ОПИСАНИЕ
Перекрестная ссылка на родственную заявку
Эта заявка заявляет на приоритет предварительной заявки на патент США с серийным номером 60/726793, поданной 14 октября 2005 года, описание которой включено в настоящее описание посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Это изобретение относится к соединениям, обладающим фармакологической активностью, композициям, содержащим эти соединения, и способу лечения, применяющего соединения и композиции. Более конкретно, это изобретение относится к некоторым производным индола и их солям и сольватам. Эти соединения изменяют активность Н3-рецептора гистамина. Это изобретение также касается фармацевтических композиций, содержащих эти соединения, и способа лечения расстройств, в которых полезно блокирование Н3-рецептора гистамина.
УРОВЕНЬ ТЕХНИКИ
Гистамин представляет собой химический мессенджер, участвующий во многих сложных биологических процессах. При высвобождении гистамин взаимодействует со специфическими макромолекулярными рецепторами на клеточной поверхности или внутри клетки-мишени, вызывая изменения многих различных функций организма. Разнообразные типы клеток, включая гладкую мускулатуру, клетки крови, клетки иммунной системы, эндокринные и экзокринные клетки, а также нейроны, откликаются на гистамин посредством модуляции формирования межклеточных сигналов, в том числе фосфатидилинозита или фермента аденилатциклазы. Очевидность того, что гистамин играет роль нейромедиатора, была обнаружена во второй половине 1970-х годов (Schwartz, 1975), Life Sci., 17: 503-518. Иммуногистохимические исследования выявили гистаминергические клеточные тела в туберомамиллярном ядре заднего гипоталамуса с распространенными проекциями в промежуточный и большой мозг (Inagaki et al., 1998), J. Comp. Neurol., 273: 283-300.
Два рецептора гистамина (Н1 и Н2) были описаны как посредники биохимических действий гистамина на нейроны. Впоследствии исследования выявили существование третьего подтипа рецептора гистамина, Н3-рецептора гистамина (Schwartz et al., 1986), TIPS, 8: 24-28. Разнообразные исследования теперь продемонстрировали, что Н3-рецепторы гистамина находятся на гистаминергических нервных окончаниях в мозге некоторых биологических видов, включая человека (Arrang et al., 1983), Nature, 302: 832-837. Н3-рецептор, найденный на гистаминергическом нервном окончании, был охарактеризован как авторецептор и мог бы непосредственно контролировать количество гистамина, выделяемого из нейронов. Гистамин, представляющий собой природное соединение, был способен стимулировать этот авторецептор, но тестирование известных агонистов и антагонистов Н1- и Н2-рецепторов привело к мысли, что Н3-рецептор имеет особый фармакологический профиль. Далее, Н3-рецепторы были идентифицированы на холинергических, серотонинергических и моноаминных нервных окончаниях в периферической нервной системе (PNS) и центральной нервной системе, включая кору головного мозга и кровеносные сосуды головного мозга. Эти наблюдения предполагают, что Н3-рецепторы размещаются единственно для модулирования высвобождения гистамина, а также прочих нейромедиаторов, и соединения, которые связывают Н3-рецепторы, могли бы быть важными медиаторами нейронной активности.
Как установлено, гистаминергические клеточные тела центральной нервной системы обнаруживаются в магноцеллюлярном ядре сосковой части гипоталамуса, и эти нейроны диффузно проецируются на большие области переднего мозга. Присутствие гистаминергических клеточных тел в туберомамиллярных ядрах заднего гипоталамуса, зоны мозга, вовлеченной в поддержание бодрствования, и их проекции на кору головного мозга свидетельствуют о роли в модулировании состояния активации или цикла сна-пробуждения. Гистаминергическая проекция на многие лимбические структуры, такие как гиппокампальная формация и амигдалоидный комплекс, свидетельствуют о роли в таких функциях, как автономная регуляция, контроль эмоций и мотивированное поведение, и процессы памяти.
Концепция, что гистамин является важным для состояния активации, на что указывает расположение гистаминергических путей, подтверждается другими типами фактов. Хорошо известно, что повреждения заднего гипоталамуса могут вызывать сон. Нейрохимические и электрофизиологические исследования также показали, что активность гистаминергических нейронов максимальна во время периодов бодрствования и подавляется барбитуратами и другими снотворными средствами. Внутрижелудочковый гистамин вызывает признаки картины пробуждения на электроэнцефалограмме кроликов и усиливает спонтанную двигательную активность, груминг и исследовательское поведение у крыс, получивших как физиологический раствор, так и пентобарбитал.
Напротив, высокоселективный ингибитор гистидиндекарбоксилазы, единственного фермента, ответственного за синтез гистамина, был показан как ухудшающий пробуждение крыс. Эти данные поддерживают гипотезу, что гистамин может быть эффективным в модулировании поведенческой активации. Была продемонстрирована роль Н3-рецептора в характеристиках сна-пробуждения (Lin et al., 1990), Brain Res., 592: 325-330. Пероральное введение препарата RAMHA, агониста Н3-рецептора, вызывало существенное усиление медленной волны глубокого сна у кошек. Наоборот, тиоперамид, антагонист/обратный агонист Н3-рецептора, усиливает бодрствование в зависимости от дозы. Тиоперамид был также показан усиливающим бодрствование и сокращающим медленную волну и быстрый сон у крыс. Эти факты согласуются с результатами исследований in vivo, демонстрирующих, что тиоперамид усиливает синтез и высвобождение гистамина. В совокупности эти данные демонстрируют, что селективные антагонисты Н3-рецептора или обратные агонисты могут быть полезными в лечении состояний активации и нарушений сна.
Было показано, что содержание как серотонина, так и гистамина и ацетилхолина в мозге снижается при болезни Альцгеймера (AD). Н3-рецептор гистамина был показан как регулятор высвобождения каждого из этих нейромедиаторов. Поэтому следовало бы ожидать, что антагонист или обратный агонист Н3-рецептора усиливал бы высвобождение этих нейромедиаторов в мозге. Поскольку было продемонстрировано, что гистамин важен для возбуждения ЦНС, антагонисты или обратные агонисты Н3-рецептора могли бы усилить возбуждение ЦНС благодаря повышению уровней высвобождения нейромедиаторов и тем самым улучшить познавательную способность. Так, применение соединений, которые подавляют функционирование Н3-рецептора при болезни Альцгеймера (AD), расстройствах концентрации внимания (ADD), связанных с возрастом нарушениях памяти и прочих расстройствах познавательных способностей, было бы обоснованным.
Антагонисты или обратные агонисты Н3-рецептора могут быть полезными в лечении некоторых прочих расстройств центральной нервной системы. Было высказано предположение, что гистамин может быть вовлечен в мозговое кровообращение, энергетический обмен и секрецию гипоталамических гормонов. Например, антагонисты или обратные агонисты Н3-рецептора были продемонстрированы влияющими на прием пищи и прирост веса тела у грызунов. Недавно полученные факты показали возможность применения антагонистов или обратных агонистов Н3-рецептора в лечении эпилепсии. Работа выявила обратную корреляцию между продолжительностью хронических конвульсий и уровнями гистамина в мозге. Тиоперамид был также показан в существенном и дозозависимом сокращении продолжительности каждой конвульсивной фазы после индуцированных электрическим током конвульсий и повышении электроконвульсивного порога.
Несмотря на свою низкую плотность, сайты связывания Н3-рецептора могут быть обнаружены вне мозга. Некоторые исследования выявили присутствие Н3-гетерорецепторов в желудочно-кишечном тракте, а также на нейронах дыхательных путей. Соответственно, соединение, связывающее Н3-рецептор, может быть полезным в лечении заболеваний и состояний, таких как астма, ринит, гиперемия дыхательных путей, воспаление, гиперкинезия и гипокинезия и секреция кислоты в желудочно-кишечном тракте. Блокирование периферических или центральных Н3-рецепторов также может содействовать изменениям кровяного давления, частоты сердечных сокращений и пропускной способности сердечно-сосудистой системы и могло бы быть использовано в лечении сердечно-сосудистых заболеваний и в лечении таких болезней или состояний, как ожирение, мигрень, воспаление, морская болезнь, боль, расстройства концентрации внимания с гиперактивностью (ADHD), слабоумие, депрессия, болезнь Паркинсона, шизофрения, эпилепсия, нарколепсия, острый инфаркт миокарда и астма.
Различные производные индола представлены в патентах США № 5631381 и 6630496 В1; WO 93/25524; WO 99/43672 и WO 2004/099192.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение, в своем главном аспекте, представляет соединения общей формулы:
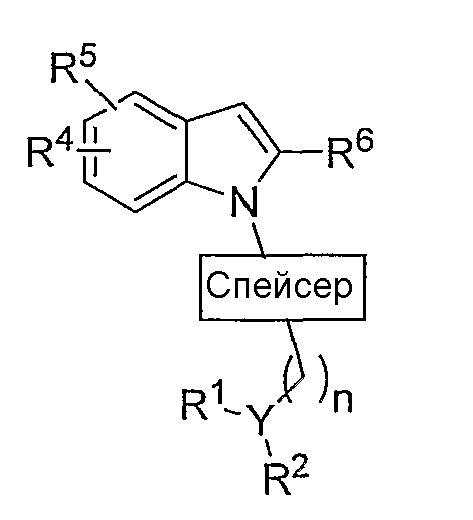
в которой
спейсер представляет собой
Y представляет собой СН или N при условии, что если Y представляет собой СН, то n равен 0-2; если Y представляет собой N, то n равен 2-4;
если Y представляет собой СН, то R1 и R2, взятые вместе, представляют собой -(CH2)a-NR11-(CH2)2-, где а равен 1-2, который вместе с Y образует пиперидиновый или пирролидиновый цикл, который, необязательно, замещен 1-3 группами, выбранными из атома фтора, фторалкильной, (С1-С4)алкильной, алкоксильной, арильной, (С3-С7)циклоалкильной, гетероциклоалкильной группы, содержащей 1-2 гетероатома, выбранных из (О, S) и (С1-С5)алкил-О-(С1-С5)алкила; и
если Y представляет собой N, то R1 и R2 независимо представляют собой (С1-С5)алкильную или (С3-С6)циклоалкильную группу, или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 5-7-членную гетероциклическую кольцевую систему с 0-1 дополнительным гетероатомом, выбранным из О и S, которая, необязательно, замещена 1-3 (С1-С5)алкильными, фторалкильными или (С3-С6)циклоалкильными группами, или R1 и R2, взятые вместе, представляют собой -(CH2)a-NR11-(CH2)2-, где а равен 2-3, который вместе с Y образует пиперазиновый или гомопиперазиновый цикл, который, необязательно, замещен 1-3 группами, выбранными из атома фтора, фторалкильной, (С1-С4)алкильной, алкоксильной, арильной, (С3-С7)циклоалкильной, гетероциклоалкильной группы, содержащей 1-2 гетероатома, выбранных из (О, S) и (С1-С5)алкил-О-(С1-С5)алкила;
R3 представляет собой 0-2 из групп, выбранных из атома галогена, (С1-С6)алкильной, (С1-С6)алкоксильной, (С3-С7)циклоалкильной, (С3-С7)циклоалкил-(С1-С6)алкильной, гетероциклоалкильной групп, содержащих 1-3 гетероатома, выбранных из (О, S) и (С1-С3)алкил-О-(С1-С5)алкила;
R4 и R5 независимо выбраны из Н, (С1-С5)алкильной, (С1-С8)алкоксильной, (С1-С5)алкил-О-(С1-С5)алкильной, (С3-С6)циклоалкильной, арильной, СF3-группы и атома галогена;
R6 представляет собой CONR7R8, -(CH2)x-O-R9, алкильную, фторалкильную группу или SO2NR7R8;
х равен 1-4;
R7 и R8 независимо представляют собой водород, (С1-С5)алкильную или (С3-С6)циклоалкильную группу или R7 и R8 вместе с атомом азота, к которому они присоединены, образуют 5-7-членную гетероциклическую кольцевую систему с 0-1 дополнительными гетероатомами, выбранными из О, S и N(R10), в которой сформированное кольцо необязательно замещается 1-3 (С1-С5)алкильными или (С3-С6)циклоалкильными группами;
R9 представляет собой водород, (С1-С5)алкильную, (С3-С7)циклоалкильную или арильную группу;
R10 представляет собой (С1-С5)алкильную, (С1-С8)алкоксильную, (С1-С5)алкил-О-(С1-С5)алкильную, (С3-С6)циклоалкильную или арильную группу; и
R11 представляет собой (С1-С5)алкильную, фторалкильную или (С3-С6)циклоалкильную группу, и их фармацевтически приемлемые соли, и их индивидуальные стереоизомеры.
Настоящее изобретение также относится к фармацевтическим композициям, включающим фармацевтически приемлемый носитель в сочетании с эффективным количеством, по меньшей мере, одного соединения формулы (I).
Настоящее изобретение также относится к способу лечения состояний, в которых модуляция Н3-рецепторов гистамина может быть терапевтически важной, таких как воспаление, мигрень, морская болезнь, боль, болезнь Паркинсона, эпилепсия, сердечно-сосудистая болезнь (т.е. повышенное или пониженное кровяное давление, острый инфаркт миокарда), желудочно-кишечные расстройства (секреция кислоты, сократительная способность) и расстройства центральной нервной системы, включая расстройства концентрации внимания или познавательной способности (т.е. болезнь Альцгеймера, расстройство концентрации внимания, связанные с возрастом нарушения памяти, удар и т.д.), психиатрические расстройства (т.е. депрессия, шизофрения, обсессивно-компульсивные расстройства и т.д.); нарушения сна (т.е. нарколепсия, апноэ во сне, бессонница, нарушение биологического и околосуточного ритма, патологическая сонливость и инсомния), и такие расстройства, как ожирение, анорексия/булимия, терморегуляция, высвобождение гормонов), включающему введение эффективного количества соединения формулы (I) пациенту, нуждающемуся в таком лечении.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
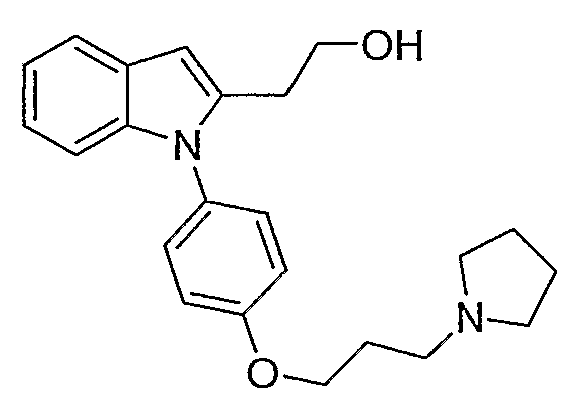
Предпочтительно для соединений формулы (I) R1-Y-R2 представляет собой 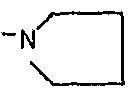 (пирролидин-1-ильный остаток); R3 представляет собой Н; R4 представляет собой Н; 5-метоксильную, 5-фтор или метильную группу; R5 представляет собой Н; и R6 представляет собой -СН2ОСН3 или -(СН2)2ОСН3.
(пирролидин-1-ильный остаток); R3 представляет собой Н; R4 представляет собой Н; 5-метоксильную, 5-фтор или метильную группу; R5 представляет собой Н; и R6 представляет собой -СН2ОСН3 или -(СН2)2ОСН3.
В настоящий момент предпочтительные соединения включают
2-метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
2-метил-1-[4-(3-пиперидин-1-илпропокси)фенил]-1Н-индол;
2-метил-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол;
1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
5-метокси-2-метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
5-метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
5-бром-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
4-хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
5-метокси-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
5-хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
2,5-диметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
6-хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
2-метил-5-фтор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
1-[3-метокси-4-(3-пирролидин-1-илпропокси)фенил]-2-метил-1Н-индол;
1-[3-хлор-4-(3-пирролидин-1-илпропокси)фенил]-2-метил-1Н-индол;
2-пропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
5-метокси-2-метил-1-[4-(4-пирролидин-1-илбут-1-инил)фенил]-1Н-индол;
(5-метокси-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)пирролидин-1-илметанон;
циклобутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
циклопентиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
циклогексиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
циклогептиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)пирролидин-1-илметанон;
2-(3-морфолин-4-илпропокси)-6,7,8,9-тетрагидропиридо[1,2-а]индол;
(1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)морфолин-4-илметанон;
бутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
изобутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
циклогексилметиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
циклогексиламид 5-метокси-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
сложный этиловый эфир 1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-карбоновой кислоты;
{1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}метанол;
2-метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
2-циклогексилоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
2-изопропоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
2-циклопентилоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
{5-метокси-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}метанол;
2-циклопропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
2-пропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол;
2-циклопропил-1-[4-(3-пирролидин-1-илпропокси)циклогексил]-1Н-индол;
2-(2-метоксиэтил)-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол и
2-{1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}этанол.
Некоторые соединения согласно изобретению могут существовать в различных изомерных (например, энантиомеры и диастереоизомеры) формах. Изобретение рассматривает все такие изомеры как в чистом виде, так и в смеси, включая рацемические смеси. Включены также енольные и таутомерные формы.
Соединения согласно изобретению могут существовать в несольватированных, а также сольватированных формах, включая гидратные формы, например полугидраты. В общем, сольватированные формы с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п., являются эквивалентными несольватированным формам в соответствии с изобретением.
Некоторые соединения согласно изобретению также образуют фармацевтически приемлемые соли, например соли присоединением кислот. Например, атомы азота могут образовывать соли с кислотами. Примерами применимых кислот для формирования солей являются соляная, серная, фосфорная, уксусная, лимонная, щавелевая, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, малеиновая, метансульфоновая и другие минеральные и карбоновые кислоты, хорошо известные специалисту в этой области. Соли готовятся путем контактирования формы свободного основания с достаточным количеством желаемой кислоты для образования соли общеупотребительным способом. Формы свободных оснований могут быть регенерированы путем обработки соли подходящим разбавленным водным раствором основания, такого как разбавленные водные растворы гидроксида, карбоната калия, аммиака и бикарбоната натрия. Формы свободных оснований до некоторой степени отличаются от своих соответствующих солевых форм некоторыми физическими свойствами, такими как растворимость в полярных растворителях, но соли кислот являются эквивалентными своим соответствующим формам свободных оснований в соответствии с изобретением (см., например, S.M. Berge, et al., “Pharmaceutical Salts”, J. Pharm. Sci., 66: 1-19 (1977), включено в описание посредством ссылки).
