ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к противораковой фармацевтической композиции, которая в качестве активного ингредиента содержит тиазолидиндионовое соединение, обладающее активирующим действием в отношении рецептора активатора пролиферации пероксисом (PPAR) γ, и к противораковой фармацевтической композиции для профилактики или лечения карциномы, саркомы или рака крови, которая в качестве активных ингредиентов содержит соединение, обладающее активирующим действием в отношении PPARγ, и ингибитор рецептора эпидермального фактора роста (EGFR), ингибитор фактора роста эндотелия сосудов (VEGFR) или ингибитор Raf-киназы.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Широко известно, что активаторы PPARγ применимы в качестве лекарственных средств для терапии сахарного диабета 2 типа, представленных такими примерами, как розиглитазон и пиоглитазон. Считается, что PPARγ выполняет различные физиологические функции, такие как индукция дифференцировки для превращения в адипоциты и корректировка энергетического обмена биогенных веществ (например, см. непатентные документы 1 и 2). С другой стороны, сообщалось, что активаторы PPARγ индуцируют дифференцировку, ингибируют клеточный цикл или индуцируют апоптоз у различных типов раковых клеток, и обусловливают ингибирование роста раковых клеток (например, см. непатентные документы 3, 4 и 5). В дополнение к этим открытиям было высказано предположение, что поскольку при раке щитовидной железы часто наблюдается хромосомная транслокация PAX8-PPARγ, и работа PPARγ инактивирована, и поскольку при раке толстой кишки наблюдается, хотя и с невысокой частотой, обусловливающая дисфункцию точечная мутация, то PPARγ действует как ингибитор онкогенной трансформации (например, см. патентные документы 6 и 7). Исходя из этих открытий, было высказано предположение о возможности использования активаторов PPARγ для лечения рака, и у больных раком были проведены немногочисленные клинические испытания с использованием розиглитазона. Однако значимого эффекта не наблюдалось (например, см. патентный документ 8). На сегодняшний день причина такого результата не установлена; тем не менее, весьма вероятно, что противораковое действие розиглитазона было недостаточно сильным. Соответственно, считается, что обнаружение активатора PPARγ, который обладает более сильным эффектом, внесет значительный вклад в лечение рака в будущем.
С другой стороны, в современных методах лечения рака предпринят подход, при котором для усиления эффективности лекарственных средств и для снижения побочных эффектов по сравнению со способом, при котором каждое из лекарственных средств вводят по отдельности, сочетанно используют несколько противораковых лекарственных средств. В качестве типов противораковых лекарственных средств, использованных при комбинированном лечении, могут быть упомянуты разрушающие раковые клетки химиотерапевтические лекарственные средства и различные молекулярно направленные лекарственные средства, которые недавно были представлены на рынке. В частности, молекулярно направленные лекарственные средства, как правило, обладают слабыми побочными эффектами по сравнению с используемыми ранее лекарственными средствами, и что касается комбинированного введения, то для предупреждения усиления побочных эффектов часто нет необходимости в снижении количества используемого ранее лекарственных средств. Поэтому, поскольку при комбинированной терапии может быть достигнута максимальная эффективность разрушающих раковые клетки химиотерапевтических лекарственных средств, и поскольку их эффект может быть усилен за счет эффективности молекулярно направленных лекарственных средств, то в настоящее время ведется активная разработка различных лекарственных средств, которые обладают молекулярно направленным действием. Примерами молекулярно направленных лекарственных средств, которые привлекают внимание в настоящее время, являются бевацизумаб (название продукта Авастин), который представляет собой лекарственное средство на основе антитела, обладающее антиангиогенетической активностью, и гефитиниб (название продукта Иресса) и эрлотиниб (название продукта Тарцева), которые представляют собой ингибиторы рецептора эпидермального фактора роста (EGFR). Кроме того, считается, что сорафениб, который обладает антиангиогенетической (ингибирующей рецептор фактора роста эндотелия сосудов (VEGFR)) активностью в сочетании с ингибирующей активностью в отношении Raf-киназы и находится в настоящее время на стадии клинических испытаний, также эффективен в клинических испытаниях и привлекает внимание. Описанное выше указание на то, что противораковые эффекты могут быть усилены путем комбинированного введения с указанными молекулярно направленными лекарственными средствами, делает возможным проведение различных вариантов лечения рака у пациента, и тем самым вносят значительный вклад в улучшение результата лечения. Так, повышение противораковой эффективности путем комбинированного введения, как правило, указывает на то, что полученный путем комбинированного введения эффект выше эффекта, полученного путем отдельного введения каждого лекарственного средства (например, см. непатентный документ 9), и считается, что клиническая значимость является высокой даже если синергический усиливающий эффект и не был достигнут.
В патенте Японии №3488099 (для других, см. патентные документы 1 и 2) раскрыто тиазолидиндионовое соединение, обладающее новым химическим строением. Представленное общей формулой (I) соединение, которое содержится в качестве активного ингредиента противораковой фармацевтической композиции по настоящему изобретению, представляет собой соединение, которое охватывается объемом соединений, относящихся к тиазолидиндионовому соединению, раскрытому в изобретении. В патенте Японии № 3488099 показано, что раскрытое в опубликованном патенте тиазолидиндионовое соединение обладает способностью активировать PPARγ и может быть использовано в качестве противоракового лекарственного средства. Тем не менее, в патенте не раскрыты какие-либо конкретные результаты испытаний, которые показывают, что тиазолидиндионовое соединение действительно обладает противораковым действием.
Кроме того, сообщалось о фармацевтических композициях, которые содержат указанное тиазолидиндионовое соединение и другое лекарственное средство.
Например, сообщалось о фармацевтической композиции, содержащей тиазолидиндионовое соединение и ингибитор MAP-киназы (см. патентные документы 3 и 4), и было раскрыто, что указанная фармацевтическая композиция применима в качестве профилактического средства, лекарственного средства или в качестве ингибитора пролиферации клеток при раке, таком как рак желудка, рак легких, рак молочной железы, рак толстой кишки, рак предстательной железы, рак поджелудочной железы, рак печени, лейкоз, рак головы и шеи или липосаркома.
Кроме того, сообщалось о фармацевтической композиции для профилактики или лечения рака, которая содержит некоторые из соединений, подпадающих под объем соединений и относящихся к упомянутому выше тиазолидиндионовому соединению, и активатор RXR (рецептора ретиноидов X) (см. патентные документы 5 и 6), и было раскрыто, что указанная фармацевтическая композиция применима в качестве лекарственного средства или в качестве профилактического средства, в особенности при раке легких, раке желудка и раке толстой кишки.
Сообщалось о фармацевтической композиции, содержащей упомянутое выше тиазолидиндионовое соединение и антиметаболит фторурацилового ряда или комплекс платины (см. патентные документы 7 и 8), и было раскрыто, что указанная фармацевтическая композиция особенно применима в качестве профилактического средства, лекарственного средства или в качестве ингибитора пролиферации клеток при раке, таком как рак желудка, рак легких, рак молочной железы, рак толстой кишки, рак предстательной железы, рак поджелудочной железы, рак печени, лейкоз, рак головы и шеи или липосаркома.
Сообщалось о фармацевтической композиции, содержащей упомянутое выше тиазолидиндионовое соединение и диуретик (см. патентные документы 9 и 10), и было раскрыто, что указанная фармацевтическая композиция может предупреждать или лечить побочные эффекты, обусловленные введением активатора PPARγ, такие как гипертрофия сердца, отек, задержка жидкости и задержка плеврального выпота, и особенно применима в качестве профилактического средства, лекарственного средства или в качестве ингибитора пролиферации клеток при раке, таком как рак желудка, рак легких, рак молочной железы, рак толстой кишки, рак предстательной железы, рак поджелудочной железы, рак печени, лейкоз, рак головы и шеи или липосаркома.
Сообщалось о фармацевтической композиции, содержащей упомянутое выше тиазолидиндионовое соединение и новое сульфамидное соединение, обладающее ингибирующей активностью в отношении MEK (см. патентные документы 11 и 12), и было раскрыто, что указанная фармацевтическая композиция особенно применима в качестве профилактического средства, лекарственного средства или в качестве ингибитора пролиферации клеток при раке, таком как рак желудка, рак легких, рак молочной железы, рак толстой кишки, рак предстательной железы, рак поджелудочной железы, рак печени, лейкоз, рак головы и шеи или липосаркома.
[Патентный документ 1] Патент США №6432993.
[Патентный документ 2] Европейский патент №1022272.
[Патентный документ 3] Заявка на выдачу патента Японии (Kokai) №2003-192592.
[Патентный документ 4] Брошюра международной публикации № WO 03/032988.
[Патентный документ 5] Заявка на выдачу патента Японии (Kokai) №2003-238406.
[Патентный документ 6] Брошюра международной публикации № WO 03/053440.
[Патентный документ 7] Заявка на выдачу патента Японии (Kokai) №2004-83558.
[Патентный документ 8] Брошюра международной публикации № WO 03/082865.
[Патентный документ 9] Заявка на выдачу патента Японии (Kokai) №2004-83574.
[Патентный документ 10] Брошюра международной публикации № WO 2004/000356.
[Патентный документ 11] Заявка на выдачу патента Японии (Kokai) №2005-162727.
[Патентный документ 12] Брошюра международной публикации № WO 2004/083167.
[Непатентный документ 1] Spiegelman B.M. PPAR-γ: Adipogenic regulator and thiazolidinedione receptor. Diabetes, 1998; 47: 507-14.
[Непатентный документ 2] Lehmann J.M., Moore L.B. et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 1995; 270: 12953-6.
[Непатентный документ 3] Mueller E., Sarraf P. et al. Terminal differentiation of the human breast cancer through PPAR gamma. Mol. Cell. 1998; 1: 465-70.
[Непатентный документ 4] Yoshizume T., Ohta T. et al. Thiazolidinedione, a peroxisome proliferator-activated receptor gamma ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. Int. J. Oncol. 2004; 25: 631-9.
[Непатентный документ 5] Ray D.M., Bernstein S.H. et al. Human multiple myeloma cells express peroxisome proliferator-activated receptor γ and undergo apoptosis upon exposure to PPARγ ligands. Clin. Immunology, 2004; 113: 203-13.
[Непатентный документ 6] Dwight T., Thoppe S.R., et al. Involvement of the PAX8/peroxisome proliferator-activated receptor gamma rearrangement in follicular thyroid tumors. J. Clin. Endocrinol. Metab. 2003; 88: 4440-5.
[Непатентный документ 7] Sarraf P., Mueller E. et al. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol. Cell. 1999; 3: 799-804.
[Непатентный документ 8] Debrock G., Vanhentenrijk V. et al. A phase II trial with rosiglitazone in liposarcoma patients. Br. J. Cancer. 2003; 89: 1409-12.
[Непатентный документ 9] Tatsuo Saito ed., Development of Drug Therapy for Cancer and Evaluation of Efficiency, Realize inc., pp. 128-138 (1985).
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Проблемы, требующие решения настоящим изобретением
Соответственно, авторы настоящего изобретения отобрали соединение, представленное общей формулой (I), представляющее собой активный ингредиент противораковой фармацевтической композиции по настоящему изобретению, из соединений, подпадающих под объем упомянутого выше тиазолидионового соединения, и исследовали противораковые эффекты соединения, представленного общей формулой (I) по настоящему изобретению, при использовании только самого соединения.
В результате было обнаружено, что соединение, представленное общей формулой (I), или его фармакологически приемлемая соль по настоящему изобретению обладает превосходным противораковым эффектом в отношении конкретного типа рака.
В результате проведения обширных исследований по обнаружению сочетания лекарственных средств, обладающих лучшим противораковым действием, авторы настоящего изобретения обнаружили, что путем введения соединения, способного активировать PPARγ (в особенности соединения, представленного общей формулой (I) по настоящему изобретению), или его фармацевтически приемлемой соли, в сочетании с ингибитором рецептора эпидермального фактора роста (EGFR), ингибитором рецептора фактора роста эндотелия сосудов (VEGFR) или ингибитором Raf-киназы, противораковые эффекты могут быть усилены по сравнению со случаем, когда они вводятся по отдельности, оформив тем самым настоящее изобретение.
