Область техники
Настоящее изобретение относится к способу конструирования рекомбинантной бактерии, принадлежащей к роду Pantoea, и к способу получения L-аминокислот с использованием этой бактерии.
Описание предшествующего уровня техники
Обычно увеличение активности генных продуктов, участвующих в биосинтезе L-аминокислот или нуклеиновых кислот, является общепринятым способом, используемым для увеличения продукции L-аминокислот или нуклеиновых кислот. Это увеличение часто достигалось путем создания мутантных штаммов, устойчивых к целевым веществам или их аналогам, путем усиления экспрессии генов биосинтеза, путем устранения чувствительности ферментов биосинтеза к ингибированию связи конечными продуктами или промежуточными продуктами биосинтеза по типу обратной, и путем создания бактериальных штаммов, дефицитных по генам, использующим предшественников целевого вещества для других путей, или путем создания бактериальных штаммов, дефицитных по генам, ответственным за деградацию целевого вещества.
Эти манипуляции обычно приводят к получению штаммов, которые неспособны расти, растут только со значительно сниженной скоростью или нуждаются в дополнительных питательных веществах, таких как аминокислоты, для своего роста. Например, усиление экспрессии некоторых генов может стать чрезмерным и может привести к значительному ингибированию роста бактерии и как результат к снижению способности бактерии к продукции целевого вещества.
Одним из подходов, позволяющих избегать вышеописанные трудности, является оптимизация экспрессии генов, которые кодируют белки, участвующие в распределении потоков углерода, азота или фосфора или в выведении целевого продукта из бактериальной клетки.
Было описано получение библиотеки синтетических промоторов различной силы для Lactococcus lactis (Jensen P.R., and Hammer K., Appl. Environ. Microbiol., 1998, 64, No.1. 82-87 Biotechnol. Bioeng., 1998, 58, 2-3, 191-5). Библиотека состоит из 38 химически синтезированных промоторных фрагментов ДНК, которые обладают рандомизированными спейсерами между консенсусными последовательностями в позициях с -35-го по -15-тый нуклеотиды. Для оценки силы полученных промоторов, олигонуклеотиды из библиотеки были клонированы в экспрессирующий вектор рАК80, который содержит β-галактозидазу. Было установлено, что большинство искусственных промоторов очень слабые (менее 500) и только три из них имели силу около 2000 относительных единиц. Однако практическое применение для этой библиотеки синтетических промоторов не было раскрыто.
Способ получения коринеформных бактерий, обладающих улучшенной продуктивностью аминокислот или нуклеиновых кислот, путем введения мутации в последовательность промотора генов биосинтеза аминокислот или нуклеиновых кислот раскрыт в Европейской патентной заявке ЕР 1033407 А1. Для каждого из следующих генов использовали до 8 различных вариантов мутантных промоторов: ген глутаматдегидрогеназы (gdh), ген цитратсинтазы (gltA), ген изоцитратдегидрогеназы (icd), ген пируватдегидрогеназы (pdhA) и ген аргининсукцинатсинтазы (argG). Несостоятельность этой техники, описанная в Европейской патентной заявке ЕР 1033407 А1, заключается в том, что каждый из описанных мутантных штаммов приготавливался отдельно, то есть один за другим. Также увеличение продуктивности L-аминокислоты во всех случаях, раскрытых в этой Европейской патентной заявке, достигался путем увеличения активности определенных ферментов, использующих ограниченное количество различных промоторов для генов, кодирующих эти ферменты. Этот подход является обычным для уровня техники приготовления бактерий-продуцентов аминокислот и бактерий-продуцентов нуклеиновых кислот и не направлен на оптимизацию или тонкую настройку промоторной активности генов, необходимых для продукции L-аминокислот или нуклеиновых кислот.
Способ создания библиотеки искусственных промоторов был раскрыт в заявке PCT WO 03089605. Этот способ включает: а) получение ДНК-кассеты для вставки, которая включает первый сайт рекомбиназы, второй сайт рекомбиназы и селективный маркерный ген, расположенный между этими сайтами; в) получение первого олигонуклеотида, который включает: i) первый фрагмент нуклеиновой кислоты, который гомологичен области, предшествующей по направлению транскрипции интересующему гену на хромосоме (upstream-область), и ii) второй фрагмент нуклеиновой кислоты, который гомологичен 5'-концу вставочной ДНК- кассеты; с) получение второго олигонуклеотида, который включает: i) третий фрагмент нуклеиновой кислоты, гомологичный 3'-концу указанной вставочной ДНК- кассеты, ii) предшественник промотора, который включает консенсусную область из 35-ти нуклеотидов (от -35-ти до -30-ти нуклеотидов), линкерную последовательность, и консенсусную область из 10-ти нуклеотидов (от-12-ти до -7-и нуклеотидов), причем линкерная последовательность включает от 14-х до 20-ти нуклеотидов и фланкируется областью из 35-ти и областью из 10-ти нуклеотидов, причем указанный предшественник промотора модифицирован таким образом, что включает по крайней мере одну измененную нуклеотидную позицию промоторного предшественника, при этом область из 35-ти и 10-ти нуклеотидов каждая включает от 4-х до 6-ти консервативных нуклеотидов промотора, и iii) четвертый фрагмент нуклеиновой кислоты, который гомологичен области, расположенной дальше по отношению к точке инициации транскрипции промотора (downstream-область); и d) смешивания первого и второго олигонуклеотидов в реакции амплификации со вставочной ДНК-кассетой для получения библиотеки двухцепочечных амплифицированных продуктов, содержащих искусственные промоторы.
В качестве способа встраивания фрагментов ДНК в хромосому бактерий семейства Enterobacteriaceae уже известны температуро-чувствительная плазмида или метод Mu-интеграции (Microbiol Res. 2006 Mar 29, J Bacteriol. 1981 Jan; 145(1):358-68).
Однако в настоящее время нет сообщений, описывающих использование Red-зависимой интеграции для встраивания целевых фрагментов ДНК в бактерии, принадлежащие к роду Pantoea. Также в настоящее время нет сообщений, описывающих оптимизацию экспрессии гена для продукции полезного метаболита, например, для продукции L-аминокислоты, путем ферментации бактерии, принадлежащей к роду Pantoea, которая модифицирована таким образом, что в ней оптимизирована экспрессия целевого гена.
Описание изобретения
Целью настоящего изобретения является предоставление способа конструирования рекомбинантной бактерии, включающего:
a) получение экзогенного фрагмента ДНК, который имеет в своем составе по крайней мере 2 сайта, как минимум по 30 п.о. в длину каждый, идентичных сайтам, расположенным в целевом гене на хромосоме бактерии, принадлежащей к роду Pantoea,
b) введение указанного линейного фрагмента ДНК в бактерию, принадлежащую к роду Pantoea, причем указанная бактерия модифицирована таким образом, что является устойчивой к продуктам генов gam, bet и exo из фага лямбда и
c) отбор рекомбинантной бактерии, в которой указанный целевой ген заменен на указанный линейный фрагмент ДНК или линейный фрагмент ДНК встроен в целевой ген.
Также целью настоящего изобретения является предоставление способа, описанного выше, при этом указанная бактерия является бактерией Pantoea ananatis SC17(0).
Также целью настоящего изобретения является предоставление способа конструирования библиотеки фрагментов ДНК в хромосоме бактерии, включающего:
a) введение набора фрагментов ДНК, которые частично идентичны целевому гену на хромосоме бактерии, принадлежащей к роду Pantoea, в указанную бактерию, причем указанная бактерия модифицирована таким образом, что является устойчивой к продуктам генов gam, bet и exo фага лямбда и
b) отбор бактерии, в которой указанный целевой ген заменен на указанные фрагменты ДНК.
Также целью настоящего изобретения является предоставление способа, описанного выше, причем указанные фрагменты ДНК содержат регуляторный элемент для экспрессии гена, выбранный из группы, состоящей их промотора, терминатора, сайта связывания рибосомы (RBS), оператора и их сочетания.
Также целью настоящего изобретения является предоставление способа, описанного выше, причем указанный регуляторный элемент содержит рандомизированную последовательность.
Также целью настоящего изобретения является предоставление способа конструирования бактерии, принадлежащей к роду Pantoea, которая обладает оптимизированным уровнем экспрессии гена, кодирующего белок, участвующий в распределении потоков углерода, азота или фосфора или в выведении целевого вещества из указанной бактерии, включающий:
1) замену природных регуляторных элементов на библиотеку фрагментов ДНК, полученную способом, описанным выше, в хромосоме указанной бактерии и
2) отбор бактерии с указанным оптимизированным уровнем экспрессии указанного гена.
Также целью настоящего изобретения является предоставление способа, описанного выше, причем указанная бактерия является бактерией-продуцентом L-аминокислоты.
Также целью настоящего изобретения является предоставление способа, описанного выше, причем указанная L-аминокислота выбрана из группы, состоящей из L-аланина, L-аргинина, L-аспарагина, L-аспарагиновой кислоты, L-цистеина, L-глутаминовой кислоты, L-глутамина, L-глицина, L-гистидина, L-изолейцина, L-лейцина, L-лизина, L-метионина, L-фенилаланина, L-пролина, L-серина, L-треонина, L-триптофана, L-тирозина и L-валина.
Также целью настоящего изобретения является предоставление способа, описанного выше, причем указанная бактерия является бактерией-продуцентом нуклеиновой кислоты.
Также целью настоящего изобретения является предоставление способа, описанного выше, причем указанная нуклеиновая кислота выбрана из группы, состоящей из инозина, гуанозина, ксантозина и аденозина.
Также целью настоящего изобретения является предоставление бактерии, принадлежащей к роду Pantoea и полученной способом, описанным выше.
Также целью настоящего изобретения является предоставление бактерии, описанной выше, причем оптимизированный уровень экспрессии гена достигается путем модификации области промотора указанного гена, состоящей из 35-ти пар оснований.
Также целью настоящего изобретения является предоставление бактерии, описанной выше, причем указанным геном является ген yhfK.
Также целью настоящего изобретения является предоставление бактерии, описанной выше, причем область из 35-ти пар оснований выбрана из группы, состоящей из SEQ ID NO: 16, SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 23, SEQ ID NO: 24, SEQ ID NO: 25, SEQ ID NO: 26, SEQ ID NO: 27, SEQ ID NO: 28 и их сочетания.
Также целью настоящего изобретения является предоставление способа получения L-аминокислоты, включающего:
a) выращивание описанной выше бактерии в питательной среде, и
b) выделение L-аминокислоты из культуральной жидкости. Также целью настоящего изобретения является предоставление способа, описанного выше, причем указанная L-аминокислота выбрана из группы, состоящей из L-аланина, L-аргинина, L-аспарагина, L-аспарагиновой кислоты, L-цистеина, L-глутаминовой кислоты, L-глутамина, L-глицина, L-гистидина, L-изолейцина, L-лейцина, L-лизина, L-метионина, L-фенилаланина, L-пролина, L-серина, L-треонина, L-триптофана, L-тирозина и L-валина.
Подробное описание наилучшего способа осуществления изобретения
1. Способ согласно настоящему изобретению
Настоящее изобретение предоставляет способ конструирования рекомбинантной бактерии, принадлежащей к роду Pantoea. Конструирование такой рекомбинантной бактерии обычно достигается путем использования бактерии, которая модифицирована таким образом, что обладает устойчивостью к продуктам генов gam, bet и ехо из фага лямбда. Именно в процессе этого конструирования было установлено, что продукты генов gam, bet и ехо фага лямбда системы Red-зависимой интеграции являются токсичными для бактерии рода Pantoea. Эта проблема была решена путем выделения мутантного штамма, который устойчив к продуктам генов gam, bet и exo фага лямбда. Этот полученный мутантный штамм использовали в качестве реципиентного штамма для интеграции фрагментов ДНК с использованием системы Red-интеграции (см. ниже Пример 1).
Настоящее изобретение также предоставляет простой и прямой одноступенчатый способ получения бактерии-продуцента L-аминокислоты или бактерии-продуцента нуклеиновой кислоты, которая обладает оптимизированным уровнем экспрессии гена, участвующего в метаболизме и биосинтезе целевого вещества или в выведении целевого вещества из указанной бактерии, и, соответственно, влияет на продукцию L-аминокислоты и нуклеиновой кислоты.
