Область техники
Настоящее изобретение относится к биарильному производному, представленному формулой (1), проявляющему ингибирующую активность в отношении диацилглицерол-ацилтрансферазы 2 (DGAT2), к фармацевтической композиции, включающей его в качестве активного ингредиента, и его применению.
Уровень техники
Повышение уровня жизни в соответствии с экономическим развитием, частое употребление продуктов быстрого приготовления и изменение привычек питания на основе мяса вызвали чрезмерное накопление тепловой энергии в организме. Эти изменения в рационе питания современных людей также привели к снижению расхода тепловой энергии из-за отсутствия физической нагрузки, что привело к серьезному распространению метаболических заболеваний, таких как ожирение, гиперлипидемия, диабет, сердечно-сосудистые заболевания и ишемическая болезнь сердца. В частности, ожирение является одним из быстрорастущих заболеваний и, как сообщается, является причиной метаболических заболеваний, таких как диабет. Привлекает внимание разработка терапевтических средств для лечения метаболических заболеваний путем контроля функций ферментов, участвующих в пути биосинтеза триглицеридов, что является основной причиной ожирения.
Нейтральные жиры, такие как триглицериды (ТГ), играют очень важную роль в функции накопления в качестве источника энергии в организме. Однако при избыточном накоплении нейтральных жиров в органах или тканях они вызывают ожирение, гипертриглицеридемию, жировой гепатоз и тому подобное, тем самым вызывая такие серьезные заболевания, как диабет, атеросклероз, метаболические нарушения и гипофункцию органов. Диацилглицерол-ацилтрансфераза, которая является ключевым ферментом в биосинтезе триглицеридов, обнаружена в различных тканях млекопитающих и представляет собой фермент, который синтезирует ТГ путем связывания жирного ацил-КоА с гидроксильной группой диацилглицерина на заключительной стадии глицеролфосфатного пути, который является основным путем синтеза триглицеридов. В настоящее время известны две изоформы DGAT1 и DGAT2. Хотя их биохимические функции схожи, существует различие в том, что DGAT1 в основном экспрессируется в тонком кишечнике и жировой ткани, а DGAT2 в основном экспрессируется в печени и жировой ткани. Кроме того, что касается семейства генов, DGAT1 принадлежит к семейству ACAT, а DGAT2 принадлежит к семейству MGAT. Таким образом, ожидается, что их роль в биосинтезе ТГ также различна.
Несколько исследований, включая исследования на животных, показали, что DGAT2 в первую очередь участвует в биосинтезе ТГ in vivo. В отличие от мышей с нокаутом DGAT2, которые почти не синтезируют ТГ и умирают вскоре после рождения из-за аномального слоя кожи, у мышей с нокаутом DGAT1 наблюдали небольшое снижение уровня ТГ и не было проблем с выживанием мышей (Stone SJ et al., 2000. Nat. Genet. 25: 87-90). Кроме того, в результате снижения уровня экспрессии DGAT1 или DGAT2 с помощью антисмыслового олигонуклеотида (ASO) на животной модели жирового гепатоза, симптомы жирового гепатоза облегчались, а скорость продукции глюкозы в печени значительно снижалась только при снижении количества DGAT2 (Choi CS et al., 2007. Hepatology. 45: 1366-74).
Лежащие в основе молекулярные механизмы полностью не выяснены, но считается, что ингибирование DGAT2 приводит к подавлению экспрессии нескольких генов, кодирующих белки, участвующие в выработке липидов, такие как белок, связывающий стеролрегулирующие элементы 1c (SREBP1c), и стеароил-КоА-десатураза 1 (SCD1). В то же время считалось, что путь окисления индуцируется увеличением количества генов, таких как карнитин-пальмитоилтрансфераза 1 (CPT1). Это изменение, в свою очередь, приводит к снижению уровней липидов DAG и TAG в печени и, таким образом, к улучшению чувствительности печени к инсулину. Кроме того, ингибирование DGAT2 ингибирует секрецию VLDL TAG в печени и снижает уровень холестерина в циркулирующей крови. Наконец, уровни аполипопротеина B (APOB) в плазме были подавлены, что, как полагали, было связано с уменьшением поступления TAG при липидировании вновь синтезированного белка APOB. То есть, при ингибировании DGAT2 наблюдалось благоприятное воздействие как на гликемический контроль, так и на профиль холестерина в плазме, что означает, что ингибирование DGAT2 можно применять для лечения метаболических нарушений.
Описание изобретения
Техническая задача
Целью настоящего изобретения является предоставление нового биарильного производного, представленного формулой (1), обладающего ингибирующей активностью в отношении диацилглицерол-ацилтрансферазы 2 (DGAT2).
Другой целью настоящего изобретения является предоставление способа получения биарильного производного.
Еще одной целью настоящего изобретения является предоставление фармацевтической композиции для лечения метаболических заболеваний, связанных с DGAT2, включающей биарильное производное в качестве активного ингредиента, и способа ее получения.
Еще одной целью настоящего изобретения является предоставление способа лечения метаболических заболеваний, связанных с DGAT2, у субъекта, в котором повышается эффективность на животных моделях заболеваний, а также эффективность и удобство приема субъектом улучшаются за счет использования биарильного производного в качестве активного ингредиента, обладающего улучшенными физическими и химическими свойствами по сравнению с обычными соединениями.
Решение задачи
Для достижения вышеуказанной цели настоящее изобретение предоставляет соединение следующей формулы (1) или его фармацевтически приемлемую соль или изомер:
[формула (1)]
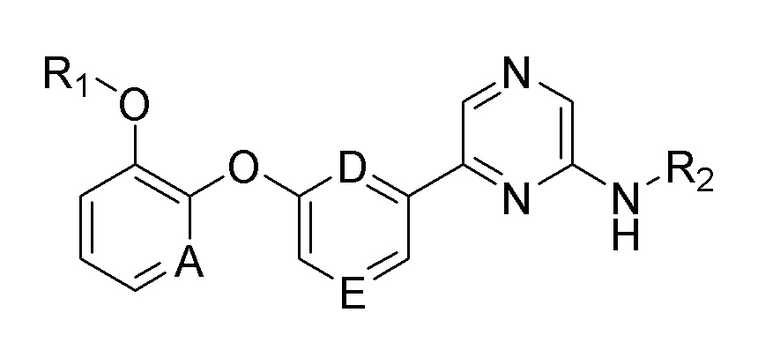
где
A, D и E, каждый независимо, представляют собой CH или N;
R1 представляет собой алкил, циклоалкил или галогеналкил;
R2 представляет собой -G-J-L;
где G представляет собой -C(=O)- или прямую связь;
J представляет собой алкилен, алкенилен, алкилен-арилен, алкенилен-арилен, алкоксиен-арилен, арилен, гетероарилен-гетероциклоалкилен, гетероарилен-арилен или гетероарилен-окси-циклоалкилен;
L представляет собой водород, галоген, амино, нитро, карбокси (-COOH), карбоксиалкил, карбоксиалкокси, циклоалкил или арил;
где алкил, алкилен, карбоксиалкил, карбоксиалкокси или арил необязательно замещен одним или несколькими заместителями, выбранными из гидрокси, галогена, алкила и алкокси; и
гетероциклоалкилен или гетероарилен включает один или несколько гетероатомов, выбранных из N, O и S.
Соединение формулы (1) по настоящему изобретению может образовывать фармацевтически приемлемую соль. Фармацевтически приемлемая соль может включать кислотно-аддитивную соль, которая образуется из неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической кислоты, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота, малеиновая кислота и салициловая кислота; или сульфоновой кислоты, такой как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота и п-толуолсульфоновая кислота, которые образуют нетоксичную кислотно-аддитивную соль, включая фармацевтически приемлемый анион. Кроме того, фармацевтически приемлемая соль карбоновой кислоты включает соль щелочного металла или щелочноземельного металла, такого как литий, натрий, калий, кальций и магний; соли с аминокислотой, такой как лизин, аргинин и гуанидин; органическую соль, такую как дициклогексиламин, N-метил-D-глюкамин, трис(гидроксиметил)метиламин, диэтаноламин, холин и триэтиламин. Соединение формулы (1) по настоящему изобретению можно превратить в ее соли общепринятыми способами.
Между тем, поскольку соединение формулы (1) по настоящему изобретению может иметь асимметричный углеродный центр и асимметричную ось или плоскость, они могут существовать в виде E- или Z-изомера, R- или S-изомера, рацемических смесей или смесей диастереоизомеров, и каждого диастереоизомера, все из которых входят в объем настоящего изобретения.
В настоящем описании, если не указано иное, термин «соединение формулы (1)» используется для обозначения всех соединений формулы (1), включая их фармацевтически приемлемые соли и изомеры.
В настоящем описании следующие понятия, определенные для заместителей, используются для определения соединения формулы (1).
Термин «галоген» или «гало» означает фтор (F), хлор (Cl), бром (Br) или йод (I).
Термин «алкил» или «алкилен» означает углеводороды с прямой или разветвленной цепью, может включать одинарную связь, двойную связь или тройную связь и предпочтительно представляет собой C1-C10 алкил или C1-C10 алкилен, или C1-C7 алкил или C1-C7 алкилен. Примеры алкила включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, ацетилен, винил, трифторметил и тому подобное.
Термин «алкенил» или «алкенилен» означает углеводороды с прямой или разветвленной цепью, имеющие по меньшей мере одну углерод-углеродную двойную связь, и предпочтительно представляет собой C2-C10 алкенил или C2-C10 алкенилен, или C2-C7 алкенил или C2-C7 алкенилен. Примеры алкенила включают, но не ограничиваются ими, винил, аллил, бутенил, изопропенил, изобутенил и тому подобное.
Термин «циклоалкил» означает частично или полностью насыщенные углеводороды с одинарным или конденсированным кольцом и предпочтительно представляет собой C3-C10-циклоалкил. Примеры циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и тому подобное.
Если не указано иное, термин «алкокси» означает алкилокси, имеющий от 1 до 10 атомов углерода.
Термин «арил» или «арилен» означает ароматические углеводороды, предпочтительно C5-C12 арил или C5-C12 арилен, более предпочтительно C6-C10 арил или C6-C10 арилен, и включает, но не ограничивается ими, фенил, нафтил и тому подобное.
Термин «гетероарил» или «гетероарилен» означает 3-12-членные, более предпочтительно 5-12-членные ароматические углеводороды, которые образуют одинарное или конденсированное кольцо, которое может быть конденсировано с бензо- или C3-C8-циклоалкилом, включая один или несколько гетероатомов, выбранных из N, O и S в качестве члена кольца. Примеры гетероарила включают, но не ограничиваются ими, пиридинил, пиримидинил, пиридазинил, пиразинил, оксадиазолил, изоксадиазолил, тетразолил, триазолил, индолил, индазолил, изоксазолил, оксазолил, тиазолил, изотиазолил, фуранил, бензофуранил, имидазолил, тиофенил, бензтиазол, бензимидазол, хинолинил, индолинил, 1,2,3,4-тетрагидроизохинолил, 3,4-дигидроизохинолинил, тиазолопиридил, 2,3-дигидробензофуран, 2,3-дигидротиофен, 2,3-дигидроиндол, бензо[1,3]диоксин, хроман, тиохроман, 1,2,3,4-тетрагидрохинолин, 4H-бензо[1,3]диоксин, 2,3-дигидробензо[1,4]диоксин, 6,7-дигидро-5H-циклопента[d]пиримидин и тому подобное.
Термин «гетероциклоалкил» или «гетероциклоалкилен» означает частично или полностью насыщенные углеводороды, которые образуют одинарное или конденсированное кольцо, включающее один или несколько гетероатомов, выбранных из N, O и S, и предпочтительно представляет собой 3-12-членный гетероциклоалкил или гетероциклоалкилен, или 5-12-членный гетероциклоалкил или гетероциклоалкилен. Примеры гетероциклоалкила или гетероциклоалкилена включают, но не ограничиваются ими, пирролидинил, пиперидинил, морфолинил, имидазолинил, пиперазинил, тетрагидрофуран, тетрагидротиофуран и тому подобное.
Согласно одному варианту осуществления настоящего изобретения в приведенной выше формуле (1)
A, D и E, каждый независимо, представляют собой CH или N;
R1 представляет собой C1-C7 алкил, C3-C10 циклоалкил или галоген-C1-C7 алкил;
R2 представляет собой -G-J-L;
где G представляет собой -C(=O)- или прямую связь;
J представляет собой C1-C7 алкилен, C2-C7 алкенилен, C1-C7 алкилен-C6-C10 арилен, C2-C7 алкенилен-C6-C10 арилен, C1-C7 алкоксиен-C6-C10 арилен, C6-C10 арилен, 5-12-членный гетероарилен-5-12-членный гетероциклоалкилен, 5-12-членный гетероарилен-C6-C10 арилен или 5-12-членный гетероарилен-окси-C3-C10 циклоалкилен;
L представляет собой водород, галоген, амино, нитро, карбокси, карбокси-C1-C7 алкил, карбокси-C1-C7 алкокси, C3-C10 циклоалкил или C6-C10 арил;
где алкил, алкилен, карбоксиалкил, карбоксиалкокси или арил необязательно замещен 1-4 заместителями, выбранными из гидрокси, галогена, C1-C7 алкила и C1-C7 алкокси; и
гетероциклоалкилен или гетероарилен включает 1-4 гетероатомов, выбранных из N, O и S.
Репрезентативные соединения формулы (1) по настоящему изобретению включают, но не ограничиваются ими, следующие соединения:
N-(6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)-3-фенилпропанамид;
метил 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)ацетат;
2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)уксусная кислота;
2-(4-(3-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-3-оксопропил)фенил)уксусная кислота;
метил 2-(4-(3-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропаноат;
этил 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-дифторацетат;
3-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановая кислота;
(R)-1-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновая кислота;
3-(3-(6-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановая кислота;
N-(6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)-3-фенилпропанамид;
2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)уксусная кислота;
2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)уксусная кислота;
2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенокси)-2-метилпропановая кислота;
2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил-2,2-дифторуксусная кислота;
3-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановая кислота;
2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропановая кислота;
(E)-2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопро-1-фен-1-ил)фенил)-2-метилпропановая кислота;
3-(4-(1-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропановая кислота;
2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенокси-2-метилпропановая кислота;
2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)фенил)уксусная кислота;
(1r,4r)-4-((2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)окси)циклогексан-1-карбоновая кислота;
N-(6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)-3-фенилпропанамид;
3-(4-(2-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановая кислота;
(R)-1-(2-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновая кислота;
3-(3-(6-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановая кислота;
2-(4-(3-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропановая кислота;
3-(4-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановая кислота;
2-(4-(3-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенокси)-2-метилпропановая кислота;
3-(4-(1-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропановая кислота;
2-(4-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенокси)-2-метилпропановая кислота;
(R)-1-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновая кислота;
3-(3-(6-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановая кислота; и
(1r,4r)-4-((2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)окси)циклогексан-1-карбоновая кислота.
Термины и сокращения, используемые в настоящем документе, сохраняют свои обычные значения, если не указано иное.
Настоящее изобретение также относится к способу получения соединения формулы (1). Далее в настоящем документе, способ получения соединения формулы (1) поясняется на примерах реакций для того, чтобы проиллюстрировать настоящее изобретение. Однако специалист в данной области может получить соединение формулы (1) различными способами, основанными на структуре формулы (1), и такие способы следует интерпретировать как входящие в объем настоящего изобретения. То есть соединение Формулы (1) может быть получено способами, описанными в настоящем документе, или комбинированием различных способов, раскрытых в предшествующем уровне техники, что следует интерпретировать как входящее в объем настоящего изобретения. Соответственно, способ получения соединения формулы (1) не ограничивается нижеследующими способами.
Соединение формулы (1) по настоящему изобретению может быть получено путем прямого введения замещенной аминогруппы в соединение (2) или введения защищенного амина в соединение (2), удаления защитной группы для получения соединения (3) и проведения реакции амидирования соединения (3) в соответствии с приведенной ниже схемой реакции 1.
[Схема реакции 1]
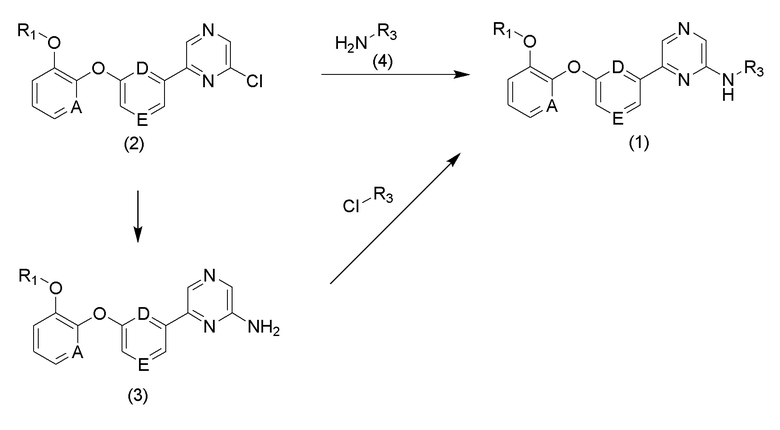
Соединение (2) может быть получено с использованием 2-этоксифенола в качестве исходного вещества в соответствии со способом, приведенным на схеме реакции 2 ниже.
[Схема реакции 2]
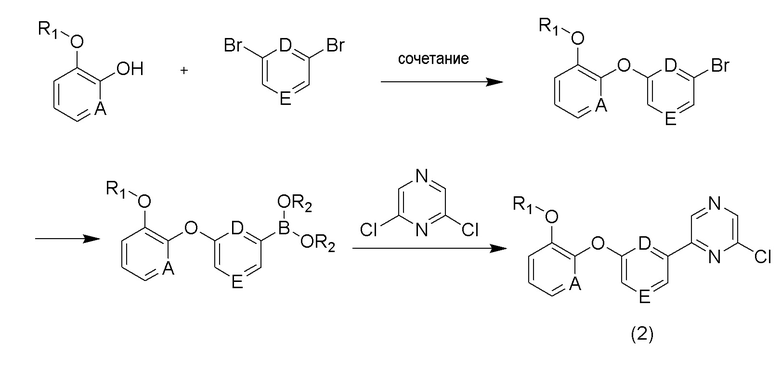
Кроме того, соединение (3) может быть получено в соответствии со способом, приведенным на схеме реакции 3 ниже.
[Схема реакции 3]
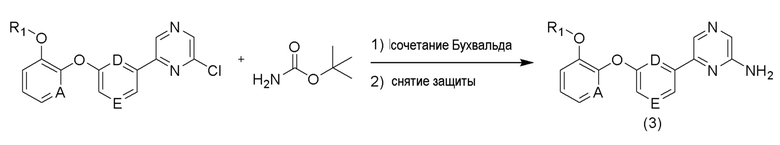
Среди соединений формулы (4) амидные производные могут быть получены обработкой тионилхлоридом или оксалилхлоридом из соответствующей кислоты с последующей обработкой водным аммиаком. Например, метил-4-(3-амино-3-оксопропил)бензоат можно получить в соответствии со способом, приведенным на схеме реакции 4 ниже. Среди соединений формулы (4) производные амина могут быть получены путем введения аминогруппы в соединение, полученное в результате реакции кросс-сочетания между промежуточным соединением диоксабороланового ядра и различными видами хлорарильных соединений для синтеза аминоарильных промежуточных соединений. Например, этил-2-(4-(2-аминопиримидин-4-ил)фенил)ацетат можно получить в соответствии со способом, приведенным на схеме реакции 5 ниже.
