Область, к которой относится изобретение
Изобретение относится к способам получения конъюгатов "мономерное цитотоксическое лекарственное средство/носитель" ("конъюгатов") с высокой нагрузкой лекарственного средства и, в основном, с низким содержанием низкоконъюгированной фракции (LCF). Более конкретно, настоящее изобретение относится к конъюгатам "анти-CD22 антитело-мономерный калихеамицин". Настоящее изобретение также относится к указанным конъюгатам настоящего изобретения, к способам очистки этих конъюгатов, к фармацевтическим композициям, содержащим указанные конъюгаты, и к использованию этих конъюгатов.
Предпосылки создания изобретения
Конъюгаты лекарственных средств, разработанные для системной фармакотерапии, представляют собой мишень-специфические цитотоксические агенты. Указанная фармакотерапия основана на связывании терапевтического агента с молекулой-носителем, обладающей специфичностью к определенной популяции клеток-мишеней. Антитела с высокой аффинностью по отношению к антигенам являются природными распознающими цель молекулами. Благодаря доступности высокоаффинных моноклональных антител использование доставляемых антителом терапевтических средств является особенно перспективным. Токсическими веществами, которые были конъюгированы с моноклональными антителами, являются токсины, низкомолекулярные цитотоксические лекарственные средства, модификаторы биологических ответов и радионуклиды. Конъюгаты "антитело-токсин" часто называют иммунотоксинами, а иммуноконъюгаты, состоящие из антител и низкомолекулярных лекарственных средств, таких как метотрексат и адриамицин, называют хемоиммуноконъюгатами. Иммуномодуляторы содержат модификаторы биологических ответов, которые, как известно, обладают регуляторными функциями, такие как лимфокины, факторы роста и комплемент-активирующий фактор яда кобры (CVF). Радиоиммуноконъюгаты, которые состоят из радиоактивных изотопов, могут быть использованы в качестве терапевтических средств для индуцирования гибели клеток под действием их излучения, либо они могут быть использованы для визуализации. Предполагается, что опосредованная антителом специфическая доставка цитотоксических лекарственных средств в опухолевые клетки не только повышает их противоопухолевую эффективность, но также и предотвращает неспецифическое поглощение этих лекарственных средств нормальными тканями, и, тем самым, повышает их терапевтические индексы.
Настоящее изобретение относится к иммуноконъюгатам, включающим антитело в качестве нацеливающего носителя и обладающим специфичностью к антигенным детерминантам, присутствующим на поверхности злокачественных клеток, конъюгированным с цитотоксическим лекарственным средством. Настоящее изобретение относится к конъюгатам "цитотоксическое лекарственное средство - антитело", где указанное антитело обладает специфичностью к антигенным детерминантам, присутствующим на клетках, ассоциированных со злокачественными В-клеточными опухолями, лимфопролиферативными расстройствами и хроническими воспалительными заболеваниями. Настоящее изобретение также относится к способам продуцирования иммуноконъюгатов и к их использованию в терапии.
Различные терапевтические средства, полученные на основе указанных антител и предназначенные для лечения различных заболеваний, включая рак и ревматоидный артрит, были апробированы для клинического применения, либо они находятся на стадии клинических испытаний по их использованию в качестве лекарственных средств для лечения злокачественных опухолей, включая В-клеточные злокачественные опухоли, такие как не-ходжскинская лимфома. Одним из таких терапевтических средств на основе антител является ритуксимаб (Rituxan™), немеченное химерное антитело, которое содержит область γ1 (+mγ1V-область) человеческого антитела, и которое является специфичным к поверхностно-клеточному антигену CD20, экспрессируемому на В-клетках. Эти терапевтические средства основаны на использовании антител и обладают опосредованной комплементом цитотоксичностью (CDCC) или антитело-зависимой клеточной цитотоксичностью (ADCC), направленной против В-клеток, либо эти терапевтические средства основаны на использовании радионуклидов, таких как 131I или 90Y, и получение и применение таких терапевтических средств связано с определенными проблемами для врачей-клиницистов и пациентов. Поэтому необходимость в получении иммуноконъюгатов, которые не имели бы недостатков современных терапевтических средств, полученных на основе антител и используемых для лечения различных злокачественных опухолей, включая гемопоэтические злокачественные опухоли, такие как не-ходжкинская лимфома (НХЛ), и которые можно было бы легко и эффективно продуцировать, а также повторно использовать без индуцирования иммунного ответа, все еще остается актуальной.
Иммуноконъюгаты, содержащие член семейства эффективных антибактериальных и противоопухолевых агентов, известных под общим названием калихеамицины или комплекс LL-E33288 (см. патент США №4970198 (1990)), были разработаны для лечения миелом. Наиболее эффективным из калихеамицинов является агент, обозначенный γ1, который в настоящем описании будет просто обозначаться гамма. Эти соединения содержат метилтрисульфид, который может быть подвергнут реакции взаимодействия с соответствующими тиолами с образованием дисульфидов и с одновременным введением функциональной группы, такой как гидразид или другой функциональной группы, которая может быть использована для присоединения производного калихеамицина к носителю (см., патент США №5053394). Использование конъюгатов "мономерное производное калихеамицина/носитель" в современной терапии рака различных типов ограничено из-за недостаточной доступности специфических нацеливающих агентов (носителей), а также из-за несовершенства технологий конъюгирования, которые, в случае увеличения количества производного калихеамицина, конъюгированного с носителем (то есть увеличения нагрузки конъюгата лекарственным средством), приводят к образованию белковых агрегатов. Поскольку более высокая нагрузка лекарственного средства приводит к увеличению внутренней эффективности конъюгата, то желательно, чтобы нагрузка лекарственным средством была как можно выше, насколько это позволяют условия для сохранения аффинности белка-носителя. Присутствие агрегированного белка, который может быть неспецифически токсичным и иммуногенным и который, поэтому, должен быть удален при терапевтическом применении, затрудняет процесс крупномасштабного производства этих конъюгатов и снижает выход продуктов. Количество калихеамицина, нагруженного на белок-носитель (нагрузка лекарственного средства), количество агрегата, образующегося в результате реакции конъюгирования, и выход получаемого в итоге очищенного мономерного конъюгата являются взаимосвязанными факторами. Поэтому компромисс между более высокой загрузкой лекарственного средства и выходом конечного мономера может быть достигнут путем выбора соответствующего количества реакционно-способного производного калихеамицина, добавляемого в реакционную смесь для конъюгирования.
Тенденция конъюгатов цитотоксического лекарственного средства, а в частности, конъюгатов калихеамицина образовывать агрегаты, является особенно проблематичной в том случае, когда реакцию конъюгирования осуществляют с использованием линкеров, как описано в патентах США №№5877296 и 5773001, которые во всей своей полноте вводятся в настоящее описание посредством ссылки. В этом случае большой процент продуцируемых конъюгатов находится в агрегированной форме, что значительно затрудняет очистку конъюгатов стандартными способами (СМА-способами) для их последующего терапевтического использования. В случае некоторых белков-носителей, конъюгаты даже с весьма умеренной нагрузкой невозможно фактически получить, за исключением разве что в лабораторных масштабах. Следовательно, разработка усовершенствованных методов получения конъюгатов цитотоксических лекарственных средств, таких как калихеамицинов, с носителями, которые обеспечивали бы минимизацию агрегации, а поэтому позволяли бы получать конъюгаты, по возможности, с большей нагрузкой лекарственным средством и в то же время с высоким выходом продукта, является крайне необходимой.
Используемые ранее методы конъюгирования для получения препаратов "мономерное производное калихеамицина/носитель" с высокой нагрузкой лекарственным средством/высоким выходом и низкой степенью агрегации были описаны в литературе (см. патенты США №5712374 и США №5714586, которые во всей своей полноте вводятся в настоящее описание посредством ссылки). И хотя эти способы позволяют получать конъюгированные препараты со значительно пониженным содержанием агрегатов, однако, позже было обнаружено, что эти способы дают возможность получать конъюгаты, имеющие неприемлемо высокие уровни (45-65% ВЭЖХ, % площади) низкоконъюгированной фракции (LCF), то есть фракции, состоящей, в основном, из неконъюгированного антитела. Присутствие LCF в данном продукте означает неэффективное использование антитела, поскольку оно не содержит цитотоксического лекарственного средства. Это антитело может также конкурировать с конъюгатом "калихеамицин-носитель" за связывание с мишенью и, тем самым, потенциально снижать степень доступности этой мишени, что приводит к снижению эффективности цитотоксического лекарственного средства. Поэтому, желательно разработать усовершенствованный способ конъюгирования, который обеспечивал бы значительное снижение уровней LCF и давал бы приемлемые минимальные уровни агрегации, но, при этом, не оказывал бы значительного влияния на физические свойства указанного конъюгата.
Краткое описание изобретения
Настоящее изобретение относится к способам получения конъюгатов "производное мономерного цитотоксического лекарственного средства/носитель" ("конъюгатов") с высокой нагрузкой лекарственным средством и, в основном, с низким содержанием низкоконъюгированной фракции (LCF). В частности, настоящее изобретение относится к получению конъюгатов "мономерное производное калихеамицина/носитель", к указанным конъюгатам, к композициям, к способу очистки таких конъюгатов и к использованию этих конъюгатов. Более конкретно, настоящее изобретение относится к способам получения конъюгата "мономерное производное калихеамицина - анти-CD22 антитело" (СМС-544).
В одном из вариантов своего осуществления настоящее изобретение относится к усовершенствованному способу получения конъюгатов, обеспечивающему значительное снижение уровней LCF (ниже 10%) но, при этом, не оказывающему значительного воздействия на физические или химические свойства указанного конъюгата. Настоящее изобретение также относится к дополнительно усовершенствованному способу конъюгирования, который, по сравнению с уже описанными способами, позволяет не только значительно снизить уровни LCF, а также значительно снизить степень агрегации по сравнению с ранее описанными способами и значительно увеличить нагрузку конъюгата лекарственным средством. Конъюгаты настоящего изобретения имеют формулу:
Pr(-X-W)m,
где Pr представляет собой белковый носитель;
X представляет собой линкер, включающий продукт реакции любой реакционно-способной группы, которая может взаимодействовать с белковым носителем;
W представляет собой цитотоксическое лекарственное средство;
m означает среднюю нагрузку для продукта очищенного конъюгата, такую, при которой указанное цитотоксическое лекарственное средство составляет 7-9% по массе конъюгата; и
(-X-W)m представляет собой производное цитотоксического лекарственного средства.
В одном из вариантов, конъюгаты настоящего изобретения получают способом настоящего изобретения, включающим стадии: (1) добавления производного цитотоксического лекарственного средства к белковому носителю, где указанное производное цитотоксического лекарственного средства составляет 4,5-11% по массе белкового носителя; (2) инкубирования цитотоксического лекарственного средства и белкового носителя в ненуклеофильном и совместимом с белком буферном растворе, имеющем рН в пределах примерно от 7 до 9, с продуцированием конъюгата "мономерное цитотоксическое лекарственное средство/носитель", где указанный раствор, кроме того, содержит (а) органический сорастворитель и (b) добавку, содержащую, по крайней мере, одну С6-С18-карбоновую кислоту или ее соль, и где указанное инкубирование осуществляют при температуре в пределах примерно от 30°С до 35°С в течение периода времени примерно от 15 минут до 24 часов; и (3) хроматографии конъюгата, полученного на стадии (2), для отделения конъюгатов "производное мономерного цитотоксического лекарственного средства/белковый носитель", имеющих нагрузку цитотоксическим лекарственным средством, составляющую в пределах 4-10 мас.%, и содержание низкоконъюгированной фракции (LCF), составляющее ниже 10%, от неконъюгированного белкового носителя, производного цитотоксического лекарственного средства и агрегированных конъюгатов.
В одном из вариантов осуществления изобретения белковый носитель указанного конъюгата выбран из группы, состоящей из гормонов, факторов роста, антител, фрагментов антител, миметиков антител и их генетически или ферментативно сконструированных аналогов.
В одном из вариантов осуществления изобретения указанным белковым носителем является антитело. В предпочтительном варианте осуществления изобретения указанное антитело выбрано из группы, состоящей из моноклонального антитела, химерного антитела, человеческого антитела, гуманизированного антитела, одноцепочечного антитела, Fab-фрагмента и F(ab)2-фрагмента.
В другом варианте осуществления изобретения указанное гуманизированное антитело направлено против антигена клеточной поверхности CD22.
В предпочтительном варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, которое включает вариабельную область легкой цепи 5/44-gLl (SEQ ID NO: 19) и вариабельную область тяжелой цепи 5/44-gH7 (SEQ ID NO: 27).
В другом предпочтительном варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, содержащее легкую цепь, имеющую последовательность, представленную в SEQ ID NO: 28.
В еще одном предпочтительном варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, содержащее тяжелую цепь, имеющую последовательность, представленную в SEQ ID NO: 30.
В другом предпочтительном варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, содержащее легкую цепь, имеющую последовательность, представленную в SEQ ID NO: 28, и тяжелую цепь, имеющую последовательность, представленную в SEQ ID NO: 30.
В другом варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, которое представляет собой вариант антитела, полученный в соответствии с протоколом осуществления аффинного созревания и обладающий повышенной специфичностью к человеческому CD22.
В другом своем аспекте цитотоксическое лекарственное средство, используемое для генерирования конъюгата "мономерное цитотоксическое лекарственное средство/носитель" настоящего изобретения, представляет собой ингибитор полимеризации тубулина, алкилирующий агент, который связывается с ДНК и разрушает ее, ингибитор синтеза белка или ингибитор тирозинкиназы.
В одном из вариантов осуществления изобретения указанное цитотоксическое лекарственное средство выбрано из калихеамицинов, тиотепы, таксанов, винкристина, даунорубицина, доксорубицина, эпирубицина, эсперамицинов, актиномицина, аутрамицина, азасеринов, блеомицинов, тамоксифена, идарубицина, доластатинов/ауристатинов, гемиастерлинов и маутанзиноидов.
В предпочтительном варианте осуществления изобретения указанным цитотоксическим лекарственным средством является калихеамицин. В особенно предпочтительном варианте осуществления изобретения указанным калихеамицином является гамма-калихеамицин или производное N-ацетил-гамма-калихеамицина.
В еще одном аспекте настоящего изобретения указанное цитотоксическое лекарственное средство имеет функциональную 3-меркапто-3-метилбутаноилгидразидную группу и конъюгировано с белковым носителем посредством гидролизуемого линкера, способного высвобождать цитотоксическое лекарственное средство из указанного конъюгата после его связывания и проникновения в клетки-мишени.
В предпочтительном варианте этого аспекта указанным гидролизуемым линкером является 4-(4-ацетилфенокси)бутановая кислота (AcBut).
В еще одном аспекте настоящего изобретения в процессе конъюгирования используется октановая кислота или ее соль или декановая кислота или ее соль в качестве добавки для снижения степени агрегации и увеличения нагрузки лекарственным средством.
В еще одном аспекте настоящего изобретения конъюгаты настоящего изобретения очищают методом хроматографического разделения.
В одном из вариантов осуществления изобретения указанным методом хроматографического разделения, используемым для разделения конъюгата "производное мономерного лекарственного средства - носитель", является эксклюзионная хроматография (SEC).
В другом варианте осуществления изобретения методом хроматографического разделения, используемым для разделения конъюгата "производное мономерного лекарственного средства - носитель", является ВЭЖХ, ЖЭХБ или хроматография на Сефакриле S-200.
В предпочтительном варианте осуществления изобретения методом хроматографического разделения, используемым для разделения конъюгата "производное мономерного лекарственного средства - носитель", является гидрофобная хроматография (ГФХ). В особенно предпочтительном варианте осуществления изобретения ГФХ осуществляют с использованием в качестве хроматографической среды фенилсефарозы 6 Fast Flow, бутилсефарозы 4 Fast Flow, октилсефарозы 4 Fast Flow, Toyopearl Ether-650M, метиловой ГФХ-среды Macro-Prep или трет-бутиловой ГФХ-среды Macro-Prep. В более предпочтительном варианте осуществления изобретения ГФХ осуществляют с использованием в качестве хроматографической среды бутилсефарозы 4 Fast Flow.
В другом своем аспекте настоящее изобретение относится к конъюгату "производное мономерного цитотоксического лекарственного средства/носитель", полученному способом настоящего изобретения. В предпочтительном варианте этого аспекта используемым цитотоксическим лекарственным средством является калихеамицин, а используемым носителем является антитело.
В другом предпочтительном варианте осуществления изобретения указанное антитело выбрано из группы, состоящей из моноклонального антитела, химерного антитела, человеческого антитела, гуманизированного антитела, одноцепочечного антитела, Fab-фрагмента и F(ab)2-фрагмента. В более предпочтительном аспекте изобретения используется гуманизированное антитело, направленное против антигена клеточной поверхности CD22.
В одном из вариантов осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, которое включает вариабельную область легкой цепи 5/44-gLl (SEQ ID NO: 19) и вариабельную область тяжелой цепи 5/44-gH7 (SEQ ID NO: 27).
В другом варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, содержащее легкую цепь, имеющую последовательность, представленную в SEQ ID NO: 28.
В предпочтительном варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, содержащее тяжелую цепь, имеющую последовательность, представленную в SEQ ID NO: 30.
В другом предпочтительном варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, содержащее легкую цепь, имеющую последовательность, представленную в SEQ ID NO: 28, и тяжелую цепь, имеющую последовательность, представленную в SEQ ID NO: 30.
В еще одном варианте осуществления изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, которое представляет собой вариант антитела, полученный в соответствии с протоколом осуществления аффинного созревания и обладающий повышенной специфичностью к человеческому CD22.
В предпочтительном варианте осуществления изобретения указанным калихеамицином является гамма-калихеамицин или N-ацетил-гамма-калихеамицин.
В одном из вариантов осуществления изобретения указанное производное калихеамицина имеет функциональную 3-меркапто-3-метилбутаноилгидразидную группу.
В другом варианте осуществления изобретения линкером, используемым для конъюгирования указанного лекарственного средства с указанным носителем, является гидролизуемый линкер, способный высвобождать цитотоксическое лекарственное средство из конъюгата после его связывания и проникновения в клетки-мишени. В предпочтительном варианте осуществления изобретения указанным гидролизуемым линкером является 4-(4-ацетилфенокси)бутановая кислота (AcBut).
В другом своем аспекте настоящее изобретение относится к конъюгату "производное мономерного калихеамицина/анти-CD22 антитело", имеющему формулу: Pr(-X-S-S-W)m, где: Pr представляет собой анти-CD22 антитело; X представляет собой гидролизуемый линкер, включающий продукт реакции любой реакционно-способной группы, которая может взаимодействовать с антителом; W представляет собой радикал калихеамицина; m означает среднюю нагрузку для очищенного продукта конъюгирования, такую, при которой указанный калихеамицин составляет 4-10% по массе конъюгата; a (-X-S-S-W)m представляет собой производное калихеамицина, полученное способом настоящего изобретения.
В одном из вариантов этого аспекта указанное антитело выбрано из группы, состоящей из моноклонального антитела, химерного антитела, человеческого антитела, гуманизированного антитела, одноцепочечного антитела, Fab-фрагмента и F(ab)2-фрагмента.
В предпочтительном варианте осуществления изобретения указанным антителом является анти-CD22 антитело, которое обладает специфичностью к человеческому CD22 и включает тяжелую цепь, где вариабельный домен содержит CDR, имеющую, по крайней мере, одну из последовательностей, представленных на фиг.1 и обозначенных H1 (SEQ ID NO: 1) для CDR-H1, Н2 (SEQ ID NO: 2), Н2′ (SEQ ID NO: 13), Н2′′ (SEQ ID NO: 15) или Н2′′′ (SEQ ID NO: 16) для CDR-H2; или Н3 (SEQ ID NO: 3) для CDR-H3; и легкую цепь, где вариабельный домен содержит CDR, имеющую, по крайней мере, одну из последовательностей, представленных на фиг.1 и обозначенных L1 (SEQ ID NO: 4) для CDR-L1, L2 (SEQ ID NO: 5) для CDR-L2 или L3 (SEQ ID NO: 6) для CDR-L3.
В другом предпочтительном варианте осуществления изобретения анти-CD22 антитело включает тяжелую цепь, где вариабельный домен содержит CDR, имеющую, по крайней мере, одну из последовательностей, представленных в SEQ ID NO: 1 для CDR-H1, SEQ ID NO: 2, SEQ ID NO: 13, SEQ ID NO: 15 или SEQ ID NO: 16 для CDR-H2, или SEQ ID NO: 3 для CDR-H3, и легкую цепь, где вариабельный домен содержит CDR, имеющую, по крайней мере, одну из последовательностей, представленных в SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 или SEQ ID NO: 6 для CDR-L3.
В еще одном предпочтительном варианте осуществления изобретения анти-CD22 антитело содержит SEQ ID NO: 1 для CDR-H1; SEQ ID NO: 2, SEQ ID NO: 13, SEQ ID NO: 15 или SEQ ID NO: 16 для CDR-H2; SEQ ID NO: 3 для CDR-H3; SEQ ID NO: 4 для CDR-L1; SEQ ID NO: 5 для CDR-L2 или SEQ ID NO: 6 для CDR-L3.
В другом варианте осуществления изобретения гуманизированным анти-CD22 антителом является CDR-привитое анти-CD22 антитело, которое содержит вариабельный домен, включающий человеческие акцепторные каркасные области и не человеческие донорные CDR.
В другом варианте осуществления изобретения гуманизированное анти-CD22 антитело имеет человеческую акцепторную каркасную область, где области вариабельного домена тяжелой цепи указанного антитела основаны на человеческой консенсусной последовательности подгруппы I и включают не человеческие донорные остатки в положениях 1, 28, 48, 71 и 93. В другом варианте осуществления изобретения, указанное гуманизированное антитело, кроме того, включает не человеческие донорные остатки в положениях 67 и 69.
В одном из предпочтительных вариантов осуществления изобретения CDR-привитое гуманизированное антитело содержит вариабельный домен легкой цепи, включающий человеческую акцепторную каркасную область, основанную на человеческой консенсусной последовательности подгруппы I, и кроме того, включающий не человеческие донорные остатки в положениях 2, 4, 37, 38, 45 и 60. В другом варианте осуществления изобретения, указанное CDR-привитое антитело, кроме того, включает не человеческий донорный остаток в положении 3.
В другом варианте осуществления изобретения CDR-привитое антитело включает вариабельную область легкой цепи 5/44-gL1 (SEQ ID NO: 19) и вариабельную область тяжелой цепи 5/44-gH7 (SEQ ID NO: 27).
В другом варианте осуществления изобретения указанное CDR-привитое антитело включает легкую цепь, имеющую последовательность, представленную в SEQ ID NO: 28, и тяжелую цепь, имеющую последовательность, представленную в SEQ ID NO: 30.
В другом предпочтительном варианте осуществления изобретения указанное CDR-привитое антитело содержит легкую цепь, имеющую последовательность, представленную в SEQ ID NO: 28, и тяжелую цепь, имеющую последовательность, представленную в SEQ ID NO: 30.
В еще одном варианте осуществления изобретения указанным CDR-привитым анти-CD22 антителом является вариант антитела, полученный в соответствии с протоколом осуществления аффинного созревания и обладающий повышенной специфичностью к человеческому CD22.
В другом варианте осуществления изобретения указанным анти-CD22 антителом является химерное антитело, содержащее последовательности вариабельных доменов легкой и тяжелой цепей моноклонального антитела, представленные в SEQ ID NO: 7 и SEQ ID NO: 8, соответственно.
В еще одном варианте осуществления изобретения указанное анти-CD22 антитело содержит гибридную CDR с усеченной донорной последовательностью CDR, где на месте отсутствующей части донорной CDR находится другая заменяющая ее последовательность, в результате чего образуется функциональная CDR.
В особенно предпочтительном варианте осуществления изобретения указанным производным цитотоксического лекарственного средства является либо гамма-калихеамицин, либо производное N-ацетил гамма-калихеамицин.
В другом своем аспекте настоящее изобретение относится к способу получения стабильной лиофилизированной композиции конъюгата "производное мономерного цитотоксического лекарственного средства/носитель". В предпочтительном варианте осуществления изобретения стабильную лиофилизированную композицию конъюгата "производное мономерного цитотоксического лекарственного средства/носитель" получают путем (а) растворения указанного конъюгата "производное мономерного цитотоксического лекарственного средства/носитель" до конечной концентрации 0,5-2 мг/мл в растворе, содержащем криозащитный агент в концентрации 1,5-5 мас.%, полимерный наполнитель в концентрации 0,5-1,5 мас.%, электролиты в концентрации 0,01-0,1М, агент, способствующий растворению в концентрации 0,005-0,05 мас.%, забуферивающий агент в концентрации 5-50 мМ, необходимой для доведения конечного рН раствора до 7,8-8,2, и воду; (b) распределения вышеуказанного раствора по сосудам при температуре от +5°С до +10°С; (с) замораживания указанного раствора при температуре замораживания от -35°С до -50°С; (d) проведения предварительной стадии лиофилизации замороженного раствора для его первичной сушки под давлением 20-80 мкПа и при температуре хранения от -10°С до -40°С в течение 24-78 часов; и (е) проведения вторичной сушки лиофилизированного продукта стадии (d) под давлением 20-80 мкПа и при температуре хранения от +10°С до +35°С в течение 15-30 часов.
В одном из вариантов осуществления изобретения, криозащитный агент, используемый для лиофилизации конъюгата "цитотоксическое лекарственное средство/носитель", выбран из альдита, маннита, сорбита, инозита, полиэтиленгликоля, альдоновой кислоты, уроновой кислоты, альдаровой кислоты, альдоз, кетоз, аминосахаров, альдитов, инозитов, глицеральдегидов, арабинозы, ликсозы, пентозы, рибозы, ксилозы, галактозы, глюкозы, гексозы, идозы, маннозы, талозы, гептозы, глюкозы, фруктозы, глюконовой кислоты, сорбита, лактозы, маннита, метил-α-глюкопиранозида, мальтозы, изоаскорбиновой кислоты, аскорбиновой кислоты, лактона, сорбозы, глюкаровой кислоты, эритрозы, треозы, арабинозы, аллозы, альтрозы, гуллозы, идозы, таллозы, эритрулозы, рибулозы, ксилулозы, псикозы, тагатозы, глюкуроновой кислоты, глюконовой кислоты, глюкаровой кислоты, галактуроновой кислоты, маннуроновой кислоты, глюкозамина, галактозамина, сахарозы, трегалозы, нейраминовой кислоты, арабинанов, фруктанов, фуканов, галактанов, галактуронанов, глюканов, маннанов, ксиланов, левана, фукоидана, карагенана, галактокаролозы, пектинов, пектиновых кислот, амилозы, пуллулана, гликогена, амилопектина, целлюлозы, декстрана, пустулана, хитина, агарозы, кератина, хондротина, дерматана, гиалуроновой кислоты, альгиновой кислоты, ксантановой камеди, крахмала, сахарозы, глюкозы, лактозы, трегалозы, этиленгликоля, полиэтиленгликоля, полипропиленгликоля, глицерина и пентаэритритола.
В предпочтительном варианте осуществления изобретения указанным криозащитным агентом является сахароза, которая присутствует в концентрации 1,5 мас.%.
В одном из вариантов осуществления изобретения указанный полимерный наполнитель, используемый в процессе лиофилизации, выбран из декстрана 40 или гидроксиэтилированного крахмала 40, и присутствует в концентрации 0,9 мас.%.
В другом варианте осуществления изобретения электролитом, используемым в растворе для лиофилизации, является хлорид натрия, который присутствует в концентрации 0,05М.
В предпочтительном варианте осуществления изобретения в процессе лиофилизации используется агент, повышающий растворимость. Указанным агентом, повышающим растворимость, предпочтительно, является поверхностно-активное вещество. В особенно предпочтительном варианте осуществления изобретения указанным поверхностно-активным веществом является полисорбат 80, который присутствует в концентрации 0,01 мас.%.
В одном из вариантов осуществления изобретения используемым забуферивающим агентом является трометамин, который присутствует в концентрации 0,02М. При этом, предпочтительно, чтобы в начале лиофилизации рН раствора составлял 8,0. Раствор, содержащий конъюгат "цитотоксическое лекарственное средство/носитель", распределяют по сосудами при температуре +5°С до начала лиофилизации.
В предпочтительном варианте осуществления изобретения раствор в сосудах замораживают при температуре -45°С; затем проводят предварительную стадию лиофилизации для первичной сушки замороженного раствора под давлением 60 мкПа и при температуре хранения -30°С в течение 60 часов; после этого проводят вторую стадию сушки лиофилизированного продукта под давлением 60 мкПа и при температуре хранения +25°С в течение 24 часов.
В другом своем аспекте настоящее изобретение относится к композиции, содержащей терапевтически эффективную дозу конъюгата "производное мономерного цитотоксического лекарственного средства/носитель", полученного способом настоящего изобретения.
В одном из вариантов осуществления изобретения указанным носителем в конъюгате "производное мономерного цитотоксического лекарственного средства/носитель" является белковый носитель, выбранный из гормонов, факторов роста, антител и миметиков антител.
В предпочтительном варианте осуществления изобретения указанным белковым носителем является человеческое моноклональное антитело, химерное антитело, человеческое антитело или гуманизированное антитело.
В предпочтительном варианте осуществления изобретения указанное гуманизированное антитело направлено против антигена клеточной поверхности CD22.
В особенно предпочтительном варианте этого аспекта изобретения указанное анти-CD22 антитело обладает специфичностью к человеческому CD22 и включает тяжелую цепь, где вариабельный домен содержит область CDR, имеющую, по крайней мере, одну из последовательностей, представленных на фиг.1 и обозначенных H1 (SEQ ID NO: 1) для CDR-H1, Н2 (SEQ ID NO: 2), Н2′ (SEQ ID NO: 13), Н2′′ (SEQ ID NO: 15) или Н2′′′ (SEQ ID NO: 16) для CDR-H2, или Н3 (SEQ ID NO: 3) для CDR-H3; и легкую цепь, где вариабельный домен содержит CDR, имеющую, по крайней мере, одну из последовательностей, представленных на фиг.1 и обозначенных L1 (SEQ ID NO: 4) для CDR-L1, L2 (SEQ ID NO: 5) для CDR-L2 или L3 (SEQ ID NO: 6) для CDR-L3.
В другом предпочтительном варианте изобретения анти-CD22 антитело включает тяжелую цепь, где вариабельный домен содержит CDR, имеющую, по крайней мере, одну из последовательностей, представленных в SEQ ID NO: 1 для CDR-H1; SEQ ID NO: 2, SEQ ID NO: 13, SEQ ID NO: 15 или SEQ ID NO: 16 для CDR-H2; или SEQ ID NO: 3 для CDR-H3, и легкую цепь, где вариабельный домен содержит CDR, имеющую, по крайней мере, одну из последовательностей, представленных в SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 или SEQ ID NO: 6 для CDR-L3.
В еще одном предпочтительном варианте изобретения анти-CD22 антитело содержит SEQ ID NO: 1 для CDR-H1; SEQ ID NO: 2, SEQ ID NO: 13, SEQ ID NO: 15 или SEQ ID NO: 16 для CDR-H2; SEQ ID NO: 3 для CDR-H3; SEQ ID NO: 4 для CDR-L1; SEQ ID NO: 5 для CDR-L2; SEQ ID NO: 6 для CDR-L3.
В особенно предпочтительном варианте изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое гуманизированное анти-CD22 антитело, которое включает вариабельную область легкой цепи 5/44-gL1 (SEQ ID NO: 19) и вариабельную область тяжелой цепи 5/44-gH7 (SEQ ID NO: 27).
В другом особенно предпочтительном варианте изобретения указанным гуманизированным анти-CD22 антителом является CDR-привитое антитело, которое обладает специфичностью к человеческому CD22 и содержит легкую цепь, имеющую последовательность, представленную в SEQ ID NO: 28, и тяжелую цепь, имеющую последовательность, представленную в SEQ ID NO: 30.
В одном из вариантов осуществления изобретения указанным CDR-привитым антителом является вариант антитела, обладающий повышенной специфичностью к человеческому CD22 и полученный в соответствии с протоколом осуществления аффинного созревания.
В одном из вариантов осуществления изобретения указанным мономерным цитотоксическим лекарственным средством является калихеамицин, предпочтительно выбранный из гамма-калихеамицина или N-ацетил-калихеамицина.
В одном из вариантов осуществления изобретения указанная композиция может, но необязательно, содержать дополнительный биологически активный агент. Таким биологически активным агентом может быть цитотоксическое лекарственное средство, фактор роста или гормон.
В еще одном своем аспекте настоящее изобретение относится к способу лечения индивидуума с пролиферативным расстройством, предусматривающему введение такому индивидууму терапевтически эффективной дозы композиции настоящего изобретения. Указанная композиция может быть введена подкожно, внутрибрюшинно, внутривенно, внутриартериально, интрамедуллярно, интратекально, трансдермально, чрескожно, интраназально, местно, внутрикишечно, интравагинально, подъязычно или ректально. В предпочтительном варианте изобретения композицию настоящего изобретения вводят внутривенно.
