Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям формулы I, фармацевтическим композициям, содержащим указанные соединения, и к применению указанных соединений в терапии. Кроме того, настоящее изобретение относится к способам получения соединений формулы I.
Предпосылки создания изобретения
Нейрокинины, также известные как тахикинины, составляют класс пептидных нейротрансмиттеров, которые найдены в периферической и центральной нервной системах. Тремя главными тахикининами являются Субстанция Р (SP), Нейрокинин А (NKA) и Нейрокинин В (NKB). По меньшей мере три типа рецепторов известны для этих трех главных тахикининов. На основании их относительной селективности с предпочтением к агонистам SP, NKA и NKB рецепторы классифицируют как рецепторы нейрокининов 1 (NK1), рецепторы нейрокининов 2 (NK2) и рецепторы нейрокининов 3 (NK3) соответственно.
Имеется необходимость в орально активном антагонисте рецептора NK для лечения, например, респираторных, сердечно-сосудистых, неврологических, болезненных, онкологических, воспалительных и/или желудочно-кишечных расстройств. Для повышения терапевтического индекса такой терапии желательно получить такое соединение, не обладающее токсичностью или обладающее минимальной токсичностью, а также селективное к указанным рецепторам NK. Кроме того, считают необходимым, чтобы указанный медикамент имел благоприятные фармакокинетические и метаболические свойства, тем самым обеспечивая улучшенный терапевтический и безопасный профиль с более низкой способностью ингибировать ферменты печени.
Хорошо известно, что определенные соединения могут нежелательным образом влиять на реполяризацию сердца у человека, что наблюдается как удлинение интервала QT на электрокардиограмме (ЭКГ). В крайних обстоятельствах это удлинение интервала QT, индуцированное лекарственным средством, может приводить к типу сердечной аритмии, называемому пируэтной аритмией («Torsades de Pointes», TdP; Vandenberg et al. hERG K+ channels: friend и foe. (К+-каналы hERG: друг и враг) Trends Pharmacol Sci 2001; 22: 240-246), в крайних случаях приводя к фибрилляции желудочков и внезапной смерти. Первичным явлением в этом синдроме является ингибирование быстрого компонента замедленного выпрямляющего калиевого тока (delayed rectifying potassium current, IKr) этими соединениями. Эти соединения связываются с белковыми альфа-субъединицами, формирующими пору канала, проводящего этот ток. Альфа-субъединицы, образующие эту пору, кодируются геном, обозначаемым как «human ether-a-go-go-related gene» (hERG). Поскольку IKr играет ключевую роль в реполяризации потенциала действия в сердце, его ингибирование замедляет реполяризацию, и это проявляется удлинением интервала QT. Хотя удлинение интервала QT само по себе не опасно, оно несет риск побочных сердечно-сосудистых эффектов, и у небольшой части людей оно может приводить к TdP с перерождением в фибрилляцию желудочков.
В частности, желательно, чтобы антагонист рецептора NK имел приемлемый баланс фармакодинамических и фармакокинетических свойств, который делал бы его терапевтически полезным. В дополнение к обладанию достаточной и селективной активностью, антагонист рецептора NK должен быть сбалансированным в отношении важных фармакокинетических свойств. Так, необходимо, чтобы антагонист рецептора NK имел: а) достаточно высокое сродство к различным рецепторам NK, b) фармакокинетические свойства (характеристики абсорбции, распределения и элиминации), которые делают лекарственное средство способным действовать на NK-рецепторы-мишени главным образом на периферии. Например, антагонист рецептора NK должен обладать достаточно высокой метаболической стабильностью, с) достаточно низким сродством к другим ионным каналам, таким как кодируемый hERG калиевый канал, для получения приемлемого профиля безопасности, и d) низким уровнем ингибирования ферментов печени (таких как CYP3A4) для предупреждения межлекарственных взаимодействий.
Кроме того, для повышения эффективности антагониста рецептора NK выгодно иметь антагонист NK с длительным конкурентным действием на рецептор.
EP 0625509, EP 0630887, WO 95/05377, WO 95/12577, WO 95/15961, WO 96/24582, WO 00/02859, WO 00/20003, WO 00/20389, WO 00/25766, WO 00/34243, WO 02/51807 и WO 03/037889 раскрывают производные пиперидинилбутиламида, которые являются антагонистами тахикининов.
"4-Амино-2-(арил)-бутилбензамиды и их конформационно заторможенные аналоги. Сильные антагонисты человеческого рецептора нейрокинина-2 (NK2)" Roderick MacKenzie, A., et al., Bioorganic & Medicinal Chemistry Letters (2003), 13, 2211-2215, раскрывает соединение N-[2-(3,4-дихлорфенил)-4-(3-морфолин-4-илазетидин-1-ил)бутил]-N-метилбензамид, который, как было найдено, обладает антагонистическими свойствами в отношении функционального рецептора NK2.
WO 96/05193, WO 97/27185 и EP 0962457 раскрывают производные азетидинилалкиллактама с активностью антагонистов тахикининов.
EP 0790248 раскрывает азетидинилалкилазапиперидоны и азетидинилалкилоксапиперидоны, которые, как утверждают, являются антагонистами тахикининов.
WO 99/01451 и WO 97/25322 раскрывают производные азетидинилалкилпиперидина, про которые заявлено, что они являются антагонистами тахикининов.
EP 0791592 раскрывает азетидинилалкилглутаримиды с антагонистическими свойствами в отношении тахикининов.
WO 2004/110344 A2 раскрывает двойные антагонисты NK1,2 и их применение.
Целью настоящего изобретения было создание новых антагонистов нейрокининов, применимых в терапии. Следующей целью было создание новых соединений, имеющих хорошо сбалансированные фармакокинетические и фармакодинамические свойства.
Описание изобретения
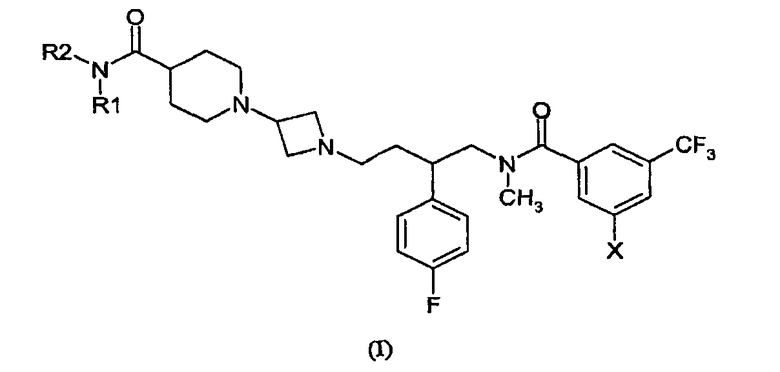
Настоящее изобретение предоставляет соединение общей формулы (I)
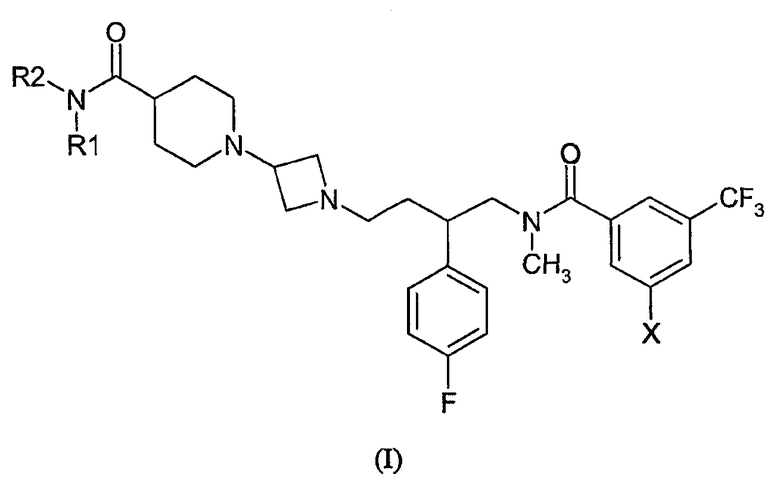
где
каждый R1 и R2 независимо выбран из водорода, метила, этила, или R1 и R2 образуют четырех-, пяти- или шестичленное кольцо вместе с амидным азотом, причем указанное кольцо необязательно содержит атом кислорода;
X представляет собой бром или хлор;
а также их фармацевтически и фармакологически приемлемые соли и энантиомеры соединения формулы I и их соли.
В одном варианте осуществления R1 и R2 вместе с амидным азотом образуют морфолиновое кольцо.
В одном варианте осуществления R1 и R2 вместе с амидным азотом образуют азетидиновое кольцо.
В одном варианте осуществления R1 и R2 вместе с амидным азотом образуют пирролидиновое кольцо.
В одном варианте осуществления R1 и R2 вместе с амидным азотом образуют изоксазолидиновое кольцо.
В одном варианте осуществления R1 и R2 вместе с амидным азотом образуют оксазолидиновое кольцо.
Настоящее изобретение относится к соединениям формулы I, как определено выше, а также к его солям. Соли для применения в фармацевтических композициях будут фармацевтически приемлемыми солями, но для получения соединений формулы I могут быть полезными другие соли.
Соединения настоящего изобретения способны образовывать соли с различными неорганическими и органическими кислотами, и такие соли также входят в объем настоящего изобретения. Примеры таких кислотно-аддитивных солей включают ацетат, адипат, аскорбат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, цитрат, циклогексилсульфамат, этансульфонат, фумарат, глутамат, гликолат, полусульфат, 2-гидроксиэтилсульфонат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, гидроксималеат, лактат, малат, малеат, метансульфонат, 2-нафталинсульфонат, нитрат, оксалат, пальмоат, персульфат, фенилацетат, фосфат, пикрат, пивалат, пропионат, хинат, салицилат, стеарат, сукцинат, сульфамат, сульфанилат, сульфат, тартрат, тозилат (п-толуолсульфонат) и ундеканоат.
Фармацевтически приемлемые соли могут быть получены из соответствующих кислот традиционным образом. Соли, не являющиеся фармацевтически приемлемыми, могут быть полезными в качестве промежуточных соединений и как таковые являются другим аспектом настоящего изобретения.
Кислотно-аддитивные соли могут быть также в форме полимерных солей, таких как полимерные сульфонаты.
Соли могут быть образованы традиционными средствами, такими как реакция свободной основной формы продукта с одним или более эквивалентов соответствующей кислоты в растворителе или среде, в которой соль плохо растворима, или в растворителе, таком как вода, который удаляют в вакууме или лиофилизацией, или обменом анионов существующей соли на другой анион на приемлемой ионообменной смоле.
Соединения формулы I имеют один хиральный центр, и следует понимать, что настоящее изобретение охватывает все оптические изомеры и энантиомеры. Соединения по формуле (I) могут быть в форме отдельных стереоизомеров, т.е. отдельного энантиомера (R-энантиомера или S-энантиомера). Соединения по формуле (I) могут также быть в форме рацемической смеси, т.е. эквимолярной смеси энантиомеров.
Эти соединения могут также существовать в виде смеси конформационных изомеров. Соединения настоящего изобретения включают как индивидуальные конформационные изомеры, так и их смеси.
Фармацевтические композиции
Согласно одному аспекту настоящего изобретения создают фармацевтическую композицию, содержащую соединение формулы I в виде отдельного энантиомера, рацемата или их смеси, в виде свободного основания или его фармацевтически приемлемой соли, для применения при предупреждении и/или лечении респираторных, сердечно-сосудистых, неврологических, болезненных, онкологических, воспалительных и/или желудочно-кишечных расстройств.