Средства решения проблем
Настоящее изобретение представляет собой
(1) противораковую фармацевтическую композицию для профилактики или лечения рака желудка, рака толстой кишки, рака легких, рака молочной железы, рака поджелудочной железы, рака почки, рака предстательной железы, медуллобластомы, рабдомиосаркомы, саркомы Юинга, липосаркомы, множественной миеломы или лейкоза, содержащую в качестве активного ингредиента соединение, представленное следующей общей формулой (I):

[в которой
R представляет собой фенильную группу, замещенную 1-5 группами, выбранными из группы заместителей α, и
X представляет собой атом кислорода или атом серы;
<группа заместителей α>: атом галогена, гидроксигруппа, C1-C6алкильная группа, галогенC1-C6алкильная группа, C1-C6алкоксигруппа, C1-C6алкилтиогруппа, аминогруппа, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, C3-C10циклоалкильная, C6-C10арильная, C7-C16аралкильная, C6-C10арилокси, C7-C16аралкилокси или C6-C10арилтио группа, которая может быть замещена 1-3 группами, выбранными из группы заместителей β, C1-C7алифатическая ацилоксигруппа, 4-7-членная насыщенная гетероциклическая группа, содержащая атом(ы) азота, 5-6-членная ароматическая гетероциклическая группа, содержащая атом(ы) азота, нитрогруппа и цианогруппа;
<группа заместителей β>: атом галогена, гидроксигруппа, C1-C6алкильная группа, галогенC1-C6алкильная группа, C1-C6алкоксигруппа, аминогруппа, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, C6-C10арильная группа и нитрогруппа;
<группа заместителей γ>: C1-C10алкильная группа, C6-C10арильная группа, C7-C16аралкильная группа, C1-C7алифатическая ацильная группа, C7-C11ароматическая ацильная группа, C8-C12ароматическоалифатическая ацильная группа, C4-C11циклоалкилкарбонильная группа и 5-6-членная ароматическая гетероциклическая карбонильная группа, содержащая атом(ы) азота], или его фармацевтически приемлемую соль,
(2) фармацевтическую композицию в соответствии с упомянутым выше п.(1), в которой
R представляет собой фенильную группу, замещенную 1-5 группами, выбранными из группы заместителей α, и
группа заместителей α представляет собой группу, состоящую из атома галогена, C1-C6алкильной группы, галогенC1-C6алкильной группы, аминогруппы, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, 4-7-членной насыщенной гетероциклической группы, содержащей атом(ы) азота, и 5-6-членной ароматической гетероциклической группы, содержащей атом(ы) азота,
(3) фармацевтическую композицию в соответствии с упомянутым выше п.(1), в которой R представляет собой фенильную группу, замещенную одной аминогруппой, которая может быть замещена 1 или 2 заместителями (заместители могут быть одинаковыми или разными, и каждый представляет собой группу, выбранную из группы, состоящей из C1-С10алкильной группы, С6-С10арильной группы и C7-С16аралкильной группы), и может быть дополнительно замещена 1-3 заместителями (каждый заместитель представляет собой группу, выбранную из группы, состоящей из атома галогена, C1-С6алкильной группы и галогенС1-С6алкильной группы),
(4) фармацевтическую композицию в соответствии с упомянутым выше п.(1), в которой R представляет собой фенильную группу, замещенную аминогруппой или моно- или ди-С1-С10алкиламиногруппой, и которая может быть дополнительно замещена 1 или 2 C1-С6алкильными группами,
(5) фармацевтическую композицию в соответствии с любым из упомянутых выше п.(1)-(4), в которой Х представляет собой атом кислорода,
(6) фармацевтическую композицию в соответствии с упомянутым выше п.(1), в которой соединение, представленное общей формулой (I), представляет собой соединение, выбранное из следующих соединений:
5-(4-(6-(3-изопропиламинофенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(3-(изобутилметиламино)фенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-(изобутилметиламино)фенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(3-(этилизопропиламино)фенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-втор-бутиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-изобутиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион и
5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
(7) фармацевтическую композицию в соответствии с упомянутым выше п.(1), в которой соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль представляет собой дигидрохлорид 5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона,
(8) фармацевтическую композицию в соответствии с упомянутым выше п.(1), в которой соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль представляет собой дигидрохлорид 5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона,
(9) противораковую фармацевтическую композицию для профилактики или лечения карциномы, саркомы или рака крови, содержащую в качестве активных ингредиентов
по меньшей мере, одно противораковое лекарственное средство, выбранное из группы, состоящей из ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и ингибитора Raf-киназы; и
по меньшей мере, одно соединение, выбранное из группы, состоящей из химических соединений, представленных следующей общей формулой (I):
[в которой
R представляет собой фенильную группу, замещенную 1-5 группами, выбранными из группы заместителей α, и
X представляет собой атом кислорода или атом серы;
<группа заместителей α>: атом галогена, гидроксигруппа, C1-C6алкильная группа, галогенC1-C6алкильная группа, C1-C6алкоксигруппа, C1-C6алкилтиогруппа, аминогруппа, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, C3-C10циклоалкильная, C6-C10арильная, C7-C16аралкильная, C6-C10арилокси, C7-C16аралкилокси или C6-C10арилтиогруппа, которая может быть замещена 1-3 группами, выбранными из группы заместителей β, C1-C7алифатическая ацилоксигруппа, 4-7-членная насыщенная гетероциклическая группа, содержащая атом(ы) азота, 5-6-членная ароматическая гетероциклическая группа, содержащая атом(ы) азота, нитрогруппа и цианогруппа;
<группа заместителей β>: атом галогена, гидроксигруппа, C1-C6алкильная группа, галогенC1-C6алкильная группа, C1-C6алкоксигруппа, аминогруппа, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, C6-C10арильная группа и нитрогруппа;
<группа заместителей γ>: C1-C10алкильная группа, C6-C10арильная группа, C7-C16аралкильная группа, C1-C7алифатическая ацильная группа, C7-C11ароматическая ацильная группа, C8-C12ароматическоалифатическая ацильная группа, C4-C11циклоалкилкарбонильная группа и 5-6-членная ароматическая гетероциклическая карбонильная группа, содержащая атом(ы) азота],
или его фармакологически приемлемую соль,
где активные ингредиенты предназначены для одновременного введения или введения по отдельности в разное время,
(10) фармацевтическую композицию в соответствии с упомянутым выше п.(9), в которой противораковое лекарство выбирают, по меньшей мере, из группы, состоящей из ингибитора рецептора эпидермального фактора роста (EGFR) (лекарственное средство представляет собой цетуксимаб, панитумумаб, гефитиниб, эрлотиниб или лапатиниб), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) (лекарственное средство представляет собой бевацизумаб, сорафениб, SU11248 или ваталаниб) и ингибитора Raf-киназы (лекарственное средство представляет собой сорафениб),
(11) фармацевтическую композицию в соответствии с упомянутым выше п.(9), в которой противораковое лекарственное средство выбирают, по меньшей мере, из группы, состоящей из гефитиниба и сорафениба,
(12) фармацевтическую композицию в соответствии с любым из упомянутых выше пп.(9)-(11), где карцинома представляет собой рак желудка, рак толстой кишки, рак легких, рак молочной железы, рак поджелудочной железы, рак почки или рак предстательной железы,
(13) фармацевтическую композицию в соответствии с любым из упомянутых выше пп.(9)-(12), где саркома представляет собой медуллобластому, рабдомиосаркому, саркому Юинга или липосаркому,
(14) фармацевтическую композицию в соответствии с любым из упомянутых выше пп.(9)-(13), где рак крови представляет собой множественную миелому или лейкоз,
(15) фармацевтическую композицию в соответствии с любым из упомянутых выше пп.(9)-(14), в которой
R представляет собой фенильную группу, замещенную 1-5 группами, выбранными из группы заместителей α, и
группа заместителей α представляет собой группу, состоящую из атома галогена, C1-C6алкильной группы, галогенC1-C6алкильной группы, аминогруппы, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, 4-7-членной насыщенной гетероциклической группы, содержащей атом(ы) азота, и 5-6-членной ароматической гетероциклической группы, содержащей атом(ы) азота,
(16) фармацевтическую композицию в соответствии с любым из упомянутых выше пп.(9)-(14), в которой R представляет собой фенильную группу, замещенную одной аминогруппой, которая может быть замещена 1 или 2 заместителями (заместители могут быть одинаковыми или разными, и каждый представляет собой группу, выбранную из группы, состоящей из C1-C10алкильной группы, C6-C10арильной группы и C7-C16аралкильной группы), и может быть дополнительно замещена 1-3 заместителями (каждый заместитель представляет собой группу, выбранную из группы, состоящей из атома галогена, C1-C6алкильной группы и галогенC1-C6алкильной группы),
(17) фармацевтическую композицию в соответствии с любым из упомянутых выше пп.(9)-(14), в которой R представляет собой фенильную группу, замещенную аминогруппой или моно- или ди-C1-C10алкиламиногруппой, и которая может быть дополнительно замещена 1 или 2 C1-C6алкильными группами,
(18) фармацевтическую композицию в соответствии с любым из упомянутых выше пп.(9)-(17), в которой X представляет собой атом кислорода,
(19) фармацевтическую композицию в соответствии с упомянутым выше п.(9), в которой соединение, представленное общей формулой (I), представляет собой соединение, выбранное из следующих соединений:
5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(3-(изобутилметиламино)фенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-(изобутилметиламино)фенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(3-(этилизопропиламино)фенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-втор-бутиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-изобутиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион и
5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
(20) фармацевтическую композицию в соответствии с упомянутым выше п.(9), в которой, по меньшей мере, одно соединение, выбранное из группы, состоящей из соединений, представленных общей формулой (I), или его фармацевтически приемлемая соль представляет собой дигидрохлорид 5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона, и
(21) фармацевтическую композицию в соответствии с упомянутым выше п.(9), в которой соединение, выбранное из группы, состоящей из соединений, представленных общей формулой (I), или его фармацевтически приемлемая соль представляет собой дигидрохлорид 5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона.
Кроме того, настоящее изобретение относится к способу профилактики или лечения рака желудка, рака толстой кишки, рака легких, рака молочной железы, рака поджелудочной железы, рака почки, рака предстательной железы, медуллобластомы, рабдомиосаркомы, саркомы Юинга, липосаркомы, множественной миеломы или лейкоза, который включает введение фармацевтической композиции, описанной в любом из упомянутых выше пп.(1)-(8), теплокровному животному (предпочтительно, человеку).
Кроме того, настоящее изобретение относится к способу профилактики или лечения карциномы (в особенности, рака желудка, рака толстой кишки, рака легких, рака молочной железы, рака поджелудочной железы, рака почки или рака предстательной железы), саркомы (в особенности, медуллобластомы, рабдомиосаркомы, саркомы Юинга или липосаркомы) или рака крови (в особенности, множественной миеломы или лейкоза), который включает одновременное введение активных ингредиентов фармацевтической композиции или введение каждого из ингредиентов в различное время, причем активные ингредиенты описаны в упомянутых выше пп.(9)-(21).
В настоящем изобретении в определении групп заместителей α и β «атом галогена» представляет собой атом фтора, атом хлора, атом брома или атом йода, предпочтительно атом фтора или атом хлора, и более предпочтительно атом фтора.
В определении групп заместителей α и β «C1-C6алкильная группа» представляет собой неразветвленную или разветвленную алкильную группу, содержащую 1-6 атомов углерода, и представляет собой, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную, изопентильную, 2-метилбутильную, неопентильную, 1-этилпропильную, гексильную, 4-метилпентильную, 3-метилпентильную, 2-метилпентильную, 1-метилпентильную, 3,3-диметилбутильную, 2,2-диметилбутильную, 1,1-диметилбутильную, 1,2-диметилбутильную, 1,3-диметилбутильную, 2,3-диметилбутильную или 2-этилбутильную группу. В отношении группы заместителей α она предпочтительно представляет собой метильную или трет-бутильную группу, а в отношении группы заместителей β она предпочтительно представляет собой C1-C4алкильную группу, и более предпочтительно метильную или этильную группу.
В определении групп заместителей α и β «галогенC1-C6алкильная группа» представляет собой группу, в которой с упомянутой выше C1-C6алкильной группой связаны от 1 до 3 из упомянутых выше атомов галогена, и представляет собой, например, трифторметильную, трихлорметильную, трибромметильную, дифторметильную, дихлорметильную, дибромметильную, фторметильную, 2,2,2-трихлорэтильную, 2,2,2-трифторэтильную, 2-бромэтильную, 2-хлорэтильную, 2-фторэтильную, 2-йодэтильную, 3-хлорпропильную, 4-фторбутильную, 6-йодгексильную или 2,2-дибромэтильную группу, предпочтительно галогенC1-C2алкильную группу, и более предпочтительно трифторметильную группу.
В определении групп заместителей α и β «C1-C6алкоксигруппа» представляет собой группу, в которой упомянутая выше C1-C6алкильная группа связана с атомом кислорода, и представляет собой, например, метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси, изопентокси, 2-метилбутокси, неопентокси, 1-этилпропокси, гексилокси, 4-метилпентокси, 3-метилпентокси, 2-метилпентокси, 3,3-диметилбутокси, 2,2-диметилбутокси, 1,1-диметилбутокси, 1,2-диметилбутокси, 1,3-диметилбутокси, 2,3-диметилбутокси или 2-этилбутокси группу, предпочтительно C1-C4алкоксигруппу, более предпочтительно C1-C2алкоксигруппу, и особенно предпочтительно метоксигруппу.
В определении группы заместителей α «C1-C6алкилтиогруппа» представляет собой группу, в которой упомянутая выше C1-C6алкильная группа связана с атомом серы, и представляет собой, например, метилтио, этилтио, пропилтио, изопропилтио, бутилтио, изобутилтио, втор-бутилтио, трет-бутилтио, пентилтио, изопентилтио, 2-метилбутилтио, неопентилтио, 1-этилпропилтио, гексилтио, 4-метилпентилтио, 3-метилпентилтио, 2-метилпентилтио, 1-метилпентилтио, 3,3-диметилбутилтио, 2,2-диметилбутилтио, 1,1-диметилбутилтио, 1,2-диметилбутилтио, 1,3-диметилбутилтио, 2,3-диметилбутилтио или 2-этилбутилтио группу, предпочтительно C1-C4алкилтиогруппу, более предпочтительно C1-C2алкилтиогруппу, и особенно предпочтительно метилтиогруппу.
В определении групп заместителей α и β «аминогруппа, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ» представляет собой аминогруппу, которая может быть замещена одной или двумя группами, которые могут быть одинаковыми или разными, причем группу выбирают из группы заместителей γ, состоящей из C1-C10алкильной группы, C6-C10арильной группы, C7-C16аралкильной группы, C1-C7алифатической ацильной группы, C7-C11ароматической ацильной группы, C8-C12ароматическоалифатической ацильной группы, C4-C11циклоалкилкарбонильной группы и 5- или 6-членной ароматической гетероциклической карбонильной группы, содержащей атом(ы) азота.
В представленном выше определении группы заместителей γ «C1-C10алкильная группа» представляет собой неразветвленную или разветвленную алкильную группу, содержащую 1-10 атомов углерода, и представляет собой, например, упомянутую выше C1-С6алкильную, гептильную, 1-метилгексильную, 2-метилгексильную, 3-метилгексильную, 4-метилгексильную, 5-метилгексильную, 1-пропилбутильную, 4,4-диметилпентильную, октильную, 1-метилгептильную, 2-метилгептильную, 3-метилгептильную, 4-метилгептильную, 5-метилгептильную, 6-метилгептильную, 1-пропилпентильную, 2-этилгексильную, 5,5-диметилгексильную, нонильную, 3-метилоктильную, 4-метилоктильную, 5-метилоктильную, 6-метилоктильную, 1-пропилгексильную, 2-этилгептильную, 6,6-диметилгептильную, децильную, 1-метилнонильную, 3-метилнонильную, 8-метилнонильную, 3-этилоктильную, 3,7-диметилоктильную или 7,7-диметилоктильную группу, и предпочтительно представляет собой неразветвленную или разветвленную алкильную группу, содержащую 1-4 атома углерода.
В представленном выше определении группы заместителей γ «C6-С10арильная группа» представляет собой ароматическую углеводородную группу, содержащую 6-10 атомов углерода, и группа может быть замещена нитрогруппой, упомянутыми выше атомами галогена, гидроксигруппой, упомянутой выше C1-С6алкильной группой, C1-С6алкилкарбонилоксигруппой или C1-С6алкоксигруппой. Такая группа представляет собой, например, фенильную, нафтильную, пара-нитрофенильную, пара-хлорфенильную, пара-фторфенильную, пара-гидроксифенильную, пара-ацетоксифенильную, пара-метилфенильную, пара-этилфенильную, пара-пропилфенильную, пара-метоксифенильную, пара-этоксифенильную или пара-пропоксифенильную группу, и
предпочтительно представляет собой фенильную, пара-нитрофенильную или пара-пропоксифенильную группу.
В представленном выше определении группы заместителей γ «C7-C16аралкильная группа» представляет собой группу, в которой упомянутая выше C6-C10арильная группа связана с упомянутой выше C1-C6алкильной группой, и представляет собой, например, бензильную, нафтилметильную, инденилметильную, дифенилметильную, 1-фенетильную, 2-фенетильную, 1-нафтилэтильную, 2-нафтилэтильную, 1-фенилпропильную, 2-фенилпропильную, 3-фенилпропильную, 1-нафтилпропильную, 2-нафтилпропильную, 3-нафтилпропильную, 1-фенилбутильную, 2-фенилбутильную, 3-фенилбутильную, 4-фенилбутильную, 1-нафтилбутильную, 2-нафтилбутильную, 3-нафтилбутильную, 4-нафтилбутильную, 5-фенилпентильную, 5-нафтилпентильную, 6-фенилгексильную или 6-нафтилгексильную группу, предпочтительно аралкильную группу, в которой фенильная группа связана с C1-C4алкильной группой, и более предпочтительно бензильную группу.
В представленном выше определении группы заместителей γ «C1-C7алифатическая ацильная группа» представляет собой группу, в которой атом водорода или насыщенная или ненасыщенная неразветвленная C1-C6углеводородная группа связана с карбонильной группой, и представляет собой, например, формильную, ацетильную, пропионильную, бутирильную, изобутирильную, валерильную, изовалерильную, пивалоильную, гексаноильную, акрилоильную, метакрилоильную или кротоноильную группу, предпочтительно ацетильную, пропионильную или пивалоильную группу, и более предпочтительно ацетильную группу.
