Область техники, к которой относится изобретение
Настоящее изобретение касается пероральных дозированных форм замедленного высвобождения (6) 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты и методов лечения или предотвращения заболеваний и расстройств, для которых габапентин является терапевтически эффективным при приеме таких лекарственных форм.
Уровень техники
1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусная кислота (1), которая является пролекарством аналога гамма аминобутановой кислоты (GABA) габапентина (2), имеет высокий уровень бионакопления в виде габапентина при введении перорально или непосредственно в толстую кишку млекопитающего (Gallop et al., International Publication No. WO 02/100346; Cundy et al., J. Pharmacol. Exp. Ther. 2004, 311:315-323; Cundy et al., J. Pharmacol. Exp. Ther. 2004, 311:324-3332). Высокий уровень бионакопления делает вещество (1) полезным компонентом пероральных лекарственных форм (включая лекарственные формы с длительным выделением), которые применяют для лечения или предотвращения эпилепсии, боли (особенно невропатической боли и мышечной или скелетной боли), депрессии, беспокойства, психоза, приступов слабости, гипокинезии, черепных заболеваний, нейродегенеративных заболеваний, паники, воспалительного заболевания (например, артрита), бессонницы, гастроинтестинальных заболеваний, приступов жара, синдрома неспокойных ног, недержания мочи и синдрома похмелья.
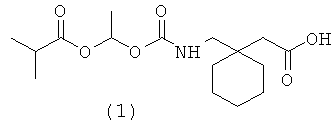
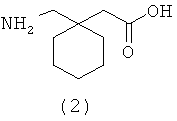
Соединение (1), полученное, как описано в Gallop et al., International Publication No. WO 02/100347, было изолировано в чистом виде в виде прозрачного стекловидного твердого вещества лиофилизацией из водного ацетонитрила. Материал, полученный этим методом, является частично или полностью аморфным, а определенные формы солей щелочных металлов являются гигроскопичными. Ранее были описаны кристаллическая форма 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты и способы ее получения (Estrada et al., International Publication No. WO 2005/037784). Кристаллическая форма вещества (1) имеет улучшенные физико-химические свойства, полезные в фармацевтическом производстве и фармацевтических композициях.
Раскрытие изобретения
Настоящее изобретение описывает пероральные дозированные формы соединения (1) замедленного высвобождения. В конкретных вариантах осуществления пероральные дозированные формы соединения (1) замедленного высвобождения при приеме на тощий желудок одним или более пациентами в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты от 1100 мг до 1300 мг дают кривую изменения концентраций габапентина в плазме, характеризующуюся Смах(Сmax) от 3 мкг/мл до 6 мкг/мл, Тмах(Тmax) от 4 часов до 7 часов и (AUC) от 30 мкг·ч/мл до 70 мкг·ч/мл; или при приеме на сытый желудок одним или более пациентами в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты от 1100 мг до 1300 мг дают кривую изменения концентраций габапентина в плазме, характеризующуюся Смах(Сmax) от 5 мкг/мл до 8 мкг/мл, Тмах(Тmax) от 6 часов до 11 часов и (AUC) от 60 мкг·ч/мл до 110 мкг·ч/мл.
Дозированная форма может быть, например, таблетками, включающими (а) от 10 мас.% до 80 мас.% вещества (1) и (в) от 1 мас.% до 50 мас.% полимера, определяющего скорость высвобождения активного компонента, где мас.% рассчитываются от общего сухого веса дозированной формы. Подходящими полимерами, определяющими скорость высвобождения активного компонента, являются: сложные эфиры глицерина, такие как глицерин моностеарат, глицерил бегенат, глицерил пальминостеарат, лаурилмакроголглицерид, стиролмакроголглицерид. Другие подходящие полимеры, определяющие скорость высвобождения активного компонента, включают: сополимеры метакрилата, сополимеры аммонийалкилметакриллата и их сополимеры или комбинации.
При пероральном приеме (т.е. когда пациент глотает таблетку), лекарственная форма может дать кривую изменения концентраций габапентина в плазме во времени, кривую, форма которой и время достижения максимальной концентрации в плазме (Тmax), показана на фигурах далее.
Краткое описание фигур
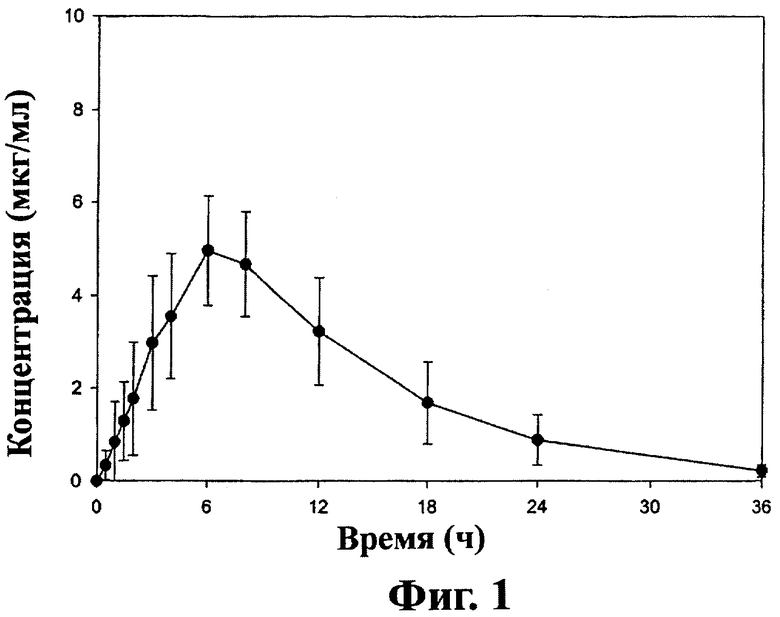
Фиг.1 - кривая средних значений концентраций ±SD (стандартное отклонение) габапентина в плазме после перорального приема таблеток замедленного высвобождения, содержащих соединение (1) (2×600 мг) пациентами на тощий желудок (терапия А), как описано в примере 3.
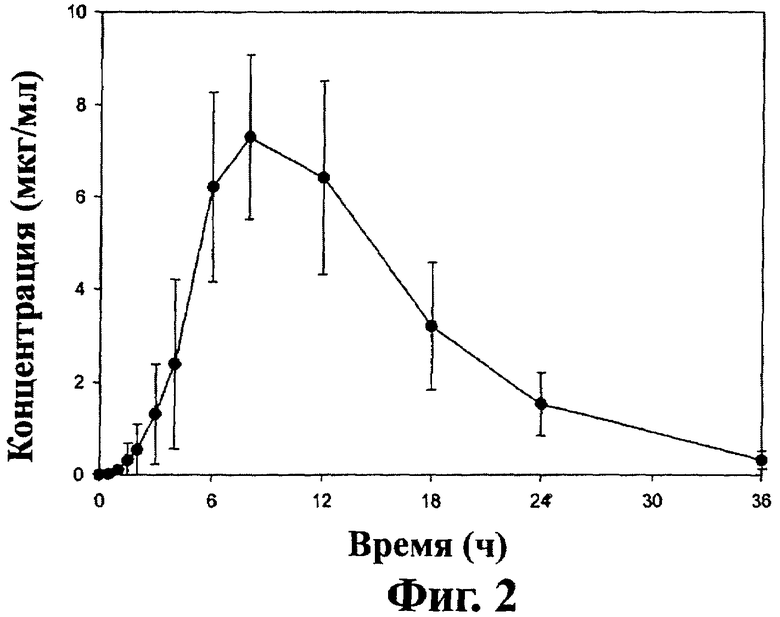
Фиг.2 - кривая средних значений концентраций ±SD габапентина в плазме после перорального приема таблеток замедленного высвобождения, содержащих соединение (1) (2×600 мг) пациентами на сытый желудок (терапия В), как описано в примере 3.
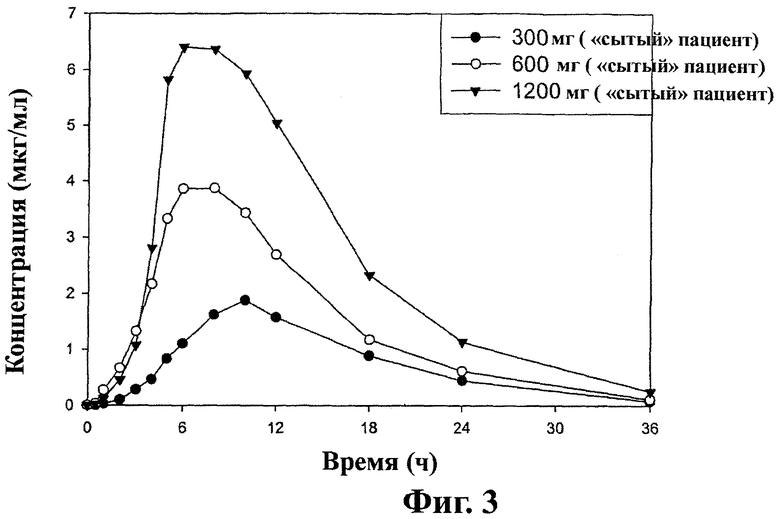
Фиг.3 - кривая средних значений концентраций ±SD габапентина в плазме после перорального приема таблеток замедленного высвобождения, содержащих соединение (1) (1×300 мг, 1×600 мг и 2×600 мг), пациентами на сытый желудок (терапия В), как описано в примере 4.
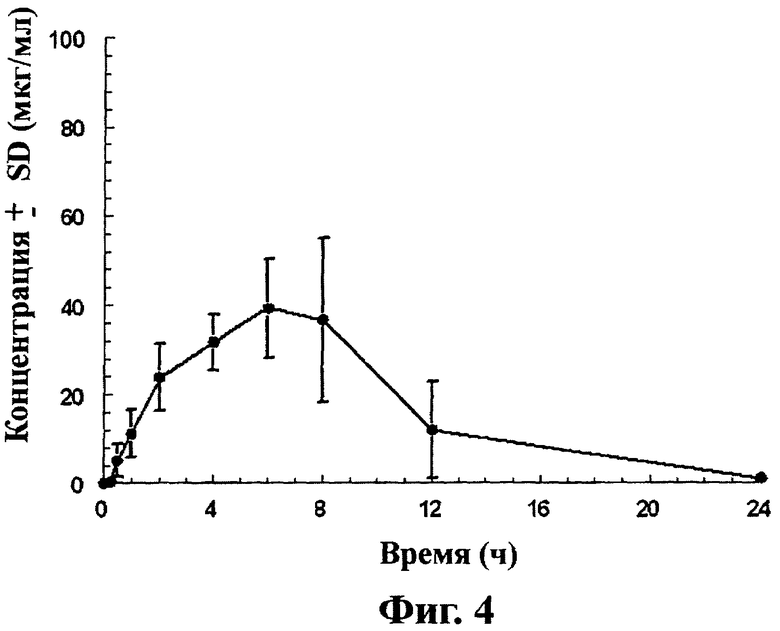
Фиг.4 - кривая средних значений концентраций ±SD габапентина в крови после перорального приема таблеток замедленного высвобождения, содержащих соединение (1) (1×600 мг), взрослым самцом Cynomologous monkeys на тощий желудок, как описано в примере 5.
