Настоящее изобретение относится к способу получения блокатора ангиотензинового рецептора (ARB; также называемый антагонистом рецептора ангиотензина II или антагонистом рецептора AT1) и его солей, к новым промежуточным соединениям и стадиям способа. ARB могут, например, использоваться для лечения гипертензии и связанных с нею заболеваний и состояний.
Настоящее изобретение относится к новому способу получения соединения формулы (IIа), раскрытому ниже в описании, включающему следующую последовательность стадий:
i) защиту тетразольной группы галидного соединения формулы (IVa) настоящего изобретения при реакции с соединением формулы (IVб) настоящего изобретения;
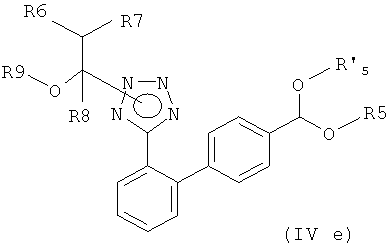
ii) катализируемую переходным металлом реакцию кросс-сочетания между полученным галоидным соединением формулы (IVв) с защищенным тетразолом настоящего изобретения и цинкорганическим соединением формулы (IVв)
настоящего изобретения; и
iii) удаление защитной группы.
В литературе приведены различные методы приготовления соединений формулы (IIа) настоящего изобретения. Например, документы уровня техники ЕР 0550313 и FR 2688503 описывают альтернативный способ, отличающийся от способа согласно настоящему изобретению защитной группой тетразольного кольца. А именно, если согласно настоящему изобретению тетразол защищен во время реакции соединением формулы (IVб) настоящего изобретения, то в описанных в ЕР 0550313 и FR 2688503 соединениях защитная группа тетразола представляет собой алкил, например t-бутил, арилалкил, например, трифенилметил, алкилстаннил, алкилсилил или группу CH2OR6, где R выбран из группы, включающей метил, фенилметил, 1,1-диметилэтил, 2,2,2-трихлорэтил, бензилоксикарбонил или 2,2,2-трихлорэтилоксикарбонил. Однако ни один из примеров, включенных в описание и направленных на поддержку соответствующего процесса в документе FR 2688503, не относится к стадии удаления защитной группы, а документ ЕР 0550313 описывает лишь удаление защитной группы у тетразола с тритильной защитной группой (Пример 7).
Класс ARB включает соединения с различными структурными особенностями, особенно предпочтительными являются непептидные соединения. Например, могут быть упомянуты соединения, выбранные из группы, состоящей из валсартана (ЕР 443983), лосартана (ЕР 253310), кандесартана (ЕР 459136), эпросартана (ЕР 403159), ирбесартана (ЕР 454511), олмесартана (ЕР 503785) и тасосартана (ЕР 539086), или, в каждом случае, их фармацевтически приемлемые соли.
Все эти ARB включают следующий общий структурный элемент:
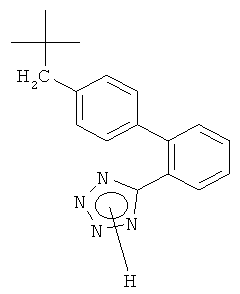
Образование тетразольного кольца является критической стадией при получении этих соединений. Способы получения ARB с такими структурными особенностями включают образование указанного тетразольного кольца исходя из соответствующих цианопроизводных, которые реагируют с НN3 или его подходящей солью щелочного металла, такой как азид натрия, или с органическим азидом олова, таким как азид трибутилолова, или с силилазидом. Применение азидов для получения тетразольной кольцевой системы требует сложной системы для обеспечения безопасности реакции при широкомасштабном производстве. Соответственно, целью является разработка альтернативного варианта способа, который бы исключил применение азидов на последних стадиях получения соответствующих ARB.
Целью настоящего изобретения является синтез соединений формулы (I), который (1) не включает стадии способа с использованием азида, (2) приводит к хорошим выходам, (3) уменьшает загрязнение окружающей среды, например, исключая органические соединения олова, (4) является экономичным благодаря использованию меньшего количества реакционных стадий для получения соединений формулы (I), (5) обеспечивает энантиомерно чистые конечные продукты и продукты высокой способности к кристаллизации. Кроме того, поскольку тетразольная кольцевая система образуется на ранней стадии реакции, (6) риск загрязнения конечного продукта (и последних промежуточных соединений) следовыми количествами компонентов олова является меньшим. Обычно, тетразольное кольцо образуется по реакции соответствующего цианопроизводного с органическим соединением олова, таким как азид трибутилолова. По экологическим причинам тяжелый металл олова и особенно органические соединения олова вводят с особой осторожностью. Кроме того, (7) другой целью настоящего изобретения является обеспечения способа, который может осуществляться в промышленном масштабе и может использоваться для соответствующего способа получения и для устранения, например, рацемизации и таким образом для отделения любых энантиомеров.
Неожиданно было обнаружено, что способ в соответствии с настоящим изобретением отвечает указанным выше целям.
Настоящее изобретение относится к способу получения соединения формулы (I)
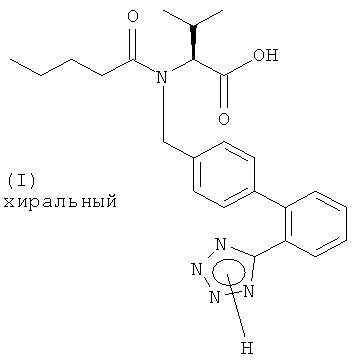
или его соли, включающему:
(а) реакцию соединения формулы (IIа)
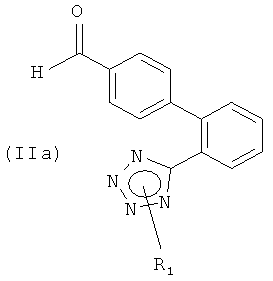
или его соли, где R1 представляет собой водород или защитную группу тетразола, с соединением формулы
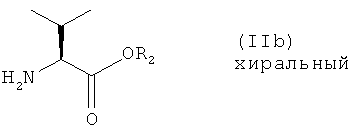
или его солью, где R2 представляет собой водород или карбоксизащитную группу, в условиях восстановительного аминирования; и
(б) ацилирование полученного соединения формулы (II с)
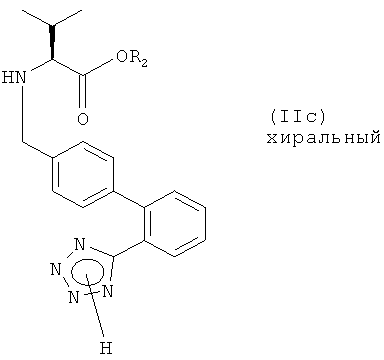
или его соли соединением формулы (IId)
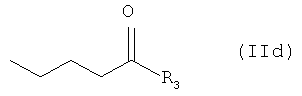
где R3 представляет собой активирующую группу; и,
(в) если R1 и/или R2 являются отличными от водорода, удаление защитной группы(групп) в полученном соединении формулы (IIе)

или его соли; и
(г) выделение полученного соединения формулы (I) или его соли; и, при необходимости, превращение полученной свободной кислоты формулы (I) в ее соль или превращение полученной соли соединения формулы (I) в свободную кислоту формулы (I) или превращение полученной соли соединения формулы (I) в различные соли.
Описанные выше и далее реакции в вариантах осуществляют, например, в отсутствие или, обычно, в присутствии подходящего растворителя или разбавителя или в из смеси, реакцию, при необходимости, проводят при охлаждении, при комнатной температуре или при нагревании, например, при температуре в области приблизительно от -80°С до температуры кипения реакционной среды, предпочтительно приблизительно от -10°С приблизительно до +200°С, и, при необходимости, в закрытом сосуде, под давлением, в атмосфере инертного газа и/или в безводных условиях.
Соединения формул (IIа), (IIb), (IIс) и (IIе), в которых один или оба R1 и R2 представляют собой водород, могут образовывать соли с основаниями, поскольку и незащищенное тетразольное кольцо, и незащищенная карбоксигруппа имеют кислотные свойства, тогда как соединения формул (IIb) и (IIе) могут также образовывать соли с кислотами.
Соответствующая тетразольная защитная группа (R1) выбирается из групп, известных из уровня техники. Особенно R1 выбирается из группы, состоящей из трет-С4-С7-алкила, такого как трет-бутил; С1-С2-алкила, который моно-, ди- или тризамещен фенилом, такого как бензил или бензгидрил или тритил, где фенильное кольцо является незамещенным или замещено одним или несколькими, например двумя или тремя, остатками, например остатками, выбранными из группы, состоящей из трет-С1-С7-алкила, гидрокси, С1-С7-алкокси, С2-С8-алканоилокси, галогена, нитро, циано и трифторметила (СF3); пиколинила; пиперонила; кумила; аллила; циннамоила; флуоренила; силила, такого как три-С1-С4-алкилсилила, например триметилсилила, триэтилсилила или трет-бутилдиметилсилила, или ди-С1-С4-алкилфенилсилила, например димстилфенилсилила; С1-С7-алкилсульфонила; арилсульфонила, такого как фенилсульфонил, где фенильное кольцо является незамещенным или замещенным одним или несколькими, например двумя или тремя, остатками, например остатками, выбранным из группы, состоящей из С1-С7-алкила, гидрокси, С1-С7-алкокси, С2-С8-алканоилокси, галогена, нитро, циано и СF3; С2-C8-алканоила, такого как ацетил или валероил; и этерифицированного карбокси, такого как С1-С7-алкоксикарбонила, например метокси-, этокси- или трет-бутилокси-карбонила; и аллилоксикарбонила. Примерами предпочтительных защитных групп являются трет-бутил, бензил, п-метоксибензил, 2-фенил-2-пропил, дифенилметил, ди(п-метоксифснил)мстил, тритил, (п-метоксифенил)дифенилметил; дифенил(4-пиридил)метил, бензилоксиметил, метоксиметил, этоксимстил, метилтиометил, 2-тетрагидропиранил, аллил, триметилсилил и триэтилсилил.
Соответствующая карбоксизащитная группа (R2) выбирается из групп, известных из уровня техники. Особенно R2 выбирается из группы, состоящей из С1-С7-алкила, такого как метил, этил или трет-C4-С7-алкил, особенно трет-бутил; C1-C2-алкила, который является моно-, ди- или тризамещенным фенилом, таким как бензил или бензгидрил, где фенильное кольцо является незамещенным или замещенным одним или несколькими, например двумя или тремя, остатками, например остатками, выбранными из группы, состоящей из С1-C7-алкила, гидрокси, С1-С7-алкокси, С2-С8-алканоилокси, галогена, нитро, циано и СF3; пиколинила; пиперонила; аллила; циннамила; тетрагидрофуранила; тетрагидропиранила; метоксиэтоксиметила и бензилоксиметила.