В представленном выше определении группы заместителей γ «C7-C11ароматическая ацильная группа» представляет собой группу, в которой C6-C10арильная группа связана с карбонильной группой, и представляет собой, например, бензоильную, 1-инданкарбонильную, 2-инданкарбонильную или 1- или 2-нафтоильную группу, и предпочтительно представляет собой бензоильную или нафтоильную группу.
В представленном выше определении группы заместителей γ «C8-C12ароматическоалифатическая ацильная группа» представляет собой группу, в которой фенильная группа связана с C2-C6алифатической ацильной группой, и представляет собой, например, фенилацетильную, 3-фенилпропионильную, 4-фенилбутирильную, 5-фенилпентаноильную или 6-фенилгексаноильную группу, и предпочтительно представляет собой фенилацетильную группу.
В представленном выше определении группы заместителей γ «C4-C11циклоалкилкарбонильная группа» представляет собой группу, в которой C3-C10циклоалкильная группа (которая представляет собой 3-10-членную насыщенную циклическую углеводородную группу, которая может быть конденсированной, и представляет собой, например, циклопропильную, циклобутильную, циклопентильную, циклогексильную, циклогептильную, норборнильную или адамантильную группу, и предпочтительно C3-C6циклоалкильную группу) связана с карбонильной группой, и представляет собой, например, циклопропаноильную, циклобутирильную, циклопентаноильную, циклогексаноильную, циклогептилкарбонильную, норборнилкарбонильную или адамантилкарбонильную группу, предпочтительно C4-C7циклоалкилкарбонильную группу, и особенно предпочтительно циклопентаноильную или циклогексаноильную группу.
В представленном выше определении группы заместителей γ «5- или 6-членная ароматическая гетероциклическая карбонильная группа, содержащая атом(ы) азота» представляет собой группу, в которой 5- или 6-членное ароматическое гетероциклическое кольцо, которое содержит, по меньшей мере, один атом азота и может дополнительно содержать гетероатом, выбранный из группы гетероатомов, состоящей из атома азота, атома кислорода и атома серы (например, пирролильная, имидазолильная, пиразолильная, триазолильная, тетразолильная, пиридильная, пиразинильная, пиримидинильная, пиридазинильная, тиазолильная, оксазолильная, оксадиазолильная или тиадиазолильная группа), связана с карбонильной группой, и представляет собой, например, пирролилкарбонильную, имидазолилкарбонильную, пиразолилкарбонильную, триазолилкарбонильную, тетразолилкарбонильную, никотиноильную, изоникотиноильную, пиразинилкарбонильную, пиримидинилкарбонильную, пиридазинилкарбонильную, тиазолилкарбонильную, оксазолилкарбонильную, оксадиазолилкарбонильную или тиадиазолилкарбонильную группу, предпочтительно пиридилкарбонильную группу, и особенно предпочтительно никотиноильную или изоникотиноильную группу.
В определении групп заместителей α и β «аминогруппа, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ» предпочтительно представляет собой аминогруппу или аминогруппу, которая замещена 1 или 2 заместителями (заместители представляют собой одинаковые или разные группы, каждую из которых выбирают из группы, состоящей из C1-C10алкильной группы, C6-C10арильной группы и C7-C16аралкильной группы), более предпочтительно аминогруппу или моно- или ди-C1-C10алкиламиногруппу, и особенно предпочтительно амино, диметиламино или изопропиламиногруппу.
В определении группы заместителей α C3-C10циклоалкильный фрагмент «C3-C10циклоалкильной группы, которая может быть замещена 1-3 группами, выбранными из группы заместителей β» имеет то же описанное выше значение и предпочтительно представляет собой C3-C10циклоалкильную группу, которая может быть замещена одной группой, выбранной из группы заместителей β, более предпочтительно C3-C10циклоалкильную группу, которая может быть замещена одной группой, выбранной из группы, состоящей из галогена, гидрокси, C1-C6алкила и галогенC1-C6алкила, еще более предпочтительно адамантильную группу, которая может быть замещена одним из фтора, хлора, гидрокси, метила, этила, трет-бутила, трифторметила, метокси, амино, метиламино или диметиламино, и особенно предпочтительно адамантильную группу.
В отношении «C6-C10арильной группы, которая может быть замещена 1-3 группами, выбранными из группы заместителей β» в определении группы заместителей α и в отношении «C6-C10арильной группы» в определении группы заместителей β C6-C10арильный фрагмент имеет то же описанное выше значение и предпочтительно представляет собой C6-C10арильную группу, которая может быть замещена одной группой, выбранной из группы заместителей β, более предпочтительно C6-C10арильную группу, которая может быть замещена одним из галогена, гидрокси, C1-C6алкила, галогенC1-C6алкила, C1-C6алкокси или амино, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, еще более предпочтительно фенильную группу, которая может быть замещена одним из фтора, хлора, гидрокси, метила, этила, трет-бутила, трифторметила, метокси, амино, метиламино или диметиламино, и особенно предпочтительно фенильную или 4-гидроксифенильную группу.
В определении группы заместителей α C7-C16аралкильный фрагмент «C7-C16аралкильной группы, которая может быть замещена 1-3 группами, выбранными из группы заместителей β» имеет то же описанное выше значение и предпочтительно представляет собой C7-C16аралкильную группу, которая может быть замещена одной группой, выбранной из группы заместителей β, более предпочтительно бензильную группу, которая может быть замещена одним из галогена, гидрокси, C1-C6алкила, галогенC1-C6алкила, C1-C6алкокси или амино, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, еще более предпочтительно бензильную группу, которая может быть замещена одним из фтора, хлора, гидрокси, метила, этила, трет-бутила, трифторметила, метокси, амино, метиламино или диметиламино, и особенно предпочтительно бензильную группу.
В определении группы заместителей α C6-C10арилокси фрагмент «C6-C10арилоксигруппы, которая может быть замещена 1-3 группами, выбранными из группы заместителей β» представляет собой группу, в которой упомянутый выше C6-C10арил связан с атомом кислорода, и представляет собой, например, фенокси, 1-инденилокси, 2-инденилокси, 3-инденилокси, 1-нафтилокси или 2-нафтилокси группу, и предпочтительно представляет собой феноксигруппу.
В определении группы заместителей α C7-C16аралкилокси фрагмент «C7-C16аралкилоксигруппы, которая может быть замещена 1-3 группами, выбранными из группы заместителей β» представляет собой группу, в которой упомянутая выше C7-C16аралкильная группа связана с атомом кислорода, и представляет собой, например, бензилокси, нафтилметокси, инденилметокси, дифенилметокси, 1-фенетилокси, 2-фенетилокси, 1-нафтилэтокси, 2-нафтилэтокси, 1-фенилпропокси, 2-фенилпропокси, 3-фенилпропокси, 1-нафтилпропокси, 2-нафтилпропокси, 3-нафтилпропокси, 1-фенилбутокси, 2-фенилбутокси, 3-фенилбутокси, 4-фенилбутокси, 1-нафтилбутокси, 2-нафтилбутокси, 3-нафтилбутокси, 4-нафтилбутокси, 5-фенилпентилокси, 5-нафтилпентилокси, 6-фенилгексилокси или 6-нафтилгексилокси группу, и предпочтительно представляет собой бензилоксигруппу.
В определении группы заместителей α C6-C10арилтио фрагмент «C6-C10арилтиогруппы, которая может быть замещена 1-3 группами, выбранными из группы заместителей β» представляет собой группу, в которой упомянутая выше C6-C10арильная группа связана с атомом серы, и представляет собой, например, фенилтио, 1-инденилтио, 2-инденилтио, 3-инденилтио, 1-нафтилтио или 2-нафтилтиогруппу, и предпочтительно представляет собой фенилтиогруппу.
В определении группы заместителей α «C1-C7алифатическая ацилоксигруппа» представляет собой группу, в которой упомянутая выше C1-C7алифатическая ацильная группа связана с атомом кислорода, и представляет собой, например, формилокси, ацетокси, пропионилокси, бутирилокси, изобутирилокси, валерилокси, изовалерилокси, пивалоилокси, гексаноилокси, акрилоилокси, метакрилоилокси или кротоилоксигруппу, и предпочтительно ацетоксигруппу.
В определении группы заместителей α «4-7-членная насыщенная гетероциклическая группа, содержащая атом(ы) азота» представляет собой 4-7-членную насыщенную гетероциклическую группу, которая содержит, по меньшей мере, один атом азота и может дополнительно содержать гетероатом(ы), выбранные из группы гетероатомов, состоящей из атома азота, атома кислорода и атома серы, и представляет собой, например, азетидинильную, пирролидинильную, имидазолидинильную, тиазолидинильную, пиразолидинильную, пиперидинильную, морфолинильную, тиоморфолинильную, пиперазинильную или гомопиперазинильную группу, предпочтительно пирролидинильную, пиперидинильную или морфолинильную группу, и более предпочтительно пирроилидин-1-ильную, пиперидин-1-ильную или морфолин-4-ильную группу.
В определении группы заместителей α «5- или 6-членная ароматическая гетероциклическая группа, содержащая атом(ы) азота» имеет то же описанное выше значение и предпочтительно представляет собой имидазолильную, тетразолильную или пиридинильную группу, и более предпочтительно пиридин-2-ильную или пиридин-3-ильную группу.
R предпочтительно представляет собой фенильную группу, замещенную 1-5 группами, выбранными из группы, состоящей из атома галогена, C1-C6алкильной группы, галогенC1-C6алкильной группы, аминогруппы, которая может быть замещена 1 или 2 группами, выбранными из группы заместителей γ, 4-7-членной насыщенной гетероциклической группы, содержащей атом(ы) азота и 5-6-членной ароматической гетероциклической группы, содержащей атом(ы) азота.
Более предпочтительно R представляет собой фенильную группу, которая замещена амино или амино, которая замещена 1 или 2 заместителями (заместители являются одинаковыми или разными, и каждый представляет собой группу, выбранную из группы, состоящей из C1-C10алкильной группы, C6-C10арильной группы и C7-C16аралкильной группы), и может быть дополнительно замещена 1-3 заместителями (каждый заместитель представляет собой группу, выбранную из группы, состоящей из атома галогена, C1-C6алкильной группы и галогенC1-C6алкильной группы).
Еще более предпочтительно R представляет собой фенильную группу, которая замещена амино или моно- или ди-C1-C10алкиламиногруппой, и может быть дополнительно замещена 1 или 2 C1-C6алкильными группами.
Соединение, представленное общей формулой (I), которое является активным ингредиентом фармацевтической композиции по настоящему изобретению, предпочтительно представляет собой
5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(3-(изобутилметиламино)фенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-(изобутилметиламино)фенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(3-(этилизопропиламино)фенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-втор-бутиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
5-(4-(6-(4-изобутиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион
или
5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион,
или его фармакологически приемлемую соль, и более предпочтительно
дигидрохлорид 5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона или
дигидрохлорид 5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона.
Настоящее изобретение относится к противораковой фармацевтической композиции для профилактики или лечения карциномы, саркомы или рака крови, причем в качестве активных ингредиентов композиция содержит, по меньшей мере, одно противораковое лекарство, выбранное из группы, состоящей из ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и ингибитора Raf-киназы, и, по меньшей мере, одно соединение, выбранное из группы, состоящей из соединения, обладающего активирующим действием в отношении рецептора активатора пролиферации пероксисом (PPAR) γ или его фармацевтически приемлемой соли, для одновременного введения активных ингредиентов или введения по отдельности в разное время.
Рецептор эпидермального фактора роста (EGFR) представляет собой рецепторный белок клеточной поверхности к эпидермальному фактору роста. Рецептор представляет собой трансмембранный белок и содержит внутриклеточный участок, обладающий тирозинкиназной активностью. Было обнаружено, что рецептор экспрессируется на поверхности многих раковых клеток, а часто наблюдается повышенная экспрессия, в особенности при раке легкого, раке молочной железы, раке поджелудочной железы, и тому подобное. Что касается лекарственных средств, которые ингибируют функцию рецептора эпидермального фактора роста (EGFR), то в качестве антител, связывающихся с внеклеточным доменом, могут быть упомянуты, например, цетуксимаб (торговое наименование Эрбитукс) и панитумумаб. Кроме того, что касается ингибиторов тирозинкиназной активности, то могут быть упомянуты гефитиниб (торговое наименование Иресса), эрлотиниб (торговое наименование Тарцева) и лапатиниб, предпочтительно, эрлотиниб (торговое наименование Тарцева).
Рецептор фактора роста эндотелия сосудов (VEGFR) представляет собой рецепторный белок клеточной поверхности к фактору роста эндотелия сосудов. Рецептор представляет собой трансмембранный белок и содержит внутриклеточный участок, обладающий тирозинкиназной активностью. Было известно, что рецептор экспрессируется, главным образом, на клетках эндотелия сосудов и способствует пролиферации клеток эндотелия сосудов при стимуляции фактором роста эндотелия сосудов, секретируемым раковыми клетками. В результате на периферии раковой ткани усиливается ангиогенез и стимулируется пролиферация раковых тканей. Что касается лекарственных средств, которые ингибируют функцию рецептора фактора роста эндотелия сосудов (VEGFR), то могут быть упомянуты бевацизумаб (торговое наименование Авастин), который представляет собой нейтрализующее антитело против собственно фактора роста эндотелия сосудов, и сорафениб, SU11248 и ваталаниб (PTK787), которые представляют собой ингибиторы тирозинкиназной активности. Предпочтительно, может быть упомянут сорафениб.
Raf-киназа представляет собой тип серинтреонинкиназы, которая широко участвует в передаче сигнала при пролиферации клеток, и известно, что она участвует в каскаде передачи сигнала к пролиферации от белка Ras, которые представляет собой низкомолекулярный G-белок, к ядру клетки. Что касается лекарственных средств, которые ингибируют киназную активность Raf, то может быть упомянут, например, сорафениб.
Обладающее активирующим действием в отношении PPARγ соединение, которое представляет собой один из активных ингредиентов упомянутой выше противораковой фармацевтической композиции по настоящему изобретению, включает любые соединения, которые активируют PPARγ человека при анализе любым доступным методом, или любые соединения, которые, как правило, известны в качестве активаторов PPARγ или в качестве агонистов PPARγ. Такой активатор PPARγ может быть двух или более подтипов активаторов PPAR. В качестве предпочтительных активаторов PPARγ могут быть упомянуты тиазолидиндионовые соединения, которые, как известно, применимы для лечения сахарного диабета, и не относящиеся к тиазолидиндионам соединения, такие как раскрытые в патенте США №6294580. Что касается предпочтительных тиазолидиндионовых соединений, то в дополнение к соединению, представленному общей формулой (I) по настоящему изобретению, могут быть упомянуты коммерчески доступные в настоящее время розиглитазон и пиоглитазон, и соединения, раскрытые в патенте Японии №2976885 (патент США №5886014), патенте Японии № 3488099 (патент США №6432993, европейский патент №1022272) и в заявке на выдачу патента Японии (Kokai) №2000-351779 (международная публикация WO 00/61581). Что касается предпочтительных не относящихся к тиазолидиндионам соединений, то могут быть упомянуты соединения, находящиеся в стадии разработки, такие как соединение GI262570 (фарглитазар, Glaxo Smith Kline), и тому подобное. Среди указанных соединений, обладающих активирующим действием в отношении PPARγ, особенно предпочтительными являются соединения, представленные общей формулой (I) по настоящему изобретению, или их фармакологически приемлемая соль.