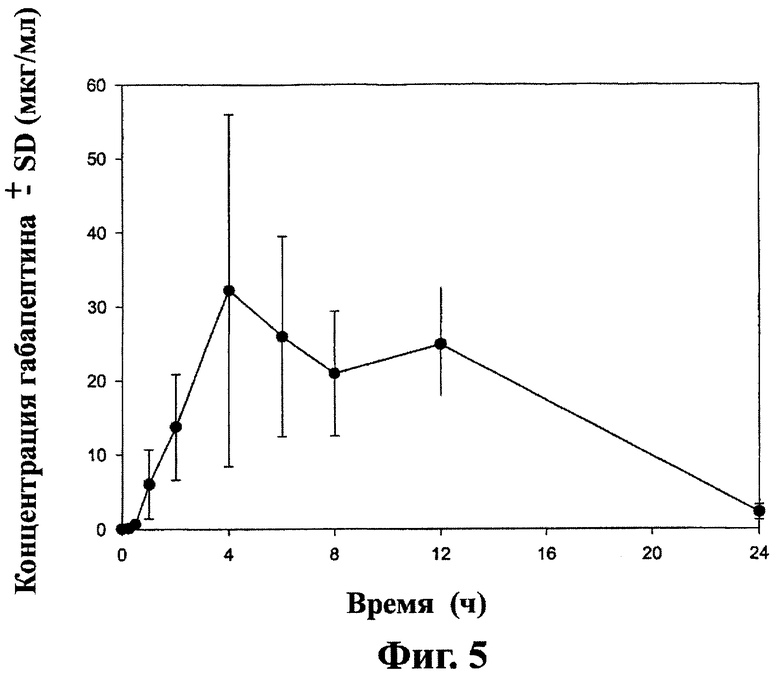
Фиг.5 - кривая средних значений концентраций ±SD габапентина в крови после перорального приема таблеток замедленного высвобождения, содержащих соединение (1) (1×600 мг), взрослым самцом Cynomologous monkeys на тощий желудок, как описано в примере 7.
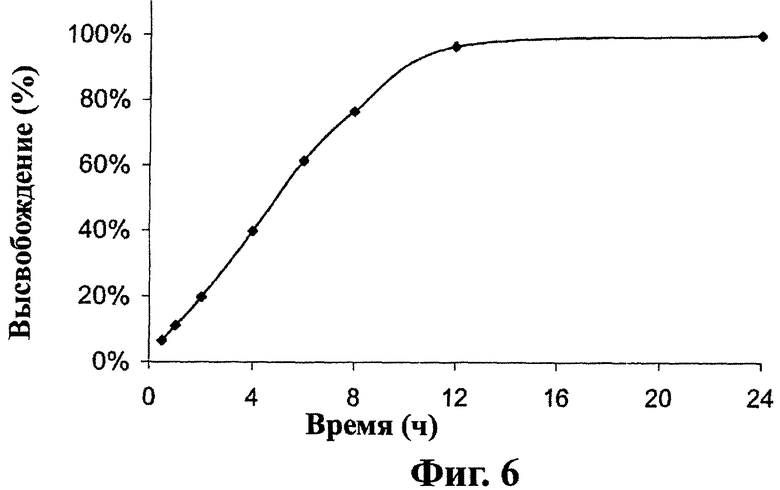
Фиг.6 - in vitro (в лабораторных условиях) кривая растворения дозированной формы настоящего изобретения, как описано в примере 1.
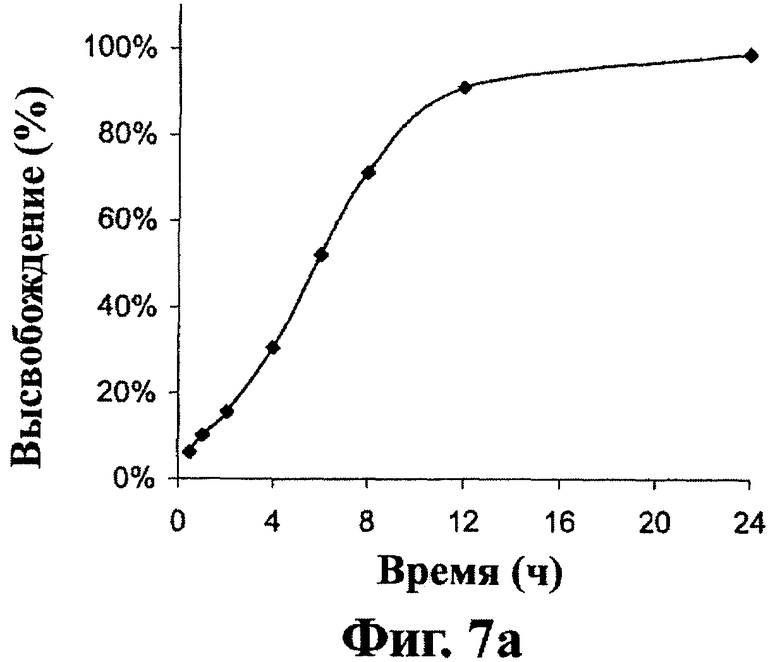
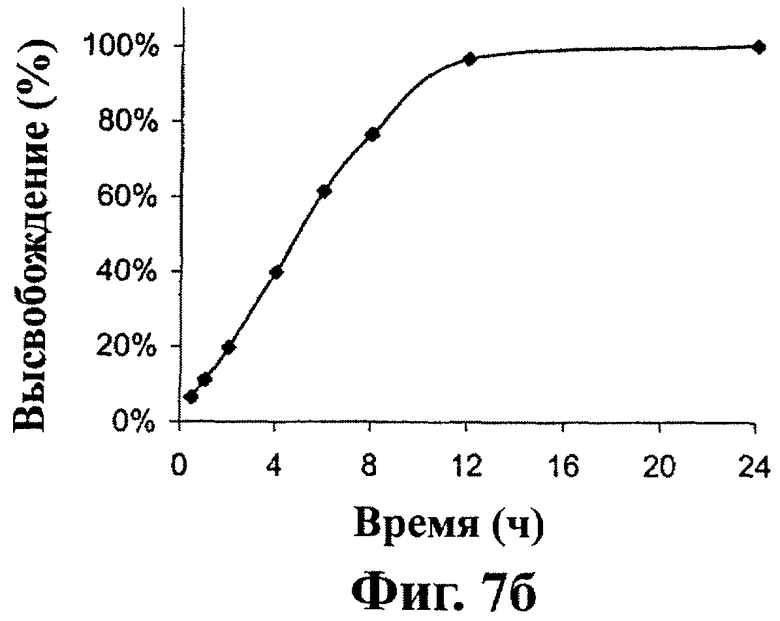
Фиг.7А и 7В - in vitro кривая растворения остальных дозированных форм настоящего изобретения, как описано в примере 2.
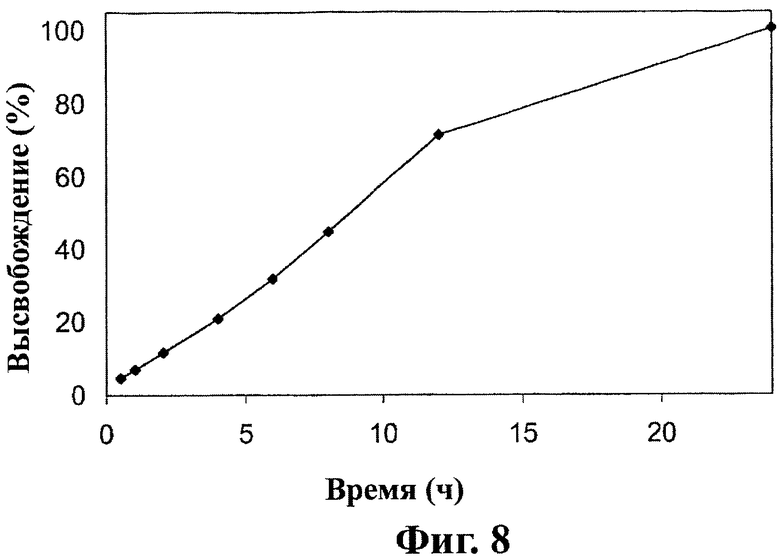
Фиг.8 - in vitro кривая растворения другой дозированной формы настоящего изобретения, как описано в примере 6.
Осуществление изобретения
Определения
"AUC" - площадь под кривой, представляющая концентрацию соединения или его метаболита в биологической жидкости пациента как функция времени, прошедшего после приема соединения пациентом. В конкретных вариантах осуществления изобретения соединение может быть пролекарством, а метаболит может быть лекарством. Примерами биологических жидкостей являются кровь и плазма. AUC определяют измерением концентрации соединения или его метаболита в биологической жидкости, такой как плазма или кровь с помощью методов, таких как жидкостная хроматография/масс/спектрометрия (LC/MS/MS), в различных временных интервалах, и расчетом площади под кривой зависимости концентрации в плазме от времени. Подходящие способы расчета AUC из кривой зависимости концентрации лекарственного средства от времени хорошо известны из литературы. Относительно данного описания изобретения, AUC для габапентина может быть определена измерением концентрации габапентина в плазме и/или крови пациента после перорального приема дозированной формы, включающей соединение (1), 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусную кислоту.
«Биологическая доступность» - скорость и количество лекарственного средства, которое достигает большого круга кровообращения пациента после приема дозированной формы или пролекарства пациентом и может быть оценена по, например, кривой зависимости концентрации соединения в плазме и/или крови от времени. Параметры, применимые для характеризации кривой зависимости концентрации соединения в плазме и/или крови от времени, включают площадь под кривой (AUC), время достижения максимальной концентрации (Тmax) и максимальную концентрацию лекарственного средства (Сmax).
«Биоэквивалентность» - эквивалентность скорости и степени всасывания лекарственного средства после приема пациентом эквивалентных доз лекарственного средства или пролекарства. В данном случае две кривые концентраций в плазме или крови являются биоэквивалентными, если 90% доверительный интервал для соотношения среднего отклика двух кривых находится в рамках от 0,8 до 1,25. Средний отклик включает по крайней мере один из характеристических параметров кривой, таких как Сmax, Tmax и AUC.
