Настоящая заявка согласно §119(е) Раздела 35 Кодекса законов США утверждает преимущество Предварительной Патентной Заявки № 60/852380, поданной 17 октября 2006 года, которая приведена здесь для сведения.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к твердым кислотным катализаторам для применения в процессах алкилирования. Твердый кислотный катализатор включает мультиметаллический (например, биметаллический, триметаллический или тетраметаллический) компонент, который производит гидрирующее действие для реактивации (или регенерации) катализатора в присутствии водорода. Изобретение также относится к процессам алкилирования с использованием твердого кислотного катализатора, имеющего мультиметаллический компонент для гидрирования.
УРОВЕНЬ ТЕХНИКИ
«Алкилирование» в общем имеет отношение к реакции углеводорода, такого как ароматический или насыщенный углеводород, с олефином. Например, в одном типе реакции, представляющей особенный интерес, разветвленный насыщенный углеводород, такой как изобутан, может подвергаться алкилированию олефином, содержащим 2-6 атомов углерода, таким как 2-бутен, с образованием алкилата, который имеет более высокое октановое число и который кипит в температурном интервале кипения бензиновой фракции. В процессах, направленных на алкилирование парафинов олефинами, получаются молекулы разветвленных углеводородов для бензиновых компонентов, таких как изомеры октана, например триметилпентаны (“TMP”), которые имеют высокие значения октанового числа. Бензин с высоким октановым числом, часто выражаемым как октановое число бензина по исследовательскому методу (“RON”), может уменьшать детонацию в двигателе, чем сокращается необходимость добавления вредных для окружающей среды антидетонационных соединений, таких как тетраэтилсвинец. Второй октановый показатель, моторное октановое число («ОЧМ», “MON”), также описывает антидетонационные свойства бензина. Моторное октановое число (MON) измеряют, когда испытуемый двигатель работает с высокой нагрузкой (на высоких оборотах), и октановое число бензина по исследовательскому методу (RON) измеряют при низкой нагрузке (на малых оборотах).
Бензин, полученный в процессе алкилирования, по существу не содержит загрязняющих примесей, таких как сера и азот, которые могут присутствовать в бензине, производимом другими способами, такими как крекинг тяжелых нефтяных фракций, например, вакуумном газойле и продуктах атмосферной отгонки. Оксиды серы (“SOx”), продукт сгорания, представляют собой первостепенную причину загрязнений. В дополнение к прямым выбросам оксидов серы (SOx) оксиды серы (SOx) могут существенно снижать работоспособность каталитических конвертеров, тем самым ухудшая эффективность поглощения выбросов оксидов серы (SOx), оксидов азота (NOx) и монооксида углерода (СО). Оксиды серы (SOx) также участвуют в формировании косвенных частиц загрязнений - комбинации воды и оксидов серы (SOx) с образованием сернистой и серной кислот. Эти косвенные частицы загрязнений, которые обычно существуют в 1-10-микронном диапазоне, представляют собой «вдыхаемые частицы», которые оказывают вредное воздействие на здоровье, особенно тех людей, которые страдают астмой или эмфиземой. Кроме того, в отличие от бензина, получаемого риформингом нафты или крекингом тяжелых нефтяных фракций, алкилат содержит мало ароматических соединений или олефинов или совсем их не содержит. Ароматические соединения, в особенности бензол, токсичны, и олефины являются реакционноспособными в фотохимических реакциях, которые формируют озон и смог.
Реакция алкилирования является кислотнокатализируемой. В процессах алкилирования обычно использовались жидкие кислотные катализаторы, такие как серная кислота или фтористоводородная (плавиковая) кислота. Применение жидких кислотных катализаторов имеет несколько недостатков. Использованные жидкие кислоты являются высококоррозивными, требуя применения более дорогостоящего оборудования специального качества. Поскольку присутствие этих кислот в конечном топливе нежелательно, любая кислота, остающаяся в алкилате, должна быть удалена. Этот процесс является сложным и дорогостоящим. В дополнение жидкие кислоты, в особенности фтористоводородная (плавиковая) кислота, опасны при попадании в окружающую среду.
Чтобы устранить эти и прочие недостатки жидких кислотных катализаторов, для применения в процессах алкилирования были разработаны твердые кислотные катализаторы. В твердых катализаторах обычно используют твердый кислотный катализатор и металл, который выполняет функцию гидрирования. Например, Патент США № 6855856 описывает катализатор, включающий твердую кислоту, такую как цеолит, и исполнение функции гидрирования. Описанная твердая кислота имеет определенный диапазон для отношения объема пор в катализаторе к удельной длине каталитических частиц.
Недостаток твердых кислотных катализаторов, которые используются в прототипах, состоит в том, что катализатор может становиться быстро дезактивируемым вследствие образования полиалкилатов (например, продуктов С12+), которые ингибируют реакции алкилирования - в некотором отношении подобно очень мягкому коксу. Как только катализатор сформирует определенный уровень содержания полиалкилатов, катализатор по существу прекращает реакции алкилирования. В реакторе с неподвижным слоем, зачастую предпочтительной конструкции, можно наблюдать дезактивирование, которое происходит как полосовидное старение, с зоной дезактивации, перемещающейся в виде полосы через слой до тех пор, пока бόльшая часть слоя не дезактивируется. Эта дезактивация катализатора приводит к необходимости периодической регенерации катализатора для обеспечения удовлетворительного выхода желаемого продукта, производимого в процессе. Регенерация катализатора обычно требует остановки процесса алкилирования на некоторый период времени. Это снижает производительность и повышает затраты в процессе алкилирования, в особенности вследствие снижения «эксплуатационного» фактора процесса.
Предпочтительным способом регенерации катализатора является гидрирование. Гидрирующее действие обычно обеспечивается металлом Группы VIII Периодической таблицы элементов, в частности благородными металлами, такими как платина (Pt) или палладий (Pd). В отличие от классического бифункционального (металл/кислота) катализатора гидрирующее действие играет малую или косвенную роль в самих реакциях алкилирования. Напротив, оно играет решающую роль в эффективной реактивации водородом (Н2) (также называемой здесь «регенерацией») дезактивированного катализатора. Гидрирующее действие важно в обеих описанных ниже регенерациях, как для так называемой низкотемпературной (“low T”), так и высокотемпературной (“high T”) регенерации.
Предпринимались разнообразные попытки разработать усовершенствованные твердые кислотные катализаторы. Например, Патентная Публикация США № 2004/0162454 описывает катализатор алкилирования, включающий нанокристаллический цеолит Y и металл для гидрирования. Размер пор нанокристаллического цеолита Y обеспечивает получение алкилата с более высокими значениями октанового числа бензина по исследовательскому методу и моторного октанового числа (RON/MON), а также более продолжительный срок действия катализатора. Катализатор на основе нанокристаллического цеолита Y также включает металл Группы VIII Периодической таблицы элементов, такой как платина (Pt) или палладий (Pd), для исполнения функции гидрирования.
Для повышения эффективности и производительности процесса алкилирования с использованием твердых кислотных катализаторов были разработаны разнообразные методы усовершенствования процесса регенерации твердых кислотных катализаторов. Например, Патент США № 7176340 описывает непрерывный процесс алкилирования с использованием в целом по меньшей мере четырех реакторов, содержащих катализатор. Однако применение нескольких реакторов повышает стоимость процесса; это возрастание стоимости может быть компенсировано, по меньшей мере отчасти, повышением общей эффективности всего процесса в целом. Патент США № 5986158 описывает процесс алкилирования, в котором катализатор периодически подвергают обработке в стадии регенерации путем контактирования с сырьевым материалом, содержащим насыщенный углеводород и водород, причем регенерацию проводят при 90%-ном или менее протекании активного каталитического цикла. В то время как эти способы регенерации улучшают общую эффективность процесса алкилирования, относительно большие количества нужных для этого твердых кислотных катализаторов и связанных с этих благородных металлов могут составить проблему, которая делает процесс алкилирования экономически неприемлемым.
Было бы желательным иметь твердый кислотный катализатор для процесса алкилирования, который обеспечивает длительный срок службы до наступления дезактивации. Также было бы желательным иметь твердый кислотный катализатор, в котором используется металл для выполнения гидрирования, который производит равное или улучшенное действие по сравнению с платиной (Pt) или палладием (Pd) и который может быть доступен по низкой цене. Настоящее изобретение преодолевает один или более этих и прочих недостатков или затруднений, вызванных использованием твердых кислотных катализаторов, в процессах алкилирования согласно прототипам.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на твердые кислотные катализаторы для алкилирования парафинов олефинами и на применение твердых кислотных катализаторов в процессе алкилирования. Твердый кислотный катализатор включает цеолит и гидрирующее действие. Цеолит может быть любым цеолитом, известным квалифицированным специалистам в этой области техники для использования в твердых кислотных катализаторах для процессов алкилирования. В предпочтительном варианте осуществления в твердом кислотном катализаторе могут быть применены цеолиты, имеющие структуру фоязита (фожазита). Расширенное семейство фоязитов может включать сорта X, Y, ZSM-20 и ЕМТ. В целях иллюстрирования настоящего изобретения описание предпочтительных вариантов осуществления будет сосредоточено на цеолите Y (включая ультрастабильный Y - “USY”, то есть имеющий размер элементарной ячейки 24,50 Å или менее (24,5×10-10 м), цеолите Х и комбинациях цеолитов Х и Y.
Гидрирующее действие часто предпочтительно обеспечивается биметаллическим или триметаллическим компонентом. Традиционно биметаллический или триметаллический компонент должен включать разнообразные благородные металлы, например платину (Pt) или палладий (Pd), в таких комбинациях, как, например, PtNi, PtCo, PtAg, PtAu, PtPdNi, PtPdAg, PtPdAu, PdNi, PdAg и PdAu. Использовались также комбинации платины (Pt) или палладия (Pd) с рутением (Ru), иридием (Ir), родием (Rh), медью (Cu) и рением (Re). В некоторых примерах могут использоваться тетраметаллические системы (например, PtPdAgAu или PtNiReIrAu).
Настоящее изобретение представляет собой самый настоящий отход от традиционного использования катализаторов на основе благородных металлов. В катализаторах согласно настоящему изобретению эффективную функцию гидрирования исполняет комбинация платины (Pt) и по меньшей мере одного 3d-металла (то есть никеля (Ni), кобальта (Co), марганца (Mn), хрома (Cr), ванадия (V), железа (Fe) или титана (Ti)). Авторы настоящего изобретения обнаружили, что эта синергическая комбинация может обеспечить превосходную регенерацию при более низких уровнях содержания платины (Pt) или палладия (Pd). Этот состав может снизить общую стоимость катализатора и упростить утилизацию отходов. В одном варианте осуществления новый катализатор может включать один или два 3d-металла вместе с одним или двумя благородными металлами при условии, что один благородный металл представляет собой платину (Pt) или палладий (Pd). В еще одном варианте осуществления катализатор включает платину (Pt) или палладий (Pd) и три 3d-металла.
