Настоящее изобретение описывает 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-он и фармацевтические композиции, приготовленные с применением указанного соединения, а также применение указанного соединения для приготовления лекарственного препарата, направленного на предотвращение или лечение воспалительных состояний.
Воспаление является важным компонентом физиологической иммунной защиты организма. Однако существует множество исследований, показывающих, что неправильная пространственно-временная регуляция воспалительной реакции играет роль при широком спектре заболеваний, включая те, в которые явным образом вовлечены лейкоциты (таких как аутоиммунные заболевания, астма или атеросклероз), а также при заболеваниях, которые традиционно не связывали с активностями лейкоцитов (такие как остеопороз или болезнь Альцгеймера).
Хемокины являются большим семейством сигнальных молекул, гомологичных интерлейкину-8, которые задействованы в регуляции перемещения лейкоцитов как при физиологических, так и при патологических состояниях. Более пятидесяти лигандов и двадцати рецепторов, задействованных в сигнальной системе с участием хемокинов, обеспечивают организацию путей передачи информации, достаточную для того, чтобы направлять лейкоциты посредством сложных иммунных регуляторных процессов из костного мозга к периферии, а затем обратно, через вторичные лимфоидные органы. Однако сложность данной системы хемокинов в первое время затрудняла развитие фармакологических подходов к модулированию воспалительных ответов через блокаду рецепторов хемокинов. Оказалось сложным определить, который из рецепторов хемокинов должен (должны) быть ингибирован(ы) для того, чтобы вызвать благоприятное терапевтическое действие при конкретном воспалительном нарушении.
Недавно было описано семейство агентов, которые одновременно блокируют передачу сигнала широким спектром хемокинов (Reckless et al., 1999 // Biochem J. 340, 803-811). Первым подобным агентом является пептид, называемый "пептид 3", для которого показано, что он ингибирует миграцию лейкоцитов, индуцированную пятью различными хемокинами, в то время как миграция, которая возникает в ответ на другие хемоаттрактанты (такие как fMLP или TGF-beta), не изменяется. Соединения данной группы, включающей данный пептид и его аналоги, такие как NR58-3.14.3 (т.е. SEQ ID NO №1, c(DCys-DGl-DIle-DTrp-DLys-DGIn-DLys-DPro-DAsp-DLeu-DCys)-NH2), были названы "Ингибиторы хемокинов широкого спектра действия" (Broad Spectrum Chemokine Inhibitors, BSCl). Grainger et al. (2003, Biochem. Pharm. 65, 1027-1034) в дальнейшем показали на ряде моделей болезней на животных, что BSCl обладают потенциально полезной противовоспалительной активностью. Интересным является тот факт, что одновременная блокада множества хемокинов не имеет очевидной связи с развитием острой или хронической токсичности, и это позволяет предположить, что данный подход может являться полезной стратегией для развития новых противовоспалительных лекарственных средств, которые будут обладать преимуществами стероидных препаратов, но обладать меньшим побочным действием.
Однако пептиды и производные пептидов, такие как NR58-3.14.3, могут не быть оптимальными при применении in vivo. Синтез данных веществ является достаточно дорогостоящим, и они характеризуются относительно неблагоприятными фармакокинетическими и фармакодинамическими свойствами. Например, соединение NR58-3.14.3 не обладает биодоступностью при оральном применении, а период полувыведения из плазмы крови после внутривенного введения составляет менее 30 минут.
Были приняты две параллельные стратегии для идентификации новых препаратов, которые сохраняют противовоспалительные свойства пептида 3 и NR58-3.14.3, но имеют улучшенные характеристики для использования в качестве фармацевтических препаратов. Во-первых, был разработан ряд аналогов пептидов, некоторые из которых характеризуются более продолжительным периодом полувыведения из плазмы, чем NR58-3.14.3, и синтез которых значительно дешевле. Во-вторых, был проведен анализ структура/активность данных пептидов для того, чтобы определить фармакофоры и предложить низкомолекулярные соединения непептидной природы, которые сохраняют полезные свойства исходного пептида.
При втором подходе было получено несколько серий различных по своей структуре веществ, которые сохраняли противовоспалительные свойства указанных пептидов, включая 16-амино- и 16-аминоалкил-производные алкалоида йохимбина, а также ряд N-замещенных 3-аминоглутаримидов (см: Fox et al., (2002) / J. Med. Chem. 45, 360-370; WO 99/12968 и WO 00/42071). Все эти вещества являются ингибиторами хемокинов широкого спектра действия, которые сохраняют избирательность по сравнению с отличными от хемокинов хемоаттрактантами, и показано, что некоторые из них блокируют острое воспаление in vivo.
Наиболее сильным и избирательным действием из указанных веществ обладает S-3-(ундек-10-еноил)-аминоглутаримид (NR58.4), который ингибирует индуцированную хемокинами миграцию in vitro с ED50 равной 5 нМ. Однако при проведении дальнейших исследований было показано, что могло происходить ферментативное расщепление аминоглутаримидного кольца в сыворотке крови. Следовательно, в некоторых случаях (например, если воспаление, которое подвергают лечению, является хроническим, как в случае аутоиммунных заболеваний) свойства данных соединений могут быть не оптимальными, и более стабильное вещество со сходными противовоспалительными свойствами может оказаться более эффективным.
В качестве подхода для идентификации подобных стабильных аналогов провели исследование стабильности в сыворотке крови различных производных S-3-(ундек-10-еноил)-аминоглутаримида. Одно из таких производных, 6-дезоксо-аналог S-3-(ундек-10-еноил)-тетрагидропиридин-2-она, полностью стабильно в сыворотке крови человека в течение периода не менее 7 дней при 37°С, но характеризуется значительно меньшей активностью по сравнению с исходной молекулой.
Одним из таких семейств стабильных ингибиторов хемокинов широкого спектра действия являются 3-амино-капролактамы с семичленным монолактамным кольцом (см., например, WO 2005/053702 и WO 2006/016152). Однако в дальнейшем полезные противовоспалительные вещества были получены также из других 3-аминолактамов с различным размером кольца (см., например, WO 2006/134385). При осуществлении других модификаций лактамного кольца, включая введение гетероатомов и конструирование систем из двух лактамных колец, также были получены вещества, обладающие активностью ингибиторов хемокинов широкого спектра действия (см., например, WO 2006/018609 и WO 2006/085096).
К настоящему времени идентификация широких классов действующих веществ, обладающих активностью ингибиторов хемокинов широкого спектра действия и, следовательно, противовоспалительными свойствами in vivo, была основана на оптимизации активности ингибиторов хемокинов широкого спектра действия. Например, по результатам предыдущих наблюдений было показано, что двойное замещение по положению 2 (по альфа- или центральному атому углерода в боковой ацильной цепи ацил-3-аминолактамов) приводит к существенному увеличению активности в качестве BSCl как in vitro, так и in vivo в моделях острого воспаления, в случае, когда двузамещенная по положению 2 ацильная группа представляет собой открытую цепь (см. WO 2005/053702), один цикл (см. WO 2006/134384) или полициклическую систему (см. WO 2006/016152).
Однако сила желаемого фармакологического действия является хотя и важным, но только одним из факторов, которые определяют, будет ли вещество полезным фармацевтическим препаратом для человека. В особенности, фармакокинетические параметры (или показатели распределения действующего вещества внутри организма) являются наиболее важными при определении полезности отдельного действующего вещества. Фармакокинетика, определяемая в самом широком смысле, как исследование действия организма на лекарственное вещество (напротив, фармакодинамика изучает воздействие лекарственного вещества на организм), зависит от сложных физиологических процессов в организме, без ограничения включая: абсорбцию, стабильность в плазме, объем распределения (и, особенно, скорость установления равновесной концентрации в тканях-мишенях), метаболические превращения (включая метаболические процессы в печени, такие как окисление под действием цитохрома Р450 и метаболические реакции фазы II, такие как сульфатирование и глюкуронирование, а также метаболические процессы, проходящие за пределами печени, такие как модификации под действием ферментов сыворотки крови) и экскрецию (такую как почечный клиренс и выведение с испражнениями). Данные процессы часто совместно называют "ADME" свойствами действующего вещества (от ADME: Absorption, Distribution, Metabolism and Excretion - «всасывание, распределение, метаболизм и выведение»).
Другим важным фактором при определении полезности действующего вещества в качестве лекарственного средства при применении на человеке является его безопасность. Многие, если не все, используемые вещества оказывают множество воздействий на организм человека, среди которых благоприятное фармакологическое действие обычно является только одним из эффектов. Другие действия могут приводить к повреждениям (токсическое действие) или неудобству (побочные явления) для пациента. Изучение данных свойств потенциальных компонентов фармацевтических препаратов называют токсикология или фармакологические исследования безопасности. Нежелательные воздействия можно, в целом, отнести к двум типам. Эффекты Класса тесно связаны с желательным фармакологическим действием и (в большей или меньшей степени) являются неизбежным следствием воздействия на выбранную молекулярную мишень. Например, вещества, предназначенные для предотвращения патологического воспаления, могут, в некоторой степени, подавлять иммунную систему организма и приводить к повышенному риску развития инфекции. Это происходит потому, что повреждение ткани вследствие воспаления и инфекция зависят от степени активности иммунной системы. В результате все молекулы, которые направлены на одну и ту же фармакологическую мишень, будут в меньшей или большей степени обладать сходными эффектами класса. Напротив, Специфичные эффекты связаны с особенной структурой соединения и обусловлены результатом взаимодействия (обычно непредвиденного) с мишенью, отличной от предполагаемой фармакологической цели. В принципе, возможно найти другую молекулу, обладающую таким же предполагаемым фармакологическим действием, но полностью лишенную специфичного побочного действия. Некоторые специфичные активности соединений являются общими (такие, как взаимодействие со hERG калиевыми каналами сердца, которое может привести к опасной пролонгации QT-интервала во время кардиостимуляции, которая приводит к потенциально фатальной сердечной аритмии), в то время как другие специфичные активности могут, по всей видимости, быть уникальными для каждого вещества.
Важно то, что, несмотря на десятилетия накопленного опыта в области разработки фармацевтических агентов, до сих пор не существует общепринятого способа предсказания, как фармакокинетических, так и токсикологических свойств агента или его фармакологической безопасности. По этой причине проведение исследований с применением модельных систем in vitro (таких, как клеточные линии, экспрессирующие hERG), затем испытания на животных и, наконец, проведение фазы I клинических исследований на человеке являются распространенным по всему миру нормативным требованием при разработке нового фармацевтического препарата.
Были описаны способы для предсказания определенных фармакокинетических свойств на основании изучения структуры молекулы и очевидно, что квалифицированные специалисты в области медицинской химии могут правильным образом исключить многие соединения, руководствуясь только теоретическими соображениями. Примером такого "эмпирического правила" является предложенное Липински "правило пяти", основанное на том наблюдении, что большинство одобренных фармацевтических препаратов отвечают определенным критериям, к которым относятся молекулярный вес, число связей, вокруг которых возможно вращение, и полярность. Аналогичным образом, хорошо известно, что молекулы с крупными гидрофобными группами с большей вероятностью окажут нежелательное действие на hERG калиевые каналы.
Такие общие предписания, даже при одновременном применении, могут быть полезными для исключения неподходящих молекул, но большое количество очень неподходящих (по разным причинам) молекул, тем не менее, не сможет быть отсеяно. В настоящее время никто не будет выбирать вероятное лекарственное вещество из класса активных соединений только на основании теоретических соображений. Поэтому выбор определенного соединения из класса веществ, которые характеризуются благоприятными ADME, фармакокинетическими и токсикологическими свойствами, а также свойствами фармакологической безопасности, требует проведения обширной практической экспериментальной работы среди перспективных соединений, обеспечивающей новую информацию, которая не может быть предсказана даже специалистами в данной области.
В данной заявке авторы настоящего изобретения описывают новое соединение - 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-он (I), которое ранее не было описано:
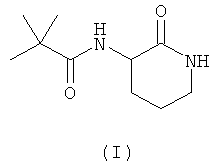
Данное соединение принадлежит к обширному классу ингибиторов хемокинов широкого спектра действия (BSCl), которые были описаны ранее (см., например, WO 2006/134385). Однако авторы настоящего изобретения при экспериментальном сравнении с другими веществами, принадлежащими к тому же классу соединений, показали, что в то время как все молекулы, принадлежащие к указанному классу, обладают BSCl активностью, соединения формулы (I) обладают значительными преимуществами при применении в качестве фармацевтического препарата для человека в результате сочетания их ADME, фармакокинетических и токсикологических свойств и параметров фармакологической безопасности.
Атом углерода в положении 3 лактамного кольца является ассиметричным, и, следовательно, данные соединения в соответствии с настоящим изобретением могут существовать в виде двух различных форм, которые представляют собой "R" и "S" конфигурации соединений. Данное изобретение охватывает обе указанные энантиомерные формы и все комбинации этих форм, включая рацемические "RS" смеси. Для простоты, когда в структурной формуле не отражено никакой специфической конфигурации, следует понимать, что представлены две указанные различные энантиомерные формы и их смеси. Поскольку структурные различия между энантиомерами не оказывают никакого влияния на ключевые ADME свойства, которые определяют преимущества соединений согласно настоящему изобретению (и, кроме того, оказывают только незначительное действие на активность исследуемого соединения в качестве BSCl), обе энантиомерные формы, а также их смеси представляют собой конкретные примеры, которые обладают существенными преимуществами по отношению к другим представителям указанного класса соединений.
Предпочтительно соединение согласно настоящему изобретению, описываемое формулой (I), представляет собой соединение формулы (I')
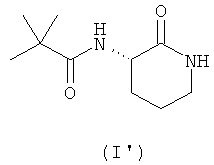
Данное соединение (I'), имеющее S-конфигурацию стереоцентра, является в 5-25 раз более активным в качестве ингибитора хемокинов широкого спектра действия, чем R-энантиомер.
Настоящее изобретение также обеспечивает фармацевтические композиции, которые в качестве действующего вещества содержат соединение, описываемое общей формулой (I) или (I'), или фармацевтически приемлемую соль данного соединения и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и/или носитель.
Под фармацевтически приемлемой солью понимают, в частности, соли, полученные присоединением неорганических кислот, такие как гидрохлориды, гидробромиды, йодгидраты, сульфаты, фосфаты, дифосфаты и нитраты, или органических кислот, такие как ацетаты, малеаты, фумараты, тартараты, сукцинаты, цитраты, лактаты, метансульфонаты, р-толуенсульфонаты, пальмоаты и стеараты. Также в объем настоящего изобретения, когда они могут быть использованы, входят соли, образованные основаниями, такими как гидроксид натрия или гидроксид калия. Другие примеры фармацевтически приемлемых солей представлены в работе: "Salt selection for basic drugs", Int. J. Pharm. (1986), 33: 201-217.
Фармацевтическая композиция может быть представлена в твердой лекарственной форме, например в виде порошков, гранул, таблеток, желатиновых капсул, липосом или суппозиториев. В качестве подходящих твердых наполнителей могут быть использованы, например, фосфат кальция, стеарат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, натрия карбоксиметилцеллюлоза, поливинилпирролидон и воск. Другие фармацевтически приемлемые вспомогательные вещества и/или наполнители известны специалистам в данной области.
Данные фармацевтические композиции в соответствии с данным изобретением также могут быть представлены в жидкой форме, например в виде растворов, эмульсий, суспензий или сиропов. В качестве подходящих жидких наполнителей можно применять воду, органические растворители, такие как глицерин или гликоли, а также их смеси с водой, приготовленные в разных пропорциях.
Данное изобретение также охватывает применение соединения формулы (I) или (I') или фармацевтически приемлемой соли указанного соединения для приготовления лекарственного средства, предназначенного для лечения воспалительного состояния.
Настоящее изобретение включает соединения, композиции и их применение, как определено в данном описании, в случае, когда указанное соединение находится в гидратированной или сольватированной форме.
Преимущество настоящего изобретения по сравнению с уровнем техники основано на том, что неожиданно оказалось, что 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-он обладает лучшими ADME свойствами по сравнению с общими классами лактамных BSCl, которые были описаны ранее (см., например, международные заявки, перечисленные выше). Хотя было известно, что подобные соединения обладают фармацевтически приемлемыми фармакодинамическими свойствами (что означает, что они обладают сильным противовоспалительным действием in vivo как результат их BSCl активности), и было сделано предположение, что одни должны обладать приемлемыми фармакокинетическими и, следовательно, ADME-свойствами, прямая оценка ADME-свойств неожиданно показала, что 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-он обладает значительными преимуществами (см. примеры, приведенные ниже).
В частности, хотя, как следует из литературных данных (см., например, Fox et al. J. Med. Chem. 2005 48:867-74), предшествующие исследования стабильности в сыворотке крови in vitro указывали на то, что лактамные BSCl были значительно лучше, чем ранее описанные имидные BSCl (см., например, WO 99/12968), теперь ясно, что многие (или, возможно, все) представители класса лактамных BSCl подвергаются нежелательному воздействию метаболических реакций in vivo. Авторы настоящего изобретения синтезировали и протестировали более двенадцати BSCl, принадлежащих к аминолактамному классу, и показали, что все протестированные к настоящему времени лактамные BSCl, кроме 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-она, подвергаются быстрому метаболическому превращению в печени (гидроксилирование при действии цитохрома Р450, и/или реакции фазы II метаболизма).
Как минимум частично вследствие сниженного метаболизма in vivo, общий клиренс 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-она является заметно более низким, чем общий клиренс других протестированных лактамных BSCl. В результате воздействие 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-она после введения единичной оральной дозы более чем в 10 раз выше. Таким образом, 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-он является более пригодным для применения в качестве фармацевтического препарата для применения у человека, чем большинство (если не все) ранее описанные лактамные BSCl, особенно в том случае, если для достижения эффективного воздействия требуется постоянное оральное применение.
Пептиды, известные из уровня техники (такие как NR58-3.14.3), характеризуются следующими недостатками: (а) высокая стоимость и необходимость проведения твердофазного синтеза (как минимум для более длинных молекул), (b) быстрое выведение через почки и (с) в целом, значительно меньшая активность (более чем в 25 раз менее сильное действие in vitro и более чем в 10000 раз in vivo).
Известные из уровня техники аминоглутаримиды являются более дешевыми, не выводятся быстро через почки и оказывают более сильное действие in vitro, НО они чрезвычайно нестабильны в сыворотке крови (как результат открывания имидного кольца под действием ферментов; см., например, Fox et al. J. Med. Chem. 2005 48:867-74). Поэтому действие аминоглутаримидных BSCl, таких как S-3-(ундек-10-еноиламино) глутаримид, как минимум в 250 раз менее сильное in vivo, чем действие 2',2'-диметилпропаноиламино)-тетрагидропиридин-2-она, даже при применении в моделях острого воспаления (таких, как эндотоксикоз, индуцированный липополисахаридами с характерной системной продукцией TNF-α, где влияние стабильности соединения и ADME свойств менее выражено).
Другие структурно родственные (но функционально сильно различающихся) соединения, которые описаны в литературных источниках, представляют собой бактериальные автоиндукторы, и, как правило, в основе их структуры лежит 6-членный гомосеринлактон, обычно с 3-оксоацильной боковой цепью (см., например, публикацию Bycroft et al. US 5,969,158, в которой заявлен широкий спектр таких веществ). Несмотря на то что подобные публикации раскрывают общие формулы, которые охватывают как лактамы и лактоны, только небольшое число примеров описанных соединений (если вообще такие имеются), обладающих свойствами автоиндукторов бактерий, имеют лактамные концевые группы. Известно, что все подобные соединения (в особенности те, которые имеют концевую лактонную и/или 3-оксоацильную группу) относительно нестабильны, что ограничивает возможности их применения в качестве медицинских препаратов.
Преимущества 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-она, описанные в данном документе, состоят в том, что синтез данного соединения является дешевым (и приведенный в данном описании способ позволяет осуществлять прямой синтез даже в количествах, измеряемых килограммами), и в том, что данное соединение характеризуется высокой метаболической стабильностью не только в изолированной сыворотке крови in vitro (свойство, которое относится ко всему классу описанных ранее лактамных BSCl), но также in vivo. Исходя из вышесказанного, соединения согласно настоящему изобретению (по сравнению с соединениями, широко исследованными к настоящему времени), по эффективности, силе воздействия и фармацевтическим свойствам, таким как ADME, фармакокинетическим, токсикологическим свойствам и параметрам фармакологической безопасности, являются уникальным образом оптимизированными для лечения воспалительных заболеваний у человека.
