Изобретение относится к способу получения 5,8-дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинона (1) - полупродукта в синтезе 2,5,6,7,8-пентагидрокси-3-этил-1,4-нафтохинона (эхинохрома А) (2), являющегося действующим началом кардиопротекгорного и офтальмологического препарата гистохром [RU 2137472 С1, 10.08.99, RU 2134107 C1, 20.09.99].

Известен способ получения целевого продукта (1), заключающийся в окислении 5,8-дигидрокси-6,7-дихлор-1,4-нафтохинона (3) диоксидом марганца (MnO2) в конц. H2SO4, свободнорадикальном С-этилировании полученного 2,5,8-тригидрокси-6,7-дихлор-1,4-нафтохинона (4) пропионилпероксидом (EtCOO)2 в кипящем трет-бутаноле, О-алкилировании образующегося при этом 2,5,8-тригидрокси-6,7-дихлор-3-этил-1,4-нафтохинона (5) кипящим триэтилортоформиатом и замещении атомов хлора в полученном 5,8-дигидрокси-6,7-дихлор-3-этил-2-этокси-1,4-нафтохиноне (6) действием системы реагентов MeOH-K(Cs)F-Al2О3 с добавлением диметиловых эфиров этиленгликоля или диэталенгликоля (схема 1) [RU 2203265 С2, 27.04.2003] или без добавления этих сорастворителей [SU 1821022 A3, 07.06.93]. В ходе последнего превращения (6→1) происходит замещение этоксирадикала в положении 2 на метоксифункцию. Выход желаемого продукта (1) составил при этом около 20%.
Схема 1
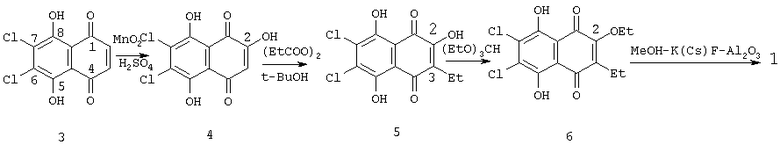
Необходимо отметить, что исходный субстрат (3) является дорогостоящим продуктом (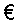 18.0 за 1 г [5, Aldrich Catalog 2003-2004, р.608]). Кроме того, недостатком этого способа является использование пропионилпероксида, технологически неприемлемого из-за своей взрывоопасности, а также дорогостоящего и пожароопасного триэтилортоформиата.
18.0 за 1 г [5, Aldrich Catalog 2003-2004, р.608]). Кроме того, недостатком этого способа является использование пропионилпероксида, технологически неприемлемого из-за своей взрывоопасности, а также дорогостоящего и пожароопасного триэтилортоформиата.
Другим способом получения целевого продукта (1) является последовательность реакций 9→5→6→1, представленная на схеме 2 [RU 1821023 A3, 07.06.93]. Исходным субстратом в этом способе является 1,2,4-триметокси-3-этилбензол (9), который вводится в реакцию циклоацилирования дихлормалеиновым ангидридом (ДХМА) (10) в расплаве AlCl3-NaCl, давая 2,5,8-тригидрокси-6,7-дихлор-3-этил-1,4-нафтохинон (5). Далее конверсия 5→6→1 осуществляется согласно способу [SU 1821022] или (на стадии 6→1) согласно способу [RU 2203265]. Полупродукт (9) не является коммерчески доступным и, в свою очередь, получается литиированием 1,2,4-триметоксибензола (7) в положение 3 действием бутиллития в абс. тетрагидрофуране (ТГФ) с последующим этилированием полученного литиевого производного (8) действием диэтилсульфата [Изв. АН, Сер. хим. 1999, N5, с.947-952]. Выход целевого продукта (1) с учетом превращения 7→8→9, составляет при этом около 21%.
