Настоящее изобретение относится к новым производным пептидов, содержащим их композициям и их применению для лечения медицинских нарушений, являющихся результатом дефицита гормона роста.
Предпосылки к созданию изобретения
Гормон роста является гормоном, который стимулирует рост всех способных расти тканей. Кроме того, известно, что гормон роста обладает рядом действий на метаболические процессы, например, стимуляцию синтеза белков и мобилизацию свободных жирных кислот, и вызывает переключение в энергетическом метаболизме из метаболизма углеводов в метаболизм жирных кислот. Дефицит гормона роста может приводить к ряду тяжелых медицинских нарушений, например карликовости.
Гормон роста секретируется из гипофиза. Секреция происходит под строгим контролем ряда гормонов и нейромедиаторов непосредственно или косвенно. Секрецию гормона роста можно стимулировать гормоном, высвобождающим гормон роста (GHRH), и ингибировать соматостатином. В обоих случаях гормоны секретируются из гипоталамуса, но их действие связано главным образом через специфические рецепторы, локализованные в гипофизе. Описаны также другие соединения, которые стимулируют секрецию гормона роста из гипофиза. Например, аргинин L-3,4-дигидроксифенилаланин (L-Дола), глюкагон, вазопрессин, PACAP (пептид, активирующий аденилилциклазу гипофиза), агонисты мускариновых рецепторов и синтетических пептид, GHRP (пептид, высвобождающий гормон роста) высвобождают эндогенный гормон роста путем непосредственного действия на гипофиз или путем воздействия на высвобождение GHRH и/или соматостатина из гипоталамуса.
В заболеваниях или состояниях, где желателен повышенный уровень гормона роста, природа белка гормона роста делает невозможным любые пути введения, кроме парантерального введения. Кроме того, другие непосредственно действующие природные усиливающие секрецию средства, например GHRH и PACAP, являются более длинными пептидами, по этой причине пероральное введение их невозможно.
Применение более коротких пептидов для повышения уровня гормона роста у млекопитающих ранее описано, например, в ЕР 18072, ЕР 83864, WO 89/07110, WO 89/1711, WO 89/10933, WO 88/9780, WO 83/02272, WO 91/18016, VO 92/01711 и WO 93/04081.
Строение пептидов или производных пептидов, высвобождающих гормон роста, важно для их активности по высвобождению гормона роста, а также их биологической доступности. Поэтому целью настоящего изобретения является предложение пептидов со свойствами высвобождения гормона роста, которые обладают улучшенными свойствами по сравнению с известными пептидами этого типа.
Краткое изложение существа изобретения
Настоящее изобретение относится к соединению общей формулы I
A-B-C-D-E-(F)p,
где p принимает значения 0 или 1;
А представляет собой имидазолил-C1-6-алкановую кислоту, имидазолил-C1-6-алкеновую кислоту, амино- C1-6-алкановую кислоту или амино-C1-6-алкеновую кислоту или L- или D- α -аминокислоту, выбранную из группы, состоящей из H-His, H-Ala, H-D-Ala, H- (β-аланина), H-Aib, саркозина и Gly;
B представляет собой D-Trp, D-2Nal или D-Phe;
C представляет собой Ala, Ser или Gly;
D представляет собой Trp, Phe, β -(2-тиенил)аланин или -аралкилглицин;
E, когда p равно 1, представляет собой D-Phe, или, когда p равно 2, E представляет собой -NH-CH(CH2-R3)-CO-R4 или -NH- CH(CH2-R3-CH2-R4, где
R3 представляет собой фенил,
и R4 представляет собой пиперазино-, морфолино-, пиперидиногруппу, -ОН или -N(R5)P, где каждый из R5 и R6 независимо представляет собой водород или низший алкил;
F, когда p принимает значение 1, представляет собой -NH-CH(R10)-(CH2)v -R7)-, где v является 0 или целым числом от 1 до 8 и
R7 представляет собой имидазолил, пиперазино-, морфолино-, пиперидиногруппу или П-(P8)-P9, где каждый из R8 и R9 независимо представляет собой водород или низший алкил, или продукт перегруппировки Амадори из аминогруппы и гексапиранозы или гексапиранозилгексапиранозы формулы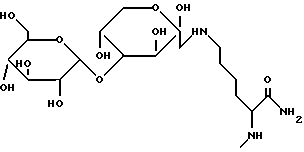
и R10 представляет собой -H, -COOH, -CO-R11, CH2-R11 или -CH2-ОН, где R11 представляет собой пиперазино-, морфолино-, пиперидиногруппу или -N(R12)-R13, где каждый из R12 и R13 независимо является водородом или низшим алкилом; с условием, что по меньшей мере одна амидная связь между А и B, В и С, С и D, D и E или, когда p является 1, E и P замещена аминометиленовой, или что, когда p является 0, E представляет собой -NH-CH(CH2-R3)-CH2-R4, или что, когда p является 1, R10 представляет собой CH2-R11;
или фармацевтически приемлемой соли этого соединения.
Полагают, что производные пептидов формулы I обладают повышенной устойчивостью к протеолитической деградации ферментами желудочно-кишечного тракта или плазмы вследствие замещения амидной связи (-CONH-) аминометиленовой (-CH2NH-) или путем введения N- аралкилглицина, Предполагается, что повышенная устойчивость к протеолитической деградации производных пептидов настоящего изобретения повышает их биологическую доступность по сравнению с биологической доступностью пептидов, предложенных в предшествующей литературе.
В настоящем контексте термин "низший алкил" предназначен для обозначения алкила с 1-6 атомами углерода, конкретно, метила, этила, пропила, изопропила, бутила, пентила или гексила.
Подробное описание изобретения
В предпочтительном варианте соединения формулы I p является 1. В другом предпочтительном варианте соединения формулы I А представляет собой His, D-Ala или имидазолилпропионовую кислоту. B предпочтительно представляет собой -Trp или D-2Nal. C предпочтительно представляет собой Ala. D в значении N-аралкилглицин предпочтительно представляет собой N-бензилглицин. D предпочтительно представляет собой Trp. E предпочтительно представляет, собой Phe. В пределах значения F, V предпочтительно является числом 3 - 6, и R7 предпочтительно является -NH2. R10 предпочтительно представляет собой -CONH2, -CH2OH.