В соответствии с настоящим изобретением воспалительные нарушения, которые можно предотвращать или лечить с применением соединений, описываемых формулами (I) или (I'), или фармацевтически приемлемых солей указанных соединений, или фармацевтических композиций, содержащих данные соединения в качестве активных компонентов, включают следующие заболевания:
- аутоиммунные заболевания, например, такие, как рассеянный склероз, ревматоидный артрит, обыкновенная волчанка, синдром раздраженного кишечника, болезнь Крона;
- заболевания сосудов, включая нарушения мозгового кровообращения (инсульт), ишемическую болезнь сердца, инфаркт миокарда, нестабильную стенокардию, атеросклероз или воспаление кровеносных сосудов, например синдром Бехчета, гигантоклеточный артериит, ревматическую полимиалгию, гранулематоз Вегенера, синдром Черджа-Стросс, пурпуру Геноха-Шенлейна и болезнь Кавасаки;
- вирусную инфекцию или репликацию вируса, например инфекции, вызываемые вирусами или размножением вирусов, включая поксвирус, вирус герпеса (например, Herpesvirus samiri), цитомегаловирус (ЦМВ), вирусы гепатита или лентивирусы (включая ВИЧ);
- астму и сопутствующие нарушения со стороны дыхательной системы, такие как аллергический ринит и хроническое неспецифическое заболевание легких;
- остеопороз (низкая плотность минерального вещества кости);
- рост опухоли;
- отторжение трансплантированных органов и/или задержку в функционировании трансплантата или органа, например, у пациентов, подвергнувшихся трансплантации почки;
- нарушения, характеризующиеся повышением уровня TNF-α;
- псориаз;
- повреждения кожи и другие фиброзные нарушения, включающие гипертрофические рубцы (келоидные образования), формирование спаек после общей или гинекологической хирургии, фиброз легкого, фиброз печени (включая алкогольную болезнь печени) или фиброз почки, неясной этиологии или получившие развитие как следствие основной болезни, такой как диабет (диабетическая нефропатия);
- нарушения, вызванные внутриклеточными паразитами, такие как малярия и туберкулез;
- нейропатическая боль (такие, как постоперативные фантомные боли в конечностях, постгерпетическая невралгия и другие);
- аллергические реакции или
- болезнь Альцгеймера.
В соответствие с настоящим изобретением к прочим воспалительным нарушениям относятся:
- боковой амиотрофический склероз;
- ответная реакция, вызванная присутствием антигенов; подавление иммунного ответа.
Данные клинические показания в широком понимании относятся к воспалительным нарушениям или нарушениям, характеризующихся повышением уровня TNF-α.
Для того чтобы исключить неправильное толкование, следует отметить, что первичной мишенью действия BSCl, включая соединения, описанные в данной заявке, является иммунная система. Следовательно, заявленные благоприятное воздействие на заболевания, такие как вирусная инфекция и/или репликация вируса и рост опухоли (состояния, которые сами по себе не являются первичными заболевания иммунной системы), является следствием модулирования иммунной системы на фоне инфекции и/или репликации вируса или роста и распространения опухоли. Поскольку BSCl, включая соединения согласно настоящему изобретению, в целом, прямым образом не влияют на репликацию вирусов или рост опухоли, следует ожидать, что они не будут оказывать никакого воздействия в изолированной системе (например, на инфицированную in vitro клеточную линию или на линию пролиферирующих опухолевых клеток), где отсутствует целостная и функционирующая иммунная система. Следовательно, известные данные, имеющие отношение к действию любых соединений в таких изолированных системах, не могут предоставить сведения для разработки BSCl, оказывающих действие на иммунную систему.
В том случае, если это разрешено законом, настоящее изобретение также можно применять для лечения, улучшения состояния или профилактики симптомов воспалительного заболевания (включая неблагоприятную воспалительную реакцию, которая возникла в ответ на любой агента) путем введения пациенту соединения, композиции или медикамента согласно настоящему изобретению в количестве, обеспечивающем противовоспалительное действие.
Введение медикамента в соответствии с настоящим изобретением можно осуществлять локальным, оральным, парентеральным путями, посредством внутримышечной инъекции и другими способами.
Вводимая доза, предполагаемая для лекарственного средства в соответствии с настоящим изобретением, составляет от 0,1 мг до 10 г в зависимости от состава препарата и способа введения.
В соответствии с настоящим изобретением соединения общей формулы (I) или (I') можно синтезировать с применением процессов, описание которых приведено ниже.
Получение соединений общей формулы (I) или (I')
Все соединения общей формулы (I) или (I') можно легко получить с применением общих способов, известных специалистам в данной области.
Соединение (I) представляет собой бесцветное кристаллическое вещество, которое может быть получено из орнитина и 2,2-диметилпропионилхлорида. Для синтеза соединения (I') используют чистый энантиомер S-орнитин. Можно осуществить замыкание кольца при воздействии на орнитин или его метиловый эфир. Данная аминокислота может быть этерифицирована в сухом метиловом спирте при in situ образовании хлористоводородной кислоты с применением триметилсилилхлорида. Альтернативно, можно осуществить замыкание кольца изолированного эфира с применением триэтиламина. Полученный неочищенный продукт можно затем ацилировать после обмена растворителя.
Ацил-лактамный продукт (I) характеризуется значительной растворимостью в воде, и вследствие этого условия ацилирования, используемые для более гидрофобных родственных продуктов (см., например, WO 2006/134385), были неудовлетворительными. Применение трех эквивалентов натрия карбоната в качестве основания привело к значительной преципитации побочного продукта натрия гидрокарбоната, несмотря на то, что использовали большое количество воды (более 4 мл/ммоль орнитина). При использовании данных концентраций экстракция продукта дихлорметаном является неэффективной. Поэтому три эквивалента натрия карбоната заменили на 2,5 эквивалента калия гидроксида (который нейтрализует 2,5 эквивалента триэтиламина гидрохлорида, получаемого на этапе замыкания кольца). С применением данного основания можно использовать значительно меньшее количество воды (менее 1 мл/ммоль орнитина) (конечное значение pH составляет от 8 до 9). При экстракции водного слоя этиловым эфиром уксусной кислоты (3×2 мл/ммоль орнитина) и повторной кристаллизации из этилацетата (0,5 мл/ммоль, горячий) и 40-60 петролейного эфира (5 мл/ммоль) первичный выход продукта составил 43% (4,25 г из 50 ммоль орнитина).
Следует отметить, что если во время выделения продукта реакции значение pH водного слоя слишком низкое, то в слой этилового эфира уксусной кислоты экстрагируются небольшие количества триэтиламина гидрохлорида. При промывании данного раствора этилацетата водой происходит экстракция значительного количества лактамного продукта (I) вместе с амина гидрохлоридом. Этого можно избежать повышением pH водного слоя до 12 (например, добавлением приблизительно одного эквивалента калия гидроксида по отношению к кислому хлориду) до проведения экстракции этиловым эфиром уксусной кислоты, тогда вместе с лактамным продуктом (I) экстрагируется только свободное основание триэтиламина, которое будет легче удалить путем выпаривания или в процессе повторной кристаллизации.
Ниже представлен следующий предпочтительный путь синтеза:
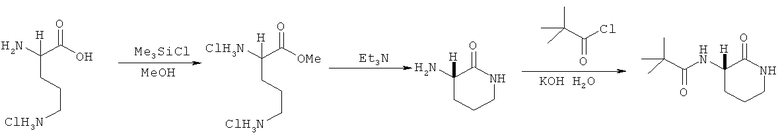
S-орнитина моногидрохлорид (50 ммоль) суспендируют в 100% метиловом спирте (100 мл) при добавлении триметилсилила хлорида (75 ммоль). Реакционную смесь нагревают в колбе с обратным холодильником в течение 24 часов. Затем добавляют триэтиламин (150 ммоль) и полученную смесь нагревают в колбе с обратным холодильником в течение 48 часов. Метиловый спирт затем удаляют при пониженном давлении (при желании можно на поздних стадиях добавить толуол, для того чтобы облегчить удаление спирта), полученный осадок растворяют в воде (20 мл) с добавлением калия гидроксида (125 ммоль).
Полученную смесь охлаждают до 0°С, затем медленно добавляют 2,2-диметилпропионилхлорид (50 ммоль) и реакционную смесь перемешивают в течение 18 часов при нагревании до комнатной температуры. Затем добавляют твердый гидроксид калия (50 ммоль) и после того, как он растворится, реакционную смесь экстрагируют этиловым эфиром уксусной кислоты (3×100 мл). Комбинированные органические слои быстро высушивают над смесью К2СО3 и Na2SO4 при пониженном давлении. Полученный твердый осадок затем подвергают повторной кристаллизации этиловым эфиром уксусной кислоты (25 мл) / 40-60 петролейного эфира (200-250 мл), чтобы получить лактам (I') в виде кристаллического твердого вещества (выход составляет более 50%).
Подлинность и чистоту (более 95%) данного продукта затем подтверждали способом протонной ЯМР-спектроскопии (δН (400 МГц, CDCl3) 6,63 (1Н, br s, NH); 6,01 (1Н, br s, NH); 4,20 (1Н, dt, J 11, 5.5, CHNH); 3,40-3,31 (2H, m, CH2NH); 2,61 (1H, dq, J 13, 4.5, CH2); 1,97-1,88 (2H, m, CH2); 1,50 (1H, dddd, J 13, 12, 9.5, 7.5, CH2); 1,22 (9H, s, 3×CH3).
ОПРЕДЕЛЕНИЯ
Термин "приблизительно" относится к промежутку вокруг рассматриваемого значения. Как использовано в приложении к данному описанию, "приблизительно X" означает интервал от значения X минус 10% от значения X до X плюс 10% от значения X и предпочтительно интервал от X минус 5% от значения X до X плюс 5% от значения X.
Использование числовых значений в данном описании направлено на то, чтобы однозначно включить в объем данного описания все индивидуальные значения в заданном диапазоне и все комбинации верхних и нижних предельных значений в границах данного интервала. Соответственно, например, промежуток от 0,1 мг до 10 г, определенный в отношении (inter alia) дозы 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-она, предназначенной для применения, введен для того, чтобы включить все дозы между 0,1 мг и 10 г и все более узкие интервалы, определяемые каждой комбинацией верхних и нижних предельных значений, вне зависимости от того, приведены примеры явным образом или нет.
В данном документе, термин "включает/содержит" следует понимать как "включающий" и как "состоящий из". Следовательно, если изобретение относится к "фармацевтической композиции, которая включает/содержит активный компонент", данная терминология предназначена для того, чтобы описать как составы, в которых могут присутствовать другие активные компоненты, так и составы, которые состоят только из одного активного компонента, как определено.
Если не определено иное, все технические и научные термины, использованные при составлении данного документа, имеют то же значение, которое обычно понимается обычным специалистом в области, к которой относится изобретение. Так же все публикации, приложения к патентам, все патенты и все прочие ссылки, упомянутые в тексте, включены в данное описание посредством ссылок (в тех случаях, когда это дозволено законом).
Следующие примеры представлены для того, чтобы проиллюстрировать вышеописанные процедуры и никаким образом не ограничивают сферу применения данного изобретения.
ФИГУРЫ
На Фиг.1 (части А-Е) представлены графики зависимости концентрации от времени для пяти исследуемых соединений от (I') до (V) после орального введения крысам единичной дозы 3 мг/кг в 1% растворе карбоксиметилцеллюлозы. Три линии для каждого соединения соответствуют трем повторениям опыта (три экспериментальных животных). На оси Y представлены концентрации в единицах измерения нг/мл (0-3000); на оси X представлено время в минутах (0-480).
На Фиг.2 показаны основные метаболиты, идентифицированные с применением способа жидкостной хроматографии/тандемной масс-спектрометрии (при сканировании в полном диапазоне) в моче, которую собирали через 24 часа у крыс, получивших при оральном введении единичной дозы в количестве 3 мг/кг в 1% растворе карбоксиметилцеллюлозы каждого из пяти исследуемых соединений от (I') до (V). Примечательно, что основной метаболит соединения (IV) был идентифицирован, но его структура не была определена при сравнении с паттернами фрагментации/перестройки в общедоступных базах данных по метаболитам. Хотя концентрации отдельных метаболитов не были определены, приведен порядок их количественного содержания в моче, в левой части каждого ряда приведены виды метаболитов, присутствующие в наибольшем количестве.
На Фиг.3 (части А-F) представлены графики зависимости тока от времени для клеток, экспрессирующих продукт гена hERG при воздействии носителя или одного из пяти соединений от (I') до (V), каждого в отдельном эксперименте. В каждом эксперименте дополнительные клетки подвергали действию соединения, взятого в качестве положительного контроля, которое полностью блокирует ток, проводимый по каналам hERG. На оси Y представлен ток в нА; по оси X представлено время в секундах. В части рисунка G показан hERG следовой ток (площадь под кривой зависимости тока от времени в частях рисунка А-F) при повторении опыта на клетках, подвергнутых действию каждого соединения или только 0,1% ДМСО в качестве носителя, или соединения, взятого в качестве положительного контроля, Е-4031 (+ носитель). В части G рисунка на оси Y гистограммы представлено в процентах ингибирование следового тока hERG по отношению к клеткам, не подвергнутым воздействию.
На Фиг.4 показана типичная кривая связывания соединения (I'). В данном эксперименте в каждую реакцию добавляли 10 нМ меченого соединения BN83250 (лактамный BSCl, который связывается с теми же рецепторами, что и соединение (I')), вместе с разными конкурирующими веществами (100 мкМ - 1 пМ). Общее специфическое связывание определяли как количество, вытесненное большим избытком (100 мкМ). Каждый столбец на графике представляет собой среднее из трех повторных определений, усы отражают стандартную ошибку среднего. На оси Y представлена радиоактивность, связавшаяся в каждом эксперименте в единицах импульсов в минуту (имп./мин). Верхняя пунктирная линия показывает общее связывание в условиях экспериментах, в то время как нижняя пунктирная линия показывает неспецифическое связывание; расстояния между пунктирными линиями показывает специфическое связывание.
На Фиг.5 показан профиль перекрестной реактивности для каждого из пяти соединений от (I') до (V) (части А: (II); В: (III); С: (V); D: (I') и Е: (IV)) против набора из 75 различных рецепторов, которые составляют набор CEREP (см. текст). Данные соединения были протестированы в единичной концентрации (10 мкМ), и описано ингибирование связывания (ось Y каждой гистограммы) для известного лиганда с каждым из 75 рецепторов (-100%, таким образом, представляет собой 2-кратное повышение связывания специфического лиганда в присутствии исследуемого соединения). Все реакции проводили в двух повторениях, и столбцы на графике представляют собой среднее значение (для упрощения графиков ошибка между повторениями не показана). При проведении данного теста связывание считали статистически и биологически значимым только при 50% ингибирования (или стимулирования) или выше (что соответствует значению средней эффективной дозы ниже 10 мкМ для взаимодействия исследуемого соединения с отдельным рецептором). Для пяти данных соединений, исследуемых в данной заявке, только в одном случае взаимодействие (взаимодействие соединения (II) с рецептором NK2, отмеченное стрелкой в части А рисунка) может быть потенциально значимым, хотя даже это взаимодействие было слабым (оценка значения средней эффективной дозы составила 5-10 мкМ).
На Фиг.6 (части А-Е) показаны репрезентативные кривые дозовой зависимости ингибирования хемокин-индуцированной миграции лейкоцитов in vitro, для каждого из указанных пяти соединений от (I') до (V) при проведении анализа по миграции клеток между лунками в системе ChemoTx™. В каждом эксперименте индуцировали миграцию клеток ТНР-1 с применением максимально эффективной дозы хемокина МСР-1 в присутствии или в отсутствие различных доз (от 10 пМ до 1 мкМ) каждого соединения. При проведении каждого эксперимента использовали надлежащий носитель в качестве контроля. В процентном отношении ингибирование миграции клеток индуцированного МСР-1 (определенное как число клеток, мигрировавших в присутствии МСР-1, минус число клеток, мигрировавших в отсутствие МСР-1 в нижней камере) при каждой концентрации каждого тестируемого соединения показано как среднее значение при трех повторениях опыта, усы на графике показывают стандартную ошибку среднего. Значение средней эффективной дозы оценивали путем линейной интерполяции представленных графиков. На оси Y каждого графика показано процентное отношение ингибирования миграции клеток, индуцированного МСР-1; на оси X показана концентрация присутствующего тестируемого соединения в нМ (0,01-1000).
На Фиг.7 показаны репрезентативные кривые дозовой зависимости ингибирования индуцированной липополисахаридами продукции TNF-α in vivo в модели сублетального эндотоксикоза на мышах. В каждом эксперименте группы из шести мышей получали предварительное лечение разными дозами пяти соединений (часть А: (II); В: (III); С: (V); D: (I') и Е: (IV)) орально (кружки) или подкожно (треугольники). Через 30-60 минут животным вводили липополисахариды внутрибрюшинно и через 3 часа из цельной крови приготавливали сыворотку. Концентрацию TNF-α в крови измеряли с применением способа иммуноферментного анализа, и степень ингибирования индуцированного липополисахаридами синтеза TNF-α (определяемой как концентрация TNF-α у мышей, подвергнутых действию липополисахаридов, минус концентрация TNF-α у мышей, которым в качестве контроля вводили физиологический раствор с фосфатно-солевым буфером, не содержащий эндотоксинов) показана на оси Y каждого графика как среднее значение по шести животным, усы показывают стандартную ошибку среднего. Концентрация TNF-α у мышей, которым вводили липополисахариды, но которые не получали лечение BSCl, составляла в среднем от 5000 до 6000 пг/мл (по сравнению с менее чем 10 пг/мл мышей, не подвергнутых воздействию). Среднюю эффективную дозу оценивали путем линейной интерполяции представленных графиков. На оси X каждого графика показаны дозы каждого соединения, которое вводили каждой мыши в группе в мг (1Е-07 до 1).
На Фиг.8 показано действие соединения (I') при воспалении легких в репрезентативном эксперименте, которое оценивали по числу клеток в бронхеоальвеолярной жидкости в модели астмы на грызунах. Столбцы показывают среднее число клеток, показанное на оси Y в единицах 106 клеток для групп из пяти животных, усы показывают стандартную ошибку среднего; (р<0,01 против "сенситизированных" с применением непарного критерия Стьюдента для одной выборки в предположении симметричного распределения с одинаковыми вариациями). Горизонтальная линия показывает среднее число лейкоцитов, присутствующих в бронхоальвеолярной жидкости из легких крыс, не подвергнутых указанному воздействию. Всех крыс в оставшихся трех группах подвергали одинаковой сенситизации и режиму воздействия, но при этом либо были подвергнуты воздействию только носителя ("сенситизированные"), либо воздействию соединения (I') в количестве 0,3 мг/кг веса тела или монтелукаста ('Singulair™') в количестве 30 мг/кг веса тела; во всех случаях введение осуществляли ежедневно оральным путем.
На Фиг.9 показано действие соединения (I') на поляризацию Т-хелперных клеток в репрезентативном эксперименте, которую оценивали способом проточной цитометрии по продукции IFN-γ (маркерный цитокин для Th1) и продукции IL-4 (маркерный цитокин Th2) CD4+ - спленоцитами в модели астмы на крысах. Полосы на графике представляют среднее значение отношения Th1/Th2, показанные на оси Y для групп из 10 животных, усы показывают стандартную ошибку среднего; (р<0,05 против необработанных крыс; † <0,05 против сенситизированных крыс и крыс, получавших препарат, в обоих случаях для оценки равных дисперсий применяли t-критерий Стьюдента для одной выборки в предположении симметричного распределения с одинаковыми вариациями). Всех крыс (кроме группы "BN-крыс", которые не получали овальбумин) подвергали такой же сенситизации и режиму применения препарата, но либо давали только носитель ("сенситизированные + носитель"), или носитель вместе с соединением (I') в количестве 0,3 мг/кг веса тела или вместе с монтелукастом ("Singulair™") в количестве 30 мг/кг веса тела; все варианты вводили ежедневно орально через зонд.
На Фиг.10 представлен график зависимости периода полувыведения из плазмы крови (в минутах) от воздействия при оральном применении для ряда разных BSCl соединений у крыс. В каждом случае оценку времени полувыведения проводили с применением стандартной однокамерной модели концентраций в плазме через 0; 0,25; 0,5; 1; 2; 4; 6 и 8 часов после внутривенного введения единичной болюсной дозы соединения в количестве 1 мг/кг растворенного в 1% растворе карбоксиметилцеллюлозы, 0,9% ДМСО в физиологическом растворе. Воздействие при оральном применении (AUC 0-t в мин·нг/мл) было определено после введения единичной оральной дозы соединения в количестве 3 мг/кг растворенной в 1% растворе карбоксиметилцеллюлозы, 0,9% ДМСО в физиологическом растворе. R-энантиомер соединения (I), который является инверсией соединения (I') и идентифицирован как "R-(I)", отмечен пустым кружком.