Предпочтительным примером защитных групп является бензил.
Активирующая группа R3 представляет собой, например, активирующую группу, которая используется в химии пептидов, такую как галоген, такой как хлор, фтор или бром; С1-С7-алкилтио, такой как метилтио, этилтио или трет-бутилтио; пиридилтио, такой как 2-пиридилтио; имидазолил, такой как 1-имидазолил; бензтиазолилокси, такой как бензтиазолил-2-окси-; бензотриазолокси, такой как бензотриазолил-1-окси-; С2-С8-алканоилокси, такой как бутироилокси или пивалоилокси; или 2,5-диоксопирролидинилокси.
Используемые здесь и далее общие термины имеют следующие значения, если не указано иное:
С1-С7-Алкил представляет собой, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил или соответствующий пентил, гексил или гептил. С1-С4-алкил, особенно метил, этил или трет-бутил, являются предпочтительными.
С1-С7-Алкокси представляет собой, например, метокси, этокси, н-пропилокси, изопропилокси, н-бутилокси, изобутилокси, втор-бутилокси, трет-бутилокси или соответствующий пентилокси, гексилокси или гептилокси. C1-C4-алкокси является предпочтительным. Особенно предпочтительным является метокси, этокси и трет-бутокси.
С2-С8-Алканоил в С2-С8-алканоилокси представляет собой ацетил, пропионил, бутирил, изобутирил или пивалоил. С2-С5-Алканоил является предпочтительным. Особенно предпочтительным является ацетил или пивалоил.
Галоген представляет собой, в частности, хлор, фтор или бром, и в широком смысле включает йод. Хлор является предпочтительным.
Стадия (а):
На стадии реакции (а) восстановительное аминирование осуществляют в присутствии восстанавливающего агента. Подходящим восстанавливающим агентом является боргидрид, который также может находиться в форме комплекса, или водород или донор водорода, в присутствии катализатора гидрирования. Кроме того, восстанавливающим агентом является подходящий селенид или силан.
Подходящим боргидридом или боргидридным комплексом является, например, боргидрид щелочного металла, такой как боргидрид натрия или боргидрид лития; боргидрид щелочно-земельного металла, такой как боргидрид кальция; цианоборгидрид щелочного металла, такой как цианоборгидрид натрия или цианоборгидрид лития, три-(С1-С7-алкокси)-боргидрид щелочного металла, такой как триметоксиэтоксиборгидрид натрия; тетра-С1-С7-алкиламмоний-(циано)боргидрид, такой как тетрабутиламмонийборгидрид или тетрабутиламмонийцианоборгидрид.
Подходящим катализатором для восстановительного аминирования водородом или донором водорода является, например, никель, такой как никель Ренея, и благородные металлы или их производные, например оксиды, такие как палладий, платина или оксид платины, которые могут применяться, при необходимости, на носителях, например, на угле или карбонате кальция, например, платина на угле. Гидрирование водородом или донором водорода предпочтительно может осуществляться под давлением от 1 и приблизительно 100 атмосфер и при комнатной температуре приблизительно от -80° приблизительно до 200°С, в частности между комнатной температурой и приблизительно 100°С.
Предпочтительным донором водорода является, например, система, включающая 2-пропанол и, при необходимости, основание, или, наиболее предпочтительно, муравьиную кислоту или ее соль, например, с щелочным металлом, или ее три-С1-С7-алкил-аммониевую соль, например, ее соль натрия или калия, при необходимости, в присутствии третичного амина, такого как триэтиламин. Другие доноры водорода включают другие спирты, такие как этанол, 2-метоксиэтанол, бензиловый спирт, бензгидрол, пентан-2-ол, 1,2-этандиол, 2,3-бутандиол или циклогександиол, гидразин, циклогексен, циклогексадиен, индан, тетралин, индолин, тетрагидрохинолин, гидрохинон, гипофосфиновая кислота или их подходящая соль, такая как соль натрия, тетрагидроборат натрия, углеводы, аскорбиновая кислота, лимонен или силаны. Донор водорода может также использоваться в качестве растворителя, особенно 2-пропанол или муравьиная кислота.
Подходящим селенидом является, например, селенофенол, который является незамещенным или замещенным. Подходящие заместители включают, например, один, два или три заместителя, выбранные, например, из галогена, трифторметила, трифторметокси, С1-С7-алкила, С1-С7-алкокси, нитро, циано, гидроксила,
С2-С12-алканоила, C1-C12-алканоилокси и карбокси. Предпочтительными являются те силаны, которые полностью растворимы в реакционной среде и которые могут производить органически растворимые побочные продукты. Особенно предпочтительными являются три-С1-С7-алкилсиланы, особенно триэтилсилан и триизопропилсилан. Предпочтительными являются коммерчески доступные селениды.
Подходящим силаном является, например, силан, который является тризамещенным заместителем, выбранным из группы, состоящей из C1-C12-алкила, особенно С1-C7-алкила, и С2-С30-ацила, особенно C1-C8-ацила. Предпочтительными являются коммерчески доступные силаны.
Восстановительное аминирование предпочтительно осуществляют в кислотных, нейтральных или предпочтительно основных условиях. Подходящее основание включает, например, гидроксид или карбонат щелочного металла, такой как гидроксид натрия, гидроксид калия или карбонат калия. Кроме того, может использоваться основание амина, например, три-С1-С7-алкиламина, такого как триэтиламин, три-н-пропиламин, трибутиламин или этилдиизопропиламин, пиперидина, такого как N-метилпиперидин, или морфолина, такого как N-метилморфолин. Предпочтительные основания включают гидроксид лития, гидроксид натрия, гидрокарбонат натрия, карбонат натрия, гидрокарбонат калия и карбонат калия. Особенно предпочтительным является гидроксид натрия, карбонат натрия или три-н-пропиламин.
Восстановительное аминирование осуществляют в подходящем инертном растворителе или смеси растворителей, включая воду. Инертные растворители обычно не реагируют с соответствующими исходными реагентами формул (IIa) и (IIb). Если в качестве восстанавливающего агента используется боргидрид щелочного металла, такой как боргидрид натрия или боргидрид лития; боргидрид щелочно-земельного металла, такой как боргидрид кальция; цианоборгидрид щелочного металла, такой как цианоборгидрид натрия или цианоборгидрид лития, например, предпочтительным является полярный растворитель, например, спирт, такой как метанол, этанол, изопропанол или 2-метоксиэтанол, или глим. Если в качестве восстанавливающего агента используется три-(С1-С7-алкокси)-боргидрид щелочного металла, такой как триметоксиэтоксиборгидрид натрия; тетра-С1-С7-алкиламмоний-(циано)боргидрид, такой как тетрабутиламмоний-боргидрид или тетрабутиламмоний-цианоборгидрид, например, углеводороды, такие как толуол, эфиры, такие как этилацетат или изопропилацетат, простые эфиры, такие как тетрагидрофуран или трет-бутилметиловый эфир, являются предпочтительными. Если в качестве восстанавливающей системы используются водород или донор водорода, каждый в присутствии катализатора гидрирования, полярный растворитель является предпочтительным. Восстановительное аминирование можно также осуществлять, например, в смеси органического растворителя в воде, в моно- и двухфазном состоянии. В двухфазной системе может добавляться катализатор переноса фаз, такой как галогенид тетрабутиламмония, например, бромид, или галогенид бензилтриметиламмония, например, хлорид.
Если R1 и R2 оба представляют собой защитную группу и если соединение формулы (IIb) является свободным основанием, присутствия основания не требуется. Однако, если R1 представляет собой водород и R2 представляет собой защитную группу, может добавляться не менее молярного эквивалента основания. Для устранения рацемизации реакцию предпочтительно осуществляют менее чем с одним эквимолярным количеством основания. Если R1 и R2 каждый представляет собой водород, рацемизации не происходит, даже если реакцию осуществляют с равным или более чем с одним эквивалентом основания в мягких условиях, предпочтительно при температурах между -10°С и 20°С.
Настоящее изобретение также относится к новым соединениям формулы (IIа), которые могут использоваться в качестве промежуточных соединений для получения соединения формулы (I).
Настоящее изобретение также относится к новым соединениям формулы (IIb), которые могут использоваться в качестве промежуточных соединений для получения соединения формулы (I).
Реакция соединения формулы (IIа) с соединением формулы (IIb) немедленно приводит к получению имина (основание Шиффа) формулы (IIс'):
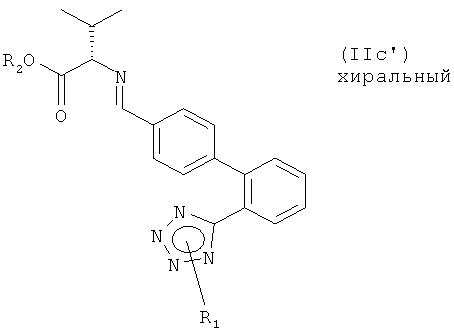
которое может, при определенных условиях реакции, выть выделено или которое может подвергаться восстановлению без выделения.
Восстановительное аминирование является двухстадийной реакцией, включающей образование имина при отщеплении молекулы воды с последующей стадией восстановления. Отщепление является обратимой реакцией, которая может проводиться для получения имина при постоянном удалении воды, например, азеотропным удалением. Кроме того, для удаления или инактивации свободной воды может использоваться приемник воды, который может действовать физическим способом, таким как абсорбция или адсорбция, или химической реакцией. Подходящий приемник воды включает, не ограничиваясь ими, ангидриды органической кислоты, алюмосиликаты, такие как молекулярные сита, другие цеолиты, тонко измельченный силикагель, тонко измельченный алюминий, ангидриды неорганических кислот, такие как ангидрид фосфора (Р2O5), неорганические сульфаты, такие как сульфат кальция, сульфат натрия и сульфат магния, и другие неорганические соли, такие как хлорид кальция.
Если стадию (а) осуществляют с получением и выделением соединения формулы (IIс'), соединение формулы (IIа) подвергают реакции с соединением формулы (IIb), возможно в присутствии основания, если R1 и/или R2 представляют собой водород. Соединения формулы (IIc') можно затем превращать в соответствующие соединения формулы (IIc) восстановлением соединений формулы (IIc') соответствующим восстанавливающим агентом, указанным выше.
Промежуточный имин формулы (IIc'), например, может быть выделен удалением растворителя, например дистилляцией, особенно при азеотропном удалении воды.
