Область техники, к которой относится изобретение
Настоящее изобретение относится к новому способу получения 4′-[2-н-пропил-4-метил-6-(1-метилбензимидазол-2-ил)бензимидазол-1-илметил]бифенил-2-карбоновой кислоты (МНН: телмисартан).
Предпосылки создания изобретения
Телмисартан представляет собой антагонист рецептора ангиотензина II, который может применяться для лечения гипертонии, а также других заболеваний, как это описано в заявке ЕР 502314 В1.
Это действующее вещество имеет следующую химическую структуру:
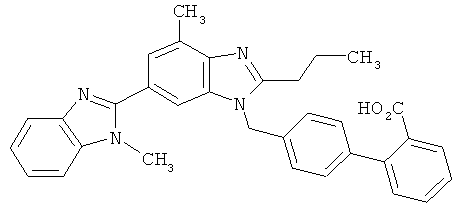
Телмисартан получают и соответственно поставляют на рынок, как правило, в виде свободной кислоты. Как указано в заявке WO 00/43370, кристаллический телмисартан существует в двух полиморфных формах, имеющих разную температуру плавления. Под воздействием повышенных температур и влажности имеющая более низкую температуру плавления полиморфная форма В необратимо превращается в полиморфную форму А с более высокой температурой плавления.
Технологически синтез телмисартана до настоящего времени осуществляли взаимодействием 2-н-пропил-4-метил-6-(1′-метилбензимидазол-2′-ил)бензимидазола (I) с трет-бутиловым эфиром 4′-бромметилбифенил-2-карбоновой кислоты (II) и последующим омылением согласно следующей схеме 1.
Схема 1
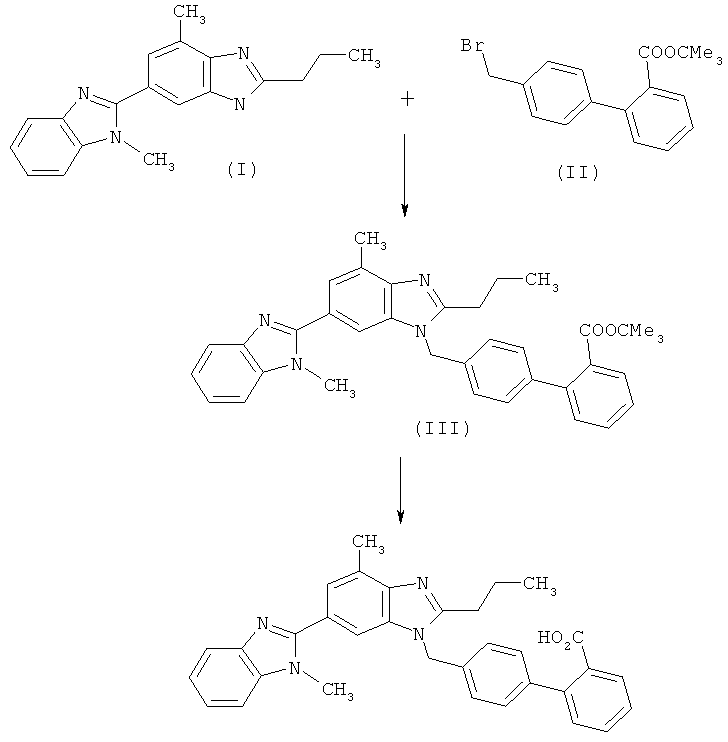
Сочетание компонентов путем нуклеофильного замещения на первой стадии реакции представлено в ЕР 502314 В1 лишь в общем, без каких-либо пояснений как способ б), а омыление трет-бутилэфирной группы в лабораторном масштабе с использованием трифторметилуксусной кислоты описано в данной заявке в примере 1. Технологически омыление до настоящего времени проводят с использованием концентрированной водной бромистоводородной кислоты. При реализации известного из указанной заявки способа синтеза в промышленном масштабе неожиданно возникли значительные трудности. Так, в частности, удовлетворительное качество получаемого предложенным способом действующего вещества удается достичь лишь после проведения нескольких технологических операций (необходимую чистоту сырой продукт имеет только лишь после двукратной перекристаллизации), при этом для выделения продукта требуется значительное время на центрифугирование и сушку. Синтезируемый согласно схеме 1 в промышленном масштабе телмисартан после соответствующей переработки получают в виде такого продукта, который для завершающей очистки приходится подвергать вторичной операции по кристаллизации. В результате этой повторной вынужденной операции морфология выкристаллизовавшегося конечного продукта обусловливала непредсказуемые трудности.
Выпадающий в виде твердого вещества в форме длинных иголок продукт лишь с трудом удается фильтровать, промывать и выделять, кроме того, из-за включений остатков растворителя для его сушки требуется продолжительное время и при этом образуются крупные, крайне твердые куски. После измельчения подобных кусков образуется сухой порошок, который проявляет заметную тенденцию к приобретению электростатического заряда и практически не обладает сыпучестью.
