ОПИСАНИЕ
Изобретение относится к способу получения карведилола. Более конкретно, настоящее изобретение относится к способу эффективного получения хирального карведилола.
Большинство медицинских препаратов, которые недавно были разработаны и являются коммерчески доступными, представляют собой хиральные продукты. Хиральность сопровождается побочными эффектами или сниженными эффективностями, проявляемыми рацемическими лекарственными средствами. Следовательно, для того, чтобы увеличить как безопасность, так и эффективность, были предприняты многочисленные исследования для разработки хиральных лекарственных средств, содержащих оптически чистые стереоизомеры. В хиральных лекарственных средствах высокая химическая чистота и высокая оптическая чистота требуются для гарантирования безопасности и эффективности.
Карведилол (название по номенклатуре IUPAC: 1-(9Н-карбазол-4-илокси)-3-[[2-(2-метоксифенокси)этил]амино]-2-пропанол) представляет собой соединение, имеющее формулу 1:
Формула 1
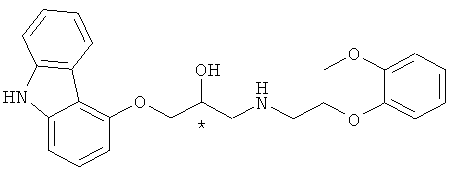
где, * обозначает хиральный центр.
Как показано в формуле 1, карведилол имеет один хиральный центр и может существовать или в виде (R)-изомера или (S)-изомера. Здесь, в качестве блокатора б1-андренорецептора, (R)-изомер и (S)-изомер проявляют почти одинаковые активности. В противоположность этому, в качестве блокатора в1-андренорецепторов, (S)-изомер имеет усиленную, превосходную активность по отношению к (R)-изомеру [ЕР 127,099; Chirality 1989, 1, 265; J. Pharm. Exp. Titer., 1992, 263, 92; Clin. Pharmacokin., 1994, 26, 335; Cardiovasc. Res., 1994, 28, 400; J. of Chromatography В. 1996, 682, 349}. Далее, карведилол в настоящее время применяется как антиоксидант, антивоспалительный агент, агент с анти-апоптическим действием [The American Journal of Cardiology, 2004, 93(9A), 3В]. По этим причинам, предоставление способа эффективного получения высоко оптически чистого хирального карведилола экономичным способом является важной задачей в разработке различных лекарственных средств, включающих в себя хиральный карведилол.
Традиционные способы получения хирального карведилола показаны на схеме реакции 1:
Схема реакции 1
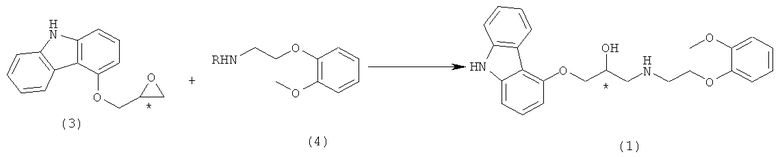
в которой R представляет собой атом водорода или бензильную группу.
Как показано на схеме реакции 1, целевой карведилол получали путем раскрытия цикла хирального эпокси-карбазола формулы 3 с этиламинным соединением формулы 4 [R = водород, патенты США №№ 4503067 и 4697022]. Однако способ ведет к образованию бис-замещенного побочного продукта, который не с трудом удаляется при очистке. Способ требует весьма сложной методики очистки, таким образом, затрудняя получение высоко оптически чистого карведилола. Кроме того, потеря целевого карведилола является неизбежной при очистке, что приводит к значительному уменьшению выхода карведилола.
Для того чтобы преодолеть или избежать недостатков, возникающих из-за образования бис-замещенного побочного продукта, предпринимались различные исследования. Для того чтобы предотвратить образование бис-замещенного побочного продукта, N-защищенное соединение формулы 4 (R = бензил) применяли в качестве исходного вещества в схеме реакции 1 и использовали для раскрытия цикла хирального эпокси-соединения формулы 3 [R = бензил, EP 918055]. Способ приводит к отсутствию бис-замещенного побочного продукта. Однако недостатком этого способа является то, что должен применяться дорогостоящий палладиевый катализатор для удаления бензильной группы.
В качестве альтернативы, этиламинное соединение формулы 4 применяли в избытке по сравнению с соединением формулы 3 для уменьшения образования бис-замещенного побочного продукта [R = водород, WO 02/00216]. Даже несмотря на то, что способ может уменьшать образование бис-замещенного побочного продукта, по-прежнему остаются проблемы, связанные с присутствием небольшого количества бис-замещенного побочного продукта. Кроме того, недостатком способа является применение избытка дорогостоящего этиламинного соединения, что повышает его стоимость.
Кроме того, этиламинное соединение формулы 4 или его бензилированная форма претерпевает разложение при воздействии воздуха и света. Следовательно, соединение формулы 4 имеет ограничение для применения в массовом производстве. Для того чтобы преодолеть данное затруднение, в качестве исходного вещества в реакции раскрытия цикла с соединением формулы 3 для увеличения стабильности [WO 2004/041783] применяли соль присоединения кислоты этиламинного соединения формулы 4 (R = водород). Преимущество способа состоит в возможности применения его в массовом производстве хирального карведилола. Однако его недостатком является образование бис-замещенного побочного продукта и избыточное применение этиламинного соединения.
Была предпринята новая попытка для того, чтобы избежать образования бис-замещенного побочного продукта.
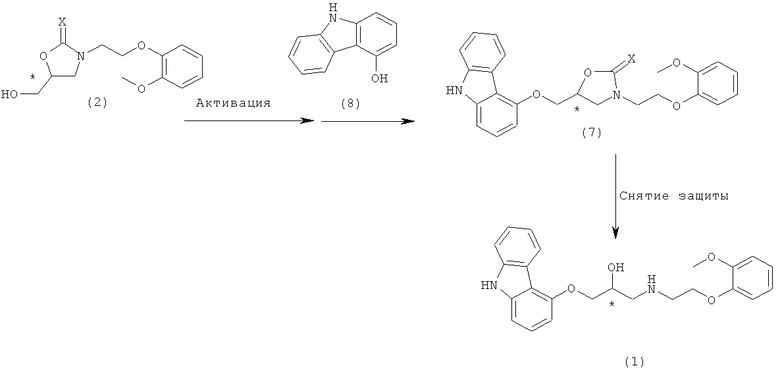
Аминогруппу соединения формулы 4 сначала защищали бензильной группой, и туда же вводили хлорпропанонильную группу. Полученный продукт алкилировали 9Н-4-гидроксикарбазолом формулы 8, затем проводили стадию восстановления и стадию дебензилирования для получения карведилола (опубликованный патент Кореи №2005-0003764). Однако способ требует сильного восстанавливающего агента, такого как боргидрид натрия или боргидрид лития и дорогостоящего палладиевого катализатора.
Следующий способ получения карведилола, известный в технике, включает в себя реакцию аминного соединения формулы 4 с карбонатным соединением для получения карбамата, имеющего две уходящие группы, с последующей реакцией циклизации для получения оксазолидинонового соединения, которое применяется в качестве промежуточного соединения для синтеза карведилола [EP 1282601 и 1367052]. В этом способе для получения карведилола, оксазолидиноновое соединение алкилировали 9H-4-гидроксикарбазолом и удаляли защитную группу.
Несмотря на то что эти два упомянутых способа эффективно снижают образование бис-замещенного побочного продукта, они не пригодны для получения хирального карведилола.
Как упомянуто выше, традиционные способы имеют одну или несколько нерешенных технических проблем для применения в массовом производстве высоко оптически чистого хирального карведилола. Следовательно, в настоящее время настоятельно необходим эффективный способ получения высоко оптически чистого хирального карведилола.
Согласно исчерпывающим исследованиям авторов настоящего изобретения было найдено, что эффективное получение высоко оптически чистого ключевого промежуточного соединения и предоставление эффективных путей химического синтеза являются решающими для эффективного массового производства хирального карведилола.
