КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым (2R)-2-фенилпропанамидам, несущим 4-сульфониламиновый заместитель в положении 4 фенильной группы, и к содержащим их фармацевтическим композициям, которые можно использовать в качестве ингибиторов хемотаксиса полиморфно-нуклеарных и мононуклеарных клеток и которые можно использовать при лечении различных расстройств, опосредуемых хемокином ELR+CXC. В частности, соединения этого изобретения можно использовать при лечении и контроле специфических CXCR2-зависимых патологий, таких как СОБ (синдром облитерирующего бронхиолита), ХОЗЛ (хроническое обструктивное заболевание легких), ангиогенез и меланома.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Хемокины составляют большое семейство цитокинов хемотаксиса, которые оказывают свое действие через взаимодействие с рецепторами, относящимися к семейству 7TM-GPCRs. Система хемокинов является ключевой для регуляции и контроля базального перемещения лейкоцитов при гомеостазе и воспалении. Функциональные следствия активации рецепторов хемокинов включают перемещение лейкоцитов, дегрануляцию, транскрипцию генов, митогенные и апоптозные эффекты. Клетки многих типов, наряду с гемопоэтическими клетками, экспрессируют рецепторы хемокинов; к ним относятся эндотелиальные клетки, клетки гладких мышц, стромальные клетки, нейроны и этителиальные клетки. Их активация распространяет эффекты активации рецепторов хемокинов на другие аспекты тканевой регуляции и гомеостаза, такие как ангиогенез и морфогенетические перемещения при органогенезе (в дополнение к развитию рака и метастазированию).
Ангиогенез, характеризующийся новообразованием кровеносных сосудов, является существенным фактором для ряда физиологических и патофизиологических явлений, таких как эмбриональное развитие, заживление ран, хроническое воспаление и рост злокачественных опухолей, и хемокины через различные механизмы влияют на все эти аспекты анагиогенеза. Первым, описанным еще в 1992 г., сильным ангиогенным хемокином был CXCL8 (также называемый IL-8) [Koch A.E. et al., Science, 258, 1798, 1992]. С патофизиологической точки зрения, регуляция ангиогенеза хемокинами представляется очень важной для образования и роста опухолей. Известны два рецептора CXCL8 (CXCR1 и CXCR2); они связывают CXCL8 с высоким сродством. CXCR1 является селективным для CXCL8, тогда как CXCR2 взаимодействует и с другими хемокинами как с естественными лигандами. В настоящее время накапливаются свидетельства того, что, например, CXCL8 играет патогенетическую роль в развитии кожной меланомы, опосредуемую CXCR2. Было показано, что образцы меланомы и линии клеток, выделенные из них, экспрессируют несколько хемокинов, включая CXCL8 и CXCL1 (а также GRO-α). CXCL8 влияет на процессы развития опухолей и метастазирование, поскольку было показано, что он является аутокринным фактором роста [Schadendorf D. et al, J. Immunol, 151, 2667, 1993], индуцирует ангиогенез [Strieter R. M. et al., Am. J. Pathol., 141, 1279, 1992] и влияет на миграцию клеток меланомы [Wang J.M. et al., Biochem. Biophys. Res. Commun., 169, 165, 1990] посредством связывания и активации своих рецепторов. Оба эти рецептора экспрессируются в клетках нескольких типов (эндотелиальных клетках и клетках меланомы) и также участвуют в ангиогенной реакции [Addison CL. et al., J. Immunol, 165, 5269, 2000], но другим образом. Сообщалось [Norgauer J. et al J. Immunol, 156, 1132, 1996], что низкую экспрессию CXCR2 можно обнаружить в нормальных меланоцитах человека, но этот рецептор стимулируется обработкой фактором TNF-α, что проявляется в усилении пролиферации в ответ на воздействие CXCL8, тогда как в том же самом эксперименте экспрессия CXCR1 не детектируется. Хорошо оценена роль CXCL8 как важного ангиогенного фактора, и было продемонстрировано, что рецептор CXCR2 является предполагаемым ангиогенным рецептором. Лишь недавно выяснилась роль специфических рецепторов CXCR1 и 2 в развитии меланомы [Varney M. L. et al, Am. J. Clin. Pathol, 125, 209, 2006]. Было продемонстрировано in vitro, что в отличие от CXCR1, который повсеместно экспрессирован в злокачественных человеческих меланомах всех уровней по Кларку, CXCR2 экспрессирован преимущественно в меланомных опухолях высших степеней и что имеются значительные различия в уровнях экспрессии CXCR2 между толстыми и тонкими меланомами, что предполагает различные роли, выполняемые CXCR2 и CXCR1 in vivo. CXCR1 и CXCR2 участвуют в механизме ангиогенной реакции и гаптотаксической миграции и хемотаксиса клеток меланомы. Несмотря на примерно одинаковое сродство к CXCL8 и приблизительно одинаковое количество этих рецепторов, хемотаксис нейтрофилов опосредуется преимущественно рецептором CXCR1 [Quan J.M. et al, Biochem. Biophys. Res. Commun., 219, 405, 1996], а экспрессия CXCL8 эндотелиальными клетками стимулирует хемотаксическую реакцию клеток меланомы, опосредуемую рецептором CXCR1. Как указывалось выше, рецептор CXCR2 считают предполагаемым рецептором, опосредующим ангиогенез, индуцированный хемокином ELR+CXC, чем подтверждается заключение о различных ролях, исполняемых CXCR1 и CXCR2 в модулировании агрессивного злокачественного фенотипа, и о том, что при развитии меланомы и метастазировании экспрессия CXCL8 взаимосвязана с экспрессией CXCR2, но не с экспрессией CXCR1 [Varney M. L. et al, Am. J. Clin. Pathol, 125, 209, 2006].
Ингибирование продуцирования и/или активности CXCL8 могло бы быть идеальной мишенью (посредством модулирования CXCR2) для воздействия на злокачественную меланому.
Уже описана возможная патогенетическая роль CXCL8 в заболеваниях легких (повреждение легких, синдром острой дыхательной недостаточности, астма, хроническое воспаление легких и муковисцидоз) и, конкретно, в патогенезе ХОЗЛ (хроническое обструктивное заболевание легких) по пути, опосредуемому CXCR2 [Hill A.T. et al, Am. J. Respir. Crit. Care med., 160, 893, 1999]. ХОЗЛ - это заболевание, характеризующееся воспалением периферических дыхательных путей, протекающим с участием многих воспалительных клеток и медиаторов. Оно сопряжено с усиленным притоком воспалительных клеток, включая увеличение числа макрофагов в дыхательных путях и в ткани. Альвеолярные макрофаги развиваются из моноцитов; они способны вызвать патологические изменения, характерные для ХОЗЛ. Сообщалось, что увеличенное количество макрофагов при ХОЗЛ является результатом привлечения моноцитов из системного кровотока. Исследования хемотаксиса мононуклеарных клеток периферической крови пациентов с ХОЗЛ показывают повышенные по сравнению с контролем хемотаксические ответы по отношению к GRO-α, но не к MCP-1, CXCL8 или NAP(ENA)-78 [Traves S. L. et al., J. Leuk. Biol, 76, 441, 2004]. Этот ответ не опосредуется различиями в экспрессии клеточных рецепторов CXCR1 и CXCR2, но у пациентов с ХОЗЛ экспрессия CXCR2 в моноцитах регулируется иначе: CXCR1 реагирует на высокие концентрации CXCL8 и отвечает за активацию нейтрофилов и выделение пероксидных анионов и нейтрофильной эластазы, тогда как CXCR2 реагирует на низкие концентрации СХС-хемокинов и участвует в хемотаксических реакциях. В настоящее время разработаны сильные низкомолекулярные ингибиторы CXCR2, такие как SB225002, в качестве блокаторов хемотаксической реакции нейтрофилов на CXCL8 и GRO-α. Этот антагонист обладает значительным ингибирующим эффектом по отношению к хемотаксической реакции на мокроту пациентов с ХОЗЛ, при которой увеличиваются концентрации GRO-α [Traves S.L., et al., Thorax, 57, 590, 2002]. Поэтому антагонисты CXCR2 могут также ослаблять хемотаксис моноцитов и накопление макрофагов у больных с ХОЗЛ. Эти результаты демонстрируют потенциал селективных низкомолекулярных антагонистов CXCR2 (в отличие от CXCR1) при лечении ХОЗЛ и для контроля поражения легких.
В последнее время высказываются предположения о возможной роли хемокинов ELR+CXC в развитии синдрома облитерирующего бронхиолита (СОБ). СОБ - это фиброзный процесс, приводящий к постепенному сужению просвета бронхиол и обструкции дыхательных путей (затрудненности дыхания). Обычно СОБ развивается после аденовирусной инфекции или инфекции Mycoplasma pneumoniae, но это заболевание также связано с хроническим отторжением трансплантированных легких, особенно с хроническим отторжением легочных аллотрансплантатов. Кумулятивная частота СОБ к 5 годам после трансплантации легких составляет 50-80%, а 5-летняя приживаемость трансплантата после начала СОБ составляет только 30-50% [Douglas, LS. et al., J. Clin. Invest. 115, 1133, 2005]. СОБ характеризуется инфильтрацией перибронхиолярных лейкоцитов, которые проникают в подслизистую оболочку, базальную мембрану и эпителий дыхательных путей и разрушают их. За повреждением ткани бронхов, опосредуемым инфильтрацией и активацией лейкоцитов, следует фибропролиферация и образование грануляционной ткани [Trulock, E. P. Am. J. Respir. Crit Care Med. 155, 789, 1997].
