ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ:
По данной заявке испрашивается приоритет на основании предварительной заявки U.S. 60/488712, зарегистрированной 18 июля 2003 г., которая включена во всей полноте посредством ссылки и является частью данной заявки.
ФЕДЕРАЛЬНОЕ СПОНСИРОВАНИЕ ИССЛЕДОВАНИЯ ИЛИ РАЗРАБОТКИ
Не проводилось
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Техническая область
Изобретение относится к способам изготовления, способам применения и композициям небольших сферических частиц активного агента. В соответствии со способом производства активный агент растворяют в водном или смешивающемся с водой растворителе, содержащем растворенный усиливающий фазовое разделение агент (УФРА), чтобы образовать раствор в единственной жидкой фазе. Раствор затем подвергают фазовому разделению жидкость-твердое, причем имеющийся активный агент содержится в твердой фазе, а УФРА и растворитель содержатся в жидкой фазе. Фазовое разделение жидкость-твердое может быть вызвано рядом способов, например, изменением температуры раствора ниже температуры фазового перехода системы. Способ является наиболее подходящим для образования небольших сферических частиц терапевтических агентов, которые могут быть потом доставлены к объекту, который нуждается в терапевтическом агенте. Способ также является наиболее подходящим для образования твердых небольших сферических частиц макромолекул, в частности, макромолекул, которые термически неустойчивы, например, протеины.
Предшествующий уровень техники
Несколько технологий были использованы в прошлом для изготовления биополимерных нано- и микрочастиц. Обычные технологии для образования частиц включают в себя распылительную сушку и измельчение и могут быть использованы для производства частиц размером 5 мкм или меньше.
В патентах U.S. 5654010 и U.S. 5667808 описано производство твердых форм рекомбинантного гормона роста человека, ГРЧ, через комплексообразование с цинком, для того чтобы создать аморфный комплекс, который затем мелко измельчают, пропуская через ультразвуковое сопло, и распыляют в жидком азоте, чтобы заморозить капли. Жидкому азоту затем дают возможность испариться при температуре -80°С, и полученный материал сушат сублимацией.
Микрочастицы, микросферы и микрокапсулы представляют собой твердые или полутвердые частицы с диаметром меньшим, чем один миллиметр, более предпочтительно, меньшим, чем 100 мкм, и наиболее предпочтительно, меньшим, чем 10 мкм, которые могут быть образованы из различных материалов, включая в себя протеины, синтетические полимеры, полисахариды и их комбинации. Микросферы были использованы по многим различным назначениям, главным образом, при разделении, диагностике и доставке лекарства.
Наиболее хорошо известные примеры микросфер, используемых в технологиях разделения, представляют собой микросферы, образованные полимерами синтетического или природного происхождения, такими как полиакриламид, гидроксиапатит или агароза. В области лекарств с контролируемой доставкой молекулы часто объединены или инкапсулированы в небольшие сферические частицы или объединены в монолитную матрицу для последующего освобождения. Ряд различных технологий регулярно используется для получения указанных микросфер из синтетических полимеров, натуральных полимеров, протеинов и полисахаридов, включая в себя фазовое разделение, испарение растворителя, коацервацию, эмульгирование и распылительную сушку. Обычно полимеры образуют структуру носителя для данных микросфер, и нужный лекарственный препарат заключается в полимерную структуру.
В настоящее время доступны частицы, приготовленные с использованием липидов, чтобы инкапсулировать целевые лекарственные препараты. Липосомы представляют собой сферические частицы, состоящие из одного или множества фосфолипидных и/или холестериновых бислоев. Липосомы имеют размер, равный 100 нанометрам или больше, и могут нести множество водорастворимых или липидорастворимых лекарственных препаратов. Например, липиды, организованные в двухслойные мембраны, окружающие множество водных отделений, чтобы образовать частицы, могут быть использованы для инкапсулирования водорастворимых лекарственных препаратов для последующей доставки, как описано в патенте U.S. 5422120, Sinil Kim.
Сферические бусины коммерчески доступны как инструмент для биохимиков в течение многих лет. Например, антитела, соединенные с бусинами, создают относительно большие частицы, которые специфически связываются с конкретными лигандами. Антитела, регулярно используемые для связывания с рецепторами на поверхности клетки для клеточной активации, привязывают к твердой фазе, чтобы образовать частицы, покрытые антителами, для иммуноаффинной очистки, и могут быть использованы для доставки терапевтических агентов, которые медленно выделяются с течением времени, используя ткань или опухолеспецифические антитела, присоединенные к частицам, чтобы доставить агент к желаемому участку.
В настоящее время имеется острая необходимость развития новых способов получения частиц, особенно тех, которые могут быть приспособлены для использования в областях доставки лекарственного препарата, разделения и диагностики. Наиболее желательными частицами с точки зрения использования, являлись бы небольшие сферические частицы со следующими характеристиками: ограниченный гранулометрический состав, по существу, сферические, по существу, состоящие только из активного агента, сохранение биохимической целостности и биологической активности активного агента. Частицы должны обеспечивать подходящую твердость, что дает дополнительную стабилизацию частиц при нанесении покрытия или при микроинкапсулировании. Кроме того, способ изготовления небольших сферических частиц должен обладать следующими желательными характеристиками: простота изготовления, по существу, водный способ, высокий выход и не требующий последующего просеивания.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам производства и способам применения небольших сферических частиц активного агента. В соответствии со способом активный агент растворяют в растворителе, содержащем усиливающий фазовое разделение агент, чтобы образовать раствор, который представляет собой единственную жидкую фазу. Растворитель представляет собой, предпочтительно, водный или смешивающийся с водой растворитель. Раствор затем подвергают фазовому разделению жидкость-твердое, причем имеющийся активный агент содержится в твердой фазе, а УФРА и растворитель содержатся в жидкой фазе. Фазовое разделение жидкость-твердое может быть вызвано рядом способов, например, изменением температуры раствора ниже температуры фазового перехода раствора.
В предпочтительном варианте осуществления настоящего изобретения используется способ фазового разделения раствора жидкость-твердое охлаждением раствора ниже температуры фазового перехода активного агента в растворе. Температура может быть выше или ниже температуры замерзания раствора. Для растворов, в которых температура замерзания выше температуры фазового перехода, раствор может включать в себя агент, понижающий температуру замерзания, например, полиэтиленгликоль или полипропиленгликоль, чтобы понизить температуру замерзания раствора и позволить произойти фазовому разделению в растворе без замерзания раствора.
Усиливающий фазовое разделение агент по настоящему изобретению усиливает или вызывает фазовое разделение жидкость-твердое активного агента в растворе, когда раствор подвергается этапу фазового изменения, в котором активный агент затвердевает, образуя суспензию небольших сферических частиц как дискретную фазу, в то время как усиливающий фазовое разделение агент остается растворенным в непрерывной фазе. То есть, усиливающий фазовое разделение агент не изменяет фазы, в то время как активный агент изменяет фазу.
Способ производства частиц по настоящему изобретению может также включать в себя дополнительный этап управления фазовым разделением частиц жидкость-твердое, чтобы регулировать размер и форму образованных частиц. Способы управления фазовым разделением включают в себя регулирование ионной силы, pH, концентрации усиливающего фазовое разделение агента, концентрации активного агента в растворе или регулирование скорости изменения температуры раствора, причем регулирование указанных параметров проводят или перед фазовым разделением или изменяют один или несколько из них для того, чтобы вызвать фазовое разделение.
В предпочтительном варианте осуществления настоящего изобретения небольшие сферические частицы отделяют от УФРА в непрерывной фазе после образования частиц. В другом предпочтительном варианте осуществления способ разделения представляет собой промывание раствора, содержащего частицы, жидкостью, в которой активный агент не растворим, в то время как усиливающий фазовое разделение агент растворим в жидкости. Промывная жидкость может содержать агент, который уменьшает растворимость активного агента в жидкости. Промывная жидкость может также содержать один или более наполнитель. Наполнитель может действовать как стабилизатор для небольших сферических частиц или для активного агента, или для несущего агента. Наполнитель может также придавать активному агенту или частице дополнительные характеристики, такие как контролируемое выделение активного агента из частиц, или модифицированное проникновение активного агента через биологические ткани.
В другом предпочтительном варианте осуществления, несмотря на то, что небольшие частицы не включают в себя УФРА, они могут быть образованы в присутствии фазы УФРА для последующих этапов обработки до отделения от фазы УФРА.
В другом предпочтительном варианте осуществления раствор представляет собой водный раствор, содержащий водный или смешивающийся с водой растворитель.
Активный агент настоящего изобретения представляет собой, предпочтительно, фармацевтически активный агент, который может быть терапевтическим агентом, диагностическим агентом, косметическим веществом, пищевой добавкой или пестицидом. В предпочтительном варианте осуществления настоящего изобретения активный агент представляет собой макромолекулу, такую как протеин, полипептид, углевод, полинуклеотид или нуклеиновая кислота. В еще одном предпочтительном варианте осуществления частицы, содержащие активный агент, являются подходящими для доставки in vivo агента к необходимому объекту соответствующим путем, таким как парентеральная инъекция, топически, орально, ректально, через легкие, вагинально, через щеку, под язык, подкожно, через слизистые оболочки, через глаза, внутри глаз или через уши.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 представляет собой двумерную фазовую диаграмму зависимости концентрации активного компонента от температуры.
Фиг.2 представляет собой профиль уменьшения температуры, иллюстрирующий влияние температуры раствора и скорости охлаждения на фазовое изменение инсулина в буферном растворе полимера. При температуре выше 60°С инсулин остается в растворе (область А). Область В представляет собой площадь оптимального образования небольших сферических частиц, ограниченную наиболее высокой и наиболее низкой скоростью изменения температуры, наблюдаемой в теплообменнике. Большие скорости охлаждения (область С) приводят к образованию очень мелких несферических частиц, в то время как низкие скорости охлаждения (область D) приводят к смеси небольших сферических частиц различных размеров, наряду с частицами неправильной формы и рыхлого осадка.
Фиг.3а представляет собой изображение исходного материала инсулина, полученное сканирующим электронным микрографом (СЭМ).
Фиг.3b представляет собой изображение (СЭМ) небольших сферических частиц инсулина (пример 4).
Фиг.4 представляет собой ВЭЖХ анализ, показывающий сохранение химической стабильности инсулина при приготовлении небольших сферических частиц, иллюстрирующий химическую стабильность во время способа изготовления микросфер инсулина.
ВЭЖХ анализ показал отсутствие увеличения высокомолекулярных соединений, связанного со способом, и увеличение (относительно исходного сырья инсулина) % димера, % А21 дезамидоинсулина, % поздно элюируемых пиков и % других соединений в пределах USP.
Фиг.5 представляет собой схематическую демонстрацию воспроизводимости от порции к порции. Фигура показывает распределение небольших сферических частиц инсулина (время-пролетные данные Aerosizer). Для всех шести полученных партий больше чем 96% частиц попали между 0,86 и 2,9 мкм, с более 60% попавшими между 1,6 и 2,5 мкм. Меньше чем 1,1% небольших сферических частиц попали за пределы размеров, охватываемых диаграммой.
Фиг.6 представляет собой схематическую демонстрацию воспроизводимости от порции к порции. Фигура показывает распределение небольших сферических частиц инсулина с использованием каскадного импактора Андерсена. Данные являются средними (означает+/-КО (квадратичное отклонение)) результатами для шести партий небольших сферических частиц инсулина, полученных на устройстве Cyclohaler при 60 ЛВМ. ЭПД для ступеней 1, 2, 3 и 4 составляло 4,4; 3,3; 2,0 и 1,1 мкм соответственно.
Фиг.7 представляет собой схематическую диаграмму непрерывного проточного способа получения небольших сферических частиц инсулина в примере 3.
Фиг.8 представляет собой изображение (сканирующий электронный микрограф (при 10 Кв и 6260Х увеличении)) небольших сферических частиц инсулина, полученных в непрерывном проточном способе примера 3.
Фиг.9 представляет собой ВЭЖХ хроматограмму растворенных небольших сферических частиц инсулина, полученных в способе с непрерывным потоком примера 3.
Фиг.10а демонстрирует влияние хлорида натрия (при 2,5 мг/мл) на растворимость инсулина в объеме пробирки (NaCl против температуры). На фигуре представлены следующие данные (подъем от 60°С):
Фиг.10b демонстрирует влияние хлорида натрия (при 5 мг/мл) на растворимость инсулина в объеме пробирки (NaCl против температуры). На фигуре представлены следующие данные (подъем от 60°С):
Фиг.10с демонстрирует влияние хлорида натрия (при 10 мг/мл) на растворимость инсулина в объеме пробирки (NaCl против температуры). На фигуре представлены следующие данные (подъем от 40°С):
Фиг.10d демонстрирует влияние хлорида натрия (при 20 мг/мл) на растворимость инсулина в объеме пробирки (NaCl против температуры). На фигуре представлены следующие данные (подъем от 40°С):
Фиг.10е-10h демонстрируют влияние солей на растворимость инсулина.
Фиг.10i представляет собой спектр КР сырого материала инсулина, инсулина, выделенного из небольших сферических частиц, и инсулина в небольших сферических частицах. Приведен КР спектр в области амидной полосы I для необработанного порошка инсулина и порошка из небольших сферических частиц, а также из соответствующих растворов.
Фиг.11 представляет собой результаты, полученные на каскадном импакторе Андерсена для радиомеченого инсулина примера 10. Результаты с каскадного импактора Андерсена для 99mТc радиомеченого порошка инсулина показывают устойчивую связь 99тТc с инсулином перед тем, как первой собаке ввели дозу, и после того, как последнему животному была доставлена доза.
Фиг.12 представляет собой гистограмму отношения P/I для примера 8, показывающую среднее значение соотношения P/I, равное 0,93, для пяти проверенных собак.
Фиг.13 представляет собой сцинтиграфический вид легкого из примера 8, где 99mТc радиомеченый инсулин был гомогенизированно распределен на периферии легких. Нет визуального подтверждения осаждения в центре легких. Это поддерживает унимодальное распределение по размеру небольших сферических частиц инсулина после введения собакам.
Фиг.14а представляет собой график кругового дихроизма (КД) для альфа-1-антитрипсина (ААТ).
Фиг.14b представляет собой график зависимости активности от времени хранения при комнатной температуре в примере 17.
Фиг.14 с представляет собой график зависимости активности от времени хранения при 4°С в примере 17.
Фиг.15 представляет собой график ДСК, показывающий первую термограмму нагрева 50 мг/мл рААТ образца в BDS растворе против BDS как эталона.
Фиг.16 представляет собой график ДСК, показывающий первую термограмму охлаждения 50 мг/мл рААТ образца в BDS растворе против BDS как эталона.
Фиг.17 представляет собой график ДСК, показывающий вторую термограмму нагрева 50 мг/мл рААТ образца в BDS растворе против BDS как эталона.
Фиг.18 представляет собой график ДСК, показывающий вторую термограмму охлаждения 50 мг/мл рААТ образца в BDS растворе против BDS как эталона.
Фиг.19 представляет собой график ДСК, показывающий вторую термограмму нагрева 45 мг/мл рААТ образца в растворе ацетата против ацетатного буфера как эталона.
Фиг.20 представляет собой график ДСК, показывающий вторую термограмму охлаждения 45 мг/мл рААТ образца в растворе ацетата против ацетатного буфера как эталона.
Фиг.21 представляет собой график ДСК, показывающий вторую термограмму нагрева 1 мг/мл рААТ образца в BDS растворе против BDS буфера как эталона. рААТ образец получен при растворении 3 небольших сферических частиц рААТ в BDS.
Фиг.22 представляет собой график ДСК, показывающий вторую термаграмму охлаждения 1 мг/мл рААТ образца в BDS растворе против BDS буфера как эталона, рААТ образец получен при растворении 3 небольших сферических частиц рААТ в BDS.
Фиг.23 представляет собой график ДСК, показывающий первую термограмму охлаждения небольших сферических частиц изготовленной партии.
Фиг.24 представляет собой график ДСК, показывающий первую термограмму нагрева небольших сферических частиц изготовленной партии.
Фиг.25a представляет собой график ДСК, показывающий вторую термограмму охлаждения небольших сферических частиц полученной партии.
Фиг.25b представляет собой график ДСК, показывающий вторую термограмму нагрева небольших сферических частиц изготовленной партии.
Фиг.26 представляет собой график данных по размеру частиц (TSI Corporation Aerosizer).
Фиг.27 представляет собой СЭМ небольших сферических частиц гормона роста человека (ГРЧ).
Фиг.28 представляет собой график, показывающий, что инсулин в форме микросферы сохраняет стабильность после хранения в газе-вытеснителе HFA 134а.
Фиг.29 представляет собой график сравнения аэродинамического качества инсулина с использованием трех устройств для ингаляции, где аэродинамические качества небольших сферических частиц инсулина сравнивали с использованием трех различных приборов для ингаляции: ИИД, Cyclohaler ИСП, Disphaler ИСП.
Фиг.30 представляет собой график данных по стабильности небольших сферических частиц инсулина в сравнении с исходным инсулином, хранящимся при 25°С, показывающий, что при хранении при температуре 25°С процент образования А21-дезамидоинсулина из исходного материала значительно больше по сравнению с образованием из небольших сферических частиц инсулина. Данный результат показывает, что небольшие сферические частицы инсулина значительно более устойчивы к химическому разрушению, чем исходный материал, без добавления стабилизирующих наполнителей.
Фиг.31 представляет собой график данных по стабильности небольших сферических частиц инсулина в сравнении с исходным инсулином, хранящимся при 37°С, показывающий, что при хранении при температуре 37°С процент образования А21-дезамидоинсулина из исходного материала значительно больше по сравнению с образованием из небольших сферических частиц инсулина. Данный результат показывает, что небольшие сферические частицы инсулина значительно более устойчивы к химическому разрушению, чем исходный материал, без добавления стабилизирующих наполнителей.
Фиг.32 представляет собой график данных по стабильности небольших сферических частиц инсулина в сравнении с исходным инсулином, хранящимся при 25°С, показывающий, что при хранении при температуре 25°С процент образования димера и олигомера инсулина из исходного материала значительно больше по сравнению с образованием из небольших сферических частиц инсулина. Данный результат показывает, что небольшие сферические частицы инсулина значительно более устойчивы к химическому разрушению, чем исходный материал, без добавления стабилизирующих наполнителей.
Фиг.33 представляет собой график данных по стабильности небольших сферических частиц инсулина в сравнении с исходным инсулином, хранящимся при 37°С, показывающий, что при хранении при температуре 37°С процент образования димера и олигомера инсулина из исходного материала значительно больше по сравнению с образованием из небольших сферических частиц инсулина. Данный результат показывает, что небольшие сферические частицы инсулина значительно более устойчивы к химическому разрушению, чем исходный материал, без добавления стабилизирующих наполнителей.
Фиг.34 представляет собой график данных по стабильности небольших сферических частиц инсулина в сравнении с исходным инсулином, хранящимся при 25°С, показывающий, что при хранении при температуре 25°С процент образования суммарных соединений, родственных инсулину, из исходного материала значительно больше по сравнению с образованием из небольших сферических частиц инсулина. Данный результат показывает, что небольшие сферические частицы инсулина значительно более устойчивы к химическому разрушению, чем исходный материал, без добавления стабилизирующих наполнителей.
Фиг.35 представляет собой график данных по стабильности небольших сферических частиц инсулина в сравнении с исходным инсулином, хранящимся при 37°С, показывающий, что при хранении при температуре 37°С процент образования суммарных соединений, родственных инсулину, из исходного материала значительно больше по сравнению с образованием из небольших сферических частиц инсулина. Данный результат показывает, что небольшие сферические частицы инсулина значительно более устойчивы к химическому разрушению, чем исходный материал, без добавления стабилизирующих наполнителей.
Фиг.36 представляет собой гистограмму аэродинамической стабильности небольших сферических частиц инсулина с использованием ингалятора сухого порошка (ИСП) Cyclohaler в каскадном импакторе Андерсена.
Фиг.37 представляет собой световую микрофотографию небольших сферических частиц ДНКазы (световой микроскоп Nikon, 100x масляная иммерсионная линза).
Фиг.38 представляет собой график ферментативной активности ДНКазы.
Фиг.39 представляет собой световую микрофотографию небольших сферических частиц SOD (световой микроскоп Nikon, 100x масляная иммерсионная линза, образец сухого порошка).
Фиг.40 представляет собой график ферментативных данных для небольших сферических частиц SOD (методика определения активности описана в Worthington Biochemical Catalogue).
Фиг.41А-В представляют собой схематические иллюстрации реактора непрерывного эмульгирования, где фиг.41А представляет собой схематическую иллюстрацию реактора непрерывного эмульгирования, когда поверхностно-активное соединение добавлено в непрерывную фазу или в дисперсную фазу перед эмульгированием, и фиг.41В представляет собой схематическую иллюстрацию реактора непрерывного эмульгирования, когда поверхностно-активное соединение добавлено после эмульгирования.
Фиг.42 иллюстрирует влияние PEG на IVR профиль PLLA-инкапсулированных частиц HSA (пример 32).
Фиг.43 иллюстрирует IVR профиль PLGA-инкапсулированных небольших сферических частиц LDS (пример 33).
Фиг.44 иллюстрирует влияние рН непрерывной фазы на IVR профиль PLGA-инкапсулированных небольших сферических частиц инсулина (пример 31).
Фиг.45 иллюстрирует IVR профиль PLGA-инкапсулированных небольших сферических частиц ГРЧ (пример 34).
Фиг.46 иллюстрирует влияние переменных микроинкапсулирования (рН непрерывной фазы и материал матрицы) на образование димеров ИНС в инкапсулированном ИНСмс (пример 35).
Фиг.47 иллюстрирует влияние переменных микроинкапсулирования (рН непрерывной фазы и материал матрицы) на образование ВМ частиц в инкапсулированном ИНСмс (пример 35).
Фиг.48 иллюстрирует in vivo выделение рекомбинантного инсулина человека из не инкапсулированных и инкапсулированных предварительно изготовленных небольших сферических частиц инсулина у крыс (пример 36).
Фиг.49 представляет собой СЭМ при 10 кВ частиц примера 27.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение включает варианты осуществления во многих различных формах. Предпочтительные варианты осуществления изобретения излагаются с пониманием того, что настоящее изложение должно рассматриваться как ряд примеров принципа изобретения без намерения ограничить широкие аспекты изобретения проиллюстрированными вариантами осуществления.
Настоящее изобретение относится к способам производства, способам применения и композиции небольших сферических частиц активного агента. В соответствии со способом производства активный агент растворяют в растворителе, содержащем растворенный агент, усиливающий фазовое разделение, чтобы образовать раствор, который представляет собой единственную непрерывную жидкую фазу. Растворитель, предпочтительно, представляет собой водный или смешивающийся с водой растворитель. Раствор далее подвергается фазовому изменению, например, понижением температуры раствора ниже температуры фазового перехода активного агента, таким образом, активный агент проходит через фазовое разделение жидкость-твердое с образованием суспензии небольших сферических частиц, составляющих дискретную фазу, в то время как усиливающий фазовое разделение агент остается в непрерывной фазе.
Фазы
Непрерывная фаза
Способ приготовления небольших сферических частиц активного агента по настоящему изобретению начинается с получения раствора, имеющего активный агент и усиливающий фазовое разделение агент, растворенный в первом растворителе в единственной жидкой фазе. Раствор может быть органической системой, содержащей органический растворитель или смесь смешивающихся органических растворителей. Раствор может также быть раствором, основанным на воде, содержащим водную среду или смешивающийся с водой органический растворитель, или смесь смешивающихся с водой органических растворителей, или их комбинации. Водная среда может быть водой, раствором соли или буферным раствором, буферным раствором соли и тому подобное. Подходящие смешивающиеся с водой органические растворители включают в себя, но не ограничиваются ими, N-метил-2-пирролидинон (N-метил-2-пирролидон), 2-пирролидинон (2-пирролидон), 1,3-диметил-2-имидазолидинон (ДМИ), диметилсульфоксид, диметилацетамид, уксусную кислоту, молочную кислоту, ацетон, метилэтилкетон, ацетонитрил, метанол, этанол, изопропанол, 3-пентанол, н-пропанол, бензиловый спирт, глицерин, тетрагидрофуран (ТГФ), полиэтиленгликоль (ПЭГ), ПЭГ-4, ПЭГ-8, ПЭГ-9, ПЭГ-12, ПЭГ-14, ПЭГ-16, ПЭГ-120, ПЭГ-75, ПЭГ-150, сложные эфиры полиэтиленгликоля, ПЭГ-4 дилаурат, ПЭГ-20 дилаурат, ПЭГ-6 изостеарат, ПЭГ-8 пальмитостеарат, ПЭГ-150 пальмитостеарат, полиэтиленгликольсорбитаны, ПЭГ-20 сорбитанизостеарат, моноалкиловые эфиры полиэтиленгликоля, ПЭГ-3 диметиловый эфир, ПЭГ-4 диметиловый эфир, полипропиленгликоль (ППГ), полипропиленальгинат, ППГ-10 бутандиол, ППГ-10 метиловый эфир глюкозы, ППГ-20 метиловый эфир глюкозы, ППГ-15 стеариловый эфир, пропиленгликоль дикаприлат/дикапрат, пропиленгликольлаурат и гликофурол (тетрагидрофурфуриловый спирт эфир пропиленгликоля), алканы, включающие в себя пропан, бутан, пентан, гексан, гептан, октан, нонан, декан или их комбинации.
Единственная непрерывная фаза может быть получена при первом приготовлении раствора усиливающего фазовое разделение агента, который или растворим, или смешивается с первым растворителем. За этим следует добавление в раствор активного агента. Активный агент может быть добавлен непосредственно в раствор или активный агент может быть сначала растворен во втором растворителе и затем прибавлен к раствору. Второй растворитель может быть тем же самым растворителем, что и первый растворитель, или это может быть другой растворитель, выбранный из перечисленных выше, и который смешивается с раствором. Предпочтительно, чтобы агент добавляли к раствору при температуре окружающей среды или ниже, что особенно важно для термолабильных молекул, таких как некоторые протеины. Температура «окружающей среды» означает температуру около комнатной температуры, от примерно 20°С до примерно 40°С. Однако система также может быть нагрета для увеличения растворимости активного агента в системе настолько, пока нагревание не вызывает значительного уменьшения активности агента.
Усиливающий фазовое разделение агент
Усиливающий фазовое разделение агент (УФРА) по настоящему изобретению усиливает или вызывает фазовое разделение жидкость-твердое активного агента из раствора, когда раствор проходит этап фазового разделения, в котором активный агент становится твердым или почти твердым с образованием суспензии небольших сферических частиц в качестве дискретной фазы, в то время как усиливающий фазовое разделение агент остается растворенным в непрерывной фазе. Усиливающий разделение фаз агент уменьшает растворимость активного агента, когда раствор находится в условиях фазового разделения. Подходящие усиливающие фазовое разделение агенты включают в себя, но не ограничиваются ими, полимеры или смеси полимеров, которые растворимы или смешиваются с раствором. Примеры подходящих полимеров включают в себя линейные или разветвленные полимеры. Указанные полимеры могут быть водорастворимыми, частично растворимыми в воде, смешивающимися с водой или нерастворимыми.
В предпочтительной форме изобретения усиливающий фазовое разделение агент является водорастворимым или смешивается с водой. Типы полимеров, которые могут быть использованы, включают в себя полимеры на основе углеводов, полиалифатические спирты, поли(винил)полимеры, полиакриловые кислоты, полиорганические кислоты, полиаминокислоты, сополимеры и блок сополимеры (например, полоксамеры, такие как Pluronics F127 или F68) трет-полимеры, полиэфиры, полимеры, встречающиеся в природе, полиимиды, поверхностно-активные вещества, сложные полиэфиры, разветвленные и циклические полимеры и полиальдегиды.
Предпочтительные полимеры представляют собой полимеры, которые допустимы как фармацевтические добавки для намеченного пути введения частиц активного агента. Предпочтительные полимеры представляют собой фармацевтически допустимые добавки, такие как полиэтиленгликоль (ПЭГ) с различным молекулярным весом, такой как ПЭГ 200, ПЭГ 300, ПЭГ 3350, ПЭГ 8000, ПЭГ 10000, ПЭГ 20000 и т.д., и полоксамеры, такие как Pluronics F127 или Pluronics F68. Другой предпочтительный полимер представляет собой поливинилпирролидон (ПВП). Еще один предпочтительный полимер представляет собой гидроксиэтилкрахмал. Другие амфифильные полимеры также могут использоваться по отдельности или в комбинациях. Усиливающий фазовое разделение агент может также не быть полимером, например, смесь пропиленгликоля и этанола.
Фазовое разделение жидкость-твердое
Фазовое разделение жидкость-твердое активного агента в растворе может быть вызвано любым способом, известным в данной области, например, изменение температуры, изменение давления, изменение pH, изменение ионной силы раствора, изменение концентрации активного агента, изменение концентрации усиливающего фазовое разделение агента, изменение осмотического давления раствора, комбинация указанных факторов и тому подобное.
В предпочтительном варианте осуществления настоящего изобретения фазовое изменение представляет собой температурно индуцированное фазовое изменение понижением температуры ниже температуры фазового перехода активного агента в растворе.
Фиг.1 представляет собой двумерную фазовую диаграмму 10 для раствора, содержащего растворитель, УФРА и активный агент. Диаграмма изображает зависимость концентрации активного агента от температуры раствора. Концентрацию УФРА поддерживают постоянной.
Диаграмма содержит кривую насыщения 12, кривую перенасыщения 14; метастабильную площадь 16 между ними; первую площадь 18 ниже кривой насыщения, где система представляет собой гомогенную единую жидкую фазу, где все компоненты находятся в жидкой фазе; и вторую площадь 20 выше кривой перенасыщения, где система представляет собой двухфазную систему, имеющую твердую фазу активного агента и жидкую фазу УФРА и растворителя. Фазовая диаграмма является полезной для определения температуры системы и относительной концентрации компонентов в чистой жидкой фазе, в фазе жидкость-твердое и условий перехода между двумя данными фазами.
Как здесь изложено, приготовление небольших сферических частиц активного агента принципиально включает в себя вымораживание из ненасыщенного раствора (точка А') с достижением насыщения в точке А, где раствор находится в равновесии с любой твердой фазой, которая может присутствовать. При дополнительном охлаждении достигается состояние, когда раствор содержит больше активного агента, чем это соответствует равновесной растворимости при данной температуре; раствор, таким образом, становится перенасыщенным. Самопроизвольного образования твердой фазы не происходит, пока не будет достигнута точка В. Данная точка В представляет собой границу метастабильной зоны. Ширина метастабильной зоны может быть выражена или по максимуму достижимого переохлаждения ∆Тмакс=T2-T1 или перенасыщению ∆Смакс=С*2-C*1. Данные два выражения термодинамически эквивалентны:
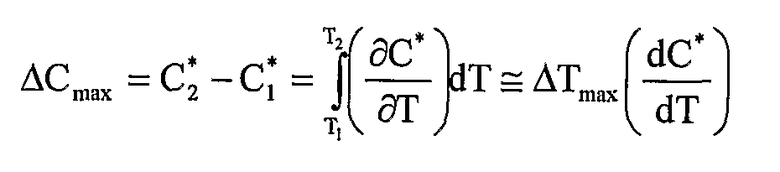
Путь А'-А-В представляет собой политермический способ получения метастабильного раствора. В изотермическом способе исходной точкой была бы А''. Увеличением концентрации при постоянной температуре насыщение будет снова достигнуто в точке А. Изотермическое увеличение концентрации (например, испарением растворителя или введением затравки/прибавлением активного агента) до точки С вызовет сдвиг раствора в метастабильное состояние, пока метастабильный предел не будет снова достигнут. Когда метастабильный предел превышен, раствор становится неустойчивым, и немедленно происходит самопроизвольное образование твердой фазы.
Значение (∆Смакс)Т=С*3-C*2, полученное изотермически, может быть отличным от соответствующего значения ∆Тмакс=T3-T2, полученного политермически. Когда приближается граница метастабильной зоны, уменьшается время, необходимое для образования твердой частицы, пока метастабильный предел не будет достигнут.
