Изобретение относится к способу реализуемого в непрерывном режиме длительного гетерогенно катализируемого частичного дегидрирования подлежащего дегидрированию углеводорода до дегидрированного углеводорода, в соответствии с которым обладающий повышенной температурой поток реакционной газовой смеси, который содержит подлежащий дегидрированию углеводород в исходном молярном количестве KW, таким образом пропускают через находящийся в реакционной зоне RZ совокупный слой катализатора, который может состоять из нескольких отдельных слоев катализатора, последовательно упорядоченных в направлении потока, и содержит суммарное массовое количество М катализатора дегидрирования, чтобы в рабочий момент t=t0 при (однократном) прохождении потока реакционной газовой смеси через первую в направлении потока треть количества М в дегидрированный углеводород превращалась выраженная в % мол. доля А молярного исходного количества KW, при (однократном) прохождении потока реакционной газовой смеси через вторую в направлении потока треть количества М в дегидрированный углеводород превращалась выраженная в % мол. доля В молярного исходного количества KW и при (однократном) прохождении потока реакционной газовой смеси через третью в направлении потока треть количества М в дегидрированный углеводород превращалась выраженная в % мол. доля С молярного исходного количества KW, при условии, что А>В>С и при (однократном) прохождении потока реакционной газовой смеси через совокупный слой катализатора суммарная степень превращения G присутствующего в этом потоке молярного исходного количества KW подлежащего дегидрированию углеводорода в дегидрированный углеводород составляет (А+В+С) % мол., причем в поток реакционной газовой смеси между его входом в совокупный слой катализатора и его выходом из совокупного слоя катализатора при необходимости вводят потоки молекулярного кислорода, молекулярного водорода, водяного пара и/или другого инертного газа, используемых в качестве вспомогательных газов дегидрирования, и деактивированию совокупного слоя катализатора, происходящему по мере его эксплуатации в интервале рабочих моментов t0<t<tR, причем tR означает рабочий момент, в который дегидрирование прекращают и выполняют первую после рабочего момента t0 регенерацию совокупного слоя катализатора, противодействуют тем, что варьируют ход температуры потока реакционной газовой смеси внутри совокупного слоя катализатора и/или расход при необходимости используемых вспомогательных газов дегидрирования.
Под дегидрированным углеводородом согласно изобретению подразумевают углеводород, молекулы которого содержат по меньшей мере на два атома водорода меньше, чем молекулы подлежащего дегидрированию углеводорода, причем технически предпочтительной является именно такая разность количеств содержащихся в указанных углеводородах атомов водорода. При этом под углеводородами подразумевают вещества, молекулы которых состоят только из углерода и водорода.
В соответствии с этим дегидрированными углеводородами прежде всего являются ациклические (неразветвленные и/или разветвленные) и циклические алифатические углеводороды, молекула которых содержит одну или несколько углерод-углеродных связей.
Примерами подобных алифатических дегидрированных углеводородов являются пропилен, изобутилен, этилен, 1-бутен, 2-бутен и бутадиен. Таким образом, к дегидрированным углеводородам прежде всего относятся однократно ненасыщенные неразветвленные алифатические углеводороды (н-алкены) или разветвленные алифатические углеводороды (например, изоалкены), а также циклоалкены. Кроме того, согласно изобретению к дегидрированным углеводородам относятся алкаполиены (например, диены и триены), молекула которых содержит более одной углерод-углеродной двойной связи. Дегидрированными углеводородами согласно изобретению являются также углеводороды, которые могут быть получены из алкилароматических соединений, таких как этилбензол или изопропил-бензол, дегидрированием соответствующего алкильного заместителя. К подобным дегидрированным углеводородам относятся, например, стирол или α-метилстирол.
Дегидрированные углеводороды в общем случае являются ценными исходными продуктами, используемыми, например, для синтеза способных к радикальной полимеризации функционализованных соединений (например, для синтеза акриловой кислоты из пропена или метакриловой кислоты из изобутилена) и получения продуктов полимеризации функционализованных соединений. Функционализованные соединения могут быть получены, например, частичным окислением дегидрированных углеводородов. Дегидрированные углеводороды пригодны также для получения таких соединений, как метил-трет-бутиловый эфир (синтезируемый из изобутена продукт, пригодный для использования, например, в качестве топливной присадки, повышающей октановое число). Кроме того, дегидрированные углеводороды сами можно использовать для полимеризации.
В качестве подлежащих дегидрированию углеводородов согласно изобретению прежде всего используют ациклические (неразветвленные и/или разветвленные) и циклические алканы, а также олефины, дегидрирование которых осуществляют с целью увеличения числа содержащихся в них углерод-углеродных двойных связей (как, например, в случае гетерогенно катализируемого частичного дегидрирования н-бутенов до бутадиена).
Таким образом, под подлежащими дегидрированию углеводородами согласно изобретению подразумевают, например, углеводороды общей формулы СnН2n+2, в которой n означает целое число от 1 до 20, углеводороды общей формулы СnH2n, в которой n означает целое число от 1 до 20, а также углеводороды общей формулы СnН2н-2, в которой n означает целое число от 2 до 20, прежде всего алканы с 2-16 атомами углерода, например, такие как этан (подвергаемый превращению в этен), пропан (подвергаемый превращению в пропен), н-бутан, изобутан (подвергаемый превращению в изобутен), н-пентан, изопентан, н-гексан, н-гептан, н-октан, н-нонан, н-декан, н-ундекан, н-додекан, н-тридекан, н-тетрадекан, н-пентадекан и н-гексадекан.
Подлежащими дегидрированию углеводородами согласно изобретению прежде всего являются алканы с 2-6 атомами углерода и в особенности углеводороды с 2-4 атомами углерода (прежде всего алканы, а среди них прежде всего пропан). Таким образом, согласно изобретению под подлежащими дегидрированию углеводородами прежде всего подразумевают этан, пропан, н-бутан и изобутан, а также 1-бутен и 2-бутен.
Под гетерогенно катализируемым частичным дегидрированием углеводорода согласно изобретению подразумевают (обычное) дегидрирование, по меньшей мере на промежуточной стадии которого образуется свободный молекулярный водород, а следовательно, стадия дегидрирования протекает с эндотермическим эффектом (последующей стадией может являться экзотермическое сгорание водорода). В отличие от этого в случае гетерогенно катализируемого частичного окислительного дегидрирования подлежащего дегидрированию углеводорода водород от последнего отрывается посредством присутствующего в системе кислорода с непосредственным образованием воды. В связи с этим стадия дегидрирования гетерогенно катализируемого частичного окислительного дегидрирования в принципе протекает с экзотермическим эффектом.
В типичном случае (обычное) гетерогенно катализируемое частичное дегидрирование подлежащего дегидрированию углеводорода (например, пропана), например, указанного в начале настоящего описания типа требует использования сравнительно высоких реакционных температур. Типичная температура дегидрирования составляет от 300 до 850°С или от 300 до 800°С, соответственно от 400 до 700°С.
Достигаемое при этом превращение подлежащего дегидрированию углеводорода обычно ограничено не кинетическими факторами, а термодинамическим равновесием реакции дегидрирования.
Поскольку гетерогенно катализируемое частичное дегидрирование углеводорода является типичной эндотермической реакцией, в результате которой, например, из моля дегидрируемого до пропена пропана дополнительно образуется моль водорода, смещению термодинамического равновесия в сторону образования дегидрированного углеводорода как целевого продукта благоприятствуют повышение температуры и удаление образующегося в качестве продукта реакции водорода, а также снижение парциального давления исходного углеводорода, реализуемое благодаря разбавлению инертным газом. При этом согласно изобретению под инертным газом (инертным разбавляющим газом) в общем случае подразумевают компонент реакционной газовой смеси, который в условиях дегидрирования ведет себя в основном химически инертно, то есть химически неизменным остается более чем 95% мол., предпочтительно более чем 97% мол., соответственно более чем 99% мол. подобного разбавляющего газа. Примерами типичных инертных разбавляющих газов являются, например, азот, диоксид углерода, водяной пар, благородные газы, такие как гелий, неон или аргон, а также смеси указанных газов.
Поскольку при гетерогенно катализируемом частичном дегидрировании подлежащего дегидрированию углеводорода (например, пропана) стадия дегидрирования, приводящая к образованию дегидрированного углеводорода, протекает с эндотермическим эффектом, для обеспечения необходимой степени дегидрирования до гетерогенно катализируемого дегидрирования и/или в процессе его осуществления следует подводить тепло.
С учетом указанного обстоятельства гетерогенно катализируемое частичное дегидрирование в наиболее простом варианте следует осуществлять в функционирующем в адиабатическом режиме (совокупном) слое катализатора.
Реакционную газовую смесь, которая содержит подлежащий дегидрированию углеводород, нагревают до начальной температуры, в типичном случае составляющей от 300 до 850°С, часто от 400 до 800°С, наиболее часто от 450 до 750°С, соответственно от 500 до 700°С или от 550 до 650°С, а затем пропускают через функционирующий в адиабатическом режиме (то есть теплоизолированный от внешней среды) (совокупный) слой катализатора. В зависимости от необходимого превращения подлежащего дегидрированию углеводорода и выбранного инертного разбавляющего газа температура реакционной газовой смеси в результате прохождения через (совокупный) слой катализатора снижается на величину, составляющую примерно от 30 до 200°С.
При этом в случае (мысленного) разделения совокупного слоя катализатора, который содержит суммарное количество М катализатора дегидрирования, на три участка, последовательно упорядоченные в направлении потока реакционной газовой смеси, каждый из которых содержит третью часть от суммарного количества М катализатора дегидрирования, причем содержащееся в потоке реакционной газовой смеси исходное молярное количество подлежащего дегидрированию углеводорода составляет KW, в начальный период указанного гетерогенно катализируемого частичного дегидрирования (то есть при введении в действие свежего, соответственно свежерегенерированного совокупного слоя катализатора) при (однократном) прохождении потока реакционной газовой смеси через первую треть М в дегидрированный углеводород будет превращаться выраженная в % мол. доля А молярного исходного количества KW, при (однократном) прохождении потока реакционной газовой смеси через вторую треть М в дегидрированный углеводород будет превращаться выраженная в % мол. доля В молярного исходного количества KW и при (однократном) прохождении потока реакционной газовой смеси через третью треть М в дегидрированный углеводород будет превращаться выраженная в % мол. доля С молярного исходного количества KW, причем для случая естественного протекания процесса обычно справедливо соотношение А>В>С.
При (однократном) прохождении через совокупный слой катализатора потока реакционной газовой смеси, который содержит подлежащий дегидрированию углеводород в молярном исходном количестве KW, суммарная степень превращения G этого углеводорода в дегидрированный углеводород составит (А+В+С) % мол.
При этом в наиболее общем варианте осуществления предлагаемого в изобретении способа (совокупный) слой катализатора может являться по меньшей мере одним стационарным слоем, по меньшей мере одним кипящим слоем, по меньшей мере одним движущимся слоем или может представлять собой комбинацию, например, последовательно упорядоченных слоев, состоящую более чем из одного подобного слоя. Согласно изобретению (совокупный) слой катализатора в общем случае предпочтительно является по меньшей мере одним стационарным слоем (все приведенные ниже данные прежде всего относятся к стационарному слою катализатора).
Таким образом, в наиболее простом варианте указанный выше адиабатический режим дегидрирования может быть реализован по меньшей мере в одном стационарном слое катализатора (совокупном стационарном слое катализатора), помещенном в адиабатический (теплоизолированный от внешней среды) шахтный реактор. При этом под шахтным реактором подразумевают реакционный объем, окруженный контактирующей с ним материальной огибающей, снабженной по меньшей мере одним первым отверстием, предназначенным для подачи в реакционный объем реакционной газовой смеси, которая содержит подлежащий дегидрированию углеводород, и по меньшей мере одним вторым отверстием, предназначенным для отбора газового потока продуктов дегидрирования из реакционного объема. В адиабатической (теплоизолированной от внешней среды) шахте (реакционном объеме) находится предпочтительно по меньшей мере один стационарный слой катализатора, через который пропускают реакционную газовую смесь. Во время контакта подлежащего дегидрированию углеводорода (например, пропана) по меньшей мере с одним стационарным слоем катализатора происходит целевое частичное дегидрирование этого углеводорода с образованием дегидрированного углеводорода. В обычном случае по меньшей мере один находящийся в адиабатическом шахтном реакторе стационарный слой катализатора является стационарным слоем, через который пропускают содержащую подлежащий дегидрированию углеводород реакционную газовую смесь, температура которой по мере прохождения через этот стационарный слой постепенно снижается, что в случае естественного протекания процесса дегидрирования ограничивает конечную степень превращения подлежащего дегидрированию углеводорода.
Хотя при осуществлении дегидрирования в указанном выше адиабатическом режиме повышение начальной температуры реакционной газовой смеси обычно и приводит к увеличению степени превращения G, однако при этом в случае естественного протекания процесса кинетически сравнительно более выгодной часто оказывается нежелательная побочная реакция термического крекинга (расщепления одинарных углерод-углеродных связей) нежели дегидрирование (расщепление по связям С-Н), причем инверсия соотношения указанных реакций требует использования катализатора, селективно катализирующего дегидрирование, и является основным принципом гетерогенно катализируемого дегидрирования подлежащих дегидрированию углеводородов.
В этой связи в случае практического осуществления гетерогенно катализируемого частичного дегидрирования в реакционную газовую смесь преимущественно всегда добавляют предпочтительно используемый в качестве инертного разбавляющего газа водяной пар. В отличие от других возможных инертных разбавляющих газов водяной пар обладает повышенной молярной теплоемкостью, поэтому при совместном использовании в реакционной газовой смеси он уменьшает указанное выше снижение температуры и способствует повышению значений G.
