Настоящее изобретение относится к миметикам пептидов, а именно к фармацевтически приемлемым солям (S)-N-[4-(1-адамантил)бензоил]-α-аминокислот, обладающим противовоспалительными, противоболевыми, противопаркинсоническими, антигипоксическими, актопротекторными и иммуномодулирующими свойствами, маловыраженным ульцерогенным действием, низкой токсичностью и не вызывающих лекарственной зависимости, а также к их способу получения.
В настоящее время миметики пептидов достаточно хорошо известны в фармакологии, благодаря чему используются в клинической практике в качестве диагностических, профилактических и терапевтических средств. Несмотря на это продолжаются исследования в области поиска миметиков, обладающих новыми свойствами при низкой токсичности, низких побочных эффектах и являющихся легко доступными с точки зрения синтеза.
Миметик пептида - это химически синтезированное соединение, которое является «функциональным эквивалентом» по отношению к пептиду и имитирует его структуру, но обладает измененными свойствами. Одним из подходов к созданию структуры миметика является введение в известный пептид или аминокислоту какого-либо липофильного фрагмента, в результате чего увеличивается способность соединения проникать через клеточные мембраны и накапливаться в определенных тканях и органах.
Одним из таких липофильных фрагментов является адамантановый фрагмент, который имеет жесткую, пространственно определенную структуру и обладает способностью эффективно адсорбироваться на клеточных мембранах. Однако на различных примерах было показано, что в результате введения в молекулу адамантана измененяется не только ее липофильность. При этом также наблюдается пролонгирование фармакологических эффектов соединений [1], повышается их устойчивость к метаболическим превращениям в организме и меняется характер взаимодействия соединений с рецепторами мембран [2-4]. Таким образом, введение адамантанового фрагмента в молекулу может приводить к появлению новых видов биологической активности.
Данный подход к синтезу миметиков был применен и описан на примере эндогенного пептида энкефалина и его аналогов, обладающих противоболевой активностью [5]. В структуру энкефалина, содержащего пять аминокислотных остатков, и его аналогов вводили фрагмент адамантана, в результате чего получали миметики с улучшенной проницаемостью через гемато-энцефалический барьер и повышенной устойчивостью к метаболическим превращениям. Основной недостаток указанных соединений заключается в том, что по механизму действия они являются агонистами-антагонистами опиатных рецепторов, что обуславливает наличие выраженных побочных эффектов, одними из которых могут быть лекарственная зависимость и развитие депрессивного состояния. К недостаткам можно также отнести сложность строения миметиков энкефалина.
Кроме этого, известны N-адамантилзамещенные амиды тетрапептидов и их соли, обладающие противоболевой активностью [6]. Данные соединения, как и в приведенном выше примере, являются аналогами энкефалина и, следовательно, могут иметь аналогичные побочные эффекты.
Задачей настоящего изобретения является получение новых эффективных миметиков пептидов, обладающих высокой способностью проникать через клеточные мембраны и низкой токсичностью, а также не проявляющих побочных эффектов и не вызывающих лекарственной зависимости.
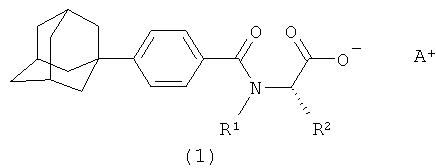
В настоящем изобретении поставленная задача была решена с помощью новых соединений, описываемых общей формулой (1)
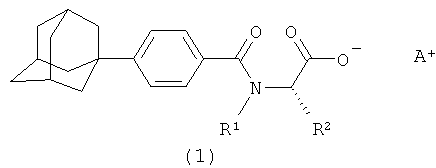
где А+ представляет собой ион щелочного металла, аммония и N+(C1-4алкил)4;
R2 при условии, что R1 представляет собой атом водорода, представляет собой атом водорода, радикалы -СН3, -СН(СН3)2, -CH2CH(СН3)2, -СН(СН3)СН2СН3, 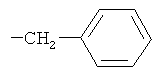 ,
, 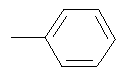 , -CH2-CH2-SH, -CH2-CH2-S-CH3,
, -CH2-CH2-SH, -CH2-CH2-S-CH3, 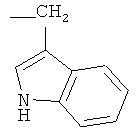 ,
, 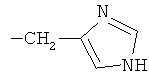 ;
;
R1 и R2 вместе представляют собой бирадикал -СН2-СН2-СН2-, замкнутый в пирролидиновый цикл.
Способ получения соединений формулы (1) в литературе не описан.
В соответствии с настоящим изобретением способ получения соединений по п.1 заключается в превращении 4-(1-адамантил)бензойной кислоты в соответствующий хлорангидрид, дальнейшей реакции ацилирования (S)-α-аминокислоты в присутствии растворителя, представляющего собой смесь воды и 1,4-диоксана, выделении (S)-N-[4-(1-адамантил)бензоил]-α-аминокислоты и ее нейтрализации водным раствором основания в присутствии этилового спирта с образованием соли.
Способ получения соединений формулы (1) осуществляют следующим образом.
4-(1-Адамантил)бензойную кислоту при взаимодействии с избытком хлористого тионила и каталитических количеств N,N-диметилформамида превращают в соответствующий хлорангидрид, после чего избыточный хлористый тионил отгоняют. Хлорангидрид 4-(1-адамантил)бензойной кислоты может быть также получен при взаимодействии с пятихлористым фосфором или другим хлорирующим агентом.
Последующую реакцию ацилирования проводят добавлением раствора хлорангидрида 4-(1-адамантил)бензойной кислоты в 1,4-диоксане к раствору (S)-α-аминокислоты, представляющему собой смесь воды и 1,4-диоксана в объемном соотношении 1:(0,5-5), при рН, равном 8-10 единиц, и при комнатной температуре. При этом соотношение исходной (S)-α-аминокислоты и общего количества растворителя составляет 1:(10-30) по массе. В рассматриваемом процессе на стадии ацилирования (S)-α-аминокислоты образуются продукты, плохо растворимые в воде, что затрудняет течение реакции. Использование смеси растворителей воды и 1,4-диоксана позволяет предотвратить это явление.
Выделение (S)-N-[4-(1-адамантил)бензоил]-α-аминокислоты осуществляют путем добавления к реакционной смеси охлажденной воды и подкислением до рН, равного 3-4 единицы, после чего выпавший осадок отфильтровывают и переосаждают.
На последней стадии растворяют (S)-N-[4-(1-адамантил)бензоил]-α-аминокислоту при комнатной температуре в этиловом спирте в качестве растворителя и нейтрализуют водным раствором основания или соли, после чего отфильтровывают выпавший осадок соединения формулы (1).