[Схема реакции 4]
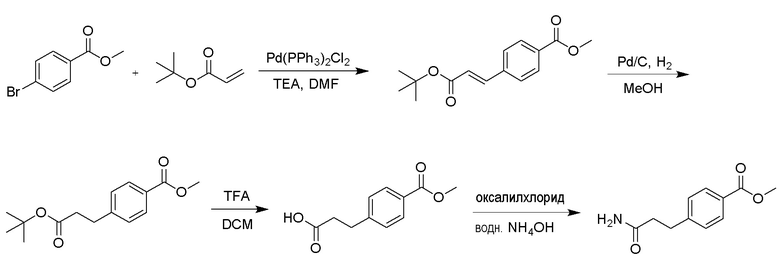
[Схема реакции 5]
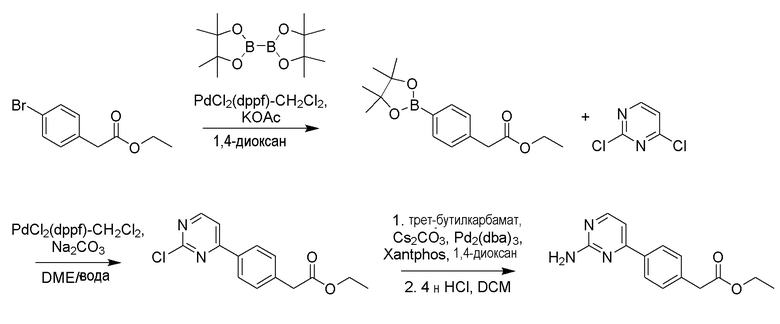
Соединение, конкретно не описанное в способе получения по настоящему описанию, представляет собой известное соединение или соединение, которое можно легко синтезировать из известного соединения с помощью известного способа синтеза или аналогичного способа.
Соединение формулы (1), полученное вышеуказанными способами, может быть выделено или очищено от продуктов реакции обычными способами, такими как перекристаллизация, ионосфера, колоночная хроматография на силикагеле или ионообменная хроматография.
Как упоминалось выше, соединения по настоящему изобретению, исходные вещества или промежуточные соединения для их получения могут быть получены различными способами, которые следует интерпретировать как входящие в объем настоящего изобретения в отношении получения соединения формулы (1).
Соединение формулы (1) по настоящему изобретению проявляет ингибирующую активность в отношении диацилглицерол-ацилтрансферазы 2 (DGAT2). Соответственно, настоящее изобретение предоставляет фармацевтическую композицию для лечения заболеваний, связанных с DGAT2, включающую соединение формулы (1) или его фармацевтически приемлемую соль или изомер вместе с фармацевтически приемлемым носителем. Различные виды пролекарств, которые превращаются в соединение формулы (1) in vivo, также входят в объем настоящего изобретения.
Типичные заболевания, связанные с DGAT2, которые можно лечить фармацевтической композицией по настоящему изобретению, включают, но не ограничиваются ими, заболевания, выбранные из группы, состоящей из жирового гепатоза, неалкогольного стеатогепатита (НАСГ), неалкогольной жировой болезни печени (НАЖБП), диабета, ожирения, гиперлипидемии, атеросклероз и гиперхолестеринемии.
В настоящем изобретении «фармацевтическая композиция» может включать другие компоненты, такие как носители, разбавители, эксципиенты и т.д., в дополнение к активному ингредиенту по настоящему изобретению. Соответственно, фармацевтическая композиция может включать фармацевтически приемлемые носители, разбавители, эксципиенты или их комбинации, если это необходимо. Фармацевтическая композиция облегчает введение соединений в организм. Различные способы введения соединений включают, но не ограничиваются ими, пероральное, инъекционное, аэрозольное, парентеральное и местное введение.
В настоящем документе «носитель» означает соединение, которое облегчает добавление соединений в клетку или ткань. Например, диметилсульфоксид (DMSO) является стандартным носителем, облегчающим введение многих органических соединений в живые клетки или ткани.
В настоящем документе «разбавитель» означает соединение, не только стабилизирующее биологически активную форму, но и разбавленное в растворителе, растворяющем соединения. Растворенная соль в буфере используется в качестве разбавителя в этой области. Традиционно используемый буфер является солевым раствором фосфатного буфера, имитирующим солевую форму в жидкости организма. Так как буферный раствор может регулировать pH раствора в низкой концентрации, буферный разбавитель не сильно изменяет биологическую активность соединений.
В настоящем документе «фармацевтически приемлемый» означает такое свойство, которое не ухудшает биологическую активность и физические свойства соединений.
Соединения согласно настоящему изобретению могут быть составлены как различные фармацевтически вводимые лекарственные формы. В получении фармацевтической композиции согласно настоящему изобретению, активный компонент, в частности, соединение формулы 1 или его фармацевтически приемлемую соль или изомер, смешивают с выбранными фармацевтически приемлемыми носителями, в зависимости от получаемой лекарственной формы. Например, фармацевтическая композиция согласно настоящему изобретению может быть составлена в форме инъекций, пероральных препаратов и т.п. в соответствии с необходимостью.
Соединение согласно настоящему изобретению может быть составлено стандартными способами с использованием известных фармацевтических носителей и эксципиентов и помещено в контейнеры, содержащие одну или несколько доз. Лекарственные формы могут представлять собой раствор, суспензию или эмульсию в масляном или водном растворителе и могут включать стандартные диспергирующие агенты, суспендирующие агенты или стабилизаторы. Кроме того, соединение может быть, например, в сухой порошковой форме, растворенной в стерилизованной апирогенной воде перед использованием. Соединение согласно настоящему изобретению может быть составлено в суппозитории с использованием стандартной основы для суппозитория, такой как масло какао или другие глицериды. Твердые формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. Капсулы и таблетки являются предпочтительными. Таблетки и пилюли предпочтительно имеют энтеросолюбильное покрытие. Твердые формы получают путем смешивания соединений согласно настоящему изобретению с по меньшей мере одним носителем, выбранным из инертных разбавителей, таких как сахароза, лактоза или крахмал, лубрикантов, таких как стеарат магния, разрыхлителей, связующих и тому подобное.
Соединение или содержащую его фармацевтическую композицию по настоящему изобретению можно вводить в комбинации с другими лекарственными средствами, например, с другими терапевтическими средствами для лечения метаболического расстройства, при необходимости.
Дозу соединения формулы (1) по настоящему изобретению определяют по назначению врача с учетом массы тела пациента, возраста и болезненного состояния. Типичная доза для взрослых находится в диапазоне примерно от 0,3 до 500 мг в день в зависимости от частоты и интенсивности введения. Типичная суточная доза для внутримышечного или внутривенного введения для взрослых находится в диапазоне примерно от 1 до 300 мг в день, которые можно вводить в виде разделенных стандартных доз. Некоторым пациентам требуется более высокая суточная доза.
В настоящем документе термин «лечение» используется для обозначения задержки, отсрочки или уменьшения интенсивности течения заболеваний у субъекта, демонстрирующего симптомы заболеваний.
Эффекты изобретения
Новое биарильное производное формулы (1) в соответствии с настоящим изобретением проявляет превосходную ингибирующую активность в отношении диацилглицерол-ацилтрансферазы 2 (DGAT2) и, таким образом, может быть эффективно использовано для профилактики, облегчения или лечения метаболических нарушений, связанных с DGAT2. Кроме того, новое биарильное производное формулы (1) согласно настоящему изобретению обладает повышенной липофильностью и селективностью в отношении печени, тем самым повышая эффективность за счет увеличения воздействия на печень, а также ожидая преимущества удобства приема, поскольку период полувыведения относительно длительный в моделях заболеваний на животных и в клинической практике.
СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее в настоящем документе, настоящее изобретение поясняется более подробно следующими примерами. Однако следует понимать, что объем правовой охраны настоящего изобретения не ограничивается примерами.
В следующих примерах М относится к молярной концентрации, а N относится к нормальной концентрации. Кроме того, описания аббревиатур и терминов, используемых в схеме реакции, примерах получения и примерах, являются следующими:
DCM: дихлорметан
DIPEA: N, N-диизопропилэтиламин
DMF: N, N-диметилформамид
DMSO: диметилсульфоксид
NMP: N-метилпирролидон
Pd(dppf)Cl2.CH2Cl2: комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) - дихлорметан (1:1)
TEA: триэтиламин
THF: тетрагидрофуран
PyBroP: бромтрипирролидинофосфония гексафторфосфат
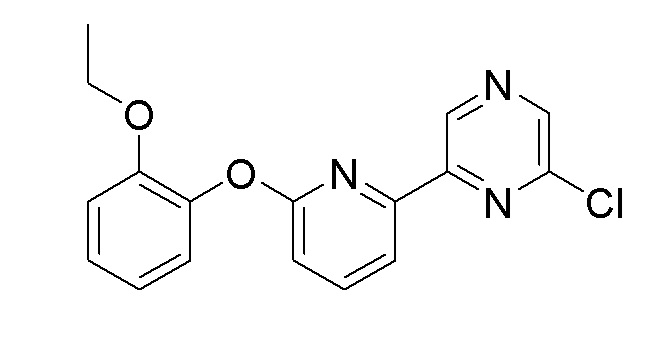
Пример получения 1: Синтез 2-хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразина
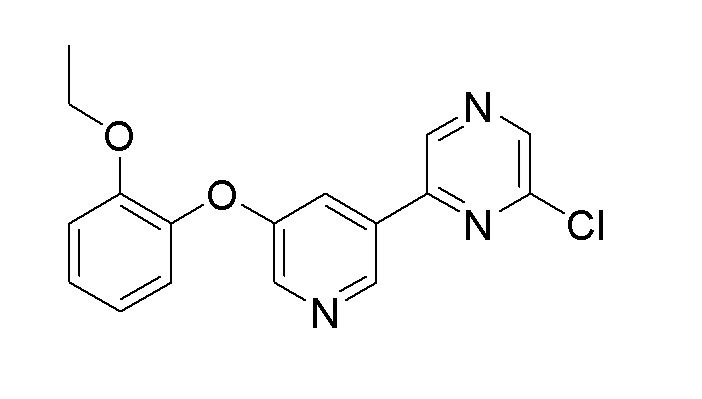
Стадия 1: Синтез 3-бром-5-(2-этоксифенокси)пиридина
60% гидрид натрия (1,82 г, 46 ммоль) добавляли к NMP (100 мл) при 0°C, и медленно по каплям добавляли 2-этоксифенол (6,1 г, 44 ммоль) в присутствии азота. После перемешивания реакционного раствора при комнатной температуре в течение 1 часа, 3,5-дибромпиридин (7,2 г, 30.4 ммоль) добавляли по каплям и перемешивали при 150°C в течение 72 часов. После завершения реакции, реакционную смесь охлаждали до комнатной температуры, разбавляли водой (120 мл), добавляли 5 н. водный раствор гидроксида натрия (15 мл), и экстрагировали эфиром. После сушки над сульфатом магния, растворитель удаляли при пониженном давлении, и очистку осуществляли с помощью колоночной хроматографии с получением целевого продукта (выход: 19,5%).
1H ЯМР (500 МГц, хлороформ-D): δ 8,32 (д, J=1,2 Гц, 1H), 8,26 (д, J=2,4 Гц, 1H), 7,28 (с, 1H), 7,20 (с, 1H), 7,09 (д, J=7,9 Гц, 1H), 7,04-6,87 (м, 2H), 4,01 (т, J=7,0 Гц, 2H), 1,24 (т, J=7,0 Гц, 3H)
Стадия 2: Синтез (5-(2-этоксифенокси)пиридин-3-ил)бороновой кислоты
3-Бром-5-(2-этоксифенокси)пиридин (1,74 г, 5,92 ммоль), полученный на стадии 1, 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (2,25 г, 8,87 ммоль, 1,5 экв.), ацетат калия (2,32 г, 23,66 ммоль) и Pd(dppf)Cl2.CH2Cl2 (48 мг, 0,06 ммоль) добавляли к толуолу (30 мл) и перемешивали при нагревании с обратным холодильником при 120°C в течение 12 часов. После завершения реакции, реакционную смесь фильтровали через слой целита, промывали толуолом, растворитель удаляли при пониженном давлении, и следующую реакцию проводили без отдельного процесса очистки.
m/z (M+H)+ рассчитано для C13H14BNO4: 259,0, найдено 260,1
Стадия 3: Синтез 2-хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразина
(5-(2-Этоксифенокси)пиридин-3-ил)бороновую кислоту (1,53 г, 5,92 ммоль), полученную на стадии 2, 2,6-дихлорпиразин (0,97 г, 6,5 ммоль, 1,1 экв.), карбонат натрия (1,25 г, 11,81 ммоль) и Pd(dppf)Cl2.CH2Cl2 (48 мг, 0,06 ммоль) добавляли к 1,4-диоксану (20 мл)/воде (1 мл) и перемешивали при нагревании с обратным холодильником при 120°C в течение 12 часов. После завершения реакции, полученный продукт фильтровали через слой целита, промывали толуолом, растворитель удаляли при пониженном давлении и очистку осуществляли с помощью колоночной хроматографии с получением целевого продукта (двухстадийный выход: 39%).
1H-ЯМР (400 МГц, хлороформ-D) δ 8,90 (с, 2H), 8,57 (с, 1H), 8,41 (д, j=4 Гц, 1H), 7,81 (д, J=4 Гц, 1H), 7,20 (м, 1H), 7,13 (м, 1H), 7,02 (м, 2H), 4,06 (кв, 2H), 1,24 (т, 3H)
Пример получения 2: Синтез 2-хлор-6-(3-(2-этоксифенокси)фенил)пиразина
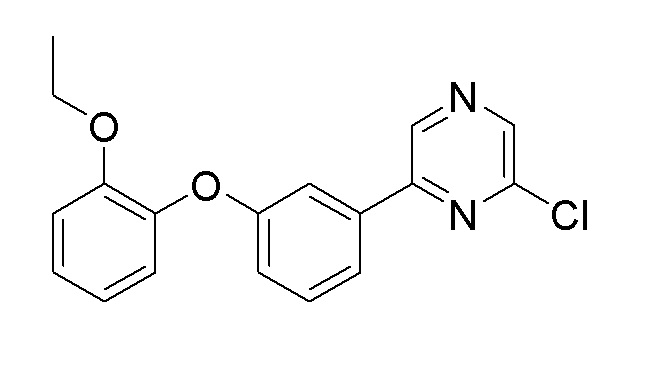
Стадия 1: Синтез 1-(3-бромфенокси)-2-этоксибензола
2-Этоксифенол (4,33 г, 31,4 ммоль), 1-бром-3-йодбензол (6 мл, 47,1 ммоль), хлорид меди(I) (1,553 г, 15,69 ммоль), 2,2,6,6-тетраметил-3,5-гептадион (1,310 мл, 6,27 ммоль) и карбонат цезия (10,22 г, 31,4 ммоль) растворяли в 70 мл NMP и нагревали до 120°C. После перемешивания в течение 16 часов, реакционную смесь охлаждали до комнатной температуры. Реакцию останавливали 1 н. водным раствором хлористоводородной кислоты, затем экстрагировали диэтиловым эфиром. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом магния, и органический растворитель удаляли при пониженном давлении. Очистку осуществляли на колонке с силикагелем (этилацетат:гексан=1:5) с получением целевого продукта (выход: 96%).
1H-ЯМР (500 МГц, хлороформ-D) δ: 7,14-7,12 (м, 3H), 7,04-7,03 (м, 2H), 7,00-6,98 (м, 1H), 6,97-6,93 (м, 1H), 6,88-6,86 (м, 1H), 4,03 (кв, 2H, J=7,35 Гц), 1,26 (т, 3H, J=7,03 Гц)
Стадия 2: Синтез 2-(3-(2-этоксифеноксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана
1-(3-Бромфенокси)-2-этоксибензол (1,74 г, 5,94 ммоль), полученный на стадии 1, 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (2,26 г, 8,90 ммоль, 1,5 экв.), ацетат калия (2,33 г, 23,74 ммоль) и Pd(dppf)Cl2.CH2Cl2 (48 мг, 0,06 ммоль) добавляли к толуолу (30 мл) и перемешивали при нагревании с обратным холодильником при 120°C в течение 12 часов. После завершения реакции, полученный продукт фильтровали через слой целита, промывали толуолом, растворитель удаляли при пониженном давлении, и очистку осуществляли с помощью колоночной хроматографии с получением целевого продукта (выход: 45%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,48 (д, 1H), 7,43 (с, 1H), 7,28 (т, 1H), 7,09 (т, 1H), 6,94 ~ 7,03 (м, 4H), 4,06 (кв, 2H), 1,32 (с, 12H), 1,28 (т, 3H)
Стадия 3: Синтез 2-хлор-6-(3-(2-этоксифенокси)фенил)пиразина
2-(3-(2-этоксифеноксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан, полученный на стадии 2 (0,9 г, 2,65 ммоль), 2,6-дихлорпиразин (0,43 г, 2,91 ммоль, 1,1 экв.), карбонат натрия (0,56 г, 5,29 ммоль) и Pd(dppf)Cl2.CH2Cl2 (22 мг, 0,03 ммоль) добавляли к 1,4-диоксану (20 мл)/воде (1 мл) и перемешивали при нагревании с обратным холодильником при 120°C в течение 12 часов. После завершения реакции, полученный продукт фильтровали через слой целита, промывали толуолом, растворитель удаляли при пониженном давлении и очистку осуществляли с помощью колоночной хроматографии с получением целевого продукта (выход: 75%).
1H-ЯМР (500 МГц, хлороформ-D) δ 8,85 (с, 1H), 8,49 (с, 1H), 7,68 (д, 1H), 7,61 (с, 1H), 7,40 (т, 1H), 7,14 (т, 1H), 7,05 (д, J=6 Гц, 1H), 7,02 (д, J=6 Гц, 2H), 6,95 (т, 1H), 4,05 (кв, 2H), 1,26 (т, 3H)
Пример получения 3: Синтез 2-хлор-6-(6-(2-этоксифенокси)пиридин-2-ил)пиразина
2-Этоксифенол (1,5 г, 10,86 ммоль) и 2,6-дибромпиридин (3,86 г, 16,28 ммоль) использовали таким же образом, как в примере получения 2, с получением целевого продукта (выход: 44,9%).
m/z (M+H)+ рассчитано для C17H14ClN3O2: 327,77, найдено 328,0
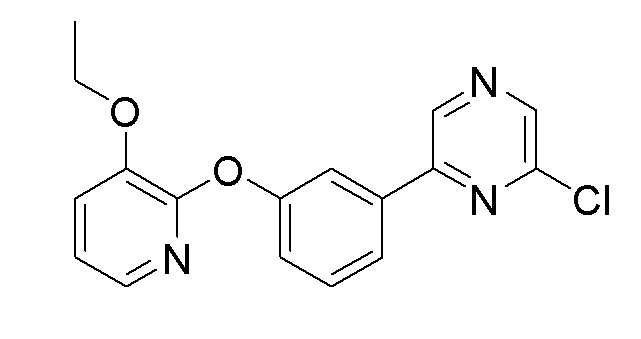
Пример получения 4: Синтез 2-хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразина
Стадия 1: Синтез 3-этоксипиридин 1-оксид
3-Этоксипиридин (1,683 г, 13,67 ммоль) растворяли в DCM (32,5 мл) и затем добавляли м-хлорпероксибензойную кислоту (3,07 г, 17,77 ммоль) при 10°C и перемешивали при комнатной температуре в течение 22 часов. Добавляли тиосульфат натрия и перемешивали при 15°C в течение 3 часов. После завершения реакции, полученный продукт экстрагировали DCM. Растворитель удаляли при пониженном давлении и очистку осуществляли с помощью колоночной хроматографии с получением целевого продукта (выход: 87%).