Необязательно, твердый кислотный каталитический компонент может включать матричный материал, такой как оксид алюминия, оксид кремния, оксид кремния-алюминия, оксид циркония, глина или их комбинации.
Катализатор может быть использован в процессе алкилирования парафина олефином, таком как алкилирование изобутана бутиленом (предпочтительно 2-бутеном), с образованием бензинового продукта, имеющего высокие значения октанового числа бензина по исследовательскому методу (RON) и моторного октанового числа (MON).
Одно преимущество твердых кислотных катализаторов согласно настоящему изобретению состоит в том, что они включают мультиметаллические материалы, которые являются менее дорогостоящими, чем катализаторы, имеющие только платину (Pt) или палладий (Pd), в то же время обеспечивая эквивалентное или превосходящее регенерационное действие. Прочие преимущества настоящего изобретения будут очевидными квалифицированным специалистам в этой области техники из подробного описания предпочтительных вариантов осуществления, изложенного ниже.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
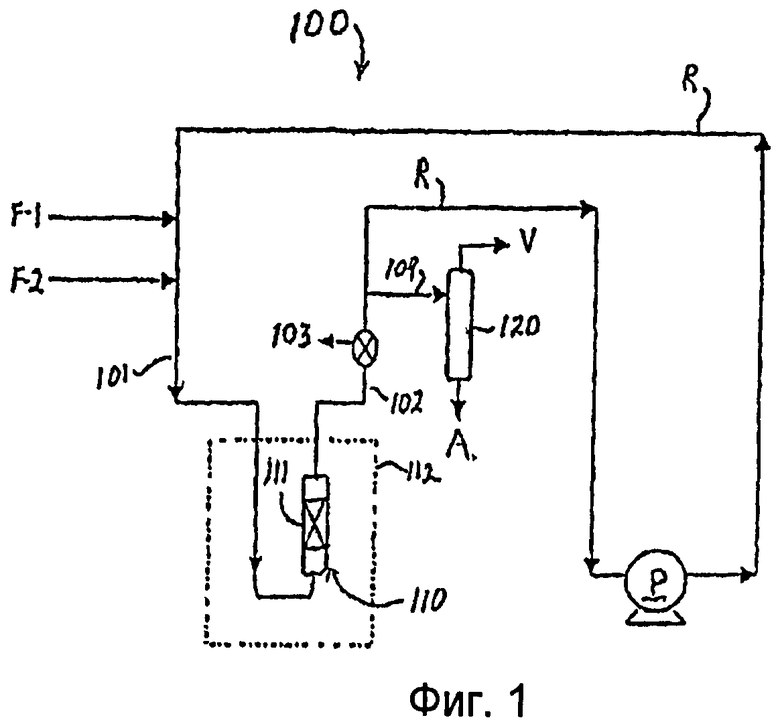
Фиг.1 представляет схематический чертеж реакторной системы, используемой в процессах, описанных в Примерах 8-14.
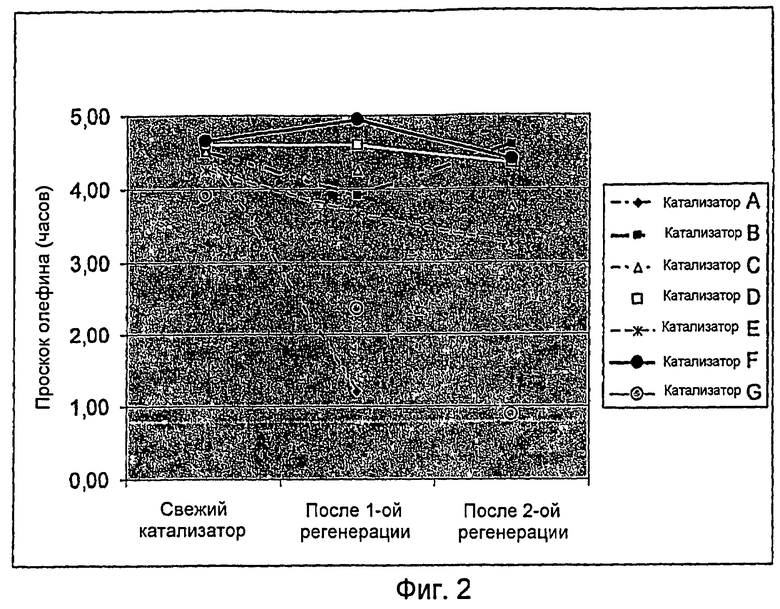
Фиг.2 представляет график, показывающий время проскока олефина для катализаторов из примеров 1-7, используемых в процессах Примеров 8-14.
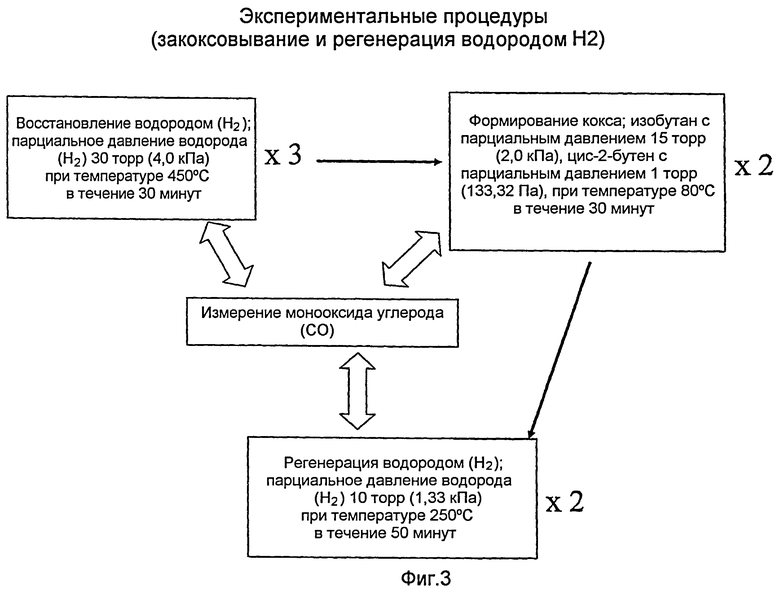
Фиг.3 представляет диаграмму, обобщающую экспериментальные методики, используемые в процедурах закоксовывания и регенерации водородом (Н2) в примерах.
Фиг.4 представляет таблицу, обобщающую характеристические полосы поглощения колебательных спектров кокса и прекурсоров кокса.
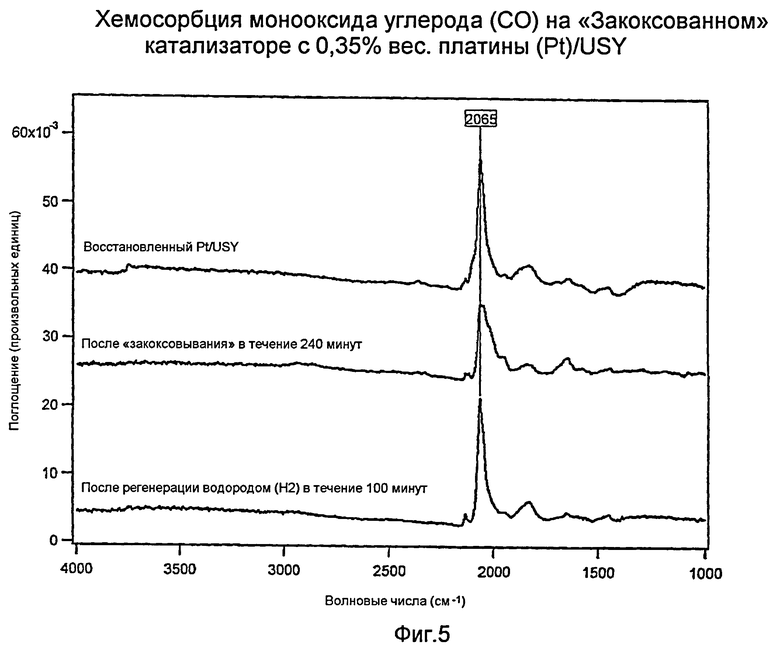
Фиг.5 представляет график хемосорбции оксида углерода (СО) на «закоксованном» USY-катализаторе (ультрастабильном Y-цеолите) с 0,35% вес. платины (Pt).
Фиг.6 представляет график, показывающий формирование «кокса» на USY-катализаторе с 0,35% вес. платины (Pt).
Фиг.7 представляет график, показывающий регенерацию водородом (Н2) USY-катализатора с 0,35% вес. платины (Pt).
Фиг.8 представляет график, показывающий формирование «кокса» на USY-катализаторе с 0,105% вес. никеля (Ni) и 0,35% вес. платины (Pt).
Фиг.9 представляет график, показывающий регенерацию водородом (Н2) USY-катализатора с 0,105% вес. никеля (Ni) и 0,35% вес. платины (Pt).
Фиг.10 представляет график, показывающий формирование «кокса» на USY-катализаторе с 0,105% вес. никеля (Ni) и 0,12% вес. платины (Pt).
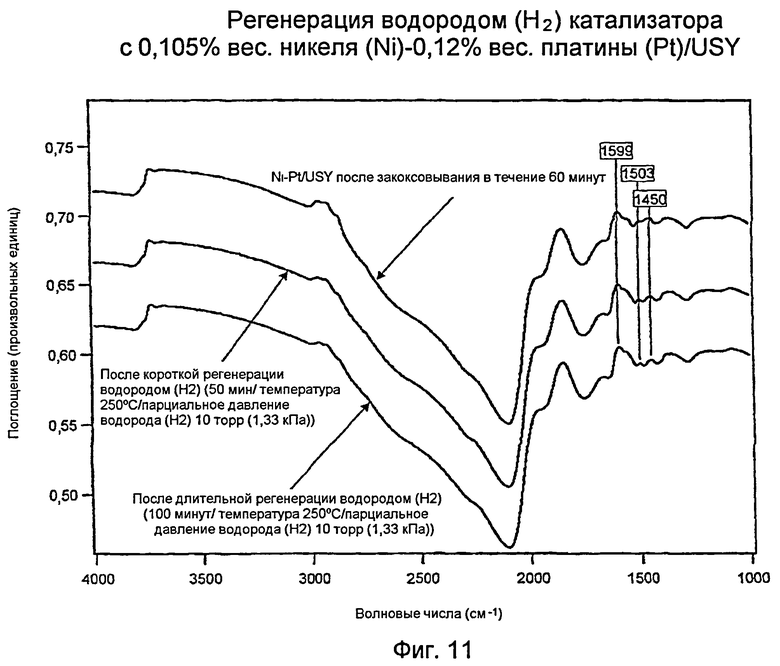
Фиг.11 представляет график, показывающий регенерацию водородом (Н2) USY-катализатора с 0,105% вес. никеля (Ni) и 0,12% вес. платины (Pt).
