Область техники
Настоящее изобретение относится к солям яблочной кислоты (малатам) и различным полиморфным формам солей яблочной кислоты и (3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты и их фармацевтическим композициям.
Известный уровень техники
Антимикробные хинолоновые соединения, (3S,5S)-7-[3-амино-5-метил-пиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновая кислота и (3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновая кислота, раскрыты в патенте США №6329391, который целиком включен в данное описание в качестве ссылки. Синтез различных хинолоновых соединений описан в литературе, например патент США №6329391; патент США №6803469; В. Ledoussal et al., "Non 6-Fluoro Substituted Quinolone Antibacterials: Structure and Activity", J. Med. Chem., Vol.35, pp.198-200 (1992); V. Cecchetti et al., "Studies on 6-Aminoquinolones: Synthesis and Antibacterial Evaluation of 6-Amino-8-methylquinolones", J. Med. Chem., Vol.39, pp.436-445 (1996); V. Cecchetti et al., "Potent 6-Desfluoro-8-methylquinoloues as New Lead Compounds in Antibacterial Chemotherapy", J. Med. Chem., Vol.39, pp.4952-4957 (1996)).
Вышеупомянутые соединения пригодны для лечения микробных инфекций. Однако остается неизвестным, какие солевые формы дают препараты, пригодные для производства фармацевтически приемлемой композиции. Таким образом, существует техническая потребность в разработке пригодных солевых форм и полиморфов этих антимикробных соединений.
Сущность изобретения
В одном аспекте изобретение относится к солям яблочной кислоты и

(3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты (тут и далее Соединение I, см. также промежуточное соединение (23) в разделе D детального описания изобретения).
В одном аспекте, изобретение относится к полиморфным солям яблочной кислоты Соединения I, в которых присутствует от примерно 0% до примерно 5 мас.% воды.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, в которой присутствует от примерно 1% до примерно 5 мас.% воды.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, в которой присутствует от примерно 0% до примерно 2 мас.% воды.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей рентгеновскую дифрактограмму, по существу соответствующую образцу по Фигуре 1.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей рентгеновскую дифрактограмму, по существу соответствующую образцу по Фигуре 2.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей рентгеновскую дифрактограмму, по существу соответствующую образцу по Фигуре 3.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей 13С ЯМР, по существу соответствующий образцу по Фигуре 4.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей твердофазовый спектр 13С ЯМР, по существу соответствующий образцу по Фигуре 5.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей твердофазовый спектр 13С ЯМР, по существу соответствующий образцу по Фигуре 6.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей твердофазовый спектр 13С ЯМР, по существу соответствующий образцу по Фигуре 7.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей твердофазовый спектр 13С ЯМР, по существу соответствующий образцу по Фигуре 8.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей инфракрасный спектр, по существу соответствующий образцу по Фигуре 9.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей инфракрасный спектр, по существу соответствующий образцу по Фигуре 10.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей инфракрасный спектр, по существу соответствующий образцу по Фигуре 11.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей инфракрасный спектр, по существу соответствующий образцу по Фигуре 12.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей инфракрасный спектр, по существу соответствующий образцу по Фигуре 13.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей характеристические пики рентгеновской дифракции при примерно 10,7, примерно 11,98 и примерно 12,5 градусов 2-тета.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей характеристические пики рентгеновской дифракции при примерно 9,3, примерно 12,1 и примерно 22,6 градусов 2-тета.
В другом аспекте, изобретение относится к полиморфной соли Соединения I, имеющей характеристические пики рентгеновской дифракции при примерно 9,5, примерно 11,7 и примерно 12,3 градусов 2-тета.
В другом аспекте, изобретение относится к полиморфной соли, выбранной из группы, состоящей из D,L-малата полугидрата, D-малата гидрата, L-малата гидрата, D-малата безводного и L-малата безводного.
В другом аспекте, изобретение относится к фармацевтической композиции, включающей безопасное и эффективное количество полиморфа, соответствующего любому из вышеописанных полиморфов, и фармацевтически приемлемого носителя.
В другом аспекте, изобретение относится к способу лечения или профилактики инфекционного расстройства человека или другого животного, нуждающегося в таком лечении, включающему идентификацию человека или другого животного, нуждающегося в лечении или профилактике инфекционного расстройства, и введение человеку или другому животному безопасного и эффективного количества Соединения по п.1.
Краткое описание чертежей
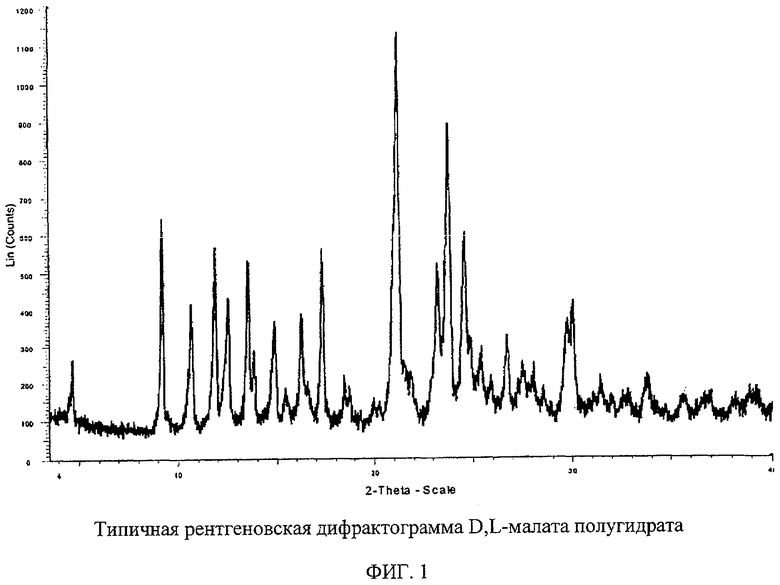
Фигура 1 изображает типичную рентгеновскую дифрактограмму полиморфной соли D,L-малата Соединения I полугидрата.
Фигура 2 изображает типичную рентгеновскую дифрактограмму полиморфной соли D-малата Соединения I гидрата.
Фигура 3 изображает типичную рентгеновскую дифрактограмму полиморфной соли L-малата Соединения I гидрата.
Фигура 4 изображает типичный твердофазовый спектр 13С ЯМР полиморфной соли D,L-малата Соединения I полугидрата.
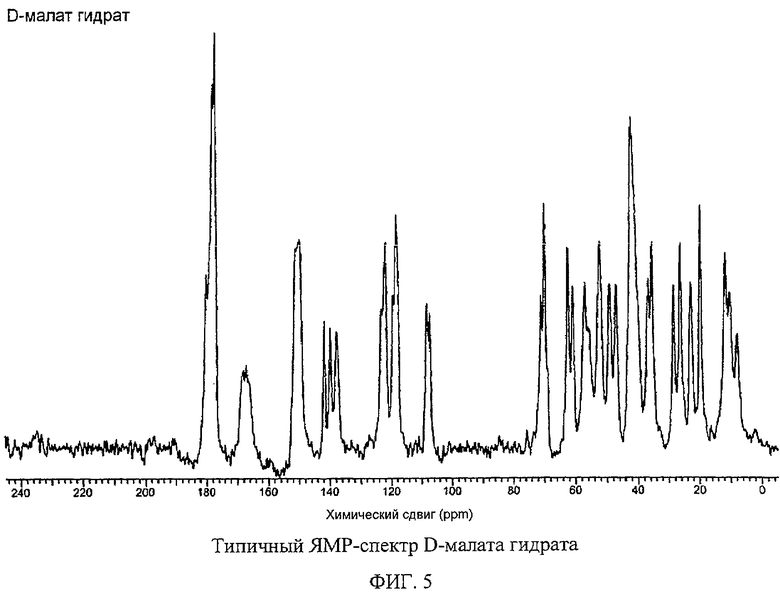
Фигура 5 изображает типичный твердофазовый спектр 13С ЯМР полиморфной соли D-малата Соединения I гидрата.
Фигура 6 изображает типичный твердофазовый спектр 13С ЯМР полиморфной соли L-малата Соединения I гидрата.
Фигура 7 изображает типичный твердофазовый спектр 13С ЯМР полиморфной соли D-малата Соединения I безводного.
Фигура 8 изображает типичный твердофазовый спектр 13С ЯМР полиморфной соли L-малата Соединения I безводного.
Фигура 9 изображает типичный инфракрасный спектр полиморфной соли D,L-малата Соединения I полугидрата.
Фигура 10 изображает типичный инфракрасный спектр полиморфной соли D-малата Соединения I гидрата.
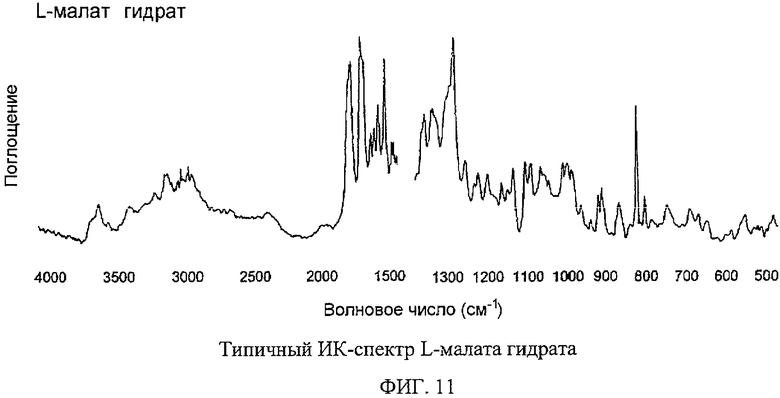
Фигура 11 изображает типичный инфракрасный спектр полиморфной соли L-малата Соединения I гидрата.
Фигура 12 изображает типичный инфракрасный спектр полиморфной соли D-малата Соединения I безводного.
