. В соответствии с этим вариантом реализации изобретения биоразлагаемый полимер, предпочтительно, но не обязательно, представляет собой блок-сополимер, соответствующий данному здесь определению, состоящий из поли(этиленгликоль)терефталата (ПЭГТ) и поли(бутилентерефталата) (ПБТ). Изобретателями было обнаружено, что описанные выше блок-полимеры могут образовывать удивительно удобную матрицу для инкорпорирования интерферонов и их последующего контролируемого высвобождения. Особенно важно, что они могут инкорпорировать большое количество интерферонов без потери их биологической активности, причем сохраняют инкорпорированные интерфероны в мономерном (не агрегированном) состоянии. Это свойство особенно важно, поскольку известно, что интерфероны чувствительны к разным полимерам и режимам обработки, будучи чрезвычайно склонными к агрегации, которая часто сопровождается их инактивацией. В отличие от этого, использование описанных здесь блок-полимеров в качестве носителей интерферонов, возможно, позволяет достичь того, что большая часть инкорпорированного интерферона высвобождается в мономерной форме.
Предпочтительно, чтобы разновидность полимера и режим обработки выбирались таким образом, чтобы обеспечить высвобождение, по меньшей мере, 80% активного ингредиента, т.е., интерферона в мономерной (не агрегированной) форме. Еще более предпочтительно, чтобы в виде мономеров высвобождалось около 90% интерферона или, согласно другим вариантам реализации изобретения, около 95%, 97% и 98%, соответственно. Эти процентные показатели соответствуют весовым отношениям в расчете на общий вес инкорпорированного активного ингредиента.
Другие предпочтительные варианты композиций в соответствии с данным изобретением описаны ниже.
Разовая доза композиции, то есть количество композиции, которое вводится в организм за один раз, предпочтительно, содержит такое количество активного соединения, которое эквивалентно 1 миллиону международных единиц (1 ММЕ) соответствующего интерферона. Точное количество инкорпорированного интерферона, разумеется, зависит от профиля высвобождения в данной композиции и от того, получает ли конкретный больной ежедневные или еженедельные дозы лекарства.
В одном из вариантов реализации изобретения композиция адаптирована для высвобождения, по меньшей мере, 5 ММЕ интерферона в течение 14 дней, имея в виду первые 14 дней после введения дозы лекарства. В другом варианте реализации изобретения доза интерферона составляет приблизительно от 10 до 150 ММЕ, а период высвобождения составляет примерно от 10 дней до 1 месяца, особенно предпочтительно, около 14 дней. Также предпочтительна композиция, которая содержит и высвобождает в указанный период времени дозу интерферона приблизительно от 20 до 100 ММЕ. Такие композиции особенно предпочтительны, если активный ингредиент это альфа-интерферон, в частности, альфа-ИФН-2b или его производные.
В расчете на средний день в течение периода высвобождения интерферона после введения дозы лекарства композиция, предпочтительно, адаптирована для высвобождения соответствующего интерферона в количестве приблизительно от 0,5 до 20 ММЕ либо от 1 до 10 ММЕ. В зависимости от очертаний профиля высвобождения возможно, чтобы количество активного ингредиента, высвобождаемое в первый день после введения дозы лекарства, превышало 10 или 20 ММЕ, однако среднесуточное высвобождение интерферона за весь период должно оставаться в предпочтительном диапазоне.
Фармацевтическая композиция, предлагаемая данным изобретением, может быть создана, составлена и изготовлена так, чтобы она была пригодна для многих вариантов терапевтического использования и способов применения, в частности, местного, орального, ректального, вагинального или глазного введения в организм, однако предпочтительным способом введения считается парентеральный. В том смысле, в котором этот термин используется здесь, парентеральное введение включает любой инвазивный способ введения, в частности, внутрикожный, подкожный, внутримышечный, локально-региональный, интратуморальный, интраперитонеальный, интерстициальный, интралезионный (внутрь участка повреждения), а также, несколько менее предпочтительно в контексте данного изобретения, внутривенный, внутриартериальный и т.д. Весьма предпочтительными способами введения лекарственной композиции являются подкожная и внутримышечная инъекция или имплантация.
Пригодность для парентерального введения означает, в частности, что композиция, предпочтительно, стерильна и соответствует требованиям действующих фармакопей в отношении содержания эндотоксинов, осмотического давления и т.д. Наполнители, преимущественно, выбираются из числа безопасных и переносимых при парентеральном введении. В следующем аспекте изобретения фармацевтическая композиция составляется таким образом, чтобы она была относительно изотонической (или изоосмотической), например, в диапазоне приблизительно от 150 до 500 мОсмоль/кг (mOsmol/kg), предпочтительно, в диапазоне приблизительно от 250 до 400 мОсмоль/кг. Кроме того, показатель pH композиции должен находиться приблизительно в физиологическом диапазоне во избежание боли и местных реакций непереносимости при инъекциях. Предпочтительно, чтобы показатель pH композиции находился в диапазоне приблизительно от 4 до 8,5 и еще более предпочтительно - от 5,0 до 7,5.
Композиция, предлагаемая данным изобретением, может быть создана и составлена таким образом, чтобы она содержала микрочастицы, которые, в свою очередь, содержат биоразлагаемый блок-сополимер и активное соединение или, по меньшей мере, значительную фракцию активного соединения, представленного в этой композиции. В данном случае лекарственная форма упомянутой композиции, которая вводится в организм, представляет собой типичную инъекционную суспензию, состоящую из микрочастиц и связывающего их жидкого носителя.
В контексте изобретения микрочастицы необходимо понимать как твердые или полутвердые частицы, имеющие диаметр порядка 0,1-500 мкм, независимо от их формы или внутренней структуры. Например, микрочастицы могут также заключать в себе микросферы и микрокапсулы. В более предпочтительном варианте реализации изобретения микрочастицы имеют диаметр приблизительно от 1 до 300 мкм. Кроме того, было обнаружено, что желательные характеристики высвобождения лучше всего достигаются, если интерфероны инкорпорированы в микрочастицы, основанные на блок-сополимерах ПЭГТ/ПБТ со среднеобъемным диаметром порядка 25-200 мкм при измерении по методу фотонной корреляционной спектроскопии. Выбор микрочастиц такого размера также позволяет хорошо набирать их суспензию в шприц для проведения простой и удобной внутримышечной или подкожной инъекции.
В пределах этого диапазона размеров можно дополнительно оптимизировать диаметр микрочастиц для введения специфических продуктов или аккомодации к специфическим интерферонам. Например, в случае применения дополнительно усеченных альфа-интерферона-2a и альфа-интерферона-2b считается наиболее предпочтительным выбор микрочастиц со средне-объемным диаметром порядка 30-175 мкм. В других предпочтительных вариантах средний диаметр микрочастиц находится в диапазоне приблизительно от 50 до 150 мкм.
Предпочтительно, чтобы микрочастицы имели относительно низкую пористость. Особенно важно отметить обнаруженный изобретателями факт, что желательный профиль контролируемого высвобождения альфа-интерферонов поддерживается лучше всего, если удается в значительной степени избежать наличия в микрочастицах крупных пор. В этом контексте крупные поры можно определить как поры с диаметром порядка 5 мкм и более. Таким образом, в одном из предпочтительных вариантов реализации изобретения большинство микрочастиц практически не имеют пор с диаметром 5 мкм и более. В другом варианте большинство микрочастиц практически не имеют пор с диаметром 2 мкм и более.
Дополнительно (необязательно) микрочастицы могут быть покрыты слоем полимера, не содержащего лекарственного вещества. Такой вариант может быть полезен для предупреждения начального взрывного высвобождения инкорпорированного активного соединения или даже для достижения заранее определенного времени задержки перед началом его высвобождения, если это желательно.
Микрочастицы основаны на блок-сополимере ПЭГТ и ПБТ, который используется в качестве носителя и агента контролируемого высвобождения. Однако было обнаружено, что не все сополимеры ПЭГТ и ПБТ одинаково полезны для изготовления микрочастиц, способных осуществлять контролируемое высвобождение всех интерферонов. Кроме того, при выборе блок-сополимера важно учитывать намеченное время высвобождения или длительность лечебного эффекта активного вещества. Применительно к альфа-интерферонам было обнаружено, что сополимер, предпочтительно, должен содержать приблизительно от 50% до 95% вес. ПЭГТ и от 5%
до 50% вес. ПБТ. В другом варианте реализации изобретения сополимер содержит приблизительно от 70% до 95% вес. ПЭБТ. В соответствии еще с одним предпочтительным вариантом реализации изобретения сополимер содержит приблизительно от 70 до 85% вес. ПЭГТ.
Важным параметром для дальнейшего уточнения химического состава сополимера является молекулярный вес сегментов ПЭГ в компоненте ПЭГТ. Было обнаружено, что альфа-интерфероны очень легко инкорпорируются в микрочастицы сополимера, для которых профиль высвобождения можно отрегулировать в необходимом диапазоне при среднем молекулярном весе ПЭГ приблизительно от 600 до 3000. Еще предпочтительнее, чтобы средний молекулярный вес ПЭГ составлял приблизительно от 1000 до 2000.
