Область изобретения
Настоящее изобретение относится к фармацевтическому средству, в частности к новому оксадиазолидиндионовому соединению или его фармацевтически приемлемой соли, которые являются полезными в качестве средства, стимулирующего секрецию инсулина или средства для профилактики/лечения диабета.
Предпосылки изобретения
Диабет представляет собой заболевание, основным симптомом которого является хронически высокий уровень глюкозы в крови, который образуется в результате абсолютной или относительной недостаточности действия инсулина. Клинически он грубо подразделяется на инсулин-зависимый сахарный диабет (IDDM) и инсулин-независимый сахарный диабет (NIDDM). При инсулин-независимом сахарном диабете (NIDDM) снижение секреции инсулина из панкреатических β-клеток является одной из основных причин проявления заболевания, и, в частности, было признано, что высокий уровень глюкозы в крови после приема пищи возникает в результате начальной стадии нарушения секреции инсулина.
В недавнее время крупномаcштабные клинические исследования подтвердили, что коррекция высокого уровня глюкозы в крови после приема пищи является важной для проявления и супрессии диабетических осложнений. Кроме того, были сообщения о том, что артериосклероз развивается на стадии только высокого уровня глюкозы в крови после приема пищи и что сохраняющийся несколько повышенный уровень глюкозы в крови после приема пищи повышает показатель смертности в результате сосудистых заболеваний и т.п. Это показывает, что повышенный уровень глюкозы в крови после приема пищи является независимым фактором риска смерти от сердечно-сосудистых заболеваний, даже когда он слегка повышен. На основании изложенной выше информации, была признана необходимость лекарственной терапии от повышенного уровня глюкозы в крови после приема пищи.
В настоящее время основным направлением в области средств, стимулирующих секрецию инсулина, являются препараты сульфонилмочевины (SU), но известно, что они способны вызывать гипогликемию и приводят к вторичной инвалидности из-за истощения поджелудочной железы в случае их долговременного введения. Кроме того, SU препараты являются эффективными для контроля уровня глюкозы в крови в процессе приема пищи, но очень трудно подавлять уровень глюкозы в крови после приема пищи.
GPR40 представляет собой связанный с G белком рецептор, который был идентифицирован как рецептор жирных кислот и имеет высокий уровень экспрессии в β-клетках поджелудочной железы, и были сообщения о том, что он связан с инсулин-секреторным действием жирной кислоты (непатентный ссылочный документ 1).
Следовательно, поскольку коррекция высокого уровня глюкозы в крови после приема пищи, как ожидается, основана на действии по стимуляции секреции инсулина, агонист GPR40 рецептора является полезным в качестве средства для профилактики/лечения инсулин-зависимого сахарного диабета (IDDM), инсулин-независимого сахарного диабета (NIDDM) и легких форм диабета пограничного типа (аномальные толерантность к глюкозе и уровни глюкозы в крови натощак).
В ссылочном патентном документе 1 сообщается, что соединение, представленное формулой (A), включающее широкий ряд соединений, обладает контролирующим GPR40 рецептор действием и является полезным в качестве средства, стимулирующего секрецию инсулина, или средства для профилактики/лечения диабета. Однако отсутствует какое-либо иллюстративное раскрытие соединения, имеющего структуру оксадиазолидиндиона.

(В представленной формуле кольцо P представляет собой ароматическое кольцо, которое может содержать заместитель, и кольцо Q представляет собой ароматическое кольцо, которое может дополнительно содержать заместитель, отличный от

X и Y спейсеры, и

группа, способная к высвобождению катиона.)
В ссылочном патентном документе 2 сообщается, что соединение, представленное формулой (B), обладает контролирующим GPR40 рецептор действием и является полезным в качестве средства, стимулирующего секрецию инсулина или средства для профилактики/лечения диабета. Однако отсутствует какое-либо иллюстративное раскрытие соединения, имеющего структуру оксадиазолидиндиона.

(Символы в представленной формуле см. в указанной публикации.)
В ссылочном патентном документе 3 сообщается, что соединение, представленное формулой (C), обладает контролирующим GPR40 рецептор действием и является полезным в качестве средства, стимулирующего секрецию инсулина или средства для профилактики/лечения диабета. Однако отсутствует какое-либо иллюстративное раскрытие соединения, имеющего структуру оксадиазолидиндиона.

(Символы в представленной формуле см. в указанной публикации.)
В ссылочном патентном документе 4 сообщается, что оксадиазолидиндионовое соединение, представленное формулой (D), обладает ингибирующим действием в отношении ингибитора активации плазминогена (PAI)-1 и является полезным для лечения тромбов, фибрилляции предсердия, ишемии миокарда, диабета и т.п. Однако отсутствует какое-либо описание его действия в отношении GPR40 рецептора.

(В представленной формуле X представляет собой
Другие символы см. в указанной публикации)
В ссылочном патентном документе 5 сообщается, что соединение, имеющее две оксадиазолидиндионовые структуры, представленное формулой (E), обладает действием усиления чувствительности к инсулину и является полезным для лечения диабета. Однако отсутствует какое-либо описание его действия на GPR40 рецептор.

(Символы в представленной формуле см. в указанной публикации.)
В ссылочном патентном документе 6 сообщается, что оксазолидиндионовое соединение, представленное формулой (F), обладает действием по снижению уровня глюкозы в крови и действием по снижению уровня липидов в крови и является полезным для лечения диабета. Однако кольцо, которое соответствует оксадиазолидиндиону по настоящему изобретению, представляет собой оксазолидиндион. Кроме того, отсутствует какое-либо описание его действия в отношении GPR40 рецептора.

(Символы в представленной формуле см. в указанной публикации.)
В ссылочном патентном документе 7 сообщается, что оксадиазолидиндионовое соединение, представленное формулой (G), обладает действием по снижению уровня глюкозы в крови и является полезным для лечения диабета. Однако кольцо, которое соответствует кольцу A по настоящему изобретению, представляет собой оксадиазольное кольцо. Кроме того, отсутствует какое-либо описание его действия на GPR40 рецептор.

(Символы в представленной формуле см. в указанной публикации.)
В ссылочном патентном документе 8 сообщается, что соединение, представленное формулой (H), обладает действием по снижению уровня глюкозы в крови и является полезным для лечения диабета. Однако отсутствует какое-либо описание его действия на GPR40 рецептор.

(Символы в представленной формуле см. в указанной публикации.)
В ссылочном патентном документе 9 сообщается, что оксадиазолидиндионовое соединение, представленное формулой (J), обладает действием по снижению уровня глюкозы в крови и является полезным для лечения диабета. Однако кольцо, которое соответствует кольцу A соединения по настоящему изобретению, представляет собой оксазол или тиазол. Кроме того, отсутствует какое-либо описание его действия на GPR40 рецептор.

(X в представленной формуле представляет собой атом кислорода или атом серы. Другие символы см. в указанной публикации.)
В ссылочном патентном документе 10 сообщается, что соединение, представленное формулой (K), является полезным от гиперлипемии, гипергликемии, ожирения и т.п. Однако кольцо, которое соответствует кольцу A соединения по настоящему изобретению, представляет собой морфолин или тиоморфолин. Кроме того, отсутствует какое-либо описание его действия на GPR40 рецептор.

(В представленной формуле представляет собой атом кислорода или атом серы. Другие символы см. в указанной публикации.)
В непатентном ссылочном документе 2 сообщается, что оксадиазолидиндионовое соединение, представленное формулой (L), обладает действием по снижению уровня глюкозы в крови и является полезным для лечения диабета. Однако кольцо, которое соответствует кольцу A соединения по настоящему изобретению, представляет собой (ди)азольное кольцо. Кроме того, отсутствует какое-либо описание его действия на GPR40 рецептор.

(В представленной формуле, X представляет собой O, S или N, Y представляет собой C или N, и n имеет значение 1 или 2. Другие символы см. в указанном ссылочном документе.)
Непатентный ссылочный документ 1: Nature, (England), 2003, vol.422, pр.173-176.
Непатентный ссылочный документ 2: European Journal of Medicinal Chemistry, (France), 2001, vol.36, pр.31-42.
Патентный ссылочный документ 1: Международная Публикация № 2004/041266.
Патентный ссылочный документ 2: Международная Публикация № 2005/063729.
Патентный ссылочный документ 3: Международная Публикация № 2005/063725.
Патентный ссылочный документ 4: Международная Публикация № 2005/030203.
Патентный ссылочный документ 5: Международная Публикация № 94/25448.
Патентный ссылочный документ 6: JP-A-2000-212174.
Патентный ссылочный документ 7: Международная Публикация № 95/30664.
Патентный ссылочный документ 8: Международная Публикация № 97/41097.
Патентный ссылочный документ 9: Патент США № 5480896.
Патентный ссылочный документ 10: JP-A-7-2848.
Раскрытие изобретения
Задачи, решаемые настоящим изобретением
Настоящее изобретение имеет целью обеспечение нового соединения, которое обладает агонистическим действием в отношении GPR40 рецептора и является полезным в качестве средства, стимулирующего секрецию инсулина или средства для профилактики/лечения диабета.
Средства для решения задач
Авторы настоящего изобретения провели всесторонние исследования соединений, обладающих агонистическим действием в отношении GPR40 рецептора, и обнаружили, что новые оксадиазолидиндионовые соединения или их соли обладают отличным агонистическим действием в отношении GPR40 рецептора. Как результат, было создано настоящее изобретение путем открытия того, что оксадиазолидиндионовые соединения обладают отличным действием по стимуляции секреции инсулина и сильно ингибируют повышение уровня глюкозы в крови после глюкозной нагрузки.
Таким образом, настоящее изобретение относится к оксадиазолидиндионовому соединению, представленному следующей формулой (I), или его фармацевтически приемлемой соли.
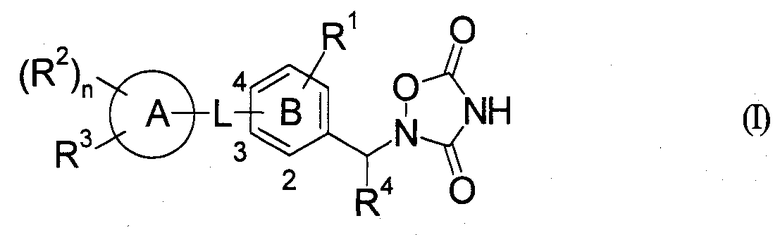
(Символы в представленной формуле представляют следующие значения,
R1: -H, галоген, -R0, галогено-низший алкил, -ORz, -S-R0 или -O-галогено-низший алкил,
R0: низший алкил,
Rz: одинаковые или отличные друг от друга, и каждый представляет собой -H или низший алкил,
L: *-низший алкилен-O-, *-низший алкилен-N(Rz)- или *-CON(Rz)-, где знак * в L означает связывание с кольцом A,
кольцо A: бензол, пиридин, тиофен, пиперидин, дигидропиридин, пиримидин или тетрагидрохинолин,
кольцо B: бензол или пиридин,
R2: соответственно одинаковые или отличные друг от друга, и каждый представляет собой -галоген,
-R0, галогено-низший алкил, -ORz, -S-R0, -O-галогено-низший алкил, -O-низший алкилен-арил или оксо,
n: 0, 1 или 2,
R3: -галоген, -R0, -галогено-низший алкил, -OR0, -S-R0, -O-галогено-низший алкил, -X-(фенил, который может быть замещен) или -X-(гетероарил, который может быть замещен),
X: простая связь, O, S или N(Rz),
R4: -H или низший алкил,
или R1 и R4 вместе могут образовывать низший алкилен,
при условии, что
2-{4-[2-(4-метил-6-оксо-2-пропилпиримидин-1(6H)-ил)этокси]бензил}-1,2,4-оксадиазолидин-3,5-дион и
2-{4-[2-(2-этил-4-метил-6-оксопилпиримидин-1(6H)-ил)этокси]бензил}-1,2,4-оксадиазолидин-3,5-дион
исключаются. То же применимо к следующему далее описанию.)
Кроме того, настоящая заявка также относится к фармацевтическому средству, в частности агонисту GPR40, в котором используют оксадиазолидиндионовое соединение, представленное общей формулой (I), или его соль в качестве активного ингредиента.
Кроме того, настоящая заявка также относится к применению соединения, представленного формулой (I), или его фармацевтически приемлемой соли для получения агониста GPR40, средства, стимулирующего секрецию инсулина или средства для профилактики и/или лечения диабета, и к способу профилактики и/или лечения диабета, который включает введение пациенту эффективного количества соединения, представленного формулой (I), или его фармацевтически приемлемой соли.
А именно представлены
(1) фармацевтическая композиция, которая содержит соединение, представленное формулой (I), или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель,
(2) фармацевтическая композиция, описанная в п.(1), которая представляет собой агонист GPR40,
(3) фармацевтическая композиция, описанная в п.(1), которая представляет собой средство, стимулирующее секрецию инсулина,
(4) фармацевтическая композиция, описанная в п.(1), которая представляет собой средство для профилактики и/или лечения диабета,
(5) применение соединения, описанного в представленной формуле (I), или его фармацевтически приемлемой соли для получения агониста GPR40, средства, стимулирующего секрецию инсулина, или средства для профилактики и/или лечения диабета,
(6) способ профилактики и/или лечения диабета, который включает введение пациенту эффективного количества соединения, описанного в представленной формуле (I), или его фармацевтически приемлемой соли.
Эффект настоящего изобретения
Фармакологическая активность соединения по настоящему изобретению была подтверждена методами испытаний, описанными ниже.
Метод испытания 1: определение агонистического действия в отношении GPR40
i) Клонирование GPR40 человека
Полноразмерную последовательность GPR40 получали путем осуществления метода ПЦР в соответствии с процедурой, представленной ниже, с использованием геномной ДНК человека (Clontech) в качестве матрицы.
Олигонуклеотид, состоящий из нуклеотидной последовательности, представленной как SEQ ID NO:1, использовали в качестве прямого праймера, а олигонуклеотид состоящий из нуклеотидной последовательности, представленной как SEQ ID NO:2 - в качестве обратного праймера. В этой связи нуклеотидную последовательность, включающую область распознавания XbaI, добавляли к соответствующему 5'-концу указанных выше прямого праймера и обратного праймера. ПЦР осуществляли в присутствии 5% диметилсульфоксида (DMSO) с использованием Taq ДНК полимеразы (Ex Taq DNA polymerase; Takara Bio) путем повторения 30 раз цикла, включающего 94°C (15 секунд)/55°C (30 секунд)/72°C (1 минута). Как результат, был амплифицирован ДНК фрагмент около 0,9 т.п.н. Этот ДНК фрагмент расщепляли при помощи XbaI и затем вставляли в XbaI сайт плазмиды pEF-BOS-dhfr (Nucleic acids Research, 18, 5322, 1990) с получением, таким образом, плазмиды pEF-BOS-dhfr-GPR40.
Нуклеотидную последовательность GPR40 гена в pEF-BOS-dhfr-GPR40 определяли методом дидезокси-терминатора с использованием ДНК секвенатора (ABI 377 DNA Sequencer, Applied Biosystems). Нуклеотидная последовательность гена GPR40 представляла собой такую же, как нуклеотидная последовательность, представленная как SEQ ID NO:3. Нуклеотидная последовательность, представленная как SEQ ID NO:3,
имеет открытую рамку считывания (ORF) из 903 оснований, и аминокислотная последовательность, выведенная из этой ORF (300 аминокислот), представляла собой такую же, как аминокислотная последовательность, представленная как SEQ ID NO:4.
ii) Получение клеток со стабильной экспрессией GPR40
В качестве клеток, экспрессирующих белок GPR, использовали клетки CHO dhfr (дигидрофолатредуктаза(dhfr)-дефицитные CHO клетки). Также в качестве плазмиды, экспрессирующей белок GPR40, использовали плазмиду pEF-BOS-dhfr-GPR40, полученную на указанной выше стадии i). Клетки CHO dhfr вносили в среду αMEM, содержащую 10% фетальную телячью сыворотку (FCS), с использованием 6-луночного планшета (Asahi Techno Glass) и культивировали в течение ночи до 80-90% конфлюентности, а затем использовали 2 мкг на лунку плазмиды pEF-BOS-dhfr-GPR40 для переноса генов с использованием реагента для трансфекции (Lipofectamine 2000; Invitrogen). После 24 часов культивирования с момента переноса генов клетки разбавляли и снова вносили в среду. В этом случае среду αMEM, содержащую 10% FCS, заменяли на среду αMEM, которая содержала 10% FCS, но не содержала нуклеиновую кислоту. После 20 дней культивирования образованные таким образом колонии клеток отдельно извлекали и культивировали с получением CHO клеток, стабильно экспрессирующих GPR40. Из них отбирали клетки, обладающие высокой реакционной способностью в отношении внутренних лигандов - олеиновой кислоты и линоленовой кислоты.
iii) Определение агонистического действия в отношении GPR40
В этом испытании осуществляли измерения при помощи FLIPR (зарегистрированная торговая марка, Molecular Device) с использованием в качестве показателя изменения внутриклеточной концентрации кальция. Этот метод испытания описан ниже.
Штамм CHO клеток, в котором происходила экспрессия GPR40 человека, вносили в 384-луночный черный планшет (Becton Dickinson) при плотности 6×103 клеток на лунку и культивировали в течение ночи в CO2 инкубаторе.
С использованием набора для анализа Calcium-3 (Molecular Device) одну колбу с фосфоресцентным пигментом растворяли в 10 мл буфера HBSS-HEPES (pH 7.4, 1 × HBSS, 20 мМ HEPES, Invitrogen). 35,68 мг пробенецида (Sigma) растворяли в 250 мкл 1M NaOH и регулировали путем добавления 250 мкл буфера HBSS-HEPES. Раствор фосфоресцентного пигмента получали путем смешивания 16 мл буфера HBSS-HEPES, 640 мкл фосфоресцентного пигмента и 32 мкл пробенецида на один планшет. Среду сливали из планшета и раствор фосфоресцентного пигмента, распределяли по 40 мкл на лунку и затем инкубировали при комнатной температуре в течение 2 часов. Каждое испытываемое соединение растворяли в DMSO и затем разводили буфером HBSS-HEPES и распределяли по 10-мкл порциям в планшет, начиная таким образом реакцию, и изменения внутриклеточной концентрации кальция измеряли при помощи FLIPR. Значение EC50 для каждого испытываемого соединения рассчитывали при помощи кривой доза-ответ по изменениям интенсивности флуоресценции через 1 минуту измерений.
Результаты испытания представлены в таблице 1. Пример представляет собой номер примера соединения, описанного ниже.
Метод испытания 2: действие по стимуляции секреции инсулина с использованием клеток MIN6
В этом испытании исследовали действие испытываемых соединений по ускорению секреции инсулина с использованием штамма β-клеток поджелудочной железы мыши, клеток MIN6. Этот метод испытания описан ниже.
MIN6 клетки распределяли при плотности 5×104 клеток/лунка (200 мл) в 96-луночный планшет. В качестве среды использовали DMEM (25 мМ глюкозы), содержащую 10% FBS, 55 мкМ 2-меркаптоэтанола, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина. Среду сливали через 2 дня с использованием аспиратора с последующей однократной промывкой 200 мкл KRB-HEPES (116 мМ NaCl, 4,7 мМ KCl, 1,2 мМ KH2PO4, 1,2 мМ MgSO4, 0,25 мМ CaCl2, 25 мМ NaHCO3, 0,005% BSA без FFA, 24 мМ HEPES (pH 7,4)), содержащим 2,8 мМ глюкозы, который был нагрет до 37°C, а затем снова инкубировали при 37°C в течение 1 часа путем добавления 200 мкл этого же буфера. После того как указанный выше буфер сливали, используя аспиратор, и снова промывали буфером (200 мкл), предварительно определенную концентрацию испытываемого соединения добавляли к KRB-HEPES, содержащему 2,8 мМ или 22,4 мМ глюкозы, и добавляли в соответствующие лунки порциями по 100 мкл и инкубировали при 37°C в течение 2 часов. Указанные выше образцы разделяли на фракции и разбавляли 100-кратно, и концентрацию инсулина определяли с использованием набора инсулин RIA (Amersham RI). Активность показана в виде значения относительной активности (%) на момент 1 мкМ каждого соединения исходя из 100% контроля (DMSO).
Результаты испытания представлены в таблице 2. В результате было подтверждено, что соединение по настоящему изобретению обладает отличным действием, стимулирующим секрецию инсулина.
Метод испытания 3: испытание толерантности к глюкозе при однократном пероральном введении здоровым мышам
В этом испытании исследовали действие испытываемых соединений по подавлению глюкозы в крови после глюкозной нагрузки с использованием здоровых мышей. Этот метод испытания описан ниже.
Самцов мышей ICR (возраст 6 недель) после 1 недели предварительного выведения подвергали голоданию в течение ночи и использовали в качестве испытываемых животных. Каждое испытываемое соединение суспендировали в 0,5% метилцеллюлозе и перорально вводили при дозе 10 мг/кг за 30 минут до глюкозной нагрузки (2 г/кг). Введение 0,5% метилцеллюлозы использовали для контрольной группы. Снижение уровня глюкозы в крови (%) через 30 минут после глюкозной нагрузки рассчитывали на основании контрольной группы.
Результаты испытания представлены в таблице 3. В результате было подтверждено, что соединение по настоящему изобретению обладает отличным действием по снижению уровня глюкозы в крови.
Как результат описанных выше соответствующих испытаний, очевидно, что соединение по настоящему изобретению обладает отличным агонистическим действием в отношении GPR40 и поэтому является полезным в качестве средства, стимулирующего секрецию инсулина или средства для профилактики/лечения заболевания, где задействован GPR40, такого как диабет (инсулин-зависимый сахарный диабет (IDDM), инсулин-независимый сахарный диабет (NIDDM) и легкие формы диабета пограничного типа (аномальные толерантность к глюкозе и уровни глюкозы в крови натощак)) и т.п.
Лучший способ осуществления изобретения
Ниже представлено подробное описание настоящего изобретения.
В представленном описании “алкил” и “алкилен” означают линейные или разветвленные углеводородные цепи.
“Низший алкил”, предпочтительно, представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода (далее указана как C1-6), более предпочтительно C1-4 алкил и еще более предпочтительно метил и этил.
“Низший алкинил”, предпочтительно, представляет собой линейную или разветвленную C2-6 алкинильную группу, и иллюстративно можно указать этинил, пропинил, бутинил, пентинил, 1-метил-2-пропинил, 1,3-бутадинил, 1,3-пентадинил или т.п. Более предпочтительным является C2-4 алкинил и особенно предпочтительным является этинил или пропинил.
“Низший алкилен” означает двухвалентную группу (C1-6 алкилен), в которой один необязательный водород удален из указанного выше “низшего алкила” и, предпочтительно, представляет собой C1-4 алкилен, более предпочтительно метилен, этилен, триметилен, пропилен или диметилметилен и еще более предпочтительно метилен или этилен.
“Галоген” означает F, Cl, Br и I.
“Галогено-низший алкил”, предпочтительно, представляет собой C1-6 алкил, замещенный, по меньшей мере, одним галогеном, более предпочтителен галогено- C1-3 алкил, еще более предпочтителен фторметил, дифторметил, трифторметил, 1,1-дифторэтил, 2,2,2-трифторэтил или 3,3,3-трифторпропил, более предпочтительно трифторметил, 1,1-дифторэтил или 2,2,2-трифторэтил.
“Циклоалкил” представляет собой C3-10 насыщенную углеводородную кольцевую группу, которая может содержать мостик. Иллюстративно, можно указать циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, адамантил или т.п. Предпочтительным является C3-6 циклоалкил, циклопропил и еще более предпочтительным является циклопропил, циклобутил, циклопентил или циклогексил.
“Циклоалкенил” представляет собой C3-15 циклоалкенил, который может содержать мостик, и включает кольцевую группу, конденсированную с бензольным кольцом на участке двойной связи. Иллюстративно можно указать циклопентенил, циклопентадиенил, циклогексенил, циклогексадиенил, 1-тетрагидронафтил, 1-инденил, 9-флуоренил или т.п. Предпочтительным является C5-10 циклоалкенил, и более предпочтительным является циклопентенил, циклогексенил, 1-инденил или 1-тетрагидронафтил.
“Арил” представляет собой C6-14 ароматический углеводородный радикал, предпочтительно, фенил, нафтил или тетрагидронафтил, и более предпочтительным является фенил.
“Гетероарил” означает группу, содержащую кольцо, выбранное из i) моноциклического 5- или 6-членного ароматического гетерокольца, содержащего от 1 до 4 гетероатомов, выбранных из O, S и N, ii) бициклического гетерокольца, в котором гетерокольца, указанные в представленном выше i), являются конденсированными по кольцу, где конденсированные кольца могут быть одинаковые или отличные друг от друга, и iii) бициклического гетерокольца, в котором гетерокольцо, указанное в представленном выше i), является конденсированным с бензольным кольцом или 5-7-членным циклоалканом. В качестве примера кольца, составляющего указанную группу, можно указать, например, i) пиридин, пиразин, пиримидин, пиридазин, имидазол, пиррол, тиофен, фуран, триазин, триазол, тиазол, тиадиазол, оксадиазол, пиразол, изотиазол, оксазол, изоксазол, ii) нафтиридин, имидазопиридин, пирролoпиримидин, тиенопиридин, тиенопирролин, iii) хинолин, бензимидазол, бензофуран, бензотиофен, бензотиадиазол, бензотиазол, бензоизотиазол, бензоксазол, бензоизоксазол, хинолин, изохинолин, 5,6,7,8-тетрагидрохинолин, 5,6,7,8-тетрагидроизохинолин, хиназолин, хиноксалин, фталазин, индол, изоиндол, тетрагидробензимидазол, хроман и индазол. Кроме того, оксидо может быть образован через окисление атомов S или N кольца. Предпочтительным является указанное выше i) моноциклическое ароматическое гетерокольцо.
“Гетерокольцо” или “гетерокольцевая группа” означает группу, содержащую кольцо, выбранное из i) моноциклического 4-8-членного, предпочтительно из 5-7-членного, насыщенного, ненасыщенного или частично ненасыщенного гетерокольца, содержащего от 1 до 4 гетероатомов, выбранных из O, S и N, ii) бициклического гетерокольца, в котором гетерокольца, указанные в представленном выше i), являются конденсированными по кольцу, где конденсированные кольца могут быть одинаковые или отличные друг от друга, и iii) бициклического гетерокольца, в котором гетерокольцо, указанное в представленном выше i), является конденсированным с бензольным кольцом или 5-7-членным циклоалканом. В качестве примера кольца, составляющего указанную группу, можно указать, например, i) азетидин, пиперидин, пирролидин, пиперазин, азепан, диазепан, морфолин, тиоморфолин, диоксан, диоксолан, пиразолин, пиперидин, пиперазин, оксетан, тетрагидрофуран, дигидропиридин, пиридин, пиразин, пиримидин, пиридазин, имидазол, пиррол, тиофен, фуран, триазин, триазол, тиазол, тиадиазол, оксадиазол, пиразол, изотиазол, оксазол, изоксазол, ii) хинуклидин, нафтиридин, имидазопиридин, пирролoпиримидин, тиенопиридин, тиенопирролин, iii) дигидробензофуран, 1,2,3,4-тетрагидрохинолин, 1,2,3,4-тетрагидроизохинолин, дигидробензофуран, бензодиоксолан, индолин, индазолин, хинолин, бензимидазол, бензофуран, бензотиофен, бензотиадиазол, бензотиазол, бензоизотиазол, бензоксазол, бензоизоксазол, изохинолин, 5,6,7,8-тетрагидрохинолин, 5,6,7,8-тетрагидроизохинолин, хиназолин, хиноксалин, фталазин, индол, изоиндол, тетрагидробензимидазол, хроман и индазол. Кроме того, оксидо или диоксидо могут быть образованы через окисление атома S или N кольца. Предпочтительным является указанное выше i) моноциклическое гетерокольцо.
Термин “может быть замещен” означает “незамещенный” или “замещенный одинаковыми или отличными друг от друга 1-5 заместителями”. “Замещенный” означает “имеющий одинаковые или отличные друг от друга 1-5 заместителей”.
Предпочтительным в качестве приемлемого заместителя “фенила, который может быть замещен”, и “гетероарила, который может быть замещен” в R3, является группа, выбранная из группы G, представленной ниже.
Группа G: галоген, -CN, -R0, галогено-низший алкил, -ORz, -O-галогено-низший алкил, -N(Rz)CO-Rz, -CO2Rz, -CON(Rz)2, -CO-гетерокольцевая группа, -CON(Rz)-низший алкинил, -CON(Rz)-циклоалкил, -CON(Rz)-циклоалкенил, -CON(циклоалкил)(гетерокольцевая группа), -CON(Rz)-гетерокольцевая группа, -S-R0, -SO2-R0, -O-S(O)2-R0, -O-S(O)2-галогено-низший алкил, низший алкилен-ORz, низший алкилен-O-CORz, низший алкилен-N(Rz)2, низший алкилен-N(Rz)CO-Rz, низший алкилен-CORz, низший алкилен-CO2Rz, низший алкилен-CON(Rz)2, -O-низший алкилен-ORz, -O-низший алкилен-O-CORz, -O-низший алкилен-N(Rz)2, -O-низший алкилен-N(Rz)CO-Rz, -O-низший алкилен-N(Rz)CO2-R0, -O-низший алкилен-CO-Rz, -O-низший алкилен-CO2-Rz, -O-низший алкилен-CON(Rz)2, -O-низший алкилен-CON(Rz)-(низший алкил, который может быть замещен группой -ORz), -O-низший алкилен-SR0, -O-низший алкилен-циклоалкил, -O-низший алкилен-CON(Rz)-циклоалкил, -O-гетерокольцевая группа, -O-низший алкилен-гетерокольцевая группа, -O-низший алкилен-CO-гетерокольцевая группа, -O-низший алкилен-CON(Rz)-гетерокольцевая группа, -N(Rz)CO-низший алкилен-ORz, -CON(Rz)-галогено-низший алкил, -CON(Rz)-(низший алкил, замещенный группой -ORz), -CON(Rz)-низший алкилен-CN, -CON(Rz)-низший алкилен-O-низший алкилен-ORz, -CON(низший алкилен-ORz)2, -CON(Rz)-низший алкилен-O-CORz, -CON(Rz)-низший алкилен-N(Rz)2, -CON(Rz)-низший алкилен-N(Rz)CO-Rz, -CON(Rz)-низший алкилен-CORz, -CON(Rz)-низший алкилен-CO2Rz, -CON(Rz)-низший алкилен-CON(Rz)2, -CON(Rz)-низший алкилен-SO2Rz, -CON(Rz)-низший алкилен-циклоалкил, -CON(Rz)-низший алкилен-O-циклоалкил, -CON(Rz)-низший алкилен-арил, -CON(Rz)-(низший алкилен, замещенный группой -N(Rz)2)-арил, -CON(Rz)-низший алкилен-O-арил, -CON(Rz)-низший алкилен-N(Rz)-арил, -CON(Rz)-низший алкилен-CO-арил, -CON(низший алкилен-ORz)-низший алкилен-арил, -CON(Rz)-низший алкилен-гетерокольцевая группа, -CON(Rz)-низший алкилен-O-гетерокольцевая группа, -CON(Rz)-низший алкилен-N(Rz)-гетерокольцевая группа, -CON(Rz)-низший алкилен-CO-гетерокольцевая группа, -CON(низший алкилен-ORz)-низший алкилен-гетерокольцевая группа, -CON(низший алкилен-CN)-низший алкилен-гетерокольцевая группа и -CON(низший алкилен-гетерокольцевая группа)2.
В этом отношении в группе G низший алкилен может быть замещен галогеном или -ORz, и циклоалкильная, циклоалкенильная, арильная и гетерокольцевая группа могут быть замещены группой, выбранной из представленной ниже группы G1.
Группа G1: галоген, циано, -R0, галогено-низший алкил, -ORz, -O-галогено-низший алкил, -N(Rz)2, -S-R0, -SO2-R0, -SO2N(Rz)2, -CO-Rz, -CON(Rz)2, -CON(Rz)-низший алкилен-ORz, -N(Rz)CO-Rz, оксо, низший алкилен-CN, низший алкилен-ORz, -арил, -(низший алкилен, который может быть замещен группой -ORz)-арил, низший алкилен-O-арил, гетерокольцевая группа и низший алкилен-гетерокольцевая группа.
В этом отношении арил и гетерокольцевая группа в группе G1 могут быть замещены группой, выбранной из представленной ниже группы G2.
Группа G2: галоген, циано, галогено-низший алкил, -ORz, -O-галогено-низший алкил и оксо.
Предпочтительной в качестве приемлемого заместителя для “фенила, который может быть замещен”, и “гетероарила, который может быть замещен” в R3, более предпочтительно, является группа из представленной ниже группы G3.
Группа G3: галоген, -R0, галогено-низший алкил, -ORz, -CON(Rz)2, -CON(Rz)-гетерокольцевая группа, -O-S(O)2-R0, -O-низший алкилен-ORz, -O-низший алкилен-O-CORz, -O-низший алкилен-N(Rz)2, -O-низший алкилен-N(Rz)CO-Rz, -O-низший алкилен-CO2R0, -O-низший алкилен-CON(Rz)2, -O-низший алкилен-CON(Rz)-(низший алкил замещен группой -ORz), -O-низший алкилен-SR0, -O-низший алкилен-циклоалкил, -O-низший алкилен-CON(Rz)-циклоалкил, -O-низший алкилен-гетерокольцевая группа и -O-низший алкилен-CON(Rz)-гетерокольцевая группа.
В этом отношении низший алкилен в группе G3 может быть замещен галогеном или -ORz, и циклоалкил, и гетерокольцевая группа могут быть замещены группой, выбранной из указанной выше группы G1.
Предпочтительной в качестве приемлемого заместителя “фенила, который может быть замещен”, и “гетероарила, который может быть замещен” в R3, еще более предпочтительно, является группа, выбранная из галогена, -R0 , -ORz, -O-галогено-низшего алкила, группы -O-низший алкилен-ORz, группы -O-низший алкилен-CON(Rz)2 и группы -O-низший алкилен-(циклоалкил, который может быть замещен группой -ORz).
Предпочтительным в качестве приемлемого заместителя для “фенила, который может быть замещен”, и “гетероарила, который может быть замещен” в R3, более предпочтительно, является -O-низший алкилен-ORz, O-низший алкилен-CON(Rz)2 или -O-низший алкилен-(циклоалкил, который может быть замещен группой -ORz).
Предпочтительный вариант воплощения настоящего изобретения представлен ниже.
(a) Предпочтительным в качестве R1 является -H, -галоген или -R0, более предпочтительно -H.
(b) Предпочтительным в качестве R2 является -галоген, -O-R0 или -R0, более предпочтительно -галоген или -R0.
(c) Предпочтительным в качестве n является 0 или 1.
(d) Предпочтительным в качестве R3 является -X-(фенил, который может быть замещен) или -X-(гетероарил, который может быть замещен), более предпочтительно - фенил или пиридил, который, соответственно, может быть замещен, еще более предпочтительно - фенил, который может быть замещен, еще более предпочтительно - фенил, который может быть замещен группой, выбранной из указанной выше группы G3, особенно предпочтительно - фенил, который замещен группой, выбранной из класса, включающего -O-низший алкилен-ORz, -O-низший алкилен-CON(Rz)2 и -O-низший алкилен-(циклоалкил, который может быть замещен группой -ORz), и может быть дополнительно замещен R0, галогеном или -OR0.
(e) Предпочтительным в качестве R4 является -H.
(f) Предпочтительным в качестве кольца A является бензольное кольцо, пиридиновое кольцо или тиофеновое кольцо, более предпочтительно бензольное кольцо.
(g) Предпочтительным в качестве кольца B является бензольное кольцо.
(h) Предпочтительным в качестве L является *-низший алкилен-O- или *-низший алкилен-NH-, более предпочтительно *-CH2-O- или *-CH2-NH- (где * означает связывание с кольцом A). Кроме того, в качестве положения замещения L в кольце B 4-положение относительно -CH(R4)-(3,5-диоксо-1,2,4-оксадиазолин-2-ил) является предпочтительным.
В качестве другого предпочтительного варианта воплощения соединение, содержащее комбинацию предпочтительных групп, описанных в представленных выше п.(a)-(h), является предпочтительным.
Также еще один предпочтительный вариант воплощения соединения по настоящему изобретению, представленного общей формулой (I), представлен ниже.
(1) Соединение, описанное общей формулой (I), где положение замещения L в кольце B представляет собой 4-положение.
(2) Соединение, описанное в п.(1), где кольцо A представляет собой бензольное кольцо.
(3) Соединение по п.(2), где R3 представляет собой фенил или пиридил, который может быть соответственно замещен.
(4) Соединение, описанное в п.(3), где L представляет собой *-CH2-O- или *-CH2-NH- (где * означает связывание с кольцом A).
(5) Соединение по п.(4), где R4 представляет собой -H.
(6) Соединение, описанное в п.(5), где R1 представляет собой -H, галоген или R0.
(7) Соединение, описанное в п.(6), где n имеет значение 0, или R2 представляет собой галоген или R0.
(8) Соединение по п.(7), где R3 представляет собой фенил, который замещен группой, выбранной из класса, включающего -O-низший алкилен-ORz, -O-низший алкилен-CON(Rz)2 и -O-низший алкилен-(циклоалкил, который может быть замещен группой -ORz), и может быть дополнительно замещен 1 или 2 группами низший алкил, галоген или -OR0.
(9) Соединение, описанное формулой (I), которое выбрано из группы, включающей
2-{[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,6-диметилбифенил-4-ил]окси}-N-метилацетамид,
2-(4-{[4'-(2-гидроксиэтокси)-2'-метилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-(4-{[4'-(3-гидрокси-3-метилбутокси)-2',6'-диметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-(4-{[4'-(3-гидрокси-3-метилбутокси)-2,2'-диметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-(4-{[4'-(3-гидрокси-3-метилбутокси)-2,2',6'-триметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-{4-[(4'-{[(3R)-3-гидроксибутил]окси}-2,2'-диметилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-дион,
2-{4-[(4'-{[(3S)-3-гидроксибутил]окси}-2,2'-диметилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-дион,
2-[4-({[4'-(3-гидрокси-3-метилбутокси)-2,2'-диметилбифенил-3-ил]метил}амино)бензил]-1,2,4-оксадиазолидин-3,5-дион,
2-(4-{[4'-(3-гидрокси-3-метилбутокси)-2'-метокси-2-метилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-{4-[(4'-{[(3R)-3-гидроксибутил]окси}-2,2'6'-триметилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-дион,
2-{4-[(4'-{[(3S)-3-гидроксибутил]окси}-2,2'6'-триметилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-дион,
2-[(6-{[4'-(3-гидрокси-3-метилбутокси)-2,2',6'-триметилбифенил-3-ил]метокси}пиридин-3-ил)метокси]-1,2,4-оксадиазолидин-3,5-дион и
2-[4-({4'-[2-(1-гидроксициклопропил)этокси]-2,2',6'-триметилбифенил-3-ил}метокси)бензил]-1,2,4-оксадиазолидин-3,5-дион,
или его фармацевтически приемлемая соль.
Существуют случаи, когда соединение по настоящему изобретению, представленное формулой (I), образует соль, и такая соль включена в соединение по настоящему изобретению при условии, что она представляет собой фармацевтически приемлемую соль. Иллюстративно можно указать в качестве примера кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, аспарагиновая кислота, глутаминовая кислота и т.п., соли с неорганическими основаниями, которые содержат металлы (например, натрий, калий, кальций, магний и т.п.), или с органическими основаниями, такими как метиламин, этиламин, этаноламин, лизин, орнитин и т.п., соли аммония и т.п.
Кроме того, соединение по настоящему изобретению в некоторых случаях может содержать асимметрический атом углерода, в зависимости от типа заместителей, и на основании этого могут присутствовать оптические изомеры. Настоящее изобретение включает все смеси и выделенные формы этих оптических изомеров. Также в некоторых случаях могут присутствовать таутомеры в соединении по настоящему изобретению, и настоящее изобретение включает разделенные формы этих изомеров или их смеси. Кроме того, настоящее изобретение также включает меченое вещество, а именно соединение, в котором, по меньшей мере, один атом соединения по настоящему изобретению замещен радиоизотопом или нерадиоактивным изотопом.
Кроме того, различные типы гидратов и сольватов и полиморфизм соединения по настоящему изобретению также включены в настоящее изобретение. В этой связи должно быть понятно, что соединение по настоящему изобретению не ограничивается соединениями, описанными в примерах, которые представлены ниже, и все соединения, представленные формулой (I), и их фармацевтически приемлемые соли включены в настоящее изобретение.
В этой связи все соединения, которые преобразуются в соединения по настоящему изобретению в живом организме, так называемые пролекарствами, также включены в соединение по настоящему изобретению. В качестве групп, которые образуют пролекарства соединений по настоящему изобретению, можно указать в качестве примера группы, описанные в “Progress in Medicine”, Lifescience Medica, 1985, vol.5, pр.2157-2161, и группы, описанные в “Iyakuhin no Kaihatsu (Development of Medicines)”, vol.7 Bunshi Sekkei (Molecular Design), pp.163-198, опубликован Hirokawa Shoten в 1990.
(Способы получения)
Соединение по настоящему изобретению и его фармацевтически приемлемую соль можно получить с использованием различных традиционно известных способов синтеза, используя характеристики, основанные на его основном скелете или типе заместителей. Типичные способы получения приведены в качестве примера ниже. В этой связи, в зависимости от типа функциональной группы, существует эффективный способ, с точки зрения технологии производства, замещения указанной функциональной группы соответствующей защитной группой, а именно группой, которая может быть легко преобразована в указанную функциональную группу, на стадии исходного вещества до промежуточного соединения. Затем желаемое соединение может быть получено путем удаления защитной группы, как это будет необходимо в каждом конкретном случае. В качестве такой функциональной группы можно указать гидроксильную группу, карбоксильную группу, аминогруппу и т.п., а в качестве их защитных групп - защитные группы, описанные, например, в “Protective Groups in Organic Synthesis” (USA) third edition, edited by Greene and Wuts, John Wiley & Sons, 1999, которые, необязательно, можно использовать в соответствии с реакционными условиями.
Способ получения 1: реакция циклизации