В предпочтительном варианте восстановительное аминирование осуществляют без выделения соединения формулы (IIc').
Восстановительное аминирование наиболее предпочтительно осуществляют без удаления свободной воды, особенно, если R1 и R2 представляют собой водород, и с основанием, таким как гидроксид натрия, в растворителе, таком как метанол, и с восстанавливающим реагентом, таким как боргидрид натрия.
Благодаря иминному структурному элементу соединения формулы (IIc') включают как соответствующий Е, так и соответствующий Z изомер. Предпочтительным является Е изомер.
Настоящее изобретение также относится к соединениям формулы (IIс'), в которых R1 представляет собой водород или защитную группу тетразола и в которых R2 представляет собой водород или карбоксизащитную группу. Соответствующие соединения могут использоваться в качестве промежуточных соединений для получения соединения формулы (I). Предпочтительными являются соединения формулы (IIc'), в которых по крайней мере один из R1 и R2 представляет собой водород или оба R1 и R2 представляют собой водород.
Соединения формул (IIа) и (IIb) являются известными и могут быть получены известными способами.
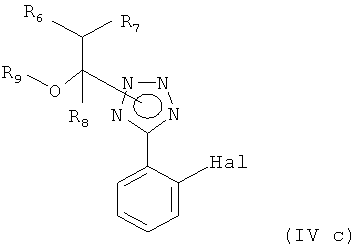
Другим вариантом осуществления настоящего изобретения является способ получения соединения формулы
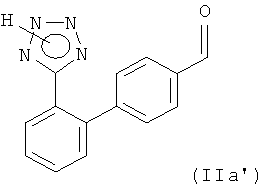
или его соли, включающий
(i) реакцию соединения формулы
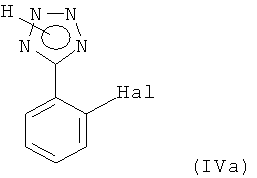
или его соли, где Hal представляет собой галоген, с соединением формулы

где R6, R7 и R8, независимо друг от друга, представляют собой водород или C1-С6-алкил, такой как метил или этил, и R9 представляет собой С1-С6-алкил, или R7 и R9 вместе образуют С2-С5-алкилен, такой как этилен, пропилен, бутилен, или R6 и R8 вместе образуют С3-С6-алкилен, в присутствии кислоты; и
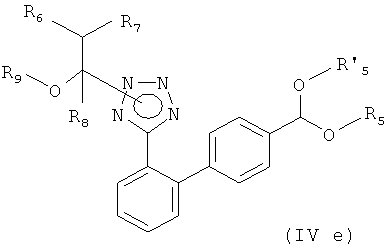
(ii) реакцию полученного соединения формулы

с соединением формулы

где Х представляет собой галоген, такой как йод, бром или хлор, и R5 и R'5, независимо друг от друга, представляет собой С1-С7-алкил, такой как метил или этил, или вместе образуют С2-С4-алкилен, такой как этилен, пропилен, бутилен или 1,2-диметилэтилен или 2,2-диметилпропилен, в присутствии катализатора переходного металла; и
(iii) снятие далее или на этой же стадии защитных групп с полученного соединения формулы
обработкой кислотой, предпочтительно в присутствии воды,
(iv) получение соединения формулы (IIа') или его соли.
Реакционные стадии (i) -(iv), описанные выше в вариантах, осуществляют, например, в отсутствие или, обычно, в присутствии подходящего растворителя или разбавителя или их смеси, реакцию, при необходимости, осуществляют при охлаждении, при комнатной температуре или при нагревании, например, при температуре в области приблизительно от -80°С до температуры кипения реакционной среды, предпочтительно приблизительно от -10°С приблизительно до +200°С, и, при необходимости, в закрытом сосуде, под давлением, в атмосфере инертного газа и/или в безводных условиях.
Стадию (i) осуществляют, например, в присутствии от 0,0001 до 0,1 эквивалентов, предпочтительно от 0,001 до 0,04 эквивалентов кислоты Бренстеда, такой как серная кислота, соляная кислота, фосфорная кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, камфор-10-сульфоновая кислота, трифторуксусная кислота, трихлоруксусная кислота, O,O'-дибензоилвинная кислота и им подобные.
Реакцию осуществляют в растворителе, который является достаточно стабильным к безводным кислотным условиям, например в этилацетате, изопропилацетате, в ароматическом растворителе, таком как толуол или ксилол, или в эфирном растворителе, таком как трет-бутилметиловый эфир, тетрагидрофуран, бутиловый эфир или 1,2-диметоксиэтан, или в нитриле, таком как ацетонитрил. Предпочтительным растворителем является толуол. Температуру реакции поддерживают между 15°С и температурой кипения реакционной среды, предпочтительно между 30 и 60°С.
Стадию (ii) осуществляют, например, с помощью обычного катализатора переходного металла, например, соответствующего обычно используемого платинового или палладиевого катализатора, такого как дихлорбис(трифенилфосфин)палладий(II).
Стадию (iii) осуществляют, например, растворением полученного соединения формулы (IVe) в воде или в смеси воды и подходящего органического растворителя и последующей обработкой кислотой при повышенной температуре. Кристаллизация продукта сопровождается отгонкой всего или части органического растворителя, добавлением воды, охлаждением смеси или объединением этих способов. Подходящими органическими растворителями являются эфиры, такие как тетрагидрофуран, 1,4-диоксан, бутиловый эфир, нитрилы, такие как ацетонитрил, спирты, такие как метанол, этанол, 1-пропанол, 2-пропанол, изопропилацетат, толуол, ксилол, уксусная кислота или муравьиная кислота. Предпочтительными растворителями являются метанол и этанол. Подходящими кислотами являются кислоты Бренстеда, такие как серная кислота, соляная кислота, фосфорная кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, бензойная кислота, уксусная кислота, муравьиная кислота. Предпочтительными кислотами являются серная кислота и соляная кислота. Кислота используется в количестве от 0,05 до 6,0 эквивалентов относительно исходного реагента, предпочтительно от 0,1 до 1,5 эквивалентов.
Стадию (iv) выделения осуществляют обычными методами выделения, такими как кристаллизация полученного соединения формулы (IIа') из реакционной смеси, если желательно или необходимо после обработки, особенно экстракции, или хроматография реакционной смеси.
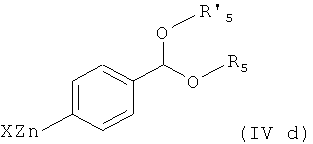
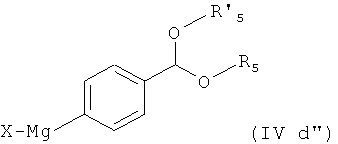
Соединения формулы (IVd) получают по реакции соединения формулы
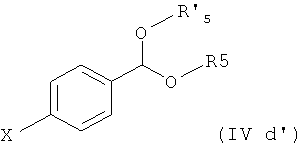
где Х представляет собой галоген, например бром, с магнием в условиях реакции Гриньяра, особенно в безводных условиях, предпочтительно в присутствии активатора, такого как 1,2-дибромэтан, с получением соединения формулы
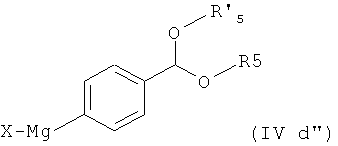
которое затем обрабатывают Zn(X)2, X представляет собой галоген, особенно хлор.
Как указано в начале описания, большинство антагонистов рецептора ангиотензина II включает в качестве структурной особенности тетразольное кольцо. В последовательности получения таких соединений для тетразольного кольца необходимы защитные группы.
Например, используется трифенилметильная группа для защиты тетразольного кольца в отношении металлоорганических реагентов. Трифенилметильная группа отщепляется позднее в кислотных условиях. Недостатком трифенилметильной группы является ее молекулярный вес. 2-Фенил-2-пропилтетразольная защитная группа используется при взаимодействии с металлом до следующей реакции. Удаление этой защитной группы требует применения коррозионных и токсичных реагентов, таких как эферат трифторида бора, или стадию снятия защиты, катализируемую переходным металлом, что является нежелательным.
Защита тетразольного кольца в отношении металлоорганических реагентов с помощью 2-метил-2-пропильной группы является другим вариантом. Для удаления этой группы необходимы жесткие кислотные условия, которые не подходят для чувствительных функциональных групп конечного соединения.
Альтернативно, используется 2-цианоэтильная защитная группа тетразола. Низкая стабильность этой защитной группы относительно большинства металлоорганических реагентов, а также образование высокотоксичных побочных продуктов во время снятия защиты являются ее недостатками.
Тетразольные кольца могут быть также защищены (фенилметил)оксиметильной группой. Однако один из двух получаемых изомеров является нестабильным в отношении металлоорганических реагентов, не только при высоких температурах.
Целью настоящего изобретения является способ синтеза соединения формулы (IIа') или его солей с помощью защитных групп, которые (I) не имеют указанных выше недостатков, (2) легко вводятся с хорошим выходом, (3) имеют низкий молекулярный вес, (4) являются стабильными в присутствии металлоорганических реагентов, таких как цинкарильные и магнийарильные соединения, (5) легко удаляются с высоким выходом в кислотных условиях, подходящих чувствительным функциональным группам, таким как формильная группа.
Неожиданно было обнаружено, что описанный выше способ отвечает указанным выше целям. Например, специалист в данной области техники не мог предположить, что соединения формулы (IVc) могут использоваться для связывания с соединением формулы (IVd) в качестве соответствующих защитных групп тетразольного кольца соединений формулы (IVd), которое не считается стабильным в такой реакции металлоорганического связывания. Специалист в данной области предположил бы, что соответствующие защитные группы тетразола формулы (IVd) отщеплялись бы. Кроме того, специалист в данной области предположил бы, что соответствующие защитные группы тетразолов формулы (IVe) отщеплялись бы легко в мягких условиях, указанных выше.
Соответственно, другим вариантом осуществления настоящего изобретения являются новые соединения формул (IVa), (IVb), (IVc), (IVd), (IVd'), (IVd") и (IVe), особенно соединения формулы (IVe).
Предпочтительный вариант осуществления этого варианта настоящего изобретения относится к соединению формулы
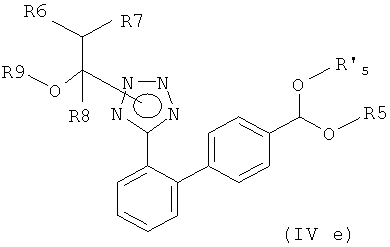
где R5 и R'5, независимо друг от друга, представляют собой С1-С7алкил, такой как метил или этил, или вместе образуют С2-С4-алкилен, такой как этилен, пропилен, бутилен или 1,2-диметилэтилен или 2,2-диметилпропилен, или где R6, R7 и R8, независимо друг от друга, представляют собой водород или C1-C7-алкил, такой как метил или этил, и R9 представляет собой С1-С7-алкил, или R7 и R9 вместе образуют С2-С5-алкилен, такой как этилен, пропилен, бутилен, или R6 и R8 вместе образуют С3-С6-алкилен.