В политермическом способе охлаждение проводят при контролируемой скорости, чтобы регулировать размер и форму частиц. Под контролируемой скоростью понимают скорость примерно от 0,2°С/минуту до примерно 50°С/минуту, и, более предпочтительно, от 0,2°С/минуту до 30°С/минуту. Скорость изменения может быть постоянной или линейной скоростью, нелинейной скоростью, неравномерной или программируемой скоростью (с наличием множества фазовых циклов).
Частицы могут быть отделены от УФРА в растворе и очищены промыванием, как это будет рассмотрено ниже.
В настоящем изобретении рассмотрено регулирование концентрации активного агента, концентрации УФРА, температуры или любой комбинации указанных параметров, чтобы вызвать фазовое изменение, при котором активный агент переходит из жидкого состояния в твердое состояние, в то время как УФРА и растворитель не проходят через фазовое изменение и остаются жидкостями. Также рассмотрено изменение pH, ионной силы, осмотического давления и тому подобного, чтобы усиливать, промотировать, контролировать или подавлять фазовое изменение. Для растворов, у которых температура замерзания относительно высока или температура замерзания выше температуры фазового перехода, раствор может включать в себя агент, понижающий температуру замерзания, например, пропиленгликоль, сахароза, этиленгликоль, спирты (например, этанол, метанол) или водные смеси агентов, понижающих температуру замерзания, чтобы понизить температуру замерзания системы и сделать возможным фазовое изменение системы без замерзания системы. Способ также может быть проведен таким образом, что температуру понижают ниже температуры замерзания системы. Способ, описанный здесь, является особенно подходящим для молекул, которые являются термолабильными (например, протеины).
Возможные наполнители
Частицы по настоящему изобретению могут включать в себя один или более наполнитель. Наполнитель может придавать активному агенту или частице дополнительные характеристики, такие как увеличение стабильности частиц активного агента или несущих агентов, контролируемое выделение активного агента из частиц или модифицирование проникновения активного агента через биологические ткани. Подходящие наполнители включают в себя, но не ограничиваются ими, углеводы (например, трегалоза, сахароза, маннит), катионы (например, Zn2+, Mg2+, Ca2+), анионы (например, SO4 2-), аминокислоты (например, глицин), липиды, фосфолипиды, жирные кислоты, поверхностно-активные вещества, триглицериды, кислоты желчи или их соли (например, холат или его соли, такие как холат натрия; дезоксихолиевая кислота или ее соли), сложные эфиры жирных кислот и полимеры, присутствующие на уровне ниже их функционирования в качестве УФРА. Когда используют наполнитель, наполнитель не влияет значительно на фазовую диаграмму раствора.
Разделение и промывка частиц
В предпочтительном варианте осуществления по настоящему изобретению небольшие сферические частицы собирают, отделяя их от усиливающего фазовое разделение агента в растворе. В еще одном предпочтительном варианте осуществления способ разделения представляет собой промывку раствора, содержащего небольшие сферические частицы, жидкой средой, в которой активный агент не растворим, в то время как усиливающий фазовое разделение агент растворим. Некоторые способы промывки могут быть диафильтрацией или центрифугированием. Жидкая среда может быть водной средой или органическим растворителем. Для активных агентов с низкой растворимостью в воде жидкая среда может быть водной средой или водной средой, содержащей агенты, которые уменьшают растворимость в воде активного агента, такие как двухвалентные катионы. Для активных агентов с высокой растворимостью в воде, таких как многие протеины, могут быть использованы органические растворители или водные растворители, содержащие агент, осаждающий белок, такие как сульфат аммония.
Примеры подходящих органических растворителей для использования в качестве жидкой среды включают в себя органические растворители, охарактеризованные выше как подходящие для непрерывной фазы, и более предпочтительны метиленхлорид, хлороформ, ацетонитрил, этилацетат, метанол, этанол, пентан и тому подобное.
Также рассмотрено использование смесей любых указанных растворителей. Одна предпочтительная смесь представляет собой метиленхлорид или 1:1 смесь метиленхлорида и ацетона. Предпочтительно, чтобы жидкая среда имела низкую температуру кипения для легкого удаления, например, лиофилизацией, упариванием или высушиванием.
Жидкая среда может также быть надкритической жидкостью, например, жидкий диоксид углерода, или жидкостью вблизи ее критической точки. Надкритические жидкости могут быть подходящими растворителями для усиливающих фазовое разделение агентов, в частности, некоторые полимеры, но не являются растворителями для частиц белка. Надкритические жидкости могут использоваться самостоятельно или с сорастворителем. Могут быть использованы следующие надкритические жидкости: жидкий СО2, этан или ксенон. Потенциальными сорастворителями могут быть ацетонитрил, дихлорметан, этанол, метанол, вода или 2-пропанол.
Жидкая среда, используемая для отделения небольших сферических частиц от УФРА, описанная здесь, может содержать агент, который понижает растворимость активного агента в жидкой среде. Наиболее желательно, чтобы частицы проявляли минимальную растворимость в жидкой среде, чтобы максимально увеличить выход частиц. Для некоторых протеинов, таких как инсулин или гормон роста человека, уменьшение растворимости может быть достигнуто добавлением двухвалентных катионов, таких как Zn2+, к протеину. Другие ионы, которые могут быть использованы для образования комплексов, включают в себя, но не ограничиваются ими, Ca2+, Cu2+, Fe2+, Fe3+ и аналогичные.
Растворимость комплексов инсулин-Zn или гормон роста-Zn достаточно низкая, что делает возможной диафильтрацию комплекса в водных растворах.
Жидкая среда может также содержать один или более наполнитель, который может снабжать активный агент или частицу дополнительными свойствами, такими как увеличение стабильности частиц и/или активного или несущего агентов, контролируемое выделение активного агента из частиц, или модификацию проникания активного агента через биологические ткани, как обсуждено ранее.
В других формах изобретения небольшие сферические частицы не отделяют от раствора, содержащего УФРА.
Способ, основанный на водной системе
В другом предпочтительном варианте осуществления способа получения настоящая система представляет собой водную систему, включающую в себя водный или смешивающийся с водой растворитель. Примеры подходящих, смешивающихся с водой растворителей включают в себя, но не ограничиваются ими, те, которые идентифицированы выше для непрерывной фазы. Преимуществом использования основанного на водной системе способа является то, что раствор может быть буферным и может содержать наполнители, которые обеспечивают биохимическую стабилизацию для защиты активных агентов, таких как протеины.
Активный агент
Активный агент по настоящему изобретению, предпочтительно, представляет собой фармацевтически активный агент, который может быть терапевтическим агентом, диагностическим агентом, косметическим средством, пищевой добавкой или пестицидом.
Терапевтический агент может быть биологическим веществом, которое включает в себя, но не ограничивается ими, протеины, полипептиды, углеводы, полинуклеотиды и нуклеиновые кислоты. Протеин может быть антителом, которое может быть поликлональным или моноклональным. Терапевтический агент может быть низкой молекулярной массы. Кроме того, терапевтические агенты могут быть выбраны из множества известных фармацевтических веществ, но не ограничены ими: анальгетики, анестезирующие средства, аналептики, адренергические агенты, адренергические блокирующие агенты, адренолитики, адренокортикоиды, адреномиметики, антихолинергичесие агенты, антихолинэстеразы, антиконвульсанты, алкилирующие агенты, алколоиды, аллостерические ингибиторы, анаболические стероиды, анорексанты, антациды, агенты против диареи, антидоты, антифолики, антипиретики, антиревматические агенты, психотерапевтические агенты, невральные блокирующие агенты, противовоспалительные агенты, антигельминтики, противоаритмические агенты, антибиотики, антикоагулянты, антидепрессанты, противодиабетические агенты, антиэпилептики, противогрибковые агенты, антигистамины, противогипертонические агенты, антимускариновые агенты, антимикобактериальные агенты, противомалярийные агенты, антисептики, антинеопластические агенты, антипротозоальные агенты, иммуносуппрессанты, иммуностимуляторы, антитиреоидные агенты, противовирусные агенты, транквилизаторы, астрингенты, бета-адреноцепторные блокирующие агенты, контрастные средства, кортикостероиды, противокашлевый агент, диагностические агенты, диагностические воспроизводящие агенты, диуретики, допаминергики, гемостатики, гематологические агенты, модификаторы гемоглобина, гормоны, гипнотики, иммуриологические агенты, антигиперлипидемики и другие агенты, регулирующие липиды, мускариники, мышечные релаксаторы, парасимпатомиметики, паратиреоидный гормон, кальцитонин, простагландины, радиофармацевтические агенты, седативные агенты, половые гормоны, антиаллергические агенты, стимуляторы, симпатомиметики, тиреоидные агенты, сосудорасширяющие агенты, вакцины, витамины и ксантины. Антинеопластик или противоопухолевые агенты включают в себя, но не ограничиваются ими, paclitaxel и производные соединения и другие антинеопластики, выбранные из группы, состоящей из алкалоидов, антиметаболитов, ингибиторов ферментов, алкилирующих агентов и антибиотиков.
Косметический агент представляет собой любой активный ингредиент, способный иметь косметическую активность. Примерами указанных активных ингредиентов могут быть, например, смягчающие агенты, увлажнители, агенты, ингибирующие свободные радикалы, противовоспалительные агенты, витамины, депигментационные агенты, противоугревые агенты, агенты против себореи, кератолитики, агенты для похудения, окрашивающие кожу агенты и солнцезащитные агенты, и, в частности, линолевая кислота, ретинол, ретиновая кислота, сложные алкиловые эфиры аскорбиновой кислоты, полиненасыщенные жирные кислоты, никотиновые сложные эфиры, токоферолникотинат, неомыляемые остатки риса, соевые или масляного дерева, керамиды, гидроксикислоты, такие как гликолевая кислота, производные селена, антиоксиданты, бета-каротин, гамма-оризанол и стеарилглицерат. Косметические средства коммерчески доступны и/или могут быть приготовлены по технологиям, хорошо известным специалистам в данной области.
Примеры пищевых добавок, рассматриваемые для использования на практике по настоящему изобретению, включают в себя, но не ограничиваются ими, протеины, углеводы, водорастворимые витамины (например, витамин С, витамины В-комплекса и подобное), жирорастворимые витамины (например, витамины A, D, E, K и подобное) и травяные экстракты. Пищевые добавки коммерчески доступны и/или могут быть приготовлены по технологиям, хорошо известным специалистам в данной области.
Под термином "пестицид" понимают в совокупности гербициды, инсектициды, акарициды, нематоциды, эктопаразитициды и фунгициды. Примеры классов соединений, к которым может принадлежать пестицид в настоящем изобретении, включают в себя мочевины, триазины, триазолы, карбаматы, сложные эфиры фосфорной кислоты, динитроанилины, морфолины, ацилаланины, пиретроиды, сложные эфиры бензиловой кислоты, дифениловые эфиры и полициклические галогенированные углеводороды. Особые примеры пестицидов каждого из указанных классов регистрируют в Pesticide Manual, 9th Edition, British Crop Protection Council. Пестициды коммерчески доступны и/или могут быть приготовлены по технологиям, хорошо известным специалистам в данной области.
В предпочтительном варианте осуществления настоящего изобретения активный агент представляет собой макромолекулу, такую как протеин, полипептид, углевод, полинуклеотид, вирус или нуклеиновая кислота. Нуклеиновые кислоты включают в себя ДНК, олигонуклеотиды, античувствительные олигонуклеотиды, аптимеры, РНК и SiРНК. Макромолекула может быть природной или синтетической. Протеин может быть антителом, которое может быть моноклоновым или поликлоновым. Протеин также может быть любым известным терапевтическим протеином, выделенным из природных источников или полученным синтетическим или рекомбинационным способами. Примеры терапевтических протеинов включают в себя, но не ограничиваются ими, протеины каскада свертывания крови (например, Фактор VII, Фактор VIII, Фактор IX и другие), субтилизин, яичный альбумин, альфа-1-антитрипсин (ААТ), ДНКаза, пероксид дисмутазы (ПОД), лизоцим, рибонуклеаза, гиалуронидаза, коллагеназа, гормон роста, эритропоетин, инсулиноподобные факторы роста или их аналоги, интерфероны, гратирамер, гранулоцит-макрофаг колониестимулирующий фактор, гранулоцит колониестимулирующий фактор, антитела, ПЭГилированные протеины, гликозилированные или сверхгликозилированные протеины, десмопрессин, LHRH агонисты, такие как лейпролид, госерелин, нафарелин, бусерелин; LHRH антагонисты, вазопрессин, циклоспорин, кальцитонин, гормон околощитовидной железы, пептиды гормона околощитовидной железы и инсулин. Предпочтительные терапевтические протеины представляют собой инсулин, альфа-1-антитрипсин, LHRH антагонисты и гормоны роста.
Примеры терапевтических молекул с низкой молекулярной массой включают в себя, но не ограничиваются ими, стероиды, бета-агонисты, противомикробные агенты, противогрибковые агенты, таксаны (антимитотические и антимикротрубочные агенты), аминокислоты, алифатические соединения, ароматические соединения и мочевины.
В предпочтительном варианте осуществления активный агент представляет собой терапевтический агент для лечения легочных расстройств. Примеры таких агентов включают в себя, но не ограничиваются ими, стероиды, бета-агонисты, противогрибковые агенты, противомикробные соединения, бронхиальные диалаторы, противоастматические агенты, нестероидные противовоспалительные агенты (НСПВАГ), альфа-1-антитрипсин и агенты для лечения пузырного фиброза. Примеры стероидов включают в себя, но не ограничиваются ими, беклометазон (включая беклометазондипропионат), флутиказон (включая флутиказондипропионат), будезонид, эстрадиол, флудрокортизон, флуцинонид, триамцинолон (включая триамцинолонацетонид) и флунизолид. Примеры бета-агонистов включают в себя, но не ограничиваются ими, сальметерол, ксинафоат, формотеролфумарат, лево-альбутерол, бамбутерол и тулобутерол.
Примеры противогрибковых агентов включают в себя, но не ограничиваются ими, интраконазол, флуконазол и амфотерицин В.
Диагностические агенты включают в себя агент, воспроизводящий рентгеновский снимок, и контрастные средства. Примеры агентов, воспроизводящих рентгеновские снимки, включают в себя WIN-8883 (этил-3,5-диацетамидо-2,4,6-трииодобензоат), также хорошо известный как сложный этиловый эфир диатразойной кислоты (ЭЭДК), WIN 67722, т.е. (6-этокси-6-оксогексил-3,5-бис(ацетамидо)-2,4,6-трииодобензоат); этил-2-(3,5-бис(ацетамидо)-2,4,6-трииодобензоилокси)бутират (WIN 16318); этилдиатризоксиацетат (WIN 12901); этил-2-(3,5-бис(ацетамидо)-2,4,6-трииодобензоилокси)пропионат (WIN 16923); N-этил-2-(3,5-бис(ацетамидо)-2,4,6-трииодобензоилокси)ацетамид (WIN 65312); изопропил-2-(3,5-бис(ацетамидо)-2,4,6-трииодобензоилокси)ацетамид (WIN 12855); диэтил-2-(3,5-бис(ацетамидо)-2,4,6-трииодобензоилокси)малонат (WIN 67721); этил-2-(3,5-бис(ацетамидо)-2,4,6-трииодобензоилокси)фенилацетат (WIN 67585); пропандикарбоновая кислота, [[3,5-бис(ацетиламино)-2,4,5-трииодобензоил]окси]бис(1-метил)сложный эфир (WIN 68165) и бензойная кислота, 3,5-бис(ацетамино)-2,4,6-трииодо-4-(этил-3-этокси-2-бутеноат)сложный эфир (WIN 68209). Предпочтительные контрастные агенты включают в себя такие, которые разлагаются относительно быстро в организме, минимизируя таким образом любую связанную с частицами воспалительную реакцию. Разложение может быть результатом ферментативного гидролиза, растворением карбоновых кислот при физиологическом pH или других механизмов. Таким образом, могут быть предпочтительны иодированные карбоновые кислоты, такие как иодипамид, диатризойная кислота и метризойная кислота вместе с гидролитически лабильными иодированными представителями, такими как WIN 67721, WIN 12901, WIN 68165 и WIN 68209 или другими.
Ряд комбинаций активных агентов может быть желательным, включая в себя, например, комбинацию стероида и бета-агониста, например, флутиказонпропионата и сальметерола, будезонида и форметерола и т.д.
Примерами углеводов являются декстраны, гетакрахмал, циклодекстрины, альгинаты, хитозаны, хондроитины, гепарины и тому подобное.
Небольшие сферические частицы
Частицы и небольшие сферические частицы по настоящему изобретению, предпочтительно, имеют средний геометрический размер частиц от примерно 0,01 мкм до примерно 200 мкм, более предпочтительно от 0,1 мкм до 10 мкм и еще более предпочтительно от примерно 0,5 мкм до примерно 5 мкм, и наиболее предпочтительно от примерно 0,5 мкм до примерно 3 мкм, как измерено способами динамического светового рассеяния (например, фотокорреляционной спектроскопией, лазерной дифракцией, лазерной дифракцией с малым углом рассеивания (ЛДМУР), лазерной дифракцией со средним углом рассеивания (ЛДСУР), способами светового затемнения (например, метод анализа Коултера) или другими способами, такими как реология или микроскопия (световая или электронная). Частицы для доставки в легкие будут иметь аэродинамический размер частиц, определенный по измерениям времени полета (например, аэрораспылитель) или измерения с помощью каскадного импактора Андерсена.
Небольшие сферические частицы являются, по существу, сферическими. «По существу, сферические» означает, что отношение длины наиболее длинной к наиболее короткой перпендикулярной оси сечения частицы меньше или равно примерно 1,5. По существу, сферичность не требует оси симметрии. Кроме того, частицы могут иметь поверхностную текстуру, такую как линии или углубления или выпуклости, которые являются небольшими по масштабу при сравнении с полным размером частицы, и еще являются, по существу, сферическими. Более предпочтительно, когда соотношение длин наиболее длинной и наиболее короткой осей частицы меньше или равно примерно 1,33. Наиболее предпочтительно, когда соотношение длин наиболее длинной и наиболее короткой осей частицы меньше или равно примерно 1,25. Поверхностный контакт минимален в микросферах, которые, по существу, сферические, что минимизирует нежелательную агломерацию частиц при хранении. Многие кристаллы или хлопья имеют плоские поверхности, что может привести к большой площади поверхностного контакта, где агломерация может происходить ионным или не ионным взаимодействием. Сфера позволяет контактировать на значительно меньшей площади.
Частицы также имеют, предпочтительно, одинаковый размер частиц. Частицы, имеющие широкое распределение по размеру, где имеются и относительно большие, и небольшие частицы, дают возможность меньшим частицам заполнить промежутки между большими частицами, создавая, таким образом, новые контактирующие поверхности. Широкое распределение по размеру может привести к большим сферам созданием большого количества возможностей контакта для связывающей агломерации. Данное изобретение создает сферические частицы с узким распределением по размеру, минимизируя таким образом возможности для контактной агломерации. «Узкое распределение по размеру» означает то, что предпочтительное распределение частиц по размеру будет иметь соотношение объемного диаметра 90го процентиля небольших сферических частиц к объемному диаметру 10го процентиля, меньшее или равное 5. Более предпочтительно, когда соотношение объемного диаметра 90го процентиля небольших сферических частиц к объемному диаметру 10го процентиля, меньшее или равное 3. Наиболее предпочтительно, когда распределение частиц по размеру будет иметь соотношение объемного диаметра 90го процентиля небольших сферических частиц к объемному диаметру 10го процентиля, меньшее или равное 2.
Геометрическое стандартное отклонение (ГСО) также может быть использовано для характеристики узкого распределения по размеру. ГСО расчеты включают в себя определение эффективного предельного диаметра (ЭПД) при накоплении меньше чем 15,9% и 84,1%. ГСО равно корню квадратному из соотношения ЭПД меньшего чем 84,17% к ЭПД меньшему чем 15,9%. ГСО имеет узкое распределение по размеру, когда ГСО<2,5, более предпочтительно, меньше чем 1,8.
В предпочтительном варианте изобретения активный агент в небольших сферических частицах представляет собой полукристаллическое или некристаллическое вещество.
Типично, когда небольшие сферические частицы, полученные способом изобретения, являются, по существу, непористыми, и имеют плотность больше чем 0,5 г/см3, более предпочтительно, больше чем 0,75 г/см3, и наиболее предпочтительно, больше чем 0,85 г/см3. Предпочтительный интервал плотности составляет от примерно 0,5 до примерно 2,0 г/см3, более предпочтительно от примерно 0,75 г/см3 до примерно 1,75 г/см3, и наиболее предпочтительно от примерно 0,85 г/см3 до примерно 1,5 г/см3.
Частицы по данному изобретению могут демонстрировать высокое содержание активного агента. Не имеется требований значительного количества объемных агентов или аналогичных наполнителей, которые требуются во многих других способах приготовления частиц. Например, небольшие сферические частицы инсулина состоят из равного или большего чем 95% мас. количества частиц. Однако в частицы могут быть включены объемные агенты или наполнители. Предпочтительно, когда активный агент представляет от 0,1% до больше чем 95% мас. частицы, более предпочтительно от примерно 30% до примерно 100% мас., еще более предпочтительно от примерно 50% до примерно 100% мас., еще более предпочтительно от примерно 75% до примерно 100% мас., и наиболее предпочтительно больше чем 90% мас. Установление здесь указанных интервалов означает, что они включают в себя любой интервал или их комбинацию.
Дополнительным аспектом данного изобретения является то, что небольшие сферические частицы сохраняют биохимическую чистоту и биологическую активность активного агента с или без добавления наполнителей.
Доставка частиц in vivo
Частицы, содержащие активный агент, по настоящему изобретению, подходят для in vivo доставки агента до необходимого объекта подходящим путем, таким как инъекция, топически, орально, ректально, через нос, через легкие, вагинально, через щеку, под язык, подкожно, через слизистые оболочки, через уши, внутри глаз, через глаза. Частицы могут быть доставлены в качестве устойчивой жидкой суспензии или рецептированы в качестве твердой формы дозы, такой как таблетки, таблетки в виде капсулы, капсулы и т.д. Предпочтительный путь доставки представляет собой инъекцию, которая включает в себя внутривенную, внутримышечную, подкожную, внутрибрюшинную, подоболочечную, эпидуральную, внутриартериальную, внутрисуставную и тому подобное. Другой предпочтительный путь доставки представляет собой легочную ингаляцию. При данном пути доставки частицы могут осаждаться глубоко в легких, в верхних дыхательных путях или где-нибудь в дыхательных путях. Частицы могут быть доставлены как сухой порошок с помощью ингалятора сухого порошка или они могут быть доставлены с помощью дозированного ингалятора или распылителя.
Лекарственные препараты системного действия, такие как инсулин, желательно осаждать в альвеолах, где имеется очень большая площадь поверхности, доступная для абсорбции при кровообращении. Если целью является осаждение препарата на определенные участки внутри легких, аэродинамический диаметр частицы может быть отрегулирован до оптимального интервала изменением фундаментальных физических характеристик частиц, таких как форма, плотность и размер частицы.
Приемлемые вдыхаемые фракции частиц лекарственного препарата при ингаляции часто достигаются добавлением наполнителя к рецептуре или введением в композицию частицы, или в виде смеси с частицами лекарственного препарата. Например, на улучшение дисперсии измельченных частиц лекарственного препарата (около 5 мкм) влияет смешивание с большими (30-90 мкм) частицами инертного носителя, такого как трегалоза, лактоза или мальтодекстрин. Большие частицы наполнителя улучшают свойства текучести порошка, что коррелирует с фармакодинамическим эффектом. При дополнительном усовершенствовании наполнители непосредственно вводят в небольшие сферические частицы, что влияет на качество аэрозоля, а также потенциально усиливает стабильность протеиновых лекарственных препаратов. В общем, наполнители выбирают так, чтобы они были предварительно одобрены FDA для ингаляции, такие как лактоза или органические молекулы, эндогенные к легким, такие как альбумин и дипальмитоил-DL-α-фосфатидилхолин (ДПФХ). Другие наполнители, такие как поли(молочная кислота-со-гликолевая кислота) (ПМГК) использовали для создания частиц с желаемыми физическими и химическими характеристиками. Однако большой опыт ингаляционного введения наполнителей, одобренных FDA, имелся с противоастматическими лекарственными препаратами, имеющими большие аэродинамические размеры частиц, чтобы осадить их в трахеобронхиальном участке, и которые не проникают существенно глубоко в легкие. По поводу вдыхаемого протеина или пептида, терапевтически доставляемых глубоко в легкие, имеется опасение, что могут появиться нежелательные долгосрочные побочные эффекты, такие как воспаление или раздражение, которые могут быть вызваны иммунологическим откликом или наполнителями, когда они доставляются в альвеолярный участок.
Для того чтобы минимизировать потенциально вредные побочные эффекты от глубокого легочного вдыхания терапевтических средств, может быть целесообразно изготавливать частицы для ингаляции, которые, по существу, состоят только из доставляемого лекарственного препарата. Такая стратегия может минимизировать альвеолярное воздействие наполнителей и уменьшить общую массу частиц, отлагаемых на альвеолярных поверхностях с каждой дозой, по возможности минимизируя раздражение при хроническом использовании вдыхаемых терапевтических препаратов. Небольшие сферические частицы с аэродинамическими свойствами, подходящими для осаждения глубоко в легком, которые, по существу, полностью состоят из терапевтического протеина или пептида, могут быть, в частности, полезны для отдельного изучения влияния хронического терапевтического дозирования на альвеолярную мембрану легкого. Влияние систематической доставки протеина или пептида в виде небольших сферических частиц при ингаляции может затем быть изучено без осложняющих факторов, введенных соответствующими наполнителями.
Требования к доставке частиц глубоко в легкие при ингаляции состоят в том, чтобы частицы имели небольшой средний аэродинамический диаметр от 0,5 до 10 мкм и узкое распределение по размеру. Изобретение также рассматривает смешивание вместе различных партий частиц, имеющих различные интервалы размера частиц. Способ по настоящему изобретению дает возможность изготовлять небольшие сферические частицы с вышеупомянутыми характеристиками.
Имеется два принципиальных подхода к образованию частиц с аэродинамическим диаметром от 0,5 до 3 мкм. Первый подход состоит в том, чтобы получить относительно большие, но очень пористые (или перфорированные) микрочастицы. Так как есть связь между аэродинамическим диаметром (Даэродинамический) и геометрическим диаметром (Дгеометрический), Даэродинамический равен Дгеометрический, умноженному на корень квадратный плотности частиц, частицы с очень низкой массовой плотностью (около 0,1 г/см3) могут иметь небольшой аэродинамический диаметр (от 0,5 до 3 мкм), в то время как обладают относительно большим геометрическим диаметром (от 5 до 10 мкм).
Альтернативный подход представляет собой получение частиц с относительно низкой пористостью, в случае по настоящему изобретению частицы имеют плотность, изложенную в интервалах выше, и обычно она близка к 1 г/см3. Таким образом, аэродинамический диаметр таких непористых плотных частиц близок к их геометрическому диаметру.
Настоящий способ образования частиц, изложенный выше, относится к образованию частиц с наполнителями или без наполнителей.
Изготовление небольших сферических частиц протеина из самого протеина без добавок предоставляет большие преимущества при использовании в легочной доставке, так как предлагает выбор для больших полезных нагрузок лекарственного препарата, увеличивает безопасность и уменьшает число необходимых ингаляций.
Микроинкапсулирование изготовленных ранее небольших сферических частиц
Небольшие сферические частицы по настоящему изобретению или небольшие частицы, приготовленные другими способами (включающие в себя микрочастицы, микросферы, наносферы, наночастицы и т.д.), могут дополнительно быть инкапсулированы в матрицы из материала, образующего стенки, для образования микроинкапсулированных частиц. Микроинкапсулирование может быть выполнено любым способом, известным в данной области. В предпочтительном варианте осуществления микроинкапсулирование небольших сферических частиц по настоящему изобретению или любых других небольших частиц выполняют способами эмульгирование/экстракция растворителем, как описано ниже. Матрица может передавать свойство продолжительного высвобождения активному агенту, приводя к скоростям выделения, которые сохраняются от минут до часов, дней или недель в соответствии с желаемым терапевтическим применением. Микроинкапсулированные частицы также могут осуществлять задержку выделения рецептур предварительно полученных небольших сферических частиц. В предпочтительном варианте осуществления предварительно изготовленные небольшие сферические частицы представляют собой частицы макромолекул. В другом предпочтительном варианте осуществления макромолекула представляет собой протеин или полипептид.
В способе эмульгирование/экстракция растворителем эмульгирование проводят смешиванием двух несмешивающихся фаз, непрерывной фазы и дискретной фазы (которая также известна как дисперсная фаза), для образования эмульсии. В предпочтительном варианте осуществления непрерывная фаза является водной фазой (или водяной фазой), и дискретная фаза является органической фазой (или масляной фазой) для образования эмульсии масло-в-воде (М/В). Дискретная фаза может дополнительно содержать дисперсию твердых частиц, присутствующих или в качестве мелкозернистой суспензии, или в качестве мелкозернистой дисперсии, образующей фазу твердое-в-масле (Т/М). Органическая фаза представляет собой, предпочтительно, не смешивающийся с водой или частично смешивающийся с водой органический растворитель. Соотношение масс органической фазы и водной фазы находится в интервале от примерно 1:99 до примерно 99:1, более предпочтительно, от 1:99 до примерно 40:60, и наиболее предпочтительно от примерно 2:98 до примерно 1:3, или в любом интервале или в комбинации указанных интервалов. В предпочтительном варианте осуществления соотношение органической и водной фазы равно примерно 1:3. Настоящее изобретение дополнительно рассматривает использование обращенных эмульсий или эмульсий вода-в-масле (В/М), где масляная фаза образует непрерывную фазу, и водная фаза образует дискретную фазу. Настоящее изобретение дополнительно рассматривает использование эмульсий, имеющих больше чем две фазы, таких как эмульсия масло-в-воде-в-масле (М/В/М) или эмульсия вода-в-масле-в-воде (В/М/В).
В предпочтительном варианте осуществления способ микроинкапсулирования, использующий способ эмульгирование/экстракция растворителем, начинается с приготовления предварительно изготовленных небольших сферических частиц способами, описанными ранее, и органической фазы, содержащей образующий стенки материал. Предварительно изготовленные небольшие сферические частицы диспергируют в органическую фазу с образующим стенки материалом для образования фазы твердое-в-масле (Т/М), содержащей дисперсию предварительно изготовленных небольших сферических частиц в масляной фазе. В предпочтительном варианте осуществления дисперсию завершают гомогенизацией смеси небольших сферических частиц и органической фазы. Водная среда образует непрерывную фазу. В данном случае эмульсионная система, образованная эмульгированием Т/М фазы с водной фазой, представляет собой эмульсионную систему твердое-в-масле-в-воде (Т/М/В).
Образующий стенки материал относится к материалам, способным к образованию структурной сущности матрицы индивидуально или в комбинации. Биоразлагающиеся образующие стенки материалы являются предпочтительными, особенно для инъекционных применений. Примеры таких материалов включают в себя, но не ограничиваются ими, семейство полилактид/полигликолид полимеров (ПЛГП), полиэтиленгликоль сопряженных ПЛГП (ПЛГП-ПЭГ) и триглицериды. В варианте осуществления, в котором используют ПЛГП или ПЛГП-ПЭГ, ПЛГП, предпочтительно, имеют соотношение полилактидов к полигликолидам от 100:0 до 0:100, более предпочтительно, от примерно 90:10 до примерно 15:85, и наиболее предпочтительно примерно 50:50. В общем, чем больше соотношение полигликолидов к полилактидам в полимере, тем более гидрофильными являются микроинкапсулированные частицы, приводя к более быстрой гидратации и к более быстрому разложению. Также могут быть использованы ПЛГП с различной молекулярной массой. В общем, при одном и том же соотношении полигликолидов и полилактидов в полимере, чем больше молекулярная масса ПЛГП, тем медленнее происходит выделение активного агента и тем шире распределение по размеру микроинкапсулированных частиц.