Настоящее изобретение также предоставляет бактерию-продуцент L-аминокислоты с оптимизированным уровнем экспрессии гена, который участвует в метаболизме и биосинтезе целевого вещества или в процессе выведения целевого вещества из указанной бактерии, а также предоставляет способ инактивации целевого гена на хромосоме, который не включен в открытые рамки считывания (ORF), генов или аллелей, которые необходимы для роста бактерий и продукции L-аминокислоты, продукции нуклеиновой кислоты.
Настоящее изобретение также предоставляет способ получения L-аминокислот, таких как L-аланин, L-аргинин, L-аспарагин, L-аспарагиновая кислота, L-цистеин, L-глутаминовая кислота, L-глутамин, L-глицин, L-гистидин, L-изолейцин, L-лейцин, L-лизин, L-метионин, L-фенилаланин, L-пролин, L-серин, L-треонин, L-триптофан, L-тирозин и L-валин; и способ получения нуклеиновой кислоты.
Настоящее изобретение было осуществлено благодаря использованию штамма, который был модифицирован таким образом, что является устойчивым к продукту генов gam, bet и ехо из фага лямбда (здесь и далее обозначаемые как "λ-Red-гены") в качестве штамма-реципиента фрагментов ДНК.
Штамм, который модифицирован таким образом, что является устойчивым к продукту λ-Red-генов (здесь и далее обозначаемый как λ-Red-устойчивый штамм), может быть получен следующим способом. Для получения λ-Red-устойчивого штамма, клетки штамма Р. ananatis SC17 (патент США 6596517) были электропорированы плазмидой, несущей λ-Red-гены. После культивирования в течение 18-ти часов было получено приблизительно 105-106 трансформантов, среди которых около 10-ти клонов имели большой размер, тогда как другие клоны были очень маленькими. После 18-ти часов культивирования большие клоны были приблизительно 2 мм в диаметре, маленькие колонии были приблизительно 0,2 мм в диаметре. В течение пролонгированного культивирования (до 24 часов), маленькие колонии больше не росли, тогда как большие продолжали расти. Штаммы P. ananatis с таким большим размером колонии являются устойчивыми к экспрессии всех трех Red-генов из фага лямбда (gam, bet и ехо). Предпочтительным для настоящего изобретения λ-Red-устойчивым штаммом является штамм Pantoea ananatis SC17(0) (VKPM B-9246).
λ-Red-устойчивый штамм может быть использован во многих разных методиках для генетической рекомбинации целевого гена, например, для инактивации гена, для интеграции гена.
Для получения рекомбинантного штамма могут быть выполнены следующие стадии:
a) приготовление экзогенного линейного фрагмента ДНК, содержащего по крайней мере 2 сайта длиной минимум по 30 п.о. каждый, идентичных сайтам, расположенным в целевом гене на хромосоме бактерии, принадлежащей к роду Pantoea,
b) введение указанного линейного фрагмента ДНК в бактерию, принадлежащую к роду Pantoea, причем указанная бактерия модифицирована таким образом, что является устойчивой к продуктам генов gam, bet и ехо из фага лямбда, и
c) отбор рекомбинантной бактерии, в которой указанный целевой ген заменен на указанный линейный фрагмент ДНК или линейный фрагмент ДНК встроен в целевой ген.
Термин «целевой ген» означает локус на хромосоме указанного λ-Red-устойчивого штамма бактерии рода Pantoea, который должен быть модифицирован. Источником чужеродного фрагмента ДНК может быть организм, другой, чем λ-Red-устойчивый штамм, или это может быть λ-Red-устойчивый штамм.
Термин «рекомбинантная бактерия» означает бактерию рода Pantoea, в ген которой встраивается интегративная кассета или часть гена которой заменяется на интегративную кассету.
Термин «рекомбинантная бактерия» означает бактерию рода Pantoea, в которой указанный линейный фрагмент ДНК встроен в целевой ген на хромосоме или заменяет некоторую часть целевого гена.
Фрагмент ДНК согласно настоящему изобретению означает фрагмент ДНК, обладающий свободным 5'-концом и 3'-концом и означает, что он не является кольцевой ДНК. Линейный фрагмент ДНК, который является частично идентичным целевой области, может быть получен путем клонирования линейного фрагмента ДНК на хромосоме бактерии рода Pantoea или другой бактерии семейства Enterobacteriaceae. Линейный фрагмент ДНК может быть, к примеру, один раз клонирован в плазмиду из ДНК хромосомы для получения рекомбинантной плазмиды и затем извлечен из рекомбинантной плазмиды с помощью рестриктаз, или он может быть получен путем прямого амплифицирования целевого гена из геномной молекулы ДНК методом ПЦР.
Ген для встраивания может быть слитым геном, состоящим из двух или больше генов или из комплекса генов, содержащего два или более генов.
Для того чтобы отобрать необходимую рекомбинантную бактерию, в линейный фрагмент ДНК могут быть включены генетические маркеры для позитивной (например, гены устойчивости к антибиотикам) или для негативной (например, ген sacB) селекции. Согласно методу red-управляемой интеграции, интегрированный ген устойчивости к антибиотику может быть удален путем введения сайтов attL и attR, которые являются сайтами прикрепления фага лямбда и продукта ПЦР, и путем комбинирования системы вырезания, происходящей из фага лямбда с методом red-управляемой интеграции. Для удаления селективного маркера, в частности, может быть использована система int-xis (заявка РСТ WO 05/010175).
Для встраивания линейного фрагмента ДНК, содержащего необходимый ген, может быть использован ген, который участвует в метаболизме и биосинтезе целевого вещества или в процессе выведения целевого вещества из бактерии рода Pantoea. Примеры таких генов включают, но не ограничиваются только ими, гены гликолиза или ассимиляции азота, гены пентозного цикла, гены цикла Кребса и так далее. Более точно, примеры включают, но не ограничиваются только глутаматдегидрогеназой, глутаматсинтазой, глутаматсинтетазой, изоцитратдегидрогеназой, аконитатгидратазой, цитратсинтазой, фософенолпируваткарбоксилазой, пируватдегидрогеназой, пируваткиназой, фосфоенолпируватсинтазой, енолазой, фосфоглицеромутазой, фосфоглицераткиназой, глицеральдегид-3-фосфатдегидрогеназой, триозофосфатизомеразой, фруктозобифосфатальдолазой, фосфофруктокиназой, глюкозофосфатизомеразой, глутамин-оксоглутаратаминотрансферазой, изопропилмалатсинтазой и так далее. Бактериальные гены, включенные в биосинтез L-аминокислот, нуклеиновых кислот и их предшественников, могут также быть использованы. Фраза «ген, кодирующий белок, участвующий в выведении целевого вещества из бактерии» означает ген, кодирующий мембранный белок, который непосредственно экскретирует целевое вещество из бактериальной клетки, или ген, кодирующий белок, который активирует мембранный белок, который экскретирует целевое вещество.
Этот основной подход был использован для точной настройки экспрессии гена yfhK в штамме Pantoea ananatis для увеличения уровня продукции L-глутаминовой кислоты. Достигнутая оптимизация, скорее, чем максимизация экспрессии гена или оперона, является актуальной проблемой при конструировании различных бактериальных штаммов, способных продуцировать биологически активные метаболиты, особенно в случаях, когда низкие уровни экспрессии целевого гена являются недостаточными для достижения конечной цели. С другой стороны, сверхэкспрессия целевого гена может привести не только к нейтральному, но и к негативному действию. Кроме того, желаемый уровень этого ключевого гена остается неизвестным. Тонкая настройка является крайне желательной для обеспечения оптимизации и поэтому должно быть протестировано огромное количество вариантов. Обычный подход для проведения этого рода тонкой настройки включает молекулярное клонирование целевого гена в рекомбинантных плазмидах и размещение их под контролем различных промоторов, с последующей оценкой полученных рекомбинантных штаммов. После отбора лучших вариантов необходимо провести большую дополнительную работу, если желателен улучшенный штамм-продуцент.
Способ согласно настоящему изобретению включает конструирование библиотеки фрагментов ДНК в хромосоме бактерии, принадлежащей к роду Pantoea, путем встраивания в хромосому указанной бактерии набора фрагментов ДНК, синтезированных in vitro, причем мутантная бактерия принадлежит к роду Pantoea и является устойчивой к продуктам генов gam, bet и ехо фага лямбда, и используется в качестве бактерии-реципиента. Предпочтительной мутантной бактерией, используемой в данном методе, является бактерия Pantoea ananatis SC17(0). Точнее, способ согласно настоящему изобретению включает конструирование библиотеки ДНК-фрагментов, которая содержит один или более регуляторных элементов для экспрессии гена, включая промоторы, сайты связывания рибосом (RBS) и операторы. Более точно, способ согласно настоящему изобретению включает конструирование библиотеки ДНК-фрагментов, которые содержат один или более регуляторных элементов, которые включают рандомизированную последовательность.
Термин «бактерия, принадлежащая к роду Pantoea» означает, что бактерия относится к роду Pantoea в соответствии с классификацией, известной специалисту в области микробиологии. Некоторые штаммы Enterobacter agglomerans были недавно переклассифицированы в Pantoea agglomerans, Pantoea ananatis, Pantoea stewartii или подобные им, на основе анализа нуклеотидной последовательности 16S rRNA, и т.д.
Фраза «набор фрагментов ДНК» означает смесь заново синтезированных фрагментов ДНК или смесь известных фрагментов ДНК, полученных из природных или мутированных микроорганизмов, библиотеки ДНК, GenBank и т.д. Набор ДНК-фрагментов может быть образован путем непосредственного смешивания заново синтезированных фрагментов ДНК с известными последовательностями, фрагментов ДНК, полученных из различных источников, указанных выше, или фрагментов ДНК, полученных путем химического синтеза, фрагментов ДНК, которые имеют область, содержащую рандомизированную последовательность.
Фраза «бактерия модифицирована таким образом, что является устойчивой к продукту гена» означает, что бактерия модифицирована таким образом, что способна расти и образовывать колонии в среде, в условиях, когда ген в бактерии экспрессируется.
Фраза «регуляторные элементы» обозначает нуклеотидные последовательности, расположенные перед, внутри и/или после кодирующей области, которые контролируют транскрипцию и/или экспрессию кодирующей области вместе с аппаратом биосинтеза белка. Эта фраза обычно используется при описании промоторов, сайтов связывания рибосом (RBS), операторов или других элементов генома и тех, которые влияют на уровень экспрессии генов.
Фраза «рандомизированная последовательность» означает, что в процессе обычного химического синтеза фрагмента ДНК, случайные нуклеотиды (обычно обозначаемые как N, где N - это аденин, гуанин, цитозин или тимин) встраиваются в определенные позиции фрагмента ДНК или в некоторую область фрагмента ДНК, имеющего случайные нуклеотидные последовательности, соответственно. Фрагменты ДНК содержат последовательности, названные «регуляторные элементы».
Способ согласно настоящему изобретению также включает получение бактерии-продуцента L-аминокислоты с оптимизированным уровнем экспрессии целевого гена, который кодирует белок, участвующий в метаболизме и биосинтезе целевого вещества, или в выведении целевого вещества из указанной бактерии и, как результат, влияет на продукцию L-аминокислоты. Способ согласно настоящему изобретению подразделяется на следующие этапы:
1) введение библиотеки искусственно созданных фрагментов ДНК, которые содержат регуляторные элементы для экспрессии указанного целевого гена, в хромосому указанной бактерии вместо природных регуляторных элементов указанного целевого гена, и
2) отбор бактерии с желаемым фенотипом.
Термин «экспрессия» в том значении, в котором он используется здесь, означает продукцию белкового продукта, кодируемого геном.
Фраза «оптимизированный уровень экспрессии» означает такую экспрессию целевого гена или нескольких генов, следствием которой является бактерия с желаемым фенотипом.
Фраза «желаемый фенотип» включает одну или более характеристик бактерии, которые являются объектами для улучшения. Предпочтительно, это может быть способность бактерии к продукции L-аминокислоты в большем количестве, чем родительский штамм; это может также быть способность расти на минимальной среде, которая не содержит добавок, которые обычно используются для комплементации ауксотрофности или другие необходимые для роста факторы или их сочетание.