«Сmax» - максимальная концентрация лекарственного средства в плазме или крови пациента после приема пациентом дозы лекарственного средства или пролекарства.
«Тmax» - время, соответствующее максимальной концентрации (Сmax) лекарственного средства в плазме или крови после приема пациентом дозы лекарственного средства или пролекарства.
«голодный (натощак) пациент» - обозначает пациента, желудок которого по существу является свободным от пищи в момент принятия пациентом дозы лекарственного средства и, по крайней мере, в течение 4 часов после такого приема. Время, в течение которого желудок пациента остается по существу свободным от пищи после приема пищи, может зависеть от ряда факторов, включающих, например, размер порции, например, количество калорий, состав пищи, например, содержание жира, состояние здоровья пациента и состояние желудочно-кишечного тракта пациента. Желудок здорового человека становится по существу свободным от пищи после 4-8 часов после приема пищи. В конкретных вариантах осуществления натощак пациент не принимает никакой еды (но может принять некоторое количество воды или чистой жидкости) в течение около 10 часов до приема и около 4 часов после приема лекарственного средства, выпивает около 250 мл воды за примерно 2 часа и за примерно 1 час до приема и около 250 мл воды через примерно 2 часа после приема лекарственного средства, съедает ленч через примерно 4 часа после приема и съедает обед через примерно 10 часов после приема лекарственного средства.
«Сытый (после еды) пациент» - обозначает пациента, желудок которого содержит пищу. В конкретных вариантах осуществления «сытый» пациент начинает принимать пробный завтрак за примерно 30 минут до и заканчивает принимать пищу за примерно 5 минут до принятия лекарственного средства, съедает ленч через примерно 4 часа после приема и съедает обед через примерно 10 часов после приема лекарственного средства. Пробный завтрак включает высокожирный (около 50% общего количества калорий в пробной пище) и высококалорийный (в сумме около 1000 калорий) завтрак, такой как, например, 2 яйца, жареных в масле, 2 полоски бекона, 2 куска пшеничного тоста с маслом, 4 унции нарезанного коричневого картофеля и 8 унций молока. Пробный завтрак содержит около 150 калорий белка, 250 калорий углеводов и около 500-600 калорий жира.
«Пациент» - млекопитающее, например человек.
«Фармацевтически приемлемый» - одобренный или утвержденный регулирующим ведомством государства или занесенный в Фармакопею США или другие общепризнанные фармакопии для использования на животных и более конкретно на человеке.
«Фармацевтически приемлемые соли» - соль соединения по настоящему изобретению, которая является фармацевтически приемлемой и обладает желаемой фармакологической активностью исходного вещества. Такие соли включают: соли (1), образующиеся присоединением кислоты, например неорганической кислоты, такой как хлороводородная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные; или образованные из органических кислот, таких как: уксусная кислота, пропановая кислота, гексановая кислота, циклопентанпропановая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил) бензойная кислота, коричная кислота, миндальная кислота метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]-окта-2-ен-1 карбоновая кислота, глюкогептановая кислота, 3-фенилпропановая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и подобные; или соли (2), образующиеся либо при замене кислотного протона исходной кислоты ионом металла, например ионом щелочного, щелочноземельного металла или ионом алюминия или при координации кислотного протона исходной кислоты с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, N-метилглюкамин и подобными.
«Предотвращение» - уменьшение риска приобретения заболевания или расстройства (то есть лекарственное средство не должно быть причиной возникновения ни одного клинического симптома заболевания, не развитого у пациента, который может быть подверженным или предрасположенным к заболеванию, но еще не чувствовать или не обнаруживать симптомы заболевания).
«Пролекарство» - производное молекулы лекарственного средства, которое требует трансформации в организме для высвобождения активного лекарственного средства. Соединение (1) является пролекарством, которое в процессе метаболизма в организме пациента, образует исходное (материнское) лекарственное средство, габапентин и, следовательно, соединение (1) является пролекарством габапентина. Соединение (1), 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусная кислота, включает фармацевтически приемлемые соли и фармацевтически приемлемые сольваты свободной кислотной формы соединения (1), также как и кристаллические формы всех вышеупомянутых соединений.
«Сольват» - молекулярный комплекс соединения с одной или более молекулами растворителя в стехиометрическом или нестехиометрическом количестве. Такими молекулами могут быть молекулы растворителей, которые традиционно используются в фармацевтической отрасли и которые являются безвредными для пациента, например вода, этанол и подобные. Молекулярный комплекс соединения или части соединения и растворителя может быть стабилизирован нековалентными внутримолекулярными силами, такими как, например, электростатические силы, Ван-дер-Ваальсовы силы или водородные связи. Термин «гидрат» обозначает комплекс, в котором присутствует одна или более молекул воды.
«Длительное выделение (замедленное высвобождение)» - высвобождение соединения (1) из дозированной формы со скоростью, эффективной для достижения терапевтической или профилактической концентрации соединения (1) или его активного метаболита в большом круге кровообращения в течение продолжительного периода времени по сравнению с периодом времени, которое требуется для препарата соединения (1) немедленного действия при пероральном приеме. В некоторых воплощениях соединение (1) высвобождается в течение, по меньшей мере, 6 часов, в некоторых воплощениях - по меньшей мере - 12 часов, в некоторых - по меньшей мере - 18 часов и в некоторых - по меньшей мере - 24 часа.
«Терапевтически эффективное количество» - количество соединения (1), которое при приеме пациентом для лечения или предотвращения заболевания является достаточным для оказания такого действия по лечению или предотвращению заболевания. «Терапевтически эффективное количество» будет варьироваться в зависимости от заболевания и его серьезности и возраста, веса и т.д. пациента, страдающего заболеванием, которое необходимо излечить или предупредить.
«Лечением» при любом заболевании или нарушении называется, в конкретных вариантах осуществления, улучшение при заболевании или нарушении (то есть приостановка или уменьшение развития заболевания или, по крайней мере, одного из его клинических симптомов). В других конкретных вариантах осуществления «лечением» называется уменьшение по крайней мере одного физического параметра, который может быть или может не быть видимым для пациента. В конкретных вариантах осуществления «лечением» называется ингибирование (подавление) заболевания или нарушения, либо физически (то есть стабилизацией видимого симптома) или физиологически (то есть стабилизацией физического параметра), или обоих. В конкретных вариантах осуществления «лечением» называется приостановка приступа болезни или нарушения.
«мас.%» - вес компонента или ингредиента по отношению к общему сухому весу композиции или дозированной формы, то есть процент по весу. Например, дозированная форма, включающая 40 мас.% соединения (1), и вес которой составляет 1000 мг, будет содержать 400 мг вещества (1). При рассмотрении соли и/или сольвата соединения (1), мас.% рассчитывается по массе, эквивалентной весу 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, образующей фармацевтически приемлемую соль и/или фармацевтически приемлемый сольват соединения (1).
Далее будут приведены конкретные варианты осуществления дозированных форм и способов. Раскрытые варианты осуществления не ограничивают притязания, отраженные в формуле изобретения. Наоборот, формула изобретения будет включать все альтернативы, модификации и эквиваленты раскрытых вариантов осуществления.
Дозированные формы замедленного высвобождения
Хотя конкретные варианты осуществления настоящего изобретения представляют дозированные формы замедленного высвобождения в форме таблеток, специалисты в области пероральных дозированных форм замедленного высвобождения определят, что также могут быть использованы другие дозированные формы, такие как: порошки, частицы, саше (маленькие пакеты), жидкие суспензии и/или капсулы. В конкретных вариантах осуществления дозированная форма может быть таблеткой, которая может иметь любую подходящую форму для перорального применения лекарственного средства, такую как: сферическую, кубовидную овальную или эллипсоидальную. Таблетка может содержать порционную дозу соединения (1); в случае мини таблетки, которая включает менее одной порционной дозы, мини таблетки могут быть помещены в капсулы для получения одной порционной дозы. В конкретных вариантах осуществления таблетка может являться многослойной таблеткой, в которой разные слои содержат частицы разного размера и/или наполнители, которые влияют на высвобождение соединения (1) из каждого слоя таблетки. Примерами таких таблеток являются: распадающиеся таблетки, быстрорастворимые таблетки, шипучие таблетки, быстроплавящиеся таблетки, жевательные таблетки, дробящиеся таблетки и мини таблетки. В общем, таблетированные лекарственные формы могут прессоваться до степени твердости в 15 килофунтов (кр) (эквивалентных 147,1 Ньютону). Дозированные формы могут быть получены описанными в литературе способами и могут, кроме того, при необходимости содержать фармацевтически приемлемые наполнители.
В некоторых вариантах осуществления дозированные форма замедленного высвобождения включает около (а) 10 мас.% - 80 мас.% соединения (1) и около (в) 1 мас.% - 30 мас.% полимера, модулирующего скорость выделения лекарственного вещества.
В некоторых вариантах осуществления лекарственная доза замедленного высвобождения включает около (а) 10 мас.% - 80 мас.% соединения (1) и около (в) 1 мас.% - 50 мас.% полимера, модулирующего скорость выделения лекарственного вещества.
В некоторых вариантах осуществления дозированная форма замедленного высвобождения включает около (а) 30 мас.% - 75 мас.% соединения (1) и около (в) 1 мас.% - 50 мас.% полимера, модулирующего скорость выделения лекарственного вещества.
В некоторых вариантах осуществления дозированная форма замедленного высвобождения включает около (а) 40 мас.% - 65 мас.% соединения (1) и около (в) 1 мас.% - 50 мас.% полимера, модулирующего скорость выделения лекарственного вещества.
В некоторых вариантах осуществления дозированная форма замедленного высвобождения включает около (а) 50 мас.% - 60 мас.% соединения (1) и около (в) 20 мас.% - 50 мас.% полимера, модулирующего скорость выделения лекарственного вещества.
Дозированные формы данного изобретения являются матричными системами, в которых соединение (1) является гомогенно распределенным в полимере, модулирующем скорость выделения лекарственного вещества, и, при необходимости, наполнителях. Матричные системы хорошо известны в литературе и описаны, например, в «Handbook of Pharmaceutical Controlled Release Technology», ed. D.L.Wise, Marcel Dekker, Inc. (2000) и «Treatise on Controlled Drug Delivery, Fundamentals, Optimization, and Applications», ed. A. Kydonieus, Marcel Dekker, Inc, (1992). Полимеры, модулирующие скорость выделения, могут тормозить выделение активного вещества из дозированной формы. Подходящие скорость модулирующие полимеры включают, но не ограничиваются рН чувствительными полимерами, рН нечувствительными полимерами, гидрофильными полимерами, которые имеют высокую степень набухаемости при контакте с водой или водными средами, полимерами, образующими гель при контакте с водой или водными средами, полимерами, которые и набухают, и образуют гели при контакте с водой или водными средами, жирные вещества, такие как смолы и биоразлагающиеся полимеры.