В одном из вариантов осуществления изобретения указанную композицию вводят индивидууму, страдающему пролиферативным расстройством, таким как рак. В предпочтительном варианте осуществления изобретения указанным раковым заболеванием является злокачественная В-клеточная опухоль. Указанной злокачественной В-клеточной опухолью может быть лейкоз или лимфома, экспрессирующая антиген клеточной поверхности CD22.
В еще одном варианте изобретения указанным раковым заболеванием является карцинома или саркома.
В другом своем аспекте настоящее изобретение относится к способу лечения В-клеточной злокачественной опухоли, предусматривающему введение пациенту с указанной злокачественной опухолью терапевтически эффективной композиции, содержащей конъюгат "цитотоксическое лекарственное средство - анти-CD22 антитело" настоящего изобретения. В предпочтительном варианте осуществления изобретения, указанной В-клеточной злокачественной опухолью является лимфома, а в частности, не-ходжкинская лимфома.
В одном из вариантов осуществления изобретения указанное цитотоксическое лекарственное средство, используемое для получения конъюгатов настоящего изобретения, выбрано из группы, состоящей из калихеамицинов, тиотепы, таксанов, винкристина, даунорубицина, доксорубицина, эпирубицина, актиномицина, аутрамицина, азасеринов, блеомицинов, тамоксифена, идарубицина, доластатинов/ауристатинов, гемиастерлинов, маитанзиноидов и эсперамицинов.
В предпочтительном варианте осуществления изобретения указанным цитотоксическим лекарственным средством является гамма-калихеамицин или N-ацетил-калихеамицин.
В другом варианте изобретения указанное лечение предусматривает введение конъюгата цитотоксического лекарственного средства настоящего изобретения в комбинации с одним или несколькими биологически активными агентами, выбранными из антител, факторов роста, гормонов, цитокинов, антигормонов, ксантинов, интерлейкинов, интерферонов и цитотоксических лекарственных средств.
В предпочтительном варианте изобретения указанным биологически активным агентом является антитело, направленное против антигена клеточной поверхности, экспрессируемого на В-клеточных злокачественных опухолях. В другом предпочтительном варианте изобретения указанное антитело, направленное против антигенов клеточной поверхности, экспрессируемых на В-клеточных злокачественных опухолях, выбрано из группы, состоящей из анти-CD19, анти-CD20 и анти-CD33 антител. Такие антитела включают анти-CD20 антитело, ритуксимаб (Rituxan™).
В другом варианте изобретения указанными биологически активными агентами являются цитокины или факторы роста, которые включают, но не ограничиваются ими, интерлейкин 2 (IL-2), TNF, CSF, GM-CSF и G-CSF.
В другом варианте изобретения указанными биологически активными агентами являются гормоны, а именно, эстрогены, андрогены, прогестины и кортикостероиды.
В еще одном варианте осуществления изобретения указанным биологически активным агентом является цитотоксическое лекарственное средство, выбранное из доксорубицина, даунорубицина, идарубицина, акларубицина, зорубицина, митоксантрона, эпирубицина, карубицина, ногаламицина, меногарила, питарубицина, валрубицина, цитарабина, гемцитабина, трифлуридина, анцитабина, эноцитабина, азацитидина, доксифлуридина, пентостатина, броксуридина, капецитабина, кладрибина, децитабина, флоксуридина, флударабина, гугеротина, пуромицина, тегафура, тиазофурина, адриамицина, цисплатина, карбоплатина, циклофосфамида, дакарбазина, винбластина, винкристина, митоксантрона, блеомицина, мехлоретамина, преднизона, прокарбазина, метотрексата, фторурацилов, этопозида, таксола, аналогов таксола и митомицина.
В предпочтительном варианте осуществления изобретения терапевтически эффективную композицию конъюгата "цитотоксическое лекарственное средство - анти-CD22 антитело" вводят вместе с одной или несколькими комбинациями цитотоксических агентов, используемых в качестве составной части курса лечения, где указанную комбинацию цитотоксических агентов выбирают из: СНОРР (циклофосфамид, доксорубицин, винкристин, преднизон и прокарбазин); CHOP (циклофосфамид, доксорубицин, винкристин и преднизон); СОР (циклофосфамид, винкристин и преднизон); САР-ВОР (циклофосфамид, доксорубицин, прокарбазин, блеомицин, винкристин и преднизон); m-BACOD (метотрексат, блеомицин, доксорубицин, циклофосфамид, винкристин, дексаметазон и лейковорин); ProMACE-МОРР (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, мехлоэтамин, винкристин, преднизон и прокарбазин); ProMACE-CytaBOM (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, цитарабин, блеомицин и винкристин); МАСОР-В (метотрексат, доксорубицин, циклофосфамид, винкристин, преднизон, блеомицин и лейковорин); МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин); ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин); МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABV (адриамицин/доксорубицин, блеомицин и винбластин); МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин); ChlVPP (хлорамбуцил, винбластин, прокарбазин и преднизон); IMVP-16 (ифосфамид, метотрексат и этопозид); MIME (метил-gag, ифосфамид, метотрексат и этопозид); DHAP (дексаметазон, высокая доза цитарабина и цисплатин); ESHAP (этопозид, метилпреднизолон, высокая доза цитарабина и цисплатин); СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин); CAMP (ломустин, митоксантрон, цитарабин и преднизон) и CVP-1 (циклофосфамид, винкристин и преднизон).
В предпочтительном варианте осуществления изобретения терапевтически эффективную композицию конъюгата "цитотоксическое лекарственное средство - анти-CD22 антитело" вводят перед введением одной или нескольких вышеуказанных комбинаций цитотоксических лекарственных средств. В другом предпочтительном варианте осуществления изобретения терапевтически эффективную композицию конъюгата "цитотоксическое лекарственное средство - анти-CD22 антитело" вводят после введения одной или нескольких вышеуказанных комбинаций цитотоксических лекарственных средств, используемых в качестве составной части курса лечения.
В другом своем аспекте настоящее изобретение относится к способу лечения агрессивных лимфом, предусматривающему введение пациенту, нуждающемуся в таком лечении, терапевтически эффективной композиции конъюгата "производное мономерного калихеамицина - анти-CD22 антитело" вместе с одним или несколькими биологически активными агентами.
В еще одном своем аспекте настоящее изобретение относится к использованию композиции настоящего изобретения для лечения индивидуума с пролиферативным расстройством, таким как рак. В частности, указанным раком является В-клеточная злокачественная опухоль, экспрессирующая антиген CD22 на клеточной поверхности. В частности, такой злокачественной В-клеточной опухолью является лейкоз или лимфома. В одном из вариантов осуществления изобретения указанным раком является карцинома или лейкоз.
В одном из вариантов осуществления изобретения терапевтически эффективную дозу указанной композиции вводят подкожно, внутрибрюшинно, внутривенно, внутриартериально, интрамедуллярно, интратекально, трансдермально, чрескожно, интраназально, местно, внутрикишечно, интравагинально, подъязычно или ректально.
В предпочтительном варианте изобретения терапевтически эффективную дозу указанной фармацевтической композиции настоящего изобретения вводят внутривенно.
В другом своем аспекте настоящее изобретение относится к использованию конъюгата "производное мономерного калихеамицина/анти-CD22 антитело" настоящего изобретения для лечения индивидуума с В-клеточной злокачественной опухолью, такой как не-ходжкинская лимфома. В одном из вариантов изобретения указанный конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" настоящего изобретения вводят вместе с одним или несколькими биологически активными агентами.
В одном из вариантов изобретения указанные биологически активные агенты выбраны из группы, состоящей из антител, факторов роста, гормонов, цитокинов, антигормонов, ксантинов, интерлейкинов, интерферонов и цитотоксических лекарственных средств.
В предпочтительном варианте осуществления изобретения указанным биологически активным агентом является антитело, направленное против антигена клеточной поверхности, экспрессируемого на В-клеточных злокачественных опухолях, такое как антитело против CD19, CD20 или CD33. В предпочтительном варианте указанным анти-CD20 антителом является ритуксимаб (Rituxan™).
В другом варианте осуществления изобретения указанными биологически активными агентами являются цитокины или факторы роста, такие как интерлейкин 2 (IL-2), TNF, CSF, GM-CSF и G-CSF, или гормоны, включая эстрогены, андрогены, прогестины и кортикостероиды.
В другом варианте осуществления изобретения биологически активным агентом является цитотоксическое лекарственное средство, выбранное из доксорубицина, даунорубицина, идарубицина, акларубицина, зорубицина, митоксантрона, эпирубицина, карубицина, ногаламицина, меногарила, питарубицина, валрубицина, цитарабина, гемцитабина, трифлуридина, анцитабина, эноцитабина, азацитидина, доксифлуридина, пентостатина, броксуридина, капецитабина, кладрибина, децитабина, флуксуридина, флударабина, гугеротина, пуромицина, тегафура, тиазофурина, адриамицина, цисплатина, карбоплатина, циклофосфамида, дакарбазина, винбластина, винкристина, митоксантрона, блеомицина, мехлоретамина, преднизона, прокарбазина, метотрексата, фторурацилов, этопозида, таксола, аналогов таксола и митомицина.
В предпочтительном варианте осуществления изобретения терапевтически эффективную дозу конъюгата "производное мономерного калихеамицина/анти-CD22 антитело" вводят вместе с одной или несколькими комбинациями цитотоксических агентов, используемых в качестве составной части курса лечения, где указанную комбинацию цитотоксических агентов выбирают из: СНОРР (циклофосфамид, доксорубицин, винкристин, преднизон и прокарбазин); CHOP (циклофосфамид, доксорубицин, винкристин и преднизон); СОР (циклофосфамид, винкристин и преднизон); САР-ВОР (циклофосфамид, доксорубицин, прокарбазин, блеомицин, винкристин и преднизон); m-BACOD (метотрексат, блеомицин, доксорубицин, циклофосфамид, винкристин, дексаметазон и лейковорин); ProMACE-МОРР (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, мехлоэтамин, винкристин, преднизон и прокарбазин); ProMACE-CytaBOM (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, цитарабин, блеомицин и винкристин); МАСОР-В (метотрексат, доксорубицин, циклофосфамид, винкристин, преднизон, блеомицин и лейковорин); МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин); ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин); МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABV (адриамицин/доксорубицин, блеомицин и винбластин); МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин); ChlVPP (хлорамбуцил, винбластин, прокарбазин и преднизон); IMVP-16 (ифосфамид, метотрексат и этопозид); MIME (метил-gag, ифосфамид, метотрексат и этопозид); DHAP (дексаметазон, высокая доза цитарабина и цисплатин); ESHAP (этопозид, метилпреднизолон, высокая доза цитарабина и цисплатин); СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин); CAMP (ломустин, митоксантрон, цитарабин и преднизон); CVP-1 (циклофосфамид, винкристин и преднизон); ESHOP (этопозид, метилпреднизолон, высокая доза цитарабина, винкристин и цисплатин); EPOCH (этопозид, винкристин и доксорубицин), вводимой в течение 96 часов с ударными дозами циклофосфамида и перорально вводимого преднизона), ICE (ифосфамид, циклофосфамид и этопозид), СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин), СНОР-В (циклофосфамид, доксорубицин, винкристин, преднизон и блеомицин), СЕРР-В (циклофосфамид, этопозид, прокарбазин и блеомицин) и P/DOCE (эпирубицин или доксорубицин, винкристин, циклофосфамид и преднизон).
В одном из предпочтительных вариантов осуществления изобретения конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" вводят перед введением одной или нескольких комбинаций цитотоксических агентов как часть курса лечения.
В другом предпочтительном варианте осуществления изобретения терапевтически эффективную дозу конъюгата "производное мономерного калихеамицина/анти-CD22 антитело" вводят после введения одной или нескольких комбинаций цитотоксических агентов как часть курса лечения.
В другом предпочтительном варианте осуществления изобретения терапевтически эффективную дозу конъюгата "производное мономерного калихеамицина/анти-CD22 антитело" вводят вместе с антителом, направленным против антигена клеточной поверхности, присутствующего на В-клеточных злокачественных опухолях, и эта терапевтически эффективная доза необязательно содержит одну или несколько комбинаций цитотоксических агентов как часть курса лечения.
В другом своем аспекте настоящее изобретение относится к использованию конъюгата "производное мономерного калихеамицина/анти-CD22 антитело" в целях изготовления лекарственного средства для лечения пролиферативного расстройства. Такое лекарственное средство может быть использовано для лечения В-клеточных пролиферативных расстройств либо отдельно, либо в комбинации с другими биологически активными агентами.
Краткое описание чертежей
На фиг.1 представлена аминокислотная последовательность CDR мышиного моноклонального антитела 5/44 (SEQ ID NO: 1-6).
На фиг.2 представлена ДНК и аминокислотная последовательность вариабельного домена (VL) легкой цепи мышиного моноклонального антитела 5/44.
На фиг.3 представлена ДНК и аминокислотная последовательность вариабельного домена (VH) тяжелой цепи мышиного моноклонального антитела 5/44.
На фиг.4 показана стратегия удаления сайта гликозилирования и реакционно-способного лизина из CDR-H2.
На фиг.5 показана привитая конструкция для последовательности легкой цепи 5/44. DPK-9 представляет собой акцепторную каркасную последовательность человеческой зародышевой линии. Вертикальные линии указывают на различия между мышиными и человеческими остатками. Подчеркнутые последовательности указывают на донорные остатки, которые были сохранены в трансплантате. CDR обозначены курсивом, набранным жирным шрифтом (для DPK-9 не показано). Трансплантат gL1 имеет 6 донорных каркасных остатков, a gL2 имеет 7 таких остатков.
На фиг.6 показана привитая конструкция для последовательности тяжелой цепи 5/44. DP7 представляет собой акцепторную каркасную последовательность человеческой зародышевой линии. Вертикальные линии указывают на различия между мышиными и человеческими остатками. Подчеркнутые последовательности указывают на донорные остатки, которые были сохранены в трансплантате. CDR обозначены курсивом, набранным жирным шрифтом (для DP7 не показано). Трансплантаты gH4 и gH6 имеют 6 донорных каркасных остатков. Трансплантаты gH5 и gH7 имеют 4 донорных каркасных остатка.
На фиг.7 представлена карта вектора pMRR14.
На фиг.8 представлена карта вектора pMRR10.1.
На фиг.9 представлены результаты Biacore-анализа химерных мутантов 5/44.
На фиг.10 представлены олигонуклеотиды для сборки генов gHl и gL1 5/44.
На фиг.11 представлена плазмидная карта промежуточного вектора pCR2.1 (544gHl).
На фиг.12 представлена плазмидная карта промежуточного вектора pCR2.1 (544gLl).
На фиг.13 представлены олигонуклеотидные кластеры, используемые для создания дополнительных трансплантатов.
На фиг.14 представлен график, который иллюстрирует анализ на конкуренцию между флуоресцентно меченным мышиным антителом 5/44 и привитыми вариантами.
На фиг.15 представлен график, который иллюстрирует анализ на конкуренцию между флуоресцентно меченным мышиным антителом 5/44 и привитыми вариантами.
На фиг.16 представлена полноразмерная ДНК и аминокислотная последовательность привитых тяжелой и легкой цепей.
На фиг.17 схематически представлен конъюгат "антитело-NAc-гамма-калихеамицин-DMH".
На фиг.18 представлен график, иллюстрирующий влияние СМС-544 на рост В-клеточной лимфомы RAMOS.
На фиг.19 представлен график, иллюстрирующий влияние СМС-544 на рост крупноклеточной В-лимфомы в модели ксенотрансплантата in vivo у "голых" мышей.
На фиг.20 представлен график, иллюстрирующий сравнение влияния СМС-544, полученного методом конъюгирования СМА-676 и методом конъюгирования СМС-544, на рост RL-лимфомы.
На фиг.21 представлен график, указывающий на то, что обработанная ритуксимабом (Rituxan™) крупноклеточная RL-лимфома является чувствительной к СМС-544-обработке.
На фит.22 представлен график, иллюстрирующий влияние ритуксимаба (Rituxan™) на цитотоксическое действие СМС-544.
На фиг.23 представлен график, иллюстрирующий влияние СМС-544, ритуксимаба (Rituxan™) и СМА-676 на выживание мышей SCID с диссеминированной ранней В-клеточной лимфомой RAMOS.
На фиг.24 представлен график, иллюстрирующий влияние СМС-544, ритуксимаба (Rituxan™) и СМА-676 на выживание мышей SCID с диссеминированной поздней В-клеточной лимфомой RAMOS.
На фиг.25 представлен график, иллюстрирующий влияние СМС-544, ритуксимаба (Rituxan™) и СМА-676 на выживание мышей SCID с диссеминированной поздней В-клеточной лимфомой RAMOS.
На фиг.26 представлен график, иллюстрирующий влияние СМС-544, ритуксимаба (Rituxan™) и СМА-676 на выживание мышей SCID с диссеминированной поздней В-клеточной лимфомой RAMOS.
На фиг.27 представлен график, иллюстрирующий влияние СМС-544, ритуксимаба (Rituxan™) и СМА-676 на выживание мышей SCID с диссеминированной поздней В-клеточной лимфомой RAMOS.
На фиг.28 представлен график, иллюстрирующий противоопухолевую активность СМС-544 с ритуксимабом (Rituxan™) или без него, направленную против не-ходжкинской RL-лимфомы.
На фиг.29 представлен график, иллюстрирующий противоопухолевую активность СМС-544 и CHOP, направленную против не-ходжкинской RL-лимфомы.
Подробное описание изобретения
Конъюгаты настоящего изобретения содержат цитотоксическое лекарственное средство, дериватизированное линкером, который включает любую реакционно-способную группу, реагирующую с белковым носителем с образованием конъюгата "производное цитотоксического лекарственного средства - белковый носитель". В частности, конъюгаты настоящего изобретения содержат цитотоксическое лекарственное средство, дериватизированное линкером, который включает любую реакционно-способную группу, реагирующую с антителом, используемым в качестве белкового носителя, с образованием конъюгата "производное цитотоксического лекарственного средства - антитело". В частности, указанное антитело реагирует с антигеном клеточной поверхности на В-клеточных злокачественных опухолях. Ниже описан усовершенствованный способ получения и очистки таких конъюгатов. Использование конкретных сорастворителей, добавок и конкретных реакционных условий в комбинации с процедурой разделения приводит к образованию конъюгата "производное мономерного цитотоксического лекарственного средства/антитело" со значительно более низким содержанием LCF. В противоположность агрегированной форме, мономерная форма имеет значительно более высокий терапевтический индекс, а минимизация LCF и значительное снижение степени агрегации позволяет использовать указанное антитело в качестве исходного материала в эффективном терапевтическом лечении благодаря предотвращению конкуренции LCF с более высоко конъюгированной фракцией (HCF).
I. Носители
Носители/нацеливающие агенты настоящего изобретения, предпочтительно, представляют собой белковые носители/нацеливающие агенты. Такими носителями/нацеливающими агентами являются гормоны, факторы роста, антитела, фрагменты антител, миметики антител и их генетически или ферментативно сконструированные аналоги, которые, независимо от того, упоминаются ли они отдельно или группой, будут далее называться "носителями" или группами "носителей". Главным свойством носителя является его способность распознавать антиген или рецептор, ассоциированный с нежелательными клетками, и связываться с ним с последующей его интернализацией. Примерами носителей, используемых в настоящем изобретении, являются носители, описанные в патенте США №5053394, который во всей своей полноте вводится в настоящее описание посредством ссылки. Носителями, предпочтительными для использования в настоящем изобретении, являются антитела и миметики антител.
Для генерирования миметиков антитела, которые связываются с антигенными детерминантами со специфичностью антитела, были использованы не иммуноглобулиновые белковые каркасные молекулы (публикация РСТ №WO 00/34784). Так, например, каркас "миниантитела", который имеет укладку, подобную иммуноглобулиновой укладке, был сконструирован путем делеции трех бета-цепей из вариабельного домена тяжелой цепи моноклонального антитела (Tramontano et al., J. Mol. Recognit. 7:9, 1994). Этот белок включает 61 остаток и может быть использован для представления двух гипервариабельных петель. Эти две петли были рандомизированы, а продукты были отобраны для связывания с антигеном, однако совершенно очевидно, что каркасная область имеет достаточно ограниченное применение из-за плохой растворимости. Другой каркасной областью, используемой для представления петель, является тендамистат, т.е. белок, который специфически ингибирует альфа-амилазы млекопитающих и представляет собой "сэндвич"-структуру из 74 остатков, состоящую из 6-цепочечных бета-складчатых слоев, связанных вместе двумя дисульфидными связями (McConnell & Hoess, J. Mol. Biol. 250:460, 1995). Этот каркас включает три петли, но в настоящее время только две из этих петель были оценены на возможность рандомизации.
Другие белки были протестированы на каркасные области и были использованы для представления рандомизированных остатков на альфа-спиральных поверхностях (Nord et al., Nat. Biotechnol. 15:772, 1997; Nord et al., Protein Eng. 8:601, 1995) и для представления петель между альфа-спиралями в пучках альфа-спиралей (Ku & Schultz, Proc. Natl. Acad. Sci. USA 92:6552, 1995) и петель, ограниченных дисульфидными мостиками, такими как дисульфидные мостики ингибиторов малых протеаз (Markland et al., Biochemistry 35:8045, 1996; Markland et al., Biochemistry 35:8058, 1996; Rottgen & Collins, Gene 164:243, 1995; Wang et al., J. Biol. Chem. 270:12250, 1995).
Примерами антител-носителей, которые могут быть использованы в настоящем изобретении, являются моноклональные антитела, химерные антитела, гуманизированные антитела, человеческие антитела и их биологически активные фрагменты. Предпочтительно, такие антитела направлены против антигенов клеточной поверхности, экспрессируемых на клетках-мишенях и/или тканях, ассоциированных с пролиферативными расстройствами, такими как рак. Примерами специфических антител, направленных против антигенов клеточной поверхности на клетках-мишенях, являются, но не ограничиваются ими, антитела против антигена CD22, который сверхэкспрессируется на большинстве В-клеточных лимфом; G5/44, гуманизированная форма мышиного анти-CD22 моноклонального антитела; антитела против антигена клеточной поверхности CD33, который преобладает на некоторых человеческих миелоидных опухолях, в частности, при остром миелоидном лейкозе; hP67.6, гуманизированная форма мышиного анти-CD33 антитела (см. патент США №5773001); антитело против антигена РЕМ, обнаруживаемого на множестве опухолей эпителиального происхождения, которое было обозначено mP67.6 (см. I.D. Bernstein et al., J. Clin. Invest. 79:1153 (1987) и I.D. Bernstein et al., J. Immunol. 128:867-881 (1992)); и гуманизированное антитело против углеродного антигена Lewis Y, сверхэкпрессированного на многих солидных опухолях, обозначенное hu3S193 (см. патент США №6310185 В1). Кроме того, в качестве носителей/агентов для доставки могут быть также использованы несколько коммерчески доступных антител, таких как ритуксимаб (Rituxan™) и трастузумаб (Herceptin™). Ритуксимаб (Rituxan™) представляет собой химерное анти-CD20 антитело, используемое для лечения различных В-клеточных лимфом, а трастузумаб (Herceptin™) представляет собой гуманизированное анти-Her2 антитело, используемое для лечения рака молочной железы.
Примером антитела, используемого в настоящем изобретении в качестве носителя, является молекула CDR-привитого гуманизированного антитела, направленная против антигена клеточной поверхности CD22 и обозначенная G5/44. Это антитело представляет собой гуманизированную форму мышиного моноклонального анти-CD22 антитела, направленного против антигена клеточной поверхности CD22, который преобладает на некоторых человеческих лимфомах. Используемый здесь термин "молекула CDR-привитого антитела" означает молекулу антитела, в которой тяжелая и/или легкая цепи содержат одну или несколько определяющих комплементарность областей (CDR), включая, если необходимо, модифицированную CDR (далее называемую просто CDR), где CDR от донорного антитела (например, мышиного моноклонального антитела) была привита к каркасной вариабельной области тяжелой и/или легкой цепей акцепторного антитела (например, человеческого антитела). Предпочтительно, чтобы такое CDR-привитое антитело имело вариабельный домен, содержащий человеческие акцепторные каркасные области, а также одну или несколько донорных CDR, определенных выше.
Если CDR являются привитыми, то может быть использована любая подходящая последовательность каркасной акцепторной вариабельной области, относящаяся к такому же классу/типу, как и донорное антитело, от которого происходят указанные CDR, включая каркасные области мыши, приматов и человека. Примерами каркасных человеческих областей, которые могут быть использованы в настоящем изобретении, являются KOL, NEWM, REI, EU, TUR, TEI, LAY и POM (Rabat et al., Seq. of Proteins of Immunol. Interest, 1:310-334 (1994)). Так, например, KOL и NEWM могут быть использованы для тяжелой цепи, REI может быть использована для легкой цепи, a EU, LAY и РОМ могут быть использованы как для тяжелой, так и для легкой цепи.
В CDR-привитом антителе настоящего изобретения в качестве акцепторного антитела предпочтительно использовать антитело, имеющее цепи, гомологичные цепям донорного антитела. Акцепторные тяжелые и легкие цепи необязательно должны происходить от одного и того же антитела, и, если это необходимо, то они могут содержать составные цепи, имеющие каркасные области, происходящие от других цепей.
Кроме того, в CDR-привитом антителе настоящего изобретения каркасные области необязательно должны иметь последовательность, точно совпадающую с последовательностью акцепторного антитела. Так, например, необычные остатки могут быть заменены остатками, более часто встречающимися в антителах, принадлежащих к такому же классу или типу, как и акцепторное антитело. Альтернативно, выбранные остатки в акцепторных каркасных областях могут быть заменены так, чтобы они соответствовали остаткам, присутствующим в том же положении донорного антитела, либо они могут быть заменены консервативным остатком, находящимся в том же положении донорного антитела. Такие условия замены должны соблюдаться, по крайней мере, для сохранения аффинности донорного антитела. Протокол отбора остатков в акцепторных каркасных областях, которые, если это необходимо, должны быть заменены, описан в публикации РСТ №WO 91/09967, которая во всей своей полноте вводится в настоящее описание посредством ссылки.
Донорными остатками являются остатки, происходящие от донорного антитела, то есть антитела, от которого происходят CDR.
Антитело настоящего изобретения может содержать тяжелую цепь, где вариабельный домен в качестве CDR-H2 (как определено Rabat et al. (см. выше)) включает область H2', в которой возможная последовательность сайта гликозилирования была удалена в целях повышения аффинности антитела к антигену.
Альтернативно или дополнительно, антитело настоящего изобретения может содержать тяжелую цепь, где вариабельный домен в качестве CDR-H2 включает (как определено Rabat et al. (см. выше)) область Н2", в которой лизиновый остаток находится в положении 60. Этот лизиновый остаток, который локализован в доступном положении в области CDR-H2 и который рассматривается как остаток, способный вступать в реакцию с агентами конъюгирования, приводящую к снижению аффинности связывания с антигеном, заменен альтернативной аминокислотой.
Кроме того, антитело настоящего изобретения может содержать тяжелую цепь, где вариабельный домен в качестве CDR-H2 (как определено Rabat et al. (см. выше)) включает область Н2′′′, в которой возможная последовательность сайта гликозилирования и лизиновый остаток в положении 60 заменены альтернативными аминокислотами.
Антитело настоящего изобретения может включать: целое антитело, имеющее полноразмерные тяжелые и легкие цепи; его биологически активный фрагмент, такой как Fab, модифицированный Fab, Fab′, F(ab′)2 или Fv-фрагмент; мономер или димер легкой цепи или тяжелой цепи, или одноцепочечное антитело, например, одноцепочечный Fv, в котором вариабельные домены тяжелой и легкой цепей связаны пептидным линкером. Аналогичным образом, вариабельные области тяжелой и легкой цепей могут быть объединены, если это необходимо, с другими доменами антитела.
Антитело настоящего изобретения может также включать модифицированный Fab-фрагмент, где указанной модификацией является добавление одной или нескольких аминокислот, необходимых для присоединения эффекторной или репортерной молекулы к С-концу его тяжелой цепи. Предпочтительно, чтобы дополнительные аминокислоты образовывали модифицированную шарнирную область, содержащую один или два цистеиновых остатка, к которым может быть присоединена эффекторная или репортерная молекула.
Домены константной области антитела настоящего изобретения, если они присутствуют, могут быть выбраны, исходя из предполагаемой функции указанного антитела, а в частности, зффекторных функций, которые могут быть, а могут и не быть, необходимыми. Так, например, в качестве доменов константной области могут служить домены человеческого IgA, IgD, IgE, IgG или IgM. В частности, если данное антитело предназначено для терапевтического использования и его эффекторные функции являются необходимыми, то могут быть использованы домены константной области человеческого IgG, особенно изотипов IgG1 и IgG3. Альтернативно, если указанное антитело предназначено для терапевтического использования, но его эффекторные функции не являются необходимыми или желательными, то могут быть использованы изотипы IgG2 и IgG4, либо Fc-область IgG1 может быть мутирована для отмены эффекторной функции.
Аффинность связывания антитела настоящего изобретения составляет, по крайней мере, 5×10-8М, предпочтительно, по крайней мере, 1×10-9М, более предпочтительно, по крайней мере, 0,75×10-10М, а наиболее предпочтительно, по крайней мере, 0,5×10-10М.
В одном из своих вариантов настоящее изобретение относится к конъюгатам иммунотоксина и к способам получения таких конъюгатов с использованием вариантов антител или миметиков антител. В предпочтительном варианте осуществления изобретения варианты антитела настоящего изобретения направлены против CD22 и обладают повышенной аффинностью по отношению к CD22. Такие варианты могут быть получены в соответствии с различными протоколами обеспечения созревания аффинности, включая мутирование CDR (Yang et al., J. Mol. Biol. 254, 392-403, 1995), перестановку цепей (Marks et al., Bio/Technology, 10, 779-783, 1992), использование штаммов-мутантов E.coli (Low et al., J. Mol. Biol., 250, 359-368, 1996), перестановку ДНК (Patten et al., Curr. Opin. Biotechnol., 8, 724-733, 1997), фаговое представление (Thompson et al., J. Mol. Biol. 256, 77-88, 1996) и ПЦР по аллельному исключению (только аллеля, соответствующего одному полу) (Crameri et al., Nature, 391, 288-291, 1998).
Для экспрессии ДНК-последовательностей, кодирующих носитель, включая антитела настоящего изобретения, может быть использована любая подходящая система клетка-хозяин/вектор. При этом могут быть использованы бактериальные системы, например, E.coli, и другие микробные системы, отчасти для экспрессии фрагментов антител, таких как Fab- и F(ab′)2-фрагменты, а особенно Fv-фрагменты и фрагменты одноцепочечного антитела, например, одноцепочечные Fv-фрагменты. Для продуцирования более крупного антитела, включая полноразмерные молекулы антител, могут быть использованы эукариотические экспрессионные системы клеток-хозяев, например, клетки млекопитающих. Подходящими клетками-хозяевами млекопитающих являются СНО, миеломные клетки, дрожжевые клетки, клетки насекомых, гибридомные клетки, клетки NSO, VERO или PER C6. Подходящими экспрессионными системами также являются трансгенные животные и растения.
II. Терапевтические агенты
Терапевтические агенты, подходящие для использования в настоящем изобретении, представляют собой цитотоксические лекарственные средства, которые ингибируют или прерывают полимеризацию тубулина; алкилирующие агенты, которые связываются с ДНК и разрушают ее, и агенты, которые ингибируют синтез белка или основных клеточных белков, таких как протеинкиназы, ферменты и циклины. Примерами таких цитотоксических лекарственных средств являются, но не ограничиваются ими, тиотепа, таксаны, винкристин, даунорубицин, доксорубицин, эпирубицин, актиномицин, аутрамицин, азасерины, блеомицины, тамоксифен, идарубицин, доластатины/ауристатины, гемиастерлины, калихеамицины, эсперамицины и маитанзиноиды. Предпочтительными цитотоксическими лекарственными средствами являются калихеамицины, которые относятся к метилтрисульфидным противоопухолевым антибиотикам. Примеры калихеамицинов, которые могут быть использованы в настоящем изобретении, описаны, например, в патентах США №№4671958, 4970198, 5053394, 5037651 и 5079233, которые во всей своей полноте вводятся в настоящее описание посредством ссылки. Предпочтительными калихеамицинами являются производные гамма-калихеамицина или производные N-ацетил-гамма-калихеамицин.
