Описание
Область техники
Настоящее изобретение относится к фармацевтическому средству, в частности новому производному хинолона или его фармацевтически приемлемой соли, которые применимы в качестве ингибиторов агрегации тромбоцитов или ингибиторов P2Y12.
Предпосылки создания изобретения
Тромбоциты после открытия их Donne et al. в 1842 году длительное время рассматривались как компонент крови, необходимый для гемостаза. В настоящее время показано, что тромбоциты не только играют ведущую роль в механизме гемостаза, но также проявляют заслуживающие внимания в плане клинического применения многофункциональные свойства, такие как связанные с проявлением артериосклероза, заболеваний органов системы кровообращения, в том числе тромботических заболеваний, метастазирования, воспаления, реакции отторжения после пересадки и иммунной реакции.
Как правило, терапии для реперфузии крови с фармацевтическими средствами или физическими методами осуществляют в случае тромботических заболеваний и ишемических заболеваний. Однако недавно обнаружен вызывающий клинические проблемы феномен, при котором активация, адгезия и агрегация тромбоцитов ускоряются после осуществления реваскуляризации из-за разрушения тканей сосудов, включая эндотелиальные клетки, или коллапса равновесия фибринолиз-свертывание, вызванных самим лекарственным средством. Например, показано, что после рециркуляции за счет лечения тромболитическими средствами с использованием t-PA или подобных средств активируются способность к фибринолизу и способность к свертыванию и нарушается общее равновесие фибринолиз-свертывание. Клинически это приводит к повторной обструкции, которая вызывает большую терапевтическую проблему (непатентная ссылка 1).
С другой стороны, РТСА-терапия или метод постоянного стента быстро популяризируются и достигают определенных результатов в случае лечения болезней, в основе которых лежит стенокардия, инфаркт миокарда, и таких, как стриктура венечной артерии и стриктура аорты. Однако, так как указанные методы лечения повреждают ткани сосудов, в том числе эндотелиальные клетки, возникают проблемы острой коронарной обструкции и другой рестриктуры, которая происходит на хронической стадии. Тромбоциты играют важную роль в различных тромболитических побочных эффектах (реобструкции и т.п.) после такой реваскуляризации. Таким образом, несмотря на ожидаемую эффективность антитромботического средства, достаточное действие обычных антитромботических средств не подтверждается.
В качестве профилактических или лечебных средств в случае таких заболеваний органов системы кровообращения используются аспирин, цилостазол, простагландин I2, простагландин Е1, тиклопидин, клопидогрель, дипиридамол и подобные ингибиторы агрегации тромбоцитов. Также в последнее время разработан антагонист GPIIb/IIIa, который ингибирует конечную стадию агрегации тромбоцитов и обладает сильной активностью ингибирования агрегации тромбоцитов, но его применение ограничивается внутривенной капельной инфузией при острой фазе тромбоза (непатентная ссылка 2).
В последние годы показано, что в отношении тиклопидина и клопидогреля, которые применяют в качестве антитромботических средств, такие средства проявляют активность ингибирования агрегации тромбоцитов через ингибирование P2Y12 как рецептора АДФ (ADP) за счет их активных метаболитов. Далее, имеются сообщения о производном триазоло[4,5-D]пиримидина (патентная ссылка 1), производных пиперазина и/или гомопиперазина (патентная ссылка 2 и патентная ссылка 3), производном пиразолидиндиона (патентная ссылка 4), производном изохинолинона (патентная ссылка 5) и подобных соединениях как соединениях, обладающих активностью ингибирования P2Y12.
С другой стороны, из патентных ссылок 6 и 7 известны производные хинолона.
Из патентной ссылки 6 известно соединение, представленное формулой (А), обладающее антимикробным действием, но сведения о его активности ингибирования агрегации тромбоцитов не приводятся. Кроме того, его структура отличается от структуры соединения по настоящему изобретению в том смысле, что группа, которая соответствует R5 в соединении по настоящему изобретению, представляет собой карбоксильную группу, сложноэфирную группу или карбамоил

(В приведенной формуле R1 представляет собой -OR9, аминогруппу или низшую алкиламиногруппу и R9 представляет собой атом водорода или карбоксизащитную группу. Значение других символов см. в указанном сообщении.)
В патентной ссылке 7 сообщается, что соединение, представленное формулой (В), обладает активностью ингибирования P2Y12. Однако его структура отличается от структуры соединения по настоящему изобретению в том смысле, что группа, которая соответствует R5 в соединении по настоящему изобретению, представляет собой карбамоил

(Значение символов в приведенной формуле см. в указанном сообщении.)
В патентной ссылке 8 сообщается, что соединение, представленное формулой (С), обладает активностью ингибирования P2Y12. Однако его структура отличается от структуры соединения по настоящему изобретению в том смысле, что группа, которая соответствует R5 в соединении по настоящему изобретению, представляет собой карбамоил
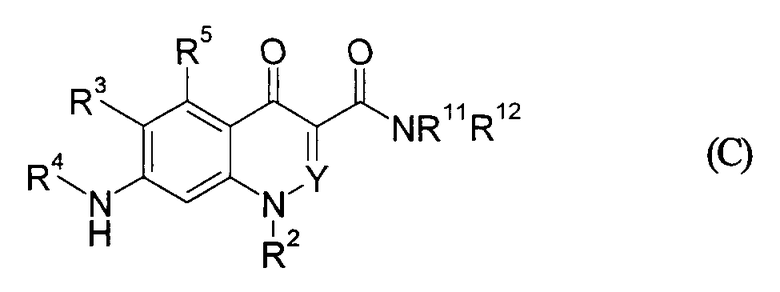
(Значение символов в приведенной формуле см. в указанном сообщении.)
Непатентная ссылка 1: “Journal of American College of Cardiology”, 1998, vol.12, p. 616-623.
Непатентная ссылка 2: “Sogo Rinsho (Synthetic Clinic)”, 2003, vol.52, p. 1516-1521.
Патентная ссылка 1: международная публикация WO 00/34283.
Патентная ссылка 2: международная публикация WO 02/098856.
Патентная ссылка 3: международная публикация WO 03/022214.
Патентная ссылка 4: международная публикация WO 05/000281.
Патентная ссылка 5: международная публикация WO 05/035520.
Патентная ссылка 6: международная публикация WO 98/23592.
Патентная ссылка 7: международная публикация WO 05/009971.
Патентная ссылка 8: международная публикация WO 06/077851.
Раскрытие изобретения
Проблемы, на решение которых направлено изобретение
В такой ситуации весьма желательна разработка антитромботического средства с профилем высокой безопасности, с ослабленным нежелательным эффектом кровотечения и с отчетливой фармацевтической эффективностью не только в острой фазе, но также в хронической фазе. Таким образом, задача изобретения состоит в разработке ингибитора агрегации тромбоцитов или ингибитора P2Y12, обладающего сильным фармакологическим действием и хорошим балансом между фармакологическим действием и профилем безопасности.
Способы решения проблем
Соответственно, авторы настоящего изобретения провели интенсивные исследования с целью преодоления вышеуказанных проблем и в результате обнаружили, что новое производное хинолона обладает превосходной активностью ингибирования агрегации тромбоцитов или активностью ингибирования P2Y12 и имеет превосходную фармакокинетику, и таким образом осуществили настоящее изобретение.
Иными словами, настоящее изобретение относится к производному хинолона, представленному приведенной далее общей формулой (I), или его фармацевтически приемлемой соли
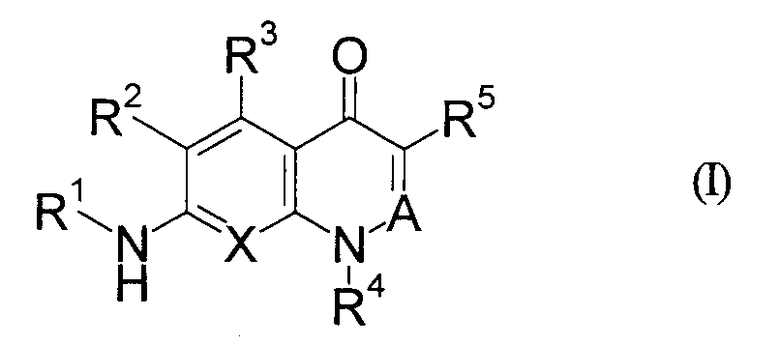
[Символы в приведенной формуле имеют значения, указанные далее.]
R1: циклоалкил или низший алкиленциклоалкил, где циклоалкил в R1 может быть замещенным,
R2: -Н или галоген,
R3: -Н, галоген, -OR0 или -О-(низший алкилен)-арил,
R0: одинаковые или отличаются один от другого, и каждый представляет собой -Н или низший алкил,
R4: низший алкил, галоген(низший алкил), низший алкиленциклоалкил, циклоалкил или гетероциклическая группа,
где указанные в R4 циклоалкил и гетероциклическая группа могут быть соответственно замещенными,
R5: -NO2, -CN, низший алкил, низший алкенил, галоген(низший алкенил), -L-Ra, -C(O)R0, -O-Rb, -N(R6)2, низший алкилен-N(R6)(Rc), -N(R6)C(O)-Rd, низший алкилен-N(R6)C(O)-Rd, низший алкилен-N(R0)C(O)О-(низший алкил), -N(R0)C(O)N(R0)-Rе, низший алкилен-N(R0)C(O)N(R0)-Rе, -N(R0)S(O)2N(R0)C(O)-Rd, -CH=NOH, циклоалкил, гетероциклическая группа, (2,4-диоксо-1,3-тиазолидин-5-илиден)метил или (4-оксо-2-тиоксо-1,3-тиазолидин-5-илиден)метил,
где указанные в R5 циклоалкил и гетероциклическая группа могут быть соответственно замещенными,
R6: Н, низший алкил, низший алкилен-СО2R0 или низший алкилен-Р(О)(ORp)2,
где указанный в R6 низший алкилен может быть замещенным,
L: низший алкилен или низший алкенилен, которые могут быть соответственно замещенными,
Rа: -OR0, -CN, -О-(низший алкилен)-арил, -О-(низший алкилен)-СО2R0, -C(O)R0, -СО2R0, -C(O)NHOH, -С(О)N(R6)2, -C(O)N(R0)-арил, -С(О)N(R0)-S(O)2-(низший алкил), -С(О)N(R0)-S(O)2-арил, -С(О)N(R0)-S(O)2-(гетероциклическая группа), -NH2OH, -ОC(O)R0, -ОC(O)-галоген(низший алкил), -Р(О)(ORp)2, арил или гетероциклическая группа,
где указанные в Rа арил и гетероциклическая группа могут быть замещенными,
Rр: R0, низший алкилен-ОС(О)-(низший алкил), низший алкилен-ОС(О)-циклоалкил, низший алкилен-ОС(О)О-(низший алкил), низший алкилен-ОС(О)О-циклоалкил или низший алкилен-(гетероциклическая группа),
где указанная в Rр гетероциклическая группа может быть замещенной,
Rb: Н, циклоалкил, арил, гетероциклическая группа, низший алкилен-Rbа или низший алкенилен-Rbа,
где указанные в Rb низший алкилен, низший алкенилен, циклоалкил, арил и гетероциклическая группа могут быть замещенными,
Rba: -OR0, -О-Si(низший алкил)3, -СО2R0, -C(O)NHOH, -С(О)N(R0)2, -С(О)N(R0)-S(O)2-(низший алкил), -С(О)N(R0)-S(O)2-арил, -C(NH2)=NOH, -C(NH2)=NO-C(O)R0, -C(NH2)=NO-C(O)-(низший алкилен)-C(O)R0, -СО2-(низший алкилен)-арил, -Р(О)(ORp)2, -C(O)R0, -С(О)-арил, циклоалкил, арил или гетероциклическая группа,
где указанные в Rba арил и гетероциклическая группа могут быть замещенными,
Rс: Н, низший алкил, низший алкилен-OR0, низший алкилен-СО2R0, низший алкилен-C(O)NHOH, низший алкилен-С(О)N(R0)2, низший алкилен-Р(О)(ORp)2, низший алкиленарил, низший алкилен-(гетероциклическая группа), арил или гетероциклическая группа,
где указанные в Rс низший алкилен, арил и гетероциклическая группа могут быть замещенными,
Rd: С1-7-алкил, низший алкенил, галоген(низший алкил), низший алкилен-Rdа, низший алкенилен-Rdа, циклоалкил, арил или гетероциклическая группа,
где указанные в Rd низший алкилен, низший алкенилен, циклоалкил, арил и гетероциклическая группа могут быть замещенными,
Rda: -CN, -OR0, -ОC(O)R0, -О-(низший алкилен)-СО2R0, -О-арил, -СО2R0, -C(O)NHOH, -С(О)N(R0)2, -СО2-(низший алкилен)-N(R0)2, -Р(О)(ORp)2, -N(R6)2, -N(R0)C(O)R0, -С(О)N(R0)-арил, -С(О)N(R0)-(низший алкилен, который может быть замещен -СО2R0)-арил, -N(R0)C(O)-арил, -N(R0)C(O)-ОR0, -N(R0)C(O)-О-(низший алкилен)-арил, -N(R0)S(O)2-арил, -S-гетероциклическая группа, -С(О)N(R0)-(гетероциклическая группа), -N(R0)C(O)-(гетероциклическая группа), циклоалкил, арил или гетероциклическая группа,
где указанные в Rda циклоалкил, арил и гетероциклическая группа могут быть замещенными,
Rе: низший алкилен-СО2R0, низший алкилен-C(O)NHOH, низший алкилен-С(О)N(R0)2, низший алкилен-(гетероциклическая группа), арил, гетероциклическая группа, -S(O)2-арил или -S(O)2-(гетероциклическая группа),
где указанные в Rе арил и гетероциклическая группа могут быть замещенными,
Х: СН или N,
A: C(R7) или N,
R7: -Н и низший алкил,
или R4 и R7 вместе могут образовывать низший алкилен, который может быть замещенным,
при условии, что
исключается 7-(циклогексиламино)-1-этил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбонитрил. То же самое будет справедливо в данном описании далее.]
Кроме того, данная заявка относится к фармацевтическому средству, в частности ингибитору рецептора P2Y12 и/или ингибитору агрегации тромбоцитов, который включает производное хинолона, представленное общей формулой (I), или его соль в качестве активного ингредиента.
Кроме того, данная заявка также относится к применению соединения, представленного общей формулой (I), или его фармацевтически приемлемой соли для получения ингибитора рецептора P2Y12 и/или ингибитора агрегации тромбоцитов и к способу лечения заболевания органов кровообращения, тесно связанного с тромбообразованием путем агрегации тромбоцитов, который включает введение пациенту эффективного количества соединения, представленного общей формулой (I), или его фармацевтически приемлемой соли. Иными словами, изобретение также относится к (1) фармацевтической композиции, включающей соединение, описанное общей формулой (I), или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель;
(2) фармацевтической композиции согласно (1), которая является ингибитором агрегации тромбоцитов;
(3) фармацевтической композиции согласно (1), которая является ингибитором P2Y12;
(4) применению соединения, описанного общей формулой (I), или его фармацевтически приемлемой соли для получения ингибитора агрегации тромбоцитов или ингибитора P2Y12.
Действие изобретения
Так как соединение по настоящему изобретению обладает превосходной активностью ингибирования агрегации тромбоцитов или активностью ингибирования P2Y12, оно применимо в качестве фармацевтического средства, в частности ингибитора агрегации тромбоцитов или ингибитора P2Y12. Соответственно, соединение по настоящему изобретению применимо в качестве средства для предупреждения и/или лечения заболевания органов кровообращения, тесно связанного с тромбообразованием путем агрегации тромбоцитов, такого как нестабильная стенокардия и острый инфаркт миокарда, и для предупреждения его повторения, повторной обструкции и рестриктуры после операции шунтирования коронарной артерии, операции РТСА или операции по установке стента, ускорения тромболизиса в коронарной артерии и предупреждения повторной обструкции и подобных ишемических болезней; такого как церебральный инфаркт при преходящем ишемическом нарушении мозгового кровообращения (TIA), субарахноидальное кровоизлияние (вазоспазм) и подобные инсульты; хроническая окклюзия артерии и подобные заболевания периферических артерий; и подобных заболеваний; и в качестве вспомогательного средства при операции на сердце или операции на сосудах.
Наилучший способ осуществления изобретения
Далее настоящее изобретение описывается подробно.
В данном описании «низший алкил», «низший алкенил», «низший алкилен» и «низший алкенилен» обозначают, соответственно, углеводородные цепи с 1-6 атомами углерода, которые могут быть прямыми или разветвленными, если не указано иное.
Соответственно, «низший алкил» обозначает С1-6-алкил, и его пояснительные примеры включают метил, этил, пропил, бутил, пентил или гексил или их структурные изомеры, такие как изопропил, трет-бутил или подобные, предпочтительно С1-5-алкил, предпочтительнее метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил или 3-пентил.
«Низший алкенил» обозначает С2-6-алкенил, и он может содержать две или большее число двойных связей. Его пояснительные примеры включают этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил и т.п., из которых предпочтительным является С2-3-алкенил, и более предпочтительным является этенил или пропенил.
«Низший алкилен» обозначает двухвалентную группу, образовавшуюся при удалении одного атома водорода из какой-либо позиции «низшего алкила», и представляет собой, как пояснение, метилен, метилметилен, этилен, пропилен, бутилен или подобную группу, предпочтительно С1-4-алкилен, предпочтительнее метилен, метилметилен, этилен или пропилен.
«Низший алкенилен» обозначает двухвалентную группу, образовавшуюся при удалении одного атома водорода из какой-либо позиции «низшего алкенила», и представляет собой, как пояснение, винилен, пропенилен, бутенилен или подобную группу, предпочтительно С2-3-алкенилен, предпочтительнее винилен, пропенилен.
«Галоген» обозначает одновалентную группу, образованную атомом галогена, и в качестве пояснений можно назвать группу фтор, хлор, бром, йод или подобные, из которых предпочтительны фтор или хлор.
«Галоген(низший алкил)» обозначает группу, в которой, по меньшей мере, один какой-либо атом водорода вышеуказанного «низшего алкила» замещен вышеуказанным «галогеном», и его пояснительные примеры включают трифторметил, трифторэтил или подобные группы, из которых предпочтительным является трифторметил.
«Галоген(низший алкенил)» обозначает группу, в которой, по меньшей мере, один какой-либо атом водорода вышеуказанного «низшего алкенила» замещен вышеуказанным «галогеном», и его пояснительные примеры включают фторвинил, хлорвинил или подобные группы.
«Циклоалкил» обозначает неароматический С3-10-углеводородный цикл, и он может образовывать мостиковый цикл или спироцикл, частично содержать ненасыщенную связь или конденсироваться с бензольным циклом. Однако, когда «циклоалкил» конденсирован с бензольным циклом, положение присоединения находится на неароматическом цикле. Пояснительные примеры «циклоалкила» включают циклопропил, циклобутил, циклопентил, циклогексил, циклооктил, циклогексенил, циклооктадиенил, адамантил, норборнил, инданил с местом присоединения в положении 1-3 и т.п. Предпочтительными являются циклопропил, циклобутил, циклопентил или циклогексил, и более предпочтительны циклопентил или циклогексил.
«Арил» обозначает моноциклический-трициклический ароматический С6-14-углеводородный цикл, и его пояснительные примеры включают фенил, нафтил или подобные группы, из которых предпочтителен фенил. Кроме того, с «арилом» может конденсироваться С5-8-циклоалкил. Однако, когда «арил» конденсирован с циклоалкилом, положение присоединения находится на ароматическом цикле. Например, «арил» может образовывать инданил с местом присоединения в положениях 4-7 или тетрагидронафталин с местом присоединения в положениях 5-8.
«Гетероцикл» является общим названием, которое включает «ароматический гетероцикл» и «неароматический гетероцикл». «Ароматический гетероцикл» обозначает моноциклический ароматический гетероцикл, который представляет собой моноциклическую 5-7-членную ароматическую группу, содержащую от 1 до 4 одинаковых или различных гетероатомов, выбранных из группы, состоящей из атомов азота, кислорода и серы, бициклический ароматический гетероцикл, в котором конденсированы моноциклические ароматические гетероциклы или моноциклический ароматический гетероцикл конденсирован с бензольным циклом, или трициклический ароматический гетероцикл, в котором бициклический ароматический гетероцикл конденсирован с моноциклическим ароматическим гетероциклом или с бензольным циклом. Его пояснительные примеры включают пирролил, фурил, тиенил, имидазолил, пиразолил, триазолил, оксазолил, тиазолил, фуразанил, пиридил, пиранил, тиопиранил, пиридазинил, пиримидинил, пиразил, индолил, изоиндолил, индолизинил, бензофурил, бензотиенил, бензоимидазолил, индазолил, бензотриазолил, бензоксазолил, бензотиазолил, бензоксадиазонил, хинолил, изохинолил, хроменил, бензотиопиранил, фталазинил, нафтиридинил, хиноксалинил, хиназолинил, циннолинил, бензодиоксолил, бензодиоксинил, бензодиоксепинил, карбазолил и подобные группы, и атом азота и/или атом серы, содержащиеся в таких циклах, могут быть окислены. Кроме того, указанные циклы могут быть частично насыщенными. Предпочтительными являются пиридил, фурил, тиенил, индолил или хинолил.
«Неароматический гетероцикл» обозначает насыщенный или частично насыщенный моноциклический 3-10-членный, предпочтительно 5-7-членный моноциклический неароматический гетероцикл, который содержит от 1 до 4 гетероатомов, выбранных из O, S и N, бициклический неароматический гетероцикл, в котором конденсированы моноциклические неароматические гетероциклы или моноциклический неароматический гетероцикл конденсирован с моноциклическим неароматическим гетероциклом, С5-8-циклоалкилом, бензольным циклом или ароматическим гетероциклом, или трициклический неароматический гетероцикл, в котором бициклический неароматический гетероцикл конденсирован с С5-8-циклоалкилом, бензольным циклом или ароматическим гетероциклом. Такие гетероциклы могут образовывать оксид или диоксид через окисление S или N как циклического атома или могут образовывать мостиковый цикл или спироцикл. Их пояснительные примеры включают гидропиридил, дигидропирролил, дигидрооксазолил, дигидротиазолил, дигидроимидазолил, пиперидил, морфолинил, тиоморфолинил, пиперазинил, пиразолидинил, имидазолидинил, пирролидинил, оксазолидинил, тиазолидинил, азепанил, гомопиперазинил, тетрагидрофуранил, тетрагидропиранил, тетрагидропиримидинил, хроманил, диоксоранил, гомоморфолинил и подобные группы. Предпочтительными являются пирролидинил, пиперидинил, морфолинил, тиоморфолинил или пиперазинил.
Термин «может быть замещенным» обозначает «незамещенный» или «замещенный одинаковыми или различными 1-5 заместителями».
В описании заместители, приемлемые как заместители в случае выражения «который может быть замещенным», достаточно включают заместители, обычно применяемые в технике как заместители для отдельных групп. Кроме того, когда присутствуют две группы или больше, подобно случаю для R0 в -N(R0)2, соответствующие группы могут быть одинаковыми или отличаться друг от друга.
В качестве приемлемого заместителя для «низшего алкилена», который может быть замещенным в случае R6, можно упомянуть предпочтительно галоген.
Предпочтительно группу, выбранную из описанной далее группы G1, можно упомянуть как приемлемый заместитель для «низшего алкилена» и «низшего алкенилена», которые могут быть замещенными в случае L; «низшего алкилена» и «низшего алкенилена», которые могут быть замещенными в случае Rb; «низшего алкилена», который может быть замещенным в случае Rc; «низшего алкилена» и «низшего алкенилена», которые могут быть замещенными в случае Rd; и образованного R4 и R7 «низшего алкилена», который может быть замещенным.
Группа G1: галоген, -OR0, -CO2R0 и -СО2-(низший алкилен)-арил.
Предпочтительно группу, выбранную из описанной далее группы G2, можно упомянуть как приемлемый заместитель для «циклоалкила», который может быть замещенным в случае R1; «циклоалкила», который может быть замещенным в случае R4; «циклоалкила», который может быть замещенным в случае R5; «циклоалкила», который может быть замещенным в случае Rb; «циклоалкила», который может быть замещенным в случае Rd; и «циклоалкила», который может быть замещенным в случае Rdа.
Группа G2: галоген, низший алкил, -OR0, -CO2R0 и -С(О)-арил.
Предпочтительно группу, выбранную из описанной далее группы G3, можно упомянуть как приемлемый заместитель для «арила», который может быть замещенным в случае Rа; «арила», который может быть замещенным в случае Rb; «арила», который может быть замещенным в Rbа; «арила», который может быть замещенным в случае Rс; «арила», который может быть замещенным в случае Rdа; и «арила», который может быть замещенным в случае Rе.
Группа G3: галоген, -CN, низший алкил, галоген(низший алкил), -OR0, -О-(галоген(низший алкил)), -CO2R0 и -О-(низший алкилен)-CO2R0.
Предпочтительно группу, выбранную из описанной далее группы G4, можно упомянуть как приемлемый заместитель для «арила», который может быть замещенным в Rd.
Группа G4: галоген, -CN, -NO2, низший алкил, галоген(низший алкил), -OR0, -О-(галоген(низший алкил)), -C(O)R0, -CO2R0, низший алкилен-CO2R0, -О-(низший алкилен)-CO2R0, -ОC(O)R0, -N(R0)2, -S(O)2-(низший алкил), арил и гетероциклическая группа. Однако арил и гетероциклическая группа в группе G4 могут быть замещены группой Q.
Группа Q: галоген, низший алкил, галоген(низший алкил), -OR0, -О-(галоген(низший алкил)), оксо и -CO2R0.
Предпочтительно группу, выбранную из описанной далее группы G5, можно упомянуть как приемлемый заместитель для «гетероциклической группы», которая может быть замещенной в случае R4; «гетероциклической группы», которая может быть замещенной в случае R5; «гетероциклической группы», которая может быть замещенной в случае Rа; «гетероциклической группы», которая может быть замещенной в случае Rb; «гетероциклической группы», которая может быть замещенной в случае Rр; «гетероциклической группы», которая может быть замещенной в случае Rbа; и «гетероциклической группы», которая может быть замещенной в случае Rе.
Группа G5: галоген, низший алкил, галоген(низший алкил), -OR0, -О-(галоген(низший алкил)), оксо, -CO2R0, низший алкилен-CO2R0 и -S(O)2-(низший алкил).
Предпочтительно группу, выбранную из описанной далее группы G6, можно упомянуть как приемлемый заместитель для «гетероциклической группы», которая может быть замещенной в случае Rс; и «гетероциклической группы», которая может быть замещенной в случае Rda.
Группа G6: галоген, низший алкил, галоген(низший алкил), -OR0, -О-(галоген(низший алкил)), оксо, -CO2R0, низший алкилен-C(O)2R0, -S(O)2-(низший алкил), арил, -S-(низший алкилен)-арил и гетероциклическая группа.
В этом отношении арил и гетероциклическая группа в группе G6 могут быть замещены группой, выбранной из вышеуказанной группы Q.
Предпочтительно группу, выбранную из описанной далее группы G7, можно упомянуть как приемлемый заместитель для «гетероциклической группы», которая может быть замещенной в случае Rd.
Группа G7: галоген, нитро, низший алкил, галоген(низший алкил), -OR0, -О-(галоген(низший алкил)), оксо, -CO2R0, низший алкилен-CO2R0, -N(R0)2, -S(O)2-(низший алкил), -S(O)2-арил, арил, низший алкиленарил, гетероциклическая группа, (низший алкилен)-(гетероциклическая группа) и -S-(низший алкилен)-CO2R0.
В этом отношении арил и гетероциклическая группа в группе G7 могут быть замещены группой, выбранной из вышеуказанной группы Q.
Предпочтительное воплощение настоящего изобретения показано далее.
(а) Предпочтительным R1 является циклогексил или циклопропилметил, предпочтительнее циклогексил.
(b) Предпочтительным R2 является -F.
(с) Предпочтительным R3 является -Н, -ОН или -F, более предпочтительным является -Н.
(d) Предпочтительным R4 является низший алкил или циклоалкил, предпочтительнее изопропил, 3-пентил или циклопентил, еще более предпочтительным является изопропил, 3-пентил или циклопентил.
(е) Предпочтительным R5 является -N(R0)С(О)-(низший алкилен)-CO2R0, -N(R0)С(О)-(низший алкенилен)-CO2R0, низший алкилен-CO2R0, низший алкенилен-CO2R0, -О-(низший алкенилен)-CO2R0, -О-(низший алкилен, который может быть замещен -CO2R0)-арил, -О-(низший алкенилен)-CO2R0, -О-(низший алкенилен, который может быть замещен -CO2R0)-арил или -О-(низший алкенилен)тетразолил, предпочтительнее -N(R0)С(О)-(низший алкилен)-CO2R0, низший алкилен-CO2R0, низший алкенилен-CO2R0, -О-(низший алкилен)-CO2R0, -О-(низший алкилен, который может быть замещен -CO2R0)-арил или -О-(низший алкенилен)-CO2R0, еще более предпочтительным низший алкенилен-CO2R0 или -О-(низший алкилен)-CO2R0.
(f) Предпочтительным Х является СН.
(g) Предпочтительным А является СН.
Также более предпочтительным является соединение, включающее сочетания вышеуказанных предпочтительных групп (а)-(g).
Также далее показано другое предпочтительное воплощение соединения по настоящему изобретению, представленного общей формулой (I).
(1) Соединение, описанное общей формулой (I), где Х представляет собой СН.
(2) Соединение, описанное в (1), где R3 представляет собой -Н, -ОН или -F.
(3) Соединение, описанное в (2), где А представляет собой СН.
(4) Соединение, описанное в (3), где R1 представляет собой циклогексил или циклопропилметил.
(5) Соединение, описанное в (4), где R2 представляет собой -F.
(6) Соединение, описанное в (5), где R4 представляет собой низший алкил или циклоалкил.
(7) Соединение, описанное в (6), где R5 представляет собой -N(R0)С(О)-(низший алкилен)-CO2R0, низший алкилен-CO2R0, низший алкенилен-CO2R0, -О-(низший алкилен)-CO2R0, -О-(низший алкилен, который может быть замещен -CO2R0)-арил или -О-(низший алкенилен)-CO2R0.
(8) Соединение, описанное общей формулой (I), которое выбрано из группы, в которую входят
4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-4-оксобутановая кислота,
5-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-5-оксопентановая кислота,
(2Е)-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]акриловая кислота,
(2S)-2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}-3-фенилпропановая кислота,
(2Е)-3-[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]акриловая кислота,
(2S)-2-{[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]окси}-3-фенилпропановая кислота,
(2S)-2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}пропановая кислота и
(2S)-2-{[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]окси}пропановая кислота,
или его фармацевтически приемлемая соль.
Также соединения по настоящему изобретению могут образовывать соли, и такие соли входят в соединения по настоящему изобретению, если они являются фармацевтически приемлемыми солями. Их пояснительные примеры включают кислотно-аддитивные соли, образованные с неорганическими кислотами (например, хлористоводородной кислотой, бромистоводородной кислотой, йодистоводородной кислотой, серной кислотой, азотной кислотой, фосфорной кислотой и т.п.) или органическими кислотами (например, муравьиной кислотой, уксусной кислотой, пропионовой кислотой, щавелевой кислотой, малоновой кислотой, янтарной кислотой, фумаровой кислотой, малеиновой кислотой, молочной кислотой, яблочной кислотой, винной кислотой, лимонной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, п-толуолсульфоновой кислотой, аспарагиновой кислотой, глутаминовой кислотой и т.п.), соли с неорганическими основаниями, в том числе соли металлов (например, натрия, калия, кальция, магния и т.п.), или соли металлов основного характера (например, метиламин, этиламин, этаноламин, лизин, орнитин и т.п.), аммониевые соли и подобные соли.
Кроме того, в некоторых случаях, в зависимости от вида заместителей соединения по настоящему изобретению могут содержать асимметричный атом углерода, и в связи с этим могут иметь место оптические изомеры. Настоящее изобретение включает все смеси и изолированные формы таких оптических изомеров. Также у соединений по настоящему изобретению в некоторых случаях могут быть таутомеры, и настоящее изобретение включает отдельные формы таких изомеров или их смеси. Кроме того, меченое вещество, а именно соединение, в котором, по меньшей мере, один атом соединения по настоящему изобретению заменен радиоактивным изотопом или нерадиоактивным изотопом, также входит в объем настоящего изобретения.
Кроме того, в изобретение также включены различные типы гидратов и сольватов и полиморфов соединения по настоящему изобретению. В связи с этим, как предмет обсуждения, соединения по настоящему изобретению не ограничиваются соединениями, описанными в примерах, которые приводятся далее, и в них включены все производные, представленные формулой (I), и их фармацевтически приемлемые соли.
В связи с этим все соединения, которые в живом организме превращаются в соединения по настоящему изобретению, представленные вышеуказанной общей формулой (I), так называемые пролекарства, также входят в соединения по настоящему изобретению. В качестве примеров групп, которые могут образовывать пролекарства соединений по настоящему изобретению, можно назвать группы, описанные в Prog. Med., 5: 2157-2161 (1985), и группы, описанные в “Iyakuhin no Kaihatsu (Development of Medicines)”, vol.7, Bunshi Sekkei (Molecular Design), pp. 163-198, публикация Hirokawa Shoten в 1990.
Способы получения
Соединение по настоящему изобретению и его фармацевтически приемлемую соль можно получить с использованием различных широко известных способов синтеза с применением особенностей, основанных на его основной структуре или виде заместителей. Примеры типичных способов получения приводятся далее. В связи с этим, в зависимости от вида функциональной группы, эффективно, с точки зрения технологии, замещать указанную функциональную группу соответствующей защитной группой, а именно группой, которую можно легко превратить в указанную функциональную группу на стадии исходного вещества - промежуточного вещества. Затем нужное соединение можно получить, удаляя защитную группу, как требует ситуация. Примеры функциональной группы включают гидроксильную группу, карбоксильную группу, аминогруппу и подобные группы, а в качестве их защитных групп можно назвать защитные группы, описанные в “Protective Groups in Organic Synthesis (third edition)”, редакторы Greene и Wuts, которые можно, необязательно, использовать в соответствии с условиями реакции.
Первый способ получения
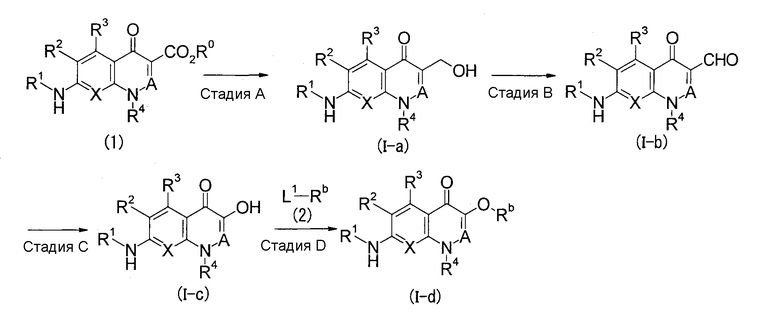
(В приведенных формулах L1 представляет собой удаляемую группу, такую как галоген, -О-метансульфонил, -О-пара-толуолсульфонил или подобную. То же самое справедливо для данного описания далее.)
Стадия А
Данная стадия является стадией, на которой соединение (I-a) по настоящему изобретению получают восстановлением соединения (1).
В качестве реакции восстановления на данной стадии можно использовать реакцию восстановления карбоновой кислоты или сложного эфира, обычно используемую специалистами в данной области техники. Например, реакцию можно осуществить при охлаждении и при кипячения с обратным холодильником с использованием эквимолярного и избыточного количества восстановителя, такого как алюмогидрид лития, гидрид диизобутилалюминия, борогидрид натрия или подобного, при взаимодействии в инертном растворителе, например ароматических углеводородах, таких как бензол, толуол, ксилол, простых эфирах, таких как диэтиловый эфир, тетрагидрофуран (ТГФ), диоксан, галогенсодержащих углеводородах, таких как дихлорметан, 1,2-дихлорэтан, хлороформ, N,N-диметилформамиде (ДМФА), N,N-диметилацетамиде (DMA), N-метилпирролидоне (NMP), диметилсульфоксиде (ДМСО), ацетонитриле, спиртах, таких как метанол, этанол, и воде. Кроме того, когда соединение (1) представляет собой карбоновую кислоту, где R0 представляет собой -Н, карбоновую кислоту также можно восстановить после превращения ее в рекционно-способное производное. В качестве реакционно-способного производного карбоновой кислоты можно назвать ацилимидазол, получаемый взаимодействием с 1,1'-карбонилдиимидазолом (CDI), смешанный ангидрид, получаемый взаимодействием с изобутилхлорформиатом, и т.д., и подобные производные.
Стадия В
Данная стадия является стадией, на которой соединение (I-b) по настоящему изобретению получают окислением соединения (I-а) по настоящему изобретению.
В реакции окисления на данной стадии можно использовать реакцию окисления спиртов, обычно используемую специалистами в данной области техники. Например, реакцию можно осуществить при температуре от комнатной до нагревания с использованием от эквивалентного до избыточного количества диоксида марганца в качестве окислителя, в растворителе, таком как вышеуказанные ароматические углеводороды, галогенсодержащие углеводороды или подобные.
Стадия С
Данная стадия является стадией, на которой соединение (I-с) по настоящему изобретению получают, подвергая соединение (I-b) по настоящему изобретению реакции окислительной перегруппировки (по Байеру-Виллигеру) и затем гидролизу.
Реакцию окислительной перегруппировки на данной стадии можно осуществить при температуре от комнатной до нагревания с использованием от эквивалентного до избыточного количества м-хлорпербензойной кислоты, перуксусной кислоты, водного пероксида водорода или подобного соединения в качестве окислителя в растворителе, инертном для реакции, таком как вышеуказанные ароматические углеводороды, галогенсодержащие углеводороды, уксусная кислота, вода или подобный растворитель.
Реакцию гидролиза на данной стадии можно осуществить с использованием реакции гидролиза сложных эфиров, обычно используемой специалистами в данной области техники. Например, реакцию можно осуществить при охлаждении и при нагревании в инертном для реакции растворителе, таком как вышеуказанные ароматические углеводороды, простые эфиры, галогенсодержащие углеводороды, спирты, ДМФА, DMA, NMP, ДМСО, пиридин, вода или подобный растворитель, в присутствии минеральной кислоты, такой как серная кислота, хлористоводородная кислота, бромистоводородная кислота или подобная кислота, или органической кислоты, такой как муравьиная кислота, уксусная кислота или подобная кислота, или в присутствии основания, такого как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия, карбонат цезия или аммиак, или подобного основания.
В зависимости от вида соединений, соединение (I-c) можно получить в некоторых случаях, осуществляя гидролиз по ходу реакции окислительной перегруппировки.
Стадия D
Данная стадия является стадией, на которой соединение (I-d) по настоящему изобретению получают, подвергая соединение (I-с) по настоящему изобретению реакции нуклеофильного замещения.
Реакцию нуклеофильного замещения на данной стадии можно осуществить с использованием от эквивалентного до избыточного количества соединения (2) от комнатной температуры до нагревания в растворителе, таком как вышеуказанные ароматические углеводороды, простые эфиры, галогенсодержащие углеводороды, ДМФА, DMA, NMP, ДМСО или подобный растворитель, в присутствии основания, такого как карбонат калия, трет-бутоксид калия, гидрид натрия, триэтиламин или подобное основание.
Второй способ получения