(В представленной формуле Lv представляет собой удаляемую группу. То же применимо к следующему далее описанию.)
Этот способ получения представляет собой способ, в котором соединение (I) по настоящему изобретению получают путем реакции циклизации соединения (1) и соединения (2). В качестве удаляемой группы Lv предпочтительной группой является галоген (например, хлор, бром или т.п.) или алкоксигруппа (например, метокси, этокси или т.п.).
Реакцию можно осуществить с использованием соединения (1) и соединения (2) в эквивалентных количествах или с использованием одного из них в избыточном количестве при охлаждении, при комнатной температуре или при нагревании, в растворителе, таком как простые эфиры (например, диэтиловый эфир, тетрагидрофуран (ТГФ), диоксан, диметоксиэтан (DME) или т.п.), галогенированные углеводороды (например, дихлорметан, 1,2-дихлорэтан, хлороформ или т.п.), ароматические углеводороды (например, бензол, толуол, ксилол или т.п.) или т.п.
Когда соединение (1) содержит гидроксильную группу, отличную от гидроксиаминогруппы, эту гидроксильную группу в некоторых случаях подвергают карбамоилированию. Удаление карбамоильной группы можно осуществить способом, обычно используемым специалистами в данной области для декарбамоилирования. Например, это можно осуществить в растворителе, таком как спирты (например, метанол, этанол или т.п.), вода или т.п., при охлаждении, при комнатной температуре или при нагревании с использованием основания, такого как метоксид натрия, натрия этоксид, гидроксид натрия или т.п.
Способ получения 2: реакция сочетания

(В представленной формуле любой один из Lv1 и Lv2 представляет собой галоген или трифторметилсульфонилоксигруппу, а другой представляет собой -B(OH)2, -B(OR00)2 или -SnR0 3, Ar представляет собой фенил или гетероарил, который может быть соответственно замещен, и R00 представляет собой низший алкил или два R00 вместе образуют низший алкилен. То же применимо к следующему далее описанию).
Этот способ получения представляет собой способ, в котором соединение (I-a) по настоящему изобретению получают путем реакции сочетания соединения (3) и соединения (4).
Реакцию можно осуществить с использованием палладиевого комплекса, такого как тетракистрифенилфосфинпалладий, ацетата палладия или т.п. в качестве катализатора и с использованием соединения (3) и соединения (4) в эквивалентных количествах или с использованием одного из них в избыточном количестве при охлаждении, при комнатной температуре или при нагревании, в растворителе, таком как простые эфиры, спирты, галогенированные углеводороды, ароматические углеводороды, вода или т.п. Кроме того, в некоторых случаях для обеспечения ровного протекания реакции выгодно осуществление реакции в присутствии основания, такого как карбонат натрия, карбонат цезия, трет-бутоксид натрия или т.п., или литиевой соли, такой как хлорид лития, бромид лития или т.п.
Способ получения 3: восстановительное аминирование
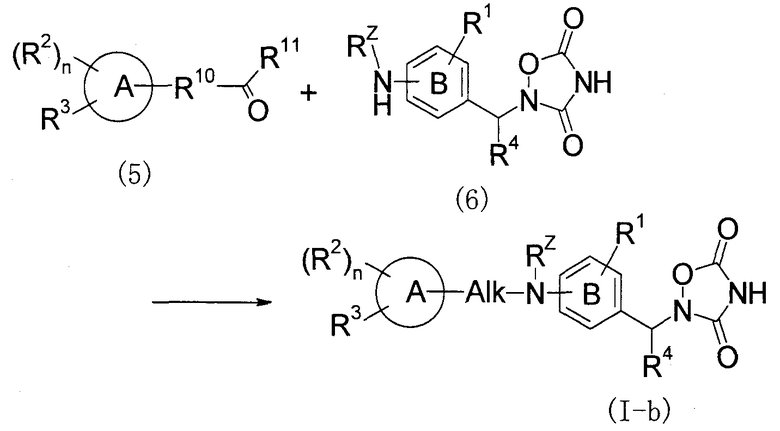
(В представленной формуле Alk представляет собой низший алкилен, и R10 представляет собой связь или C1-5 алкилен, и R11 представляет собой H или C1-5 алкил. Однако количество атомов углерода в R10 и R11 в целом составляет от 0 до 5. То же применимо к следующему далее описанию.)
Этот способ получения представляет собой способ, в котором соединение (I-b) по настоящему изобретению получают, подвергая соединение (5) и соединение (6) восстановительному аминированию.
Реакцию осуществляют с использованием соединения (5) и соединения (6) в эквивалентных количествах или с использованием одного из них в избыточном количестве и при перемешивании в присутствии восстановителя и в реакционно-инертном растворителе при температуре от -45°C до температуры кипения с обратным холодильником, предпочтительно при температуре от 0°C до комнатной температуры, обычно в течение времени от 0,1 часа до 5 дней. В качестве растворителя в этом случае, например, можно указать спирты, простые эфиры или их смеси. В качестве восстановителя можно указать цианоборгидрид натрия, триацетоксиборгидрид натрия, боргидрид натрия и т.п. В некоторых случаях предпочтительно осуществление реакции в присутствии дегидрирующего агента, такого как молекулярные сита или т.п., или кислоты, такой как уксусная кислота, хлористоводородная кислота, комплекс изопропоксида титана(IV) или т.п. В зависимости от реакции, когда иминовое соединение, которое образуется в реакционной системе в качестве промежуточного соединения, можно выделять стабильным образом, реакцию восстановления можно осуществлять отдельно после получения указанного иминового соединения.
Способ получения 4: амидирование

Этот способ получения представляет собой способ, в котором соединение (I-c) по настоящему изобретению получают, подвергая соединение (7) и соединение (6) амидированию.
Вместо карбоновокислотного соединения (7) также можно использовать его реакционно-способное производное. Реакцию можно осуществить с использованием карбоновокислотного соединения (7) или его реакционно-способного производного и аминосоединения (6) в эквивалентных количествах или с использованием одного из них в избыточном количестве при охлаждении, при комнатной температуре или при нагревании, в растворителе, таком как ароматические углеводороды, галогенированные углеводороды, простые эфиры, N,N-диметилформамид (ДМФА), N,N-диметилацетамид (DMA), 1-метилпирролидин-2-он (NMP), диметилсульфоксид (DMSO), этилацетат, пиридин, ацетонитрил или т.п.
Когда используют карбоновокислотное соединение (7), предпочтительно использовать N,N'-дициклогексилкарбодиимид (DCC), PS-карбодиимид (Argonaut, USA), 1-[3-(диметиламино)пропил]-3-этилкарбодиимид (WSC), 1,1'-карбонилбисимидазол (CDI), N,N'-дисукцинимидилкарбонат, Bop реагент (Aldrich, USA), 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетрафторборат тетраметилурония (TBTU), гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU), азид дифенилфосфорной кислоты (DPPA), 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорид (DMT-MM) или т.п. в качестве агента конденсации и, в некоторых случаях дополнительно использовать 1-гидроксибензотриазол ((HOBt), N-гидроксисукцинимид (HONSu), 1-гидрокси-7-азабензотриазол ((HOAt) или т.п. в качестве добавочного агента.
В качестве реакционно-способного производного карбоновокислотного соединения (7) можно указать в качестве примера галогенангидрид кислоты (хлорангидрид кислоты, бромангидрид кислоты или т.п.), ангидрид кислоты (смешанный ангидрид кислоты, полученный путем взаимодействия с этилхлоркарбонатом, бензилхлоркарбонатом, фенилхлоркарбонатом, п-толуолсульфоновой кислотой, изовалериановой кислотой и т.п., или симметрический ангидрид кислоты), активный сложный эфир (сложный эфир, полученный с использованием фенола, HOBt, HONSu или т.п., который может быть замещен электрон-акцепторной группой, такой как нитрогруппа, атом фтора или т.п.), низший алкиловый эфир, азид кислоты и т.п. Эти реакционно-способные производные можно получить общими способами.
В зависимости от типа реакции для обеспечения ровного протекания реакции иногда выгодно осуществление реакции в присутствии основания, такого как триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин (DMAP) или т.п.
Способ получения 5: другие способы получения
Кроме того, некоторые соединения, представленные формулой (I), также могут быть получены из соединений по настоящему изобретению, полученных, как указано выше, при необязательном сочетании традиционно известных способов амидирования, окисления, гидролиза и т.п., которые обычно могут использовать специалисты в данной области. Например, можно использовать следующие реакции.
5-1: амидирование
Амидирование можно осуществить таким же способом, как в способе получения 4.
5-2: окисление
Сульфоксидное соединение или сульфоновое соединение можно получить путем окисления S атома сульфидного соединения различными агентами окисления. Реакцию можно осуществить, например, при охлаждении, при комнатной температуре или при нагревании с использованием эквивалентного количества или избыточного количества м-хлорпербензойной кислоты, перуксусной кислоты, водного раствора пероксида водорода, реагента Dess-Martin (1,1,1-триацетокси-1,1-дигидро-1,2-бензоиодоксол-3(1H)-он) или т.п. в качестве окислителя, в растворителе, таком как галогенированные углеводороды, уксусная кислота, вода или т.п.
5-3: гидролиз
Соединение, содержащее карбоксильную группу, можно получить путем гидролиза соединения, содержащего сложноэфирную группу. Например, это можно осуществить при температуре от охлаждения до нагревания в реакционно-инертном растворителе, таком как ароматические углеводороды, простые эфиры, галогенированные углеводороды, спирты, ДМФА, DMA, NMP, DMSO, пиридин, вода или т.п., в присутствии минеральной кислоты, такой как серная кислота, хлористоводородная кислота, бромистоводородная кислота или т.п., или органической кислоты, такой как муравьиная кислота, уксусная кислота или т.п., или т.п.; или в присутствии основания, такого как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия, карбонат цезия, аммиак или т.п.
(Способы получения исходных соединений)
Исходные вещества для использования при получении соединений по настоящему изобретению можно получить, например, с использованием нижеследующих способов, способов, описанных в примерах получения, которые описаны далее, традиционно известных способов или способов, очевидных для специалистов в данной области, или способов, представляющих собой модификацию таких способов.
Синтез исходных веществ
Синтез исходных веществ 1: O-алкилирование
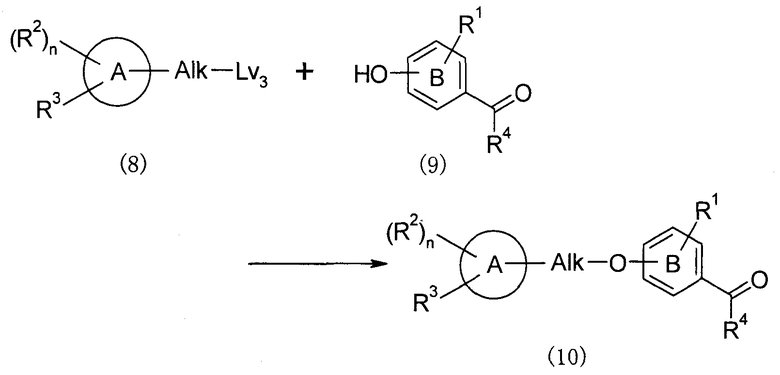
(В представленной формуле Lv3 представляет собой -OH или удаляемую группу, такую как галоген, метансульфонилокси, п-толуолсульфонилокси или т.п. То же применимо к следующему далее описанию.)
Этот способ получения представляет собой способ, в котором соединение (10) получают путем O-алкилирования соединения (8) при помощи соединения (9).
Когда используют соединение (8), в котором Lv3 представляет собой -OH, синтез можно осуществить с использованием общего способа реакции Мицунобу, который обычно используют специалисты в данной области. Например, его можно осуществить с использованием активирующего агента, полученного из соединения фосфора (например, трибутилфосфина, трифенилфосфина или т.п.) и азодикарбонильного соединения (например, диэтилазодикарбоксилата, 1,1'-(азодикарбонил)дипиперидина или т.п.), или с использованием цианометилентрибутилфосфоранового или т.п. реагента в растворителе, таком как галогенированные углеводороды, простые эфиры, ароматические углеводороды или т.п. при охлаждении, при комнатной температуре или при нагревании.
Когда используют соединение (8), в котором Lv3 представляет собой удаляемую группу, такую как галоген, метансульфонилокси, п-толуолсульфонилокси или т.п., синтез, например, можно осуществить с использованием соединения (8) и соединения (9) в эквивалентных количествах или с использованием одного из них в избыточном количестве в присутствии основания, такого как карбонат калия, карбонат цезия, метоксид натрия, гидрид натрия или т.п., в растворителе, таком как галогенированные углеводороды, простые эфиры, ароматические углеводороды или т.п., ДМФА или т.п., при охлаждении, при комнатной температуре или при нагревании.
Синтез исходных веществ 2