III. Конъюгаты "производное цитотоксического лекарственного средства/носитель"
Конъюгаты настоящего изобретения имеют формулу Pr(-X-W)m,
где Pr представляет собой белковый носитель;
X представляет собой линкер, включающий продукт реакции любой реакционно-способной группы, которая может взаимодействовать с белковым носителем;
W представляет собой цитотоксическое лекарственное средство;
m означает среднюю нагрузку для очищенного продукта конъюгирования, такую, при которой указанный калихеамицин составляет 4-10% по массе конъюгата; и
(-X-W)m представляет собой цитотоксическое лекарственное средство.
Предпочтительно, X имеет формулу:
(CO-Alk1-Sp1-Ar-Sp2-Alk2-C(Z1)=Q-Sp),
где Alk1 и Alk2 независимо представляют собой связь или разветвленную или неразветвленную (C1-С10)алкиленовую цепь;
Sp1 представляет собой связь, -S-, -O-, -CONH, -NHCO-, -NR′-, -N(CH2CH2)2N- или -X-Ar1-Y-(CH2)n-Z, где X, Y и Z независимо представляют собой связь, -NR′-, -S- или -O-, при условии, что если n=0, то, по крайней мере, один из Y и Z должен представлять собой связь, а Ar1 представляет собой 1,2-, 1,3- или 1,4-фенилен, необязательно замещенный одной, двумя или тремя группами, выбранными из (С1-С5)алкила, (C1-C4)алкокси, (C1-С4)тиоалкокси, галогена, нитро, -COOR′, -CONHR′, -(СН2)nCOOR′, -S(CH2)nCOOR′, -O(CH2)nCONHR′ или -S(CH2)nCONHR′, при условии, что если Alk1 представляет собой связь, то Sp1 также представляет собой связь;
n равно целому числу от 0 до 5;
R′ представляет собой разветвленную или неразветвленную (С1-С5)цепь, необязательно замещенную одной или двумя группами, выбранными из -ОН, (C1-C4)алкокси, (C1-C4)тиоалкокси, галогена, нитро, (C1-C3)диалкиламино или (C1-С3)триалкиламмоний-А-, где А- представляет собой фармацевтически приемлемый анион, образующий соль;
Ar представляет собой 1,2-, 1,3- или 1,4-фенилен, необязательно замещенный одной, двумя или тремя группами, выбранными из (C1-C6)алкила, (C1-C5)алкокси, (C1-C4)тиоалкокси, галогена, нитро, -COOR′, -CONHR′, -О(СН2)nCOOR′, -S(CH2)nCOOR′, -O(CH2)nCONHR′ или -S(CH2)nCONHR′, где n и R′ определены выше, либо Ar представляет собой 1,2-, 1,3-, 1,4-, 1,5-, 1,6-, 1,7-, 1,8-, 2,3-, 2,6- или 2,7-нафтилиден, или
где каждый нафтилиден или фенотиазин необязательно замещены одной, двумя, тремя или четырьмя группами, выбранными из (C1-C6)алкила, (C1-C5)алкокси, (C1-C4)тиоалкокси, галогена, нитро, -COOR′, -CONHR′, -O(CH2)nCOOR′, -S(CH2)nCOOR′ или -S(CH2)nCONHR′, где n и R′ определены выше, при условии, что если Ar представляет собой фенотиазин, то Sp1 представляет собой связь, присоединенную только к азоту;
Sp2 представляет собой связь, -S- или -O-, при условии, что если Alk2 представляет собой связь, то Sp2 также представляет собой связь;
Z1 представляет собой Н, (С1-С5)алкил или фенил, необязательно замещенный одной, двумя или тремя группами, выбранными из (С1-С5)алкила, (C1-C5)алкокси, (С1-С4)тиоалкокси, галогена, нитро, -COOR′, -ONHR′, -О(СН2)nCOOR′, -S(CH2)nCOOR′, -О(CH2)nCONHR′ или -S(CH2)nCONHR′, где n и R′ определены выше;
Sp представляет собой прямой или разветвленный двухвалентный или трехвалентный (C1-C18)радикал, двухвалентный или трехвалентный арильный или гетероарильный радикал, двухвалентный или трехвалентный (С1-C18)циклоалкильный или гетероциклоалкильный радикал, двухвалентный или трехвалентный арил- или гетероариларил(C1-C18)радикал, двухвалентный или трехвалентный циклоалкил- или гетероциклоалкилалкил(C1-C18)радикал или двухвалентный или трехвалентный ненасыщенный (С2-С18)алкильный радикал, где гетероарил, предпочтительно, представляет собой фурил, тиенил, N-метилпирролил, пиридинил, N-метилимидазолил, оксазолил, пиримидинил, хинолил, изохинолил, N-метилкарбазоил, аминокумаринил или феназинил, и где, в том случае, если Sp представляет собой трехвалентный радикал, то Sp может быть, кроме того, замещен низшей (С1-С5)диалкиламиногруппой, низшей (C1-C5)алкоксигруппой, гидроксигруппой или низшей (C1-С5)алкилтиогруппой; и
Q представляет собой =NHNCO-, =NHNCS-, =NHNCONH-, =NHNCSNH- или =NHO-.
Alk1 предпочтительно представляет собой разветвленную или неразветвленную (C1-С10)алкиленовую цепь; Sp′ представляет собой связь, -S-, -O-, -CONH-, -NHCO- или -NR′, где R′ определен выше, при условии, что если Alk1 представляет собой связь, то Sp1 также представляет собой связь;
Ar представляет собой 1,2-, 1,3- или 1,4-фенилен, необязательно замещенный одной, двумя или тремя группами, выбранными из (С1-С6)алкила, (C1-C5)алкокси, (C1-C4)тиоалкокси, галогена, нитро, -COOR′, -CONHR′, -О(СН2)nCOOR′, -S(CH2)nCOOR′, -O(CH2)nCONHR′ или -S(CH2)nCONHR′, где n и R′ определены выше, или Ar представляет собой 1,2-, 1,3-, 1,4-, 1,5-, 1,6-, 1,7-, 1,8-, 2,3-, 2,6- или 2,7-нафтилиден, каждый из которых необязательно замещен одной, двумя, тремя или четырьмя группами, выбранными из (C1-C6)алкила, (C1-C5)алкокси, (C1-C4)тиоалкокси, галогена, нитро, -COOR′, -CONHR′, -O(CH2)nCOOR′, -S(CH2)nCOOR′, -О(СН2)nCONHR′ или -S(CH2)nCONHR′.
Z1 представляет собой (С1-С5)алкил или фенил, необязательно замещенный одной, двумя или тремя группами, выбранными из (C1-С5)алкила, (C1-C4)алкокси, (C1-C4)тиоалкокси, галогена, нитро, -COOR′, -CONHR′, -O(CH2)nCOOR′, -S(CH2)nCOOR′, -О(СН2)nCONHR′ или -S(CH2)nCONHR′; Alk2 и Sp2, взятые вместе, образуют связь, a Sp и Q являются такими, как они были определены выше.
В патенте США №5773001, который во всей своей полноте вводится в настоящее описание посредством ссылки, описаны линкеры, которые могут быть использованы вместе с нуклеофильными производными, а в частности, гидразидами и родственными нуклеофилами, полученными из калихеамицинов. Эти линкеры являются особенно подходящими в тех случаях, когда наилучшая активность достигается при образовании связи между лекарственным средством и гидролизуемым линкером. Эти линкеры содержат две функциональные группы. Одной из таких групп обычно является карбоновая кислота, используемая для реакции взаимодействия с носителем. Такая кислотная функциональная группа, если она является соответствующим образом активированной, может образовывать амидную связь со свободной аминогруппой указанного носителя, такой как амин боковой цепи лизина антитела или другого белкового носителя. Другой функциональной группой обычно является карбонильная группа, то есть альдегид или кетон, которые реагируют с соответствующим образом модифицированным терапевтическим агентом. Эти карбонильные группы могут реагировать с гидразидной группой на указанном лекарственном средстве с образованием гидразоновой связи. Такая связь является гидролизуемой, что позволяет терапевтическому агенту высвобождаться из конъюгата после связывания с клетками-мишенями.
Наиболее предпочтительным бифункциональным линкером для использования в настоящем изобретении является 4-(4-ацетилфенокси)бутановая кислота (AcBut), которая образует предпочтительный конъюгат, состоящий из β-калихеамицина, γ-калихеамицина или N-ацетил-γ-калихеамицина, функционализированного посредством реакции взаимодействия с 3-меркапто-3-метилбутаноилгидразидом, AcBut-линкером, и из человеческого или гуманизированного антитела IgG, используемого в качестве нацеливающего носителя.
IV. Мономерное конъюгирование
Гидрофобная природа многих цитотоксических лекарственных средств, включая калихеамицины, создает определенные трудности в получении конъюгатов мономерного лекарственного средства с высокой нагрузкой лекарственным средством и с приемлемыми выходами, необходимыми для его терапевтического применения. Эта проблема еще более осложняется увеличением гидрофобности связи, сообщаемой линкерами, такими как AcBut-линкер, как описано в патенте США №5773001, а также увеличением длины ковалентной связи, разделяющей терапевтический агент и носитель (антитело).
Агрегация конъюгатов "производное цитотоксического лекарственного средства/носитель" с повышенной нагрузкой лекарственным средством обусловлена гидрофобной природой лекарственных средств. Нагрузка лекарственным средством часто имеет ограничения и не позволяет получать приемлемые количества мономерного продукта. В некоторых случаях, например, в случае конъюгатов, описанных в патенте США №5877296, получение конъюгата с нужным выходом и с нужной нагрузкой терапевтического средства в реакционных условиях, описанных в патенте США №5053394, часто представляет определенные трудности из-за избыточной агрегации. Эти реакционные условия предусматривают использование ДМФ в качестве сорастворителя в реакции конъюгирования. Поэтому необходимо разработать способы, которые позволяли бы увеличить нагрузку лекарственным средством с получением более высокого выхода, но без агрегации и, соответственно, без потери материала.
Методы снижения агрегации описаны в патентах США №№5712374 и 5714586, которые во всей своей полноте вводятся в настоящее описание посредством ссылки. В этих патентах описаны белковые носители, включая, но не ограничиваясь ими, белки, такие как человеческие или гуманизированные антитела, используемые для доставки цитотоксических терапевтических агентов, такие как, например, hP67.6 и другие описанные там гуманизированные антитела. Как указывается в этих патентах, было обнаружено, что использование ненуклеофильного, совместимого с белком забуференного раствора, содержащего (i) пропиленгликоль в качестве сорастворителя и (ii) добавку, включающую, по крайней мере, одну С6-С18карбоновую кислоту, обычно позволяет продуцировать конъюгаты "производное мономерного цитотоксического лекарственного средства/носитель" с более высокой нагрузкой лекарственным средством, с более высоким выходом и с пониженной степенью агрегации, обладающие превосходной активностью. Описанными в этих патентах кислотами, предпочтительно, являются С7-С12-кислоты, а наиболее предпочтительной кислотой является октановая кислота (такая как каприловая кислота) или ее соли. Предпочтительными забуференными растворами для конъюгатов, полученных из N-гидроксисукцинимидоэфиров (OSu) или других аналогичным образом активированных сложных эфиров, являются забуференный фосфатом физиологический раствор (PBS) или N-2-гидроксиэтилпиперазин-N′-2-этансульфоновая кислота (буфер HEPES). Забуференный раствор, используемый в таких реакциях конъюгирования, не может содержать свободных аминов или нуклеофилов. Буферы, подходящие для конъюгатов других типов, могут быть легко определены специалистом. Альтернативно, было обнаружено, что использование ненуклеофильного, совместимого с белком, забуференного раствора, содержащего трет-бутанол и не содержащего каких-либо других добавок, позволяет продуцировать конъюгаты "производное мономерного калихеамицина/носитель" с более высокой нагрузкой лекарственным средством, с более высоким выходом и с пониженной степенью агрегации.
Количество сорастворителя, необходимого для образования мономерного конъюгата, до некоторой степени варьируется в зависимости от используемого белка и может быть определено любым специалистом без излишнего экспериментирования. Количество добавок, необходимых для эффективного образования мономерного конъюгата, также варьируется в зависимости от используемого антитела. Такое количество также может быть определено любым специалистом без излишнего экспериментирования. Как описано в патентах США №№5712374 и 5714586, добавление пропиленгликоля в количестве от 10% до 60%, предпочтительно, от 10% до 40%, а наиболее предпочтительно примерно 30% по объему всего раствора, и добавки, содержащей, по крайней мере, одну С6-С18карбоновую кислоту или ее соль, а предпочтительно, каприловую кислоту или ее соль в количестве от 20 мМ до 100 мМ, предпочтительно, от 40 мМ до 90 мМ, а наиболее предпочтительно, примерно от 60 мМ до 90 мМ в реакционную смесь для конъюгирования позволяет продуцировать конъюгаты "производное мономерного цитотоксического лекарственного средства/носитель" с более высокой нагрузкой лекарственным средством, с более высоким выходом и с пониженной степенью агрегации. Могут быть также использованы и другие совместимые с белком органические сорастворители, не являющиеся пропиленгликолем, такие как этиленгликоль, этанол, ДМФ, ДМСО и т.п. Некоторые из этих органических сорастворителей или все указанные сорастворители были использованы для переноса лекарственного средства в смесь для конъюгирования.
Альтернативно, как описано в указанных патентах, концентрация С6-С18карбоновой кислоты или ее соли может быть увеличена до 150-300 мМ, а концентрация сорастворителя может быть снижена до 1-10%. В одном из вариантов указанной карбоновой кислотой является октановая кислота или ее соль. В предпочтительном варианте карбоновой кислотой является декановая кислота или ее соль. В другом предпочтительном варианте указанной карбоновой кислотой является каприловая кислота или ее соль, где указанная каприловая кислота присутствует в концентрации 200 мМ вместе с 5% пропиленгликолем или этанолом.
В другом альтернативном варианте, описанном в этих патентах, для получения конъюгатов "производное мономерного цитотоксического лекарственного средства/носитель" с более высокой нагрузкой лекарственным средством, с более высоким выходом и с пониженной степенью агрегации, в реакционную смесь для конъюгирования может быть добавлен трет-бутанол в концентрациях от 10% до 25%, а предпочтительно, 15% по объему всего раствора.
Эти установленные условия конъюгирования были использованы для получения конъюгата СМА-676 (Gemtuzumab Ozogamicin), который в настоящее время имеется в продаже под торговым знаком Милотарг™ (Mylotarg™). При разработке этого способа лечения острого миелоидного лейкоза (ОМЛ), было проведено исследование с помощью ионообменной хроматографии, которое показало, что калихеамицин не обнаруживает однородного распределения по всему антителу. Большая часть калихеамицина локализуется, приблизительно, на одной половине антитела, а другая его часть присутствует в LCF, которая содержит лишь небольшие количества калихеамицина. Следовательно, необходимость в разработке усовершенствованных способов конъюгирования цитотоксических лекарственных средств, таких как калихеамицины, с носителями, которые минимизировали бы степень агрегации и позволяли бы достичь более высокой однородной нагрузки лекарственным средством со значительно более высоким выходом продукта-конъюгата, остается крайне актуальной.
Конкретным примером может служить конъюгат G5/44-NAc-гамма-калихеамицин-DMH-AcBut, который будет далее называться СМС-544 и который, в общих чертах, показан на фиг.17. Для продуцирования СМС-544 желательно снижение количества LCF до <10% от общего количества антитела, и при этом рассматриваются различные варианты такого снижения уровней LCF. Конечный выбор параметров не должен влиять на другие свойства указанного иммуноконъюгата, такие как связывание с антигеном и цитотоксичность. Рассматриваемыми параметрами являются генетическая или физическая модификация указанного антитела, методы хроматографического разделения или изменение условий реакции.
Реакция антитела G5/44 с NAc-гамма-калихеамицин-DMH-AcBut-OSu с использованием старых реакционных условий (условий получения СМА-676) приводила к получению продукта, имеющего физические свойства (нагрузка лекарственным средством, LCF и агрегация), аналогичные свойствам СМА-676. Однако, в этом случае, после конъюгирования наблюдалось образование нежелательных высоких уровней LCF (50-60%). Оптимальные реакционные условия были определены с помощью методики планирования статистического эксперимента, где оценивались ключевые параметры реакции, такие как температура, рН, исходное количество производного калихеамицина и концентрация добавки. Анализ этих экспериментов продемонстрировал, что исходное количество калихеамицина и концентрация добавки оказывали очень большое влияние на уровень низкоконъюгированной фракции LCF и образование агрегатов, но гораздо меньшее влияние на температуру и рН. В дополнительных экспериментах было также продемонстрировано, что концентрации белка-носителя (антитела) и сорастворителя (этанола) в равной степени оказывали меньшее влияние (по сравнению с исходным количеством калихеамицина и концентрацией добавки) на регуляцию уровней LCF и агрегата. Для снижения LCF до уровня <10%, количество исходного производного калихеамицина увеличивали с 3% до 8,5% (мас./мас.) по сравнению с количеством антитела в данной реакционной смеси. Октановую кислоту или ее соль, используемую в качестве добавки с концентрацией 200 мМ (способ СМА-676), заменяли на декановую кислоту или ее соль в концентрации 37,5 мМ. Реакция конъюгирования проходила более эффективно при слегка повышенной температуре (30-35°С) и при рН 8,2-8,7. Реакционные условия, обеспечивающие осуществление таких замен, позволяют снизить LCF до уровня ниже 10% и при этом увеличить нагрузку калихеамицином, и такие условия далее будут называться условиями технологии получения СМС-54 4 или "новыми" условиями данной технологии. Сравнение результатов, полученных в условиях, применяемых в способах продуцирования СМА-676 и СМС-544, проиллюстрировано в таблице 1.
Увеличение количества исходного калихеамицина приводит к увеличению нагрузки лекарственным средством с 2,5-3,0 мас.% до 7,0-9,0 (а в основном, 7,5-8,5) мас.% и не приводит к увеличению уровня агрегации белка в реакционной смеси. Благодаря снижению уровней агрегата и LCF, условия способа получения СМС-54 4 позволяют получать более гомогенный продукт.Эта новая процедура конъюгирования позволяет получать воспроизводимые уровни СМС-544 в мультиграммовом масштабе.
В вышеописанных реакциях, концентрация антитела может варьироваться в пределах от 1 до 15 мг/мл, а концентрация производного калихеамицина, например, соединения N-ацетил-гамма-калихеамицин-DMH-AcBut-OSu-эфир (используемая для получения конъюгатов, представленных на фиг.17), составляет в пределах примерно от 4,5 до 11% по массе антитела. Сорастворителем является этанол, использование которого дало хорошие результаты при концентрациях от 6 до 11,4% (по объему.). Реакции осуществляли в PBS, HEPES, N-(2-гидроксиэтил)пиперазин-N′-(4-бутансульфоновая кислота) (HEPBS) или в другом совместимом буфере при рН 8-9, при температуре примерно от 30°С до 35°С и в течение периода времени от 15 минут до 24 часов. Специалист может самостоятельно легко определить приемлемые пределы рН для других типов конъюгатов. Было установлено, что для различных антител незначительные изменения в комбинации вышеупомянутых добавок приводят к увеличению нагрузки лекарственным средством и увеличению выхода мономерного конъюгата, при этом следует отметить, что для достижения оптимальных результатов при использовании любого конкретного белка-носителя может потребоваться внесение небольших изменений в конкретные условия или выбор добавок.
V. Очистка и выделение конъюгата
После конъюгирования, мономерные конъюгаты могут быть отделены от неконъюгированных реагентов (таких как белковый носитель и свободное цитотоксическое лекарственное средство/калихеамицин) и/или от агрегированной формы указанных конъюгатов стандартными методами, такими как, например, эксклюзионная хроматография (ЭХ), гидрофобная хроматография
(ГФХ), ионообменная хроматография (ИОХ) или хроматографическое фокусирование (ХФ). Очищенные конъюгаты являются мономерными и обычно содержат от 4 до 10 мас.% цитотоксического лекарственного средства/калихеамицина. В предпочтительном варианте указанные конъюгаты очищают гидрофобной хроматографией (ГФ). В способах, используемых ранее для промышленного производства конъюгатов "цитотоксическое лекарственное средство/калихеамицин - антитело" (способ СМА-676), после конъюгирования проводили одну стадию разделения с помощью эксклюзионной хроматографии (ЭХ). Хотя эта стадия является достаточно эффективной как для удаления агрегированного конъюгата, так и для замены буфера для препарата, однако эта стадия оказалась неэффективной для снижения содержания LCF. Следовательно, для регуляции содержания LCF в конечном продукте, ЭХ-способ основан исключительно на химической реакции конъюгирования. Другим недостатком ЭХ является ограничение объема реакционной смеси для конъюгирования, подаваемой на колонку (обычно не выше 5% по объему слоя на колонке в данном процессе). Это в значительной степени ограничивает размер партии (а следовательно, и пропускную способность), который может поддерживаться в данной продуктивной зоне. И наконец, очистка с помощью ЭХ также приводит к значительному разведению раствора конъюгата, что накладывает определенные ограничения на концентрацию белка, которая должна быть достигнута в данном препарате.
Если цитотоксическое лекарственное средство, например, такое как производное калихеамицина, имеет в высокой степени гидрофобную природу и используется в конъюгате, то для осуществления эффективного разделения конъюгированного и неконъюгированного антитела гидрофобная хроматография (ГФХ) является предпочтительным выбором. По сравнению с ЭХ, ГФХ имеет три важных преимущества: (1) она является эффективной для снижения содержания LCF, а также уровня агрегата; (2) загрузочная емкость колонки для ГФХ гораздо выше; и (3) она позволяет избежать чрезмерного разбавления продукта.
Различные среды для высокоэффективной ГФХ, подходящие для использования в промышленном производстве, например, такие как бутил-, фенил и октилсефароза 4 Fast Flow (Amersham Biosciences Piscataway, NJ), могут обеспечивать эффективное отделение неконъюгированных компонентов и агрегатов конъюгата от мономерных конъюгированных компонентов после завершения конъюгирования.
VI. Композиции и препараты
Настоящее изобретение также относится к способу получения терапевтической или диагностической композиции/терапевтического или диагностического препарата, предусматривающему смешивание конъюгата "производное мономерного цитотоксического лекарственного средства/носитель" настоящего изобретения с фармацевтически приемлемым наполнителем, разбавителем или носителем.
Конъюгат "производное мономерного цитотоксического лекарственного средства/носитель" может быть единственным активным ингредиентом в терапевтической или диагностической композиции/терапевтическом или диагностическом препарате, либо он может быть использован в сочетании с другими активными ингредиентами, включая другие антитела, например, антитела против CD19, CD20, CD33, Т-клеток, IFNγ или ЛПС, или ингредиенты, не относящиеся к антителам, такие как цитокины, факторы роста, гормоны, антигормоны, цитотоксические лекарственные средства и ксантины.
Цитокинами и факторами роста, которые могут быть использованы для лечения пролиферативных расстройств, таких как рак, и которые могут быть использованы вместе с конъюгатами "производное цитотоксического лекарственного средства/носитель" настоящего изобретения, являются интерфероны, интерлейкины, такие как интерлейкин 2 (IL-2), TNF, CSF, GM-CSF и G-CSF.
Гормонами, обычно используемыми для лечения пролиферативных расстройств, таких как рак, и которые могут быть использованы вместе с конъюгатами "производное цитотоксического лекарственного средства/носитель" настоящего изобретения, являются эстрогены, такие как диэтилстильбестрол и эстрадиол, андрогены, такие как тестостерон и галотестин, прогестины, такие как Megace и Provera, и кортикостероиды, такие как преднизон, дексаметазон и гидрокортизон.
Для лечения пролиферативных расстройств, таких как рак, обычно применяются антигормоны, такие как антиэстрогены, то есть тамоксифен, антиандрогены, то есть, флутамид и антиадреналовые агенты, и эти антигормоны могут быть использованы вместе с конъюгатом "производное цитотоксического лекарственного средства/носитель" настоящего изобретения.
Химиотерапевтическими/противоопухолевыми агентами, которые обычно используются для лечения пролиферативных расстройств, таких как рак, и которые могут быть использованы вместе с конъюгатом "производное цитотоксического лекарственного средства/носитель" настоящего изобретения, являются, но не ограничиваются ими, адриамицин, цисплатин, карбоплатин, винбластин, винкристин, блеомицин, метотрексат, доксорубицин, фторурацилы, этопозид, таксол и его различные аналоги, и митомицин.
Указанные композиции должны предпочтительно содержать терапевтически эффективное количество конъюгата настоящего изобретения. Используемый здесь термин "терапевтически эффективное количество" означает количество терапевтического агента, необходимое для лечения, ослабления или предупреждения данного заболевания или состояния, или для продуцирования детектируемого терапевтического или профилактического эффекта. Для любого конъюгата терапевтически эффективная доза может быть предварительно оценена либо в анализах клеточных культур, либо на животных с моделью заболевания, обычно на грызунах, кроликах, собаках, свиньях или приматах. Животное с моделью заболевания может быть также использовано для определения соответствующих интервалов концентраций и способа введения. Такая информация может быть затем использована для определения подходящих доз и способов их введения человеку.
Конкретное эффективное количество для введения человеку будет зависеть от тяжести заболевания, общего состояния здоровья индивидуума, его возраста, веса и пола, режима питания, времени и частоты введения, комбинации(й) лекарственных средств, аллергической реакции и толерантности/восприимчивости к данной терапии. Это количество может быть определено с помощью рутинного экспериментирования и исходя из опыта врача-клинициста. В основном, эффективная доза будет составлять от 0,1 мг/м2 до 50 мг/м2, предпочтительно, от 0,4 мг/м2 до 30 мг/м2, а более предпочтительно, от 2 мг/м2 до 9 мг/м2, где указанная доза вычислена в расчете на белковый носитель.
Композиции могут быть введены пациенту либо отдельно, либо в комбинации с другими агентами, лекарственными средствами или гормонами. Доза вводимого конъюгата "производное мономерного цитотоксического лекарственного средства/антитело" настоящего изобретения зависит от природы состояния, подвергаемого лечению, стадии развития злокачественной лимфомы или лейкоза и от цели введения данного конъюгата, где такой целью может быть предупреждение заболевания или лечение уже имеющегося заболевания.
Частота введения дозы будет зависеть от времени полужизни данного конъюгата и продолжительности его действия. Если данный конъюгат имеет короткое время полужизни (например, 2-10 часов), то он может быть введен в виде одной или нескольких доз в день. Альтернативно, если данная молекула конъюгата имеет продолжительное время полужизни (например, от 2 до 15 дней), то в этом случае может оказаться достаточным введение одной дозы в день, одной дозы в неделю или даже одной дозы через каждые 1 или 2 месяца.
Композиция может также содержать фармацевтически приемлемый носитель для введения указанного конъюгата антитела. Такой носитель сам по себе не должен индуцировать вырабатывание антител, негативно влияющих на индивидуума, принимающего указанную композицию, и не должен быть токсичным. Подходящими носителями могут быть крупные макромолекулы с замедленным метаболизмом, такие как белки, полипептиды, липосомы, полисахариды, полимолочные кислоты, полигликолевые кислоты, полимерные аминокислоты, сополимеры аминокислот и неактивные вирусные частицы.
При этом могут быть использованы фармацевтически приемлемые соли, например, соли минеральных кислот, такие как гидрохлориды, гидробромиды, фосфаты и сульфаты, или соли органических кислот, такие как ацетаты, пропионаты, малонаты и бензоаты.
Фармацевтически приемлемые носители в этих композициях могут, кроме того, содержать жидкости, такие как вода, физиологический раствор, глицерин и этанол. Кроме того, в таких композициях могут присутствовать добавки, такие как смачивающие или эмульгирующие агенты или забуферивающие вещества для коррекции рН. Указанные носители позволяют изготавливать композиции в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей или суспензий для их введения пациенту.
Предпочтительными формами являются формы, подходящие для парентерального введения, например, путем инъекции или вливания, а именно, путем инъекции ударной дозы или непрерывного вливания. Если данный продукт предназначен для инъекции или вливания, то он может быть приготовлен в форме суспензии, раствора или эмульсии в масляном или водном носителе, и может содержать технологические добавки, такие как суспендирующие агенты, консерванты, стабилизаторы и/или диспергирующие агенты.
Хотя стабильность забуференных растворов конъюгата может обеспечить кратковременную стабильность конъюгата, однако она является недостаточной для обеспечения долговременной стабильности. Для повышения стабильности конъюгата и увеличения его срока действия, конъюгат "антитело - лекарственное средство" может быть лиофилизирован с получением сухой формы для последующего разведения в подходящей стерильной жидкости перед его использованием. Проблемы, связанные с лиофилизацией раствора белка, хорошо описаны в литературе. Во время замораживания и сушки может происходить разрушение вторичной, третичной и четвертичной структуры. А поэтому могут быть добавлены криозащитные агенты, которые будут действовать как аморфный стабилизатор конъюгата и будут сохранять структурную целостность белка в процессе лиофилизации. В одном из вариантов осуществления изобретения криозащитным агентом, используемым в настоящем изобретении, является спирт ряда сахаров, такой как альдит, маннит, сорбит, инозит, полиэтиленгликоль и их комбинации. В другом варианте осуществления изобретения указанным криозащитным агентом является сахарная кислота, включая альдоновую кислоту, уроновую кислоту, альдаровую кислоту и их комбинации.
Криозащитным агентом настоящего изобретения может быть также углевод. Подходящими углеводами являются альдегидные или кетоновые соединения, содержащие две или более гидроксильные группы. Эти углеводы могут быть циклическими или прямыми, и такими углеводами являются, например, альдозы, кетозы, аминосахара, альдиты, инозиты, альдоновые кислоты, уроновые кислоты, альдаровые кислоты или их комбинации. Таким углеводом может быть также моно-, ди- или полиуглевод, такой как, например, дисахарид или полисахарид. Подходящими углеводами являются, например, глицеральдегиды, арабиноза, ликсоза, пентоза, рибоза, ксилоза, галактоза, глюкоза, гексоза, идоза, манноза, талоза, гептоза, глюкоза, фруктоза, глюконовая кислота, сорбит, лактоза, маннит, метил-α-глюкопиранозид, мальтоза, изоаскорбиновая кислота, аскорбиновая кислота, лактон, сорбоза, глюкаровая кислота, эритроза, треоза, арабиноза, аллоза, альтроза, гулоза, идоза, талоза, эритрулоза, рибулоза, ксилулоза, псикоза, тагатоза, глюкуроновая кислота, глюконовая кислота, глюкаровая кислота, галактуроновая кислота, маннуроновая кислота, глюкозамин, галактозамин, сахароза, трегалоза, нейраминовая кислота или их производные. Подходящими полиуглеводами являются, например, арабинаны, фруктаны, фуканы, галактаны, галактуронаны, глюканы, маннаны, ксиланы (такие как, например, инулин), леван, фукоидан, каррагенан, галактокаролоза, пектины, пектиновые кислоты, амилоза, пуллулан, гликоген, амилопектин, целлюлоза, декстран, пустулан, хитин, агароза, кератин, хондроитин, дерматан, гиалуроновая кислота, альгиновая кислота, ксантановая камедь или крахмал. Из них наиболее подходящими углеводами являются сахароза, глюкоза, лактоза, трегалоза и их комбинации. Особенно предпочтительным криозащитным агентом является сахароза.
Предпочтительным криозащитным агентом настоящего изобретения является углевод или спирт ряда "сахаров", которым может быть многоатомный спирт. Многоатомными спиртами являются соединения, содержащие более чем одну гидроксильную группу. Предпочтительными многоатомными соединениями являются соединения с прямой цепью. Подходящими многоатомными соединениями являются, например, гликоли, такие как этиленгликоль, полиэтиленгликоль и полипропиленгликоль, глицерин или пентаэритрит, или их комбинации.
В некоторых предпочтительных вариантах осуществления изобретения указанным криозащитным агентом является сахароза, трегалоза, маннит или сорбит.
Композиции настоящего изобретения, после их приготовления, могут быть непосредственно введены индивидууму. Индивидуумами, подвергаемыми лечению, могут быть животные. Однако предпочтительно, чтобы указанные композиции были адаптированы для введения человеку.
Композиции настоящего изобретения могут быть введены любыми способами введения, включая, но не ограничиваясь ими, пероральное, внутривенное, внутримышечное, внутриартериальное, интрамедуллярное, интратекальное, интравентрикулярное, трансдермальное, чрескожное (см. публикацию РСТ №WO 98/20734), подкожное, внутрибрюшинное, интраназальное, внутрикишечное, местное, подъязычное, интравагинальное или ректальное введение. Для введения композиций настоящего изобретения могут быть также использованы безыгольные шприцы. Обычно указанные композиции могут быть получены в виде препаратов для инъекций либо в виде жидких растворов, либо в виде суспензий. Могут быть также получены твердые формы, подходящие для приготовления растворов или суспензий в жидких носителях перед их инъекцией.