Фигура 13 изображает типичный инфракрасный спектр полиморфной соли L-малата Соединения I безводного.
Детальное описание изобретения
Настоящее изобретение описывает различные соли яблочной кислоты и разные полиморфы соли яблочной кислоты. Выбор фармацевтически приемлемой соли с желательными характеристиками, например растворимостью, стабильностью, удобством составления композиций, требует проведения оценки многих солей и образуемых ими полиморфов (см. Handbook of Pharmaceutical Salts, Properties, Selection and Use. Edited by P.H.Stahl, C.G.Wermuth (Wiley-VCH, Zurich, 2002)).
Твердые вещества существуют в аморфных или кристаллических формах. В случае кристаллических форм молекулы расположены в узлах 3-мерной решетки. Когда соединение кристаллизуется из раствора или суспензии, оно может кристаллизоваться с различными конфигурациями пространственной решетки, причем такая способность называется "полиморфизмом", а разные кристаллические формы индивидуально называются "полиморфами". Разные полиморфные формы данного вещества могут отличаться друг от друга по одному или больше физическим свойствам, таким как растворимость и скорость растворения, абсолютная плотность, форма кристаллов, поведение при сжатии, текучесть и/или стабильность в твердом состоянии.
Кристаллизация
В промышленных масштабах кристаллизация производится путем осуществления манипуляций с раствором таким образом, чтобы превысить предел растворимости для требуемого соединения. Это может быть достигнуто различными способами, например путем растворения соединения при относительно высокой температуре и затем охлаждения раствора до состояния ниже предела насыщения. Альтернативно объем жидкости может быть уменьшен путем кипячения, выпаривания при давлении окружающей среды, вакуумным высушиванием или другими средствами. Растворимость соединения, представляющего интерес, может быть уменьшена путем прибавления антирастворителя или растворителя, в котором соединение проявляет пониженную растворимость, или смеси таких растворителей. Другим вариантом может быть регулировка рН для снижения растворимости. Детальное описание кристаллизации приведено в Crystallization, 3rd edition, J.W.Mullens, Butterworth-Heineman Ltd, 1993, ISBN 0750611294.
Если желательно, чтобы образование соли протекало параллельно кристаллизации, то прибавление соответствующей кислоты или основания может приводить к прямой кристаллизации желательной соли, если соль является менее растворимой в реакционной среде, чем исходный материал. Аналогично завершение реакции синтеза в среде, в которой конечная желательная форма является менее растворимой, чем реагенты, может обеспечить возможность прямой кристаллизации конечного продукта.
Оптимизация кристаллизации может включать затравливание среды кристаллизации кристаллами желательной формы. Дополнительно, многие процессы кристаллизации используют комбинации вышеописанных стратегий. Примером может быть растворение соединения, представляющего интерес, в растворителе при высокой температуре, с последующим контролируемым прибавлением антирастворителя в объеме, достаточном для того, чтобы привести систему в состояние ниже уровня насыщения. После этого могут быть добавлены затравочные кристаллы желательной формы и, при сохранении затравочных кристаллов в интактном виде, система охлаждается для протекания кристаллизации.
Фармацевтические композиции и способы использования
Данное изобретение также предусматривает способы лечения или профилактики инфекционного расстройства человека или другого животного субъекта путем введения безопасного и эффективного количества соли или полиморфа указанному субъекту. В используемом тут значении "инфекционное расстройство" означает любое расстройство, характеризующееся присутствием микробной инфекции. Предпочтительными способами по настоящему изобретению являются способы лечения бактериальных инфекций. Такие инфекционные расстройства включают (например) инфекции центральной нервной системы, инфекции наружного уха, инфекции среднего уха (такие как острый средний отит), инфекции черепных синусов, глазные инфекции, инфекции полости рта (такие как инфекции зубов, десен и слизистой), инфекции верхних дыхательных путей, инфекции нижних дыхательных путей, включая пневмонию, инфекции мочеполовой системы, желудочно-кишечные инфекции, гинекологические инфекции, септицемию, сепсис, перитонит, инфекции костей и суставов, инфекции кожи и кожных структур, бактериальный эндокардит, ожоги, антибактериальную профилактику при хирургии и антибактериальную профилактику у послеоперационных пациентов или у пациентов с ослабленным иммунитетом (таких как пациенты, получающие противоопухолевую химиотерапию или пациенты с пересадкой органов).
Соли или полиморфы по изобретению могут быть введены для лечения или для предотвращения различных микробных болезней. Фармацевтическая композиция может включать
(a) безопасное и эффективное количество соли или полиморфа по изобретению и
(b) фармацевтически приемлемый носитель.
Термин "лечение" используется в данном изобретении для обозначения того, что введение соединения по настоящему изобретению облегчает болезнь или расстройство у хозяина. Таким образом, термин "лечение" включает предотвращение возникновения расстройства у хозяина, особенно, если хозяин имеет предрасположенность к развитию болезни, но диагноз болезни еще не был установлен; ингибирование расстройства и/или облегчение или обращение расстройства. Постольку поскольку способы по настоящему изобретению направлены на предотвращение расстройства, следует понимать, что термин "предотвращать" не требует полного прекращения болезненного состояния (см. Webster's Ninth Collegiate Dictionary). Скорее, в используемом тут значении, термин предотвращение охватывает способность квалифицированного специалиста идентифицировать популяцию, восприимчивую к расстройствам, таким образом, чтобы введение соединений по настоящему изобретению могло происходить до начала болезни. Термин не подразумевает, что болезненного состояния можно будет полностью избежать. Соединения, идентифицированные способами скрининга по настоящему изобретению, могут быть введены в сочетании с другими соединениями.
Безопасность и терапевтическая эффективность идентифицированных соединений могут быть определены с помощью стандартных процедур, использующих технологии in vitro или in vivo. Соединения, проявляющие достаточные терапевтические показатели, могут быть предпочтительными, хотя соединения с недостаточными в других отношениях терапевтическими показателями также могут оказаться полезными. Данные, полученные с помощью токсикологических и фармакологических методик in vitro и in vivo, могут быть использованы для составления композиций с количественным диапазоном доз. Эффективность соединения может быть дополнительно оценена на животных моделях или в клинических испытаниях на пациентах.
"Безопасное и эффективное количество" соединения по изобретению представляет собой количество, эффективно ингибирующее микробный рост в очаге инфекции, подвергаемому лечению у хозяина, с приемлемыми побочными эффектами (такими как токсичность, раздражение или аллергическая реакция). Конкретная величина "безопасного и эффективного количества" будет меняться в зависимости от таких факторов, как конкретное состояние, лечение которого проводится, физическое состояние пациента, продолжительность лечения, характер сопутствующей терапии (если она проводится), конкретный вид используемой дозированной формы, используемый эксципиент(ы) и схема введения доз, желательная для композиции.
В используемом тут значении "фармацевтически приемлемый носитель" должен включать растворители, дисперсионные среды, покрытия, антибактериальные и антигрибковые агенты, изотонические средства и агенты замедления всасывания и т.д., совместимые с фармацевтическим введением. Использование таких сред и агентов для фармацевтически активных веществ известено из уровня техники. За исключением любой возможной обычной несовместимости среды или агента с соединением, такие среды могут быть использованы в композициях по изобретению. Дополнительные соединения также могут быть включены в композиции. Фармацевтическая композиция по изобретению составляется так, чтобы она была совместимой с ее предполагаемым путем введения. Примеры путей введения включают парентеральное (например, внутривенное, интрадермальное, подкожное, внутримышечное), пероральное, с помощью ингаляции, трансдермальное (местное), через слизистую и ректальное введение. Растворы или суспензии, используемые для парентерального, интрадермального или подкожного применения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфид натрия; хелатообразующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. рН может быть отрегулирован с помощью пригодных кислот или оснований. Препарат для парентерального введения может быть заключен в ампулы, разовые шприцы или флаконы с многократными дозами, изготовленные из стекла или пластика.
Фармацевтические композиции, пригодные для инъекционного применения, включают стерильные водные растворы (в случае водорастворимых компонентов) или дисперсии и стерильные порошки для приготовления предназначенных для немедленного приема стерильных инъецируемых растворов или дисперсий. Для внутривенного введения пригодные носители включают физиологический раствор, Cremophor ELTM (BASF, Parsippany, NJ) или фосфатно-солевой буферный раствор (PBS). Композиция может быть стерильной и быть жидкой в такой степени, чтобы существовала возможность простого введения шприцем. Она должна быть стабильной в условиях производства и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и полиэтиленгликоль) и их пригодные смеси. Текучесть может поддерживаться, например, путем использования покрытий, таких как лецитин, путем обеспечения требуемого размера частиц, в случае дисперсий, и путем использования поверхностно-активных веществ. Предотвращение микробного роста может быть достигнуто с помощью различны антибактериальных и антигрибковых агентов, например парабенов, хлорбутанола, фенола, аскорбиновой кислоты, тимеросала. В состав могут входить изотонические агенты, например, сахара многоатомные спирты, такие как маннит, сорбит, и хлорид натрия. Замедленное всасывание композиций для инъекций может быть достигнуто путем включения в композиции агента, замедляющего всасывание, например алюминия моностеарата и желатина.
Стерильные растворы для инъекций могут быть приготовлены путем включения соединения в требуемом количестве в соответствующий растворитель с одним или комбинацией ингредиентов, перечисленных выше, с последующей стерилизацией фильтрацией. Дисперсные среды могут быть приготовлены путем включения соединения в стерильный носитель, который может содержать основную дисперсионную среду и другие ингредиенты. В случае стерильных порошков для приготовления стерильных растворов для инъекций предпочтительные способы приготовления включают вакуумное высушивание и лиофилизацию, которые позволяют получить порошок соединения плюс любые дополнительные желательные ингредиенты из их предварительно стерилизованных фильтрацией растворов.