1H-ЯМР (500 МГц, хлороформ-D) δ 7,96 (т, J=2,0 Гц, 1H), 7,92-7,83 (м, 1H), 7,15 (дд, J=8,7, 6,3 Гц, 1H), 6,86 (дд, J=8,5, 2,1 Гц, 1H), 4,05 (кв, J=7,0 Гц, 2H), 1,44 (т, J=6,9 Гц, 3H)
Стадия 2: Синтез 2-(3-бромфенокси)-3-этоксипиридина
3-Этоксипиридин 1-оксид (825 мг, 5,93 ммоль), полученный на стадии 1, и 3-бромфенол (1,02 г, 5,93 ммоль) растворяли в THF (19 мл), и добавляли DIPEA (3,83 мл, 21,94 ммоль) и PyBroP (3,59 г, 7,71 ммоль) и перемешивали при комнатной температуре в течение 17 часов. После завершения реакции, полученный продукт концентрировали при пониженном давлении, разбавляли DCM, и органический слой промывали 1 н. водным раствором гидроксида натрия. Полученный продукт сушили над сульфатом магния, органический растворитель удаляли при пониженном давлении, и очистку осуществляли на колонке с силикагелем с получением целевого продукта (выход: 88%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,73 (дд, J=5,0, 1,4 Гц, 1H), 7,28 (тд, J=3,4, 1,8 Гц, 2H), 7,24-7,19 (м, 2H), 7,12-7,02 (м, 2H), 7,02-6,91 (м, 1H), 6,82-6,66 (м, 1H), 4,13 (кв, J=7,0 Гц, 2H), 1,46 (тд, J=7,1, 4,6 Гц, 3H)
Стадия 3: Синтез 2-хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразина
2-(3-Бромфенокси)-3-этоксипиридин (1,54 г, 5,24 ммоль), полученный на стадии 2, использовали таким же образом, как на стадиях 2 и 3 примера получения 2, с получением целевого продукта (выход: 24,4%).
1H-ЯМР (400 МГц, хлороформ-D): δ 8,90 (с, 1H), 8,49 (с, 1H), 7,84 (дд, J=8,7, 1,4 Гц, 2H), 7,71 (дд, J=4,8, 1,6 Гц, 1H), 7,52 (т, J=7,8 Гц, 1H), 7,33-7,26 (м, 1H), 7,24-7,15 (м, 1H), 6,98 (дд, J=7,8, 5,0 Гц, 1H), 4,17 (кв, J=7,0 Гц, 2H), 1,48 (т, J=7,1 Гц, 3H)
Пример получения 5: Синтез 3-фенилпропанамида
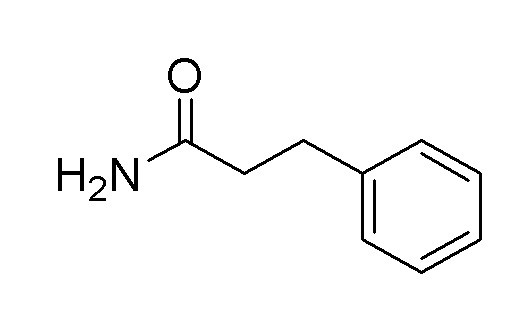
3-Фенилпропаноилхлорид (4,5 мл, 30,3 ммоль), растворенный в THF (46 мл), добавляли по каплям к водному раствору аммиака (189 мл) при 0°C, затем перемешивали в течение 1 часа. Органический растворитель удаляли при пониженном давлении, разбавляли водой и экстрагировали этилацетатом. Органический растворитель сушили над сульфатом магния и удаляли при пониженном давлении. Очистку осуществляли на колонке с силикагелем с получением целевого продукта (выход: 100%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,29-7,25 (м, 2H), 7,22-7,20 (м, 3H), 5,44 (с, 1H), 5,35 (с, 1H), 2,97 (т, J=7,6 Гц, 2H), 2,53 (т, J=7,6 Гц, 2H)
Пример получения 6: Синтез метил 2-(4-(2-амино-2-оксоэтил)фенил)ацетата
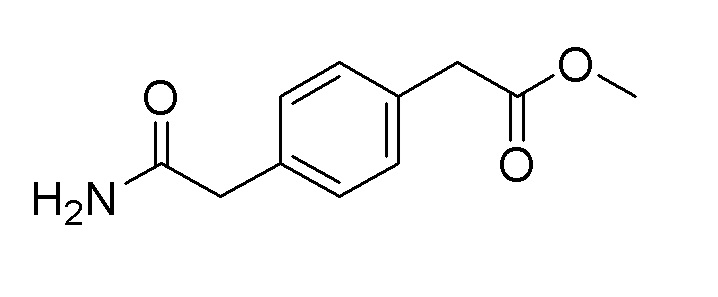
Стадия 1: Синтез диметил 2,2'-(1,4-фенилен)диацетата
Ацетилхлорид (2,9 мл, 40,8 ммоль) медленно по каплям добавляли к метанолу (20 мл) при 0°C. Затем, растворяли 1,4-фенилендиуксусную кислоту (4,0 г, 20,6 ммоль) и реакционную смесь перемешивали при нагревании с обратным холодильником в течение 5 часов. После подтверждения завершения реакции с помощью TLC, полученный продукт охлаждали до комнатной температуры, и органический растворитель удаляли при пониженном давлении. Продукт реакции разбавляли 100 мл этилацетата, промывали водным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушили над сульфатом магния и органический растворитель удаляли при пониженном давлении с получением целевого продукта.
1H-ЯМР (500 МГц, хлороформ-D): δ 7,24 (с, 2H), 3,68 (с, 3H), 3,61 (с, 2H)
Стадия 2: Синтез 2-(4-(2-метокси-2-оксоэтил)фенил)уксусной кислоты
Диметил 2,2'-(1,4-фенилен)диацетат (4,58 г, 20,6 ммоль), полученный на стадии 1, растворяли в THF (30 мл) и метаноле (10 мл), и добавляли 10 мл 2 н. гидроксида натрия медленно по каплям и перемешивали при комнатной температуре в течение 3 часов. Органический растворитель удаляли при пониженном давлении, разбавляли водой и подкисляли 2 н. раствором хлористоводородной кислоты. После экстракции этилацетатом, органический растворитель сушили над сульфатом магния и удаляли при пониженном давлении. Осуществляли перекристаллизацию с получением целевого продукта (выход: 30%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,25 (д, J=4,9 Гц, 4H), 3,68 (с, 3H), 3,66-3,62 (2H), 3,61 (с, 2H)
Стадия 3: Синтез метил 2-(4-карбамоилфенокси)-2-метилпропаноата
2-(4-(2-Метокси-2-оксоэтил)фенил)уксусную кислоту (1,0 г, 4,8 ммоль), полученную на стадии 2, растворяли в 30 мл дихлорметана и медленно по каплям добавляли тионилхлорид (0,7 мл, 9,6 ммоль) при комнатной температуре. После перемешивания при комнатной температуре в течение 4 часов, органический растворитель удаляли при пониженном давлении. Полученный продукт растворяли в 5 мл THF и затем медленно по каплям добавляли к 25% водному раствору аммиака при 0°C. После перемешивания в течение 1 часа, полученное твердое вещество фильтровали с получением целевого продукта (выход: 74%).
1H-ЯМР (500 МГц, DMSO-D6): δ 7,42 (с, 1H), 7,15 (дд, J=12,2, 7,9 Гц, 4H), 6,83 (с, 1H), 3,60 (с, 2H), 3,57 (д, J=4,3 Гц, 3H), 3,30 (с, 2H)
Пример получения 7: Синтез метил 2-(4-(3-амино-3-оксопропил)фенил)ацетата
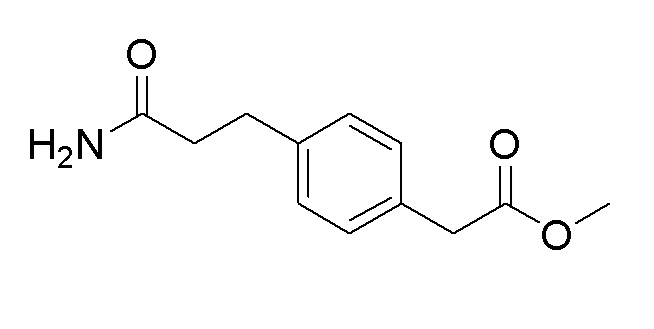
Стадия 1: Синтез трет-бутил (E)-3-(4-(2-метокси-2-оксоэтил)фенил)акрилата
Метил 2-(4-бромфенил)ацетат (16,4 г, 71,56 ммоль), трет-бутил акрилат (18,0 г, 143,0 ммоль) и триэтиламин (50 мл, 0,35 моль) растворяли в 200 мл диметилформамида. После удаления растворенного кислорода барботированием азота, добавляли по каплям бис(трифенилфосфин)палладия дихлорид (2,5 г, 3,58 ммоль) и перемешивали при 75°C в течение 12 часов. Органический растворитель удаляли при пониженном давлении, разбавляли этилацетатом, промывали насыщенным солевым раствором, сушили над сульфатом магния, и затем органический растворитель удаляли при пониженном давлении. Очистку осуществляли на колонке с силикагелем (этилацетат:гексан=1:3) с получением целевого продукта (выход: 79%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,56 (д, J=15,9 Гц, 1H), 7,46 (д, J=8,7 Гц, 2H), 7,28 (д, J=7,9 Гц, 2H), 6,34 (д, J=15,9 Гц, 1H), 3,70 (с, 3H), 3,64 (с, 2H), 1,53 (с, 9H)
Стадия 2: Синтез трет-бутил 3-(4-(2-метокси-2-оксоэтил)фенил)пропаноата
Трет-бутил (E)-3-(4-(2-метокси-2-оксоэтил)фенил)акрилат (5,0 г, 18,0 ммоль), полученный на стадии 1, растворяли в 50 мл метанола, и добавляли по каплям палладированный уголь (0,5 г, 0,452 ммоль). Реакцию восстановления проводили с помощью водорода из баллона. После подтверждения завершения реакции, полученный продукт фильтровали через слой целита, и органический растворитель удаляли при пониженном давлении с получением целевого продукта (выход: 93%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,17 (дд, J=18,3, 7,9 Гц, 4H), 3,68 (с, 3H), 3,59 (с, 2H), 2,88 (т, J=7,9 Гц, 2H), 2,52 (т, J=7,6 Гц, 2H), 1,41 (с, 9H)
Стадия 3: Синтез 3-(4-(2-метокси-2-оксоэтил)фенил)пропановой кислоты
Трет-бутил 3-(4-(2-метокси-2-оксоэтил)фенил)пропаноат (4,67 г, 16,8 ммоль), полученный на стадии 2, растворяли в 100 мл раствора 20% трифторуксусной кислоты/дихлорметана и перемешивали при комнатной температуре в течение 2 часов. После подтверждения завершения реакции, органический растворитель удаляли при пониженном давлении и проводили перекристаллизацию с получением целевого продукта (выход: 100%).
1H-ЯМР (500 МГц, хлороформ-D): δ 9,58 (с, 2H), 7,18 (дд, J=19,0, 7,9 Гц, 4H), 3,70 (с, 3H), 3,61 (с, 2H), 2,95 (т, J=7,6 Гц, 2H), 2,69 (т, J=7,9 Гц, 2H)
Стадия 4: Синтез метил 2-(4-(3-амино-3-оксопропил)фенил)ацетата
3-(4-(2-Метокси-2-оксоэтил)фенил)пропановую кислоту (3,73 г, 16,8 ммоль), полученную на стадии 3, использовали таким же образом, как на стадии 3 примера получения 6, с получением целевого продукта (выход: 65%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,18 (кв, J=7,7 Гц, 4H), 5,41 (с, 2H), 3,66 (д, J=15,9 Гц, 3H), 3,59 (с, 2H), 3,02-2,87 (2H), 2,51 (т, J=7,6 Гц, 2H)
Пример получения 8: Синтез метил 2-(4-(3-амино-3-оксопропил)фенил-2-метилпропаноата
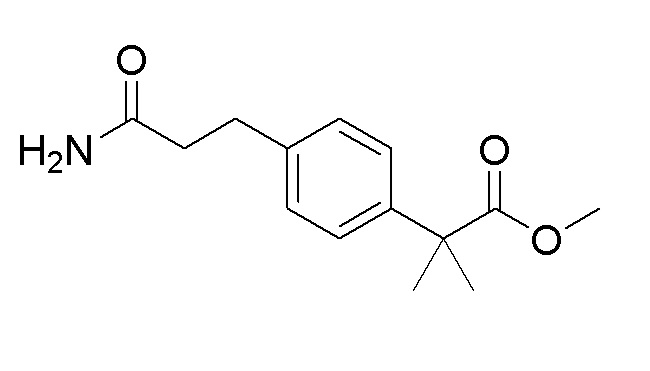
Стадия 1: Синтез трет-бутил (E)-3-(4-(1-метокси-2-метил-1-оксопропан-2-ил)фенил)акрилата
Метил 2-(4-бромфенил)-2-метилпропаноат (1,0 г, 3,89 ммоль) и трет-бутил акрилат (0,98 г, 7,8 ммоль) использовали таким же образом, как на стадии 1 примера получения 7, с получением целевого продукта (выход: 79%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,56 (дд, J=15,9, 4,3 Гц, 1H), 7,51-7,42 (2H), 7,41-7,31 (м, 2H), 6,34 (дд, J=15,9, 4,9 Гц, 1H), 3,66 (д, J=4,9 Гц, 3H), 1,58 (д, J=4,9 Гц, 6H), 1,53 (д, J=4,9 Гц, 9H)
Стадия 2: Синтез метил 2-(4-(3-(трет-бутокси)-3-оксопропил)фенил)-2-метилпропаноата
Трет-бутил (E)-3-(4-(1-метокси-2-метил-1-оксопропан-2-ил)фенил)акрилат (0,93 г, 3,06 ммоль), полученный на стадии 1, использовали таким же образом, как на стадии 2 примера получения 7, посредством реакции восстановления с получением целевого продукта (выход: 96%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,23 (с, 2H), 7,15 (д, J=7,9 Гц, 2H), 3,63 (с, 3H), 2,87 (т, J=7,9 Гц, 2H), 2,52 (т, J=7,9 Гц, 2H), 1,55 (с, 6H), 1,40 (с, 9H)
Стадия 3: Синтез 3-(4-(1-метокси-2-метил-1-оксопропан-2-ил)фенил)пропановой кислоты
Метил 2-(4-(3-(трет-бутокси)-3-оксопропил)фенил)-2-метилпропаноат (0,90 г, 2,92 ммоль), полученный на стадии 2, использовали таким же образом, как на стадии 3 примера получения 7, с получением целевого продукта (выход: 96%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,26 (д, J=7,3 Гц, 2H), 7,16 (д, J=7,9 Гц, 2H), 3,66 (с, 3H), 3,03-2,84 (2H), 2,82-2,55 (2H), 1,56 (с, 6H)
Стадия 4: Синтез метил 2-(4-(3-амино-3-оксопропил)фенил)-2-метилпропаноата
3-(4-(1-метокси-2-метил-1-оксопропан-2-ил)фенил)пропановую кислоту (0,7 г, 2,8 ммоль), полученную на стадии 3, использовали таким же образом, как на стадии 3 примера получения 6, посредством реакции амидирования с получением целевого продукта (выход: 99%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,25 (дд, J=6,4, 2,1 Гц, 2H), 7,17 (д, J=7,9 Гц, 2H), 5,36 (с, 2H), 3,64 (с, 3H), 3,00-2,90 (2H), 2,52 (т, J=7,6 Гц, 2H), 1,56 (д, J=4,3 Гц, 6H)
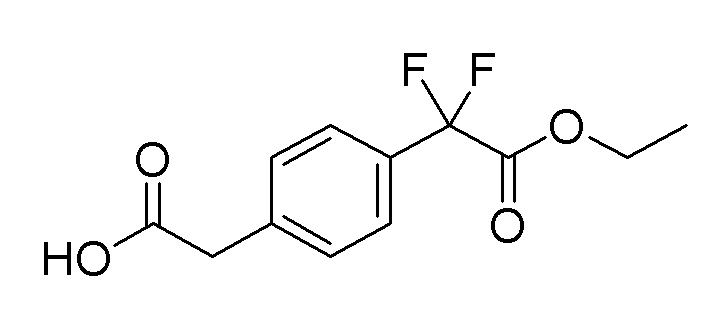
Пример получения 9: Синтез 2-(4-(2-этокси-1,1-дифтор-2-оксоэтил)фенил)уксусной кислоты
Стадия 1: Синтез трет-бутил 2-(4-йодофенил)ацетата
Трет-бутанол (130 мл) добавляли к 2-(4-йодофенил)уксусной кислоте (13,0 г, 49,6 ммоль) и перемешивали при барботировании азотом до тех пор, пока смесь не становилась прозрачной. Добавляли ди-трет-бутилдикарбонат (10,83 г, 49,6 ммоль) и перемешивали до растворения, затем добавляли 4-диметиламинопиридин (6,06 г, 49,6 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Органический растворитель концентрировали при пониженном давлении, и очистку осуществляли на колонке с силикагелем (этилацетат:н-гексан=1:9) с получением целевого продукта (выход: 68,9%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,63 (д, J=7,95 Гц, 2H), 7,01 (д, J=8,55 Гц, 2H), 3,45 (с, 2H), 1,42 (с, 9H)
Стадия 2: Синтез этил 2-(4-(2-(трет-бутокси)-2-оксоэтил)фенил)-2,2-дифторацетата
Трет-бутил 2-(4-йодофенил)ацетат (8,4 г, 26,4 ммоль), полученный на стадии 1, и 2-бром-2,2-дифторацетат (5,36 г, 26,4 ммоль) добавляли к порошку активированной меди (4,37 г, 68,6 ммоль), растворенному в DMSO (80 мл). После перемешивания при 60°C в течение 12 часов, полученный продукт выливали на лед и водный раствор хлорида аммония, затем экстрагировали диэтиловым эфиром. Органический слой промывали водным раствором хлорида аммония и насыщенным солевым раствором, и затем сушили над сульфатом магния. Органический слой концентрировали при пониженном давлении и очищали на колонке с силикагелем (этилацетат:н-гексан=1:9) с получением целевого продукта (выход: 60%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,56 (д, J=8 Гц, 2H), 7,36 (д, J=8 Гц, 2H), 4,31 (кв, J=8 Гц, 2H), 1,44 (с, 9H), 1,30 (т, J=8 Гц, 3H)
Стадия 3: Синтез 2-(4-(2-этокси-1,1-дифтор-2-оксоэтил)фенил)уксусной кислоты
Этил 2-(4-(2-(трет-бутокси)-2-оксоэтил)фенил)-2,2-дифторацетат (5 г, 15,91 ммоль), полученный на стадии 2, растворяли в DCM (10 мл), и затем добавляли трифторуксусную кислоту (15 мл), растворенную в DCM (50 мл), и перемешивали при комнатной температуре в течение 1 часа. После добавления толуола и удаления растворителя при пониженном давлении целевой продукт получали без дополнительной очистки (выход: 100%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,58 (д, J=7,95 Гц, 2H), 7,37 (д, J=7,95 Гц, 2H), 4,28 (кв, J=6,7 Гц, 2H), 3,69 (с, 2H), 1,30 (т, J=7,03 Гц, 3H)
Пример получения 10: Синтез трет-бутил 3-(4-(2-амино-2-оксоэтил)фенил)-2,2-диметилпропаноата
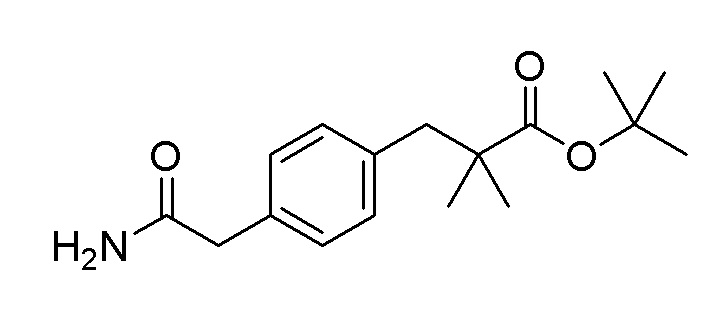
Стадия 1: Синтез 2-(4-(3-трет-бутокси-2,2-диметил-3-оксопропил)фенил)уксусной кислоты
Диизопропиламин (14,0 мл, 98 ммоль) добавляли к безводному тетрагидрофурану (164 мл) и к этому медленно по каплям добавляли 2,5 M н-бутиллития (39,3 мл, 98 ммоль) при -78°C. Реакционный раствор перемешивали при той же температуре в течение 20 минут. После повышения температуры до комнатной температуры и перемешивания в течение 10 минут температуру реакционного раствора снова понижали до -78°C и перемешивали в течение 10 минут. К реакционному раствору, добавляли по каплям трет-бутилизобутират (14,16 г, 98 ммоль), растворенный в безводном тетрагидрофуране (163 мл). Реакционный раствор перемешивали при -78°C в течение 1 часа и медленно добавляли по каплям к 2-(4-(бромметил)фенил)уксусной кислоте (7,5 г, 32,7 ммоль), растворенной в безводном тетрагидрофуране (163 мл). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 20 минут. Реакцию останавливали, добавляя к реакционному раствору 1 н. водную хлористоводородную кислоту (100 мл), затем экстрагировали диэтиловым эфиром. Органический слой концентрировали при пониженном давлении и очищали на колонке с силикагелем (метанол:дихлорметан=1:9) с получением целевого продукта (выход: 92%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,20 (д, J=7,9 Гц, 2H), 7,14 (д, J=7,9 Гц, 2H), 3,64 (с, 2H), 2,83 (с, 2H), 1,63-1,40 (м, 9H), 1,18-1,06 (6H)
Стадия 2: Синтез трет-бутил 3-(4-(2-амино-2-оксоэтил)фенил)-2,2-диметилпропаноата
2-(4-(3-Трет-бутокси-2,2-диметил-3-оксопропил)фенил)уксусную кислоту (6,82 г, 23,33 ммоль), полученную на стадии 1, использовали таким же образом, как на стадии 3 примера получения 6, с получением целевого продукта (выход: 52,7%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,20-7,09 (м, 4H), 5,33 (д, J=36,1 Гц, 2H), 3,54 (с, 2H), 2,80 (с, 2H), 1,42 (с, 9H), 1,11 (с, 6H)
Пример получения 11: Синтез этил (R)-1-(2-аминопиримидин-4-ил)пиперидин-3-карбоксилата
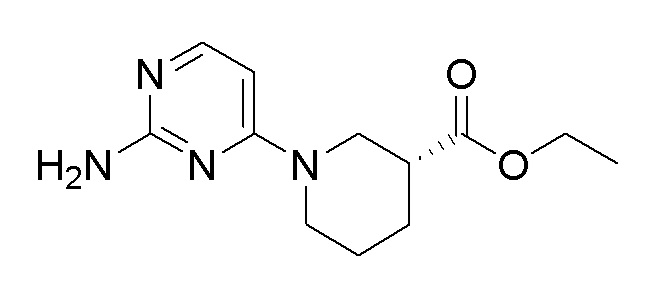
Стадия 1: Синтез этил (R)-1-(2-хлорпиримидин-4-ил)пиперидин-3-карбоксилата
2,4-Дихлорпиримидин (0,5 г, 3,36 ммоль) растворяли в этаноле (6,71 мл), и добавляли этил (R)-пиперидин-3-карбоксилат (0,621 мл, 4,03 ммоль) и TEA (0,187 мл, 1,343 ммоль). Реакционную смесь перемешивали при 85°C в течение 3 часов. После удаления растворителя при пониженном давлении, полученный продукт растворяли в этилацетате и промывали водой. Очистку осуществляли на колонке с силикагелем с получением целевого продукта (выход: 86%).