Фармацевтические композиции настоящего изобретения вводят стандартным образом для состояния заболевания, которое желают лечить, например пероральным, местным, парентеральным, буккальным, интраназальным, интравагинальным или ректальным введением или ингаляцией или вдуванием. Для этих целей соединения настоящего изобретения могут быть приготовлены средствами, известными в данной области, например, в форме таблеток, гранул, капсул, водных или масляных растворов, суспензий, эмульсий, кремов, мазей, гелей, интраназальных аэрозолей, суппозиториев, тонко измельченных порошков или аэрозолей или распыленных материалов для ингаляции и стерильных водных или масляных растворов или суспензий или стерильных эмульсий для парентерального применения (включая внутривенное, внутримышечное или инфузию).
В дополнение к соединениям настоящего изобретения фармацевтическая композиция настоящего изобретения может также содержать или ее можно вводить (одновременно или последовательно) вместе с одним или более фармакологических агентов, способных терапевтически воздействовать на одно или более состояний заболеваний, указанных в настоящем документе.
Фармацевтические композиции настоящего изобретения можно нормально вводить людям в дневной дозе соединения формулы I от 0,01 до 25 мг/кг массы тела. В качестве альтернативы вводят дневную дозу соединения формулы I от 0,1 до 5 мг/кг массы тела. Эту дневную дозу можно при необходимости давать разделенными дозами, причем точное количество вводимого соединения и путь введения зависят от массы тела, возраста и пола пациента, принимающего лечение, и от конкретного состояния заболевания, подвергающегося лечению по правилам, известным в данной области.
Типичные единичные дозированные формы будут содержать примерно от 1 мг до 500 мг соединения настоящего изобретения. Например, таблетка или капсула для перорального введения может содержать до 250 мг (обычно 5-100 мг) соединения формулы I или его фармацевтически приемлемой соли. В другом примере, для введения ингаляцией, соединение формулы (I) или его фармацевтически приемлемую соль можно вводить в диапазоне дневных доз от 5 до 100 мг в одной дозе или разделенным на две-четыре дневные дозы. В следующем примере, для введения внутривенной или внутримышечной инъекцией или инфузией, можно применять стерильный раствор или суспензию, содержащую до 10% по массе (обычно 5% по массе) соединения формулы (I) или его фармацевтически приемлемой соли.
Медицинское и фармацевтическое применение
Настоящее изобретение предоставляет способ лечения или предупреждения состояния заболевания, при котором полезен антагонизм в отношении тахикининов, действующих на рецепторы NK, причем этот способ включает в себя введение субъекту эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение также обеспечивает применение соединения формулы (I) или его фармацевтически приемлемой соли при получении медикамента для применения при состоянии заболевания, при котором полезен антагонизм в отношении тахикининов, действующих на рецепторы NK.
Соединения формулы (I) или их фармацевтически приемлемые соли или сольваты можно применять для изготовления медикамента для применения при предупреждении или лечении респираторных, сердечно-сосудистых, неврологических, болезненных, онкологических и/или желудочно-кишечных расстройств.
Примерами таких расстройств являются астма, аллергический ринит, заболевания легких, кашель, простуда, воспаление, хроническое обструктивное заболевание легких, реактивность дыхательных путей, крапивница, гипертензия, ревматоидный артрит, отек, ангиогенез, боль, мигрень, головная боль при повышенном давлении, психозы, депрессия, тревожность, болезнь Альцгеймера, шизофрения, болезнь Гентингтона, гипермотильность мочевого пузыря, недержание мочи, расстройства пищевого поведения, маниакальная депрессия, зависимость от психоактивных веществ, расстройство движения, расстройство сознания, ожирение, стрессовые расстройства, расстройства мочеиспускания, мания, гипомания и агрессивность, биполярное расстройство, рак, карцинома, фибромиалгия, некардиогенная боль в груди, желудочно-кишечная гипермотильность, желудочная астма, болезнь Крона, расстройства опорожнения желудка, язвенный колит, синдром раздраженной толстой кишки (IBS), воспалительная болезнь кишечника (IBD), рвота, желудочная астма, расстройства мотильности желудка, гастроэзофагеальная рефлюксная болезнь (GERD) или функциональная диспепсия.
Способы получения
В другом аспекте настоящее изобретение предоставляет способ получения соединения формулы (I) или его солей, включающий в себя:
а) реакцию соединения формулы (II) с соединением формулы (III):
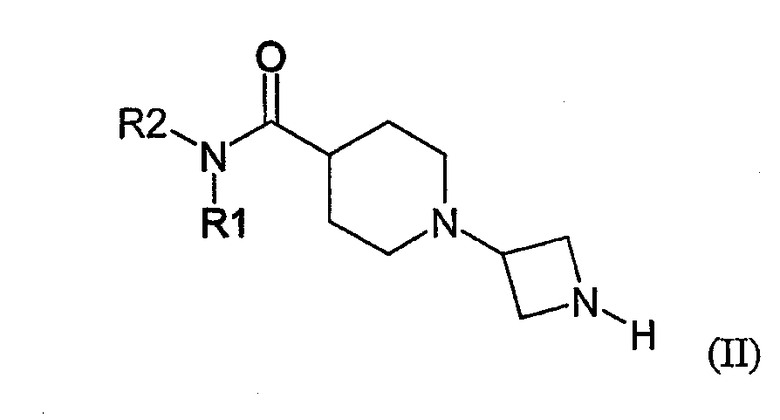
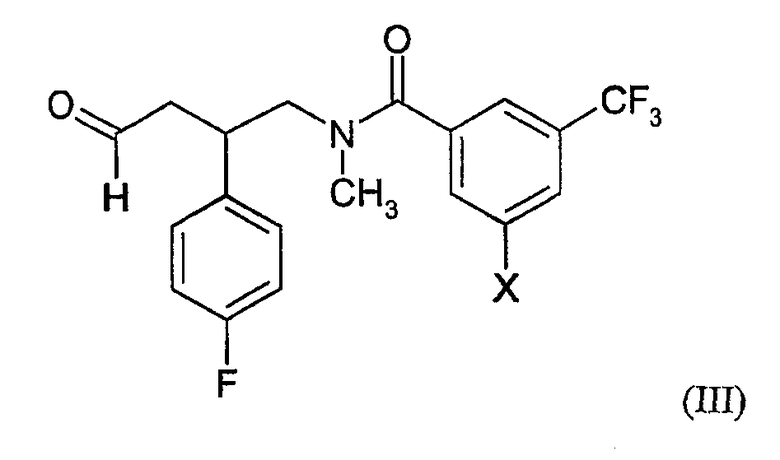
где R1, R2 и X определены выше в настоящем документе; и условия такие, что восстановительное алкилирование соединения формулы (II) образует связь N-C между атомом азота соединения формулы (II) и атомом углерода альдегидной группы соединения формулы (III); или
b) реакцию соединения формулы (II) с соединением формулы (IV):
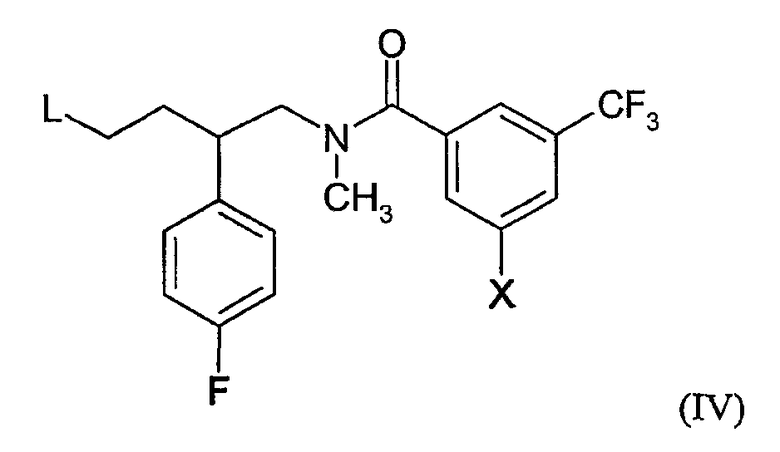
где X определен выше в настоящем документе; и L является такой группой, что алкилирование соединения формулы (II) образует связь N-C между атомом азота азетидиновой группы соединения формулы (II) и атомом углерода соединений формулы (IV), который является соседним с группой L; или
c) реакцию соединения формулы (V) с соединением формулы (VI):
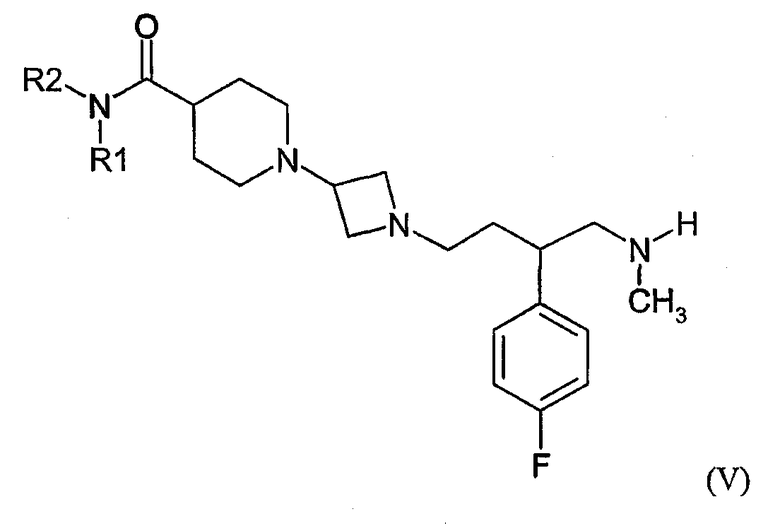
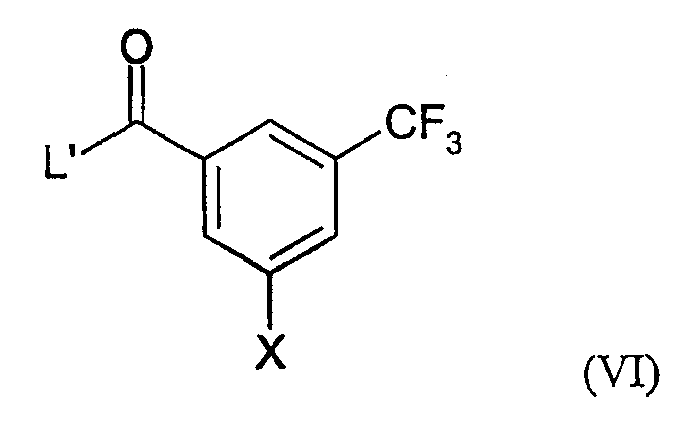
где R1, R2 и X определены выше в настоящем документе; и L' является уходящей группой; и необязательно образование фармацевтически приемлемой соли.
Соединения формул (II) и (III) реагируют в условиях восстановительного алкилирования. Эту реакцию обычно проводят при неэкстремальной температуре, например при 0-10°С, в существенно инертном растворителе, например дихлорметане. Типичные восстановители включают боргидриды, такие как цианоборгидрид натрия.
Соединения формул (II) и (IV) реагируют в условиях алкилирования. Обычно в соединениях формулы (IV) L является уходящей группой, такой как галоген или алкилсульфонилоксигруппа. Реакцию обычно проводят при повышенной температуре, например 30-130°С, в существенно инертном растворителе, например DMF.