Подавление функций CXCR2 антителами, специфичными к этому рецептору, ингибировало раннюю инфильтрацию полиморфно-нуклеарных лейкоцитов в экспериментальной модели СОБ у мышей [Belperio, J.A., et al., J Clin Invest. 115, 1150, 2005]. Считают также, что ангиогенез является ключевым фактором развития фиброза при СОБ и что хемокины ELR+ непосредственно вовлечены в механизм ангиогенеза при этом синдроме. Ангиогенная активность в бронхоальвеолярной лаважной жидкости у пациентов с СОБ является следствием, прежде всего, присутствия хемокинов ELR+CXC. Кроме того, исследования, выполненные на модели СОБ у мышей, также продемонстрировали усиленное ремоделирование сосудов, происходившее параллельно с экспрессией хемокинов ELR+CXC в аллотрансплантате трахеи. Взятые вместе эти данные подтверждают гипотезу о том, что патофизиологическая роль, исполняемая хемокинами ELR+CXC при развитии СОБ, должна быть двоякой: на первой фазе хемокины ELR+CXC влияют на привлечение полиморфно-нуклеарных лейкоцитов (т.е. на стадии ишемического/реперфузионного поражения), а на поздней хронической стадии они участвуют в ремоделировании сосудов и ангиогенезе (т.е. на фибропролиферативной стадии). Это означает, что блокада активности хемокинов ELR+CXC могла бы быть действенным терапевтическим средством при лечении этого синдрома.
Молекулы, являющиеся предметом этого изобретения, представляют собой новые терапевтические агенты для лечения и контроля специфических заболеваний, связанных с CXCL8, особенно патологий, при которых хорошо установлена явная патофизиологически ключевая роль CXCR2 (таких как СОБ, ХОЗЛ и развитие опухолей). Хорошо известно, что контроль движения, активации и дифференцировки лейкоцитов определяет систему цитокинов с ее главной ролью и при иммунной реакции хозяина на вторжение патогенов. Это подтверждается тем, что вирусы индуцируют или кодируют хемокины, рецепторы хемокинов или белки, связывающие хемокины, которые различным образом воздействуют на иммунную систему [Murphy, PM., Nature Immunol., 2, 116, 2001]. Установлено, что чувствительность к CXCL8 необходима уже на самой ранней стадии иммунной реакции хозяина на инфекцию [McCoIl SR. et al., J. Immunol, 163, 2829, 1999; Moore TA et al. J. Immunol, 164, 908, 2000] и что CXCR1 является преобладающим подтипом рецепторов CXCL8, экспрессируемых на нейтрофилах человека. В недавней статье [Hess C, et al. Blood, 104, 3463, 2004] рецептор CXCR1 описан как система, способная определять подмножество «быстро реагирующих» Т-клеток (подобных Т-клеткам типа CD8+), которые заполняют промежуток между врожденными и приобретенными иммунными реакциями, не создавая на ранней стадии высокоцитотоксичную антиген-специфичную эффекторную функцию, действующую в очаге инфекции еще до появления новых эффекторных клеток. Более того, вследствие строгого контроля уровней CXCR1 на нейтрофилах и Т-клетках типа CD8+, важной особенностью тонкой настройки иммунной реакции является различная чувствительность к агонистам и антагонистам CXCR1. В заключение можно высказать гипотезу о том, что при лечении хронических заболеваний с четко выявленной главной ролью CXCR2 [т.е. при онкологических (меланома) и легочных (ХОЗЛ, СОБ) заболеваниях], не требуется блокада рецептора CXCR1 (осуществляемая большинством известных модуляторов CXCL8); более того, вследствие ненужной аномальной иммунной реакции, являющейся результатом необходимого длительного лечения, такая блокада была бы вредной.
Недавно были описаны новые классы «2R-арилпропиониламидов» (WO 02/58858) и «2-арилпропионовых кислот» (WO 03/043625), которые можно использовать для ингибирования хемотаксической активации полиморфно-нуклеарных лейкоцитов посредством взаимодействия CXCL8 с CXCR1 и CXCR2. Было заявлено, что соединения класса 2-арилпропионовых кислот обладают биологической активностью по отношению к обоим рецепторам CXCL8; кроме того, были описаны соединения с селективной активностью по отношению к рецептору CXCR2. Соединения класса амидов не проявляли заметной селективности по отношению к подтипам рецепторов CXCR1 и CXCR2. Химическая трансформация подмножества 2-арилпропионовых кислот в амиды усиливала селективность по отношению к CXCR2 у соединений, которые без этого преобразования являются двойными ингибиторами CXCR1/2. Отмеченная селективность и новые физико-химические характеристики делают это подмножество амидов предпочтительными соединениями этого изобретения, особо полезными для лечения специфических CXCR2-зависимых патологий в онкологии (меланома) и пульмонологии (ХОЗЛ и СОБ).
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении описан новый класс (2R)-2-фенилпропанамидов, несущих 4-сульфониламиновый заместитель в положении 4 фенильной группы, и содержащие их фармацевтические композиции, которые можно использовать как ингибиторы хемотаксиса полиморфно-нуклеарных и мононуклеарных клеток и которые можно использовать для лечения различных заболеваний, опосредуемых хемокинами ELR+CXC, подобными острым воспалительным заболеваниям, таким как ХОЗЛ, или ангиогенез, опосредуемый хемокинами ELR+CXC, который может привести к туморигенезу, как при злокачественной меланоме. Эти соединения характеризуются хорошей растворимостью в воде вследствие химических особенностей R'-заместителя в кольце и независимо от природы остатка R. Примерами таких заместителей являются алкилсульфониламино-, арилсульфониламино- и гетероарилсульфониламиногруппы. Некоторые из описанных амидов являются производными уже описанных 2-арилпропионовых кислот, проявляющих хорошую специфичность к GROα-индуцированному хемотаксису. Неожиданно оказалось, что химическое модифицирование этих кислот в амиды позволило получить новые соединения, не проявляющие активности по отношению к рецептору CXCR1, но с повышенной активностью по отношению к CXCR2. Селективность новых описанных амидов по отношению к CXCR2 оценивали в экспериментах по ингибированию миграции трансфектантов CXCR1/L1.2 и CXCR2/L1.2 в ответ на воздействие CXCL8. Данные, приведенные в Таблице 1, показывают, что эти соединения являются сильными ингибиторами CXCL1-индуцированного хемотаксиса человеческих полиморфно-нуклеарных лейкоцитов с IC50 около 10-8 M. В отличие от этого, те же самые соединения при концентрации 10-7 M не оказывают значительного ингибирующего эффекта по отношению к CXCL8-индуцированному хемотаксису человеческих полиморфно-нуклеарных лейкоцитов. Эти результаты согласуются с главной ролью, исполняемой CXCR1 в стимуляции хемотаксиса, индуцированного CXCL8. В то же время исследования селективности показали, что эти соединения не проявляют значительной ингибирующей активности по отношению к миграции трансфектантов CXCR1/L1.2 в ответ на воздействие CXCL8 в концентрациях до 10-6 M. Более того, у соединений этого класса было подтверждено полное отсутствие активности циклооксигеназного пути. На основании материала, описанного выше во введении, очевидна потенциальная роль этого нового класса соединений в лечении CXCR2-зависимых патологий в области онкологии (конкретно, меланомы) и в области пульмонологии (ХОЗЛ, СОБ).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
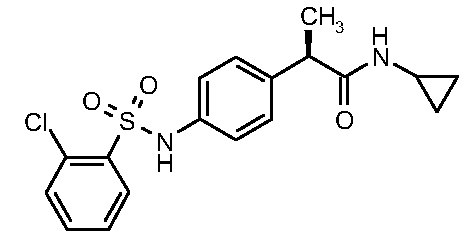
В настоящее время обнаружен новый класс (2R)-2-фенилпропанамидов как ингибиторов хемотаксиса полиморфно-нуклеарных и монуклеарных клеток. В частности, соединения этого изобретения являются сильными ингибиторами хемотаксиса нейтрофилов, индуцированного CXCL1, с улучшенными фармакокинетическими и фармакологическими профилями активности. Таким образом, настоящее изобретение предоставляет (2R)-2-фенилпропанамидные производные формулы (I):
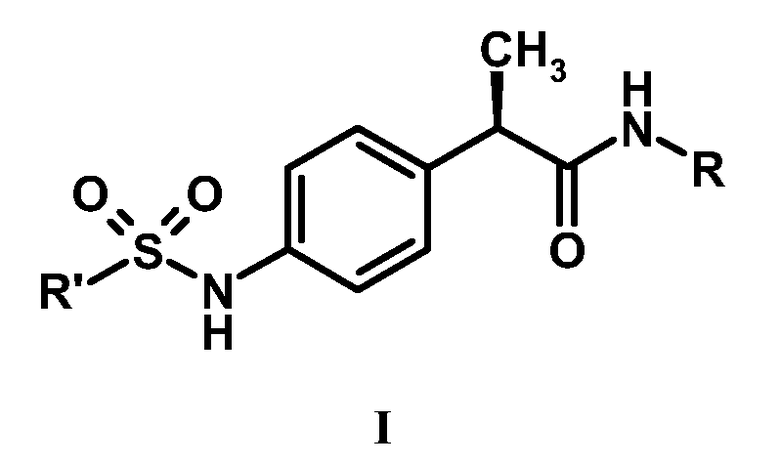
и их фармацевтически приемлемые соли,
где R выбран из
- H, OH, C1-C5-алкила, C3-C6-циклоалкила, C2-C5-алкенила, C1-C5-алкоксигруппы и фенила;
- гетероарильной группы, выбранной из замещенных и незамещенных пиррола, тиофена, фурана, индола, имидазола, тиазола, оксазола, пиридина и пиримидина;
- остатка формулы -CH2-CH2-O-(CH2-CH2O)nR", где R" представляет собой H или C1-C5-алкил, n равно целому числу от 0 до 2;
или R, вместе с NH-группой, к которой он присоединен, является группой-радикалом первичных амидов природных аминокислот, таких как (2S)-2-аминопропанамид, (2S)-2-амино-3-фенилпропанамид, (2S)-2-амино-3-гидроксипропанамид, (2S)-2-амино-3-карбоксипропанамид, (2S)-2,6-диаминогексанамид. NH-группа, упомянутая выше, как часть группы-радикала первичных амидов природных аминокислот, представляет собой аминогруппу природной аминокислоты.
