Родственные заявки
Настоящая заявка претендует на приоритет заявки на патент США № 10/881160, поданной 1 июля 2004 года, которая преобразована в предварительную заявку, описание которой включается сюда в качестве ссылки во всей ее полноте.
Область техники
Здесь описываются новые пептиды и новые защищенные пептиды, полученные из пептидов полимиксина и октапептина, включая, например, колистин, циркулин A, полимиксин A, полимиксин B, полимиксин D, октапептин B, октапептин C и [Ile7]полимиксин B1. Новые пептиды и новые защищенные пептиды имеют антибактериальные свойства. Также описываются фармацевтические композиции, содержащие новые пептиды и новые защищенные пептиды, а также способы получения новых пептидов и новых защищенных пептидов.
Уровень техники
Грамотрицательные бактерии, которые устойчивы к аминогликозидным, β-лактамным и фторхинолоновым антибиотикам, становятся все более распространенными. Эти бактерии часто являются чувствительными к полимиксинам и родственным пептидам, имеющим антибактериальные свойства (ссылки 1, 10, 23). В результате представляет интерес использование полимиксинов против устойчивых к множеству лекарственных средств грамотрицательных бактериальных инфекций у людей (ссылка 23).
Пептиды, такие как полимиксин B и родственный ему колистин (полимиксин E), вводятся людям в качестве антибактериальных агентов. Однако их использование ранее было ограниченным из-за их токсичности. Эти пептиды включают в себя циклический пептид из семи аминокислот, присоединенный к экзоциклической цепи из трех аминокислот, в которой N-концевой амин экзоциклической цепи связан с "боковой цепью" или "хвостом". Чаще всего, хвост представляет собой ацильную группу.
При рекомендуемой дозировке полимиксина B пациентам наблюдается некоторая почечная токсичность. Нейротоксичность наблюдается также у пациентов с ослабленной функцией почек, в среднем в 7,3% случаев, как сообщается в одном из больших исследований с колистином (ссылка 1). Ацильная экзоциклическая цепь и соседний N-концевой остаток 2,4-диаминобутановой кислоты (Dab) может ферментативно отщепляться от полимиксина, тем самым давая соответствующий нонапептид. Токсичность in vivo нонапептида полимиксина B значительно меньше, чем у самого полимиксина B (ссылка 16). Токсичность нонапептида в культуре клеток уменьшается примерно в 100 раз, по отношению к полимиксину B; однако антибактериальная активность нонапептида также уменьшается примерно в 2-64 раза, по отношению к полимиксину B (ссылка 11).
Делаются попытки химической модификации полимиксина и колистина для получения пептидов с улучшенными антибактериальными свойствами. Например, общий синтез полимиксина B и четырех аналогов осуществлялся ранее посредством объединения твердофазного синтеза пептида для получения линейных структур, с последующим отщеплением от смолы и конденсирования в растворе при сильном разбавлении, с получением структуры циклического пептида (ссылка 7). Производные, однако, были менее активны, чем полимиксин B. Более поздний общий синтез полимиксина B и нескольких близко родственных соединений осуществляют только посредством твердофазного пептидного синтеза (ссылки 15, 26). Хотя оба этих полностью твердофазных подхода к синтезу могут дать новые производные полимиксина, эти способы выглядят ограниченными, поскольку количества полученного антибиотика являются малыми и требуются большие количества аминокислотных предшественников. Любое масштабирование этих способов для клинических исследований, как показано, может быть сложным и дорогостоящим.
Соответственно, остается возрастающая потребность в новых пептидных соединениях, имеющих антибактериальные свойства, и в новых способах получения таких соединений.
Сущность изобретения
Здесь описываются новые пептиды, такие как пептидные антибиотики, и/или другие пептиды, имеющие антибактериальные свойства, и способы получения пептидов. Описанные здесь соединения могут обеспечить структурное разнообразие в экзоциклической области (экзоциклические аминокислоты и хвост) циклического пептида.
Определения
"Ацил", как здесь используется, относится к карбонильному радикалу, присоединенному к алкильной, алкенильной, алкинильной, циклоалкильной, гетероциклильной, арильной или гетероарильной группе. Пример ацилов включают в себя, но, не ограничиваясь этим: (1) "незамещенный алканоил", который определяется как карбонильный радикал, присоединенный к незамещенной алкильной группе; (2) "незамещенный алкеноил", который определяется как карбонильный радикал, присоединенный к незамещенной алкенильной группе; (3) "замещенный алканоил", который определяется как карбонильный радикал, присоединенный к замещенной алкильной группе, в которой один или несколько атомов водорода заменены группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила; амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо; и (4) "замещенный алкеноил", который определяется как карбонильный радикал, присоединенный к замещенной алкенильной группе, в которой один или несколько атомов водорода заменены группой-заместителем, как описано выше. Неограничивающие примеры ацилов включают в себя радикалы, такие как ацетил, н-октаноил, н-нонаноил, бензоил и изоникотиноил.
"Ациламино", как здесь используется, относится к аминогруппе, соединенной с ацильной группой.
"Ацилокси", как здесь используется, относится к кислородному радикалу, замещенному ацильной группой. В некоторых вариантах осуществления, ацилокси является замещенной ацильной, ациламино, ацилокси, алкенильной, алкокси, алкильной, алкинильной, амино, арильной, арилокси, карбамоильной, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формильной, гуанидино, галогеновой, гетероарильной, гетероциклильной, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинильной, сульфонамино, сульфонильной, тио, тиоациламино, тиоуреидо или уреидо группой.
"Реагент присоединения", как здесь используется, представляет собой соединение, которое может взаимодействовать с аминогруппой, такой как N-концевая группа пептида, при этом химически модифицируя аминогруппу посредством связывания всего реагента присоединения или его компонента с аминогруппой. Например, реагент присоединения может представлять собой ациламино реагент, такой как R'-(C=O)-LG или сульфонирующий реагент, такой как R'-SO2-LG, где LG представляет собой уходящую группу, которая может взаимодействовать с аминогруппой, с образованием ациламиногруппы или сульфонаминогруппы соответственно. Реагент присоединения может также представлять собой, например, изоцианат, изотиоцианат, сложный активированный эфир, хлорангидрид, сульфонилхлорид, активированный сульфонамид, активированный гетероцикл, активированный гетероарил, хлорформиат, цианоформиат, сложный тиоациловый эфир, фосфорилхлорид, фосфорамидат, имидат или лактон. Реагент присоединения может также представлять собой альдегид или кетон, который взаимодействует с амином при восстановительных условиях, с образованием алкилированного амина. Реагент присоединения может также представлять собой активированную аминокислоту или аминокислоту и пептидный конденсирующий реагент, такой, например, как PyBOP® (бензотриазол-1-ил-окси-трис-пирролидинофосфоний гексафторфосфат), HBtU (2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат), HBtU/HOBt (2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат/N-гидроксибензотриазол) или DCC (дициклогексилкарбодиимид).
"Алкенил", как здесь используется, относится к линейным или разветвленным радикалам, имеющим 2-20 атомов углерода, например, 2-12, 2-10 или 2-6 атомов углерода, и содержащим, по меньшей мере, одну двойную связь углерод-углерод. Один или несколько атомов водорода могут также заменяться группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо. Часть (части) с двойной связью ненасыщенной углеводородной цепи может находиться либо в цис, либо в транс конфигурации. Неограничивающие примеры алкенилов включают в себя "незамещенный алкенил", который определяется как алкенильная группа, которая не несет группы-заместителя. Другие неограничивающие примеры алкенильных групп включают в себя этенил, 2-фенил-1-этенил, проп-1-ен-2-ил, проп-2-ен-1-ил (аллил), проп-1-ен-1-ил, циклопроп-1-ен-1-ил; циклопроп-2-ен-1-ил, бут-1-ен-1-ил, бут-1-ен-2-ил, 2-метил-проп-1-ен-1-ил, бут-2-ен-1-ил, бут-2-ен-2-ил, бута-1,3-диен-1-ил, бута-1,3-диен-2-ил, циклобут-1-ен-1-ил, циклобут-1-ен-3-ил, циклобут-1,3-диен-1-ил.
"Алкокси", как здесь используется, относится к кислородному радикалу, замещенному алкильной, алкенильной, алкинильной, циклоалкильной или гетероциклильной группой. Неограничивающие примеры включают в себя метокси, трет-бутокси, бензилокси и циклогексилокси.
"Алкил", как здесь используется, относится к линейным или разветвленным насыщенным радикалам, имеющим, по меньшей мере, один атом углерода, например, 1-20 атомов углерода, 1-12, 1-10, или 1-6 атомов углерода, или, по меньшей мере, 6 атомов углерода, по меньшей мере, 7 атомов углерода, по меньшей мере, 8 атомов углерода, по меньшей мере, 9 атомов углерода или, по меньшей мере, 10 атомов углерода, если не указано иного. "Низший алкил" определяется как алкильная группа, содержащая 1-4 атома углерода. Один или несколько атомов водорода могут также заменяться группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинил, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо. Неограничивающие примеры алкильных групп включают в себя метил, бутил, трет-бутил, изопропил, трифторметил, нонил, ундецил, октил, додецил, метоксиметил, 2-(2'-аминофенацил), 3-индолилметил, бензил и карбоксиметил. Другие примеры алкилов включают в себя, но, не ограничиваясь этим: (1) "незамещенный алкил", который определяется как алкильная группа, которая не несет группы-заместителя; и (2) "замещенный алкил", который обозначает алкильный радикал, в котором один или несколько атомов водорода заменяются группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо. Примеры алкильных групп включают в себя, но, не ограничиваясь этим, метил, этилы, такие как этанил (этил), пропилы, такие как пропан-1-ил (н-пропил), пропан-2-ил (изопропил), бутилы, такие как бутан-1-ил (н-бутил), бутан-2-ил (втор-бутил), 2-метил-пропан-1-ил (изо-бутил), 2-метил-пропан-2-ил (трет-бутил), трифторметил, нонил, ундецил, октил, додецил, метоксиметил, 2-(2'-аминофенацил), 3-индолилметил, бензил и карбоксиметил.
"Алкинил", как здесь используется, относится к линейным и разветвленным радикалам, имеющим 2-10 атомов углерода и содержащим, по меньшей мере, одну тройную связь углерод-углерод. Один или несколько атомов водорода могут также заменяться группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо. Примеры алкинильных групп включают в себя, но, не ограничиваясь этим, этинил, проп-1-ин-1-ил, проп-2-ин-1-ил, бут-1-ин-1-ил, бут-1-ин-3-ил и бут-3-ин-1-ил.
"Амино", как здесь используется, относится к радикалу NR1R2, в котором R1 и R2 могут выбираться из гидридо, ацила, ацилокси, алкенила, алкокси, алкила, алкинила, арила, арилокси, карбамоила, карбоалкокси, карбокси, формила, гетероарила, гетероциклила, гидрокси, имино, нитро, оксо, сульфинила, сульфонила и тио. Монозамещенная амино относится к радикалу NR1R2, где R1 представляет собой гидридо и R2 выбирают из ацила, ацилокси, алкенила, алкокси, алкила, алкинила, арила, арилокси, карбамоила, карбоалкокси, карбокси, формила, гетероарила, гетероциклила, гидрокси, имино, нитро, оксо, сульфинила, сульфонила и тио. Дизамещенная амино относится к радикалу NR1R2, где R1 и R2, каждый, независимо, выбирают из ацила, ацилокси, алкенила, алкокси, алкила, алкинила, арила, арилокси, карбамоила, карбоалкокси, карбокси, формила, гетероарила, гетероциклила, гидрокси, имино, нитро, оксо, сульфинила, сульфонила и тио.
"Аминокислота", как здесь используется, относится к соединению, содержащему группу карбоновой кислоты и аминогруппу и имеющему формулу H2N[C(R)(R')]n-C(O)OH, где n представляет собой целое число, которое больше или равно единице, и R и R' независимо выбираются из водорода и аминокислотных боковых цепей. Например, когда n равен единице, аминокислота формулы H2N[C(R)(R')]C(O)OH представляет собой альфа аминокислоту, а когда n равен двум, аминокислота формулы H2N-C(R1)(R1')-C(R2)(R2')-C(O)OH представляет собой бета-аминокислоту, где R1, R1', R2 и R2', каждый, независимо, выбирают из аминокислотных боковых цепей. "Аминокислотный остаток", как здесь используется, относится к аминокислоте, которая представляет собой часть пептида или белка и имеет формулу -N(H)C(R)(R')C(O)-. "Аминокислотная боковая цепь", как здесь используется, относится к любой боковой цепи из встречающейся в природе или синтетической аминокислоты. Например, метил может упоминаться как аланиновая боковая цепь, а 2-амино-1-этил может упоминаться как боковая цепь 2,4-диаминобутановой кислоты.
Примеры аминокислот могут выбираться из двенадцати кодированных аминокислот и их производных, а также, например, из других α-аминокислот, β-аминокислот, γ-аминокислот, δ-аминокислот и ω-аминокислот. Аминокислота может иметь R или S хиральность на любом хиральном атоме. Аминокислота может выбираться, например, из аланина, β-аланина, α-аминоадипиновой кислоты, 2-аминобутановой кислоты, 4-аминобутановой кислоты, 1-аминоциклопентанкарбоновой кислоты, 6-аминогексановой кислота, 2-аминогептандионовой кислоты, 7-аминогептановой кислоты, 2-аминоизомасляной кислоты, аминометилпирролкарбоновой кислоты, 8-амино-3,6-диоксаоктановой кислоты, аминопиперидинкарбоновой кислоты, 3-аминопропионовой кислоты, аминосерина, аминотетрагидропиран-4-карбоновой кислоты, аргинина, аспарагина, аспарагиновой кислоты, азетидинкарбоновой кислоты, бензотиазолилаланина, бутилглицина, карнитина, 4-хлорфенилаланина, цитруллина, циклогексилаланина, циклогексилстатина, цистеина, 2,4-диаминобутановой кислоты, 2,3-диаминопропионовой кислоты, дигидроксифенилаланина, диметилтиазолидинкарбоновой кислоты, глутаминовой кислоты, глютамина, глицина, гистидина, гомосерина, гидроксипролина, изолейцина, изонипекотиновой кислоты, лейцина, лизина, метанопролина, метионина, норлейцина, норвалина, орнитина, п-аминобензойной кислоты, пеницилламина, фенилаланина, фенилглицина, пиперидинилаланина, пиперидинилглицина, пролина, пирролидинилаланина, саркозина, селеноцистеина, серина, статина, тетрагидропиранглицина, тиенилаланина, треонина, триптофана, тирозина, валина, алло-изолейцина, алло-треонина, 2,6-диамино-4-гексановой кислоты, 2,6-диаминопимелиновой кислоты, 2,3-диаминопропионовой кислоты, дикарбоксидина, гомоаргинина, гомоцитрулина, гомоцистеина, гомоцистина, гомофенилаланина, гомопролина и 4-гидразинобензойной кислоты.
N-защищенные α-аминокислоты для пептидного синтеза, имеющие L- или D-конфигурацию на Cα, являются коммерчески доступными, например, от Novabiochem® (San Diego, CA) и Bachem (Bubendorf, Switzerland). Синтез хиральных α-аминокислот и других аминокислот также хорошо известен специалистам в данной области, и описывается, например, в Arnstein Synthesis of amino acids and proteins, University Park Press, 1975; Enantioselective Synthesis of Beta-Amino Acids, Juaristi et al., Eds., Wiley-VCH: New York, 2005; и Williams Synthesis of optically active α-amino acids, Pergamon Press, 1989.
"Аминозащитная группа", как здесь используется, относится к любому заместителю, который может использоваться для предотвращения участия аминогруппы молекулы в химической реакции, в то время как осуществляется химическое изменение в другой части молекулы. Аминозащитная группа может удаляться при соответствующих химических условиях. Многочисленные аминозащитные группы известны специалистам в данной области, и примеры аминозащитных групп, способов их введения, и способов их удаления можно найти в "Protective Groups in Organic Synthesis" by Theodora W. Greene, John Wiley and Sons, New York, 1991, описание которой включается сюда в качестве ссылки. Неограничивающие примеры аминозащитных групп включают в себя фталимидо, трихлорацетил, STA-основание, бензилоксикарбонил, трет-бутоксикарбонил, трет-амилоксикарбонил, изоборнилоксикарбонил, адамантилоксикарбонил, хлорбензилоксикарбонил и нитробензилоксикарбонил. Другие примеры аминозащитных групп включает в себя "карбаматные аминозащитные группы", которые определяются как карбонил-содержащая защитная группа, которая когда связывается с аминогруппой, образует карбамат. Неограничивающие примеры карбаматных аминозащитных групп включают в себя 9-флуоренилметоксикарбонильную (Fmoc), аллилоксикарбонильную (Alloc), карбобензилокси (CBZ) и трет-бутоксикарбонильную (Boc) защитные группы. Другие примеры защитных групп включают в себя 9-флуоренилметоксикарбонил (Fmoc) замещенный кислотными заместителями, такой как 2-сульфо-9-флуоренилметоксикарбонилкарбонил, 2-карбоксиметил-9-флуоренилметоксикарбонил и 4-карбокси-9-флуоренилметоксикарбонил.
"Реагенты для защиты аминогрупп", как здесь используется, относятся к реагентам присоединения, которые могут взаимодействовать с аминогруппой, такой как N-концевая группа пептида, при этом, химически модифицируя указанную аминогруппу посредством присоединения аминозащитной группы.
"Арил", как здесь используется, относится к моно-, би- или другой мульти-карбоциклической ароматической кольцевой системе. Арильная группа может быть необязательно сконденсирована с одним или несколькими кольцами, выбранными из арилов, циклоалкилов и гетероциклилов. Арилы могут иметь от 5 до 14 элементов кольца, например, от шести до десяти элементов кольца. Один или несколько атомов водорода также могут заменяться группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, азидо, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, циклоалкила, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо. Неограничивающие примеры арильных групп включают в себя фенил, нафтил, бифенил и антраценил.
"Арилокси", как здесь используется, относится к кислородному радикалу, замещенному арильной или гетероарильной группой. Примеры арилокси включают в себя, но, не ограничиваясь этим, фенокси.
"Карбамоил", как здесь используется, относится к азотному радикалу формулы -N(Rx2)-C(O)-ORx3, где Rx2 выбирают из гидридо, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила, и Rx3 выбирают из алкила, арила, циклоалкила, гетероарила и гетероциклила.
"Карбоалкокси", как здесь используется, относится к карбонильному радикалу, соединенному с алкокси- или арилоксигруппой.
"Карбокси», как здесь используется, относится к радикалу COOH.
"Карбоксиамино", как здесь используется, относится к радикалу CONH2.
"Карбоксиамидо", как здесь используется, относится к карбонильному радикалу, соединенному с монозамещенной амино или дизамещенной аминогруппой.
"Циклоалкил", как здесь используется, относится к насыщенному или частично ненасыщенному карбоциклическому кольцу в одинарной или конденсированной карбоциклической кольцевой системе, имеющей от трех до двенадцати элементов кольца, такой как кольцевая система, имеющая от трех до семи элементов кольца. Один или несколько атомов водорода могут также заменяться группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинил, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, циклоалкила, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо. Неограничивающие примеры циклоалкильных групп включают в себя циклопропил, циклобутил, циклогексил и циклогептил.
"Fmoc" представляет собой 9-флуоренилметоксикарбонильную группу.
"Галоген", как здесь используется, относится к радикалу брома, хлора, фтора или йода.
"Гетероарил", как здесь используется, относится к ароматическому радикалу, имеющему от одного до четырех гетероатомов или гетерогрупп, выбранных из O, N, NH, S или SO, в одинарной или конденсированной гетероциклической кольцевой системе, имеющей от пяти до пятнадцати элементов кольца, такой как гетероарильная кольцевая система, имеющая от шести до десяти элементов кольца. Один или несколько атомов водорода также могут заменяться группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, циклоалкила, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо. Неограничивающие примеры гетероарильных групп включают в себя индолильную, пиридинильную, тиазолильную, тиадиазолильную, изохинолинильную, пиразолильную, оксазолильную, оксадиазолильную, триазолильную и пирролильную группы.
"Гетероциклил" или "гетероциклическое соединение", как здесь используется, относится к насыщенному или частично ненасыщенному кольцу, содержащему от одного до четырех гетероатомов или гетерогрупп, выбранных из O, N, NH, N-(алкила, такого как низший алкил), S, SO или SO2, в отдельной или конденсированной гетероциклической кольцевой системе, имеющей от трех, до двенадцати элементов кольца, такой как гетероциклильная кольцевая система, имеющая от трех до семи элементов кольца. Один или несколько атомов водорода также могут заменяться группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, циклоалкила, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо. Неограничивающие примеры гетероциклильных групп включают в себя морфолинил, пиперидинил, пирролидинил и сукцинимидил.
"Гидрокси", как здесь используется, относится к -OH.
"Иминоамино", как здесь используется, относится к -N(H)C(=NRx26)Rx27, где Rx26 и Rx27 выбираются из гидридо, алкила, арила, циклоалкила, гетероарила и гетероциклила.
"Фосфонамино", как здесь используется, относится к 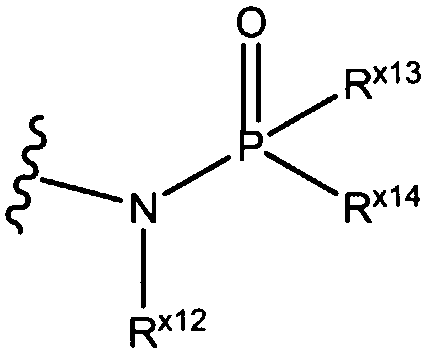 , где Rx13 и Rx14 независимо выбирают из алкокси, алкила, амино, арила, арилокси, циклоалкила, дизамещенной амино, галогена, гетероарила, гетероциклила, гидрокси, монозамещенной амино и тио.
, где Rx13 и Rx14 независимо выбирают из алкокси, алкила, амино, арила, арилокси, циклоалкила, дизамещенной амино, галогена, гетероарила, гетероциклила, гидрокси, монозамещенной амино и тио.
"Сульфинил", как здесь используется, относится к -S(=O)OH.
"Сульфо", как здесь используется, относится к -SO3H.
"Сульфонамино", как здесь используется, относится к амино радикалу формулы 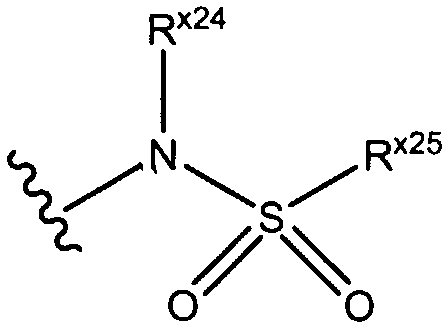 , где каждый Rx24 выбирают из гидридо, алкила, циклоалкила, арила, гетероарила и гетероциклила, и Rx25 выбирают из алкила, арила, циклоалкила, гетероарила и гетероциклила.
, где каждый Rx24 выбирают из гидридо, алкила, циклоалкила, арила, гетероарила и гетероциклила, и Rx25 выбирают из алкила, арила, циклоалкила, гетероарила и гетероциклила.
"Сульфонил", как здесь используется, относится к шестивалентному радикалу серы, замещенному двумя оксо заместителями, а третий заместитель выбирают из алкила, циклоалкила, гетероциклила, арила и гетероарила.
"Тио", как здесь используется, относится к радикалу, содержащему группы-заместители, независимо выбранные из гидридо, алкила, циклоалкила, гетероциклила, арила и гетероарила, присоединенному к двухвалентному атому серы, такому как метилтио и фенилтио.
"Тиоацил", как здесь используется, относится к тиокарбонильному радикалу, присоединенному к алкильной, алкенильной, алкинильной, циклоалкильной, гетероциклильной, арильной или гетероарильной группе.
"Тиоациламино", как здесь используется, относится к амино радикалу, соединенному с тиоацильной группой.
"Сложный тиоациловый эфир", как здесь используется, относится к тиокарбонильному радикалу, присоединенному к алкоксигруппе.
"Тиоуреидо", как здесь используется, относится к азотному радикалу формулы -N(Rx5)-C(S)-N(Rx6)(Rx7), где каждый из Rx5 и Rx6 независимо выбирают из гидридо, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила; и Rx7 выбирают из алкила, арила, циклоалкила, гетероарила и гетероциклила.
"Уреидо", как здесь используется, относится к азотному радикалу формулы -N(Rx21)-C(O)-NRx22Rx23, где каждый из Rx21 и Rx22 независимо выбирают из гидридо, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила; и Rx23 выбирают из алкила, арила, циклоалкила, гетероарила и гетероциклила.
Соединения по настоящему изобретению могут использоваться в форме солей или фармацевтически приемлемых солей, полученных от неорганических или органических кислот. Настоящее изобретение включает в себя все такие соли и все кристаллические формы таких солей. Под "фармацевтически приемлемой солью" подразумеваются такие соли, которые, с точки зрения специалиста в области медицины, являются пригодными для использования в контакте с тканями людей и низших животных без излишней токсической, раздражительной, и аллергической реакции и являются совместимыми с разумным отношением выгода/риск. Фармацевтически приемлемые соли хорошо известны в данной области. Например, S.M. Berge, et al. описывает фармацевтически приемлемые соли в J. Pharm. Sci., 1977, 66:1-19. Все эти соли могут быть получены с помощью обычных средств из соответствующего соединения по настоящему изобретению посредством обработки соединения, например, соответствующей кислотой или основанием.
Соли или фармацевтически приемлемые соли могут быть получены in situ во время конечного выделения и очистки соединений по настоящему изобретению, или отдельно, посредством взаимодействия функциональной группы свободного основания с соответствующей кислотой. Например, аддитивные соли оснований могут быть получены in situ во время конечного выделения и очистки соединений по настоящему изобретению посредством объединения группы, содержащей карбоновую кислоту, с соответствующим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, или с аммонием или с органическим первичным, вторичным или третичным амином.
Неограничивающие примеры органических кислот могут выбираться из алифатических, циклоалифатических, ароматических, арильных, гетероциклических, карбоновых и сульфоновых классов органических кислот, примеры которых включают в себя, без ограничения, муравьиную, уксусную, пропионовую, янтарную, гликолевую, глюконовую, малеиновую, эмбоновую (памовую), метансульфоновую, этансульфоновую, 2-гидроксиэтансульфоновую, пантотеновую, бензолсульфоновую, толуолсульфоновую, сульфаниловую, метансульфоновую, циклогексиламиносульфоновую, стеариновую, альгеновую, β-гидроксимасляную, малоновую, галактоновую и галактуроновую кислоту. Репрезентативные аддитивные соли органических кислот включают в себя ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, глицерофосфат, хемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат (изетионат), лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, фосфат, глютамат, бикарбонат, п-толуолсульфонат и ундеканоат. Примеры кислот, которые могут использоваться для получения фармацевтически приемлемых аддитивных солей кислот, включают в себя такие неорганические кислоты как хлористоводородная кислота, бромистоводородная кислота, серная кислота и фосфорная кислота, и такие органические кислоты, как щавелевая кислота, малеиновая кислота, янтарная кислота и лимонная кислота.
Также, основные азотсодержащие группы могут кватернизоваться с помощью таких агентов как низшие алкилгалогениды (например, метил-, этил-, пропил- и бутилхлориды, -бромиды и -йодиды); диалкилсульфаты (например, диметил-, диэтил-, дибутил- и диамилсульфаты); длинноцепочечные галогениды (например, децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -йодиды); или арилалкилгалогениды (например, бензил- и фенетилхлориды, -бромиды и -йодиды) и другие. При этом получаются водо- или маслорастворимые или диспергируемые продукты.
Например, пригодные для использования фармацевтически приемлемые аддитивные соли оснований и соединений по настоящему изобретению включает в себя, но, не ограничиваясь этим, соли металлов, полученные из алюминия, кальция, лития, магния, калия, натрия и цинка, или органические соли, полученные из N,N'-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, N-метилглюкамина, лизина и прокаина. Фармацевтически приемлемые аддитивные соли оснований включают в себя катионы на основе щелочных металлов или щелочноземельных металлов, такие как соли лития, натрия, калия, кальция, магния, и алюминия, и нетоксичные катионы четвертичного аммония и амина, включая аммоний, тетраметиламмоний, тетраэтиламмоний, метиламмоний, диметиламмоний, триметиламмоний, триэтиламмоний, диэтиламмоний и этиламмоний. Другие репрезентативные органические амины, пригодные для получения аддитивных солей оснований, включают в себя этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин.
Соединения по настоящему изобретению могут обладать одним или несколькими асимметричными атомами углерода и являются, таким образом, способными проявлять оптическую активность. Соединения по настоящему изобретению могут существовать в энантиомерной и/или диастереомерной формах, а также в форме их рацемических или нерацемических смесей. Соединения, описанные здесь, могут использоваться в настоящем изобретении в виде отдельного изомера или в виде смеси стереохимических изомерных форм.
Диастереомеры могут разделяться с помощью обычных средств, таких как хроматография, дистилляция, кристаллизация или сублимация. Энантиомеры могут быть получены посредством разделения рацемических смесей в соответствии с обычными способами, например, посредством образования диастереомерных солей путем обработки оптически активной кислотой или основанием. Неограничивающие примеры пригодных для использования кислот включают в себя винную, диацетилвинную, дибензоилвинную, дитолуоилвинную и камфорсульфоновую кислоту. Смесь диастереомеров может разделяться посредством кристаллизации с последующим высвобождением оптически активных оснований из оптически активных солей. Альтернативный способ разделения энантиомеров включает в себя использование хиральной хроматографической колонки, оптимально выбранной для доведения до максимума разделения энантиомеров. Другой способ включает в себя синтез ковалентных диастереомерных молекул посредством обработки соединений по настоящему изобретению активированной формой энантиомерно обогащенной кислоты или энантиомерно обогащенным изоцианатом. Синтезированные диастереомеры могут разделяться с помощью обычных средств, таких как хроматография, дистилляция, кристаллизация или сублимация, а затем гидролизоваться с получением энантиомерно обогащенного соединения. Подобным же образом, оптически активные соединения могут быть получены посредством использования оптически активных исходных материалов. Эти изомеры могут находиться в форме свободной кислоты, свободного основания, сложного эфира или соли.
Геометрические изомеры могут также существовать в соединениях по настоящему изобретению. Настоящее изобретение охватывает различные геометрические изомеры и их смеси, связанные с расположением заместителей вокруг двойной связи углерод-углерод или расположением заместителей вокруг карбоциклического кольца. Заместители вокруг двойной связи углерод-углерод обозначаются как находящиеся в "Z" или "E" конфигурации, где термины "Z" и "E" используются в соответствии со стандартами IUPAC. Заместители вокруг двойной связи углерод-углерод альтернативно могут упоминаться как "цис" или "транс", где "цис" представляет собой заместители по одну и ту же сторону от двойной связи, а "транс" представляет собой заместители по обе стороны от двойной связи.
Расположение заместителей вокруг карбоциклического кольца также обозначается как "цис" или "транс". Термин "цис" представляет собой заместители по одну и ту же сторону от плоскости кольца, а термин "транс" представляет собой заместители по обе стороны от плоскости кольца. Смеси соединений, где заместители располагаются как по одну, так и по обе стороны от плоскости кольца, обозначаются "цис/транс".
Выделенное, чистое или очищенное соединение относится к композиции, содержащей, по меньшей мере, 10%, например, по меньшей мере, 20%, по меньшей мере, 50%, по меньшей мере, 80%, или, по меньшей мере, 90% соединения. В одном из вариантов осуществления, ее фармацевтически приемлемая соль или фармацевтическая композиция, содержащая любое из соединений, описанных здесь, демонстрирует детектируемую (например, статистически значимую) противомикробную активность, когда она исследуется в обычных биологических анализах, таких как те, которые здесь описываются.
Термин "фармацевтически приемлемые пролекарства", как здесь используется, представляет собой те пролекарства соединений по настоящему изобретению, которые являются, с точки зрения специалиста в области медицины, пригодными для использования при контактах с тканями человека и низших животных без излишней токсической, раздражительной, аллергической реакции, совместимыми с разумным отношением выгода/риск и эффективными при их предполагаемом использовании, а также цвиттерионные формы, где это возможно, соединений по настоящему изобретению.
Термин "пролекарсто", как здесь используется, представляет собой соединения, которое быстро преобразуется in vivo в исходное соединение с формулами, описанными здесь, например, посредством гидролиза в крови. Обсуждение приводится в T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of ACS Symposium Series, и в Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, обе они включаются сюда в качестве ссылок.
Подробное описание
Здесь описываются новые пептиды и способы получения пептидов. Пептиды могут найти применение в качестве антибактериальных агентов.
Один из вариантов осуществления описывает новые пептиды, включающие в себя циклический гептапептид, присоединенный к цепи и хвосту экзоциклического пептида. В одном из вариантов осуществления, новые пептиды получают из встречающихся в природе пептидов, имеющих следующую структуру:
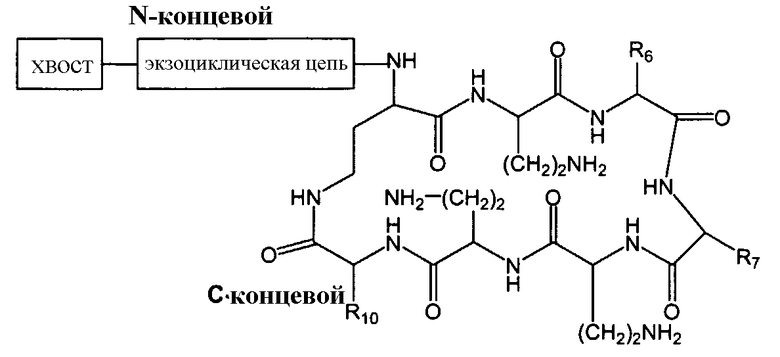
Эти встречающиеся в природе пептиды, имеют экзоциклическую пептидную цепь, присоединенную к N-концевой "боковой цепи" или "хвосту". В одном из вариантов осуществления, такие пептиды относятся к встречающимся в природе пептидам, включая полимиксины или октапептины. Эти пептиды имеют общую структуру, показанную выше, с модификацией аминокислот 6, 7, и 10 (нумерация для полимиксина), где R6 изображает боковую цепь аминокислоты в положении 6, R7 изображает боковую цепь аминокислоты в положении 7, и R10 изображает боковую цепь аминокислоты в положении 10. В одном из вариантов осуществления, R6, R7 и R10 могут, каждый, независимо, выбираться из изо-пропила (с получением Val), бензила (с получением Phe), изо-бутила (с получением Leu), фтор-бутила (с получением Ile), 1-гидрокси-1-этила (с получением Thr) и гидроксиметила (с получением Ser). В другом варианте осуществления, R6 и R7, каждый, независимо, выбирают из изо-пропила, бензила, изо-бутила, втор-бутила, 1-гидрокси-1-этил, и гидроксиметила, и R10 выбирают из изо-пропила, изо-бутила, втор-бутила, 1-гидрокси-1-этила и гидроксиметила. Экзоциклическая цепь и хвост также включают модификации. В одном из вариантов осуществления, экзоциклическая цепь содержит 1-3 аминокислотных остатков.
В одном из вариантов осуществления, экзоциклическая цепь включает в себя цепь из аминокислотных остатков. В другом варианте осуществления, экзоциклическая цепь представлена -(Y1)x(Y2)y(Y3)z-, где Y1, Y2 и Y3, каждый, независимо, выбирают из аминокислотных остатков, включая некодируемые аминокислотные остатки, и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1.
В одном из вариантов осуществления, хвост изображается как "T", где часть "хвоста-экзоциклической цепи" имеет формулу T(Y1)x(Y2)y(Y3)z-. В одном из вариантов осуществления, T выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила и атома водорода, где R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила.