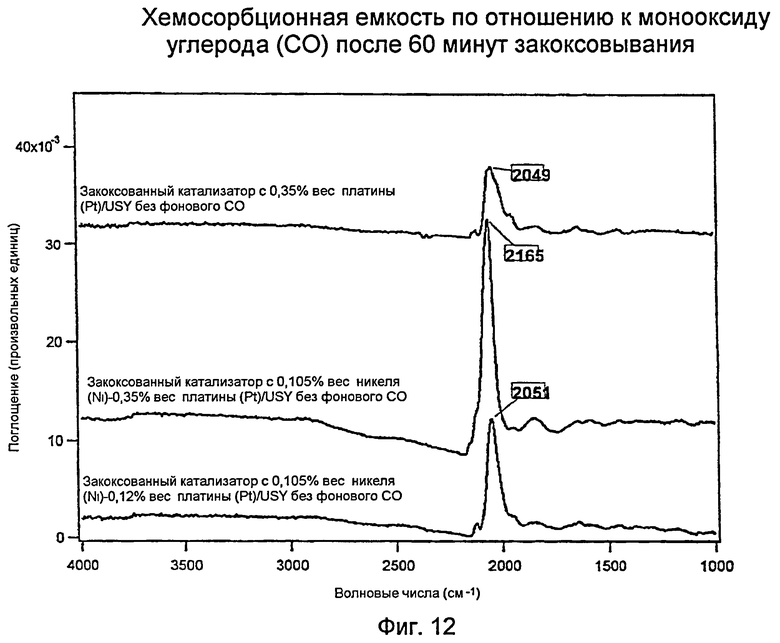
Фиг.12 представляет график, показывающий хемосорбционную емкость для монооксида углерода (СО) на различных катализаторах после 60-минутного закоксовывания.
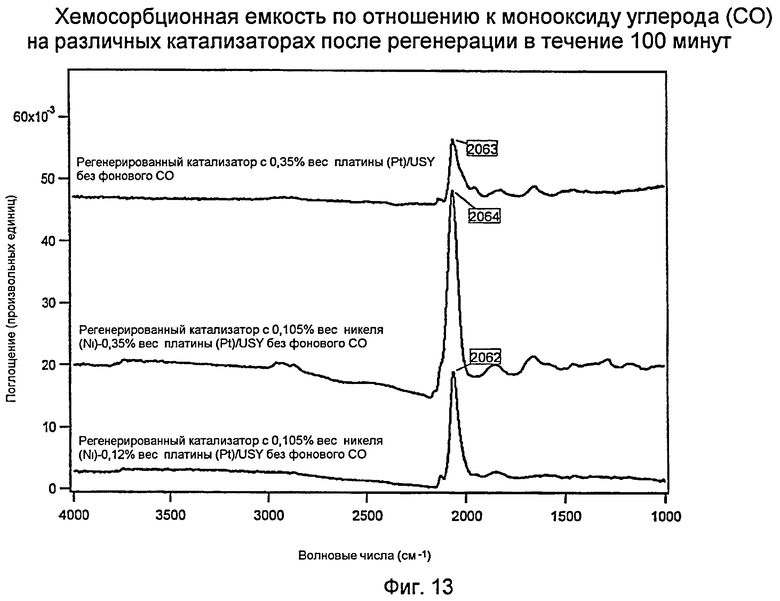
Фиг.13 представляет график, показывающий хемосорбционную емкость для монооксида углерода (СО) на различных катализаторах после 100-минутной регенерации.
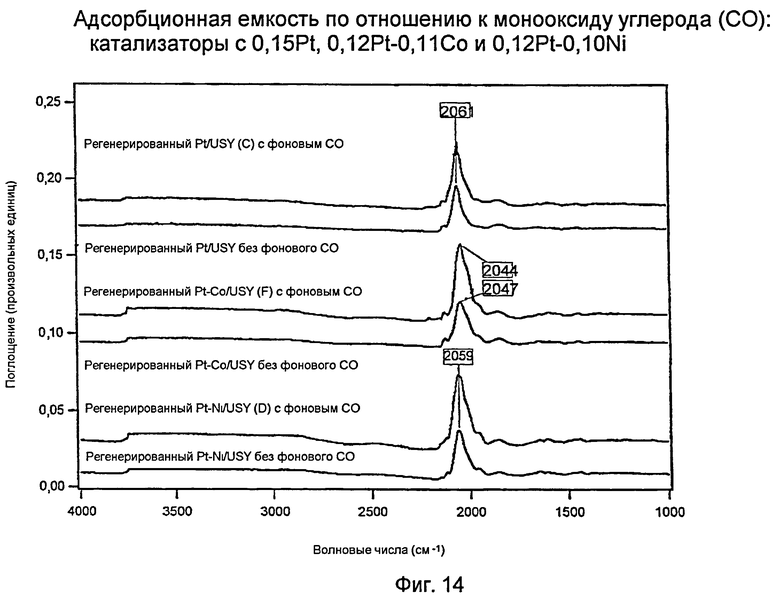
Фиг.14 представляет график, показывающий хемосорбционную емкость для монооксида углерода (СО) катализаторов с 0,15% вес. платины (Pt), 0,12% вес. платины (Pt) и 0,11% вес. кобальта (Co) и 0,12% вес. платины (Pt) и 0,10% вес. никеля (Ni).
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Настоящее изобретение относится к катализатору для применения в процессах алкилирования парафина (например, изобутана) олефином (например, 2-бутеном) для получения бензинового продукта. Катализатор включает твердый кислотный цеолит и гидрирующее действие. Катализатор также может включать матричный или связующий материал. Гидрирующее действие обеспечивается мультиметаллическим компонентом, предпочтительно биметаллическим или триметаллическим компонентом, который внедрен главным образом в структуру цеолита. Однако изобретение не ограничивается в этом отношении, и некоторая доля упомянутого гидрирующего действия может исполняться связующим материалом, а также наружной поверхностью цеолита. В нижеследующем описании предпочтительных вариантов осуществления, в общем, применяется термин «биметаллический» в описании катализатора, но следует понимать, что изобретение не ограничивается применением биметаллов и что любой мультиметаллический материал или комбинация мультиметаллических материалов, которые обеспечивают гидрирующее действие, могут быть введены в твердый кислотный катализатор.
В технике для использования в процессах алкилирования известно множество твердых кислотных цеолитов. В настоящем изобретении любой из этих цеолитов может быть использован в качестве твердого кислотного катализатора. Предпочтительным цеолитом для применения в настоящем изобретении является цеолит, имеющий структуру фоязита, такой как цеолит Y или цеолит Х. В особенности предпочтителен цеолит Y. В особенно предпочтительном варианте осуществления применяется нанокристаллический цеолит Y, например, такой как нанокристаллический цеолит Y, описанный в Патенте США № 6793911, и в Патентной Публикации Соединенных Штатов № 2004/0162454, которые приведены здесь для сведения.
Как описано в Патентной Публикации Соединенных Штатов № 2004/0162454, нанокристаллический цеолит Y является преимущественным при использовании в процессе алкилирования. Цеолит имеет размер кристаллической элементарной ячейки не более чем около 100 нанометров (нм). Максимальное расстояние диффузии многих реактантов и, что более важно, продуктов, ограничено. Закоксовывание и связанная с ним дезактивация катализатора снижены обеспечением возможности того, что 1) предпочтительные С8-продукты покидают катализатор до протекания последующих реакций алкилирования, то есть образования более тяжелых продуктов С12+, и 2) прекурсоры кокса (например, более тяжелые продукты С12+) покидают катализатор до того, как подвергнутся ретрогрессивным реакциям конденсации. Катализаторы на основе нанокристаллического цеолита Y также проявляют более высокую активность, чем цеолитные катализаторы, имеющие более крупные кристаллические элементарные ячейки, благодаря улучшенному коэффициенту эффективности.
Гидрирующее действие в твердых кислотных катализаторах предпочтительно обеспечивается каталитически активным биметаллическим или триметаллическим компонентом. Биметаллический или триметаллический компонент предпочтительно включает по меньшей мере один металл из Группы VIII Периодической Таблицы элементов. Предпочтительны мультиметаллические компоненты, которые включают платину (Pt) или палладий (Pd) в качестве одного из металлов, такие как PtNi, PtCo, PtAg, PtAu, PdNi, PdAg и PdAu, или PtPdNi, PtPdAg, PtPdAu. Также могут быть использованы комбинации платины (Pt) или палладия (Pd) с рутением (Ru), иридием (Ir), родием (Rh), медью (Cu) и рением (Re). Изобретение не ограничивается в этом отношении, и может быть применен любой мультиметаллический (в особенности биметаллический и триметаллический) компонент, который будет обеспечивать гидрирующее действие в реакции алкилирования. Например, металлы Группы VIB, включающие, например, молибден (Mo) и ванадий (V), могут быть использованы в мультиметаллическом компоненте и могут быть применены мультиметаллические материалы, которые не включают ни платины (Pt), ни палладия (Pd), например, такие как комбинация TiNiHf.
Мультиметаллический (в особенности биметаллический или триметаллический) компонент вводят в цеолит с использованием общеупотребительных способов, известных квалифицированным специалистам в этой области техники, например, таких как ионный обмен, импрегнирование цеолита или внедрение мультиметаллического материала в синтетический материал, из которого изготавливают цеолит. Предпочтительным способом внедрения мультиметаллического материала является ионный обмен. Поскольку цеолиты являются высокоселективными, для усиления поглощения мультиметаллического компонента может быть выбрана соль мультиметалла. Например, тетраамминные соли (например, хлориды, нитраты или гидроксиды) мультиметаллов могут быть приготовлены и использованы для введения биметаллического компонента в цеолит. Мультиметалл может быть также введен с использованием последовательного процесса, иногда называемого как «двойное погружение», в котором цеолит обрабатывают раствором соли с последующим высушиванием и кальцинацией для фиксации мультиметалла в цеолите, и процесс повторяют, пока в цеолит не будет введено желаемое количество мультиметалла.
Твердый кислотный катализатор предпочтительно содержит от около 0,01% вес. до около 2,0% вес. мультиметаллического (например, биметаллического или триметаллического) компонента, более предпочтительно содержит от около 0,02% вес. до около 1,0% вес. мультиметаллического компонента и еще более предпочтительно от около 0,05% вес. до около 0,5% вес. мультиметаллического компонента.