1H-ЯМР (500 МГц, хлороформ-D): δ 8,05 (д, J=6,1 Гц, 1H), 6,47 (д, J=6,4 Гц, 1H), 4,51-4,11 (м, 3H), 4,06 (с, 1H), 3,43 (дд, J=13,4, 9,5 Гц, 1H), 3,36-3,21 (м, 1H), 2,67-2,46 (м, 1H), 2,22-2,03 (м, 1H), 1,96-1,79 (м, 2H), 1,69-1,59 (м, 1H), 1,33-1,23 (м, 3H)
Стадия 2: Синтез этил (R)-1-(2-((трет-бутоксикарбонил)амино)пиримидин-4-ил)пиперидин-3-карбоксилата
После растворения этил (R)-1-(2-хлорпиримидин-4-ил)пиперидин-3-карбоксилата (0,78 г, 2,89 ммоль), полученного на стадии 1, трет-бутилкарбамата (0,407 г, 3,47 ммоль), карбоната цезия (2,36 г, 7,23 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантина (0,201 г, 0,347 ммоль) и трис(дибензилиденацетон)дипалладия(0) (0,265 г, 0,289 ммоль) в 50 мл 1,4-диоксана, растворенный кислород удаляли барботированием азота при перемешивании, и затем перекрывали поступление наружного воздуха в герметичном контейнере. Реакционную смесь перемешивали при 145°C в течение 6 часов и затем охлаждали до комнатной температуры. После фильтрации через слой целита и удаления органического растворителя при пониженном давлении, полученный продукт растворяли в этилацетате и промывали насыщенным солевым раствором. Органический растворитель сушили над сульфатом магния и удаляли при пониженном давлении. Очистку осуществляли на колонке с силикагелем с получением целевого продукта (выход: 11,8%).
m/z (M+H)+ рассчитано для C17H26N4O4: 350,42, найдено 351,2
Стадия 3: Синтез этил (R)-1-(2-аминопиримидин-4-ил)пиперидин-3-карбоксилата
После растворения этил (R)-1-(2-((трет-бутоксикарбонил)амино)пиримидин-4-ил)пиперидин-3-карбоксилата (0,120 г, 0,342 ммоль), полученного на стадии 2, в DCM (3 мл), добавляли трифторуксусную кислоту (0,3 мл), растворенную в DCM, и перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя при пониженном давлении, полученный продукт растворяли в DCM и промывали водой. Очистку осуществляли на колонке с силикагелем с получением целевого продукта (выход: 58,3%).
1H-ЯМР (500 МГц, хлороформ-D): δ 7,68 (д, J=6,7 Гц, 1H), 6,49-6,21 (1H), 6,07 (д, J=6,7 Гц, 1H), 4,31 (д, J=13,1 Гц, 1H), 4,16 (кв, J=7,1 Гц, 2H), 4,01 (д, J=13,1 Гц, 1H), 3,43-3,30 (1H), 3,30-3,16 (1H), 2,63-2,47 (м, 1H), 2,19-2,03 (м, 1H), 1,91-1,76 (м, 2H), 1,65-1,46 (м, 1H), 1,26 (т, J=7,0 Гц, 3H)
Пример получения 12: Синтез трет-бутил 3-(3-(6-аминопиридин-2-ил)фенил)-2,2-диметилпропаноата
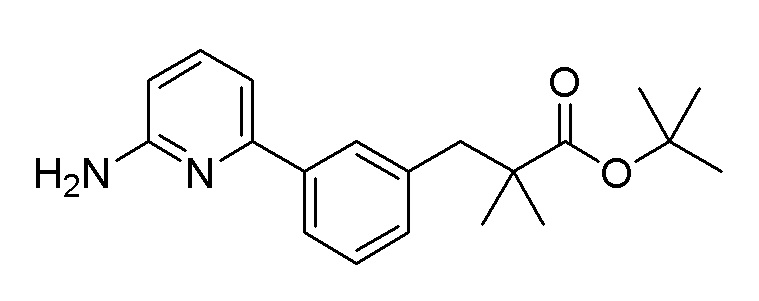
Стадия 1: Синтез трет-бутил 3-(3-бромфенил)-2,2-диметилпропаноата
1-Бром-3-(бромметил)бензол (20,0 г, 80 ммоль) использовали таким же образом, как на стадии 1 получения 10, с получением целевого продукта (выход 77%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,37-7,30 (м, 2H), 7,16-7,04 (м, 2H), 2,78 (с, 2H), 1,44 (с, 9H), 1,13 (с, 6H)
Стадия 2: Синтез трет-бутил 2,2-диметил-3-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил-пропаноата
После растворения трет-бутил 3-(3-бромфенил)-2,2-диметилпропаноата (19,3 г, 61,6 ммоль), полученного на стадии 1, 4,4,4,4,5,5,5,5-октаметил-2,2-би(1,3,2-диоксаборолан) (18,78 г, 73,9 ммоль), ацетата калия (18,14 г, 185 ммоль) и Pd(dppf)Cl2.CH2Cl2 (2,52 г, 3,08 ммоль) в 616 мл 1,4-диоксана, растворенный кислород удаляли барботированием азота при перемешивании, и затем перекрывали поступление наружного воздуха в герметичном контейнере. Реакционную смесь перемешивали при 110°C в течение 16 часов и затем охлаждали до комнатной температуры. После фильтрации через слой целита и удаления органического растворителя при пониженном давлении, очистку осуществляли на колонке с силикагелем (этилацетат:гексан) с получением целевого продукта (выход: 69,8%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,72-7,55 (м, 2H), 7,26-7,17 (м, 2H), 2,83 (с, 2H), 1,45 (с, 9H), 1,33 (с, 12H), 1,13 (с, 6H)
Стадия 3: Синтез трет-бутил 3-(3-(6-аминопиридин-2-ил)-фенил)-2,2-диметилпропаноата
После растворения 6-хлорпиридин-2-амина (5,53 г, 43 ммоль), трет-бутил 2,2-диметил-3-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил-пропаноата (15,5 г, 43 ммоль), полученного на стадии 2, 2 М водного раствора карбоната натрия (64,5 мл, 129 ммоль) и бис(трифенилфосфино)дихлорпалладия (3,02 г, 4,30 ммоль) в 358 мл диметоксиэтана, растворенный кислород удаляли барботированием азота при перемешивании, и затем перекрывали поступление наружного воздуха в герметичном контейнере. Реакционную смесь перемешивали при 100°C в течение 16 часов и охлаждали до комнатной температуры. После фильтрации через слой целита и удаления органического растворителя при пониженном давлении, полученный продукт растворяли в этилацетате и промывали насыщенным солевым раствором. Органический растворитель сушили над сульфатом магния и удаляли при пониженном давлении. Очистку осуществляли на колонке с силикагелем (этилацетат:гексан) с получением целевого продукта (выход: 41,6%).
m/z (M+H)+ рассчитано для C20H26N2O2: 326,44, найдено 327,2
Пример получения 13: Синтез трет-бутил 2-(4-(3-амино-3-оксопропил)фенокси)-2-метилпропаноата
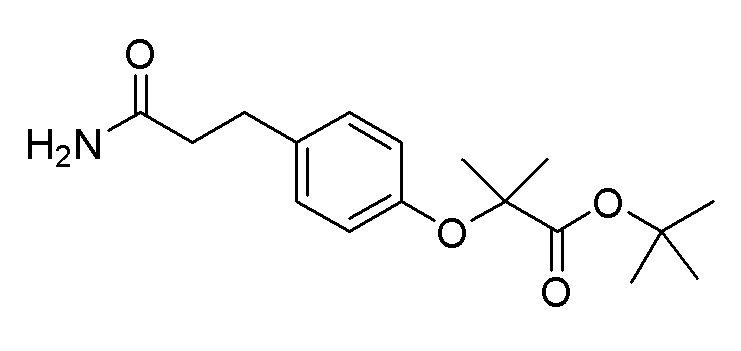
Стадия 1: Синтез трет-бутил 2-(4-(3-метокси-3-оксопропил)фенокси)-2-метилпропаноата
Метил 3-(4-гидроксифенил)пропаноат (2,17 г, 12,04 ммоль), сульфат магния (0,29 г, 2,41 ммоль) и карбонат калия (6,66 г, 48,2 ммоль) растворяли в DMF (30,1 мл), и после добавляли трет-бутил 2-бром-2-метилпропаноат (9,40 г, 42,1 ммоль). Реакционную смесь перемешивали при 75°C в течение 16 часов и затем охлаждали до комнатной температуры. После фильтрации через слой целита и удаления органического растворителя при пониженном давлении, полученный продукт растворяли в этилацетате и промывали насыщенным солевым раствором. Органический растворитель сушили над сульфатом магния и удаляли при пониженном давлении. Очистку осуществляли на колонке с силикагелем (этилацетат:гексан) с получением целевого продукта (выход: 62%).
Стадия 2: Синтез 3-(4-((1-(трет-бутокси)-2-метил-1-оксопропан-2-ил)окси)фенил)пропановой кислоты
После растворения трет-бутил 2-(4-(3-метокси-3-оксопропил)фенокси)-2-метилпропаноата (2,4 г, 7,44 ммоль) в THF (15 мл) и метаноле (15 мл), далее добавляли 1 н. гидроксида натрия (15 мл) и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь подкисляли водным раствором хлористоводородной кислоты, экстрагировали этилацетатом и промывали насыщенным солевым раствором. Органический растворитель сушили над сульфатом магния и удаляли при пониженном давлении (выход: 100%).
Стадия 3: Синтез трет-бутил 2-(4-(3-амино-3-оксопропил)фенокси)-2-метилпропаноата
3-(4-((1-(Трет-бутокси)-2-метил-1-оксопропан-2-ил)окси)фенил)пропановую кислоту (2,30 г, 7,46 ммоль) растворяли в DCM (37 мл), и далее добавляли оксалилхлорид (1,31 мл, 14,92 ммоль) и DMF (0,058 мл, 0,75 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, растворитель удаляли при пониженном давлении, и затем добавляли THF (19 мл). Температуру снижали до 0°C, и медленно по каплям добавляли 25% гидроксид аммония (8,71 мл, 224 ммоль). После удаления органического растворителя при пониженном давлении, реакционную смесь экстрагировали добавлением этилацетата и промывали насыщенным солевым раствором. Органический растворитель сушили над сульфатом магния и удаляли при пониженном давлении с получением целевого продукта (выход: 87%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,11-7,01 (м, 2H), 6,77 (дт, J=9,3, 2,5 Гц, 2H), 5,33 (с, 2H), 2,89 (т, J=7,5 Гц, 2H), 2,53-2,44 (м, 2H), 1,53 (с, 6H), 1,43 (с, 9H)
Пример получения 14: Синтез бензил 2-(4-(3-амино-3-оксопропил)фенил)-2-метилпропаноата
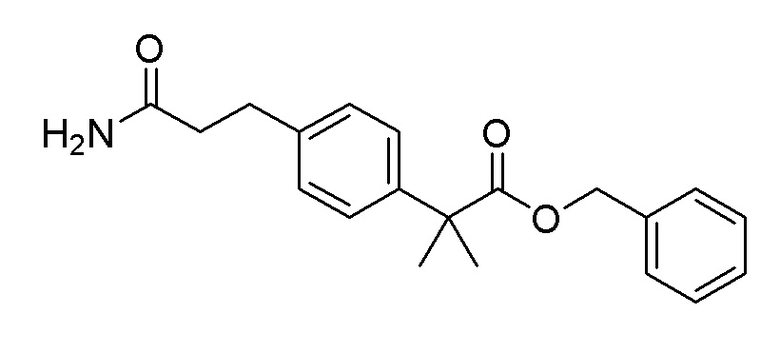
Используя 2-(4-бромфенил)-2-метилпропановую кислоту (5,00 г, 20,57 ммоль) и бензилбромид (4,22 г, 24,68 ммоль), последовательно проводили способы, аналогичные стадии 1 примера получения 13 и стадиям 1, 3 и 4 примера получения 7, с получением целевого продукта (выход: 60%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,66-7,59 (м, 1H), 7,54-7,40 (м, 2H), 7,40-7,25 (м, 5H), 7,19-7,10 (м, 2H), 6,47-6,40 (м, 1H), 5,61 (с, 2H), 5,09 (с, 2H), 1,59 (с, 6H)
Пример получения 15: Синтез трет-бутил 3-(4-(1-амино-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропаноата
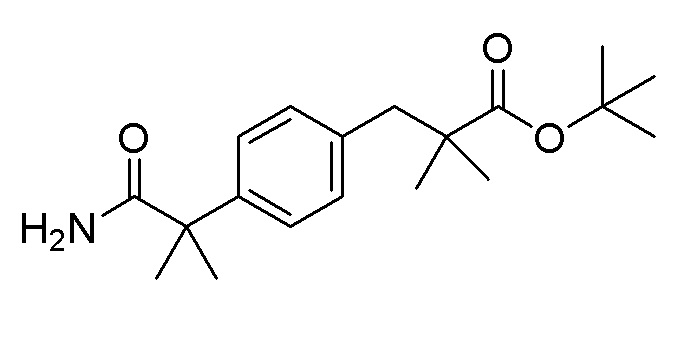
Стадия 1: Синтез трет-бутил 3-(4-(1-метокси-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропаноата
Метил 2-(4-(бромметил)фенил)-2-метилпропаноат (5,40 г, 19,91 ммоль) и трет-бутилизобутират (3,45 г, 23,90 ммоль) использовали таким же образом, как на стадии 1 примера получения 10, с получением целевого продукта (выход: 78%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,20 (д, J=8,2 Гц, 2H), 7,09 (д, J=8,2 Гц, 2H), 3,63 (с, 3H), 2,78 (с, 2H), 1,54 (с, 6H), 1,41 (с, 9H), 1,11 (с, 6H)
Стадия 2: Синтез трет-бутил 3-(4-(1-амино-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропаноата
Используя трет-бутил 3-(4-(1-метокси-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропаноат (0,50 г, 1,50 ммоль), полученный на стадии 1, последовательно проводили способы, аналогичные стадиям 2 и 3 примера получения 13, с получением целевого продукта (выход: 76%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,25 (д, J=8,2 Гц, 2H), 7,11 (д, J=8,2 Гц, 2H), 5,85 (с, 1H), 5,31 (с, 1H), 2,78 (с, 2H), 1,53 (с, 6H), 1,41 (с, 9H), 1,10 (с, 6H)
Пример получения 16: Синтез трет-бутил 2-(4-(2-амино-2-оксоэтил)фенокси)-2-метилпропаноат
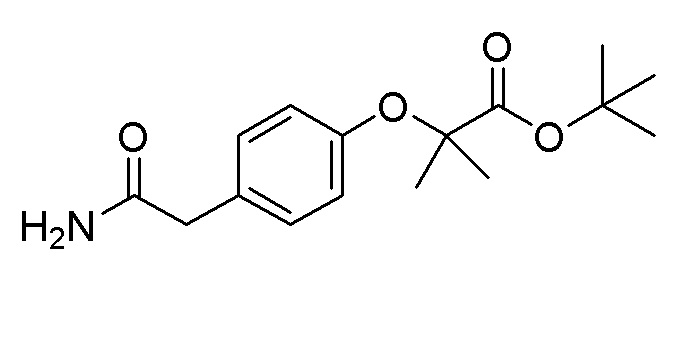
Метил 2-(4-гидроксифенил) ацетат (2,00 г, 12,04 ммоль) и трет-бутил 2-бром-2-метилпропаноат (9,40 г, 42,1 ммоль) использовали таким же образом, как в примере получения 13, с получением целевого продукта (выход: 54%).