Органический растворитель в органической фазе (масляная фаза) масло-в-воде (М/В) или эмульсии твердое-в-масле-в-воде (Т/М/В) может быть не смешивающимся с водой или частично не смешивающимся с водой. Под термином «не смешивающийся с водой растворитель» понимаются такие растворители, которые образуют граничный мениск при объединении с водным раствором в соотношении 1:1 (М/В). Подходящие не смешивающиеся с водой растворители включают в себя, но не ограничиваются ими, замещенные или незамещенные, линейные, разветвленные или циклические алканы с числом атомов углерода от 5 и выше, замещенные или незамещенные, линейные, разветвленные или циклические алкены с числом атомов углерода от 5 и выше, замещенные или незамещенные, линейные, разветвленные или циклические алкины с числом атомов углерода от 5 и выше, ароматические углеводороды, полностью или частично галогенированные углеводороды, эфиры, сложные эфиры, кетоны, моно-, ди- или триглицериды, природные масла, спирты, альдегиды, кислоты, амины, линейные или циклические силиконы, гексаметилдисилоксан или любая комбинация указанных растворителей. Галогенированные углеводороды включают в себя, но не ограничиваются ими, тетрахлорид углерода, метиленхлорид, хлороформ, тетрахлорэтилен, трихлорэтилен, триххлорэтан, фторуглеводороды, хлорированный бензол (моно, ди, три), трихлорфторметан. Особенно подходящими растворителями являются метиленхлорид, хлороформ, диэтиловый эфир, толуол, ксилол и этилацетат. Под термином «частично смешивающиеся с водой растворители» понимаются такие растворители, которые не смешиваются с водой при одной концентрации и смешиваются с водой при другой более низкой концентрации. Примерами частично смешивающихся с водой растворителей являются тетрагидрофуран (ТГФ), пропиленкарбонат, бензиловый спирт и этилацетат.
Может быть добавлено поверхностно-активное соединение, например, для увеличения смачивающих свойств органической фазы. Поверхностно-активное соединение может быть добавлено перед эмульгированием к водной фазе, к органической фазе, к обеим и к водной среде и к органическому раствору или к эмульсии после эмульгирования. Использование поверхностно-активного соединения может уменьшить число неинкапсулированных или частично инкапсулированных небольших сферических частиц, приводя к уменьшению начального выброса активного агента во время выделения. Поверхностно-активное соединение может быть добавлено к органической фазе или к водной фазе, или к обеим - и к органической, и к водной фазам, в зависимости от растворимости соединения.
Под термином «поверхностно-активные соединения» понимают соединения, такие как анионные, катионные, амфотерные, неионные поверхностно-активные вещества или биологические поверхностно-активные молекулы. Поверхностно-активное соединение может присутствовать в количестве по массе от водной фазы, или от органической фазы, или от эмульсии в зависимости от применения, меньшем чем примерно, от 0,01% до примерно 30%, более предпочтительно, от примерно 0,01% до примерно 10%, или в любой комбинации указанных интервалов.
Подходящие анионные поверхностно-активные вещества включают в себя, но не ограничиваются ими, лаурат калия, лаурилсульфат натрия, додецилсульфат натрия, алкилполиоксиэтиленсульфаты, альгинат натрия, диоктилсульфосукцинат натрия, фосфатидилхолин, фосфатидилглицерин, фосфатидилинозин, фосфатидилсерин, фосфатидиновая кислота и ее соли, глицериновые сложные эфиры, натрий карбометилцеллюлоза, холиевая кислота и другие кислоты желчи (например, холиевая кислота, дезоксихолиевая кислота, гликохолиевая кислота, таурохолиевая кислота, гликодезоксихолиевая кислота) и их соли (например, дезоксихолат натрия и т.д.).
Подходящие катионные поверхностно-активные вещества включают в себя, но не ограничиваются ими: четвертичные аммониевые соединения, такие как хлорид бензалкония, бромид цетилтриметиламмония, хлорид лаурилдиметилбензиламмония, гидрохлориды ацилкарнитина или галогениды алкилпиридиния. В качестве анионных поверхностно-активных веществ могут быть использованы фосфолипиды. Подходящие фосфолипиды включают в себя, например, фосфатидилхолин, фосфатидилэтаноламин, фосфатидилсерин, фосфатидилинозит, фосфатидилглицерин, фосфатидиновая кислота, лизофосфолипиды, фосфолипиды яиц или соевых бобов, или их комбинация. Фосфолипиды могут быть солевые или бессолевые, гидрированные или частично гидрированные, или природные, полусинтетические или синтетические.
Подходящие неионные поверхностно-активные вещества включают в себя: эфиры полиоксиэтиленового жирного спирта (Macrogol и Brij), полиоксиэтиленовые сложные эфиры сорбитана и жирной кислоты (Polysorbates), сложные эфиры полиоксиэтиленовой жирной кислоты (Myrj), сложные эфиры сорбитана (Span), моностеарат глицерина, полиэтиленгликоли, полипропиленгликоли, цетиловый спирт, цетостеариловый спирт, стеариловый спирт, арилалкилполиэфировые спирты, сополимеры полиоксиэтилен-полиоксипропилен (полоксомеры), полаксамины, поливиниловый спирт, поливинилпирролидон и полисахариды (включая крахмал и производные крахмала, такие как гидроксиэтилкрахмал (ГЭК), метилцеллюлоза, гидроксицеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза и некристаллическая целлюлоза). В предпочтительной форме изобретения неионное поверхностно-активное вещество представляет собой сополимер полиоксиэтилена и полиоксипропилена и, предпочтительно, блоксополимер полипропиленгликоля и этиленгликоля. Такие полимеры продаются под торговой маркой POLOXAMER также иногда относятся к PLURONIC®, и продаются несколькими поставщиками, такими как Spectrum Chemical и Ruger. Среди сложных эфиров полиоксиэтиленовой жирной кислоты используются такие, которые имеют короткие алкильные цепи. Одним примером такого поверхностно-активного вещества является SOLUTOL® HS 15, полиэтилен-660-гидроксистеарат, производимый BASF Aktiengesellschaft.
Поверхностно-активные биологические молекулы включают в себя такие молекулы, как альбумин, казеин, гепарин, гирудин, гетакрахмал и другие подходящие биохимические агенты.
В предпочтительном варианте изобретения водная фаза включает в себя протеин в качестве поверхностно-активного соединения. Предпочтительным протеином является альбумин. Протеин также может функционировать в качестве наполнителя. В вариантах осуществления, в которых протеин не является поверхностно-активным соединением, другие наполнители могут быть включены в эмульсию добавлением или до, или после эмульгирования. Подходящие наполнители включают в себя, но не ограничиваются ими, сахариды, дисахариды и спирты сахаров. Предпочтительным дисахаридом является сахароза, и предпочтительным спиртом сахара является маннит.
Кроме того, использование каналообразующих агентов, таких как полиэтиленгликоль (ПЭГ), может увеличить скорость водопроницаемости конечного продукта, что приводит к изменению начальной кинетики выделения активного агента из матрицы, а также к изменению скорости распада матрицы и зависящей от разложения кинетики выделения при изменении скорости гидратации. Использование ПЭГ в качестве каналообразующего агента во время инкапсулирования может быть выгодным в смысле исключения части процесса промывки во время изготовления небольших сферических частиц, в котором ПЭГ используют как усиливающий фазовое разделение агент. Кроме того, изменение pH непрерывной фазы с использованием буферов может значительно увеличить смачивание между поверхностью частицы и органической фазой, следовательно, результатом будет значительное уменьшение начального выброса инкапсулированного терапевтического агента из матрицы микроинкапсулированных частиц. Свойства непрерывной фазы также могут быть модифицированы, например, увеличением ее солености добавлением такой соли, как NaCl, что уменьшает смешиваемость двух фаз.
После диспергирования небольших сферических частиц в органической фазе (масляная фаза) непрерывная фаза водной среды (водяная фаза) энергично перемешивается, например, гомогенизацией или ультразвуковой обработкой, с дискретной фазой органической фазы для образования эмульсии, содержащей эмульгированные капли зародышевых микроинкапсулированных частиц. Непрерывная водная фаза может быть насыщена органическим растворителем, используемым в органической фазе, до смешивания водной фазы и органической фазы для того, чтобы минимизировать быстрое выделение органического растворителя из эмульгированных капель. Эмульгирование может быть проведено при любой температуре, при которой смесь может сохранять свои свойства жидкости. Стабильность эмульсии является функцией концентрации поверхностно-активного соединения в органической фазе или в водной фазе, или в эмульсии, если поверхностно-активное соединение добавляют к эмульсии после эмульгирования. Это является одним из факторов, который определяет размер капель в эмульсионной системе (эмбриональные микроинкапсулированные частицы) и размер и распределение по размеру микроинкапсулированных частиц. Другие факторы, влияющие на распределение по размеру микроинкапсулированных частиц, представляют собой вязкость непрерывной фазы, вязкость дискретной фазы, силу сдвига во время эмульгирования, тип и концентрацию поверхностно-активного соединения и соотношение масло/вода.
После эмульгирования эмульсию затем переводят в среду затвердевания. Среда затвердевания экстрагирует растворитель дискретной фазы из зародышевых микроинкапсулированных частиц, приводя к образованию твердых микроинкапсулированных частиц, имеющих твердую полимерную матрицу вокруг предварительно изготовленных небольших сферических частиц в окрестности эмульгированных капель. В варианте осуществления с системами М/В или Т/М/В среда затвердевания представляет собой водную среду, которая может содержать поверхностно-активные соединения или агенты, создающие объем, или другие наполнители. Микроинкапсулированные частицы являются, предпочтительно, сферическими с размером частиц от примерно 0,6 до примерно 300 мкм, и более предпочтительно, от примерно 0,8 до примерно 60 мкм. Кроме того, микроинкапсулированные частицы, предпочтительно, имеют узкое распределение по размеру. Чтобы уменьшить время экстракции дискретной фазы, к среде затвердевания могут быть применены нагрев или пониженное давление. Скорость экстракции дискретной фазы из зародышевых микроинкапсулированных частиц представляет собой важный фактор в степени пористости конечных микроинкапсулированных частиц, поскольку быстрое удаление, например, при испарении (эффект кипения), дискретной фазы приводит к деструкции целостности матрицы.
В предпочтительном варианте осуществления эмульгирование проводят непрерывным способом, а не периодическим. Фиг.41 изображает конструкцию реактора непрерывного эмульгирования.
В другом предпочтительном варианте осуществления затвердевшие, образующие стенку полимерные матрицы, инкапсулирующие небольшие сферические частицы активного агента, дополнительно собирают центрифугированием и/или фильтрацией (включая в себя диафильтрацию) и промывают водой. Остающаяся жидкая фаза может быть дополнительно удалена таким способом, как лиофилизация или испарение.
А. Небольшие сферические частицы инсулина
Пример 1
Общий способ получения небольших сферических частиц инсулина
Готовили буферный раствор с рН 5,65 (буфер 0,033М ацетат натрия), содержащий 16,67% ПЭГ 3350. Добавляли к данному раствору при перемешивании концентрированную взвесь кристаллического инсулин-цинка. Концентрация инсулина в конечном растворе составляла 0,83 мг/мл. Раствор нагревали до примерно от 85 до 90°С. Кристаллы инсулина полностью растворялись в данном температурном интервале в пределах пяти минут. Небольшие сферические частицы инсулина начинали образовываться при температуре около 60°С, когда температуру раствора уменьшали при контролируемой скорости. Выход увеличивался с увеличением концентрации ПЭГ. Указанным способом получали небольшие сферические частицы с различным распределением по размеру, при среднем размере 1,4 мкм.
Полученные небольшие сферические частицы инсулина отделяли от ПЭГ промыванием микрочастиц диафильтрацией при условиях, в которых небольшие сферические частицы не растворялись. Небольшие сферические частицы инсулина отмывали от суспензии, используя водный раствор, содержащий Zn2+. Zn2+ ион понижает растворимость инсулина и препятствует растворению, которое понижает выход и вызывает агломерацию небольших сферических частиц.
Пример 2
Периодический процесс без перемешивания для получения небольших сферических частиц инсулина
20,2 мг кристаллического инсулин-цинка суспендировали в 1 мл деионизированной воды при комнатной температуре. К инсулину добавляли 50 микролитров 0,5н. HCl. 1 мл деионизированной воды добавляли для образования раствора 10 мг/мл цинка в кристаллическом инсулине. 12,5 г полиэтиленгликоля 3350 (Sigma) и 12,5 г поливинилпирролидона (Sigma) растворяли в 50 мл 100 миллимолярного буфера из ацетата натрия, pH 5,7. Объем раствора полимера доводили до 100 мл буфером из ацетата натрия. К 800 мкл раствора полимера в пробирке Эппендорфа добавляли 400 мкл раствора инсулина 10 мг/мл. Раствор инсулин/полимер при смешивании стал непрозрачным. Контрольный опыт готовили при использовании воды вместо раствора полимера. Пробирки Эппендорфа нагревали на водяной бане при 90°С в течение 30 минут без перемешивания или взбалтывания, затем удаляли и помещали в лед на 10 минут. Раствор инсулин/полимер становился прозрачным при удалении из водяной бани при 90°С, но начинал мутнеть при охлаждении. Контрольный опыт без полимера оставался прозрачным весь эксперимент. Частицы инсулин/полимер выделяли центрифугированием пробирки с последующим двукратным промыванием для удаления полимера. Последнюю суспензию в воде высушивали лиофильной сушкой для получения сухого порошка. СЭМ анализ высушенных лиофильной сушкой частиц инсулин/полимер из пробирок показал однородное распределение небольших сферических частиц около 1 мкм диаметром. Анализ светового рассеяния по Коултеру показал узкое распределение частиц по размеру со средним размером частицы, равным 1,413 мкм, 95% интервалом достоверности 0,941-1,88 мкм и стандартным отклонением, равным 0,241 мкм. Инсулиновый контрольный опыт без полимера и этапов промывки, но иначе обработанный и прошедший лиофильную сушку тем же способом, показал только хлопья (отсутствие частиц) по анализу СЭМ, аналогичные по внешнему виду тому, что обычно получают после лиофильной сушки протеинов.
Пример 3
Непрерывный проточный способ получения небольших сферических частиц инсулина
36,5 мг инсулина отвешивали и суспендировали в 3 мл деионизированной воды. Добавляли 30 мкл 1н. HCl для растворения инсулина. Конечный объем раствора доводили до 3,65 мл деионизированной водой. 7,3 мл раствора ПЭГ/ПВП (25% ПЭГ/ПВП pH 5,6 в 100 мМ буфера из NaOАс) добавляли к раствору инсулина до конечного суммарного объема раствора инсулина, равного 10,95 мл. Раствор затем встряхивали для образования гомогенной суспензии инсулина и ПЭГ/ПВП.
Суспензию инсулина подсоединяли к перистальтическому насосу BioRad, прокачивающему со скоростью 0,4 мл/мин через Teflon® трубку (TFE 1/32" внутренний диаметр гибкой трубки). Трубку после насоса погружали в водяную баню, поддерживаемую при 90°С, перед вводом в сборную трубу, опущенную в лед. Небольшие сферические частицы инсулина образовывались, когда температура раствора инсулина уменьшалась от 90°С в водяной бане до примерно 4°С в сборной трубке. Фиг.7 представляет собой схематическую диаграмму указанного способа. Суммарное время данного процесса составило 35 минут для объема, равного 10,95 мл. После сбора небольших сферических частиц сборную трубку центрифугировали при 3000 об/мин в течение 20 минут на центрифуге Beckman J6B. Вторичную промывку водой заканчивали и шарики небольших сферических частиц центрифугировали при 2600 об/мин в течение 15 минут. После конечной промывки водой центрифугировали при 1500 об/мин в течение 15 минут. Аликвоту отбирали для анализа размера частиц. Небольшие сферические частицы замораживали при -80°С и высушивали лиофильной сушкой в течение 2 дней.
Размер частицы, определенный по объему, оказался равным 1,397 мкм, по площади поверхности 1,119 мкм и по количеству 0,691 мкм, как было определено с помощью счетчика частиц Beckman Coulter LS 230. Сканирующий электронный микроснимок показал равенство размеров и отсутствие агломерации небольших сферических частиц инсулина (фиг.8).
Использование непрерывного проточного способа, в котором раствор инсулина выдерживали при 90°С в течение короткого времени, давало возможность получать небольшие сферические частицы. Указанный способ приводил к конечной композиции, которая содержала 90% протеина, как было определено высокоэффективной жидкостной хроматографией (ВЭЖХ) (Фиг.9). ВЭЖХ анализ также показал, что растворенные небольшие сферические частицы инсулина имели время элюирования около 4,74 минуты, незначительно отличающиеся от времени стандарта инсулина или исходного материала природного инсулина, показывая этим сохранение биохимической чистоты инсулина после изготовления в виде небольших сферических частиц.
Пример 4
Периодический способ получения небольших сферических частиц инсулина в теплообменнике
Кристаллический инсулин-цинк человека суспендировали в минимальном количестве деионизированной воды с ультразвуковым перемешиванием для гарантии полной дисперсии. Суспензию инсулина добавляли к перемешиваемому, содержащему буфер раствору полимера (pH 5,65 при 25°С), предварительно нагретому до 77°С, так что конечная концентрация растворенного составила 0,83% кристаллического инсулин-цинка, 18,5% полиэтиленгликоля 3350, 0,7% хлорида натрия в 0,1М растворе буфера из ацетата натрия. Непрозрачная вначале смесь просветлела в течение трех минут, когда растворился кристаллический инсулин. Немедленно после просветления раствор перенесли в стеклянную, с водяной рубашкой хроматографическую колонку, которую использовали как теплообменник (колонка с внутренним диаметром: 25 мм, длиной: 600 мм; Ace Glass Incorporated, Vineland, NJ). Стеклянную колонку располагали вертикально, и теплоноситель вводили в водяную рубашку снизу колонки и выводили сверху. Для того чтобы документировать теплообменные свойства системы, термопары (Type J, Cole Parmer) располагали в центре инсулиновой жидкости препарата сверху и снизу колонки, и профиль понижения температуры получали во время предварительного испытания. Термопары удаляли на время шести циклов проведения указанного эксперимента, чтобы не вводить инородных переменных поверхности.
Теплообменник предварительно нагревали до 65°С и раствор инсулин-полимер с буфером перемещали таким способом, чтобы температура раствора не упала ниже 65°С и пузыри воздуха не попали в раствор. Затем прозрачный раствор оставляли на четыре минуты для достижения равновесия при 65°С в теплообменнике, подавали теплоноситель с температурой, изменяемой от 65°С до 15°С. Препарат инсулина в теплообменнике оставляли при 15°С на двадцать минут для достижения равновесия. Небольшие сферические частицы инсулина образовывались, когда температура опускалась от 60 до 55°С с образованием однородной стабильной кремово-белой суспензии.
Небольшие сферические частицы инсулина отделяли от полиэтиленгликоля диафильтрацией (картридж для ультрафильтрации А/G Technologies, 750000 MWCO) с пятью объемами 0,16% ацетата натрия - 0,026% хлорида цинка в качестве буфера, pH 7,0, с последующим концентрированием до одной пятой первоначального объема. Суспензию небольших сферических частиц инсулина дополнительно промывали диафильтрацией с пятью объемами деионизированной воды, с последующей лиофилизацией для удаления воды. Были приняты меры, препятствующие агломерации небольших сферических частиц во время диафильтрации (от поляризационного уплотнения частиц на поверхности мембраны) и во время лиофилизации (от осаждения небольших сферических частиц до замораживания). Высушенные небольшие сферические частицы свободно струились и были готовы для использования без необходимости деагломерации и просеивания.
Небольшие сферические частицы инсулина
Выше описан способ получения сферических частиц одинакового размера из кристаллического инсулин-цинка без добавления наполнителей. Небольшие сферические частицы, приготовленные указанным способом, имеют превосходные аэродинамические свойства, как определено измерениями времени пролета (AerosizerTM) и измерениями с помощью каскадного импактора Андерсена, с высокими вдыхаемыми фракциями, указывающими на доставку глубоко в легкие, с доставкой простым, широко используемым ингалятором сухого порошка (CyclohalerTM). При использовании инсулина в качестве модельного протеина авторы также смогли изучить влияние способа на химическую чистоту протеина, используя установленные U.S.P. способы.
Небольшие сферические частицы сухого порошка инсулина отображали с помощью микроскопии в поляризованном свете (Leica EPISTAR®, Buffalo, NY) и с помощью сканирующего электронного микроскопа (AMRAY 1000, Bedford, MA). Анализ размера частиц проводили с использованием системы измерения размера частиц Aerosizer® Model 3292, которая включает в себя Aero-Disperser® Dry Powder Disperser Model 3230 для введения порошка в прибор (TSI Incorporated, St. Paul, MN). Индивидуальные размеры частиц подтверждали сравнением результатов, полученных с помощью Aerosizer, с результатами электронных микроснимков.
Химическую чистоту инсулина перед и после реализации способа определяли с помощью ВЭЖХ в соответствии с USP монографией Инсулин человеческого организма (USP 26). Содержание инсулина и протеина высокой молекулярной массы измеряли, используя изократический способ SEC ВЭЖХ с УФ детектированием при 276 нм. Для измерения инсулина, А-21 дезамидоинсулина и других, родственных инсулину веществ, образец анализировали, используя градиентную ВЭЖХ с обращенной фазой. Содержание инсулина измеряли с использованием УФ детектора при 214 нм. Для высокомолекулярного протеина, дезамидоинсулина и других, родственных инсулину веществ, проводили количественную оценку любого химического разложения, вызванного способом.
Аэродинамические характеристики небольших сферических частиц инсулина проверяли с использованием прибора Aerosizer®. Измерения распределения по размеру сухого порошка инсулина проводили с использованием приспособления AeroDisperser с небольшим усилием сдвига, средней скоростью подачи и нормальной деагломерацией. Приборное программное обеспечение превращает время-пролетные данные в размер и располагает их в логарифмическом масштабе. Число частиц, определенных для каждого набора размеров, использовали для статистического анализа, а также как суммарный объем частиц, определенных в каждом наборе размеров. Объемное распределение выделяет большие частицы лучше, чем числовое распределение, и, поэтому, является более чувствительным для определения агломератов не диспергированных частиц, а также больших частиц.
Комплект каскадного импактора Андерсена состоит из пред-сепаратора, девяти ступеней, восьми пластин сбора и резервного фильтра. Ступени пронумерованы -1, 0, 1, 2, 3, 4, 5, 6 и F. Ступень -1 представляет собой только сопло. Ступень F содержит пластину сбора для ступени 6 и резервный фильтр. Пластины для сбора из нержавеющей стали покрывали тонким слоем пищевой марки силикона, чтобы препятствовать «отскоку» частиц. Простой регулятор потока воздуха 60 ЛВМ через образец использовали для анализа. Точно взвешенный образец, примерно 10 мг, взвешивали в каждой крахмальной капсуле (Vendor) с порошком, доставленным как аэрозоль, из Cyclohaler в течение четырех секунд. Количество порошка инсулина, отложенного на каждой пластине, определяли с помощью ВЭЖХ с обращенной фазой детектированием при 214 нм, согласно USP 26 анализу для инсулина человека.
Средний массовый аэродинамический диаметр (СМАД) рассчитывали по компьютерной программе Sigma plot, используя пробит подбор при накоплении меньшем, чем массовый процент против эффективного предельного диаметра (ЭПД). Эмитируемую дозу (ЭД) определяли как суммарную наблюдаемую массу инсулина, осевшую в каскадном импакторе. Это отображали как процент массы небольших сферических частиц инсулина, загруженных в капсулу Cyclohaler.
Результаты демонстрируют, что при тщательном контроле параметров способа в соединении с фазовым изменением рецептуры можно получить: 1) преимущественно сферические частицы диаметром около 2 мкм; 2) узкое распределение по размеру; 3) воспроизводимые аэродинамические свойства от партии к партии и 4) небольшие сферические частицы, состоящие из более чем 95% активного лекарственного препарата (инсулин человека), исключая остаточную влагу. Изобретатели определили, что растворимость кристаллического инсулин-цинка может регулироваться температурой раствора, pH, концентрацией полимера и ионной силой. Изобретатели также нашли, что регулирование скорости охлаждения во время периода фазового изменения представляет собой важный параметр, который дает возможность образования преимущественно сферических частиц с узким интервалом размера.
Фиг.2 представляет собой профиль понижения температуры для способа, соответствующего данному примеру. Профиль измеряли с использованием хроматографической колонки с водяной рубашкой, расположенной вертикально, и теплообменным флюидом, входящим в водяную рубашку снизу колонки и выходящим сверху. Две термопары располагали в колонке в контакте с раствором. Одна термопара располагалась сверху колонки и вторая снизу колонки. Температурные кривые делят график время-температура на отдельные области, в которых предварительные эксперименты по оптимизации, обусловленные индуцированным фазовым изменением выше или ниже оптимальной скорости изменения температуры, приводят к уширению интервала размера частиц и несферической форме. При температуре выше 60°С инсулин остается растворимым в буферном растворе полимера (область А; фиг.2). Когда температура уменьшается при скоростях примерно от 8,6°С/минуту до 26,5°С/минуту, это благоприятно для оптимального образования одинаковых по размеру сферических частиц (область В; фиг.2). Если к рецептуре применять скорость понижения температуры больше чем 26,5°С/мин, имеется тенденция к образованию очень мелких (меньше чем 0,5 мкм) несферических частиц инсулина, легко образующих агломерат (область С; фиг.2). Скорости охлаждения меньшие чем 8,6°С/мин приводят к уширению распределения по размеру небольших сферических частиц инсулина, а также к несферической форме и аморфному, хлопьевидному осадку (область D; фиг.2).
Когда температура инсулин-буферсодержащего раствора полимера в пределах теплообменника падает в пределах области В фиг.2, происходит фазовое изменение, приводящее к молочно-белой устойчивой суспензии небольших сферических частиц инсулина. Фазовое разделение, показывающее образование микросфер, начинается, когда температура падает ниже 60°С, и заканчивается, когда температура достигает 40°С. Никаких дополнительных изменений суспензии не наблюдалось, когда рецептуру охладили до 15°С перед промыванием диафильтрацией для удаления полимера ПЭГ.
В то время как СЭМ исходного сырья кристаллического инсулин-цинка человека показал неоднородность размера и кристаллических форм с размерами частиц примерно от 5 до 40 мкм, СЭМ картины, полученный для одной из партий данного примера, показал сферическую форму и одинаковые размеры небольших сферических частиц инсулина (фиг.3В). Форма и размер частицы, показанные СЭМ, являются характерными для других пяти партий, полученных в данном примере.
После диафильтрации для отделения от полимера, содержащего буфер, и последующего промывания и лиофилизации суспензии с деионизированной водой сухой порошок небольших сферических частиц инсулина был относительно свободно текучим и легко взвешенным и обрабатываемым. Содержание влаги в небольших сферических частицах инсулина находилось в пределах от 2,1 до 4,4%, по сравнению с 12% в исходном сырье кристаллического инсулин-цинка. Химический анализ небольших сферических частиц инсулина с помощью ВЭЖХ показал очень незначительное разложение инсулина в результате способа (фиг.4) при отсутствии увеличения соединений с высокой молекулярной массой. Хотя имелось увеличение (над исходным сырьем инсулина) в % димера, % А21 дезамидоинсулина, % поздно элюирующихся пиков и % других соединений, результаты для всех шести партий находились в USP пределах. Удерживание эффективности инсулина составило от 28,3 до 29,9 IU/мг, по сравнению с 28,7 IU/мг для исходного сырья. Остаточные уровни полимера, используемого в способе (полиэтиленгликоль), были ниже 0,13% и до недетектируемого уровня, показывая, что полимер не является значительным компонентом в небольших сферических частицах инсулина.
Воспроизводимость аэродинамических свойств в сопоставлении различных партий небольших сферических частиц инсулина
Наблюдалась отличная воспроизводимость аэродинамических свойств при сопоставлении шести отдельных партий полученных небольших сферических частиц инсулина, что продемонстрировано данными, полученными с помощью Aerosizer и каскадного импактора Андерсена. Для всех шести партий данные Aerosizer показали, что больше 99,5% частиц имеют размер в интервале от 0,63 до 3,4 мкм, минимум 60% небольших сферических частиц инсулина имеют размер в узком интервале от 1,6 до 2,5 мкм (фиг.5). Статистически данные показывают, что с вероятностью 95%, по меньшей мере, 99% небольших сферических частиц в полученных партиях инсулина имеют, по меньшей мере, 96,52% частиц с размерами в интервале от 0,63 до 3,4 мкм (от 68,5% до 70% с диаметром около 2 мкм).
Данные каскадного импактора Андерсена хорошо соответствуют данным Aerosizer, за исключением того, что в среднем 17,6% от дозы, доставляемой из Cuclohaler, осаждалось во входном отверстии и в пред-сепаратор/горловине аппаратуры (фиг.6). Данные показывают, что эффективность дисперсии порошка c использованием прибора Aerosizer выше, чем с использованием устройства Cyclohaler. Однако средняя эмитируемая доза для шести партий составила 71,4% от Cyclohaler, с 72,8% эмитируемой дозы, осевшей на ступени 3 импактора. Если установлено, что вдыхаемая фракция для глубокой легочной доставки должна быть фракцией с ЭПД между 1,1 и 3,3 мкм, в среднем 60,1% небольших сферических частиц вдыхаемого инсулина может быть доступно для глубокой легочной доставки и последующего системного поглощения. Отличная воспроизводимость способа показана в таблице 1, где значения стандартного отклонения для СМАД и ГСО средние для шести отдельных партий, являются чрезвычайно низкими. Это показывает, что переменные способа находятся под жестким контролем, приводя к одинаковым аэродинамическим свойствам от партии к партии.
В таблице 1 представлены аэродинамические свойства небольших сферических частиц инсулина. Результаты (среднее±КО) рассчитывали из анализа отдельных партий небольших сферических частиц инсулина (N=6) на каскадном импакторе Андерсена. Очень хорошая воспроизводимость способа демонстрируется чрезвычайно низкими значениями квадратных отклонений для СМАД и ГСО.
Небольшие сферические частицы инсулина, полученные способом охлаждения, показали незначительную тенденцию к агломерации, что видно из аэродинамических данных таблицы 1.
Пример 5
Способ получения небольших сферических частиц инсулина в перемешиваемом сосуде
2880 мл буферного раствора полимера (18,5% полиэтиленгликоля 3350, 0,7% хлорида натрия в 0,1 М ацетате натрия в качестве буфера с pH 5,65 при 2°С) загружали в 3-литровый стеклянный сосуд с рубашкой, заполненной водой, с перемешиванием и предварительно нагревали до 75°С. 2,4 грамма кристаллического инсулин-цинка человека суспендировали в 80 мл буферного раствора полимера с применением ультразвука для гарантии полноты диспергирования. Суспензию инсулина добавляли к перемешиваемому, предварительно нагретому буферному раствору полимера и перемешивали еще 5 минут. Смесь становилась прозрачной в течение данного времени, показывая, что кристаллический инсулин-цинк растворился. Воду из холодильника, установленную при 10°С, прокачивали через рубашку сосуда, пока инсулин в растворе полимера не охладился до 15-20°С. Полученную суспензию диафильтровали с пятью объемами буферного раствора 0,16% ацетата натрия - 0,026% хлорида цинка, pH 7,0, с последующими пятью объемами деионизированной воды и с последующей лиофилизацией для удаления воды. СЭМ анализ лиофилизованного порошка показал однородные небольшие сферические частицы со средним аэродинамическим диаметром, равным 1,433 мкм, с помощью время-пролетного анализа TSI Aerosizer. Анализ с помощью каскадного импактора Андерсена показал в 73% эмитированной дозы, осевшей на фильтре ступени 3, СМАД 2,2 мкм и ГСО 1,6 мкм; все указывает на превосходные аэродинамические свойства порошка.