Первая стадия: образование оксима
Это стадия, на которой соединение (12) получают, подвергая соединение (11) реакции образования оксима.
Что касается образования оксима, можно использовать способ образования оксима, обычно используемый специалистами в данной области. Например, это можно осуществить с использованием соединения (11) и гидроксиламина или его соли в эквивалентных количествах или с использованием одного из них в избыточном количестве в растворителе, таком как спирты, уксусная кислота, пиридин, вода или т.п., при охлаждении, при комнатной температуре или при нагревании. В зависимости от типа соединения в некоторых случаях для обеспечения развития реакции выгодно добавление ацетата натрия, п-толуолсульфоновой кислоты или т.п.
Вторая стадия: восстановление
Это - стадия, на которой соединение (1) получают путем восстановления соединения (12).
Что касается реакции восстановления оксима, можно использовать способ восстановления оксима, обычно используемый специалистами в данной области. Например, это можно осуществить с использованием соединения (12) и восстановителя, такого как комплекс боран-пиридин, цианоборгидрид натрия или т.п., в эквивалентных количествах или с использованием одного из них в избыточном количестве в растворителе, таком как простые эфиры, спирты, ароматические углеводороды, уксусная кислота или т.п., при охлаждении, при комнатной температуре или при нагревании.
Соединение по настоящему изобретению, полученное таким способом, выделяют и очищают непосредственно как таковое или в виде соли такого соединения, используя для этого обычную обработку для образования соли. Выделение и очистку осуществляют с использованием общих химических процедур, таких как экстракция, концентрирование, выпаривание, кристаллизация, фильтрование, перекристаллизация, различные типы хроматографии и т.п.
Различные типы изомеров можно выделять обычным способом с использованием различий в физико-химических свойствах между изомерами. Например, рацемическую смесь можно преобразовать в оптически чистый изомер при помощи общего способа разделения рацемических смесей, такого как, например, способ, в котором такие соединения преобразовывают в диастереомерные соли с использованием оптически активных кислот, таких как винная кислота или т.п., и затем подвергают оптическому разделению. Также диастереомерную смесь можно разделить, например, при помощи фракционированной кристаллизации или различных типов хроматографии. Кроме того, оптически активное соединение также может быть получено с использованием соответствующего оптически активного соединения в качестве исходного вещества.
Фармацевтическую композицию, которая содержит одно или несколько соединений по настоящему изобретению или их фармацевтически приемлемых солей в качестве активного ингредиента, получают в виде таблеток, порошков, тонкоизмельченных веществ, гранул, капсул, пилюль, растворов, препаратов для инъекций, суппозиториев, мазей, пластырей и т.п. с использованием носителей, наполнителей и других добавочных веществ, обычно используемых для получения фармацевтических препаратов, и вводят перорально или парентерально.
Клиническую дозу соединения по настоящему изобретению для человека, необязательно, определяют с учетом симптомов, возраста, пола и т.п. каждого пациента, но в случае перорального введения суточная доза обычно составляет от около 0,0001 до 50 мг/кг, предпочтительно от около 0,001 до 10 мг/кг, еще более предпочтительно от 0,01 до 1 мг/кг, и ее вводят в виде одной дозы или разделенной на 2-4 части. В случае внутривенного введения суточная доза в расчете на массу тела составляет от около 0,0001 до 1 мг/кг, предпочтительно от около 0,0001 до 0,1 мг/кг, и ее вводят в виде одной дозы или разделенной на два или несколько введений в день. Поскольку доза варьирует в зависимости от различных условий, в некоторых случаях удовлетворительный эффект получают при использовании более низких количеств, чем указанные выше пределы доз.
В качестве твердой композиции для перорального введения согласно настоящему изобретению используют таблетки, порошки, гранулы и т.п. В таких твердых композициях одно или несколько активных веществ смешивают с, по меньшей мере, одним инертным разбавителем, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон, алюминий-магниевый силикат или т.п. Обычно композиция может содержать другие добавки, помимо инертного разбавителя, такие как смазывающие вещества (например, стеарат магния или т.п.), разрыхлитель (например, гликолят кальцийцеллюлозы или т.п.), стабилизатор, солюбилизирующее вещество и т.п. Если необходимо, на таблетки или пилюли может быть нанесено покрытие, такое как сахарное покрытие или пленочное покрытие из гастро- или энтеросолюбильного вещества, такого как сахароза, желатин, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы или т.п.
Жидкая композиция для перорального введения включает фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и т.п. и содержит обычно используемый инертный разбавитель, такой как дистиллированная вода или этанол (EtOH). Помимо инертного разбавителя, такая композиция может содержать смачивающее вещество, суспендирующее вещество и подобные вспомогательные вещества, а также подсластители, отдушки, ароматизаторы и антисептики.
В качестве препаратов для инъекций для парентерального введения включены в изобретение асептические водные или неводные растворы, суспензии и эмульсии. В качестве водных растворов и суспензий, например, включены дистиллированная вода для инъекций и физиологический раствор. В качестве неводных растворов и суспензий, например, используют пропиленгликоль, полиэтиленгликоль, растительное масло (например, оливковое масло или т.п.), спирты (например, EtOH или т.п.), полисорбат 80 и т.п. Такая композиция может дополнительно содержать вспомогательные вещества, такие как антисептик, смачивающее вещество, эмульгатор, диспергирующее вещество, стабилизатор, солюбилизирующее вещество или т.п. Их стерилизуют, например, путем фильтрования через удерживающий бактерии фильтр, смешивания с бактерицидным веществом или облучения. Их также можно использовать путем получения твердых композиций и растворения их в стерильной воде или стерильном растворителе для инъекций перед применением.
В качестве наружных препаратов включены мази, твердые кремы, кремы, желе, припарки, спреи, лосьоны, глазные капли, глазные мази и т.п. Такие препараты включают обычно используемые основу для мази, основу для лосьона, водные или неводные растворы, суспензии, эмульсии и т.п. Например, в качестве основы для мази или лосьона можно указать в качестве примера полиэтиленгликоль, пропиленгликоль, вазелин, белый пчелиный воск, полиоксиэтилен-гидрогенированное касторовое масло, глицеринмоностеарат, стеариловый спирт, цетиловый спирт, лауромакроголь, сорбитансесквиолеат и т.п.
Препараты для ингаляций, препараты для введения через слизистую, такие как препараты для введения через нос и т.п., используют в твердой, жидкой или полутвердой форме и их можно получить в соответствии с традиционно известными способами. Например, можно добавить, но необязательно, традиционно известный наполнитель и, кроме того, агент, регулирующий pH, антисептик, поверхностно-активное вещество, лубрикант, стабилизатор, загуститель и т.п. Для введения можно использовать соответствующее устройство для ингаляции или вдувания. Например, с использованием мерного устройства для введения путем ингаляции или подобного традиционно известного устройства или спреевого устройства соединение можно вводить отдельно, или в виде порошкообразной сформулированной смеси, или в виде раствора или суспензии путем комбинирования с приемлемым, с медицинской точки зрения, носителем. Ингалятор сухого порошка или т.п. может быть предназначен для разового или многократного применения, и можно использовать порошок или содержащую порошок капсулу. Альтернативно, это может быть находящийся под давлением аэрозольный спрей или подобная форма, в которой используют хлорфторалкан, гидрофторалкан или диоксид углерода или подобный подходящий газ.
ПРИМЕРЫ
Ниже следует иллюстративное описание настоящего изобретения на основе примеров, но настоящее изобретение не ограничивается этими примерами. В этой связи, поскольку новые вещества включены в исходные соединения для использования в этих примерах, способы получения таких исходных соединений описаны как примеры получения.
В связи с этим используют следующие аббревиатуры в примерах и таблицах. Пр.пол.: номер примера получения, Пр.: номер примера, №: номер соединения, Стр.: структурная формула (Когда в структурной формуле присутствует HCl, это означает, что соединение представляет собой гидрохлорид.), Синт.: способ получения (В случае, когда указаны только цифры, они показывают номер примера, в котором соединение получают таким же образом, и когда перед цифрой присутствует R, это указывает номер примера получения, в котором соединение получают таким же образом соответственно). Данные: физико-химические данные (ЯМР1: δ (м.д.) - данные 1H ЯМР в DMSO-d6, ЯМР2: δ (м.д.) -данные 1H ЯМР в CDCl3, FAB: FAB-MS (катион), FAB-N; FAB-MS (анион), ESI: ESI-MS (катион), ESI-N: ESI-MS (анион), EI: EI-MS (катион), CI: CI-MS (катион)), Me: метил, Et: этил, Ac: ацетил, TBS: трет-бутилдиметилсилил, Boc: трет-бутоксикарбонил, Ts: п-толуолсульфонил.
Пример получения 1
Путем добавления тионилхлорида и ДМФА к 1-(2,6-диметилфенил)-6-оксо-1,6-дигидропирролидин-3-карбоновой кислоте и при перемешивании реакционной смеси при 60°C в течение 2 часов получали 1-(2,6-диметилфенил)-6-оксо-1,6-дигидропирролидин-3-карбонилхлорид. Полученный 1-(2,6-диметилфенил)-6-оксо-1,6-дигидропирролидин-3-карбонилхлорид растворяли в ДМФА и добавляли боргидрид натрия при охлаждении льдом с последующим перемешиванием в течение 0,5 часов с получением 1-(2,6-диметилфенил)-5-(гидроксиметил)пиридин-2(1H)она.
Пример получения 2
В атмосфере азота раствор 1,0 M диизобутилалюминийгидрида в ТГФ добавляли по каплям при -78°C к раствору в ТГФ метил 1-(2,6-диметилфенил)-6-оксо-1,6-дигидропиридин-3-карбоксилата. После перемешивания при -78°C в течение 2 часов температуру повышали до 0°C с последующим перемешиванием при 0°C в течение 1,5 часов. Реакционную смесь нагревали до комнатной температуры с последующим перемешиванием при комнатной температуре в течение 2 часов. Раствор 1,0 M диизобутилалюминийгидрида в ТГФ добавляли по каплям к реакционной смеси при 0°C с последующим нагреванием до комнатной температуры и перемешиванием в течение 1 часа с получением метил 1-(2,6-диметилфенил)-6-оксо-1,4,5,6-тетрагидропиридин-3-карбоксилата. В атмосфере азота литийалюминийгидрид добавляли к полученному метил 1-(2,6-диметилфенил)-6-оксо-1,4,5,6-тетрагидропиридин-3-карбоксилату при одновременном охлаждении на бане лед-метанол. Затем при перемешивании реакционной смеси в течение 2 часов при одновременном нагревании до температуры кипения с обратным холодильником получали [1-(2,6-диметилфенил)пиперидин-3-ил]метанол.
Пример получения 3
Путем добавления трет-бутил(диметил)силилхлорида к раствору в ДМФА 4'-гидрокси-2',6'-диметилбифенил-3-карбальдегида и имидазола и при перемешивании при комнатной температуре в течение 10 часов получали 4'-{[трет-бутил(диметил)силил]окси}-2',6'-диметилбифенил-3-карбальдегид.
Пример получения 4
В атмосфере азота н-бутиллитий (раствор в гексане) добавляли при -75°C к раствору в ТГФ (4-бром-3-метоксифенокси)(трет-бутил)диметилсилана с последующим перемешиванием при -75°C в течение 1 часа. К реакционной смеси добавляли триизопропилборат с последующим перемешиванием при комнатной температуре в течение 30 минут. Реакционную смесь обрабатывали хлористоводородной кислотой с получением (4-{[трет-бутил(диметил)силил]окси}-2-метоксифенил)бороновой кислоты.
Пример получения 5
В атмосфере азота смесь метил 3-бром-2-метилбензоата, бис(пинаколат)дибора, дихлорида бис(трифенилфосфин)палладия(II), трифенилфосфина, трикалийфосфата и диоксана перемешивали при 100°C в течение 3 дней с получением, таким образом, метил 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоата.
Пример получения 6
В атмосфере азота смесь (2,6-диметилфенил)бороновой кислоты, этил 3-бромбензоата, тетракис(трифенилфосфин)палладия, водного раствора 1 M карбоната натрия, толуола и этанола перемешивали при нагревании при 80°C с получением, таким образом, этил 2',6'-диметилбифенил-3-карбоксилата. Смесь этил 2',6'-диметилбифенил-3-карбоксилата, водного раствора 1 M гидроксида натрия и этанола перемешивали при нагревании при 60°C с получением, таким образом, 2',6'-диметилбифенил-3-карбоновой кислоты.
Пример получения 7
В атмосфере азота тетракис(трифенилфосфин)палладий добавляли к смеси 2-бром-1,3-диметилбензола, (5-формил-2-метоксифенил)бороновой кислоты, водного раствора 1М карбоната натрия, этанола и диметоксиэтана с последующим перемешиванием при 80°C в течение 25 часов с получением 6-метокси-2',6'-диметилбифенил-3-карбальдегида.
Пример получения 8
Трифторметансульфоновый ангидрид добавляли по каплям при охлаждении льдом к смеси 4-гидрокси-3,5-диметилбензонитрила, пиридина и дихлорметана с последующим перемешиванием при комнатной температуре в течение 2 часов с получением 4-циано-2,6-диметилфенилтрифторметансульфоната. В атмосфере азота смесь 4-циано-2,6-диметилфенилтрифторметансульфоната, (3-формилфенил)бороновой кислоты, ацетата палладия, дициклогексил(2',6'-диметоксибифенил-2-ил)фосфина, трикалийфосфата, толуола и воды перемешивали при комнатной температуре в течение 6 часов с получением 3'-формил-2,6-диметилбифенил-4-карбонитрила.
Пример получения 9
В атмосфере азота смесь метил 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоата, 4-бром-3,5-диметилфенола, ацетата палладия, дициклогексил(2',6'-диметоксибифенил-2-ил)фосфина, трикалийфосфата, толуола и воды перемешивали в течение 14,5 часов при нагревании при 60°C с получением метил 4'-гидрокси-2,2',6'-триметилбифенил-3-карбоксилата.
Пример получения 10
В атмосфере азота смесь (4-{[трет-бутил(диметил)силил]окси}-2,6-диметилфенил)бороновой кислоты, 6-бромпиридин-2-карбальдегида, ацетата палладия, 2'-(дициклогексилфосфино)-N,N-диметилбифенил-2-амина, трикалийфосфата, толуола и воды перемешивали в течение 20 часов при нагревании при 60°C с получением, таким образом, 6-(4-{[трет-бутил(диметил)силил]окси}-2,6-диметилфенил)пиридин-2-карбальдегида.
Пример получения 11
В атмосфере азота при охлаждении льдом боргидрид натрия добавляли к этанольному раствору 4'-хлор-2'-метилбифенил-3-карбальдегида с последующим перемешиванием в течение 1 часа с получением (4'-хлор-2'-метилбифенил-3-ил)метанола.
Пример получения 12
В атмосфере азота при охлаждении льдом метил 4'-(метилсульфонил)бифенил-3-карбоксилат добавляли к суспензии в ТГФ литийалюминийгидрида с последующим перемешиванием в течение 20 минут с получением [4'-(метилсульфонил)бифенил-3-ил]метанола.
Пример получения 13
В атмосфере азота смесь метил 3-бром-4-хлорбензоата, (2,6-диметилфенил)бороновой кислоты, хлорида лития, карбоната натрия, воды, этанола, диметоксиэтана и тетракис(трифенилфосфин)палладия перемешивали при 90°C в течение 15 часов с получением, таким образом, метил 6-хлор-2',6'-диметилбифенил-3-карбоксилата. Литийалюминийгидрид добавляли при охлаждении льдом к раствору в ТГФ полученного метил 6-хлор-2',6'-диметилбифенил-3-карбоксилата с последующим нагреванием до комнатной температуры и при перемешивании в течение 2 часов с получением (6-хлор-2',6'-диметилбифенил-3-ил)метанола.
Пример получения 14
В атмосфере азота тетракистрифенилфосфинпалладий добавляли к смеси 2-бром-1,3-диметилбензола, 2-фтор-5-формилфенилбороновой кислоты, водного раствора 1М карбоната натрия, этанола и толуола с последующим перемешиванием при 80°C в течение 8 часов с получением 6-фтор-2',6'-диметилбифенил-3-карбальдегида. При охлаждении на бане лед-метанол, боргидрид натрия добавляли небольшими порциями к этанольному раствору полученного 6-фтор-2',6'-диметилбифенил-3-карбальдегида и реакционную смесь перемешивали при этой же температуре в течение 1 часа с получением (6-фтор-2',6'-диметилбифенил-3-ил)метанола.
Пример получения 15
Тионилхлорид добавляли к (4'-хлор-2'-метилбифенил-3-ил)метанолу при охлаждении с использованием бани лед-метанол с последующим перемешиванием при комнатной температуре в течение 1 часа с получением 4-хлор-3'-(хлорметил)-2-метилбифенила.
Пример получения 16
Карбонат калия добавляли к раствору в ДМФА 4-хлор-3'-(хлорметил)-2-метилбифенила и 4-гидроксибензальдегида с последующим перемешиванием при комнатной температуре в течение 20 часов с получением 4-[(4'-хлор-2'-метилбифенил-3-ил)метокси]бензальдегида.
Пример получения 17
Трибутилфосфин и 1,1'-(азодикарбонил)дипиперидин добавляли к раствору в ТГФ (4-{[трет-бутил(диметил)силил]окси}-2',6'-диметилбифенил-3-ил)метанола и 4-гидроксибензальдегида с последующим перемешиванием при комнатной температуре в течение 14 часов с получением 4-[(4-{[трет-бутил(диметил)силил]окси}-2',6'-диметилбифенил-3-ил)метоксибензальдегида.
Пример получения 18
В атмосфере азота гидрид натрия добавляли при охлаждении льдом к раствору в ТГФ (2',6'-диметилбифенил-3-ил)метанола с последующим перемешиванием при указанной температуре в течение 15 минут. Затем к реакционной смеси добавляли 6-хлорникотинонитрил при охлаждении льдом с последующим нагреванием до комнатной температуры и при перемешивании в течение 3 часов с получением 6-[(2',6'-диметилбифенил-3-ил)метокси]никотинонитрила.
Пример получения 19
В атмосфере азота раствор 1,0 M диизопропилалюминийгидрида в толуоле добавляли по каплям при -78°C к толуольному раствору 6-[(2',6'-диметилбифенил-3-ил)метокси]никотинонитрила с последующим перемешиванием при -78°C в течение 1,5 часов с получением 6-[(2',6'-диметилбифенил-3-ил)метокси]никотинальдегида.
Пример получения 20
В атмосфере азота раствор в ТГФ метил 4-[(4'-хлор-2'-метилбифенил-3-ил)метокси]-2-фторбензоата добавляли по каплям к суспензии в ТГФ литийалюминийгидрида при охлаждении на бане лед-метанол с последующим перемешиванием при комнатной температуре в течение 1 часа с получением 4-[(4'-хлор-2'-метилбифенил-3-ил)метокси]-2-фторбензилового спирта. При добавлении диоксида марганца к раствору в ТГФ полученного 4-[(4'-хлор-2'-метилбифенил-3-ил)метокси]-2-фторбензилового спирта и при перемешивании при 40°C в течение 17 часов получали 4-[(4'-хлор-2'-метилбифенил-3-ил)метокси]-2-фторбензальдегид.
Пример получения 21
В атмосфере азота боргидрид натрия добавляли к метанольному раствору 4-{[2',6'-диметил-4'-(2-оксопропокси)бифенил-3-ил]метокси}бензальдегида при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 2 часов с получением 1-[(-3'-{[4-(гидроксиметил)фенокси]метил}-2,6-диметилбифенил-4-ил)окси]пропан-2-ола. При добавлении хлороформа и диоксида марганца к полученному соединению и при перемешивании при 60°C в течение 5 часов получали 4-{[4'-(2-гидроксипропокси)-2',6'-диметилбифенил-3-ил]метокси}бензальдегид.
Пример получения 22
Метансульфонилхлорид добавляли по каплям при охлаждении льдом к смеси 4-[(4'-гидрокси-2',6'-диметилбифенил-3-ил)метокси]бензальдегида, триэтиламина и этилацетата с последующим перемешиванием при 0°C в течение 2 часов с получением 3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-илметансульфоната.
Пример получения 23
Смесь 4-[(4'-гидрокси-2',6'-диметилбифенил-3-ил)метокси]бензальдегида, 2-бромэтилацетата, карбоната цезия и ДМФА перемешивали при 60°C в течение 21 часов с получением 2-({3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-ил}окси)этилацетата.
Пример получения 24
В атмосфере азота смесь 2-гидроксиэтилацетата, гидрида натрия и ДМФА перемешивали при комнатной температуре в течение 15 минут и затем добавляли 1-бром-4-фтор-2-(трифторметил)бензол с последующим перемешиванием при комнатной температуре в течение 1,5 часов с получением 2-[4-бром-3-(трифторметил)фенокси]этанола.
Пример получения 25
Метансульфонилхлорид добавляли по каплям к смеси 1-(3-гидроксипропил)пирролидин-2-она, триэтиламина и этилацетата при охлаждении льдом с последующим перемешиванием при 0°C в течение 2 часов с получением бесцветного масла. 4-[(4'-Гидрокси-2',6'-диметилбифенил-3-ил)метокси]бензальдегид, карбонат цезия и ДМФА добавляли к полученному маслу с последующим перемешиванием при нагревании при 60°C в течение 19 часов с получением 4-({2',6'-диметил-4'-[3-(2-оксопирролидин-1-ил)пропокси]бифенил-3-ил}метокси)бензальдегида.
Пример получения 26
В атмосфере азота раствор в ТГФ метилмагнийиодида добавляли по каплям при охлаждении льдом к раствору в ТГФ 1-[(3'-{[4-(гидроксиметил)фенокси]метил}-2,6-диметилбифенил-4-ил)окси]ацетона с последующим перемешиванием при комнатной температуре в течение 30 минут с получением 1-[(3'-{[4-(гидроксиметил)фенокси]метил}-2,6-диметилбифенил-4-ил)окси]-2-метилпропан-2-ола.
Пример получения 27
Смесь 1-[(3'-{[4-(гидроксиметил)фенокси]метил}-2,6-диметилбифенил-4-ил)окси]-2-метилпропан-2-ола, диоксида марганца и хлороформа перемешивали при нагревании при 50°C в течение 20 часов с получением 4-{[4'-(2-гидрокси-2-метилпропокси)-2',6'-диметилбифенил-3-ил]метокси}бензальдегида.
Пример получения 28
Смесь 4-{[4'-(3-гидроксипропокси)-2',6'-диметилбифенил-3-ил]метокси}бензальдегида, ацетилхлорида, триэтиламина и дихлорметана перемешивали при комнатной температуре в течение 3,5 часов с получением 3-({3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-ил}окси)пропилацетата.
Пример получения 29
Смесь 4-{[4'-(3-гидрокси-3-метилбутокси)-2',6'-диметилбифенил-3-ил]метокси}бензальдегида, уксусного ангидрида, пиридина, DMAP и хлороформа перемешивали при комнатной температуре в течение 2 дней с получением 3-({3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-ил}окси)-1,1-диметилпропилацетата.
Пример получения 30
Раствор 4 M хлористого водорода в этилацетате добавляли по каплям при охлаждении льдом к раствору в этилацетате трет-бутил [2-({3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-ил}окси)этил]карбамата с последующим перемешиванием при 0°C в течение 2 часов. Дихлорметан, ацетилхлорид и триэтиламин добавляли к полученному соединению с последующим перемешиванием при комнатной температуре в течение 12 часов с получением N-[2-({3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-ил}окси)этил]ацетамида.
Пример получения 31
Трифторметансульфоновый ангидрид добавляли по каплям при охлаждении льдом к смеси 4-[(4'-гидрокси-2,2'-диметилбифенил-3-ил)метокси]бензальдегида, пиридина и дихлорметана с последующим перемешиванием при 0°C в течение 1 часа с получением 3'-[(4-формилфенокси)метил]-2,2'-диметилбифенил-4-илтрифторметансульфоната.
Пример получения 32
Гидрохлорид гидроксиламина и водный раствор ацетата натрия добавляли к этанольному раствору 4-[(4'-хлор-2'-метилбифенил-3-ил)метокси]бензальдегида с последующим перемешиванием при комнатной температуре в течение 18 часов с получением 4-[(4-{[трет-бутил(диметил)силил]окси}-2',6'-диметилбифенил-3-ил)метокси]бензальдегидоксима. Цианоборгидрид натрия добавляли к смешанному раствору в метаноле-ТГФ полученного 4-[(4-{[трет-бутил(диметил)силил]окси}-2',6'-диметилбифенил-3-ил)метокси]бензальдегидоксима и затем к смеси добавляли по каплям раствор 4 M хлористого водорода в диоксане с последующим перемешиванием при комнатной температуре в течение 1 часа с получением N-{4-[(4-{[трет-бутил(диметил)силил]окси}-2',6'-диметилбифенил-3-ил)метокси]бензил)гидроксиламина.
Пример получения 33
В атмосфере азота комплекс трифторида серы и диэтиламина добавляли по каплям при -75°C к раствору в дихлорметане 4-(4-бром-3-метилфенокси)-2-метилбутан-2-ола и температуру повышали до комнатной температуры с получением 1-бром-4-(3-фтор-3-метилбутокси)-2-метилбензола.
Пример получения 34
В атмосфере азота смесь 1-бром-4-(3-фтор-3-метилбутокси)-2-метилбензола, метил 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоата, ацетата палладия, дициклогексил(2',6'-диметоксибифенил-2-ил)фосфина, трикалийфосфата, толуола и воды перемешивали при 80°C в течение 12 часов с получением метил 4'-(3-фтор-3-метилбутокси)-2,2'-диметилбифенил-3-карбоксилата. Литийалюминийгидрид добавляли к раствору в ТГФ полученного метил 4'-(3-фтор-3-метилбутокси)-2,2'-диметилбифенил-3-карбоксилата при охлаждении льдом с последующим нагреванием до комнатной температуры и при перемешивании в течение 1 часа с получением [4'-(3-фтор-3-метилбутокси)-2,2'-диметилбифенил-3-ил]метанола.
Пример получения 35
В атмосфере азота гидрид натрия добавляли при охлаждении льдом к смеси 5-бром-4-метилпиридин-2-ола и ДМФА с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем к смеси добавляли 3-гидрокси-3-метилбутил 4-метилбензолсульфонат с последующим перемешиванием при 40°C в течение 14 часов с получением 4-[(5-бром-4-метилпиридин-2-ил)окси]2-метилбутан-2-ола и 5-бром-1-(3-гидрокси-3-метилбутил)-4-метилпиридин-2(1H)-она.
Пример получения 36
В атмосфере азота смесь (4-{[трет-бутил(диметил)силил]окси}-2-метилфенил)бороновой кислоты, 6-[(3-бром-2-метилбензил)окси]никотинальдегида, ацетата палладия, дициклогексил(2',6'-диметоксибифенил-2-ил)фосфина, трикалийфосфата, толуола и воды перемешивали при нагревании при 60°C в течение 2 дней с получением 6-[(4'-{[трет-бутил(диметил)силил]окси}-2,2'-диметилбифенил-3-ил)метокси]никотинальдегида.
Пример получения 37
В атмосфере азота смесь трет-бутил[3,5-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]диметилсилана, 6-[(3-бром-2-метилбензил)окси]никотинальдегида, ацетата палладия, дициклогексил(2',6'-диметоксибифенил-2-ил)фосфина, трикалийфосфата, толуола и воды перемешивали при нагревании при 60°C в течение 3 дней с получением 6-[(4'-{[трет-бутил(диметил)силил]окси}-2,2',6'-триметилбифенил-3-ил)метокси]никотинальдегида.
Пример получения 38
В атмосфере азота трет-бутоксид калия добавляли при охлаждении льдом к смеси циклобутанона, этилхлорацетата и ТГФ в течение 40 минут с последующим перемешиванием при 0°C в течение 2 часов, повышая температуру до комнатной температуры, и при перемешивании при комнатной температуре в течение 1 дня с получением, таким образом, этил 1-оксаспиро[2,3]гексан-2-карбоксилата. Раствор полученного этил 1-оксаспиро[2,3]гексан-2-карбоксилата в диэтиловом эфире добавляли к суспензии литийалюминийгидрида в ТГФ при охлаждении льдом в атмосфере азота с последующим перемешиванием при комнатной температуре в течение 7 часов с получением 1-(2-гидроксиэтил)циклобутанола. Смесь полученного 1-(2-гидроксиэтил)циклобутанола, 4-метилбензолсульфонилхлорида, триэтиламина и ТГФ перемешивали при комнатной температуре в течение 16 часов с получением 2-(1-гидроксициклобутил)этил 4-метилбензолсульфоната.
Таким же способом, как в описанных выше способах примеров получения 1-38, получали соединения примеров получения 39-299 с использованием соответствующих исходных веществ соответственно. Структуры соединений примеров получения представлены в таблицах 4-44, а способы получения и физико-химические данные - в таблицах 45-52.
Пример 1
Хлоркарбонилизоцианат (0,10 мл) добавляли по каплям при охлаждении на бане лед-метанол к раствору в ТГФ (10 мл) N-{4-[(4'-хлор-2'-метилбифенил-3-ил)метокси]бензил}гидроксиламина (430 мг) и температуру повышали до комнатной температуры с последующим перемешиванием в течение 1 часа. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты (30 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Путем очистки полученного остатка при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) получали бесцветное пенообразное вещество. Полученное пенообразное вещество растворяли в этаноле (5 мл) и к смеси добавляли водный раствор 1 М гидроксида натрия (1,06 мл) с последующим концентрированием при пониженном давлении. Путем перекристаллизации полученного остатка из воды-изопропанола получали натрий 2-{4-[(4'-хлор-2'-метилбифенил-3-ил)метокси]бензил}-3,5-диоксо-1,2,4-оксадиазолидин-4-ид (347 мг) в виде бесцветных кристаллов.
Пример 2
Хлоркарбонилизоцианат (0,14 мл) добавляли по каплям при охлаждении на бане лед-метанол к раствору в ТГФ (15 мл) 4-({[3'-({4-[(гидроксиамино)метил]фенокси}метил)-2,6-диметилбифенил-4-ил]окси}метил)тетрагидро-2H-тиопиран-4-ола (792 мг) с последующим повышением температуры до комнатной температуры и затем перемешиванием в течение 1 часа. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты (40 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Путем очистки полученного остатка при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) получали бесцветное пенообразное вещество (777 мг). Метоксид натрия (50 мг) добавляли к метанольному раствору (10 мл) полученного пенообразного вещества (116 мг) с последующим перемешиванием при комнатной температуре в течение 30 минут. Затем к смеси добавляли метоксид натрия (200 мг) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционную смесь нагревали до 60°C, перемешивали в течение 2 часов и затем спонтанно охлаждали до комнатной температуры. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты (10 мл) и воду (20 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в метаноле (5 мл)-ТГФ (10 мл) и добавляли водный раствор 1 М гидроксида натрия (0,20 мл) с последующим концентрированием при пониженном давлении. Путем промывки полученного остатка смесью изопропанол-диэтиловый эфир получали натрий 2-[4-({4'-[(4-гидрокситетрагидро-2H-тиопиран-4-ил)метокси]-2',6'-диметилбифенил-3-ил}метокси)бензил]-3,5-диоксо-1,2,4-оксадиазолидин-4-ид (80 мг) в виде бледно-желтого твердого вещества.
Пример 3
Хлоркарбонилизоцианат (0,14 мл) добавляли по каплям при охлаждении на бане лед-метанол к раствору в ТГФ (15 мл) 4-({[3'-({4-[(гидроксиамино)метил]фенокси}метил)-2,6-диметилбифенил-4-ил]окси}метил)тетрагидро-2H-тиопиран-4-ола (792 мг) с последующим повышением температуры до комнатной температуры и последующим перемешиванием в течение 1 часа. К реакционной смеси добавляли раствор 1M хлористоводородной кислоты (40 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Путем очистки полученного остатка при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) получали бесцветное пенообразное вещество (777 мг). При охлаждении на бане лед-метанол м-хлорпербензойную кислоту (630 мг) добавляли к раствору в хлороформе (20 мл) полученного пенообразного вещества (600 мг) с последующим перемешиванием в течение 30 минут. К реакционной смеси добавляли воду (20 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Путем очистки полученного остатка при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) получали бесцветное пенообразное вещество (510 мг). Полученное пенообразное вещество (510 мг) промывали диизопропиловым эфиром-этилацетатом-гексаном и сушили при пониженном давлении с получением слегка желтоватого твердого вещества (432 мг). Метоксид натрия (800 мг) добавляли к метанольному раствору (30 мл) полученного слегка желтоватого твердого вещества (387 мг) с последующим перемешиванием при 60°C в течение 2 часов и затем спонтанным охлаждением до комнатной температуры. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты (30 мл) и воду (50 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в метаноле (5 мл)-ТГФ (15 мл), добавляли водный раствор 1 М гидроксида натрия (0,63 мл) с последующим концентрированием при пониженном давлении. Путем промывки полученного остатка смесью изопропанол-диэтиловый эфир получали натрий 2-[4-({4'-[(4-гидрокси-1,1-диоксотетрагидро-2H-тиопиран-4-ил)метокси]-2',6'-диметилбифенил-3-ил}метокси)бензил]-3,5-диоксо-1,2,4-оксадиазолидин-4-ид (252 мг) в виде бесцветного твердого вещества.
Пример 4
Смесь 2-{4-[(3-бромбензил)окси]бензил}-1,2,4-оксадиазолидин-3,5-диона (500 мг), 2,6-дифтор-4-метоксифенилбороновой кислоты (325 мг), тетракистрифенилфосфинпалладия (80 мг), хлорида лития (6 мг), водного раствора карбоната натрия (562 мг/5 мл), этанола (5 мл) и 1,2-диметоксиэтана (25 мл) перемешивали при 90°C в течение 5 часов в атмосфере азота. К реакционной смеси снова добавляли 2,6-дифтор-4-метоксифенилбороновую кислоту (325 мг) с последующим перемешиванием при 90°C в течение 13 часов. К реакционной смеси добавляли следующее дополнительное количество 2,6-дифтор-4-метоксифенилбороновой кислоты (325 мг) с последующим перемешиванием при 90°C в течение 2 часов. К реакционной смеси добавляли следующее дополнительное количество 2,6-дифтор-4-метоксифенилбороновой кислоты (325 мг) с последующим перемешиванием при 90°C в течение 5 часов и спонтанным охлаждением до комнатной температуры. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты (50 мл) с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния и фильтровали. К фильтрату добавляли силикагель (3 г) с последующим концентрированием при пониженном давлении. Продукт, полученный таким образом, очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) с получением желтоватого пенообразного вещества (614 мг). Полученное пенообразное вещество (614 мг) растворяли в ТГФ (5 мл)-этаноле (5 мл), добавляли водный раствор 1 М гидроксида натрия (1,32 мл) с последующим концентрированием при пониженном давлении. Путем перекристаллизации полученного остатка из изопропанола-воды получали натрий 2-{4-[(2',6'-дифтор-4'-метоксибифенил-3-ил)метокси]бензил}-3,5-диоксо-1,2,4-оксадиазолидин-4-ид (366 мг) в виде бесцветного твердого вещества.
Пример 5