1H-ЯМР (400 МГц, хлороформ-D): δ 7,12 (дд, J=11,4, 2,7 Гц, 2H), 6,83 (тд, J=5,7, 3,7 Гц, 2H), 5,43 (д, J=26,5 Гц, 2H), 3,50 (с, 2H), 1,57-1,50 (м, 6H), 1,45-1,37 (м, 9H)
Пример получения 17: Синтез этил 2-(4-(2-аминопиримидин-4-ил)фенил)ацетата
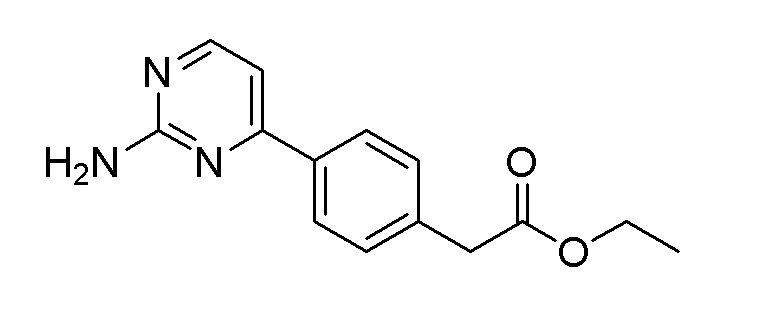
Этил 2-(4-бромфенил)ацетат (27,6 г, 114 ммоль) использовали таким же образом, как на стадиях 2 и 3 примера получения 12, и стадиях 2 и 3 примера получения 11, с получением целевого продукта (выход: 17,2%).
m/z (M+H)+ рассчитано для C14H15N3O2: 257,29, найдено 258,1
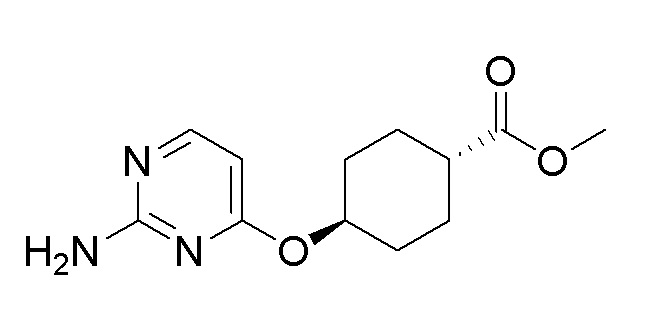
Пример получения 18: Синтез метил (1r,4r)-4-((2-аминопиримидин-4-ил)окси)циклогексан-1-карбоксилата
Стадия 1: Синтез метил (1r,4r)-4-гидроксициклогексан-1-карбоксилата
После растворения (1r,4r)-4-гидроксициклогексан-1-карбоновой кислоты (0,300 г, 2,081 ммоль) в метаноле (10 мл), добавляли серную кислоту (0,017 мл, 0,312 ммоль). После перемешивания при 60°C в течение 16 часов, органический растворитель удаляли при пониженном давлении, и очистку осуществляли на колонке с силикагелем с получением целевого продукта (выход: 100%).
1H-ЯМР (400 МГц, хлороформ-D): δ 8,25 (д, J=5,9 Гц, 1H), 6,64-6,50 (м, 1H), 5,16-5,05 (1H), 3,73-3,62 (м, 3H), 2,35 (тт, J=11,4, 3,7 Гц, 1H), 2,18 (дт, J=12,8, 3,5 Гц, 2H), 2,07 (дд, J=14,2, 3,7 Гц, 2H), 1,74-1,56 (м, 2H), 1,49 (ддд, J=23,0, 12,7, 3,5 Гц, 2H)
Стадия 2: Синтез метил (1r,4r)-4-((2-хлорпиримидин-4-ил)окси)циклогексан-1-карбоксилата
После растворения 2,4-дихлорпиримидина (0,28 г, 1,91 ммоль) в DMF (10 мл), добавляли метил (1r,4r)-4-гидроксициклогексан-1-карбоксилат (0,33 г, 2,10 ммоль), полученный на стадии 1, и карбонат цезия (2,56 г, 4,78 ммоль). После перемешивания при 80°C в течение 3 часов, реакционную смесь разбавляли диэтиловым эфиром и промывали водой. Органический растворитель сушили над сульфатом магния, и очистку осуществляли на колонке с силикагелем с получением целевого продукта (выход: 39,8%).
1H-ЯМР (400 МГц, хлороформ-D): δ 8,25 (д, J=5,9 Гц, 1H), 6,64-6,50 (м, 1H), 5,16-5,05 (1H), 3,73-3,62 (м, 3H), 2,35 (тт, J=11,4, 3,7 Гц, 1H), 2,18 (дт, J=12,8, 3,5 Гц, 2H), 2,07 (дд, J=14,2, 3,7 Гц, 2H), 1,74-1,56 (м, 2H), 1,49 (ддд, J=23,0, 12,7, 3,5 Гц, 2H)
Стадия 3: Синтез метил (1r,4r)-4-((2-аминопиримидин-4-ил)окси)циклогексан-1-карбоксилата
Метил (1r,4r)-4-((2-хлорпиримидин-4-ил)окси)циклогексан-1-карбоксилат (0,21 г, 0,76 ммоль), полученный на стадии 2, использовали таким же образом, как на стадии 2 примера получения 11, с получением целевого продукта (выход: 69%).
1H-ЯМР (400 МГц, хлороформ-D) δ 7,98 (д, J=5,9 Гц, 1H), 6,01 (д, J=5,5 Гц, 1H), 5,02-4,89 (м, 1H), 4,82 (с, 2H), 3,68 (дд, J=7,3, 2,7 Гц, 5H), 2,44-2,26 (м, 1H), 2,20-1,99 (м, 4H), 1,71-1,57 (м, 2H), 1,52 (с, 1H), 1,41 (дд, J=12,6, 3,4 Гц, 1H)
Пример 1: Синтез N-(6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)-3-фенилпропанамида
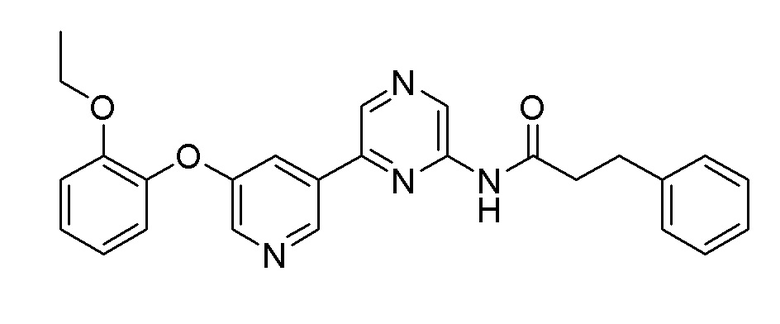
После растворения 2-хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразина (0,1 г, 0,305 ммоль), полученного в примере получения 1, 3-фенилпропанамида, полученного в примере получения 5 (0,055 г, 0,366 ммоль), карбоната цезия (0,249 г, 0,763 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантина (21 мг, 0,037 ммоль) и трис(дибензилиденацетон)дипалладия(0) (28 мг, 0,031 ммоль) в 15 мл 1,4-диоксана, растворенный кислород удаляли барботированием азота при перемешивании, и затем перекрывали поступление наружного воздуха в герметичном контейнере. Реакционную смесь перемешивали при 110°C в течение 16 часов и затем охлаждали до комнатной температуры. После фильтрации через слой целита и удаления органического растворителя при пониженном давлении, полученный продукт растворяли в этилацетате и промывали насыщенным солевым раствором. Органический растворитель сушили над сульфатом магния и удаляли при пониженном давлении. Очистку осуществляли на колонке с силикагелем (этилацетат:гексан=1:2) с получением целевого продукта (выход: 67%).
m/z (M+H)+ рассчитано для C26H24N4O3: 440,50, найдено 441,1
Пример 2: Синтез метил 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)ацетата
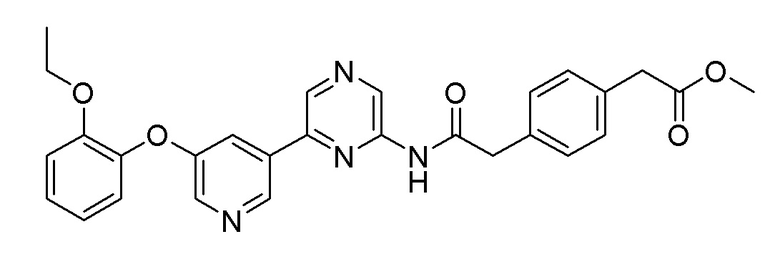
2-Хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин (0,32 г, 0,976 ммоль), полученный в примере получения 1, и метил 2-(4-(2-амино-2-оксоэтил)фенил)ацетат (0,243 г, 1,172 ммоль), полученный в примере получения 6, использовали таким же образом, как в примере 1, с получением целевого продукта (выход: 22,6%).
1H ЯМР (500 МГц, хлороформ-D): δ 9,50 (с, 1H), 8,86 (с, 1H), 8,67 (с, 1H), 8,61 (с, 1H), 8,33 (с, 1H), 7,67 (д, J=1,8 Гц, 1H), 7,27 (4H), 7,17 (1H), 7,08 (д, J=7,3 Гц, 1H), 7,00-6,94 (м, 2H), 3,99 (кв, J=6,7 Гц, 2H), 3,76 (с, 2H), 3,67 (с, 3H), 3,62 (с, 2H), 1,19 (т, J=6,7 Гц, 3H)
Пример 3: Синтез 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)уксусной кислоты
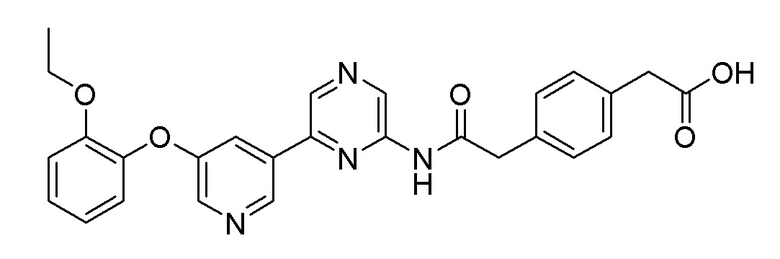
Метил 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)ацетат (110 мг, 0,221 ммоль), полученный в примере 2, растворяли в THF (6 мл) и метаноле (2 мл). Добавляли гидроксид натрия (44 мг, 1,103 ммоль), растворенный в воде (2 мл), затем перемешивали при комнатной температуре в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры, реакционную смесь титровали до рН 4,5 с использованием 1 н. водного раствора хлористоводородной кислоты, разбавляли этилацетатом, и удаляли водный слой. Полученный продукт сушили над сульфатом магния, и органический растворитель удаляли при пониженном давлении. Очистку осуществляли на колонке с силикагелем (этилацетат:гексан=1:1) с получением целевого продукта (выход: 33,6%).
m/z (M+H)+ рассчитано для C27H24N4O5: 484,51, найдено 485,1
Пример 4: Синтез 2-(4-(3-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-3-оксопропил)фенил)уксусной кислоты
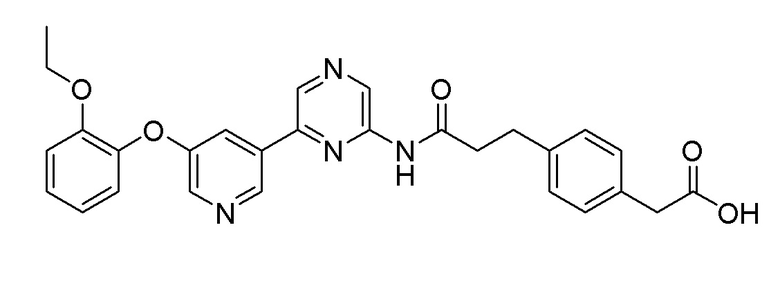
Стадия 1: Синтез метил 2-(4-(3-((6-(5-(2-этоксифенокси)пиридин-3-ил)амино)-3-оксопропил)фенил)ацетата
2-Хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин (0,20 г, 0,61 ммоль), полученный в примере получения 1, и метил 2-(4-(3-амино-3-оксопропил)фенил)ацетат (0,14 г, 0,61 ммоль), полученный в примере получения 7, использовали таким же образом, как в примере 1, с получением целевого продукта (выход: 35%).
1H-ЯМР (400 МГц, хлороформ-D): δ 9,51 (с, 1H), 8,87 (с, 1H), 8,70 (с, 1H), 8,36 (д, J=10 Гц, 2H), 7,68 (с, 1H), 7,25 (м, 5H), 7,10 (д, 1H), 6,94 ~ 7,02 (м, 2H), 4,02 (кв, 2H), 3,66 (с, 3H), 3,58 (с, 2H), 3,06 (т, 2H), 2,77 (т, 2H), 1,21 (т, 3H)
Стадия 2: Синтез 2-(4-(3-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-3-оксопропил)фенил)уксусной кислоты
Сложноэфирное соединение (0,11 г, 2,39 ммоль), полученное на стадии 1, гидролизовали таким же образом, как в примере 3, с получением целевого продукта (выход: 28%).
1H-ЯМР (400 МГц, DMSO-D6): δ 10,87 (с, 1H), 9,34 (с, 1H), 9,00 (с, 2H), 8,33 (с, 1H), 7,89 (с, 1H), 7,17 (м, 7H), 7,02 (т, 1H), 4,03 (т, 2H), 3,51 (с, 2H), 2,92 (т, 2H), 2,78 (т, 2H), 1,10 (т, 3H)
Пример 5: Синтез метил 2-(4-(3-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропаноата
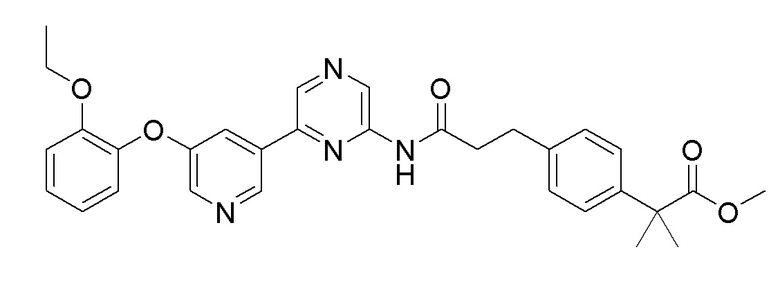
2-Хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин (0,25 г, 0,76 ммоль), полученный в примере получения 1, и метил 2-(4-(3-амино-3-оксопропил)фенил-2-метилпропаноат (0,19 г, 0,76 ммоль), полученный в примере получения 8, использовали таким же образом, как в примере 1, с получением целевого продукта (выход: 51%).
1H ЯМР (400 МГц, хлороформ-D): δ 9,51 (с, 1H), 8,86 (с, 1H), 8,72 (с, 1H), 8,37 (д, J=4 Гц, 1H), 7,99 (с, 1H), 7,70 (с, 1H), 7,25 (м, 3H), 7,20 (м, 2H), 7,12 (м, 1H), 7,03 (м, 2H), 4,03 (кв, 2H), 3,06 (т, 2H), 2,78 (т, 2H), 1,55 (с, 6H), 1,22 (т, 3H)
Пример 6: Синтез этил 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-дифторацетата
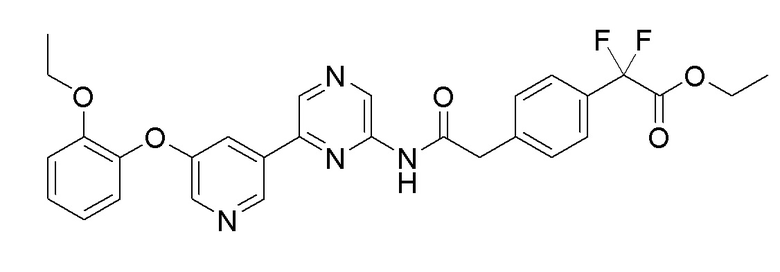
Стадия 1: Синтез 6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-амина
Указанное в заголовке соединение получали в качестве побочного продукта в процессе получения 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)уксусной кислоты в примере 3.
m/z (M+H)+ рассчитано для C17H16N4O2: 308,3, найдено 309,1
Стадия 2: Синтез этил 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-дифторацетата
После растворения 2-(4-(2-этокси-1,1-дифтор-2-оксоэтил)фенил)уксусной кислоты (0,02 г, 0,077 ммоль), полученной в примере получения 9, в DCM (0,4 мл), добавляли оксалилхлорид (0,02 г, 0,155 ммоль), и добавляли 1 каплю DMF. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и растворитель удаляли при пониженном давлении. После растворения концентрата в THF (0,2 мл), температуру снижали до 0°C, 6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-амин, полученный на стадии 1 (0,02 г, 0,065 ммоль), растворяли в THF (0,2 мл), и добавляли TEA (0,022 г, 0,216 ммоль). После перемешивания при комнатной температуре в течение 16 часов добавляли воду и реакционную смесь экстрагировали этилацетатом. После промывки водой и насыщенным солевым раствором, органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Очистку осуществляли с помощью колоночной хроматографии с получением целевого продукта (выход: 13%).
1H-ЯМР (500 МГц, хлороформ-D): δ 9,49 (с, 1H), 8,84 (с, 1H), 8,74 (с, 1H), 8,38 (с, 1H), 7,91 (с, 1H), 7,67 (с, 1H), 7,66 (д, J=7,9 Гц, 2H), 7,46 (д, J=7,9 Гц, 2H), 7,21 (1H), 7,11 (1H), 7,03-6,99 (2H), 4,30 (кв, J=7,95 Гц, 2H), 4,03 (кв, J=6,15 Гц, 2H), 3,86 (с, 2H), 1,31 (т, J=6,15 Гц, 3H), 1,22 (т, J=7,95 Гц, 3H)
Пример 7: Синтез 3-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановой кислоты
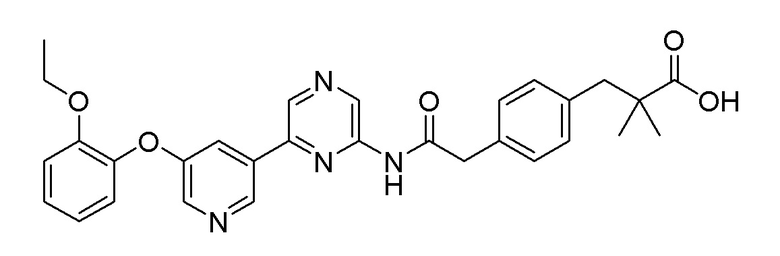
2-Хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин (0,1 г, 0,305 ммоль), полученный в примере получения 1, и трет-бутил 3-(4-(2-амино-2-оксоэтил)фенил)-2,2-диметилпропаноат (0,081 г, 0,277 ммоль), полученный в примере получения 10, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 12%).