Соответственно, один из вариантов осуществления предусматривает новые пептиды, получаемые из пептидов, встречающихся в природе, где пептиды, встречающиеся в природе, имеют структуру формулы (A):
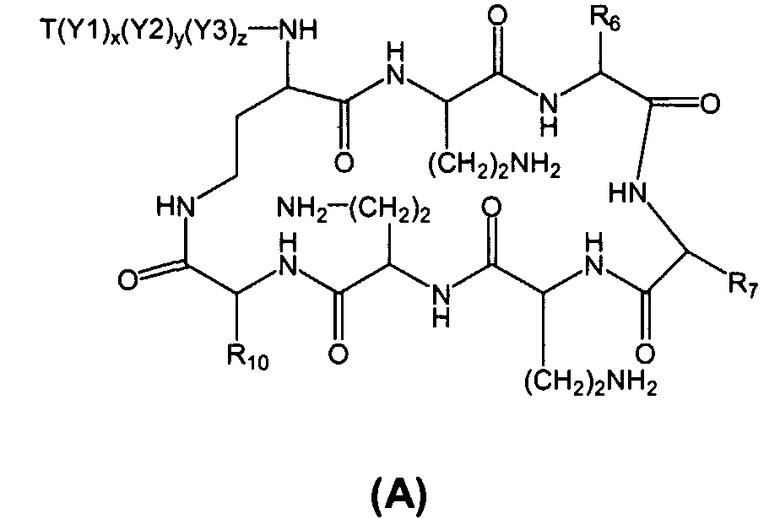
Также описываются способы получения новых пептидов, содержащие либо свободные аминогруппы, либо защищенные аминогруппы. В одном из вариантов осуществления, новые пептиды относятся, например, к пептидам, встречающимся в природе, описанным здесь. В одном из вариантов осуществления, новые пептиды имеют структуру формулы (B):
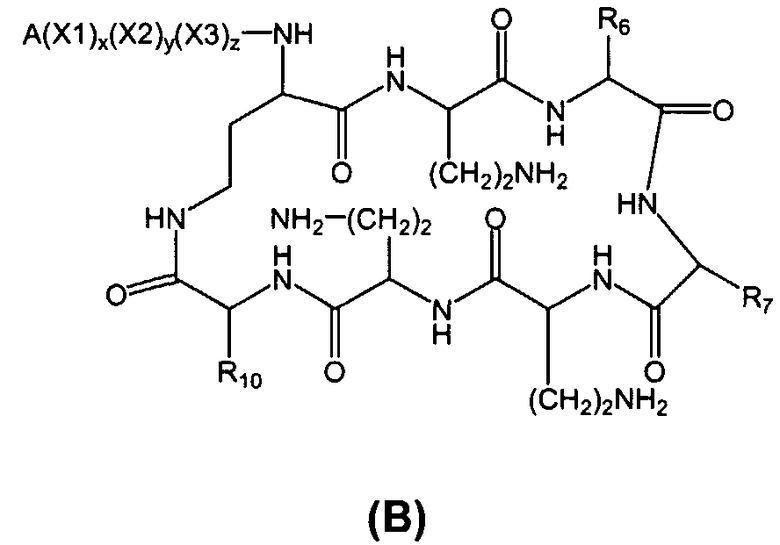
В одном из вариантов осуществления, экзоциклическая цепь представляет -(X1)x(X2)y(X3)z-, где X1, X2 и X3, каждый, независимо, выбирают из аминокислотных остатков, включая любой остаток, описанный здесь, и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1, и "A" представляет собой хвост, где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила и атома водорода, где R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила.
Один из вариантов осуществления, описанный здесь, представляет собой способ получения соединения, включающий в себя:
(a) обработку пептида, имеющего, по меньшей мере, одну боковую цепь, содержащую аминогруппу, с реагентом для защиты аминогруппы, с образованием защищенного пептида,
где пептид включает в себя циклический гептапептид, присоединенный к экзоциклической пептидной цепи, содержащей ацильную группу, и защитная группа содержит, по меньшей мере, один кислотный заместитель; и
(b) обработку защищенного пептида деацилирующим агентом, с образованием защищенного деацилированного пептида.
В одном из вариантов осуществления, пептид, который должен обрабатываться реагентом защитной группы в (a), выбирают из пептидов, встречающихся в природе, имеющих структуру формулы (A), таких как полимиксины и октапептины. Репрезентативные полимиксины и октапептины включают в себя полимиксин A (PA), полимиксин B (PB), [Ile7]-полимиксин B1 (IL), полимиксин D, полимиксин E, полимиксин F, полимиксин M (маттацин), полимиксин S, полимиксин T, циркулин A, полимиксин D (PD), октапептин A, октапептин B (OB), октапептин C (OC) и октапептин D; полимиксин E обычно называют колистин. Буквы (например, полимиксин A, полимиксин B, полимиксин C, и тому подобное), как правило, относится к полимиксинам, имеющим изменения в аминокислотной последовательности. Численные нижние индексы (например, октапептин A1, октапептин A2, октапептин A3, и тому подобное, или полимиксин B1, полимиксин B2, полимиксин B3, и тому подобное), как правило, относятся к изменениям в хвосте. Полимиксины и октапептины могут выделяться из природных источников с различными липидными хвостами, как показано в Таблице 1 для полимиксина B. Одним из исключений является колистин (полимиксин E): колистин A (полимиксин E1) и колистин B (полимиксин E2), отличающиеся по липидному хвосту.
Полимиксин B2
Полимиксин B3
Полимиксин B4
Полимиксин B5
Полимиксин B6
6-метилгептаноил
октаноил
гептаноил
нонаноил
3-гидрокси-6-метилоктаноил
Полимиксин E2 (колистин B)
6-метилгептаноил
Таблица 2 показывает пептидные последовательности нескольких пептидов, описанных здесь.
4-5-6-7-8-9-10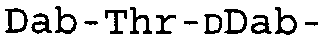

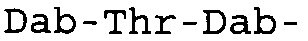

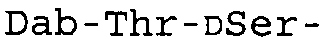
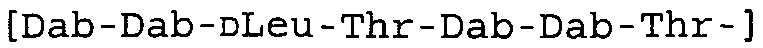
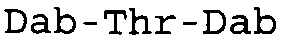
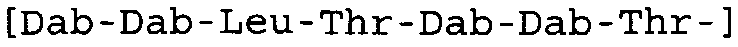
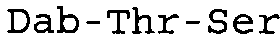
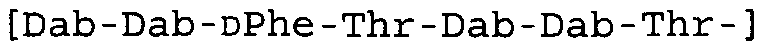
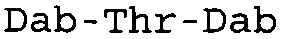

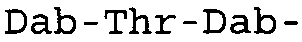

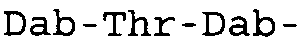
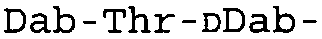

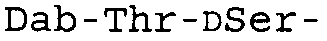



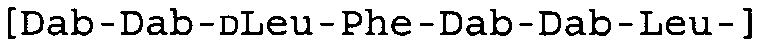


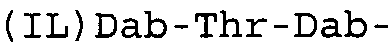
*Все аминокислоты представляют собой L-изомеры, если только не обозначаются как D-изомер. Dab представляют собой 2,4-диаминобутановую кислоту.
В одном из вариантов осуществления, пептид, который подлежит обработке, выбирают из полимиксина A, полимиксина B (например, полимиксина B1, полимиксина B2 и полимиксина B3), [Ile7]-полимиксина B1, полимиксина C, полимиксина D, полимиксина E (также называемый колистин), полимиксина F, полимиксина M (также называемый маттацин), полимиксина P, полимиксина S, полимиксина T (например, полимиксина T1), колистина (например, колистина A и колистина B), циркулина A, октапептина A (например, октапептина A1, октапептина A2 и октапептина A3) октапептина B (например, октапептина B1, октапептина B2 и октапептина B3), октапептина C (например, октапептина C1) и октапептина D. Известен также полимиксин F, где композиция аминокислот представляет собой Dab:Thr:Ser:Ile:Leu 5:1:1:1:2.
В одном из вариантов осуществления, пептид, который подлежит обработке, выбирают из полимиксина B, полимиксина A, полимиксина D, [Ile7]-полимиксина B1, колистина, циркулина A, октапептина B и октапептина C.
В одном из вариантов осуществления, полученный защищенный деацилированный пептид может дополнительно модифицироваться, с образованием новых защищенных пептидных соединений. Затем новые пептидные соединения могут образовываться после снятия защиты.
Схема I, ниже, показывает один из вариантов осуществления способа защиты и деацилирования пептида. Для иллюстративных целей, способ изображается с полимиксином B1. Однако этот способ является применимым ко всем пептидам, имеющим общую гептапептидную циклическую основную структуру формулы (A), таким как полимиксины и октапептины.
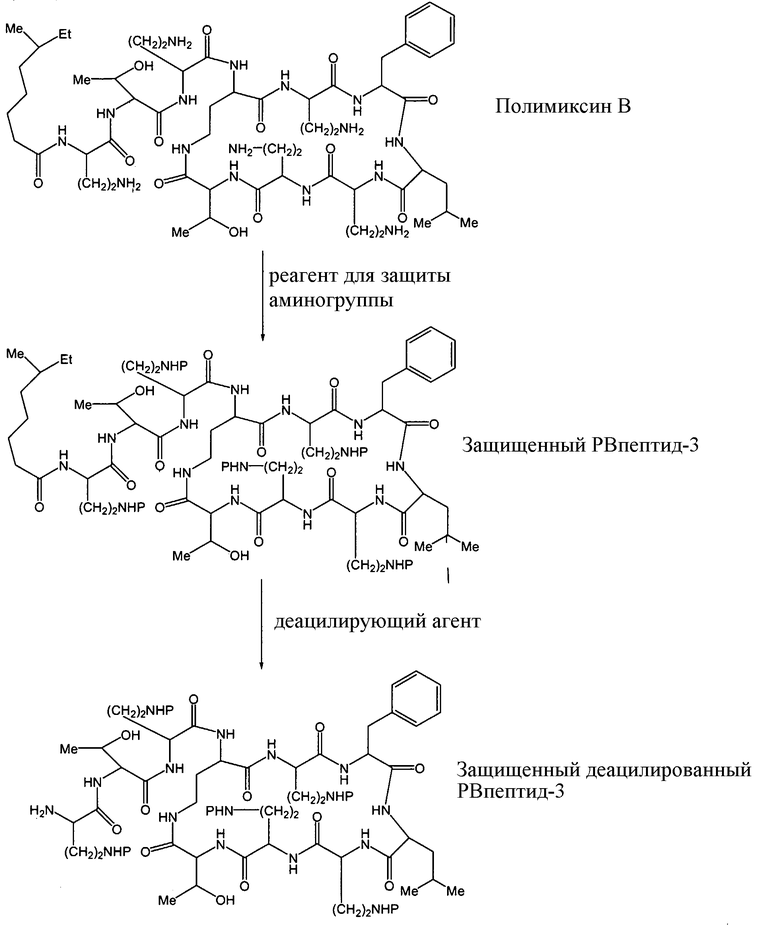
Схема I
На Схеме I, "P" изображает защитную группу, содержащую, по меньшей мере, один кислотный заместитель. Пептид Схемы I (полимиксин B или PB) имеет три аминокислоты в экзоциклической цепи, и таким образом, определяется как "PBпептид-3".
Хотя Схема I изображает защиту всех аминогрупп полимиксина B, настоящее изобретение охватывает также защиту одной или нескольких аминогрупп. Специалист в данной области легко заметит, что способ, изображенный на Схеме I, может таким же образом применяться к любому пептиду, описанному здесь.
В одном из вариантов осуществления, защитная группа содержит, по меньшей мере, один "кислотный заместитель", который, как здесь используется, относится к заместителю, содержащему отдаваемый атом водорода. Примеры кислотных заместителей включают в себя кислотную или солевую форму сульфо, сульфата, сульфоната, карбокси, карбоксилата, фосфоната и фосфата. В одном из вариантов осуществления, защитная группа содержит арил или гетероарил, замещенный кислотным заместителем.
В одном из вариантов осуществления, защитную группу выбирают из кислотной или солевой формы сульфо, сульфатных, сульфонатных, карбокси, карбоксилатных, фосфонатных и фосфатных производных аминозащитных групп, таких как мочевинные аминозащитные группы. Примеры карбоматных аминозащитных групп включают в себя, но, не ограничиваясь этим, защитные группы, описанные в "Protected Groups in Organic Synthesis" by Theodora W. Greene, John Wiley and Sons, New York, 1991 at pp. 315-348, описание которой включается сюда в качестве ссылки. Неограничивающие примеры карбоматных аминозащитных групп включают в себя 9-(2,7-дибром)флуоренилметилкарбомат, 2,7-ди-трет-бутил-[9-(10,10-диоксо-10,10,10,10-тетрагидротиоксантил)]-метилкарбомат (DBD-Tmoc), 4-метоксифенацилкарбомат (phenoc), 2-фенилэтилкарбомат (hZ), 1-метил-1-(4-бифенилил)этилкарбомат (Bpoc), 1-(3,5-ди-трет-бутилфенил)-1-метилэтилкарбомат (t-bumeoc), 2-(2'- и 4'-пиридил)этилкарбомат (pyoc), 8-хинолилкарбомат, бензилкарбомат (Cbz- или Z), п-нитробензилкарбомат, п-бромбензилкарбомат, п-хлорбензилкарбомат, 2,4-дихлорбензилкарбомат, 4-метилсульфинилбензилкарбомат (Msz), 9-антрилметилкарбомат, дифенилметилкарбомат, 2-(п-толуолсульфонил)этилкарбомат, 4-метилтиофенилкарбомат (Mtpc), 2-трифенилфосфониоизопропилкарбомат (Ppoc), м-хлор-п-ацилоксибензилкарбамат, п-(дигидроксиборил)бензилкарбомат (Dobz), 5-бенизоксазолилметилкарбомат (Bic), 2-(трифторметил)-6-хромонилметилкарбомат (Tcroc), м-нитрофенилкарбомат, 3,5-диметоксибензилкарбомат, o-нитробензилкарбомат, 3,4-диметокси-6-нитробензилкарбомат, фенил(o-нитрофенил)метилкарбомат, фенотиазинил-(10)карбонильное производное, N'-п-толуолсульфониламинокарбонильное производное, трет-амилкарбамат, п-цианобензилкарбамат, п-децилоксибензилкарбамат, o-(N,N-диметилкарбоксамидо)бензилкарбамат, ди(2-пиридил)метилкарбамат, 2-фуранилметилкарбамат, п-(п'-метоксифенилазо)бензилкарбамат, 1-метил-1-(3,5-диметоксифенил)этилкарбамат, 1-метил-1-(п-фенилазофенил)этилкарбамат, 1-метил-1-фенилэтилкарбамат, 1-метил-1-(4-пиридил)этилкарбамат, фенилкарбамат, п-(фенилазо)бензилкарбамат, 2,4,6-три-трет-бутилфенилкарбамат, 4-(триметиламмоний)бензилкарбамат и 2,4,6-триметилбензилкарбамат.
Другой пример защитной группы включает в себя 9-флуоренилметоксикарбонил (Fmoc), замещенный кислотными заместителями, или его соли. Ниже изображены примеры защитных групп, замещенных кислотными заместителями, для пептидов полимиксина и других родственных пептидов, где 2-(сульфо)-9-флуоренилметоксикарбонил сокращенно обозначается как HSO3-Fmoc, а его натриевая соль обозначается как NaSO3-Fmoc:
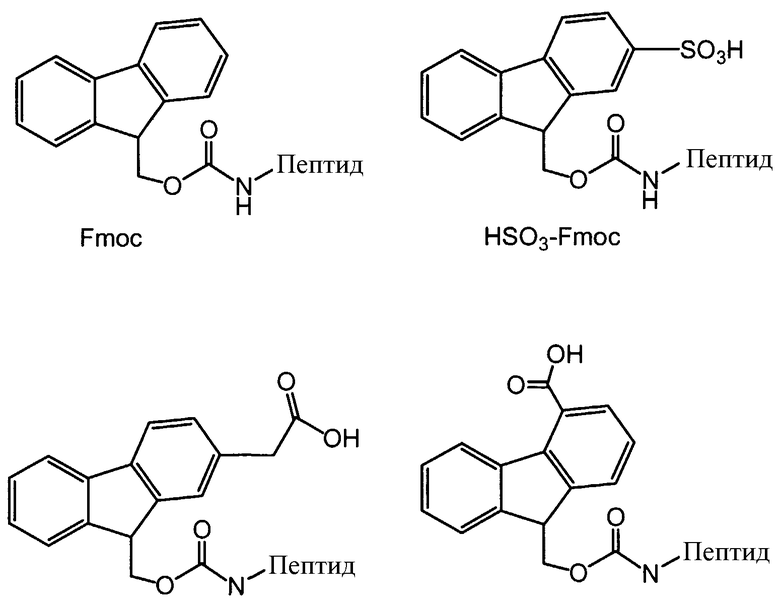
Соединения карбоновых кислот и Fmoc, пригодные в качестве реагентов для защиты аминогрупп для защиты пептидов по настоящему изобретению, могут быть получены в соответствии со ссылками 8 и 9, описания которых включаются сюда в качестве ссылок. Сульфатированные производные Fmoc могут быть получены в соответствии со ссылками 28, 29, 32 и 33, описания которых включаются сюда в качестве ссылок.
В одном из вариантов осуществления, защищенный пептид является водорастворимым. Растворимость в воде может дать возможность защищенному пептиду (например, защищенному пептиду PBпептид-3 перед деацилированием на Схеме I), для взаимодействия с деацилазой на биологической основе, например, с ферментом, в водной системе. В одном из вариантов осуществления, защищенный пептид является полианионным, водорастворимым в виде соли и может взаимодействовать в водном или частично водном растворе с ферментом деацилазой.
В другом варианте осуществления, защищенный пептид является водорастворимым и может подвергаться опосредуемому ферментом преобразованию, иному, чем деацилирование, или в дополнение к нему.
В одном из вариантов осуществления, "водорастворимый" относится к пептиду, являющемуся по существу полностью водорастворимым. В другом варианте осуществления, "водорастворимый" относится к пептиду, являющемуся водорастворимым в достаточной степени, так что любая реакция, которая использует пептид в водной системе, дает возможность ранее нерастворимой части пептида для растворения в воде, и таким образом, может вызвать доведение реакции до завершения. В одном из вариантов осуществления, защитная группа имеет количество и/или тип кислотных заместителей, достаточные для того, чтобы придать водорастворимость защищенному пептиду.
В одном из вариантов осуществления, защищенный пептид является водорастворимым и может вводиться в виде пролекарства (смотри ниже).
Новые пептидные соединения могут быть получены из полимиксина A, полимиксина B (например, полимиксина B1, полимиксина B2, и полимиксина B3), [Ile7]-полимиксина B1, полимиксина C, полимиксина D, полимиксина E (также называемого колистин), полимиксина F, полимиксина M (также называемого маттацин), полимиксина P, полимиксина S, полимиксина T (например, полимиксина T1), циркулина A, октапептина A (например, октапептина A1, октапептина A2 и октапептина A3), октапептина B (например, октапептина B1, октапептина B2 и октапептина B3), октапептина C (например, октапептина C1) и октапептина D.
В одном из вариантов осуществления, пептид является встречающимся в природе. Пептиды, имеющие общую гептапептидную циклическую сердцевину структуры (A), могут выделяться из Bacillus spp. (например, Bacillus circulans, Bacillus polymyxa, Bacillus colistinus), Aerohacter aerogenes, Paenibacillus kobensis M, и других видов бактерий (ссылки 30-31). Например, полимиксины могут выделяться из ферментативной смеси Bacillus polymyxa в соответствии с процедурами, описанными в ссылке 14.
В другом варианте осуществления, пептид синтезируется химически. Химический синтез пептидов хорошо известен специалистам в данной области и описывается, например, в Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Chan et al., Eds., Oxford University Press: New York, 2000; Bodanszky Principles of Peptide Synthesis, Springer Verlag: New York, 1993; Lloyd-Williams et al. Chemical Approaches to the Synthesis of Peptides and Proteins, CRC Press: Boca Raton, FL, 1997; и Novabiochem® (San Diego, CA) Catalog.
В одном из вариантов осуществления, деацилирующий агент представляет собой ферментативный деацилирующий агент. Один из примеров фермента, пригодного для деацилирования защищенного пептида, производится определенными микроорганизмами семейства Actinoplanaceae. Некоторые из известных видов и подвидов этого семейства включают в себя Actinoplanes philippinensis, Actinoplanes armeniacus, Actinoplanes utahensis, Actinoplanes missouriensis, Spirillospora albida, Streptosporangium roseum, Streptosporangium vulgare, Streptosporangium roseum var hollandensi, Streptosporangium album, Streptosporangium viridialbum, Amorphosporangium auranticolor, Ampullariella regularis, Ampullariella campanulata, Ampullariella lobata, Ampullariella, digitata, Pilimelia terevasa, Pimelia anulata, Planomonospora parontospora, Planomonospora venezuelensis, Planobispora longispora, Planobispora rosea, Dactylosporangium aurantiacum и Dactylosporangium thailandende. Все природные и искусственные варианты и мутанты, которые получают из семейства Actinoplanacea, и которые производят фермент, могут использоваться в настоящем изобретении.
Соответствующие способы ферментативного деацилирования можно найти в патентах США № 4524135, 4537717, 4482487, RE 32310, RE 32311, 5039789 и 5028590, в публикациях заявок на Международный патент №№ WO 03/014147, WO 01/44272, WO 01/44274 и WO 01/44271, описания которых включаются сюда в качестве ссылок.
Фермент деацилаза может быть получен в виде водорастворимого, полученного сушкой вымораживанием твердого продукта. В одном из вариантов осуществления, деацилаза получается посредством ферментирования Actinoplanes utahensis, отделения клеток от ферментационной среды, промывки клеток водой, экстрагирования клеток основным буфером при pH 8-11 в течение примерно 20 минут, доведения pH экстракта до 7-8 и сушки вымораживанием. Порошкообразная форма фермента, получаемого в результате этого процесса, может быть относительно стабильной и может легко повторно растворяться в воде для использования. Дополнительная очистка может быть получена посредством гель-фильтрации или других типов хроматографии. Этот фермент может деацилировать, например, N-[(2-сульфо-9-флуоренилметоксикарбонил)]5-полимиксин B, с получением защищенного декапептида полимиксина B (защищенный деацилированный PBпептид-3). В других вариантах осуществления, фермент из Actinoplanes utahensis может использоваться в качестве цельного бульона от ферментирования или в виде промытых клеток.
Фермент от Actinoplanes utahensis может также использоваться в виде солюбилизированного в воде препарата. Солюбилизированный в воде препарат фермента может быть получен посредством относительно сильного щелочного экстрагирования промытых клеток, с последующим доведением pH прозрачного экстракта до pH 7-8. Этот солюбилизированный в воде препарат фермента может сушиться вымораживанием до твердой формы.
Деацилирование защищенного пептида дает свободный N-конец, который может быть модифицирован посредством взаимодействия с реагентом присоединения (Схема IIa).
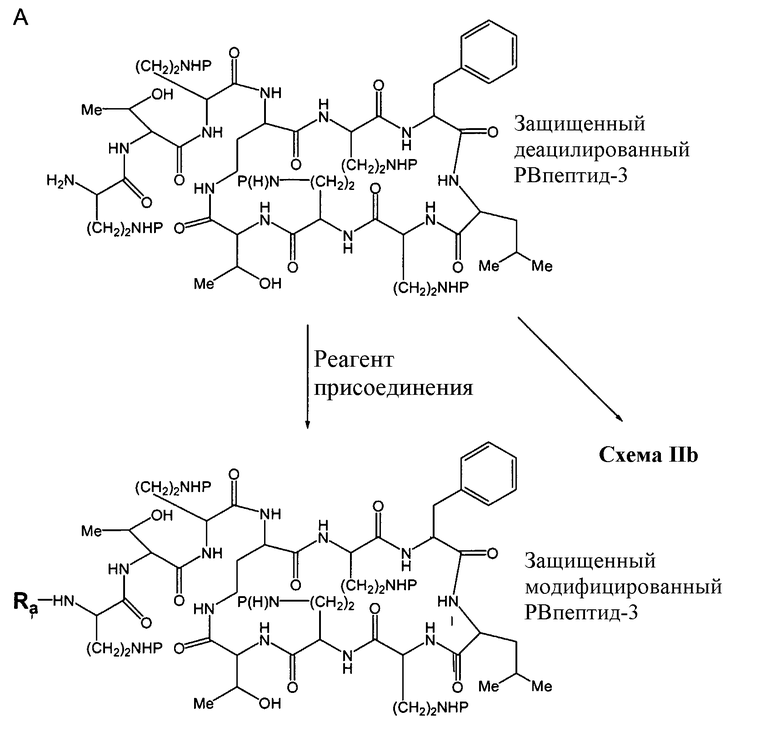
Схема IIa
Схема IIa иллюстрирует непосредственную модификацию деацилированного PBпептида-3. Обработка деацилированного PBпептида-3 агентом присоединения приводит к образованию соединения защищенного модифицированного PBпептида-3. Взаимодействие амина с реагентом присоединения, как здесь определено, хорошо известно специалистам в данной области. Например, обработка деацилированного защищенного PBпептида-3 изоцианатом приводит к получению соединений, в которых Ra-NH- представляет собой уреидо. Подобным же образом, обработка деацилированного защищенного PBпептида-3 активированным эфиром, лактоном или хлорангидридом, приводит к получению соединений, в которых Ra-NH- представляет собой ациламино. Обработка деацилированного защищенного PBпептида-3 сульфонилхлоридом или активированным сульфонамидом приводит к получению соединений, в которых Ra-NH- представляет собой сульфонамино. Обработка деацилированного защищенного PBпептида-3 активированным гетероциклом приводит к получению соединений, в которых Ra-NH- представляет собой гетероциклиламино. Обработка деацилированого защищенного PBпептида-3 активированным гетероарилом приводит к получению соединений, в которых Ra-NH- представляет собой гетероариламино. Обработка деацилированного защищенного PBпептида-3 карбонатом, хлорформиатом или цианоформиатом приводит к получению соединений, в которых Ra-NH- представляет собой карбамат. Обработка деацилированного защищенного PBпептида-3 сложным тиоациловым эфиром приводит к получению соединений, в которых Ra-NH- представляет собой тиоациламино. Обработка деацилированного защищенного PBпептида-3 фосфорилхлоридом или фосфорамидатом приводит к получению соединений, в которых Ra-NH- представляет собой фосфонамино. Обработка деацилированного защищенного PBпептида-3 имидатом или кетенамином (R"2C=C=NH) приводит к получению соединений, в которых Ra-NH- представляет собой иминоамино. Обработка деацилированного защищенного PBпептида-3 изоцианатом приводит к получению соединений, в которых Ra-NH- представляет собой уреидо. Обработка деацилированного защищенного PBпептида-3 тиоизоцианатом приводит к получению соединений, в которых Ra-NH- представляет собой тиоуреидо. Обработка деацилированного защищенного PBпептида-3 альдегидом или кетоном при восстановительных условиях приводит к получению соединений, в которых Ra-NH- представляет собой монозамещенную аминогруппу или дизамещенную аминогруппу. Неограничивающие примеры восстанавливающих агентов включает в себя H2/Ni (катализатор), Zn/HCl, NaBH3CN, NaBH(OAc)3, NaBH4, BH3-пиридин и муравьиную кислоту. Обработка деацилированного защищенного PBпептида-3 гуанидинилирующим агентом, таким как
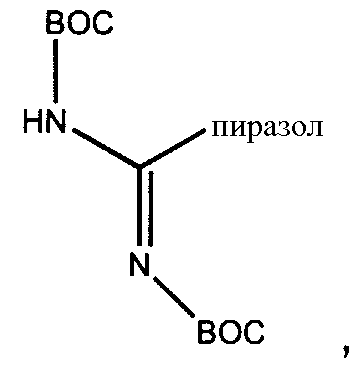
приводит к получению соединений, в которых Ra-NH- представляет собой гуанидино.
Специалисту в данной области будет понятно, что, если агент присоединения содержит заместители, которые несовместимы с условиями реакции, при которых образуется защищенный модифицированный PBпептид, указанные заместители могут защищаться перед использованием в реакции. Соответствующие защитные группы и способы их получения могут быть найдены в Greene (смотри выше).
В другом варианте осуществления, одна или несколько аминокислот N-конца могут отщепляться от экзоциклической цепи деацилированного защищенного пептида. Например, последовательность аминокислот N-конца гидролизируется, например, деградация Эдмана, модифицированная деградация Эдмана или ферментативные реакции (например, катализируемые аминопептидазой) могут использоваться для получения защищенных форм других пептидов, обозначаемых как "пептид-0", "пептид-1" или "пептид-2", которые содержат ни одной, одну или две аминокислот, соответственно, в экзоциклической цепи.
Деградация Эдмана представляет собой хорошо изученную реакцию, известную специалистам в данной области (смотри, например, P. Edman, 1950, Acta Chem. Scan. 4: 283-93 и P. Эдмана, 1956, Acta Chem. Scan. 10: 761-768). Примеры способов осуществления гидролиза аминокислот N-окончаний описаны в Voet et al., Biochemistry, 2nd Edition, John Wiley & Sons, New York, 1995, pp. 107-109, Creighton et al., Proteins, 2nd Edition, W.H. Freeman, New York, 1993, pp. 31-35, и Loudon et al., Organic Chemistry, 2nd Edition, Benjamin/Cummings, Menlo Park, CA, 1988, pp. 1154-1161, описания которых включаются сюда в качестве ссылок.
Деградация Эдмана может осуществляться при различных условиях. На первой стадии деградации Эдмана, изотиоцианат взаимодействует с концевым амином при условиях, от нейтральных до умеренно щелочных (pH < 9,5), в растворителях, таких как, но, не ограничиваясь этим, тетрагидрофуран, N,N'-диметилформамид, дихлорметан, диоксан или этанол, с образованием тиомочевинного производного пептида. Могут использоваться разнообразные изотиоцианаты (смотри K. K. Han et al. Biochemie 1977, 59: 557-576).
При обработке кислотой или основанием, тиомочевинный пептид подвергается реакции циклизации, с получением тиогидантоина и пептида, укороченного на один аминокислотный остаток. Последующая циклизация и расщепление могут осуществляться при разнообразных условиях. Как правило, используются безводная трифторуксусная кислота, гептафтормасляная кислота (смотри, например, W. F. Brandt et al., 1976, Z. Physiol. Chem. 357: 1505-1508) или концентрированная хлористоводородная кислота (смотри, например, G. E. Tarr, 1977, Methods Enzymol. 47: 335-337). Умеренно щелочные условия, такие как триэтиламин или N,N-диметилаллиламин/уксусная кислота (pH ~9) также могут использоваться (смотри G.C. Barrett et al., 1985, Tetrahedron Lett. 26(36): 4375-4378). Обзор этой реакции (смотри K.K. Han, 1985, Int. J. Biochem. 17(4): 429-445). В одном из вариантов осуществления, циклизация и расщепление осуществляются при щелочных условиях.
В другом варианте осуществления, Схема IIa иллюстрирует модификацию защищенного диацилированного PBпептида-3 непосредственно, например, когда реагент присоединения представляет собой ациламино реагент, выбранный из R'-(C=O)-LG и R'-SO2-LG, где LG представляет собой уходящую группу. В некоторых вариантах осуществления, реагент присоединения может представлять собой активированную аминокислоту или реагент конденсации аминокислоты и пептида, такой, например, как PyBOP® (бензотриазол-1-ил-окси-трис-пирролидинофосфоний гексафторфосфат), HBtU (2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат), HBtU/HOBt (2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат/N-гидроксибензотриазол) или DCC (дициклогексилкарбодиимид). В одном из вариантов осуществления, R' и R" выбираются из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила. Неограничивающие примеры R'-(C=O)-LG включает в себя ацилгалогениды, такие как ацилхлориды, ацилцианиды, сложные эфиры (например, сложные сукцинимидиловые эфиры), ацилазиды, лактоны и ангидриды. Неограничивающие примеры R'-SO2-LG включает в себя сульфонилхлориды. Когда Ra представляет собой имин, R'-(C=NH)-, модифицированный PBпептид-3, содержащий иминоаминогруппу, может образовываться посредством взаимодействия деацилированного PBпептида-3 с кетенамином R"2C=C=NH, где R" выбирают из R' и атома водорода. Когда Ra представляет собой амид R'-NH-(C=O)- или тиоамид R'-NH-(C=S)-, модифицированный PBпептид-3, содержащий мочевину или тиомочевину, может образовываться посредством взаимодействия деацилированного PВпептида-3 с изоцианатом или изотиоцианатом, соответственно. Когда Ra представляет собой замещенный алкил, R'-алкил-, модифицированный PBпептид-3, содержащий замещенный алкил, может образовываться посредством взаимодействия деацилированного PВпептида-3 с R'C(O)R" и восстанавливающим агентом. Неограничивающие примеры восстанавливающих агентов включают в себя H2/Ni (катализатор), Zn/HCl, NaBH3CN, NaBH(OAc)3, NaBHu, BH3·пиридин и муравьиную кислоту.
Схемы IIb, III и IV, схематически показывают, как могут быть получены версии полимиксина B "пептид-0", "пептид-1" или "пептид-2" (смотри ниже). Схема IIb показывает удаление аминокислоты с преобразованием защищенного деацилированного PBпептида-3 в защищенный деацилированный PBпептид-2. Затем защищенный деацилированный PBпептид-2 может химически модифицироваться с помощью процедур, описанных для Ra выше, для установки заместителя Rb.
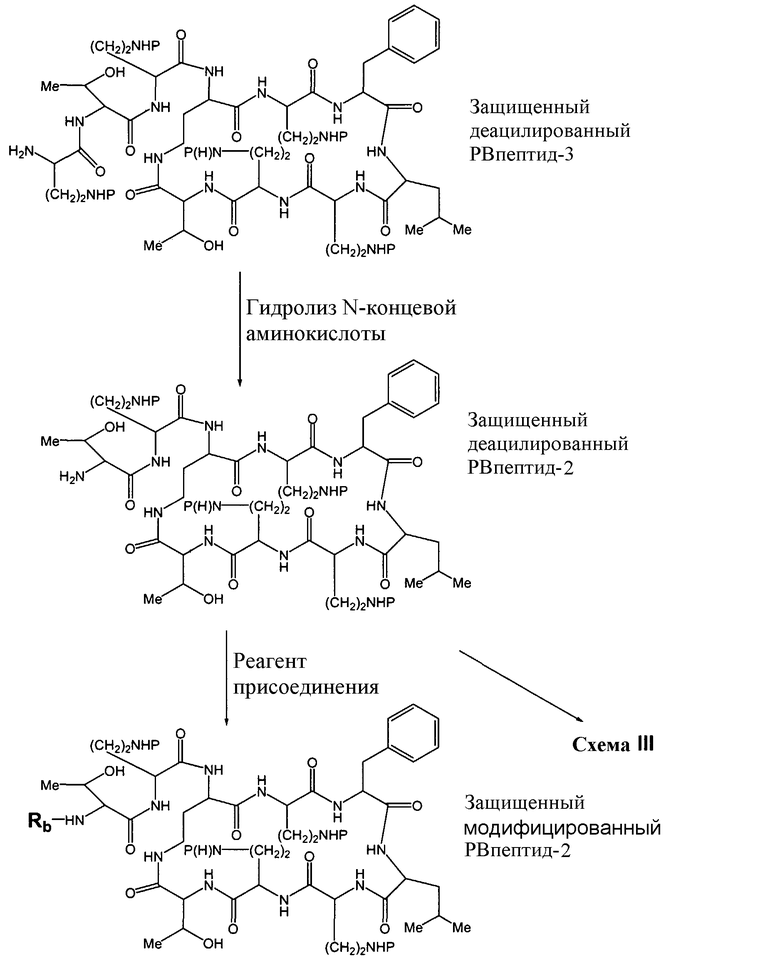
Схема IIb
Схема III подобным же образом показывает преобразование защищенного диацилированного PBпептида-2 в защищенный деацилированный PBпептид-1, который затем может химически модифицироваться с помощью реагента присоединения, как описано выше, для установки заместителя Rc.
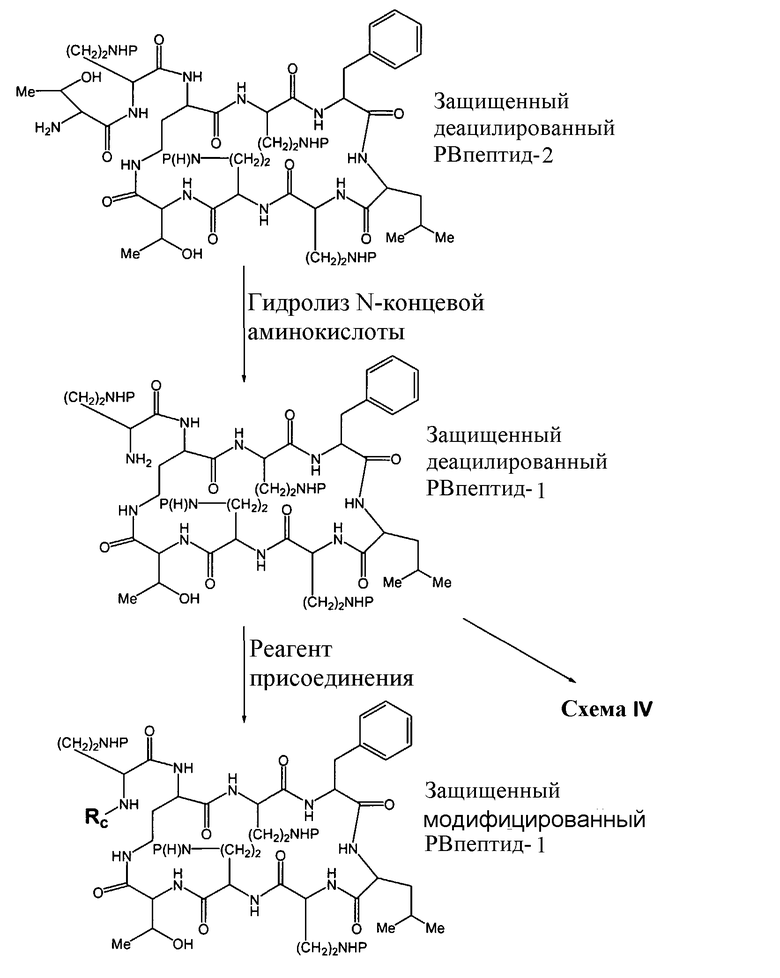
Схема III
Альтернативно, деацилированный защищенный PBпептид-1 может подвергаться другой реакции гидролиза N-концевой аминокислоты (Схема IV), с образованием деацилированного защищенного PBпептид-0, который не содержит экзоциклической пептидной цепи.