Композиции для ухода за полостью рта могут включать инертный разбавитель или съедобный носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для перорального введения агенты могут быть заключены в энтеросолюбильные формы для прохождения через желудок или на них может быть нанесено дополнительное покрытие или приготовлены смеси для высвобождения в конкретном участке желудочно-кишечного тракта известными способами. Для перорального терапевтического введения соединение может быть объединено с эксципиентами и использовано в форме таблеток, лепешек или капсул. Композиции для ухода за полостью рта также могут быть приготовлены с использованием текучего носителя для применения в качестве жидкости для полоскания рта, в которой соединение в текучем носителе наносится орально и используется для полоскания и отхаркивания или проглатывается. Фармацевтически совместимые связующие агенты и/или вспомогательные материалы могут быть включены в состав композиций. Таблетки, пилюли, капсулы, лепешки и т.д. могут содержать любые из следующих ингредиентов или соединений аналогичного характера: связующее, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиент, такой как крахмал или лактоза, распушивающий агент, такой как альгиновая кислота, PrimogelTM или кукурузный крахмал; смазывающее вещество, такое как стеарат магния; скользящее вещество, такое как коллоидная двуокись кремния; подсластитель, такой как сахароза или сахарин или вкусовой агент, такой как ароматизатор со вкусом перечной мяты, апельсина, или метилсалицилат.
Для введения путем ингаляции соединения могут быть доставлены в форме аэрозольного спрея из контейнера или дозатора, находящихся под давлением, которые содержат пригодный вытеснитель, например газ, такой как двуокись углерода, или с помощью распылителя.
Системное введение также может быть осуществлено трансмукозальными или трансдермальными средствами. Для трансмукозального или трансдермального введения в композиции могут быть использованы пенетранты, соответствующие барьеру, через который необходимо пройти. Такие пенетранты хорошо известны специалистам и включают, например, для трансмукозального введения поверхностно-активные вещества, соли желчных кислот и производные фусидовой кислоты. Трансмукозальное введение может быть осуществлено с помощью назальных спреев или суппозиториев. Для трансдермального введения соединения могут быть изготовлены в виде жидких мазей, мазей, гелей или кремов, как хорошо известно специалистам.
Соединения также могут быть приготовлены в форме суппозиториев (например, с обычными основами суппозиториев, такими как масло какао и другие глицериды) или удерживающих клизм для ректальной доставки.
В одном варианте исполнения соединения готовят с носителями, которые будут защищать соединение от быстрого выведения из организма, такими как композиции с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Могут быть использованы биоразлагаемые, биосовместимые полимеры, такие как этилен-винилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы приготовления таких композиций понятны специалистам. Липосомальные суспензии также могут быть использованы в качестве фармацевтически приемлемых носителей.
Может быть предпочтительным изготовление композиций для перорального или парентерального введения в виде дозированных форм для простоты введения и однородности доз. "Дозированная лекарственная форма" в используемом тут значении относится к физически дискретным единицам, пригодным для использования в качестве разовых доз для субъекта, лечение которого проводится, причем каждая доза содержит предварительно определенное количество соединения, рассчитанное на получение желательного терапевтического эффекта, в сочетании с фармацевтическим носителем. Характеристики дозированных лекарственных форм по изобретению могут определяться и могут зависеть от характеристик соединения, конкретного терапевтического эффекта, который должен быть достигнут, и ограничений, присущих технологиям приготовления таких соединений для лечения животных.
Примеры
Пример 1. Синтез (3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты и ее малатной соли.
А. Синтез трет-бутилового сложного эфира (3S,5S)-(5-метилпиперидин-3-ил)карбаминовой кислоты (8).

2-Метиловый сложный эфир (2S)-1-(1,1-диметилэтил)-5-оксо-1,2-пирролидиндикарбоновой кислоты (2). Загружают в реактор на 50 л соединение (1) (5,50 кг, 42,60 моль), метанол (27 л) и охлаждают до 10-15°С. Добавляют тионилхлорид (10,11 кг, 2,0 экв.) через капельную воронку в течение 65 мин с наружным охлаждением для поддержания температуры <30°С. Полученный раствор перемешивают при 25°С + 5°С в течение 1,0 часа, после чего метанол отгоняют при пониженном давлении. Полученную вязкую маслянистую жидкость подвергают азеотропной перегонке с этилацетатом (3×2,5 л) для удаления остатков метанола. Остаток растворяют в этилацетате (27,4 л), загружают в реактор на 50 л и нейтрализуют прибавлением триэтиламина (3,6 кг) из капельной воронки в течение 30 минут. Температуру нейтрализации поддерживают ниже 30°С с помощью внешнего охлаждения. Полученную суспензию триэтиламина гидрохлорида удаляют фильтрацией и осветленный маточный раствор загружают в реактор на 50 л вместе с DMAP (0,53 кг). Прибавляют ди-трет-бутилдикарбонат (8,43 кг) через подогреваемую горячей водой капельную воронку на протяжении периода 30 мин с внешним охлаждением для поддержания температуры примерно 20-30°С. Реакция заканчивается через 1 час по результатам анализа методом ТСХ. Органическую фазу промывают охлажденной льдом IN HCl (2×7,5 л), насыщенным раствором бикарбоната натрия (1×7,5 л) и осушают над сульфатом магния. Смесь фильтруют через нутч-фильтр и этилацетат удаляют при пониженном давлении, получая суспензию кристаллического вещества, которую тритурируют с МТВЕ (10,0 л) и фильтруют, получая промежуточное соединение (2) в виде белого твердого вещества (5,45 кг, 52,4%). Анализ. Рассчитано для C11H17NO5: С 54,3; Н 7,04; N 5,76. Найдено: С 54,5; Н 6,96; N 5,80. HRMS (масс-спектроскопия высокого разрешения) (ESI+): ожидается для C11H18NO5, [M+H] 244,1185; найдено: 244,1174; 1Н ЯМР (СDСl3, 500 МГц): δ=4,54 (дд, J=3,1, 9,5 Гц, 1Н), 3,7 (с, 3Н), 2,58-2,50 (м, 1Н), 2,41 (ддд, 1Н, J=17,6, 9,5, 3,7), 2,30-2,23 (м, 1Н), 1,98-1,93 (м, 1Н), 1,40 (с, 9Н); 13С ЯМР (CDCl3, 125,70 МГц): δ 173,3, 171,9, 149,2, 83,5, 58,8, 52,5, 31,1,27,9, 21,5. Т.пл. 70,2°С.
2-Метиловый сложный эфир (2S,4Е)-1-(1,1-диметилэтил)-4-[(диметиламино)метилен]-5-оксо-1,2-пирролидиндикарбоновой кислоты (3). Загружают в реактор на 50 л промежуточное соединение (2) (7,25 кг, 28,8 моль), DME (6,31 кг) и реагент Бредерека (Bredereck) (7,7 кг, 44,2 моль). Раствор перемешивают и нагревают до 75°С±5°С в течение по меньшей мере трех часов. Ход реакции контролируют методом ВЭЖХ. Реакцию охлаждают до 0°С±5°С в течение часа, за это время образуется осадок. Смесь выдерживают при 0°С±5°С в течение одного часа, фильтруют через нутч-фильтр и продукт высушивают в вакуумной печи в течение по меньшей мере 30 часов при 30°С±5°С, получая промежуточное соединение (3) в виде белого кристаллического твердого вещества (6,93 кг, 77,9%). Анализ. Рассчитано для C14H22N2O5: С 56,4; Н 7,43; N 9,39. Найдено: С 56,4; Н 7,32; N 9,48; HRMS (ЕSI+): Ожидается для C14H22N2O5, [M+H] 299,1607. Найдено: 299,1613; 1Н ЯМР (СDСl3, 499,8 МГц) δ=7,11 (с, 1Н), 4,54 (дд, 1Н, J=10,8, 3,6), 3,74 (с, 3Н), 3,28-3,19 (м, 1Н), 3,00 (с, 6Н), 2,97-2,85 (м, 1Н), 1,48 (с, 9Н); 13С ЯМР (CDCl3, 125,7 МГц) δ=172,6, 169,5, 150,5, 146,5, 90,8, 82,2, 56,0, 52,3, 42,0, 28,1, 26,3. Т.пл. 127,9°С.
2-Метиловый сложный эфир (2S,4S)-1-(1,1-диметилэтил)-4-метил-5-оксо-1,2-пирролидиндикарбоновой кислоты (4). Реактор Pfaudler емкостью 10 галлонов пассивируют азотом и загружают 5% порошкообразным палладием на угле ESCAT 142 (50% во влажном состоянии, 0,58 кг во влажном состоянии), промежуточным соединением (3) (1,89 кг, 6,33 моль) и изопропанолом (22,4 кг). Реакционную смесь перемешивают под атмосферой водорода с давлением 45 psi при 45°С в течение 18 ч. Реакционную смесь затем охлаждают до комнатной температуры и фильтруют через слой целита (0,51 кг) на нутч-фильтре для удаления катализатора. Маточный раствор упаривают при пониженном давлении, получая вязкую маслянистую жидкость, которая со временем кристаллизуется, получают соединение 4 (1,69 кг, 100%) в виде 93:7 диастереомерной смеси. Образец смеси продукта очищают методом препаративной ВЭЖХ, получая материал для аналитических данных. Анализ. Рассчитано для C12H19NO5: С 56,0; Н 7,44; N 5,44. Найдено: С 55,8; Н 7,31; N 5,44; MS (ESI+): Ожидается для C12H19NO5, [M+H] 258,1342. Найдено: 258,1321; 1Н ЯМР (СDСl3, 499,8 МГц) δ=4,44 (м, 1Н), 3,72 (с, 3Н), 2,60-2,48 (м, 2Н), 1,59-1,54 (м, 1Н), 1,43 (с, 9Н), 1,20 (д, J=6,8 Гц, 3Н); 13С ЯМР (CDCl3, 125,7 МГц) δ=175,7, 172,1, 149,5, 83,6, 57,4, 52,5, 37,5, 29,8, 27,9, 16,2. Т.пл. 89,9°С.