ПРИМЕРЫ
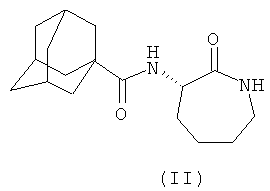
В каждом из следующих примеров от 1 до 6 проводили сравнение S-3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-она (соединение I') с рядом других лактамных BSCl, которые были выбраны так, чтобы быть репрезентативными для различных подклассов. Например, 3-(адамантан-1-карбониламино)-капролактам (II) был выбран в качестве типичного представителя подкласса полициклоациллактамных BSCl (таких, как ранее описанные в WO 2006/016152)
Аналогично, 3-(1'-метилциклогексилкарбониламино)-капролактам (III) был выбран в качестве типичного представителя подкласса моноциклоацил лактам BSCl (таких, как описанные ранее WO 2006/134384)
Соединение 3-(1',1'-диметилэтилсульфониламино)-капролактам (IV) было выбрано в качестве типичного представителя подкласса BSCl с простыми (нециклическими) алкильными боковыми цепями (такими, как описанные ранее в WO 2005/053702), так же как соединения с сульфониламинным линкером (в противоположность карбамидному линкеру в оставшихся выбранных соединениях)
Последнее выбранное соединение 3-(3'-гидроксиадамантил-1-карбониламино)-капролактам (V) было типичным для BSCl с замещенной ацильной боковой цепью (простой линейной, разветвленной, моно- или полицикличной структурой)
Важно отметить, что все данные BSCl соединения от (II) до (V) были непосредственно описаны ранее, и все обладают сильным BSCl действием in vitro (средняя эффективная доза составила менее 1 нМ при ингибировании миграции ТНР-1 клеток, вызванной МСР-1). Все соединения характеризуются отличной стабильностью в сыворотке крови, in vitro и в перспективе теоретически являются замечательными веществами-кандидатами для развития фармацевтических препаратов, обладающих противовоспалительными свойствами, для использования у человека in vivo.
Все описанные выше соединения (I', II, III, IV и V) были протестированы в S-конфигурации лактамного стереоцентра.
Пример 1 Фармакокинетические параметры после введения единичной дозы
Соединения вводили в единичной дозе (в количестве 1 мг/кг веса тела в 5% ДМСО внутривенно или в количестве 3 мг/кг в 1% растворе карбоксиметилцеллюлозы перорально) трем взрослым крысам (с применением разных животных для каждого соединения и каждого способа введения).
Затем отбирали образцы крови в разные моменты времени (в том числе непосредственно перед введением дозы) до 24 часов после введения дозы и оценивали уровень различных соединений с применением валидированного анализа способом жидкостной хроматографии/тандемной масс-спектрометрии. Вкратце, 3-5 мкл депротеинизированного образца наносили на колонку для хроматографии в обращенной фазе Waters Atlantis (С18 20×2,1 мм, размер гранул 3 мкм), уравновешенную 0,1% раствором метановой кислоты в смеси воды:ацетонитрила в соотношении 95:5. После 3.5 минут связавшийся материал подвергали градиентной элюции 0,1% раствором метановой кислоты в смеси вода:ацетонитрил в соотношении 5:95, затем пошаговой элюции 0,1% раствором метановой кислоты в смеси вода:ацетонитрил в соотношении 95:5. Полученный элюент затем исследовали способом тандемной масс-спектрометрии с применением масс-спектрометрического детектора Applied Biosystems API 4000/3200 QTrap, снабженного источником ионов Turbolonspray™, действующего в режиме положительно заряженных ионов. Температуру поверхности устанавливали 650°С, время удерживания составило 40 мс для каждого MRM-перехода при мониторинге следующих ионов:
В качестве внутреннего стандарта при каждом определении использовали родственное соединение S-3-(2',2'-диметилпропаноиламино)-капролактам, который вводили в образец до проведения депротеинизации. Значение нижнего предела количественного определения (НПКО) для данного способа анализа составило 2,4 нг/мл для каждого соединения, кроме (I'), для которого нижний предел количественного определения составил 38,1 нг/мл.
После проведения анализа каждого отобранного образца способом жидкостной хроматографии/тандемной масс-спектрометрии моделирование фармакокинетических параметров соединения проводили с применением программного обеспечения Kinetica - хорошо известного пакета программ для решения подобных задач.
Результаты
Графики зависимости отдельных концентраций от времени для каждой крысы, которую подвергали воздействию каждого соединения при оральном введении, показаны на Фиг.1. Очевидно, что из данных пяти различающихся по структуре лактамных BSCl только соединения (IV), (V) и (I') характеризуются каким-либо действием при оральном применении и что из этих соединений (I') значительно лучше, чем другие.
Параметры простой однокамерной фармакокинетической модели представлены в Таблице 1. Во-первых, данные параметры показывают, что воздействие соединения (I') приблизительно в 20 раз сильнее, чем следующего соединения с наилучшими показателями - (V). Причина указанного лучшего воздействия (которое определяли как площадь под кривой зависимости концентрации от времени в мин·нг/мл) также очевидна: клиренс соединения (I'), который определяли как теоретический объем крови, который полностью очищается от лекарственного вещества каждую минуту в мл/мин/кг, более чем в 10 раз ниже, чем у других лактамных BSCl.
Таблица 1. Фармакокинетические параметры отличающихся по структуре лактамных BSCl. Биодоступность при оральном применении (биодоступность, %), основной период полувыведения из плазмы (t1/2, минуты), клиренс (мл/мин/кг), объем распределения (объем распределения в равновесном состоянии, л/кг) и воздействие (AUC 0→∞, мин·нг/мл) при простой однокамерной фармакокинетической модели; для каждого соединения усреднены значения, полученные для трех крыс. * 24 минуты - это основной период полувыведения соединения (V), когда клиренс составил более 95% введенной дозы; второстепенное значение t1/2 составило 110 минут. Во всех случаях значение Cmax было достигнуто в течение 30 минут в соответствии с оптимальной абсорбцией.
Клиренс соединений (II) и (III) приближается к скорости потока крови в печени крысы, что убедительно свидетельствует в пользу того, что оба данных соединения подвергаются практически полному метаболическому превращению при первом прохождении через печень. Аналогично, клиренс обоих соединений, (IV) и (V), превышает скорость потока крови в почках у крысы приблизительно в 3-4 раза, что снова свидетельствует в пользу того, что предположительно основная часть метаболически обусловленного клиренса также осуществляется в печени. Напротив, клиренс соединения (I') составляет 2,6 мл/мин/кг, что меньше половины скорости потока крови через почки (обычно сообщают о 7-9 мл/мин/кг), что позволяет предположить минимальное влияние метаболического клиренса. Поскольку соединение (I') исключительно хорошо растворяется в воде, значение объема распределения соответствует равновесному значению свободного распределения во всем объеме воды, содержащейся в теле (0,7 л/кг), похоже, что значение клиренса ниже скорости потока крови через почки представляет собой повторную абсорбцию с водой в дистальных почечных канальцах (более вероятное объяснение, чем, например, сниженное воздействие на почки по причине изоляции соединения в липофильных компартментах).
В соответствии со значительно более низким значением клиренса соединения (I') по сравнению с другими соединениями соединение (I') характеризуется значительно более длительным периодом полувыведения из плазмы (более 3 часов для (I') и менее 30 минут для остальных четырех соединений).
Не все BSCl характеризуются биодоступностью при оральном применении, даже при том, что все пять выбранных соединений обладают биоактивностью при оральном применении в конечной стадии острого воспаления. Скорее всего, это отражает быстрое метаболическое превращение соединений (II) и (III) в печени, которые эффективно абсорбируются, но при первом прохождении через печень превращаются в метаболиты, которые сохраняют некоторую активность в качестве BSCl (см. пример 2).
На основании фармакокинетического анализа для специалистов в данной области очевидно, что, несмотря на сходную химическую стабильность и стабильность in vitro в изолированной сыворотке для данных пяти соединений, так же как и сходство их теоретически предсказанных свойств, соединение (I') существенно превосходит все остальные соединения. В особенности, клиренс данного соединения существенно ниже, что, вероятно, отражает более низкую подверженность данного соединения метаболическим реакциям в печени, что приводит к в 10 раз более длительному периоду полувыведения данного соединения из плазмы и почти в 20 раз лучшему воздействию при оральном применении, чем для следующего лучшего по свойствам исследованного соединения.
При проведении отдельного эксперимента соединение (I') сравнивали со вторым лучшим по свойствам соединением (V) по фармакокинетическим параметрам у другого вида животных (не грызунов) - у собак. Единичные дозы (1 мг/кг в 5% ДМСО внутривенно или 3 мг/кг в 1% растворе карбоксиметилцеллюлозы перорально) вводили одной собаке по одному соединению в соответствии с простым перекрестным планом эксперимента, в котором был предусмотрен период вымывания между двумя способами введения продолжительностью 1 неделя.
Результаты представлены в Таблице 2.
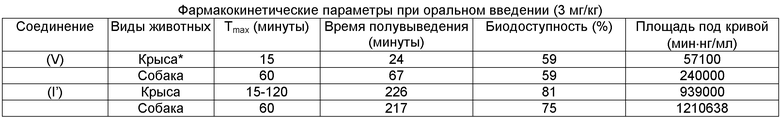
Таблица 2. Сравнение фармакокинетических параметров двух соединений лактамных BSCl с наилучшими свойствами у крыс и собак. Фармакокинетические параметры у собак, в целом, сходны с параметрами у крыс. У обоих видов соединение (I'), характеризующееся значительно более низким клиренсом, более длительным периодом полувыведения из плазмы и, следовательно, большим воздействием (AUC в мин·нг/мл). В каждом случае основной период полувыведения (более быстрый t1/2α) отвечает за выведение более чем 95% введенной дозы.
Результаты данных наблюдений указывают на то, что проявление преимущественных фармакокинетических свойств соединения (I') не является видоспецифичным и, следовательно, скорее всего, аналогичные свойства будут наблюдаться у человека.
Пример 2 Определение первичных метаболитов
У крыс, содержавшихся в метаболических клетках и подвергнутых воздействию единичной оральной дозы (3 мг/кг в 1% растворе карбоксиметилцеллюлозы) каждого соединения, в течение 24 часов собирали мочу. Объединенный образец мочи затем подвергали полному анализу способом масс-спектрометрии с применением тех же условий проведения жидкостной хроматографии/масс-спектрометрии, как указано выше при описании примера 1. Затем проводили дальнейший анализ ионных продукта способом тандемной масс-спектрометрии и оценивали возможную фрагментацию/перестройку соединений по публично доступной базе данных метаболитов.
Для основных метаболитов предсказанные структуры были подтверждены путем синтеза аутентичных образцов с применением хорошо известных в данной области способов, которые затем были подвергнуты анализу способом жидкостной хроматографии/тандемной масс-спектрометрии в тех же условиях, что и образцы мочи.
Заметим, что исследования по определению идентичности метаболитов обеспечивают только количественную оценку относительных количеств различных присутствующих метаболитов и потребуется проведение отдельных количественных анализов с применением валидированных способов и надлежащих внутренних стандартов для того, чтобы определить количество каждого вида метаболитов.
Результаты
На Фиг.2 представлены метаболиты, которые были определены для пяти проанализированных соединений в порядке обнаруженных концентраций. Важно отметить, что данная методология, использованная здесь, не обязательно является исчерпывающей и дополнительные (в особенности второстепенные) метаболиты могут также присутствовать в количествах ниже предела обнаружения использованных при указанном анализе способов. В качестве общего правила можно предположить, что будут обнаружены метаболиты, составляющие 10% или более от введенной дозы (хотя не обязательно структурно идентифицированные использованными здесь способами).
Для соединений (II) и (III) основным путем метаболизма является гидроксилирование, опосредованное цитохромом Р450, что согласуется со значениями быстрого клиренса при скорости, приближающейся к скорости потока крови в печени (см. пример 1 выше). Главной мишенью, гидроксилирования для обоих соединений является циклоалкильная концевая группа, второе (более медленное) гидроксилирование происходит в лактамной концевой группе. Заметим, что продукты гидроксилирования лактамов появлялись при определении способом тандемной масс-спектрометрии при -2 а.е.м. (в противоположность +16 а.е.м.), по причине нестабильности 7-гидроксипроизводных в источнике электрораспыления.
Продукт дигидроксилирования соединения (II) присутствовал в количестве, достаточном для обнаружения в моче, в то время как для соединения (III) не было обнаружено никакого продукта дигидроксилирования. В обоих случаях, похоже, что также формировались дополнительные второстепенные продукты в количествах ниже предела обнаружения использованного способа (например, 3,5-дигидрокси- и 3,5,7-тригидроксиадамантил-производные соединения (II), и глюкуронированные производные как гидрокси-(II), так и гидрокси-(III), особенно поскольку было обнаружено глюкуронированное производное соединения (V)).
Для соединения (V) глюкуронированное производное являлось главным метаболитом, хотя у крыс только второстепенная фракция глюкуроната выводилась с мочой, основная масса при этом выводилась из организма с фекалиями (у человека, напротив, данное глюкуронированное производное будет в первую очередь выводиться с мочой). Возможно, также образовывались другие метаболиты фазы II (такие, как 3'-О-сульфат), но только в количествах (как минимум в моче), слишком низких, чтобы быть детектированными с применением описанных в данной заявке способов. Для соединений (II) и (III) также определили небольшое количество продукта, гидроксилированного по лактамной концевой группе (в основном как ионный продукт - 2 а.е.м.).
Основной метаболит соединения (IV) не мог быть идентифицирован, хотя убыль основного соединения (см. Таблицу 3) ясно соответствовала формированию неидентифицированного метаболита (менее 10% введенной дозы соединения (IV) было обнаружено без изменений). При условии, что соединение (IV) было единственным, содержащим сульфонамидную связь, вероятно, но на данный момент не доказано, что происходило метаболическое расщепление (или другая модификация) данной связи. Также наблюдали небольшое количество продукта гидроксилирования лактамной концевой группы.
В отличие от всех других соединений никаких значимых метаболитов соединения (I') не было обнаружено в моче, что соответствует появлению большего количества введенной дозы в моче в неизменном виде (см. Таблицу 3), и значения скорости клиренса совпадают или ниже значения скорости почечного кровотока (см. пример 1 выше). Отсутствие формирования метаболитов является основным и неожиданным преимуществом соединения (I') по сравнению с другими протестированными здесь соединениями для разработки лекарственного средства для применения у человека и является, как минимум частично, объяснением преимущественных фармакокинетических свойств, приведенных в описании примера 1 выше.
Для того чтобы обеспечить количественную оценку степени метаболических превращений каждого из исследуемых соединений, количество соединений, не претерпевших изменений, измеряли с применением того же валидированного способа анализа путем жидкостной хроматографии/масс-спектрометрии, приведенного в описании примера 1, с применением S-3-(2',2'-диметилпропаноиламино)-капролактама в качестве внутреннего стандарта. Полученные результаты представлены в Таблице 3. Кроме того, определяли концентрацию исследуемых соединений в различных тканях-мишенях.
но = не определено; НПКО = 2,4 нг/г
Таблица 3. Распределение соединений в различных тканях в течение 24 часов после введения единичной дозы у крыс. Только соединения (I'), (V) и (IV) могли быть обнаружены в моче через 24 часа после введения единичной оральной дозы (3 мг/кг в 1% растворе карбоксиметилцеллюлозы). Из них соединение (I') претерпело существенно меньше метаболических превращений (более 60% от введенной дозы было обнаружено в моче). Более того, только соединение (I') можно было обнаружить в других тканях через 24 часа после введения единичной оральной дозы. Полученные данные, скорее всего, отражают преимущества распределения и повышенное воздействие соединения (I') по сравнению с другими исследуемыми здесь соединениями.
Значительно более низкий уровень метаболизма соединения (I') по сравнению с другими соединениями показывает, что неожиданным образом соединение (I') значительно превосходит и широкий спектр лактамных BSCl, ранее описанных для разработки лекарственных средств для применения у человека. Этот сниженный уровень метаболизма (и, следовательно, улучшенные ADME свойства), видимо, является объяснением улучшенных фармакокинетических свойств, приведенных выше в описании примера 1. Более того, поскольку BSCl предназначены для разработки противовоспалительных лекарственных средств, направленных против нежелательного присутствия лейкоцитов в разных тканях, неожиданное открытие, что соединение (I') обнаруживается во всех тканях тела при проведении теста через 24 часа после введения единичной дозы, в то время как все остальные исследуемые лактамные BSCl не присутствовали, ясно указывает на особую полезность данного нового соединения.
Пример 3. Фармакологические исследования безопасности
Указанные пять соединений были подвергнуты стандартному тесту Эймса для того, чтобы провести оценку их возможной генотоксичности. Три ауксотрофных по гистидину штамма S.typhinurium (ТА102, ТА98 и ТА100) были обработаны каждым из пяти исследуемых соединений в пяти концентрациях (до 5 мг/мл) в присутствие и в отсутствие микросомальной метаболической системы крыс S9. Затем определяли число ревертантных колоний путем высевания на минимальную среду, содержащую следовые количества гистидина.
Полученные результаты (Таблица 4) показывают, что ни одно из пяти исследуемых соединений не усиливает значительным образом формирование ревертантной колонии (с или без метаболической активации) для любых протестированных штаммов.
Таблица 4. Формирование ревертантных колоний при проведении теста Эймса. Ни одно из протестированных соединений не привело к значительному повышению формирования ревертантных колоний при любой из использованных концентраций (приведены данные, полученные только при применении самой высокой дозы). Заметим, что соединение (II) привело к подавлению роста бактериального газона при концентрации 5 мг/мл.
При проведении отдельного эксперимента все пять исследуемых соединений были протестированы на взаимодействие с белком ионного канала hERG. Применение соединений, которые взаимодействуют с hERG, является рискованным, так как они могут вызывать пролонгацию QT-интервала и потенциально фатальную сердечную аритмию. Соединения, которые подавляют следовой ток hERG более чем на 50% при концентрации 10 мкМ, в целом, считаются высокорискованными для разработки лекарственных препаратов для применения у человека.
Клетки НЕК239, подвергнутые стабильной трансфекции для экспрессии белка hERG, промывали жидкостью, содержащей исследуемые соединения в концентрации 10 мкМ (0,1% ДМСО). Затем записывали значения hERG следового тока от трех клеток способом фиксации мембранного потенциала после деполяризации до +20 мВ в течение 5 с. Силу взаимодействия с hERG любых соединений, для которых была показана значительная модуляция при концентрации 10 мкМ, затем определяли с применением кривой дозовой зависимости, построенной по 4 точкам.
Полученные результаты (Фиг.3) показали, что ни одно из пяти исследуемых соединений в концентрации 10 мкМ не вступало в значительное взаимодействие с белком hERG канала.
Авторы сделали заключение, что с точки зрения фармакологии безопасности все пять исследуемых соединений, включая (I'), являются равным образом пригодными для разработки лекарственных средств, предназначенных для применения у человека. В частности, значительно преимущественным ADME и фармакокинетическим свойствам соединения (I'), которые приведены выше в описании Примера 1 и Примера 2, не соответствуют худшие данные по фармакологической безопасности.
Пример 4. Общая фармакология
Оценку общих фармакологических параметров указанных исследуемых пяти соединений проводили как при взаимодействии со специфическим рецептором-мишенью, так и при реакции с широким спектром других рецепторов, многие из которых сходны по структуре с рецептором-мишенью. Связывание со специфическим рецептором-мишенью оценивали по конкурентному связыванию [3H]-BN83250 (BN83250 - это S-3-(2',2'-диметилдодеканоиламино)-капролактам; Fox et al. J Med Chem. 200; 48(3): 867-74; вещество, которое связывается с тем же рецептором-мишенью, что и лактамные BSCl, описанные в данном документе). Связывание с рецепторами, которые не являются мишенью, оценивали по конкурентному связыванию с различными радиоактивно меченными лигандами, специфичными к другим рецепторам, хорошо известными специалистам в данной области.
Для специфичного связывания с клетками линии миеломоноцитов человека клетки ресуспендировали в буфере для связывания (20 мМ HEPES, 150 мМ NaCl; pH 7,4; 106 клеток на реакцию) при 4°С в присутствие 10 нМ [3H]-BN83250 (приготовленного из 1 мкМ стокового раствора в 100% этиловом спирте; 30 Ки/ммоль) и различных агентов для конкурентного связывания (1% ДМСО максимальная концентрация носителя). Реакции проводили в течение 2 часов на льду, затем фильтруют через GF/C-фильтры, предварительно насыщенные 0,5% полиэтиленимином. Несвязавшийся материал удаляли 5-кратным промыванием 5 мл ледяного буфера для промывания (20 мМ HEPES, 150 мМ NaCl; pH 7,4) при слабом вакууме. Было ранее показано, что при использовании данных условий достигается равновесное связывание, при этом не менее 80% связывания является специфичным (способным к конкуренции с 10 мкМ немеченого BN83250).
Конкурентное специфическое связывание [3H]-BN83250 и соединений (I'), (II) и (V) затем определяли при различных концентрациях от 1 пМ до 10 мкМ. Соединения (III) и (IV) не исследовали при проведении данных экспериментов. Типичная кривая конкурентного связывания для соединения (I') показана на Фиг.4.
Затем к кривым конкурентного связывания для различных соединений применяли нелинейное моделирование для того, чтобы сравнить их свойства в качестве агентов, связывающихся с рецептором-мишенью (определены в участке специфического взаимодействия с соединением BN83250). Параметры полученных моделей представлены в Таблице 5.
эффективной дозы против МСР-1 (пМ)
Таблица 5. Нелинейное моделирование кривых конкурентного связывания.