1H-ЯМР (400 МГц, МЕТАНОЛ-D4): δ 9,34 (с, 1H), 8,89 (с, 1H), 8,85-8,74 (м, 1H), 8,23 (д, J=2,7 Гц, 1H), 7,91 (кв, J=1,4 Гц, 1H), 7,33-7,19 (м, 3H), 7,19-7,06 (м, 4H), 7,01 (т, J=7,5 Гц, 1H), 3,99 (кв, J=7,0 Гц, 2H), 3,72 (с, 2H), 2,82 (с, 2H), 1,18-1,01 (м, 9H)
Пример 8: Синтез (R)-1-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновой кислоты
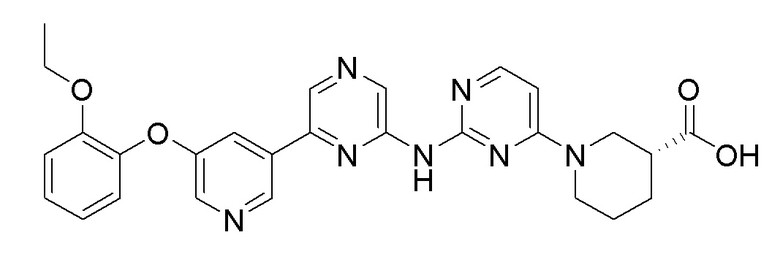
2-Хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин (0,1 г, 0,305 ммоль), полученный в примере получения 1, и этил (R)-1-(2-аминопиримидин-4-ил)пиперидин-3-карбоксилат (0,069 г, 0,277 ммоль), полученный в примере получения 11, использовали таким же образом, как в примере 1 и примере 3, с получением целевого продукта (выход: 49%).
1H-ЯМР (400 МГц, метанол-D4): δ 9,45 (с, 1H), 8,88 (д, J=1,8 Гц, 1H), 8,64 (с, 1H), 8,21 (д, J=2,7 Гц, 1H), 7,96 (д, J=6,4 Гц, 1H), 7,92 (т, J=2,3 Гц, 1H), 7,31-7,21 (м, 1H), 7,21-7,16 (1H), 7,16-7,09 (м, 1H), 7,06-6,95 (м, 1H), 6,42 (д, J=6,4 Гц, 1H), 4,50-4,06 (1H), 4,01 (кв, J=7,0 Гц, 2H), 3,53-3,33 (м, 1H), 3,23 (с, 1H), 2,62-2,40 (1H), 2,19-2,01 (1H), 1,81 (д, J=12,3 Гц, 2H), 1,68-1,44 (1H), 1,14 (т, J=6,9 Гц, 3H)
Пример 9: Синтез 3-(3-(6-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановой кислоты
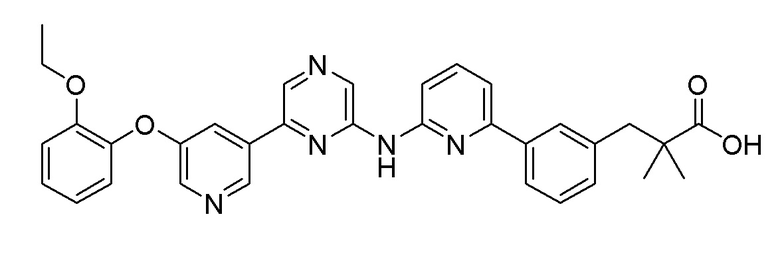
2-Хлор-6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин (0,1 г, 0,305 ммоль), полученный в примере получения 1, и трет-бутил 3-(4-(2-амино-2-оксоэтил)фенил)-2,2-диметилпропаноат (0,091 г, 0,277 ммоль), полученный в примере получения 10, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 26,9%).
1H-ЯМР (400 МГц, метанол-D4): δ 9,36 (с, 1H), 8,89 (д, J=1,8 Гц, 1H), 8,65-8,50 (1H), 8,25 (д, J=2,7 Гц, 1H), 7,99-7,88 (м, 2H), 7,84 (д, J=7,8 Гц, 1H), 7,65 (т, J=7,8 Гц, 1H), 7,40 (д, J=7,3 Гц, 1H), 7,34 (дд, J=8,0, 5,7 Гц, 2H), 7,30-7,24 (м, 1H), 7,20 (дд, J=8,0, 1,6 Гц, 2H), 7,15 (д, J=8,2 Гц, 1H), 7,11-6,95 (м, 1H), 4,04-3,91 (2H), 2,95 (с, 2H), 1,19 (с, 6H), 1,13 (т, J=7,1 Гц, 3H)
Пример 10: Синтез N-(6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)-3-фенилпропанамида
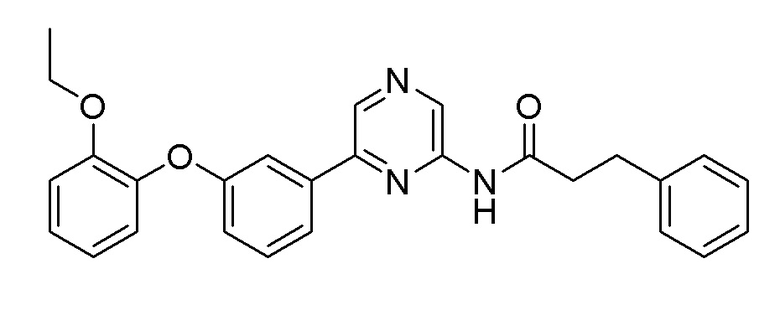
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,1 г, 0,306 ммоль), полученный в примере получения 2, и 3-фенилпропанамид, полученный в примере получения 5 (0,055 г, 0,367 ммоль), использовали таким же образом, как в примере 1, с получением целевого продукта (выход: 60%).
1H ЯМР (500 МГц, хлороформ-D): δ 9,44 (с, 1H), 8,70 (с, 1H), 8,00 (с, 1H), 7,57 (1H), 7,50 (с, 1H), 7,36 (1H), 7,28-7,25 (2H), 7,21-7,19 (3H), 7,10 (1H), 7,05-6,98 (3H), 6,90 (1H), 4,30 (кв, J=7,3 Гц, 2H), 3,04 (т, J=7,65 Гц, 2H), 2,69 (т, J=7,95 Гц, 2H), 1,23 (3H)
Пример 11: Синтез 2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)уксусной кислоты
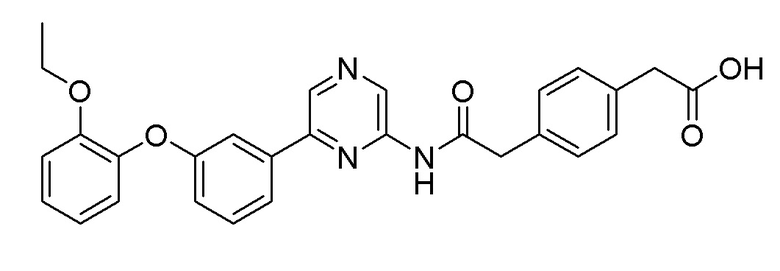
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,250 г, 0,765 ммоль), полученный в примере получения 2, и метил 2-(4-(2-амино-2-оксоэтил)фенил) ацетат (0,190 г, 0,918 ммоль), полученный в примере получения 6, использовали таким же образом, как в примере 1 и примере 3, последовательно с получением целевого продукта (выход: 34,1%).
1H ЯМР (300 МГц, Метанол-D): δ 9,35 (с, 1H), 8,61 (с, 1H), 8,52 (с, 1H), 7,45 (1H), 7,40 (д, J=1,25 Гц, 1H), 7,26 (1H), 7,13 (м, 4H), 7,02 (1H), 6,94-6,89 (3H), 6,81 (1H), 3,95 (2H), 3,59 (с, 2H), 3,39 (с, 2H), 1,18 (3H)
Пример 12: Синтез 2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)уксусной кислоты
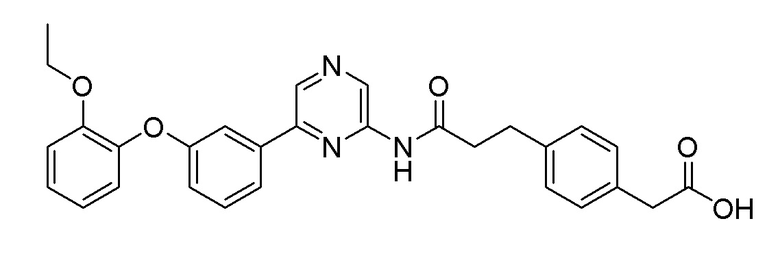
Стадия 1: Синтез метил 2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)ацетата
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,10 г, 0,31 ммоль), полученный в примере получения 4, и метил 2-(4-(3-амино-3-оксопропил)фенил)ацетат (0,07 г, 0,31 ммоль), полученный в примере получения 11, использовали таким же образом, как в примере 1, с получением целевого продукта (выход: 75%).
1H-ЯМР (400 МГц, хлороформ-D): δ 9,45 (с, 1H), 8,71 (с, 1H), 8,11 (с, 1H), 7,51 (д, J=12 Гц, 1H), 7,51 (с, 1H), 7,36 (т, 1H), 7,16 (д, J=8 Гц, 2H), 7,14 (д, J=8 Гц, 2H), 7,09 (т, 1H), 6,90 ~ 6,98 (м, 3H), 6,89 (т, 1H), 4,06 (т, 2H), 3,67 (с, 3H), 3,59 9s, 2H), 3,01 (т, 2H), 2,65 (т, 2H), 1,24 (т, 3H)
Стадия 2: Синтез 2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)уксусной кислоты
Сложноэфирное соединение (0,11 г, 2,39 ммоль), полученное на стадии 1, гидролизовали таким же образом, как в примере 3, с получением целевого продукта (выход: 69%).
1H-ЯМР (400 МГц, хлороформ-D): δ 9,49 (с, 1H), 8,91 (с, 1H), 8,67 (с, 1H), 7,47 (д, 1H), 7,43 (с, 1H), 7,35 9t, 1H), 7,11 ~ 7,26 (м, 5H), 6,97 ~ 7,05 (м, 3H), 6,91 (т, 1H), 4,06 (т, 2H), 3,61 (с, 2H), 3,00 (т, 2H), 2,68 (т, 2H), 1,26 (т, 3H)
Пример 13: Синтез 2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенокси)-2-метилпропановой кислоты
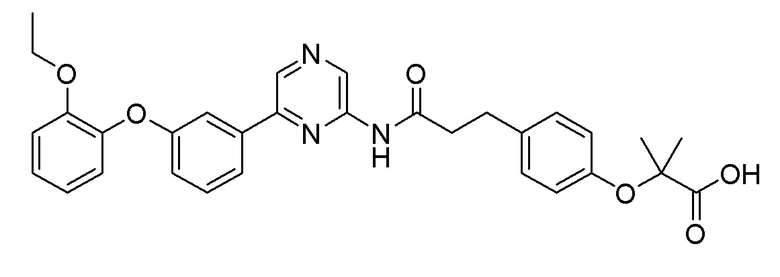
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,080 г, 0,245 ммоль), полученный в примере получения 2, и трет-бутил 2-(4-(3-амино-3-оксопропил)фенокси)-2-метилпропаноат (0,075 г, 0,245 ммоль), полученный в примере получения 13, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 64%).
1H-ЯМР (400 МГц, хлороформ-D): δ 9,43 (с, 1H), 8,65-8,52 (м, 2H), 7,51-7,41 (м, 2H), 7,36-7,27 (м, 1H), 7,10-6,91 (м, 6H), 6,91-6,76 (м, 3H), 4,01 (кв, J=6,9 Гц, 2H), 2,93 (т, J=6,9 Гц, 2H), 2,63 (д, J=5,0 Гц, 2H), 1,56 (с, 6H), 1,23 (т, J=7,1 Гц, 3H)
Пример 14: Синтез 2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил-2,2-дифторуксусной кислоты
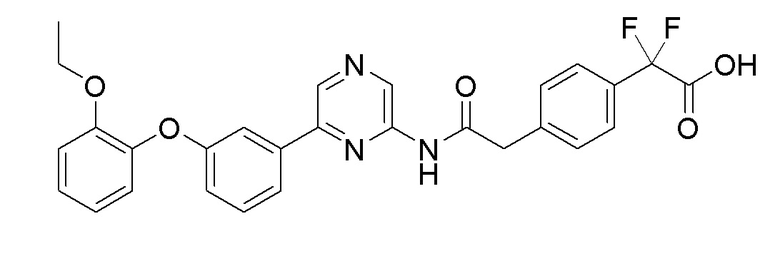
Стадия 1: Синтез 6-(3-(2-этоксифенокси)фенил)пиразин-2-амина
Указанное в заголовке соединение получали в качестве побочного продукта в процессе получения 2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)уксусной кислоты в примере 11.
m/z (M+H)+ рассчитано для C18H17N3O2: 307,3, найдено 308,1
Стадия 2: Синтез 2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил-2,2-дифторуксусной кислоты
2-(4-(2-Этокси-1,1-дифтор-2-оксоэтил)фенил)уксусную кислоту (0,046 г, 0,178 ммоль), полученную в примере получения 9, и 6-(3-(2-этоксифенокси)фенил)пиразин-2-амин (0,061 г, 0,199 ммоль), полученный на стадии 1, использовали таким же образом, как в примере 6 и примере 3, последовательно с получением целевого продукта (выход: 5,7%).
1H-ЯМР (500 МГц, метанол-D4): δ 9,19 (с, 1H), 8,66 (с, 1H), 7,65 (1H), 7,54-7,52 (3H), 7,35-7,33 (3H), 7,10 (1H), 7,04-6,98 (2H), 6,90-6,88 (2H), 3,94 (кв, J=7,3 Гц, 2H), 3,74 (с, 1H), 1,10 (т, J - 6,7 Гц, 3H)
Пример 15: Синтез 3-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановой кислоты
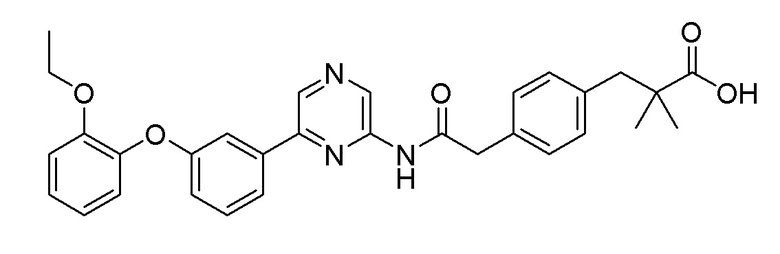
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,10 г, 0,306 ммоль), полученный в примере получения 2, и трет-бутил 3-(4-(2-амино-2-оксоэтил)фенил)-2,2-диметилпропаноат (0,089 г, 0,306 ммоль), полученный в примере получения 10, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 74%) .
1H ЯМР (400 МГц, хлороформ-D): δ 9,43 (с, 1H), 8,68 (с, 1H), 8,19 (с, 1H), 7,56-7,47 (м, 2H), 7,34 (т, J=8,0 Гц, 1H), 7,18 (тд, J=7,8, 5,6 Гц, 4H), 7,13-7,05 (м, 1H), 7,05-6,93 (м, 3H), 6,90 (т, J=7,8 Гц, 1H), 4,03 (кв, J=7,0 Гц, 2H), 3,76-3,69 (м, 2H), 2,88 (с, 2H), 1,29-1,16 (м, 9H)
Пример 16: Синтез 2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропановой кислоты
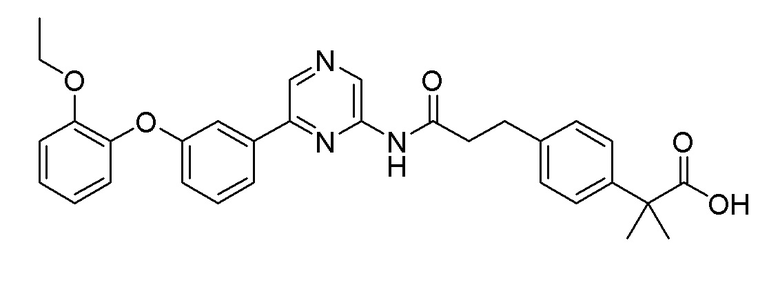
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,10 г, 0,306 ммоль), полученный в примере получения 2, и бензил 2-(4-(3-амино-3-оксопропил)фенил)-2-метилпропаноат (0,099 г, 0,306 ммоль), полученный в примере получения 14, использовали таким же образом, как в примере 1 и на стадии 2 примера получения 7, с получением целевого продукта (выход: 31%).
1H ЯМР (400 МГц, хлороформ-D): δ 9,44 (с, 1H), 8,68 (с, 1H), 8,34 (с, 1H), 7,58-7,45 (м, 2H), 7,39-7,27 (м, 3H), 7,17 (д, J=8,2 Гц, 2H), 7,10 (тд, J=7,8, 1,5 Гц, 1H), 7,06-6,94 (м, 3H), 6,94-6,85 (м, 1H), 4,03 (кв, J=7,0 Гц, 2H), 3,10-2,95 (м, 2H), 2,70 (т, J=7,5 Гц, 2H), 1,57 (с, 6H), 1,24 (т, J=6,6 Гц, 3H)
Пример 17: Синтез (E)-2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопро-1-фен-1-ил)фенил)-2-метилпропановой кислоты
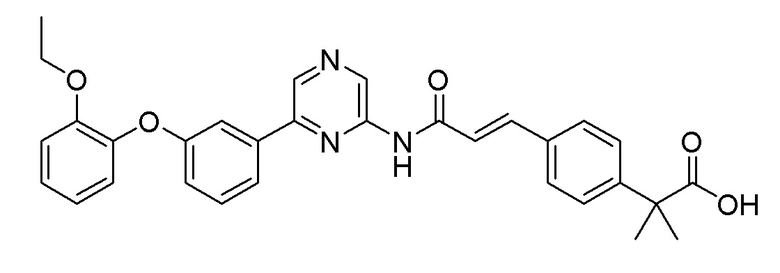
Указанное в заголовке соединение получали в качестве побочного продукта в процессе получения Примера 16 (выход: 41%).
1H ЯМР (400 МГц, хлороформ-D): δ 9,60 (с, 1H), 8,92 (д, J=31,1 Гц, 1H), 8,70 (д, J=0,9 Гц, 1H), 7,84-7,72 (м, 1H), 7,61-7,42 (м, 6H), 7,38 (тд, J=7,9, 2,4 Гц, 1H), 7,19-7,10 (м, 1H), 7,10-6,97 (м, 3H), 6,97-6,87 (м, 1H), 6,59 (д, J=15,6 Гц, 1H), 4,08-3,99 (м, 2H), 1,63 (с, 6H), 1,33-1,19 (м, 3H)
Пример 18: Синтез 3-(4-(1-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропановой кислоты
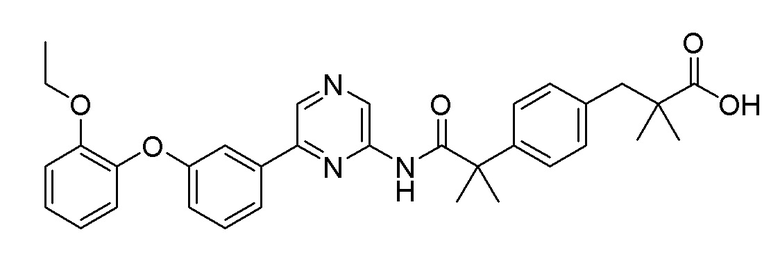
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,10 г, 0,306 ммоль), полученный в примере получения 2, и трет-бутил 3-(4-(1-амино-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропаноат (0,098 г, 0,306 ммоль), полученный в примере получения 15, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 77%).