(В приведенных формулах R10 и R11 обозначают -Н, галоген, -CO2R0 или низший алкил или арил, который может быть, соответственно, замещен, и R20 обозначает остальную часть реагента Хорнера-Эммонса (4), R21 обозначает остальную часть соли фосфония (5), Ха- обозначает Cl-, Br- или подобный противоион, и R22 обозначает остальную часть илида соединения (6)).
Стадия Е
Данная стадия является стадией, на которой соединение (I-е) по настоящему изобретению получают, подвергая соединение (I-b) по настоящему изобретению реакции восстановительного алкилирования.
Как реакцию восстановительного алкилирования на данной стадии можно использовать реакцию восстановительного алкилирования, обычно используемую специалистами в данной области техники. Например, можно упомянуть способ, описанный в “Jikken Kagaku Koza (Experimental Chemistry Course)”, редакция The Chemical Society of Japan, Vol.20 (1992) (Maruzen), или подобные способы. Взаимодействие желательно осуществлять при охлаждении и до кипячения с обратным холодильником с использованием восстановителя, такого как боргидрид натрия, триацетоксиборгидрид натрия, или подобного восстановителя в отсутствие растворителя или в инертном для реакции растворителе, таком как вышеуказанные ароматические углеводороды, простые эфиры, спирты, сложные эфиры, в том числе этилацетат или подобные эфиры, уксусная кислота или подобный растворитель. В зависимости от соединения в некоторых случаях выгодно осуществлять взаимодействие в присутствии органической кислоты, такой как серная кислота, хлористоводородная кислота, бромистоводородная кислота или подобная минеральная кислота, муравьиной кислоты, уксусной кислоты или подобной органической кислоты или кислоты Льюиса, такой как хлорид титана(IV), тетраизопропилортотитанат или подобная кислота. Кроме того, взаимодействие также можно осуществить при комнатной температуре и нагревании в атмосфере водорода при обычном давлении и при повышенном давлении с использованием в качестве катализатора, например палладия-на-угле, родия-на-угле, никеля Ренея, платины или подобного, в инертном для реакции растворителе, таком как вышеуказанные ароматические углеводороды, сложные эфиры, простые эфиры, галогенсодержащие углеводороды, ДМФА, DMA, NMP, ацетонитрил, уксусная кислота или подобный растворитель. В зависимости от соединения в некоторых случаях для гладкого протекания реакции выгодно осуществлять взаимодействие в присутствии кислоты (предпочтительно хлористоводородной кислоты, уксусной кислоты или подобной кислоты).
Стадия F
Данная стадия является стадией, на которой соединение (I-f) по настоящему изобретению получают, подвергая соединение (I-b) по настоящему изобретению реакции Хорнера-Эммонса или Виттига.
Для реакции Хорнера-Эммонса или Виттига на данной стадии можно использовать способ, обычно используемый специалистами в данной области техники. Например, когда используют реагент Хорнера-Эммонса (4) или соль фосфония (5), взаимодействие можно осуществить при охлаждении и при нагревании с использованием карбоната калия, трет-бутоксида калия, гидрида натрия, н-бутиллития или подобного алкиллития или подобного соединения в качестве основания, в растворителе, таком как вышеуказанные ароматические углеводороды, простые эфиры, галогенсодержащие углеводороды, ДМФА, DMA, NMP, ДМСО, ацетонитрил или подобный растворитель. Также, когда используют илид соединения (6), взаимодействие можно осуществить при охлаждении и при нагревании в растворителе, таком как вышеуказанные ароматические углеводороды, простые эфиры, галогенсодержащие углеводороды, ДМФА, DMA, NMP, ДМСО, ацетонитрил или подобный растворитель.
Стадия G
Данная стадия является стадией, на которой соединение (I-g) по настоящему изобретению получают восстановлением двойной связи соединения (I-f) по настоящему изобретению.
Для реакции восстановления на данной стадии можно использовать способ, обычно используемый специалистами в данной области техники. Например, реакцию также можно осуществлять от комнатной температуры до нагревания в атмосфере водорода при обычном давлении и повышенном давлении с использованием в качестве катализатора палладия-на-угле, никеля Ренея, платины или подобного соединения, в инертном для реакции растворителе, таком как вышеуказанные ароматические углеводороды, сложные эфиры, простые эфиры, галогенсодержащие углеводороды, ДМФА, DMA, NMP, уксусная кислота или подобный растворитель. В зависимости от соединения в некоторых случаях для гладкого протекания реакции выгодно осуществлять реакцию в присутствии кислоты (предпочтительно хлористоводородной кислоты, уксусной кислоты или подобной кислоты).
Третий способ получения

(В приведенных формулах L2 представляет собой удаляемую группу, такую как галоген, -О-метансульфонил, -О-пара-толуолсульфонил или подобную. То же самое применяется в данном описании далее.)
Стадия Н
Данная стадия является стадией, на которой соединение (I-h) по настоящему изобретению получают, подвергая соединение (7) реакции нуклеофильного замещения.
Реакцию нуклеофильного замещения на данной стадии можно осуществить с использованием соединения (7) и соединения (8) в эквимолярных количествах или одного из них в избыточном количестве в интервале температур от комнатной температуры до нагревания в отсутствие растворителя или в растворителе, таком как вышеуказанные ароматические углеводороды, простые эфиры, галогенсодержащие углеводороды, ДМФА, ДМСО, сложные эфиры, включая этилацетат и подобные эфиры, ацетонитрил, спирты или подобный растворитель. В зависимости от соединений, в некоторых случаях осуществлять реакцию выгодно в присутствии органического основания (подходящим является триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин или подобное основание), или основания, образованного солью металла (карбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия, гидрид натрия, трет-бутокси калий и др.).
Стадия I
Данная стадия является стадией, на которой соединение (I-i) по настоящему изобретению получают восстановлением соединения (I-h) по настоящему изобретению.
Для реакции восстановления нитрогруппы на данной стадии можно использовать способ, обычно используемый специалистами в данной области техники. Например, реакцию также можно осуществить в интервале температур от комнатной температуры до нагревания в атмосфере водорода при обычном давлении и повышенном давлении с использованием в качестве катализатора палладия-на-угле, никеля Ренея, платины или подобного соединения, в инертном для реакции растворителе, таком как вышеуказанные ароматические углеводороды, сложные эфиры, простые эфиры, галогенсодержащие углеводороды, ДМФА, DMA, NMP, уксусная кислота или подобный растворитель. В зависимости от соединения в некоторых случаях для гладкого протекания реакции выгодно осуществлять реакцию в присутствии кислоты (предпочтительно хлористоводородной кислоты, уксусной кислоты или подобной кислоты).
Четвертый способ получения

Стадия J
Данная стадия является стадией, на которой соединение (I-j) по настоящему изобретению получают дегидратацией соединения (9).
Для реакции дегидратации на данной стадии можно использовать способ, обычно используемый специалистами в данной области техники. Например, реакцию можно осуществить в интервале температур от комнатной температуры до нагревания с использованием в качестве дегидратирующего агента пентоксида дифосфора, оксихлорида фосфора, трифторуксусного ангидрида или подобного соединения, в отсутствие растворителя или в инертном для реакции растворителе, таком как ароматические углеводороды, галогенсодержащие углеводороды, простые эфиры или подобные растворители. Однако, когда в качестве дегидратирующего агента используют трифторуксусный ангидрид, аминогруппу в положении 7 хинолона в некоторых случаях трифторацетилируют, в зависимости от вида соединения, и это является случаем, когда для последующей обработки требуется гидролиз. При гидролизе можно использовать способ, который специалисты в данной области техники обычно используют при гидролизе амидов.
Стадия К
Данная стадия является стадией, на которой соединение (I-k) по настоящему изобретению получают восстановлением соединения (I-j) по настоящему изобретению.
Реакцию восстановления нитрильной группы на данной стадии также можно осуществить в интервале температур от комнатной температуры до нагревания в атмосфере водорода при обычном давлении и повышенном давлении с использованием в качестве катализатора палладия-на-угле, никеля Ренея, платины или подобного соединения, в инертном для реакции растворителе, таком как вышеуказанные ароматические углеводороды, сложные эфиры, простые эфиры, галогенсодержащие углеводороды, ДМФА, DMA, NMP, уксусная кислота или подобный растворитель. В зависимости от соединения в некоторых случаях для гладкого протекания реакции выгодно осуществлять реакцию в присутствии кислоты (предпочтительно хлористоводородной кислоты, уксусной кислоты или подобной кислоты).
Пятый способ получения
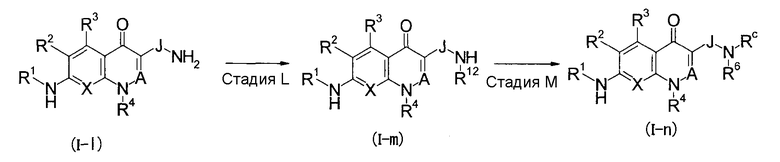
(В приведенных формулах J представляет собой простую связь или низший алкилен и R12 представляет собой R6 или Rc. То же самое будет применяться в данном описании далее.)
Стадия L
Данная стадия является стадией, на которой соединение (I-m) по настоящему изобретению получают, подвергая соединение (I-l) по настоящему изобретению реакции нуклеофильного замещения или реакции восстановительного алкилирования.
Реакцию нуклеофильного замещения и реакцию восстановительного алкилирования на данной стадии можно осуществить, соответственно, таким же способом, как на стадии D и на стадии Е.
Стадия М
Данная стадия является стадией, на которой соединение (I-n) по настоящему изобретению получают, подвергая соединение (I-m) по настоящему изобретению реакции нуклеофильного замещения или реакции восстановительного алкилирования.
Реакцию нуклеофильного замещения и реакцию восстановительного алкилирования на данной стадии можно осуществить, соответственно, таким же способом, как на стадии D и на стадии Е.
Шестой способ получения
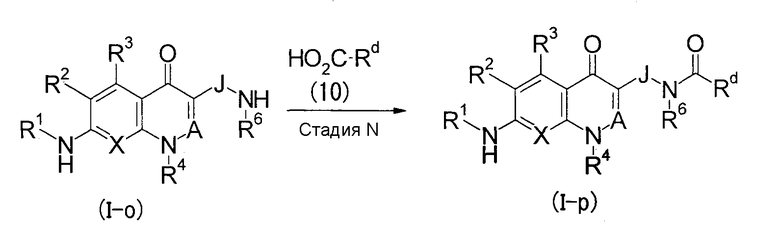
Стадия N
Данная стадия является стадией, на которой соединение (I-р) по настоящему изобретению получают, подвергая соединение (I-о) по настоящему изобретению реакции амидирования соединением (10) или его реакционно-способным производным.
Для реакции амидирования на данной стадии можно использовать амидирование, которое обычно могут использовать специалисты в данной области техники. В частности, подходящим является способ, в котором используют агент конденсации, такой как карбонилдиимидазол (CDI), гидрохлорид 1-этил-3-(диметиламинопропил)карбодиимида (WSC·HCl), дициклогексилкарбодиимид, дифенилфосфорилазид, диэтилфосфорилцианид или подобное соединение, способ, который осуществляют с помощью смешанного ангидрида кислоты с использованием изобутилхлорформиата, этилхлорформиата и т.п., и способ, который осуществляют с помощью галогенидангидрида с использованием тионилхлорида, оксихлорида фосфора или подобного соединения. Условия реакции можно выбрать произвольно в зависимости от используемых реакционно-способного производного и агента конденсации, и, как правило, реакцию осуществляют при охлаждении, в интервале температур от охлаждения до комнатной температуры или от комнатной температуры до нагревания в инертном для реакции растворителе, таком как галогенсодержащие углеводороды, ароматические углеводороды, простые эфиры, ДМФА, ДМСО или подобный растворитель. В зависимости от реакции в некоторых случаях ее выгодно осуществлять в присутствии органического основания (подходящим является триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин или подобное основание) или основания, образованного солью металла (подходящим является карбонат калия, карбонат цезия или подобное).
Седьмой способ получения

(В приведенной формуле L3 представляет собой удаляемую группу, такую как -О-(низший алкил), -О-пара-нитрофенил или подобную.)
Стадия О
Данная стадия является стадией, на которой соединение (I-r) по настоящему изобретению получают путем образования мочевины из соединения (I-q) по настоящему изобретению.
Реакцию образования мочевины можно осуществить от комнатной температуры до нагревания с использованием эквивалентных количеств соединения (I-q) и соединения (11) или избыточного количества одного из них, в инертном для реакции растворителе, таком как ароматические углеводороды, галогенсодержащие углеводороды, простые эфиры, ДМФА, ДМСО или подобный растворитель. В зависимости от реакции, в некоторых случаях ее выгодно осуществлять в присутствии органического основания (подходящим является триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, 1,8-диазабицикло[5.4.0]-7-ундецен или подобное основание) или соли металла основного характера (подходящими являются карбонат калия, карбонат цезия или подобная соль).
Кроме того, некоторые соединения, представленные формулой (I), также можно получить из соединений, полученных вышеописанными способами, произвольно комбинируя стадии, такие как широко известные алкилирование, ацилирование, реакция замещения, окисление, восстановление, гидролиз и т.п., которые вообще могут использовать специалисты в данной области техники. В частности, соединения (I-a), (I-b), (I-c), (I-h), (I-i) и (I-j) по настоящему изобретению также применимы в качестве промежуточных соединений для синтеза соединений по настоящему изобретению.
Синтез исходных соединений
Исходные соединения для применения при получении соединения (I) по настоящему изобретению можно синтезировать с использованием описанных далее способов, широко известных способов или таких модифицированных способов.
Синтез 1 исходных веществ