Схема 2
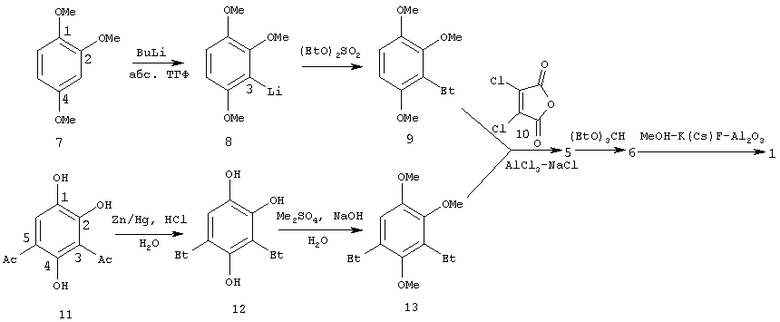
Недостатком этого способа является использование полупродукта (9), при получении которого применяется раствор бутиллития в абс. ТГФ. Известно, что растворы алкиллития в абсолютных органических растворителях являются чрезвычайно пожароопасными реагентами, и их приготовление сопряжено с известными трудностями [Уэйкфилд Б. Методы синтеза с использованием литийорганических соединений. М.: Мир. 1991. 183 с.] Кроме того, в указанном способе используется дорогостоящий и пожароопасный триэтилортоформиат.
Недавно предложен способ получения 2,5,8-тригидрокси-6,7-дихлор-3-этил-1,4-нафтохинона 5 (схема 2, реакции 11→12→13→5), который во многом свободен от недостатков, присущих описанному выше способу, в части получения этого полупродукта (схема 2, реакции 7→8→9→5) [RU 2193550 С1, 27.11.2002]. Исходным субстратом в этом способе является 3,5-диацетил-1,2,4-тригидроксибензол (11), который восстанавливается до 1,2,4-тригидрокси-3,5-диэтилбензола (12) действием амальгамы цинка в водном растворе соляной кислоты, последний метилируется действием диметилсульфата в водном растворе NaOH, давая 1,2,4-триметокси-3,5-диэтилбензол (13). Полупродукт 13 вводится в реакцию циклоацилирования ДХМА (10) в расплаве А1С13-NaCl, давая 2,5,8-тригидрокси-6,7-дихлор-3-этил-1,4-нафтохинон (5). В итоге выход целевого продукта (1) с учетом последующей конверсии 5→6→1 составил 21%. В качестве прототипа выбран последний способ получения целевого продукта 1 (схема 2, реакции 11→12→13→5→6→1).
Недостатком способа-прототипа является использование дорогостоящего и пожароопасного триэтилортоформиата.
Задача изобретения - разработка способа получения 5,8-дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинона (1), в котором используют исходное вещество, получаемое способом, не требующим применения пожароопасного триэтилортоформиата; повышение выхода целевого продукта.
Суть решения задачи заключается в том, что в предлагаемом способе получения 5,8-дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинона (1) кислородсодержащая функция (МеО) в положении 2, как и метоксигруппы в положениях 6,7, формируются на последней стадии получения целевого продукта, что делает ненужным использование триэтилортоформиата.
Схема 3
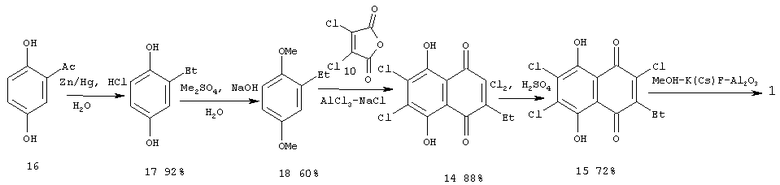
Задача решена тем, что в способе получения 5,8-дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинона замещением атомов хлора метоксигруппами в хлорированном производном 1,4-нафтохинона действием реагента МеОН-CsF-Al2О3 или MeOH-CsF-Al2О3 в качестве производного 1,4-нафтохинона используют 5,8-дигидрокси-2,6,7-трихлор-3-этил-1,4-нафтохинон, получаемый путем восстановления 2-ацетил-1,4-дигидроксибензола, метилирования образующегося 1,4-дигидрокси-2-этилбензола, циклоацилирования образующегося при этом 1,4-диметокси-2-этилбензола дихлормалеиновым ангидридом с последующим хлорированием полученного 5,8-дигидрокси-6,7-дихлор-3-этил-1,4-нафтохинона.