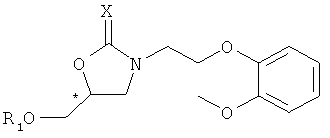
В результате интенсивных исследований авторов изобретения, предоставлен способ получения хирального карведилола, в котором хиральный оксазолидинон, имеющий формулу 2, применяется в качестве ключевого промежуточного продукта. Ключевой промежуточный продукт формулы 2 может быть эффективно синтезирован путем взаимодействия производного высоко оптически чистого хирального глицидола с N-защищенным 2-(2-метоксифенокси)этиламином, оба из которых являются коммерчески доступными и производятся в промышленном масштабе.
Формула 2
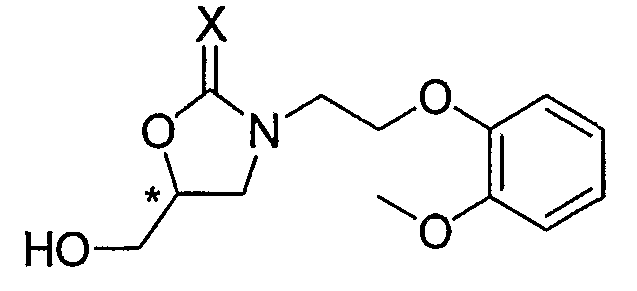
где, * обозначает хиральный центр, и X представляет собой кислород или серу.
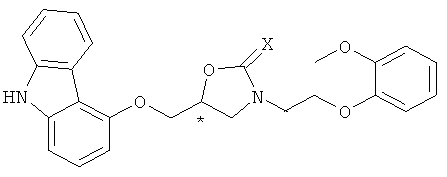
Получение хирального карведилола из ключевого промежуточного соединения формулы 2 включает в себя a) взаимодействие соединения формулы 2 с галогенирующим агентом, сульфонилирующим агентом или реагентом Мицунобу для активации гидрокси-группы соединения формулы 2, с последующей реакцией нуклеофильного замещения с 9H-4-гидроксикарбазолом для получения соединения формулы 7; и b) снятие защиты с полученного соединения формулы 7 в присутствии неорганического основания для получения целевого хирального карведилола.
Конкретно, соединение формулы 2 получают реакцией раскрытия цикла и последующей in situ реакцией внутримолекулярной циклизации между аминным соединением формулы 4 и хиральным глицидолом формулы 5, с последующей реакцией снятия защиты, и затем хиральный карведилол с высокой оптической чистотой образуется из соединения формулы 2. Здесь аминное соединение формулы 4 эффективно предотвращает образование бис-замещенного побочного продукта и продуцирует оксазолидин-2-он или оксазолидин-2-тион формулы 2 посредством реакции раскрытия цикла с соединением формулы 5 и последующей in situ реакцией внутримолекулярной циклизации. Затем, гидроксильная группа соединения формулы 2, полученная таким образом, активируется с помощью галогенирующего агента, сульфонилирующего агента или реагента Мицунобу, с последующей реакцией нуклеофильного замещения с 9H-4-гидроксикарбазолом для получения соединения формулы 7 (5-(9H-карбазол-4-илоксиметил)-3-[2-(2-метоксифенокси)этил]оксазолидин-2-он или (5-(9H-карбазол-4-илоксиметил)-3-[2-(2-метоксифенокси)этил]оксазолидин-2-тион). Соединение формулы 7 раскрывает цикл в присутствии неорганического основания для получения хирального карведилола формулы 1.
Способ по настоящему изобретению являются безопасным и промышленно применимым и предоставляет хиральный карведилол в высоко оптически чистой форме.
Настоящее изобретение относится к способу для эффективного получения хирального карведилола, включающему a) взаимодействие соединения формулы 2 с галогенирующим агентом, сульфонилирующим агентом или реагентом Мицунобу для активации гидроксильной группы соединения формулы 2, с последующей реакцией нуклеофильного замещения с 9H-4-гидроксикарбазолом для получения соединения формулы 7, и b) снятие защиты с полученного соединения формулы 7 в присутствии неорганического основания для получения целевого хирального карведилола. Способ обобщен на схеме реакции 2:
Схема реакции 2
На схеме реакции 2, * обозначает хиральный центр, и X представляет собой кислород или серу.
Как показано на схеме реакции 2, в настоящем изобретении в качестве хирального ключевого промежуточного продукта используются хиральный оксазолидин-2-он или оксазолидин-2-тион, имеющие формулу 2, в которой атом азота замещен 2-(2-метоксифенокси)этильной группой.
Ключевой промежуточный продукт образуется путем раскрытия цикла хирального глицидольного соединения формулы 5 под действием аминного соединения формулы 4 и последующей in situ внутримолекулярной циклизацией, продолженной затем снятием защиты с гидроки-группы соединения формулы 6.
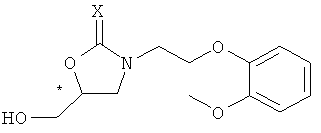
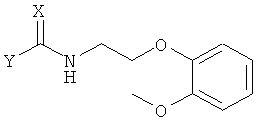
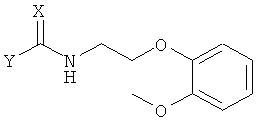
Ниже представлен конкретный пример аминного соединения формулы 4, применяемого при получении хирального ключевого промежуточного соединения формулы 2:
Формула 4
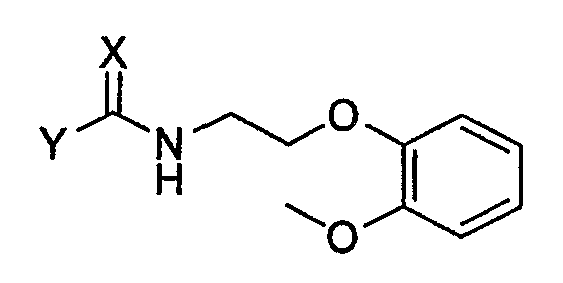
в которой X представляет собой кислород или серу, и Y обозначает уходящую группу.
Соединение формулы 4 может быть получено известной методикой с применением соответствующего первичного аминного соединения. В частности, если X представляет собой атом кислорода, оно может быть получено путем реакции первичного аминного соединения с карбоновой кислотой, эфиром карбоновой кислоты, соединением карбонилгалогенида, ангидридом карбоновой кислоты или галогенформиатом.
Когда X представляет собой атом серы, соединение формулы 4 может быть получено путем реакции первичного аминного соединения с тиоэфиром карбоновой кислоты [J. Chem. Soc. C, 1969, 2631; Chem. Ber. 1971, 104, 3146], или его соответствующим изотиоцианатом [Chem. Ber. 1914, 47, 1255; J. Am. Chem. Soc., 1968, 90, 6008; J. Chem. Eng. Data, 1980, 25, 176]. Предпочтительными примерами уходящей группы Y являются C1-C10-алкокси-группа, C6-C10-арилокси-группа, аллилокси-группа, C7-C14-алкиларилокси-группа.