Соединение (I-a) можно получить с использованием способа, описанного в патентной ссылке 7 или такого модифицированного способа.
Стадия Р
Данная стадия является стадией, на которой соединение (9) получают амидированием соединения (I-a).
Что касается реакции амидирования на указанной стадии, то ее можно осуществить, например, способом, описанным на стадии N.
Синтез 2 исходных веществ
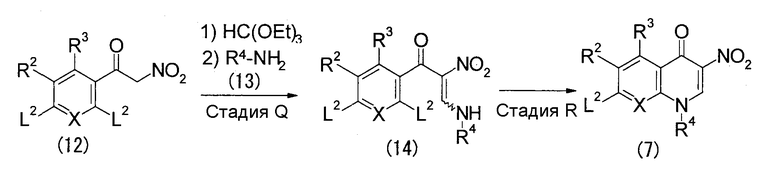
Стадия Q
Данная стадия является стадией, на которой соединение (14) получают конденсацией соединения (12) со сложным эфиром ортомуравьиной кислоты и последующей реакцией присоединения-элиминирования соединением (13).
Реакцию конденсации на данной стадии эфиром ортомуравьиной кислоты можно осуществить в интервале температур от комнатной температуры до нагревания с использованием в качестве растворителя реагента, который захватывает спирты, образовавшиеся из эфира ортомуравьиной кислоты, такого как уксусный ангидрид, или с использованием реагента, который захватывает спирты, образовавшиеся из эфира ортомуравьиной кислоты, в инертном для реакции растворителе, таком как галогенсодержащие углеводороды, ароматические углеводороды, ДМФА, ДМСО, сложные эфиры, ацетонитрил или подобный растворитель.
Реакцию присоединения-элиминирования после вышеуказанной реакции конденсации можно осуществить при охлаждении, комнатной температуре или нагревании в инертном для реакции растворителе, таком как спирты, галогенсодержащие углеводороды, простые эфиры, ароматические углеводороды, ДМФА, ДМСО или подобный растворитель. В этой связи реакцию также можно осуществить с использованием избытка соединения (13). В зависимости от соединений, в некотором случае реакцию выгодно осуществлять в присутствии органического основания (подходящим является триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин или подобное основание) или основания, образованного солью металла (подходящими являются карбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия, гидрид натрия, трет-бутоксид калия или подобное соединение).
Стадия R
Данная стадия является стадией, на которой соединение (7) получают реакцией внутримолекулярной циклизации аминогруппы соединения (14).
Реакцию внутримолекулярной циклизации на данной стадии можно осуществить при охлаждении, комнатной температуре или нагревании в инертном для реакции растворителе, таком как галогенсодержащие углеводороды, простые эфиры, ароматические углеводороды, ДМФА, ДМСО или подобный растворитель. В зависимости от соединений, в некотором случае реакцию выгодно осуществлять в присутствии органического основания (подходящим является триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, 1,8-диазабицикло[5.4.0]-7-ундецен или подобное основание) или соли металла основного характера (подходящими являются карбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия, гидрид натрия, трет-бутоксид калия или подобное соединение).
Соединение по настоящему изобретению, полученное таким способом, извлекают и очищают непосредственно в свободном виде или в виде его соли, применяя обработку для образования соли обычным способом. Извлечение и очистку осуществляют с использованием обычных химических операций, таких как экстракция, концентрирование, выпаривание, кристаллизация, фильтрация, перекристаллизация, различные типы хроматографии и т.п.
Различные типы изомеров можно извлечь обычным способом, используя различие в физико-химических свойствах между изомерами. Например, рацемическую смесь можно превратить в оптически чистые изомеры обычным способом расщепления рацематов, включающих, например, превращение в соли диастереомеров с оптически активной кислотой, такой как винная кислота, и последующее расщепление оптических изомеров. Также смесь диастереомеров можно разделить, например, фракционной перекристаллизацией или различными типами хроматографии. Кроме того, оптически активное соединение также можно получить с использованием соответствующего оптически активного соединения в качестве исходного вещества.
Фармацевтическую композицию, которая содержит одно или несколько соединений по настоящему изобретению или их фармацевтически приемлемые соли в качестве активного ингредиента, получают с использованием носителей и наполнителей и других добавок, обычно используемых при получении фармацевтических препаратов.
Вводить фармацевтическую композицию можно или оральным способом с помощью таблеток, пилюль, капсул, гранул, порошков, растворов и т.п., или парентеральным способом с помощью внутривенных, внутримышечных или подобных инъекций, суппозиториев, чрескожных препаратов, трансназальных препаратов, ингаляций и т.п. Дозу композиции определяют произвольно с учетом симптомов, возраста, пола и т.п. субъекта, которого лечат, в каждом случае, но в случае орального введения доза, как правило, составляет от 0,001 мг/кг до 100 мг/кг в сутки на взрослого субъекта, и такую дозу вводят в виде одной порции или разделяя на 2-4 порции. Также в случае внутривенного введения дозу вводят в интервале от 0,0001 мг/кг до 10 мг/кг для взрослого субъекта один или два или большее число раз в сутки. Кроме того, в случае трансназального введения дозу вводят в интервале от 0,0001 мг/кг до 10 мг/кг для взрослого субъекта один или два или большее число раз в сутки.
Как твердую композицию для орального введения по настоящему изобретению используют таблетки, порошки, гранулы и т.п. В такой твердой композиции одно или несколько активных веществ смешаны с, по меньшей мере, одним инертным разбавителем, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон, алюмосиликат магния, или подобным разбавителем. Согласно обычной практике композиция может содержать добавки, иные, чем инертный разбавитель, такие как смазывающее вещество (например, стеарат магния или подобное вещество), вещество, способствующее рассыпанию (например, кальцийгликолят целлюлозы или подобное вещество), стабилизатор, солюбилизатор и т.п. При необходимости на таблетки или пилюли может быть нанесено сахарное покрытие или пленка гастро- или энтеросолюбильного вещества, такого как сахароза, желатин, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы, или подобного вещества.
Жидкая композиция для перорального введения включает фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и т.п. и содержит обычно используемый разбавитель, такой как дистиллированная вода или этанол (EtOH). Кроме инертного разбавителя такая композиция может содержать смачивающее вещество, суспендирующее вещество и подобные вспомогательные вещества, а также подслащивающие средства, корригенты, ароматизаторы и антисептики.
Инъекции для парентерального введения включают асептические водные или неводные растворы, суспензии и эмульсии. Водные растворы и суспензии, например, включают дистиллированную воду для инъекций и физиологический раствор. В неводные растворы и суспензии включены, например, пропиленгликоль, полиэтиленгликоль, оливковое масло или подобное растительное масло, EtOH или подобные спирты, полисорбат 80 и т.п. Такая композиция также может содержать вспомогательные вещества, такие как антисептик, смачивающее вещество, эмульгатор, диспергирующее вещество, стабилизатор, солюбилизатор или подобное вещество. Такие композиции стерилизуют, например, фильтрацией через фильтр, задерживающий бактерии, подмешиванием бактерицида или облучением. Их также можно использовать, получая стерильные твердые композиции и затем растворяя их в стерильной воде или стерильном растворителе для инъекции перед применением.
Фармакологическую активность соединений по настоящему изобретению подтверждают описанными далее испытаниями.
Метод испытаний (1). Испытание с измерением активности ингибирования агрегации тромбоцитов человека
У здоровых добровольцев (взрослые мужчины) берут образцы крови с использованием шприца, содержащего 1/10 объема 3,8% раствора цитрата натрия, и центрифугируют при 160×g в течение 10 минут, причем таким образом отделяют от супернатанта обогащенную тромбоцитами плазму (PRP). Кровь, остающуюся после сбора PRP, центрифугируют при 1800×g в течение 10 минут и отделяют плазму, обедненную тромбоцитами (РРР). Число тромбоцитов в PRP измеряют с помощью автоматического счетчика форменных элементов крови (МЕК-6258, Nihon Kodhen Corp.), и затем число тромбоцитов доводят до 3×108/мл, добавляя РРР-PRP, и используют в следующем испытании. АДФ как индуктор агрегации тромбоцитов закупают у МС Medical. Агрегацию тромбоцитов измеряют с использованием агрегометра (МСМ Hematracer 212; МС Medical). А именно, 80 мкл PRP с 3×108 тромбоцитов/мл и 10 мкл раствора испытываемого соединения или растворителя (10% ДМСО или 10% ДМСО-9% гидроксипропил-β-циклодекстрина-4,5% d-маннита) инкубируют при 37°С в течение 1 минуты, затем добавляют к смеси 10 мкл раствора АДФ (50 мкМ) для того, чтобы вызвать агрегацию тромбоцитов, и регистрируют изменения проходящего света в течение 5 минут. Степень ингибирования вычисляют с использованием в качестве показателя площади под кривой агрегации тромбоцитов. Результаты при концентрации соединений по настоящему изобретению 10 мкМ (конечная концентрация) приводятся в таблице 1.
В этой связи номер с С.пр. представляет номер ссылочного примера, и с Пр. - номер примера. Кроме того, ссылочные примеры 1 и 2 являются примерами соединений, описанных в вышеуказанной патентной ссылке 7, и их получают согласно способу, описанному в указанной патентной ссылке.
Ссылочный пример 1 (пример 467 в патентной ссылке 7)
4-({[7-(Циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]карбонил}амино)бутановая кислота
Ссылочный пример 2 (пример 6 в патентной ссылке 7)
({[7-(Циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]карбонил}амино)уксусная кислота
Метод испытаний (2). Испытание на замещение для связывания человеческого P2Y12 с 2-метилтио-АДФ (2-MeS-ADP)
Клетки С6-15 инокулируют в 10-см чашку Петри до плотности 1×106 клеток с использованием среды DMEM и культивируют в течение 1 дня и затем генотрансфицируют 8 мкг плазмиды человеческого P2Y12 pEF-BOS-dfhr и 0,8 мкг pEF-BOS-neo (Nucleic Acid Res., 18, 5322, 1990) с использованием реагента трансфекции (LipofectAMINE 2000; изг. GIBCO BRL).
Через 24 часа после указанной операции переноса генотрансфицированные клетки извлекают, суспендируют в среде DMEM с добавлением 0,6 мг/мл G 418 (изг. GIBCO BRL) и затем серийно разводят и снова инокулируют в 10-см чашке Петри. Колонии, появившиеся через 2 недели, получают отдельно и используют в последующем испытании как клетки С6-15, экспрессирующие белок P2Y12 (WO 02/36631, Mol. Pharmacol., 60, 432, 2001).
После культивирования клеток С6-15, экспрессирующих белок P2Y12, клетки извлекают. Клетки промывают PBS и затем суспендируют в 20 мМ трис-HCl (рН 7,4), содержащем 5 ммоль/л ЭДТК и набор коктейля ингибиторов протеаз Complete™ (изг. Boehringer-Mannheim), и гомогенизируют с использованием Polytron. После осуществления ультрацентрифугирования преципитат суспендируют в 50 мМ трис-HCl (рН 7,4), содержащем 1 мМ ЭДТК, 100 мМ NaCl и Complete™, и полученную смесь используют как мембранную фракцию.
Часть в 100 мкл полученной выше мембранной фракции клеток С6-15, экспрессирующих белок P2Y12 (100 мкг/мл), смешивают с 1,5 мкл раствора испытываемого соединения и 50 мкл 0,75 нМ [3H]-2-MeS-ADP (80 Ки/ммоль, изг. Amersham Pharmacia Biotech) или 0,75 нМ [33Р]-2-MeS-ADP (2100 Ки/ммоль, изг. Perkin Elmer), инкубируют при комнатной температуре в течение 1 часа в 50 мМ трис-HCl (рН 7,4), содержащем 100 мМ NaCl и 50 мМ MgCl2, и затем извлекают на стеклянном фильтре с использованием клеточного харвестера. На стеклянный фильтр добавляют микросцинтиллятор и измеряют радиоактивность с использованием жидкостного сцинтилляционного счетчика. Образцы, где в вышеуказанном испытании в одно и то же время добавляют один растворитель и 1,5 мкл 250-мкМ АДФ, рассматривают как показывающие общее связывание и неспецифическое связывание и измеряют их радиоактивность. Принимая общее связывание и неспецифическое связывание за степень ингибирования 0% и 100% соответственно, вычисляют степень ингибирования (%) для каждого испытываемого соединения. Результаты при концентрации соединений по настоящему изобретению 30 нМ (конечная концентрация) приводятся в таблице 2.
Метод испытаний (3). Испытание на ингибирование агрегации тромбоцитов крысы и измерение концентрации испытываемого соединения в плазме
Добавляя водный раствор гидроксида натрия к соединению по настоящему изобретению, получают водный раствор или суспензию с 0,5% метилцеллюлозы. Полученную таким образом жидкость вводят орально самцам крыс SD (в возрасте 5-7 недель) с использованием зонда через 12 или более часов после кормления. Через 2 часа после введения соединения берут образцы крови с использованием шприца, содержащего 1/10 объема 3,8% раствора цитрата натрия. Таким же способом, как в методе испытаний (1), получают РРР и PRP с 3×108 тромбоцитов/мл. Порцию PRP с 3×108 тромбоцитов/мл в 90 мкл инкубируют при 37°С в течение 1 минуты и затем к ней добавляют 10 мкл АДФ (50 мкМ) для того, чтобы вызвать агрегацию тромбоцитов, и регистрируют изменения в проходящем свете в течение 5 минут. Степень ингибирования вычисляют с использованием в качестве показателя площади под кривой агрегации тромбоцитов.
Концентрацию в плазме измеряют с использованием РРР, полученной выше. Для того чтобы получить стандартную кривую, также выделяют РРР крыс SD, которым соединение не вводили, и также получают образцы с соединением по настоящему изобретению, разведенные серийно такой РРР (от 30 мкМ до 0,0003 мкМ в конечной концентрации: выбор произвольный по реакции на каждое соединение). Порцию в 100 мкл РРР крысы, которой вводили соединение по настоящему изобретению, и РРР, содержащей разведенное соединение по настоящему изобретению, смешивают с таким же объемом дистиллированной воды, добавляют в смесь еще 5% трифторуксусную кислоту и перемешивают. После выстаивания на льду в течение 10 минут извлекают супернатант операцией центрифугирования. Супернатант нейтрализуют, добавляя к нему 3 мкл 2 М трис-основания и перемешивая. Порцию в 150 мкл мембранной фракции клеток С6-15, экспрессирующих белок P2Y12 (200 мкг/мл), смешивают с 50 мкл такой обработанной трихлоруксусной кислотой РРР (в зависимости от соединения, используют РРР, разбавленную 50 мМ трис-HCl (рН 7,4), содержащим 100 мМ NaCl и 50 мМ MgCl2). Затем к смеси добавляют 50 мкл 0,75 нМ [3H]-2-MeS-ADP (80 Ки/ммоль, изг. Amersham Pharmacia Biotech) или 0,75 нМ [33Р]-2-MeS-ADP (2100 Ки/ммоль, изг. Perkin Elmer), инкубируют при комнатной температуре в течение 1 часа в 50 мМ трис-HCl (рН 7,4), содержащем 100 мМ NaCl и 50 мМ MgCl2, и затем извлекают на стеклянном фильтре с использованием клеточного харвестера. На стеклянный фильтр добавляют микросцинтиллятор и измеряют радиоактивность с использованием жидкостного сцинтилляционного счетчика. С использованием в качестве стандартной кривой ингибирования связывания, рассчитанной из результатов измерения, полученных с РРР, содержащей серийно разведенное соединение по настоящему изобретению, концентрацию соединения по настоящему изобретению в РРР преобразуют из результатов измерений, полученных для крыс, которым вводили соединение по настоящему изобретению.
Результаты показаны в таблице 3. В результате оценки с помощью вышеописанного метода получают, что соединения по настоящему изобретению показывают хорошую активность ингибирования агрегации тромбоцитов при оральном введении и также показывают хорошую фармакокинетику.
ПРИМЕРЫ
Настоящее изобретение описывается для пояснения на основе примеров, но настоящее изобретение не ограничивается приведенными примерами. В этой связи, так как новые вещества включены в исходные соединения, используемые в примерах, способы получения из таких исходных веществ описываются как примеры получения.
В этой связи символы в примерах получения и примерах имеют значения, приведенные далее (то же применимо в данном описании далее).
Пр.пол. (Rf): номер примера получения, пр.: номер примера, №: номер соединения, данные: физические данные (соль (отсутствие такого обозначения означает, что это свободная форма, и цифра перед кислотным компонентом показывает пропорцию в составе. Например, когда написано 2HCl, это показывает, что соединение представляет собой дигидрохлорид. Окса: оксалат, ТФК: трифторацетата)), ЯМР: δ (м.д.) характеристического пика в 1Н-ЯМР, EI: EI-МC (М+, если не указано иное), FAB: FAB-MC (полож.) (М+ + 1, если не указано иное), ESI: ESI-MC (полож.) (М+ + 1, если не указано иное), ACPI: ACPI-МС (полож.) (М+ + 1, если не указано иное), ESI (отр.): ESI-МС (отр.) (М- - 1, если не указано иное), FAB (отр.): FAB-MC (отр.) (М- - 1, если не указано иное), Ме - метил, Et - этил, н-Pr - н-пропил, изо-Pr - изопропил, cPr - циклопропил, н-Bu - н-бутил, изо-Bu - изобутил, трет-Bu - трет-бутил, cBu - циклобутил, cPen - циклопентил, сНех - циклогексил, Ph - фенил, Bn - бензил, Вос - трет-бутоксикарбонил, Ас - ацетил, Bz - бензоил, TBDMS - трет-бутилдиметилсилил. Синт.: способ получения (цифра показывает, что, подобно соединению примера, имеющего номер такой же, как и номер примера, в котором его получают, его осуществляют с использованием соответствующего исходного вещества; когда перед цифрой добавлено «пр.пол.», это показывает, что, подобно соединению примера получения, имеющего тот же номер, что и пример получения, в котором его получают, его осуществляют с использованием соответствующего исходного вещества; когда написаны две или больше цифр, это показывает, что его осуществляют, выполняя соответствующие способы получения исходя из вещества под первой цифрой). Синт.пр.пол.: способ получения (цифра показывает, что, подобно соединению примера получения (пр.пол.), имеющего номер такой же, как и номер примера получения, в котором его получают, его осуществляют с использованием соответствующего исходного вещества; когда перед цифрой добавлено «пр», это показывает, что, подобно соединению примера, имеющего тот же номер, что и пример, в котором его получают, его осуществляют с использованием соответствующего исходного вещества).
Пример получения 1
Добавляют 2,6 г 1,1'-карбонилдиимидазола к суспензии 4,0 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты в 30 мл ДМФА и затем смесь перемешивают при 100°С в течение 13,5 часов. К смеси при охлаждении льдом добавляют 10 мл 28% водного аммиака и затем смесь перемешивают при охлаждении льдом в течение 75 минут и при комнатной температуре в течение 5 часов. После выпаривания растворителя при пониженном давлении добавляют этанол и осуществляют кипячение с обратным холодильником. После охлаждения до комнатной температуры нерастворимое вещество собирают фильтрацией и сушат, и получают 3,7 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксамида.
Пример получения 2
При 0°С добавляют 0,87 мл триэтиламина и 0,4 мл изобутилхлорформиата к раствору 1,0 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты в 20 мл дихлорметана и затем смесь перемешивают при 0°С в течение 30 минут. Затем к смеси добавляют 315 мг гидрохлорида N,O-диметилгидроксиламина и затем смесь перемешивают при комнатной температуре в течение 1 часа. К реакционной смеси добавляют хлороформ и водный насыщенный раствор хлорида аммония, слои разделяют и промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия и последующей фильтрации растворитель выпаривают при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 950 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-N-метокси-N-метил-4-оксо-1,4-дигидрохинолин-3-карбоксамида.
Пример получения 3
Растворяют 5 г 2-нитро-1-(2,4,5-трифторфенил)этанона в 100 мл уксусного ангидрида, к раствору при комнатной температуре добавляют 4,0 мл триэтилортоформиата, затем смесь перемешивают при 130°С в течение 3 часов и концентрируют при пониженном давлении. Полученный остаток растворяют в 100 мл дихлорметана, добавляют при охлаждении льдом раствор 2,5 мл циклопентиламина в 50 мл дихлорметана и смесь перемешивают при комнатной температуре в течение 3 часов. Затем добавляют воду и затем смесь экстрагируют хлороформом. Органический слой сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Полученный остаток растворяют в 80 мл 1,4-диоксана, добавляют при комнатной температуре раствор 3,6 мл 1,8-диазабицикло[5.4.0]-7-ундецена в 20 мл диоксана и затем смесь перемешивают при комнатной температуре в течение 3 часов. Выливая полученную реакционную смесь в охлаждаемую льдом воду и собирая нерастворимое вещество фильтрацией, получают 1,8 г 1-циклопентил-6,7-дифтор-3-нитрохинолин-4(1Н)-она.
Пример получения 4
При охлаждении льдом 11,5 г триацетоксиборгидрида натрия добавляют небольшими порциями к раствору 4,0 г 3,4,5-трифторанилина и 3,6 мл циклопентанона в 150 мл дихлорэтана и 3,1 мл уксусной кислоты и после повышения температуры до комнатной смесь перемешивают в течение 3,5 часов. К смеси добавляют водный насыщенный раствор гидрокарбоната натрия, затем смесь экстрагируют хлороформом и затем органический слой сушат над безводным сульфатом натрия. После фильтрации растворитель выпаривают при пониженном давлении, полученный остаток очищают колоночной хроматографией на силикагеле и получают 5,4 г N-циклопентил-3,4,5-трифторанилина.
Пример получения 5
Добавляют 3,2 мл диэтил(этоксиметилен)малоната к 3,3 г N-циклопентил-3,4,5-трифторанилина и затем смесь перемешивают при 130°С в течение 4 часов. Очисткой колоночной хроматографией на силикагеле получают 2,2 г диэтил{[циклопентил(3,4,5-трифторфенил)амино]метилен}малоната.
Пример получения 6
Добавляют 5,7 г полифосфорной кислоты к 2,2 г диэтил{[циклопентил(3,4,5-трифторфенил)амино]метилен}малоната и затем смесь перемешивают при 140°С в течение 40 минут. Реакционную смесь выливают в смесь воды со льдом и нерастворимое вещество собирают фильтрацией. Полученное вещество растворяют в хлороформе, промывают водой и водным насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия. После фильтрации растворитель выпаривают и получают 1,4 г этил-1-циклопентил-5,6,7-трифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилата.
Пример получения 7
Добавляют 42% борфтористоводородную кислоту к 1,1 г этил-1-циклопентил-5,6,7-трифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилата и затем смесь греют при 90°С в течение 20 часов. К реакционной смеси добавляют воду, полученное таким образом нерастворимое вещество собирают фильтрацией и сушат, и получают 1,4 г соединения бора. К 1,4 г полученного таким образом соединения бора добавляют 15 мл ДМСО и 0,97 мл циклогексиламина и затем смесь перемешивают при комнатной температуре в течение 30 минут. К реакционной смеси добавляют воду и нерастворимое вещество собирают фильтрацией. После сушки к полученному веществу добавляют 30 мл этанола и 15 мл водного 1 М раствора гидроксида натрия и затем смесь перемешивают при 80°С в течение 1,5 часов. По завершении реакции нарастворимое вещество удаляют фильтрацией, к фильтрату добавляют воду и диэтиловый эфир, осуществляют разделение слоев и к водному слою добавляют 1 М соляную кислоту. Образовавшееся выпавшее в осадок вещество собирают фильтрацией и сушат, и получают 1,0 г 7-(циклогексиламино)-1-циклопентил-5,6-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты.
Пример получения 8
При охлаждении льдом 3,2 мл 1,60 М раствора н-бутиллития в гексане добавляют к раствору 0,58 мл бензилового спирта в 2,4 мл ТГФ и затем смесь перемешивают в течение 1 часа. Растворитель выпаривают при пониженном давлении и затем добавляют 8,0 мл толуола для получения суспензии. Полученную суспензию добавляют к полученной в отдельной емкости суспензии 400 мг 7-(циклогексиламино)-1-циклопентил-5,6-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты в толуоле и затем смесь перемешивают при комнатной температуре в течение 6 часов. Затем к реакционной смеси добавляют 1 М соляную кислоту, смесь экстрагируют хлороформом и органический слой промывают насыщенным водным раствором хлорида натрия. После сушки над безводным сульфатом натрия и последующей фильтрации растворитель выпаривают при пониженном давлении. Перекристаллизацией полученного остатка с использованием этилацетата получают 400 мг 5-(бензилокси)-7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты.
Пример получения 9
Растворяют 900 мг этил-1-циклопентил-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксилата в 6,4 мл уксусной кислоты, добавляют 0,8 мл 6 М соляной кислоты и затем смесь перемешивают в течение ночи при 120°С. Полученную реакционную смесь охлаждают до комнатной температуры, нерастворимое вещество собирают фильтрацией и промывают водой, и получают 710 мг 1-циклопентил-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты.
Пример получения 10
При охлаждении льдом 1,02 г гидрохлорида 1-циклобутилэтиламина и 1,05 мл триэтиламина добавляют к раствору 2,0 г этил-2-(2-хлор-4,5-дифторбензоил)-3-этоксиакрилата в 15 мл ТГФ, после чего смесь перемешивают в течение ночи при комнатной температуре. К полученной реакционной смеси добавляют воду, затем смесь экстрагируют эфиром и органический слой промывают водой и водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом магния осуществляют концентрирование при пониженном давлении. При охлаждении льдом 315 мг 55% гидрида натрия добавляют к раствору полученного остатка в 30 мл диоксана и затем смесь перемешивают в течение ночи при 80°С. Реакционную смесь выливают в 1 М соляную кислоту, затем смесь экстрагируют хлороформом и органический слой промывают водой и водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия и последующего концентрирования при пониженном давлении полученный остаток очищают колоночной хроматографией на силикагеле и получают 1,13 г этил-1-(1-циклобутилэтил)-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилата.
Таким же способом, как в примерах получения 1-10, с использованием соответствующих исходных веществ получают соединения примеров получения 11-27, указанные в таблицах 4-9. Структуры и физико-химические свойства соединений примеров получения приводятся в таблицах 4-9.
Пример 1
Растворяют 250 мг 3-амино-7-(циклогексиламино)-1-циклопентил-6-фторхинолин-4(1Н)-она и 127 мг 4-этокси-4-оксобутановой кислоты в 20 мл ДМФА, добавляют 170 мг гидрохлорида N-[3-(диметиламино)пропил]-N'-этилкарбодиимида и 160 мг 1-гидроксибензотриазола и затем смесь перемешивают в течение ночи при комнатной температуре. Добавляя воду к реакционной смеси и затем собирая нерастворимое вещество фильтрацией, получают 220 мг этил-4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-4-оксобутаноата.
Пример 2
Растворяют 200 мг этил-4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-4-оксобутаноата в 2,0 мл ТГФ и 2,0 мл этанола, добавляют 1,3 мл водного 1 М раствора гидроксида натрия и затем смесь перемешивают при комнатной температуре в течение 4 часов. После добавления к смеси 1 М соляной кислоты и воды нерастворимое вещество собирают фильтрацией и получают 180 мг 4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-4-оксобутановой кислоты.
Пример 3
Растворяют 200 мг диэтил-{(Е)-2-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]винил}фосфоната в 2,0 мл хлороформа, добавляют 0,4 мл бромтриметилсилана и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют этанол и затем смесь концентрируют при пониженном давлении. К полученному остатку добавляют этилацетат, нерастворимое вещество собирают фильтрацией и получают 120 мг гидробромида {(Е)-2-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]винил}фосфоновой кислоты.
Пример 4
При перемешивании добавляют 169 мг триацетоксиборгидрида натрия к раствору 142 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида и 66 мг 4-аминофенола в 10 мл 1,2-дихлорэтана и 0,05 мл уксусной кислоты и затем смесь перемешивают в течение 24 часов. К смеси добавляют водный насыщенный раствор гидрокарбоната натрия и затем смесь экстрагируют хлороформом. После сушки над безводным сульфатом натрия и последующей фильтрации осуществляют концентрирование при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и затем кристаллизуют из этилацетата, и получают 46 мг [7-(циклогексиламино)-1-циклопентил-6-фтор-3-{[(4-гидроксифенил)амино]метил}хинолин-4(1Н)-она.
Пример 5
Растворяют 250 мг гидрохлорида 3-(аминометил)-7-(циклогексиламино)-1-(1-этилпропил)-6-фторхинолин-4(1Н)-она в 25 мл ТГФ, добавляют 0,11 мл диэтил(2-оксопропил)фосфоната и 123 мг триацетоксиборгидрида натрия, 0,16 мл триэтиламина и 1,25 мл уксусной кислоты в указанном порядке и затем смесь перемешивают в течение ночи при комнатной температуре. Добавляют воду, нерастворимое вещество собирают фильтрацией и затем очищают колоночной хроматографией на силикагеле, и получают 135 мг диэтил[2-({[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}амино)пропил]фосфоната.
Пример 6
Растворяют 170 мг этил-4-({[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}амино)бутаноата в 2,0 мл пиридина, добавляют 0,040 мл уксусного ангидрида и затем смесь перемешивают в течение ночи при комнатной температуре. После концентрирования реакционной смеси при пониженном давлении к полученному остатку добавляют воду и затем смесь экстрагируют хлороформом. Органический слой сушат над безводным сульфатом натрия и затем фильтруют и концентрируют при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 165 мг этил-4-(ацетил{[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}амино)бутаноата.
Пример 7
Растворяют 180 мг 4-нитрофенилхлорформиата в 3,0 мл дихлорметана, добавляют 140 мг гидрохлорида этил-3-аминопропаноата и 0,15 мл пиридина и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют воду и затем смесь экстрагируют хлороформом. Органический слой промывают водным насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия, фильтруют и затем концентрируют при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 180 мг этил-3-{[(4-нитрофенокси)карбонил]амино}пропаноата. Растворяют 180 мг этил-3-{[(4-нитрофенокси)карбонил]амино}пропаноата в 2,0 мл дихлорметана, добавляют 220 мг 3-амино-7-(циклогексиламино)-1-(1-этилпропил)-6-фторхинолин-4(1Н)-она и 0,15 мл пиридина и затем смесь перемешивают в течение ночи при комнатной температуре. К смеси добавляют воду и затем смесь экстрагируют хлороформом. Органический слой промывают водным насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия и затем фильтруют и концентрируют при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 120 мг этил-3-[({[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}карбонил)амино]пропаноата.
Пример 8
Растворяют 287 мг этил[(5-хлор-2-тиенил)сульфонил]карбамата в 5,0 мл толуола, добавляют 250 мг 3-амино-7-(циклогексиламино)-6-фтор-1-изопропилхинолин-4(1Н)-она и затем смесь перемешивают в течение ночи при 110°С. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении. Затем добавляют этилацетат и нерастворимое вещество собирают фильтрацией и посредством этого получают 280 мг 5-хлор-N-({[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]амино}карбонил)тиофен-2-сульфонамида.
Пример 9
Растворяют 224 мг гидрохлорида 2-амино-N-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]ацетамида в 5,0 мл ДМФА, добавляют 228 мг карбоната калия и 0,18 мл этилбромацетата и затем смесь перемешивают в течение ночи при 60°С. Реакционную смесь охлаждают до комнатной температуры, добавляют воду, нерастворимое вещество собирают фильтрацией и получают 35 мг диэтил-2,2'-[(2-{[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-2-оксоэтил)имино]диацетата.
Пример 10
Растворяют 150 мг этил-{[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}ацетата в 3,0 мл ТГФ, добавляют 0,060 мл триэтиламина и 0,060 мл этил-5-хлор-5-оксопентаноата и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют воду и затем смесь экстрагируют хлороформом. Полученный органический слой промывают водным насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 199 мг этил-5-[[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил](2-этокси-2-оксоэтил)амино]-5-оксопентаноата.
Пример 11
Растворяют 200 мг этил-(2Е)-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]акрилата в 4,0 мл этанола, добавляют 50 мг палладия-на-угле и затем смесь перемешивают в течение ночи при комнатной температуре в атмосфере водорода. Реакционную смесь фильтруют с использованием целита и концентрируют при пониженном давлении и получают 200 мг этил-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]пропаноата.
Пример 12
При 0°С 213 мкл диизопропилазодикарбоксилата добавляют к раствору 263 мг бензил-(2R)-2-гидрокси-3-фенилпропаноата и 270 мг трифенилфосфина в 5,0 мл дихлорметана и затем смесь перемешивают в течение 15 минут. Затем к смеси добавляют 177 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(гидроксиметил)хинолин-4(1Н)-она и затем смесь перемешивают при комнатной температуре в течение 4 часов. К реакционной смеси добавляют воду, затем смесь экстрагируют EtOAc и органический слой промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия и последующего упаривания при пониженном давлении остаток очищают колоночной хроматографией на силикагеле и получают 160 мг бензил-(2S)-2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}-3-фенилпропаноата.
Пример 13
Добавляют 690 мг карбоната калия и 363 мг 4-фторбензонитрила к раствору 344 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-гидроксихинолин-4(1Н)-она в 10 мл ДМФА и затем смесь перемешивают в течение ночи при 80°С. После завершения реакции и последующего охлаждения до комнатной температуры к реакционной смеси добавляют водный насыщенный раствор хлорида аммония и затем смесь экстрагируют этилацетатом. После сушки над безводным сульфатом натрия и последующего упаривания при пониженном давлении остаток очищают колоночной хроматографией на силикагеле и получают 100 мг 4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}бензонитрила.
Пример 14
Добавляют 5,0 мл этанола и 1,5 мл водного 6 М раствора гидроксида натрия к 93 мг 4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}бензонитрила и затем смесь кипятят с обратным холодильником в течение 2 суток. После охлаждения реакционную систему нейтрализуют 1 М соляной кислотой. Добавляют воду и твердое выпавшее в осадок вещество собирают фильтрацией. Кристаллизацией полученного твердого вещества из этилацетата-гексана получают 65 мг 4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}бензойной кислоты.
Пример 15
Растворяют 840 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 40 мл хлороформа, при охлаждении льдом добавляют 0,47 мл триметилсилилцианида и 0,05 мл триэтиламина и затем смесь перемешивают при комнатной температуре в течение 5,5 часов. После перемешивания при комнатной температуре в течение 1,5 часов после добавления еще 0,06 мл триметилсилилцианида к смеси добавляют еще 0,06 мл триметилсилилцианида и затем смесь перемешивают при комнатной температуре в течение 2 суток. Полученное выпавшее в осадок вещество отфильтровывают и промывают хлороформом, и получают твердое вещество. Полученное твердое вещество растворяют в 13 мл концентрированной соляной кислоты и затем перемешивают при 100°С в течение 2,5 часов. После охлаждения до комнатной температуры добавляют воду, затем смесь экстрагируют хлороформом, и органический слой сушат над безводным сульфатом натрия, и затем концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и получают сырой продукт реакции [7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил](гидрокси)уксусную кислоту. Полученный сырой продукт промывают смесью вода:метанол (1:2) и этилацетатом. К полученному твердому веществу добавляют этилацетат и водный насыщенный раствор гидрокарбоната натрия и осуществляют операцию разделения слоев. К водному слою добавляют 1 М соляную кислоту, затем экстрагируют этилацетатом и органический слой концентрируют при пониженном давлении. К полученному остатку добавляют смешанный растворитель из ТГФ и воды, нерастворимое вещество собирают фильтрацией и получают 149 мг [7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил](гидрокси)уксусной кислоты.
Пример 16
Растворяют 52 мг [7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил](гидрокси)уксусной кислоты в 10 мл метанола, добавляют 0,4 мл концентрированной серной кислоты и затем смесь перемешивают при комнатной температуре в течение 1 часа. К реакционной смеси добавляют водный насыщенный раствор гидрокарбоната натрия, затем смесь экстрагируют этилацетатом, и органический слой промывают водным насыщенным раствором хлорида натрия, и концентрируют при пониженном давлении. Полученный остаток перекристаллизовывают из водного метанола и получают 53 мг метил-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил](гидрокси)ацетата.
Пример 17
Растворяют 146 мг метил-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил](гидрокси)ацетата в 10 мл ТГФ, при охлаждении льдом добавляют 46 мг 60% гидрида натрия и затем смесь перемешивают при комнатной температуре в течение 30 минут. Затем к реакционной смеси добавляют 58 мкл этилбромацетата и затем смесь перемешивают при комнатной температуре в течение 5 часов. При охлаждении льдом добавляют еще 46 мг 60% гидрида натрия и 10 мл ТГФ и затем смесь перемешивают при комнатной температуре в течение 2 часов. Затем к смеси добавляют 58 мкл этилбромацетата и затем смесь перемешивают при комнатной температуре в течение 17 часов. К реакционной смеси добавляют водный насыщенный раствор гидрокарбоната натрия и затем смесь экстрагируют этилацетатом. Затем органический слой промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия осуществляют концентрирование при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и получают 66 мг метил-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил](2-этокси-2-оксоэтокси)ацетата.
Пример 18
Растворяют 13 г диэтил-{(E)-2-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]винил}фосфоната в 150 мл хлороформа, добавляют 27,2 мл бромтриметилсилана и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют этанол и затем смесь концентрируют при пониженном давлении. К полученному остатку добавляют водный 1 М раствор гидроксида натрия и эфир и осуществляют операцию разделения слоев. К водному слою добавляют концентрированную соляную кислоту и затем смесь перемешивают при комнатной температуре в течение 2 часов. Затем нерастворимое вещество собирают фильтрацией и промывают водой, и получают 10,32 г {(E)-2-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]винил}фосфоновой кислоты.