Ниже приводятся примеры получения соединений, заявленных в настоящем изобретении. Для подтверждения структуры указанных соединений снимались 1Н-ЯМР спектры в диметилсульфоксиде на приборе Bruker DRX 500 с рабочей частотой 500 МГц.
Пример 1
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]этановая кислота
К 5,0 г (0,02 моль) 4-(1-адамантил)бензойной кислоты добавляют 19,7 мл (0,10 моль) хлористого тионила и 2-3 капли N,N-диметилформамида. Полученную смесь кипятят в течение 3 ч, после чего отгоняют под вакуумом избыток хлористого тионила. Получают 5,1 г хлорангидрида 4-(1-адамантил)бензойной кислоты, который используют без предварительной очистки в реакции ацилирования.
Для проведения реакции ацилирования 1,5 г (0,02 моль) глицина растворяют в 10,0 мл (0,02 моль) 2 н. NaOH и добавляют 5,0 мл 1,4-диоксана. В полученный раствор при перемешивании вносят порциями раствор полученного ранее 5,1 г (0,02 моль) хлорангидрида 4-(1-адамантил)бензойной кислоты в 30,0 мл 1,4-диоксана. По мере протекания реакции для поддержания первоначального значения рН 9-10 в смесь добавляют 4 н. водный раствор NaOH. Время добавления хлорангидрида 1 ч. После этого продолжают перемешивание еще 1 ч при 20°С, а затем реакционную смесь разбавляют в 2 раза водой и подкисляют HCl до рН, равного 3. Выпавший осадок отфильтровывают, промывают водой до нейтрального значения рН. Очистка проводится переосаждением через натриевую соль, после чего продукт высушивают и получают 4,9 г (85%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]этановой кислоты с т.пл. 245-246°С. 1Н-ЯМР, δ, м.д.: 8,15 (д, 1Н, J=6,4 Гц), 7,80 (д, 2Н, J=7,8 Гц), 7,38 (д, 2Н, J=7,8 Гц), 4,60 (м, 2Н), 2,12 (м, 3Н), 1,93 (м, 6Н), 1,79 (м, 6Н).
4-(1-адамантил)фенилкарбоксамидоацетат натрия (1а)
1,0 г (3,2·10-3 моль) (2S)-[4-(1-адамантил)фенилкарбоксамидо]этановой кислоты растворяют в 10 мл этилового спирта и добавляют 0,8 мл (3,2·10-3 моль) 4 н. NaOH. Выпавшую в осадок соль отфильтровывают и промывают на фильтре этиловым спиртом. Получают 0,96 г (90%) (4-(1-адамантил)фенилкарбоксамидоацетата натрия. Т.пл. 208-210°С. Рассчитано для C19H22NO3Na, %: С 68,12; Н 6,57; N 4,18. Найдено, %: С 68,00; Н 6,41; N 4,10.
Пример 2
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]пропионовая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 0,7 г (7,8·10-3 моль) (S)-аланина, следуя методике, описанной в примере 1, получают 1,9 г (75%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]пропионовой кислоты с т.пл. 203-205°С. 1Н-ЯМР, δ, м.д.: 8,15 (д, 1Н, J=6,4 Гц), 7,80 (д, 2Н, J=7,8 Гц), 7,38 (д, 2Н, J=7,8 Гц), 4,45 (м, 1Н), 2,12 (м, 3Н), 1,93 (м, 6Н), 1,79 (м, 6Н), 1,43 (д, 3Н, J=8,0 Гц).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]пропионат натрия (1б)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]пропионовой кислоты, следуя методике, описанной в примере 1, получают 0,9 г (85%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]пропионата натрия. Т.пл. 195-197°С. Рассчитано для C20H24NO3Na, %: С 68,79; Н 6,87; N 4,01. Найдено, %: С 68,65; Н 6,90; N 4,08.
Пример 3
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-метилбутановая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 0,9 г (7,8·10-3 моль) (S)-валина, следуя методике, описанной в примере 1, получают 2,5 г (90%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-метилбутановой кислоты с т.пл. 215-218°С. 1Н-ЯМР, δ, м.д.: 7,80 (д, 2Н, J=7,8 Гц), 7,74 (м, 1Н), 7,37 (д, 2Н, J=7,8 Гц), 4,40 (дд, 1Н, J=6,7, J=6,4 Гц), 2,22 (м, 1Н), 2,12 (м, 3Н), 1,92 (м, 6Н), 1,78 (м, 6Н), 1,00 (д, 6Н, J=6,0 Гц).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-метилбутират натрия (1в)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-метилбутановой кислоты, следуя методике, описанной в примере 1, получают 0,9 г (82%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-метилбутирата натрия. Т.пл. 169-172°С. Рассчитано для C22H28NO3Na, %: С 70,05; Н 7,42; N 3,71. Найдено, %: С 70,10; Н 7,53; N 3,69.
Пример 4
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-4-метилпентановая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 1,0 г (7,8·10-3 моль) (S)-лейцина, следуя методике, описанной в примере 1, получают 2,3 г (80%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-4-метилпентановой кислоты с т.пл. 113-114°С. 1Н-ЯМР, δ, м.д.: 8,20 (м, 1Н), 7,82 (д, 2Н, J=7,8 Гц), 7,38 (д, 2Н, J=7,8 Гц), 4,50 (м, 1Н), 2,12 (м, 3Н), 1,92 (м, 6Н), 1,78 (м, 6Н), 1,75 (м, 2Н), 1,65 (м, 1Н), 0,95 (д, 6Н, J=9,3 Гц).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-4-метилпентаноат натрия (1 г)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]-4-метилпентановой кислоты, следуя методике, описанной в примере 1, получают 0,8 г (78%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-4-метилпентаноата натрия. Т.пл. 151-153°С. Рассчитано для C23H30NO3Na, %: С 70,61; Н 7,67; N 3,58. Найдено, %: С 70,40; Н 7,73; N 3,51.
Пример 5
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-метилпентановая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 1,0 г (7,8·10-3 моль) (S)-изолейцина, следуя методике, описанной в примере 1, получают 2,2 г (76%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-метилпентановой кислоты с т.пл. 154-155°С. 1Н-ЯМР, δ, м.д.: 8,20 (м, 1Н), 7,82 (д, 2Н, J=7,8 Гц), 7,38 (д, 2Н, J=7,8 Гц), 4,50 (м, 1Н), 2,12 (м, 3Н), 1,92 (м, 6Н), 1,78 (м, 6Н), 1,65 (м, 1Н), 1,55 (м, 2Н), 0,95 (д, 3Н, J=9,0 Гц), 0,85 (м, 3Н).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-метилпентаноат натрия (1д)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-метилпентановой кислоты, следуя методике, описанной в примере 1, получают 0,8 г (78%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-метилпентаноата натрия. Т.пл. 141-142°С. Рассчитано для C23H30NO3Na, %: С 70,61; Н 7,67; N 3,58. Найдено, %: С 70,49; Н 7,75; N 3,52.