Водный раствор 1 М гидроксида натрия (5 мл) добавляли к смеси метил 3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-4-бифенилкарбоксилата (196 мг), метанола (5 мл) и ТГФ (5 мл) с последующим перемешиванием в течение 1 часа при нагревании при 60°C. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты (7 мл) с последующим перемешиванием при комнатной температуре. Осажденное твердое вещество собирали фильтрованием и сушили путем нагревания при пониженном давлении с получением 3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-4-бифенилкарбоновой кислоты (176 мг) в виде белого твердого вещества.
Пример 6
WSC гидрохлорид (163 мг) добавляли к смеси 3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-4-бифенилкарбоновой кислоты (293 мг), (2-этоксиэтил)амина (0,11 мл), HOBt (142 мг) и ДМФА (10 мл) с последующим перемешиванием при комнатной температуре в течение 27 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли хлороформ/метанол (4/1) с последующей промывкой водой и насыщенным водным раствором хлорида аммония. Растворитель выпаривали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ/метанол), полученное пенообразное вещество затем кристаллизовали путем добавления диэтилового эфира, и полученные кристаллы перекристаллизовывали из метанола с получением 3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-N-(2-этоксиэтил)-4-бифенилкарбоксамида (135 мг) в виде белых кристаллов.
Пример 7
DMT-MM (653 мг) добавляли к охлажденной льдом смеси 3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-4-бифенилкарбоновой кислоты (329 мг), 2-аминоэтанола (0,14 мл), ТГФ (20 мл) и метанола (4 мл) с последующим перемешиванием при комнатной температуре в течение 25 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией хлороформом/метанолом (4/1). Растворитель выпаривали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол). Полученное бледно-желтое твердое вещество (276 мг) растворяли в ТГФ (5 мл)-метаноле (5 мл) и добавляли водный раствор 1 М гидроксида натрия (0,79 мл) с последующим перемешиванием при комнатной температуре в течение 10 минут. Осажденное твердое вещество собирали фильтрованием и сушили путем нагревания при пониженном давлении с получением натрий 2-{4-[(4'-{[(2-гидроксиэтил)амино]карбонил}бифенил-3-ил)метокси]бензил}-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (188 мг) в виде белого твердого вещества.
Пример 8
Раствор 1,0 M тетрабутиламмонийфторида (TBAF) в ТГФ (1,94 мл) добавляли по каплям к охлажденной льдом смеси 2-(4-{[4'-(2-{[трет-бутил(диметил)силил]окси}этокси)бифенил-3-ил]метокси}бензил-1,2,4-оксадиазолидин-3,5-диона (532 мг) и ТГФ (10 мл) с последующим постепенным повышением температуры до комнатной температуры и последующим перемешиванием в течение 13 часов. Реакционную смесь разбавляли хлороформом/метанолом (4/1) и промывали насыщенным водным раствором хлорида аммония и насыщенным водным раствором хлорида натрия. Затем растворитель выпаривали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) и полученное твердое вещество перекристаллизовывали из этилацетата-гексана-диэтилового эфира с получением 2-(4-{[4'-(2-гидроксиэтокси)-3-бифенил]метокси}бензил-1,2,4-оксадиазолидин-3,5-диона (171 мг) в виде белых кристаллов.
Пример 9
Гидрохлорид гидроксиламина (12,85 г) и водный раствор ацетата натрия (19,22 г/110 мл) добавляли к этанольной (800 мл) суспензии 4-[(3-бромбензил)окси]бензальдегида (17,94 г) с последующим перемешиванием при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении и к остатку добавляли воду (100 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении с получением бесцветного твердого вещества (19,94 г). К раствору в метаноле (350 мл)-ТГФ (350 мл) полученного бесцветного твердого вещества (19,94 г) добавляли цианоборгидрид натрия (19,36 г). Затем раствор 4 M хлористого водорода в диоксане (160 мл) медленно добавляли по каплям при охлаждении льдом. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли водный раствор 1 М гидроксида натрия (700 мл) при охлаждении льдом с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением желтоватого твердого вещества (25,53 г). Хлоркарбонилизоцианат (5,00 мл) добавляли по каплям к раствору в ТГФ (380 мл) полученного желтоватого твердого вещества (25,53 г) при охлаждении на бане лед-метанол с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты (400 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Полученный остаток промывали этилацетатом и затем сушили при 50°C при пониженном давлении с получением 2-{4-[(3-бромбензил)окси]бензил}-1,2,4-оксадиазолидин-3,5-диона (6,08 г) в виде бесцветного твердого вещества.
Пример 10
Смесь 2-(4-аминобензил)-1,2,4-оксадиазолидин-3,5-диона (500 мг), 4'-хлор-2'-метилбифенил-3-карбальдегида (668 мг), уксусной кислоты (0,33 мл) и ТГФ (40 мл) перемешивали при комнатной температуре в течение 24 часов. К реакционной смеси добавляли триацетоксиборгидрид натрия (767 мг) с последующим перемешиванием при комнатной температуре в течение 15 минут. Растворитель выпаривали при пониженном давлении и к остатку добавляли воду с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия, растворитель выпаривали при пониженном давлении, к остатку добавляли толуол и растворитель снова выпаривали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) и добавляли ТГФ (5 мл), метанол (5 мл) и водный раствор 1 М гидроксида натрия (1,47 мл) к полученному бледно-желтому пенообразному веществу (620 мг) с последующим перемешиванием при комнатной температуре в течение 5 минут. Растворитель выпаривали при пониженном давлении, остаток очищали при помощи ODS колоночной хроматографии (вода-ацетонитрил) и отверждали путем добавления диэтилового эфира. Это твердое вещество собирали фильтрованием и затем сушили путем нагревания при пониженном давлении с получением натрий 2-(4-{[(4'-хлор-2'-метилбифенил-3-ил)метил]амино}бензил)3,5-диоксо-1,2,4-оксадиазолидин-4-ида (160 мг) в виде белого твердого вещества.
Пример 11
Смесь 2-(4-аминобензил)-1,2,4-оксадиазолидин-3,5-диона (365 мг), 2-[(3'-формил-2,6-диметилбифенил-4-ил)окси]этилацетата (660 мг), уксусной кислоты (0,3 мл), ТГФ (20 мл) и молекулярных сит 4A (1 г) перемешивали при комнатной температуре в течение 22 часов. К реакционной смеси добавляли триацетоксиборгидрид натрия (560 мг) с последующим перемешиванием при комнатной температуре в течение 22 часов. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, к остатку добавляли толуол, растворитель снова выпаривали при пониженном давлении и затем остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат). Метанол (10 мл) и метоксид натрия (52 мг) добавляли к полученному бледно-желтому пенообразному веществу (406 мг) с последующим перемешиванием в течение 2 часов при одновременном нагревании при 60°C. Растворитель выпаривали при пониженном давлении и к остатку добавляли хлороформ с последующей промывкой водой и насыщенным водным раствором хлорида натрия и сушкой над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и ТГФ (5 мл), метанол (5 мл) и водный раствор 1 М гидроксида натрия (0,81 мл) добавляли к полученному бледно-желтому пенообразному веществу (373 мг) с последующим перемешиванием при комнатной температуре в течение 5 минут. Растворитель выпаривали при пониженном давлении, остаток очищали при помощи ODS колоночной хроматографии (вода-ацетонитрил), полученное бледно-желтое пенообразное вещество преобразовывали в твердое вещество путем добавления диэтилового эфира. Это твердое вещество собирали фильтрованием и затем сушили путем нагревания при пониженном давлении с получением натрий 2-[4-({[4'-(2-гидроксиэтокси)-2',6'-диметилбифенил-3-ил]метил}амино)бензил]-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (226 мг) в виде бледно-желтого твердого вещества.
Пример 12
Смесь 3-[(3'-формил-2,2'-диметилбифенил-4-ил)окси]-1,1-диметилпропилацетата (479 мг), 2-(4-аминобензил)-1,2,4-оксадиазолидин-3,5-диона (340 мг) и уксусной кислоты (6 мл) перемешивали при комнатной температуре в течение 20 часов. К реакционному раствору добавляли триацетоксиборгидрид натрия (573 мг) с последующим перемешиванием при комнатной температуре в течение 2 часов. После выпаривания растворителя при пониженном давлении к остатку добавляли воду с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) и смесь полученного бледно-желтого пенообразного вещества (719 мг), ТГФ (5 мл), метанола (5 мл) и водного раствора 1 М гидроксида натрия (4 мл) перемешивали при 50°C в течение 4 часов. pH доводили до 4-5 путем добавления раствора 1 M хлористоводородной кислоты с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) и водный раствор 1 М гидроксида натрия (0,89 мл) добавляли к смеси полученного бледно-желтого масла (448 мг), ТГФ (3 мл) и метанола (3 мл) с последующим перемешиванием в течение 10 минут. После выпаривания растворителя при пониженном давлении полученный остаток промывали диэтиловым эфиром с получением натрий 2-[4-({[4'-(3-гидрокси-3-метилбутокси)-2,2'-диметилбифенил-3-ил]метил}амино)бензил]-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (398 мг) в виде белого твердого вещества.
Пример 13
Смесь 3-({3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-ил}окси)пропилацетата (675 мг), гидрохлорида гидроксиламина (217 мг), ацетата натрия (307 мг), этанола (15 мл) и воды (4 мл) перемешивали при комнатной температуре в течение 18 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли воду с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и к остатку добавляли уксусную кислоту (5 мл) и цианоборгидрид натрия (196 мг) с последующим перемешиванием при комнатной температуре в течение 7 часов. Реакционную систему подщелачивали путем добавления насыщенного водного раствора карбоната натрия с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем растворитель выпаривали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) и ТГФ (10 мл) добавляли к полученному бесцветному маслу (256 мг) с последующим охлаждением льдом. К смеси добавляли по каплям хлоркарбонилизоцианат (0,05 мл) с последующим перемешиванием в течение 15,5 часов при комнатной температуре. Растворитель выпаривали при пониженном давлении и к остатку добавляли хлороформ с последующей промывкой раствором 1 M хлористоводородной кислоты и насыщенным водным раствором хлорида натрия. Растворитель выпаривали при пониженном давлении, остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) и к полученному бесцветному маслу (242 мг) добавляли метанол (10 мл) и метоксид натрия (92 мг) с последующим перемешиванием при нагревании при 60°C в течение 2 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли воду с последующей экстракцией хлороформом и промывкой насыщенным водным раствором хлорида натрия. Растворитель выпаривали при пониженном давлении, остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) и к полученному бесцветному маслу (152 мг) добавляли ТГФ (5 мл), метанол (5 мл) и водный раствор 1 М гидроксида натрия (0,33 мл) с последующим перемешиванием при комнатной температуре в течение 5 минут. Растворитель выпаривали при пониженном давлении, остаток очищали при помощи ODS колоночной хроматографии (вода-ацетонитрил), полученное бесцветное масло преобразовывали в твердое вещество путем добавления диэтилового эфира, и это твердое вещество собирали фильтрованием и затем сушили путем нагревания при пониженном давлении с получением натрий 2-(4-{[4'-(3-гидроксиpropокси)-2',6'-диметилбифенил-3-ил]метокси}бензил)-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (126 мг) в виде белого твердого вещества.
Пример 14
Смесь 2-({3'-[(4-формилфенокси)метил]-2-метилбифенил-4-ил}окси)этилацетата (935 мг), гидрохлорида гидроксиламина (480 мг), водного раствора ацетата натрия (760 мг/3 мл) и этанола (15 мл) перемешивали при комнатной температуре в течение 1,5 часов. После выпаривания растворителя при пониженном давлении к остатку добавляли воду (20 мл) с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении с получением бесцветного масла (1,07 г). Цианоборгидрид натрия (430 мг) и уксусную кислоту (1 мл) добавляли в указанном порядке при охлаждении льдом к смеси полученного масла, метанола (10 мл) и ТГФ (10 мл) с последующим перемешиванием в течение 5 минут. При постепенном нагревании реакционной жидкости до комнатной температуры к смеси подходящим образом добавляли раствор 4 M хлористого водорода в диоксане (всего 1 мл) с последующим перемешиванием в течение 5 часов. К реакционной жидкости добавляли насыщенный водный раствор бикарбоната натрия (20 мл) с последующей экстракцией хлороформом. Затем органический слой сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) с получением бесцветного масла (0,50 г). Хлоркарбонилизоцианат (190 мг) добавляли к смеси полученного масла и ТГФ (5 мл) с последующим перемешиванием при комнатной температуре в течение 15 минут, затем давая смеси выстояться в течение ночи. К реакционной жидкости добавляли воду (10 мл) с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Путем выпаривания растворителя при пониженном давлении получали бесцветное масла (413 мг). Смесь этого масла с водным раствором 1 М гидроксида натрия (3 мл), метанолом (3 мл) и ТГФ (6 мл) перемешивали при 60°C в течение 3 часов. К реакционной жидкости добавляли раствор 1 M хлористоводородной кислоты (3,5 мл) с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) с получением бесцветного масла (331 мг). ТГФ (10 мл), метанол (1 мл) и водный раствор 1 М гидроксида натрия (0,7 мл) добавляли к полученному маслу и растворитель выпаривали при пониженном давлении. К полученному остатку добавляли диэтиловый эфир и твердое вещество собирали фильтрованием и сушили при 60°C при пониженном давлении с получением натрий 2-(4-{[4'-(2-гидроксиэтокси)-2'-метилбифенил-3-ил]метокси}бензил)-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (243 мг) в виде бесцветного твердого вещества.
Пример 15
Смесь {[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,6-диметилбифенил-4-ил]окси}уксусной кислоты (482 мг), гидрохлорида диметиламина (165 мг), WSC гидрохлорида (388 мг), триэтиламина (0,56 мл) и ДМФА (10 мл) перемешивали при комнатной температуре в течение 22,5 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли раствор 1 M хлористоводородной кислоты с последующей экстракцией хлороформом/метанолом (4/1). Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат и хлороформ-метанол). Метанол (5 мл), ТГФ (5 мл) и водный раствор 1 М гидроксида натрия (0,36 мл) добавляли к полученному бледно-желтому маслу (178 мг) с последующим перемешиванием при комнатной температуре в течение 5 минут. Растворитель выпаривали при пониженном давлении и остаток очищали при помощи ODS колоночной хроматографии (вода-ацетонитрил). Полученное бесцветное пенообразное вещество отверждали путем добавления диэтилового эфира и это твердое вещество собирали фильтрованием и затем сушили путем нагревания при пониженном давлении с получением натрий 2-[4-({4'-[2-(диметиламино)-2-оксоэтокси]-2',6'-диметилбифенил-3-ил}метокси)бензил]-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (52 мг) в виде белого твердого вещества.
Пример 16
Оксалилдихлорид (0,15 мл) добавляли к раствору в ТГФ (10 мл) 2',6'-диметилбифенил-3-карбоновой кислоты (277 мг) с последующим перемешиванием при комнатной температуре в течение 5 минут. Затем добавляли ДМФА (1 капля) с последующим перемешиванием при этой же температуре в течение 1 часа. Растворитель выпаривали при пониженном давлении и раствор полученного остатка в ТГФ (10 мл) добавляли по каплям к смеси 2-(4-аминобензил-1,2,4-оксадиазолидин-3,5-диона (380 мг) и насыщенного водного раствора бикарбоната натрия (10 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной жидкости добавляли раствор 1 M хлористоводородной кислоты (20 мл) с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Метанол (5 мл), ТГФ (5 мл) и водный раствор 1 М гидроксида натрия (1,2 мл) добавляли к полученному остатку и растворитель выпаривали при пониженном давлении. К полученному остатку добавляли ТГФ-гексан и растворитель выпаривали при пониженном давлении с последующей сушкой при 50°C при пониженном давлении с получением, таким образом, натрий 2-(4-{[(2',6'-диметилбифенил-3-ил)карбонил]амино}бензил)-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (530 мг) в виде желтого твердого вещества.
Пример 17
Смесь {[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,6-диметилбифенил-4-ил]окси}уксусной кислоты (500 мг), водного раствора 12 M этиламина (0,175 мл), WSC гидрохлорида (302 мг), HOAt (214 мг) и ДМФА (10 мл) перемешивали при комнатной температуре в течение 21,5 часов. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты и воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол и гексан-этилацетат). Метанол (3 мл), ТГФ (3 мл) и водный раствор 1 M гидроксида калия (0,611 мл) добавляли к полученному бесцветному пенообразному веществу (308 мг) с последующим перемешиванием при комнатной температуре в течение 10 минут. Растворитель выпаривали при пониженном давлении, к остатку добавляли этилацетат, растворитель снова выпаривали при пониженном давлении. Осажденное твердое вещество собирали фильтрованием и затем сушили путем нагревания при пониженном давлении с получением калий 2-[4-({4'-[2-(этиламино)-2-оксоэтокси]-2',6'-диметилбифенил-3-ил}метокси)бензил]-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (300 мг) в виде белого твердого вещества.
Пример 18
Смесь 2,2,2-трифтор-1-[({3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-ил}окси)метил]этилацетата (590 мг), гидрохлорида гидроксиламина (253 мг), ацетата натрия (378 мг), этанола (15 мл) и воды (4 мл) перемешивали при комнатной температуре в течение 21 часа. Растворитель выпаривали при пониженном давлении и к остатку добавляли воду с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) и к полученному бесцветному маслу (310 мг) добавляли уксусную кислоту (8 мл) и цианоборгидрид натрия (127 мг) с последующим перемешиванием при комнатной температуре в течение 4 часов. Растворитель выпаривали при пониженном давлении и остаток подщелачивали путем добавления водного раствора 1 М гидроксида натрия с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол). К полученному бесцветному пенообразному веществу (266 мг) добавляли ТГФ (10 мл) с последующим охлаждением льдом. Затем к смеси добавляли по каплям этоксикарбонилизоцианат (0,065 мл) с последующим перемешиванием при 0°C в течение некоторого времени, а затем перемешиванием при комнатной температуре в течение 4 дней. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем растворитель выпаривали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат) и ТГФ (5 мл) и к полученному бесцветному пенообразному веществу добавляли водный раствор 1 М гидроксида натрия (0,36 мл) с последующим перемешиванием при комнатной температуре в течение 6 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли раствор 1 M хлористоводородной кислоты с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат). Метанол (4 мл), ТГФ (4 мл) и водный раствор 1 M гидроксида калия (0,31 мл) добавляли к полученному бесцветному пенообразному веществу (164 мг) с последующим перемешиванием при комнатной температуре в течение 10 минут. Растворитель выпаривали при пониженном давлении, к остатку добавляли этилацетат и растворитель снова выпаривали при пониженном давлении. К остатку добавляли диэтиловый эфир с последующим перемешиванием при комнатной температуре. Осажденное твердое вещество собирали фильтрованием и затем сушили путем нагревания при пониженном давлении с получением калий 2-(4-{[2',6'-диметил-4'-(3,3,3-трифтор-2-гидроксипропокси)бифенил-3-ил]метокси}бензил)-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (163 мг) в виде белого твердого вещества.
Пример 19
Смесь 2-(4-{[4'-(2-гидроксипpопокси)-2',6'-диметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-диона (240 мг), 1,1,1-триацетокси-1,1-дигидро-1,2-бензоиодоксол-3(1H)-она (320 мг) и дихлорметана(10 мл) перемешивали при комнатной температуре в течение 1,5 часов. К реакционной смеси добавляли воду с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол и гексан-этилацетат) и к полученному бесцветному пенообразному веществу (196 мг) добавляли метанол (3 мл), ТГФ (3 мл) и водный раствор 1 М гидроксида натрия (0,41 мл) с последующим перемешиванием при комнатной температуре в течение 10 минут. Растворитель выпаривали при пониженном давлении и остаток очищали при помощи ODS колоночной хроматографии (вода-ацетонитрил). Полученное бесцветное пенообразное вещество отверждали путем добавления диэтилового эфира и это твердое вещество собирали фильтрованием и сушили путем нагревания при пониженном давлении с получением натрий 2-(4-{[2',6'-диметил-4'-(2-оксопропокси)бифенил-3-ил]метокси}бензил)-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (70 мг) в виде белого твердого вещества.
Пример 20
Смесь трет-бутил[2-({3'-[(4-формилфенокси)метил]-2,6-диметилбифенил-4-ил}окси)этил]карбамата (505 мг), гидрохлорида гидроксиламина (221 мг), ацетата натрия (331 мг), этанола (15 мл) и воды (4 мл) перемешивали при комнатной температуре в течение 24 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли воду с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и к остатку добавляли уксусную кислоту (10 мл) и цианоборгидрид натрия (167 мг) с последующим перемешиванием при комнатной температуре в течение 4 часов. Реакционную смесь подщелачивали путем добавления водного раствора 1 М гидроксида натрия с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и к полученному бесцветному пенообразному веществу (528 мг) добавляли ТГФ (8 мл) с последующим охлаждением льдом. К реакционной смеси добавляли по каплям хлоркарбонилизоцианат (0,094 мл) с последующим перемешиванием при комнатной температуре в течение 14,5 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией хлороформом. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (гексан-этилацетат). К полученному бесцветному пенообразному веществу добавляли этилацетат (3 мл) с последующим охлаждением льдом. К реакционной смеси добавляли по каплям раствор 4 M хлористого водорода в этилацетате (12 мл) с последующим перемешиванием при 0°C в течение 2 часов. Растворитель выпаривали при пониженном давлении и к полученному бледно-желтому пенообразному веществу добавляли диэтиловый эфир-гексан с последующим перемешиванием при комнатной температуре. Это твердое вещество собирали фильтрованием и затем сушили путем нагревания при пониженном давлении с получением гидрохлорида 2-(4-{[4'-(2-аминоэтокси)-2',6'-диметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-диона (200 мг) в виде бледно-желтого твердого вещества.
Пример 21
Смесь этил 2-({3'-[(4-формилфенокси)метил]-2,2'-диметилбифенил-4-ил}окси)-2-метилпропаноата (1,87 г), гидрохлорида гидроксиламина (378 мг), ацетата натрия (515 мг), этанола (36 мл) и воды (9 мл) перемешивали при комнатной температуре в течение 3 часов. После выпаривания растворителя при пониженном давлении к остатку добавляли воду с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Цианоборгидрид натрия (790 мг) добавляли к раствору в уксусной кислоте (20 мл) полученного бледно-желтого пенообразного вещества (1,95 г) с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционный раствор подщелачивали путем добавления насыщенного водного раствора бикарбоната натрия и карбоната натрия с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) и к раствору полученного бесцветного пенообразного вещества (1,17 г) в ТГФ (15 мл) добавляли хлоркарбонилизоцианат (0,234 мл) при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 24 часов. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Смесь полученного бесцветного пенообразного вещества (1,43 г), ТГФ (15 мл), метанола (15 мл) и водного раствора 1 М гидроксида натрия (15 мл) перемешивали при 60°C в течение 1 часа. Растворитель выпаривали при пониженном давлении и к полученному остатку добавляли раствор 1 M хлористоводородной кислоты с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении с получением 2-{[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,2'-диметилбифенил-4-ил]окси}-2-метилпропановой кислоты (1,29 г) в виде бесцветного пенообразного вещества. К смеси полученной 2-{[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,2'-диметилбифенил-4-ил]окси}-2-метилпропановой кислоты (291 мг), ТГФ (3 мл) и метанола (3 мл) добавляли водный раствор 1 М гидроксида натрия (1,15 мл) с последующим перемешиванием в течение 10 минут. Затем растворитель выпаривали при пониженном давлении. Путем перекристаллизации полученного остатка из этанола-воды получали динатрий 2-{[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,2'-диметилбифенил-4-ил]окси}-2-метилпропаноат (149 мг) в виде белых кристаллов.
Пример 22
При охлаждении льдом раствор 4 M хлористого водорода в диоксане (15 мл) добавляли по каплям к смеси трет-бутил (3-{[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,2'-диметилбифенил-4-ил]окси}пропил)карбамата (1,95 г) и этилацетата (5 мл) с последующим перемешиванием при 0°C в течение некоторого времени и затем перемешиванием при комнатной температуре в течение 1,5 часов. Растворитель выпаривали при пониженном давлении и осажденное твердое вещество собирали фильтрованием и сушили путем нагревания при пониженном давлении с получением гидрохлорида 2-(4-{[4'-(3-аминопропокси)-2,2'-диметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-диона (1,53 г) в виде белого твердого вещества.
Пример 23
Смесь натрий 2-{4-[(4'-{[(4R)-2,2-диметил-1,3-диоксолан-4-ил]метокси}-2,2'-диметилбифенил-3-ил)метокси]бензил}-3,5-диоксо-1,2,4-оксадиазолидин-4-ида (467 мг), 1 M хлористоводородной кислоты (5 мл) и ТГФ (5 мл) перемешивали при 50°C в течение 2 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду (10 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Водный раствор 1 М гидроксида натрия (0,977 мл) добавляли к раствору полученного остатка в ТГФ (5 мл) с последующим концентрированием при пониженном давлении. Путем промывки полученного остатка диэтиловым эфиром получали натрий 2-{[(4'-{[(2S)-2,2-дигидроксипропил]окси}-2,2'-диметилбифенил-3-ил)метокси]бензил}-3,5-диоксо-1,2,4-оксадиазолидин-4-ид (392 мг) в виде белого твердого вещества.
Пример 24
Смесь 2,2-дифтор-2-({3'-[(4-формилфенокси)метил]-2,2'-диметилбифенил-4-ил}окси)-N-метилацетамида, гидрохлорида гидроксиламина (122 мг), ацетата натрия (167 мг), этанола (12 мл) и воды (3 мл) перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли воду с последующей экстракцией хлороформом. После сушки органического слоя над безводным сульфатом магния растворитель выпаривали при пониженном давлении. Цианоборгидрид натрия (257 мг) добавляли к раствору в метаноле (5 мл)-ТГФ (5 мл) полученного бесцветного пенообразного вещества (594 мг) и затем при охлаждении льдом к реакционной смеси медленно добавляли по каплям раствор 4 M хлористого водорода в диоксане (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли водный раствор 1 М гидроксида натрия (7 мл) при охлаждении льдом с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-метанол), к полученному бесцветному маслу добавляли ТГФ (6 мл) (559 мг) с последующим охлаждением льдом. К реакционной смеси добавляли по каплям этоксикарбонилизоцианат (0,152 мл) с последующим перемешиванием при 0°C в течение 30 минут и затем перемешиванием при комнатной температуре в течение 1 часа. К реакционной смеси добавляли водный раствор 1 М гидроксида натрия (3 мл) с последующим перемешиванием при комнатной температуре в течение 12 часов. К реакционной смеси добавляли раствор 1 M хлористоводородной кислоты (4 мл) с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали при пониженном давлении. Путем очистки полученного остатка при помощи колоночной хроматографии на силикагеле (хлороформ-метанол) получали 2-{[3'-({4-[(3,5-диоксо-1,2,3-оксадиазолидин-2-ил)метил]фенокси}метил)-2,2'-диметилбифенил-4-ил]окси}-2,2-дифтор-N-метилацетамид (76 мг) в виде бесцветного пенообразного вещества и {[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,2'-диметилбифенил-4-ил]окси}(дифтор)уксусную кислоту (217 мг) в виде бесцветного пенообразного вещества. К раствору в ТГФ (5 мл) полученной {[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,2'-диметилбифенил-4-ил]окси}(дифтор)уксусной кислоты добавляли водный раствор 1 М гидроксида натрия (0,847 мл) с последующим концентрированием при пониженном давлении. Путем промывки полученного остатка диэтиловым эфиром получали динатрий {[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-4-ид-2-ил)метил]фенокси}метил)-2,2'-диметилбифенил-4-ил]окси}(дифтор)ацетат (203 мг) в виде белого твердого вещества.
Пример 25
3'-({4-[(3,5-Диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2-метилбифенил-4-карбоновую кислоту (10,8 мг) растворяли в смешанном растворе ТГФ-метанола [1 мл, 4:1 (об/об)] и раствор добавляли к пирролидину (3,2 мг). Добавляли DMT-MM (12 мг) с последующим перемешиванием в течение ночи при комнатной температуре. Затем к реакционной жидкости добавляли хлороформ и органический слой промывали раствором 1 M хлористоводородной кислоты. Органический слой концентрировали и остаток очищали при помощи фракционированной ВЭЖХ (Waters, наименование продукта: Waters SunFireTM Prep C18OBDTM (19×100 мм, 5 мкм)) с получением 2-(4-{[2'-метил-4'-(пирролидин-1-илкарбонил)бифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-диона (8,2 мг).
Пример 26
3'-({4-[(3,5-Диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2-метилбифенил-4-карбоновую кислоту (10,8 мг) растворяли в смешанном растворе ТГФ-метанола [1 мл, 4:1 (об/об)] и раствор добавляли к гидрохлориду 4-(метоксиметил)пиперидина (7,5 мг). Добавляли DMT-MM (12 мг) и триэтиламин (20 мкл) с последующим перемешиванием в течение ночи при комнатной температуре. Затем к реакционной жидкости добавляли хлороформ и органический слой промывали раствором 1 M хлористоводородной кислоты. Органический слой концентрировали и остаток очищали при помощи фракционированной ВЭЖХ (Waters, наименование продукта: Waters SunFireTM Prep C18OBDTM (19×100 мм, 5 мкм)) с получением 2-{4-[(4'-{[4-(метоксиметил)пиперидин-1-ил]карбонил}-2'-метилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-диона (9,0 мг).
Пример 27
3'-({4-[(3,5-Диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2-метилбифенил-4-карбоновую кислоту (10,8 мг) растворяли в смешанном растворе ТГФ-метанола [1 мл, 4:1 (об/об)] и раствор добавляли к 1-этилпиперидин-3-амину (5,8 мг). Добавляли DMT-MM (12 мг) с последующим перемешиванием в течение ночи при комнатной температуре. Затем к реакционной жидкости добавляли хлороформ и органический слой промывали водой. Органический слой концентрировали и остаток очищали при помощи фракционированной ВЭЖХ (Waters, наименование продукта: Waters SunFireTM Prep C18OBDTM (19×100 мм, 5 мкм)) с получением 3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-N-(1-этилпиперидин-3-ил)-2-метилбифенил-4-карбоксамида (3,4 мг).
Таким же способом, как в способах примеров 1-27, получали соединения примеров 28-407, представленные в таблицах ниже, с использованием соответствующих исходных веществ соответственно. Структуры соединений примеров представлены в таблицах 53-113, а способы получения и физико-химические данные в таблицах 114-135.
Кроме того, структуры других соединений по настоящему изобретению представлены в таблицах 136-138. Их можно легко синтезировать с использованием указанных выше способов получения, способов, описанных в примерах, и способов, очевидных для специалистов в данной области, или способов, представляющих собой модификацию таких способов.
Промышленная применимость
Поскольку соединение по настоящему изобретению обладает отличным агонистическим действием в отношении GPR40, оно является полезным в качестве средства, стимулирующего секрецию инсулина или средства для профилактики/лечения заболевания, с которым связан GPR40, такого как диабет (инсулин-зависимый сахарный диабет (IDDM), инсулин-независимый сахарный диабет (NIDDM), легкая форма диабета пограничного типа (аномальные толерантность к глюкозе и уровни глюкозы в крови натощак) и т.п.
Отдельно представленный список последовательностей
Объяснение “Искусственной последовательности” представлено под цифровым обозначением <223> в представленном далее списке последовательностей. Иллюстративно, нуклеотидная последовательность, представленная как SEQ ID NO:1 в списке последовательностей, является нуклеотидной последовательностью искусственно синтезированного праймера. Также нуклеотидная последовательность, представленная как SEQ ID NO:2 в списке последовательностей, является нуклеотидной последовательностью искусственно синтезированного праймера.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИН-1-ОНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТА ВВ2 | 2008 |
|
RU2479578C2 |
| ПОЛИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2012 |
|
RU2621039C1 |
| ПРОИЗВОДНОЕ ХИНОЛОНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 2007 |
|
RU2440987C2 |
| ПРОИЗВОДНЫЕ ТРИАЗИНОНА, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ СОРНЯКОВ | 1989 |
|
RU2047296C1 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ АМИДА | 2010 |
|
RU2563639C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ АМИДА | 2010 |
|
RU2609021C2 |
| ПРОИЗВОДНОЕ ПИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2002 |
|
RU2285002C2 |
| СОЕДИНЕНИЕ СУЛЬФОНАМИДА ИЛИ ЕГО СОЛЬ | 2007 |
|
RU2425029C2 |
| ПРОИЗВОДНЫЕ НАФТИРИДИНА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2240322C2 |
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА ИЛИ ЕГО СОЛЬ | 2005 |
|
RU2347776C2 |