В ряде вариантов осуществления полимер, модулирующий скорость высвобождения, может быть рН чувствительным полимером, таким как полимеры и сополимеры акриловой кислоты и метакриловой кислоты, сополимеры метил метакрилата, этоксиэтил метакрилата, цианоэтил метакрилата, поли(акриловой кислоты), поли(метакриловой кислоты), сополимеры алкиламида метилакриловой кислоты, поли(метилметакрилата), полиметакрилата, сополимеры поли(метилметакрилата), полиактиламида, полиакриламид, сополимер аминоалкил метакрилата, поли(ангидрид метакриловой кислоты), сополимеры глицидил метакрилата, сополимеры аммонийалкил метакрилата и комбинаций всех вышеперечисленных. В конкретных вариантах осуществления рН-зависимый полимер может быть сополимером, полученным из диэтиламиноэтил метакрилата и других нейтральных метакриловых эфиров, также известных как сополимеры метакриловой кислоты или полимер метакрилаты, коммерчески доступные под торговой маркой Eudragit (Rohm Pharma).
В конкретных вариантах осуществления рН-нечувствительный полимер является сополимером аммонийалкил метакрилата, таким как Eudragit RS и Eudragit RL, которые являются акриловыми резинами, включающими сополимеры эфиров акриловой и метакриловой кислот с низким содержанием четвертичных аммонийных групп.
Примеры гидрофильных модулирующих скорость высвобождения полимеров, имеющих высокую степень набухания, включают сшитую натрий карбоксиметилцеллюлозу, сшитую гидроксипропилцеллюлозу, высокомолекулярную гидроксипропилметилцеллюлозу, карбоксиметиламид, сополимер калий метакрилатдивинилбензола, полиметилметакрилат, поливинилпирролидон, высокомолекулярные поливиниловые спирты, метилцеллюлозы, сополимеры винилацетата и подобных.
Примеры модулирующих скорость высвобождения полимеров, образующих гель при контакте с водой, включают метилцеллюлозу, карбоксиметилцеллюлозу, низкомолекулярную гидроксипропилметилцеллюлозу, низкомолекулярные поливиниловые спирты, полиэтиленгликоли, несшитые поливинилпирролидоны, ксантановую смолу и подобные.
Примеры модулирующих скорость высвобождения полимеров, которые и набухают, и образуют гели при контакте с водой, включают средневязкие гидроксипропилметил целлюлозы и средне вязкие поливиниловые спирты.
В конкретных вариантах осуществления полимером, модулирующим скорость высвобождения лекарства, является глицериловый эфир, такой как глицерил моностеарат, глицерил бехенат, глицерил пальмитостеарат, лаурилмакроголглицерид, стиролмакроголглицерид или комбинации всех вышеперечисленных. В конкретных вариантах осуществления полимером, модулирующим скорость высвобождения лекарства, является глицерил бехенат. Другие жирные и/или воскообразные скорость модулирующие полимеры включают лауриловый спирт, миристиловый спирт, стеариловый спирт, цетиловый спирт, цитостеариловый спирт, пальмитоиловый спирт, урицирный воск, гидрированное растительное масло, канделильский воск, эспартовый воск, стеариновую кислоту, твердый парафин, пчелиный воск, глико-воск, гидрированное касторовое масло и карнубский воск.
Примеры биоразлагающихся полимеров включают: коллаген, желатин, поливиниловые спирты, сложные полиортоэфиры, полиацетилы, полиортокарбонаты, полиамиды, полиаминокислоты, сложные полиэфиры, полимолочные кислоты, полигликолевые кислоты, полиуглеводы, сложные полиэфиры, полиортокарбонаты, полиацетилы, полиангидриды, полидегидропираны, полидиоксиноны и подобные.
Другие приемлемые скорость модулирующие полимеры, которые могут быть включены в дозированную форму настоящего изобретения, включают гидроколлоиды, такие как натуральные или синтетические клеи, вещества на основе углеводов, такие как камедь, трагакантовая камедь, смола плодоворожкового дерева, гуаровая смола, агар, пектин, карагенин, растворимые и нерастворимые альгинаты, карбоксиполиметилен, казеин, зеин, полиэтиленоксид, сополимеры малеинового ангидрида/метилвинилового эфира, и белкоподобные вещества, такие как желатин.
Скорость модулирующий полимер может быть использован один или в комбинации с одним или более другими скорость модулирующими полимерами и/или может быть сополимером более чем одного скорость модулирующего полимера.
Препарат соединения (1) и одного или более полимеров, модулирующих скорость выделения лекарственного вещества, может быть приготовлен с использованием стандартных способов, хорошо известных из литературы, таких как влажное гранулирование, жидкослойное гранулирование, сухое гранулирование и прямое прессование (см. «Remington′s Pharmaceutical Sciences» Lippincott Williams&Wilkins, 889-928, (2005)). Например, матричный препарат может быть приготовлен сухим смешиванием модулирующего скорость выделения полимера, соединения (1) и наполнителей, за которым следует гранулирование смеси с использованием спирта до получения надлежащего гранулирования. Гранулирование может быть проведено способами, известными в литературе. Влажные гранулы могут быть высушены в жидкослойной сушилке, просеяны и загрунтованы до подходящего размера. Смазки могут быть смешаны с сухими гранулами для получения конечного препарата. В конкретных вариантах осуществления такие препараты могут быть спрессованы в таблеточные дозированные формы методами, хорошо известными из литературы.
В некоторых вариантах осуществления количество соединения (1) в дозированной форме может колебаться от 50 мг до 800 мг, в некоторых вариантах осуществления - от 100 мг до 800 мг и в некоторых вариантах осуществления - от 300 мг до 700 мг. Для дозированной формы, включающей фармацевтически приемлемую соль и/или сольват соединения (1), количество соединения (1) в дозированной форме рассчитывается из массы, эквивалентной весу соединения (1). Количество или загрузка соединения (1), включенного в дозированную форму, может зависеть от конкретного условия приема и от количества габапентина, генерируемого из пролекарства в процессе последующего всасывания.
Кроме соединения (1) и описанных здесь модулирующих скорость выделения полимеров дозированная форма может также включать один или несколько фармацевтически приемлемых наполнителей, таких как поверхностно-активные вещества, смазки, разбавители, антиадгезивы, вещества, способствующие скольжению (лубриканты), буферы, красители, увлажнители, эмульгаторы, рН-буферы, стабилизаторы, загустители, дезинтегранты и окрашивающие вещества. Такие наполнители включают крахмал, сахара, желатин, солод, рис, муку, мел, силикагель, стеарат натрия, моностеарат глицерина, тальк, хлорид натрия, глицерин, пропиленгликоль, воду, этанол и подобные.
Разбавители могут прибавляться для увеличения объема, для придания практического размера лекарственной форме для дальнейшего прессования. Примерами используемых разбавителей могут быть: двухосновный фосфат кальция, двухосновный фосфат кальция дигидрат, сульфат кальция, дифосфат кальция, трифосфат кальция, лактоза, целлюлоза, включающая микрокристаллическую целлюлозу, каолин, маннит, натрий хлорид, сухой крахмал, прежелатинизированный крахмал, сжатый сахар, комбинации всех перечисленных. В конкретных вариантах осуществления разбавители выбирают из двухосновного фосфата кальция и микрокристаллической целлюлозы. В некоторых вариантах осуществления, где разбавителем является двухосновный фосфат кальция, дозированная форма может включать количество разбавителя около 30 мас.% - 50 мас.% и в некоторых вариантах осуществления около 35 мас.% - 45 мас.%. В некоторых вариантах осуществления, где разбавителем является микрокристаллическая целлюлоза, дозированная форма может включать около 5 мас.% - 20 мас.% разжижителя и в некоторых вариантах осуществления около 10 мас.% - 16 мас.%.
Вещества, способствующие скольжению (лубриканты), могут быть включены в дозированную форму в настоящем изобретении для уменьшения эффекта слипания в процессе обработки, образования пленки и/или высушивания. Примерами используемых веществ, способствующих скольжению, могут быть тальк, стеарат магния, моностеарат глицерина, коллоидный диоксид кремния, осажденный диоксид кремния и комбинации всех перечисленных. В некоторых вариантах осуществления в качестве вещества, способствующего скольжению, используется коллоидный диоксид кремния. Дозированная форма может включать менее примерно 2 мас.% вещества, способствующего скольжению, и в некоторых вариантах осуществления менее примерно 1 мас.% вещества, способствующего скольжению.
Смазки и антиадгезивы могут быть включены в дозированную форму по настоящему изобретению для облегчения процесса обработки. Примерами используемых смазок и/или антиадгезивов могут быть стеарат кальция, бегенат глицерина, моностеарат глицерина, стеарат магния, минеральное масло, полиэтиленгликоль, натрия стеарил фумарат, натрия лаурил сульфат, натрия додецил сульфат, стеариновая кислота, тальк, гидрированное растительное масло, стеарат цинка и их комбинация. В некоторых вариантах осуществления в качестве смазки используется моностеарат глицерина. В некоторых вариантах осуществления в качестве смазки используется стеарат магния. Дозированная форма может включать количество смазки и антиадгезива около 1 мас.% - 13 мас.% и в некоторых вариантах осуществления около 4 мас.% - 10 мас.%.
Примерами поверхностно-активных веществ, используемых в дозированных формах настоящего изобретения, могут быть фармацевтически приемлемые анионные поверхностно-активные вещества, катионные поверхностно-активные вещества, амфотерные (амфипатические/амфифильные) поверхностно-активные вещества, неионные поверхностно-активные веществ, сложные или простые эфиры полиэтиленгликоля и комбинации всех перечисленных. Примерами используемых фармацевтически допустимых анионных поверхностно-активных веществ могут быть моновалентные алкил карбоксилаты, ацил лактилаты, алкилэфирные карбоксилаты, N-ацил саркосинаты, поливалентные алкил карбонаты, N-ацил глютаматы, полипептидные конденсаты жирных кислот, сложные эфиры серной кислоты, алкил сульфаты, такие как натрия лаурил сульфат и натрия додецил сульфат, этоксилированные алкил сульфаты, сложноэфирные сшитые сульфонаты, такие как натрия докузат и диоктил натрия сукцинат, альфа олефин сульфонаты или фосфаты этоксилированных спиртов. Примерами используемых фармацевтически допустимых катионных поверхностно-активных веществ могут быть моноалкильные четвертичные аммонийные соли, диалкильные четвертичные аммонийные вещества, амидоамины и аминимиды. Примерами используемых фармацевтически допустимых амфотерных поверхностно-активных веществ могут быть N-замещенные алкил амиды, N-алкилбетаины, сульфобетаины и N-алкил-6-аминопропионаты. Примерами используемых фармацевтически допустимых сложных или простых эфиров полиэтиленгликоля могут быть полиэтоксилированное касторовое масло, полиэтоксилированное гидрированное касторовое масло и гидрированное касторовое масло. В некоторых вариантах осуществления поверхностно-активное вещество выбирают из натрия лаурил сульфата и натрия додецил сульфата. В некоторых вариантах осуществления дозированная форма может включать менее примерно 3 мас.% поверхностно-активного вещества и в некоторых вариантах осуществления менее примерно 2 мас.% поверхностно-активного вещества.