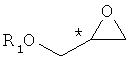
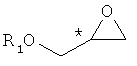
Конкретный пример хирального глицидольного соединения формулы 5, применяемого при получении хирального ключевого промежуточного соединения формулы 2, представлен ниже:
Формула 5
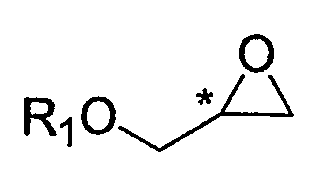
В формуле 5, * обозначает хиральный центр, и R1 представляет собой защитную группу для гидрокси-группы. Предпочтительные примеры R1 включают в себя C1-C10-алкильную группу, C2-C10-алкенильную группу, C2-C10-алкинильную группу, C1-C10-алкокси группу, (C1-C10)-алкокси-карбонильную группу, C6-C10-арильную группу, C3-C10-циклоалкильную группу, C4-C10-циклоалкенильную группу, гетероциклическую или полициклическую группу, C2-C10-карбонильную группу, C2-C10-карбоксильную группу, силильную группу, группу простого эфира, тиоэфирную группу, селеноэфирную группу, кетонную группу, альдегидную группу, сложноэфирную группу, фосфорильную группу, фосфонатную группу, фосфиновую группу, сульфонильную группу или -(CH2)k-R2 (где R2 представляет собой C2-C10-алкенильную группу, C2-C10-алкинильную группу, C1-C10-алкокси группу, (C1-C10)-алкоксикарбонильную группу, C6-C10-арильную группу, C3-C10-циклоалкильную группу, C4-C10-циклоалкенильную группу, гетероциклическую или полициклическую группу, C2-C10-карбонильную группу, C2-C10-карбоксильную группу, силильную группу, группу простого эфира, тиоэфирную группу, селеноэфирную группу, кетонную группу, альдегидную группу, сложноэфирную группу, фосфорильную группу, фосфонатную группу, фосфиновую группу, сульфонильную группу, и k представляет собой целое число от 1 до 8).
Хиральный глицидол формулы 5 является коммерчески доступным и может быть легко получен по известной методике. Конкретно, хиральный глицидол может быть получен путем реакции асимметрического эпоксидирования аллильного спирта [патенты США №№ 4946974, 5153338 и 5344947], из хирального 3-хлорпропандиола [JP 7-165743, США 5965753 и 2248635 и DE 1226554], или путем асимметрической каталитической реакции с применением фермента или металлического катализатора [J. Am. Chem. Soc. 1984, 106, 7250; Tetrahedron Asymmetry 1991, 2, 481; Enzyme Microb. Technol. 1991, 13, 306; Biotech.Tech, 1998, 12, 225; Tetrahedron 1994, 40, 8885; Biotech. Bioeng, 1996, 49, 70; Acta Chem, Scand. 1996, 50, 249; Tetrahedron Asymmetry 1997, 8, 639; Biotech. Tech, 1998, 12, 225; US 6720434; WO 01/89690; JP 2003-534117; EP 289655; US 2004-0054201; JP 2004-515356; WO 02/48162, KR 2002-01219; US 6262278; US 6448414; US 6693236; US 6800766 и WO 00/09463].
Получение хирального оксазолидин-2-она или оксазолидин-2-тиона формулы 2, ключевого промежуточного соединения для синтеза хирального карведилола, из соединения формулы 4 и соединения формулы 5, обобщено на схеме реакции 3:
Схема реакции 3
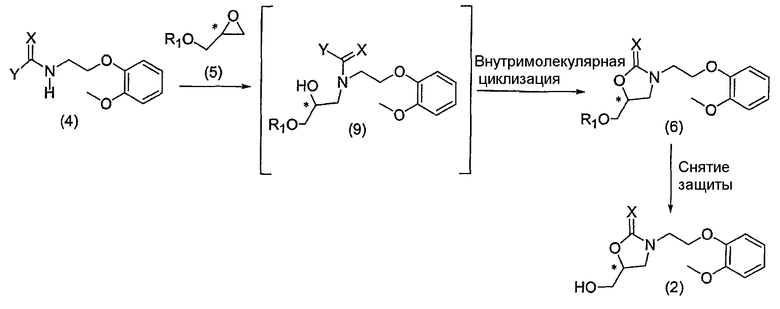
На схеме реакции 3, * обозначает хиральный центр, и X, Y и R1 определены выше.
Как показано на схеме реакции 3, N-защищенное соединение формулы 4 участвует в раскрытии цикла хирального глицидола формулы 5. В результате реакции образуется промежуточное соединение, представленное формулой 9. Промежуточное соединение, полученное таким образом, in situ претерпевает реакцию внутримолекулярной циклизации, таким образом приводя к гидрокси-защищенному хиральному оксазолидин-2-ону или оксазолидин-2-тиону формулы 6.
Здесь соединение формулы 5, основываясь на соединении формулы 4, добавляли в количестве 0,8-5 эквивалентов, предпочтительно 1-1,5 эквивалентов. Реакцию раскрытия цикла и последующую in situ реакцию внутримолекулярной циклизации проводили в присутствии основания. Применяемое основание включает в себя неорганическое или органическое основание. Например, в качестве основания может быть применена соль щелочного металла, такая как метоксид натрия, метоксид лития, карбонат натрия, бикарбонат натрия, карбонат калия, гидроксид натрия или гидроксид калия, имидазол, 2,6-лутидин, N,N-диметиламинопиридин и их соли, третичный амин и его гидратированная форма. Применяемый органический растворитель не является специально ограниченным. В качестве органического растворителя могут быть применены N,N-диметилформамид, алифатический или ароматический углеводородный растворитель, галогенированный углеводород и простой эфир. Конкретно, могут быть применены ароматический органический растворитель, такой как толуол или бензол, галогеналкан, такой как хлористый метилен или хлороформ, или простой эфир, такой как диэтиловый эфир, тетрагидрофуран или диоксан. Температура реакции предпочтительно устанавливается в диапазоне 0-150°C, более предпочтительно 80-100°C.
Соединение формулы 6, полученное таким образом, используется в следующей реакции снятия защиты без какой-либо сложной очистки (например, фракционной дистилляции или перекристаллизации). Конкретно, после полного использования исходных веществ и промежуточных соединений, участвующих в реакции раскрытия цикла и последующей in situ реакции внутримолекулярной циклизации, добавляли удаляющий защитную группу реагент, зависящий от защитной группы, для гидрокси-группы, в тот же реактор для получения хирального ключевого промежуточного соединения формулы 2 [Protecting Groups, Thieme Medical Publishers Inc,, New York, 1994; Protective Groups in Organic Synthesis, John Wiley and Sons, Inc, 1991].
Соединение формулы 2 также применяется непосредственно, без какой-либо очистки, в реакции алкилирования с соединением формулы 8. Другими словами, гетероциклическое соединение формулы 2, имеющее гидрокси-группу, получают в высоко чистой форме, так что оно применяется непосредственно, в качестве исходного вещества, в алкилировании и последующих стадиях снятия защиты для синтеза целевого карведилола.
Получение соединения предшественника, имеющего формулу 7, из соединения формулы 2, достигается путем реакции соединения, имеющего формулу 2, с галогенирующим агентом, сульфонилирующим агентом или реагентом Мицунобу для активации гидроксильной группы соединения, имеющего формулу 2, с последующей реакцией нуклеофильного замещения с 9Н-4-гидроксикарбазолом. Этот путь обобщен на схеме реакции 4, где активация под действием галогенирующего агента или сульфонилирующего агента представлена как путь (1) и активация под действием реагента Мицунобу как путь (2).
Схема реакции 4
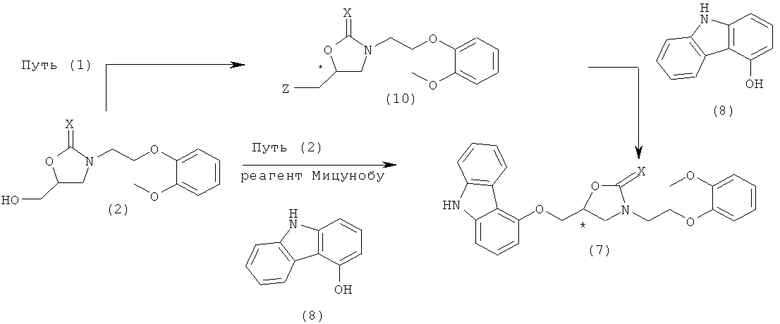
На схеме реакции 4, * обозначает хиральный центр, и Z представляет собой галогеновую группу или сульфонатную группу, и X, Y и R1 определены выше.
