Предпосылки создания изобретения
Область техники, к которой относится изобретение
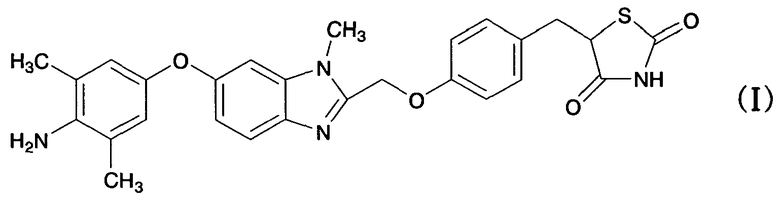
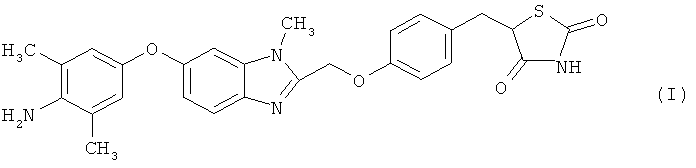
Настоящее изобретение относится к кристаллическим формам соединения тиазолидиндиона, обладающим значительной способностью активировать γ-рецептор, активируемый пролифератором пероксисом (PPAR), значительной противораковой активностью и свойствами, благоприятными для производства лекарственных средств, высокочистым, легко обрабатываемым и высокостабильным при хранении, способам их получения и лекарственным средствам, содержащим в качестве активного компонента кристаллические формы соединения тиазолидиндиона (в частности, PPARγ-активаторы или противораковые фармацевтические композиции).
Описание предшествующего уровня техники
Японский патент № 3488099 (выложенная международная заявка на патент № 99/18081, патент США № 6432993, европейский патент № 1022272) (Патентный документ 1), выложенный японский патент № 2003-238406 (выложенная международная заявка на патент № WO 03/053440) (Патентный документ 2), выложенный японский патент № 2004-083574 (WO 2004/000356) (Патентный документ 3), выложенный японский патент № 2005-162727 (WO 2004/083167) (Патентный документ 4) и выложенная международная заявка на патент № 2007/091622 (Патентный документ 5) раскрывают соединение тиазолидиндиона, представленное указанной выше формулой (I) (далее в настоящем документе сокращенно называемое соединением (I)). Соединение (I) обладает значительной способностью активировать γ-рецептор, активируемый пролифератором пероксисом (PPAR), и ожидают, что оно окажется полезным в качестве активатора PPARγ или противораковой фармацевтической композиции.
Как правило, вещества, применяемые в фармацевтических продуктах, должны быть высокочистыми во избежание непредсказуемых побочных эффектов, вызываемых загрязняющими примесями в этих веществах. Такие примеси включают побочные продукты (аналогичные вещества), образуемые при получении самих фармацевтических ингредиентов, сырье и растворители, применяемые при производстве фармацевтических ингредиентов, и т.п. Кроме того, эти вещества должны иметь физические и химические свойства, более благоприятные для производства лекарственных средств, такие как кристаллические формы фармацевтических ингредиентов, устойчивые на производственной стадии тепловой обработки и т.п., или высокая растворимость, способствующая повышению абсорбируемости фармацевтических ингредиентов, благодаря чему их эффекты могут проявляться при более низких дозах. Кроме того, важно, чтобы фармацевтические ингредиенты можно было хранить в течение продолжительных периодов времени с сохранением качества. Кроме того, крупномасштабные холодильные установки должны поддерживать качество фармацевтических ингредиентов, если их следует хранить при низких температурах. Поэтому в промышленном отношении важно найти стабильные кристаллические формы, которые можно хранить при комнатной температуре или даже при более высоких температурах.
Как указано выше, необходим промышленный и крупномасштабный способ получения высокочистых фармацевтических ингредиентов, обладающих более благоприятными свойствами для производства лекарственных средств и долго сохраняющихся, при стабильном сохранении их чистоты и свойств в течение продолжительных периодов времени.
[Патентный документ 1] Японский патент № 3488099 (выложенная международная заявка на патент № 99/18081, спецификация патента США № 6432993, спецификация европейского патента № 1022272)
[Патентный документ 2] Выложенный японский патент № 2003-238406 (выложенная международная заявка на патент № WO 03/053440)
[Патентный документ 3] Выложенный японский патент № 2004-083574 (№ WO 2004/000356)
[Патентный документ 4] Выложенный японский патент № 2005-162727 (№ WO 2004/083167)
[Патентный документ 5] Выложенная международная заявка на патент № 2007/091622
Авторы настоящего изобретения провели серьезное исследование соединения (I), которое обладает значительной способностью активировать γ-рецептор, активируемый пролифератором пероксисом (PPAR), от которого ожидают, что оно окажется полезным в качестве активатора PPARγ или противораковой фармацевтической композиции. В результате авторы настоящего изобретения нашли новые кристаллические формы гидратов дигидрохлорида соединения (I), обладающие значительными физическими и химическими свойствами в качестве фармацевтических ингредиентов, превосходно сохраняющие стабильность при комнатной температуре, а также имеющие высокую чистоту, и способ их получения. На основании этих найденных данных авторы создали настоящее изобретение.
Сущность изобретения
Настоящее изобретение относится к следующему:
(1) кристаллической форме гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, представленного следующей формулой (I); [формула 1]
(2) кристаллической форме гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, представленного общей формулой (I) согласно приведенному выше п.(1), в котором кристаллическая форма показывает главные пики при межплоскостных расстояниях, равных 7,06, 5,79, 5,43, 4,44, 4,18, 3,97, 3,91, 3,68, 3,61, 3,48, 3,24 и 2,97 ангстрем в порошковой рентгенограмме, полученной с Kα-линией излучения Cu (длина волны λ=1,54 ангстрем) (кристаллическая форма A);
(3) кристаллической форме гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, представленного общей формулой (I) согласно приведенному выше п.(1), в котором кристаллическая форма показывает главные пики при межплоскостных расстояниях, равных 10,42, 5,85, 5,52, 3,84, 3,46 и 2,95 ангстрем в порошковой рентгенограмме, полученной с Kα-линией излучения Cu (длина волны λ=1,54 ангстрем) (кристаллическая форма В);
(4) кристаллической форме согласно приведенному выше п.(2), в которой содержание примесей, измеренное ВЭЖХ, составляет 2,00% или менее;
(5) кристаллической форме согласно приведенному выше п.(2), в которой содержание примесей, измеренное ВЭЖХ, составляет 1,50% или менее;
(6) фармацевтической композиции, содержащей в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), согласно любому из приведенных выше пп.(1)-(5);
(7) PPARγ-активатору, содержащему в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), согласно любому из приведенных выше пп.(1)-(5);
(8) противораковой фармацевтической композиции, содержащей в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), согласно любому из приведенных выше пп.(1)-(5);
(9) фармацевтической композиции для предупреждения или лечения диабета, содержащей в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), согласно любому из приведенных выше пп.(1)-(5);
(10) фармацевтической композиции для предупреждения или лечения рака, сопровождаемого диабетом, содержащей в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), согласно любому из приведенных выше пп.(1)-(5);
(11) способу получения кристаллической формы согласно приведенному выше п.(2), отличающемуся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион преобразуют в его водный раствор, и затем к нему по каплям добавляют хлористоводородную кислоту;
(12) способу получения кристаллической формы согласно приведенному выше п.(2), отличающемуся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион преобразуют в его водный раствор и затем к нему по каплям добавляют хлористоводородную кислоту для получения кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, который опять растворяют или суспендируют в воде, и затем к нему по каплям добавляют хлористоводородную кислоту; и
(13) способу получения кристаллической формы согласно приведенному выше п.(2), отличающемуся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион преобразуют в его водный раствор, и затем к нему по каплям добавляют хлористоводородную кислоту для получения кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, который опять растворяют или суспендируют в воде, и затем к нему добавляют основание для получения раствора или суспензии, и затем к нему по каплям добавляют хлористоводородную кислоту;
(14) способу получения кристаллической формы согласно приведенному выше п.(2), отличающемуся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион преобразуют в его водный раствор, и затем добавляют затравку кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, одновременно к нему по каплям добавляя хлористоводородную кислоту;
(15) способу получения кристаллической формы согласно приведенному выше п.(2), отличающемуся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион преобразуют в его водный раствор, и затем к нему по каплям добавляют хлористоводородную кислоту для получения кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, который опять растворяют или суспендируют в воде, и затем добавляют затравку кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, одновременно к нему по каплям добавляя хлористоводородную кислоту;
(16) способу получения кристаллической формы согласно приведенному выше п.(2), отличающемуся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион преобразуют в его водный раствор, и затем к нему по каплям добавляют хлористоводородную кислоту для получения кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, который опять растворяют или суспендируют в воде, и затем к нему добавляют основание для получения раствора или дисперсии, и затем добавляют затравку кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, одновременно к нему по каплям добавляя хлористоводородную кислоту;
(17) способу получения кристаллической формы согласно приведенному выше п.(3), отличающемуся тем, что гидрат дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона кристаллизуют из метанола.
Кристаллическая форма гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, представленная выше формулой (I) (далее в настоящем документе может использоваться сокращенно как «кристаллическая форма по настоящему изобретению»), является твердым веществом, внутренняя структура которого состоит из атомов (или их групп) в виде регулярно структурированных 3-мерных повторов. Кристаллическая форма отличается от аморфного твердого вещества, которое не имеет такой регулярно структурированной внутренней структуры.
Даже одно соединение может быть преобразовано в ряд различных кристаллических форм, имеющих различные внутренние структуры и физические и химические свойства (кристаллические полиморфы) в зависимости от условий кристаллизации. Кристаллическая форма по настоящему изобретению может быть любым из кристаллических полиморфов или смесью двух или более кристаллических полиморфов.
Кристаллическая форма по настоящему изобретению может абсорбировать воду и содержать воду, присоединенную из воды, остающейся в атмосфере. Кристаллическая форма по настоящему изобретению, когда ее смешивают с органическим растворителем, может также абсорбировать этот растворитель с образованием сольвата. Кроме того, кристаллическая форма по настоящему изобретению может образовывать полугидрат и даже ее безводную форму, например, при ее нагревании между 25°С и 150°С в нормальных атмосферных условиях.