Пример 19
Растворяют 428 мг хлорида (метоксиметил)трифенилфосфония в 5 мл ТГФ, при охлаждении льдом в атмосфере аргона добавляют 1,2 мл 1,6 М раствора н-бутиллития в гексане и затем смесь перемешивают при той же температуре в течение 30 минут. К смеси при охлаждении льдом добавляют раствор 178 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 5 мл ТГФ, затем смесь перемешивают при той же температуре в течение 15 минут и затем перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь выливают в смесь воды со льдом, затем экстрагируют этилацетатом, и органический слой сушат над безводным сульфатом натрия, и затем концентрируют при пониженном давлении. Полученный остаток растворяют в 10 мл диоксана, добавляют 5 мл 4 М раствора хлористого водорода в диоксане и затем перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь выливают в охлаждаемый льдом водный насыщенный раствор гидрокарбоната натрия, затем экстрагируют этилацетатом, и органический слой сушат над безводным сульфатом натрия, и затем концентрируют при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 239 мг сырого продукта реакции [7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]ацетальдегида. Полученный сырой продукт растворяют в 10 мл этанола, добавляют 75 мг борогидрида натрия и затем смесь перемешивают при комнатной температуре в течение 1 часа. К реакционной смеси добавляют воду, затем экстрагируют этилацетатом, органический слой сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и кристаллизуют из этилацетата, и получают 18 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(2-гидроксиэтил)хинолин-4(1Н)-она.
Пример 20
Растворяют 856 мг хлорида (метоксиметил)трифенилфосфония в 10 мг ТГФ, при охлаждении льдом в атмосфере аргона добавляют 1,8 мл 1,6 М раствора н-бутиллития в гексане и затем смесь перемешивают при той же температуре в течение 30 минут. К смеси при охлаждении льдом добавляют раствор 356 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 10 мл ТГФ и затем смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь выливают в смесь воды со льдом, затем экстрагируют этилацетатом, сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и получают 552 мг сырого продукта реакции 7-(циклогексиламино)-1-циклопентил-6-фтор-3-[2-метоксивинил]хинолин-4(1Н)-она. Растворяют 159 мг полученного сырого продукта в 14 мл диоксана, добавляют 7 мл 4 М раствора хлористого водорода в диоксане и затем смесь перемешивают при комнатной температуре в течение 0,5 часов. Реакционную смесь концентрируют при пониженном давлении и полученный остаток растворяют в 6 мл 2-метил-2-пропанола, 1 мл ацетонитрила и 2 мл воды. Затем при охлаждении льдом добавляют 0,26 мл 2-метил-2-бутена, 78 мг дигидрата дигидрофосфата натрия и 228 мг 79% водного раствора хлорита натрия и затем смесь перемешивают при комнатной температуре в течение 14 часов. К реакционной смеси добавляют воду, затем смесь экстрагируют хлороформом, и органический слой сушат над безводным сульфатом натрия, и затем концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и кристаллизуют из этилацетата, и получают 5 мг [7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]уксусной кислоты.
Пример 21
Растворяют 199 мг 7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-3-(4-гидроксибутил)хинолин-4(1Н)-она в 11 мл 1,2-дихлорэтана, добавляют при комнатной температуре 257 мг трифенилфосфина и 405 мг тетрабромида углерода и затем смесь перемешивают в течение 15 минут. К реакционной смеси добавляют водный насыщенный раствор гидрокарбоната натрия, затем смесь экстрагируют хлороформом, органический слой промывают водным насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Полученный остаток очищают хроматографией и получают 78 мг 3-(4-бромбутил)-7-(циклогексиламино)-1-(1-этилпропил)-6-фторхинолин-4(1Н)-она.
Пример 22
К 557 г 3-(4-бромбутил)-7-(циклогексиламино)-1-(1-этилпропил)-6-фторхинолин-4(1Н)-она добавляют 5 мл триэтилфосфита и затем смесь перемешивают при 160°С в течение 4 часов. Реакционную смесь концентрируют при пониженном давлении, полученный остаток очищают колоночной хроматографией и получают 240 мг диэтил-{4-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]бутил}фосфоната.
Пример 23
К 2 мл 2 М раствора изопропилмагнийхлорида в ТГФ при -78°С добавляют 2 мл ТГФ и 0,71 мл диэтил[бром(дифтор)метил]фосфоната и затем смесь перемешивают при той же температуре в течение 5 минут. К реакционной смеси добавляют по каплям раствор 358 мг 7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 10 мл ТГФ и после постепенного повышения температуры до комнатной температуры полученную смесь перемешивают в течение 2,5 часов. К реакционной смеси добавляют водный насыщенный раствор хлорида натрия и затем смесь экстрагируют хлороформом и этилацетатом. Органический слой сушат над безводным сульфатом магния и затем концентрируют при пониженном давлении. К остатку добавляют метанол, нерастворимое вещество отфильтровывают и полученный фильтрат упаривают при пониженном давлении. Очисткой полученного остатка колоночной хроматографией получают 257 мг диэтил-{2-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]-1,1-дифтор-2-гидроксиэтил}фосфоната.
Пример 24
Растворяют 1,0 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 20 мл ДМФА, добавляют 2,0 г карбоната калия и 2,8 мл этил(диэтоксифосфорил)ацетата и затем смесь перемешивают в течение ночи при 60°С. Полученную реакционную смесь охлаждают до комнатной температуры, добавляют воду, затем нерастворимое вещество собирают фильтрацией и получают 1,2 г этил-(2Е)-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]акрилата.
Пример 25 и пример 26
Растворяют 500 мг {(Е)-2-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]винил}фосфоновой кислоты в 10 мл ацетонитрила, добавляют 86 мг иодида натрия, 0,51 мл 1,8-диазабицикло[5.4.0]-7-ундецена, 194 мг гидросульфата тетрабутиламмония и 0,53 мл хлорметилпивалата в указанном порядке и затем смесь перемешивают в течение ночи при 80°С. К реакционной смеси добавляют водный насыщенный раствор хлорида аммония и затем смесь экстрагируют хлороформом. Органический слой промывают водным насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия и затем упаривают при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на силикагеле получают 400 мг бис{[(2,2-диметилпропаноил)окси]метил}{(E)-2-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]винил}фосфоната (пример 25) и 190 мг {[{(E)-2-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидро-3-хинолинил]винил}(гидрокси)фосфорил]окси}метилпивалата (пример 26).
Пример 27
К раствору 144 мг 7-(циклогексиламино)-6-фтор-3-гидрокси-1-изопропилхинолин-4(1Н)-она в 5,0 мл ДМФА добавляют 313 мг карбоната калия и 100 мкл этилбромацетата в указанном порядке и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют водный насыщенный раствор хлорида аммония и затем смесь экстрагируют этилацетатом. После сушки над безводным сульфатом натрия и последующего упаривания при пониженном давлении остаток очищают колоночной хроматографией на силикагеле и получают 159 мг этил {[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]окси}ацетата.
Пример 28
К 2,9 мл раствора трет-бутил-(2-{[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-2-оксоэтил)карбамата в диоксане добавляют 3,0 мл 4 М раствора хлористого водорода в диоксане, затем смесь перемешивают в течение ночи при комнатной температуре. Нерастворимое вещество собирают фильтрацией и получают 550 мг гидрохлорида 2-амино-N-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]ацетамида.
Пример 29
К раствору 15,0 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты в 90 мл ДМФА добавляют 9,9 г 1,1'-карбонилдиимидазола и затем смесь перемешивают при 80°С в течение 24 часов. После охлаждения реакционную смесь выливают в смесь воды со льдом и выпавшее в осадок твердое вещество собирают фильтрацией. Затем при 0°С добавляют 1,9 г боргидрида натрия к раствору полученного твердого вещества в смеси 200 мл ТГФ и 100 мл воды и затем смесь перемешивают при той же температуре в течение 2 часов. Добавляют воду, растворитель выпаривают при пониженном давлении, нерастворимое вещество собирают фильтрацией и получают 13,8 г 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(гидроксиметил)хинолин-4(1Н)-она.
Пример 30
После добавления при -78°С 0,32 мл ДМСО к раствору 0,20 мл оксалилдихлорида в 7,0 мл дихлорметана и перемешивания в течение 30 минут добавляют при -78°С раствор 330 мг N-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]-2-(гидроксиметил)бутанамида в дихлорметане и затем смесь перемешивают в течение 30 минут. Затем к смеси добавляют 1,2 мл триэтиламина и повышают температуру от -78°С до комнатной температуры в течение 2 часов. К реакционной смеси добавляют водный насыщенный раствор хлорида натрия, затем смесь экстрагируют этилацетатом, органический слой сушат над безводным сульфатом натрия и упаривают при пониженном давлении, и получают посредством этого 320 мг сырого продукта реакции N-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]-2-формилбутанамида. К раствору 320 г полученного сырого продукта в 6,4 мл дихлорметана добавляют 290 мг метил(трифенилфосфоранилиден)ацетата и затем смесь перемешивают в течение ночи при комнатной температуре. Упариванием реакционной смеси при пониженном давлении и очисткой полученной смеси колоночной хроматографией на силикагеле получают 220 мг метил-(2Е)-4-({[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}карбонил)гекс-2-еноата.
Пример 31
К раствору 400 мг этил-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]пропаноата в 8 мл ТГФ при 0°С добавляют 40 мг алюмогидрида лития и затем смесь перемешивают в течение 2 часов. К реакционной смеси добавляют воду и затем фильтруют через целит. После упаривания при пониженном давлении полученный остаток очищают колоночной хроматографией на силикагеле и получают 288 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(3-гидроксипропил)хинолин-4(1Н)-она.
Пример 32
К раствору 300 мг {[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}ацетонитрила в 5 мл 1,4-диоксана добавляют 0,8 мл азида трибутилолова и затем смесь кипятят с обратным холодильником в течение 2 суток. После охлаждения до комнатной температуры добавляют водный 1 М раствор гидроксида натрия и эфир и затем проводят операцию разделения слоев. К водному слою добавляют 1 М соляную кислоту, затем экстрагируют хлороформом и органический слой промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия растворитель выпаривают при пониженном давлении. Добавляя эфир к полученному остатку и собирая нерастворимое вещество фильтрацией, получают 70 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(1Н-тетразол-5-илметокси)хинолин-4(1Н)-она.
Пример 33
К суспензии 3,69 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксамида в 30 мл дихлорметана при -78°С добавляют 7,0 мл триэтиламина и раствор 4,0 мл трифторуксусного ангидрида в 10 мл дихлорметана. После постепенного повышения температуры смесь перемешивают при комнатной температуре в течение 2 суток. После добавления воды смесь экстрагируют хлороформом и затем органический слой сушат над безводным сульфатом натрия. После фильтрации растворитель выпаривают при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле. После добавления к полученному твердому веществу смешанного растворителя из 30 мл ТГФ, 30 мл метанола и 10 мл воды к смеси при охлаждении льдом добавляют 2,3 г карбоната калия. После перемешивания при комнатной температуре в течение 15 часов к смеси добавляют 1,0 г карбоната калия и перемешивают при комнатной температуре в течение 4 суток. После выпаривания растворителя при пониженном давлении добавляют воду и затем проводят экстракцию хлороформом. После сушки над безводным сульфатом натрия и последующей фильтрации растворитель выпаривают при пониженном давлении. После промывки полученного остатка этилацетатом получают 2,62 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбонитрила.
Пример 34
Промывают 10 мл никеля Ренея этанолом три раза. Добавляют к нему 30 мл этанола, 3 мл водного аммиака и 2,5 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбонитрила и затем смесь перемешивают в течение ночи в атмосфере водорода. После добавления хлороформа и последующей фильтрации с использованием целита растворитель выпаривают при пониженном давлении. Полученный остаток растворяют в 20 мл ТГФ, добавляют при охлаждении льдом раствор 1,8 г ди-трет-бутилдикарбоната в 10 мл ТГФ и затем смесь перемешивают в течение ночи при комнатной температуре. К смеси при охлаждении льдом добавляют раствор 1,0 г ди-трет-бутилдикарбоната в 10 мл ТГФ и затем смесь перемешивают при комнатной температуре в течение 3 суток. К смеси при охлаждении льдом добавляют раствор 1,0 г ди-трет-бутилдикарбоната в 10 мл ТГФ и затем перемешивают в течение ночи при комнатной температуре. После выпаривания растворителя при пониженном давлении и последующей очистки колоночной хроматографией на силикагеле полученное твердое вещество перекристаллизовывают из гексана-этилацетата и получают 1,22 г трет-бутил {[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}карбамата.
Пример 35
К 5,50 г 7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбонитрила добавляют 50 мл этанола, 3,0 мл концентрированной соляной кислоты и 0,60 г оксида платины и затем смесь перемешивают в течение ночи в атмосфере водорода. После добавления воды осуществляют фильтрацию через целит и выпаривают растворитель при пониженном давлении. Полученный остаток растворяют, добавляя 30 мл воды и 20 мл ТГФ, добавляют при охлаждении льдом 4,0 г гидрокарбоната натрия и 4,5 г ди-трет-бутилдикарбоната и затем смесь перемешивают при охлаждении льдом в течение 1 часа и в течение ночи при комнатной температуре. После выпаривания растворителя при пониженном давлении добавляют воду, затем смесь экстрагируют хлороформом и органический слой сушат над безводным сульфатом натрия. После фильтрации растворитель выпаривают при пониженном давлении, полученный остаток очищают колоночной хроматографией на силикагеле и получают 5,72 г трет-бутил-{[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}карбамата.
Пример 36
К 0,31 г гидрохлорида 7-(циклогексиламино)-6-фтор-4-оксо-1-(пирролидин-3-ил)-1,4-дигидрохинолин-3-карбонитрила добавляют 5 мл этанола, 0,2 мл концентрированной соляной кислоты и 0,10 г оксида платины и затем смесь перемешивают в течение ночи в атмосфере водорода. После добавления воды осуществляют фильтрацию через целит и выпаривают растворитель при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на ODS получают 256 мг гидрохлорида 3-(аминометил)-7-(циклогексиламино)-6-фтор-1-пирролидин-3-илхинолин-4(1Н)-она.
Пример 37
К раствору 2,0 г 1-циклопентил-6,7-дифтор-3-нитрохинолин-4(1Н)-она в 40 мл ДМСО добавляют 2,3 мл циклогексиламина и затем смесь перемешивают в течение ночи при 90°С. Реакционную смесь охлаждают до комнатной температуры и выливают в воду, охлаждаемую льдом, и затем нерастворимое вещество собирают фильтрацией. Перекристаллизацией полученного твердого вещества из этанола получают 2,5 г 7-(циклогексиламино)-1-циклопентил-6-фтор-3-нитрохинолин-4(1Н)-она.
Пример 38
К раствору 220 мг 3-амино-7-(циклогексиламино)-1-(1-этилпропил)-6-фторхинолин-4(1Н)-она в 4 мл этанола добавляют 105 мг 1Н-1,2,3-бензотриазол-1-илметанола и затем смесь перемешивают в течение ночи при комнатной температуре. Затем к реакционной смеси добавляют 48 мг боргидрида натрия и затем смесь перемешивают в течение 3 часов. Добавляя воду к полученной реакционной смеси и собирая нерастворимое вещество фильтрацией, получают 100 мг 7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-3-(метиламино)хинолин-4(1Н)-она.
Пример 39
К раствору 13,8 г 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(гидроксиметил)хинолин-4(1Н)-она в 100 мл дихлорметана при комнатной температуре добавляют 67,0 г диоксида марганца и затем смесь перемешивают в течение ночи. После завершения реакции и последующей фильтрации с использованием целита фильтрат упаривают при пониженном давлении. Кристаллизацией полученного твердого вещества из этилацетата получают 13,0 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида.
Пример 40
К раствору 8,0 г 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 100 мл дихлорметана при комнатной температуре постепенно добавляют 6,1 г метахлорпербензойной кислоты и затем смесь перемешивают в течение 2 часов. К реакционной смеси добавляют водный насыщенный раствор гидрокарбоната натрия и водный раствор гидротиосульфата натрия, смесь перемешивают в течение 30 минут и затем экстрагируют хлороформом. После сушки над безводным сульфатом натрия и последующего упаривания при пониженном давлении остаток очищают колоночной хроматографией на силикагеле и получают 7,7 г 7-(циклогексиламино)-1-циклопентил-6-фтор-3-гидроксихинолин-4(1Н)-она.
Пример 41
К раствору 150 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 2,0 мл уксусной кислоты добавляют 60 мг 2-тиоксо-1,3-тиазолин-4-она и 40 мг ацетата натрия в указанном порядке и затем смесь перемешивают в течение ночи при 100°С. Реакционную смесь охлаждают до комнатной температуры и упаривают при пониженном давлении. Добавляют этилацетат, нерастворимое вещество собирают фильтрацией и получают 173 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-[(Z)-(4-оксо-2-тиоксо-1,3-тиазолидин-5-илиден)метил]хинолин-4(1Н)-она.
Пример 42
К раствору 762 мг 3-(3-{[трет-бутил(диметил)силил]окси}пропокси)-7-(циклогексиламино)-1-циклопентил-6-фторхинолин-4(1Н)-она в 15 мл ТГФ при комнатной температуре добавляют 1,5 мл 1 М раствора фторида тетрабутиламмония в ТГФ и затем смесь перемешивают в течение 2 часов. К реакционной смеси добавляют воду, затем смесь экстрагируют этилацетатом и промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия осуществляют упаривание при пониженном давлении. Очисткой остатка колоночной хроматографией на силикагеле получают 273 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(3-гидроксипропокси)хинолин-4(1Н)-она.
Пример 43
К раствору 500 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-N-метокси-N-метил-4-оксо-1,4-дигидрохинолин-3-карбоксамида в 10 мл ТГФ добавляют 1,2 мл 1 М раствора метиллития в ТГФ и затем смесь перемешивают при комнатной температуре в течение 3 суток. К реакционной смеси добавляют воду и осуществляют фильтрацию через целит. После упаривания при пониженном давлении полученный остаток очищают колоночной хроматографией на силикагеле и получают 150 мг 3-ацетил-7-(циклогексиламино)-1-циклопентил-6-фторхинолин-4(1Н)-она.
Пример 44
К раствору 500 мг 7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 5,0 мл дихлорметана при -40°С добавляют 0,23 мл диэтилфосфита и 0,22 мл 1,8-диазабицикло[5.4.0]-7-ундецена и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют водный насыщенный раствор хлорида аммония, смесь экстрагируют хлороформом и затем промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия и последующего упаривания при пониженном давлении полученный остаток очищают колоночной хроматографией на силикагеле и получают 400 мг диэтил[[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил](гидрокси)метил]фосфоната.
Пример 45
К раствору 160 мг этил{[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}ацетата в 3,2 мл ДМФА добавляют 0,05 мл бензилбромида и 75 мг карбоната калия и затем смесь перемешивают в течение ночи при комнатной температуре. Добавляя воду к реакционной смеси и собирая нерастворимое вещество фильтрацией, получают 200 мг этил{бензил[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}ацетата.
Пример 46
К раствору 210 мг (2Е)-3-{7-[(циклопропилметил)амино]-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил}акриловой кислоты в 4,2 мл ТГФ добавляют 120 мг 1,1'-карбонилдиимидазола и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют воду и нерастворимое вещество собирают фильтрацией. Полученное твердое вещество растворяют в 4,2 мл ДМФА, добавляют 0,11 мл 1,8-диазабицикло[5.4.0]-7-ундецена и 150 мг 5-хлортиофен-2-сульфонамида и затем смесь перемешивают в течение ночи при 80°С. Добавляя воду к полученной реакционной смеси и собирая нерастворимое вещество фильтрацией, получают 145 мг (2Е)-N-[(5-хлор-2-тиенил)сульфонил]-3-{7-[(циклопропилметил)амино]-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил}акриламида.
Пример 47
При охлаждении льдом 2,0 мл трифторуксусной кислоты добавляют к раствору 0,20 г трет-бутил{[7-(циклогексиламино)-6-фтор-4-оксо-1-(тетрагидрофуран-3-ил)-1,4-дигидрохинолин-3-ил]метил}карбамата в 5 мл дихлорметана. После перемешивания в течение 1,5 часов при охлаждении льдом и при комнатной температуре в течение ночи растворитель выпаривают при пониженном давлении. Очисткой полученного остатка колоночной хроматографией на ODS получают 184 мг трифторацетата 3-(аминометил)-7-(циклогексиламино)-6-фтор-1-(тетрагидрофуран-3-ил)хинолин-4(1Н)-она.
Пример 48
К раствору 240 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(3-гидроксипропил)хинолин-4(1Н)-она в 4,8 мл ДМСО добавляют 300 мг комплекса триоксида серы и пиридина и 0,8 мл триэтиламина и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют воду, затем смесь экстрагируют хлороформом и затем органический слой промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия и последующего упаривания при пониженном давлении полученный остаток очищают колоночной хроматографией на силикагеле и получают 140 мг альдегида. К раствору 140 мг альдегида в 2,8 мл ДМФА добавляют 141 мг карбоната калия и 414 мг этил(диэтоксифосфорил)ацетата и затем смесь перемешивают в течение ночи при 60°С. Реакционную смесь охлаждают до комнатной температуры, добавляют воду и нерастворимое вещество собирают фильтрацией. Очисткой полученного нерастворимого вещества колоночной хроматографией на силикагеле получают 57 мг этил-(2Е)-5-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]пент-2-еноата.
Пример 49
Добавляют 3 мл этилацетата и 0,35 мл 1 М раствора хлористого водорода в этилацетате к 0,13 г этил({[7-(циклогексиламино)-1-этил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}амино)ацетата, полученного таким же способом, как в примере 9. После выпаривания растворителя при пониженном давлении и последующего добавления эфира нерастворимое вещество собирают фильтрацией и получают 97 мг гидрохлорида этил({[7-(циклогексиламино)-1-этил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}амино)ацетата.
Пример 50
Добавляют 10 мл этилацетата, 45 мг щавелевой кислоты и 10 мл этанола к 440 мг этил({[7-(циклогексиламино)-6-фтор-4-оксо-1-(тетрагидрофуран-3-ил)-1,4-дигидрохинолин-3-ил]метил}амино)ацетата, полученного таким же способом, как в примере 4. После выпаривания растворителя при пониженном давлении и последующего добавления этилацетата нерастворимое вещество собирают фильтрацией и получают 349 мг оксалата этил({[7-(циклогексиламино)-6-фтор-4-оксо-1-(тетрагидрофуран-3-ил)-1,4-дигидрохинолин-3-ил]метил}амино)ацетата.
Пример 51
К 0,25 г оксалата этил({[7-(циклогексиламино)-6-фтор-4-оксо-1-(тетрагидрофуран-3-ил)-1,4-дигидрохинолин-3-ил]метил}амино)ацетата добавляют воду и карбонат калия и затем смесь экстрагируют хлороформом. После сушки над безводным сульфатом натрия и последующей фильтрации растворитель выпаривают при пониженном давлении. К раствору полученного остатка в 10 мл этанола при охлаждении льдом добавляют 0,60 мл водного 1 М раствора гидроксида натрия и затем смесь перемешивают при охлаждении льдом в течение 1 часа и при комнатной температуре в течение ночи. После выпаривания растворителя при пониженном давлении добавляют воду и трифторуксусную кислоту. Очисткой колоночной хроматографией на ODS получают 251 мг трифторацетата ({[7-(циклогексиламино)-6-фтор-4-оксо-1-(тетрагидрофуран-3-ил)-1,4-дигидрохинолин-3-ил]метил}амино)уксусной кислоты.
Пример 52
К раствору 155 мг диэтил[2-(ацетил{[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}амино)-1,1-дифторэтил]фосфоната в 2,0 мл хлороформа добавляют 0,27 мл бромтриметилсилана и затем смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь упаривают при пониженном давлении, к полученному остатку добавляют водный 1 М раствор гидроксида натрия, затем очищают колоночной хроматографией на ODS, промывают этилацетатом и получают 100 мг динатрий[2-(ацетил{[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}амино)-1,1-дифторэтил]фосфоната.
Пример 53
К раствору 280 мг этил-(2Е)-4-{[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]окси}бут-2-еноата в 8,0 мл этанола при комнатной температуре добавляют 28 мг родия-на-угле (10%) и затем смесь перемешивают в течение 2 часов в атмосфере водорода. После фильтрации с использованием целита фильтрат упаривают при пониженном давлении, остаток очищают колоночной хроматографией на силикагеле и получают 202 мг этил 4-{[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]окси}бутаноата.
Пример 54
К раствору 130 мг N-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]-2-[(4R)-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]ацетамида в смешанном растворителе (1,0 мл ТГФ и 1,0 мл метанола) добавляют 0,3 мл 1 М соляной кислоты и затем смесь перемешивают в течение ночи при комнатной температуре. Затем добавляют 0,8 мл водного 1 М раствора гидроксида натрия и смесь перемешивают в течение ночи при комнатной температуре. Полученную реакционную смесь нейтрализуют 1 М соляной кислотой, затем нерастворимое вещество собирают фильтрацией и очищают колоночной хроматографией на силикагеле, и получают 11 мг (2R)-4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-2-гидрокси-4-оксобутановой кислоты.
Пример 55
К раствору 100 мг (2Е)-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]акриловой кислоты в 2,0 мл ТГФ добавляют 60 мг 1,1'-карбонилдиимидазола и затем смесь перемешивают в течение ночи при комнатной температуре. Добавляя воду к полученной реакционной смеси и собирая нерастворимое вещество фильтрацией, получают ацилимидазол. К раствору полученного ацилимидазола в 2,0 мл ДМФА добавляют 0,5 мл водного аммиака и затем смесь перемешивают в течение ночи при 60°С. Полученную реакционную смесь охлаждают до комнатной температуры, добавляют воду, нерастворимое вещество собирают фильтрацией и получают 58 мг (2Е)-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]акриламида.
Пример 56
К раствору 500 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 5,0 мл этанола добавляют 215 мг гидрохлорида этиламиноацетата и затем смесь перемешивают при комнатной температуре в течение 2 часов. Затем к смеси добавляют 50 мг палладия-на-угле и смесь перемешивают при комнатной температуре в течение 4 часов в атмосфере водорода. Реакционную смесь фильтруют через целит и затем упаривают при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле. К раствору полученного соединения в 3,6 мл диоксана добавляют 4,0 мл 4 М раствора хлористого водорода в диоксане и затем смесь перемешивают в течение ночи при комнатной температуре. Нерастворимое вещество собирают фильтрацией и получают 320 мг гидрохлорида этил-({[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}амино)ацетата.
Пример 57
Добавляют 2,0 мл ТГФ и 0,071 мл хлортриметилсилана к 73 мг цинка и затем смесь перемешивают при комнатной температуре в течение 15 минут. Затем к смеси добавляют 200 мг этил (2Е)-4-бромбут-2-еноата и затем смесь перемешивают при комнатной температуре в течение 30 минут. К реакционной смеси добавляют 7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегид и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют воду и затем смесь экстрагируют хлороформом. Органический слой промывают водным насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия и затем упаривают при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и получают 30 мг этил-(2Е)-5-[7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]-5-гидроксипент-2-еноата.
Пример 58
К раствору 119 мг 60% гидрида натрия в 5 мл ТГФ при 0°С добавляют 598 мкл этил(диэтоксифосфорил)ацетата и затем смесь перемешивают в течение 30 минут. К смеси при той же температуре добавляют раствор 400 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(2-оксопропокси)хинолин-4(1Н)-она в 5 мл ТГФ и затем смесь перемешивают при комнатной температуре в течение 2 часов. К реакционной смеси добавляют водный насыщенный раствор хлорида аммония и затем смесь экстрагируют этилацетатом. Органический слой промывают водным насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и получают 200 мг этил-(2Е)-4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}-3-метилбут-2-еноата.
Пример 59
К суспензии 1081 мг бромида (3-бензилоксипропил)трифенилфосфония в 20 мл ТГФ добавляют 258 мг трет-бутоксида калия и затем смесь перемешивают в течение 1,5 часов. К смеси добавляют раствор 358 мг 7-(циклогексиламино)-1-(1-этилпропил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида в 10 мл ТГФ и затем смесь перемешивают в течение 1 часа. К реакционной смеси добавляют водный насыщенный раствор хлорида аммония, затем смесь экстрагируют этилацетатом и промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия и последующего концентрирования при пониженном давлении остаток очищают колоночной хроматографией на силикагеле и получают 488 мг 3-[4-(бензилокси)бут-1-ен-1-ил]-7-(циклогексиламино)-1-(1-этилпропил)-6-фторхинолин-4(1Н)-она.
Пример 60
К раствору 100 мг 5-хлортиофен-2-карбоновой кислоты в 2 мл дихлорметана добавляют 0,55 мл хлорсульфонилизоцианата и затем смесь перемешивают в течение ночи при 40°С. Растворитель выпаривают при пониженном давлении и полученный остаток растворяют в 1,5 мл дихлорметана. Затем добавляют 150 мг 3-амино-7-(циклогексиламино)-1-циклопентил-6-фторхинолин-4(1Н)-она и 0,91 мл триэтиламина и затем смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют воду и затем смесь экстрагируют хлороформом. Органический слой промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия и последующей фильтрации растворитель выпаривают при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и получают 64 мг 5-хлор-N-({[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}сульфонил)тиофен-2-карбоксамида.
Пример 61
К суспензии 200 мг 60% гидрида натрия в 6 мл ДМСО добавляют 1,1 г иодида триметилсульфоксония и затем смесь перемешивают в течение 30 минут. К реакционной смеси добавляют 242 мг этил-(2Е)-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]акрилата и затем смесь перемешивают при комнатной температуре в течение 1 часа и при 60°С в течение 1 часа. К реакционной смеси добавляют воду и затем смесь экстрагируют диэтиловым эфиром. Органический слой промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия растворитель выпаривают при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и получают 55 мг этил-2-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]циклопропанкарбоксилата.
Пример 62
К раствору 1,5 г {[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}ацетонитрила в 30 мл метанола добавляют 1,1 мл триэтиламина и 540 мг гидрохлорида гидроксиламина и затем смесь кипятят с обратным холодильником в течение 27 часов. Растворитель выпаривают при пониженном давлении, полученный остаток очищают колоночной хроматографией на силикагеле и получают 850 мг 2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}-N'-гидроксиэтанимидамида.
Пример 63
При охлаждении льдом 40 мкл дикетена добавляют по каплям к раствору 800 мг 2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}-N'-гидроксиэтанимидамида в 8 мл хлороформа и затем смесь перемешивают при охлаждении льдом в течение 6 часов. Выпариванием растворителя при пониженном давлении получают 180 мг N'-(ацетоацетилокси)-2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}этанимидамида.
Пример 64 и пример 65
Добавляют 5 мл толуола и 41 мг 60% гидрида натрия к 180 мг N'-(ацетоацетилокси)-2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}этанимидамида и затем смесь кипятят с обратным холодильником в течение 24 часов. Растворитель выпаривают при пониженном давлении, к полученному остатку добавляют разбавленную соляную кислоту, затем смесь экстрагируют этилацетатом и промывают водой и водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия растворитель выпаривают при пониженном давлении, полученный остаток очищают колоночной хроматографией на силикагеле и получают 10 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-[(5-оксо-4,5-дигидро-1,2,4-оксадиазол-3-ил)метокси]хинолин-4(1Н)-она (пример 64) и 30 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-{[5-(2-оксопропил)-1,2,4-оксадиазол-3-ил]метокси}хинолин-4(1Н)-она (пример 65).
Пример 66
При -50°С 0,14 мл N-(хлоркарбонил)изоцианата добавляют по каплям к раствору 110 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-[(гидроксиамино)метил]хинолин-4(1Н)-она в 4 мл ТГФ и затем смесь перемешивают при комнатной температуре в течение 1 часа. К реакционной смеси добавляют 1 М соляную кислоту, затем смесь экстрагируют хлороформом и промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия растворитель выпаривают при пониженном давлении, полученный остаток очищают колоночной хроматографией на силикагеле и получают 45 мг 2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]метил}-1,2,4-оксадиазолидин-3,5-диона.
Пример 67
К раствору 310 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-(4-гидроксибутил)хинолин-4(1Н)-она в 4 мл ДМСО добавляют 0,7 мл триэтиламина и 620 мг комплекса триоксида серы и пиридина и затем смесь перемешивают при комнатной температуре в течение 24 часов. После добавления 1 М соляной кислоты и воды нерастворимое вещество собирают фильтрацией и получают 290 мг 4-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]бутаналя.
Пример 68
К 285 мг 4-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]бутаналя добавляют 9 мл толуола и 250 мг метил(трифенилфосфоранилиден)ацетата и затем смесь перемешивают при 80°С в течение 14 часов. Растворитель выпаривают при пониженном давлении, полученный остаток очищают колоночной хроматографией на силикагеле и получают 260 мг метил-(2Е)-6-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]гекс-2-еноата.
Пример 69
К 500 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида добавляют 5 мл этанола, 460 мг ацетата натрия и 290 мг гидрохлорида гидроксиламина и затем смесь перемешивают при комнатной температуре в течение 15 часов и при 70°С в течение 12 часов. Растворитель выпаривают при пониженном давлении, к полученному остатку добавляют воду, затем смесь экстрагируют хлороформом и промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия растворитель выпаривают при пониженном давлении и получают 300 мг оксима 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида.
Пример 70
Добавляют 15 мл метанола, 15 мл ТГФ и 250 мг цианоборгидрида натрия к 300 мг оксима 7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбальдегида. К смеси при охлаждении льдом добавляют 2 мл 4 М раствора хлористого водорода в диоксане и затем смесь перемешивают при комнатной температуре в течение 3 часов. К смеси при охлаждении льдом добавляют водный 1 М раствор гидроксида натрия, затем смесь экстрагируют хлороформом и затем промывают водным насыщенным раствором хлорида натрия. После сушки над безводным сульфатом натрия растворитель выпаривают при пониженном давлении, полученный остаток очищают колоночной хроматографией на силикагеле и получают 130 мг 7-(циклогексиламино)-1-циклопентил-6-фтор-3-[(гидроксиамино)метил]хинолин-4(1Н)-она.
Пример 71
К суспензии 1,02 г 9-(циклогексиламино)-8-фтор-6-оксо-2,3,4,6-тетрагидро-1Н-пиридо[1,2-a]хинолин-5-карбоновой кислоты в 50 мл ТГФ при охлаждении льдом добавляют 0,5 мл триэтиламина и 0,4 мл изобутилхлорформиата и затем смесь перемешивают при охлаждении льдом в течение 1 часа. К смеси при -78°С добавляют по каплям водный раствор (4 мл) 431 мг боргидрида натрия и затем смесь перемешивают при -15°С в течение 15 минут и при охлаждении льдом в течение 30 минут. К смеси добавляют водный насыщенный раствор хлорида аммония, затем смесь экстрагируют этилацетатом и затем органический слой сушат над безводным сульфатом натрия. Растворитель выпаривают при пониженном давлении, полученный остаток очищают колоночной хроматографией на силикагеле и получают 495 мг 9-(циклогексиламино)-8-фтор-5-(гидроксиметил)-1,2,3,4-тетрагидро-6Н-пиридо[1,2-a]хинолин-6-она.
Таким же способом, как в описанных выше примерах 1-71, с использованием соответствующих исходных веществ, получают соединения примеров, указанные в приведенных далее таблицах 10-73. Данные по МС соединений примеров приводятся в приведенных далее таблицах 10-73, а данные по ЯМР некоторых соединений примеров - в таблицах 74 и 75.
Структуры других соединений по настоящему изобретению показаны в таблицах 76-83. Их можно легко получить, используя описанные выше способы получения и способы, описанные в примерах, или способы, очевидные для специалистов в данной области техники, или их модификации.
Промышленная применимость
Так как производные хинолона по настоящему изобретению или их соли обладают превосходной активностью ингибирования агрегации тромбоцитов или активностью ингибирования P2Y12, они применимы в качестве фармацевтических средств, в частности ингибиторов агрегации тромбоцитов или ингибиторов P2Y12. Соответственно, соединения по настоящему изобретению применимы в качестве средств для предупреждения и/или лечения заболеваний органов кровообращения, тесно связанных с тромбообразованием путем агрегации тромбоцитов, таких как нестабильная стенокардия, острый инфаркт миокарда, и для предупреждения его повторения, повторной обструкции и рестриктуры после операции шунтирования коронарной артерии, операции РТСА или операции по установке стента, ускорения тромболизиса в коронарной артерии и предупреждения повторной обструкции и подобных ишемических болезней; таких как церебральный инфаркт при преходящем ишемическом нарушении мозгового кровообращения (TIA), субарахноидальное кровоизлияние (вазоспазм) и подобные инсульты; хроническая окклюзия артерии и подобные заболевания периферических артерий; и подобных заболеваний; и в качестве вспомогательного средства при операции на сердце или операции на сосудах.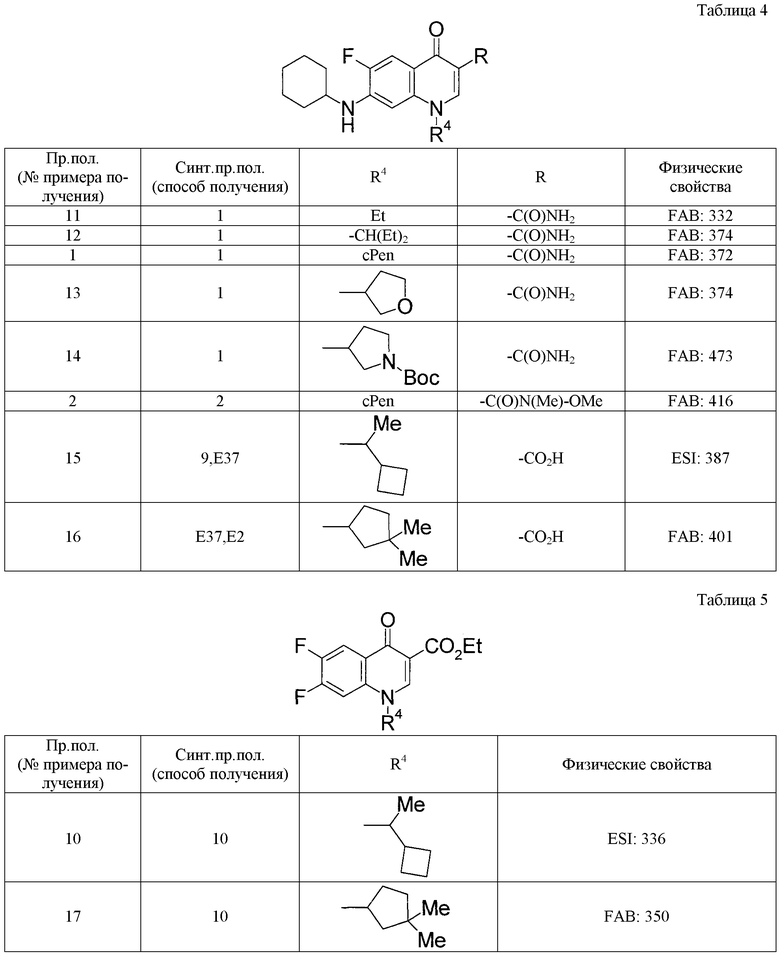