Среди соединений, представленных общей формулой (I) по настоящему изобретению, соединение, обладающее активирующим действием в отношении PPARγ, ингибитор рецептора эпидермального фактора роста (EGFR), ингибитор рецептора фактора роста эндотелия сосудов (VEGFR) и ингибитор Raf-киназы, которые представляют собой активные ингредиенты по настоящему изобретению и которые способны образовывать соли, каждое из таких соединений может быть преобразовано в соль в соответствии с общими методами, и такие соли также охватываются настоящим изобретением.
Среди таких солей в качестве кислотно-аддитивной соли могут быть упомянуты, например, соль неорганической кислоты, такая как соль соляной кислоты, соль бромисто-водородной кислоты, соль серной кислоты, соль азотной кислоты и соль фосфорной кислоты; соль карбоновой кислоты, такая как соль уксусной кислоты, соль фумаровой кислоты, соль малеиновой кислоты, соль щавелевой кислоты, соль малоновой кислоты, соль янтарной кислоты, соль лимонной кислоты и соль яблочной кислоты; соль сульфоновой кислоты, такая как соль метансульфоновой кислоты, соль этансульфоновой кислоты, соль бензолсульфоновой кислоты и соль толуолсульфоновой кислоты; и соль аминокислоты, такая как соль глутаминовой кислоты и соль аспарагиновой кислоты.
В качестве основно-аддитивной соли могут быть упомянуты, например, соль щелочного металла, такая как соль лития, соль натрия и соль калия; соль щелочноземельного металла, такая как соль кальция и соль магния; или соль органического основания, такая как соль аммония, соль триэтиламина, соль диизопропиламина и соль циклогексиламина.
Существуют случаи, когда каждое из соединений, представленных общей формулой (I), причем соединение обладает активирующим действием в отношении PPARγ, ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и ингибитора Raf-киназы, каждое из которых представляет собой активный ингредиент фармацевтической композиции по настоящему изобретению, имеет оптический изомер, и все такие изомеры и их смесь охватываются настоящим изобретением. Такой оптический изомер может быть получен либо путем синтеза с использованием исходного материала для каждого изомера, или путем выделения синтезированного соединения с использованием при желании общего способа выделения или способа разделения.
Существуют случаи, когда каждое из соединений, представленных общей формулой (I), причем соединение обладает активирующим действием в отношении PPARγ, ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и ингибитора Raf-киназы, каждое из которых представляет собой активный ингредиент фармацевтической композиции по настоящему изобретению, существует в виде гидрата или в виде сольвата, и все такие формы и их смесь охватываются настоящим изобретением.
Настоящее изобретение относится к способу профилактики или лечения карциномы (в особенности, рака желудка, рака толстой кишки, рака легких, рака молочной железы, рака поджелудочной железы, рака почки или рака предстательной железы), саркомы (в особенности, медуллобластомы, рабдомиосаркомы, саркомы Юинга или липосаркомы) или рака крови (в особенности, множественной миеломы или лейкоза), причем способ профилактики или лечения по настоящему изобретению осуществляют с использованием только соединения, представленного упомянутой выше общей формулой (I), или его фармакологически приемлемой соли, или путем использования сочетания соединения, обладающего активирующим действием в отношении PPARγ (предпочтительно соединения, представленного упомянутой выше общей формулой (I)), или его фармакологически приемлемой соли с ингибитором рецептора эпидермального фактора роста (EGFR), ингибитором рецептора фактора роста эндотелия сосудов (VEGFR) или ингибитором Raf-киназы.
В настоящем изобретении выражение «использование сочетания» означает использование двух или более типов лекарственных средств, и могут быть упомянуты форма введения, при которой каждое из лекарственных средств вводят одновременно, форма введения, при которой каждое из них вводят по отдельности с некоторым интервалом, и форма введения, при которой они смешаны и вводятся в виде физически однородной композиции.
В настоящем изобретении термин «одновременное введение» не имеет какого-либо конкретного ограничения при условии, что это форма введения, допускающая по существу одновременное введение; тем не менее, предпочтительным является введение в виде однородной композиции.
Кроме того, термин «введение по отдельности с некоторым интервалом» не имеет какого-либо конкретного ограничения при условии, что это форма введения, допускающая введение по отдельности в разное время. Например, может быть упомянута форма введения, когда первым вводят ингибитор рецептора эпидермального фактора роста (EGFR), ингибитор рецептора фактора роста эндотелия сосудов (VEGFR) или ингибитор Raf-киназы, а через заданное время вводят соединение, обладающее активирующим действием в отношении PPARγ, или его фармацевтически приемлемую соль.
ЭФФЕКТЫ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Противораковая фармацевтическая композиция по настоящему изобретению, которая в качестве активного ингредиента содержит соединение, представленное общей формулой (I), применима в качестве средства для профилактики или лечения рака желудка, рака толстой кишки, рака легких, рака молочной железы, рака поджелудочной железы, рака почки, рака предстательной железы, медуллобластомы, рабдомиосаркомы, саркомы Юинга, липосаркомы, множественной миеломы или лейкоза.
Противораковая фармацевтическая композиция по настоящему изобретению, которая содержит, по меньшей мере, одно противораковое лекарственное средство, выбранное из группы, состоящей из ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и ингибитора Raf-киназы, и, по меньшей мере, одно соединение, выбранное из группы, состоящей из соединения, обладающего активирующим действием в отношении PPARγ, и его фармацевтически приемлемой соли, в качестве активных ингредиентов, которые предназначены для одновременного введения или введения по отдельности в разное время, применима в качестве противоракового лекарственного средства (противоракового лекарственного средства для профилактики или лечения карциномы, такой как рак желудка, рак толстой кишки, рак легких, рак молочной железы, рак поджелудочной железы, так почки и рак предстательной железы, саркомы, такой как медуллобластома, рабдомиосаркома, саркома Юинга и липосаркома, и рака крови, такого как множественная миелома и лейкоз).
НАИЛУЧШИЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Соединение, представленное общей формулой (I), которое представляет собой активный ингредиент фармацевтической композиции по настоящему изобретению, может быть легко получено в соответствии со способом, описанным в патенте Японии № 3488099.
Розиглитазон может быть легко получен в соответствии со способом, раскрытым в патенте США №5741803, а пиоглитазон - в соответствии с патентом США №4687777.
Что касается тиазолидиндионовых соединений, обладающих активирующим действием в отношении PPARγ, которые раскрыты в патенте Японии №2976885 (патент США №5886014), патенте Японии № 3488099 (патент США №6432993, европейский патент №1022272) и заявке на выдачу патента Японии (Kokai) №2000-351799 (международная публикация № WO 00/61581), то способ производства также раскрыт в каждой из опубликованных заявок, и соединения могут быть легко получены в соответствии со способами, раскрытыми в каждой из опубликованных заявок.
Фарглитазар может быть легко получен в соответствии со способом, раскрытым в международной публикации № WO 00/08002.
В качестве ингибитора эпидермального фактора роста (EGFR), который представляет собой активный ингредиент противораковой фармацевтической композиции по настоящему изобретению, причем в качестве активных ингредиентов композиция содержит соединение, обладающее активирующим действием в отношении PPARγ, такое как соединение, представленное общей формулой (I), или его фармакологически приемлемую соль, и другое противораковое средство(а), доступным является гефитиниб производства AstraZeneca.
Цетуксимаб может быть легко получен в соответствии со способом, раскрытым в европейским патентом №359282, панитумумаб - в соответствии с международной публикацией № WO 96/33735, эрлотиниб - в соответствии с международной публикацией № WO 96/30347, лапатиниб - в соответствии с международной публикацией № WO 99/35146, а среди ингибиторов рецептора фактора роста эндотелия сосудов (VEGFR) бевацизумаб - в соответствии с европейским патентом №1325932, сорафениб - в соответствии с международной публикацией № WO 99/35146, SU11248 - в соответствии с международной публикацией № WO 2001/060814, и ваталаниб - в соответствии с международной публикацией № WO 98/035958.
В том случае, когда соединение, представленное общей формулой (I), или его фармакологически приемлемая соль, которое представляет собой активный ингредиент фармацевтической композиции по настоящему изобретению, используется в качестве терапевтического средства, средства для улучшения качества жизни или в качестве профилактического средства, соединение или его фармакологически приемлемая соль отдельно или в смеси с наполнителем, разбавителем, и тому подобное, которые являются фармацевтически приемлемыми, могут вводиться орально в виде таблетки, капсулы, гранул, порошков или сиропа, или парентерально путем инъекции или введения суппозитория.
Указанные фармацевтические препараты приготавливают в соответствии с известным способом приготовления с использованием добавок, включая наполнители (могут быть упомянуты, например, органические наполнители, такие как производные сахаров, например, лактоза, сахароза, глюкоза, маннит или сорбит; производные крахмала, например, кукурузный крахмал, картофельный крахмал, α-крахмал или декстрин; производные целлюлозы, например, кристаллическая целлюлоза; аравийская камедь; декстран; или пуллулан, и неорганические наполнители, такие как производные силикатов, например, светлый кремневый ангидрид, синтетический алюмосиликат, силикат кальция, алюмометасиликат магния; фосфаты, например, моногидрофосфат кальция; карбонаты, например, карбонат кальция; и соли серной кислоты, такие как сульфат кальция), смазки (могут быть упомянуты, например, стеариновая кислота, соли металлов и стеариновой кислоты, такие как стеарат кальция или стеарат магния; тальк; коллоидный кремнезем; воски, такие как прополис или спермацет; борная кислота; адипиновая кислота; сульфаты, такие как сульфат натрия; гликоль; фумаровая кислота; бензоат натрия; D,L-лейцин; жирнокислая соль натрия; лаурилсульфаты, такие как лаурилсульфат натрия или лаурилсульфат магния; кремневые кислоты, такие как кремневый ангидрид или гидросиликат; и упомянутые выше производные крахмала), связующие вещества (могут быть упомянуты, например, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, поливинилпирролидон, макрогол и соединения, сходные с упомянутым выше наполнителем), разрыхлители (могут быть упомянуты, например, производные целлюлозы, такие как низкозамещенная гидроксипропилцеллюлоза, карбоксиметилцеллюлоза, карбоксиметилцеллюлоза кальция или поперечно-сшитая карбоксиметилцеллюлоза натрия; или химически модифицированные крахмалы или целлюлозы, такие как карбоксиметилкрахмал, карбоксиметилкрахмал натрия или поперечно-сшитый поливинилпирролидон), стабилизаторы (могут быть упомянуты, например, сложные эфиры пара-гидроксибензойной кислоты, такие как метилпарабен или пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт или фенилэтиловый спирт, хлорид бензалкония; фенолы, такие как фенол и крезол; тимерозал; дегидроацетовая кислота; и сорбиновая кислота) и вкусоароматические корригенты (могут быть упомянуты, например, часто используемые подсластители, подкислители или ароматизаторы) или разбавители.
Хотя дозировка в значительной мере варьирует в зависимости от условий, таких как активность лекарственного средства, симптомы, возраст, масса, и тому подобное, пациента (теплокровного животного, в особенности человека), в случае орального введения, например, желательно введение дозы от 0,0005 мг/кг массы тела до 50 мг/кг массы тела, а в случае внутривенного введения, желательно введение дозы от 0,0005 мг/кг массы тела до 50 мг/кг массы тела, однократно или несколько раз в сутки в зависимости от симптомов.
Кроме того, соединение, обладающее активирующим действием в отношении PPARγ (предпочтительно соединение, представленное общей формулой (I)), или его фармакологически приемлемая соль и ингибитор рецептора эпидермального фактора роста (EGFR), ингибитор рецептора фактора роста эндотелия сосудов (VEGFR) или ингибитор Raf-киназы могут быть, каждый, включены в состав отдельно вводимой формы или могут быть включены в состав физически однородной вводимой формы путем их смешивания.
Если используется каждая из такой отдельно вводимой формы или однородной вводимой формы, то каждое из соединения, обладающего активирующим действием в отношении PPARγ, или его фармакологически приемлемой соли, ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и ингибитора Raf-киназы или смесь с наполнителем, разбавителем, и тому подобное, которые являются фармакологически приемлемыми, могут вводиться орально в виде таблетки, капсулы, гранул, порошков или сиропа, или парентерально путем инъекции или введения суппозитория.
Указанные фармацевтические препараты приготавливают в соответствии с известным способом приготовления с использованием добавок, включая наполнители (могут быть упомянуты, например, органические наполнители, такие как производные сахаров, например, лактоза, сахароза, глюкоза, маннит или сорбит; производные крахмала, например, кукурузный крахмал, картофельный крахмал, α-крахмал или декстрин; производные целлюлозы, например, кристаллическая целлюлоза; аравийская камедь; декстран; или пуллулан, и неорганические наполнители, такие как производные силикатов, например, светлый кремневый ангидрид, синтетический алюмосиликат, силикат кальция, алюмометасиликат магния; фосфаты, например, моногидрофосфат кальция; карбонаты, например, карбонат кальция; и соли серной кислоты, такие как сульфат кальция), смазки (могут быть упомянуты, например, стеариновая кислота, соли металлов и стеариновой кислоты, такие как стеарат кальция или стеарат магния; тальк; коллоидный кремнезем; воски, такие как пчелиный воск или спермацет; борная кислота; адипиновая кислота; сульфаты, такие как сульфат натрия; гликоль; фумаровая кислота; бензоат натрия; D,L-лейцин; лаурилсульфаты, такие как лаурилсульфат натрия или лаурилсульфат магния; кремневые кислоты, такие как кремневый ангидрид или гидросиликат; и упомянутые выше производные крахмала), связующие вещества (могут быть упомянуты, например, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, поливинилпирролидон, макрогол и соединения, сходные с упомянутым выше наполнителем), разрыхлители (могут быть упомянуты, например, производные целлюлозы, такие как низкозамещенная гидроксипропилцеллюлоза, карбоксиметилцеллюлоза, карбоксиметилцеллюлоза кальция или поперечно-сшитая карбоксиметилцеллюлоза натрия; или химически модифицированные крахмалы или целлюлозы, такие как карбоксиметилкрахмал, карбоксиметилкрахмал натрия или поперечно-сшитый поливинилпирролидон), эмульгаторы (например, коллоидные глины, такие как бентонит и прополис; гидроксиды металлов, такие как гидроксид магния и гидроксид алюминия; анионные поверхностно-активные вещества, такие как лаурилсульфат натрия и стеарат кальция; катионные поверхностно-активные вещества, такие как хлоридбензалкония; и неионные поверхностно-активные вещества, такие как полиоксиэтиленалкиловый эфир, сложный эфир полиоксиэтиленсорбитана и жирной кислоты и сложный эфир сахарозы и жирной кислоты), стабилизаторы (могут быть упомянуты, например, сложные эфиры пара-гидроксибензойной кислоты, такие как метилпарабен или пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт или фенилэтиловый спирт, хлорид бензалкония; фенолы, такие как фенол и крезол; тимерозал; дегидроацетовая кислота; и сорбиновая кислота) и вкусоароматические корригенты (могут быть упомянуты, например, часто используемые подсластители, подкислители или ароматизаторы) или разбавители.
