Область изобретения
Настоящее изобретение относится к производным 2-пиразинона, способам их получения, фармацевтическим композициям, содержащим их, и их применению в терапии.
Предпосылки изобретения
Эластазы являются, возможно, наиболее разрушительными ферментами в организме, обладая способностью разрушать практически все компоненты соединительной ткани. Неконтролируемый протеолитический распад под действием эластаз вовлечен в ряд патологических состояний. Человеческая нейтрофильная эластаза (hNE), член химотрипсинового суперсемейства сериновых протеаз, представляет собой 33-кДа фермент, который находится в азурофильных гранулах нейтрофилов. В нейтрофилах концентрация NE превышает 5 мМ, и ее общее клеточное количество, как подсчитано, составляет вплоть до 3 пг. При активации NE быстро высвобождается из гранул во внеклеточное пространство, а некоторая часть остается связанной с мембраной плазмы нейтрофилов (см. Kawabat et al. 2002, Eur. J. Pharmacol. 451, 1-10). Главная внутриклеточная физиологическая функция NE заключается в деградации чужеродных органических молекул, поглощенных нейтрофилами путем фагоцитоза, тогда как главной мишенью внеклеточной эластазы является эластин (Janoffand Scherer, 1968, J. Exp.Med. 128, 1137-1155). По сравнению с другими протеазами (например, протеиназой 3), NE уникальна тем, что обладает способностью разрушать практически все белки внеклеточного матрикса и ключевые белки плазмы (см. Kawabat et al., 2002, Eur. J. Pharmacol. 451, 1-10). Она разрушает широкий спектр внеклеточных матриксных белков, таких как эластин, коллагены типа 3 и типа 4, ламинин, фибронектин, цитокины и т.д. (Ohbayashi, Н, 2002, Expert Opin. Investig. Drugs, 11, 965-980). NE является главным общим медиатором многих патологических изменений, наблюдаемых при хронической болезни легких, включая разрушение эпителия (Stockley, R.A. 1994, Am. J. Resp. Crit. Care Med. 150, 109-113).
Деструктивная роль NE была установлена почти 40 лет назад, когда Laurell и Eriksson сообщили о связи хронической обструкции дыхательных путей и эмфиземы с дефицитом сывороточного α1-антитрипсина (Laurell and Eriksson, 1963, Scand. J. Clin. Invest. 15, 132-140). Впоследствии было определено, что α1-антитрипсин является наиболее важным эндогенным ингибитором человеческой NE. Дисбаланс между человеческой NE и эндогенной антипротеазой, как полагают, является причиной избытка человеческой NE в легочных тканях, что считается главным патогенным фактором при хронической обструктивной болезни легких (COPD). Избыток человеческой NE демонстрирует заметный деструктивный профиль и активно принимает участие в разрушении нормальных легочных структур с последующим необратимым расширением дыхательных путей, что наблюдается главным образом при эмфиземе. Имеет место увеличение рекрутмента нейтрофилов в легкие, что связано с увеличенной эластазной нагрузкой в легких и эмфиземой у мышей с дефицитом ингибитора α1-протеиназы (Cavarra et al., 1996, Lab. Invest. 75, 273-280). Индивидуумы с более высокими уровнями комплекса NE-ингибитор α1-протеазы в бронхоальвеолярном лаваже показывают значительно ускоренное ухудшение функции легких по сравнению с теми, у кого эти уровни ниже (Betsuyaku et al. 2000, Respiration, 67, 261-267). Инстилляция человеческой NE через трахею у крыс вызывает легочное кровотечение, аккумуляцию нейтрофилов в течение острой фазы и эмфизематозные изменения в течение хронической фазы (Karaki et al., 2002, Am. J. Resp. Crit. Care Med., 166, 496-500). Исследования показали, что острая фаза легочной эмфиземы и легочного кровотечения, вызванные NE у хомяков, могут быть ингибированы предварительным лечением ингибиторами NE (Fujie et al., 1999, Inflamm. Res. 48, 160-167).
Нейтрофил-доминирующее воспаление дыхательных путей и слизистая обструкция дыхательных путей являются главными патологическими признаками COPD, включая кистозный фиброз и хронический бронхит.NE нарушает выработку муцина, что приводит к слизистой обструкции дыхательных путей. Сообщается, что NE усиливает экспрессию главного гена респираторного муцина, MUC5AC (Fischer, B.M & Voynow, 2002, Am. J. Respir. Cell Biol., 26, 447-452). Аэрозольное введение NE морским свинкам вызывает обширное повреждение эпителия в течение 20 минут контакта (Suzuki et al., 1996, Am. J. Resp. Crit. Care Med., 153, 1405-1411). Более того, NE снижает частоту пульсации ресничек человеческого респираторного эпителия in vitro (Smallman et al., 1984, Thorax, 39, 663-667), что согласуется со сниженным клиренсом реснитчатого эпителия, который наблюдается у больных COPD (Currie et al., 1984, Thorax, 42, 126-130). Инстилляция NE в дыхательные пути приводит к гиперплазии слизистой железы у хомячков (Lucey et al., 1985, Am. Resp.Crit. Care Med., 132, 362-366). NE также играет роль в гиперсекреции слизи при астме. В модели острой астмы на сенсибилизированных аллергеном морских свинках ингибитор NE предотвращал дегрануляцию бокаловидных клеток и гиперсекрецию слизи (Nadel et al., 1999, Eur. Resp. J., 13,190-196).
Также было показано, что NE играет роль в патогенезе легочного фиброза. Комплекс NE-ингибатор α1-протеиназы повышен в сыворотке у пациентов с фиброзом легких, что коррелирует с клиническими параметрами этих пациентов (Yamanouchi et al., 1998, Eur. Resp.J. 11, 120-125). В мышиной модели человеческого фиброза легких NE ингибитор уменьшает фиброз легких, индуцированный блеомицином (Taooka et al., 1997, Am. J. Resp.Crit. Care Med., 156, 260-265). Более того, исследователи показали, что мыши с дефицитом NE являются резистентными к фиброзу легких, индуцированному блеомицином (Dunsmore et al., 2001, Chest, 120, 35S-36S). Уровень NE в плазме, как обнаружено, повышен у пациентов, у которых развился ARDS (респираторный дистресс-синдром взрослых), что подразумевает важность NE в раннем патогенезе ARDS (Donnelly et al., 1995, Am. J. Res. Crit. Care Med., 151, 428-1433). Антипротеазы и NE в комплексе с антипротеазой повышены в области рака легких (Marchandise et al., 1989, Eur. Resp.J. 2, 623-629). Недавние исследования показали, что полиморфизм в промоторной области гена NE ассоциирован с развитием рака легких (Taniguchi et al., 2002, Clin. Cancer Res., 8, 1115-1120).
С повышенными уровнями NE ассоциировано острое повреждение легких, вызванное эндотоксином у экспериментальных животных (Kawabata, et al., 1999, Am. J. Resp.Crit. Care, 161, 2013-2018). Как было показано, при остром воспалении легких, вызванном интратрахеальной инъекцией липополисахарида у мышей, повышена активность NE в бронхоальвеолярном лаваже, которая в значительной степени ингибируется ингибитором NE (Fujie et al., 1999, Eur. J. Pharmacol., 374, 117-125; Yasui, et al., 1995, Eur. Resp. J., 8, 1293-1299). NE также играет важную роль в индуцированном нейтрофилами повышении легочной капиллярной проницаемости, наблюдаемой в модели острого легочного повреждения, вызванного фактором некроза опухоли α (TNFa) и форбол-миристат-ацетатом (РМА) в изолированных перфузированных легких кролика (Miyazaki et al., 1998, Am. J. Respir. Crit. Care Med., 157, 89-94).
Также предположили, что NE играет роль в уплотнении легочной сосудистой стенки, индуцированной монокротолином, и сердечной гипертрофии (Molteni et al., 1989, Biochemical Pharmacol. 38, 2411-2419). Ингибитор сериновой эластазы реверсирует легочную гипертензию, индуцированную монокроталином, и ремоделирование в легочных артериях крыс (Cowan et al., 2000, Nature Medicine, 6, 698-702). Последние исследования показали, что сериновая эластаза, то есть NE или сосудистая эластаза, играют важную роль в мускуляризации небольших легочных артерий, вызванной сигаретным дымом у морских свинок (Wright et al., 2002, Am. J. Respir. Crit. Care Med., 166, 954-960).
NE играет ключевую роль в экспериментальном церебральном ишемическом повреждении (Shimakura et al., 2000, Brain Research, 858, 55-60), ишемическо-реперфузионном повреждении легких (Kishima et al., 1998, Ann. Thorac. Surg. 65, 913-918) и ишемии миокарда в сердце крыс (Tiefenbacher et al., 1997, Eur. J. Physiol., 433, 563-570). Уровни человеческой NE в плазме значительно повышены по сравнению с нормой при воспалительном заболевании кишечника, например болезни Крона или неспецифическом язвенном колите (Adeyemi et al., 1985, Gut, 26, 1306-1311). Кроме того, было высказано предположение, что NE вовлечена в патогенез ревматоидного артрита (Adeyemi et al., 1986, Rheumatol. Int., 6, 57). Развитие коллаген-индуцированного артрита у мышей подавляется ингибитором NE (Kakimoto et al., 1995, Cellular Immunol. 165, 26-32).
Таким образом, человеческая NE известна как одна из самых разрушительных сериновых протеаз и она вовлечена в ряд воспалительных заболеваний. Важным эндогенным ингибитором человеческой NE является α1-антитрипсин. Дисбаланс между человеческой NE и антипротеазой, как полагают, вызывает избыток человеческой NE, что в результате приводит к неконтролируемому разрушению ткани. Баланс протеаза/антипротеаза может быть нарушен сниженной доступностью α1-антитрипсина либо посредством инактивации окислителями, такими как сигаретный дым, либо в результате генетической неспособности производить достаточные сывороточные уровни. Человеческая NE вовлечена в активацию или обострение ряда заболеваний, таких как легочная эмфизема, фиброз легких, респираторный дистресс-синдром взрослых (ARDS), ишемическо-реперфузионное повреждение, ревматоидный артрит и легочная гипертензия.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с настоящим изобретением предложено соединение формулы (I)
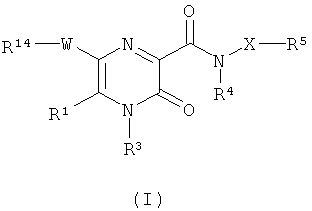
где
R1 представляет собой водород или С1-С6алкил;
W представляет собой 5-членное гетероциклическое кольцо, содержащее по меньшей мере один кольцевой гетероатом, выбранный из азота, кислорода и серы, где по меньшей мере один из кольцевых атомов углерода возможно может быть заменен карбонильной группой; и где гетероциклическое кольцо возможно замещено по меньшей мере одним заместителем, выбранным из галогена, С1-С4алкила, С1-С4алкокси, CN, ОН, NO2, C1-С3алкила, замещенного одним или более атомами F, C1-С3алкокси, замещенного одним или более атомами F, групп NR10R11, C≡CR15, CONR16R17, CHO, С2-С4алканоила, групп S(O)xR18 и OSO2R19;
R14 представляет собой фенил или 6-членное гетероароматическое кольцо, содержащее от 1 до 3 кольцевых атомов азота; причем указанное кольцо возможно замещено по меньшей мере одним заместителем, выбранным из галогена, С1-С4алкила, С1-C4алкокси, CN, ОН, NO2, C1-С3алкила, замещенного одним или более атомами F, C1-С3алкокси, замещенного одним или более атомами F, групп NR12R13, C≡CR30, CONR31R32, CHO, С2-С4алканоила, групп S(O)pR33 и OSO2R34;
R10, R11, R12 и R13 независимо представляют собой Н, С1-С6алкил, формил или С2-С6алканоил; либо группа -NR10R11 или -NR12R13 в совокупности представляет собой 5-7-членное азациклическое кольцо, возможно включающее один дополнительный гетероатом, выбранный из О, S и NR26;
R15 и R30 независимо представляют собой Н, С1-С3алкил или Si(СН3)3;
R18, R19, R33 и R34 независимо представляют собой Н или C1-С3алкил; причем указанный алкил возможно замещен одним или более атомами F;
R3 представляет собой фенил либо пяти- или шестичленное гетероароматическое кольцо, содержащее от 1 до 3 гетероатомов, независимо выбранных из О, S и N; причем указанное кольцо возможно замещено по меньшей мере одним заместителем, выбранным из галогена, С1-С6алкила, циано, C1-С6алкокси, нитро, метилкарбонила, NR35R36, C1-С3алкила, замещенного одним или более атомами F, или С1-С3алкокси, замещенного одним или более атомами F;
R35 и R36 независимо представляют собой Н или С1-С3алкил; причем указанный алкил возможно дополнительно замещен одним или более атомами F;
R4 представляет собой водород или С1-С6алкил, возможно замещенный по меньшей мере одним заместителем, выбранным из фторо, гидроксила и С1-С6алкокси;
Х представляет собой простую связь, О, NR24 или группу -С1-С6алкилен-Y-, где Y представляет собой простую связь, атом кислорода, NR24 или S(O)w; и указанный алкилен возможно дополнительно замещен ОН, галогеном, CN, NR37R38, С1-С3алкокси, CONR39R40, CO2R66, SO2R41 и SO2NR42R43;
либо R4 и Х соединены вместе так, что группа -NR4X в совокупности представляет собой 5-7-членное азациклическое кольцо, возможно включающее один дополнительный гетероатом, выбранный из О, S и NR44; причем указанное кольцо возможно замещено С1-С6алкилом или NR45R46; и указанный алкил возможно дополнительно замещен ОН;
либо R5 представляет собой моноциклическую кольцевую систему, выбранную из
1) фенокси,
2) фенила,
3) 5- или 6-членного гетероароматического кольца, содержащего по меньшей мере один кольцевой гетероатом, выбранный из азота, кислорода и серы,
4) насыщенного или частично ненасыщенного С3-С6циклоалкильного кольца, или
5) насыщенного или частично ненасыщенного 4-7-членного гетероциклического кольца, содержащего по меньшей мере один кольцевой гетероатом, выбранный из кислорода, S(O)r и NR20, где по меньшей мере один из кольцевых атомов углерода возможно может быть заменен карбонильной группой,
либо R5 представляет собой бициклическую кольцевую систему, в которой эти два кольца независимо выбраны из моноциклических кольцевых систем, определенных в (2), (3), (4) и (5) выше, где эти два кольца либо конденсированы вместе, либо соединены непосредственно друг с другом, либо отделены друг от друга линкерной группой, выбранной из кислорода, S(O)t или С1-С6алкилена, возможно содержащего один или более внутренних или концевых гетероатомов, выбранных из кислорода, серы и NR27, и возможно замещенного по меньшей мере одним заместителем, выбранным из гидроксила, оксо и С1-С6алкокси,
где моноциклическая или бициклическая кольцевая система возможно замещена по меньшей мере одним заместителем, выбранным из кислорода, CN, ОН, C1-С6алкила, C1-С6алкокси, галогена, NR47R48, NO2, OSO2R49, CO2R50, C(=NH)NH2, C(O)NR51R52, C(S)NR53R54, SC(=NH)NH2, NR55C(=NH)NH2, S(O)vR21, SO2NR56R57, C1-С3алкокси, замещенного одним или более атомами F, и C1-С3алкила, замещенного SO2R58 или одним или более атомами F; причем указанный С1-С6алкил возможно дополнительно замещен по меньшей мере одним заместителем, выбранным из циано, гидроксила, С1-С6алкокси, С1-С6алкилтио и -C(O)NR22R23;
либо R5 также может представлять собой Н;
R20 представляет собой водород, С1-С6алкил, С1-С6алкилкарбонил или C1-С6алкоксикарбонил;
R21 представляет собой водород, C1-С6алкил или С3-С6циклоалкил; причем указанная алкильная или циклоалкильная группа возможно дополнительно замещена одним или более заместителями, независимо выбранными из ОН, CN, C1-С3алкокси и CONR59R60;
R37 и R38 независимо представляют собой Н, C1-С6алкил, формил или С2-С6алканоил;
R47 и R48 независимо представляют собой Н, C1-С6алкил, формил, С2-С6алканоил, S(O)qR61 или SO2NR62R63; причем указанная алкильная группа возможно дополнительно замещена галогеном, CN, С1-С4алкокси или CONR64R65;
R41 и R61 независимо представляют собой Н, С1-С6алкил или С3-С6циклоалкил;
р равен 0, 1 или 2;
q равен 0, 1 или 2;
r равен 0, 1 или 2;
t равен 0, 1 или 2;
w равен 0, 1 или 2;
х равен 0, 1 или 2;
v равен 0, 1 или 2;
каждый из R16, R17, R22, R23, R24, R26, R27, R31, R32, R39, R40, R42, R43, R44, R45, R46, R49, R50, R51, R52, R53, R54, R55, R56, R57, R58, R59, R60, R62, R63, R64, R65 и R66 независимо представляет собой водород или С1-С6алкил;
или его фармацевтически приемлемая соль.