1H ЯМР (400 МГц, хлороформ-D): δ 9,46 (с, 1H), 8,69-8,60 (м, 1H), 7,57 (с, 1H), 7,53-7,44 (м, 2H), 7,36-7,26 (м, 3H), 7,19 (д, J=8,7 Гц, 2H), 7,15-7,06 (м, 1H), 6,98 (ддд, J=8,0, 5,0, 1,6 Гц, 2H), 6,94-6,85 (м, 2H), 4,01 (кв, J=7,0 Гц, 2H), 2,88 (с, 2H), 1,66 (с, 6H), 1,28-1,13 (м, 9H)
Пример 19: Синтез 2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенокси-2-метилпропановой кислоты
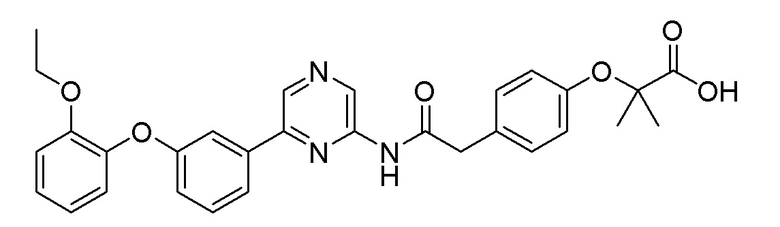
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,08 г, 0,245 ммоль), полученный в примере получения 2, и трет-бутил 2-(4-(2-амино-2-оксоэтил)фенокси)-2-метилпропаноат (0,072 г, 0,245 ммоль), полученный в примере получения 16, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 62%) .
1H-ЯМР (400 МГц, хлороформ-D): δ 9,42 (с, 1H), 8,67 (с, 1H), 8,40 (д, J=12,3 Гц, 1H), 7,54-7,42 (м, 2H), 7,32 (тд, J=7,9, 2,0 Гц, 1H), 7,20 (кв, J=4,0 Гц, 2H), 7,16-7,05 (м, 1H), 7,04-6,84 (м, 6H), 4,02 (квд, J=7,0, 1,5 Гц, 2H), 3,69 (д, J=2,7 Гц, 2H), 1,59 (д, J=15,1 Гц, 6H), 1,30-1,16 (м, 3H)
Пример 20: Синтез 2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)фенил)уксусной кислоты
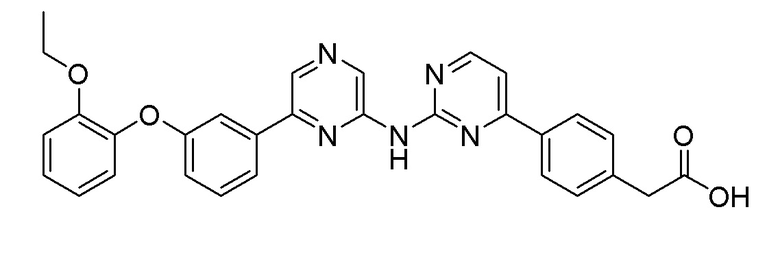
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,140 г, 0,428 ммоль), полученный в примере получения 2, и этил 2-(4-(2-аминопиримидин-4-ил)фенил)ацетат (0,1 г, 0,389 ммоль), полученный в примере получения 17, использовали таким же образом, как в примере 1 и примере 3, с получением целевого продукта (выход: 0,69%).
m/z (M+H)+ рассчитано для C30H25N5O4: 519,56, найдено 520,1
Пример 21: Синтез (1r,4r)-4-((2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)окси)циклогексан-1-карбоновой кислоты
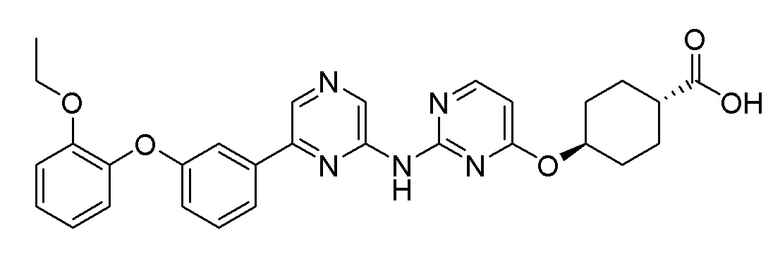
2-Хлор-6-(3-(2-этоксифенокси)фенил)пиразин (0,100 г, 0,306 ммоль), полученный в примере получения 2, и метил (1r,4r)-4-((2-аминопиримидин-4-ил)окси)циклогексан-1-карбоксилат (0,085 г, 0,337 ммоль), полученный в примере получения 18, использовали таким же образом, как в примере 1 и примере 3, последовательно с получением целевого продукта (выход: 22,8%).
1H-ЯМР (400 МГц, DMSO-D6): δ 10,09 (с, 1H), 9,38 (с, 1H), 8,75 (с, 1H), 8,25 (д, J=5,9 Гц, 1H), 7,78 (д, J=8,2 Гц, 1H), 7,70 (т, J=2,1 Гц, 1H), 7,41 (т, J=8,0 Гц, 1H), 7,21-7,10 (м, 2H), 7,08-7,01 (м, 1H), 7,00-6,91 (м, 1H), 6,85 (дд, J=7,8, 2,3 Гц, 1H), 6,43-6,33 (м, 1H), 4,95 (дд, J=10,3, 4,3 Гц, 1H), 4,00 (кв, J=6,9 Гц, 2H), 2,18 (с, 1H), 2,11 (д, J=7,8 Гц, 2H), 1,93 (д, J=9,6 Гц, 2H), 1,55-1,34 (м, 4H), 1,12 (т, J=7,1 Гц, 3H)
Пример 22: Синтез N-(6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)-3-фенилпропанамида
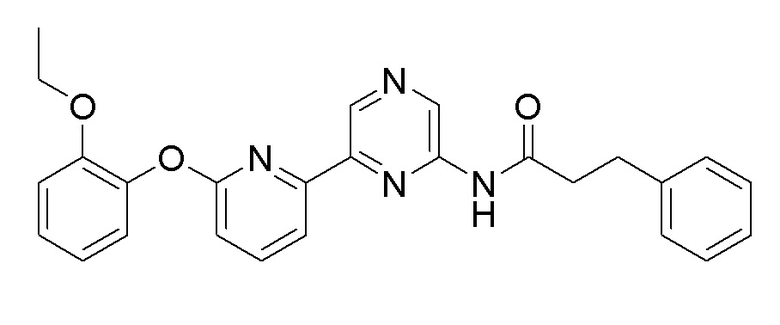
2-Хлор-6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин (0,1 г, 0,305 ммоль), полученный в примере получения 3, и 3-фенилпропанамид (0,059 г, 0,397 ммоль), полученный в примере получения 5, использовали таким же образом, как в примере 1, с получением целевого продукта (выход: 52,1%).
m/z (M+H)+ рассчитано для C26H24N4O3: 440,50, найдено 441,1
Пример 23: Синтез 3-(4-(2-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановой кислоты
2-Хлор-6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин (0,070 г, 0,214 ммоль), полученный в примере получения 3, и трет-бутил 3-(4-(2-амино-2-оксоэтил)фенил)-2,2-диметилпропаноат (0,056 г, 0,194 ммоль), полученный в примере получения 10, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 74,4%).
1H-ЯМР (500 МГц, метанол-D4): δ 9,32 (с, 1H), 8,75 (с, 1H), 8,05 (д, J=7,6 Гц, 1H), 7,99-7,85 (1H), 7,30 (д, J=8,2 Гц, 2H), 7,28-7,23 (м, 1H), 7,23-7,16 (3H), 7,13 (д, J=6,7 Гц, 1H), 7,10-6,96 (м, 2H), 3,99 (кв, J=6,9 Гц, 2H), 3,79 (с, 2H), 2,87 (с, 2H), 1,17 (с, 7H), 1,09 (т, J=6,9 Гц, 4H)
Пример 24: Синтез (R)-1-(2-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновой кислоты
2-Хлор-6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин (0,070 г, 0,214 ммоль), полученный в примере получения 3, и этил (R)-1-(2-аминопиримидин-4-ил)пиперидин-3-карбоксилат (0,048 г, 0,194 ммоль), использовали таким же образом, как в примере 1 и примере 3, с получением целевого продукта (выход: 20%).
1H-ЯМР (400 МГц, метанол-D4): δ 9,40 (с, 1H), 8,54 (с, 1H), 8,00 (д, J=7,8 Гц, 1H), 7,93 (д, J=6,4 Гц, 1H), 7,85 (т, J=7,8 Гц, 1H), 7,19 (т, J=8,0 Гц, 1H), 7,15 (дд, J=7,8, 1,4 Гц, 1H), 7,07 (д, J=6,9 Гц, 1H), 6,98 (т, J=7,1 Гц, 1H), 6,93 (д, J=8,2 Гц, 1H), 6,36 (д, J=6,4 Гц, 1H), 4,61-4,32 (1H), 4,29-4,04 (1H), 3,94 (кв, J=7,0 Гц, 2H), 3,26-3,01 (м, 2H), 2,41 (т, J=3,9 Гц, 1H), 2,08 (т, J=4,8 Гц, 1H), 1,87-1,68 (м, 2H), 1,52 (д, J=12,8 Гц, 1H), 1,04 (т, J=6,9 Гц, 3H)
Пример 25: Синтез 3-(3-(6-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановой кислоты
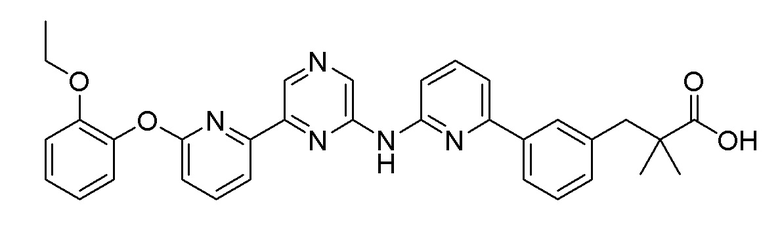
2-Хлор-6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин (0,140 г, 0,427 ммоль), полученный в примере получения 3, и трет-бутил 3-(3-(6-аминопиридин-2-ил)фенил)-2,2-диметилпропаноат (0,127 г, 0,388 ммоль), полученный в примере получения 12, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 16%).
1H-ЯМР (400 МГц, МЕТАНОЛ-D4) δ 9,40 (с, 1H), 8,57-8,44 (1H), 8,06 (д, J=6,9 Гц, 1H), 7,98-7,89 (м, 2H), 7,89-7,82 (м, 1H), 7,76 (т, J=8,0 Гц, 1H), 7,43 (дд, J=10,5, 7,8 Гц, 2H), 7,35 (т, J=7,5 Гц, 1H), 7,30-7,20 (м, 2H), 7,20-7,14 (м, 1H), 7,11 (д, J=8,2 Гц, 1H), 7,01 (т, J=7,5 Гц, 1H), 6,96 (д, J=8,2 Гц, 1H), 4,54 (с, 1H), 3,97 (кв, J=7,0 Гц, 2H), 3,46 (с, 0H), 2,95 (с, 2H), 1,19 (с, 7H), 1,06 (т, J=6,9 Гц, 3H)
Пример 26: Синтез 2-(4-(3-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропановой кислоты
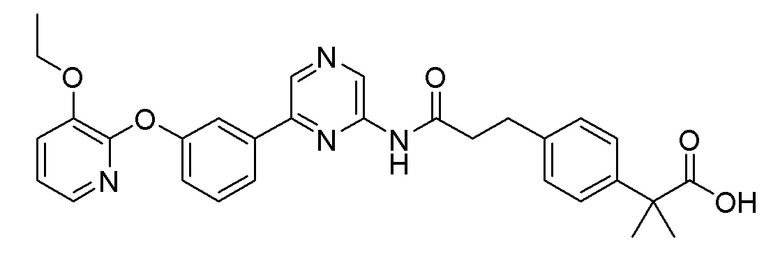
2-Хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин (0,08 г, 0,244 ммоль), полученный в примере получения 4, и бензил 2-(4-(3-амино-3-оксопропил)фенил)-2-метилпропаноат (0,079 г, 0,244 ммоль), полученный в примере получения 14, использовали таким же образом, как в примере 1 и на стадии 2 примера получения 7, с получением целевого продукта (выход: 16%).
1H-ЯМР (400 МГц, хлороформ-D): δ 9,41 (с, 1H), 8,68 (с, 1H), 8,25 (д, J=8,2 Гц, 1H), 7,73 (д, J=5,0 Гц, 1H), 7,64 (кв, J=2,1 Гц, 2H), 7,48-7,38 (м, 1H), 7,30-7,09 (м, 6H), 6,97 (дд, J=7,8, 5,0 Гц, 1H), 4,19-4,11 (м, 2H), 3,01 (т, J=7,5 Гц, 2H), 2,71 (т, J=7,5 Гц, 2H), 1,52 (с, 6H), 1,46 (т, J=7,1 Гц, 3H)
Пример 27: Синтез 3-(4-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановой кислоты
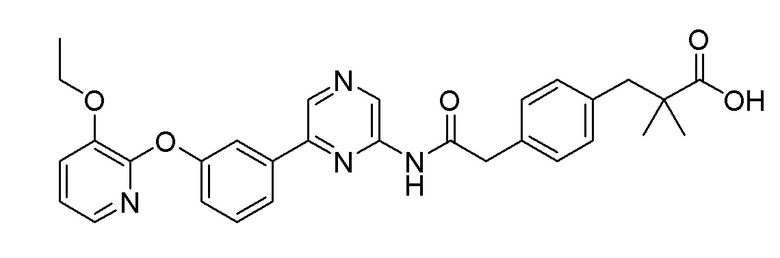
2-Хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин (0,08 г, 0,244 ммоль), полученный в примере получения 4, и трет-бутил 3-(4-(2-амино-2-оксоэтил)фенил)-2,2-диметилпропаноат (0,071 г, 0,244 ммоль), полученный в примере получения 10, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 62%).
1H-ЯМР (400 МГц, хлороформ-D): δ 9,42 (с, 1H), 8,70 (с, 1H), 8,37 (с, 1H), 7,76-7,69 (м, 1H), 7,68-7,61 (м, 2H), 7,43 (т, J=8,2 Гц, 1H), 7,23-7,10 (м, 6H), 6,97 (дд, J=7,8, 5,0 Гц, 1H), 4,18-4,07 (м, 2H), 3,72 (с, 2H), 2,85 (с, 2H), 1,49-1,41 (м, 3H), 1,19 (с, 6H)
Пример 28: Синтез 2-(4-(3-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенокси)-2-метилпропановой кислоты
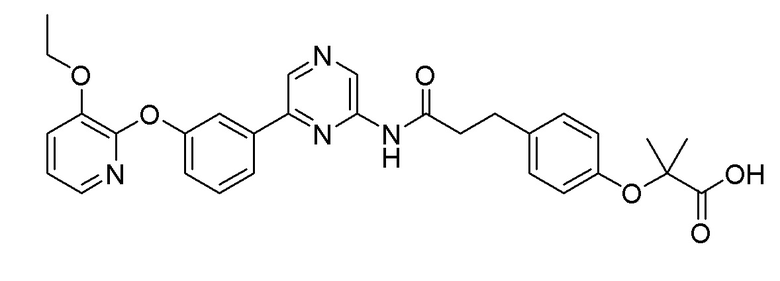
2-Хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин (0,08 г, 0,244 ммоль), полученный в примере получения 4, и трет-бутил 2-(4-(3-амино-3-оксопропил)фенокси)-2-метилпропаноат (0,075 г, 0,244 ммоль), полученный в примере получения 13, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 56%).
1H-ЯМР (400 МГц, DMSO-D6): δ 10,74 (с, 1H), 9,26 (с, 1H), 8,93 (с, 1H), 7,92 (д, J=7,8 Гц, 1H), 7,81 (т, J=1,8 Гц, 1H), 7,60 (дд, J=5,0, 1,4 Гц, 1H), 7,56-7,41 (м, 2H), 7,18 (дд, J=7,5, 2,1 Гц, 1H), 7,13-7,00 (м, 3H), 6,78-6,65 (м, 2H), 4,11 (кв, J=7,0 Гц, 2H), 2,87-2,76 (м, 2H), 2,69 (т, J=7,5 Гц, 2H), 1,42 (с, 6H), 1,33 (т, J=7,1 Гц, 3H)
Пример 29: Синтез 3-(4-(1-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропановой кислоты
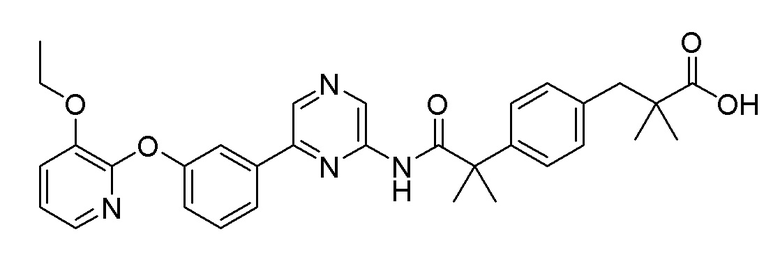
2-Хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин (0,08 г, 0,244 ммоль), полученный в примере получения 4, и трет-бутил 3-(4-(1-амино-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропаноат (0,078 г, 0,244 ммоль), полученный в примере получения 15, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 59%).
1H ЯМР (400 МГц, хлороформ-D): δ 9,43 (с, 1H), 8,68 (с, 1H), 7,72-7,59 (м, 3H), 7,56 (с, 1H), 7,41 (т, J=8,2 Гц, 1H), 7,29 (д, J=8,2 Гц, 2H), 7,23-7,07 (м, 4H), 7,00-6,88 (м, 1H), 4,18-4,05 (м, 2H), 2,84 (с, 2H), 1,65 (с, 6H), 1,44 (т, J=6,9 Гц, 3H), 1,16 (с, 6H)
Пример 30: Синтез 2-(4-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенокси)-2-метилпропановой кислоты
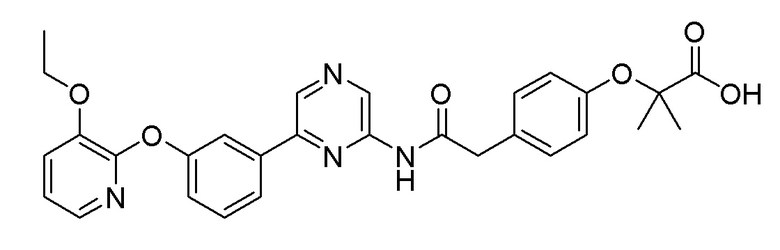
2-Хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин (0,08 г, 0,244 ммоль), полученный в примере получения 4, и трет-бутил 2-(4-(2-амино-2-оксоэтил)фенокси)-2-метилпропаноат (0,072 г, 0,244 ммоль), полученный в примере получения 16, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 35%).