Однако, во-первых, по мере повышения содержания водяного пара в реакционной газовой смеси возрастает ее объем (что обусловливает увеличение капитальных затрат), а во-вторых, повышение содержания водяного пара в реакционной газовой смеси способствует протеканию другой нежелательной побочной реакции, а именно парового риформинга углеводородов, продуктами которого являются монооксид углерода, диоксид углерода и водород. Количество вводимого в исходную реакционную газовую смесь водяного пара, в отдельных случаях еще допустимое с учетом вероятного протекания нежелательного парового риформинга углеводородов, в первую очередь определяется абсолютным давлением, под которым выполняют гетерогенно катализируемое дегидрирование (оно, как правило, составляет от 0,2 до 10 бар, соответственно от 0,5 до 6 бар или до 3 бар), а также типом используемого для дегидрирования катализатора.
Снижению температуры, огранивающему степень дегидрирования G, в принципе можно дополнительно противодействовать путем внешнего управления температурой. Согласно изобретению под внешним управлением температурой в общем случае подразумевают регулирование температуры за счет косвенного теплообмена. Подобное внешнее управление температурой при осуществлении гетерогенно катализируемого дегидрирования углеводородов можно легко выполнять, например, благодаря наружному нагреванию реактора, содержащего (совокупный) слой катализатора, а следовательно, и самого (совокупного) слоя катализатора. Примеры подобного внешнего управления температурой приведены в заявке США на патент US-A 5235121.
В другом варианте в принципе можно использовать также функционирующий в адиабатическом режиме совокупный слой катализатора, который находится, например, на нескольких последовательно упорядоченных полках или помещен в несколько последовательно соединенных реакторов дегидрирования, причем реакционную газовую смесь после прохождения через каждый функционирующий в адиабатическом режиме отдельный слой катализатора подвергают нагреванию, реализуемому путем косвенного теплообмена, то есть внешнего управления температурой.
Таким образом, в особенно предпочтительном варианте гетерогенно катализируемое дегидрирование углеводорода (например, пропана) можно осуществлять в адиабатическом шахтном реакторе в том случае, если он обладает полочной конструкцией.
В шахте подобного реактора (снабженном огибающей в реакционном объеме) находятся последовательно упорядоченные (отдельные) стационарные слои катализатора дегидрирования, число которых составляет, например, более одного. Количество (отдельных) стационарных слоев катализатора дегидрирования может составлять, например, от 1 до 20, в целесообразном варианте от 2 до 8, соответственно от 3 до 6. При этом слои катализатора, как правило, последовательно упорядочены в радиальном или осевом направлении.
В особенно легко выполнимом конструктивном варианте (отдельные) стационарные слои катализатора последовательно располагаются в адиабатически теплоизолированной шахте вдоль ее оси, совпадающей с направлением потока реакционной газовой смеси. (Отдельные) стационарные слои катализатора могут располагаться также в кольцевых зазорах, образуемых помещенными в шахту центрически вставленными друг в друга цилиндрическими колосниковыми решетками. Кроме того, кольцевые зазоры в шахте могут быть расположены друг над другом в сегментах, причем реакционный газ последовательно проходит в радиальном направлении через один сегмент, а затем через следующий сегмент, расположенный выше или ниже первого.
В этом случае реакционную газовую смесь на пути от одного (отдельного) стационарного слоя катализатора к другому (отдельному) стационарному слою катализатора можно подвергать косвенному промежуточному нагреванию (то есть осуществлять внешнее управление температурой), например, благодаря ее пропусканию над соответствующими поверхностями обогреваемых горячими газами и/или жидкостями косвенных теплообменников (например, над их ребрами, пластинами или пучками трубок), помещенных внутрь имеющей адиабатическую конструкцию шахты между полками со стационарными слоями, и/или благодаря пропусканию реакционной газовой смеси через подобные теплообменники.
Для обеспечения степеней превращения G, которые при однократном прохождении реакционного газа (прежде всего при совместном использовании водяного пара в качестве инертного разбавляющего газа и катализаторов, например, описанных в немецких заявках на патент DE-A 1020050449216 и DE-A 19937107, прежде всего в соответствующих примерах), например, в случае дегидрирования пропана в пропилен достигают 50% мол., часто 40% мол., как правило, оказывается достаточным ввести в шахту полочного реактора указанной выше конструкции (то есть в окруженный огибающей, теплоизолированный от внешней среды реакционный объем, в котором находятся (отдельные) стационарные слои катализатора) реакционную газовую смесь, содержащую подлежащий дегидрированию углеводород, которая предварительно нагрета до температуры, составляющей от 350, 400 или 450 до 550°С (предпочтительно от 400 до 500°С), и выдержать ее внутри шахты (внутри снабженного полками реакционного объема) по меньшей мере в указанном температурном интервале благодаря косвенному теплообмену (внешнему управлению температурой). При этом технически целесообразное давление потока исходной газовой смеси на входе в первый в направлении потока (отдельный) стационарный слой катализатора составляет от 1 до 10 бар, предпочтительно от 1,5 до 5 бар. Давление указанного потока в принципе может составлять также менее 1 бар. Так, например, оно может находиться также в интервале от 0,2 до 1 бар.
Однако указанное выше промежуточное нагревание технически проще реализовать непосредственно (речь при этом идет о внутреннем управлении температурой). С этой целью к реакционной газовой смеси (реакционному газу) в соответствующих промежутках на пути от одного (отдельного) стационарного слоя к другому (отдельному) стационарному слою катализатора можно добавлять ограниченное количество содержащего молекулярный кислород газа, в результате чего образуется соответствующий поток реакционной газовой смеси, содержащий молекулярный кислород, молекулярный водород и углеводород, а также обычно используемый водяной пар.
В случае, если (отдельный) стационарный слой катализатора, следующий в направлении перемещения подобного потока реакционной газовой смеси, благодаря выбору соответствующей активной массы выполнен таким образом, что катализатор дегидрирования катализирует также взаимодействие молекулярного водорода с молекулярным кислородом, сопровождаемое образованием воды (то есть сгорание молекулярного водорода) и/или взаимодействие содержащегося в потоке реакционной газовой смеси углеводорода, сопровождаемое образованием оксидов углерода и воды (то есть сгорание углеводорода), или в случае добавления к присутствующему в (отдельном) стационарном слое катализатору дегидрирования катализаторов, селективно катализирующих указанное сгорание, при прохождении реакционной газовой смеси через подобный (отдельный) стационарный слой при необходимости до гетерогенно катализируемого дегидрирования и/или в дополнение к нему происходит экзотермическое сгорание ограниченного количества содержащегося в реакционной газовой смеси молекулярного водорода и/или углеводорода, то есть их взаимодействие с молекулярным кислородом, сопровождаемое образованием воды, соответственно воды и оксидов углерода. Выделяющееся при этом тепло может расходоваться при последующем и/или одновременно протекающем эндотермическом дегидрировании. Образующиеся продукты сгорания, такие как диоксид углерода, вода, а также азот, при необходимости сопровождающий необходимый для сгорания молекулярный кислород (если в качестве источника кислорода используют, например, воздух) образуют предпочтительные для гетерогенно катализируемого дегидрирования инертные разбавляющие газы. Баланс между сгоранием водорода и сгоранием углеводородов, в первую очередь, зависит от выбранного катализатора. Катализаторы, которые сравнительно селективно катализируют сгорание молекулярного водорода и/или углеводорода, приведены, например, в заявках США на патент US-A 4788371, US-A 4886928, US-A 5430209, US-A 5530171, US-A 5527979 и US-A 5563314.
Предпочтительным обычно является преимущественное сгорание водорода нежели преимущественное сгорание углеводорода, поскольку это обусловливает повышенную селективность образования дегидрированного углеводорода, а также повышенную степень дегидрирования (в расчете на однократное прохождение реакционной газовой смеси через полочный реактор). Преимущественное сгорание водорода, как правило, имеет место в том случае, если соответствующий (отдельный) стационарный слой катализатора содержит только катализатор дегидрирования, прежде всего рекомендованный в немецкой заявке на патент DE-A 19937107 (прежде всего приведенный в соответствующих примерах), поскольку этот катализатор, как правило, способен катализировать не только дегидрирование подлежащего дегидрированию углеводорода (например, пропана), но и сгорание молекулярного водорода и углеводородов. При этом сгорание водорода на указанных катализаторах, как правило, протекает гораздо быстрее как по сравнению с дегидрированием подлежащего дегидрированию углеводорода (например, пропана), так и по сравнению со сгоранием углеводорода, например, в случае соответствующей конкурентной ситуации (то есть в данных условиях они, как правило, обусловливают минимальную энергию активации сгорания молекулярного водорода).
Интегральный тепловой эффект (то есть общий тепловой брутто-эффект) при однократном прохождении реакционной газовой смеси через полочный реактор в зависимости от полноты реакции сгорания (то есть также в зависимости от количества подаваемого молекулярного кислорода) может быть эндотермическим (отрицательным), автотермическим (преимущественно нулевым) или экзотермическим (положительным).
При этом в результате сгорания молекулярного водорода выделяется примерно в два раза больше тепловой энергии, чем расходуется на образование такого же количества водорода в рамках реакции дегидрирования.
Для регулирования температуры между двумя (отдельными) слоями катализатора, очевидно, можно использовать принцип как внутреннего, так и внешнего управления температурой. Кроме того, изотермию гетерогенно катализируемого частичного дегидрирования углеводорода в этом случае или дополнительно можно оптимизировать благодаря тому, что в пустое реакционное пространство между (отдельными) стационарными слоями катализатора и/или в сами слои помещают герметично замкнутые встроенные элементы (например, в виде труб), которые перед заполнением целесообразно, но не обязательно подвергнуть вакуумированию. Подобные встроенные элементы, как указано выше, могут быть помещены также в соответствующий (отдельный) стационарный слой катализатора. Подобные встроенные элементы содержат соответствующие твердые вещества или жидкости, которые испаряются или плавятся выше определенной температуры и при этом потребляют тепло, а в зонах с более высокой температурой по сравнению с точкой их испарения или плавления конденсируются и при этом выделяют тепло. В принципе для оптимизации изотермического режима снаружи материальной огибающей шахтного реактора дополнительно могут циркулировать текучие (газообразные и/или жидкие) теплоносители. Однако этот вариант связан со значительными производственными издержками, поэтому предпочтительным, как правило, является теплоизолированное адиабатическое конструктивное исполнение реактора.
Рассмотренные выше технические мероприятия по внутреннему управлению температурой используют также для нагревания до необходимой температуры реакционной газовой смеси, подаваемой в первый по ее ходу слой катализатора. С этой целью соответствующее количество молекулярного кислорода добавляют уже к направляемой в шахтный реактор реакционной газовой смеси, которая содержит подлежащий дегидрированию углеводород. Для нагревания за счет выделяющегося тепла к реакционной газовой смеси перед поступлением в первый (стационарный) слой катализатора дополнительно можно добавлять молекулярный водород. От подобной подачи водорода можно также отказаться. В этом случае внутреннее управление температурой с целью нагревания реакционной газовой смеси до реакционной температуры осуществляют в основном лишь благодаря сгоранию углеводорода. Тепло к реакционной газовой смеси между (отдельными) слоями катализатора можно подводить также благодаря добавлению к реакционному газу перегретого водяного пара. Температуру реакционной газовой смеси, подаваемой в первый отдельный (стационарный) слой катализатора, часто повышают до температуры реакции также другими методами. Так, например, исходные газовые потоки, из которых компонуют подаваемую в первый (стационарный) слой катализатора реакционную газовую смесь, уже могут обладать соответствующей температурой. Указанные исходные газовые потоки могут также уже содержать молекулярный кислород, что позволяет избежать проблемы дозирования содержащих молекулярный кислород газов в реакционную газовую смесь, подаваемую в первый по направлению потока (стационарный) слой катализатора или осуществлять косвенный теплообмен подаваемой в реактор дегидрирования реакционной газовой смеси с выходящей из реактора дегидрирования горячей смесью продуктов.
Рассмотренные выше варианты гетерогенно катализируемого частичного дегидрирования подлежащего дегидрированию углеводорода до дегидрированного углеводорода в принципе известны (см., например, немецкие заявки на патент DE-A 102005061626, DE-A 102005057197, DE-A 102005052923, DE-A 102005052917, DE-A 102005022798, DE-A 102005009885, DE-A 102005010111, DE-A 102004032129, DE-A 102005013039, DE-A 10211275, DE-A 102004054657, DE-A 102006029790 и DE-A 102006024901, международные заявки WO 01/96270, WO 2004/039920, WO 03/076370 и WO 2006/050957, а также цитируемый в этих публикациях уровень техники).
Независимо от конструктивного оформления процесса гетерогенно катализируемого частичного дегидрирования и метода управления температурой общая особенность всех указанных выше вариантов его осуществления состоит в том, что на начальной стадии (то есть при использовании свежего, соответственно свежерегенерированного совокупного слоя катализатора) степени дегидрирования в (отдельных) (воображаемых) слоях катализатора, как правило, находятся в соотношении А>В>С, причем ни на одном из этих слоев не удается полностью исключить протекающий в виде побочной реакции нежелательный термический крекинг углеводородов. Таким образом, во всех случаях образуются незначительные количества нежелательных побочных продуктов в виде труднокипящих высокомолекулярных органических соединений (продуктов термической деструкции), вплоть до элементарного углерода, которые оседают на поверхности катализатора и, следовательно, обусловливают деактивирование катализатора по мере его эксплуатации. Хотя водяной пар, совместно используемый в реакционной газовой смеси в качестве инертного разбавляющего газа, и способен частично удалять осажденный на поверхности катализатора углерод в соответствии с принципом газификации угля, однако полностью избежать осаждения углерода не удается. Аналогичным образом хотя добавление молекулярного водорода к реакционной газовой смеси, поступающей к первому (отдельному) стационарному слою катализатора (соответственно к прочим (отдельным) слоям катализатора), всегда удлиняет срок службы катализаторов дегидрирования, однако также не позволяет предотвратить их постепенное деактивирование.