Пример 6
Уменьшение образования продуктов разложения инсулина при регулировании ионной силы состава для получения небольших сферических частиц
Инсулин также может быть растворен в растворе при более низких начальных температурах, например 75°С, без увеличения периода времени или подкисления среды, но в результате происходит значительная агрегация при добавлении к раствору NaCl.
Улучшенный способ получения небольших сферических частиц завершали с использованием следующей методики. Концентрированную взвесь кристаллического инсулин-цинка (при комнатной температуре) прибавляли (при перемешивании) к 16,7% раствору полиэтиленгликоля в 0,1М ацетате натрия, pH 5,65, нагретому предварительно до, приблизительно, 85-90°С. Кристаллы инсулина полностью растворялись в данном температурном интервале в пределах пяти минут. Небольшие сферические частицы инсулина образовывались при понижении температуры раствора.
Значительное образование А21 дезамидоинсулина и димеров инсулина в результате химических реакций происходило при подъеме температуры от начальной температуры, равной 85-90°С. Однако, при 75°С, на это требуется значительный период времени. Длительное время также приводит к значительному разложению инсулина. Предварительное растворение инсулина в кислой среде также вызывает нежелательную конверсию большого процента инсулина в продукт разложения А21 дезамидоинсулин.
В эксперименте хлорид натрия добавляли к реакционной смеси буферного полимера, чтобы химическими средствами уменьшить образование димеров инсулина. Хотя добавленный хлорид натрия незначительно уменьшал образование дезамидо- или димеров инсулина, являющихся продуктами разложения, добавление хлорида натрия значительно уменьшало образование олигомеров (высокомолекулярных продуктов инсулина) (таблица 2).
Кроме того, кристаллический инсулин-Zn растворяется гораздо быстрее в присутствии NaCl, чем контрольный без NaCl. Это приводит к предположению, что добавление хлорида натрия увеличивает скорость растворения инсулина и дает возможность уменьшить температуру начала растворения кристаллов инсулин-цинка. Данная гипотеза была подтверждена в эксперименте, который продемонстрировал, что добавление 0,7% NaCl к составу дает возможность сырью кристаллического инсулин-цинка растворяться при 75°С в течение пяти минут, значительно более низкой температуре чем 87°С, требуемой ранее без добавления NaCl. При 75°С в отсутствие NaCl инсулин за 13 минут растворяется не полностью.
Серии экспериментов демонстрируют, что увеличение концентрации NaCl (2,5 мг/мл, 5,0 мг/мл, 10 мл/мл и 20 мг/мл) дополнительно понижает температуру, при которой растворяются кристаллы инсулина, и также понижает температуру, при которой начинают образовываться небольшие сферические частицы (фиг.10a-d). Дополнительно было определено, что увеличение концентрации NaCl в рецептуре быстро растворяет высокие концентрации кристаллического инсулин-Zn. Таким образом, было подтверждено, что растворимость инсулина при данной температуре может тщательно контролироваться регулированием уровня хлорида натрия в начальной непрерывной фазе. Это дает возможность проводить способ при температурах, которые менее благоприятны для образования продуктов разложения.
Для того чтобы определить, обладает ли хлорид натрия уникальными химическими свойствами, которые дают возможность уменьшать температуру растворения инсулина, эквимолярные концентрации хлорида аммония и сульфата натрия сравнивали для контроля с хлоридом натрия. И NH4Cl, и Na2SO4 одинаково понижали температуру, необходимую для растворения сырья кристаллического инсулин-цинка. По-видимому, увеличение ионной силы увеличивает растворимость инсулина в составе для получения микросфер без влияния на способность образовывать небольшие сферические частицы при понижении температуры раствора.
Пример 7
Изучение влияния концентрации ПЭГ на выход и концентрацию инсулина и на размер небольших сферических частиц инсулина
Данные по титрованию полиэтиленгликоля (3350) показали, что увеличение ПЭГ-3350 также увеличивает выход небольших сферических частиц. Однако, когда концентрация ПЭГ слишком высока, частицы теряют свою сферическую форму, что сводит на нет небольшое увеличение выхода.
Данные по концентрации инсулина показывают направление, противоположное ПЭГ, при котором увеличение концентрации инсулина приводит к уменьшению выхода небольших сферических частиц.
Авторы видят общую тенденцию в том, что более высокие концентрации инсулина приводят к образованию небольших сферических частиц большего диаметра. В данном эксперименте более высокие концентрации также приводят к смеси несферических частиц и небольших сферических частиц.
Пример 8
Изучение небольших сферических частиц инсулина на собаках
Целью данного экспериментального изучения было проведение количественного и визуального эксперимента с осаждением аэрозоля порошка инсулина в легких гончих собак. 99mTc меченые частицы инсулина получали по способам, изложенным здесь. Отложения аэрозоля инсулина в легких оценивали с использованием гамма-топографии.
Пять собак использовали в данном изучении, и каждой собаке был введен аэрозоль 99mTc меченных радиоактивным изотопом частиц инсулина. Идентификационные номера собак были 101, 102, 103, 104 и 105.
До введения аэрозоля животных подвергали анестезии пропофолом через линию вливания для анестезии, и в каждое животное помещали эндотрахеальную трубку для доставки аэрозоля.
Каждую собаку помещали в камеру “Spangler box” для ингаляции меченным радиоактивным изотопом аэрозолем. Сразу же после введения меченного радиоактивным изотопом аэрозоля, получали компьютерное изображение гамма-камеры для передней, а также для задней грудной области. Два in-vitro сбора каскадного импактора оценивали один перед введением аэрозоля первому животному (101) и следующий после воздействия на последнее животное (105), чтобы определить стабильность 99mTc меченного радиоактивным изотопом порошка инсулина.
Результаты представлены на фиг.11. Сборы каскадного импактора в обоих случаях показали унимодальное распределение.
Фиг.12 показывает результаты вычисления соотношения P/I для всех животных. Соотношение P/I представляет собой меру пропорции 99mTc порошка инсулина, который осел в периферической части легких, то есть глубоко в легких. Типичное соотношение P/I будет, вероятно, около 0,7. Соотношения P/I большие чем 0,7 указывают на значительное осаждение в периферической части легких по сравнению с центральной частью легких или бронхиальной областью.
Сцинтиграфическое изображение на фиг.13 показывает места отложения инсулина в респираторной системе и является совместимым с данными P/I (фиг.12). Сцинтиграфическое изображение для собаки 101 является характерным для всех пяти собак в данном исследовании.
Сцинтиграфическое изображение для собаки 101 показывает небольшие трахейные или бронхиальные отложения с очевидным увеличением отложения в периферической части легких. Радиоактивность вне легких обусловлена быстрой абсорбцией 99mTc из отложения аэрозольного порошка глубоко в легких.
Соотношение P/I и данные по изображению показывают, что инсулин, меченный радиоактивным изотопом 99mTc, оседает преимущественно глубоко в легких. Количество меченного радиоактивным изотопом инсулина, осевшего на периферии легких, указывало на низкие уровни агломерации частиц.
Пример 9
Диафильтрация с цинксодержащим буфером для удаления полимера из небольших сферических частиц инсулина
Для последующего получения небольших сферических частиц инсулина из УФРА раствора было желательно удалить весь УФРА из суспензии до лиофилизации. Даже несколько процентов оставшегося УФРА могут действовать как связующее, образуя нерыхлые агломераты небольших сферических частиц. Данные агломераты могут неблагоприятно влиять на эмитируемую дозу и аэродинамические свойства порошка, поставляемого из ИСП устройства. Кроме того, в тканях легких под повторяющимися воздействиями доз УФРА могут усилиться токсикологические последствия.
Три методики рассматривались для отделения небольших сферических частиц от УФРА до лиофилизации. Фильтрация могла быть использована для сбора небольших количеств частиц. Однако большие количества небольших сферических частиц быстро блокируют поры фильтрационной среды, делая промывание и возврат более чем нескольких миллиграмм непрактичным.
Центрифугирование для сбора частиц с последующими несколькими циклами промывания, включающими в себя ресуспендирование в водный растворитель и рецентрифугирование, было успешно применено для удаления УФРА. Деионизированную воду использовали в качестве водного растворителя, поскольку небольшие сферические частицы растворяются плохо, и УФРА остается в растворе. Одним неудобством центрифугирования было то, что небольшие сферические частицы были плотно сжаты в шарики высокими ускорениями, необходимыми для осаждения частиц вращением. С каждым последующим промыванием становилось все более трудно ресуспендировать шарики в дискретные частицы. Агломерация частиц инсулина часто была нежелательным побочным эффектом способа центрифугирования.
Диафильтрацию с использованием полых волокнистых ампул применяли в качестве альтернативы центрифугированию для промывания небольших сферических частиц инсулина. В обычной установке с аппаратурой для диафильтрации буферную суспензию УФРА/частицы инсулина помещали в запечатанный контейнер, и суспензия рециркулировала через волокна с достаточным противодавлением, чтобы заставить фильтрат проходить через пустоты волокнистой мембраны. Скорость рециркуляции и противодавление оптимизировали, чтобы препятствовать закупорке (поляризации) пор мембраны. Объем фильтрата, удаленный из суспензии, непрерывно пополнялся сифонированием растворителя для промывания в перемешиваемый запечатанный контейнер. Во время процесса диафильтрации концентрация УФРА в суспензии постепенно уменьшалась, и суспензия небольших сферических частиц инсулина была, по существу, свободна от УФРА после пяти-, семикратного обмена исходного объема суспензии с растворителем для промывания в течение периода часа или около.
Хотя способ диафильтрации очень эффективен для удаления полимера и очень подходящий для увеличения коммерческих количеств, небольшие сферические частицы инсулина медленно растворялись в деионизированной воде, первоначально используемой как растворитель для промывания. Эксперименты показали, что инсулин постепенно терялся в фильтрате, и частицы инсулина полностью растворялись после обмена деионизированной воды в количестве, эквивалентном двадцатикратному исходному объему суспензии. Хотя было найдено, что небольшие сферические частицы инсулина плохо растворимы в деионизированной воде, из-за высокой эффективности способа диафильтрации постоянно удаляются растворенный инсулин, и, вероятно, ионы цинка из суспензии. Поэтому равновесие между нерастворенным и концентрацией растворенного инсулина в данном объеме деионизированной воды не устанавливается при диафильтрации, условие, которое благоприятствует растворению инсулина.
Таблица 3 показывает различные растворы, которые оценивались в качестве потенциальной промывной среды. Десять миллиграмм сухих небольших сферических частиц инсулина суспендировали в 1 мл каждого раствора и слабо перемешивали в течение 48 часов при комнатной температуре. Процент растворенного инсулина измеряли через 24 и 48 часов. Было найдено, что инсулин плохо растворяется в деионизированной воде с равновесием, достигаемым при 1% от суммарной массы растворенного инсулина, менее чем за 24 часа. Однако, как отмечено ранее, высокая эффективность диафильтрации непрерывно удаляет растворенный инсулин (и цинк), так что это равновесие никогда не достигается, и небольшие сферические частицы инсулина постоянно растворяются. Поэтому растворимость инсулина в идеальном промывном растворе должна быть ниже, чем в воде. Поскольку инсулин менее всего растворим вблизи его изоэлектрической точки, проверили двумолярный ацетатный буфер и pH 5,65. Было найдено, что растворимость инсулина зависит от молярности буфера и сравнима с растворимостью в воде при низкой молярности. Этанол значительно понижает растворимость инсулина, но только вблизи безводных концентраций. Фактическое увеличение растворимости инсулина при смешивании этанола с водным раствором использовалось в суспензиях УФРА/небольшие сферические частицы инсулина на ранних ступенях диафильтрации.
Буферные растворы, используемые в коммерческих суспензиях для инъекций, содержащих кристаллический инсулин-цинк, также содержали цинк в растворе. Два из данных растворов проверяли с небольшими сферическими частицами инсулина, и нашли, что они значительно понижают растворимость инсулина по сравнению с деионизированной водой. Согласно литературе, кристаллический инсулин-цинк должен иметь от 2 до 4 ионов Zn, связанных с каждым гексамером инсулина. Кристаллический инсулин-цинк, полученный с различным числом ионов цинка на гексамер в интервале от 1,93 до 2,46, использовали как сырье для изготовления небольших сферических частиц инсулина. Это соответствует от 0,36 до 0,46% цинка на данную массу сырья кристаллического инсулин-цинка. После образования небольших сферических частиц инсулина и диафильтрации с деионизированной водой от 58% до 74% цинка были потеряны во время обработки. Потеря цинка из частиц инсулина вызывает увеличение растворимости инсулина и потерю во время диафильтрации.
Диафильтрация небольших сферических частиц инсулина с 0,16% ацетата натрия - 0,027% ZnCl2, pH 7,0 фактически исключила потерю инсулина в фильтрате. Неожиданно, однако, содержание цинка в небольших сферических частицах инсулина увеличилось до почти 2%, что значительно выше 0,46%, измеренных для исходного сырья кристаллического инсулин-цинка. Другим неожиданным результатом диафильтрации с цинксодержащим буфером стало значительное улучшение в эмитируемой дозе, наблюдаемой из Cyclohaler ИСП аппаратуры (68% диафильтрованного с деионизированной водой в сравнении с 84-90% после диафильтрации с цинковым буфером) и уменьшение количества частиц инсулина, отложенных в горловине каскадного импактора Андерсена. Диафильтрация с цинковым буфером улучшает способность небольших сферических частиц сухого порошка инсулина к диспергированию и уменьшает агломерацию частиц, приводя к уменьшению СМАД и большему осаждению на низких ступенях импактора. Это приводило к предположению, что диафильтрация с цинковым буфером и более высокое содержание цинка в небольших сферических частицах инсулина может улучшить процент дозы, отложенной глубоко в легких.
При суспендировании в газ-вытеснитель ГФА(гидрофторалкан)-134a без добавления наполнителей для использования в ИИД (ингалятор измеряемой дозы) применении не было видимой необратимой агломерации промытых цинковым буфером небольших сферических частиц инсулина. Частицы инсулина, действительно, выпадают хлопьями из суспензии меньше, чем за минуту, но легко переходят в суспензию при встряхивании перед использованием. Встряхивание ИИД контейнера непосредственно перед использованием является нормальной частью инструкций, выдаваемых для использования любого ИИД продукта. Фактически, потеря выпавших хлопьями частиц, которые располагаются на дне ИИД контейнера, может, фактически, ингибировать долгосрочную агломерацию частиц инсулина (в добавление к минимальному контакту из-за их сферической формы), поскольку частицы не располагаются в плотноупакованном слое на дне ИИД контейнера, находящегося под давлением. Поэтому свойства, приобретаемые небольшими сферическими частицами инсулина при диафильтрации с цинковым буфером, могут улучшить срок длительного хранения и дисперсионную способность ИИД препаратов инсулина и других, связанных с цинком соединений.
Поскольку было найдено с помощью анализа порошковой рентгеновской дифракцией, что небольшие сферические частицы инсулина являются некристаллическими, связанный цинк не ассоциируется с координацией иона цинка с мономерами инсулина для образования гексамера. Поэтому, неспецифическое связывание ионов и полученные потенциальные выгоды могут распространяться на ионы, другие чем цинк. Различные протеины, которые не связывают цинк, могут связать другие ионы, которые будут уменьшать растворимость в способе диафильтрации и оказывать аналогичное благоприятное влияние.
Небольшие сферические частицы суспендировали в газе-вытеснителе гидрофторалкане (ГФА) 134а с концентрацией, равной 10 мг/мл. Химическую стабильность инсулина после хранения в ГФА 134а оценивали при времени, равном 0, и через один месяц. Данные, представленные на фиг.28, показывают сохранение микросфер инсулина по мономеру инсулина, димеру инсулина, олигомерам инсулина, главному пику инсулина и А21-дезамидоинсулину.
В следующем исследовании небольшие сферические частицы инсулина, полученные по способам примера 4, сравнивали по их качеству в трех различных аппаратах для ингаляции с использованием способа каскадного импактора Андерсена. Устройство Cyclohaler представляет собой коммерческий ингалятор сухого порошка, Disphaler представляет собой другой ингалятор сухого порошка и ингалятор измеряемой дозы (ИИД) представляет собой устройство, в котором микросферы суспендируют в ГФА 134а, как описано в данном примере, и проталкивают через 100-микролитровый или другого размера измерительный клапан. Результаты на фиг.29 ясно показали, что небольшие сферические частицы, прошедшие ступени устройства каскадного импактора Андерсена, откладываются на ступенях 3 и 4. Это является показателем высокой воспроизводимости качества небольших сферических частиц вне зависимости от устройства, используемого в качестве ингалятора. Единственное главное отличие между ИСП и ИИД устройствами состоит в значительно большем количестве небольших сферических частиц, отложенных в горловине каскадного импактора Андерсена, при использовании ИИД. Высокая скорость, с которой ИИД устройство проталкивает небольшие сферические частицы через горловину импактора Андерсена, объясняет более высокую пропорцию отложений микросфер инсулина по сравнению с ИСП устройством. Специалисты в данной области могут допустить, что ИИД устройство с уменьшенной или модифицированной скоростью выхода может быть использовано для уменьшения числа небольших сферических частиц, отложенных в горловине. Для дополнительных измерений могут быть использованы разделительные устройства на конце ИИД.
Небольшие сферические частицы инсулина (партия номер YQ010302) изготовляли из лиофилизованного исходного материала инсулина по способам, описанным в данном примере. Стабильность при хранении в течение года небольших сферических частиц инсулина сравнивали с лиофилизованным исходным материалом инсулина при 25°С и 37°С. Стабильность инсулина сравнивали при исследовании всех родственных инсулину соединений, димеров инсулина, олигомеров инсулина и А21-дезамидоинсулина.
Фиг. 30-35 показывают, что после периода, равного одному году, небольшие сферические частицы инсулина показали значительно меньшие количества димеров инсулина, олигомеров инсулина, А21-дезамидоинсулина и всех родственных инсулину соединений при сравнении с исходным материалом инсулина, хранящимся в таких же условиях. Это указывает на то, что инсулин в виде микросферы является значительно более устойчивым к химическим изменениям, чем исходный материал.
Небольшие сферические частицы инсулина тестировали с использованием каскадного импактора Андерсена во время, равное 0, и через 10 месяцев после изготовления. Устройство Cyclohaler ИСП использовали для определения аэродинамической стабильности после долгосрочного хранения. Фиг.36 показывает, что аэродинамическое качество остается в высшей степени постоянным после 10 месяцев хранения.
Исследование с помощью спектроскопии КР было предпринято, чтобы объяснить структурные различия между необработанным образцом инсулина и инсулином в небольших сферических частицах, приготовленных в данном примере. Было показано, что инсулин в небольших сферических частицах обладает значительно более высоким содержанием β-листа, а впоследствии более низким содержанием α-спирали, чем исходный необработанный образец инсулина. Данные обнаружения совпадают с образованием агрегатов микроволокнистых структур в небольших сферических частицах. Однако при растворении в водной среде спектры показывают в значительной степени идентичные структуры протеинов, полученные или из необработанных микросфер, или из инсулина, указывающие, что любые структурные изменения в микросферах являются полностью обратимыми при растворении.
Две партии инсулина были проверены с использованием спектроскопии КР: А) необработанный инсулин USP (Intergen, Cat №.4502-10, Lot# XDH 1350110) и B) инсулин в небольших сферических частицах (JKPL072502-2 NB 32: P.64). Образцы в виде порошка или растворы инсулина (около 15 мг/мл в 0,01М HCl) помещали в стандартные стеклянные капилляры и термостатировали при 12°С для анализа КР. Обычно 2-15 мкл аликвоты было достаточно, чтобы заполнить образцом часть капилляра, подвергающуюся лазерному освещению. Спектры возбуждали при 514,5 нм аргоновым лазером (Coherent Innova 70-4 Argon Ion Laser, Coherent Inc., Santa Clara, CA) и записывали на сканирующем двойном спектрометре (Ramalog V/VI, Spex Industries, Edison, NJ) с фотоно-считающим детектором (Model R928P, Hamamatsu, Middlesex, NJ). Данные с интервалом в 1,0 см-1 собирали с временем интегрирования 1,5 сек и спектральной щелью шириной 8 см-1. Образцы сканировали повторно, и индивидуальные изображения воспроизводили и изучали до усреднения. Обычно собирали, по меньшей мере, 4 изображения каждого образца. Спектрометр калибровали с помощью индена и четыреххлористого углерода. Спектры сравнивали различными численными методами, используя SpectraCalc и GRAMS/AI Version 7 программное обеспечение (Thermo Galactic, Salem, NH). Спектры корректировали с учетом растворителя (если имелся) и фона. Спектры растворов корректировали полученным спектром 0,01М HCl при идентичных условиях и подгоняли к серии пяти перекрывающихся функций Гаусса-Лорентца для спадающего фона [S.-D. Yeo, P.G. Debenedetti, S.Y.Patro, T.M.Przybycien, J. Pharm. Sci., 1994, 83, 1651-1656]. Подгонку осуществляли в области 1500-1800 см-1.
Спектры КР получали и для порошковых образцов инсулина, и для их соответствующих растворов (фиг.10i). Спектр необработанного образца очень хорошо соответствует ранее описанным спектрам образцов коммерческого инсулина [S.-D. Yeo, P.G. Debenedetti, S.Y. Patro, T.M. Przybycien, J. Pharm. Sci., 1994, 83, 1651-1656; J.L. Lippert, D. Tuminski, P.J. Desmueles, J. Amer. Chem. Soc., 1976, 98, 7076-7080]. Образец небольших сферических частиц показал заметный сдвиг (примерно, от +10 до +15 см-1) в состояние амида I, указывающее на значительные изменения во вторичной структуре протеина. Примечательно, однако, что спектр коммерческого порошка и спектр небольших сферических частиц были фактически идентичны при растворении образцов в водной среде, показывая, что изменения во вторичной структуре при обработке были полностью обратимы.
Вторичные структурные параметры устанавливали с использованием компьютерных алгоритмов, которые включали в себя сглаживание, вычитание флюоресценции и ароматического фона, и деконволюцию полос амида I. Экспоненциальное угасание флуоресценции вычиталось в значительной степени, как это описано в другом месте [S.-D. Yeo, P.G. Debenedetti, S.Y. Patro, T.M. Przybycien, J. Pharm. Sci., 1994, 83, 1651-1656]. Установленные структурные параметры собраны в таблице 4.
Пример 10
Приготовление небольших сферических частиц инсулина человека изотермическим способом
Инсулин человека USP (Intergen) диспергировали в растворе NaCl и ПЭГ (ММ 3350, спектр Lot# RP0741) c образованием конечной концентрации инсулина, равной 0,86 мг/мл, и концентраций NaCl и ПЭГ, равных 0,7% мас. и 8,3% мас. pH доводили до 5,65 добавлением мельчайших количеств растворов ледяной уксусной кислоты и 1М NaOH. После нагревания до T1=77°С получали прозрачные растворы протеина с конечной концентрацией инсулина Ceq. Затем растворы охлаждали с определенной заранее скоростью до температуры T2=37°C. При Т2 наблюдали осаждение протеина. Осадки удаляли центрифугированием (13000×g, 3 мин) при температуре 37°С, и концентрация инсулина (С*) в полученной надосадочной жидкости, определенная бицинхониновым анализом протеина, составила 0,45 мг/мл. Полученный таким образом раствор инсулина, который держали при 37°С, обозначили раствор А.
Раствор В готовили растворением инсулина человека в 0,7% мас. NaCl/8,3% мас. ПЭГ (pH довели до примерно 2,1 добавлением HCl) с конечной концентрацией инсулина, равной 2 мг/мл. Раствор выдерживали при 37°С с перемешиванием в течение 7 час и последующей обработкой ультразвуком в течение 2 мин. Аликвоты полученного раствора В добавляли к раствору А, получая суммарную концентрацию инсулина 1 мг/мл. Полученную смесь энергично перемешивали при 37°С в течение ночи, получая осадок инсулина, который осторожно удаляли из жидкости с применением мембранного фильтра (эффективный диаметр пор 22 мкм). Полученные микрочастицы протеина затем быстро замораживали жидким азотом и подвергали лиофилизации.
В. Небольшие сферические частицы альфа-1-антитрипсина (ААТ)
Настоящее изобретение также может быть использовано для получения небольших сферических частиц ААТ, которые практически удобны для доставки в легкие.
Пример 11
Приготовление небольших сферических частиц ААТ периодическим способом в колонке с рубашкой (масштаб 10-300 мг)
Буферный раствор при pH 6,0 с 10 мМ ацетата аммония, содержащий 16% ПЭГ 3350 и 0,02% Pluronic F-68, перемешивали магнитной мешалкой в химическом стакане с рубашкой и нагревали до 30°С. Температуру в стакане контролировали, используя циркуляцию воды водяной бани. Концентрированный раствор рекомбинантного ААТ (рААТ) добавляли к данному раствору при перемешивании и доводили pH до 6,0. Концентрация рААТ в конечном растворе составила 2 мг/мл. рААТ полностью растворился при указанной температуре в данной композиции раствора. Все содержимое сосуда перемещали в колонку с рубашкой и нагревали до 25-30°С. Циркуляционную водяную баню для колонки устанавливали на снижение температуры до -5°С. Колонку и ее содержимое охлаждали примерно на 1°С/мин до температуры примерно 4°С. Небольшие сферические частицы рААТ образовывались во время этапа охлаждения. Суспензию микрочастиц замораживали в стеклянном кристаллизаторе и подвергали лиофилизации для удаления воды и буфера.
Для того чтобы экстрагировать ПЭГ из небольших сферических частиц протеина после лиофилизации, ПЭГ/протеин осадок промывали метиленхлоридом (МеCl2). Другие используемые промывные жидкости представляли собой метиленхлорид:ацетон 1:1 или метиленхлорид:пентан 1:1. Процедуру промывания повторяли суммарно 3 раза первоначальным объемом промывки. Полученные гранулы ресуспендировали в небольшом объеме ацетона или пентана и сушили или непосредственно в газообразном азоте, или с помощью роторного испарения.
Пример 12
Приготовление небольших сферических частиц ААТ периодическим способом в колонке с рубашкой (масштаб 200-2000 мг)
Данный тип приготовления осуществляли, используя ту же самую композицию рецептуры, как и в колонке с рубашкой, но он приспособлен для больших объемов и более подходит для увеличения масштаба производства. В данном случае рецептуру смешивали при 75 об/мин с помощью А-образного импеллера лопастного типа в сосуде с рубашкой, обычно 500-1000 мл, и нагревали до 30°С. Температуру в сосуде регулировали, используя циркуляционную водяную баню. Раствор оставляли в том же сосуде, и переключали источник водяной бани с 30°С в бане до 2°С в бане. Сосуд и содержимое охлаждали со скоростью примерно 1°С/мин до температуры 4°С. Небольшие сферические частицы рААТ образовывались во время этапа охлаждения. Температуру контролировали с использованием термопары, и когда суспензия достигала 4°С, ее держали вблизи данной температуры еще 30 мин. После этапа выдерживания небольшие сферические частицы суспензии концентрировали диафильтрацией при примерно 4°С для удаления приблизительно 75% полимера и объема. Суспензию оставшихся небольших сферических частиц замораживали в тонком слое в предварительно охлажденном поддоне для лиофилизации, чтобы удалить воду и оставшийся буфер.
Небольшие сферические частицы протеина отделяли от оставшегося сухого полимера или центрифугированием с органическими растворителями (как описано в примере 10), или экстракцией надкритической жидкостью (НКЖ). Для экстракции НКЖ высушенный материал переносили в экстракционную камеру высокого давления, которое поднимали до 2500 фунт/кв.дюйм (при комнатной температуре) с помощью СО2. Когда достигали рабочего давления, во вход потока жидкости вводили этанол как смесь 70:30 СО2:этанол. Данная НКЖ растворяла полимер, оставляя небольшие сферические частицы. В завершение способа систему промывали этанолом и медленно декомпрессировали.
Пример 13
Выход по способу - % конверсии рAAT в небольшие сферические частицы
Небольшие сферические частицы изготавливали, как описано в примерах 10 и 11. После завершения процесса охлаждения небольшую аликвоту суспензии удаляли и фильтровали через 0,2 мкм шприц-фильтр для удаления небольших твердых сферических частиц. Оптическую плотность фильтрата, который представлял собой рAAT, оставшийся в растворе, определяли при 280 нм с помощью УФ спектрофотометра. Концентрацию рAAT вычисляли затем по стандартной кривой. % конверсии вычисляли как:
(начальная концентрация рAAT-концентрация рAAT фильтрата)
-----------------------------------------------------------*100%=%конверсии
начальная концентрация рAAT
Как показано в вышеприведенной таблице, высокий процент ААТ протеина превращался в небольшие сферические частицы независимо от масштаба способа.
Пример 14
Распределение размера частиц ААТ при различных масштабах способа
Aerosizer данные
Образец небольших сферических частиц конечного сухого порошка ААТ анализировали в TSI Aerosizer 3225, который измеряет размер частиц с помощью время-пролетных измерений. Из указанных измерений вычисляли различные соотношения объемных диаметров для демонстрации распределения размера небольших сферических частиц ААТ и использовали для сравнения с частицами, полученными способами, иными, чем способ настоящего изобретения.
Andersen данные
5-10 мг образец взвешивали в гелевой капсуле и вводили в каскадный импактор Андерсена, используя Cyclohaler ингалятор сухого порошка при скорости потока 60 литров в минуту (ЛВМ). Небольшие сферические частицы собирали со всех ступеней импактора, растворяли в 0,2М Tris-НСl буфере при рН 8,0 и количественно анализировали, используя ВЭЖХ с обращенной фазой. Данные анализировали и вычисляли геометрическое стандартное отклонение (ГСО), как описано в United States Pharmacopeia (USP). Результаты демонстрировали узкое распределение размера.
Все параметры распределения, показанные выше, демонстрировали отличное распределение размера частиц, что является результатом способа получения по настоящему изобретению.
Пример 15
Сохранение биоактивности ААТ
Для определения удельной активности небольшие сферические частицы рААТ растворяли в 0,2М Tris-HCl рН 8,0 при комнатной температуре. Полученный раствор анализировали методом, который измеряет возможность рААТ ингибировать способность свиной панкреатической эластазы (СПЭ) гидролизовать синтетические пептиды, которые содержат п-нитроанилидную группу на их С-конце. Тот же раствор небольших сферических частиц рААТ затем анализировали на концентрацию протеина, используя метод бицинхониновой кислоты (БЦК). Контрольный раствор рААТ исходного материала также анализировали обоими методами. Так как анализ активности разрабатывали для определения активности на основе концентрации 1 мг/мл протеина на образец, величину активности корректировали на основе действительной концентрации протеина, определенной с помощью БЦК, получая величину удельной активности:
величина активности образца
------------------------------------- = удельная активность образца
действительная концентрация протеина
Таким образом, удельная активность демонстрирует сохранение биоактивности после изготовления ААТ в виде небольших сферических частиц.
Пример 16
Сохранение структурной чистоты ААТ
Одно из главных отличий технологии контролируемого фазового разделения (КФР) представляет собой образование частиц в мягких условиях, используя водные системы во время образования частиц и избегая других вызывающих напряжения условий, таких как увеличенная температура, сдвиг и т.д. В области технологии частиц основными проблемами являются стабильность протеинов во время изготовления и стабильность при хранении. Полагают, что основные пути разложения, такие как окисление, деамидирование и, особенно, агрегация протеинов, ответственны за побочные эффекты протеиновых рецептур, включая иммуногенность. Поэтому проблемы регуляции требуют исключительно низкого уровня продуктов разложения в конечных рецептурах частиц. Чтобы определить, действительно ли происходит модификация протеина во время образования, использовали ВЭЖХ, физико-химические характеристики, такие как КД или ДСК.
Круговой дихроизм (КД) является наиболее обычно используемым способом оценки структурных изменений в протеине, подвергшемся возмущению, или сравнения структуры изготавливаемого протеина с родоначальным протеином. Способ КД определяет конформацию протеина и вторичную и третичную структуру протеина.
Вторичную структуру можно определять с помощью КД-спектроскопии в "дальней УФ" спектральной области (190-250 нм). При данных длинах волн хромофор представляет собой пептидную связь, когда она находится в обычном конформационном окружении. Альфа-спираль, бета-лист и случайные спиральные структуры, каждая, дают увеличение характеристической формы и интенсивности КД-спектра. Приблизительная доля каждого типа вторичной структуры, которая присутствует в любом протеине, может быть, таким образом, определена путем анализа его дальнего УФ КД-спектра как суммы относительных вкладов таких справочных спектров для каждого структурного типа.
КД-спектр белка в "ближней УФ" спектральной области (250-350 нм) может быть чувствительным к определенным аспектам четвертичной структуры. При данных длинах волн хромофоры представляют собой ароматические аминокислоты и дисульфидные связи, и КД-сигналы, которые они производят, чувствительны ко всей четвертичной структуре протеина. Сигналы в области от 250 до 270 нм могут быть отнесены к фенилаланиновым остаткам, сигналы от 270 до 290 нм могут быть отнесены к тирозину и сигналы от 280 до 300 нм могут быть отнесены к триптофану. Дисульфидные связи дают слабые широкие сигналы по всему ближнему УФ-спектру.
Дальние УФ КД-спектры исходного раствора рААТ и ААТ, выделенного из небольших сферических частиц, в фосфатном буфере (рН 7,4, Т=25°С, концентрация протеина 0,05 мг/мл), показаны на фиг.13. Каждый спектр усреднен по 10 сканированиям.
Дальние УФ КД-спектры неразличимы, демонстрируя, что изготовление ААТ в небольших сферических частицах с их последующим выделением приводит к молекулам ААТ со структурой, идентичной структуре исходного материала ААТ.
ОФ-ВЭЖХ
Небольшие сферические частицы растворяли в 0,2М Tris-HCl при рН 8,0 и анализировали с помощью ВЭЖХ с обращенной фазой. При сравнении с контрольным раствором исходного рААТ протеина заметной разницы в хроматограммах не обнаружено.
ВЭЖХ система:
ВЭЖХ колонка - Pheomenex Jupiter, 5 мкм, С4, 300А, 250×4,6 мм
Waters Alliance 2965 насос/автоматический пробоотборник
Длина волны - 280 нм
Объем пробы - 75 мкл
Градиент концентрации:
Подвижная фаза 1: 0,1% ТГФ в воде
Подвижная фаза 2: 0,085% ТГФ в 90% (c/v) ацетонитриле в воде
Время опыта - 60 мин
Объемный расход - 1,0 мл/мин
ДСК
Были получены графики ДСК. См. фиг. 15-25b.
Пример 17
Стабильность хранения небольших сферических частиц ААТ относительно стабильности хранения исходного материала ААТ
Небольшие сферические частицы анализировали на сохранение биоактивности (используя анализ, описанный в примере 15) после хранения при комнатной температуре и 4°С в течение 1 недели, 1 месяца, 2 месяцев, 3 месяцев, 6 месяцев и 12 месяцев (фиг. 14b и 14с.) Твердый материал представляет собой исходный раствор рААТ, который был диализован и затем лиофилизован. Для каждого момента времени и условий хранения имелись дубликатные образцы, которые анализировали параллельно.
С. Небольшие сферические частицы гормона роста человека (ГРЧ)
Настоящее изобретение также может быть использовано для приготовления небольших сферических частиц ГРЧ.
Пример 18
Приготовление небольших сферических частиц ГРЧ
в пробирке периодическим способом (масштаб от 20 до 50 мг)
Буферный раствор с рН 5,6 (50 мМ ацетата аммония/50 мМ бикарбоната аммония), содержащий 18% ПЭГ 3350, с конечной концентрацией ГРЧ в растворе 1 мг/мл смешивали в 50-мл конической пробирке и нагревали в стационарной водяной бане до 58°С. ГРЧ растворяли в данном растворе при данных условиях. Пробирку извлекали из водяной бани и охлаждали в ледяной бане до достижения раствором 10°С. Скорость охлаждения поддерживали от 4 до 6°С/мин. Небольшие сферические частицы протеина ГРЧ образовывались во время этапа охлаждения. Небольшие сферические частицы начинали образовываться, когда температура раствора достигала примерно 40°С. После образования частиц небольшие сферические частицы протеина ГРЧ отделяли от ПЭГ одним из двух способов, которые описаны ниже.
Промывка органическим растворителем требует, чтобы после этапа охлаждения и образования частиц суспензию небольших сферических частиц быстро заморозили жидким азотом и лиофилизовали для удаления воды и буфера. Чтобы отделить небольшие сферические частицы от ПЭГ после лиофилизации, брикет ПЭГ/протеин суспендировали в метиленхлориде (МеCl2). ПЭГ растворим в МеCl2, тогда как небольшие сферические частицы протеина нерастворимы. Суспензию перемешивали при комнатной температуре в течение 5 минут. Так как плотность небольших сферических частиц ГРЧ близка к плотности МеCl2 (d=1,335 г/мл), необходим второй растворитель, чтобы снизить плотность жидкости для успешного центрифугирования. Ацетон, который смешивается с МеCl2, добавляли в объеме, равном объему МеCl2. Суспензию небольших сферических частиц затем центрифугировали при 3300 об/мин в течение 5 минут при комнатной температуре. Надосадочную жидкость декантировали, и гранулы ресуспендировали в МеCl2 и опять перемешивали в течение 5 минут при комнатной температуре. Данную промывочную процедуру полностью повторяли для 5 промывок. После конечной промывки гранулы повторно суспендировали в небольшом объеме МеCl2 и сушили роторным испарением, получая конечный порошок небольших сферических частиц ГРЧ.
Промывка цинковым буфером требовала, чтобы после этапа охлаждения и образования частиц суспензию небольших сферических частиц центрифугировали при 4000 об/мин в течение 10 минут при 4°С для отделения небольших сферических частиц от ПЭГ. Надосадочную жидкость удаляли, и гранулы ресуспендировали в холодном буфере, содержащем 50 мМ ацетата цинка, в объеме, равном объему удаленной надосадочной жидкости. Ион Zn2+ снижал растворимость ГРЧ и предотвращал растворение во время промывки. Промывочный буфер хранили на льду. Суспензию затем немедленно центрифугировали при 3000 об/мин в течение 5 минут при 4°С. Надосадочную жидкость удаляли, и промывку цинковым буфером полностью повторяли 3 раза. После 3-разовой промывки цинковым буфером гранулы промывали 2 раза водой и центрифугировали при 3000 об/мин в течение 5 минут при 4°С для удаления избыточного цинка. После конечной водной промывки гранулы ресуспендировали в небольшом количестве воды и быстро замораживали, используя жидкий азот. Замороженные гранулы затем лиофилизовали для удаления воды, получая конечный порошок небольших сферических частиц ГРЧ.
Пример 19
Приготовление небольших сферических частиц ГРЧ в сосуде с рубашкой периодическим способом (100 мг масштаб)
Данный способ приготовления проводится, используя состав, подобный примеру 18, но может вмещать большие объемы и более пригоден для увеличенного масштаба.
Буферный раствор с рН 6,1 (80 мМ ацетата аммония/10 мМ бикарбоната аммония), содержащий 18% ПЭГ 3350 и 0,02% Pluronic F-68, перемешивали в химическом стакане с рубашкой посредством верхнего импеллера и нагревали до 58°С. Температуру смеси регулировали с помощью водяной бани с циркуляцией. Концентрированный раствор ГРЧ добавляли к данному раствору при перемешивании. Конечная концентрация ГРЧ в растворе составляла 1 мг/мл. ГРЧ полностью растворим при данной температуре в композиции данного раствора. Сосуд и содержимое затем охлаждали со скоростью 8°С/мин до температуры приблизительно 10°С. Небольшие сферические частицы ГРЧ образовывались во время этапа охлаждения. Небольшие сферические частицы начинали образовываться около 40°С, и данный процесс продолжался по мере дальнейшего охлаждения суспензии. После этапа охлаждения небольшие сферические частицы отделяли от ПЭГ одним из двух способов, описанных в примере 20а.
Пример 20
Сохранение чистоты ГРЧ
Протеиновую чистоту ГРЧ в небольших сферических частицах оценивали на следующих стадиях способа: после образования частиц, после экстракции ПЭГ и после удаления растворителя или после сушки. Измерение химической чистоты ГРЧ после изготовления в виде небольших сферических частиц определяли, используя ВЭЖХ анализ (размерная эксклюзионная хроматография (РЭХ), обращенная фаза (ОФ)) для количественного анализа продуктов агломерации и разложения. Результаты продемонстрировали отсутствие существенного накопления агломератов или других родственных веществ во время процесса разработки рецептуры небольших сферических частиц.
а. Промывка органическим растворителем
Агломерация ГРЧ с помощью размерного исключения: увеличение агломерации по сравнению с исходным материалом
Вещества, родственные ГРЧ, с помощью обращенной фазы: увеличение разложения по сравнению с исходным материалом
b. Промывка цинковым буфером
Агломерация ГРЧ с помощью размерного исключения: увеличение агломерации по сравнению с исходным материалом
Вещества, родственные ГРЧ, с помощью обращенной фазы: увеличение разложения по сравнению с исходным материалом
частицах
Пример 21
Распределение размера частиц небольших сферических частиц ГРЧ
Снятие характеристик распределения размеров частиц небольших сферических частиц проводили с помощью аэродинамических время-пролетных измерений, используя TSI Aerosizer (фиг.26) и сканирующую электронную микроскопию (фиг.27).
Пример 22
Кинетика растворения небольших сферических частиц ГРЧ
Сравнивали кинетику растворения небольших сферических частиц ГРЧ, подвергнутых двум различным экстракционным процедурам.
Небольшие сферические частицы ГРЧ, промытые органическим растворителем, растворяли непосредственно в водной среде подобно исходному материалу ГРЧ.
Когда небольшие сферические частицы ГРЧ промывали цинковым буфером, растворимость снижалась (фиг.28). Растворение небольших сферических частиц ГРЧ осуществляли в 10 мМ Tris, 154 мМ NaCl, 0,05% Brij 35, pH 7,5 при 37°С. Более полное выделение протеина достигалось в других средах in vitro. Кинетика растворения демонстрировала, что приблизительно 30% всего ГРЧ выделялось в первые 15 минут и приблизительно 50% выделялось в первые 24 часа. Выделение протеина завершалось полностью за 1 месяц. Тот факт, что растворение небольших сферических частиц происходило двухфазным образом, может приводить к некоторой задержке выделения in vivo.
D. Небольшие сферические частицы лизоцима
Пример 23
Приготовление небольших сферических частиц лизоцима
Раствор: 1,6 мг/мл лизоцима, 13,2% ПЭГ 3350, 55 мМ ацетата аммония, рН 9,5, 53 мМ сульфата аммония, 263 мМ хлорида натрия, 26 мМ хлорида кальция.
ПЭГ и буфер нагревали до 40°С (рН 9,55). Полученную суспензию быстро замораживали в жидком азоте и лиофилизовали в многократном лиофилизаторе. Получали небольшие сферические частицы.
Е. Небольшие сферические частицы дезоксирибонуклеазы (ДНКазы)
Пример 24
Приготовление небольших сферических частиц ДНКазы
Пример рецептуры: раствор: 0,18 мг/мл ДНКазы (из сырья 1 мг/мл), 18,2% ПЭГ 3350 (из сырья 25%), 9 мМ ацетата аммония, рН 5,15 (из сырья 1М).
Данную суспензию охлаждали в морозильнике при -80°С и после охлаждения лиофилизовали в многократном лиофилизаторе, и далее промывали центрифугированием с MeCl2/ацетон.
Проверенные исходные концентрации равнялись 0,1 мг/мл ДНКазы и 20% ПЭГ 3350. Но после попытки охлаждения от 37°С до 0°С и отсутствия осадка добавили другое количество ДНКазы, чтобы получить более высокие концентрации. Данный раствор охлаждали в морозильнике при -80°С и после замораживания лиофилизовали в многократном лиофилизаторе. Промывали центрифугированием с MeCl2/ацетоном (фиг. 37, 38).
Активность (анализ на ДНКазу-I с использованием ДНК-метилового зеленого, полученного от Sigma).
Теоретическая активность исходного материала составляет 775 Ки/мг протеина. В растворе сырья определили 0,145 мг/мл протеина. Данную концентрацию разбавили 5 мл для конечной концентрации 0,0199 мг/мл. Активность должна составлять 775 Ки/мг * 0,0199 мг/мл = 15,46 Ки/мл.
Ки/мл=-0,0004×40×1/-0,0011=14,55 Ки/мл
Сравнение с теоретической:
небольшие сферические частицы/теоретическая*100%=% активность
14,55 Ки/мл / 15,46 Ки/мл * 100% = 94,1%
F. Небольшие сферические частицы супероксидной дисмутазы
Пример 25
Приготовление небольших сферических частиц супероксидной дисмутазы
Раствор 0,68 мг/мл ПОД (из сырья 5 мг/мл), 24,15% ПЭГ 3350 (из сырья 31,25%), 9,1 мМ ацетата аммония (из сырья 1М), конечный рН 4,99, установленный гидроксидом аммония и уксусной кислотой. Раствор охлаждали от 40°С до 0°С в течение 50 минут (~0,8°С/мин), и осаждение начиналось около 25°С. Суспензию быстро замораживали в жидком азоте и лиофилизовали в многократном лиофилизаторе, и затем промывали центрифугированием с MeCl2/ацетоном (фиг. 39, 40).
Охлаждали от 40°С до 0°С в течение 50 минут (~0,8°С/мин). Осаждение начиналось около 25°С. Быстро замораживали в жидком азоте и лиофилизовали в многократном лиофилизаторе. Промывали центрифугированием с MeCl2/ацетоном. Образовывались небольшие сферические частицы, и большая часть ацетона удерживалась.
G. Небольшие сферические частицы субтилизина
Пример 26
Небольшие сферические частицы субтилизина с использованием не полимерных усиливающих фазовое разделение агентов
Непрерывная фаза исходной системы может содержать не полимерный усиливающий фазовое разделение агент, чтобы вызывать отделение фазы протеина во время охлаждения. Небольшие сферические частицы субтилизина могут быть образованы по настоящему изобретению с использованием смеси пропиленгликоля и этанола без использования любых других полимеров. Пропиленгликоль служит агентом снижения температуры замерзания, а этанол служит усиливающим фазовое разделение агентом в данной системе. Пропиленгликоль также способствует образованию сферической формы небольших сферических частиц.
Готовили 20 мг/мл раствор субтилизина в 35% пропиленгликоле - 10% формиате - 0,02% CaCl2. Раствор 35% пропиленгликоля-субтилизина затем вносили в 67% этанол при перемешивании. Раствор оставался прозрачным при комнатной температуре. Однако при охлаждении до -20°С в течение одного часа образовывалась суспензия частиц. После центрифугирования для сбора частиц и промывания 90% этанолом проводили Coulter анализ размера частиц с абсолютным этанолом в качестве суспендирующей жидкости. Частицы, дающие Coulter результаты, состоят из дискретных частиц, имеющих средний диметр 2,2 мкм, и 95% указанных частиц были от 0,46 до 3,94 мкм. Оценка оптической микроскопии подтверждает данные результаты, показывая существенно сферические частицы. СЭМ анализ частиц подтвердил Coulter результаты.
Сохранение ферментативной активности субтилизина после образования небольших сферических частиц
Сохранение ферментативной активности после превращения субтилизина в растворе в небольшие сферические частицы субтилизина подтверждали колориметрическим анализом. Теоретические суммарные единицы активности небольших сферических частиц вычисляли вычитанием суммарных единиц, найденных в надосадочной жидкости (после отделения частиц субтилизина), от суммарных единиц субтилизина, найденных в этанол-субтилизин-пропиленгликольном растворе до охлаждения. Действительные суммарные единицы, найденные для небольших сферических частиц субтилизина, разделенные на теоретические единицы и выраженные в процентах, отражают сохранение активности субтилизина после образования частиц. Согласно данному вычислению 107% теоретической активности субтилизина сохранялось после образования небольших сферических частиц субтилизина.
Н. Небольшие сферические частицы углевода
Пример 27
Образование небольших сферических частиц углевода
Настоящее изобретение можно использовать для приготовления небольших сферических частиц углевода. Можно вызвать фазовое разделение между фазой ПЭГ и фазой декстрана во время охлаждения системы. Можно использовать декстраны с различной молекулярной массой, например, 5К, 40К, 144К и 500К. Смесь 5 мг/мл декстрана 40К в 30% ПЭГ 300 приводили к равновесию при 35°С, затем смесь охлаждали до 0°С и лиофилизовали. Частицы собирали промыванием смеси метиленхлоридом:ацетоном (1:1) и центрифугированием. Как видно из фиг.49, образовывались небольшие сферические частицы. Другие углеводы, такие как крахмал, гидроксиэтилкрахмал, трегалоза, лактоза, маннит, сорбит, гилоза, сульфат декстрана и т.д., могут быть получены в виде небольших сферических частиц, используя данный способ.
I. Микроинкапсулирование предварительно изготовленных небольших сферических частиц
Пример 28
Приготовление PLGA-инкапсулированных небольших сферических частиц предварительно изготовленного инсулина
а) 20% (мас./об.) раствор полимера (8 мл) готовили растворением 1600 мг полилактид-со-гликолида (ПМГК, ММ 35k) в метиленхлориде. К данному раствору добавляли 100 мг небольших сферических частиц инсулина (ИНСмс) и получали гомогенную суспензию энергичным перемешиванием среды с помощью ротор/статор гомогенизатора при 11k об/мин. Непрерывная фаза состояла из 0,02% водного раствора метилцеллюлозы (24 мл), насыщенного метиленхлоридом. Непрерывную фазу перемешивали при 11k об/мин с помощью того же гомогенизатора, и описанную суспензию постепенно вводили в среду для генерации зародышевых микроинкапсулированных частиц органической фазы. Данная эмульсия имела соотношение М/В 1:3. Эмульгирование продолжали в течение 5 минут. Затем эмульсию немедленно переносили в среду затвердевания, состоящую из 150 мл деионизированной (ДИ) воды, причем среду перемешивали при 400 об/мин. Органические растворители удаляли в течение одного часа при пониженном давлении -0,7 бар. Затвердевшие микроинкапсулированные частицы собирали фильтрованием и промывали водой. Промытые микроинкапсулированные частицы лиофилизовали для удаления избыточной воды. Полученные микроинкапсулированные частицы имели средний размер частиц около 30 мкм, причем большая часть частиц была меньше 90 мкм, и содержали 5,7% (мас./мас.) инсулина.
b) 30% (мас./об.) раствор полимера (4 мл) готовили растворением 1200 мг 50:50 полилактид-со-гликолида (ПМГК, ММ 35k) в метиленхлориде. Затем суспензию 100 мг ИНСмс в растворе желаемого полимера готовили с помощью гомогенизатора. Данную суспензию использовали для генерации М/В эмульсии в 12 мл 0,02% водного раствора метилцеллюлозы, как описано в примере 28 (В/М соотношение = 1:3). Те же процедуры, как в примере 28, использовали для приготовления конечных микроинкапсулированных частиц. Полученные микроинкапсулированные частицы имели средний размер частиц 25 мкм, меняясь от 0,8 до 60 мкм. Содержание инсулина в данных микроинкапсулированных частицах равнялось 8,8% (мас./мас.).
Альтернативно, 10% (мас./об.) раствор полимера использовали для осуществления способа микроинкапсулирования в тех же описанных условиях. Данный способ приводил к микроинкапсулированным частицам со средним размером частиц около 12 мкм, с большей частью частиц менее чем 50 мкм и содержанием инсулина 21,1% (мас./мас.).
Способ in vitro выделения
In vitro выделение (IVВ) инсулина из микроинкапсулированных частиц достигали добавлением 10 мл выделяющего буфера (10 мМ Tris, 0,05% Brij 35, 0,9% NaCl, pH 7,4) в стеклянные ампулы, содержащие 3 мг эквивалента инкапсулированного инсулина, выдерживаемые при 37°С. Через назначенные интервалы времени 400 мкл IVВ среды переносили в трубу микроцентрифуги и центрифугировали в течение 2 мин при 13k об/мин. Верхние 300 мкл надосадочной жидкости удаляли и сохраняли при -80°С до анализа. Взятый объем замещали 300 мкл свежей среды, которую использовали для воссоздания твердого остатка вместе с оставшейся надосадочной жидкостью (100 мкл). Суспензию переносили обратно в соответствующую in vitro выделяющую среду.
Пример 29
Методика для микроинкапсулирования предварительно изготовленных небольших сферических частиц инсулина в матричной системе сплава ПМГК/ПМК
30% (мас./об.) раствор ПМГК/ПМК сплава готовили в метиленхлориде (4 мл). Сплав состоял из 50:50 ПМГК (ММ 35k), D,L-полимолочной кислоты (ПМК, ММ 19k) и поли L-ПМК (PLLA, ММ 180k) с 40, 54 и 6% (0,48, 0,68 и 0,07 г) соответственно. Для приготовления конечных микроинкапсулированных частиц следовали тем же процедурам, как в примере 28b. Примеры микроинкапсулированных частиц имели диапазон размера частиц от 0,8 до 120 мкм, среднее при 40 мкм, с наибольшим количеством частиц меньших чем 90 мкм.
Пример 30
Методика микроинкапсулирования предварительно изготовленных небольших сферических частиц инсулина в ПМГК матричной системе с использованием ПЭГ и в непрерывной и в дискретной фазах
Раствор 4 мл 10% 50:50 ПМГК (0,4 г) и 25% полиэтиленгликоля (ПЭГ, ММ 8k) готовили в метиленхлориде. Используя ротор/статор гомогенизатор, 100 мг ИНСмс суспендировали в данном растворе при 11k об/мин. Непрерывная фаза состояла из водного раствора (12 мл) 0,02% (мас./об.) метилцеллюлозы и 25% ПЭГ (ММ 8k), насыщенного метиленхлоридом. Непрерывную фазу перемешивали при 11k об/мин, используя тот же гомогенизатор, и описанную суспензию постепенно вводили в среду для генерации зародышевых микроинкапсулированных частиц органической фазы. Данная эмульсия имела соотношение М/В 1:3. Эмульгирование проводили в течение 5 минут. Затем эмульсию немедленно переносили в среду затвердевания, состоящую из 150 мл ДИ-воды, причем среду перемешивали при 400 об/мин. Органический растворитель удаляли в течение одного часа при пониженном давлении -0,7 бар. Затвердевшие микроинкапсулированные частицы собирали фильтрованием и промывали водой. Промытые микроинкапсулированные частицы лиофилизовали для удаления избыточной воды. Микроинкапсулированные частицы данного примера имели средний размер частиц 30 мкм в диапазоне от 2 до 90 мкм, причем большая часть частиц была меньше чем 70 мкм. Содержание инсулина в данных микросферах равнялось 16,0% (мас./мас.).
Пример 31
Методика для микроинкапсулирования предварительно изготовленных небольших сферических частиц инсулина в ПМГК матричной системе при различных рН непрерывной фазы с использованием фосфатного буфера
Раствор 4 мл 20% 50:50 35 кD ПМГК (0,8 г) готовили в метиленхлориде. Используя гомогенизатор ротор/статор, 100 мг ИНСмс суспензировали в данном растворе при 11k об/мин. Непрерывная фаза состояла из водного раствора 0,1% (мас./об.) метилцеллюлозы и 50 мМ фосфатного буфера при рН 2,5, 5,4 и 7,8. Микроинкапсулирование проводили, используя непрерывную установку (фиг.41А). Непрерывную фазу перемешивали при 11k об/мин и подавали в камеру эмульгирования при 12 мл/мин. Диспергированную фазу вводили в данную камеру при 2,7 мл/мин для генерации зародышевых микроинкапсулированных частиц. Полученную эмульсию удаляли из камеры и переносили в баню затвердевания непрерывным образом. Среду затвердевания перемешивали при 400 об/мин. Органический растворитель удаляли в течение одного часа при пониженном давлении -0,4 бар. Затвердевшие микроинкапсулированные частицы собирали фильтрованием и промывали водой. Промытые микроинкапсулированные частицы лиофилизовали для удаления избыточной воды.
Содержание инсулина в полученных микроинкапсулированных частицах, приготовленных при рН 2,5, 5,4 и 7,8, было оценено как 12,5, 11,5 и 10,9, соответственно. Результаты анализа распределения размера микроинкапсулированных частиц суммированы в таблице 5.
Способ in vitro выделения
In vitro выделение инсулина из микроинкапсулированных частиц достигали добавлением 10 мл выделяющего буфера (10 мМ Tris, 0,05% Brij 35, 0,9% NaCl, pH 7,4) в стеклянные ампулы, содержащие 3 мг эквивалента инкапсулированного инсулина, выдерживаемые при 37°С. Через назначенные интервалы времени 400 мкл IVВ среды переносили в трубу микроцентрифуги и центрифугировали в течение 2 мин при 13k об/мин. Верхние 300 мкл надосадочной жидкости удаляли и сохраняли при -80°С до анализа. Взятый объем замещали 300 мкл свежей среды, которую использовали для воссоздания твердого остатка вместе с оставшейся надосадочной жидкостью (100 мкм). Суспензию переносили обратно в соответствующую in vitro выделяющую среду.
Результаты in vitro выделения (IVВ) вышеописанных приготовлений показаны на фиг.44 и демонстрируют существенное влияние рН непрерывной фазы на кинетику выделения инсулина из рецептур.
Пример 32
Методика микроинкапсулирования предварительно изготовленных небольших сферических частиц альбумина сыворотки человека в матричной системе PLLA или PLLA/PEG
Раствор 2 мл 25% (мас./об., 500 мг) ПЭГ (ММ 3k или 8k) готовили в метиленхлориде. Раствор ПЭГ или 2 мл метиленхлорида использовали для образования суспензии 50 мг предварительно изготовленных небольших сферических частиц сыворотки альбумина человека (САЧ) с помощью ротор/статор гомогенизатора при 11k об/мин. К данной суспензии добавляли 2 мл 4% PLLA (80 мг, ММ 180k) в метиленхлориде, и среду гомогенизировали при 11-27k об/мин с получением органической фазы. Непрерывная фаза состояла из 12 мл 0,02% водного раствора метилцеллюлозы, насыщенной метиленхлоридом. Эмульгирование инициировали энергичным перемешиванием непрерывной фазы при 11k об/мин с последующим постепенным введением органической фазы. Среду эмульгировали в течение 5 минут, затем эмульсию переносили в 150 мл ДИ-воды, перемешиваемой при 400 об/мин. Все описанные процедуры выполняли при 4°С. Среду затвердевания затем перемещали в комнатную температуру, и органический растворитель удаляли в течение одного часа при пониженном давлении -0,7 бар. Затвердевшие микроинкапсулированные частицы собирали фильтрованием и промывали водой. Промытые микроинкапсулированные частицы лиофилизовали для удаления избыточной воды. Каналообразующее влияние ПЭГ на IVВ САЧ из вышеописанных рецептур показано на фиг.42.
Способ in vitro выделения
In vitro выделение (IVВ) САЧ из инкапсулированных микроинкапсулированных частиц достигали добавлением 15 мл выделяющего буфера (20 мМ HEPES, 0,01% Tween-80, 0,1M NaCl, 1 мM CaCl2, pH 7,4) в 15-мл полипропиленовые трубки для центрифугирования, содержащие 2,5 мг эквивалента инкапсулированного САЧ, выдерживаемые при 37°С. Процедура отбора проб описана в примере 31.
Пример 33
Приготовление PLGA-инкапсулированных предварительно изготовленных небольших сферических частиц лейпролид/сульфат декстрана
30% (мас./об.) раствор полимера (4 мл) готовили растворением 1200 мг 50:50 полилактид-со-гликолида (ПМГК, ММ 35к) в метиленхлориде. Затем 65,9 мг предварительно изготовленных небольших сферических частиц лейпролид/сульфат декстрана (ЛСД), содержащих 50 мг лейпролида, суспендировали в описанном растворе полимера с помощью гомогенизатора. Данную суспензию использовали для генерации М/В эмульсии в 12 мл 0,02% водного раствора метилцеллюлозы, как описано в примере 28 (В/М соотношение=1:3). Те же процедуры, как в примере 28b, использовали для приготовления конечных микроинкапсулированных частиц.
Микроинкапсулированные частицы имели средний размер частиц 20 мкм с большинством частиц менее чем 50 мкм. Результаты IVВ лейпролида из микроинкапсулированных частиц показаны на фиг.43.
Способ in vitro выделения
In vitro выделение (IVВ) лейпролида из микроинкапсулированных частиц достигали добавлением 15 мл выделяющего буфера (10 мМ Na-фосфатного буфера, 0,01% Tween-80, 0,9% NaCl, 0,04% NaN3 pH, 7,4) в 15-мл полипропиленовые пробирки для центрифугирования, содержащие 2,5 мг эквивалента инкапсулированного лейпролида, выдерживаемые при 37°С. Процедура отбора проб описана в примере 28.
Пример 34
Приготовление PLGA-инкапсулированных предварительно изготовленных небольших сферических частиц рекомбинанта гормона роста человека
10% (мас./об.) раствор полимера (4 мл) готовили растворением 0,4 г PLGA-PEG в метиленхлориде. Затем 100 мг предварительно полученных небольших сферических частиц рекомбинанта гормона роста человека (ГРЧ) суспендировали в описанном растворе полимера, используя гомогенизатор. Непрерывная фаза состояла из водного раствора 0,1% (мас./об.) метилцеллюлозы и 50 мМ фосфатного буфера при рН 7,0. Микроинкапсулирование проводили, используя непрерывную установку (фиг.41А), как описано в примере 31. Средний размер данных микроинкапсулированных частиц составлял 25 мкм с диапазоном от 1 до 60 мкм. IVВ профиль ГРЧ из полимерной матрицы показан на фиг.45.
Способ in vitro выделения
IVВ ГРЧ из микроинкапсулированных частиц достигали, как описано в примере 28.
Пример 35
Определение чистоты микроинкапсулированных предварительно изготовленных небольших сферических частиц инсулина
Чтобы оценить влияние процесса микроинкапсулирования на чистоту инкапсулированых предварительно изготовленных небольших сферических частиц инсулина, полимерные микроинкапсулированные частицы, содержащие предварительно изготовленный ИНСмс, разложили, используя способ двухфазной двойной экстракции. Взвешенный образец инкапсулированного ИНСмс суспендировали в метиленхлориде и осторожно перемешивали для растворения полимерной матрицы. Для экстракции протеина добавляли 0,01н. HCl, и две фазы перемешивали, получая эмульсию. Затем две фазы разделяли, водную фазу удаляли и заменяли тем же раствором, и процесс экстракции повторяли. Чистоту экстрагированного инсулина определяли с помощью размерной эксклюзионной хроматографии (РЭХ). Данный способ идентифицирует содержание мономера, димера и высокомолекулярных (ВМ) частиц ИНС в экстрагированной среде. Соответствующие проверки применяли, чтобы идентифицировать влияние процесса разложения на чистоту ИНС. Результаты не показали существенного влияния данного процесса на чистоту ИНС.
Инкапсулированные ИНСмс содержали от 97,5 до 98,94% мономеров протеина в зависимости от условий и содержания процесса микроинкапсулирования по сравнению с 99,13% содержания мономера в исходном ИНСмс (неинкапсулированном). Содержание частиц димера в инкапсулированном ИНСмс менялось от 1,04% до 1,99% по сравнению с 0,85% в исходном ИНСмс. Содержание ВМ в инкапсулированном ИНСмс менялось от 0,02% до 0,06% против 0,02% в исходном ИНСмс. Результаты суммированы в таблице 6. Влияние полимерной матрицы изображено на фиг. 46 и 47.
ИНСмс
Пример 36
In vivo выделение инсулина из микроинкапсулированных предварительно изготовленных небольших сферических частиц инсулина
In vivo выделение инсулина из микроинкапсулированных предварительно изготовленных небольших сферических частиц инсулина исследовали на Spraguе Dawley (SD) крысах. Животные получали исходную подкожную дозу 1 IU/кг неинкапсулированных или инкапсулированных, предварительно изготовленных небольших сферических частиц инсулина. ELISA использовали для определения уровней рекомбинанта сыворотки инсулина человека (рчИНС) в собранных образцах. Результаты показаны на фиг.48.
Хотя были проиллюстрированы и описаны конкретные варианты осуществления, могут быть предложены многочисленные модификации без нарушения духа изобретения, и рамки ограждения ограничиваются только рамками сопровождающей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОРАЗЛАГАЕМЫЕ ПОЛУКРИСТАЛЛИЧЕСКИЕ ТЕРМОПЛАСТИЧНЫЕ МУЛЬТИБЛОЧНЫЕ СОПОЛИМЕРЫ С РАЗДЕЛЕННЫМИ ФАЗАМИ ДЛЯ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ | 2012 |
|
RU2662818C2 |
| ЛЕКАРСТВЕННЫЕ СОСТАВЫ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ, ОСНОВАННЫЕ НА БЛОК-СОПОЛИМЕРАХ | 2006 |
|
RU2409348C2 |
| КОМПОЗИЦИЯ ИНСУЛИНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2010 |
|
RU2556340C2 |
| КОМПОЗИЦИЯ СТАБИЛИЗИРОВАННОГО ГОРМОНА РОСТА И СПОСОБ ЕЕ ПРИГОТОВЛЕНИЯ | 1997 |
|
RU2191029C2 |
| МИКРОЧАСТИЦА И ЕЕ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2490009C2 |
| КОМПОЗИЦИЯ ДЛЯ УЛУЧШЕНИЯ СОСТОЯНИЯ КОЖИ, СОДЕРЖАЩАЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА ГОРМОНЫ РОСТА ЧЕЛОВЕКА | 2005 |
|
RU2401099C2 |
| СПОСОБ ОЧИСТКИ ИНСУЛИНА-СЫРЦА, ПОЛУЧАЕМОГО ИЗ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ СВИНЕЙ | 1996 |
|
RU2126690C1 |
| КОМПОЗИЦИИ, КОРРИГИРУЮЩИЕ ВКУС КОНТРАСТНЫХ СРЕДСТВ | 2014 |
|
RU2683568C1 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ И/ИЛИ ЦЕЛЕВЫЕ ДЕНДРИМЕРНЫЕ КОНЪЮГАТЫ | 1995 |
|
RU2127125C1 |
| ПЭГ-ИЛИРОВАННЫЕ НАНОЧАСТИЦЫ | 2005 |
|
RU2400215C2 |
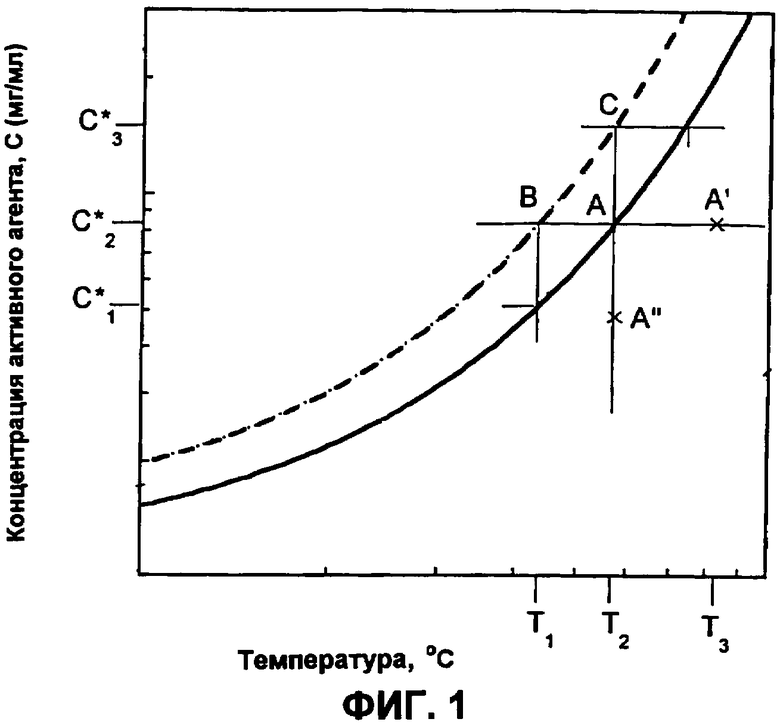
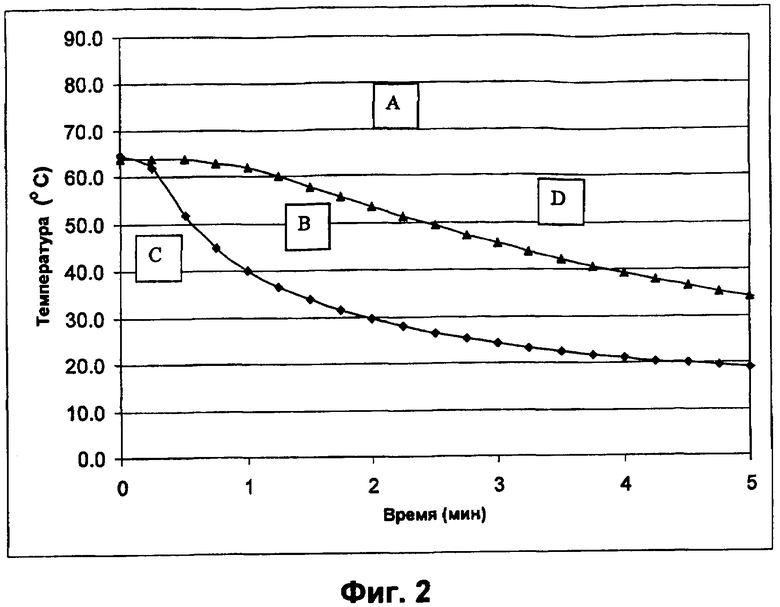
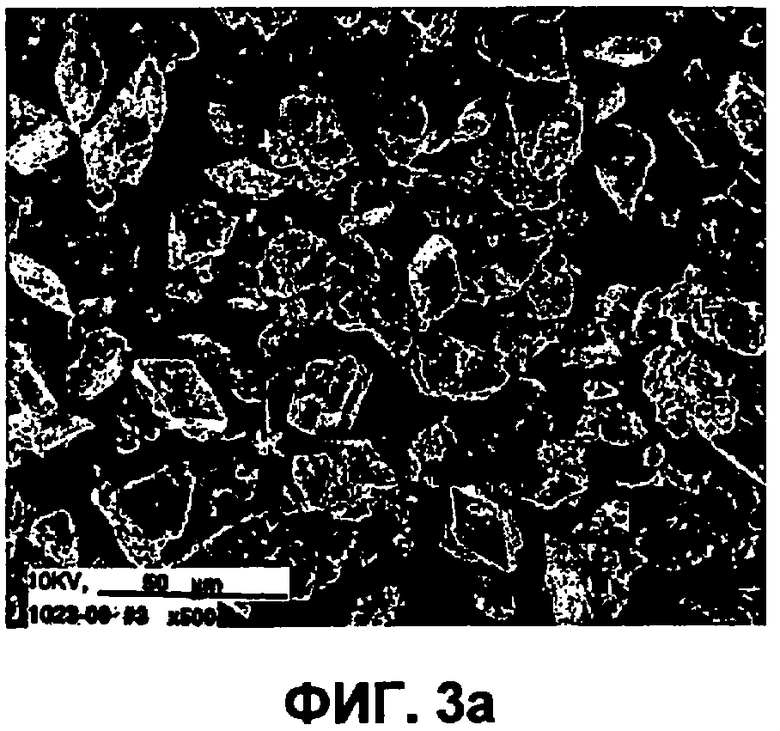
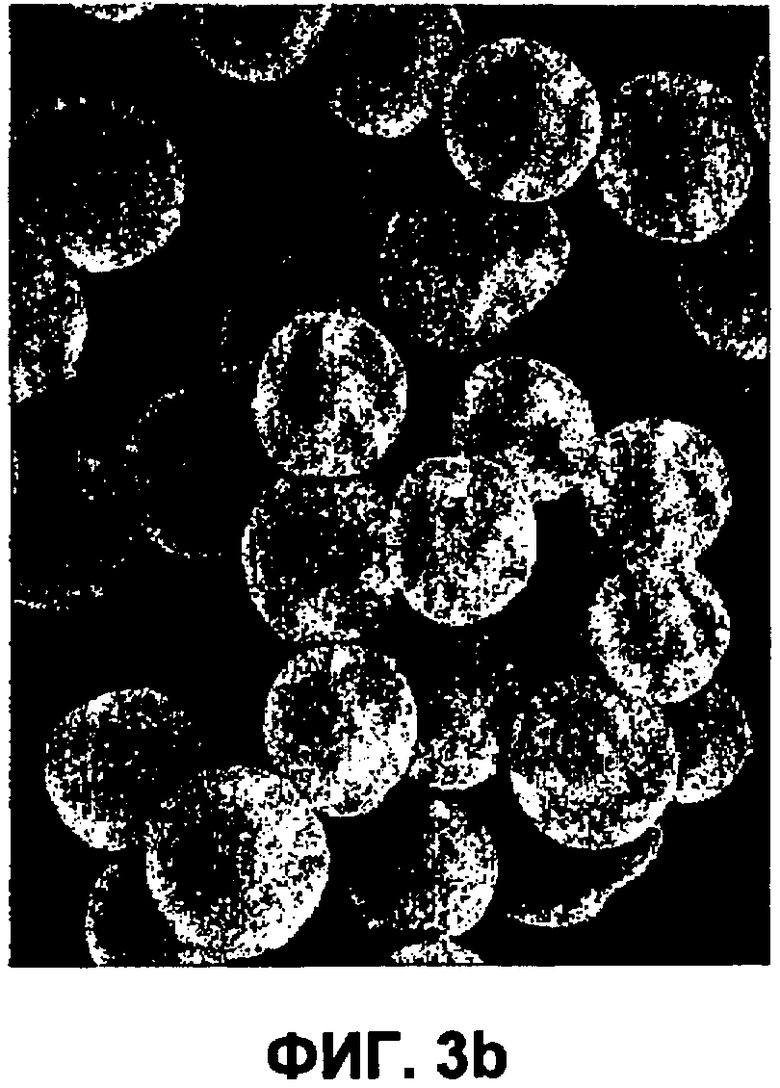

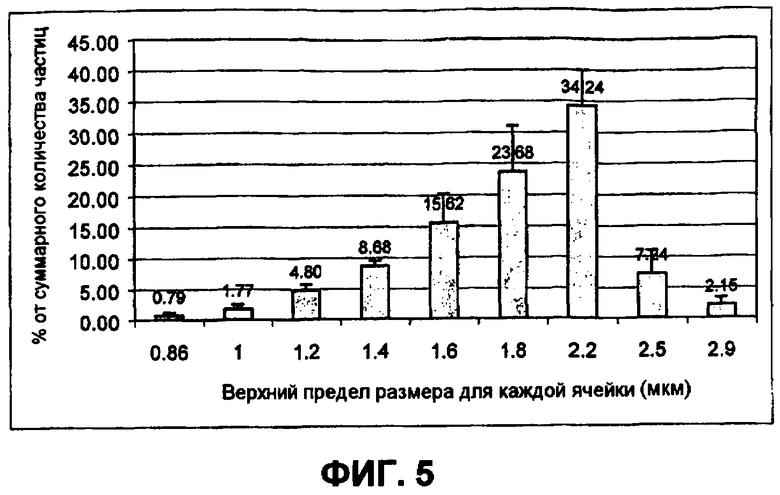
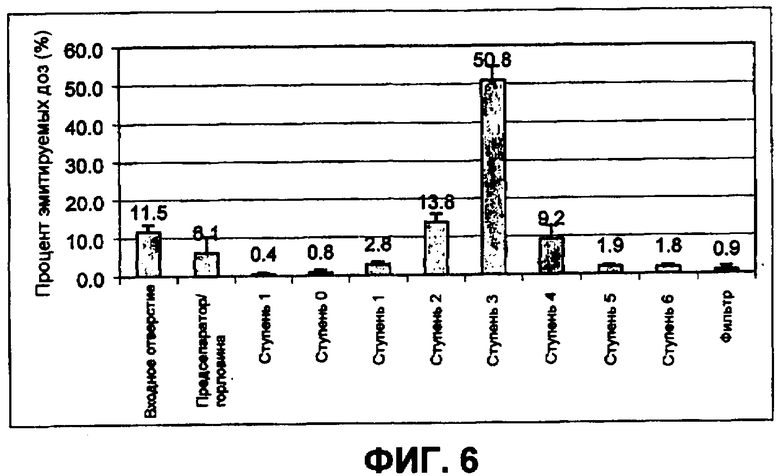
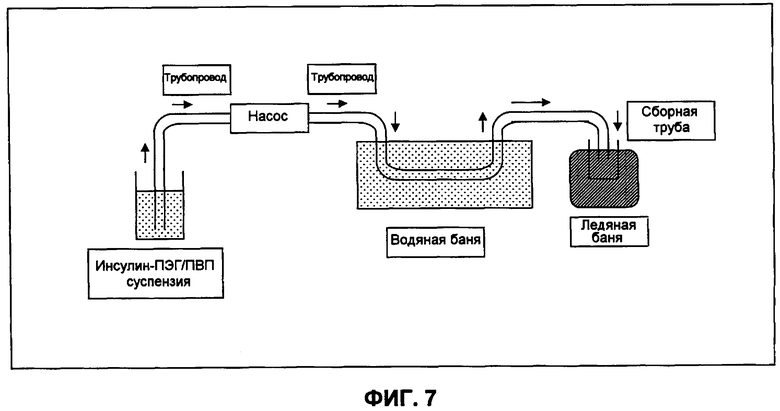
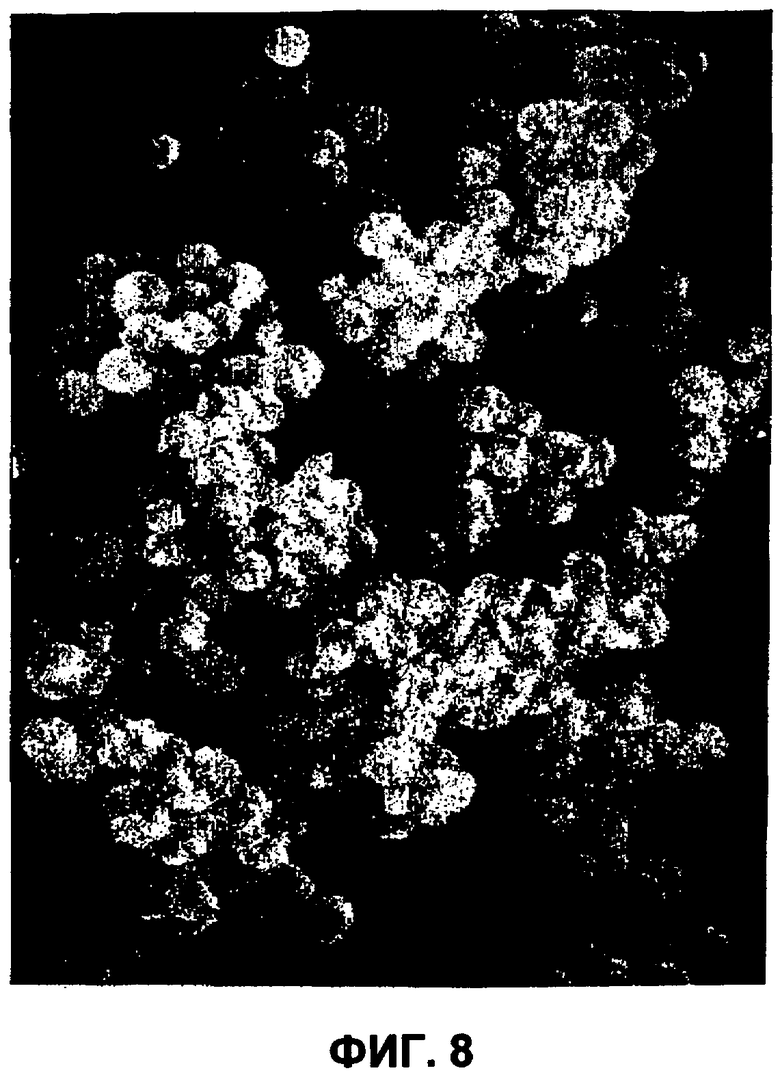
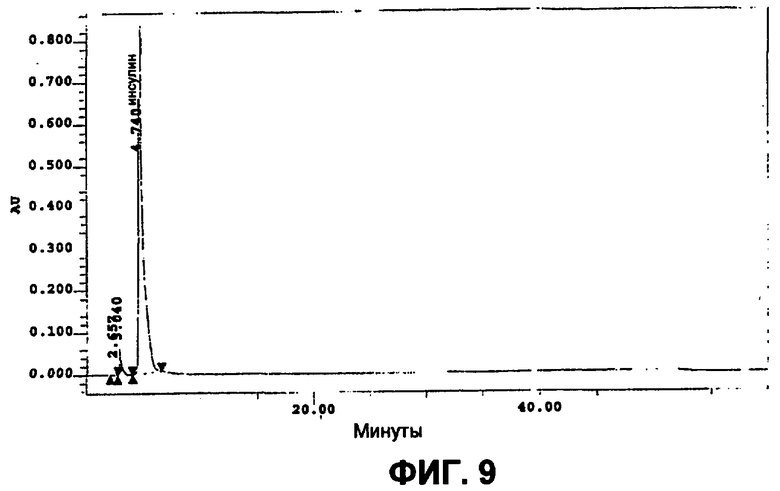
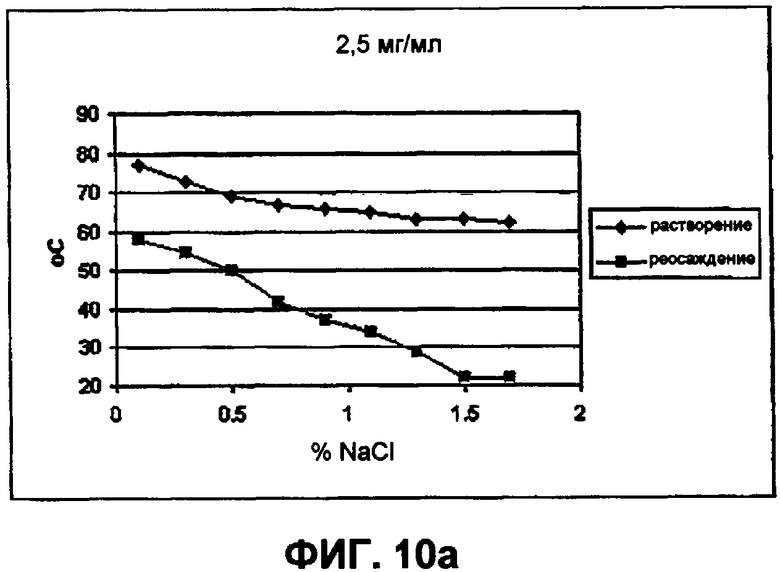
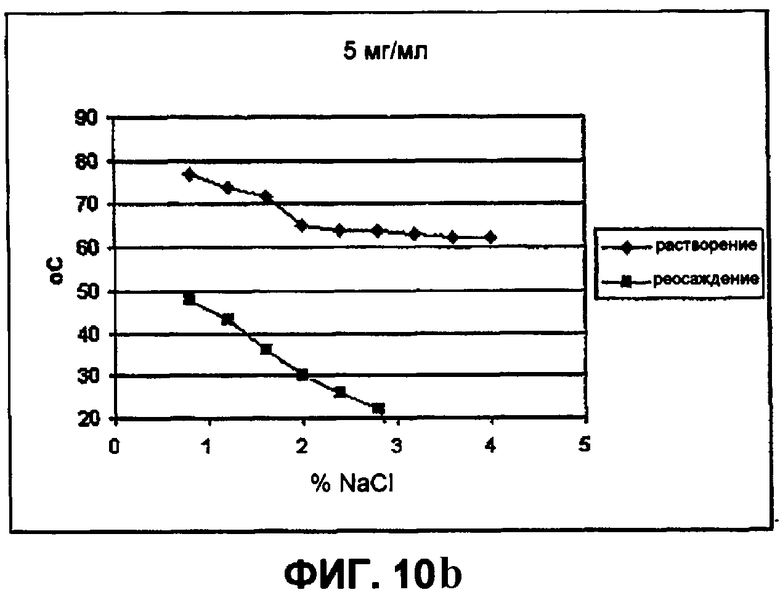
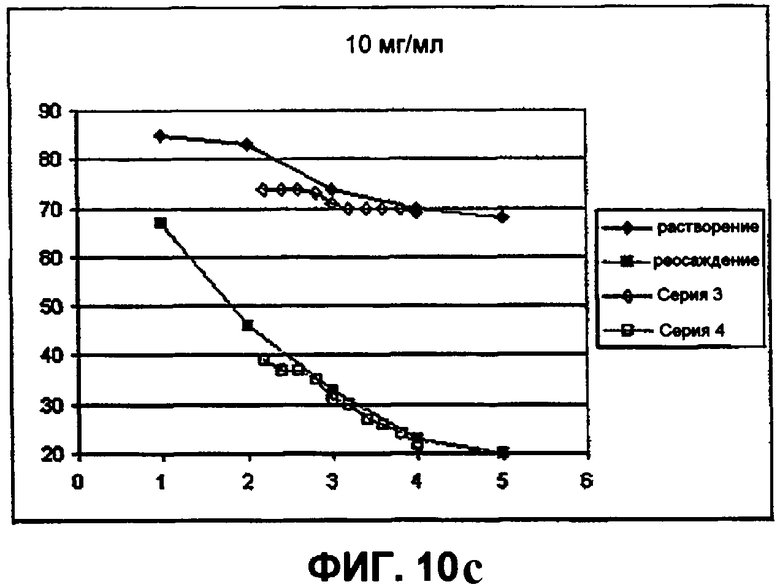
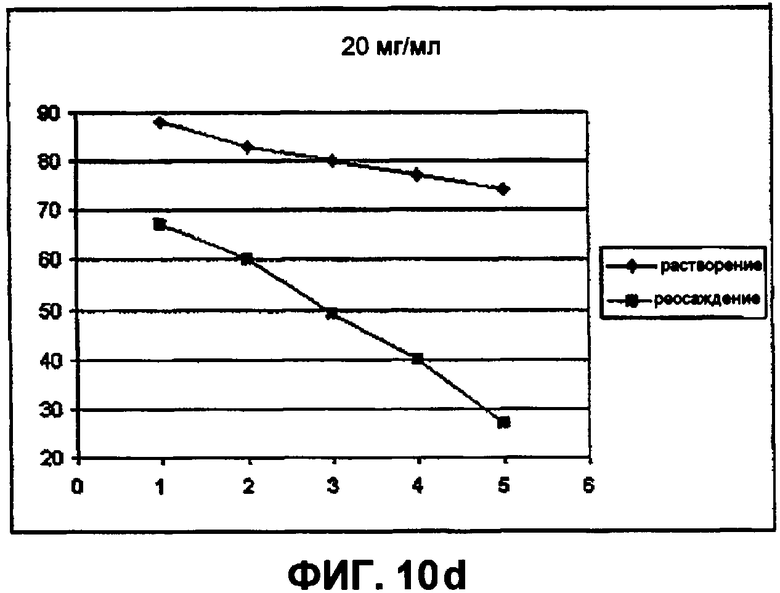
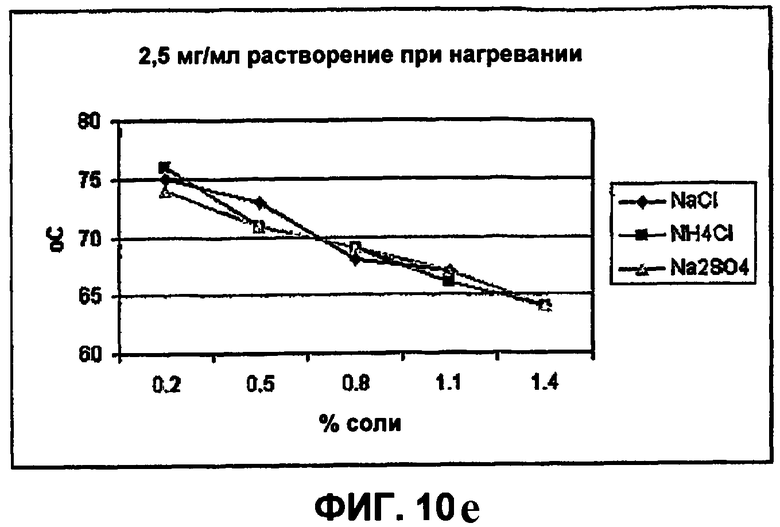
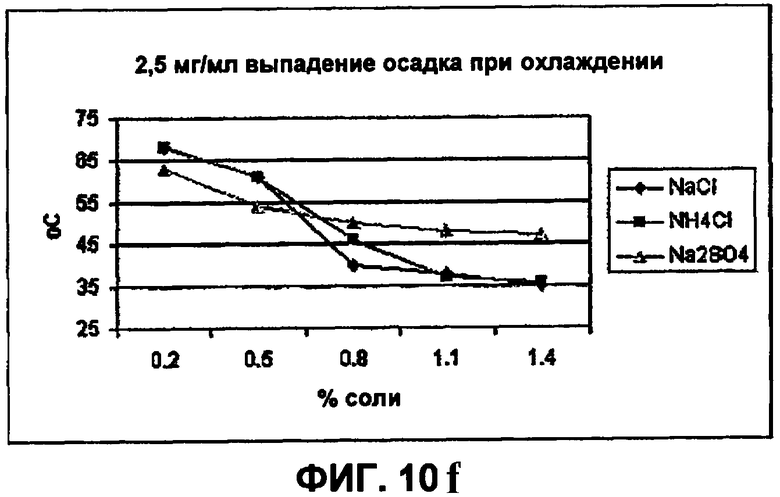
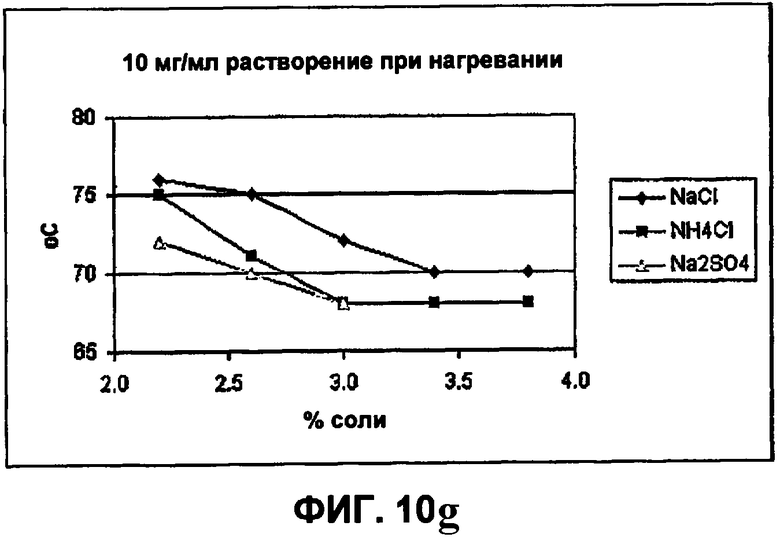
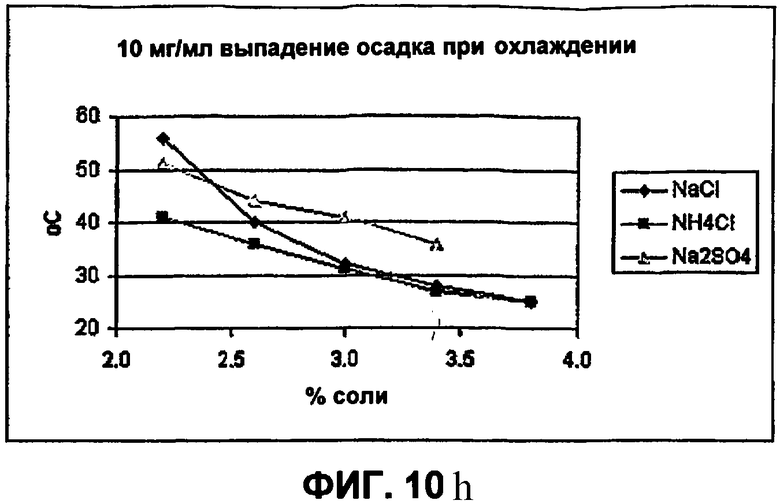
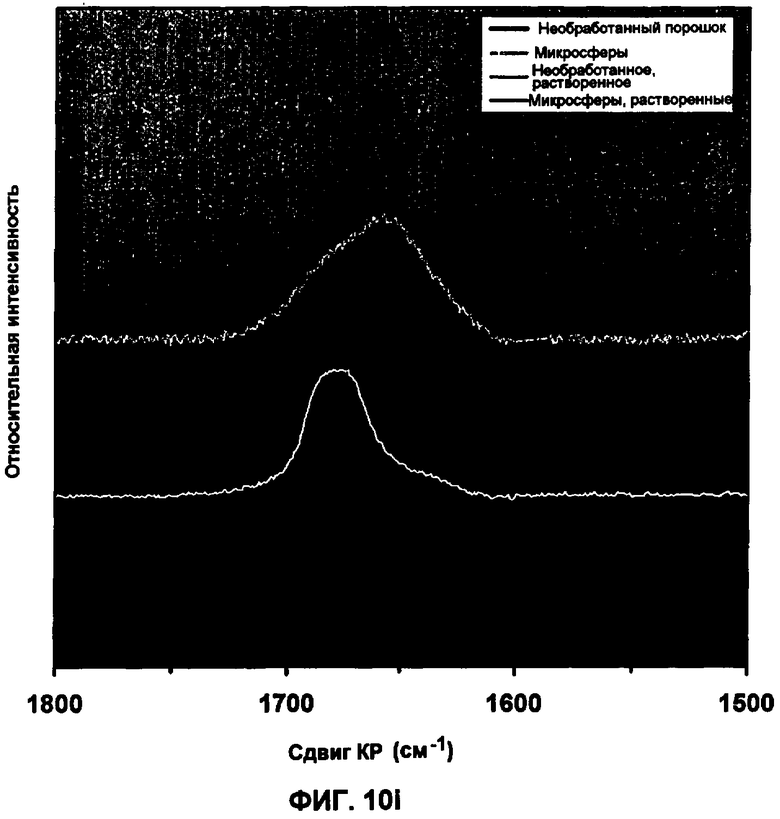
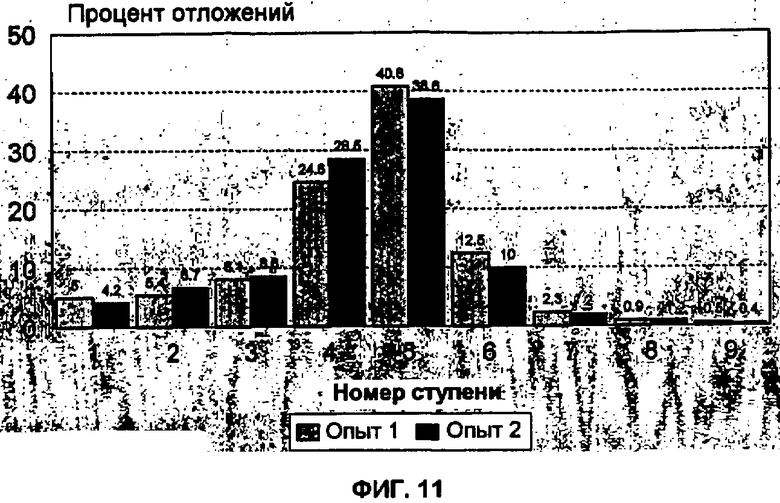


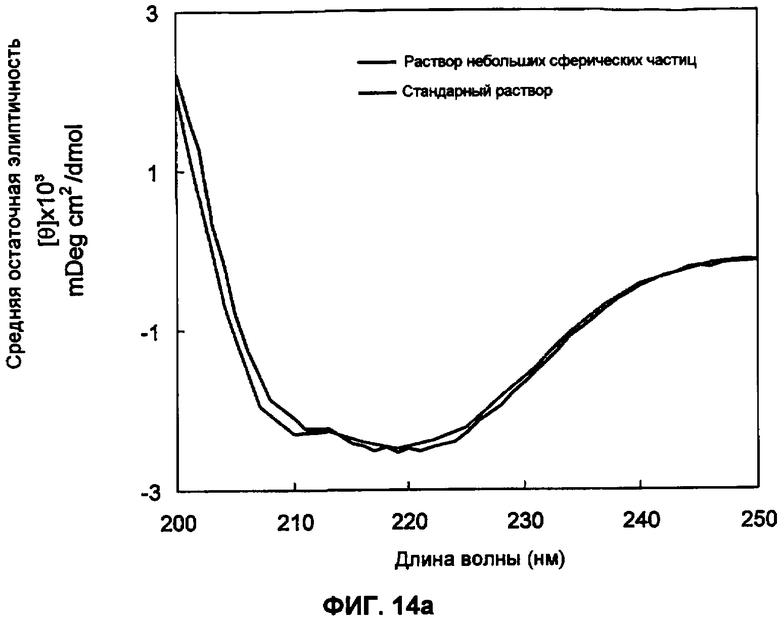
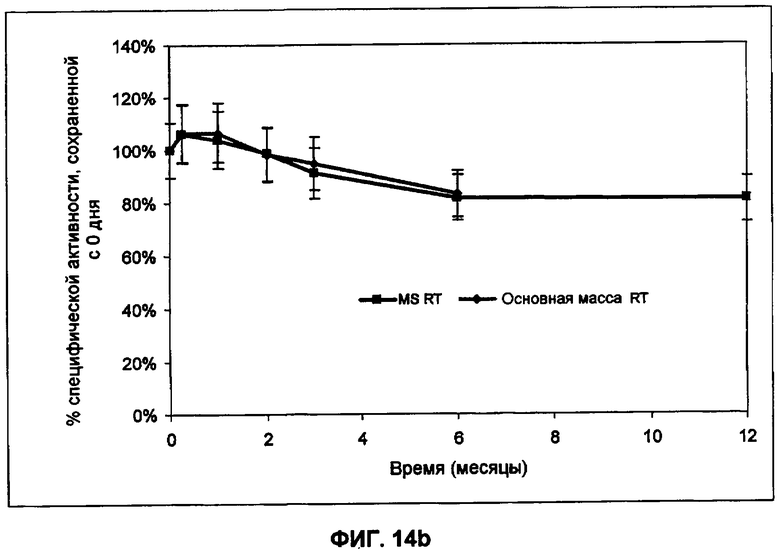
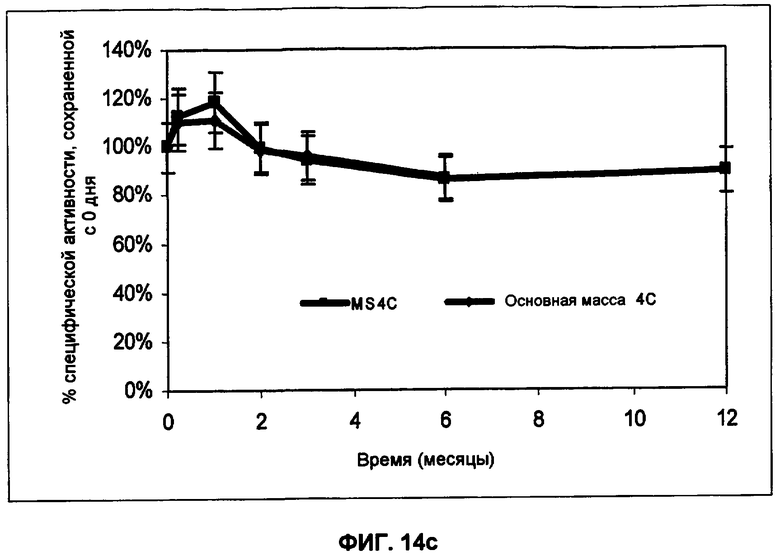
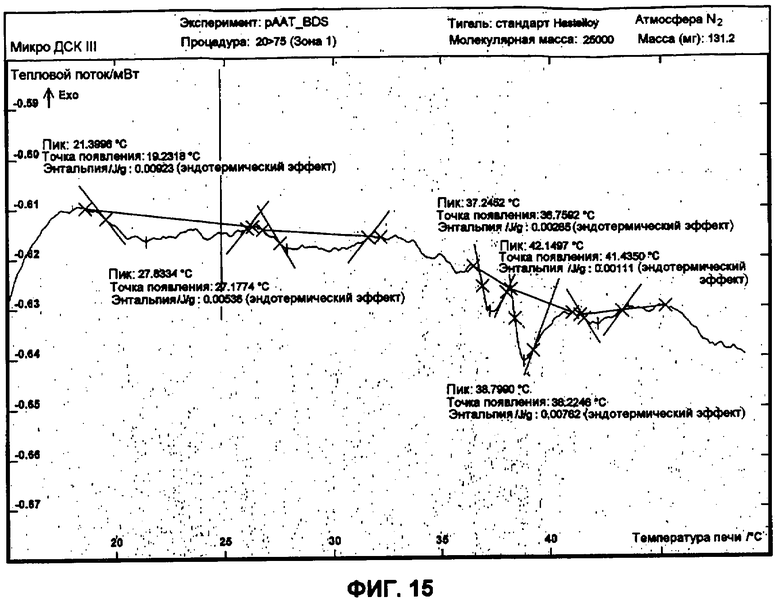
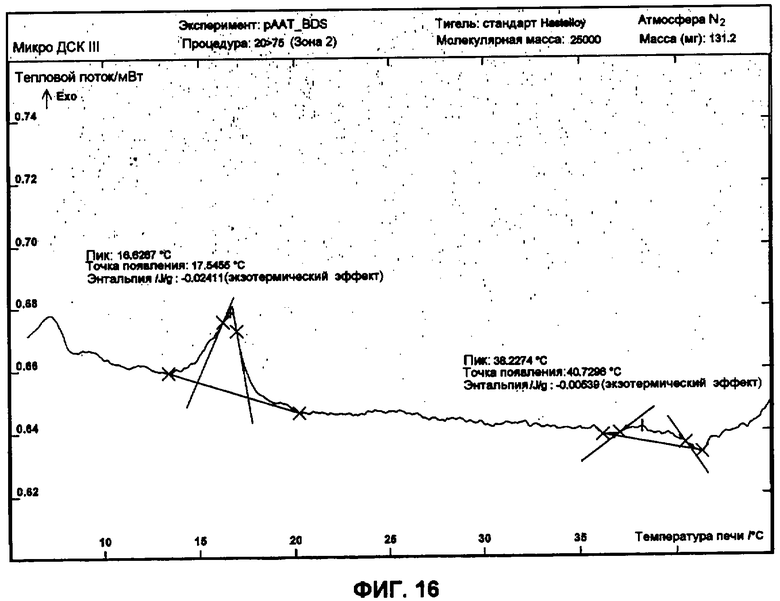
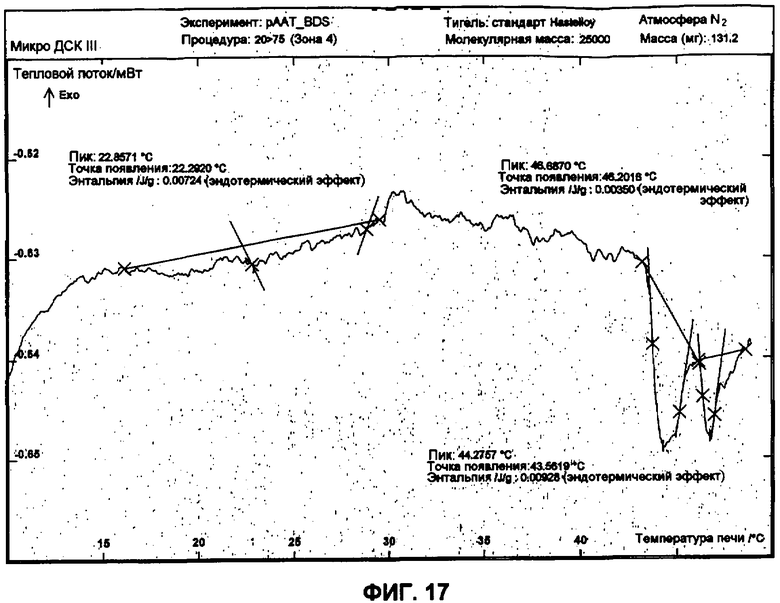
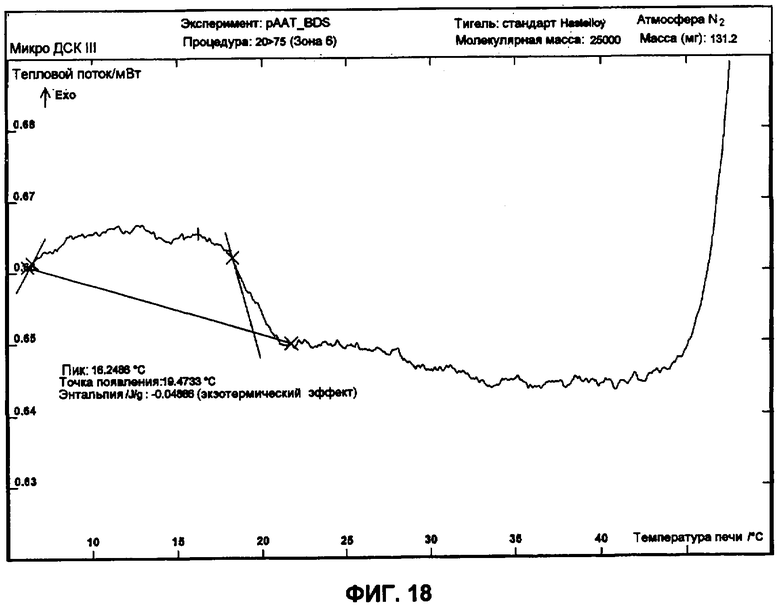
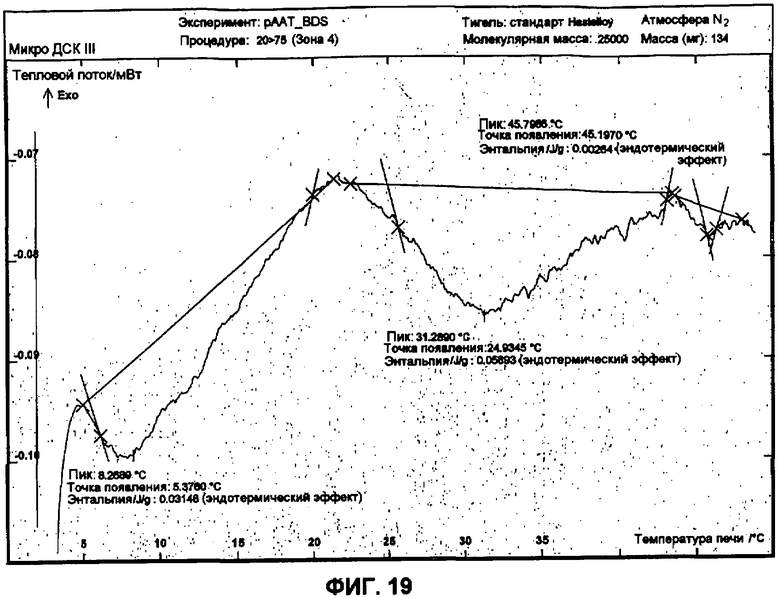
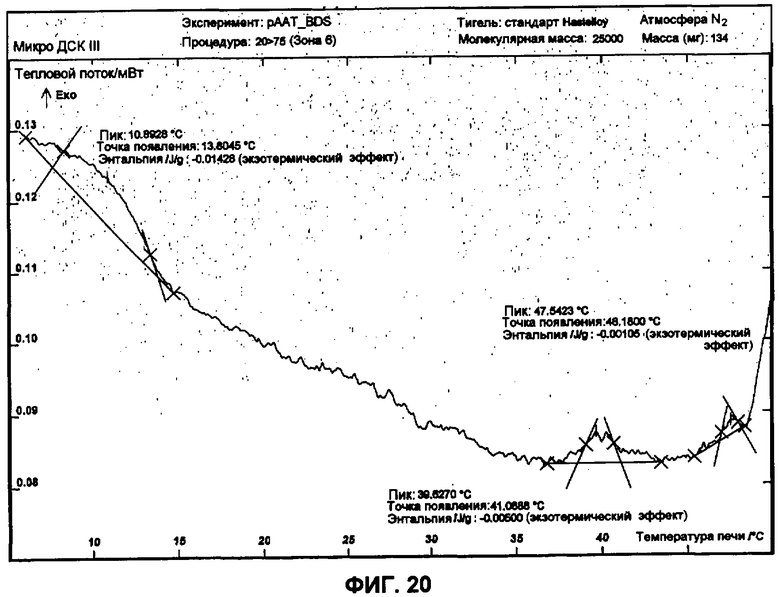
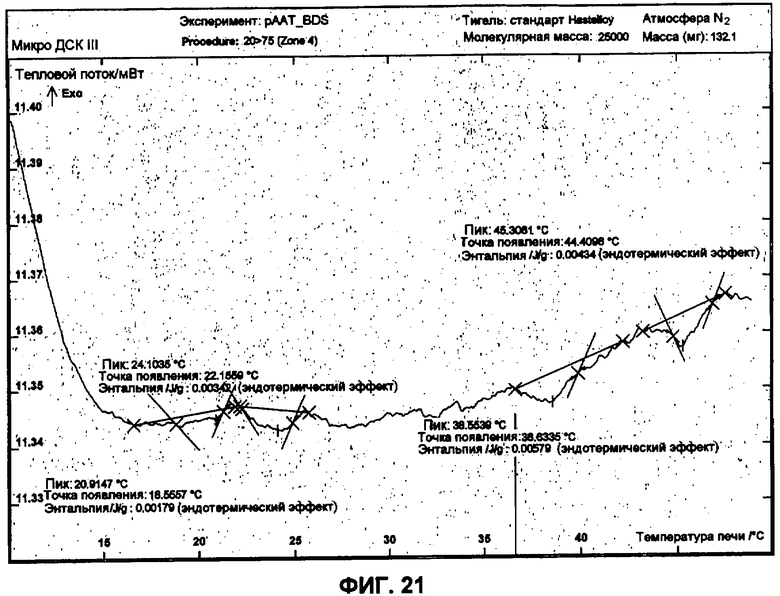
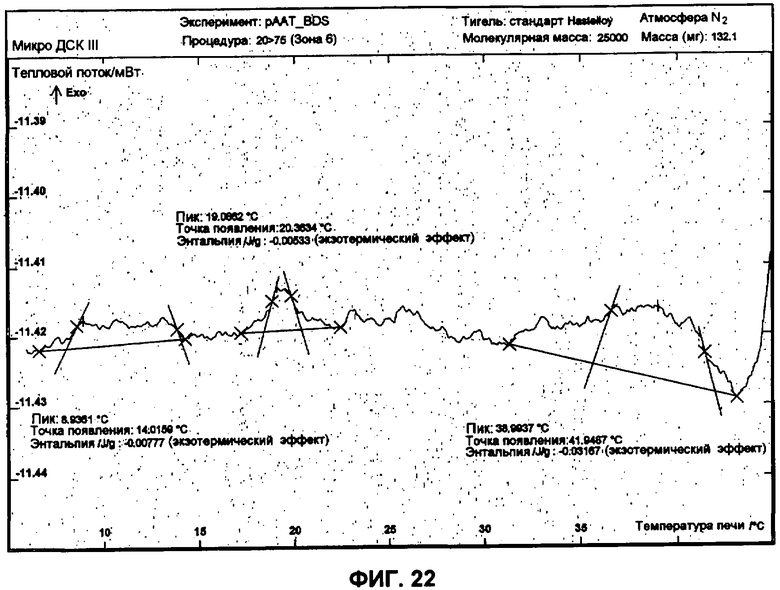
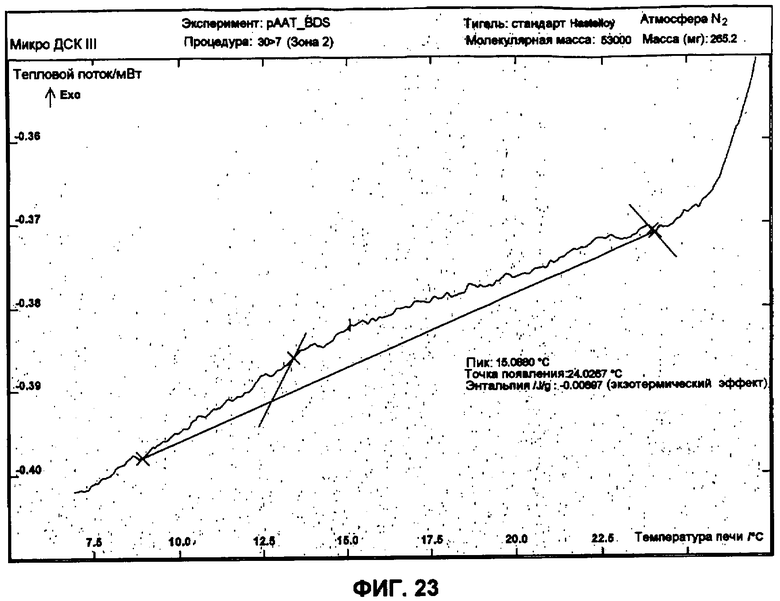
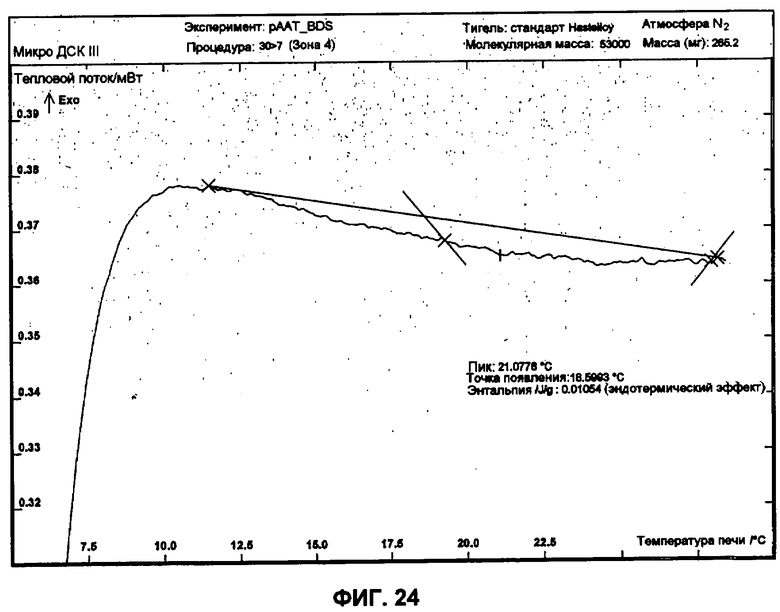
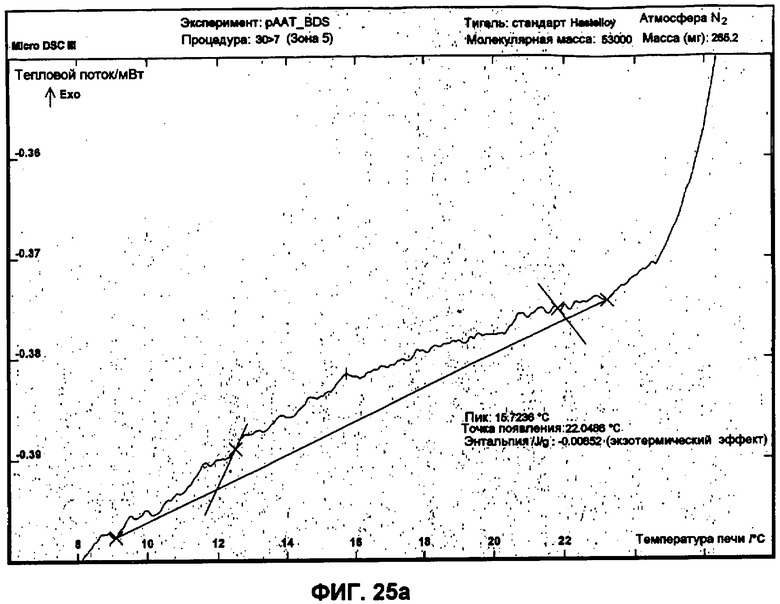
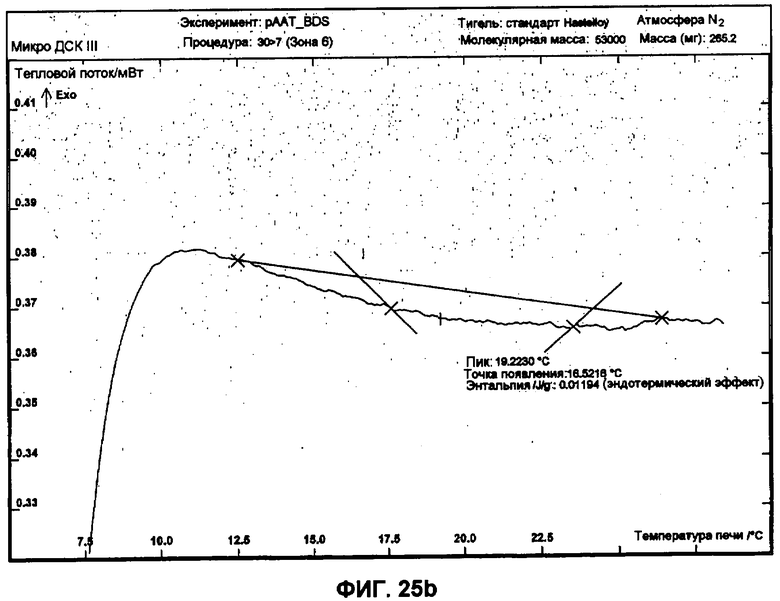

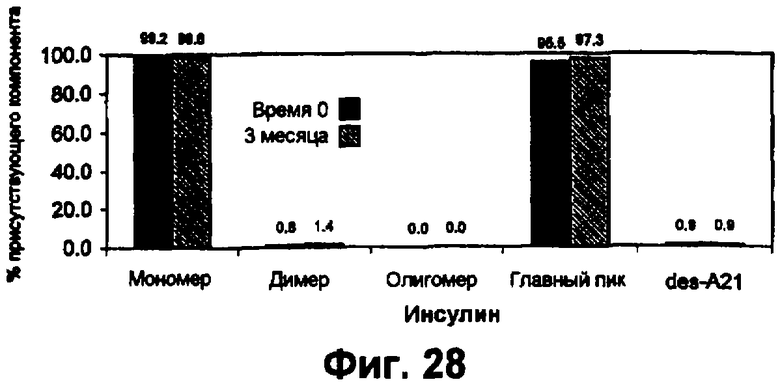
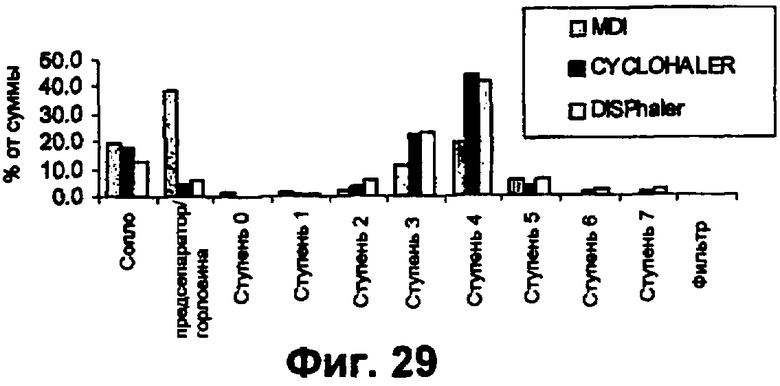
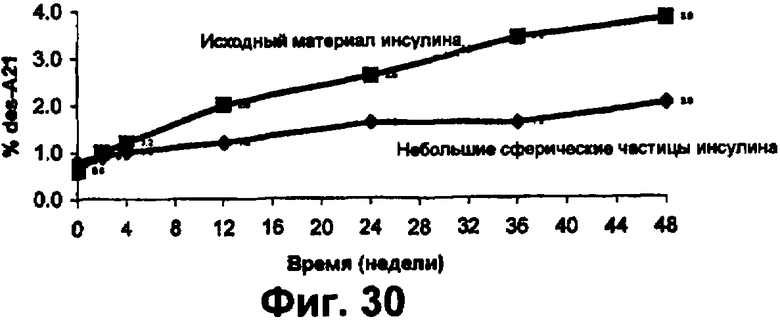
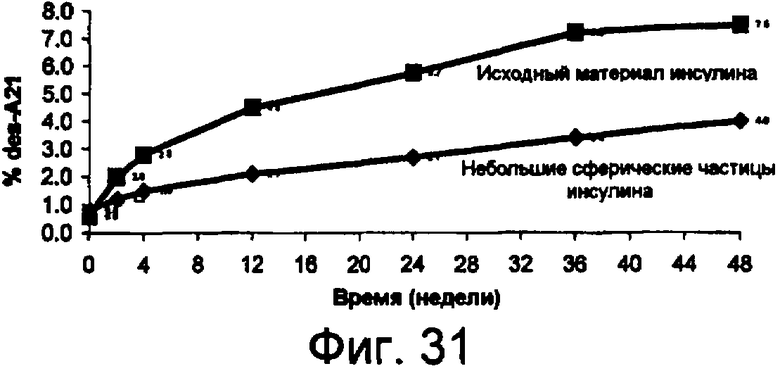
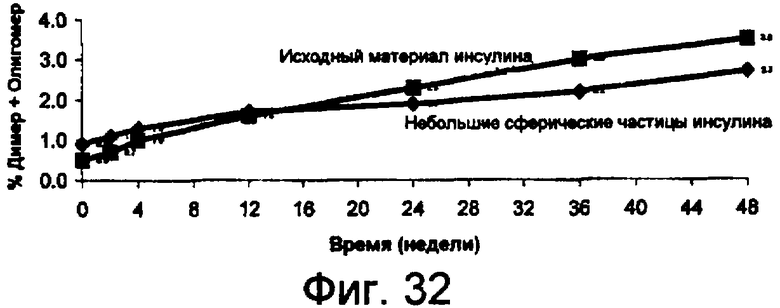
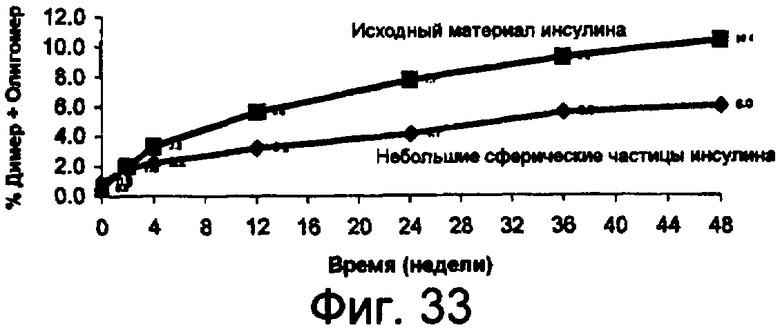
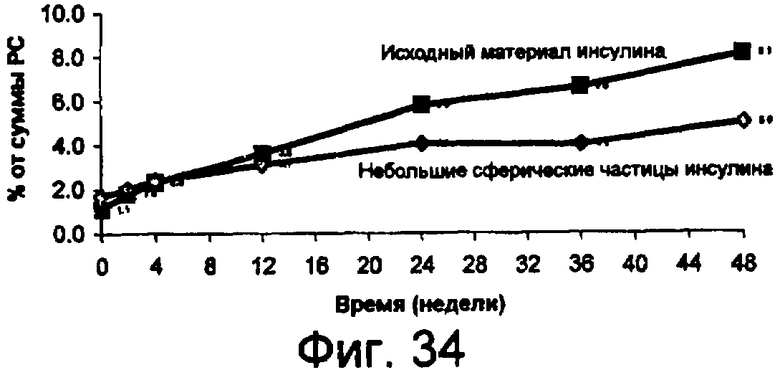
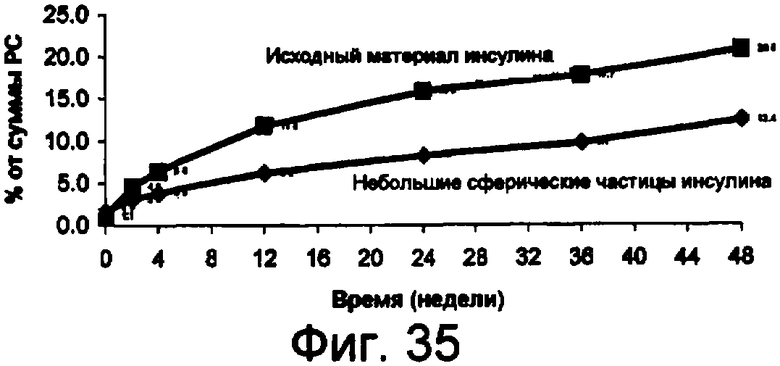
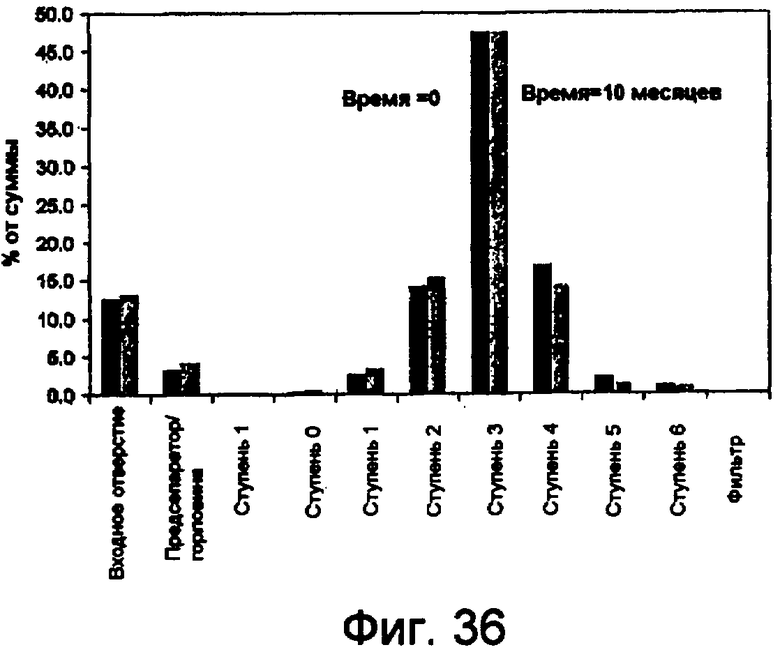

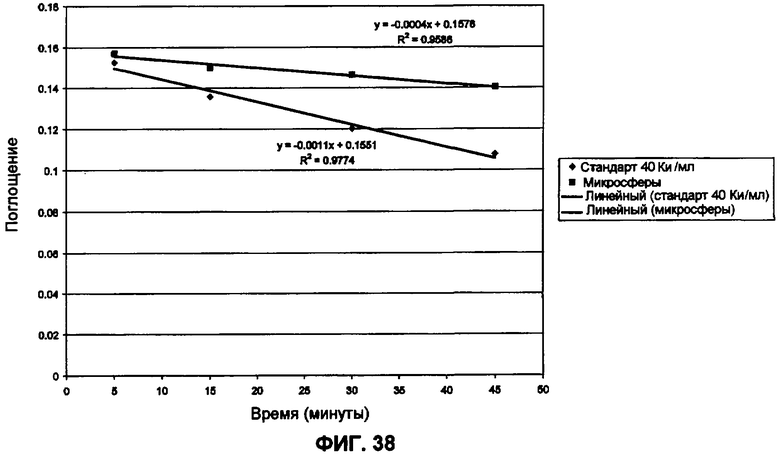

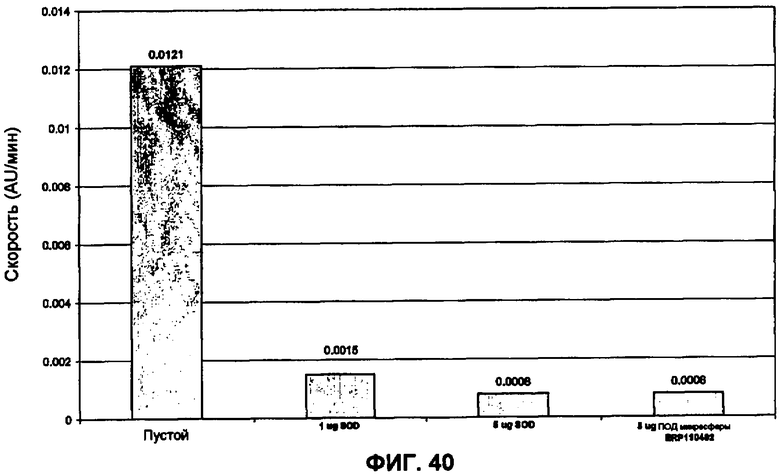
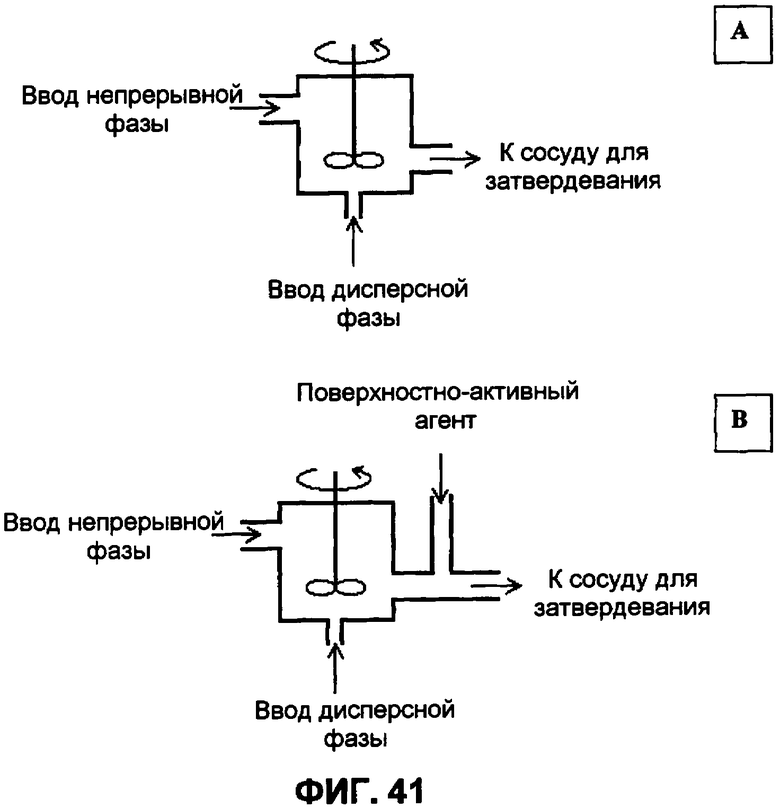
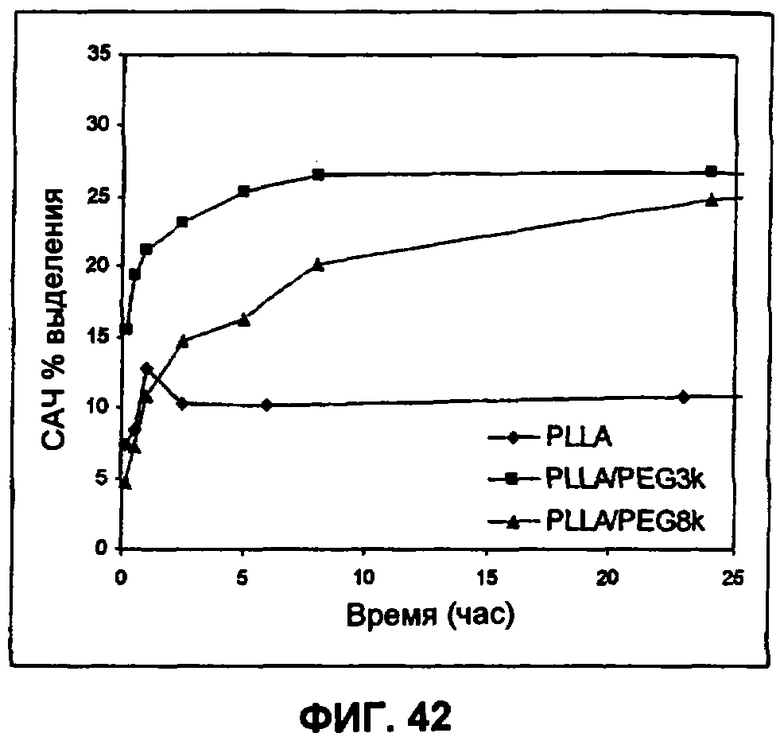
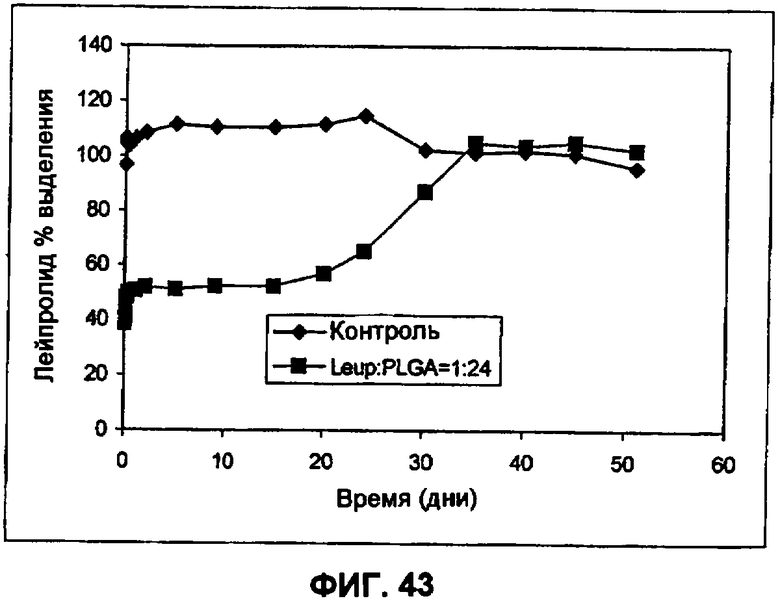
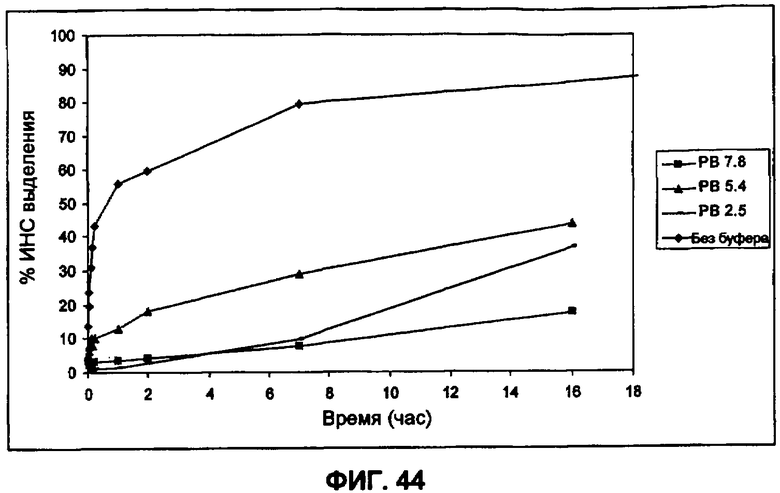
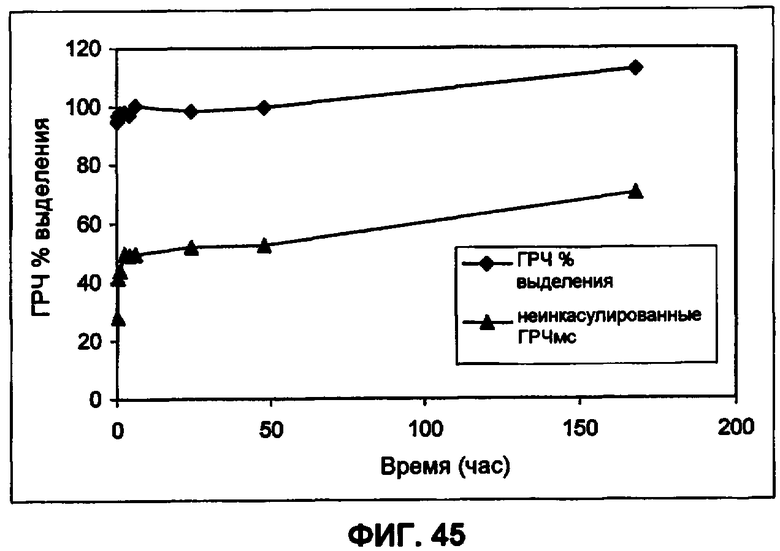
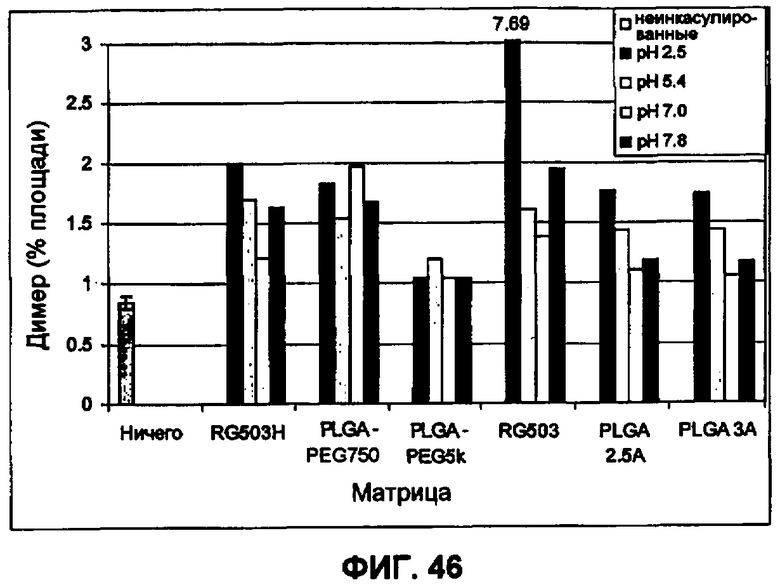
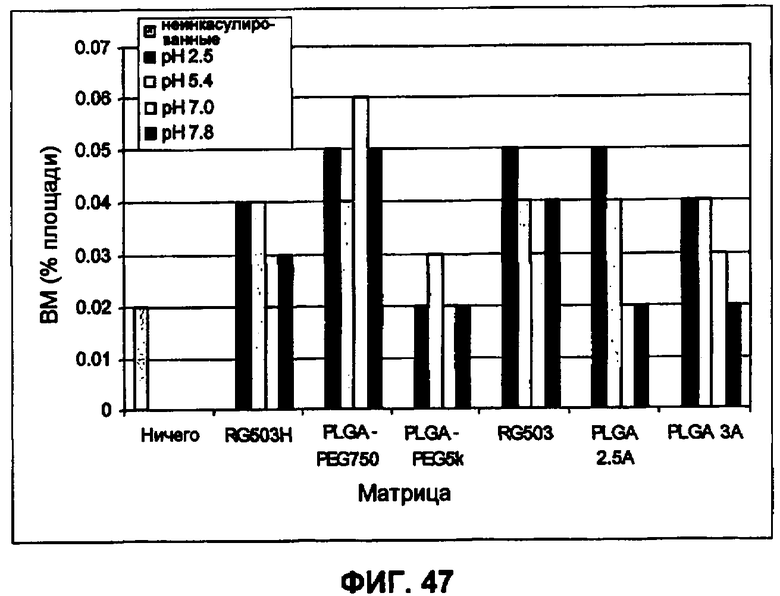
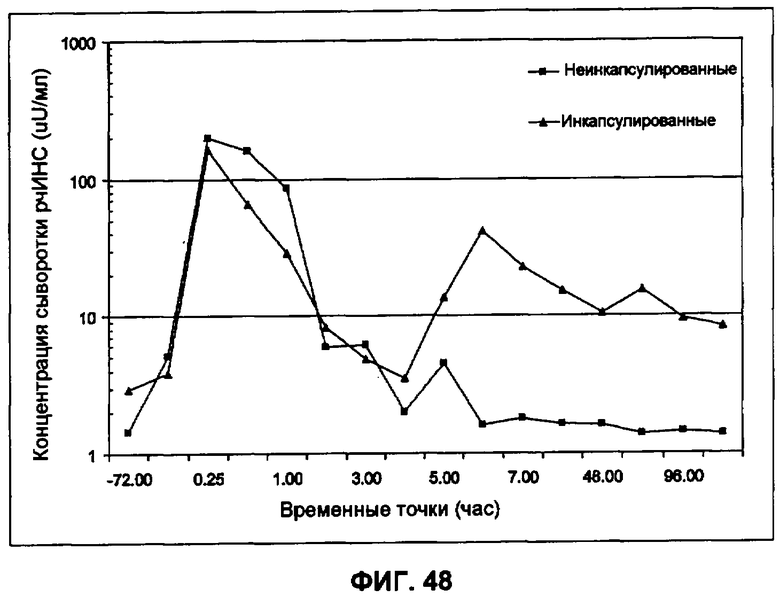
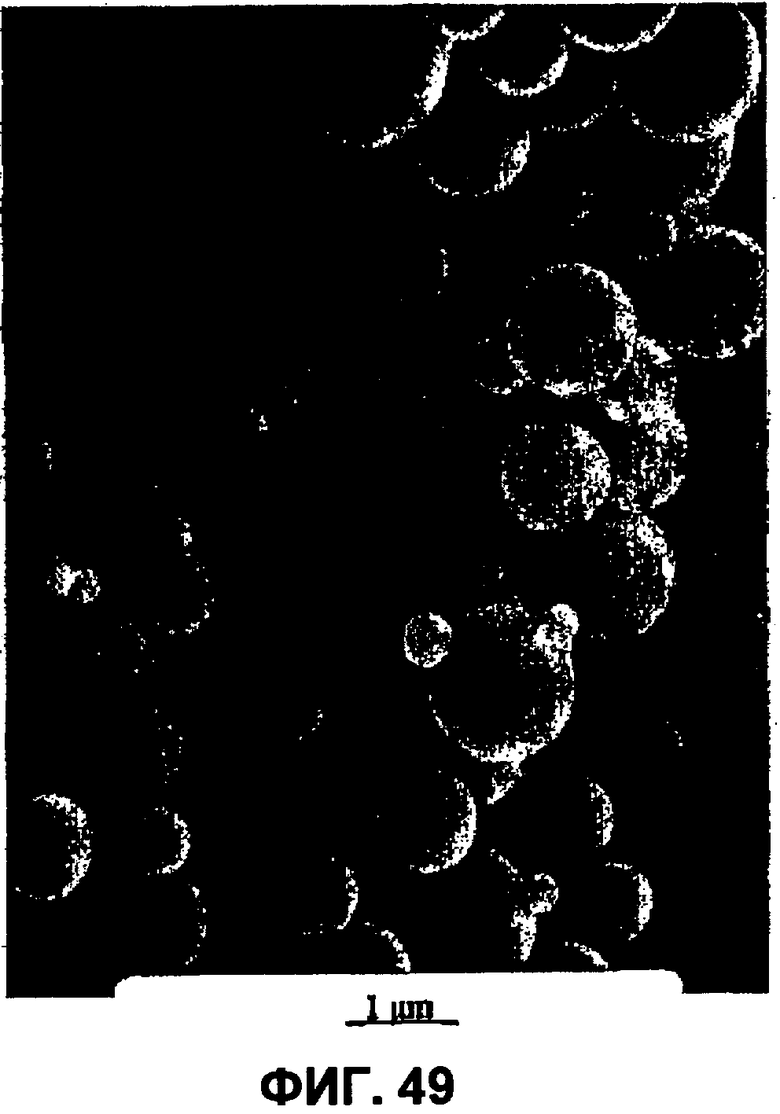
Изобретение относится к способу приготовления небольших сферических частиц активного агента при обеспечении раствора в единственной жидкой фазе. Единственная жидкая фаза содержит активный агент, усиливающий фазовое разделение агент и первый растворитель. Фазовое изменение индуцируется при контролируемой скорости в растворе, чтобы вызвать фазовое разделение жидкость-твердое активного агента и образовать жидкую фазу и твердую фазу, причем указанное индуцирование включает в себя охлаждение раствора. Твердая фаза содержит небольшие сферические частицы активного агента. Жидкая фаза содержит усиливающий фазовое разделение агент и растворитель. Небольшие сферические частицы являются, по существу, сферическими и имеют размер от, примерно, 0,01 мкм до, примерно, 200 мкм. 3 н. и 74 з.п. ф-лы, 49 ил., 6 табл.
1. Способ приготовления небольших сферических частиц активного агента, включающий
получение раствора в виде единственной жидкой фазы, содержащего активный агент, усиливающий фазовое разделение агент и первый растворитель, и
индуцирование фазового изменения с регулируемой скоростью в данном растворе для возникновения фазового разделения жидкость-твердое активного агента с образованием твердой фазы и жидкой фазы, причем твердая фаза содержит твердые небольшие сферические частицы активного агента, и жидкая фаза содержит усиливающий фазовое разделение агент и первый растворитель, небольшие, по существу сферические частицы, причем указанное индуцирование включает в себя охлаждение раствора.
2. Способ по п.1, в котором раствор имеет температуру фазового перехода, первую температуру и вторую температуру, и этот раствор охлаждают от первой температуры до второй температуры, причем первая температура находится выше температуры указанного фазового перехода раствора, а вторая температура находится ниже температуры указанного фазового перехода раствора.
3. Способ по п.1 или 2, дополнительно включающий этап, выбранный из группы, состоящей из регулирования концентрации активного агента, регулирования концентрации усиливающего фазовое разделение агента, регулирования ионной силы раствора, регулирования рН и регулирования осмоляльности раствора.
4. Способ по п.1 или 2, дополнительно включающий изменение концентрации активного агента.
5. Способ по п.1 или 2, дополнительно содержащий изменение концентрации усиливающего фазовое разделение агента.
6. Способ по п.2, в котором вторая температура находится выше температуры замерзания раствора.
7. Способ по п.2, в котором вторая температура находится ниже температуры замерзания раствора.
8. Способ по п.1 или 2, в котором этап охлаждения проводят с регулируемой скоростью, составляющей от 0,2 до 50°С/мин.
9. Способ по п.8, в котором регулируемая скорость находится в интервале от примерно 0,2 до примерно 30°С/мин.
10. Способ по п.1 или 2, в котором этап получения раствора включает растворение усиливающего фазовое разделение агента в первом растворителе с образованием смеси; и добавление активного агента к смеси с образованием раствора.
11. Способ по п.10 дополнительно содержит этап растворения активного агента в первом растворителе или втором растворителе, который смешивают с первым растворителем перед добавлением активного агента к смеси.
12. Способ по п.6, в котором раствор дополнительно содержит понижающий температуру замерзания агент для снижения температуры замерзания раствора.
13. Способ по п.12, в котором понижающий температуру замерзания агент выбирают из группы полиэтиленгликоля и пропиленгликоля.
14. Способ по п.1 или 2, в котором усиливающий фазовое разделение агент представляет собой водорастворимый или смешивающийся с водой агент.
15. Способ по п.1 или 2, в котором усиливающий фазовое разделение агент выбирают из группы, состоящей из линейных или разветвленных полимеров, полимеров на основе углеводов, полиалифатических спиртов, поливиниловых полимеров, полиакриловых кислот, полиорганических кислот, полиаминокислот, сополимеров и блоксополимеров, трет-полимеров, простых полиэфиров, природных полимеров, полиимидов, поверхностно-активных веществ, сложных полиэфиров, разветвленных и циклополимеров, полиальдегидов, крахмалов, замещенных крахмалов, полиэтиленгликоля, поливинилпирролидона, полоксамеров, этанола, ацетона и изопропанола.
16. Способ по п.1 или 2, в котором усиливающий фазовое разделение агент представляет собой полиэтиленгликоль (PEG).
17. Способ по п.1 или 2, в котором небольшие сферические частицы дополнительно содержат наполнитель для увеличения стабильности небольших сферических частиц, для обеспечения регулируемого выделения активного агента из небольших сферических частиц, или для усиления проникновения активного агента через биологические ткани.
18. Способ по п.17, в котором наполнитель выбирают из группы, состоящей из углеводов, катионов, анионов, аминокислот, липидов, жирных кислот, поверхностно-активных веществ, триглицеридов, желчных кислот или их солей, сложных эфиров жирных кислот и полимеров.
19. Способ по п.18, в котором катион выбирают из группы, состоящей из Zn2+, Mg2+ и Са2+.
20. Способ по п.18, в котором желчная кислота представляет собой холевую кислоту или ее соль.
21. Способ по п.1 или 2, дополнительно содержащий этап сбора небольших сферических частиц.
22. Способ по п.21, в котором этап сбора небольших сферических частиц осуществляют промыванием частиц жидкой средой при температуре, при которой активный агент не растворим в данной жидкой среде, а усиливающий фазовое разделение агент растворим в данной жидкой среде.
23. Способ по п.22, в котором этап промывания осуществляют диафильтрацией или центрифугированием.
24. Способ по п.22, в котором жидкая среда является водной или органической.
25. Способ по п.22, в котором жидкая среда представляет собой надкритическую жидкость или смесь надкритической жидкости и растворителя, смешивающегося с надкритической жидкостью.
26. Способ по п.24, в котором органическую жидкую среду выбирают из группы, состоящей из метиленхлорида, хлороформа, ацетонитрила, этилацетата, этанола и пентана.
27. Способ по п.22, в котором жидкая среда дополнительно содержит агент, который уменьшает растворимость активного агента в жидкой среде.
28. Способ по п.27, в котором агент, снижающий растворимость активного агента в жидкой среде, содержит комплексообразующий ион.
29. Способ по п.28, в котором комплексообразующий агент представляет собой катион, выбранный из группы, состоящей из: Zn2+, Са2+, Fe2+, Mg2+, Mn2+, Na+ и NH4 +.
30. Способ по п.22 дополнительно включающий этап удаления жидкой среды.
31. Способ по п.30, в котором этап удаления жидкой среды осуществляют лиофилизацией, сушкой или испарением.
32. Способ по п.22, в котором жидкая среда дополнительно содержит наполнитель.
33. Способ по п.32, в котором наполнитель увеличивает стабильность небольших сферических частиц, обеспечивает регулируемое выделение активного агента из небольших сферических частиц, или усиливает проникновение активного агента через биологические ткани.
34. Способ по п.33, в котором наполнитель выбирают из группы, состоящей из углеводов, катионов, анионов, аминокислот, липидов, жирных кислот, поверхностно-активных веществ, триглицеридов, желчных кислот или их солей, сложных эфиров жирных кислот и полимеров.
35. Способ по п.34, в котором катион выбирают из группы, состоящей из Zn2+, Mg2+ и Са2+.
36. Способ по п.34, в котором наполнитель представляет собой холевую кислоту или ее соль.
37. Способ по п.36, в котором усиливающий фазовое разделение агент выбран из группы, состоящей из полоксамеров, полиэтиленгликолей и их смесей.
38. Способ по п.1 или 2, в котором раствор содержит водный или смешивающийся с водой растворитель.
39. Способ по п.38, в котором смешивающийся с водой растворитель выбирают из группы, состоящей из N-метил-2-пирролидинона (N-метил-2-пирролидона), 2-пирролидинона (2-пирролидона), 1,3-диметил-2-имидазолидинона (ДМИ), диметилсульфоксида, диметилацетамида, уксусной кислоты, молочной кислоты, метанола, этанола, изопропанола, 3-пентанола, н-пропанола, бензилового спирта, глицерина, полиэтиленгликоля (ПЭГ), ПЭГ-4, ПЭГ-8, ПЭГ-9, ПЭГ-12, ПЭГ-14, ПЭГ-16, ПЭГ-120, ПЭГ-75, ПЭГ-150, сложных эфиров полиэтиленгликоля, ПЭГ-4 дилаурата, ПЭГ-20 дилаурата, ПЭГ-6 изостеарата, ПЭГ-8 пальмитостеарата, ПЭГ-150 пальмитостеарата, полиэтиленгликольсорбитанов, ПЭГ-20 сорбитанизостеарата, моноалкиловых эфиров полиэтиленгликоля, ПЭГ-3 диметилового эфира, ПЭГ-4 диметилового эфира, полипропиленгликоля (ППГ), полипропиленальгината, ППГ-10 бутандиола, ППГ-10 метилового эфира глюкозы, ППГ-20 метилового эфира глюкозы, ППГ-15 стеарилового эфира, дикаприлата/дикапрата пропиленгликоля, лаурата пропиленгликоля и гликофурола (эфира тетрагидрофурфурилового спирта и полиэтиленгликоля) или их комбинаций.
40. Способ по п.1 или 2, в котором активный агент представляет собой фармацевтически активный агент.
41. Способ по п.40, в котором фармацевтически активное соединение выбирают из группы, состоящей из терапевтических агентов, диагностических агентов, косметических средств, пищевых добавок и пестицидов.
42. Способ по п.1 или 2, в котором активный агент представляет собой макромолекулу.
43. Способ по п.42, в котором макромолекулу выбирают из группы, состоящей из протеинов, полипептидов, углеводов, полинуклеотидов, вирусов и нуклеиновых кислот.
44. Способ по п.43, в котором протеин выбирают из группы, состоящей из протеина свертывания потока крови, фактора VII, фактора VIII, фактора IX, субтилизина, овальбумина, альфа-1-антитрипсина, ДНКазы, супероксидной дисмутазы, лизоцима, рибонуклеазы, гиалуронидазы, коллагеназы, гормона роста, эритропоэтина, инсулино-подобных факторов роста или их аналогов, интерферонов, глатирамера, фактора стимуляции колоний гранулоцитов/макрофагов, фактора стимуляции колоний гранулоцитов, антител, моноклональных антител, поликлональных антител, Fab-фрагментов, одноцепочечных антител, ПЭГилированных протеинов, гликозилированных или гипергликозилированных протеинов, десмопрессина, LHRH агонистов, таких как лейпролид, госерелин, нафарелин, бусерелин, LHRH антагонисты, вазопрессин, циклоспорин, кальцитонин, гормон околощитовидной железы, пептиды гормона околощитовидной железы и инсулин.
45. Способ по п.1 или 2, в котором частицы пригодны для in vivo доставки к объекту, нуждающемуся в активном агенте.
46. Способ по п.45, в котором способ доставки выбирают из группы, состоящей из способов впрыскивания, вдыхания, парентерального, местного, орального, ректального, носового, легочного, вагинального, буккального, через кожу, через слизистую оболочку, подъязычного, ушного, глазного, внутриглазного и офтальмического.
47. Способ по п.46, в котором способ доставки представляет собой доставку в легкие.
48. Способ по п.47, в котором частицы пригодны для отложения в центральной или периферийной области легких.
49. Способ по п.47, в котором частицы доставляют устройством, выбранным из группы, состоящей из ингалятора сухого порошка, ингалятора измеренной дозы и распылителя.
50. Способ по п.45, в котором частицы доставляют в виде стабильной жидкой суспензии.
51. Способ по п.1 или 2, в котором частицы имеют, по существу, одинаковый размер частиц.
52. Способ по п.1 или 2, в котором частицы имеют средний размер частиц от примерно 0,01 до примерно 200 мкм.
53. Способ по п.1 или 2, в котором частицы имеют средний размер частиц от примерно 0,5 до примерно 10 мкм.
54. Способ по п.1 или 2, в котором активный агент составляет от примерно 0,1 до примерно 100% частицы по массе.
55. Способ по п.1 или 2, в котором активный агент составляет от примерно 75 до примерно 100% частицы по массе.
56. Способ по п.1 или 2, в котором содержание активного агента равно или превышает 90% частицы по массе.
57. Способ по п.1 или 2, в котором небольшие сферические частицы имеют узкое распределение размера.
58. Способ по п.57, в котором соотношение объемного диаметра 90-го процентиля небольших сферических частиц к объемному диаметру 10-го процентиля, меньшее или равное примерно 5.
59. Способ по п.1 или 2, в котором небольшие сферические частицы являются полукристаллическими или не кристаллическими.
60. Способ приготовления небольших сферических частиц активного агента, причем данный способ содержит этапы:
растворения активного агента и усиливающего фазовое разделение агента в водном или смешивающемся с водой растворителе с образованием раствора единственной непрерывной фазы, и
индуцирования фазового изменения, посредством чего активный агент претерпевает фазовое разделение жидкость-твердое с образованием твердой фазы, содержащей твердые небольшие сферические частицы активного агента, и жидкой фазы усиливающего фазовое разделение агента, причем указанное индуцирование включает в себя охлаждение раствора с регулируемой скоростью.
61. Способ по п.60, в котором раствор имеет температуру фазового превращения, первую температуру и вторую температуру, и этап доведения раствора до фазового изменения осуществляют охлаждением раствора от первой температуры до второй температуры, где первая температура находится выше температуры фазового превращения раствора и вторая температура находится ниже температуры фазового превращения раствора.
62. Способ по п.60, в котором регулируемая скорость находится в интервале от примерно 0,2 до примерно 50°С/мин.
63. Способ по п.1, в котором указанное индуцирование включает охлаждение раствора с регулируемой скоростью до температуры, выше температуры замерзания указанного раствора.
64. Способ по п.63, в котором указанная регулируемая скорость выше 25,6°С/мин.
65. Способ по п.1, в котором указанный усиливающий фазовое разделение агент включает смесь полоксамера и полиэтиленгликоля.
66. Способ по п.1, в котором первым растворителем является вода.
67. Способ по п.1, в котором процент превращения активного агента из исходного раствора в частицы составляет по меньшей мере 90%.
68. Способ по п.1, в котором активный агент, высвобождаемый из частиц, сохраняет биологическую активность активного агента в исходном растворе, которая определена на основе его особых свойств.
69. Способ по п.1, в котором активный агент, высвобождаемый из частиц, сохраняет структурную чистоту активного агента в исходном растворе, которая определена спектроскопией кругового дихроизма.
70. Способ по п.1, в котором небольшие сферические частицы имеют геометрическое стандартное отклонение менее чем 2,5.
71. Способ по п.70, в котором геометрическое стандартное отклонение менее 1,8 и соотношение объемного диаметра 90-го процентиля указанных частиц к объемному диаметру 10-го процентиля указанных частиц составляет менее 3.
72. Способ приготовления твердых, небольших сферических частиц активного агента, включающий:
получение водного раствора в виде единственной жидкой фазы, и содержащего активный агент и усиливающий фазовое разделение агент;
охлаждение водного раствора с регулируемой скоростью до температуры ниже фазового перехода активного агента в растворе;
с получением таким образом суспензии, содержащей твердые небольшие частицы активного агента, суспендированные в жидкой фазе, содержащей усиливающий фазовое разделение агент и воду, причем небольшие сферические частицы, по существу, сферические.
73. Способ по п.72, в котором охлаждение водного раствора проводят с постоянной или линейной скоростью, нелинейной скоростью, неравномерной или программируемой скоростью.
74. Способ по п.72, в котором охлаждение водного раствора проводят со скоростью более 0,2°С/мин.
75. Способ по п.72, в котором водный раствор дополнительно содержит агент, понижающий температуру замерзания.
76. Способ по п.72, в котором указанное охлаждение включает охлаждение раствора до температуры выше температуры замерзания раствора.
77. Способ по п.75, в котором агент, понижающий температуру замерзания, содержит полиэтиленгликоль.
| ВЫСУШЕННЫЕ РАСПЫЛЕНИЕМ МИКРОЧАСТИЦЫ КАК ТЕРАПЕВТИЧЕСКИЕ НОСИТЕЛИ | 1995 |
|
RU2147226C1 |
| US 5300464 A, 05.04.1994 | |||
| US 6395302 B1, 28.05.2002 | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 5849884 A, 15.12.1998. | |||