Как показано выше, соединение формулы 7 может быть получено путем введения соединения формулы 2 в реакцию галогенирования или сульфонилирования для получения соединения формулы 10, с последующей затем нуклеофильной атакой 9Н-4-гидроксикарбазола. Альтернативно, оно может быть получено по реакции Мицунобу соединения формулы 2 с 9Н-4-гидроксикарбазолом.
Более конкретно, как показано в пути (1) схемы реакции 4, соединение формулы 2 образует соединение формулы 10 реакцией с галогенирующим агентом. Здесь примеры галогенирующего агента включают тионилхлорид, тионилбромид, оксалилхлорид, трибромид фосфора и трихлорид фосфора. Галогенирующий агент добавляли обычно в количестве 0,8-10 эквивалентов, предпочтительно 1,1-2,0 эквивалентов.
Далее, соединение формулы 10 может быть также получено путем реакции соединения формулы 2 с сульфонилирующим агентом (сульфонилгалогенид) путем превращения гидрокси-группы соединения 2 в соответствующую сульфонатную группу. Здесь примеры сульфонилирующего агента включают в себя метансульфонилхлорид (кратко, MsCl), пара-толуолсульфонилхлорид (кратко, TsCl), бензолсульфонилхлорид, трифторметансульфонилхлорид (кратко, TfCl) и нитробензолсульфонилхлорид. Сульфонилирующий агент добавляли обычно в количестве 0,8-5 эквивалентов, предпочтительно 1,1-2,0 эквивалентов.
Реакцию галогенирования или сульфонилирования проводили в присутствии органического основания. В качестве примера органического основания, могут быть упомянуты имидазол, 2,6-лутидин, N,N-диметиламинопиридин и их соли, или третичный амин и его гидратированная форма. Предпочтительными являются триалкиламины, включая триметиламин, триэтиламин и диизопропилэтиламин. Основание добавляли в количестве 0,8-10 эквивалентов, предпочтительно 1,0-3,0 эквивалентов. Реакцию проводили в присутствии органического растворителя. Иногда, реакция может быть проведена без добавления растворителя. Применяемый органический растворитель не является специально ограниченным. В качестве органического растворителя могут быть применены N,N-диметилформамид, алифатический или ароматический углеводородный растворитель, галогенированный углеводород и простой эфир. Конкретно, могут быть применены ароматический органический растворитель, такой как толуол или бензол, галогеналкан, такой как хлористый метилен или хлороформ, или простой эфир, такой как диэтиловый эфир, тетрагидрофуран или диоксан. Температура реакции предпочтительно устанавливается в диапазоне 0-100°С, более предпочтительно 0-20°С.
В реакции соединение формулы 10 образуется в высоко чистой форме посредством типичной процедуры выделения. Полученное соединение может быть непосредственно подвергнуто последующей реакции алкилирования, без какой-либо сложной очистки. Это упрощает получение хирального карведилола и увеличивает выход реакции.
Соединение формулы 10 приводит к соединению формулы 7, которое является предшественником карведилола, посредством реакции с 9Н-4-гидроксикарбазолом формулы 8. 9Н-4-Гидроксикарбазол формулы 8 является коммерчески доступным или может быть промышленно получен посредством хорошо известной методики [DE 2240599 и US 4273711].
Специфические условия реакции между соединением формулы 10 и 9Н-4-гидроксикарбазолом формулы 8 описаны ниже. 9Н-4-гидроксикарбазол формулы 8 добавляли, основываясь на соединении формулы 10, в диапазоне 0,5-2,0 эквивалентов, предпочтительно, 1,0-1,1 эквивалентов. Сначала соединение формулы 10 и 9Н-4-гидроксикарбазол формулы 8 растворяли в органическом растворителе. К раствору добавляли 0,1-10 эквивалентов основания (предпочтительно, 0,5-2,0 эквивалентов). Устанавливалась температура реакции 30-150°C, предпочтительно 70-100°C. Применяемое основание включает в себя неорганическое или органическое основание. В качестве примера неорганического основания, могут быть упомянуты карбонат натрия, бикарбонат натрия, карбонат калия, гидроксид натрия, гидроксид калия или алкоксид металла. Предпочтительным является карбонат калия или карбонат натрия. В качестве органического основания, предпочтительным является соединение триалкиламина, например, триметиламин, триэтиламин или диизопропилэтиламин. Применяемый органический растворитель не является специально ограниченным. В качестве органического растворителя могут быть применены N,N-диметилформамид, диметилсульфоксид, алифатический или ароматический углеводородный растворитель, галогенированный углеводород, простой эфир или спирт. Предпочтительным примером спирта является C1-C4-спирт, такой как метанол, этанол, пропанол, изопропанол, бутанол, изобутанол или трет-бутанол.
Полученное соединение может быть непосредственно подвергнуто одностадийной реакции снятия защиты, без какой-либо сложной очистки. Это соответствует тому факту, что реакция протекает по существу без примесей, и что небольшое количество примесей может быть легко удалено при очистке на последующей стадии снятия защиты.
Согласно настоящему изобретению, соединение формулы 7 может быть также получено при непосредственной комбинации соединения формулы 2 с 9H-4-гидроксикарбазолом формулы 8.
То есть, соединение формулы 2 может быть непосредственно преобразовано в соединение 7 посредством комбинации с соединением формулы 8, без использования каких-либо методик для увеличения уходящей способности гидрокси-группы, достигаемых путем галогенирования или сульфонилирования. Такая методика представлена как путь (2) на схеме реакции 4. В результате пути (2), карведилол может быть получен через уменьшенное количество стадий по сравнению с путем (1).
Реакция Мицунобу может быть применена для непосредственной комбинации соединения формулы 2 с соединением формулы 8, через путь (2) схемы реакции 4. В реакции Мицунобу, активация гидрокси-группы соединения 2 и in situ нуклеофильное замещение проводятся как одностадийная реакция [Advanced Organic Chemistry 3rd Ed. Part B., Plenum Press, 1993; Advanced Organic Chemistry 4th Ed. A Wiley-Interscience Publication, 1992].
Получение соединения формулы 7 через путь (2) объяснено детально. Гетероциклическое соединение формулы 2, имеющее гидрокси-группу, растворяли в органическом растворителе, и реагент Мицунобу, включающий в себя соединение фосфина формулы 11 и диалкилазокарбоксилат формулы 12, добавляли для активации гидрокси-группы. К раствору добавляли 9H-4-гидроксикарбазол формулы 8. Нуклеофильная атака 9H-4-гидроксикарбазола формулы 8 на активированную гидрокси-группу приводит к соединению формулы 7.
Конкретные примеры соединения формулы 11 и соединения формулы 12 представлены ниже:
Формула 11
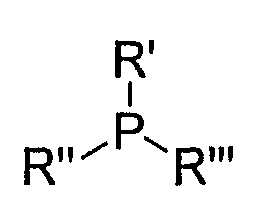
где R', R'' и R''' представляют собой заместители. Предпочтительно, R', R'' и R''', каждый, независимо представляют собой C1-C6-алкильную группу, C3-C6-циклоалкильную группу, C2-C6-алкенильную группу, C2-C6-алкинильную группу, C1-C6-алкокси-группу, C6-C10-арильную группу или (CH2)L-R3 (где R3 представляет собой C3-C6-циклоалкильную группу, C2-C6-алкенильную группу, C2-C6-алкинильную группу, C1-C6-алкокси-группу или C6-C10-арильную группу, и L представляет собой целое число от 1 до 8).
Формула 12
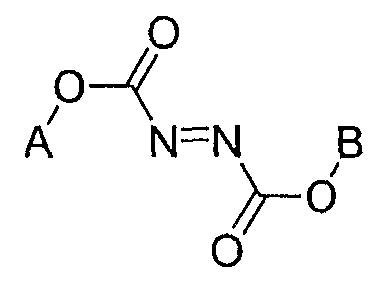
в которой A и B, каждый, независимо представляют собой C1-C6-алкильную группу, C3-C6-циклоалкильную группу, C2-C6-алкенильную группу или C2-C6-алкинильную группу.