Прямая доставка данных композиций может быть осуществлена, в основном, путем подкожной, внутрибрюшинной, внутривенной или внутримышечной инъекции, либо такая доставка может быть осуществлена в интерстициальное пространство ткани. Данные композиции могут быть также введены в область поражения. Схема лечения может предусматривать введение разовой дозы или дробных доз.
Следует отметить, что активным ингредиентом, входящим в состав данной композиции, является конъюгат "цитотоксическое лекарственное средство/белковый носитель". Этот конъюгат может подвергаться разложению в желудочно-кишечном тракте. Поэтому, если указанную композицию вводят через желудочно-кишечный тракт, то необходимо, чтобы указанная композиция содержала агенты, которые защищали бы данный конъюгат от разложения в желудочно-кишечном тракте, но при этом обеспечивали бы высвобождение указанного конъюгата сразу после его абсорбции в желудочно-кишечном тракте.
Детальное обсуждение фармацевтически приемлемых носителей можно найти в Remington′s Pharmaceutical Sciences (Mack Publishing Company, N.J., 1991).
Более конкретно, настоящее изобретение относится к конъюгату "производное мономерного калихеамицина/гуманизированное анти-CD22 антитело (G5/44), СМС-544", предназначенному для лечения пролиферативных расстройств, характеризующихся экспрессией антигена CD22 на клеточной поверхности.
Настоящее изобретение, кроме того, относится к использованию СМС-544 в целях изготовления композиции или лекарственных средств для лечения пролиферативного расстройства, характеризующегося экспрессией CD22 на клеточной поверхности.
СМС-544 может быть также использован в любой терапии, при которой может оказаться желательным снижение уровня экспрессии CD22 на клеточной поверхности у индивидуума, подвергаемого лечению описанными здесь композициями или лекарственными средствами. В частности, указанную композицию или лекарственное средство используют для лечения человека или животных с пролиферативными расстройствами, а именно, с лимфомой и лейкозом, при которых на клеточной поверхности экспрессируется антиген CD22. Эти CD22-экспрессирующие клетки могут циркулировать в организме, либо они могут присутствовать в нежелательно большом количестве в конкретном участке организма.
СМС-544 может быть также, что предпочтительно, использован для лечения В-лимфоцитарных злокачественных опухолей, включая лимфомы и лейкозы, а наиболее предпочтительно/ не-ходжкинскую лимфому (НХЛ), ' множественную миелому, острый лимфоцитарный лейкоз (ОЛЛ) и хронический лимфоцитарный лейкоз (ХЛЛ). СМС-544 может быть использовано отдельно или в комбинации с другими биологически активными агентами для лечения индивидуумов, страдающих В-клеточными злокачественными опухолями.
Биологически активными агентами обычно являются факторы роста, цитокины и цитотоксические лекарственные средства. Цитотоксическими лекарственными средствами, которые обычно используются для лечения пролиферативных расстройств, таких как рак, и такими средствами, которые могут быть также использованы в комбинации с CMC-544, являются антрациклин, такой как доксорубицин, даунорубицин, идарубицин, акларубицин, зорубицин, митоксантрон, эпирубицин, карубицин, ногаламицин, меногарил, питарубицин и валрубицин, в течение периода времени до трех дней; и пиримидиновый или пуриновый нуклеозид, такой как цитарабин, гемцитабин, трифлуридин, анцитабин, эноцитабин, азацитидин, доксифлуридин, пентостатин, броксуридин, капецитабин, кладрибин, децитабин, флоксуридин, флударабин, гугеротин, пуромицин, тегафур и тиазофурин. Другими химиотерапевтическими/противоопухолевыми агентами, которые могут быть введены в комбинации с СМС-544, являются адриамицин, цисплатин, карбоплатин, циклофосфамид, дакарбазин, винбластин, винкристин, митоксантрон, блеомицин, мехлоретамин, преднизон, прокарбазин, метотрексат, фторурацилы, этопозид, таксол и различные его аналоги, и митомицин. СМС-544 может быть введено одновременно с одним или несколькими из указанных терапевтических агентов. Альтернативно, СМС-544 может быть введено поочередно с одним или несколькими из указанных терапевтических агентов.
СМС-544 может быть также введено отдельно, одновременно или поочередно с комбинацией других биологически активных агентов, таких как факторы роста, цитокины, стероиды, антитела, такие как анти-CD20 антитело, ритуксимаб (Rituxan™), и химиотерапевтических средств, используемых как составная часть схемы лечения. Разработанные схемы лечения злокачественных лимфопролиферативных заболеваний включают: СНОРР (циклофосфамид, доксорубицин, винкристин, преднизон и прокарбазин); CHOP (циклофосфамид, доксорубицин, винкристин и преднизон); СОР (циклофосфамид, винкристин и преднизон); CAP-ВОР (циклофосфамид, доксорубицин, прокарбазин, блеомицин, винкристин и преднизон); m-BACOD (метотрексат, блеомицин, доксорубицин, циклофосфамид, винкристин, дексаметазон и лейковорин); ProMACE-МОРР (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, мехлоэтамин, винкристин, преднизон и прокарбазин); ProMACE-CytaBOM (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, цитарабин, блеомицин и винкристин); МАСОР-В (метотрексат, доксорубицин, циклофосфамид, винкристин, фиксированная доза преднизона, блеомицин и лейковорин); МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин); ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин); МОРР поочередно с ABV (адриамицин/доксорубицин, блеомицин и винбластин); МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин) поочередно с ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин); и ChlVPP (хлорамбуцил, винбластин, прокарбазин и преднизон). Применяемая терапия может включать индуктивную фазу терапии, консолидированную фазу терапии и поддерживающую фазу терапии. СМС-544 может быть также введено отдельно, одновременно или поочередно с любой из комбинаций, используемых в вышеуказанных схемах терапии в качестве составной части индуктивной фазы терапии, консолидированной фазы терапии и поддерживающей фазы терапии.
Конъюгаты настоящего изобретения могут быть также введены вместе с другими биологически активными и химиотерапевтическими агентами, используемыми в качестве составной части комбинированной схемы терапии, применяемой для лечения рецидивирующих, агрессивных лимфом. Такие схемы лечения предусматривают использование комбинаций IMVP-16 (ифосфамид, метотрексат и этопозид); MIME (метил-gag, ифосфамид, метотрексат и этопозид); DHAP (дексаметазон, высокая доза цитарабина и цисплатин); ESHAP (этопозид, метилпреднизолон, высокая доза цитарабина и цисплатин); EPOCH (этопозид, винкристин и доксорубицин, вводимые в течение 96 часов вместе с ударными дозами циклофосфамида и с перорально вводимым преднизоном), СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин); CAMP (ломустин, митоксантрон, цитарабин и преднизон); CVP-1 (циклофосфамид, винкристин и преднизон); CHOP-В (циклофосфамид, доксорубицин, винкристин, преднизон и блеомицин), СЕРР-В (циклофосфамид, этопозид, прокарбазин и блеомицин) и P/DOCE (эпирубицин или доксорубицин, винкристин, циклофосфамид и преднизон). Дополнительные схемы лечения агрессивных лимфом могут включать, в фазе 1, первичную терапию комбинацией CHOP (циклофосфамид, доксорубицин, винкристин и преднизон)-ритуксимаб (Rituxan™)-CMC-544, а затем, в фазе 2 и в фазе 3, комбинацией СНОР-ритуксимаб (Rituxan™)-СМС-544, СНОР-СМС-544 или СНОР-ритуксимаб (Rituxan™)-СМС-544. Альтернативно, лечение в фазе 1 может предусматривать первичную терапию комбинацией СОР (циклофосфамид, винкристин и преднизон)-ритуксимаб (Rituxan™)-СМС-544, а затем, в фазе 2 и в фазе 3, комбинацией СОР-ритуксимаб (Rituxan™), СОР-СМС-544 или СОР-ритуксимаб (Rituxan™)-СМС-544. В другом варианте осуществления изобретения лечение агрессивных лимфом может предусматривать первичную или вторичную терапию, в фазе 1, с использованием конъюгата СМС-544 "антитело - лекарственное средство", а затем, в фазе 2 и в фазе 3, комбинаций СМС-544 и CHOP (циклофосфамид, доксорубицин, винкристин и преднизон), СМС-544 и СОР (циклофосфамид, винкристин и преднизон) или СМС-544 и ритуксимаба (Rituxan™), или одного ритуксимаба (Rituxan™). В еще одном варианте осуществления изобретения, лечение агрессивных лимфом в фазе 1 может предусматривать первичную терапию конъюгатом СМС-544 "антитело - лекарственное средство", а затем, в фазе 2 и в фазе 3, одним СМС-544 или в комбинации с другими схемами лечения, включая, но не ограничиваясь ими, ESHOP (этопозид, метилпреднизолон, высокая доза цитарабина, винкристин и цисплатин); EPOCH (этопозид, винкристин и доксорубицин, вводимые в течение 96 часов с ударными дозами циклофосфамида и с перорально вводимым преднизоном), IMVP-16 (ифосфамид, метотрексат и этопозид), ASHAP (адриамицин, солюмедрол, Ara-С и цисплатин), MIME (метил-gag, ифосфамид, метотрексат и этопозид) и ICE (ифосфамид, циклофосфамид и этопозид). Подробное описание различных цитотоксических лекарственных средств, используемых в химиотерапии злокачественных опухолей, включая комбинированную химиотерапию, схему введения доз и т.п., описанные в настоящей заявке, можно найти в работах Cancer Principles and Practice of Oncology, Eds. Vincent T. DeVita, Samuelo Hellman, Steven A. Rosenberg, 6th Edition, Publishers: Lippincott, Williams & Wilkins (2001) and Physician′s Cancer Chemotherapy Drug Manual, Eds. Edward Chu and Vincent T. DeVita, Publishers: Jones and Bartlett (2002).
Настоящее изобретение также относится к способу лечения человека или животного, страдающего пролиферативным заболеванием, характеризующимся экспрессией CD22 на клеточной поверхности, либо предупреждения пролиферативного заболевания у человека или животного, подверженного риску развития такого заболевания, где указанный способ предусматривает введение указанному индивидууму эффективного количества СМС-544 настоящего изобретения.
Настоящее изобретение более подробно описано на нижеследующих примерах, которые приводятся в иллюстративных целях и не должны рассматриваться как ограничение объема изобретения.
Пример 1
Генерирование антител-кандидатов
Панель антител против CD22 была отобрана из гибридом в соответствии со следующими критериями: связывание с клетками Дауди, интернализация на клетках Дауди, связывание с мононуклеарными клетками периферической крови (МКПК), интернализация на МКПК, аффинность (более чем 10-9 М), продуцирование мышиного γ1 и уровень его продуцирования. В качестве предпочтительного антитела было выбрано антитело 5/44.
I. Клонирование генов и экспрессия химерной молекулы антитела 5/44
a) Получение клеток гибридомы 5/44 и выделение РНК из этих клеток
Гибридому 5/44 генерировали в соответствии со стандартной гибридомной техникой после иммунизации мышей человеческим белком CD22. РНК выделяли из клеток гибридомы 5/44 с использованием набора RNEasy kit (Qiagen, Crawley, UK; Catalogue №74106). Полученную РНК подвергали обратной транскрипции в кДНК, как описано ниже.
b) Распределение CD22 на опухолях НХЛ
Иммуностереохимическое исследование было проведено в целях оценки сферы распространения и распределения окраски с использованием моноклональных антител анти-CD22 антител 5/44. В качестве контроля в исследовании, проводимом для подтверждения областей, пораженных В-клеточными опухолями, были включены антитела против CD20 и против CD79a.
Было исследовано всего 50 опухолей, и они были классифицированы с использованием рабочей разработки и систем классификации REAL следующим образом:
- 7 В-лимфобластных лейкозов/лимфом (High/I)
- 4 B-CLL/мелкоклеточных лимфоцитарных лимфом (Low/A)
- 3 лимфоплазмацитоидов/иммуноцитом (Low/A)
- 1 клетка коры головного мозга (Int/F)
- 14 фолликулярных лимфом (Low-Int/D)
- 13 диффузных крупноклеточных лимфом (Int-Low/G,H)
- 6 неклассифицированных клеток (К)
- 2 Т-клеточных лимфом
Сорок В-клеточных лимфом были положительными на антиген CD22, при этом антитело 5/44 в концентрации 0,1 мкг/мл, а другие шесть лимфом стали положительными при увеличении концентрации до 0,5 мкг/мл. Что касается остальных двух В-клеточных опухолей, которые были отрицательными при 0,1 мкг/мл, то количества тканей для их тестирования при более высокой концентрации оказалось недостаточным. Однако параллельное тестирование с другим анти-CD22 антителом, обозначенным 6/13 (Celltech, Slough, UK), которое давало более интенсивное окрашивание, чем 5/44, показало, что все 48 В-клеточных лимфом давали положительную окраску на CD22.
Таким образом, можно сделать вывод, что антиген CD22 широко экспрессируется на В-клеточных лимфомах, а поэтому он представляет собой подходящую мишень для иммунотерапии НХЛ.
с) ПЦР-клонирование VH и VL антитела 5/44
кДНК-последовательности, кодирующие вариабельный домен тяжелой и легкой цепей антитела 5/44, синтезировали с использованием обратной транскриптазы для продуцирования одноцепочечных кДНК-копий мРНК, присутствующей в полноразмерной РНК. Затем эти последовательности использовали в качестве матрицы для амплификации последовательностей мышиной V-области с использованием специфических олигонуклеотидных праймеров с помощью полимеразной цепной реакции (ПЦР).
(i) Синтез кДНК
кДНК синтезировали в реакционном объеме 20 мкл, содержащем следующие реагенты: 50 мМ Трис-HCl, рН 8,3, 75 мМ KCl, 10 мМ дитиотреитола, 3 мМ MgCl2, 0,5 мМ dATP, dTTP, dCTP и dGTP, 20 единиц РНКазина, 75 нг рандомизированного гексануклеотидного праймера, 2 мкг РНК 5/44 и 200 единиц обратной транскриптазы вируса мышиного лейкоза Молони. После инкубирования в течение 60 минут при 42°С, реакцию прекращали путем нагревания при 95°С в течение 5 минут.
(ii) ПЦР
Аликвоты кДНК подвергали ПЦР с использованием комбинаций праймеров, специфичных для тяжелых и легких цепей. Затем пулы вырожденных праймеров, сконструированные для отжига с консервативными последовательностями сигнального пептида, использовали в качестве прямых праймеров. Все эти последовательности содержали, в указанном порядке,
рестрикционный сайт (SfuI для VL; HindIII для VH), начиная с 7 нуклеотида от их 5′-конца; последовательность GCCGCCACC (SEQ ID NO: 50) для оптимальной трансляции полученных мРНК; инициирующий кодон и 20-30 нуклеотидов на основе лидерных пептидных последовательностей известных мышиных антител (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Edition, 1991, U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health).
3′-праймеры конструировали так, чтобы они включали стык J-C каркасной области 4 указанного антитела и содержали рестрикционный сайт фермента BsiWI для облегчения клонирования ПЦР-фрагмента VL. 3′-праймеры тяжелой цепи представляют собой смесь, полученную так, чтобы она охватывала стык J-C указанного антитела. 3′-праймер включал рестрикционный ApaI-сайт для облегчения клонирования. 3′-область указанных праймеров содержит смешанную последовательность, полученную на основе известных последовательностей мышиных антител (Kabat et al., 1991, см. выше).
Комбинации праймеров, описанных выше, позволяют получить ПЦР-продукты для VH и VL, клонируемых непосредственно в подходящий экспрессирующий вектор (см. ниже) для продуцирования химерных ("мышь-человек") тяжелых и легких цепей, и для их генов, экспрессируемых в клетках млекопитающих для продуцирования химерных антител нужного изотипа.
Инкубирование (100 мкл) для ПЦР проводили следующим образом. Каждая реакционная смесь содержала 10 мМ Трис-HCl, рН 8,3, 1,5 мМ MgCl2, 50 мМ KCl, 0,01% мас./об. желатина, 0,25 мМ dATP, dTTP, dCTP и dGTP, 10 пмоль 5′-праймерной смеси, 10 пмоль 3′-праймера, 1 мкл кДНК и 1 единицу полимеразы Taq. Реакционную смесь инкубировали при 95°С в течение 5 минут, а затем проводили циклы: 94°С, 1 минута, 55°С, 1 минута и 72°С, 1 минута. После проведения 30 циклов, аликвоты каждой реакционной смеси анализировали путем электрофореза на агарозном геле.
Для V-области тяжелой цепи амплифицированный ДНК-продукт получали лишь в том случае, когда праймерный пул сигнального пептида был заменен праймерным пулом, гибридизующимся в начале каркасной области I. Фрагменты клонировали в ДНК-секвенирующие векторы. ДНК-последовательность определяли и подвергали трансляции с получением выведенной аминокислотной последовательности. Эту выведенную последовательность подтверждали путем сравнения с экспериментально определенной N-концевой последовательностью белка. На фиг.1 показана аминокислотная последовательность CDR мышиного моноклонального антитела 5/44. На фиг.2 и 3 показана последовательность ДНК/белка V-областей зрелых легкой и тяжелой цепей мышиного моноклонального антитела 5/44, соответственно.
(iii) Молекулярное клонирование ПЦР-фрагментов
V-области мышиных последовательностей клонировали в экспрессирующие векторы pMRR10.1 и pMRR14 (фиг.7 и 8). Эти векторы предназначены для экспрессии ДНК легкой и тяжелой цепи, кодирующей константные области человеческой легкой цепи-каппа и человеческой тяжелой цепи гамма-4. VL-область субклонировали в экспрессирующий вектор путем рестрикционного гидролиза и лигирования из секвенирующего вектора с использованием рестрикционных SfuI- BsiWI-сайтов, в результате чего получали плазмиду pMRR10 (544cL) (фиг.8). ДНК тяжелой цепи амплифицировали с помощью ПЦР с использованием 5′-праймера для введения сигнального пептида, поскольку он не был получен при осуществлении стратегии клонирования, где использовали лидерную последовательность тяжелой цепи мышиного антитела, происходящую от другой внутривидовой гибридомы (обозначенной 162). 5′-праймер имел следующую последовательность: 5′ GCGCGCAAGCTTGCCGCCACCATGGACTTCGGATTCTCTCTCGTGTTCCTGGCACTCATTCT CAAGGGAGTGCAGTGTGAGGTGCAGCTCGTCGAGTCTGG 3′ (SEQ ID NO: 51).
Обратный праймер был идентичен праймеру, используемому в клонировании исходного гена VH. Полученный ПЦР-продукт гидролизовали ферментами HindIII и ApaI, субклонировали и его ДНК-последовательность подтверждали, в результате чего получали плазмиду pMRR14 (544 сН) (фиг.7). Кратковременная ко-трансфекция обоих экспрессирующих векторов в клетки СНО приводила к генерированию химерного антитела с5/44. Это было достигнуто с использованием реагента липофектамина в соответствии с протоколами, представленными производителями (InVitrogen: Life Technology, Groningen, The Netherlands. Catalogue №11668-027).
II. Удаление сайта гликозилирования и реакционно-способного лизина
Потенциальная последовательность сайта N-связанного гликозилирования обнаруживалась в CDR-H2, имеющей аминокислотную последовательность N-Y-T (фиг.3). Электрофорез в ДСН-ПААГ, Вестерн-блоттинг и углеводное окрашивание гелей для антитела 5/44 и его фрагментов (включая Fab) показали, что этот сайт был действительно гликозилированным (не показано). Кроме того, лизиновый остаток наблюдался в экспонируемом положении в области CDR-H2, что, вероятно, снижает аффинность связывания с антителом благодаря присутствию дополнительного сайта для конъюгирования с агентом, с которым может быть конъюгировано указанное антитело.
Для введения аминокислотных замен в последовательность CDR-Н2 в целях удаления сайта гликозилирования и/или реакционно-способного лизина использовали ПЦР-стратегию, как показано на фиг.4. Прямые праймеры, кодирующие мутации N55Q, Т57А или T57V, использовали для удаления сайта гликозилирования (фиг.4), а четвертый прямой праймер, содержащий замену K60R, генерировали для удаления реакционно-способного лизинового остатка (фиг.4). В каждой из этих ПЦР-амплификаций использовали обратный праймер каркасной области 4. ПЦР-продукты гидролизовали ферментами XbaI и ApaI и встраивали в pMRR14 (544 сН) (также расщепленную ферментами XbaI и ApaI), в результате чего получали экспрессионные плазмиды, кодирующие эти мутанты. Мутации N55Q, Т57А и T57V удаляют сайт гликозилирования путем замены аминокислотной последовательности в консенсусной последовательности N-X-T/S, а мутация K60R приводит к замене потенциального реакционно-способного лизина аналогичным положительно заряженным аргининовым остатком. Полученные плазмидные варианты сН ко-трансфицировали плазмидой cL, в результате чего получали экспрессированные варианты химерного антитела.
III. Оценка активностей химерных генов
Активности химерных генов оценивали после кратковременной трансфекции в клетки СНО и определения константы аффинности с помощью анализа BiaCore.
Аффинности химерного 5/44 или его вариантов, у которых был удален их сайт гликозилирования или реакционно-способный лизин, исследовали с использованием BIA-технологии на связывание с конструкциями CD22-mFc. Результаты приводятся на фиг.9. Все измерения по связыванию осуществляли на приборах BIACore™ 2000 (Pharmacia Biosensor AB, Uppsala, Sweden). Этот анализ был осуществлен путем захвата CD22mFc с помощью иммобилизованного антитела против мышиного Fc. Это антитело находилось в растворимой фазе. Поверх иммобилизованного антитела против мышиного Fc инъецировали образцы, стандарты и контроль (50 мкл), а затем антитело в растворимой фазе. После каждого цикла поверхность восстанавливали добавлением 50 мкл 40 мМ HCl при концентрации 30 мкл/мин. Кинетический анализ проводили с использованием программы BIAevaluation 3.1 (Pharmacia).
Удаление сайта гликозилирования в конструкции Т57А приводило к небольшому ускорению ассоциации и к значительному замедлению диссоциации по сравнению с химерным антителом 5/44, что обеспечивало приблизительно 5-кратное увеличение аффинности. Мутация N55Q не влияла на аффинность. Этот результат был неожиданным, поскольку предполагалось, что удаление самого углевода не будет оказывать заметного влияния на связывание (как в случае замены N55Q). Увеличение аффинности наблюдалось только в случае замены Т57А. Это можно объяснить тем, что, независимо от присутствия углевода, треонин в положении 57, который не был удален после замены треонина на аланин, оказывает негативное влияние на связывание. Предположение о том, что небольшой размер аланина играет важную роль, и что негативное действие треонина связано с его размером, подтверждается результатом, полученным с использованием мутации T57V, то есть замена валином в положении 57 не давала положительного эффекта (данные не приводятся).
Удаление лизинового остатка посредством мутации K60R имело нейтральный эффект в отношении аффинности, то есть введение аргининового остатка приводило к удалению потенциального реакционно-способного сайта, но при этом не оказывало негативного влияния на аффинность.
Поэтому введение мутации для удаления сайта гликозилирования и для удаления реакционно-способного лизина было предусмотрено в протоколе "гуманизации".
Пример 2
CDR-прививка антитела 5/44
Молекулярное клонирование генов вариабельных областей тяжелой и легкой цепей антитела 5/44 и их использование для продуцирования химерных (мышь/человек) антител 5/44 было описано выше. Нуклеотидные и аминокислотные последовательности доменов VL и VH мышиного антитела 5/44 показаны на фиг.2 и 3 (SEQ ID NO: 7 и 8), соответственно. В этом примере показано, что введение CDR в человеческие каркасные области антитела 5/44, проводимое в соответствии с методом Adair et al. (заявка РСТ №WO 91/09967), приводит к снижению потенциальной иммуногенности этого антитела у человека.
I. CDR-прививка легкой цепи антитела 5/44
Сопоставление первичной последовательности белка с консенсусными последовательностями V-области человеческой легкой цепи-каппа подгруппы I указывало на 64%-ную идентичность этих последовательностей. Поэтому для конструирования CDR-привитой легкой цепи акцепторные каркасные области были выбраны так, чтобы они соответствовали областям последовательности человеческой VK подгруппы I зародышевой линии O12, DPK9. Акцепторная последовательность каркасной области 4 происходила от последовательности человеческой J-области зародышевой линии JK1.
Сравнение аминокислотных последовательностей каркасных областей мышиного антитела 5/44 и акцепторной последовательности проиллюстрировано на фиг.5, и это сравнение показало, что между донорными и акцепторными цепями имеется 27 отличий. Для того чтобы определить, может ли мышиный остаток в каждом положении или в области стыка VH/VL прямо или опосредованно участвовать в связывании с антигеном, проводили анализ на воздействие этого остатка на упаковку. Если подтверждалось, что данный мышиный остаток имеет важную функцию и значительно отличается от человеческого остатка по размеру, полярности или заряду, то такой мышиный остаток был сохранен. Исходя из этого анализа были сконструированы два варианта CDR-привитой легкой цепи, имеющие последовательности, представленные в SEQ ID NO: 19 и SEQ ID NO: 20 (фиг.5).
II. CDR-прививка тяжелой цепи антитела 5/44
CDR-прививку тяжелой цепи антитела 5/44 осуществляли в соответствии с той же самой стратегией, описанной для легкой цепи. Было обнаружено, что V-домен тяжелой цепи антитела 5/44 был гомологичен V-домену человеческих тяжелых цепей, принадлежащих к подгруппе I (70%-ная идентичность последовательностей), а поэтому последовательность человеческой каркасной области VH1-3, DP7 подгруппы I зародышевой линии была использована в качестве акцепторной каркасной области. Акцепторные последовательности каркасной области 4 происходили от последовательности человеческой J-области JH4 зародышевой линии.
Сравнение тяжелой цепи антитела 5/44 с каркасными областями показано на фиг.6, где можно видеть, что тяжелая цепь антитела 5/44 отличается от акцепторной последовательности в 22 положениях. Для того чтобы определить, какую роль играет любая из указанных областей в связывании с антигеном, было сконструировано 5 вариантов CDR-привитых тяжелых цепей, имеющих последовательности, представленные в SEQ ID NO: 23, SEQ ID NO: 24, SEQ ID NO: 25, SEQ ID NO: 26 и SEQ ID NO: 27 (фиг.6).
III. Конструирование генов для привитых последовательностей
Были сконструированы гены, кодирующие привитые последовательности gH1 и gL1, а также были разработаны и сконструированы серии перекрывающихся олигонуклеотидов (фиг.10). Для конструирования генов CDR-привитой V-области использовали технику ПЦР-сборки. Была получена реакционная смесь в объеме 100 мкл, содержащая 10 мМ Трис-HCl, рН 8,3, 1,5 мМ MgCl2, 50 мМ KCl, 0,001% желатина, 0,25 мМ dATP, dTTP, dCTP и dGTP, 1 пмоль каждого из "внутренних" праймеров (T1, T2, Т3, B1, B2, В3), 10 пмоль каждого из "внешних" праймеров (F1, R1) и 1 единицу полимеразы Taq (AmpliTaq, Applied BioSystems, № по каталогу N808-0171). 30 циклов ПЦР проводили в следующем режиме: 94°С, 1 минута, 55°С, 1 минута и 72°С, 1 минута. Затем продукты реакции подвергали электрофорезу в 1,5% агарозном геле, вырезали и выделяли на центрифужных колонках QIAGEN (набор для гель-экстракции QIAquick, № по каталогу 28706). ДНК элюировали в объеме 30 мкл. Затем аликвоты (1 мкл) ДНК для gH1 и gL1 клонировали в клонирующий вектор InVitrogen ТОРО ТА, pCR2.1 ТОРО (№ по каталогу К4500-01) в соответствии с инструкциями производителей. Этот неэкспрессирующий вектор служил в качестве промежуточного клонирующего вектора для облегчения секвенирования большого числа клонов. Затем, для идентификации "правильных" клонов, содержащих gHl и gL1, проводили ДНК-секвенирование с использованием вектор-специфических праймеров, в результате чего получали плазмиды pCR2.1 (544gH1) и pCR2.1 (544gL1) (фиг.11 и 12).
Для создания гуманизированных трансплантатов gH4, 5, 6 и 7 и gL2 использовали метод замены олигонуклеотидных кластеров. На фиг.13 проиллюстрировано конструирование олигонуклеотидных кластеров. Для конструирования каждого варианта, вектор pCR2.1 (544gH1) или pCR2.1 (544 gL1) разрезали рестриктирующими ферментами (XmaI/SacII для тяжелой цепи, XmaI/BstEII для легкой цепи). Большой фрагмент вектора подвергали очистке на агарозном геле и использовали для лигирования с олигонуклеотидным кластером. Эти кластеры состояли из 2 комплементарных олигонуклеотидов (как показано на фиг.13), смешанных в концентрации 0,5 пмоль/мкл в объеме 200 мкл 12,5 мМ Трис-HCl, рН 7,5, 2,5 мМ MgCl2, 25 мМ NaCl, 0,25 мМ дитиоэритритола. Отжиг проводили путем нагревания до 95°С в течение 3 минут в водяной бане (в объеме 500 мл), а затем реакционную смесь оставляли для медленного охлаждения до комнатной температуры. Затем гибридизованный олигонуклеотидный кластер десятикратно разводили в воде, а после этого лигировали в соответствующим образом разрезанный вектор. Для подтверждения получения нужной последовательности при конструировании плазмид pCR2.1 (5/44-gH4-7) и pCR2.1 (5/44-gL2) проводили ДНК-секвенирование. Затем подтвержденные привитые последовательности субклонировали в экспрессирующие векторы pMRR14 (тяжелая цепь) и pMR10.1 (легкая цепь).
IV. Активность связывания CDR-привитых последовательностей с CD22
Векторы, кодирующие привитые варианты, ко-трансфицировали в клетки СНО в различных комбинациях вместе с исходными цепями химерных антител. Активность связывания сравнивали в анализе на конкурентное связывание исходного мышиного антитела 5/4 4 с клетками Ramos (полученными из АТСС, лимфобластная клеточная линия человеческой лимфомы Беркитта, экспрессирующая на своей поверхности CD22). Этот анализ рассматривался как наилучший способ сравнения способности трансплантатов к связыванию с CD22 клеточной поверхности. Результаты представлены на фиг.14 и 15. Как можно видеть, эти трансплантаты имели очень незначительные отличия, и все они оказались более эффективными, чем химерные антитела, при конкуренции с мышиным родительским антителом. Введение 3 дополнительных человеческих остатков в конец CDR-H3 (gH5 и gH7) не оказывало какого-либо влияния на связывание.
Была отобрана комбинация трансплантатов с наименьшим числом мышиных остатков gL1gH7. Трансплантат легкой цепи gL1 имеет 6 донорных остатков. Остатки V2, V4, L37 и Q45, возможно, играют важную роль в упаковке. Остаток Н38 находится в области стыка VH/VL. Остаток D60 представляет собой поверхностный остаток, близкий к области CDR-L2 и может непосредственно участвовать в связывании с антигеном. Из этих остатков остатки V2, L37, Q45 и D60 были обнаружены в зародышевой линии в последовательностях генов для человеческих каппа-цепей других подгрупп. Трансплантат тяжелой цепи gH7 имел 4 донорных каркасных остатка (остаток R28 считался частью CDR-H1, как было установлено при определении структуры, используемой для CDR-прививки (см. Adair et al. (1991), заявка РСТ №WO 91/09967)). Остатки Е1 и А71 представляют собой поверхностные остатки, близкие к CDR. Остаток I48 является остатком, который, возможно, участвует в упаковке. Остаток Т93 присутствует в области стыка VH/VL. Из этих остатков остатки Е1 и А71 находятся в других генах подгруппы I человеческой зародышевой линии. Остаток I48 находится в подгруппе 4 человеческой зародышевой линии, а Т73 находится в подгруппе 3 человеческой зародышевой линии.
Последовательности полноразмерной ДНК и белка для тяжелой и легкой цепей, включая приблизительное положение интронов в генах константной области, присутствующих в векторах, показаны на фиг.16, и представлены в SEQ ID NO: 29 и SEQ ID NO: 28, соответственно, для легкой цепи, и в SEQ ID NO: 31 и SEQ ID NO: 30, соответственно, для тяжелой цепи.
Из этих векторов вырезали ДНК генов, кодирующих указанные легкие и тяжелые цепи. ДНК тяжелой цепи гидролизовали в 5′-концевом HindIII-сайте, а затем обрабатывали фрагментом Кленова ДНК-полимеразы I E.coli для затупления 5′-конца. Расщепление в 3′-EcoRI-сайте приводило к образованию фрагмента тяжелой цепи, который выделяли из агарозных гелей. Аналогичным образом был продуцирован фрагмент легкой цепи, который затупляли в 5′-SfuI-сайте и в 3′-EcoRI-сайте. Оба фрагмента клонировали в экспрессирующие векторы на основе DHFR и использовали в клетках СНО для генерирования стабильных клеточных линий.