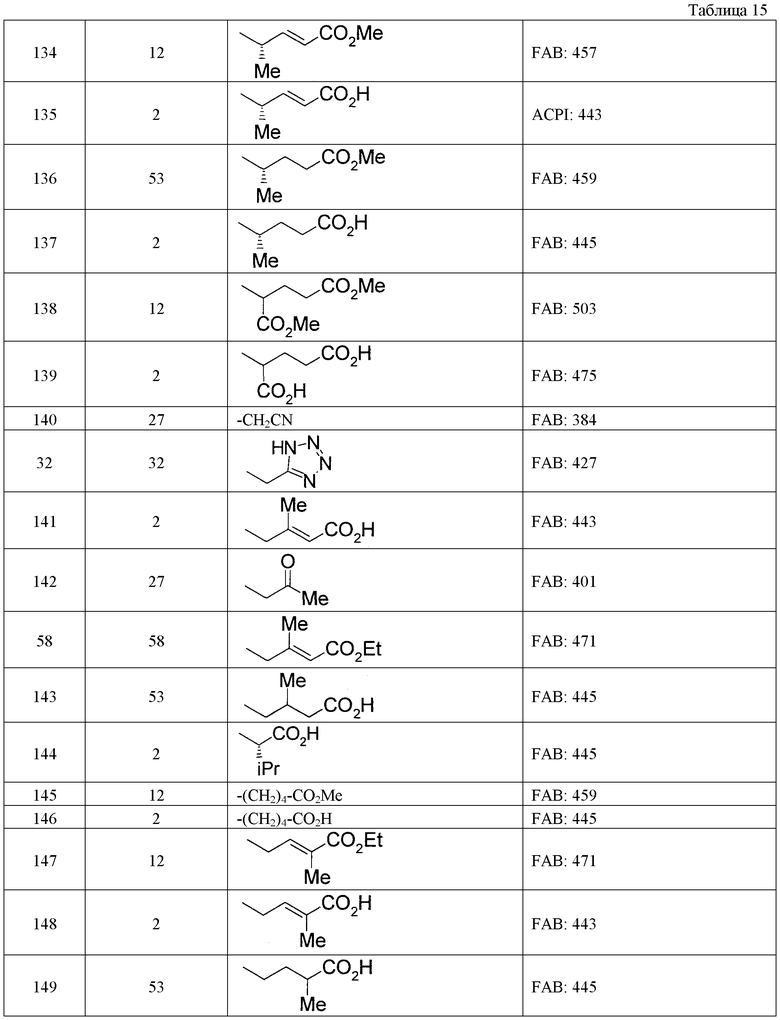

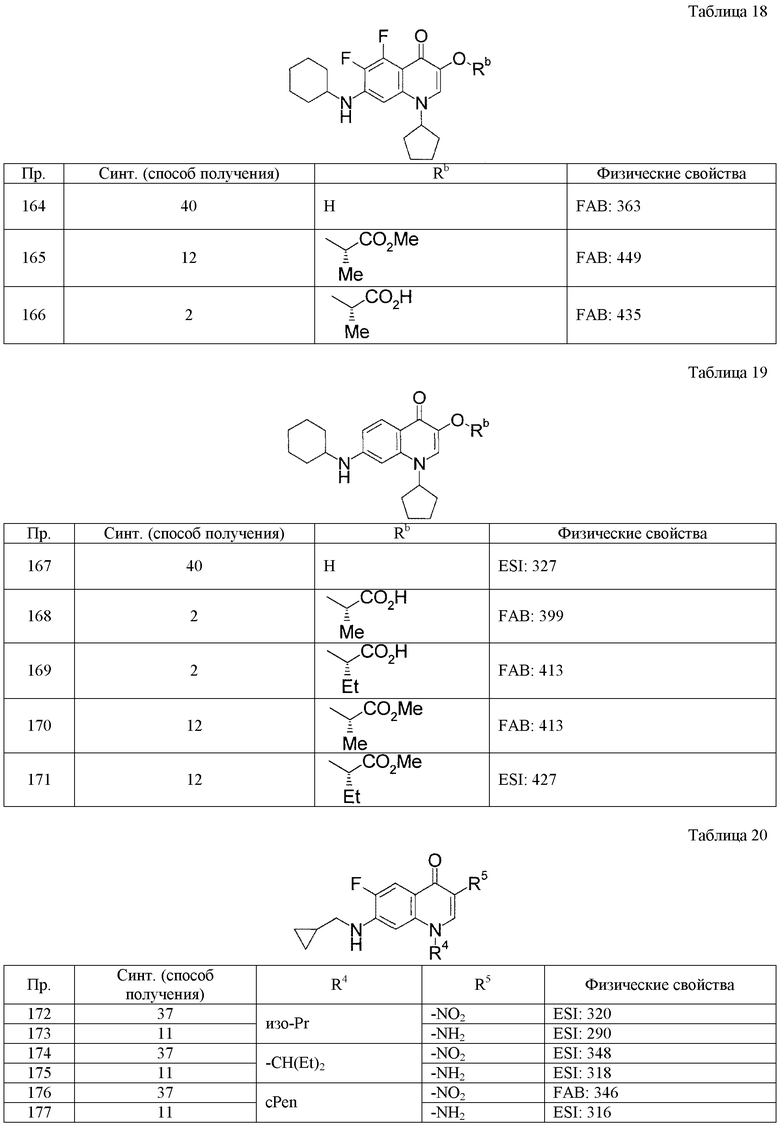
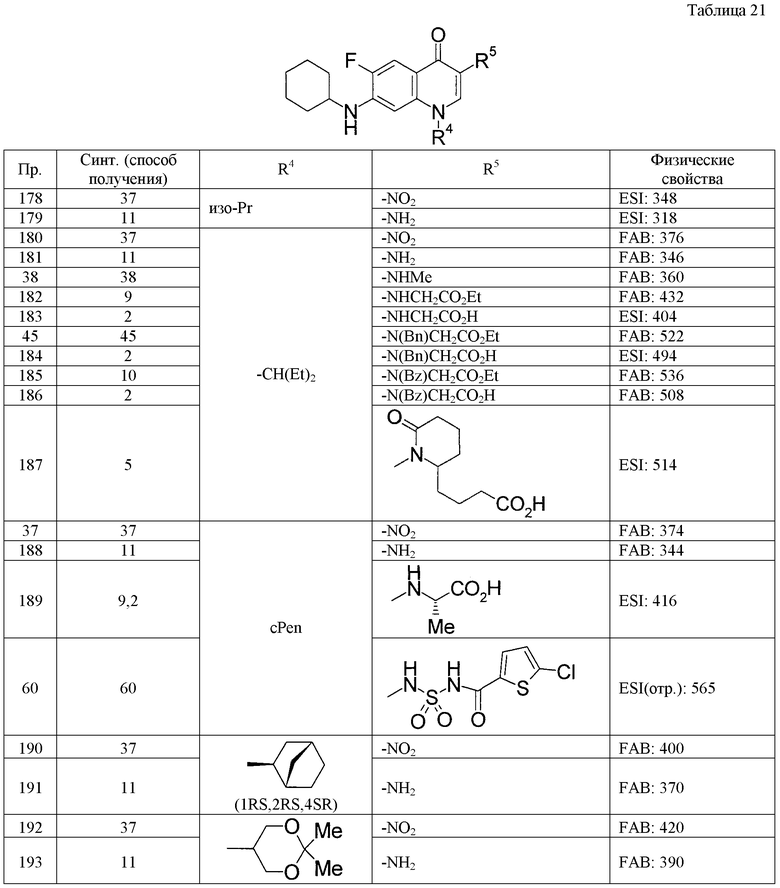
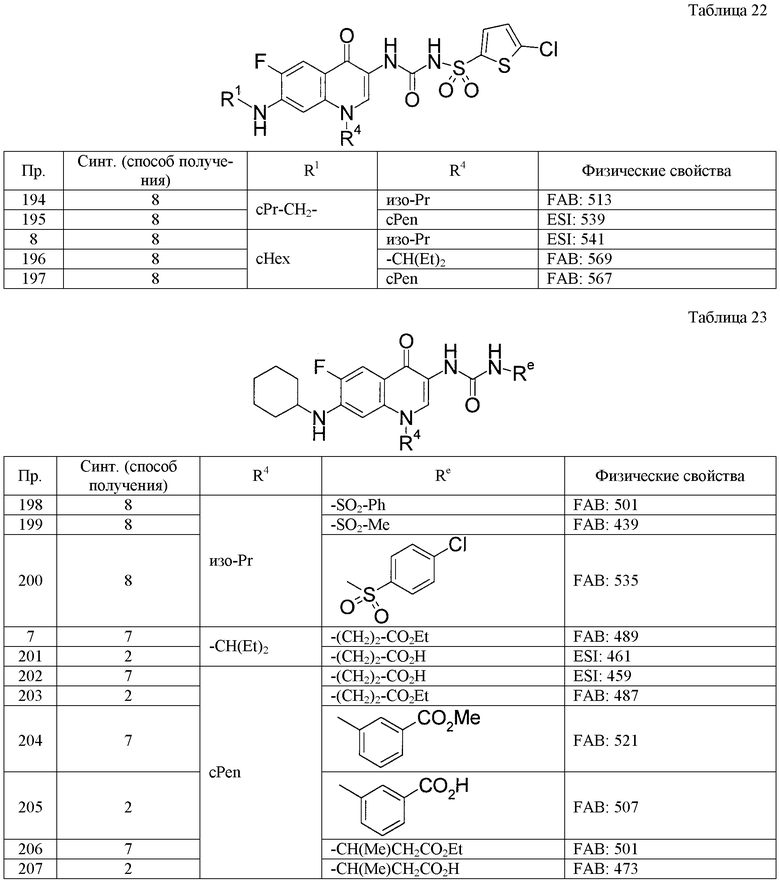