Важно отметить, что для соединения (I') в противоположность лактамным BSCl (II) и (V) было показано идеальное и предсказуемое связывание с рецептором-мишенью. В частности, определенное значение сродства при связывании с рецептором было сходно с функциональным значением средней эффективной дозы при проведении анализа по ингибированию миграции. Похожим образом, значение коэффициента Хилла составило приблизительно -1,0 (теоретически ожидаемое значение для модели простого некооперативного конкурентного связывания), в то время как при изучении других лактамных BSCl были определены существенно менее значительные коэффициенты Хилла. Причина отличий от идеального связывания с рецептором-мишенью для соединений (II) и (V) неизвестна, но данное различие свойств лежит в основе неожиданного превосходства соединения (I').
Связывание с рецепторами, не являющимися мишенью, оценивали с применением сходных протоколов, с применением специфических радиоактивно меченных лигандов для каждого рецептора, которые хорошо известны специалистам в данной области. Каждое соединение исследовали на конкурентное специфическое связывание с каждым лигандом только при одной концентрации (10 мкМ). Если ингибирование связывания было между 20% и 80%, определяли значение Ка для данного взаимодействия. Если ингибирование было менее 20%, считали, что такое соединение не обладает способностью к конкурентному связыванию с рецептором. Если ингибирование составляло более 80%, считали, что Ка для данного взаимодействия составляет менее 1 мкМ. Детальное описание использованных в данном исследовании рецепторов, радиоактивно меченных лигандов и типов клеток находится по адресу
Полученные результаты (Фиг.5) указывают на то, что все протестированные лактамные BSCl соединения свободны от главных перекрестных реактивностей на основании изучения данного набора из 75 рецепторов (не было обнаружено никаких взаимодействий, характеризующихся Ка<1 мкМ). Была обнаружена только одна слабая (но статистически значимая) перекрестная реакция (соединение (II) и рецептор NK2). На основании этого соединение (I') было признано косвенно более специфичным при взаимодействии с рецептором-мишенью, чем соединение (II), но при этом все протестированные лактамные BSCl соединения были пригодны для разработки фармацевтических препаратов, предназначенных для использования у человека на основании того, что они не обладали способностью связываться с ложными мишенями, как было показано при проведении данного обладающего высокой пропускной способностью аналитического скрининга.
Пример 5. Ингибирование хемокинов широкого спектра действия in vitro
Биологическая активность соединения, описываемого в данном изобретении, может быть показана с применением любого из широкого спектра способов функционального исследования миграции лейкоцитов in vitro, включая (но не ограничиваясь) камеру Бойдена и родственные эксперименты по миграции между лунками, эксперименты по миграции под слоем агарозы и применение камер для прямой визуализации, таких как камера Данна.
Например, для того чтобы показать ингибирование миграции лейкоцитов, возникшей в ответ на хемокины (но не другие хемоаттрактанты), использовали систему для проведения анализа по миграции между лунками, состоящую из 96 микролунок ChemoTx™, произведенную Neuroprobe (Gaithersburg, MD, USA). В целом, в данном анализе используют две камеры, разделенные пористой мембраной. Хемоаттрактант помещают в нижний компартмент, а клетки помещают в верхний компартмент. После инкубации в течение некоторого времени при 37°С клетки передвигаются в направлении хемоаттрактанта и число клеток в нижнем компартменте становится пропорциональным активности исследуемого хемоаттрактанта (относительно результатов серии контрольных экспериментов).
Данный способ анализа можно использовать с применением широкого спектра популяций лейкоцитов. Например, могут быть использованы свежеприготовленные лейкоциты из периферической крови человека. В ином случае, могут быть приготовлены отдельные разновидности лейкоцитов, включая полиморфноядерные клетки или лимфоциты, или моноциты с применением способов, хорошо известных специалистам в данной области, такие как центрифугирование в градиенте плотности или разделение на магнитных бусинах. Также можно использовать иммортализованные клеточные линии, которые были широко используют в качестве моделей лейкоцитов периферической крови человека, включая (но не ограничиваясь), клетки ТНР-1 в качестве модели моноцитов или клетки Jurkat в качестве модели наивных Т-клеток.
Хотя многие условия являются приемлемыми при проведении экспериментов для демонстрации подавления миграции лейкоцитов, вызванной хемокинами (см., например, рекомендации Frow et al. Med Res Rev. 2004; 24(3): 276-98 по условиям, требуемым для интерпретации результатов, полученных при проведении экспериментов по изучению миграции клеток in vitro), в данной заявке приведено описание условий, использованных в данном специфическом случае.
Материалы
Системы для изучения миграции клеток между лунками произведены Neuroprobe, Gaithersburg, MD, USA. Использованные при проведении экспериментов планшеты: планшеты ChemoTx™ (Neuroprobe 101-8) и прозрачные плашки 30 мкл (Neuroprobe МР30).
Сбалансированный солевой раствор Гейса (ССР) приобретали в компании Sigma (Sigma G-9779). БСА, не содержащий жирных кислот, приобретали в компании Sigma (Sigma А-8806). МТТ, например, 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия бромид, приобретали в компании Sigma (Sigma М-5655). RPMI-1640, не содержащий феноловый красный, приобретали в компании Sigma (Sigma R-8755).
Клеточную линию ТНР-1 (Европейская коллекция клеточных культур) использовали в качестве популяции лейкоцитов.
Протокол проведения теста
Следующую процедуру использовали для проведения тестирования соединения согласно настоящему изобретению, для определения его способности специфически блокировать миграцию лейкоцитов, индуцированную хемокинами:
Во-первых, приготавливали суспензию клеток, которую затем помещали в верхний компартмент. Клетки ТНР-1 осаждали центрифугированием (770 g; 4 мин) и промывали сбалансированным солевым раствором Гейса, содержащим 1 мг/мл БСА (ССР+БСА). Это промывание затем повторяли и клетки повторно осаждали перед тем, как ресуспендировать их в небольшом объеме ССР+БСА для проведения подсчета, например, с применением стандартной счетной камеры.
Объем ССР + БСА затем подбирали в зависимости от числа присутствующих клеток таким образом, чтобы конечная плотность клеток составляла 4,45×106 клеток на 1 мл ССР + БСА. При этом в каждых 25 мкл раствора будет содержаться 100000 клеток ТНР-1, которые будут помещены в верхнюю камеру планшета.
Для того чтобы протестировать отдельное соединение на его способность подавлять миграцию лейкоцитов, индуцированную хемокинами, необходимо приготовить две серии клеток. Суспензию клеток ТНР-1 с плотностью 4,45×106 клеток/мл помещают в две емкости. В одну емкость добавляют исследуемый ингибитор до требуемой конечной концентрации в подходящем носителе (например, при концентрации 1 мкМ в не более чем 1% ДМСО). Во вторую емкость добавляют равный объем ССР + БСА плюс носитель как следует (например, не более 1% ДМСО) в качестве контроля.
Затем приготавливают раствор хемоаттрактанта, который затем помещают в нижний компартмент. Например, МСР-1 разбавляют в ССР + БСА до конечной концентрации 25 нг/мл. Это количество разделяют между двумя емкостями, как суспензию клеток. В одну емкость добавляют тестируемое соединение до той же конечной концентрации, в то время как в другую емкость добавляют равный объем ССР + БСА плюс подходящий носитель (например, не более чем 1% ДМСО). В ином случае, можно применять другие хемокины (SDF-1α в концентрации 7,5 нг/мл; RANTES в концентрации 50 нг/мл; IL-8 в концентрации 10 нг/мл с применением нейтрофилов в качестве популяции клеток-мишеней). В каждом случае важно определить (в отдельном эксперименте) концентрацию каждого хемокина, который приводит к максимальной стимуляции миграции выбранной популяции лейкоцитов-мишеней. Данную максимальную концентрацию затем следует применять при проведении экспериментов для тестирования ингибиторной активности данного соединения, которое является предметом данного изобретения. Поскольку хемокины обычно индуцируют миграцию лейкоцитов, которая описывается колоколобразной кривой дозовой зависимости, применение суб- или супра-максимальной концентрации хемокина может привести к артефактным результатам (например, соединение, которое является ингибитором хемокинов, может привести к парадоксальной стимуляции миграции, если для проведения эксперимента была некорректно выбрана супрамаксимальная концентрация хемоаттрактанта. Дополнительные свидетельства важности данного фактора в планировании экспериментов по миграции лейкоцитов in vitro были предоставлены в работе Frow и сотрудников (Med Res Rev. 2004; 24(3): 276-98). Кроме того, хемоаттрактанты, не относящиеся к хемокинам, также могут быть использованы для демонстрации избирательности биологической активности изучаемого соединения, которое является предметом данного изобретения, в отношении хемокинов (например, С5а в концентрации 25 нг/мл с применением нейтрофилов в качестве популяции клеток-мишеней или TGF-β1 в концентрации 10 нг/мл с применением ТНР-1 в качестве популяции клеток-мишеней).
Заметим, что объем жидкости, который добавляют в составе раствора тестируемого соединения, нужно учитывать при выведении конечной концентрации МСР-1 в растворе для нижнего компартмента и конечной концентрации клеток в верхнем компартменте.
Когда растворы хемоаттрактанта для нижних ячеек и растворы клеток для верхних компартментов приготовлены, следует собрать камеру для миграции. Помещают 29 мкл раствора нужного хемоаттрактанта в нижнюю емкость камеры. Эксперименты следует проводить не менее чем в трех повторениях для каждого условия. Когда все нижние камеры заполнены, в камеру помещают пористую мембрану в соответствии с инструкциями производителя. Наконец, в каждую верхнюю камеру помещают по 25 мкл подходящего раствора клеток. Помещают пластиковую крышку поверх всего приспособления для того, чтобы избежать испарения.
Подготовленную камеру выдерживают при 37°С, 5% СО2 в течение 2 часов. Суспензию клеток в ССР + БСА также инкубируют в идентичных условиях в пробирке: данные клетки будут использовать для построения стандартной кривой для определения числа клеток, которые мигрировали в нижнюю камеру при указанных условиях.
При окончании инкубирования жидкую суспензию клеток осторожно удаляют из верхней камеры, добавляют в верхнюю камеру 20 мкл ледяного 20 мМ ЭДТА в фосфатно-солевом буфере и выдерживают приспособление при 4°С в течение 15 минут. Данная процедура приводит к тому, что все клетки, которые прикрепились к нижней части мембраны, падают внутрь нижней камеры.
После проведения данного инкубирования фильтр осторожно промывают раствором ССР + БСА для того, чтобы удалить ЭДТА, затем фильтр удаляют.
Число клеток, которые мигрировали в нижнюю камеру при указанных условиях, может быть определено при применении некоторого числа способов, включая прямой подсчет, мечение флуоресцентными или радиоактивными маркерами или путем использования красителей для прижизненной окраски клеток. Обычно для прижизненной окраски клеток используют МТТ. 3 мкл стокового раствора МТТ добавляли в каждую лунку, затем планшет инкубировали при 37°С в течение 1-2 часов; в течение этого времени ферменты дегидрогеназы, содержащиеся внутри клеток, превращают растворимый МТТ в нерастворимый продукт синий формазан, который может быть количественно определен спектрофотометрическим способом.
Параллельно, проводят построение стандартной кривой по 8 точкам. Клетки добавляют в плашку в 25 мкл вместе с 3 мкл стокового раствора МТТ, начиная с числа клеток, добавленных в каждую верхнюю камеру (100000), и снижают количество клеток, выполняя 2-кратные серийные разбавления раствором ССР + БСА. Планшет для стандартной кривой инкубируют рядом с планшетом для исследования миграции клеток.
После окончания данного инкубирования жидкость осторожно удаляют из нижних камер, стараясь не взмутить выпавший в осадок продукт реакции, который представляет собой формазан. После кратковременного высушивания на воздухе в каждую нижнюю камеру добавляют 20 мкл ДМСО для того, чтобы растворить краску синего цвета и измеряют оптическую плотность при 595 нм с применением считывающего устройства для 96-луночного планшета. Затем проводят оценку числа клеток в каждой нижней камере путем интерполяции полученных значений оптической плотности для каждой лунки с применением стандартной кривой.
Миграцию, которая стимулируется хемоаттрактантом, определяют вычитанием среднего числа клеток, которые достигли нижнего компартмента в лунках, куда не добавляли никакого хемоаттрактанта, из среднего числа клеток, которые достигли нижнего компартмента, где присутствовал хемоаттрактант.
Влияние исследуемого соединения определяют путем сравнения миграции, вызванной действием хемоаттрактанта, которое случилось в присутствие или в отсутствие различных концентраций тестируемых соединений. Обычно подавление миграции выражают как процентное отношение от общей миграции, вызванной действием хемоаттрактанта, которая была ингибирована в присутствии данного соединения. Для большинства соединений построение графика дозовой зависимости выполняют путем определения подавления миграции, индуцированной действием хемоаттрактанта, которое происходит при разных концентрациях соединения (обычно концентрацию варьируют от 1 нМ до 1 мкМ или выше в случае низкой активности соединения). Ингибиторную активность каждого соединения затем выражают как концентрацию соединения, которая требуется для того, чтобы снизить миграцию, вызванную действием хемоаттрактанта на 50% (концентрация, соответствующая средней эффективной дозе). Обычно, миграцию клеток ТНР-1, вызванную действием МСР-1, использовали в качестве стандартизованной тест-системы для сравнения биологической активности широкого спектра соединений (см., например, Reckless & Grainger Biochem J. 1999 Jun 15;340 (Pt 3): 803-11; Reckless et al. Immunology. 2001 Jun; 103(2): 244-54; Fox et al. J Med Chem. 2002 Jan 17; 45(2): 360-70; Fox et al. J Med Chem. 2005 Feb 10; 48(3): 867-74; the International applications supra). Соединения, которые ингибируют миграцию лейкоцитов, индуцированную более чем одним хемокином, но не хемоаттрактантами, не относящимися к хемокинам (такими, как TGF-βе или С5а), определяют как ингибиторы хемокинов широкого спектра действия (Broad-spectrum Chemokine Inhibitors, BSCl; см., например, Grainger & Reckless Biochem Pharmacol. 2003 Apr 1; 65(7): 1027-34; Grainger et al. Mini Rev Med Chem. 2005 Sep; 5(9): 825-32).
Результаты
На Фиг.6 показана типичная кривая дозовой зависимости для соединение (I'), которое подавляет миграцию ТНР-1 клеток, вызванную МСР-1, вместе со сравнимой кривой дозовой зависимости для других выбранных лактамных BSCl, для которых известно, что они характеризуются особенно сильным (то есть менее 1 нМ) действием. Активность соединения (I') при действии против различных хемокинов и хемоаттрактантов, которые не относятся к хемокинам, выраженную в значениях средней эффективной дозы, показана в Таблице 6, и проведено сравнение с другими ранее описанными лактамными BSCl.
Из анализа этих данных ясно, что соединение (I') может быть классифицировано как BSCl (поскольку оно сильно и активно подавляет миграцию лейкоцитов, индуцированную рядом хемокинов, но не оказывает никакого действия на миграцию лейкоцитов, индуцированную хемоаттрактантом, которые не относятся к хемокинам, в данном случае, С5а анафилотоксином). Более того, очевидно, что соединение (I') является как минимум таким же сильнодействующим и активным, в качестве BSCl in vitro, как выбранные лактамные BSCl, которые были описаны ранее (например, соединение (II) в WO 2006/016152 или соединение (IV) в WO 2005/053702). Все исследуемые здесь лактамные BSCl являются значительно более активными, чем любой из нелактамных BSCl, которые были описаны к настоящему времени (включая имиды, такие как NR58.4, йохимбамиды, лизергамиды и пептид 3 и родственные по структуре соединения, такие как NR58-3.14.3). Более того, соединение (I') является более сильнодействующим в качестве BSCl in vitro (как минимум, для подавления миграции, индуцированной МСР-1), чем любое другое соединение, открытое или описанное ранее. Хотя данное действие в качестве BSCl является количественно превосходным по сравнению с известными ранее прототипами BSCl (хотя и в небольшой степени), это не то свойство, которое главным образом выделяет соединение (I) как обладающее неожиданным преимуществом по сравнению с известными ранее прототипами ингибиторов хемокинов широкого спектра действия. Напротив, это показывает, что данные неожиданные и значительно преимущественные ADME и фармакокинетические свойства соединения (I) по сравнению с широким спектром ранее описанных BSCl были достигнуты без какого-либо снижения силы действия или активности данного соединения в качестве ингибиторов хемокинов широкого спектра действия in vitro.
Таблица 6. Действие выбранных лактамных BSCl на миграцию лейкоцитов in vitro. В каждом случае указана доза соединения (в пМ), которая требуется для подавления миграции лейкоцитов в ответ на введение максимальной дозы утвержденного хемоаттрактанта на 50% (средняя эффективная доза). Если не указано обратное, данные, включенные в отчет, получены для клеточных линий для ТНР-1. Было показано, что ни одно из протестированных соединений даже в самой высокой концентрации (1 мкМ) нисколько не подавляло миграцию нейтрофилов, индуцированную С5а.
Пример 6. Противовоспалительная активность in vivo
Мы использовали модель сублетального эндотоксикоза, индуцированного липополисахаридами, для того, чтобы продемонстрировать генерализованные противовоспалительные свойства ранее описанных BSCl in vivo (см., например, Fox et al. J Med Chem. 2002; 45(2): 360-70; Fox et al. J Med Chem. 2005; 48(3): 867-74). При проведении данного исследования мыши были подвергнуты неспецифическому провоспалительному воздействию бактериального эндотоксина (липополисахариды), и степень развития системной воспалительной ответной реакции (реакцию количественно оценивали по концентрации провоспалительного цитокина TNF-α в сыворотке, который в норме отсутствует в крови, но его уровень быстро повышается в ответ на широкий спектр провоспалительных стимулов). Заявители выбрали данную модель, несмотря на то, что она не является близкой к условиям какого-либо воспалительного заболевания у человека, поскольку известно, что концентрация TNF-α важна при многих заболеваниях (включая ревматоидный артрит, аутоиммунные заболевания, болезнь Крона, атеросклероз, астму и многие другие). Поэтому активные вещества, подавляющие синтез TNF-α, уже используют в клинике (например, Enbrel™ и другие антитела против TNF-α) для лечения широкого спектра подобных заболеваний. Таким образом, в данной модели была показана активность, подавляющая TNF-α, что очень эффективно для предсказания противовоспалительного действия, клинически полезного при широком спектре заболеваний.
Мышей (в каждой группе содержалось по шесть животных) подвергали предварительному воздействию различных доз каждого соединения, которые вводили подкожно за 30 минут до введения липополисахаридов или принудительно перорально за 60 минут до введения липополисахаридов. Данным мышам затем вводили внутрибрюшинно 750 мкг бактериальных липополисахаридов и умерщвляли через 3 часа. Сыворотку крови приготавливали из цельной крови и определяли концентрацию TNF-α способом иммуноферментного анализа (R&D Systems). В каждом эксперименте группа из шести мышей, которую использовали в качестве отрицательного контроля, не получала никаких липополисахаридов, вторая группа мышей получала только липополисахариды (без какого-либо ингибитора). Концентрацию TNF-α в сыворотке крови данных животных, которые получали липополисахариды без предварительного лечения с применением лекарственных средств, принимали за 100% (обычно данная концентрация составляла приблизительно 6000 пг/мл по сравнению в концентрациями менее 10 пг/мл у животных из группы отрицательного контроля). Ранее заявители показали, что синтетический кортикостероид дексаметазон (хорошо известный противовоспалительный медицинский препарат, который используют при широком спектре воспалительных заболеваний) в данной модели подавляет синтез TNF-α, индуцированный действием липополисахаридами, как минимум на 90%, в то время как талидомид (другой известный ингибитор продукции TNF-α, который действует скорее на уровне внутриклеточного синтеза TNF-α, чем в качестве ингибитора привлечения лейкоцитов, как описанные в данной заявке BSCl) подавляет синтез TNF-α, индуцированный действием липополисахаридов, приблизительно на 60%.
Действие соединения (I') при применении в различных дозах, а также других выбранных лактамных BSCl показано на Фиг.7. В соответствии с ожиданиями данное соединение сильно подавляло синтез TNF-α, индуцированного действием липополисахаридов, при подкожном (кружки) или пероральном (треугольники) применении. При применении данного соединения в количествах, превышающих 1 мкг/мышь, концентрации TNF-α, индуцированного действием липополисахаридов, обычно снижались более чем на 90%, что было сравнимо с действием кортикостерида дексаметазона.