Используемые в описании и прилагаемых пунктах формулы изобретения термины имеют следующие значения:
Термин «алкил», как используется в описании, относится к радикалам с линейными или разветвленными цепями насыщенных углеводородов, при удалении одного атома водорода. Примерами алкильных групп являются метильная, этильная, н-пропильная, изопропильная, н-бутильная, втор-бутильная, изобутильная, трет-бутильная группы и т.п.
Термин «циклоалкил», как используется в описании, относится к алифатической циклической системе, имеющей от 3 до 10 атомов углерода и от 1 до 3 циклов, включая, но не ограничиваясь таковыми, циклопропильную, циклопентильную, циклогексильную, норборнильную и адамантильную группы, среди прочих. Циклоалкильные группы могут быть незамещенными или замещенными одним, двумя или тремя заместителями, независимо выбранными из низших алкильных, галогеналкильных, алкоксильных, тиоалкоксильных групп, аминогрупп, алкиламиногрупп, диалкиламиногрупп, гидроксильной группы, атомов галогенов, меркаптогруппы, нитрогруппы, карбоксальдегидной группы, карбоксильной группы, алкоксикарбонильных и карбоксимидных групп.
Термин «циклоалкил» включает цис- и транс-формы. Далее, заместители могут быть либо в эндо-, либо в экзоположениях в мостиковых бициклических системах.
Термин «галоген», как используется в настоящем описании, относится к атомам I, Br, Cl или F.
Термин «гетероатом», как используется в настоящем описании, относится, по меньшей мере, к одному из атомов N, О или S.
Термин «гетероциклический», как используется в настоящем описании, по отдельности или в сочетании, касается неароматического 3-10-членного цикла, содержащего, по меньшей мере, один эндоциклический атом N, О или S. Необязательно, гетероцикл может быть конденсированным с арильным кольцом. Гетероцикл также, необязательно, может быть замещен, по меньшей мере, одним заместителем, который независимо выбран из группы, состоящей из атома водорода, атомов галогенов, гидроксильной группы, аминогруппы, нитрогруппы, трифторметильной, трифторметоксильной, алкильной, арилалкильной, алкенильной, алкинильной, арильной групп, цианогруппы, карбоксильной, карбалкоксильной, карбоксиалкильной групп, оксогруппы, арилсульфонильной и арилалкиламинокарбонильной группы, среди прочих.
Как используется в описании, термин «композиция» предполагает продукт, включающий специфические ингредиенты в специфических количествах, а также любой продукт, который получается, непосредственно или косвенно, из комбинации специфических ингредиентов в специфических количествах.
Соединения согласно настоящему изобретению могут быть использованы в форме фармацевтически приемлемых солей, производных из неорганических или органических кислот. «Фармацевтически приемлемая соль» означает такие соли, которые, в объеме тщательного медицинского обследования, пригодны для применения в контакте с тканями человека и низших животных без чрезмерной токсичности, раздражения, аллергической реакции и т.п. и являются соразмерными с благоразумным соотношением польза/риск. Фармацевтически приемлемые соли хорошо известны в уровне техники. Например, S.M. Berge et al. подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 1977, 66: с.1 и след. Соли могут быть приготовлены in situ во время окончательного выделения и очистки соединений согласно изобретению, или отдельно с помощью реакции функциональной группы свободного основания с подходящей органической кислотой. Представителями солей как продуктов присоединения кислоты являются, но не ограничиваются таковыми, ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат (изотионат), лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмитат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, фосфат, глутамат, бикарбонат, пара-толуолсульфонат и ундеканоат. Также, основные азотсодержащие группы могут быть кватернизованы действием таких средств, как низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, -бромиды и -иодиды; диалкилсульфаты типа диметил-, диэтил-, дибутил- и диамилсульфатов; длинноцепочечные галогениды, такие как децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -иодиды; арилалкилгалогениды типа бензил- и фенетилбромидов и других. Таким образом получают продукты, растворимые или диспергируемые в воде или масле. Примеры кислот, которые могут быть применены для образования фармацевтически приемлемых солей как продуктов присоединения кислот, включают такие неорганические кислоты, как соляная кислота, бромистоводородная кислота, серная кислота и фосфорная кислота, и такие органические кислоты, как щавелевая кислота, малеиновая кислота, янтарная кислота и лимонная кислота.
Соли, получаемые присоединением оснований, могут быть приготовлены in situ во время окончательного выделения и очистки соединений согласно этому изобретению посредством реакции фрагмента, содержащего остаток карбоновой кислоты, с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого металлического катиона, или аммиак, или органический первичный, вторичный или третичный амин. Фармацевтически приемлемые соли включают, но не ограничиваются таковыми, катионы, производные щелочных металлов или щелочноземельных металлов, таких как соли лития, натрия, калия, кальция, магния и алюминия и т.п., и нетоксичные четвертичные аммониевые и аминные катионы, включая аммоний, тетраметиламмоний, тетраэтиламмоний, метиламмоний, диметиламмоний, триметиламмоний, триэтиламмоний, диэтиламмоний и этиламмоний, среди прочих. Другими примерами органических аминов, используемых для формирования солей как продуктов присоединения оснований, являются этилендиамин, этаноламин, диэтаноламин, пиперидин, пиперазин и т.п.
Лекарственные формы для местного применения соединения согласно этому изобретению включают порошки, спреи, мази и средства для ингаляции. Активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми требуемыми консервантами, буферами или пропеллентами, при необходимости. Офтальмические составы, глазные мази, порошки и растворы также рассматриваются в объеме настоящего изобретения.
Уровни дозировки активных ингредиентов в фармацевтических композициях согласно настоящему изобретению могут варьировать так, чтобы получить количество активного соединения, которое является эффективным для достижения желаемого терапевтического ответа для конкретного пациента, композиций и способа введения. Выбранный уровень дозировки будет зависеть от активности конкретного соединения, пути введения, серьезности состояния, которое лечат, и состояния и предшествующей истории болезни пациента, которого лечат. Однако специалисту в этой области свойственно начинать дозировку соединения с уровней, более низких, чем требуется для достижения желаемого терапевтического эффекта, и постепенно увеличивать дозировку до появления желаемого эффекта.
При использовании в вышеперечисленных или прочих видах лечения терапевтически эффективное количество одного из соединений согласно настоящему изобретению может быть использовано в чистой форме или, где такие формы существуют, в форме фармацевтически приемлемой соли, сложного эфира или пролекарства. Альтернативно, соединение может быть введено как фармацевтическая композиция, содержащая конкретное соединение в комбинации с одним или более фармацевтически приемлемыми наполнителями. Термин «терапевтически эффективное количество» соединения согласно изобретению означает количество соединения, достаточное для излечения заболеваний, при разумном соотношении польза/риск, применимом к любому медицинскому воздействию. Однако должно быть понятно, что общая суточная дозировка соединений и композиций согласно настоящему изобретению будет определяться лечащим врачом в пределах рамок тщательного медицинского обследования. Конкретный уровень терапевтически эффективной дозы для любого конкретного пациента будет зависеть от множества факторов, включающих излечиваемое расстройство и серьезность расстройства; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и режим питания пациента; время введения, путь введения и скорость выведения конкретного применяемого соединения; продолжительность лечения; лекарственные средства, употребляемые в комбинации или совпадающие с конкретным применяемым соединением; и подобных факторов, хорошо известных квалифицированному медицинскому работнику. Например, специалисту в этой области изначально свойственно начинать дозировку соединения с доз, более низких, чем требуется для достижения желаемого терапевтического эффекта, и постепенно увеличивать дозировку до появления желаемого эффекта.
Общая суточная доза соединений согласно настоящему изобретению, вводимая человеку или низшему животному, может варьировать от около 0,0001 до около 1000 мг/кг/день. Для перорального введения более предпочтительные дозы могут находиться в интервале от около 0,001 до около 5 мг/кг/день. При необходимости эффективная суточная доза может быть разделена на несколько доз для целей введения; следовательно, композиции разовой дозировки могут содержать такие количества или целые доли их, чтобы составить суточную дозу.
Настоящее изобретение также относится к фармацевтическим композициям, которые включают соединения согласно настоящему изобретению, введенные в состав вместе с одним или более нетоксичными фармацевтически приемлемыми носителями. Фармацевтические композиции могут быть специально составлены для перорального введения в твердой или жидкой форме, для парентеральной инъекции или для ректального введения.
Фармацевтические композиции согласно этому изобретению могут быть введены людям или прочим млекопитающим перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонеально, местно (в виде порошков, мазей или капель), буккально или в виде орального или назального спрея. Термин «парентерально», как используется в описании, относится к методам введения, которые включают внутривенную, внутримышечную, интраперитонеальную, внутригрудинную, подкожную и интраартикулярную инъекцию и инфузию.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, включающей компонент согласно настоящему изобретению и физиологически приемлемый разбавитель. Настоящее изобретение включает одно или более соединений, описанных выше, введенных в состав композиций вместе с одним или более нетоксичными физиологически толерантными или приемлемыми разбавителями, носителями, вспомогательными лекарственными веществами или средами, которые в совокупности называются разбавители, для парентеральной инъекции, для интраназального введения, для перорального введения в твердой или жидкой форме, для ректального или местного введения, среди прочих.
Композиции могут быть также введены через катетер для местной доставки в назначенное место, через внутрикоронарный стент (трубчатое устройство, состоящее из тонкой проволочной сетки), или с помощью биоразлагаемого полимера. Соединения также могут образовывать комплекс с лигандами, такими как антитела, для целевой доставки.
Соединения, пригодные для парентеральной инъекции, могут включать физиологически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для разбавления в стерильные пригодные для инъекции растворы или дисперсии. Примеры пригодных водных и неводных носителей, разбавителей, растворителей или сред включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), растительные масла (такие как оливковое масло), пригодные к инъекции органические сложные эфиры, такие как этилолеат, и их применимые смеси.
Эти композиции также могут содержать вспомогательные лекарственные вещества, такие как консерванты, смачивающие средства, эмульгаторы и диспергаторы. Защита от микроорганизмов может быть обеспечена с помощью разнообразных бактерицидных средств и противогрибковых препаратов, например парабенов, хлорбутанола, фенола, сорбиновой кислоты и т.п. Может быть также желательным включить изотонические агенты, например сахара, хлорид натрия и т.п. Пролонгированная абсорбция пригодной к инъекции фармацевтической формы может быть реализована применением агентов, замедляющих абсорбцию, например моностеарата алюминия или желатина.
Суспензии, в дополнение к активным соединениям, могут содержать суспендирующие средства, например, такие как этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, или их смеси, или подобные.
В некоторых случаях, чтобы пролонгировать действие лекарственного средства, желательно замедлить абсорбцию лекарственного средства при подкожной или внутримышечной инъекции. Это может быть выполнено с помощью жидкой суспензии кристаллического или аморфного материала с низкой растворимостью в воде. Скорость абсорбции лекарственного средства, кроме того, зависит от скорости его растворения, которая, в свою очередь, может зависеть от величины кристаллов или кристаллической формы. Альтернативно, замедленная абсорбция введенной парентерально лекарственной формы достигается растворением или суспендированием лекарственного средства в масляном наполнителе.
Пригодные для инъекции депо-формы изготавливаются формированием микроинкапсулированных матриц лекарственного средства в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от отношения лекарственного средства к полимеру и природы конкретного применяемого полимера скорость высвобождения лекарственного средства может контролироваться. Примеры прочих биоразлагаемых полимеров включают полиортоэфиры и полиангидриды. Пригодные для инъекции депо-составы также готовят включением лекарственного средства в липосомы или микроэмульсии, которые являются совместимыми с тканями организма.
Составы для инъекций могут быть стерилизованы, например, фильтрованием через задерживающий бактерии фильтр, или введением стерилизующих средств в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или иной стерильной инъецируемой среде непосредственно перед употреблением.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение может быть смешано, по меньшей мере, с одним инертным, фармацевтически приемлемым наполнителем или носителем, таким как цитрат натрия или дикальцийфосфат, и/или а) наполнителями или добавками, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; b) связующими средствами, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик; с) увлажнителями, такими как глицерин; d) диспергаторами, такими как агар-агар, карбонат кальция, крахмал из картофеля или маниоки, альгиновая кислота, некоторые силикаты и карбонат натрия; е) замедлителями растворения, такими как парафин; f) ускорителями абсорбции, такими как четвертичные аммониевые соединения; g) смачивателями, такими как цетиловый спирт и моностеарат глицерина; h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими средствами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае применения капсул, таблеток и пилюль лекарственные формы также могут включать буферные средства.
Твердые композиции подобного типа могут быть также применены в качестве наполнителей в желатиновых капсулах мягкого или твердого наполнения с использованием таких формообразующих средств, как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой, и т.п.
Твердые лекарственные формы в виде таблеток, драже, капсул, пилюль и гранул могут быть приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и прочие покрытия, хорошо известные в области приготовления фармацевтических составов. Необязательно, они могут содержать контрастные вещества и также могут состоять из композиции так, что они высвобождают только активный(е) ингредиент(ы), или предпочтительно таковые, в некоторых частях кишечного тракта, необязательно, в замедленном режиме. Примеры покровных композиций, которые могут быть использованы, включают полимерные субстанции и воски.
Активные соединения также могут быть в микроинкапсулированной форме, если это приемлемо, с одним или более вышеназванными наполнителями.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям, жидкие лекарственные формы могут содержать инертные разбавители, обычно употребляемые в уровне техники, такие как, например, вода или прочие растворители, солюбилизаторы и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в особенности хлопковое, арахисовое, кукурузное, масло проростков, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, и их смеси.
Наряду с инертными разбавителями, композиции для перорального введения также могут включать вспомогательные вещества, такие как смачиватели, эмульгаторы и суспендирующие средства, подсластители, вкусо-ароматические добавки и отдушки.
Композиции для ректального или вагинального введения представляют собой предпочтительно суппозитории, которые могут быть приготовлены смешиванием соединений согласно настоящему изобретению с пригодными нераздражающими наполнителями или носителями, такими как масло какао, полиэтиленгликоль или суппозиторный воск, которые являются твердыми при комнатной температуре, но разжижаются при температуре тела и поэтому расплавляются в прямой кишке или вагинальной полости и высвобождают активное соединение.
Соединения согласно настоящему изобретению также могут быть введены в форме липосом. Как известно в уровне техники, липосомы в общем являются производными фосфолипидов или прочих липидных субстанций. Липосомы формируются моно- или мультиламеллярными гидратированными жидкими кристаллами, которые диспергированы в водной среде. Могут быть использованы любые нетоксичные, физиологически приемлемые и разлагаемые в организме липиды, способные к формированию липосом. Настоящие композиции в липосомной форме могут содержать, в дополнение к соединению согласно настоящему изобретению, стабилизаторы, консерванты, наполнители и т.п. Предпочтительными липидами являются натуральные и синтетические фосфолипиды и фосфатидилхолины (лецитины), применяемые по отдельности или в совокупности.
Методы формирования липосом известны в уровне техники. См., например, Prescott, Ed., Methods in Cell Biology, vol. XIV, Academic Press, New York, N.Y. (1976), с.33 и след.
Термин «фармацевтически приемлемое пролекарство», как используется в описании, представляет такие пролекарства из соединений согласно настоящему изобретению, которые, в пределах тщательного медицинского обследования, пригодны для применения в контакте с тканями людей и низших животных без чрезмерной токсичности, раздражения, аллергической реакции и т.п., соответствующие разумному отношению польза/риск и эффективные для применения в своем предназначении, а также цвиттерионные формы, где это возможно, соединений согласно изобретению. Пролекарства согласно настоящему изобретению могут быть быстро преобразованы in vivo в родительское соединение вышеприведенной формулы, например, гидролизом в крови. Подробное обсуждение представлено в T. Higuchi and V. Stella Prodrugs as Novel Delivery Systems, vol.14, A.S.C. Symposium Series (Доклады симпозиумов Американского Химического Общества) и Edward B., ed., Bioreversible Carriers in Drug Design, Roche, American Pharmaceutical Association and Pergamon Press (1987), включенные в настоящее описание посредством ссылки.
Соединения согласно настоящему изобретению, которые образуются путем преобразования in vivo иного соединения, которое было введено млекопитающему, предполагаются быть включенными в объем настоящего изобретения.
Соединения согласно настоящему изобретению могут существовать как стереоизомеры, в которых присутствуют асимметричные или хиральные центры. Такие стереоизомеры обозначают как “R” или “S” в зависимости от конфигурации заместителей при хиральном атоме углерода. Настоящее изобретение рассматривает разнообразные стереоизомеры и их смеси. Стереоизомеры включают энантиомеры и диастереомеры и смеси энантиомеров или диастереомеров. Индивидуальные стереоизомеры соединений согласно настоящему изобретению могут быть приготовлены синтетическим путем из имеющихся в продаже исходных материалов, которые содержат асимметричные или хиральные центры, или путем получения рацемических смесей с последующим разделением, хорошо известным квалифицированному специалисту в этой области методом. Эти методы разделения иллюстрируются (1) присоединением смеси энантиомеров к хиральному вспомогательному веществу, разделением полученной смеси диастереомеров с помощью перекристаллизации или хроматографии и высвобождением оптически чистого продукта из вспомогательного вещества, или (2) прямым разделением смеси оптически активных энантиомеров на хроматографических колонках с хиральным сорбентом.
Соединения согласно изобретению могут существовать в несольватированных, а также сольватированных формах, включая гидратные формы, такие как полугидраты. В общем, сольватированные формы с фармацевтически приемлемыми растворителями, такими как вода и этанол, среди прочих, являются эквивалентными несольватированным формам в соответствии с изобретением.
Изобретение может быть иллюстрировано нижеследующими схемой и примерами.