трет-Бутиловый сложный эфир (1S,3S)-(4-гидроксил-1-гидроксиметил-3-метил6утил)карбаминовой кислоты (5). Загружают в реактор на 50 л промежуточное соединение (4) (3,02 кг, 11,7 моль), абсолютный этанол (8,22 кг) и МТВЕ (14,81 кг). Раствор перемешивают и охлаждают до 0°С±5°С и добавляют борогидрид натрия (1,36 кг, 35,9 моль) маленькими порциями для поддержания температуры реакции на уровне 0°С±5°С. Наблюдается незначительное газовыделение. Реакционную смесь нагревают до 10°С±5°С и добавляют кальция хлорид дигидрат (2,65 кг) порциями с малой скоростью на протяжении часа, чтобы поддерживать температуру реакции на уровне 10°С±5°С. Реакции позволяют нагреться до 20°С±5°С в течение одного часа и перемешивают дополнительно в течение 12 часов при 20°С±5°С. Реакцию охлаждают до -5°С±5°С, добавляют охлажденную на льду 2N НСl (26,9 кг) с такой скоростью, чтобы поддерживать температуру реакции на уровне 0°С±5°С. Перемешивание прекращают, позволяя фазам разделиться. Нижнюю водную фазу (рН=1) удаляют. Загружают в реактор водный насыщенный бикарбонат натрия (15,6 кг) в течение пяти минут. Перемешивание прекращают, позволяя фазам разделиться. Нижнюю водную фазу (рН=8) удаляют. Загружают в реактор сульфат магния (2,5 кг) и перемешивают в течение по меньшей мере 10 минут. Смесь фильтруют через нутч-фильтр и конденсируют при пониженном давлении, получая промежуточное соединение (5) (1,80 кг, 66%). Анализ. Рассчитано для С11Н23НO4: С 56,6 Н 9,94; N 6,00. Найдено: С 56,0; Н 9,68; N 5,96; HRMS (ESF+): ожидается для C11H23NO4, [M+H] 234,1705. Найдено: 234,1703; 1Н ЯМР (CDCl3, 500 МГц) δ=6,34 (д, J=8,9 Гц, 1Н, NH), 4,51 (т, J=5,8, 5,3 Гц, 1Н, NHCHCH2OH), 4,34 (т, J=5,3, 5,3 Гц, 1Н, СН3СНСН2ОН), 3,46-3,45, (м, 1Н, NHCH), 3,28 (дд, J=10,6, 5,3 Гц, NHCHCHHOH), 3,21 (дд, J=10,2, 5,8 Гц, 1Н, СН3СНСННОН), 3,16 (дд, J=10,2, 6,2 Гц, 1Н, NHCHCHHOH), 3,12 (дд, J=10,6, 7,1 Гц, 1Н, СН3СНСННОН), 1,53-1,50 (м, 1Н, СН3СНСННОН), 1,35 (с, 9Н, O(СН3)3, 1,30 (ддд, J=13,9, 10,2, 3,7 Гц, 1Н, NHCHCHHCH), 1,14 (ддд, J=13,6, 10,2, 3,4 Гц, 1Н, NHCHCHHCH), 0,80 (д, J=6,6 Гц, 3Н, СН3); 13С ЯМР (СDСl3, 125,7 МГц) δ 156,1, 77,9, 50,8, 65,1,67,6, 65,1, 35,6, 32,8, 29,0, 17,1. Т.пл. 92,1°С.
2-трет-Бутоксикарбониламино-5-метансульфонилокси-4-метилпентиловый сложный эфир (2S,4S)-метансульфоновой кислоты (6). Загружают в реактор на 50 л раствор промежуточного соединения (5) (5,1 кг) в изопропилацетате (i-PrOAc) 11,8 кг с последующим промыванием дополнительными 7,9 кг i-PrOAc. Реакцию охлаждают до 15°С±5°С и добавляют триэтиламин (TEA) (7,8 кг), поддерживая заданную температуру. Реактор далее охлаждают до 0°С±5°С и добавляют к реакционному раствору метансульфонилхлорид (MsCl) (6,6 кг), поддерживая заданную температуру. Реакцию перемешивают в течение нескольких часов и контролируют завершение методом ВЭЖХ или ТСХ. Реакцию гасят прибавлением насыщенного водного раствора бикарбоната и полученную изолированную органическую фазу промывают последовательно холодным 10% водным раствором триэтиламина, холодным водным раствор НСl, холодным насыщенным водным раствором бикарбоната и наконец насыщенным водным солевым раствором. Органическую фазу высушивают, фильтруют и концентрируют под вакуумом при температуре ниже 55°С±5°С до получения суспензии твердого вещества/жидкости, содержащей промежуточное соединение (6). Суспензию используют в неочищенном виде для следующей реакции без проведения дополнительных анализов.
трет-Бутиловый сложный эфир (3S,5S)-(1-бензил-5-метилпиперидин-3-ил)-карбаминовой кислоты (7). Загружают в реактор на 50 л 9,1 кг неразбавленного бензиламина. Реактор доводят до температуры 55°С и добавляют в реактор раствор промежуточного соединения (6) (8,2 кг) в 1,2-диметоксиэтане (DME) (14,1 кг), поддерживая температуру 60°С±5°С. После завершения прибавления этого раствора реакцию перемешивают при 60°С±5°С в течение нескольких часов и контролируют завершение методами ТСХ или ВЭЖХ. Реакцию охлаждают до температуры окружающей среды и летучие вещества (DME) удаляют на роторном испарителе под вакуумом. Остаток разводят 11,7 кг 15% (об./об.) раствора этилацетат/гексаны и обрабатывают, при перемешивании, 18,7 кг 20% (мас.) водным раствором карбоната калия. После отстаивания получают трехфазную смесь. Донную водную фазу удаляют и среднюю фазу отставляют. Верхнюю органическую фазу собирают и сохраняют для объединения с экстрактами, полученными при дополнительном экстрагировании. Изолированную среднюю фазу экстрагируют дважды порциями по 11,7 кг 15% (об./об.) раствора этилацетат/гексаны, каждый раз объединяя экстракты с первоначальной органической фазой. Объединенные органические экстракты переносят в роторный испаритель и растворитель удаляют под вакуумом до получения маслянистого остатка. Остаток затем очищают методом крупномасштабной препаративной хроматографии, получая очищенное промежуточное соединение (7) в виде маслянистой жидкости.
трет-Бутиловый сложный эфир (3S,5S)-(5-метилпиперидин-3-ил)карбаминовой кислоты (8). Сосуд высокого давления на 40 л загружают 0,6 кг 50% влажным твердым палладием на угле (Е101, 10 мас.%) в потоке азота. Раствор 3,2 кг промежуточного соединения (7) в 13,7 кг абсолютного этанола затем загружают в реактор под азотом. Реактор продувают азотом и затем закачивают водородом до давления 45 psi. Реакцию затем нагревают до 45°С, поддерживая давление водорода, равное 45 psi. Реакцию контролируют методом ТСХ или ЖХ до завершения. Реакцию охлаждают до температуры окружающей среды, сбрасывают давление и продувают азотом. Содержимое реактора фильтруют через слой целита и твердые вещества промывают 2,8 кг абсолютного этанола. Фильтрат концентрируют с помощью роторного испарителя под вакуумом до воскообразного твердого вещества, получая промежуточное соединение (8): ТСХ Rf (Silica F254, 70:30 об./об. этилацетат-гексаны, окрашивание KМnO4)=0,12; 1Н ЯМР (300 МГц, CDCl3) δ 5,31 (шир.с, 1Н), 3,80-3,68 (м, 1Н), 2,92 (д, J=11,4 Гц, 1Н), 2,77 (АВ кварт, JAB=12,0 Гц, Δν=50,2 Гц, 2Н), 2,19 (т, J=10,7 Гц, 1Н), 1,82-1,68 (м, 2Н), 1,54 (шир.с, 1Н), 1,43 (с, 9Н), 1,25-1,15 (м, 1Н), 0,83 (д, J=6,6 Гц, 3Н); 13С ЯМР (75 МГц, СDСl3) δ 155,3, 78,9, 54,3, 50,8, 45,3, 37,9, 28,4, 27,1, 19,2; MS (ESI+) m/z 215 (M+H), 429 (2M+H).
В. Синтез 1-циклопропил-7-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (19):

Промежуточное соединение (12). Загружают в реактор раствор промежуточного соединения (11) (1,2 кг, 7,7 моль, 1,0 экв.) в безводном толуоле (12 л) с последующим добавлением этиленгликоля (1,8 л, 15,7 моль, 4,2 экв.) и твердой п-толуолсульфоновой кислоты (120 г, 10 мас.%). Реакционную смесь перемешивают при температуре окружающей среды в течение по меньшей мере 30 минут и затем нагревают до кипения с обратным холодильником, собирая азеотропную смесь вода/толуол в аппарат ловушки типа Дина-Старка до завершения реакции по результатам анализа методом ТСХ (15% EtOAc/гексаны об./об.). После завершения, реакцию охлаждают до температуры окружающей среды и выливают в водный раствор бикарбоната натрия (6 л). Органическую фазу толуола удаляют и промывают насыщенным раствором бикарбоната натрия (6 л), дистиллированной водой (2×6 л) и насыщенным водным рассолом (6 л). Органическую фазу удаляют и осушают над MgSO4, фильтруют и упаривают при пониженном давлении, получая промежуточное соединение (12) в виде маслянистой жидкости (1,3 кг, 86%). Материал используется без дополнительной очистки на последующих стадиях реакции.