R' выбран из
- линейного или разветвленного C1-C5-алкила, C3-C6-циклоалкила, C2-C5-алкенила и трифторметила;
- замещенного или незамещенного фенила;
- замещенного или незамещенного бензила;
- гетероарильной группы, выбранной из замещенных и незамещенных пиридина, пиримидина, пиррола, тиофена, фурана, индола, тиазола и оксазола.
Далее, настоящее изобретение предоставляет соединения формулы (I) для применения в качестве лекарственных средств. В частности, такие лекарственные средства являются ингибиторами CXCL1-индуцированного хемотаксиса полиморфно-нуклеарных и мононуклеарных клеток.
Соединения настоящего изобретения относятся к химическому классу (2R)-2-(4-сульфонил)аминофенилпропанамидов. Соединения формулы (I) по своей природе включены в общие формулы соединений, описанных прежде в WO 01/58852, но их характеристики отличаются от предпочтительных соединений вышеупомянутых изобретений значительными преимуществами.
Неожиданно оказалось, что этот класс соединений обладает повышенной селективностью по отношению к рецептору CXCR2, как показано при определении активности по отношению к рецептору CXCR1 в исследовании хемотаксиса, что позволяет использовать этот класс соединений в качестве лекарственных средств для лечения различных хронических или острых CXCR2-зависимых патологических состояний, особенно неопластических расстройств, таких как меланома. Фактически было продемонстрировано, что CXCR2 экспрессируется преимущественно меланомными опухолями и метастазами высоких степеней и что имеются значительные различия в уровнях экспрессии CXCR2 между толстыми и тонкими меланомами, что предполагает различные роли для CXCR2 и CXCR1 также и in vivo [Varney M. L. et al, Am. J. Clin. Pathol, 125, 209, 2006]. Кроме того, антагонисты CXCR2 находят особенно полезное терапевтическое применение при лечении важных легочных заболеваний, таких как ХОЗЛ [Hay D.W.P. et al., Current Opinion in Pharmacology, 1, 242, 2001].
Предпочтительными R-группами являются:
H, C1-C5-алкил, C3-C6-циклоалкил, L-2-амино-1-метил-2-оксоэтил; гетероарильная группа, выбранная из замещенных и незамещенных тиазола, оксазола, пиридина.
Предпочтительными R'-группами являются:
линейный или разветвленный C1-C5-алкил, C3-C6-циклоалкил, трифторметил, бензил; фенил, незамещенный или замещенный группой, выбранной из галогена, C1-C4-алкила, C1-C4-алкоксигруппы, трифторметила, тиофена.
Особо предпочтительными соединениями этого изобретения являются:
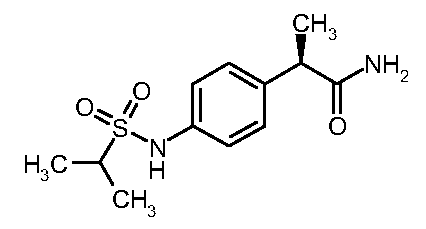
1 - (2R)-2-{4-[(изопропилсульфонил]амино}фенил)пропанамид;
2 - (2R)-2-{4-[(изопропилсульфонил]амино}фенил)пропанамида, натриевая соль;
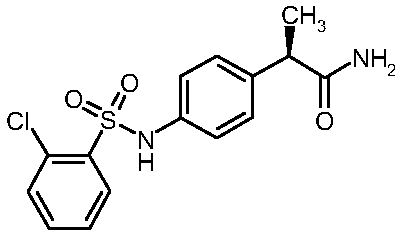
3 - (2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил) пропанамид;
4 - (2R)-2-{4-{[(2,6-дихлорфенил)сульфонил]амино}фенил) пропанамид;
5 - (2R)-2-{4-[(метилсульфонил)амино]фенил}пропанамид;
6 - (2R)-2-{4-[(фенилсульфонил)амино]фенил}пропанамид;
7 - (2R)-2-{4-{[(4-метилфенил)сульфонил]амино}фенил) пропанамид;
8 - (2R)-2-{4-{[(4-метоксилфенил)сульфонил]амино}фенил) пропанамид;
9 - (2R)-2-(4-[(бензилсульфонил]амино}фенил)пропанамид;
10 - (2R)-2-(4-{[(4-хлорфенил)сульфонил]амино}фенил) пропанамид;
11 - (2R)-2-(4-{[(4-(трифторметил)фенил]сульфонил}амино) фенил]пропанамид;
12 - (2R)-2-{4-[(тиен-2-илсульфонил)амино]фенил}пропанамид;
13 - (2R)-2-{4-[(циклопентилсульфонил)амино]фенил} пропанамид;
14 - (2R)-2-(4-{[(трифторметил)сульфонил]амино} фенил)пропанамид;
15 - (2R)-2-{4-[(изопропилсульфонил]амино}фенил)-N-метилпропанамид;
16 - (2R)-N-[(1S)-2-амино-1-метил-2-оксоэтил]-2-{4-[(изопропилсульфонил]амино}фенил)пропанамид;
17 - (2R)-2-{4-[(изопропилсульфонил]амино}фенил)-N-[4-(трифторметил)-1,3-тиазол-2-ил]пропанамид;
18 - (2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-[4-(трифторметил)-1,3-тиазол-2-ил] пропанамид;
19 - (2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-[2-(2-гидроксиэтокси)этил] пропанамид;
20 - (2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-циклопропилпропанамид.
Наиболее предпочтительным соединением в этом списке является соединение 1 и соответствующая натриевая соль.
Соединения этого изобретения являются мощными ингибиторами хемотаксиса человеческих полиморфно-нуклеарных лейкоцитов, индуцированного CXCL1. Соединения этого изобретения формулы (I) обычно выделяют в форме их аддитивных солей с органическими и неорганическими фармацевтически приемлемыми основаниями.
Примерами таких оснований являются гидроксид натрия, гидроксид калия, гидроксид кальция, (D,L)-лизин, L-лизин, трометамин.
Соединения этого изобретения формулы (I) оценивали in vitro в отношении их способности ингибировать хемотаксис полиморфно-нуклеарных лейкоцитов (далее обозначаемых как PMNs) и моноцитов, индуцированных фракциями IL-8 и GRO-α. Для этой цели, чтобы выделить PMNs, из гепаринизованной человеческой крови, полученной от здоровых взрослых добровольцев, удалили мононуклеары (седиментацией на декстране, согласно процедуре, раскрытой у WJ. Ming et al., J. Immunol, 138, 1469, 1987) и красные кровяные клетки (гипотоническим раствором). Жизнеспособность клеток рассчитывали по исключению трипанового синего, а отношение циркулирующих полиморфно-нуклеарных клеток оценивали в центрифугате после окрашивания красителем Diff Quick.
При исследовании CXCL8-индуцированного хемотаксиса использовали рекомбинантный человеческий CXCL8 (Pepro Tech) в качестве стимулятора в экспериментах по хемотаксису: лиофилизованный белок растворяли в объеме HBSS, содержавшем 0,2% бычьего сывороточного альбумина (БСА), чтобы получить запасной раствор с концентрацией 10-5 M, который разбавляли в HBSS до концентрации 10-8 M, которую использовали при исследовании хемотаксиса.
Ингибирование хемотаксиса, индуцированного GRO-α, оценивали в аналогичных тестах.