Пример 6
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-фенилпропионовая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 1,3 г (7,8·10-3 моль) (S)-β-фенилаланина, следуя методике, описанной в примере 1, получают 2,5 г (80%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-фенилпропионовой кислоты с т.пл. 108-110°С. 1Н-ЯМР, δ, м.д.: 8,12 (м, 1Н), 7,73 (д, 2Н, J=7,8 Гц), 7,36 (д, 2Н, J=7,8 Гц), 7,23 (м, 2Н), 7,28 (м, 2Н), 7,15 (м, 1Н), 4,65 (м, 1Н), 3,20 (дд, 1Н, J=14,8 Гц, J=4,6 Гц), 3,10 (дд, 1Н, J=14,8 Гц, J=10,0 Гц), 2,12 (м, 3Н), 1,93 (м, 6Н), 1,78 (м, 6Н).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-фенилпропионат натрия (1е)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-фенилпропионовой кислоты, следуя методике, описанной в примере 1, получают 0,9 г (86%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-фенилпропионата натрия. Т.пл. 205-207°С. Рассчитано для C26H28NO3Na, %: С 73,43; Н 6,58; N 3,29. Найдено, %: С 73,55; Н 6,63; N 3,31.
Пример 7
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-2-фенилэтановая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 1,2 г (7,8·10-3 моль) (S)-фенилглицина, следуя методике, описанной в примере 1, получают 2,7 г (89%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-2-фенилэтановой кислоты с т.пл. 180-185°С. 1Н-ЯМР, δ, м.д.: 8,40 (м, 1Н), 7,86 (д, 2Н, J=7,8 Гц), 7,49 (м, 2Н), 7,38 (д, 2Н, J=7,8 Гц), 7,33 (м, 2Н), 7,27 (м, 1Н), 5,58 (д, 1Н, J=5,0 Гц), 2,12 (м, 3Н), 1,92 (м, 6Н), 1,78 (м, 6Н).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-2-фенилацетат натрия (1ж)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]-2-фенилэтановой кислоты, следуя методике, описанной в примере 1, получают 0,97 г (92%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-2-фенилацетата натрия. Т.пл. 208-210°С. Рассчитано для C25H26NO3Na, %: С 73,01; Н 6,32; N 3,40. Найдено, %: С 73,11; Н 6,40; N 3,46.
Пример 8
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-4-сульфанилбутановая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 0,9 г (7,8·10-3 моль) (S)-цистеина, следуя методике, описанной в примере 1, получают 2,3 г (83%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-4-сульфанилбутановой кислоты с т.пл. 166-167°С. 1Н-ЯМР, δ, м.д.: 8,20 (м, 1Н), 7,80 (д, 2Н, J=7,8 Гц), 7,40 (д, 2Н, J=7,8 Гц), 4,55 (м, 1Н), 2,56 (м, 2Н), 2,12 (м, 3Н), 2,10 (м, 2Н), 1,92 (м, 6Н), 1,78 (м, 6Н).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-4-сульфанилбутират натрия (1з)
Исходя из 1,0 г (2S)-[4-(1-Адамантил)фенилкарбоксамидо]-4-сульфанилбутановой кислоты, следуя методике, описанной в примере 1, получают 0,9 г (86%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-4-сульфанилбутирата натрия. Т.пл. 201-203°С. Рассчитано для C21H26NO3SNa, %: С 63,81; Н 6,58; N 3,54. Найдено, %: С 63,70; Н 6,61; N 3,46.
Пример 9
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-4-метилсульфанилбутановая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 1,2 г (7,8·10-3 моль) (S)-метионина, следуя методике, описанной в примере 1, получают 2,7 г (88%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-4-метилсульфанилбутановой кислоты с т.пл. 139-140°С. 1Н-ЯМР, δ, м.д.: 8,20 (м, 1H), 7,80 (д, 2Н, J=7,8 Гц), 7,40 (д, 2Н, J=7,8 Гц), 4,55 (м, 1Н), 2,56 (м, 2Н), 2,12 (м, 3Н), 2,10 (м, 2Н), 2,08 (с, 3Н), 1,92 (м, 6Н), 1,78 (м, 6Н).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-4-метилсульфанилбутират натрия (1и)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]-4-метилсульфанилбутановой кислоты, следуя методике, описанной в примере 1, получают 0,95 г (90%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-4-метилсульфанилбутирата натрия. Т.пл. 174-177°С. Рассчитано для C22H28NO3SNa, %: С 64,56; Н 6,84; N 3,42. Найдено, %: С 64,70; Н 6,71; N 3,46.
Пример 10
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-(1H-3-индолил)пропионовая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 1,6 г (7,8·10-3 моль) (S)-триптофана, следуя методике, описанной в примере 1, получают 3,1 г (91%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-(1H-3-индолил)пропионовой кислоты с т.пл. 160-162°С. 1Н-ЯМР, δ, м.д.: 10,60 (с, 1Н), 8,05 (д, 1Н, J=6,4 Гц), 7,74 (д, 2Н, J=7,8 Гц), 7,58 (д, 1Н, J=8,4 Гц), 7,34 (д, 2Н, J=7,8 Гц), 7,30 (д, 1Н, J=8,4 Гц), 7,13 (д, 1Н, J=1,0 Гц), 7,03 (дд, 1Н, J=8,7 Гц, J=8,4 Гц), 6,95 (дд, 1Н, J=8,7 Гц, J=8,4 Гц), 4,72 (м, 1Н), 3,32 (дд, 1Н, J=14,8 Гц, J=4,6 Гц), 3,22 (дд, 1Н, J=14,8 Гц, J=10,0 Гц), 2,12 (м, 3Н), 1,90 (м, 6Н), 1,78 (м, 6Н).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-(1H-3-индолил)пропионат натрия (1к)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-(1H-3-индолил)пропионовой кислоты, следуя методике, описанной в примере 1, получают 1,0 г (95%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-(1H-3-индолил)пропионата натрия. Т.пл. 210-211°С. Рассчитано для C28H29N2O3Na, %: С 72,43; Н 6,25; N 6,03. Найдено, %: С 72,30; Н 6,21; N 6,06.