Выбор среднего молекулярного веса ПЭГ также может быть сопряжен с особым вниманием к среднему размеру микрочастиц. Если, например, по соображениям технологии выбираются частицы относительно маленького размера, в частности менее 100 мкм и даже менее 75 мкм, предпочтительно остановить выбор на блок-полимере с относительно низкой степенью гидрофильности, т.е. имеющем относительно низкий средний молекулярный вес ПЭГ (около 1500 и менее или около 1000 и менее), особенно, если желательная продолжительность высвобождения составляет две недели и более. В альтернативном или дополнительном варианте низкой степени гидрофильности также можно достичь выбором относительно низкого содержания сегментов ПЭГТ, например, не более 75% вес.
И наоборот, могут возникнуть соображения для выбора частиц с относительно большим средним размером (порядка 100 мкм), например, в интересах технологии или для достижения желательного поведения in vivo. В таком случае предпочтительно выбирать средний молекулярный вес ПЭГ в диапазоне приблизительно от 1000 до 3000 или, по меньшей мере, порядка 1500 и/или относительно низкое содержание ПЭГТ, например, порядка 75% вес.
Кроме того, может принести пользу комбинирование двух или более разных блок-сополимеров ПЭГТ/ПБТ для изготовления микрочастиц с оптимизированным профилем высвобождения. Два или более блок-сополимера могут отличаться, например, или по относительному содержанию ПЭГТ, или по среднему молекулярному весу ПЭГ, или по обоим упомянутым параметрам. В частности, полезные смеси полимеров для изготовления микрочастиц с инкорпорацией альфа-интерферона в качестве активного агента могут включать два полимера, одинаковых по содержанию ПЭГТ порядка 80% вес., но отличающихся по среднему молекулярному весу ПЭГ (1000 и 2000, соответственно). Еще один вариант полезной смеси включает два полимера с содержанием ПЭГТ около 80% вес. и средним молекулярным весом ПЭГ около 1000 и 1500, соответственно. Два разных полимера (и более) можно смешивать в различных соотношениях, например, 50:50, 75:35 или 75:25.
Было обнаружено, что лекарственные композиции, предлагаемые изобретением, пригодны для инкорпорирования альфа-интерферонов с их последующим высвобождением на протяжении приблизительно от 1 до 8 недель. Например, подобрав соответствующие блок-сополимеры, профиль высвобождения можно отрегулировать таким образом, чтобы лечебный эффект активного вещества проявлялся на протяжении отрезка времени длительностью от 10 дней или 2 недель приблизительно до 4 недель, что в настоящее время считается наиболее предпочтительным временем высвобождения. Время высвобождения или длительность высвобождения следует понимать как отрезок времени, в течение которого высвобождается, по меньшей мере, около 80% вес., и более предпочтительно не менее 90 или 95% вес. инкорпорированного активного соединения. Профили высвобождения не демонстрируют сколько-либо заметного взрывного эффекта, т.е. начальное высвобождение (в первые 4 часа) составляет не более 10% инкорпорированной дозы, более предпочтительно, не более 7% инкорпорированной дозы.
Использование блок-сополимеров, как это описано выше, возможно, позволяет изготовить микрочастицы, инкорпорирующие терапевтически полезное количество интерферонов. Например, было обнаружено, что полимеры, выбранные в соответствии с предпочтительными вариантами реализации изобретения, могут инкорпорировать альфа-интерферон в количестве приблизительно от 0,1 до 20% вес. в соотношении к общему весу микрочастиц. Более предпочтительно, чтобы весовое содержание интерферона в микрочастицах составляло приблизительно от 0,2 до 10% вес. или от 0,5 до 5% вес., соответственно. В этих пределах интерферон совместим с полимерной матрицей, проявляя незначительную тенденцию к агрегации или вообще не проявляя такой тенденции. В то же время концентрация активного вещества достаточно велика для того, чтобы обеспечить удобное введение относительно небольшого объема суспензии микрочастиц при помощи инъекции.
В типичном случае доза альфа-интерферона в расчете на одну инъекцию составляет приблизительно от 3 до 2400 миллионов международных единиц (ММЕ) в зависимости от таких факторов как состояние больного, тип и тяжесть заболевания и, в особенности, длительность высвобождения из микрочастиц. Если микрочастицы созданы для высвобождения интерферона в течение приблизительно 2 или 4 недель, соответственно, то доза интерферона обычно будет находиться в диапазоне от 10 до 150 ММЕ. Фактически, в одном из предпочтительных вариантов реализации изобретения предлагаемая композиция содержит альфа-интерферон-2a, альфа-интерферон-2b или его фрагмент с силой действия в диапазоне приблизительно от 10 до 150 ММЕ в расчете на введенный с помощью инъекции объем лекарства. В соответствии с более предпочтительным вариантом композиция имеет силу воздействия, эквивалентную диапазону приблизительно от 20 до 100 ММЕ в расчете на одну инъекцию.
Ради удобства для больного объем инъекции должен быть не слишком большим, например, не более 3 мл (с учетом предпочтительного способа введения - внутримышечной или внутрикожной инъекции). В случае подкожного введения более предпочтительно, чтобы объем инъекции не превышал 2 мл. С другой стороны, высоко концентрированные инъекции в очень малых объемах трудно точно дозировать, и по этим соображениям предпочтительно, чтобы объем одной инъекции составлял, по меньшей мере, 0,1 мл или, более предпочтительно, по меньшей мере, 0,3 мл. В настоящее время наиболее предпочтительным объемом инъекции считается приблизительный диапазон от 0,5 мл до 2 мл.
Несмотря на то, что внутримышечные и/или подкожные инъекции лекарственных композиций с микрочастицами считаются предпочтительным способом их введения в организм, разумеется, вполне возможны, а для некоторых больных и при некоторых заболеваниях даже полезны другие способы введения. В более типичном случае такие способы связаны с парентеральным путем введения, но могут также подразумевать пульмональный, назальный, орально-слизистый (сублингвальный или буккальный), а также другие варианты. К числу полезных парентеральных способов введения таких лекарств, помимо внутримышечных и внутрикожных инъекций, можно отнести, особенно, интратуморальные, интралезионные (внутрь участка повреждения), локально-региональные, артериальные, интерстициальные и интраперитонеальные инъекции.
Микрочастицы и их инъекционные суспензии адаптированы для парентерального введения, а это означает, что они составлены и обработаны таким образом, чтобы соответствовать требованиям к парентеральным лекарственным формам. Такие требования, в частности, изложены в главных фармакопеях. В одном аспекте фармацевтическая композиция или ее предварительные смеси, или наборы, из которых она готовится, перед введением в организм должны быть стерильными. В другом аспекте наполнители необходимо выбирать из числа безопасных и хорошо переносимых при парентеральном введении. В следующем аспекте изобретения композиции составляются таким образом, чтобы они были относительно изотоническими (или изоосмотическими), например, в диапазоне приблизительно от 150 до 500 мОсмоль/кг, предпочтительно, в диапазоне приблизительно от 250 до 400 мОсмоль/кг. Кроме того, показатель pH композиции должен находиться приблизительно в физиологическом диапазоне во избежание боли и местных реакций непереносимости при инъекциях. Предпочтительно, чтобы показатель pH композиции находился в диапазоне приблизительно от 4 до 8,5 и еще более предпочтительно - от 5,0 до 7,5.
Микрочастицы обычно приобретают инъекционные свойства при их суспендировании в соответствующем биологически приемлемом жидком носителе, который, предпочтительно, основан на воде, хотя в нем могут присутствовать и другие биологически приемлемые растворители, в частности, этанол, глицерин, пропиленгликоль, полиэтиленгликоль или другие органические растворители. В более предпочтительном варианте реализации изобретения жидкая составляющая жидкого носителя имеет водную природу и в достаточной степени свободна от органических растворителей. С другой стороны, инкорпорирование других фармацевтических наполнителей может оказаться полезным или необходимым для оптимизации свойств лекарственного состава, в частности, переносимости, производительности (в смысле высвобождения активного вещества) и стабильности. Это может относиться как к микрочастицам, так и к жидкому носителю. Любая фаза может содержать одну и более физиологически переносимых добавок.
Обычно микрочастицы ресуспендируют в жидком носителе для образования суспензии с содержанием твердых частиц приблизительно от 1 до 20% вес., и, предпочтительнее, от 3 до 10% вес. Размер частиц и вязкость жидкого носителя, предпочтительно, выбирают таким образом, чтобы можно было делать инъекции относительно тонкой иглой, например, калибра от 20 до 22 G. В другом предпочтительном варианте реализации изобретения размер частиц и вязкость жидкого носителя адаптированы для выполнения подкожных и внутримышечных инъекций иглами калибра от 23 до 25 G.
Желательно микрочастицы создавать в таком виде, чтобы их можно было восстанавливать при помощи стерильного изотонического раствора натрия хлорида для инъекций.
Может оказаться полезной и стабилизация интерферона при помощи стабилизирующего наполнителя или комбинации наполнителей, в частности, одной или более солей, сахаров, сахарных спиртов, аминокислот, пептидов, протеинов, полимеров, сурфактантов, криопротектантов, осмотических агентов, буферных солей, кислот или оснований. Некоторые из этих наполнителей также могут быть полезны по другим фармацевтическим соображениям, например, для улучшения переносимости микрочастиц или их суспензии. Для модулирования свойств полимерного носителя или улучшения его стабильности может оказаться полезным дополнительное инкорпорирование одного или более пластификаторов, порообразователей, модификаторов высвобождения или антиоксидантов.