Твердый кислотный катализатор обычно будет включать матричные (также называемые связующими) компоненты, в частности, для придания физической целостности (например, прочности на раздавливание, образования тонкодисперсных частиц и т.д.), а также макропористости. Будучи используемыми, матричные компоненты могут быть скомбинированы с цеолитом до или после процесса, в котором мультиметаллический компонент вводят в катализатор. Материалы, которые могут быть применены в качестве матричных компонентов, в общем представляют собой неорганические оксиды, такие как оксиды алюминия, оксиды кремния, оксиды кремния-алюминия, оксиды циркония, глины и т.д. Матрица может быть в форме золя, гидрогеля или геля, и она может быть каталитически активной или инертной. Если используется матричный материал, матричный материал может содержать от около 2% до около 98% вес. матричного материала в расчете на объединенный вес матричного материала и цеолита. Количество матричного материала, включенного в твердый кислотный катализатор, выбирается для достижения желаемой прочности на раздавливание, в то же время сохраняя достаточную каталитическую активность, принимая во внимание разбавление цеолита матричным компонентом. Предпочтительно матричный материал будет содержаться в количестве от около 5% вес. до около 70% вес. в расчете на объединенный вес матричного материала и цеолита, и более предпочтительно от около 10% вес. до около 50% вес. В особенно предпочтительном варианте осуществления матричный материал содержится в количестве от около 15% вес. до около 30% вес. в расчете на объединенный вес матричного материала и цеолита.
Мультиметаллический твердый кислотный катализатор предпочтительно имеет диаметр экструдата от около 0,08 мм до около 2,5 мм. При употреблении в реакторе с неподвижным слоем диаметр экструдата предпочтительно составляет по меньшей мере около 0,5 мм, с верхним пределом около 1,8 мм. Меньшие диаметры могут быть использованы в реакторах с псевдоожиженным слоем или суспензионных реакторах. Катализатор предпочтительно имеет средний диаметр микропор около 7,4Å (7,4×10-10 м), когда применяют цеолит Х или Y.
Мультиметаллический твердый кислотный катализатор может быть использован в нескольких компоновках процесса алкилирования для катализирования реакции парафина и олефина с образованием бензина, имеющего высокие значения октанового числа бензина по исследовательскому методу (RON) и моторного октанового числа (MON). Процесс алкилирования может быть выполнен в любой пригодной форме реакционной системы, известной квалифицированным специалистам в этой области техники, такой как процессы с увлеченным псевдоожиженным слоем, процессы с фиксированным псевдоожиженным слоем, реакторы с кипящим слоем, суспензионные процессы и процессы с неподвижным слоем. Например, процесс алкилирования может представлять собой такой процесс, который описан в Патенте Соединенных Штатов № 6844479 или Патентной Публикации Соединенных Штатов № 2004/0162454, которые приведены здесь для сведения.
Обычно процесс алкилирования проводят в таких условиях, что по меньшей мере часть алкилирующего реагента и алкилируемого соединения будут находиться в жидкой фазе или в суперкритической фазе. В общем, процесс проводят при температуре в диапазоне от около -40° до около 250°С, предпочтительно в диапазоне от около 50° до около 150°С, более предпочтительно в диапазоне от около 70° до около 100°С и давлении от около 1 до около 100 бар (0,1-10 МПа), предпочтительно от около 10 до около 40 бар (1-4 МПа) и более предпочтительно от около 15 до около 30 бар (1,5-3 МПа). Молярное отношение алкилируемого соединения к алкилирующему реагенту в общей массе сырьевого материала в реакторе предпочтительно составляет выше чем около 5:1 и более предпочтительно выше чем около 50:1. Более высокие молярные соотношения рассматриваются как предпочтительные из соображений производительности, поскольку они в общем обеспечивают повышение октанового числа продукта и стабильности катализатора. Верхний предел для этого соотношения определяется типом используемого процесса и экономическими факторами процесса. Верхний предел молярного соотношения не является критически важным и может достигать максимально 5000:1. В общем, из экономических соображений являются предпочтительными молярные соотношения, например, на уровне около 1000:1 или ниже. Во многих современных вариантах применения молярное отношение алкилируемого соединения к алкилирующему реагенту на уровне 150-750:1 рассматривается как наиболее предпочтительное. Скорость подачи сырьевого материала (WHSV, среднечасовая скорость подачи сырья) алкилирующего реагента в общем варьирует в диапазоне от около 0,01 до около 5, предпочтительно в диапазоне от около 0,05 до около 0,5 и более предпочтительно в диапазоне от около 0,1 до около 0,3 грамма алкилирующего реагента на грамм катализатора в час. Значение среднечасовой скорости подачи сырья (WHSV) алкилируемого насыщенного углеводорода предпочтительно варьирует в диапазоне от около 0,1 до около 500. Следует понимать, что применение твердого кислотного катализатора согласно настоящему изобретению не ограничивается какими-либо конкретными условиями реакции, и вышеописанные условия являются примерными.
Катализатор согласно изобретению в особенности пригоден для использования в алкилировании изоалканов, имеющих 4-10 атомов углерода, таких как изобутан, изопентаны или изогексаны, или их смеси, олефинами, имеющими 2-10 атомов углерода, предпочтительно 2-6 атомов углерода и более предпочтительно 3-5 атомов углерода. Алкилирование изобутана бутаном или смесью бутенов представляет собой в особенности предпочтительный вариант осуществления. Изобретение не ограничено в этом отношении, и в процессе алкилирования для получения желаемого продукта может быть использован любой подходящий парафин или олефин.
Катализатор согласно настоящему изобретению может быть также использован для других типов процессов алкилирования, таких как процессы, в которых употребляются циклоалканы или арилалканы. Например, катализаторы могут быть использованы в процессах для корректирования цетанового числа некоторых потоков дистиллятов. Например, легкий рецикловый гайзоль (“LCO”), продукт отгонки процесса Каталитического Крекинга (“FCC”), содержит незамещенный цикл и в низкой степени алкилированные ароматические циклы; для как такового, его цетановое число является довольно низким - обычно 10-30. Гидрирование легкого рециклового гайзоля (LCO) лишь незначительно повышает цетановое число. Обычно гидрированными компонентами являются в низкой степени алкилированные циклоалканы, такие как метилэтилциклогексан. В одном варианте осуществления катализатор может быть использован в процессе для сочетания гидрированного легкого рециклового гайзоля (LCO) с потоком черновых С4-С6-олефинов для получения высококачественного дизельного топлива.
Регенерация катализатора может быть проведена с использованием низкотемпературного способа или высокотемпературного способа. Низкотемпературный способ часто выполняют задолго до того, как в составе продукта обнаруживаются олефины, предпочтительно в состоянии менее около 20% времени активного цикла катализатора. Активный цикл катализатора определяется как время от начала подачи алкилирующего реагента до момента, когда по сравнению с показателями на входе в содержащую катализатор секцию реактора около 20% алкилирующего реагента выходят из содержащей катализатор секции реактора непрореагировавшими, не считая изомеризации внутри молекулы. Низкотемпературная регенерация может быть выполнена наиболее практично путем отключения подачи олефина и введения водорода в обогащенный изобутаном углеводородный сырьевой материал, при реакционной температуре от около 70° до около 100°С, для удаления С12+ тяжелых углеводородов и кокса. Более жесткую высокотемпературную реактивацию обычно проводят после большого числа низкотемпературных регенераций, при температурах от около 175° до около 350°С. В высокотемпературной реактивации останавливают потоки как парафина, так и олефина, и газообразный водород пропускают над катализатором для удаления кокса и тяжелых углеводородов.
Нижеследующие примеры иллюстрируют признаки настоящего изобретения. В Примерах 8-14 используется реакторная система 100, показанная в Фиг.1. В реакторной системе используют рециркуляционный поток R, в котором объединены сырьевые потоки F-1 (олефин) и F-2 (изопарафин). Поток олефина F-1 включает цис-2-бутен, и поток изопарафина включает изобутан. Объединенные потоки через трубопровод 101 направляются в реактор 110 для алкилирования, который содержит неподвижный слой 111 катализатора согласно изобретению. Реактор для алкилирования погружен в масляную баню 112 для поддержания заранее заданной реакционной температуры. Образец для газохроматографического (GC) анализа может быть отобран в канале 103 из отходящего потока 102 из реактора 110 для алкилирования. Отходящий поток разделяют на рециркуляционный поток R, который циркулирует с помощью насоса Р обратно в реактор 110 для алкилирования после добавления свежего сырья F-1 и F-2, и поток 104, который направляется в разделительный барабан 120, из которого пары V выводятся из верхней части, и алкилат А как продукт (например, изомерные триметилпентаны (TMP)) выводится из донной части. Реактор 110 работает как рециркуляционный реактор с неподвижным слоем для поддержания высокого значения отношения изобутана к бутену и моделирования реактора постоянного перемешивания (CSTR). Высокие значения отношений «изобутан/бутен» помогают свести к минимуму образование кокса и высококипящих компонентов, которые дезактивируют катализатор. Может быть использован реактор с неподвижным слоем, имеющий несколько каналов для впрыскивания бутена в местах, расположенных на разной высоте неподвижного слоя, чтобы поддерживать желаемое соотношение «изобутан/бутен» в любом данном месте и в целом по всему слою катализатора. Реакционный продукт представлял собой смесь разнообразных компонентов и/или изомеров. Предпочтительные компоненты алкилирования представляют собой изомеры триметилпентановых (ТМР) разветвленных С8-углеводородов, каждый из которых имеет высокое значение октанового числа бензина по исследовательскому методу (“RON”). Например, 2,2,4-триметилпентан (изооктан) имеет значение октанового числа бензина по исследовательскому методу (RON), равное 100. Общее октановое число бензина по исследовательскому методу (RON) полученного алкилата в примерах было получено суммированием произведения весовой доли каждого компонента (определенной с помощью газохроматографического (GC) анализа), умноженной на октановое число компонента. Эксперименты продолжали до тех пор, пока в реакционном продукте не появлялись олефины (отключение при 0,012% вес. было определено как момент проскока). В этот момент олефиновые пики в газохроматографическом анализе показывали дезактивацию катализатора.
ПРИМЕРЫ
ПРИМЕР 1
Контрольный катализатор (не содержащий металлов)
В приведенных здесь в качестве примеров катализаторах использовали обычную цеолитную основу, а именно имеющийся в продаже ультрастабильный цеолит Y (“USY”) под наименованием CBV500, производимый фирмой PQ Corp. Продукт CBV500 имеет приблизительно 80% вес. цеолита и 20% оксида алюминия. Катализатор просеяли через сито с размером ячеек 18/25 меш для использования в испытаниях производительности в лабораторном масштабе (описанных ниже). Готовый катализатор был обозначен как катализатор «А».
ПРИМЕР 2
Получение катализатора, содержащего 0,5% вес. платины (Pt)
Платиновый (Pt) контрольный катализатор (то есть без второго или третьего добавленного металла) приготовили с имеющимся в продаже ультрастабильным цеолитом Y (“USY”) под наименованием CBV500, описанным в Примере 1. Платину (Pt) добавили с помощью раствора азотнокислой соли тетраамминоплатины, введенного общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли платины (Pt), достаточное для того, чтобы конечный катализатор содержал 0,5% вес. платины (Pt). Полученный катализатор просеяли через сито с размером ячеек 18/25 меш для использования в испытаниях производительности в лабораторном масштабе (описанных ниже). Готовый катализатор был обозначен как катализатор «В».