Схема 1
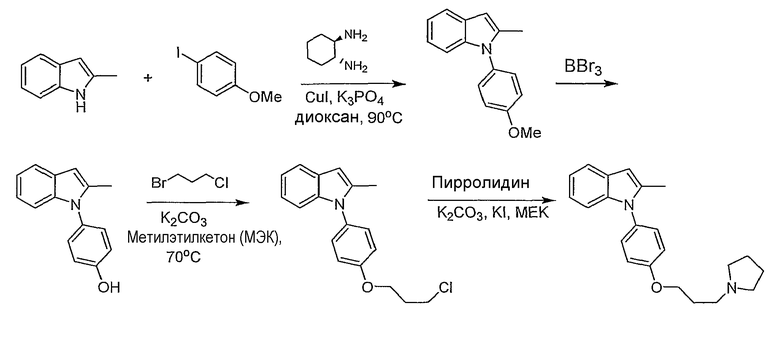
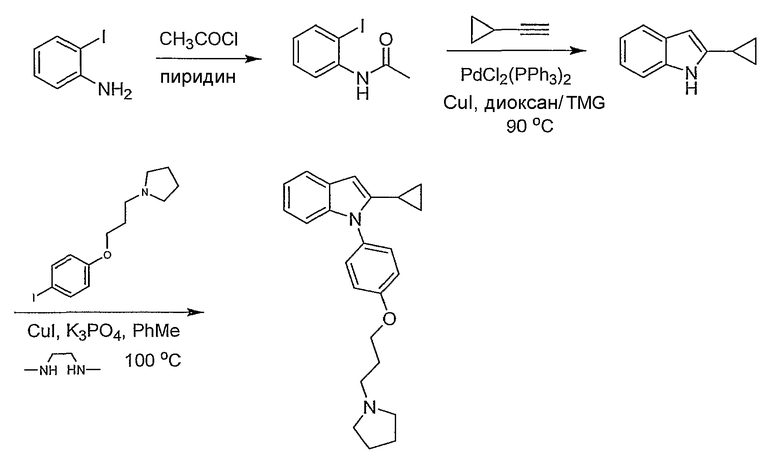
Пример 1
2-Метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
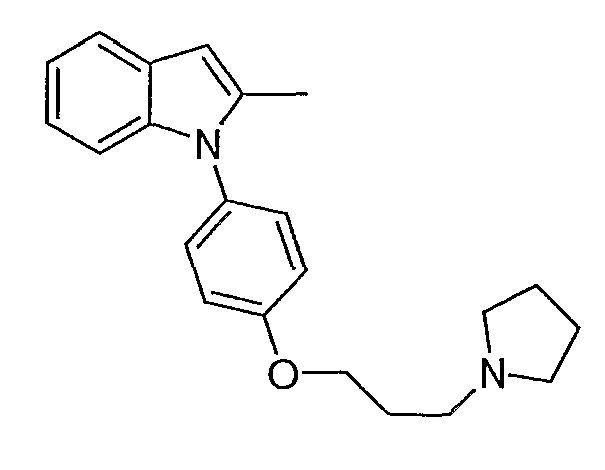
1-(4-Метоксифенил)-2-метил-1Н-индол. 1-(4-Метоксифенил)-2-метил-1Н-индол синтезировали согласно методике Buchwald et al., J. Am. Chem. Soc., 2001, 123, 7727. Смесь 2-метилиндола (157 мг, 1,2 ммол), 4-йоданизола (234 мг, 1 ммол), иодида меди(I) (2 мг, 0,01 ммол), транс-1,2-диаминоциклогексана (11,4 мг, 0,1 ммол) и трехосновного фосфата калия (446 мг, 2,1 ммол) перемешивали в диоксане (1 мл) при 90°С в течение ночи. Реакционную смесь фильтровали через слой диоксида кремния, промывали этилацетатом. Хроматография на SiO2 со смесью 5-20% этилацетата/гексана дала 175 мг желаемого продукта (выход 74%). LC-MS (С16Н15NO, рассчитано 237), m/z 238 (M+H).
4-(2-Метилиндол-1-ил)фенол. 1-(4-Метоксифенил)-2-метил-1Н-индол (175 мг, 0,73 ммол) растворяли в дихлорметане (2 мл) и охлаждали до 0°С. Трибромид бора (2,19 мл, 1М раствор в дихлорметане, 2,19 ммол) добавляли по каплям, и реакционную смесь перемешивали в течение 2 часов. Реакционную смесь гасили добавлением насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном, потом этилацетатом. Органические экстракты высушивали над MgSO4 и концентрировали. Выход реакции был принят количественным. LC-MS (C15H13NO, рассчитано 223), m/z 224 (М+Н).
1-[4-(3-Хлорпропокси)фенил]-2-метил-1Н-индол. 4-(2-Метилиндол-1-ил)фенол (0,36 ммол) нагревали при 70°С в 2-бутаноне (3 мл) с 1-бром-3-хлорпропаном (0,107 мл, 1,08 ммол) и карбонатом калия (0,15 г, 1,08 ммол) в течение ночи. Растворитель выпаривали. Полученный остаток разбавляли этилацетатом и промывали насыщенным раствором хлорида аммония. Водный слой снова экстрагировали этилацетатом. Органические экстракты сушили над MgSO4 и концентрировали. Хроматография на SiO2 со смесью 5-20% этилацетата/гексанов дала желаемый продукт в виде бесцветного масла, 60 мг (выход 56% на две стадии). LC-MS (С18Н18ClNO, рассчитано 299), m/z 300, 302 (M+H).
2-Метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол. 1-[4-(3-Хлорпропокси)фенил]-2-метил-1Н-индол (20 мг, 0,067 ммол) растворяли в N-метилпирролидиноне (0,5 мл) и добавляли пирролидин (0,017 мл, 0,2 ммол), карбонат калия (46 мг, 0,34 ммол) и каталитическое количество иодида калия. Реакционную смесь нагревали в течение ночи при 70°С. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические экстракты высушивали над MgSO4 и концентрировали. Остаток очищали с помощью полупрепаративной LC-MS с получением 9,6 мг желаемого продукта (выход 43%). LC-MS (C22Н26N2O, рассчитано 334), m/z 335 (М+Н); 1Н ЯМР (300 МГц, CDCl3) δ 7,58-7,53 (м, 1Н), 7,26-7,25 (м, 3Н), 7,12-6,99 (м, 4Н), 6,37 (с, 1Н), 4,10 (т, J=6 Гц, 2Н), 2,99-2,90 (м, 6Н), 2,24 (с, 3Н), 2,22-2,14 (м, 2Н), 2,00-1,90 (м, 6Н).
Пример 2
2-Метил-1-[4-(3-пиперидин-1-илпропокси)фенил]-1Н-индол
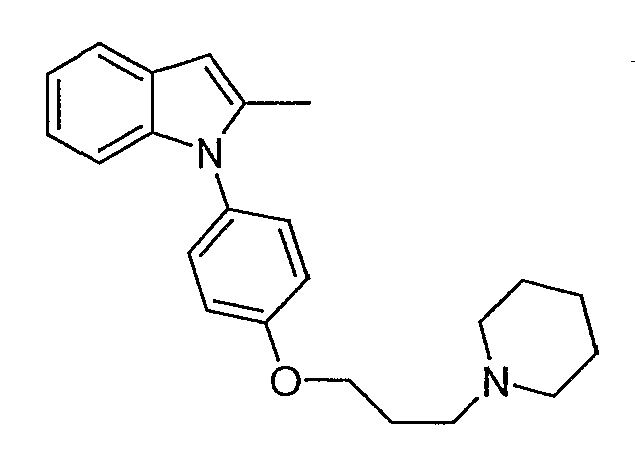
2-Метил-1-[4-(3-пиперидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 1, с использованием пиперидина вместо пирролидина на конечной стадии. LC-MS (C23H28N20, рассчитано 348), m/z 349 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,58-7,52 (м, 1H), 7,23-7,21 (м, 3H), 7,12-6,99 (м, 4H), 6,37 (с, 1H), 4,09 (т, J=6 Гц, 2H), 2,79-2,68 (м, 6H), 2,27 (c, 3H), 2,20-2,09 (м, 2H), 1,77-1,69 (м, 4H), 1,56-1,52 (м, 2H).
Пример 3
2-Метил-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол
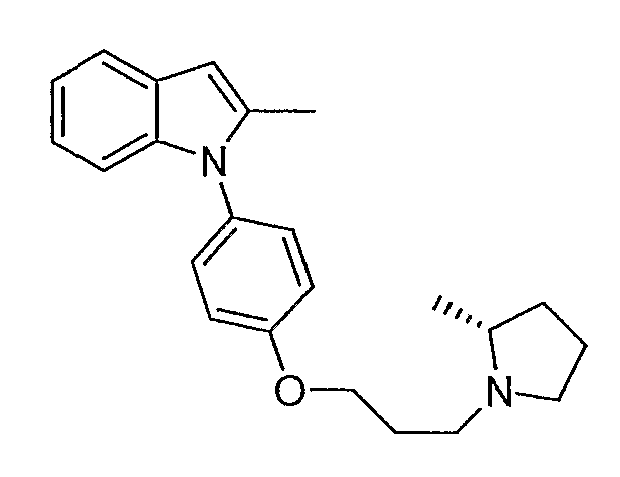
2-Метил-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол синтезировали способом, аналогичным использованному в примере 1, с использованием (R)-2-метилпирролидина вместо пирролидина на конечной стадии. LC-MS (C23H28N2O, рассчитано 348), m/z 349 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,58-7,52 (м, 1H), 7,26-7,25 (м, 3H), 7,12-7,00 (м, 4H), 6,37 (с, 1H), 4,16-4,05 (м, 2H), 3,46-3,39 (м, 1H), 3,22-3,13 (м, 1H), 2,75-2,63 (м, 1H), 2,57-2,42 (м, 2H), 2,27 (c, 3H), 2,21-1,55 (м, 6H), 1,25 (д, J=6,3 Гц, 3H).
Схема 2
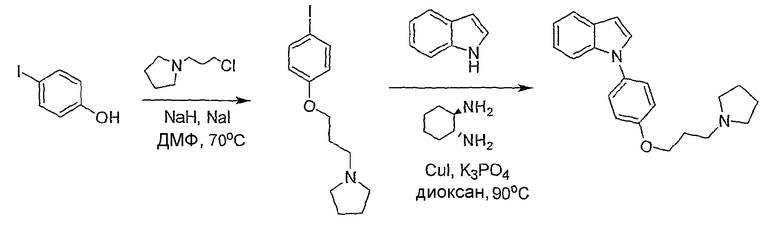
Пример 4
1-[4-(3-Пирролидин-1-илпропокси)фенил]-1Н-индол
1-[3-(4-Йодфенокси)пропил]пирролидин. 4-йодфенол (2,2 г, 10 ммол) растворяли в N,N-диметилформамиде (30 мл) в токе азота и порциями добавляли гидрид натрия (0,48 г, 60%-я дисперсия в минеральном масле, 12 ммол). Добавляли 1-(3-хлорпропил)пирролидин (1,77 г, 12 ммол) и иодид натрия (1,8 г, 12 ммол) и реакционную смесь перемешивали в течение ночи при 70°С. Реакционную смесь разбавляли этилацетатом и промывали водой. Этилацетатный раствор промывали 1н. HCl (дважды). Кислые экстракты подщелачивали 2н. раствором NaOH, затем экстрагировали этилацетатом (дважды). Все этилацетатные экстракты объединяли, высушивали над MgSO4 и концентрировали с получением желтого масла, 2,98 г (выход неочищенного продукта 90%). LC-MS (C13H18INO, рассчитано 331), m/z 332 (М+Н).
1-[4-(3-Пирролидин-1-илпропокси)фенил]-1Н-индол. Смесь 1-[3-(4-йодфенокси)пропил]пирролидина (66 мг, 0,2 ммол), индола (28 мг, 0,24 ммол), иодида меди(I) (0,4 мг, 0,002 ммол), транс-1,2-диаминоциклогексана (0,0024 ммол, 0,02 ммол) и трехосновного фосфата калия (89 мг, 0,42 ммол) перемешивали при 90°С в диоксане в течение ночи. Реакционную смесь фильтровали через целит и промывали дихлорметаном. Фильтрат концентрировали и полученный остаток очищали с помощью полупрепаративной LC-MS с получением 22,4 мг желаемого продукта (выход 35%). LC-MS (C21H24N2O, рассчитано 320), m/z 321 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,67 (д, J=6,9 Гц, 1H), 7,45 (д, J=8,4 Гц, 1H), 7,40-7,35 (м, 2H), 7,27 (д, J=3 Гц, 1H), 7,22-7,10 (м, 2H), 7,04-6,99 (м, 2H), 6,64 (д, J=3 Гц, 1H), 4,08 (т, J=6,3 Гц, 2H), 2,66 (т, J=12 Гц, 2H), 2,57-2,52 (м, 4H), 2,09-2,00 (м, 2H), 1,83-1,78 (м, 4H).
Пример 5
5-Метокси-2-метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
5-Метокси-2-метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 5-метокси-2-метилиндола вместо индола на конечной стадии. LC-MS (C23H28N2O2, рассчитано 364), m/z 365 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,23-7,20 (м, 2H), 7,03 (д, J=2,4 Гц, 1H), 7,02-6,97 (м, 2H), 6,93 (д, J=8,7 Гц, 1H), 6,72 (дд, J=9 Гц, 2,4 Гц, 1H), 6,30 (с, 1H), 4,10 (т, J=6 Гц, 2H), 3,85 (с, 3H), 3,03-2,96 (м, 4H), 2,25 (с, 3H), 2,24-2,16 (м, 2H), 1,99-1,94 (м, 4H).
Пример 6
5-Метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
5-Метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 5-метилиндола вместо индола на конечной стадии. LC-MS (C22H26N2O, рассчитано 334), m/z 335 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,46 (с, 1H), 7,40-7,33 (м, 2H), 7,23 (д, J=3,3 Гц, 1H), 7,23 (д, J=3,3 Гц, 1H), 7,04-6,98 (м, 2H), 6,56 (д, J=3,3 Гц, 1H), 4,08 (т, J=6,3 Гц, 2H), 2,80-2,70 (м, 6H), 2,17-2,06 (м, 2H), 1,88-1,84 (м, 4H).
Пример 7
5-Бром-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
5-Бром-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 5-броминдола вместо индола на конечной стадии. LC-MS (C21H23BrN2O, рассчитано 399), m/z 400, 402 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,79 (с, 1H), 7,37 (д, J=4,8 Гц, 2H), 7,29-7,26 (м, 3H), 7,01 (д, J=8,7 Гц, 2H), 6,58 (д, J=3,3 Гц, 1H), 4,09 (т, J=6,3 Гц, 2H), 2,92-2,82 (м, 6H), 2,20-2,11 (м, 2H), 1,94-1,89 (м, 4H).
Пример 8
4-Хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
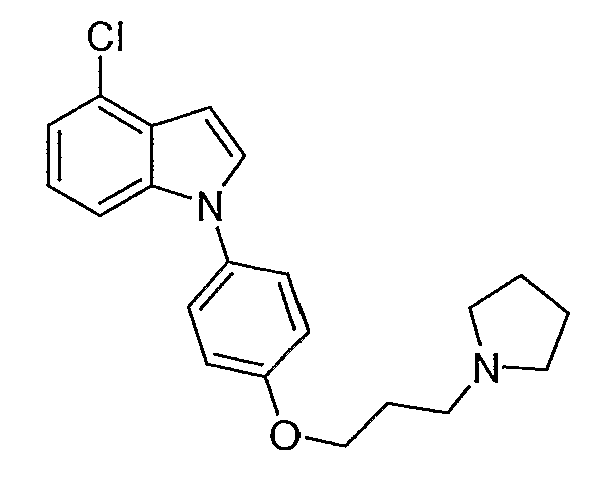
4-Хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 4-хлориндола вместо индола на конечной стадии. LC-MS (C21H23ClN2O, рассчитано 354), m/z 355, 357 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,39-7,30 (м, 4H), 7,17-7,08 (м, 2H), 7,04-6,99 (м, 2H), 6,75 (д, J=3,3 Гц, 1H), 4,09 (т, J=6 Гц, 2H), 2,97-2,89 (м, 6H), 2,22-2,13 (м, 2H), 1,96-1,92 (м, 4H).
Пример 9
5-Метокси-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
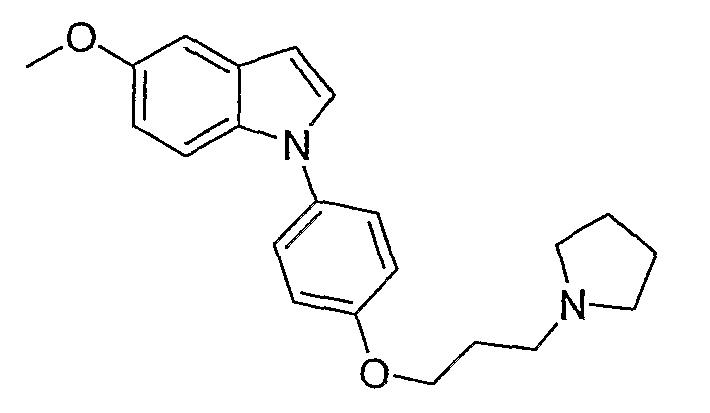
5-Метокси-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 5-метоксииндола вместо индола на конечной стадии. LC-MS (C22H26N2O2, рассчитано 350), m/z 351 (М+Н).
Пример 10
5-Хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
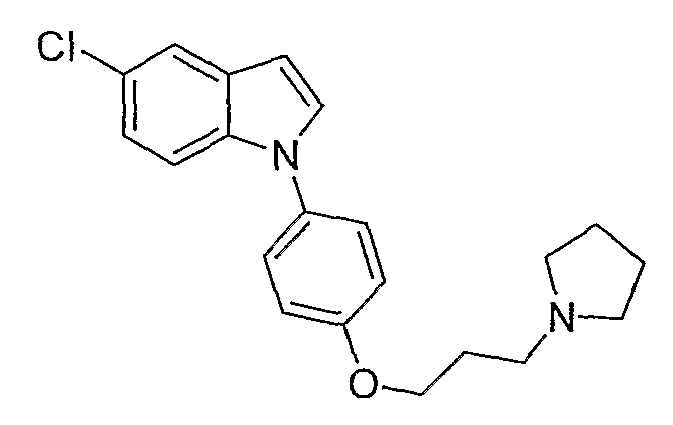
5-Хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 5-хлориндола вместо индола на конечной стадии. LC-MS (C21H23ClN2O, рассчитано 354), m/z 355, 357 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,63 (д, J=2,1 Гц, 1H), 7,37-7,32 (м, 3H), 7,28 (д, J=3 Гц, 1H), 7,14 (дд, J=8,7 Гц, 1,8 Гц, 1H), 7,04-6,99 (м, 2H), 6,58 (д, J=3 Гц, 1H), 4,09 (т, J=6,3 Гц, 2H), 2,83-2,73 (м, 6H), 2,16-2,07 (м, 2H), 1,90-1,85 (м, 4H).