Предлагаемый способ получения целевого продукта 1 представлен на схеме 3.
В качестве исходного субстрата авторы используют соединение (15), а именно 5,8-дигидрокси-2,6,7-трихлор-3-этил-1,4-нафтохинон, которое впервые получено путем восстановления ацетилгидрохинона (16), метилирования образующегося при этом этилгидрохинона (17), циклоацилирования полученного диметилового эфира этилгидрохинона (18) дихлормалеиновым ангидридом с последующими хлорированием полученного при этом 5,8-дигидрокси-6,7-дихлор-3-этил-1,4-нафтохинона (14).
Выход желаемого продукта (1) составляет при этом 24%.
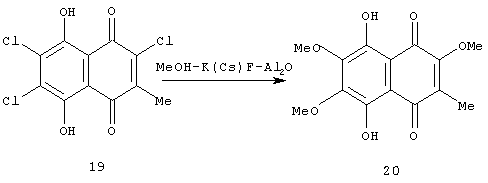
Необходимо отметить, что выход 5,8-дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинона - (1) в реакции нуклеофильного замещения атомов хлора в предлагаемом способе, при действии системы реагентов MeOH-CsF-Al2O3 на 5,8-дигидрокси-2,6,7-трихлор-3-этил-1,4-нафтохинон (15), составляет 68% и практически равен выходу того же продукта при действии MeOH-CsF-Al2O3 на 5,8-дигидрокси-6,7-дихлор-3-этил-2-этокси-1,4-нафтохинон (6) (72%), как описано в способе-прототипе. Однако этот результат не является очевидным, поскольку действие того же реагента на ближайший аналог полупродукта (15) - 5,8-дигидрокси-2,6,7-трихлор-3-метил-1,4-нафтохинон (19), дает соответствующий триметиловый эфир (20) с выходом лишь 8% (схема 4).
Схема 4
Технический результат, обеспечиваемый изобретением, заключается в снижении пожароопасности способа, т.к. предлагаемый способ исключает использование триэтилортоформиата; в упрощении способа, т.к. большинство реакций проводится в водных растворах (16→17→18; 14→15), при обработке реакционных смесей и выделении полупродуктов (17, 18, 14, 15) используются водные растворы или негорючие растворители, что также снижает пожароопасность всего процесса; в результате оптимизации условий на каждой стадии достигнуты достаточно высокие выходы полупродуктов, что в итоге приводит к относительному повышению выхода целевого продукта (на 11,5%) в расчете на исходный субстрат. Большинство используемых реагентов относительно дешевы. Достигнута высокая степень унификации схемы получения целевого продукта (1) по используемым реагентам и оборудованию, что позволяет снизить затраты на его производство.
Сведения, подтверждающие возможность осуществления изобретения:
1,4-Дигидрокси-2-этилбензол (17). В 3-литровую колбу, снабженную механической мешалкой и обратным холодильником, помещают 600 г твердой амальгамы цинка [Корякин Ю.В., Ангелов И.И. Чистые химические вещества. М.: Химия. 1974. С.398], 30 г (0.2 моль) 2-ацетил-1,4-дигидроксибензола (16) [Dictionary of Organic Compounds. Ed.J.Buckingham, 5th ed. New York - London - Toronto: Chapman and Hall. 1982. V.2. P.1093] и 375 мл конц. HCl. Энергично перемешивая, реакционную смесь нагревают до кипения и, спустя 30 мин, порциями, добавляют еще 30 г (0.2 моль) субстрата 16 и 375 мл конц. HCl. Смесь кипятят при перемешивании 3 ч, горячий раствор декантируют и оставляют на ночь. Образовавшийся на поверхности раствора при охлаждении верхний твердый слой и выпавший белый осадок отделяют фильтрованием, промывают небольшим (30 мл) количеством ледяной воды и высушивают в вакуумном эксикаторе до постоянного веса. Получают 51 г (92%) 1,4-дигидрокси-2-этилбензола (17). ИК-спектр (CDCl3), ν/см-1: 3604 (ОН), 1602,1504 (C=С). Спектр ЯМР 1H (ацетон-d6): 1.16 т (3Н, J 7.6, СН3), 2.57 к (2Н, J 7.6, СН2), 6,49 д.д (1Н, J 8.5,. J 2.9, Н(5)), 6.61 д (1H,. J 2.9, Н(3)), 6.65 д (1Н, J 8.5, Н(6)), 7.59 с (1Н, ОН), 7.64 с (1Н, ОН). Масс-спектр (ЭУ), m/z (Iотн (%)): 138 [M]+ (100), 123 (51). Полученный сырой продукт 17 используют для получения 1,4-диметокси-2-этилбензола (18) без очистки.