Во избежание агломерации микрочастиц при их суспендировании в водном носителе можно добавлять в водный носитель один или более биологически приемлемых сурфактантов (ПАВ). Фактически, в зависимости от реального состава лекарственной формы необходимый наполнитель, в частности, сурфактант, можно инкорпорировать или в водный носитель, или в сухую композицию, содержащую микрочастицы. Выбор подходящего сурфактанта также помогает добиться быстрого и легкого восстановления микрочастиц, например, не более чем за 3 минуты, предпочтительнее, приблизительно за 60 секунд, и еще предпочтительнее, не более чем за 30 секунд. Примерами потенциально полезных сурфактантов могут служить полоксамеры (поверхностно-активный полимер), полисорбаты, фосфолипиды и витамин E-TPGS.
В другом варианте реализации изобретение предлагает фармацевтический набор, включающий описанные выше микрочастицы. В этом контексте фармацевтический набор можно определить как комплект, включающий, по меньшей мере, две композиции, которые необходимо объединить для специального использования в лечебных, профилактических или диагностических целях. В данном случае набор содержит первый и второй герметически упакованные отделения, которые могут быть заключены в общую или в разные первичные упаковки. Первое отделение содержит композицию, соответствующую пункту 1 формулы изобретения, главным образом, в сухой форме, а второе отделение содержит жидкий водный носитель для восстановления этой сухой композиции в инъекционную суспензию микрочастиц. По выбору набор может содержать два или более комплекта первого и второго отделений.
Обычно сухая композиция, содержащаяся в первом отделении, соответствует разовой дозе инъекции, а второе отделение обычно содержит такой объем жидкого носителя, который необходим для восстановления содержимого первого отделения. Менее предпочтительными считаются отделения, содержащие более одной разовой дозы для введения посредством инъекции. Предпочтительно, чтобы содержание интерферона в первом отделении составляло приблизительно от 10 до 150 ММЕ, а объем жидкого носителя во втором отделении, который можно извлечь с помощью иглы, составлял приблизительно от 0,3 мл до 3 мл, в особенности, приблизительно от 0,5 мл до 2 мл.
Кроме того, набор имеет вторичную упаковку, которая используется для того, чтобы поместить в нее комплект или комплекты первого и второго отделений.
Первое и второе отделения могут представлять собой разные полости единого устройства или общую первичную упаковку. Например, это могут быть две полости двухкамерного шприца. Преимущество предварительно заполненного двухкамерного шприца заключается в том, что подготовка рабочей смеси и ее введение безопасны и удобны, поскольку не требуют манипуляций с разными контейнерами в асептических условиях. Один из недостатков таких шприцов это их относительная дороговизна, кроме того, они не всегда обеспечивают полное и надежное восстановление рабочей смеси.
В альтернативном варианте два отделения набора могут содержаться в двух разных первичных контейнерах или упаковках. Например, первое отделение, содержащее по существу сухую композицию микрочастиц, может иметь форму герметически упакованного пузырька или флакона из подходящего стекла или пластика, а жидкий водный носитель может поставляться в отдельном пузырьке, флаконе или ампуле. В следующем варианте реализации изобретения первое отделение набора представляет собой полость шприца, а второе отделение - пузырек, флакон или ампулу.
Один из контейнеров может быть сконструирован как картридж для автоматического инжектора. Полученная при комбинировании сухой композиции и жидкого водного носителя готовая к использованию жидкая суспензия хранится в картридже, который может быть вставлен в автоматический инжектор.
И вновь следует подчеркнуть, что, либо по существу сухая композиция в первом отделении набора, либо жидкий водный носитель, либо оба компонента могут содержать один или более дополнительных наполнителей, в частности, присадок, веществ, увеличивающих объем, сурфактантов, консервантов, кислот, оснований, солей, сахаров, сахарных спиртов, аминокислот, стабилизаторов, антиоксидантов, полимеров, буферов, многоатомных спиртов, протеинов, например, таких, как сывороточный альбумин человека, и пластификаторов.
Сухая композиция, содержащая микрочастицы, и жидкий водный носитель адаптированы для получения восстановленной суспензии, которая пригодна для инъекции, т.е. стерильна, относительно изотонична и изоосмотична, а также в достаточной степени свободна от токсических ингредиентов при парентеральном введении. Вязкость этой суспензии должна быть достаточно низкой, чтобы можно было проводить инъекцию иглой 17-го или более высокого калибра, предпочтительнее, иглой 20-го или более высокого калибра, даже иглой 22-го калибра. В этом документе способность суспензии быть введенной в организм означает реологические свойства, которые позволяют выполнить инъекцию иглой указанного типа без приложения усилия более чем приблизительно 25 N. В более предпочтительном варианте реологические свойства суспензии адаптированы, а размер иглы подобран так, чтобы инъекцию можно было выполнить с усилием не более чем приблизительно 20 N, еще предпочтительнее, не более чем приблизительно 15 N, причем инъекцию могли бы выполнить врачи, медицинские сестры и больные, даже не обладающие большой физической силой. Разумеется, еще одно предварительное условие, связанное с такими размерами инъекционных игл, обусловлено достаточно малым диаметром микрочастиц, а также тем, что после восстановления суспензии микрочастицы не агрегируются. Как упоминалось выше, диаметр большинства микрочастиц среднего веса не должен превышать 200 мкм, и, более предпочтительно, чтобы он находился в диапазоне приблизительно от 30 до 175 мкм.
Предлагаемые изобретением микрочастицы могут быть изготовлены любым известным способом получения микрочастиц из амфифильных полимеров, в частности, распылительной сушкой, коацервацией, акустическим образованием капелек с последующей десольватацией, распылительной сублимационной сушкой и т.д. Однако более предпочтительно, чтобы микрочастицы были изготовлены способом, основанным на эмульсиях, который включает следующие этапы: (a) изготовление эмульсии с водной внутренней фазой, содержащей активный ингредиент, и органической внешней фазой, содержащей биоразлагаемый полимер и, по меньшей мере, один органический растворитель; (b) затвердевание биоразлагаемого полимера и его превращение в микрочастицы при удалении, по меньшей мере, фракции органического растворителя из эмульсии, полученной на этапе (a), и (c) сбор и высушивание микрочастиц, сформированных на этапе (b). Схема основного процесса описана, например, JM. Bezemer et al. в издании J. Control Release 2000, 67 (2-3), 233-248 и 249-260, а также в J. Control Release 2000, 66 (2-3), 307-320, которые раскрыты и включены в этот документ в качестве ссылки.
Говоря в общем смысле, микрочастицы формируются из органического раствора полимера, который рассеивается на капельки в водной или гидрофильной фазе. Чтобы могло произойти затвердение капелек в микрочастицы, необходимо, по меньшей мере, частично удалить из дисперсной фазы органический растворитель. Это достигается посредством экстракции или выпаривания растворителя, либо комбинированием обоих способов. Экстракция растворителя означает, что непрерывная водная фаза модифицируется настолько, что позволяет разрушить или извлечь из дисперсной фазы значительную часть органического растворителя. Например, если органический растворитель имеет умеренную способность смешиваться с водой, то разбавление эмульсии или увеличение объема водной фазы уже позволяет существенно экстрагировать органическую фазу. В альтернативном варианте можно модифицировать состав внешней фазы, добавив в нее один или более органических растворителей, способных смешиваться с водой, но при том условии, что они могут действовать как сорастворители, растворяя и экстрагируя основной органический растворитель из дисперсной фазы. В качестве таких сорастворителей можно использовать, в частности, этанол, метанол, ацетон, изопропиловый спирт.
С другой стороны, выпаривание растворителя не требует добавлять какие-либо компоненты для того, чтобы прямо повлиять на состав и свойства органической фазы, а в типичном случае использует резкое увеличение давления паров органического растворителя в дисперсной фазе по сравнению с водной фазой. При воздействии вакуума или тепла можно выпарить органический растворитель. Когда концентрация полимера в органической фазе достигает определенного уровня, происходит его затвердевание и образование микрочастиц. Важно отметить, что любое выпаривание растворителя из дисперсной фазы обычно также подразумевает присутствие механизма экстракции растворителя.
Для того чтобы инкорпорировать в микрочастицы гидрофильные активные соединения, может оказаться нецелесообразным прямое наполнение органической фазы активным ингредиентом. Во-первых, это может привести к недостаточно эффективной инкорпорации, поскольку гидрофильные соединения при образовании эмульсии будут перераспределяться в водную фазу. Во-вторых, многие соединения, входящие в сферу интересов, особенно пептиды и протеины, в частности, интерфероны, которые, согласно данному изобретению, должны инкорпорироваться в микрочастицы, довольно чувствительны к органическим растворителям и могут терять свою активность. Поэтому предпочтительно, чтобы интерферон инкорпорировался в виде водного раствора, эмульгированного в органическом растворе блок-сополимера с образованием эмульсии "вода-в-масле", которая впоследствии эмульгируется в другой водной фазе, образуя двойную эмульсию типа "вода-в-масле-в-воде» (в/м/в). При проведении этапа экстракции растворителя или выпаривания растворителя, как это было описано выше, внутренняя водная фаза, содержащая интерферон, становится инкапсулированной в полимерных микрочастицах.