Пример 11
2,5-Диметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
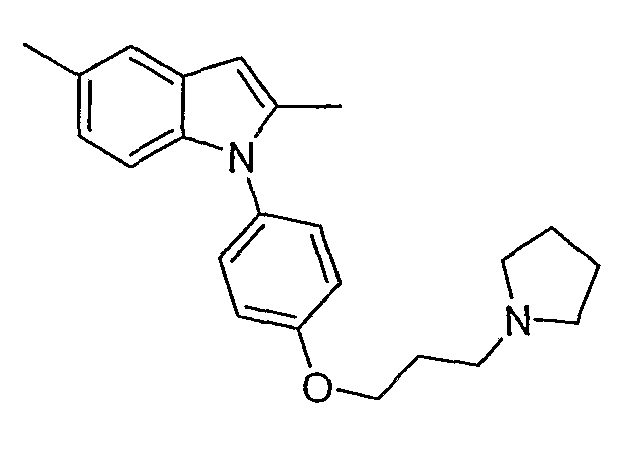
2,5-Диметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 2,5-диметилиндола вместо индола на конечной стадии. LC-MS (C23H28N2O, рассчитано 348), m/z 349 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,40 (с, 1H), 7,23-7,19 (м, 2H), 7,04-6,99 (м, 2H), 6,95-6,90 (м, 2H), 6,28 (с, 1H), 4,10 (т, J=6,3 Гц, 2H), 2,68 (т, J=7,2 Гц, 2H), 2,57 (м, 4H), 2,43 (с, 3H), 2,25 (с, 3H), 2,12-1,99 (м, 2H), 1,84-1,80 (м, 4H).
Пример 12
6-Хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
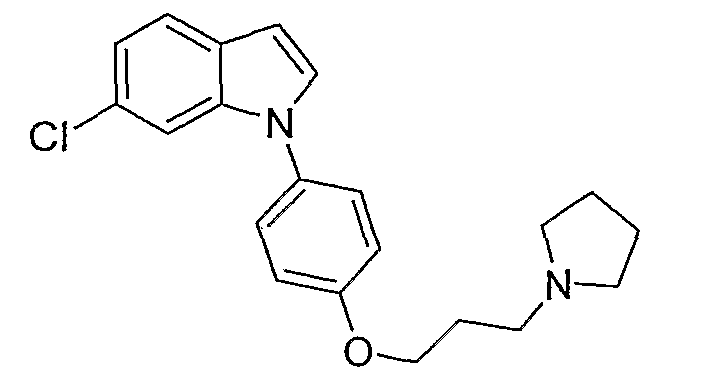
6-Хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 6-хлориндола вместо индола на конечной стадии. LC-MS (C21H23ClN2O, рассчитано 354), m/z 355, 357 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,57 (д, J=8,4 Гц, 1H), 7,38 (с, 1H), 7,37-7,34 (м, 2H), 7,26-7,25 (м, 1H), 7,11 (дд, J=8,4 Гц, 1,8 Гц, 1H), 7,06-7,01 (м, 2H), 6,62 (д, J=3,3 Гц, 1H), 4,10 (т, J=6,3 Гц, 2H), 2,69 (т, J=7,5 Гц, 2H), 2,59 (м, 4H), 2,12-2,03 (м, 2H), 1,88-1,78 (м, 4H).
Пример 13
2-Метил-5-фтор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
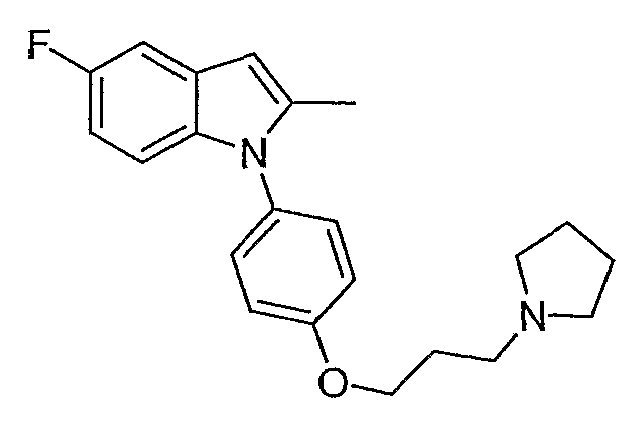
2-Метил-5-фтор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 5-фтор-2-метилиндола вместо индола на конечной стадии. LC-MS (C22H25FN2O, рассчитано 352), m/z 353 (М+Н).
Пример 14
1-[3-Метокси-4-(3-пирролидин-1-илпропокси)фенил]-2-метил-1Н-индол
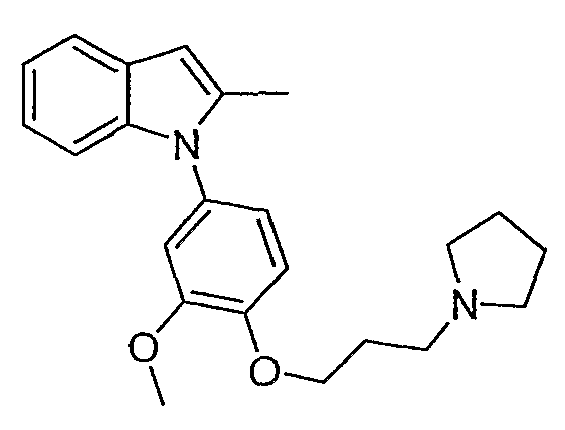
1-[3-Метокси-4-(3-пирролидин-1-илпропокси)фенил]-2-метил-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 4-бромгваякола в первой стадии вместо 4-йодфенола и 2-метилиндола на конечной стадии. LC-MS (C23H28N2O2, рассчитано 364), m/z 365 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,58-7,55 (м, 1H), 7,14-7,07 (м, 3H), 6,99 (д, J=8,7 Гц, 1H), 6,88 (дд, J=8,4 Гц, 2,4 Гц, 2H), 6,83 (д, J=2,4 Гц, 1H), 6,38 (с, 1H), 4,17 (т, J=6,3 Гц, 2H), 3,83 (с, 3H), 3,02-2,94 (м, 6H), 2,29 (с, 3H), 2,27-2,20 (м, 2H), 1,97-1,93 (м, 4H).
Пример 15
1-[3-Хлор-4-(3-пирролидин-1-илпропокси)фенил]-2-метил-1Н-индол
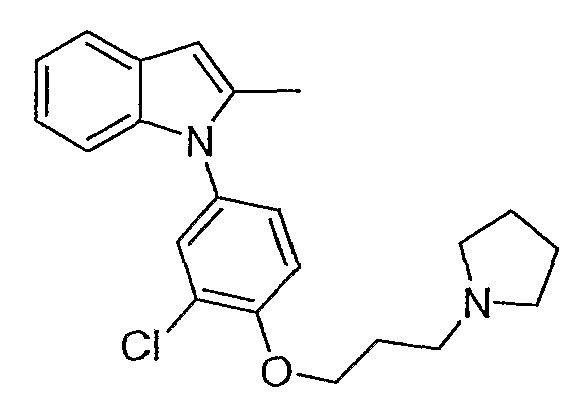
1-[3-Хлор-4-(3-пирролидин-1-илпропокси)фенил]-2-метил-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 4-бром-2-хлорфенола в первой стадии, вместо 4-йодфенола, и 2-метилиндола на конечной стадии. LC-MS (C22H25ClN2O, рассчитано 368), m/z 369, 371 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,58-7,54 (м, 1H), 7,37 (д, J=2,4 Гц, 1H), 7,20 (дд, J=8,7 Гц, 2,7 Гц, 1H), 7,11-7,02 (м, 4H), 6,38 (с, 1H), 4,19 (т, J=6,3 Гц, 2H), 2,88 (т, J=7,2 Гц, 2H), 2,77 (м, 4H), 2,24-2,15 (м, 2H), 1,91-1,87 (м, 4H).
Пример 16
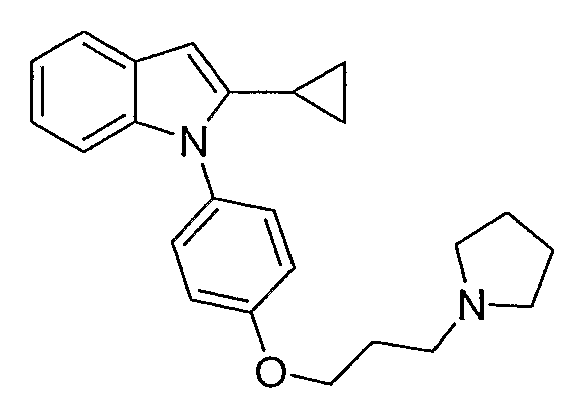
2-Пропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
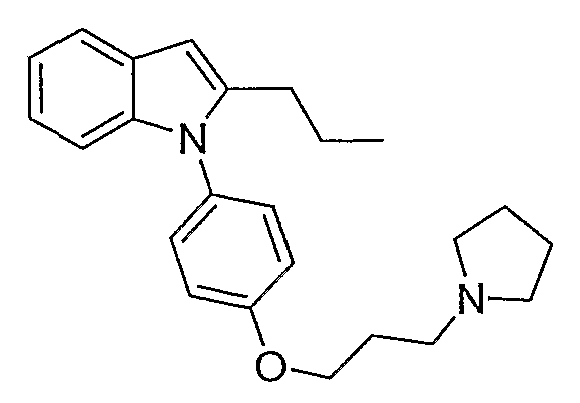
2-Пропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали способом, аналогичным использованному в примере 4, с использованием 2-пропилиндола вместо индола на конечной стадии. 2-Пропилиндол приготовили согласно Kuyper et al. (J. Med. Chem., 1996, 39, 892). LC-MS (C24H30N2O, рассчитано 362), m/z 363 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,58 (м, 1H), 7,25-7,21 (м, 2H), 7,12-6,99 (м, 5H), 6,38 (с, 1H), 4,10 (т, J=6 Гц, 2H), 3,01-2,93 (м, 6H), 2,55 (т, J=7,8 Гц, 2H), 2,25-2,15 (м, 2H), 1,98-1,91 (м, 4H), 1,66-1,54 (м, 2H), 0,91 (т, J=7,5 Гц, 3H).
Схема 3
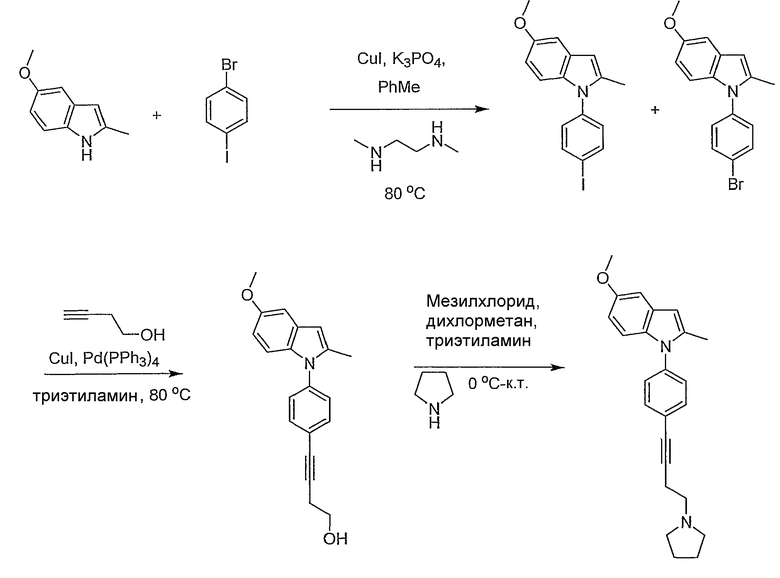
Пример 17
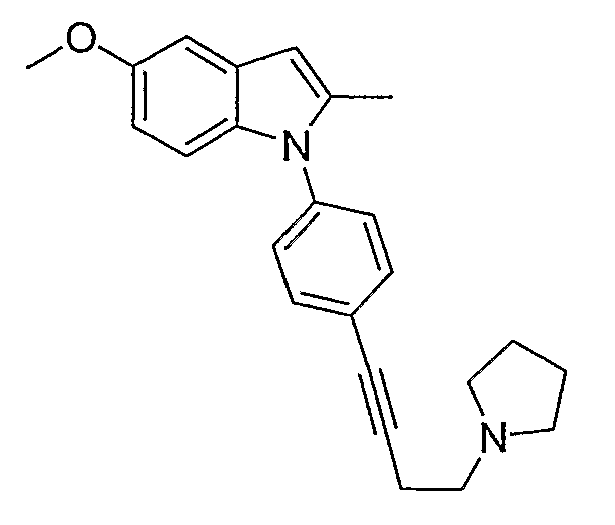
5-Метокси-2-метил-1-[4-(4-пирролидин-1-илбут-1-инил)фенил]-1Н-индол
1-(4-Бромфенил)-5-метокси-2-метил-1Н-индол и 1-(4-йодфенил)-5-метокси-2-метил-1Н-индол. 5-Метокси-2-метилиндол (500 мг, 3,1 ммол) и 1-бром-4-йодбензол (877 мг, 3,1 ммол) растворяли в толуоле (6 мл). К полученному раствору добавляли иодид меди(I) (12 мг, 0,062 ммол), трехосновный фосфат калия (1,32 г, 6,2 ммол) и N,N'-диметилэтилендиамин (6,6 мкл, 0,062 ммол). Смесь нагревали при 80°С в течение ночи, оставляли охлаждаться до комнатной температуры и фильтровали через слой диоксида кремния. Полученный раствор концентрировали с получением смеси как бромированного, так и иодированного галогенфенильных индолов, которые использовали без дальнейшей очистки (количественный выход).
4-[4-(5-Метокси-2-метилиндол-1-ил)фенил]бут-3-ин-1-ол. К раствору вышеописанной смеси индолов (50 мг) в триэтиламине (1 мл) добавляли иодид меди(I) (6 мг, 0,03 ммол) и тетракис(трифенилфосфино)палладий(0) (17 мг, 0,015 ммол). После этого добавляли 3-бутин-1-ол (15 мкл, 0,20 ммол), полученную смесь нагревали в течение ночи при 80°С. Реакционную смесь оставляли остывать и затем фильтровали через слой силикагеля. Силикагель промывали этилацетатом. Концентрирование дало желаемый спирт, который использовали без дальнейшей очистки (количественный выход).
5-Метокси-2-метил-1-[4-(4-пирролидин-1-илбут-1-инил)фенил]-1Н-индол. К раствору 4-[4-(5-метокси-2-метилиндол-1-ил)фенил]бут-3-ин-1-ола (60 мг, 0,19 ммол) в метиленхлориде (1 мл) при 0°С добавляли триэтиламин (54 мкл, 0,39 ммол) и метансульфонилхлорид (18 мкл, 0,39 ммол). После перемешивания раствора при 0°С в течение 2 часов добавляли пирролидин (163 мкл, 1,95 ммол) и реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. После того как реакционную смесь гасили водой, органический слой высушивали над MgSO4 и концентрировали. Остаток очищали с помощью ВЭЖХ с получением желаемого индола (2,8 мг). LC-MS (C24H26N2O, рассчитано 358), m/z 359 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,53 (д, J=8,4 Гц, 2H), 7,26 (д, J=8,4 Гц, 2H), 7,03 (д, J=2,4 Гц, 1H), 6,99 (д, J=9,0 Гц, 1H), 6,73 (дд, J=2,7, 9,0 Гц, 1H), 6,32 (с, 1H), 3,85 (с, 3H), 2,92-2,83 (м, 4H), 2,76 (м, 4H), 2,28 (с, 3H), 1,88 (м, 4H).
Схема 4
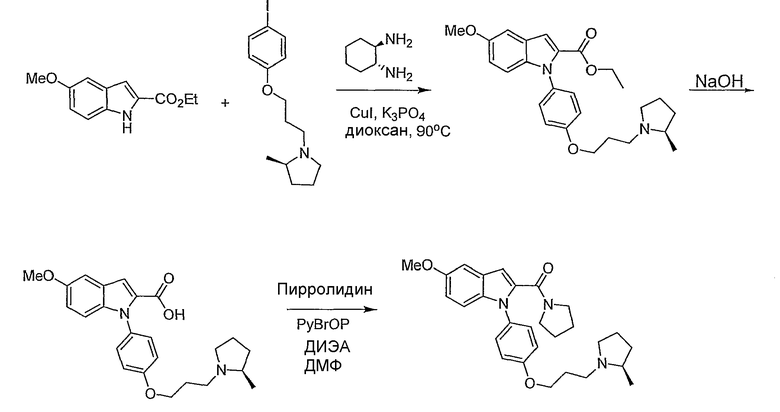
Пример 18
(5-Метокси-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)пирролидин-1-илметанон
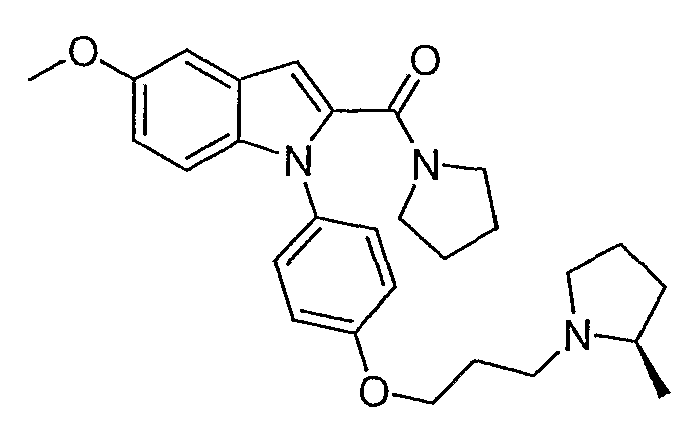
Этиловый эфир 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 4, исходя из этил 5-метоксииндол-2-карбоксилата. Этиловый эфир 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты (0,43 ммол) растворяли в тетрагидрофуране (ТГФ) (2,4 мл), метаноле (1,2 мл) и воде (0,4 мл) и добавляли гидроксид натрия (103 мг, 2,58 ммол). Реакционную смесь нагревали в течение ночи при 50°С. 1н. раствор HCl добавляли для подкисления смеси до величины рН 7 и растворители удаляли выпариванием. Часть остатка (примерно 0,2 ммол) растворяли в N,N-диметилформамиде (1 мл) и добавляли пирролидин (0,017 мл, 0,2 ммол), PyBrOP (0,14 г, 0,3 ммол) и диизопропилэтиламин (0,104 мл, 0,6 ммол). Реакционную смесь перемешивали в течение ночи и затем растворитель упаривали. Остаток очищали с помощью полупрепаративной LC-MS с получением желаемого продукта и побочного продукта, производного от PyBrOp. Затем остаток очищали хроматографически на SiO2 с использованием в качестве элюента сначала этилацетата, затем 10%-го раствора метанола в этилацетате, затем 2% триэтиламин и 10% метанол/этилацетат с получением желаемого продукта, 15,1 мг. LC-MS (C28H35N3O3, рассчитано 461), m/z 462 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,30 (д, J=9 Гц, 2H), 7,17 (д, J=9 Гц, 1H), 7,10 (д, J=2,4 Гц, 1H), 6,97 (д, J=8,7 Гц, 2H), 6,88 (дд, J=9 Гц, 2,4 Гц, 1H), 6,76 (с, 1H), 4,09-4,04 (м, 2H), 3,86 (с, 3H), 3,49 (т, J=6,6 Гц, 2H), 3,36 (т, J=6,2 Гц, 2H), 3,22 (дт, J=2,7 Гц, 8,7 Гц, 1H), 3,07-2,98 (м, 1H), 2,36-1,65 (м, 12H), 1,51-1,39 (м, 1H), 1,12 (д, J=6 Гц, 3H).