Кристаллическая форма по настоящему изобретению включает кристаллическую форму с присоединенной водой, кристаллическую форму, состоящую из ее гидрата или сольвата, кристаллическую форму, содержащую ее полугидрат, и кристаллическую форму, содержащую ее безводную форму. Среди этих кристаллических форм в качестве кристаллической формы по настоящему изобретению предпочтительной является кристаллическая форма моногидрата дигидрохлорида соединения (I).
В качестве одной из форм кристаллической формы по настоящему изобретению может быть включена кристаллическая форма А, которая показывает главные пики при межплоскостных расстояниях, равных 7,06, 5,79, 5,43, 4,44, 4,18, 3,97, 3,91, 3,68, 3,61, 3,48, 3,24 и 2,97 ангстрем в порошковой рентгенограмме, полученной с Kα-линией излучения Cu (длина волны λ=1,54 ангстрем), в которой главными пиками являются пики, которые имеют относительные интенсивности, большие чем 30%, считая за 100 интенсивность пика при межплоскостном расстоянии d=7,06 ангстрем, и пики, которые не наблюдают в указанной ниже кристаллической форме В по настоящему изобретению.
Межплоскостное расстояние d рассчитывают согласно уравнению: 2 dsinθ=nλ, в котором n равно 1.
Другой формой кристаллической формы по настоящему изобретению является кристаллическая форма В, которая показывает главные пики при межплоскостных расстояниях, равных 10,42, 5,85, 5,52, 3,84, 3,46 и 2,95 ангстрем в порошковой рентгенограмме, полученной с Kα-линией излучения Cu (длина волны λ=1,54 ангстрем), в которой главными пиками являются пики, которые имеют относительные интенсивности, большие чем 20%, считая за 100 интенсивность пика при межплоскостном расстоянии d=3,46 ангстрем.
Кроме того, указанные выше относительные интенсивности главных пиков могут варьировать согласно различиям во фронтах роста кристаллических форм (в габитусах кристаллов). Такие кристаллические формы признают идентичными в отношении кристаллической формы, и поэтому они включены в настоящее изобретение.
По настоящему изобретению уровень загрязнений, содержащихся в кристаллической форме, можно определять традиционными способами аналитической химии, такими как высокоэффективная жидкостная хроматография (далее может быть сокращенной как ВЭЖХ), в % по массе и, предпочтительно, по отношению площадей пиков, определенных ВЭЖХ. Условия измерений для ВЭЖХ можно выбирать произвольно. Предпочтительные условия измерений описаны ниже.
Условия ВЭЖХ-измерений (1)
Детектор: УФ-абсорбциометр (длина волны: 230 нм)
Колонка: Waters Corporation, XTerra RP18 (4,6 мм × 150 мм)
Температура колонки: 40°C
Подвижная фаза: аммоний-ацетатный буфер (0,01 моль/мл) с ацетонитрилом (65:35)
Скорость потока: 1 мл/мин (в данных условиях соединение (I) показывало время удерживания, составлявшее приблизительно 25 минут)
Инъецируемое количество: 10 мкл
Диапазон измерения площадей пиков: 70 минут после начала инъецирования
Условия ВЭЖХ-измерений (2)
Детектор: УФ-абсорбциометр (длина волны: 230 нм)
Колонка: Waters Corporation, XTerra RP18 (4,6 мм × 150 мм)
Температура колонки: 40°C
Подвижная фаза: аммоний-ацетатный буфер (0,01 моль/мл) с ацетонитрилом (56:44)
Скорость потока: 1 мл/мин (в данных условиях соединение (I) показывало время удерживания, составлявшее приблизительно 8 минут)
Инъецируемое количество: 10 мкл
Диапазон измерения площадей пиков: 70 минут после пика, который элюируется вслед за пиком, время удерживания которого относительно соединения (I) равно 1,48
При ВЭЖХ-измерениях в условиях (1) измеряют отношения площадей пиков соединения (I) и соединений, которые являются загрязняющими примесями, детектированных от минуты 0 до минуты 70. При ВЭЖХ-измерениях в условиях (2) измеряют отношения площадей пиков соединения (I) и соединений, являющихся загрязняющими примесями, детектированных в течение 70 минут после пика, который элюируется вслед за пиком, время удерживания которого относительно соединения (I) составляет 1,48.
Термин «пик» для соединений, являющихся загрязняющими примесями, в настоящем описании относится ко всем пикам, отношение площадей пиков которых измерено на уровне 0,01% или более, исключая пик соединения (I) и пик, который детектируют, когда инъецируют один растворитель.
По настоящему изобретению термин «отношение содержания загрязняющих примесей, измеренное ВЭЖХ», представляет собой отношение интегрированных площадей для всех пиков, отношения площадей которых измерены на уровне 0,01% или более, исключая пик соединения (I) и пик, который детектируют, когда инъецируют один растворитель, к площади пика соединения (I) в указанных выше условиях с использованием ВЭЖХ.
Среди кристаллических форм по настоящему изобретению для кристаллической формы А отношение содержания загрязняющих примесей, измеренное ВЭЖХ, предпочтительно составляет 2% или менее и, более предпочтительно, 1,50% или менее.
Кристаллические формы по настоящему изобретению являются очень чистыми, их цветовой тон белый, они характеризуются превосходной стабильностью при хранении, обрабатываемостью и т.п. Особенно кристаллическая форма А по настоящему изобретению имеет низкое количество остаточного растворителя и является очень чистой, белой по своему цветовому тону, и характеризуется превосходной стабильностью при хранении в форме гидрата при комнатной температуре, поскольку температура дегидратации гидрата является высокой. Кристаллическая форма В по настоящему изобретению имеет более высокую растворимость, чем известное соединение (I). Взятые вместе, кристаллические формы по настоящему изобретению являются полезными в качестве материалов для производства лекарственных средств в массовом производстве (в частности, PPARγ-активаторов, средств предупреждения и/или лечения различных форм рака, средств предупреждения и/или лечения диабета или средств предупреждения и/или лечения различных форм рака, когда они сопровождаются диабетом).
Кроме того, способ получения кристаллических форм по настоящему изобретению является пригодным для получения материалов для лекарственных средств, производимых в массовом масштабе в промышленности, поскольку этот способ может снизить количество остаточного растворителя, производить высокочистую кристаллическую форму и производить белую кристаллическую форму посредством обесцвечивания кристаллической формы.
Краткое описание чертежей
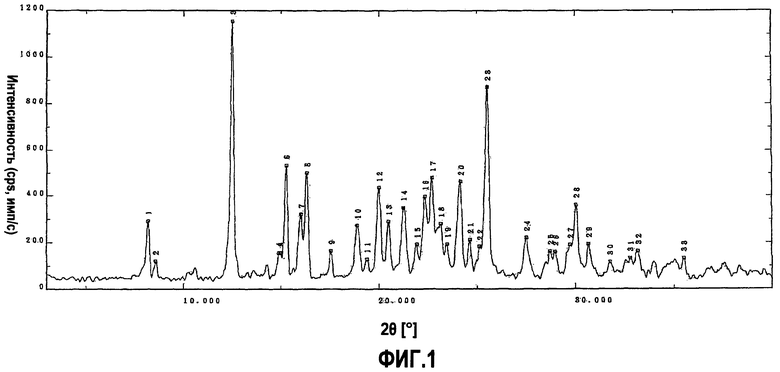
Фиг.1 является порошковой рентгенограммой для кристаллической формы А, полученной в примере 1, на которой вертикальная ось показывает интенсивность дифракции в единицах импульсов, посчитанных в секунду (cps), а горизонтальная ось показывает значения угла дифракции 2θ;
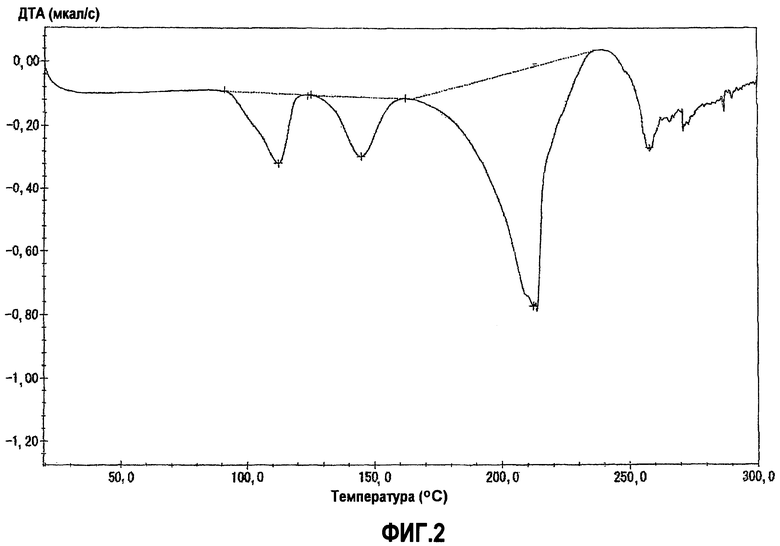
фиг.2 является графиком дифференциального термического анализа (ДТА) для кристаллической формы А, полученной в примере 1, когда температуру повышали со скоростью 5°С в минуту, на котором вертикальная ось показывает тепловой эффект в секунду (мкал/с) (или эндотермический тепловой эффект, когда эта величина отрицательна), а горизонтальная ось показывает температуру (°С);
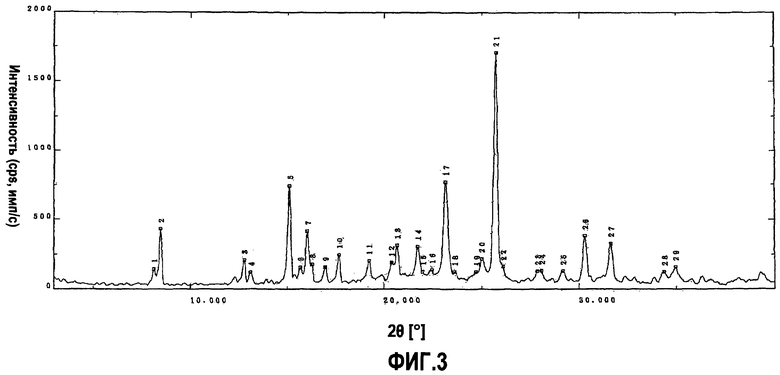
фиг.3 является порошковой рентгенограммой для кристаллической формы В, полученной в примере 3, на которой вертикальная ось показывает интенсивность дифракции в единицах импульсов, посчитанных в секунду (cps), а горизонтальная ось показывает значения угла дифракции 2θ;
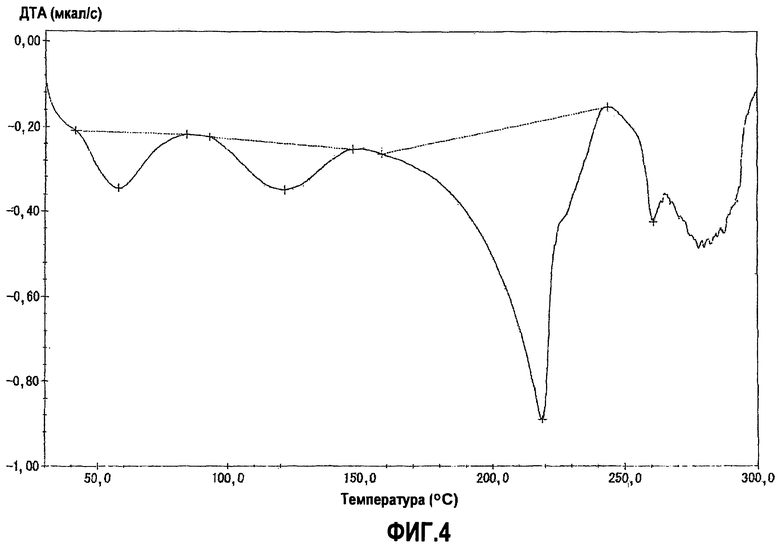
фиг.4 является графиком дифференциального термического анализа (ДТА) для кристаллической формы В, полученной в примере 3, когда температуру повышали со скоростью 5°С в минуту, на котором вертикальная ось показывает тепловой эффект в секунду (мкал/с) (или эндотермический тепловой эффект, когда эта величина отрицательна), а горизонтальная ось показывает температуру (°С);