Вышеназванные отрицательные свойства продукта постоянно обусловливают при получении соответствующего соединения в промышленном масштабе существенные трудности, преодоление которых в условиях роста производства при обеспечении высокой чистоты продукта требует значительных усилий или дополнительных, высоких материально-технических затрат.
Исходя из вышеизложенного в основу настоящего изобретения была положена задача разработать и предложить альтернативный способ получения телмисартана, который можно было бы осуществлять в промышленном масштабе и который обеспечивал бы простую переработку, очистку и выделение телмисартана без указанных выше недостатков.
Краткое изложение сущности изобретения
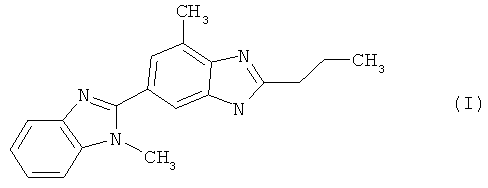
При создании изобретения неожиданно было установлено, что с технологической точки зрения взаимодействие 2-н-пропил-4-метил-6-(1′-метилбензимидазол-2′-ил)бензимидазола формулы (I)
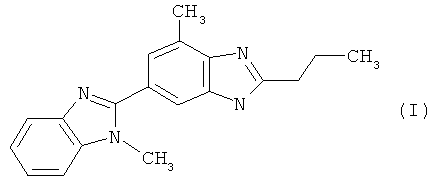
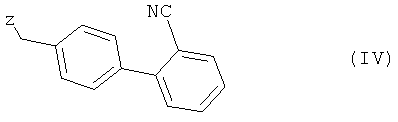
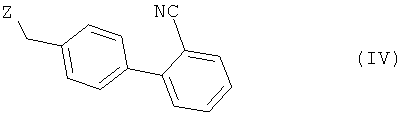
с соединением общей формулы (IV)
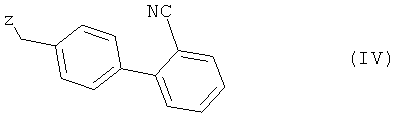
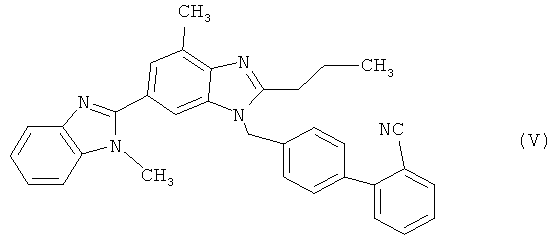
в которой Z представляет собой уходящую группу, такую как атом галогена, например атом хлора, брома либо иода, или замещенную сульфонилоксигруппу, например метансульфонилокси-, фенилсульфонилокси- или п-толуолсульфонилоксигруппу, с получением при этом соединения 2-циано-4′-[2′′-н-пропил-4′′-метил-6′′-(1′′′-метилбензимидазол-2′′′-ил)бензимидазол-1′′-илметил]бифенила формулы (V)
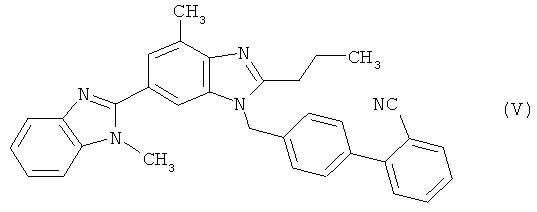
которое при необходимости подвергают переработке (стадия (а)) и последующему гидролизу нитрильной функциональной группы с образованием кислотной функциональной группы (стадия (б)) и которое при необходимости в процессе переработки переводят в гидрохлорид, позволяет добиться значительных преимуществ в сравнении с представленным на схеме 1 синтезом, прежде всего заключающихся в том, что удается избежать вышеуказанных недостатков, присущих традиционному способу при его осуществлении в промышленном масштабе.
Подробное описание изобретения
Стадия (а)
Взаимодействие соединения формулы (I) с соединением общей формулы (IV), в которой Z предпочтительно представляет собой атом галогена, прежде всего атом брома, целесообразно проводить в соответствующем растворителе либо в смеси растворителей, таком(их) как метиленхлорид, диэтиловый эфир, тетрагидрофуран, диоксан, диметилсульфоксид, диметилформамид, диметилацетамид, диметилформамид/трет-бутанол, диметилацетамид/трет-бутанол, толуол или бензол, необязательно в присутствии связывающего кислоту агента, такого как карбонат натрия, карбонат калия, гидроксид натрия, гидроксид калия, метилат натрия, метилат калия, трет-пентилат калия, трет-бутилат калия, н-бутилат калия, гидрид натрия, триэтиламин или пиридин, причем оба последних из числа указанных могут использоваться также в качестве растворителей, например, при температуре в интервале от 0 до 100°С. Основание при этом, если оно представляет собой твердое вещество, предпочтительно применять в порошкообразном виде.