Пример 19
Циклобутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты
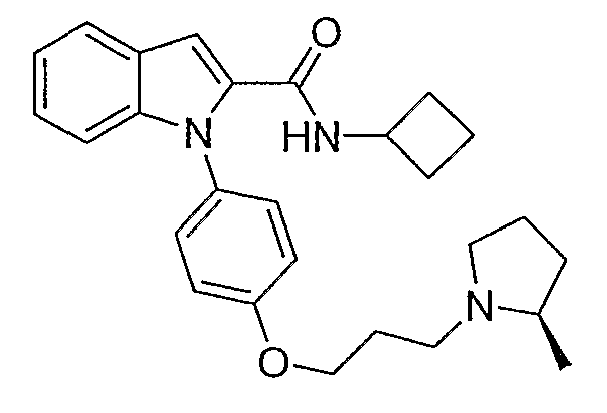
Циклобутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 18, с использованием циклобутиламина вместо пирролидина. LC-MS (C27H33N3O2, рассчитано 431), m/z 432 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,68 (д, J=7,8 Гц, 1H), 7,29-7,02 (м, 8H), 5,91 (д, J=7,8 Гц, 1H), 4,51-4,37 (м, 1H), 4,10 (т, J=6,3 Гц, 2H), 3,25-3,14 (м, 4H), 3,07-2,98 (м, 1H), 2,40-1,60 (м, 11H), 1,51-1,38 (м, 1H), 1,12 (д, J=6 Гц, 3H).
Пример 20
Циклопентиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты
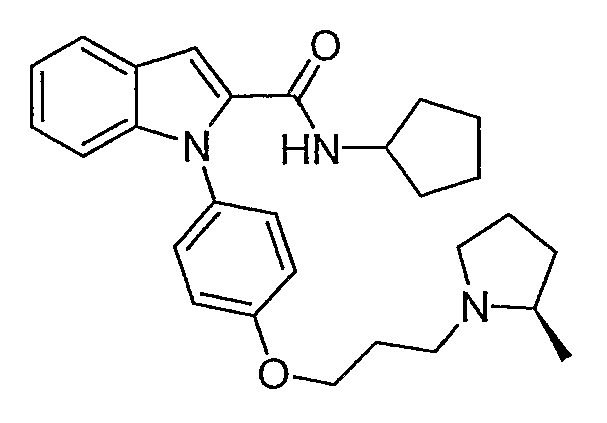
Циклопентиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 18, с использованием циклопентиламина вместо пирролидина. LC-MS (C28H35N3O2, рассчитано 445), m/z 446 (М+Н).
Пример 21
Циклогексиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты
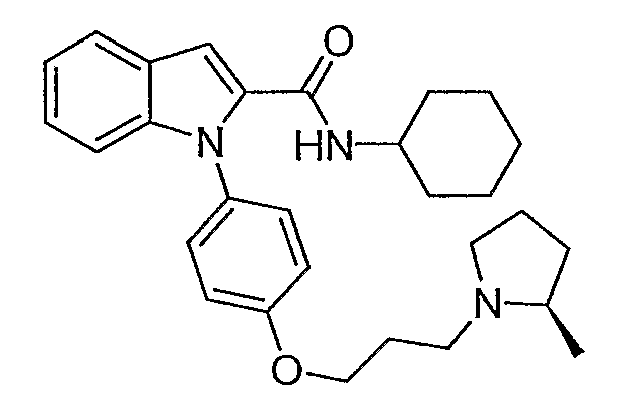
Циклогексиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 18, с использованием циклогексиламина вместо пирролидина. LC-MS (C29H37N3O2, рассчитано 459), m/z 460 (М+Н).
Пример 22
Циклогептиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты
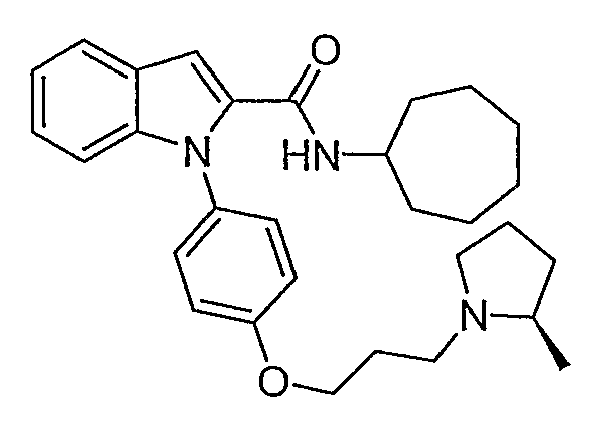
Циклогептиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 18, с использованием циклогептиламина вместо пирролидина. LC-MS (C30H39N3O2, рассчитано 473), m/z 474 (М+Н).
Пример 23
(1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)пирролидин-1-илметанон
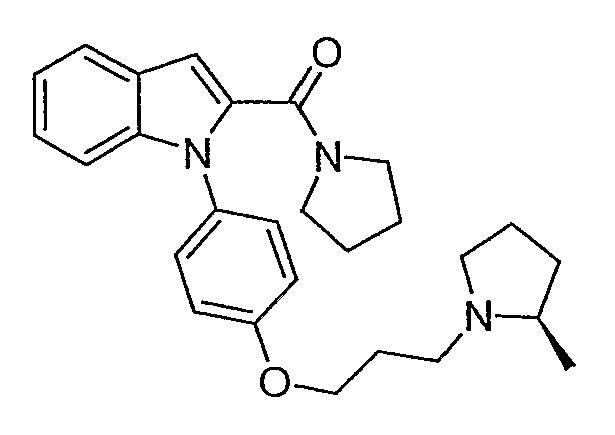
(1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)пирролидин-1-илметанон синтезировали по способу, аналогичному использованному для примера 18. LC-MS (C27H33N3O2, рассчитано 431), m/z 432 (М+Н).
Пример 24
2-(3-Морфолин-4-илпропокси)-6,7,8,9-тетрагидропиридо[1,2-а]индол
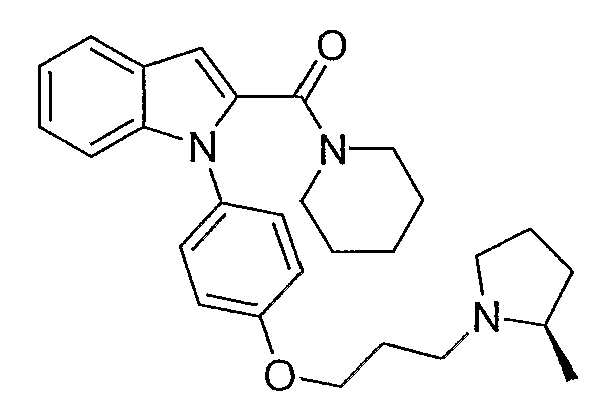
(1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)пиперидин-1-илметанон синтезировали по способу, аналогичному использованному для примера 18, с использованием пиперидина вместо пирролидина. LC-MS (C28H35N3O2, рассчитано 445), m/z 446 (М+Н).
Пример 25
(1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)морфолин-4-илметанон
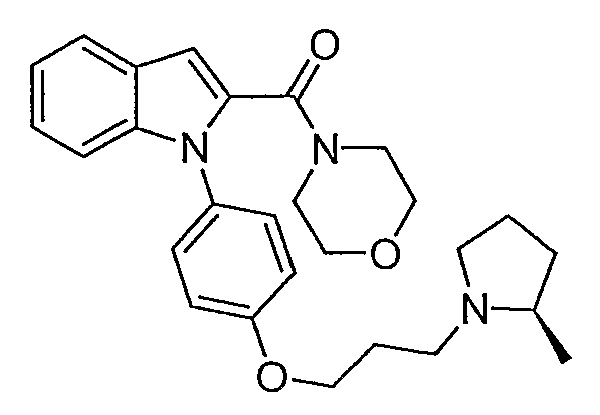
(1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)морфолин-4-илметанон синтезировали по способу, аналогичному использованному для примера 18, с использованием морфолина вместо пирролидина. LC-MS (C27H33N3O3, рассчитано 447), m/z 448 (М+Н).
Пример 26
Бутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты
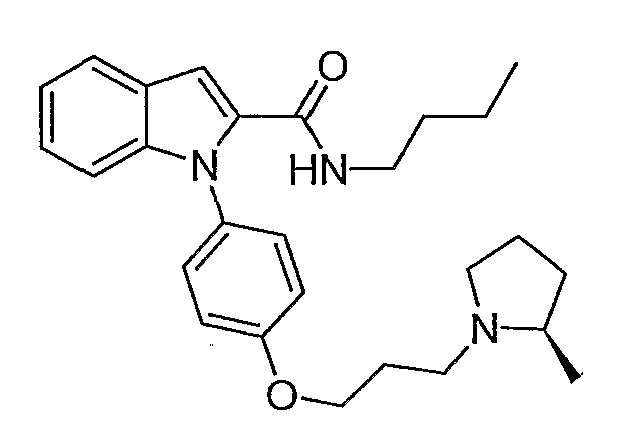
Бутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 18, с использованием бутиламина вместо пирролидина. LC-MS (C27H35N3O2, рассчитано 433), m/z 434 (М+Н).
Пример 27
Изобутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты
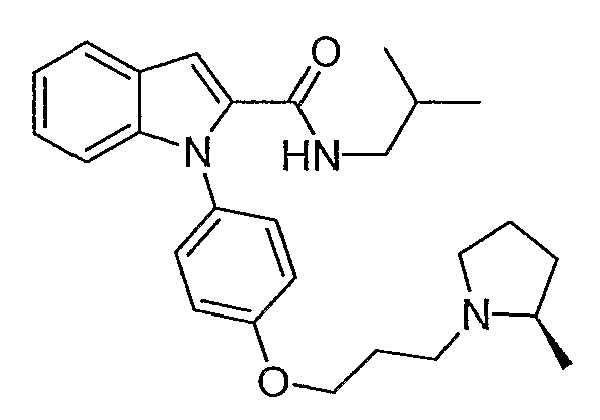
Изобутиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 18, с использованием изобутиламина вместо пирролидина. LC-MS (C27H35N3O2, рассчитано 433), m/z 434 (М+Н).
Пример 28
Циклогексилметиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты
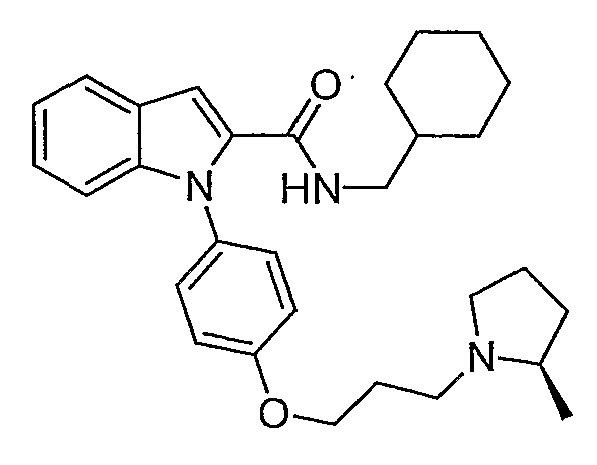
Циклогексилметиламид 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 18, с использованием циклогексилметиламина вместо пирролидина. LC-MS (C30H39N3O2, рассчитано 473), m/z 474 (М+Н).
Пример 29
Циклогексиламид 5-метокси-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты
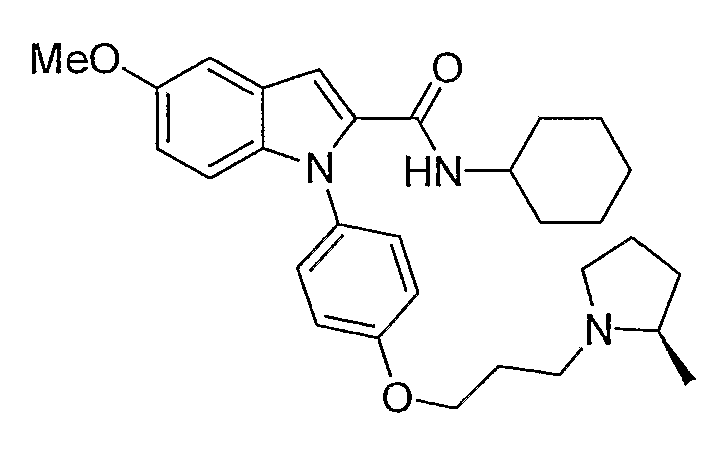
Циклогексиламид 5-метокси-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 18, с использованием циклогексиламина вместо пирролидина. LC-MS (C30H39N3O3, рассчитано 489), m/z 490 (М+Н).
Схема 5
Пример 30
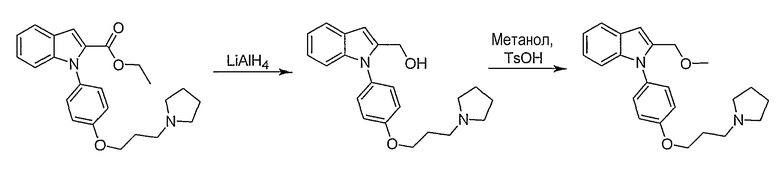
Этиловый эфир 1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-карбоновой кислоты
Этиловый эфир 1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-карбоновой кислоты синтезировали по способу, аналогичному использованному для примера 4, исходя из этил индол-2-карбоксилата. LC-MS (C24H28N2O3, рассчитано 392), m/z 393 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,72 (д, J=7,8 Гц, 1H), 7,43 (с, 1H), 7,29-7,15 (м, 4H), 7,06 (д, J=8,4 Гц, 1H), 6,99 (д, J=8,7 Гц, 2H), 4,23 (кв., J=7,2 Гц, 2H), 4,10 (т, J=6 Гц, 2H), 3,06-3,01 (м, 4H), 2,27-2,17 (м, 2H), 2,01-1,96 (м, 6H), 1,26 (т, J=7,2 Гц, 3H).
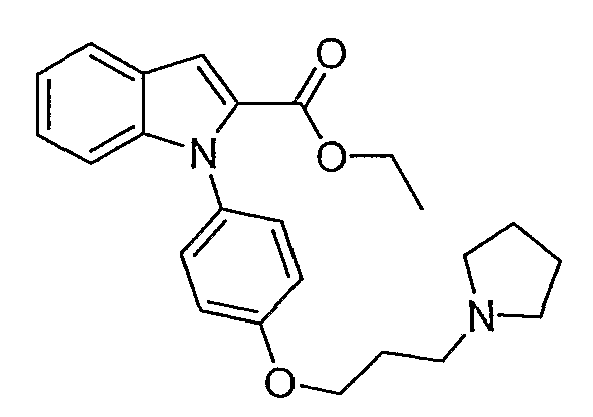
Пример 31
{1-[4-(3-Пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}метанол
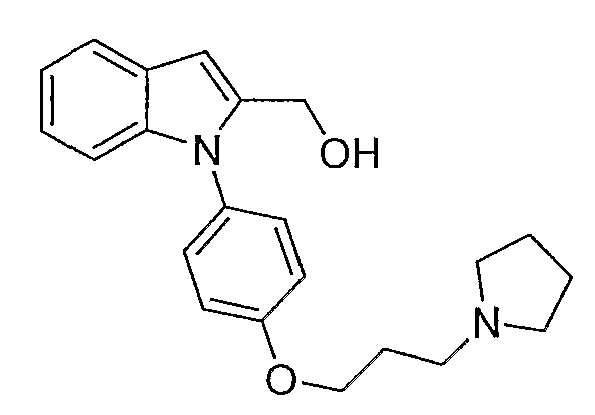
Этиловый эфир 1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-карбоновой кислоты (0,5 г, 1,27 ммол) растворяли в тетрагидрофуране (ТГФ) (10 мл) и раствор добавляли по каплям к алюмогидриду лития (1,53 мл, 1М раствор в ТГФ, 1,53 ммол) в ТГФ (10 мл). Реакционную смесь перемешивали при 60°С в течение 2 часов. Добавляли воду (0,3 мл), 2н. раствор NaOH (0,3 мл) и воду (0,9 мл) и растворитель упаривали. Полученный остаток разбавляли водой и экстрагировали дихлорметаном. Дихлорметановые экстракты высушивали над MgSO4 и концентрировали с получением белого твердого вещества, 0,36 г. Небольшое количество продукта очищали с помощью полупрепаративной LC-MS с получением 3,5 мг чистого желаемого продукта. LC-MS (C22H26N2O2, рассчитано 350), m/z 351 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,65-7,61 (м, 1H), 7,35 (д, J=8,7 Гц, 2H), 7,16-7,08 (м, 3H), 7,01 (д, J=8,7 Гц, 2H), 6,65 (с, 1H), 4,64 (с, 2H), 4,10 (т, J=6 Гц, 2H), 2,93-2,86 (м, 6H), 2,21-2,12 (м, 2H), 1,94-1,90 (м, 4H).