Предпочтительными соединениями формулы (IVe) являются соединения, где R5 и R'5, независимо друг от друга, представляют собой С1-С4-алкил, такой как метил или этил, или вместе образуют С2-С4-алкилен, такой как этилен, пропилен, бутилен или 1,2-диметилэтилен или 2,2-диметилпропилен, или где R6, R7 и R8, независимо друг от друга, представляет собой водород или С1-С4-алкил, такой как метил или этил, и R9 представляет собой С1-С4-алкил, или R7 и R9 вместе образуют С2-С5-алкилен, такой как этилен, пропилен, бутилен.
Еще более предпочтительными соединениями формулы (IVe) являются соединения, где R5 и R'5, независимо друг от друга, представляют собой С1-С3-алкил, такой как метил, этил или пропил, или где R6, R7 и R8 представляет собой водород, и R9 представляет собой С1-С4-алкил.
Наиболее предпочтительными соединениями формулы (IVe) являются соединения, где R5 и R'5, независимо друг от друга, представляют собой С1-С3-алкил, такой как метил, этил или пропил, и где R6 и R8 представляют собой водород, и R7 и R9 вместе образуют С2-С3-алкилен, такой как этилен или пропилен.
Особенно предпочтительными являются соединения формулы (IVe), которые конкретно описаны в примерах.
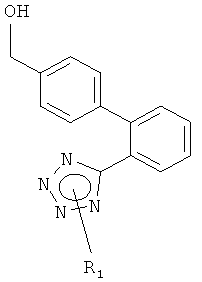
В другом варианте осуществления изобретения реакционная стадия (а) может быть объединена с образованием соединения формулы (IIа) обычным окислением соответствующего гидроксиметильного производного формулы
обычным восстановлением соответствующего производного карбоновой кислоты формулы
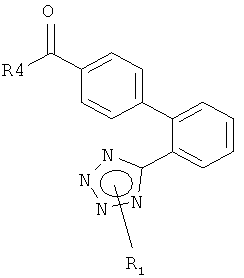
где R4 представляет собой, например, гидрокси, С1-С7-алкокси или галоген, такой как хлор; или обычным гидролизом ацеталя формулы
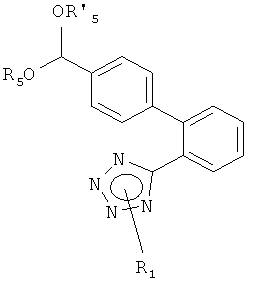
где R5 и R5', независимо друг от друга, представляют собой С1-С7-алкил, такой как метил или этил, или вместе образуют С2-С4-алкилен, такой как этилен, пропилен или бутилен или 1,2-диметилэтилен.
Настоящее изобретение также относится к реакционной стадии (а), особенно к стадии восстановления восстановительным аминированием. Если реакцию осуществляют, например, с боргидридом и при основных условиях в полярном растворителе, необязательно, в присутствии воды, предпочтительно в низшем (особенно безводном) алканоле, таком как метанол, этанол, изопропанол или глим, полученное соединение формулы (IIс) или (IIc'), соответственно, может неожиданно быть получено по существу в энантиомерно чистой форме. Предполагается, что в основных условиях, обычно происходит по крайней мере частичная рацемизация. В противоположность этому, неожиданно, например, может быть получен энантиомерный избыток (ее) соединений формулы (IIc) или (IIc'), соответственно, ≥95%, предпочтительно ≥98% и наиболее предпочтительно ≥99%.
Стадию (а) предпочтительно осуществляют в мягких условиях, особенно при температуре в области приблизительно от -10°С приблизительно до комнатной температуры, предпочтительно в области приблизительно от -5°С до +5°С.
Стадия (б):
На стадии реакции (б) осуществляют ацилирование, например, в отсутствие или в присутствии подходящего основания.
Подходящими основаниями являются, например, гидроксиды или карбонаты щелочных металлов, морфолиновые или пиперидиновые амины, незамещенные или замещенные пиридины, анилины, нафталеновые амины, три-С1-С7-алкиламины, основные гетероциклы или гидроксиды тетра-С1-С7-алкиламмония. Примерами являются гидроксид натрия, карбонат калия, триэтиламин, трипропиламин, трибутиламин или этилдиизопропиламин, N-метил-морфолин или N-метилпиперидин, диметиланилин или диметиламинонафтален, лютидин, коллидин или гидроксид бензилтриметиламмония. Предпочтительным основанием является три-С1-С4-алкиламин, такой как этилдиизопропиламин, или пиридин.
Ацилирование осуществляют в подходящем инертном растворителе или в смеси растворителей. Специалист в данной области техники способен выбрать подходящий растворитель или систему растворителей. Например, ароматический углеводород, такой как толуол, сложный эфир, такой как этилацетат или смесь этилацетата и воды, галогенированный углеводород, такой как метиленхлорид, нитрил, такой как ацетонитрил или проприонитрил, простой эфир, такой как тетрагидрофуран или диоксан, 1,2-диметоксиэтан, амид, такой как диметилформамид, или углеводород, такой как толуол, могут использоваться в качестве растворителя.
В процессе ацилирования соединения формулы (IIc), если R2 представляет собой водород, карбоксильная группа может быть ацилирована с получением смешанного ангидрида. Этот промежуточный продукт сильно подвергается рацемизации, главным образом в основных условиях. Рацемизация однако может устраняться сначала добавлением соединения формулы (IId), особенно галогенида, к соединению формулы (IIс) в подходящем инертном растворителе (например, диметоксиэтане, тетрагидрофуране или ацетонитриле), затем медленным добавлением субстехиометрического количества основания, особенно пиридина, относительно соединения формулы (IId). Небольшие количества воды реакционной смеси, предпочтительно два эквивалента, могут дополнительно понизить рацемизацию.
Реакцию можно также осуществлять одновременным или альтернативным добавлением соединения формулы (IId) и основания, такого как пиридин, все время поддерживая кислотность реакционной смеси.
Изобретение также относится к соединению формулы (IIc), где R1 представляет собой водород или тетразольную защитную группу и R2 представляет собой водород или карбоксизащитную группу, исключая соединение формулы (IIc), где R1 представляет собой этил и R2 представляет собой тритил; которое может использоваться, например, в качестве промежуточного соединения для получения соединения формулы (I).
Изобретение также относится к реакционной стадии (б). Полученное соединение формулы (IIе) может быть получено по существу в энантиомерно чистой форме. Например, может быть получен энантиомерный избыток (ее) соединений формулы (IIс) или (IIс'), соответственно, ≥95%, предпочтительно ≥98% и наиболее предпочтительно ≥99%.
Если R2 представляет собой защитную группу и R1 представляет собой водород или защитную группу, например, добавляют два эквивалента относительно соединения формулы (IId), например, соответствующего галогенида, и основания, например, этилдиизопропиламина или три-н-пропиламина к соответствующему соединению формулы (IIе), растворенному в подходящем растворителе, например толуоле. Неожиданно, рацемизация не происходит.
Обычно в соответствующих соединениях формулы (IIc), где R2 представляет собой водород или защитную группу, можно ожидать по крайней мере частичную рацемизацию, главным образом в присутствии основания или кислоты и при повышенной температуре. Однако рацемизации не происходит в условиях, приведенных в соответствии с изобретением.
Обычно в соответствующих соединениях формулы (IIc), где R2 представляет собой водород, ожидается рацемизация. Однако в присутствии основания рацемизации не происходит.
Если R1 представляет собой водород и R2 представляет собой защитную группу, тетразольное кольцо может быть также ацилировано. Когда, однако, реакционную смесь нейтрализуют, например, водой или спиртом, таким как метанол, может быть получено соответствующее соединение, где R1 представляет собой водород.
Соединения формулы (IId) являются известными или могут быть получены известными способами.
Стадия (в):
Удаление защитных групп, как тетразольной, так и карбоксизащитной группы, может осуществляться способами, известными из уровня техники.
Например, бензиловый эфир может быть превращен в соответствующую кислоту особенно гидрированием в присутствии подходящего катализатора гидрирования. Подходящий катализатор включает, например, никель, такой как никель Ренея, и благородные металлы или их производные, например, оксиды, такие как оксид палладия или платины, которые могут применяться, при необходимости, на носителях, например, на угле или карбонате кальция. Гидрирование предпочтительно может осуществляться под давлением от 1 и приблизительно до 100 атм и при комнатной температуре приблизительно от -80° приблизительно до 200°С, в частности от комнатной температуры и приблизительно до 100°С.
Удаление тритильной или трет-бутильной группы, соответственно, может достигаться обработкой соответствующих защищенных соединений кислотой, особенно в мягких условиях.
Стадия (г):
Стадию (г) выделения соединения формулы (I) осуществляют в соответствии с обычными способами выделения, такими как кристаллизация полученного соединения формулы (I) из реакционной смеси, при желании или необходимости после обработки, особенно экстракции, или хроматография реакционной смеси.
Превращение кислоты формулы (I) в соль осуществляют известным способом. Так, например, соль с основанием соединений формулы (I) получают обработкой кислотной формы основанием. Соли с основанием могут, с другой стороны, быть преобразованы в кислоту (свободное соединение) обычным способом, и соли с основанием могут быть преобразованы, например, обработкой подходящим кислотным агентом.
Настоящее изобретение также относится к новым соединениям, как описано в разделе примеров.
Следующие примеры иллюстрируют описанное выше изобретение; однако они никоим образом не предназначены для ограничения его объема.
Примеры
Пример 1
а) Получение 3-метил-2{[1-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил]-мет-(Е/Z)-илиден]-амино-бутановой кислоты
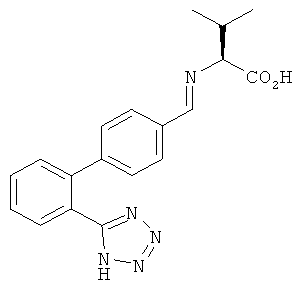
Водный 30% раствор гидроксида натрия (4,2 мл; 31,5 ммоль) добавляли к перемешиваемой суспензии L-валина (2, 43 г; 20,8 ммоль) и 2'-(1Н-тетразол-5-ил)-бифенил-4-карбальдегида (5 г; 19,6 ммоль) в воде (20 мл) при комнатной температуре до достижения рН 11. Полученный раствор перемешивали при комнатной температуре в течение 15 минут. Прозрачный раствор упаривали при 60°С в вакууме и оставшуюся воду азеотропно удаляли с 10 мл 1-бутанола.