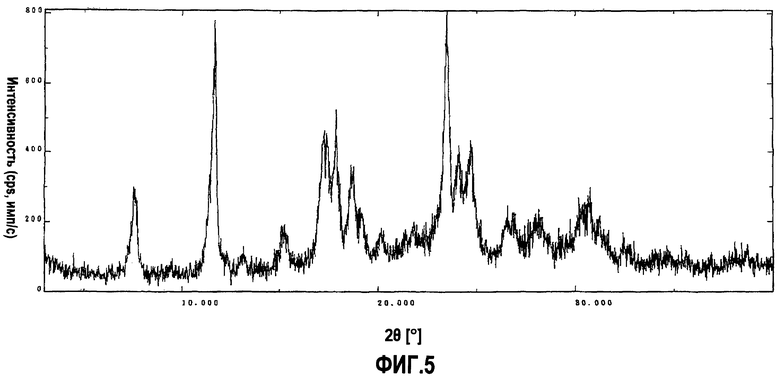
фиг.5 является порошковой рентгенограммой для соединения, полученного в сравнительном примере 1. На этой фигуре вертикальная ось показывает интенсивность дифракции в единицах импульсов, посчитанных в секунду (cps), а горизонтальная ось показывает значения угла дифракции 2θ.
Подробное описание предпочтительных вариантов осуществления
Указанное выше соединение (I) можно получать способом, раскрытым в японском патенте № 3488099, или способом, аналогичным этому способу.
Кристаллическую форму по настоящему изобретению можно получать путем растворения соединения (I) или различных солей соединения (I), или его различных сольватов, дигидрохлорида соединения (I), или различных его сольватов, или гидрата дигидрохлорида соединения (I) в соответствующем растворителе, с последующим обессоливанием (нейтрализацией), добавлением хлористого водорода или хлористоводородной кислоты, концентрированием раствора, охлаждением, смешиванием с хорошим растворителем и плохим растворителем и т.п., что ведет к условиям перенасыщенности и дает возможность осадить гидрат дигидрохлорида соединения (I) с последующим выделением осажденной кристаллической формы. Кроме того, раствор синтетических неочищенных продуктов, содержащий соединение (I), описанное выше, можно применять в качестве раствора, в котором растворены соединение (I) или различные соли соединения (I), или его различные сольваты, дигидрохлорид соединения (I) или его различные сольваты, или сам гидрат дигидрохлорида соединения (I).
Осаждение кристаллической формы может быть инициировано спонтанно в реакционном контейнере, но оно также может быть инициировано или стимулировано приданием механического стимула, такого как добавление затравки кристаллической формы, стимуляция ультразвуком и трение поверхности реактора.
Полученная кристаллическая форма может быть подвергнута перекристаллизации и суспензионной очистке для дальнейшего улучшения ее чистоты и качества.
В тех случаях, когда применяют различные соли соединения (I) или его различные сольваты, можно проводить обессоливание (нейтрализацию) для обеспечения получения дигидрохлорида соединения (I). В таких случаях для обессоливания обычно применяют основание. Примеры основания не ограничены, пока они осуществляют обессоливание, и они включают гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия, гидроксид калия или гидроксид цезия, гидроксиды щелочноземельных металлов, такие как гидроксид магния или гидроксид кальция, или амины, такие как аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин или трибутиламин. Предпочтительно, основание является гидроксидом щелочного металла или амином. Более предпочтительно, основание является гидроксидом лития, гидроксидом натрия, гидроксидом калия, трибутиламином или аммиаком.
Основание можно добавлять либо непосредственно, либо после растворения в различных растворителях.
Количество добавляемого основания не ограничено, но обычно оно находится в диапазоне от 1 до 4 эквивалентов на эквивалент различных солей соединения (I).
В тех случаях, когда для кристаллизации гидрата дигидрохлорида соединения (I) добавляют хлористый водород или хлористоводородную кислоту, их количество предпочтительно является большим, чем количество, требуемое для образования дигидрохлорида соединения (I) и осаждения в виде его кристаллической формы. Кроме того, количество добавляемых хлористого водорода или хлористоводородной кислоты обычно находится в диапазоне от 0,1 до 20 эквивалентов и, более предпочтительно, в диапазоне от 2 до 10 эквивалентов на эквивалент соединения (I) для снижения растворимости гидрата дигидрохлорида соединения (I) в растворителях.
Примеры способов концентрирования раствора соединения (I) или его гидрата дигидрохлорида включают способ концентрирования, который позволяет испарить растворитель посредством нагревания при атмосферном давлении или при пониженном давлении с использованием роторного испарителя или т.п., или способ концентрирования с использованием мембраны обратного осмоса. Мембрану обратного осмоса, применяемую для конденсирования раствора, можно выбирать, например, из мембраны полиакрилонитрильного типа, мембраны поливинилспиртового типа, мембраны полиамидного типа, мембраны целлюлозоацетатного типа и т.п.
Температура, при которой кристаллизуют гидрат дигидрохлорида соединения (I), обычно находится в диапазоне от -70 до 150°С и предпочтительно от -70 до 100°С.
Примеры хороших растворителей, применимых в производстве кристаллической формы по настоящему изобретению, включают, например, воду, спирты, такие как метанол или этанол, кетоны, такие как ацетон или метилэтилкетон, простые эфиры, такие как тетрагидрофуран или диоксан, нитрилы, такие как ацетонитрил или пропионитрил, сложные эфиры, такие как метилацетат или этилацетат, амиды, такие как диметилформамид, диметилацетамид или триамид гексаметилфосфорной кислоты, сульфоксиды, такие как диметисульфоксид, или растворители, смешанные из них. Предпочтительно применяют метанол, тетрагидрофуран или эти растворители, смешанные с водой.
Плохие растворители, применяемые для получения кристаллической формы по настоящему изобретению, выбирают соответственно растворителям, применяемым в качестве хороших растворителей. Примеры плохих растворителей включают, например, воду, C2-C4-спирты, такие как этанол, пропанол и бутанол, кетоны, такие как ацетон или метилэтилкетон, простые эфиры, такие как диэтиловый эфир, сложные эфиры, такие как этилацетат и пропилацетат, и нитрилы, такие как ацетонитрил, пропионитрил и бутиронитрил.
В тех случаях, когда получают кристаллическую форму А, эту кристаллическую форму А можно очищать посредством образования водного раствора соединения (I) (предпочтительно, смешанного раствора с тетрагидрофураном и водой), к которому затем по каплям добавляют хлористоводородную кислоту для повышения чистоты кристаллической формы.
После растворения или суспендирования полученной кристаллической формы А в воде к ней можно по каплям добавлять хлористоводородную кислоту для дальнейшего повышения ее чистоты.
В таких случаях эффект очистки можно повысить, в некоторых случаях добавляя основание до начала капельного добавления хлористоводородной кислоты. В тех случаях, когда добавляют основание, примеры оснований не ограничены, но обычно они включают неорганические основания, такие как гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид магния и гидроксид кальция; и органические основания, такие как триметиламин, триэтиламин, диизопропилэтиламин и трибутиламин. Предпочтительно применяют гидроксид натрия или гидроксид калия.
Температура растворения или температура суспендирования в воде не ограничены, но обычно они находятся в диапазоне от 0 до 100°С (температура кипячения с обратным холодильником), предпочтительно в диапазоне от 20 до 100°С (температура кипячения с обратным холодильником) и, более предпочтительно, в диапазоне от 30 до 100°С (температура кипячения с обратным холодильником).
Продолжительность растворения или суспендирования не ограничена, но обычно она находится в диапазоне от 5 минут до 12 часов и, предпочтительно, в диапазоне от 10 минут до 6 часов.
В случае суспендирования капельное добавление хлористоводородной кислоты после суспендирования можно проводить либо после сушки, либо без сушки кристаллической формы, которую выделяют после суспендирования или непосредственно после суспендирования без выделения. Возможен любой вариант.