Пример 32
2-Метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
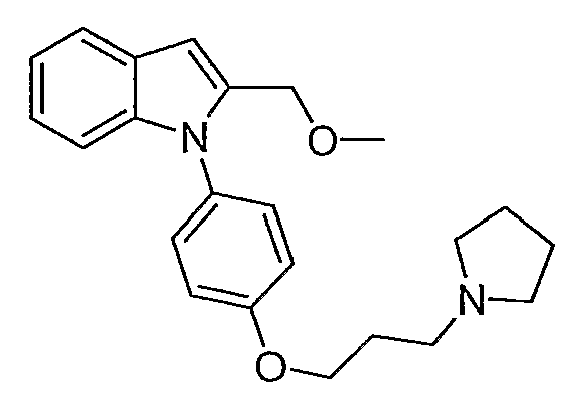
{1-[4-(3-Пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}метанол (15 мг) растворяли в смеси метанола, ацетонитрила и 1н. HCl. После отстаивания при комнатной температуре в течение 2 часов раствор очищали с помощью полупрепаративной LC-MS с получением 1,1 мг желаемого продукта. LC-MS (C23H28N2O2, рассчитано 364), m/z 365 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,66-7,61 (м, 1H), 7,33 (д, J=9 Гц, 2H), 7,20-6,75 (м, 5H), 6,66 (с, 1H), 4,40 (с, 2H), 4,10 (т, J=6,3 Гц, 2H), 3,28 (с, 3H), 2,82-2,71 (м, 6H), 2,17-2,08 (м, 2H), 1,87 (м, 4H).
Пример 33
2-Циклогексилоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
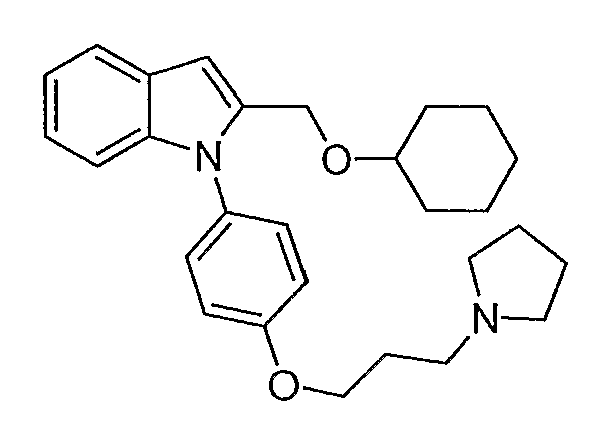
2-Циклогексилоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали по способу, аналогичному использованному для примера 32, с использованием циклогексанола вместо метанола. LC-MS (C28H36N2O2, рассчитано 432), m/z 433 (М+Н).
Пример 34
2-Изопропоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
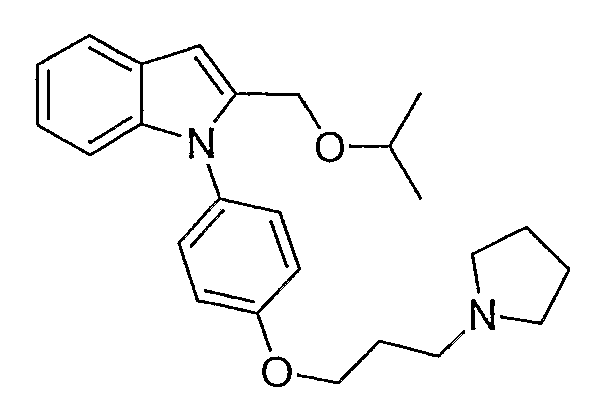
2-Изопропоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали по способу, аналогичному использованному для примера 32, с использованием изопропанола вместо метанола. LC-MS (C25H32N2O2, рассчитано 392), m/z 393 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,63-7,61 (м, 1H), 7,35-7,32 (м, 2H), 7,14-7,07 (м, 3H), 7,01 (д, J=8,7 Гц, 2H), 6,64 (с, 1H), 4,42 (с, 2H), 4,10 (т, J=6,3 Гц, 2H), 3,58-3,48 (м, 1H), 2,76-2,38 (м, 6H), 2,08-2,02 (м, 2H), 1,83-1,71 (м, 4H), 1,08 (д, J=6 Гц, 6H).
Пример 35
2-Циклопентилоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
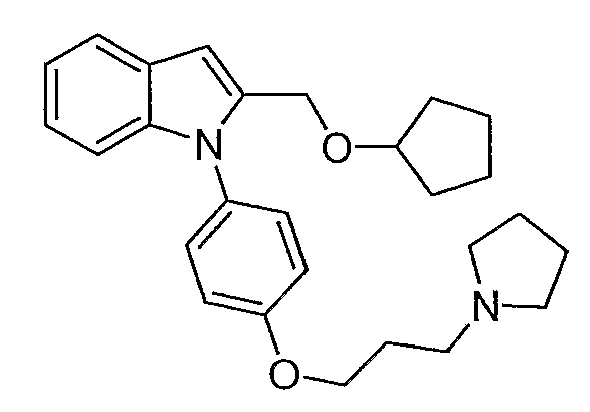
2-Циклопентилоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали по способу, аналогичному использованному для примера 32, с использованием циклопентанола вместо метанола. LC-MS (C27H34N2O2, рассчитано 418), m/z 419 (М+Н).
Пример 36
{5-Метокси-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}метанол
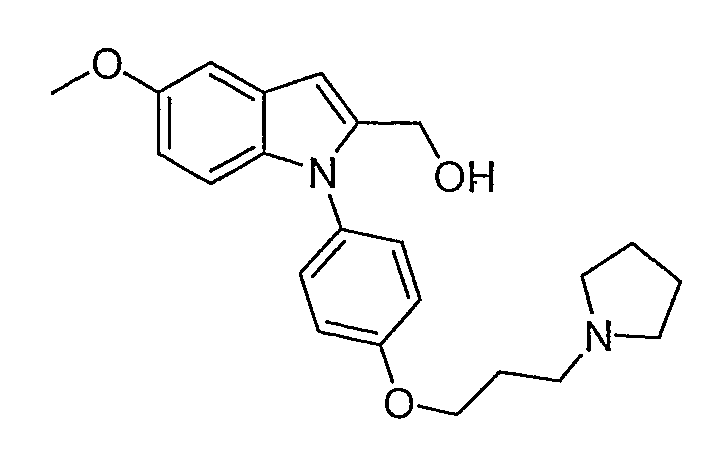
{5-Метокси-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}метанол синтезировали по способу, аналогичному использованному для примера 31, исходя из этил 5-метоксииндол-2-карбоксилата. LC-MS (C23H28N2O3, рассчитано 380), m/z 381 (М+Н). 1H ЯМР (300 МГц, CDCl3) δ 7,33 (д, J=8,1 Гц, 2H), 7,08 (с, 1H), 6,99 (д, J=8,4 Гц, 3H), 6,80 (д, J=8,7 Гц, 1H), 6,56 (c, 1H), 4,61 (с, 2H), 4,07 (м, 2H), 3,85 (с, 3H), 2,83 (м, 6H), 2,14 (м, 2H), 1,91 (м, 4H).
Схема 6
Пример 37
2-Циклопропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
N-(2-Йодфенил)ацетамид. 2-Йоданилин (1,00 г, 4,56 ммол) растворяли в пиридине (5 мл) и раствор охлаждали до 0°С. После этого добавляли ацетилхлорид (314 мкл, 5,94 ммол), реакционную смесь перемешивали при 0°С в течение 1 часа и затем при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли 1н. HCl и экстрагировали эфиром. Органический слой высушивали (MgSO4) и концентрировали с получением желаемого ацетамида (количественный выход), который использовали в следующей реакции без дальнейшей очистки.
2-Циклопропил-1Н-индол. К раствору N-(2-йодфенил)ацетамида (100 мг, 0,38 ммол) в диоксане (750 мл) и 1,1,3,3-тетраметилгуанидине (750 мл) добавляли циклопропилацетилен (41 мл, 0,49 ммол), хлорид бис(трифенилфосфин)палладия(II) (35 мг, 0,05 ммол) и иодид меди(I) (10 мг, 0,05 ммол). Реакционную смесь перемешивали в течение ночи при 80°С. Раствор охлаждали и разбавляли водой и продукт экстрагировали из воды метиленхлоридом. Органический слой высушивали (MgSO4) и концентрировали с получением нециклизованного продукта реакции связывания по Соногашира. Добавляли диоксан (750 мл) и 1,1,3,3-тетраметилгуанидин (750 мл) и реакционную смесь перемешивали в течение ночи при 90°С. Раствор снова разбавляли водой и экстрагировали метиленхлоридом. Органический слой высушивали (MgSO4) и концентрировали с получением желаемого индола, который использовали без дальнейшей очистки.
2-Циклопропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол. К раствору 2-циклопропил-1Н-индола (17 мг, 0,11 ммол) и 1-[3-(4-йодфенокси)пропил]пирролидина (36 мг, 0,11 ммол) в толуоле (0,2 мл) добавляли иодид меди (0,2 мг), фосфат калия (47 мг, 0,22 ммол) и N,N-диметилэтилендиамин (1,2 мкл, 0,11 ммол). Реакционную смесь перемешивали в течение ночи при 100°С. После охлаждения до комнатной температуры реакционную смесь фильтровали через слой диоксида кремния. Реакционную смесь концентрировали и очищали с помощью препаративной ВЭЖХ с получением 1,6 мг желаемого индола. LC-MS (C24H28N2O, рассчитано 360), m/z 361 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,55-7,52 (м, 1H), 7,35-7,32 (м, 2H), 7,10-7,02 (м, 5H), 6,16 (с, 1H), 4,11 (т, J=6,3 Гц, 2H), 2,70 (т, J=7,5 Гц, 2H), 2,59 (м, 4H), 2,08 (квинт., J=7,0 Гц, 1H), 1,83 (м, 2H), 1,64 (м, 4H), 0,88-0,81 (м, 2H), 0,79-0,73 (м, 2H).
Пример 38
2-Пропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
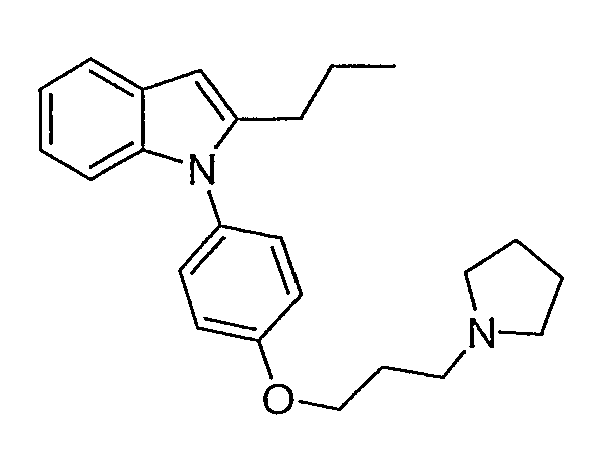
2-Пропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали по способу, аналогичному использованному для примера 37, с использованием 1-пентина во второй стадии. LC-MS (C24H30N2O, рассчитано 362), m/z 363 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,61-7,54 (м, 1H), 7,25-7,21 (м, 2H), 7,09-6,99 (м, 5H), 6,38 (с, 1H), 4,10 (т, J=6 Гц, 2H), 3,01-2,93 (м, 6H), 2,55 (т, J=7,8 Гц, 2H), 2,25-2,15 (м, 2H), 1,98-1,93 (м, 4H), 1,60 (секстет, J=7,5 Гц, 2H), 0,91 (т, J=7,5 Гц, 3H).
Схема 7
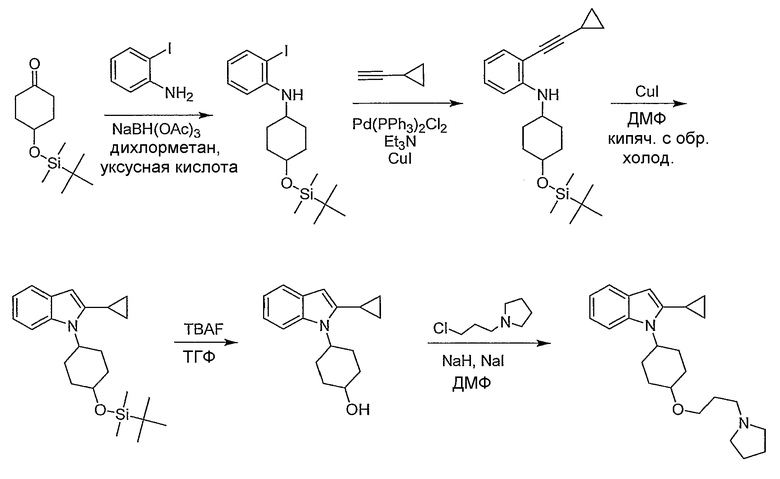
Пример 39
2-Циклопропил-1-[4-(3-пирролидин-1-илпропокси)циклогексил]-1Н-индол
4-(трет-Бутилдиметилсиланилокси)циклогексанон. Синтезирован согласно Carreсo, M.C.; Urbano, A.; Di Vitta C. J. Org. Chem., 1998, 63, 8320.
[4-(трет-Бутилдиметилсиланилокси)циклогексил]-(2-йодфенил)амин. К раствору 2-йоданилина (2,5 г, 10,9 ммол) в дихлорметане (160 мл) добавляли 4-(трет-бутилдиметилсиланилокси)циклогексанон (2,39 г, 10,9 ммол) и уксусную кислоту (8 мл). После перемешивания реакционной смеси в течение 1 часа при комнатной температуре добавляли триацетоксиборгидрид натрия (3,47 г, 16,4 ммол) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь гасили насыщенным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органический раствор высушивали (MgSO4) и концентрировали с получением 4,47 г желаемого амина, который использовали без дальнейшей очистки. LC-MS (C18H30INOSi, рассчитано 431), m/z 432 (М+Н).
[4-(трет-Бутилдиметилсиланилокси)циклогексил]-(2-циклопропилэтинилфенил)амин. К раствору [4-(трет-бутилдиметилсиланилокси)циклогексил]-(2-йодфенил)амина (4,47 г, 10,4 ммол) в триэтиламине (70 мл) добавляли иодид меди(I) (198 мг, 1,04 ммол), затем хлорид бис(трифенилфосфино)палладия(II) (730 мг, 1,04 ммол) и циклопропилацетилен (1,73 мл, 20,8 ммол). Реакционную смесь перемешивали в токе азота при комнатной температуре в течение ночи. После этого реакционную смесь упаривали, остаток растворяли в эфире и раствор фильтровали через Целит. Концентрирование дало неочищенный продукт с количественным выходом, который использовали без дальнейшей очистки. LC-MS (C23H35NOSi, рассчитано 369), m/z 370 (М+Н).
1-[4-(трет-Бутилдиметилсиланилокси)циклогексил]-2-циклопропил-1Н-индол. К раствору [4-(трет-бутилдиметилсиланилокси)циклогексил]-(2-циклопропилэтинилфенил)амина (3,84 г, 10,4 ммол) в N,N-диметилформамиде (60 мл) добавляли иодид меди(I) (100 мг, 0,525 ммол). После кипячения реакционной смеси с обратным холодильником в течение 48 часов ее оставляли остывать до комнатной температуры и растворитель удаляли в вакууме. Остаток разбавляли водой и продукт экстрагировали дихлорметаном. Дихлорметановый раствор высушивали (MgSO4) и концентрировали с получением темного остатка, который использовали на следующей стадии без дальнейшей очистки. LC-MS (C23H35NOSi, рассчитано 369), m/z 370 (М+Н).
4-(2-Циклопропилиндол-1-ил)циклогексанол. Неочищенный 1-[4-(трет-Бутилдиметилсиланилокси)циклогексил]-2-циклопропил-1Н-индол, описанный выше (10,4 ммол), растворяли в тетрагидрофуране (150 мл) и добавляли фторид тетрабутиламмония (21 мл, 1М раствор ТГФ, 21 ммол). После перемешивания реакционной смеси в течение 72 часов ее концентрировали и остаток разбавляли водой и экстрагировали этилацетатом. Органический слой высушивали (MgSO4), концентрировали и остаток очищали колоночной хроматографией на SiO2 (10-50% этилацетат/гексан) с получением двух (цис/транс)-изомеров (355 мг более полярного изомера, 681 мг менее полярного изомера) желаемого спирта. LC-MS (C17H21NO, рассчитано 255), m/z 256 (М+Н).
2-Циклопропил-1-[4-(3-пирролидин-1-илпропокси)циклогексил]-1Н-индол. К раствору 4-(2-циклопропилиндол-1-ил)циклогексанола (25 мг, 0,098 ммол, более полярный изомер) в N,N'-диметилформамиде (2 мл) добавляли иодид натрия (8 мг) и гидрид натрия (6 мг, 60%-я дисперсия в минеральном масле, 0,15 ммол). После перемешивания реакционной смеси при комнатной температуре в течение 5 минут добавляли 1-(3-хлорпропил)пирролидин (22 мг, 0,15 ммол) и реакционную смесь перемешивали при 85°С в течение 3 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, разбавляли водой и экстрагировали дихлорметаном. Органический слой высушивали (MgSO4) и концентрировали. Остаток очищали препаративной LC-MS с получением 8,0 мг желаемого амина. 1H ЯМР (300 МГц, CDCl3) δ 7,50 (д, J=7,2 Гц, 1H), 7,42 (д, J=8,1 Гц, 1H), 7,12-6,99 (м, 2H), 6,14 (с, 1H), 4,64-4,53 (м, 3H), 3,58 (т, J=6,2 Гц, 2H), 3,42 (тт, J=3,9, 10,8 Гц, 1H), 2,91-2,82 (м, 4H), 2,42 (м, 2H), 2,24 (м, 2H), 2,00-1,79 (м, 9H), 1,45 (м, 2H), 0,95 (м, 2H), 0,74 (м, 2H); LC-MS (C24H34N2O, рассчитано 366), m/z 367 (M+H).
Спектральные данные для продукта, образованного из менее полярного изомера 4-(2-циклопропилиндол-1-ил)циклогексанола: 1H ЯМР (300 МГц, CDCl3) δ 7,56-7,48 (м, 2H), 7,10-6,96 (м, 2H), 6,12 (с, 1H), 4,58 (тт, J=4,2, 12,6 Гц, 1H), 3,65 (с, 1H), 3,55 (т, J=6,0 Гц, 2H), 2,78-2,62 (м, 7H), 2,15 (д, J=14,7 Гц, 2H), 2,00-1,83 (м, 8H), 1,69-1,25 (м, 4H), 0,984-0,873 (м, 2H), 0,764-0,713 (м, 2H); LC-MS (C24H34N2O, рассчитано 366), m/z 367 (M+H).