ПРИМЕР 3
Получение катализатора, содержащего 0,15% вес. платины (Pt)
Второй катализатор, содержащий только платину (Pt) (то есть без второго или третьего добавленного металла), приготовили с имеющимся в продаже ультрастабильным цеолитом Y (“USY”) под наименованием CBV500, описанным в Примере 1. Платину (Pt) добавили с помощью раствора азотнокислой соли тетраамминоплатины, введенного общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли платины (Pt), достаточное для того, чтобы конечный катализатор содержал 0,15% вес. платины (Pt). Полученный катализатор просеяли через сито с размером ячеек 18/25 меш для использования в испытаниях производительности в лабораторном масштабе (описанных ниже). Готовый катализатор был обозначен как катализатор «С».
ПРИМЕР 4
Получение платино-никелевого (Pt/Ni) сравнительного катализатора
Платино-никелевый (Pt/Ni) катализатор приготовили с имеющимся в продаже ультрастабильным цеолитом Y (“USY”) под наименованием CBV500, описанным в Примере 1. Платину (Pt) добавили с помощью раствора азотнокислой соли тетраамминоплатины, введенного общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли платины (Pt), достаточное для того, чтобы конечный катализатор содержал 0,12% вес. платины (Pt). Стадию введения второго металла использовали для добавления никеля (Ni). Никель (Ni) добавили с помощью раствора азотнокислой соли никеля, внедренной общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли никеля (Ni), достаточное для того, чтобы конечный катализатор содержал 0,10% вес. никеля (Ni). Полученный катализатор просеяли через сито с размером ячеек 18/25 меш для использования в испытаниях производительности в лабораторном масштабе (описанных ниже). Готовый катализатор был обозначен как катализатор «D».
ПРИМЕР 5
Получение платино-никелевого (Pt/Ni) сравнительного катализатора (с низким уровнем содержания)
Платино-никелевый (Pt/Ni) катализатор приготовили с имеющимся в продаже ультрастабильным цеолитом Y (“USY”) под наименованием CBV500, описанным в Примере 1. Платину (Pt) добавили с помощью раствора азотнокислой соли тетраамминоплатины, введенного общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли платины (Pt), достаточное для того, чтобы конечный катализатор содержал 0,06% вес. платины (Pt). Стадию введения второго металла использовали для добавления никеля (Ni). Никель (Ni) добавили с помощью раствора азотнокислой соли никеля, внедренной общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли никеля (Ni), достаточное для того, чтобы конечный катализатор содержал 0,05% вес. никеля (Ni). Полученный катализатор просеяли через сито с размером ячеек 18/25 меш для использования в испытаниях производительности в лабораторном масштабе (описанных ниже). Готовый катализатор был обозначен как катализатор «Е».
ПРИМЕР 6
Получение платино-кобальтового (Pt/Со) сравнительного катализатора
На основе неожиданных и синергических преимущественных свойствах платино-никелевого (Pt/Ni) катализатора приготовили также платино-кобальтовый (Pt/Со) катализатор с имеющимся в продаже ультрастабильным цеолитом Y (“USY”) под наименованием CBV500, описанным в Примере 1. Платину (Pt) добавили с помощью раствора азотнокислой соли тетраамминоплатины, введенного общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли платины (Pt), достаточное для того чтобы конечный катализатор содержал 0,12% вес. платины (Pt). Стадию введения второго металла использовали для добавления кобальта (Со). Кобальт (Со) добавили с помощью раствора азотнокислой соли кобальта, внедренной общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли кобальта (Со), достаточное для того, чтобы конечный катализатор содержал 0,11% вес. кобальта (Со). Полученный катализатор просеяли через сито с размером ячеек 18/25 меш для использования в испытаниях производительности в лабораторном масштабе (описанных ниже). Готовый катализатор был обозначен как катализатор «F».
ПРИМЕР 7
Получение катализатора, содержащего 0,5% вес. никеля (Ni) (без включения платины (Pt))
Катализатор, содержащий только никель (Ni), приготовили с имеющимся в продаже ультрастабильным цеолитом Y (“USY”) под наименованием CBV500, описанным в Примере 1. Никель (Ni) добавили с помощью раствора азотнокислой соли никеля (Ni), введенного общеупотребительным способом первоначального смачивания. Катализатор высушили при температуре 110°С на воздухе с последующей кальцинацией при температуре 400°С в воздушной атмосфере. Использовали количество соли никеля (Ni), достаточное для того, чтобы конечный катализатор содержал 0,5% вес. никеля (Ni). Полученный катализатор просеяли через сито с размером ячеек 18/25 меш для использования в испытаниях производительности в лабораторном масштабе (описанных ниже). Готовый катализатор был обозначен как катализатор «G».
ПРИМЕР 8
Испытание производительности алкилирования и регенерации катализатора: катализатор В
Испытание алкилирования в лабораторном масштабе проводили в реакторной системе 100, иллюстрированной в Фиг.1, при температуре 80°С и общем давлении 400 psig (2,757 МПа (избыточных)). Реактант представлял собой смесь 2-бутена («олефин» или «О») и изобутана (“I”), с общим молярным соотношением “I/O”, равным 16. Благодаря рециркуляции изобутана внутреннее соотношение “I/O” составляло приблизительно 750. Катализатор В предварительно обработали нагреванием от комнатной температуры до 300°С со скоростью повышения температуры 1°С/мин в потоке воздуха (с расходом 75 мл/мин/грамм катализатора), выдерживанием в течение 2 часов при этой температуре, охлаждением обратно до комнатной температуры, с последующим переключением на поток водорода с расходом 20 мл/мин/грамм катализатора, в то же время с нагреванием до температуры 275°С со скоростью повышения температуры 1°С/мин, выдерживанием в течение 2 часов и охлаждением до комнатной температуры. В каждом испытании использовали 4 части катализатора и 0,27 части/мин вышеупомянутой смеси реактантов. Состав продукта отслеживали с помощью газовой хроматографии (GC) и рассчитывали октановое число (С5+)-бензина (октановое число бензина по исследовательскому методу (RON)). Экспериментальные условия отражают таковые для промышленного производства, где начальная конверсия олефина составляет 100% при начале процесса.
В этом процессе катализатор подвергается «полосовидному» старению, то есть накопление «кокса» (тяжелых углеводородов) происходит от фронтальной части реактора к задней части реактора. Дезактивация катализатора происходит, когда конверсия олефина становится неполной. Этот эффект называется «проскок олефина» или просто «проскок». Продолжительность цикла определяется временем проскока, после которого катализатор должен быть регенерирован. В этом эксперименте проскок определяют как время, когда выход олефина достигает 0,012% вес. от количества продукта и рециркулирующего изобутана.
После проскока катализатор регенерировали с помощью потока водорода со скоростью подъема температуры 1°С/мин до достижения температуры 275°С и затем выдерживали при температуре в течение 2 часов.
После регенерации катализатор испытывали опять в таких же условиях, как в первом испытательном цикле, и отслеживали проскок олефина. По достижении конца цикла катализатор опять регенерировали и испытывали производительность третьего цикла.
Результаты производительности показаны в Фиг.2. Катализатор В проявил времена проскока 4,6 часа, 3,9 часа и 4,7 часа для трех испытательных циклов. Полное восстановление производительности показывает, что процедура регенерации водородом является весьма эффективной.
ПРИМЕР 9
Испытание производительности алкилирования и Регенерации Катализатора: Катализатор А
Катализатор А, контрольный катализатор без содержания металлов, испытывали подобным образом, как в Примере 8, за исключением того, что его испытывали только в двух циклах. Свежий катализатор имел время проскока 4,5 часа, показывая производительность, эквивалентную катализатору В. Этот результат вместе с эквивалентным октановым продуктом демонстрирует, что металл не играл существенной роли для производительности алкилирования.
После высокотемпературной регенерации катализатор испытали для второго цикла, и наступление проскока олефина было довольно быстрым - 1,2 часа. Этот результат демонстрирует решающую роль металла для регенерации водородом. Поскольку производительность второго цикла была плохой, проведение третьего цикла посчитали нецелесообразным.
ПРИМЕР 10
Испытание производительности алкилирования и регенерации катализатора: катализатор С
Катализатор С, катализатор с низким содержанием платины (Pt), испытывали эквивалентным образом, как катализатор В (пример 8). Свежий катализатор имел первый цикл продолжительностью 4,5 часа перед проскоком олефина - идентично производительности катализатора В. На основе его производительности во втором и третьем цикле, от цикла к циклу имела место некоторая деградация примерно на 10% в каждом цикле. Чтобы достигнуть полной регенерации, эти результаты показывают, что критический уровень содержания платины (Pt) лежит выше 0,15% вес., когда не присутствуют никакие другие металлы.
ПРИМЕР 11
Испытание производительности алкилирования и регенерации катализатора: катализатор D
Катализатор D, катализатор с содержанием 0,12% вес. платины (Pt) и 0,10% вес. никеля (Ni), испытывали эквивалентным образом, как катализатор В (пример 8). Свежий катализатор имел первый цикл продолжительностью 4,6 часа перед проскоком олефина - идентично производительности катализатора В. Его второй цикл также имел время проскока 4,6 часа, с последующим временем проскока 4,4 часа в его третьем цикле. Эти результаты демонстрируют, что биметаллический катализатор имел необычно хорошую регенерацию производительности от цикла к циклу.
ПРИМЕР 12
Испытание производительности алкилирования и регенерации катализатора: катализатор Е
Катализатор Е, катализатор с содержанием 0,06% вес. платины (Pt) и 0,05% вес. никеля (Ni), испытывали эквивалентным образом, как катализатор В (пример 8). Свежий катализатор имел первый цикл продолжительностью 4,3 часа перед проскоком олефинов - почти идентично производительности катализатора В. Однако от цикла к циклу имела место некоторая деградация производительности (второй цикл имел время проскока 3,7 часа, с последующим временем проскока 3,3 часа в его третьем цикле). Эти результаты демонстрируют, что биметаллический катализатор имел сохранение производительности от незначительного, низкого до плохого уровня, но явно превосходил не содержащий металлов катализатор.
ПРИМЕР 13
Испытание производительности алкилирования и регенерации катализатора: катализатор F
Катализатор F, катализатор с содержанием 0,12% вес. платины (Pt) и 0,11: вес. кобальта (Со), испытывали эквивалентным образом, как катализатор В (пример 8). Свежий катализатор имел первый цикл продолжительностью 4,7 часа перед проскоком олефинов - идентично производительности катализатора В. Его второй цикл имел время проскока 4,9 часа, с последующим временем проскока 4,4 часа в его третьем цикле. Эти результаты демонстрируют, что платино-кобальтовый (PtCo) биметаллический катализатор также имел хорошее восстановление производительности от цикла к циклу.