Взаимодействие соединения формулы (I) с соединением общей формулы (IV) предпочтительно проводить в растворителе либо в смеси растворителей, выбранном(ых) из группы, включающей диметилсульфоксид, диметилформамид, диметилацетамид, диметилформамид/трет-бутанол и диметилацетамид/трет-бутанол, в присутствии гидроксида натрия, гидроксида калия или трет-бутилата калия при температуре в интервале от 0 до 30°С. Особенно предпочтительно взаимодействие соединения формулы (I) с соединением общей формулы (IV) проводить в диметилацетамиде или в смеси диметилацетамида и трет-бутанола в присутствии гидроксида калия при температуре в интервале от 0 до 20°С.
Переработка
По завершении реакции растворитель удаляют, например, в вакууме, создаваемом водоструйным насосом, остаток обрабатывают растворителем, в котором нитрил (V) проявляет лишь ограниченную растворимость, а под тепловым воздействием - соответственно умеренную растворимость, например обрабатывают спиртом, таким как метанол, этанол, н-пропанол, изопропанол, н-бутанол или изобутанол, ароматическим углеводородом, таким как бензол или толуол, эфирным растворителем, таким как диэтиловый эфир, тетрагидрофуран, диоксан или трет-бутилметиловый эфир, предпочтительны при этом эфирные растворители и прежде всего трет-бутилметиловый эфир, или же также водой, выпавшие при определенных условиях после охлаждения до 10-20°С кристаллы отделяют вакуум-фильтрацией и после этого промывают сначала используемым растворителем, а затем водой. В случае необходимости продукт сушат при повышенной температуре, например при 50-100°С, в вакуумном сушильном шкафу. Нитрил (V) получают, как правило, с высоким выходом (от 80 до 90% от теории) и отличного качества (чистота согласно данным ЖХВР-анализа выше 99,5%).
Стадия (б)
Последующий гидролиз нитрильной функциональной группы с образованием карбоксигруппы целесообразно проводить в воде, в органическом растворителе либо в смеси из органического растворителя и воды, при этом органическим растворителем может служить, например, метанол, этанол, н-пропанол, изопропанол, тетрагидрофуран, диоксан, этиленгликоль, пропиленгликоль, диглим, диметилсульфоксид или монометиловый эфир диэтиленгликоля, в присутствии кислоты, такой как трифторуксусная кислота, трихлоруксусная кислота, соляная кислота, серная кислота или фосфорная кислота, либо в присутствии основания, такого как гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид цезия или гидроксид кальция, или же соответствующих ангидридов при температурах в интервале от 80 до 200°С, при этом требуемая для реакции вода может быть также компонентом одного из используемых реагентов, например одной из вышеназванных кислот, или же ее можно получать в условиях реакции из реагентов, например из одного из вышеуказанных гидроксидов щелочных металлов.
Гидролиз нитрильной функциональной группы предпочтительно проводить в системе высококипящих растворителей, выбранных из группы, включающей этиленгликоль/воду и пропиленгликоль/воду, в присутствии соответствующего основания, особенно предпочтительно гидроксида калия, при температурах в интервале от 140 до 200°С, прежде всего при температуре в интервале от 155 до 185°С.
Переработка
По завершении реакции растворитель удаляют, например, отгонкой в вакууме, создаваемом водоструйным насосом, остаток разбавляют водой и растворяют с помощью соляной кислоты, например 5-32%-ной (концентрированной) соляной кислоты, предпочтительно 5-20%-ной соляной кислоты, при этом телмисартан выкристаллизовывается в виде гидрохлорида. Суспензию кристаллов при необходимости охлаждают до 10-25°С и при этой температуре ее можно перемешивать в течение определенного промежутка времени, например в течение 3 часов. После отделения кристаллов вакуум-фильтрацией их промывают водой и при необходимости сушат в вакуумном сушильном шкафу при повышенной температуре, например в интервале от 50 до 120°С.
Из гидрохлорида телмисартана обычным путем можно высвобождать телмисартан в кислотной форме, например, путем титрования с использованием водного раствора гидроксида щелочного металла. Процесс высвобождения телмисартана в кислотной форме можно осуществлять, в частности, способом, аналогичным описанному в заявке WO 00/43370 (см. стр.3, строка 6 - стр.4, строка 38, а также примеры 1-3).