Фраза «ген, кодирующий белок, участвующий в метаболизме или биосинтезе целевого вещества» означает ген, кодирующий белок, который вовлечен в метаболические пути углерода, азота или фосфора. Примеры таких генов включают, но не ограничиваются только ими, гены гликолиза или ассимиляции азота, гены пентозного цикла, гены цикла Кребса и т.д. Более точно, примеры включают, но не ограничиваются только глутаматдегидрогеназой, глутаминсинтетазой, глутаматсинтазой, изоцитратдегидрогеназой, аконитатгидратазой, ситратсинтазой, фосфоенолпируваткарбоксилазой, пируватдегидрогеназой, пируваткиназой, фосфоенолпируватсинтазой, енолазой, фосфоглицеромутазой, фосфоглицераткиназой, глицеральдегид-3-фосфатдегидрогеназой, триозофосфатизомеразой, фруктозобифосфатальдолазой, фосфофруктокиназой, глюкозофосфатизомеразой, глутамин-оксоглутаратаминотрансферазой, изопропилмалатсинтазой и т.д. Бактериальные гены, участвующие в биосинтезе L-аминокислот, нуклеиновых кислот и их предшественников могут также быть включены.
Фраза «ген, кодирующий белок, участвующий в выведении целевого вещества из бактерии» означает ген, кодирующий мембранный белок, который непосредственно выводит целевое вещество из бактериальной клетки, или ген, кодирующий белок, который активирует мембранный белок, который выводит целевое вещество.
Смесь фрагментов ДНК, содержащих регуляторные элементы, встраивается в хромосому бактерии вместо природного регуляторного элемента, приводя к возникновению популяции бактериальных клеток с различными уровнями экспрессии интересующих генов. Отбор бактерии с желаемым фенотипом может быть осуществлен путем прямой оценки количества наработанной L-аминокислоты в стандартной минимальной среде или другими методами, пригодными для установления характеристик, необходимых для желаемого фенотипа.
Термин «бактерия-продуцент L-аминокислоты» в том значении, в котором он используется здесь, означает бактерию, которая способна к продукции целевой L-аминокислоты и вызывает накопление L-аминокислоты в ферментационной среде в больших количествах, по сравнению с природным или родительским штаммом, и предпочтительно означает, что указанный микроорганизм способен накапливать в среде целевую L-аминокислоту в количестве не менее чем 0.5 г/л, более предпочтительно, не менее чем 1.0 г/л.
Определение уровня ферментативной активности и отбор регуляторных элементов последовательностей, оптимальных для определенных условий, требует от специалиста глубоких знаний в данной области техники; отбор регуляторных элементов последовательностей, основанный на научном предположении или интуиции, может не привести к желаемому или оптимальному результату. Кроме того, приготовление даже ограниченного количества мутантов с различными уровнями ферментативной активности является трудоемким и требует больших затрат времени, так как такие мутанты обычно приготавливаются один за другим. Однако способ согласно настоящему изобретению преимущественно предлагает простую и прямую одноступенчатую процедуру интеграции природного или искусственного фрагмента ДНК в хромосому, генерирующую популяцию бактериальных клеток с широким спектром уровней экспрессии целевого гена. К примеру, рандомизация четырех нуклеотидов в определенной области химически синтезированного регуляторного элемента обеспечивает 44, или 256 теоретически возможных вариантов этой области.
Кроме того, обычным является увеличение активности целевого фермента путем встраивания копии гена, кодирующего фермент, в плазмиду бактерии.
Однако уровень экспрессии гена, интегрированного в хромосому бактерии, и уровень экспрессии того же гена, встроенного в плазмиду, заселяющую бактерию, может быть не одним и тем же. Поэтому другим преимуществом метода, согласно настоящему изобретению, является то, что метод позволяет оценить и отобрать бактерию в условиях, оптимальных для экспрессии интересующего гена, с последующим прямым использованием бактерии для продукции L-аминокислоты без дополнительных манипуляций.
Методами получения плазмидной ДНК, разрезания и лигирования ДНК, трансформации, выбора олигонуклеотидов в качестве праймеров и подобными им могут являться обычные методы, хорошо известные специалисту в данной области, Эти методы описаны, например, в книге Sambrook, J., Fritsch, E.F., and Maniatis, Т., "Molecular Cloning A Laboratory Manual, Second Edition", Cold Spring Harbor Laboratory Press (1989).
Бактерия согласно настоящему изобретению может быть бактерией-продуцентом L-аминокислоты и нуклеиновой кислоты с оптимальным уровнем экспрессии гена, который кодирует белок, участвующий или в распределении углерода, азота и фосфора, или в выведении целевого вещества из указанной бактерии, при этом оптимизация экспрессии приводит к увеличенной продукции L-аминокислоты. Такая бактерия может быть получена методом согласно настоящему изобретению.
Более точно, бактерия согласно настоящему изобретению продуцирует L-аминокислоту, такую как L-аланин, L-аргинин, L-аспарагин, L-аспарагиновая кислота, L-цистеин, L-глутаминовая кислота, L-глицин, L-гистидин, L-изолейцин, L-лейцин, L-лизин, L-метионин, L-фенилаланин, L-пролин, L-серин, L-треонин, L-триптофан, L-тирозин и L-валин. Также бактерия согласно настоящему изобретению включает бактерию-продуцент L-глутаминовой кислоты, имеющую оптимизированный уровень экспрессии гена, который влияет на продукцию L-глутаминовой кислоты. Кроме того, L-глутаминовая кислота играет значительную роль в биосинтезе L-лейцина, L-лизина, L-изолейцина, L-валина, L-гистидина, L-аспартата, L-аланина, L-тирозина и L-фенилаланина в качестве донора аминогруппы. И бактерия согласно настоящему изобретению является бактерией-продуцентом L-лейцина с оптимизированным уровнем экспрессии гена, который влияет на продукцию L-лейцина.
Поэтому улучшенное снабжение азотом для гена, кодирующего аминотрансферазу, является полезным для продукции других аминокислот, таких как L-лейцин, L-лизин, L-изолейцин, L-валин, L-гистидин, L-аспартат, L-аланин, L-тирозин и L-фенилаланин.
Примеры бактерий-продуцентов L-глутаминовой кислоты включают мутантные штаммы, принадлежащие к роду Pantoea, которые дефицитны по активности α-кетоглутаратдегидрогеназы или имеют сниженную активность α-кетоглутаратдегидрогеназы, и могут быть получены как описано выше. Такие штаммы включают штамм Pantoea ananatis AJ13356 (патент США 6331419). Штамм Pantoea ananatis AJ13356 был депонирован в Национальном Институте Биологических Наук и Человеческих Технологий, Агенство Промышленной Науки и Технологии, Министерство Международной Торговли и Промышленности (National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology, Ministry of International Trade and Industry) (в настоящее время называющийся Национальный Институт Прогрессивной Промышленной Науки и Технологии, Международный Депозитарий Организмов для Целей Патентования, Централ 6, 1-1, Хигаши 1-Чоме, Тсукуба-ши, Ибараки-кен, 305-8566, Япония - National Institute of Advanced Industrial Science and Technology, International Patent Organism Depositary, Central 6, 1-1, Higashi 1-Chome, Tsukuba-shi, Ibaraki-ken, 305-8566, Japan, 19 февраля 1998 и получивший инвентарный номер FERM P-16645). Затем было произведено международное депонирование этого штамма согласно условиям Будапештского Договора от 11 января 1999 г., и штамм получил инвентарный номер FERM ВР-6615. Штамм Pantoea ananatis AJ13356 не имеет α-KGDH активности в результате инактивации гена αKGDH-E1 субъединицы (sucA). Вышеупомянутый штамм при выделении был идентифицирован как Enterobacter agglomerans и депонирован как штамм Enterobacter agglomerans AJ13356. Тем не менее, позднее он был классифицирован как Pantoea ananatis на основе нуклеотидной последовательности 16S pPHK и других доказательств. Несмотря на то что штамм АJ13356, был депонирован в указанный выше депозитарий как Enterobacter agglomerans, для целей данного описания он будет упоминаться как Pantoea ananatis.
Стратегия, описанная выше для получения L-глутаминовой кислоты, может быть использована для получения бактерии, продуцирующей другие L-аминокислоты.
Бактерия согласно настоящему изобретению является бактерией-продуцентом нуклеиновой кислоты, обладающей оптимизированным уровнем экспрессии гена, кодирующей белок, участвующий в распределении потоков углерода, азота или фосфора, или в выведении целевого вещества из указанной бактерии, которое приводит к увеличению продукции нуклеиновой кислоты. Нуклеиновая кислота выбирается из группы, состоящей из инозина, ксантозина, гуанозина и аденозина. Такая бактерия может быть получена способом согласно настоящему изобретению.
2. Способ получения L-аминокислоты
Способом получения L-аминокислоты является способ, включающий стадии выращивания бактерии согласно настоящему изобретению в питательной среде с целью продукции и накопления L-аминокислоты в культуральной жидкости и выделения L-аминокислоты из культуральной жидкости. Более точно, способ получения L-аминокислоты включает способ получения L-глутаминовой кислоты, который включает этапы выращивания бактерии согласно настоящему изобретению в питательной среде с целью продукции и накопления L-глутаминовой кислоты в культуральной жидкости и выделения и очистки L-глутаминовой кислоты из культуральной жидкости.
Согласно настоящему изобретению выращивание, выделение и очистка L-аминокислоты из культуральной или подобной ей жидкости может быть осуществлена способом, подобным традиционным способам ферментации, в которых аминокислота продуцируется с использованием бактерии.
Питательная среда, используемая для выращивания, может быть как синтетической, так и натуральной, при условии, что указанная среда содержит источники углерода, азота, минеральные добавки и, если необходимо, соответствующее количество питательных добавок, необходимых для роста микроорганизмов. К источникам углерода относятся различные углеводы, такие как глюкоза и сахароза, а также различные органические кислоты. В зависимости от характера ассимиляции используемого микроорганизма могут использоваться спирты, такие как этанол и глицерин. В качестве источника азота могут использоваться различные неорганические соли аммония, такие как аммиак и сульфат аммония, другие соединения азота, такие как амины, природные источники азота, такие как пептон, гидролизат соевых бобов, ферментолизат микроорганизмов. В качестве минеральных добавок могут использоваться фосфат калия, сульфат магния, хлорид натрия, сульфат железа, сульфат марганца, хлорид кальция и подобные им соединения. В качестве витаминов могут использоваться тиамин, дрожжевой экстракт и подобные им соединения.
Выращивание осуществляется предпочтительно в аэробных условиях, например при перемешивании культуральной жидкости на качалке, взбалтывании с аэрацией, при температуре в пределах от 20 до 40°С, предпочтительно в пределах от 30°С до 38°С. pH среды поддерживают в пределах от 5 до 9, предпочтительно от 6.5 до 7.2. pH среды можно доводить аммиаком, карбонатом кальция, различными кислотами, основаниями и буферными растворами. Обычно выращивание в течение от 1-го до 5-ти дней приводит к накоплению целевой L-аминокислоты в культуральной жидкости.
После выращивания твердые остатки, такие как клетки, могут быть удалены из культуральной жидкости методом центрифугирования или фильтрацией через мембрану, а затем L-аминокислота может быть выделена и очищена методами ионообменной хроматографии, концентрирования и/или кристаллизации.
Краткое описание чертежей
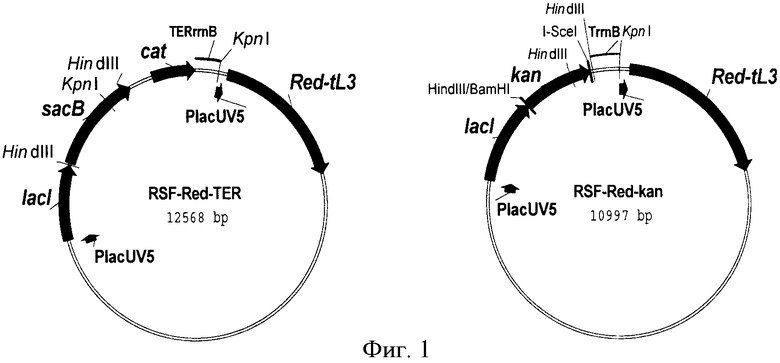
На Фиг.1 изображены генетические карты вспомогательных («хелперных») плазмид, которые индуцируют экспрессию λ-Red-генов.