Другие протестированные лактамные BSCl также подавляли синтез TNF-α, индуцированного действием липополисахаридов, дозозависимым способом (Фиг.7), хотя активность соединения (I') in vivo была выше, чем у любого другого протестированного соединения (и действительно выше, чем активность других ранее описанных где-либо лактамных BSCl, которые были протестированы при проведении данного анализа). Данное количественное (хотя и небольшое) повышение активности не является главной причиной того, что заявители настоящим утверждают, что соединение (I) обладает неожиданным преимуществом по сравнению с ранее известными прототипами BSCl. Напротив, это показывает, что неожиданные и значительно преимущественные ADME и фармакокинетические свойства соединения (I) по сравнению с широким спектром раннее описанных BSCl были получены без потери активности или силы действия в качестве противовоспалительного агента in vivo. Кроме того, эти данные ясно показывают полезность соединения (I) при применении в качестве противовоспалительного агента in vivo в модели воспаления, которая указывает на полезность применения при широком спектре воспалительных заболеваний, которые сопровождаются повышением уровня синтеза TNF-α как компонента патогенного механизма.
Важно отметить, что сверхострое воспаление, которое наблюдали в данной модели, особенно нечувствительно к ADME и фармакокинетические свойствам протестированных противовоспалительных агентов. Поскольку стимуляцию липополисахаридами осуществляли только через 30 минут после введения лекарственного препарата, даже агенты, обладающие очень короткими временами удержания в плазме крови (такие, как соединения (II) и (III)), оставались в плазме в достаточных концентрациях для того, чтобы оказать сильное противовоспалительное действие. Таким образом, во время проведения данного теста не было подчеркнуто превосходство заявленного соединения над известными ранее прототипами, что тем не менее показывает полезность данного соединения.
Полезность заявленного соединения была показана далее во время проведения исследований на животной модели астмы человека (сверхострая ответная воспалительная реакция, которую наблюдали в ответ на введение липополисахаридов, может не быть типичной для какого-либо заболевания человека, хотя ясно, что она является полезной моделью для изучения острого воспаления в целом). При проведении данных исследований грызуны (обычно крысы) получали овальбумин в соответствии с планом эксперимента:
Взрослых крыс Brown Norway (вес тела 200-300 г; число животных = 10 в группе) подвергали сенситизации путем единичной внутрибрюшинной инъекции 0,1 мг овальбумина в 0-й день. Затем каждой крысе вводили эндотрахеально 1% раствора овальбумина (в отношении веса к объему) на 8-й день и 2% раствора овальбумина (в отношении веса к объему) в дни 15-й, 18-й и 21-й. Данных животных затем умерщвляли через 3 часа после последнего введения на 21-й день. Заметим, что при проведении экспериментов использовали овальбумин (Sigma; самая высокая степень очистки), который дополнительно очищали от эндотоксинов на колонках EndoTrap™ Red (приобретали в Cambrex; использовали в соответствии с инструкциями производителя), и было показано с применением ЛАЛ-системы для количественного определения эндотоксинов (QCL-1000™; Cambrex; тест проводили в соответствии с инструкциями производителя; 1 мг стандартного эндотоксина содержит ~900000 ЕЭ/мг), что концентрация эндотоксинов составляла менее 5 ЕЭ/мг белка. Полученные данные показывают, что ответная реакция на воспаление в легких скорее является результатом аллергической реакции на овальбумин, чем на непреднамеренную стимуляцию липополисахаридами, которая развивается даже с применением коммерческих препаратов овальбумина самой высокой степени очистки; таким образом, подтвердили, что при применении данной модели лучше воспроизводится молекулярная патология, лежащая в основе развития астмы у человека.
Одна группа мышей (которую использовали в качестве основного контроля) не получала овальбумин, но во всем остальном была подвергнута идентичному воздействию. Мыши из второй группы (положительный контроль) получали овальбумин, но не получали лечение лекарственными средствами. Мыши из третьей группы были обработаны идентично, но получали ежедневную дозу соединения (I') в количестве 0,3 мг/кг орально принудительно в дни с 8-го по 21-й за один час до любого последующего введения овальбумина, приготовленного в тот же день. Соединение (I') вводили в виде стерильного раствора в свободном от эндотоксинов фосфатно-солевом буферном растворе. Мыши из четвертой группы получали монтелукаст (активный компонент коммерческого лекарственного препарата, используемого при лечении астмы, произведенного Singulair™) в количестве 30 мг/кг перорально по схеме лечения, идентичной использованию соединения (I').
После забивания животных проводили оценку общего участия лейкоцитов легких путем анализа бронхоальвеолярных смывов с введением через трахеальные канюли 4 серий по 3 мл стерильного фосфатно-солевого буферного раствора. Для каждого животного полученные бронхоальвеолярные смывы объединяли и определяли общее число клеток (с применением счетной камеры). Кроме того, проводили определение типов присутствующих лейкоцитов с применением проточного цитометра в соответствии со способами, хорошо известными специалистам в данной области.
Также отбирали из каждой мыши селезенку и помещали в раствор питательной среды RPMI + 10% сыворотки (FCS) + антибиотики. Затем селезенку продавливали через нейлоновую сетку с мелкими (100 мкМ) отверстиями в стерильной емкости для фильтрования, которую помещали в стерильные чашки Петри для получения суспензий, состоящих из отдельных клеток. Полученные суспензии клеток затем центрифугировали (328 g; 5 минут) и промывали питательной средой RPMI + 10% сыворотки + антибиотики, перед применением проводили ресуспендирование в свежей среде и определяли количество клеток в счетной камере.
Общее количество спленоцитов 4×106 (за исключением красных кровяных клеток) культивировали в течение ночи при 37°С; 5% СО2 в питательной среде RPMI + 10% сыворотки + антибиотики в присутствие 2 Ед/мл (10 нг/мл) мышиного ИЛ-2 в 4 лунках 96-луночного планшета (объем 100 мкл на лунку / 1×106 клеток/лунку) из каждой мыши. Приблизительно через 24 часа 4 лунки разделяли на две группы из 2 ячеек: две лунки не обрабатывали, в две другие лунки добавляли 500 нг/мл иономицина и 50 нг/мл РМА и выдерживали при 37°С в течение 4 часов. В течение двух последних часов инкубации в одну лунку из каждой группы добавляли 10 мкг/мл брефелдина А (стоковый раствор 1 мг/мл в этиловом спирте). Брефелдин А блокирует транспорт белков в аппарат Гольджи и, таким образом, способствует накоплению белков в эндоплазматическом ретикулуме.
Лунки, в которые не добавляли брефелдин А, инкубировали далее в течение 48 часов при 37°С. После окончания инкубации суспензии клеток центрифугировали (328 g; 5 минут) и полученный супернатант исследовали с применением способа иммуноферментного анализа на присутствие мышиного IL-4 (маркер Th2-клеток) и мышиного интерферона-γ (IFN-γ; маркер Th1-клеток) (R&D Systems; анализ проводили в соответствии с инструкциями производителя).
Лунки, в которые добавляли брефелдин А, окрашивали на внутриклеточный IL-4 и IFN-γ сразу после окончания 4-часовой инкубации следующим образом: проводили окрашивание клеток CD4-FITC-антителами (eBioscience Rat lgG2b, Cat. Code. 11-0041) в течение 30 минут на льду, затем промывали фосфатно-солевым буферным раствором Дульбекко и фиксировали 2% раствором параформальдегида (конечная концентрация) в фосфатно-солевом буферном растворе Дульбекко в течение 20 минут. После фиксации клетки обрабатывали 1% БСА/0,5% сапонином в фосфатно-солевом буферном растворе Дульбекко (Sigma S7900) в течение 10 минут при комнатной температуре для того, чтобы клетки стали проницаемыми. Затем клетки из каждой лунки распределяли в три отдельные пробирки для проведения флуоресцентной сортировки клеток и добавляли:
- IFN-g-PE (eBioscience Rat IgG1, Cat. Code. 12-7311-82, 100 мкг)
или
- IL-4-PE (eBioscience Rat IgG1, Cat. Code. 12-7041-82, 100 мкг)
или
- Изотипные контроли (смесь Rat IgG2b-FITC, eBioscience Cat. Code 11-4031 и Rat IgG1-PE, eBioscience Cat. Code 12-4301)
и инкубировали в течение 30 минут при комнатной температуре. Затем клетки промывали (дважды раствором БСА/сапонина в фосфатно-солевом буфере и затем раствором БСА в фосфатно-солевом буфере без сапонина для того, чтобы мембранные поры закрылись) и ресуспендировали в фосфатно-солевом буферном растворе Дульбекко для проведения анализа способом проточной цитометрии.
Клетки, специфично окрашенные на CD4 по FITC (что определяет их как Т-хелперные клетки), были проанализированы на присутствие специфического окрашивания на IL-4 или на IFN-γ по РЕ. Соотношение CD4+ клеток, положительно окрашенных на IFN-γ к числу CD4+ клеток, окрашенных положительно на IL-4, затем включали в отчет как отношение Th1/Th2. Крысы линии Brown Norway, не подвергнутые воздействию, характеризовались отношением Th1/Th2 в селезенке приблизительно равным 2,7 (это означает, что в селезенке приблизительно в 2,7 раза больше CD4+ клеток синтезируют INF-γ, чем IL-4). После сенситизации и повторной обработки овальбумином данное отношение уменьшилось до <1,5, что отражает значительную Th2-поляризацию, которая сопровождает изменения при астме как у крыс, так и у людей (более низкое отношение Th1/Th2 указывает на относительную Th2 поляризацию, в то время как увеличение отношения Th1/Th2 указывает на относительную Th1-поляризацию).
Ежедневное применение соединения (I') и используемого в качестве положительного контроля монтелукаста значительно снижало число лейкоцитов в бронхоальвеолярных смывах (снижение на 70% при применении соединения (I'); р<0,01; непарный критерий Стьюдента; Фиг.8). Полученные данные однозначно показывают, что тестируемое соединение, которое является предметом данного изобретения, обладает полезным противовоспалительным действием в модели астмы человека, что следует из способности данного соединения подавлять миграцию лейкоцитов, индуцированную хемокинами. Величина полученного эффекта как минимум сравнима с действием коммерческих препаратов, предназначенных для лечения астмы у человека (таких как Singular™), а преимущественные фармакокинетические параметры и биораспределение соединения (I') определяют значительно большую силу действия соединения (I') по сравнению с монтелукастом (для того, чтобы вызвать такое снижение числа лейкоцитов в бронхоальвеолярной жидкости, которое происходит при введении только 0,3 мг/кг соединения (I'), требуется доза монтелукаста 30 мг/кг).
Ежедневное применение соединения (I'), при отсутствии введения используемого в качестве положительного контроля монтелукаста, значительным образом изменяло в обратную сторону Th2-поляризацию (Фиг.9), которая считается главной движущей силой патогенеза астмы как в использованной при проведении данного исследования модели воспаления легких, которое вызывали введением овальбумина, так и при астме у человека. При лечении с применением соединения (I'), даже с введением низких доз, таких как 0,3 мг/кг перорально, полностью прекращается поляризация Th2, вызванная хроническим воздействием аллергенов, таких как овальбумин, таким образом, что по отношению Th1/Th2 животные, которые получали соединение (I'), практически не отличаются от крыс Brown Norway, которых не подвергали действию аллергена.
Интересно отметить, что другие хронические воспалительные заболевания, такие как атеросклероз, связаны с Th1-поляризацией (в противоположность Th2-поляризации). При заболеваниях обоих типов нарушение баланса цитокинов, вырабатываемых Т-хелперными клетками, описано как основная патогенетическая причина развития хронического воспалительного компонента заболевания. В моделях заболевания, связанного с Th1-поляризацией (таких как атеросклероз), заявители ранее наблюдали значительный сдвиг в сторону Th2 при лечении с применением BSCl, таких как соединение (I'), описанное в данном документе. У мышей с гомозиготной делецией гена, кодирующего ароЕ (ароЕ-/-mice), развивается несколько липидных очагов повреждения внутри сосудов, даже при нормальном питании, и такие животные характеризуются отношением Th1/Th2 приблизительно равным 8 (по сравнению с 3,2 у используемых в качестве контроля мышей дикого типа линии C57BI6). Однако последующее лечение с применением BSCl в течение 3 месяцев (в возрасте от 12 до 24 недель - период жизни, в течение которого происходит развитие большинства липидных очагов поражений сосудов) нормализует отношение Th1/Th2 (и в данной модели при очень высоких дозах даже вызывает Th2-поляризацию). Указанные данные и данные, полученные при изучении модели воспаления легких, которое вызывали введением овальбумина, совместно показывают, что применение BSCl может нормализовать или восстановить баланс синтеза цитокинов Т-хелперными клетками независимо от того, какой дефект лежит в основе патологии: Th2-поляризация (как при астме) или Тh1-поляризация (как при атеросклерозе). По мнению заявителя, в настоящее время ингибиторы хемокинов широкого спектра действия являются единственными агентами, которые характеризуются подобным "восстанавливающим баланс" действием на популяцию Т-хелперных клеток. Указанные механистические представления в дальнейшем могут подтвердить (вместе с данными по эффективности, которые были получены при изучении многочисленных моделей различных заболеваний на животных с воспалительным компонентом) наше утверждение о том, что BSCl, и в особенности соединение (I), заявленное в данном документе (как результат неожиданно превосходных фармакокинетических параметров и биораспределения данного соединения), являются полезными в качестве медицинских препаратов для лечения необычно широкого спектра состояний, ассоциированных с воспалительным компонентом.
Пример 7. Сравнение вещества (I') и близких по структуре аналогов
После проведения идентификации соединения (I) как обладающего неожиданно превосходными фармацевтическими свойствами, особенно в отношении его фармакокинетических параметров по сравнению с другими ациламинолактамными BSCl, затем заявители провели прямое сравнение данного соединения с наиболее близкими структурными аналогами, которые были описаны ранее.
Исходный набор соединений, которые сравнивали в примерах 1-6, составляли так, чтобы они были структурно как можно более различными, в рамках ранее описанных ациламинолактам BSCl. На основании строения соединения (I), другие соединения, имеющие 6-членные лактамные концевые группы, были включены в исследование, хотя нет никакой особенной причины считать, что указанные соединения в целом обладают преимуществом (или, напротив, меньшей эффективностью) по сравнению с соединениями, содержащими 7-членное лактамное кольцо (см. WO 2006/134385 для широкого сравнительного анализа ациламинолактамных аналогов с разными размерами кольца в отношении их эффективности в качестве BSCl in vitro). Подобным образом, в набор исследуемых соединений включили единственный пример соединения с сульфонамидной связью (IV) как отличающегося от остальных молекул с амидной связью.
В отношении концевых групп (tailgroups) в список исследуемых соединений были включены две простые алкильные группы (обе пивоил) и три циклические алкильные группы (метилциклогексильная группа и две группы адамантана, один из которых был замещен). Все пять соединений имели хорошо установленное подтвержденное двойное замещение в положении 2'2 тетраэдрическими угловыми связями у центрального атома углерода (в позиции 2 относительно карбонила амидного кардонила, 4 атомами углерода, связанными с данным центральным атомом углерода в тетраэдрической струтктуре). Было ранее показано (WO 2006/016152), что наличие данного двойного замещения в позиции 2'2' у центрального атома углерода наделяет BSCl дополнительной эффективностью по сравнению с линейными алкильными концевыми группами или другими структурами, у которых нет четырех атомов углерода, связанных с данным центральным атомом углерода именно в тетраэдрической струткуре.
В результате указанные концевые группы в исходном наборе соединений были выбраны с условием наличия данного двойного замещения в положении 2',2', для того, чтобы обеспечить надлежащую эффективность in vitro, но также с требованием наличия сходных приемлемых физических свойств результирующих молекул. Например, ожидали, что присутствие адамантановой, циклогексильной группы и короткой цепи разветвленных в положении 2'2 алкильных групп приведет к тому, что соединения будут находиться в кристаллической (или полукристаллической) твердой форме и обладать приемлемой растворимостью в воде. Напротив, другие возможные концевые группы, такие как диметилдодеканоильная группа в положении 2',2' (которая иным образом соответствует желаемой конфигурации атомов углерода у центрального атома углерода в положении 2), были исключены из списка на основании того, что в твердом состоянии представляют собой воскообразные, аморфные вещества, не обладающие растворимостью в физиологических буферах. Эти свойства являются причиной, по которой данные соединения становятся менее пригодными для фармацевтической разработки даже до определения их ADME и токсикологических свойств, потому что очевидны трудности в обращения с данными соединениями, например, при составлении формулы препарата или таблетировании, и наличие подобных свойств повышает вероятность проявления нежелательных ADME показателей, таких как накопление в жировой ткани при постоянном применении или недостаточная биодоступность при оральном применении.
Таким образом, данные пять соединений, исследованные в примерах 1-6, имели приблизительно равные шансы на то, чтобы быть пригодными для фармацевтической разработки. До определения ADME свойств не было возможности предсказать, которое из данных соединений является лучшим.
Когда исследование, описанное в примерах 1-6, было завершено, однако стало ясно, что соединение (I) значительно и неожиданно превосходило другие исследованные соединения. Поскольку соединение (I) было единственным соединением в данном наборе, у которого имелось 6-членное кольцо, было возможно, что преимущественные свойства были обусловлены присутствием указанного лактамного кольца. Для того чтобы подтвердить или опровергнуть это, авторы настоящего изобретения провели обзор примеров всех ранее описанных ациламинолактамных соединений, обладающих активностью ингибиторов хемокинов широкого спектра действия, и выбрали два соединения, наиболее сходные по структуре с соединением (I). Из всех ранее известных соединений, обладающих BSCl активностью, данные два соединения характеризовались наиболее близкой структурой к соединению (I).
Выбранные соединения представляли собой 3-(2',2'-диметилдодеканоиламино)-тетрагидропиридин-2-он (пример 3 из WO 2006/134385; здесь - соединение VI) и 3-(адамантан-1-карбониламино)-тетрагидропиридин-2-он (пример 1 из WO 2006/134385; здесь - соединение VII):
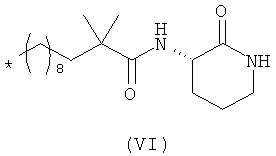
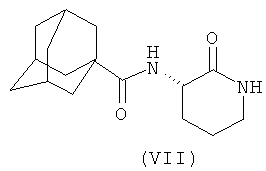
Данные соединения, так же как и соединение (I), содержат 6-членное лактамное кольцо, амидный линкер, двойное замещение в положении 2',2' с 4 атомами углерода, связанными с центральным атомом углерода во 2-й позиции относительно амидной карбонильной группы, которые формируют важную тетраэдрическую структуру.
Провели фармакокинетический анализ соединений (VI) и (VII) на крысах с применением точно таких же протоколов, которые применяли ранее при изучении соединений (I)-(V) в Примере 1. Следует отметить, что при проведении данных экспериментов использовали S-энантиомеры обоих соединений (VI) и (VII), поскольку ожидали, что они будут более активными энантиомерами, как и у других описанных к настоящему времени ациламинолактамных BSCl.
Результаты
К сожалению, соединение (VI) представляло собой воскообразное трудно растворимое твердое вещество. Было трудно получить препарат соединения (VI), не содержащий растворителя, но со временем такой препарат был получен путем значительного высушивания в течение многих недель. Однако полученный образец был полностью нерастворим в 5% ДМСО в физиологическом растворе, который использовали в качестве носителя для внутривенного введения при исследовании остальных соединений. Поэтому при изучении соединения (VI) было необходимо использовать полностью отличающиеся носители для того, чтобы приготавливать растворы, пригодные для внутривенного введения. Соединение (VI) растворяли в 50% ПЭГ 400/20% solutol/30% растворе натрия хлорида для внутривенного введения. Все соединения, включая (VI), суспендировали (или растворяли) в 1% растворе карбоксиметилцеллюлозы в физиологическом растворе для орального дозирования. Хотя сравнение значений биодоступности при оральном применении, полученных при проведении экспериментов с применением различных носителей, обычно не рекомендуется (поскольку носитель может оказывать значительное влияние на абсорбцию соединения в кишечнике), тем не менее, маловероятно, чтобы выбор носителя существенно влиял на значения периода полувыведения после внутривенного введения соединения (VI) при сравнении с другими соединениями, такими как (I).
Параметры простой однокамерной фармакокинетической модели приведены в Таблице 7. Данные для соединения (I') из Таблицы 1 выше приведены для сравнения. Очевидно, что оба соединения (VI) и (VII) являются значительно менее эффективными по сравнению с соединением (I'): клиренс более чем в 10 раз выше и действие при оральном применении в 25-50 раз ниже, чем при применении соединения (I')
Таблица 7. Фармакокинетические параметры выбранных ациламинолактамных BSCl с шестью кольцами. Определяли биодоступность при оральном применении (биодоступность, %), основной период полувыведения из плазмы (t1/2, минут) после внутривенного введения, клиренс (мл/мин/кг), объем распределения (объем распределения в равновесном состоянии, л/кг) и воздействие при оральном применении (AUC 0-t, мин·нг/мл) из простой однокомпартментной фармакокинетической модели для каждого соединения, представлены средние данные, полученные для трех крысам. Для соединения (VII) Cmax достигалась в течение 15 минут в соответствии с оптимальной абсорбцией, как и у соединения (I'). Гидрофобное соединение (VI) абсорбировалось более медленно, однако значение Tmax составило между 60 и 120 минутами.