Недостаток подобного деактивирования катализатора состоит в том, что по мере деактивирования при прочих неизменных условиях дегидрирования наблюдается снижение степени превращения подлежащего дегидрированию углеводорода в дегидрированный углеводород при однократном прохождении реакционной газовой смеси через совокупный слой катализатора, что проявляется в уменьшении выхода дегидрированного углеводорода в расчете на единицу объема в единицу времени.
Указанный недостаток прежде всего проявляется в том случае, если непосредственно после гетерогенно катализируемого частичного дегидрирования подлежащего дегидрированию углеводорода осуществляют, например, гетерогенно катализируемое частичное окисление полученного дегидрированного углеводорода (например, пропилена в акролеин и/или акриловую кислоту) предпочтительно в присутствии непревращенного подлежащего дегидрированию углеводорода (например, пропана) в качестве инертного газа, поскольку в этом случае одновременно со снижением выхода дегидрированного углеводорода в расчете на единицу объема в единицу времени снижается и рассчитываемый аналогичным образом выход продукта частичного окисления.
В связи с этим ищут такой способ гетерогенно катализируемого частичного дегидрирования, который позволял бы максимально ограничить указанное выше деактивирование совокупного слоя катализатора.
В немецкой заявке на патент DE-A 102005013039 предложен способ гетерогенно катализируемого частичного дегидрирования, согласно которому совокупный слой катализатора разделен на три отдельных слоя, находящихся в трех последовательно соединенных адиабатических реакторах, которые совместно образуют реакционную зону RZ (в каждом из реакторов находится отдельный слой катализатора). Перед подачей реакционной газовой смеси в соответствующий отдельный слой катализатора ее можно нагреть путем косвенного теплообмена в устройстве для предварительного нагревания до необходимой температуры входа (в отдельный слой катализатора).
К реакционной газовой смеси перед входом в соответствующий отдельный слой катализатора дополнительно добавляют воздух. Реакционная газовая смесь, поступающая в первый по ходу газового потока отдельный слой катализатора, также уже содержит молекулярный кислород. Используемый для дегидрирования катализатор одновременно катализирует сгорание молекулярного водорода и углеводорода. В начальный период дегидрирования (то есть при использовании свежего или свежерегенерированного совокупного слоя катализатора) справедливо соотношение А>В>С.
С целью предотвращения деактивирования совокупного слоя катализатора в немецкой заявке на патент DE-A 102005013039 рекомендуется регулировать температуру реакционной газовой смеси таким образом, чтобы степень дегидрирования G при однократном прохождении реакционной газовой смеси через совокупный слой катализатора и соответствующий выход дегидрированного углеводорода в расчете на единицу объема в единицу времени оставались постоянными.
Недостаток предложенного в заявке DE-A 102005013039 варианта предотвращения деактивирования катализатора состоит в том, что хотя повышение соответствующей температуры реакционной газовой смеси при соотношении А>В>С и противодействует деактивированию катализатора, однако по мере повышения температуры интенсифицируется причина деактивирования. Поэтому уже по истечении сравнительно кратковременного рабочего периода наступает момент, когда дальнейшее повышение соответствующей температуры реакционной газовой смеси не позволяет больше сохранять постоянство выхода дегидрированного углеводорода в расчете на единицу объема в единицу времени.
В этом случае вынуждены прекращать дегидрирование и осуществлять регенерацию совокупного слоя катализатора. Регенерация, как правило, состоит в том (см. немецкую заявку на патент DE-A 10028582), что через совокупный слой катализатора при повышенной температуре пропускают кислородсодержащий газ, благодаря чему происходит квазисгорание осажденного на поверхности катализатора углерода. Завершающей стадией регенерации обычно является последующая восстанавливающая обработка катализатора молекулярным водородом.
Однако проблема состоит в том, что деактивирование катализатора всегда включает обратимую и необратимую фазы.
Другой недостаток предложенного в заявке DE-A 102005013039 варианта, в соответствии с которым деактивированию катализатора следует противодействовать перед его регенерацией, состоит в сравнительно быстром наступлении указанной необратимой фазы. Вероятно, чтобы избежать этого, в международной заявке WO 01/96008 деактивированию катализатора вообще не противодействуют до регенерации.
Исходя из вышеизложенного в основу настоящего изобретения была положена задача предложить способ реализуемого в непрерывном режиме длительного гетерогенно катализируемого частичного дегидрирования указанного в начале описания типа, в соответствии с которым деактивированию совокупного слоя катализатора дегидрирования противодействуют таким образом, чтобы указанные выше недостатки проявлялись лишь в незначительной мере.
Указанная задача решается благодаря способу реализуемого в непрерывном режиме длительного гетерогенно катализируемого частичного дегидрирования подлежащего дегидрированию углеводорода до дегидрированного углеводорода, в соответствии с которым обладающий повышенной температурой поток реакционной газовой смеси, который содержит подлежащий дегидрированию углеводород в исходном молярном количестве KW, таким образом пропускают через находящийся в реакционной зоне RZ совокупный слой катализатора, который может состоять из нескольких отдельных слоев катализатора, последовательно упорядоченных в направлении потока, и содержит суммарное массовое количество М катализатора дегидрирования, чтобы в рабочий момент t=t0 при (однократном) прохождении потока реакционной газовой смеси через первую в направлении потока треть количества М в дегидрированный углеводород превращалась выраженная в % мол. доля А молярного исходного количества KW, при (однократном) прохождении потока реакционной газовой смеси через вторую в направлении потока треть количества М в дегидрированный углеводород превращалась выраженная в % мол. доля В молярного исходного количества KW и при (однократном) прохождении потока реакционной газовой смеси через третью в направлении потока треть количества М в дегидрированный углеводород превращалась выраженная в % мол. доля С молярного исходного количества KW, при условии, что А>В>С и при (однократном) прохождении потока реакционной газовой смеси через совокупный слой катализатора суммарная степень превращения G присутствующего в этом потоке молярного исходного количества KW подлежащего дегидрированию углеводорода в дегидрированный углеводород составляет (А+В+С) % мол., причем в поток реакционной газовой смеси между его входом в совокупный слой катализатора и его выходом из совокупного слоя катализатора при необходимости вводят потоки молекулярного кислорода, молекулярного водорода, водяного пара и/или другого инертного газа, используемых в качестве вспомогательных газов дегидрирования, и деактивированию совокупного слоя катализатора, происходящему по мере его эксплуатации в интервале рабочих моментов t0<t<tR, причем tR означает рабочий момент, в который дегидрирование прекращают и выполняют первую после рабочего момента t0 регенерацию совокупного слоя катализатора, противодействуют тем, что варьируют ход температуры потока реакционной газовой смеси внутри совокупного слоя катализатора и/или расход при необходимости используемых вспомогательных газов дегидрирования, отличающемуся тем, что указанное варьирование выполняют таким образом, чтобы по мере протекания рабочего процесса, то есть по мере возрастания t, доля А снижалась, доля В проходила через максимум и доля С возрастала.
В случае промышленной реализации предлагаемого в изобретении способа частичные превращения А, В и С по различным причинам, очевидно, могут быть подвержены известным колебаниям. В подобном случае определяют фактические частичные превращения А, В и С в зависимости от продолжительности дегидрирования и на основании полученных данных, используя метод наименьших квадратов Лежандра и Гаусса, строят соответствующие сглаженные кривые. В случае если в соответствии с полученной сглаженной кривой частичные превращения А в интервале рабочих моментов t0<t<tR характеризуются тенденцией к снижению, частичные превращения С характеризуется тенденцией к увеличению, а сглаженная кривая для частичных превращений В характеризуется максимумом, это означает, что дегидрирование протекает в соответствии с предлагаемым в изобретении способом. Перерывы в реализации предлагаемого в изобретении способа на сглаженных кривых не учитываются.
По мере возрастания t в указанном интервале рабочих моментов частичные степени превращения А на соответствующей сглаженной кривой либо непрерывно уменьшаются, либо хотя и характеризуются общей тенденцией к снижению, но при необходимости временно сохраняются постоянными.
По мере возрастания t в указанном интервале рабочих моментов частичные степени превращения С на соответствующей сглаженной кривой либо непрерывно возрастают, либо хотя и характеризуются общей тенденцией к возрастанию, но при необходимости временно сохраняются постоянными.
Сглаженная кривая частичных степеней превращения В в указанном интервале рабочих моментов характеризуется максимумом (абсолютным) в области рабочего момента tmax, причем, во-первых, первая производная в соответствующей точке этой кривой равна нулю, а во-вторых, значения В для рабочих моментов t<tmax ниже значения В для рабочего момента tmax, a значения В для рабочих моментов t>tmax ниже значения В для рабочего момента tmax и/или равны этому значению.
В соответствии с предлагаемым в изобретении способом гетерогенно катализируемого частичного дегидрирования рабочему моменту t=t0 в целесообразном варианте соответствует момент эксплуатации свежего или свежерегенерированного совокупного слоя катализатора дегидрирования, при котором преимущественно впервые достигают стационарного рабочего режима, обычно характеризующегося тем, что в дегидрированный углеводород превращается G % мол. содержащегося в реакционной газовой смеси молярного исходного количества KW подлежащего дегидрированию углеводорода (в расчете на однократное прохождение реакционной газовой смеси через совокупный слой катализатора дегидрирования). Однако согласно предлагаемому в изобретении способу рабочему моменту t0 в принципе может соответствовать также более поздний период эксплуатации катализатора дегидрирования.
Согласно изобретению в производственно-техническом отношении целесообразно, если в соответствии с предлагаемым в изобретении способом рабочему моменту t=t0 соответствует частичное превращение А, составляющее от 45 до 80% (или до 75%), частичное превращение В, составляющее от 20 до 40%, и частичное превращение С, составляющее от 0 до 5% или до 15%, в пересчете на суммарную степень превращения G=(А+В+С).
Согласно изобретению предпочтительно, если в соответствии с предлагаемым в изобретении способом рабочему моменту t=t0 соответствует частичное превращение А, составляющее от 55 до 70%, частичное превращение В, составляющее от 25 до 35%, и частичное превращение С, составляющее от 7 до 13%, в пересчете на суммарную степень превращения G=(А+В+С).
Согласно изобретению особенно предпочтительно, если в соответствии с предлагаемым в изобретении способом рабочему моменту t=t0 соответствует частичное превращение А, составляющее от 55 до 65%, частичное превращение В, составляющее от 27 до 33%, и частичное превращение С, составляющее от 8 до 12%, в пересчете на суммарную степень превращения G=(А+В+С).
В соответствии с предлагаемым в изобретении способом значение суммарной степени превращения G (то есть выраженного в % мол. общего количества подлежащего дегидрированию углеводорода, превращенного в дегидрированный углеводород) в пересчете на (однократное) прохождении потока реакционной газовой смеси через совокупный слой катализатора и в пересчете на содержащееся в указанном потоке молярное исходное количество KW подлежащего дегидрированию углеводорода остается постоянным в интервале рабочих моментов t0<t<tR и обычно составляет G±5% мол., предпочтительно G±4% мол. или G±3% мол., особенно предпочтительно G±2% мол. или G±1% мол., еще более предпочтительно G±0,75% мол. или G±0,50% мол. и наиболее предпочтительно G±0,25% мол. или G±0,1% мол.
Кроме того, согласно изобретению благоприятно, чтобы частичное превращение А уменьшалось в интервале рабочих моментов t0<t<tR, становясь ниже частичного превращения С, то есть чтобы по истечении определенного времени оказывалось справедливым соотношение С>А. Во многих случаях даже предпочтительно, чтобы в соответствии с предлагаемым в изобретении способом в указанном интервале рабочих моментов происходило изменение соотношения между частичными превращениями А, В и С, то есть чтобы по истечении определенного времени становилось справедливым соотношение С>В>А, а не А>В>С.
Однако в соответствии с предлагаемым в изобретении способом значения частичного превращения А, как правило, отличаются от значения частичного превращения А в рабочий момент t=t0 не более чем на 20%, предпочтительно не более чем на 30%. Таким образом, перед переходом через соответствующий количественный рубеж дегидрирование обычно прекращают и совокупный слой катализатора подвергают регенерации (подобным образом необратимое деактивирование катализатора дегидрирования обычно удерживают на минимальном уровне). Кроме того, в соответствии с предлагаемым в изобретении способом обусловленная указанными выше причинами разница между значениями частичного превращения В и частичного превращения С и значением частичного превращения А в рабочий момент t=t0 обычно, как правило, составляет не более 95%, предпочтительно не более 85%.
Однако в соответствии с предлагаемым в изобретении способом значения как частичного превращения В, так и частичного превращения С отличаются от значения частичного превращения А в рабочий момент t=t0, как правило, не более чем на 50%.
В соответствии с предлагаемым в изобретении способом суммарная степень превращения G обычно составляет от 5 до 10 или до 60% мол., часто от 10 до 50% мол., наиболее часто от 15 до 40% мол., от 15 до 30% мол. или от 15 до 25% мол.
Кроме того, предлагаемый в изобретении способ можно использовать для реализации любых процессов гетерогенно катализируемого частичного дегидрирования углеводорода, указанных в начале настоящего описания.
Таким образом, в качестве катализатора дегидрирования, используемого в предназначенном для реализации предлагаемого в изобретении способа совокупном слое, в принципе пригодны любые известные из уровня техники катализаторы гетерогенно катализируемого дегидрирования. Подобные катализаторы грубо можно разделить на две группы. Одну из них образуют катализаторы оксидного типа (например, оксид хрома и/или оксид алюминия), тогда как в состав другой группы входят катализаторы, которые содержат по меньшей мере один, как правило, сравнительно благородный металл (например, элемент платиновой группы, например, платину и/или палладий) на носителе оксидного типа (например, оксиде циркония, оксиде алюминия, диоксиде кремния, диоксиде титана, оксиде магния, оксиде лантана и/или оксиде церия). В частности, можно использовать любые катализаторы дегидрирования, рекомендованные в международной заявке WO 01/96270, европейской заявке на патент ЕР-А 731077, немецких заявках на патент DE-A 10211275 и DE-A 10131297, международной заявке WO 99/46039, заявке США на патент US-A 4788371, европейской заявке на патент ЕР-А 705136, международной заявке WO 99/29420, заявках США на патент US-A 4220091, US-A 5430220 и US-A 5877369, европейской заявке на патент ЕР-А 117146, немецких заявках на патент DE-A 19937196 и DE-A 19937105, заявках США на патент US-A 3670044 и US-A 6566573 и международной заявке WO 94/29021. Другие катализаторы дегидрирования, пригодные для реализации предлагаемого в изобретении способа, приведены в международной заявке WO 01/83405.