Среди преимуществ предлагаемого в изобретении способа при его реализации в промышленном масштабе прежде всего можно назвать следующие:
- соединения формулы (IV), прежде всего 4′-бромметил-2-цианобифенил, получают в промышленном масштабе и в качестве коммерчески доступных продуктов они могут приобретаться по вполне умеренным ценам;
- компоненты формул (I) и (IV) удается подвергать сочетанию между собой согласно стадии (а) при их высокой концентрации и соответственно высоком расходе, при этом в результате переработки, прежде всего при использовании трет-бутилметилового эфира, нитрил (V) получают в виде легко фильтруемого и промываемого осадка, вследствие чего отпадает необходимость в проведении дальнейших затратных операций по очистке;
- нитрил (V) получают с высоким выходом (от 80 до 90% от теории) и отличного качества (чистота согласно данным ЖХВР-анализа выше 99,5%);
- омыление нитрила (V) на стадии (б) также обеспечивает высокий выход продукта, составляющий более 95% от теории;
- конечный продукт - телмисартан - можно выделять либо в виде цвиттер-иона, либо, что предпочтительно, путем осаждения с помощью соляной кислоты в виде легко фильтруемого и тем самым без проблем очищаемого гидрохлорида.
Согласно изобретению особенно предпочтительно работать следующим образом.
Стадия (а): все количества указаны в пересчете на исходные применяемые количества соединения формулы (I), равные 0,1 моля, и при изменении этой величины умножаются на соответствующий коэффициент.
В реакционный сосуд соответствующей емкости из расчета на 0,1 моля соединения (I) предварительно помещают 50-200 мл растворителя, предпочтительно 80-120 мл, затем соединение (I) суспендируют в этом растворителе и при перемешивании порциями добавляют 0,1-0,2, предпочтительно 0,102-0,12 моля основания, при этом температуру поддерживают в интервале от 10 до 50°С, предпочтительно в интервале от 15 до 30°С, и по завершении экзотермической реакции перемешивание при этой температуре можно продолжать еще в течение 3 часов. Далее смесь охлаждают до температуры порядка 0-10°С, например до приблизительно 5°С, после чего при температуре в интервале от 10 до 30°С, предпочтительно при приблизительно 20°С, по каплям добавляют смесь из 0,1-0,2 моля, предпочтительно 0,100-0,12 моля, соединения общей формулы (IV) и 50-200 мл растворителя (из расчета на 0,1 моля соединения (IV)). Температуру реакционной смеси, при необходимости при охлаждении ледяной баней, поддерживают в интервале от 0 до 20°С, предпочтительно от 5 до 10°С. После этого можно проводить промывку несколькими миллилитрами растворителя и затем продолжать перемешивание еще в течение примерно 3 часов при 0-20°С.
В одном из других вариантов осуществления изобретения основание предварительно помещают в 30-100 мл растворителя, предпочтительно диметилформамида, диметилацетамида, диметилформамида/трет-бутанола или диметилацетамида/трет-бутанола, затем при температуре в интервале от 10 до 30°С, например при температуре порядка 20°С, перемешивают в течение примерно 1 ч, после чего при этой температуре медленно добавляют суспензию соединения (I) в 30-100 мл растворителя. Все последующие операции проводят аналогично описанному выше варианту. Указанные смеси растворителей используют при объемном соотношении между амидом и трет-бутанолом от 10:1 до 2,5:1, например 5:1.
Переработка
Растворитель целесообразно предельно полностью отгонять при пониженном давлении, например в вакууме, создаваемом водоструйным насосом, при этом происходит выкристаллизовывание продукта. После охлаждения остатка до температуры в интервале от 40 до 80°С, предпочтительно до примерно 60°С, его разбавляют 100-300 мл растворителя (из расчета на 0,1 моля соединения (I)), предпочтительно трет-бутилметилового эфира, и вплоть до 5 часов перемешивают без подвода какой-либо энергии. Затем смесь охлаждают до 0-30°С, предпочтительно до 15-20°С, и повторно вплоть до 5 часов перемешивают при этой температуре. Кристаллы отделяют вакуум-фильтрацией и порциями промывают сначала 50-150 мл растворителя, а затем 200-300 мл воды. В завершение продукт сушат в вакуумном сушильном шкафу при температуре в интервале от 50 до 100°С, предпочтительно при температуре порядка 60°С.
Стадия (б): все количества, если не указано иное, указаны в пересчете на исходные применяемые количества соединения формулы (V), равные 0,05 моля, и при изменении этой величины умножаются на соответствующий коэффициент.
0,05 моля соединения (V), 200-300 мл органического растворителя, 0,5-5 мл воды и 0,3-0,5 моля основания совместно помещают в реакционный сосуд и нагревают до температуры кипения используемой системы растворителей, в случае предпочтительного использования смеси этиленгликоля и воды нагревают до 140-200°С, предпочтительно до 155-185°С. При этой температуре смесь перемешивают вплоть до 24 часов.