Схема 8
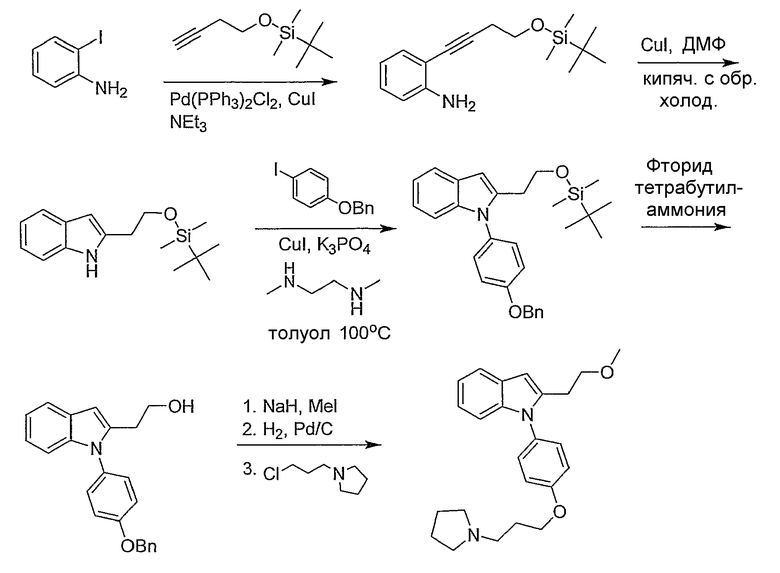
Пример 40
2-(2-Метоксиэтил)-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
2-[4-(трет-Бутилдиметилсиланилокси)бут-1-инил]фениламин. 2-Йоданилин (1,76 г, 8 ммол) растворяли в триэтиламине (50 мл) и поместили в атмосферу азота. Добавляли трет-бутилбут-3-инилоксидиметилсилан (2,58 г, 14 ммол), затем хлорид бис(трифенилфосфино)палладия(II) (30 мг, 0,042 ммол) и иодид меди(I) (7 мг, 0,036 ммол) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Триэтиламин выпаривали и остаток разбавляли простым эфиром и фильтровали через Целит. Фильтрат концентрировали и остаток очищали колоночной хроматографией на SiO2 (5-20% этилацетат/гексаны) с получением желаемого продукта. Выход реакции количественный.
2-[2-(трет-Бутилдиметилсиланилокси)этил]-1Н-индол. 2-[4-(трет-бутилдиметилсиланилокси)бут-1-инил]фениламин (8 ммол) и иодид меди(I) (5 мг, 0,026 ммол) в N,N-диметилформамиде (30 мл) нагревали с обратным холодильником в течение 3 часов. Растворитель упаривали и остаток разбавляли эфиром и фильтровали через Целит. Фильтрат концентрировали и остаток очищали колоночной хроматографией на SiO2 (5-20% этилацетат/гексаны) с получением желаемого продукта, 0,88 г. 1H ЯМР (300 МГц, CDCl3) δ 8,62 (ушир., 1H), 7,53 (д, J=7,5 Гц, 1H), 7,27 (д, J=7,8 Гц, 1H), 7,13-7,03 (м, 2H), 6,22 (с, 1H), 3,92 (т, J=5,7 Гц, 2H), 2,95 (т, J=5,7 Гц, 2H), 0,95 (с, 9H), 0,08 (с, 6H).
1-(4-Бензилоксифенил)-2-[2-(трет-бутилдиметилсиланилокси)этил]-1Н-индол. 2-[2-(трет-Бутилдиметилсиланилокси)этил]-1Н-индол (0,44 г, 1,6 ммол) и (4-бензилокси)йодбензол (0,6 г, 1,92 ммол) растворяли в толуоле (1,6 мл) и добавляли N,N-диметилэтилендиамин (0,034 мл, 0,32 ммол), иодид меди(I) (16 мг, 0,08 ммол) и фосфат калия (0,72 г, 3,36 ммол). Смесь нагревали при 100°С в течение ночи, затем фильтровали через слой диоксида кремния, промывали простым эфиром. Фильтрат концентрировали и остаток очищали колоночной хроматографией на SiO2 (0-10% этилацетат/гексаны) с получением желаемого продукта, 0,62 г. LC-MS (C29H35NO2Si, рассчитано 457), m/z 458 (М+Н).
2-[1-(4-Бензилоксифенил)-1Н-индол-2-ил]этанол. 1-(4-Бензилоксифенил)-2-[2-(трет-бутилдиметилсиланилокси)этил]-1Н-индол (0,62 г, 1,35 ммол) растворяли в тетрагидрофуране (6 мл) в токе азота и добавляли фторид тетрабутиламмония (1,49 мл, 1М раствор в тетрагидрофуране, 1,49 ммол). Реакционную смесь перемешивали в течение 2 часов, затем гасили насыщенным раствором ацетата аммония. Смесь экстрагировали этилацетатом, сушили над MgSO4 и концентрировали. Остаток смешивали с этилацетатом и смесь пропускали через слой диоксида кремния. Фильтрат концентрировали с получением желаемого продукта. Выход реакции количественный. LC-MS (C23H21NO2, рассчитано 343), m/z 344 (М+Н).
1-(4-Бензилоксифенил)-2-(2-метоксиэтил)-1Н-индол. 2-[1-(4-Бензилоксифенил)-1Н-индол-2-ил]этанол (0,675 ммол) растворяли в тетрагидрофуране (5 мл) в токе азота и добавляли гидрид натрия (81 мг, 60%-я дисперсия в минеральном масле, 2,03 ммол). Реакционную смесь кипятили с обратным холодильником, в течение этого времени добавляли йодметан (0,42 мл, 6,75 ммол). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 3 часов, затем осторожно гасили водой. Смесь экстрагировали этилацетатом, экстракт высушивали над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией на SiO2 (5-20% этилацетат/гексан) с получением желаемого продукта, 0,14 г. LC-MS (C24H23NO2, рассчитано 357), m/z 358 (М+Н).
4-[2-(2-Метоксиэтил)индол-1-ил]фенол. 1-(4-Бензилоксифенил)-2-(2-метоксиэтил)-1Н-индол (0,14 г, 0,39 ммол) растворяли в тетрагидрофуране (2 мл) и метаноле (1 мл). Добавляли каталитическое количество палладия-на-углероде (влажный, на основе 10%-го сухого реагента) и колбу продували азотом и водородом. Реакционную смесь перемешивали в течение ночи при давлении водорода 1 атм. Смесь фильтровали через Целит и фильтрат концентрировали с получением желаемого продукта. Выход реакции количественный. LC-MS (C17H17NO2, рассчитано 267), m/z 266 (М-Н).
2-(2-Метоксиэтил)-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол. 4-[2-(2-Метоксиэтил)индол-1-ил]фенол (0,39 ммол) растворяли в N,N-диметилформамиде (4 мл) и добавляли 1-(3-хлорпропил)пирролидин (58 мг, 0,39 ммол), гидрид натрия (19 мг, 60%-я дисперсия в минеральном масле, 0,47 ммол) и иодид натрия (59 мг, 0,39 ммол). Реакционную смесь нагревали при 70°С в течение 1,5 часов, затем осторожно гасили насыщенным раствором бикарбоната натрия. Смесь экстрагировали этилацетатом, экстракт высушивали над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией на SiO2 с получением 70 мг желаемого продукта. LC-MS (C24H30N2O2, рассчитано 378), m/z 379 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,60-7,56 (м, 1H), 7,25-7,21 (м, 2H), 7,10-7,00 (м, 5H), 6,45 (с, 1H), 4,11 (т, J=6,3 Гц, 2H), 3,57 (т, J=7,2 Гц, 2H), 3,30 (с, 3H), 2,89 (т, J=7,2 Гц, 2H), 2,74 (т, J=7,5 Гц, 2H), 2,65 (м, 4H), 2,16-2,05 (м, 2H), 1,90-1,81 (м, 4H).
Схема 9
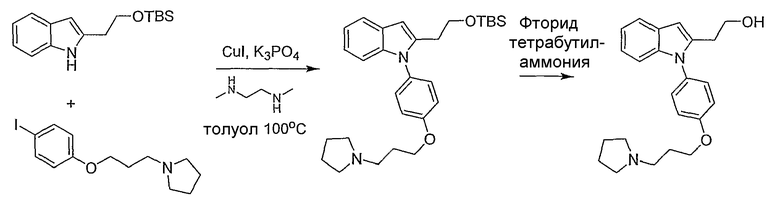
Пример 41
2-{1-[4-(3-Пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}этанол
2-[2-(трет-Бутилдиметилсиланилокси)этил]-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол 2-[2-(трет-Бутилдиметилсиланилокси)этил]- 1Н-индол (0,11 г, 0,4 ммол) и 1-[3-(4-йодфенокси)пропил]пирролидин (0,16 г, 0,48 ммол) растворяли в толуоле (0,4 мл) и добавляли N,N-диметилэтилендиамин (0,017 мл, 0,16 ммол), иодид меди(I) (30 мг, 0,16 ммол) и фосфат калия (0,18 г, 0,84 ммол). Реакционную смесь нагревали при 100°С в течение ночи. Смесь фильтровали через Целит, затем промывали дихлорметаном. Фильтрат концентрировали и остаток использовали далее без очистки (неочищенный продукт содержит некоторое количество исходного материала). LC-MS (C29H42N2O2Si, рассчитано 478), m/z 479 (М+Н).
2-{1-[4-(3-Пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}этанол. 2-[2-(трет-Бутилдиметилсиланилокси)этил]-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол (0,4 ммол) растворяли в тетрагидрофуране (2 мл) в токе азота и добавляли фторид тетрабутиламмония (0,44 мл, 1М раствор в тетрагидрофуране, 0,44 ммол). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем гасили насыщенным раствором хлорида аммония. Смесь разбавляли насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические экстракты высушивали над MgSO4 и концентрировали. Остаток очищали с помощью полупрепаративной LC-MS с получением желаемого продукта, 15,8 мг. LC-MS (C23H28N2O2, рассчитано 364), m/z 365 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,61-7,59 (м, 1H), 7,24-7,20 (м, 2H), 7,13-7,07 (м, 2H), 7,03-7,00 (м, 3H), 6,48 (с, 1H), 4,09 (т, J=6,3 Гц, 2H), 3,75 (т, J=6,6 Гц, 2H), 2,89 (т, J=6,6 Гц, 2H), 2,66 (т, J=7,5 Гц, 2H), 2,60-2,49 (м, 4H), 2,10-2,01 (м, 2H), 1,85-1,76 (м, 4H).
Схема 10
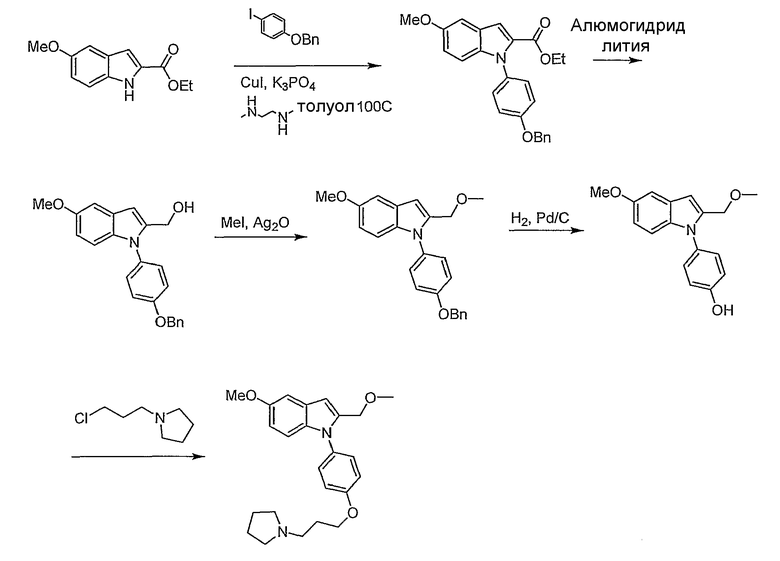
Пример 42
5-Метокси-2-метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
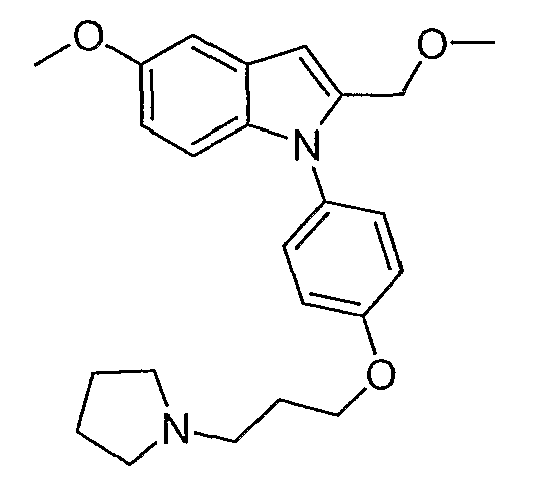
Этиловый эфир 1-(4-бензилоксифенил)-5-метокси-1Н-индол-2-карбоновой кислоты. Смесь 2-этилового эфира 5-метоксииндола (0,59 г, 2,7 ммол), 1-бензилокси-4-йодбензола (1 г, 3,23 ммол), N,N-диметилэтилендиамина (0,057 мл, 0,54 ммол), иодида меди(I) (0,1 г, 0,54 ммол) и трехосновного фосфата калия (1,2 г, 5,67 ммол) нагревали в толуоле при 100°С в течение 24 часов. Смесь фильтровали через слой диоксида кремния, затем промывали этилацетатом и фильтрат концентрировали. Хроматография на SiO2 со смесью 5-20% этилацетат/гексаны дала желаемый продукт (0,51 г, выход 57%), вместе с некоторым количеством смешанных фракций (0,36 г), которые сохраняли для последующей очистки. LC-MS (C25H23NO4, рассчитано 401), m/z 402 (М+Н).
[1-(4-Бензилоксифенил)-5-метокси-1Н-индол-2-ил]метанол. Алюмогидрид лития (1,53 мл, 1М раствор в тетрагидрофуране, 1,53 ммол) разбавляли тетрагидрофураном (5 мл) в токе азота и по каплям добавляли раствор этилового эфира 1-(4-бензилоксифенил)-5-метокси-1Н-индол-2-карбоновой кислоты (0,51 г, 1,27 ммол) в тетрагидрофуране (5 мл). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 2 часов, затем охлаждали до комнатной температуры. С осторожностью добавляли воду (0,3 мл), затем 2н. раствор NaOH (0,3 мл) и воду (0,9 мл). Растворитель удаляли выпариванием и остаток разбавляли водой и экстрагировали этилацетатом. Органический раствор отделяли, сушили над MgSO4 и концентрировали с получением 0,41 г (выход 88%) неочищенного продукта. LC-MS (C23H21NO3, рассчитано 359), m/z 360 (М+Н).
1-(4-Бензилоксифенил)-5-метокси-2-метоксиметил-1Н-индол. [1-(4-Бензилоксифенил)-5-метокси-1Н-индол-2-ил]метанол (0,2 г, 0,56 ммол) растворяли в ацетонитриле (2 мл) и добавляли йодметан (0,35 мл, 5,6 ммол) и оксид серебра(I) (0,39 г, 1,68 ммол). Смесь перемешивали в течение ночи при 40°С, затем охлаждали до комнатной температуры и фильтровали через слой Целита. Фильтрат концентрировали. Хроматография на SiO2 с 3-50%-й смесью этилацетат/гексаны дала 0,16 г (выход 77%) желаемого продукта. LC-MS (C24H23NO3, рассчитано 373), m/z 374 (М+Н).
4-(5-Метокси-2-метоксиметилиндол-1-ил)фенол. 1-(4-Бензилоксифенил)-5-метокси-2-метоксиметил-1Н-индол (0,16 г, 0,43 ммол) растворяли в метаноле (3 мл) и тетрагидрофуране (1 мл). Добавляли палладий-на-углероде (0,32 г, влажный 10%-й) и формиат аммония (0,14 г, 2,14 ммол) и реакционную смесь перемешивали при кипячении с обратным холодильником в течение 2 часов. Смесь охлаждали до комнатной температуры и фильтровали через слой Целита. Фильтрат концентрировали и неочищенный материал использовали без очистки. LC-MS (C17H17NO3, рассчитано 283), m/z 284 (М+Н).
5-Метокси-2-метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол. 4-(5-Метокси-2-метоксиметилиндол-1-ил)фенол (0,43 ммол) растворяли в N,N-диметилформамиде в токе азота. Добавляли 1-(3-хлорпропил)пирролидин (74 мг, 0,5 ммол), гидрид натрия (20 мг, 60%-я дисперсия в минеральном масле, 0,5 ммол) и иодид натрия (75 мг, 0,5 ммол) и смесь нагревали при 70°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Органические экстракты высушивали над MgSO4 и концентрировали. Очистка с помощью полупрепаративной LC-MS дала желаемый продукт, 67,4 мг (выход 40%, две стадии). LC-MS (C24H30N2O3, рассчитано 394), m/z 395 (M+H); 1Н ЯМР (300 МГц, CDCl3) δ 7,34-7,29 (м, 2H), 7,09 (д, J=2,4 Гц, 1H), 7,04-6,98 (м, 3H), 6,82 (д, J=2,4 Гц, 1H), 6,58 (с, 1H), 4,37 (с, 2H), 4,10 (т, J=6,3 Гц, 2H), 2H), 3,86 (с, 3H), 3,28 (с, 3H), 2,69-2,64 (м, 2H), 2,58-2,54 (м, 4H), 2,11-2,01 (м, 2H), 1,83-1,79 (м, 4H).
Пример 43
5-Метил-2-метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
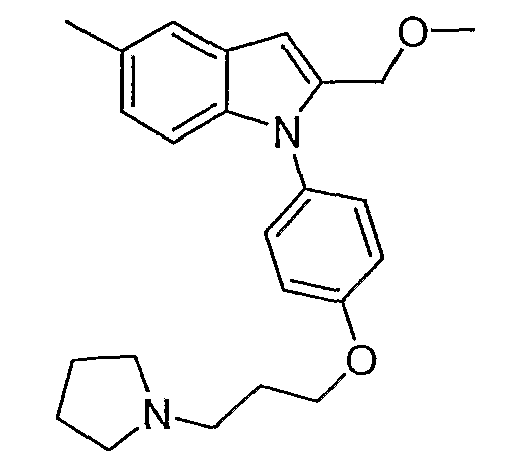
5-Метил-2-метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали по способу, аналогичному использованному для примера 42. LC-MS (C24H36N2O2, рассчитано 378), m/z 379 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,42-7,41 (м, 1H), 7,34-7,29 (м, 2H), 7,04-6,95 (м, 4H), 6,57 (с, 1H), 4,38 (с, 2H), 4,09 (т, J=6,3 Гц, 2H), 3,27 (с, 3H), 2,69-2,64 (м, 2H), 2,58-2,53 (м, 4H), 2,44 (с, 3H), 2,10-2,01 (м, 2H), 1,83-1,79 (м, 4H).