В контексте настоящего описания, если не указано иного, алкильная, алкенильная или алкинильная группа-заместитель или алкильная группировка в группе-заместителе может быть линейной или разветвленной. Аналогично, алкиленовая группа может быть линейной или разветвленной.
В определении W, 5-членная гетероциклическая кольцевая система может обладать алициклическими или ароматическими свойствами и может, таким образом, представлять собой насыщенную кольцевую систему, или частично ненасыщенную кольцевую систему, или полностью ненасыщенную кольцевую систему.
R1 представляет собой водород или C1-С6алкил (например, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, трет-бутил, н-пентил или н-гексил).
В одном воплощении изобретения R1 представляет собой С1-С4- или С1-С2алкильную группу, в частности метильную группу.
W представляет собой 5-членное гетероциклическое кольцо, содержащее по меньшей мере один кольцевой гетероатом, выбранный из азота, кислорода и серы, где по меньшей мере один из кольцевых атомов углерода возможно может быть заменен карбонильной группой; и где это гетероциклическое кольцо возможно замещено по меньшей мере одним заместителем, выбранным из галогена (например, фтора, хлора, брома или иода), С1-C4алкила (например, метила, этила, н-пропила, изо-пропила, н-бутила, изо-бутила или трет-бутила), С1-С4алкокси (например, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси или трет-бутокси), циано, ОН, NO2, С1-С3алкила, замещенного одним или более атомами F (например, CH2F, CHF2, CF3, CH2CH2F, CH2CF3, CF2CF3, СН(CF3)2 и CH2CH2CF3), С1-С3алкокси, замещенного одним или более атомами F (например, OCH2F, OCHF2, OCF3, OCH2CH2F, OCH2CF3, OCF2CF3, ОСН(CF3)2 и OCH2CH2CF3), NR10R11, C≡CR15, -C(O)NR16R17, СНО, С2-С4алканоила (например, метилкарбонила (ацетила), этилкарбонила, н-пропилкарбонила или изо-пропилкарбонила), -S(O)xR18 и OSO2R19.
В одном воплощении группа R14 и пиразиноновое кольцо присоединены к 5-членному кольцу W в положениях 1, 2.
В одном воплощении W представляет собой 5-членное гетероароматическое кольцо, в частности незамещенное 5-членное гетероароматическое кольцо.
Примерами 5-членных гетероциклических кольцевых систем, которые могут быть использованы и которые могут насыщенными, или частично ненасыщенными, или полностью ненасыщенными, включают любые из следующего: пирролидинил, тетрагидрофуранил, пирролин, имидазолидинил, имидазолинил, пиразолидинил, пиразолинил, пирролидинонил, имидазолидинонил, оксазолил, пиразолил, тиазолидинил, тиенил, изоксазолил, изотиазолил, тиадиазолил, пирролил, фуранил, тиазолил, имидазолил, фуразанил, триазолил и тетразолил.
Предпочтительные кольцевые системы для группы W включают пиразолил, тиазолил, оксазолил и имидазолил.
В одном воплощении W представляет собой пиразолил, триазолил, тиазолил, оксазолил или имидазолил.
В одном воплощении W представляет собой пиразолил или триазолил.
R14 представляет собой фенил или 6-членное гетероароматическое кольцо, содержащее от 1 до 3 (например, один, два или три) кольцевых атома азота; причем указанное кольцо возможно замещено по меньшей мере одним (например, одним, двумя, тремя или четырьмя) заместителем, выбранным из галогена (например, фтора, хлора, брома или иода), С1-С4алкила (например, метила, этила, н-пропила, изо-пропила, н-бутила, изо-бутила или трет-бутила), С1-С4алкокси (например, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси или трет-бутокси), CN, ОН, NO2, С1-С3алкила, замещенного одним или более атомами F (например, CH2F, CHF2, CF3, CH2CH2F, CH2CF3, CF2CF3, СН(CF3)2 и CH2CH2CF3), С1-С3алкокси, замещенного одним или более атомами F (например, OCH2F, OCHF2, OCF3, OCH2CH2F, OCH2CF3, OCF2CF3, ОСН(CF3)2 и OCH2CH2CF3), NR12R13, C≡CR30, CONR31R32, СНО, С2-С4алканоила (например, метилкарбонила (ацетила), этилкарбонила, н-пропилкарбонила или изо-пропилкарбонила), S(O)pR33 и OSO2R34.
Примеры 6-членного гетероароматического кольца, содержащего от 1 до 3 кольцевых атомов азота, включают пиридинил, пиримидинил, пиридазинил, пиразинил и триазинил. Предпочтительной кольцевой системой является пиридинил.
В одном воплощении один заместитель на ароматическом кольце группы R14 должен находиться в 4-(пара)-положении относительно группы W.
В одном воплощении изобретения R14 представляет собой фенил или 6-членное гетероароматическое кольцо, содержащее от 1 до 3 кольцевых атома азота; причем указанное кольцо возможно замещено по меньшей мере одним заместителем, выбранным из F, Cl, CN и CF3.
В одном воплощении изобретения R14 представляет собой фенил или пиридинил; причем указанное кольцо возможно замещено по меньшей мере одним заместителем, выбранным из F, Cl, CN и CF3.
В одном воплощении изобретения R14 представляет собой фенильную или пиридинильную группу, возможно замещенную одним или двумя заместителями, независимо выбранными из F, Cl, CN и CF3.
В одном воплощении изобретения R14 представляет собой фенил или пиридинил; причем указанное кольцо замещено в 4-(пара)-положении F, Cl или CN и возможно дополнительно замещено.
В одном воплощении изобретения R14 представляет собой фенил или пиридинил; причем указанное кольцо замещено в 4-(пара)-положении F, Cl или CN.
R3 представляет собой фенил либо пяти- или шестичленное гетероароматическое кольцо, содержащее от 1 до 3 (например, один, два или три) гетероатома, независимо выбранных из О, S и N; причем указанное кольцо возможно замещено по меньшей мере одним (например, одним, двумя, тремя или четырьмя) заместителем, выбранным из галогена (например, фтора, хлора, брома или иода), С1-С6алкила (например, метила, этила, н-пропила, изо-пропила, н-бутила, изо-бутила, трет-бутила, н-пентила или н-гексила), циано, C1-С6алкокси (например, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, трет-бутокси, н-пентокси или н-гексокси), нитро, метилкарбонила, NR35R36, С1-С3алкила, замещенного одним или более атомами F (например, CH2F, CHF2, CF3, CH2CH2F, CH2CF3, CF2CF3, СН(CF3)2 и CH2CH2CF3), и С1-С3алкокси, замещенного одним или более атомами F (например, OCH2F, OCHF2, OCF3, OCH2CH2F, OCH2CF3, OCF2CF3, ОСН(CF3)2 и OCH2CH2CF3).
В одном воплощении R3 представляет собой фенильное или пиридинильное кольцо, замещенное по меньшей мере одним заместителем (например, одним, двумя или тремя заместителями), независимо выбранным из галогена, циано, нитро, метила, трифторметила и метилкарбонила.
В одном воплощении R3 представляет собой фенильную группу, замещенную одним или двумя заместителями, независимо выбранными из фтора, хлора, циано, нитро и трифторметила.
В другом воплощении R3 представляет собой фенильную группу, замещенную одним или двумя заместителями, независимо выбранными из фтора, хлора и трифторметила.
В еще одном воплощении R3 представляет собой фенильную группу, замещенную трифторметильным заместителем (предпочтительно в мета-положении).
В еще одном воплощении R3 представляет собой фенильную группу, замещенную в метаположении Br, Cl, CF3 или CN.
R4 представляет собой водород или С1-С6алкил (например, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, трет-бутил, н-пентил или н-гексил), возможно замещенный по меньшей мере одним заместителем (например, одним или двумя заместителями), независимо выбранным из фторо, гидроксила и С1-С6алкокси (например, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, трет-бутокси, н-пентокси или н-гексокси).
В одном воплощении R4 представляет собой водород или С1-С4алкил, возможно замещенный одним или двумя заместителями, независимо выбранными из гидроксила и С1-С4алкокси.
В другом воплощении R4 представляет собой водород.
В одном воплощении изобретения Х представляет собой простую связь или группу -С1-С6алкилен-Y-, где Y представляет собой простую связь, атом кислорода, NR24 или S(O)w; причем указанный алкилен возможно дополнительно замещен ОН, галогеном, CN, NR37R38, С1-С3алкокси, CONR39R40, CO2R66, SO2R41 и SO2NR42R43.
В одном воплощении изобретения Х представляет собой простую связь или группу -С1-С6алкилен-Y-, где Y представляет собой простую связь, атом кислорода, NR24 или S(O)w; причем указанный алкилен возможно дополнительно замещен ОН, галогеном, CN, NR37R38, С1-С3алкокси, CONR39R40, SO2R41 и SO2NR42R43.
В одном воплощении изобретения Х представляет собой группу -С1-С6алкилен-Y-, и Y представляет собой простую связь, а алкиленовая группировка представляет собой линейный или разветвленный С1-С6-, или С1-С4-, или С1-С2алкилен, возможно замещеный ОН, галогеном, CN, CO2R66 или С1-С3алкокси.
В одном воплощении изобретения Х представляет собой группу -С1-С6алкилен-Y-, и Y представляет собой простую связь, а алкиленовая группировка представляет собой линейный или разветвленный С1-С6-, или С1-С4-, или С1-С2алкилен, возможно замещенный ОН, галогеном, CN или С1-С3алкокси.
В другом воплощении изобретения Х представляет собой незамещенный С1-C2алкилен, в частности метилен.
В другом воплощении изобретения Х представляет собой простую связь.
В одном воплощении изобретения R4 и Х соединены вместе так, что группа -NR4X в совокупности представляет собой 5-7-членное азациклическое кольцо, возможно включающее один дополнительный гетероатом, выбранный из О, S и NR44; причем указанное кольцо возможно замещено С1-С6алкилом или NR45R46; и указанный алкил возможно дополнительно замещен ОН.
Примеры 5-7-членного азациклического кольца, возможно включающего один дополнительный гетероатом, выбранный из О, S и NR44, включают пирролидин, пиперидин, пиперазин, морфолин и пергидроазепин.
R5 представляет собой моноциклическую кольцевую систему, выбранную из
(1) фенокси,
(2) фенила,
(3) 5- или 6-членного гетероароматического кольца, содержащего по меньшей мере один кольцевой гетероатом (например, один, два, три или четыре кольцевых гетероатома), независимо выбранный из азота, кислорода и серы,
(4) насыщенного или частично ненасыщенного С3-С6циклоалкильного кольца или
(5) насыщенного или частично ненасыщенного 4-7-членного гетероциклического кольца, содержащего по меньшей мере один кольцевой гетероатом (например, один, два, три или четыре кольцевых гетероатома), независимо выбранный из кислорода, S(O)r и NR20, где по меньшей мере один из кольцевых атомов углерода возможно может быть заменен карбонильной группой,
либо R5 представляет собой бициклическую кольцевую систему, в которой эти два кольца независимо выбраны из моноциклических кольцевых систем, как определено в (2), (3), (4) и (5) выше, где эти два кольца либо конденсированы вместе, либо соединены непосредственно друг с другом, либо отделены друг от друга линкерной группой, выбранной из кислорода, S(O)t или С1-С6алкилена, возможно содержащего один или более (например, один или два) внутренних или концевых гетероатомов, выбранных из кислорода, серы и NR27, и возможно замещены по меньшей мере одним заместителем (например, одним или двумя заместителями), независимо выбранным из гидроксила, оксо и С1-С6алкокси (например, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, трет-бутокси, н-пентокси или н-гексокси);
причем эта моноциклическая или бициклическая кольцевая система возможно замещена (по кольцевому атому) по меньшей мере одним заместителем (например, одним, двумя или тремя заместителями), независимо выбранным из кислорода (например, с образованием N-оксида), CN, ОН, С1-С6алкила (например, метила, этила, н-пропила, изо-пропила, н-бутила, изо-бутила, трет-бутила, н-пентила или н-гексила), С1-С6алкокси (например, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, трет-бутокси, н-пентокси или н-гексокси), галогена (например, фтора, хлора, брома или иода), NR47R48, NO2, OSO2R49, CO2R50, C(=NH)NH2, C(O)NR51R52, C(S)NR53R54, SC(=NH)NH2, NR55C(=NH)NH2, -S(O)vR21, SO2NR56R57, С1-С3алкокси, который замещен одним или более атомами F (например, OCH2F, OCHF2, OCF3, OCH2CH2F, OCH2CF3, OCH2CF3, ОСН(CF3)2 и OCH2CH2CF3), и С1-С3алкила, который замещен SO2R58 или одним или более атомами F (например, CH2SO2R58, CH2CH2SO2R58, CH(SO2R58)CH3, CH2F, CHF2, CF3, CH2CH2F, CH2CF3, CF2CF3, СН(CF3)2 и CH2CH2CF3); где указанный С1-С6алкил возможно дополнительно замещен по меньшей мере одним заместителем, выбранным из циано, гидроксила, С1-Свалкокси (например, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, трет-бутокси, н-пентокси или н-гексокси), С1-С6алкилтио (например, метилтио, этилтио, н-пропилтио, изо-пропилтио, н-бутилтио, изо-бутилтио, трет-бутилтио, н-пентилтио или н-гексилтио) и -C(O)NR22R23;
либо R5 может также представлять собой водород.
Примеры 5- или 6-членного гетероароматического кольца включают фуранил, тиенил, пирролил, оксазолил, 1,2,4-оксадиазолил, 1,3,4-оксадиазолил, изоксазолил, имидазолил, пиразолил, тиазолил, триазолил, тетразолил, тиадиазолил, пиридинил, пиримидинил и пиразинил. Предпочтительные гетероароматические кольца включают изоксазолил, пиридинил, имидазолил и триазолил.
Если не указано иного, "насыщенное или частично ненасыщенное С3-С6циклоалкильное кольцо" означает 3-6-членное неароматическое циклоалкильное кольцо, возможно включающее одну или более двойных связей, примеры которого включают циклопропил, циклобутил, циклопентил, циклогексил, циклопентенил и циклогексенил. Предпочтительным циклоалкильным кольцом является циклопропил.