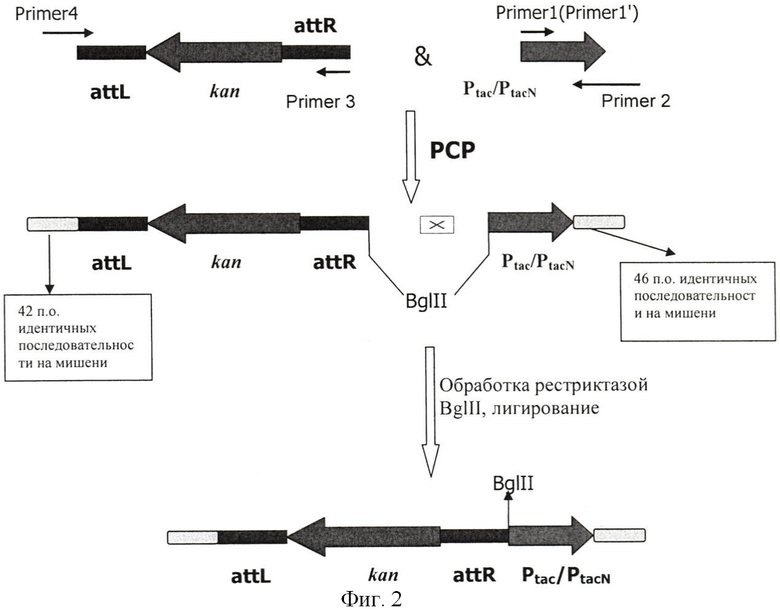
На Фиг.2 изображено конструирование библиотеки промоторов in vitro.
На Фиг.3 изображена интеграция ПЦР фрагментов ДНК в хромосому Р. ananatis.
На Фиг.4 изображены 2 варианта модификации природного гена yhfK. А) Фрагмент хромосомы природного штамма Р. ananatis, несущей ген yhfK. В) Нуклеотидная последовательность фрагмента хромосомы, содержащего upstream-область гена yhfK. С) Первый вариант модификации: встраивание кассеты, содержащей терминатор Tthr, вырезаемый маркер KmR и промотор Ptac сразу после стоп-кодона гена crp. D) Второй вариант модификации: замена upstream-области гена yhfK на интегративную кассету, содержащую терминатор Tthr, вырезаемый маркер KmR рандомизированный промотор PtacN из полученной библиотеки промоторов и консенсусную SD-последовательность AGGAGG.
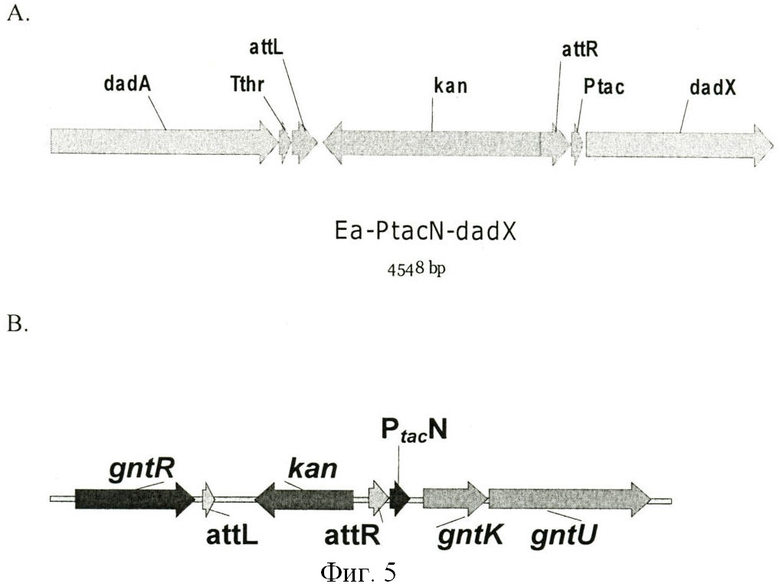
На Фиг.5 показана структура хромосомы перед генами dadX(A) и gntK (В), содержащими замененные участки с промоторами.
На Фиг.6 показана структура плазмиды RSFPlacsacB.
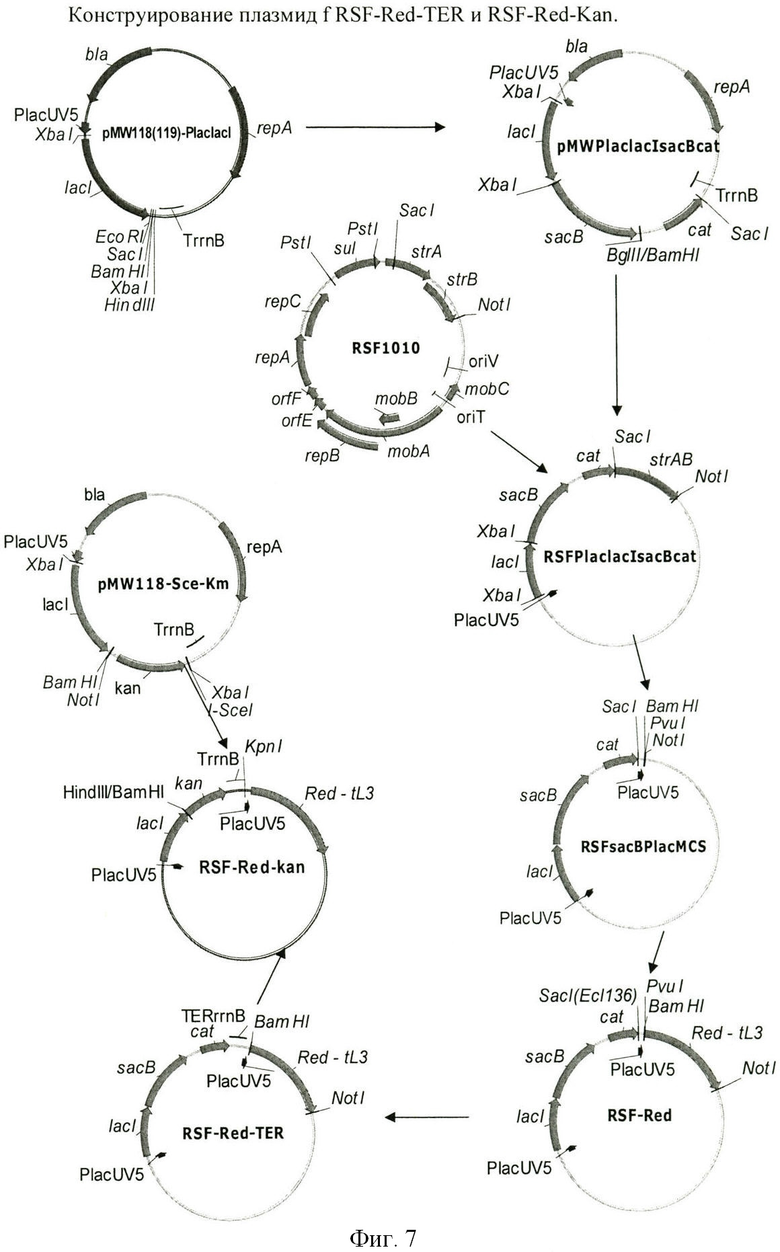
На Фиг.7 показана схема конструирования плазмид RSF-Red-TER и RSF-Red-Kan.
Примеры
Настоящее изобретение будет более подробно описано ниже со ссылкой на следующие неограничивающие настоящее изобретение Примеры.
Пример 1. Отбор штамма-реципиента для высокоэффективного λ-Red-опосредованного рекомбинантного конструирования штамма P. ananatis.
λ-Red-зависимую рекомбинацию использовали для интеграции фрагментов ДНК, полученных методом ПЦР, с фланкирующими участками размером 40 п.о., которые гомологичны участкам в геноме Р. ananatis. Для этой цели конструировали две новые хелперные плазмиды RSF-Red-TER, RSF-Red-Kan, которые экспрессируют гены λ gam, bet и ехо (здесь и далее обозначенные как «λ-Red-гены») (Фиг.1). Детальная конструкция этих плазмид описана ниже в Примере-ссылке. Обе плазмиды могут быть использованы для широкого круга бактерий-хозяев, имеющих различные генотипы, потому что: 1) они имеют репликон плазмиды RSF1010 для широкого круга хозяев, который способен быть стабильно удерживаемым во многих грамотрицательных и грамположительных бактериях и даже в растениях (Scholz, et al., 1989; Buchanan-Wollaston et al,, 1987); 2) λ-Red-гены gam, bet и ехо находятся под контролем промотора PlacUV5, который распознается многими бактериальными РНК-полимеразами (см., например, Brunschwig, Е. and Darzins, A., Gene, 111, 1, 35-41 (1992); Dehio, M. et al., Gene, 215, 2, 223-229 (1998)); 3) автоматически регулируемый элемент PlacUV5-lacI и ρ-независимый терминатор транскрипции оперона rrnB из Е.coli (TrrnB) обеспечивает низкий уровень базальной экспрессии λ-Red-генов (Skorokhodova, A.Yu et al., Biotekhnologiya (Rus), 5, 3-21 (2004)). Кроме того, плазмида RSF-Red-TER содержит ген левансахарозы (sacB), который позволяет извлекать эту плазмиду из клеток на среде, содержащей сахарозу. В клетках Е. coli частота интеграции фрагментов ДНК, полученных методом ПЦР, с короткой фланкирующей областью обеспеченная плазмидами RSF-Red-TER и RSF-Red-Kan, была такой же высокой, как для хелперной плазмиды pKD46 (Datsenko, K.A., Wanner, B.L., Proc. Natl. Acad. Sci. USA, 97, 6640-6645, (2000)). Однако экспрессия λ-Red генов была токсичной для клеток Р. ananatis. Клетки, трансформированные хелперными плазмидами RSF-Red-TER или RSF-Red-Kan, имели очень низкую скорость роста на LB-среде, содержащей ИПТГ (1 мМ) и соответствующий антибиотик (хлорамфиникол, 25 мг/мл, или канамицин, 40 мг/мл), и чрезвычайно низкую частоту λ-Red-зависимой рекомбинации (10-8), в случае, когда она вообще происходила.
Были отобраны мутантные штаммы Р. ananatis, устойчивые к экспрессии всех трех Red-генов из фага лямбда (gam, bet и ехо). Для этого клетки штамма Р. ananatis SC17 (патент США 6596517) были электропорированы плазмидой RSP-Red-TER. Приблизительно 106 трансформантов было получено после культивирования в течение 18-ти часов и до 10 клонов имели большой размер, тогда как все другие клоны были очень маленькие. Большие колонии были приблизительно 2 мм в диаметре, маленькие колонии были приблизительно 0.2 мм после 18-ти часового культивирования. В течение продолженного культивирования (до 24 часов) маленькие колонии больше не росли, тогда как большие колонии продолжали расти. Одну из мутантных колоний большого размера штамма Р. ananatis, которая была устойчивой к экспрессии всех трех Red-генов из фага лямбда (gam, bet и ехо), отобрали для дальнейшего анализа.
ДНК плазмиды RSF-Red-TER из этой большой колонии и из нескольких маленьких колоний выделяли и ретрансформировали в Е. coli MG1655 для проверки способности плазмиды обеспечивать синтез активных продуктов генов Red-системы. Контрольный эксперимент Red-зависимой интеграции в полученных трансформантах показал, что только плазмида, очищенная из большой колонии, обеспечивает экспрессию λ-Red-генов, необходимую для Red-зависимой интеграции.