Пример 3
Конъюгирование NAc-гамма-калихеамицин-DMH-AcBut с гуманизированным анти-CD22 антителом (G5/44)
В типичной реакции конъюгирования гуманизированное анти-CD22 антитело (G5/44) конъюгировали с N-ацетил-гамма-калихеамицин-DMH-AcBut-OSu (производным калихеамицина) (см. фиг.17), где концентрация нужного белка составляла 7,5 мг/мл, а нагрузка нужным производным калихеамицина составляла 8,5% по массе белка. рН нужной реакционной смеси составлял 8,5±0,2, а нужные концентрации других реакционных компонентов составляли: 5 0 мМ N-(2-гидроксиэтил)пиперазин-N′-4-бутансульфоновой кислоты (HEPBS), 37,5 мМ деканоата натрия и 9 об.% этанола. Реакцию проводили при 33±2°С в течение одного часа. Результаты анализа этой типичной реакции, проводимой перед очисткой, были следующими: белок - 7,34 мг/мл; нагрузка калихеамицином - 82,7 мкг/мл; агрегация - 93,25% и неконъюгированный белок (LCF) - 1,07% (% УФ-площади ВЭЖХ).
Было проведено тестирование действия различных поверхностно-активных добавок и их концентраций на выход и чистоту продукта в целях определения их влияния на уровень продуцирования конъюгированного мономера (см. таблицу 2). Для этого проводили реакции, в которых все параметры сохранялись постоянными, за исключением конкретной добавки и ее концентрации. Конъюгаты, полученные в таких реакциях, анализировали на концентрацию белка, нагрузку калихеамицином, содержание агрегатов и LCF. Хотя все N-карбоновые кислоты, имеющие от 6 атомов С (гексаноат) до 12 атомов С (додеканоат), давали приемлемые результаты, однако, наилучшие результаты (низкое содержание LCF, низкое содержание агрегатов и высокий выход мономерного конъюгата) были получены с использованием деканоата в концентрации от 30 мМ до 50 мМ.
Пример 4
Способ хроматографической очистки
I. Способы хроматографического разделения
Хотя бутилсефароза 4 Fast Flow была идентифицирована как наилучшая ГФХ-среда, однако приемлемые результаты могли быть получены с небольшими изменениями хроматографических условий, которые заключались в использовании других смол, таких как октилсефароза Fast Flow, PPG-600C (Tosoh Biosep), Fractogel EMD Propyl (EM Processing) и Source 15ISO (Amersham Biosciences Piscataway, NJ).
Исходным материалом для очистки была реакционная смесь для конъюгирования, содержащая 7,2 мг/мл белка при нагрузке производным калихеамицина 83 мкг/мг; при этом содержание агрегатов составляло 10,1% (процент площади ВЭЖХ), а содержание LCF составляло 5,6% (процент площади ВЭЖХ).
После завершения реакции конъюгирования, реакционную смесь четырехкратно разводили путем добавления раствора фосфата калия до конечной концентрации фосфата 0,7 М (рН 8,2). После смешивания, раствор фильтровали через фильтры 0,45 микрон. Разведенный раствор загружали на колонку с бутилсефарозой 4 Fast Flow. Общее количество белка, загруженного на эту колонку, составляло 29 мг на один мл объема слоя. После промывки 0,7 мМ фосфатом калия колонку элюировали ступенчатым градиентом 0,7 М - 4 мМ фосфата калия, рН 8,2. Фракции, элюированные в ступенчатом градиенте, объединяли для последующей обработки, при этом полученный пул состоял из мономерного конъюгата, где содержание агрегата и LCF составляло менее чем 1 процент по площади. Этот пул загружали на обессоливающую колонку с сефадексом G-25 (Amersham Biosciences) для замены на буфер, подходящий для данного препарата и содержащий 20 мМ Трис-HCl и 100 мМ хлорида натрия при рН 8,0. Очищенный с заменой буфера препарат СМС-544 обладал следующими свойствами: нагрузка калихеамицином составляла 81 мкг/мг; содержание агрегата - 0,4% (процент площади ВЭЖХ); а LCF - 0,8% (процент площади ВЭЖХ).
Пример 5
Анализ на связывание иммуноконъюгата NAc-гамма-калихеамицин-DMH-AcBut-G5/44 (СМС-544)
Иммуноконъюгат гуманизированного анти-CD22 антитела (G5/44) с калихеамицином (СМС-544), полученный вышеописанным способом конъюгирования, анализировали в тесте на связывание, который проводили для того, чтобы определить, оказывает ли конъюгат, полученный усовершенствованным способом, какое-либо негативное влияние на связывание с антигеном. В таблице 3 показано, что указанная процедура конъюгирования не влияет на аффинность связывания указанного антитела с антигеном. Иммуноконъюгат СМС-544, полученный либо старым, либо новым способом конъюгирования, связывался с антигеном-мишенью с аналогичными аффинностями, которые не отличались от аффинности неконъюгированого антитела G5/44.
Биосенсорные анализы проводили с использованием BIAcore 200 (BIACore AB, Uppsala, Sweden). CD22mFc подвергали ковалентной иммобилизации на биосенсорном чипе, покрытом карбоксиметилированным декстраном, активированным N-гидроксисукцинимидом, (СМ5), с использованием стандартного метода химического связывания с амином при плотности белка примерно 2000 резонансных единиц. Образцы CMC-544 или G5/44 разводили в буфере HBS (10 мМ HEPES, рН 7,4, содержащем 150 мМ NaCl, 3 мМ EDTA и 0,005% полисорбата 20 (об./об.)) и наносили в концентрации 1-100 нМ на CD22 mFc-сенсибилизированную поверхность биосенсорного чипа при скорости потока 30 мкл/мин в течение 3 минут для осуществления связывания. По окончании стадии связывания проводили мониторинг диссоциации связанного антитела путем промывки чипа буфером HBS в течение 15 минут. Антигенную поверхность регенерировали путем промывки биосенсорного чипа 15 мкл регенерирующего буфера (10 мМ NaOH и 200 мМ NaCl) в течение 30 секунд, с последующей стабилизацией в течение 2 минут, а затем проводили следующий цикл. Кинетические константы вычисляли с помощью нелинейного регрессионного анализа методом наименьших квадратов с использованием 1:1-модели Langmuir для построения кривой связывания с использованием программы BIAevaluation (version 3.0, BIAcore). Связывание СМС-544 с антигеном оценивали с помощью анализа методом поверхностного плазмонного резонанса с использованием CD22mFc, ковалентно иммобилизованного на биосенсорном чипе. Результаты кинетических анализов на связывание СМС-544 и G5/44 с CD22mFc показали, что на основе данных, полученных исходя из 1:1-модели связывания Langmuir с учетом переноса массы, можно сделать вывод, что как СМС-544, так и неконъюгированное антитело G544 связывались с CD22 с аналогичной аффинностью (СМС-544:CD22, KD=200 пМ; G5/44:CD22, KD=235 пМ). Конъюгирование с калихеамицином не влияло на способность G5/44 эффективно связываться с CD22mFc.
Связывание СМС-544 и G5/44 с CD22, экспрессируемым на поверхности В-клеточных лимфом, также оценивали с помощью проточной цитометрии. В этом способе оценки были использованы анти-CD3 mAb, антитело гемтузумаб (hP67.6) и его конъюгат с калихеамицином СМА-676 (gemtuzumab ozogamicin) в качестве контроля, соответствующего каждому изотипу. В качестве позитивного контроля использовали ритуксимаб (Rituxan™), химерное антитело "человеческий IgG1 - mAb против человеческого CD20". В качестве негативного контроля также использовали очищенное человеческое поликлональное IgG1 и IgG4. Связывание СМС-544 и G5/44 с CD22 на клетках Ramos или RL-BCL было аналогичным и отличалось от связывания человеческого поликлонального IgG4. Клетки RL-BCL обнаруживали меньший уровень экспрессии CD22 на своей поверхности, чем клетки BCL Ramos. В противоположность этому, связывание СМА-676 или gL1gH7 с любыми BCL было аналогично связыванию человеческого поликлонального IgG4, что соответствовало отсутствию у них экспрессии CD33 (данные не приводятся). Те же самые клетки продемонстрировали высокие уровни связывания с анти-CD20 ритуксимабом (Rituxan™). В отличие от hP67.6 и СМА-676, ни СМС-544, ни G5/44 не обнаруживали какого-либо связывания с CD22-, CD33+-клетками лейкоза HL-60 (данные не приводятся). Эти результаты дают основание предположить, что конъюгирование G5/44 с калихеамицином не влияет на его специфичность к антигену. CMC-544 специфически распознает CD22 на В-клетках человека, но не на В-клетках мышей, крыс, собак, свиней или приматов (собакоподобных обезьян и макак-резусов)(данные не приводятся).
Пример 6
Анализ in vitro и in vivo эффектов СМС-544
I. In vitro-цитотоксичность
Было проведено сравнение влияния СМС-544, полученного с использованием способов СМА-676 и СМС-544, на in vitro рост клеточных линий CD22+-В-клеточной лимфомы, RL, Daudi, Raji и Ramos. Для определения неспецифического связывания данного конъюгата с антигеном использовали конъюгат соответствующего изотипа, нацеленного на человеческий CD33 (СМА-676). Использование неконъюгированного N-Ас-гамма-калихеамицин-DMH (лекарственное средство, высвобождающееся из конъюгата после гидролиза кислотой) в данном анализе показало, что каждая из этих клеточных линий была чувствительна к летальным эффектам калихеамицина. В таблице 4 представлены результаты этих анализов, где полученные оценки выражены исходя из эквивалентности калихеамицина; а в таблице 5 приводятся результаты, выраженные как концентрации конъюгированного белка антитела. CD22-опосредованная доставка калихеамицина к CD22+-клеткам была, по крайней мере, в 10 раз более эффективной в отношении цитолиза клеток-мишеней, чем само неконъюгированное лекарственное средство. Контрольные конъюгаты соответствующего изотипа (СМА-676) обнаруживали цитотоксичность, которая была меньше, чем цитотоксичность неконъюгированного производного калихеамицина или аналогична данной цитотоксичности. Из таблицы 4 видно, что конъюгат, полученный способом конъюгирования СМС-544, может генерировать эквивалентный цитотоксический эффект при меньших концентрациях антитела, чем конъюгат, полученный способом конъюгирования СМА-676.
Цитотоксичность in vivo. Конъюгат СМС-544, полученный способом СМС-544, был затем оценен в ксенотрансплантатах В-клеточной лимфомы. В этих исследованиях использовали две опухоли В-клеточной лимфомы RAMOS и RL. RL-лимфома представляла собой клеточную линию, происходящую от НХЛ и не относящуюся к лимфоме Беркитта, а лимфома RAMOS происходила от лимфомы Беркитта. В репрезентативном эксперименте, проиллюстрированном на фиг.18, было показано, что СМС-544 и его мышиное антитело-аналог являются эффективными ингибиторами дозозависимого роста В-клеточной лимфомы RAMOS.
Было показано, что конъюгат гуманизированного антитела является более эффективным, чем его мышиный аналог. В этом исследовании наименьшая доза конъюгата калихеамицина, которая дает значительное ингибирование роста лимфомы, включала 10 мкг/кг конъюгированного NAc-гамма-калихеамицин-DMH. В противоположность этому, неконъюгированное антитело G5/44 при концентрации 10 мг/кг, вводимое внутрибрюшинно по такой же схеме, как и конъюгаты, не оказывало никакого воздействия на рост опухоли.
Аналогичные исследования проводили с использованием модели RL-лимфомы. В таблице 6 проиллюстрированы комбинированные анализы трех независимых экспериментов, в которых оценивали противоопухолевое действие СМС-544 на RL-опухоли НХЛ, развивающиеся у "голых" мышей и достигшие размера 300-400 мг. Введение СМС-544, в зависимости от дозы, приводило к обратному развитию опухоли в течение 3-недельного периода. Минимально эффективная доза СМС-544 для модели RL-лимфомы была установлена исходя из статистических анализов в этих исследованиях и составляла 20 мкг/кг в расчете на содержание калихеамицина. В любом из этих трех исследований летального исхода не наблюдалось. Более высокие дозы (60-320 мкг/кг) СМС-544 приводили почти к полному исчезновению RL-лимфомы. В целом, результаты, полученные для этих двух моделей В-клеточных лимфом, явно продемонстрировали способность СМС-544 вызывать обратное развитие опухоли.
Были также проведены исследования способности СМС-544, полученного в соответствии с новой процедурой, к ингибированию роста крупных укоренившихся трансплантатов В-клеточной лимфомы, с использованием моделей лимфомы RAMOS и RL. Эти опухоли росли и развивались до опухолевой массы 1,5 или 2 г, после чего СМС-544 или конъюгат негативного контроля, с антителом соответствующего изотипа, (СМА-676) внутрибрюшинно вводили при дозе конъюгированого калихеамицина 160 мкг/кг в соответствии с исходной схемой введения доз в дни 1, 5 и 9. Ранее было показано, что та же самая схема введения доз приводила к очень длительной ремиссии опухолей небольших размеров (см. таблицу 6). Как показано на фиг.19, введение СМС-544 мышам, имеющим большие лимфомы RAMOS, приводило к постепенному уменьшению массы уже имеющейся лимфомы, а на день 20, у 3 из 4 мышей с опухолью эти опухоли исчезали. Мониторинг мышей, у которых отсутствовали опухоли, вплоть до 50-го дня не обнаруживал какого-либо рецидива лимфомы RAMOS. В противоположность этому, контроль соответствующего изотипа, СМА-676, не оказывал какого-либо воздействия на рост опухоли. Четыре из пяти СМА-676 - обработанных мышей с большими опухолями были умерщвлены на 15-й день, поскольку масса этих опухолей достигала почти 15% от их массы тела.
Аналогичный эксперимент с использованием СМС-544 был проведен на модели RL-лимфомы. Внутрибрюшинное введение СМС-544 в дозе 160 мкг/кг по схеме, аналогичной описанной выше, приводило к >90% снижению массы уже имеющейся RL-лимфомы через 30 дней. Однако на 45 день у 2 мышей этой группы с почти исчезнувшими лимфомами наблюдался рецидив опухоли. Эти результаты показали, что СМС-544 может вызывать обратное развитие небольших, а также крупных укоренившихся лимфом. В небольшом числе исследований, не описанных в настоящей заявке, RL-лимфомы, которые снова обнаруживали спорадический рост после начальной СМС-544 - индуцированной регрессии, снова подвергали обработке СМС-544. Эти исследования показали, что RL-опухоли оставались еще восприимчивыми ко второму курсу лечения, проводимому с использованием СМС-544, и снова обнаруживали обратное развитие. Таким образом, обработка конъюгатом СМС-544 может быть эффективной против В-клеточных лимфом как небольшого, так и крупного размера, при этом может быть проведена повторная терапия.
II. Сравнение in vivo конъюгата, полученного способами конъюгирования СМА-676 и СМС-544
На фиг.20 представлены результаты репрезентативного эксперимента, в котором мышам, имеющим диссеминированную RL-лимфому, вводили две различные дозы (80 и 320 мкг/кг конъюгированного калихеамицина) СМС-544, полученного с использованием способа конъюгирования СМА-676 и способа конъюгирования СМС-544, в соответствии со стандартной схемой введения доз. Как и ожидалось, наблюдаемая противоопухолевая эффективность была дозозависимой, и при этом не наблюдалось никакого различия в эффективностях двух указанных препаратов СМС-544. В противоположность этому, неконъюгированный N-ацетил-гамма-калихеамицин-DMH, вводимый внутрибрюшинно в концентрации 160 мкг/кг, был неактивным. Однако необходимо подчеркнуть, что для каждой дозы конъюгированного калихеамицина количество белка антитела, вводимого в форме конъюгата СМС-544, полученного способом СМА-676, в 4 раза превышало количество СМС-544, полученного способом СМС-544. Поскольку за продуцирование противоопухолевого эффекта, в основном, ответственно содержание калихеамицина в данном конъюгате, то для доставки требуемого количества калихеамицина с помощью конъюгата, полученного по новой технологии, может потребоваться гораздо меньшее количество нацеливающего антитела. Фактически увеличение нагрузки конъюгата, полученного способом СМС-544, стало возможным благодаря отсутствию значительного количества низкоконъюгированной фракции (LCF).
III. Обработка опухолей, резистентных к ретуксимабу (Rituxan™)
Кроме того, необходимо ответить на следующий вопрос: остаются ли растущие В-клеточные лимфомы, после прекращения их обработки коммерчески доступным анти-CD22 антителом ритуксимаб (Rituxan™), восприимчивыми к обработке СМС-544. Для этой цели развивающиеся (недиссеминированные) RL-лимфомы обрабатывали ритуксимабом (Rituxan™) в течение трех недель. При продолжении терапии ритуксимабом (Rituxan™) рост RL-лимфомы ингибировался. После прекращения терапии ритуксимабом (Rituxan™) наблюдался быстрый рост RL-лимфом до достижения ими массы ~1 г, и на этой стадии эти лимфомы внутрибрюшинно обрабатывали дозой СМС-544, составляющей 160 мкг/кг. Как показано на фиг.21 и 22, эти RL-лимфомы оставались еще восприимчивыми к СМС-544, при этом 80% мышей не имели опухолей на 60-й день. Таким образом, СМС-544, при его введении в трех дозах, мог приводить к исчезновению В-клеточных лимфом, которые лишь подавлялись при непрерывном введении доз ритуксимаба (Rituxan™).
Пример 8
In vitro и in vivo эффект СМС-544
I. Исследования на связывание и токсичность
СМС-544 оценивали на его связывание с CD22, а также на его активность в in vitro и in vivo-моделях. СМС-544 также сравнивали с СМА-676, с контрольным конъюгатом hP67.6 для соответствующего изотипа (IgG4), в котором калихеамицин был связан с AcBut, и с ритуксимабом (Rituxan™), то есть химерным антителом IgG1-анти-CD20 mAb (IDEC Pharmaceuticals, San Diego, CA), которое является коммерчески доступным и было закуплено у Medworld Pharmacy (Chestnut Ridge, NY). В исследованиях на связывание домена G5/44 были использованы следующие антитела: BU12 (Celltech, Slough, UK); BLCAM, HD239 (Santa Cruz Biotech, Santa Cruz, CA); RFB-4 (Ancell Corp, Bayport, MN); SHCL-1, Leu 14 (Becton Dickinson, Franklin Lakes, NJ); 4KB128 и То15 (Dako Corp, Carpintera, CA); М6/13 и М5/44 (Celltech, Slough, UK). Другими антителами, используемыми в исследованиях по блокированию, являются SJ10 (Immunotech, Fullerton, CA) и М17.1.1, М19.1.1, М38.1.1 (Celltech, Slough, UK). Клеточные линии, используемые для исследований, включая клеточную линию (CRL-1923) лимфомы Беркитта Ramos и клеточную линию не-ходжкинской лимфомы (НХЛ) RL (CRL-2261), были взяты из Американской коллекции типовых культур. Эти клеточные линии не содержали микоплазмы, как было определено с помощью анализа на детекцию микоплазмы путем проведения полимеразной цепной реакции (АТСС, Manassas, VA). Клеточные линии поддерживали в виде суспензионных культур в среде RPMI, содержащей 10% FBS, 10 мМ HEPES, 1 мМ пирувата натрия, 0,2% глюкозы, 100 ед./мл натрий-содержащего пенициллина G и 100 мкг/мл сульфата стрептомицина.
Для того чтобы определить, может ли антитело G5/44 ингибировать связывание обладающих известной специфичностью мышиных mAb с CD22, проводили BIAcore-анализ с использованием FC-CD22, иммобилизованного на чипе СМ5 BIAcore. Было проведено сравнение единиц поверхностного плазмонного резонанса (RU), полученных без предварительного насыщения иммобилизованным Fc-CD22 антителом G5/44. Анализ на биомолекулярное взаимодействие проводили с использованием BIACORE 2000. Антитела пропускали над поверхностью "слепого" контроля (проточная кювета 1, служащая в качестве контроля, несвязанный белок) и над тест-поверхностью FC-CD22 (проточная кювета 2), иммобилизованного на сенсорном чипе СМ5 посредством химического связывания с амином до уровня 9,042 RU. Полученная в результате сенсорограмма представляет собой ответ (RU) на проточной кювете 2 минус ответ (RU) на проточной кювете 1. Вторую сенсорограмму получали путем предварительного насыщения проточной кюветы антителом G5/44 (100 мкг/мл) перед введением мышиных mAb против CD22, которые были предварительно охарактеризованы на их связывание. Сразу после измерения G5/44-ответа, мышиные анти-CD22 mAb были отдельно введены путем перфузии без удаления G544. Также регистрировали второй комбинированный ответ, продуцируемый связыванием мышиного анти-CD22 антитела с G5/44-сенсибилизированным CD22. Если мышиное антитело связывается с CD22 в сайтах, не относящихся к сайтам, занятым G5/44, то такие комбинированные ответы должны быть аддитивными. Если связывание G5/44 с CD22 препятствует связыванию или предотвращает связывание со "вторым" антителом, то такие комбинированные ответы не должны быть аддитивными. Каждое из измерений второго комбинированного ответа было скорректировано на "диссоциацию" взаимодействия G5/44:CD22.
G5/44 блокировало связывание только тех антител, которые связывались с эпитопом A/Ig-подобным доменом 1 CD22 (SHCL1 Leu 14 и HD239), что указывало на то, что антитело G5/44 также связывалось в этом домене с CD22. Антитела, которые связывались с эпитопом B/Ig-подобным доменом 3 CD22 (RFB-4), с эпитопом C/Ig-подобным доменом 4 CD22 (То 15) и с Ig-подобным доменом 2 CD22 (4КВ128), не блокировались антителом G5/44. Эти результаты показали, что С5/44-связывающий сайт на CD22 расположен в первом Ig-подобном домене, поскольку он предотвращал связывание тех анти-СВ22 mAb, которые распознавали первый Ig-подобный домен CD22 (эпитоп А). Другое анти-CD22 антитело, М6/13 (Celltech, Slough, UK), с неизвестной субспецифичностью, также блокировалось антителом G5/44 (Celltech, Slough, UK), на что указывало картирование сайта связывания Мб/13 с эпитопом A/Ig-подобным доменом 1 CD22. Антитело М5/44, мышиное родительское антитело G5/44, которое обладало такой же специфичностью, как и G5/44, ингибировало связывание с G5/44 и служило в качестве позитивного контроля. В этих исследованиях, анти-CD19 антитело BU12 служило в качестве негативного контроля. Результаты систематизированы в Таблице 7.
С использованием мышиных mAb, имеющих отдельные домены с известной специфичностью к CD22, были проведены исследования на способность G5/44 блокировать связывание этих антител с В-клетками. Кроме того были также проведены исследования на способность mAb блокировать связывание G5/44 с В-клетками. В этих исследованиях 1×105 клеток RAMOS было сначала обработано мышиным анти-CD22 антителом (10 мкг/мл гуманизированного G5/44 или мышиного моноклонального антитела против CD22) в течение 1 часа при 4°С, а затем эти клетки обрабатывали антителом G5/44 (10 мкг/мл). Клетки инкубировали еще 1 час при 4°С. После обработки антителом, В-клетки осаждали и промывали PBS-1% BSA, после чего добавляли соответствующее "второе" антитело (либо ФИТЦ-меченое козье античеловеческое антитело (тяжелая и легкая цепь), либо ФИТЦ-меченое козье антимышиное антитело (тяжелая и легкая цепь)) при концентрации 100 мкл, разведенное 1:100 в PBS-1% BSA, в течение 30 минут при 4°С. Клетки снова осаждали, промывали, ресуспендировали в PBS-1% BSA и добавляли в пробирку, содержащую 250 мкл PBS-1% формальдегида. Интенсивность флуоресценции, ассоциированную с клетками, измеряли с помощью проточной цитометрии на проточном цитометре BD FACSort.
Результаты показали, что предварительная обработка CD22+-B-клеток антителом G5/44 приводила к значительному ингибированию последующего связывания анти-CD22 mAb M5/44 и М6/13. В противоположность этому, связывание анти-CD22 mAb RFB4, То15, HD239 и 4KB с В-клетками, не ингибировалось антителом G5/44. С помощью проточной цитометрии было установлено отсутствие значимого ингибирования антителом G5/44 связывания HD239 с В-клетками, что оказалось довольно неожиданным, особенно потому, что анализ BIAcore указывал на способность G5/44 блокировать связывание HD239 и CD22. Отсутствие сильного ингибирования связывания HD239 антителом G5/44 можно объяснить различиями их относительных аффинностей по отношению к CD22. Исследование на способность вышеуказанных анти-CD22 mAb ингибировать связывание G5/44 с СВ22+-В-клетками, показало, что SHCL1 и М6/13, но не другие анти-CD22 mAb, ингибировали связывание G5/44. Эпитопы связывания HD239 и SHCL1 были картированы по первому Ig-подобному домену CD22. Однако эпитопы, распознаваемые М6/13 или М5/44, не были картированы. Исследования по блокированию, подробно описанные выше, показали, что вышеуказанные антитела распознают эпитопы, локализованные на первом Ig-подобном домене CD22, которые известны под общим названием эпитоп А.
Двадцать тысяч клеток Ramos инкубировали с различными дозами СМС-544 в присутствии и в отсутствие ритуксимаба (Rituxan™) в течение 96 часов. Через 96 часов жизнеспособность клеток определяли по вытеснению иодида пропидия в анализе, проводимом с помощью проточной цитометрии. Затем вычисляли среднюю жизнеспособность клеток в 3-6 лунках, и для каждой обработки вычисляли дозозависимое ингибирование жизнеспособности клеток. Фоновое ингибирование жизнеспособности клеток вычисляли исходя из нулевой концентрации СМС-544. Для того чтобы определить, оказывает ли СМС-544 статистически значимое дозозависимое ингибирующее действие на рост клеток Ramos при дозе порядка от 0,01 до 3 нг калихеамицин-DMH/мл, проводили логистический регрессионный анализ. Логистическую регрессию также использовали для того, чтобы определить, является ли взаимодействие СМС-544 с ритуксимабом (Rituxan™) статистически значимым. Были также вычислены средние ингибирующие концентрации (IC50) и были зарегистрированы эффективности каждой обработки по сравнению с обработкой одним антителом СМС-544. Статистический анализ проводили с использованием программы PROBIT в статистической аналитической системе (SAS, version 8.2).
Результаты исследований показали, что СМС-544 оказывает дозозависимое ингибирующее действие на рост клеток Ramos при дозе порядка от 0,01 до 3 нг калихеамицин-DMH/мл. Средние ингибирующие концентрации (IC50) одного СМС-544 составляют 0,029 нг/мл. Для того чтобы определить, является ли взаимодействие ритуксимаба (Rituxan™) с цитотоксически активным СМС-544 статистически значимым, к СМС-544 - обработанным клеткам добавляли ритуксимаб (Rituxan™) в концентрациях 2, 20 и 200 мкг/мл. Ритуксимаб (Rituxan™), добавленный отдельно без СМС-544 в концентрации 20 и 200 мкг/мл, не оказывал значимого воздействия на рост клеток (111,7% и 94,0%, рост клеток и носитель, соответственно). В комбинации с СМС-544, все три концентрации ритуксимаба (Rituxan™) продуцировали статистические значимые (р<0,05) сдвиги влево наклона кривой и отсекаемого отрезка дозозависимой кривой для СМС-544. Комбинация с концентрациями 2 и 200 мкг/мл ритуксимаба продуцировала наибольшие сдвиги на дозозависимой кривой. Эти 2 кривые статистически не отличались друг от друга, но имели значимые отличия (р<0,05) по сравнению с кривой, построенной для комбинации с дозой 20 мкг/мл. Второе исследование (данные не приводятся) подтвердило результаты, полученные в первом исследовании. Средние ингибирующие концентрации для комбинаций 2, 20 и 200 мкг/мл ритуксимаба (Rituxan™) плюс СМС-544 составляли 0,0072, 0,0081 и 0,0072 нг/мл, соответственно. Средние ингибирующие концентрации СМС-544 плюс ритуксимаб (Rituxan™) были приблизительно в четыре раза более эффективными, чем IС50 для одного СМС-544.
II. Противоопухолевая активность in vivo против подкожных ксенотрансплантатов и системно диссеминированной В-клеточной лимфомы у мышей SCID
Самки бестимусных "голых" мышей весом 18-22 г получали общую дозу облучения 400 рад. Это облучение подавляло иммунную систему мышей и усиливало развитие опухоли. Через три дня после облучения мышам в правый задний бок подкожно инъецировали 107 RL-клеток в матригеле (Matrigel) (Collaborative Biomedical Products, Belford, MA, разведенном 1:1 в среде RPMI). Когда опухоли достигали соответствующего размера (0,3 г, обычно через 21 день), мышам, i.p., вводили СМС-544, ритуксимаб (Rituxan™) или комбинацию CHOP (см. ниже) в стерильном физиологическим растворе, 0,2 мл/мышь. Первый день введения лекарственного средства считался днем 1. Две дополнительные дозы вводили на 5-й и 9-й день (обработка = q4Dx3). Затем проводили терапию комбинацией CHOP, включающей циклофосфамид (С) (Cytoxan™, Bristol-Meyers Squibb Co., Princeton, NJ) - 40 мг/кг, i.p.; доксорубицин-HCl (H) (Sigma-Aldrich, Co., St. Louis, MO) 3,3 мг/кг, i.p; винкристин (О) (GensiaSicor Pharmaceuticals, Irvine, CA) - 0,5 мг/кг, i.p; и преднизон (Р) (Roxane Labs., Columbia, OH), 0,2 мг/кг, р. о. СНО вводили по такой же схеме, как СМС-544 и ритуксимаб (Rituxan™) (q4Dx3), а преднизон вводили перорально через день, всего 5 доз (q2Dx5). По крайней мере, через неделю опухоли измеряли и размер опухоли выражали как массу опухоли (г) = 0,5 (толщина опухоли/2)(длина опухоли). Затем вычисляли среднее ср.кв.от. для тест-группы и сравнивали со значениями, полученными для обработанной носителем группы, после чего определяли статистическую значимость с использованием Т-критериев множественного сравнения. Среднее значение для группы регистрировали в течение 50 дней включительно или до тех пор, пока мышь не погибала (что не позволяло определить среднее для данной группы), либо пока ее опухоль не становилась слишком большой (>3,5 г), и в этом случае мышь подвергали эвтаназии. По истечении этого времени регистрировали массу опухоли только для каждой отдельной мыши во всех группах обработки. В конце каждого исследования для каждой группы обработки регистрировали также число мышей, не имеющих опухолей.
Для определения действия СМС-544, взятого отдельно или в комбинации с другими биологически активными агентами, на диссеминированные лимфомы использовали мышиную модель SCID. Самцам мышей SCID (CB17 SCID), 20-25 г, в хвостовую вену инъецировали 106 клеток Ramos (0,2 мл). Через 3 или 9 дней после инъекции клеток мышам вводили, i.p., носитель, конъюгаты (СМС-544 или СМС-676) или ритуксимаб (Rituxan™), всего 3 дозы. Для схемы обработки с введением дозы на 3-й день, мышам вводили дозы на дни 3, 7 и 11. Для схемы обработки с введением дозы на 9-й день, мышам вводили дозы на дни 9, 13 и 17. При схеме обработки на 9-й день, также вводили комбинации СМС-544 и ритуксима (Rituxan™), как описано ниже. При этом проводили ежедневный мониторинг мышей на наличие паралича задних лап, и если он обнаруживался, то мышей умерщвляли. Для группы обработки использовали от 7 до 10 мышей. Для этой группы вычисляли среднее время выживания (±ср.кв.от.), а также медиану, минимум и максимум выживаемости. Различие в распределении выживаемости между группами определяли с использованием непараметрического логрангового критерия, при этом значимость регистрировали на уровне 0,05. Кривые выживаемости строили с использованием метода Каплана-Майера.
В предварительном исследовании оценивали влияние двух различных схем введения доз на выживаемость мышей SCID с диссеминированной лимфомой. В первом исследовании отслеживали начало действия дозы лекарственного средства, введенной через 3 дня после внутривенной инъекции опухолевых клеток (модель развивающейся опухоли), а во втором исследовании введение дозы лекарственного средства начинали только с 9-го дня после инъекции опухолевых клеток (модель укоренившейся опухоли). В каждом исследовании вводили, i.p., 3 дозы СМС-544 (160 мкг/кг), СМА-676 (160 мкг/кг), или ритуксимаба (Rituxan™) (20 мг/кг), через 4 дня (Q4Dx3). Для модели развивающейся опухоли среднее время выживания мышей, обработанных носителем, составляло 27 дней (фиг.23, таблица 8). СМА-676, то есть контроль, соответствующий изотипу СМС-544, не давал значимого увеличения времени выживания (р>0,05). СМС-544 давал значимое увеличение времени выживания до 41 дня, а ритуксимаб давал превосходный эффект, то есть увеличение времени выживания до >125 дней (что значительно превышало действие СМС-544, р<0,05). Задержка введения дозы до тех пор, пока опухолевые клетки не будут иметь возможность проникать в кровоток (эффект "хоминга") и накапливаться в ткани-мишени (укоренившаяся модель), приводила к изменению результатов для СМС-54 4 и ритуксимаба (Rituxan™). И снова, СМА-676 не оказывал значимого влияния на выживаемость (фиг.24, таблица 8). Введение ритуксимаба (Rituxan™) приводило к увеличению средней выживаемости до 62,6 дней, а введение СМС-544 приводило к увеличению средней выживаемости до 83,5 дней. В данной модели укоренившейся опухоли такое различие между действием СМС-544 и ритуксимабом (Rituxan™) не было значимым.