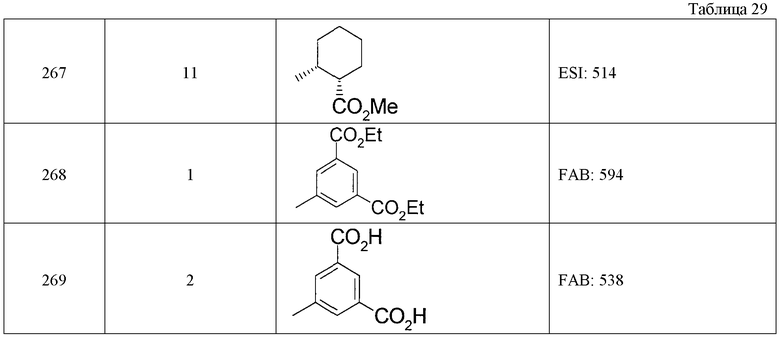


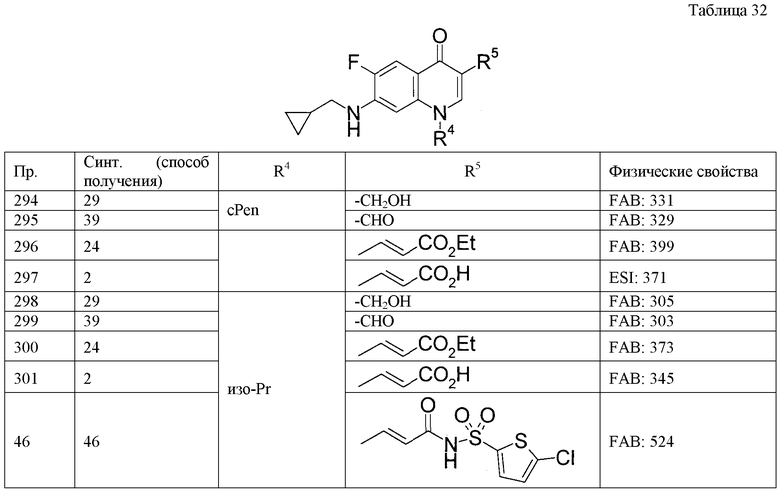
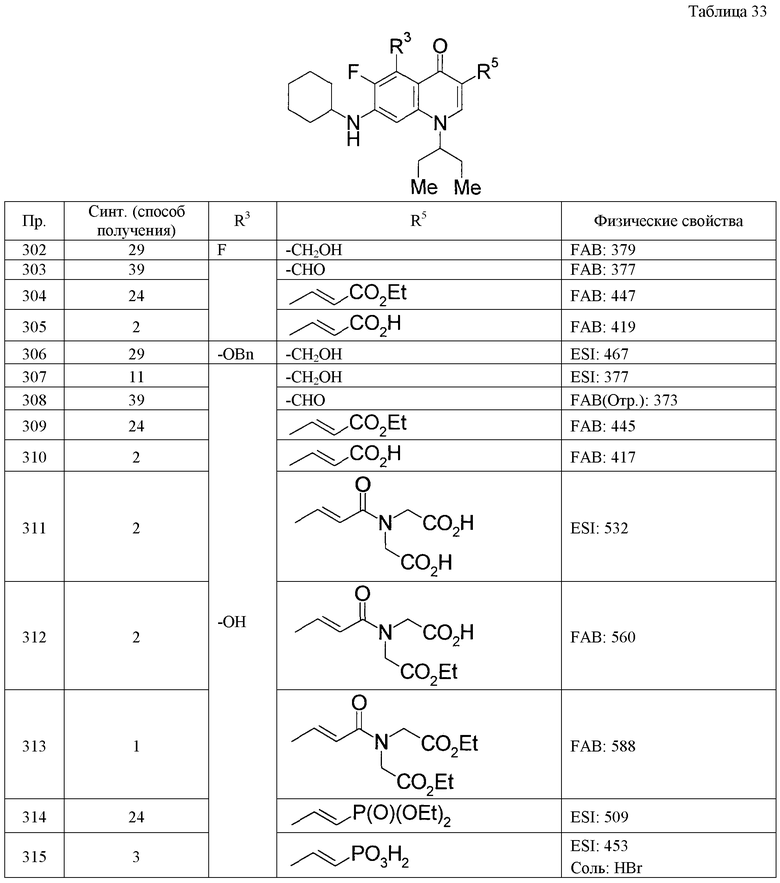
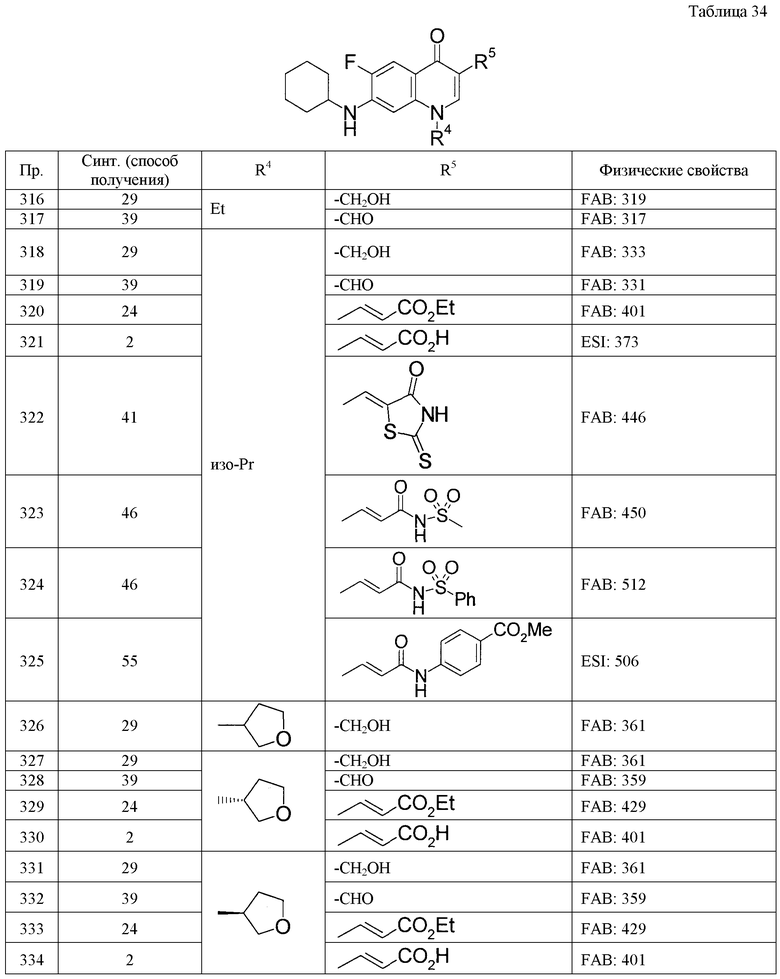


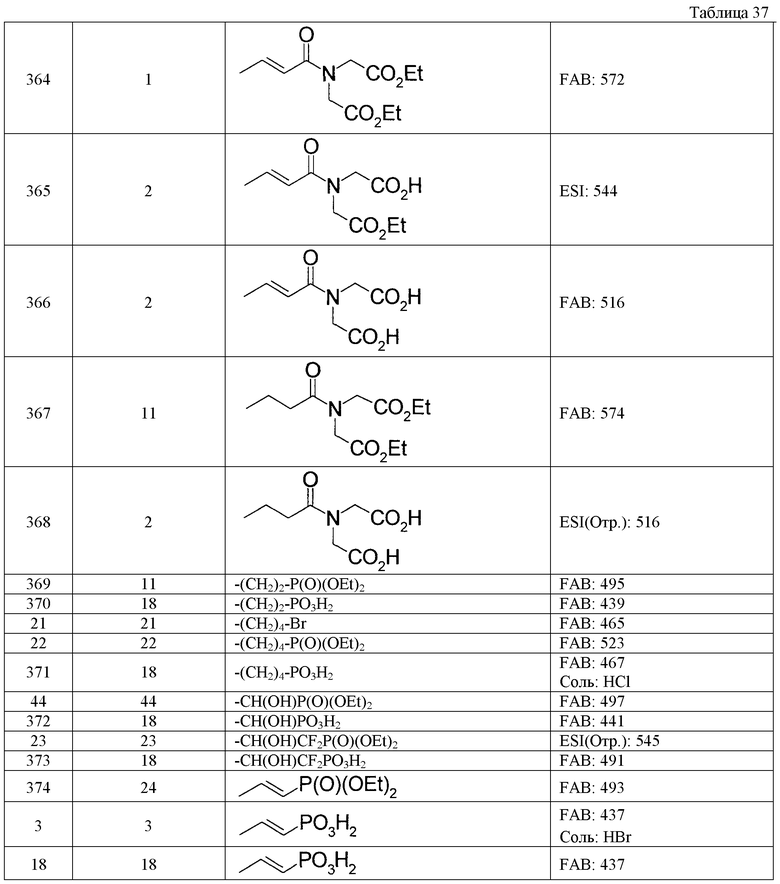

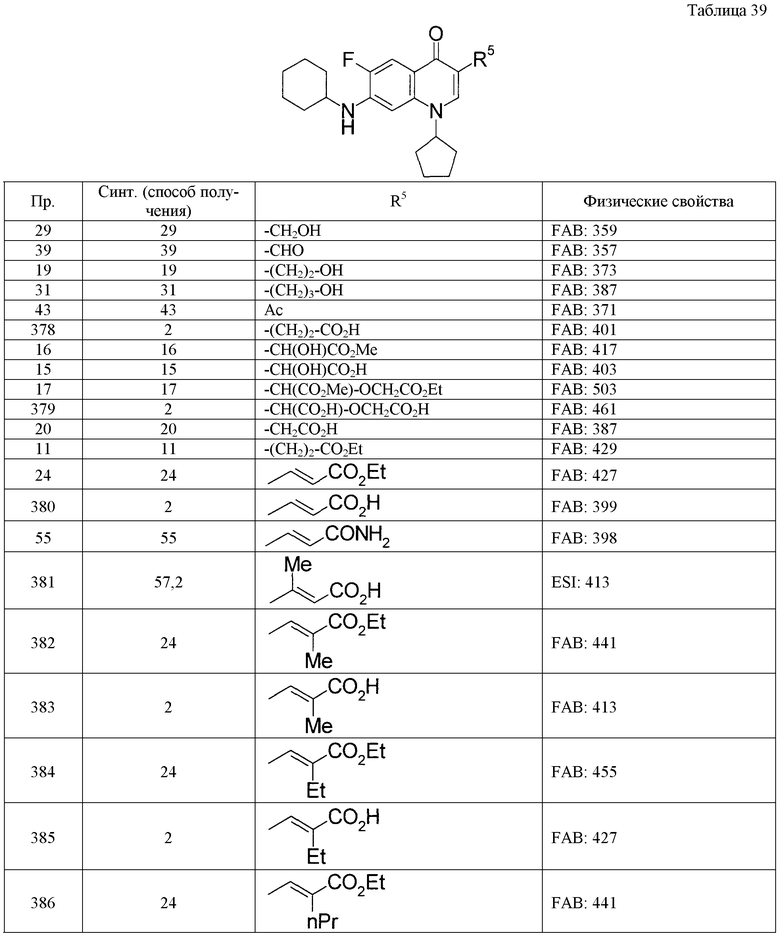


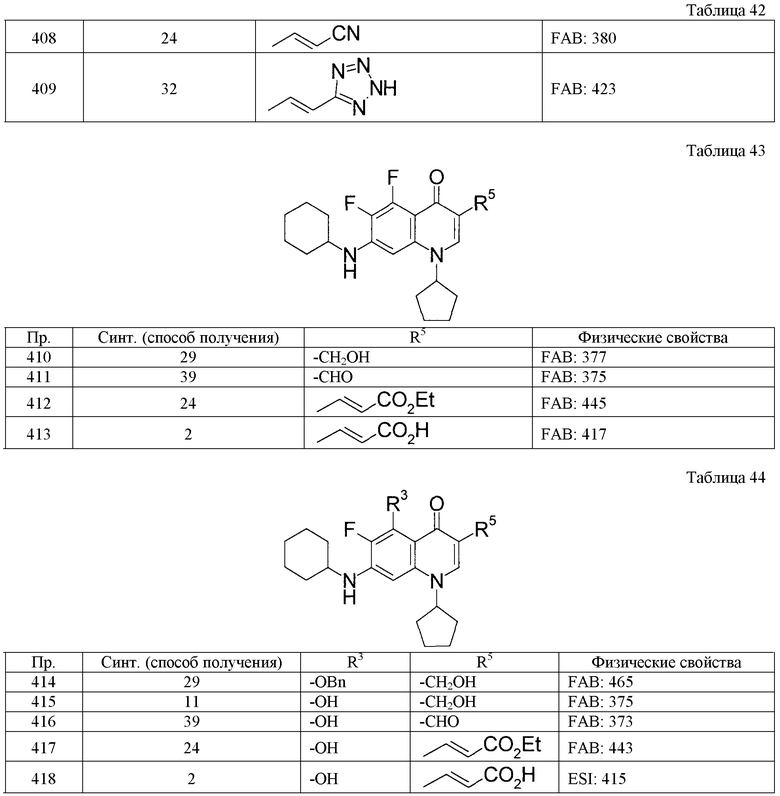

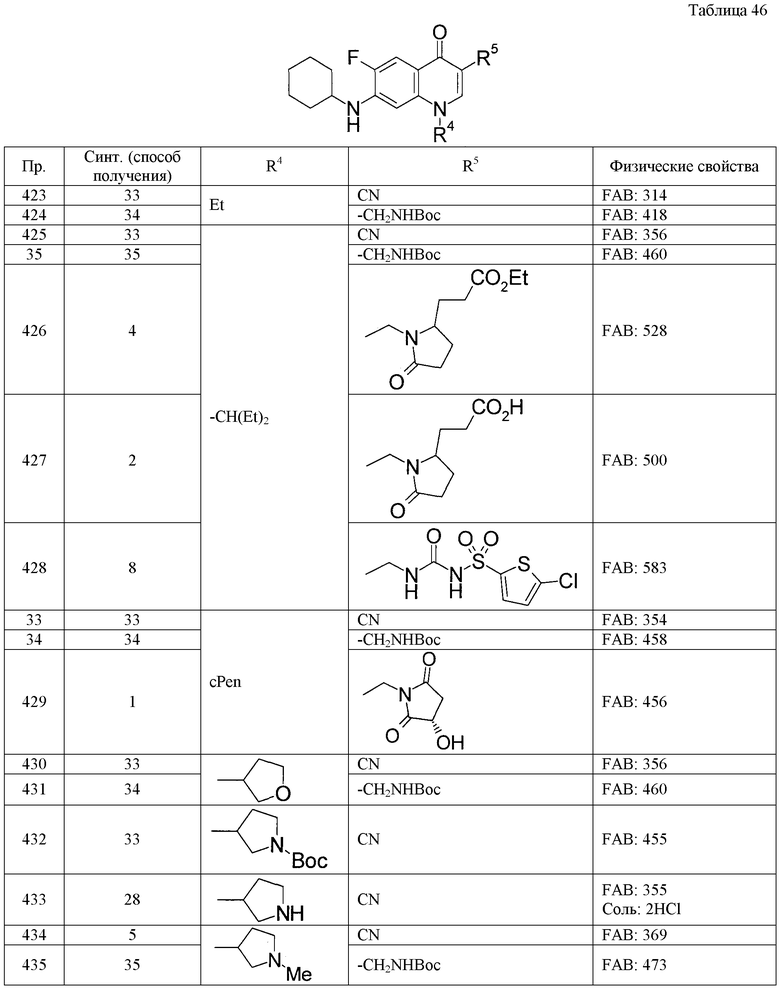
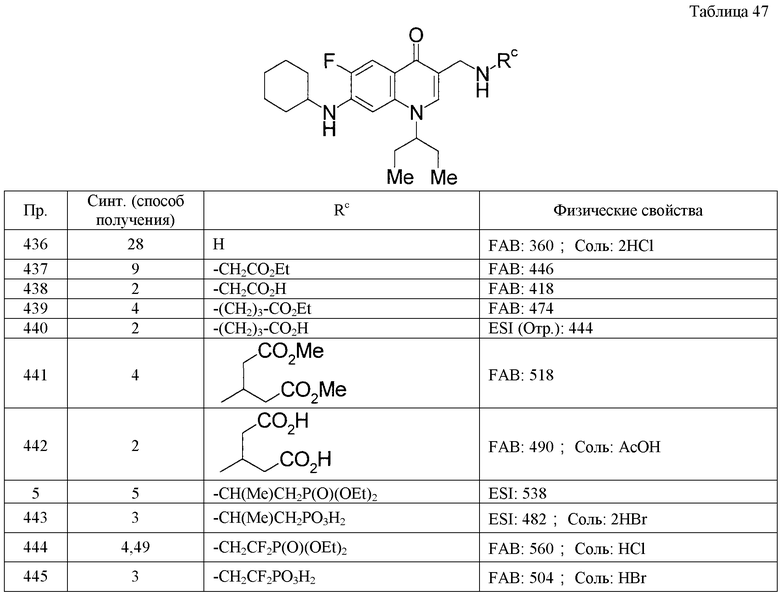
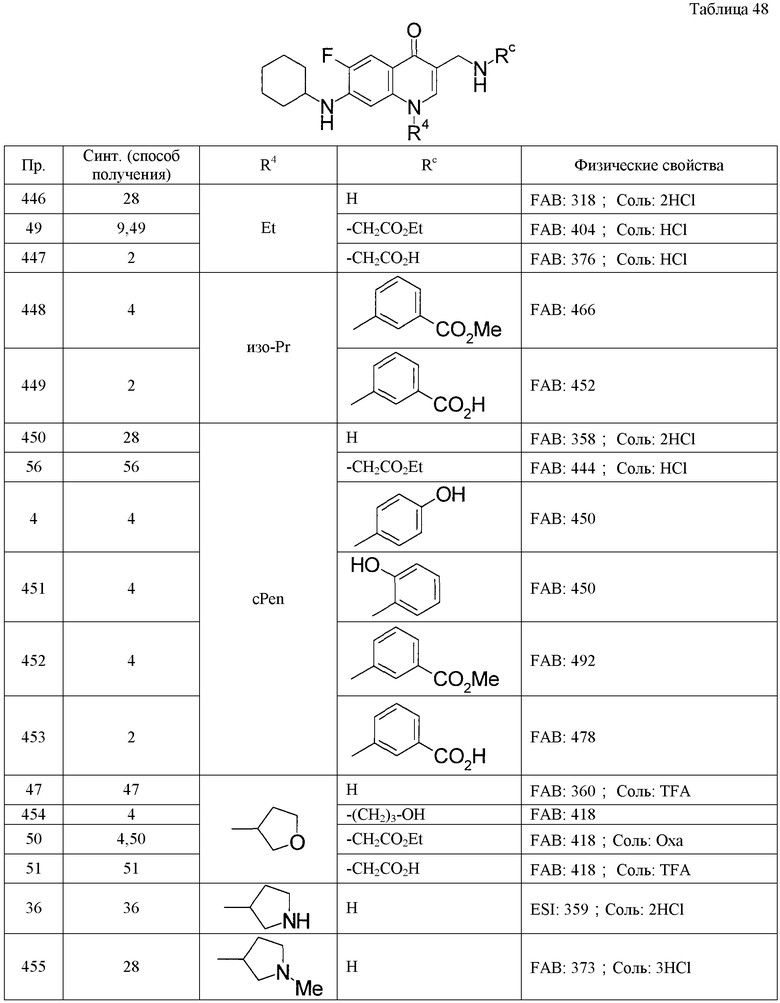
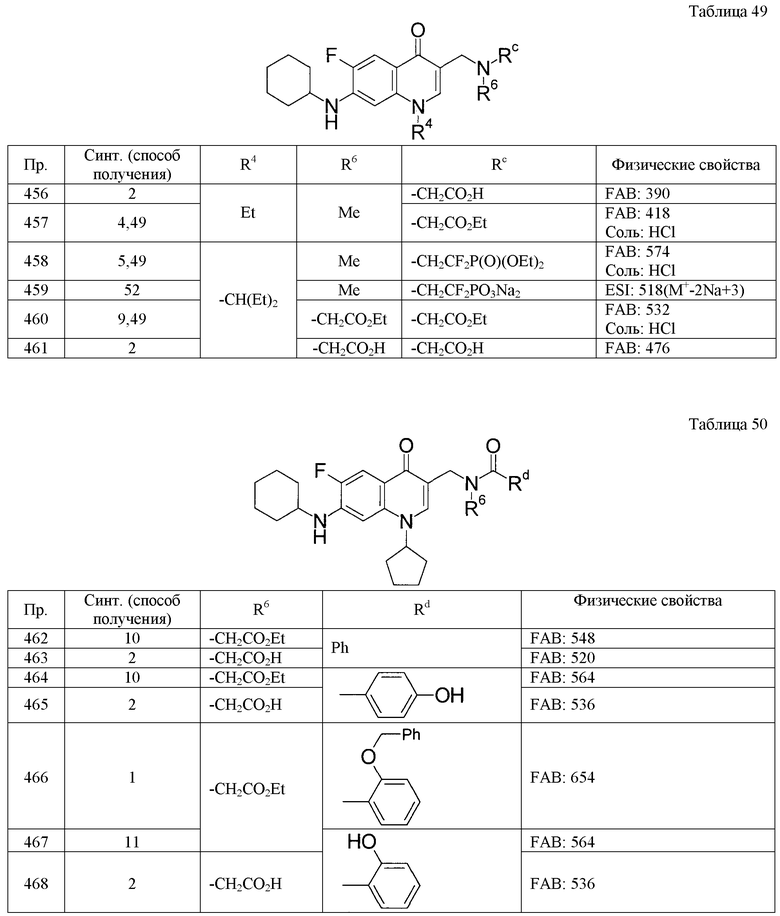
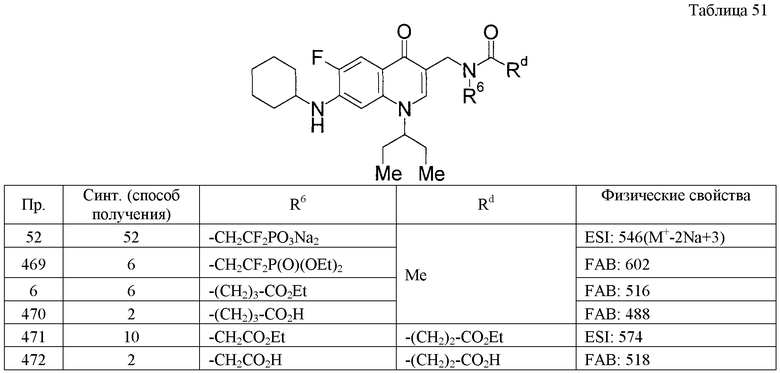