Действительно, из сравнения данных Таблицы 7 и Таблицы 1 очевидно, что по силе действия соединения (VI) и (VII) не так хороши, даже как соединения (IV) и (V). Несмотря на то что данные соединения содержат 6-членное лактамное кольцо, так же как и соединение (I), соединения (VI) и (VII) обладают значительно менее выраженными ADME свойствами и не лучше, чем широкий спектр других ациламинолактамных BSCl соединений, которые были описаны ранее.
В соответствии с более высокими значениями клиренса для соединений (VI) и (VII) по сравнению с соединением (I') данные соединения характеризуются значительно более коротким периодом времени полувыведения из плазмы крови, чем соединение (I') (менее 1 часа по сравнению с более чем 3 часами).
На основании результатов, полученных при проведении данного фармакокинетического анализа, специалистам в данной области очевидно, что, несмотря на сходные показатели химической стабильности, а также на сходные теоретически предсказанные свойства и сходство структуры, тем не менее, соединение (I') обладает значительным преимуществом по сравнению с соединениями (VI) и (VII), так как обладает в 25-50 раз лучшим действием при оральном применении, чем любой из данных аналогов 6-членного лактамного кольца. Заявители сделали вывод о том, что неожиданно преимущественные ADME свойства соединения (I') не могут объясняться ни наличием 6-членного лактамного кольца (поскольку другие соединения, имеющие 6-членное лактамное кольцо, такие как (VI) и (VII), не обладают таким же благоприятными ADME характеристиками), ни наличием амидного линкера (поскольку другие соединения, имеющие амидный линкер, включая соединения (II), (III) и (V), не обладают таким же благоприятными ADME характеристиками), ни наличием концевой группы пивоила (поскольку соединение (IV) имеет данную группу и не обладает такими же выдающимися ADME характеристиками, как соединение (I')). Напротив, неожиданные и значительно преимущественные ADME характеристики соединения (I') зависят от особенного сочетания его структурных особенностей, которое не может быть предсказано.
Пример 8. Попытка рационального конструирования ациламинолактамных BSCl. обладающего АDМЕ свойствами соединения (I'). Во время ииследования ADME свойства соединения (I') заявители провели анализ фармакокинетических свойств некоторого числа различных ациламинолактамных соединений, обладающих активностью в качестве BSCl in vitro в концентрациях, непревышающих наномоли. Заявители ожидали в результате понять, какие факторы определяют ADME свойства молекул среди данного структурного класса соединений. Заявители, таким образом, попытались использовать полученный опыт для того, чтобы выбрать второе ациламинолактамное соединение с BSCl активностью из соединений, по своей структуре относящихся к классам веществ, у которых были ранее описаны с ADME свойствами, насколько возможно близкими к свойствам соединения (I').
Относительно быстрый клиренс соединения (IV) по сравнению с клиренсом соединения (I') (см. Таблицу 1 выше) позволяет предположить, что наличие амидного линкера может коррелировать с благоприятными ADME свойствами. Заявители, таким образом, ограничились выбором соединений с амидными линкерами.
Соединение (II) характеризовалось очень хорошей абсорбцией, но подвергалось быстрому превращению в соединение (V) in vivo, предположительно под действием изофермента (изоферментов) цитохрома Р450 в печени. Соединение (V), видимо, не подвергалось дальнейшему окислению (вероятно, в результате наличия электроноакцепторного заместителя на адамантиновом кольце), но, тем не менее, выводилось из организма относительно быстро. Отсутствие детектируемых первичных метаболитов в моче является сильным аргументом в пользу того, что быстрый клиренс был опосредован формированием конъюгата в фазе II, такого как глюкуронид, который у крыс экскретируется с фекалиями. Конъюгаты фазы II уже сформированы на гидроксильных группах, такие как 3-гидроксиадамантан в соединении (V). В результате заявители предположили, что производное соединения (II), которое имеет стабильный электроноакцепторный заместитель (для того, чтобы предотвратить гидроксилирование адамантинового кольца при действии цитохрома Р450 или других гидроксилаз или оксидаз печени), которое было неспособно формировать конъюгаты фазы II, такие как галозаместители, могут обладать оптимальными ADME свойствами, которые искали авторы настоящего изобретения.
В результате было синтезировано и протестировано соединение (VIII), приведенное ниже. Заявители выбрали хлор-замещенный аналог (вместо фтор-замещенного аналога, например) потому, что это было известное соединение (см. пример 8 в WO 2006/016152), для которого уже был доступен путь синтеза. Более того, введение алифатических C-F связей является сложной задачей с точки зрения организации синтеза и может повысить стоимость товаропродукта из-за стоимости активного фармацевтического компонента на порядок или более
Как и ранее (см. Пример 7), было принято решение подготовить и провести оценку S-энантиомера данного соединения, поскольку известно, что, в целом, S-энантиомеры ациламинолактамных соединений характеризуются более высокой BSCl активностью in vitro.
Очевидное беспокойство, которое было связано с данной молекулой, была стабильность C-Cl связи. Поэтому заявители исследовали стабильность соединения (VIII) in vitro как в физиологическом солевом растворе, так и в препарате микросом печени крысы. Были получены оптимистичные данные, так как менее 2% соединения (VIII) было подвергнуто гидролизу (определяли способом жидкостной хроматографии/масс-спектрометрии с проведением двойной ионной детекции продукта гидролиза соединения (V) и исходного соединения (VIII)) даже через 24 часа при 37°С. Аналогичным образом, при инкубации соединения (VIII) в концентрации 1 мкМ совместно с препаратом микросом крыс в течение 1 часа при 37°С было показано, что менее 10% соединения подверглось деградации по сравнению с контрольным исследованием, которое проводили "в отсутствие НАДФ·Н". В результате заявители сделали заключение, что соединение (VIII) характеризуется сходной стабильностью in vitro и собственным клиренсом по сравнению с соединением (I').
Таким образом, на основании теоретических рассуждений и с применением опыта, полученного при исследовании соединения (I'), а также общих специальных знаний для предсказания ADME свойств, а также с применением общепринятых способов скрининга in vitro, таких как исследование стабильности in vitro и собственного клиренса в препаратах микросом печени, ожидается, что соединение (VIII) будет обладать ADME свойствами, сравнимыми со свойствами соединения (I').
Соответственно, соединение (VIII) подвергли фармакокинетическому анализу у крыс с применением протоколов, которые использовали при исследовании соединений (I')-(VII) в примерах 1 и 7.
Результаты
Параметры простой однокамерной фармакокинетической модели представлены в Таблице 8. Данные для соединение (I') из Таблицы 1 выше приведены для сравнения. Очевидно, что соединение (VIII) является значительно менее эффективным по сравнению с соединением (I'): скорость клиренса более чем в 20 раз выше, и не было обнаружено никакой биодоступности при оральном применении.