Примерами конкретных соединений изобретения являются H-His
ψ (CH2NH)-D-Trp-Ala-Trp-D-Phe-Iys-NH2
H-Нis-D-Trp ψ (CH2NH)-Ala-Trp-D-Phe-lye-NH2
H-His-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-lye-NH2
H-His-D-Trp-Ala-Trp ψ (CH2NH)-D-Phe-lys-NH2
H-His-D-Trp-Ala-Trp-D-Phe ψ (CH2NH)-lys-NH2
H-D-Ala-D-2Nal-Ala ψ (CH2NH)-Trp-D-Phe-lys-NH2
H-D-Ala-D-2Nal-Ala-Trp-D-Phe ψ (CH2NH-lys-NH2
(3-(4-имидазолил)пропионил-D-2Nal-Ala-Trp-D-Phe ψ (CH2NH)-lys-OH
(3-(4-имидазолил)пропионил-D-2Nal-Ala-Trp-D-Phe ψ (CH2NH)-lys-NH2
(3-(4-имидазолил)акрилоил-D-2Nal-Ala-Trp-D-Phe ψ (CH2NH)-lys-NH2
H-D-Ala-D-Phe-Ala-Trp-D-Phe ψ (CH2NH)-lys-NH2
(2R)-(H-D-Ala-D-Phe-Ala-Trp-NH)-3-фенилпропиламин
(2S)-(H-D-Ala-D-2Nal-Ala ψ (CH2NH)-Trp-D-Phe-NH)-6-аминогексанол
H-D-Ala-D-2Nal-Ala ψ (CH2NH)-Trp-D-Phe-NH2
4-(Н-D-Ala-D-2Nal-Ala ψ (CH2NH)-Trp-D-Phe-NH)бутиламин
(2R)-(H-D-Ala-D-2Nal-Ala-Trp-NH)-3-фенилпропиламин
((2R)-(H-D-Ala-D-2Nal-Ala-Trp-NH)-3-фенилпропиламино) гексиламин
(2R)-(H-D-2Nal-Ala-N-Bzl-Gly-NH)-3-фенилпропиламин
(2R)-(H-D-Ala-D-2Nal-Ala-N-Bzl-Gly-NH)-3-фенилпропиламин
H-Aib-D-2Nal-Ala-N-Bzl-Gly-D-Pne ψ (CH2NH)-lys-NH2
(2S)-((3-(4-имидазолил)пропионил ψ (CH2NH)-D-Phe-Ala-Trp-D-Phe-NH)-6-аминогексанол
(2S)-((3-(4-имидазолил)пропионил)-D-Phe ψ (CH2NH)-Ala-Trp-D-Phe-NH)-6-аминогексанол
(2S)-((3-(4-имидазолил)пропионил)-D-Phe-Ala ψ (CH2NH)-Trp-D-Phe-NH)-6-аминогексанол
(2S)-((3-(4-имидазолил)пропионил)-D-Phe-Ala-Trp ψ (CH2NH)-Phe-NH)-6-аминогексанол
(2S)-(2R)-)(3-(4-имидазолил)пропионил)-D-Phe-Ala-Trp-NH)- 3-фенилпропиламино)-6-аминогексанол
3-((3-(4-имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-NH)-6-пропиламин
(2S)-((3-(4-имидазолил)пропионил)-D-Phe-Ala-Trp-D-Phe ψ (CH2NH)-NH)-6-аминогексанол
(2S)-((3-(4-имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-NH)-6-аминогексанол
3-((3-(4-имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH-Trp-D-Phe-NH)пропиламин
H-D-Ala-D-2Nal-Ala-N-Bzl-Gly-D-Pne ψ (CH2NH)-lys-NH2
H-Aib-D-2Nal-Ala-N-Bzl-Gly-D-Pne ψ (CH2NH2).
Применяемая аббревиатура:
D-2Nal:D-2-нафтилаланин
N-Bzl: N-бензилглицин
H-Aib: H-аминоизомасляная кислота
Соединения формулы I можно получить обычными способами синтеза пептидов в растворе или твердой фазе. Например, твердофазный синтез можно проводить по существу, как описано Stewart and Voung, Solid Phase Peptid Synthesis, 2 nd. Ed. , Rockford, lilinois, USA, 1976. Синтез пептидов в растворе можно, например, проводить способом, описанным Bodansky et al., Peptide Synthesis, 2 nd. Ed., New York, USA, 1976.
Аминометиленовую связь в качестве замещения амидной связи можно ввести в соответствии со способом, описанным Y. Sasaki and D.H.Coy, Peptides 8(1), 1987, p. 119 - 121. Производные пептидов, содержащие моно- или дигексапиранозолзамещенную аминогруппу, можно получить перегруппировкой Амадори при помощи способа, описанного R. Alberti et al., Life Sciences 53, 1993, p. 517-525. Примерами подходящих моно- или дигексапираноз являются глюкоза, галактоза, мальтоза, лактоза или целлобиоза. Производные, применяемые в качестве исходных соединений синтеза, можно получить коммерческим путем, и в них, если требуется, можно ввести подходящие защитные группы.
Фармацевтически приемлемые соли соединений формулы I с кислотами включают соли, полученные реакцией пептида с неорганическими или органическими кислотами, например, соляной, бромистоводородной, серной, уксусной, фосфорной, молочной, малеиновой, фталевой, лимонной, глутаровой, глюконовой, метансульфокислотой, салициловой, янтарной, винной, толуолсульфокислотой, трифторуксусной, сульфаминовой и фумаровой кислотой.
В другой особенности настоящее изобретение относится к фармацевтической композиции, содержащей в качестве активного компонента соединение общей формулы I или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем или разбавителем.
Фармацевтические композиции, содержащие соединение настоящего изобретения, можно получить обычными методиками, например, как описано в Remington's Pharmaeсutical Sciencts, 1985. Композиции могут быть в обычных лекарственных формах, например, в форме капсул, таблеток, аэрозолей, растворов, суспензий или местных аппликаций.
Применяемый фармацевтический носитель или разбавитель может быть обычным твердым или жидким носителем. Примеры твердых носителей включают лактозу, terra alba, сахарозу, циклодекстрин, тальк, желатин, агар, пектин, аравийскую камедь, стеарат магния, стеариновую кислоту или низшие алкиловые эфиры целлюлозы. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло, фосфолипиды, жирные кислоты, амиды жирных кислот, полиоксиэтилен и вода.