Соединение формулы 7, полученное путем (2) схемы реакции 2, может быть также применено в последующей реакции снятия защиты без использования какой-либо методики очистки, поскольку фосфиноксид, образующийся в качестве побочного продукта, может быть легко удален при очистке в последующей реакции снятия защиты. То есть, соединение формулы 7 может быть непосредственно использовано, без какой-либо очистки, в реакции снятия защиты как одностадийной реакции. Это обеспечивает простоту способа и увеличение выхода.
Получение карведилола из соединения формулы 7 проиллюстрировано на схеме реакции 5:
Схема реакции 5
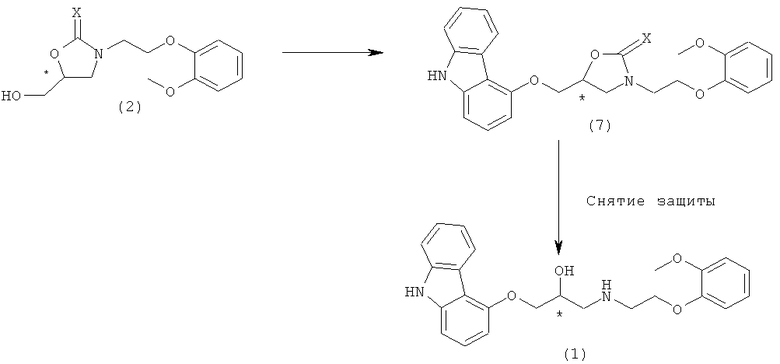
Как упомянуто выше, превращение соединения формулы 7 в карведилол достигается путем добавления основания, необязательно в сочетании с реакционным растворителем, в реактор, внутри которого образуется соединение формулы 7, как показано на схеме реакции 4. В целом, соединение оксазолидин-2-она или оксазолидин-2-тиона претерпевает реакцию гидролиза в основных условиях, таким образом приводя к образованию аминоспирта [гидролиз оксазолидин-2-она: J. Org. Chem., 1986, 51, 713; J. Org. Chem., 1988, 53, 3865; Tetrahedron Lett, 1990, 51, 7407; Tetrahedron 1998, 54, 1221; гидролиз оксазолидин-2-тиона: J. Org. Chem., 1992, 57, 4331; J. Am. Chem. Soc, 1994, 116, 5607]. Согласно настоящему изобретению, соединение формулы 7, как было найдено, является применимым в реакции снятия защиты для получения целевого карведилола. Как показано на схеме реакции 5, после полного взаимодействия исходного вещества схемы реакции 4, добавляли основание или комбинацию основания и растворителя в тот же реактор при перемешивании для завершения реакции снятия защиты. Согласно предпочтительному варианту осуществления настоящего изобретения основание добавляли при перемешивании к реакционной смеси реакции нуклеофильного замещения между соединением формулы 2 и соединением формулы 8, с последующим добавлением растворителя, такого как вода, спирт и их смесь. При этом температура реакции обычно находится в диапазоне 0-150°C, предпочтительно 30-70°C.
Основание добавляли, основываясь на соединении формулы 2, в количестве 0,8-10 эквивалентов, предпочтительно 1,0-3,0 эквивалентов. В качестве применяемого неорганического основания могут быть упомянуты неорганический карбонат (например, карбонат натрия, карбонат калия, карбонат цезия или бикарбонат натрия) или неорганический гидроксид (например, гидроксид натрия, гидроксид калия или гидроксид лития).
В качестве добавленного растворителя в смеси с основанием обычно использовали воду, спирт или их смесь. Предпочтительным примером спирта является C1-C4-спирт, такой как метанол, этанол, пропанол, изопропанол, бутанол, изо-бутанол или трет-бутанол.
Как упомянуто выше, настоящее изобретение четко отличается от предшествующего уровня техники в том, что целевой хиральный карведилол формулы 1 эффективно получают из хирального ключевого промежуточного соединения формулы 2 с высокой оптической чистотой и в мягких условиях, применимых для промышленного массового производства. Далее, хиральное ключевое промежуточное соединение образуется, без какого-либо изменения стереоконфигурции, из оптически чистого хирального глицидильного производного.
В противоположность этому, согласно традиционным способам получения хирального карведилола, как показано на схеме реакции 1, исходное вещество, 4-(2,3-эпоксипропил)карбазол формулы 3, не может быть получено в высоко оптически чистой форме. Конкретно, соединение формулы 3 получают реакцией нуклеофильного замещения хирального глицидила мета-нитробензолсульфоната с 9H-4-гидроксикарбазолом формулы 8. Даже, несмотря на то, что реакция, как описано, проходит стереоселективным образом, так что стереоконфигурация продукта сохраняется [J. Org. Chem., 1989, 54, 1295; Tetrahedron: Asymmetry 1992, 3, 539], Дубоис и соавторы (Dubois et al.) сообщали, что оптическая чистота соединения формулы 3 значительно уменьшается в некоторых случаях [J Med. Chem., 1996, 39, 3256]. Далее, согласно исследованиям авторов настоящего изобретения, оптическая чистота продукта, как было найдено, является очень чувствительной к условиям реакции. Следовательно, способ, как предполагается, требует строго определенных условий реакции. В результате способ не применим для промышленного массового производства.
Согласно настоящему изобретению, ключевое промежуточное соединение формулы 2 с высокой оптической чистотой образуется из оптически чистого хирального производного глицидола, без какого-либо снижения оптической чистоты. Далее, снижение оптической чистоты не имеет места при получении хирального карведилола из ключевого промежуточного соединения формулы 2. Следовательно, по сравнению с традиционными методиками, способ по настоящему изобретению ведет к образованию карведилола в высоко оптически чистой форме.
В добавление, способ по настоящему изобретению не требует какой-либо сложной очистки промежуточных продуктов, участвующих в образовании целевого хирального карведилола. Это подразумевает, что способ по настоящему изобретению является очень простым и экономичным. Кроме того, способ по настоящему изобретению проводится в мягких условиях, так что не требуется ни энергичных условий реакции, ни сильного окисляющего или восстанавливающего агента. В результате, способ по настоящему изобретению пригоден для применения в промышленном массовом производстве.
В конечном итоге, предложен способ получения высоко оптически чистого карведилола из хирального соединения формулы 2, промышленно применимым способом без какого-либо снижения оптической чистоты.
Далее настоящее изобретение будет более полно проиллюстрировано со ссылкой на примеры. Однако необходимо понимать, что эти примеры предложены только для иллюстрации и не должны обсуждаться как ограничивающие объем настоящего изобретения. Многочисленные модификации могут быть сделаны без отклонения от объема и сущности изобретения.
ПРИМЕРЫ
Пример 1: Получение (S)-3-[2-(2-метоксифенокси)этил]-5-(гидроксиметил)оксазолидин-2-она [Формула 2, X = кислород]
Изобутил-2-(2-метоксифенокси)этилкарбамат 106,9 г (0,4 моль) и трет-бутоксид лития 6,40 г (0,08 моль) растворяли в N,N-диметилформамиде (200 мл) и перемешивали при комнатной температуре в течение 10 мин. К смеси добавляли 69,5 г (0,44 моль) (S)-2-оксиранилметокситетрагидропирана и перемешивали при 80°C в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры и приводили к pH 1 с применением 20% раствора серной кислоты в метиловом спирте. Далее реакционную смесь перемешивали при комнатной температуре в течение 5 часов и нейтрализовали с применением триэтиламина. К раствору добавляли воду (400 мл) и хлористый метилен (1000 мл). Затем органический слой отделяли от смешанного раствора, сушили над безводным сульфатом магния и фильтровали. Упаривание при пониженном давлении давало целевое соединение формулы 2 в жидком виде. Полученный продукт вводили без какой-либо очистки в следующую реакцию снятия защиты.