В другом варианте осуществления изобретения 0,361 моля нитрила (V) предварительно помещают в 1,5-2 л органического растворителя, предпочтительно этиленгликоля, затем добавляют 25-50 мл воды и 2,5-3 моля основания и при перемешивании нагревают в течение примерно 24 ч до 140-200°С, предпочтительно до 155-185°С. Все последующие операции проводят аналогично описанному выше варианту.
Переработка
Количества указаны в пересчете на исходные применяемые количества соединения формулы (V), равные 0,05 моля.
Растворитель целесообразно удалять при пониженном давлении, например путем отгонки в вакууме, создаваемом водоструйным насосом, остаток разбавляют 30-100 мл, предпочтительно примерно 5 мл, воды и вмешивают в смесь из 100-150 мл воды (предпочтительно примерно 125 мл) и 40-60 мл (предпочтительно примерно 50 мл) концентрированной соляной кислоты (примерно 32%-ной), после чего можно проводить промывку небольшим количеством воды. Выкристаллизовывающийся в виде гидрохлорида телмисартан охлаждают до 10-25°С и перемешивают при этой температуре в течение приблизительно 3 часов. После отделения кристаллов вакуум-фильтрацией их промывают 50-200 мл воды и сушат в вакуумном сушильном шкафу при температуре в интервале от 50 до 120°С.
Ниже изобретение более подробно поясняется на примерах вариантов осуществления предлагаемого в нем способа получения телмисартана, которые (примеры) при этом не ограничивают объем изобретения.
Пример 1
2-Циано-4′-[2′′-н-пропил-4′′-метил-6′′-(1′′′-метилбензимидазол-2′′′-ил)бензимидазол-1′′-илметил]бифенил
32,24 г 2-н-пропил-4-метил-6-(1′-метилбензимидазол-2′-ил)бензимидазола • Н2О предварительно помещают в 100 мл диметилацетамида (ДМА), затем при температуре примерно 20°С при перемешивании порциями добавляют 11,8 г трет-бутилата калия и в течение 1 часа перемешивают при той же температуре порядка 20°С. После этого смесь охлаждают до 5°С и затем в течение примерно 30 мин по каплям добавляют смесь из 28,6 г 4-бромметил-2′-цианобифенила и 95 мл ДМА (раствор приготовлен при температуре порядка 20°С). Температуру реакционной смеси при охлаждении ледяной баней поддерживают в интервале от 5 до 10°С. После этого промывают 5 мл ДМА и в течение последующих 1,5 ч перемешивают при 5-10°С. Растворитель практически полностью отгоняют в вакууме, создаваемом водоструйным насосом, при этом происходит выкристаллизовывание продукта. Остаток охлаждают до 60°С, разбавляют 230 мл трет-бутилметилового эфира и в течение 1 часа перемешивают без подвода какой-либо энергии, затем охлаждают до 15-20°С и при этой температуре продолжают перемешивание еще в течение 1 часа. Кристаллы отделяют вакуум-фильтрацией, промывают порциями сначала 100 мл трет-бутилметилового эфира, а затем 250 мл воды и в завершение сушат в вакуумном сушильном шкафу при 80°С.
Выход 43,3 г (87,5% от теории), tпл 196-197°С, чистота согласно данным ЖХВР-анализа 99,9%.
Пример 2
2-Циано-4′-[2′′-н-пропил-4′′-метил-6′′-(1′′′-метилбензимидазол-2′′′-ил)бензимидазол-1′′-илметил]бифенил
32,4 г 2-н-пропил-4-метил-6-(1′-метилбензимидазол-2′-ил)бензимидазола • Н2О предварительно помещают в 100 мл ДМА, затем при перемешивании при температуре примерно 20°С порциями добавляют 6,9 г гидроксида калия (в виде порошка) и в течение 1 часа перемешивают при температуре порядка 20-25°С. После этого смесь охлаждают до 5°С и затем в течение примерно 30 мин по каплям добавляют 28,6 г 4-бромметил-2′-цианобифенила в 95 мл ДМА (раствор приготовлен при температуре порядка 20°С). Температуру реакционной смеси при охлаждении ледяной баней поддерживают в интервале от 5 до 10°С. После этого промывают 5 мл ДМА и в течение последующих 1,5 ч перемешивают при 5-10°С. Растворитель практически полностью отгоняют в вакууме, создаваемом водоструйным насосом, при этом происходит выкристаллизовывание продукта. Остаток охлаждают до 60°С, разбавляют 225 мл трет-бутилметилового эфира и в течение 1 часа перемешивают без подвода какой-либо энергии, затем охлаждают до 15-20°С и при этой температуре продолжают перемешивание еще в течение 1 часа. Кристаллы отделяют вакуум-фильтрацией, промывают порциями сначала 100 мл трет-бутилметилового эфира, а затем 250 мл воды и в завершение сушат в вакуумном сушильном шкафу при 80°С.