1,4-Диметокси-2-этилбензол (18) В 0.5 литровую колбу, снабженную механической мешалкой, газоподводящей трубкой и капельной воронкой, помещают 50.0 г (0.36 моль) этилгидрохинона (17), 95.4 г (0.76 моль) диметилсульфата, 43 мл воды и при интенсивном перемешивании в атмосфере аргона в течение 4 ч по каплям добавляют 80 мл (0.5 моль) 25% раствора NaOH, поддерживая температуру реакционной смеси в интервале 20-25°С. Перемешивают еще 1 ч при комнатной температуре и 1 ч нагревают реакционную смесь на кипящей водяной бане. Охлаждают, подкисляют серной кислотой до рН 3, отделяют верхний органический слой, водную фазу экстрагируют 4×100 мл диэтиловым эфиром. Объединяют эфирные вытяжки и органический слой, промывают 2×100 мл насыщенным раствором NaCl и сушат безвод. CaCl2. Отгоняют растворитель, остаток перегоняют в вакууме. Выход продукта 18 составил 35.9 г (60%), т.кип. 98-100°С (7 мм рт.ст.) ИК-спектр (CDCl3), ν/см-1: 1590, 1501 (С=С). Спектр ЯМР 1Н (CDCl3): 1.19 т (3Н, J 7.8, СН3,), 2.64 к (2Н, J 7.8, СН2), 3.77 с (3Н, ОСН3), 3.76 с (3Н, ОСН3) 6.67 д.д (1Н, J1 8.8, J2 2.9, Н(5)), 6.75 д (1H, J 2.9, Н(3)), 6.76 д (1Н, J 8.8, Н(6)). Масс-спектр (ЭУ), m/z (Iотн. (%)): 166 [M]+ (40), 153 (10), 152 (100), 151 (17), 137 (41).
5,8-Дигидрокси-6,7-дихлор-3-этил-1,4-нафтохинон (14). В расплав 195 г (1.46 моль) безводного AlCl3 и 37 г (0.63 моль) NaCl при 140°С вносят при перемешивании смесь 23.5 г (0.14 моль) дихлормалеинового ангидрида и 10 г (0.06 моль) диметилового эфира этилгидрохинона (18). Температуру повышают до 190°С, и расплав перемешивают еще 3 мин. Реакционную смесь охлаждают и гидролизуют 5% раствором HCl. Выделившийся за 12 ч осадок отделяют, промывают 1 л горячей воды и сушат. Горячей экстракцией хлороформом извлекают сырой продукт 14, который кристаллизуют из системы гексан-ацетон. Получают 15 г (88%) продукта 14, т.пл. 123-125°С. ИК-спектр (CDCl3), ν/см-1: 1617 (С=O), 1575, 1562 (С=С). Спектр ЯМР 1H (CDCl3): 1.27 т (3Н, J 7.3, СН3), 2.73 д.к (2Н, J1 7.3, J2 1.4, CH2), 7.06 т (1Н, J 1.4, Н(7)), 12.56 с (1Н, α-ОН), 12.90 с (1Н, α-ОН). Масс-спектр (ЭУ), m/z (Iотн. (%)): 286/288/290 [М]+ (100), 285/287/289 [M-1]+ (21), 271/273/275 [М-СН3]+ (7), 268/270/272 [M-H2O]+ (8), 258/260/262 [M-CO]+ (12), 257/259/261 (5).