Выход: 104,6 г (98%)
1H-ЯМР (300 МГц, CDCl3): δ 2,32 (ушир.с, 1H), 3,65-3,74 (м, 4H), 3,80-3,92 (м, 2H), 3,85 (с, 3H), 4,18 (т, J= 7,8 Гц, 2H), 4,60 (м, 1H), 6,89-6,99 (м, 4H).
Пример 2: Получение (R)-3-[2-(2-метоксифенокси)этил]-5-(гидроксиметил)оксазолидин-2-она [Формула 2, X = кислород]
Используя раствор в N,N-диметилформамиде изобутил-2-(2-метоксифенокси)этилкарбамата 53,46 г (0,2 моль), трет-бутоксида лития 3,20 г (0,04 моль) и (R)-2-оксиранилметокси-тетрагидропирана 34,76 г (0,22 моль), для получения целевого продукта проводили методики, как описано в Примере 1.
Выход: 51,2 г (96%)
Пример 3: Получение (S)-3-[2-(2-метоксифенокси)этил]-5-(гидроксиметил)оксазолидин-2-она [Формула 2, X = кислород]
Используя раствор в N,N-диметилформамиде изобутил-2-(2-метоксифенокси)этилкарбамата 26,7 г (0,1 моль), трет-бутоксида лития 1,60 г (0,02 моль) и (S)-2-трет-бутоксиметилоксирана, 14,3 г (0,11 моль), для получения целевого продукта проводили методики, как описано в Примере 1.
Выход: 25,6 г (96%)
Пример 4: Получение (R)-3-[2-(2-метоксифенокси)этил]-5-(гидроксиметил)оксазолидин-2-она [Формула 2, X = кислород]
Используя раствор в N,N-диметилформамиде изобутил-2-(2-метоксифенокси)этилкарбамата 53,46 г (0,2 моль), трет-бутоксида лития 3,2 г (0,04 моль) и (R)-2-трет-бутоксиметил-оксирана 28,6 г (0,22 моль), для получения целевого продукта проводили методики, как описано в Примере 1.
Выход: 50,7 г (95%)
Пример 5: Получение (S)-3-[2-(2-метоксифенокси)этил]-5- (гидроксиметил)оксазолидин-2-тиона [Формула 2, X = сера]
Используя раствор в N,N-диметилформамиде этил-2-(2-метоксифенокси)этилтиокарбамат 25,5 г (0,1 моль) [J. Chem. Soc, 1952, 2076; J. Chem. Soc, 1952, 2079; Tetrahedron Lett., 1969, 3631], трет-бутоксида лития 1,6 г (0,02 моль) и (S)-2-оксиранилэтокси-тетрагидропирана 17,38 г (0,11 моль), для получения целевого продукта проводили методики, как описано в Примере 1.
Выход: 21,5 г (76%)
Пример 6: Получение (R)-3-[2-(2-метоксифенокси)этил]-5-(гидроксиметил)оксазолидин-2-тиона [Формула 2, X = сера]
Используя раствор в N,N-диметилформамиде этил-2-(2-метоксифенокси)этилтиокарбамата 51,0 г (0,2 моль), трет-бутоксида лития 3,2 г (0,04 моль) и (R)-2-оксиранилметокси-тетрагидропирана 34,76 г (0,22 моль), для получения целевого продукта проводили методики, как описано в Примере 1.
Выход: 42,5 г (75%)
Пример 7: Получение (S)-{3-[2-(2-метоксифенокси)этил]-2-оксо-оксазолидин-5-ил}метилметансульфоната [Формула 10, Х = кислород, Z = метансульфонат]
Соединение формулы 2, полученное в Примере 1, 53,4 г (0,2 моль), и триэтиламин 30,36 г (0,3 моль) добавляли к хлористому метилену (300 мл), и полученный раствор охлаждали до 0°С. К раствору по каплям добавляли метансульфонилхлорид 25,2 г (0,22 моль) при перемешивании. Реакционный раствор перемешивали в течение 3 часов и затем добавляли воду (300 мл). Затем органический слой отделяли от смешанного раствора, сушили над безводным сульфатом магния и фильтровали. Упаривание при пониженном давлении давало целевое соединение формулы 7 в жидком виде. Полученный продукт вводили без какой-либо очистки в следующую реакцию.
Выход: 68,4 г (99%)
Пример 8: Получение (R)-{3-[2-(2-метоксифенокси)этил]-2-оксооксазолидин-5-ил}метилметансульфоната [Формула 10, Х = кислород, Z = метансульфонат]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 2, 80,1 г (0,3 моль), триэтиламина 45,5 г (0,45 моль) и метансульфонилхлорида 37,8 г (0,33 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 102,5 г (99%)
Пример 9: Получение (S)-{3-[2-(2-метоксифенокси)этил]-2-оксотиооксазолидин-5-ил}метилметансульфоната [Формула 10, Х = сера, Z = метансульфонат]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 5, 28,33 г (0,1 моль), триэтиламина 15,18 г (0,15 моль) и метансульфонилхлорида 12,6 г (0,11 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 35,7 г (99%).
Пример 10: Получение (R)-{3-[2-(2-метоксифенокси)этил]-2-оксотиооксазолидин-5-ил}метилметансульфоната [Формула 10, Х = сера, Z = метансульфонат]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 6, 42,45 г (0,15 моль), триэтиламина 22,8 г (0,225 моль) и метансульфонилхлорида 18,9 г (0,165 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 53,67 г (99%)
Пример 11: Получение (S)-{3-[2-(2-метоксифенокси)этил]-2-оксооксазолидин-5-ил}метилтолуолсульфоната [Формула 10, Х = кислород, Z = толуолсульфонат]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 1, 26,7 г (0,1 моль), триэтиламина 15,18 г (0,15 моль) и пара-толуолсульфонилхлорида 21,0 г (0,11 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 38,7 г (92%)
Пример 12: Получение (R)-{3-[2-(2-метоксифенокси)этил]-2-оксооксазолидин-5-ил}метилтолуолсульфоната [Формула 10, Х = кислород, Z = толуолсульфонат]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 2, 42,7 г (0,16 моль), триэтиламина 24,3 г (0,24 моль) и пара-толуолсульфонилхлорида 33,6 г (0,176 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 62,6 г (93%)
Пример 13: Получение (S)-{3-[2-(2-метоксифенокси)этил]-2-оксотиооксазолидин-5-ил}метилтолуолсульфоната [Формула 10, X = сера, Z = толуолсульфонат]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 5, 28,3 г (0,1 моль), триэтиламина 15,18 г (0,15 моль) и пара-толуолсульфонилхлорида 21,0 г (0,11 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 40,64 г (93%)
Пример 14: Получение (R)-{3-[2-(2-метоксифенокси)этил]-2-оксотиооксазолидин-5-ил}метилтолуолсульфоната [Формула 10, Х = сера, Z = толуолсульфонат]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 6, 25,5 г (0,09 моль), триэтиламина 13,66 г (0,135 моль) и пара-толуолсульфонилхлорида 19,0 г (0,1 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 35,4 г (90%)
Пример 15: Получение (S)-5-хлорометил-3-[2-(2-метоксифенокси)этил]оксазолидин-2-она [Формула 10, Х = кислород, Z = хлорид]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 1, 26,7 г (0,1 моль), триэтиламина 10,1 г (0,1 моль) и тионилхлорида 35,7 г (0,3 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 26,3 г (92%)
Пример 16: Получение (S)-5-хлорметил-3-[2-(2-метоксифенокси)этил]оксазолидин-2-тиона [Формула 10, Х = сера, Z = хлорид]
Используя раствор в хлористом метилене соединения формулы 2, полученного в Примере 5, 28,3 г (0,1 моль), триэтиламина 10,1 г (0,1 моль) и тионилхлорида 35,7 г (0,3 моль), для получения целевого продукта проводили методики, как описано в Примере 7.