Выход 40,45 г (81,7% от теории), tпл 196-197°C, чистота согласно данным ЖХВР-анализа 99,9%.
Пример 3
2-Циано-4′-[2′′-н-пропил-4′′-метил-6′′-(1”'′-метилбензимидазол-2′′′-ил)бензимидазол-1′′-илметил]бифенил
6,9 г гидроксида калия (в виде порошка) предварительно помещают в 50 мл ДМА, перемешивают в течение 15 мин при 20-25°С и затем при той же температуре 20-25°С добавляют суспензию 32,24 г 2-н-пропил-4-метил-6-(1′-метилбензимидазол-2′-ил)бензимидазола • Н2О в 50 мл ДМА. По завершении процесса добавления реакционные сосуды промывают 10 мл ДМА и продолжают перемешивание еще в течение 1 часа при 20-25°С. Далее смесь охлаждают до 5°С и затем добавляют 28,6 г 4-бромметил-2′-цианобифенила в 95 мл ДМА (раствор приготовлен при температуре порядка 20°С). Температуру реакционной смеси поддерживают при охлаждении ледяной баней в интервале от 5 до 10°С. После этого промывают 5 мл ДМА и в течение еще 1 часа перемешивают при 5-10°С. Растворитель практически полностью отгоняют в вакууме, создаваемом водоструйным насосом, при этом происходит выкристаллизовывание продукта. Остаток охлаждают до 60°С, разбавляют 250 мл трет-бутилметилового эфира и в течение 2 часов перемешивают без подвода какой-либо энергии. Кристаллы отделяют вакуум-фильтрацией, промывают порциями сначала 100 мл трет-бутилметилового эфира, а затем 250 мл воды и в завершение сушат в вакуумном сушильном шкафу при 80°С.
Выход 43,37 г (87,5% от теории), tпл 196-198°С, чистота согласно данным ЖХВР-анализа 99,1%.
Пример 4
2-Циано-4′-[2′′-н-пропил-4′′-метил-6′′-(1′′′-метилбензимидазол-2′′′-ил)бензимидазол-1′′-илметил]бифенил
53,4 г гидроксида калия (в виде порошка) предварительно помещают в 385 мл ДМА и затем при температуре 20-25°С добавляют суспензию 248,25 г 2-н-пропил-4-метил-6-(1′-метилбензимидазол-2′-ил)бензимидазола • Н2О в 385 мл ДМА. По завершении процесса добавления реакционные сосуды промывают 77 мл ДМА и перемешивают еще в течение 1 часа при 20-25°С. Далее смесь охлаждают до 5°С и затем добавляют 209,5 г 4-бромметил-2′-цианобифенила в 731,5 мл ДМА (раствор приготовлен при температуре порядка 20°С). Температуру реакционной смеси поддерживают при охлаждении ледяной баней на уровне примерно 5-10°С. После этого промывают 38,5 мл ДМА и перемешивают еще в течение 1 часа при 5-10°С. Растворитель практически полностью отгоняют в вакууме, создаваемом водоструйным насосом, при этом происходит выкристаллизовывание продукта. Остаток охлаждают до 60°С, разбавляют 1925 мл трет-бутилметиловото эфира и в течение 2 часов перемешивают без подвода какой-либо энергии. Кристаллы отделяют вакуум-фильтрацией, промывают сначала 770 мл смеси трет-бутилметилового эфира и ДМА (в соотношении 9:1), а затем 1925 мл воды и дважды порциями по 250 мл трет-бутилметилового эфира, после чего сушат в вакуумном сушильном шкафу при 80°С.
Выход 322,15 г (84,4% от теории), tпл 197-198,5°С, чистота согласно данным ЖХВР-анализа 99,6%.
Пример 5
Телмисартан • HCl
25 г 2-циано-4′-[2′′-н-пропил-4′′-метил-6′′-(1′′′-метилбензимидазол-2′′′-ил)бензимидазол-1′′-илметил]бифенила, 250 мл этиленгликоля, 0,9 мл воды и 24,75 г едкого кали (>85%-ного) совместно загружают в реакционный сосуд и нагревают при перемешивании до 160°С. При этой температуре смесь перемешивают в течение 13,5 ч. Затем растворитель практически полностью отгоняют в вакууме, создаваемом водоструйным насосом, остаток охлаждают до 100°С, разбавляют 50 мл воды и вмешивают в смесь из 125 мл воды и 50 мл концентрированной соляной кислоты (примерно 32%-ной), после чего промывают 50 мл воды. Выкристаллизовывающийся в виде гидрохлорида телмисартан охлаждают до 15-20°С и при этой температуре перемешивают в течение приблизительно 1 часа. После отделения кристаллов вакуум-фильтрацией их промывают 100 мл воды и сушат в вакуумном сушильном шкафу при 100°С.