Дозированные формы настоящего изобретения, такие как таблетированные лекарственные формы, могут кроме этого включать одно или более покрытий (оболочек). Целью одного или более покрытий может быть физическая защита, эстетика, легкость проглатывания, идентификации и/или дальнейшее облегчение процесса обработки частиц. В то время как конкретные покрытия могут применяться для модификации или воздействия на процесс высвобождения соединения (1) из дозированной формы в желудочно-кишечном тракте, другие покрытия могут не иметь такого эффекта. Покрытия могут быть влагонепроницаемыми или влагопроницаемыми. Влагопроницаемая внешняя оболочка таблетки может быть использована для поддержания низкого содержания влаги в лекарственной форме, которая пакуется в присутствии осушителя и может, таким образом, улучшить, например, стойкость лекарственной формы при хранении. Эти дополнительные покрытия могут наноситься на лекарственные формы настоящего изобретения способами, хорошо известными специалистам в данной области. Примерами материалов, используемых в покрытиях для физической защиты, могут быть проницаемые или растворимые материалы, такие как гидроксипропил метилцеллюлоза, гидроксипропил целлюлоза, гидроксипропил этилцеллюлоза и ксантановая смола. Примерами материалов, применяемых в покрытиях для облегчения дальнейшего процесса обработки, могут быть тальк, коллоидный диоксид кремния, поливиниловый спирт, диоксид титана, тонкоизмельченный диоксид кремния, коллоидный диоксид кремния, моностеарат глицерина, трисиликат магния и стеарат магния. Покрытие может включать один материал или комбинацию более чем одного материала, включая любой из описанных в данной работе.
Дозированная форма в настоящем изобретении может, в основном, не содержать примесей побочных лактамовых продуктов, которые образуются внутримолекулярной циклизацией соединения (1) и/или габапентина. Дозированная форма предпочтительно является стабильной при продолжительном хранении (более предпочтительно в течение более 1 года) без значительного образования лактама (предпочтительно менее 0,5% лактама по весу, более предпочтительно менее чем 0,2% лактама по весу, наиболее предпочтительно менее 0,1% лактама по весу).
В некоторых вариантах осуществления соединение (1) в дозированной форме является кристаллической формой, описанной в Estrada et al., U.S. Patent Application Publication US 2005/0154057 от 14 июля 2005.
При пероральном назначении пациенту (то есть, когда пациент проглатывает таблетку) дозированная форма настоящего изобретения может давать кривую изменения концентрации габапентина в плазме или крови во времени. Для дозированной формы, имеющей состав и загрузку, описанные в примере 1, кривая изменения концентрации габапентина в плазме имеет форму, величину и AUC, представленные на фиг.1 и 2 для «голодного» и «сытого» пациентов-людей соответственно после приема соединения (1) пациентом. Эти кривые отличаются от кривых, полученных после приема одного габапентина. Одно важное отличие - время (Тmax) для достижения максимальной концентрации в крови (Сmax). Для «голодного» пациента Тmax для оральной дозированной формы замедленного высвобождения настоящего изобретения составляет более 4 часов. Для «сытого» пациента Тmax для пероральной дозированной формы замедленного высвобождения настоящего изобретения составляет более 6 часов. Для сравнения Тmax после приема габапентина для «голодного» и «сытого» пациента составляет около 2-4 часов. Другое важное преимущество пероральных дозированных форм замедленного высвобождения настоящего изобретения состоит в биодоступности метаболита габапентина. При ударной дозе в 1200 мг соединения (1) лекарственная форма настоящего изобретения может дать, по крайней мере, на 20% больше биодоступности габапентина, и в некоторых вариантах осуществления, по крайней мере, на 25% больше биодоступности для «голодных» пациентов по сравнению с приемом эквимолярной дозы габапентина. При ударной дозе 1200 мг соединения (1) пероральная дозированная форма замедленного высвобождения по настоящему изобретению может дать, по крайней мере, на 50% больше биодоступности габапентина и в некоторых вариантах осуществления по крайней мере на 100% больше биодоступности габапентина для «сытого» пациента по сравнению с приемом эквимолярной дозы габапентина.
В некоторых вариантах осуществления пероральная дозированная форма замедленного высвобождения 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, при приеме одним или более «голодным» пациентом в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты около 1100-1300 мг, может обеспечить кривую изменения концентрации габапентина в плазме Смах около 3-6 мкг/мл при Тмах примерно 4-7 часов и AUC 30-70 мкг·час/мл.
В некоторых вариантах осуществления пероральная дозированная форма замедленного высвобождения 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, при приеме одним или более «сытым» пациентом в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты от около 1100 мг - 1300 мг, может обеспечить кривую изменения концентрации габапентина в плазме с примерно Смах от 5 мкг/мл до 8 мкг/мл при Тмах от примерно 6 часов до 11 часов и AUC примерно от 60 мкг·час/мл до 110 мкг·час/мл.
В некоторых вариантах осуществления пероральная дозированная форма замедленного высвобождения 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, при приеме группой «голодных» пациентов в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты около 1100-1300 мг, может обеспечить кривую изменения концентрации габапентина в плазме с примерно Смах от примерно 3 мкг/мл до 6 мкг/мл при Тмах от примерно 4 часов до 7 часов и AUC примерно от 30 мкг·час/мл до 70 мкг·час/мл; и при приеме группой «сытых» пациентов в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты около 1100 мг - 1300 мг, может обеспечить кривую изменения концентрации габапентина в плазме с примерно Смах примерно от 5 мкг/мл до 8 мкг/мл при Тмах от примерно 6 часов до 11 часов и AUC примерно от 60 мкг·час/мл до 110 мкг·час/мл.
В некоторых вариантах осуществления пероральный прием двух дозированных форм замедленного высвобождения, каждая по 600 мг 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, одним или более «голодным» пациентом-человеком может обеспечить кривую изменения концентрации габапентина в плазме с примерно Смах от 3 мкг/мл до 6 мкг/мл при Тмах от примерно 4 часов до 7 часов и AUC примерно от 30 мкг·час/мл до 70 мкг·час/мл.
В некоторых вариантах осуществления пероральный прием двух дозированных форм замедленного высвобождения, каждая по 600 мг 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, одним или более сытым пациентом-человеком может обеспечить кривую изменения концентрации габапентина в плазме с примерно Смах от 5 мкг/мл до 8 мкг/мл при Тмах примерно от 6 часов до 11 часов и AUC от примерно 60 мкг·час/мл до 110 мкг·час/мл.
В некоторых вариантах осуществления пероральный прием двух дозированных форм замедленного высвобождения, каждая по 600 мг 1-{[(α-изобутаноилоксиэтокси)-карбонил]аминометил}-1-циклогексан уксусной кислоты, группой «голодных» пациентов-людей может обеспечить кривую изменения концентрации габапентина в плазме с Смах от около 3 мкг/мл до 6 мкг/мл при Тмах от около 4 часов до около 7 часов и AUC от около 30 мкг·час/мл до около 70 мкг·час/мл; и пероральный прием двух дозированных форм замедленного высвобождения, каждая по 600 мг 1-{[(α-изобутаноилоксиэтокси)-карбонил]аминометил}-1-циклогексан уксусной кислоты, группой «сытых» пациентов-людей может обеспечить кривую изменения концентрации габапентина в плазме с Смах от примерно 5 мкг/мл до около 8 мкг/мл при Тмах от около 6 часов до около 11 часов и AUC от около 60 мкг·час/мл до около 110 мкг·час/мл.
Дозированные формы настоящего изобретения включают лекарственные формы, которые являются биоэквивалентными дозированным формам, описанным в данном изобретении в показателях скорости и степени всасывания, например, как обговаривается в U.S. Food and Drug Administration и обсуждается в «Guidance for Industry - Bioavailability and Bioequivalence Studies for Orally Administered Drug Products» (2003).
В некоторых вариантах осуществления пероральная дозированная форма замедленного высвобождения 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, при приеме одним или более «голодным» пациентом-человеком в дозе около 1100-1300 мг обеспечивает кривую изменения концентрации габапентина в плазме, биоэквивалентную кривой на фиг.1, или при приеме одним или более «сытым» пациентом-человеком обеспечивает кривую изменения концентрации габапентина в плазме, биоэквивалентную кривой на фиг.2.
В некоторых вариантах осуществления пероральная дозированная форма замедленного высвобождения 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, при приеме одним или более «голодным» пациентом-человеком в дозе около 1100-1300 мг обеспечивает кривую изменения концентрации габапентина в плазме, биоэквивалентную кривой на фиг.1, или при приеме одним или более «сытым» пациентом-человеком обеспечивает кривую изменения концентрации габапентина в плазме, биоэквивалентную кривой на фиг.2.
В некоторых вариантах осуществления при приеме двух пероральных дозированных форм замедленного высвобождения, каждая дозированная форма включает по 600 мг 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, одним или более «голодным» пациентом-человеком обеспечивает кривую изменения концентрации габапентина в плазме, биоэквивалентную кривой на фиг.1, или при приеме той же дозированной формы одним или более «сытым» пациентом обеспечивает кривую изменения концентрации габапентина в плазме, биоэквивалентную кривой на фиг.2.
В некоторых вариантах осуществления при приеме двух пероральных дозированных форм замедленного высвобождения каждая дозированная форма включает по 600 мг 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, одним или более «голодным» пациентом-человеком обеспечивает кривую изменения концентрации габапентина в плазме, биоэквивалентную кривой на фиг.1, или при приеме той же дозированной формы одним или более «сытым» пациентом обеспечивает кривую изменения концентрации габапентина в плазме, биоэквивалентную кривой на фиг.2.