Количество хлористоводородной кислоты, применяемой после растворения или суспендирования в воде, не ограничено, пока оно больше или равно количеству, необходимому соединению (I) для образования его дигидрохлорида и осаждения его кристаллической формы. Хлористоводородную кислоту обычно добавляют до достижения получаемым раствором значения рН в диапазоне от 2 до 0,5 для понижения растворимости кристаллической формы А в воде.
Кристаллическую форму А можно добавлять в качестве затравки кристаллической формы либо до начала капельного добавления хлористоводородной кислоты, или во время ее капельного добавления. В тех случаях когда добавляют затравку кристаллической формы, количество добавляемой затравки кристаллической формы не ограничено, но обычно оно находится в диапазоне от 0,0001 до 20%, предпочтительно от 0,001 до 10%, и более предпочтительно, от 0,01 до 5% относительно количества очищаемой кристаллической формы А.
Температура, при которой добавляют по каплям хлористоводородную кислоту, обычно находится в диапазоне от 0 до 100°С (температура кипячения с обратным холодильником), предпочтительно в диапазоне от 20 до 100°С (температура кипячения с обратным холодильником) и, более предпочтительно, в диапазоне от 50 до 100°С (температура кипячения с обратным холодильником).
В тех случаях когда получают кристаллическую форму В по настоящему изобретению, дигидрохлорид соединения (I) или его гидрат кристаллизуют из метанола (предпочтительно безводного метанола).
В качестве исходного материала для производства кристаллической формы А по настоящему изобретению можно применять соединение (I), различные соли соединения (I), его различные сольваты, дигидрохлорид соединения (I) или различные сольваты дигидрохлорида соединения (I), или уже выделенный гидрат дигидрохлорида соединения (I). В качестве альтернативы можно применять раствор синтетических неочищенных продуктов, содержащий соединение (I), так как продукт можно очистить кристаллизацией. В качестве исходного материала для производства кристаллической формы В по настоящему изобретению применяют дигидрохлорид соединения (I) или его гидрат.
Осажденные кристаллические формы А или В можно выделить, например, фильтрованием, центрифугированием или декантацией и т.п. Выделенные кристаллические формы при необходимости можно промыть соответствующим растворителем. Для промывания кристаллической формы по настоящему изобретению можно применять растворители, например воду; спирты, такие как метанол, этанол и изопропанол; кетоны, такие как ацетон; сложные эфиры, такие как метилформиат, этилформиат, метилацетат и этилацетат; ароматические углеводороды, такие как толуол и ксилол; нитрилы, такие как ацетонитрил; простые эфиры, такие как диэтиловый эфир и тетрагидрофуран, и растворители, смешанные из них. Предпочтительно, применяют воду, метанол, тетрагидрофуран или смеси растворителей.
Выделенную кристаллическую форму обычно сушат в диапазоне от 0 до 150°С и, предпочтительно, в диапазоне от 20 до 90°С до тех пор, пока масса не остается почти постоянной. Сушку кристаллической формы можно проводить в присутствии осушителей, таких как силикагель или хлорид кальция, или при пониженном давлении, если это необходимо. При пониженном давлении кристаллическую форму можно сушить без дегидратации воды кристаллической формы, регулируя температуру и давление. В таких случаях давление регулируют, устанавливая его относительно высоко для высокой температуры сушки. Например, в случаях когда сушат кристаллическую форму А по настоящему изобретению, давление устанавливают в диапазоне от 0,7 до 50 кПа и, предпочтительно, в диапазоне от 1,8 до 11 кПа для температуры сушки 50°С.
В случаях когда высушенную кристаллическую форму дегидратируют сушкой без регулирования температуры и давления, высушенную кристаллическую форму можно подвергать абсорбции для удаления влаги в основном температурном диапазоне от 0 до 50°С и в диапазоне относительной влажности от 10 до 100% и, предпочтительно, в температурном диапазоне от 10 до 40°С и в диапазоне относительной влажности от 20 до 100% до тех пор, пока масса не останется почти постоянной. Полученную кристаллическую форму можно подвергать перекристаллизации и суспензионной очистке для дальнейшего повышения чистоты и качества кристаллической формы.
Перекристаллизацию кристаллической формы по настоящему изобретению осуществляют способами, обычно применяемыми в синтетической органической химии, такими как (1) растворение при нагревании с последующим охлаждением, (2) способ концентрирования с применением отгонки растворителя после растворения и (3) осаждение кристаллической формы посредством растворения в хорошем растворителе с последующим добавлением к нему плохого растворителя.
В случаях когда кристаллическую форму растворяют в растворителе для перекристаллизации, может происходить дегидрохлорирование. В таких случаях кристаллическую форму по настоящему изобретению можно получить, добавляя хлористый водород или хлористоводородную кислоту.
Суспензионная очистка является способом очистки, при котором кристаллическую форму соединения суспендируют в соответствующем растворителе и снова собирают при взбалтывании суспензии.
Примеры растворителей, применимых для суспензионной очистки кристаллической формы А по настоящему изобретению, включают сложные эфиры, такие как метилацетат и этилацетат; галогенированные углеводороды, такие как метиленхлорид и хлороформ; ароматические углеводороды, такие как толуол и ксилол; этанол; воду; алифатические углеводороды, такие как гексан; простые эфиры, такие как диизопропиловый эфир, диэтиловый эфир и тетрагидрофуран; кетоны, такие как ацетон и метилэтилкетон; нитрилы, такие как ацетонитрил, и смеси растворителей. Предпочтительно применяют воду, метанол, тетрагидрофуран или растворители, смешанные из них. Более предпочтительно, применяют воду.
В случаях когда кристаллическую форму суспендируют в растворителе для суспензионной очистки, может происходить дегидрохлорирование. В таких случаях кристаллическую форму А можно получить, добавляя хлористый водород или хлористоводородную кислоту.
Примеры растворителей, применимых для суспензионной очистки кристаллической формы В по настоящему изобретению, включают кетоны, такие как ацетон и метилэтилкетон; сложные эфиры, такие как метилацетат и этилацетат; нитрилы, такие как ацетонитрил; галогенированные углеводороды, такие как метиленхлорид и хлороформ; ароматические углеводороды, такие как толуол и ксилол; спирты, такие как этанол и изопропанол; амиды, такие как N,N-диметилформамид, воду; алифатические углеводороды, такие как гексан; простые эфиры, такие как тетрагидрофуран, диизопропиловый эфир и диэтиловый эфир, и смеси растворителей. Предпочтительно, применяют воду, метанол, тетрагидрофуран или смеси растворителей. Более предпочтительно, применяют воду.
В случаях когда кристаллическую форму суспендируют в растворителе для суспензионной очистки, может происходить дегидрохлорирование. В таких случаях кристаллическую форму В можно получить, добавляя хлористый водород или хлористоводородную кислоту.
Кристаллическую форму, которую получают перекристаллизацией и суспензионной очисткой, можно выделять и сушить способами, аналогичными способам, указанным выше.
Известно, что соединение (I) и его фармакологически приемлемые соли (особо предпочтительно, его гидрохлоридная соль) обладают значительной способностью активировать γ-рецептор, активируемый пролифератором пероксисом (PPAR), как раскрыто в японском патенте № 3488099 (WO 99/18081, патент США № 6432993, европейский патент № 1022272) (Патентный документ 1), выложенном японском патенте № 2003-238406 (WO 03/053440) (Патентный документ 2), выложенном японском патенте № 2004-083574 (WO 2004/000356) (Патентный документ 3), выложенном японском патенте № 2005-162727 (WO 2004/083167) (Патентный документ 4), WO 2007/091622 (Патентный документ 5) и т.п.
В частности, WO 2007/091622 (Патентный документ 5) раскрывает, что соединение (I) и его гидрохлоридная соль полезны в качестве противораковых фармацевтических композиций для предупреждения или лечения рака желудка, рака толстой кишки, рака легкого, рака молочной железы, рака поджелудочной железы, рака почки, рака предстательной железы, медуллобластомы, рабдомиосаркомы, саркомы Эвинга, липосаркомы, множественной миеломы и лейкоза.
Более конкретно, тестовый пример 1 в WO 2007/091622 (Патентный документ 5) наряду с экспериментальными данными раскрывает, что дигидрохлорид соединения (I) проявляет значительную активность в подавлении пролиферации любых раковых клеток, включая клетки рака желудка человека, клетки рака молочной железы человека, клетки мелкоклеточного рака легкого, клетки рака поджелудочной железы, клетки рака предстательной железы, клетки рака почки, медуллобластомы, клетки саркомы человека (рабдомиосаркомы, саркомы Эвинга, липосаркомы) и множественной миеломы.
Кроме того, тестовый пример 2 в том же документе (Патентный документ 5) наряду с экспериментальными данными раскрывает, что дигидрохлорид соединения (I) значительно ингибирует пролиферацию, подавляя активность человеческих лейкозных клеток.
В дополнение, тестовый пример 3 в WO 2007/091622 (Патентный документ 5) раскрывает, что дигидрохлорид соединения (I) проявляет значительную противораковую активность in vivo против линий клеток рака толстой кишки человека.
Кроме того, тестовый пример 4 в том же документе (Патентный документ 5) раскрывает, что введение дигидрохлорида соединения (I) в сочетании с ингибитором рецептора эпидермального фактора роста (EGFR) проявляет синергическую активность в подавлении пролиферации в раковых клетках.
В том же документе (Патентный документ 5) тестовый пример 5 раскрывает, что дигидрохлорид соединения (I) проявляет противоопухолевую активность против немелкоклеточного рака легкого человека и что его введение в сочетании с ингибитором рецептора эпидермального фактора роста (EGFR) проявляет повышенную противоопухолевую активность.
Кроме того, тестовый пример 6 в том же документе (Патентный документ 5) раскрывает, что введение дигидрохлорида соединения (I) в сочетании с ингибитором рецептора сосудистого эндотелиального фактора роста (VEGFR) или ингибитором Raf-киназы проявляет синергическую активность в подавлении пролиферации в раковых клетках.
Далее, тестовый пример 7 в том же документе (Патентный документ 5) раскрывает, что дигидрохлорид соединения (I) проявляет противоопухолевую активность против рака почки человека и что его введение в сочетании с ингибитором рецептора сосудистого эндотелиального фактора роста или ингибитором Raf-киназы проявляет повышенную противоопухолевую активность.