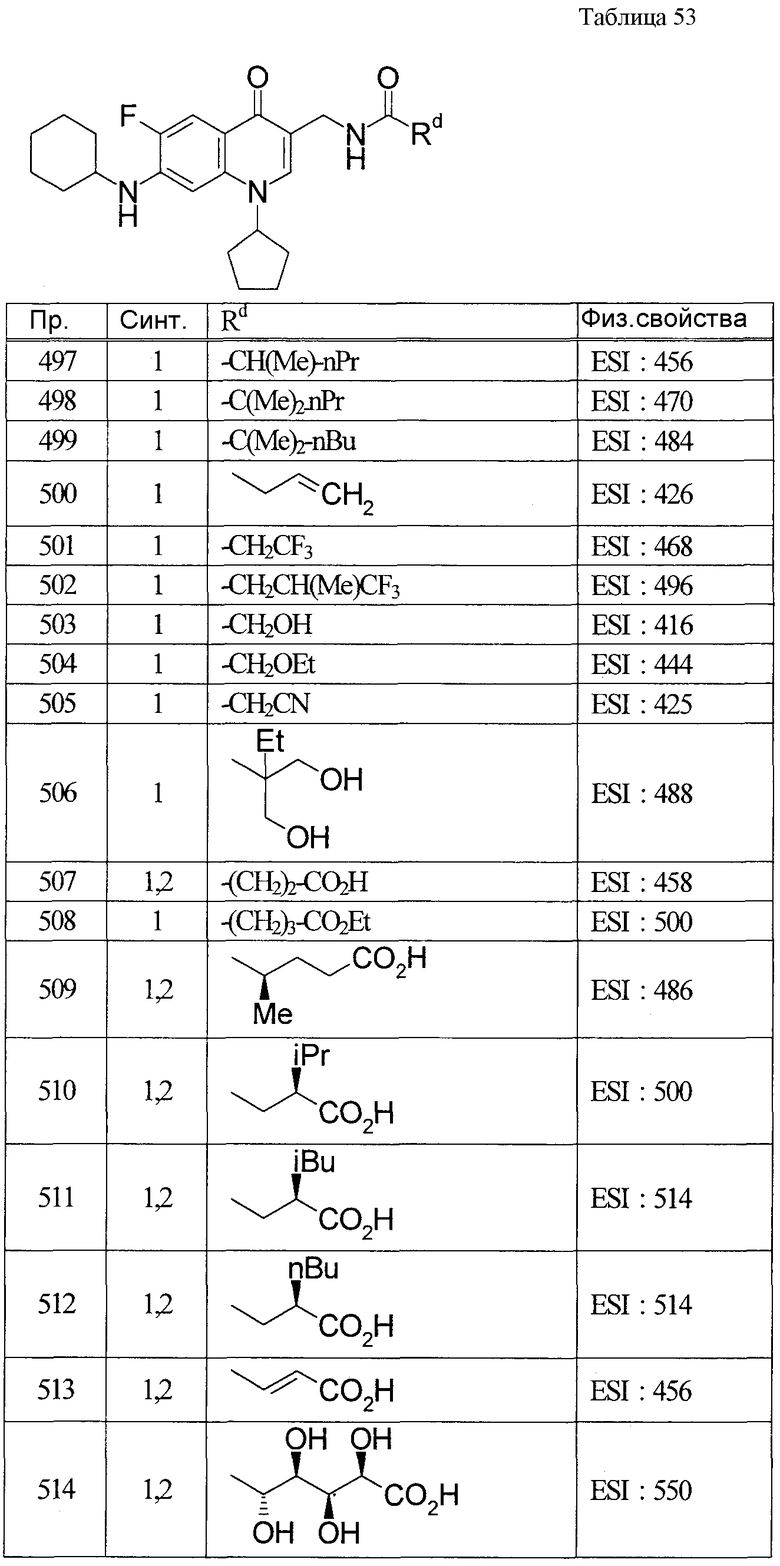

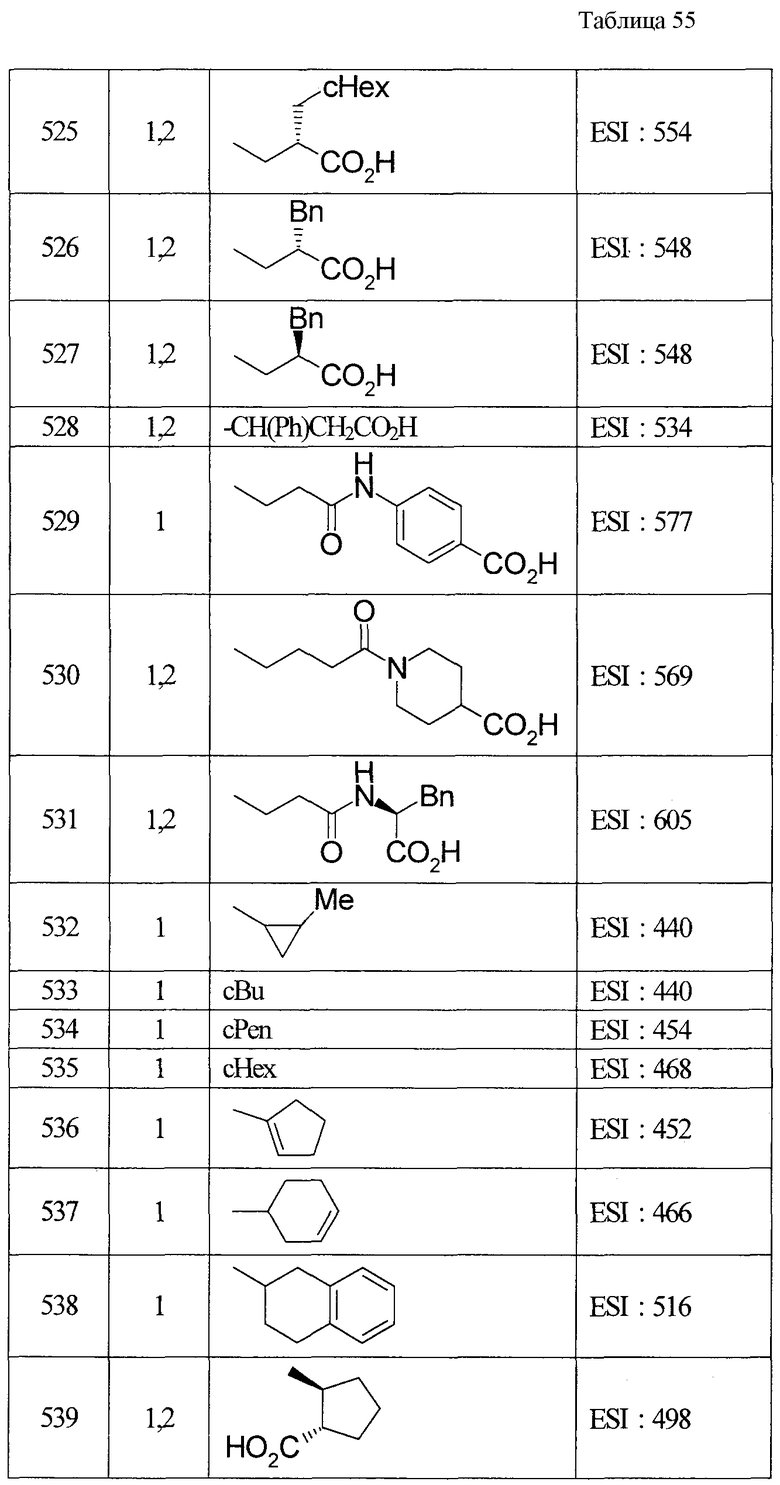

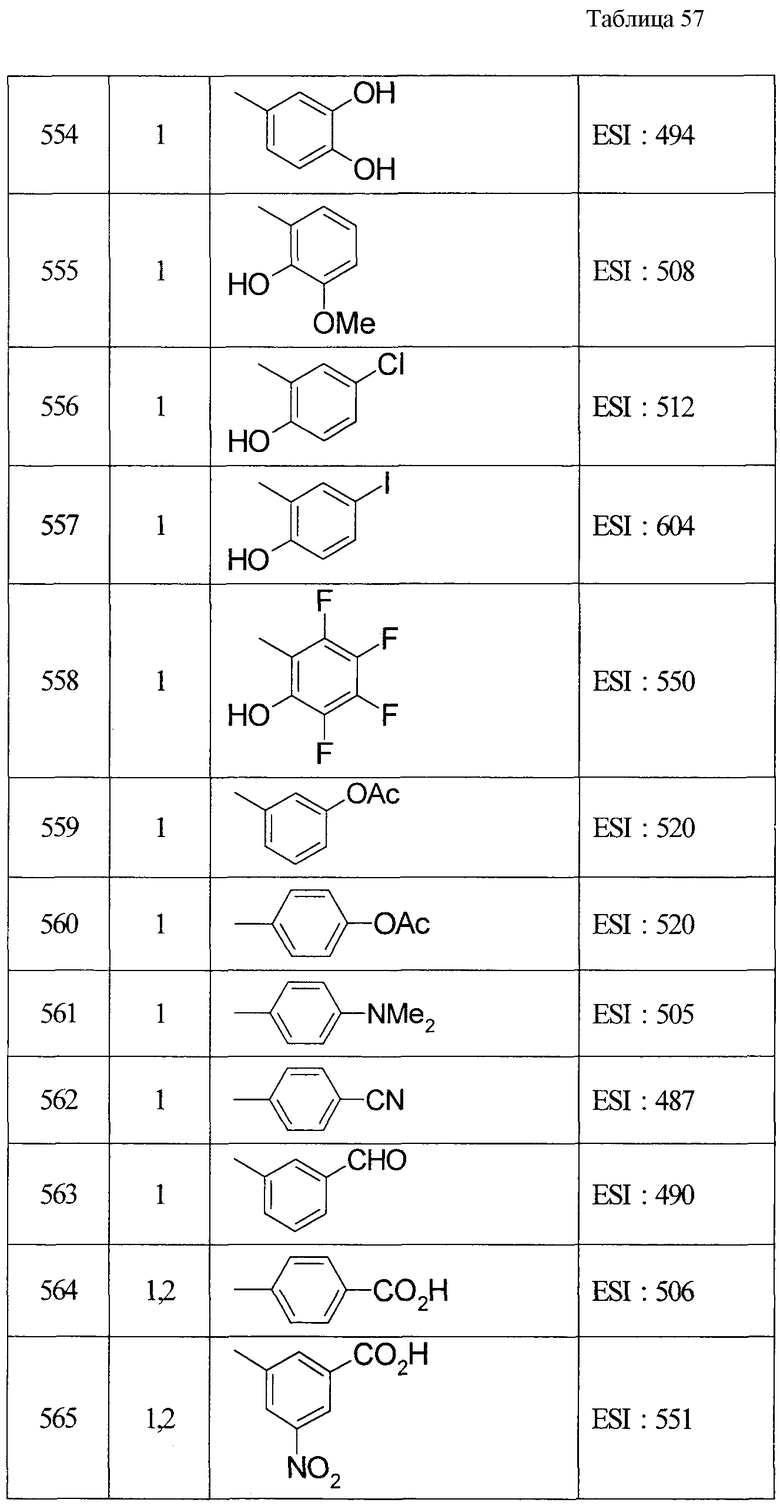

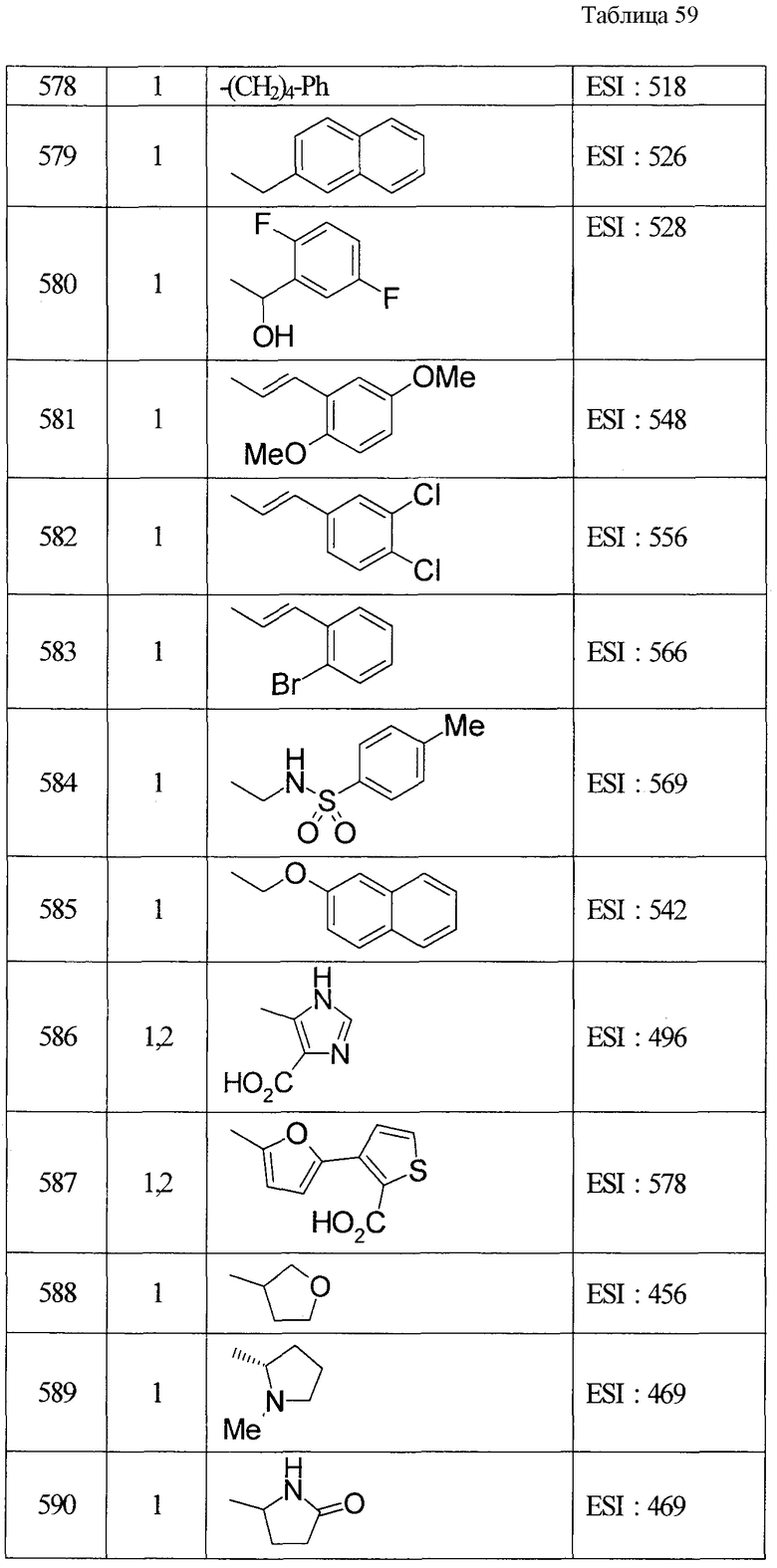
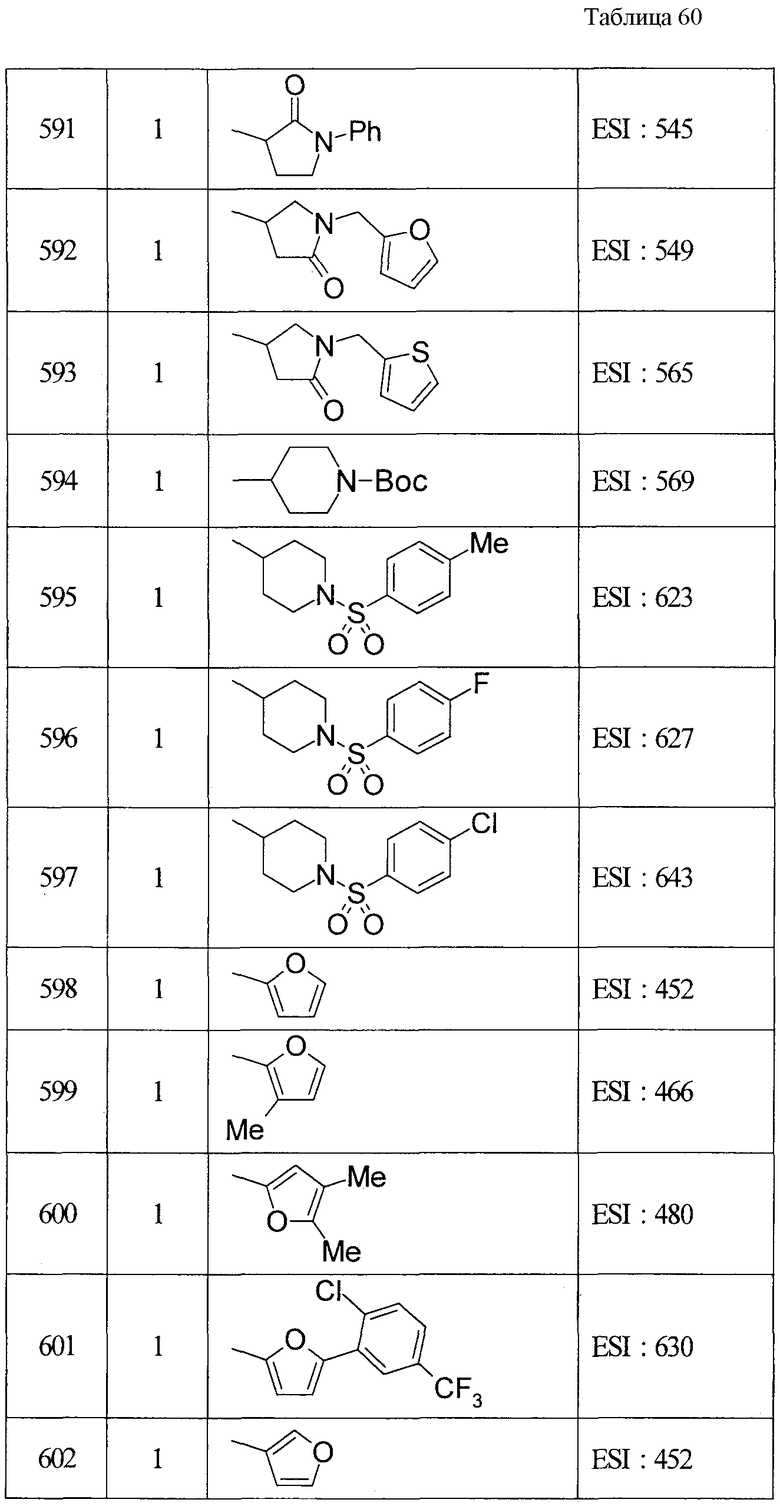

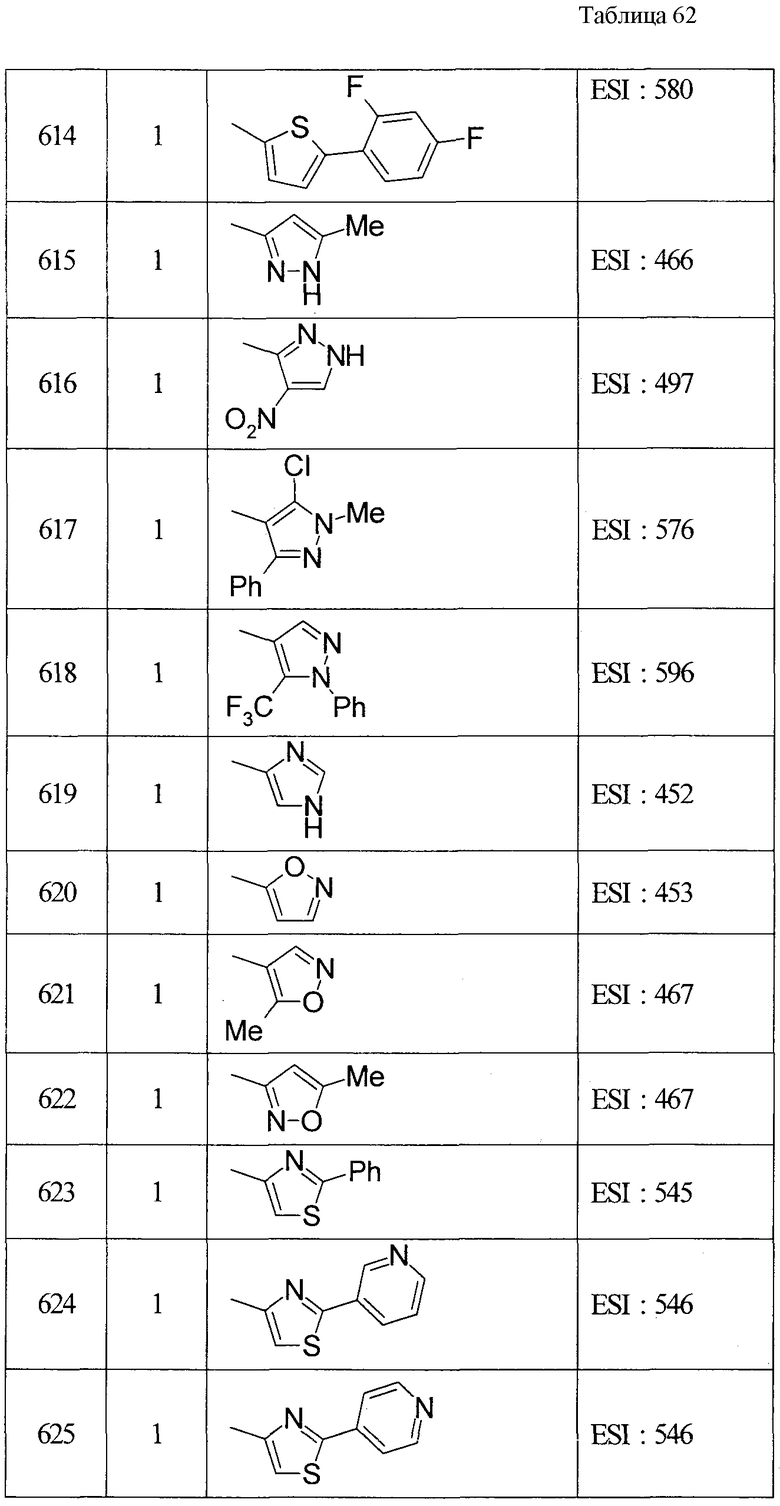
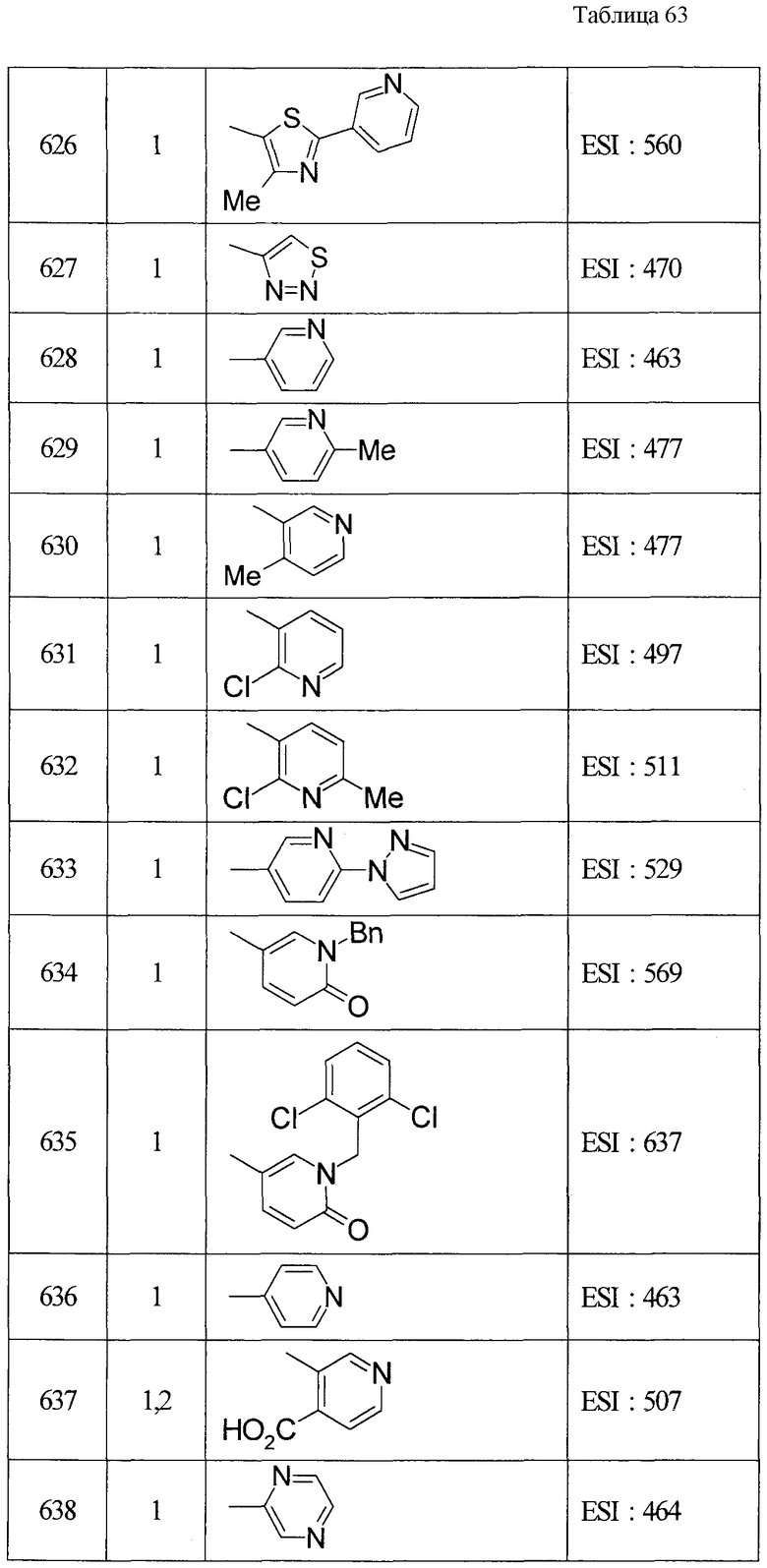
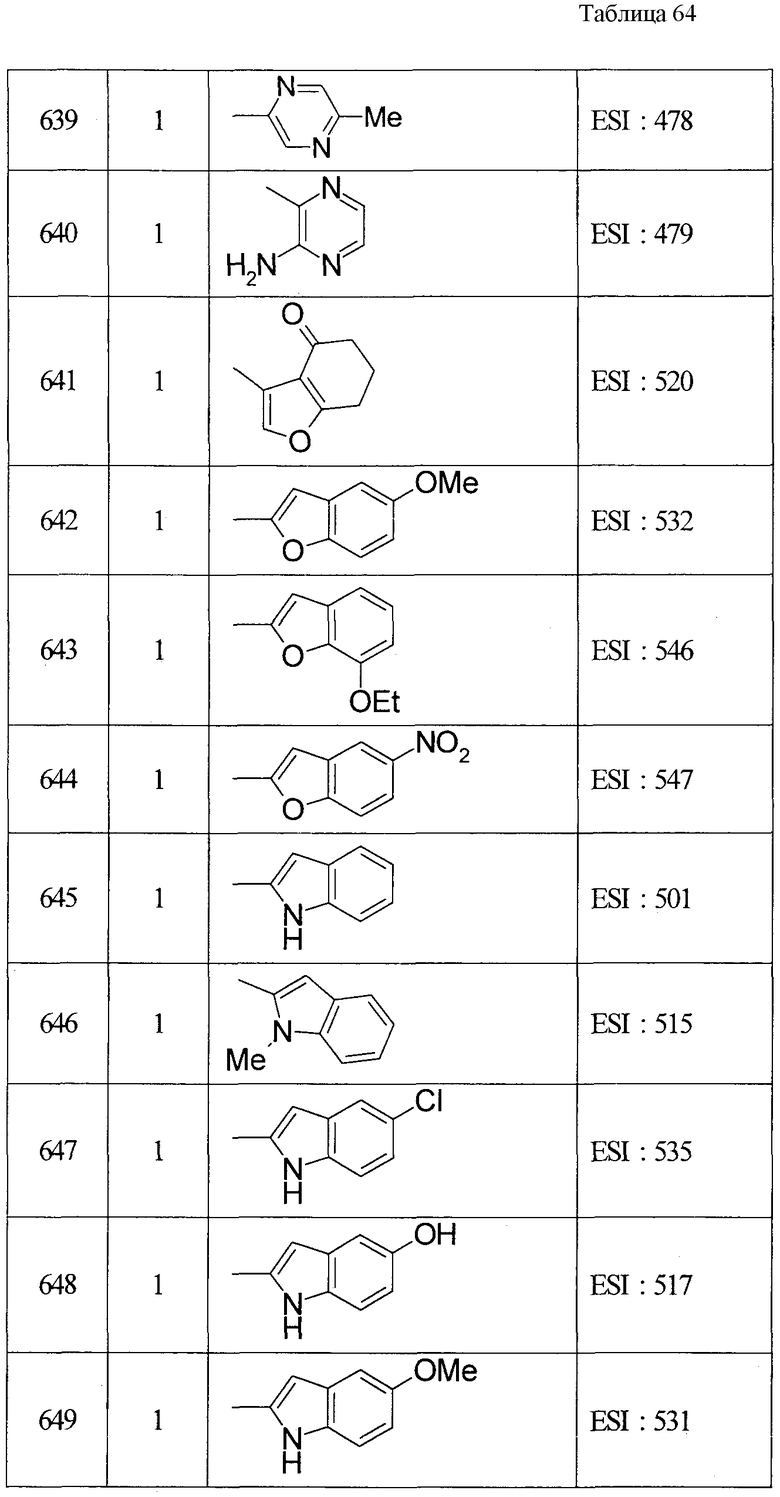
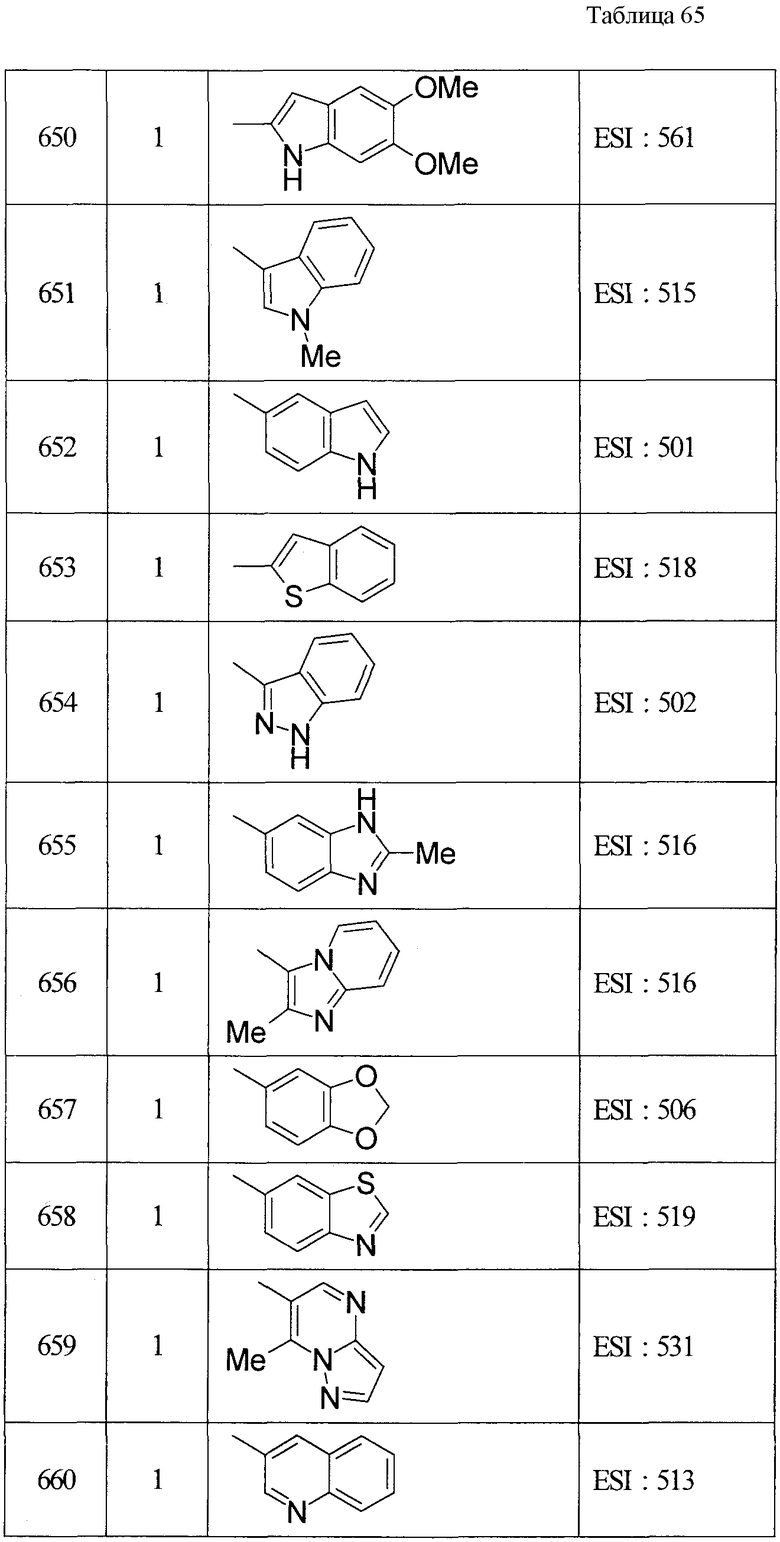