Как совокупный слой, так и отдельные слои катализатора в принципе могут состоять только из катализатора дегидрирования. Однако как совокупный слой, так и отдельные слои катализатора, очевидно, могут содержать также катализатор дегидрирования, разбавленный инертным материалом.
Поскольку совокупный слой катализатора в соответствии с предлагаемым в изобретении способом предпочтительно является стационарным слоем, причем все приведенные в настоящей заявке данные относятся прежде всего к варианту, предусматривающему использование стационарного слоя катализатора (в других вариантах можно использовать, например, псевдоожиженный или движущий слой), то согласно изобретению под катализатором дегидрирования прежде всего подразумевают формованные тела, продольный размер которых L (наибольшее расстояние между двумя находящимися на поверхности формованного тела точками) составляет от 0,1 или 1 до 30 мм, предпочтительно от 1 до 20 мм, особенно предпочтительно от 1 до 10 мм или от 1 до 5 мм, и которые, как показано в приведенных ниже примерах, обеспечивают дегидрирование по меньшей мере 5% мол. содержащегося в реакционном газе пропана до пропилена в пересчете на однократное прохождение реакционной газовой смеси через реакционную трубку.
Реакционную трубку из стали марки 1.4835 (согласно EN) длиной 80 см с внутренним диаметром 35 мм и толщиной стенок 2 мм заполняют следующим образом.
В среднюю часть реакционной трубки помещают 50 мл соответствующего катализатора дегидрирования. Внутреннее пространство реакционной трубки выше и ниже загруженных формованных тел катализатора заполняют шариками из стеатита (инертного материала) диаметром от 1,5 до 2,5 мм. Общий загруженный в реакционную трубку слой опирается на колосниковую решетку. Наружную температуру реакционной трубки по всей длине поддерживают на уровне 550°С. Через реакционную трубку пропускают смесь пропана с водяным паром в объемном соотношении 2:1, причем часовой расход пропана через единицу объема слоя катализатора составляет 1000 нл/л·ч. Направляемый в реакционную трубку поток реакционной газовой смеси предварительно нагревают до температуры 550°С. Для реализации предлагаемого в изобретении способа особенно предпочтительным является использование катализатора дегидрирования, на котором в указанных выше условиях из пропана образуется не более 5% моль побочных продуктов (этана, этена и метана).
Согласно изобретению под загруженностью слоя катализатора дегидрирования реакционным газом в общем случае подразумевают количество реакционного газа в нормальных литрах (нл), то есть приведенный к нормальным условиям (температура 0°С, давление 1 атм) объем соответствующего количества реакционного газа, пропускаемого в течение часа через литр слоя катализатора. Загруженность может относиться только к одному компоненту реакционной газовой смеси. В этом случае под загруженностью подразумевают количество этого компонента в нл/л·ч, пропускаемое через литр слоя катализатора (без учета загруженного в реакционную трубку инертного материала). При использовании смеси чистого катализатора с разбавляющим его инертным материалом загруженность может относиться только к содержащемуся в подобной смеси катализатору.
Предлагаемый в изобретении способ в принципе может быть реализован при загруженности совокупного слоя катализатора (в пересчете на общее количество М содержащегося в нем катализатора дегидрирования) как реакционным газом, так и содержащимся в ней подлежащим дегидрированию углеводородом (например, пропаном), составляющей от 100 до 10000 нл/л·ч (сокращенно ч-1), часто от 300 до 5000 ч-1, наиболее часто от 500 до 3000 ч-1 (в основном независимо от требуемой степени превращения подлежащего дегидрированию углеводорода).
Пригодными инертными материалами, используемыми для разбавления катализатора дегидрирования, присутствующего в совокупном и/или отдельном слое, являются, например, обожженные глины (алюмосиликаты), стеатит (например, продукт марки С 220 фирмы CeramTec) или другие (предпочтительно в основном не содержащие пор) высокотемпературные химикаты, такие как оксиды алюминия, диоксид кремния, диоксид титана, оксид магния, оксид лантана, оксид церия, оксид цинкаалюминия, диоксид тория, диоксид циркония, карбид кремния, или прочие силикаты, такие как силикат алюминия и/или силикат магния, а также смеси указанных материалов. В соответствии с предлагаемым в изобретении способом состоящие из указанных выше материалов формованные тела можно использовать не только для разбавления совокупного стационарного слоя катализатора и/или отдельных стационарных слоев катализатора, но и в качестве закрывающих и/или при необходимости завершающих инертных насыпных слоев совокупного стационарного слоя катализатора и/или отдельных стационарных слоев катализатора.
В соответствии с предлагаемым в изобретении способом подобный закрывающий и/или завершающий насыпной слой из инертных формованных тел предпочтительно следует формировать таким образом, чтобы при пропускании через него потока реакционной газовой смеси в дегидрированный углеводород превращалось не более 3% мол., лучше не более 2% мол., еще лучше не более 1% мол. или 0% мол. подлежащего дегидрированию углеводорода, содержащегося в указанном потоке.
Для осуществления предлагаемого в изобретении способа в качестве катализатора дегидрирования в совокупном слое катализатора можно использовать, в частности, катализаторы из примеров 1-4, приведенных в немецкой заявке на патент DE-A 19937107.
Речь при этом идет о катализаторах дегидрирования, которые содержат от 10 до 99,9% мас. диоксида циркония, от 0 до 60% мас. оксида алюминия, диоксида кремния и/или диоксида титана и от 0,1 до 10% мас. по меньшей мере одного элемента первой или второй главной группы, элемента третьей побочной группы, элемента восьмой побочной группы периодической системы элементов, лантана и/или олова, при условии, что сумма указанных количеств составляет 100% мас.
В предпочтительном варианте указанные катализаторы дегидрирования содержат по меньшей мере один элемент VIII-й побочной группы, по меньшей мере один элемент I-й и II-й главных групп, по меньшей мере один элемент III-й и/или IV-й главных групп и по меньшей мере один элемент III-й побочной группы, включая лантаниды и актиниды. В качестве элементов VIII-й побочной группы активная масса указанных катализаторов дегидрирования предпочтительно содержит платину и/или палладий, особенно предпочтительно платину. В качестве элементов I-й и II-й главных групп активная масса указанных катализаторов дегидрирования предпочтительно содержит калий и/или цезий. В качестве элементов III-й побочной группы, включая лантаниды и актиниды, активная масса указанных катализаторов дегидрирования предпочтительно содержит лантан и/или хром. В качестве элементов III-й и/или IV-й главной групп активная масса указанных катализаторов дегидрирования предпочтительно содержит один или несколько элементов, выбранных из группы, включающей бор, галлий, кремний, германий, индий, олово и свинец, особенно предпочтительно олово. Еще более предпочтительно активная масса указанных катализаторов дегидрирования содержит по меньшей мере один элемент, являющийся представителем указанных выше групп элементов.
В общем случае в качестве катализатора дегидрирования в составе совокупного и/или отдельного стационарного слоя используют цилиндрики с типичным диаметром от 0,1 или 1 до 10 мм, предпочтительно от 1,5 до 5 мм и типичной длиной от 1 до 20 мм, предпочтительно от 3 до 10 мм, таблетки, предпочтительно обладающие аналогичными размерами, и/или кольца, типичные диаметр и длина которых составляют соответственно от 2 до 30 или до 10 мм, а типичная толщина стенок в целесообразном варианте составляет от 1 до 10,5 или 3 мм.
Какие-либо ограничения относительно геометрической конфигурации используемых в стационарном слое катализаторов (прежде всего катализаторов на носителе) и инертных формованных тел в принципе отсутствуют. Наиболее часто подобные катализаторы представляют собой сплошные цилиндры, полые цилиндры (кольца), шарики, конусы, пирамиды и кубики, а также стержни, колесики, звезды и монолиты.
При этом продольный размер формованных тел катализатора и инертного материала (максимальное расстояние между двумя находящимися на поверхности формованных тел точками) может составлять от 0,5 до 100 мм, часто от 1,5 до 80 мм и наиболее часто от 3 до 50 или 20 мм.
В качестве особенно пригодного катализатора дегидрирования, используемого в предлагаемом в изобретении совокупном и/или отдельном стационарном слое, можно использовать также катализатор, приведенный в немецкой заявке на патент с регистрационным номером 102005044916.
Регенерацию рекомендуемых для осуществления предлагаемого в изобретении способа катализаторов дегидрирования прежде всего можно осуществлять, используя технологию, на первой стадии которой через общий (стационарный) слой катализатора пропускают разбавленный молекулярным азотом и/или (предпочтительно) водяным паром воздух с температурой на входе, составляющей от 300 до 600°С (при необходимости также до 750°С), часто также от 400 до 450°С. При этом загруженность катализатора регенерирующим газом может составлять, например, от 50 до 10000 ч-1 (в пересчете на общее количество М катализатора дегидрирования), а содержание кислорода в регенерирующем газе может находиться в интервале от 0,1 или 0,5 до 20% об. На следующей дополнительной стадии регенерации в качестве регенерирующего газа можно использовать воздух, сохраняя прочие условия регенерации неизменными. В производственно-техническом отношении перед регенерацией целесообразно осуществлять продувку общего (стационарного) слоя катализатора, содержащего катализатор дегидрирования, инертным газом (например, азотом, в частности техническим азотом, содержащим до 1% об. кислорода, влаги или их смеси). В заключение, как правило, рекомендуется выполнять дополнительную регенерацию совокупного слоя катализатора чистым молекулярным водородом со степенью чистоты более 99% об. или молекулярным водородом, который разбавлен инертным газом (предпочтительно водяным паром и/или азотом), причем содержание водорода в соответствующей смеси не должно составлять более 1% об., сохраняя прочие условия регенерации неизменными.
В соответствии с предлагаемым в изобретении способом реакционная зона RZ в принципе может состоять только из одного или же из нескольких реакторов дегидрирования. Можно использовать последовательно и/или параллельно соединенные реакторы дегидрирования. Совокупный слой катализатора в соответствующем варианте может находиться только в одном реакторе дегидрирования или его можно распределить на несколько отдельных слоев, помещенных в несколько реакторов. В случае, если совокупный слой катализатора находится в единственном реакторе дегидрирования, речь в принципе идет о сплошном слое катализатора.
Однако в подобном случае сплошность активной массы совокупного слоя катализатора в направлении потока газовой смеси может быть нарушена, например, в связи с наличием замыкающих и/или предварительных слоев инертного материала, например насыпных формованных тел. Совокупный слой катализатора может состоять также, например, из отдельных, последовательно упорядоченных в радиальном или осевом направлении стационарных слоев, между которыми находятся не занятые формованными телами (свободные) промежуточные пространства. Подобная ситуация имеет место, например, в случае если под реакционной зоной RZ подразумевают полочный реактор. Реакционная зона, очевидно, может состоять также из нескольких полочных реакторов. Кроме того, реакционная зона может состоять из нескольких соединенных между собой реакторов, один или несколько из которых являются реакторами дегидрирования, содержащими единственный отдельный слой катализатора дегидрирования (например, отдельный стационарный слой катализатора дегидрирования), а один или несколько являются реакторами дегидрирования, содержащими более одного отдельного слоя катализатора дегидрирования (например, в виде полок с отдельными стационарными слоями катализатора).
Согласно изобретению для предотвращения деактивирования совокупного слоя катализатора, происходящего по мере его эксплуатации в интервале рабочих моментов t0<t<tR, в первую очередь можно использовать любые указанные в начале описания методы внешнего и внутреннего управления температурой реакционной газовой смеси на ее пути через совокупный слой катализатора, включая регулирование температуры поступающего в совокупный слой катализатора потока реакционной газовой смеси, содержащего молярное исходное количество KW подлежащего дегидрированию углеводорода. Однако в принципе можно, например, изменять также парциальное давление, например, благодаря варьированию степени разбавления потока реакционной газовой смеси инертным газом. Часто совместно используют также разные возможные методы управления температурой реакционной газовой смеси. К ним относится также удаление образующегося в процессе дегидрирования водорода.
Простой возможный вариант достижения цели настоящего изобретения, например, состоит в том, что в соответствии с предлагаемым в изобретении способом первая, вторая и последняя трети общего количества катализатора М имеют адиабатическое конструктивное исполнение, однако с возможностью изменения теплосодержания реакционной газовой смеси перед входом в каждую из этих адиабатически выполненных третей.
С технической точки зрения в подобном случае целесообразно, чтобы в рабочий момент t0 температура T1 реакционной газовой смеси на входе в первую по ходу газового потока треть общего количества катализатора М превышала температуру Т2 реакционной газовой смеси на ее входе во вторую треть общего количества катализатора М, а температура Т2 была выше температуры Т3 реакционной газовой смеси на входе в последнюю треть общего количества катализатора М.
В соответствии с этим простым методом противодействия деактивированию совокупного слоя катализатора, происходящему в интервале рабочих моментов t0<t<tR, может являться целенаправленное взаимное согласование скоростей приращения как температуры T1, так и температур Т2 и Т3. При этом скорость приращения температуры Т3 обычно всегда превышает скорость приращения температуры T1. В отличие от этого скорость приращения температуры T2 вначале процесса дегидрирования обычно превышает скорость приращения температуры Т3, а по мере протекания этого процесса снижается, становясь ниже скорости приращения температуры Т3 и все более приближаясь к более низкой скорости приращения температуры T1.