ПРИМЕР 14
Испытание производительности алкилирования и регенерации катализатора: катализатор G
Катализатор G, катализатор с содержанием 0,50% вес. никеля (Ni), испытывали эквивалентным образом, как катализатор В (пример 8). Свежий катализатор имел первый цикл продолжительностью 3,9 часа перед проскоком олефинов - на 15% ниже, чем производительность Катализатора В. Его второй цикл имел время проскока 2,4 часа, с последующим временем проскока 0,9 часа в его третьем цикле. Эти результаты демонстрируют, что катализатор, содержащий только никель (Ni), проявил очень плохое восстановление производительности от цикла к циклу, показывая, что для целей регенерации требуется присутствие по меньшей мере одного благородного металла.
ПРИМЕР 15
Методология охарактеризования платиновых (Pt) и биметаллических катализаторов алкилирования
Серию катализаторов приготовили для охарактеризования хемосорбции монооксида углерода (СО) и снятия инфракрасных спектров с преобразованием Фурье (FTIR). Приготовили образец с 0,35% вес. платины (Pt) на ультрастабильном цеолите Y (“USY”), обозначенный как «Катализатор Н». Второй образец, обозначенный как «Катализатор I», включал 0,105% вес. никеля (Ni) и 0,35% вес. платины (Pt) на USY. Третий образец, обозначенный как «Катализатор J», включал 0,105% вес. никеля (Ni) и 0,12% вес. платины (Pt) на USY.
Разнообразные лабораторные обработки этих трех катализаторов показаны в Фиг.3. Таковыми являются:
Восстановление водородом (Н2): обработка при температуре 450°С и парциальном давлении водорода (H2) 30 торр (30 мм рт.ст., 4,0 кПа) в течение 30 минут. Это восстановление проводили с периодическим повторением до трех раз.
Образование кокса: Катализатор подвергали воздействию изобутана (парциальное давление 15 торр) (2,0 кПа) и цис-2-бутена (парциальное давление 1 Торр) (133,32 Па) при температуре 80°С в течение 30 минут, с периодическим повторением до двух раз.
Регенерация водородом (Н2): Закоксованный катализатор подвергали воздействию водорода (Н2) с парциальным давлением 10 торр (1,33 кПа) при температуре 250°С в течение 50 минут, с периодическим повторением до двух раз.
Измерение монооксида углерода (СО): стандартная хемосорбция монооксида углерода (СО) характеризовала различные обработанные катализаторы. Дисперсия платины (Pt) могла быть оценена с использованием стехиометрического отношения «Pt:СО» на уровне 1:1. Кроме того, катализатор прослеживали с помощью инфракрасной спектроскопии с преобразованием Фурье (FTIR), где можно было определить дисперсные частицы платины (Pt).
Фиг.4 показывает характеристические частоты поглощения углерод-углеродных связей в колебательных спектрах кокса и прекурсоров кокса.
ПРИМЕР 16
Хемосорбция монооксида углерода (СО) на «закоксованном» Катализаторе Н: 0,35% вес. платины (Pt)/USY
Фиг.5 показывает измерения с помощью инфракрасной спектроскопии с преобразованием Фурье (FTIR) катализатора Н в трех состояниях: (а) восстановленное, (b) после «закоксовывания» в течение 240 минут и (с) после регенерации водородом (Н2) в течение 100 минут. Острый одиночный пик при волновом числе 2065 см-1 для (а) и (с) показывает, что регенерация водородом (Н2) восстанавливает платину (Pt) до ее четко определенного активного состояния. Широкий пик для (b) свидетельствует о том, что металлическая функция платины (Pt) ухудшалась.
ПРИМЕР 17
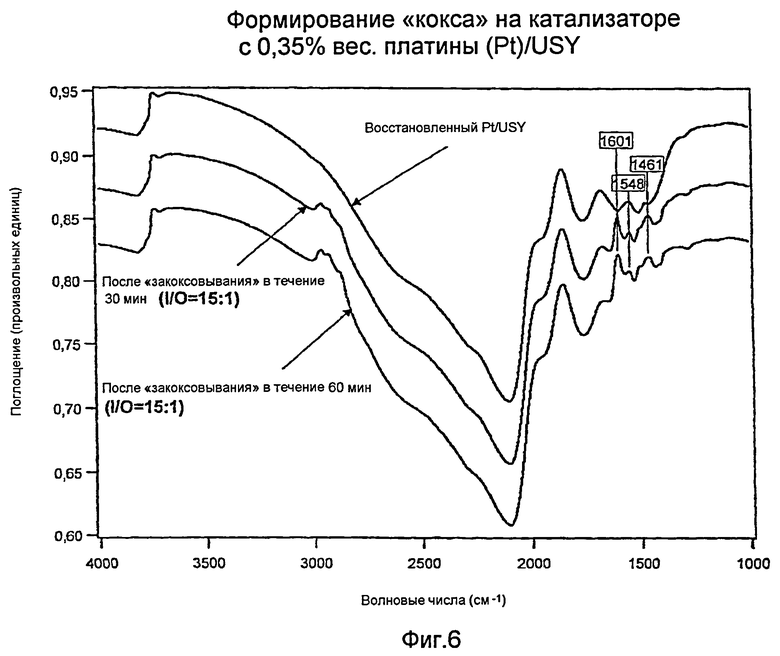
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): Образование кокса на катализаторе Н: 0,35% вес. платины (Pt)/USY
Фиг.6 показывает три инфракрасных спектра с Фурье-преобразованием (“FTIR”) для катализатора с 0,35% вес. платины (Pt)/USY («катализатор Н») и углеводородного «кокса». Сюда входят не содержащий углеводородов, восстановленный катализатор (верхняя кривая); катализатор, закоксованный в течение тридцати минут (средняя кривая); и катализатор, закоксованный в течение шестидесяти минут (нижняя кривая). Ключевая область интереса находится вблизи области волновых чисел 3000 см-1, области, которая соответствует алифатическим (то есть парафиновым) углеводородам. Восстановленный катализатор показывает по существу отсутствие углеводорода, тогда как оба закоксованных катализатора обнаруживают наличие алифатических углеводородов. Закоксованный в течение более длительного времени катализатор (нижняя кривая) имел существенно более высокое содержание углеводородного материала.
ПРИМЕР 18
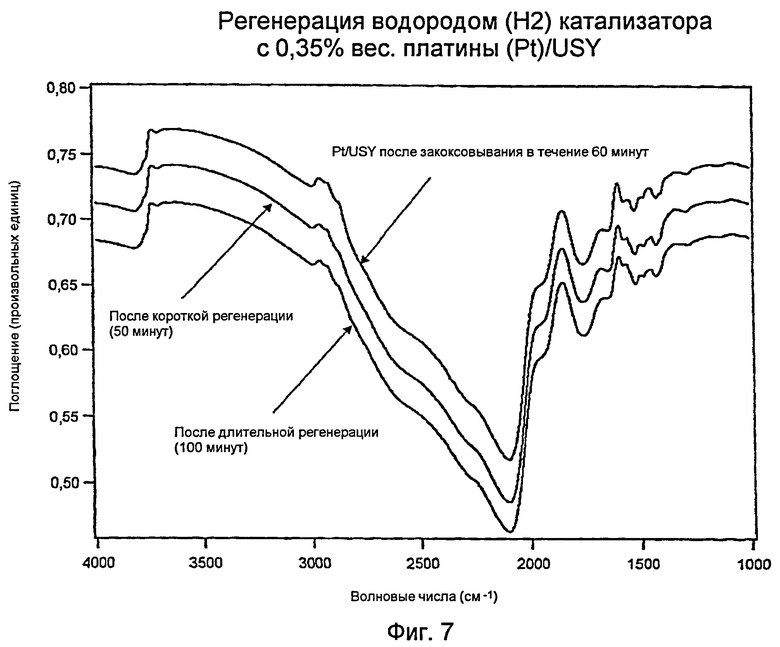
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): регенерированный водородом (Н2) катализатор Н: 0,35% вес. платины (Pt)/USY
Фиг.7 показывает три инфракрасных спектра с Фурье-преобразованием (“FTIR”) для катализатора с 0,35% вес. платины (Pt)/USY («Катализатор Н») и углеводорода. Верхняя кривая (такая же, как нижняя кривая для Фигуры 6) соответствует шестидесятиминутному закоксовыванию Pt/USY-образца. Этот катализатор был регенерирован в водороде (Н2) в течение 50 минут (средняя кривая) и 100 минут (нижняя кривая). В то время как действенными были обе регенерации, увеличенная продолжительность была более эффективной для удаления большей части кокса.
ПРИМЕР 19
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): Образование кокса на катализаторе I: 0,105% вес. никеля (Ni) - 0,35% вес. платины (Pt)/USY
Фиг.8 показывает три инфракрасных спектра с Фурье-преобразованием (“FTIR”) для катализатора I (0,105% вес. никеля (Ni) - 0,35% вес. платины (Pt)/USY) и углеводородного «кокса». Сюда входят не содержащий углеводородов, восстановленный катализатор (верхняя кривая); катализатор, закоксованный в течение тридцати минут (средняя кривая); и катализатор, закоксованный в течение шестидесяти минут (нижняя кривая). Ключевая область интереса находится вблизи области волновых чисел 3000 см-1, области, которая соответствует алифатическим (то есть парафиновым) углеводородам. Восстановленный катализатор показывает по существу отсутствие углеводорода, тогда как оба закоксованных катализатора обнаруживают наличие алифатических углеводородов. Закоксованный в течение более длительного времени катализатор (нижняя кривая) имел существенно более высокое содержание углеводородного материала. Главный факт можно увидеть при сравнении нижних кривых в Фиг.6 и 8: при одинаковых условиях закоксовывания биметаллический катализатор I (Фиг.8) имел меньшее накопление углеводорода, чем Катализатор Н «только с платиной (Pt)» (Фиг.6), как показано в области волновых чисел 3000 см-1. Этот неожиданный результат свидетельствует о том, что биметаллический катализатор имеет более высокую активность при гидрировании, чем катализатор «только с платиной (Pt)».