Выход: 28,0 г (93%)
Пример 17: Получение (S)-карведилола
51,75 г (0,15 моль) (S)-{3-[2-(2-метоксифенокси)этил]-2-оксооксазолидин-5-ил}метилметансульфоната, полученного в Примере 7, и 27,45 г (0,15 моль) 9Н-4-гидроксикарбазола растворяли в 450 мл безводного этилового спирта. К раствору добавляли 31,10 г (0,225 моль) карбоната калия и кипятили с обратным холодильником в течение 16 часов при перемешивании. После того как соединение формулы 10 было полностью израсходовано, температуру реакции доводили до комнатной температуры. К смеси добавляли 150 мл 4N водного раствора KOH и затем кипятили с обратным холодильником в течение 6 часов при перемешивании. Реакционную смесь охлаждали до комнатной температуры, и этиловый спирт упаривали при пониженном давлении. К остатку добавляли воду (200 мл) и хлористый метилен (500 мл) и перемешивали в течение 30 мин. Органический слой отделяли, сушили над безводным сульфатом магния и фильтровали. Упаривание при пониженном давлении давало твердый остаток. К полученному остатку добавляли этилацетат 150 мл и перемешивали. После фильтрования и промывки получали целевой (S)-карведилол формулы 1.
Выход: 44,5 г (73%)
1H-ЯМР (300 МГц, CDCl3): δ 1,85 (ушир.с, 1H), 2,97 (м, 1H), 3,10 (м, 3H), 3,83 (с, 3H), 4,15 (т, J=7,7 Гц, 2H), 4,18-4,29 (м, 3H), 6,66 (д, J=8,1 Гц, 1H), 6,85-6,97 (м, 4H), 7,04 (д, J=8,1 Гц, 1H), 7,25-7,38 (м, 3H), 8,19 (ушир.с, 1H), 8,26 (д, J=7,8 Гц, 1H).
Оптическая чистота: > 99% ee [ВЭЖХ: Chirolsil SCA(-), элюент: смесь растворителей, ацетонитрил:метиловый спирт = 2:1, содержащий 0,1% триэтиламина, скорость элюента = 1 мл/мин, УФ-детектор: 254 нм, время удержания (S)-изомера, ts = 23,2 мин, время удержания (R)-изомера, tR = 20,6 мин]
Пример 18: Получение (R)-карведилола
Используя 34,5 г (0,1 моль) (R)-{3-[2-(2-метоксифенокси)этил]-2-оксооксазолидин-5-ил}метилметансульфоната, полученного в Примере 8, 18,3 г (0,1 моль) 9H-4-гидроксикарбазола и 20,7 г (0,15 моль) карбоната калия, для получения целевого (R)-карведилола проводили методики, как описано в Примере 17.
Выход: 28,8 г (71%)
Пример 19: Получение (S)-карведилола
Используя 36,1 г (0,1 моль) (S)-{3-[2-(2-метоксифенокси)этил]-2-оксотиооксазолидин-5-ил]метилметансульфоната, полученного в Примере 9, 18,3 г (0,1 моль) 9H-4-гидроксикарбазола и 20,7 г (0,15 моль) карбоната калия, для получения целевого (S)-карведилола проводили методики, как описано в Примере 17.
Выход: 28 г (69%).
Пример 20: Получение (R)-карведилола
Используя 43,3 г (0,12 моль) (R)-{3-[2-(2-метоксифенокси)этил]-2-оксотиооксазолидин-5-ил}метилметансульфоната, полученного в Примере 10, 22,0 г (0,12 моль) 9H-4-гидроксикарбазола и 24,8 г (0,18 моль) карбоната калия, для получения целевого (R)-карведилола проводили методики, как описано в Примере 17.
Выход: 32,6 г (67%)
Пример 21: Получение (S)-карведилола
Используя 42,1 г (0,1 моль) (S)-{3-[2-(2-метоксифенокси)этил]-2-оксооксазолидин-5-ил}метилтолуолсульфоната, полученного в Примере 11, 18,3 г (0,1 моль) 9H-4-гидроксикарбазола и 20,7 г (0,15 моль) карбоната калия, для получения целевого (S)-карведилола проводили методики, как описано в Примере 17.
Выход: 26,4 г (65%)
Пример 22: Получение (S)-карведилола
Используя 56,8 г (0,13 моль) (S)-{3-[2-(2-метоксифенокси)этил]-2-оксотиооксазолидин-5-ил}метилтолуолсульфоната, полученного в Примере 13, 23,8 г (0,13 моль) 9H-4-гидроксикарбазола и 26,9 г (0,195 моль) карбоната калия, для получения целевого (S)-карведилола проводили методики, как описано в Примере 17.
Выход: 32,7 г (62%)
Пример 23: Получение (S)-карведилола
Используя 28,6 г (0,1 моль) (S)-5-хлорметил-3-[2-(2-метоксифенокси)этил]оксазолидин-2-она, полученного в Примере 15, 18,3 г (0,1 моль) 9H-4-гидроксикарбазола, 0,17 г (0,001 моль) иодида калия и 20,7 г (0,15 моль) карбоната калия, для получения целевого (S)-карведилола проводили методики, как описано в Примере 17.
Выход: 26,8 г (66%)
Пример 24: Получение (S)-карведилола
Используя 42,8 г (0,15 моль) (S)-5-хлорметил-3-[2-(2-метоксифенокси)этил]оксазолидин-2-тиона, полученного в примере 16, 27,45 г (0,15 моль) 9H-4-гидроксикарбазола, 0,249 г (0,0015 моль) иодида калия и 31,1 г (0,225 моль) карбоната калия, для получения целевого (S)-карведилола проводили методики, как описано в Примере 17.
Выход: 37,8 г (62%)
Пример 25: Получение (S)-карведилола
26,7 г (0,1 моль) (S)-3-[2-(2-метоксифенокси)этил]-5-(гидроксиметил)оксазолидин-2-она растворяли в тетрагидрофуране (100 мл), затем к раствору последовательно добавляли трифенилфосфин (31,44 г, 0,12 моль) и диизопропилазодикарбоксилат (24,2 г, 0,12 моль). Смешанный раствор перемешивали в течение 1 часа при комнатной температуре. К смешанному раствору по каплям добавляли раствор тетрагидрофурана (50 мл), содержащий 18,3 г (0,1 моль) 9H-4-гидроксикарбазола, и перемешивали в течение 12 часов при комнатной температуре. После полного взаимодействия исходного вещества, соединения формулы 2, реакционный раствор упаривали при пониженном давлении. К оставшемуся остатку добавляли 300 мл этилового спирта и 100 мл 4N водного раствора KOH, смесь кипятили с обратным холодильником в течение 6 часов при перемешивании. Реакционную смесь охлаждали до комнатной температуры, и этиловый спирт упаривали при пониженном давлении. К остатку добавляли воду (200 мл) и хлористый метилен (300 мл), и смесь перемешивали в течение 30 мин. Органический слой отделяли, сушили над безводным сульфатом магния и фильтровали. Упаривание при пониженном давлении давало твердый остаток. К полученному остатку добавляли этилацетат (150 мл) и перемешивали. После фильтрования и промывки получали целевой (S)-карведилол формулы 1.
Выход: 21,1 г (52%).
Пример 26: Получение (R)-карведилола
Используя (S)-3-[2-(2-метоксифенокси)этил]-5-(гидроксиметил)оксазолидин-2-тион (32 г, 0,12 моль), полученный в Примере 5, трифенилфосфин (37,7 г, 0,144 моль), диизопропилазодикарбоксилат (29,0 г, 0,144 моль) и 9H-4-гидроксикарбазол (22,0 г, 0,12 моль), для получения целевого (R)-карведилола проводили методики, как описано в Примере 25.