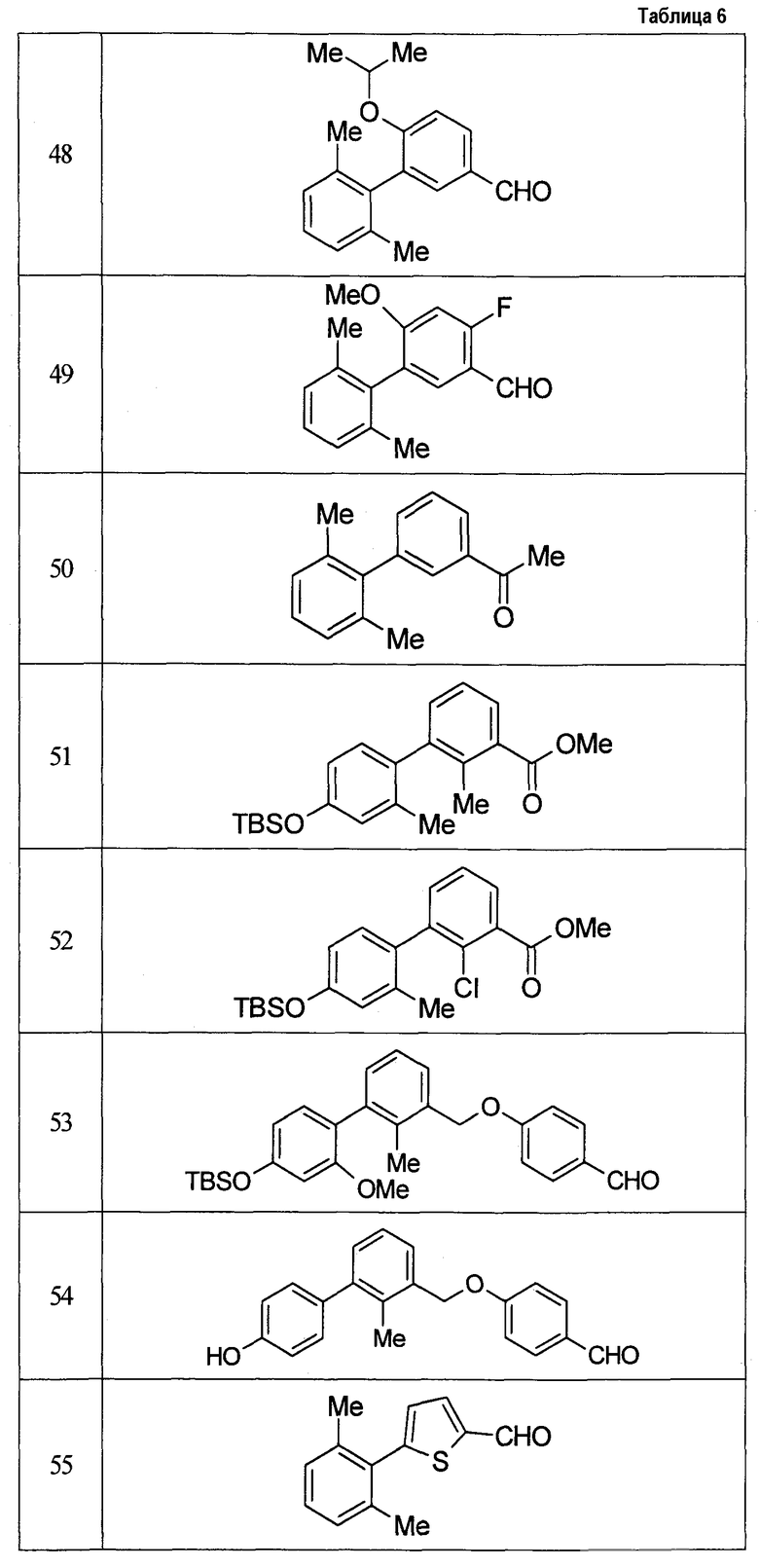

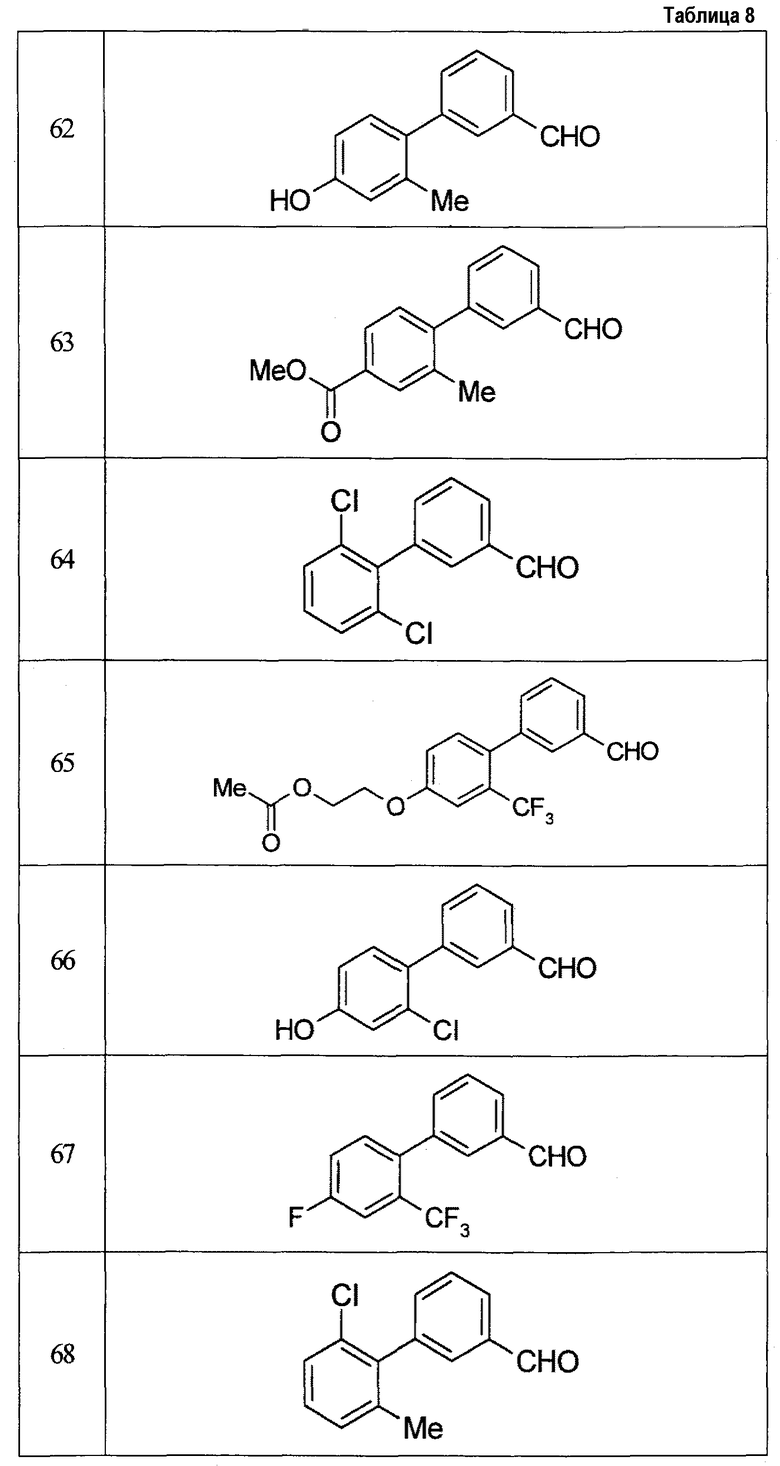
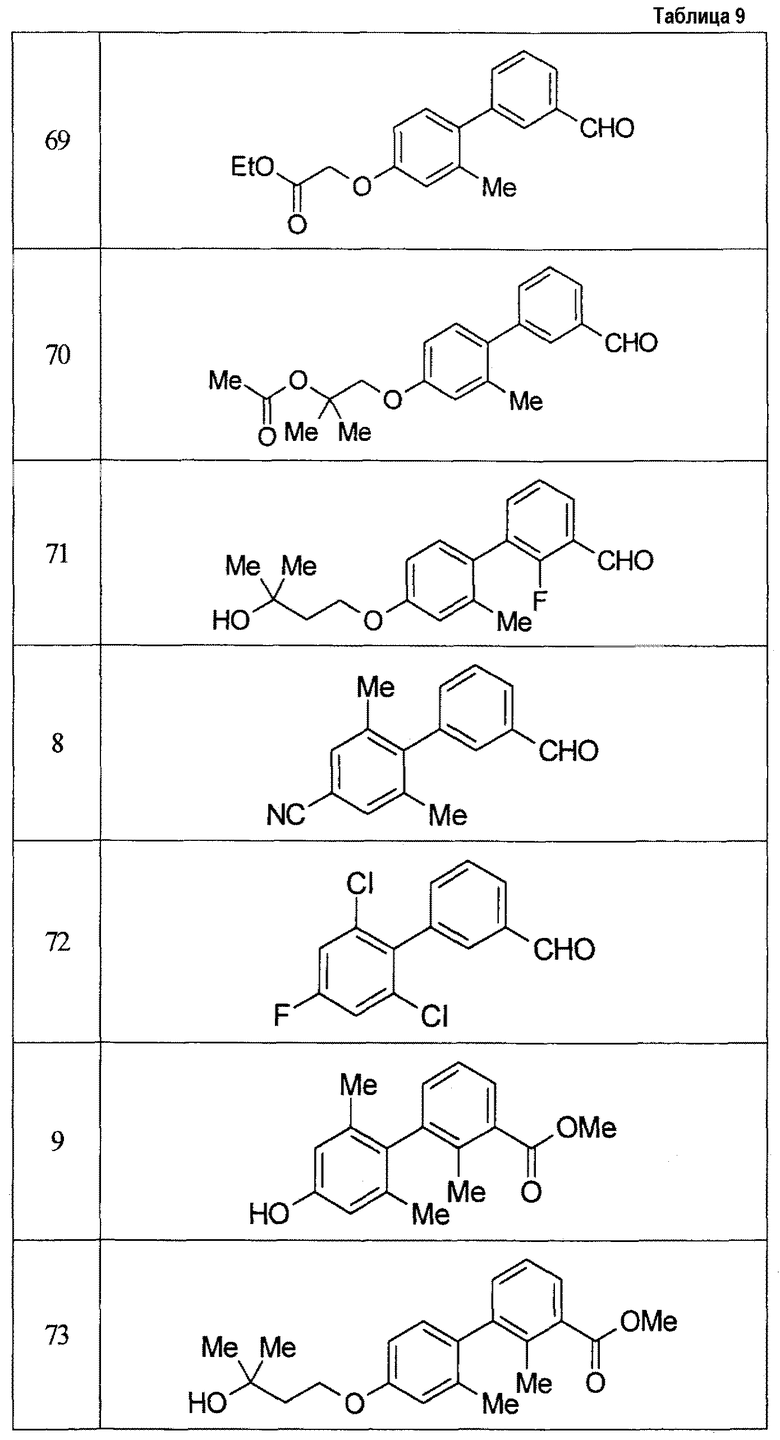