ПРИМЕР 20
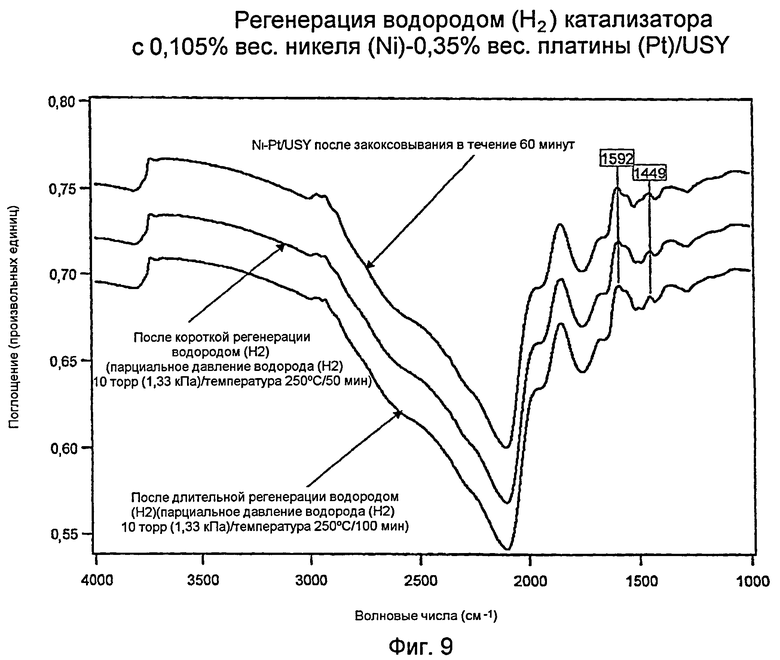
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): регенерированный водородом (Н2) Катализатор I: 0,105% вес. никеля (Ni) - 0,35% вес. платины (Pt)/USY
Фиг.9 показывает три инфракрасных спектра с Фурье-преобразованием (“FTIR”) для катализатора 0,105% вес. никеля (Ni)/0,35% вес. платины (Pt)/USY («катализатор I») и углеводородного остатка («кокса»). Верхняя кривая (такая же, как нижняя кривая для Фигуры 8) соответствует шестидесятиминутному закоксовыванию Pt/USY-образца. Этот катализатор был регенерирован в водороде (Н2) в течение 50 минут (средняя кривая) и 100 минут (нижняя кривая). В то время как действенными были обе регенерации, увеличенная продолжительность была более эффективной для удаления большей части кокса. Главный факт можно увидеть при сравнении нижних кривых в Фиг.7 и 9: при одинаковых условиях регенерации в водороде (Н2) биметаллический катализатор I (Фиг.9) имел меньшее накопление углеводорода, чем катализатор I «только с платиной (Pt)» (Фиг.7), как показано в области волновых чисел 3000 см-1. Этот неожиданный результат свидетельствует о том, что биметаллический катализатор имеет более высокую активность при гидрировании, чем катализатор «только с платиной (Pt)». Поскольку биметаллический катализатор также формирует меньшее количество кокса (см. пример 19), не ясно, состоит ли преимущество биметаллического материала в (а) пониженном формировании кокса или (b) как в пониженном формировании кокса, так и в лучшей регенерации в водороде (Н2). Независимо от механизма (а) или (b), любой из них представляет собой очевидно превосходный и неожиданный результат.
ПРИМЕР 21
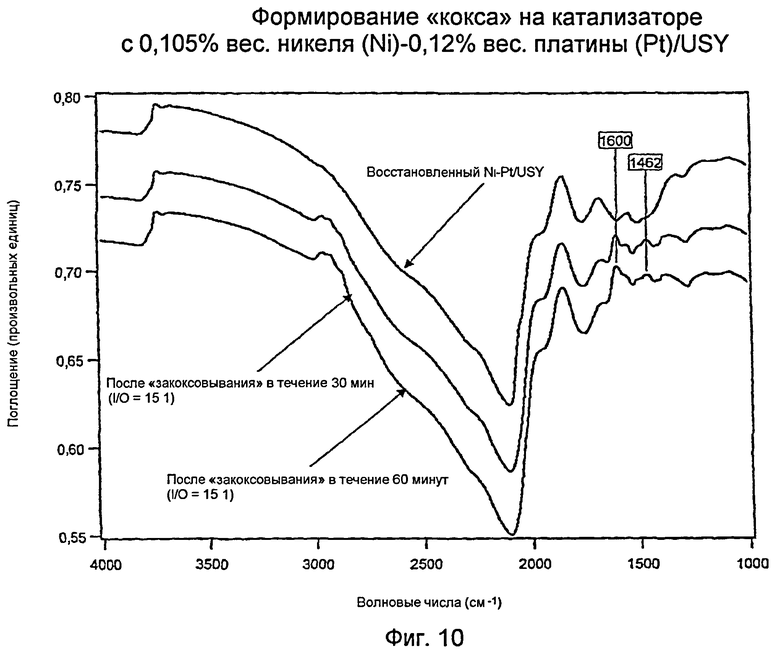
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): Образование кокса на катализаторе J: 0,105% вес. никеля -(Ni) - 0,12% вес. платины (Pt)/USY
Фиг.10 показывает три инфракрасных спектра с Фурье-преобразованием (“FTIR”) для катализатора с 0,105% вес. никеля (Ni)/0,12% вес. платины (Pt)/USY) («катализатор I») и углеводородного «кокса». Сюда входят не содержащий углеводородов, восстановленный катализатор (верхняя кривая); катализатор, закоксованный в течение тридцати минут (средняя кривая); и катализатор, закоксованный в течение шестидесяти минут (нижняя кривая). Ключевая область интереса находится вблизи области волновых чисел 3000 см-1, области, которая соответствует алифатическим (то есть парафиновым) углеводородам. Восстановленный катализатор показывает по существу отсутствие углеводорода, тогда как оба закоксованных катализатора обнаруживают наличие алифатических углеводородов. Закоксованный в течение более длительного времени катализатор (нижняя кривая) имел существенно более высокое содержание углеводородного материала. Главный факт можно увидеть при сравнении нижних кривых в Фиг.6 и 8: при одинаковых условиях закоксовывания биметаллический катализатор (Фиг.8) имел меньшее накопление углеводорода, чем катализатор Н «только с платиной (Pt)» (Фиг.6), как показано в области волновых чисел 3000 см-1. Этот неожиданный результат свидетельствует о том, что биметаллический катализатор имел более высокую активность при гидрировании, чем катализатор Н «только с платиной (Pt)». Более того, как показано в Фиг.10, количество алифатического кокса было больше для катализатора с 0,105% вес. никеля -(Ni)/0,12% вес. платины (Pt)/USY), катализатора J. Этот факт свидетельствует о том, что либо (а) уровень содержания платины (Pt) был ниже критического уровня для эффективности, либо что (b) молярное отношение “Ni/Pt” и уровни содержания металлов были слишком низкими для обеспечения того, что большинство атомов платины (Pt) были окружены достаточным количеством никеля (Ni) для достижения синергического эффекта, наблюдаемого для катализатора I.
ПРИМЕР 22
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): регенерированный водородом (Н2) катализатор J: 0,105% вес. никеля -(Ni)/0,12% вес. платины (Pt)/USY
Фиг.11 показывает три инфракрасных спектра с Фурье-преобразованием (“FTIR”) для катализатора 0,105% вес. никеля (Ni) - 0,12% вес. платины (Pt)/USY («катализатор J») и углеводородного «кокса». Верхняя кривая (такая же, как нижняя кривая для Фиг.8) соответствует шестидесятиминутному закоксовыванию катализатора J. Этот катализатор был регенерирован в водороде (Н2) в течение 50 минут (средняя кривая) и 100 минут (нижняя кривая). В то время как действенными были обе регенерации, увеличенная продолжительность была более эффективной для удаления большей части кокса. Главный факт можно увидеть при сравнении нижних кривых в Фиг.7 и 9: при одинаковых условиях регенерации в водороде (Н2) биметаллические катализаторы (Фиг.9 и 11) имели меньшее накопление углеводорода, чем катализатор «только с платиной (Pt)» (Фиг.7), как показано в области волновых чисел 3000 см-1. Этот неожиданный результат свидетельствует о том, что биметаллический катализатор имел более высокую активность при гидрировании, чем катализатор «только с платиной (Pt)». Поскольку биметаллический катализатор J Примера 21 также не формирует меньшего количества кокса, чем катализатор I с 0,105% вес. никеля (Ni) - 0,35% вес. платины (Pt) (см. пример 19), ясно, что преимущество биметаллических катализаторов с меньшим содержанием металла состоит в лучшей регенерации в водороде (Н2). Это очевидно представляет превосходный и неожиданный результат.
ПРИМЕР 23
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): Хемосорбционная емкость в отношении монооксида углерода (СО) трех закоксованных катализаторов
Фиг.12 показывает инфракрасные спектры с Фурье-преобразованием (FTIR) для трех катализаторов (Н: 0,35% вес. платины (Pt)/USY [ВЕРХ], I: 0,105% вес. никеля (Ni) - 0,35% вес. платины (Pt)/USY [СЕРЕДИНА], и J: 0,105% вес. никеля (Ni) - 0,12% вес. платины (Pt)/USY [НИЗ]) после 60 минут закоксовывания. Следует отметить, что закоксованный катализатор Н «только с платиной (Pt) на USY» имел более широкий пик платины (Pt) меньшей интенсивности. Это означает, что платина (Pt) очевидно менее диспергирована, и имело место более низкое содержание восстановленной платины (Pt). Кривые для двух биметаллических катализаторов показали, что (а) пик платины (Pt) был более интенсивным, и (b) более четко сформированным (более узким). Эти результаты показывают, что при закоксовывании биметаллические катализаторы сохраняют дисперсность платины (Pt) и действие восстановленной платины (Pt) лучше, чем катализатор «только с платиной (Pt)». Опять же это представляет собой превосходный и неожиданный результат.
ПРИМЕР 24
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): хемосорбционная емкость в отношении монооксида углерода (СО) трех регенерированных водородом (Н2) катализаторов
Фиг.13 показывает инфракрасные спектры с Фурье-преобразованием (FTIR) для трех катализаторов (Н: 0,35% вес. платины (Pt)/USY [ВЕРХ], I: 0,105% вес. никеля (Ni) - 0,35% вес. платины (Pt)/USY [СЕРЕДИНА], и J: 0,105% вес. никеля (Ni) - 0,12% вес. платины (Pt)/USY [НИЗ]) после 100 минут регенерации водородом (Н2). Следует отметить, что регенерированный водородом (Н2) катализатор Н «только с платиной (Pt) на USY» имел более широкий пик платины (Pt) меньшей интенсивности. Это означает, что платина (Pt) очевидно менее диспергирована и имела меньше восстановленной платины (Pt). Кривые для двух биметаллических катализаторов показали, что (а) пик платины (Pt) был более интенсивным и (b) более четко сформированным (более узким). Эти результаты показывают, что при регенерации водородом (Н2) биметаллические катализаторы сохраняют высокую дисперсность платины (Pt) и функцию восстановленной платины (Pt) по сравнению с катализатором «только с платиной (Pt)». Опять же это представляет собой превосходный и неожиданный результат.