5,8-дигидрокси-2,6,7-трихлор-3-этил-1,4-нафтохинон (15). К раствору 6.0 г (0.02 моль) этилдихлорнафтазарина (14) в 200 мл ледяной уксусной кислоты при перемешивании добавляют 40 мл конц. соляной кислоты и 3.8 г (0.04 моль) диоксида марганца и кипятят в течение 1.5 ч. Затем к реакционной смеси добавляют еще 20 мл конц. соляной кислоты и 1.8 г (0.02 моль) диоксида марганца и кипятят в течение 1 ч. Смесь охлаждают, выливают в холодную воду (400 мл), выпавший осадок отфильтровывают, промывают водой, высушивают в вакуумном эксикаторе. После кристаллизации из ацетона получают продукт 15, 4.8 г (72%), в виде бордовых игл, т.пл. 132-134°С. ИК-спектр (CDCl3), ν/см-1: 1640, 1619 (С=O), 1587, 1566 (С=С). Спектр ЯМР 1Н (CDCl3): 1.20 т (3Н, J 7.6, СН3), 2.91 к (2Н, J 7.6, СН2), 12.92 с (1Н, α-ОН), 13.05 с (1Н, α-ОН). Масс-спектр (ЭУ), m/z (Iотн. (%)): 321/323/325/327 [M+1]+ (16), 320/322/324/326 [M]+ (100), 302/304/306/308 [М-Н2O]+ (7), 292/294/296/298 [М-СО]+ (6), 285/287/289 [М-Cl]+ (39).
5,8-Дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинон (1). Смесь 1.0 г (0.003 моль) хорошо высушенного 5,8-дигидрокси-2,6,7-трихлор-3-этил-1,4-нафтохинона (15), 4.2 г (0.028 моль) безводного CsF, 3.1 г (0.03 моль) активированного нейтрального Al2О3 марки «Реахим», 150 мл абс. МеОН, продутую сухим аргоном, перемешивают при 80-85°С в автоклаве в течение 30 мин, затем продолжают реакцию при 95(±1)°С в течение 7.5 ч. После окончания процесса реакционную смесь охлаждают, сорбент отделяют фильтрованием, промывают ацетоном (50 мл) с добавлением небольшого количества 10% HCl (5 мл). Объединенный фильтрат концентрируют в вакууме, к остатку добавляют воду (200 мл) и обрабатывают CHCl3. Органический слой промывают водой, конц. раствором NaCl, сушат безв. Na2SO4 и упаривают. Методом колоночной хроматографии на силикагеле при элюировании системой петролейный эфир-ацетон, 40:1, выделяют хроматографически однородный продукт 1, 0.65 г (68%), т.пл. 129-131°С (из ацетона). Спектр ЯМР 1Н (CDCl3): 1.15 т (3Н, J 7.5, СН3), 2.71 к (2Н, J 7.5, СН2), 4.06 с (3Н, ОСН3), 4.09 с (3Н, ОСН3), 4.12 с (3Н, ОСН3), 12.97 с (1Н, α-ОН), 13.12 с (1Н, α-ОН). Масс-спектр (ЭУ), m/z (Iотн. (%)): 308 [М]+ (100).