В экспериментах по хемотаксису PMNs инкубировали с соединениями этого изобретения, имеющими формулу (I), в течение 15 мин при 37°С в атмосфере, содержавшей 5% CO2.
Во время теста на хемотаксис [W. Falket et al., J. Immunol. Methods, 33, 239, 1980] использовали фильтры без PVP с 5-мкм порами и микрокамеры, пригодные для репликации.
Соединения этого изобретения, имеющие формулу (I), испытывали при концентрации в диапазоне между 10-6 и 10-10 M; для этого их добавляли в этой концентрации к нижним и верхним порам микрокамеры. Способность соединений этого изобретения, имеющих формулу (I), ингибировать хемотаксис человеческих моноцитов оценивали согласно раскрытому способу [Van Damme J. et al.,Eur. J. Immunol, 19, 2367, 1989].
Соединения формулы (I) испытывали для оценки их селективности в миграционном тесте, используя клетки L1.2, трансфецированные CXCR1 и CXCR2. Анализ проводили, используя фильтры Transwell с порами размером 5 мкм и следуя описанной процедуре [Imai T. et al., J. Biol. Chem, 273, 1764, 1998]. Клетки L1.2 - это мышиные пре-Т-лимфоциты, трансфецированные вектором (pc-DNA-CXCR1 или CXCR2), содержащим ген, кодирующий специфический белок (CXCR1 или CXCR2). В тестах, проведенных ex vivo с цельной кровью согласно процедуре, раскрытой Patrignani et al., in J. Pharmacol. Exper. Ther., 271, 1705, 1994, было найдено, что соединения формулы (I) полностью неэффективны как ингибиторы фермента циклооксигеназы (COX).
В большинстве случаев соединения формулы (I) в концентрациях 10-5-10-7 M не мешают продуцированию PGE2, индуцированному в мышиных макрофагах стимуляцией липополисахаридами (LPS, 1 мкг/мл). Ингибирование продуцирования PGE2, которое можно зарегистрировать, в большинстве случаев находится на пределе статистической значимости, чаще всего ниже 15-20% базального значения. Пониженная эффективность ингибирования COX является выгодной для терапевтического применения соединения этого изобретения, поскольку ингибирование синтеза простагландинов стимулирует макрофаги увеличивать синтез TNF-α (индуцированный LPS или пероксидом водорода), который является важным медиатором активации нейтрофилов и стимулятором продуцирования такого цитокина, как интерлейкин-8.
Ингибиторы активации CXCR2 находят полезное применение, как подробно изложено выше, особенно при лечении хронических воспалительных патологий, при которых предполагается, что рецепторы CXCL8 и GRO-α играют ключевую патофизиологическую роль в развитии заболевания. Конкретно, предполагают, что активация CXCR2 является существенной при опосредовании ангиогенной активности хемокинов ELR+CXC, опосредованной CXCL8 пролиферации эпидермальных клеток, ангиогенезе и меланоме в моделях на животных [Keane M. P. et al. J. Immunol, 172, 2853, 2004] и у пациентов с различными уровнями злокачественной меланомы [Varney M. L. et al, Am. J. Clin. Pathol, 2006, 125, 209].
Кроме того, антагонисты CXCR2 находят особенно полезное терапевтическое применение при лечении таких важных пульмонологических заболеваний, как хроническое обструктивное заболевание легких (ХОЗЛ) (D. WP Hay и H.M. Sarau., Current Opinion in Pharmacology 2001, 1:242-247) и синдром облитерирующего бронхиолита (СОБ) [Trulock, E. P. Am. J. Respir. Crit Care Med. 155, 789, 1997].
Поэтому следующим предметом настоящего изобретение является создание соединений для использования при лечении ангиогенеза, меланомы, хронического обструктивного заболевания легких (ХОЗЛ) и синдрома облитерирующего бронхиолита (СОБ), а также использование таких соединений при изготовлении лекарственного средства и для лечения описанных выше заболеваний.
В объем настоящего изобретения также входят фармацевтические композиции, включающие соединение этого изобретения и его подходящий носитель.
Соединения этого изобретения, вместе с традиционно применяемыми адъювантом, носителем, разбавителем или эксципиентом, можно фактически поместить в форму фармацевтических композиций и их единичных лекарственных форм и в такой форме их можно применять в твердом виде, таком как таблетки или наполненные капсулы, или в виде жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или капсулы, заполненные этими жидкостями, или в виде, предназначенном для перорального применения, или в форме стерильных инъецируемых растворов для парентерального (включая подкожное) использования. Такие фармацевтические композиции и их единичные лекарственные формы могут включать в себя ингредиенты в традиционных пропорциях, совместно с дополнительными активными соединениями или факторами или без них, и такие единичные лекарственные формы могут содержать любое допустимое эффективное количество активного ингредиента, соизмеримое с предполагаемым диапазоном применяемой суточной дозы.
При применении в качестве фармацевтических средств соединения этого изобретения обычно вводят в форме фармацевтической композиции. Такие композиции можно приготовить в форме, хорошо известной в фармацевтике и включающей в себя по меньшей мере одно активное соединение. Обычно соединения этого изобретения вводят в фармацевтически эффективном количестве. Количество реально вводимого соединения обычно определяют на основании существенных обстоятельств, включая то патологическое состояние, которое подвергается лечению, выбранный путь введения, природу вводимого соединения, возраст, массу тела и реакцию конкретного пациента, тяжесть симптомов у этого пациента и т.п.
Фармацевтические композиции этого изобретения можно вводить различными путями, включая пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный. В зависимости от предполагаемого пути доставки эти соединения готовят предпочтительно в виде инъецируемых или пероральных композиций. Композиции для перорального введения могут принимать форму нерасфасованных жидких растворов или суспензий или нерасфасованных порошков. Однако более обычно эти композиции предоставляют в виде готовых форм, расфасованных на единичные дозы, для облегчения точного дозирования при приеме. Термин «единичные лекарственные формы» относится к физически дискретным единицам, применимым в качестве единичных доз для людей и других животных, причем каждая единица содержит предопределенное количество активного материала, рассчитанного на создание желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим эксципиентом. Типичные единичные лекарственные формы включают предварительно наполненные и предварительно измеренные ампулы или шприцы с жидкими композициями или, в случае твердых композиций, пилюли, таблетки, капсулы и т.п. В таких композициях кислотное соединение обычно является меньшим по количеству компонентом (составляющим от примерно 0,1 до примерно 50% по массе или, предпочтительно, от примерно 1 до примерно 40% по массе), а остальное количество составляют различные наполнители или носители со вспомогательными средствами, облегчающими технологическую обработку и формирование желаемой готовой формы.
Жидкие формы, применимые для перорального введения, могут включать в себя приемлемый водный или неводный носитель с буферными веществами, суспендирующими и диспергирующими средствами, красителями, отдушками и т.п. Жидкие формы, включая инъецируемые композиции, описанные здесь ниже, всегда хранят в темноте во избежание каталитического эффекта света, такого как образование гидропероксида или пероксида. Твердые формы могут включать в себя, например, любой из следующих ингредиентов или соединения похожей природы: связующее вещество, такое как микрокристаллическая целлюлоза, камедь трагаканта или желатин; эксципиент, такой как крахмал или лактоза; дезинтегратор, такой как альгиновая кислота, примогель или кукурузный крахмал; смазывающее вещество, такое как стеарат магния; глидант, такой как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или отдушка, такая как перечно-мятная, метилсалицилатная или апельсиновая отдушка.
Инъецируемые композиции обычно основаны на инъецируемом стерильном физиологическом растворе соли или физиологическом растворе соли с фосфатным буфером или на других инъекционных носителях, известных в данной области. Как указано выше, кислое производное формулы (I) в таких композициях обычно является меньшим по количеству компонентом, часто составляющим 0,05-10% по массе, а остальное количество приходится на инъецируемый носитель и т.п. Средняя суточная доза будет зависеть от различных факторов, таких как серьезность заболевания и состояние пациента (возраст, пол и масса тела). Обычно доза варьируется от 1 мг или нескольких мг до 1500 мг соединения формулы (I) в день, необязательно, разделенных на неоднократные приемы. Благодаря малой токсичности соединений этого изобретения можно также вводить более высокие дозы в течение длительных периодов времени.
Описанные выше компоненты для перорально вводимых или инъецируемых композиций являются лишь типичными примерами. Дополнительные материалы, а также технологические способы и т.п., описаны в Части 8 монографии "Remington's Pharmaceutical Sciences Handbook", 18th Edition, 1990, Mack Publishing Company, Easton, Pennsylvania, которая включена сюда в качестве ссылки. Соединения этого изобретения можно также вводить в формах с продолжительным высвобождением или из систем доставки лекарственных средств с продолжительным высвобождением. Описание типичных материалов с продолжительным высвобождением можно также найти в материалах, включенных в руководство Ремингтона, указанное выше.
Настоящее изобретение проиллюстрировано следующими примерами, которые не должны рассматриваться как ограничивающие объем этого изобретения.