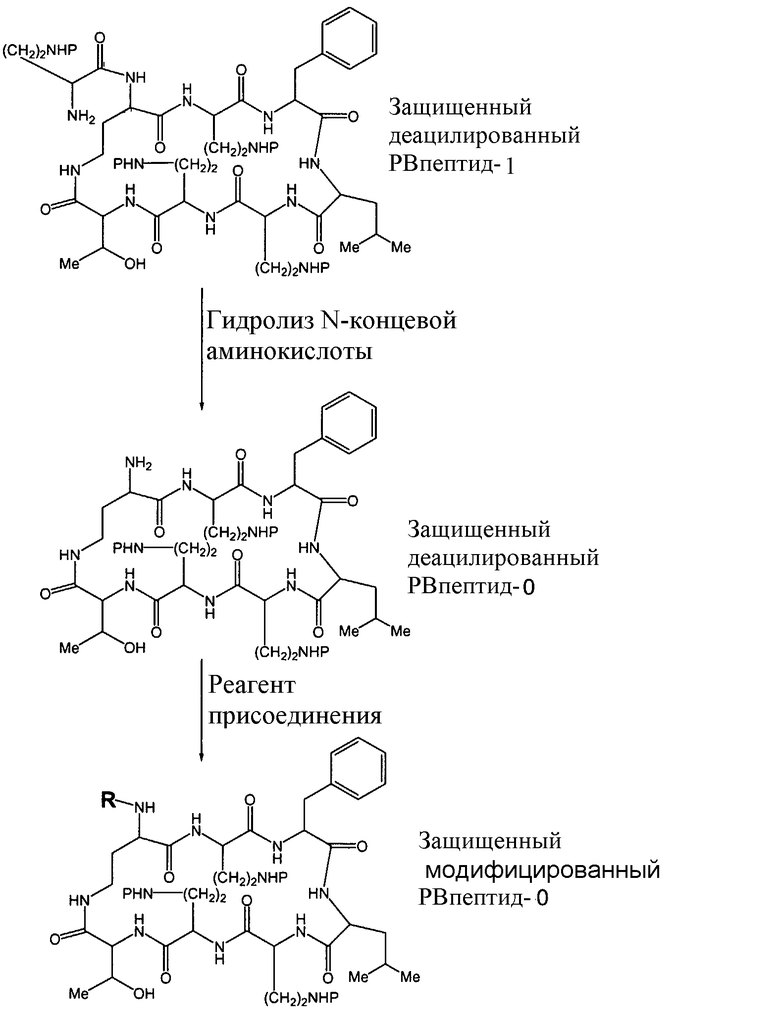
Схема IV
Хотя Схемы выше иллюстрируют реакции, включающие в себя производные полимиксина B, специалист в данной области может заметить, что эти способы являются в равной степени применимыми к любому из пептидов, описанных здесь.
Специалист в данной области может заметить, что стадии гидролиза/химического модифицирования не должны осуществляться строго в отдельных реакциях. Например, стадии гидролиза и химического модифицирования могут осуществляться почти одновременно или могут осуществляться в реакционной смеси в одной и той же емкости.
Каждый из защищенных пептидов, таких как защищенный деацилированный PBпептид-2, может использоваться для получения новых пептидов, имеющих антибактериальную активность. Например, реагенты, которые образуют хвост, например, алкил или ароматические кислоты, могут преобразовываться в активированные частицы, а затем соединяться с защищенными пептидами, с последующим снятием защиты, с получением новых пептидов, таких как соединения 1, 2, 6, 7, 11 и 12 в Таблице 3. Защищенные пептиды также могут обрабатываться непосредственно алкильными или ароматическими изоцианатами или изотиоцианатами, с получением соответствующих мочевин и тиомочевин, и с продуктов снимается защита, с получением других новых рядов новых пептидов, как представлено соединением 35. Мочевины также могут быть получены посредством обработки соответствующих аминов N-карбонилоксисукцинимидильными производными. Последние производные легко получают из реакции аминов и дисукцинимидилкарбонатов.
Один из вариантов осуществления предусматривает способ получения соединения, включающий в себя:
(a) обработку пептида, имеющего, по меньшей мере, одну боковую цепь, содержащую аминогруппу, реагентом для защиты аминогруппы, с образованием защищенного пептида,
где пептид содержит циклический гептапептид, присоединенный к экзоциклической пептидной цепи, содержащей ацильную группу, и защитная группа содержит, по меньшей мере, один кислотный заместитель; и
(b) обработку защищенного пептида деацилирующим агентом, с образованием защищенного деацилированного пептида.
В одном из вариантов осуществления, пептид на стадии (a) имеет структуру формулы (A):
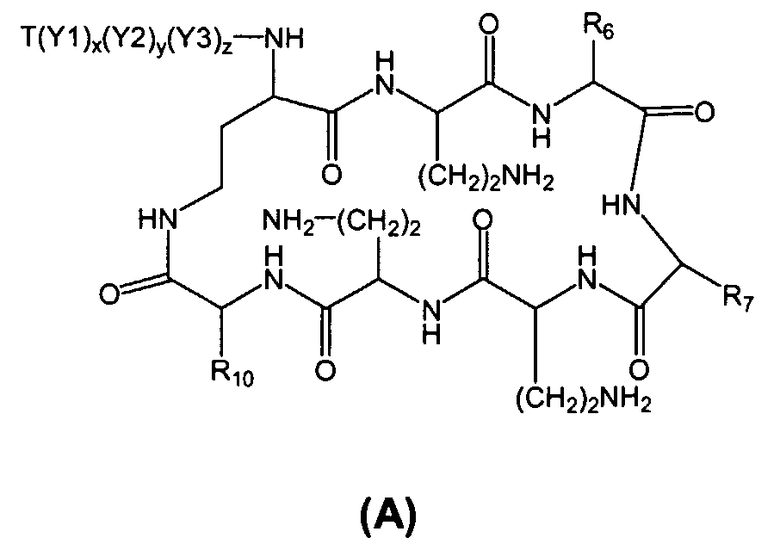
где
Y1, Y2, и Y3, каждый, независимо, выбирают из аминокислотных остатков; T выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; и R6, R7 и R10, каждый, независимо, выбирают из изопропила, бензила, изобутила, втор-бутила, 1-гидрокси-1-этила и гидроксиметила. В другом варианте осуществления, R6 и R7, каждый, независимо, выбирают из изопропила, бензила, изобутила, втор-бутила, 1-гидрокси-1-этила и гидроксиметила, и R10 выбирают из изопропила, изобутила, втор-бутила, 1-гидрокси-1-этила и гидроксиметила. В другом варианте осуществления, Y1, Y2, и Y3, каждый, независимо, выбирают из остатка 2,4-диаминобутановой кислоты, треонинового остатка и серинового остатка. В дополнительном варианте осуществления, T выбирают из 6-метилоктаноила, 6-метилгептаноила, октаноила, гептаноила, нонаноила и 3-гидрокси-6-метилоктаноила.
В другом варианте осуществления, пептид на стадии (a) выбирают из полимиксина A, полимиксина B, [Ile7]-полимиксина B, полимиксина C, полимиксина D, колистина, полимиксина F, полимиксина M, полимиксина P, полимиксина S, полимиксина T, циркулина A, октапептина A, октапептина B, октапептина C и октапептина D. В другом варианте осуществления, пептид на стадии (a) выбирают из полимиксина B, полимиксина A, полимиксина D, [Ile7]-полимиксина B, колистина, циркулина A, октапептина B и октапептина C. В дополнительном варианте осуществления, пептид на стадии (a) выбирают из полимиксина A, полимиксина D, [Ile7]-полимиксина B, колистина, циркулина A, октапептина B и октапептина C. В другом варианте осуществления, пептид на стадии (a) представляет собой полимиксин B.
В одном из вариантов осуществления, по меньшей мере, один кислотный заместитель выбирают из карбокси, карбоксилата, сульфо, сульфата, фосфоната и их солей. Другой вариант осуществления предусматривает защитную группу, содержащую арил или гетероарил, замещенный, по меньшей мере, одним кислотным заместителем. В дополнительном варианте осуществления, защитная группа представляет собой сульфоновую кислоту 9-флуоренилметоксикарбонила, такую как 2-сульфо-9-флуоренилметоксикарбонил.
В одном из вариантов осуществления, деацилирующий агент представляет собой фермент. В другом варианте осуществления, источник фермента представляет собой Actinoplanes utahensis.
В одном из вариантов осуществления, способ получения соединения дополнительно включает в себя стадию (c): образования из защищенного деацилированного пептида соединений, имеющих следующие формулы:
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1; и P представляет собой защитную группу, содержащую, по меньшей мере, один кислотный заместитель.
В другом варианте осуществления, получение включает в себя обработку защищенного пептида реагентом, имеющим формулу A-LG, где LG представляет собой уходящую группу. В другом варианте осуществления, образование включает в себя обработку защищенного пептида реагентом, выбранным из изоцианата, тиоизоцианата, лактона, активированного гетероцикла, активированного гетероарила, имидата, кетенамина, альдегида и восстанавливающего агента, и кетона, и восстанавливающего агента. В дополнительном варианте осуществления, получение включает в себя обработку защищенного пептида ацилирующим реагентом, выбранным из ацилгалогенидов, ацилцианидов, сложных эфиров, лактонов и ангидридов. В другом варианте осуществления, получение включает в себя обработку защищенного пептида сульфонирующим реагентом, выбранным из сульфонилхлорида и активированных сульфонамидов.
В одном из вариантов осуществления, образование включает в себя воздействие на защищенный деацилированный пептид реакции гидролиза аминокислоты N-конца, так что x представляет собой 0, и каждый из y и z независимо представляет собой 1. В другом варианте осуществления, образование включает в себя воздействие на защищенный деацилированный пептид второй реакции гидролиза аминокислоты N-конца, так что каждый из x и y независимо представляет собой 0, и z представляет собой 1. Другой вариант осуществления включает в себя воздействие на защищенный деацилированный пептид третьей реакции гидролиза аминокислоты N-конца, так что каждый из x, y и z представляет собой 0.
Один из вариантов осуществления описывает способ получения соединения, включающий в себя:
(a) обработку пептида, имеющего, по меньшей мере, одну боковую цепь, содержащую аминогруппу, реагентом для защиты аминогруппы, с образованием защищенного пептида,
где пептид содержит циклический гептапептид, присоединенный к экзоциклической пептидной цепи, содержащей ацильную группу, и защищенный пептид является водорастворимым; и
(b) обработку защищенного пептида деацилирующим агентом, с образованием защищенного деацилированного пептида.
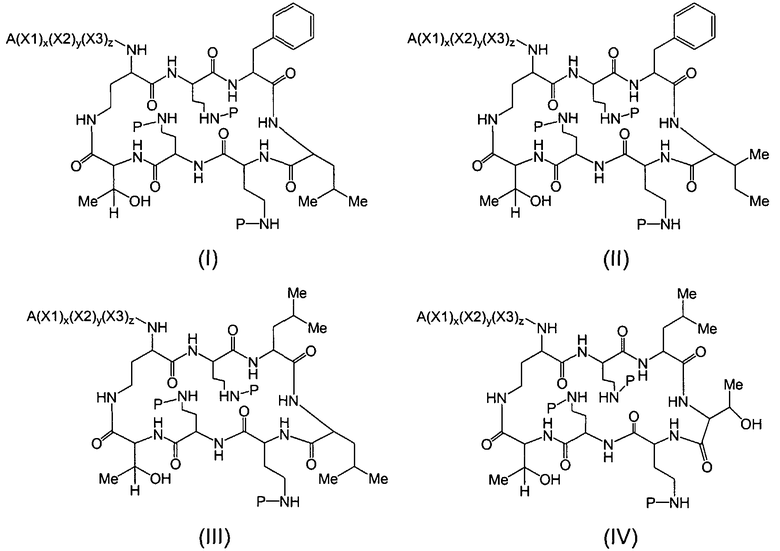
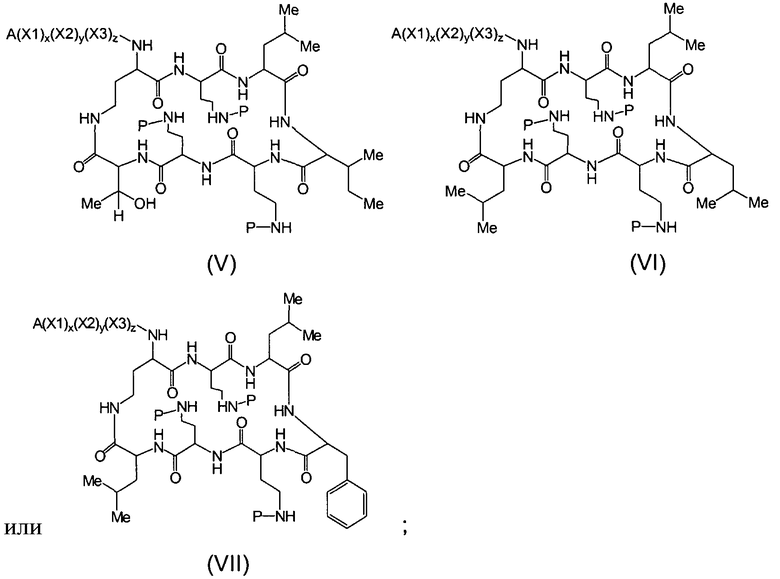
Один из вариантов осуществления предусматривает новые защищенные пептиды, имеющие структуру, выбранную из формул (I) - (VII):
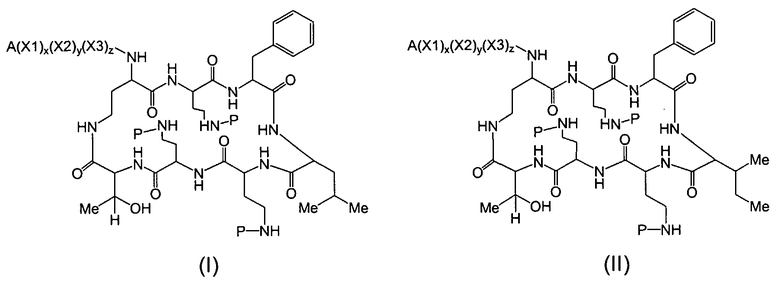
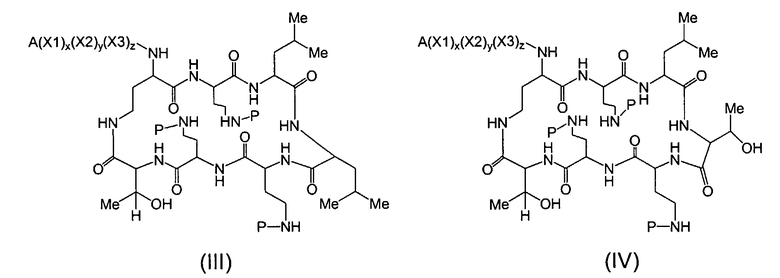
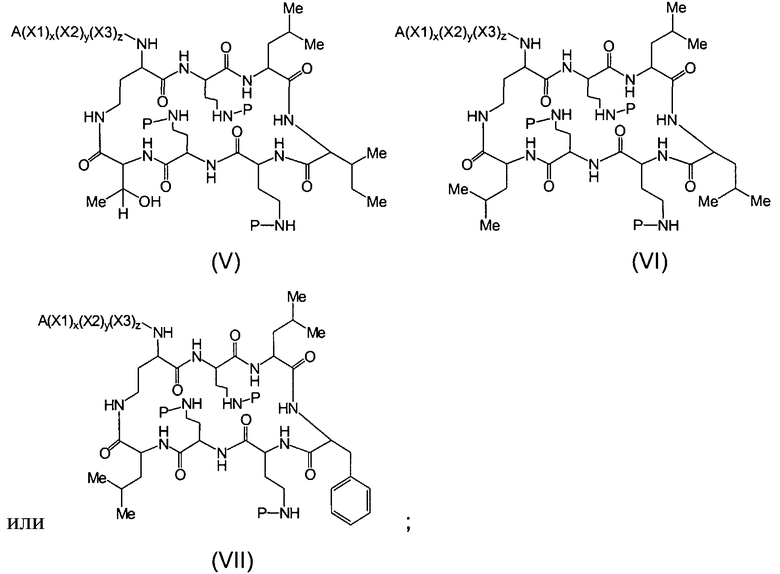
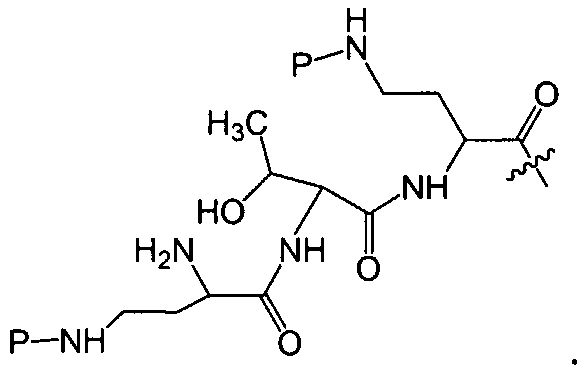
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(С=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1; и P представляет собой защитную группу, содержащую, по меньшей мере, один кислотный заместитель. В другом варианте осуществления, P представляет собой 9-флуоренилметоксикарбонильную группу, замещенную, по меньшей мере, одним кислотным заместителем, такую как 2-сульфо-9-флуоренилметоксикарбонильная группа. Другой вариант осуществления описывает соединение, где x и y, каждый, независимо, представляет собой 0, z представляет собой 1, и X3 представляет собой:
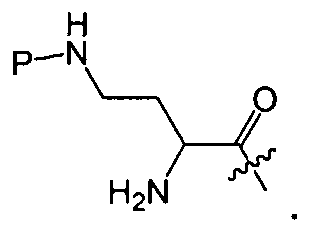
Дополнительный вариант осуществления включает в себя соединение, где x представляет собой 0, каждый из y и z независимо представляет собой 1, и X2-X3 представляет собой:
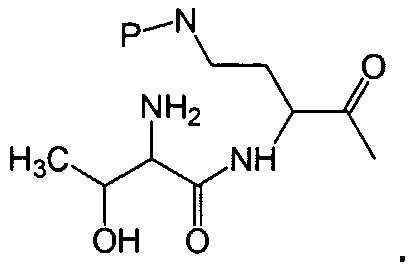
Другой вариант осуществления предусматривает соединение, где x, y и z, каждый, независимо, равны 1, и X1-X2-X3 представляет собой:
В дополнительном варианте осуществления, каждый из x, y, и z независимо представляет собой 0, и A представляет собой атом водорода. В другом варианте осуществления, соединение представляет собой пролекарство.
Один из вариантов осуществления описывает соединение, имеющее структуру, выбранную из формул (I)-(VII):
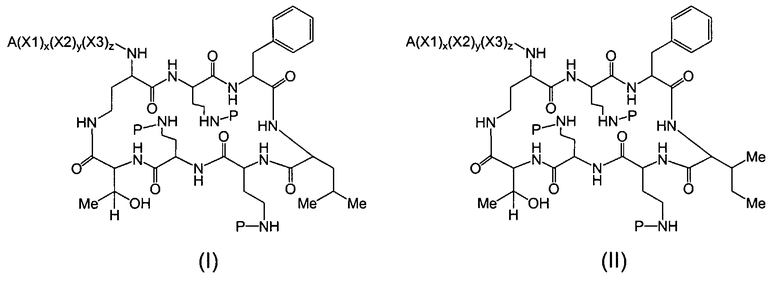
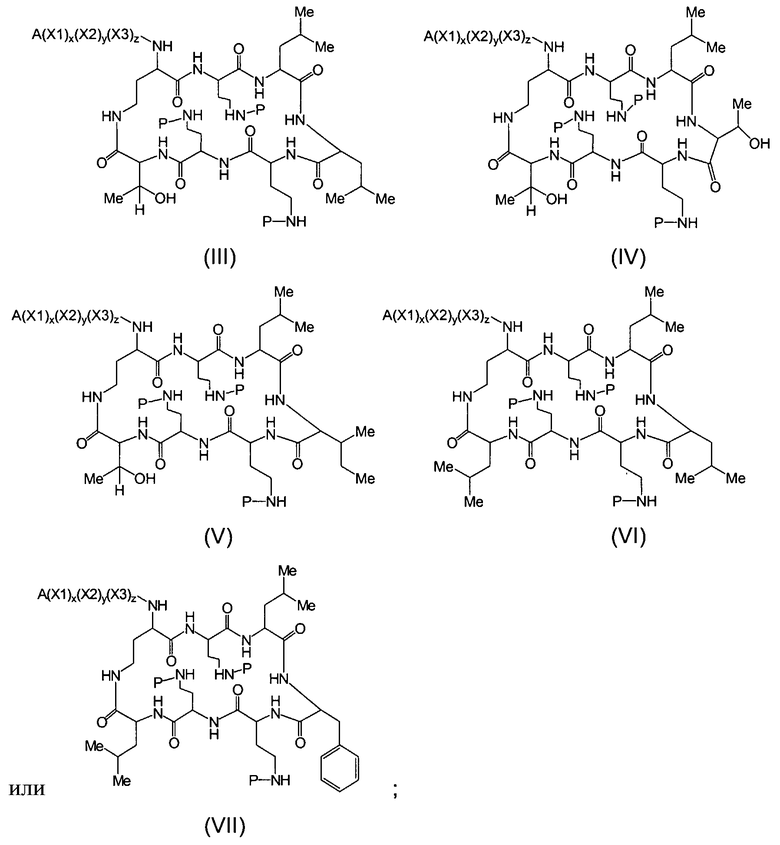
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1; P представляет собой защитную группу; и соединение, имеющее структуру, выбранную из формул (I)-(VII) является водорастворимым.
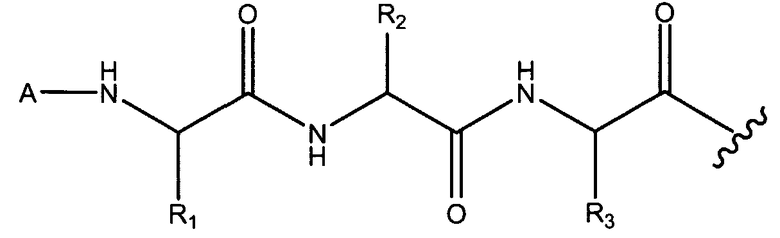
Неограничивающие примеры аминокислотных остатков для X1, X2, и X3 включают в себя те, которые получены из двадцати кодирующих аминокислот и их производных, других α-аминокислот, β-аминокислот, γ-аминокислот, δ-аминокислот и ω-аминокислот. X1, X2, и X3 могут иметь либо R, либо S хиральность на любом хиральном атоме. В одном из вариантов осуществления, X1, X2, и X3 выбираются из аланина, β-аланина, α-аминоадипиновой кислоты, α-аминобутановой кислоты, γ-аминобутановой кислоты, ε-аминокапроновой кислоты, 1-аминоциклопентанкарбоновой кислоты, ε-аминогексановой кислоты, 2-аминогептандионовой кислоты, 7-аминогептановой кислоты, α-аминоизомасляной кислоты, аминометилпирролкарбоновой кислоты, 8-амино-3,6-диоксаоктановой кислоты, аминопиперидинкарбоновой кислоты, 3-аминопропионовой кислоты, аминосерина, аминотетрагидропиран-4-карбоновой кислоты, аргинина, аспарагина, аспарагиновой кислоты, азетидинкарбоновой кислоты, бензотиазолилаланина, бутилглицина, карнитина, 4-хлорфенилаланина; цитрулина, циклогексилаланина, циклогексилстатина, цистеина, диаминобутановой кислоты, диаминопропионовой кислоты, дигидроксифенилаланина, диметилтиазолидинкарбоновой кислоты, глутаминовой кислоты, глутамина, глицина, гистидина, гомосерина, гидроксипролина, изолейцина, изонипекотиновой кислоты, лейцина, лизина, метанопролина, метионина, норлейцина, норвалина, орнитина, п-аминобензойной кислоты, пеницилламина, фенилаланина, фенилглицина, пиперидинилаланина, пиперидинилглицина, пролина, пирролидинилаланина, саркозина, селеноцистеина, серина, статина, тетрагидропиранглицина, тиенилаланина, треонина, триптофана, тирозина, валина, алло-изолейцина, алло-треонина, 2,6-диамино-4-гексановой кислоты, 2,6-диаминопимелиновой кислоты, 2,3-диаминопропионовой кислоты, дикарбоксидина, гомоаргинина, гомоцитрулина, гомоцистеина, гомоцистина, гомофенилаланина, гомопролина и 4-гидразинобензойной кислоты.
X1 и X2; X2 и X3; и X3 и остаток N-конца циклического гептапептида могут соединяться посредством амидных связей. Например, в варианте осуществления, где X1, X2 и X3 представляют собой α-аминокислоты, X1, X2 и X3 (имеющие боковые цепи R1, R2 и R3, соответственно) могут соединяться, как изображено ниже.
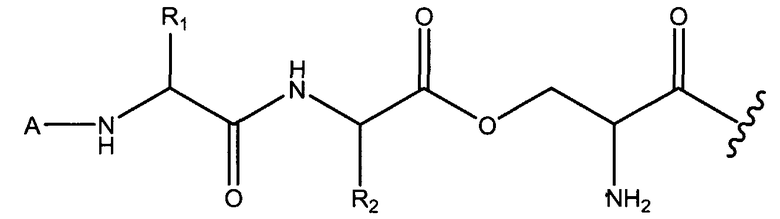
В некоторых вариантах осуществления, X1 и X2 и/или X2 и X3 могут соединяться сложноэфирными связями, как изображено ниже в варианте осуществления, где X3 представляет собой серин.
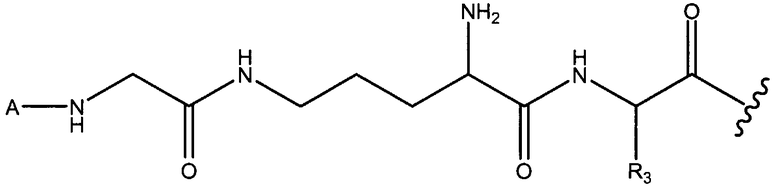
В вариантах осуществления, где X1 или X2 имеют аминогруппу боковой цепи, X1 и X2 и/или X2 и X3 могут соединяться через амид боковой цепи, как изображено ниже в варианте осуществления, где X2 представляет собой орнитин.
В одном из вариантов осуществления, хвост нового пептида изображается как "AB", где часть "хвоста-экзоциклической цепи" имеет формулу AB(X1)x(X2)y(X3)z-. В одном из вариантов осуществления, AB выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-алкила и атома водорода, где R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила.
В одном из вариантов осуществления, новые соединения могут образовываться посредством удаления защитных групп защищенного пептида, то есть, посредством снятия защиты, с помощью способов, описанных в "Protective Groups in Organic Synthesis" by Theodora W. Greene, John Wiley and Sons, New York, 1991, описание которой включается сюда в качестве ссылки.
Удаление аминозащитных групп может осуществляться в соответствии с процедурами, описанными в Greene (смотри выше). Как могут увидеть специалисты в данной области, выбор аминозащитной группы, используемой на первой стадии способа, будет диктовать реагенты и процедуры, используемые при удалении указанной аминозащитной группы.
Когда реагент для химического модифицирования содержит одну или несколько защитных групп, эти защитные группы также должны удаляться. Выбор защитной группы (групп), используемой в заместителе (заместителях) химического модифицирующего агента будет диктовать реагенты и процедуры, используемые при удалении указанной защитной группы (групп). Когда защитная группа (группы), используемая на заместителе (заместителях) модифицирующего агента, и защитная группа, используемая для защищенного пептида, являются совместимыми, защитные группы могут удаляться на одной стадии. Однако, когда защитная группа (группы) являются несовместимыми, может потребоваться множество стадий для удаления всех защитных групп.
В одном из вариантов осуществления, пептиды со снятой защитой очищают с помощью гель-фильтрации, хроматографии или ВЭЖХ с обращенной фазой.
Один из вариантов осуществления включает в себя соединение формулы (1):
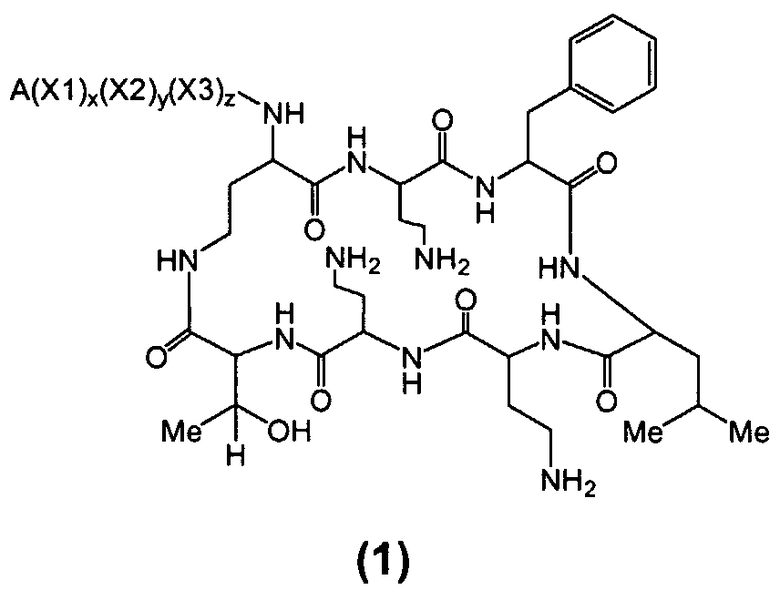
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2 и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что:
1) A не содержит, по меньшей мере, одного аминокислотного остатка;
2) если X3 представляет собой 2,4-диаминобутановую кислоту, каждый из x и y независимо выбирают из 0 и 1, и A представляет собой R'-(C=O)-, тогда R' выбирают из незамещенного алкила, имеющего, по меньшей мере, 9 атомов углерода, циклоалкила, алкенила, арила, гетероарила, гетероциклила, и
замещенного алкила, где, по меньшей мере, один из атомов водорода заменяется группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо,
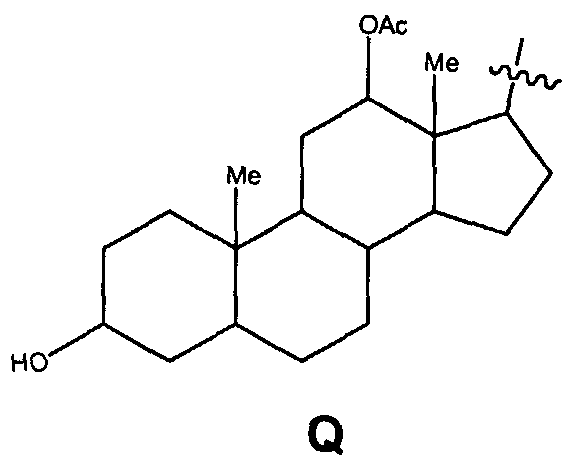
при условии, что замещенный алкил не является выбранным из алкил-CHOH-CH2-, фенил-CH2-, адамантил-CH2-, замещенной арилокси-CH2- и CH3-CHQ-CH2-CH2-, где Q представляет собой структуру:
3) если x, y и z, каждый, представляют собой 1, X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-(C=O)- и R' представляет собой арил, тогда арил не является 6-членным кольцом, имеющим три гидрокси заместителя; и
4) если каждый из x, y и z независимо представляет собой 0, и A представляет собой R'-(C=O)-, тогда R' выбирают из C8-20-алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила.
В другом варианте осуществления, если X3 представляет собой 2,4-диаминобутановую кислоту, x и y независимо выбираются из 0 и 1 и A представляет собой R'-(C=O)-, тогда R' выбирают из C9-20 незамещенного алкила, циклоалкила, алкенила, арила, гетероарила, гетероциклила и замещенного алкила, где, по меньшей мере, один из атомов водорода заменяется группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкинила, амино, замещенного арила, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо.
Один из вариантов осуществления предусматривает соединение формулы (2):
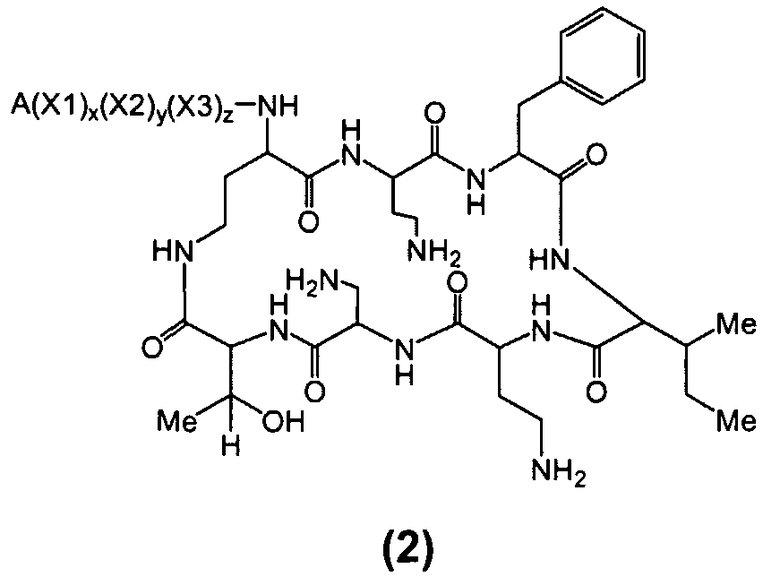
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что:
1) A не содержит, по меньшей мере, одного аминокислотного остатка; и
2) если каждый из x, y и z независимо представляет собой 1, X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' не является разветвленным C8-алкилом.
Один из вариантов осуществления описывает соединение, выбранное из формул (I) -(VII), (1) и (2), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-O-(C=O)- и R' представляет собой фенил. Другой вариант осуществления предусматривает соединение, выбранное из формул (I)-(VII), (1) и (2), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-(C=O)- и R' представляет собой н-C9-алкил. Другой вариант осуществления предусматривает соединение, выбранное из формул (I) - (VII), (1) и (2), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-(C=O)- и R' представляет собой алкил, замещенный группой, выбранной из N-(C1-10-алкил)-4-аминофенильной и бензилокси группы. Дополнительный вариант осуществления включает в себя соединение, выбранное из формул (I)-(VII), (1) и (2), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-NH-(C=O)- и R' выбирают из н-C8-алкила, C2-алкила, замещенного 3-индолильной группой, циклогексила, незамещенного фенила, бензила, 4'-бифенила и фенила, замещенного группой, выбранной из 4-C10-алкила, 4'-фенилокси и 4-хлора.
Другой вариант осуществления предусматривает соединение, выбранное из формул (I)- (VII), (1) и (2), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-NH-(C=S)- и R' представляет собой фенил. Другой вариант осуществления описывает соединение, выбранное из формул (I) - (VII), (1) и (2), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-(C=O)- и R' выбирают из фенила, 4-пиридинила и алкила, замещенного 2-нафтокси группой. Дополнительный вариант осуществления предусматривает соединение, выбранное из формул (I) - (VII), (1) и (2), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-SO2- и R' представляет собой 4-метилфенил. Другой вариант осуществления включает в себя соединение, выбранное из формул (I) - (VII), (1) и (2), где x представляет собой 0, каждый из y и z независимо представляет собой 1, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-NH-(C=O)- и R' представляет собой н-C8-алкил.
Другой вариант осуществления предусматривает соединение, выбранное из формул (I) - (VII), (1) и (2), где x представляет собой 0, y и z, каждый, равны 1, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-NH-(C=S)- и R' представляет собой фенил. Дополнительный вариант осуществления включает в себя соединение, выбранное из формул (I) - (VII), (1) и (2), где каждый из x и y независимо представляет собой 0, z представляет собой 1, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-NH-(C=O)- и R' представляет собой н-C9-алкил. Другой вариант осуществления описывает соединение, выбранное из формул (I) - (VII), (1) и (2), где X1 представляет собой глицин, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-SO2- и R' представляет собой н-C10-алкил. Другой вариант осуществления предусматривает соединение, выбранное из формул (I) - (VII), (1) и (2), где X1 представляет собой лизин, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-(C=O)- и R' представляет собой н-C9-алкил. Дополнительный вариант осуществления включает в себя соединение, выбранное из формул (I) - (VII), (1) и (2), где X1 представляет собой фенилаланин, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-(C=O)- и R' представляет собой н-C9-алкил.
Один из вариантов осуществления предусматривает соединение формулы (3):
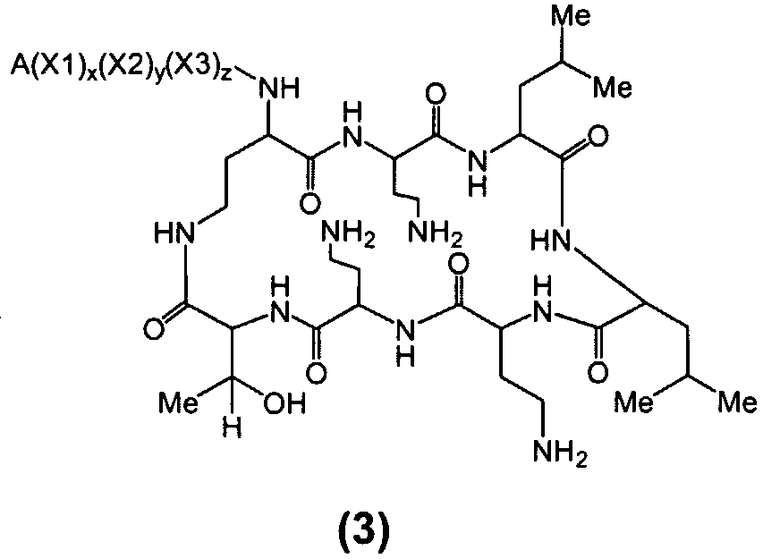
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O) -, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что:
1) A не содержит, по меньшей мере, одного аминокислотного остатка;
2) если x выбирают из 0 или 1, каждый из y и z независимо представляет собой 1, X1 представляет собой аминокислотный остаток, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' выбирают из неразветвленного незамещенного C1-4-алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила, при условии, что: a) алкенил не представляет собой фуранил-CH=CH-, b) арил не является выбранным из нафтила и 4-нитрофенила и c) гетероарил не представляет собой 2-тиофенил; и
3) если x выбирают из 0 или 1, каждый из y и z независимо представляет собой 1, X1 представляет собой аминокислотный остаток, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-SO2-, тогда R' выбирают из C1-7-алкила, C9-20-алкила, циклоалкила, алкенила, гетероарила и гетероциклила.