В настоящее время одним из наиболее предпочтительных органических растворителей для разбавления блок-сополимера и создания органической фазы эмульсии м/в или двойной эмульсии в/м/в считается дихлорметан. Содержание полимера в органической фазе может варьироваться в зависимости от специфического состава полимера и фактически используемого органического растворителя (растворителей), находясь в диапазоне приблизительно от 1 до 300 мг/мл. Если в качестве растворителя используется дихлорметан, то более предпочтительно, чтобы содержание полимера находилось в диапазоне приблизительно от 50 до 250 мг/мл или даже приблизительно от 100 до 150 мг/мл.
Активный ингредиент, т.е. интерферон, предпочтительно, инкорпорируется в виде водного раствора, который эмульгируется в органическом растворе полимера. Водный раствор интерферона можно стабилизировать наполнителями, в частности, кислотами, основаниями или буферными солями, чтобы достичь определенного уровня pH и поддерживать его, либо осмотическими агентами, в частности, одной или более солей, сахаров, сахарных спиртов, аминокислот и т.д. Некоторые из таких наполнителей также могут оказаться ценными средствами не только в отношении осмотического давления, но и в плане эффектов стабилизации. Однако было обнаружено, что интерферон, и, в особенности, альфа-интерфероны могут легко инкорпорироваться с применением методики двойной эмульсии в/м/в, если использовать простой водный раствор интерферона как самую глубокую фазу эмульсии, не содержащую никаких добавок и наполнителей.
Содержание интерферона во внутренней водной фазе, очевидно, будет влиять на содержание интерферона в микрочастицах и, следовательно, должно выбираться в соответствии с желательными свойствами микрочастиц. Например, в случае альфа-интерферонов, содержание может варьироваться в диапазоне приблизительно от 1 до 100 мг/мл и, предпочтительнее, приблизительно от 10 до 50 мг/мл.
Соотношение объемов внутренней водной фазы и органической фазы также будет влиять на содержание активного ингредиента в микрочастицах. Кроме того, оно может повлиять на другие важные свойства частиц, в частности, на их пористость и профиль высвобождения. Поэтому упомянутое соотношение надо тщательно подбирать в каждом отдельном случае, исходя из желательных характеристик продукта. Если характеристики внутренней водной и органической фаз выбираются с учетом вышеупомянутых предпочтений, то полезным считается их объемное соотношение приблизительно от 1:3 до 1:15 (внутренняя водная фаза:органическая фаза). Согласно одному из предпочтительных вариантов реализации изобретения, объемное соотношение составляет приблизительно от 1:5 до 1:10.
Для стабилизации двойной эмульсии в/м/в может оказаться полезным инкорпорировать в наружную водную фазу один или более стабилизаторов, обладающих свойствами сурфактантов. Полезными стабилизаторами могут быть маленькие амфифильные молекулы, в частности, ионные или неионные сурфактанты, либо детергенты, либо поверхностно-активные полимеры. Например, было обнаружено, что поливиниловый спирт это полезная добавка, способная стабилизировать эмульсию, не оказывая каких-либо пагубных влияний на способ изготовления или на свойства конечного продукта. Полезные поливиниловые спирты могут иметь средний молекулярный вес в диапазоне приблизительно от 10000 до 1 миллиона и степень гидрофильности приблизительно от 80 до 99%, предпочтительнее, от 85 до 90%. В альтернативных вариантах могут быть использованы поливинилпирролидон или поверхностно-активные полисахариды. Содержание стабилизаторов во внешней фазе зависит от их химической природы, а также от природы и относительного объема дисперсной органической фазы. Например, в случае использования поливиниловых спиртов, он может варьироваться приблизительно от 0,1 до 10% вес., предпочтительнее, от 0,5 до 5% вес. При использовании поливинилпирролидона полезный диапазон варьируется приблизительно от 1 до 30% вес. и, предпочтительнее, от 5 до 25% вес.
Наружная водная фаза может также содержать другие наполнители, в частности буферные агенты, осмотические агенты или сорастворители. Такие сорастворители как этанол или метанол можно использовать для того, чтобы модулировать гидрофильность водной фазы и улучшить этап экстракции любого растворителя в процессе изготовления микрочастиц. Например, осмотические агенты можно выбрать из группы, включающей соли, сахара, сахарные спирты, олигосахариды, гликоли, другие спирты и аминокислоты. В одном из предпочтительных вариантов реализации изобретения в качестве осмотического агента используется хлорид натрия. Следует отметить, что любая буферная система, присутствующая во внешней фазе, будет индуцировать некоторое осмотическое давление.
По-видимому, полезно регулировать осмотическое давление внешней фазы до такой величины, которая равна осмотическому давлению самой глубокой водной фазы или превышает его. При этом можно в значительной степени избежать диффузии воды из внешней водной фазы во внутреннюю водную фазу под воздействием осмотического давления. Было обнаружено, что такой процесс диффузии может увеличивать пористость микрочастиц, образующихся при экстракции и/или выпаривании растворителя на следующем этапе. Более предпочтительно, чтобы осмотическое давление внешней водной фазы было отрегулировано так, чтобы оно значительно превышало таковое в самой глубокой водной фазе, в частности, посредством инкорпорирования хлорида натрия на уровне приблизительно от 3 до 6% вес.
Относительный объем внешней фазы необходимо выбирать так, чтобы он превышал минимальный объем, необходимый для инкорпорирования двух других фаз, то есть также в зависимости от природы и состава всех фаз, в особенности, органической фазы и внешней водной фазы. Фактический объем внешней водной фазы, превышающий минимальный объем, важен, прежде всего, в отношении последующих этапов процесса, т.е. экстракции и/или выпаривания растворителя. Обычно объем внешней водной фазы превышает объем эмульсии в/м, которая будет инкорпорирована в нее. Например, он может превышать объем эмульсии в/м, по меньшей мере, вдвое. Более предпочтительно, чтобы объем внешней водной фазы превышал объем эмульсии в/м приблизительно от 5 до 40 или 50 раз.
Если активный ингредиент относительно устойчив по отношению к усилию сдвига, то подготовку внутренней эмульсии в/м можно осуществлять с использованием традиционного оборудования высокого сдвига, в частности, высокоскоростных роторно-статорных устройств, например, типа Ultra-Turrax. Для создания такой эмульсии в водной фазе, содержащей поверхностно-активное соединение, могут оказаться ненужными большой сдвиг или сильное встряхивание. Вполне достаточным может оказаться использование обычного оборудования типа мешалок. Приготовление эмульсий типа в/м и в/м/в, предпочтительно, проводят при комнатной температуре или при температуре несколько ниже комнатной, в частности, в диапазоне от 0°C приблизительно до 25°C, а также при нормальном давлении. Очевидно, что использованный метод эмульгирования будет влиять на результат в отношении среднего диаметра и распределения частиц дисперсной фазы, а также на размеры и распределение микрочастиц. Другие факторы, способные повлиять на упомянутые параметры, это состав соответствующих фаз а также, в особенности, природа органического растворителя и содержание поверхностно-активного стабилизатора во внешней фазе.
Затвердение полимера, растворенного в органической фазе, для образования микрочастиц можно индуцировать выпариванием растворителя (в качестве основного механизма). Это может сопровождаться повышением температуры двойной эмульсии в/м/в при ее перемешивании и/или при воздействии вакуума.
Однако более предпочтительно, чтобы образование микрочастиц индуцировалось тем этапом, который включает экстракцию растворителя. Для того чтобы это сделать, внешнюю фазу двойной эмульсии в/м/в разбавляют дополнительным водным растворителем, который, по выбору, может быть сходным по составу или даже идентичным растворителю внешней водной фазы. Если содержание стабилизатора во внешней водной фазе эмульсии достаточно велико, то водный раствор, добавляемый для стимуляции процесса экстракции растворителя не обязательно должен содержать какой-либо дополнительный стабилизатор. С другой стороны, рекомендуется, чтобы добавляемый водный раствор содержал осмотически активный ингредиент, в частности, одну или более солей, сахаров, сахарных спиртов, олигосахаридов, гликолей, других спиртов и аминокислот, чтобы поддерживать осмотический градиент между внутренней и внешней водными фазами двойной эмульсии и чтобы избежать диффузии воды во внутреннюю фазу. По выбору, добавляемый водный раствор также может содержать сорастворитель, в частности, метанол или этанол, либо буферный агент.
Объем водного раствора, добавляемого к двойной эмульсии, в типичном случае вдвое превышает объем эмульсии до проведения этапа экстракции растворителя. Более предпочтительно, чтобы он превышал объем двойной эмульсии приблизительно от 1 до 5 раз. Может оказаться разумным медленное добавление раствора при постоянном перемешивании во избежание местной негомогенности в пределах сосуда. По выбору, можно поднимать температуру и/или прикладывать некоторый вакуум для удаления некоторой части экстрагированного органического растворителя. После добавления водного раствора на некоторое время можно продолжить перемешивание для более обширной экстракции растворителя из органической фазы, а также, возможно, для достижения диффузии воды из внутренней водной фазы эмульсии во внешнюю фазу.