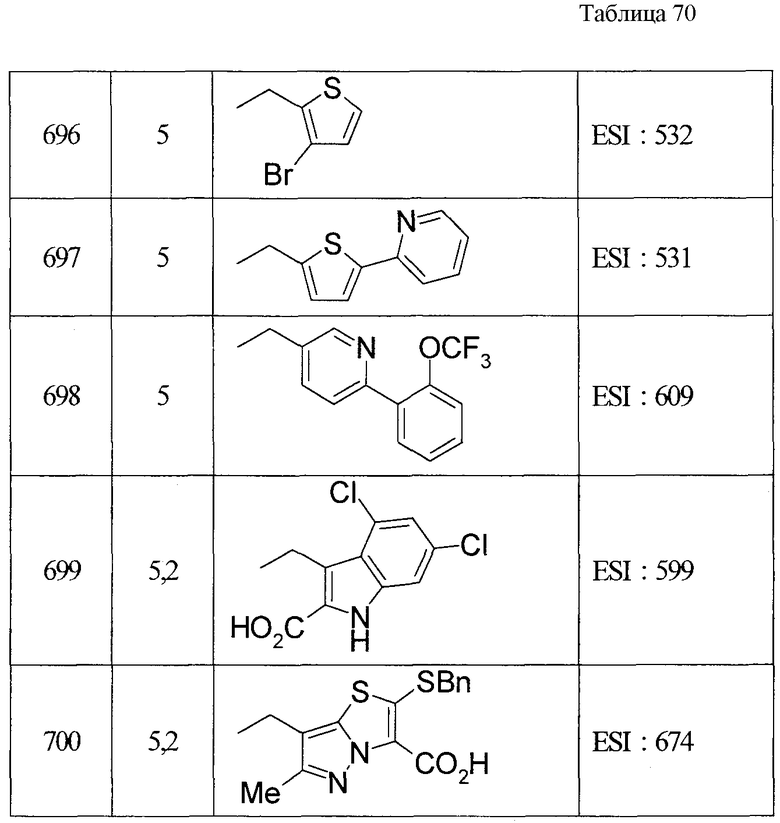

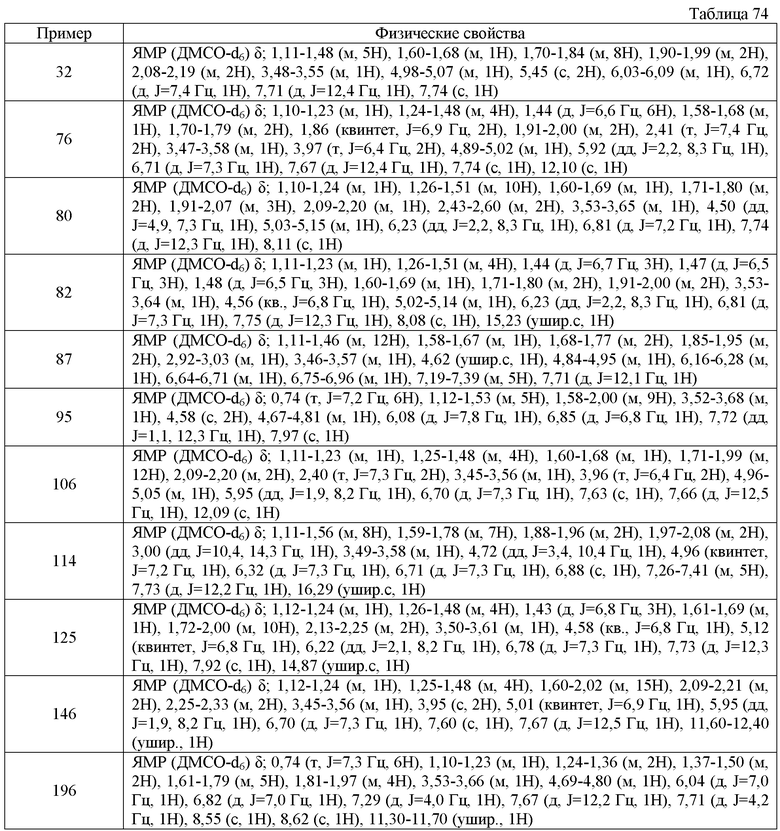

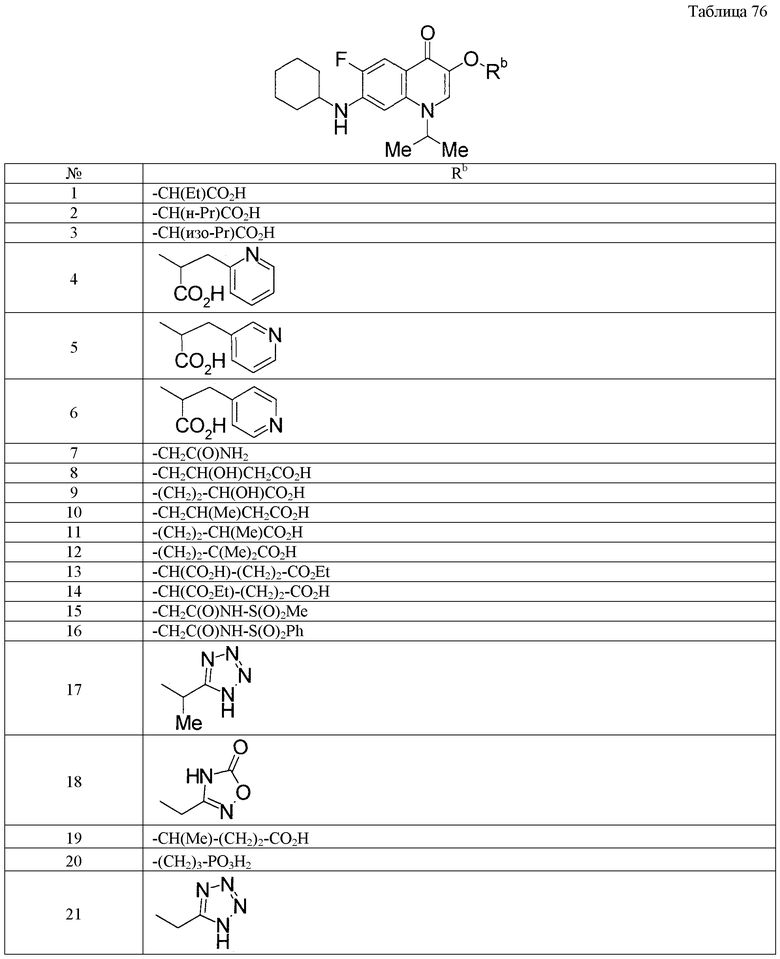

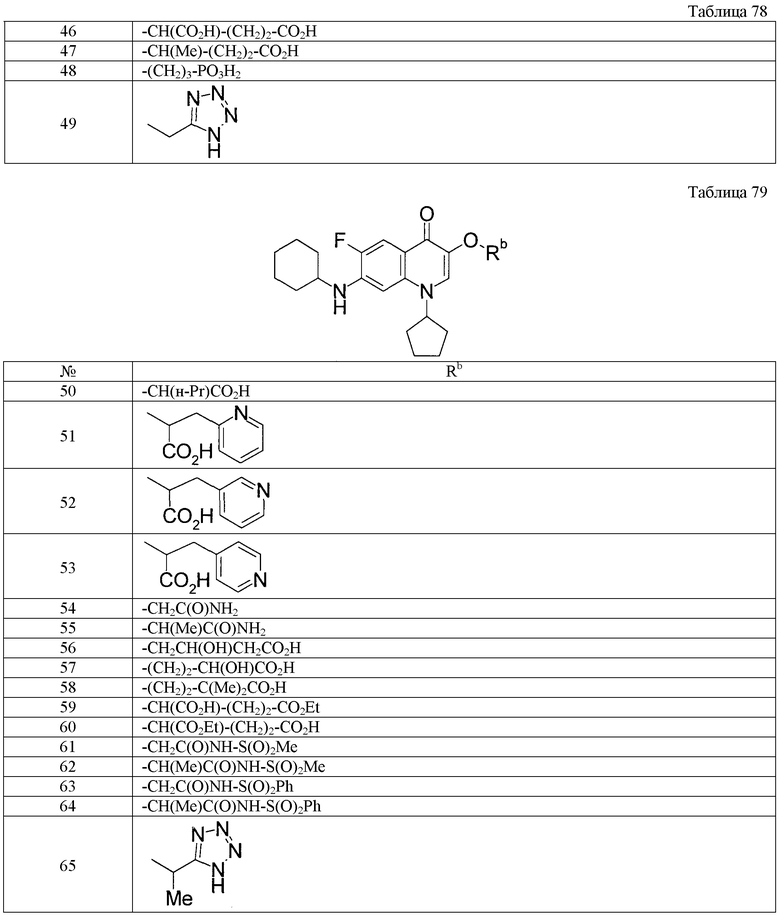
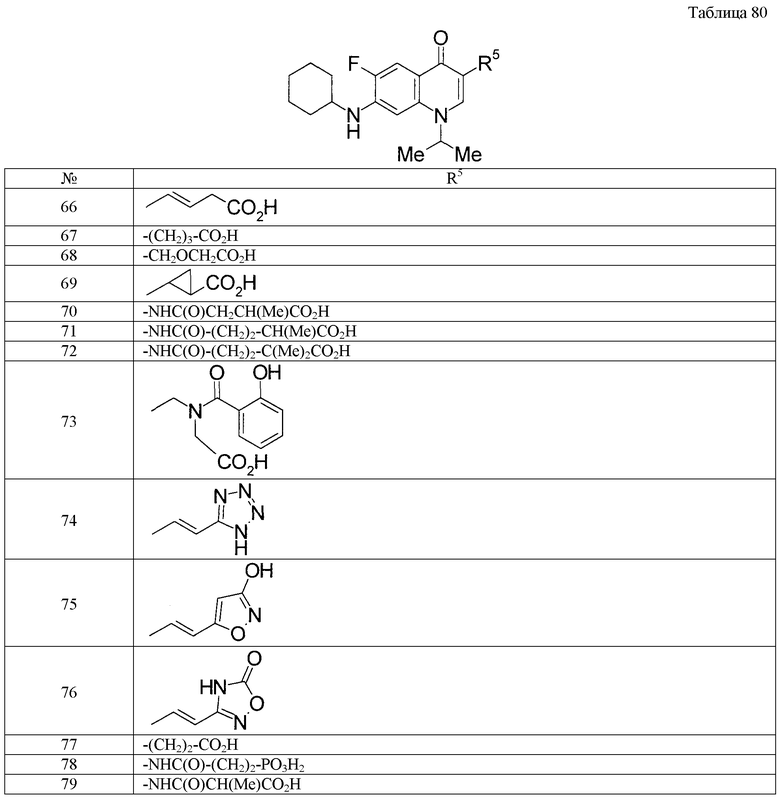
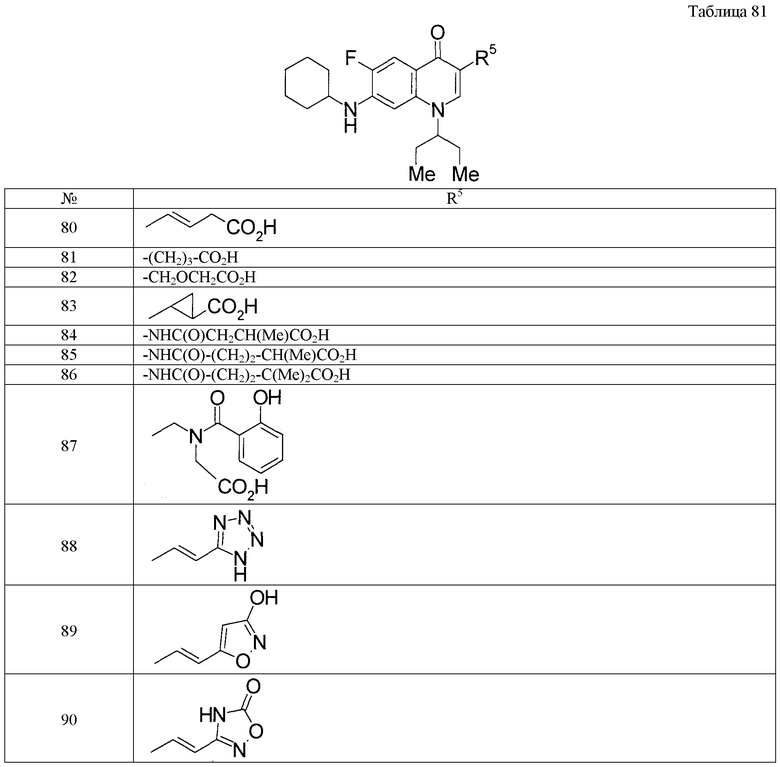
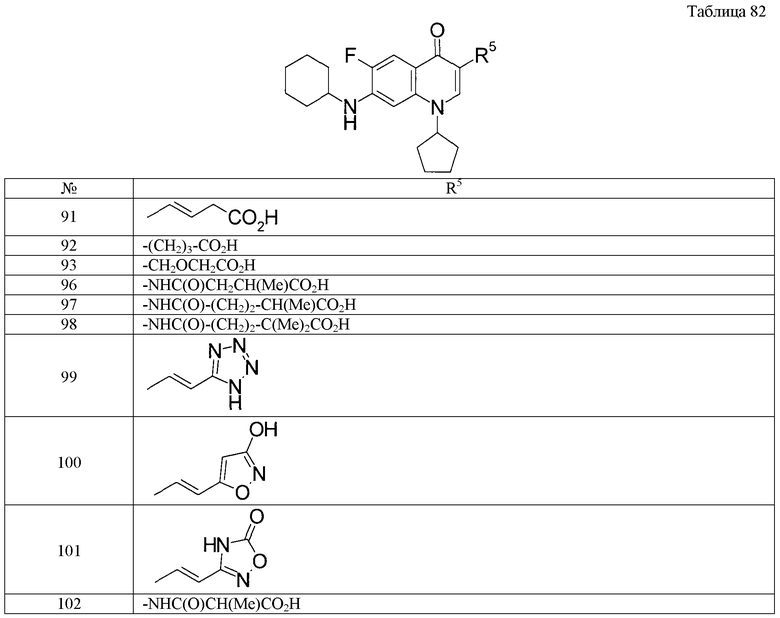

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИН-1-ОНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТА ВВ2 | 2008 |
|
RU2479578C2 |
| СОЕДИНЕНИЕ СУЛЬФОНАМИДА ИЛИ ЕГО СОЛЬ | 2007 |
|
RU2425029C2 |
| ПРОИЗВОДНОЕ ПИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2002 |
|
RU2285002C2 |
| ПРОИЗВОДНЫЕ НАФТИРИДИНА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2240322C2 |
| ОКСАДИАЗОЛИДИНДИОНОВОЕ СОЕДИНЕНИЕ | 2007 |
|
RU2440994C2 |
| ПРОИЗВОДНЫЕ АМИНОХИНОЛОНА, ЗАМЕЩЕННЫЕ ФЕНИЛЬНОЙ ИЛИ ГЕТЕРОАРОМАТИЧЕСКОЙ ГРУППОЙ | 1993 |
|
RU2124510C1 |
| ПРОИЗВОДНЫЕ БЕНЗАМИДА ИЛИ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ УКАЗАННОГО ПРОИЗВОДНОГО, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2333198C2 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА | 2004 |
|
RU2330841C2 |
| ПРОИЗВОДНЫЕ 8-ТРИФТОРМЕТИЛХИНОЛИНКАРБОНОВОЙ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ВИРУСА ИММУНОДЕФИЦИТА | 1995 |
|
RU2140919C1 |
| СПОСОБ ЛЕЧЕНИЯ ШИЗОФРЕНИИ | 2012 |
|
RU2648474C2 |
Настоящее изобретение относится к новым производным хинолона общей формулы (I), где R1: С3-6циклоалкил или низший алкиленС3-6циклоалкил, R2: -Н или галоген, R3: -Н, галоген, -OR0 или -О-(низший алкилен)-фенил, R0: одинаковые или отличаются один от другого, и каждый представляет собой -Н или низший алкил, R4: низший алкил, галоген(низший алкил), низший алкиленС3-6циклоалкил, С3-7циклоалкил или гетероциклическая группа, где указанные в R4 циклоалкил и гетероциклическая группа могут быть соответственно замещенными, R5: -NO2, -CN, -L-Ra, -C(O)R0, -O-Rb, -N(R6)2, низший алкилен-N(R6)(Rc), -N(R6)C(O)-Rd, низший алкилен-N(R6)C(O)-Rd, низший алкилен-N(R0)C(O)O-(низший алкил), -N(R0)C(O)N(R0)-Re, низший алкилен-N(R0)C(O)N(R0)-Re, -N(R0)S(O)2N(R0)C(O)-Rd, -CH=NOH, С3-6циклоалкил, (2,4-диоксо-1,3-тиазолидин-5-илиден)метил или (4-оксо-2-тиоксо-1,3-тиазолидин-5-илиден)метил, где указанный в R5 циклоалкил может быть соответственно замещенным, R6: Н, низший алкил, низший алкилен-CO2R0 или низший алкилен-P(O)(ORp)2, где указанный в R6 низший алкилен может быть замещенным, L: низший алкилен или низший алкенилен, которые могут быть соответственно замещенными, Ra: -OR0, -О-(низший алкилен)-фенил, -О-(низший алкилен)-CO2R0, -CO2R0, -C(O)NHOH, -C(O)N(R6)2, -C(O)N(R0)-S(O)2-(низший алкил), -C(O)N(R0)-S(O)2-фенил, -С(О)N(R0)-S(О)2-(гетероциклическая группа), -NH2OH, -OC(O)R0, -ОС(О)-(галоген(низший алкил)), -P(O)(ORp)2, фенил или гетероциклическая группа, где указанные в Ra фенил и гетероциклическая группа могут быть замещенными, Rp: R0, низший алкилен-ОС(О)-(низший алкил), низший алкилен-ОС(О)-С3-6циклоалкил, низший алкилен-ОС(О)О-(низший алкил), Rb: H, низший алкилен-Rba или низший алкенилен-Rba, где указанные в Rb низший алкилен и низший алкенилен могут быть замещенными, Rba: -OR0, -CO2R0, -C(O)N(R0)2, -C(O)N(R0)-S(O)2-(низший алкил), -C(O)N(R0)-S(O)2-фенил, -C(NH2)=NOH, -C(NH2)=NO-C(O)-(низший алкилен)-С(О)R0, -СО2-(низший алкилен)-фенил, -P(O)(ORp)2, -C(O)R0, -С(О)-фенил, С3-6циклоалкил, фенил или гетероциклическая группа, где указанные в Rba фенил и гетероциклическая группа могут быть замещенными, Rc: H, низший алкилен-OR0, низший алкилен-CO2R0, низший алкилен-Р(О)(ORp)2, фенил, где указанные в Rc низший алкилен и фенил могут быть замещенными, Rd: С1-7-алкил, низший алкенил, галоген(низший алкил), низший алкилен-Rda, низший алкенилен-Rda, С3-6циклоалкил, фенил, нафтил или гетероциклическая группа, где указанные в Rd низший алкилен, циклоалкил, фенил, нафтил и гетероциклическая группа могут быть замещенными, Rda: -CN, -OR0, -О-(низший алкилен)-CO2R0, -О-нафтил, -CO2R0, -СО2-(низший алкилен)-N(R0)2, -P(O)(ORp)2, -N(R6)2, -C(O)N(R0)-фенил, -C(O)N(R0)-низший алкилен, который может быть замещен -CO2R0)-фенил, -N(R0)C(O)-фенил, -N(R0)C(O)-OR0, -N(R0)C(O)-O-(низший алкилен)-фенил, -N(R0)S(O)2-фенил, C3-6циклоалкил, фенил, нафтил или гетероциклическая группа, где указанные в Rda фенил, нафтил и гетероциклическая группа могут быть замещенными, Re: низший алкилен-CO2R0, фенил, -S(О)2-фенил или -S(О)2-(гетероциклическая группа), где указанные в Re фенил и гетероциклическая группа могут быть замещенными, X: СН, A: C(R7), R7: -Н, или R4 и R7 вместе могут образовывать низший алкилен, где замещенные группы имеют заместители, указанные в п.1, и при условии, что исключается 7-(циклогексиламино)-1-этил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбонитрил. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I) и применению соединения формулы (I) для получения ингибитора агрегации тромбоцитов или ингибитора P2Y12. Технический результат: получены новые производные хинол-4-она, обладающие полезными биологическими свойствами. 4 н. и 7 з.п. ф-лы, 83 табл.
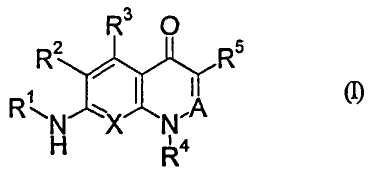
1. Производное хинолона, представленное формулой (I), или его фармацевтически приемлемая соль
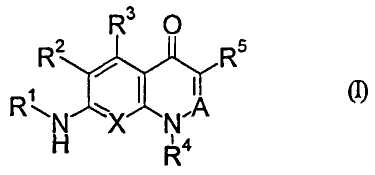
где символы в приведенной формуле имеют следующие значения:
R1 - С3-6циклоалкил или низший алкиленС3-6циклоалкил,
R2 - Н или галоген,
R3 - Н, галоген, -OR0 или -О-(низший алкилен)-фенил,
R0 - одинаковые или отличаются один от другого, и каждый представляет собой -Н или низший алкил,
R4 - низший алкил, галоген(низший алкил), низший алкиленС3-6циклоалкил, С3-7циклоалкил или гетероциклическая группа,
где указанные в R4 циклоалкил и гетероциклическая группа могут быть соответственно замещенными,
R5 - -NO2, -CN, -L-Ra, -C(O)R0, -O-Rb, -N(R6)2, низший алкилен-N(R6)(Rc), -N(R6)C(O)-Rd, низший алкилен-N(R6)С(О)-Rd, низший алкилен-N(R0)C(O)O-(низший алкил), -N(R0)C(O)N(R0)-Re, низший алкилен-N(R0)C(O)N(R0)-Re, -N(R0)S(O)2N(R0)C(O)-Rd, -CH=NOH, C3-6циклоалкил, (2,4-диоксо-1,3-тиазолидин-5-илиден)метил или (4-оксо-2-тиоксо-1,3-тиазолидин-5-илиден)метил,
где указанный в R5 циклоалкил может быть соответственно замещенным,
R6 - Н, низший алкил, низший алкилен-CO2R0 или низший алкилен-P(O)(ORp)2,
где указанный в R6 низший алкилен может быть замещенным,
L - низший алкилен или низший алкенилен, которые могут быть соответственно замещенными,
Ra - -OR0, -О-(низший алкилен)-фенил, -О-(низший алкилен)-CO2R0, -CO2R0, -C(O)NHOH, -C(O)N(R6)2, -C(O)N(R0)-S(O)2-(низший алкил), -C(O)N(R0)-S(O)2-фенил, -С(О)N(R0)-S(О)2-(гетероциклическая группа), -NH2OH, -OC(O)R0, -ОС(О)-(галоген(низший алкил)), -P(O)(ORp)2, фенил или гетероциклическая группа,
где указанные в Ra фенил и гетероциклическая группа могут быть замещенными,
Rp - R0, низший алкилен-ОС(О)-(низший алкил), низший алкилен-ОС(О)-С3-6циклоалкил, низший алкилен-ОС(О)О-(низший алкил),
Rb - H, низший алкилен-Rba или низший алкенилен-Rba,
где указанные в Rb низший алкилен и низший алкенилен могут быть замещенными,
Rba - -OR0, -CO2R0, -C(O)N(R0)2, -C(O)N(R0)-S(O)2-(низший алкил), -C(O)N(R0)-S(O)2-фенил, -C(NH2)=NOH, -С(NH2)=NO-С(О)-низший алкилен)-C(O)R0, -СО2-(низший алкилен)-фенил, -P(O)(ORp)2, -C(O)R0, -С(О)-фенил, С3-6циклоалкил, фенил или гетероциклическая группа,
где указанные в Rba фенил и гетероциклическая группа могут быть замещенными,
Rc - Н, низший алкилен-OR0, низший алкилен-CO2R0, низший алкилен-P(O)(ORp)2, фенил,
где указанные в Rc низший алкилен и фенил могут быть замещенными,
Rd - C1-7-алкил, низший алкенил, галоген(низший алкил), низший алкилен-Rda, низший алкенилен-Rda, С3-6циклоалкил, фенил, нафтил или гетероциклическая группа,
где указанные в Rd низший алкилен, циклоалкил, фенил, нафтил и гетероциклическая группа могут быть замещенными,
Rda - -CN, -OR0, -О-(низший алкилен)-CO2R0, -О-нафтил, -CO2R0, -СО2-(низший алкилен)-N(R0)2, -P(O)(ORp)2, -N(R6)2, -C(O)N(R0)-фенил, -C(O)N(R0)-(низший алкилен, который может быть замещен -CO2R0)-фенил, -N(R0)C(O)-фенил, -N(R0)C(O)-OR0, -N(R0)C(O)-O-(низший алкилен)-фенил, -N(R0)S(O)2-фенил, С3-6циклоалкил, фенил, нафтил или гетероциклическая группа,
где указанные в Rda фенил, нафтил и гетероциклическая группа могут быть замещенными,
Re - низший алкилен-CO2R0, фенил, -S(О)2-фенил или -S(O)2-(гетероциклическая группа),
где указанные в Re фенил и гетероциклическая группа могут быть замещенными,
Х - СН,
A - C(R7),
R7 - Н,
или R4 и R7 вместе могут образовывать низший алкилен,
где термин «гетероциклическая группа» включает «ароматическую гетероцикличесмкую группу» и «неароматическую гетероциклическую группу»,
где термин «ароматическая гетероциклическая группа» обозначает моноциклическую 5-6-членную ароматическую группу, содержащую от 1 до 4 одинаковых или различных гетероатомов, выбранных из группы, состоящей из атомов азота, кислорода и серы, бициклический ароматический гетероцикл, в котором конденсированы моноциклические ароматические гетероциклы или моноциклический ароматический гетероцикл конденсирован с бензольным циклом, или трициклический ароматический гетероцикл, в котором бициклический ароматический гетероцикл конденсирован с моноциклическим ароматическим гетероциклом или с бензольным циклом, и где атом азота и/или атом серы, содержащиеся в таких циклах, могут быть окислены, и указанные циклы могут быть частично насыщенными; и
где термин «неароматическая гетероциклическая группа» обозначает насыщенный или частично насыщенный моноциклический 3-6-членный неароматический гетероцикл, который содержит от 1 до 4 гетероатомов, выбранных из О, S или N, бициклический неароматический гетероцикл, в котором моноциклический неароматический гетероцикл конденсирован с бензольным циклом или указанным выше ароматическим гетероциклом, и где такие неароматические гетероциклы могут образовывать оксид или диоксид через окисление S или N как циклического атома или могут образовывать мостиковый цикл или спироцикл;
термин «может быть замещенным» обозначает «незамещенный» или «замещенный одинаковыми или различными 1-5 заместителями», где
приемлемый заместитель для «низшего алкилена», за исключением «низшего алкилена» в определении R6, и для «низшего алкенилена» означает группу G1;
приемлемый заместитель для «низшего алкилена» в определении R6 означает галоген;
приемлемый заместитель для циклоалкила означает группу G2;
приемлемый заместитель для фенила и нафтила, за исключением фенила и нафтила в определении Rd, означает группу G3;
приемлемый заместитель для фенила и нафтила в определении Rd, означает группу G4;
приемлемый заместитель для гетероциклической группы, за исключением гетероциклической группы в определении Rda и Rd, означает группу G5;
приемлемый заместитель для гетероциклической группы в определении Rda означает группу G6;
приемлемый заместитель для гетероциклической группы в определении Rd означает группу G7; где
G1 выбирают из галогена, -OR0, -CO2R0 и -СО2-низший алкилен-фенил;
G2 выбирают из галогена, низшего алкила, -OR0, -CO2R0 и -С(О)-фенила;
G3 выбирают из галогена, -CN, низшего алкила, галоген-низшего алкила, -OR0, -О-галоген-низшего алкила, -CO2R0 и -О-низший алкилен-CO2R0;
G4 выбирают из галогена, -CN, низшего алкила, -OR0, -C(O)R0, -CO2R0, -N(R0)2, -S(O)2-низшего алкила и фенила;
G5 выбирают из галогена, низшего алкила, галоген-низшего алкила, -OR0, -О-галоген-низшего алкила, оксо, -CO2R0, низшего алкилена-С(О)R0, низшего алкилена-CO2R0 и -S(O)2-низшего алкила;
G6 выбирают из галогена, низшего алкила, галоген-низшего алкила, -OR0, -О-галоген-низшего алкила, оксо, -CO2R0, низшего алкилена-С(О)2R0, -S(O)2-низшего алкила, фенила и -S-низшего алкилена-фенила; и
G7 выбирают из галогена, нитро, низшего алкила, галоген-низшего алкила, -OR0, -О-галоген-низшего алкила, оксо, -CO2R0, низшего алкилена-CO2R0, -N(R0)2, -S(O)2-низшего алкила, -S(О)2-фенила, фенила, низшего алкилен-фенила и -S-низшего алкилена-CO2R0;
и при условии, что
исключается 7-(циклогексиламино)-1-этил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбонитрил.
2. Соединение по п.1, где R3 представляет собой -Н, -ОН или -F.
3. Соединение по п.2, где А представляет собой СН.
4. Соединение по п.3, где R1 представляет собой циклогексил или циклопропилметил.
5. Соединение по п.4, где R2 представляет собой -F.
6. Соединение по п.5, где R4 представляет собой низший алкил или циклоалкил.
7. Соединение по п.6, где R5 представляет собой -N(R6)C(O)-(низший алкилен)-CO2R0, где R6 означает -Н или низший алкил, низший алкилен-CO2R0, низший алкенилен-CO2R0, -О-(низший алкилен)-CO2R0, -О-(низший алкилен, который может быть замещен -CO2R0)-фенил или -О-(низший алкенилен)-CO2R0.
8. Соединение по п.1, которое выбрано из группы, состоящей из
4-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-4-оксобутановой кислоты,
5-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]амино}-5-оксопентановой кислоты,
(2Е)-3-[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]акриловой кислоты,
(2S)-2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}-3-фенилпропановой кислоты,
(2Е)-3-[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]акриловой кислоты,
(2S)-2-{[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]окси}-3-фенилпропановой кислоты,
(2S)-2-{[7-(циклогексиламино)-1-циклопентил-6-фтор-4-оксо-1,4-дигидрохинолин-3-ил]окси}пропановой кислоты и
(2S)-2-{[7-(циклогексиламино)-6-фтор-1-изопропил-4-оксо-1,4-дигидрохинолин-3-ил]окси}пропановой кислоты,
или его фармацевтически приемлемая соль.
9. Фармацевтическая композиция, которая является ингибитором агрегации тромбоцитов, включающая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
10. Фармацевтическая композиция, которая является ингибитором P2Y12, включающая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
11. Применение соединения по п.1 или его фармацевтически приемлемой соли для получения ингибитора агрегации тромбоцитов или ингибитора P2Y12.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| АЗАБИЦИКЛО-ХИНОЛОН-КАРБОНОВЫЕ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1989 |
|
RU2049777C1 |
| Способ получения нафтиридинхинолин-или бензоксазинкарбоновых кислот или их фармацевтически допустимых солей присоединения кислоты | 1983 |
|
SU1360584A3 |
| ПРОИЗВОДНЫЕ ХИНОЛОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2193558C2 |