Указанный вариант позволяет в течение максимально длительного времени поддерживать преимущественное постоянство выраженной в % мол. от KW молярной доли G содержащегося в молярном исходном количестве KW подлежащего дегидрированию углеводорода, превращенного в дегидрированный углеводород при (однократном) прохождении потока реакционной газовой смеси через совокупный слой катализатора, а также в течение сравнительно долго реализуемого в этом режиме рабочего периода обеспечивать преимущественно обратимое деактивирование катализатора благодаря возможности преимущественно полного отказа от последующего использования описанного выше метода регенерации.
Согласно изобретению важно, чтобы в соответствии с предлагаемым в изобретении способом (прежде всего в соответствии с рассмотренным выше вариантом способа) реальная степень разбавления инертным материалом разных третей общего количества М катализатора дегидрирования в реальном совокупном слое катализатора могла быть как идентичной, так и разной. В принципе все трети М могут находиться в составе совокупного слоя катализатора также в неразбавленном состоянии. Степень разбавления катализатора дегидрирования, очевидно, может также быть переменной в пределах той или иной трети общего количества М катализатора.
В одном из вариантов предлагаемый в изобретении способ осуществляют таким образом, чтобы обусловленный произвольно малым частичным количеством М вклад в G, нормированный к этому частичному количеству, при условии максимального значения этого вклада был реализован таким образом, чтобы место этого нормированного максимального вклада в G в течение предлагаемой в изобретении длительной работы в интервале рабочих моментов t0<t<tR по мере возрастания t перемещалось от входа потока реакционной газовой смеси в совокупный слой катализатора до выхода потока газовой смеси продуктов реакции из совокупного слоя катализатора.
Рассмотренный выше предлагаемый в изобретении долговременный рабочий режим особенно просто реализовать в адиабатическом полочном реакторе. При этом соответствующий отдельный слой катализатора может находиться в реакционном объеме в виде псевдоожиженного, движущегося или стационарного слоя. В реакционном объеме, очевидно, может находиться также комбинация псевдоожиженного слоя катализатора, например, со стационарным слоем, движущегося слоя катализатора со стационарным слоем или любая иная комбинация указанных слоев. В технически целесообразном варианте поток реакционной газовой смеси на пути от одной полки с отдельным слоем катализатора к следующей полке с отдельным слоем катализатора подвергают промежуточному нагреванию, необходимому для реализации этого варианта предлагаемого в изобретении способа и осуществляемому, например, благодаря пропусканию реакционной газовой смеси над ребрами обогреваемых горячими газами или водяным паром теплообменников, через обогреваемые горячими топочными газами или водяным паром трубы или обогреваемые горячими газами пластины теплообменников, находящихся в адиабатически сконструированном остальном реакционном объеме. В соответствующем варианте можно также варьировать температуру потока содержащей исходное количество KW реакционной газовой смеси на входе в совокупный слой катализатора. В особенно простом варианте полочный реактор включает три идентичные полки с катализатором. Однако полочный реактор в принципе может содержать также шесть, девять, двенадцать или иное количество идентичных или по-разному загруженных катализатором полок. При этом промежуточное нагревание можно осуществлять между всеми последовательно упорядоченными полками или только между некоторыми из них.
В производственно-техническом отношении особенно целесообразным полочным реактором указанного выше типа является полочный реактор с чисто стационарным слоем катализатора. При этом в наиболее простом варианте отдельные стационарные слои катализатора на полках, количество которых соответствует количеству отдельных стационарных слоев, пространственно упорядочены в реакционном объеме вдоль оси в направлении потока реакционной газовой смеси или в кольцевых зазорах между центрически вставленными друг в друга цилиндрическими колосниковыми решетками. Кроме того, кольцевые зазоры могут быть расположены в реакционном объеме друг над другом в сегментах, причем поток реакционной газовой смеси последовательно проходит в радиальном направлении через один сегмент, а затем через следующий сегмент, расположенный выше или ниже первого. В наиболее простом варианте отдельные полки со (стационарными) слоями катализатора расположены внутри адиабатического реактора типа шахтной печи.
Согласно изобретению в полочном реакторе со стационарными слоями катализатора, используемом для осуществления предлагаемого в изобретении способа, как перед отдельным стационарным слоем катализатора, так и после него предпочтительно находится насыпной слой из инертных формованных тел. Необходимость использования подобных слоев обусловлена тем, что максимальный размер формованных тел катализатора дегидрирования нередко меньше размера ячеек соответствующей (опорной) колосниковой решетки. Использование насыпного слоя из инертных формованных тел с соответствующими размерами позволяет решить эту проблему.
Загрузка насыпного слоя из инертных формованных тел перед катализатором дегидрирования (в направлении потока реакционных газов) благоприятна, в частности, в связи с тем, что это позволяет обеспечить максимальную однородность как температуры, так и состава потока реакционной газовой смеси. Прежде всего это относится к случаю, если вводимый в отдельный стационарный слой катализатора поток реакционной газовой смеси сформирован из отдельных потоков разных газов.
Последнее обстоятельство имеет особенно большое значение, например, в том случае, если косвенное управление температурой между отдельными слоями катализатора дегидрирования, подлежащее использованию в соответствии с описанным выше вариантом осуществления предлагаемого в изобретении способа, по меньшей мере частично заменяют прямым управлением температурой. В соответствии с технически предпочтительным вариантом предлагаемый в изобретении способ осуществляют исключительно методом прямого управления температурой между двумя отдельными слоями катализатора.
С этой целью к потоку реакционной газовой смеси на его реализуемом согласно изобретению пути через реакционную зону RZ, например, после прохождения первого в направлении потока отдельного слоя катализатора и при необходимости между последующими отдельными слоями катализатора в течение рабочего периода реализации предлагаемого в изобретении способа добавляют варьируемые ограниченные количества содержащего молекулярный кислород газа.
Речь при этом может идти, например, о чистом молекулярном кислороде или смеси инертного газа с молекулярным кислородом (например, о воздухе). В зависимости от используемого катализатора дегидрирования вызывают указанное в начале настоящего описания, соответствующим образом варьируемое ограниченное сгорание содержащихся в реакционной газовой смеси углеводородов, при необходимости уже осажденного на поверхности катализатора угля или углеподобных соединений и/или образующихся во время обычного гетерогенно катализируемого дегидрирования (например, дегидрирования пропана) и/или добавляемого к реакционному газу водорода [может быть также технически целесообразным добавление в реакционный объем полочного реактора (в реакционную зону RZ) слоев, которые содержат катализатор, способный катализировать специфическое (селективное) сгорание водорода (и/или углеводорода) (подобные катализаторы приведены например, в заявках США на патент US-A 4788371, US-A 4886928, US-A 5430209, US-A 5530171, US-A 5527979 и US-A 5563314; слои подобных катализаторов можно, например, чередовать в реакционном объеме полочного реактора со слоями, содержащими катализатор дегидрирования)]. В этом случае варьирование выделяющегося в результате ограниченного сгорания тепла, реализуемое благодаря варьированию количества дозируемого молекулярного кислорода, обусловливает необходимое согласно изобретению варьирование степени дегидрирования. В отличие от этого в пределах предлагаемого в изобретении рабочего периода обычно технически целесообразно поддерживать преимущественно на постоянном уровне содержание кислорода в исходном количестве KW потока реакционной газовой смеси, содержащего подлежащий дегидрированию углеводород, и варьировать только температуру этого (исходного) потока реакционной газовой смеси (как правило, путем косвенного теплообмена). Однако технические мероприятия, используемые для обеспечения прямого управления температурой, можно реализовать не только в случае использования полочного реактора в качестве реакционной зоны RZ. Часто они могут быть реализованы также при наличии в реакционной зоне RZ единственного непрерывного слоя катализатора дегидрирования. Простая техническая реализация подобных мероприятий возможна, например, благодаря тому, что, например, в стационарный слой катализатора на различных уровнях вводят дозирующие трубки, через которые в поток реакционной газовой смеси можно направлять содержащий молекулярный кислород газ.
В наиболее простом варианте цель настоящего изобретения может быть достигнута также благодаря тому, что в соответствии с предлагаемым в изобретении способом первая, вторая и последняя трети общего количества катализатора М (в направлении потока реакционной газовой смеси) имеют адиабатическое конструктивное исполнение, однако с возможностью изменения теплосодержания реакционной газовой смеси перед входом в первую адиабатически выполненную треть путем косвенного управления температурой, а на входе во вторую, соответственно третью адиабатически выполненную треть общего количества катализатора М путем прямого управления температурой (например, путем варьирования расхода реакционной газовой смеси на входе во вторую, соответственно третью адиабатически выполненную треть этого, соответственно содержащего молекулярный кислород газа).
В этом случае технически целесообразно осуществлять технологический процесс в рабочий момент t0 таким образом, чтобы максимальная температура T1* потока реакционной газовой смеси при его прохождении через первую треть общего количества катализатора М превышала максимальную температуру T2* потока реакционной газовой смеси при его прохождении через вторую треть общего количества катализатора М, и температура Т2* превышала максимальную температуру Т3* потока реакционной газовой смеси при его прохождении через последнюю треть общего количества катализатора М.
Температурой T1* можно легко управлять путем регулирования теплосодержания потока реакционной газовой смеси и содержания в ней молекулярного кислорода при входе в первую треть общего количества М катализатора. В этом случае соотношение между температурами T2* и Т3*, а также соотношение между температурой Т2* и температурой Т3*, соответственно T1*, можно устанавливать путем дозирования в поток реакционной газовой смеси содержащего молекулярный кислород газа между первой и второй третями М, соответственно между второй и последней (третьей) третями М. В обоих случаях для этого используют один и тот же содержащий молекулярный кислород газ, например чистый молекулярный кислород со степенью чистоты более 99% мол., воздух или смесь инертного газа с молекулярным кислородом. В этом случае необходимое соотношение между температурами Т2* и Т3* в рабочий момент t=t0 обычно особенно легко обеспечить, если в этот рабочий момент использовать идентичный расход дозируемого молекулярного кислорода.
В этом случае деактивированию совокупного слоя катализатора в интервале рабочих моментов t0<t<tR согласно изобретению можно противодействовать простым методом, состоящим в том, что взаимно согласованным образом повышают теплосодержание (температуру) потока реакционной газовой смеси на его входе в первую треть общего количества М катализатора, и между первой и второй третями М (в поток 1), соответственно между второй и последней (третьей) третями М (в поток 2) дозируют молекулярный кислород (соответственно содержащий молекулярный кислород газ).
Вначале выбирают в основном идентичные скорости приращения потоков молекулярного кислорода, дозируемых в потоки 1 и 2. В дальнейшем скорость приращения потока молекулярного кислорода, дозируемого в поток 2, превышает скорость приращения потока молекулярного кислорода, дозируемого в поток 1, а затем скорость приращения потока молекулярного кислорода, дозируемого в поток 1, превышает скорость приращения потока молекулярного кислорода, дозируемого в поток 2, то есть расход молекулярного кислорода, дозируемого в поток 2, снижается медленнее расхода молекулярного кислорода, дозируемого в поток 1. Совокупную подачу молекулярного кислорода в потоки 1 и 2 (соответственно в пересчете на чистый кислород) в интервале рабочих моментов t0<t<tR повышают по сравнению с температурой потока реакционной газовой смеси на входе в первую треть общего количества М катализатора таким образом, чтобы при этом скорость приращения температуры Т3* в основном всегда превышала скорость приращения температуры T1*. В отличие от этого скорость приращения температуры Т2* при этом вначале обычно превышает скорость приращения температуры Т3*, а в дальнейшем становится ниже скорости приращения температуры Т3*, все более приближаясь к низкой скорости приращения температуры T1*.
Рассмотренный выше вариант предлагаемого в изобретении способа непрерывного дегидрирования особенно просто можно реализовать в адиабатически выполненном полочном реакторе, прежде всего в полочном реакторе со стационарным слоем катализатора. Согласно изобретению полочный реактор со стационарным слоем катализатора в оптимальном варианте содержит три предпочтительно идентичные полки с соответствующими стационарными слоями (отдельными стационарными слоями) катализатора. Промежуточную подачу кислорода можно осуществлять между двумя соседними полками. Отдельные стационарные слои катализатора дегидрирования могут последовательно располагаться вдоль оси реактора или в кольцевых зазорах центрически вставленных друг в друга цилиндрических колосниковых решеток. В особенно простом варианте в этом случае используют адиабатически выполненный реактор типа шахтной печи, описанный, например, в немецких заявках на патент с регистрационными номерами 102006015235, 102006017623 и 102006029790, а также в DE-А 102005051401.
В случае подачи кислорода в виде соответствующих промежуточных потоков для обеспечения преимущественного постоянства общего расхода кислорода в течение рабочего периода дозируемый газ, содержащий молекулярный кислород, технически предпочтительно разбавлять используемым в качестве инертного разбавляющего газа водяным паром. В этом случае повышение расхода кислорода в предпочтительном варианте сопровождается снижением расхода водяного пара, что позволяет поддерживать преимущественно постоянный расход соответствующего содержащего кислород газа в течение рабочего периода.
Согласно изобретению промежуточную подачу содержащего молекулярный кислород газа предпочтительно осуществляют таким образом, чтобы при однородном перемешивании последнего с потоком реакционной газовой смеси в месте дозирования газа могла образоваться новая реакционная газовая смесь, в которой молярное отношение молекулярного кислорода к молекулярному водороду составляло бы менее 1:2. Соблюдение этого рамочного условия позволяет получать целевой продукт с повышенной селективностью.