1H-ЯМР (400 МГц, DMSO-D6): δ 10,94 (с, 1H), 9,23 (с, 1H), 8,94 (с, 1H), 7,93 (д, J=8,2 Гц, 1H), 7,83 (т, J=1,8 Гц, 1H), 7,61 (дд, J=5,0, 1,4 Гц, 1H), 7,58-7,43 (м, 2H), 7,19 (д, J=8,7 Гц, 3H), 7,07 (дд, J=8,0, 4,8 Гц, 1H), 6,74 (д, J=8,7 Гц, 2H), 4,20-4,07 (м, 2H), 3,66 (с, 2H), 1,45 (с, 6H), 1,33 (т, J=7,1 Гц, 3H)
Пример 31: Синтез (R)-1-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновой кислоты
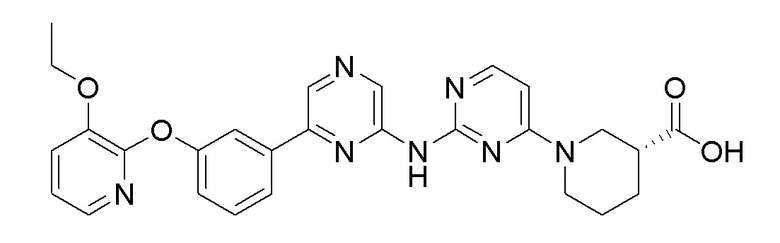
2-Хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин (0,08 г, 0,244 ммоль), полученный в примере получения 4, и этил (R)-1-(2-аминопиримидин-4-ил)пиперидин-3-карбоксилат (0,055 г, 0,222 ммоль), полученный в примере получения 11, использовали таким же образом, как в примере 1 и примере 3, с получением целевого продукта (выход: 20%) ).
1H-ЯМР (400 МГц, МЕТАНОЛ-D4): δ 9,36 (с, 1H), 8,61 (с, 1H), 7,96 (д, J=6,4 Гц, 1H), 7,86 (д, J=7,8 Гц, 1H), 7,77 (т, J=2,1 Гц, 1H), 7,66 (дд, J=4,8, 1,6 Гц, 1H), 7,51 (д, J=7,8 Гц, 1H), 7,46 (дд, J=8,5, 2,1 Гц, 1H), 7,11 (дд, J=8,0, 4,8 Гц, 2H), 6,41 (д, J=6,4 Гц, 1H), 4,48-4,29 (1H), 4,29-4,17 (1H), 4,13 (кв, J=7,0 Гц, 2H), 3,35 (д, J=9,1 Гц, 1H), 3,23-2,96 (1H), 2,52-2,39 (1H), 2,07 (с, 1H), 1,80 (д, J=10,5 Гц, 2H), 1,57 (д, J=3,7 Гц, 1H), 1,37 (т, J=7,1 Гц, 3H)
Пример 32: Синтез 3-(3-(6-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановой кислоты
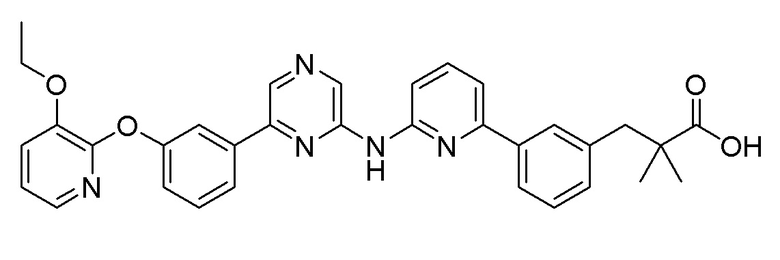
2-Хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин (0,08 г, 0,244 ммоль), полученный в примере получения 4, и трет-бутил 3-(3-(6-аминопиридин-2-ил)фенил)-2,2-диметилпропаноат (0,072 г, 0,222 ммоль), полученный в примере получения 12, использовали таким же образом, как в примере 1 и на стадии 3 примера получения 11, с получением целевого продукта (выход: 26%).
1H-ЯМР (400 МГц, МЕТАНОЛ-D4): δ 9,40 (с, 1H), 8,62-8,47 (1H), 7,91 (с, 1H), 7,86 (т, J=8,5 Гц, 2H), 7,80 (д, J=2,3 Гц, 1H), 7,76-7,59 (м, 2H), 7,58-7,43 (м, 2H), 7,43-7,27 (м, 3H), 7,21 (д, J=8,2 Гц, 1H), 7,13 (тд, J=5,1, 2,7 Гц, 2H), 4,22-4,10 (м, 2H), 2,96 (с, 2H), 1,36 (т, J=6,9 Гц, 3H), 1,20 (с, 7H)
Пример 33: Синтез (1r,4r)-4-((2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)окси)циклогексан-1-карбоновой кислоты
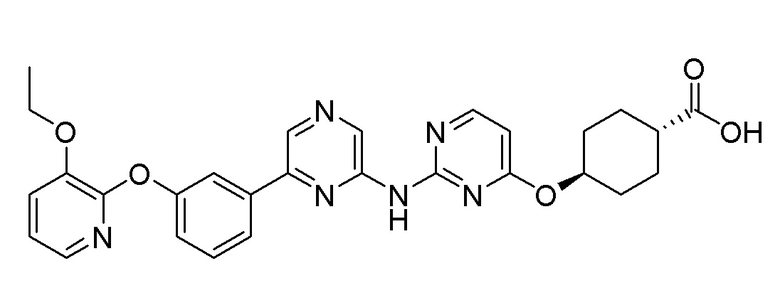
2-Хлор-6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин (0,10 г, 0,305 ммоль), полученный в примере получения 4, и метил (1r,4r)-4-((2-аминопиримидин-4-ил)окси)циклогексан-1-карбоксилат (0,084 г, 0,336 ммоль), полученный в примере получения 18, использовали таким же образом, как в примере 1 и примере 3, последовательно с получением целевого продукта (выход: 42,2%).
1H-ЯМР (400 МГц, DMSO-D6): δ 10,09 (с, 1H), 9,40 (с, 1H), 8,82 (с, 1H), 8,26 (д, J=5,5 Гц, 1H), 7,94 (д, J=7,8 Гц, 1H), 7,86 (т, J=2,1 Гц, 1H), 7,61 (дд, J=5,0, 1,4 Гц, 1H), 7,55-7,43 (м, 2H), 7,16 (дд, J=7,5, 2,1 Гц, 1H), 7,07 (дд, J=8,0, 4,8 Гц, 1H), 6,36 (д, J=5,9 Гц, 1H), 4,96 (т, J=4,8 Гц, 1H), 4,12 (кв, J=7,0 Гц, 2H), 2,24-2,05 (м, 3H), 1,93 (д, J=9,6 Гц, 2H), 1,56-1,37 (4H), 1,33 (т, J=7,1 Гц, 3H)
Экспериментальный пример: Измерение ингибирующего эффекта в отношении активности фермента DGAT2
Ингибирующий эффект в отношении активности фермента DGAT2 исследовали путем проведения следующего эксперимента с соединениями формулы (1) по настоящему изобретению.
1. Получение экспрессирующего вектора DGAT2
Для получения pBacPAK9-DGAT2, который представляет собой экспрессирующий вектор DGAT2, ген DGAT2 человека, амплифицированный с помощью полимеразной цепной реакции (ПЦР), клонировали в сайты EcoR1 и Xho1 вектора pBacPAK9 (clonctech). Нуклеотидная последовательность праймеров, используемых в ПЦР, представляла собой прямой праймер 5' CTATAAATACGGATCCCGGGAATTCATGGACTACAAGGACGACGATGACAAGCTTAAGACCCTCATAGCCGCC и обратный праймер 5' TAAGCGGCCGCCCTGCAGGCCTCGAGTCAGTTCACCTCCAGGAC. Композиция реакционного раствора содержала 50 нг клона кДНК (OriGene), 200 мкМ dATP, dCTP, dTTP, dGTP, 200 нМ каждого праймера, 1 единицу Tag ДНК-полимеразы (Toyobo), 1x ПЦР-буфер, и конечный объем доводили до 20 мкл. Условия реакции денатурировали при 95°C в течение 5 минут, затем 30 раз при 94°C в течение 20 секунд, 60°C в течение 20 секунд и 72°C в течение 90 секунд с последующей реакцией при 72°C в течение 7 минут.
2. Экспрессия DGAT2 и получение мембранного белка
Рекомбинантный человеческий белок DGAT2 экспрессировали в клетках Sf-21, которые представляют собой клетки насекомых, с использованием бакуловирусной системы экспрессии BacPack (Clontech). Кратко способ получения выглядит следующим образом. Сначала экспрессирующий вектор pBacPAK9-DGAT2 трансфицировали ДНК вируса BacPAK6 (гидролиз Bsu36I) в клетки sf21 с использованием Bacfectin для получения рекомбинантного бакуловируса, экспрессирующего DGAT2. Полученный таким образом бакуловирус инфицировали клетками Sf-21 при 10 MOI (множественность заражения), и через 72 часа собирали инфицированные клетки насекомых и выделяли мембранные белки. Для разделения мембранных белков клеточный осадок растворяли в растворе сахарозы, содержащем 250 мМ сахарозы, 10 мМ Tris (pH 7,4), и 1 мМ этилендиаминтетрауксусной кислоты (EDTA), и затем гомогенизировали с помощью гомогенизатора Даунса, и супернатант отбирали путем центрифугирования при 600×g в течение 15 минут и центрифугировали при 100000×g в течение 1 часа, чтобы удалить супернатант, и оставшийся осадок ресуспендировали в 20 мМ буфере HEPES (pH 7,4). Полученный сверхэкспрессирующий мембранный белок DGAT2 распределяли по 100 мкл и хранили при -80°C до использования. Концентрацию белка определяли количественно с помощью набора для анализа белка BCA Protein Assay Kit (Thermo Scientific).
3. Измерение ингибирующего эффекта в отношении активности фермента DGAT2
Анализ DGAT2 in vitro осуществляли с использованием планшета Phospholipid Flash Plate (PerkinElmer) на основе принципа SPA (сцинтилляционный анализ сближения). Сначала соединения, ингибирующие DGAT2, серийно разведенные в 5 раз от 3 нМ до 10 мкМ (конечная концентрация, 1% DMSO), смешивали в буферном растворе, содержащем 2 мкг DGAT2-мембранного белка, и 20 мМ HEPES, 20 мМ MgCl2, 1 мг/мл BSA, 50 мкМ 1,2 sn-олеоилглицерина (Sigma), помещали в 96-луночный flash-планшет (FlashPlate) и подвергали взаимодействию при 37°C в течение 20 минут, и затем 1 мкМ [14C] ole oil CoA (PerkinElmer, NEC651050UC) добавляли до конечного объема 100 мкл и далее подвергали взаимодействию при 37°C течение 15 минут. После завершения ферментативной реакции добавляли 100 мкл изопропанола, планшет герметично закрывали пленкой и медленно встряхивали на шейкере для планшетов. На следующий день измеряли амплифицированный сцинтилляционный сигнал (имп/мин) в Topcounter (Packard) для измерения степени образования [14C]-меченого триацилглицерина (ТГ) в качестве продукта реакции. Измеренное значение, когда соединение не обрабатывали, использовали в качестве положительного контроля, а измеренное значение для группы, получавшей соединение, рассчитывали как относительный % для измерения ингибирующего эффекта соединения на образование ТГ. Значение IC50, представляющее собой концентрацию соединения, которая ингибирует образование ТГ на 50%, определяли путем обработки значения ответа в зависимости от концентрации соединения с помощью кривой нелинейной регрессии с использованием PRISM (Graphpad Inc.).
В результате измерения ингибирующего эффекта на действие фермента DGAT2 для соединения формулы (1) конкретные значения IC50 отдельных соединений примеров были такими, как показано в таблице 1 ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ, ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ДИАЦИЛГЛИЦЕРОЛ O-АЦИЛТРАНСФЕРАЗЫ 2, И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2810064C1 |
| НОВОЕ АМИНОАРИЛЬНОЕ ПРОИЗВОДНОЕ, ПРИГОДНОЕ В КАЧЕСТВЕ ИНГИБИТОРА ДИАЦИЛГЛИЦЕРОЛАЦИЛТРАНСФЕРАЗЫ 2, И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2799819C1 |
| НОВОЕ СОЕДИНЕНИЕ, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ЭНТЕРОПЕПТИДАЗЫ | 2019 |
|
RU2768755C1 |
| СОЕДИНЕНИЕ ДЛЯ РАЗРУШЕНИЯ АНДРОГЕНОВОГО РЕЦЕПТОРА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2817356C1 |
| СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ ОКСАДИАЗОЛ, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2789456C2 |
| БИАРИЛЬНОЕ ПРОИЗВОДНОЕ В КАЧЕСТВЕ АГОНИСТА GPR120 | 2015 |
|
RU2683648C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
| СОЕДИНЕНИЯ ИНДОЛА И ИНДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА НЕКРОЗА КЛЕТКИ | 2008 |
|
RU2437883C1 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 2021 |
|
RU2817736C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРИДИНА И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2017 |
|
RU2722594C1 |
Группа изобретений относится к органической химии и включает соединение формулы (1), его фармацевтически приемлемую соль, стереоизомер и фармацевтическую композицию, его содержащую. В формуле (1) A, D и E каждый независимо представляет собой CH или N; R1 представляет собой C1-C7 алкил, C3-C10 циклоалкил или галоген- C1-C7 алкил; R2 представляет собой -G-J-L; где G представляет собой -C(=O)- или прямую связь; J представляет собой C1-C7 алкилен, C2-C7 алкенилен, C1-C7 алкилен-C6-C10 арилен, C2-C7 алкенилен-C6-C10 арилен, C1-C7 алкоксиен-C6-C10 арилен, C6-C10 арилен, 5-12-членный гетероарилен-5-12-членный гетероциклоалкилен, 5-12-членный гетероарилен-C6-C10 арилен или 5-12-членный гетероарилен-окси-C3-C10 циклоалкилен; L представляет собой водород, галоген, амино, нитро, карбокси, карбокси- C1-C7 алкил, карбокси- C1-C7 алкокси, C3-C10 циклоалкил или C6-C10 арил; где алкил, алкилен, карбоксиалкил, карбоксиалкокси или арил необязательно замещен 1-4 заместителями, выбранными из гидрокси, галогена, C1-C7 алкила и C1-C7 алкокси; и гетероциклоалкилен или гетероарилен включает 1-4 гетероатома, выбранных из N, O и S. Технический результат – соединение формулы (1), которое проявляет активность ингибитора диацилглицерол-ацилтрансферазы 2 (DGAT2). 2 н. и 2 з.п. ф-лы, 1 табл., 34 пр.
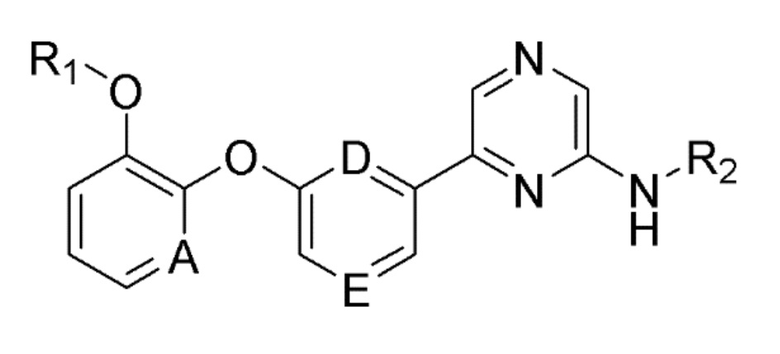
формула (1)
1. Соединение следующей формулы (1) или его фармацевтически приемлемая соль или стереоизомер:
[Формула (1)]
 ,
,
где
A, D и Е, каждый независимо, представляют собой СН или N;
R1 представляет собой C1-C7 алкил, С3-С10 циклоалкил или галоген- C1-C7 алкил;
R2 представляет собой -G-J-L;
где G представляет собой -С(=O)- или прямую связь;
J представляет собой C1-C7 алкилен, С2-С7 алкенилен, С1-С7 алкилен-С6-С10 арилен, С2-С7 алкенилен-С6-С10 арилен, C1-C7 алкоксиен-С6-С10 арилен, С6-С10 арилен, 5-12-членный гетероарилен-5-12-членный гетероциклоалкилен, 5-12-членный гетероарилен-С6-С10 арилен или 5-12-членный гетероарилен-окси-C3-C10 циклоалкилен;
L представляет собой водород, галоген, амино, нитро, карбокси, карбокси- С1-С7 алкил, карбокси- C1-C7 алкокси, С3-С10 циклоалкил или С6-С10 арил;
где алкил, алкилен, карбоксиалкил, карбоксиалкокси или арил необязательно замещен 1-4 заместителями, выбранными из гидрокси, галогена, C1-C7 алкила и C1-C7 алкокси; и
гетероциклоалкилен или гетероарилен включает 1-4 гетероатома, выбранных из N, О и S.
2. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 1, где соединение выбрано из следующей группы:
N-(6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)-3-фенилпропанамид;
метил 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)ацетат;
2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)уксусная кислота;
2-(4-(3-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-3-оксопропил)фенил)уксусная кислота;
метил 2-(4-(3-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропаноат;
этил 2-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-дифторацетат;
3-(4-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановая кислота;
(R)-1-(2-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновая кислота;
3-(3-(6-((6-(5-(2-этоксифенокси)пиридин-3-ил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановая кислота;
N-(6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)-3-фенилпропанамид;
2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)уксусная кислота;
2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)уксусная кислота;
2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенокси)-2-метилпропановая кислота;
2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил-2,2-дифторуксусная кислота;
3-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановая кислота;
2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропановая кислота;
(Е)-2-(4-(3-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-3-оксопро-1-фен-1-ил)фенил)-2-метилпропановая кислота;
3-(4-(1-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропановая кислота;
2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенокси-2-метилпропановая кислота;
2-(4-(2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)фенил)уксусная кислота;
(1R,4R)-4-((2-((6-(3-(2-этоксифенокси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)окси)циклогексан-1-карбоновая кислота;
N-(6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)-3-фенилпропанамид;
3-(4-(2-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановая кислота;
(R)-1-(2-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновая кислота;
3-(3-(6-((6-(6-(2-этоксифенокси)пиридин-2-ил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановая кислота;
2-(4-(3-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенил)-2-метилпропановая кислота;
3-(4-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенил)-2,2-диметилпропановая кислота;
2-(4-(3-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-3-оксопропил)фенокси)-2-метилпропановая кислота;
3-(4-(1-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-метил-1-оксопропан-2-ил)фенил)-2,2-диметилпропановая кислота;
2-(4-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)-2-оксоэтил)фенокси)-2-метилпропановая кислота;
(R)-1-(2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)пиперидин-3-карбоновая кислота;
3-(3-(6-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиридин-2-ил)фенил)-2,2-диметилпропановая кислота; и
(1R,4R)-4-((2-((6-(3-((3-этоксипиридин-2-ил)окси)фенил)пиразин-2-ил)амино)пиримидин-4-ил)окси)циклогексан-1-карбоновая кислота.
3. Фармацевтическая композиция для лечения заболеваний, связанных с диацилглицерол-ацилтрансферазой 2 (DGAT2), включающая соединение формулы (1) или его фармацевтически приемлемую соль или стереоизомер по п. 1 или 2 в качестве активного ингредиента вместе с фармацевтически приемлемым носителем.
4. Фармацевтическая композиция по п. 3, где заболевание, связанное с DGAT2, выбрано из группы, состоящей из жирового гепатоза, неалкогольного стеатогепатита (НАСГ), неалкогольной жировой болезни печени (НАЖБП), диабета, ожирения, гиперлипидемии, атеросклероза и гиперхолестеринемии.
| KR 1020190035897 A, 03.04.2019 | |||
| WO 2010069504 A1, 24.06.2010 | |||
| KR 1020160115997 A, 06.10.2016 | |||
| KR 101464429 B1, 27.11.2014 | |||
| WO 2011031628 A1, 17.03.2011 | |||
| RU 2008143057 A, 10.05.2010 | |||
| WO 2013056679 A1, 25.04.2013. |