Соединения формулы (II) можно получить традиционным образом, например по реакции соединения формулы VII:
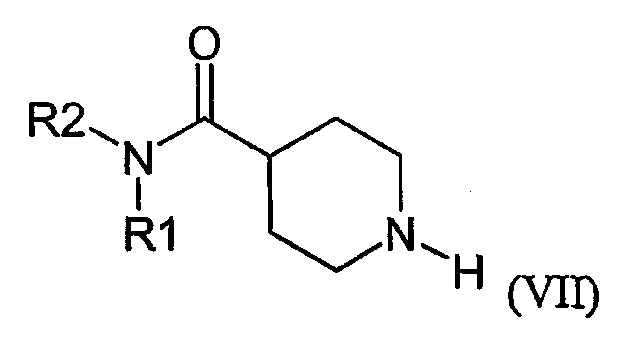
с соединением формулы (VIII):
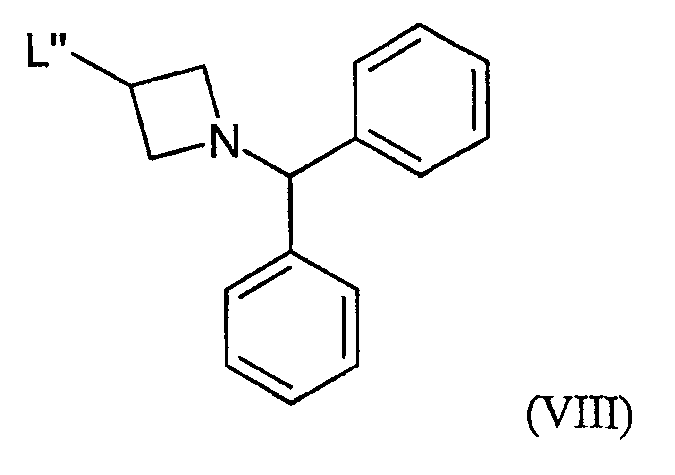
где R1 и R2 определены выше в настоящем документе; и L" является такой группой, что алкилирование соединения формулы (VII) образует связь N-C между атомом азота пиперидиновой группы соединения формулы (VII) и атомом углерода соединения формулы (VIII), который является соседним к группе L"; и последующим удалением защитной группы (-CH(Ph)2), например, реакцией каталитического гидрирования.
Соединения формулы (III) можно получить, например, по реакции соединения формулы (IX) с соединением формулы (VI):
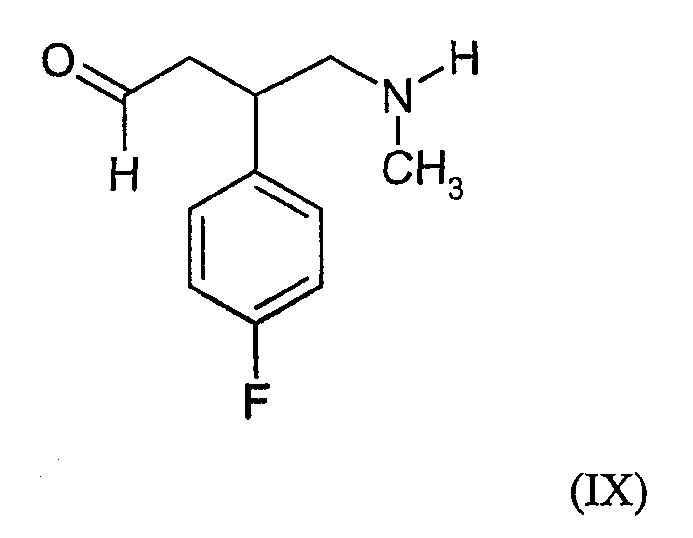
где R1 определен выше в настоящем документе, в обычных условиях ацилирования.
Соединения формулы (IV) можно получить, например, по реакции соединения формулы (VI) с соединением формулы (Х):
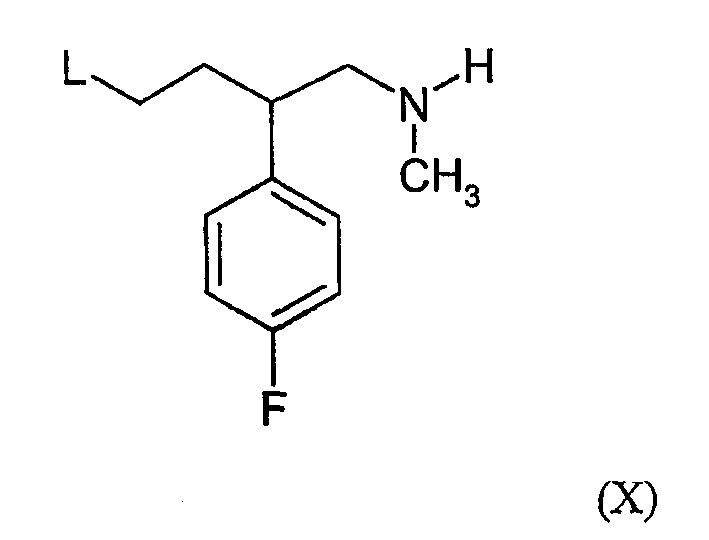
где L определена выше в настоящем документе, в обычных условиях ацилирования.
Соединения формул (V) и (VI) могут реагировать в обычных условиях ацилирования, где
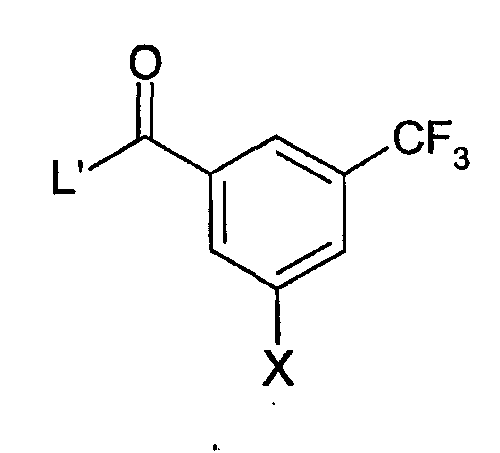
является кислотой или активированным производным кислоты. Такие активированные производные кислоты хорошо известны в литературе. Они могу быть образованы in situ из кислоты или их можно получить, выделить и затем ввести в реакцию. Обычно L' является хлором и поэтому образует кислый хлорид. Обычно реакцию ацилирования проводят в присутствии ненуклеофильного основания, например N,N-диизопропилэтиламина, в существенно инертном растворителе, таком как дихлорметан, при неэкстремальной температуре.
Соединения формул (VII) и (VIII) известны или могут быть получены традиционным образом.
Примеры
Следует подчеркнуть, что соединения настоящего изобретения чаще всего показывают сложные спектры ЯМР вследствие существования конформационных изомеров. Считают, что это является результатом медленного вращения вокруг амидной и/или арильной связи. При представлении данных ЯМР этих соединений использованы следующие сокращения: с - синглет; д - дублет; т - триплет; квт - квартет; квн - квинтет; м - мультиплет; шир. - широкий; слож. м - сложный мультиплет, который может включать в себя широкие пики.
Следующие примеры описывают, но не ограничивают настоящее изобретение.
В экспериментальной части применяли следующие сокращения: DIPEA (N,N-диизопропилэтиламин), TBTU (тетрафторборат N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)урония), DMF (N,N-диметилформамид), THF (тетрагидрофуран) и RT (room temperature, комнатная температура).
Пример 1
N-[(2S)-4-{3-[4-(Азетидин-1-илкарбонил)пиперидин-1-ил]азетидин-1-ил}-2-(4-фторфенил)бутил]-3-бром-N-метил-5-(трифторметил)бензамид
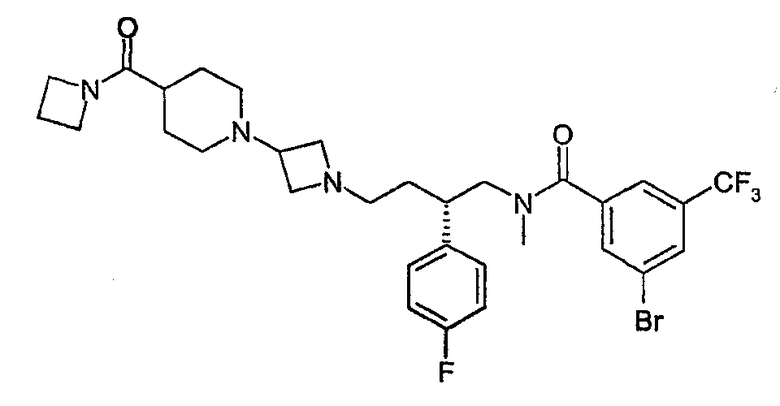
3-Бром-N-[(2S)-2-(4-фторфенил)-4-оксобутил]-N-метил-5-(трифторметил)бензамид (см. способ 1; 0,16 г, 0,36 ммоль) и 1-азетидин-3-ил-4-(азетидин-1-илкарбонил)пиперидин (см. способ 2; 0,10 г, 0,47 ммоль) растворяли в метиленхлориде (10 мл) вместе с небольшим количеством сухого метанола (0,2 мл). К полученному раствору добавляли DIPEA (0,14 г, 1,08 ммоль) и триацетоксиборгидрид натрия (0,15 г, 0,72 ммоль). Смесь 4 ч перемешивали в атмосфере азота при комнатной температуре. Смесь разбавляли метиленхлоридом и дважды промывали насыщенным водным NaHCO3 и затем раствором соли. Органическую фазу фильтровали через фазоразделитель и растворитель удаляли выпариванием. Продукт очищали хроматографией на силикагеле (метанол-метиленхлорид, 10:1). Получено 0,14 г (59%) соединения, указанного в заголовке, в виде белого пенообразного вещества. 1H ЯМР (500 МГц, CDCl3): δ 1,4-1,8 (слож.м, 6H), 2,1 (м, 1H) 2,2 (квн, 2H), 2,3-2,4 (слож.м, 2H), 2,5-3,5 (слож.м, 14H), 3,6 (д, 1H), 3,9 (т, 2H), 4,1 (т, 2H), 6,8-7,4 (слож.м, 6H), 7,7 (с, 1H); LCMS: m/z 654 (M+1)+.
Пример 2
Диформиат 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}-N,N-диметилпиперидин-4-карбоксамида
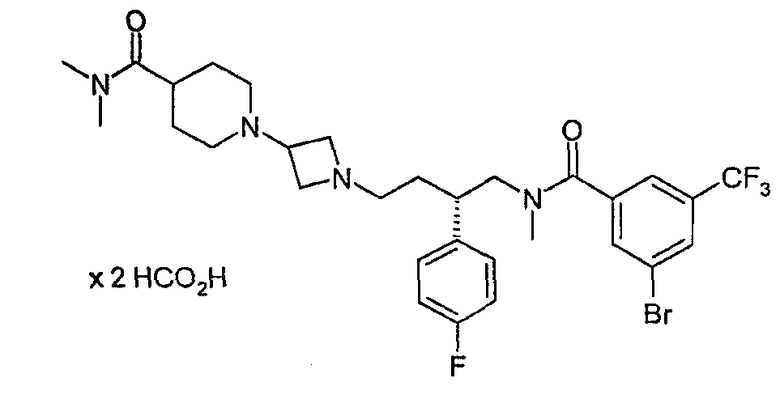
Смесь 3-бром-N-[(2S)-2-(4-фторфенил)-4-оксобутил]-N-метил-5-(трифторметил)бензамида (см. способ 1; 0,178 г, 0,40 ммоль), 1-азетидин-3-ил-N,N-диметилпиперидин-4-карбоксамида (см. способ 3; 0,084 г, 0,40 ммоль), уксусной кислоты (0,3 мл), цианоборгидрида (полистирилметил)триметиламмония (0,098 г, 0,52 ммоль) и метанола 6 ч перемешивали при комнатной температуре. Смолу отфильтровывали и промывали метанолом. Растворитель фильтрата удаляли выпариванием и продукт очищали обращенно-фазовой хроматографией (С8), применяя ацетонитрил и водный раствор формиата аммония с муравьиной кислотой (0,1 M NH4CO2H, 0,1 M HCO2H, pH 4) в качестве элюента. Получено 0,23 г (77%) соединения, указанного в заголовке. 1H ЯМР (500 МГц, CD3OD): δ 1,6-2,0 (слож.м, 6H), 2,6-4,2 (слож.м, 24H), 7,0-8,0 (слож.м, 6H), 8,4 (с, 1H); LCMS: m/z 642 (M+1)+.