После перорального приема дозированные формы настоящего изобретения могут обеспечивать терапевтическую или профилактическую концентрацию габапентина в крови и/или плазме пациента в течение, по крайней мере, около 6 часов, в некотор7ых вариантах осуществления в течение по крайней мере около 12 часов, в некоторых вариантах осуществления в течение по крайней мере около 18 часов и в некоторых вариантах осуществления в течение по крайней мере около 24 часов. Терапевтически или профилактически эффективные концентрации габапентина в крови и/или плазме пациента могут зависеть от ряда факторов, например заболевания, которое требует лечения, интенсивности заболевания, веса пациента, здоровья пациента и так далее.
В некоторых вариантах осуществления дозированные лекарственные формы настоящего изобретения могут приниматься два раза в день и в некоторых вариантах осуществления - один раз в день.
Терапевтическое применение
Дозированные формы замедленного высвобождения по настоящему изобретению могут применяться пациентом, страдающим от любого заболевания или нарушения, при котором родоначальное лекарственное средство, габапентин, как уже известно или будет открыто, является терапевтически эффективным. Условия, которые были установлены для габапентина, и при которых, следовательно, дозированные лекарственные формы данного изобретения также будут эффективными, включают: эпилепсию, депрессию, беспокойство, психоз, cognition, шизофрению, приступы слабости, гипокинезию, черепные заболевания, нейродегенеративные заболевания, панику, боль (особенно невропатическую боль (например, пост-герпетическую невралгию), мышечную или скелетную боль), синдром усталых ног, приступы жара, недержание мочи, воспалительные заболевания (например, артрит), бессонницу, гастроинтестинальные заболевания, алкогольная/кокаиновая зависимости, синдром отмены этанола, вульводинию, преждевременную эякуляция и как глютаматергик. Лекарственные формы по изобретению также применяются для пациентов в качестве превентивных мер против вышеперечисленных заболеваний и нарушений. Так, дозированные формы могут применяться в качестве превентивных мер для пациентов, имеющих предрасположенность к эпилепсии, депрессии, беспокойству, психозу, приступам слабости, гипокинезии, черепным заболеваниям, нейродегенеративным заболеваниям, панике, боли (особенно невропатической боли, мышечной или скелетной боли), воспалительным заболеваниям (например, артриту), бессоннице, гастроинтестинальным заболеваниям, синдрому отмены этанола, преждевременной эякуляции и вульводинии. Таким образом, дозированные формы могут применяться для предотвращения одного заболевания или нарушения и одновременно для лечения другого заболевания или нарушения (например, предотвращения психоза при лечении гастроинтестинальных заболеваний; предотвращения невропатической боли при лечении синдрома отмены этанола). Лекарственные формы по изобретению могут применяться в комбинации с другими лекарственными средствами, такими как противовирусные лекарства при первоначальной вирусной инфекции для предотвращения или уменьшения возникших позже невропатических заболеваний. Кроме того, дозированные лекарственные формы могут применяться в комбинации с другими лекарственными средствами, которые сами являются причиной возникновения невропатических заболеваний в качестве побочного эффекта, таким образом, предотвращая или уменьшая появление таких побочных процессов.
Применимость дозированных лекарственных форм для лечения или предотвращения вышеперечисленных заболеваний или нарушений может быть определена методами, описанными в литературе ((See, e.g., Satzinger et al, United States Patent No. 4,024,175; Satzinger et al, United States Patent No. 4,087,544; Woodruff, United States Patent No. 5,084,169; Silverman et al, United States Patent No. 5,563,175; Singh, United States Patent No. 6,001,876; Horwell et al, United States Patent No. 6,020,370; Silverman et al, United States Patent No. 6,028,214; Horwell et al, United States Patent No. 6,103,932; Silverman et al, United States Patent No. 6,117,906; Silverman, International Publication No. WO 92/09560; Silverman et al, International Publication No. WO 93/23383; Horwell et al, International Publication No. WO 97/29101, Horwell et al. International Publication No. WO 97/33858; Horwell et al, International Publication No. WO 97/33859; Bryans et al, International Publication No. WO 98/17627; Guglietta et al, International Publication No. WO 99/08671; Bryans et al, International Publication No. WO 99/21824; Bryans et al, International Publication No. WO 99/31057; Magnus-Miller et al, International Publication No. WO 99/37296; Bryans et al, International Publication No. WO 99/31075; Bryans et al, International Publication No. WO 99/61424; Pande, International Publication No. WO 00/23067; Bryans, International Publication No. WO 00/31020; Bryans et al, International Publication No. WO 00/50027; Bryans et al, International Publication No. WO 02/00209; Tran, U.S. Application Serial No. 60/711,477 filed Aug. 5, 2005; and Tran, U.S. Application Serial No. 60/710,963 filed Aug. 5, 2005)
Дозировка
Количество соединения (1), которое будет эффективным для лечения конкретного заболевания, нарушения или патологического состояния, раскрытое в данной работе, будет зависеть, по крайней мере частично, от природы заболевания или условий, и может быть определено стандартными клиническими способами, известными из литературы как ранее описанные. Дополнительно, in vitro и in vivo анализы могут, при необходимости, быть использованы идентификации оптимальных пределов дозы. Количество назначаемого пролекарства будет, конечно, зависеть от, среди многих факторов, объекта, имеющего заболевания, нуждающегося в лечении, веса объекта, интенсивности заболевания, способа приема лекарственного средства и заключения, прописываемого врачом.
В некоторых конкретных вариантах осуществления пероральные дозированные лекарственные формы замедленного высвобождения адаптируются для приема пациентом от 1 до 3 раз в день. В других вариантах осуществления изобретения пероральные дозированные лекарственные формы замедленного высвобождения адаптируются для приема пациентом 1-2 раза в день. Доза может быть назначена индивидуально или в комбинации с другими лекарствами, прием может продолжаться так долго, как требуется для эффективного лечения или предотвращения заболевания или нарушения.
Подходящие пределы дозы для перорального приема габапентина обычно составляют около 100-3600 мг/день, и доза фармацевтически приемлемых солей или фармацевтически приемлемых сольватов должна быть выверена для предоставления эквивалентного количества габапентина. Пределы дозы могут быть легко определены методами, известными специалистам в данной области.
Следующие примеры более подробно иллюстрируют пероральные дозированные лекарственные формы замедленного высвобождения, способы и материалы, используемые для их производства и результаты, полученные при их приеме пациентами.
Примеры
Пример 1
Таблетки лекарственной формы замедленного высвобождения, содержащие соединение (1), были произведены из ингредиентов, приведенных в таблице 1:
Категория
лаурил сульфат, NF
Таблетки готовят в несколько стадий. Соединение (1), двухосновный фосфат кальция, бегенат глицерина, тальк и коллоидный диоксид кремния взвешивают, просеивают через сито 20 меш и смешивают в V-блендере в течение 15 минут. Агрегированную порцию натрия лаурил сульфата взвешивают и просеивают через сито 30 меш. Агрегированную порцию стеарата магния взвешивают и просеивают через сито 40 меш. Просеянные натрий лаурил сульфат и стеарат магния помещают в V-блендер и перемешивают в течение 5 минут. Смесь выгружают и прессуют в формы примерно по 400 мг в прессе для таблеток. Формы затем пропускают через Comil 194 Ultra мельницу (Quadro Engineering, Inc., Millburn, NJ) для получения раздробленного материала для дальнейшего прессования. Таблеточные порции натрий лаурил сульфата взвешивают и пропускают через сито 30 меш. Таблеточные порции стеарата магния взвешивают и пропускают через сито 40 меш. Раздробленный материал и таблеточные порции натрий лаурил сульфата и стеарата магния помещают в V-блендер и перемешивают в течение 3 минут. Смесь выгружают и прессуют в форме таблеток с общим весом 1300 мг и загрузкой вещества (1) 600 г (45,8 мас.%). Таблетки имеют среднюю конечную твердость от 16,1 до 22,2 кр (158-218 Ньютонов).
Пример 2
Пероральную лекарственную таблетированную форму, имеющую ингредиенты, показанные в табл.1, готовят с использованием другого метода, в несколько стадий. Соединение (1), двухосновный фосфат кальция, бегенат глицерина, тальк и коллоидный диоксид кремния взвешивают, просеивают через сито 20 меш и смешивают в V-блендере в течение 15 минут. Уплотненную порцию натрия лаурил сульфата взвешивают и просеивают через сито 30 меш. Уплотненную порцию стеарата магния взвешивают и просеивают через сито 40 меш. Просеянные натрий лаурил сульфат и стеарат магния помещают в V-блендер и перемешивают в течение 5 минут. Смесь выгружают и прессуют в уплотненные формы на Chilsonator (роликовый уплотнитель, FitzPatrick, Elmhurst, IL) машине. Полученные формы затем пропускают через бильную мельницу (FitzPatrick, Elmhurst, IL) для получения раздробленного материала для дальнейшего прессования. Таблеточные порции натрий лаурил сульфата взвешивают и пропускают через сито 30 меш. Таблеточные порции стеарата магния взвешивают и пропускают через сито 40 меш. Раздробленный материал и таблеточные порции натрий лаурил сульфата и стеарата магния помещают в V-блендер и перемешивают в течение 3 минут. Смесь выгружают и прессуют в форме таблеток с общим весом 1310 мг с содержанием вещества (1) 600 г (45,8 мас.%). Смесь также прессуют в 655 мг таблетки с содержанием вещества (1) 300 г. Таблетки имеют среднюю конечную твердость от 15,7 до 18,9 кр и от 11,1 до 13,7 кр соответственно.
Пример 3
Проводят рандомизированное, перекрестное, с приемом на сытый/на тощий желудок изучение безопасности, толерантности и фармакокинетики одиночной дозы оральной лекарственной формы соединения (1) с длительным выделением на здоровых взрослых пациентах. Используют оральную лекарственную форму длительного действия из примера 1. Изучение выстраивают для оценки поведения этого препарата на людях по сравнению с коммерческим препаратом, капсулами габапентина (Neurontin). Двенадцать здоровых взрослых добровольцев участвуют в исследовании (7 мужчин и 5 женщин). Средний вес тела составляет 75,6 кг. Все пациенты проходят две различных терапии в случайном порядке с недельным промыванием (кишечника) между лечениями. Две терапии состоят в: А) одна оральная доза из примера 1, таблетки (2×600 мг вещества (1)) на тощий желудок с утра; и В) одна оральная доза из примера 1, таблетки (2×600 мг вещества (1)) после высокожирного завтрака.
Образцы плазмы и крови отбирают у всех пациентов до приема лекарства и через 0,5, 1, 1.5, 2, 3, 4, 6, 8, 12, 18, 24 и 36 часов после приема лекарства. Образцы мочи отбирают у всех пациентов до приема лекарства и полные сборы мочи были получены в часовых интервалах 0-4, 4-8, 8-12, 12-18, 18-24 и 24-36 после приема лекарства. Образцы крови сразу разбавили метанолом и заморозили при <=70°С. Отобрали аликвотные образцы для проведения анализа габапентина и вещества (1) с использованием специфических методов LC/MS/MS.