1Н ЯМР (CD3OD, 300МГц): δ=8,21 (CH=N, s), 7,67 (С6Н5-СН, d), 7,40-7,60 (4 С6Н5-СН, m), 7,18 (С6Н5-СН, d), 3,42 (CH, d), 2,31 (CH, m), 0,98 (СН3, d), 0,82 (CH3, d).
б1) Получение (S)-3-метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-илметил)-амино)-бутановой кислоты
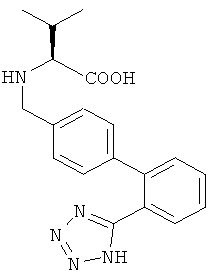
Водный 2,0 М раствор гидроксида натрия (приблизительно 100 мл; 200 ммоль) добавляли к перемешиваемой суспензии L-валина (11,8 г; 100 ммоль) и 2'-(1Н-тетразол-5-ил)-бифенил-4-карбальдегида (25,1 г; 100 ммоль) в воде (100 мл) при комнатной температуре, до достижения рН 11. Полученный чистый раствор упаривали при 60°С в вакууме и оставшуюся воду азеотропно удаляли с 1-бутанолом. Остаток (имин в виде твердой пены) растворяли в абсолютном этаноле (300 мл) и к раствору при 0-5°С порциями добавляли боргидрид натрия (3,78 г; 100 ммоль). Реакционную смесь перемешивали в течение 30 мин при 0-5°С и после окончания реакции (ВЭЖХ) гасили добавлением воды (100 мл) и соляной кислоты 2,0 М (80 мл; 160 ммоль). Органический растворитель (этанол) удаляли из прозрачного раствора (рН 7) при 50°С в вакууме. Оставшийся водный концентрат доводили до рН 2 медленным добавлением 2,0 М соляной кислоты (приблизительно 70 мл; 140 ммоль) при 40°С. В процессе добавления осаждался желаемый продукт. Его собирали фильтрацией, промывали водой и сушили в вакууме. Сырой продукт суспендировали в метаноле при 50°С и мутную смесь охлаждали до комнатной температуры. (S)-3-Метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил)-амино)-бутановую кислоту собирали фильтрацией и затем сушили в вакууме.
б2) Альтернативно, (S)-3-метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил)-амино)-бутановая кислота может быть получена, например, следующим образом:
Водный 10 М раствор гидроксида натрия (приблизительно 41 мл; 410 ммоль) добавляли к перемешиваемой суспензии L-валина (24,8 г; 210 ммоль) и 2'-(1Н-тетразол-5-ил)-бифенил-4-карбальдегида (50 г; 200 ммоль) в воде (200 мл) при комнатной температуре, до достижения рН 11. Полученный прозрачный раствор упаривали при 60°С в вакууме и оставшуюся воду азеотропно удаляли с 1-бутанолом. Остаток (имин в виде твердой пены) растворяли в метаноле (600 мл) и к раствору при 0-5°С порциями добавляли боргидрид натрия (3,13 г; 80 ммоль). Реакционную смесь перемешивали в течение 30 мин при 0-5°С и после окончания реакции (ВЭЖХ) гасили добавлением воды (300 мл) и соляной кислоты 2,0 М (160 мл; 320 ммоль). Органический растворитель (метанол) удаляли из прозрачного раствора (рН 7) при 50°С в вакууме. Оставшийся водный концентрат доводили до рН 2 медленным добавлением 2,0 М соляной кислоты (приблизительно 90 мл) при 40°С. В процессе добавления осаждался желаемый продукт. Его собирали фильтрацией, промывали водой и сушили в вакууме. Сырой продукт суспендировали в метаноле при 50°С и перемешивали несколько минут. Затем мутную смесь охлаждали до комнатной температуры. (S)-3-Метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил)-амино)-бутановую кислоту собирали фильтрацией и затем сушили в вакууме.
Энантиомерный избыток (по данным ВЭЖХ): ее>99,9%.
б3) Альтернативно, (S)-3-метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил)-амино)-бутановая кислота может быть получена, например, следующим образом:
Гидроксид натрия (1,71 г; 41,89 ммоль) добавляли порциями к перемешиваемой суспензии L-валина (2,48 г; 21 ммоль) в 15 мл метанола. Смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 2'-(1Н-тетразол-5-ил)-бифенил-4-карбальдегид (5 г; 20 ммоль). Смесь становилась прозрачным раствором через несколько минут. Смесь затем охлаждали до -5°С и к раствору порциями добавляли боргидрид натрия (0,315 г; 8 ммоль). Температуру в процессе добавления поддерживали между 0-5°С. Полученную смесь перемешивали в течение 2 часов при 0°С, за завершением реакции наблюдали по данным ВЭЖХ, затем гасили добавлением воды (10 мл) и соляной кислоты 37% (5,3 г) до рН 2-2,5. Последующую обработку и кристаллизацию проводили в соответствии с примером 1 б2).
Энантиомерный избыток (по данным ВЭЖХ): ее>99,9%.
б4) Альтернативно, (S)-3-метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил)-амино)-бутановая кислота может быть получена, например, следующим образом:
В стальной автоклав на 50 мл под аргоном загружали 3-метил-2{[1-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил]-мет-(Е/Z)-илиден]-амино}-бутановую кислоту (1,5 г; 3,2 ммоль) и 5% Pt/C (7,5 мг, 5% вес.). Затем добавляли 15 мл метанола, автоклав закрывали и подавали аргон и водород. Давление поддерживали равным 5 бар и реакционную смесь перемешивали при комнатной температуре. За завершением реакции следили по данным ВЭЖХ. Затем в автоклав подавали аргон и катализатор отфильтровывали. Последующую обработку и кристаллизацию проводили аналогично примеру 1 б2).
б5) Альтернативно, (S)-3-метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил)-амино)-бутановая кислота может быть получена, например, следующим образом:
2'-(1Н-Тетразол-5-ил)-бифенил-4-карбальдегид (0,79 г; 3,2 ммоль) и L-валин (0,4 г; 3,4 ммоль) суспендировали в 15 мл метанола. Затем добавляли гидроксид натрия (0,27 г; 6,72 ммоль) и реакционную смесь перемешивали при комнатной температуре до получения прозрачного раствора. Добавляли 5% Pt/C (15,8 мг; 2% вес.). Автоклав закрывали и подавали аргон и водород. Давление поддерживали равным 5 бар и реакцию перемешивали при 60°С. За завершением реакции следили по данным ВЭЖХ. Затем в автоклав подавали аргон и катализатор отфильтровывали. Последующую обработку и кристаллизацию проводили аналогично примеру 1 б2).
Энантиомерный избыток (по данным ВЭЖХ): ее>99,9%.
в) Получение (S)-3-метил-2-{(пентаноил-5-ил)-[2'-(тетразол-5-ил)-бифенил-4-илметил]-амино}-бутановой кислоты
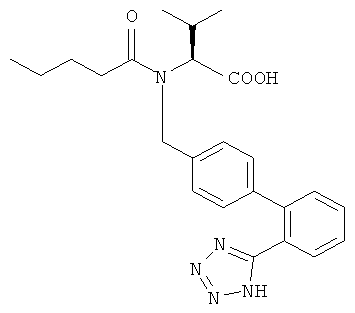
Суспензию (S)-3-метил-2-((2'-(1H-тетразол-5-ил)-бифенил-4-илметил)-амино)-бутановой кислоты (17,6 г; 50,0 ммоль) в 1,2-диметоксиэтане (116 г) охлаждали до -5°С и добавляли валероилхлорид (9,9 мл; 80 ммоль), затем медленно добавляли пиридин (6,0 мл; 75 ммоль), разбавленный 1,2-диметоксиэтаном (60 мл). [1] После окончания реакции реакционную смесь гасили метанолом (18 мл). В конце добавляли воду (50 мл) при комнатной температуре и после перемешивания в течение 1 ч рН смеси доводили до 7,5 добавлением водного 10% раствора карбоната натрия (~116 мл, 120 ммоль) при 0°С. Органические растворители удаляли при 50°С в вакууме. К оставшемуся водному концентрату добавляли этилацетат (125 мл) и рН двухфазной системы доводили до 2 при 0-5°С добавлением 2,0 М НСl (~98 мл). Органическую фазу отделяли и концентрировали при 45°С в вакууме (воду азеотропно удаляли). Кристаллизацию продукта начинали при 45°С и после добавления циклогексана (102 мл) заканчивали охлаждением до -5°С. Твердый остаток собирали фильтрацией и после высушивания при 50°С оставалась (S)-3-метил-2-{пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-илметил]-амино}-бутановая кислота в виде белого порошка.
Температура плавления: 108-110°С.
Энантиомерный избыток (по данным ВЭЖХ): ее>99,5%.
[1] Альтернативно пиридин и валероилхлорид могут добавляться следующим образом: Суспензию (S)-3-метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-илметил)-амино)-бутановой кислоты (25,5 г; 72,6 ммоль) в 1,2-диметоксиэтане (126 г) охлаждали до -10°С и добавляли валероилхлорид (8,75 г; 72,6 ммоль) в течение 15 мин, с последующим медленным добавлением смеси (7,16 г) пиридина (5,6 г) и воды (1,5 г) в течение 61 мин. После перемешивания в течение 30 мин добавляли валероилхлорид (5,3 г; 43,5 ммоль) в течение 8 мин, затем медленно добавляли в течение 30 мин смесь (4,3 г) пиридина (3,4 г) и воды (0,9 г). После каждого добавления пиридина рН контролировали отбором образцов (гидролиз в воде). рН образцов всегда должен быть ниже 2,5. Реакцию перемешивали в течение 25 мин, затем добавляли воду (25,6 г) в течение 30 мин. Смесь перемешивали еще в течение 30 мин, затем нагревали до 23°С в течение 30 мин и перемешивалиеще в течение 2 часов. Регулирование рН, удаление органических растворителей дистилляцией, дальнейшую обработку и кристаллизацию осуществляли, как описано в примере 1в) выше.