Другой вариант осуществления включает в себя соединение формулы (3), где, если x выбирают из 0 или 1, y и z, каждый, равны 1, X1 представляет собой аминокислотный остаток, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' выбирают из неразветвленного незамещенного C1-4 алкила, циклоалкила и гетероциклила. Другой вариант осуществления предусматривает соединение формулы (3), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-NH-(C=O)- и R' представляет собой н-C8-алкил. Дополнительный вариант осуществления описывает соединение формулы (3), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-SO2- и R' представляет собой 4-метилфенил.
Один из вариантов осуществления предусматривает соединение формулы (4):
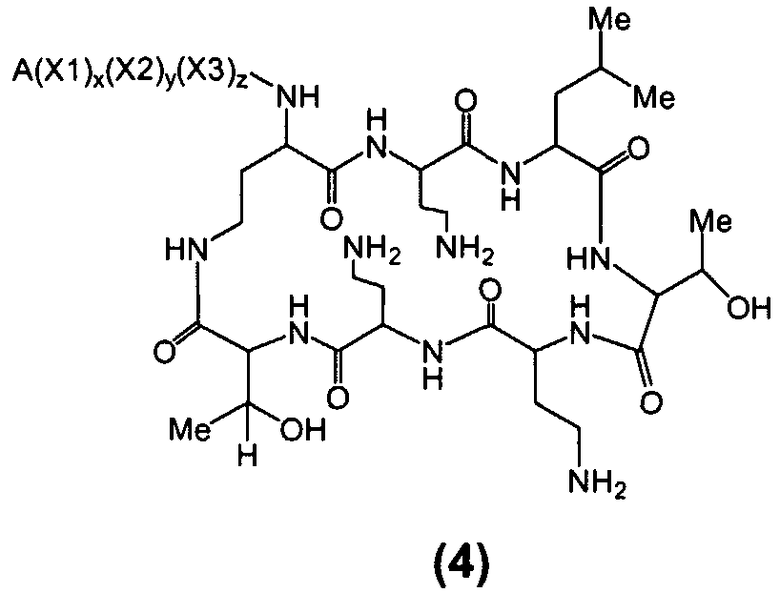
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что если x, y и z, каждый, равны 1, X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' не является разветвленным C8-9-алкилом. В другом варианте осуществления, A не содержит, по меньшей мере, одного аминокислотного остатка.
Один из вариантов осуществления включает в себя соединение формулы (5):
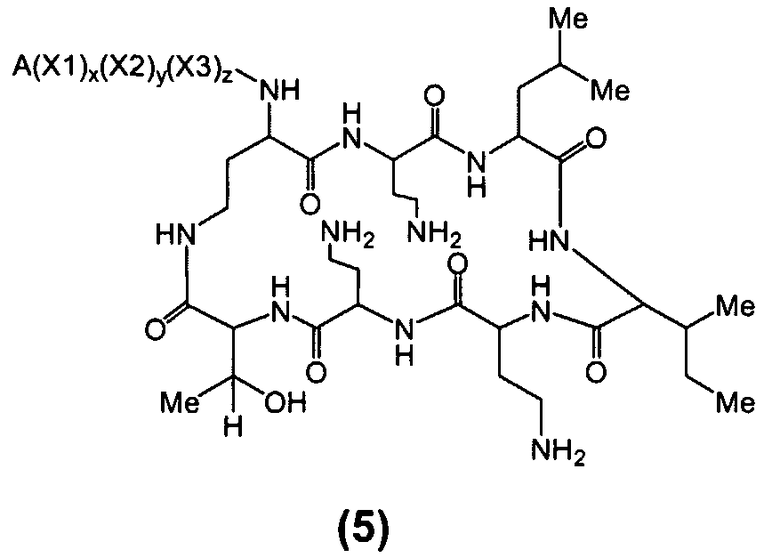
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что если x, y и z, каждый, равны 1, X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' не является разветвленным C7-8-алкилом. В другом варианте осуществления, A не содержит, по меньшей мере, одного аминокислотного остатка.
Один из вариантов осуществления предусматривает соединение формулы (6):
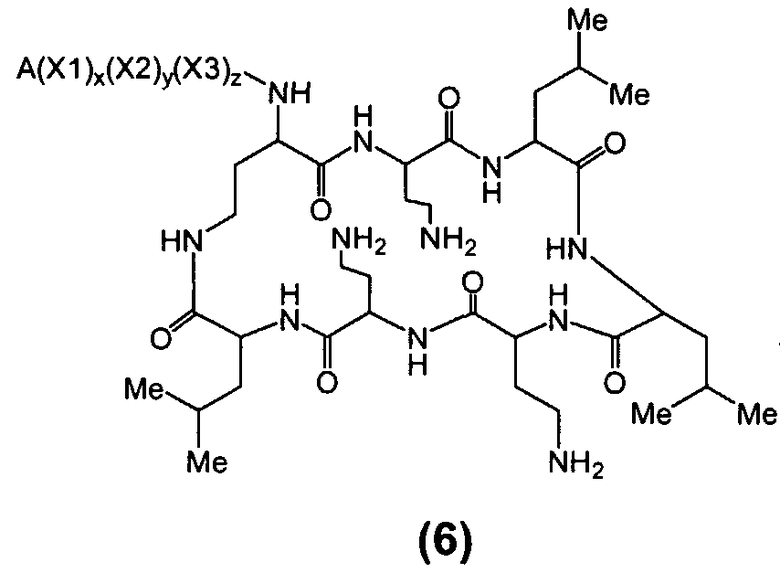
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что если каждый из x и y независимо представляет собой 0, z представляет собой 1, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' выбирают из замещенного алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила, при условии, что замещенный алкил a) не является замещенным оксазолидином или b) не представляет собой формулу алкил-CHOH-CH2-. В другом варианте осуществления, A не содержит, по меньшей мере, одного аминокислотного остатка.
Один из вариантов осуществления описывает соединение формулы (6):
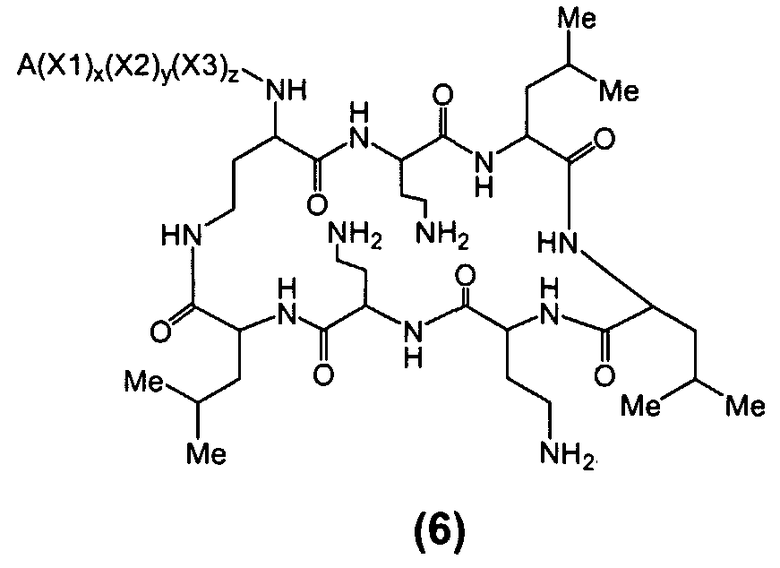
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1.
при условии, что если каждый из x и y независимо представляет собой 0, z представляет собой 1, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' выбирают из замещенного алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила, при условии, что замещенный алкил a) не является замещенным оксазолидином или b) не представляет собой формулу алкил-CHOH-CH2-. В другом варианте осуществления, A не содержит, по меньшей мере, одного аминокислотного остатка. В дополнительном варианте осуществления, если каждый из x и y независимо представляет собой 0, z представляет собой 1, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' выбирают из замещенного C1-7-алкила, замещенного C12-20-алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила.
Один из вариантов осуществления предусматривает соединение формулы (7):
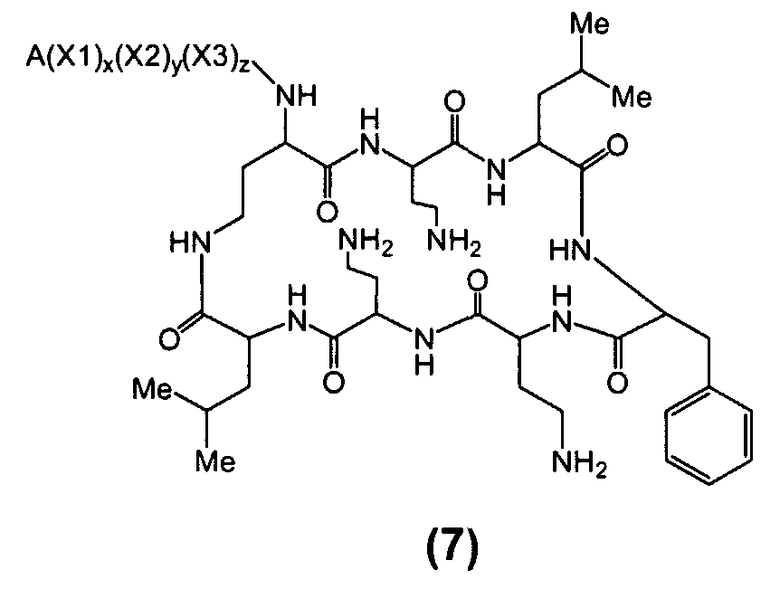
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2 и X3, каждый, независимо, выбирают из аминокислотных остатков; и каждый из x, y, и z представляет собой целое число независимо, выбранное из 0 и 1,
при условии, что если каждый из x и y независимо представляет собой 0, z представляет собой 1, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' не является разветвленным C9-11-алкилом с гидроксильным заместителем. В другом варианте осуществления, A не содержит, по меньшей мере, одного аминокислотного остатка.
Один из вариантов осуществления включает в себя соединение формулы (1):
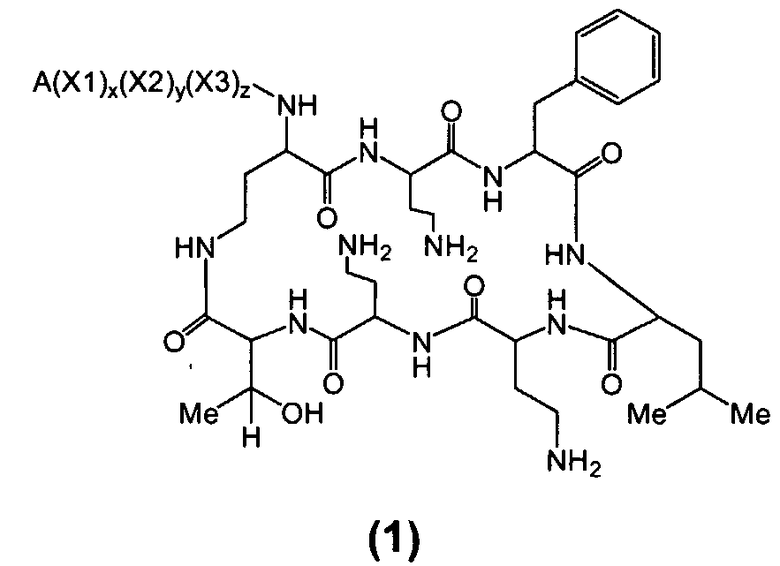
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что:
1) если X3 представляет собой 2,4-диаминобутановую кислоту, каждый из x и y независимо выбирают из 0 и 1 и A представляет собой R'-(C=O)-, тогда R' выбирают из незамещенного алкила, имеющего, по меньшей мере, 9 атомов углерода, циклоалкила, алкенила, арила, гетероарила, гетероциклила, и
замещенного алкила, где, по меньшей мере, один из атомов водорода заменяется группой-заместителем, выбранной из ацила, ациламино, ацилокси, алкенила, алкокси, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенной амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенной амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо и уреидо,
при условии, что замещенный алкил не является выбранным из алкил-CHOH-CH2-, фенил-CH2-, адамантил-CH2-, замещенной арилокси-CH2- и CH3-CHQ- CH2-CH2-, где Q представляет собой структуру:
2) если x, y и z, каждый, равны 1, X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-(C=O)- и R' представляет собой арил, тогда арил не является 6-членным кольцом, имеющим три гидрокси заместителя; и
3) если каждый из x, y, и z независимо представляет собой 0, тогда A выбирают из C8-20-алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила.
Другой вариант осуществления предусматривает соединение, выбранное из формул (I) - (VII) и (1), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой Nα-(н-C9-алканоил)лизин.
Один из вариантов осуществления включает в себя соединение формулы (2):
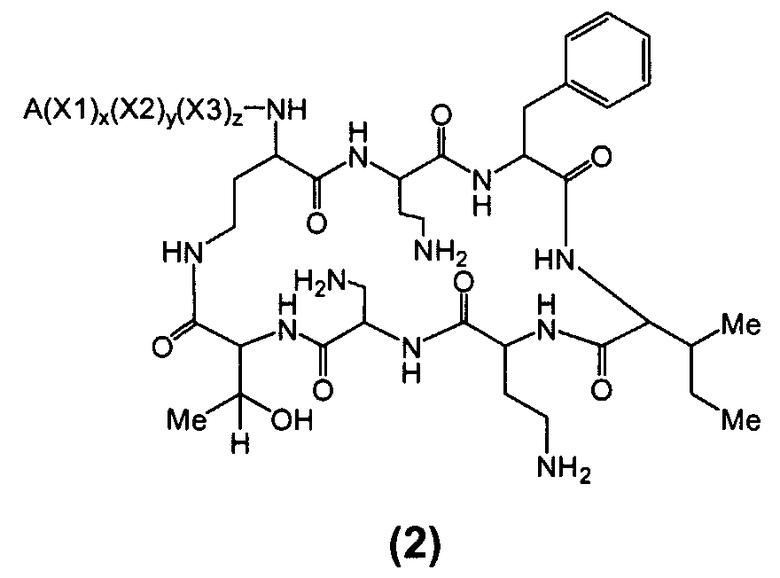
где A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2, и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что если каждый из x, y и z независимо представляет собой 1, X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' не является разветвленным C8-алкилом.
Один из вариантов осуществления описывает соединение формулы (3):
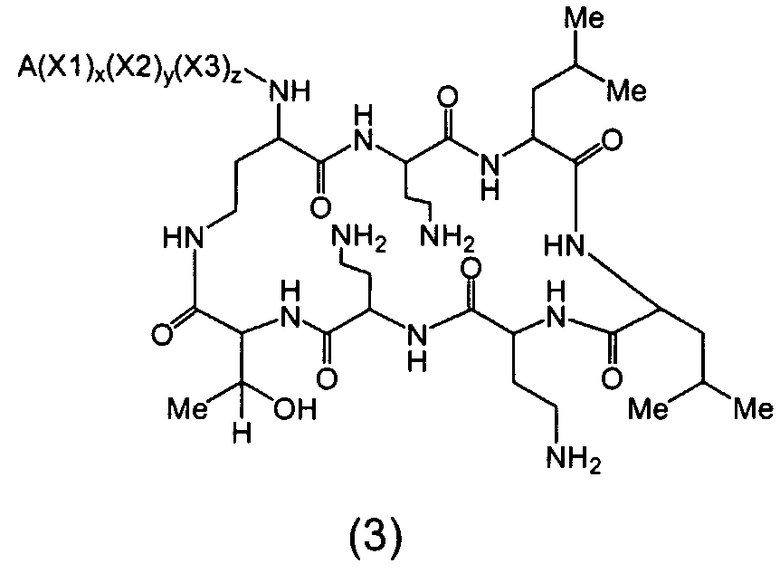
где
A выбирают из R'-(C=O)-, R'-SO2-, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, R'-NH-(C=NH)-, R'-O-(C=O)-, R'-O-(C=S)-, R'-P(O)OH-, R'-(C=S)-, R'-алкила-, R'- и атома водорода; R' выбирают из алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила; X1, X2 и X3, каждый, независимо, выбирают из аминокислотных остатков; и x, y, и z представляют собой целые числа, независимо выбранные из 0 и 1,
при условии, что:
1) если x выбирают из 0 или 1, каждый из y и z независимо представляет собой 1, X1 представляет собой аминокислотный остаток, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-, тогда R' выбирают из Nα-алканоилфенилаланина, Nα-алкеноилфенилаланина, Nα-арилкарбонилфенилаланина, Nα-гетероарилкарбонилфенилаланина;
2) если x выбирают из 0 или 1, каждый из y и z независимо представляет собой 1, X1 представляет собой аминокислотный остаток, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-(C=O)-, тогда R' выбирают из неразветвленного незамещенного C1-4-алкила, циклоалкила, алкенила, арила, гетероарила и гетероциклила, при условии, что: a) алкенил не представляет собой фуранил-CH=CH-, b) арил не является выбранным из нафтила и 4-нитрофенила и c) гетероарил не представляет собой 2-тиофенил; и
3) если x выбирают из 0 или 1, каждый из y и z независимо представляет собой 1, X1 представляет собой аминокислотный остаток, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту и A представляет собой R'-SO2-, тогда R' выбирают из C1-7-алкила, C9-20-алкила, циклоалкила, алкенила, гетероарила и гетероциклила.
Другой вариант осуществления включает в себя соединение, выбранное из формул (I)- (VII) и (1)-(3), где X1 представляет собой 2,4-диаминобутановую кислоту, X2 представляет собой треонин, X3 представляет собой 2,4-диаминобутановую кислоту, A представляет собой R'-(C=O)- и R' представляет собой Nα-(н-C9-алканоил)фенилаланин.
Один из вариантов осуществления включает в себя способ лечения инфекции у субъекта посредством введения терапевтически эффективного количества фармацевтической композиции, содержащей соединение, выбранное из формул (I)-(VII) и (1)-(7) и фармацевтически приемлемый носитель.
Антибактериальная активность
Антибактериальные активности определенных новых соединений указаны в Таблице 4 как их минимальная ингибиторная концентрация (MIC) против Escherichia coli, Staphylococcus aureus и Pseudomonas aeruginosa. MIC могут определяться посредством условий, описанных в Таблице 4, а также в Jarolmen, H. et al., "Activity of Minocycline Against R-Factor Carrying Enterobacteriaceae», Infectious Immunity, Vol. 1, № 4, pp. 321-326, 1970, описание которой включается сюда в качестве ссылки.
Пролекарства
В одном из вариантов осуществления, предусматриваются пролекарства описанных пептидов. Пептиды, имеющие гептапептидную структуру формулы (B) и защищенные HSO3-Fmoc и/или другими аминозащитными группами, содержащими, по меньшей мере, одну кислотную группу, представляют собой антибактериальные пролекарства. Группа HSO3-Fmoc отщепляется после введения животному, такому, например, как млекопитающее, включая человека, высвобождая биологически активный пептид. Введение биологического активного пептида в виде защищенного пролекарства может приводить к возникновению механизма замедленного высвобождения для антибактериального соединения. Обсуждение пролекарств приводится в ссылках 32-33.
Фармацевтические композиции
Также описываются фармацевтические композиции или препараты, содержащие соединения, описанные здесь, или их соли.
Фармацевтические композиции могут приготавливаться для перорального, внутривенного, внутримышечного, подкожного или парентерального введения, для терапевтического или профилактического лечения заболеваний, таких как бактериальные инфекции.
Фармацевтические препараты, описанные здесь, могут приготавливаться в соответствии со стандартными процедурами и вводятся при дозах, которые выбираются для уменьшения, предотвращения или устранения инфекции (смотри, например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, и Goodman and Gilman's "Pharmaceutical Basis of Therapeutics», Pergamon Press, New York, NY, содержание которой включается сюда в качестве ссылки, для общего описания способов введения различных противомикробных агентов для терапии человека).
Фармацевтические композиции могут содержать одно или несколько соединений, описанных здесь в сочетании с одним или несколькими нетоксичными фармацевтически приемлемыми носителями и/или разбавителями, и/или вспомогательными веществами, и/или наполнителями. Как здесь используется, фраза "фармацевтически приемлемый носитель" относится ко всем растворителям, дисперсионным средам, покрытиям, антибактериальным и противогрибковым агентам, изотоническим и замедляющим поглощение агентам, и тому подобное, и к любому из них, которые являются совместимыми с введением фармацевтического препарата. Использование таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области. Неограничивающие примеры носителей и наполнителей включают в себя кукурузный крахмал или желатин, лактозу, сахарозу, микрокристаллическую целлюлозу, каолин, маннит, дикальций фосфат, хлорид натрия и альгиновую кислоту. Композиции могут содержать натрий кросс-кармелозу, микрокристаллическую целлюлозу, кукурузный крахмал, крахмал глюколят натрия и альгиновую кислоту.
Связующие таблеток, которые могут включаться, представляют собой смолу акации, метилцеллюлозу, натрий карбоксиметилцеллюлозу, поливинилпирролидон (повидон), гидроксипропилметилцеллюлозу, сахарозу, крахмал и этилцеллюлозу.
Смазывающие материалы, которые могут использоваться, включают в себя стеарат магния или другие стеараты металлов, стеариновую кислоту, жидкий силикон, тальк, воски, масла и коллоидную окись кремния.
Также могут использоваться ароматизирующие агенты, такие как ароматы перечной мяты, масло грушанки, вишни или что-либо подобное. Может также быть желательным добавление окрашивающего агента, чтобы сделать дозированную форму более эстетичной на вид или чтобы помочь в идентификации продукта.
Для перорального или парентерального введения, соединения по настоящему изобретению могут смешиваться с обычными фармацевтическими носителями и наполнителями и использоваться в форме таблеток, капсул, эликсиров, суспензий, сиропов, пластинок и тому подобное. Композиции, содержащие соединение по настоящему изобретению, могут содержать примерно от 0,1% примерно до 99 мас.% активного соединения, например, примерно от 10% примерно до 30%.
Для перорального введения пригодными являются твердые препараты, такие как таблетки и капсулы. Препараты с замедленным высвобождением или ентеросолюбильным покрытием также могут предусматриваться. Для педиатрических и гериатрических применений, один из вариантов осуществления предусматривает суспензии, сиропы и жевательные таблетки. Для перорального введения, фармацевтические композиции находятся в форме, например, таблетки, капсулы, суспензии или жидкости.
Фармацевтические композиции могут изготавливаться в форме стандартной дозированной формы, содержащей терапевтически эффективное количество активного ингредиента. Примеры таких стандартных дозированных форм представляют собой таблетки и капсулы. Для терапевтических целей, таблетки и капсулы могут содержать, в дополнение к активному ингредиенту, обычные носители, такие как связывающие агенты, например смолу акации, желатин, поливинилпирролидон, сорбитол или смолу трагаканта; начинку, например фосфат кальция, глицин, лактозу, кукурузный крахмал, сорбит или сахарозу; смазывающие вещества, например стеарат магния, полиэтиленгликоль, окись кремния или тальк; разрыхлители, например картофельный крахмал, ароматизирующие или окрашивающие агенты, или приемлемые смачивающие агенты. Жидкие препараты для перорального введения, как правило, находятся в форме водных или масляных растворов, суспензий, эмульсий, сиропов или эликсиров, препараты по настоящему изобретению могут содержать обычные добавки, такие как суспендирующие агенты, эмульсифицирующие агенты, неводные агенты, консерванты, окрашивающие агенты и ароматизирующие агенты. Неограничивающие примеры добавок для жидких препаратов включают в себя смолу акации, миндальное масло, этиловый спирт, фракционированное кокосовое масло, желатин, сироп глюкозы, глицерин, гидрированные съедобные жиры, лецитин, метилцеллюлозу, метил или пропил пара-гидроксибензоат, пропиленгликоль, сорбит или сорбиновую кислоту.
Для внутривенного (IV) использования, фармацевтическая композиция может растворяться или суспендироваться в любой из повсеместно используемых внутривенных жидкостей и вводиться посредством вливания. Внутривенные жидкости включают в себя, без ограничения, физиологический раствор или раствор Рингера. Внутривенное введение может осуществляться посредством использования, без ограничения, шприца, мининасоса или внутривенной трубки.
Фармацевтические композиции по настоящему изобретению для парентеральной инъекции включают в себя фармацевтически приемлемые водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для разбавления в виде стерильных растворов или дисперсий для инъекций непосредственно перед использованием.
Примеры пригодных для использования водных и неводных носителей, разбавителей, растворителей или наполнителей включают в себя воду, этанол, бензиловый спирт, полиолы (такие как глицерин, пропиленгликоль и полиэтиленгликоль), и соответствующие их смеси, растительные масла (такие как кукурузное масло или оливковое масло) и органические сложные эфиры для инъекций, такие как этилолеат. Соответствующая текучесть может поддерживаться, например, посредством использования материалов для покрытия, таких как лецитин, посредством поддержания требуемого размера частиц, в случае дисперсий, и посредством использования поверхностно-активных веществ. Композиции могут содержать различные буферы.
Эти композиции могут также содержать вспомогательные вещества, такие как консерванты, смачивающие агенты, эмульгирующие агенты и диспергирующие агенты. Они могут также содержать маркировочные или другие агенты для защиты от подделки, которые хорошо известны в данной области. Предотвращение действия микроорганизмов может обеспечиваться посредством включения различных антибактериальных и противогрибковых агентов, например парабена, хлорбутанола и фенолсорбиновой кислоты. Также может быть желательным включение изотонических агентов, таких как сахара и хлорид натрия. Пролонгированное поглощение фармацевтической формы для инъекций может быть получено посредством включения агентов, которые замедляют поглощение, таких как моностеарат алюминия и желатин.
Формы депо для инъекций могут быть изготовлены посредством образования микроинкапсулирующих матриц для лекарственного средства в биологически деградируемых полимерах, таких как полилактид-полигликолид. В зависимости от отношения лекарственного средства к полимеру и природы конкретного используемого полимера, может контролироваться скорость высвобождения лекарственного средства. Примеры других биологически деградируемых полимеров включают в себя сложные поли(ортоэфиры) и поли(ангидриды). Препараты депо для инъекций могут также быть получены посредством захвата лекарственного средства в липосомах или микроэмульсиях, которые являются совместимыми с телесными тканями.
Препараты для инъекций могут стерилизоваться, например, посредством фильтрования через бактериально-удерживающий фильтр, или посредством включения стерилизующих агентов в форме стерильных твердых композиций, которые могут растворяться или диспергироваться в стерильной воде или другой стерильной среде для инъекций непосредственно перед использованием.
Твердые дозированные формы для перорального введения включают в себя капсулы, таблетки, пилюли, порошки и гранулы. Такие формы могут включать в себя формы, которые быстро растворяются или разрыхляются в оральной окружающей среде. В таких твердых дозированных формах, активное соединение может смешиваться, по меньшей мере, с одним инертным, фармацевтически приемлемым наполнителем или носителем. Пригодные для использования наполнители включают в себя, например, (a) заполнители или препараты для увеличения объема, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (b) связующие, такие как целлюлоза и производные целлюлозы (такие как гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза и карбоксиметилцеллюлоза), альгинаты, желатин, поливинилпирролидон, сахароза и смола акации; (c) увлажнители, такие как глицерин; (d) разрыхляющие агенты, такие как натрий крахмал глюколят, кросс-кармелоза, агар-агар, карбонат кальция, крахмал из картофеля или тапиоки, альгиновая кислота, определенные силикаты и карбонат натрия; (e) агенты, удерживающие раствор, такие как парафин; (f) ускорители поглощения, такие как соединения четвертичного аммония; (g) смачивающие агенты, такие как цетиловый спирт и глицерин моностеарат, сложные эфиры жирных кислота и сорбитана, полоксамеры и полиэтиленгликоли; (h) поглотители, такие как каолин и бентонитовая глина; (i) смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурил сульфат натрия, и их смеси; и (j) вещества, способствующие скольжению, такие как тальк и двуокись кремния. Другие пригодные для использования наполнители включают в себя, например, цитрат натрия или дикальций фосфат. Дозированные формы могут также содержать буферные агенты.
Твердые дозированные формы, включая формы таблеток, драже, капсул, пилюль и гранул, могут приготавливаться с покрытиями и оболочками, такими как функциональные и эстетические ентеральные покрытия и другие покрытия, хорошо известные в области фармацевтических препаратов. Они могут необязательно содержать замутняющие агенты и красители. Они также могут находиться в форме, приспособленной для контролируемого или замедленного высвобождения. Примеры композиций для погружения, которые могут использоваться для таких целей, включают в себя полимерные вещества и воски.
Фармацевтические композиции могут доставляться с использованием систем доставки с контролируемым (например, капсулы) или замедленным высвобождением (например, биологически эродируемые матрицы). Примеры систем доставки с замедленным высвобождением для доставки лекарственных средств, которые пригодны для введения фармацевтических композиций, описываются в патентах США №№ 4452775 (выдан Kent), 5239660 (выдан Leonard), и 3854480 (выдан Zaffaroni).
В некоторых случаях, для пролонгирования действия лекарственного средства, может быть желательным замедление поглощения лекарственного средства после подкожной или внутримышечной инъекции. Это может достигаться посредством использования жидкой суспензии кристаллического или аморфного материала с плохой растворимостью в воде. Аморфный материал может использоваться сам по себе или вместе со стабилизаторами, по необходимости. Скорость поглощения лекарственного средства зависит тогда от его скорости растворения, которая, в свою очередь, может зависеть от размера кристалла и формы кристалла.
Альтернативно, замедленное поглощение парентерально вводимой лекарственной формы может достигаться посредством растворения или суспендирования лекарственного средства в масляном носителе.
Для внутримышечных препаратов, стерильный препарат соединения по настоящему изобретению или соответствующая растворимая солевая форма соединения, например, гидрохлоридная соль, может растворяться и вводиться в фармацевтическом разбавителе, таком как вода для инъекций (WFI), физиологический раствор или 5% глюкоза. Соответствующая нерастворимая форма соединения может получаться и вводиться в виде суспензии в водной основе или фармацевтически приемлемой масляной основе, например в сложном эфире длинноцепочечной жирной кислоты, таком как этилолеат.
Доза внутривенного, внутримышечного или парентерального препарата соединения по настоящему изобретению может вводиться в виде болюса или посредством медленного вливания. Болюс представляет собой дозу, которая вводится менее чем за 30 минут. В одном из вариантов осуществления, болюс вводится менее чем за 15 или менее чем за 10 минут. В другом варианте осуществления, болюс вводится менее чем за 5 минут. Еще в одном варианте осуществления, болюс вводится за одну минуту или менее. Вливание представляет собой дозу, которая вводится со скоростью 30 минут или больше. В одном из вариантов осуществления, вливание составляет один час или больше. В другом варианте осуществления, вливание является по существу постоянным.
Для местного использования, фармацевтические композиции также могут приготавливаться в соответствующих формах, для нанесения на кожу или слизистые мембраны носа и глотки, и могут принимать форму кремов, мазей, жидких спреев или препаратов для ингаляции, лепешек или препаратов для смазывания горла. Такие препараты для местного введения, кроме того, могут содержать химические соединения, такие как диметилсульфоксид (DMSO), для облегчения проникновения активного ингредиента через поверхность.
Для применения для глаз или ушей, фармацевтическая композиция может быть представлена в жидкой или полужидкой форме, приготовленной в гидрофобных или гидрофильных основах как мази, кремы, лосьоны, препараты для смазывания или порошки.
Для ректального введения, фармацевтические композиции могут вводиться в форме суппозиториев, смешанных с обычными носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, или другой глицерид, которые являются твердыми при комнатной температуре, но жидкими при температуре тела, и по этой причине, расплавляются в прямой кишке или вагинальной полости и высвобождают активное соединение.
Альтернативно, фармацевтические композиции могут находиться в порошкообразной форме для разбавления в соответствующем фармацевтически приемлемом носителе во время доставки. В другом варианте осуществления, стандартная дозированная форма соединения может представлять собой раствор соединения или его соли в соответствующем разбавителе, в стерильных, герметично изолированных ампулах или стерильных шприцах. Концентрация соединения в стандартной дозированной форме может изменяться, например, примерно от 1% примерно до 50%, в зависимости от используемого соединения и его растворимости, и дозы, желаемой врачом. Если композиции содержатся в стандартных дозированных формах, каждая стандартная дозированная форма может содержать 1-500 мг активного материала. Для лечения взрослого человека, используемая доза может находиться в пределах от 5 мг до 10 г в день, в зависимости от способа и частоты введения.
Фармацевтические композиции, описанные здесь, могут помещаться в фармацевтически приемлемый носитель и доставляться принимающему их субъекту (например, человеку) в соответствии с известными способами доставки лекарственных средств. Как правило, способы доставки фармацевтических композиций in vivo используют известные в данной области протоколы для доставки агента, при этом единственная существенная модификация процедуры представляет собой замену соединениями по настоящему изобретению лекарственных средств в протоколах, известных в данной области. Подобным образом, способы использования заявляемых композиций для обработки клеток в культуре, например, для устранения или уменьшения уровня бактериального загрязнения культуры клеток, используют известные в данной области протоколы для обработки культур клеток антибактериальным агентом (агентами), при этом единственная существенная модификация процедуры представляет собой замену соединениями по настоящему изобретению лекарственных средств в протоколах, известных в данной области.
Способы применения
В одном из вариантов осуществления, настоящее изобретение предусматривает способ лечения инфекции у субъекта посредством введения терапевтически эффективного количества соединения или композиции по настоящему изобретению. В одном из вариантов осуществления, способ включает в себя введение субъекту, нуждающемуся в этом, фармацевтической композиции, содержащей, по меньшей мере, одно из соединений, описанных здесь. В одном из вариантов осуществления, фармацевтическая композиция может содержать любое из соединений, описанных здесь, в виде единственного активного соединения или в сочетании с другим соединением, композицией или биологическим материалом.
Термины "лечение", "терапевтический метод" и родственные им слова относятся как к терапевтическому лечению, так и к профилактическим/превентивным мерам. Те, кто нуждается в лечении, могут включать в себя индивидуумов, уже имеющих конкретное медицинское заболевание, а также тех, у кого имеется риск заболевания (то есть, тех, кто вероятно, в конечном счете, получат расстройство). Терапевтический способ приводит к предотвращению или ослаблению симптомов, или к иным образом желательному биологическому результату и может оцениваться по улучшению клинических признаков, замедлению наступления заболевания, пониженным/повышенным уровням лимфоцитов и/или антител и тому подобное.
В одном из вариантов осуществления, фармацевтический препарат является терапевтически эффективным для людей, хотя и не обладает перекрестной устойчивостью с аминогликозидами, β-лактамами и фторхинолонами.
Примеры процедур для доставки антибактериального агента описаны в патенте США № 5041567 и PCT заявке на Европейский патент номер EP94/02552 (публикации заявки на Международный патент № WO 95/05384), описания которых включается сюда в качестве ссылок во всей их полноте. Как здесь используется, фразы "терапевтически эффективная доза" и "терапевтически эффективное количество" относятся к количеству соединения по настоящему изобретению, которое предотвращает наступление, ослабляет симптомы, прекращает развитие бактериальной инфекции или приводит к получению другого желаемого биологического результата, такого, например, как улучшение клинических симптомов или понижение/повышение уровней лимфоцитов и/или антител. Термин "лечение" определяется как введение субъекту терапевтически эффективного количества соединения по настоящему изобретению, как для предотвращения возникновения инфекции, так и для контроля или устранения инфекции. Термин "субъект", как здесь используется, относится к млекопитающему, растению, низшему животному или клеточной культуре. В одном из вариантов осуществления, субъект представляет собой человека или другое животное, пациента, нуждающегося в антибактериальном лечении.
Настоящее изобретение также предусматривает способы введения соединения, описанного здесь, или его фармацевтической композиции субъекту, нуждающемуся в этом, в количестве, которое является эффективным при уменьшении или устранении бактериальной инфекции. Фармацевтические композиции по настоящему изобретению могут вводиться людям и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местным образом (например, посредством порошков, мазей или капель), буккально, в виде перорального или назального спрея, или посредством имплантированного резервуара, внешнего насоса или катетера. Композиции также могут вводиться через легкие посредством ингаляции. Термин "парентеральное введение" как здесь используется, относится к способам введения, которые включают в себя внутривенную, внутримышечную, внутрибрюшинную, интрацистернальную, подкожную и интраартикулярную инъекцию и вливание. Соединение или композиция могут приготавливаться для офтальмологических или аэрозольных применений.
В одном из вариантов осуществления, фармацевтическая композиция представляет собой антибактериальный агент, антибиотик или противогрибковый агент.
В одном из вариантов осуществления, способ по настоящему изобретению может использоваться для лечения субъекта, имеющего бактериальную инфекцию, у которого инфекция вызывается или обостряется посредством любого типа бактерий, таких как грамотрицательные или грамположительные бактерии. В одном из вариантов осуществления, бактериальная инфекция может вызываться или обостряться грамотрицательными бактериями. Эти грамотрицательные бактерии включают в себя, но, не ограничиваясь этим, Acinetobacter spp. (включая Acinetobacter baumannii), Citrobacter spp., Enterobacter spp., Escherichia spp. (включая Escherichia coli), Haemophilus influenzae, Morganella morganii, Pseudomonas aeruginosa, Klebsiella spp. (включая Klebsiella pneumoniae), Salmonella spp., Shigella spp., Yersinia pseudotuberculosis, и все виды Enterobacter, Pasteurella, Brucella, Bordetella, Proteus, Serratia, Providencia и Edwardsiella.
В одном из вариантов осуществления, бактериальная инфекция может вызываться или обостряться грамположительными бактериями. Эти грамположительные бактерии включают в себя, но, не ограничиваясь этим, метициллин-чувствительные и метициллин-устойчивые стафилококки (включая Staphylococcus aureus, S. epidermidis, S. haemolyticus, S. hominis, S. saprophyticus и коагулаза-отрицательные стафилококки), интермедиарно-чувствительные к гликопептидам S. aureus (GISA), ванкомицин-устойчивые Staphylococcus aureus (VRSA), пенициллин-чувствительные и пенициллин-устойчивые стрептококки (включая Streptococcus pneumoniae, S. pyogenes, S. agalactiae, S. avium, S. bovis, S. lactis, S. sangius и Streptococci Group C, Streptococci Group G и виридановые стрептококки), энтерококки (включая ванкомицин-чувствительные и ванкомицин-устойчивые штаммы, такие как Enterococcus faecalis и E. faecium), Clostridium difficite, C. clostridiiforme, C. innocuum, C. perfringens, C. ramosum, Haemophilus influenzae, Listeria monocytogenes, Corynebacterium jeikeium, Bifidobacterium spp., Eubacteriurn aerofaciens, E. lentum, Lactobacillus acidophilus, L. casei, L. plantarum, Lactococcus spp., Leuconostoc spp., Pediococcus spp., Peptostreptococcus anaerobius, P. asaccarolyticus, P.magnus, P. micros, P. prevotii, P. productus, Propionibacterium acnes, Actinomyces spp. и Moraxella spp. (включая M. catarrhalis).
В одном из вариантов осуществления, антибактериальная активность соединений, описанных здесь, против классических "устойчивых" штаммов сравнима с активностью против классически "чувствительных" штаммов в экспериментах in vitro. В одном из вариантов осуществления, соединение в соответствии с настоящим изобретением или его фармацевтическая композиция вводится в соответствии со способами по настоящему изобретению пациенту, нуждающемуся в быстро действующей антибиотической терапии.
Способ по настоящему изобретению может использоваться для лечения любой бактериальной инфекции любого органа или ткани в организме. В одном из вариантов осуществления, бактериальная инфекция вызывается грамотрицательными бактериями. Эти органы или ткани включают в себя, без ограничения, скелетные мышцы, кожу, кровоток, почки, сердце, лёгкие и кости. Способ по настоящему изобретению может использоваться для лечения, без ограничения, инфекций кожи и мягких тканей, бактериемии и инфекций мочевого тракта. Способ по настоящему изобретению может использоваться для лечения общественно значимых респираторных инфекций, включая, без ограничения, воспаление среднего уха, синусит, хронический бронхит и пневмонию, включая пневмонию, вызванную устойчивыми к лекарственным средствам S. pneumoniae или H. influenzae. Способ по настоящему изобретению также может использоваться для лечения смешанных инфекций, которые включают в себя различные типы грамотрицательных бактерий или которые включают в себя как грамположительные, так и грамотрицательные бактерии. Эти типы инфекций включают в себя интраабдоминальные инфекции и обстетрические/гинекологические инфекции. Способ по настоящему изобретению также может использоваться для лечения инфекции, включающей в себя, без ограничения, эндокардит, нефрит, септический артрит, интраабдоминальный сепсис, инфекции костей и суставов и остеомиелит. В одном из вариантов осуществления, любое из описанных выше заболеваний может лечиться с использованием соединения в соответствии с настоящим изобретением или их фармацевтических композиций.
Способ по настоящему изобретению может также осуществляться, в то же время, одновременно вводя один или несколько других противомикробных агентов, таких как антибактериальные агенты (антибиотики) или противогрибковые агенты. В одном из аспектов, способ может осуществляться посредством введения более одного соединения в соответствии с настоящим изобретением. В другом варианте осуществления, способ может осуществляться посредством введения соединения в соответствии с настоящим изобретением вместе с другим пептидным соединением, описанным здесь, или пептидными соединениями описанными, например, в заявках на Международный патент WO 01/44272; WO 01/44274; и WO 01/44271.
Антибактериальные агенты и их классы, которые могут вводиться совместно с соединением по настоящему изобретению, включают в себя, без ограничения, пенициллины и родственные лекарственные средства, карбапенемы, цефалоспорины и родственные лекарственные средства, аминогликозиды, бацитрацин, грамицидин, мупироцин, хлорамфеникол, тиамфеникол, натрий фузидат, линкомицин, клиндамицин, макролиды, новобиоцин, полимиксины, рифамицины, спектиномицин, тетрациклины, ванкомицин, тейкопланин, стрептограмины, антифолиатные агенты, включая сульфонамиды, триметоприм и его сочетания и пириметамин, синтетические антибактериальные средства, включая нитрофураны, метенамин манделат и метенамин гиппурат, нитроимидазолы, хинолоны, фторхинолоны, изониазид, этамбутол, пиразинамид, пара-аминосалициловую кислоту (PAS), циклосерин, капреомицин, этионамид, протионамид, тиацетазон, виомицин, эверниномицин, гликопептид, глицилциклин, кетолиды, оксазолидинон; имипенен, амикацин, нетилмицин, фосфомицин, гентамицин, цефтриаксон, ZIRACIN® (56-деацетил-57-деметил-45-O-де(2-метил-1-оксопропил)-12-O-(2,3,6-тридезокси-3-C-метил-4-O-метил-3-нитро-альфа-L-арабино-гексопиранoзил)фламбамицин), LY 333328, CL 331002, HMR 3647 ZYVOX® (линезолид), SYNERCID® (дальфопристин-хинупристин), Азтреонам, Метронидазол, эпироприм, OCA-983, GV-143253, Натрий санфетринем, CS-834, Биапенем, A-99058.1, A-165600, A-179796, KA 159, Динемицин A, DX8739, DU 6681; Цефлупренам, ER 35786, Цефоселиз, Санфетринем целексетил, HGP-31, Цефпиром, HMR-3647, RU-59863, Мерсацидин, KP 736, Рифалазил; Козан, AM 1732, MEN 10700, Ленапенем, BO 2502A, NE-1530, K130, OPC 20000, OPC 2045, Венеприм, PD 138312, PD 140248, CP 111905, Сулопенем, ритипенам ацоксил, RO-65-5788, Циклотиалидин, Sch-40832, SEP-132613, микакоцидин A, SB-275833, SR-15402, SUN A0026, TOC 39, карумонам, Цефозопран, Цефетамет пивоксил и T 3811.
Противогрибковые агенты, которые могут вводиться совместно с соединением в соответствии с настоящим изобретением, включают в себя, без ограничения, каспофунген, вориконазол, сертаконазол, IB-367, FK-463, LY-303366, Sch-56592, ситафлоксацин, DB-289, полиены, такие как амфотерицин, нистатин, примарицин; азолы, такие как флуконазол, итраконазол и кетоконазол; аллиламины, такие как нафтифин и тербинафин; и антиметаболиты, такие как флуцитозин. Другие противогрибковые агенты включают в себя, без ограничения, те, которые описаны в Fostel et al., Drug Discovery Today 5:25-32 (2000), включаемой сюда в качестве ссылки. Fostel et al. описывает противогрибковые соединения, включая коринекандин, Mer-WF3010, фузакандины, артрихитин/LL 15G256, сордарины, циспентацин, азоксибациллин, ауреобазидин и кафрефунгин.
Соединения по настоящему изобретению могут вводиться в виде аэрозоля для лечения пневмонии или других инфекций, связанных с легкими. В одном из вариантов осуществления, носитель для доставки аэрозоля представляет собой безводный или сухой порошок для ингаляций. Соединения или его фармацевтические композиции могут также непосредственно вводиться в виде инъекции или вводиться в абсцесс, вентрикулу или сустав. Парентеральное введение включает в себя подкожную, внутривенную, внутримышечную, интраартикулярную, интрасиновиальную, цистернальную, интратекальную, внутрипеченочную, интралесиональную и интракраниальную инъекцию или вливание. В другом варианте осуществления, соединения по настоящему изобретению вводятся внутривенно, подкожно или перорально. Еще в одном варианте осуществления, соединение или композиция вводится в культуру клеток, например, посредством введения в питательную среду.
Дозировка
Реальные уровни дозировки активных ингредиентов в фармацевтических композициях по настоящему изобретению могут изменяться с тем, чтобы получить терапевтически эффективное количество активного соединения (соединений) для достижения желаемой терапевтической реакции конкретного пациента, композиции и способы введения. Эффективное количество может определяться, как здесь описано. Выбранный уровень дозировки будет зависеть от активности конкретного соединения, способа введения, тяжести состояния, которое лечится, и состояния и предыдущей медицинской истории пациента, которого лечат. Однако в рамках знаний в данной области находится способ начинать дозировку соединения при уровнях, более низких, чем требуется для достижения желаемого терапевтического воздействия, и постепенно увеличивать дозировку, пока не будет достигнуто желаемое воздействие. В одном из вариантов осуществления, данные, полученные из анализов, могут использоваться при определении диапазона дозировок для использования на людях.
Способ может также включать в себя введение субъекту эффективной дозы соединения по настоящему изобретению. Эффективная доза может находиться в пределах примерно от 0,1 примерно до 100 мг/кг соединения по настоящему изобретению или его фармацевтически приемлемой соли. В одном из вариантов осуществления, доза находится в пределах примерно от 0,1 примерно до 50 мг/кг соединения по настоящему изобретению или его фармацевтически приемлемой соли. В другом варианте осуществления, доза находится в пределах примерно от 1 до 25 мг/кг соединения по настоящему изобретению или его фармацевтически приемлемой соли. Эффективная доза для культуры клеток может находиться в пределах от 0,1 до 1000 пг/мл, например, от 0,1 до 200 мкг/мл.
Как правило, уровни доз примерно от 0,1 мкг/кг примерно до 50 мг/кг, например, уровень в пределах примерно от 5 примерно до 20 мг активного соединения на килограмм массы тела в день, могут вводиться местным образом, перорально или внутривенно пациенту млекопитающему. Другие уровни доз находятся в пределах примерно от 1 мкг/кг примерно до 20 мг/кг, примерно от 1 мкг/кг примерно до 10 мг/кг, примерно от 1 мкг/кг примерно до 1 мг/кг, от 10 мкг/кг до 1 мг/кг, от 10 мкг/кг до 100 мкг/кг, от 100 мкг до 1 мг/кг и примерно от 500 мкг/кг примерно до 5 мг/кг в день. Если это желательно, эффективная дневная доза может разделяться на множество доз для целей введения, например от двух до четырех раздельных доз в день. В одном из вариантов осуществления, фармацевтическая композиция может вводиться один раз в день.
Композиции, содержащие соединения по настоящему изобретению, могут вводиться в виде одной ежедневной дозы или как множество доз в день. Режим лечения может требовать введения в течение продолжительных периодов времени, например, в течение нескольких дней или в течение от двух до четырех недель. Количество на введенную дозу или общее введенное количество будет зависеть от таких факторов как природа и тяжесть инфекции, возраст и общее состояние здоровья пациента, толерантность пациента к соединению и микроорганизм или микроорганизмы, вовлеченные в инфекцию.
Соединения по настоящему изобретению могут также вводиться в пище или корме пациента или животного. Если оно вводится как часть общего приема пищи, используемое количество соединения может быть меньшим чем 1 мас.% от пищи, например не больше чем 0,5 мас.%. Пища для животных может представлять нормальный корм, в который может добавляться соединение, или оно может добавляться к полуфабрикату.
Другие варианты осуществления, описанные здесь, включают в себя:
1. Способ получения промежуточного соединения для использования при синтезе нового пептидного антибиотика, включающий в себя стадии:
(a) Защиты аминогрупп полимиксина B, колистина, [Ile7]-полимиксина B1, циркулина и октапептина с помощью (2-сульфо)-9-флуоренилметоксикарбонильного или другого кислотного производного 9-флуоренилметоксикарбонила;
(b) Обработки продукта из реакций на стадии (a) деацилазой, с получением защищенного пептидного промежуточного соединения; и
(c) Использования модифицированного способа деградации Эдмана или ферментативной реакции пептидазы для получения другого защищенного промежуточного соединения посредством уменьшения, на одну- три аминокислоты боковой цепи защищенного пептидного промежуточного соединения; и
(d) Очистки защищенного пептидного промежуточного соединения с помощью хроматографии.
Другой вариант осуществления описывает:
2. Способ получения антибиотика, активного против грамотрицательных и грамположительных бактерий, включая штаммы, устойчивые к клинически используемым антибиотикам, включающий в себя стадии:
(a) Защиты аминогрупп полимиксинов или других родственных антибиотиков, выбранных из группы, состоящей из колистина, [Ile7]-полимиксина B1, циркулина и октапептина, с помощью (2-сульфо)-9-флуоренилметоксикарбонила или другого кислотного производного 9-флуоренилметоксикарбонила;
(b) Обработки продукта из реакции на стадии (a) деацилазой, с получением защищенного пептидного промежуточного соединения;
(c) Использования модифицированного способа деградации Эдмана или ферментативной реакции пептидазы для получения другого защищенного промежуточного соединения пептида посредством уменьшения размера, на одну-три аминокислоты, экзоциклической пептидной боковой цепи защищенного пептида.
(d) Химического модифицирования промежуточного соединения, с получением защищенного антибактериального производного; и
(e) Удаления кислотных защитных групп с получением антибиотика.
Другой вариант осуществления описывает:
3. Промежуточное соединение, которое представляет собой химически защищенную форму пептида, полученную из полимиксинов, [Ile7]-полимиксина B1, октапептинов, колистина, циркулинов или родственных антибиотиков и выбирают из группы, состоящей из следующих групп или соответствующих их солей:
1) H-(X1)(X2)(X3)-пептид-[(2-сульфо)-9-Fmoc]n
2) H-(X2)(X3)-пептид-[(2-сульфо)-9-Fmoc]n
3) H-(X3)-пептид-[(2-сульфо)-9-Fmoc]n
4) H-пептид-[(2-сульфо)-9-Fmoc]3,
где для случая 1) H-(X1)(X2)(X3)-пептид-[(2-сульфо)Fmoc]n: H представляет собой атом водорода, X1 представляет собой L-Dab или другую аминокислоту, X2 представляет собой L-Thr или другую аминокислоту, X3 представляет собой L-Dab или D-Dab или другую аминокислоту и n=3-6;
для случая 2) H-(X2)(X3)-пептид-[(сульфо)-9-Fmoc]n: H представляет собой атом водорода, X2 представляет собой L-Thr или другую аминокислоту, X3 представляет собой L-Dab или D-Dab или другую аминокислоту, n=3-5;
для случая 3) H-(X3)-[пептид-[(2-сульфо)-9-Fmoc]n: H представляет собой атом водорода, X3 представляет собой L-Dab или D-Dab или другую аминокислоту и n=3-4; и
для случая 4) H-пептид-[(сульфо)-9-Fmoc]n: H представляет собой атом водорода.
Другой вариант осуществления описывает:
4. Кислотно защищенные пептидные промежуточные соединения, полученные из соответствующего защищенного полимиксина B, которые могут использоваться для синтеза новых пептидных антибиотиков или их пролекарств, где защитная группа предпочтительно представляет собой (2-сульфо)-9-Fmoc и защищенные пептидные промежуточные соединения имеют структуру:
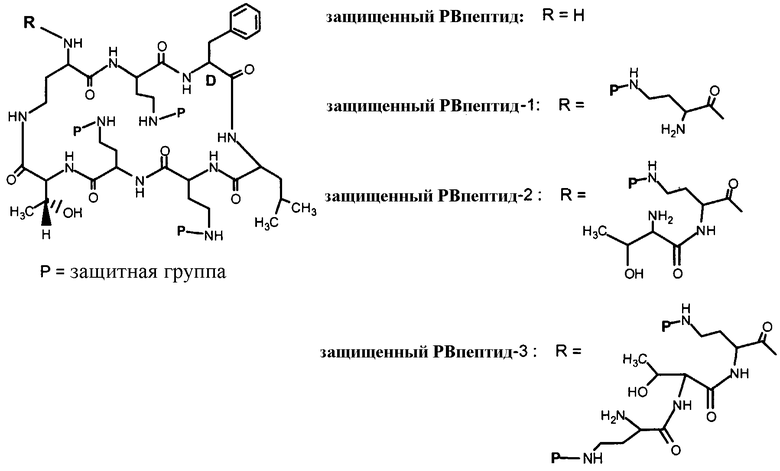
Другой вариант осуществления описывает:
5. Кислотно защищенные пептидные промежуточные соединения, полученные из соответствующего защищенного [Ile7]-полимиксина B1, которые могут использоваться для синтеза новых пептидных антибиотиков, или их пролекарства, где защитная группа предпочтительно представляет собой (2-сульфо)-9-Fmoc и защищенные пептидные промежуточные соединения имеют структуру:
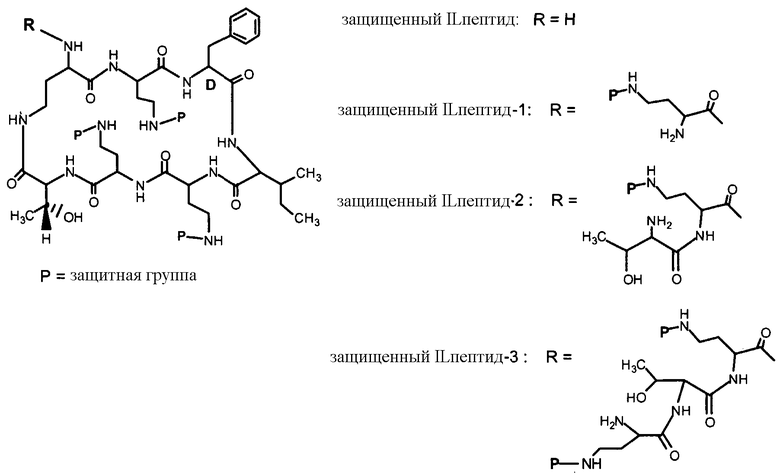
Другой вариант осуществления описывает:
6. Кислотно защищенные пептидные промежуточные соединения, полученные из соответствующего защищенного колистина, которые могут использоваться для синтеза новых пептидных антибиотиков, или их пролекарства, где защитная группа предпочтительно представляет собой (2-сульфо)-9-Fmoc и защищенные пептидные промежуточные соединения имеют структуру:
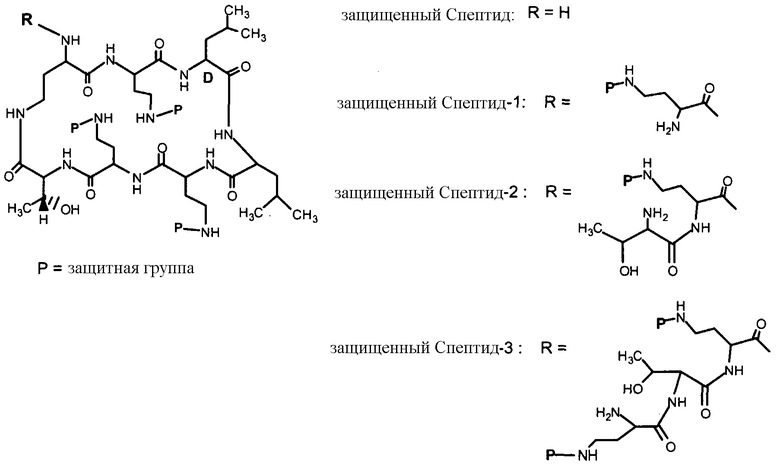
Другой вариант осуществления описывает:
7. Кислотно защищенные пептидные промежуточные соединения, полученные из соответствующего защищенного циркулина A, которые могут использоваться для синтеза новых пептидных антибиотиков, или их пролекарства, где защитная группа предпочтительно представляет собой (2-сульфо)-9-Fmoc и защищенные пептидные промежуточные соединения имеют структуру:
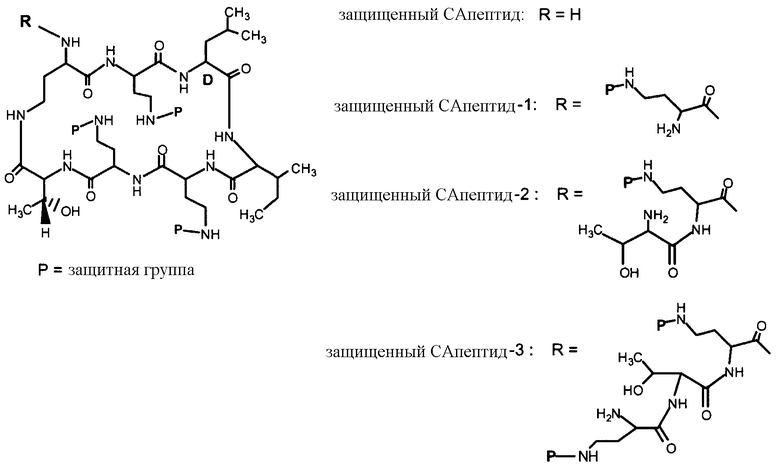
Другой вариант осуществления описывает:
8. Кислотно защищенные пептидные промежуточные соединения, полученные из соответствующего защищенного октапептина, которые могут использоваться для синтеза новых пептидных антибиотиков, или их пролекарства, где защитная группа предпочтительно представляет собой (2-сульфо)-9-Fmoc и защищенные пептидные промежуточные соединения имеют структуру:
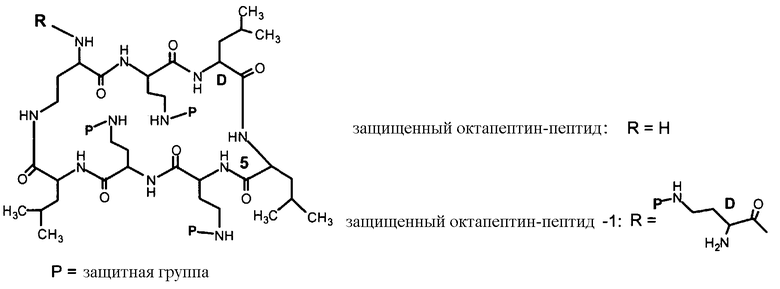
Другой вариант осуществления описывает:
9. Защищенное пептидное промежуточное соединение, полученное из соответствующего октапептина, где другой компонент октапептинового антибиотика содержит L-фенилаланин вместо L-лейцина в 5-положении, и где компонент образует сходное, но альтернативное защищенное пептидное промежуточное соединение.
Другой вариант осуществления описывает:
10. Антибактериальное соединение или защищенное соединение, полученное из химически защищенной формы PBпептидов, Cпептидов или ILпептидов, имеющее следующую структуру, где P равно защитной группе (2-сульфо)-9-Fmoc или атому водорода:
Таблица 3
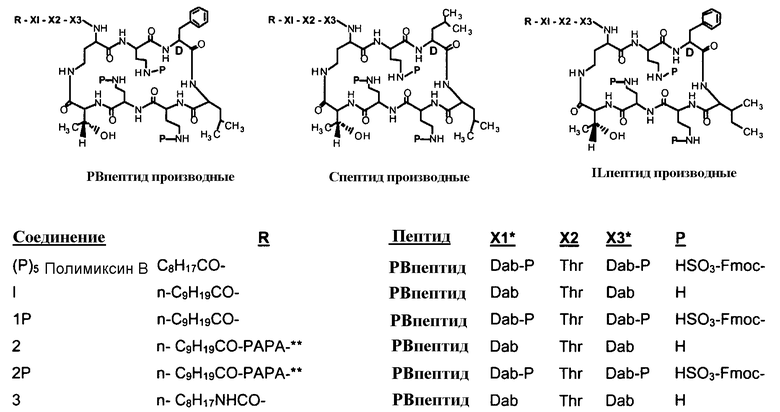
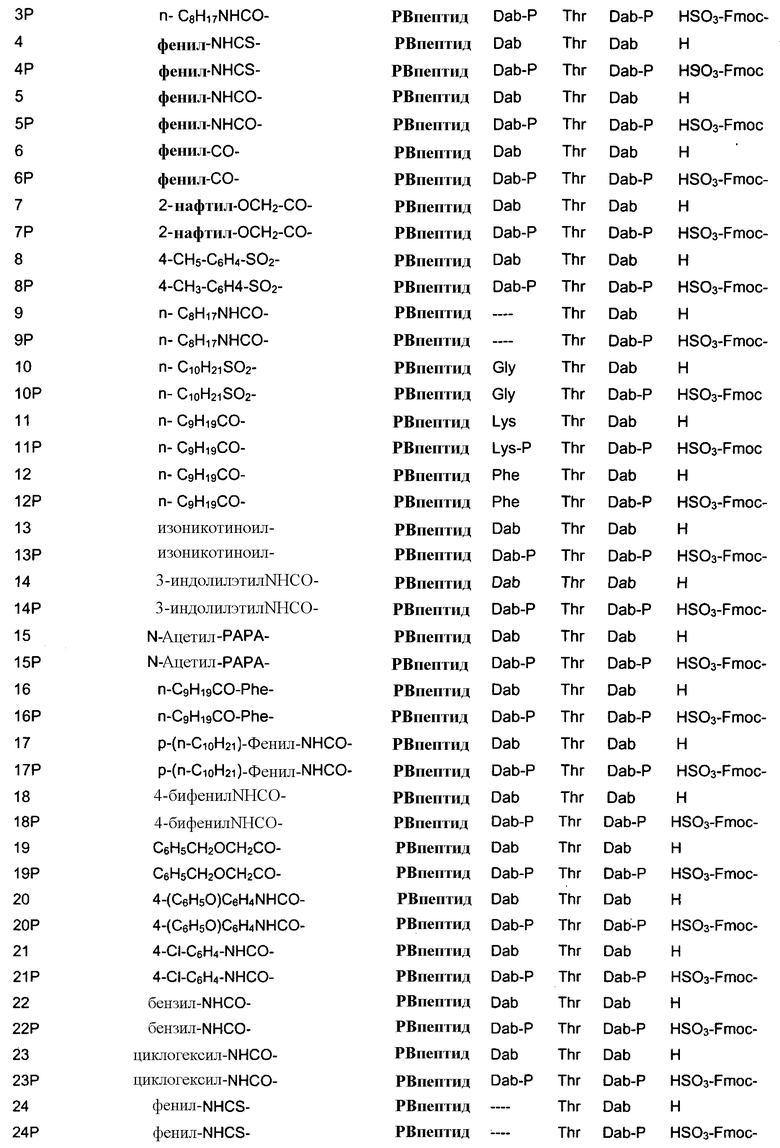
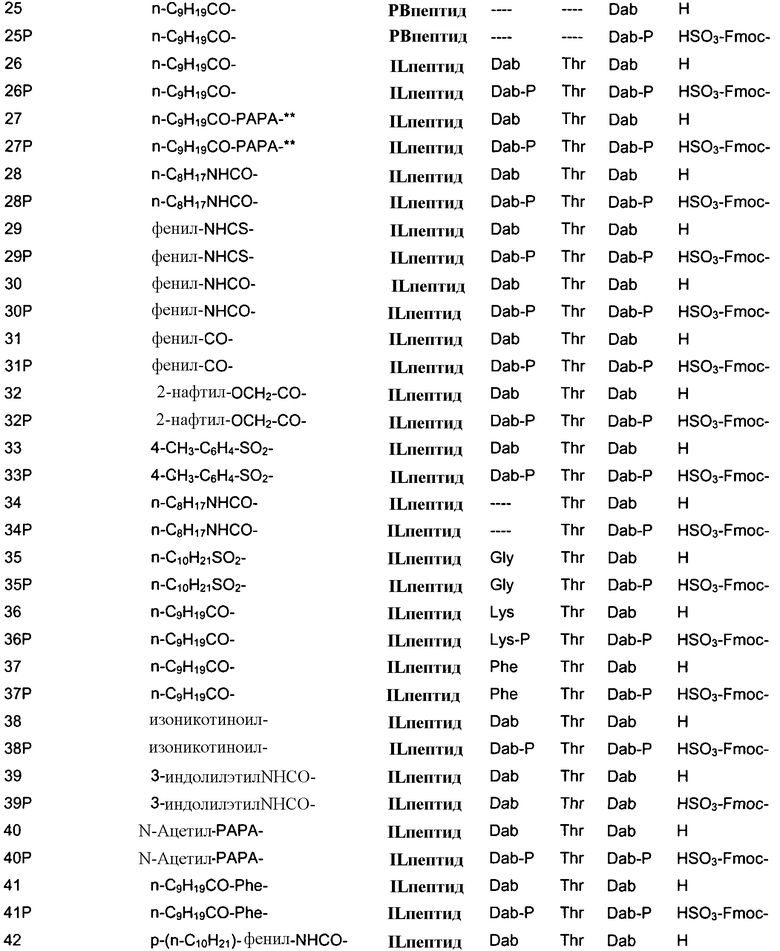


* Dab-P представляет собой 4-N-(HSO3-Fmoc)-диаминобутирил, Lys-P представляет собой 6-N-(HSO3-Fmoc)лизил
** PAPA представляет собой п-аминофенилацетил
Все аминокислоты представляют собой L-изомеры, если не указано иного.
Другой вариант осуществления описывает:
11. Антибиотик, полученный из промежуточного соединения, который представляет собой химически защищенную форму пептида, полученного из полимиксинов, октапептинов, колистина, [Ile7]полимиксина B1, указанный антибиотик выбирают из группы, состоящей из следующих соединений или соответствующих их солей:
где для случая 1) A-(X1)(X2)(X3)-Пептид: A=R'-(C=O)-, R'-SO2, R'-(C=NH)-, R'-NH-(C=S)-, R'-NH-(C=O)-, где R' представляет собой алкил, циклоалкил, алкенил, арил, гетероарил или гетероциклил и X1 представляет собой L-Dab или другую аминокислоту, X2 представляет собой L-Thr или другую аминокислоту и X3 представляет собой L-Dab или другую аминокислоту;
для случая 2) A-(X2)(X3)-Пептид: "A" является таким же, как описано для случая 1, X2 представляет собой L-Thr или другую аминокислоту и X3 представляет собой L-Dab или другую аминокислоту;
для случая 3) A-(X3)-пептид: "A" является таким же, как описано для случая 1 и X3 представляет собой L-Dab или другую аминокислоту;
для случая 4) A-пептид: "A" является таким же, как описано для случая 1.
Другой вариант осуществления описывает:
12. Пептидные антибиотики, имеющие следующую структуру и минимальные ингибиторные концентрации для использования против грамотрицательных и грамположительных бактерий:
Таблица 4
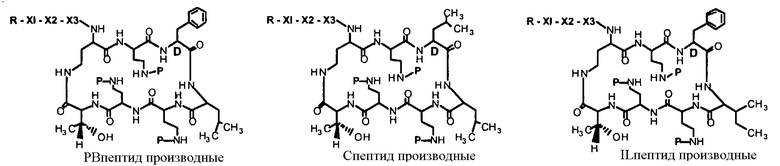
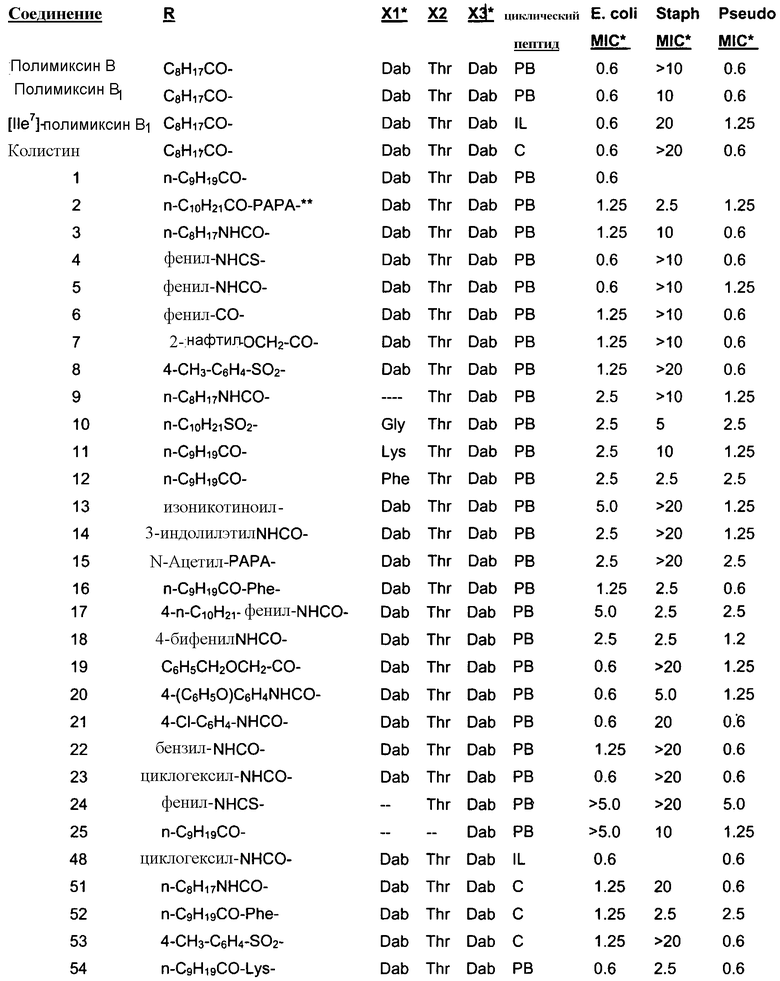
* Значения MIC определяют посредством способа последовательного двукратного разбавления бульона с использованием Escherichia coli, ATCC #26, Staphylococcus aureus Smith и Pseudomonas aeruginosa, ATCC 27853, в качестве организмов для анализа, которые выращивают в бульоне Мюллера-Хинтона.
** PAPA = п-аминофенилацетил
Другой вариант осуществления описывает:
Способ получения водорастворимой, стабильной, твердой формы фермента деацилазы, включающий в себя стадии:
(a) Ферментирования штаммов Actinoplanes utahensis с получением клеток организма.
(b) Промывки клеток водой для удаления примесей.
(c) Экстрагирования промытых клеток водным раствором основания, pH 8-11.
(d) Доведение экстракта до pH 7-8 и сушки вымораживанием раствора, с получением твердой формы фермента.
Другой вариант осуществления описывает:
14. Способ, дополнительно включающий в себя стадию (e):
(e) после этого, дополнительную очистку указанного фермента посредством использования хроматографии.
Ряд новых антибиотиков представлен сульфонильными производными (таблица 3, соединения 8 и 10). Боковые цепи от этих соединений не являются присоединенными к PBпептидам посредством ацильной группы, но вместо этого, посредством сульфонильной группы. Другие линкеры, такие как мочевины или тиомочевины, были получены, и они приводят к получению соединений с хорошей бактериальной активностью. Сильная активность показана также для ароматических ацильных экзоциклических цепей и ароматических групп, соединенных посредством мочевинных и тиомочевинных связей (соединения 4 и 5), как показано в Таблице 3.
Другие варианты осуществления по настоящему изобретению будут ясны специалистам в данной области из рассмотрения описания и осуществления изобретения, описанного здесь. Предполагается, что описание и примеры должны использоваться только в качестве иллюстрации, при этом истинные рамки и дух изобретения указывается посредством следующей далее формулы изобретения.
Примеры
Полимиксин B1 и [Ile7]-полимиксин B1 (таблица 3) выделяют из комплекса полимиксина B. Новые пептиды, полученные из ILпептида-3, могут быть получены посредством очистки [Ile7]-полимиксина B1 из комплекса полимиксина B и защиты этого пептида, деацилирования этого защищенного продукта, присоединения обратно новой боковой цепи и снятия защиты. Альтернативная процедура включает в себя защиту комплекса полимиксина B, деацилирование, с получением смеси защищенного PBпептида-3 и ILпептида-3, присоединения новой боковой цепи, снятия защиты, а затем разрешения смеси, с получением новых производных обоих пептидов. Сводка времен удерживания ВЭЖХ, наблюдаемых для новых пептидов, полученных из PBпептида и Ilпептида, приводится в таблице 5.
% Ацетонитрила
Элюент B: 0,05 M (NH4)2SO4 + 0,005 M H2SO4 pH ~2,44
Элюент C: 85% ацетонитрила и 15% воды
Анализ, с помощью ВЭЖХ, пептидов, защищенных, по меньшей мере, одной (2-сульфо)-9-флуоренилметоксикарбонильной группой, может осуществляться с использованием колонки с обращенной фазой C8, с использованием градиента от 35% до 75% MeCN/0,2M (NH4)3PO4 при pH 7,2. Анализ с помощью ВЭЖХ пептидов со снятой защитой может осуществляться с использованием условий, приведенных в таблице.
Если не указано иного, N-гидроксисукцинимидные реагенты получают в соответствии со способами, описанными в Gershonov, E., Goldwaser, I., Fridkin, M., Schecter, Y, 2000, "A Novel Approach for Water-Soluble Long-Acting Insulin ProDrug: Design, Preparation, and Analysis of [(2-Sulfo)-9-Fluorenylmethoxycarbonyl]3-insulin», Journal of Medicinal Chemistry 43: (13), 2530-2537, and Schechter, Y.Tsudbery, H., Fridkin, M., 2002, N-[(2-Sulfo)-9-Fluorenylmethoxycarbonyl)3-Gentamicin Is Long-Acting Prodrug Derivative», Journal of Medicinal Chemistry 45: (19), 4264-4270.
Пример 1. N-[(2-Сульфо)-9-флуоренилметоксикарбонил)]5- полимиксин B
Полимиксин B сульфат (1,0 г, 0,841 ммоль) растворяют в растворе из 25 мл насыщенного бикарбоната натрия, 25 мл воды и 25 мл тетрагидрофурана. Полимиксин B сульфат является коммерчески доступным, например, от Sigma (Milwaukee, WI). Раствор (2-сульфо)-9-флуоренилметокси-N-гидроксисукцинимида (2,0 г, 4,8 ммоль) в 25 мл тетрагидрофурана добавляют несколькими порциями в течение 45 мин. Реакционную смесь перемешивают при комнатной температуре в течение ночи и разбавляют 50 мл воды, затем подкисляют с помощью 25 мл 6 н хлористоводородной кислоты, с получением маслянистого осадка. Смесь быстро охлаждают, и водный слой декантируют, и маслянистый остаток растворяют в 100 мл этанола. Этанол выпаривают в вакууме (35°C) и полученный твердый продукт растирают с этилацетатом, фильтруют и сушат, с получением 1,74 г продукта. ВЭЖХ с градиентом на колонке с обращенной фазой C8 показывает один пик, N-[2-(сульфо)-9-флуоренилметоксикарбонил)]5-полимиксин B, C131H150N16O38S5, когда элюент колонки отслеживают при 215 нм. ESIMS: вычислено m/z для C131H152N16O38S5, (M+2H)+2 = 1358,4. Найдено 1358,5.
Указанная выше процедура может использоваться для свободного основания полимиксина и 2(сульфо)-9-флуоренилметоксикарбонилхлорида со сходными результатами.
Пример 2. Получение деацилазы
Деацилазу получают посредством культивирования Actinoplanes utahensis NRRL 12052 в условиях поверхностного суспензионного аэробного ферментирования. Используемый протокол ферментирования известен (Boeck, L.D. et al., Journal of Antibiotics 41:(8), 1085-1092). Исходную культуру варианта NRRL 12052, консервируемую в 20% глицерина при -70°C, вводят в пробирку 25x150 мм, с помощью стеклянного стержня и пробки Мортона, содержащую 10 мл среды, состоящей из сахарозы, 2,0%, предварительно сваренной овсянки, 2,0%, зерен и растворимых продуктов из перегонного аппарата, 0,5%, экстракта дрожжей, 0,25%, K2HPO4, 0,1%, KCl, 0,05%, MgSO4·7H2O, 0,05%, и FeSO4·7H2O, 0,0002%, в деионизованной воде. После инкубирования при 30°C в течение 72 часов на роторном планетарном шейкере, при 250 об/мин, полученную мицелиальную суспензию переносят в 50 мл среды PM3 в 250-мл колбе Эрленмайера. Эта среда содержит сахарозу, 2,0%, арахисовую муку, 1,0%, K2HPO4, 0,12%, KH2PO4, 0,05% и MgSO4·7H2O, 0,025%, в водопроводной воде. Колбу инкубируют при температуре 30°C в течение периода от 60 до 90 часов. Время сбора клеток из культуры определяют с помощью анализа, который включает в себя ВЭЖХ анализы деацилирования (2-сульфо-9-флуоренилметоксикарбонил)]5 полимиксина B посредством цельного бульона при различных временах во время ферментирования.
Поскольку изоляты отдельных колоний из лиофилизата культуры являются гетерогенными, как по морфологии, так и по способности производить фермент, осуществляют селекцию для извлечения стабильного, высокопроизводительного варианта. Сначала, множество ферментирований осуществляют с использованием посевного материала, полученного из штамма 12052. Вегетативный пророст из колбы, дающей наилучшую активность деацилирования, наносят на дифференциальный агар (СМ). СМ агар содержит кукурузный экстракт, 0,5%, пептон Bacto, 0,5%, растворимый крахмал, 1,0%, NaCl, 0,05%, CaCl2·2H2O, 0,05% и агар Bacto, 2,0%. Затем выбирают колонии для дальнейшей оценки. Изолят № 18 выбирают как тип с малыми колониями и, как показано, он представляет собой самый лучший продуцент деацилазы из всех выбранных колоний. Сравнение основывается на преобразовании защищенного полимиксина B в деацилированный защищенный полимиксин B, как определяется с помощью ВЭЖХ. Этот изолят рутинно используют для производства фермента деацилазы.
Пример 3. Деацилирование защищенного полимиксина B ферментом в клетках
Способ осаждения: Клетки от 450 мл фермента деацилазы из исходного ферментирования в соответствии с примером 2 промывают 3X водой, затем опять доводят до исходного объема с помощью 0,02 M аммонийфосфатного буфера и доводят pH до 8,0. Добавляют N-[(2-Сульфо)-9-флуоренилметоксикарбонил)]5-полимиксин B, 897 мг, и смесь помещают в шейкер при 174 об/мин и выдерживают при 30°C. Через пять часов смесь разделяют посредством центрифугирования. Прозрачный супернатант декантируют и доводят pH до 2,3 с помощью 1 н раствора HCl, чтобы вызвать осаждение, и оставляют стоять при комнатной температуре. Осадок отделяют из смеси, суспендируют в 80 мл воды, доводят до pH 6,5, с получением прозрачного раствора, и сушат вымораживанием, с получением 400 мг соли защищенного пептида, N-[(2-сульфо)-9-флуоренилметоксикарбонила]5-полимиксина пептида [(NaSO3-Fmoc)5-PBP-3] в виде порошка желтовато-коричневого цвета. Клетки, оставшиеся после центрифугирования, экстрагируют метанолом/водой для получения дополнительного материала.
Твердофазный способ: Клетки от 1 литра фермента деацилазы, от исходного ферментирования в соответствии с примером 2, промывают 3X водой, доводят обратно до исходного объема с помощью 0,02 M аммонийфосфатного буфера и объединяют с 2,0 г N-(2-сульфо)-9-флуоренилметоксикарбонил)]5-полимиксина B. Смесь помещают в шейкер при 175 об/мин и выдерживают при 30°C в течение 17 часов. Затем смесь отделяют посредством центрифугирования и супернатант декантируют и объединяют с 20 мл смолы Amberchrom® CG-161m. Смолу промывают 150 мл воды, 100 мл 10% CH3CN:H2O (3X), и 100 мл 20% CH3CN:H2O (2x). Затем пептид элюируют 2X 30% CH3CN:H2O, выпаривают, для удаления CH3CN, и сушат вымораживанием, с получением 283 мг порошка защищенного пептид-(HSO3-Fmoc)5-PBP-3. Вторую порцию пептида затем элюируют 3X 100 мл 50% CH3CN:H2O. Элюаты объединяют, выпаривают и сушат вымораживанием, с получением 460 мг защищенного пептида в виде порошка.
Третью порцию пептида экстрагируют из клеток, оставшихся после центрифугирования, с использованием MeOH:H2O, 6 x 100 мл каждый раз. Экстракты объединяют, доводят объем до четырех литров с помощью воды, доводят pH до 2,1 с помощью серной кислоты и объединяют со смолой Amberchrom® CG-161m. Смолу промывают водой, а затем элюируют пептид 100 мл 35% CH3CN:H2O, который выпаривают и сушат вымораживанием, с получением 94 мг очищенного пептида. Оставшийся пептид элюируют 50% CH3CN:H2O, выпаривают, для удаления CH3CN, и сушат вымораживанием, с получением 539 мг защищенного пептида. Защищенный пептид [(2-сульфо)-9-флуоренилметоксикарбонил)]5-полимиксин B пептид[(HSO3-Fmoc)5-PBP-3)] выделяют из этой процедуры в виде порошка желтовато-коричневого цвета, C122H134N16O37S5. Материалы, полученные как с помощью способа со смолой, так и с помощью способа осаждения, имеют чистоту примерно 75%, когда их анализируют с помощью ВЭЖХ, с помощью колонки, элюент отслеживают на 215 нм. ESIMS: вычислено m/z для C122H136N16O37S5: (M+2H)+2=1288,4; найдено: 1288.
Пример 4. Деацилирование защищенного полимиксина B с помощью солюбилизированного фермента
Промытые водой клетки от 250 мл ферментирования Actinoplanes utahensis, как осуществляется в соответствии с примером 2, объединяют с 125 мл 0,02 M аммонийфосфатного буфера, доводят pH до 10,1 и перемешивают тридцать минут. Раствор, содержащий фермент, разделяют с помощью центрифуги и доводят pH до 8,0. Этот раствор используют непосредственно для деацилирования или сушат вымораживанием, с получением порошкообразной формы для хранения. Супернатант после декантирования, содержащий солюбилизированный фермент при pH 8,0, объединяют с 100 мг (HSO3-Fmoc)5-полимиксина B, растворенного в 10 мл CH3CN:H2O (1:1), и помещают в шейкер при 175 об/мин, 84°C. Через два часа смесь, после завершения реакции, удаляют из шейкера и доводят pH до 2,0. Осадок смешивают с 40 мл метанола, и растворимый продукт отделяют от темного осадка. Раствор, содержащий 80 мг (HSO3-Fmoc)5-PBP-3, выпаривают до 2 мл и добавляют к 10 мл EtOAc, с осаждением (HSO3-Fmoc)5-PBP-3 в виде порошка желтовато-коричневого цвета.
Пример 5. Очистка (NaSO3-Fmoc)5-PBP-3 (защищенного декапептида)
(NaSO3-Fmoc)5-PBP-3, 143 мг, получают в соответствии с процедурой примера 3. Защищенный декапептид растворяют в 40 мл 20% CH3CN, 0,05M, в натрийфосфатном буфере, при pH 6,7. Нерастворимые продукты удаляют посредством центрифугирования. Супернатант декантируют и наносят на картридж из стирол-дивинилбензольной смолы (Supelco EnviChrom-P®, 25 x 35 мм), который представляет собой суспензию, набитую и промытую 20 мл 20% CH3CN буфера, 0,05M, pH 6,7. Скорость потока равна примерно 2 мл/мин при комнатной температуре. Картридж элюируют при ступенчатом увеличении концентрации CH3CN примерно от 0,05M в натрийфосфатном буфере, при pH 6,7. Собранные фракции оценивают с помощью аналитической ВЭЖХ. Желаемый продукт элюируют элюентами, 33% и 40% CH3CN. Фракции, содержащие продукт, собирают, и CH3CN удаляют в вакууме. Пул продукта обессоливают посредством поглощения на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®), который затем промывают четырьмя 1,0 мл порциями дистиллированной воды. Продукт элюируют из картриджа с помощью 16 мл 67% CH3CN, растворитель выпаривают в вакууме, pH полученного водного раствора доводят примерно до 5,8 с помощью разбавленного NaOH, затем продукт сушат вымораживанием. Выход: 72 мг твердого продукта светлого желтовато-коричневого цвета, (NaSO3-Fmoc)5-PBP-3, C122H134N16O37S5. ESIMS: вычислено m/z для C122H136N16O37S5: (M+2H)+2=1288,4. Найдено: 1288,2.
Пример 6. Получение N-фенилтиокарбамил-(NHaSO3-Fmoc)5-PBP-3 (Соединение 4P)
Очищенный (NaSO3-Fmoc)5-PBP-3, 3,9 мг, получают в соответствии с процедурой примера 5. Защищенный пептид растворяют в 0,20 мл 75% MeOH, 0,25 M, в калийборатном буфере, pH 8,8, и добавляют 0,003 мл фенилизотиоцианата, и перемешивают при комнатной температуре. Фенилизотиоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). Примерно через 90 мин, реакционную смесь разбавляют 4 мл 0,4 M аммонийсульфатного буфера при pH 7,2. Раствор продукта наносят на 0,5 г картридж из стирол-дивинилбензольной смолы (EnviChrom-P®), который промывают 6 мл 20% CH3CN 0,10 M, в натрийфосфатном буфере, при pH 6,7. Продукт элюируют с использованием 6 мл 40% CH3CN 0,05 M в натрийфосфатном буфере, при pH 6,7. Растворитель выпаривают в вакууме, и продукт обессоливают с использованием процедуры примера 5. Выход: 3,5 мг белого твердого продукта, N-фенилтиокарбамил-(NaSO3-Fmoc)5-PBP-3, C129H137N17O37S6. ESIMS: вычислено m/z для C129H139N17O37S6: (M+2H)+2=1355,4. Найдено: 1355,6.
Пример 7. Получение N-фенилтиокарбамил-PBP-3 (PTC-PBP-3, Соединение 4)
N-Фенилтиокарбамил-(NHaSO3-Fmoc)5-PBP-3, 8,4 мг, получают в соответствии с процедурой примера 6. Защищенный пептид растворяют в 0,20 мл диметилформамида (DMF), добавляют 0,010 мл пиперидина и перемешивают при комнатной температуре в течение 60 мин. Реакционную смесь разбавляют 4 мл 0,10 M ацетата аммония - 0,050 M уксусной кислоты (pH 5,05), 0,006 мл уксусной кислоты и 4 мл MeOH. Прозрачный раствор наносят на колонку СМ-Sepharose® (10 x 20 мм, объемом примерно 2 мл), которую кондиционируют с помощью 50% MeOH 0,05 M в аммонийацетатном буфере, при pH 5,0. Нагруженную образцом колонку промывают 4 мл 50% MeOH - 0,05 M аммоний ацетата, pH 5,0, затем 4 мл 0,05 M аммонийацетатного буфера, pH 5,0. Продукт элюируют 8 мл 0,27 M сульфата натрия при pH 2,3. Продукт дополнительно очищают посредством нанесения на 0,5 г стирол-дивинилбензольный картридж (EnviChrom-P®), который элюируют при ступенчатом увеличении концентрации CH3CN от 0,05M, в натрийсульфатном буфере, pH 2,3; продукт элюируют 20% CH3CN. Растворитель удаляют в вакууме из пула фракций, содержащих продукт, который затем обессоливают с использованием процедуры примера 5, pH доводят до 6,3, затем сушат вымораживанием. Выход: 2,7 мг белого твердого продукта, PTC-PBP-3, C54H87N17O12S. FABMS: вычислено для C54H88N17O12S: (M+H)+=1198,7. Найдено: 1198,5 (M+H)+, 1220,4 (M+Na)+.
Пример 8. (NHaSO3-Fmoc)4-PBP-2 (защищенный нонапептид)
N-Фенилтиокарбамил-(NHaSO3-Fmoc)5-PBP-3, 2,9 мг, получают в соответствии с процедурой примера 6. Защищенный пептид растворяют в 0,30 мл безводной трифторуксусной кислоты (TFA) и нагревают при 50°C на водяной бане в течение 15 мин. TFA выпаривают в потоке сухого азота, и остаток растворяют в 12 мл 0,20M аммонийсульфатного буфера при pH 7,2, и 6 мл CH3CN, содержащем 49 мг триглицина (в качестве поглотителя кислорода ацилирующего агента). Для удаления потенциально реакционноспособных промежуточных соединений, раствор продукта сначала наносят на 0,5 г картридж из стирол-дивинилбензольной смолы (EnviChrom-P®), который элюируют 10 мл 40% CH3CN, 0,04M, в аммонийфосфатном буфере, при pH 7,2. Фракции, содержащий продукт, собирают (~18 мл), разбавляют 12 мл дистиллированной воды, затем наносят на свежий картридж 0,5 г EnviChrom-P® из смолы, который элюируют при ступенчатом увеличении концентрации CH3CN примерно от 0,05M в аммонийфосфатном буфере, при pH 7,2. Фракции, содержащие продукт, собирают (~12 мл), добавляют 8 мл 0,54M натрий сульфатного буфера, pH 2,3, и продукт обессоливают с использованием процедуры примера 5. pH раствора доводят до 5,9, и образец сушат вымораживанием. Выход: 1,8 мг белого твердого продукта, (NaSO3-Fmoc)4-PBP-2, C103H116N14031S4. ESIMS: вычислено m/z для C103H118N14031S4 (M+2H)+2 = 1087,3. Найдено: 1087,0.
Пример 9. н-Деканоил-PBP-3 (Соединения 1 и 1P)
(NaSO3-Fmoc)5-PBP-3, 21,8 мг, получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,20 мл DMF, 0,020 мл дистиллированной воды и 0,030 мл насыщенного раствора NaHCO3 (pH ~8,9); добавляют 4,5 мг н-деканоил-N-гидроксисукцинимида (активированного сложного эфира) и перемешивают при комнатной температуре в течение 55 мин (примерно 82% преобразования, согласно ВЭЖХ). Добавляют дополнительные 1,8 мг активированного сложного эфира; через 20 мин при комнатной температуре, преобразование в Соединение 1P составляет, по меньшей мере, 95%. К реакционной смеси добавляют 0,010 мл пиперидина. Через 35 мин при комнатной температуре реакционную смесь разбавляют 4 мл 0,25 M аммонийсульфатного буфера при pH 2,3, с получением очень мутной беловатой смеси при pH 3,0, которую экстрагируют 4 мл этилацетата; водную фазу, содержащую продукт, разбавляют 4 мл дистиллированной воды. Продукт сначала выделяют с помощью эксклюзионной хроматографии на колонке Sephadex® G-25 (2,5 x 40 см), элюируют 0,10 M аммонийсульфатным буфером при pH 2,3. Фракции, содержащие продукт, собирают и дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) посредством элюирования при ступенчатом увеличении концентрации CH3CN примерно от 0,05M в натрийфосфатном буфере, при pH 6,7; продукт элюируют 25% CH3CN. Продукт обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 2,5 мг белого твердого продукта, Соединение 1, C57H100N16O13. FABMS: вычислено для C57H101N16O13, (M+H)+=1217,8. Найдено: 1217(M+H)+, 1239(M+Na)+.
Пример 10. N-(н-Деканоил)-п-аминофенилацетил-PBP-3 (Соединения 2 и 2P)
(NHaSO3-Fmoc)5-PBP-3, 18,5 мг, получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,20 мл DMF и 0,030 мл насыщенного раствора NaHCO3 (pH ~8,9); добавляют 5,8 мг N-(н-деканоил)-п-аминофенилацетил-N-гидроксисукцинимида (C10-PAPA-OSu) и перемешивают при комнатной температуре в течение 50 мин (примерно 69% преобразование, согласно ВЭЖХ). Добавляют дополнительные 2,5 мг C10-PAPA-OSu; через 30 мин при комнатной температуре преобразование в Соединение 2P составляет примерно 87%. К реакционной смеси добавляют 0,010 мл пиперидина. После 25 мин при комнатной температуре реакционную смесь разбавляют 5 мл 0,20 M аммонийсульфатного буфера при pH 2,3, с получением очень мутной беловатой смеси при pH 2,8, которую экстрагируют 5 мл этилацетата. Продукт сначала выделяют с помощью эксклюзионной хроматографии на колонке Sephadex® G-25 (2,5 x 40 см), элюируют 0,10 M аммонийсульфатного буфера при pH 2,3. Фракции, содержащие продукт, собирают и дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) посредством элюирования при ступенчатом увеличении концентрации CH3CN примерно от 0,10 M в сульфате аммония при pH 2,3; продукт элюируют 30% CH3CN. Дополнительная очистка достигается при использовании эксклюзионной хроматографии на колонке Sephadex® LH-20 (2,5 x 40 см), элюируют 0,026 M ацетатом аммония/0,053 M уксусной кислотой в MeOH. Фракции, содержащие продукт, выпаривают в вакууме почти досуха, остаток растворяют в 10 мл дистиллированной воды, затем обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 2,5 мг белого твердого продукта, Соединение 2, C65H107N17O14. FABMS: вычислено для C65H108N17O14, (M+H)+=1350,8. Найдено: 1351 (M+H)+, 1373 (M+Na)+.
Пример 11. н-Октаноилкарбамил-PBP-3 (Соединения 3 и 3P)
(NaSO3-Fmoc)5-PBP-3, 10,2 мг, получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,20 мл DMF и 0,020 мл дистиллированной воды; добавляют 0,003 мл октилизоцианата и перемешивают при комнатной температуре в течение 60 мин (примерно 31% преобразование согласно ВЭЖХ). Октилизоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). К реакционной смеси добавляют 0,020 мл насыщенного раствора NaHCO3 pH 8,9; через 20 мин при комнатной температуре преобразование в Соединение 3P составляет примерно 91%. Еще через 35 мин при комнатной температуре добавляют 0,010 мл пиперидина. После 30 мин при комнатной температуре реакционную смесь разбавляют 4 мл 0,026 M ацетата аммония/0,053 M уксусной кислоты в MeOH, и продукт выделяют с помощью эксклюзионной хроматографии на колонке Sephadex® LH-20, как описано в примере 10. Фракции, содержащие продукт, выпаривают в вакууме почти досуха, остаток растворяют в 8 мл дистиллированной воды, затем обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 2,3 мг белого твердого продукта, Соединение 3, C56H99N17O13. FABMS: вычислено для C56H100N17O13, (M+H)+=1218,8. Найдено: 1219(M+H)+, 1241 (M+Na)+.
Пример 12. Фенилкарбамил-PBP-3 (Соединения 5 и 5P)
(NaSO3-Fmoc)5-PBP-3, 19,8 мг, (чистотой примерно 85%) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,20 мл DMF и 0,020 мл насыщенного раствора NaHCO3 pH 8,7; добавляют 0,005 мл фенилизоцианата и перемешивают при комнатной температуре, с получением Соединения 5P. Фенилизоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). Через 45 мин добавляют 0,020 мл пиперидина для удаления защитных групп. После 45 мин при комнатной температуре реакционную смесь разбавляют 4 мл 0,10 M ацетата аммония - 0,05 M уксусной кислоты, 0,012 мл уксусной кислоты и 4 мл MeOH, с получением прозрачного раствора, при видимом pH 6,2. Продукт выделяют на колонке СМ-Sepharose®, как описано в примере 7. Продукт дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) с использованием ступенчатого увеличения концентрации CH3CN от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 20% CH3CN. Фракции, содержащие продукт, обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 4,4 мг белого твердого продукта, Соединение 5, C54H87N17O13. FABMS: вычислено для C54H88N17O13, (M+H)+=1182,7. Найдено: 1183(M+H)+, 1205 (M+Na)+, 1221 (M+K)+.
Пример 13. Бензоил-PBP-3 (Соединения 6 и 6P)
(NaSO3-Fmoc)5-PBP-3, 20,4 мг; (ca. 85% чист) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,20 мл DMF и 0,020 мл насыщенного раствора NaHCO3 pH 8,0; добавляют 8,4 мг бензоил-N-гидроксисукцинимида и перемешивают при 29°C, с получением Соединения 6P. Через 60 мин добавляют 0,020 мл пиперидина. После 20 мин при комнатной температуре реакционную смесь разбавляют 4 мл 0,10 M ацетата аммония - 0,05 M уксусной кислоты, 0,013 мл уксусной кислоты и 4 мл MeOH, с получением прозрачного раствора. Продукт выделяют на колонке СМ-Sepharose®, как описано в примере 7. Продукт дополнительно очищают с помощью препаративной ВЭЖХ с использованием колонки Delta-Pak® C18 (25 x 210 мм, Waters Corp.), элюируют с помощью градиента изопропанола (18%-23% в течение 100 мин, линейно, при 5 мл/мин) в 0,05M натрийсульфатном буфере, при pH 2,5. Фракции, содержащие продукт, собирают, растворитель удаляют в вакууме, продукт обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 1,6 мг белого твердого продукта, Соединение 6, C54H86N16O13. FABMS: вычислено для C54H87N16O13, (M+H)+=1166,7. Найдено: 1167(M+H)+, 1189(M+Na)+.
Пример 14. 2-Нафтаоксиацетил-PBP-3 (Соединения 7 и 7P)
(NaSO3-Fmoc)5-PBP-3, 19,0 мг, получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,40 мл 75% MeOH 0,25 M в калийборатном буфере, pH 8,8; добавляют 6,3 мг 2-нафтаоксиацетил-N-гидроксисукцинимида и перемешивают при комнатной температуре, с получением Соединения 7P. Через 35 мин добавляют 0,020 мл пиперидина. После 30 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose®, как в примере 7. Продукт дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) с использованием ступенчатого увеличения концентрации CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 25% CH3CN. Фракции, содержащие продукт, собирают, разбавляют равным объемом дистиллированной воды, продукт обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 4,7 мг белого твердого продукта, Соединение 7, C59H90N16O14.
FABMS: вычислено для C59H91N16O14, (M+H)+=1247,7. Найдено: 1247 (M+H)+, 1269 (M+Na)+.
Пример 15. п-Толуолсульфонил-PBP-3 (Соединения 8 и 8P)
(NaSO3-Fmoc)5-PBP-3, 21,7 мг, получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,50 мл 75% MeOH 0,25 M в калийборатном буфере, pH 8,8; добавляют 5,1 мг п-толуолсульфонилхлорида и перемешивают при комнатной температуре, с получением Соединения 8P. п-Толуолсульфонилхлорид является коммерчески доступным, например, Aldrich (Milwaukee, WI). Через 60 мин добавляют 0,020 мл пиперидина. После 30 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose®, как описано в примере 7. Продукт дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) с использованием ступенчатого увеличения концентрации CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 20% CH3CN. Фракции, содержащие продукт, собирают, разбавляют равным объемом дистиллированной воды, и продукт обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 4,9 мг белого твердого продукта, Соединение 8, C54H88N16O14S. FABMS: вычислено для C54H89N16O14S (M+H)+=1217,6, Найдено: 1217 (M+H)+, 1239(M+Na)+.
Пример 16. н-Октаноилкарбамил-PBP-2 (Соединения 9 и 9P)
(NaSO3-Fmoc)4-PBP-2, 10,1 мг, получают в соответствии с процедурой примера 8. Защищенный пептид растворяют в 0,20 мл DMF и 0,020 мл насыщенного раствора NaHCO3 pH 8,7; добавляют 0,003 мл октилизоцианата и перемешивают при комнатной температуре, с получением Соединения 9P. Октилизоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). Через 45 мин при комнатной температуре добавляют 0,020 мл пиперидина. После 40 мин при комнатной температуре реакционную смесь разбавляют 10 мл 20% CH3CN, содержащего 0,014 мл H2SO4. Продукт выделяют на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) с использованием ступенчатого увеличения концентрации CH3CN примерно от 0,05M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 25% и 30% CH3CN. Фракции, содержащие продукт, собирают, растворитель удаляют в вакууме, добавляют 2 мл 1,0 M аммонийацетатного буфера pH 5,0, pH доводят до 5,0 посредством добавления примерно 0,6 мл 1,5 M NH4OH, а затем разбавляют равным объемом MeOH. Продукт выделяют посредством хроматографии на СМ-Sepharose®, как описано в примере 7. Продукт дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже, как выше, но используя элюенты при pH 9,9 (0,10 M NH4OH - 0,01 M (NH4)2SO4); продукт элюируют 30% CH3CN. Фракции, содержащие продукт, собирают, растворитель удаляют в вакууме, продукт обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 2,7 мг белого твердого продукта, C52H92N16O11. FABMS: вычислено для C52H93N16O11 (M+H)+ = 1118,7, Найдено: 1119(M+H)+, 1141 (M+Na)+, 1157(M+K)+.
Пример 17. N-(н-Децилсульфонил)глицил-PBP-2 (Соединения 10 и 10P)
Очищенный (NaSO3-Fmoc)4-PBP-2, 9,7 мг, получают в соответствии с процедурой примера 8. Защищенный пептид растворяют в 0,20 мл DMF и 0,020 мл насыщенного раствора NaHCO3 pH 8,7; добавляют 5,2 мг н-децилсульфонамидоглицил-N-гидроксисукцинимида и перемешивают при комнатной температуре, с получением Соединения 10P. Через 45 мин добавляют 0,020 мл пиперидина. После дополнительных 40 мин при комнатной температуре, реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose®, как описано в примере 7. Продукт дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) с использованием ступенчатого увеличения концентрации CH3CN примерно от 0,05M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 33% CH3CN. Фракции, содержащие продукт, собирают, растворитель удаляют в вакууме, продукт обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 1,5 мг белого твердого продукта, Соединение 10, C55H97N15O14S. FABMS: вычислено для C55H98N15O14S, (M+H)+=1224,7. Найдено: 1224,5 (M+H)+, 1246,5 (M+Na)+.
Пример 18. 2-N-(н-Деканоил)лизил-PBP-2 (Соединения 11 и 11P)
Очищенный (NaSO3-Fmoc)4-PBP-2, 10,3 мг, получают в соответствии с процедурой примера 8. Защищенный пептид растворяют в 0,20 мл DMF и 0,020 мл насыщенного раствора NaHCO3 pH 8,3; добавляют 8,7 мг сложного 6-N-Fmoc-2-N-(н-деканоил)лизил-N-гидроксисукцинимидного эфира и перемешивают при комнатной температуре, с получением Соединения 11P. Через 50 мин добавляют 0,020 мл пиперидина для удаления защитных групп. После 45 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose®, как описано в примере 7. Продукт дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) с использованием ступенчатого увеличения концентрации CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 25% CH3CN. Фракции, содержащие продукт, собирают, разбавляют равным объемом дистиллированной воды, продукт обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 2,0 мг белого твердого продукта, Соединение 11, C59H104N16O13. FABMS: вычислено для C59H105N16O13, (M+H)+=1245,8, Найдено: 1245 (M+H)+, 1267 (M+Na)+.
Пример 19. N-(н-Деканоил)фенилаланил-PBP-2 (Соединения 12 и 12P)
Примерно 10 мг очищенного (NaSO3-Fmoc)4-PBP-2 получают в соответствии с процедурой примера 8. Защищенный пептид растворяют в 0,40 мл 75% MeOH 0,25M в калийборатном буфере, pH 8,8; добавляют 6 мг сложного N-(н-деканоил)фенилаланил-N-гидроксисукцинимидного эфира и перемешивают при комнатной температуре, с получением Соединения 12P. Через 50 мин добавляют 0,020 мл пиперидина. После 45 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose®, как в примере 7. Продукт дополнительно очищают на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P®) с использованием ступенчатого увеличения концентрации CH3CN примерно от 0,05M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 33% CH3CN. Фракции, содержащие продукт, собирают, разбавляют равным объемом дистиллированной воды, продукт обессоливают и сушат вымораживанием с использованием процедуры примера 5. Выход: 3,0 мг белого твердого продукта, Соединение 12, C62H101N15O13. FABMS: вычислено для C62H102N15O13 (M+H)+=1264,8. Найдено: 1265 (M+H)+, 1287 (M+Na)+.
Пример 20. N-Фенилтиокарбамил-(NaSO3-Fmoc)4PBP-2 (Соединение 24P)
Очищенный (NaSO3-Fmoc)4-PBP-2, 90,2 мг, получают в соответствии с процедурой примера 8. Защищенный пептид растворяют в 2,0 мл реакционного растворителя, как в примере 23; добавляют 0,050 мл фенилизотиоцианата. Фенилизотиоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). После 87 мин перемешивания при комнатной температуре реакционную смесь разбавляют 50 мл 20% CH3CN 0,1 M, в натрийфосфатном буфере, pH 6,76. Этот раствор наносят на 2,5 г колонку EnviChrom-P®, которую элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05M в натрийфосфатном буфере, при pH 6,76; продукт элюируют 30-35% CH3CN. Продукт обессоливают на 3,1 г колонке EnviChrom-P® и сушат вымораживанием с использованием процедуры примера 23. Выход: 85 мг белого твердого продукта, Соединение 24P.
Пример 21. (NaSO3-Fmoc)4PBP-1 (защищенный октапептид)
N-фенилтиокарбамил-(NaSO3-Fmoc)4-PBP-2 (70,0 мг) получают в соответствии с процедурой примера 20. Защищенный пептид растворяют в 0,70 мл безводной трифторуксусной кислоты (TFA) и нагревают при 50°C на водяной бане в течение 15 мин. TFA удаляют с использованием потока сухого азота. Остаток растворяют в 40 мл 25% CH3CN, 0,05 M в натрийсульфатном буфере, при pH 2,3. Продукт очищают посредством нанесения на 2,06 г колонку EhviChrom-P®, которую элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 40% CH3CN. Фракции, содержащие продукт, собирают и CH3CN удаляют в вакууме при 35°C. Продукт, обессоливают посредством поглощения на 3,1 г колонке EnviChrom-P®, которую затем промывают 8,8 и 4 мл порциями дистиллированной воды. Продукт извлекают из колонки с помощью 40 мл растворителя, 60% CH3CN, выпариваемого в вакууме, и водный раствор сушат вымораживанием. Выход: 60 мг белого твердого продукта, защищенного октапептида, C99H109N13O29S4. ESIMS: вычислено m/z для (M+2H)+2=1036,8. Найдено: 1036,4(M+2H)+2.
Пример 22. (NaSO3-Fmoc)3PBP (защищенный гептапептид)
(NaSO3-Fmoc)4-PBP-2 (0,35 мг) получают в соответствии с процедурой примера 8. Защищенный пептид растворяют в 0,40 мл буфера (0,02M цитрата натрия, 0,006M бета-меркаптоэтиламина, доводят pH до 5,5 с помощью HCl).
Катепсин C (Sigma product code C8511, дипептидил аминопептидаза, EC 3.4.14.1) растворяют в таком же буфере, при концентрации 10 единиц/мл. Затем раствор катепсина C (0,10 мл) добавляют к 0,40 мл раствора (NaSO3-Fmoc)4-PBP-2 и смесь инкубируют при 37°C. Реакцию отслеживают с помощью ВЭЖХ через 3,0 часа (примерно 43% преобразования в защищенный гептапептид) и через 24 часа (примерно 90% преобразования). Относительное время удерживания и ультрафиолетовый спектр поглощения пика продукта согласуется с продуцированием защищенного гептапептида; зависимый от времени внешний вид пика с коротким временем удерживания, с характерным спектром поглощения Fmoc согласуется с высвобождением дипептида N-конца H2N-Thr-Dab(Fmoc)-CO2H из защищенного нонапептида.
Пример 23. Изоникотиноил-PBP-3 (Соединения 13 и 13P)
(NaSO3-Fmoc)5-PBP-3 (20,5 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,40 мл 75% MeOH, 0,25M, в калийборатном буфере, pH 8,8; добавляют 8,7 мг сложного изоникотиноил-N-гидроксисукцинимидного эфира. Раствор перемешивают 63 мин при комнатной температуре, с образованием Соединения 13P, а затем добавляют 0,030 мл пиперидина для удаления защитных групп. После перемешивания 45 мин при комнатной температуре реакционную смесь разбавляют 10 мл 60% MeOH 0,04 M в аммонийацетатном буфере, pH 5,0 и 0,019 мл уксусной кислоты. Прозрачный раствор наносят на колонку СМ-Sepharose (10 x 20 мм, объем примерно 2 мл), которую кондиционируют посредством промывки 20 мл 60% MeOH 0,04M в аммонийацетатном буфере, pH 5,0. Нагруженную образцом колонку промывают 10 мл 60% MeOH 0,04M в аммонийацетатном буфере, pH 5,0, а затем 4 мл водном растворе 0,05 M аммонийацетатного буфера, pH 5,0. Продукт элюируют 8 мл 0,27 M натрийсульфатного буфера, при pH 2,3. Продукт дополнительно очищают посредством нанесения на 0,5 г стирол-дивинилбензольный картридж (EnviChrom-P), который элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 15% CH3CN. Фракции, содержащие продукт, собирают и CH3CN удаляют в вакууме при 35°C. Раствор продукта обессоливают посредством поглощения на 0,5 г стирол-дивинилбензольном картридже (EnviChrom-P), который затем промывают четырьмя 1,0 мл порциями дистиллированной воды. Продукт извлекают из картриджа с помощью 12 мл 67% CH3CN, растворитель выпаривают в вакууме и водный раствор сушат вымораживанием. Выход: 9,4 мг белого твердого продукта, Соединение 13, C53H85N17O13. FABMS: вычислено для C53H85N17O13Na (M+Na)+=1190,7. Найдено: 1190 (M+Na)+.
Пример 24. 3-Индолилэтилкарбамил-PBP-3 (Соединения 14 и 14P)
(NaSO3-Fmoc)5-PBP-3 (22,5 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,30 мл коллидина/уксусной кислоты/воды (40:1:4); добавляют 18,6 мг 3-индолилэтилкарбамил-N-гидроксисукцинимида, полученного в соответствии с процедурой примера 43, в двух порциях. Раствор перемешивают 130 мин при комнатной температуре, с образованием Соединения 14P, реакционную смесь продувают аргоном, добавляют 0,050 мл пиперидина для удаления защитных групп и смесь опять продувают аргоном. После перемешивания 25 мин при комнатной температуре реакционную смесь разбавляют 10 мл 60% MeOH 0,04 M в аммонийацетатном буфере, pH 5,0, и 0,042 мл уксусной кислоты. Продукт выделяют из прозрачной разбавленной реакционной смеси на колонке СМ-Sepharose, как в примере 23. Продукт дополнительно очищают посредством нанесения на 0,5 г стирол-дивинилбензольный картридж (EnviChrom-P), который элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 20% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 7,3 мг белого твердого продукта, Соединение 14, C58H92N18013. FABMS: вычислено для C58H93N18O13 (M+H)+=1249,7. Найдено: 1250 (M+H)+.
Пример 25. N-Ацетил-п-аминофенилацетил-PBP-3 (Соединения 15 и 15P)
(NaSO3-Fmoc)5-PBP-3 (20,7 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют как в примере 23; добавляют 5,8 мг N-ацетил-п-аминофенилацетил-N-гидроксисукцинимида. Раствор перемешивают 48 мин при комнатной температуре, с получением Соединения 15P, а затем добавляют 0,020 мл пиперидина для удаления защитных групп. После перемешивания 30 мин при комнатной температуре реакционную смесь разбавляют 10 мл 50% MeOH 0,05 M, в аммонийацетатном буфере, pH 5,0, и 0,013 мл уксусной кислоты. Продукт выделяют из прозрачной разбавленной реакционной смеси на колонке СМ-Sepharose, подобным же образом, как в примере 23. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, который элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 15% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 3,2 мг белого твердого продукта, Соединение 15, C57H91N17O14. FABMS: вычислено для C57H92N17O14(M+H)+=1238,7 Найдено: 1238(M+H)+.
Пример 26. N-(н-Деканоил)фенилаланил-PBP-3 (Соединения 16 и 16P)
(NaSO3-Fmoc)5-PBP-3 (24,0 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют как в примере 24; добавляют 12,9 мг N-(н-деканоил)фенилаланил-N-гидроксисукцинимида. Раствор перемешивают 48 мин при комнатной температуре, с образованием Соединения 16P, а затем добавляют 0,050 мл пиперидина для удаления защитных групп. После перемешивания 30 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose, как в примере 24. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 30% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 7,5 мг белого твердого продукта, Соединение 16, C66H109N17O14. FABMS: вычислено для C66H110N17O14, (M+H)+=1364,8. Найдено: 1365 (M+H)+.
Пример 27. 4-н-Децилфенилкарбамил-PBP-3 (Соединения 17 и 17P)
(NaSO3-Fmoc)5-PBP-3 (12,1 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,40 мл коллидина/уксусной кислоты/воды (20:1:2); добавляют 9,7 мг 4-н-децилфенилкарбамил-N-гидроксисукцинимида, полученного в соответствии с процедурой примера 43, в двух добавлениях, вместе с 0,030 мл диизопропилэтиламина, и перемешивают примерно 2 часа при комнатной температуре, с образованием Соединения 17P.
Добавляют пиперидин, 0,055 мл, для удаления блокирующих групп. После перемешивания 40 мин при комнатной температуре реакционную смесь разбавляют 10 мл 60% MeOH 0,04 M в аммонийацетатном буфере, pH 5,0, и 0,060 мл уксусной кислоты. Продукт выделяют из фильтрата разбавленной реакционной смеси (мембрана PVDF) на колонке СМ-Sepharose, как в примере 23. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 5,0 мг белого твердого продукта, Соединение 17, C64H105N17O13. FABMS: вычислено для C64H106N17O13 (M+H)+=1320,8. Найдено: 1322 (M+H)+.
Пример 28. 4-Бифенилкарбамил-PBP-3 (Соединения 18 и 18P)
(NaSO3-Fmoc)5-PBP-3 (11,9 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,30 мл коллидина/уксусной кислоты/воды (20:1:2); добавляют 7,0 мг 4-бифенилизоцианата (коммерчески доступного, например, от Aldrich (Milwaukee, WI)), в двух добавлениях. После перемешивания примерно 2 часа при комнатной температуре, с образованием Соединения 18P, добавляют 0,050 мл пиперидина для удаления защитных групп. После перемешивания 32 мин при комнатной температуре реакционную смесь разбавляют 10 мл 60% MeOH, 0,04M, в аммонийацетатном буфере, pH 5,0, и 0,035 мл уксусной кислоты. Продукт выделяют из фильтрата разбавленной реакционной смеси (мембрана PVDF) на колонке СМ-Sepharose, как в примере 23. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 5,1 мг белого твердого продукта, Соединение 18, C60H91N17O13. FABMS: вычислено для C60H92N17O13 (M+H)+=1258,7. Найдено: 1258 (M+H)+.
Пример 29. Бензилоксиацетил-PBP-3 (Соединения 19 и 19P)
(NaSO3-Fmoc)5-PBP-3 (23,5 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют, как в примере 24; добавляют 12,5 мг сложного бензилоксиацетил-N-гидроксисукцинимидного эфира и раствор перемешивают 56 мин при комнатной температуре, с образованием Соединения 19P. Затем добавляют пиперидин, 0,050 мл, для удаления защитных групп. После перемешивания 30 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose, как в примере 24. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 17,5% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 7,7 мг белого твердого продукта, Соединение 19, C56H90N16O14. FABMS: вычислено для C56H91N16O14, (M+H)+=1211,7. Найдено: 1211 (M+H)+.
Пример 30. 4-Фенилоксифенилкарбамил-PBP-3 (Соединения 20 и 20P)
(NaSO3-Fmoc)5-PBP-3 (16,6 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют, как в примере 24; добавляют 7,7 мг 4-фенилоксифенилизоцианата и раствор перемешивают 85 мин при комнатной температуре, с образованием Соединения 20P. 4-фенилоксифенилизоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). Пиперидин, 0,040 мл, добавляют для удаления защитных групп. После перемешивания 15 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose подобным же образом, как в примере 24. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 25% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 5,1 мг белого твердого продукта, Соединение 20, C60H91N17O14. FABMS: вычислено для C60H92N17O14, (M+H)+=1274,7. Найдено: 1274 (M+H)+.
Пример 31. 4-Хлорфенилкарбамил-PBP-3 (Соединения 21 и 21P)
(NaSO3-Fmoc)5-PBP-3 (12,0 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют, как в примере 24; добавляют примерно 8,2 мг 4-хлорфенилизоцианата в двух добавлениях. 4-Хлорфенилизоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). Раствор перемешивают 80 мин при комнатной температуре, с образованием Соединения 21P, а затем добавляют 0,040 мл пиперидина для удаления защитных групп. Реакционную смесь перемешивают 27 мин при комнатной температуре и разбавляют, как в примере 23. Продукт выделяют из фильтрата разбавленной реакционной смеси (мембрана PVDF) на колонке СМ-Sepharose, как в примере 23. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 25% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 3,6 мг белого твердого продукта, Соединение 21, C54H86N17O13Cl. FABMS: вычислено для C54H87N17O13Cl, (M+H)+=1216,6. Найдено: 1216(M+H)+.
Пример 32. Бензилкарбамил-PBP-3 (Соединения 22 и 22P)
(NaSO3-Fmoc)5-PBP-3 (12,2 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют как в примере 24; добавляют примерно 0,004 мл бензилизоцианата (коммерчески доступный, например, от Aldrich (Milwaukee, WI)). Раствор перемешивают 45 мин при комнатной температуре, с образованием Соединения 22P, а затем добавляют 0,040 мл пиперидина для удаления защитных групп. После перемешивания 35 мин при комнатной температуре реакционную смесь разбавляют, как в примере 23, и продукт выделяют из разбавленной реакционной смеси на колонке СМ-Sepharose, как в примере 23. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 5,5 мг белого твердого продукта, Соединение 22, C55H89N17O13. FABMS: вычислено для C55H90N17O13, (M+H)+=1196,7. Найдено: 1196(M+H)+.
Пример 33. Циклогексилкарбамил-PBP-3 (Соединения 23 и 23P)
(NaSO3-Fmoc)5-PBP-3 (19,3 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют, как в примере 24; добавляют 0,008 мл циклогексилизоцианата. Циклогексилизоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). Раствор перемешивают 30 мин при комнатной температуре, с образованием Соединения 23P, а затем добавляют 0,050 мл пиперидина для удаления защитных групп. Реакционную смесь перемешивают 35 мин при комнатной температуре и разбавляют. Продукт выделяют на колонке СМ-Sepharose, как в примере 24, а затем дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 18% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 5,1 мг белого твердого продукта, Соединение 23, C54H93N17O13, FABMS: вычислено для C54H94N17O13 (M+H)+=1188,7. Найдено: 1189 (M+H)+.
Пример 34. Фенилтиокарбамил-PBP-2 (Соединение 24)
Очищенный фенилтиокарбамил-(NaSO3-Fmoc)4-PBP-2 (13,7 мг, соединение 24P из примера 20) растворяют в 0,20 мл DMF и 0,010 мл пиперидина. После перемешивания 15 мин при комнатной температуре реакционную смесь разбавляют 8 мл 50% MeOH, 0,05M, в аммонийацетатном буфере, pH 5,0, и 0,006 мл уксусной кислоты. Продукт выделяют из разбавленной реакционной смеси на колонке СМ-Sepharose подобным же образом, как в примере 23. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 6,9 мг белого твердого продукта, Соединение 24, C50H80N15O11S FABMS: вычислено для C50H81N15O11S (M+H)+=1098,6. Найдено: 1099 (M+H)+.
Пример 35. н-Деканоил-PBP-1 (Соединения 25 и 25P)
(NaSO3-Fmoc)4-PBP-1 (16,2 мг) получают в соответствии с примером примера 20. Защищенный пептид растворяют, как в примере 24; добавляют 12,6 мг сложного н-деканоил-N-гидроксисукцинимидного эфира, в двух добавлениях. Раствор перемешивают примерно 2 часа при комнатной температуре, с образованием Соединения 25P, а затем добавляют 0,050 мл пиперидина для удаления защитных групп. После перемешивания 37 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose, как в примере 24. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 24% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 2,6 мг белого твердого продукта, Соединение 25, C49H85N13O10. FABMS: вычислено для C49H86N13O10,(M+H)+=1016,7. Найдено: 1017 (M+H)+.
Пример 36. н-Октилкарбамил-колистин декапептид (Соединения 51 и 51P)
(NaSO3-Fmoc)5-колистин декапептид (20,2 мг), полученный в соответствии с процедурой примера 45, растворяют, как в примере 24; добавляют 0,006 мл н-октилизоцианата. Оцилизоцианат является коммерчески доступным, например, от Aldrich (Milwaukee, WI). Раствор перемешивают 48 мин при комнатной температуре, с образованием Соединения 51P, а затем добавляют 0,050 мл пиперидина для удаления защитных групп. После перемешивания в течение 25 мин при комнатной температуре реакционную смесь разбавляют и фильтруют через мембрану PVDF. Продукт выделяют из фильтрата на колонке СМ-Sepharose, как в примере 24. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 22% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 3,0 мг белого твердого продукта, Соединение 51, C53H101N17O13. FABMS: вычислено для C53H102N17O13, (M+H)+=1184,8. Найдено: 1185 (M+H)+.
Пример 37. N-(н-Деканоил)фенилаланил-колистин декапептид (Соединения 52 и 52P)
(NaSO3-Fmoc)5-колистин декапептид (20,7 мг), полученный в соответствии с процедурой примера 45, растворяют, как в примере 24; добавляют 9,5 мг сложного N-(n-деканоил)фенилаланил-N-гидроксисукцинимидного эфира. Раствор перемешивают 46 мин при комнатной температуре, с образованием Соединения 52P, а затем добавляют 0,050 мл пиперидина для удаления защитных групп. Реакционную смесь перемешивают 25 мин при комнатной температуре и разбавляют. Продукт выделяют на колонке СМ-Sepharose, как в примере 24, и дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 30,5% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 2,5 мг белого твердого продукта, Соединение 52, C63H111N17O14. FABMS: вычислено для C63H112N17O14. (M+H)+=1330,9. Найдено: 1331(M+H)+.
Пример 38. P-Толуолсульфонил-колистин декапептид (Соединения 53 и 53Р)
(NaSO3-Fmoc)5-колистин декапептид (20,8 мг), полученный в соответствии с процедурой примера 45, растворяют, как в примере 24; добавляют 6,8 мг п-толуолсульфонилхлорида (коммерчески доступный, например, от Aldrich (Milwaukee, WI)). Реакционную смесь перемешивают 51 мин при комнатной температуре, с образованием Соединения 53P, а затем добавляют 0,050 мл пиперидина для удаления защитных групп. После перемешивания 47 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose, как в примере 24. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05 M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 20% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 1,1 мг белого твердого продукта, Соединение 53, C51H90N16O14S. FABMS: вычислено для C51H91N16O14S, (M+H)+=1183,7. Найдено: 1184 (M+H)+.
Пример 39. Выделение [Ile7]-полимиксина B1 и [Leu7]-полимиксина B1
Коммерческий полимиксин B сульфат (46 мг) растворяют в 10 мл 0,05 M натрийсульфатного буфера pH 2,3. Примерно 9 мл раствора образца инжектируют на колонку Waters Delta-Pak® C-18 Radial-Pak (2,5 x 21 см), которую элюируют при комнатной температуре, при 5,0 мл/мин, линейным градиентом от 22% до 32% CH3CN в течение 120 мин; элюенты дополняют буфером, примерно 0,04 M сульфата натрия, при pH 2,3. УФ поглощение элюата отслеживают при 225 нм. [Ile7] вариант элюируют примерно 65 мин и главный компонент, полимиксин B1 ([Leu7]-полимиксин B1), элюируют примерно 77 мин. Как определено с помощью аналитической ВЭЖХ, фракции, содержащие желаемые продукты, собирают отдельно, CH3CN выпаривают в вакууме при 35°C, водные растворы обессоливают и сушат вымораживанием, как в примере 23. Выходы: 3,8 мг белого твердого продукта, [Ile7]-полимиксина B1, C56H98N16O13. FABMS: вычислено для C56H99N16O13, (M+H)+=1203,8. Найдено: 1204 (M+H)+; 18 мг белого твердого продукта, [Leu7]-полимиксина B1, C56H98N16O13. FABMS: вычислено для C56H99N16O13, (M+H)+ =1203,8. Найдено: 1204 (M+H)+. Примерно 2 мг каждого из продуктов отдельно растворяют в 1,0 мл 6M HCl и нагревают примерно при 120°C в течение 22 часов. Растворитель удаляют посредством выпаривания в вакууме при 50°C и остатки растворяют в 1,0 мл дистиллированной воды. Образцы дериватизируют Fmoc хлоридом в водном растворе CH3CN с натрийборатным буфером, pH 10,1, в течение 5 мин при 50°C. Аминокислотные производные Fmoc отделяют при pH 6,7 на аналитической колонке с обращенной фазой (C18, 15 см), элюируя в градиенте CH3CN при 40°C. Площади пиков интегрируют на 264 нм и аминокислоты идентифицируют посредством соответствующих времен удерживания эталонных Fmoc аминокислот.
Пример 40. Циклогексилкарбамил-[Ile7]-полимиксин декапептид (Соединения 48 и 48P)
(NaSCO3-Fmoc)5-PBP-3 (55,8 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют в 0,60 мл реакционного растворителя, как в примере 24; добавляют 0,012 мл циклогексилизоцианата. Циклогексилизоцианат является коммерчески доступным, например, от Aldrich. Раствор перемешивают 50 мин при комнатной температуре, с образованием Соединения 48P, а затем добавляют 0,10 мл пиперидина для удаления защитных групп. После перемешивания 30 мин при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose подобным же образом, как в примере 24. Фракции, содержащие продукт, собирают (8 мл) и инжектируют на колонке Waters Delta-Pak®C-18 Radial-Pak (2,5 x 21 см), которую элюируют при комнатной температуре, при 5,0 мл/мин, в линейном градиенте CH3CN, от 15% до 25%, в течение 120 мин; элюенты дополняют натрийсульфатным буфером, примерно 0,04 M, при pH 2,3. УФ поглощение элюата отслеживают при 225 нм. Циклогексилкарбамил-[Ile7] вариант (Соединение 48) элюируют примерно при 83 мин, и главный компонент, циклогексилкарбамил-[Leu7]-полмиксин декапептид (Соединение 23), элюируют примерно при 93 мин. Фракции продукта собирают, обессоливают и сушат вымораживанием, как в примере 39. Выходы: 2,6 мг белого твердого продукта, Соединение 48, C54H93N17O13. FABMS: вычислено для C54H94N17O13, (M+H)+=1188,7. Найдено: 1189 (M+H)+; и 20,1 мг белого твердого продукта, Соединение 23. Анализы аминокислот осуществляют на обоих образцах подобным же образом, как в примере 39.
Пример 41. Получение 2-N-(н-деканоил)лизил-PBP-3 (Соединения 54 и 54P)
(NaSO3-Fmoc)5-PBP-3 (21,7 мг) получают в соответствии с процедурой примера 3. Защищенный пептид растворяют, как в примере 24; добавляют 11,8 мг 6-N-Fmoc-2-N-(н-деканоил)лизил-N-гидроксисукцинимида. Раствор перемешивают 45 мин при комнатной температуре, с образованием Соединения 54P, а затем добавляют 0,050 мл пиперидина для удаления защитных групп. После 30 мин перемешивания при комнатной температуре реакционную смесь разбавляют и продукт выделяют на колонке СМ-Sepharose, как в примере 24. Продукт дополнительно очищают на 0,5 г картридже EnviChrom-P, элюируют при ступенчатом увеличении концентраций CH3CN примерно от 0,05M в натрийсульфатном буфере, при pH 2,3; продукт элюируют 22,5% CH3CN. Продукт обессоливают и сушат вымораживанием, как в примере 23. Выход: 7,2 мг белого твердого продукта, Соединение 54, C63H112N18O14. FABMS: вычислено для C63H113N18O14 (M+H)+=1345,9. Найдено: 1346 (M+H)+.
Пример 42. 4-н-Децилфенилкарбамил-N-гидроксисукцинимид
Смесь п-дециланилина (0,4504 г, 1,93 ммоль), дисукцинимидил карбоната (0,5840 г, 2,28 ммоль) и триэтиламина (0,26 мл, 1,93 ммоль) в 10 мл ацетонитрила перемешивают при комнатной температуре в течение ночи. Малое количество нерастворимого твердого продукта отфильтровывают и фильтрат выпаривают досуха. Остаток извлекают в этилацетате и экстрагируют 0,5 н хлористоводородной кислотой, водой, насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушат над сульфатом магния и выпаривают до получения продукта, C21H3ON2O4, бежевого твердого продукта, выход 0,3784 г, FABMS: вычислено для C21H31N2O4, (M+H)+ = 375,2. Найдено 375 (M+H)+. Это промежуточное соединение используют в примере 27 для получения Соединения 17 и 17P.
Пример 43. 3-Индолилэтилкарбамил-N-гидроксисукцинимид
Раствор триптамина (0,1730 г, 1,08 ммоль) и дисукцинимидил карбоната (0,5213 г, 2,03 ммоль) в 20 мл ацетонитрила перемешивают при комнатной температуре в течение ночи. Реакционную смесь выпаривают досуха и суспендируют в этилацетате, и отфильтровывают от нерастворимого твердого продукта. Выпаривание этилацетата дает стеклообразный твердый продукт, который очищают с помощью градиентной хроматографии на силикагеле с использованием этилацетата:гексана. Продукт элюируют этилацетатом:гексаном (8:2), с получением 0,1402 г желаемого соединения, C15H15N3O4, FABMS: вычислено для C15H15N3O4, M+=301,1, Найдено 301(M)+. Это промежуточное соединение используют для получения Соединений 14 и 14P в примере 24.
Пример 44. N-[(2-Сульфо)-9-флуоренилметоксикарбонил]5-колистин [(HSO3-Fmoc)5-колистин]
Колистин сульфат (полимиксин E) (0,2481 г, 0,1956 ммоль) растворяют в растворе 7,0 мл насыщенного раствора бикарбоната натрия, 7,0 мл воды и 7,0 мл тетрагидрофурана. Колистин сульфат является коммерчески доступным от Sigma (Milwaukee, WI). Раствор (2-сульфо)-9-флуоренилметокси-N-гидроксисукцинимида (0,5017 г, 1,203 ммоль) в 5 мл тетрагидрофурана добавляют несколькими порциями в течение 45 мин. Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем разбавляют 20 мл воды и подкисляют 10 мл 6 н хлористоводородной кислотой, с получением масляного осадка. Смесь быстро охлаждают, водный слой декантируют и масляный остаток растворяют в 100 мл этанола. Этанол выпаривают в вакууме (35°C) и полученный твердый продукт перетирают с этилацетатом, фильтруют и сушат, с получением 0,431 г продукта ВЭЖХ с градиентом на колонке с обращенной фазой, показывающего один главный пик, N-[2-(сульфо)-9-флуоренилметоксикарбонил)]5-колистин, [C128H152N16O38S5], ESIMS: вычислено m/z для C128H154N16O38S5 (M+2H)+2=1341,4. Найдено: 1341,6.
Указанная выше процедура может использоваться для 2(сульфо-9-)-флуоренилметоксикарбонилхлорида, с подобными же результатами.
Пример 45. (NaSO3-Fmoc)5 -колистин декапептид
Солюбилизированный фермент деацилазу, 940 мг, представляющий собой 400 мл исходного объема ферментирования фермента, полученного в соответствии с примером 2, объединяют с 320 мл 0,02 молярным аммонийфосфатным буфером, pH 7,2, 80 мл этанола, 2,96 грамм динатриевой соли EDTA и 200 мг (HSO3-Fmoc)5-колистина, полученного в соответствии с процедурой примера 44. Смесь доводят pH до 8 и помещают в шейкер при 140 об/мин, 30°C. Ход деацилирования отслеживают с помощью ВЭЖХ. После трех часов полностью прореагировавшую реакционную смесь удаляют из шейкера и дают ей возможность постоять при комнатной температуре в течение одного часа перед тем, как pH доводят до 2,0 с помощью 1 н HCl. Осадок смешивают с 50 мл воды, растворяют посредством доведения pH до 6,46 и дают возможность постоять при 4°C в течение ночи, что приводит к появлению малого количества осадка защищенного декапептида. Осадок объединяют с метанолом, который затем выпаривают досуха, повторно растворяют в воде и сушат вымораживанием, с получением 12,5 мг порошка светлого желтовато-коричневого цвета, C122H134N16O37S5. ESIMS: вычислено m/z для C122H135N16O37S5Na: (M+H+Na)+2=1282,4. Найдено: 1282,2. Оставшийся декантированный супернатант сушат вымораживанием, с получением дополнительных 192 мг порошка желтовато-коричневого цвета защищенного колистин декапептида.
Пример 46. Другие пептиды
Другие новые пептиды и новые защищенные пептиды могут быть получены с помощью способов, описанных выше. Любой пептид, имеющий общую гептапептидную циклическую сердцевинную структуру полимиксинов и октапептинов, включая, например, полимиксин A, полимиксин B (например, полимиксин B1, полимиксин B2 и полимиксин B3), [Ile7]-полимиксин B1 полимиксин C, полимиксин D, полимиксин E (также называемый колистин), полимиксин F, полимиксин M (также называемый маттацин), полимиксин P, полимиксин S, полимиксин T (например, полимиксин T1), колистин, циркулин A, октапептин A (например, октапептин A1, октапептин A2 и октапептин A3) октапептин B (например, октапептин B1, октапептин B2 и октапептин B3), октапептин C (например, октапептин C1) и октапептин D, могут иметь аминозащитные группы, как описано в примере 1, деацилироваться, как описано в примерах 3 и 4, очищаться, как в примере 5, модифицироваться, как описано в примерах 7 и 9, и подвергаться деградации Эдмана, как описано в примере 8.
Ссылки
1. Evans, M.E., Feola, D.J., Rapp, R.P. 1999. Polymyxin B Sulfate and Colistin: Old Antibiotics for Emerging Multiresistant Gram-Negative Bacteria. Ann. Pharmacother. 33:960-967.
2. McCallister, S.M., Alpar H.O., Brown, M.R.W. 1999. Antimicrob. Chemother. 43:203-210.
3. Chihara, S., Ito, A., Yahata, M., Tobita, T., Koyama, Y. 1974. Chemical Synthesis and Characterization of n-Fattyacyl Mono-Aminoacyl Derivatives of Colistin Nonapeptide. Agr. Biol. Chem. 38: (10), 1767-1777.
4. Chihara, S., Ito, A., Yahata, M., Tobita, T., Koyama, Y. 1973. Chemical Synthesis and Characterization of a -N-Octanoyl and Other a-N-Acyl Nonapeptide Derivatives. Agr. Biol. Chem. 37: (12), 2709-2717.
5. Chichara.S., Ito, A., Yahata, M., Tobita, T., Koyama, Y. 1974. Chemical Synthesis, Isolation and Characterization of n-N-Fattyacyl Colistin Nonapepetide with Special Reference to the Correlation Between Antimicrobial Activity and Carbon Number of Fattyacyl Moiety. Agr. Biol. Chem. 38: (3), 521-529.
6. Weinstein, J., Afonso, A., Moss, E.JR., Miller, G.H. 1988. Selective Chemical Modifications of Polymyxin B. Biorg. Med. Chem. Lett. 8:3991-3996.
7. Tsubery, H., Ofek, I., Cohen, S., Fridkin, M. 2001. N-Terminal Modifications of Polymyxin B Nonapeptide and Their Effect on Antibacterial Activity. Peptides. 22: 1675-1681.
8. Mutter, M., and Bellof, D. 1984. A New Base-labile Anchoring Group for, Polymer-supported peptide synthesis, Helv. Chim. Acta, 67, 2009.
9. Liu, Y.-Z., Ding, S.H., Chu, J.-Y. and Felix, A.M. 1990. A Novel Fmoc-based Anchorage for the Synthesis of protected Peptide on Solid Phase, Int. J. Pept. Protein Res., 35, 95.
10. Markou, N., Apostolakos, H., Koumoudiou, C, Anthanasiou, M., Koutsoukou, A., Alamanos, I., and Gregorakos, L. 2003. Intravenous colistin in the treatment of sepsis from multiresistant Gram- negative bacilli in critically ill patients, Critical Care 7, R78-R83.
11. Duwe, A.K., Rupar, C.A., Horsman, G.B., and Vas, S.I., 1986. In Vitro Cytotoxicity and Antibiotic Activity of Polymyxin B Nonapeptide, Antimicrob. Agents Chemother, 30:340-341.
12. Kurihara T., Takeda, H., and Ito, H. 1972. Compounds related to colistin. V. Synthesis and pharmacological activity of colistin analogs, Yakugaku Zasshi 92:129-34.
13. Parker, W.L., Rathnum, M.L., 1975. EM49, A New Peptide Antibiotic IV. The Structure of EM49. J. Antibiot. 28: (5), 379-389.
14. Hausmann, W., et al.,1954. Polymyxin B1. Fractionation, molecular-weight determination, amino acid and fatty acid composition, J. Am. Chem. Soc. 76, 4892-4896.
15. DeVisser Kriek, N.M.A.J., van Hooft, P.A.V., Van Schepdael A., Fillipov, D.V., van der Marel, G.A., Overcleeft, H.S., van Boom, J.H., and Noort, D.M. 2003. Synthesis of polymyxin B and analogues, J. Peptide Res. 61, 298-306.
16. Kimura, Y., Matsunaga, and Vaara, M., 1992. Polymyxin B Octapeptide and Polymyxin B Heptapeptide are Potent Outer Membrane Permeability-Increasing Agents. J. Antibiot, 45, 742-749.
17. Boeck, L.D., Fukuda, D.S., Abbott,, B.J., Debono, M. 1988. Deacylation of A21978C, an acidic Lipopeptide Antibiotic Complex, by Actinoplanes utahensis. J. Antibiot. 4.1: (8), 1085-1092.
18. Kreuzman, A.J., Hodges, R.L., Swartling, J.R., Ghag, S.K., Baker, P.J., McGilvray, D., Yeh, W.K. 2000. Membrane-Associated Echinocandin B Deacylase of Actinoplanes utahensis: Purification, Characterization, Heterologous Cloning.
19. Boeck, L.D., Fukuda, D.S., Abbott, B.J., Debono, M. 1989. Deacylation of Echinocandin B by Actinoplanes utahensis. J. Antibiot. 42: (3), 382-388.
20. Borders, D. B. Curran, W. V., Fantini, A. A., Francis, N.D., Jarolmen, H., Leese, R. A., 2003. Derivatives of Laspartomycin and Preparation and Use Thereof, U.S. Patent No. 6511962.
21. Elverdam, I., Larsen, P., Lund, E. 1981. Isolation and Characterization of Three New Polymyxins in Polymyxins B and E by High-Performance Liquid Chromatography. J. Chromatogr. 218: 653-661.
22. Sakura, N., Itoh, T., Uchida, Y., Ohki, K., Okimura, K., Chiba, K., Sato, Y., Sawanishi, H. 2004. The Contribution of the N-Terminal Structure of Polymyxin B Peptides to Antimicrobial and Lipopolysaccharide Binding Activity, Bull. Chem. Soc. Jpn. 77: 1915-1924.
23. Falagas, M. E., Kasiakou, S. K. 2005. Colistin: The Revival of Polymyxins for the Management of Multidrug-Resistant Gram-Negative Bacterial Infections. Rev. Anti-lnfect. Agents. 40:1333-1341.
24. Salem, E. M., El-Gammal, A. A. 1980. Synthesis of Pelargonoyl-Cyclic Decapeptide Analog of the Antibiotic Polymyxin B 1 . Pharmazie 35: 540-541.
25. Srinivasa, B. R., Ramachandran, L. K. 1980. Essential Amino Groups of Polymyxin B. Indian J. Biochem. Biophys. 17: 112-118.
26. Kline, T., Holub, D., Therrien, J., Leung, T., Ryckman, D. 2001. Synthesis and characterization of the colistin peptide polymyxin E1 and related antimicrobial peptides. J. Pept. Res., 57: 175-187.
27. Bouchaudon, J., Jolles, G. 1973. Cyclopeptides Derived From Polymyxins and their Preparation. U.S. Patent No. 3753970.
28. Shechter, Y., Preciado-Patt, L, Schreiber, G., Fridkin, M. 2001. Prolonging the half-life of human interferon-α2 in circulation: Design, preparation, and analysis of (2-sulfo-9-fluorenylmethoxycarbonyl)7-interferon-α2. Proc. Natl. Acad. Sci., U.S.A. 98: 1212-1217.
29. Merrifield, R. B., Bach, A. E. 1978. 9-(2-Sulfp)fluorenylmethyloxycarbonyl chloride, a new reagent for the purification of synthetic peptides. J. Org. Chem., 43: 4808-4816.
30. Kleinkauf, H., van Dohren, H. 1990. Nonribosomal synthesis of peptide antibiotics. Eur. J. Biochem. 192: 1-15.
31. Martin, N. I., Hu, H., Moakes, M. M., Churey, J. J., Whittal, R., Worobo, R. W., Vederas, J. C. 2003. Isolation, structural characterization, and properties of mattacin (polymyxin M), a cyclic peptide antibiotic produced by Paenibacillus kobensis M. J. Biol. Chem. 278: 13124-13132.
32. Gershonov, E., Goldwaser, I., Fridkin, M., Schecter, Y, 2000, "A Novel Approach for a Water-Soluble Long-Acting Insulin ProDrug: Design, Preparation, and Analysis of [(2-Sulfo)-9-Fluorenylmethoxycarbonyl]3-lnsulin," J. Med. Chem. 43: (13), 2530-2537, and
33. Schechter, Y., Tsudbery, H., Fridkin, M., 2002, N-[(2-Sulfo)-9-Fluorenylmethoxycarbonyl]3-Gentamicin Is a Long-Acting Prodrug Derivative," J. Med. Chem. 45: (19), 4264-4270.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПОЛИМИКСИНА С КОРОТКОЦЕПОЧЕЧНЫМ ЖИРНОКИСЛОТНЫМ ХВОСТОМ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2492179C2 |
| Ингибиторы фактора VIIa | 1999 |
|
RU2223967C2 |
| АНАЛОГИ ОКСИТОЦИНА | 2009 |
|
RU2496788C2 |
| Агонисты V1а-рецепторов | 2013 |
|
RU2634617C2 |
| ИНГИБИТОРЫ IAP | 2008 |
|
RU2491276C2 |
| РАДИОФАРМАЦЕВТИЧЕСКИЕ СРЕДСТВА, РАДИОВИЗУАЛИЗИРУЮЩИЕ СРЕДСТВА И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2804297C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2014 |
|
RU2676324C2 |
| АНАЛОГИ ЦИКЛИЧЕСКОГО СОМАТОСТАТИНА | 1999 |
|
RU2242481C2 |
| СОЕДИНЕНИЯ, СПОСОБСТВУЮЩИЕ ВЫСВОБОЖДЕНИЮ ГОРМОНА РОСТА | 1994 |
|
RU2167881C2 |
| ИНГИБИТОРЫ ПРЕДСТАВЛЕНИЯ АНТИГЕНОВ МОЛЕКУЛАМИ МНС II КЛАССА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2333219C2 |
Раскрыты новые пептиды и защищенные пептиды, которые получают из пептидов, встречающихся в природе, таких как полимиксин В. Также раскрыты фармацевтические композиции, содержащие новые пептиды, и способы получения новых пептидов и защищенных пептидов. 12 н. и 46 з.п. ф-лы.
1. Способ получения соединения формулы (I),
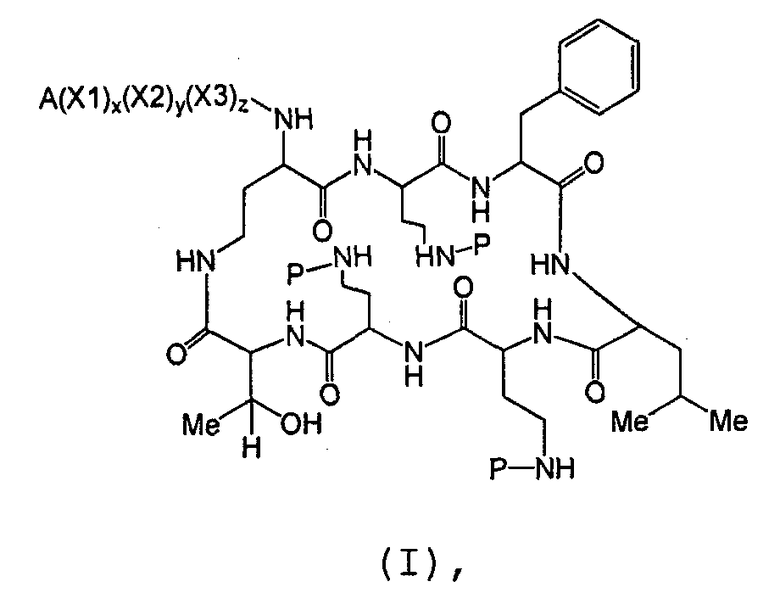
где А выбирают из
R'-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 9-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-SO2-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-NH-(C=S)-, где R' представляет собой арил, имеющий от 5 до 14 элементов кольца, или
R'-NH-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, арил, имеющий от 5 до 14 элементов кольца, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца;
X1, Х2 и Х3, каждый, независимо, выбирают из аминокислотных остатков;
х, у и z представляют собой целые числа, независимо выбранные из 0 и 1;
и Р представляет собой водород;
включающий:
(а) обработку пептида формулы
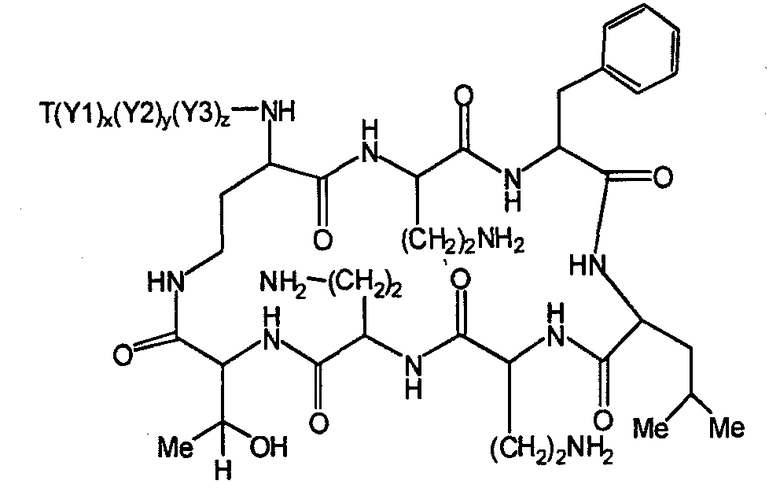
реагентом для защиты аминогруппы, с образованием защищенного пептида,
где Y1, Y2 и Y3, каждый, независимо, выбирают из аминокислотных остатков;
Т представляет собой R'-(C=O)-, где R' представляет собой алкил, имеющий 1-20 атомов углерода, арил, имеющий от 5 до 14 элементов кольца, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца; и
х, у и z представляют собой целые числа, независимо выбранные из 0 и 1;
где пептид включает циклический гептапептид, присоединенный к экзоциклической пептидной цепи, содержащей ацильную группу, и защитная группа содержит, по меньшей мере, один кислотный заместитель; и
(b) обработку защищенного пептида деацилирующим агентом с образованием защищенного деацилированного пептида А1
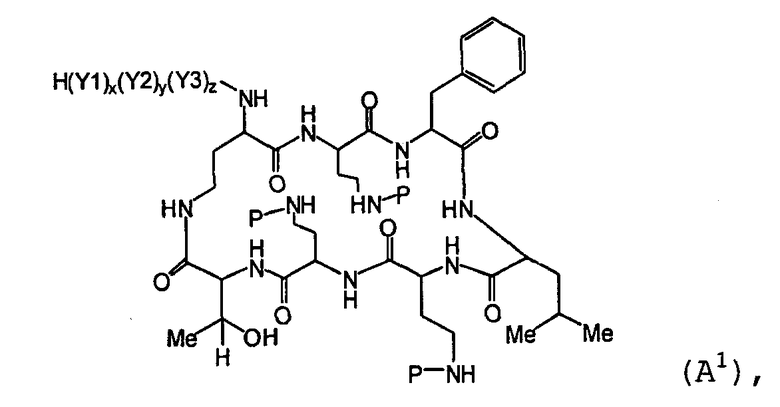
где Y1, Y2 и Y3, каждый, независимо, выбирают из аминокислотных остатков;
х, у и z представляют собой целые числа, независимо выбранные из 0 и 1; и
Р представляет собой защитную группу; и
(с) образование соединения формулы (I) из деацилированного защищенного пептида формулы А1.
2. Способ по п.1, в котором Y1, Y2 и Y3, каждый, независимо, выбирают из остатка 2,4-диаминобутановой кислоты, треонинового остатка и серинового остатка.
3. Способ по п.1, в котором Т выбирают из 6-метилоктаноила, 6-метилгептаноила, октаноила, гептаноила, нонаноила и 3-гидрокси-6-метилоктаноила.
4. Способ по п.1, в котором пептид на стадии (а) представляет собой полимиксин В.
5. Способ по п.1, в котором R' представляет собой арил, имеющий от 6 до 10 элементов кольца.
6. Способ по п.1, в котором защитная группа представляет собой сульфоновую кислоту 9-флуоренилметоксикарбонила.
7. Способ по п.1, в котором защитная группа представляет собой 2-сульфо-9-флуоренилметоксикарбонил.
8. Способ по п.1, в котором каждую аминогруппу пептида на стадии (а) защищают защитной группой.
9. Способ по п.1, в котором деацилирующий агент представляет собой фермент.
10. Способ по п.9, в котором источник фермента представляет собой Actinoplanes utahensis.
11. Способ по п.1, в котором А представляет собой R'-NH-(C=O)-.
12. Способ по п.1, в котором образование соединений включает обработку защищенного пептида реагентом, выбранным из изоцианата, тиоизоцианата и лактона.
13. Способ по п.1, в котором образование соединений включает обработку защищенного пептида ацилирующим реагентом, выбранным из ацилгалогенидов, ацилцианидов, сложных эфиров, лактонов и ангидридов.
14. Способ по п.1, в котором образование соединений включает обработку защищенного пептида сульфонирующим реагентом, выбранным из сульфонилхлорида и активированных сульфонамидов.
15. Способ по п.1, в котором образование соединений включает проведение реакции гидролиза N-концевой аминокислоты защищенного деацилированного пептида, так что х представляет собой 0 и каждый из у и z представляет собой 1.
16. Способ по п.1, в котором образование соединений включает проведение реакции гидролиза второй N-концевой аминокислоты защищенного деацилированного пептида, так что каждый из х и у представляет собой 0, и z представляет собой 1.
17. Способ по п.1, в котором образование соединений включает проведение реакции гидролиза третьей N-концевой аминокислоты защищенного деацилированного пептида, так что каждый из х, у и z представляет собой 0.
18. Способ по п.1, в котором А выбирают из R'-(C=O)- и R'-NH-(C=O)-.
19. Способ получения деацилированного пептида формулы (I)
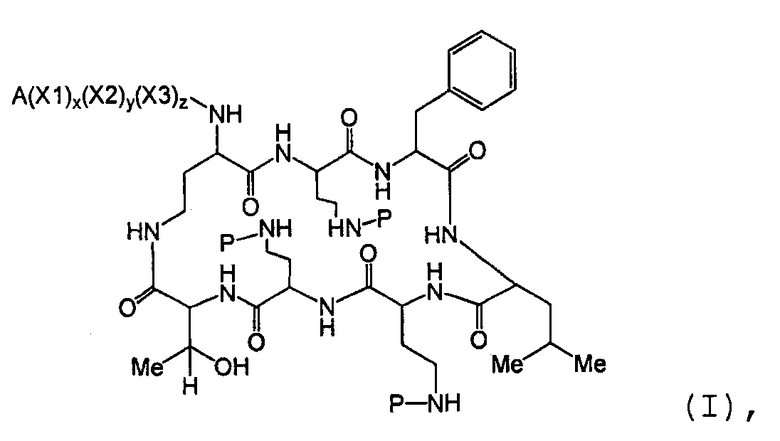
где А выбирают из R'-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 9-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца, R'-SO2-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, или арил, имеющий от 6 до 10 элементов кольца, R'-NH-(C=S)-, где R' представляет собой арил, имеющий от 6 до 10 элементов кольца, или R'-NH-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, арил, имеющий от 6 до 10 элементов кольца, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца;
X1, Х2 и Х3, каждый, независимо, выбирают из аминокислотных остатков;
х, у и z представляют собой целые числа, независимо выбранные из 0 и 1; и
Р представляет собой водород или сульфоновую кислоту 9-флуоренилметоксикарбонила;
включающий:
(а) обработку пептида формулы
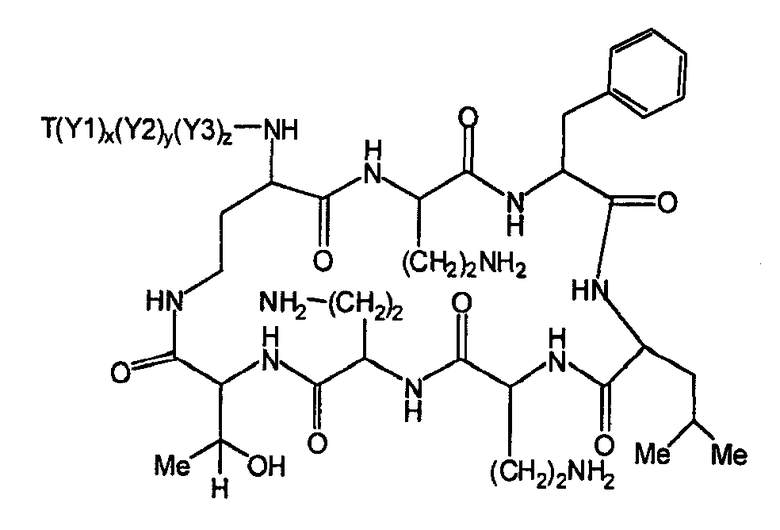
реагентом для защиты аминогруппы, с образованием водорастворимого защищенного пептида,
где Y1, Y2 и Y3, каждый, независимо, выбирают из аминокислотных остатков;
Т представляет собой R'-(C=O)-, где R' представляет собой алкил, имеющий 1-20 атомов углерода, арил, имеющий от 5 до 14 элементов кольца; и
х, у и z представляют собой целые числа, независимо выбранные из 0 и 1;
и где пептид содержит циклический гептапептид, присоединенный к экзоциклической пептидной цепи, содержащей ацильную группу, и защитная группа содержит, по меньшей мере, один кислотный заместитель;
(b) обработку защищенного пептида деацилирующим агентом, с образованием защищенного деацилированного пептида; и
(c) образование защищенного деацилированного пептида формулы (I) из деацилированного защищенного пептида формулы со стадии (b).
20. Способ по п.19, в котором водорастворимый защищенный пептид обрабатывают деацилирующим агентом в водной среде.
21. Способ по п.20, в котором каждый из х, у, и z равен 1.
22. Способ по п.21, в котором Р представляет собой сульфоновую кислоту 9-флуоренилметоксикарбонила и деацилирующий агент включает фермент.
23. Способ по п.22, в котором Y1 и Y3 оба представляют собой 2,4-диаминомасляную кислоту, а Y2 представляет собой треонин.
24. Соединение, имеющее структуру формулы (I):
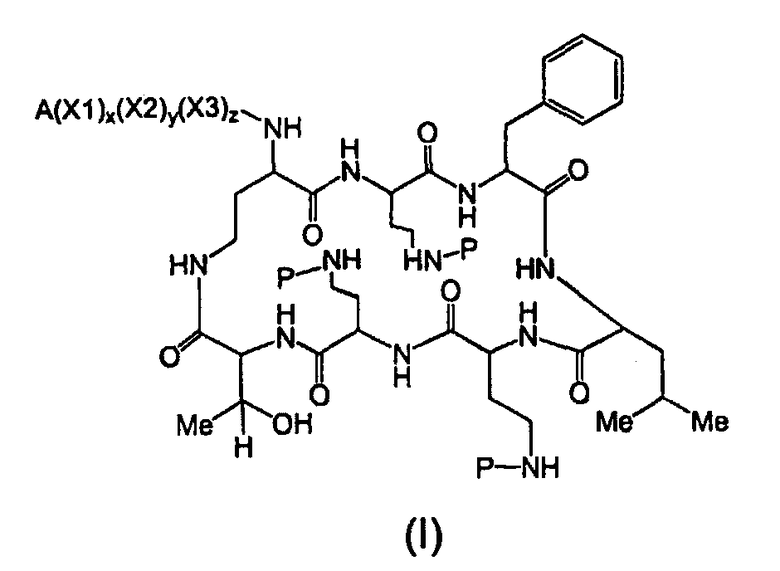
где А выбирают из
R'-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 9-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-SO2-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-NH-(C=S)-, где R' представляет собой арил, имеющий от 5 до 14 элементов кольца, или
R'-NH-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, арил, имеющий от 5 до 14 элементов кольца, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца;
X1, Х2 и Х3, каждый, независимо, выбирают из аминокислотных остатков;
х, у и z представляют собой целые числа, независимо выбранные из 0 и 1;
и Р представляет собой водород.
25. Соединение по п.24, в котором А представляет собой R'-(C=O)-.
26. Соединение по п.25, в котором R' представляет собой арил, имеющий от 6 до 10 элементов кольца.
27. Водорастворимое соединение, имеющее структуру формулы (I):
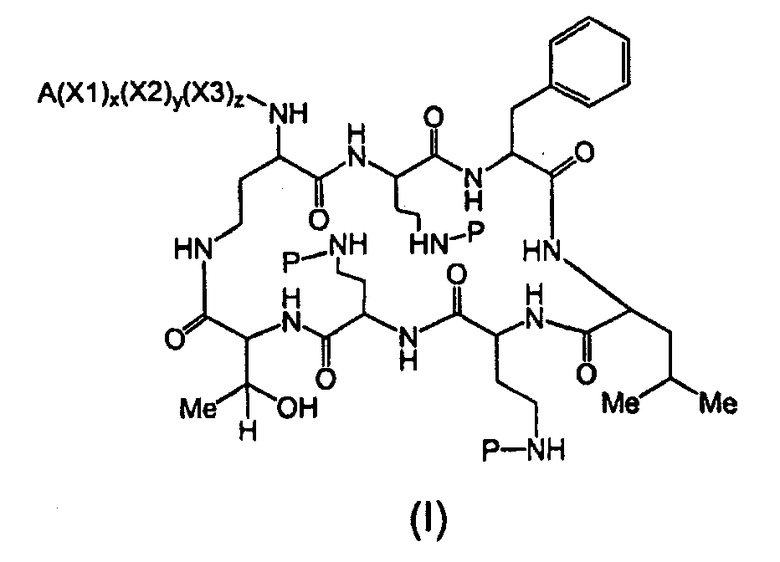
где:
А выбирают из
R'-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 9-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-SO2-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-NH-(C=S)-, где R' представляет собой арил, имеющий от 5 до 14 элементов кольца, или
R'-NH-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, арил, имеющий от 5 до 14 элементов кольца, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца;
X1, Х2 и Х3, каждый, независимо, выбирают из аминокислотных остатков;
х, у и z представляют собой целые числа, независимо выбранные из 0 и 1;
Р представляет собой 2-сульфо-9-флуоренилметоксикарбонильную группу.
28. Соединение формулы (I):
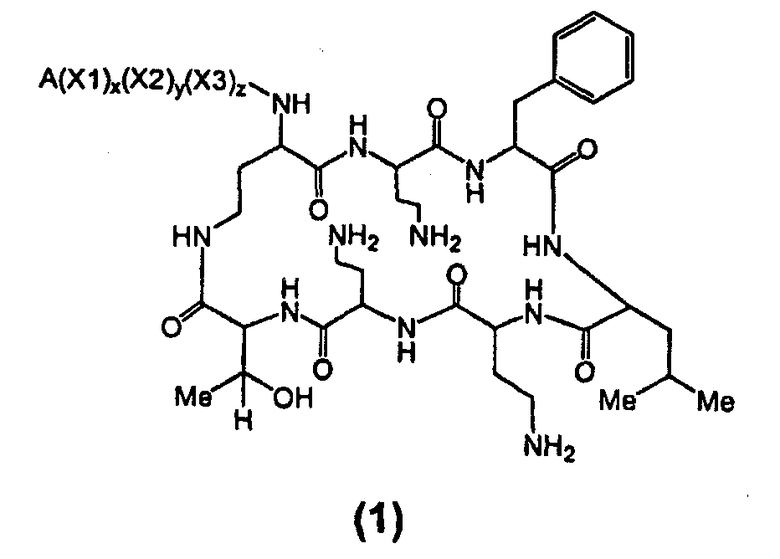
где А выбирают из
R'-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 9-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-SO2-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-NH-(C=S)-, где R' представляет собой арил, имеющий от 5 до 14 элементов кольца, или
R'-NH-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, арил, имеющий от 5 до 14 элементов кольца, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца;
X1, Х2 и Х3, каждый, независимо, выбирают из аминокислотных остатков;
и х, у и z представляют собой целые числа, независимо выбранные из 0 и 1.
29. Соединение по п.28, в котором А представляет собой R'-NH-(C=O)-.
30. Соединение по п.29, в котором X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту.
31. Соединение по п.28, в котором X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-(C=O)-.
32. Соединение по п.28, в котором X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-SO2-.
33. Соединение по п.28, в котором X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-NH-(C=O)- и R' представляет собой арил, имеющий от 6 до 10 элементов кольца.
34. Соединение по п.28, где X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-NH-(C=S)-.
35. Соединение по п.28, в котором X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин, и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-NH-(C=O)- и R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца.
36. Соединение по п.28, в котором х представляет собой 0, у и z, каждый, равен 1, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-NH-(C=O)- и R' представляет собой н-C8-алкил.
37. Соединение по п.28, в котором х представляет собой 0, каждый из у и z независимо представляет собой 1, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-NH-(C=S)- и R' представляет собой фенил.
38. Соединение по п.28, в котором каждый из х и у независимо представляет собой 0, z представляет собой 1 и Х3 представляет собой 2, 4-диаминобутановую кислоту, и где А представляет собой R'-NH-(C=O)- и R' представляет собой н-С9-алкил.
39. Соединение по п.28, в котором X1 представляет собой глицин, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-SO2- и R' представляет собой н-С10-алкил.
40. Соединение по п.28, в котором X1 представляет собой лизин, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-(С=O)-и R' представляет собой н-С9-алкил.
41. Соединение по п.28, в котором X1 представляет собой фенилаланин, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-(C=O)-.
42. Соединение по п.24 или 28, в котором R' представляет собой арил, имеющий 6 элементов кольца.
43. Соединение по п.42, в котором R' представляет собой фенил.
44. Соединение формулы (I):
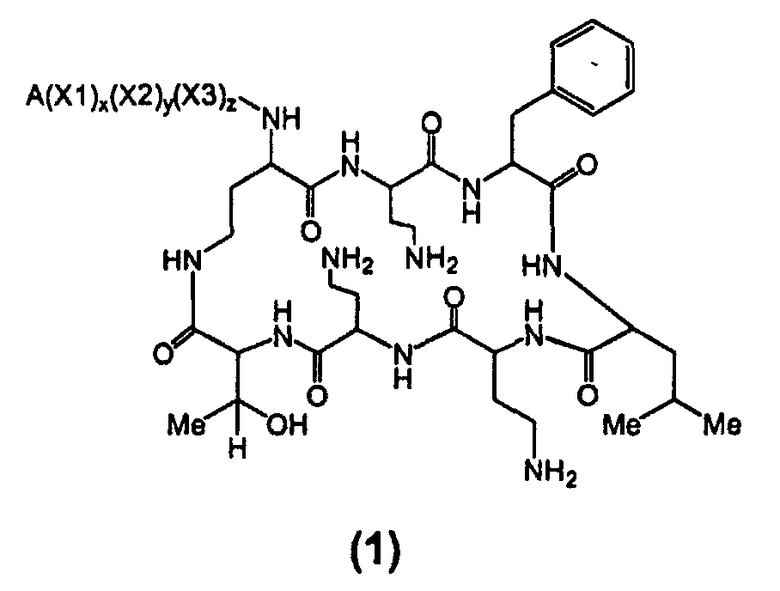
где А выбирают из
R'-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 9-20 атомов углерода, или арил, имеющий от 6 до 10 элементов кольца, или R'-NH-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 8-14 атомов углерода, арил, имеющий от 6 до 10 элементов кольца, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца;
X1, Х2 и Х3, каждый, независимо, выбирают из аминокислотных остатков и
х, у и z представляют собой целые числа, независимо выбранные из 0 и 1.
45. Соединение по п.44, в котором X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту.
46. Соединение по п.44, в котором X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту, и где А представляет собой R'-(C=O)-.
47. Соединение по любому из пп.24, 27, 28 или 44, в котором А представляет собой R'-NH-(C=O)-, где R' представляет собой арил, имеющий 6 элементов кольца.
48. Применение терапевтически эффективного количества соединения по любому из пп.24-47 и фармацевтически приемлемого носителя для получения антибактериальной фармацевтической композиции.
49. Способ получения промежуточного соединения для применения при синтезе новых пептидных антибиотиков, включающий стадии:
(a) защиты аминогрупп полимиксина В с помощью (2-сульфо)-9-флуоренилметоксикарбонила или другого кислотного производного 9-флуоренилметоксикарбонила;
(b) обработки продукта реакций стадии (а) деацилазой, с получением защищенного пептидного промежуточного соединения; и
(c) очистки защищенного пептидного промежуточного соединения с помощью хроматографии;
(d) обработки защищенного пептидного промежуточного соединения реагентом формулы A-LG, где LG представляет собой уходящую группу или A-LG представляет собой изоцианат;
(e) снятия защиты аминогрупп со стадии (а) с образованием соединения формулы (1)
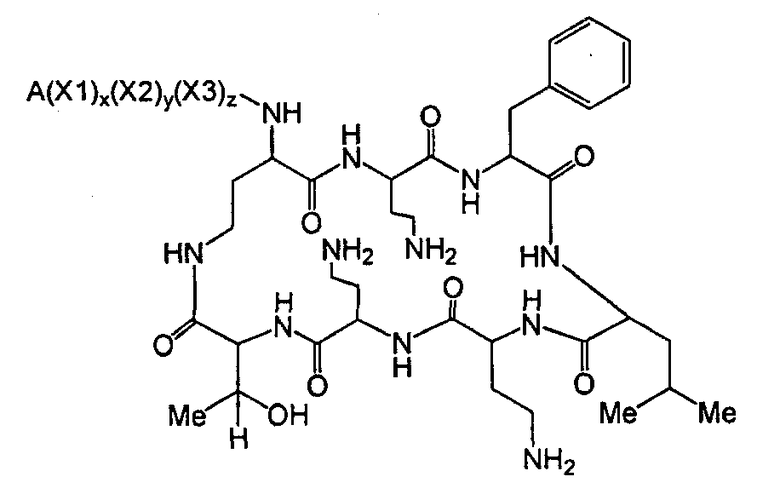
(1)
где А выбирают из
R'-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 9-20 атомов углерода, или арил, имеющий от 5 до 14 элементов кольца,
R'-SO2-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, или арил, имеющий от 6 до 10 элементов кольца,
R'-NH-(C=S)-, где R' представляет собой арил, имеющий от 5 до 14 элементов кольца, или
R'-NH-(C=O)-, где R' представляет собой незамещенный алкил, имеющий 8-20 атомов углерода, арил, имеющий от 5 до 14 элементов кольца, или циклоалкил, имеющий от 3 до 12 элементов карбоциклического кольца;
X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту; и каждый х, у и z представляет собой 1.
50. Способ по п.49, дополнительно включающий после стадии (b) и до стадии (с) использование модифицированного способа деградации Эдмана или пептидазной ферментативной реакции для получения другого защищенного промежуточного соединения посредством уменьшения количества аминокислот боковой цепи защищенного пептидного промежуточного соединения.
51. Кислотно защищенное пептидное промежуточное соединение, имеющее следующую формулу:
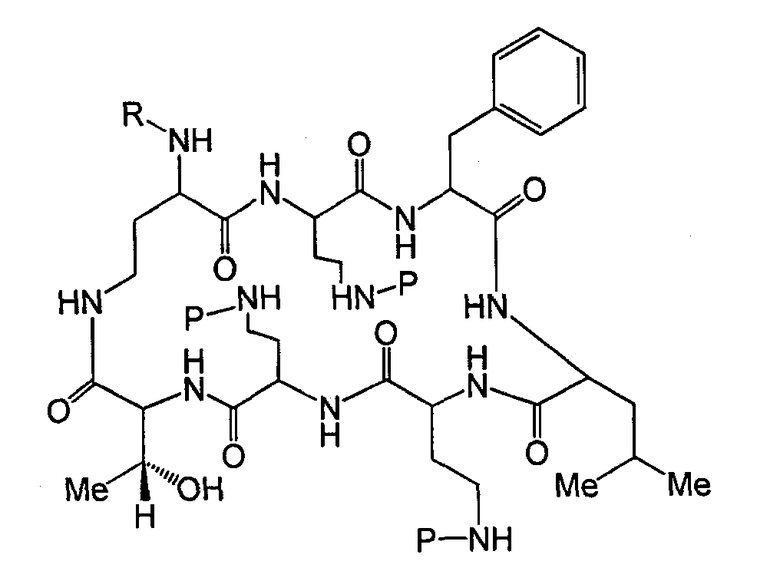
где Р представляет собой сульфоновую кислоту 9-флуоренилметоксикарбонила; и
R выбирают из атома водорода, 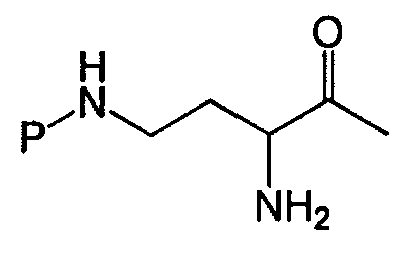 ,
, 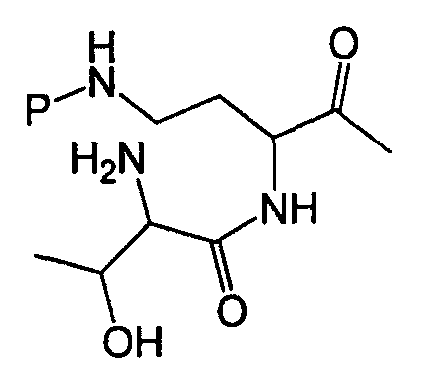 и
и 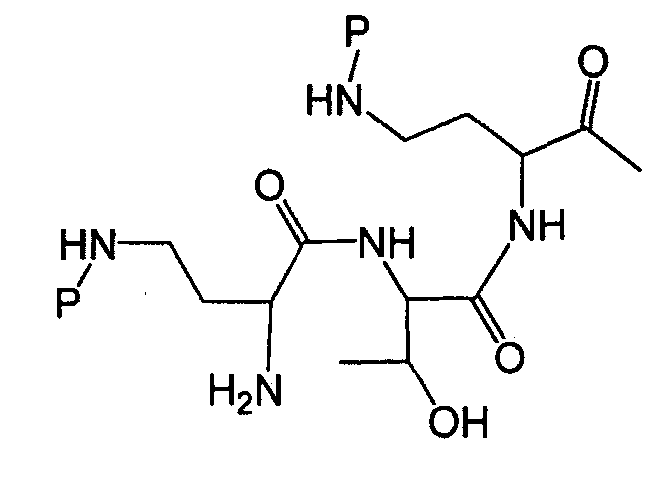 , и
, и
кислотно защищенное пептидное промежуточное соединение получено из полимиксина В.
52. Антибактериальное соединение или защищенное соединение, имеющее формулу:
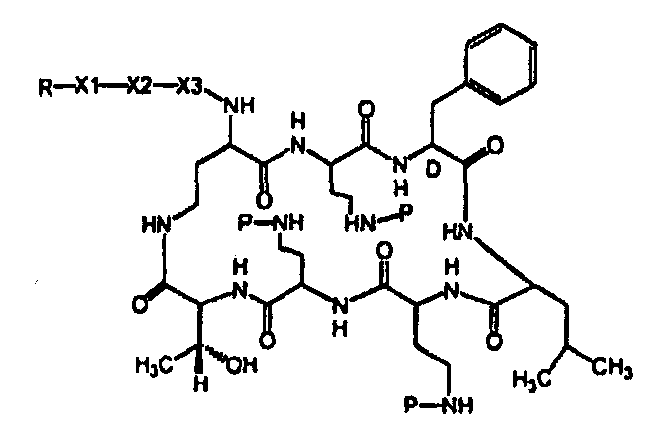
где R, X1, Х2, Х3 и Р выбирают из указанных ниже:
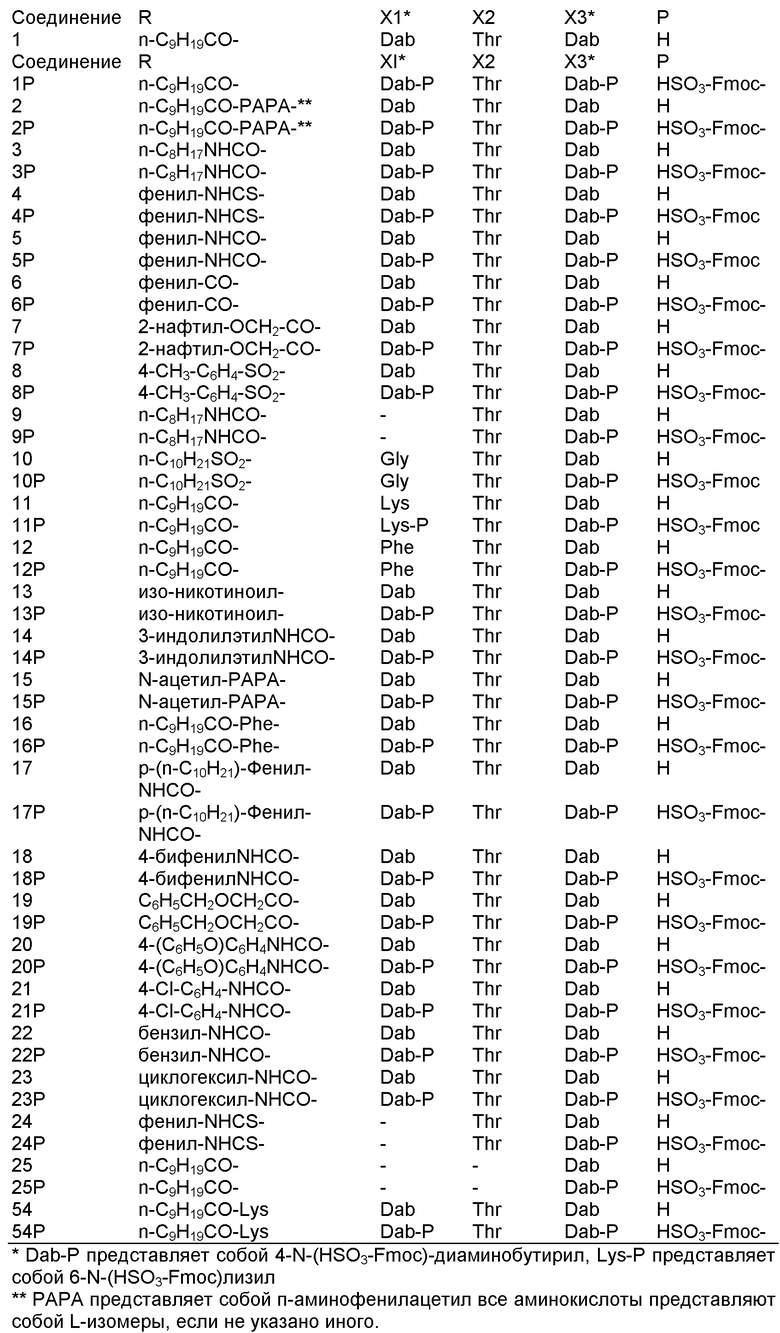
53. Пептидные антибиотики, имеющие следующую структуру
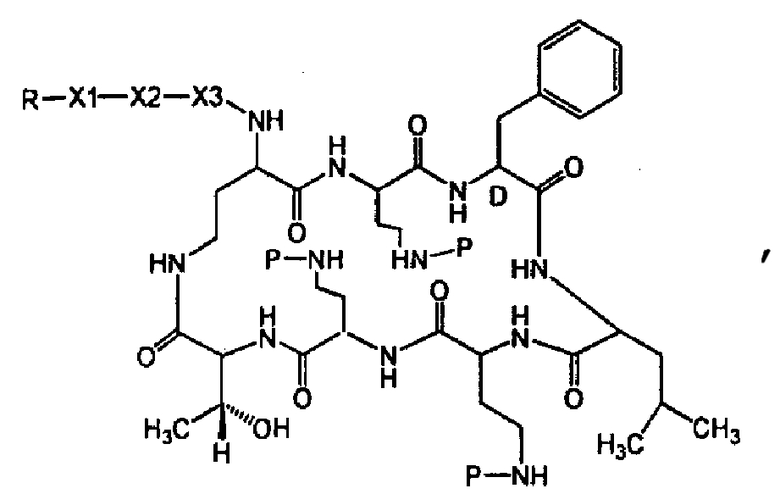
для использования против грамотрицательных и грамположительных бактерий, где R, X1, Х2 и Х3 выбирают из указанных ниже:
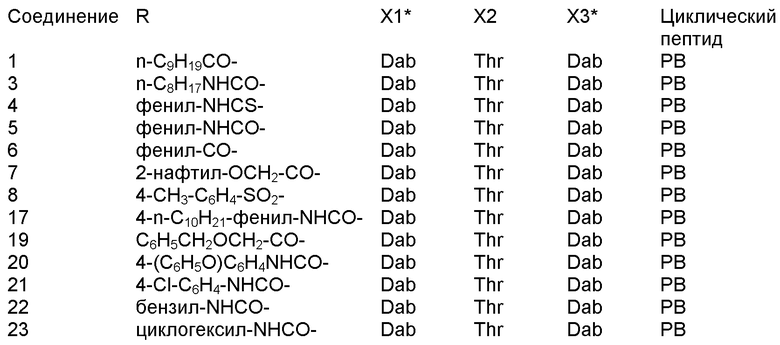
54. Способ получения соединения формулы (I):
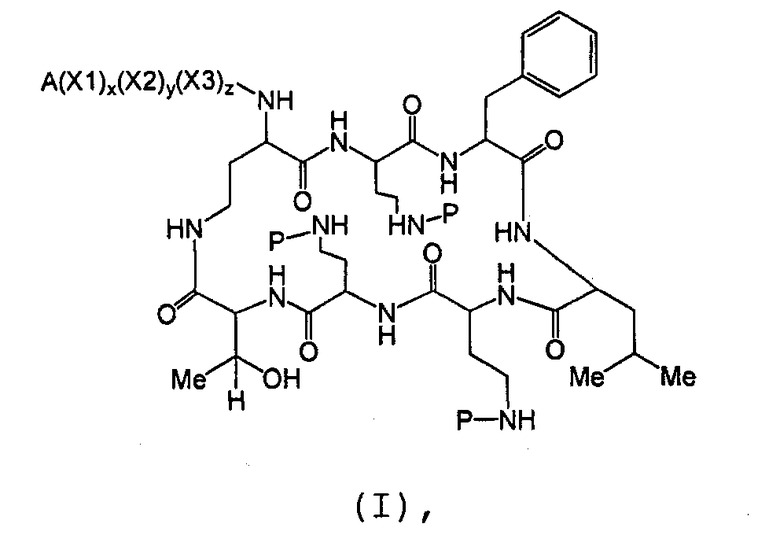
где А представляет собой R'-NH-(C=O)-;
R' выбирают из незамещенного алкила, имеющего 8-20 атомов углерода, циклоалкила, имеющего от 3 до 12 элементов карбоциклического кольца, или арила, имеющего от 5 до 14 элементов кольца;
X1 представляет собой 2,4-диаминобутановую кислоту, Х2 представляет собой треонин и Х3 представляет собой 2,4-диаминобутановую кислоту; и
каждый х, у и z представляет собой 1; и Р представляет собой водород;
включающий:
(а) обработку пептида формулы
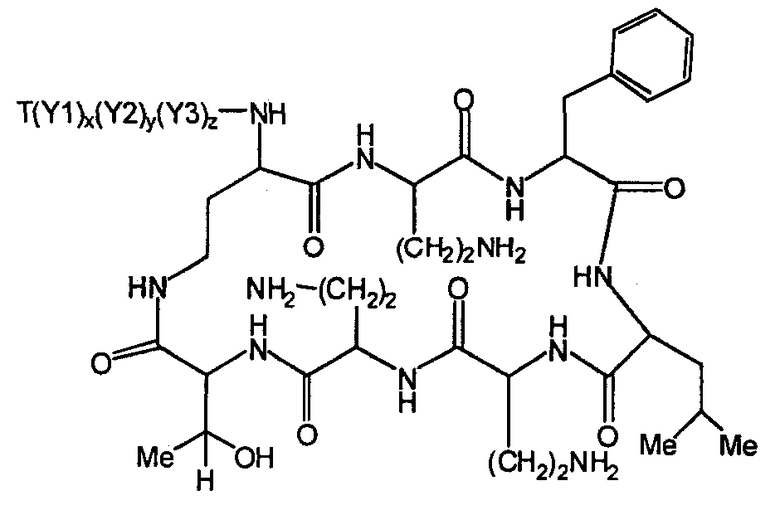
реагентом для защиты аминогруппы, с образованием защищенного пептида,
где Y1 и Y3 оба представляют собой 2,4-диаминомасляную кислоту;
и Y2 представляет собой треонин;
Т представляет собой R'-(C=O)-; где R' представляет собой алкил, имеющий 1-20 атомов углерода; и
каждый из х, у и z равен 1;
и где пептид содержит циклический гептапептид, присоединенный к экзоциклической пептидной цепи, содержащей ацильную группу, и защитная группа содержит, по меньшей мере, один кислотный заместитель;
(b) обработку защищенного пептида деацилирующим агентом, который представляет собой фермент, с образованием защищенного деацилированного пептида формулы А1:
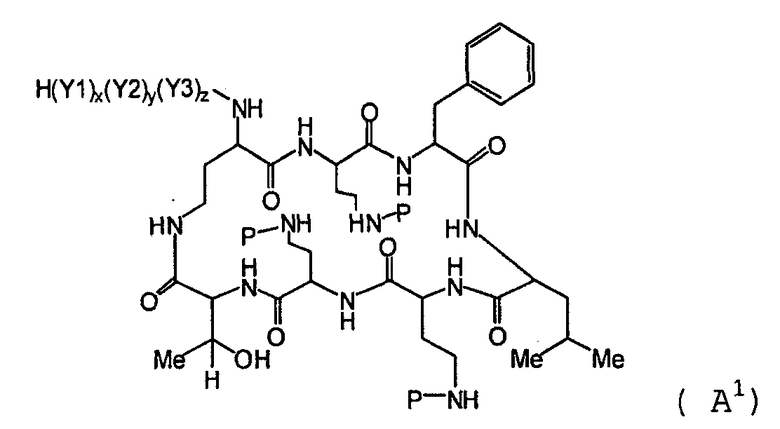
где Y1 и Y3 оба представляют собой 2,4-диаминомасляную кислоту;
и Y2 представляет собой треонин;
каждый из х, у и z равен 1; и
(с) обработку защищенного деацилированного пептида А1 реагентом формулы A-LG, где LG представляет собой уходящую группу или A-LG представляет собой изоцианат, с образованием соединения формулы (I).
55. Способ по п.54, в котором A-LG представляет собой изоцианат.
56. Способ по п.54, в котором защищенный пептид является водорастворимым.
57. Способ по п.54, в котором источником фермента является Actinoplanes utahensis.
58. Способ по п.57, в котором реагент для защиты аминогруппы представляет собой 2-сульфо-9-флуоренилметоксикарбонил.
| WO 02055543 A2, 18.07.2002 | |||
| US 2003224475 A1, 04.12.2003 | |||
| 0 |
|
SU205837A1 | |
| US 4399067 A, 16.08.1983 | |||
| US 5039789 A, 13.08.1991 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛОПЕПТИДНЫХ ПРОИЗВОДНЫХполимиксинов | 0 |
|
SU360766A1 |
| STORM D R; ROSENTHAL К S; SWANSON P E ANNUAL REVIEW OF BIOCHEMISTRY, 1977 Vol:46, Page(s): 723-763 | |||
| VISSER DE P C; ET AL JOURNAL OF PEPTIDE RESEARCH, 2003 Vol:61, Nr:6, Page(s): 298-306. | |||