Пример 3
1-{1-[(3S)-4-[[3-Бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамид
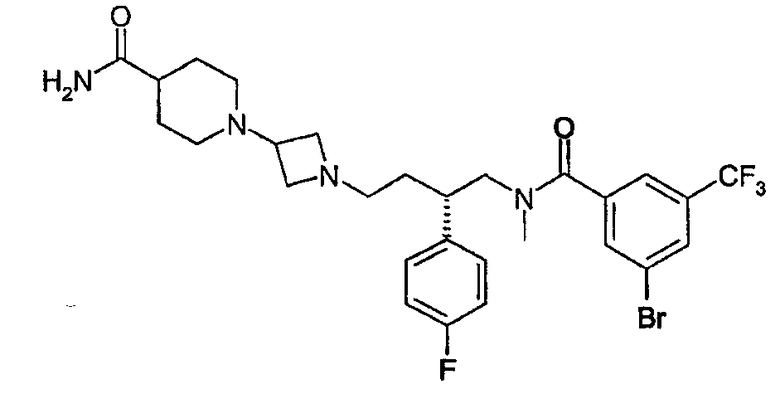
3-Бром-N-[(2S)-2-(4-фторфенил)-4-оксобутил]-N-метил-5-(трифторметил)бензамид (см. способ 1; 1,00 г, 2,24 ммоль) и 1-азетидин-3-илпиперидин-4-карбоксамид (см. способ 4; 0,49 г, 2,69 ммоль) и триэтиламин (1,24 мл, 9,0 ммоль) растворяли в метиленхлориде (30 мл) вместе с метанолом (5 мл). К полученному раствору добавляли цианоборгидрид натрия (0,21 г, 3,36 ммоль) и смесь 20 мин перемешивали при комнатной температуре. Растворитель удаляли выпариванием и остаток распределяли между метиленхлоридом и насыщенным водным NaHCO3. Органическую фазу фильтровали через фазоразделитель и растворитель удаляли выпариванием. Продукт очищали хроматографией на силикагеле (смесь метанола, насыщенного аммиаком, и метиленхлорида, 1-20% метанола). Получено 0,24 г (17%), соединения, указанного в заголовке. 1H ЯМР (500 МГц, CDCl3): δ 1,4-3,8 (слож.м, 23H), 5,7 (шир., 1H), 5,8 (шир., 1H), 6,8-7,4 (слож.м, 6H), 7,7 (с, 1H); LCMS: m/z 614 (M+1)+.
Пример 3a
Для получения дополнительной информации о существующей твердой форме провели суспензионную кристаллизацию при комнатной температуре в различных растворителях. Спустя приблизительно 2 недели твердую форму исследовали с помощью XRPD. Образцы, суспендированные в метаноле, этаноле, изопропаноле, ацетоне и хлороформе, показывают очень сходные картины при XRPD, отличные от исходного образца. Новая форма обладает заметно лучшей кристалличностью. Материал, суспендированный в этилметилкетоне, показывает совершенно уникальную картину. Высокотемпературные XRPD, проведенные на образце, суспендированном в изопропаноле, показывают, что порошковая рентгенограмма изменяется выше 120°С. Новая картина также отлична от исходного образца.
Малеатная соль 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида
1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамид (2,0 г, 3,26 ммоль) растворяли в горячем ацетоне (20 мл). Малеиновую кислоту (0,74 г, 6,4 ммоль) растворяли в горячем метаноле (4 мл) и этот раствор затем добавляли к первому раствору. Объединенные растворы оставляли при комнатной температуре на ночь, однако не смогли выделить никакого полезного осадка. Смесь разбавляли метанолом и растворитель удаляли выпариванием. Остаток прибавляли к смеси толуола (17 мл) и 2-пропанола (50 мл). Добавляли метанол (20 мл) и смесь нагревали до получения прозрачного раствора. Раствор охлаждали до комнатной температуры и затем выдерживали в холодильнике в течение ночи. Белый осадок выделяли фильтрацией и затем сушили при пониженном давлении в течение 48 ч. Получено 2,4 г соединения, указанного в заголовке, в виде белого порошка. 1H-ЯМР-анализ продукта показывает, что образец состоит из приблизительно 1,5-2 молей малеиновой кислоты на моль 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида. 1H ЯМР (500 МГц, D2O): δ 1,2 (д, 1,6 H), 1,8-2,2 (слож.м, 5,8H), 2,6-2,7 (м, 1H), 2,7 (с, 1H), 2,8-3,2 (слож.м, 5,1H), 3,2-3,3 (м, 1H), 3,3-3,5 (слож.м, 2,3H), 3,5-3,8 (м, 1,4H), 3,9-4,1 (м, 0,6H), 4,2-4,7 (слож.м, 4,6H), 6,3 (с, 2,9H), 6,9-7,3 (м, 4,2H), 7,4 (м, 1H), 8,0 (с, 0,6H).
Малеатная соль 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида характеризуется тем, что она дает порошковую рентгеновскую дифрактограмму, демонстрирующую существенно следующие главные пики со значениями d (значение d - расстояние между последовательными параллельными hkl-плоскостями в кристаллической решетке):
Пики, идентифицированные по значениям d, рассчитанным по формуле Брэгга, и интенсивностям, вычитали из дифрактограммы малеата 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида. Относительные интенсивности менее надежны, и вместо численных значений применяли следующие определения:
Исходную форму также получали суспендированием в воде, н-гептане, ацетонитриле, изооктане, THF и метилизобутилкетоне при комнатной температуре.
Форма малеатной соли 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамид после суспендирования
Образцы, суспендированные в метаноле, этаноле, изопропаноле, ацетоне и хлороформе, показывают очень сходные картины при XRPD, которые отличаются от исходного образца. Эта форма характеризуется тем, что она дает порошковую рентгеновскую дифрактограмму, демонстрирующую существенно следующие главные пики со значениями d (значение d - расстояние между последовательными параллельными hkl-плоскостями в кристаллической решетке):
Пики, идентифицированные по значениям d, рассчитанным по формуле Брэгга, и интенсивностям, вычитали из дифрактограммы малеата 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида. Относительные интенсивности менее надежны, и вместо численных значений применяли следующие определения:
Форма малеата 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида после высокотемпературной XRPD
XRPD, проведенные на образце, суспендированном в изопропаноле, показывают, что порошковая рентгенограмма изменяется выше 120°С. Новая картина также отлична от исходного образца.
Эта форма характеризуется тем, что она дает порошковую рентгеновскую дифрактограмму, демонстрирующую существенно следующие главные пики со значениями d (значение d - расстояние между последовательными параллельными hkl-плоскостями в кристаллической решетке):
Пики, идентифицированные по значениям d, рассчитанным по формуле Брэгга, и интенсивностям, вычитали из дифрактограммы малеата 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида после высокотемпературной XRPD. Относительные интенсивности менее надежны, и вместо численных значений применяли следующие определения:
Форма малеата 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида после суспендирования в метилэтилкетоне
Следующая форма 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида была получена после суспендирования в метилэтилкетоне. Эта форма характеризуется тем, что она дает порошковую рентгеновскую дифрактограмму, демонстрирующую существенно следующие главные пики со значениями d (значение d - расстояние между последовательными параллельными hkl-плоскостями в кристаллической решетке):
Пики, идентифицированные по значениям d, рассчитанным по формуле Брэгга, и интенсивностям, вычитали из дифрактограммы малеата 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида после суспендирования в метилэтилкетоне. Относительные интенсивности менее надежны, и вместо численных значений применяли следующие определения:
Рентгеновская порошковая дифрактометрия (XRPD)
Эксперименты с XRPD проводили на дифрактометре D8 Advance (Bruxer AXS GmbH, Karlsruhe, Германия) с геометрией Брэгга-Брентано, оснащенном позиционно-чувствительным детектором (PSD) VANTEC-1. Применяли Kα-излучение Cu, отфильтрованное никелем. Образцы, приблизительно 10 мг, помещали на держатель с нулевым фоном (кристалл кремния). Данные собирали, применяя режим непрерывного сканирования в диапазоне 1-50° 2θ с угловым шагом в 0,017° и временным шагом в 0,5 с. Применяли переменную (V20) вертикальную щель и приемную щель 12 мм, соответствующую окну детектора шириной 3,47°.
Высокотемпературную XRPD проводили на приборе, описанном выше, применяя сходные настройки, с MRI-камерой (Bruxer AXS GmbH, Karlsruhe, Германия), соединенной с контроллером температуры Ansyco.
Пример 4
3-Бром-N-((2S)-2-(4-фторфенил)-4-{3-[4-(морфолин-4-илкарбонил)пиперидин-1-ил]азетидин-1-ил}бутил)-N-метил-5-(трифторметил)бензамид
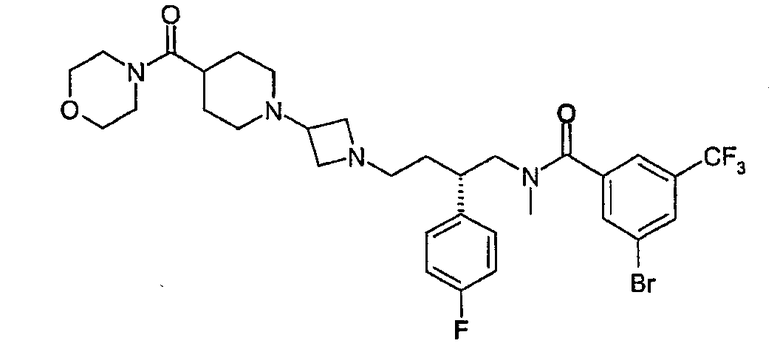
3-Бром-N-[(2S)-2-(4-фторфенил)-4-оксобутил]-N-метил-5-(трифторметил)бензамид (см. способ 1; 0,14 г, 0,31 ммоль) и 4-[(1-азетидин-3-илпиперидин-4-ил)карбонил]морфолин (см. способ 5; 0,11 г, 0,42 ммоль) растворяли в метиленхлориде (10 мл) вместе с сухим метанолом (0,2 мл). К полученному раствору добавляли DIPEA (0,12 г, 0,94 ммоль) и триацетоксиборгидрид натрия (0,13 г, 0,63 ммоль). Смесь 4 ч перемешивали в атмосфере азота при комнатной температуре. Смесь разбавляли метиленхлоридом и дважды промывали насыщенным водным NaHCO3 и затем раствором соли. Органическую фазу фильтровали через фазоразделитель и растворитель удаляли выпариванием. Продукт очищали хроматографией на силикагеле (метанол-метиленхлорид, 10:1). Получено 0,11 г (53%) соединения, указанного в заголовке, в виде белого пенообразного вещества. 1H ЯМР (500 МГц, CDCl3): δ 1,4-3,8 (слож.м, 32H), 6,8-7,4 (слож.м, 6H), 7,7 (с, 1H); LCMS: m/z 684 (M+1)+.
Получение исходных материалов
Исходные материалы для вышеописанных примеров являются либо коммерчески доступными, либо могут быть легко получены стандартными способами из известных материалов. Например, следующие реакции являются иллюстрацией, но не ограничением, некоторых из исходных материалов.