ПРИМЕРЫ
Алкил- и арилсульфонилхлориды, использованные в качестве реагентов при синтезе соединений формулы (I), являются известными продуктами, обычно доступными коммерчески, или их можно приготовить способами, описанными в литературе.
(2R)-2-(4-Аминофенил)пропанамид
(2R)-2-(4-Нитрофенил)пропановую кислоту (6 г, 30,6 ммоль) растворяли в сухом CH2Cl2 (80 мл) и добавляли 1,1-карбонилдиимидазол (5,58 г, 34,41 ммоль) и полученный раствор 2 ч перемешивали при комнатной температуре. Через этот раствор барботировали газообразный аммиак до полного исчезновения промежуточного продукта, определенного ИК-анализом (8 ч). К органическому раствору добавляли насыщенный раствор NH4Cl (20 мл) и разделяли две фазы. Органическую фазу опять промывали водой (2×25 мл), осушали над Na2SO4, профильтровывали, выпаривали под вакуумом и получали (2R)-2-(4-нитрофенил)пропанамид в виде белого твердого вещества (5,1 г, 26,15 ммоль).
(2R)-2-(4-Нитрофенил)пропанамид (4,9 г, 25,2 ммоль) растворяли в смеси THF (30 мл) и CH3OH (30 мл) и полученный раствор охлаждали до T=0-5°C. Добавляли формиат аммония (8 г, 126 ммоль), затем порциями осторожно добавляли 10% Pd/C (1,6 г). Полученную смесь оставляли перемешиваться при комнатной температуре в течение ночи, до полного исчезновения исходного материала (по данным ТСХ). После фильтрования под вакуумом через слой целита и отгонки растворителя при пониженном давлении выделяли чистый (2R)-2-(4-аминофенил)пропанамид в виде белого порошка (4 г, 24,24 ммоль). Выход 96,2%. т.пл. 110-112°C; [α]D 25 (c=0,6, CH3OH): -1,9°; 1H-ЯМР (CDCl3) δ 7,10 (д, 2H, J=7 Гц), 6,65 (д, 2H, J=7 Гц), 5,35 (ушир.с, 2H, CONH2), 3,52 (м, 1H), 1,50 (д, 3H, J=7 Гц).
(2R)-2-{4-[(Изопропилсульфонил]амино}фенил)пропанамид (1)
(2R)-2-(4-Аминофенил)пропанамид (0,5 г, 3,05 ммоль) растворяли в пиридине (2 мл) и добавляли 2-пропансульфонилхлорид (0,53 мл, 3,66 ммоль). Полученный раствор 4 ч кипятили с обратным холодильником и оставляли на ночь при комнатной температуре. После полного исчезновения исходного амида раствор разбавили Et2O (10 мл) и органический слой промывали 1 н HCl (2×5 мл) и H2O (2×5 мл), сушили над Na2SO4, профильтровывали, выпаривали под вакуумом и получали (2R)-2-{(4-[(изопропилсульфонил) амино]фенил}пропанамид в виде бледно-желтого твердого вещества (667 мг, 2,47 ммоль). Выход 81%, т.пл. 125-127°C; [α]D 25 (c=0,3, CH3OH): -12,7°; 1H-ЯМР (DMSO-d6) δ 9,65 (ушир.с, 1H, SO2NH), 7,40 (ушир.с, 1H, CONH2), 7,25 (д, 2H, J=7 Гц), 7,12 (д, 2H, J=7 Гц), 6,80 (ушир.с, 1H, CONH2), 3,52 (кв, 1H, J=7 Гц), 3,15 (м, 1H), 1,22 (д, 3H, J=7 Гц ), 1,18 (д, 6H, J=7 Гц).
Следуя вышеописанной процедуре и исходя из соответствующих сульфонилхлоридов, приготовили следующие амиды:
(2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)пропанамид (2); воскообразное твердое вещество; [α]D 25 (c=0,5, CH3OH): -8,3°; 1H-ЯМР (CDCl3) δ 8,10 (д, 1H, J=7 Гц), 7,52-7,45 (м, 2H+NH), 7,32-7,27 (м, 1H), 7,13 (д, 2H, J=7 Гц), 7,05 (д, 2H, J=7 Гц), 5,55 (ушир.с, 1H, CONH2), 5,28 (ушир.с, 1H, CONH2), 3,48 (кв, 1H, J=7 Гц), 1,42 (д, 3H, J=7 Гц).
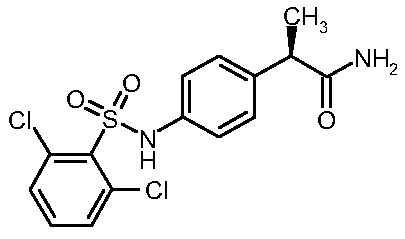
(2R)-2-{4-{[(2,6-дихлорфенил)сульфонил]амино}фенил) пропанамид (3); воскообразное твердое вещество; [α]D 25 (c=0,5, CH3OH): -10°; 1H-ЯМР (CDCl3) δ 7,52 (ушир.с, 1H, NH), 7,35-7,20 (м, 3H), 7,13 (д, 2H, J=7 Гц), 7,05 (д, 2H, J=7 Гц), 5,55 (ушир.с, 1H, CONH2), 5,28 (ушир.с, 1H, CONH2), 3,48 (кв, 1H, J=7 Гц), 1,42 (д, 3H, J=7 Гц).
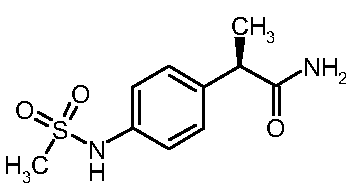
(2R)-2-{4-[(метилсульфонил)амино]фенил}пропанамид (4); воскообразное твердое вещество; [α]D 25 (C=0,5, CH3OH): -12,5°; 1H-ЯМР (DMSO-d6) δ 9,65 (ушир.с, 1H, NH), 7,40 (ушир.с, 1H, CONH2), 7,25 (д, 2H, J=7 Гц), 7,12 (д, 2H, J=7 Гц), 6,80 (ушир.с, 1H, CONH2), 3,64 (c, 3H), 3,52 (кв, 1H, J=7 Гц), 1,22 (д, 3H, J=7 Гц).
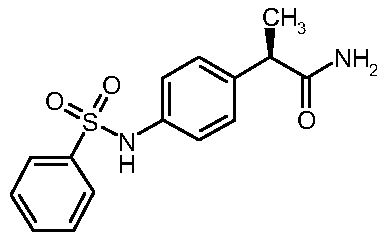
(2R)-2-{4-[(фенилсульфонил)амино]фенил}пропанамид (5); белый порошок; т.пл. 152-153°C; [α]D 25 (c=0,5, CH3OH): -13,5°; 1H-ЯМР (DMSO-d6) δ 7,92 (м, 2H), 7,74-7,62 (м, 3H+NH), 7,40 (ушир.с, 1H, CONH2), 7,30 (д, 2H, J=7 Гц), 7,15 (д, 2H, J=7 Гц), 6,88 (ушир.с, 1H, CONH2), 3,60 (кв, 1H, J=7 Гц), 1,40 (д, 3H, J=7 Гц).
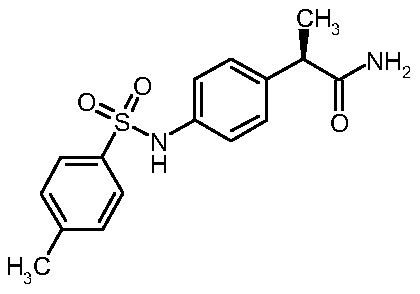
(2R)-2-{4-{[(4-метилфенил)сульфонил]амино}фенил)пропанамид (6); белый порошок; т.пл. 138-140°C; [α]D 25 (c=0,2, CH3OH): -7,1°; 1H-ЯМР (CDCl3) δ 7,65 (д, 2H, J=7 Гц), 7,28-7,15 (м, 4H), 7,05 (д, 2H, J=7 Гц), 6,45 (ушир.с, 1H, NH), 5,25 (ушир.с, 1H, CONH2), 3,52 (кв, 1H, J=7 Гц), 2,38 (c, 3H), 1,47 (д, 3H, J=7 Гц).
(2R)-2-{4-{[(4-метоксилфенил)сульфонил]амино}фенил) пропанамид (7); белый порошок; т.пл. 118-120°C; [α]D 25 (c=0,2, CH3OH): -3,6°; 1H-ЯМР (CDCl3) δ 7,70 (д, 2H, J=7 Гц), 7,22 (д, 2H, J=7 Гц), 7,05 (д, 2H, J=7 Гц), 6,90 (д, 2H, J=7 Гц), 6,52 (ушир.с, 1H, NH), 5,25 (ушир.с, 2H, CONH2), 3,85 (c, 3H), 3,55 (кв, 1H, J=7 Гц), 1,45 (д, 3H, J=7 Гц).