5,8-Дигидрокси-3-метил-2,6,7-триметокси-1,4-нафтохинон (20) получают из 1.0 г (0.003 моль) 5,8-дигидрокси-2,6,7-трихлор-3-метил-1,4-нафтохинона (19) действием комплексного реагента МеОН-CsF-Al2О3 по вышеописанной методике. Методом колоночной хроматографии на силикагеле при элюировании системой петролейный эфир-ацетон, 30:1, выделяют хроматографически однородный продукт 20, 0.08 г (8%), т.пл. 148-150°С. Спектр ЯМР 1H (CDCl3): 2.21 с (3Н, J 7.5, СН3), 4.05 с (3Н, ОСН3), 4.10 с (3Н, ОСН3), 4.14 с (3Н, ОСН3), 12.94 с (1Н, α-ОН), 13.10 с (1Н, α-ОН). Масс-спектр (ЭУ), m/z (Iотн.(%)): 294 [M]+ (100).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2,5,8-ТРИГИДРОКСИ-6,7-ДИХЛОР-3-ЭТИЛ-1,4 НАФТОХИНОНА | 2001 |
|
RU2193550C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5,8-ДИГИДРОКСИ-2,3,6-ТРИМЕТОКСИ-7-ЭТИЛ-1,4-НАФТОХИНОНА | 2001 |
|
RU2203265C2 |
| Способ получения спинохрома D | 2016 |
|
RU2612265C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТАХИНОНОВ | 1990 |
|
RU2022959C1 |
| Способ получения 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D) и промежуточные соединения, используемые в этом способе | 2016 |
|
RU2632668C2 |
| СПОСОБ ПОЛУЧЕНИЯ 6,7-ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТОХИНОНОВ (СПИНАЗАРИНОВ) | 2012 |
|
RU2478607C1 |
| СПОСОБ ПОЛУЧЕНИЯ СПИНОХРОМА Е | 2014 |
|
RU2561280C1 |
| НОВЫЕ ОПТИЧЕСКИ АКТИВНЫЕ 4-ГИДРОКСИ-2-АЗА-9,10-АНТРАХИНОНЫ, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2010 |
|
RU2436775C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2024 |
|
RU2839611C1 |
| АНГИДРИДЫ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ ТАКСОЛА ИЛИ ТАКСОТЕРА | 1993 |
|
RU2104274C1 |
Изобретение относится к органической химии, конкретно к способу получения 5,8-дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинона, который является полупродуктом в синтезе 2,5,6,7,8-пентагадрокси-3-этил-1,4-нафтохинона (эхинохрома А), действующего начала кардиопротекторного и офтальмологического препарата гистохром. 5,8-Дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинон получают замещением атомов хлора метоксигруппами в хлорированном производном 1,4-нафтохинона под действием реагента МеОН-CsF-Al2O3 или МеОН-KF-Al2О3. В качестве производного 1,4-нафтохинона используют 5,8-дигидрокси-2,6,7-трихлор-3-этил-1,4-нафтохинон, получаемый путем восстановления ацетилгидрохинона, метилирования образующегося 1,4-дигидрокси-2-этилбензола, циклоацилирования полученного 1,4-диметокси-2-этилбензола дихлормалеиновым ангидридом с последующим хлорированием полученного 5,8-дигидрокси-6,7-дихлор-3-этил-1,4-нафтохинона. Технический результат - упрощение способа, снижение его пожароопасности, повышение выхода целевого продукта.
Способ получения 5,8-дигидрокси-2,6,7-триметокси-3-этил-1,4-нафтохинона замещением атомов хлора метоксигруппами в хлорированном производном 1,4-нафтохинона действием реагента MeOH-CsF-Al2O3 или MeOH-KF-Al2O3, отличающийся тем, что в качестве производного 1,4-нафтохинона используют 5,8-дигидрокси-2,6,7-трихлор-3-этил-1,4-нафтохинон, получаемый путем восстановления 2-ацетил-1,4-дигидроксибензола, метилирования образующегося 1,4-дигидрокси-2-этилбензола, циклоацилирования образующегося при этом 1,4-диметокси-2-этилбензола дихлормалеиновым ангидридом с последующим хлорированием полученного 5,8-дигидрокси-6,7-дихлор-3-этил-1,4-нафтохинона.
| СПОСОБ ПОЛУЧЕНИЯ 2,5,8-ТРИГИДРОКСИ-6,7-ДИХЛОР-3-ЭТИЛ-1,4 НАФТОХИНОНА | 2001 |
|
RU2193550C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5,8-ДИГИДРОКСИ-2,3,6-ТРИМЕТОКСИ-7-ЭТИЛ-1,4-НАФТОХИНОНА | 2001 |
|
RU2203265C2 |
| RU 1822549 A3, 20.08.1996 | |||
| ХИНОНОВЫЕ ПРОИЗВОДНЫЕ | 1992 |
|
RU2054412C1 |