Концентрация габапентина в плазме ±1 SD после перорального приема пероральной лекарственной формы замедленного высвобождения, полученной в примере 1, на тощий и сытый желудок взрослыми здоровыми пациентами показана на фиг.1 и 2 соответственно.
Среднее значение Смах±SD габапентина в плазме после перорального приема таблеток на тощий желудок составляет 4,21±1,15 мкг/мл. При приеме таблеток после высокожирного завтрака, Смах увеличивается до 6,24±1,55 мкг/мл. Среднее значение AUC±SD габапентина в плазме после перорального приема таблеток на тощий желудок составляет 54,5±12,2 мкг·ч/мл. При приеме таблеток после высокожирного завтрака AUC увеличивается до 83,0±21,8 мкг·ч /мл. В присутствии пищи выделение габапентина после орального приема таблеток увеличивается еще на 52% по сравнению с приемом на тощий желудок.
Время достижения пиковой концентрации габапентина в плазме (Тмах) значительно задерживается после перорального приема таблеток. Для голодных пациентов пероральный прием таблеток дает Тмах габапентина 5,08±1,62 ч. Это соответствует типичному Тмах для габапентина немедленного действия, которое составляет 2-4 часа. Достижение Тмах габапентина в присутствии пищи еще более задерживается до 8,40±2,07 ч. Видимый период окончательного выведения половины габапентина из плазмы оказался одинаковым для всех типов лечения и составил 6,47±0,77 ч для таблеток у голодных пациентов и 5,38±0,80 ч для таблеток у сытых пациентов.
После перорального применения таблеток процент дозы габапентина, выделенного из мочи, составил 46,5±15,8% для «голодных» пациентов и 73,7±7,2% для «сытых» пациентов.
Содержание незатронутого пролекарства в плазме после перорального приема таблеток было низким. После перорального приема дозы таблеток «голодным» пациентом концентрация незатронутого соединения (1) в плазме достигает максимума 0,040 мкг/мл, примерно 1,0% соответствующего пика концентрации габапентина. Аналогично, AUC соединения (1) в плазме у этих пациентов составляет 0,3% соответствующей AUC габапентина в плазме. После перорального приема дозы таблеток «сытым» пациентом концентрация незатронутого соединения (1) в плазме достигает максимума 0,018 мкг/мл, примерно 0,3% соответствующего пика концентрации габапентина. Аналогично, AUC вещества (1) в плазме у этих пациентов составляет <0,1% соответствующей AUC габапентина в плазме.
Пример 4
Средняя концентрация габапентина в плазме после принятия пероральной дозированной формы замедленного высвобождения, полученной в примере 2, «сытым» пациентом показана на фиг.3. Среднюю концентрацию габапентина у 12 пациентов-людей определяют в соответствии с методом, описанным в примере 3 после приема (а) одной таблетки, включающей 300 мг соединения (1); (b) одной таблетки, включающей 600 мг соединения (1); и (с) двух таблеток, каждая из которых включает по 600 мг соединения (1).
Пример 5
Концентрация габапентина в крови ±SD после приема дозы оральной лекарственной формы длительного действия, полученной в примере 1 Cynomologous monkey показана на фиг.4. Концентрацию габапентина в крови Cynomologous monkey определяют следующим способом.
Протокол введения
Таблетки, включающие соединение (1) (1×600 мг соединения (1) на одну таблетку), вводят перорально группе из 4 взрослых Cynomologous (Macaca fascicularis) monkeys (вес около 3 кг). Каждой обезьяне вводят одну таблетку. Животным не дают пищи в течение ночи перед введением и 4 часа после введения. Образцы крови (1 мл) получают из бедренной вены с интервалами в течение 24 ч после введения лекарства. Кровь немедленно разбавляют метанолом и замораживают при -20°С до анализа. Тестовые вещества вводят обезьянам с интервалами минимум 72 часа на промывание (кишечника).
Приготовление образца поглощенного вещества
300 мкл метанола помещают в 1,5 мл пробирку Эппендорфа для приготовления образцов и стандартов.
Приготовление образца: кровь получают в разные моменты времени и немедленно 100 мкл крови помещают в 1,5 мл пробирку Эппендорфа, содержащую 300 мкл метанола, и интенсивно смешивают.
Стандартная процедура: 90 мкл крови прибавляют к 300 мкл метанола в 1,5 мл пробирке Эппендорфа. 10 мкл стандартного раствора габапентина (0,04, 0,2, 1, 5, 25 и 100 мг/мл) прибавляют в каждую пробирку для создания конечных калибровочных растворов (0,04, 0,2, 1, 5, 25 и 100 мг/мл). 20 мкл пара-хлорфенилаланина прибавляют ко всем образцам и стандартам. Образцы интенсивно перемешиваются и центрифугируются при 14000 об/мин в течение 20 минут. Суспензию анализируют методом LC/MS/MS.
LC/MS/MS анализ
Концентрацию габапентина в крови обезьяны определяют с использованием API 2000 LC/MS/MS прибора, оборудованного Shimadzu SCL-10AVP и LЕАР автосамлером. Колонка Zorbax C8×DB 4,6×150 мм, комнатная температура. Мобильная фаза (А) 0,1% муравьиной кислоты в воде, и (В) 0,1% муравьиной кислоты в ацетонитриле. Градиентные условия: 2% В 3,5 мин, возрастание В до 95% за 3,5 мин и поддерживание В 2 мин, затем уменьшение до 2% В за 5,6 мин и поддерживание 2,3 мин. 30 мкл Образца вкалывают в колонку. Используют Turbo-IonSpray генератор, габапентин детектируют в режиме определения положительных ионов как MRM переход 172/173. Пики интегрировали с использованием Analyst 1.2 quantitative software.
Пример 6
Таблетки по изобретению, содержащие соединение (1), были произведены из ингредиентов, приведенных в таблице 2.
Категория
Таблетки готовят в несколько стадий. Соединение (1), микрокристаллическую целлюлозу (MCC PH113), бегенат глицерина, тальк и коллоидный диоксид кремния, натрий додецил сульфат (SDS) (первая измельченная порция) и стеарат магния (первая измельченная порция) взвешивают, просеивают через сито 20 меш и смешивают в V-блендере в течение 7 минут (Maxiblend Lab Blender MB-1 (Globepharma)). Измельченные ингредиенты выливают в питающую воронку роликового уплотнителя (ВO50OН Compactor cropped roll/closed end, 3,9 дюйма диаметр ролика, 1,5 дюйма толщина ролика, 11,6 кН сила, 12 об/мин скорость роликов и 7 об/мин скорость горизонтальной подачи шнековым питателем). Уплотненный материал затем пропускают через Quadro Underdriven Comil Model U5 мельницу (Quadro Engineering, Inc., Millburn, NJ, 0,079 in grater hole side, 1607 impeller style, 2500 об/мин) для получения раздробленного материала для дальнейшего прессования. Раздробленный материал помещают в блендер (Maxiblend Lab Blender MB-1 (Globepharma) 25 об/мин) и перемешивают 5 минут. Дополнительные количества SDS (вторая измельченная порция) и/или стеарата магния (вторая измельченная порция) прибавляют при необходимости для создания конкретных количеств. Смесь выгружают и прессуют в форме таблеток с общим весом 1100 мг и загрузкой соединения (1) 600 г (54,55 мас.%). Таблетки имеют среднюю конечную твердость от 14 до 17 кр (137-214 Ньютонов).
Пример 7
Концентрация габапентина в крови ±SD после принятия пероральной дозы дозированной лекарственной формы замедленного высвобождения (1×600 мг), полученной в примере 6, Cynomologous monkeys показана на фиг.5. Концентрацию габапентина в крови Cynomologous monkeys определяли в соответствии со способом в примере 5.
Пример 8
Следующие шаги предпринимают для получения in vitro кривой растворения лекарственных форм, полученных в примерах 1, 2 и 6. Лекарственную форму помещают в сосуд для растворения, содержащий 900 мл 10 мМ моноосновного калий фосфатного буфера (К2НРO4, рН 7,4) и 1% (вес/объем) натрий лаурил сульфата при 37°С. Среду для растворения перемешивают со скоростью 50 об/мин (USP, Type II, мешалка с лопастью). Образцы отбирают через 0,5, 1, 2, 4, 6, 8, 12 и 24 часа и определяют содержание вещества (1) в растворе методом обратнофазовой HPLC с использованием С 18 колонки, смесь фосфатного буфера/ацетонитрила/воды в качестве изократической мобильной фазы с фотодиодным детектированием при 210 нм.
Как показано на фиг.6, лекарственные формы, полученные в примере 1, высвобождают около 20% соединения (1) после 2 часов, 50% после 5 часов и около 80% после 8 часов перемешивания. Как показано на фиг.7А, лекарственные формы, включающие 300 мг соединения (1), полученные в примере 2, высвобождают около 20% соединения (1) после 2 часов, 50% после 6 часов и около 80% после 10 часов перемешивания. Как показано на фиг.7В, лекарственные формы, включающие 600 мг соединения (1), полученные в примере 2, высвобождают около 20% соединения (1) после 2 часов, 50% после 5 часов и около 80% после 8 часов перемешивания. Как показано на фиг.8, лекарственные формы, полученные в примере 6, высвобождают около 30% соединения (1) после 5 часов, 60% после 10 часов и около 80% после 15 часов перемешивания.
В заключение необходимо заметить, что существуют альтернативные пути выполнения настоящего изобретения. Соответственно настоящие варианты осуществления должны рассматриваться в качестве иллюстрации, а не ограничения, и изобретение не должно быть ограничено деталями, приведенными здесь, но может быть модифицировано в соответствии с областью применения и эквивалентами, заявленными в формуле изобретения.
Все публикации и патенты, упомянутые здесь, включают посредством ссылки на их полный текст.
Изобретение относится к лекарственным средствам и касается таблетки для перорального приема замедленного высвобождения, содержащей: (а) 10-80 мас.% 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, и (b) 1-30 мас.% жирного соединения, такого как сложный эфир глицерина, лауриловый спирт, миристиловый спирт, стеариловый спирт, цетиловый спирт, цитостеариловый спирт, пальмитоиловый спирт, урицирный воск, гидрированное растительное масло, канделильный воск, эспартовый воск, стеариновая кислота, твердый парафин, пчелиный воск, глико-воск, гидрированное касторовое масло и карнаубский воск или их комбинация, где величина мас.% рассчитана на общий сухой вес дозированной лекарственной формы, которая при приеме натощак одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг, обеспечивает профиль концентрации габапентина в плазме Смах 3-6 мкг/мл при Тмах 4-7 часов и AUC 30-70 мкг·час/мл; или при приеме после еды одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг, обеспечивает профиль концентрации габапентина в плазме Смах 5-8 мкг/мл при Тмах 6-11 часов и AUC 60-110 мкг·час/мл. Также раскрыто применение 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты для производства таблетки замедленного высвобождения для лечения синдрома беспокойных ног и постгерпетической невралгии. Таблетки по изобретению обладают улучшенным фармакокинетическим профилем габапентина. 7 н. и 26 з.п. ф-лы, 8 ил., 2 табл.