Были проведены предварительные исследования для определения влияния ритуксимаба (Rituxan™), позитивного или негативного, на выживаемость при введении СМС-544. СМС-544 (160 мкг/кг) вводили в отсутствие или в присутствии ритуксимаба (Rituxan™) (комбинация 20 мг/кг ритуксимаба с высокой дозой лекарственного средства была отмечена как HD). Кроме того, более низкие дозы СМС-544 (80 мкг/кг) вводили вместе с более низкими дозами ритуксимаба (Rituxan™) (10 мг/кг). Эти соединения не вводили отдельно в соответствующих дозах 80 мкг/кг или 10 мг/кг из-за ограниченного числа исследуемых мышей. Комбинация СМС-544 (80 мкг/кг) с ритуксимабом (Rituxan™) (10 мг/кг) была отмечена как комбинация со средней дозой (MD) и эта комбинация была использована в исследовании для оценки влияния комбинаций с более низкими дозами лекарственных средств на выживаемость мышей SCID. Отдельное введение СМС-544 (160 мкг/кг) и ритуксимаба (Rituxan™) (20 мг/кг) осуществляли, как описано выше для модели укоренившейся опухоли. При этом среднее время выживания увеличивалось до 58,5 и 50,5 дней, соответственно (фиг.25, таблица 9). При введении их комбинации с высокими дозами, среднее время выживания немного увеличивалось (хотя и не было статистически значимым, р>0,05) до 64,4 дня. Комбинации со средними дозами 80 мкг/кг СМС-544 и 10 мг/кг ритуксимаба (Rituxan™) давала значимое увеличение (р<0,05, по сравнению с группой, обработанной носителем) выживаемости в среднем до 92,4 дней. Эти результаты дают основание предположить, что могут быть также использованы комбинации СМС-544 и ритуксимаба (Rituxan™) и в более низких дозах.
Были проведены дополнительные исследования с использованием комбинации СМС-544 с ритуксимабом (Rituxan™). Использовали нижеследующие группы обработки: СМС-544 в концентрации 40, 80 и 160 мкг/кг; ритуксимаб (Rituxan™) в концентрации 5, 10 и 20 мг/кг и СМС-544 в концентрации 40 мкг/кг плюс ритуксимаб (Rituxan™) в концентрации 5 мг/кг; СМС-544 в концентрации 80 мкг/кг плюс ритуксимаб (Rituxan™) в концентрации 10 мг/кг и СМС-544 в концентрации 160 мкг/кг плюс ритуксимаб (Rituxan™) в концентрации 20 мг/кг. Все дозы ритуксимаба (Rituxan™) давали небольшое увеличение среднего времени выживания до 33-40 дней (все дозы давали значимое увеличение выживаемости, р<0,05, по сравнению со средней выживаемостью для контрольной группы (носитель), которая составляла 25,8, фиг.26, таблица 10). Высокая доза СМС-544, 160 мкг/кг, давала увеличение средней выживаемости до 85 дней, что соответствовало результатам, полученным в двух более ранних исследованиях. Комбинация СМС-544 с ритуксимабом (Rituxan™) не давала значимого увеличения времени выживания (фиг.27, таблица 10). Каждая из двух более низких доз СМС-544 (80 и 40 мкг/кг) давала значимое увеличение (р<0,05) средней выживаемости по сравнению с высокой дозой СМС-544. Для доз СМС-544 40 и 80 мкг/кг, у 90% и 80% мышей, соответственно, время выживания все еще составляло 125 дней. Для групп, которым вводили обе комбинации лекарственного средства, 100% мышей оставались живыми на 125-й день. Более низкие дозы СМС-544 были более эффективными, чем высокая доза 160 мкг/кг.
Ритуксимаб (Rituxan™), в комбинации с СМС-544, не оказывал какого-либо видимого влияния на активность СМС-544 у мышей SCID с моделью диссеминированной В-клеточной лимфомы в тестируемых дозах (см. выше). Для того чтобы определить, приводит ли введение СМС-544 вместе с ритуксимабом (Rituxan™) к усилению или ингибированию противоопухолевой активности, было также проведено исследование с использованием модели ксенотрансплантата В-клеточной RL-лимфомы, подкожно инъецированного "голым" мышам Balb/c. В подкожной модели В-клеточной лимфомы опухоли достигали средних масс 300 мг, и на этом этапе были введены, i.p., два тестируемых терапевтических препарата. СМС-544 вводили в дозах 20 или 80 мкг/кг, Q4Dx3, в отсутствие или в присутствии ритуксимаба (Rituxan™) (20 мг/кг, Q4Dx3). Совместное введение с ритуксимабом (Rituxan™) не приводило к какому-либо значимому увеличению или ингибированию (р>0,05) терапевтической эффективности СМС-544 (фиг.28). В этом исследовании ритуксимаб (Rituxan™), вводимый отдельно, ингибировал рост RL-B-лимфомы (57%-ное ингибирование роста опухоли на 20-й день, р<0,05, по сравнению с группой, обработанной носителем), и аналогичные результаты были получены при более низкой дозе СМС-544.
Схема комбинированной химиотерапии CHOP (циклофосфамид, доксорубицин, винкристин и преднизон) является наиболее распространенным методом лечения пациентов с не-ходжкинской лимфомой. Противоопухолевое действие CHOP сравнивали с действием СМС-544 в укоренившихся ксенотрансплантатах В-клеточной RL-лимфомы. Отдельные компоненты схемы CHOP были использованы в их соответствующих максимально допустимых дозах, оцененных на "голых" мышах (данные не приводятся), а именно: циклофосфамид (С) - 40 мг/кг, i.p.; доксорубицин (Н) - 3,3 мг/кг, i.p.; винкристин (O) - 0,5 мг/кг, i.p.; и преднизон (Р) - 0,2 мг/кг, р.о. СНО вводили Q4Dx3, a P вводили р.о., Q2Dx5. СМС-544 вводили i.p., Q4Dx3, в дозе 160 мкг/кг эквивалентов калихеамицина. Схема лечения CHOP вначале приводила к значимому ингибированию роста В-клеточной RL-лимфомы (фиг.29). Однако через 3 недели опухоль снова начинала расти с такой же скоростью, как и опухоль у группы, обработанной носителем. В противоположность этому, противоопухолевый эффект СМС-544 был полным и не менялся на протяжении всего эксперимента. Эти результаты позволяют предположить, что СМС-544, вводимый в дозе значительно более низкой, чем максимально допустимая (нелетальная) доза для "голых" мышей, был более эффективным, чем схема химиотерапии CHOP.
Эти исследования показали, что ритуксимаб (Rituxan™), добавленный к СМС-544, давал значимое увеличение цитотоксической активности СМС-544, наблюдаемой в отношении клеток В-лимфомы Ramos. Недавно также сообщалось о синергическом действии ритуксимаба (Rituxan™) и глюкокортикоидов в клетках Ramos. Кроме того, синергическое ингибирование роста 4 из 8 дополнительных клеточных линий наблюдалось в случае введения ритуксимаба (Rituxan™) в комбинации с 10 мкМ дексаметазона.
Сообщалось, что сам ритуксимаб (Rituxan™) в концентрации 0,4-10 мкг/мл дает значимое, хотя и небольшое (максимум 18%) ингибирование роста клеток Ramos. Кроме того, при инкубировании в концентрации 10 мкг/мл (48 часов инкубирования) он обладал активностью по отношению к 6 из 8 В-клеточных линий не-ходжкинской лимфомы. Ghetie et al. показали, что ритуксимаб (Rituxan™), 10 мкг/мл, давал 6,2%-ное увеличение уровня апоптоза (в отличие от 3,5% для клеток, обработанных носителем) после 24-часового инкубирования с клетками Ramos. В недавно проводимых исследованиях ритуксимаб (Rituxan™), вводимый отдельно в дозах 20 и 200 мкг/мл, не оказывал какого-либо воздействия на рост клеток Ramos. У мышей с моделью диссеминированной опухоли или с подкожным ксенотрансплантатом не наблюдалось какого-либо взаимодействия между СМС-544 и ритуксимабом (Rituxan™). Тестируемые комбинации лекарственных средств не оказывали влияние друг на друга и не давали усиленного эффекта. При этом необходимо было определить, каким образом снижение доз каждого лекарственного средства, вводимых в диссеминированную модель, может изменять полученный результат.
Модель диссеминированной В-клеточной лимфомы Ramos была описана Flavell и др. Сообщалось, что медиана выживаемости мышей, обработанных носителем, составляла 34-36 дней. У мышей развивался паралич задних конечностей, который прогрессировал и вскоре приводил к агонии. Гистологический анализ органов выявил, что, в данном случае, наиболее часто поражаемыми органами являются надпочечник, селезенка и подпаутинное пространство. Инфильтрат в подпаутинном пространстве, в большинстве случаев, распространяется на головной мозг. Ритуксимаб (Rituxan™) оказывал хорошее действие при его введении в ранней фазе патологического процесса у мышей SCID с диссеминированной опухолью (фиг.23), но его действие становилось менее явным при введении на 9-й день в опухолевую модель на стадии укоренения (фиг.24). По всей вероятности, ритуксимаб (Rituxan™), относящийся к изотипу IgGl, действует по эффекторному механизму у мышей-хозяев. Такими механизмами являются опосредованная комплементом цитотоксичность и/или антитело-зависимая клеточная цитотоксичность, реализуемая посредством "рекрутинга" природных клеток-киллеров, присутствующих у мышей SCID. Инъецированные опухолевые клетки Ramos, вероятно, являются более восприимчивыми к более ранним иммунным механизмам хозяина, которые активируются ритуксимабом (Rituxan™), до того, как клетки могут приобретать способность к инфильтрации в пораженные органы. Неконъюгированное антитело G5/44 (нацеливающая молекула в СМС-544) пока не было проанализировано у мышей SCID с моделью диссеминированной опухоли, но при введении в подкожные ксенотрансплантаты оно не давало какого-либо эффекта. При этом не следует ожидать, что G5/44, относящееся в изотипу IgG4, будет активировать эффекторные механизмы хозяина, а поэтому оно необязательно будет обладать противоопухолевой активностью.
G5/44, конъюгированное с калихеамицином (СМС-544), оказывает действие, противоположное действию ритуксимаба (Rituxan™), и дает лучшие результаты при введении в установившейся фазе заболевания. Причина такого улучшенного действия СМС-544 в установившейся фазе не совсем ясна, но, по всей вероятности, эта установившаяся фаза представляет собой клиническую ситуацию. СМА-676 соответствующего изотипа, т.е. несвязывающийся контрольный конъюгат, не обнаруживал значимого влияния на среднюю выживаемость. Результаты, полученные для мышей SCID с моделью диссеминированной опухоли, позволяют предположить, что для определения максимально эффективной дозы (MED) необходимо уменьшить дозы СМС-544. Доза 160 мкг/кг была менее эффективной, чем более низкие дозы в 80 и 40 мкг/кг. Причина, по которой это происходит, не ясна, но доза 160 мкг/кг значительно больше, чем MED. Для изучения этого результата планируются дополнительные исследования.
Мохаммед и др. использовали схему терапии CHOP (циклофосфамид (С) - 40 мг/кг, i.v.; доксорубицин (Н) 3,3 мг/кг, i.v.; винкристин (О) - 0,5 мг/кг, i.v.; преднизон (Р) 0,2 мг/кг, р.о.) в модели подкожных ксенотрансплантатов клеточной линии диффузной крупноклеточной лимфомы, DLCL. Для доз, используемых в СНОР-терапии, определяли максимально допустимую дозу. Схему СНО-терапии проводили путем внутривенного и перорального ежедневного введения в течение 5 дней, где оценку "активная" доза давали при продуцировании Т/С 25,8%. При этом какого-либо уменьшения опухолей не наблюдалось. Результаты с использованием модели, описанной Мохаммедом и др., были, очевидно, аналогичны результатам, полученным при терапии с использованием комбинации CHOP (вводимой i.p., Q4Dx3) на RL-модели, описанной в настоящей заявке. В отличие от СМС-544, ни в одном исследовании СНОР-терапия не давала долговременного эффекта.
Пример 9
Стабильные композиции СМС-544
Стабильные композиции СМС-544 для введения in vivo получали путем добавления разбавителей, наполнителей, носителей и стабилизаторов. После ГФХ-хроматографии, хроматографические фракции анализировали с помощью ЭХ-ВЭЖХ и УФ-анализа в длинном УФ-диапазоне. Исходя из вышеуказанных анализов, в которых была получена информация о содержании агрегатов, концентрации белка и нагрузки калихеамицином, отбирали соответствующие фракции для получения пула. Для стабилизации раствора добавляли эксципиенты, стабилизаторы, наполнители и забуферивающие агенты. Поскольку СМС-544 может подвергаться деградации под действием ряда различных механизмов разложения, то физическую нестабильность данного конъюгата необходимо учитывать в стадии разработки технологии его приготовления. Одной из возможных мер, которые необходимо предпринять в данном случае, является минимизация скорости гидролитического отделения калихеамицина от антитела при сохранении физической и химической целостности анти-CD22 антитела. Кроме того, должно быть минимизировано осаждение конъюгата "калихеамицин-антитело", которое может происходить при некоторых значениях рН и условиях концентрирования.
При разработке технологии приготовления конъюгата "производное мономерного калихеамицина - антитело", рН данного препарата имеет важное значение, поскольку изменение рН позволяет минимизировать деградацию и физическую нестабильность данного конъюгата. Для минимизации гидролиза калихеамицина и поддержания адекватной растворимости конъюгата, рН должен составлять 8,0. Дополнительные данные, полученные с использованием электрофореза в ДСН-ПААГ и анализа ELISA на связывание с антигеном, показали, что значительная структурная целостность и специфичность антитела поддерживается при рН 8,0. Поэтому, для поддерживания рН при 8,0, в качестве забуферивающего агента должен быть выбран трометамин. Альтернативный буфер может включать двухосновный натрий или фосфат калия. Концентрация буфера может составлять от 5 до 50 мМ. При этом предполагается, что для оптимальной стабильности/растворимости рН должен составлять, предпочтительно, от 7,5 до 8,5. В соответствии с современными техническими требованиями рН конечного продукта должен составлять 7,0-9,0.
Хотя стабильность забуференных растворов конъюгата является адекватной в течение короткого промежутка времени, однако, они не обладают долговременной стабильностью. Для увеличения срока действия конъюгатов используют лиофилизацию. Проблемы, связанные с лиофилизацией белкового раствора, хорошо известны и заключаются в том, что в процессе замораживания и сушки происходит разрушение вторичной, третичной и четвертичной структуры. Для поддержания структурной целостности антитела в процессе замораживания и сушки, в препарат включают сахарозу, которая действует как аморфный стабилизотор данного конъюгата. Сахарозу обычно используют в концентрациях 1,5-5,0% мас./об. Кроме того, для улучшения внешнего вида и физической прочности лиофилизированных остатков, в них может быть включен полимерный наполнитель, такой как декстран 40 или гидроксиэтилированный крахмал в концентрации 0,5-1,5 мас.%. Эти материалы, при относительно низких концентрациях, образуют лиофилизированные остатки и могут быть использованы для минимизации содержания твердых веществ в лиофилизированном препарате, что позволяет осуществлять более быструю сушку. В исследуемых препаратах использовали декстран 40 в концентрации 0,9% мас.
Для повышения растворимости конъюгата используют полисорбат 80. Обычно полисорбат 80 используется в количестве 0,005-0,05%, а его предпочтительная концентрация составляет 0,01%. В указанном препарате также используется твин в концентрации 0,91-0,05% об.
В данном препарате для улучшения эффективности конечного процесса очистки может быть также использован электролит. В качестве электролита обычно используется хлорид натрия в концентрации от 0,01М до 0,1М. Вместо хлорида натрия могут быть также использованы дополнительные электролиты, такие как сульфат натрия, поскольку он легче лиофилизируется. Оптимально, конечный раствор СМС-544 содержит 1,5% сахарозы (по массе), 0,9% декстрана 40 (по массе), 0,01% твина 80, 50 мМ хлорида натрия, 0,01% полисорбата 80 (по массе) и 20 мМ трометамина.
Предпочтительная формула для раствора до его лиофилизации является следующей: 0,5 мг/мл СМС-544, 1,5 мас.% сахарозы, 0,9 мас.% декстрана 40, 0,05М хлорида натрия, 0,01-0,05 об.% твина, 0,01 мас.% полисорбата 80, 0,02М трометамина, рН 8,0 и вода. Раствор разливали по сосудам из янтарного стекла при температуре от +5°С до 10°С (оптимальная температура +5°С); затем раствор замораживали при температуре замораживания от -35°С до -50°С (оптимально при -45°С); замороженный раствор подвергали предварительной стадии лиофилизации для первичной сушки раствора под давлением 20-80 микропаскаль (оптимально 60 микропаскаль); лиофилизированный продукт выдерживали при температуре хранения от -10°С до -40°С (оптимально -30°С), в течение 24-72 часов (оптимально в течение 60 часов); и наконец, лиофилизированный продут подвергали второй стадии сушки под давлением 20-80 микропаскаль (оптимально, 60 микропаскаль) при температуре хранения от +10°С до +35°С (оптимально +25°С) в течение 15-30 часов (оптимально в течение 24 часов). Для определения времени завершения первичной сушки проводили тест с повышением давления. После завершения цикла лиофилизации сосуды снова наполняли азотом и закрывали пробкой.
В таблице 11 показаны различия в технологии приготовления, применяемой для получения препарата СМА-544 и препарата СМА-676. Основными различиями между препаратом СМА-676 и препаратом СМС-544 являются пониженная концентрация белка в новом препарате (0,5 мг/мл), использование трометанина в качестве буфера и присутствие 0,01% твина 80. В результате этого препарат СМС-544, полученный по новой технологии, был прозрачным, тогда как препарат, полученный по технологии СМА-676, был мутным (см. таблицы 12 и 13).