Подобным образом носитель или разбавитель может включать любой известный в настоящей области материал, постепенно высвобождающий активный компонент, например, глицерилмоностеарат или глицерилдистеарат, возможно в смеси с воском.
Если твердый носитель применяют для перорального введения, препарат можно таблетировать, помещать в виде порошка или шариков в твердые желатиновые капсулы или препарат может быть в форме пастилки или лепешки. Количество твердого носителя будет широко изменяться, но обычно оно будет от около 25 мг до около 1 г.
Типичная таблетка, которую можно получить обычными техниками таблетирования, может содержать:
Сердцевина:
Активный компонент (в виде свободного соединения или его соли) - 100 мг
Коллоидный диоксид кремния (Aerosil) - 1,5 мг
Целлюлозу, микрокристаллическую (Avicel) - 70 мг
Модифицированную целлюлозную смолу (Ac-Di-Sol) - 7,5 мг
Стеарат магния
Оболочка:
HPMC (около) - 9 мг
*"Мywacett" 9-40 Т (около) - 0,9 мг
* Ацилированный моноглицерид, применяемый в качестве пластификатора для пленочной оболочки.
Если применяют жидкий носитель, препарат может быть в форме сиропа, эмульсии, мягкой желатиновой капсулы или стерильной инъецируемой жидкости, например, водной или неводной жидкой суспензии или раствора.
Для назального введения в виде аэрозоля препарат может содержать соединение формулы I, растворенное или суспендированное в жидком носителе, в частности водном носителе. Носитель может содержать добавки, такие как солюбилизирующие агенты, например пропиленгликоль, поверхностно-активные вещества, например соли желчных кислот или полиоксиэтилированные высшие спирты, средства, повышающие поглощение, например лецитин (фосфатидилхолин) или циклодекстрин, или консерванты, например парабены.
Обычно соединения настоящего изобретения готовят в виде унифицированной лекарственной формы, содержащей 0,0001-100 мг активного компонента вместе с фармацевтически приемлемым носителем в каждой унифицированной дозе.
Подходящая доза соединений в соответствии с настоящим изобретением составляет 1 - 500 мг/день, например около 100 мг на дозу, при введении пациентам, например людям, в качестве лекарственного средства.
Показано, что соединения общей формулы I обладают способностью высвобождать эндогенный гормон роста in vivo. Следовательно, эти соединения можно применять для лечения состояний, для которых требуется повышенный уровень гормона роста в плазме крови, например, у имеющих дефицит гормона роста людей или пожилых пациентов или домашнего скота.
Таким образом, в конкретной особенности настоящее изобретение относится к фармацевтической композиции для стимулирования секреции гормона роста из гипофиза, причем данная композиция содержит в качестве активного компонента соединение общей формулы I или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем или разбавителем.
В следующей особенности настоящее изобретение относится к способу стимулирования секреции гормона роста из гипофиза, причем данный способ предусматривает введение субъекту, нуждающемуся в этом, эффективного количества соединения общей формулы I или его фармацевтически приемлемой соли.
Еще в одной особенности настоящее изобретение относится к применению соединения общей формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для стимулирования секреции гормона роста из гипофиза.
Тем, кто является специалистом настоящей области, хорошо известно, что современное и возможное применение гормона роста для людей разнообразно и многочисленно. Предполагается, что соединения формулы I можно вводить для целей стимулирования секреции гормона роста из гипофиза, и они в таком случае могли бы иметь действия или применения, аналогичные тем, которые имеет сам гормон роста. Применения гормона роста можно суммировать следующим образом: стимулирование секреции гормона роста у пожилых пациентов; предотвращение катаболических побочных эффектов глюкокорикоидов, лечение остеопороза, стимулирование иммунной системы, ускорение заживления ран, ускорение заживления перелома костей, лечение задержки роста, лечение почечной недостаточности или недостаточности, являющейся результатом задержки роста, лечение физиологического маленького роста, включая дефицит гормона роста у детей и маленький рост, связанный с хроническим заболеванием, лечение ожирения и задержки роста, связанной с ожирением, лечение задержки роста, связанной с синдромом Прадера-Вилли и синдромом Turner's; ускорение выздоровления и уменьшение госпитализации обожженных пациентов; лечение внутриутробной задержки роста, скелетной десплазии, гиперкортизолизма и синдрома Кушинга; индуцирование пульсирующей секреции гормона роста; восполнение гормона роста у пациентов со стрессовым состоянием, лечение остеохондродисплазии, синдрома Нунана, шизофрении, депрессий, болезни Альцгеймера, замедленного заживления раны и психосоциальной депривации, лечение легочной дисфункции и зависимости от искусственной вентиляции легких, ослабление белковых катаболических реакций после "большой" операции, снижение кахексии и белковой потери вследствие хронического заболевания, например, рака или СПИДа; лечение гиперинсулинемии, включая гиперплазию панкреатических островков, адъювантное лечение для индуцирования овуляции; стимулирование развития тимуса и предупреждения связанного с возрастом снижения функции тимуса, лечение пациентов с иммунодепрессией, укрепление повышения подвижности мышц, поддерживание толщины кожи, метаболического гомеостаза, почечного гомеостаза у ослабленных пожилых людей, стимулирование остеобластов, костной коррекции и роста хрящей, стимулирование иммунной системы у домашних животных и лечение нарушения из-за старения у домашних животных, стимулятора роста у скота и стимулирование роста шерсти у овец.
Для указанных выше показаний дозировка будет изменяться в зависимости от применяемого соединения формулы I, способа введения и желаемой терапии. Однако обычно пациентам и животным вводят дозу между 0,0001 и 100 мг/кг массы тела ежедневно для достижения эффективной секреции эндогенного гормона роста. Обычно лекарственные формы, пригодные для перорального или назального введения, содержат от около 0,0001 мг до около 100 мг, предпочтительно от около 0,001 мг до около 50 мг соединений формулы I в смеси с фармацевтически приемлемым носителем или разбавителем.
Соединения формулы I можно вводить в форме фармацевтически приемлемой соли с кислотой или в тех случаях, когда целесообразно, соли щелочного металла или щелочноземельного металла или (низший алкил) аммония. Считают, что такие соли обладают приблизительно такой же степенью активности, как и свободные основания.
Фармацевтическая композиция изобретения может содержать соединение формулы I в сочетании с одним или несколькими соединениями, обладающими другой активностью, например антибиотической, или другими фармакологически активными материалами. Им может быть другое усиливающее секрецию средство, например GHEP (1 или 6) или GHRP или его аналоги, гормон роста или его аналог, или соматомедины, например 1GF-1 или 1GF-2.