Способ 1
3-Бром-N-[(2S)-2-(4-фторфенил)-4-оксобутил]-N-метил-5-(трифторметил)бензамид
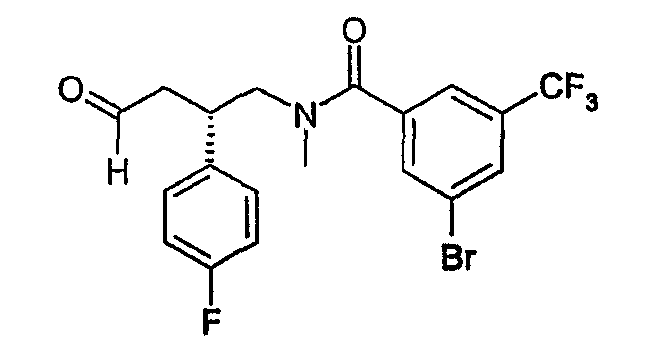
(a) 3-Бром-N-[(2S)-2-(4-фторфенил)пент-4-ен-1-ил]-N-метил-5-(трифторметил)бензамид
К раствору [(2S)-2-(4-фторфенил)пент-4-ен-1-ил]метиламина (см. Bioorg. Med. Chem. Lett; 2001; 265-270; 0,54 г, 2,8 ммоль) и 3-бром-5-трифторметилбензойной кислоты (0,81 г, 3,0 ммоль) в DMF (7 мл) добавляли TBTU (0,96 г, 3,0 ммоль) и DIPEA (1,41 г, 10,9 ммоль). Реакционную смесь перемешивали в атмосфере азота в течение ночи при комнатной температуре и затем распределяли между этилацетатом и водным раствором NaHCO3. Водную фазу трижды экстрагировали этилацетатом. Объединенные органические растворы трижды промывали водой и затем осушали на колонке фазоразделителя. Растворитель удаляли выпариванием и продукт очищали хроматографией на силикагеле (этилацетат-гептан от 10% до 17%). Получено 0,86 г (68%) 3-бром-N-[(2S)-2-(4-фторфенил)пент-4-ен-1-ил]-N-метил-5-(трифторметил)бензамида. 1H ЯМР (500 МГц, CDCl3): 2,1-3,8 (слож.м, 8H), 4,9-5,1 (м, 2H), 5,5-5,8 (м, 1H), 6,8-7,4 (слож.м, 6H), 7,8 (с, 1H). LCMS: m/z 445 (M+1)+.
(b) 3-Бром-N-[(2S)-2-(4-фторфенил)-4-оксобутил]-N-метил-5-(трифторметил)бензамид
К раствору 3-бром-N-[(2S)-2-(4-фторфенил)пент-4-ен-1-ил]-N-метил-5-(трифторметил)бензамида (0,86 г, 1,9 ммоль) в ацетоне (45 мл) добавляли OsO4 (2,5% в трет-бутиловом спирте, 0,49 мл, 0,039 ммоль) и 4-метилморфолин-4-оксид (0,41 г, 3,5 ммоль). Раствор перемешивали в атмосфере азота при комнатной температуре в течение ночи и затем добавляли водный раствор NaHSO3 (39%, 45 мл). Смесь перемешивали в течение 2 ч, разбавляли водой и затем дважды экстрагировали метиленхлоридом. Объединенные органические растворы разделяли с помощью колонки фазоразделителя, растворитель удаляли выпариванием. Остаток (1,08 г) растворяли в THF (18 мл) и воде (4,5 мл) и к полученному раствору добавляли NaIO4 (0,73 г, 3,4 ммоль). Смесь перемешивали в атмосфере азота в течение ночи при комнатной температуре. Смесь распределяли между метиленхлоридом и водой. Водную фазу экстрагировали метиленхлоридом и затем объединенные органические растворы промывали раствором соли и отделяли с помощью колонки фазоразделителя. Растворитель удаляли выпариванием. Получено 0,78 г (90%) соединения, указанного в заголовке. 1H ЯМР (500 МГц, CDCl3): 2,4-4,4 (слож.м, 8H), 6,2-8,2 (слож.м, 7H), 9,8 (с, 1H); LCMS: m/z 447 (M+1)+.
Способ 2
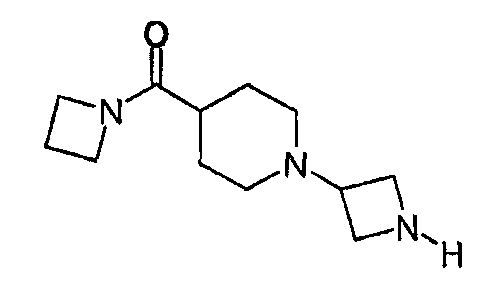
1-Азетидин-3-ил-4-(азетидин-1-илкарбонил)пиперидин
(a) трет-Бутил-4-(азетидин-1-илкарбонил)пиперидин-1-карбоксилат
1-(трет-Бутоксикарбонил)пиперидин-4-карбоновую кислоту (0,40 г, 1,75 ммоль) растворяли в сухом DMF (5 мл) и к раствору добавляли DIPEA (1,22 мл, 7,0 ммоль), TBTU (0,67 г, 2,1 ммоль) и азетидин (0,12 г, 2,1 ммоль). Реакционную смесь 12 ч перемешивали при комнатной температуре. Смесь разбавляли метиленхлоридом и затем промывали водным раствором HCl (2 M) и затем насыщенным водным раствором NaHCO3. Фазы разделяли с помощью колонки фазоразделителя и растворитель удаляли выпариванием. Получено 0,50 г (100%) трет-бутил-4-(азетидин-1-илкарбонил)пиперидин-1-карбоксилата в виде неочищенного твердого вещества. 1H ЯМР (500 МГц, CDCl3): 1,4-1,5 (с, 9H), 1,6-1,9 (м, 5H), 2,2-2,4 (м, 3H), 2,6-2,8 (м, 2H), 3,9-4,2 (м, 5H).
(b) 4-(Азетидин-1-илкарбонил)пиперидин
трет-Бутил-4-(азетидин-1-илкарбонил)пиперидин-1-карбоксилат (0,50 г, 1,86 ммоль) растворяли в метиленхлориде (10 мл) и к раствору добавляли трифторуксусную кислоту (2,12 г, 18,6 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и затем растворитель удаляли выпариванием. Остаток растворяли в небольшом количестве метанола и THF и раствор затем загружали на катионообменный сорбент (Isolute® SCX-2; 10 г). Колонку дважды промывали THF и затем продукт элюировали метанолом, насыщенным аммиаком. Растворитель удаляли выпариванием. Получено 0,32 г (100%) 4-(азетидин-1-илкарбонил)пиперидина. 1H ЯМР (500 МГц, CDCl3): 1,4-1,5 (м, 4H), 2,0-2,2 (м, 3H), 2,4-2,5 (м, 2H), 2,9-3,0 (д, 2H), 3,7-3,8 (т, 2H), 3,9 (с, 1H), 4,0 (т, 2H).
(c) 4-(Азетидин-1-илкарбонил)-1-[1-(дифенилметил)азетидин-3-ил]пиперидин
К смеси 4-(азетидин-1-илкарбонил)пиперидина (0,34 г, 2,0 ммоль) и 1-(дифенилметил)азетидин-3-она (см. Bioorg. Med. Chem. Lett.; 13; 2003; 2191-2194, 0,37 г, 1,6 ммоль), метанола (5 мл) и уксусной кислоты (0,1 мл) добавляли цианоборгидрид (полистирилметил)триметиламмония (4,1 ммоль/г, 0,61 г). Реакционную смесь 10 мин нагревали при 120°С, применяя микроволновое одноузловое нагревание, и затем фильтровали через фазоразделитель. Растворитель удаляли выпариванием и продукт очищали хроматографией на силикагеле (метанол-метиленхлорид, 5:95). Получено 0,42 г (70%) 4-(азетидин-1-илкарбонил)-1-[1-(дифенилметил)азетидин-3-ил]пиперидина в виде бесцветного пенообразного вещества. 1H ЯМР (500 МГц, CDCl3): 1,6-1,7 (м, 2H), 1,7-1,8 (м, 4H), 2,0-2,1 (м, 1H), 2,2 (квн, 2H), 2,7-2,8 (м, 2H), 2,8-3,0 (м, 3H), 3,4 (т, 2H), 4,0 (т, 2H), 4,1 (т, 2H), 4,4 (с, 1H), 7,1-7,2 (т, 2H), 7,2-7,3 (т, 4H), 7,4 (д, 4H); LCMS: m/z 390 (M+1)+.
(d) 1-Азетидин-3-ил-4-(азетидин-1-илкарбонил)пиперидин
4-(Азетидин-1-илкарбонил)-1-[1-(дифенилметил)азетидин-3-ил]пиперидин (0,42 г, 1,1 ммоль) растворяли в этаноле и к полученному раствору добавляли гидроксид палладия на углероде (0,15 г) и формиат аммония (0,28 г, 4,4 ммоль). Реакционную смесь нагревали 4 мин при 120°С, применяя микроволновое одноузловое нагревание. Катализатор отфильтровывали с помощью фазоразделителя и осадок на фильтре промывали этанолом. Растворитель удаляли выпариванием и остаток растворяли в метаноле (1 мл) и THF (10 мл). Раствор загружали на катионообменный сорбент (Isolute® SCX-2; 10 г). Колонку промывали THF и затем продукт элюировали метанолом, насыщенным аммиаком. Растворитель удаляли выпариванием, получая 0,25 г соединения, указанного в заголовке, в виде бесцветного масла. 1H ЯМР (500 МГц, CD3OD): 2,0-2,2 (м, 4H), 2,3-2,4 (м, 2H), 2,6-2,8 (м, 3H), 3,2-3,3 (д, 2H), 3,8 (квн, 1H), 4,3 (т, 2H), 4,4 (м, 2H), 4,5 (м, 2H), 4,6 (т, 2H); LCMS: m/z 224 (M+1)+.
Способ 3
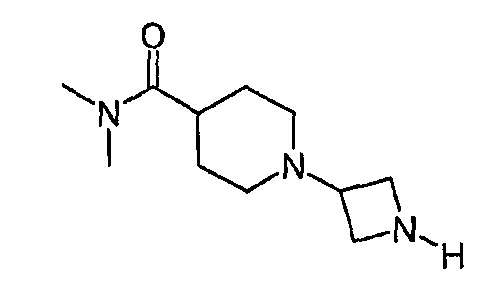
1-Азетидин-3-ил-N,N-диметилпиперидин-4-карбоксамид
(a) 1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-карбоновая кислота
К смеси пиперидин-4-карбоновой кислоты (0,13 г, 1,0 ммоль) и 1-(дифенилметил)азетидин-3-она (см. Bioorg. Med. Chem. Lett.; 13; 2003; 2191-2194, 0,24 г, 1,0 ммоль), метанола (3 мл) и уксусной кислоты (0,3 мл) добавляли цианоборгидрид (полистирилметил)триметиламмония (4,1 ммоль/г, 0,25 г). Реакционную смесь нагревали 5 мин при 120°С, применяя микроволновое одноузловое нагревание. Добавляли метанол и затем отфильтровывали смолу. Растворитель удаляли выпариванием. Получено 0,35 г (100%) 1-[1-(дифенилметил)азетидин-3-ил]пиперидин-4-карбоновой кислоты. 1H ЯМР (500 МГц, CDCl3): 1,6-1,8 (м, 2H), 1,9-2,0 (м, 4H), 2,3-2,4 (м, 1H), 2,7-2,8 (м, 2H), 2,9-3,0 (м, 3H), 3,4 (т, 2H), 4,4 (с, 1H), 7,2 (т, 2H), 7,2-7,3 (т, 4H), 7,4 (д, 4H); LCMS: m/z 351 (M+1)+.