Если не указано иного, "насыщенное или частично ненасыщенное 4-7-членное гетероциклическое кольцо", как оно уточнено выше, означает 4-7-членное неароматическое гетероциклическое кольцо, возможно включающее одну или более двойных связей и возможно включающее карбонильную группу, примеры которого включают тетрагидрофуранил, тетраметиленсульфонил, тетрагидропиранил, 4-оксо-4Н-пиранил (4H-пиран-4-онил), пирролидинил, 3-пирролинил, имидазолидинил, 1,3-диоксоланил (1,3-диоксациклопентанил), пиперидинил, пиперазинил, морфолинил, пергидроазепинил (гексаметилениминил), пирролидонил и пиперидонил. Предпочтительным насыщенным или частично ненасыщенным 4-7-членным гетероциклическим кольцом является пирролидонил.
Примеры бициклических кольцевых систем, в которых два кольца либо конденсированы вместе, либо соединены непосредственно друг с другом, либо отделены друг от друга линкерной группой, включают бифенил, тиенилфенил, пиразолилфенил, феноксифенил, фенилциклопропил, нафтил, инданил, хинолил, тетрагидрхинолил, бензофуранил, индолил, (изо-индолил, индолинил, бензофуранил, бензотиенил, индазолил, бензимидазолил, бензтиазолил, пуринил, изо-хинолил, хроманил, инденил, хиназолил, хиноксалил, хроманил, узо-хроманил, 3Н-индолил, 1H-индазолил, хинуклидил, тетрагидронафтил, дигидробензофуранил, морфолин-4-илфенил, 1,3-бензодиоксолил, 2,3-дигидро-1,4-бензодиоксинил, 1,3-бензодиоксинил и 3,4-дигидро-изо-хроменил.
В одном воплощении изобретения R5 представляет собой замещенную моноциклическую кольцевую систему, как определено выше.
В другом воплощении изобретения R5 представляет собой замещенную бициклическую кольцевую систему, как определено выше.
В другом воплощении изобретения R5 представляет собой Н.
В дополнительном воплощении изобретения R5 представляет собой моноциклическую кольцевую систему, выбранную из
(1) фенокси,
(2) фенила,
(3) 5- или 6-членного гетероароматического кольца, содержащего один или два кольцевых гетероатома, независимо выбранных из азота, кислорода и серы,
(4) насыщенного или частично ненасыщенного С3-С6циклоалкильного кольца или
(5) насыщенного или частично ненасыщенного 4-7-членного гетероциклического кольца, содержащего один или два кольцевых гетероатома, независимо выбранных из кислорода, S(O)r и NR20, где по меньшей мере один из кольцевых атомов углерода возможно может быть заменен карбонильной группой;
либо R5 представляет собой бициклическую кольцевую систему, в которой эти два кольца независимо выбраны из моноциклических кольцевых систем, как определено в (2), (3), (4) и (5) выше, где эти два кольца либо конденсированы вместе, либо соединены непосредственно друг с другом, либо отделены друг от друга линкерной группой, выбранной из кислорода, метилена и S(O)t;
причем моноциклическая или бициклическая кольцевая система замещена одним или двумя заместителями, независимо выбранными из ОН, -S(O)vR21 и С1-C4алкила.
В еще одном дополнительном воплощении изобретения R5 представляет собой моноциклическую кольцевую систему, выбранную из фенила либо 5- или 6-членного гетероароматического кольца, содержащего один или два кольцевых гетероатома, независимо выбранных из азота и кислорода, которая замещена одним или двумя заместителями, независимо выбранными из ОН, -S(O)vR21 и С1-C4алкила.
В еще одном дополнительном воплощении изобретения R5 представляет собой фенил или пиридинил, замещенный -S(O)vR21, где v представляет собой целое число 2.
В еще одном дополнительном воплощении изобретения R5 представляет собой фенил, замещенный одним или двумя заместителями, независимо выбранными из ОН, -S(O)vR21 и С1-С4алкила.
В еще одном дополнительном воплощении изобретения R5 представляет собой Н.
В еще одном дополнительном воплощении изобретения R5 представляет собой незамещенное С3-С6циклоалкильное кольцо, в частности циклопропил.
В одном воплощении х равно 2.
В одном воплощении р равно 2.
В одном воплощении R10 и R11 независимо представляют собой Н, С1-С3алкил или С2-С3алкилкарбонил.
В одном воплощении R12 и R13 независимо представляют собой Н, С1-С3алкил или С2-С3алкилкарбонил.
В дополнительном воплощении R20 представляет собой водород, метил, этил, метилкарбонил (ацетил), этилкарбонил, метоксикарбонил или этоксикарбонил.
В одном воплощении v равно 2.
R21 представляет собой водород, C1-С6алкил (например, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, трет-бутил, н-пентил или н-гексил) или С3-С6циклоалкил (циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил); причем указанная алкильная или циклоалкильная группа возможно дополнительно замещена одним или более заместителями, выбранными независимо из ОН, CN, С1-С3алкокси и CONR59R60.
В еще одном дополнительном воплощении изобретения R21 представляет собой С1-С4алкил или С3-С6циклоалкил.
В другом воплощении R21 представляет собой С1-С3алкил (в частности метил, этил или изо-пропил) или циклопропил.
В другом воплощении R41 представляет собой С1-С3алкил (в частности метил, этил или изо-пропил) или циклопропил.
В одном воплощении изобретения каждый из R15, R16, R17, R18, R19, R30, R33, R34, R35, R36, R37, R38, R47, R48, R61, R22, R23, R24, R26, R27, R31, R32, R39, R40, R42, R43, R44, R45, R46, R49, R50, R51, R52, R53, R54, R55, R56, R57, R58, R59, R60, R62, R63, R64, R65 и R66 независимо представляет собой водород или С1-С3алкил, в частности метил, этил, 1-пропил или 2-пропил.
В одном воплощении изобретения каждый из R15, R16, R17, R18, R19, R30, R33, R34, R35, R36, R37, R38, R47, R48, R61, R22, R23, R24, R26, R27, R31, R32, R39, R40, R42, R43, R44, R45, R46, R49, R50, R51, R52, R53, R54, R55, R56, R57, R58, R59, R60, R62, R63, R64, R65 и R66 независимо представляет собой водород или метил.
В одном воплощении изобретения R66 представляет собой водород.
В одном воплощении изобретения R1 представляет собой метил;
W представляет собой 5-членное гетероароматическое кольцо, и группа R14 и 2-пиразиноновое кольцо присоединены к 5-членному кольцу W в 1,2-положениях;
R14 представляет собой фенил или пиридинил; причем указанное кольцо возможно замещено по меньшей мере одним заместителем, выбранным из F, Cl, CN и CF3;
R3 представляет собой фенильную группу, замещенную одним или двумя заместителями, независимо выбранными из фтора, хлора, циано, нитро или трифторметила;
R4 представляет собой водород;
Х представляет собой незамещенный С1-С2алкилен, в частности метилен; и
R5 представляет собой фенил, замещенный одним или двумя заместителями, независимо выбранными из ОН, -S(O)vR21 и С1-С4алкила, где v представляет собой целое число 2.
В одном воплощении изобретения
R1 представляет собой метил;
W представляет собой 5-членное гетероароматическое кольцо, и группа R14 и 2-пиразиноновое кольцо присоединены к 5-членному кольцу W в 1,2-положениях;
R14 представляет собой фенил или пиридинил; причем указанное кольцо возможно замещено по меньшей мере одним заместителем, выбранным из F, Cl, CN и CF3;
R3 представляет собой фенильную группу, замещенную одним или двумя заместителями, независимо выбранными из фтора, хлора, циано, нитро или трифторметила;
R4 представляет собой водород;
Х представляет собой незамещенный С1-C2алкилен, в частности метилен; и
R5 представляет собой Н.
В одном воплощении изобретения
R1 представляет собой метил;
W представляет собой пиразолильное или триазолильное кольцо, и группа R14 и 2-пиразиноновое кольцо присоединены к 5-членному кольцу W в 1,2-положениях;
R14 представляет собой фенил или пиридинил; причем указанное кольцо замещено в 4-(пара)-положении F, Cl или CN;
R3 представляет собой фенильную группу, замещенную мета-положении Br, Cl, CF3 или CN;
R4 представляет собой водород;
Х представляет собой линейный или разветвленный С1-С4алкилен, возможно замещенный ОН, галогеном, CN, CO2R66 или С1-С3алкокси; и
R5 представляет собой Н.
Примеры соединений по изобретению включают:
6-[2-(4-цианофенил)-2H-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты метиламид;
6-[2-(4-цианофенил)-2H-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты (5-метансульфонил-пиридин-2-илметил)-амид;
6-[2-(4-цианофенил)-2H-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты этиламид;
6-[1-(4-цианофенил)-1Н-1,2,3-триазол-5-ил]-N,5-диметил-3-оксо-4-[3-(трифторметил)-фенил]-3,4-дигидропиразин-2-карбоксамид;
трет-бутил-2-[[6-[2-(4-цианофенил)пиразол-3-ил]-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбонил]амино]ацетат;
6-[3-(4-хлорфенил)-3H-[1,2,3]триазол-4-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты метиламид;
6-[2-(4-хлорфенил)-2Н-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты метиламид;
6-[1-(4-цианофенил)-1H-пиразол-5-ил]-N-(2-метоксиэтил)-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид;
6-[1-(4-цианофенил)-1Н-пиразол-5-ил]-N-(2-гидрокси-1,1-диметилэтил)-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид;
6-[1-(4-цианофенил)-1H-пиразол-5-ил]-N,N,5-триметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид;
6-[1-(4-цианофенил)-1H-пиразол-5-ил]-N-циклопропил-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид;
6-[1-(6-цианопиридин-3-ил)-1H-пиразол-5-ил]-N-циклопропил-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид;
6-[1-(6-цианопиридин-3-ил)-1H-пиразол-5-ил]-N,5-диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид;
6-[1-(5-цианопиридин-2-ил)-1H-пиразол-5-ил]-N,5-диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид;
6-[1-(5-цианопиридин-2-ил)-1H-пиразол-5-ил]-N-циклопропил-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид и
2-[[6-[2-(4-цианофенил)пиразол-3-ил]-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидро-пиразин-2-карбонил]амино]уксусную кислоту;
и фармацевтически приемлемые соли любого из вышеперечисленного.
В настоящем изобретении дополнительно предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, который включает
(а) взаимодействие соединения формулы (II)
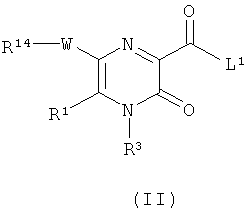
где L1 представляет собой уходящую группу (такую как галоген или гидроксил), и R1, R3, R14 и W являются такими, как определено в формуле (I), с соединением формулы
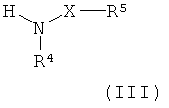
где X, R4 и R5 являются такими, как определено в формуле (I); или
(б) взаимодействие соединения формулы (IV)
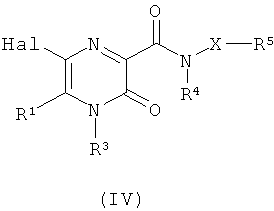
где Hal представляет собой атом галогена, и X, R1, R3, R4 и R5 являются такими, как определено в формуле (I),
с нуклеофилом R14-W-M, где R14 и W являются такими, как определено в формуле (I), и М представляет собой оловоорганическую группу или группу органобороновой кислоты; или
(в) когда W представляет собой тиазолил или оксазолил, взаимодействие соединения формулы (V)
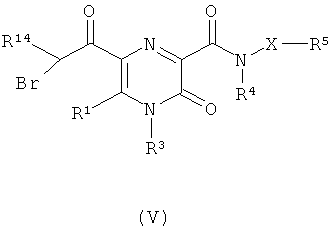
где X, R1, R3, R4, R5 и R14 являются такими, как определено в формуле (I), с тиомочевиной или формамидом, соответственно;
и, возможно, после стадий (а), (б) или (в) проводят одну или более из следующих стадий:
- превращение полученного соединения в другое соединение по изобретению,
- получение фармацевтически приемлемой соли соединения.
В способе (а) взаимодействие может легко быть осуществлено в органическом растворителе, таком как дихлорметан или N-метилпирролидинон, при температуре, например, в диапазоне от 0°С до точки кипения растворителя. Если необходимо или желательно, может быть добавлено основание и/или агент сочетания, такие как HATU (O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат), НОАТ (1-гидрокси-7-азабензотриазол), НОВТ (1-гидроксибензотриазол гидрат) или DIEA (N,N-диизопропилэтиламин).
В способе (б) взаимодействие может легко быть осуществлено в органическом растворителе, таком как DMF, NMP (N-метилпирролидон) или толуол, или в их смеси при повышенной температуре (т.е. выше температуры окружающей среды, 20°С), например в диапазоне от 50°С до 150°С, и в присутствии подходящего катализатора на основе переходного металла, такого как бис(три-трет-бутилфосфин)палладий. Если необходимо или желательно, может быть добавлено основание, такое как карбонат калия.
В способе (в) взаимодействие может легко быть осуществлено путем совместного нагревания двух исходных веществ в подходящем органическом растворителе, таком как ацетонитрил, при температуре, например, в диапазоне от 50°С до 150°С.
Конкретные способы получения соединений формулы (I) раскрыты в разделе «Примеры» настоящего описания изобретения. Такие способы составляют один из аспектов настоящего изобретения.
Необходимые исходные вещества либо коммерчески доступны, либо известны из литературы, либо могут быть получены с использованием известных методик. Конкретные способы получения определенных ключевых исходных веществ раскрыты в разделе «Примеры» настоящего описания изобретения, и такие способы составляют один из аспектов настоящего изобретения.
Соединения формулы (I) могут быть превращены в другие соединения формулы (I) с использованием стандартных процедур.
Некоторые промежуточные соединения формул (II), (IV) и (V) являются новыми. Эти новые промежуточные соединения составляют другой аспект изобретения.
Специалисту в данной области техники будет понятно, что в способах по настоящему изобретению определенные функциональные группы, такие как гидроксильные или аминогруппы, может быть необходимо защитить с помощью защитных групп. Таким образом, получение соединений формулы (I) может включать в себя, на соответствующей стадии, введение и/или удаление одной или более защитных групп.
Защита и снятие защиты с функциональных групп описаны в "Protective Groups in Organic Chemistry', edited by J.W.F.McOmie, Plenum Press (1973) и 'Protective Groups in Organic Synthesis', 3rd edition, T.W.Greene and P.G.M.Wuts, Wiley-lnterscience (1999).
Соединения формулы (I) выше могут быть превращены в их фармацевтически приемлемую соль, предпочтительно в соль присоединения кислоты, такую как гидрохлорид, гидробромид, сульфат, фосфат, ацетат, фумарат, малеат, тартрат, лактат, цитрат, пируват, сукцинат, оксалат, метансульфонат или пара-толуолсульфонат.
Соединения формулы (I) способны к существованию в стереоизомерных формах. Понятно, что данное изобретение охватывает применение всех геометрических и оптических изомеров (включая атропизомеры) соединений формулы (I) и их смесей, включая рацематы. Применение таутомеров и их смесей также составляет аспект настоящего изобретения. Особенно желательными являются энантиомерно чистые формы.
Соединения формулы (I) и их фармацевтически приемлемые соли обладают активностью в качестве фармацевтических средств, в частности в качестве модуляторов сериновых протеаз, таких как протеиназа 3 и панкреатическая эластаза, и особенно человеческой нейтрофильной эластазы, и таким образом могут быть полезны в лечении или профилактике воспалительных заболеваний и состояний.
Соединения формулы (I) и их фармацевтически приемлемые соли можно использовать в лечении заболеваний респираторного тракта, таких как обструктивные заболевания дыхательных путей, включая астму, в том числе бронхиальную, аллергическую, наследственную, приобретенную, индуцированную физической нагрузкой, индуцированную приемом лекарственных средств (включая индуцированную приемом аспирина и NSAID (нестероидные противовоспалительные лекарственные средства)) и индуцированную пылью астму, как интермиттирующую, так и персистирующую, и всех степеней тяжести, и другие случаи гиперреактивности дыхательных путей; хроническая обструктивная болезнь легких (COPD); бронхит, в том числе инфекционный и эозинофильный бронхит; эмфизема; бронхоэктаз; цистический фиброз; саркоидоз; экзогенный аллергический альвеолит и родственные заболевания; гиперчувствительный пневмонит; пневмофиброз, в том числе криптогенный фиброзный альвеолит, идиопатическая интерстициальная пневмония, фиброз, являющийся осложнением противоопухолевой терапии и хронической инфекции, включая туберкулез и аспергиллез и другие грибковые инфекции; осложнения легочной трансплантации; васкулитоподобные и тромботические расстройства легочной сосудистой сети и легочная гипертензия; противокашлевая активность, в том числе лечение хронического кашля, ассоциированного с воспалительными и секреторными состояниями дыхательных путей, и ятрогенного кашля; острый и хронический ринит, в том числе медикаментозный ринит и вазомоторный ринит; круглогодичный и сезонный аллергический ринит, в том числе нервный ринит (сенная лихорадка); назальный полипоз; острая вирусная инфекция, в том числе простуда, и инфекция, вызванная респираторно-синцитиальным вирусом, вирусом гриппа, коронавирусом (включая атипичную пневмонию (SARS)) и аденовирусом.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний костей и суставов, таких как артритиды, ассоциированные с остеоартритом/остеоартрозом или включающие остеоартрит/остеоартроз, как первичный, так и вторичный по отношению, например, к врожденной дисплазии тазобедренного сустава; цервикальный и поясничный спондилит и боль в нижней области спины и шеи; ревматоидный артрит и болезнь Стилла; серонегативные спондилоартропатии, в том числе анкилозирующий спондилит, псориатический артрит, реактивный артрит и недифференцируемая спондилоартропатия; септический артрит и другие ассоциированные с инфекцией артропатии и заболевания кости, такие как туберкулез, в том числе болезнь Потта и полиартрит Понсе; острый и хронический индуцируемый отложением кристаллов синовит, в том числе уратная подагра, заболевание вследствие отложения пирофосфата кальция и воспаление сухожилий, синовиальной сумки и синовиальной жидкости, ассоциированное с апатитом кальция; болезнь Бехчета; первичный и вторичный синдром Шегрена; системный склероз и ограниченная склеродермия; системная красная волчанка, заболевание соединительной ткани смешанного типа и недифференцируемое заболевание соединительной ткани; воспалительные миопатии, в том числе дерматомиозит и полимиозит; ревматическая полимиалгия; юношеский артрит, в том числе идиопатические воспалительные артритиды суставов любой локализации и ассоциированные синдромы, ревматическая лихорадка и ее системные осложнения; васкулитиды, в том числе гигантоклеточный артериит, синдром Такаясу, синдром Чурга-Штраусса (Churg-Strauss), полиартериит нодозный, микроскопический полиартериит и васкулитиды, ассоциированные с вирусной инфекцией, реакциями гиперчувствительности, криоглобулинами и парапротеинами; боль в нижней части спины; семейная средиземноморская лихорадка, синдром Мукла-Уэльса (Muckle-Wells), семейная ирландская лихорадка, болезнь Кикучи (Kikuchi); индуцированные приемом лекарственных средств артралгии, поражения кожи при тендините и миопатии.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении боли и перестройки соединительной ткани при скелетно-мышечных нарушениях из-за повреждения (например, спортивной травмы) или следующих заболеваний: артритиды (например, ревматоидный артрит, остеоартрит, подагра или вызванная кристаллами артропатия), другое заболевание суставов (такое как дегенерация межпозвоночного диска или дегенерация височно-челюстного сустава), заболевание с перестройкой кости (такое как остеопороз, болезнь Педжета или остеонекроз), полихондриты, склеродермия, смешанное нарушение соединительной ткани, спондилоартропатии или заболевание периодонта (такое как периодонтит).
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний кожи, таких как псориаз, атопический дерматит, контактный дерматит или другие экзематозные поражения кожи и реакции гиперчувствительности замедленного типа; фито- и фотодерматит; себорейная экзема, герпетиформная экзема, красный плоский лишай, склеротический и атрофический лишай, гангренозная пиодермия, кожный саркоид, дискоидная красная волчанка, пузырчатка, пемфигоид, врожденный буллезный эпидермолиз, крапивница, ангионевротические отеки, васкулитиды, токсические эритемы, кожные эозинофилии, гнездная алопеция, облысение по мужскому типу, синдром Свита (Sweet), болезнь Вебера-Крисчена, множественная эритема; целлюлит, как инфекционный, так и неинфекционный; панникулит; кожные лимфомы, немеланомный рак кожи и другие диспластические поражения; индуцированные приемом лекарственных средств расстройства, в том числе стойкая лекарственная сыпь.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний глаз, таких как блефарит; конъюнктивит, в том числе круглогодичный и весенний аллергический конъюнктивит; иритит; передний и задний увеит; хореоидит; аутоиммунная реакция; дегенеративные или воспалительные расстройства, влияющие на сетчатку; офтальмия, в том числе симпатическая офтальмия; саркоидоз; инфекции, в том числе вирусная, грибковая и бактериальная.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний желудочно-кишечного тракта, такого как глоссит, гингивит, периодонтит; эзофагит, в том числе рефлюкс-эзофагит; эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона, колит, в том числе неспецифический язвенный колит, проктит, зуд заднего прохода; глютеновая болезнь, синдром раздраженного кишечника, диарея невоспалительного характера и пищевые аллергии, которые проявляют действие, не связанное с кишечником (например, мигрень, ринит и экзема).
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний сердечно-сосудистой системы, таких как атеросклероз, влияющий на коронарное и периферическое кровообращение; перикардит; миокардит, воспалительные и аутоиммунные кардиомиопатии, в том числе миокардиальный саркоид; ишемические реперфузионные поражения; эндокардит, вальвулит и аортит, в том числе инфекционный (например, сифилитический); васкулитиды; заболевания проксимальных и периферических вен, в том числе флебит и тромбоз, включая тромбоз глубоких вен и осложнения варикозных вен.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении онкологических заболеваний, например в лечении обычных видов рака, в том числе опухолей предстательной железы, молочной железы, легкого, яичников, поджелудочной железы, кишечника и толстой кишки, желудка, кожи, и опухолей головного мозга, и злокачественных новообразований, влияющих на костный мозг (включая лейкозы) и лимфопролиферативные системы, например лимфомы Ходжкина или неходжкинских лимфом; включая предупреждение и лечение метастатического заболевания и опухолевых рецидивов и паранеопластических синдромов.
Соединения формулы (I) и их фармацевтически приемлемые соли можно использовать в лечении респираторного дистресс-синдрома взрослых (ARDS), цистического фиброза, легочной эмфиземы, бронхита, включая хронический бронхит, бронхоэктаза, хронического обструктивного заболевания легких (COPD), легочной гипертензии, астмы, включая рефрактивную астму, ринита, псориаза, ишемическо-реперфузионного повреждения, ревматоидного артрита, синдрома системного воспалительного ответа (SIRS), длительно не заживающих ран, рака, атеросклероза, пептических язв, болезни Крона, неспецифического язвенного колита и повреждения слизистой оболочки желудка.
В частности, соединения формулы (I) и их фармацевтически приемлемые соли можно использовать в лечении хронического обструктивного заболевания легких (COPD), астмы и ринита.
Еще более конкретно, соединения формулы (I) и их фармацевтически приемлемые соли можно использовать в лечении хронического обструктивного заболевания легких (COPD).
Таким образом, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь выше, для применения в терапии.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в терапии.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для лечения заболеваний или состояний человека, при которых полезно модулирование активности нейтрофильной эластазы.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в лечении воспалительного заболевания или состояния.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в лечении респираторного дистресс-синдрома взрослых (ARDS), цистического фиброза, легочной эмфиземы, бронхита, включая хронический бронхит, бронхоэктаза, хронического обструктивного заболевания легких (COPD), легочной гипертензии, астмы, включая рефрактивную астму, ринита, псориаза, ишемическо-реперфузионного повреждения, ревматоидного артрита, синдрома системного воспалительного ответа (SIRS), длительно не заживающих ран, рака, атеросклероза, пептических язв, болезни Крона, неспецифического язвенного колита и повреждения слизистой оболочки желудка.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в лечении хронического обструктивного заболевания легких (COPD).
В контексте настоящего описания термин «терапия» также включает «профилактику», если нет конкретных указаний на противоположное. Термины «терапевтический» и «терапевтически» следует понимать соответственно.
Профилактика представляется особенно подходящей для лечения субъектов, которые пострадали от предыдущего эпизода или иным образом считаются имеющими повышенный риск данного заболевания или состояния. Субъекты, имеющие риск развития конкретного заболевания или состояния, обычно включают в себя тех, кто имеет семейную историю этого заболевания или состояния, или тех, кто был идентифицирован генетическим тестированием или скринингом как особенно подверженный развитию этого заболевания или состояния.
В данном изобретении также дополнительно предложен способ лечения или снижения риска заболевания или состояния, при котором полезно ингибирование активности нейтрофильной эластазы, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше.
В данном изобретении также дополнительно предложен способ лечения или снижения риска воспалительного заболевания или состояния, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше.
В данном изобретении также дополнительно предложен способ лечения или снижения риска респираторного дистресс-синдрома взрослых (ARDS), цистического фиброза, легочной эмфиземы, бронхита, включая хронический бронхит, бронхоэктаза, хронического обструктивного заболевания легких (COPD), легочной гипертензии, астмы, включая рефрактивную астму, ринита, псориаза, ишемическо-реперфузионного повреждения, ревматоидного артрита, остеоартрита, синдрома системного воспалительного ответа (SIRS), длительно не заживающих ран, атеросклероза, пептических язв, болезни Крона, неспецифического язвенного колита и повреждения слизистой оболочки желудка, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше.
В данном изобретении также дополнительно предложен способ лечения или снижения риска хронического обструктивного заболевания легких (COPD), включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше.
Для указанных терапевтических применений вводимые дозировки будут, конечно, варьировать в зависимости от используемого соединения, способа введения, желаемого лечения и указанного расстройства. Суточная дозировка соединения по этому изобретению может находиться в диапазоне от 0,05 мкг/кг до 100 мкг/кг.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы сами по себе, но обычно их будут вводить в форме фармацевтической композиции, в которой соединение/соль формулы (I) (активный ингредиент) находится в сочетании с фармацевтически приемлемым адъювантом, разбавителем или носителем. Общепринятые способы выбора и изготовления подходящих фармацевтических композиций раскрыты, например, в "Pharmaceuticals - The Science of Drug Design", M.E.Aulton, Churchill Livingstone, 1988.
В зависимости от способа введения, фармацевтическая композиция будет предпочтительно содержать от 0,05 до 99 масс.% (процентов по массе), более предпочтительно от 0,05 до 80 масс.%, еще более предпочтительно от 0,10 до 70 масс.% и, даже более предпочтительно, от 0,10 до 50 масс.% активного ингредиента, причем все проценты по массе даны в расчете на общую массу композиции.
В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь выше, в сочетании с фармацевтически приемлемым адъювантом, разбавителем или носителем.
В этом изобретении дополнительно предложен способ получения фармацевтической композиции по изобретению, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Фармацевтические композиции можно вводить местно (например, на кожу или в легкое и/или дыхательные пути) в форме, например, кремов, растворов, суспензий, гептафторалкановых (HFA) аэрозолей и сухих порошковых препаратов, например препаратов в ингаляторном устройстве, известном как Turbuhaler®; или системно, например, пероральным введением в форме таблеток, капсул, сиропов, порошков или гранул; или парентеральным введением в форме растворов или суспензий; или подкожным введением; или ректальным введением в форме суппозиториев; или чрескожным путем.
Сухие порошковые препараты и HFA-аэрозоли соединений по изобретению под давлением можно вводить ингаляцией в ротовую полость или нос. Для ингаляции соединение тонко измельчают желаемым образом. Тонко измельченное соединение предпочтительно имеет среднемассовый диаметр менее чем 10 мкм, и его можно суспендировать в пропелленте в смеси со вспомогательным диспергирующим веществом, таким как С8-С20 жирная кислота или ее соль (например, олеиновая кислота), соль желчной кислоты, фосфолипид, алкилсахарид, перфторированное или полиэтоксилированное поверхностно-активное вещество или другое фармацевтически приемлемое диспергирующее вещество.
Соединения по изобретению можно также вводить посредством ингалятора сухого порошка. Ингалятор может представлять собой одно- или многодозовый ингалятор и может представлять собой срабатывающий при вдохе ингалятор сухого порошка.
Одной из возможностей является смешивание тонко измельченного соединения по изобретению с веществом-носителем, например моно-, ди- или полисахаридом, сахарным спиртом или другим полиолом. Подходящими носителями являются сахара, например лактоза, глюкоза, раффиноза, мелезитоза, лактит, мальтит, трегалоза, сахароза, маннит и крахмал. Альтернативно, тонко измельченное соединение можно покрывать другим веществом. Порошковую смесь можно также распределить в твердые желатиновые капсулы, каждая из которых содержит желаемую дозу активного соединения.
Другой возможностью является получение тонко измельченного порошка в сферах, которые разрываются во время процедуры ингаляции. Этим сферонизированным порошком можно заполнять резервуар многодозового ингалятора для лекарственного средства, например, известного как Turbuhaler®, в котором дозирующий блок отмеряет желаемую дозу, которая затем ингалируется пациентом. Используя эту систему, активный ингредиент доставляют пациенту с веществом-носителем или без него.
Для перорального введения соединение по изобретению можно смешивать с адъювантом или носителем, например лактозой, сахарозой, сорбитом, маннитом, крахмалом, например картофельным крахмалом, кукурузным крахмалом или амилопектином; производным целлюлозы; связывающим веществом, например желатином или поливинилпирролидоном; и/или смазывающим веществом, например стеаратом магния, стеаратом кальция, полиэтиленгликолем, воском, парафином и тому подобным, и затем прессовать в таблетки. Если требуются таблетки с оболочкой, ядра, полученные как описано выше, можно покрывать концентрированным раствором сахара, который может содержать, например, аравийскую камедь, желатин, тальк и диоксид титана. Альтернативно, таблетку можно покрывать подходящим полимером, растворенным в легколетучем органическом растворителе.
Для получения мягких желатиновых капсул соединение по изобретению можно смешивать, например, с растительным маслом или полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы соединения с использованием любых указанных эксципиентов для таблеток. Также твердые желатиновые капсулы можно заполнять жидкими или полутвердыми препаратами соединения по изобретению.
Жидкие препараты для перорального применения могут быть в форме сиропов или суспензий, например растворов, содержащих соединение по изобретению, причем остальное представляет собой сахар и смесь этанола, воды, глицерина и пропиленгликоля. Возможно, такие жидкие препараты могут содержать красящие агенты, корригенты, сахарин и/или карбоксиметилцеллюлозу в качестве загущающего агента или другие эксципиенты, известные специалистам в данной области техники.
Соединения по изобретению можно также вводить в сочетании с другими соединениями, используемыми для лечения указанных выше состояний.
Таким образом, данное изобретение дополнительно относится к комбинированным терапиям, где соединение по изобретению или его фармацевтически приемлемую соль, или фармацевтическую композицию либо препарат, содержащие соединение по изобретению, вводят одновременно или последовательно либо в виде комбинированного препарата с другим терапевтическим агентом или агентами для лечения одного или более чем одного из перечисленных выше состояний.
В частности, для лечения воспалительных заболеваний, таких как (но без ограничения ими) ревматоидный артрит, остеоартрит, астма, аллергический ринит, хроническое обструктивное заболевание легких (COPD), псориаз и воспалительное заболевание кишечника, соединения по изобретению можно объединять с агентами, перечисленными ниже.
Нестероидные противовоспалительные агенты (далее NSAID), в том числе неселективные ингибиторы циклооксигеназ СОХ-1 и СОХ-2, применяемые как местно, так и системно (такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флурбипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, азапропазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин); селективные ингибиторы СОХ-2 (такие как мелоксикам, целекоксиб, рофекоксиб, валдекоксиб, лумарококсиб, парекоксиб и эторикоксиб); ингибирующие циклооксигеназу доноры оксида азота (CINOD); глюкокортикостероиды (вводимые как местным, так и пероральным, внутримышечным, внутривенным или внутрисуставным путем); метотрексат; лефлуномид; гидроксихлороквин; d-пеницилламин; ауранофин или другие парентеральные или пероральные препараты золота; анальгетики; диацереин; внутрисуставные лекарства, такие как производные гиалуроновой кислоты; и пищевые добавки, такие как глюкозамин.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с цитокином или агонистом либо антагонистом функции цитокинов (включая агенты, которые действуют на цитокиновые сигнальные пути, такие как модуляторы системы супрессоров цитокиновых сигнальных путей, SOCS), включая альфа-, бета- и гамма-интерфероны; инсулиноподобный фактор роста типа I (IGF-1); интерлейкины (IL), в том числе IL1-23, и антагонисты или ингибиторы интерлейкинов, такие как анакинра; ингибиторы фактора некроза опухолей альфа (TNF-α), такие как моноклональные антитела против TNF (например, инфликсимаб, адалимумаб и CDP-870) и антагонисты рецепторов TNF, в том числе иммуноглобулиновые молекулы (такие как этанерцепт) и низкомолекулярные агенты, такие как пентоксифиллин.
В дополнение, это изобретение относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с моноклональным антителом, направленным на В-лимфоциты (таким как CD20 (ритуксимаб)), MRA-alLI6R, или Т-лимфоциты (CTLA4-Ig, HuMax II-15).
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с модулятором функции рецепторов хемокинов, таким как антагонист CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства С-С); CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства С-Х-С) и CX3CR1 для семейства С-Х3-С.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором матриксных металлопротеаз (ММР), то есть стромелизинов, коллагеназ и желатиназ, а также аггреканазы; особенно коллагеназы-1 (ММР-1), коллагеназы-2 (ММР-8), коллагеназы-3 (ММР-13), стромелизина-1 (ММР-3), стромелизина-2 (ММР-10) и стромелизина-3 (ММР-11) и ММР-9 и ММР-12, в том числе такими агентами, как доксициклин.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором биосинтеза лейкотриенов, ингибитором 5-липоксигеназы (5-LO) или антагонистом белка, активирующего 5-липоксигеназу (FLAP), таким как: зилейтон; АВТ-761; фенлейтон; тепоксалин; Abbott-79175; Abbott-85761; N-(5-замещенный)-тиофен-2-алкилсульфонамид; 2,6-ди-трет-бутилфенол-гидразоны; метокситетрагидропираны, такие как Zeneca ZD-2138; соединение SB-210661; соединение, представляющее собой пиридинил-замещенный 2-цианонафталин, такое как L-739010; соединение, представляющее собой 2-цианохинолин, такое как L-746530; или индольное или хинолиновое соединение, такое как МК-591, МК-886 и BAY×1005.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом рецепторов лейкотриенов (LT) B4, LTC4, LTD4 и LTE4, выбранным из группы, состоящей из фенотиазинов-3-1, таких как L-651392; амидиновых соединений, таких как CGS-25019C; бензоксаламинов, таких как онтазоласт; бензокарбоксиимидаминов, таких как BIIL 284/260; и таких соединений, как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст (МК-679), RG-12525, Ro-245913, иралукаст (CGP 45715A) и BAY×7195.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором фосфодиэстеразы (PDE), таким как метилксантин, включая теофиллин и аминофиллин; селективным ингибитором изофермента PDE, включая ингибитор PDE4, ингибитор изоформы PDE4D или ингибитор PDE5.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом гистаминовых рецепторов типа 1, таким как цетиризин, лоратадин, дезлоратадин, фексофенадин, акривастин, терфенадин, астемизол, азеластин, левокабастин, хлорфенирамин, прометазин, циклизин или мизоластин, применяемым перорально, местно или парентерально.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором протонного насоса (таким как омепразол) или гастропротективным антагонистом гистаминовых рецепторов типа 2.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом гистаминовых рецепторов типа 4.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с сосудосуживающим симпатомиметиком, представляющим собой агонист адренорецепторов альфа-1/альфа-2, таким как пропилгекседрин, фенилэфрин, фенилпропаноламин, эфедрин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид, трамазолина гидрохлорид или этилнорэпинефрина гидрохлорид.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антихолинергическими агентами, в том числе антагонистом мускаринового рецептора (М1, М2 и М3), таким как атропин, хиосцин, гликопирролат, ипратропия бромид, тиотропия бромид, окситропия бромид, пирензепин или телензепин.
Настоящее изобретение также относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли и агониста бета-адренорецептора (включая бета-рецепторы подтипов 1-4), такого как изопреналин, сальбутамол, формотерол, сальметерол, тербуталин, орципреналин, битолтерола мезилат или пирбутерол, либо их хиральный энантиомер.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с хромоном, таким как хромогликат натрия или недохромил натрия.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с глюкокортикоидом, таким как флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат, циклезонид или мометазона фуроат.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом, который модулирует ядерные рецепторы гормона, такие как рецепторы, активируемые пролифератором пероксисом (PPAR).
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с иммуноглобулином (Ig) или препаратом Ig либо антагонистом или антителом, модулирующим функционирование Ig, таким как анти-IgE (например, омализумаб).
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с другим системным или местно наносимым противовоспалительным агентом, таким как талидомид или его производное, ретиноид, дитранол или кальципотриол.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с комбинациями аминосалицилатов и сульфапиридина, такого как сульфасалазин, месалазин, бальсалазид и олсалазин; и иммуномодуляторными агентами, такими как тиопурины, и кортикостероидами, такими как будесонид.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с антибактериальным агентом, таким как производное пенициллина, тетрациклин, макролид, бета-лактам, фторхинолон, метронидазол, ингалируемый аминогликозид; противовирусным агентом, включая ацикловир, фамцикловир, валацикловир, ганцикловир, цидофовир, амантадин, римантадин, рибавирин, занамавир и оселтамавир; ингибитором протеазы, таким как индинавир, нелфинавир, ритонавир и саквинавир; нуклеозидным ингибитором обратной транскриптазы, таким как диданозин, ламивудин, ставудин, залцитабин или зидовудин; или ненуклеозидным ингибитором обратной транскриптазы, таким как невирапин или эфавиренц.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с сердечно-сосудистым агентом, таким как блокатор кальциевых каналов, блокатор бета-адренорецепторов, ингибитор ангиотензин-конвертирующего фермента (АСЕ), антагонист рецепторов ангиотензина-2; агентом снижения липидов, таким как статин или фибрат; модулятором морфологии клеток крови, таким как пентоксифиллин; тромболитиком или антикоагулянтом, таким как ингибитор агрегации тромбоцитов.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом, действующим на ЦНС, таким как антидепрессант (такой как сертралин), лекарственное средство против болезни Паркинсона (такое как депренил, L-ДОФА, ропинирол, прамипексол, ингибитор моноаминооксидазы В (МАОВ), такой как селегин и расагилин, ингибитор соединения Р (comP), такой как тасмар, ингибитор А-2, ингибитор обратного захвата дофамина, антагонист N-метил-D-аспартата (NMDA), никотиновый агонист, дофаминовый агонист или ингибитор нейрональной синтазы оксида азота), или лекарственное средство против болезни Альцгеймера, такое как донепезил, ривастигмин, такрин, ингибитор СОХ-2, пропентофиллин или метрифонат.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом для лечения острой или хронической боли, таким как действующий центрально или периферически анальгетик (например, опиоид или его производное), карбамазепин, фенитоин, вальпроат натрия, амитриптилин или другие антидепрессанты, парацетамол или нестероидный противовоспалительный агент.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с парентерально или местно применяемым (в том числе ингалируемым) локальным обезболивающим средством, таким как лигнокаин или его производное. Соединение по настоящему изобретению или его фармацевтически приемлемую соль можно также использовать в комбинации с агентом против остеопороза, включая гормональный агент, такой как ралоксифен или бифосфонат, такой как алендронат.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с: (1) ингибитором триптазы; (2) антагонистом фактора активации тромбоцитов (PAF); (3) ингибитором интерлейкин-превращающего фермента (ICE); (4) ингибитором инозинмонофосфатдегидрогеназы IMPDH; (5) ингибитором молекул адгезии, в том числе антагонистами VLA-4 (очень поздний антиген); (6) катепсином; (7) ингибитором киназы, таким как ингибитор тирозинкиназы (такой как Btk, Itk, Jak3 или MAP, например, гефитиниб или мезилат иматиниба), серин/треонинкиназы (таким как ингибитор митоген-активируемой протеинкиназы (МАР-киназы), такой как р38, JNK, протеинкиназа А, В или С, или киназа ингибиторной киназы (IKK)), или киназы, участвующей в регуляции клеточного цикла (такой как циклин-зависимая киназа); (8) ингибитором глюкозо-6-фосфат-дегидрогеназы; (9) антагонистом рецепторов кинин-B1 либо - В2; (10) средством против подагры, например, колхицином; (11) ингибитором ксантин-оксидазы, например, аллопуринолом; (12) средством, способствующим выведению мочевой кислоты, например, пробенецидом, сульфинпиразоном или бензбромароном; (13) стимулятором секреции гормона роста; (14) трансформирующим фактором роста (TGFβ); (15) тромбоцитарным фактором роста (PDGF); (16) фактором роста фибробластов, например, основным фактором роста фибробластов (bFGF); (17) гранулоцитарно-макрофагальным колониестимулирующим фактором (GM-CSF); (18) капсаициновым кремом; (19) антагонистом рецепторов тахикинина NK1 или NK3, выбранным из группы, состоящей из NKP-608C, SB-233412 (талнетант) или D-4418; (20) ингибитором эластазы, выбранным из группы, состоящей из UT-77 или ZD-0892; (21) ингибитором THF-альфа-конвертирующего фермента (ТАСЕ); (22) ингибитором индуцибельной синтазы оксида азота (iNOS); (23) рецептор-гомологичной молекулой для хемоаттрактанта, экспрессированной на ТН2-клетках (такой как антагонист CRTH2); (24) ингибитором Р38; (25) агентом модулирования функционирования Toll-подобных рецепторов (TLR); (26) агентом модулирования активности пуринергических рецепторов, таких как Р2Х7; (27) ингибитором фактора активации транскрипции, такого как NF-κВ, API или STATS.
Соединение по изобретению или его фармацевтически приемлемая соль могут также быть использованы в комбинации с существующим терапевтическим агентом для лечения рака, причем подходящие агенты включают, например:
(1) антипролиферативное/антинеопластическое лекарственное средство или их комбинацию, используемые в медицинской онкологии, такое как алкилирующий агент (например, цисплатин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан или нитрозомочевина); антиметаболит (например, антифолат, такой как фторпиримидин, например 5-фторурацил или тегафур, ралтитрексед, метотрексат, цитозина арабинозид, гидроксимочевина, гемцитабин или паклитаксел); противоопухолевый антибиотик (например, антрациклин, такой как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин или митрамицин); антимитотический агент (например, алкалоид барвинка, такой как винкристин, винбластин, виндезин или винорелбин, или таксоид, такой как таксол или таксотере); или ингибитор топоизомеразы (например, эпиподофиллотоксин, такой как этопозид, тенипозид, амсакрин, топотекан или камптотецин);
(2) цитостатический агент, такой как антиэстроген (например, тамоксифен, торемифен, ралоксифен, дролоксифен или иодоксифен), обратный регулятор эстрогенового рецептора (например, фулвестрант), антиандроген (например, бикалутамид, флутамид, нилутамид или ципротерона ацетат), антагонист LHRH (лютеинизирующий гормон роста) или агонист LHRH (например, гозерелин, леупрорелин или бузерелин), прогестоген (например, мерестрола ацетат), ингибитор ароматазы (например, анастрозол, летрозол, воразол или экземестан) или ингибитор баредуктазы, такой как финастерид;
(3) агент, который ингибирует инвазию раковых клеток (например, ингибитор металлпротеиназ, такой как маримастат, или ингибитор рецепторного функционирования урокиназного активатора плазминогена);
(4) ингибитор функционирования ростовых факторов, например: антитело к ростовому фактору (например, анти-erbb2 антитело трастузумаб или анти-erbb1 антитело цетуксимаб [С225]), ингибитор фарнезилтрансферазы, ингибитор тирозинкиназ или ингибитор серин/треонинкиназ, ингибитор семейства эпидермальных факторов роста (например, ингибитор EGFR семейства тирозинкиназ, такой как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, AZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) или 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)-хиназолин-4-амин (CI 1033)), ингибитор семейства факторов роста тромбоцитарного происхождения или ингибитор семейства факторов роста гепатоцитарного происхождения;
(5) антиангиогенный агент, такой как агент, который ингибирует эффекты васкулярно-эндотелиального фактора роста (например, антитело против васкулярно-эндотелиального клеточного фактора роста бевацизумаб, соединение, описанное в WO 97/22596, WO 97/30035, WO 97/32856 или WO 98/13354), или соединение, которое действует по другому механизму (например, линомид, ингибитор функционирования интегрина αvβ3 или ангиостатин);
(6) агент, поражающий сосуды, такой как комбретастатин А4, или соединение, описанное в WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 или WO 02/08213;
(7) агент, используемый в антисмысловой терапии, например агент, нацеленный на одну из мишеней, указанных выше, такой как ISIS 2503, анти-ras антисмысловой агент;
(8) агент, используемый в методологии генной терапии, например в подходах, заключающихся в замене аберрантных генов, таких как подходы в отношении аберрантного р53 или аберрантного BRCA1 или BRCA2, GDEPT (нацеленная на гены ферментная пролекарственная терапия), такие как подходы с использованием цитозиндезаминазы, тимидинкиназы или бактериальной азотредуктазы, и подходы, состоящие в увеличении устойчивости пациента к химиотерапии или радиотерапии, такие как генная терапия множественной лекарственной устойчивости; или
(9) агент, используемый в иммунотерапевтическом подходе, например ex-vivo и in-vivo методы для увеличения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, методы снижения вялости Т-клеток, методы с использованием трансфицированных иммунных клеток, таких как цитокин-трансфицированные дендритные клетки, методы с использованием цитокин-трансфицированных опухолевых клеточных линий и методы с использованием антиидиотипических антител.
В частности, соединения по изобретению могут быть введены в сочетании со вторым активным ингредиентом, который выбран из:
(а) ингибитора PDE4, включая ингибитор изоформы PDE4D;
(б) агониста (β-адренорецептора, такого как метапротеренол, изопротеренол, изопреналин, альбутерол, сальбутамол, формотерол, сальметерол, тербуталин, орципреналин, битолтерол мезилат, пирбутерол или индакатерол;
(в) антагониста мускариновых рецепторов (например, М1-, М2- или М3-антагониста, такого как селективный М3-антагонист), такого как ипратропия бромид, тиотропия бромид, окситропия бромид, пирензепин, телензепин;
(г) модулятора функционирования хемокинового рецептора (такого как антагонист CCR1- или CCRS-рецепторов);
(д) ингибитора функционирования киназы;
(е) агониста рецептора нестероидного глюкокортикоида;
(ж) агониста рецептора стероидного глюкокортикоида; и
(з) ингибитора протеазы (такого как ММР12 или ММР9 ингибитор).
Настоящее изобретение далее будет дополнительно проиллюстрировано следующими репрезентативными примерами.
Общие способы
1H-ЯМР и 13С-ЯМР спектры записывали на приборе Varian Inova при 400 МГц или Varian Mercury-VX при 300 МГц. Центральные пики хлороформа-d (δH 7,27 м.д.), диметилсульфоксида-d6 (δH 2,50 м.д.), ацетонитрила-d3 (δH 1,95 м.д.) или метанола-d4 (δH 3,31 м.д.) использовали как внутренние стандарты. Колоночную хроматографию осуществляли с использованием силикагеля (0,040-0,063 мм, Merck). Если не указано иначе, исходные вещества имелись в продаже. Все растворители и коммерческие реагенты были лабораторного качества и были использованы в том виде, как они были получены.
Следующий способ использовали для анализа жидкостной хроматографией/масс-спектрометрией (LC/MS).
Прибор Agilent 1100; колонка Waters Symmetry 2,1×30 мм; масс-спектрометрия с химической ионизацией при атмосферном давлении (APCI); скорость потока 0,7 мл/мин; длина волны 254 нм; растворитель А: вода + 0,1% трифторуксусная кислота (TFA); растворитель Б: ацетонитрил + 0,1% TFA; градиент: 15-95%/Б 8 мин, 95% Б 1 мин.
Аналитическую хроматографию проводили на колонке Symmetry С18, 2,1×30 мм, с размером частиц 3,5 мкм. Мобильная фаза ацетонитрил/вода/0,1% трифторуксусная кислота в градиенте от 5% до 95% ацетонитрила в течение 8 минут при токе 0,7 мл/мин.
Сокращения или термины, используемые в примерах, имеют следующие значения:
Пример 1
6-[2-(4-Циано-фенил)-2H-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифтор-метил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты метиламид
6-Бром-N,5-диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид (ИВ2, 0,05 г, 0,128 ммоль), 1-(4-цианофенил)-1Н-пиразол-5-бороновую кислоту (ИВ4, 0,062 г, 0,256 ммоль), Cs2CO3 (0,125 г, 0,384 ммоль) и DME (3 мл) добавляли в стеклянную пробирку для синтеза с использованием микроволнового излучения. Смесь дегазировали азотом и добавляли Pd(PBut 3)2 (0,010 г). Пробирку запаивали и нагревали с перемешиванием при 110°С (150 Вт) в микроволновой печи в течение 10 минут. Смесь разбавляли EtOAc (5 мл) и фильтровали. Раствор концентрировали в вакууме и очищали посредством хроматографии на диоксиде кремния с получением достаточно чистого вещества, которое дополнительно очищали посредством препаративной HPLC (высокоэффективная жидкостная хроматография) с получением 0,012 г (20%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO-D6) δ 8.70 (m, 1H), 7.97-7.84 (m, 6H), 7.77 (d, J=7,8 Гц, 1H), 7.67 (d, J=8,8 Гц, 2Н), 6.76 (d, J=1,8 Гц, 1H), 2.72 (d, J=4,8 Гц, 3H), 1.86 (s, 3H);
APCI-MS m/z: 479,3 [MH+].
Примеры 2 и 3
Аналогично Примеру 1 были синтезированы следующие соединения.
ИВ4
Пример 4
6-[1-(4-Циано-фенил)-1Н-1,2,3-триазол-5-ил]-N,5-диметил-3-оксо-4-[3-(трисрторметил)-фенил]-3,4-дигидропиразин-2-карбоксамид
а) 4-[1,2,3]Триазол-1-ил-бензонитрил
4-Фторбензонитрил (0,847 г, 7 ммоль), 1Н-[1,2,3]триазол (0,483 г, 7 ммоль), Cs2CO3 (2,27 г, 7 ммоль) и DMF (8 мл), а также магнитную мешалку помещали в сосуд. Смесь нагревали с перемешиванием в течение 3 ч при 80°С. Экстрактивная обработка (EtOAc/H2O) и последующая сушка (Na2SO4) давали сырой продукт, который очищали на диоксиде кремния с получением 0,55 г (46%) указанного в заголовке промежуточного соединения.
1H ЯМР (400 МГц, DMSO-D6) δ 9.00 (d, J=1,2 Гц, 1H), 8.18 (d, J=8,8 Гц, 2Н), 8.11 (d, J=8,8 Гц, 2Н), 8.05 (d, J=1,2 Гц, 1Н).
б) 4-(5-Трибутилстаннил-Г1,2.31 триазол-1-ил)-бензонитрил
4-[1,2,3]Триазол-1-ил-бензонитрил (0,105 г, 0,6 ммоль) и сухой THF (6 мл), а также магнитную мешалку помещали в колбу. Колбу заполняли аргоном и хранили в инертной атмосфере и охлаждали до -78°С. При этой температуре по каплям добавляли трет-BuLi (0,36 мл, 1,7 М, 0,6 ммоль) в течение 1-2 минут. Смесь перемешивали при этой температуре в течение 15 минут и в течение 1 минуты добавляли Bu3SnCl (0,19 г, 0,6 ммоль), затем смеси давали медленно нагреться до КТ. Сырую смесь непосредственно очищали на диоксиде кремния (гептан:EtOAc, 4:1) с получением 0,12 г (43%) указанного в заголовке станнана.
APCI-MS m/z: 460 [МН+].
в) 6-[1-(4-Циано-фенил)-1H-1,2.3-триазол-5-ил]-N,5-диметил-3-оксо-4-[3-(трифторметил)-сренил]-3,4-дигидропиразин-2-карбоксамид
4-(5-Трибутилстаннил-[1,2,3]триазол-1-ил)-бензонитрил (0,15 г, 0,32 ммоль), 6-бром-N,5-диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид (ИВ2, 0,054 г, 0,14 ммоль), Pd(PBut 3)2 (10 мг) и DME (2 мл) помещали в пробирку для синтеза с использованием микроволнового излучения. Смесь дегазировали и нагревали в микроволновом реакторе для синтеза (СЕМ) при 100°С (максимум 150 Вт) в течение 10 минут. Растворитель удаляли в вакууме с получением сырого продукта, который очищали на диоксиде кремния и затем дополнительно очищали посредством препаративной HPLC. Чистые фракции сушили замораживанием с получением 27 мг (41%) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-D6) δ 8.64-8.57 (m, 1Н), 8.18 (s, 1H), 8.02 (d, J=8,4 Гц, 2H), 7.98-7.92 (m, 2H), 7.88 (t, J=8,0 Гц, 1H), 7.83 (d, J=8,4 Гц, 2H), 7.77 (d, J=8,0 Гц, 1H), 2.70 (d, J=4,9 Гц, 3H), 1.94 (s, 3H).
APCI-MS m/z: 480,0 [MH+].
Примеры 5-9
Аналогично Примеру 4 были синтезированы следующие соединения.
Пример 10
6-[1-(4-Цианофенил)-1H-пиразол-5-ил]-N,N,5-триметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид
а) 5-Метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоновая кислота
NaOH (1 M, 6 мл) добавляли к метил-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксилату (0,60 г, 1,92 ммоль), растворенному в EtOH (12 мл) и смесь перемешивали в течение 15 минут. Водную фазу нейтрализовали добавлением HCl (1 M, 7 мл) до рН от 6 до 7 и экстрагировали этилацетатом (3×15 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали. Никакой дополнительной очистки не проводили.
APCI-MS т/г: 299,0 [МН+].
б) N,N,5-Триметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид
Смесь 5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоновой кислоты (0,21 г, 0,7 ммоль), HATU (0,266 г, 0,7 ммоль) и Et3N (0,293 г, 2,9 ммоль) в DMF (2 мл) подвергали взаимодействию с диметиламином HCl. Спустя 2 ч реакционную смесь разбавляли водой и экстрагировали этилацетатом (3×5 мл). Органическую фазу сушили (MgSO4), фильтровали и выпаривали. Никакой дополнительной очистки не проводили.
APCI-MS m/z: 326,0 [MH+].
в) 6-Бром-N,N,5-триметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид
N,N,5-Триметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид (0,14 г, 0,43 ммоль) во флаконе растворяли под аргоном в DMF (2 мл). Добавляли N-бромсукцинимид (0,089 г, 0,5 ммоль). Флакон запаивали и осуществляли перемешивание в течение 30 минут.Сырую смесь очищали посредством препаративной HPLC с получением 0,100 г (57%) указанного в заголовке соединения в виде твердого вещества.
APCI-MS m/z: 403,9 [MH+].
г) 6-(3,3-Диэтоксипроп-1-инил)-N,N,5-триметил-3-оксо-4-[3-(трифтор-метил)фенил]-3,4-дигидропиразин-2-карбоксамид
6-Бром-N,N,5-триметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид (0,10 г, 0,247 ммоль), диэтилацеталь пропаргилового альдегида (0,48 мг, 0,370 ммоль), иодид меди(I) (0,001 мг, 0,005 ммоль) и Et3N (1 мл) в THF (1 мл) помещали в стеклянную пробирку для микроволнового синтеза. Смесь дегазировали аргоном и добавляли Pd(Cl2) (PPh3)2 (0,007 г). Пробирку запаивали и нагревали с перемешиванием при 60°С (150 Вт) в микроволновой печи в течение 20 минут. Смесь разбавляли EtOAc (5 мл) и фильтровали. Раствор концентрировали в вакууме и затем очищали на диоксиде кремния с получением указанного в заголовке соединения (28 мг, 25%).
д) 6-[1-(4-Цианофенил)-1H-пиразол-5-ил]-N,N,5-триметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид
6-(3,3-Диэтоксипроп-1-инил)-N,N,5-триметил-3-оксо-4-[3-(трифторметил)-фенил]-3,4-дигидропиразин-2-карбоксамид (0,028 г, 0,062 ммоль) растворяли в DMF (1 мл) в сосуде для микроволнового синтеза. Добавляли 4-цианофенилгидразин гидрохлорид (0,013 г, 0,074 ммоль). Сосуд герметизировали и нагревали с перемешиванием до 120°С в течение 5 минут.Сырую смесь очищали посредством препаративной HPLC с получением 9 мг (29%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (399,99 МГц, CD3CN) δ 7.88 (d, J=7,8 Гц, 1Н), 7.79 (m, 5H), 7.64 (m, 3H), 6.64 (d, J=1,8 Гц, 1Н), 2.90 (d, J=8,8 Гц, 3H), 2.70 (s, 3H), 1.97 (s, 3H).
APCI-MS m/z: 493,0 [MH+].
Пример 11
Аналогично Примеру 10 было синтезировано следующее соединение.
Пример 12
6-[1-(6-Цианопиридин-3-ил)-1H-пиразол-5-ил]-N-циклопропил-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид
а) Метил-6-иод-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксилат
Метил-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксилат (описан в синтезе ИВ2, 1,5 г, 4,8 ммоль), сухой DCM (7,0 мл), трифторуксусную кислоту (3,0 мл) и N-иодсукцинимид (1,0 г, 4,5 ммоль) смешивали и перемешивали при КТ в темноте (колбу накрывали алюминиевой фольгой). Через 5 ч добавляли воду (5 мл) и смесь концентрировали с помощью роторного испарителя. Добавляли еще воду (3 мл) и смесь концентрировали, как описано выше. Полученную смесь разбавляли ацетонитрилом до общего объема 50 мл. Очистка посредством препаративной HPLC со смесью ацетонитрил-вода в качестве элюента (нейтральный элюент) давала 0,905 г (выход 46%) указанного в заголовке соединения в виде желтого кристаллического твердого вещества.
1H ЯМР (400 МГц, DMSO-D6) δ 7.93 (brs, 1Н), 7.92 (d, J=7,6 Гц, 1Н), 7.84 (t, J=7,6 Гц, 1Н), 7.75 (d, J=7,6 Гц, 1Н), 3.82 (s, 3H), 2.14 (s, 3H).
APCI-MS m/z 438,8 (MH+).
б) Метил-6-(3,3-диэтоксипроп-1-инил)-5-метил-3-оксо-4-[3-(трифтор-метил)фенил]-3,4-дигидропиразин-2-карбоксилат
Метил-6-иод-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксилат (1,2 г, 2,8 ммоль), димер аллилпалладий(II)хлорид (0,0072 г), 10% по массе три(трет-бутил)фосфина в гексане (2,1 мл) и безводный DMF (3,0 мл) перемешивали до получения прозрачного раствора. Добавляли диэтилацеталь пропаргилового альдегида (0,44 мл, 3,1 ммоль) в безводном DMF (2,3 мл), затем небольшими порциями 1,4-диазабицикло[2.2.2]октан (0,63 г, 5,6 ммоль). Красный раствор продували сухим аргоном в течение 5 минут и затем перемешивали под аргоном при KT. Через 4 ч растворитель выпаривали с использованием масляного насоса. Остаток переносили в ацетонитрил (10 мл), фильтровали через стеклянную вату и затем концентрировали с помощью диоксида кремния. Хроматография на диоксиде кремния со смесями этилацетат-гептаны (1:10 и 1:2) в качестве элюентов давала 0,46 г (37%) указанного в заголовке соединения в виде желтого масла.
1H ЯМР (400 МГц, CD2Cl2) δ 7.84 (d, J=8,8 Гц, 1Н), 7.77 (d, J=8,0 Гц, 1Н), 7.49 (brs, 1Н), 7.43 (d, J=8,4 Гц, 1Н), 5.47 (s, 1Н), 3.92 (s, 3H), 3.80-3.71 (m, 2H), 3.68-3.58 (m, 2H), 2.20 (s, 3H), 1.23 (t, J=7,2 Гц, 6Н).
APCI-MS m/z 439 (МН+), 393 (М-45).
в) Метил-6-[1-(6-цианопиридин-3-ил)-1H-пиразол-5-ил]-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксилат
Метил-6-(3,3-диэтоксипроп-1-инил)-5-метил-3-оксо-4-[3-(трифтор-метил)-фенил]-3,4-дигидропиразин-2-карбоксилат (0,073 г, 0,17 ммоль) и 5-гидразинопиридин-2-карбонитрил трифторацетат (0,050 г, 0,20 ммоль) в диоксане (3 мл) помещали в сосуд. Добавляли 2 M HCl (0,188 мл) и смесь перемешивали при 55°С в течение 15 минут. После охлаждения добавляли NaHCO3 (0,048 г) и смесь экстрагировали DCM и водой. Объединенные органические фазы промывали водой, рассолом, сушили (Na2SO4) и выпаривали. Остаток растворяли в уксусной кислоте (10 мл) и сосуд герметизировали. Раствор перемешивали при 90°С в течение 10 ч. После выпаривания остаток очищали посредством препаративной HPLC с получением 0,023 г (28%) указанного в заголовке соединения.
APCI-MS m/z: 481,0 [MH+].
г) 6-[1-(6-Цианопиридин-3-ил)-1H-пиразол-5-ил]-N-циклопропил-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид
Метил-6-[1-(6-цианопиридин-3-ил)-1Н-пиразол-5-ил]-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксилат (0,026 г, 0,054 ммоль) в ацетонитриле (0,33 мл) и этаноле (0,074 мл) помещали в сосуд. Добавляли циклопропиламин (0,14 мл, 2,0 ммоль), сосуд герметизировали и перемешивали при 60°С в течение 10 ч. После выпаривания остаток очищали посредством препаративной HPLC с получением 0,010 г (37%) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-D6) δ 8.89 (d, J=2,3 Гц, 1Н), 8.69 (d, J=4,5 Гц, 1H), 8.18-8.09 (m, 2H), 8.01-7.93 (m, 3H), 7.88 (t, J=8,0 Гц, 1Н), 7.79 (d, J=7,9 Гц, 1Н), 6.82 (d, J=1,8 Гц, 1Н), 2.79-2.69 (m, 1Н), 2.03 (s, 3H), 0.71-0.62 (m, 2H), 0.45-0.36 (m, 2H).
APCI-MS m/z: 506,0 [MH+].
Примеры 13-15
Аналогично Примеру 12 были синтезированы следующие соединения.
Пример 16
2-[[6-[2-(4-Цианофенил)пиразол-3-ил]-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидро-пиразин-2-карбонил]амино]уксусная кислота
Указанное в заголовке соединение получали из соединения из Примера 5 после кислотного расщепления трет-бутилового эфира и очистки посредством HPLC.
1H ЯМР (400 МГц, DMSO-D6) δ 12.71 (s, 1Н), 9.25 (t, J=5,6 Гц, 1Н), 7.96 (d, J=6,2 Гц, 2H), 7.94 (d, J=1,8 Гц, 1H), 7.92-7.85 (m, 3H), 7.80 (d, J=8,0 Гц, 1H), 7.68 (d, J=9,3 Гц, 2H), 6.78 (d, J=1,8 Гц, 1Н), 3.94 (d, J=5,5 Гц, 2H), 1.88 (s, 3H).
APCI-MS m/z: 523,3 [MH+].
Получение исходных веществ
Исходные вещества для вышеописанных примеров либо имеются в продаже, либо легко получаются посредством стандартных способов из известных веществ. Например, следующие взаимодействия представляют собой неограничивающие иллюстрации получения некоторых из исходных веществ.
Исходное вещество ИВ1
3-Бром-6-метил-1-[3-(трифторметил)фенил]пиразин-2(1H)-он
3-Трифторметиланилин (5,0 г, 31 ммоль) и триэтиламин (3,54 г, 35 ммоль) растворяли в DCM (60 мл, высушенный). Смесь охлаждали на льду и к перемешиваемому раствору по каплям добавляли раствор этилоксалилхлорида (4,36 г, 32 ммоль) в DCM (15 мл). После завершения добавления реакционную смесь оставляли стоять в течение 10 минут. Реакционную смесь промывали водой (50 мл), затем промывали рассолом (30 мл) и органическую фазу сушили над Na2SO4. После фильтрования и выпаривания получали 8,04 г (99%) этил-оксо{[3-(трифторметил)фенил]амино}ацетата в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-D6) δ 11.09 (s, 1H), 8.19 (s, 1H), 8.03 (d, J=8,0 Гц, 1H), 7.61 (t, J=8,1 Гц, 1H), 7.51 (d, J=7,8 Гц, 1H), 4.32 (q, J=7,5 Гц, 2Н), 1.32 (t=7,0 Гц, 3H);
APCI-MS m/z: 262,0 [МН+].
Этил-оксо{[3-(трифторметил)-фенил]амино}ацетат (8,04 г, 30,7 ммоль) растворяли в этаноле (50 мл, 99,5%). К перемешиваемому раствору одной порцией добавляли 1-амино-2-пропанол (рацемический, 2,32 г, 31 ммоль) и смесь нагревали до температуры дефлегмации в течение 90 минут. Смеси давали охладиться и выпаривали до сухости, получая 8,80 г (99%) N-(2-гидроксипропил)-N'-[3-(трифторметил)-фенил]этандиамид в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-D6) δ 10.99 (bs, 1H), 8.77 (t, J=6,3 Гц, 1H), 8.29 (s, 1H), 8.11 (d, J=8,2 Гц, 1H), 7.60 (t, J=8,1 Гц, 1H), 7.49 (d, J=7,5 Гц, 1H), 4.91 (d, J=4,9 Гц, 1H), 3.78 (p, J=5,7 Гц, 1H), 3.20-3.12 (m, 2Н), 1.05 (d, J=6,3 Гц, 3H);
APCI-MS m/z: 273,1 [MH+].
N-(2-Гидроксипропил)-N'-[3-(трифторметил)фенил]-этандиамид (2,2 г, 7,58 ммоль) растворяли в CH3CN (50 мл) и воде (7 мл). К перемешиваемому раствору добавляли NaBrO3 (1,15 г, 7,58 ммоль) и раствор RuCl3×H2O в CH3CN (3 мл). Смесь перемешивали в течение 1 ч и за реакцией следили посредством LC-MS (сочетание жидкостной хроматографии и масс-спектрометрии) и TLC (тонкослойная хроматография). Органический растворитель удаляли в вакууме и остаток распределяли между DCM (200 мл) и водой (200 мл). Органическую фазу сушили с помощью Na2SO4 и после фильтрования и выпаривания получали 2,0 г (91%) N-(2-оксопропил)-N'-[3-(трифторметил)фенил]-этандиамида в виде грязно-белого твердого вещества.
1H ЯМР (300 МГц, DMSO-D6) δ 11.04 (s, 1H), 9.08 (t, J=6,0 Гц, 1H), 8.29 (s, 1Н), 8.12 (d,J=8,1 Гц, 1Н), 7.61 (t, J=8,1 Гц, 1Н), 7.50 (d, J=7,9 Гц, 1H), 4.09 (d, J=6,0 Гц, 2H), 2.14 (s, 3H).
N-(2-Оксопропил)-N'-[3-(трифторметил)фенил]этандиамид (1,6 г, 5,5 ммоль) и ледяную уксусную кислоту (15 мл) помещали в сосуд (20 мл). К этому раствору добавляли концентрированную серную кислоту (40 капель) и колбу герметизировали и нагревали с перемешиванием до 100°С в течение 90 минут. Добавляли еще 40 капель серной кислоты и реакции давали протекать еще в течение 90 минут. Реакционной смеси давали охладиться и уксусную кислоту удаляли в вакууме. Остаток распределяли между EtOAc (60 мл) и водой (40 мл). Водную фазу нейтрализовали с помощью раствора NaOH до рН 6-7. Органическую фазу сушили и после фильтрования и выпаривания получали сырой продукт, который очищали на диоксиде кремния с получением 1,1 г (74%) 6-метил-1-[3-(трифторметил)фенил]-1,4-дигидропиразин-2,3-диона в виде желтоватого твердого вещества.
1H ЯМР (400 МГц, DMSO-D6) δ 11.24 (bs, 1Н), 7.87-7.81 (m, 2H), 7.77 (t, J=7,8 Гц, 1H), 7.67 (d, J=7,8 Гц, 1H), 6.30 (d, J=5,2 Гц, 1H), 1.61 (d, J=1,1 Гц, 3H);
APCl-MS m/z: 271,0 [MH+].
6-Метил-1-[3-(трифторметил)фенил]-1,4-дигидропиразин-2,3-дион (0,52 г, 1,92 ммоль) и 1,2-дихлорэтан (10 мл) помещали в сосуд (20 мл). К полученной суспензии осторожно добавляли оксалилбромид (0,53 мл, 1,24 г, 5,75 ммоль). В процессе добавления образовывалась пена, и как только эта пена оседала, начинали перемешивание. Добавляли DMF (3 капли) и сосуд герметизировали и смесь перемешивали в течение ночи. Добавляли еще одну порцию оксалилбромида (0,2 мл, 0,46 г, 2,23 ммоль) и DMF (3 капли) и реакцию проводили еще в течение 24 ч. Смесь распределяли между DCM (20 мл) и водой (20 мл) и органическую фазу сушили. В результате фильтрования и выпаривания получали сырой продукт, который очищали на диоксиде кремния, получая 0,59 г (93%) 3-бром-6-метил-1-[3-(трифторметил)фенил]пиразин-2(1Н)-она.
1H ЯМР (400 МГц, DMSO-D6) δ 7.96 (s, 1Н), 7.92 (d, J=7,5 Гц, 1Н), 7.83 (t, J=7,5 Гц, 1H), 7.77 (d, J=7,5 Гц, 1H), 7.27 (s, 1H), 1.84 (s, 3H);
APCI-MS m/z: 232,9 и 234,9 [MH+].
Исходное вещество ИВ2
6-Бром-N,5-диметил-3-оксо-4-[3-(трисрторметил)фенил]-3,4-дигидро-пиразин-2-карбоксамид
В стальной реактор высокого давления (Парра) с подводом газообразного СО загружали 3-бром-6-метил-1-[3-(трифторметил)фенил]-пиразин-2(1Н)-он (ИВ1, 0,25 г, 0,75 ммоль), Pd(OAc)2 (0,015 г, 0,067 ммоль), PPh3 (0,030 г, 0,11 ммоль) и метанол (25 мл). К этой смеси добавляли триэтиламин (0,5 мл, 0,36 г, 3,6 ммоль) и магнитную мешалку. Реактор продували СО и в системе создавали давление СО, равное 6 атмосферам (прибл. 600 кПа). Реактор нагревали с перемешиванием до 90°С, смесь интенсивно перемешивали и реакции давали протекать в течение 4 ч. Летучие вещества удаляли в вакууме и сырой продукт очищали на диоксиде кремния с получением 0,11 г (47%) метил-5-метил-3-оксо-4-[3-(трифторметил)-фенил]-3,4-дигидропиразин-2-карбоксилат в виде твердого вещества.
1H ЯМР (400 МГц, DMSO-D6) δ 7.97 (s, 1H), 7.92 (d, J=7,5 Гц, 1Н), 7.83 (t, J=7,5 Гц, 1H), 7.77 (d, J=7,5 Гц, 1H), 7.52 (s, 1 H), 3.80 (s, 3H), 1.94 (s, 3H);
APCI-MS m/z: 310,0 [МН+].
Метил-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксилат (0,11 г, 0,35 ммоль) растворяли в растворе метиламина (33% в этаноле, 5 мл) в сосуде. Сосуд герметизировали и нагревали с перемешиванием при 50°С в течение 30 минут. Летучие вещества удаляли в вакууме с получением сырого продукта, который очищали для аналитических целей посредством препаративной HPLC с получением 0,079 г (73%) N,5-диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамида в виде твердого вещества.
1H ЯМР (400 МГц, DMSO-D6) δ 8.95 (m, 1H), 7.98-7.90 (m, 2H), 7.84 (t, J=7,7 Гц, 1H), 7.76 (d, J=7,7 Гц, 1H), 7.64 (s, 1H), 2.78 (d, J=4,7 Гц, 3H), 1.98 (s, 3H);
APCI-MS m/z: 312,0 [МН+].
N,5-Диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид (0,079 г, 0,25 ммоль) растворяли в DMF (1,5 мл) в сосуде. Добавляли N-бромсукцинимид (0,066 г, 0,38 ммоль). Сосуд герметизировали и нагревали с перемешиванием до 50°С в течение 90 минут. Сырую смесь по каплям добавляли к воде (20 мл) при перемешивании с помощью магнитной мешалки. Осадок отделяли фильтрованием с получением 0,078 г (80%) 6-бром-N,5-диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамида в виде твердого вещества.
1H ЯМР (400 МГц, DMSO-D6) δ 8.96 (m, 1H), 7.98-7.91 (m, 2H), 7.86 (t, J=8,2 Гц, 1H), 7.77 (d, J=8,2 Гц, 1H), 2.79 (d, J=4,8 Гц, 3H), 2.12 (s, 3H);
APCI-MS m/z: 389,9 и 391,9 [MH+].
Исходное вещество ИВ3
6-Бром-5-метил-N-{[5-(метилсульфонил)пиридин-2-ил]метил}-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид
В стальной реактор высокого давления (Парра) с подводом газообразного СО загружали 3-бром-6-метил-1-[3-(трифторметил)фенил]-пиразин-2(1Н)-он (ИВ1, 0,30 г, 0,89 ммоль), Pd(OAc)2 (0,035 г, 0,16 ммоль), PPh3 (0,070 г, 0,26 ммоль) и 5-метансульфонил-пиридин-2-иламин (0,46 г, 1,79 ммоль) в метаноле (25 мл). К этой смеси добавляли триэтиламин (1,5 мл, 1,09 г, 10 ммоль) и магнитную мешалку. Реактор продували СО и в системе создавали давление СО, равное 6 атмосферам (прибл. 600 кПа). Реактор нагревали до 90°С, смесь интенсивно перемешивали и реакции давали протекать в течение 4 ч, затем давали охладиться. Летучие примеси удаляли в вакууме и сырой продукт очищали посредством препаративной HPLC, что давало 0,22 г (53%) 5-метил-N-{[5-(метилсульфонил)-пиридин-2-ил]метил}-3-оксо-4-[3-(трифторметил)-фенил]-3,4-дигидропиразин-2-карбоксамида в виде белого твердого вещества после сушки замораживанием чистых фракций.
1H ЯМР (400 МГц, DMSO-D6) δ 9.81 (t, J=5,8 Гц, 1H), 8.99 (d, J=2,15 Гц, 1H), 8.29 (dd, J=8,3 и 2,3 Гц, 1H), 7.98 (s, 1H), 7.94 (d, J=7,9 Гц, 1H), 7.86 (t, J=7,9 Гц, 1H), 7.80 (d, J=7,9 Гц, 1H), 7.71 (s, 1H), 7.61 (d, J=8,2 Гц, 1H), 4.71 (d, J=5,8 Гц, 2H), 3.29 (s, 3H), 2.01 (s, 3H);
APCI-MS m/z: 467,0 [MH+].
5-Метил-N-{[5-(метилсульфонил)-пиридин-2-ил]метил}-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамид (0,097 г, 0,21 ммоль) растворяли в DMF (2 мл) в сосуде. К этой смеси добавляли N-бромсукцинимид (0,055 г, 0,31 ммоль), сосуд герметизировали и смесь нагревали при 50°С с перемешиванием в течение 1 ч. Сырую смесь по каплям добавляли к воде (40 мл) при перемешивании с помощью магнитной мешалки. Осадок отделяли фильтрованием с получением 0,105 г (92%) 6-бром-5-метил-N-{[5-(метилсульфонил)пиридин-2-ил]метил}-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамида в виде твердого вещества.
1H ЯМР (400 МГц, DMSO-D6) δ 9.80 (t, J=6,0 Гц, 1Н), 8.99 (d, J=2,10 Гц, 1H), 8.29 (dd, J=8,2 и 2,3 Гц, 1Н), 8.00-7.94 (m, 2H), 7.88 (t, J=8,3 Гц, 1Н), 7.79 (d, J=8,3 Гц, 1Н), 7.61 (d, J=8,3 Гц, 1Н), 4.72 (d, J=5,7 Гц, 2H), 3.30 (s, 3H), 2.15 (s, 3H);
APCI-MS m/z: 544,9 и 546,9 [МН+].