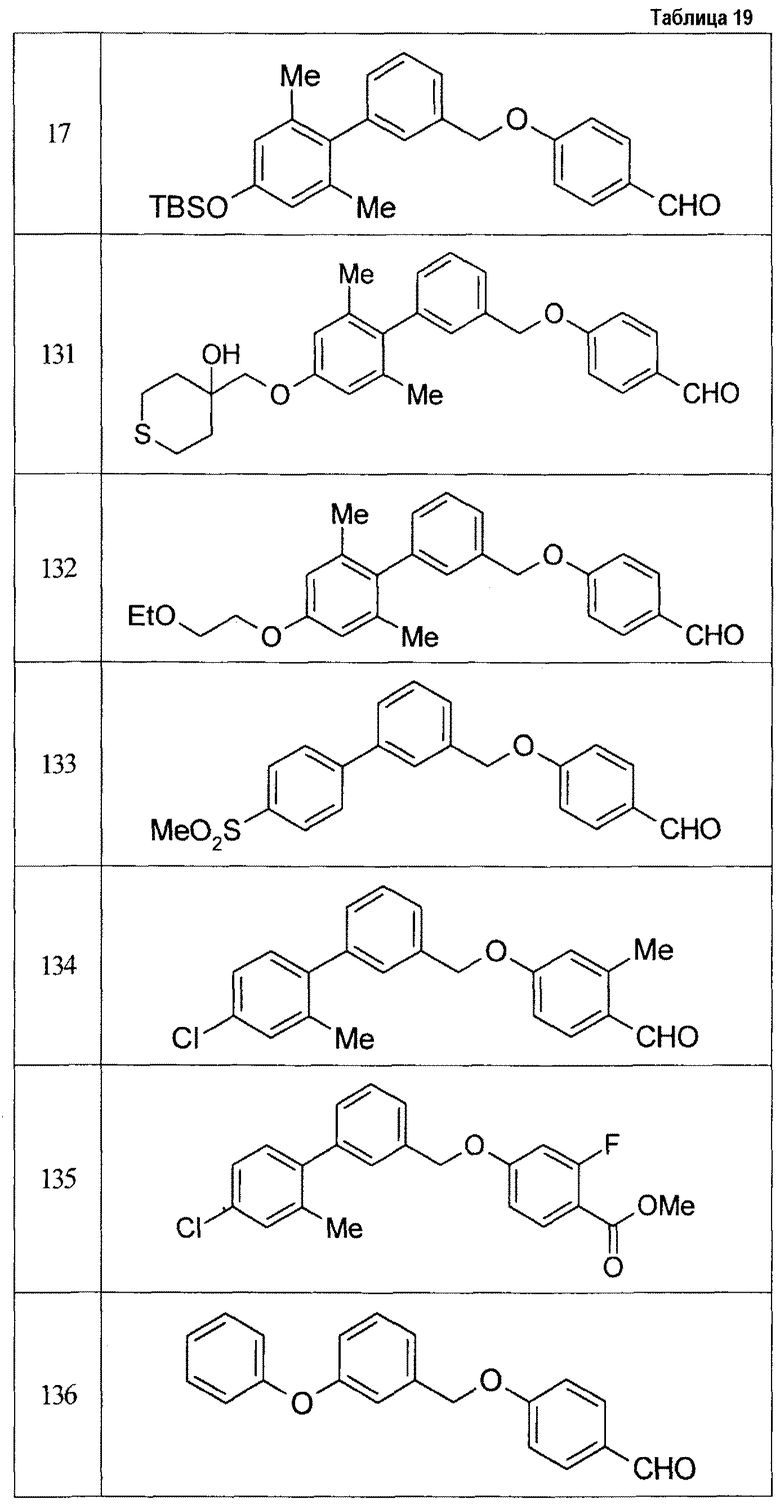






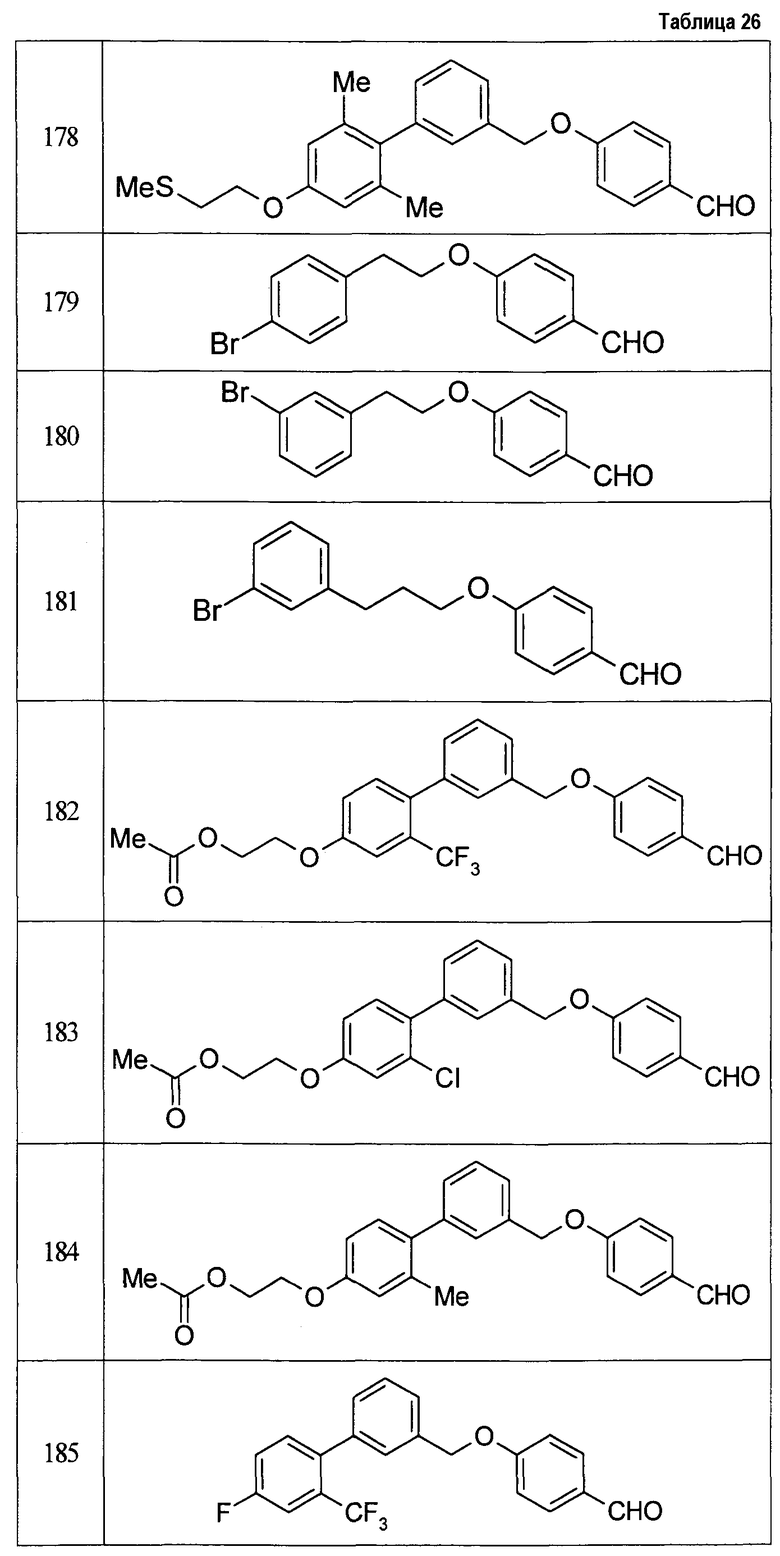




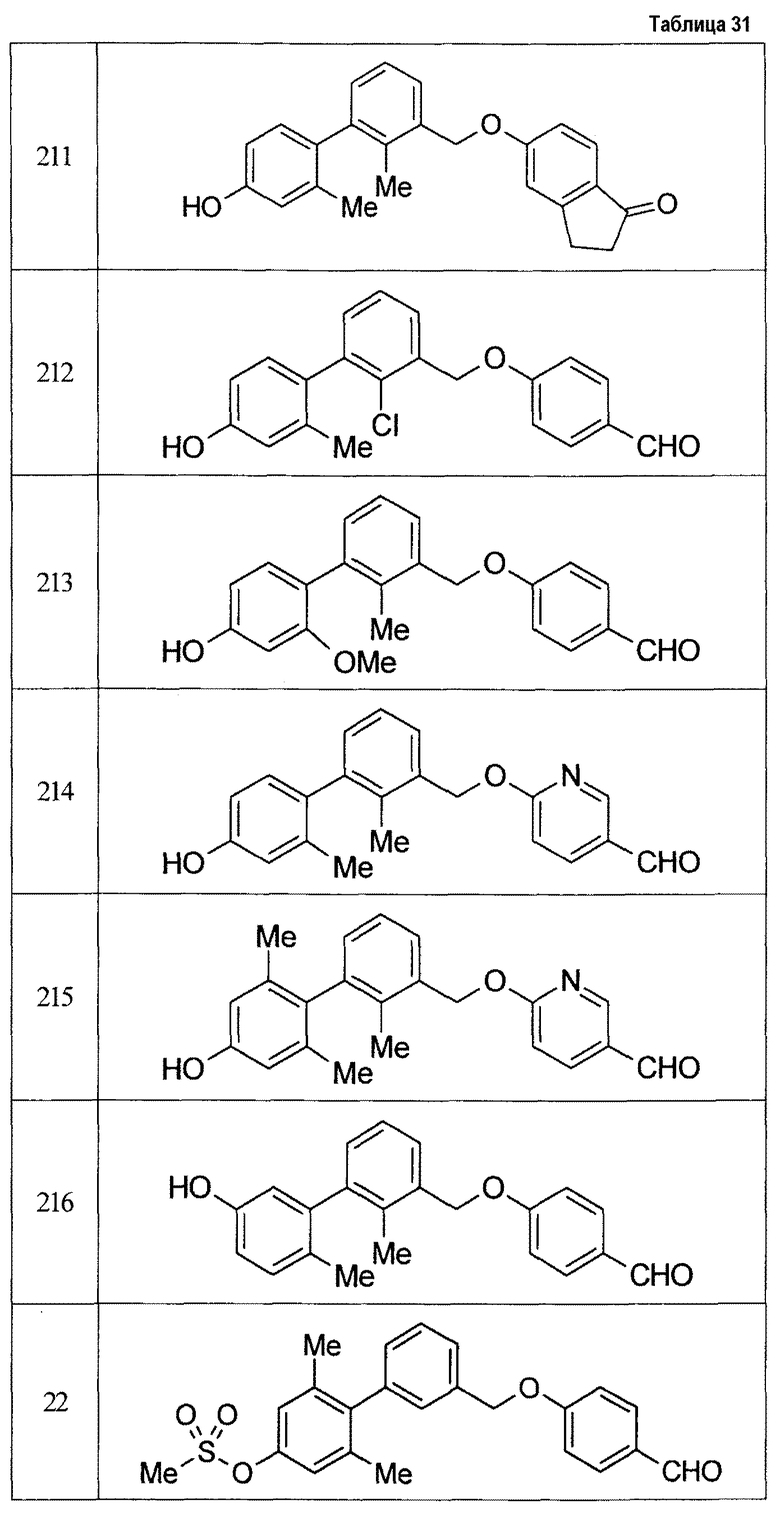



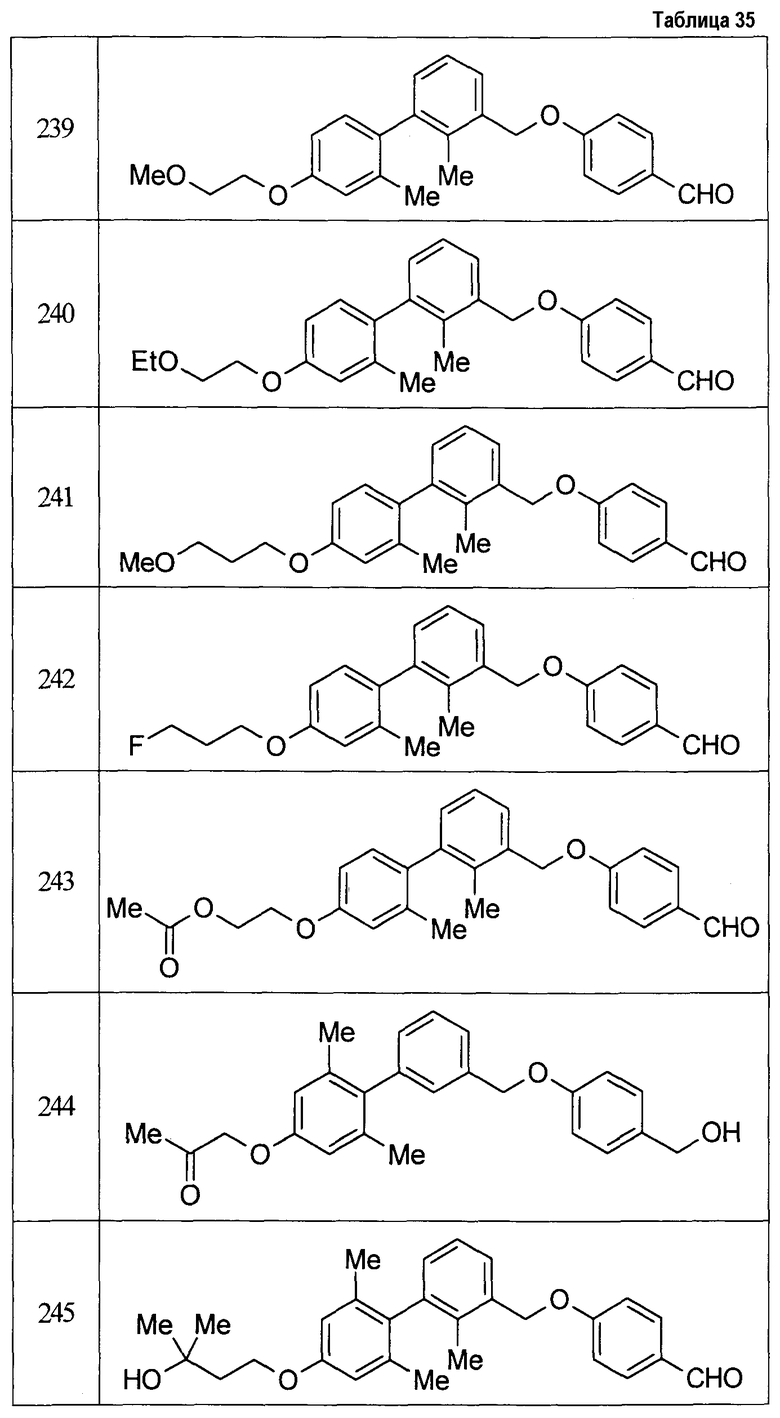








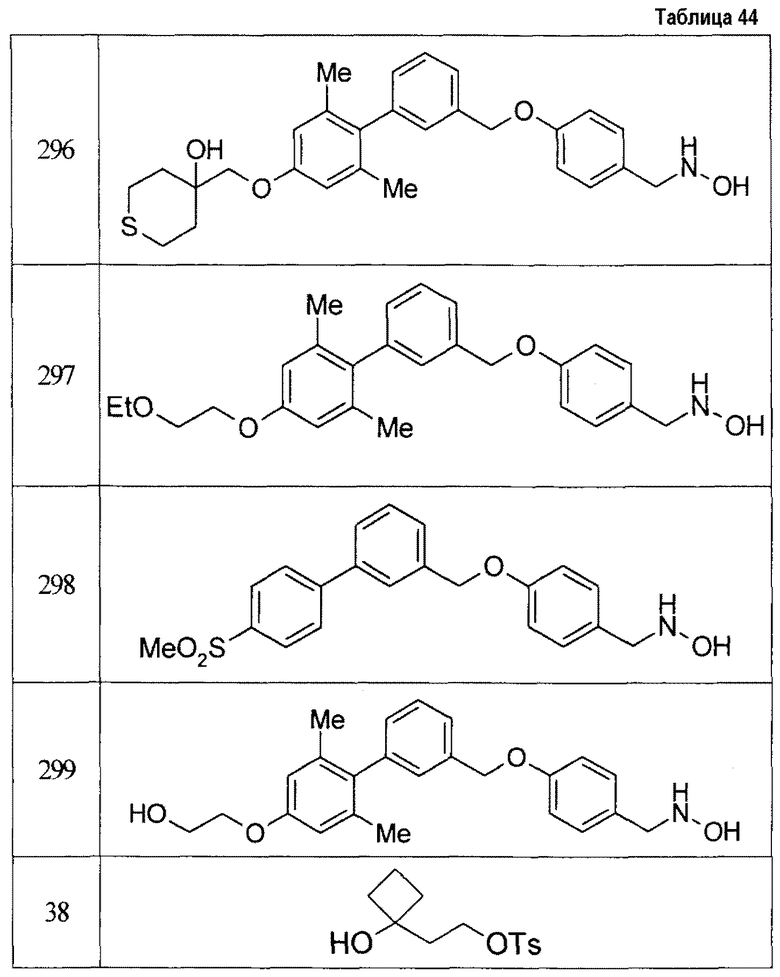









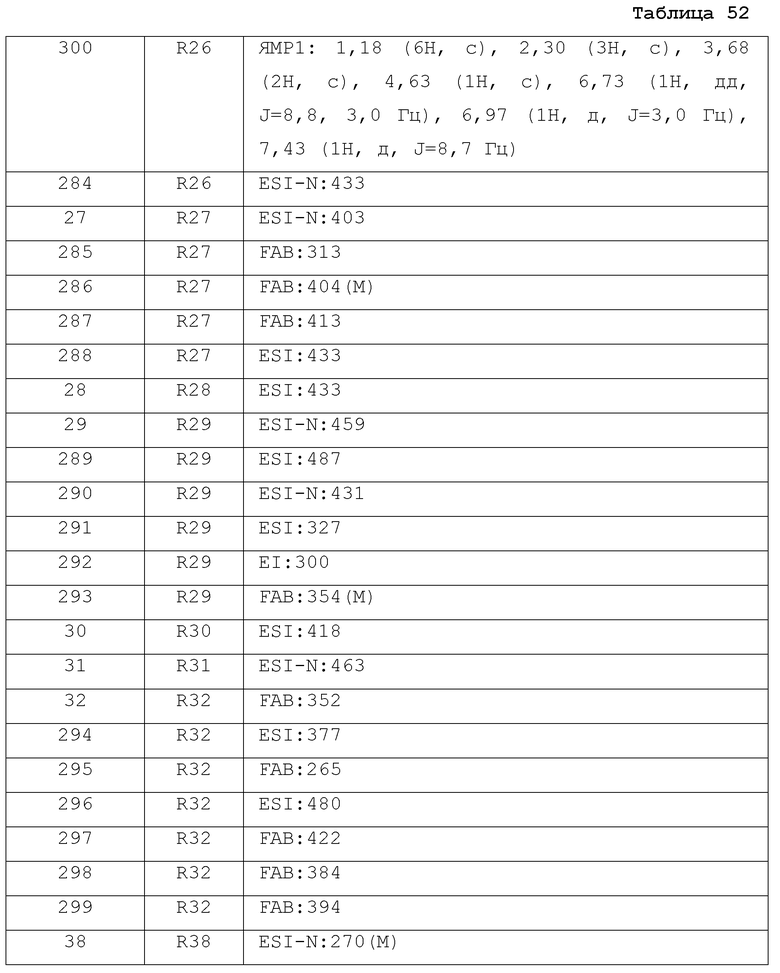


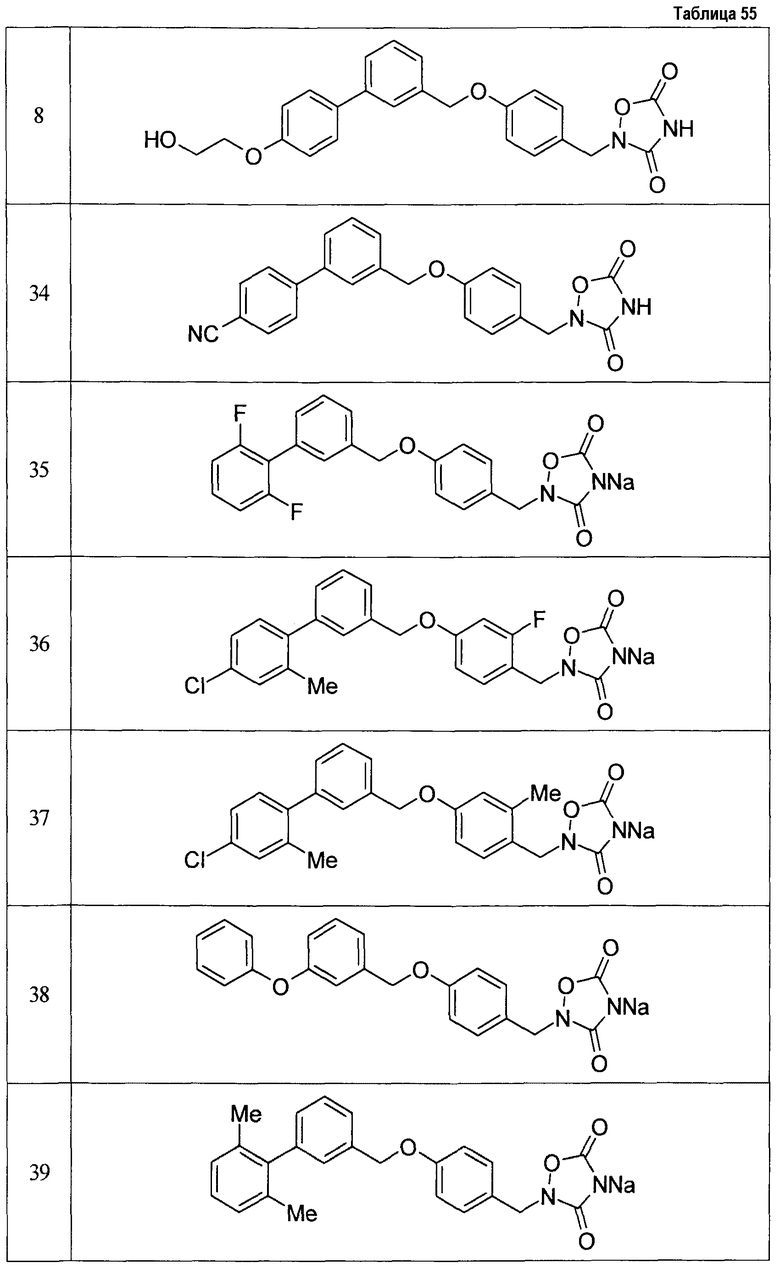



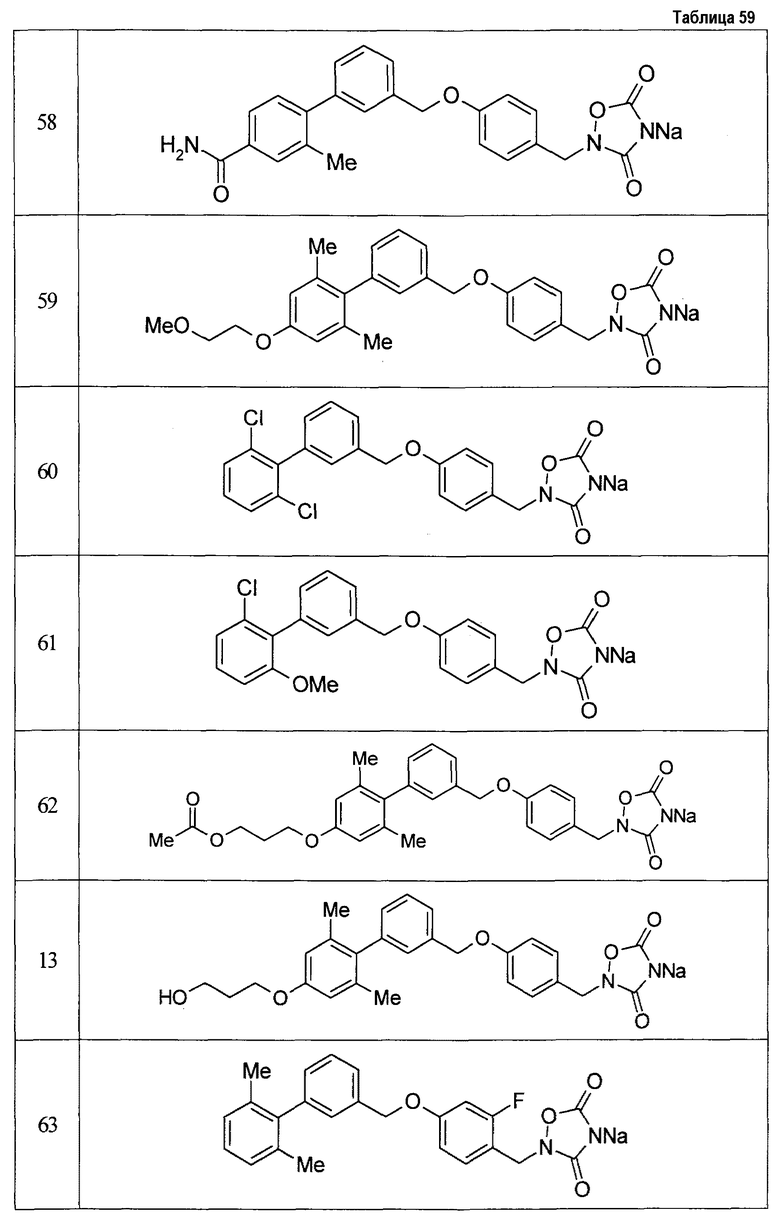


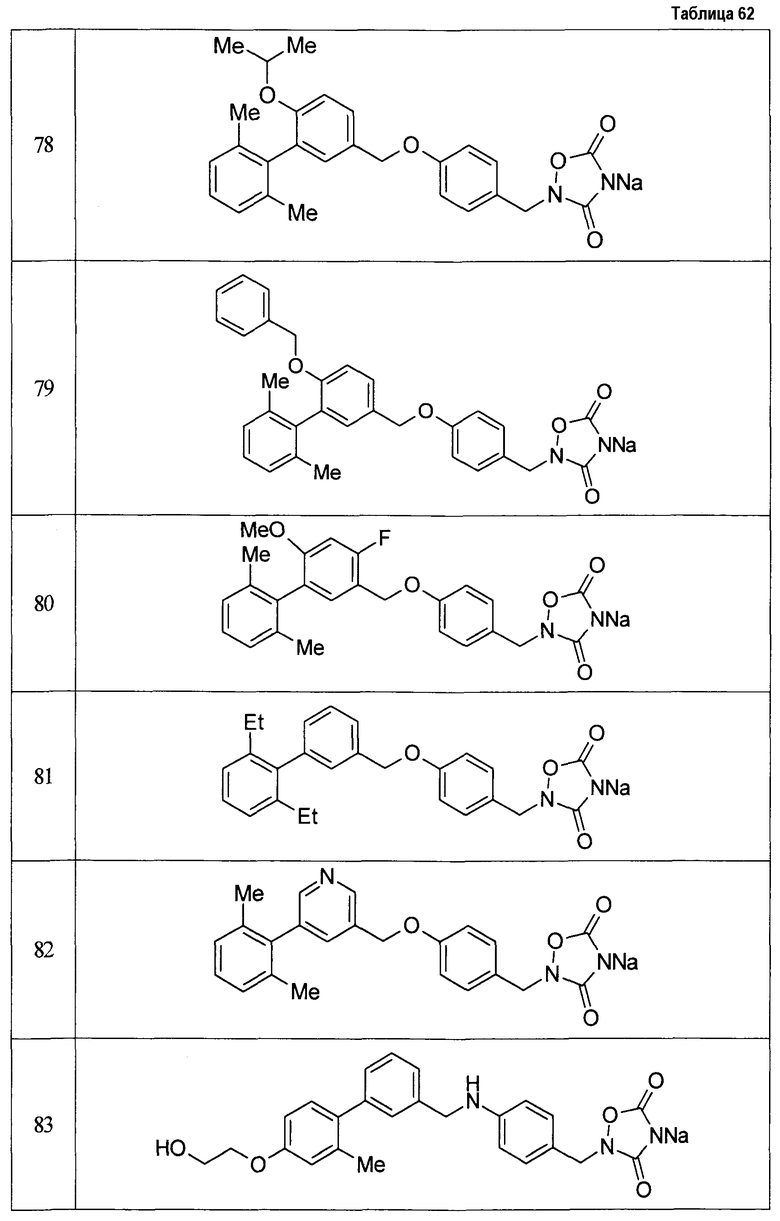
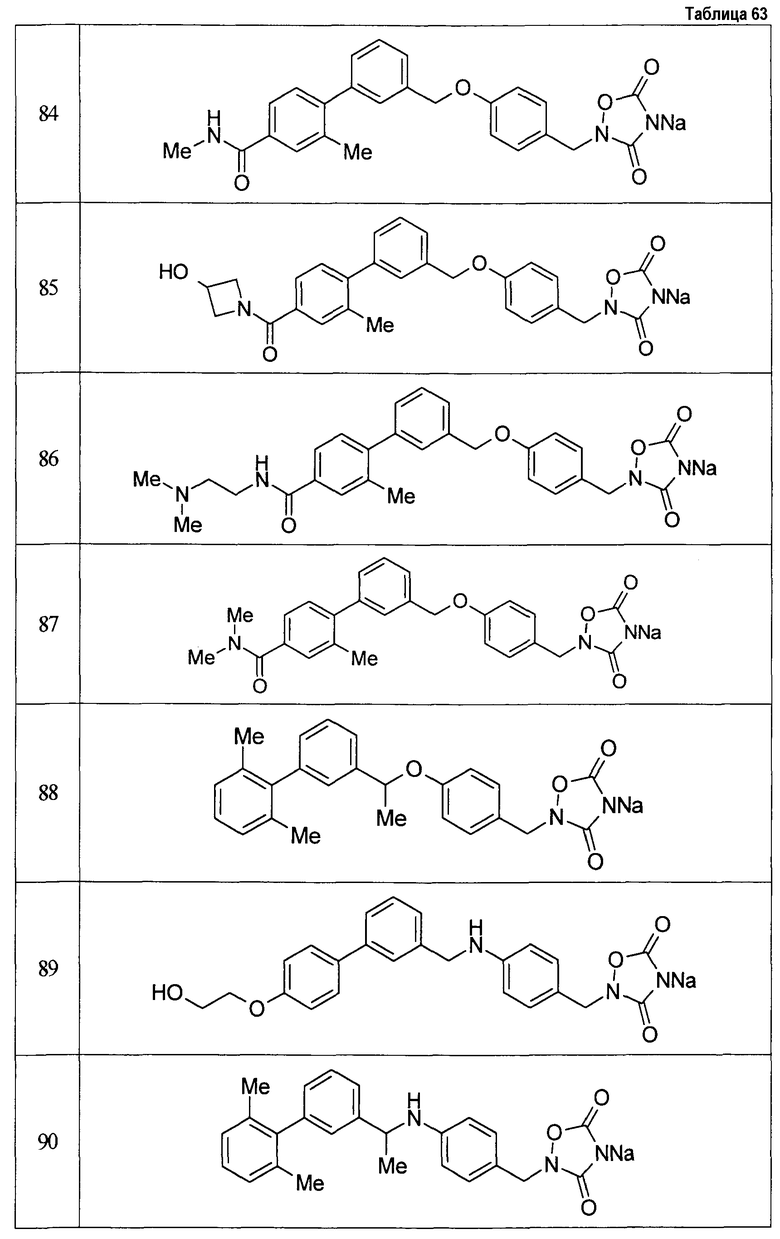