После того как микрочастицы затвердеют, их можно будет собрать, например, центрифугированием, фильтрацией или просеиванием. Для удаления практически всех остатков органических растворителей и всех растворимых соединений, присутствие которых в микрочастицах нежелательно, необходимо повторно провести центрифугирование, фильтрацию или просеивание после ресуспендирования микрочастиц в каком-нибудь свежем, например, в буферном водном растворе. По выбору, микрочастицы можно скринировать просеиванием для выделения фракции нужного размера.
После отмывания микрочастицы можно высушить для хранения. Предпочтительный метод высушивания это сублимационная сушка. Например, микрочастицы можно заморозить в жидком азоте, а затем высушить под вакуумом для сублимации остаточной воды. Обычно процесс высушивания включает первую фазу сушки, которая проводится при температуре ниже 0°C с последующей второй фазой сушки при окружающей или даже более высокой температуре.
Для получения конечной композиции, соответствующей данному изобретению, высушенные микрочастицы можно смешивать с другими наполнителями по выбору, как это было описано выше. Например, композиция, соответствующая пункту 1 формулы изобретения, может представлять собой порошковую смесь, которая содержит микрочастицы и один или более твердых наполнителей, выбранных из группы сурфактантов, ресуспендирующих агентов, осмотических агентов и буферных агентов. Предпочтительно, чтобы микрочастицы и наполнители были представлены в стерильном виде, а смешивание компонентов происходило в асептических условиях. Такую порошковую смесь можно асептически расфасовывать в пузырьки или флаконы. Как было упомянуто выше, содержимое пузырьков или флаконов в составе фармацевтических наборов можно соединять с жидким водным носителем для восстановления порошка.
В следующем варианте реализации изобретения предлагаемая композиция существует в виде инъекционной жидкой лекарственной формы. В этом варианте интерферон и блок-сополимер растворены и диспергированы в жидком носителе, который должен быть биологически приемлемым. При парентеральном введении раствор полимера или дисперсная (коллоидная) жидкость образует в мышцах или подкожной ткани депо, из которого медленно высвобождается интерферон. Этот вариант основан на том открытии, что блок-сополимеры в композиции, предлагаемой изобретением, действительно, способны образовывать в физиологической окружающей среде микроскопические гели.
Предпочтительно, чтобы состав жидкой лекарственной формы был адаптирован для образования геля после инъекции. Определение геля основано на его реологических свойствах. В этом документе термин гель означает полутвердый материал, который ведет себя как твердый при небольшом усилии сдвига, но как вязкая жидкость, если усилие сдвига превышает порог, который принято называть пределом текучести. Другими словами, гель это система с ограниченным, обычно довольно небольшим, пределом текучести.
Инъекционные гели и гели, образующиеся in-situ, как лекарственные формы с контролируемым высвобождением, были описаны в работе A. Hatefi et al., J. Control. Rel. 80 (2002), 9-28, которая упоминается здесь только в качестве ссылки. Существует несколько общих подходов к технологии приготовления инъекционных гелей, причем в большинстве они основаны на использовании гелеобразующих полимерных носителей. Например, некоторые полимеры могут образовывать гели, реагирующие на определенные условия окружающей среды, например, на pH или температуру. Например, были описаны системы золь-гель, которые при относительно низких значениях pH и комнатной температуре представляют собой золи (вязкие и жидкие коллоидные растворы). После инъекции pH медленно забуферивается физиологическими жидкостями до величины, более близкой к нейтральной, что приводит к затвердеванию раствора и образованию геля. В системе, реагирующей на температуру, гелеобразование происходит тогда, когда после инъекции температура повышается до физиологического уровня.
Однако более предпочтительно, чтобы инъекционный раствор содержал неводный, биологически приемлемый органический растворитель или сорастворитель, который in vitro представляет собой жидкий раствор или суспензию, но после инъекции медленно диффундирует, отдаляясь от блок-сополимера, нерастворимого, но способного образовывать гель в водной среде.
Органический растворитель или сорастворитель можно выбрать из тех органических растворителей, которые способны растворять блок-сополимер(ы) и считаются биологически совместимыми с учетом предназначенных объема и частоты введения. Примеры таких растворителей включают бензиловый спирт, бензилбензоат, диацетин, трибутирин, триэтилцитрат, трибутилцитрат, ацетилтриэтилцитрат, ацетилтрибутилцитрат, триэтилглицериды, триэтилфосфат, диэтилфталат, диэтилтартрат, полибутилен, глицерин, этиленгликоль, полиэтиленгликоль, октанол, этиллактат, пропиленгликоль, пропиленкарбонат, этиленкарбонат, бутиролактон, этиленоксид, пропиленоксид, N-метил-2-пирролидон, 2-пирролидон, формаль глицерина, метилацетат, этилацетат, метилэтилкетон, диметилформамид, диметилсульфоксид, тетрагидрофуран, капролактам, децилметилсульфоксид, олеиновую кислоту, 1-додецилазациклогептан-2-он и их смеси.
В одном из предпочтительных вариантов реализации изобретения неводный растворитель представляет собой один или более членов из группы, в состав которой входят DMSO (диметилсульфоксид), NMP (N-метилпирролидон), бензиловый спирт, тетрагидрофуран, этилацетат и бензилбензоат.
Типичное содержание блок-сополимера в жидкой инъекционной композиции составляет приблизительно от 5 до 60% вес. и, прежде всего, зависит от того, какой именно полимер (полимеры) фактически используется. Более предпочтительно, чтобы содержание полимера составляло приблизительно от 15 до 45% вес.
Особенно подходящие блок-сополимеры для реализации этого аспекта изобретения содержат в среднем довольно много ПЭГТ, например, приблизительно от 70 до 98% вес. или, предпочтительнее, приблизительно от 75 до 95% вес. В настоящее время наиболее предпочтительными считаются блок-сополимеры со средним содержанием ПЭГТ приблизительно от 80 до 90% вес.
Типичный средний молекулярный вес сегментов ПЭГ в блоках ПЭГТ составляет приблизительно от 300 до 6000 или, более предпочтительно, приблизительно от 600 до 2000.
По выбору, композиция может содержать более двух или более одного блок-сополимера, отличающихся по содержанию ПЭГТ, по молекулярному весу сегментов ПЭГ или по обоим упомянутым параметрам. В предпочтительном варианте реализации изобретения композиция содержит один или два блок-сополимера.
К тому же композиция может содержать один или более добавочных наполнителей, в частности, сорастворителей, сурфактантов, консервантов, кислот, оснований, солей, сахаров, сахарных спиртов, аминокислот, стабилизаторов, антиоксидантов, осмотических агентов и полимеров. Логическое обоснование для введения этих наполнителей может быть таким же, как это обсуждалось выше в контексте композиций на основе микрочастиц. В альтернативном варианте наполнитель может выполнять любую функцию, типичную для жидких инъекционных лекарственных форм.
Типичный объем жидкой лекарственной формы составляет приблизительно от 0,3 до 3 мл на разовую дозу инъекции, или, предпочтительнее, приблизительно от 0,5 до 2 мл.
Инъекционные лекарственные формы, предлагаемые изобретением, в типичном случае предназначены для внутримышечной или подкожной инъекции. Эти способы введения требуют соблюдения определенных условий, связанных с качеством, которые обычно характерны для парентеральных продуктов, например, стерильность. Таким образом, предпочтительно, чтобы инъекционная жидкая лекарственная форма была стерильна и соответствовала всем требованиям, предъявляемым к парентеральным лекарствам, которые перечислены в основных фармакопеях, в частности, в действующей редакции фармакопеи США (USP).
Инъекционную жидкую лекарственную форму можно получить, растворяя блок-сополимер(ы) в неводном биологически совместимом растворителе, по выбору, при повышенной температуре. Активное соединение, т.е. интерферон, можно добавлять к этому полимерному раствору в виде сухого, например, сублимированного порошка при перемешивании. Во избежание присутствия воды в лекарственной композиции предпочтительно, чтобы интерферон инкорпорировался не в виде водного раствора.
В следующем варианте реализации изобретения на основе интерферона создается композиция, соответствующая пункту 1 формулы изобретения, которая имеет форму макроскопического твердого имплантата. Имплантат можно определить как твердую, по существу сухую лекарственную форму, которая отличается от микрочастиц тем, что типичный имплантат содержит разовую дозу активного ингредиента в одном модуле или лишь в нескольких модулях. Обычно самые крупные имплантаты имеют размер порядка нескольких миллиметров или более, тогда как микрочастицы, вводятся во множественном количестве и имеют размеры меньше нижней границы миллиметровой шкалы.
В одном из предпочтительных вариантов реализации изобретения имплантат сформирован в виде стержня. Это создает дополнительные преимущества с позиций “менее инвазивного” введения, позволяющего в значительной степени избежать тканевой травмы. Кроме того, полимерные изделия в виде стержней можно эффективно изготовлять методом плавильной экструзии с последующим нарезанием полученной заготовки на стержни нужного размера. Для того чтобы осуществить такую экструзию, блок-сополимер(ы), интерферон и добавочные наполнители должны быть представлены в виде сухого порошка или гранул с последующим гомогенным перемешиванием компонентов. После перемешивания смесь вводится в экструзионный пресс, например, в одинарный или двойной червячный экструдер и выдавливается в виде единой твердой нити, которую затем можно разрезать на отдельные стержни.