Промежуточное соединение (13). Загружают в реактор раствор промежуточного соединения (12) (1,2 кг, 6,0 моль, 1,0 экв.) в безводном тетрагидрофуране (12 л) и добавляют н-бутиллитий (2,5 М в гексанах, 2,6 л, 6,6 моль, 1,1 экв.) при -40°С, поддерживая эту температуру на протяжении добавления. Реакцию перемешивают в течение по меньшей мере одного часа при -40°С и прибавляют к смеси триметилборат (0,9 л, 7,8 моль, 1,3 экв.), поддерживая температуру равной или ниже -40°С. Реакционную смесь перемешивают в течение по меньшей мере одного часа при -40°С до завершения реакции по результатам анализа методом ТСХ (30% ЕtOАс/гексаны об./об.). Реакцию слегка нагревают до -30°С и медленно прибавляют уксусную кислоту (3 л). После завершения прибавления в реакцию добавляют воду (0,5 л) и смеси позволяют быстро нагреться до температуры окружающей среды, оставляя ее при перемешивании в течение ночи. Органический растворитель удаляют из реакции перегонкой при пониженном давлении при 45°С. К остатку реакции прибавляют 3-4 объема воды (6 л) и, медленно, 30% перекись водорода (0,7 л, 1,0 экв.) при температуре окружающей среды при охлаждении для контроля экзотермической реакции. Реакцию перемешивают в течение по меньшей мере часа при температуре окружающей среды до завершения при определении методом ТСХ (15% ЕtOАс/гексаны об./об.). Реакционную смесь охлаждают до 0-5°С и избыток пероксида гасят добавлением 10% водного раствора бисульфита натрия (2 л). Смесь тестируют для подтверждения отрицательного результата определения пероксида и реакцию подкисляют прибавлением 6N НСl (водн.) (1,2 л). Реакцию перемешивают до завершения реакции гидролиза при определении методом ТСХ или ЯМР-анализа. Полученные твердые вещества собирают вакуумной фильтрацией, получая промежуточное соединение (13) в виде твердого вещества желтого цвета (1,0 кг, 79%).
Промежуточное соединение (14). Загружают в реактор промежуточное соединение (13) (0,53 кг, 3,0 моль, 1,0 экв.) и растворяют в сухом толуоле (2,7 кг, 3,1 л). К этому раствору добавляют диметилсульфат (0,49 кг, 3,9 моль, 1,30 экв.), а затем твердый карбонат калия (0,58 кг, 4,2 моль, 1,4 экв.). Реакционную смесь нагревают до кипения с обратным холодильником и выдерживают в течение по меньшей мере 1 часа до завершения реакции при определении методом ВЭЖХ. На протяжении этого периода времени наблюдается энергичное выделение газа. Реакцию затем охлаждают до температуры окружающей среды и разводят дистиллированной водой (3,2 л) вместе с 30% NaOH (водн.) (0,13 кг, 0,33 экв.). Водную фазу отделяют и остальную толуольную фазу экстрагируют еще два раза дистиллированной водой (3,2 л), объединенной с 30% NaOH (водн.) (0,13 кг, 0,33 экв.), каждый раз удаляя водную фазу. Верхнюю органическую фазу концентрируют перегонкой под вакуумом (<100 мбар) при приблизительно 40°С до получения концентрированного толуольного раствора. Полученный раствор охлаждают до температуры окружающей среды, проверяют качество и выход методом ВЭЖХ и используют на следующей стадии синтеза без дополнительной очистки (предполагая теоретический выход промежуточного соединения (14), равный 0,56 кг).
Промежуточное соединение (15а,b). Загружают в реактор 1,8 кг (2,1 л) безводного толуола вместе с гидридом натрия (0,26 кг, 6,6 моль, 2,20 экв.) в виде 60 мас.% дисперсии в минеральном масле. К этой смеси добавляют (0,85 кг, 7,2 моль, 2,4 экв.) диэтилкарбоната при нагревании реакционной смеси до 90°С в течение 1 часа. Раствор промежуточного соединения (14) (-1,0 экв.) в толуоле с предшествующей стадии добавляют к реакции, поддерживая температуру 90°С±5°С. Во время этого прибавления может наблюдаться выделение газа. После завершения прибавления реакцию перемешивают в течение по меньшей мере 30 минут или до завершения при определении методом ВЭЖХ. После завершения смесь охлаждают до температуры окружающей среды и разводят 10 мас.% водной серной кислотой (3,8 кг, 3,9 моль, 1,3 экв.) при перемешивании. Фазам позволяют разделиться и нижнюю водную фазу удаляют. Оставшуюся органическую фазу концентрируют под вакуумом (<100 мбар) при приблизительно 40°С до получения концентрированного толуольного раствора. Полученный раствор охлаждают до температуры окружающей среды и используют на следующей стадии синтеза без дополнительной очистки (предполагают теоретический выход промежуточного соединения (15а,b), равный 0,85 кг).
Промежуточные соединения (16a,b; 17a,b). Загружают в реактор раствор промежуточного соединения (15а,b) (0,85 кг, ~3,0 моль, ~1,0 экв.) в толуоле с предшествующей стадии. В реактор затем прибавляют диметилформамид-диметилацеталь (0,54 кг, 4,5 моль, 1,5 экв.) и полученный раствор нагревают до температуры кипения с обратным холодильником (~95-105°С). Позволяют выпариться низкокипящему растворителю (метанол из реакции), поддерживая температуру >90°С. Нагревание продолжают в течение по меньшей мере 1 часа или до завершения реакции по результатам анализа методом ВЭЖХ. После завершения реакцию, содержащую смесь промежуточного соединения (16а, b), охлаждают до температуры окружающей среды и прибавляют к реакции толуол (1,8 кг, 2,1 л) вместе с циклопропиламином (0,21 кг, 3,6 моль, 1,2 экв.). Реакцию перемешивают при температура окружающей среды в течение по меньшей мере 30 минут до завершения при определении методом ВЭЖХ. После завершения реакцию разводят 10 мас.% водной серной кислотой (2,9 кг, 3,0 моль, 1,0 экв.) при перемешивании и фазам затем позволяют разделиться. Водную фазу удаляют и органическую фазу концентрируют перегонкой при пониженном давлении (<100 мбар) при приблизительно 40°С. При достижении желательной концентрации раствор охлаждают до температуры окружающей среды и толуольный раствор, содержащий смесь промежуточного соединения (17а,b), используют на следующей стадии синтеза без дополнительной очистки (принимают теоретический выход промежуточного соединения (17а,b), равный ~1,1 кг).
Промежуточное соединение (18). Загружают в реактор раствор смеси промежуточного соединения (17а,b) (~4,7 кг, ~3,0 моль) при температуре окружающей среды. В реактор добавляют N,O-бис(триметилсилил)ацетамид (0,61 кг, 3,0 моль, 1,0 экв.) и реакцию нагревают до температуры кипения с обратным холодильником (~105-115°С) в течение по меньшей мере 30 минут или до завершения по результатам анализа методом ВЭЖХ. Если реакция не завершилась, прибавляют к реакции дополнительное количество N,O-бис(триметилсилил)ацетамида (0,18 кг, 0,9 моль, 0,3 экв.) до ее завершения. После завершения реакцию охлаждают до температуры ниже 40°С и органический растворитель удаляют при пониженном давлении (<100 мбар) при приблизительно 40°С перегонкой до образования осадка. Реакцию охлаждают до температуры окружающей среды и осадок твердого вещества отделяют вакуумной фильтрацией и дважды промывают дистиллированной водой (1×1,8 л, 1×0,9 л). Твердое вещество высушивают, получая промежуточное соединение (18) в виде белого твердого вещества (0,76 кг, 82%). Материал используется без дополнительной очистки на следующей стадии реакции.
Промежуточное соединение (19). Загружают в реактор твердое промежуточное соединение (18) (0,76 кг, ~2,5 моль, ~1,0 экв.) при температуре окружающей среды с последующим добавлением этанола (5,3 кг, 6,8 л) и 32 мас.% водной хлористоводородной кислоты (1,1 кг, 10 моль). Реакционную смесь доводят до температуры кипения с обратным холодильником (76-80°С), причем за это время смесь сначала становится гомогенной, а позднее становится гетерогенной. Смесь нагревают до кипения с обратным холодильником в течение по меньшей мере 5 часов или до завершения по результатам анализа методом ТСХ (15% EtOAc/гексаны об./об.). После завершения реакцию охлаждают до 0°С±5°С и осадок твердого вещества выделяют методом фильтрации, промывают дистиллированной водой (1,7 кг), а затем этанолом (1,7 кг). Собранное твердое вещество высушивают, получая промежуточное соединение (19) в виде белого твердого вещества (0,65 кг, ~95%). 1Н ЯМР (CDCl3, 300 МГц) δ (млн-1): 14,58 (с, 1Н), 8,9 (с, 1Н), 8,25 (м, 1Н), 7,35 (м, 1Н), 4,35 (м, 1Н), 4,08 (с, 3Н), 1,3 (м, 2Н), 1,1 (м, 2Н). 19F ЯМР (CDCl3+CFCl3, 292 МГц) δ (млн-1): -119. ВЭЖХ: 99,5% по площади.
С.Синтез хелата боронового сложного эфира 1-циклопропил-7-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (20).

Загружают в реактор оксид бора (2,0 кг, 29 моль) с последующим разведением ледяной уксусной кислотой (8,1 л, 142 моль) и уксусным ангидридом (16,2 л, 171 моль). Полученную смесь нагревают до температуры кипения с обратным холодильником в течение по меньшей мере 2 часов. Содержимое реакции охлаждают до 40°С и добавляют к реакционной смеси твердое промежуточное соединение (19) 7-фторхинолоновую кислоту (14,2 кг, 51 моль). Смесь снова нагревают до температуры кипения с обратным холодильником в течение по меньшей мере 6 часов. Ход реакции контролируют методами ВЭЖХ и ЯМР. Смесь охлаждают до приблизительно 90°С и прибавляют к реакции толуол (45 л). Реакцию дальше охлаждают до 50°С и прибавляют к реакционной смеси трет-бутилметиловый эфир (19 л) для того, чтобы вызвать осаждение продукта. Смесь затем охлаждают до 20°С и твердый продукт 19 выделяют методом фильтрации. Выделенные твердые вещества затем промывают трет-бутилметиловым эфиром (26 л) перед высушиванием в вакуумной печи при 40°С (50 торр). Выход продукта, полученный для промежуточного соединения (20) в этой реакции, составляет 86,4%. Рамановский спектр (см-1): 3084,7, 3022,3, 2930,8, 1709,2, 1620,8, 1548,5, 1468,0, 1397,7, 1368,3, 1338,5, 1201,5, 955,3, 653,9, 580,7, 552,8, 384,0, 305,8. ЯМР (СDСl3, 300 МГц) δ (млн-1): 9,22 (с, 1Н), 8,38-8,33 (м, 1Н), 7,54 (т, J=9,8 Гц, 1Н), 4,38-4,35 (м, 1Н), 4,13 (с, 3Н), 2,04 (с, 6Н), 1,42-1,38 (м, 2Н), 1,34-1,29 (м, 2Н). ТСХ (Whatman MKC18F Silica, 60Å, 200 мкм). Подвижная фаза: 1:1 (об./об.) СН3СN: 0,5N NaCl (водн.), УФ (254/366 нм) визуализация; Rf=0,4-0,5.
D. Реакция сочетания 1-циклопропил-7-фтор-8-метокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (20) с трет-бутиловым сложным эфиром (3S,5S)-(5-метилпиперидин-3-ил)кар6аминовой кислоты (8) и синтез малата (3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты (25):
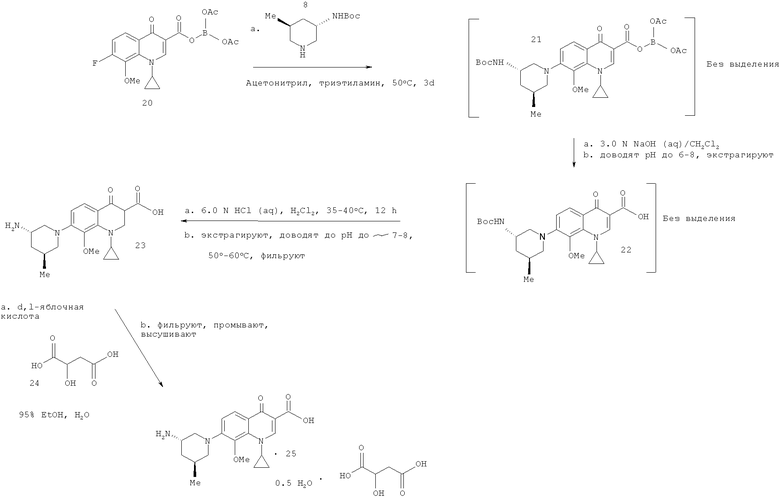
Загружают в реактор твердое промежуточное соединение (20) (4,4 кг, 10,9 моль) с последующим разведением раствором триэтиламина (TEA) (2,1 л, 14,8 моль) и промежуточное соединение с пиперидиновой боковой цепью (8) (2,1 кг, 9,8 моль) в ацетонитриле (33,5 л, 15,7 л/кг) при комнатной температуре. Полученную смесь нагревают до приблизительно 50°С до тех пор, пока реакция не может считаться законченной. Ход реакции контролируют методом ВЭЖХ или обращенно-фазовой ТСХ. После завершения реакцию охлаждают до приблизительно 35°С и объем реакции уменьшают приблизительно наполовину перегонкой ацетонитрила под вакуумом в интервале 0-400 торр. Реактор затем загружают 28,2 кг 3,0 N раствора NaOH (водн.) и температуру повышают до приблизительно 40°С. Перегонку под вакуумом продолжают в течение 1-4 часов или до тех пор, пока не перестанет наблюдаться образование дистиллятов. Реакцию затем охлаждают до комнатной температуры и реакцию гидролиза контролируют методом ВЭЖХ или обращено-фазовой ТСХ. После завершения реакционную смесь нейтрализуют до рН в интервале значений 6-8 путем добавления ~4-5 кг ледяной уксусной кислоты. В реактор затем загружают 12,7 кг (9,6 л) дихлорметана в качестве экстракционного растворителя, смесь перемешивают, фазам позволяют разделиться и органическую дихлорметановую фазу удаляют. Процесс экстракции повторяют еще два раза, используя 12,7 кг (9,6 л) дихлорметана, каждый раз собирая нижнюю органическую фазу. Водную фазу отбрасывают и органические экстракты объединяют в одном реакторе. Содержимое реактора нагревают до 40°С и объем реакции уменьшают приблизительно наполовину перегонкой. В реактор затем загружают 20,2 кг 6,0 N раствора НСl (водн.), температуру доводят до 35°С и перемешивают в течение по меньшей мере 12 часов для обеспечения протекания реакции удаления защитных Вос-групп. Реакцию контролируют методом ВЭЖХ или обращено-фазовой ТСХ. После завершения перемешивание прекращают и фазам позволяют разделиться. Нижнюю органическую фазу удаляют и сохраняют. Реактор затем загружают 12,7 кг (9,6 л) дихлорметана в качестве экстракционного растворителя, смесь перемешивают, фазам позволяют разделиться и органическую дихлорметановую фазу удаляют. Органические экстракты объединяют и отбрасывают. Оставшуюся водную фазу разводят 18,3 кг дистиллированной воды и температуру повышают до приблизительно 50°С. Проводят дистилляцию под вакуумом (100-400 торр) для удаления из реакции остаточного дихлорметана. рН реакции затем доводят до значения в интервале 7,8-8,1 с помощью примерно 9,42 кг 3,0 N раствора NaOH (водн.), поддерживая температуру реакции ниже 65°С. Реакцию охлаждают до 50°С и осадок твердых веществ выдерживают в течение по меньшей мере часа перед охлаждением смеси до комнатной температуры. Твердые вещества изолируют вакуумной фильтрацией и дважды промывают порциями 5,2 кг дистиллированной воды. Твердые вещества высушивают в течение по меньшей мере 12 часов с отсасыванием и затем дополнительно в течение 12 часов в конвекционной печи при 55°С. Выход, полученный для промежуточного соединения (23) в данном примере, составляет 3,2 кг (79%). Загружают в реактор 3,2 кг твердого промежуточного соединения (23) и твердые вещества суспендируют в 25,6 кг 95% этанола в качестве растворителя. В реактор затем добавляют 1,1 кг твердой D,L-яблочной кислоты (24) и смесь нагревают до температуры кипения с обратным холодильником (~80°С). Прибавляют к реакции дистиллированную воду (~5,7 л) до полного растворения и прибавляют 0,2 кг активированного угля. Реакционную смесь пропускают через фильтр для осветления, охлаждают до 45°С и выдерживают в течение периода по меньшей мере 2 часов для протекания кристаллизации. Реакционную смесь далее охлаждают до 5°С и суспендированные твердые вещества выделяют вакуумной фильтрацией. Твердые вещества затем промывают 6,6 кг 95% этанола и высушивают в течение по меньшей мере 4 часов с вакуумным отсосом. Твердые вещества затем далее сушат в конвекционной печи в течение по меньшей мере 12 часов при 45°С, получая 3,1 кг промежуточного соединения (24) (70%). ЯМР (D2O, 300 МГц) δ (млн-1): 8,54 (с, 1Н), 7,37 (д, J=9,0 Гц, 1Н), 7,05 (д, J=9,0 Гц, 1Н), 4,23-4,18 (м, 1Н), 4,10-3,89 (м, 1Н), 3,66 (шир.с, 1Н), 3,58 (с, 3Н), 3,45 (д, J=9,0 Гц, 1Н), 3,34 (д, J=9,3 Гц, 1Н), 3,16 (д, J=12,9 Гц, 1Н), 2,65 (дд, J=16,1, 4,1 Гц, 1Н), 2,64-2,53 (м, 1Н), 2,46 (дд, J=16,1, 8,0 Гц, 1Н), 2,06 (шир.с, 1Н), 1,87 (д, J=14,4 Гц, 1Н), 1,58-1,45 (м, 1Н), 1,15-0,95 (м, 2Н), 0,91 (д, J=6,3 Гц, 3Н), 0,85-0,78 (м, 2Н). ТСХ (Whatman MKC18F Silica, 60Å, 200 мкм). Подвижная фаза: 1:1 (об./об.) CН3CN: 0,5 N NaCl (водн.), УФ (254/366 нм) визуализация. ВЭЖХ: подвижная фаза Н2О с 0,1% муравьиной кислоты/ацетонитрил с 0,1% муравьиной кислоты, градиентное элюирование от 88% Н2O/муравьиная кислота до 20% Н2O/муравьиная кислота, колонка Zorbax SB-C8 4,6 мм × 150 мм. Part №883975,906, объемный расход 1,5 мл/мин, время прогона 20 мин, 292 нм, детектор Model G1314A, S/N JP72003849, четырехкомпонентный насос Model G1311A, S/N US72102299, автоматический пробоотборник Model G1313A, S/N DE14918139, дегазатор Model G1322A, S/N JP73007229; приблизительное время удержания промежуточного соединения (19): 13,0 мин; приблизительное время удержания промежуточного соединения (20): 11,6 мин; приблизительное время удержания промежуточного соединения (21): 16,3 мин; приблизительное время удержания промежуточного соединения (22): 18,2 мин; приблизительное время удержания промежуточного соединения (23): 8,6 мин; приблизительное время удержания соединения (25): 8,6 мин.
Пример 2. Приготовление соли и анализ солевых форм
Скрининг солей проводят с использованием 100 мг свободного основания. Изолированные солевые формы анализируют методами ЯМР, элементного анализа, ТГ-ДТА, рентгенографическим и ВЭЖХ. Таблица 1 содержит физические и химические характеристики этих солевых форм. Как видно, соли яблочной кислоты могут обеспечить баланс между желательной растворимостью, стабильностью и простотой выделения. Дополнительно, использование солей яблочной кислоты может облегчить хиральную очистку. Далее, D,L-малатная, D-малатная или L-малатная соли могут обеспечивать различные преимущества в зависимости от природы хиральной примеси, которую необходимо удалить. Гидратированные формы могут обеспечивать лучшую влагостойкость и стабильность в твердом состоянии, а также большую простоту выделения, использование безводных форм может улучшить кажущуюся растворимость и скорость растворения. Таким образом, малаты Соединения 1 обладают определенными преимуществами, включая простоту выделения, пониженную гигроскопичность, большую растворимость в воде, большую стабильность и простоту составления рецептур.
(мг/мл)
полугидрат,безводный
Пример 3. Получение полугидрата D,L-малатной соли Соединения 1
А. Синтез D,L-малатной соли Соединения 1 из свободного основания. Десять грамм свободного основания Соединения 1 и один эквивалент D,L-яблочной кислоты нагревают в 105 мл 95% этанола до кипения с обратным холодильником (приблизительно 78°С). Добавляют 15 мл воды, поддерживая температуру около 78°С. Перемешивание и нагревание продолжают до полного растворения. Дополнительное количество воды может быть добавлено для обеспечения полного растворения. Раствор медленно охлаждают (по меньшей мере 3 часа) до комнатной температуры при перемешивании для инициирования кристаллизации. Если осаждается маслянистая или воскообразная масса (или фаза, отличающаяся от полугидрата), раствор снова нагревают для полного растворения осадка и охлаждают более медленно. Кристаллические твердые вещества затем фильтруют и промывают небольшим объемом 95% этанола. Кристаллы высушивают при давлении окружающей среды, комнатной температуре и относительной влажности 25-75%.
В. Кристаллизация существующей малатной соли Соединения I: 10 грамм D,L-малатной соли Соединения 1 нагревают в 105 мл 95% этанола до кипения с обратным холодильником (приблизительно 78°С). Добавляют 15 мл воды, поддерживая температуру около 78°С. Перемешивание и нагревание продолжают до полного растворения. Дополнительное количество воды может быть добавлено для обеспечения полного растворения. Раствор медленно охлаждают (по меньшей мере 3 часа) до комнатной температуры при перемешивании для инициирования кристаллизации. Если осаждается маслянистая или воскообразная масса (или фаза, отличающаяся от полугидрата), раствор снова нагревают для полного растворения осадка и охлаждают более медленно. Если осаждается маслянистая или воскообразная масса (или фаза, отличающаяся от полугидрата), раствор снова нагревают для полного растворения осадка и охлаждают более медленно.
Пример 4. Получение гидрата D-малатной соли Соединения 1.
А. Синтез D-малатной соли Соединения 1 из свободного основания. 10 грамм свободного основания Соединения 1 и один эквивалент D-яблочной кислоты нагревают в 75 мл 95% этанола до кипения с обратным холодильником (приблизительно 78°С). Добавляют 25 мл воды, поддерживая температуру около 78°С. Перемешивание и нагревание продолжают до полного растворения. Может быть добавлено дополнительное количество воды для обеспечения полного растворения. Раствор медленно охлаждают (по меньшей мере 3 часа) до комнатной температуры при перемешивании для инициирования кристаллизации. Если образуется маслянистая или воскообразная масса (или фаза, отличающаяся от гидрата), раствор снова нагревают для полного растворения осадка и охлаждают более медленно. Кристаллические твердые вещества затем фильтруют и промывают небольшим объемом 95% этанола. Кристаллы высушивают при давлении окружающей среды, комнатной температуре и относительной влажности 25-75%.
В. Кристаллизация существующей D-малатной соли Соединения I. Десять грамм D-малатной соли Соединения I нагревают в 75 мл 95% этанола до кипения с обратным холодильником (приблизительно 78°С). Добавляют 25 мл воды, поддерживая температуру около 78°С. Перемешивание и нагревание продолжают до полного растворения соли. Дополнительное количество воды может быть добавлено для обеспечения полного растворения. Раствор медленно охлаждают (по меньшей мере 3 часа) до комнатной температуры при перемешивании для инициирования кристаллизации. Если осаждается маслянистая или воскообразная масса (или фаза, отличающаяся от гидрата), раствор снова нагревают до полного растворения осадка и охлаждают более медленно. Кристаллические твердые вещества затем фильтруют и промывают малым объемом 95% этанола. Кристаллы высушивают при давлении окружающей среды, комнатной температуре и относительной влажности 25-75%.
Пример 5. Получение гидрата L-малата Соединения 1.
А. Синтез L-малатной соли Соединения I из свободного основания. 10 грамм свободного основания Соединения I и один эквивалент L-яблочной кислоты нагревают в 75 мл 95% этанола до кипения с обратным холодильником (приблизительно 78°С). Добавляют 25 мл воды, поддерживая температуру около 78°С. Перемешивание и нагревание продолжают до полного растворения. Дополнительное количество воды может быть добавлено для обеспечения полного растворения. Раствор медленно охлаждают (по меньшей мере 3 часа) до комнатной температуры при перемешивании для инициирования кристаллизации. Если осаждается маслянистая или воскообразная масса (или фаза, отличающаяся от гидрата), раствор снова нагревают до полного растворения осадка и охлаждают более медленно. Кристаллически твердые вещества затем фильтруют и промывают маленьким объемом 95% этанола. Кристаллы высушивают при давлении окружающей среды, комнатной температуре и относительной влажности 25-75%.
В. Кристаллизация существующей L-малатной соли Соединения I. 10 грамм L-малатной соли Соединения I нагревают в 75 мл 95% этанола до кипения с обратным холодильником (приблизительно 78°С). Добавляют 25 мл воды, поддерживая температуру около 78°С. Перемешивание и нагревание продолжают до полного растворения соли. Дополнительное количество воды может быть добавлено для обеспечения полного растворения. Раствор медленно охлаждают (по меньшей мере 3 часа) до комнатной температуры при перемешивании для инициирования кристаллизации. Если осаждается маслянистая или воскообразная масса (или фаза, отличающаяся от гидрата), раствор снова нагревают до полного растворения осадка и охлаждают более медленно. Кристаллически твердые вещества затем фильтруют и промывают малым объемом 95% этанола. Кристаллы высушивают при давлении окружающей среды, комнатной температуре и относительной влажности 25-75%.
Пример 6. Получение Соединения I D-малатной безводной соли.
Нагревают 280 мг полугидрата D-малатной соли Соединения I в 5 мл сухого метанола до 70°С. Нагревание и перемешивание продолжают до полного растворения соли. Раствору затем позволяют медленно остыть до комнатной температуры при перемешивании (охлаждение занимает по меньшей мере примерно 3 часа). Кристаллы фильтруют и высушивают при продувке сухим азотом для защиты образца от влаги в процессе сушки.
Пример 7. Получение безводной L-малатной соли Соединения I. Нагревают 200 мг полугидрата L-малатной соли Соединения I в 2 мл сухого метанола до 70°С. Нагревание и перемешивание продолжают до полного растворения соли. Раствору позволяют очень медленно остыть до комнатной температуры. Раствор перемешивают в течение длительного периода времени до протекания кристаллизации или раствор упаривают сухим азотом для индуцирования более быстрой кристаллизации для защиты материала от увеличения водосодержания на стадиях кристаллизации и выделения.
Пример 8. Анализ полиморфов.
Различные полиморфы, которые могут быть получены описанными выше способами, могут быть далее охарактеризованы с использованием описанных ниже методик.
Водосодержание определяется термогравиметрическим анализом (ТГ). Для проведения анализа на воду используется Perkin-Elmer TGA-7. Образцы (5-12 мг) анализируют в сухом азоте в открытых алюминиевых чашечках для образцов при скорости сканирования 5°С/минуту.
Влагосодержание для полугидрата и гидратов, сразу после получения, составляет от 1,5% до 3,0%. Гидраты и полугидраты могут быть высушены до более низкого водосодержания и могут при этом сохранять спектроскопические и рентгенографические характеристики полностью гидратированных материалов. Влагосодержание для безводных материалов колеблется в интервале от недектируемого количества до 1,0%.
Рентгенодифракционный анализ. Рентгеновскую дифракцию образцов порошка проводят с помощью рентгеновского дифрактометра Bruker D5000. D5000 оснащен 2,2 кВт Сu-анодной рентгеновской трубкой, низкотемпературным предметным столиком Anton Parr TTK-1 и высокоскоростным позиционно-чувствительным детектором (PSD). Для получения рентгенограмм порошка используется Cu-K излучение (=1,5418 Å). Сдвоенный фильтр из никелевой фольги помещают на приемном пути рентгеновских лучей для устранения К β-излучения. Материал закрепляют и анализируют на фронтальном держателе образцов. Сканы осуществляют в диапазоне углов 3,5-40 2-тета, при величине шага 0,02 и 0,2 секундах на шаг.
Анализ методом твердофазового ядерного магнитного резонанса (SSNMR). Все данные регистрируют с помощью спектрометра Varian 300 Unity Inova, оснащенного зондом 7 мм CPMAS, вращающимся с частотой 5 кГц.
Спектры 75,4 МГц 13С регистрируют при кроссполяризации с вращением под магическим углом (CP/MAS) TOSS (полное подавление боковых полос), не размалывают, а непосредственно набивают в 7 мм роторы из нитрида кремния.