Выход 27,3 г (98,2% от теории), чистота согласно данным ЖХВР-анализа 99,9%.
Пример 6
Телмисартан • HCl
179 г 2-циано-4′-[2′′-н-пропил-4′′-метил-6′′-(1′′′-метилбензимидазол-2′′′-ил)бензимидазол-1′′-илметил]бифенила предварительно помещают в 1611 мл этиленгликоля, затем добавляют 32,5 мл воды и 178,7 г гидроксида калия (в виде порошка) и при перемешивании нагревают до 150-160°С. При этой температуре смесь перемешивают в течение примерно 15 ч, после чего охлаждают до 100°С. Растворитель практически полностью отгоняют в вакууме, создаваемом водоструйным насосом, остаток охлаждают до 100°С, разбавляют 358 мл воды и вмешивают в смесь из 716 мл воды и 358 мл концентрированной соляной кислоты (примерно 32%-ной), после чего промывают 179 мл воды. Выкристаллизовывающийся в виде гидрохлорида телмисартан перемешивают в течение 1 часа при 60°С, охлаждают до 15-20°С и приблизительно в течение еще одного часа перемешивают при этой температуре. После отделения кристаллов вакуум-фильтрацией их промывают 716 мл воды и сушат в вакуумном сушильном шкафу при 100°С.
Выход 192,1 г (96,5% от теории), чистота согласно данным ЖХВР-анализа 99,9%.
Пример 7
Телмисартан
5,51 г телмисартана • HCl растворяют в 50 мл 40%-ной уксусной кислоты при кипячении с обратным холодильником. Затем коричневый раствор фильтруют в горячем состоянии через 1,1 г угля, промывают 2,5 мл 40%-ной уксусной кислоты и светло-коричневый фильтрат при перемешивании при 80-90°С по каплям смешивают с 2,5 мл 4 н. NaOH. Телмисартан выкристаллизовывается, суспензию разбавляют 30 мл воды и медленно охлаждают до комнатной температуры. Телмисартан отделяют вакуум-фильтрацией и промывают 50 мл воды. В завершение телмисартан сушат в вакуумном сушильном шкафу при 80°С.
Выход 4,80 г (93,3% от теории).
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С НАТРИЕВОЙ СОЛЬЮ ТЕЛМИСАРТАНА | 2004 |
|
RU2372918C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТИАЗОЛА, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1997 |
|
RU2191775C2 |
| ЗАМЕЩЕННОЕ КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ФАРМАКОЛОГИЧЕСКОГО ВОЗДЕЙСТВИЯ, СПОСОБ ИНГИБИРОВАНИЯ 5-ЛИПОКСИГЕНАЗЫ, ИНГИБИРОВАНИЯ ПРОДУКЦИИ ЛИПИДНЫХ ПЕРОКСИДОВ ИЛИ СНИЖЕНИЯ УРОВНЯ САХАРА В КРОВИ | 1998 |
|
RU2196141C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕНЕЛИГЛИПТИНА | 2013 |
|
RU2654069C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С АНТАГОНИСТИЧЕСКОЙ В ОТНОШЕНИИ АНГИОТЕНЗИНА АКТИВНОСТЬЮ НА ИХ ОСНОВЕ | 1995 |
|
RU2139869C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ ГЕТЕРОЦИКЛОМ ПРОИЗВОДНЫХ ПИРИДИНА | 2008 |
|
RU2474581C2 |
| СПОСОБ ПОЛУЧЕНИЯ БЛОКАТОРА АНГИОТЕНЗИНОВОГО РЕЦЕПТОРА | 2003 |
|
RU2412173C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВАЛСАРТАНА | 2003 |
|
RU2348619C2 |
| МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИДАЗИНОНА | 2014 |
|
RU2660435C2 |
| НОВЫЕ ИНГИБИТОРЫ РАССАСЫВАНИЯ КОСТЕЙ И АНТАГОНИСТЫ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2195460C2 |
Изобретение относится к способу получения телмисартана или его гидрохлорида. Способ осуществляют путем взаимодействия 2-н-пропил-4-метил-6-(1′-метилбензимидазол-2′-ил)бензимидазола с соединением общей формулы (IV), в которой Z представляет собой уходящую группу. Полученное таким путем соединение при необходимости подвергают соответствующей переработке. Цианогруппу полученного таким путем соединения 2-циано-4′-[2″-н-пропил-4″-метил-6″-(1″′-метилбензимидазол-2″′-ил)бензимидазол-1″-илметил]бифенила формулы (V) переводят затем посредством гидролиза в кислотную функциональную группу при температуре в интервале от 140 до 200°С в присутствии основания в системе высококипящих растворителей и полученный таким образом телмисартан при необходимости переводят в процессе переработки в гидрохлорид. Технический результат - упрощение переработки, очистки и выделения телмисартана. 11 з.п. ф-лы.