1. Таблетка для перорального приема замедленного высвобождения, содержащая:
(a) 10-80 мас.% 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, и
(b) 1-30 мас.% жирного соединения, такого как сложный эфир глицерина, лаурилового спирта, миристилового спирта, стеарилового спирта, цетилового спирта, цитостеарилового спирта, пальмитоилового спирта, урицирного воска, гидрированного растительного масла, канделильного воска, эспартового воска, стеариновой кислоты, твердого парафина, пчелиного воска, глико-воска, гидрированного касторового масла и карнаубского воска или их комбинация, где
величина мас.% рассчитана на общий сухой вес дозированной лекарственной формы,
которая при приеме натощак одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме Смах 3-6 мкг/мл при Тмах 4-7 ч и AUC 30-70 мкг·ч/мл; или
при приеме после еды одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме Смах 5-8 мкг/мл при Тмах 6-11 ч и AUC 60-110 мкг·ч/мл.
2. Таблетка для перорального приема замедленного высвобождения, содержащая:
(a) 10-80 мас.% 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты, и
(b) 1-30 мас.% жирного соединения, такого как сложный эфир глицерина, лаурилового спирта, миристилового спирта, стеарилового спирта, цетилового спирта, цитостеарилового спирта, пальмитоилового спирта, урицирного воска, гидрированного растительного масла, канделильного воска, эспартового воска, стеариновой кислоты, твердого парафина, пчелиного воска, глико-воска, гидрированного касторового масла и карнаубского воска или их комбинация,
где величина мас.% рассчитана на общий сухой вес дозированной лекарственной формы, которая:
при приеме натощак одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме, показанный на фиг.1; или
при приеме после еды одним или более пациентом-человеком в дозе 1 - {[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты около 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме биоэквивалентный кривой на фиг.2.
3. Таблетка по любому из пп.1 и 2, которая прессуется до степени твердости в 15 килофунтов.
4. Таблетка по любому из пп.1-3, в которой жирным соединением является сложный эфир глицерина.
5. Таблетка по п.4, где сложный эфир глицерина выбирают из моностеарата глицерина, бегената глицерина, пальмитостеарата глицерина, лаурилмакроголглицерида, стироилмакроголглицерида и их комбинации.
6. Таблетка по п.5, где сложным эфиром глицерина является бегенат глицерина.
7. Таблетка по любому из пп.1-3, где жирное соединение выбрают из лаурилового спирта, миристилового спирта, стеарилового спирта, цетилового спирта, цитостеарилового спирта, пальмитоилового спирта, урицирного воска, гидрированного растительного масла, канделильного воска, эспартового воска, стеариновой кислоты, твердого парафина, пчелиного воска, глико-воска, гидрированного касторового масла и карнаубского воска.
8. Таблетка по любому из пп.1 и 2, включающая 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусную кислоту в количестве 300-700 мг.
9. Таблетка по любому из пп.1 и 2, включающая 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусную кислоту в кристаллической форме.
10. Таблетка по любому из пп.1 и 2, дополнительно включающая один или несколько фармацевтически приемлемых эксципиентов, которые выбирают из разбавителей, смазок, антиадгезивов, глидантов, поверхностно-активных веществ, дезинтегрантов и их комбинацию.
11. Таблетка по п.10, где разбавитель выбирают из двухосновного фосфата кальция и микрокристаллической целлюлозы.
12. Таблетка по п.11, где разбавитель представляет собой двухосновный фосфат кальция, а его количество составляет 30-50 мас.%.
13. Таблетка по любому из пп.1 и 2, которая содержит 60 мг 1-{[(α-изобутаноилокси-этокси)карбонил]аминометил}-1-пиклогексан уксусной кислоты, а доза включает 2 таблетки.
14. Таблетка по любому из пп.1 и 2, содержащая покрытие (оболочку).
15. Таблетка по п.1, которая при приеме натощак одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме Смах 3-6 мкг/мл при Тмах 4-7 ч и AUC 30-70 мкг·ч/мл; и
при приеме после еды одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме Смах 5-8 мкг/мл при Тмах 6-11 ч и AUC 60-110 мкг·ч/мл.
16. Таблетка по п.1, которая при приеме натощак популяцией указанного пациента-человека в дозе 1-{[(α-изобутаноилоксиэтокси)-карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме Смах 3-6 мкг/мл при Тмах 4-7 ч и AUC 30-70 мкг·ч/мл; и
при приеме после еды группой названных пациентов-людей в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме Смах 5-8 мкг/мл при Тмах 6-11 ч и AUC 60-110 мкг·ч/мл.
17. Таблетка по п.2, которая при приеме натощак одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме, биоэквивалентный профилю на фиг.1; и
при приеме после еды одним или более пациентом-человеком в дозе 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты 1100-1300 мг обеспечивает профиль концентрации габапентина в плазме, биоэквивалентный профилю на фиг.2.
18. Таблетка по любому из пп.1 и 2, где
при приеме натощак один или несколько пациентов не принимают никакой пищи в течение 10 ч до приема и 4 ч после приема лекарственного средства, выпивают 250 мл воды за 2 ч и за 1 ч до приема и 250 мл воды через 2 ч после приема лекарственного средства, съедают ленч через 4 ч после приема и съедают обед через 10 ч после приема лекарственного средства; а
при приеме после еды один или несколько пациентов начинают принимать пробный завтрак за 30 мин до и заканчивает принимать пищу за 5 мин до принятия лекарственного средства, съедает ленч через 4 ч после приема и съедает обед через 10 ч после приема лекарственного средства, где пробный завтрак включает калорий, из которых 500 включают жировые калории.
19. Таблетка для перорального приема замедленного высвобождения, содержащая:
(a) 10-80 мас.% 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил} -1 -циклогексан уксусной кислоты, и
(b) 1-30 мас.% жирного соединения, такого как сложный эфир глицерина, лаурилового спирта, миристилового спирта, стеарилового спирта, цетилового спирта, цитостеарилового спирта, пальмитоилового спирта, урицирного воска, гидрированного растительного масла, канделильного воска, эспартового воска, стеариновой кислоты, твердого парафина, пчелиного воска, глико-воска, гидрированного касторового масла и карнаубского воска или их комбинация, где
величина мас.% рассчитана на общий сухой вес таблетки,
и эта дозированная форма будучи помещенной в 10 мМ моноосновной натрий фосфатный буфер и 1% (вес/объем)лаурил сульфат натрия при рН 7,4 и 37°С при перемешивании со скоростью 50 об/мин (USP, Type II (Американская фармакопия. Тип II)) высвобождает 20% 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан
уксусной кислоты в течение 2 ч, 50% в течение 5 ч и 80% в течение 8 ч.
20. Таблетка по п.19, содержащая 1-{[(α-изобутаноилоксиэтокси)-карбонил]аминометил}-1-циклогексан уксусную кислоту в количестве 500-700 мг.
21. Таблетка по п.19, дополнительно включающая двухосновный фосфат кальция.
22. Таблетка для перорального приема замедленного высвобождения, содержащая:
(а) 10-80 мас.% 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты; и
(в) 1-30 мас.% жирного соединения, такого как сложный эфир глицерина, лаурилового спирта, миристилового спирта, стеарилового спирта, цетилового спирта, цитостеарилового спирта, пальмитоилового спирта, урицирного воска, гидрированного растительного масла, канделильного воска, эспартового воска, стеариновой кислоты, твердого парафина, пчелиного воска, глико-воска, гидрированного касторового масла и карнаубского воска или их комбинации,
где величина мас.% рассчитаны на общий сухой вес таблетки, которая будучи помещенной в 10 мМ моноосновной натрий фосфатный буфер и 1% (вес/объем) лаурилсульфат натрия при рН 7,4 и 37°С при перемешивании со скоростью 50 об/мин (USP, Type II) высвобождает 30% 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты в течение 5 ч, 60% в течение 10 ч и 80% в течение 15 ч.
23. Таблетка по п.22, содержащая 1-{[(α-изобутаноилоксиэтокси)-карбонил]аминометил}-1-циклогексан уксусную кислоту в количестве 500-700 мг.
24. Таблетка по п.22, дополнительно содержащая микрокристаллическую целлюлозу.
25. Таблетка для перорального приема замедленного высвобождения, содержащая:
600 мг - 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты;
518,26 мг - дигидрофосфата кальция;
60,05 мг - бегенат глицерина;
80,02 мг - талька;
5,43 мг - коллоидного диоксида кремния;
24,00 мг - лаурилсульфата натрия и
22,22 мг - стеарата магния,
которая при необходимости имеет оболочку.
26. Таблетка по п.25, содержащая 300-700 мг 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты.
27. Таблетка по любому из пп.1-26 для применения в способе лечения человека или животного.
28. Таблетка по любому из пп.1-26 для применения в способе лечения синдрома беспокойных ног.
29. Таблетка по любому из пп.1-26 для применения в способе лечения постгерпетической невралгии.
30. Применение 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты для производства таблетки замедленного высвобождения по любому из пп.1-26 для лечения синдрома беспокойных ног.
31. Применение 1-{[(α-изобутаноилоксиэтокси)карбонил]аминометил}-1-циклогексан уксусной кислоты для производства таблетки замедленного высвобождения по любому из пп.1-26 для лечения постгерпетической невралгии.
32. Таблетка по любому из пп.19-24, в которой сложный эфир глицерина выбран из моностеарата глицерина, бегената глицерина, пальмитостеарата глицерина, лаурилмакроголглицерида, стироилмакроголглицерида и их комбинации.
33. Таблетка по п.32, в которой сложный эфир глицерина представляет собой бегенат глицерина.
| СПОСОБ ПОЛУЧЕНИЯ ДИНИТРОТОЛУОЛА | 1995 |
|
RU2100347C1 |
| WO 2004089289 A2, 21.10.2004 | |||
| Cundy R.C | |||
| Предохранительное приспособление в пробочных сверлильных станках | 1928 |
|
SU13512A1 |
| Improved Oral Bioavailability, Dose Proportionality, and Colonic Absorption Compared with Gabapentin in Rats and Monkeys" JPET October 2004 | |||