Пример 11
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-(1H-5-имидазолил)пропионовая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 1,2 г (7,8·10-3 моль) (S)-гистидина, следуя методике, описанной в примере 1, получают 2,7 г (88%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-(1H-5-имидазолил)пропионовой кислоты с т.пл. 277-279°С. 1Н-ЯМР, δ, м.д.: 8,60 (с, 1Н), 7,96 (с, 1Н), 7,88 (д, 1Н, J=6,4 Гц), 7,79 (д, 2Н, J=7,8 Гц), 7,42 (д, 2Н, J=7,8 Гц), 7,00 (с, 1Н), 4,64 (м, 1Н), 3,12 (м, 2Н), 2,12 (м, 3Н), 1,93 (м, 6Н), 1,78 (м, 6Н).
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-(1H-5-имидазолил)пропионат натрия (1л)
Исходя из 1,0 г (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-(1H-5-имидазолил)пропионовой кислоты, следуя методике, описанной в примере 1, получают 0,9 г (86%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-(1H-5-имидазолил)пропионата натрия. Т.пл. 212-214°С. Рассчитано для C23H26N3O3Na, %: С 66,52; Н 6,26; N 10,11. Найдено, %: С 66,69; Н 6,21; N 10,16.
Пример 12
1-[4-(1-Адамантил)бензоил]-(2S)-пирролидинкарбоновая кислота
Исходя из 2,0 г (7,8·10-3 моль) 4-(1-адамантил)бензойной кислоты и 0,9 г (7,8·10-3 моль) (S)-пролина, следуя методике, описанной в примере 1, получают 2,5 г (90%) 1-[4-(1-адамантил)бензоил]-(2S)-пирролидинкарбоновой кислоты с т.пл. 222-224°С. 1Н-ЯМР, δ, м.д.: 12,20 (с, 1Н), 7,45 (д, 2Н, J=7,8 Гц), 7,38 (д, 2Н, J=7,8 Гц), 4,41…4,30 (м, 1Н), 3,60 (м, 2Н), 2,30 (м, 1Н), 2,12 (м, 3Н), 2,0 (м, 2Н), 1,92 (м, 6Н), 1,85 (м, 1Н), 1,78 (м, 6Н).
1-[4-(1-Адамантил)бензоил]-(2S)-пирролидинкарбоксилат натрия (1 м)
Исходя из 1,0 г 1-[4-(1-адамантил)бензоил]-(2S)-пирролидинкарбоновой кислоты, следуя методике, описанной в примере 1, получают 1,05 г (97%) 1-[4-(1-адамантил)бензоил]-(2S)-пирролидинкарбоксилата натрия. Т.пл. 175-178°С. Рассчитано для C22H26NO3Na, %: С 70,42; Н 6,93; N 3,73. Найдено, %: С 70,42; Н 6,91; N 3,76.
Пример 13
4-(1-Адамантил)фенилкарбоксамидоацетат аммония (1н)
1,0 г (3,2·10-3 моль) (2S)-[4-(1-Адамантил)фенилкарбоксамидо]этановой кислоты, полученной согласно примеру 1, растворяют в 10 мл этилового спирта и добавляют 0,26 мл (3,5·10-3 моль) раствора аммиака с массовой долей 25%. Полученный раствор частично упаривают до образования осадка соли, который отфильтровывают и сушат. Получают 0,85 г (80%) (4-(1-адамантил)фенилкарбоксамидоацетата аммония. Т.пл. 149-150°С. Рассчитано для C19H26N2O3, %: С 69,11; Н 7,87; N 8,48. Найдено, %: С 69,00; Н 7,81; N 8,39.
Пример 14
(2S)-[4-(1-Адамантил)фенилкарбоксамидо]-3-метилбутират аммония (1о)
1,0 г (2,8·10-3 моль) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-метилбутановой кислоты, полученной согласно примеру 3, растворяют в 10 мл этилового спирта и добавляют 0,23 мл (3,1·10-3 моль) раствора аммиака с массовой долей 25%. Полученный раствор частично упаривают до образования осадка соли, который отфильтровывают и сушат. Получают 0,86 г (82%) (2S)-[4-(1-адамантил)фенилкарбоксамидо]-3-метилбутирата аммония. Т.пл. 139-141°С. Рассчитано для C22H32N2O3, %: С 70,98; Н 8,60; N 7,52. Найдено, %: С 70,85; Н 8,61; N 7,44.
Пример 15
4-(1-Адамантил)фенилкарбоксамидоацетат тетраэтиламмония (1п)
1,0 г (3,2·10-3 моль) (2S)-[4-(1-адамантил)фенилкарбоксамидо]этановой кислоты, полученной согласно примеру 1, растворяют в 10 мл этилового спирта и добавляют 2,09 мл (3,2·10-3 моль) раствора гидроксида тетраэтиламмония с массовой долей 25%. Полученный раствор частично упаривают до образования осадка соли, который отфильтровывают и сушат. Получают 1,06 г (75%) (4-(1-адамантил)фенилкарбоксамидоацетата тетраэтиламмония. Т.пл. 129-131°С. Рассчитано для C27H42N2O3, %: С 73,32; Н 9,50; N 6,33. Найдено, %: С 73,18; Н 9,61; N 6,39.
Технический результат заключается в получении новых эффективных миметиков пептидов, обладающих высокой способностью проникать через клеточные мембраны и низкой токсичностью, а также не проявляющих побочных эффектов и не вызывающих лекарственной зависимости.
Согласно настоящему изобретению соединения формулы (1) могут примененяться в качестве эффективных противовоспалительных, противоболевых, противопаркинсонических, антигипоксических, актопротекторных и иммуномодулирующих средств. Преимуществом данных соединений является то, что они обладают более широким фармакологическим профилем по сравнению с приведенными аналогами, а также обладают маловыраженным ульцерогенным действием, низкой токсичностью и не вызывают лекарственной зависимости.
Исследование фармакологической активности соединений, заявляемых в настоящем изобретении, проводилось методом биологического тестирования. В работе использовались 500 белых беспородных крыс-самцов весом 180-200 г и 765 белых беспородных мышей-самцов весом 18-20 грамм.
С лабораторными животными работали в соответствии с действующими "Правилами проведения работ с использованием экспериментальных животных" и "Международными рекомендациями по проведению медико-биологических исследований с использованием животных" [7]. Все животные содержались в виварии в одинаковых условиях обитания и кормления (стандартный брикетированный корм). Количество животных в каждой экспериментальной группе в зависимости от модели колебалось в пределах от 10 до 23 особей.
Соединения формулы (1) вводились животным в дозе 20 мг/кг, если это не указано особо. Дозы препаратов сравнения в каждом случае рассчитывались с помощью коэффициента Т.А.Гуськовой, равного 5,9 для крыс и 11,8 для мышей [8]. Животным контрольной группы в эквивалентном объеме вводилась дистиллированная вода (0,5 мл).