Для проверки того, имеет ли место Red-зависимая интеграция в выбранном большом клоне, была произведена электропорация линейного фрагмента ДНК, полученного методом ПЦР, содержащего маркер KmR и фланкирующую область размером 40 п.о., которая гомологична гену hisD, и синтезированного таким образом, чтобы быть интегрированным в сайт распознавания SmaI гена hisD из Р. ananatis; два маленьких клона использовали в качестве контроля. Нуклеотидная последовательность гена hisD из Р. ananatis показана на SEQ ID NO: 42. Олигонуклеотиды, показанные на SEQ ID NO: 43 и 44, использовали в качестве праймеров, и плазмиду pMW118-(λattL-Kmr-λattR) использовали в качестве матрицы в ПЦР; два маленьких клона, которые не были устойчивы к λ-Red-генам, использовали в качестве контроля. Конструирование плазмиды pMW118-(λattL-Kmr-λattR) описано ниже в Примере-ссылке 2. Плазмида RSF-Red-TER способна индуцибельно экспрессировать Red-гены, что связано с наличием гена lacI на плазмиде. Проверяли два условия индукции: в первой группе ИПТГ (1 мМ) добавляли за один час до электропорации, во второй группе ИПТГ добавляли в момент начала культивирования для приготовления электрокомпетентной культуры. Скорость роста клеток, несущих плазмиду RSF-Red-TER, происходящих от большой колонии, не была значительно ниже, чем скорость роста штамма без плазмиды SC17. Добавление ИПТГ вызывало только слабое торможение скорости роста этих культур. В то же время, потомство маленьких колоний росло очень медленно без ИПТГ и индукция приводила практически к остановке их роста. После электропорации культур клеток-потомков большой колонии многие KmR клоны (18 клонов с коротким временем индукции и около 100 клонов с пролонгированным временем индукции) выросли. Все 100 протестированных колоний имели фенотип His-; ПЦР-верификация 20-ти клонов подтвердила ожидаемую структуру хромосомы этих клеток. С другой стороны, после электропорации клеток-потомков маленьких клонов интегранты не были получены.
Полученный таким образом большой клон выращивали на чашках, содержащих 7% сахарозы для избавления от плазмиды, и ретрансформировали его плазмидой RSF-Red-TER. Бесплазмидный штамм назвали SC17(0) и депонировали во Всероссийской Коллекции Промышленных Микроорганизмов (ВКПМ) (1-й Дорожный проезд, 1, Москва, 117545, РФ) 21 сентября 2005 года под инвентарным номером ВКПМ В-9246. Все клоны, выросшие после этой ретрансформации, имели большой размер, как и родительский клон SC17(0). Провели эксперимент по Red-опосредованной интеграции в штамм SC17(0), который был ретрансформирован плазмидой RSF-Red-TER. Проверили три независимо полученных трансформанта; использовали тот же фрагмент ДНК, который использовали в предыдущем эксперименте. Применяли короткое время индукции (1 час до электропорации). В каждом триплете выросло более чем 100 KmR клонов. Все протестированные клоны имели фенотип His-. Поэтому было сделано заключение о том, что отобрали мутантный штамм, названный SC17(0), который устойчив к экспрессии λ-Red-генов. Этот штамм можно использовать в качестве реципиента для Red-зависимой интеграции в хромосому Р. ananatis.
Аналогичный эксперимент провели с хелперной плазмидой RSF-Red-Kan. После электротрансформации SC17 этой плазмидой, все клоны, которые росли на твердой LB-среде, имели тот же размер, что и родительский штамм SC17, однако значительное замедление роста наблюдалось после индукции. Для того чтобы воспроизвести отбор λ-Red-устойчивого штамма, используя хелперную плазмиду RSF-Red-Kan, клетки после электропорации помещали на твердую LB-среду с добавлением ИПТГ в различных концентрациях. Отсутствие роста наблюдалось на чашке с 1 мМ ИПТГ; только несколько больших клонов выросло на чашке с 50 мкМ ИПТГ; и много маленьких и несколько больших клонов выросло на чашке, содержащей 20 мкМ ИПТГ. Одна из больших колоний, которая выросла на среде с 20 мкМ ИПТГ, как было установлено, являлась реципиентом для λ-Red-зависимой интеграции. В этом эксперименте частота интеграции для этого клона была такой же, как и для штамма SC17(0). Процедура, разработанная в последнем эксперименте, может быть успешно применена для отбора λ-Red-устойчивых производных других бактериальных штаммов или штаммов, для которых экспрессия λ-Red-генов является токсической.
Пример 2. Дизайн и характеристика библиотеки промоторов, пригодной для оптимизации уровня экспрессии интересующего гена.
Для разработки дизайна группы промоторов с различной силой в Р. ananatis применяли подход, реализованный ранее для Е. coli (заявка РСТ WO 2004080386, WO 2005/010175).
Получение сгенерированной методом ПЦР библиотеки промоторов.
Библиотеку промоторов, пригодную для оптимизации уровня экспрессии интересующего гена, конструировали путем рандомизации четырех интернальных нуклеотидов области из 35-ти нуклеотидов («-35») инициирующего промотора Ptac. Библиотеку интегрировали перед геном lacZ на хромосоме штамма Р. ananatis SC17(0) и определяли активность LacZ в каждом трансформанте. Структура области хромосомы Р. ananatis перед геном lacZ показана на Фиг.3. Нуклеотидные последовательности генов yghU, scrK, lacZ из Р. ananatis показаны на SEQ ID NO: 45, 46 и 47 соответственно.
Область «-35» промотора Ptac показана на SEQ ID NO: 16. Для получения промоторов различной силы фрагмент ДНК с группой Ptac-подобных промоторов, отличных от промотора области «-35», конструировали методом ПЦР с использованием плазмиды pDR540 ("Pharmacia", Швеция) в качестве матрицы. Для этой цели в качестве 5'-праймеров использовали или праймер «I» (SEQ ID NO: 1), полностью соответствующий последовательности Ptac, или праймер «1'», «рандомизированный» праймер (SEQ ID NO: 2). Оба содержали сайт распознавания BglII на 5'-конце. Четыре из шести позиций в праймере «1'» из консенсусной области «-35» рандомизировали в процессе химического синтеза, и они содержали комбинацию любых четырех нуклеотидов, обозначенных как «n» в SEQ ID NO: 2. Праймер «2» (SEQ ID NO: 3), который использовался как праймер на 3'-конце, содержит 46 нуклеотидов из 3'-конца Ptaс, SD-последовательность и начало кодирующей области гена lacZ. Фрагменты ДНК, полученные методом ПЦР, которые содержали или оригинальный промотор Ptaс, или группу рандомизированных tac-подобных промоторов, использовали в дальнейшем для дизайна.
Одновременно проводили амплификацию фрагмента ДНК с вырезаемым геном устойчивости Km, фланкированным сайтами attL и attR фага лямбда, с использованием плазмиды pMW118-(λattL-Kmr-λattR) в качестве матрицы и праймеров «3» (SEQ ID NО: 4) и «4» (SEQ ID NO: 5). Полученный фрагмент ДНК имел сайт распознавания на одном конце для рестриктазы BglII, который полезен для лигирования с фрагментами tac-подобных промоторов, и сайт из 42-х п.о., гомологичный хромосоме Р. ananatis и расположенный перед геном scrK для того, чтобы обеспечить последующую интеграцию в бактериальный геном (см. Фиг.3), с другого конца. Два ПЦР-генерированных фрагмента обрабатывали BglII и лигировали in vitro с помощью Т4 ДНК-лигазы.
Интеграция сгенерированной методом ПЦР библиотеки промоторов в хромосому Р. ananatis
Полученные лигазные смеси использовали для λ-Red-зависимой интеграции в хромосому Р. ananatis. Хелперную плазмиду использовали в качестве носителя λ-Red-генов фага. Для получения электрокомпетентных клеток Р. ananatis штамм SC17(0), трансформированный плазмидой RSF-Red-TER, выращивали в течение ночи при температуре 34°С на LB-среде с добавлением хлорамфиникола до концентрации 50 мкг/мл. Затем культуру разводили в 100 раз свежей LB-средой, содержащей 50 мкг/мл хлорамфиникола, и выращивали до достижения ею оптической плотности OD600 приблизительно равной 0.3 при температуре 34°C с аэрацией. После этого добавляли ИПТГ до концентрации 1 мМ и продолжали культивирование до достижения оптической плотности OD600=0.7. Образцы клеток объемом 10 мл отмывали 3 раза равными количествами деионизованной ледяной воды и ресуспендировали в 40 мкл 10% ледяного глицерина. Непосредственно перед электропорацией, 100-200 нг амплифицированных in vitro фрагментов ДНК растворяли в 5 мкл деионизованной воды и добавляли к суспензии клеток. Процедуру проводили с использованием устройства для электротрансформации бактерий ("BioRad", USA, catalog number 165-2089, version 2-89). Применяемые параметры пульсации были следующими: интенсивность электрического поля составляла 20 кВ/см, время пульсации 5 мс. После электропорации 1 мл LB-среды, обогащенной глюкозой (0.5%) немедленно добавляли к клеточной суспензии. Затем клетки выращивали с аэрацией при температуре 34°С в течение 2-х часов и рассевали на чашки с твердой LB-средой, содержащей 40 мкг/мл канамицина с последующей ночной инкубацией при температуре 34°С. Отобранные KmR - интегранты рассевали на чашки с LB-средой с добавлением ИПТГ (1 мМ) и сахарозы (5 г/л) при температуре 34°С и выращивали для формирования единичных колоний. Выделяли KmR, CmS-варианты таким образом, что хелперная плазмида RSF-Red-TER удалялась из интегрантов.
ПЦР-верификация.
Для того чтобы подтвердить идентичность полученной хромосомной структуры отобранных KmR, CmS колоний, проводили ПЦР с использованием праймеров «5» (SEQ ID NO: 6), который является гомологом сайта attR на 5'-конце и «6» (SEQ ID NO: 7), чей сайт отжига локализован в структурной части гена lacZ.
ДНК-секвенирование полученных tac-подобных промоторов.
Первоначально провели ПЦР-амплификацию геномных фрагментов ДНК, выделенных из отобранных клонов с помощью набора для выделения геномной ДНК ("Sigma", США). Для секвенирования полученных фрагментов ДНК использовали метод Сэнгера и праймеры «5» (SEQ ID NO: 6) и «7» (SEQ ID NO: 8), как для ПЦР, так и для секвенирования.
Тест на β-галактозидазу.
Все клеточные культуры выращивали ночь на LB-среде с добавлением 0.5×М9 солевого раствора, глюкозы (5 г/л) и 40 мкг/мл канамицина, затем разводили в 100 раз свежей средой, и выращивали при температуре 34°C с аэрацией в течение 3 часов. Активность β-галактозидазы измеряли по обычной методике (Miller, 1972). Представленные данные (табл.1) являются средним значением от трех независимо выросших культур каждого клона. Стандартная ошибка не превышала 30%.
Как видно из табл.1, значения активностей в репрезентативных клонах библиотеки с Ptas-подобными промоторами отличаются в два раза. Также можно видеть, что выбранная процедура позволяет конструировать библиотеку мутантных промоторов только с рандомизированной областью «-35».
Пример 3. Увеличение продукции глутаминовой кислоты путем тонкой настройки уровня экспресии гена yhfK.
Ген yhfK кодирует предположительно переносчик Glu. Было раскрыто, что его амплификация на плазмиде увеличивает продуктивность штамма-продуцента L-глутаминовой кислоты (патентная заявка США 2005/0196846). Для тонкой настройки уровня экспрессии гена yhfK использовали Red-зависимую интеграцию, как описано выше, для модификации 5'-концевой области гена в его природной локализации. Структура 5'-концевой области гена yhfK в хромосоме Р. ananatis представлена на Фиг.4А. Нуклеотидная последовательность области показана на SEQ ID NO: 48.
В хромосоме Р. ananatis ген yhfK расположен через 42 п.о. после гена crp (см. Фиг.4А). В 5'-концевой области гена yhfK не были найдены регуляторные элементы (терминатор, промотор, или SD-последовательность) (Фиг.4В). Поэтому было предложено, что введение сильного промотора и, может быть, SD-последовательности перед этим геном может усилить его уровень экспрессии значительно. В то же время, для обеспечения стабильности мRNA гена crp в штаммах с различными модификациями области, прилегающей к 3'-концу гена crp, не нарушая его характер экспрессии, сразу после стоп-кодона этого гена ввели терминатор лидерного пептида треонинового оперона Е. coli Tthr (см. Пример-ссылку 2). Структуры полученных модификаций представлены на Фиг.4С и D.