Таблица 8. Фармакокинетические параметры для выбранных лактамных BSCl. Биодоступность при оральном применении (биодоступность, %), основной период полувыведения из плазмы (t1/2, минут) после внутривенного введения, клиренс (мл/мин/кг), объем распределения (объем распределения в равновесном состоянии, л/кг) и воздействие при оральном применении (AUC 0-t, мин·нг/мл) из простой однокамерной фармакокинетической модели для каждого соединения приведены средние значения, полученные для трех крыс. Для соединения (VIII) концентрация аналита была ниже предела количественного определения (~2 нг/мл) во все моменты времени после орального применения.
В заключение соединение (VIII), которое заявители выбрали на основании теоретических соображений и результатов тестов, проведенных in vitro, с оптимальными ADME свойствами, тем не менее, находилось среди наименее пригодных ациламинолактамных соединений, как минимум среди тех, для которых были определены фармакокинетические параметры для разработки фармацевтически активного вещества. Для данного соединения характерен быстрый клиренс, что является причиной короткого периода полувыведения из плазмы крови, кроме того, было показано отсутствие какой бы то ни было биодоступности при оральном применении.
Степень превосходства соединения (I') над другими соединениями показана на Фиг.10. На фигуре приведен график для двух фармакокинетических параметров (период полувыведения из плазмы крови и воздействие при оральном применении) для каждого BSCl соединения, для которого были проанализированы фармакокинетические параметры у крыс. Соединение (I') лежит вдали от кластера, представляющего общие неблагоприятные ADME свойства соединений в данном структурном пространстве (и не является нетипичным для многих в целом неродственных классов соединений кандидатов для разработки лекарственных средств), что указывает на то, что соединение (I') неожиданным и необычным образом обладает преимуществами.
Пример 9. Сравнение S- и R-энантиомеров вещества (I)
Было показано, что соединение (I') обладает неожиданными и значительно превосходящими ADME свойствами (см. пример 1). Данное соединение имеет ассиметричный атом углерода (в месте соединения кольца с амидным линкером) и, следовательно, может существовать в форме двух стереоизомеров: S-энантиомер, обозначенный как соединение (I'), и противоположный R-энантиомер, обозначенный как соединений R-(I).
Как и у всех других описанных к настоящему времени ациламинолактамных BSCI S-энантиомер соединения (I) является более эффективным при применении в качестве BSCI in vitro, чем соответствующий R-энантиомер. Именно по этой причине во многих экспериментах изучали соединение (I'), а не изолированный R-энантиомер или смесь энантиомеров. Однако непохоже, что в данном случае ADME свойства связаны с первичной фармакологической активностью. Действительно, для многих соединений ADME свойства пар энантиомеров очень сходны, поскольку (во многих случаях) физические свойства данных соединений особенно доминируют при определении распределения in vivo, и физические свойства энантиомерных пар идентичны.
Поэтому заявители проверили, обладает ли R-энантиомер особенными преимущественными ADME свойствами соединения (I'). Поскольку полезность любого особенного соединения зависит от силы его первичного фармакологического действия (действий) и от распределения молекул данного соединения in vivo (поскольку, для того, чтобы соединение было эффективным, молекула активного вещества должна присутствовать в предназначенном участке-мишени (участках-мишенях) в достаточной концентрации для того, чтобы проявить фармакологическое действие), поэтому соединение, которое характеризуется меньшей активностью, но более преимущественными ADME свойствами, может быть более эффективным (или обладать более действием in vivo), чем соединение, взятое в качестве препарата сравнения, которое обладает большей силой действия, но менее предпочтительными ADME свойствами.
Таким образом, заявители синтезировали изолированный R-энантиомер соединения (I) (точно, как описано здесь для изолированного S-энантиомера, но замещение изначального материала S-орнитина проводили с применением коммерческого препарата R-орнитина высокой стереохимической чистоты) и определили фармакокинетические свойства данного активного соединения на крысах с применением тех же способов анализа, приведенных выше (см. Примеры 1, 7 и 8).
Результаты
Параметры простой однокамерной фармакокинетической модели показаны в Таблице 9. Данные для соединения (I') из Таблицы 1 приведены для сравнения. Очевидно, что соединение R-(I) обладает, в большой степени, неожиданно превосходными ADME свойствами, показанными соединения (I'). Как и в случае (I'), соединение R-(I) также характеризуется существенной количественной биодоступностью при оральном применении и оказывает воздействие после введения единичной оральной дозы в количестве 3 мг/кг более, которая является более чем в 5 раз более высокой, чем доза следующего лучшего соединения, исследованного к настоящему времени (см. вещество (V) в примере 1; Таблица 1).
Таблица 9. Фармакокинетические параметры изолированных R- и S-энантиомеров соединения (I). Биодоступность при оральном применении (биодоступность, %), основной период полувыведения из плазмы (t1/2, минут) после внутривенного введения дозы, клиренс (мл/мин/кг), объем распределения (объем распределения в равновесном состоянии, л/кг) и воздействие при оральном применении (AUC 0-t, мин·нг/мл) из простой однокамерной фармакокинетической модели для каждого соединения, представлены средние значения, полученные по трем крысам. В обоих случаях, максимальную концентрацию в плазме обычно наблюдали через 30 минут, что соответствует хорошей абсорбции.
Хотя соединение R-(I) разделяет в значительной степени необычно преимущественные ADME свойствами соединения (I'), тем не менее имеются некоторые статистически значимые различия. Наиболее выраженным из данных различий является повышенный плазматический клиренс (~3 раза выше у соединения R-(I) по сравнению с (I')), что приводит к тому, что период полувыведения из плазмы после внутривенного введения в 2 раза короче, а воздействие после применения единичной оральной дозы в 3 раза ниже.
Клиренс соединения R-(I) приближается к скорости гломерулярной фильтрации у крыс (обычно сообщают о величине 9 мл/мин/кг), в то время как клиренс соединения (I') значительно более низкий. Одно из возможных объяснений полученных данных состоит в том, что соединение (I') подвергается активной реабсорбции, возможно, в дистальных канальцах нефрона, и данная повторная абсорбция является избирательной по отношению к S-энантиомеру по сравнению с R-энантиомером. Если, что вероятно, причиной неожиданных и высокопреимущественных ADME свойств соединения (I') частично является обратный захват в нефроне через транспортер, способный различить S-энантиомер и R-энантиомер, тогда данное объяснение подчеркивает непредсказуемость ADME свойств и далее демонстрирует то, что открытие, описываемое в данном документе, не является очевидным.