Композицию блок-сополимера можно выбрать таким образом, как это обсуждалось выше в контексте микрочастиц. Тип наполнителя, особенно полезный в имплантатах, - это пластификатор, который способен снижать температурный интервал плавления или температуру стеклования полимера (полимеров) до такого уровня, когда исчезает негативное влияние на стабильность инкорпорированного интерферона. К числу потенциально полезных пластификаторов относятся глицерин, пропиленгликоль и полиэтиленгликоль.
Независимо от того, реализована ли композиция в виде лекарственной формы на основе микрочастиц, инъекционной жидкости, геля или твердого имплантата, для фармацевтического использования в целях борьбы с определенными заболеваниями и состояниями, которые можно лечить, а прогрессирование которых можно предотвращать или замедлять, должен быть изготовлен препарат для введения в организм интерферона, и, наиболее предпочтительно, альфа-интерферона. Примеры таких заболеваний и состояний включают острый и хронический гепатит B, острый и хронический гепатит C, лейкоз с волосковыми клетками, острый и хронический миелолейкоз, множественную миелому, фолликулярную лимфому, карциноидную опухоль, злокачественную меланому, остроконечную кондилому, SARS (тяжелый острый респираторный синдром) и саркому Капоши, в частности, ее форму, связанную со СПИДом.
Композиция, предлагаемая изобретением, имеет преимущество перед традиционными препаратами интерферона для инъекций, которое заключается в том, что частоту инъекций можно значительно снизить, благодаря характеристикам контролируемого высвобождения, например, делать одну инъекцию через каждые 2 или 4 недели вместо нескольких инъекций в неделю. В связи с этим повышается комфорт для больных и улучшается дисциплина в соблюдении врачебных назначений, тогда как расходы, связанные с частыми инъекциями, потенциально уменьшаются. В отношении других полимерных систем с контролируемым высвобождением лекарств, предназначенных для инъекции или имплантации, данное изобретение обеспечивает блестящую совместимость с интерферонами, улучшенный контроль высвобождения без взрывного эффекта, демпинга дозы или аутокаталитической деградации и эрозии полимера. Более того, системы доставки, предлагаемые изобретением, физиологически хорошо переносимы и не вызывают каких-либо значительных побочных эффектов, связанных с носителем.
Не пытаясь установить связь с какой-либо фундаментальной теорией, мы можем высказать мысль о том, что хороший профиль высвобождения лекарственного вещества системой, предлагаемой данным изобретением, по-видимому, связан с тем фактом, что активное соединение высвобождается, главным образом, через диффузию, а не через эрозию, характерную для многих ныне известных систем доставки, основанных на поли(лактидах)-и/или-(гликолидах). При использовании амфифильных блок-сополимеров не происходит их аутокаталитического разрушения, участвующего в процессе высвобождения. В отличие от известных систем доставки блок-сополимеры не создают кислотной микросреды, враждебной для чувствительных биологических соединений. С другой стороны, гидрофильные блоки блок-сополимеров, вероятно, создают гидрофильную микросреду, которая в условиях in-situ повышает стабильность таких чувствительных биологических соединений. В особенности можно думать о том, что интерфероны, особенно, принадлежащие к семейству альфа-интерферонов, в микросреде, создаваемой амфифильными блок-сополимерами из систем доставки, предлагаемых данным изобретением, стабилизируются в неагрегированном состоянии.
Особенно в случае использования микрочастиц можно полагать, что относительно низкая пористость микрочастиц, образованных блок-сополимерами, является одной из причин весьма незначительного взрывного эффекта, наблюдаемого при использовании композиций, предложенных данным изобретением.
Дальнейшие варианты реализации, приложения и преимущества данного изобретения будут более очевидно продемонстрированы следующими примерами, не устанавливающими ограничений, или достаточно просто могут быть выведены на основе этого описания специалистами, компетентными в области доставки лекарств.
Пример 1: Изготовление двойной эмульсии типа в/м/в, содержащей альфа-интерферон-2b
Негликозилированный рекомбинантный альфа-интерферон-2b (α-ИФН-2b), протеин, состоящий из 165 аминокислот с молекулярным весом приблизительно 19000 дальтон и изоэлектрической точкой около 6,0 был получен в виде водного раствора с концентрацией протеина порядка 10 мг/мл. Блок-сополимер с весовым соотношением 80% ПЭГТ и 20% ПБТ и сегментами ПЭГ, имеющими средний молекулярный вес 1500, был получен от компании IsoTis, Билтховен, Голландия. Был приготовлен раствор, содержащий 1 г полимера в 7 мл дихлорметана. Для приготовления эмульсии в/м 1 мл раствора альфа-ИФН-2b добавили к раствору полимера при перемешивании с последующей гомогенизацией в диспергаторе ultra turrax в течение приблизительно 30 секунд на скорости 19000 оборотов в минуту.
Две разные двойные эмульсии в/м/в были приготовлены разливом двух эмульсий в/м, приготовленных по вышеуказанному описанию, отдельно друг от друга в 50 мл (a) фосфатно-солевого водного буфера, содержащего 4% ПВА (вес./об.) и имеющего молекулярный все приблизительно 130000 при степени гидролиза приблизительно 87%, или (b) водного раствора хлорида натрия (5% вес./об.), также содержащего 4% ПВА (вес./об.), при перемешивании на скорости 700 оборотов в минуту.
Пример 2: Изготовление микрочастиц посредством экстракции и выпаривания растворителя
Далее двойные эмульсии, приготовленные согласно примеру 1, подвергли дополнительной обработке для получения микрочастиц. К каждой из двух двойных эмульсий при постоянном перемешивании на скорости 700 оборотов в минуту медленно добавляли 100 мл фосфатно-солевого водного буфера. Добавленный буферный раствор приводил к объемному увеличению внешней водной фазы двойных эмульсий. Далее перемешивание продолжали примерно еще один час для экстрагирования значительной части дихлорметана во внешнюю водную фазу и затвердения полимера в органической фазе. Затем отвердевшие микрочастицы центрифугировали на скорости 250 оборотов в минуту при комнатной температуре. Надосадочную жидкость сливали и удаляли в отходы, а осадок ресуспендировали в свежем фосфатно-солевом буферном растворе для повторного центрифугирования. Эту процедуру повторяли три раза. Наконец, микрочастицы замораживали в жидком азоте и лиофилизировали в течение приблизительно 12-24 часов. Эффективность инкапсулирования оценили на уровне около 85% для микрочастиц из двойной эмульсии в/м/в, внешняя фаза которой содержала 5% хлористого натрия, и приблизительно 25% для другой порции эмульсии. Полученные микрочастицы исследовали при помощи электронной микроскопии (SEM), которая показала, что они имеют почти сферическую форму и преобладающий размер приблизительно от 50 до 120 мкм.
Пример 3: Высвобождение альфа-интерферона-2b из микрочастиц in vitro
Для проверки характеристик высвобождения приблизительно 15 мг каждой партии микрочастиц, изготовленных согласно примеру 2, взвешивали в трех образцах (во флаконах емкостью 1,5 мл). В каждый флакон добавляли 1 мл фосфатно-солевого водного буфера. Флаконы выдерживали в водной бане при температуре 37°C. Для получения образцов микрочастицы центрифугировали при комнатной температуре на скорости 1000 оборотов в минуту в течение 2 минут. Из флаконов извлекали образцы объемом 700 мкл, замещая их свежим фосфатно-солевым водным буфером. Количество альфа-ИФН в каждом образце определяли по методу микроанализа общего содержания протеина с бихинной кислотой.
Было обнаружено, что обе партии отчетливо демонстрировали стабильные характеристики высвобождения. Партия, полученная из двойной эмульсии в/м/в, внешняя фаза которой содержала 5% хлористого натрия, продемонстрировала начальный взрывной эффект менее 10%, а другая партия - взрывной эффект около 20%. Обе партии высвобождали 50% содержащегося в микрочастицах интерферона примерно в течение 3-4 дней, а 75% - примерно в течение 7-8 дней. После 14 дней было высвобождено приблизительно 85-90% инкорпорированной дозы.
Пример 4: Изготовление композиции, содержащей микрочастицы с инкорпорированным усеченным альфа-ИФН-2b
Композиция на основе микрочастиц была приготовлена в асептических условиях следующим образом. Из стерильного блок-сополимера, содержащего 77% вес. ПЭГТ и 23% вес. ПБТ, с сегментами ПЭГ, имеющими средний молекулярный вес 1500, была взята навеска в количестве 6 г, которую растворили в 54 г стерильного дихлорметана. Органический раствор полимера соединили с 5,5 мл стерильного водного раствора, содержащего смесь усеченного на N-конце альфа-ИФН-2b, молекулы которого имели среднюю длину около 158 аминокислотных остатков, причем специфическая активность этого интерферона составляла приблизительно от 0,25 до 0,35 ММЕ на мкм, а его содержание в растворе - около 10 мг/мл. Для получения гомогенной эмульсии типа “вода в масле” было использовано диспергирующее устройство ultraturrax.