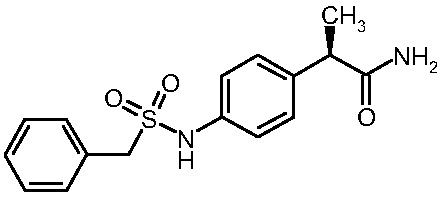
(2R)-2-(4-[(бензилсульфонил]амино}фенил)пропанамид (8); белый порошок; т.пл. 68-70°C; [αD 25 (c=0,2, CH3OH): -2,5°; 1H-ЯМР (CDCl3) δ7,40-7,35 (м, 3H), 7,30-7,25 (м, 4H), 7,15 (д, 2H, J=7 Гц), 6,21 (ушир.с, 1H, NH), 5,31 (ушир.с, 2H, CONH2), 4,35 (c, 2H), 3,58 (кв, 1H, J=7 Гц), 1,57 (д, 3H, J=7 Гц).
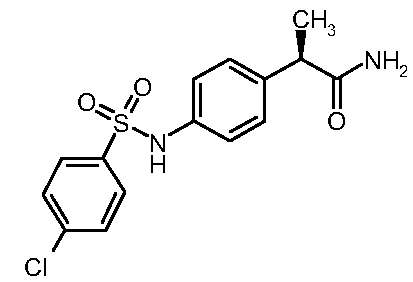
(2R)-2-(4-{[(4-хлорфенил)сульфонил]амино}фенил)пропанамид (9); белый порошок; т.пл. 150-153°C; [α]D 25 (c=0,2, CH3OH): -3,6°; 1H-ЯМР (CDCl3) δ 7,75 (д, 2H, J=7 Гц), 7,45 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,05 (д, 2H, J=7 Гц), 6,68 (ушир.с, 1H, NH), 5,28 (ушир.с, 2H, CONH2), 3,55 (кв, 1H, J=7 Гц), 1,50 (д, 3H, J=7 Гц).
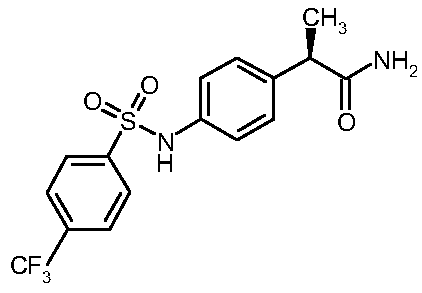
(2R)-2-(4-{[(4-(трифторметил)фенил]сульфонил}амино)фенил] пропанамид (10); белый порошок; т.пл. 178-180°C; [α]D 25 (c=0,2, CH3OH): -2,5°; 1H-ЯМР (CDCl3) δ 9,20 (ушир.с, 1H, NH), 7,90 (д, 2H, J=7 Гц), 7,68 (д, 2H, J=7 Гц), 7,15 (д, 2H, J=7 Гц), 7,05 (д, 2H, J=7 Гц), 5,45- 5,30 (ушир.с, 2H, CONH2), 3,48 (кв, 1H, J=7 Гц), 1,45 (д, 3H, J=7 Гц).
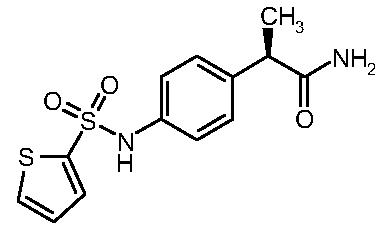
(2R)-2-{4-[(тиен-2-илсульфонил)амино]фенил}пропанамид (11); белый порошок; т.пл. 58-60°C; [α]D 25 (c=0,2, CH3OH): -3,5°; 1H-ЯМР (CDCl3) δ 7,58 (д, 1H, J=2 Гц ), 7,52 (д, 1H, J=2 Гц), 7,25 (д, 2H, J=7 Гц), 7,10 (д, 2H, J=7 Гц), 7,05 (д, 1H, J=2 Гц, 6,75 (ушир.с, 1H, NH), 5,35 (ушир.с, 2H, CONH2), 3,58 (кв, 1H, J=7 Гц), 1,48 (д, 3H, J=7 Гц).
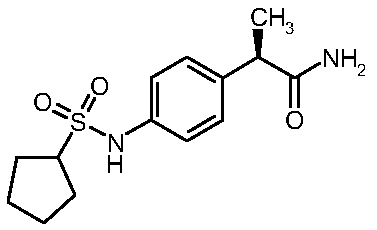
(2R)-2-{4-[(циклопентилсульфонил)амино]фенил}пропанамид (12); воскообразное твердое вещество; [α]D 25 (C=0,5, CH3OH): -10,2°; 1H-ЯМР (DMSO-d6) δ7,75 (ушир.с, 1H, NH), 7,40 (ушир.с, 1H, CONH2), 7,30 (д, 2H, J=7 Гц), 7,15 (д, 2H, J=7 Гц), 6,88 (ушир.с, 1H, CONH2), 3,60 (кв, 1H, J=7 Гц), 3,34 (м, 1H), 2,08-1,97 (м, 2H), 1,85-1,75 (м, 2H), 1,60-1,50 (м, 4H), 1,40 (д, 3H, J=7 Гц).
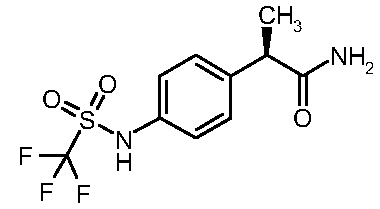
(2R)-2-(4-{[(трифторметил)сульфонил]амино}фенил)пропанамид (13); воскообразное твердое вещество; [α]D 25 (c=0,5, CH3OH): -24,5°; 1H-ЯМР (DMSO-d6) δ 9,60 (ушир.с, 1H, NH), 7,65 (д, 2H, J=7 Гц), 7,40 (ушир.с, 1H, CONH2), 7,12 (д, 2H, J=7 Гц), 6,85 (ушир.с, 1H, CONH2), 3,52 (кв, 1H, J=7 Гц), 1,40 (д, 3H, J=7 Гц).
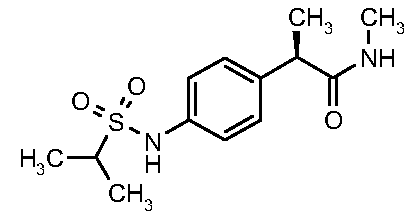
(2R)-2-{4-[(изопропилсульфонил]амино}фенил)-N-метилпропанамид (14)
(2R)-2-{[4-(изопропилсульфониламино)фенил]}пропановую кислоту, приготовленную как описано в WO 03/042625, (0,65 г, 2,4 ммоль) растворяли в CH2Cl2 (8 мл); добавляли гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (WSC) (0,46 г, 2,4 ммоль) и 1-гидроксибензотриазолгидрат (HOBT) (0,324 г, 2,4 ммоль) и полученную смесь оставляли перемешиваться при комнатной температуре в течение 30 мин. Затем по каплям добавляли смесь гидрохлорида метиламина (0,155 г, 2,43 ммоль) и триэтиламина (0,33 мл, 2,4 ммоль) в CH2Cl2 (2 мл) и полученную смесь оставляли на ночь при комнатной температуре. Эту смесь разбавили CH2Cl2 (10 мл) и органический слой промывали 1 н. HCl (2×10 мл) и H2O (2×10 мл), осушали над Na2SO4, профильтровывали, выпаривали под вакуумом и получали (2R)-2-{4-[(изопропилсульфонил]амино}фенил)-N-метилпропанамид в виде воскообразного твердого вещества (0,63 г, 2,23 ммоль). Выход 93%. [α]D 25 (c=1, CH3CH2OH): -20,5°; 1H-ЯМР (CDCl3) δ 9,65 (ушир.с, 1H, SO2NH), 7,25 (д, 2H, J=7 Гц), 7,12 (д, 2H, J=7 Гц), 5,30 (ушир.с, 1H, NH), 3,52 (кв, 1H, J=7 Гц), 3,15 (м, 1H), 2,78 (д, 3H, J=3 Гц), 1,22 (д, 3H, J=7 Гц ), 1,18 (д, 6H, J=7 Гц).
Следуя вышеописанной процедуре и исходя из соответствующих коммерческих гидрохлоридов аминов и пропановых кислот формулы (II)
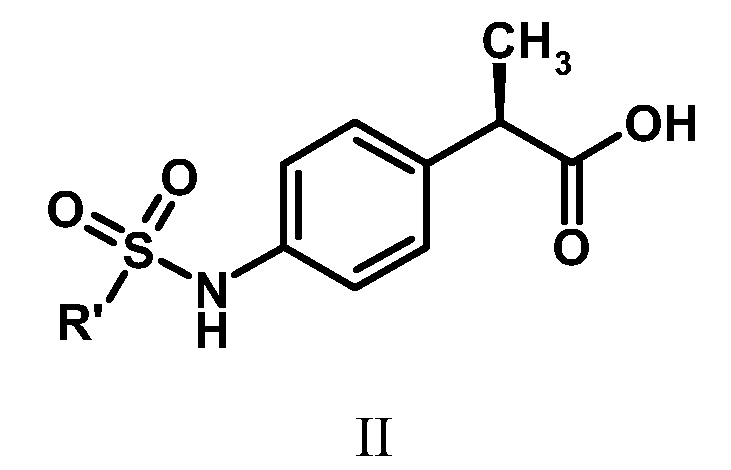
где R' определен выше, приготовили следующие амиды:
(2R)-N-[(1S)-2-амино-1-метил-2-оксоэтил]-2-{4-[(изопропилсульфонил]амино}фенил)пропанамид (15); белый порошок; т.пл. 132-135°C; [α]D 25 (c=1, CH3OH): -22,5°; 1H-ЯМР (DMSO-d6) δ 9,65 (ушир.с, 1H, SO2NH), 8,35 (ушир.с, 1H, NH), 7,70 (д, 2H, J=7 Гц), 7,62 (д, 2H, J=7 Гц), 7,50-7,35 (ушир.с, 1H, CONH2), 7,15-7,05 (ушир.с, 1H, CONH2), 4,45-4,32 (м, 1H), 4,05 (кв, 1H, J=7 Гц), 3,15 (м, 1H), 1,55 (д, 3H, J=7 Гц), 1,35 (д, 3H, J=7 Гц), 1,18 (д, 6H, J=7 Гц).