Выход: 23,4 г (48%)
Настоящее изобретение предоставляет эффективный способ получения целевого хирального карведилола формулы 1, исходя из соединения формулы 2, которое представляет собой ключевой промежуточный продукт для синтеза хирального карведилола, или посредством увеличения уходящей способности гидрокси-группы, алкилирования с 9H-4-гидроксикарбазолом и последующего снятия защиты, или посредством непосредственной комбинации с 9H-4-гидроксикарбазолом и последующего снятия защиты.
В указанном выше способе, ключевой промежуточный продукт формулы 2 может быть без труда получен в высоко оптически чистой форме из коммерчески доступных исходных веществ, что является одним из преимуществ настоящего изобретения. То есть, хиральный глицидол или его производные формулы 5 и N-защищенное аминное соединение формулы 4, которые являются исходными веществами для ключевого промежуточного продукта формулы 2, являются коммерчески доступными или легко получаются в промышленном масштабе. Далее, получение ключевого промежуточного продукта формулы 2 из исходных веществ проводили без какого-либо снижения оптической чистоты, так что ключевой промежуточный продукт может быть получен в высоко оптически чистой форме.
Кроме того, согласно настоящему изобретению, способ не включает в себя какой-либо методики, которая снижает оптическую чистоту, в течение превращения из ключевого промежуточного продукта формулы 2 в карведилол. Способ проводится просто, в мягких условиях и без какой-либо сложной очистки, таким образом, предоставляя экономичный способ получения хирального карведилола.
Следовательно, настоящее изобретение предоставляет эффективный способ получения хирального карведилола с высокой оптической чистотой, используя соединение формулы 2 в качестве ключевого промежуточного продукта для синтеза хирального карведилола, без какого-либо изменения хиральности и способом, пригодным для промышленного массового производства.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ БИАРИЛЬНОЕ ПРОИЗВОДНОЕ, ПРИМЕНЯЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ДИАЦИЛГЛИЦЕРОЛ-АЦИЛТРАНСФЕРАЗЫ 2, И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2808433C1 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ГИДРОКСИМЕТИЛ ЗАМЕЩЕННЫХ ОКСАЗОЛИДИНОНОВ, СПОСОБ ПОЛУЧЕНИЯ 5-АМИНОМЕТИЛ ЗАМЕЩЕННЫХ ОКСАЗОЛИДИНОНОВЫХ АМИНОВ, ОКСАЗОЛИДИНОНСУЛЬФОНАТ | 1997 |
|
RU2176643C2 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1,4-ДИФЕНИЛАЗЕТИДИНОНА | 2005 |
|
RU2380361C2 |
| СПОСОБ ДЕАЦЕТИЛИРОВАНИЯ α-АМИНОАЦЕТАЛЕЙ | 2008 |
|
RU2477270C2 |
| Хиральные γ-кетосульфанильные производные пинановой структуры и способ их получения | 2021 |
|
RU2783164C1 |
| НОВОЕ ПРОИЗВОДНОЕ 3-(4-(БЕНЗИЛОКСИ)ФЕНИЛ)ГЕКС-4-ИНОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ МЕТАБОЛИЧЕСКОГО ЗАБОЛЕВАНИЯ, ВКЛЮЧАЮЩАЯ ЕГО В КАЧЕСТВЕ ЭФФЕКТИВНОГО ИНГРЕДИЕНТА | 2014 |
|
RU2628077C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОГО ПРОДУКТА - КАРВЕДИЛОЛА И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2216539C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА С ЦИКЛИЧЕСКИМ АМИДОКСИМОМ ИЛИ ЦИКЛИЧЕСКИМ АМИДРАЗОНОМ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2468023C1 |
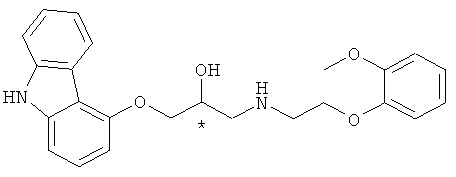
Изобретение относится к способу получения высоко оптически чистого хирального карведилола формулы I, включающему взаимодействие аминного соединения формулы 4 с хиральным глицидольным соединением формулы 5, для получения соединения формулы 6, через раскрытие цикла соединения формулы 5 под действием соединения формулы 4 и последующей in situ внутримолекулярной циклизацией для получения соединения формулы 6; удаление защитной группы для гидрокси-группы соединения формулы 6 для получения соединения формулы 2; взаимодействие соединения формулы 2 с галогенирующим агентом, сульфонилирующим агентом или реагентом Мицунобу для активации гидроксильной группы соединения формулы 2, с последующей реакцией нуклеофильного замещения с 9Н-4-гидроксикарбазолом для получения соединения формулы 7; и снятие защиты с полученного соединения формулы 7 в присутствии неорганического основания для получения целевого хирального карведилола формулы 1;
Формула 1
Формула 2
Формула 4
Формула 5
Формула 6
Формула 7
где * обозначает хиральный центр, Х представляет собой кислород или серу, Y представляет собой уходящую группу, и R1 представляет собой защитную группу для гидрокси-группы. способ не требует сложной методики очистки, не вызывает снижения оптической чистоты. 3 з.п. ф-лы.
1. Способ получения высоко оптически чистого хирального карведилола, включающий:
взаимодействие аминного соединения формулы 4 с хиральным глицидольным соединением формулы 5, для получения соединения формулы 6, через раскрытие цикла соединения формулы 5 под действием соединения формулы 4 и последующей in situ внутримолекулярной циклизацией для получения соединения формулы 6;
удаление защитной группы для гидроксигруппы соединения формулы 6 для получения соединения формулы 2;
взаимодействие соединения формулы 2 с галогенирующим агентом, сульфонилирующим агентом или реагентом Мицунобу для активации гидроксильной группы соединения формулы 2 с последующей реакцией нуклеофильного замещения с 9Н-4-гидроксикарбазолом для получения соединения формулы 7; и
снятие защиты с полученного соединения формулы 7 в присутствии неорганического основания для получения целевого хирального карведилола формулы 1;
Формула 1
Формула 2
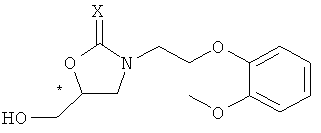
Формула 4
Формула 5
Формула 6
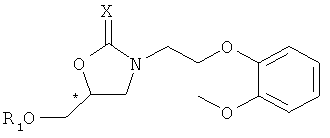
Формула 7
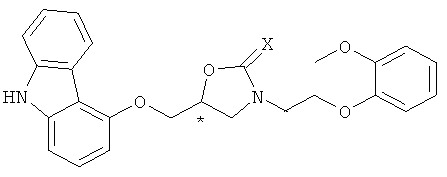
где * обозначает хиральный центр, Х представляет собой кислород или серу, Y представляет собой уходящую группу, и R1 представляет собой защитную группу для гидроксигруппы.
2. Способ по п.1, в котором галогенирующий агент выбирают из группы, состоящей из тионилхлорида, тионилбромида, оксалилхлорида, трибромида фосфора и трихлорида фосфора.
3. Способ по п.1, в котором сульфонилирующий агент выбирают из группы, состоящей из метансульфонилхлорида, паратолуолсульфонилхлорида, бензолсульфонилхлорида, трифторметансульфонилхлорида и нитробензолсульфонилхлорида.
4. Способ по п.1, в котором реакцию снятия защиты на стадии проводят в присутствии неорганического основания.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Устройство защиты импульсно-кодового модулятора | 1983 |
|
SU1142873A1 |
| Сортировочный цилиндр для зерна | 1959 |
|
SU127099A1 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОГО ПРОДУКТА - КАРВЕДИЛОЛА И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2216539C2 |