ПРИМЕР 25
Измерения с использованием инфракрасной спектроскопии с преобразованием Фурье (FTIR): хемосорбционная емкость в отношении монооксида углерода (СО) трех регенерированных водородом (Н2) катализаторов
Фиг.14 показывает два инфракрасных спектра с Фурье-преобразованием (FTIR) для каждого из трех катализаторов (С: 0,15% вес. платины (Pt)/USY [ВЕРХ], F: 0,11% вес. кобальта (Со)-0,12% вес. платины (Pt)/USY [СЕРЕДИНА] и D: 0,10% вес. никеля (Ni) - 0,12% вес. платины (Pt)/USY [НИЗ]) после 100 минут регенерации водородом (Н2). Для каждого катализатора приведен спектр с фоновой по монооксиду углерода (СО) атмосферой (1 торр) (133,32 Па) и без таковой. Три спектра без фона СО проявляются довольно похожими, показывая только прочное связывание монооксида углерода (CO) с платиной (Pt). Поскольку все три катализатора имеют приблизительно эквивалентные уровни содержания платины (Pt), эти результаты согласуются друг с другом. Однако если рассматривать три спектра с фоновым монооксидом углерода (СО), PtCo- и PtNi-катализаторы сорбируют больше монооксида углерода (CO), чем катализатор «только с платиной (Pt)». Эти результаты свидетельствуют о том, что биметаллы проявляют усиленную металлическую функцию, чем катализатор «только с платиной (Pt)», что согласуется с улучшенной производительностью, продемонстрированной биметаллическими материалами.
Как будет очевидно квалифицированным специалистам в этой области техники на основе приведенных здесь указаний, многообразные изменения и модификации вышеописанного и прочих вариантов осуществления изобретения могут быть сделаны без выхода за пределы его области, определенной в прилагаемых пунктах формулы изобретения. Соответственно этому это подробное описание предпочтительных вариантов осуществления должно толковаться как иллюстративное, но не имеющее ограничительного смысла.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ риформинга бензиновых фракций | 2018 |
|
RU2672882C1 |
| НЕПРЕРЫВНЫЙ СПОСОБ БЕСКИСЛОРОДНОЙ КОНВЕРСИИ МЕТАНА | 2008 |
|
RU2467993C2 |
| УСТРОЙСТВО ДЛЯ АЛКИЛИРОВАНИЯ ИЗОБУТАНА ОЛЕФИНАМИ НА ТВЕРДОМ КАТАЛИЗАТОРЕ | 2015 |
|
RU2622294C2 |
| Катализатор для гидроизомеризации углеводородных фракций и способ его применения | 2018 |
|
RU2667920C1 |
| КАТАЛИЗАТОР ДЛЯ ПИРОЛИЗА СЫРЬЯ | 2015 |
|
RU2684108C2 |
| КАТАЛИЗАТОР ДЛЯ ЛЕГКИХ ОЛЕФИНОВ И LPG В ПСЕВДООЖИЖЕННЫХ КАТАЛИТИЧЕСКИХ УСТАНОВКАХ И СПОСОБ КАТАЛИТИЧЕСКОГО КРЕКИНГА | 2005 |
|
RU2412760C2 |
| СПОСОБ АЛКИЛИРОВАНИЯ ПРИ ПРИМЕНЕНИИ КАТАЛИЗАТОРА, СОДЕРЖАЩЕГО ЦЕОЛИТЫ, ИМЕЮЩИЕ В СВОЕМ СОСТАВЕ РЕДКОЗЕМЕЛЬНЫЕ ЭЛЕМЕНТЫ, ПРИ ВЫСОКОМ СОДЕРЖАНИИ ЦЕРИЯ, И ГИДРОГЕНИЗИРУЮЩИЙ МЕТАЛЛ | 2015 |
|
RU2694894C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2009 |
|
RU2514915C2 |
| СПОСОБ АЛКИЛИРОВАНИЯ С УЛУЧШЕННЫМ ОКТАНОВЫМ ЧИСЛОМ | 2018 |
|
RU2762589C2 |
| Катализатор изомеризации н-алканов в процессе риформинга гидроочищенных бензиновых фракций (варианты) | 2016 |
|
RU2626747C1 |


Изобретение относится к катализаторам алкилирования. Описан твердый кислотный катализатор для применения в процессах алкилирования парафинов олефинами, включающий: (а) цеолит, выбранный из группы, состоящей из цеолита X, цеолита Y, ZSM-20, ЕМТ и их комбинаций; (b) мультиметаллический материал, введенный в цеолит, в котором, по меньшей мере, одним из металлов является Pt или Pd, а, по меньшей мере, вторым из металлов является Ni, Со, Мn, Сr, V, Ti, Fe, Сu. Описан катализатор, включающий указанные выше компоненты и дополнительно включающий связующий материал выбранных из группы, состоящей из оксидов алюминия, оксидов кремния, оксидов кремния-алюминия, оксидов циркония и глин. Описан также способ алкилирования парафинов олефинами, включающий стадии: (а) предоставления твердого кислотного катализатора, описанного выше, и (b) смешивания одного или более алкилируемых углеводородов-парафинов с одним или более алкилирующими реагентами-олефинами в присутствии указанного катализатора в условиях, которые приводят к реакции алкилирования с образованием продукта алкилирования; и регенерацию катализатора в условиях Н2. Технический результат - описан активный катализатор в процессах алкилирования парафинов олефинами, обеспечивающий длительный срок службы до его дезактивации. 3 н. и 13 з.п. ф-лы, 14 ил.
1. Твердый кислотный катализатор для применения в процессах алкилирования парафинов олефинами, включающий:
(a) цеолит, выбранный из группы, состоящей из цеолита X, цеолита Y, ZSM-20, ЕМТ и их комбинаций;
(b) мультиметаллический материал, введенный в цеолит, в котором, по меньшей мере, одним из металлов является Pt или Pd, а, по меньшей мере, вторым из металлов является Ni, Со, Мn, Сr, V, Ti, Fe, Cu.
2. Твердый кислотный катализатор по п.1, дополнительно включающий связующий материал.
3. Твердый кислотный катализатор по п.1, в котором цеолит имеет структуру фоязита.
4. Твердый кислотный катализатор по п.1, в котором мультиметаллический материал выбирается из группы, состоящей из PtNi, PtCo, PtMn, PtCr, PtV, PtTi, PtFe, PtCu, PtNiAg, PtNiAu, PtNiRu, PtNiIr, PtNiRh, PtNiRe, PdNi, PdCo, PdCu, PdMn, PdCr, PdV, PdTi, PdFe, PtPdCo, PtPdNi, PtPdMn, PtPdCr, PtPdV, PtPdTi, PtPdFe, PtPdCu, PdNiAg, PdNiAu, PdNiRu, PdNiIr, PdNiRh, PdNiRe, PtNiCo, PdNiCo, PtPdNiCo, PtNiCoFe и их комбинаций.
5. Твердый кислотный катализатор по п.4, в котором мультиметаллический материал содержится от около 0,01 вес.% до около 2,0 вес.% от веса твердого кислотного катализатора.
6. Твердый кислотный катализатор по п.2, в котором связующий материал содержится от около 5 вес.% до около 70 вес.% от веса твердого кислотного катализатора.
7. Твердый кислотный катализатор по п.6, в котором связующий материал выбирается из группы, состоящей из оксидов алюминия, оксидов кремния, оксидов кремния-алюминия, оксидов циркония, глин и их комбинаций.
8. Твердый кислотный катализатор по п.3, в котором цеолит представляет собой нанокристаллический цеолит Y.
9. Твердый кислотный катализатор по п.3, в котором цеолит представляет собой ультрастабильный Y ("USY").
10. Твердый кислотный катализатор для применения в процессах алкилирования парафинов олефинами, включающий:
(a) нанокристаллический цеолит Y;
(b) один или более мультиметаллических материалов, введенных в цеолит, выбранных из группы, состоящей из PtNi, PtCo, PtMn, PtCr, PtV, PtTi, PtFe, PtCu, PtNiAg, PtNiAu, PtNiRu, PtNiIr, PtNiRh, PtNiRe, PdNi, PdCo, PdCu, PdMn, PdCr, PdV, PdTi, PdFe, PtPdCo, PtPdNi, PtPdMn, PtPdCr, PtPdV, PtPdTi, PtPdFe, PtPdCu, PdNiAg, PdNiAu, PdNiRu, PdNiIr, PdNiRh, PdNiRe, PtNiCo, PdNiCo, PtPdNiCo, PtNiCoFe, в котором мультиметаллические материалы содержатся от около 0,01 вес.% до около 2,0 вес.% от веса твердого кислотного катализатора; и
(c) один или более связующих материалов, выбранных из группы, состоящей из оксидов алюминия, оксидов кремния, оксидов кремния-алюминия, оксидов циркония и глин, в котором связующие материалы содержатся от 5 вес.% до 70 вес.% от веса твердого кислотного катализатора.
11. Способ алкилирования парафинов олефинами, включающий стадии:
(a) предоставления твердого кислотного катализатора, включающего цеолит, выбранный из группы, состоящей их цеолита X, цеолита Y, ZSM-20, ЕМТ и их комбинаций и мультиметаллический материал, введенный в цеолит, в котором, по меньшей мере, одним из металлов является Pt или Pd, а, по меньшей мере, вторым из металлов является Ni, Co, Mn, Cr, V, Ti, Fe, Сu; и
(b) смешивания одного или более алкилируемых углеводородов-парафинов с одним или более алкилирующими реагентами-олефинами в присутствии указанного катализатора в условиях, которые приводят к реакции алкилирования с образованием продукта алкилирования; и регенерацию катализатора в условиях H2.
12. Способ по п.11, в котором цеолит имеет структуру фоязита, и металлический материал выбирается из группы, состоящей из PtNi, PtCo, PtMn, PtCr, PtV, PtTi, PtFe, PtCu, PtNiAg, PtNiAu, PtNiRu, PtNiIr, PtNiRh, PtNiRe, PdNi, PdCo, PdCu, PdMn, PdCr, PdV, PdTi, PdFe, PtPdCo, PtPdNi, PtPdMn, PtPdCr, PtPdV, PtPdTi, PtPdFe, PtPdCu, PdNiAg, PdNiAu, PdNiRu, PdNiIr, PdNiRh, PdNiRe, PtNiCo, PdNiCo, PtPdNiCo, PtNiCoFe и их комбинациий.
13. Способ по п.12, в котором цеолит представляет собой цеолит Y.
14. Способ по п.12, в котором алкилируемый углеводород представляет собой парафин, и алкилирующий реагент представляет собой олефин.
15. Способ по п.14, в котором парафин представляет собой изобутан, и олефин представляет собой бутилен или смесь бутиленов.
16. Способ по п.15, в котором парафин включает изобутан и изопентаны, и олефин включает смесь С3-С5-олефинов.
| US 5198597 А, 30.03.1993 | |||
| US 2004162454 A1, 19.08.2004 | |||
| SU 1309383 A1, 20.10.1996 | |||
| US 2002155946 A1, 24.10.2002 | |||
| US 2002013216 A1, 31.01.2002 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