Неожиданно выяснилось, что несмотря на указанное рамочное условие управление температурой указанным выше методом прямого управления, необходимое для реализации предлагаемого в изобретении долговременного рабочего режима, можно осуществлять в основном в отсутствие снижения селективности образования целевого продукта также без необходимости обязательной подачи внешнего молекулярного водорода. В случае необходимости соблюдения повышенного содержания молекулярного водорода для этого в технически целесообразном варианте можно воспользоваться также описанным в международной заявке WO 03/076370 (соответственно в немецкой заявке на патент DE-A 10211275) методом рециркуляции, согласно которому выводимый из реакционной зоны RZ газообразный поток продуктов разделяют на два отдельных потока идентичного состава, один из которых в качестве оборотного газа дегидрирования возвращают на вход в реакционную зону RZ. Гетерогенно катализируемое частичное дегидрирование, предусматривающее использование метода рециркуляции, опубликовано также в международной заявке WO 2006/050957. Однако в соответствии с предлагаемым в изобретении способом к потоку реакционной газовой смеси в любое время при необходимости можно добавлять также внешний молекулярный водород. В случае, если большое значению придают повышенному сроку службы совокупного слоя катализатора, согласно изобретению предпочтительным прежде всего является добавление молекулярного водорода уже в направляемый в реакционную зону RZ поток реакционной газовой смеси. Это относится главным образом к ситуации, в соответствии с которой указанный поток реакционной газовой смеси содержит молекулярный кислород. Подобная ситуация, как правило, имеет место прежде всего в том случае, если непосредственно после реализуемого предлагаемым в изобретении способом дегидрирования осуществляют гетерогенно катализируемое частичное окисление полученного при дегидрировании углеводорода (например, частичное окисление пропилена в акролеин и/или акриловую кислоту), причем на стадию частичного окисления направляют исходную реакционную смесь, которая в качестве инертного газа содержит непревращенный на стадии дегидрирования подлежащий дегидрированию углеводород (например, пропан).
В этом случае сначала выведенный из реакционной зоны RZ газообразный поток продуктов дегидрирования как таковой или после выделения из него по меньшей мере частичного количества компонентов (например, водорода, воды, азота и так далее), отличающихся от дегидрированного углеводорода (например, пропилена) и (непревращенного) подлежащего дегидрированию углеводорода (например, пропана), используют для подачи на вход по меньшей мере одного реактора окисления, причем содержащийся в соответствующей исходной газовой смеси дегидрированный углеводород (например, пропилен) подвергают селективному гетерогенно катализируемому частичному газофазному окислению молекулярным кислородом, получая газовую смесь продуктов частичного окисления, содержащую целевой продукт (продукт частичного окисления), например акролеин, акриловую кислоту или их смесь, а также, как правило, непревращенный подлежащий дегидрированию углеводород (например, пропан), избыточный молекулярный кислород и при необходимости непревращенный дегидрированный углеводород (например, пропилен).
В последующей зоне разделения выделяют содержащийся в газовой смеси продуктов частичного окисления целевой продукт (например, акролеин, акриловую кислоту или их смесь) и от полученного при этом остаточного газа, содержащего непревращенный подлежащий дегидрированию углеводород (например, пропан), молекулярный кислород, а также при необходимости непревращенный дегидрированный углеводород (например, пропилен), отбирают отдельный поток, содержащий по меньшей мере один непревращенный подлежащий дегидрированию углеводород (например, пропан), большую часть непревращенного молекулярного кислорода, а также при необходимости непревращенный дегидрированный углеводород (например, пропилен), и возвращают этот отдельный поток в качестве оборотного газа частичного окисления на стадию осуществления предлагаемого в изобретении способа (как правило, в качестве компонента направляемого в реакционную зону RZ потока реакционной газовой смеси, который содержит исходное количество KW подлежащего дегидрированию углеводорода).
В случае, если под предлагаемым в изобретении способом (а также иным способом) подразумевают, например, гетерогенно катализируемое частичное дегидрирование пропана в пропилен, который непосредственно после этого подвергают частичному окислению до акролеина, акриловой кислоты или их смеси, направляемый в реакционную зону RZ поток реакционной газовой смеси может обладать, например, преимущественно следующим составом:
Указанный поток может содержать также незначительное количество уксусной кислоты (например, сравнимое с возможным содержанием акриловой кислоты).
Выделение целевого продукта (например, акриловой кислоты) из газовой смеси продуктов частичного окисления обычно осуществляют переводом целевого продукта (например, акриловой кислоты) в конденсированную фазу. Подобный перевод можно осуществлять абсорбционным и/или конденсационным (охлаждение) методами. В случае выделения в качестве целевого продукта акриловой кислоты в качестве абсорбента используют, например, воду, водные растворы или высококипящие, прежде всего гидрофобные органические растворители, точка кипения которых при атмосферном давлении превышает точку кипения акриловой кислоты. Перевод акриловой кислоты в конденсированную фазу особенно предпочтительно осуществляют методом фракционирующей конденсации газовой смеси продуктов частичного окисления. Абсорбционный и/или конденсационный перевод акриловой кислоты из газовой смеси продуктов частичного окисления в конденсированную фазу предпочтительно осуществляют в колоннах с повышающими эффективность разделения встроенными элементами, газовую смесь продуктов частичного окисления через которые обычно пропускают в направлении снизу вверх. Абсорбент, как правило, подают в верхнюю часть колонны, из которой обычно осуществляют отбор остаточного газа.
Дальнейшее выделение акриловой кислоты необходимой степени чистоты из конденсированной фазы, как правило, осуществляют благодаря использованию по меньшей мере одного метода термического разделения. Речь при этом идет о технологии, в соответствии с которой создают по меньшей мере две отличающиеся друг от друга материальные фазы (например, жидкость/жидкость, газ/жидкость, твердое вещество/жидкость, газ/твердое вещество и так далее), после чего реализуют их взаимный контакт. Поскольку между обеими фазами существуют градиенты, происходят соответствующий теплообмен и массообмен, которые и обусловливают необходимое разделение (выделение). Указанные методы называют термическими, поскольку для формирования материальных фаз необходимо как отводить, так и подводить тепло, и/или поскольку отведение или подведение тепловой энергии благоприятствует материальному обмену или обеспечивает его.
Согласно изобретению по меньшей мере один метод термического разделения предпочтительно включает по меньшей мере одну операцию кристаллизационного выделения из жидкой фазы. Согласно изобретению по меньшей мере одной операцией кристаллизационного выделения акриловой кислоты в целесообразном варианте является суспензионная кристаллизация, причем суспензионный кристаллизат предпочтительно промывают расплавленным кристаллизатом, предварительно выделенным и промытым в гравиметрической, механической или гидравлической промывочной колонне (последняя согласно изобретению является предпочтительной). Однако для термического разделения можно использовать, например, экстракционный, десорбционный, кристаллизационный, ректификационный, азетропно-дистилляционный, азеотропно-ректификационный, дистилляционный и/или отпарной методы. Для выделения чистой акриловой кислоты, как правило, используют комбинацию из разных указанных выше термических методов разделения.
Непосредственно после рассмотренного выше выделения акриловой кислоты можно осуществлять процесс ее радикальной полимеризации (прежде всего с целью производства суперабсорбирующих воду полиакриловых кислот и/или соответствующих частично или полностью нейтрализованных солей щелочных металлов (предпочтительно натрия), в соответствии с которым выделенную акриловую кислоту превращают в полимеризаты методом радикальной полимеризации.
Кроме того, непосредственно после рассмотренного выше выделения акриловой кислоты можно осуществлять синтез сложных эфиров акриловой кислоты, в соответствии с которым выделенную акриловую кислоту этерифицируют спиртами, предпочтительно алканолами, особенно предпочтительно алканолами с 1-12 атомами углерода, как правило, в присутствии используемых в качестве катализатора кислот.
В свою очередь, непосредственно после этерификации можно осуществлять процесс радикальной полимеризации полученных сложных эфиров акриловой кислоты.
Без учета специфики предлагаемого в изобретении долговременного дегидрирования процессы реализуемого согласно изобретению получения пропилена из пропана в качестве источника пропилена для частичного окисления в акролеин и/или акриловую кислоту известны из уровня техники, включая рециркуляцию оборотного газа частичного окисления и при необходимости оборотного газа дегидрирования. Так, например, подобные многостадийные способы описаны в немецкой заявке на патент DE-A 102005022798, немецкой заявке с регистрационным номером 102006024901.1, немецких заявках на патент DE-A 10246119, DE-A 10245585, DE-A 102005049699, DE-A 102004032129, DE-A 102005013039, DE-A 102005010111, DE-A 102005009891, DE-A 10211275, европейской заявке на патент ЕР-А 117146, патенте США US 3161670, немецкой заявке на патент DE-A 3313573, международной заявке WO 01/96270, немецких заявках на патент DE-A 10316039, DE-A 102005009885, DE-A 102005052923, DE-A 102005057197, международной заявке WO 03/076370, немецких заявках на патент DE-A 10245585, DE-A 2213573, патенте США US 3161670 и цитируемом в указанных публикациях уровне техники. Соответствующий принцип согласно немецкой заявке на патент DE-A 10219686 используют для получения метакролеина и/или метакриловой кислоты.
Адсорбционные и/или конденсационные методы перевода акриловой кислоты из газовой смеси продуктов частичного окисления в конденсированную фазу также подробно описаны в уровне техники. В этой связи следует сослаться на немецкие заявки на патент DE-A 10336386, DE-A 19631645, DE-A 19501325, европейскую заявку на патент ЕР-А 982289, немецкую заявку на патент DE-A 19838845, международную заявку WO 02/076917, европейские заявки на патент ЕР-А 695736, ЕР-А 778225, ЕР-А 1041062, ЕР-А 982287, ЕР-А 982288, заявку США на патент US-А 2004/0242826, европейские заявки на патент ЕР-А 792867, ЕР-А 784046, ЕР-А 695736 (прежде всего абсорбционный метод), международную заявку WO 04/035514 и немецкие заявки на патент DE-A 19924532, DE-A 19814387, DE-A 19740253, DE-A 19740252 и DE-A 19627847 (прежде всего конденсационный метод).
Кроме того, подобные абсорбционные и/или конденсационные методы выделения акриловой кислоты из газовой смеси продуктов частичного окисления описаны в европейских заявках на патент ЕР-А 1338533, ЕР-А 1388532, немецкой заявке на патент DE-A 10235847, международной заявке WO 98/01415, европейских заявках на патент ЕР-А 1015411, ЕР-А 1015410, международных заявках WO 99/50219, WO 00/53560, WO 02/09839, немецкой заявке на патент DE-A 10235847, международной заявке WO 03/041833, немецких заявках на патент DE-A 10223058, DE-A 10243625, DE-A 10336386, европейской заявке на патент ЕР-А 854129, патенте США US 4317926, немецких заявках на патент DE-A 19837520, DE-A 19606877, DE-A 19501325, DE-A 10247240, DE-A 19740253, европейских заявках на патент ЕР-А 695736, ЕР-А 1041062, ЕР-А 117146, немецких заявках на патент DE-A 4308087, DE-A 4335172, DE-A 4436243, DE-A 10332758 и DE-A 19924533. В принципе выделение акриловой кислоты можно осуществлять также в соответствии с немецкими заявками на патент DE-A 10336386, DE-A 10115277, DE-A 19606877, европейскими заявками на патент ЕР-А 920408, ЕР-А 1068174, ЕР-А 1066239, ЕР-А 1066240, международными заявками WO 00/53560, WO 00/53561, немецкой заявкой на патент DE-A 10053086, международной заявкой WO 01/96271, немецкой заявкой на патент DE-A 102004032129, международными заявками WO 04/063138, WO 04/35514, немецкими заявками на патент DE-A 10243625 и DE-A 10235847.
В случае если углеводородом, подлежащим дегидрированию предлагаемым в изобретении способом, является пропан, то в соответствии с немецкой заявкой на патент DE-A 10245585 его вводят в реакционную зону RZ в качестве компонента потока реакционной газовой смеси предпочтительно в виде сырого пропана.
Содержание подлежащего дегидрированию углеводорода в направляемом в реакционную зону RZ потоке реакционной газовой смеси в общем случае составляет по меньшей мере 5% об. Соответствующая объемная доля подлежащего дегидрированию углеводорода нередко составляет более 10% об., часто более 15% об. и в большинстве случаев более 20, 25 или 30% об. Однако, как правило, она составляет менее 90% об., в большинстве случаев менее 80% об. и часто менее 70% об. Указанные данные прежде всего относятся к пропану в качестве подлежащего дегидрированию углеводорода и пропилену в качестве дегидрированного углеводорода. Однако они, очевидно, относятся также к изобутану в качестве подлежащего дегидрированию углеводород и изобутилену в качестве дегидрированного углеводорода.
Кроме того, указанный поток реакционной газовой смеси может содержать:
a) азот и воду,
b) азот, кислород и воду,
c) азот, кислород, воду и водород,
d) азот, кислород, воду, водород и диоксид углерода,
e) азот, кислород, воду, водород, диоксид углерода и монооксид углерода.
В оптимальных случаях поток реакционной газовой смеси, который в соответствии с предлагаемым в изобретении способом направляют в реакционную зону RZ, образуется вследствие совмещения, например, сырого пропана, молекулярного водорода и оборотного газа частичного окисления. В этом случае технически целесообразным являются предварительное совмещение сырого пропана с молекулярным водородом и нагревание полученной при этом смеси до реакционной температуры, которая обычно составляет более 300°С, предпочтительно более 350°С, чаще всего более 400°С. Оборотный газ частичного окисления также нагревают до реакционной температуры. Предварительное нагревание указанных газовых потоков можно осуществлять путем косвенного теплообмена с горячей газовой смесью получаемых предлагаемым в изобретении способом продуктов. После этого оба предварительно нагретых газовых потока совмещают, получая направляемый в реакционную зону RZ поток реакционной газовой смеси. Повышение температуры направляемого в реакционную зону RZ потока реакционной газовой смеси, при необходимости осуществляемое в соответствии с предлагаемым в изобретении долговременным режимом, может быть реализовано, например, повышением интенсивности косвенного теплообмена.