Поэтому кристаллическая форма по настоящему изобретению эффективна в качестве лекарственного средства, особенно в качестве активатора PPARγ, и эффективна в качестве средства (противораковой фармацевтической композиции) лечения или предупреждения различных форм рака, как описано выше.
В дополнение, японский патент № 3488099 (WO 99/18081, патент США № 6432993, европейский патент № 1022272) (Патентный документ 1) раскрывает, что соединение (I) и его фармацевтически приемлемые соли обладают значительной способностью активировать γ-рецептор, активируемый пролифератором пероксисом, проявляют значительное действие в отношении улучшения инсулиновой резистентности, а также являются эффективными средствами лечения или предупреждения диабета (в частности, диабета типа 2). Поэтому кристаллическая форма по настоящему изобретению эффективна в качестве фармацевтической композиции для предупреждения или лечения диабета (в частности, диабета типа 2).
Кроме того, кристаллическая форма по настоящему изобретению эффективна в качестве фармацевтической композиции для предупреждения или лечения различных форм рака, сопровождаемых диабетом типа 2, поскольку, как описано выше, она является противораковой фармацевтической композицией.
В случаях когда кристаллическую форму по настоящему изобретению применяют в качестве лекарственного средства, в частности в качестве активатора PPARγ, средства для лечения или предупреждения различных форм рака или средства для лечения или предупреждения диабета, кристаллическую форму можно вводить либо самостоятельно, либо в смеси с соответствующим и фармацевтически приемлемым разбавляющим агентом или агентом разбавления и т.п. с образованием, например, таблеток, капсул, гранул, порошков или сиропов для перорального введения или с образованием, например, растворов для инъекций или суппозиториев для парентерального введения.
Препараты получают известными способами, применяя такие добавки, как разбавляющие агенты (например, сахара, такие как лактоза, сахароза, глюкоза и сорбит; производные крахмала, такие как кукурузный крахмал, картофельный крахмал, α-крахмал, декстрин и карбоксиметилкрахмал; производные целлюлозы, такие как кристаллическая целлюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза, кальций-карбоксиметилцеллюлоза и внутренне поперечно-сшитая натрий-карбоксиметилцеллюлоза; гуммиарабик; декстран; пуллулан; силикаты, такие как синтетический алюмосиликат и алюмометасиликат магния; фосфаты, такие как фосфат кальция; карбонаты, такие как карбонат кальция; и гидросульфаты, такие как сульфат кальция), связующие (например, указанные выше разбавляющие агенты; желатин; поливинилпирролидон; и макрогол), дезинтеграторы (например, указанные выше разбавляющие агенты; химически модифицированные производные крахмала или целлюлозы, такие как натрий-кроскармеллоза, натрий-карбоксиметилкрахмал, поперечно-сшитый поливинилпирролидон), смазывающие средства (например, тальк; стеариновая кислота; стеараты металлов, такие как стеарат кальция и стеарат магния; коллоидный кремнезем; вигам; воски, такие как пчелиный воск и китовый воск; борная кислота; гликоль; карбоновые кислоты, такие как фумаровая кислота и адипиновая кислота; натриевые соли карбоновых кислот, такие как бензоат натрия; гидросульфаты, такие как сульфат натрия; лейцин; лаурилсульфаты, такие как лаурилсульфат натрия и лаурилсульфат магния; силикаты, такие как легкая безводная кремниевая кислота и гидраты кремниевой кислоты; производные крахмала, как описано в вышеуказанных разбавляющих агентах), фиксирующие агенты (например, сложные эфиры пара-гидроксибензоата, такие как метилпарабен и пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт и фенилэтиловый спирт; хлорид бензалкония; фенолы, такие как фенол и крезол; тимеросал; ангидрид уксусной кислоты; и сорбат), корригенты (например, обычно применяемые подсластители, подкислители и ароматические материалы), суспендирующие агенты (например, полисорбат 80 и натрий-карбоксиметилцеллюлоза), агенты разбавления и растворители для приготовления (например, вода, этанол и глицерин).
Применяемое количество кристаллической формы по настоящему изобретению может быть различным в зависимости от степени выраженности симптомов, массы тела и возраста пациентов (млекопитающих, в частности людей), которым ее вводят, способа введения и т.п. Например, рекомендуемая доза находится в диапазоне от минимума, составляющего 0,001 мг/кг массы тела (предпочтительно, 0,01 мг/кг массы тела), до максимума, составляющего 500 мг/кг массы тела (предпочтительно, 50 мг/кг массы тела), для дозы, вводимой перорально, и в диапазоне от минимума, составляющего 0,005 мг/кг массы тела (предпочтительно 0,05 мг/кг массы тела), до максимума, составляющего 50 мг/кг массы тела (предпочтительно 5 мг/кг массы тела), для дозы, вводимой внутривенно. Предпочтительно агенты вводят от одного раза до нескольких раз в день соответственно степени выраженности симптомов.
[ПРИМЕРЫ]
Далее в данном документе настоящее изобретение будет описано более конкретно примерами, тестовыми примерами и примерами препаратов.
Пример 1: Кристаллическая форма A
(1-1)
4,0 г гидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, полученного способом, аналогичным способу, описанному в примере 8 японского патента № 3488099, суспендировали в смеси тетрагидрофурана (40 мл) и воды (12 мл) при комнатной температуре в атмосфере азота и к нему по каплям добавляли 2,4 г 25%-го водного раствора гидроксида натрия для образования раствора. Полученный раствор по каплям добавляли к суспензии активированного угля в тетрагидрофуране (12 мл), полученной в атмосфере азота (0,4 г), и смесь перемешивали в течение 20 минут при той же температуре. После отфильтровывания активированного угля активированный уголь промывали 12 мл тетрагидрофурана. Фильтрат и промывной раствор объединяли и к ним добавляли 12 мл воды. К полученному раствору по каплям добавляли смешанный раствор 38%-й хлористоводородной кислоты (3,2 г) и тетрагидрофурана (12 мл). Реакционную смесь перемешивали в течение 45 минут. Смесь охлаждали до 0°С и перемешивали еще в течение 2 часов. Полученную кристаллическую форму отфильтровывали и сушили в течение 12 часов при давлении, равном приблизительно 80 Па, и при 50°С. Кристаллическую форму оставляли в данной атмосфере на 3 часа, что давало 3,64 г кристаллической формы моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона.
(1-2)
2,0 г кристаллической формы, полученной в (1-1), суспендировали в 40 мл воды и суспензию перемешивали в течение 20 минут при 80°С. К ней в течение 5 минут по каплям добавляли смесь 38%-й хлористоводородной кислоты (1,1 г) и воды (8,4 мл) при той же температуре. Затем реакционную смесь перемешивали в течение 1 часа и охлаждали до 40°С. Кристаллическую форму отфильтровывали и промывали 6 мл воды, получая гигроскопическую кристаллическую форму моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона. Полученную кристаллическую форму сушили в течение 14 часов при давлении, равном приблизительно 80 Па и при 50°С. Кристаллическую форму оставляли на 3 часа в атмосфере, что давало 1,83 г белой кристаллической формы моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, причем кристаллическая форма имела форму кристаллов, показывающих дифракционную картину, полученную при порошковой рентгенографии, описанную на фиг.1 (кристаллическая форма А).
(1-3)
Фиг.1 показывает дифракционную картину, полученную при порошковой рентгенографии (Cu Kα, λ=1,54 ангстрем) для кристаллической формы, полученной в (1-2). Таблица 1 показывает пики, которые имеют относительные интенсивности, большие или равные 10, если за 100 принята интенсивность наибольшего пика дифракционной картины, описанной на фиг.1. Цифры на фиг.1 соответствуют номерам пиков в таблице 1.
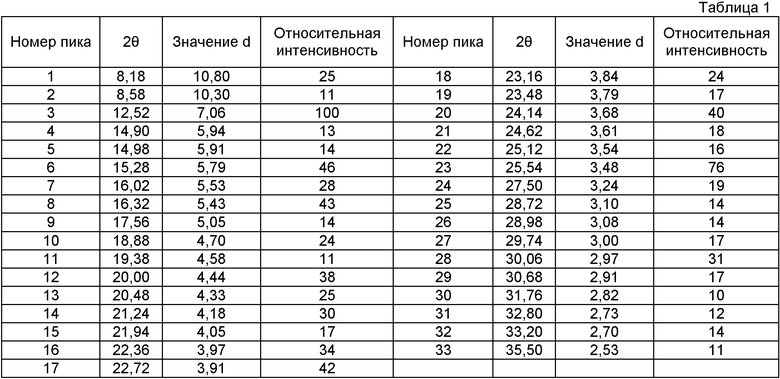
Среди этих пиков особенно характерными для кристаллической формы А являются пики при межплоскостных расстояниях (значениях d), равных 7,06, 5,79, 5,43, 4,44, 4,18, 3,97, 3,91, 3,68, 3,61, 3,48, 3,24 и 2,97 ангстрем.
(1-4)
Фиг.2 показывает график дифференциального термического анализа (ДТА).
Пример 2: Кристаллическая форма A
(2-1)
2,0 г гидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, полученного способом, аналогичным способу, описанному в примере 8 японского патента № 3488099, суспендировали в смеси тетрагидрофурана (20 мл) и воды (6 мл) при комнатной температуре в атмосфере азота и к нему по каплям добавляли 1,2 г 25%-го водного раствора гидроксида натрия для образования раствора. Полученный раствор по каплям добавляли к суспензии активированного угля (0,2 г) в тетрагидрофуране (6 мл), приготовленной в атмосфере азота, и смесь перемешивали в течение 20 минут при той же температуре. После отфильтровывания активированного угля активированный уголь промывали 6 мл тетрагидрофурана. Фильтрат и промывной раствор объединяли и к ним добавляли 6 мл воды. К полученному раствору по каплям добавляли смешанный раствор 38%-й хлористоводородной кислоты (1,6 г) и тетрагидрофурана (6 мл). Реакционную смесь перемешивали в течение 45 минут. Смесь охлаждали до 0°С и перемешивали еще в течение 2 часов. Полученную кристаллическую форму отфильтровывали и промывали водой, получая влажную кристаллическую форму моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона.