Соотношение вводимого соединения, обладающего активирующим действием в отношении PPARγ, или его фармакологически приемлемой соли и ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) или ингибитора Raf-киназы может варьировать в зависимости от различных условий, таких как активность конкретных лекарственных средств и симптомы, возраст, масса пациента.
Хотя дозировка варьирует в зависимости от активности конкретных лекарственных средств и симптомов, возраста, массы, и тому подобное, пациента (теплокровного животного, в особенности человека), в случае орального введения, например, желательно введение дозы от 0,1 мг/кг массы тела до 100 мг/кг массы тела (предпочтительно 20 мг/кг массы тела), а в случае внутривенного введения, например, желательно введение дозы от 0,01 мг/кг массы тела до 100 мг/кг массы тела (предпочтительно 10 мг/кг массы тела), 1-6 раз в сутки в зависимости от симптомов, одновременно или по отдельности через некоторое время.
Кроме того, соотношение дозы соединения, обладающего активирующим действием в отношении PPARγ, и ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) или ингибитора Raf-киназы может в значительной степени варьировать; тем не менее, соотношение дозы соединения, обладающего активирующим действием в отношении PPARγ, или его фармакологически приемлемой соли и ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) или ингибитора Raf-киназы может находиться в интервале от 1:1000 до 1000:1 по массе.
ПРИМЕРЫ
Здесь и далее в настоящем описании настоящее изобретение будет более детально описано со ссылкой на примеры получения, примеры испытаний и примеры приготовления; тем не менее, это не имеет своей целью ограничить объем настоящего изобретения.
ПРИМЕР ПОЛУЧЕНИЯ 1
Дигидрохлорид 5-(4-(6-(3-изопропиламинофенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона
Смесь 0,74 г сложного трет-бутилового эфира N-(2-амино-5-(3-изопропиламинофенокси)фенил)-N-метилкарбаминовой кислоты, полученного в справочном примере 2, 0,70 г 4-(2,4-диоксотиазолидин-5-илметил)феноксиуксусной кислоты (заявка на выдачу патента Японии (Kokai) №Hei 11-193276), 0 41 г диэтилцианофосфоната, 0,25 г триэтиламина и 30 мл безводного тетрагидрофурана перемешивали при комнатной температуре в течение 4,5 часов. Реакционную смесь концентрировали, затем добавляли воду и экстрагировали этилацетатом. После сушки экстракта над безводным сульфатом натрия отгоняли растворитель, и очищали полученный остаток по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 2/3) с получением сложного трет-бутилового эфира N-(5-(3-изопропиламинофенокси)-2-(4-(2,4-диоксотиазолидин-5-илметил)феноксиацетиламино)фенил)-N-метилкарбаминовой кислоты в качестве промежуточного продукта. После этого промежуточный продукт растворяли в 50 мл смеси 4н. соляной кислоты/1,4-диоксана, оставляли смесь отстаиваться при комнатной температуре в течение 16 часов, осажденный продукт фильтровали и промывали этилацетатом с получением указанного в заголовке соединения (0,76 г, выход 64%).
1Н-ЯМР (ДМСО-d6) δ: 1,21 (6Н, д, J=6, 4 Гц), 3,11 (1Н, дд, J=14 и 9,0 Гц), 3,34 (1Н, дд, J=14 и 4,4 Гц), 3,57-3,65 (1Н, м), 3,95 (3Н, с), 4,91 (1Н, дд, J=9,0 и 4,4 Гц), 5,63 (2Н, с), 6,70-7,20 (3Н, м), 7,14 (2Н, д, J=8,7 Гц), 7,25 (2Н, д, J=8,7 Гц), 7,25 (1Н, д, J=3,3 Гц), 7,35-7,45 (1Н, м), 7,68 (1Н, д, J=l,9 Гц), 7,83 (1Н, д, J=8,9 Гц), 12,05 (1Н, с; исчезал вследствие добавления оксида дейтерия).
ПРИМЕР ПОЛУЧЕНИЯ 2
Дигидрохлорид 5-(4-(6-(3-(изобутилметиламино)фенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона
Вместо сложного трет-бутилового эфира N-(2-амино-5-(3-изопропиламинофенокси)фенил)-N-метилкарбаминовой кислоты из примера получения 1 использовали сложный трет-бутиловый эфир N-(2-амино-5-(3-(изобутилметиламино)фенокси)фенил)-N-метилкарбаминовой кислоты, полученный в справочном примере 5, с получением указанного в заголовке соединения путем, сходным с описанным в примере получения 1.
1H-ЯМР (ДМСО-d6) δ: 0,86 (6H, д, J=6,7 Гц), 1,90-1,99 (1H, м), 2,91 (3H, с), 3,08-3,14 (3H, м), 3,34 (1H, дд, J=14 и 4,4 Гц), 3,94 (3H, с), 4,91 (1H, дд, J=9,0 и 4,4 Гц), 5,65 (2H, с), 6,21 (1H, ушир.), 6,39 (1H, ушир.), 6,53 (1H, ушир.), 7,15-7,27 (6H, м), 7,62 (1H, д, J=2,1 Гц), 7,80 (1H, д, J=8,9 Гц), 12,04 (1H, ушир.; исчезал вследствие добавления оксида дейтерия).
Пример получения 3
Дигидрохлорид 5-(4-(6-(4-(изобутилметиламино)фенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона
Вместо сложного трет-бутилового эфира N-(2-амино-5-(3-изопропиламинофенокси)фенил)-N-метилкарбаминовой кислоты из примера получения 1 использовали N-(2-метил-5-(4-(изобутилметиламино)фенокси)фенил)метиламин, полученный в справочном примере 8, с получением указанного в заголовке соединения путем, сходным с описанным в примере получения 1.
1H-ЯМР (ДМСО-d6) δ: 0,90 (6H, д, J=4,4 Гц), 1,75-2,05 (1H, м), 1,99 (3H, с), 2,90-3,10 (2H, м), 3,11 (1H, дд, J=14 и 8,9 Гц), 3,34 (1H, дд, J=14 и 4,4 Гц), 3,92 (3H, с), 4,91 (1H, дд, J=8,9 и 4,4 Гц), 5,62 (2H, с), 6,65-7,20 (5H, м), 7,13 (2H, д, J=8,7 Гц), 7,25 (2H, д, J=8,7 Гц), 7,45-7,60 (1H, м), 7,78 (1H, д, J=8,9 Гц), 12,05 (1H, с; исчезал при добавлении оксида дейтерия).
Пример получения 4
5-(4-(6-(3-(Этилизопропиламино)фенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион
Смесь 620 мг дигидрохлорида 5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона, полученного в примере получения 1, 66 мг ацетальдегида, 90 мг уксусной кислоты, 318 мг триацетоксиборгидрида натрия и 15 мл безводного тетрагидрофурана перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали, затем добавляли воду и экстрагировали этилацетатом. После сушки экстракта над безводным сульфатом натрия отгоняли растворитель и очищали полученный остаток по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 1/1) с получением указанного в заголовке соединения (260 мг, выход 48%).
1H-ЯМР (ДМСО-d6) δ: 1,06 (3H, т, J=7,0 Гц), 1,11 (6H, д, J=6,6 Гц), 3,05 (1H, дд, J=14 и 9,2 Гц), 3,18 (2H, кв, J=7,0 Гц), 3,31 (1H, дд, J=14 и 4,3 Гц), 3,79 (3H, с), 3,94-4,04 (1H, м), 4,87 (1H, дд, J=9,2 и 4,3 Гц), 5,63 (2H, с), 6,11 (1H, дд, J=7,9 и 2,0 Гц), 6,34 (1H, т, J=2,2 Гц), 6,46 (1H, дд, J=8,5 и 2,3 Гц), 6,92 (1H, дд, J=8,8 и 2,2 Гц), 7,06-7,11 (3H, м), 7,19 (1H, д, J=8,7 Гц), 7,28 (1H, д, J=2,3 Гц), 7,63 (1H, д, J=8,7 Гц), 12,02 (1H, с; исчезал вследствие добавления оксида дейтерия).
Пример получения 5
5-(4-(6-(4-Изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион
Вместо дигидрохлорида 5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона из примера получения 4 использовали дигидрохлорид 5-(4-(6-(4-аминофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона (заявка на выдачу патента Японии (Kokai) № Hei 11-193276), а вместо ацетальдегида использовали ацетон, с получением указанного в заголовке соединения путем, сходным с описанным в примере получения 4.
1H-ЯМР (ДМСО-d6) δ: 1,13 (6H, д, J=6,3 Гц), 3,05 (1H, дд, J=14 и 9,1 Гц), 3,31 (1H, дд, J=14 и 4,3 Гц), 3,45-3,52 (1H, м), 3,75 (3H, с), 4,87 (1H, дд, J=9,1 и 4,3 Гц), 5,24 (1H, ушир.; исчезал вследствие добавления оксида дейтерия), 5,34 (2H, с), 6,56 (2H, дд, J=12 и 3,3 Гц), 6,81 (2H, д, J=8,6 Гц), 6,83 (1H, дд, J=8,2 и 2,3 Гц), 7,04-7,07 (3H, м), 7,19 (2H, д, J=8,6 Гц), 7,57 (1H, д, J=8,8 Гц), 12,02 (1H, ушир.; исчезал вследствие добавления оксида дейтерия).
Пример получения 6
5-(4-(6-(4-втор-бутиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион
Вместо дигидрохлорида 5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона из примера получения 4 использовали дигидрохлорид 5-(4-(6-(4-аминофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона, а вместо ацетальдегида использовали метилэтилкетон с получением указанного в заголовке соединения путем, сходным с описанным в примере получения 4.
1H-ЯМР (ДМСО-d6) δ: 0,90 (3H, т, J=7,4 Гц), 2,17 (3H, д, J=6,4 Гц), 1,34-1,46 (1H, м), 1,48-1,59 (1H, м), 3,06 (1H, дд, J=14 и 9,2 Гц), 3,24-3,34 (2H, м), 3,75 (3H, с), 4,87 (1H, дд, J=9,2 и 4,3 Гц), 5,23 (1H, ушир.; исчезал вследствие добавления оксида дейтерия), 5,34 (2H, с), 6,57 (2H, д, J=8,7 Гц), 6,81 (2H, д, J=8,9 Гц), 6,84 (1H, дд, J=8,8 и 2,2 Гц), 7,01-7,09 (3H, м), 7,19 (2H, д, J=8,7 Гц), 7,57 (1H, д, J=8,8 Гц), 12,01 (1H, ушир.; исчезал вследствие добавления оксида дейтерия).
Пример получения 7
5-(4-(6-(4-Изобутиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион
Вместо дигидрохлорида 5-(4-(6-(3-изопропиламинофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона из примера получения 4 использовали дигидрохлорид 5-(4-(6-(4-аминофенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона, а вместо ацетальдегида использовали изобутиловый альдегид с получением указанного в заголовке соединения путем, сходным с описанным в примере получения 4.
1H-ЯМР (ДМСО-d6) δ: 0,94 (6H, д, J=6,7 Гц), 1,77-1,88 (1H, м), 2,78-2,81 (2H, м), 3,05 (1H, дд, J=14 и 9,3 Гц), 3,31 (1H, дд, J=14 и 4,3 Гц), 3,74 (3H, с), 4,86 (1H, дд, J=9,3 и 4,3 Гц), 5,34 (2H, с), 5,50 (1H, с; исчезал вследствие добавления оксида дейтерия), 6,57 (2H, дд, J=6,8 и 2,0 Гц), 6,81 (2H, д, J=8,8 Гц), 6,83 (1H, дд, J=8,6 и 2,4 Гц), 7,04-7,07 (3H, м), 7,19 (2H, д, J=8,6 Гц), 7,56 (1H, д, J=8,8 Гц), 12,01 (1H, с; исчезал вследствие добавления оксида дейтерия).
Пример получения 8
Дигидрохлорид 5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1H-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона
Вместо сложного трет-бутилового эфира N-(2-амино-5-(3-изопропиламинофенокси)фенил)-N-метилкарбаминовой кислоты из примера получения 1 использовали сложный трет-бутиловый эфир N-(2-амино-5-(4-трет-бутоксикарбониламино-3,5-диметилфенокси)фенил)-N-метилкарбаминовой кислоты, полученный в справочном примере 11, с получением указанного в заголовке соединения путем, сходным с описанным в примере получения 1.
1H-ЯМР(ДМСО-d6) δ: 2,34 (6H, с), 3,10 (1H, дд, J=14 и 9,0 Гц), 3,34 (1H, дд, J=14 и 4,4 Гц), 3,93 (3H, с), 4,91 (1H, дд, J=4,4 и 9,0 Гц), 5,62 (2H, с), 6,80 (2H, с), 7,14 (2H, д, J=8,7 Гц), 7,18 (1H, дд, J=8,9 и 2,2 Гц), 7,25 (2H, д, J=8,7 Гц), 7,61 (1H, д, J=2,2 Гц), 7,81 (1H, д, J=8,9 Гц), 12,1 (1H, ушир.; исчезал вследствие добавления оксида дейтерия).
Справочный пример 1
Трет-бутиловый эфир N-(5-(3-аминофенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты
К 80 мл безводной суспензии N,N-диметилформамида, содержащей 2,18 г гидрида натрия (55 мас.%), добавляли 5,45 г 3-аминофенола, и перемешивали смесь при комнатной температуре в течение 20 минут. Затем малыми количествами добавляли 14,3 г сложного трет-бутилового эфира N-(5-хлор-2-нитрофенил)-N-метилкарбаминовой кислоты (заявка на выдачу патента Японии (Kokai) № Hei 11-193276), и перемешивали смесь при 100°C в течение 6 часов. Реакционную смесь концентрировали, затем добавляли воду и нейтрализовали с использованием 3н соляной кислоты и порошка бикарбоната натрия. Осажденный нерастворимый продукт фильтровали, промывали водой, а затем сушили в условиях пониженного давления с получением указанного в заголовке соединения (16,6 г, выход 92%).
1H-ЯМР (ДМСО-d6) δ: 1,23 и 1,42 (9H всего, с каждый), 3,18 (3H, с), 5,38 (2H, с; исчезал вследствие добавления оксида дейтерия), 6,25 (1H, дд, J=7,6 и 2,4 Гц), 6,31 (1H, с), 6,46 (1H, дд, J=8,1 и 1,0 Гц), 6,88 (1H, дд, J=9,0 и 2,1 Гц), 7,09 (1H, т, J=8,0 Гц), 7,16 (1H, с), 8,00 (1H, д, J=9,0 Гц).