Все работы и патенты, цитируемые выше, вводятся в настоящее описание посредством ссылки. В настоящее изобретение могут быть внесены различные модификации и изменения, которые являются очевидными для каждого специалиста. Такие модификации и изменения, вносимые в указанный способ конъюгирования, в конъюгаты, полученные таким способом, и в композиции/препараты, содержащие такие конъюгаты, входят в объем формулы изобретения.




| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ "ПРОИЗВОДНОЕ КАЛИХЕАМИЦИНА-НОСИТЕЛЬ" | 2016 |
|
RU2678818C2 |
| КОНЪЮГАТЫ "ПРОИЗВОДНОЕ КАЛИХЕАМИЦИНА-НОСИТЕЛЬ" | 2011 |
|
RU2602878C2 |
| КОНЪЮГАТЫ АНТИ-РТК7 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2015 |
|
RU2708075C2 |
| АНТИТЕЛА ПРОТИВ 5Т4 И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2487889C2 |
| ЛЕЧЕНИЕ В-КЛЕТОЧНЫХ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ С ИСПОЛЬЗОВАНИЕМ АНТИТЕЛ ПРОТИВ CD40L В КОМБИНАЦИИ С АНТИТЕЛАМИ ПРОТИВ CD20 И/ИЛИ ХИМИОТЕРАПИЕЙ И ЛУЧЕВОЙ ТЕРАПИЕЙ | 2000 |
|
RU2305561C2 |
| ПРИМЕНЕНИЯ АНТИ-CD40-АНТИТЕЛ | 2008 |
|
RU2491095C2 |
| СПОСОБ ЛЕЧЕНИЯ ВАСКУЛИТА | 2005 |
|
RU2411956C2 |
| СПОСОБ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА | 2005 |
|
RU2384345C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ НА ОСНОВЕ АФУКОЗИЛИРОВАННОГО АНТИТЕЛА К CD20 В СОЧЕТАНИИ С КОНЪЮГАТОМ АНТИТЕЛО К CD22 - ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2014 |
|
RU2700092C2 |
| АНТИ-MN АНТИТЕЛА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2427590C2 |



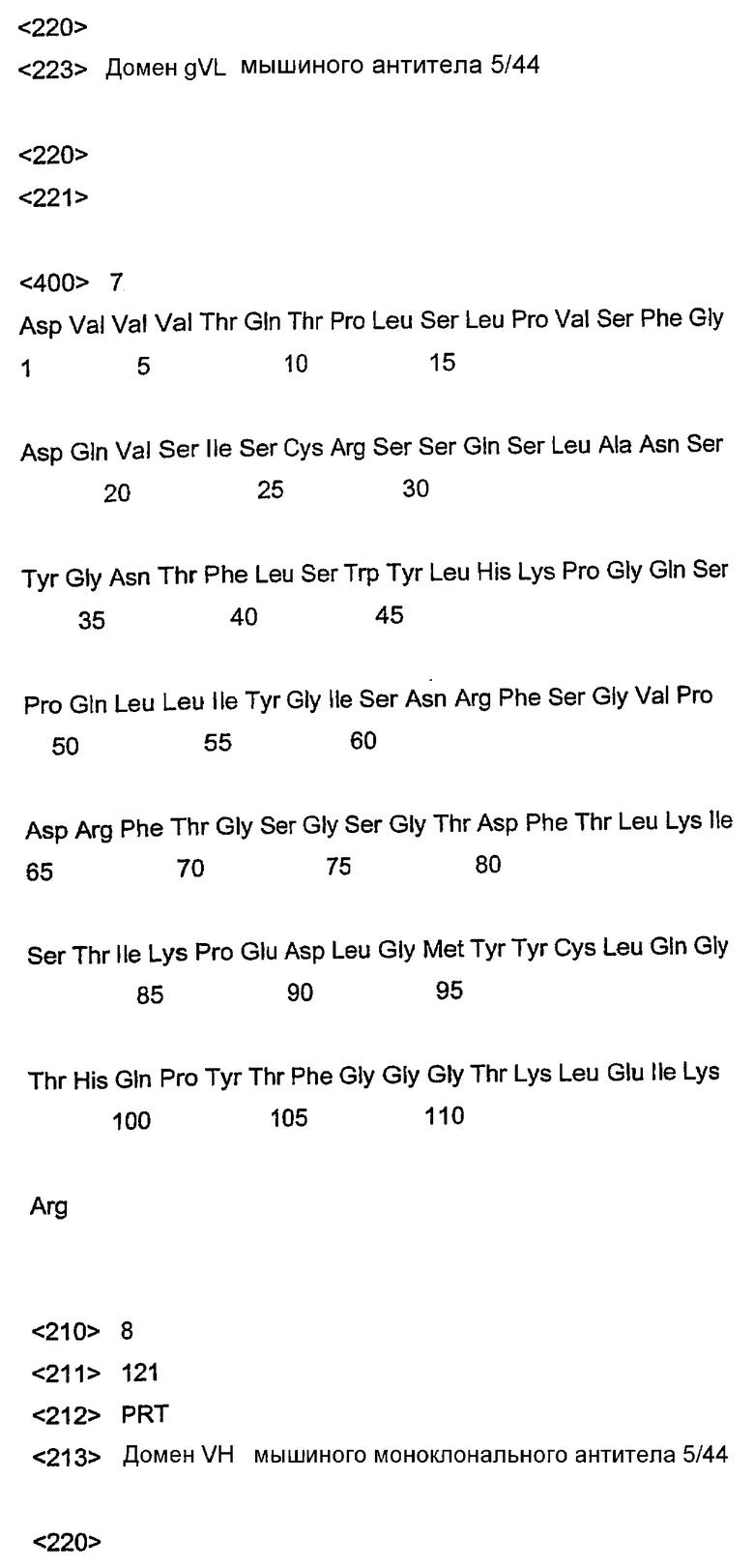
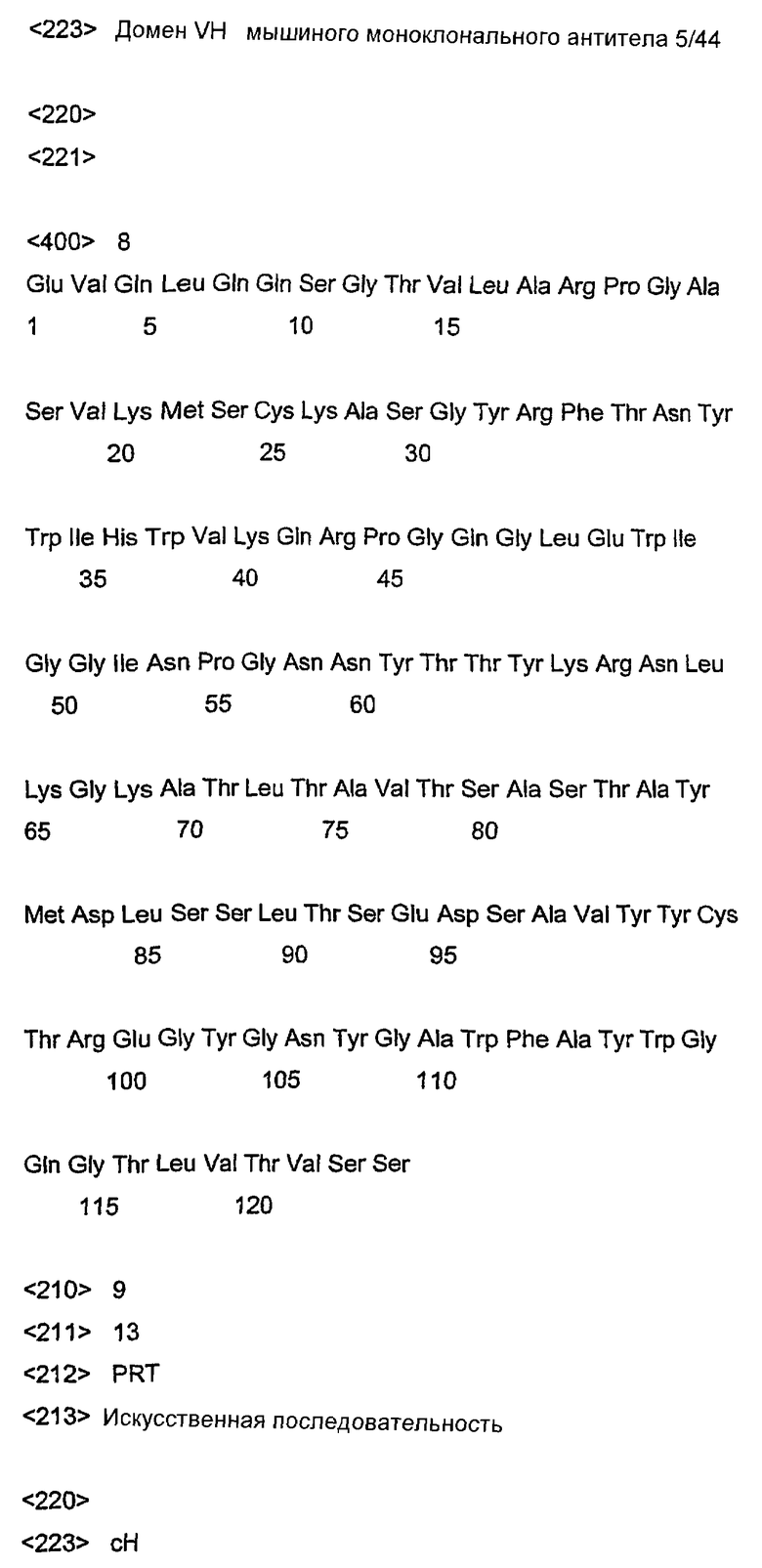






























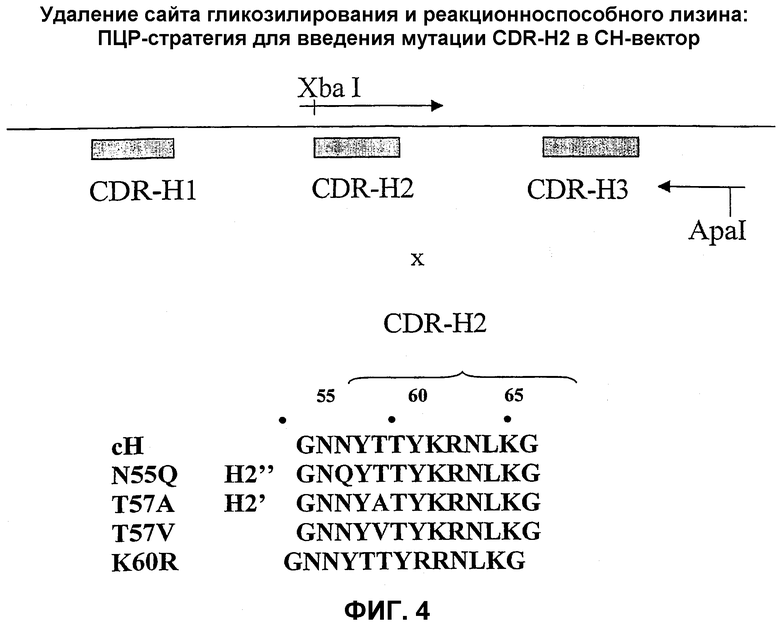
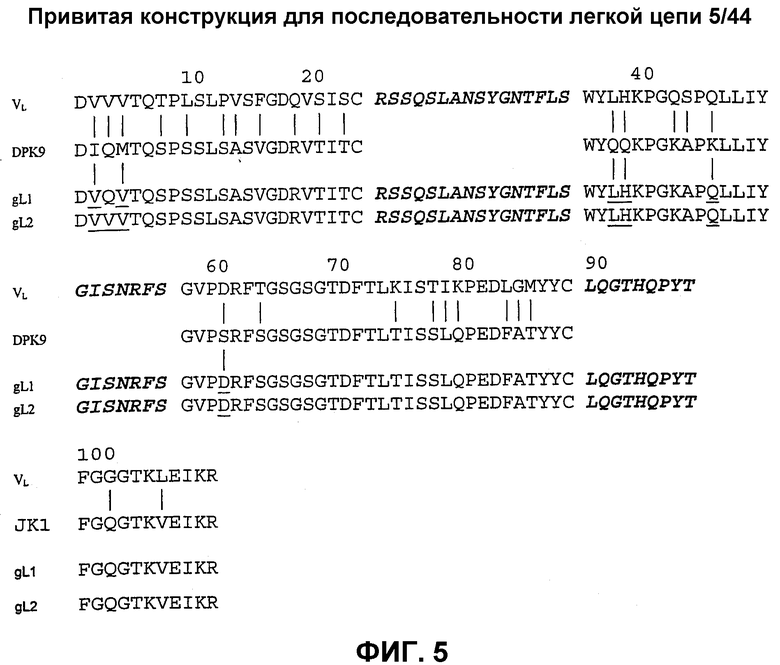





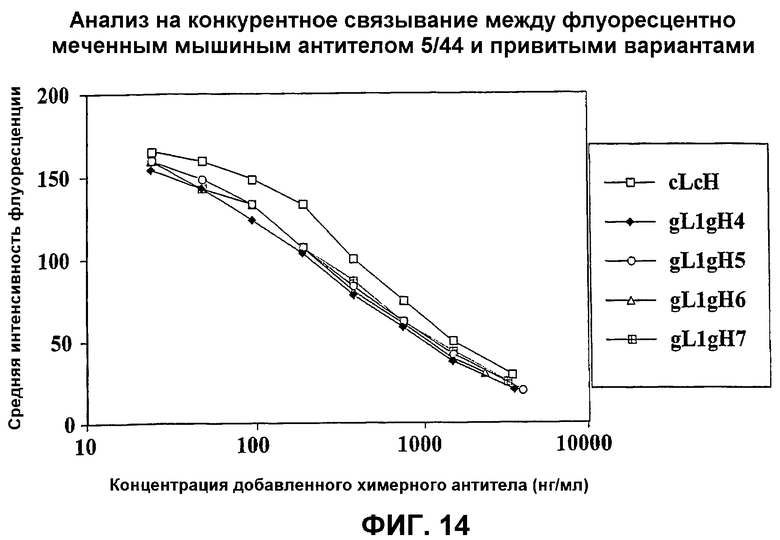
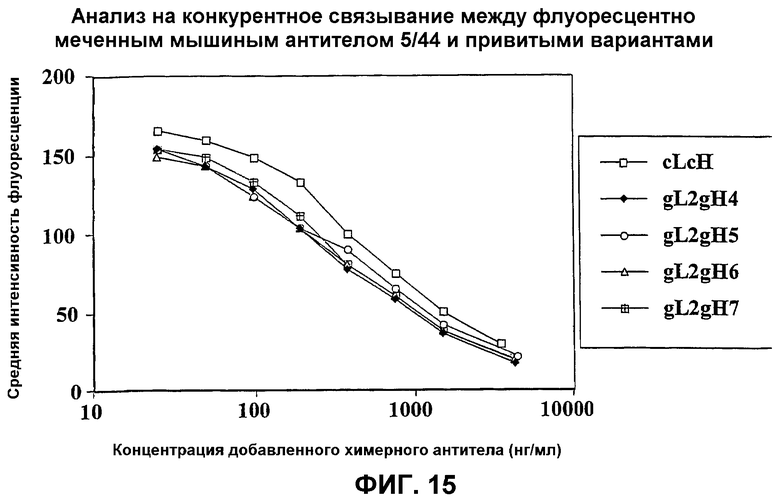




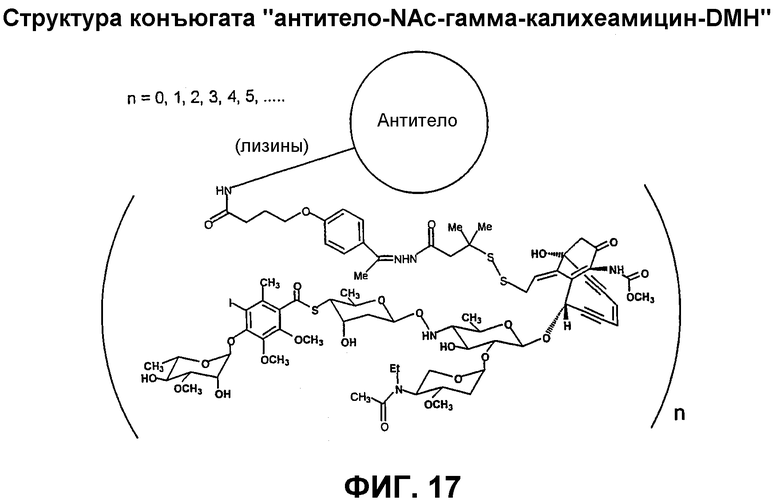
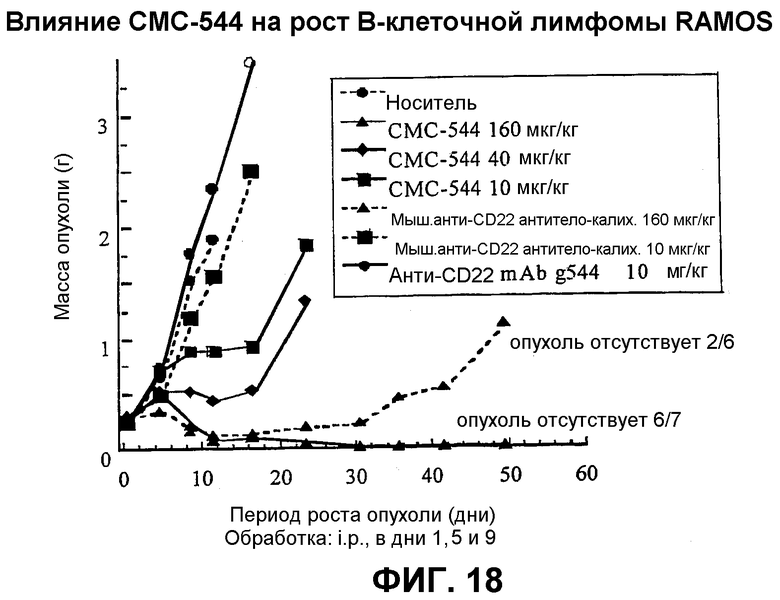
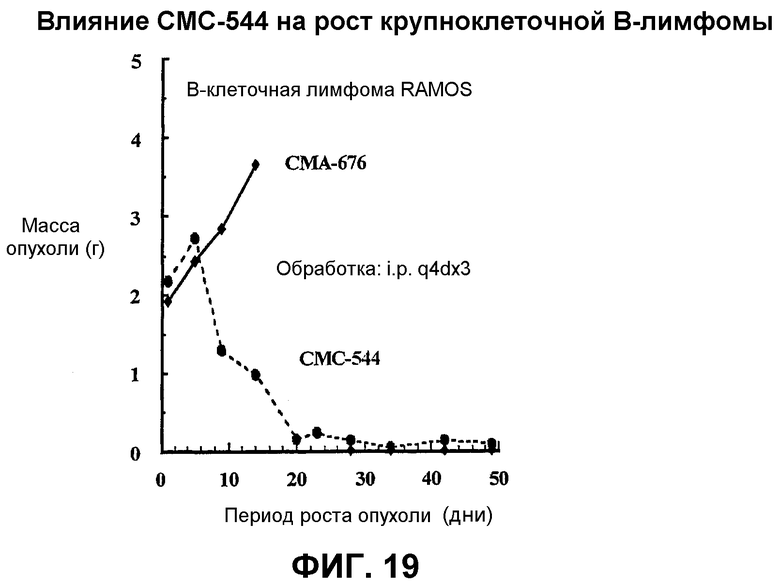
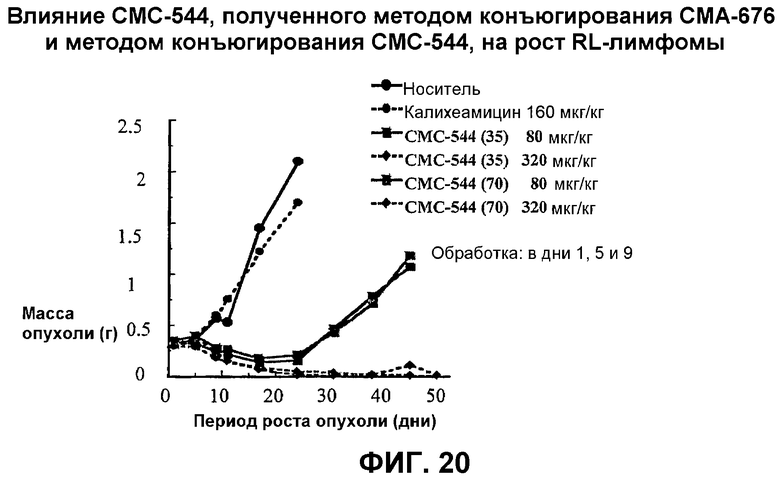
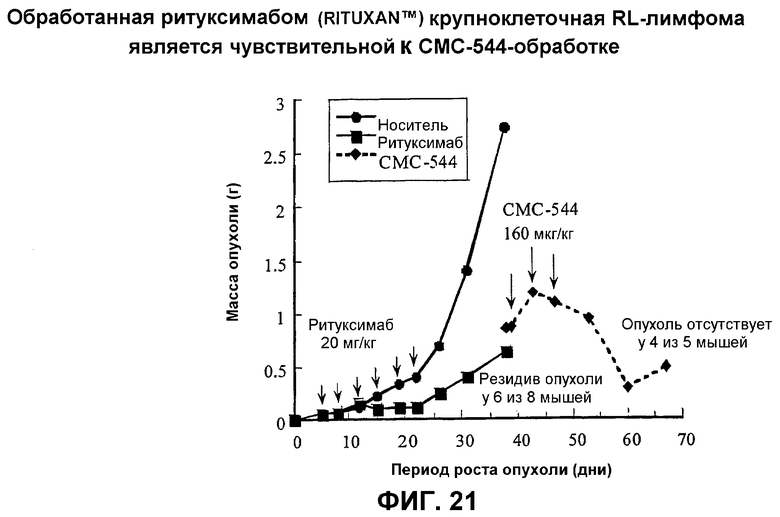
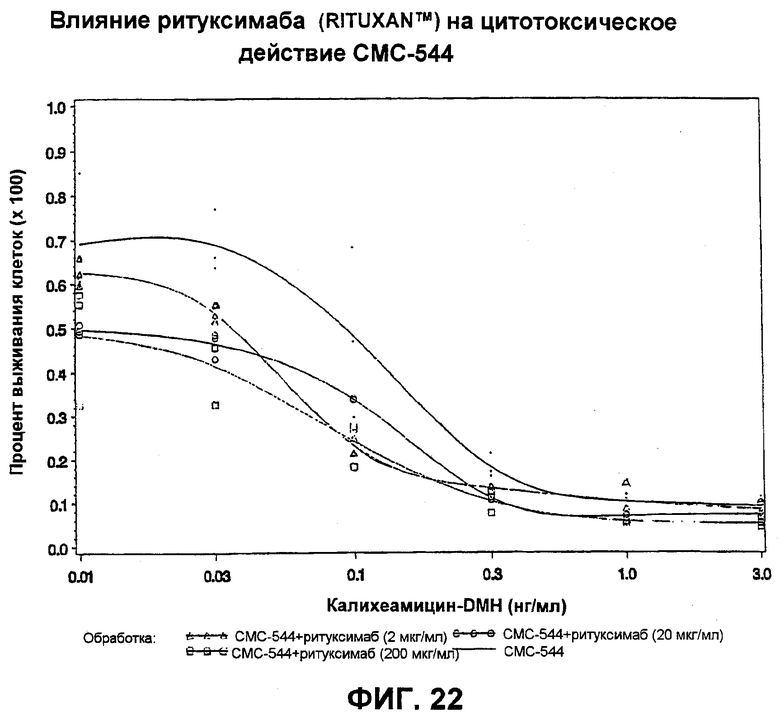
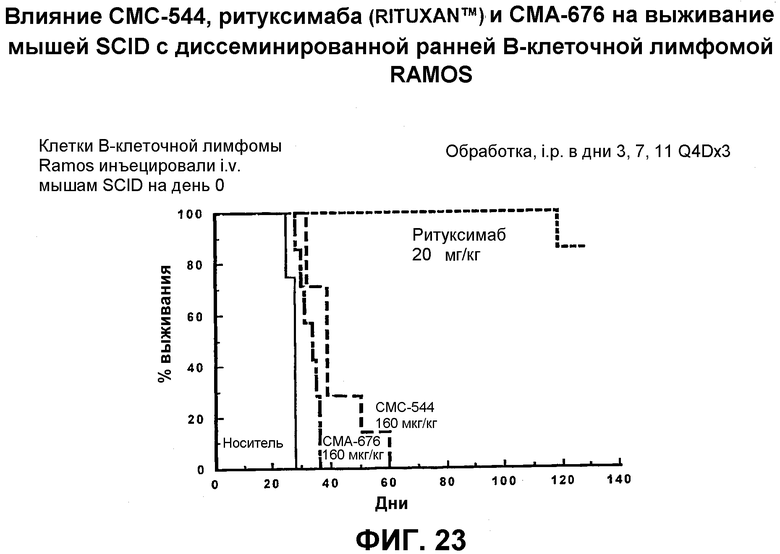
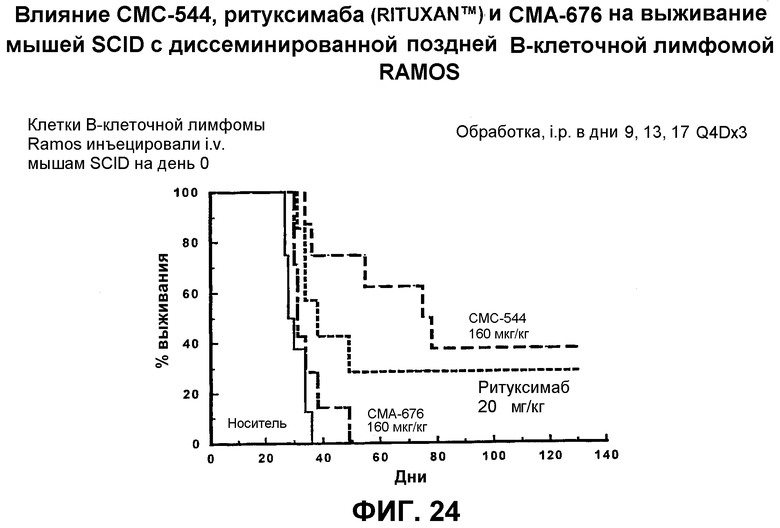
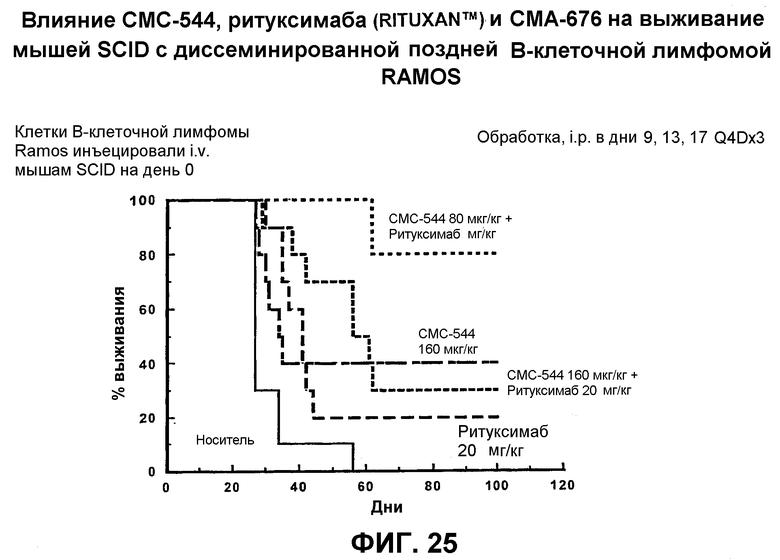
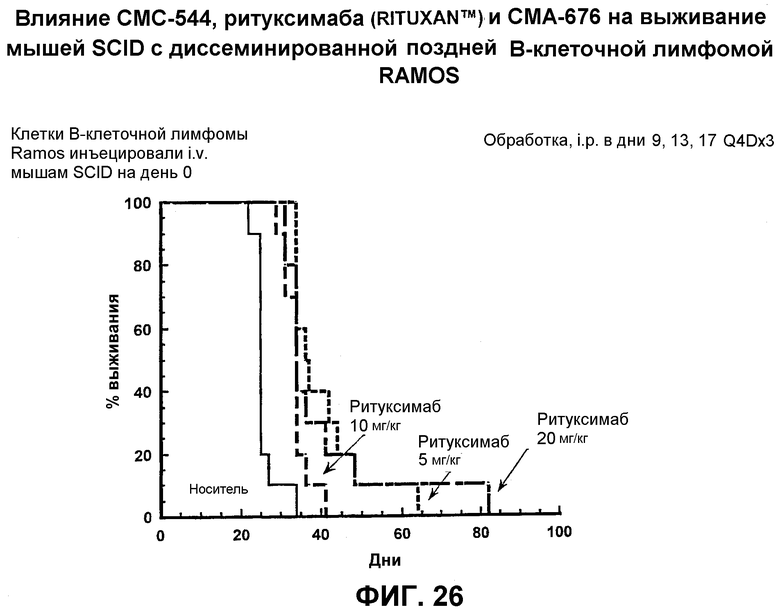
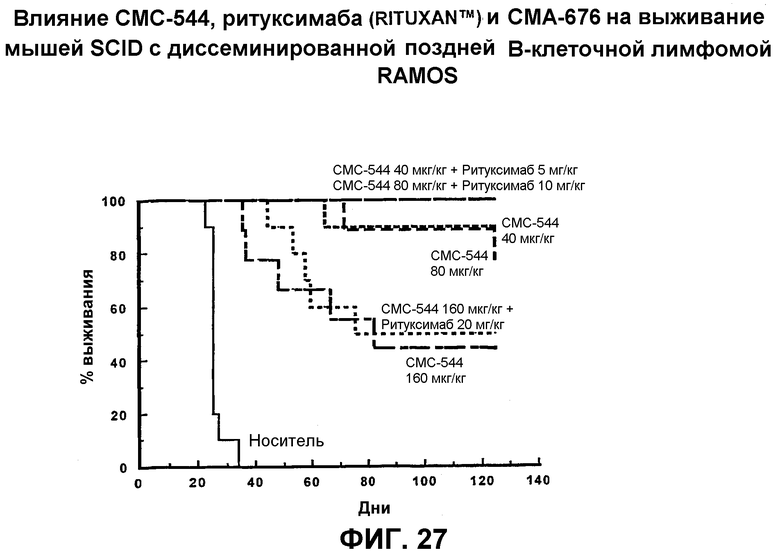
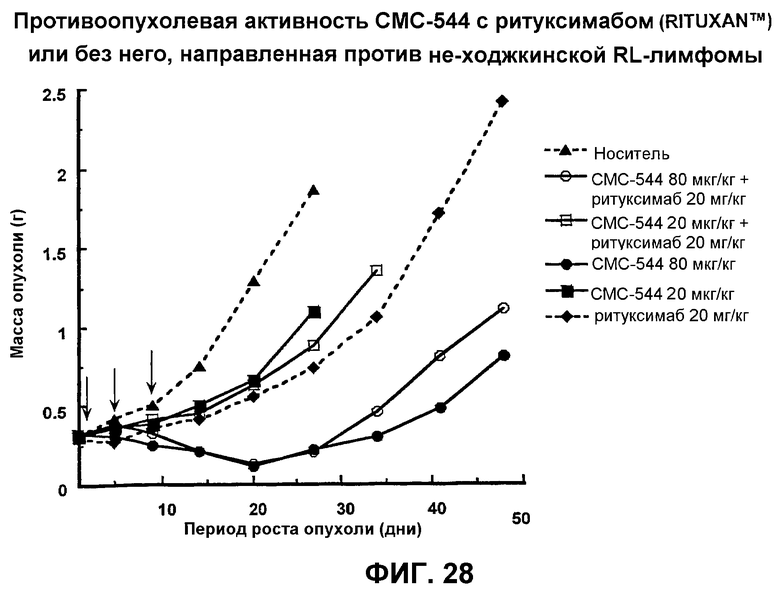
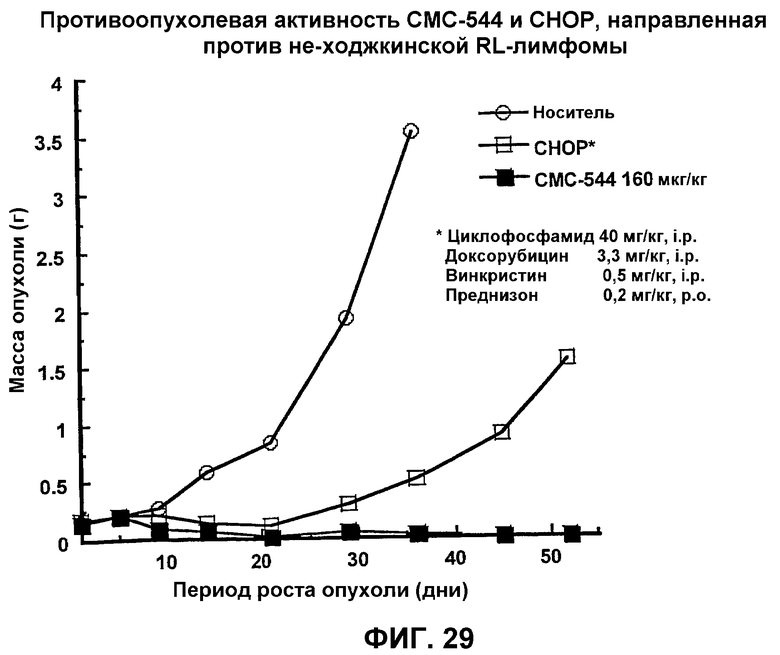
Изобретение относится к области биотехнологии и касается конъюгатов "производное калихеамицина-носитель". Сущность изобретения включает способы получения конъюгатов "мономерное цитотоксическое лекарственное средство - калихеамицин/носитель" со значительно более высокой нагрузкой лекарственным средством, чем в ранее описанных способах, с низкой степенью агрегации и с низким содержанием низкоконъюгированной фракции (LCF), а также конъюгаты "производное цитотоксического лекарственного средства- калихеамицина/ анти- CD22-антитело". Изобретение также включает композиции, содержащие конъюгат с анти - CD22 антителом, и использование таких конъюгатов. Преимущество изобретения заключается в создании коньюгата с высокой нагрузкой цитотоксического лекарственного средства. 6 н. и 175 з.п. ф-лы, 13 табл., 29 ил.
1. Способ получения конъюгатов «производное мономерного калихеамицина/антитело» с пониженным количеством низкоконъюгированной фракции (LCF), составляющей менее 10%, имеющих формулу
Pr(-X-S-S-W)m,
где Pr представляет собой антитело;
X представляет собой гидролизуемый линкер, способный высвобождать калихеамицин из указанного конъюгата после его связывания и проникновения в клетки-мишени;
W представляет собой калихеамицин;
m означает среднюю нагрузку очищенного продукта конъюгата, при которой калихеамицин составляет 4-10% по весу конъюгата; и
(-X-S-S-W)m представляет собой производное калихеамицина,
где указанный способ включает стадии:
(1) добавления производного калихеамицина к антителу, где производное калихеамицина составляет 4,5-11% по массе антитела;
(2) инкубирования производного калихеамицина и антитела в ненуклеофильном и совместимом с белком буферном растворе с рН от 7 до 9 с получением конъюгата "производное мономерного калихеамицина/антитело", где раствор, кроме того, содержит (а) органический сорастворитель и (b) добавку, содержащую по крайней мере одну C6-C18 карбоновую кислоту или ее соль, и где инкубирование проводят при температуре от примерно 30 до примерно 35°С в течение периода времени от примерно 15 мин до 24 ч; и (3) проведения процесса разделительной хроматографии композиции, полученной на стадии (2), для отделения конъюгатов "производное калихеамицина/антитело" с нагрузкой в пределах от 4 до 10% по массе калихеамицина и с содержанием низкоконъюгированной фракции (LCF), составляющей ниже 10% неконъюгированного антитела, производного калихеамицина и агрегированных конъюгатов.
2. Способ по п.1, где антитело выбрано из группы, состоящей из моноклонального антитела, химерного антитела, антитела человека, гуманизированного антитела, одноцепочечного антитела и биологически активного фрагмента антитела, где биологически активный фрагмент представляет собой Fab, модифицированный Fab, Fab′, F(ab′)2 или Fv или мономер или димер тяжелой цепи.
3. Способ по п 2, где гуманизированное антитело направлено против антигена клеточной поверхности CD22.
4. Способ по п.1, где антитело содержит вариабельный домен легкой цепи, содержащий последовательность SEQ ID NO: 19, и вариабельный домен тяжелой цепи, содержащий последовательность SEQ ID NO: 27.
5. Способ по п.1, где антитело является гуманизированным антителом и содержит легкую цепь, содержащую последовательность SEQ ID NO: 28.
6. Способ по п.1, где антитело является гуманизированным антителом и содержит тяжелую цепь, содержащую последовательность SEQ ID NO: 30.
7. Способ по п.1, где антитело содержит легкую цепь, содержащую последовательность SEQ ID NO: 28, и тяжелую цепь, содержащую последовательность SEQ ID NO: 30.
8. Способ по п.1, где антитело содержит последовательность SEQ ID NO: 1 для CDR-H1, SEQ ID NO: 2, или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, SEQ ID NO: 3 для CDR-H3, SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 и SEQ ID NO: 6 для CDR-L3.
9. Способ по п.8, где антитело включает последовательность GINPGNNYATYRRKFQG для CDR-H2.
10. Способ по п.3, где гуманизированное анти-CD22 антитело представляет собой CDR-привитое антитело, которое представляет собой вариант антитела, полученный в соответствии с протоколом осуществления аффинного созревания и обладающий повышенной специфичностью к CD22 человека.
11. Способ по п.1, где калихеамицином представляет собой гамма-калихеамицин или N-ацетил-гамма-калихеамицин.
12. Способ по п.1 или 11, где калихеамицин функционализирован 3-меркапто-3-метилбутаноилгидразидом.
13. Способ по п.1, где указанным гидролизуемым линкером является 4-(4-ацетилфенокси)бутановая кислота (AcBut).
14. Способ по п.1, где добавкой, используемой в стадии (2)(b), является октановая кислота или ее соль.
15. Способ по п.1, где добавкой, используемой в стадии (2)(b), является декановая кислота или ее соль.
16. Способ по п.1, где добавку, используемую в стадии (2)(b), вводят в концентрации менее чем 200 мМ.
17. Способ по п.16, где добавку вводят в концентрации менее чем 100 мМ.
18. Способ по п.17, где добавку вводят в концентрации менее чем 50 мМ
19. Способ по п.1, где процессом разделительной хроматографии на стадии (3) является эксклюзионная хроматография (SEC).
20. Способ по п.1, где процессом разделительной хроматографии на стадии (3) является ВЭЖХ, ЖЭХБ или хроматография на Сефакриле S-200.
21. Способ по п.1, где процессом разделительной хроматографии на стадии (3) является гидрофобная хроматография (ГФХ).
22. Способ по п.21, где гидрофобную хроматографию (ГФХ) осуществляют с использованием в качестве хроматографической среды фенилсефарозы 6 Fast Flow, бутилсефарозы 4 Fast Flow, октилсефарозы 4 Fast Flow, Toyopearl Ether-650M, метиловой ГФХ-среды Macro-Prep или трет-бутиловой ГФХ-среды Macro-Prep.
23. Способ по п.21, где указанную гидрофобную хроматографию (ГФХ) осуществляют с использованием в качестве хроматографической среды бутилсефарозы 4 Fast Flow.
24. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" с пониженным количеством низкоконъюгированной фракции (LCF), составляющей менее 10%, где конъюгат имеет формулу
Pr(-X-S-S-W)m,
где Pr представляет собой анти-CD22 антитело;
X представляет собой гидролизуемый линкер, способный высвобождать калихеамицин из указанного конъюгата после его связывания и проникновения в клетки-мишени;
W представляет собой калихеамицин;
m означает среднюю нагрузку очищенного продукта конъюгата, при
которой калихеамицин составляет 4-10% по весу конъюгата; и
(-X-S-S-W)m представляет собой производное калихеамицина.
25. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где анти-CD22 антитело выбрано из группы, состоящей из моноклонального антитела, химерного антитела, антитела человека, гуманизированного антитела, одноцепочечного антитела и биологически активного фрагмента антитела, где биологически активный фрагмент представляет собой Fab, модифицированный Fab, Fab′, F(ab′)2 или Fv или мономер или димер тяжелой цепи.
26. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где анти-CD22 антитело содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 19, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 27.
27. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где анти-CD22 антитело представляет собой гуманизированное антитело и содержит легкую цепь, имеющую последовательность SEQ ID NO: 28.
28. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где анти-CD22 антитело представляет собой гуманизированное антитело и содержит тяжелую цепь, имеющую последовательность SEQ ID NO: 30.
29. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где анти-CD22 антитело содержит легкую цепь, содержащую последовательность SEQ ID NO: 28, и тяжелую цепь, содержащую последовательность SEQ ID NO: 30.
30. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где указанное анти-CD22 антитело обладает специфичностью к CD22 человека и содержит тяжелую цепь, где ее вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей, представленных как HI (SEQ ID NO: 1) для CDR-H1, как Н2 (SEQ ID NO: 2), или Н2′ (SEQ ID NO: 13), или Н2′′ (SEQ ID NO: 15), или Н2′′′ (SEQ ID NO: 16) для CDR-H2, или как Н3 (SEQ ID NO: 3) для CDR-H3, и содержит легкую цепь, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей, представленных как LI (SEQ ID NO: 4) для CDR-L1, как L2 (SEQ ID NO: 5) для CDR-L2 или как L3 (SEQ ID NO: 6) для CDR-L3.
31. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где антитело содержит тяжелую цепь, где ее вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей SEQ ID NO: 1 для CDR-H1, SEQ ID NO: 2, или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, или SEQ ID NO: 3 для CDR-H3, и содержит легкую цепь, где вариабельный домен содержит CDR, имеющую по меньшей мере одну из последовательностей SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2, или SEQ ID NO: 6 для CDR-L3.
32. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где анти-CD22 антитело содержит SEQ ID NO: 1 для CDR-H1, SEQ ID NO: 2, или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, SEQ ID NO: 3 для CDR-H3, SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 и SEQ ID NO: 6 для CDR-L3.
33. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.32, где анти-CD22 антитело включает последовательность GINPGNNYATYRRKFQG для CDR-H2.
34. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.25, где гуманизированное анти-CD22 антитело представляет собой CDR-привитое анти-CD22 антитело.
35. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.34, где гуманизированным анти-CD22 антителом является CDR-привитое антитело, которое представляет собой вариант антитела, полученный в соответствии с протоколом осуществления аффинного созревания и обладающий повышенной специфичностью к CD22 человека.
36. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где анти-CD22 антитело содержит вариабельный домен, содержащий акцепторные каркасные области человека и донорные CDR, не являющиеся человеческими.
37. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.36, где акцепторные каркасные области человека вариабельного домена тяжелой цепи антитела основаны на SEQ ID NO: 21 и 22 и включают донорные остатки в положениях 1, 28, 48, 72 и 97 в SEQ ID NO:8.
38. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.37, где анти-CD22 антитело дополнительно содержит донорные остатки в положениях 68 и 70 в SEQ ID NO: 8.
39. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.36, где анти-CD22 антитело содержит вариабельный домен легкой цепи, содержащий акцепторную каркасную область человека, основанную на SEQ ID NO: 17 и 18, и включает донорные остатки в положениях 2, 4, 42, 43, 50 и 65 в SEQ ID NO: 7.
40. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.39, где анти-CD22 антитело дополнительно содержит донорный остаток в положении 3 в SEQ ID NO: 7.
41. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где анти-CD22 антитело содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 7, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 8.
42. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.25, где анти-CD22 антитело содержит гибрид CDR, содержащий усеченную донорную последовательность CDR, где пропущенная часть донорного CDR замещена другой последовательностью и образует функциональный CDR.
43. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где калихеамицин представляет собой гамма, калихеамицин или N-ацетил гамма-калихеамицин.
44. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24 или 43, где калихеамицин функционализирован 3-меркапто-3-метилбутаноилгидразидом.
45. Конъюгат "производное мономерного калихеамицина/анти-CD22 антитело" по п.24, где гидролизуемый линкер представляет собой 4-(4-ацетилфенокси)масляную кислоту (AcBut).
46. Способ получения стабильной лиофилизованной композиции конъюгата «производное мономерного калихеамицина/антитело, где способ включает:
(a) растворение конъюгата "производное мономерного калихеамицина/анти-CD22 антитело" по п.24 до конечной концентрации 0,5-2 мг/мл в растворе, содержащем криозащитный агент в концентрации 1,5-5% по массе, полимерный наполнитель в концентрации 0,5-1,5% по массе, электролиты в концентрации 0,01 - 0,1 М, агент, способствующий растворению, в концентрации 0,005-0,05% по массе, буферный агент в концентрации 5-50 мМ, необходимой для доведения конечного значения рН раствора до 7,8-8,2, и воду;
(b) распределение вышеуказанного раствора по сосудам при температуре от 5 до 10°С;
(c) замораживание указанного раствора при температуре замораживания от -35 до -50°С;
(d) проведение предварительной стадии лиофилизации замороженного раствора для его первичной сушки под давлением 20-80 мкПа и при температуре хранения от -10 до -40°С в течение 24-78 ч; и
(e) проведение вторичной стадии сушки лиофилизированного продукта стадии (d) под давлением 20-80 мкПа и при температуре хранения от 10 до 35°С в течение 15-30 ч.
47. Способ по п.46, где калихеамицин производного калихеамицина представляет собой гамма-калихеамицин или N-ацетилкалихеамицин.
48. Способ по п.46, где антитело выбрано из группы, состоящей из моноклонального антитела, химерного антитела, антитела человека, гуманизированного антитела, одноцепочечного антитела и биологически активного фрагмента антитела, где биологически активный фрагмент антитела представляет собой Fab, модифицированный Fab, Fab′, F(ab′)2 или Fv или мономер или димер тяжелой цепи.
49. Способ по п.48, где антитело представляет собой моноклональное антитело человека.
50. Способ по п.48, где антитело представляет собой химерное антитело.
51. Способ по п.48, где антитело представляет собой антитело человека.
52. Способ по п.48, где антитело представляет собой гуманизированное антитело.
53. Способ по п.46, где антитело содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 19, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 27.
54. Способ по п.46, где гуманизированное антитело представляет собой CDR-привитое антитело, обладающее специфичностью в отношении CD22 человека, и содержит легкую цепь с последовательностью SEQ ID NO: 28.
55. Способ по п.46, где гуманизированное антитело представляет собой CDR-привитое антитело, обладающее специфичностью в отношении CD22 человека, и содержит тяжелую цепь с последовательностью SEQ ID NO: 30.
56. Способ по п.46, где антитело содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 28, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 30.