(2-2)
Влажную кристаллическую форму, полученную в (2-1), суспендировали в 40 мл воды и суспензию перемешивали в течение 20 минут при 80°С. К ней по каплям в течение 5 минут добавляли смесь 38%-й хлористоводородной кислоты (1,1 г) и воды (8,4 мл) при той же температуре. Затем реакционную смесь перемешивали в течение 1 часа и охлаждали до 40°С. Кристаллическую форму отфильтровывали и промывали 6 мл воды, получая влажную кристаллическую форму моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона. Полученную влажную кристаллическую форму сушили в течение 12 часов при давлении 4,3 кПа и при 50°С, получая белую кристаллическую форму моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, причем эта кристаллическая форма имела форму кристаллов, описанную в п.2 формулы изобретения. Порошковая рентгенограмма и график дифференциального термического анализа этой кристаллической формы совпадали с такими же данными кристаллической формы А, полученной в примере 1.
Пример 3: Кристаллическая форма А
(3-1)
2,0 г моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, полученного способом, аналогичным способу, описанному в примере 1-1, суспендировали в 100 мл воды. Полученную суспензию кипятили с обратным холодильником в течение 2 часов. Реакционную смесь охлаждали до 0°С и перемешивали в течение 1 часа. Полученную кристаллическую форму отфильтровывали и промывали водой. Полученную влажную кристаллическую форму сушили в течение 14 часов при давлении, равном приблизительно 80 кПа, и при 50°С, получая кристаллическую форму, в которой соотношение 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона и хлористоводородной кислоты составляло приблизительно от 1 до 1,1.
(3-2)
Кристаллическую форму, полученную в (3-1), суспендировали в 40 мл воды. К ней в течение 5 минут при 80°С по каплям добавляли смесь 38%-й хлористоводородной кислоты (1,1 г) и воды (8,4 мл). Затем реакционную смесь перемешивали в течение 1 часа и охлаждали до 0°С. Кристаллическую форму отфильтровывали и промывали 6 мл воды, получая влажную кристаллическую форму моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона. Полученную влажную кристаллическую форму сушили в течение 14 часов при давлении, равном приблизительно 80 Па и при 50°С. Эту кристаллическую форму оставляли на 3 дня в атмосфере, получая 1,83 г белой кристаллической формы моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, причем эта кристаллическая форма имела форму кристаллов, описанную в п.2 формулы изобретения. Порошковая рентгенограмма и график дифференциального термического анализа этой кристаллической формы совпадали с такими же данными кристаллической формы А, полученной в примере 1.
Пример 4: Кристаллическая форма A
4,0 г моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, полученного способом, аналогичным способу, описанному в примере (1-1), суспендировали в 160 мл воды в атмосфере азота и к нему по каплям добавляли 1,08 г 25%-го водного раствора гидроксида натрия при 80°С. Полученную смесь перемешивали в течение 1 часа. Затем смесь охлаждали до 65°С и к ней при той же температуре по каплям добавляли смешанный раствор 38%-й хлористоводородной кислоты (0,65 г) и воды (4 мл). После добавления к ней 0,2 г кристаллической формы А полученный раствор перемешивали в течение 1 часа и к нему в течение 1 часа по каплям добавляли смешанный раствор 38%-й хлористоводородной кислоты (5,11 г) и воды (31,6 мл) при той же температуре. Затем реакционную смесь перемешивали в течение 30 минут при той же температуре и охлаждали до 40°С. Полученную кристаллическую форму отфильтровывали и промывали смешанным раствором 38%-й хлористоводородной кислоты (0,31 г) и воды (12 мл). Полученную кристаллическую форму сушили в течение 17 часов при давлении, равном приблизительно 4,3 кПа, и при 50°С, что давало 3,98 г кристаллической формы А. Порошковая рентгенограмма и график дифференциального термического анализа этой кристаллической формы совпадали с такими же данными кристаллической формы А, полученной в примере 1.
Пример 5: Кристаллическая форма A
5,0 г моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, полученного способом, аналогичным способу, описанному в примере (1-1), суспендировали в 300 мл воды в атмосфере аргона и к нему по каплям добавляли 1,94 г 38% хлористоводородной кислоты. Полученную смесь перемешивали при 95°С для образования раствора. К нему при той же температуре по каплям добавляли смешанный раствор 38%-й хлористоводородной кислоты (0,81 г) и воды (5 мл). После добавления к нему 0,25 г кристаллической формы А полученную смесь перемешивали в течение 30 минут и к ней в течение 2 часов при той же температуре добавляли смешанный раствор 38%-й хлористоводородной кислоты (6,14 г) и воды (38 мл). Затем реакционную смесь перемешивали в течение 30 минут и охлаждали до 40°С. Полученную кристаллическую форму отфильтровывали и промывали смешанным раствором 38%-й хлористоводородной кислоты (0,39 г) и воды (15 мл). Полученную кристаллическую форму сушили в течение 16 часов при давлении, равном приблизительно 4,3 кПа, и при 50°С, что давало 5,01 г кристаллической формы А. Порошковая рентгенограмма и график дифференциального термического анализа этой кристаллической формы совпадали с такими же данными кристаллической формы А, полученной в примере 1.
Пример 6: Кристаллическая форма B
(6-1)
2,5 г кристаллической формы моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, полученного тем же способом, что и способ, описанный в примере 1, суспендировали в 50 мл метанола и растворяли при 60°С. Полученный раствор охлаждали до 0°С и перемешивали в течение 26 часов при той же температуре. Полученную кристаллическую форму сушили в течение 16 часов при давлении, равном приблизительно 80 Па, и при 50°С, получая приблизительно полугидрат дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона.
(6-2)
Кристаллическую форму приблизительно полугидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, полученного в (6-1), оставляли на 19 часов при комнатной температуре при относительной влажности, равной приблизительно 100%, что давало 1,6 г белой кристаллической формы моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, причем эта кристаллическая форма имела форму кристаллов, показывающую картину дифракции, полученную при порошковой рентгенографии, описанную на фиг.3 (кристаллическая форма В).
(6-3)
Фиг.3 показывает картину дифракции, полученную при порошковой рентгенографии (Cu Kα, λ=1,54 ангстрем) для кристаллической формы, полученной в (6-2). Таблица 2 показывает пики, которые имеют относительные интенсивности, большие или равные 7, если за 100 принята интенсивность наибольшего пика картины дифракции, описанной на фиг.3. Цифры на фиг.3 соответствуют номерам пиков в таблице 2.
ние d
Среди этих пиков особенно характерными для кристаллической формы В являются пики при межплоскостных расстояниях (значениях d), равных 10,42, 5,85, 5,52, 3,84, 3,46 и 2,95 ангстрем.
(6-4)
Фиг.4 показывает график дифференциального термического анализа (ДТА).
Сравнительный пример 1
Гидрохлорид 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона получали способом, описанным в примере 8 японского патента № 3488099. Полученное соединение имело слабый красно-пурпурный цвет. Фиг.5 показывает картину дифракции, полученную при порошковой рентгенографии (Cu Kα, λ=1,54 ангстрем) для этого соединения.
[ТЕСТОВЫЕ ПРИМЕРЫ]
Тестовый пример 1: Измерение содержания
Содержание кристаллической формы А, полученной в примере 1, и соединения, полученного в сравнительном примере 1 (гидрохлоридной соли 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, полученной способом, аналогичным способу, описанному в примере 8 японского патента № 3488099, который применяли в качестве материала, как описано в примере 1, что также справедливо и для тестовых примеров 2-4) измеряли описанным ниже способом анализа с применением ВЭЖХ.
Аммоний-ацетатный буфер (0,01 моль/мл) получали, добавляя водный раствор ацетата аммония (0,01 моль/мл) к водному раствору уксусной кислоты (0,01 моль/мл) и устанавливая рН=4,5.
Воду, ацетонитрил и метанол смешивали в соотношении 55:40:5 по объему, получая раствор для растворения образцов.
0,2 г Изоамил-4-гидроксибензоата растворяли в растворе для растворения образцов, доводя общий объем до 200 мл, получая раствор внутреннего стандарта.
Точно отмеряли приблизительно 0,03949 г стандарта моногидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, переносили в 200-мл мерную колбу и растворяли в растворе для растворения образцов, доводя общий объем до 200 мл. Точно отмеряли 5 мл полученного раствора, переносили в 50-мл мерную колбу и туда же точно добавляли 10 мл раствора внутреннего стандарта. Туда же дополнительно добавляли раствор для растворения образцов, доводя общий объем до 50 мл, получая стандартный раствор.
Точно отмеряли приблизительно 0,01 г образца, предназначенного для определения содержания, переносили в 10-мл мерную колбу и туда же добавляли приблизительно 2,5 мл диметилсульфоксида, вводя его в раствор. Туда же дополнительно добавляли раствор для растворения образцов, доводя общий объем до 10 мл. Точно отмеряли 2 мл полученного раствора, переносили в 100-мл мерную колбу и туда же точно добавляли 20 мл раствора внутреннего стандарта. Туда же дополнительно добавляли раствор для растворения образцов, доводя общий объем до 100 мл, получая раствор образца.
Содержание измеряли при условиях, описанных ниже.
Детектор: УФ-абсорбциометр (длина волны: 290 нм)
Колонка: Waters Corporation, Symmetry C18 (4,6 мм × 100 мм)
Температура колонки: 40°С
Подвижная фаза: аммоний-ацетатный буфер (0,01 моль/мл) с ацетонитрилом (3:2)
Скорость потока: 1 мл/мин (В этих условиях 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион показывал время удерживания, равное приблизительно 8 минутам)
Инъецируемое количество раствора стандарта и раствора образца: 10 мкл
Диапазон измерения площадей пиков: приблизительно 20 минут после начала инъекции.