Ниже приводятся примеры биологического тестирования соединений формулы (1), иллюстрирующие их фармакологические свойства.
Пример 16
Острая токсичность
Для соединений формулы (1) по методике Тейтнера-Миллера определяли LD50 при тестировании на мышах-самцах [9]. Для этого готовился водный раствор соединения, который затем вводился внутрижелудочно и внутрибрюшинно животному. LD50 рассчитывалась путем оценки смертности животных в течение последующих двух недель. Полученные данные для заявляемых соединений и известного препарата диклофенака натрия, который далее использовался в качестве препарата сравнения при исследовании противовоспалительной и противоболевой активности, приведены в таблице 1.
Результаты токсикологического исследования свидетельствуют о том, что согласно ГОСТу 12.1.007-76 "Вредные вещества. Классификация и общие требования безопасности" [10] соединения формулы (1) относятся к третьему классу опасности, то есть являются малоопасными соединениями, так как их среднесмертельные дозы при внутрижелудочном введении входят в интервал от 151 до 5000 мг/кг. При внутрибрюшинном введении заявляемые соединения обладают более низкой токсичностью в 5-9 раз по сравнению с известным противовоспалительным препаратом диклофенаком натрия.
Пример 17
Противовоспалительная активность
Противовоспалительную активность соединений формулы (1) исследовали на моделях острой экссудативной реакции у мышей и формалинового отека у крыс [11]. В качестве препарата сравнения был выбран диклофенак натрия.
Острую экссудативную реакцию (уксуснокислый перитонит) у мышей вызывали внутрибрюшинным введением 1% раствора уксусной кислоты. Раствор уксусной кислоты вводили из расчета 1 мл на 100 г массы тела животного через час после внутрижелудочного введения исследуемого соединения или препарата сравнения. Через 3 ч животных забивали, вскрывали брюшную полость и собирали экссудат. Выраженность антиэкссудативного эффекта соединений определяли по проценту снижения средней массы экссудата по сравнению с контролем.
Результаты экспериментов по определению антиэкссудативной активности заявляемых соединений на модели уксуснокислого перитонита у мышей свидетельствуют о том, что все они обладали антиэкссудативным действием, выраженность которого составляет от 33% до 70% (таблица 2). Наиболее эффективные соединения 1к и 1в достоверно снижали экссудативную реакцию на 55,0% и 69,6% соответственно, превосходя по эффективности препарат сравнения диклофенак натрия, уменьшавший экссудацию на 45,6%.
Метод формалинового отека у крыс использовался в качестве дополнительного метода оценки противовоспалительной активности. Формалиновый отек моделировался путем субплантарного введения 0,1 мл 2% раствора формалина. Исследуемые соединения вводились внутрижелудочно за час до проведения эксперимента. В таблице 2 представлены данные по угнетению воспалительной реакции, которое оценивали через 3 ч после индукции воспаления по изменению объема лапы и рассчитывали по формуле (1):

где Jк и Jo - объем лапки крысы в контрольной и опытной группах [12-14].
Результаты тестирования на обеих моделях показали наличие у соединений формулы (1) выраженной противовоспалительной активности, сопоставимой или превосходящей активность диклофенака натрия, при более низкой токсичности в 5-9 раз.
Пример 18
Противоболевая активность
Противоболевую активность соединений формулы (1) оценивали по их способности уменьшать число специфических болевых реакций, «корчей» у мышей при внутрибрюшинном введении 0,75% раствора уксусной кислоты (1 мл на 100 г массы тела животного) [9]. Соединения формулы (1) вводились внутрижелудочно за час до проведения эксперимента. Количество «корчей» подсчитывали в течение последующих 15 минут после инъекции раствора уксусной кислоты для каждого животного. Противоболевую активность оценивали по проценту угнетения болевой реакции по сравнению с контрольной группой [12, 15].
Эксперименты по изучению противоболевой активности обнаружили, что исследуемые соединения обладали примерно одинаковым противоболевым действием, уменьшая «корчи» у мышей на 58,6%-72,6% (таблица 3). Их активность сопоставима с активностью препарата сравнения диклофенака натрия, но заявляемые соединения обладают более низкой токсичностью в 5-9 раз.
Анализировалось влияние соединений, заявляемых в настоящем изобретении, на опиатные рецепторы. Для этого на модели уксуснокислых «корчей» оценивалась выраженность их анальгетических свойств на фоне введения животным налоксона - антагониста µ-опиатных рецепторов в дозе 1 мг/кг [16].
На фоне введения налоксона не наблюдалось снижение выраженности анальгетической активности, то есть исследуемые вещества уменьшали среднее количество «корчей» на 54,3%-93,3% по сравнению с интактным контролем (таблица 4). Это свидетельствует об отсутствии активирующего влияния соединений формулы (1) на опиатные рецепторы, а значит они не могут вызывать наркотической зависимости и депрессивных состояний.
Пример 19
Жаропонижающее действие
Жаропонижающее действие соединений формулы (1) исследовали на модели дрожжевой лихорадки у крыс, которую вызывали путем подкожного введения 20% суспензии пекарских дрожжей одновременно с внутрибрюшинным введением исследуемых соединений [9]. Препаратом сравнения был выбран метамизол натрия. Оценку выраженности жаропонижающего действия проводили, сравнивая изменение температуры на фоне введения исследуемых соединений через 1 и 2 ч после их введения с контрольной, интактной группами и группой препарата сравнения. Далее для веществ, обладающих наиболее выраженным жаропонижающим действием, оценивалась их способность снижать температуру на протяжении 6 ч после введения пирогена, и по этим данным строились температурные кривые.
На модели дрожжевой лихорадки у крыс было установлено, что под воздействием пирогена их температура тела достоверно повышалась во всех группах по сравнению с температурой тела интактных животных - на 0,6°С в течение 1 ч и на 1,11°С в 2 ч эксперимента (таблица 5). Препарат сравнения снижал лихорадочную реакцию крыс на 0,88°С в течение 1 ч и на 1,25°С в течение 2 ч после введения суспензии пекарских дрожжей. Среди исследуемых соединений достоверное выраженное уменьшение ректальной температуры по сравнению с контрольной группой на протяжении 2 ч наблюдалось при введении субстанции 1а, которая угнетала лихорадочную реакцию на 1,28°С в течение 1 ч и на 1,71°С в течение 2 ч.
Для соединения 1а, обладающего наиболее выраженной жаропонижающей активностью среди изучаемых веществ, ниже приведена температурная кривая, показывающая его влияние на развитие дрожжевой лихорадки у крыс в течение семи часов после введения суспензии пекарских дрожжей в сравнении с контролем и эталонным препаратом метамизолом натрия (чертеж).
В контрольной группе на фоне введения суспензии пекарских дрожжей ректальная температура крыс поднималась в течение 7 ч. Через 1 ч после введения пирогена она повысилась на 0,72°С, а к 7 ч наблюдения достигла 37,45°С. У животных, которым вводили соединение 1а и метамизол натрия, в течение первых 2 ч после введения суспензии пекарских дрожжей температура тела не превышала измеренной перед введением пирогена температуры (Р<0,05). В течение последующего времени она постепенно поднималась и к концу эксперимента достигла уровня контрольной группы (±0,3°С).
Пример 20
Ульцерогенное действие
Ульцерогенное действие соединений формулы (1) исследовалось на крысах, лишенных пищи за 16 ч до опыта, при однократном внутрижелудочном введении соединений в дозе 0,2 LD50 [9]. Оценку его выраженности проводили через 3 ч после введения опытных субстанций по 4-балльной шкале:
0 - отсутствие повреждений;
0,5 - гиперемия;
1 - единичные незначительные повреждения (1 или 2 точечных кровоизлияния);
2 - множественные повреждения (эрозии, точечные кровоизлияния);
3 - значительные и множественные повреждения слизистой (эрозии, кровоизлияния);
4 - грубые повреждения, охватывающие всю поверхность слизистой (массивные кровоизлияния, эрозии, перфорации).
Препаратами сравнения были выбраны диклофенак натрия и индометацин, которые вводились животным в дозе 0,2 LD50.
При оценке ульцерогенного действия в качестве препаратов сравнения были выбраны индометацин как обладающее сильным ульцерогенным влиянием лекарственное средство (Индекс ингибирования ЦОГ-1/ЦОГ-2=30) и диклофенак натрия, для которого характерно более слабое повреждающее действие (Индекс ингибирования ЦОГ-1/ЦОГ-2=2,2). Результаты исследования показали, что соединения формулы (1) обладают маловыраженным или практически отсутствующим в случае соединений 1и и 1к ульцерогенным действием по сравнению с диклофенаком натрия и индометацином (таблица 6).
Пример 21
Противопаркинсоническая активность
Противопаркинсоническая активность веществ изучалась по двум методикам: галоперидоловой каталепсии у крыс и амфетаминовой стереотипии у мышей [9].
Галоперидоловую каталепсию у крыс вызывали внутрибрюшинным введением галоперидола в дозе 1 мг/кг. В качестве препаратов сравнения, используемых в опытах по изучению противопаркинсонической активности, были выбраны: леводопа, более эффективная в отношении акинето-ригидной формы паркинсонического синдрома, и циклодол, являющийся холинолитиком центрального действия и преимущественно используемый на ранних стадиях заболевания и в комплексной терапии при ригидно-дрожательной форме. Исследуемые соединения и препараты сравнения вводили внутрибрюшинно одновременно с галоперидолом. Для определения продолжительности каталепсии использовался метод оценки каталепсии с использованием параллельных стенок по времени пребывания крысы в неподвижном состоянии при помещении животного на параллельные стенки таким образом, чтобы спина оставалась прямой. Длительность каталептогенного состояния отмечали через 90, 120 и 180 мин. Также оценивался промежуток времени, спустя который после введения галоперидола у крыс наблюдается каталепсия.
Оценка степени ригидности, возникшей на фоне введения галоперидола, производилась с использованием симптома «горбатости», выраженность которого определялась по укорочению расстояния от шеи до хвоста за счет сгорбленности животного и измерялась в сантиметрах через 30, 60 и 90 минут после введения галоперидола.
Среди исследуемых соединений сопоставимой с леводопой антикаталептогенной активностью обладали вещества 1и, 1к, 1в и 1м. Наиболее выраженное противопаркинсоническое действие проявляли соединения 1к и 1в, которые с 60 по 180 мин наблюдения достоверно снижали продолжительность каталептогенного состояния животных по сравнению с контрольной группой (таблица 7).
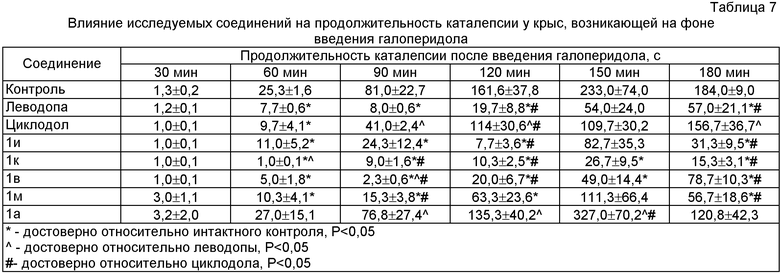
Было обнаружено, что исследуемые соединения и препараты сравнения обладали способностью влиять на время, спустя которое после введения галоперидола начиналась каталепсия (таблица 8). Изучаемые соединения 1и, 1к, 1м и 1а достоверно отдаляли начало проявления данного симптома в 1,3-2,7 раза.
Нейротоксическое действие галоперидола проявлялось не только наличием каталепсии, но и развитием у животных мышечной ригидности, о которой судили по выраженности симптома «горбатости». Соединения 1и, 1к и 1м снижали степень его проявления, при этом наибольшую эффективность, превосходящую эффективность препаратов сравнения, обнаружило вещество 1м (таблица 9).
Стереотипное поведение животных моделировалось по методике потенцирования эффектов амфетамина. Стереотипию у мышей вызывали подкожным введением амфетамина в дозе 5 мг/кг. Исследуемые вещества и препараты сравнения - леводопу и циклодол - вводили за 30 мин до введения амфетамина внутрибрюшинно. Интенсивность и выраженность стереотипии оценивали каждые 5 мин в течение 15 мин через 15 мин после введения амфетамина в баллах:
- 1 балл - отдельные стереотипные движения (например, непостоянные принюхивания);
- 2 балла - интенсивная непродолжительная стереотипия (в том числе лизание, грызение);
- 3 балла - постоянная интенсивная стереотипия.
В качестве модели, позволяющей оценить влияние соединений на дрожательную форму паркинсонического синдрома, использовалась методика потенцирования эффектов амфетамина. Из снижающих выраженность стереотипии веществ 1и, 1к, 1в и 1м максимальную активность проявляли соединения 1и и 1в, чья эффективность была сопоставима с эффективностью препарата сравнения циклодола (таблица 10).
Результаты исследований по обеим методикам показали наличие противопаркинсонической активности у соединений формулы (1).
Пример 22
Антйгипоксическая и актопротекторная активность
Антигипоксические свойства соединений исследовали на модели нормобарической нормокапнической гипоксической гипоксии помещением крыс в гермокамеру объемом 1,5 л [17; 18]. Сопротивляемость организма к действию предельных мышечных нагрузок (актопротекторная активность) исследовали на модели принудительного плавания крыс с грузом, равным 10% от массы тела до полного утомления [19]. Изучаемые соединения вводились однократно внутрибрюшинно за 1 ч до проведения экспериментов.
По антигипоксической активности наиболее эффективным было соединение 1а, которое повышало продолжительность жизни крыс в условиях гипоксии на 26,0% (таблица 11). При введении субстанций 1к, 1м и 1а наблюдалось увеличение продолжительности переносимости мышечных нагрузок в 1,4, 1,8 и 3,0 раза соответственно.

Таким образом, наиболее эффективными актопротекторными средствами являются соединения 1к, 1м и 1а. Соединение 1а проявляет антигипоксическую активность.
Пример 23
Иммунологическая активность
Соединения формулы (1) тестировались на способность влиять на фагоцитарную активность нейтрофилов in vitro и in vivo как на компонент неспецифического клеточного иммунитета и на активность лизоцима in vitro и in vivo как на компонент неспецифического гуморального иммунитета. Для экспериментов in vivo все изучаемые субстанции вводились мышам в виде водных растворов внутрижелудочно ежедневно в течение двух недель.
Оценку фагоцитарной активности нейтрофилов in vitro и in vivo проводили микрометодом [20]. Учет результатов осуществлялся микроскопически. В мазке при увеличении 90 с иммерсионной системой подсчитывали процент фагоцитирующих нейтрофилов - процент фагоцитоза и среднее количество поглощенных бактерий в каждом из ста нейтрофилов пробы - фагоцитарный индекс.
Исследование влияния изучаемых соединений на ферментативную (антибактериальную) активность лизоцима in vitro и in vivo проводилось нефелометрическим методом определения активности лизоцима по В.Г.Дорофейчук [21; 22].
Исследование соединений in vitro показало, что субстанции 1и, 1к и 1в вызывали повышение интенсивности фагоцитоза, достоверно увеличивая в течение первого часа опыта фагоцитарный индекс в 2,7-4,0 раза (таблица 12). К двадцать четвертому часу наблюдения фагоцитарный индекс на фоне введения изучаемых соединений 1к, 1в и 1м также был достоверно выше в 1,5-2,2 раза, чем значение данного параметра в контрольной группе.
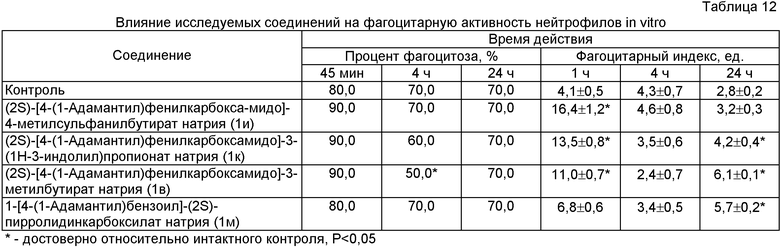
При исследовании влияния опытных соединений на фагоцитарную активность in vivo было обнаружено, что вещество 1к достоверно увеличивало процент фагоцитоза на 23,3% (таблица 13).
Стимулирующее действие изучаемых веществ на функциональную активность лизоцима in vitro было наиболее выражено на начальных этапах эксперимента. Достоверное усиление лизоцимной активности наблюдалось при введении соединений 1в и 1м, которые увеличивали ее на 8,3% и 56,0% соответственно (таблица 14). При исследовании влияния изучаемых субстанций на данный параметр неспецифического иммунитета in vivo только субстанция 1к, не проявившая таковой активности в эксперименте in vitro, увеличивала активность лизоцима в 2,2 раза.
Результаты исследований показывают, что исследуемые соединения способствуют усилению клеточного и гуморального иммунитета.
Таким образом, весь комплекс использованных экспериментальных моделей свидетельствует о значительной противовоспалительной, противоболевой, противопаркинсонической, антигипоксической, актопротекторной и иммуномодулирующей активности соединений общей формулы (1), обладающих также маловыраженным ульцерогенным действием, низкой токсичностью в отличие от использованных препаратов сравнения и не вызывающих лекарственной зависимости.
Список использованной литературы
1. Морозов И.С. Фармакология адамантанов. / И.С.Морозов, В.И.Петров, С.А.Сергеева. - Волгоград: Волгоградская медицинская академия, 2001. - 320 с.
2. Бис-1-(N-адамантил)пиперазины алифатических дикарбоновых кислот и их фармакологическая активность. / Л.И.Дуракова [и др.]. // Хим.-фармац. журн. - 1980. - Т.14, №5. - С.26-30; 53.
3. Литвинов В.П. Гетериладамантаны: синтетические исследования последних лет, биологическая активность и другие аспекты практического использования (обзор). // Химия гетероцикл. соединений. - 2002. - №1. - С.12-39; 74.
4. О курареподобной активности четвертичных аммониевых соединений, содержащих 2-адамантильные радикалы. / Д.А.Харкевич [и др.]. // Фармакология и токсикология. - 1981. - №6. - С.670.
5. Pat. 5652335, US, C07C 103/52. Physiologically active substance well capable of permeating biomembrane / Kouki, K. [и др.]; Teikoku Seiyaku Kabushiki Kaisha (Japan). - №227997, заявл. 15.04.1994; опубл. 29.07.1997, Chem. Abstr. 117 (05), 049260h.
6. Pat. 4273704, US, C07C 103/52. N-Adamantane-substituted tetrapeptide amides / Mazur R.H.; Searle & Co (US). - №99765, заявл. 03.12.1979; опубл. 16.06.1981, Chem. Abstr. 096 (09), 069439z.
7. Международные рекомендации по проведению медико-биологических исследований с использованием животных. // Хроника ВОЗ. - 1985. - Т.39, №3. - С.3-9.
8. Гуськова Т.А. Оценка безопасности лекарственных средств на стадии доклинического изучения. / Т.А.Гуськова. // Хим.-фармац. журн. - 1990. - Т.24, №7. - С.10-15.
9. Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ. / В.П.Фисенко [и др.]. - М.: Бионт, 2000. - 400 с.
10. ГОСТ 12.1.007-76 Система стандартов безопасности труда. Вредные вещества. Классификация и общие требования безопасности. - М.: ИПК Изд-во стандартов, 1999. - 7 с.
11. Колла В.Э. Дозы лекарственных средств и химических соединений для лабораторных животных. / В.Э.Колла, Б.Я.Сыропятов. - М.: «Медицина», 1998. - 264 с.
12. Мусил Я. Основы биохимии патологических процессов. / Я.Мусил: пер. с англ. - М.: Медицина, 1985 - 432 с.
13. Наточкин Ю.В. Основы физиологии почек. / Ю.В.Наточкин. // В кн.: Диурез и диуретики. - Л.: Медицина, 1982. - С.116-120.
14. Тиклеева У.М. Экспериментальное исследование противогипоксической и антиоксидантной активности гетероциклических производных 3-оксипиридина. / У.М.Тиклеева, Т.А.Воронин, В.И.Кузьмин. // Фармакология и токсикология. - 1989. - №2, - С.79-83.
15. Koster, R. Acetic acid for analgetic screening / R.Koster, M.Anderson, E.J.De Beer // Fed. Proc. - 1959. - Vol.18, №1. - Р.412.
16. Женило В.М. Нейромедиаторные механизмы анальгетического действия некоторых анестетиков. / В.М.Женило, М.В.Женило. // Съезд фармакологов России (3; 2007; Санкт-Петербург): материалы… - СПб., 2007. - С.1-1692-1-1693.
17. Бобков Ю.Г. Методологические подходы к поиску фармакологических средств, эффективных при гипоксии и ишемии мозга. / Ю.Г.Бобков, И.А.Иванова. // Патол. физиология и эксперим. терапия. - 1987. - №6. - С.13-19.
18. Бобков Ю.Г. Методологические подходы к поиску фармакологических средств, эффективных при гипоксии и ишемии мозга. / Ю.Г.Бобков, И.А.Иванова. // Патол. физиология и эксперим. терапия. - 1987. - №6. - С.13-19.
19. Русин В.Я. Сопротивляемость организма к неблагоприятным воздействиям при нарушении функции некоторых желез внутренней секреции. / В.Я.Русин, С.С.Полтырев. // В сб.: Адапт. человека и животных в норме и патологии. - Ярославль, 1976. - С.3-13.
20. Иммунологические аспекты аллотрансплантации. / Ю.М.Зарецкая [и др.]. - M.: Медицина, 1974. - 236 с.
21. Баранов А.А. Лизоцим: теория и практика. / А.А.Баранов, В.Г.Дорофейчук. - М.: Нижний Новгород: Информсвязьиздат, 1999. - 126 с.
22. Дорофейчук В.Г. Определение активности лизоцима нефелометрическим методом. / В.Г.Дорофейчук. // Лаборатор. дело. - 1968. - №1. - С.28-30.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных тиазола или их аддитивных солей с кислотами | 1986 |
|
SU1597102A3 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РВОТЫ | 1999 |
|
RU2258068C2 |
| ПРОИЗВОДНЫЕ 2-ПИПЕРИДИНО-1-АЛКАНОЛА | 1991 |
|
RU2029769C1 |
| Способ получения производных 4(3Н)-оксо-5,6,7,8-тетрагидропиридо(2,3- @ )пиримидина или их таутомерных форм | 1987 |
|
SU1581222A3 |
| ПРОИЗВОДНЫЕ БЕНЗОКСАЗОЛОНА, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2024512C1 |
| 11b-(ГEТ)АРИЛ-2,3,6,11b-ТЕТРАГИДРООКСАЗОЛО[2',3':2,1]ПИРРОЛО[3,4-b]ХИНОЛИН-5,11-ДИОНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2381229C2 |
| Способ получения 4,5-диарил-2-нитроимидазолов | 1979 |
|
SU940647A3 |
| ПРОИЗВОДНЫЕ АРОИЛПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2258702C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |
| 2-О-Сульфонаты метил 5-О-бензил-3-фтор-3-дезокси-Д-арабинозы в качестве промежуточных продуктов в синтезе биологически активных 3 @ -фтор-3 @ -дезоксирибонуклеозидов | 1987 |
|
SU1521739A1 |

Изобретение относится к новым соединениям - фармацевтически приемлемым солям (S)-N-[4-(1-адамантил)бензоил]-α-аминокислот общей формулы (1), где А+ представляет собой ион щелочного металла, аммония и N+(C1-4алкил)4; R2 при условии, что R1 представляет собой атом водорода, представляет собой атом водорода, радикалы -СН3, -СН(СН3)2, -СН2СН(СН3)2, -СН(СН3)СН2СН3, CH2-CH2-SH, -СН2-СН2-S-СН3,
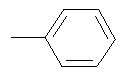 ,
,  ,
, 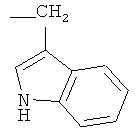 ,
, 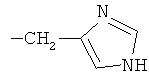 ;
;
R1 и R2 вместе представляют собой бирадикал -СН2-СН2-СН2-, замкнутый в пирролидиновый цикл, а также к способу их получения. 2 н.п. ф-лы, 14 табл., 1 ил.
1. Фармацевтически приемлемые соли (S)-N-[4-(1-адамантил)бензоил]-α-аминокислот общей формулы (I):
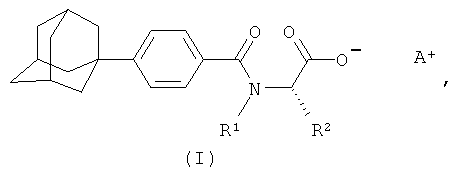
где A+ представляет собой ион щелочного металла, аммония и N+(C1-C4алкил)4;
R2 при условии, что R1 представляет собой атом водорода, представляет собой атом водорода, радикалы -СН3, -СН(СН3)2, -CH2CH(СН3)2, -СН(СН3)СН2СН3, -CH2-CH2-SH, -СН2-СН2-S-СН3,
, , , ;
R1 и R2 вместе представляют собой бирадикал -СН2-СН2-СН2-, замкнутый в пирролидиновый цикл.
2. Способ получения соединений по п.1, заключающийся в превращении 4-(1-адамантил)бензойной кислоты в соответствующий хлорангидрид, дальнейшей реакции ацилирования (S)-α-аминокислоты в присутствии растворителя, представляющего собой смесь воды и 1,4-диоксана, выделении (S)-N-[4-(1-адамантил)бензоил]-α-аминокислоты и ее нейтрализации водным раствором основания в присутствии этилового спирта с образованием соли.
| СПОСОБ ПОЛУЧЕНИЯ АДАМАНТИЛФЕНИЛКАРБОНОВЫХ КИСЛОТ | 2000 |
|
RU2183620C2 |
| КРАСНИКОВ С.В | |||
| «Синтез», Известия высших учебных заведений | |||
| Химия и химическая технология, 47(6), с.110-112 2004 | |||
| КРАСНИКОВ С.В | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Химия и химическая технология, 50(4), с.72-75, 2007 | |||
| МЕЛЬНИКОВА Н.Б. | |||