Исходное вещество ИВ4
[1-(4-Цианофенил)-1H-пиразол-5-ил]бороновая кислота
4-(1Н-Пиразол-1-ил)бензонитрил (Eur. J. Org. Chem. 2004, 695-709) (1,5 г, 8,87 ммоль) в сухом THF (50 мл) под аргоном перемешивали при -78°С, пока в течение 20 минут добавляли диизопропиламид лития (1,8 М раствор в смеси THF/гексан/этилбензол; 5,2 мл, 9,32 ммоль). Перемешивание и охлаждение продолжали в течение 1 ч, по каплям в течение 30 минут добавляли триизопропилборат (8 мл, 34,5 ммоль) и затем температуре давали подниматься в течение ночи до КТ. Величину рН реакционной смеси доводили до 5 с помощью 1 М HCl и смесь затем концентрировали до минимального объема и экстрагировали этилацетатом (200 мл) и рассолом (3×100 мл). Органическую фазу собирали, сушили (Na2SO4), фильтровали и выпаривали до коричневого твердого вещества (1,32 г), которое использовали на следующей стадии без дополнительной очистки.
APCI-MS m/z: 214,1 [МН+].
Анализ с человеческой нейтрофильной эластазой с использованием переноса энергии угасающего флуоресцентного резонанса (Quenched-FRET)
В анализе используют человеческую нейтрофильную эластазу (HNE), очищенную от сыворотки (Calbiochem art. 324681; Ref. Baugh, R.J. et al., 1976, Biochemistry. 15, 836-841). HNE хранили в 50 мМ ацетата натрия (NaOAc), 200 мМ хлорида натрия (NaCl), рН 5,5, с добавлением 30% глицерина при -20°С. Использовали протеазный субстрат Elastase Substrate V Fluorogenic, MeOSuc-AAPV-AMC (Calbiochem art. 324740; Ref. Castillo, M.J. et al., 1979, Anal. Biochem. 99, 53-64). Субстрат хранили в диметилсульфоксиде (DMSO) при -20°С. Добавления в анализе осуществляли следующим образом: тестируемые соединения и контроли добавляли в черные 96-луночные планшеты с плоским дном (Greiner 655076), 1 мкл в 100% DMSO, далее 30 мкл HNE в опытном буфере с 0,01% Triton (товарный знак) Х-100 детергентом. Состав опытного буфера: 100 мМ трис(гидроксиметил)аминометана (TRIS) (рН 7,5) и 500 мМ NaCl. Фермент и соединения инкубировали при комнатной температуре в течение 15 минут. Затем добавляли 30 мкл субстрата в опытный буфер. Опытную смесь инкубировали в течение 30 минут при комнатной температуре. Концентрация фермента HNE и субстрата во время инкубирования составляла 1,7 нМ и 100 мкМ соответственно. Анализ затем останавливали добавлением 60 мкл раствора для остановки (140 мМ уксусной кислоты, 200 мМ монохлорацетата натрия, 60 мМ ацетата натрия, рН 4,3). Измеряли флуоресценцию на приборе Wallac 1420 Victor 2 при следующих установках: возбуждение 380 нм, эмиссия 460 нм. Значения IC50 (50%-ная ингибирующая концентрация) определяли с помощью аппроксимации кривой XIfit, используя модель 205.
При тестировании с помощью вышеописанного скрининга, соединения из Примеров демонстрировали значения IC50 в отношении ингибирования активности человеческой нейтрофильной эластазы менее чем 30 мкМ (микромоль), что указывает на то, что соединения по изобретению, как полагают, обладают полезными терапевтическими свойствами. Конкретные результаты представлены в следующей Таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИДЕНТИФИКАЦИЯ МУТАЦИИ LKB1 В КАЧЕСТВЕ ПРОГНОСТИЧЕСКОГО БИОМАРКЕРА ЧУВСТВИТЕЛЬНОСТИ К ИНГИБИТОРАМ TOR-КИНАЗЫ | 2011 |
|
RU2565034C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| ЗАМЕЩЕННЫЕ АМИДЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, ЧУВСТВИТЕЛЬНОГО К Btk, СПОСОБ ПОВЫШЕНИЯ ЧУВСТВИТЕЛЬНОСТИ РАКОВЫХ КЛЕТОК К ХИМИОТЕРАПИИ, СПОСОБ УМЕНЬШЕНИЯ ОШИБОК ПРИ ПРИЕМЕ ЛЕКАРСТВА И УЛУЧШЕНИЯ СОБЛЮДЕНИЯ СХЕМЫ ЛЕЧЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ ГИДРОЛИЗА АТФ, СПОСОБ ОПРЕДЕЛЕНИЯ ПРИСУТСТВИЯ Btk В ОБРАЗЦЕ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ В-КЛЕТОК | 2008 |
|
RU2470923C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2666538C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[3, 4-b]ПИРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), ПРИМЕНЕНИЕ (ВАРИАНТЫ), КОМПОЗИЦИЯ (ВАРИАНТЫ) | 2003 |
|
RU2357967C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| НОВЫЕ АНТИГЕЛЬМИНТНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2828007C2 |
| НОВЫЕ СОЕДИНЕНИЯ - АНТАГОНИСТЫ РЕЦЕПТОРА НЕЙРОКИНИНА 1 | 2013 |
|
RU2631319C2 |
| N-((ГЕТ)АРИЛМЕТИЛ)-ГЕТЕРОАРИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПЛАЗМЕННОГО КАЛЛИКРЕИНА | 2015 |
|
RU2821520C2 |
Изобретение относится к соединениям формулы (I)
,
где R1 представляет собой C1-С6алкил; W представляет собой пиразолил, триазолил или имидазолил; R14 представляет собой фенил или 6-членное гетероароматическое кольцо, содержащее от 1 до 3 кольцевых атомов азота, которое возможно замещено по меньшей мере одним заместителем, выбранным из F, Cl, CN и СF3; R3 представляет собой фенил, замещенный трифторметильным заместителем; R4 представляет собой водород или C1-С6алкил; Х представляет собой группу -C1-С6алкилен-Y- и Y представляет собой простую связь, а алкиленовая группировка представляет собой линейный или разветвленный C1-С6алкилен, возможно замещенный ОН, CO2R66 или C1-С3алкокси; R5 представляет собой фенил или пиридинил, замещенный -S(O)vR21; или R5 представляет собой незамещенное С3-С6циклоалкильное кольцо; или R5 также может представлять собой Н; R21 представляет собой водород, C1-С6алкил или С3-С8циклоалкил; v равен 1 или 2; и R66 представляет собой водород или C1-С6алкил; или к его фармацевтически приемлемым солям. Изобретение также относится к способу получения указанных соединений, к промежуточным соединениям и к фармацевтической композиции для лечения или снижения риска заболевания или состояния, при котором полезно ингибирование активности нейтрофильной эластазы, на основе соединений формулы (I). Технический результат - получены новые соединения, которые могут найти применение в медицине для лечения или снижения риска заболевания или состояния, при котором полезно ингибирование активности нейтрофильной эластазы, например, таких как респираторный дистресс-синдром взрослых (ARDS), цистический фиброз, легочная эмфизема, бронхит и др. 5 н. и 14 з.п. ф-лы, 1 табл., 16 пр.
1. Соединение формулы (I)
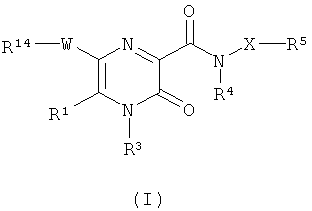
где R1 представляет собой C1-С6алкил;
W представляет собой пиразолил, триазолил или имидазолил;
R14 представляет собой фенил или 6-членное гетероароматическое кольцо, содержащее от 1 до 3 кольцевых атомов азота, которое возможно замещено по меньшей мере одним заместителем, выбранным из F, Cl, CN и СF3;
R3 представляет собой фенил, замещенный трифторметильным заместителем;
R4 представляет собой водород или С1-С6алкил;
Х представляет собой группу -С1-С6алкилен-Y-, и Y представляет собой простую связь, а алкиленовая группировка представляет собой линейный или разветвленный C1-С6алкилен, возможно замещенный ОН, CO2R66 или C1-С3алкокси;
R5 представляет собой фенил или пиридинил, замещенный -S(O)vR21; или
R5 представляет собой незамещенное С3-С6циклоалкильное кольцо;
или R5 также может представлять собой Н;
R21 представляет собой водород, C1-С6алкил или С3-С8циклоалкил;
v равен 1 или 2; и
R66 представляет собой водород или C1-С6алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где группа R14 и пиразиноновое кольцо соединены с 5-членным кольцом W в положениях 1, 2.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где R14 представляет собой фенильную или пиридинильную группу, возможно замещенную одним или двумя заместителями, независимо выбранными из F, Сl, СN и СF3.
4. Соединение по п.1 или его фармацевтически приемлемая соль, где Х представляет собой незамещенный С1-С2алкилен.
5. Соединение по п.1 или его фармацевтически приемлемая соль, где R5 представляет собой Н.
6. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 представляет собой метил;
W представляет собой пиразолильное или триазолильное кольцо, и группа R14 и 2-пиразиноновое кольцо присоединены к 5-членному кольцу W в 1, 2 положениях;
R14 представляет собой фенил или пиридинил; причем указанное кольцо замещено в 4-(пара)-положении F, Cl или CN;
R3 представляет собой фенильную группу, замещенную в мета-положении СF3;
R4 представляет собой водород;
Х представляет собой линейный или разветвленный С1-С4алкилен, возможно замещенный ОН, CO2R66 или С1-С3алкокси;
R66 представляет собой водород или метил; и
R5 представляет собой Н.
7. Соединение формулы (I) по п.1, выбранное из:
6-[2-(4-цианофенил)-2Н-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты метиламида;
6-[2-(4-цианофенил)-2Н-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты (5-метансульфонил-пиридин-2-илметил)-амида;
6-[2-(4-цианофенил)-2Н-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты этиламида;
6-[1-(4-цианофенил)-1Н-1,2,3-триазол-5-ил]-N,5-диметил-3-оксо-4-[3-(трифторметил)-фенил]-3,4-дигидропиразин-2-карбоксамида;
трет-бутил-2-[[6-[2-(4-цианофенил)пиразол-3-ил]-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидро-пиразин-2-карбонил]амино]ацетата;
6-[3-(4-хлор-фенил)-3Н-[1,2,3]триазол-4-ил]-5-метил-3-оксо-4-(3-трифторметил)-фенил)-3,4-дигидропиразин-2-карбоновой кислоты метиламида;
6-[2-(4-хлорфенил)-2Н-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты метиламида;
6-[1-(4-цианофенил)-1Н-пиразол-5-ил]-N-(2-метоксиэтил)-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамида;
6-[1-(4-цианофенил)-1Н-пиразол-5-ил]-N-(2-гидрокси-1,1-диметилэтил)-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамида;
6-[1-(4-цианофенил)-1Н-пиразол-5-ил]-N,N,5-триметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамида;
6-[1-(6-цианопиридин-3-ил)-1Н-пиразол-5-ил]-N,5-диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамида;
6-[1-(5-цианопиридин-2-ил)-1Н-пиразол-5-ил]-N,5-диметил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидропиразин-2-карбоксамида; и
2-[[6-[2-(4-цианофенил)пиразол-3-ил]-5-метил-3-оксо-4-[3-(трифторметил)фенил]-3,4-дигидро-пиразин-2-карбонил]амино]уксусной кислоты;
и фармацевтически приемлемых солей любого из вышеперечисленного.
8. Соединение формулы (I) по п.1, которое представляет собой 6-[2-(4-цианофенил)-2Н-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты метиламид, или его фармацевтически приемлемая соль.
9. Соединение формулы (I) по п.1, которое представляет собой 6-[2-(4-цианофенил)-2Н-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты (5-метансульфонил-пиридин-2-илметил)-амид, или его фармацевтически приемлемая соль.
10. Соединение формулы (I) по п.1, которое представляет собой 6-[2-(4-цианофенил)-2Н-пиразол-3-ил]-5-метил-3-оксо-4-(3-трифторметил-фенил)-3,4-дигидропиразин-2-карбоновой кислоты этиламид, или его фармацевтически приемлемая соль.
11. Соединение формулы (I) по п.1, которое представляет собой 6-[1-(4-цианофенил)-1Н-1,2,3-триазол-5-ил]-N,5-диметил-3-оксо-4-[3-(трифторметил)-фенил]-3,4-дигидропиразин-2-карбоксамид, или его фармацевтически приемлемая соль.
12. Способ получения соединения формулы (I) или его фармацевтически приемлемой соли по п.1, который включает (а) взаимодействие соединения формулы (II)
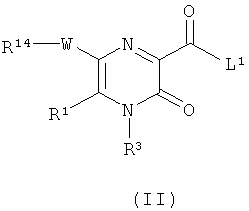
где L1 представляет собой уходящую группу (такую как галоген или гидроксил), и R1, R3, R14 и W являются такими, как определено в формуле (I),
с соединением формулы
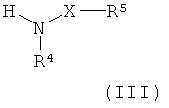
где X, R4 и R5 являются такими, как определено в формуле (I); или (б) взаимодействие соединения формулы (IV)
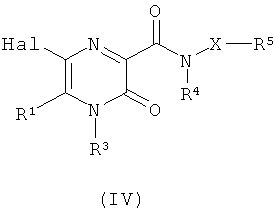
где Hal представляет собой атом галогена, и X, R1, R3, R4 и R5 являются такими, как определено в формуле (I),
с нуклеофилом R14-W-M, где R14 и W являются такими, как определено в формуле (I), и М представляет собой оловоорганическую группу или группу органобороновой кислоты;
и, возможно, после стадий (а) или (б) проведение одной или более из следующих стадий:
- превращение полученного соединения в другое соединение по изобретению,
- получение фармацевтически приемлемой соли соединения.
13. Промежуточное соединение формулы (IV)
где Hal представляет собой атом галогена, и X, R1, R3, R4 и R5 являются такими, как определено по п.1.
14. Фармацевтическая композиция для лечения или снижения риска заболевания или состояния, при котором полезно ингибирование активности нейтрофильной эластазы, содержащая соединение формулы (I) или его фармацевтически приемлемую соль по любому из пп.1-11 совместно с фармацевтически приемлемым адъювантом, разбавителем или носителем.
15. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-11 для применения в терапии.
16. Способ лечения или снижения риска заболевания или состояния, при котором полезно ингибирование активности нейтрофильной эластазы, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-11.
17. Способ по п.16, где заболевание или состояние представляет собой респираторный дистресс-синдром взрослых (ARDS), цистический фиброз, легочную эмфизему, бронхит, включая хронический бронхит, бронхоэктаз, легочную гипертензию, астму, включая рефрактивную астму, ринит, псориаз, ишемическо-реперфузионное повреждение, ревматоидный артрит, остеоартрит, синдром системного воспалительного ответа (SIRS), длительно незаживающие раны, рак, атеросклероз, пептические язвы, болезнь Крона, неспецифический язвенный колит или повреждение слизистой оболочки желудка.
18. Способ по п.17, где заболевание или состояние представляет собой цистический фиброз.
19. Способ по п.17, где заболевание или состояние представляет собой бронхоэктаз.
| WO 2005026123 A1, 24.03.2005 | |||
| WO 2005026124 A1, 24.03.2005 | |||
| WO 2004043924 A1, 27.05.2004 | |||
| WO 2005021512 A1, 10.03.2005 | |||
| RU 2005113168 A, 20.01.2006. |