Способ введения может быть выбран тот, с помощью которого переносится активное соединение в подходящее или желаемое место действия, например, пероральным, назальным или парентеральным путем. Предпочтителен пероральный путь.
Кроме фармацевтического применения соединения формулы I можно использовать in vitro в способах исследования регуляции секреции гормона роста.
Соединения формулы I могут быть пригодны in vivo в способах оценки способности гипофиза секретировать гормон роста. Например, пробы сыворотки крови, взятые до и после введения этих соединений людям, можно анализировать на гормон роста. Сравнение содержания гормона роста в каждой пробе сыворотки позволит непосредственно определить способность гипофиза пациентов секретировать гормон роста.
Соединения формулы I можно вводить имеющим коммерческое значение животным для увеличения скорости и степени их роста и для повышения надоя молока.
Фармакологические методы
Соединения формулы I можно оценить in vitro на их эффективность и активность для секреции гормона роста в первичных соматотрофных клетках крыс.
Первичные соматотрофные клетки крыс можно получить, как описано ранее (Chen et al., Endocrinology 1991, 129, 3337 - 3342 и Chen et al., Endocrinology 1989, 124, 2791-2798). Кратко, крыс убивают декапитацией. Быстро удаляют гипофиз. Гипофиз ферментируют 0,2% коллагеназы и 0,2% гуалуриназы в сбалансированном солевом растворе Хэнка. Клетки ресуспендируют в модифицированной по способу Дульбекко среде Игла, содержащей 0,37% NaHCO3, 10% лошадиной сыворотки, 2,5% фетальной телячьей сыворотки, 1% не являющихся незаменимыми аминокислот, 1% глутамина и 1% пенициллина/стрептомицина, и доводят содержание из до 1,5 х 105 клеток/мл. 1 мл этой суспензии помещают в каждую лунку планшетов из 24 лунок и оставляют на 2 - 3 дня до проведения экспериментов по секреции.
В первый день экспериментов клетки промывают два раза указанной выше средой, содержащей 25 мМ HEPES, pH 7,4. Секрецию гормона роста инициируют путем добавления среды, содержащей 25 мМ HEPES и испытываемое соединение. Инкубирование проводят в течение 15 мин при 27oC. После инкубирования гормон роста, секретированный в среду, измеряют стандартным радиоиммуноанализом (РИА).
Соединения формулы I можно оценить на их влияние на секрецию гормона роста у анестезированных пентобарбиталом самок крыс, как описано ранее (Bercu et al. , Endocrinology 1991, 129, 2592-2598). Коротко, половозрелых самцов крыс Spraque- Dawley анестезируют 50 мг/кг пентобарбитала внутрибрюшинно. После достижения полной анестезии крысам имплантируют трахотомическую трубку, катетеры в сонную артерию и яремную вену. После 15 минутного восстановления нормального состояния крыс, отбирают пробу крови во время О. Внутренне вводят средства, усиливающее секрецию гормона роста из гипофиза, и пробы артериальной крови помещают на лед на 15 мин и затем центрифугируют 2 мин при скорости вращения 12000 х C. Сыворотку декантируют и количество гормона роста определяют стандартным РИА.
Изобретение далее иллюстрируется следующими примерами, которые не ограничивают объема изобретения.
В тексте описания применяют следующую аббревиатуру.
Аббревиатура для аминокислотных остатков: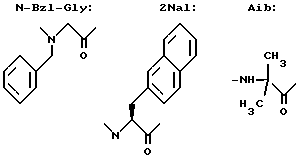
Аббревиатура, применяемая для замещения пептидной связи: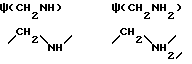
Аббревиатура, применяемая для защитных групп:

Пример 1
H-His-D-Trp ψ(CH2NH)-Ala-Trp-D-Phe-lys-NH2
Пептид на смоле формулы H-Ala-Trp-D-Phe-lys-смола синтезировали в соответствии с F oc-стратегией на синтезаторе пептидов Applied Biosystems 431 А в количестве 0,25 ммоля с использованием поставляемых производителем протоколов FastMoc UV, которые применяют медиированные HBTU 2-(1H-бензотриазол-1-ил)-1,1,3,3- тетраметилуронигексафторофосфат) связывания (соединения) в NМР (N-метилпирролидоне) и УФ-мониторинг удаления Fmoc-защитной группы. В качестве исходной для синтеза смолы применяли 470 мг 4-(2', 4'-диметоксифенил-Fmос-аминометил)феноксисмолы (Novabiocdhem AG Switzerland, cat. # : 01-64-0013), имеющей замещение 0,53 ммоля/г. Применяемыми защищенными производными аминокислот были Fmoc-lys(Boc)-OH,
Fmoc-D-Phe-OH, Fmoc-Trp-OH или Fmoc-Ala-OH.
Изостер -CH2-NH-пептидной связи вводили в соответствии с Sasaki, Y. and Coy, D.H., PEPTIDES 8(1) 119- 121, (1987).
Fmoc-D-Trp-альдегид получали из 399 мг соответствующего N,O-диметилгидроксамата в соответствии с Fehrentz, A. and Castro, B., SYNTESIS 676-678, 1983. Неочищенный альдегид растворяли в 8 мл 1%-ной уксусной кислоты в ДМФ (Диметилфомамид) и разделяли на две порции. Первую порцию добавляли в перемешиваемую суспензию 400 мг (приблизительно 0,2 ммоля) H-Ala-Trp-D-Phe-lys(Boc)-смолы в 8 мл 1%-ной уксусной кислоты в ДМФ при комнатной температуре. Затем 40 мг ПаСПВН3 (чистота 85%), растворенного в 1 мл ДМФ, добавляли в течение 60 мин и перемешивание продолжали еще 60 мин. После этого смолу выделяли и промывали 1%-ной уксусной кислотой в ДМФ на фильтровальной воронке. Смолу снова суспендировали в 5 мл 1%-ной уксусной кислоты в ДМФ и в суспензию добавляли вторую порцию Fmoc-Ala-альдегида. Снова добавляли 40 мг ПаСПВН3 (чистота 85%), растворенного в 1 мл ДМФ, при комнатной температуре в течение 60 мин и смесь перемешивали в течение 18 час.