Тем не менее, R-(I) также является преимущественным соединением в соответствии с данным изобретением (как показано открытым кругом на Фиг.10, который иллюстрирует то, что оно ближе по ADME свойствам к соединению (I'), чем к общему классу ациламинолактамных соединений). В заключение таким образом, показано, что оба соединения R-(I) и (I') и вследствие этого более общее соединение (I) характеризуются неожиданными и значительными преимущественными ADME свойствами по сравнению с другими ациламинолактамными BSCl, которые были ранее описаны. Более того, соединение (I') слегка, но значительно в целом превосходит соединение (I), и таким образом, является более предпочтительным.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВОВОСПАЛИТЕЛЬНЫЕ АГЕНТЫ | 2005 |
|
RU2410376C2 |
| ПРОТИВОВОСПАЛИТЕЛЬНЫЕ СРЕДСТВА | 2004 |
|
RU2365585C2 |
| НОВЫЙ ИНГИБИТОР ГЛУТАМИНИЛЦИКЛАЗ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2791703C2 |
| НОВЫЙ ИНГИБИТОР ГЛУТАМИНИЛЦИКЛАЗ И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2665633C1 |
| НОВЫЙ ИНГИБИТОР ГЛУТАМИНИЛЦИКЛАЗ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЛЕГКИХ И ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2017 |
|
RU2662559C1 |
| ЛИГАНДЫ ДЛЯ G-БЕЛОК СОПРЯЖЕННЫХ РЕЦЕПТОРОВ | 2005 |
|
RU2454416C2 |
| КОМПОЗИЦИЯ ДЛЯ МОДУЛИРОВАНИЯ ЭКСПРЕССИИ МОЛЕКУЛ КЛЕТОЧНОЙ АДГЕЗИИ | 2007 |
|
RU2503463C2 |
| ПИПЕРАЗИНДИОНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ОКСИТОЦИНА | 2005 |
|
RU2382038C2 |
| ЛИГАНДЫ ДЛЯ РЕЦЕПТОРОВ, СВЯЗАННЫХ С G-БЕЛКОМ | 2006 |
|
RU2471022C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К CRTH2 | 2007 |
|
RU2458918C2 |
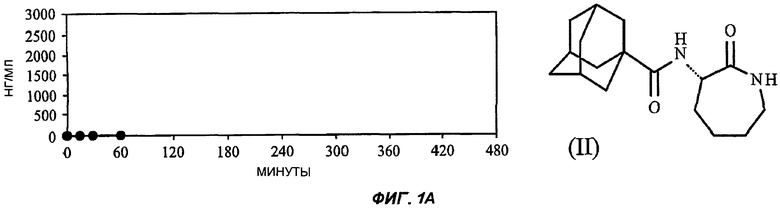
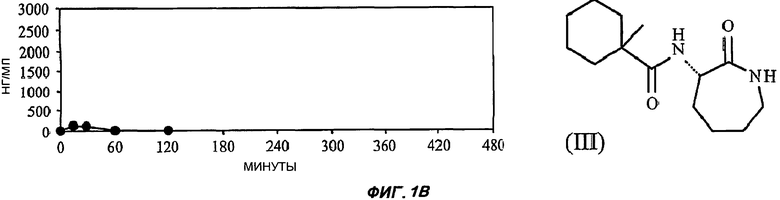
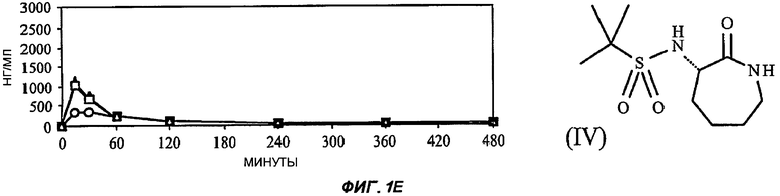
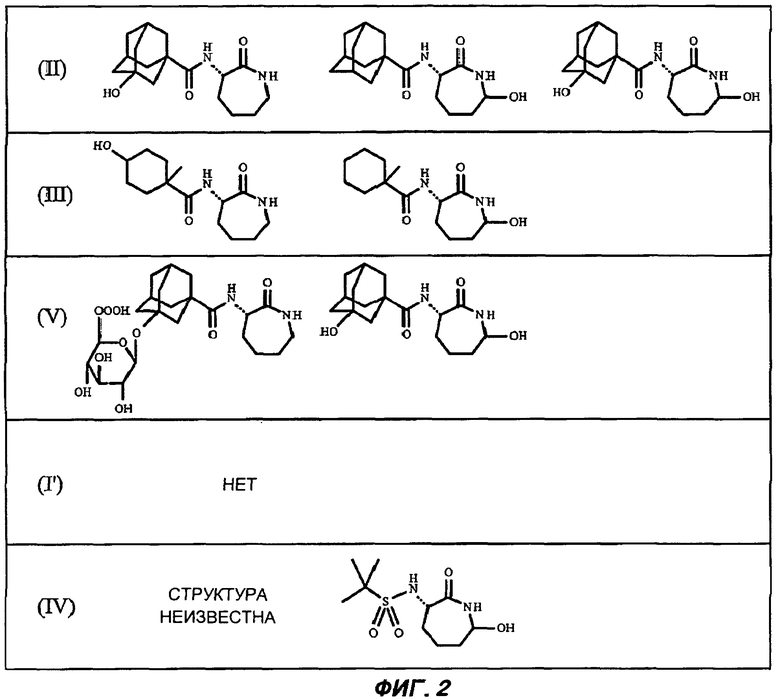
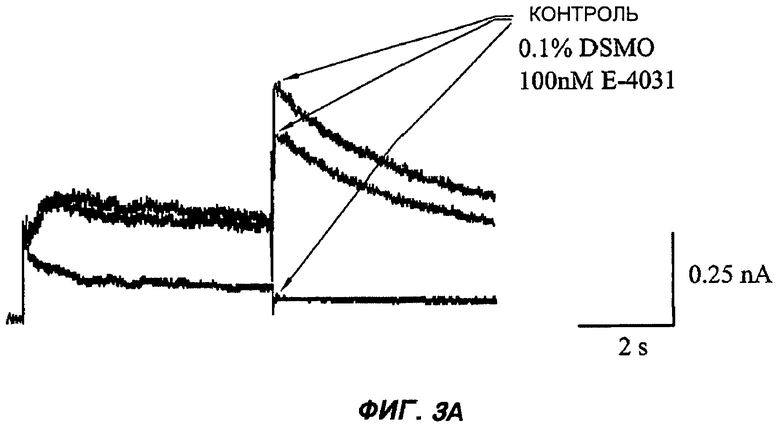
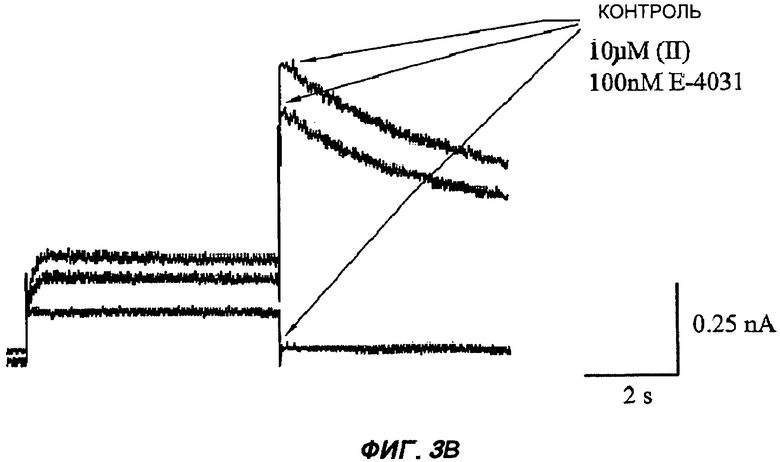
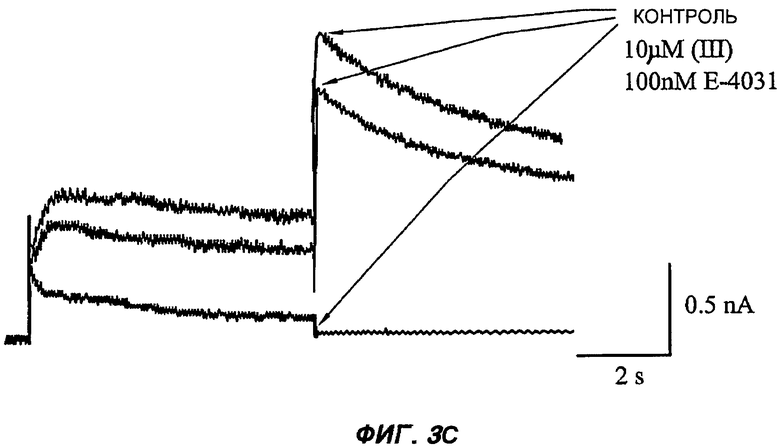
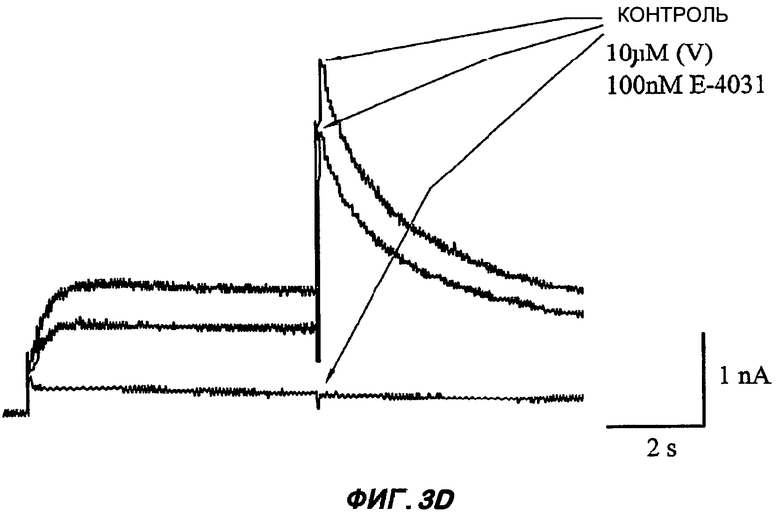
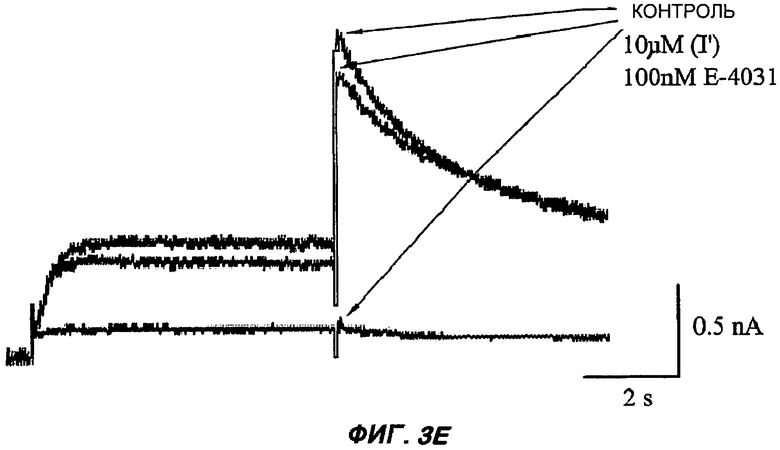
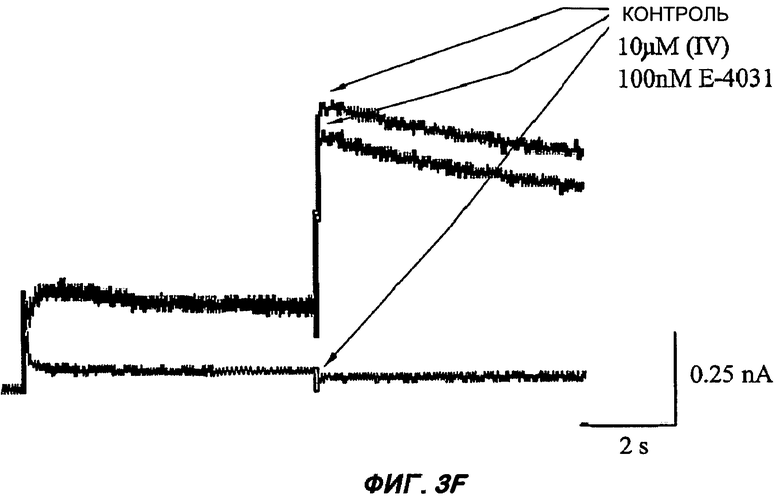
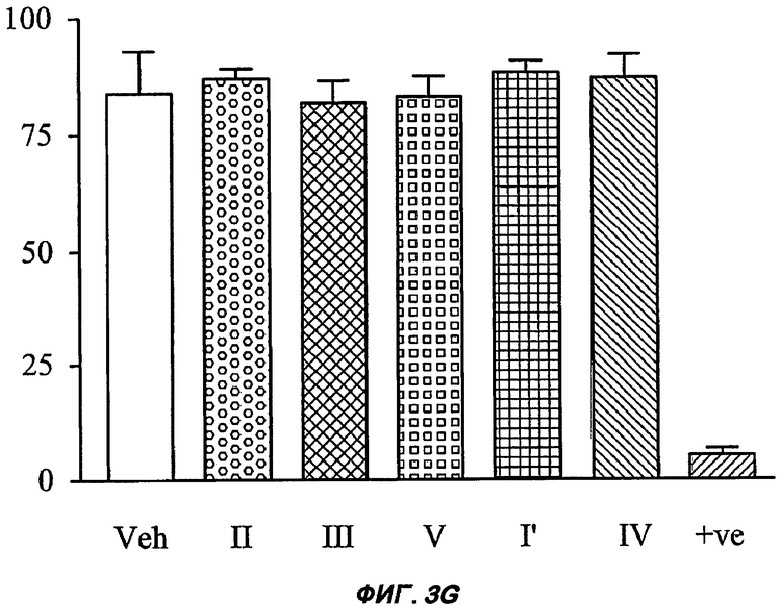
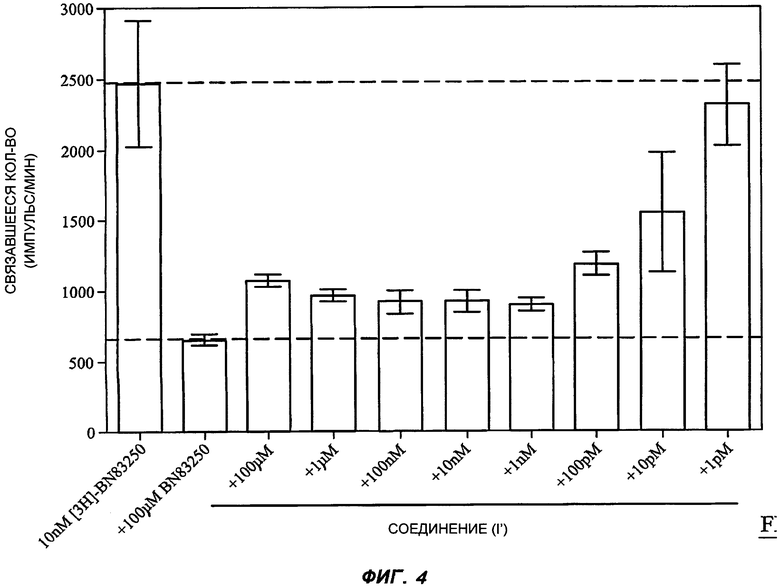
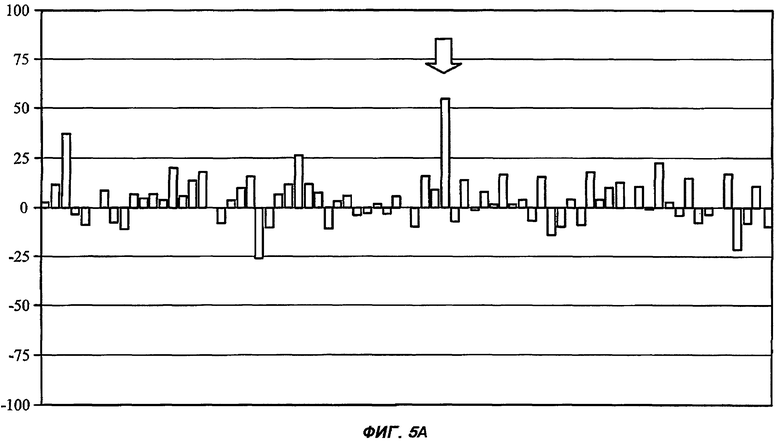
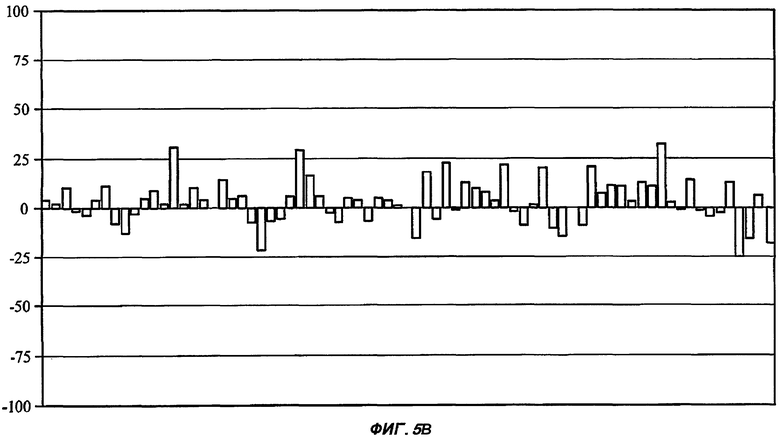
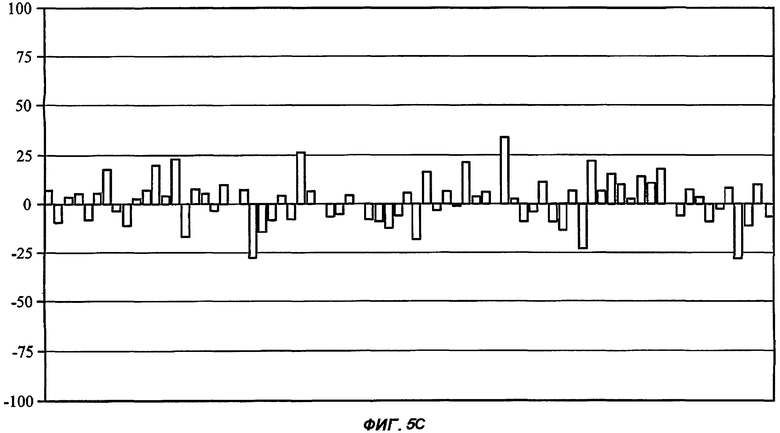
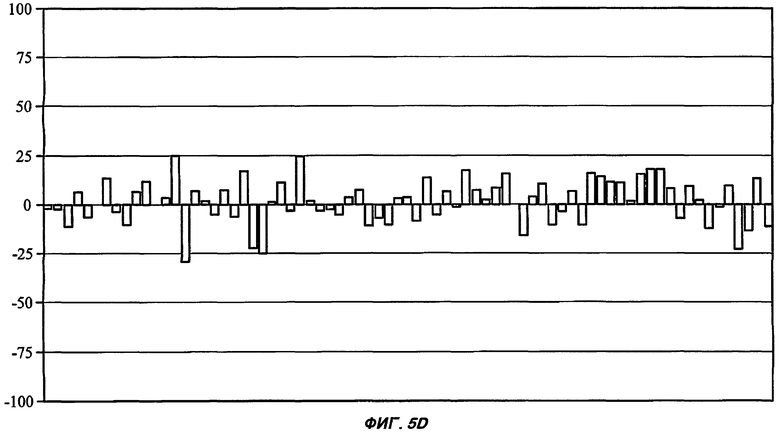
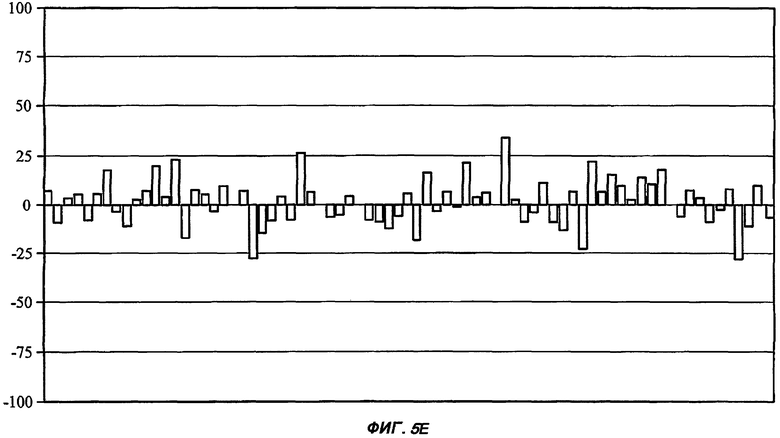
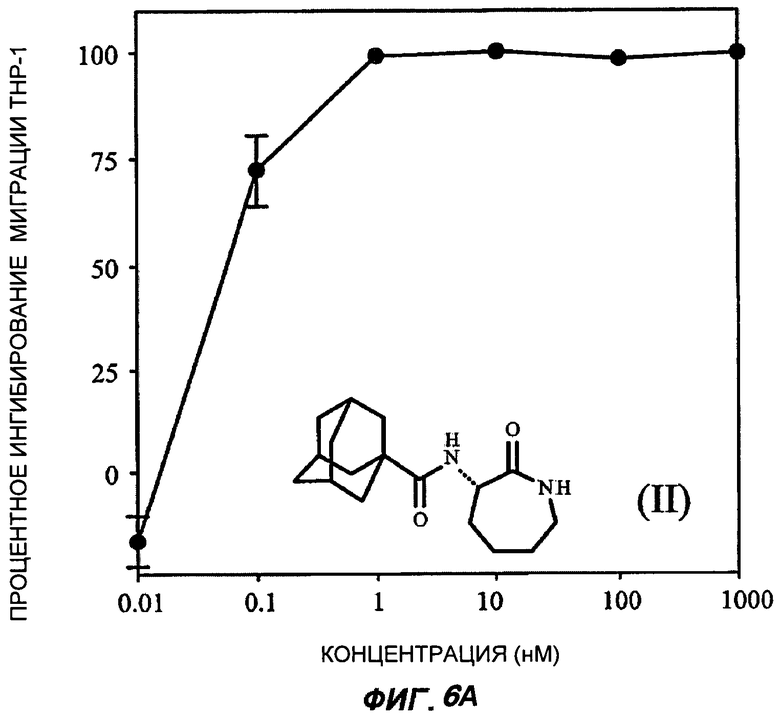
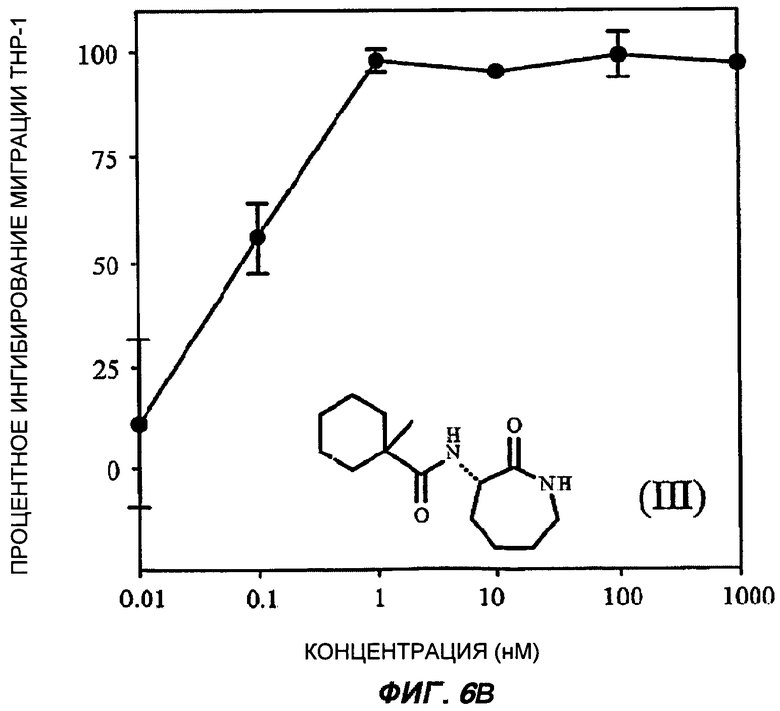
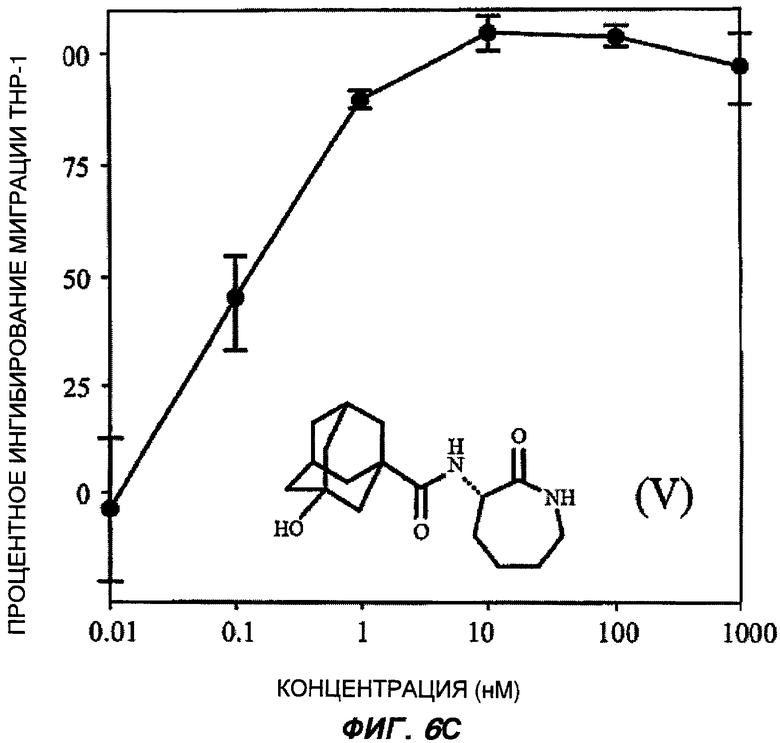
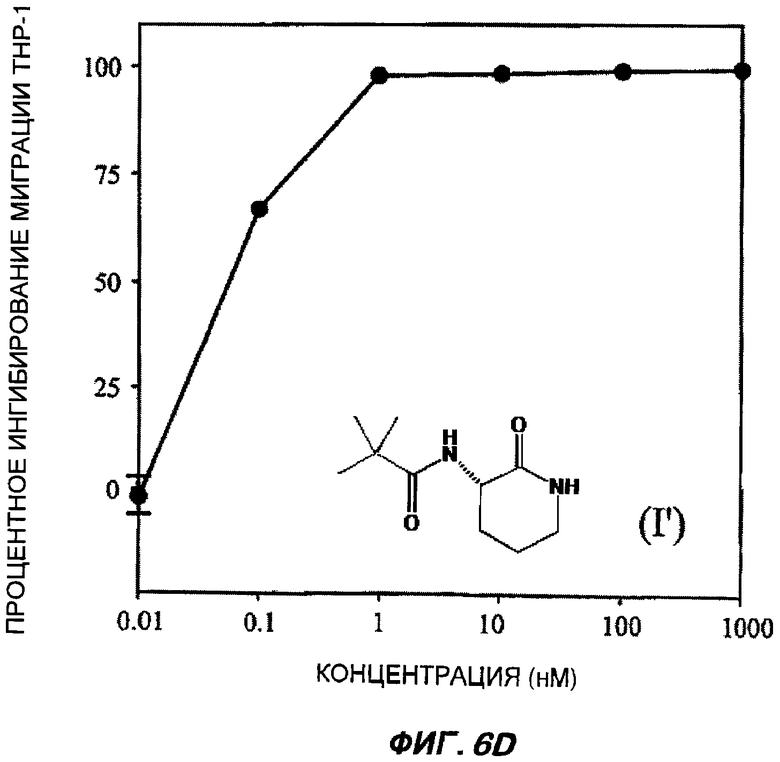
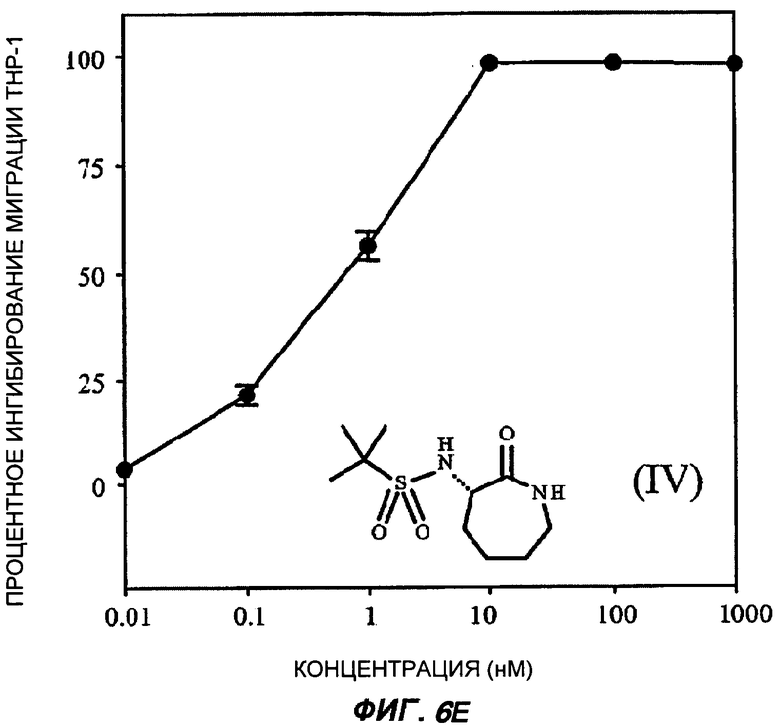
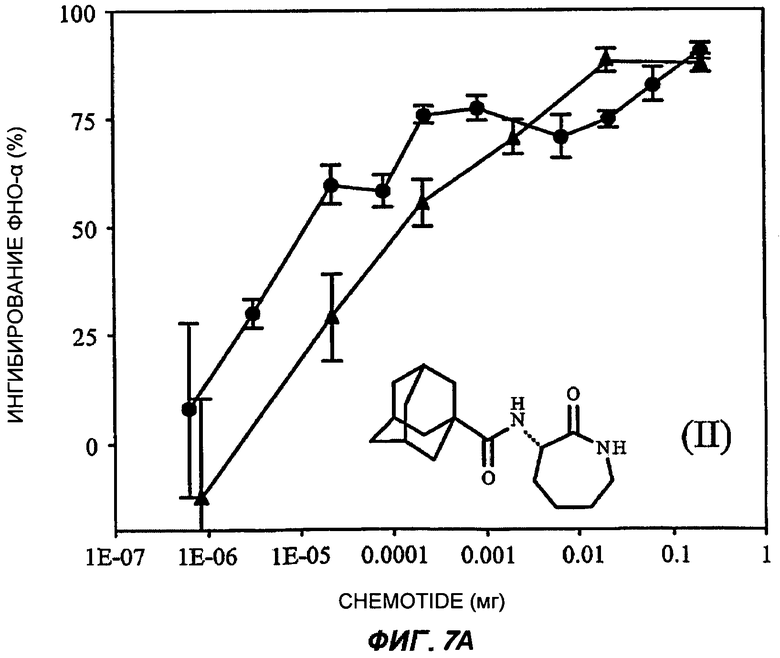
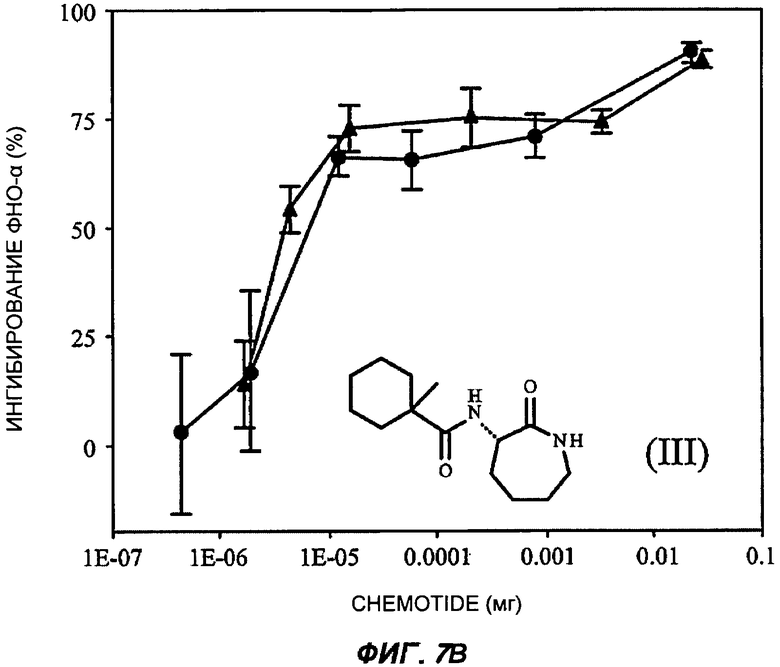
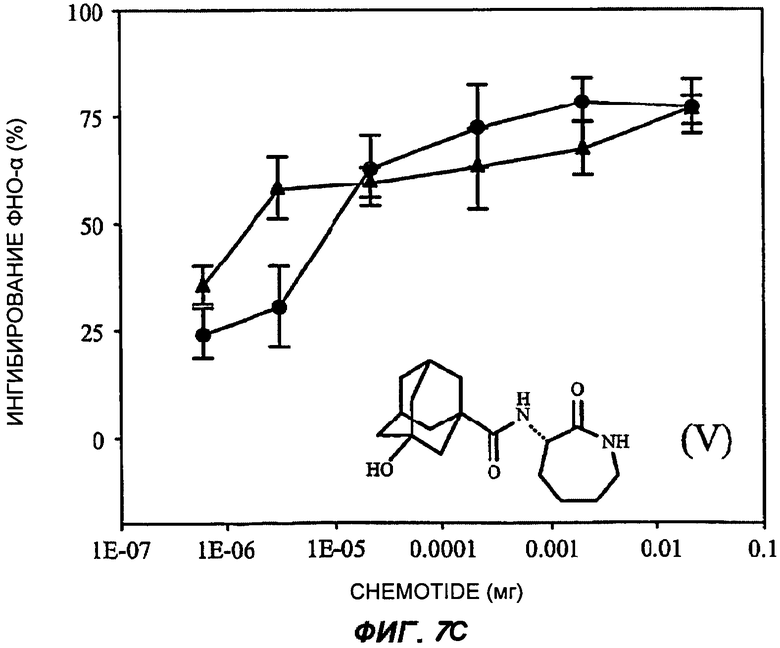
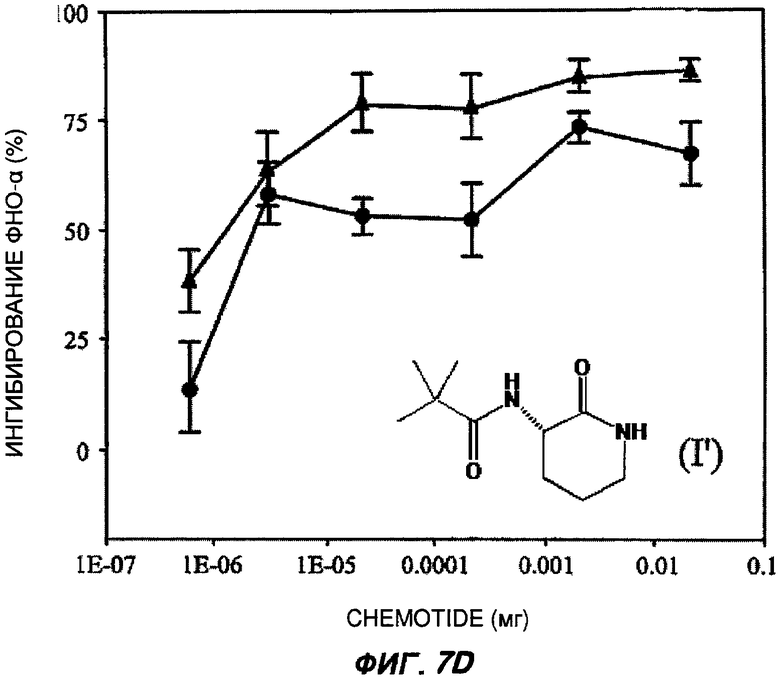
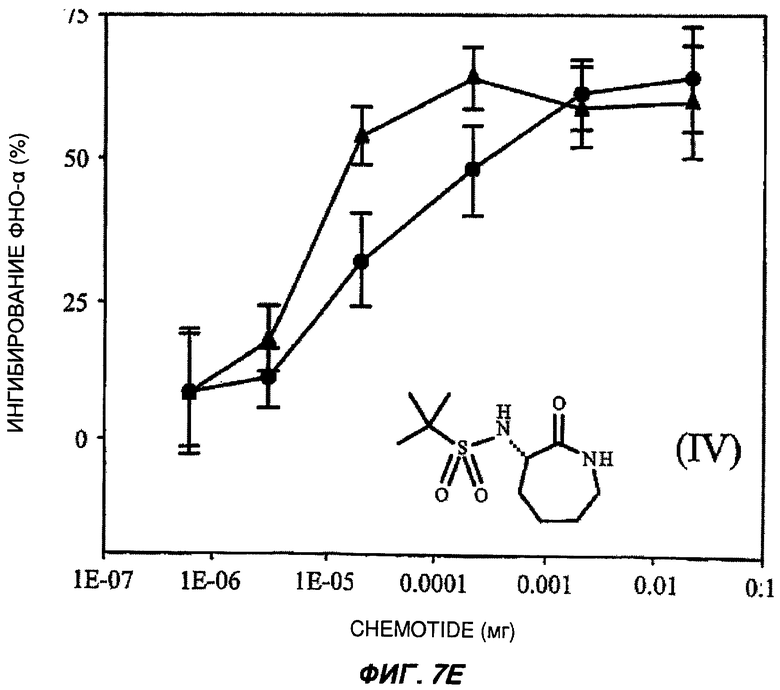
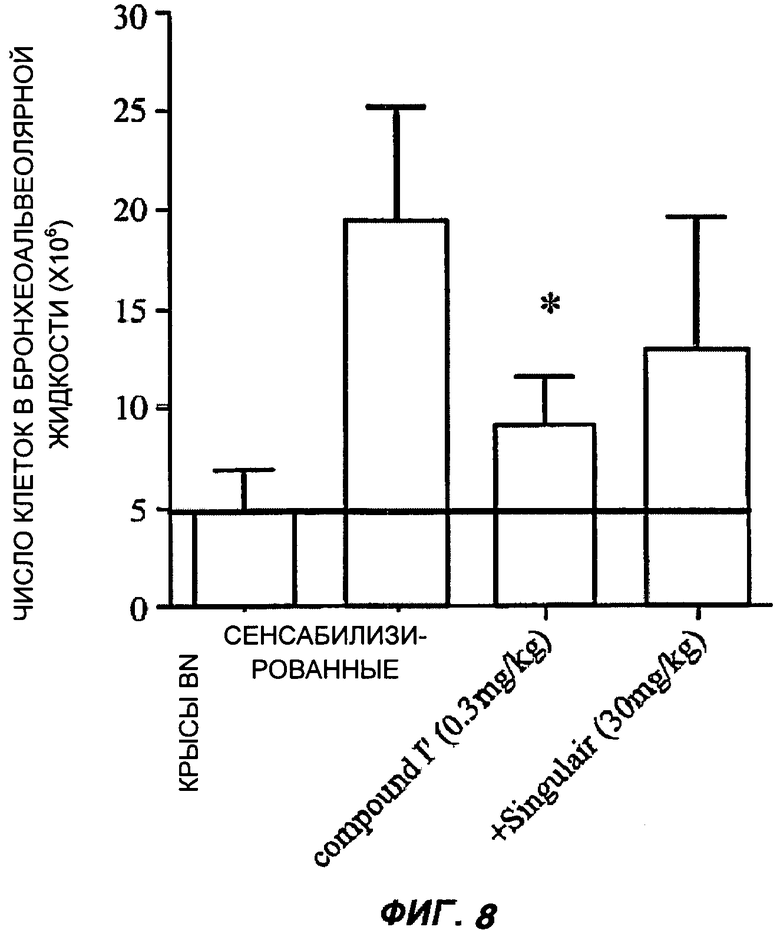
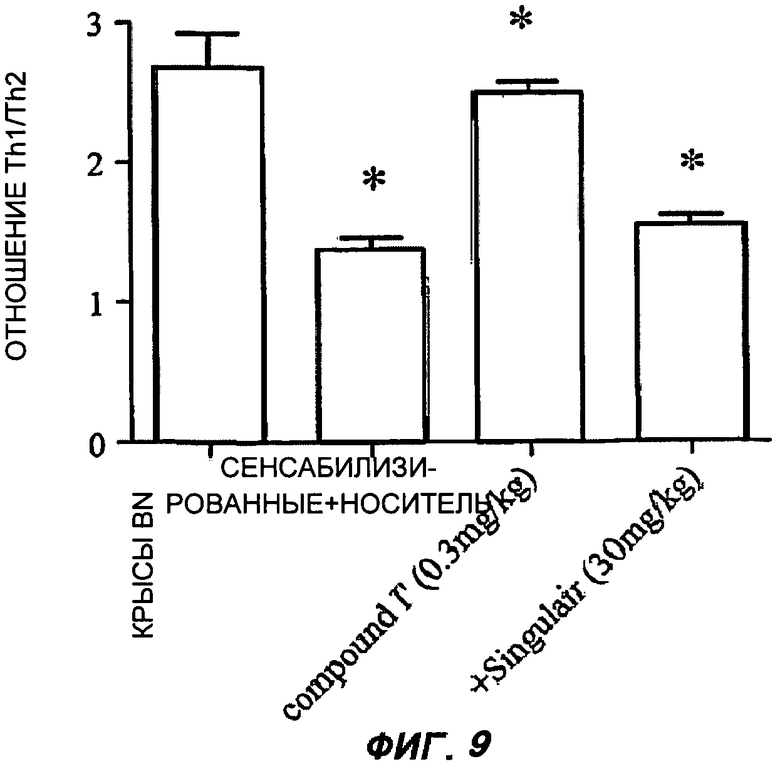
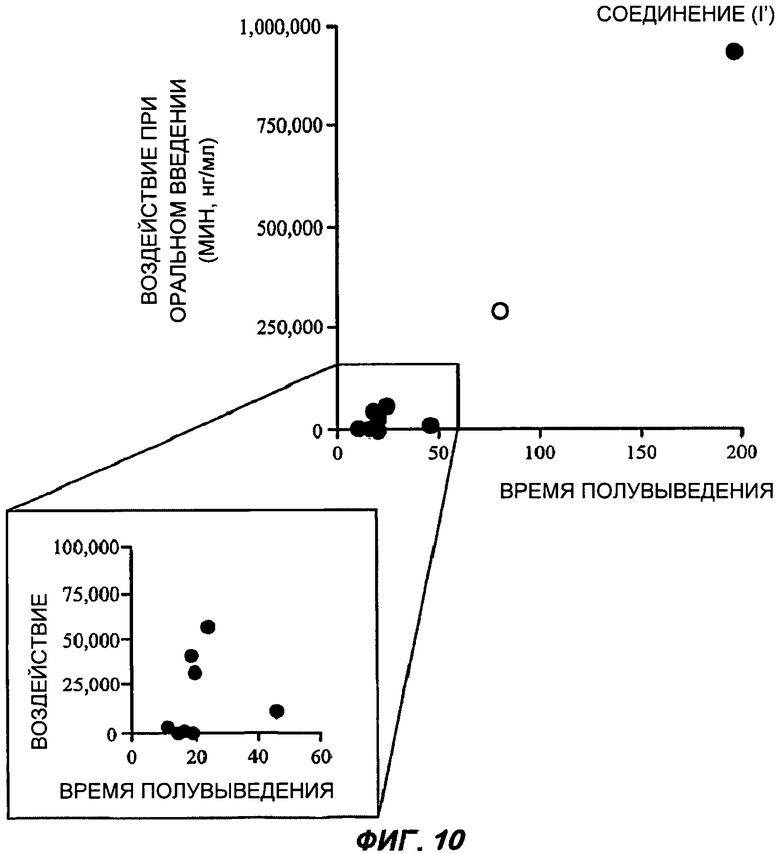
Предложены 3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-он (I), (S)-3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-он (I') и фармацевтические композиции, приготовленные с применением указанного соединения или его изомера, а также их применение для изготовления лекарственного средства, направленного на предотвращение или лечение воспалительных состояний, и соответствующий способ лечения. Показано преимущество соединений по отношению к другим ингибиторам хемокинов широкого спектра действия (BSCI) в отношении противовоспалительной активности, фармакокинетических, токсикологических свойств и параметров фармакологической безопасности: (S)-3-(2',2'-диметилпропаноиламино)-тетрагидропиридин-2-он в 5-25 раз более активен, чем (R)-изомер. 7 н. и 6 з.п. ф-лы, 10 ил., 9 табл.
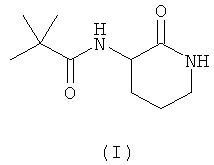
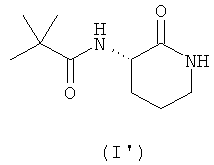
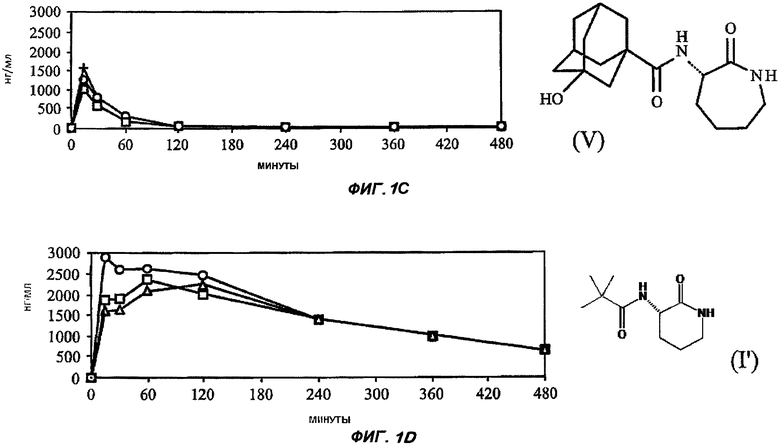
1. Применение соединения формулы (I) или фармацевтически приемлемой соли указанного соединения для приготовления лекарственного средства, предназначенного для лечения воспалительного состояния:
2. Применение соединения формулы (I') или фармацевтически приемлемой соли указанного соединения для приготовления лекарственного средства, предназначенного для лечения воспалительного состояния:
3. Фармацевтически приемлемая композиция для лечения воспалительного состояния, которая содержит в качестве активного вещества соединение формулы (I) или фармацевтически приемлемую соль указанного соединения и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и/или носитель:
4. Фармацевтически приемлемая композиция для лечения воспалительного состояния, которая содержит в качестве активного вещества соединение формулы (I') или фармацевтически приемлемую соль указанного соединения и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и/или носитель:
5. Соединение общей формулы (I):
6. Соединение общей формулы (I'):
7. Применение соединения формулы (I) или (I') по любому из пп.1 или 2, согласно которому воспалительное состояние выбрано из группы, включающей: аутоиммунные заболевания, нарушения работы сосудов, инфекцию или репликацию вируса, астму, остеопороз (низкую плотность минерального вещества кости), рост опухоли, отторжение трансплантированного органа и/или задержки функционирования трансплантата или органа, нарушение, характеризующееся повышенным уровнем TNF-α, псориаз, повреждения кожи, нарушения, вызванные внутриклеточными паразитами, невропатическую боль, аллергии, болезнь Альцгеймера, ответную реакцию, вызванную антигеном, подавление иммунного ответа, ревматоидный артрит, рассеянный склероз, боковой амио-трофический склероз, фиброз и формирование спаек.
8. Применение соединения формулы (I) или (I') по п.7, согласно которому указанное воспалительное состояние представляет собой астму, аллергический ринит или хроническое неспецифическое заболевание легких.
9. Применение соединения формулы (I) или (I') по п.7, согласно которому указанное воспалительное состояние представляет собой формирование спаек после гинекологической или общей хирургии.
10. Применение соединения формулы (I) или (I') по п.7, согласно которому указанное воспалительное состояние представляет собой поражение почек, включая диабетическую нефропатию.
11. Способ лечения, уменьшения тяжести состояния или профилактики развития симптомов воспалительного состояния (включая неблагоприятную воспалительную реакцию на действие любого агента), согласно которому пациенту вводят соединение, композицию или лекарственное средство согласно любому из пп.1-10 в количестве, обладающем противовоспалительным действием.
12. Фармацевтически приемлемая композиция по п.3 или 4 для применения при лечении, уменьшения тяжести или профилактики симптомов воспалительного состояния.
13. Фармацевтически приемлемая композиция по п.12, отличающаяся тем, что указанное состояние выбрано из состояний, перечисленных в любом из пп.7-10.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Бестрансформаторный преобразователь напряжения источника питания радиоприемника | 1986 |
|
SU1374365A1 |
| WO 2002047701 А1, 12.12.2001, Реферат [on line] [Найдено из базы данных PatSearch] | |||
| ГЕТЕРОАЛКИЛАМИНОЗАМЕЩЕННЫЕ БИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ р38 | 2000 |
|
RU2265606C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ ВЕЩЕСТВА | 2000 |
|
RU2220956C2 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| FOX DL | |||
| et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