Содержание дает следующая формула:
Содержание (%)=(QT×WS×FP)ч(QS×WT),
где
отвешенное количество стандарта во время приготовления раствора стандарта (г): WS;
отвешенное количество образца во время приготовления раствора образца (г): WT;
коэффициент очистки стандарта: FP;
значение площади пика стандарта, деленное на площадь пика внутреннего стандарта на хроматограмме раствора стандарта: QS; и
значение площади пика образца, деленное на площадь пика внутреннего стандарта на хроматограмме раствора образца: QT.
Полученные результаты измерений показаны в таблице 3.
Показано, что кристаллическая форма А по настоящему изобретению имеет значительно более высокую чистоту по сравнению с гидрохлоридом 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-H-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона (соединение сравнительного примера 1), который получали традиционным способом, и в то же время способ получения кристаллической формы А по настоящему изобретению имеет высокий эффект очистки.
Тестовый пример 2: Загрязняющие примеси
Отношение содержания загрязняющих примесей в кристаллической форме А, полученной в примере 1, и в соединении, полученном в сравнительном примере 1, измеряли описанным ниже способом анализа с использованием ВЭЖХ.
В настоящем документе термин «отношение содержания загрязняющих примесей» представляет собой отношение интегрированных площадей для всех пиков, отношения площадей пиков которых измерены равными или большими 0,01%, за исключением пика для соединения (I) и пика, который детектируют, когда инъецируют один растворитель, к площади пика для соединения (I) в описанных ниже условиях измерений.
Кроме того, термин «индивидуальная загрязняющая примесь» представляет собой отношение площади пика для пика, отношение площади пика которого измерено равным или большим 0,01%, за исключением пика для соединения (I) и пика, который детектируют, когда инъецируют один растворитель.
Аммоний-ацетатный буфер (0,01 моль/мл) получали, добавляя водный раствор ацетата аммония (0,01 моль/мл) к водному раствору уксусной кислоты (0,01 моль/мл) и устанавливая рН=4,5.
Воду и ацетонитрил смешивали в соотношении 3:2 по объему, получая раствор для растворения образцов.
Точно отмеряли 0,01 г измеряемого образца, переносили в 20-мл коричневую мерную колбу и туда же добавляли приблизительно 1 мл диметилсульфоксида для образования раствора. Затем общий объем доводили до 20 мл, получая раствор образца.
Точно отмеряли 1 мл раствора образца, переносили в 100-мл коричневую мерную колбу. Туда же добавляли раствор для растворения образцов, доводя общий объем до 100 мл, получая стандартный раствор.
Измерения проводили при условиях, описанных ниже.
Условия ВЭЖХ-измерений (1)
Детектор: УФ-абсорбциометр (длина волны: 230 нм)
Колонка: Waters Corporation, XTerra RP18 (4,6 мм × 150 мм)
Температура колонки: 40°С
Подвижная фаза: аммоний-ацетатный буфер (0,01 моль/мл) с ацетонитрилом (65:35)
Скорость потока: 1 мл/мин (В этих условиях соединение (I) показывало время удерживания, равное приблизительно 25 минутам)
Инъецируемое количество стандартного раствора и раствора образца: 10 мкл
Диапазон измерения площадей: 70 минут после начала инъекции
Условия ВЭЖХ-измерений (2)
Детектор: УФ-абсорбциометр (длина волны: 230 нм)
Колонка: Waters Corporation, XTerra RP18 (4,6 мм × 150 мм)
Температура колонки: 40°С
Подвижная фаза: аммоний-ацетатный буфер (0,01 моль/мл) с ацетонитрилом (56:44)
Скорость потока: 1 мл/мин (В этих условиях соединение (I) показывало время удерживания, равное приблизительно 8 минутам)
Инъецируемое количество стандартного раствора и раствора образца: 10 мкл
Диапазон измерения площадей: 70 минут после пика, который элюируется вслед за пиком, время удерживания которого относительно соединения (I) составляет 1,48
Отношения содержания загрязняющих примесей рассчитывали по следующей формуле:
Отношение содержания загрязняющей примеси (%)
= [Сумма индивидуальных загрязняющих примесей, больших 0,01%, измеренных в условиях ВЭЖХ-измерений (1)]+[Сумма индивидуальных загрязняющих примесей, больших 0,01%, измеренных в условиях ВЭЖХ-измерений (2)],
где
сумма индивидуальных загрязняющих примесей, больших 0,01%, измеренных в условиях ВЭЖХ-измерений (1) (%)=Ai1/AS1; и
сумма индивидуальных загрязняющих примесей, больших 0,01%, измеренных в условиях ВЭЖХ-измерений (2) (%)=Ai2/AS2,
где в вышеуказанной формуле
площадь пика для соединения (I) в стандартном растворе, измеренная в условиях ВЭЖХ-измерений (1): AS1;
площадь пика для индивидуальных загрязняющих примесей, больших 0,01%, измеренных в условиях ВЭЖХ-измерений (1): Ai1;
площадь пика для соединения (I) в стандартном растворе, измеренная в условиях ВЭЖХ-измерений (2): AS2; и
площадь пика для индивидуальных загрязняющих примесей, больших 0,01%, измеренных в условиях ВЭЖХ-измерений (2): Ai2.
Полученные результаты измерений показаны в таблице 4.
Показано, что способ получения кристаллической формы А по настоящему изобретению имеет высокий эффект удаления загрязняющих примесей.
Тестовый пример 3: Остаточный растворитель
Остаточные растворители в кристаллической форме А и кристаллической форме В, полученных в примерах, и в соединении, полученном в сравнительном примере 1, измеряли посредством газовой хроматографии согласно способу анализа, описанному ниже.
(1) Способ получения образцов кристаллической формы А и кристаллической формы В
Для получения разбавленного раствора смешивали диметилформамид и воду в соотношении по объему 7:3.
Точно отмеряли 1 мл трет-бутилового спирта, переносили в 100-мл мерную колбу и растворяли в разбавленном растворе, доводя общий объем до 100 мл. Точно отмеряли 10 мл полученного раствора, переносили в 500-мл мерную колбу и туда же добавляли разбавленный раствор, доводя общий объем до 500 мл, получая раствор внутреннего стандарта.
Точно отмеряли 2 мл тетрагидрофурана, 2 мл диизопропилового эфира, 2 мл метанола, 2 мл этилацетата, 2 мл уксусной кислоты и 2 мл 1,4-диоксана, переносили в 250-мл мерную колбу и туда же добавляли раствор внутреннего стандарта, доводя общий объем до 250 мл. Точно отмеряли 1 мл полученного раствора, переносили в 100-мл мерную колбу и туда же добавляли раствор внутреннего стандарта, доводя общий объем до 100 мл. Точно отмеряли 6 мл из 100 мл полученного раствора и переносили в 20-мл флакон-контейнер со свободным пространством над содержимым (флакон для статического парофазного анализа). Флакон-контейнер закрывали резиновой пробкой и плотно укупоривали, закручивая и закрывая алюминиевый колпачок, получая стандартный раствор.
Точно отмеряли 0,1 г образца для измерения, переносили в 20-мл флакон для статического парофазного анализа и туда же добавляли точно 6 мл раствора внутреннего стандарта. Флакон-контейнер закрывали резиновой пробкой и плотно укупоривали, закручивая и закрывая алюминиевый колпачок. Образец полностью растворяли при встряхивании в водяной бане при температуре в диапазоне от 60 до 70°С, получая раствор образца.
(2) Способ получения образца соединения в сравнительном примере 1
Точно отмеряли 1 мл трет-бутилового спирта, переносили в 100-мл мерную колбу и растворяли в хлорбензоле, доводя общий объем до 100 мл. Точно отмеряли 10 мл полученного раствора, переносили в 500-мл мерную колбу и туда же добавляли хлорбензол, доводя общий объем до 500 мл, получая раствор внутреннего стандарта.
Точно отмеряли 2 мл тетрагидрофурана, 2 мл диизопропилового эфира, 2 мл метанола, 2 мл этилацетата, 2 мл уксусной кислоты и 2 мл 1,4-диоксана, переносили в 250-мл мерную колбу и туда же добавляли раствор внутреннего стандарта, доводя общий объем до 250 мл. Точно отмеряли 1 мл полученного раствора, переносили в 100-мл мерную колбу и туда же добавляли раствор внутреннего стандарта, доводя общий объем до 100 мл. Точно отмеряли 6 мл из 100 мл полученного раствора и переносили в 20-мл флакон-контейнер для статического парофазного анализа. Флакон-контейнер закрывали резиновой пробкой и плотно укупоривали, закручивая и закрывая алюминиевый колпачок, получая стандартный раствор.
Точно отмеряли 0,1 г образца для измерения, переносили в 20-мл флакон-контейнер для статического парофазного анализа и туда же точно добавляли 6 мл раствора внутреннего стандарта и 100 мкл трибутиламина. Флакон-контейнер закрывали резиновой пробкой и плотно укупоривали, закручивая и закрывая алюминиевый колпачок. Образец полностью растворяли при встряхивании в водяной бане при температуре в диапазоне от 60 до 70°С, получая раствор образца.
(3) Условия теста
Остаточные уровни растворителей измеряли в условиях теста, описанных ниже.
Детектор: водородный пламенно-ионизационный детектор
Колонка: J&W Inc., DB-624 (0,53 мм × 30 м)
Температура колонки: 40°С (выдержать в течение 5 минут) → подъем температуры со скоростью 10°С/мин → 260°С (выдержать в течение 3 минут)
Температура камеры испарения образца: 250°С
Температура детектора: 300°С
Газ-носитель: гелий
Скорость потока в колонке: 5 мл/мин (скорость потока в колонке отрегулировать так, чтобы время удерживания тетрагидрофурана составляло приблизительно 7 минут)
Сплит (сброс части образца в инжекторе): 1:10
Способ инжекции образца: способ со сплитом (сбросом части образца в инжекторе)
Диапазон измерения площадей: 20 минут
(4) Рабочие условия для парофазного аппарата
Равновесная температура внутри флакона (температура печи): 85°С
Время уравновешивания внутри флакона: 15 минут
Температура линии вкола
Температура петли образца: 95°С
Температура линии переноса: 110°С
Газ-носитель: гелий
Время компрессии флакона: 0,20 минут
Давление флакона: приблизительно 10 кПа
Время заполнения петли образца: 0,15 минут
Время уравновешивания петли образца: 0,05 минут
Время инъекции: 1,0 минуты
Инъецируемое количество образца: 1 мл
(5) Способ расчета остаточного растворителя
Остаточный уровень каждого растворителя дает следующая формула:
Остаточный уровень каждого растворителя (ppm, ч.млн.)=(2×D×QT×6×10000000)÷(QS×25000×W),
где
весовое количество образца (г): W;
плотность каждого растворителя (г/мл): D;
отношение площадей для каждого растворителя в стандартном растворе к площади пика для вещества внутреннего стандарта: QS; и
отношение площадей для каждого растворителя в растворе образца к площади пика для вещества внутреннего стандарта: QT.
Полученные результаты измерений показаны в таблице 5.
Показано, что в кристаллической форме А и кристаллической форме В присутствуют крайне низкие количества только одного вида остаточного растворителя.
Тестовый пример 4: Цветовой тон
Приблизительно 1 г испытуемого вещества помещали на лист белой бумаги для определения его цветового тона. Результаты теста показаны в таблице 6.
Показано, что способ получения кристаллической формы А по настоящему изобретению имеет высокий обесцвечивающий эффект.
Тестовый пример 5: Тест на растворимость
Растворимость кристаллической формы А и кристаллической формы В соединения (I) в искусственной желудочной жидкости измеряли способом, описанным ниже (в расчете на свободное основание).
Для получения искусственной желудочной жидкости десятикратно сконцентрированную жидкость 1, применяемую в Японской Фармакопее для испытания на разложение (приобретенную у Kanto Chemical Co., Inc.), разбавляли в 10 раз очищенной водой. Точно отмеряли приблизительно 80 мг кристаллической формы, подлежащей измерению, переносили в 25-мл мерную колбу и растворяли в метаноле, доводя общий объем до 25 мл. 2 мл полученного раствора переносили в 50-мл мерную колбу и туда же добавляли искусственную желудочную жидкость, доводя общий объем до 50 мл, получая стандартный раствор.
Применяя машину для испытания вымывания NTR-6100A (Toyama Sangyo Co., Ltd.), приблизительно 100 мг испытуемой кристаллической формы добавляли к 500 мл искусственной желудочной жидкости при 37°С и перемешивали лопастной мешалкой (250 об/мин). Спустя 60 минут образцы извлекали из тестового раствора. Измеряли поглощение образцов стандартного раствора и тестового раствора при 289 нм и 360 нм с использованием спектрофотометра видимой области спектра.
Растворимость рассчитывали по следующей формуле:
Растворимость (мкг/мл)={(AS289-AS360)×WS×1000×502,58}÷{(AT289-AT360)×625×593,52},
где
весовое количество кристаллической формы, использованной для получения стандартного раствора (мг): WS;
поглощение стандартного раствора при 289 нм: AS289;
поглощение стандартного раствора при 360 нм: AS360;
поглощение тестового раствора при 289 нм: AT289;
поглощение тестового раствора при 360 нм: AT360;
молекулярная масса моногидрата дигидрохлорида соединения (I): 593,52; и
молекулярная масса свободного основания соединения (I): 502,58.
Полученные результаты теста показаны в таблице 7.
Кристаллическая форма А и кристаллическая форма В по настоящему изобретению показали достаточную растворимость в качестве фармацевтических ингредиентов.
[ПРИМЕРЫ ПРЕПАРАТОВ]
Пример препарата 1: Капсула
Для получения капсул смешивали 5 г кристаллической формы А, полученной в примере 1, 115 г лактозы, 58 г кукурузного крахмала и 2 г стеарата магния в смешивающей машине типа V и 180 мг этой смеси вносили в капсулу № 3.
Пример препарата 2: Таблетка
После смешивания 5 г кристаллической формы А, полученной в примере 1, с 90 г лактозы, 34 г кукурузного крахмала, 20 г кристаллической целлюлозы и 1 г стеарата магния в смешивающей машине типа V смесь прессовали в таблетирующей машине, получая таблетки массой по 150 мг каждая.
Пример препарата 3: Суспензия
Дисперсионную среду получали, пропитывая и растворяя метилцеллюлозу в очищенной воде. Кристаллическую форму А, полученную в примере 1, после взвешивания помещали в ступку и тщательно растирали, добавляя к ней небольшие порции вышеуказанной дисперсионной среды. Добавляли очищенную воду для получения 100 г суспензии.
Изобретение относится к кристаллическим формам гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, представленного формулой (I). Изобретение также относится к способам их получения и к фармацевтическим композициям на их основе, обладающим свойством активировать PPARγ. Указанные формы соединения (I) являются более стабильными и могут найти свое применение в медицине для получения лекарственных средств для лечения диабета, рака, сопровождаемого диабетом. 14 н. и 2 з.п. ф-лы, 7 табл., 5 ил.
1. Кристаллическая форма гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1 -метил-1 -Н-бензимидазол-2-ил]метокси} бензил)-1,3-тиазолидин-2,4-диона, представленного следующей формулой (I)
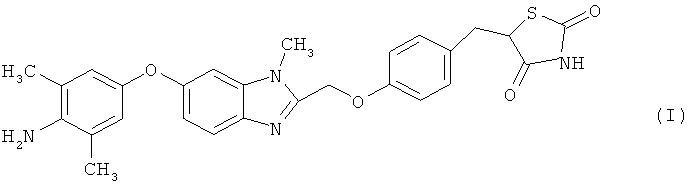 ,
,
в котором кристаллическая форма показывает главные пики при межплоскостных расстояниях, равных 7,06, 5,79, 5,43, 4,44, 4,18, 3,97, 3,91, 3,68, 3,61, 3,48, 3,24 и 2,97 ангстрем, в порошковой рентгенограмме, полученной с Кα-линией излучения Cu (длина волны λ=1,54 ангстрем).
2. Кристаллическая форма гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, представленного общей формулой (I)
,
в котором кристаллическая форма показывает главные пики при межплоскостных расстояниях, равных 10,42, 5,85, 5,52, 3,84, 3,46 и 2,95 ангстрем, в порошковой рентгенограмме, полученной с Кα-линией излучения Cu (длина волны λ=1,54 ангстрем).
3. Кристаллическая форма по п.1, в которой содержание примесей, измеренное ВЭЖХ, составляет 2,00% или менее.
4. Кристаллическая форма по п.1, в которой содержание примесей, измеренное ВЭЖХ, составляет 1,50% или менее.
5. Фармацевтическая композиция, обладающая свойством активировать PPARγ, содержащая в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), по любому из пп.1-4.
6. PPARγ-активатор, содержащий в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), по любому из пп.1-4.
7. Противораковая фармацевтическая композиция, содержащая в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), по любому из пп.1-4.
8. Фармацевтическая композиция для предупреждения или лечения диабета, содержащая в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), по любому из пп.1-4.
9. Фармацевтическая композиции для предупреждения или лечения рака, сопровождаемого диабетом, содержащая в качестве активного компонента кристаллическую форму гидрата дигидрохлорида соединения тиазолидиндиона, представленного общей формулой (I), по любому из пп.1-4.
10. Способ получения кристаллической формы по п.1, отличающийся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион переводят в водный раствор и затем к нему по каплям добавляют хлористоводородную кислоту.
11. Способ получения кристаллической формы по п.1, отличающийся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион переводят в водный раствор и затем к нему по каплям добавляют хлористоводородную кислоту с получением кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона с последующим растворением или суспендированием его в воде, и добавлением к нему по каплям хлористоводородной кислоты.
12. Способ получения кристаллической формы по п.1, отличающийся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион переводят в водный раствор и затем к нему по каплям добавляют хлористоводородную кислоту с получением кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, с последующим растворением или суспендированием его в воде, и добавлением к нему основания с получением раствора или суспензии, и затем к нему по каплям добавляют хлористоводородную кислоту.
13. Способ получения кристаллической формы по п.1, отличающийся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион переводят в водный раствор и затем добавляют затравку кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона с последующим добавлением по каплям хлористоводородной кислоты.
14. Способ получения кристаллической формы по п.1, отличающийся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион переводят в водный раствор и затем к нему по каплям добавляют хлористоводородную кислоту с получением кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона с последующим растворением или суспендированием его в воде, и затем добавляют затравку кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона с последующим добавлением к нему по каплям хлористоводородной кислоты.
15. Способ получения кристаллической формы по п.1, отличающийся тем, что 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-дион переводят в водный раствор и затем к нему по каплям добавляют хлористоводородную кислоту с получением кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, который далее растворяют или суспендируют в воде, с последующим добавлением к нему основания с получением раствора или дисперсии, и затем добавляют затравку кристаллической формы гидрата дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона, и последующим добавлением к нему по каплям хлористоводородной кислоты.
16. Способ получения кристаллической формы по п.2, отличающийся тем, что гидрат дигидрохлорида 5-(4-{[6-(4-амино-3,5-диметилфенокси)-1-метил-1-Н-бензимидазол-2-ил]метокси}бензил)-1,3-тиазолидин-2,4-диона кристаллизуют из метанола.
| ЗАМЕЩЕННОЕ КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ФАРМАКОЛОГИЧЕСКОГО ВОЗДЕЙСТВИЯ, СПОСОБ ИНГИБИРОВАНИЯ 5-ЛИПОКСИГЕНАЗЫ, ИНГИБИРОВАНИЯ ПРОДУКЦИИ ЛИПИДНЫХ ПЕРОКСИДОВ ИЛИ СНИЖЕНИЯ УРОВНЯ САХАРА В КРОВИ | 1998 |
|
RU2196141C2 |
| WO 03053440 A1, 03.07.2003 | |||
| WO 2004083167 A1, 30.09.2004 | |||
| WO 2004000356 A1, 31.12.2003 | |||
| 5-[4-(6-МЕТОКСИ-1-МЕТИЛ-1Н-БЕНЗИМИДАЗОЛ-2-ИЛМЕТОКСИ)БЕНЗИЛ]-ТИАЗОЛИДИН-2,4 -ДИОН ГИДРОХЛОРИД, ОБЛАДАЮЩИЙ ГИПОГЛИКЕМИЧЕСКОЙ АКТИВНОСТЬЮ | 2000 |
|
RU2214410C2 |