57. Способ по п.46, где антитело обладает специфичностью в отношении CD22 человека и содержит тяжелую цепь, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей, представленных как HI (SEQ ID NO: 1) для CDR-H1, как Н2 (SEQ ID NO: 2), или H2′ (SEQ ID NO: 13), или Н2′′ (SEQ ID NO: 15), или Н2′′′ (SEQ ID NO: 16) для CDR-H2, или как Н3 (SEQ ID NO: 3) для CDR-H3, и содержит легкую цепь, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей, представленных как LI (SEQ ID NO: 4) для CDR-L1, как L2 (SEQ ID NO: 5) для CDR-L2 или как L3 (SEQ ID NO: 6) для CDR-L3.
58. Способ по п.46, где антитело содержит вариабельный домен тяжелой цепи, содержащий CDR, имеющую по крайней мере одну из последовательностей SEQ ID NO: 1 для CDR-H1, SEQ ID NO: 2 или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, или SEQ ID NO: 3 для CDR-H3, и легкой цепи, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 или SEQ ID NO: 6 для CDR-L3.
59. Способ по п.46, где антитело содержит SEQ ID NO:1 для CDR-H1, SEQ ID NO: 2, или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, SEQ ID NO: 3 для CDR-H3, SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 и SEQ ID NO: 6 для CDR-L3.
60. Способ по п.59, где антитело содержит последовательность GINPGNNYATYRRKFQG для CDR-H2.
61. Способ по п.46, где гуманизированное антитело представляет собой CDR-привитое антитело, которое представляет собой вариант антитела, имеющий повышенную специфичность в отношении CD22 человека, где вариант антитела получен в соответствии с протоколом осуществления аффинного созревания.
62. Способ по п.46, где антитело содержит вариабельный домен, содержащий акцепторные каркасные области человека и донорные CDR, не являющиеся человеческими.
63. Способ по п.62, где акцепторные каркасные области человека вариабельного домена тяжелой цепи анти-CD22 антитела основаны на SEQ ID NO: 21 и 22 и включают донорные остатки в положениях 1, 28, 48, 72 и 97 в SEQ ID NO: 8.
64. Способ по п.63, где антитело дополнительно содержит донорные остатки в положениях 68 и 70 в SEQ ID NO: 8.
65. Способ по п.62, где антитело содержит вариабельный домен легкой цепи, содержащий акцепторную каркасную область человека, основанную на SEQ ID NO: 17 и 18, и включает донорные остатки в положениях 2, 4, 42, 43, 50 и 65 в SEQ ID NO: 7.
66. Способ по п.65, где антитело дополнительно содержит донорный остаток в положении 3 в SEQ ID NO: 7.
67. Способ по п.46, где антитело содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 7, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 8.
68. Способ по п.46, кроме того необязательно содержащий биоактивный агент в терапевтически эффективном количестве.
69. Способ по п.68, где биоактивным агентом является цитотоксическое средство.
70. Способ по п.68, где биоактивным агентом является фактор роста.
71. Способ по п.68, где биологическим агентом является гормон.
72. Способ по п.68, где биологическим агентом является антитело.
73. Способ по п.46, где криозащитный агент выбран из группы, содержащей альдит, маннит, сорбит, инозит, полиэтиленгликоль, альдоновую кислоту, уроновую кислоту, альдаровую кислоту, альдозы, кетозы, аминосахара, альдиты, инозиты, глицеральдегиды, арабинозу, ликсозу, пентозу, рибозу, ксилозу, галактозу, глюкозу, гексозу, идозу, маннозу, талозу, гептозу, глюкозу, фруктозу, глюконовую кислоту, сорбит, лактозу, маннит, метил-α-глюкопиранозид, мальтозу, изоаскорбиновую кислоту, аскорбиновую кислоту, лактон, сорбозу, глюкаровую кислоту, эритрозу, треозу, арабинозу, аллозу, альтрозу, гулозу, идозу, талозу, эритрулозу, рибулозу, ксилулозу, псикозу, тагатозу, глюкуроновую кислоту, глюконовую кислоту, глюкаровую кислоту, галактуроновую кислоту, маннуроновую кислоту, глюкозамин, галактозамин, сахарозу, трегалозу, нейраминовую кислоту, арабинаны, фруктаны, фуканы, галактаны, галактуронаны, глюканы, маннаны, ксиланы, леван, фукоидан, каррагенан, галактокаролозу, пектины, пектиновые кислоты, амилозу, пуллулан, гликоген, амилопектин, целлюлозу, декстран, пустулан, хитин, агарозу, кератин, хондротин, дерматан, гиалуроновую кислоту, альгиновую кислоту, ксантановую камедь, крахмал, сахарозу, глюкозу, лактозу, трегалозу, этиленгликоль, полиэтиленгликоль, полипропиленгликоль, глицерин и пентаэритрит.
74. Способ по п.73, где криозащитным агентом является сахароза.
75. Способ по п.74, где сахароза присутствует в концентрации 1,5% по массе.
76. Способ по п.46, где полимерным наполнителем является декстран 40, присутствующий в концентрации 0,9% по массе.
77. Способ по п.46, где полимерным наполнителем является гидроксиэтилированный крахмал 40, присутствующий в концентрации 0,9% по массе.
78. Способ по п.46, где электролитом является хлорид натрия, присутствующий в концентрации 0,05М.
79. Способ по п.46, где агентом, повышающим растворимость, является поверхностно-активное вещество.
80. Способ по п.79, где указанным поверхностно-активным веществом является полисорбат 80, присутствующий в концентрации 0,01% по массе.
81. Способ по п.46, где указанным забуферивающим агентом является трометамин, присутствующий в концентрации 0,02 М.
82. Способ по п.46, где рН раствора стадии (а) составляет 8,0.
83. Способ по п.46, где указанный раствор стадии (b) распределяют по сосудам при температуре 5°С.
84. Способ по п.46, где на стадии (с) замораживание указанного раствора в сосудах осуществляют при температуре -45°С.
85. Способ по п.46, где на стадии (d) замороженный раствор подвергают предварительной стадии лиофилизации для первичной сушки под давлением 60 мкПа и при температуре хранения -30°С в течение 60 ч.
86. Способ по п.46, где на стадии (е) лиофилизированный продукт стадии (d) подвергают второй стадии сушки под давлением 60 мкПа и при температуре хранения 25°С в течение 24 ч.
87. Композиция, содержащая терапевтически эффективную дозу конъюгата «производное мономерного калихеамицина/анти-CD22 антитело» по п.24, полученная:
(a) растворением конъюгата «производное мономерного калихеамицина/анти-CD22 антитело» до конечной концентрации 0,5-2 мг/мл в растворе, содержащем криозащитный агент в концентрации 1,5-5% по массе, полимерный наполнитель в концентрации 0,5-1,5% по массе, электролиты в концентрации 0,01 - 0,1 М, агент, способствующий растворению, в концентрации 0,005-0,05% по массе, забуферивающий агент в концентрации 5-50 мМ, необходимой для доведения конечного значения рН раствора до 7,8-8,2, и воду;
(b) распределением вышеуказанного раствора по сосудам при температуре от 5 до 10°С;
(c) замораживанием указанного раствора при температуре замораживания от -35 до -50°С;
(d) проведением предварительной стадии лиофилизации замороженного раствора для его первичной сушки под давлением 20-80 мкПа и при температуре хранения от -10 до -40°С в течение 24-78 ч; и
(е) проведением вторичной стадии сушки лиофилизированного продукта стадии (d) под давлением 20-80 мкПа и при температуре хранения от 10 до 35°С в течение 15-30 ч.
88. Композиция по п.87, где анти-CD22 антитело выбрано из группы, состоящей из моноклонального антитела, химерного антитела, антитела человека, гуманизированного антитела, одноцепочечного антитела и биологически активного фрагмента антитела, где биологически активный фрагмент представляет собой Fab, модифицированный Fab, Fab′, F(ab′)2 или Fv или мономер или димер тяжелой цепи.
89. Композиция по п.88, где анти-CD22 антитело представляет собой моноклональное антитело человека.
90. Композиция по п.88, где анти-CD22 антитело представляет собой химерное антитело.
91. Композиция по п.88, где анти-CD22 антитело представляет собой антитело человека.
92. Композиция по п.88, где анти-CD22 антитело представляет собой гуманизированное антитело.
93. Композиция по п.87, где анти-CD22 антитело содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 19, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 27.
94. Композиция по п.87, где гуманизированное анти-CD22 антитело представляет собой CDR-привитое антитело, обладающее специфичностью в отношении CD22 человека, и содержит легкую цепь с последовательностью SEQ ID NO: 28.
95. Композиция по п.87, где гуманизированное анти-CD22 антитело представляет собой CDR-привитое антитело, обладающее специфичностью в отношении CD22 человека, и содержит тяжелую цепь с последовательностью SEQ ID NO: 30.
96. Композиция по п.87, где анти-CD22 антитело содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 28, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 30.
97. Композиция по п.87, где анти-CD22 антитело обладает специфичностью в отношении CD22 человека и содержит тяжелую цепь, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей, представленных как HI (SEQ ID NO: 1) для CDR- Hl, как Н2 (SEQ ID NO: 2), или Н2′ (SEQ ID NO: 13), или Н2′′ (SEQ ID NO: 15), или H2′′′ (SEQ ID NO: 16) для CDR-H2 или как Н3 (SEQ ID NO: 3) для CDR-H3, и содержит легкую цепь, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей, представленных как LI (SEQ ID NO: 4) для CDR-L1, как L2 (SEQ ID NO: 5) для CDR-L2 или как L3 (SEQ ID NO: 6) для CDR-L3.
98. Композиция по п.87, где антитело содержит вариабельный домен тяжелой цепи, содержащий CDR, имеющую по крайней мере одну из последовательностей SEQ ID NO: 1 для CDR-H1, SEQ ID NO: 2, или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, или SEQ ID NO: 3 для CDR-H3, и легкой цепи, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 или SEQ ID NO: 6 для CDR-L3.
99. Композиция по п.87, где анти-CD22 антитело содержит SEQ ID NO: I для CDR-H1, SEQ ID NO: 2, или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, SEQ ID NO: 3 для CDR-H3, SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 и SEQ ID NO: 6 для CDR-L3.
100. Композиция по п.99, где анти-CD22 антитело содержит последовательность GINPGNNYATYRRKFQG для CDR-H2.
101. Композиция по п.88, где гуманизированное анти-CD22 антитело представляет собой CDR-привитое антитело, которое представляет собой вариант антитела, имеющий повышенную специфичность в отношении CD22 человека, где вариант антитела получен в соответствии с протоколом осуществления аффинного созревания.
102. Композиция по п.87, где анти-CD22 антитело содержит вариабельный домен, содержащий акцепторные каркасные области человека и донорные CDR, не являющиеся человеческими.
103. Композиция по п.102, где акцепторные каркасные области человека вариабельного домена тяжелой цепи анти-CD22 антитела основаны на SEQ ID NO: 21 и 22 и включают донорные остатки в положениях 1, 28, 48, 72 и 97 в SEQ ID NO: 8.
104. Композиция по п.103, где анти-CD22 антитело дополнительно содержит донорные остатки в положениях 68 и 70 в SEQ ID NO: 8.
105. Композиция по п.102, где анти-CD22 антитело содержит вариабельный домен легкой цепи, содержащий акцепторную каркасную область человека, основанную на SEQ ID NO: 17 и 18, и включает донорные остатки в положениях 2, 4, 42, 43, 50 и 65 в SEQ ID NO: 7.
106. Композиция по п.105, где анти-CD22 антитело дополнительно содержит донорный остаток в положении 3 в SEQ ID NO: 7.
107. Композиция по п.87, где анти-CD22 антитело содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 7, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 8.
108. Композиция по п.87, где калихеамицин в производном калихеамицина представляет собой гамма калихеамицин или N-ацетилкалихеамицин.
109. Композиция по п.87, кроме того, необязательно содержащая биоактивный агент.
110. Композиция по п.109, где биоактивным агентом является цитотоксическое средство.
111. Композиция по п.109, где биоактивным агентом является фактор роста.
112. Композиция по п.109, где биологическим агентом является гормон.
113. Композиция по п.109, где биологическим агентом является антитело.
114. Способ лечения индивидуума, страдающего пролиферативным заболеванием, при котором экспрессируется CD22, включающий введение терапевтически эффективной дозы композиции, содержащей коньюгат «производное мономерного калихеамицина/анти-CD22 антитело» по п.24.
115. Способ по п.114, где терапевтически эффективную дозу указанной композиции вводят подкожно, внутрибрюшинно, внутривенно, внутриартериально, интрамедуллярно, интратекально, трансдермально, чрескожно, интраназально, местно, внутрикишечно, интравагинально, подъязычно или ректально.
116. Способ по п.114, где терапевтически эффективную дозу указанной композиции вводят внутривенно.
117. Способ по п.114, где индивидуумом является человек, а указанным пролиферативным расстройством является рак.
118. Способ по п.117, где раковым заболеванием является злокачественная В-клеточная опухоль.
119. Способ по п.118, где злокачественной В-клеточной опухолью является лейкоз.
120. Способ по п.119, где клетки указанного лейкоза экспрессируют антиген клеточной поверхности CD22.
121. Способ по п.118, где злокачественной В-клеточной опухолью является лимфома.
122. Способ по п.121, где клетки указанной лимфомы экспрессируют антиген клеточной поверхности CD22.
123. Способ по п.118, где В-клеточной злокачественной опухолью является не-ходжкинская лимфома.
124. Способ по п.117, где раковым заболеванием является карцинома.
125. Способ по п.117, где раковым заболеванием является саркома.
126. Способ по п.114, где калихеамицином в производном калихеамицина в конъюгате «производное мономерного калихеамицина/анти-CD22 антитело» является гамма-калихеамицин или N-ацетилкалихеамицин.
127. Способ по п.114, включающий введение терапевтически эффективной дозы композиции, содержащей «коньюгат производное мономерного калихеамицина/анти-CD22 антитело» по п.24, с одним или несколькими биологически активными агентами.
128. Способ по п.127, где один или несколько биологически активных агентов выбраны из группы, состоящей из антител, факторов роста, гормонов, цитокинов, антигормонов, ксантинов, интерлейкинов, интерферонов и цитотоксических лекарственных средств.
129. Способ по п.128, где биологически активным агентом является антитело.
130. Способ по п.129, где антитело направлено против антигена клеточной поверхности, экспрессируемого на В-клеточных злокачественных опухолях.
131. Способ по п.130, где антитело, направленное против антигена клеточной поверхности, экспрессируемого на В-клеточных злокачественных опухолях, выбрано из группы, состоящей из анти-CD19, анти-CD20 и анти-CD33 антител.
132. Способ по п.131, где анти-CD20 антителом является ритуксимаб.
133. Способ по п.128, где цитокины или факторы роста выбраны из группы, состоящей из интерлейкина 2 (IL-2), TNF, CSF, GM-CSF и G-CSF.
134. Способ по п.128, где гормоном является стероидный гормон, выбранный из эстрогенов, андрогенов, прогестинов и кортикостероидов.
135. Способ по п.128, где цитотоксическое лекарственное средство выбрано из группы, состоящей из доксорубицина, даунорубицина, идарубицина, акларубицина, зорубицина, митоксантрона, эпирубицина, карубицина, ногаламицина, меногарила, питарубицина, валрубицина, цитарабина, гемцитабина, трифлуридина, анцитабина, эноцитабина, азоцитидина, доксифлуридина, пентостатина, броксуридина, капецитабина, кладрибина, децитабина, флоксуридина, флударабина, гугеротина, пуромицина, тегафура, тиазофурина, адриамицина, цисплатина, карбоплатина, циклофосфамида, дакарбазина, винбластина, винкристина, митоксантрона, блеомицина, мехлоретамина, преднизона, прокарбазина, метотрексата, фторурацилов, этопозида, таксола, аналогов таксола и митомицина.
136. Способ по п.128, где терапевтически эффективную дозу композиции, содержащей конъюгат «производное мономерного калихеамицина/анти-CD22 антитело» по п.24, вводят вместе с одной или несколькими комбинациями цитотоксических агентов, используемых в качестве составной части курса лечения, где указанная комбинация цитотоксических агентов выбрана из:
A. СНОРР (циклофосфамид, доксорубицин, винкристин, преднизон и прокарбазин);
B. CHOP (циклофосфамид, доксорубицин, винкристин и преднизон);
C. СОР (циклофосфамид, винкристин и преднизон);
D. CAP-ВОР (циклофосфамид, доксорубицин, прокарбазин, блеомицин, винкристин и преднизон);
E. m-BACOD (метотрексат, блеомицин, доксорубицин, циклофосфамид, винкристин, дексаметазон и лейковорин);
F. ProMACE-МОРР (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, мехлоэтамин, винкристин, преднизон и прокарбазин);
G. ProMACE-CytaBOM (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, цитарабин, блеомицин и винкристин);
Н. МАСОР-В (метотрексат, доксорубицин, циклофосфамид, винкристин, преднизон, блеомицин и лейковорин);
I. MOPP (мехлоэтамин, винкристин, преднизон и прокарбазин);
J. ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин);
K. МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABV (адриамицин/доксорубицин, блеомицин и винбластин);
L. МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин);
М. ChlVPP (хлорамбуцил, винбластин, прокарбазин и преднизон);
N. IMVP-16 (ифосфамид, метотрексат и этопозид);
О. MIME (метил-gag, ифосфамид, метотрексат и этопозид);
P. DHAP (дексаметазон, высокая доза цитарабина и цисплатин);
Q. ESHAP (этопозид, метилпреднизолон, высокая доза цитарабина и цисплатин);
R. СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин);
S. CAMP (ломустин, митоксантрон, цитарабин и преднизон);
Т. CVP-1 (циклофосфамид, винкристин и преднизон);
U. ESHOP (этопозид, метилпреднизолон, высокая доза цитарабина, винкристин и цисплатин);
V. EPOCH (этопозид, винкристин и доксорубицин, вводимой в течение 96 часов с ударными дозами циклофосфамида и перорально вводимого преднизона);
W. ICE (ифосфамид, циклофосфамид и этопозид);
X. СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин);
Y. СНОР-В (циклофосфамид, доксорубицин, винкристин, преднизон и блеомицин); и
Z. P/DOCE (эпирубицин или доксорубицин, винкристин, циклофосфамид и преднизон).
137. Способ по п.128, где терапевтически эффективную дозу композиции, содержащей коньюгат «производное мономерного калихеамицина/анти-CD22 антитело» по п.24, вводят перед введением одной или нескольких комбинаций цитотоксических агентов, используемых в качестве составной части курса лечения, где комбинация цитотоксических агентов выбрана из:
A. СНОРР (циклофосфамид, доксорубицин, винкристин, преднизон и прокарбазин);
B. CHOP (циклофосфамид, доксорубицин, винкристин и преднизон);
C. СОР (циклофосфамид, винкристин и преднизон);
D. CAP-ВОР (циклофосфамид, доксорубицин, прокарбазин, блеомицин, винкристин и преднизон);
E. m-BACOD (метотрексат, блеомицин, доксорубицин, циклофосфамид, винкристин, дексаметазон и лейковорин);
F. ProMACE-МОРР (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, мехлоэтамин, винкристин, преднизон и прокарбазин);
G. ProMACE-CytaBOM (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, цитарабин, блеомицин и винкристин);
Н. МАСОР-В (метотрексат, доксорубицин, циклофосфамид, винкристин, преднизон, блеомицин и лейковорин);
I. MOPP (мехлоэтамин, винкристин, преднизон и прокарбазин);
J. ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин);
K. МОРР, (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABV (адриамицин/доксорубицин, блеомицин и винбластин);
L. MOPP (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин);
М. ChlVPP (хлорамбуцил, винбластин, прокарбазин и преднизон);
N. IMVP-16 (ифосфамид, метотрексат и этопозид);
О. MIME (метил-gag, ифосфамид, метотрексат и этопозид);
P. DHAP (дексаметазон, высокая доза цитарабина и цисплатин);
Q. ESHAP (этопозид, метилпреднизолон, высокая доза цитарабина и цисплатин);
R. СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин);
S. CAMP (ломустин, митоксантрон, цитарабин и преднизон);
Т. CVP-1 (циклофосфамид, винкристин и преднизон);
U. ESHOP (этопозид, метилпреднизолон, высокая доза цитарабина, винкристин и цисплатин);
V. EPOCH (этопозид, винкристин и доксорубицин, вводимой в течение 96 часов с ударными дозами циклофосфамида и перорально вводимого преднизона);
W. ICE (ифосфамид, циклофосфамид и этопозид);
X. СЕРР (В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин);
Y. СНОР-В (циклофосфамид, доксорубицин, винкристин, преднизон и блеомицин); и
Z. P/DOCE (эпирубицин или доксорубицин, винкристин, циклофосфамид и преднизон).
138. Способ по п.128, где терапевтически эффективную дозу композиции, содержащей коньюгат «производное мономерного калихеамицина/анти-CD22 антитело» по п.24, вводят после введения одной или нескольких комбинаций цитотоксических агентов, используемых в качестве составной части курса лечения, где указанная комбинация биологически активных агентов выбрана из:
A. СНОРР (циклофосфамид, доксорубицин, винкристин, преднизон и прокарбазин);
B. CHOP (циклофосфамид, доксорубицин, винкристин и преднизон);
C. СОР (циклофосфамид, винкристин и преднизон);
D. CAP-ВОР (циклофосфамид, доксорубицин, прокарбазин, блеомицин, винкристин и преднизон);
E. m-BACOD (метотрексат, блеомицин, доксорубицин, циклофосфамид, винкристин, дексаметазон и лейковорин);
F. ProMACE-МОРР (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, мехлоэтамин, винкристин, преднизон и прокарбазин);
G. ProMACE-CytaBOM (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, цитарабин, блеомицин и винкристин);
Н. МАСОР-В (метотрексат, доксорубицин, циклофосфамид, винкристин, точная доза преднизона, блеомицин и лейковорин);
I. MOPP (мехлоэтамин, винкристин, преднизон и прокарбазин);
J. ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин);
K. МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABV (адриамицин/доксорубицин, блеомицин и винбластин);
L. МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин);
М. ChlVPP (хлорамбуцил, винбластин, прокарбазин и преднизон);
N. IMVP-16 (ифосфамид, метотрексат и этопозид);
О. MIME (метил-gag, ифосфамид, метотрексат и этопозид);
P. DHAP (дексаметазон, высокая доза цитарабина и цисплатин);
Q. ESHAP (этопозид, метилпреднизолон, высокая доза цитарабина и цисплатин);
R. СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин);
S. CAMP (ломустин, митоксантрон, цитарабин и преднизон);
Т. CVP-1 (циклофосфамид, винкристин и преднизон);
U. ESHOP (этопозид, метилпреднизолон, высокая доза цитарабина, винкристин и цисплатин);
V. EPOCH (этопозид, винкристин и доксорубицин, вводимой в течение 96 часов с ударными дозами циклофосфамида и перорально вводимого преднизона);
W. ICE (ифосфамид, циклофосфамид и этопозид);
X. СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин);
Y. СНОР-В (циклофосфамид, доксорубицин, винкристин, преднизон и блеомицин); и
Z. P/DOCE (эпирубицин или доксорубицин, винкристин, циклофосфамид и преднизон).
139. Способ по п.128, где терапевтически эффективную дозу композиции, содержащей конъюгат «производное мономерного калихеамицина/анти-CD22 антитело» по п.24, вводят вместе с антителом, направленным против антигена клеточной поверхности, присутствующего на злокачественных В-клеточных опухолях, и где способ предусматривает введение, но необязательно, одной или нескольких комбинаций цитотоксических агентов, используемых в качестве составной части курса лечения, где указанная комбинация цитотоксических агентов выбрана из:
A. СНОРР (циклофосфамид, доксорубицин, винкристин, преднизон и прокарбазин);
B. CHOP (циклофосфамид, доксорубицин, винкристин и преднизон);
C. СОР (циклофосфамид, винкристин и преднизон);
D. CAP-ВОР (циклофосфамид, доксорубицин, прокарбазин, блеомицин, винкристин и преднизон);
E. m-BACOD (метотрексат, блеомицин, доксорубицин, циклофосфамид, винкристин, дексаметазон и лейковорин);
F. ProMACE-МОРР (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, мехлоэтамин, винкристин, преднизон и прокарбазин);
G. ProMACE-CytaBOM (преднизон, метотрексат, доксорубицин, циклофосфамид, этопозид, лейковорин, цитарабин, блеомицин и винкристин);
Н. МАСОР-В (метотрексат, доксорубицин, циклофосфамид, винкристин, точная доза преднизона, блеомицина и лейковорина);
I. MOPP (мехлоэтамин, винкристин, преднизон и прокарбазин);
J. ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин);
K. МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABV (адриамицин/доксорубицин, блеомицин и винбластин);
L. МОРР (мехлоэтамин, винкристин, преднизон и прокарбазин), вводимой поочередно с ABVD (адриамицин/доксорубицин, блеомицин, винбластин и дакарбазин);
М. ChlVPP (хлорамбуцил, винбластин, прокарбазин и преднизон);
N. IMVP-16 (ифосфамид, метотрексат и этопозид);
О. MIME (метил-gag, ифосфамид, метотрексат и этопозид);
P. DHAP (дексаметазон, высокая доза цитарабина и цисплатин);
Q. ESHAP (этопозид, метилпреднизолон, высокая доза цитарабина и цисплатин);
R. СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин);
S. CAMP (ломустин, митоксантрон, цитарабин и преднизон);
Т. CVP-1 (циклофосфамид, винкристин и преднизон);
U. ESHOP (этопозид, метилпреднизолон, высокая доза цитарабина, винкристин и цисплатин);
V. EPOCH (этопозид, винкристин и доксорубицин, вводимой в течение 96 часов с ударными дозами циклофосфамида и перорально вводимого преднизона);
W. ICE (ифосфамид, циклофосфамид и этопозид);
X. СЕРР(В) (циклофосфамид, этопозид, прокарбазин, преднизон и блеомицин);
Y. СНОР-В (циклофосфамид, доксорубицин, винкристин, преднизон и блеомицин); и
Z. P/DOCE (эпирубицин или доксорубицин, винкристин, циклофосфамид и преднизон).
140. Способ по п.139, где антитело, направленное к антигену клеточной поверхности, экспрессируемого на В-клеточных злокачественных опухолях, выбрано из группы, состоящей из анти-CD19, анти-CD20 и анти-CD33 антител.
141. Способ по п.140, где анти-CD20 антитело представляет собой ритуксимаб.
142. Способ по п.114, где анти-CD22 антитело конъюгата «производное мономерного калихеамицина/анти-CD22 антитело» содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 19, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 27.
143. Способ по п.114, где анти-CD22 антитело представляет собой гуманизированное антитело и содержит легкую цепь с последовательностью SEQ ID NO: 28.
144. Способ по п.114, где анти-CD22 антитело представляет собой гуманизированное антитело и содержит тяжелую цепь с последовательностью SEQ ID NO: 30.
145. Способ по п.114, где анти-CD22 антитело конъюгата «производное мономерного калихеамицина/анти-CD22 антитело» содержит вариабельный домен легкой цепи, содержащий SEQ ID NO: 28, и вариабельный домен тяжелой цепи, содержащий SEQ ID NO: 30.
146. Способ по п.114, где анти-CD22 антитело обладает специфичностью в отношении CD22 человека и содержит тяжелую цепь, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей, представленных как HI (SEQ ID NO: 1) для CDR-H1, как Н2 (SEQ ID NO: 2), или Н2′ (SEQ ID NO: 13), или Н2′′ (SEQ ID NO: 15), или Н2′′′ (SEQ ID NO: 16) для CDR-H2 или как НЗ (SEQ ID NO: 3) для CDR-H3, и содержит легкую цепь, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей, представленных как LI (SEQ ID NO: 4) для CDR-L1, как L2 (SEQ ID NO: 5) для CDR-L2 или как L3 (SEQ ID NO: 6) для CDR-L3.
147. Способ по п.114, где антитело содержит вариабельный домен тяжелой цепи, содержащий CDR, имеющую по крайней мере одну из последовательностей SEQ ID NO: 1 для CDR-H1, SEQ ID NO: 2, или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, или SEQ ID NO: 3 для CDR-H3, и легкой цепи, где вариабельный домен содержит CDR, имеющую по крайней мере одну из последовательностей SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 или SEQ ID NO: 6 для CDR-L3.
148. Способ по п.114, где анти-CD22 антитело конъюгата «производное мономерного калихеамицина/анти-CD22 антитело» содержит SEQ ID NO: 1 для CDR-H1, SEQ ID NO: 2, или SEQ ID NO: 13, или SEQ ID NO: 15, или SEQ ID NO: 16, или последовательность GINPGNNYATYRRKFQG, или последовательность GINPGNQYTTYKRNLKG для CDR-H2, SEQ ID NO: 3 для CDR-H3, SEQ ID NO: 4 для CDR-L1, SEQ ID NO: 5 для CDR-L2 и SEQ ID NO: 6 для CDR-L3.
149. Способ по п.114, где анти-CD22 антитело конъюгата «производное мономерного калихеамицина/анти-CD22 антитело» содержит GINPGNNYATYRRKFQG для CDR-H2.
150. Способ лечения агрессивных лимфом, включающий введение пациенту терапевтически эффективной композиции, содержащей конъюгат «производное мономерного калихеамицина/анти-CD22 антитело» по п.24 и одного или нескольких биологически активных агентов.
151. Способ по п.150, где указанным конъюгатом "производное мономерного калихеамицина/анти-CD22 антитело" является СМС-544.
152. Способ по п.1, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от аспарагина, в положении 55 по Кабату.
153. Способ по п.152, где аминокислотным остатком в положении 55 по Кабату является глутамин.
154. Способ по п.1, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от треонина, в положении 57 по Кабату.
155. Способ по п.154, где аминокислотным остатком в положении 57 по Кабату является аланин или валин.
156. Способ по п.1, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от лизина, в положении 60 по Кабату.
157. Способ по п.156, где аминокислотным остатком является аргинин.
158. Конъюгат по п.24, где антитело содержит последовательность CDR-Н2, содержащую аминокислотный остаток, отличный от аспарагина, в положении 55 по Кабату.
159. Конъюгат по п.158, где аминокислотным остатком в положении 55 по Кабату является глутамин.
160. Конъюгат по п.24, где антитело содержит последовательность CDR-Н2, содержащую аминокислотный остаток, отличный от треонина, в положении 57 по Кабату.
161. Конъюгат по п.150, где аминокислотным остатком в положении 57 по Кабату является аланин или валин.
162. Конъюгат по п.24, где антитело содержит последовательность CDR-Н2, содержащую аминокислотный остаток, отличный от лизина, в положении 60 по Кабату.
163. Конъюгат по п.162, где аминокислотным остатком является аргинин.
164. Способ по п.46, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от аспарагина, в положении 55 по Кабату.
165. Способ по п.164, где аминокислотным остатком в положении 55 по Кабату является глутамин.
166. Способ по п.46, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от треонина, в положении 57 по Кабату.
167. Способ по п.166, где аминокислотным остатком в положении 57 по Кабату является аланин или валин.
168. Способ по п.46, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от лизина, в положении 60 по Кабату.
169. Способ по п.168, где аминокислотным остатком в положении 60 по Кабату является аргинин.
170. Композиция по п.87, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от аспарагина, в положении 55 по Кабату.
171. Композиция по п.170, где аминокислотным остатком в положении 55 по Кабату является глутамин.
172. Композиция по п.87, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от треонина, в положении 57 по Кабату.
173. Композиция по п.172, где аминокислотным остатком в положении 57 по Кабату является аланин или валин.
174. Композиция по п.87, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от лизина, в положении 60 по Кабату.
175. Композиция по п.174, где аминокислотным остатком в положении 60 по Кабату является аргинин.
176. Способ по п.114, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от аспарагина, в положении 55 по Кабату.
177. Способ по п.176, где аминокислотным остатком в положении 55 по Кабату является глутамин.
178. Способ по п.114, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от треонина, в положении 57 по Кабату.
179. Способ по п.178, где аминокислотным остатком в положении 57 по Кабату является аланин или валин.
180. Способ по п.114, где антитело содержит последовательность CDR-H2, содержащую аминокислотный остаток, отличный от лизина, в положении 60 по Кабату.
181. Способ по п.180, где аминокислотным остатком в положении 60 по Кабату является аргинин.
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| US 5714586 A, 03.02.1998 | |||
| US 5712474 A, 27.01.1998 | |||
| US 6183744 B1, 06.02.2001 | |||
| Огнетушитель | 0 |
|
SU91A1 |
| Экономайзер | 0 |
|
SU94A1 |
| YELTON D.E | |||
| et | |||
| al., Affinity mutaration of the BR-96 anti-carcinoma antibody by codon-based mutagenesis, The Journal of Immunology, 1995, v.155, pp.1994-2004. | |||