В заключение следует еще раз подчеркнуть, что технические мероприятия, реализуемые при прямом и косвенном управлении температурой с целью обеспечения предлагаемого в изобретении долговременного рабочего режима, можно применять по отдельности или комбинированно. Предлагаемый в изобретении способ может быть особенно просто реализован в полочных реакторах с адиабатическим конструктивным исполнением, в которых необходимое варьирование управления температурой и расхода при необходимости вводимых вспомогательных газов можно выполнять между двумя соседними полками. При этом количество используемых полок можно варьировать в широком диапазоне. Катализатор дегидрирования технически целесообразно загружать на полки идентичным образом. В оптимальном варианте количество полок составляет три или соответствующее целочисленное кратное (например, шесть, девять, двенадцать). Вместо одного полочного реактора можно использовать также несколько последовательно соединенных отдельных адиабатических полочных реакторов, число которых соответствует числу полок, и выполнять управление между этими реакторами. Предлагаемый в изобретении способ в принципе можно осуществлять также в единственном непрерывном (сплошном) слое катализатора.
В общем случае продолжительность осуществления процесса гетерогенно катализируемого частичного дегидрирования с использованием предлагаемого в изобретении способа и свежерегенерированного катализатора достигает по меньшей мере 20 часов, а во многих случаях 100 часов и более.
Примеры и сравнительные примеры
а) Реактор дегидрирования
В качестве реактора дегидрирования использовали трубку длиной 2070 мм с толщиной стенок 1 мм и внутренним диаметром 36,4 мм из специальной стали номер 1.4841 согласно DIN. Поток реакционной газовой смеси пропускали через трубчатый реактор в направлении сверху вниз. Нижняя часть реактора была снабжена выполненной из той же специальной стали контактной конструкцией высотой 115 мм, которая служила опорой для совокупного стационарного слоя катализатора (включающего три отдельные стационарные слоя катализатора с примерно одинаковым насыпным весом), причем совокупный слой обладал следующей структурой (ниже указана высота соответствующих слоев в направлении сверху вниз):
790 мм - насыпной слой (инертных) шариков из стеатита С-220 фирмы СеramТес диаметром от 4 до 5 мм (в качестве слоя для внутреннего подогрева);
195 мм - насыпной слой катализатора дегидрирования, представляющий собой сплав платины с оловом, промотированный цезием, калием и лантаном в виде соответствующих оксидов, и нанесенный на внешние и внутренние поверхности цилиндриков из смешанного оксида ZrO2·SiO2 со средней длиной 6 мм (гауссово распределение в интервале от 3 до 12 мм с максимумом около 6 мм) и диаметром 2 мм, причем массовому соотношению компонентов катализатора, включая подложку, соответствует формула Pt0,3Sn0,6La3,0Cs0,5K0,2(ZrO2)88,3(SiO2)7,1 (получение исходных продуктов для катализатора и его активирование осуществляли в соответствии с примером 4 из немецкой заявки на патент DE-А 10219879);
145 мм - насыпной слой шариков из стеатита С-220 фирмы СеrаmТес диаметром от 4 до 5 мм;
190 мм - насыпной слой указанного выше катализатора дегидрирования;
145 мм - насыпной слой шариков из стеатита С-220 фирмы СеrаmТес диаметром от 4 до 5 мм;
195 мм - насыпной слой указанного выше катализатора дегидрирования;
20 мм - насыпной слой шариков из стеатита С-220 фирмы СеrаmТес диаметром от 1,5 до 2,5 мм и
210 мм - насыпной слой шариков из стеатита С-220 фирмы СеrаmТес диаметром от 4 до 5 мм.
Над используемым для внутреннего подогрева верхним насыпным слоем в реакционной трубке оставалось свободное пространство.
Трубчатый реактор находился внутри устройства для предварительного нагревания, представляющего собой два полуцилиндра из меди с толщиной стенок 25 мм, которые обеспечивали равномерное распределение подводимой от окружающего полуцилиндры ленточного электронагревателя тепловой энергии, причем внутри образованного медными полуцилиндрами цилиндра находился самый верхний участок трубчатого реактора длиной 400 мм с находящимся внутри него насыпным слоем для внутреннего подогрева. Ленточный электронагреватель был снабжен наружной обмоткой из теплоизолирующего материала.
Трубчатый реактор окружен двумя парами полуцилиндров с толщиной стенок 25 мм из теплоизоляционного материала MPS-Super G фирмы Microtherm (Германия), повернутых относительно друг друга на 90°, причем внутри образованной указанными теплоизолирующими элементами конструкции находился нижний участок трубчатого ректора длиной 1530 мм (нижнему концу этого участка соответствовала точка, расположенная несколько ниже поверхности прилегания контактной опоры к реакционной трубке). Теплоизолирующие полуцилиндры окружала выполненная из специальной стали цилиндрическая огибающая с наружным диаметром 168 мм и внутренним диаметром 154 мм, которая, в свою очередь, была помещена внутрь отражательной печи высотой 1600 мм, назначением которой являлся сопровождающий обогрев трубчатого реактора. Благодаря этому удавалось минимизировать воздействие на адиабатическое устройство тепловых потоков от окружающей среды к реакционной трубке и в обратном направлении.
По центру реакционной трубки дополнительно введена гильза с наружным диаметром 6 мм и внутренним диаметром 4 мм, внутри которой находилась многопозиционная термопара диаметром 3,2 мм с 14 контролируемыми точками, находящимися на расстоянии 8 см друг от друга, начиная с нижнего конца реакционной трубки.
Кроме того, в трубчатый реактор введены две пары дозирующих трубок. Одна пара дозирующих трубок была введена в верхнюю часть трубчатого реактора, а другая пара - в его нижнюю часть. Прилегающие друг к другу дозирующие трубки каждой из пар позиционировали в пространстве между наружными стенками гильзы для термопары и внутренними стенками реакционной трубки таким образом, чтобы трубки находились посередине поперечного сечения указанного пространства. Обе пары дозирующих трубок располагались на проекции поперечного сечения трубчатого реактора напротив друг друга. Наружный диаметр дозирующих трубок, выполненных из специальной стали номер 1.4841 согласно DIN, составлял 3,17 мм, а их внутренний диаметр 2,17 мм. Дозирующие трубки каждой из пар обладали разной длиной. Через одну из дозирующих трубок соответствующей пары дозировали воздух, а через другую дозирующую трубку (называемую аналитической трубкой) осуществляли отбор реакционной газовой смеси для анализа. Первая в направлении газового потока аналитическая трубка (LI) с ходом 20 мм была помещена позади первого отдельного стационарного слоя катализатора. Соответствующая дозирующая трубка (ZI) с ходом 100 мм была помещена перед вторым отдельным стационарным слоем катализатора. Вторая в направлении газового потока аналитическая трубка (LII) с ходом 20 мм была помещена позади второго отдельного стационарного слоя катализатора. Соответствующая дозирующая трубка (ZII) с ходом 20 мм была помещена позади третьего отдельного стационарного слоя катализатора.
Реакционная газовая смесь перед поступлением в трубчатый реактор проходила через используемую в качестве нагревателя, присоединенную к реактору стальную трубку длиной 1300 мм, заполненную шариками из стеатита С-220 фирмы СеrаmТес диаметром от 4 до 5 мм. В указанном нагревателе выполняли предварительное нагревание потока реакционной газовой смеси до температуры входа в трубчатый реактор и одновременно обеспечивали идеальное перемешивание компонентов газового потока. С этой целью указанную трубку (специальная сталь номер 1.4841 согласно DIN, толщина стенок 3,6 мм, внутренний диаметр 53,1 мм) по длине 1200 мм подвергали электрообогреву посредством манжеты фирмы Horst (Гейдельберг, Германия). Нагреватель присоединяли к трубчатому реактору посредством обогреваемой переходной трубки длиной 300 мм из специальной стали номер 1.4841 (наружный диаметр 14 мм, внутренний диаметр 10 мм), изолированной обычными теплоизоляционными материалами.
b) Начальный момент предлагаемого в изобретении долговременного дегидрирования (рабочий момент t0)
Совокупный слой катализатора содержал свежий катализатор дегидрирования. Поток реакционной газовой смеси, направляемый к совокупному стационарному слою катализатора, представлял собой смесь сырого пропана, водяного пара и оборотного газа гетерогенно катализируемого двухстадийного частичного окисления пропилена в акриловую кислоту. Из газовой смеси продуктов дегидрирования выделяли отличающиеся от пропана и пропилена компоненты, для чего использовали абсорбционную/десорбционную технологию (отпаривание воздухом), приведенную в сравнительном примере 1 немецкой заявки на патент с регистрационным номером 102005013039. Частичное окисление пропилена до акриловой кислоты осуществляли, как предложено в немецкой заявке на патент с регистрационным номером 102005013039. Вышесказанное относится и к оборотному газу частичного окисления.
Поток реакционной газовой смеси с температурой около 200°С создавали, как указано в заявке №102005013039, в испарителе и направляли в нагреватель (соединение испарителя с нагревателем конструктивно выполнено аналогично соединению нагревателя с трубчатым реактором).
Электрообогрев нагревателя регулировали таким образом, чтобы поступающий из испарителя в нагреватель поток реакционной газовой смеси выходил из нагревателя с температурой около 400°С. Затем реакционную газовую смесь пропускали через трубчатый реактор, подвергая ее дополнительному нагреванию в соответствующем устройстве для предварительного нагревания. Через дозирующие трубки ZI и ZII поступал воздух с расходом соответственно около 30 нл/л, температура которого на входе в соответствующую дозирующую трубку составляла 25°С. По истечении одного часа (t0=1 ч) в первый раз достигали в основном (квази)стационарного рабочего режима.
Состав исходной реакционной газовой смеси, поступающей в трубчатый реактор с расходом 2807 нл/л, а также состав реакционной газовой смеси на выходе из первого, второго и третьего отдельных стационарных слоев приведен в таблице (все количественные данные согласно изобретению основаны на результатах анализа, выполненного методом газовой хроматографии).
При прохождении потока реакционной газовой смеси через совокупный слой катализатора в дегидрированный углеводород превращалось в общей сложности G=(А+В+С) % мол. содержащегося в реакционной газовой смеси исходного молярного количества KW подлежащего дегидрированию углеводорода. При этом А означает вклад в G, реализуемый в первом отдельном стационарном слое катализатора, В означает вклад в G, реализуемый во втором отдельном стационарном слое катализатора, и С означает вклад в G, реализуемый в третьем отдельном стационарном слое катализатора.
Рабочему моменту t0 соответствовали следующие численные значения параметров G, А, В и С (соответственно в пересчете на KW):
А 12% мол.,
В 5,4% мол.,
С 1,8% мол. и
G 19,2% мол.
В рабочий момент t0 температура T1 реакционной газовой смеси непосредственно после ее прохождения через находящийся в трубчатом реакторе насыпной слой для внутреннего нагревания составляла 408°С. Расход воздуха MI и МII, поступающего по соответствующим дозирующим трубкам ZI и ZII, в рабочий момент t0 составлял соответственно около 33 нл/л.
с) Реализация долговременного рабочего режима в соответствии с изобретением
Для противодействия происходящему в процессе дегидрирования деактивированию совокупного стационарного слоя катализатора, как показано на фиг.1, по мере работы постепенно повышали как T1, так и MI и МII (на фиг.1 М1, соответственно и М2) (на левой оси ординат отложена температура T1 в °С, на правой оси ординат отложен расход воздуха в нл/л, началу координат соответствует t0, по оси абсцисс отложено время в часах). Подобным образом удавалось в интервале рабочих моментов t0<t≤50 ч стабильно сохранять значение G на уровне 19,2±0,2% мол. Полученные при этом зависимости отдельных степеней превращения А, В, С от времени в интервале рабочих моментов показаны на фиг.2 (по оси абсцисс отложено время в часах, на оси ординат отложены степени превращения в % мол. KW).
На фиг.3 приведены соответствующие кривые изменения температуры реакционной газовой смеси (ось ординат, °С) в зависимости от времени (ось абсцисс, часы, причем началу координат соответствует t0:
кривая 1 - зависимость температуры от времени в точке первого отдельного слоя катализатора, находящейся неподалеку от входа реакционной газовой смеси в этот слой (максимальная температура в первом отдельном слое катализатора),
кривая 2 - зависимость температуры от времени для середины первого отдельного слоя катализатора,
кривая 3 - зависимость температуры реакционной газовой смеси на ее выходе из первого отдельного слоя катализатора,
кривая 4 - зависимость температуры от времени в точке второго отдельного слоя катализатора, находящейся неподалеку от входа реакционной газовой смеси в этот слой (максимальная температура во втором отдельном слое катализатора),
кривая 5 - зависимость температуры от времени для середины второго отдельного слоя катализатора,
кривая 6 - зависимость температуры реакционной газовой смеси на ее выходе из второго отдельного слоя катализатора,
кривая 7 - зависимость температуры от времени в точке третьего отдельного слоя катализатора, находящейся неподалеку от входа реакционной газовой смеси в этот слой (максимальная температура в третьем отдельном слое катализатора),
кривая 8 - зависимость температуры реакционной газовой смеси на ее выходе из третьего отдельного слоя катализатора.
Затем процесс дегидрирования прекращали и совокупный стационарный слой катализатора подвергали регенерации, осуществляемой в соответствии с немецкой заявкой на патент DE-A 10028582. После этого возобновляли дегидрирование. По истечении примерно 1 часа устанавливался (квази)стационарный режим дегидрирования. При этом в случае использования исходных рабочих условий вновь достигали первоначального значения G. Процесс дегидрирования в соответствии с предлагаемым в изобретении способом долговременной эксплуатации катализатора продолжали до его следующей регенерации и так далее.
d) Реализация долговременного рабочего режима не в соответствии с изобретением
Процесс дегидрирования начинали подобно тому, как описано в пунктах а) и b). Для противодействия происходящему в процессе дегидрирования деактивированию совокупного стационарного слоя катализатора температуру T1 и расход воздуха MI и МII по мере работы повышали таким образом, чтобы отдельные степени превращения А, В и С, а следовательно, суммарная степень превращения G оставались постоянными.