Справочный пример 2
Сложный трет-бутиловый эфир N-(2-амино-5-(3-изопропиламинофенокси)фенил)-N-метилкарбаминовой кислоты
Смесь 14,4 г сложного трет-бутилового эфира N-(5-(3-аминофенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты, 2,90 г ацетона, 3,00 г уксусной кислоты, 10,6 г триацетоксиборгидрида натрия и 200 мл безводного тетрагидрофурана перемешивали при комнатной температуре в течение 4 суток. Реакционную смесь концентрировали, затем добавляли воду и экстрагировали этилацетатом. После сушки экстракта над безводным сульфатом натрия отгоняли растворитель и очищали полученный остаток по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 2/3) с получением сложного трет-бутилового эфира N-(5-(3-изопропиламинофенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты в качестве промежуточного продукта. Указанный промежуточный продукт растворяли в 200 мл метанола, затем добавляли 2,02 г 10% палладированного угля, и энергично перемешивали смесь в атмосфере водорода при комнатной температуре в течение 2,5 часов. После завершения реакции катализатор фильтровали, и отгоняли растворитель с получением указанного в заголовке соединения (12,0 г, выход 81%).
1H-ЯМР (ДМСО-d6) δ: 1,08 (6H, д, J=6,4 Гц), 1,29 (9H, с), 2,98 (3H, с), 3,40-3,47 (1H, м), 4,78 (2H, с; исчезал вследствие добавления оксида дейтерия), 5,45 (1H, д, J=7,8 Гц; исчезал вследствие добавления оксида дейтерия), 5,96 (1H, д, J=7,2 Гц), 6,07 (1H, т, J=2,2 Гц), 6,20 (1H, дд, J=8,1 и 1,9 Гц), 6,60 (1H, с), 6,71 (2H, с), 6,93 (1H, т, J=8,1 Гц).
Справочный пример 3
Сложный трет-бутиловый эфир N-(5-(3-бромфенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты
К 50 мл безводной суспензии N,N-диметилформамида, содержащей 2,5 г гидрида натрия (55 мас.%), добавляли 10,0 г 3-бромфенола, и перемешивали смесь при охлаждении на льду в течение 15 минут. Затем к смеси по каплям добавляли раствор 16,6 г сложного трет-бутилового эфира N-(5-хлор-2-нитрофенил)-N-метилкарбаминовой кислоты, растворенного в 70 мл безводного N,N-диметилформамида, и перемешивали смесь при 100°C в течение 3 часов. Реакционную смесь концентрировали, затем добавляли воду, нейтрализовали с использованием 3н. соляной кислоты и экстрагировали этилацетатом. Экстракт промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Из экстракта отгоняли этилацетат, осажденный нерастворимый продукт промывали гексаном и фильтровали, а затем сушили в условиях пониженного давления, с получением указанного в заголовке соединения (20,2 г, выход 83%).
1H-ЯМР (CDCl3) δ: 1,24 (9H, с), 3,19 (3H, с), 6,97 (1H, дд, J=9,0 и 2,4 Гц), 7,22 (1H, д, J=7,9 Гц), 7,29 (1H, д, J=1,7 Гц), 7,42-7,51 (3H, м), 8,03 (1H, д, J=9,0 Гц).
Справочный пример 4
Сложный трет-бутиловый эфир N-(5-(3-(изобутилметиламино)фенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты
700,0 мг сложного трет-бутилового эфира N-(5-(3-бромфенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты, полученной в справочном примере 3, 0,24 мл изобутилметиламина, 151,0 мг трис(дибензилиденацетон)дипалладия, 115,7 мг 2-(дициклогексилфосфино)бифенила и 277,7 мг трет-бутоксида калия суспендировали в 4 мл безводного толуола, и перемешивали смесь при 100°C в течение 1,5 часов. К реакционной смеси добавляли воду, а затем экстрагировали этилацетатом. После промывки экстракта насыщенным солевым раствором и сушки над безводным сульфатом натрия отгоняли растворитель и очищали полученный остаток по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 1/7) с получением указанного в заголовке соединения (204,2 мг, выход 29%).
1H-ЯМР (CDCl3) δ: 0,80 (6H, д, J=6,6 Гц), 1,25 (9H, с), 1,88-2,01 (1H, м), 2,85 (3H, с), 2,95 (2H, д, J=7,3 Гц), 3,14 (3H, с), 6,20-6,27 (2H, м), 6,43 (1H, дд, J=8,8 и 2,2 Гц), 6,72-6,83 (2H, м), 7,11 (1H, т, J=8,1 Гц), 7,81 (1H, д, J=9,5 Гц).
Справочный пример 5
Сложный трет-бутиловый эфир N-(2-амино-5-(3-(изобутилметиламино)фенокси)фенил)-N-метилкарбаминовой кислоты
204,2 мг сложного трет-бутилового эфира N-(5-(3-(изобутилметиламино)фенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты, полученного в справочном примере 4, растворяли в 10 мл этанола, затем добавляли 100,0 мг 10% палладированного угля, и энергично перемешивали смесь в атмосфере водорода при комнатной температуре в течение 2,5 часов. После завершения реакции катализатор фильтровали и отгоняли растворитель. Полученный остаток очищали по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 1/4→1/3) с получением указанного в заголовке соединения (145,4 мг, выход 77%).
1H-ЯМР (CDCl3) δ: 0,90 (6H, д, J=6,6 Гц), 1,57 (9H, с), 1,98-2,09 (1H, м), 2,92 (3H, с), 3,06 (2H, д, J=7,3 Гц), 3,13 (3H, с), 3,64 (2H, с; исчезал вследствие добавления оксида дейтерия), 6,30 (1H, т, J=2,2 Гц), 6,35 (1H, дд, J=8,1 и 2,2 Гц), 6,70-6,88 (3H, м), 7,08 (1H, т, J=8,2 Гц), 7,25-7,31 (1H, м).
Справочный пример 6
(4-(Изобутилметиламино)фенокси)-трет-бутилдиметилсилан
В 40 мл безводного толуола суспендировали 5 мл (4-бромфенокси)-трет-бутилдиметилсилана, 2,9 мл изобутилметиламина, 458,0 мг ацетата палладия, 1,2 г 2-(ди-трет-бутилфосфино)бифенила и 2,9 г трет-бутоксида натрия, и перемешивали смесь при 100°C в течение 1,5 часов. Катализатор фильтровали, затем добавляли воду и экстрагировали этилацетатом. После промывки экстракта насыщенным солевым раствором и сушки над безводным сульфатом натрия отгоняли растворитель и очищали полученный остаток по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 1/40→1/20) с получением указанного в заголовке соединения (3,83 г, выход 64%).
1H-ЯМР (CDCl3) δ: 0,16 (6H, с), 0,91 (6H, д, J=6,6 Гц), 0,97 (9H, с), 1,94-2,05 (1H, м), 2,87 (3H, с), 2,98 (2H, д, J=7,3 Гц), 6,57 (2H, д, J=8,8 Гц), 6,72 (2H, д, J=8,8 Гц).
Справочный пример 7
N-(5-(4-(Изобутилметиламино)фенокси)-2-нитрофенил)метиламин
В 20 мл безводного тетрагидрофурана растворяли 3,83 г (4-(изобутилметиламино)фенокси)-трет-бутилдиметилсилана, полученного в справочном примере 6, затем добавляли 20 мл 1M раствора фторида тетра-н-бутиламмония в тетрагидрофуране и перемешивали смесь при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали, затем добавляли воду и экстрагировали этилацетатом. После промывки экстракта насыщенным солевым раствором и сушки над безводным сульфатом натрия отгоняли растворитель и очищали полученный остаток по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 1/5). Полученный продукт растворяли в 4н. соляной кислоте-1,4-диоксане, и перемешивали смесь при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали и промывали диэтиловым эфиром с получением моногидрохлорида 4-изобутилметиламинофенола, который является промежуточным продуктом. При комнатной температуре в течение 15 минут перемешивали 20 мл безводной суспензии N,N-диметилформамида, содержащей 500,0 мг указанного промежуточного продукта и 2,6 г карбоната калия. Затем к смеси добавляли 664,7 мг сложного трет-бутилового эфира N-(5-хлор-2-нитрофенил)-N-метилкарбаминовой кислоты и перемешивали смесь при 150°C в течение 3 часов. Реакционную смесь концентрировали, затем добавляли воду и экстрагировали этилацетатом. После промывки экстракта насыщенным солевым раствором и сушки над безводным сульфатом натрия отгоняли растворитель и очищали полученный остаток по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/толуол = 1/30) с получением указанного в заголовке соединения (256,1 мг, выход 34%).
1H-ЯМР (CDCl3) δ: 0,96 (6H, д, J=6,8 Гц), 2,00-2,13 (1H, м), 2,92 (3H, д, J=5,9 Гц), 2,98 (3H, с), 3,12 (2H, д, J=7,8 Гц), 6,19-6,23 (2H, м), 6,68 (2H, д, J=8,8 Гц), 6,96 (2H, д, J=8,8 Гц), 8,13 (1H, д, J=9,8 Гц).
Справочный пример 8
N-(2-Метил-5-(4-(изобутилметиламино)фенокси)фенил)метиламин
Вместо сложного трет-бутилового эфира N-(5-(3-(изобутилметиламино)фенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты из справочного примера 5 использовали N-(5-(4-(изобутилметиламино)фенокси)-2-нитрофенил)метиламин, полученный в справочном примере 7, с получением указанного в заголовке соединения путем, сходным с описанным в справочном примере 5.
1H-ЯМР (CDCl3) δ: 0,88 (6H, д, J=6,6 Гц), 1,94-2,04 (1H, м), 2,77 (3H, с), 2,87 (3H, с), 2,99 (2H, д, J=7,3 Гц), 3,22 (2H, с), 6,16 (1H, дд, J=8,1 и 2,5 Гц), 6,33 (1H, д, J=2,5 Гц), 6,57 (1H, д, J=8,1 Гц), 6,59 (2H, д, J=8,8 Гц), 6,87 (2H, д, J=8,8 Гц).
Справочный пример 9
Сложный трет-бутиловый эфир N-(5-(4-амино-3,5-диметилфенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты
Вместо 3-аминофенола из справочного примера 1 использовали 4-амино-3,5-диметилфенол, проводили обработку путем, сходным с описанным в справочном примере 1, а затем очищали по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 1/2) с получением указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6) δ: 1,23 и 1,41 (9H всего, с каждый), 2,10 (6H, с), 3,17 (3H, с), 4,66 (2H, ушир.; исчезал вследствие добавления оксида дейтерия), 6,69 (2H, с), 6,75 (1H, дд, J=9,1 и 2,5 Гц), 7,07 (1H, с), 7,96 (1H, д, J=9,1).
Справочный пример 10
Сложный трет-бутиловый эфир N-(5-(4-трет-бутоксикарбониламино-3,5-диметилфенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты
Смесь 2,27 г сложного трет-бутилового эфира N-(5-(4-амино-3,5-диметилфенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты, 1,28 г ди-трет-бутилбикарбоната, 0,59 г триэтиламина и 20 мл безводного тетрагидрофурана нагревали с обратным холодильником в течение 6 часов. Реакционную смесь концентрировали, затем добавляли воду и экстрагировали этилацетатом. После сушки экстракта над безводным сульфатом натрия отгоняли растворитель и очищали полученный остаток по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 1/10) с получением указанного в заголовке соединения (1,74 г, выход 61%).
1H-ЯМР (ДМСО-d6) δ: 1,24 и 1,42 (9H всего, с каждый), 1,46 (9H, с), 2,17 (6H, с), 3,19 (3H, с), 6,84 (1H, дд, J=9,0 и 2,7), 6,90 (2H, с), 7,21 (1H, с), 8,00 (1H, д, J=9,0), 8,42 (1H, с; исчезал вследствие добавления оксида дейтерия).
Справочный пример 11
Сложный трет-бутиловый эфир N-(2-амино-5-(4-трет-бутоксикарбониламино-3,5-диметилфенокси)фенил)-N-метилкарбаминовой кислоты
Вместо сложного трет-бутилового эфира N-(5-(3-(изобутилметиламино)фенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты из справочного примера 5 использовали сложный трет-бутиловый эфир N-(5-(4-трет-бутоксикарбониламино-3,5-диметилфенокси)-2-нитрофенил)-N-метилкарбаминовой кислоты, полученный в справочном примере 10, осуществляли процесс, аналогичный описанному в справочном примере 5, а затем очищали по методу колоночной хроматографии на силикагеле (элюирующий раствор: этилацетат/н-гексан = 1/2) с получением указанного в заголовке соединения.
1H-ЯМР(ДМСО-d6) δ: 1,30 и 1,37 (9H всего, с каждый), 1,44 (9H, с), 2,07 (6H, с), 2,98 (3H, с), 4,83 (2H, ушир.; исчезал вследствие добавления оксида дейтерия), 6,54 (2H, с), 6,58-6,74 (3H, м), 8,22 (1H, с; исчезал вследствие добавления оксида дейтерия).
ТЕСТОВЫЙ ПРИМЕР 1
Оценка ингибирования пролиферации раковых клеток
В тесте использовали приобретенную у Immuno-Biological Laboratories Co., Ltd. клеточную линию рака желудка человека (MKN74, MKN28) и клеточную линию рака молочной железы человека (ZR-75-1), клеточную линию мелкоклеточного рака легких (SBC-1), клеточную линию рака поджелудочной железы (AsPC-1), клеточную линию рака предстательной железы (DU-145), клеточную линию рака почки (ACHN), клеточную линию медуллобластомы (D341 Med), клеточную линию саркомы человека (клеточную линию рабдомиосаркомы А-204, клеточную линию саркомы Юинга RD-ES, клеточную линию липосаркомы SW872) и клеточную линию множественной миеломы (U266), которые приобретали у American Tissue Culture Collection. Клеточные линии MKN74, MKN28, ZR-75-1, SBC-1 и AsPC-1 выращивали с использованием среды RPMI (Invitrogen Corporation) с добавлением 10% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.), клеточную линию RD-ES выращивали с использованием среды RPMI (Invitrogen Corporation) с добавлением 15% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.), клеточную линию D341 Med выращивали с использованием среды МЕМα (Invitrogen Corporation) с добавлением 10% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.), клеточную линию ACHN выращивали с использованием среды Е-МЕМ (Invitrogen Corporation) с добавлением 10% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.), клеточную линию SW872 выращивали с использованием среды L15 (Invitrogen Corporation) с добавлением 10% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.), клеточную линию A-204 выращивали с использованием среды McCoy's 5A (Invitrogen Corporation) с добавлением 10% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.) и клеточную линию U266 выращивали с использованием среды RPMI (Invitrogen Corporation) с добавлением 0,5% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.) и 2 нг/мл IL-6 (Genzyme Corporation).