(b) 1-[1-(Дифенилметил)азетидин-3-ил]-N,N-диметилпиперидин-4-карбоксамид
1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-карбоновую кислоту (0,35 г, 0,9 ммоль) растворяли в DMF (8 мл) и к раствору добавляли TBTU (0,39 г, 1,2 ммоль), DIPEA (0,21 мл, 1,2 ммоль) и раствор диметиламина (3,0 мл, 2M в THF, 6 ммоль). Смесь 14 ч перемешивали при комнатной температуре. Добавляли водный раствор NaHCO3 и смесь три раза экстрагировали метиленхлоридом. Объединенные органические слои промывали раствором соли и осушали над MgSO4. Растворитель удаляли выпариванием и продукт очищали обращенно-фазовой хроматографией (С8), применяя ацетонитрил и водный раствор ацетата аммония (0,1 М) в качестве элюента. Получено 0,20 г (59%) 1-[1-(дифенилметил)азетидин-3-ил]-N,N-диметилпиперидин-4-карбоксамида. 1H ЯМР (500 МГц, CDCl3): δ 1,6-2,0 (сл.м, 6H), 2,4-2,5 (м, 1H), 2,8 (м, 2H), 2,9-3,0 (м, 5H), 3,1 (с, 3H), 3,4 (т, 2H), 4,4 (с, 1H), 7,2 (т, 2H), 7,3 (т, 4H), 7,4 (д, 4H); LCMS: m/z 378 (M+1)+.
(c) 1-Азетидин-3-ил-N,N-диметилпиперидин-4-карбоксамид
Гидроксид палладия на угле (0,10 г) помещали в 5-мл флакон, предназначенный для микроволнового синтеза. Добавляли 1-[1-(дифенилметил)азетидин-3-ил]-N,N-диметилпиперидин-4-карбоксамид (0,20 г, 0,53 ммоль), растворенный в метаноле (3 мл) и уксусной кислоте (0,3 мл). Смесь перемешивали четыре дня в атмосфере водорода (1,6 бар) при комнатной температуре. Смесь фильтровали через слой Celite®. Растворитель удаляли выпариванием, получая 0,11 г (53%) соединения, указанного в заголовке.
Способ 4
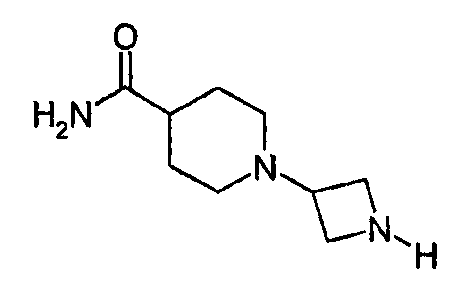
1-Азетидин-3-илпиперидин-4-карбоксамид
(a) 1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-карбоксамид
К смеси пиперидин-4-карбоксамида (1,05 г, 8,2 ммоль), 1-(дифенилметил)азетидин-3-она (см. Bioorg. Med. Chem. Lett.; 13; 2003; 2191-2194, 1,94 г, 8,2 ммоль), метанола (30 мл) и уксусной кислоты (3 мл) добавляли цианоборгидрид (полистирилметил)триметиламмония (4,1 ммоль/г, 1,9 г). Реакционную смесь нагревали 5 мин при 120°С, применяя микроволновое одноузловое нагревание. Смолу отфильтровывали и растворитель удаляли выпариванием. Получено 2,85 г (99%) 1-[1-(дифенилметил)азетидин-3-ил]пиперидин-4-карбоксамида. 1H ЯМР (500 МГц, CDCl3): 1,6-1,9 (м, 6H), 2,1-2,2 (м, 1H), 2,7-2,8 (д, 2H), 2,9-3,0 (м, 3H), 3,4 (т, 2H), 4,4 (с, 1H), 5,7-5,8 (шир, 1H), 6,2 (шир, 1H), 7,2 (т, 2H), 7,2-7,3 (т, 4H), 7,4 (д, 4H); LCMS: m/z 350 (M+1)+.
(b) Дигидрохлорид 1-азетидин-3-илпиперидин-4-карбоксамида
1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-карбоксамид (1,4 г, 4,1 ммоль), формиат аммония (0,77 г, 12 ммоль) и этанол (15 мл) загружали в 25-мл флакон, предназначенный для микроволнового синтеза. Добавляли гидроксид палладия на угле (0,55 г) и реакционную смесь 2 мин нагревали при 120°С, применяя микроволновое одноузловое нагревание. Смесь, которая все еще содержала исходный материал, фильтровали и к фильтрату добавляли другую порцию гидроксида палладия на угле вместе со смесью уксусной кислоты и этанола (1:10). Реакционную смесь перемешивали 4 ч в атмосфере водорода (5 бар) при комнатной температуре и затем фильтровали через слой Celite®. Растворитель удаляли выпариванием и остаток распределяли между толуолом и разбавленной соляной кислотой. Водную фазу высушивали из замороженного состояния и липкий остаток выпаривали вместе с толуолом, повторно растворяли в воде и затем высушивали из замороженного состояния. Получено 1,35 г (65%) соединения, указанного в заголовке. 1H ЯМР (500 МГц, CD3OD): δ 1,6-2,0 (сл.м, 6H), 2,2-2,3 (м, 1H), 2,8 (м, 2H), 3,4 (м, 1H), 3,9-4,1 (м, 4H).
Способ 5
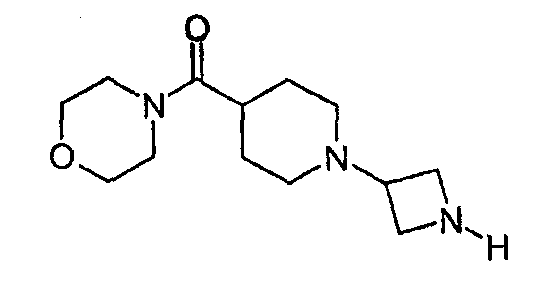
4-[(1-Азетидин-3-илпиперидин-4-ил)карбонил]морфолин
(a) 4-({1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-ил}карбонил)морфолин
4-(Пиперидин-4-илкарбонил)морфолин (0,30 г, 1,26 ммоль) и 1-(дифенилметил)азетидин-3-он (см. Bioorg. Med. Chem. Lett.; 13; 2003; 2191-2194, 0,30 г, 1,5 ммоль) растворяли в смеси метанола (5 мл) и уксусной кислоты (0,1 мл). Добавляли цианоборгидрид (полистирилметил)триметиламмония (4,1 ммоль/г, 0,38 г) и реакционную смесь нагревали 10 мин при 120°С, применяя микроволновое одноузловое нагревание. Смесь фильтровали через фазоразделитель и смолу промывали метанолом. Растворитель удаляли выпариванием и остаток растворяли в метиленхлориде. Раствор дважды промывали насыщенным раствором NaHCO3. Органическую фазу фильтровали через фазоразделитель и растворитель удаляли выпариванием. Продукт очищали хроматографией на силикагеле (метанол/метиленхлорид, 5% метанола). Получено 0,44 г (83%) 4-({1-[1-(дифенилметил)азетидин-3-ил]пиперидин-4-ил}карбонил)морфолина в виде бесцветного масла. 1H ЯМР (500 МГц, CDCl3): 1,6-1,7 (м, 2H), 1,7-1,9 (м, 4H), 2,2 (м, 1H), 2,7-2,8 (м, 2H), 2,8-3,0 (м, 4H), 3,3-3,5 (м, 3H), 3,5-3,7 (м, 6H), 4,4 (с, 1H), 7,1-7,2 (т, 2H), 7,2-7,3 (т, 4H), 7,4 (д, 4H); LCMS: m/z 420 (M+1)+.
(b) 4-[(1-Азетидин-3-илпиперидин-4-ил)карбонил]морфолин
4-({1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-ил}карбонил)морфолин (0,44 г, 1,0 ммоль) растворяли в метаноле и к полученному раствору добавляли гидроксид палладия на угле (0,15 г) и формиат аммония (0,27 г, 4,2 ммоль). Реакционную смесь нагревали 2 мин при 120°С, применяя микроволновое одноузловое нагревание. Катализатор отфильтровывали с помощью фазоразделителя и осадок на фильтре промывали этанолом. Растворитель удаляли выпариванием и остаток растворяли в метаноле (1 мл) и THF (10 мл). Раствор загружали на катионообменный сорбент (Isolute® SCX-2; 10 г). Колонку промывали THF и затем продукт элюировали метанолом, насыщенным аммиаком. Растворитель собранных фракций удаляли выпариванием, получая 0,11 г соединения, указанного в заголовке, в виде бесцветного масла. 1H ЯМР (500 МГц, CD3OD): 1,7-1,8 (м, 4H), 1,9-2,0 (м, 2H), 2,6-2,9 (м, 4H), 3,3-3,4 (м, 1H), 3,5-3,7 (м, 8H), 3,8-4,0 (м, 3H); LCMS: m/z 254 (M+1)+.
Фармакология
Трансфекция и культивирование клеток, применяемых в исследованиях FLIPR и связывания
Клетки яичника китайского хомячка (СНО) K1 (полученные из ATCC), стабильно трансфецировали человеческим рецептором NK2 (кДНК hNK2R в pRc/CMV, Invitrogen) или человеческим рецептором NK3 (hNK3R в пкДНК 3,1/Hygro (+)/IRES/CD8, вектор Invitrogen модифицированный в AstraZeneca EST-Bio UK, Alderley Park). Клетки затем трансфецировали катионным липидным реагентом LIPOFECTAMINETM (Invitrogen) и проводили отбор с генетицином (G418, Invitrogen) при 1 мг/мл для hNK2R-трансфецированных клеток и с гигромицином (Invitrogen) при 500 мкг/мл для hNK3R-трансфецированных клеток. Отдельные клеточные клоны собирали с помощью Fluorescence Activated Cell Sorter (FACS), проверенного на функциональность в тесте FLIPR (см. ниже), размножали в культуре и сохраняли в замороженном виде для будущего применения. Клетки CHO, стабильно трансфецированные человеческими рецепторами NK1 происходят из AstraZeneca R&D, Wilmington USA. кДНК человеческого рецептора NK1 (полученная из РНК-ПЦР из ткани легкого) субклонировали в pRcCMV (Invitrogen). Трансфекцию проводили с фосфатом кальция, и отбор выполняли с 1 мг/мл G418.
Клетки CHO, стабильно трансфецированные hNK1R, hNK2R и hNK3R, культивировали во влажном инкубаторе в атмосфере 5% CO2, в Nut Mix F12 (HAM) с глутамаксом I, 10% фетальной коровьей сыворотки (FBS), 1% пенициллина/стрептомицина (PEST) с добавлением 200 мкг/мл генетицина для клеток, экспрессирующих hNK1R и hNK2R, и 500 мкг/мл гигромицина для клеток, экспрессирующих hNK3R. Клетки выращивали во флаконах T175, с пассажем при 70-80% конфлюэнтности, до 20-25 пассажей.
Оценка активности избранных испытуемых соединений при ингибировании активации человеческих рецепторов NK 1 /NK 2 /NK 3 (FLIPR-тест)
Активность соединения настоящего изобретения при ингибировании активации рецепторов NK1/NK2/NK3, измеренной как опосредуемое NK1/NK2/NK3 увеличение внутриклеточного Са2+, оценивали, применяя следующую процедуру:
Клетки CHO, стабильно трансфецированные человеческими рецепторами NK1, NK2 или NK3, высевали в 96-луночные планшеты (Costar 3904), лунки которых имели черные стенки и прозрачное дно, в количестве 3,5×104 клеток на лунку и выращивали приблизительно 24 ч в нормальных ростовых средах при 37°С в CO2-инкубаторе.