После рабочего периода 17 часов сохранять достигаемую в этом случае суммарную степень превращения G на уровне 19,2±1% мол. больше не удавалось. В связи с этим дегидрирование прекращали и совокупный стационарный слой катализатора подвергали регенерации, как описано в немецкой заявке на патент DE-A 10028582. Затем возобновляли дегидрирование. Однако достичь первоначального значения суммарной степени превращения G в первоначальных условиях дегидрирования больше не удавалось.
Приведенные выше пример и сравнительный пример, очевидно, могли быть выполнены совершенно аналогичным образом и в полочном реакторе с тремя идентичными стационарными слоями катализатора. В этом случае подачу воздуха осуществляют между первой и второй, а также между второй и третьей полками.
Предварительная заявка на патент США 60/833776 от 28.07.2006 включена в настоящую заявку в качестве ссылки.
Возможны многочисленные изменения и отклонения от указанных в настоящей заявке технических решений.
В связи с вышесказанным следует исходить из того, что осуществление изобретения в соответствии с представленной ниже формулой может отличаться от приведенного выше описания.
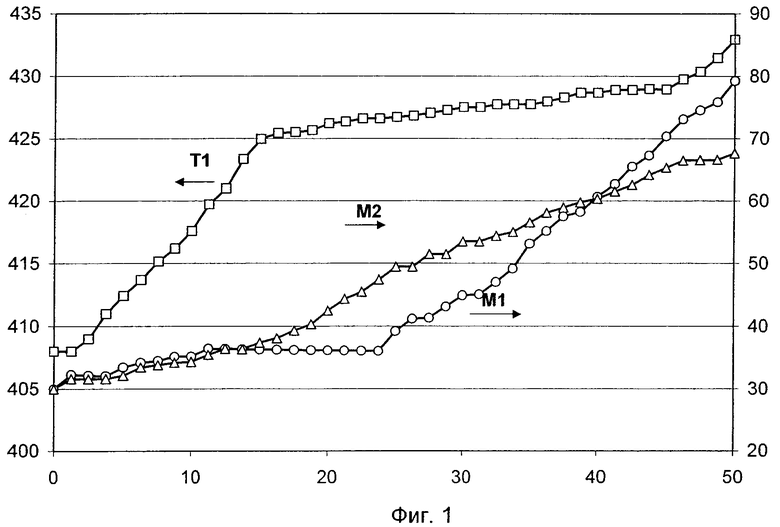
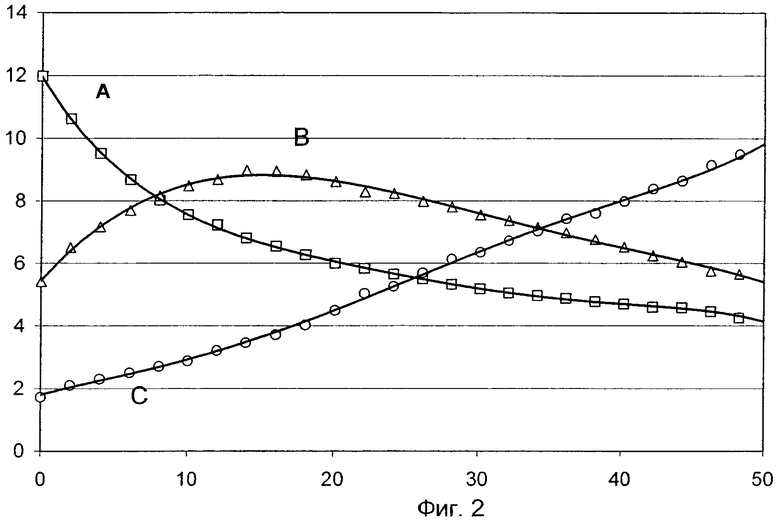

Изобретение относится к способу гетерогенно катализируемого частичного дегидрирования углеводорода. Способ заключается в том, что поток реакционной газовой смеси, который содержит углеводород в исходном молярном количестве KW, пропускают через совокупный слой катализатора, состоящий из нескольких отдельных слоев, в суммарном количестве М, так чтобы в рабочий момент t0 при прохождении потока через первую треть количества М в дегидрированный углеводород превращалась доля А исходного количества KW, через вторую треть превращалась доля В и через третью треть превращалась доля С, при условии, что А>В>С. Причем в поток между его входом в совокупный слой катализатора и выходом из него при необходимости вводят вспомогательные газы дегидрирования. Деактивированию совокупного слоя катализатора, происходящему по мере его эксплуатации, противодействуют тем, что варьируют ход температуры потока внутри совокупного слоя катализатора и/или расход при необходимости используемых вспомогательных газов дегидрирования. При этом варьирование выполняют таким образом, чтобы по мере протекания рабочего процесса доля А снижалась, доля В проходила через максимум и доля С возрастала. Изобретение позволяет максимально ограничить деактивирование совокупного слоя катализатора. 31 з.п. ф-лы, 3 ил., 1 табл.
1. Способ реализуемого в непрерывном режиме длительного гетерогенно катализируемого частичного дегидрирования подлежащего дегидрированию углеводорода до дегидрированного углеводорода, в соответствии с которым обладающий повышенной температурой поток реакционной газовой смеси, который содержит подлежащий дегидрированию углеводород в исходном молярном количестве KW, таким образом пропускают через находящийся в реакционной зоне RZ совокупный слой катализатора, который может состоять из нескольких отдельных слоев катализатора, последовательно упорядоченных в направлении потока, и содержит суммарное массовое количество М катализатора дегидрирования, чтобы в рабочий момент t=t0 при прохождении потока реакционной газовой смеси через первую в направлении потока треть количества М в дегидрированный углеводород, превращалась выраженная в % мол. доля А молярного исходного количества KW, при прохождении потока реакционной газовой смеси через вторую в направлении потока треть количества М в дегидрированный углеводород превращалась выраженная в % мол. доля В молярного исходного количества KW и при прохождении потока реакционной газовой смеси через третью в направлении потока треть количества М в дегидрированный углеводород превращалась выраженная в % мол. доля С молярного исходного количества KW, при условии, что А>В>С, и при прохождении потока реакционной газовой смеси через совокупный слой катализатора суммарная степень превращения G присутствующего в этом потоке молярного исходного количества KW подлежащего дегидрированию углеводорода в дегидрированный углеводород составляет (А+В+С) % мол., причем в поток реакционной газовой смеси между его входом в совокупный слой катализатора и его выходом из совокупного слоя катализатора при необходимости вводят потоки молекулярного кислорода, молекулярного водорода, водяного пара и/или другого инертного газа, используемых в качестве вспомогательных газов дегидрирования, и деактивированию совокупного слоя катализатора, происходящему по мере его эксплуатации в интервале рабочих моментов t0<t<tR, причем tR означает рабочий момент, в который дегидрирование прекращают и выполняют первую после рабочего момента t0 регенерацию совокупного слоя катализатора, противодействуют тем, что варьируют ход температуры потока реакционной газовой смеси внутри совокупного слоя катализатора и/или расход при необходимости используемых вспомогательных газов дегидрирования, отличающийся тем, что указанное варьирование выполняют таким образом, чтобы по мере протекания рабочего процесса, то есть по мере возрастания t, доля А снижалась, доля В проходила через максимум и доля С возрастала.
2. Способ по п.1, отличающийся тем, что подлежащим дегидрированию углеводородом является алкан с 2-6 атомами углерода, а дегидрированным углеводородом является алкен с 2-6 атомами углерода.
3. Способ по п.1, отличающийся тем, что подлежащим дегидрированию углеводородом является н-пропан, а дегидрированным углеводородом является пропилен.
4. Способ по п.1, отличающийся тем, что поток реакционной газовой смеси, содержащий молярное исходное количество KW подлежащего дегидрированию углеводорода, дополнительно содержит водяной пар.
5. Способ по п.1, отличающийся тем, что поток реакционной газовой смеси, содержащий молярное исходное количество KW подлежащего дегидрированию углеводорода, дополнительно содержит молекулярный кислород.
6. Способ по п.1, отличающийся тем, что поток реакционной газовой смеси, содержащий молярное исходное количество KW подлежащего дегидрированию углеводорода, дополнительно содержит молекулярный водород.
7. Способ по п.1, отличающийся тем, что суммарная степень превращения G в рабочий момент t0 составляет от 10 до 60% мол.
8. Способ по п.1, отличающийся тем, что суммарная степень превращения G в рабочий момент t0 составляет от 15 до 40% мол.
9. Способ по п.1, отличающийся тем, что суммарная степень превращения G в рабочий момент t0 составляет от 15 до 30% мол.
10. Способ по п.1, отличающийся тем, что в рабочий момент t=t0 частичное превращение А составляет от 45 до 80%, частичное превращение В составляет от 20 до 40% и частичное превращение С составляет от 0 до 15% соответственно в пересчете на общую степень превращения G=(A+B+C).
11. Способ по п.1, отличающийся тем, что в рабочий момент t=t0 частичное превращение А составляет от 55 до 70%, частичное превращение В составляет от 20 до 35% и частичное превращение С составляет от 7 до 13% соответственно в пересчете на общую степень превращения G=(A+B+C).
12. Способ по п.1, отличающийся тем, что по мере протекания рабочего процесса в интервале рабочих моментов t0<t<tR частичное превращение А уменьшается, оказываясь ниже частичного превращения С, в результате чего становится справедливым соотношение С>А.
13. Способ по п.1, отличающийся тем, что по мере протекания рабочего процесса в интервале рабочих моментов t0<t<tR происходит обращение соотношения частичных превращений А, В и С А>В>С в соотношение С>В>А.
14. Способ по п.1, отличающийся тем, что значения А в интервале рабочих моментов t0<t<tR не опускаются ниже уровня, которому соответствует 20% от значения А в рабочий момент t=t0.
15. Способ по п.1, отличающийся тем, что значения В и С в интервале рабочих моментов t0<t<tR не возрастают выше уровня, которому соответствует 95% от значения А в рабочий момент t=t0.
16. Способ по п.1, отличающийся тем, что реакционная зона RZ выполнена в виде адиабатического полочного реактора с одновременной подачей между полками содержащих молекулярный кислород потоков вспомогательного газа дегидрирования.
17. Способ по п.1, отличающийся тем, что реакционная зона RZ выполнена в виде по меньшей мере двух последовательно соединенных адиабатических реакторов дегидрирования с одновременной подачей содержащих молекулярный кислород потоков вспомогательного газа дегидрирования между двумя непосредственно соединенными друг с другом адиабатическими реакторами дегидрирования.
18. Способ по п.1, отличающийся тем, что совокупный слой катализатора является стационарным слоем.
19. Способ по п.1, отличающийся тем, что катализатором дегидрирования является металл, осажденный по меньшей мере на одном оксидном носителе.
20. Способ по п.19, отличающийся тем, что по меньшей мере одним металлом является элемент платиновой группы.
21. Способ по п.19, отличающийся тем, что по меньшей мере одним металлом является платина.
22. Способ по одному из пп.1-21, отличающийся тем, что из реакционной зоны RZ отбирают газообразный поток продуктов, содержащий образовавшийся в этой зоне дегидрированный углеводород и непревращенный в этой зоне подлежащий дегидрированию углеводород, причем указанный газообразный поток как таковой или после отделения по меньшей мере частичного количества содержащихся в нем компонентов, отличающихся от дегидрированного углеводорода и подлежащего дегидрированию углеводорода, используют для подачи по меньшей мере в один реактор окисления, в котором содержащийся в указанном газообразном потоке дегидрированный углеводород подвергают селективному гетерогенно катализируемому частичному газофазному окислению молекулярным кислородом, получая содержащую продукт частичного окисления газовую смесь продуктов частичного окисления.
23. Способ по п.22, отличающийся тем, что подлежащим дегидрированию углеводородом является пропан, дегидрированным углеводородом является пропилен, а продуктом частичного окисления является акролеин, акриловая кислота или их смесь.
24. Способ по п.22, отличающийся тем, что в последующей зоне разделения селективного гетерогенно катализируемого частичного газофазного окисления из газовой смеси продуктов частичного окисления выделяют продукт частичного окисления, причем по меньшей мере частичное количество, содержащее непревращенный подлежащий дегидрированию углеводород, остающегося при этом остаточного газа, содержащего непревращенный подлежащий дегидрированию углеводород, молекулярный кислород, а также при необходимости непревращенный дегидрированный углеводород, возвращают в реакционную зону RZ в качестве оборотного газа частичного окисления.
25. Способ по п.24, отличающийся тем, что продукт частичного окисления выделяют из газовой смеси продуктов частичного окисления в зоне разделения путем перевода этого продукта в конденсированную фазу.
26. Способ по п.25, отличающийся тем, что продуктом частичного окисления является акриловая кислота, которую переводят в конденсированную фазу адсорбционным и/или конденсационным методом.
27. Способ по п.26, отличающийся тем, что акриловую кислоту выделяют из конденсированной фазы с использованием по меньшей мере одного термического метода разделения.
28. Способ по п.27, отличающийся тем, что по меньшей мере один термический метод разделения включает кристаллизационное выделение акриловой кислоты из жидкой фазы.
29. Способ по п.28, отличающийся тем, что кристаллизационным выделением является суспензионная кристаллизация.
30. Способ по п.27, отличающийся тем, что за выделением акриловой кислоты следует процесс радикальной полимеризации, в соответствии с которым выделенную акриловую кислоту подвергают радикальной полимеризации с целью получения продуктов полимеризации.
31. Способ по п.27, отличающийся тем, что за выделением акриловой кислоты следует процесс синтеза сложных эфиров акриловой кислоты, в соответствии с которым выделенную акриловую кислоту этерифицируют спиртом.
32. Способ по п.31, отличающийся тем, что за процессом синтеза сложных эфиров акриловой кислоты следует процесс радикальной полимеризации, в соответствии с которым полученные сложные эфиры акриловой кислоты подвергают полимеризации.
| DE 10028582 A1, 20.12.2001 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| СПОСОБ КАТАЛИТИЧЕСКОГО ДЕГИДРИРОВАНИЯ УГЛЕВОДОРОДОВ | 2000 |
|
RU2178399C1 |