Затем эмульсию при постоянном перемешивании соединили с 445 г стерильного водного раствора поливинилового спирта (4% вес./об.), который также содержал хлорид натрия (5% вес./об.). Таким способом была получена двойная эмульсия в/м/в, в которой внешняя водная фаза была представлена раствором поливинилового спирта.
На следующем этапе происходило образование микрочастиц и их затвердение при удалении растворителя из органической фазы, которое носило характер комбинации экстрагирования растворителя и выпаривания растворителя. Частичная экстракция растворителя была осуществлена при добавлении к непрерывной фазе двойной эмульсии стерильного фосфатно-солевого водного буфера, а другая часть дихлорметана была выпарена при продувании всей поверхности двойной эмульсии стерильным азотом в течение приблизительно 24 часов со скоростью потока около 5-10 литров в минуту.
Микрочастицы были собраны и промыты стерильным раствором маннитола (26,7 г/л) с последующим ресуспендированием в соответствующем объеме раствора маннитола для подгонки осмотического давления до уровня физиологической переносимости и для адекватного формирования осадка при лиофилизации. Аликвотные пробы суспензии были перенесены в стерильные стеклянные флаконы и подвергнуты сублимационной сушке, в результате которой были получены белые лиофилизаты. Флаконы закрыли пластмассовыми пробками и алюминиевыми крышками.
Аналитическое тестирование показало, что средний диаметр микрочастиц составлял приблизительно 83 мкм, а по содержанию интерферона был сделан вывод о том, что эффективность инкапсулирования превышала 90%. Уровень остаточного дихлорметана был значительно ниже 600 ч/млн. Электронные микрофотографии микрочастиц отражали их малую пористость: большинство микрочастиц не имели пор с диаметром более 2-5 мкм.
Пример 5: Тестирование in vivo микрочастиц, содержащих усеченный альфа-ИФН-2b
Микрочастицы, содержащие композиции, приготовленные по аналогии с примером 4, были протестированы на рабочие характеристики in vivo у хомяков и обезьян. Твердые лиофилизированные композиции были суспендированы в стерильном водном растворе карбоксиметилцеллюлозы (0,1% вес./об.), в который по выбору дополнительно вводили маннитол для регулировки осмотического давления жидкой фазы. Количество водного раствора определяли расчетным способом, основываясь на содержании активного ингредиента и намеченной для введения дозе с таким расчетом, чтобы объем разовой инъекции составлял от 0,5 до 1,0 мл. Каждый из десяти хомяков получал дозу активного соединения 0,99 мг/кг, которую вводили посредством подкожной инъекции через каждые 7 дней, а другая группа из 10 хомяков каждые 7 дней получала дозу 3,46 мг/кг. С определенным интервалом у животных получали образцы сыворотки, которые хранили в замороженном виде, а затем анализировали на содержание интерферона. Оказалось, что все животные хорошо переносят лекарство.
Профиль высвобождения активного вещества in vivo определяли расчетным способом на основе профиля интерферона в сыворотке. В другом эксперименте определяли профиль высвобождения in vitro по той же схеме, как это было описано в примере 3. Сравнение профилей высвобождения in vivo и in vitro показало, что между ними существует выраженная корреляция, как по внешнему виду кривой, так и по длительности высвобождения, причем характер высвобождения in vitro является хорошим предиктором поведения композиции in vivo. Не наблюдалось сколько-нибудь значительного взрывного эффекта ни in vivo, ни in vitro.
Фиг.1 наглядно демонстрирует расчетные средние профили высвобождения in vivo в группах хомяков, получавших низкую и высокую дозы интерферона, а также соответствующий профиль высвобождения in vitro, нормализованный в расчете на общее 100% высвобождение.
В другой серии экспериментов образцы той же композиции вводили посредством подкожных инъекций самцам и самкам обезьян. Доза активного ингредиента составляла 180 мкм на животное, а для диспергирования композиции до объема от 0,5 до 1,0 мл на разовую инъекцию была использована та же восстанавливающая жидкость. Образцы сыворотки от животных были получены с определенным интервалом в течение 14 дней, начиная с момента инъекции. И вновь для расчета профиля высвобождения in vivo были использованы данные о концентрации активного вещества в сыворотке, причем расчетный профиль высвобождения in vivo сравнивали с профилем высвобождения in vitro, который определяли по другим образцам той же композиции в соответствии с методом, описанным в примере 3.
Полученная в результате корреляция между соответствующими профилями высвобождения оказалась довольно впечатляющей. Как in vitro, так и in vivo композиция плавно высвобождала интерферон в течение 14 дней без какого-либо взрывного эффекта.
Фиг.2 показывает расчетный средний профиль высвобождения in vivo, а также соответствующий профиль высвобождения in vitro, нормализованный в расчете на общее 100% высвобождение.
Пример 6: Чистота высвобождаемого интерферона
Образцы высвобожденного интерферона, полученные при оценке профиля высвобождения in vitro, как это было описано в примерах 4 и 5, были проанализированы методом высокоразрешающей хроматографии по размеру молекул для определения фракции интерферона, высвобождаемой в мономерной форме. Примечательно, что по результатам анализа менее 1% высвобождаемого активного вещества было представлено в виде димеров или более крупных агрегатов, хотя хорошо известно, что альфа-интерфероны проявляют сильную тенденцию к образованию агломератов. Таким образом, композиция на основе микрочастиц явно давала вклад в значительную стабилизацию интерферона.
Пример 7: Изготовление пленочной композиции, включающей блок-сополимер и бета-интерферон
Из блок-сополимера, содержащего 80% вес. ПЭГТ и 20% вес. ПБТ, с сегментами ПЭГ, имеющими средний молекулярный вес 2000, была взята навеска в количестве 0,5 г, которую растворили в 3,5 мл дихлорметана. Затем 1,94 мг лиофилизированного бета-интерферона диспергировали в растворе при помощи диспергатора ultraturrax. Дисперсия была распределена по стеклянным пластинкам при помощи регулируемого аппликатора пленок. После выпаривания дихлорметана были получены пленки, которые снимали со стеклянных пластинок. Эти пленки высушивали под вытяжным колпаком в течение нескольких часов.
Образцы размером приблизительно 1,77 см2, вырезанные из пленок, инкубировали в растворе водного ацетатного буфера с pH 3,5 (1 мл) при 37°C в водной бане со встряхиванием. Через каждые 24 часа инкубации весь объем среды для высвобождения меняли на свежий, продолжая инкубировать образцы. Аликвотные пробы извлеченного буфера использовали как материал для анализа методом высокоразрешающей хроматографии по размеру молекул, а результаты этого анализа показали, что приблизительно 83% инкорпорированного бета-интерферона высвобождалось в мономерной неагрегированной форме. Длительность высвобождения зависела от толщины пленок.
Пример 8: Изготовление и характеристики высвобождения саможелирующегося раствора блок-сополимера, содержащего альфа-ИФН-2a
Альфа-интерферон-2a и блок-сополимер ПЭГТ/ПБТ, содержащий 85% вес. ПЭГТ с сегментами ПЭГ, имеющими среднюю молекулярную массу около 1000, были получены в сухом виде. Полимер был разведен в смеси бензилбензоата и бензилового спирта (98:2) в концентрации 20% вес. Интерферон добавили в виде порошка в концентрации 4% вес. и тщательно перемешали с раствором полимера. Полученной смесью наполнили шприц с иглой и впрыснули ее в фосфатно-солевой буферный раствор с температурой 37°C. После впрыскивания постепенно образовывался осадок неравномерного геля. Полученный гель поддерживали при температуре 37°C и непрерывном перемешивании. С определенным интервалом из геля извлекали образцы, замещая их свежим фосфатно-солевым буферным раствором. Образцы анализировали на содержание альфа-ИФН-2b, и результаты анализа подтвердили, что продолжительность высвобождения превышает 14 дней (при 90% высвобождении интерферона).