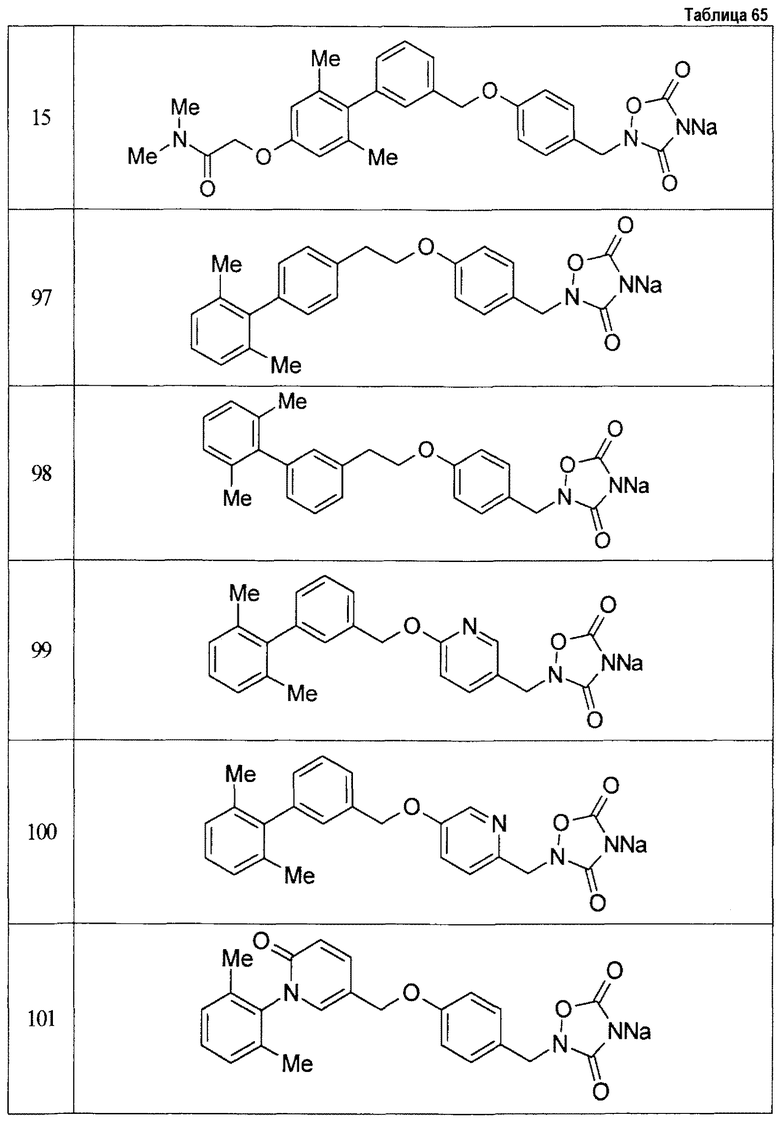


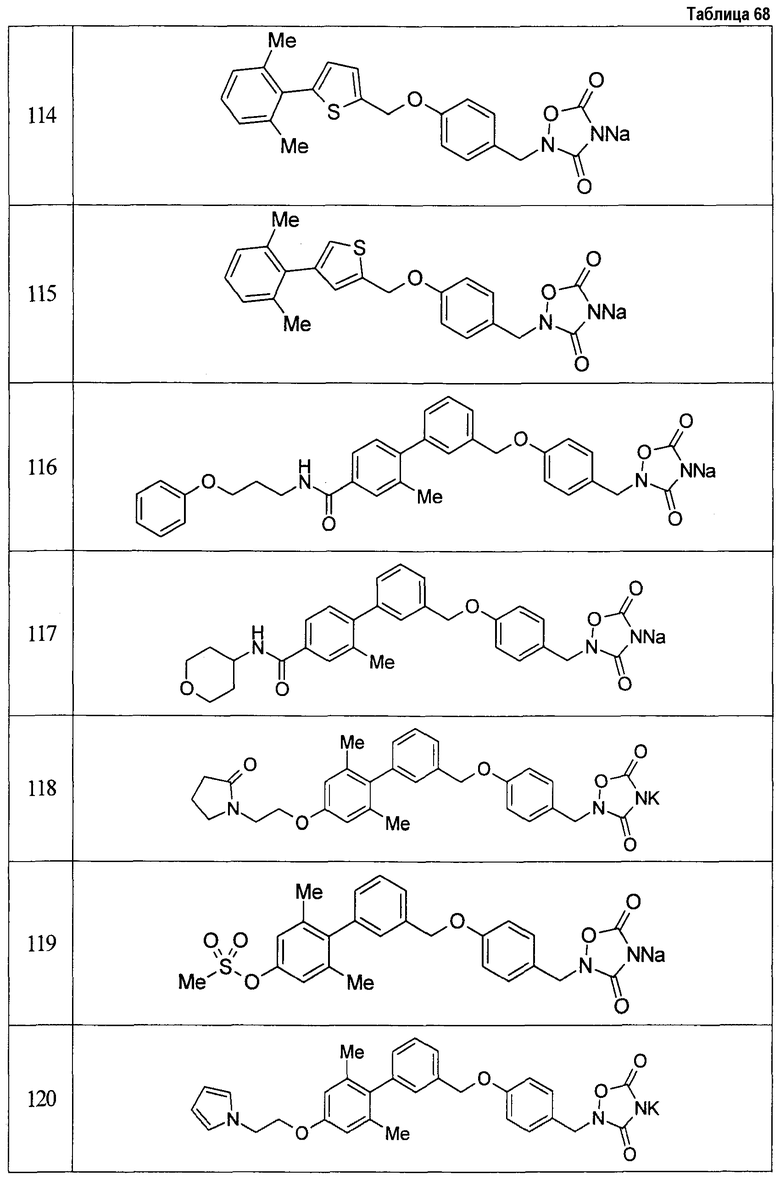






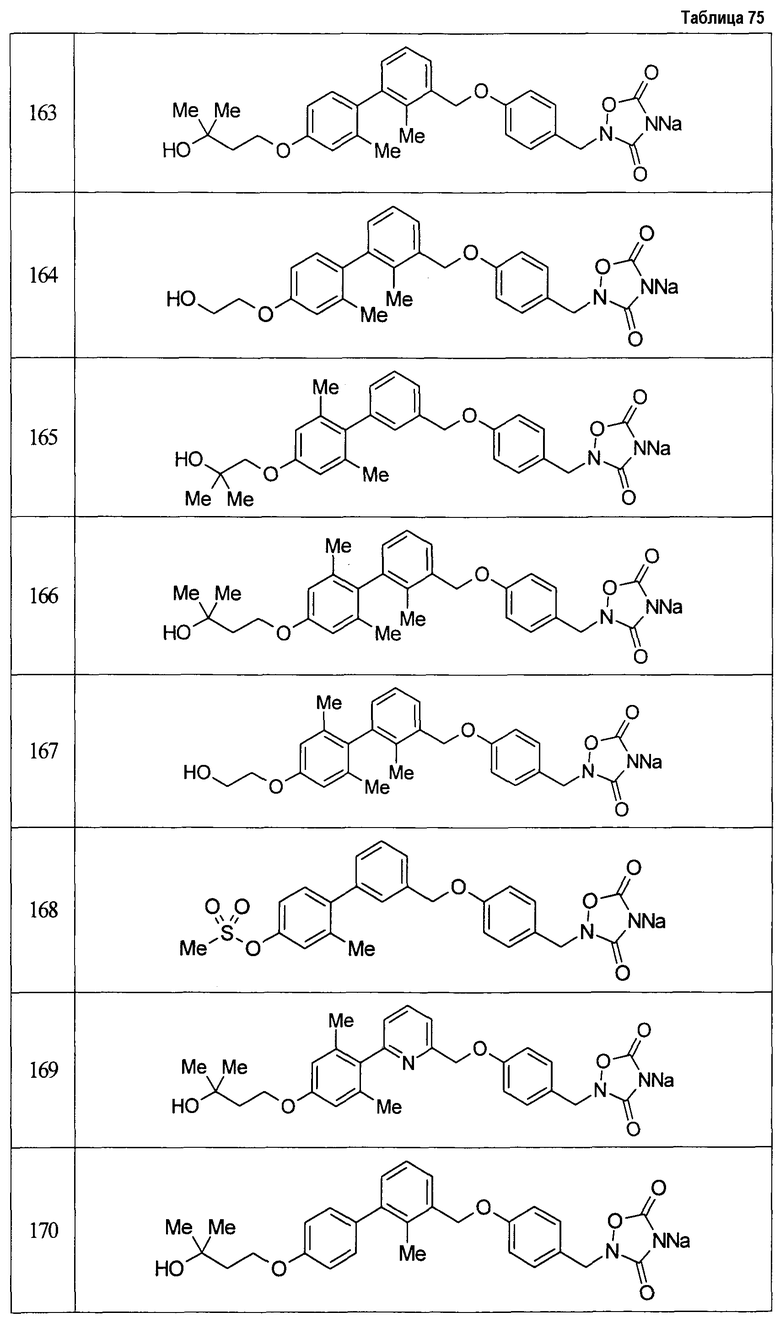








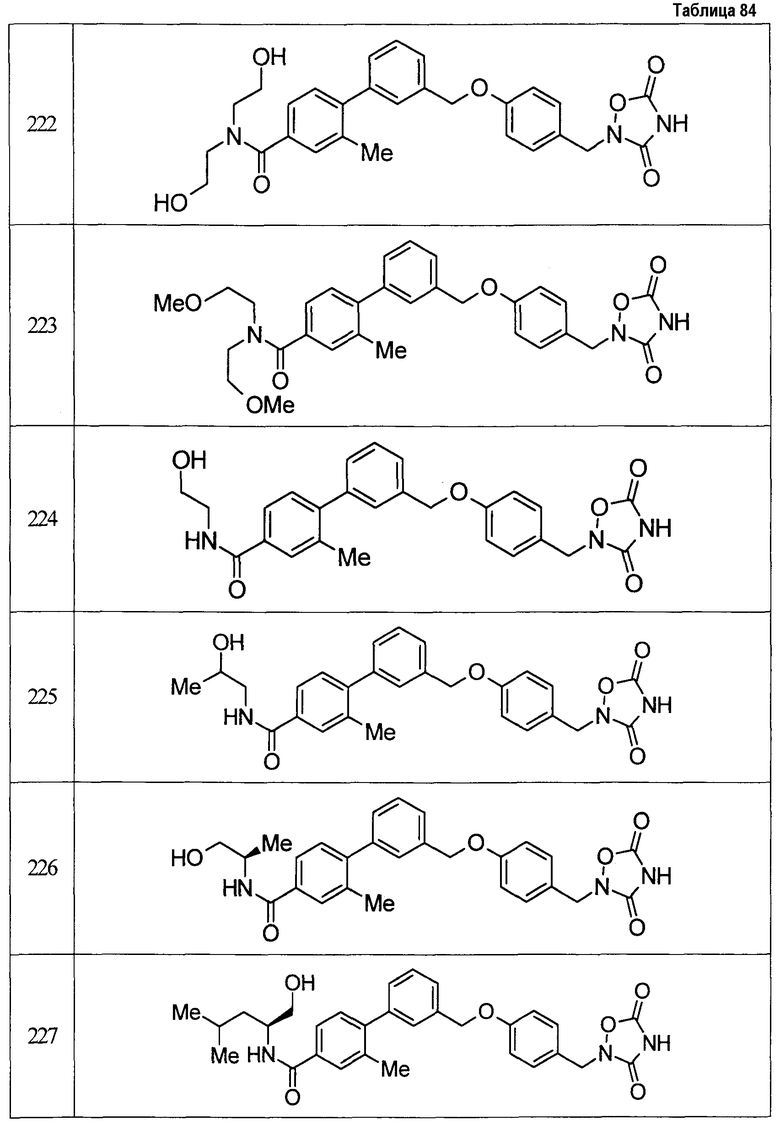

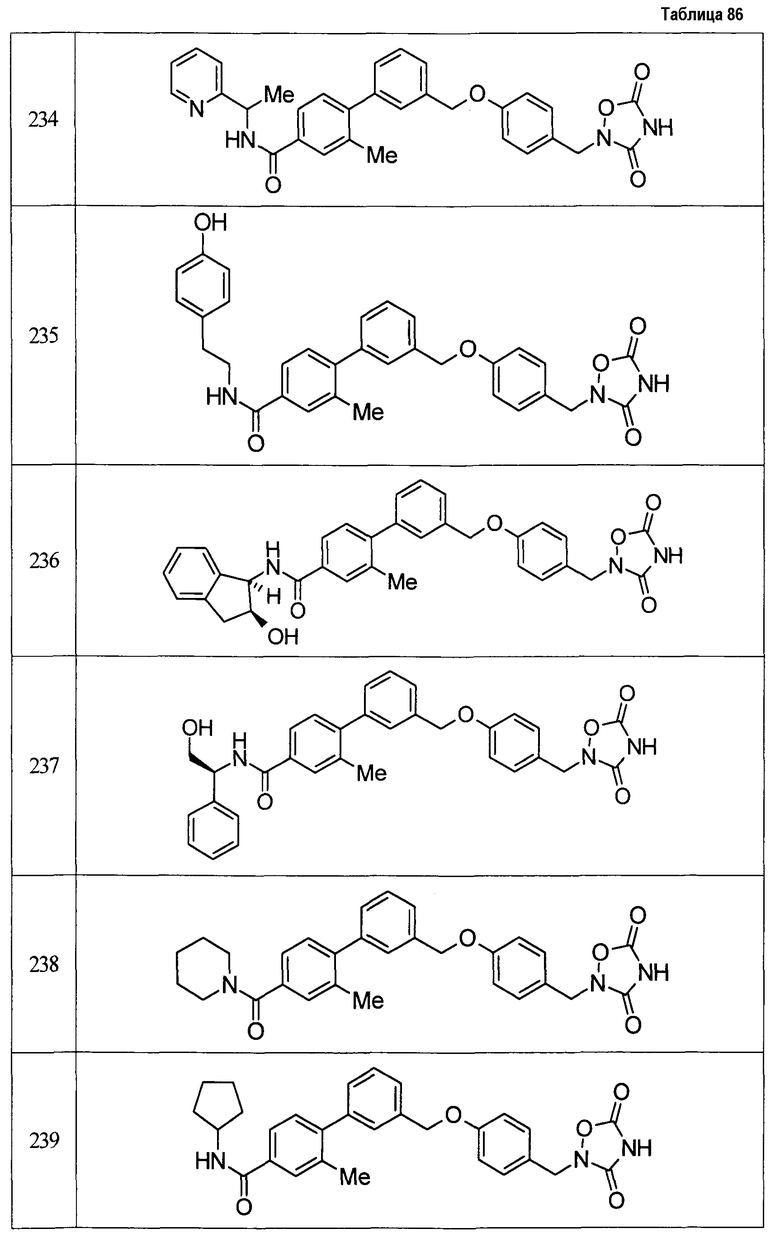
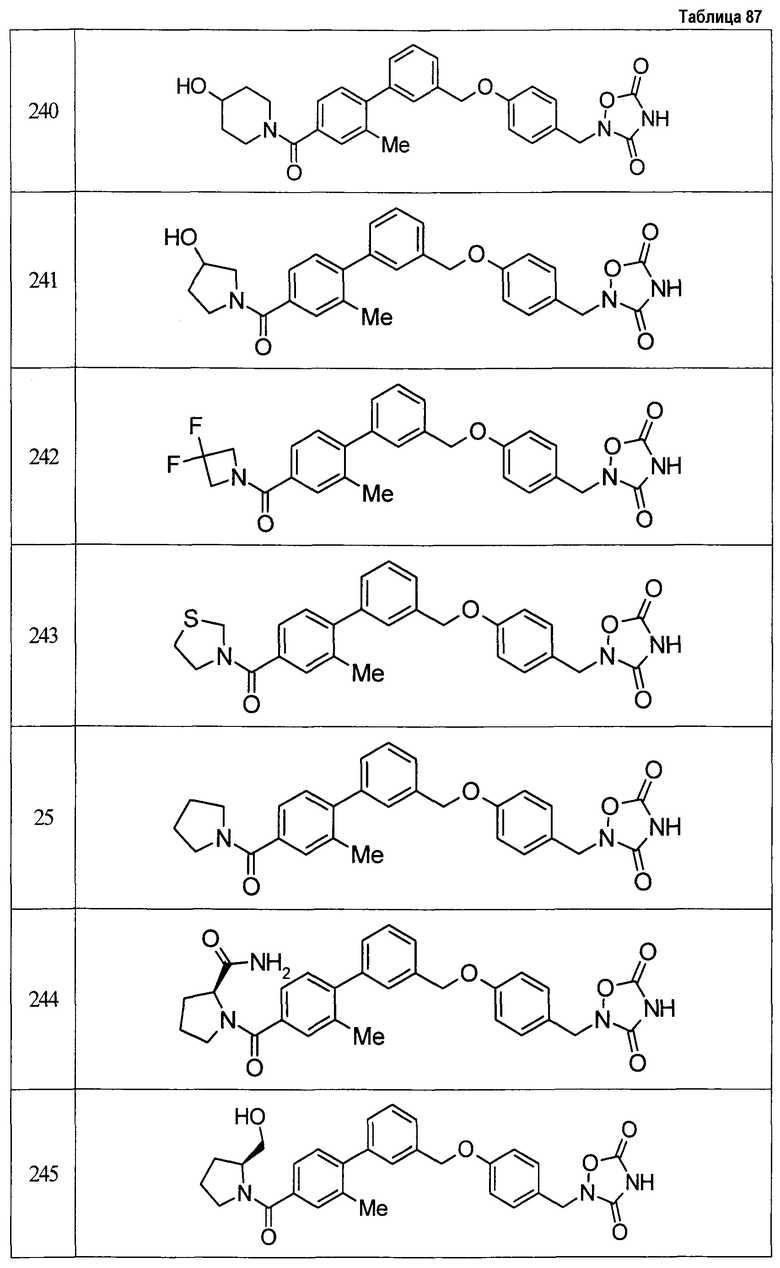


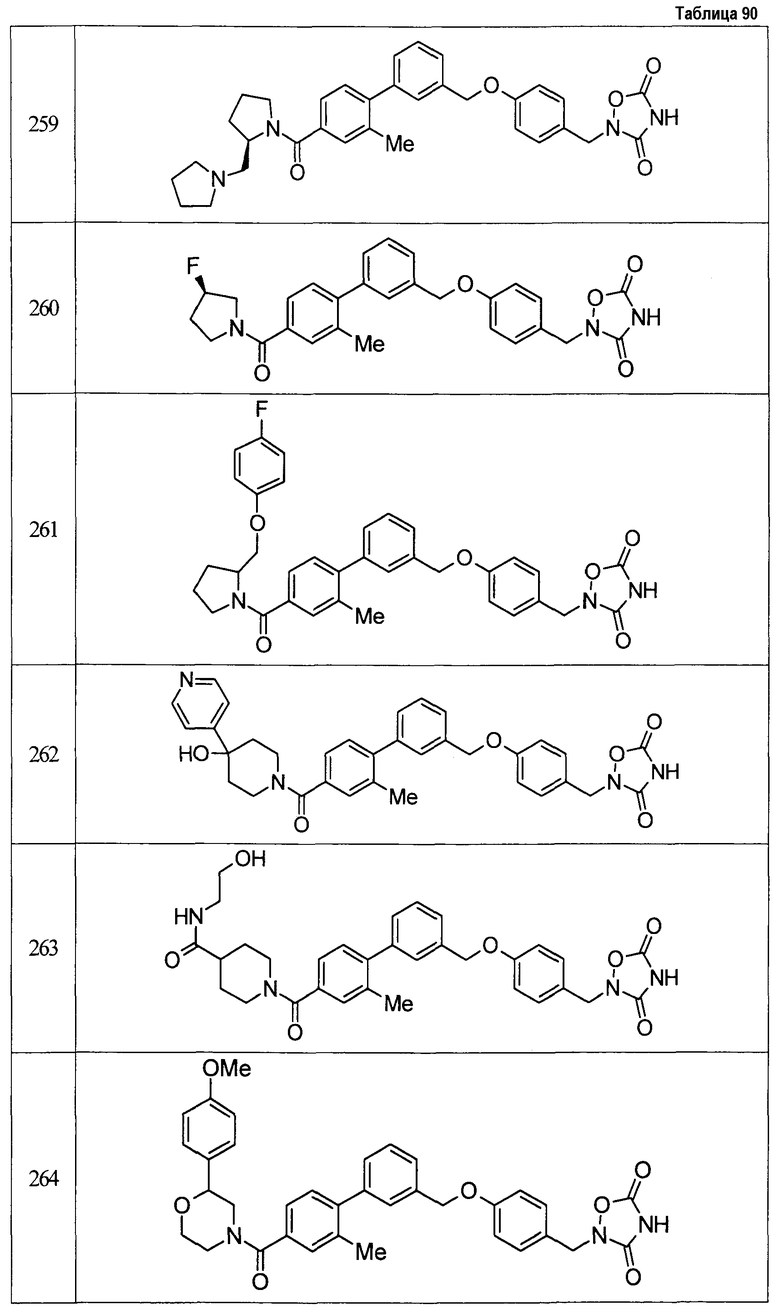
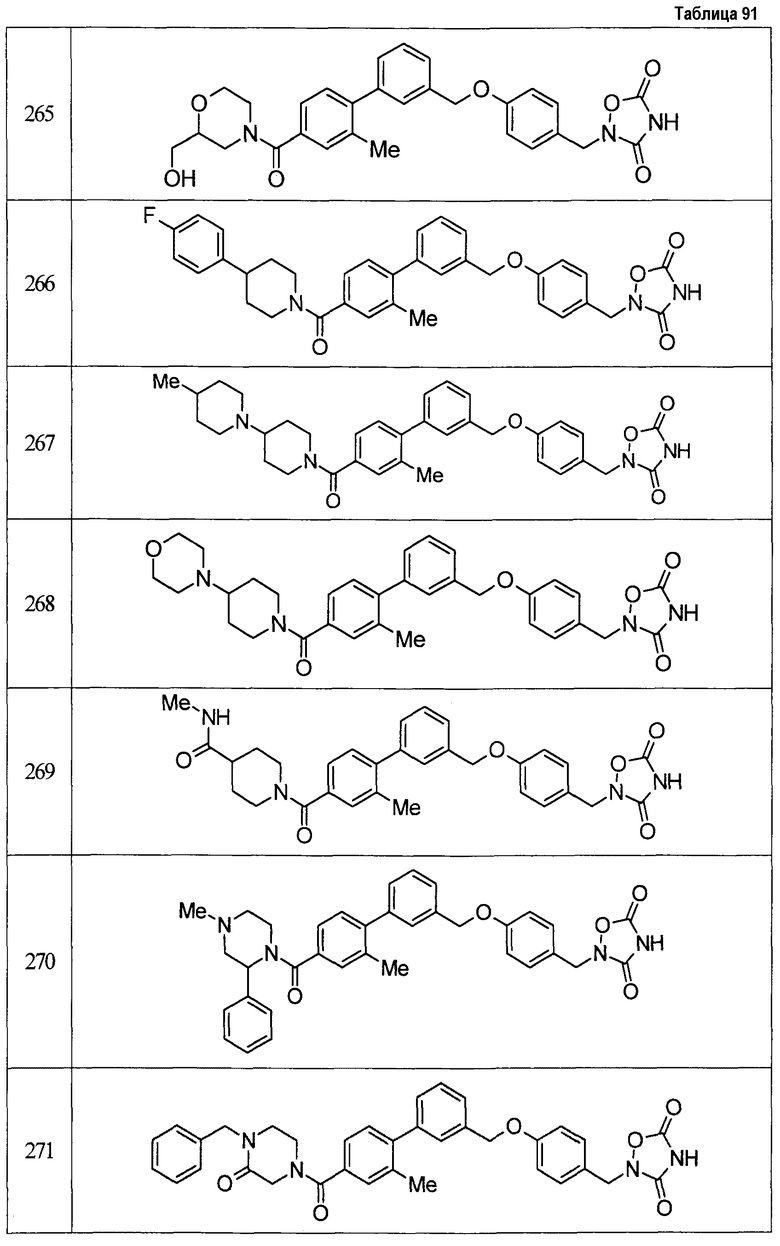
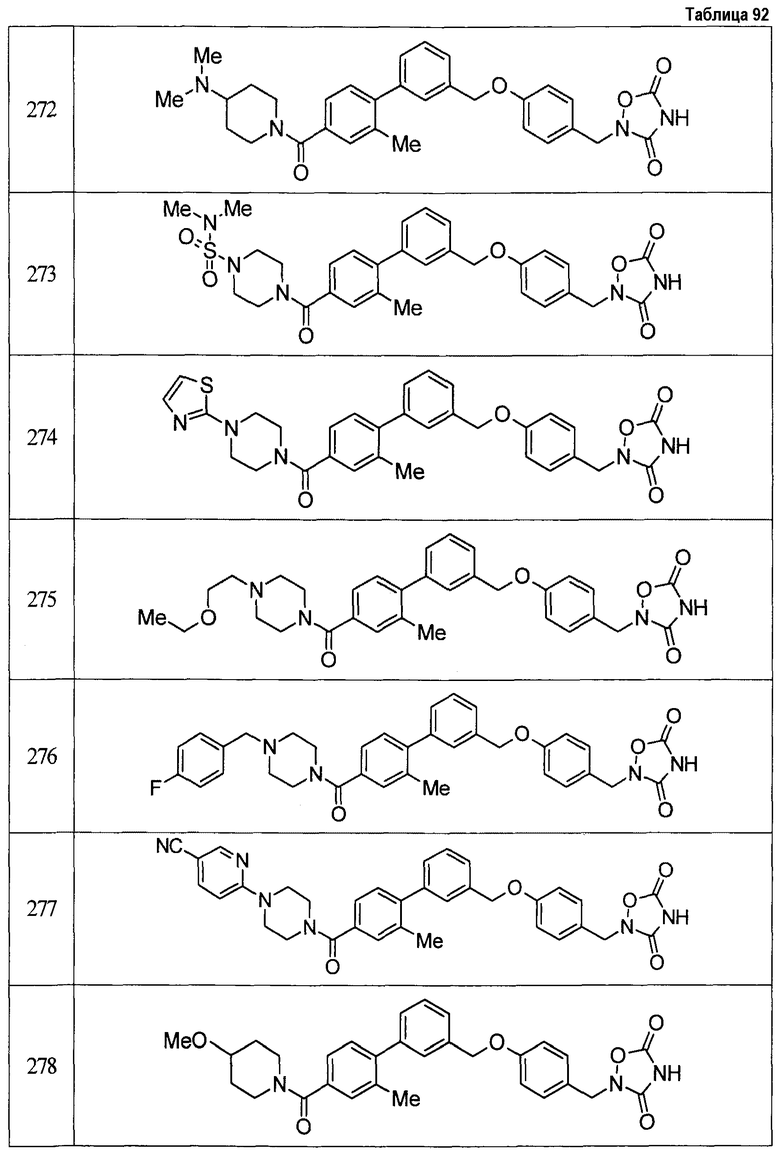