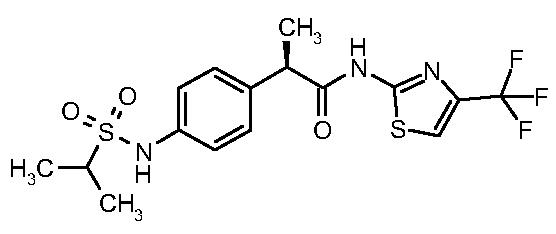
(2R)-2-{4-[(изопропилсульфонил]амино}фенил)-N-[4-(трифторметил)-1,3-тиазол-2-ил]пропанамид (16); стеклообразное твердое вещество; [α]D 25 (c=0,5, CH3OH): -8,4°; 1H-ЯМР (CDCl3) δ 9,65 (ушир.с, 1H, SO2NH), 8,75 (ушир.с, 1H, NH), 7,45 (д, 2H, J=7 Гц), 7,30 (д, 2H, J=7 Гц), 7,25 (c, 1H), 3,82 (кв, 1H, J=7 Гц), 3,15 (м, 1H), 1,24 (д, 3H, J=7 Гц ), 1,15 (д, 6H, J=7 Гц).
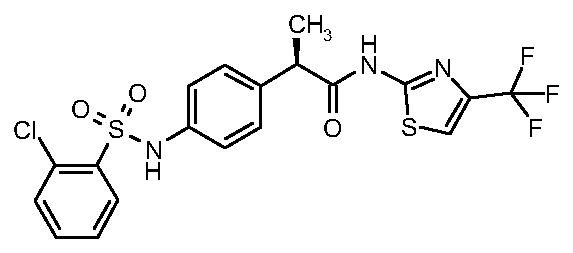
(2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-[4-(трифторметил)-1,3-тиазол-2-ил]пропанамид (17); воскообразное твердое вещество; [α]D 25 (c=0,5, CH3OH): -5,5°; 1H-ЯМР (CDCl3) δ 9,50 (ушир.с, 1H, SO2NH), 8,72 (ушир.с, 1H, NH), 8,10 (д, 1H, J=7 Гц), 7,50-7,48 (м, 2H), 7,32-7,27 (м, 1H), 7,20 (c, 1H), 7,13 (д, 2H, J=7 Гц), 7,05 (д, 2H, J=7 Гц), 3,48 (кв, 1H, J=7 Гц), 1,42 (д, 3H, J=7 Гц).
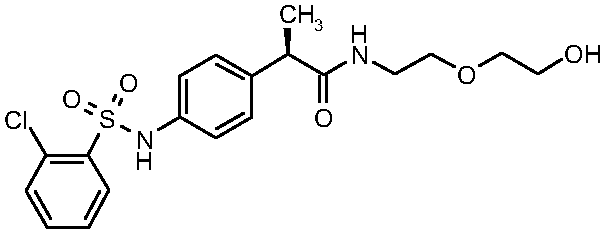
(2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-[2-(2-гидроксиэтокси)этил]пропанамид (18); бесцветное масло; [α]D 25 (c=0,5, CH3OH): -4,5°; 1H-ЯМР (CDCl3) δ 9,50 (ушир.с, 1H, SO2NH), 8,10 (д, 1H, J=7 Гц), 7,50-7,48 (м, 2H), 7,32-7,27 (м, 1H), 7,15 (д, 2H, J=7 Гц), 7,08 (д, 2H, J=7 Гц), 6,10 (ушир.с, 1H, NH), 3,70-3,60 (м, 3H), 3,55-3,40 (м, 6H), 2,05 (ушир.с, 1H, OH), 1,52 (д, 3H, J=7 Гц).
(2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-циклопропилпропанамид (19); бесцветное масло; [α]D 25 (c=0,5, CH3OH): -11,5°; 1H-ЯМР (CDCl3) δ 9,50 (ушир.с, 1H, SO2NH), 8,10 (д, 1H, J=7 Гц), 7,50-7,48 (м, 2H), 7,32-7,27 (м, 1H), 7,15 (д, 2H, J=7 Гц), 7,08 (д, 2H, J=7 Гц), 5,45 (ушир.с, 1H, NH), 3,50 (кв, 1H, J=7 Гц), 2,75-2,62 (м, 1H), 1,52 (д, 3H, J=7 Гц), 0,8 (м, 2H), 0,42 (м, 2H).
(2R)-2-{4-[(Изопропилсульфонил]амино}фенил)пропанамид, натриевая соль
(2R)-2-{(4-[(Изопропилсульфонил)амино]фенил}пропанамид (1) (500 мг, 1,85 ммоль) растворяли в CH3OH (15 мл). По каплям добавляли NORMEX 1 н NaOH (1,85 мл, 1,85 ммоль) и полученный раствор оставляли перемешиваться при комнатной температуре в течение 2 ч. После испарения растворителя добавляли воду (3 мл) и прозрачный раствор замораживали, лиофилизовали и получали натриевую соль (2R)-2-{4-[(изопропилсульфонил]амино}фенил) пропанамида (541 мг, 1,85 ммоль) в виде бледно-желтого порошка. Количественный выход. [α]D 25 (c=0,4, CH3OH): -11,75°; 1H-ЯМР (D2O) δ 7,32 (д, 2H, J=7 Гц), 7,15 (д, 2H, J=7 Гц), 3,82 (кв, 1H, J=7 Гц), 3,35 (м, 1H), 1,54 (д, 3H, J=7 Гц ), 1,35 (д, 6H, J=7 Гц).
Натриевые соли соединений 2-19 были приготовлены по той же вышеописанной процедуре.
Таблица содержит данные о биологической активности типичных соединений настоящего изобретения
10-8 M)
10-7 M)
[(изопропилсульфонил]амино}фенил)
пропанамид
(1)
хлорфенил)сульфонил]амино}фенил)
пропанамид
(2)
дихлорфенил)сульфонил]амино}фенил)
пропанамид
(3)
(4)
[(фенилсульфонил)амино]фенил}
пропанамид
(5)
метилфенил)сульфонил]амино}фенил)
пропанамид
(6)
метоксилфенил)сульфонил]амино}фенил)
пропанамид (7)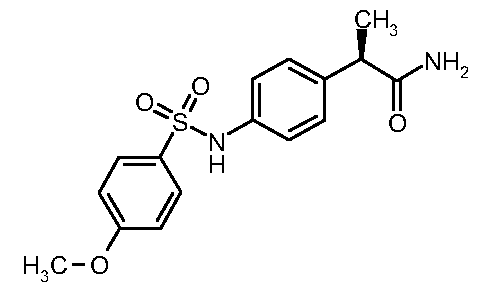
[(бензилсульфонил]амино}фенил)
пропанамид
(8)
хлорфенил)сульфонил]амино}фенил)
пропанамид
(9)
(трифторметил)фенил]сульфонил}амино)
фенил]пропанамид
(10)
амино]фенил}пропанамид
(11)
амино]фенил}пропанамид
(12)
амино}фенил)пропанамид
(13)
амино}фенил)-N-метилпропанамид
(14)
фенил)пропанамид (15)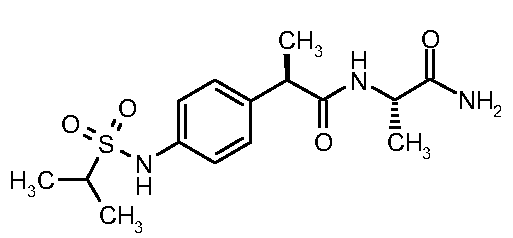
[(изопропилсульфонил]амино}фенил)-N-
[4-(трифторметил)-1,3-тиазол-2-ил]
пропанамид
(16)
амино}фенил)-N-[4-(трифторметил)-
1,3-тиазол-2-ил]пропанамид
(17)
N-[2-(2-гидроксиэтокси)этил]
пропанамид
(18)
амино}фенил)-N-циклопропилпропанамид
(19)
| название | год | авторы | номер документа |
|---|---|---|---|
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И ПРОИЗВОДНЫЕ, И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2520212C2 |
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2002 |
|
RU2317075C2 |
| (R)-4-(ГЕТЕРОАРИЛ)ФЕНИЛЭТИЛЬНЫЕ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2008 |
|
RU2475486C2 |
| 2-АРИЛУКСУСНЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2356887C2 |
| (R)-АРИЛАЛКИЛАМИНОПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2006 |
|
RU2458051C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |
| ИНГИБИТОРЫ CXCL8 ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ COVID-19 | 2021 |
|
RU2830584C1 |
| ПРОИЗВОДНЫЕ 2-АРИЛПРОПИОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ ВКЛЮЧАЮЩИЕ | 2005 |
|
RU2410372C2 |
| ПРОИЗВОДНЫЕ 2-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2005 |
|
RU2375347C2 |
| СУЛЬФОНОВЫЕ КИСЛОТЫ, ПРОИЗВОДНЫЕ УКАЗАННЫХ КИСЛОТ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2345063C2 |
Настоящее изобретение относится к новым (2R)-2-фенилпропанамидным производным формулы (I) или к их фармацевтически приемлемым солям, обладающим способностью ингибировать CXCL1-индуцированный хемотаксис человеческих полиморфно-нуклеарных лейкоцитов (PMNs), к фармацевтической композиции на их основе, к их применению для приготовления лекарственного средства и к способам их получения.