 ,
,
 ,
,
1. Способ получения телмисартана или его гидрохлорида, заключающийся в том, что
(а) 2-н-пропил-4-метил-6-(1′-метилбензимидазол-2′-ил)бензимидазол формулы (I)
подвергают взаимодействию с соединением общей формулы (IV)
 ,
,
в которой Z представляет собой уходящую группу, и полученное таким путем соединение при необходимости подвергают соответствующей переработке,
(б) цианогруппу полученного таким путем соединения 2-циано-4′-[2″-н-пропил-4″-метил-6″-(1″′-метилбензимидазол-2″′-ил)бензимидазол-1″-илметил]бифенил формулы (V)
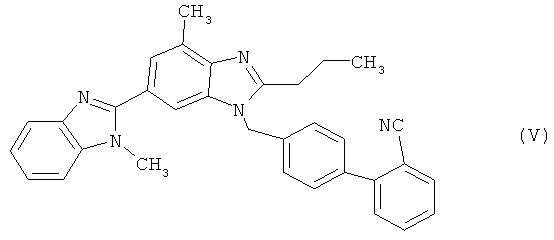 ,
,
переводят затем посредством гидролиза в кислотную функциональную группу при температуре в интервале от 140 до 200°С в присутствии основания в системе высококипящих растворителей, и полученный таким образом телмисартан при необходимости переводят в процессе переработки в гидрохлорид.
2. Способ по п.1, отличающийся тем, что в качестве системы высококипящих растворителей используют систему высококипящих растворителей, выбранных из группы, включающей этиленгликоль/воду и пропиленгликоль/воду, или органический растворитель, выбранный из группы, включающей этиленгликоль, пропиленгликоль, диглим и монометиловый эфир диэтиленгликоля.
3. Способ по п.1 или 2, отличающийся тем, что гидролиз проводят в присутствии основания, выбранного из группы, включающей гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид церия и гидроксид кальция, либо одного из соответствующих ангидридов.
4. Способ по п.1, отличающийся тем, что Z представляет собой атом галогена, предпочтительно атом брома, или замещенную сульфонилоксигруппу.
5. Способ по п.1, отличающийся тем, что стадию (а) проводят в растворителе либо в смеси растворителей, выбранного(-ых) из группы, включающей метиленхлорид, диэтиловый эфир, тетрагидрофуран, диоксан, диметилсульфоксид, диметилформамид, диметилацетамид, диметилформамид/трет-бутанол, диметилацетамид/трет-бутанол, толуол и бензол, при необходимости в присутствии связывающего кислоту агента.
6. Способ по п.5, отличающийся тем, что связывающий кислоту агент выбран из группы, включающей карбонат натрия, карбонат калия, гидроксид натрия, гидроксид калия, метилат натрия, метилат калия, трет-пентилат калия, трет-бутилат калия, н-бутилат калия, гидрид натрия, триэтиламин и пиридин.
7. Способ по п.5, отличающийся тем, что стадию (а) проводят при температуре в интервале от 0 до 30°С.
8. Способ по одному из пп.1-7, отличающийся тем, что на стадии (а) взаимодействие соединения формулы (I) с соединением общей формулы (IV) проводят в растворителе либо в смеси растворителей, выбранного(-ых) из группы, включающей диметилсульфоксид, диметилформамид, диметилацетамид, диметилформамид/трет-бутанол и диметилацетамид/трет-бутанол, в присутствии гидроксида натрия, гидрида натрия, гидроксида калия или трет-бутоксида калия.
9. Способ по одному из пп.1-8, отличающийся тем, что по завершении стадии (а) растворитель удаляют, остаток обрабатывают растворителем, в котором нитрил (V) проявляет лишь ограниченную растворимость, а под тепловым воздействием - соответственно умеренную растворимость, выпавшие при определенных условиях после охлаждения кристаллы отделяют вакуум-фильтрацией и при необходимости промывают и продукт при необходимости сушат при повышенной температуре.
10. Способ по п.9, отличающийся тем, что в качестве растворителя для обработки остатка используют спирт, ароматический углеводород, простой эфир или воду.
11. Способ по одному из пп.1-10, отличающийся тем, что для переработки по завершении стадии (б) растворитель удаляют, остаток необязательно разбавляют водой и растворяют с помощью водной соляной кислоты, выкристаллизовывающийся в виде гидрохлорида телмисартан при необходимости охлаждают, затем отделяют вакуум-фильтрацией и при определенных условиях сушат.
12. Способ по п.11, отличающийся тем, что полученный в виде гидрохлорида телмисартан переводят в кислотную форму.
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АНГИОТЕНЗИНА, НА ИХ ОСНОВЕ | 1992 |
|
RU2053229C1 |
| Ries U.J | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |