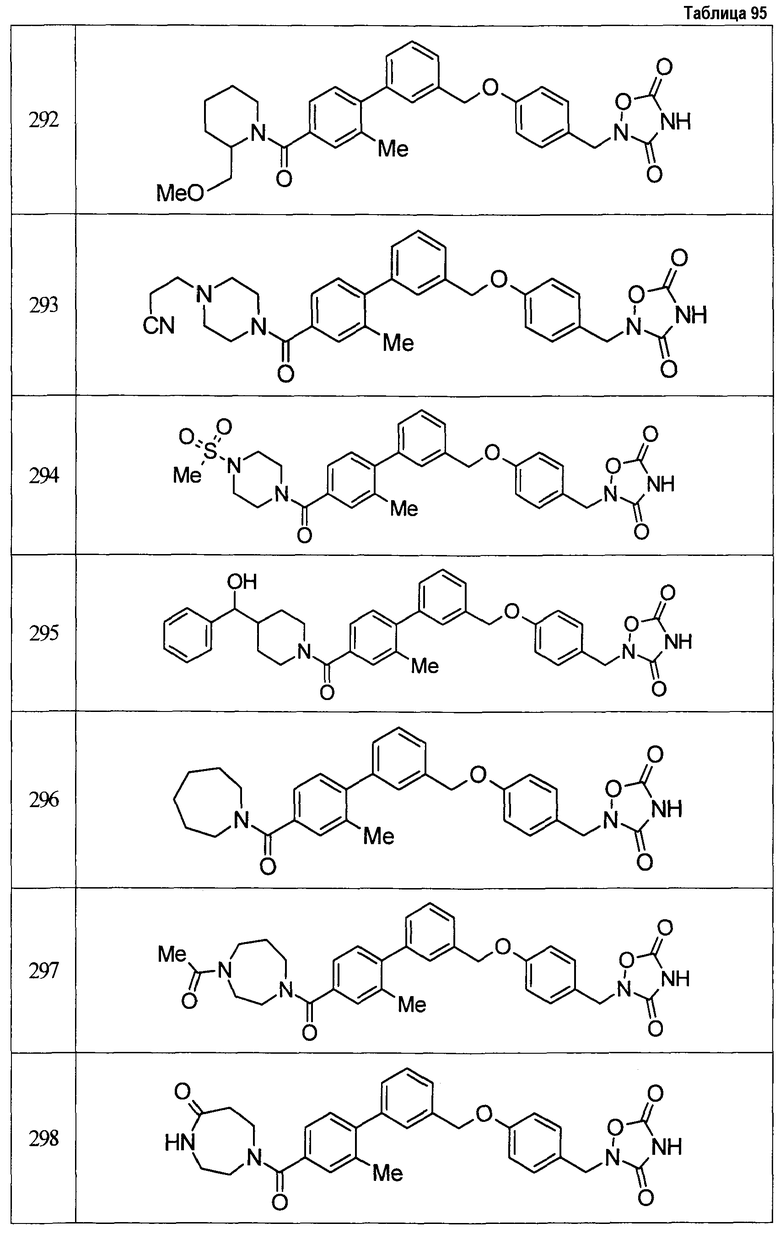



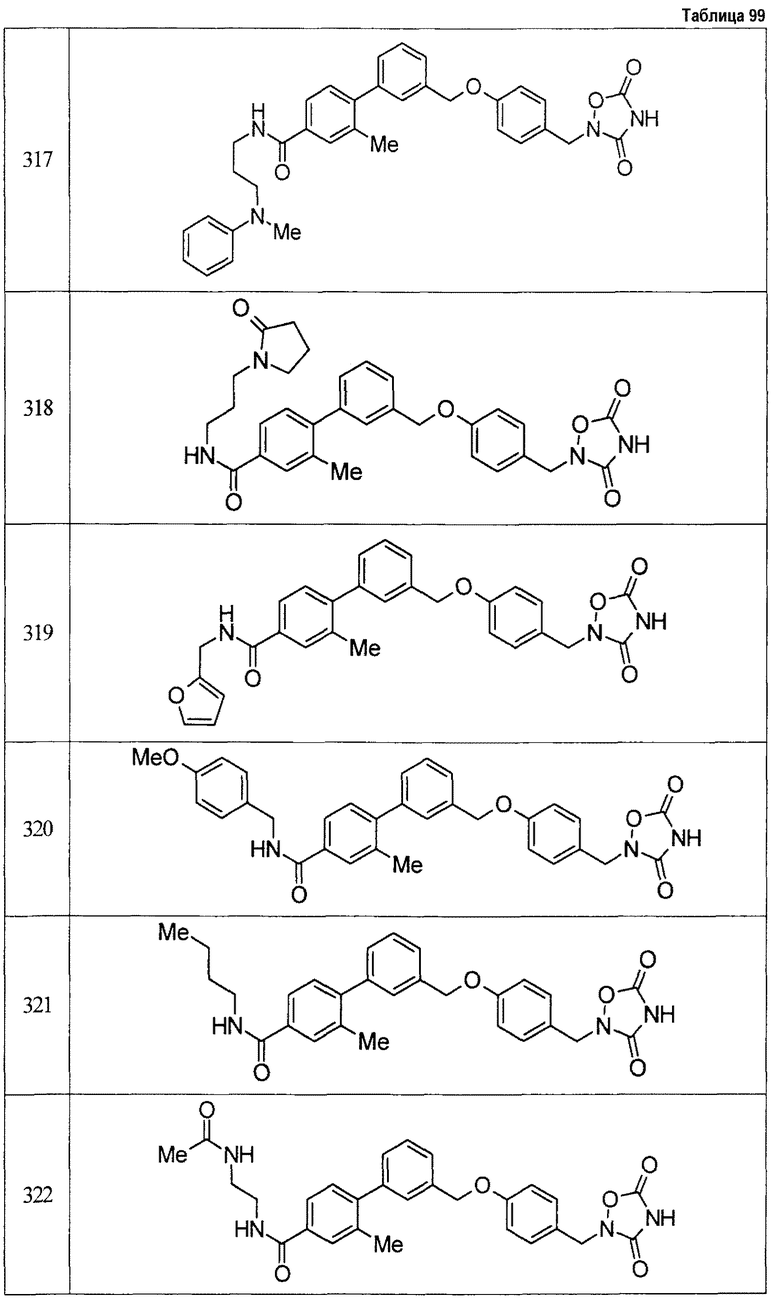



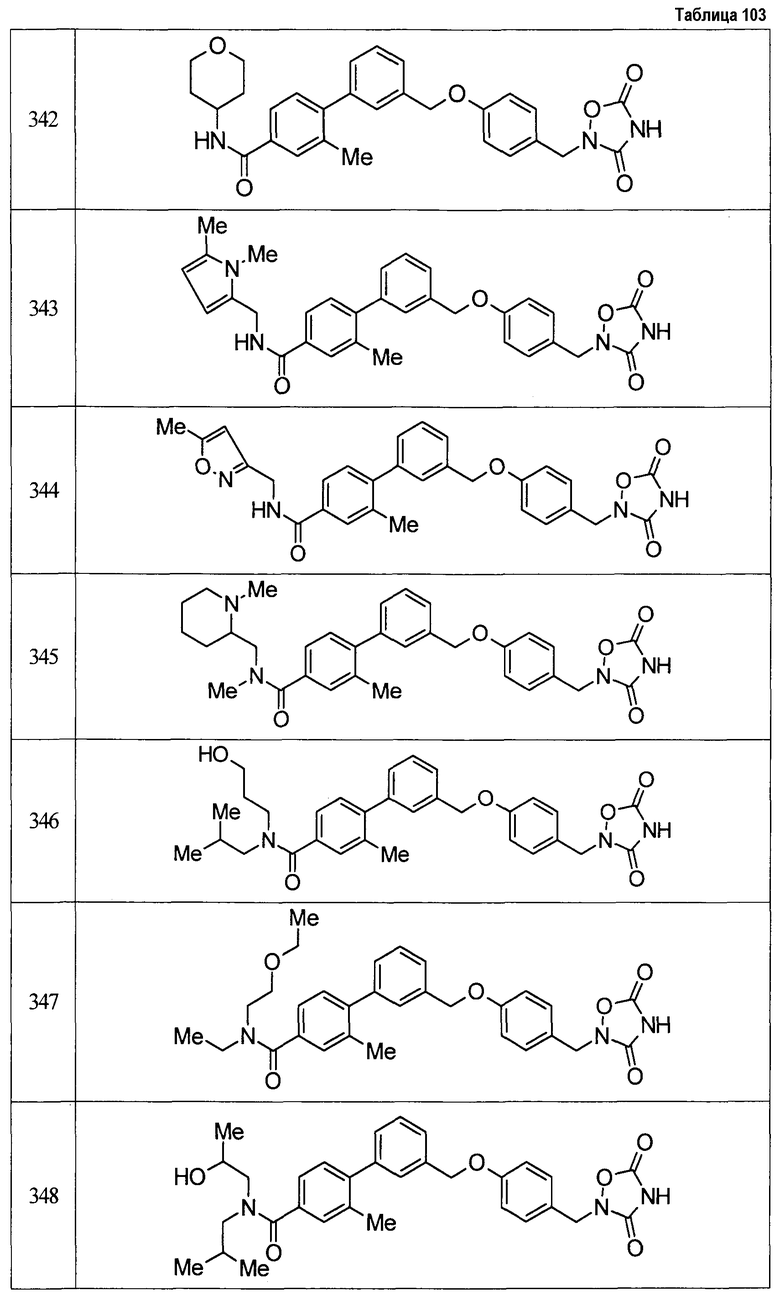










































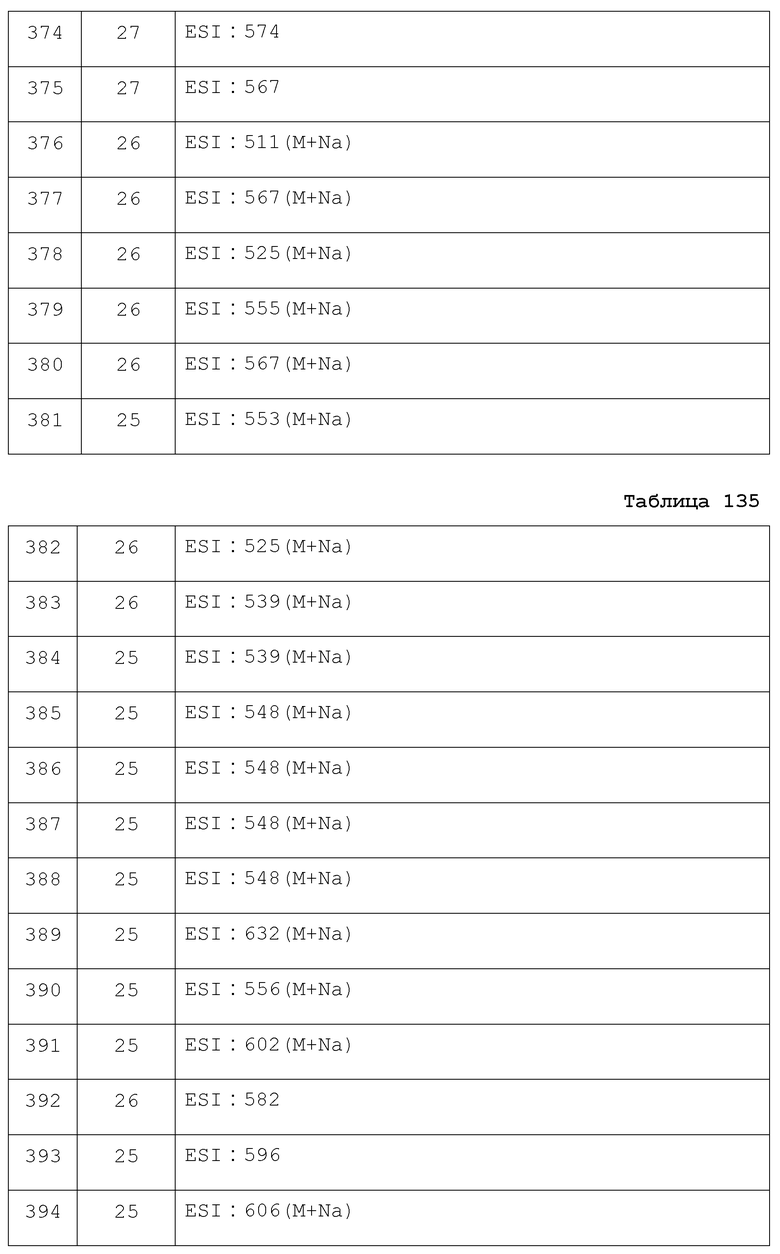







Изобретение относится к оксадиазолидиндионовым соединениям, представленным следующей формулой (I), или к их фармацевтически приемлемым солям,
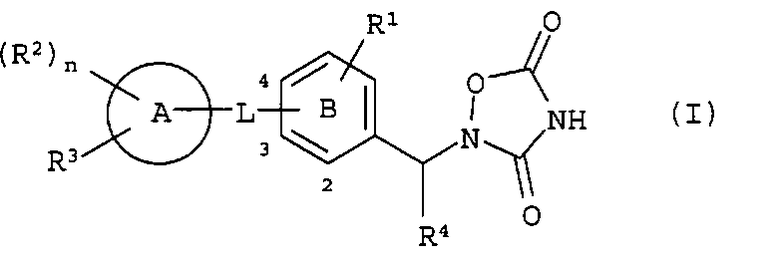
(символы в представленной формуле представляют следующие значения, R1: -Н, R0: низший алкил, Rz: одинаковые или отличные друг от друга, и каждый представляет собой -Н или низший алкил, L: *-СН2-O- или *-CH2-NH-, где знак * в L означает связывание с кольцом А и положение замещения группы L в кольце В представляет собой 4-положение, кольцо А: бензол, кольцо В: бензол или пиридин, R2: соответственно одинаковые или отличные друг от друга, и каждый представляет собой -галоген или -R0, n: 0 или 1, R3: фенил, который может быть замещен группой, выбранной из группы G3, группа G3: галоген, -R0, галогено-низший алкил, -ORz, -CON(Rz)2, -CON(Rz)-гетерокольцевая группа, -O-S(O)2-R0, -О-низший алкилен-ORz, -О-низший алкилен-O-CORz, -О-низший алкилен-N(Rz)2, -О-низший алкилен-N(Rz)CO-Rz, -О-низший алкилен-CO2Rz, -О-низший алкилен-CON(Rz)z, -О-низший алкилен-CON(Rz)-(низший алкил замещен группой -ORz), -О-низший алкилен-SR0, -О-низший алкилен-циклоалкил, -О-низший алкилен-CON(Rz)-циклоалкил, -О-низший алкилен-гетерокольцевая группа и -О-низший алкилен-CON(Rz)-гетерокольцевая группа, где низший алкилен в группе G3 может быть замещен галогеном или -ORz, и циклоалкил и гетерокольцевая группа в группе G3 могут быть замещены группой, выбранной из группы G1, группа G1: галоген, циано, -R0, -ORz, -N(Rz)2, -S-R0, -SO2-R0, -SO2N(Rz)2, -CO-Rz, -CON(Rz)2, -CON(Rz)-низший алкилен-ORz, -N(Rz)CO-Rz, оксо, -(низший алкилен, который может быть замещен группой -ORz)-арил, гетерокольцевая группа и низший алкилен-гетерокольцевая группа, где арил и гетерокольцевая группа в группе G1 могут быть замещены группой, выбранной из следующей группы G2, группа G2: галоген, циано, где "гетерокольцевая группа" означает группу, содержащую кольцо, выбранное из i) моноциклического 5-7-членного насыщенного или ненасыщенного гетерокольца, содержащего от 1 до 3 гетероатомов, выбранных из О, S и N, ii) бициклического гетерокольца, в котором гетерокольца, указанные в представленном выше i), являются конденсированными по кольцу, где конденсированные кольца могут быть одинаковые или отличные друг от друга, и iii) бициклического гетерокольца, в котором гетерокольцо, указанное в представленном выше i), является конденсированным с бензольным кольцом или 5-7-членным циклоалканом, R4: -Н. Изобретение также относится к фармацевтической композиции, к применению соединений по п.1, а также к способу профилактики и/или лечения диабета. Технический результат - получение новых биологически активных соединений, представляющих собой агонист GPR40, средство, стимулирующее секрецию инсулина. и/или средство для профилактики и/или лечения диабета. 4 н. и 5 з.п.ф-лы, 138 табл.
1. Оксадиазолидиндионовое соединение, представленное следующей формулой (I), или его фармацевтически приемлемая соль

символы в представленной формуле представляют следующие значения:
R1 - -H,
R0 - низший алкил,
Rz - одинаковые или отличные друг от друга и каждый представляет собой -Н или низший алкил,
L - *-СН2-O- или *-CH2-NH-, где знак * в L означает связывание с кольцом А и положение замещения группы L в кольце В представляет собой 4-положение,
кольцо А - бензол,
кольцо В - бензол или пиридин,
R2 - соответственно одинаковые или отличные друг от друга, и каждый представляет собой -галоген или -R0,
n - 0 или 1,
R3 - фенил, который может быть замещен группой, выбранной из группы G3,
группа G3 - галоген, -R0, галогено-низший алкил, -ORz, -CON(Rz)2, -CON(Rz)-гетерокольцевая группа, -O-S(O)2-R0, -О-низший алкилен-ORz, -О-низший алкилен-O-CORz, -О-низший алкилен-N(Rz)2, -О-низший алкилен-N(Rz)CO-Rz, -О-низший алкилен-CO2Rz, -О-низший алкилен-CON(RZ)2, -О-низший алкилен-CON(Rz)-(низший алкил замещен группой -ORz), -О-низший алкилен-SR0, -О-низший алкилен-циклоалкил, -О-низший алкилен-CON(Rz)-циклоалкил, -О-низший алкилен-гетерокольцевая группа и -О-низший алкилен-CON(Rz)-гетерокольцевая группа,
где низший алкилен в группе G3 может быть замещен галогеном или -ORz, и циклоалкил и гетерокольцевая группа в группе G3 могут быть замещены группой, выбранной из группы G1,
группа G1 - галоген, циано, -R0, -ORz, -N(Rz)2, -S-R0, -SO2-R0, -SO2N(Rz)2, -CO-Rz, -CON(Rz)2, -CON(Rz)-низший алкилен-OR2, -N(Rz)CO-Rz, оксо, - (низший алкилен, который может быть замещен группой -ORz)-арил, гетерокольцевая группа и низший алкилен-гетерокольцевая группа,
где арил и гетерокольцевая группа в группе G1 могут быть замещены группой, выбранной из следующей группы G2,
группа G2 - галоген, циано,
где "гетерокольцевая группа" означает группу, содержащую кольцо, выбранное из i) моноциклического 5-7-членного, насыщенного или ненасыщенного гетерокольца, содержащего от 1 до 3 гетероатомов, выбранных из О, S и N, ii) бициклического гетерокольца, в котором гетерокольца, указанные в представленном выше i), являются конденсированными по кольцу, где конденсированные кольца могут быть одинаковые или отличные друг от друга, и iii) бициклического гетерокольца, в котором гетерокольцо, указанное в представленном выше i), является конденсированным с бензольным кольцом или 5-7-членным циклоалканом,
R4 - -Н.
2. Соединение по п.1, где R3 представляет собой фенил, который замещен группой, выбранной из класса, включающего -О-низший алкилен-ORz, -О-низший алкилен-СО(Rz)2 и -О-низший алкилен-(циклоалкил, который может быть замещен группой -ORz), и может быть дополнительно замещен 1 или 2 группами низший алкил, галоген или -OR0.
3. Соединение по п.1, которое выбрано из группы, включающей
2-{[3'-({4-[(3,5-диоксо-1,2,4-оксадиазолидин-2-ил)метил]фенокси}метил)-2,6-диметилбифенил-4-ил]окси}-N-метилацетамид,
2-(4-{[4'-(2-гидроксиэтокси)-2'-метилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-(4-{[4'-(3-гидрокси-3-метилбутокси)-2',6'-диметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-(4-{[4'-(3-гидрокси-3-метилбутокси)-2,2'-диметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-(4-{[4'-(3-гидрокси-3-метилбутокси)-2,2',6'-триметилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-{4-[(4'-{[(3R)-3-гидроксибутил]окси}-2,2'-диметилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-дион,
2-{4-[(4'-{[(3S)-3-гидроксибутил]окси}-2,2'-диметилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-дион,
2-[4-({[4'-(3-гидрокси-3-метилбутокси)-2,2'-диметилбифенил-3-ил]метил}амино)бензил]-1,2,4-оксадиазолидин-3,5-дион,
2-(4-{[4'-(3-гидрокси-3-метилбутокси)-2'-метокси-2-метилбифенил-3-ил]метокси}бензил)-1,2,4-оксадиазолидин-3,5-дион,
2-{4-[(4'-{[(3R)-3-гидроксибутил]окси}-2,2'6'-триметилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-дион,
2-{4-[(4'-{[(3S)-3-гидроксибутил]окси}-2,2'6'-триметилбифенил-3-ил)метокси]бензил}-1,2,4-оксадиазолидин-3,5-дион,
2-[(6-{[4'-(3-гидрокси-3-метилбутокси)-2,2',6'-триметилбифенил-3-ил]метокси}пиридин-3-ил)метокси]-1,2,4-оксадиазолидин-3,5-дион и
2-[4-({4'-[2-(1-гидроксициклопропил)этокси]-2,2',6'-триметилбифенил-3-ил}метокси)бензил]-1,2,4-оксадиазо лидин-3,5-дион,
или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, где фармацевтическая композиция представляет собой агонист GPR40, средство, стимулирующее секрецию инсулина и/или средство для профилактики и/или лечения диабета.
5. Фармацевтическая композиция по п.4, которая представляет собой агонист GPR40.
6. Фармацевтическая композиция по п.4, которая представляет собой средство, стимулирующее секрецию инсулина.
7. Фармацевтическая композиция по п.4, которая представляет собой средство для профилактики и/или лечения диабета.
8. Применение соединения по п.1 или его фармацевтически приемлемой соли для получения агониста GPR40, средства, стимулирующего секрецию инсулина, или средства для профилактики и/или лечения диабета.
9. Способ профилактики и/или лечения диабета, который включает введение пациенту эффективного количества соединения по п.1 или его соли.
| Устройство для детектирования сигналов с фазовой и относительной фазовой манипуляцией | 1988 |
|
SU1559422A1 |
| WO 2005030203 А1, 07.04.2005 | |||
| WO 9857941 А1, 23.12.1998 | |||
| ПРОИЗВОДНОЕ БИСОКСАДИАЗОЛИДИНДИОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2135487C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ АЗОЛИДИНДИОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, СПОСОБ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ, СПОСОБ СНИЖЕНИЯ ГЛЮКОЗЫ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1997 |
|
RU2200161C2 |