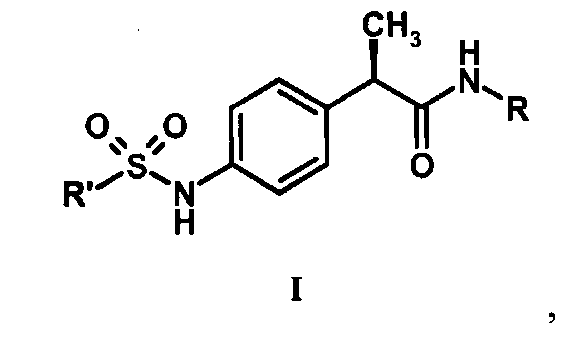
где R выбран из Н, С1-С5-алкила, С3-С6-циклоалкила, тиазола, замещенного трифторметилом, остатка формулы -CH2-CH2-O-(CH2-CH2O)nR", в которой R" представляет собой Н или С1-С5-алкил, n равно целому числу от 0 до 2, или R, вместе с NH-группой, к которой он присоединен, является группой-радикалом первичных амидов природной аминокислоты, такой как (2S)-2-аминопропанамид; R' выбран из линейного или разветвленного С1-С5-алкила, С3-С6-циклоалкила, трифторметила, фенила, необязательно замещенного группой, выбранной из галогена, С1-С5-алкила, С1-С5-алкокси и трифторметила; незамещенного бензила; тиофена. 5 н. и 4 з.п. ф-лы, 1 табл.
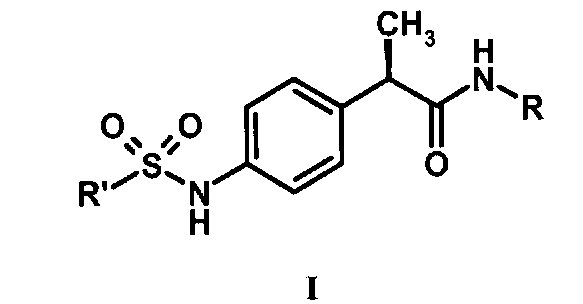
1. (2R)-2-фенилпропанамидные производные формулы (I):
и их фармацевтически приемлемые соли,
в которых
R выбран из
- Н, С1-С5-алкила и С3-С6-циклоалкила;
- тиазола, замещенного трифторметилом;
- остатка формулы -CH2-CH2-O-(CH2-CH2O)nR'', в которой R'' представляет собой Н или С1-С5-алкил, n равно целому числу от 0 до 2;
или R вместе с NH-группой, к которой он присоединен, является группой-радикалом первичных амидов природной аминокислоты, такой как (2S)-2-аминопропанамид;
R' выбран из
- линейного или разветвленного С1-С5-алкила, С3-С6-циклоалкила и трифторметила;
- фенила, необязательно замещенного группой, выбранной из галогена, С1-С5-алкила, C1-C5-алкокси и трифторметила;
- незамещенного бензила;
- тиофена.
2. Соединения по п.1, в которых
R выбран из
Н, С1-С5-алкила, С3-С6-циклоалкила, L-2-амино-1-метил-2-оксоэтила; тиазола, замещенного трифторметилом; R' выбран из
линейного или разветвленного С1-С5-алкила, С3-С6-циклоалкила, трифторметила, бензила; фенила, незамещенного или замещенного группой, выбранной из галогена, С1-С4-алкила и трифторметила; тиофена.
3. Соединения по п.1, выбранные из:
(2R)-2-{4-[(изопропилсульфонил]амино}фенил)пропанамида;
натриевой соли (2R)-2-{4-[(изопропилсульфонил]амино}
фенил)пропанамида;
(2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)пропанамида;
(2R)-2-{4-{[(2,6-дихлорфенил)сульфонил]амино}фенил)пропанамида;
(2R)-2-{4-[(метилсульфонил)амино]фенил}пропанамида;
(2R)-2-{4-[(фенилсульфонил)амино]фенил}пропанамида;
(2R)-2-{4-{[(4-метилфенил)сульфонил]амино}фенил)пропанамида;
(2R)-2-{4-{[(4-метоксилфенил)сульфонил]амино}фенил)пропанамида;
(2R)-2-(4-[(бензилсульфонил]амино}фенил)пропанамида;
(2R)-2-(4-{[(4-xлopфeнил)cyльфoнил]aминo}фенил)пропанамида;
(2R)-2-(4-{[(4-(трифторметил)фенил]сульфонил}амино)фенил]пропанамида;
(2R)-2-{4-[(тиeн-2-илcyльфoнил)aминo]фeнил}пpoпaнaмидa;
(2R)-2-{4-[(циклoпeнтилcyльфoнил)aминo]фeнил}пропанамида;
(2R)-2-(4-{[(тpифтopмeтил)cyльфoнил]aминo}фенил)пропанамида;
(2R)-2-{4-[(изoпpoпилcyльфoнил]aминo}фeнил)-N-метилпропанамида;
(2R)-N-[(1S)-2-амино-1-метил-2-оксоэтил]-2-(4-[(изопропилсульфонил]амино}фенил)пропанамида;
(2К)-2-{4-[(изопропилсульфонил]амино}фенил)-N-[4-(трифторметил)-1,3-тиазол-2-ил]пропанамида;
(2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-[4-(трифторметил)-1,3-тиазол-2-ил]пропанамида;
(2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-[2-(2-гидроксиэтокси)этил]пропанамида;
(2R)-2-{4-{[(2-хлорфенил)сульфонил]амино}фенил)-N-циклопропилпропанамида.
4. Соединения по п.1, которые являются (2R)-2-{4-[(изопропилсульфонил]амино}фенил)пропанамидом и его натриевой солью.
5. Соединения по любому из пп.1-4, предназначенные для применения в качестве лекарственных средств, ингибирующего CXCL1-индуцированный хемотаксис человеческих полиморфно-нуклеарных лейкоцитов (PMNs).
6. Применение соединений по любому из пп.1-4 для приготовления лекарственного средства для лечения заболеваний, в которые вовлечен индуцированный CXCL1 хемотаксис человеческих PMNs, где указанные заболевания выбраны из меланомы, ангиогенеза, хронического обструктивного заболевания легких (ХОЗЛ) и синдрома облитерирующего бронхиолита (СОБ).
7. Фармацевтические композиции, ингибирующие индуцированный CXCL1 хемотаксис человеческих PMNs, включающие фармацевтически эффективное количество соединения по любому из пп.1-4 в смеси с подходящим для них носителем.
8. Способ получения соединений формулы (I) по п.1, включающий взаимодействие соединения формулы (II)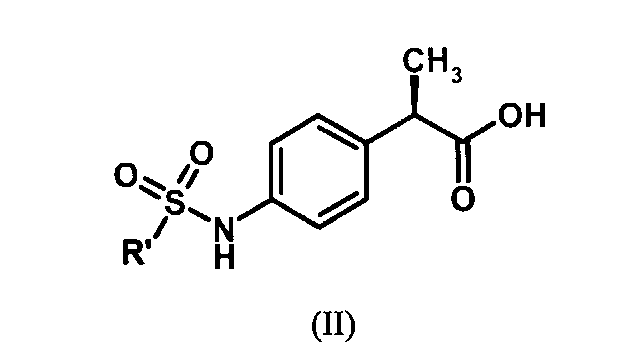
в которой R' имеет значение, определенное в п.1, с амином формулы NHR, в которой R имеет значение, определенное в п.1.
9. Способ получения соединений формулы (I) по п.1, включающий взаимодействие (2R)-2-(4-aминoфeнил)пpoпaнaмидa с сульфонилхлоридами формулы R'SO2Cl, в которой R' имеет значение, определенное в п.1.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ СОЧЕТАНИЕ ПРАВОВРАЩАЮЩЕГО И ЛЕВОВРАЩАЮЩЕГО ИЗОМЕРОВ СОТАЛОЛА | 1999 |
|
RU2213560C2 |