Клетки каждой линии высевали в 96-луночные культивационные микропланшеты (Nalge Nunc International K.K.) в количестве 1000-10000 клеток/лунка, и в то же самое время в каждую лунку добавляли соединение из примера получения 8, обладающее активирующим действием в отношении PPARγ (здесь и далее в настоящем описании называемое соединением X, и обозначаемое на фиг. 1, 2 и 3 как соединение X) и растворенное в диметилсульфоксиде (ДМСО, Dojindo Laboratories), так что концентрация ДМСО составляла 0,1%, а концентрация соединения X составляла 0,1, 1 или 10 мкМ (в случае клеточной линии AsPC-1 - 0,05, 0,5 или 5 мкМ). К контрольной группе добавляли только ДМСО, так что его концентрация составляла 0,1%. Затем выращивали клетки в культивационном микропланшете в присутствии 5% углекислого газа при 37°C в течение 7 суток. В случае клеточной линии U266 клетки выращивали в течение 4 суток. После завершения выращивания клеток в культивационную среду добавляли 50% раствор трихлоруксусной кислоты (Wako Pure Chemical Industries, Ltd.), так что ее конечная концентрация составляла 10%, и оставляли микропланшет отстаиваться при 4°C в течение 1 часа для иммобилизации клеток. Затем каждую лунку 5 раз промывали дистиллированной водой, затем в каждую лунку добавляли 100 мкл раствора 0,4% сульфородамина B (Molecular Probes) в 1% уксусной кислоте, и оставляли микропланшет отстаиваться в течение 30 минут для окрашивания клеток. Затем каждую лунку 5 раз промывали 1% раствором уксусной кислоты и сушили на воздухе. В каждую лунку с иммобилизированными и окрашенными клетками добавляли 10 мМ Трис в количестве 150 мл/лунка, и измеряли в каждой лунке поглощение при 490 нм с использованием микропланшетного ридера модели 3550 (Bio-Rad Laboratories, Inc.). Среднее значение поглощения в каждой из групп, клетки в которых были обработаны соответствующей концентрацией соединения X, выражали в процентах, принимая за 100% среднее значение поглощения в контрольной группе, клетки в которой обрабатывали ДМСО. Соответственно, оценивали ингибирование пролиферации раковых клеток соединением X.
Результаты представлены на фиг. 1, 2 и 3. На каждом графике ось ординат представляет собой поглощение [%], и ось абсцисс представляет собой концентрацию соединения X [мкМ].
Как видно на фиг. 1, 2 и 3, соединение X обладает значительной ингибирующей пролиферацию активностью в отношении всех раковых клеток. Соответственно, это убедительно указывает на возможность того, что соединение X обладает противораковой активностью в отношении клеток рака желудка человека, клеток рака молочной железы человека, мелкоклеточного рака легких, клеток рака поджелудочной железы, клеток рака предстательной железы, клеток рака почки, медуллобластомы, клеток саркомы человека (рабдомиосаркомы, саркомы Юинга и липосаркомы) и множественной миеломы.
Тестовый пример 2
Оценка ингибирования пролиферации клеток лейкоза человека
Ингибирование пролиферации клеток лейкоза человека описанным в примере получения 8 соединением, обладающим активирующим действием в отношении PPARγ (здесь и далее в настоящем описании называемым соединением X), изучали с использованием клеточных линий лейкоза человека HL-60 и THP-1, которые приобретали у American Tissue Culture Collection. Указанные клеточные линии выращивали с использованием среды RPMI (Invitrogen Corporation) с добавлением 10% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.). Клетки линии HL-60 и клетки линии THP-1 высевали в 96-луночные микропланшеты в количестве 2×103 клеток/лунка и в то же самое время добавляли растворенные в ДМСО соединения в различных концентрациях, так что концентрация ДМСО составляла 0,1%. Изучали эффект соединения X в концентрациях 10, 25 и 50 мкМ (n=4). После добавления соединения клетки выращивали в присутствии 5% углекислого газа при 37°C в течение 5 суток. Затем в каждую лунку добавляли Cell Titer 96 Aqueous One Solution Reagent (Promega Corp.) в количестве 40 мкл/лунка и инкубировали в течение 2 часов. В каждой лунке измеряли поглощение при 490 нм с использованием микропланшетного ридера модели 3550 (Bio-Rad Laboratories, Inc.). Среднее значение поглощения в каждой из групп, клетки в которых были обработаны соответствующей концентрацией соединения X, выражали в процентах, принимая за 100% среднее значение поглощения в контрольной группе, клетки в которой обрабатывали ДМСО. Соответственно, оценивали ингибирование пролиферации раковых клеток соединением Х путем вычитания полученного значения из 100%.
Результаты представлены в таблице 1.
Из представленных в таблице 1 результатов видно, что соединение Х значительно ингибирует пролиферативную активность в отношении клеток лейкоза человека.
ТЕСТОВЫЙ ПРИМЕР 3
Оценка противораковой активности in vivo в отношении клеток рака толстой кишки
Противораковую активность in vivo в отношении клеток рака толстой кишки исследовали с использованием описанного в примере получения 8 соединения, обладающего активирующим действием в отношении PPARγ (здесь и далее в настоящем описании называемым соединением X), и описанного в примере получения 1 соединения, обладающего активирующим действием в отношении PPARγ (здесь и далее в настоящем описании называемым соединением Y). В эксперименте линию клеток рака толстой кишки человека WiDr (приобретенную у American Type Culture Collection) с доказанным отсутствием патогенных для мышей микроорганизмов после карантина прививали подкожно в подмышечную область «голым» мышам линии BALB/cA Jcl-nu (CLEA Japan, Inc.) для субкультивирования опухоли. Опухоль делили на мелкие кусочки размером 5 мм и прививали подкожно в правую подмышечную область «голым» мышам линии BALB/cA Jcl-nu с использованием troker (CLEA Japan, Inc.). Требуемые количества соединений Х и У взвешивали и растворяли в N,N-диметилацетамиде (DMA, Wako Pure Chemical Industries, Ltd.), а затем к смеси в небольших количествах добавляли 5% раствор Emulphor 620 (GAF Corporation) в солевом растворе (Otsuka Pharmaceutical Co., Ltd.), так что концентрации соединений составляли 0,2, 1 или 5 мг/мл. Конечную концентрацию DMA корректировали до 2,5%. Начиная с суток, последующих после прививки опухоли, каждый из растворов вводимых соединений перорально вводили «голым» мышам с привитой опухолью WiDr при помощи зонда (Fuchigami Kikaiten) один раз в сутки 5 раз в неделю до 32 суток после прививки в количестве 0,1 мл на 10 г массы мыши. Дважды в неделю при помощи цифрового ручного микрометра (MAX-CAL МАХ-15, Nihon Sokutei Kougu Kabusiki Kaisha) измеряли длинную ось и короткую ось привитой опухоли и оценивали ингибирование пролиферации опухоли по следующей расчетной формуле и выражали в виде степени ингибирования объема опухоли.
Средний объем опухоли в каждой тестируемой группе (мм3) = 1/2 × (короткая ось)2 (мм2) × (длинная ось) (мм)
Степень ингибирования объема опухоли (%) = {1 - (средний объем опухоли в группе с введением лекарственного средства/средний объем опухоли в контрольной группе без введения лекарственного средства)} × 100
Оценку противоопухолевой активности в каждой группе с введением лекарственного средства проводили по степени ингибирования объема опухоли. Кроме того, через 38 суток после прививки опухоли путем оценки критерия Стьюдента определяли статистическое различие объема опухоли в контрольной группе без введения лекарственного средства и в группе с введением лекарственного средства. Так было обнаружено, что при значении p<0,05 между двумя группами существует достоверное различие.
Результаты представлены в таблице 2.
Из представленных в таблице 2 результатов очевидно, что соединения X и Y обладают значительной противоопухолевой активностью in vivo в отношении линии клеток рака толстой кишки человека.
Тестовый пример 4
Оценка ингибирования пролиферации in vitro клеток линии немелкоклеточного рака легких путем сочетанного введения соединения, обладающего активирующим действием в отношении PPARγ, и ингибитора рецептора эпидермального фактора роста (EGFR)
Эффекты сочетанного введения соединения, описанного в примере получения 8 и обладающего активирующим действием в отношении PPARγ (здесь и далее в настоящем описании называемого соединением X), и ингибитора рецептора эпидермального фактора роста (EGFR) изучали на клетках линии A549 немелкоклеточного рака легких человека, используя в качестве индикатора ингибирование пролиферации клеток in vitro.
Клетки линии A549 немелкоклеточного рака легких человека (приобретенные у American Tissue Culture Collection) выращивали с использованием среды RPMI (Invitrogen Corporation) с добавлением 10% фетальной бычьей сыворотки (Hyclone Laboratories, Inc.). Клетки линии A549 высевали в 96-луночные микропланшеты в количестве 5×102 клеток/лунка, и в то же самое время добавляли растворенные в ДМСО соединения в различных концентрациях, так что концентрация ДМСО составляла 0,1%. Соединение X изучали в концентрации 10 мкМ, а используемый в качестве ингибитора EGFR гефитиниб (синтезированный Sankyo Company, Limited) - в двух концентрациях, 0,1 и 0,5 мкМ (n=4). После добавления лекарственных средств клетки выращивали в присутствии 5% углекислого газа при 37°C в течение 7 суток. Затем в каждую лунку добавляли 40 мкл Cell Titer 96 Aqueous One Solution Reagent (Promega Corp.), а затем выращивали клетки в течение 2 часов. В каждой лунке измеряли поглощение при 490 нм с использованием микропланшетного ридера (Bio-Rad Laboratories, Inc.). Среднее значение поглощения в каждой из групп, клетки в которых были обработаны соединением X и гефитинибом в соответствующих концентрациях, выражали в процентах, принимая за 100% среднее значение поглощения в необработанной лекарственными средствами контрольной группе (клетки обрабатывали только ДМСО). Соответственно, оценивали ингибирование пролиферации раковых клеток соединением X, гефитинибом и их сочетанием путем вычитания полученного значения из 100%.
Результаты представлены в таблице 3.
Из представленных в таблице 3 результатов видно, что лечение только соединением X (10 мкМ) приводит к ингибированию пролиферации на 12%, а лечение только гефитинибом (0,5 мкМ) приводит к ингибированию пролиферации на 22%; тем не менее, сочетанное введение обоих лекарственных средств приводит к ингибированию пролиферации на 50%. Из представленных результатов видно, что при сочетанном введении соединение X и ингибитор рецептора эпидермального фактора роста (EGFR) проявляют синергическое воздействие, ингибируя пролиферацию раковых клеток.
Тестовый пример 5
Оценка противораковой активности in vivo в отношении клеток немелкоклеточного рака легких
Противораковую активность in vivo в отношении клеток немелкоклеточного рака легких исследовали для соединения, описанного в примере получения 8 (здесь и далее в настоящем описании называемого соединением X).
В эксперименте линию клеток немелкоклеточного рака легких человека A549 (приобретенную у American Type Culture Collection) с доказанным отсутствием патогенных для мышей микроорганизмов после карантина прививали подкожно в подмышечную область «голым» мышам линии BALB/cA Jcl-nu (CLEA Japan, Inc.) для субкультивирования опухоли. Опухоль делили на мелкие кусочки размером 5 мм и прививали подкожно в правую подмышечную область «голым» мышам линии BALB/cA Jcl-nu с использованием troker (CLEA Japan, Inc.). Требуемые количества соединения X и используемого в качестве ингибитора EGFR гефитиниба (синтезированного Sankyo Company, Limited) взвешивали и приготавливали растворы путем суспендирования соединения, описанного в примере получения 8 и обладающего активирующим действием в отношении PPARγ (здесь и далее в настоящем описании называемого соединением X), в 0,5% растворе метилцеллюлозы с получением конечной концентрации 1 мг/мл, и гефитиниба в 0,05% растворе Tween 80 (Tokyo Chemical Industry Co., Ltd.) с получением конечной концентрации 10 мг/мл. Начиная с 14-х суток после прививки опухоли, каждый из растворов вводимых соединений перорально вводили «голым» мышам с привитой опухолью А549 при помощи зонда (Fuchigami Kikaiten) один раз в сутки 5 раз в неделю до 60-х суток после прививки в количестве 0,1 мл на 10 г массы мыши. Дважды в неделю при помощи цифрового ручного микрометра (MAX-CAL МАХ-15, Nihon Sokutei Kougu Kabusiki Kaisha) измеряли длинную ось и короткую ось привитой опухоли и оценивали ингибирование пролиферации опухоли по следующей расчетной формуле и выражали в виде степени ингибирования объема опухоли.
Средний объем опухоли в каждой тестируемой группе (мм3) = 1/2 × (короткая ось)2 (мм2) × (длинная ось) (мм)
Степень ингибирования объема опухоли (%) = {1 - (средний объем опухоли в группе с введением лекарственного средства/средний объем опухоли в контрольной группе без введения лекарственного средства)} × 100
Оценку противоопухолевой активности в каждой группе с введением лекарственного средства проводили по степени ингибирования объема опухоли. Кроме того, через 63 суток после прививки опухоли путем оценки критерия Стьюдента определяли статистическое различие объема опухоли в контрольной группе без введения лекарственного средства и в группе с введением лекарственного средства. Так было обнаружено, что при значении p<0,05 между двумя группами существует достоверное различие.
Результаты представлены на фиг.4.
Как видно на фиг.4, соединение X обладает значительной противораковой активностью, ингибируя объем опухоли на 33%. Кроме того, гефитиниб обладал значительной противораковой активностью, ингибируя объем опухоли на 56%. С другой стороны, если соединение X и гефитиниб вводили сочетанно, то по сравнению с группами, в которых вводили только соединение X и только гефитиниб, наблюдалось значимо различимое усиление противораковой активности с ингибированием объема опухоли на 73%. Из представленных результатов видно, что соединение X обладает противораковой активностью в отношении немелкоклеточного рака легких человека, и что его противораковая активность может быть усилена путем его сочетанного введения с ингибитором рецептора эпидермального фактора роста (EGFR).
Тестовый пример 6
Оценка ингибирования пролиферации in vitro клеток линии немелкоклеточного рака легких путем сочетанного введения соединения, обладающего активирующим действием в отношении PPARγ, и ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и Raf-киназы
Изучали эффекты сочетанного введения соединения, описанного в примере получения 8 и обладающего активирующим действием в отношении PPARγ (здесь и далее в настоящем описании называемого соединением X), и ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и Raf-киназы в отношении клеточной линии немелкоклеточного рака легких человека, используя в качестве индикатора ингибирование пролиферации клеток in vitro.
Клетки линии A549 немелкоклеточного рака легких человека высевали в 96-луночные микропланшеты в количестве 5×102 клеток/лунка и в то же самое время добавляли растворенные в ДМСО соединения в различных концентрациях, так что концентрация ДМСО составляла 0,1%. Соединение X изучали в концентрации 10 мкМ, а используемый в качестве ингибитора VEGFR и Raf-киназы сорафениб (синтезированный Sankyo Company, Limited) - в концентрации 5 мкМ (n=4). После добавления лекарственных средств клетки выращивали в присутствии 5% углекислого газа при 37°C в течение 6 суток. Затем в каждую лунку добавляли 40 мкл Cell Titer 96 Aqueous One Solution Reagent (Promega Corp.), а затем измеряли в каждой лунке поглощение при 490 нм с использованием микропланшетного ридера (Bio-Rad Laboratories, Inc.). Среднее значение поглощения в каждой из групп, клетки в которых были обработаны соединением X и сорафенибом в соответствующих концентрациях, выражали в процентах, принимая за 100% среднее значение поглощения в необработанной лекарственными средствами контрольной группе (клетки обрабатывали только ДМСО). Соответственно, оценивали ингибирование пролиферации раковых клеток соединением X, сорафенибом и их сочетанием путем вычитания полученного значения из 100%.
Результаты представлены в таблице 4.
Из представленных в таблице 3 результатов видно, что лечение только соединением X (10 мкМ) приводит к ингибированию пролиферации на 15%, а лечение только сорафенибом (5 мкМ) приводит к ингибированию пролиферации на 47%; тем не менее, сочетанное введение обоих лекарственных средств приводит к ингибированию пролиферации на 71%. Так, было показано статистическое различие между ингибированием пролиферации в группе с сочетанным введением лекарственных средств по сравнению с группами с введением только одного лекарственного средства. Из представленных результатов видно, что при сочетанном введении соединение X и ингибитор рецептора фактора роста эндотелия сосудов (VEGFR) и Raf-киназы проявляют синергическое воздействие, ингибируя пролиферацию раковых клеток.