Инфракрасный (IR) анализ. Образцы анализируют по методу раздельного помола с использованием спектрометра BioRad FTS-3000 FTIR с KВr расщепителем пучка. Шестнадцать сканов сравнения и образца получают для каждого образца при разрешении по волновому числу 4. Подготовка образцов заключается в смешении примерно 1% образца с соответствующим мелющим агентом (например, фторированная смазка fluorolube для волновых чисел 4000-1350, медицинское масло nujol для волновых чисел 1350-450) с помощью агатовых ступки и пестика. Образцы не размалывают до смешения с мелющим агентом. Сканы сравнения получают с использованием соответствующих дисков KВr, между которыми заключают измельченный образец для анализа образцов.
Пример 9. Характеристики различных солевых форм.
Малатные соли 7-[3S-амино-5S-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты могут быть получены и выделены в условиях практического производства. Использование хирального малата для получения соли (в виде рацемической смеси или в хирально чистых формах) может, в некоторых случаях, помочь хиральной очистке 7-[3S-aминo-5S-мeтилпипepидинил]-1-циклoпpoпил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты. Как класс соли яблочной кислоты являются от растворимых до труднорастворимых в воде (согласно определению Фармакопеи США 28) и обладают подходящей химической стабильностью. Гидратированные формы проявляют фазовую стабильность при относительной влажности до 75%, по результатам измерений методами динамической сорбции пара и статических анализов в камере влажности. Показано, при использовании этих же методов испытаний, что безводные формы набирают влагу и спонтанно превращаются в соответствующую гидратированную форму при воздействии влаги.
Полугидрат D,L-малатной соли Соединения I
Структура D,L-малата полугидрата однозначно подтверждается методом рентгеновской дифракции монокристалла. Наименьшая ячейка данного материала состоит из двух молекул 7-[3S-амино-5S-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты, одной молекулы D-яблочной кислоты, одной молекулы L-яблочной кислоты и одной молекулы воды. Гидратация водой имеет канальный характер, что приводит к некоторым колебаниям содержания воды при изменении относительной влажности.
Гидрат D-малатной и гидрат L-малатной солей Соединения I
D-Малат гидрат и L-малат гидрат могут быть легко выделены в виде кристаллических твердых веществ из систем водных растворителей. Успешное выделение требует использования хирально чистой кислоты. Как и для D,L-малата полугидрата, вода гидратации, по-видимому, находится в каналах, и содержание воды несколько зависит от относительной влажности.
Безводная D-малатная и безводная L-малатная соли Соединения I
Ни одна из безводных форм не была выделена с кристаллитами достаточного размера для получения высококачественных рентгеновских дифрактограмм. Выделение безводных веществ часто приводит к получению маслянистых или воскообразных материалов, которые медленно кристаллизуются в материал с высокой площадью поверхности. Безводные материалы дают порошковые рентгенограммы, соответствующие нанокристаллическому материалу. Полученные рентгеновские дифрактограммы имеют очень низкий уровень сигнала и неразрешимые пики. При воздействии влаги нанокристаллические безводные материалы с высокой площадью поверхности превращаются в соответствующие гидратные формы.
Если не указано иное, все количества, включая численные величины, проценты, части и соотношения, следует рассматривать как модифицированные словом "примерно" и количества не следует понимать как указывающие значащие цифры.
Если не указано иное, неопределенные и определенный артикли "a", "an" и "the" означают "один или больше".
Все документы, цитируемые в детальном описании изобретения, в их соответствующей части, включены в настоящее изобретение в качестве ссылок; ссылка на любой документ не должна истолковываться как допущение того, что он является известным уровнем техники по отношению к настоящему изобретению. В той степени, в которой любое значение или определение термина в данном письменном документе противоречит любому значению или определению термина в документе, включенном в качестве ссылки, главенствующим считается значение или определение, приведенное для термина в данном письменном документе.
Хотя были проиллюстрированы и описаны конкретные варианты исполнения настоящего изобретения, специалистам будет понятно, что различные другие изменения и модификации могут быть выполнены без нарушения сущности и объема изобретения. Таким образом, предполагается, что приложенная формула изобретения охватывает все такие изменения и модификации, входящие в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ | 2010 |
|
RU2551852C2 |
| ТВЕРДЫЕ ФОРМЫ (2S,3R,4R,5S,6R)-2-(4-ХЛОР-3-(4-ЭТОКСИБЕНЗИЛ)ФЕНИЛ)-6-(МЕТИЛТИО)ТЕТРАГИДРО-2Н-ПИРАН-3,4,5-ТРИОЛА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2505543C2 |
| СПОСОБ И ПРОЦЕСС ПРИГОТОВЛЕНИЯ И ПРОИЗВОДСТВА ДЕЙТЕРИРОВАННОЙ ОМЕГА-ДИФЕНИЛМОЧЕВИНЫ | 2011 |
|
RU2527037C2 |
| СИНТЕЗ И НОВЫЕ СОЛЕВЫЕ ФОРМЫ (R)-5-((E)-2-(ПИРРОЛИДИН-3-ИЛВИНИЛ)ПИРИМИДИНА | 2009 |
|
RU2533819C2 |
| СИНТЕЗ И НОВЫЕ СОЛЕВЫЕ ФОРМЫ (R)-5-((Е)-2-ПИРРОЛИДИН-3-ИЛВИНИЛ)ПИРИМИДИНА | 2014 |
|
RU2700796C2 |
| СОЛЬ ОМЕКАМТИВА МЕКАРБИЛА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2014 |
|
RU2663663C2 |
| ТАРТРАТ 3-((1R,3R)-1-(2,6-ДИФТОР-4-((1-(3-ФТОРПРОПИЛ)АЗЕТИДИН-3-ИЛ)АМИНО)ФЕНИЛ)-3-МЕТИЛ-1,3,4,9-ТЕТРАГИДРО-2H-ПИРИДО[3,4-b]ИНДОЛ-2-ИЛ)-2,2-ДИФТОРПРОПАН-1-ОЛА, ЕГО ТВЕРДЫЕ ФОРМЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2809220C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-[(3-АМИНООКСЕТАН-3-ИЛ)МЕТИЛ]-2-(1,1-ДИОКСО-3,5-ДИГИДРО-1,4-БЕНЗОТИАЗЕПИН-4ИЛ)-6-МЕТИЛ-ХИНАЗОЛИН-4-АМИНА | 2015 |
|
RU2664643C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (3R,3aS,6aR) ГЕКСАГИДРОФУРО[2,3-b] ФУРАН-3-ОЛА | 2005 |
|
RU2421458C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3,5-ДИФТОР-БЕНЗИЛАМИДА (S)-3-ГИДРОКСИ-1-(1H-ИНДОЛ-5-ИЛ)-2-ОКСО-ПИРРОЛИДИН-3-КАРБОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2826010C2 |










Изобретение относится к малатным солям (3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты и ее полиморфам. Изобретение также относится к фармацевтической композиции, включающей описанные соли и полиморфы, которые могут найти свое применение в медицине для лечения или профилактики инфекционных расстройств. 4 н. и 12 з.п. ф-лы, 13 ил., 1 табл.
1. Малатная соль (3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты.
2. Малатная соль по п.1, которая представляет собой полиморфную соль, содержащую от 0 до 5% воды по весу.
3. Малатная соль по п.2, в которой присутствует от 1 до 5% воды по весу.
4. Малатная соль по п.2, в которой присутствует от 0 до 2% воды по весу.
5. Малатная соль по п.3, имеющая рентгеновскую дифрактограмму, по существу, соответствующую образцу по одной из Фигур 1, 2 или 3.
6. Малатная соль по п.3, имеющая твердофазовый спектр 13С ЯМР, по существу, соответствующий образцу по одной из Фигур 4, 5 или 6.
7. Малатная соль по п.4, имеющая твердофазовый спектр 13С ЯМР, по существу, соответствующий образцу по одной из Фигур 7 или 8.
8. Малатная соль по п.3, имеющая картину инфракрасного спектра, по существу, соответствующую рентгенограмме по одной из Фигур 9, 10 или 11.
9. Малатная соль по п.4, имеющая картину инфракрасного спектра, по существу, соответствующую рентгенограмме по одной из Фигур 12 или 13.
10. Малатная соль по п.3, имеющая характеристические пики рентгеновской дифракции при примерно 10,7, примерно 11,98 и примерно 12,5° 2-тета.
11. Малатная соль по п.3, имеющая характеристические пики рентгеновской дифракции при примерно 9,3, примерно 12,1 и примерно 22,6° 2-тета.
12. Малатная соль по п.3, имеющая характеристические пики рентгеновской дифракции при примерно 9,5, примерно 11,7 и примерно 12,3° 2-тета.
13. Малатная соль по п.1, представляющая собой полиморфную соль, выбранную из D,L-малата полугидрата, D-малата гидрата, L-малата гидрата, D-малата безводного и L-малата безводного.
14. Полугидрат D,L-малатной полиморфной соли (3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8-метокси-4-оксо-3-хинолин-карбоновой кислоты.
15. Фармацевтическая композиция для лечения или профилактики инфекционного расстройства, включающая:
а. безопасное и эффективное количество малатной соли по любому из предшествующих пунктов формулы и
b. фармацевтически приемлемый носитель.
16. Использование малатной соли в соответствии с любым из пп.1-14 для производства лекарственного средства для использования при лечении или профилактике инфекционного расстройства.
| US 6329391 B1, 11.12.2001 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| EP 0641793 A1, 14.04.1995 | |||
| ПРОИЗВОДНЫЕ 5-АМИНО-8-МЕТИЛ-7-ПИРРОЛИДИНИЛХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1994 |
|
RU2130932C1 |