После стадии восстановительного алкилирования продукт пептид-смола выделяли и промывали 1%-ной уксусной кислотой в ДМФ на фильтровальной воронке и удлинение цепи завершали с использованием синтезатора пептидов в соответствии с описанными выше методиками, применяя защищенное аминокислотное производное Fmoc-His(Trt)-OH.
Пептид отщепляли от 240 мл продукта пептид-смола путем перемешивания в течение 240 мин при комнатной температуре со смесью 3 мл TFA (трифторуксусной кислоты), 225 мг фенола, 75 мкл этандитиола, 150 мкл тиоанизола и 150 мкл H2O. Смесь после расщепления фильтровали, и фильтрат концентрировали до образования масла с применением струи азота. Неочищенный пептид осаждали из этого масла 45 мл диэтилового эфира и промывали 3 раза 45 мл диэтилового эфира.
Неочищенный пептид очищали полупрепаративной ЖХВР на колонке 25 мм х 250 мм, заполненной C-18-диоксидом кремния, 7 мк. Колонку уравновешивали 21%-ным раствором CH3CN в 0,05 М (NH4)2SO4, pH которого устанавливал 2,5 при помощи 4 М H2SO4. После сушки неочищенный пептид растворяли в 5 мл 70% CH3CN/O,1% TFA в H2O и разбавляли водой до достижения 50 мл. 20 мл этого раствора разбавляли до 90 мл и вводили в колонку, которую затем элюировали при градиенте 21%-31% CH3CN в 0,05 М (NH4)2SO4, pH 2,5 и скорости 10 мл/мин в течение 47 мин при 40oC. Содержащие пептид фракции собирали и разбавляли 3 объемами воды и пропускали через патрон Sep-Pak C18 (Waters part. #: 51910), который уравновешивали 0,1%-ной TFA. Патрон затем элюировали 70%-ным CH3CN, содержащим 0,1% TFA, и очищенный пептид выделяли лиофилизацией после разбавления элюата водой. Выход был 6,55 мг.
Полученный конечный продукт характеризовали аминокислотным анализом (содержание пептида и аминокислотный состав), аналитической RP-ЖХВР (время удерживания) и PDMS (плазменная десорбционная масс-спектрометрия) (молекулярная масса). Результаты аминокислотного анализа и PDMS были в согласии с предполагаемым строением в пределах экспериментальной ошибки методики (PDMS: ± 0,9 атомной единицы массы, аминокислотный анализ: ± 10%).
Анализ RP-ЖХВР проводили с применением УФ-детектора при 214 нм и колонки 4,6 мм х 250 мм, Vydac 218ТР54, с C18-диоксидом кремния, 5 мк (The Separations Group, Hesperia, USA). Элюирование проводили со скоростью 1 мл/мин при 42oC. Применяли два различных условия элюирования:
A1: Уравновешивание колонки 5%-ный CH3CN в буфере, содержащем 0,1 М (NH4)2SO4, pH которого устанавливали 2,5 при помощи концентрированной H2SO4, и элюирование с градиентом 5%-60% CH3CN в том же буфере в течение 50 мин.
B1: Уравновешивание колонки 5% CH3CN/0,1% TFA/H2O и элюирование с градиентом от 5% CH3CN/0,1% TFA/H2O до 60% CH3CN/0,1% TFA/H2O в течение 50 минут.
Время удерживания с использованием условий элюирования A1 и B1 было 22,02 мин и 23,08 мин соответственно.
Пример 2
H-His-D-Trp-Ala-Trp-D-Phe ψ (CH2NH)-lys-NH2
Продукт N-lys(2-Cl-Z)-смола синтезировали из 660 мг смолы 4-метил-BHA (Bissendorf Biochemicals, Hannover, bermany, cat. # RMIS 50), имеющей замещение 0,72 ммоля/г и BOC-lys (Cl-Z)-OH в соответствии с BOC-стратегией на синтезаторе пептидов Applied Biosystems 430A в количестве 0,5 ммоля с использованием поставляемых производителем протоколов отдельных связываний (соединений), которые применяют отдельные связывания с предварительно полученными симметричными ангидридами в ДМФ. Протоколы модифицировали для достижения времени связывания 60 мин.
Изостер связи -CH2NH-пептид вводили с использованием методики, аналогичной описанной в примере 1. Применяли 1,675 мг H-lys (Cl-Z)-смолы и BOC-D-Phe-альдегид, полученный из 616 мг соответствующего П,O-диметилгидроксамата.
После этой стадии восстановительного алкилирования смолу выделяли и промывали 1%-ной уксусной кислотой в ДМФ на фильтровальной воронке и удлинение цепи завершали с использованием синтезатора пептидов в соответствии с описанными выше методиками с применением защищенных аминокислотных производных Boc-Trp(For)OH, Boc-Ala-OH, Boc-D-Trp(For)-OH и Boc-His(Bom)-OH.
Пептид отщепляли от 391 мг продукта пептид-смола перемешиванием его в течение 75 мин со смесью 5 мл HF и 500 мкл м- крезола. HF испаряли при 0oC струей азота. Пептид осаждали из оставшегося масла вместе со смолой 50 мл диэтилового эфира и промывали 2 раза 50 мл диэтилового эфира и экстрагировали из смолы 2 х 2 мл TFA и осаждали из объединенного экстракта в TFA 50 мл диэтилового эфира и промывали 2 раза 50 мл диэтилового эфира.
После сушки формильные группы у триптофановых остатков отщепляли путем растворения и перемешивания пептида в 64 мл 6 М гидрохлорида гуанидиния, содержащего 4 мл этаноламина, при 0oC в течение 5 мин. После этого смесь нейтрализовали добавлением 4 мл уксусной кислоты и затем разбавляли 140 мл H2O.
Неочищенный пептид очищали полупрепаративной ЖХВР непосредственным введением реакционной смеси в колонку с применением методики, аналогичной описанной в примере 1. Выход был 20,6 мг.
Конечный продукт характеризовали, как описано в примере 1. Анализ RP-ЖХВР в условиях A1 и B1 дал время удерживания продукта 21,28 мин и 23,17 мин соответственно.
Примеры 3 - 5 (см. табл. 1)
Пример 6
(2R)-Н-D-Ala-D-2Nal-Ala-Trp-NH)-3-фенилпропиламин
Fmoc-D-Phe-альдегид получали из 385 мг соответствующего N,O-диметилгидроксамата в соответствии с (Ferentz, J.A. and Castro, B., SYNTHESIS 676-678, 1983). Неочищенный альдегид растворяли в 20 мл 1%-ной уксусной кислоты в ДМФ и разделяли на две порции.