Тестовый пример 7
Оценка противораковой активности in vivo в отношении клеток рака почки человека путем сочетанного введения соединения, обладающего активирующим действием в отношении PPARγ, и ингибитора рецептора фактора роста эндотелия сосудов (VEGFR) и Raf-киназы
Противораковую активность in vivo в отношении клеток рака почки человека исследовали для соединения, описанного в примере получения 8, обладающего активирующим действием в отношении PPARγ (здесь и далее в настоящем описании называемого соединением X).
В эксперименте линию клеток немелкоклеточного рака легких человека SN12-PM6 (предоставленную профессором Seiji Naito из Kyushu University) с доказанным отсутствием патогенных для мышей микроорганизмов после карантина прививали подкожно в подмышечную область «голым» мышам линии BALB/cA Jcl-nu (CLEA Japan, Inc.) для субкультивирования опухоли. Опухоль делили на мелкие кусочки размером 5 мм и прививали подкожно в правую подмышечную область «голым» мышам линии BALB/cA Jcl-nu с использованием troker (CLEA Japan, Inc.). Требуемые количества соединения X и используемого в качестве ингибитора VEGFR и Raf-киназы сорафениба (синтезированного Sankyo Company, Limited) взвешивали, и приготавливали растворы путем суспендирования соединения X в 0,5% растворе метилцеллюлозы, а сорафениба - в смеси (50%/50%) этанола (Kanto Chemical Co., Inc.) и кремофора (Sigma). Затем добавляли дистиллированную воду (Otsuka Pharmaceutical Factory, Inc.), так что конечные концентрации этанола и кремофора составляли 12,5%. Приготавливали растворы, так что конечные концентрации соединения X и сорафениба составляли 0,3 мг/мл и 10 мг/мл, соответственно. Начиная с 10-х суток после прививки опухоли, каждый из растворов вводимых соединений орально вводили «голым» мышам с привитой опухолью SN12-PM6 при помощи зонда (Fuchigami Kikaiten) один раз в сутки 5 раз в неделю до 56-х суток после прививки в количестве 0,1 мл на 10 г массы мыши. Дважды в неделю при помощи цифрового ручного микрометра (MAX-CAL МАХ-15, Nihon Sokutei Kougu Kabusikikaisha) измеряли длинную ось и короткую ось привитой опухоли, и оценивали ингибирование пролиферации опухоли по следующей расчетной формул и выражали в виде степени ингибирования объема опухоли.
Средний объем опухоли в каждой тестируемой группе (мм3) = 1/2 × (короткая ось)2 (мм2) × (длинная ось) (мм)
Степень ингибирования объема опухоли (%)={1 - (средний объем опухоли в группе с введением лекарственного средства/средний объем опухоли в контрольной группе без введения лекарственного средства)} × 100
Оценку противоопухолевой активности в каждой группе с введением лекарственного средства проводили по степени ингибирования объема опухоли. Кроме того, через 59 суток после прививки опухоли путем оценки критерия Стьюдента определяли статистическое различие объема опухоли в контрольной группе без введения лекарственного средства и в группе с введением лекарственного средства. Так было обнаружено, что при значении p<0,05 между двумя группами существует достоверное различие.
Результаты представлены на фиг.5.
Как видно на фиг.5, введение только соединения Х приводило к значительному ингибированию пролиферации клеток рака почки человека SN12-PM6 (степень ингибирования объема опухоли 37%). Кроме того, введение только сорафениба также приводило к значительному ингибированию пролиферации (степень ингибирования объема опухоли 43%). С другой стороны, если соединение Х и сорафениб вводили сочетанно, то по сравнению с группами, в которых вводили только соединение X и только сорафениб, наблюдалось значимо различимое усиление противораковой активности с ингибированием объема опухоли на 65%. Из представленных результатов очевидно, что соединение X обладает противораковой активностью в отношении рака почки человека и что его противораковая активность может быть усилена путем его введения вместе с ингибитором рецептора фактора роста эндотелия сосудов (VEGFR) и Raf-киназы.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 - на графике представлена зависимость ингибирования пролиферации раковых клеток от концентрации соединения X, где по оси ординат отложено поглощение (%) в каждой культуре клеток после добавления к каждой культуре раковых клеток соединения X, а по оси абсцисс - концентрация (мкМ) добавленного соединения X.
Фиг.2 - на графике представлена зависимость ингибирования пролиферации раковых клеток от концентрации соединения X, где по оси ординат отложено поглощение (%) в каждой культуре клеток после добавления к каждой культуре раковых клеток соединения X, а по оси абсцисс - концентрация (мкМ) добавленного соединения X.
Фиг.3 - на графике представлена зависимость ингибирования пролиферации раковых клеток от концентрации соединения X, где по оси ординат отложено поглощение (%) в каждой культуре клеток после добавления к каждой культуре раковых клеток соединения X, а по оси абсцисс - концентрация (мкМ) добавленного соединения X.
Фиг.4 - на графике представлена зависимость объема опухоли от времени после трансплантации, где по оси ординат отложен объем опухоли (мм3) для случаев, когда против трансплантированных раковых клеток использовали только соединение X, только гефитиниб или сочетание обоих лекарственных средств, а по оси абсцисс - время после трансплантации (в сутках).
Фиг.5 - на графике представлена зависимость объема опухоли от времени после трансплантации, где по оси ординат отложен объем опухоли (мм3) для случаев, когда против трансплантированных раковых клеток использовали только соединение X, только сорафениб или сочетание обоих лекарственных средств, а по оси абсцисс - время после трансплантации (в сутках).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ ТИАЗОЛИДИНДИОНА СО СНИЖЕННЫМ СРОДСТВОМ К PPAR ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ | 2010 |
|
RU2564661C2 |
| НОВОЕ СОЕДИНЕНИЕ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПОВЫШЕНИЯ ПРОТИВОРАКОВОЙ АКТИВНОСТИ | 2019 |
|
RU2781282C1 |
| РАПП-ПОДДЕРЖИВАЮЩИЕ ТИАЗОЛИДИНДИОНЫ И ИХ КОМБИНАЦИИ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2010 |
|
RU2570424C2 |
| АНТИДИАБЕТИЧЕСКИЕ ОКСАЗОЛИДИНДИОНЫ | 2005 |
|
RU2355682C2 |
| СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ НЕОПЛАЗМ | 2000 |
|
RU2219928C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА СОЕДИНЕНИЙ ТИАЗОЛИДИНДИОНА | 2011 |
|
RU2593370C2 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНИЛАНТРАЦИКЛИНА | 2015 |
|
RU2715902C2 |
| ПРОИЗВОДНОЕ ТИОФЕНА И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2781643C2 |
| НОВОЕ ПИПЕРИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581834C1 |
| СИНТЕЗ СОЕДИНЕНИЙ ТИАЗОЛИДИНДИОНА | 2011 |
|
RU2594725C2 |

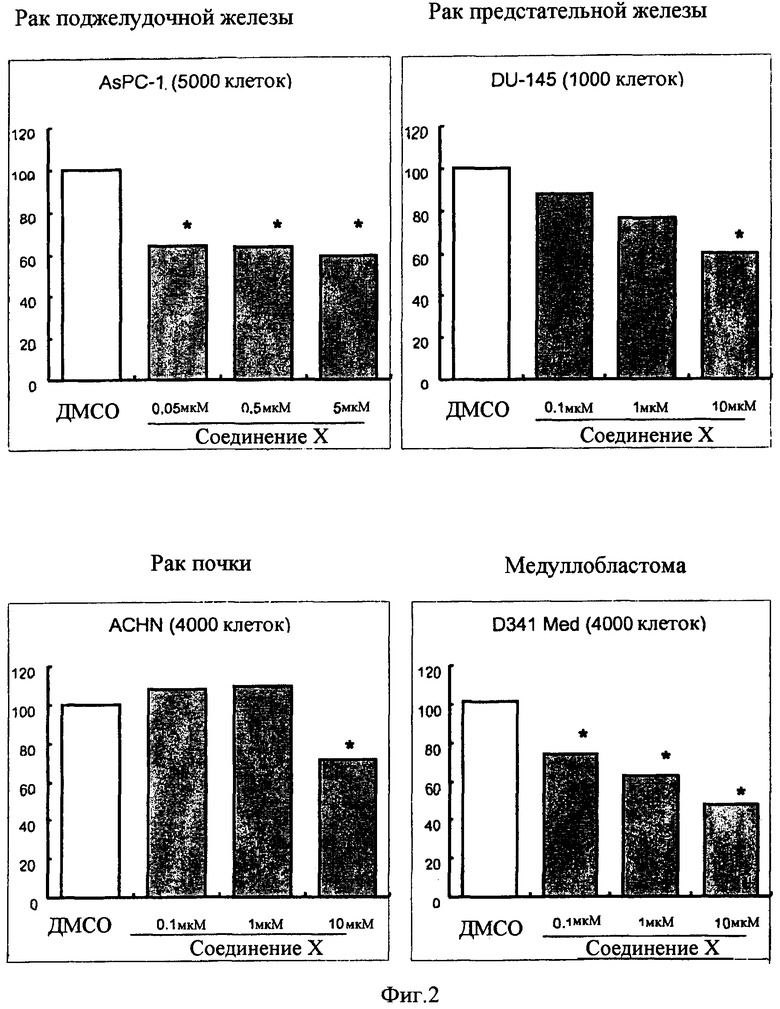
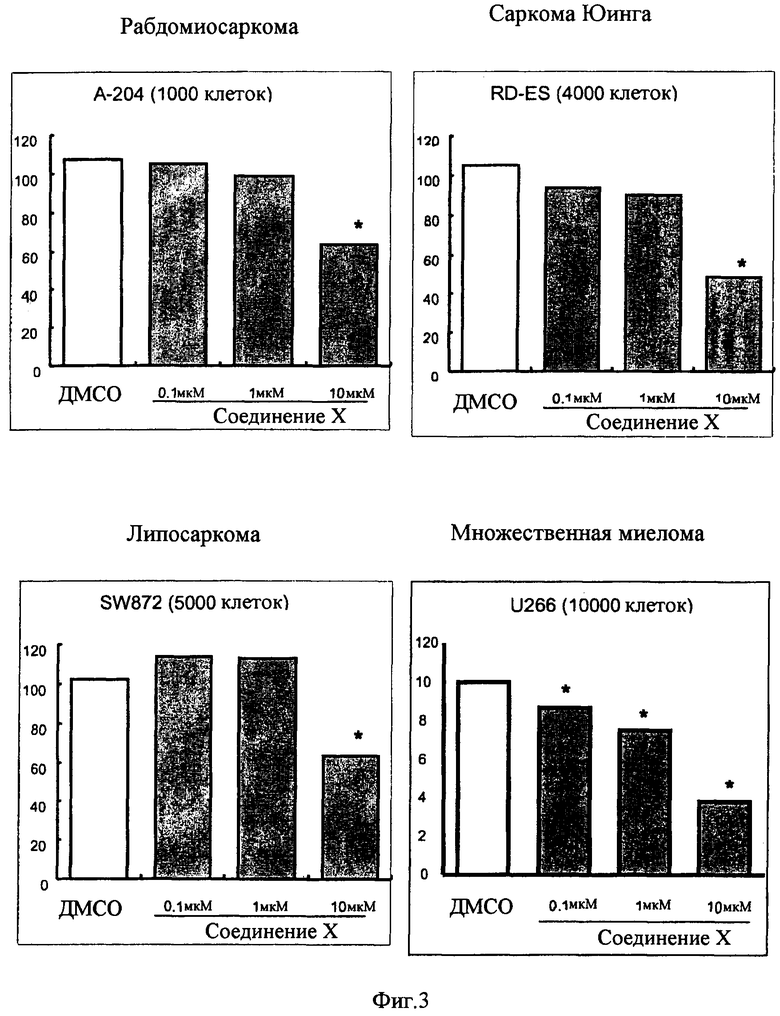


Изобретение относится к области фармацевтики и медицины и касается противораковой фармацевтической композиции или комбинации для профилактики или рака легкого, или почки, содержащая в качестве активных ингредиентов лекарственное средство, выбранное из гефитиниба, эрлотиниба и сорафениба, и дигидрохлорид 5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-дион. Изобретение обеспечивает повышенную эффективность лечения. 2 н. и 14 з.п. ф-лы, 5 ил., 4 табл.
1. Противораковая фармацевтическая композиция для профилактики или лечения рака легкого или почки, содержащая в качестве активных ингредиентов,
по меньшей мере, одно противораковое лекарственное средство, выбранное из группы, состоящей из гефитиниба, эрлотиниба и сорафениба; и
дигидрохлорид 5-(4-(6-(4-амино-З,5-диметилфенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона или его гидрат.
2. Фармацевтическая композиция по п.1, в которой противораковое лекарственное средство выбирают, по меньшей мере, из группы, состоящей из гефитиниба и сорафениба.
3. Фармацевтическая композиция по п.1, предназначенная для профилактики или лечения рака легкого.
4. Фармацевтическая композиция по п.3, где противораковое лекарственное средство является гефитинибом.
5. Фармацевтическая композиция по п.3, где противораковое лекарственное средство является сорафенибом.
6. Фармацевтическая композиция по п.3, где противораковое лекарственное средство является ерлотинибом.
7. Фармацевтическая композиция по п.1, для профилактики или лечения рака почки, где противораковое лекарственное средство является сорафенибом.
8. Фармацевтическая композиция по п.1, включающая дигидрохлорид 5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона.
9. Противораковая фармацевтическая комбинация для профилактики или лечения рака легкого или почки, содержащая в качестве активных ингредиентов,
по меньшей мере, одно противораковое лекарственное средство, выбранное из группы, состоящей из гефитиниба, эрлотиниба и сорафениба; и
дигидрохлорид 5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона или его гидрат, причем активные ингредиенты вводят одновременно или раздельно в различные времена.
10. Фармацевтическая комбинация по п.9, в которой противораковое лекарственное средство выбирают, по меньшей мере, из группы, состоящей из гефитиниба и сорафениба.
11. Фармацевтическая комбинация по п.9, предназначенная для профилактики или лечения рака легкого.
12. Фармацевтическая комбинация по п.11, где противораковое лекарственное средство является гефитинибом.
13. Фармацевтическая комбинация по п.11, где противораковое лекарственное средство является сорафенибом.
14. Фармацевтическая комбинация по п.11, где противораковое лекарственное средство является ерлотинибом.
15. Фармацевтическая комбинация по п.9, для профилактики или лечения рака почки, где противораковое лекарственное средство является сорафенибом.
16. Фармацевтическая комбинация по п.9, включающая дигидрохлорид 5-(4-(6-(4-амино-3,5-диметилфенокси)-1-метил-1Н-бензимидазол-2-илметокси)бензил)тиазолидин-2,4-диона.
| US 6432993 B1, 13.08.2002 | |||
| Способ получения замещенных тиазолидиниловых эфиров фосфорной кислоты | 1984 |
|
SU1318168A3 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