Пример 2
Этот пример иллюстрирован следующей схемой реакции:

а) Получение бензилового эфира (S)-3-метил-2-{[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты
Тозилат L-валин-бензиловый эфир (6,38 г, 16,8 ммоль) в толуоле (40 мл) экстрагировали раствором карбоната натрия (2,36 г, 22,0 ммоль) в воде (40 мл). Органическую фазу, содержащую L-валинбензиловый эфир в виде свободного основания, отделяли и при комнатной температуре добавляли 2'-(1Н-тетразол-5-ил)-бифенил-4-карбальдегид (4,13 г, 16,0 ммоль) и три-н-пропиламин (3,20 мл, 16,8 ммоль). Полученный раствор упаривали при 50°С в вакууме (воду удаляли азеотропно). Оставшееся масло, содержащее промежуточный имин, растворяли в абсолютном этаноле (40 мл) и порциями добавляли боргидрид натрия (0,68 г, 17,6 ммоль) в течение 10 минут (мин) при 0-5°С. Полученный раствор перемешивали в течение 30 мин при 0-5°С. После окончания реакции реакционную смесь гасили водой (10 мл) и доводили до рН 6-7 добавлением соляной кислоты 2 М (16 мл, 32 ммоль) при комнатной температуре. Этанол отгоняли из реакционной смеси при 50° в вакууме и оставшуюся водную смесь экстрагировали толуолом (60 мл). Органическую фазу концентрировали при 50°С в вакууме приблизительно до 50% от начального объема дистилляцией (воду и этанол азеотропно удаляли). Полученный концентрат (35 мл), содержащий бензиловый эфир (S)-3-метил-2-{[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты использовали в качестве исходного реагента для последующей стадии ацилирования.
б) Получение бензилового эфира (S)-3-метил-{2-пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты
Раствор бензилового эфира (S)-3-метил-2-{[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты (приблизительно 7,0 г, 16,0 ммоль) в толуоле (35 мл) с предыдущей стадии разбавляли толуолом (35 мл). Прозрачный раствор охлаждали до 0-5°С и в безводных условиях добавляли N-этилдиизопропиламин (6,1 мл, 35,2 моль) и валероилхлорид (4,1 мл, 33,6 ммоль) при этой температуре. Реакционную смесь нагревали до 50°С в течение 30 мин и перемешивали при 50°С в течение приблизительно 1 ч и после окончания реакции гасили добавлением метанола (10 мл) при 50°С. Прозрачный раствор перемешивали в течение приблизительно 30 мин при 50°С и в конце охлаждали до комнатной температуры. Добавляли воду (30 мл) и рН полученной двухфазной системы доводили до 2 добавлением 2,0 М соляной кислоты (приблизительно 11 мл, 22 ммоль). Оганическую фазу отделяли, экстрагировали водой (30 мл) и концентрировали при 50°С в вакууме приблизительно до 50% от начального объема дистилляцией (воду и метанол азеотропно удаляли). В полученный концентрат в толуоле (40 мл) добавляли затравку при 40°С для начала кристаллизации и перемешивали при этой температуре приблизительно в течение 1 часа (ч). Суспензию постепенно охлаждали до 0°С в течение 6-10 ч. Твердое вещество отделяли фильтрацией, промывали холодным толуолом (30 мл) и сушили в вакууме при 50°С с получением бензилового эфира (S)-3-метил-{2-пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты.
Температура плавления: 115-116°С.
Энантиомерный избыток (по данным ВЭЖХ): ее>99,8%.
в) Получение (S)-3-метил-2-{пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты
Раствор бензилового эфира (S)-3-метил-{2-пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты (10,6 г; 20,0 ммоль) в этилацетате (43 мл) гидрировали при 4 бар / 50°С в присутствии катализатора 5% сырого палладия на угле (1,12 г, содержание 50% воде). После окончания реакции (прекращение поглощения водорода) катализатор удаляли фильтрацией и фильтрат концентрировали при 45°С в вакууме (воду азеотропно удаляли). Кристаллизацию продукта инициировали при 45°С и после добавления циклогексана (102 мл) завершали охлаждением до -5°С. Твердое вещество собирали фильтрацией и после высушивания при 50°С (S)-3-метил-2-{пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановую кислоту получали в виде белого порошка.
Температура плавления: 108-110°С.
Энантиомерный избыток (по данным ВЭЖХ): ее>99,5%.
Пример 3
а) Получение трет-бутилового эфира (S)-3-метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил)-амино)-бутановой кислоты

К суспензии гидрохлорида трет-бутилового эфира L-валина (419,4 мг; 2 ммоль) в 5 мл изопропилацетата добавляли карбонат натрия (265 мг; 2,5 ммоль) в 5 мл воды. После окончания растворения две фазы отделяли немедленно. Водный слой промывали один раз 4 мл изопропилацетата. Объединенные органические слои промывали 5 мл воды. Бесцветный органический слой сушили над сульфатом натрия, отфильтровывали, упаривали в вакууме и сушили в высоком вакууме с получением бесцветного масла. Масло растворяли в 4 мл метанола. После добавления 2'-(1Н-тетразол-2-ил)-бифенил-4-карбальдегида (515 мг; 2 ммоль) и триэтиламина (0,278 мл; 2 ммоль), желтый раствор перемешивали в течение 5 минут перед упариванием в вакууме с получением желтого масла. После растворения в 4 мл этанола раствор охлаждали до 0°С. Добавляли боргидрид натрия (78 мг; 2 ммоль) 4 порциями при перемешивании до исчезновения имина (ВЭЖХ). Желтоватый раствор подкисляли с рН 11 до рН 6 с помощью 3,2 мл 1,0 М раствора НСl. Упаривание этанола приводило к получению смеси желтого масла в воде. Эту смесь экстрагировали изопропилацетатом. Объединенные органические слои сушили над сульфатом натрия, отфильтровывали, упаривали в вакууме и сушили в высоком вакууме с получением трет-бутилового эфира (S)-3-метил-2-((2'-(1H-тетразол-5-ил)-бифенил-4-илметил)-амино)-бутановой кислоты в виде масла.
б) Получение трет-бутилового эфира (S)-3-метил-{2-пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты
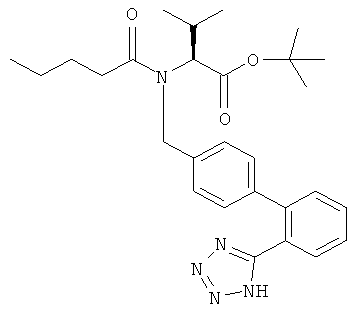
Трет-бутиловый эфир (S)-3-метил-2-((2'-(1Н-тетразол-5-ил)-бифенил-4-илметил)-амино)-бутановой кислоты (8,5 г; ~16,0 ммоль) растворяли в толуоле (63 мл) и N-этилдиизопропиламине (6,1 мл; 35,2 ммоль) и добавляли валероилхлорид (4,1 мл; 33,6 ммоль) при комнатной температуре. Прозрачный раствор нагревали до 50°С и перемешивали при этой температуре в течение 60 мин. После окончания реакции реакционную смесь гасили метанолом (10 мл) при 50°С и в конце добавляли воду при комнатной температуре. рН Двухфазной системы доводили до 2 добавлением 2,0 М НСl (~5 мл). Органическую фазу отделяли и концентрировали при 50°С в вакууме (оставшуюся воду удаляли азеотропно). При охлаждении до комнатной температуры продукт кристаллизовался из толуола. Трет-бутиловый эфир (S)-3-метил-{2-пентаноил-[2-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты получали в виде белого порошка после фильтрации и высушивания в вакууме.
Температура плавления: 153,4°С.
Энантиомерный избыток (по данным ВЭЖХ): ее>99,8%.
Пример 4
а) Получение (S)-2-((2-(2"-трет-бутил-тетразол-5"-ил)-бифенил-4-илметил)-амино)-3-метил-бутановой кислоты

Раствор углекислого натрия (1 моль/л; 1,0 мл, 1,0 ммоль) добавляли к L-валину (117,15 мг; 1,0 ммоль). После окончания растворения реакцию упаривали. К белому твердому веществу добавляли 2'-(1Н-трет-бутил-тетразол-2-ил)-бифенил-4-карбальдегид (306,4 мг, 1 ммоль) и 4 мл метанола. После окончания растворения реакционную смесь упаривали и желтоватое масло сушили в высоком вакууме. Имин растворяли в 4 мл этанола и охлаждали до 0°С перед добавлением боргидрида натрия (38 мг; 1,0 ммоль) 2 порциями при перемешивании до исчезновения имина. Желтоватый раствор подкисляли с помощью 1,8 мл 1N раствора НСl до рН 6-7. Упаривание в вакууме приводило к получению белого твердого вещества. Добавляли 10 мл изопропилацетата и 10 мл воды. Белый осадок отфильтровывали, промывали водой и сушили с получением 2-((2'-(2"-трет-бутил-тетразол-5"-ил)-бифенил-4-ил-метил)-амино)-3-метил-бутановой кислоты.
Температура плавления: 189,7°С.
Пример 5
а) Получение бензилового эфира (S)-2-{[2'-(2-бензил-2Н-тетразол-5-ил)-бифенил-4-илметил]-амино}-3-метил-бутановой кислоты
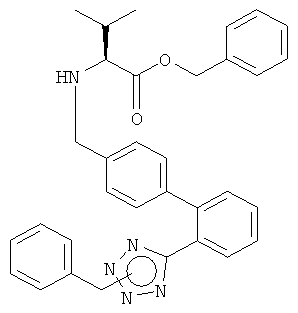
Тозилат бензилового эфира L-валина (0,97 ммоль, 368 мг) суспендировали в изопропилацетате (4 мл). К этой суспензии добавляли раствор карбоната натрия (1,21 ммоль, 128 мг) в воде (2 мл) при комнатной температуре. Полученную смесь перемешивали в течение 2 минут, переносили в отдельную воронку и фазы разделяли. Органическую фазу сушили над сульфатом натрия, отфильтровывали и концентрировали в вакууме с получением свободного основания в виде бесцветного масла. 2'-(1Н-Бензил-тетразол-2-ил)-бифенил-4-карбальдегид (0,88 ммоль, 300 мг) растворяли в 1,2 диметоксиэтане (4 мл) при комнатной температуре и полученный раствор добавляли к остатку свободного основания. Через 8 часов растворитель удаляли в вакууме и остаток растворяли в этаноле (4 мл). К реакционной смеси добавляли боргидрид натрия (1,1 ммоль, 41,6 мг). Полученный непрозрачный раствор перемешивали при комнатной температуре более 2 часов и затем концентрировали в вакууме для удаления этанола. Добавляли воду (20 мл) и дихлорметан (20 мл) и рН водной фазы доводили до 1 добавлением 1N HCl. Фазы отделяли и водную фазу экстрагировали снова дихлорметаном (10 мл). Объединенные органические фазы промывали водой (10 мл), сушили над безводным сульфатом натрия, отфильтровывали и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветного масла.
Пример 6
а) Получение трет-бутилового эфира (S)-2-{[2'-(2-трет-бутил-2Н-тетразол-5-ил)-бифенил-4-илметил]-амино}-3-метил-бутановой кислоты
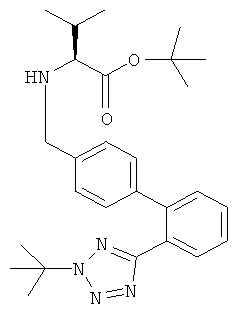
Гидрохлорид трет-бутилового эфира L-валина (1,32 ммоль, 278 мг) суспендировали в изопропилацетате (5 мл). К этой суспензии добавляли раствор карбоната натрия (1,65 ммоль, 175 мг) в воде (5 мл) при комнатной температуре. Полученную смесь перемешивали в течение 2 минут, переносили в отдельную воронку и фазы разделяли. Органическую фазу сушили над сульфатом натрия, отфильтровывали и концентрировали в вакууме с получением свободного основания в виде бесцветного масла.
2'-(1Н-Трет-бутил-тетразол-2-ил)-бифенил-4-карбальдегид (1,2 ммоль, 367,2 мг) растворяли в этаноле (5 мл) при комнатной температуре и полученный раствор добавляли к остатку свободного основания. Через 90 минут к реакционной смеси добавляли боргидрид натрия (1,5 ммоль, 56,7 мг). Полученный непрозрачный раствор перемешивали при комнатной температуре в течение 2 часов и затем концентрировали в вакууме для удаления этанола. Добавляли воду (20 мл) и дихлорметан (20 мл) и рН водной фазы доводили до 1 добавлением 1N HCl. Фазы разделяли и водную фазу экстрагировали снова дихлорметаном (10 мл). Объединенные органические фазы промывали водой (10 мл), сушили над безводным сульфатом натрия, отфильтровывали и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветного масла.
1Н ЯМР (CD3OD, 400 МГц):
δ=7,86 (1Н, d, J=8 Гц), 7,41-7,68 (3H, m), 7,44 (2H, d, J=8 Гц), 7,24 (2H, d, J=8 Гц), 4,17 (1Н, d, J=13 Гц), 4,08 (1Н, d, J=13 Гц), 3,56 (1Н, d, J=2 Гц), 2,27 (1Н, m), 1,12 (3H, d, J=7 Гц) и 1,06 (3H, d, J=7 Гц).
Пример 7
а) Получение бензилового эфира (S)-3-метил-2-{[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты
Тозилат бензилового эфира L-валина (20,1 г, 53 ммоль) в толуоле (90 мл) экстрагировали раствором карбоната натрия (7,3 г, 69 ммоль) в воде (125 мл). Органическую фазу (содержащую свободное основание бензилового эфира L-валина) отделяли и добавляли 2'-(1Н-тетразол-5-ил)-бифенил-4-карбальдегид (12,5 г, 50 ммоль) и N-этилдиизопропиламин (9,0 мл, 52 ммоль) при комнатной температуре. Полученный раствор полностью упаривали при 50°С в вакууме (воду удаляли азеотропно). Оставшееся масло (содержащее промежуточный имин) растворяли в метаноле (160 мл) и порциями добавляли боргидрид натрия (0,84 г, 22 ммоль) в течение 10 мин при 0-5°С. Полученный раствор перемешивали в течение 30 мин при 0-5°С. После окончания превращения реакционную смесь гасили добавлением 1,0 М соляной кислоты (приблизительно 42 мл, 42 ммоль) при 0-5°С и рН доводили до 6-7. Метанол отгоняли из реакционной смеси при 50°С в вакууме и полученную водную смесь экстрагировали толуолом (180 мл). Органическую фазу концентрировали при 50°С в вакууме приблизительно до 50% от начального объема дистилляцией (воду и метанол азеотропно удаляли). Полученный концентрат (приблизительно 80 г), содержащий бензиловый эфир (S)-3-метил-2-{[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты использовали в качестве исходного сырья для последующей стадии ацилирования.
б) Получение бензилового эфира (S)-3-метил-{2-пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты
Раствор бензилового эфира (S)-кислоты в толуоле (приблизительно 80 г, 48-50 ммоль) с предыдущей стадии разбавляли толуолом (85 мл). В безводных условиях медленно добавляли N-этилдиизопропиламин (24,0 мл, 140 моль) и валероилхлорид (17,3 мл, 140 ммоль) при температуре внутри сосуда 20°С. Реакционную смесь перемешивали приблизительно в течение 30 мин и после окончания превращения гасили добавлением метанола (31 мл) при 20°С. Прозрачный раствор перемешивали в течение 30 мин при 20°С, затем добавляли воду (78 мл) и полученную двухфазную систему доводили до рН 2 добавлением 2,0 М соляной кислоты (приблизительно 10 мл, 20 ммоль). Органическую фазу отделяли, экстрагировали водой (78 мл) и концентрировали при 50°С в вакууме приблизительно до 50% от начального объема дистилляцией (воду и метанол азеотропно удаляли). В полученный концентрат в толуоле (~94 г) добавляли затравку при 40°С для инициирования кристаллизации и перемешивали при этой температуре в течение приблизительно 1 ч. Суспензию постепенно охлаждали до 0°С в течение 6-10 ч. Твердое вещество отделяли фильтрацией, промывали холодным толуолом (60 мл) и сушили в вакууме при 50°С с получением бензилового эфира (S)-3-метил-{2-пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты.
Температура плавления: 115-116°С.
Энантиомерный избыток (по данным ВЭЖХ): ее>99,8%.
в) Получение (S)-3-метил-2-{пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты
Раствор бензилового эфира (S)-3-метил-{2-пентаноил-[2-(1H-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановой кислоты (10,6 г; 20,0 ммоль) в этилацетате (43 мл) гидрировали при 4 бар/ 50°С в присутствии катализатора 5% сырого палладия на угле (1,12 г, содержание 50% воде). После окончания реакции (прекращение поглощения водорода) катализатор удаляли фильтрацией и фильтрат концентрировали при 45°С в вакууме (воду азеотропно удаляли). Кристаллизацию продукта инициировали при 45°С и после добавления циклогексага (102 мл) полностью охлаждали до -5°С. Твердое вещество собирали фильтрацией и после высушивания при 50°С получали (S)-3-метил-2-{пентаноил-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-амино}-бутановую кислоту в виде белого порошка.
Температура плавления: 108-110°С.
Энантиомерный избыток (по данным ВЭЖХ): ее>99,5%.
Пример 8
5-(2-Хлорфенил)-2-(тетрагидропиран-2-ил)-2Н-тетразол и 5-(2-хлорфенил)-1-(тетрагидропиран-2-ил)-1 Н-тетразол

Метансульфоновую кислоту (0,141 г; 1,44 ммоль) добавляли к суспензии 5-(2-хлорфенил)-1Н-тетразола (88,46 г; 480,0 ммоль) в толуоле (660 мл). Полученную смесь нагревали до 50°С и добавляли раствор 3,4-дигидро-2Н-пирана (42,88 мл; 494 ммоль) в толуоле (60 мл) в течение 90 минут. Смесь далее перемешивали при 50°С в течение 90 минут. Полученный раствор промывали дважды 0,5N водным раствором гидроксида натрия (96 мл каждый) и дважды водой (96 мл каждый). Полученную мутную органическую фазу концентрировали в вакууме с помощью лопастной мешалки с получением смеси 5-(2-хлорфенил)-2-(тетрагидропиран-2-ил)-2Н-тетразола (N2-изомер) и 5-(2-хлорфенил)-1-(тетрагидропиран-2-ил)-1Н-тетразола (N1-изомер) в соотношении приблизительно 95:5 (в соответствии с 1Н-ЯМР) в виде желтой жидкости.
1Н-ЯМР N2-изомера (400МГц, CDCl3): 1,72-1,84 (m, 3 H), 2,16-2,25 (m, 2 H), 2,46-2,55 (m, 1 H), 3,80-3,86 (m, 1 H), 4,02-4,07 (m, 1 H), 6,12-6,14 (m, 1 H), 7,36-7,44 (m, 2 H), 7,52-7,56 (m, 1 H), 7,96-7,98 (m, 1 H).
1Н-ЯМР N1-изомера (400 МГц, CDCl3): 5,44-5,47 (m, 1H). Характерный сигнал, который не присутствует в сигналах N2-изомера.
Пример 9
5-(2-Бромфенил)-2-(тетрагидропиран-2-ил)-2Н-тетразол и 5-(2-бромфенил)-1-(тетрагидропиран-2-ил)-1Н-тетразол

Суспензию 5-(2-бромфенил)-1Н-тетразола (4,50 г; 20,0 ммоль) в трет-бутилметиловой эфире (40 мл) нагревали до 45°С и добавляли метансульфоновую кислоту (0,058 г; 0,60 ммоль). К полученной смеси добавляли раствор 3,4-дигидро-2Н-пирана (1,90 мл; 21 ммоль) в трет-бутилметиловом эфире (21 мл) в течение 1 часа при 45°С. Смесь затем перемешивали в течение 6 часов при 45°С. Полученный раствор охлаждали приблизительно до 0°С и добавляли раствор гидрокарбоната натрия (2,4 г) в воде (30 мл). Водную фазу отделяли и экстрагировали трет-бутилметиловым эфиром (10 мл). Объединенные органические фазы промывали дважды 1N раствором КОН (10 мл каждый) и один раз раствором 10% вес. хлоридом натрия в воде (10 мл). Полученную органическую фазу сушили над безводным сульфатом натрия, отфильтровывали и упаривали в вакууме с получением смеси 5-(2-бромфенил)-2-(тетрагидропиран-2-ил)-2Н-тетразола (N2-изомер) и 5-(2-бромфенил)-1-(тетрагидропиран-2-ил)-1Н-тетразола (N1-изомер) в соотношении приблизительно 93:7 (в соответствии с 1Н-ЯМР) в виде оранжевого масла.
1Н-ЯМР N2-изомера (400 МГц, CDCl3): 1,72-1,85 (m, 3 H), 2,18-2,26 (m, 2 H), 2,45-2,54 (m, 1 H), 3,80-3,86 (m, 1 H), 4,01-4,07 (m, 1 H), 6,12-6,15 (m, 1 H), 7,31-7,35 (m, 1 H), 7,41-7,45 (m, 1 H), 7,73-7,75 (m, 1 H), 7,87-7,90 (m, 1 H).
Пример 10
5-(4'-Диэтоксиметил-бифенил-2-ил)-2-(тетрагидропиран-2-ил)-2Н-тетразол
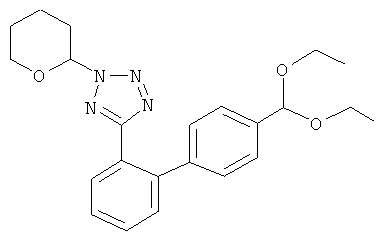
К суспензии магниевой стружки (5,11 г) в безводном тетрагидрофуране (40 мл) добавляли 1,2-дибромэтана (0,106 мл; 1,2 ммоль). Суспензию охлаждали до 12°С и добавляли 6 мл раствора 1-бром-4-(диэтоксиметил)бензола (53,6 г; 200 ммоль) в безводном тетрагидрофуране (120 мл). После начала реакции добавляли остаток раствора 1-бром-4-(диэтоксиметил)бензола в течение 90 минут. Полученную смесь затем перемешивали при 20-25°С в течение 2,5 часов. Смесь разбавляли безводным тетрагидрофураном до общего объема 250 мл, получая раствор соответствующего бромида арилмагния концентрацией приблизительно 0,78 M. В безводных условиях 15,0 мл указанного 0,78 М раствора бромида арилмагния (11,7 ммоль) охлаждали приблизительно до 0°С и добавляли 0,5 М раствор хлорида цинка в тетрагидрофуране (23,4 мл; 11,7 ммоль) в течение 15 минут. Полученную суспензию перемешивали при комнатной температуре в течение 30 минут для окончания образования соответствующего арилцинкового реагента. В другой колбе раствор смеси 5-(2-бромфенил)-2-(тетрагидропиран-2-ил)-2Н-тетразола и 5-(2-бромфенил)-1-(тетрагидропиран-2-ил)-1H-тетразола (2,78 г; 9,0 ммоль) в тетрагидрофуране (9 мл) добавляли к дихлорбис(трифенилфосфин)палладию(II) (0,253 г; 0,36 ммоль) в безводных условиях. При тщательном перемешивании полученную желто-оранжевую суспензию добавляли при комнатной температуре к указанной суспензии арилцинкового реагента в течение 40 минут. Смесь далее перемешивали при комнатной температуре в течение 17 часов. Затем добавляли раствор гидрокарбоната натрия (1,2 г) в воде (15 мл) и этилацетате (20 мл). Водную фазу отделяли и экстрагировали этилацетатом (60 мл). Объединенные органические фазы промывали дважды раствором гидрокарбоната натрия (1,2 г) в воде (15 мл каждый) и дважды водой (15 мл каждый) и упаривали в вакууме. Полученное желто-оранжевое масло растворяли в небольшом объеме трет-бутилметилового эфира, отфильтровывали через фильтр, упаривали в вакууме и очищали колоночной хроматографией на силикагеле, элюируя смесью 1:4 этилацетата и гексана с получением основного изомера (N2-изомер) 5-(4'-диэтоксиметил-бифенил-2-ил)-2-(тетрагидропиран-2-ил)-2Н-тетразола в виде бесцветного масла.
1Н-ЯМР N2-изомера (400 МГц, CDCl3): 1,24 (t, J=7,2 Гц, 6 Н), 1,61-1,66 (m, 3 Н), 1,88-2,03 (m, 2 Н), 2,11-2,18 (m, 1 Н), 3,50-3,71 (m, 6 Н), 5,49 (s, 1 Н), 5,97-5,99 (m, 1 Н), 7,18-7,20 (m, 2 Н), 7,38-7,40 (m, 2 Н), 7,43-7,56 (m, 3 Н), 7,90-7,92 (m, 1 Н).
Масс-спектр (ES+): m/z=409 [M+H]+.
Пример 11
2'-(2Н-Тетразол-5-ил)бифенил-4-карбальдегид
К 5-(4' -диэтоксиметил-бифенил-2-ил)-2-(тетрагидропиран-2-ил)-2Н-тетразолу (0,408 г; 1,00 ммоль) добавляли 94% этанол (2,5 мл) и 2N водный раствор соляной кислоты (0,5 мл; 1,0 ммоль). Полученный раствор нагревали при 45°С в течение 3 часов. После добавления воды (приблизительно 2 мл) смесь охлаждали до комнатной температуры и затем перемешивали при 0-5°С в течение 30 минут. Полученную суспензию отфильтровывали и твердые остатки промывали небольшим количеством воды, сушили в вакууме при 40°С с получением 2'-(12Н-тетразол-5-ил)бифенил-4-карбальдегида в виде белого кристаллического порошка.
Температура плавления: 187,5-190,0°С.
Масс-спектр высокого разрешения (ES+): обнаружено: m/z=251,0928 [М+Н]+; рассчитано: m/z=251,0927.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВАЛСАРТАНА | 2003 |
|
RU2348619C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-БИФЕНИЛ-4-АМИНО-2-МЕТИЛПЕНТАНОВОЙ КИСЛОТЫ | 2008 |
|
RU2530900C2 |
| СОЛИ 2'-(1Н-ТЕТРАЗОЛ-5-ИЛ)-1, 1'-БИФЕНИЛ-4-КАРБОКСАЛЬДЕГИДА С МЕТАЛЛАМИ | 2006 |
|
RU2435761C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АНГИОТЕНЗИНА, НА ИХ ОСНОВЕ | 1992 |
|
RU2053229C1 |
| ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУЧЕНИЯ ИНГИБИТОРОВ НЕЙТРАЛЬНОЙ ЭНДОПЕПТИДАЗЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2564024C2 |
| РЕАКЦИИ СОЧЕТАНИЯ, КОТОРЫЕ МОГУТ БЫТЬ ИСПОЛЬЗОВАНЫ ПРИ ПОЛУЧЕНИИ ПРОИЗВОДНЫХ (1Н-ТЕТРАЗОЛ-5-ИЛ)БИФЕНИЛА | 2005 |
|
RU2426728C2 |
| ПРОИЗВОДНЫЕ ТЕТРАЗОЛА | 1992 |
|
RU2091376C1 |
| ПРОИЗВОДНЫЕ 4-ПИРИМИДИНОНОВ ИЛИ ИХ ОРГАНИЧЕСКИЕ ИЛИ МИНЕРАЛЬНЫЕ ФАРМАЦЕВТИЧЕСКИЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2073675C1 |
| Производные бензо[d]изоксазола и их применение | 2016 |
|
RU2638155C1 |
| Производные бензимидазола, их изомеры, смеси изомеров, гидраты или их физиологически переносимые соли, обладающие антагонистическими в отношении ангиотензина свойствами | 1991 |
|
SU1836357A3 |
Настоящее изобретение относится к способу получения соединения формулы (IIа)
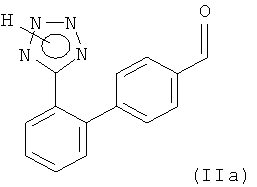
и его солей, включающему применение защитных групп, которые легко вводятся в состав соединения, обладают небольшой массой, стабильны в присутствии металлоорганических реагентов и легко и с высоким выходом удаляются в кислой среде, а также к новым промежуточным соединениям, используемым на стадиях этого способа общей формулы (IVe)

где R5 и R'5, независимо друг от друга, представляют собой С1-С7-алкил, R6, и R8 представляет собой водород, R7 и R9 вместе образуют С2-С5-алкилен. 2 н. и 6 з.п. ф-лы.
1. Способ получения соединения формулы
или его соли, включающий
(i) реакцию соединения формулы
или его соли, где Hal представляет собой галоген, в присутствии кислоты с соединением формулы

где R6, R7 и R8 независимо друг от друга выбирают из водорода и C1-C6-алкила, и R9 представляет собой C1-С6-алкил, или R7 и R9 вместе образуют С2-С5-алкилен, или R6 и R8 вместе образуют С3-С6-алкилен,
(ii) реакцию полученного соединения формулы
в присутствии катализатора переходного металла с соединением формулы
где Х представляет собой галоген, a R5 и R'5, независимо друг от друга, представляют собой С1-С7-алкил, или вместе образуют С2-С4-алкилен; и
(iii) снятие последовательно или на этой же стадии защитных групп с полученного соединения формулы
обработкой кислотой,
(iv) выделение соединения формулы (IIа') или его соли.
2. Способ по п.1, где стадию (i) осуществляют в присутствии от 0,0001 до 0,1 эквивалентов кислоты Бренстеда.
3. Способ по п.1, где стадию (ii) осуществляют с использованием платинового или палладиевого катализатора.
4. Способ по п.2, где стадию (ii) осуществляют с использованием платинового или палладиевого катализатора.
5. Способ по любому из пп.1-4, где соединение формулы (IVd) получают (i) взаимодействием соединения формулы

где X представляет собой галоген, с магнием в условиях реакции Гриньяра, с получением соединения формулы
(ii) обработкой полученного соединения формулы (IVd") с помощью Zn(X)2, где Х представляет собой галоген.
6. Способ по п.5, где реакцию соединения формулы (IVd') с магнием в условиях реакции Гриньяра проводят в присутствии 1,2-дибромэтана.
7. Соединение формулы
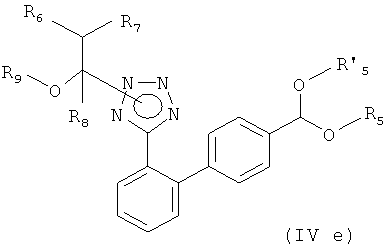
где R5 и R'5, независимо друг от друга, представляют собой С1-С7-алкил,
R6 и R8 представляют собой водород,
R7 и R9 вместе образуют С2-С5-алкилен.
8. Соединение по п.7, обладающее формулой
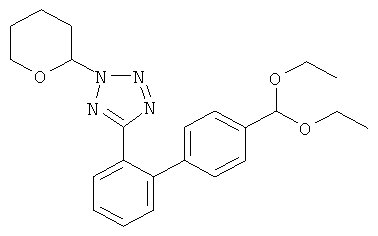
| Электромагнитный гусеничный механизм устройства для передвижения по произвольно ориентированным в пространстве стальным поверхностям | 1975 |
|
SU550313A1 |
| СОЕДИНИТЕЛЬНОЕ УСТРОЙСТВО ВЕЛОСИПЕДНОГО КОЛЕСА (ВАРИАНТЫ) | 2015 |
|
RU2688503C2 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ПРОИЗВОДНЫЕ ТЕТРАЗОЛА | 1992 |
|
RU2091376C1 |