Пример 44
5-Фтор-2-метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол
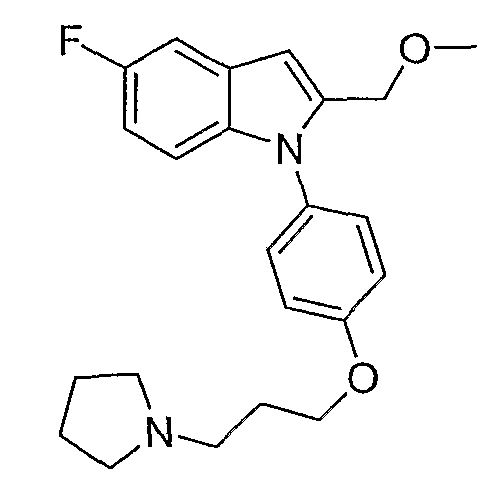
5-Фтор-2-метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол синтезировали по способу, аналогичному использованному для примера 42. LC-MS (C23H27FN2O2, рассчитано 382), m/z 383 (M+H); 1H ЯМР (300 МГц, CDCl3) δ 7,34-7,24 (м, 3H), 7,03-6,99 (м, 3H), 6,88 (тд, J=9 Гц, 2,4 Гц, 1H), 6,61 (с, 1H), 4,36 (с, 2H), 4,10 (т, J=6,3 Гц, 2H), 3,28 (c, 3H), 2,69-2,64 (м, 2H), 2,57-2,53 (м, 4H), 2,10-2,01 (м, 2H), 1,83-1,79 (м, 4H).
Показательные соединения согласно настоящему изобретению, которые были приготовлены в соответствии с методиками примеров 1-41, были оценены в анализах связывания с клетками, экспрессирующими человеческий Н3-рецептор, по следующей методике.
Клеточная культура
Материалы
[125I]-меченый йодпроксифан (2000 кюри/ммол) был приобретен у фирмы Amersham Bioscience Piscataway, NJ). [3Н]Nα-метилгистамин (85 кюри/ммол) был приобретен у фирмы Perkin Elmers Life Science (Boston, MA). Набор красителей Calcium 3 был получен от фирмы Molecular Deviсes (Sunnyvalе, CA). Все прочие химические вещества были приобретены либо у фирмы Sigma-Aldrich (St. Louis, MO), либо в Tocris Cookson Inc. (Ellisville, MO).
RAGE-методология
Человеческий Н3-рецептор гистамина был устойчиво экспрессирован в НТ1080-клетках фибросаркомы, содержащих химерный G-белок, Gqαi5 (Coward et al., Anal. Biochem., 1999; 270: 242-248). НТ1080-клетки Gqαi5 были выращены в альфа-модифицированной минимальной питательной среде (МЕМ), содержащей 10% фетальную телячью сыворотку и 7 мкг/мл бластицидина при 37°С в 5% СО2/95%-й атмосфере. Клетки (4,8·109) были подвергнуты облучению с дозой 50 рад от 137Cs-источника, и RAGE-вектор pFG8-HH3 (RAGE - рандомная активация экспрессии генов; см. Harrington et al., Nature Biotechnology, 2001; том 19: 440-445) был впоследствии интегрирован в клетки с помощью электропорации (250 В, 600 мкF, 50 Ом). RAGE-вектор pFG8-HH3 содержал последовательность комплементарной ДНК, кодирующую первый экзон (83 аминокислоты) человеческого Н3-рецептора. После электропорации клетки были помещены в Т75-флаконы и выращены в альфа-модифицированной МЕМ. Культуральную среду через 48 часов после электропорации заменяли на альфа-модифицированую МЕМ с 10% фетальной телячьей сывороткой, 500 мкг/мл гигромицина В и 3 мкг/мл пуромицина. Среду заменяли каждые четыре дня во время размножения клеток. Для идентификации RAGE-активированных клеток, экспрессирующих Н3-рецептор, пулы примерно 10000 колоний (в целом 5·107-1,5·108 клеток) были скринированы с помощью полимеразной цепной реакции (ПЦР) на желаемый генный продукт (с использованием праймеров, специфичных к RAGE-вектору и экзону 2 Н3-рецептора). Пулы, которые были найдены содержащими приемлемый транскрипт, как подтверждалось установлением последовательности, были субклонированы в пулы из 100 клеток на лунку. Позитивные пулы 100 клеток/лунка были идентифицированы с помощью ПЦР, подтверждены установлением последовательности и затем субклонированы до 0,8 клеток/лунка. Как только клоны, экспрессирующие Н3-рецептор, были идентифицированы с помощью ПЦР-анализа, были выполнены анализы [FLIPR (флуориметрический анализатор изображений плашек) или радиолигандное связывание] для подтверждения, что активированный ген продуцирует функциональный белок. Экспрессия белка в начальных клонах, полученных из RAGE-библиотеки, нарастала в присутствии метотрексата. Поскольку интегрированный RAGE-вектор содержит DHFR-ген, такая обработка выбирает клетки, которые усиливали генетический локус, содержащийся в RAGE-инсерте. Субклоны, полученные после амплификации метотрексатом, были протестированы на функциональную активность в FLIPR-анализах для идентификации клона, который был наиболее пригоден для HTS (высокопроизводительного скрининга соединений). Конечный HT1080-Gqαi5-клон (RAGE-H3), экспрессирующий человеческий Н3-рецептор гистамина, был выращен в альфа-модифицированной МЕМ, содержащей 10% фетальную телячью сыворотку, 3 мкг/мл пуромицина, 500 мкг/мл гигромицина В, 3,2 мкМ метотрексата при 37°С в 5% СО2/95% атмосфере.
Приготовление мембраны
RAGE-H3-клетки (109) были дважды промывали холодным фосфатно-солевым буфером (PBS), соскоблены с плашек и центрифугированы при 1000×g в течение 5 минут. Клетки были ресуспендированы в охлажденном во льду буфере 10 мM Tris HCl, рН 7,4, содержащем 5 мМ EDTA и таблетки коктейля ингибитора протеаз (Roche Molecular Biochemicals). После инкубации на льду в течение 10 минут клетки гомогенизировали с помощью гомогенизатора Даунса или гомогенизатора ткани типа «Политрон» и центрифугировали при 1000×g в течение 10 минут при 4°С. Полученную надосадочную жидкость центрифугировали при 32000×g в течение 30 минут при 4°С. Мембранные осадки в центрифужных пробирках ресуспендировали в буфере 50 мM Tris HCl, рН 7,4 и хранили при -80°С перед употреблением. Концентрацию белка определяли по методу Бредфорда (Bio-Rad Laboratories, CА).
Анализы радиолигандного связывания
Анализы связывания были проведены в 96-луночных полипропиленовых плашках в 50 мM Tris HCl, рН 7,4, содержащем 1 мМ EDTA. Реакционные смеси, содержащие 100 мкл мембранной суспензии, 50 мкл 4%-го ДМСО и 50 мкл нарастающих количеств [125I]-меченого йодпроксифана (конечная концентрация 0,0005-1,8 нМ для анализа насыщающего связывания человеческого Н3-рецептора). Неспецифичное связывание определяли добавлением к реакционным смесям 10 мкМ клобенпропита. Анализы конкурентного связывания выполняли в реакционной смеси, содержащей 100 мкл мембранной суспензии (~20 мкг белка/лунка), 50 мкл [125I]-йодпроксифана (конечная концентрация ~0,15 нМ) и 50 мкл испытуемого соединения. Соединения растворяли в ДМСО и затем разбавляли 4% ДМСО; конечная максимальная концентрация ДМСО в анализах связывания составляла 1%. Инкубации выполняли в течение 1,5 часов при комнатной температуре и реакции останавливали быстрым фильтрованием через стекловолоконные фильтры GF/С (Perkin Elmer, MA) с использованием коллектора клеток Брендела. Фильтры предварительно пропитывали в 0,3%-м растворе полиэтиленимина в течение 30 минут и промывали 500 мл охлажденного льдом буфера 50 мM Tris HCl, рН 7,4. Фильтры высушивали, пропитывали восковым сцинтиллятором Meltilex (Perkin Elmer, MA) и обсчитывали с помощью сцинтилляционного счетчика Betaplate (Perkin Elmer, MA).
Анализы мобилизации кальция
Клетки RAGE-H3 или НТ1080-mH3 высевали в черные 384-луночные плашки и инкубировали в течение ночи при 37°С в 5% СО2/95% атмосфере. После удаления среды клетки обрабатывали CsCl-буфером Рингера (136 мМ CsCl, 5,4 мМ KCl, 5,5 мМ D-глюкозы, 20 мМ HEPES, рН 7,4, 2,1 мМ MgCl2, 1,2 мМ CaCl2), содержащим краситель Сalcium 3 (Molecular Device, CA) и пробенецид (3,75 мМ), в течение 60 минут, согласно инструкции производителя. Соединения разбавляли в CsCl-буфере Рингера, содержащем 0,2% бычьего сывороточного альбумина и 1,0% ДМСО. Зависимость доза-отклик стимулированного (R)-α-метилгистамином Са2+-потока измеряли с помощью флуорометрического анализатора изображений плашек (FLIPR, Molecular Devices, CA) и концентрацию (R)-α-метилгистамина для стимулирования 75% максимального отклика использовали для испытания ингибирующего действия соединений.
Анализ данных
Все данные проанализировали с помощью аппроксимации кривых в нелинейном методе наименьших квадратов с использованием программы Prism 4.0. Величины KD и Bmax для [125I]-йодпроксифана выводили из уравнения RL=RtL/(KD+L), где RL представляет собой концентрацию рецепторсвязывающего лиганда при равновесии, L представляет собой концентрацию свободного лиганда и Rt представляет собой общую концентрацию лиганда (т.е. Bmax). Для экспериментов конкурентного связывания значения IC50 (концентрация соединения, проявляющая 50%-е ингибирование специфичного связывания) и коэффициенты Хилла (nH) были выведены из аппроксимации данных к четырехпараметровому логистическому уравнению. Кажущиеся значения Ki рассчитывали с использованием уравнения Чена-Пруссова Ki=IC50/(1+(L/KD)), где L представляет собой концентрацию лиганда. Стимуляцию агонистом и ингибирование антагонистом в FLIPR аппроксимировали до сигмоидальной кривой доза-отклик с использованием уравнения Y=Bottom+(Top-Bottom)/(1+10^(LogEC50-X)), где Х представляет собой логарифм концентрации соединений и Y представляет собой флуоресцентный отклик. Значения Z' [15] были выведены для оценки качества анализов. Фигуры являются показательными для двух-трех экспериментов, выполненных в трипликатах или квадрупликатах.
Результаты этого анализа приведены в нижеследующей Таблице.
Выбранные примеры
Пример 1
Пример 2
Пример 3
Пример 4
Пример 5
Пример 6
Пример 7
Пример 8
Пример 9
Пример 10
Пример 11
Пример 12
Пример 13
Пример 14
Пример 15
Пример 16
Пример 17
Пример 18
Пример 19
Пример 20
Пример 21
Пример 22
Пример 23
Пример 24
Пример 25
Пример 26
Пример 27
Пример 28
Пример 29
Пример 30
Пример 31
Пример 32
Пример 33
Пример 34
Пример 35
Пример 36
Пример 37
Пример 38
Пример 39
Пример 40
Пример 41
Пример 42
Пример 43
Пример 44
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА | 2006 |
|
RU2417985C2 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ 6,7-ДИЗАМЕЩЕННЫХ 3-ХИНОЛИНКАРБОКСАМИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ ДЛЯ ИЗГОТОВЛЕНИЯ ЛЕКАРСТВА | 2002 |
|
RU2281940C2 |
| ФЕНИЛПИРАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2008 |
|
RU2480456C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОПРОПИЛАМИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ H-ГИСТАМИНОВОГО РЕЦЕПТОРА | 2007 |
|
RU2449989C2 |
| ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2008 |
|
RU2455994C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
Изобретение относится к соединениям формулы:
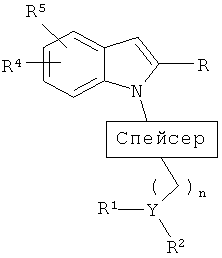
где спейсер представляет собой
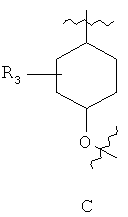
Y представляет собой N;
R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 5-6-членную гетероциклическую кольцевую систему, которая, необязательно, замещена 1-3 (С1-С5)алкильными группами; n равен 2-4; R3 представляет собой 0-2 группы, выбранные из атома галогена, (С1-С6)алкильной, (С1-С6)алкоксильной групп;
R4 и R5 независимо выбраны из Н, (С1-С5)алкильной, (C1-С8)алкоксильной группы, атома галогена;
R6 представляет собой Н, CONR7R8, -(СН2)х-О-R9, (С1-С4)алкильную группу, СООС2Н5, циклопропил;
х равен 1-4;
R7 и R8 независимо представляют собой водород, (С1-С5)алкильную группу, (С3-С7)-циклоалкильную группу, -СН2-циклогексил или
R7 и R8 вместе с атомом азота, к которому они присоединены, образуют 5-6-членную гетероциклическую кольцевую систему с 0-1 дополнительным гетероатомом, выбранным из О;
R9 представляет собой водород, (С1-С5)алкильную, (С3-С7)циклоалкильную группу;
и его фармацевтически приемлемым солям, и его индивидуальным стереоизомерам.
Соединения ингибируют Н3-рецептор гистамина, что позволяет использовать их для получения фармацевтических композиций. 4 н. и 2 з.п. ф-лы, 1 табл.
1. Соединение формулы:
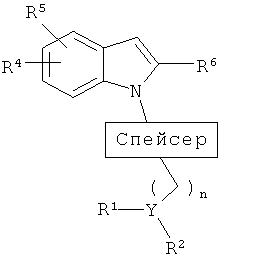
в которой спейсер представляет собой
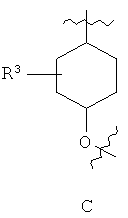
Y представляет собой N;
R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 5-6-членную гетероциклическую кольцевую систему, которая, необязательно, замещена 1-3 (С1-С5)алкильными группами; n равен 2-4;
R3 представляет собой 0-2 группы, выбранные из атома галогена, (C1-С6)алкильной, (С1-С6)алкоксильной групп;
R4 и R5 независимо выбраны из Н, (С1-С5)алкильной, (C1-С8)алкоксильной группы, атома галогена;
R6 представляет собой Н, CONR7R8,-(CH2)x-O-R9, (С1-С4)алкильную группу, СООС2Н5, циклопропил;
х равен 1-4;
R7 и R8 независимо представляют собой водород, (С1-С5)алкильную группу, (С3-С7)-циклоалкильную группу, -СН2-циклогексил или
R7 и R8 вместе с атомом азота, к которому они присоединены, образуют 5-6-членную гетероциклическую кольцевую систему с 0-1 дополнительным гетероатомом, выбранным из О;
R9 представляет собой водород, (С1-С5)алкильную, (С3-С7)циклоалкильную группу;
и его фармацевтически приемлемая соль, и его индивидуальные стереоизомеры.
2. Соединение по п.1, в котором R1-Y-R2 представляет собой 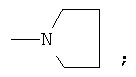
R3 представляет собой Н; R4 представляет собой Н; 5-метоксильную, 5-фтор или метильную группу; R5 представляет собой Н; и R6 представляет собой-СН2OСН3 или-(СН2)2OСН3.
3. Соединение по п.1, выбранное из группы, состоящей из 2-метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1H-индола;
2-метил-1-[4-(3-пиперидин-1-илпропокси)фенил]-1H-индола;
2-метил-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1H-индола;
1-[4-(3-пирролидин-1-илпропокси)фенил]-1H-индола;
5-метокси-2-метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1H-индола;
5-метил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
5-бром-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
4-хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
5-метокси-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
5-хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
2,5-диметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
6-хлор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
2-метил-5-фтор-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
1-[3-метокси-4-(3-пирролидин-1-илпропокси)фенил]-2-метил-1Н-индола;
1-[3-хлор-4-(3-пирролидин-1-илпропокси)фенил]-2-метил-1Н-индола;
2-пропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
5-метокси-2-метил-1-[4-(4-пирролидин-1-илбут-1-инил)фенил]-1Н-индола;
(5-метокси-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)пирролидин-1-илметанона;
циклобутиламида 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
циклопентиламида 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1H-индол-2-карбоновой кислоты;
циклогексиламида 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1H-индол-2-ил)пирролидин-1-илметанона;
2-(3-морфолин-4-илпропокси)-6,7,8,9-тетрагидропиридо[1,2-а]индола;
(1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-ил)морфолин-4-илметанона;
бутиламида 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
изобутиламида 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
циклогексиламида 5-метокси-1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
{1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}метанола;
2-метоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1H-индола;
2-циклогексилоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
2-изопропоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
2-циклопентилоксиметил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
{5-метокси-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}метанола;
2-пропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола;
2-(2-метоксиэтил)-1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индола и
2-{1-[4-(3-пирролидин-1-илпропокси)фенил]-1Н-индол-2-ил}этанола.
4. Производные индола, выбранные из
циклогептиламида 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
циклогексилметиламида 1-{4-[3-(2R-метилпирролидин-1-ил)пропокси]фенил}-1Н-индол-2-карбоновой кислоты;
сложного этилового эфира 1-[4-(3-пирролидин-1-илпропокси)фенил]-1H-индол-2-карбоновой кислоты;
2-циклопропил-1-[4-(3-пирролидин-1-илпропокси)фенил]-1H-индола;
2-циклопропил-1-[4-(3-пирролидин-1-илпропокси)циклогексил]-1Н-индола;
и их фармацевтически приемлемые соли, и индивидуальные стереоизомеры.
5. Фармацевтическая композиция, ингибирующая Н3-рецептор гистамина, включающая, по меньшей мере, одно соединение по п.1 или 4 в комбинации с фармацевтически приемлемым носителем.
6. Способ ингибирования Н3-рецептора гистамина, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества, по меньшей мере, одного соединения по п.1 или 4.
| US 3799943, 26.03.1974 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АЛКИЛСУЛЬФОНАМИД-АГОНИСТ 5HT, ДЛЯ РЕКТАЛЬНОГО ВВЕДЕНИЯ | 1993 |
|
RU2118527C1 |