Перед FLIPR-тестом клетки каждого 96-луночного планшета нагружали Ca2+-чувствительным красителем Fluo-3 (TEFLABS 0116) при 4 мкМ в нагрузочных средах, состоящих из Nut Mix F12 (HAM) с глутамаксом I, 22 мМ HEPES, 2,5 мМ пробеницида (Sigma P-8761) и 0,04% плюроника F-127 (Sigma P-2443) в течение 1 ч в темноте при 37°C в CO2-инкубаторе. Затем клетки три раза промывали тестовым буфером (сбалансированный солевой раствор Хэнкса, содержащий 20 мМ HEPES, 2,5 мМ пробеницида и 0,1% БСА), применяя многоканальную пипетку и оставляя их в 150 мкл в конце последней промывки. Серийные разбавления испытываемых соединений в тестовом буфере (конечную концентрацию ДМСО поддерживали ниже 1%) автоматически пипетировали с помощью FLIPR (Fluorometric Imaging Plate Reader, флуориметрического планшет-ридера) в каждую тестируемую лунку и записывали интенсивность флуоресценции (возбуждение при 488 нм, испускание при 530 нм) камерой FLIPR CCD в течение 2-мин периода преинкубации. 50 мкл раствора агониста - субстанции Р (NK1-специфичной), NKA (NK2-специфичного) или Pro-7-NKB (NK3-специфичного) - (в конечной концентрации, эквивалентной приблизительной концентрации EC60) добавляли с помощью FLIPR в каждую лунку, уже содержавшую 200 мкл тестового буфера (содержавшего испытываемое соединение или плацебо), и проводили непрерывный мониторинг в течение следующих 2 мин. Измеряли реакцию в виде относительной флуоресценции пика после добавления агониста и рассчитывали значения IC50 по десятиточечным кривым «концентрация-ответ» для каждого соединения. Значения IC50 затем переводили в значения pKB по следующей формуле:
KB = IC50 / 1+ (конц. EC60 агониста, применявшегося в тесте / EC50 агониста)
pKB = -log KB
Определение константы диссоциации (Ki) соединений для человеческих рецепторов NK 1 /NK 2 /NK 3 (исследование связывания)
Мембраны получали из клеток СНО, стабильно трансфецированных человеческими рецепторами NK1, NK2 или NK3 следующим способом.
Клетки снимали раствором Accutase®, центрифугированием собирали в PBS, содержавшем 5% фетальной коровьей сыворотки, дважды промывали в PBS и ресуспендировали в концентрации 1×108 клеток/мл в 50 мМ Трис-HCl, 300 мМ KCl, 10 мМ EDTA-N2, pH 7,4 (4°C). Суспензии клеток гомогенизировали в UltraTurrax 30 с при 12000 об/мин. Гомогенаты центрифугировали при 38000×g (4°C) и осадок ресуспендировали в 50 мМ Трис-HCl, pH 7,4. Гомогенизацию повторяли один раз, и гомогенаты 45 мин инкубировали на льду. Гомогенаты опять центрифугировали как описано выше и ресуспендировали в 50 мМ Трис-HCl, pH 7,4. Эту стадию центрифугирования повторяли всего 3 раза. После последней стадии центрифугирования осадок ресуспендировали в 50 мМ Трис-HCl и гомогенизировали 10 ударами Dual Potter до получения гомогенного раствора, аликвоту которого удаляли для определения белка. Мембраны разделяли на аликвоты и замораживали при -80°С до применения.
Исследование связывания радиоактивного лиганда проводили при комнатной температуре в 96-луночных микротитрационных планшетах (планшеты с несвязывающей поверхностью, No-binding Surface Plates, Coming 3600) в конечном объеме 200 мкл буфера инкубации в лунке (50 мМ Трис-буфер (pH 7,4 при комнатной температуре), содержавший 0,1% БСА, 40 мг/л бацитрацина, таблетки свободного от ЭДТА набора ингибиторов протеаз (20 гранул на литр, Roche) и 3 мМ MnCl2). Кривые конкурентного связывания строили, добавляя возрастающие количества испытываемого соединения. Испытываемые соединения растворяли и серийно разбавляли в ДМСО при 1,5% конечной концентрации ДМСО в тесте. Для измерения неспецифического связывания добавляли 50 мкл немеченого ZD 6021 (неселективный антагонист NK, в конечной концентрации 10 мкМ). Для общего связывания применяли 50 мкл 1,5% ДМСО (конечная концентрация). В экспериментах по связыванию на hNK1r применяли 4 нМ (конечная концентрация) [3H-Sar,Met(O2)-субстанции P]. В экспериментах по связыванию на hNK2r применяли 3 нМ (конечная концентрация) [3H-SR48968], а в экспериментах по связыванию на hNK3r применяли 3 нМ (конечная концентрация) [3H-SR142801]. 50 мкл радиолиганда, 3 мкл испытываемого соединения, разбавленного в ДМСО, и 47 мкл буфера инкубации, смешивали с 5-10 мкг клеточных мембран в 100 мкл буфера инкубации и 30 мин инкубировали при комнатной температуре на микропланшетном шейкере.
Затем мембраны собирали быстрой фильтрацией на Filtermat B (Wallac), предварительно пропитанном 0,1% БСА и 0,3% полиэтиленимина (Sigma P-3143), применяя Micro 96 Harvester (Skatron Instruments, Норвегия). Фильтры промывали в последнем аппарате ледяным холодным буфером промывки (50 мМ Трис-HCl, pH 7,4 при 4°C, содержащий 3 мМ MnCl2) и 30-60 мин сушили при 50°C. Сцинтилляционные пластины Meltilex расплавляли на фильтрах, применяя Microsealer (Wallac, Finland), и фильтры просчитывали в жидкостном β-сцинтилляционном счетчике (1450 Microbeta, 30 Wallac, Финляндия).
Значение Ki для немеченого лиганда рассчитывали, применяя уравнение Ченга-Прусоффа (Biochem. Pharmacol. 22:3099-3108, 1973): L является концентрацией радиоактивного лиганда и Ki является сродством радиоактивного лиганда к рецептору, определенным при насыщающем связывании.
Данные аппроксимировали четырехпараметрическим уравнением с применением Excel Fit.
Ki = IC50/(1+(L/Kd))
Результаты
В общем случае испытанные соединения настоящего изобретения демонстрировали статистически достоверную антагонистическую активность в отношении рецептора NK1 в диапазоне 7-8 для pKB. Для рецептора NK2 диапазон для pKB был 7-9. Обычно антагонистическая активность в отношении рецептора NK3 составляла 7-9 для pKB.
В общем случае испытанные соединения настоящего изобретения демонстрировали статистически достоверное ингибирование CYP3A4 на низком уровне. Значения IC50, определенные согласно Bapiro et al.; Drug Metab. Dispos. 29, 30-35 (2001), обычно были больше 10 мкМ.
Активность против hERG
Активность соединений формулы I против кодируемого hERG калиевого канала можно определять согласно Kiss L, et al. Assay Drug Dev Technol. 1 (2003), 127-35: "High throughput ion-channel pharmacology: planar-array-based voltage clamp" («Высокопроизводительная фармакология ионных каналов: вольт-кламп, основанный на планарном массиве»).
В общем случае испытанные соединения настоящего изобретения демонстрировали статистически достоверную активность hERG на низком уровне. Значения IC50, определенные как описано выше, обычно были больше 8 мкМ.
Метаболическая стабильность
Метаболическую стабильность соединения формулы I можно определить как описано ниже:
Скорость биотрансформации можно измерить либо как образование метаболита (одного или более), либо как скорость исчезновения исходного соединения. Схема эксперимента включает инкубацию при низких концентрация субстрата (обычно 1,0 мкМ) с микросомами печени (обычно 0,5 мг/мл) и отбор аликвот в различные моменты времени (обычно 0,5, 10, 15, 20, 30, 40 мин). Испытываемое соединение обычно растворяют в ДМСО. Концентрация ДМСО в инкубационной смеси обычно составляет 0,1% или менее, поскольку большее количество растворителя может резко понизить активности некоторых форм CYP450. Инкубации проводят в 100 мМ калий-фосфатном буфере, pH 7,4, при 37°C. Для остановки реакции применяют ацетонитрил или метанол. Исходное соединение анализируют с помощью ВЭЖХ-МС. По рассчитанным значениям времени полужизни, t1/2, оценивают собственный клиренс, Clint, учитывая концентрацию микросомального белка и массу печени.
В общем случае, соединения настоящего изобретения in vitro показывали высокий уровень метаболической стабильности. Значения собственного клиренса, определенные как указано выше, обычно были менее 25 мкл/мин на мг белка.
Следующая таблица иллюстрирует свойства соединений настоящего изобретения:
N-[(2S)-4-{3-[4-(Азетидин-1-илкарбонил)пиперидин-1-ил]азетидин-1-ил}-2-(4-фторфенил)бутил]-3-бром-N-метил-5-(трифторметил)бензамид (Пример 1):
Биологическая оценка
Стук лап песчанки (NK1-специфичный модельный тест)
Самцов монгольских песчанок (60-80 г) приобретали у Charles River, Германия. После поступления их размещали группами по десять особей со свободным доступом к корму и воде в комнатах с контролируемой температурой и влажностью. Животным давали не менее 7 дней для акклиматизации к условиям жизни до экспериментов. Каждое животное использовали только однократно и непосредственно после эксперимента подвергали эвтаназии посредством сердечной пункции или летальной дозой пентобарбитала натрия.
Песчанок анестезировали изофлураном. Потенциально проникающие в ЦНС антагонисты рецептора NK1 вводили внутрибрюшинно, внутривенно или подкожно. Соединения вводили в различные моменты времени (обычно за 30-120 минут) до стимуляции агонистом.
Песчанкам давали легкую анестезию изофлуораном и делали небольшой разрез кожи над брегмой. Интрацеребровентрикулярно вводили 10 пмоль ASMSP, селективного агониста рецептора NK1, в объеме 5 мкл, применяя гамильтоновский шприц с иглой длиной 4 мм. Рану зажимали, животное помещали в небольшую пластмассовую клетку и давали пробудиться. Клетку помещали на кусок пластмассовой трубки, заполненной водой и соединенной с компьютером через датчик давления. Записывали количество ударов лап.
Хромодакриорейная модель (NK1-специфичный модельный тест)
Действие антагонистов на периферийные рецепторы NK1 можно оценить in vivo на песчанках, используя так называемую хромодакриорейную модель (Bristow LJ, Young L. Chromodacryorrhea and repetitive hind paw tapping: models of peripheral and central tachykinin NK1 receptor activation in gerbils (Хромодакриорея и повторные удары задних лап: модели активации периферических и центральных тахикининовых рецепторов NK1 у песчанок). Eur J Pharmacol 1994; 253: 245-252). В кратком изложении: системное (внутривенное) введение агонистов рецепторов NK1 анестезированным песчанкам приводит к профузной секреции красных или коричневых слез вследствие секреции порфирина из гардериановой железы. Антагонисты рецептора NK1 блокируют хромодакриорею, вызванную агонистом NK1.
Выделение фекального пеллета (NK2-специфичный модельный тест)
Эффект in vivo (NK2) соединения формулы I можно определить, измеряя выделение фекального пеллета, индуцированного агонистом рецептора NK2, у песчанок, как описано, например, в The Journal of Pharmacology и Experimental Therapeutics (2001), pp.559-564.
Модель с колоректальным растяжением
Колоректальное растяжение (CRD) у песчанок проводили, как описано ранее для крыс и мышей (Tammpere A, Brusberg M, Axenborg J, Hirsch I, Larsson H, Lindstrom E. Evaluation of pseudo-affective responses to noxious colorectal distension in rats by manometric recordings (Оценка псевдо-аффективной реакции на патологическое колоректальное растяжение у крыс по манометрическим данным). Pain 2005; 116: 220-226; Arvidsson S, Larsson M, Larsson H, Lindstrom E, Martinez V. Assessment of visceral pain-related pseudo-affective responses to colorectal distension in mice by intracolonic manometric recordings (Оценка висцеральной болезненной псевдо-аффективной реакции на колоректальное растяжение у мышей по внутритолстокишечным манометрическим данным). J Pain 2006; 7: 108-118), с небольшими изменениями. В кратком изложении: песчанок приучали к клеткам Боллманна по 30-60 минут ежедневно в течение трех последовательных дней до экспериментов, чтобы уменьшить двигательные артефакты, обусловленные стрессом ограниченной иммобилизации. Полиэтиленовый баллон размером 2 см (собственного изготовления) с соединительным катетером вводили в дистальный отдел толстой кишки, в 2 см от основания баллона до ануса, под легкой анестезией изофлураном (Forene®, Abbott Scandinavia AB, Solna, Швеция). Катетер лентой фиксировали на хвосте. Баллоны соединяли с датчиками давления (P-602, CFM-k33, 100 мм ртутного столба, Bronkhorst HI-TEC, Veenendal, Нидерланды). Песчанкам давали успокоиться после помещения в клетки Боллманна в течение 15 минут до начала экспериментов.