Для конструирования интегративных кассет фрагменты ДНК с вырезаемым маркером KmR и природным промотором Ptaс или рандомизированными промоторами амплифицировали методом ПЦР с использованием геномных фрагментов ДНК, которые были выделены из клонов библиотеки промоторов с использованием праймеров: прямого праймера «9» (SEQ ID NO: 11) и обратного праймера «8» (SEQ ID NО: 9) или «8'»(SEQ ID NO: 10). Обратный праймер «8» позволяет амплифицировать природную upstream-область ген yhfK, праймер «8'» позволяет амплифицировать 5'-концевую область гена yhfK с добавочной SD-последовательностью. Полученные фрагменты ДНК содержали сайт распознавания XbaI на 5'-концах и были гомологичны на протяжении 40-а или 42-х п.о. желаемому сайту интеграции на 3'-концах. Параллельно фрагмент ДНК, содержащий Tthr, амплифицировали методом ПЦР с использованием плазмиды pML-ter_thrL (см. Пример-ссылку 2) в качестве матрицы и праймеров «10» (SEQ ID NO: 12) и «II» (SEQ ID NO: 13). Полученный второй фрагмент содержал сайт распознавания XbaI на 3'-конце и был гомологичен на протяжении 43-х п.о. желаемому сайту интеграции на 5'-конце. Все ПЦР-генерированные фрагменты ДНК обрабатывали рестриктазой XbaI и фрагменты, содержащие различные промоторы лигировали с фрагментом, содержащим терминатор Tthr.
Полученные лигированные смеси осаждали спиртом, растворяли в деионизованной воде и использовали для Red-зависимой интеграции в штамм SC17(0)/RSF-Red-TER с использованием той же процедуры, которую использовали для интеграции библиотеки промоторов. Факт интеграции подтверждали методом ПЦР с использованием праймеров «12» (SEQ ID NO: 14) и «13» (SEQ ID NO: 15).
Для переноса этих модификаций в штамм-продуцент L-глутаминовой кислоты AJ 13601 (Европейская патентная заявка 1078989), геномную ДНК выделяли из полученных штаммов с использованием набора «Genomic DNA Isolation Kit» ("Sigma", CШA). После электропорации этих фрагментов ДНК в штамм AJ13601 получали штаммы, имеющие ген yhfK под контролем промотора Ptaс или консенсусной последовательности SD и промотора PtaсN соответственно. Промотор PtaсN в общем случае означает промотор с рандомизированной областью. Процедуру проводили с использованием устройства для электротрансформации бактерий ("BioRad", США, catalog number 165-2089, version 2-89). Применяемые параметры пульсации были следующими: интенсивность электрического поля 12.5 кВ/см, время импульса 10 мс. Фрагменты промотора, используемые в этих экспериментах, содержали только первые 11 п.о. (из 21 п.о.) лактозного оператора O1, поэтому они не репрессировались LacI.
Ночную культуру выращивали при температуре 34°С на LB-среде с добавлением MES (0,1М, рН 7.0), глюкозы (5 г/л), тетрациклина (10 мкг/мл) и хлорамфиникола (25 мкг/мл). Затем культуру разводили в 100 раз свежей средой, содержащей глюкозу (40 г/л), MgSO4·7H2O (0.5 г/л), (NH4)2SO4 (16 г/л), KH2PO4 (0.3 г/л), KCl (1 г/л), MES (10 г/л), бетаин (1 г/л), FeSO4·7H2O (10 мг/л), MnSO4·5H2O (10 мг/л), лизина (100 мг/л), метионина (100 мг/л), DAP (100 мг/л), пантотенат кальция (10 мг/л), CaCO3 (30 г/л), 10 мкг/мл тетрациклин и 50 мкг/мл хлорамфиникол, и выращивали в течение 26-ти часов при температуре 34°С в условиях аэрации. Продукцию и накопление глутамата измеряли методом тонкослойной хроматографии (ТСХ). Представлены усредненные данные результатов выращивания трех независимых культур.
Отбор проводили по продукции L-глутаминовой кислоты. Один из лучших клонов, имеющий высокий уровень продукции L-глутаминовой кислоты (№11), и один из клонов, имеющий сниженную скорость роста и продукцию L-глутаминовой кислоты (№52), отобрали и секвенировали 5'-концевую область гена yhfK. Активность LacZ, экспрессируемую под контролем этих Ptaс-подобных промоторов измеряли как описано выше (данные представлены в табл.1).
Как можно видеть из табл.2, в этих экспериментальных условиях, наилучшие результаты получали при комбинировании высокоэффективной консенсусной SD-последовательности (SD-последовательность, AGGAGG, включена в праймер «8'» (SEQ ID NO: 10) как его составная часть) и относительного слабого промотора Ptaс11 (штамм AJ13601 Ptac11SDyhfK). Выход глутамата, продуцируемого этим штаммом, был выше ((Δ1% по выходу) по сравнению с исходным штаммом AJ13601. Использование более эффективного промотора Ptaс52 в сочетании с природной 5'-концевой областью гена yhfK приводило, как правило, к худшим результатам.
Описанные выше результаты служат доказательством того, что использование промоторной библиотеки, как описано выше (в сочетании с другими регуляторными элементами, такими как SD-последовательность, сайты связывания репрессоров и активаторов, и так далее), представляют рабочие инструменты для тонкой настройки уровня экспрессии интересующего гена.
Пример-ссылка 1. Конструирование хелперных плазмид RSF-Red-TER и RSF-Red-Kan.
Схема конструирования хелперных плазмид представлена на Фиг.5.
На первой стадии конструирования хелперных плазмид RSF-Red-TER и RSF-Red-Kan был сконструирован вектор RSFsacBPlacMCS. Для этой цели фрагменты ДНК, содержащие ген cat из плазмиды pACYC184, и структурную часть гена sacB из В. subtillis амплифицировали методом ПЦР с использованием олигонуклеотидов SEQ ID NO: 49, 50, 51 и 52 соответственно. Эти олигонуклеотиды содержат следующие подходящие сайты рестрикции на 5'-конце, которые необходимы для последующего клонирования: BglII, SaсI, XbaI и BamHII соответственно. Полученный фрагмент гена sacB размером 1.5 тыс.п.о. клонировали в сайты XbaI-BamHI ранее полученного вектора pMW119-PlaclacI. Этот вектор конструировали тем же способом, как описано для вектора pMW118-PlaclacI (Skorokhodova, A.Yu et al., Biotekhnologiya (Rus), 5, 3-21 (2004)), однако он содержит полилинкерный сайт из плазмиды pMW219, вместо плазмиды pMW218. После этого, ранее описанный фрагмент гена cat размером 1.0 тыс.п.о. обрабатывали рестриктазами BglII и SacI и клонировали в сайты BamHI-SacI плазмиды RSF-PlaclacIsacB, которая была получена на предыдущей стадии. Полученная плазмида pMW-PlaclacIsacBcat содержала фрагмент PlacUV5-lacI-sacB-cat. Для субклонирования этого фрагмента в вектор RSF1010 плазмиду pMWPlaclacIsacBcat расщепляли рестриктазой BglII, обрабатывали фрагментом Klenow ДНК-полимеразы I для образования липкого конца, и затем разрезали SacI. Фрагмент BglII-SacI размером 3.8 тыс.п.о. плазмиды pMWPlaclacIsacBcat элюировали из 1%-ного агарозного геля и лигировали с RSF1010, которая содержала вектор, обработанный рестриктазой PstI, фрагментом Klenow ДНК-полимеразы I и SacI. Лигированную смесь трансформировали в штамм Е. coli TG1 и высевали на LB-агар, содержащий хлорамфиникол (50 мг/л). После рестрикционного анализа плазмид, выделенных из выросших клонов, получили плазмиду RSFsacB. Для конструирования вектора RSFsacBPlacMCS фрагмент ДНК, содержащий промотор PlacUV5, амплифицировали методом ПЦР с использованием олигонуклеотидов SEQ ID NO: 53 и 54 в качестве праймеров и плазмиды pMW119-PlaclacI в качестве матрицы. Полученный фрагмент размером 146 п.о. расщепляли рестриктазами SacI и NotI и лигировали с большим фрагментом SacI-NotI из плазмиды RSFsacB.
После этого фрагмент ДНК размером 2,3 тыс.п.о., содержащий λ-Red-αβγ-гены и терминатор транскрипции tL3, амплифицировали методом ПЦР с использованием олигонуклеотидов SEQ ID NO: 55 и 56 в качестве праймеров и плазмиды pKD46 в качестве матрицы (Datsenko, K.A., Wanner, B.L., Proc. Natl. Acad. Sci. USA, 97, 6640-6645, (2000)). Полученный фрагмент клонировали в сайты PvuI-NotI вектора RSFsacBPlacMCS. Таким образом была сконструирована плазмида RSFRed. Для того чтобы исключить сквозное прочитывание генов Red с промотора гена cat, ρ-независимый терминатор транскрипции оперона rrnB из Е. coli вставили между геном cat и промотором PlacUV5. Для этой цели фрагменты ДНК, содержащие промотор PlacUV5 и терминатор TrrnB, амплифицировали методом ПЦР с использованием олтигонуклеотидов SEQ ID NO: 57 и 54 в качестве праймеров и плазмиды pMW119-PlaclacI в качестве матрицы и олигонуклеотидов SEQ ID NO: 58 и 59 в качестве праймеров и хромосомы штамма Е. coli BW3350 в качестве матрицы соответственно. Полученные фрагменты обрабатывали рестриктазой KpnI и лигировали. После этого фрагмент ДНК размером 0.5 тыс.п.о., содержащий как PlacUV5, так и d TrrnB, амплифицировали методом ПЦР с использованием олигонуклеотидов SEQ ID NO: 54 и 59 в качестве праймеров. Полученный фрагмент ДНК расщепляли рестриктазой EcoRI, обрабатывали фрагментом Klenow ДНК-полимеразы I для образования липких концов, разрезали рестриктазой BamHI и лигировали с большим фрагментом Ecl136II-BamHI вектора RSFsacBPlacMCS. Полученную плазмиду назвали RSF-Red-TER.
Для получения плазмиды RSF-Red-Kan фрагмент ДНК размером 1.3 тыс.п.о., содержащий ген kan и терминатор ТrrnB амплифицировали методом ПЦР используя ранее полученную плазмиду pMW118-SceKm в качестве матрицы и олигонуклеотиды SEQ ID NO: 58 и 60 в качестве праймеров. Для конструирования плазмиды pMW-SceKm фрагмент ДНК, содержащий ген kan, фланкированный желаемыми сайтами распознавания рестрикционных эндонуклеаз (включая I-SceI-мегануклеазный сайт распознавания), генерировали методом ПЦР с использованием олигонуклеотидов SEQ ID NO: 60 и 61 в качестве праймеров и плазмиды pUC4K в качестве матрицы. Полученный фрагмент обрабатывали XbaI и BamHI и встраивали в сайты XbaI-BamHI на плазмиде pMW118-PlaclacI.
Полученный фрагмент размером 1,3 тыс. п.о. разрезали рестрикционной эндонуклеазой BamHI, обрабатывали фрагментом Klenow ДНК-полимеразы I для образования липкого конца и расщепляли KpnI. Параллельно, плазмиду RSF-Red-TER последовательно обрабатывали рестриктазой НindIII, фрагментом Klenow ДНК-полимеразы I и рестриктазой KpnI, и большой фрагмент элюировали из 1% агарозного геля. После лигирования полученных фрагментов реакционной смесью, полученной после лигирования, трансформировали штамм TG1 и клетки высевали на LB-arap, содержащий 50 мг/л канамицина. Плазмиду RSF-Red-Kan отбирали после рестрикционного анализа плазмид, выделенных из растущих клонов.
Пример-ссылка 2. Конструирование плазмиды pMW118-(λattL-Kmr-λattR).
Плазмиду pMW118-(λattL-Kmr-λattR) конструировали из плазмиды pMW118-attL-Tc-attR (WO 2005/010175) путем замены маркерного гена устойчивости к тетрациклину на маркерный ген устойчивости к канамицину из плазмиды pUC4K (Vieira, J. and Messing, J., Gene, 19(3): 259-68 (1982)).
Для этой цели большой фрагмент EcoRI-HindIII плазмиды pMW118-attL-Tc-attR лигировали с двумя фрагментами плазмиды pUC4K: фрагментом HindIII-PstI (676 п.о.) и фрагментом EcoRI-HindIII (585 п.о.).
Основную плазмиду pMW118-attL-Tc-attR получали путем лигирования следующих четырех ДНК-фрагментов:
1) фрагмент BglII-EcoRI (114 п.о.), несущий сайт attL (SEQ ID NO: 29), который получали методом ПЦР амплификации соответствующей области хромосомы штамма Е. coli W3350 (содержащего профаг λ) с использованием праймеров Р1 и Р2 (SEQ ID NOS: 30 и 31). Эти праймеры содержали дополнительные сайты узнавания для эндонуклеаз BglII и EcoRI;
2) фрагмент PstI-HindIII (182 п.о.), несущий сайт attR (SEQ ID NO: 32), который получали методом ПЦР-амплификации соответствующей области хромосомы Е. coli W3350 (содержащей профаг λ) с использованием праймеров Р3 и Р4 (SEQ ID NOS: 33 и 34). Эти праймеры содержали дополнительные сайты узнавания для эндонуклеаз PstI и HindIII;
3) большой фрагмент BglII-HindIII (3916 п.о.) из плазмиды pMW118-ter_rrnB. Плазмиду pMW118-ter_rrnB получали лигированием нижеследующих трех фрагментов ДНК:
- большой фрагмент ДНК (2359 п.о,), несущий фрагмент AatII-EcoRI плазмиды pMW118, который получали следующим образом: плазмиду pMW118 расщепляли рестрикционной эндонуклеазой EcoRI, обрабатывали фрагментом Klenow ДНК-полимеразы I и расщепляли рестрикционной эндонуклеазой AatII;
- маленький фрагмент AatII-BglII (1194 п.о.) плазмиды pUC19, несущий ген bla для устойчивости к ампицилину (ApR) получали методом ПЦР-амплификации соответствующей области плазмиды pUC19 с использованием праймеров Р5 и Р6 (SEQ ID NOS: 35 и 36). Эти праймеры содержали дополнительные сайты узнавания эндонуклеазами AatII и BglII;
- маленький фрагмент BglII-PstIpol (363 п.о.) терминатора транскрипции ter_rrnB получали методом ПЦР-амплификации соответствующей области хромосомы Е. coli MG1655 с использованием праймеров Р7 и Р8 (SEQ ID NOS: 37 и 38). Эти праймеры содержали дополнительные сайты узнавания для эндонуклеаз BglII и PstI;
4) маленький фрагмент EcoRI-PstI (1388 п.о.) (SEQ ID NO: 39) плазмиды pML-Tc-ter_thrL, несущий ген устойчивости к тетрациклину и терминатор транскрипции; плазмиду pML-Tc-ter_thrL получали в два этапа:
- плазмиду pML-ter_thrL получали путем расщепления плазмиды pML-MCS (Mashko, S.V. et al., Biotekhnologiya (in Russian), 2001, no. 5, 3-20) эндонуклеазами рестрикции XbaI и BamHI с последующим лигированием большого фрагмента (3342 п.о.) с фрагментом XbaI-BamHI (68 п.о.), который содержит терминатор ter_thrL, полученный методом ПЦР-амплификации соответствующей области хромосомы Е. coli MG1655 с использованием праймеров Р9 и Р10 (SEQ ID NOS: 40 и 41), Эти праймеры содержали дополнительные сайты узнавания для эндонуклеаз XbaI и BamHI;
- плазмиду pML-Tc-ter_thrL получали путем расщепления плазмиды pML-ter_thrL рестрикционными эндонуклеазами KpnI и XbaI с последующей обработкой фрагментом Klenow ДНК-полимеразы I и лигированием с маленьким фрагментом EcoRI-Van91I (1317 п.о.) плазмиды pBR322, несущей ген устойчивости к тетрациклину (pBR322 расщепляли рестрикционными эндонуклеазами EcoRI и Van91I и затем обрабатывали фрагментом Klenow ДНК-полимеразы I).
Пример 4. Использование библиотеки рандомизированных промоторов.
С использованием процедуры, описанной в Примере 2, возможно получение в одну стадию библиотеки клонов с различными уровнями экспрессии интересующих генов.
С другой стороны, библиотека созданных промоторов может быть использована для конструирования набора штаммов с различными уровнями экспрессии интересующих генов, для которых соотношение между указанными уровнями экспрессии может быть приблизительно рассчитано a priori.
Для подтверждения указанного заявления несколько промоторов из библиотеки созданных промоторов интегрировали в хромосому штамма Р. ananatis SC17(0) перед геном dadX (SEQ ID NO: 64), кодирующим аланинрацемазу. Для этой цели из штаммов AJ13601Ptac11SDyhfK, AJ13601Ptac52SDyhfK и AJ13601PtacSDyhfK (см. Пример 3) выделяли геномную ДНК с использованием Genomic DNA Isolation Kit ("Sigma", USA). Полученные образцы ДНК использовали в качестве матриц для ПЦР с праймерами PdadA (SEQ ID NO: 65) и PdadX (SEQ ID NO: 66) для получения in vitro интегративной кассеты, содержащей Ptac-подобный промотор, соединенный с вырезаемым маркером устойчивости к канамицину и терминатором транскрипции Tthr. Кроме того, ген gntK (SEQ ID NO: 67), кодирующий глюконаткиназу, модифицировали подобным образом. В этом случае, в качестве матриц для ПЦР использовали хромосомные ДНК, выделенные из клонов, содержащих промоторы Ptac, Ptac49 и Ptac11, из библиотеки промоторов, и праймеры attL-gntK (SEQ ID NO: 68) и PtacN-gntK (SEQ ID NO: 69) для получения in vitro интегративной кассеты, содержащей Ptac-подобный промотор, соединенный с вырезаемым маркером устойчивости к канамицину. Указанные олигонуклеотиды содержат необходимые для интеграции участки, гомологичные нужным участкам гена dadX или gntK на их 5'-концах, и участки, гомологичные концевым участкам амплифицированного фрагмента ДНК на их 3'-концах. Полученные таким образом интегративные кассеты интегрировали в хромосому штамма SC17(0)/RSFRedTER с помощью процедуры, описанной выше.
Полученные в хромосоме структуры приведены на Фиг.5. Подтверждение правильности конструкции в штаммах проводили методом ПЦР с использованием праймеров dadX-t1 (SEQ ID NO: 70), dadX-t2 (SEQ ID NO: 71) и gntR (SEQ ID NO: 72), gntK (SEQ ID NO: 73) соответственно. В сконструированных штаммах оценивали специфические активности аланинрацемазы или глюконаткиназы. Активность аланинрацемазы оценивали по устойчивости к D-циклосерину на чашках с LB-агаром (Peter, A., Bron, Appl. Environ. Microbiol., Vol.68, No. 11, Nov. 2002, p.5663-5670).
Специфическую активность глюконаткиназы оценивали спектрофотометрически при 340 нм по появлению НАДФ-Н в связанной реакции с 6-фосфоглюконатдегидрогеназой. Для этого штаммы выращивали на минимальной среде М9 при рН=6.0 с добавлением глюкозы (5 г/л) до конечной оптической плотности А595=1.0. Клетки отмывали один раз 1 мл 50 мМ буфера Трис-HCl (рН 7.5) и ресуспендировали в растворе, содержащем 50 мМ буфера Трис-HCl (рН 7.5), 2 мМ DTT, 2 мМ MgCl2. Клетки разрушали с помощью ультразвука. Раствор для проверки активности содержал 50 мМ буфера Трис-HCl (рН 8.0), 4 мМ НАДФ, 2 мМ MgCl2, 4 мМ АТФ, 4 мМ глюконата и 2 ед./мл 6-фосфоглюконатдегидрогеназы в качестве фермента для связанной реакции. Специфическую активность определяли исходя из молярного коэффициента экстинкции НАДФ-Н, равного 6.2·103.
Как показано в Таблице 3, в конструированных штаммах наблюдалась явная корреляция между относительными активностями ферментов, определяемых как соотношение специфической активности, которой обладал штамм, содержащий один из Ptac-подобных промоторов, к специфической активности, которой обладал штамм, содержащий промотор Ptac перед соответствующим геном.
Пример 5. Использование комбинированной системы рекомбинации λRed-λInt/Xis для введения в хромосому множественных модификаций.
Проверяли возможность повторной интеграции различных кассет, фланкированных сайтами attL и attR фага λ, в хромосому Р. ananatis с использованием Red-зависимой рекомбинации линейных фрагментов с плечами гомологии длиной около 40 п.н. С этой целью несколько генов на хромосоме заменяли на ген устойчивости к тетрациклину. Использованные в экспериментах праймеры представлены в Таблице 4.
Представленные в Таблице 5 результаты свидетельствуют о том, что в неселективных условиях частота повторной интеграции в ту же точку (когда att-сайты размером 120 и 150 п.н. служат плечами гомологии) в 10-30 раз выше, чем в требуемую точку интеграции (с использованием плеч гомологии 36-40 п.н.). Но все равно возможно отобрать клоны, содержащие две модификации. В случае электропорации хромосомы (метод описан в Примере 3) все повторные интеграции кассеты, фланкированной att сайтами фага λ, имели место в требуемой точке.
Пример 6. Процедура двухстадийной λ Red-зависимой рекомбинации для введения немаркированной точечной мутации в хромосому Р. ananatis.
Была разработана процедура двухстадийной λ Red-зависимой интеграции для введения немаркированной точечной мутации в хромосому Р. ananatis. Процедура включает следующие стадии: а) введение в бактерию, устойчивую к продуктам генов фага λ gam, bet и ехо, фрагмента экзогенной линейной ДНК, который включает двойной селективный/контраселективный маркер, фланкированный 2 сайтами длиной минимум 30 п.н., идентичными сайтам, локализованным рядом с требуемой точкой на хромосоме бактерии; б) замена с использованием λ Red-зависимой интеграции маркера коротким фрагментом ДНК, содержащим требуемую точечную мутацию, фланкированную сайтами, идентичными сайтам, локализованным рядом с требуемой точкой на хромосоме; и в) отбор бактерии с немаркированной точечной мутацией с использованием контрселекции.
Любой подходящий маркер устойчивости к антибиотику может быть использован в качестве селективного маркера в рамках способа согласно настоящему изобретению. Например, маркеры устойчивости к хлорамфениколу, ампициллину, канамицину, тетрациклину, стрептомицину и подобные им.
В качестве контрселективного маркера (маркера негативной селекции) могут быть использованы ген sacB из В. subtilis, кодирующий левансукразу, экспрессия которой является летальной для бактерии, содержащей этот ген и растущей на питательной среде, содержащей сахарозу (Gay, P. et al., J. Bacteriol., 164, 918-921 (1985)); ген tetA, позволяющий проводить контрселекцию в присутствии фузаровой кислоты (Bochner, B.R. et al., J. Bacteriol., 143, 926-933 (1980)); ген rpsL, придающий чувствительность к стрептомицину (Russel, С. В. and Dahlquist, F.W., J. Bacteriol., 171, 2614-2618 (1989)) и подобные им.
Чтобы проиллюстрировать процедуру введения точечной мутации, осуществляли замену нативного сайта узнавания SmaI, локализованного внутри гена hisD бактерии Р. ananatis, сайтом узнавания XhoI.
Конструирование двойного селективного/контраселективного маркера.
Плазмиду RSFsacB, описанную Примере-ссылке 1, обрабатывали рестрикционной эндонуклеазой XbaI и лигировали при низкой концентрации ДНК (1 нг/мкл). Затем смесью, полученной после лигирования, трансформировали штамм Е. coli TG1. После выделения и рестрикционного анализа плазмидных ДНК из CmR клонов, выросших после трансформации, была отобрана плазмида RSFPlacsacB с отсутствующим геном lacI (Фиг.6).
Введение маркера PlacUV5-sacB-cat в хромосому Р. ananatis.
Плазмиду RSFPlacsacB использовали в качестве матрицы для ПЦР-амплификации кассеты PlacUV5-sacB-cat. Для этой реакции использовали праймеры HisD-Plac-5 (SEQ ID NO: 90) и HisD-cat-3 (SEQ ID NO: 91). Праймеры содержали сайты, гомологичные областям хромосомы, фланкирующим сайт узнавания SmaI, локализованный внутри гена hisD бактерии Р. ananalis на его 5'-конце.
Полученный в этой реакции фрагмент ДНК использовали для электропорации в штамм SC17(0)/RSFRedkan как описано выше (условия см. в Примере 1). После электропорации клетки высевали на чашки в LB-агаром с добавлением хлорамфеникола (20 мг/л). Для проверки выросших интегрантов проводили ПЦР с использованием праймеров HisD-L5 (SEQ ID NO: 62) и HisD-R3 (SEQ ID NO: 63) (условия см. в Примере 1).
Замена маркера коротким фрагментом ДНК, содержащим требуемую мутацию с использованием λ Red-зависимой интеграции.
Короткий, полученный с использованием ПЦР фрагмент ДНК, содержащий требуемую мутацию, получали следующим образом. Сначала гибридизовали два комплементарных праймера, XhoI-F (SEQ ID NO: 92) и XhoI-Rev (SEQ ID NO: 93). В результате получали короткий двухцепочечный фрагмент ДНК, содержащий в центре требуемую мутацию (замена сайта узнавания SmaI на XhoI). Для того чтобы увеличить фланговые области, гомологичные хромосоме, проводили ПЦР-амплификацию полученного фрагмента с использованием праймеров His-SL (SEQ ID NO: 94) и His-SR (SEQ ID NO: 95). Полученный фрагмент ДНК содержал в центре сайт узнавания XhoI, фланкированный участками длиной 83 п.н., гомологичными соответствующим участкам на хромосоме Р. ananatis.
Указанный фрагмент ДНК использовали для электропорации в описанный выше штамм SC17(0)hisD::PlacUV5-sacB-cat со вспомогательной плазмидой RSFRedkan. Клетки для электропорации культивировали в 10 мл среды LB с добавлением 40 мг/л канамицина и 1 мМ ИПТГ до достижения OD595=0.5. Клетки собирали с использованием центрифугирования, трижды отмывали ледяной водой и один раз - 10% глицерином и ресуспендировали в 50 мкл 10% глицерина. После добавления 100 нг фрагмента интегративной ДНК осуществляли электропорацию (Е=20 кВ/см, время импульса 5 мс). После электропорации клетки высевали на чашки с LB-агаром, содержащим 20% сахарозы. Проверяли 100 выросших устойчивых к сахарозе клонов. Около 25% проверенных клонов имели фенотип SucRCnSHis+. Из этих клонов выделяли геномную ДНК и амплифицировали фрагмент хромосомы, содержащий в центре ожидаемую мутацию, с помощью ПЦР с использованием праймеров HisD-L5 и HisD-R3. Рестрикционный анализ амплифицированных фрагментов показал присутствие требуемой мутации (замена сайта узнавания SmaI на XhoI) во всех отобранных клонах.
Хотя указанное изобретение описано в деталях со ссылкой на наилучший способ осуществления изобретения, для специалиста в указанной области техники очевидно, что могут быть совершены различные изменения и произведены эквивалентные замены, и такие изменения и замены не выходят за рамки настоящего изобретения.
Каждому из упомянутых выше документов соответствует ссылка, и все цитируемые документы являются частью описания настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ L-ЦИСТЕИНА С ИСПОЛЬЗОВАНИЕМ БАКТЕРИИ СЕМЕЙСТВА ENTEROBACTERIACEAE | 2010 |
|
RU2458981C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-АМИНОКИСЛОТ С ИСПОЛЬЗОВАНИЕМ БАКТЕРИЙ, ПРИНАДЛЕЖАЩИХ К РОДУ ESCHERICHIA | 2005 |
|
RU2304615C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-АМИНОКИСЛОТ С ИСПОЛЬЗОВАНИЕМ БАКТЕРИЙ СЕМЕЙСТВА ENTEROBACTERIACEAE | 2010 |
|
RU2460793C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-ЦИСТЕИНА, L-ЦИСТИНА, S-СУЛЬФОЦИСТЕИНА ИЛИ ТИАЗОЛИДИНОВОГО ПРОИЗВОДНОГО L-ЦИСТЕИНА, ИЛИ ИХ СМЕСИ С ИСПОЛЬЗОВАНИЕМ БАКТЕРИИ СЕМЕЙСТВА ENTEROBACTERIACEAE | 2010 |
|
RU2458982C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-ТРЕОНИНА И L-АРГИНИНА С ИСПОЛЬЗОВАНИЕМ БАКТЕРИИ, ПРИНАДЛЕЖАЩЕЙ К РОДУ ESCHERICHIA, В КОТОРОЙ ИНАКТИВИРОВАН ГЕН cpxR | 2005 |
|
RU2320723C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-ТРЕОНИНА И L-АРГИНИНА С ИСПОЛЬЗОВАНИЕМ БАКТЕРИИ, ПРИНАДЛЕЖАЩЕЙ К РОДУ ESCHERICHIA, В КОТОРОЙ ИНАКТИВИРОВАН ГЕН ybiV | 2005 |
|
RU2320719C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-АМИНОКИСЛОТ С ИСПОЛЬЗОВАНИЕМ БАКТЕРИИ, ПРИНАДЛЕЖАЩЕЙ К РОДУ ESCHERICHIA | 2005 |
|
RU2311454C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-АМИНОКИСЛОТЫ СЕМЕЙСТВА ГЛУТАМАТА ИЛИ L-ВАЛИНА С ИСПОЛЬЗОВАНИЕМ БАКТЕРИИ, ПРИНАДЛЕЖАЩЕЙ К РОДУ Escherichia | 2009 |
|
RU2418064C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-АМИНОКИСЛОТ С ИСПОЛЬЗОВАНИЕМ БАКТЕРИИ СЕМЕЙСТВА ENTEROBACTERIACEAE | 2007 |
|
RU2364628C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-ТРЕОНИНА С ИСПОЛЬЗОВАНИЕМ БАКТЕРИИ, ПРИНАДЛЕЖАЩЕЙ К РОДУ ESCHERICHIA, В КОТОРОЙ ИНАКТИВИРОВАН ГЕН hipA | 2005 |
|
RU2320718C2 |


































Изобретение относится к области биотехнологии. Изобретение раскрывает способ конструирования рекомбинантной бактерии, принадлежащей к роду Pantoea, способ конструирования рекомбинантной бактерии, принадлежащей к роду Pantoea - продуцента L-аминокислоты, бактерию, принадлежащую к роду Pantoea, полученную указанным способом, а также способ получения L-аминокислоты с использованием указанной бактерии. Изобретение позволяет повысить продуцирование L-аминокислот. 5 н. и 7 з.п. ф-лы, 7 ил., 5 табл.
1. Способ конструирования рекомбинантной бактерии, принадлежащей к роду Pantoea, включающий:
a) получение экзогенного линейного фрагмента ДНК, который имеет в своем составе, по крайней мере, 2 участка минимум по 30 п.о. в длину каждый, идентичных участкам, расположенным в целевом участке на хромосоме указанной бактерии,
b) введение указанного линейного фрагмента ДНК в указанную бактерию, при этом указанная бактерия модифицирована таким образом, что является устойчивой к продуктам генов gam, bet, and exo из фага лямбда, и
c) отбор рекомбинантной бактерии, в которой целевой участок заменен на указанный линейный фрагмент ДНК или линейный фрагмент ДНК встроен в целевой участок.
2. Способ по п.1, отличающийся тем, что указанная бактерия является бактерией Pantoea ananatis SC17(0).
3. Способ по п.1, отличающийся тем, что указанный целевой участок в рекомбинантной бактерии модифицирован за счет инактивации или удаления.
4. Способ конструирования бактерии, принадлежащей к роду Pantoea, содержащей библиотеку фрагментов ДНК, включающий:
а) получение набора экзогенных фрагментов ДНК, каждый из которых имеет в своем составе, по крайней мере, 2 участка минимум по 30 п.о. в длину каждый, идентичных участкам, расположенным в целевом участке на хромосоме указанной бактерии,
b) введение указанного набора линейных фрагментов ДНК в указанную бактерию, при этом указанная бактерия модифицирована таким образом, что является устойчивой к продуктам генов gam, bet и ехо из фага лямбда, и
b) отбор рекомбинантных бактерий, в которых целевой участок заменен на указанный линейный фрагмент ДНК или линейный фрагмент ДНК встроен в целевой участок.
5. Способ по п.4, отличающийся тем, что указанные фрагменты ДНК содержат регуляторный элемент для экспрессии гена, выбранный из группы, состоящей из промотора, сайта связывания рибосомы (RBS), оператора и их сочетания.
6. Способ по п.5, отличающийся тем, что указанный регуляторный элемент содержит в своем составе рандомизированную последовательность.
7. Способ конструирования рекомбинантной бактерии, принадлежащей к роду Pantoea, - продуцента L-аминокислоты, включающий:
a) замену природных регуляторных элементов целевого участка на экзогенные фрагменты ДНК способом по п.1 и
b) отбор бактерии с оптимизированным уровнем экспрессии гена в целевом участке хромосомы бактерии, отличающийся тем, что указанный ген кодирует белок, который участвует в биосинтезе и метаболизме L-аминокислоты или в выведении L-аминокислоты из указанной бактерии.
8. Способ по п.7, отличающийся тем, что указанная L-аминокислота выбрана из группы, состоящей из L-аланина, L-аргинина, L-аспарагина, L-аспарагиновой кислоты, L-цистеина, L-глутаминовой кислоты, L-глутамина, L-глицина, L-гистидина, L-изолейцина, L-лейцина, L-лизина, L-метионина, L-фенилаланина, L-пролина, L-серина, L-треонина, L-триптофана, L-тирозина, и L-валина.
9. Бактерия, принадлежащая к роду Pantoea и полученная способом по п.7, - продуцент L-аминокислоты, выбранной из группы, состоящей из L-глутаминовой кислоты, L-лейцина, L-лизина, L-изолейцина, L-валина, L-гистидина, L-аспаратата, L-аланина, L-тирозина и L-фенилаланина.
10. Бактерия по п.9, отличающаяся тем, что указанные природные регуляторные элементы содержат в целевом участке фрагмент размером в 35 пар оснований из промотора гена, выбранный из группы, состоящей из SEQ ID NO: 16, SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 23, SEQ ID NO; 24, SEQ ID NO: 25, SEQ ID NO: 26, SEQ ID NO: 27, SEQ ID NO: 28 и их сочетания.
11. Бактерия по п.10, отличающаяся тем, что указанным геном является ген yhfK.
12. Способ получения L-аминокислоты, выбранной из группы, состоящей из L-глутаминовой кислоты, L-лейцина, L-лизина, L-изолейцина, L-валина, L-гистидина, L-аспаратата, L-аланина, L-тирозина и L-фенилаланина включающий:
a) выращивание бактерии по п.9 в питательной среде и
b) выделение указанной L-аминокислоты из культуральной жидкости.
| US 6596517 В2, 22.07.2003 | |||
| MURPHY K.C | |||
| Use of Bacteriophage λ Recombination Functions to Promote Gene Replacement in Escherichia coli | |||
| Journal of Bacteriology, 1998 April, Vol.180 (8), pp.2063-2071 | |||
| POTEETE A.R | |||
| What makes the bacteriophage lambda Red system useful for genetic engineering: molecular mechanism and biological function | |||
| FEMS Microbiol | |||
| Lett., 2001 July, Vol.201 (1), pp.9-14 | |||
| СПОСОБ ПОЛУЧЕНИЯ L-ГЛУТАМИНОВОЙ КИСЛОТЫ | 1999 |
|
RU2188236C2 |