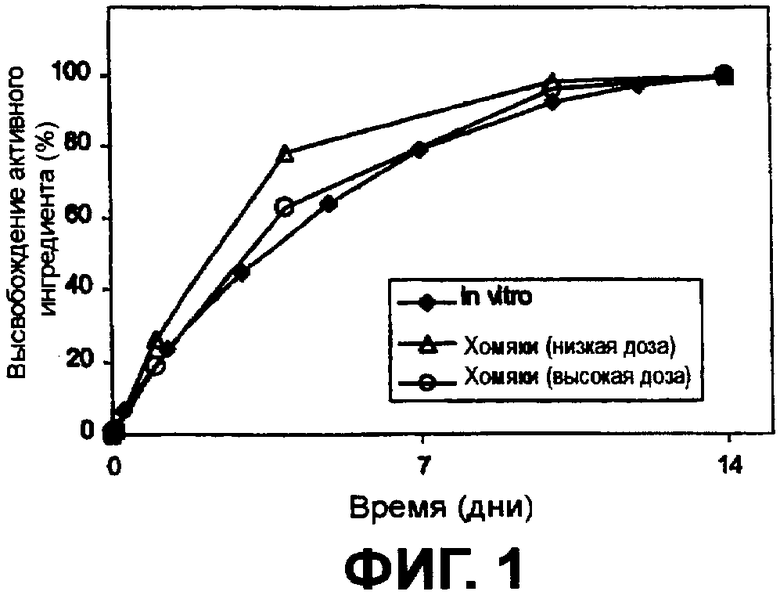
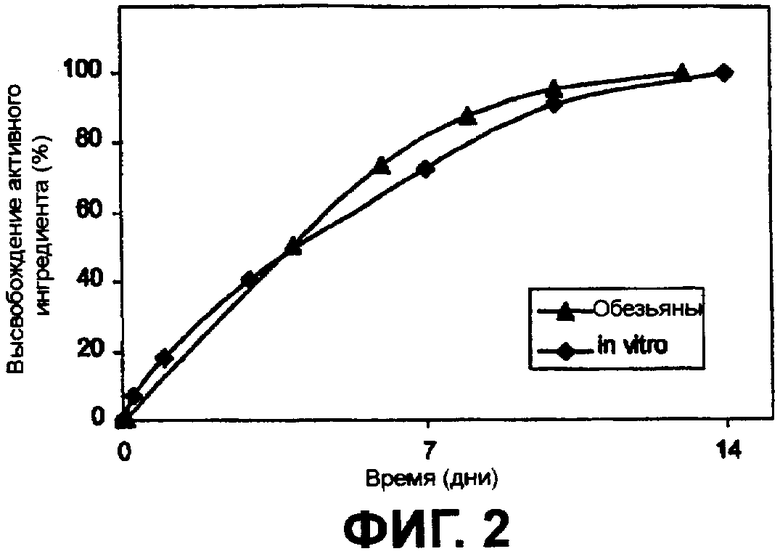
Изобретение раскрывает фармацевтическую композицию для контролируемого высвобождения относительно токсичных активных соединений, в особенности биоактивных протеинов из класса интерферонов. Композиция содержит биоразлагаемый блок-сополимер, состоящий из поли(этиленгликоль)терефталата (ПЭГТ) в количестве от 50 до 95% вес. и поли(бутилентерефталата) (ПБТ). Композиция может быть представлена в виде инъекционных микрочастиц, инъекционной жидкости, обладающей способностью самостоятельного гелеобразования или твердого имплантата. Кроме того, изобретение предлагает фармацевтический набор, включающий упомянутую композицию, способы приготовления композиции и фармацевтические варианты ее применения. Изобретение обеспечивает первоначальное высвобождение в течение 4 часов не более приблизительно 10% включенного количества одного или более альфа-интерферонов и, по меньшей мере, 80% одного или более альфа-интерферонов высвобождается в мономерной неагрегированной форме. 30 з.п. ф-лы, 2 ил.
1. Фармацевтическая композиция для контролируемого высвобождения,
включающая микрочастицы, которые состоят из полимерного носителя и одного или более альфа-интерферонов, инкорпорированных в нем без потери активности, где полимерный носитель, по существу, состоит из одного или более блок-сополимеров, состоящих из поли(этиленгликоль)терефталата (ПЭГТ) и поли(бутилентерефталата) (ПБТ), где
большинство микрочастиц практически не имеет пор с диаметром более приблизительно 5 мкм,
средняя молекулярная масса полиэтиленгликоля (ПЭГ) составляет от 1000 до 2000 Да, блок-сополимер содержит приблизительно от 50 до 95 вес.% ПЭГТ,
где композиция содержит дозу α-интерферона, по меньшей мере, 20 ММЕ, которая высвобождается за перид времени от приблизительно 10 дней до около 1 месяца,
первоначальное высвобождение в течение 4 ч составляет не более приблизительно 10% включенного количества одного или более альфа-интерферонов, и
где, по меньшей мере, 80% одного или более альфа-интерферонов высвобождается в мономерной неагрегированной форме.
2. Композиция по п.1, в которой большинство микрочастиц практически не имеет пор с диаметром более приблизительно 2 мкм.
3. Композиция по п.1, в которой доза композиции сформирована для высвобождения одного или более альфа-интерферонов в течение 14 дней, причем упомянутый период начинается с момента введения дозы.
4. Композиция по п.1 стерильна и адаптирована для парентерального введения, в частности для подкожной или внутримышечной инъекции либо имплантации.
5. Композиция по п.1, в которой часть полиэтиленгликоля (ПЭГ) имеет среднюю молекулярную массу около 1500 Да.
6. Композиция по п.5, в которой отношение ПЭГТ к ПБТ составляет, вес.%, 77 к 23.
7. Композиция по п.6, в которой интерфероном является усеченный интерферон альфа-2b.
8. Композиция по п.1, в которой средний диаметр микрочастиц составляет приблизительно от 25 до 200 мкм и предпочтительно от 50 до 150 мкм.
9. Композиция по п.1, которая дополнительно содержит один или более наполнителей, выбранных из группы, в которую входят присадки, вещества, увеличивающие объем, сурфактанты, консерванты, кислоты, основания, соли, сахара, сахарные спирты, аминокислоты, стабилизаторы, антиоксиданты, полимеры, буферы, многоатомные спирты, протеины, например сывороточный альбумин человека, и пластификаторы.
10. Композиция по п.1, в которой один или более альфа-интерферонов и один или более блок-сополимеров растворены или диспергированы в жидком носителе, образуя фармацевтическую композицию для инъекций.
11. Композиция по п.10, в которой объем фармацевтической композиции для инъекций составляет приблизительно от 0,5 до 3 мл и предпочтительно от 1 до 2 мл.
12. Композиция по п.10, в которой жидкий носитель содержит неводный биологически совместимый растворитель.
13. Композиция по п.12, в которой неводный биологически совместимый растворитель выбирается из группы, в которую входят диметилсульфоксид, N-метилпирролидон, бензилбензоат, тетрагидрофуран, этилацетат, бензиловый спирт и их смесь в любой комбинации.
14. Композиция по п.10, в которую дополнительно введены один или более наполнителей, выбранных из группы, в которую входят сурфактанты, консерванты, кислоты, основания, соли, сахара, сахарные спирты, аминокислоты, стабилизаторы, антиоксиданты и полимеры.
15. Композиция по п.10, способная образовать гель после внутримышечной или подкожной инъекции млекопитающему.
16. Композиция по п.1, используемая в качестве лекарственного средства.
17. Композиция по п.1, которая содержит дозу интерферона от приблизительно 20 до приблизительно 100 ММЕ, которая высвобождается за период времени от приблизительно 10 дней до около 1 месяца.
18. Композиция по п.1, в которой, по меньшей мере, 90% одного или более альфа-интерферонов высвобождается в мономерной неагрегированной форме.
19. Композиция по п.1, в которой, по меньшей мере, 95% одного или более альфа-интерферонов высвобождается в мономерной неагрегированной форме.
20. Композиция по п.1, в которой, по меньшей мере, 97% одного или более альфа-интерферонов высвобождается в мономерной неагрегированной форме.
21. Композиция по п.1, в которой, по меньшей мере, 98% одного или более альфа-интерферонов высвобождается в мономерной неагрегированной форме.
22. Композиция по п.1, в которой соотношение ПЭГТ к ПБТ составляет, вес.%, 77 к 23 и PEG имеет среднюю молекулярную массу 1500 Да.
23. Композиция по п.1, в которой одним или более альфа-интерфероном является интерферон альфа-2b.
24. Композиция по п.23, в которой соотношение ПЭГТ к ПБТ составляет, вес.%, 77 к 23 и PEG имеет среднюю молекулярную массу 1500 Да.
25. Композиция по п.23, в которой, по меньшей мере, 90% интерферон альфа-2b высвобождается в мономерной неагрегированной форме.
26. Композиция по п.23, в которой, по меньшей мере, 95% интерферон альфа-2b высвобождается в мономерной неагрегированной форме.
27. Композиция по п.23, в которой, по меньшей мере, 97% интерферон альфа-2b высвобождается в мономерной неагрегированной форме.
28. Композиция по п.23, в которой, по меньшей мере, 98% интерферон альфа-2b высвобождается в мономерной неагрегированной форме.
29. Композиция по п.1, в которой интерферон представляет собой рекомбинантный продукт, полученный из генетически сконструированных клеток или организмов, причем эти клетки или организмы предпочтительно выбираются из клеток или организмов млекопитающих, насекомых, бактерий, дрожжей, грибов и высших растений или организмов.
30. Композиция по п.4 стерильна и адаптирована для подкожной или внутримышечной инъекции либо имплантации.
31. Композиция по п.8, в которой средний диаметр микрочастиц составляет приблизительно от 50 до 1500 мкм.
| ЕР 0830859 А2, 25.03.1998 | |||
| WO 03041689 A1, 22.05.2003 | |||
| Устройство для резки проката | 1978 |
|
SU990440A1 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ДИАГНОСТИКИ И ЛЕЧЕНИЯ РАКА | 1996 |
|
RU2174409C2 |
| WO 03035030 A1, 01.05.2003 | |||
| ГЛАЗНЫЕ КАПЛИ ДЛЯ ЛЕЧЕНИЯ ЭНДОТЕЛИАЛЬНО-ЭПИТЕЛИАЛЬНОЙ ДИСТРОФИИ РОГОВИЦЫ | 2001 |
|
RU2191012C1 |
| Справочник биохимика: пер | |||
| с англ | |||
| / Досон Р | |||
| и др | |||
| - М.: Мир, 1991, с.350 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: ООО «Новая Волна», Издатель С.Б.Дивов, 2002, т.2, с.323-324. | |||