Специально изготовленный баростат (AstraZeneca, Molndal, Швеция) использовали для управления подачей воздуха и регулирования давления в баллоне. Специально разработанную компьютерную программу (PharmLab on-line 4.0), выполнявшуюся на стандартном компьютере, применяли для управления баростатом и сбора данных. Применявшаяся модель растяжения состоит из 12 повторных фазовых растяжений при 80 мм ртутного столба, с продолжительностью пульса, равной 30 с, и с пятиминутными интервалами. Соединения или соответствующее плацебо вводили внутрибрюшинными инъекциями до CRD эксперимента. Каждая песчанка получает как плацебо, так и соединение в различные моменты с интервалами между экспериментами длительностью не менее двух дней. Таким образом, каждая песчанка служит своим собственным плацебо-контролем.
Аналоговые входные каналы дискретизировали с индивидуальными частотами дискретизации и сигналы подвергали цифровой фильтрации. Сигналы давления в баллоне дискретизировали при частоте 50 значений в секунду. Для отделения изменений давления, вызванных сокращениями, от медленно меняющегося давления, создаваемого баростатом, применяли фильтр верхних частот на 1 Гц. Сопротивление в потоке воздуха между компрессором и датчиком давления дополнительно повышает колебания давления, вызванные брюшными сокращениями животного. Специально разработанную компьютерную программу (PharmLab off-line 4.0) применяли для расчета величины сигналов давления баллона после фильтрации верхних частот. Среднее по модулю значение (ARV) сигналов давления баллона после фильтрации верхних частот рассчитывали для 30 с до пульса (базальная реакция) и для продолжительности пульса. При расчете величины сигналов давления баллона после фильтрации верхних частот из каждого пульса исключали первую и последнюю секунду, поскольку они отражают сигналы-артефакты, производимые баростатом при нагнетании и выпуске воздуха и не исходят от животного.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ АЗЕТИДИНОВЫЕ СОЕДИНЕНИЯ | 2004 |
|
RU2356888C2 |
| 3-АЗЕТИДИНИЛАЛКИЛПИПЕРИДИНЫ ИЛИ ПИРРОЛИДИНЫ В КАЧЕСТВЕ АНТАГОНИСТОВ ТАХИКИНИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2158264C2 |
| ОКСИИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ МОТИЛИНОВОГО РЕЦЕПТОРА | 2010 |
|
RU2533116C2 |
| ПРОИЗВОДНЫЕ (АЗЕТИДИН-1-ИЛАЛКИЛ)ЛАКТАМОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ ЧЕЛОВЕКА, ПРИ КОТОРОМ ЗАБОЛЕВАНИЕ ЛЕЧАТ ПОСРЕДСТВОМ ПРОДУЦИРОВАНИЯ АНТАГОНИСТИЧЕСКОГО ДЕЙСТВИЯ НА ТАХИКИНИН, ДЕЙСТВУЮЩИЙ В ЧЕЛОВЕЧЕСКОМ NК-, NК- И NК-РЕЦЕПТОРЕ ИЛИ В ИХ СОЧЕТАНИИ | 1995 |
|
RU2150468C1 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРРОЛИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ | 2003 |
|
RU2355679C2 |
| 1,3,4,-ОКСАДИЗОЛАМИДНОЕ ПРОИЗВОДНОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2700696C2 |
| ПРОИЗВОДНЫЕ ИЗОКСАЗОЛИНА В КАЧЕСТВЕ ПРОТИВОПАРАЗИТАРНЫХ АГЕНТОВ | 2011 |
|
RU2551354C2 |
| ХИНОКСАЛИНЫ И АЗАХИНОКСАЛИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА CRTH | 2011 |
|
RU2589709C2 |
| ТИАЗОЛОПИРИМИДИНОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРОВ NMDA | 2014 |
|
RU2703273C2 |
| ИНДОЛЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-HT | 2007 |
|
RU2449990C2 |
Изобретение относится к соединениям формулы (I)
где каждый R1 и R2 независимо выбран из водорода, метила, этила, или R1 и R2 образуют четырех-, пяти- или шестичленное кольцо вместе с амидным азотом, причем указанное кольцо необязательно содержит атом кислорода; Х представляет собой бром или хлор; а также к их фармацевтически и фармакологически приемлемым солям и энантиомерам соединения формулы I и их солям. Изобретение также относится к применению соединения по любому из пп.1-21, к фармацевтической композиции, а также к соединениям, выбранным из группы. Технический результат - получение новых биологически активных соединений, обладающих антагонистической активностью в отношении рецептора NK1/NK2/NK3. 6 н. и 21 з.п. ф-лы.
1. Соединение формулы (I)
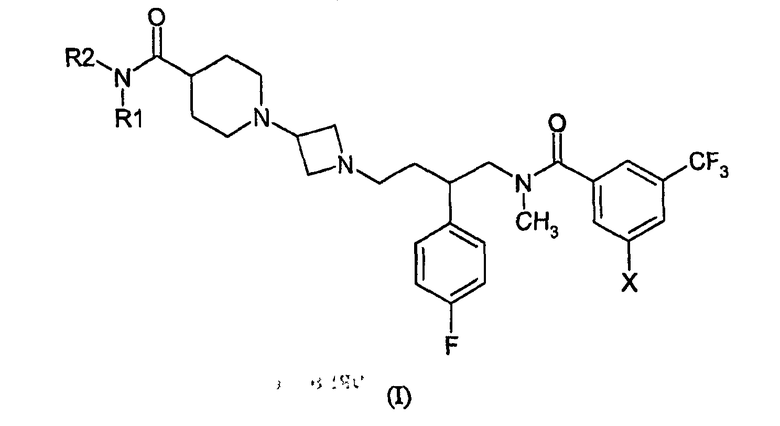
где каждый R1 и R2 независимо выбран из водорода, метила, этила, или R1 и R2 образуют четырех-, пяти- или шестичленное кольцо вместе с амидным азотом, причем указанное кольцо необязательно содержит атом кислорода;
Х представляет собой бром или хлор;
а также их фармацевтически и фармакологически приемлемые соли и энантиомеры соединения формулы I и их соли.
2. Соединение по п.1, где R1 является водородом.
3. Соединение по п.1, где R1 является метилом.
4. Соединение по п.1, где R1 является этилом.
5. Соединение по любому из пп.1-4, где R2 является водородом.
6. Соединение по любому из пп.1-4, где R2 является метилом.
7. Соединение по любому из пп.1-4, где R2 является этилом.
8. Соединение по п.1, где R1 и R2 вместе с амидным азотом образуют морфолиновое кольцо.
9. Соединение по п.1, где R1 и R2 вместе с амидным азотом образуют азетидиновое кольцо.
10. Соединение по п.1, где R1 и R2 вместе с амидным азотом образуют пирролидиновое кольцо.
11. Соединение по п.1, где R1 и R2 вместе с амидным азотом образуют изоксазолидиновое кольцо.
12. Соединение по п.1, где R1 и R2 вместе с амидным азотом образуют оксазолидиновое кольцо.
13. Соединение по п.1, где соединение является (S)-энантиомером.
14. Соединение по п.1, являющееся малеатом 1-{1-[(3S)-4-[[3-бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида.
15. Соединение по п.14, характеризующееся тем, что дает картину порошковой рентгеновской дифракции, демонстрирующей существенно следующие главные пики со значениями d:
16. Соединение по п.14, являющееся сольватом изопропанола.
17. Соединение по п.16, характеризующееся тем, что дает картину порошковой рентгеновской дифракции, демонстрирующей существенно следующие главные пики со значениями d:
18. Соединение по п.14, характеризующееся тем, что дает картину порошковой рентгеновской дифракции, демонстрирующей существенно следующие главные пики со значениями d:
19. Соединение по п.14, являющееся сольватом метилэтилкетона.
20. Соединение по п.19, характеризующееся тем, что дает картину порошковой рентгеновской дифракции, демонстрирующей существенно следующие главные пики со значениями d:
21. Соединение по п.1, выбранное из
N-[(2S)-4-{3-[4-(Азетидин-1-илкарбонил)пиперидин-1-ил]азетидин-1-ил}-2-(4-фторфенил)бутил]-3-бром-N-метил-5-(трифторметил)бензамида;
1-{1-[(3S)-4-[[3-Бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}-N,N-диметилпиперидин-4-карбоксамида;
1-{1-[(3S)-4-[[3-Бром-5-(трифторметил)бензоил](метил)амино]-3-(4-фторфенил)бутил]азетидин-3-ил}пиперидин-4-карбоксамида; и
3-Бром-N-((2S)-2-(4-фторфенил)-4-{3-[4-(морфолин-4-илкарбонил)пиперидин-1-ил]азетидин-1-ил}бутил)-N-метил-5-(трифторметил)бензамида.
22. Соединение по п.1 для применения при предупреждении или лечении респираторного, сердечно-сосудистого, неврологического, болезненного, онкологического, воспалительного и/или желудочно-кишечного расстройства.
23. Применение соединения по любому из пп.1-21 для получения лекарственного средства для лечения функционального желудочно-кишечного расстройства.
24. Применение соединения по любому из пп.1-21 для получения лекарственного средства для лечения синдрома раздраженной толстой кишки.
25. Применение соединения по любому из пп.1-21 для получения лекарственного средства для лечения функциональной диспепсии.
26. Фармацевтическая композиция, обладающая антагонистической активностью в отношении рецептора NK1/NK2/NK3, содержащая соединение по любому из пп.1-21 в качестве активного ингредиента и фармацевтически приемлемый носитель или разбавитель.
27. Соединение, выбранное из
1-Азетидин-3-ил-4-(азетидин-1-илкарбонил)пиперидина;
трет-Бутил-4-(азетидин-1-илкарбонил)пиперидин-1-карбоксилата;
4-(Азетидин-1-илкарбонил)пиперидина;
4-(Азетидин-1-илкарбонил)-1-[1-(дифенилметил)азетидин-3-ил]пиперидина;
1-Азетидин-3-ил-N,N-диметилпиперидин-4-карбоксамида;
1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-карбоновой кислоты;
1-[1-(Дифенилметил)азетидин-3-ил]-N,N-диметилпиперидин-4-карбоксамида;
1-Азетидин-3-илпиперидин-4-карбоксамида;
1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-карбоксамида;
1-Азетидин-3-илпиперидин-4-карбоксамид дигидрохлорида;
4-[(1-Азетидин-3-илпиперидин-4-ил)карбонил]морфолина и
4-({1-[1-(Дифенилметил)азетидин-3-ил]пиперидин-4-ил}карбонил)морфолина.
| WO 2004110344 A2, 23.12.2004 | |||
| ПРОИЗВОДНЫЕ (АЗЕТИДИН-1-ИЛАЛКИЛ)ЛАКТАМОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ ЧЕЛОВЕКА, ПРИ КОТОРОМ ЗАБОЛЕВАНИЕ ЛЕЧАТ ПОСРЕДСТВОМ ПРОДУЦИРОВАНИЯ АНТАГОНИСТИЧЕСКОГО ДЕЙСТВИЯ НА ТАХИКИНИН, ДЕЙСТВУЮЩИЙ В ЧЕЛОВЕЧЕСКОМ NК-, NК- И NК-РЕЦЕПТОРЕ ИЛИ В ИХ СОЧЕТАНИИ | 1995 |
|
RU2150468C1 |