С 580 мг 4-(2'-4'-диметоксифенил-Fm oc-аминометил) феноксисмолы (Novobiochem AG Switrerland, cat. #: 01-64-0013), имеющей замещение 0,43 ммоля была снята защита обработкой 20%-ным пиперидином в ДМФ в течение 20 мин и последующей промывкой ДМФ и 1%-ной уксусной кислотой в ДМФ.
Первую порцию Fmoc-D-Phe-альдегида и 58 мг NaCNBH3 (чистота 85%), растворенного в 1 мл ДМФ, добавляли в освобожденную от защитной группы смолу и суспензию перемешивали при комнатной температуре в течение 75 мин. После этого смолу отделяли на фильтровальной воронке и промывали 1%-ной уксусной кислотой в ДМФ. Вторую порцию Fmoc-D-Phe- альдегида вместе с 58 мг NаСNВН3 (чистота 85%), растворенного в 1 мл ДМФ, добавляли в смолу, смесь перемешивали при комнатной температуре в течение 18 час, смолу отделяли на фильтровальной воронке и промывали 1%-ной уксусной кислотой в ДМФ, ДМФ, смесью ДХМ/метанол (6:4) и ДХМ (дихлорметан).
С применением этой смолы удлинение цепи завершали, используя синтезатор пептидов в соответствии с методикой, описанной в примере 1, и защищенные аминокислотные производные Fmoc-Trp-OH, Fmoc-Ala-OH, Fmoc-D-2Nal-OH и Fmoc-D-Ala-OH.
Пептид отщепляли от 600 мг полученного продукта пептидсмола. Полученный неочищенный пептид растворяли в 50 мл H2O и 25 мл раствора очищали полупрепаративной ЖХВР и характеризовали с применением методик, аналогичных описанным в примере 1. Выход был 10,9 мг. Анализ продукта RP-ЖХВР в условиях A1 и B2 дал время удерживания 27,98 мин и 29,45 мин соответственно.
Примеры 7-12 (см. табл 2)
Пример 13
(2S)-((3-(4-имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-NH)-6-аминогексанол
Продукт пептида и смолы 3-(1-Adoc-4-имидазолил)пропионил- - Trp-Ala ψ (CH2NH)-Trp-D-Phe-lys(Boc)-смола Sasrin (Aooc является аббревиатурой 1-адамантилоксикарбонила) синтезировали в количестве 1 ммоля по методике, аналогичной описанной в примере 1, за исключением того, что применяли 1020 мг смолы Sasrin (смола 2-метокси-4-алкоксибензилового спирта) (Bachem, Bubendorf, Switzerland cat. # D-1295) со способностью замещения 0,87 ммоля/г и что протокол, применяемый для взаимодействия первого аминокислотного остатка со смолой, был поставляемым изготовителем протоколом катализируемого 4-диметиламинопиридином взаимодействия предварительно полученного симметричного ангидрида и последующей защиты остаточных гидроксигрупп на смоле бензойным ангидридом.
Защищенный пептид, (2S)-(3-(1-Adoc-4-имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-NH)-6-(Boc-амино)гексанол, отщепляли от 1800 мг 3-(1-Adoc-4-имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-lys(Boc)-смолы Sasrin путем перемешивания этого продукта в течение 24 час при комнатной температуре со смесью 10,8 мл ТГФ (тетрагидрофурана), 1,8 мл этанола, 211 мг LiBr и 92 мг NaBH4. Затем по каплям добавляли 2 мл H2О и 2 мл уксусной кислоты. Шарики смолы удаляли фильтрованием и промывали 25 мл этанола. Фильтрат концентрировали в вакууме и полученное масло разбавляли 50 мл Hp20 и лиофилизовали. Полученный порошок подвергали расщеплению при помощи TFA, как описано в примере 1. 1/5 часть полученного неочищенного пептида очищали, как описано в примере 1. Выход был 28,54 мг.
Конечный продукт характеризовали, как описано в примере 1. Время удерживания его в условиях A1 было 20,4 мин.
Пример 14
3-((3-(4-Имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-NH)-пропиламин
Продукт пептида и смолы, 3-(1-Adoc-4-имидазолил)пропионил)- D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-смола синтезировали в количестве 1 ммоля по методике, аналогичной описанной в примере 1, за исключением того, что применяли 1000 мг смолы Sasrin (смола 2-метокси-4-алкоксибензилового спирта) (Bachem, Bubendorf, Switzerland cat. # D-1295) со способностью замещения 0,87 ммоля/г и что протокол, применяемый для взаимодействий первого аминокислотного остатка со смолой, был поставляемым изготовителем протокол-катализируемого 4-диметиламинопиридином взаимодействия предварительно полученного симметричного ангидрида и последующей защиты остаточных гидроксигрупп на смоле бензойным ангидридом.
Пептид, 3-((-3-(1-Adoc-4-имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe-NH)-пропиламин, отщепляли от 1000 мг (3-(1-Aooc-4-имидазолил)пропионил)-D-Trp-Ala ψ (CH2NH)-Trp-D-Phe смолы Sasrin путем перемешивания этого продукта в течение 20 час при комнатной температуре с 10 мл 1,3-диаминопропана. Отработанную смолу фильтровали и экстрагировали 5 мл ДМФ. Объединенные фильтрат и экстракт медленно добавляли в 240 мл 1 М соляной кислоты при перемешивании. Смесь затем разбавляли 500 мл H2О и фильтровали.
Неочищенный продукт очищали полупрепаративной ЖХВР в 9 пробегах (последовательных операциях) путем непосредственного введения в колонку 9 х 1/9 этого фильтрата с использованием методики, аналогичной описанной в примере 1. Выход был 73,53 мг.
Конечный продукт характеризовали, как описано в примере 1. Время удерживания его в условиях A1 и B1 было 21,5 мин и 22,7 мин соответственно.
Примеры 15-18 (см. табл. 3)
Представленные соединения изобретения имеют следующее строение:
3-((3-(4-Имидазолил)пропионил)-D-Trp-Alaψ(CH2NH)-Trp-D-Phe-NH)-пропиламин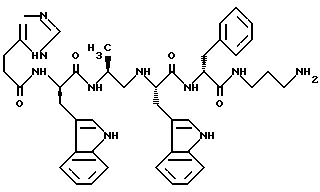
(2S)-((2R)-((3-(4-Имидазолил)пропионил-D-Phe-Ala-Trp-NH)- 3-фенилпропиламино)-6-аминогексанол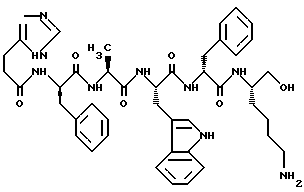
H-Aib-D-2Nal-Ala-N-Bzl-Gly-D-Phe 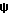 (CH2NH2)
(CH2NH2)
Пример 19
Для изучения влияния различных средств, усиливающих секрецию гормона роста, был разработан анализ in vitro с применением клеток гипофиза крыс. Смешанную клеточную культуру гипофиза выделяли из передней дозы гипофиза самцов крыс и культивировали в течение 3 дней. После промывания клетки стимулировали в течение 15 мин и в супернатанте культуры измеряли количество секретированного гормона роста.
Выделение клеток гипофиза крыс проводили по модификации Sartor, D., et al. , Endocrinology 116, 1985, p. 952-957. Гипофиз отделяли у самцов крыс Sprague-Dawley массой 250 г после декапитации. Нейропромежуточные доли удаляли и оставшуюся часть помещали в среду Грейса, дополненную 0,25% глюкозы, двумя не являющимися незаменимыми аминокислотам 1% BSA (буфер выделения). Железы делили на небольшие кусочки и переносили в пробирку, содержащую 3 мл буфера выделения + 11,5 мг трипсина и 1000 мкг ДНК-азы, и инкубировали в течение 35 мин при 37oC и атмосфере 95% O2 и вращении 70 оборотов в мин. Фрагменты промывали 3 раза путем седиментации в буфере выделения и аспирировали в отдельные клетки при помощи пипетки Пастера. После диспергирования клетки фильтровали через найлоновый фильтр (160 мкм) для удаления непереваренной ткани. Клетки промывали 3 раза буфером выделения, дополненным ингибитором трипсина (0,75 мг/мл) и снова суспендировали в культуральной среде (MEM, дополненный 25 мМ HEPES, 4 мМ глутамина, 0,75% бикарбоната натрия, 2,5% FC (фетальная телячья сыворотка), 3% конской сыворотки, 10% крысиной сыворотки, 1 нМ T3 и 40 мкг/л дексаметазона) до плотности 2 • 105 клеток/мл. Клетки засевали в титрационных микропланшетах, 200 мкл/лунку, и культивировали в течение 3 дней при 37oC и 8% CO2.
После периода культивирования клетки промывали два раза буфером стимулирования (HBSS, дополненный 1% BSA, 0,25% D-глюкозы и 25 мМ HEPES) и преинкубировали в течение 1 часа. Буфер затем удаляли и добавляли новый буфер стимулирования, содержащий соединение изобретения, и микропланшеты инкубировали 15 мин при 37oC и 5% CO2. Буфер собирали и анализировали на содержание гормона роста крыс (rGH) сцинтилляционным проксимальным анализом (SPA, по существу как описано в US 4568649, Hart and Greеnwalt, Mol. Immunol. 16,1979, p. 265-269, или Underfriend et al., Proc. Nate Acad. Sсi. USA 82, 1985, p. 8672- 8676).
Анализ rGH проводили в Opti-планшетах (планшеты с 96 лунками), пригодными для непосредственного подсчета в Packards Top Count (счетчик β-сцинтилляции). Протокол анализа:
40 мкл буфера
10 мкл пробы (инкубированный буфер стимуляции)
50 мкл 125l-rGH
50 мкл кроличьего анти-rGH
50 мкл SPA-реагента (антикроличьи антитела, соединенные с флуомикросферами)
Планшеты герметизировали и помещали на планшетный шейкер на 30 мин, затем проводили инкубирование в течение 10 час, осаждение при 10 - 15oC и подсчет.
В SPA rGH, связанный с кроличьими антителами (первичными антителами) против CH (первичные антитела), реагирует со вторичными антителами, связанными с флуомикросферами (SPA Type II РИА, доступный от Amersham). Любой радиомеченый rGH, который связан с первичными антителами, будет иммобилизован на флуомикросферах, которые будут затем продуцировать свет. Измерения в счетчике β-сцинтилляции делают возможным подсчет количества радиомеченого rGH. Количество радиомеченого rGH, связанного с флуомикросферами, снижается с повышением содержания rGH в пробе (см. табл. 4).



Описывается новое соединение общей формулы I: A-B-C-D-E(-F)p, где р принимает значение 0 или 1; А представляет собой имидазолил-С1-6-алкановую кислоту, имидазолил-С1-6-алкеновую кислоту, амино-С1-6-алкановую кислоту, L- или D-α-аминокислоту, выбранную из группы, состоящей из H-His, H-D-Ala, H-Aib; В представляет собой D-Trp, D-2Nal или D-Phe; С представляет собой Аlа или Gly; D представляет собой Trp или N-аралкилглицин, Е, когда p равно 1, представляет собой D-Phe, или, когда Р равно 0, Е представляет собой -NН-СН(СH2R3)-СО-R4 или -NH-CH(CH2R3)-CH2-R4, где R3 представляет собой фенил, и R4 представляет собой -N(R5)6, где каждый из R5 и R6 независимо представляет собой водород или низший алкил, F, когда р равно 1, представляет собой -NH-CH(R10)-(CH2)v-R7)-, где v приравнивается к целому числу от 1 до 8, и R7 представляет собой -N(R8)-R9), где каждый из R8 и R9 независимо представляет собой водород, R10 представляет собой -Н, -СООН, CO-R11 или СH2-ОН, где R11 представляет собой -N(R12)-(R13), где каждый из R12 и R13 независимо является водородом, при условии, что по меньшей мере одна амидная связь между А и В, В и С, С и D, D и Е или, когда р является 1, между К и F, замещена аминометиленом, или что когда р является О, Е представляет собой -NН-CH(CH2-R3)CH2-R4, или что когда р является 1, R10 представляет собой СН2-R11, или фармацевтически приемлемая соль этого соединения. Соединения обладают повышенной устойчивостью к протеолитической деградации под действием ферментов вследствие замещения амидной связи (-CO-NH-) аминометиленовой (-CH2NH-) или путем введения N-аралкилглицина. Описывается также фармацевтическая композиция для стимулирования секреции гормона роста из гипофиза. 5 с. и 10 з.п. ф-лы, 4 табл.
A-B-C-D-E(-F)p-;
где p принимает значение 0 или 1;
А представляет собой имидазолил-С1-6-алкановую кислоту, имидазолил-С1-6-алкеновую кислоту, амино-С1-6-алкановую кислоту, L- или D-α-аминокислоту, выбранную из группы, состоящей из H-His, Н-Аlа, Н-Aib, H-D-Ala;
В представляет собой D-Trp, D-2Nal или D-Phe;
С представляет собой А1a или Gly;
D представляет собой Trp или N-аралкилглицин;
Е, когда p равно 1, представляет собой D-Phe, или, когда p равно О, Е представляет собой -NH-CH(CH2R3)-CO-R4 или -NH-CH(CH2R3)-CH2-R4, где R3 представляет собой фенил, и R4 представляет собой -N(R5)R6, где каждый из R5 и R6 независимо представляет собой водород,
F, когда p равно 1, представляет собой - NH-CH(R10)-(CH2)v-R7)-, где v приравнивается к целому числу от 1 до 8, и R7 представляет собой -N(R8)-R9 где каждый из R8 и R9 независимо представляет собой водород, R10 представляет собой -H, -СООН, CO-R11 или СН2-ОН, где R11 представляет собой -N(R12)-(R13), где каждый из R12 и R13 независимо является водородом,
при условии, что по меньшей мере одна амидная связь между А и В, В и С, С и D, D и Е или, когда p является 1, между Е и F, замещена аминометиленом, или когда p является О, Е представляет собой -NH-CH(CH2-R3)-CH2-R4, или что когда p является 1, R10 представляет собой CH2-R11;
или фармацевтически приемлемая соль этого соединения.
H-Hisψ(CH2NH)D-Trp-Ala-Trp-D-Phe-Lys-NH2
H-His-D-Trpψ(CH2NH)Ala-Trp-D-Phe-Lys-NH2
H-His-D-Trp-Alaψ(CH2NH)Trp-D-Phe-Lys-NH2
H-His-D-Trp-Ala-Trpψ(CH2NH)D-Phe-Lys-NH2
H-His-D-Trp-Ala-Trp-D-Рheψ(CH2NH)Lys-NH2
H-D-Ala-D-2Nal-Alaψ(CH2NH)Trp-D-Phe-Lys-NH2
H-D-Ala-D-2Nal-Ala-Trp-D-Pheψ(CH2NH)Lys-NH2
(3-(4-имидазолил)пропионил-D-2Nal-Ala-Trp-D-Pheψ(CH2NH)Lys-OH
(3-(4-имидазолил)пропионил-D-2Nal-Ala-Trp-D-Pheψ(CH2NH)Lys-NH2
(3-(4-имидазолил)акрилоил-D-2Nal-Ala-Trp-D-Pheψ(CH2NH)Lys-NH2
H-D-Ala-D-Phe-Ala-Trp-D-Pheψ(CH2NH)Lys-NH2
(2R)-(Н-D-А1а-D-Рhе-А1а-Тrр-NН)-3-фенилпропиламин
(2S)-(H-D-Ala-D-2Nal-Alaψ(CH2NH)Trp-D-Phe-NH)-6-аминогексанол
(H-D-Ala-D-2Nal-Alaψ(CH2NH)Trp-D-Phe-NH2
4-(H-D-Ala-D-2Nal-Alaψ(CH2NH)Trp-D-Phe-NH)-бутиламин
(2R)-(H-D-Ala-D-2Nal-Ala-Trp-NH)-3-фенилпропиламин
((2R)-(H-D-Ala-D-2Nal-Ala-Trp-NH)-3-фeнилпpoпилaминo)гeкcилaмин
(2R)-(H-D-2Nal-Ala-N-Bzl-Gly-NH)-3-фенилпропиламин
(2R)-(H-D-Ala-D-2Nal-Ala-N-Bzl-Glу-NH)-3-фeнилпpoпилaмин
(H-Aib-D-2Nal-Ala-N-Bzl-Gly-D-Pheψ(CH2NH)Lys-NH2
(2S)-((3-(4-имидaзoлил)пpoпиoнил)ψ(CH2NH)D-Phe-Ala-Trp-D-Phe-NH)-6-аминогексанол
(2S)-((3-(4-имидaзoлил)пpoпиoнил)-D-Pheψ(CH2NH)Ala-Trp-D-Phe-NH)-6-аминогексанол
(2S)-((3-(4-имидaзoлил)пpoпиoнил)-D-Phe-Alaψ(CH2NH)Trp-D-Phe-NН)-6-аминогексанол
(2S)-((3-(4-имидaзoлил)пpoпиoнил)-D-Phe-Ala-Trpψ(CH2NH)D-Phe-NH)-6-аминогексанол
(2S)(2R)((3-(4-имидазолил)пропионил)-D-Рhe-А1а-Тrр-NH)-3-фенилпропиламино)-6-аминогексанол
3-((3-(4-имидазолил)пропионил)-D-Trp-Alaψ(CH2NH)Trp-D-Phe-NН)пропиламин
(2S)-((3-(4-имидазолил)пропионил)-D-Рhе-А1а-Тrр-Рheψ(СН2NН)NH)-6-аминогексанол
(2S)-((3-(4-имидазолил)пропионил)-D-Тrр-А1аψ(CH2NH)Trp-D-Phe-NН)-6-аминогексанол
3-((3-(4-имидазолил)пропионил)-D-Trp-Alaψ(CH2NH)Trp-D-Phe-NН)пропиламин
H-D-Ala-D-2Nal-Ala-N-BzL-Gly-D-Pheψ(CH2NH)Lys-NH2
H-Aib-D-2Nal-Ala-N-BzL-Gly-D-Pheψ(CH2NH2).
Приоритет по пунктам:
23.12.93 по пп.1, 2 - 5, 6, 7, 8, 10, 11 - 16;
17.01.94 по пп.9, 10;
30.06.94 по пп.1, 6, 10;
07.10.94 по пп.1, 10.
| SU 622807 A, 05.09.1978 | |||
| 0 |
|
SU257742A1 | |
| HOCART SIMON | |||
| J | |||
| et | |||
| al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Med.Chem | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |

