ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к замещенным производным имидазопиридина, применяемым в качестве модуляторов рецептора меланокортина-4. В зависимости от структуры и пространственного строения, модуляторы рецептора меланокортина-4 являются либо агонистами, либо антагонистами. Соединения, предлагаемые согласно настоящему изобретению, представляют собой селективные антагонисты рецептора меланокортина-4 человека (МК-4Р). Указанные антагонисты пригодны для лечения таких нарушений и заболеваний, как раковая кахексия (истощение), мышечная атрофия, анорексия, боковой амиотрофический склероз (болезнь Шарко) (БАС), тревожность и депрессия.
УРОВЕНЬ ТЕХНИКИ
Меланокортины (МК) образуются из про-опиомеланокортина (ПОМК) путем протеолитического расщепления. В состав этих пептидов, адренокортикотропного гормона (АКТГ), α-меланоцит-стимулирующего гормона (интермедина) (α-МСГ), β-МСГ и γ-МСГ, входят от 12 до 39 аминокислот. По-видимому, наиболее важным эндогенным агонистом для активации МК-4Р в центральной нервной системе является тридекапептид α-МСГ. Известно, что α-МСГ, принадлежащий к меланокортинам, выполняет в тканях мозга функцию нейротрансмиттера или нейромодулятора. Пептиды МК, в частности, α-МСГ, имеют широкий спектр воздействия на биологические функции, включающие пищевое поведение, пигментацию и экзокринную функцию. Биологическое воздействие α-МСГ опосредуется подсемейством 7-трансмембранных рецепторов, сопряженных с G-белком, называемых рецепторами меланокортина (МК-Рц). Активация любого из указанных МК-Рц приводит к стимуляции выработки цАМФ (циклического аденозинмонофосфата).
В настоящее время для МК известны пять различных подтипов рецепторов (от МК-1Р до МК-5Р), которые, как было обнаружено, экспрессируются в различных тканях.
Первым в меланоцитах был обнаружен рецептор МК-1Р. Было показано, что встречающиеся в природе в организмах животных неактивные варианты МК-1Р приводят к изменениям в пигментации и проявлению более светлой окраски за счет контролирования превращения феомеланина в эумеланин при участии тирозиназы. Эти и другие исследования показывают, что МК-1Р является важным регулятором выработки меланина, обуславливающим окраску шерсти животных и кожи человека. МК-2Р экспрессируется надпочечниками и представляет собой рецептор АКТГ. МК-2Р не является рецептором α-МСГ, но представляет собой рецептор адренокортикотропного гормона I (АКТГ I).
МК-3Р экспрессируется в тканях мозга (в основном локализованных в гипоталамусе) и периферийных тканях, таких как кишечник и плацента; исследования с использованием генного нокаута показали, что действие МК-3Р может обусловливать изменения в пищевом поведении, изменения массы тела и термогенеза.
МК-4Р в основном экспрессируется в тканях мозга. Многочисленные данные подтверждают роль МК-4Р в гомеостазе энергии. Опыты на животных с использованием генного нокаута и фармакологического воздействия на МК-4Р показали, что агонистическое воздействие на МК-4Р вызывает снижение массы, а антагонистическое воздействие на рецептор МК-4Р приводит к повышению массы тела (A.Kask, et al., "Selective antagonist for the melanocortin-4 receptor (HS014) increases food intake in free-feeding rats," Biochem. Biophys. Res. Commun., 245:90-93 (1998)).
MK-5P экспрессируется повсеместно во многих периферических тканях, включая жировую ткань и плаценту; кроме того, низкий уровень экспрессии этого рецептора также обнаружен в тканях мозга. Тем не менее, максимально этот рецептор экспрессируется в экзокринных железах. Генный нокаут в отношении указанного рецептора у мышей приводит к изменению регулирования функций экзокринной железы, приводящему к изменениям в процессах водоотталкивания и терморегуляции. Кроме того, у МК-5Р-нокаутных мышей (с «выключенным» соответствующим геном) отмечается пониженная выработка липидов сальными железами (Chen et al., Cell, 91:789-798 (1997)).
Исследование модуляторов МК-3Р и МК-4Р и их применения в лечении нарушений, связанных с изменением массы тела, например, ожирения и анорексии, вызывает большой интерес. Тем не менее, было показано, что кроме регулирования пигментации, пищевого поведения и экзокринной функции, МК пептиды могут оказывать и другие важные физиологические воздействия. В частности, ранее было показано, что α-МСГ оказывают сильное противовоспалительное действие как при острых, так и при хронических воспалительных процессах, включающих воспалительную болезнь кишечника, почечную ишемию/реперфузионное повреждение и гепатит, вызываемый эндотоксинами. Введение α-МСГ при лечении таких нарушений приводит к значительному снижению повреждений ткани, вызываемых воспалением, существенному снижению лейкоцитарной инфильтрации и резкому понижению повышенных уровней цитокинов и других медиаторов воспаления до показателей, близких к нормальным. Недавние исследования показали, что медиаторами противовоспалительного действия α-МСГ являются МК-1Р. Вероятно, что механизм, в соответствии с которым агонистическое воздействие на МК-1Р приводит к противовоспалительному процессу, включает ингибирование активатора провоспалительной транскрипции, NF-кВ. Активатор NF-кВ представляет собой главный компонент провоспалительного каскада, и его активация представляет собой основной процесс инициации многих воспалительных заболеваний. Кроме того, противовоспалительное действие α-МСГ частично может быть опосредовано агонистическим воздействием на рецепторы МК-3Р и/или МК-5Р.
До настоящего времени пока еще не был идентифицирован конкретный специфический МК-Рц, который контролирует развитие ожирения, хотя были получены данные, свидетельствующие о том, что сигнализация МК-4Р играет важную роль в опосредовании пищевого поведения (S.Q.Giraudo et al., "Feeding effects of hypothalamic injection of melanocortin-4 receptor ligands," Brain Research, 80:302-306 (1998)). Другие свидетельства участия МК-Рц в развитии ожирения включают следующие данные: 1) мыши агути (Avy), у которых эктопически экспрессируются антагонисты МК-1Р, МК-3Р и МК-4Р, страдают ожирением, что свидетельствует о том, что блокировка действия указанных трех МК-РЦ может приводить к развитию булимии и метаболических нарушений; 2) фенотип МК-4Р нокаутных мышей (D.Huszar et al., Cell, 88:131-141 (1997)) идентичен фенотипу мышей агути, и эти мыши также страдают ожирением; 3) интрацеребровентрикулярное (ИЦВ) введение циклического гептапептида меланотанина II (MT-II) (неселективного агониста МК-1Р, -3Р, -4Р и -5Р) грызунам уменьшает потребление ими пищи в некоторых исследованиях режимов питания животных (NPY, ob/ob, агути, голодающие грызуны), в то время как ИЦВ-введенный SHU-9119 (антагонист МК-3Р и 4Р; агонист МК-1Р и -5Р) приводит к противоположному действию и может вызывать развитие булимии; 4) сообщали, что хроническое внутрибрюшинное введение страдающим ожирением крысам Цукера производного α-NDP-МСГ (HP-228) активирует МК-1Р, -3Р, -4Р и -5Р и приводит в норму потребление пищи и прирост массы тела в течение 12 недель (I.Corcos et аl., "HP-228 is a potent agonist of melanocortin receptor-4 and significantly attenuates obesity and diabetes in Zucker fatty rats," Society for Neiroscience Abstracts, 23:673 (1997)).
По-видимому, МК-4Р также влияет и на другие физиологические функции, контролируя груминг, эрекцию и кровяное давление. Эректильная дисфункция означает медицинское состояние, определяющее неспособность к эрекции полового члена, достаточной для совершения успешного полового акта. Для описания указанного распространенного состояния часто используют термин «импотенция». Было обнаружено, что синтетические агонисты рецепторов меланокортина способствуют возникновению эрекции у мужчин, страдающих психогенной эректильной дисфункцией (Н.Wessells et al., "Synthetic Melanotropic Peptide Initiates Erections in Men With Psychogenic Erectile Dysfunction: Double-Blind, Placebo Controlled Crossover Study," J.Urol., 160:389-393, 1998). Активация рецепторов меланокортина, находящихся в тканях мозга, по-видимому, вызывает нормальную стимуляцию полового возбуждения. Данные об участии МК-РЦ в развитии половых дисфункций у особей мужского и/или женского пола рассмотрены в заявке WO 00/74679.
Диабет представляет собой заболевание, при котором способность организма млекопитающего к регулированию концентрации глюкозы в крови нарушена вследствие того, что в организме млекопитающего понижается способность к превращению глюкозы в гликоген, который откладывается в мышечной ткани и в клетках печени. При диабете I типа указанная пониженная способность к такому накоплению глюкозы вызвана пониженной выработкой инсулина. Диабет II типа или «сахарный неинсулинозависимый диабет» (СНИЗД) является формой диабета, обусловленной значительным снижением чувствительности к стимулирующей или регуляторной функции инсулина в отношении метаболизма глюкозы и липидов основных инсулинозависимых тканей, мышечной ткани, клеток печени и жировой ткани. Такая резистентность к воздействию инсулина приводит к пониженной инсулиновой активации потребления глюкозы, ее окисления и накопления в мышечной ткани, к неадекватному инсулиновому подавлению липолиза в жировой ткани и неадекватной выработке и секреции глюкозы в печени. При понижении чувствительности указанных клеток к инсулину, организм стремится скомпенсировать этот процесс, вырабатывая ненормально высокие концентрации инсулина, что приводит к развитию гиперинсулинемии. Гиперинсулинемия сопровождается гипертонией и повышением массы тела. Поскольку инсулин способствует проникновению глюкозы, аминокислот и триглицеридов, находящихся в крови, в инсулинозависимые клетки, то нечувствительность к инсулину может привести к повышению концентраций триглицеридов и липопротеинов низкой плотности (ЛПНП), которые представляют собой факторы риска в развитии сердечно-сосудистых заболеваний. Ряд симптомов, которые включают гиперинсулинемию в сочетании с гипертонией, повышенной массой тела, повышенными концентрациями триглицеридов и повышенными уровнями ЛПНП, известен под названием синдрома X. Агонисты МК-4Р могут быть пригодны для лечения СНИЗД и синдрома X.
Среди субтипов рецепторов МК, также вызывают интерес рецепторы МК4, поскольку они связаны с реакцией на стресс и регулированием эмоционального поведения, что показывают следующие полученные данные. Стресс инициирует сложный каскад ответных реакций, которые включают эндокринные, биохимические и поведенческие процессы. Многие из этих ответных реакций инициируются высвобождением кортиколиберина (кортикотропин-рилизинг фактора) (КРФ) (Owen M.J., Nemeroff C.B. (1991) Physiology and pharmacology of corticotrophin releasing factor. Pharmacol Rev. 43:425-473). Кроме активации КРФ системы головного мозга, имеются некоторые свидетельства того, что меланокортины (МК), которые вырабатываются из про-опиомеланокортина в результате ферментных реакций, являются медиаторами важных поведенческих и биохимических ответных реакций на стресс и, следовательно, участвуют в развитии вызываемых стрессом нарушений, таких как тревожность и депрессия (Anxiolytic-Like and Antidepressant-Like Activities of MKL 0129 (1-[(S)-2-(4-Fluorophenyl)-2-(4-isopropylpiperadin-1-yl)ethyl]-4-[4-(2-methoxynaphthalen-1-yl)butyl]piperazine), a Novel and Potent Nonpeptide Antagonist of the Melanocortin-4 Receptor; Shigeyuki Chaki et al, J.Pharm. Exp. Ther. (2003) 304(2), 818-26).
Хронические заболевания, например, злокачественные опухоли и инфекции, часто сопровождаются истощением, возникающим при одновременном понижении аппетита и потере безжировой массы тела. Быстрое снижение безжировой массы тела часто инициируется воспалительным процессом и обычно связано с повышенными уровнями цитокинов (например, TNF-α) в плазме крови, которые вызывают повышение выработки α-МСГ в тканях мозга. Активация рецепторов МК4 в гипоталамусе при воздействии α-МСГ снижает аппетит и повышает расход энергии. Эксперименты, проводимые на мышах, имеющих опухоли, показали, что истощение может быть предотвращено или вылечено генетическим нокаутом в отношении рецептора МК4 или блокировкой рецептора МК4. Повышение массы тела у мышей, подверженных указанному воздействию, относят на счет повышения безжировой массы тела, которая в основном состоит из скелетной мускулатуры (Marks D.L. et al. Role of the central melanocortin system in cachexia. Cancer Res. (2001) 61:1432-1438).
Клинические наблюдения показывают, что может быть найдена взаимосвязь между развитием бокового амиотрофического склероза (БАС) и потерей массы тела (например, Ludolph A.C., Neuromuscul Disord. (2006) 16 (8):530-8). Соответственно, ингибиторы МК-4Р могут быть использованы в лечении пациентов, страдающих БАС.
Модуляторы рецепторов меланокортина-4 описаны в опубликованной литературе. Например, были синтезированы замещенные производные фенилпиперидина, которые исследовали на проявление как агонистической, так и антагонистической активности по отношению к рецептору МК-4Р.
Ввиду имеющихся недостатков в области лечения различных заболеваний и нарушений, представленных выше, задача настоящего изобретения состоит в обеспечении новых соединений, обладающих повышенной способностью к прониканию через гематоэнцефалический барьер и которые могут быть использованы в качестве антагонистов рецепторов меланокортина-4 для лечения раковой кахексии, мышечной атрофии, анорексии, бокового амиотрофического склероза (БАС), тревожности, депрессии и других заболеваний, в развитии которых участвует МК-4Р.
Неожиданно было обнаружено, что новые имидазопиридины, соответствующие Формуле (I), представленной ниже, отвечают задаче, поставленной в настоящем изобретении.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к замещенным производным имидазопиридина, соответствующим структурной формуле (I)
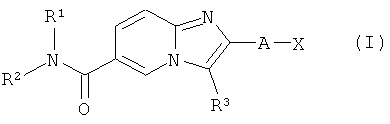
в которой R1, R2, R3, А и Х определены ниже.
Производные имидазопиридина, соответствующие структурной формуле (I), представляют собой эффективные модуляторы рецепторов меланокортина, и, в частности, они эффективны в качестве селективных антагонистов рецепторов меланокортина-4 (МК-4Р). Таким образом, они могут быть использованы для лечения нарушений, которые включают инактивацию МК-4Р. Указанные антагонисты пригодны для лечения таких нарушений и заболеваний, как раковая кахексия, мышечная атрофия, анорексия, боковой амиотрофический склероз, тревожность и депрессия.
Таким образом, настоящее изобретение относится к соединениям, соответствующим формуле (I), предназначенным для лечения и/или профилактики раковой кахексии, мышечной атрофии, анорексии, бокового амиотрофического склероза (БАС), тревожности и депрессии.
Другой аспект изобретения относится к применению соединения, соответствующего формуле (I), для приготовления медикамента, предназначенного для лечения и/или профилактики раковой кахексии, мышечной атрофии, анорексии, бокового амиотрофического склероза (БАС), тревожности и депрессии.
Настоящее изобретение также относится к фармацевтическим композициям, включающим соединения, предлагаемые согласно настоящему изобретению, и фармацевтически приемлемый носитель.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к замещенным производным имидазопиридина, пригодным для применения в качестве модуляторов рецепторов меланокортина, в частности, селективных антагонистов МК-4Р.
Замещенные N-бензил-N-метил-2-фенил-5-диэтиламидо-3-метиламино-имидазо[1,2-a]пиридины описаны в заявке WO-A-02/066478, в которой рассмотрены антагонисты гонадолиберина (гонадотропин-рилизинг гормона). Настоящее изобретение относится к новым имидазопиридинам, которые могут быть использованы в качестве антагонистов МК-4Р.
Соединения, предлагаемые согласно настоящему изобретению, представлены структурной формулой (I)
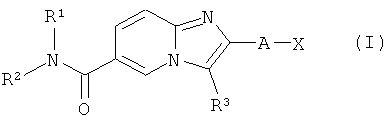
и включают энантиомеры, диастереомеры, таутомеры, сольваты и фармацевтически приемлемые соли указанных соединений,
в которых
А представляет собой -NH-, -СН2-, -СН2-СН2- или связь;
Х представляет собой
Н,
фенил,
фенил, конденсированный с насыщенным гетероциклическим 5- или 6-членным кольцом, при этом гетероциклическое кольцо может содержать один или два гетероатома, выбранных из О и N, и при этом гетероциклическое кольцо дополнительно возможно замещено оксогруппой,
насыщенный или ненасыщенный 4-8-членный гетероциклил, содержащий один или два гетероатома, выбранных из N, О и S,
-5-6-членный гетероарил, содержащий один или два гетероатома, выбранных из N, О и S, или
-C(O)-R6,
и при этом каждый фенил, гетероциклил и гетероарил возможно замещен одним, двумя или тремя заместителями R14 и/или одним заместителем R4b, и/или одним заместителем R5;
R1 и R2 независимо друг от друга выбирают из следующих групп:
Н,
C1-6-алкила,
С1-6-алкилен-O-С1-6-алкила,
С1-3-алкилен-гетероциклила,
С1-6-алкилен-С3-7-циклоалкила, и
при этом каждый алкил, алкилен, гетероциклил и циклоалкил возможно замещен группой ОН, или
R1 и R2 вместе с атомом азота, к которому они присоединены, образуют 5-6-членное кольцо, которое дополнительно может содержать в кольце один атом кислорода и возможно замещено одним или более заместителями, выбранными из ОН, C1-6-алкила, O-C1-6-алкила, С0-3-алкилен-С3-5-циклоалкила, С1-6-алкилен-O-С1-6-алкила или (СН2)0-3-фенила;
R4a представляет собой атом галогена,
CN,
C1-6-алкил, возможно замещенный одним или более атомами галогена,
O-C1-6-алкил, возможно замещенный одним или более атомами галогена, или
OH;
R4b представляет собой C(O)NH2,
С(O)ОН,
C(O)NH-C1-6-алкил,
С(O)N-(С1-6-алкил)2,
SO2-C1-6-алкил,
C(O)NH-SO2-C1-6-алкил,
оксогруппу, и при этом цикл по меньшей мере частично насыщен,
NH2,
NH-C1-6-алкил,
N-(C1-6-алкил)2,
NH-SO2-CH3, или
NH-SO2-CF3;
R5 представляет собой
5-6-членный насыщенный или ненасыщенный гетероциклил, содержащий от одного до трех гетероатомов, выбранных из N, О и S, или
5-6-членный гетероарил, содержащий от одного до трех гетероатомов, выбранных из N, О и S, и
при этом гетероциклил и гетероарил возможно замещен одним или двумя заместителями R14;
R6 представляет собой
Н,
С1-6-алкил, возможно замещенный одним или более атомами галогена,
фенил, или
4-8-членный насыщенный или ненасыщенный гетероциклил, содержащий от одного до трех гетероатомов, выбранных из N, О и S, и
при этом каждый фенил или гетероциклил возможно замещен одним, двумя или тремя заместителями R14 и/или одним заместителем R5;
R3 представляет собой -(CR8R9)n-T;
R8 и R9 независимо друг от друга выбирают из следующих групп:
Н,
ОН,
атома галогена,
C1-6-алкила и
O-C1-6-алкила;
n равен 1, 2, 3, 4, 5 или 6;
Т представляет собой
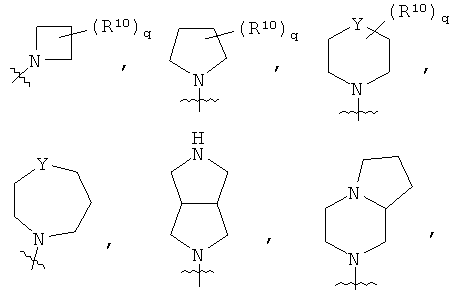
или NR12R13;
R10 представляет собой
H,
NH2,
ОН,
C1-6-алкил, возможно имеющий один или более заместителей, выбранных из атома галогена, ОН и O-C1-6-алкила,
O-C1-6-алкил, в котором алкил возможно замещен одним или более заместителями, выбранными из атома галогена, ОН и O-C1-6-алкила,
атом галогена,
NH(C1-6-алкил),
N(С1-6-алкил)2,
фенил или гетероарил, и
при этом фенил и гетероарил возможно замещены одним, двумя или тремя заместителями R4a;
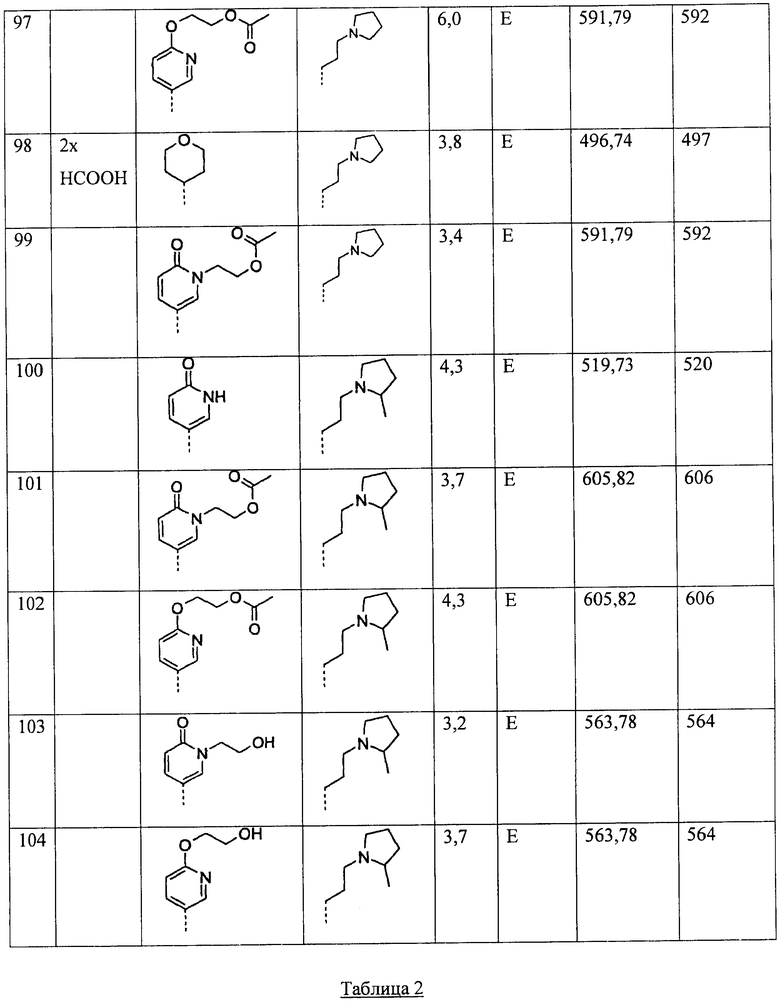
q равен 1 или 2;
Y представляет собой СН2, NR11 или О;
R11 представляет собой
H,
C1-6-алкил, или
(СН2)0-6-С3-7-циклоалкил;
R12 и R13 независимо друг от друга выбирают из следующих групп:
H,
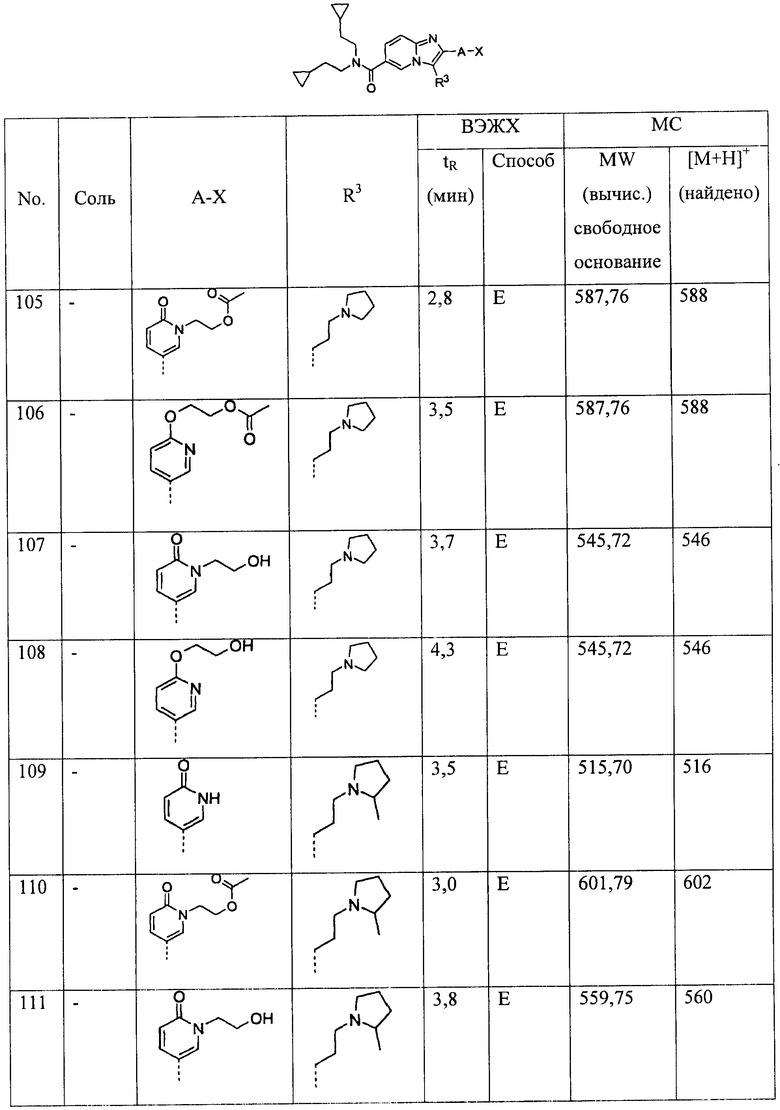
C1-6-алкила,
С2-6-алкенила,
С2-6-алкинила,
(СН2)0-2-С3-7-циклоалкила, и
С1-6-алкилен-O-С1-6-алкил,
при этом C1-6-алкил, C1-6-алкилен и С3-7-пиклоалкил возможно замещены одним, двумя или тремя заместителями R14;
R14 представляет собой
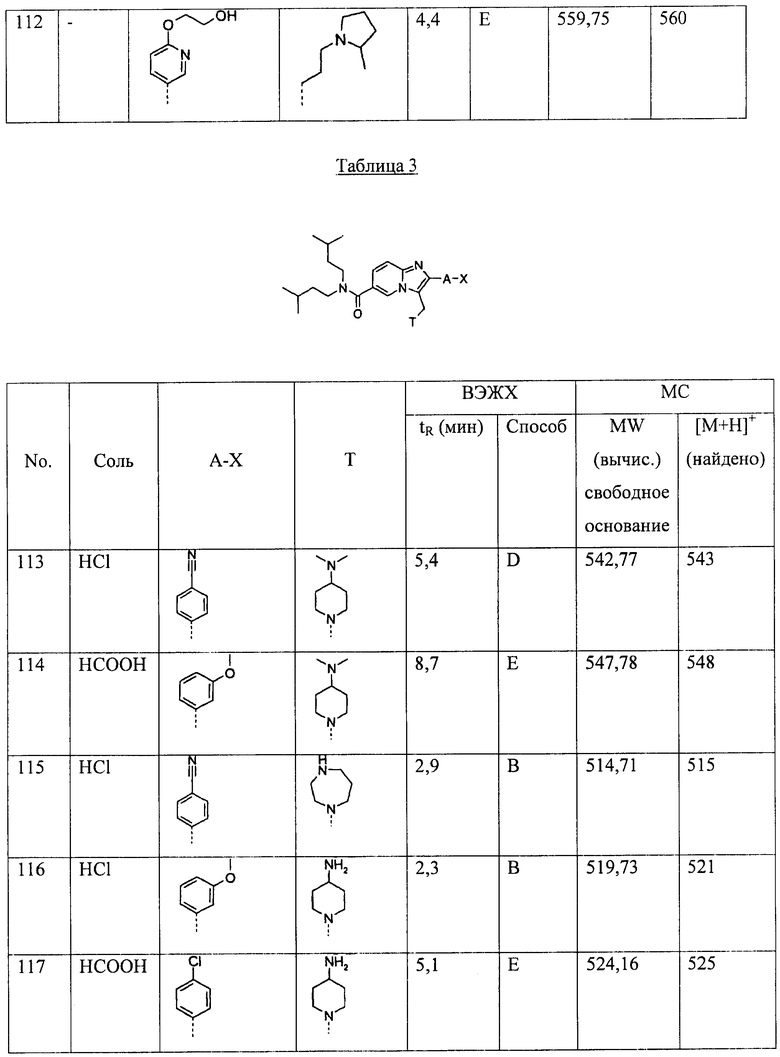
атом галогена,
CN,
C1-6-алкил, возможно замещенный одним или более заместителями, выбранными из атома галогена, ОН, O-C1-6-алкила, О-С3-7-пиклоалкила, O-С(O)С1-6-алкила, O-С(O)С3-7-циклоалкила,
O-C1-6-алкил, возможно замещенный одним или более заместителями, выбранными из атома галогена, ОН, O-C1-6-алкила, О-С3-7-пиклоалкила, O-С(O)С1-6-алкила, O-С(O)С3-7-циклоалкила, или
ОН.
В одном из предпочтительных примеров осуществления А представляет собой -NH- или связь. Более предпочтительно, А представляет собой связь.
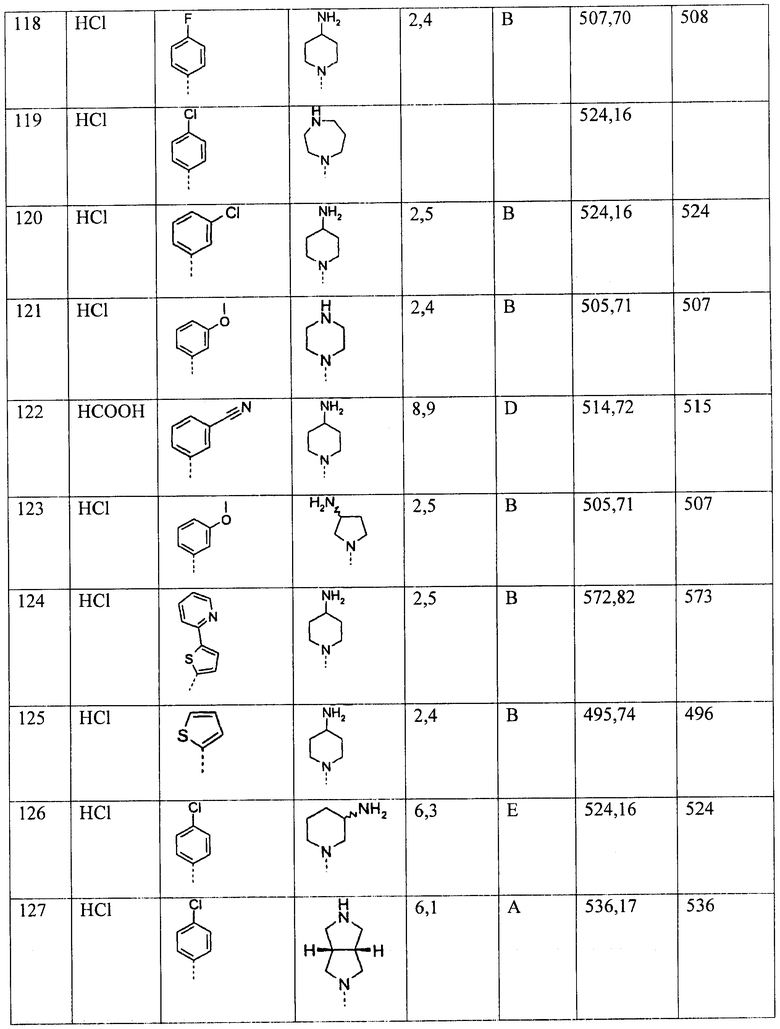
Далее, предпочтительно, если R1 и R2 независимо друг от друга представляют собой С3-6-алкил, или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют 5-6-членное кольцо, которое дополнительно может содержать в цикле один атом кислорода и возможно замещено одним или более заместителями, выбранными из ОН, C1-6-алкила, С0-3-алкилен-С3-5-пиклоалкила, O-C1-6-алкила, С1-6-алкилен-O-С1-6-алкила или (СН2)0-3-фенила. Более предпочтительно, если R1 и R2 независимо друг от друга представляют собой С3-6-алкил.
В одном из предпочтительных примеров осуществления Т представляет собой NR12R13. В этом примере, R12 и R13 предпочтительно независимо друг от друга выбирают из группы, включающей Н, C1-3-алкил или (СН2)0-2-С3-6-циклоалкил, и при этом алкил и циклоалкил возможно замещены одним, двумя или тремя заместителями R14.
В альтернативном предпочтительном примере осуществления, Т выбирают из следующих групп:
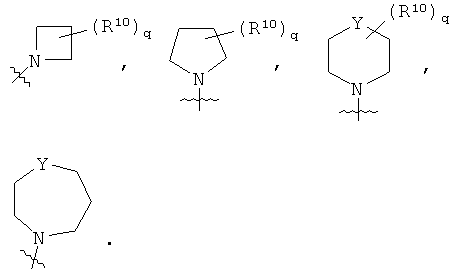
Предпочтительно, Y представляет собой СН2 или NR11. Предпочтительно, R11 представляет собой атом водорода.
Далее, предпочтительно, R10 выбирают из группы, включающей Н, NH2, C1-6-алкил, NH(C1-6-алкил) или N(C1-6алкил)2. Более предпочтительно, R10 представляет собой Н, NH2 или C1-6-алкил.
Что касается X, то указанная группа предпочтительно представляет собой Н; фенил, который конденсирован с насыщенным гетероциклическим 6-членным кольцом, и при этом гетероциклическое кольцо может содержать один или два гетероатома, выбранных из О и N, и при этом гетероциклическое кольцо дополнительно возможно замещено оксогруппой, или Х представляет собой 4-8-членный насыщенный или ненасыщенный гетероциклил, содержащий один или два гетероатома, выбранных из N, О и S, и при этом каждый фенил и гетероциклил возможно замещен одним, двумя или тремя заместителями R14 и/или одним заместителем R4b и/или одним заместителем R5.
В аналогичном предпочтительном примере осуществления, Х представляет собой фенил или 4-8-членный гетероарил, содержащий один или два гетероатома, выбранных из N, О и S, и при этом каждый фенил и гетероарил возможно замещен одним, двумя или тремя заместителями R14 и/или одним заместителем R4b и/или R5. Более предпочтительно, Х представляет собой фенил или пиридил, наиболее предпочтительно Х представляет собой фенил.
Настоящее изобретение также включает соединения, соответствующие формуле (I), в которых некоторые или все группы представляют собой указанные предпочтительные или более предпочтительные фрагменты.
Используемые в настоящем описании термины имеют значения, представленные ниже:
Алкил представляет собой линейный или разветвленный алкил, включающий один, два, три, четыре, пять или шесть атомов углерода, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил или гексил.
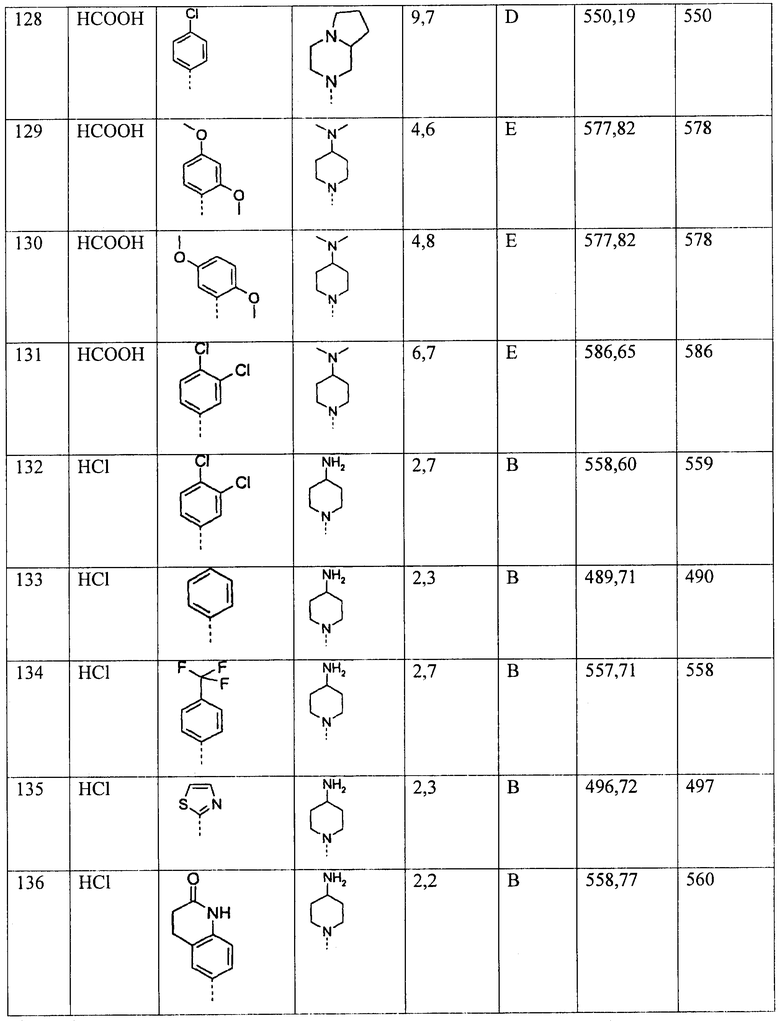
Алкенил представляет собой линейный или разветвленный алкил, включающий два, три, четыре, пять или шесть атомов углерода и от одной до трех двойных связей, предпочтительно одну или две двойные связи, наиболее предпочтительно одну двойную связь. Предпочтительные примеры С2-6-алкенильных групп включают этенил, проп-1-енил, проп-2-енил, изопроп-1-енил, н-бут-1-енил, н-бут-2-енил, н-бут-3-енил, изобут-1-енил, изобут-2-енил, н-пент-1-енил, н-пент-2-енил, н-пент-3-енил, н-пент-4-енил, н-пент-1,3-енил, изопент-1-енил, изопент-2-енил, неопент-1-енил, н-гекс-1-енил, н-гекс-2-енил, н-гекс-3-енил, н-гекс-4-енил, н-гекс-5-енил, н-гекс-1,3-енил, н-гекс-2,4-енил, н-гекс-3,5-енил и н-гекс-1,3,5-енил. Более предпочтительные примеры С2-6-алкенильных групп включают этенил и проп-1-енил.
Алкинил представляет собой линейный или разветвленный алкил, включающий два, три, четыре, пять или шесть атомов углерода и от одной до трех тройных связей, предпочтительно одну или две тройные связи, наиболее предпочтительно одну тройную связь. Предпочтительные примеры С2-6-алкинильных групп включают этинил, проп-1-инил, проп-2-инил, н-бут-1-инил, н-бут-2-инил, н-бут-3-инил, н-пент-1-инил, н-пент-2-инил, н-пент-3-инил, н-пент-4-инил, н-пент-1,3-инил, изопент-1-инил, неопент-1-инил, н-гекс-1-инил, н-гекс-2-инил, н-гекс-3-инил, н-гекс-4-инил, н-гекс-5-инил, н-гекс-1,3-инил, н-гекс-2,4-инил, н-гекс-3,5-инил и н-гекс-1,3,5-инил. Более предпочтительные примеры С2-6-алкинильных групп включают этинил и проп-1-инил.
Циклоалкил представляет собой циклический алкил, предпочтительно включающий три, четыре, пять, шесть или максимально семь атомов углерода, например, циклопропил, циклобутил, циклопентил, циклогексил или циклогептил, более предпочтительно, включающий три, четыре, пять или шесть атомов углерода.
Гетероарил представляет собой ароматическую группу, включающую один, два, три, четыре или пять атомов углерода и по меньшей мере один гетероатом, выбранный из О, N и/или S, и предпочтительно представляет собой группу, выбранную из тиенила, пирролила, имидазолила, пиразолила, пиридила, пиразинила, пиримидинила, пиридазинила, изотиазолила, изоксазолила, фуранила и имидазолила, более предпочтительно, тиенила, фуранила, имидазолила, пиридила и пиримидинила.
Гетероциклил представляет собой насыщенное или ненасыщенное кольцо, содержащее по меньшей мере один гетероатом, выбранный из О, N и/или S, и один, два, три, четыре, пять, шесть или семь атомов углерода. Предпочтительно, гетероциклил представляет собой 4-8-членное кольцо, и предпочтительно представляет собой группу, выбираемую из тетрагидрофуранила, азетидинила, пирролидинила, пиперидинила, пиранила, морфолинила, тиоморфолинила, более предпочтительно, пиперидинила и пйрролидинила.
Галоген представляет собой атом галогена, выбранный из F, Cl, Br и I, предпочтительно из F, Cl и Br.
Соединения, соответствующие структурной формуле (I), представляют собой эффективные модуляторы рецепторов меланокортина и, в частности, применимы в качестве селективных модуляторов МК-4Р. Они пригодны для лечения и/или предотвращения нарушений, чувствительных к процессу инактивации МК-4Р, например, раковой кахексии, мышечной атрофии, анорексии, бокового амиотрофического склероза, тревожности, депрессии и других заболеваний, развивающихся при участии МК-4Р.
ОПТИЧЕСКИЕ ИЗОМЕРЫ - ДИАСТЕРЕОМЕРЫ - ГЕОМЕТРИЧЕСКИЕ ИЗОМЕРЫ - ТАУТОМЕРЫ
Соединения, соответствующие структурной формуле (I), содержат один или более асимметрических центров и могут существовать в виде рацематов и рацемических смесей, отдельных энантиомеров, диастереомерных смесей и отдельных диастереомеров. Настоящее изобретение включает все указанные изомерные формы соединений, соответствующих структурной формуле (I).
Соединения, соответствующие структурной формуле (I), могут быть разделены на отдельные диастереоизомеры, например, фракционной кристаллизацией из подходящего растворителя, например, метанола или этилацетата или смеси указанных растворителей, или посредством хиральной хроматографии с использованием оптически активной неподвижной фазы. Абсолютная стереохимическая конфигурация соединений может быть определена при помощи рентгеновского кристаллографического исследования кристаллических продуктов или кристаллических промежуточных соединений, при необходимости получаемых с использованием реагента, содержащего асимметрический центр известной абсолютной конфигурации.
В альтернативном случае, любой стереоизомер соединения, соответствующего общей формуле (I), может быть получен посредством стереоспецифического синтеза с использованием оптически чистых исходных материалов или реагентов, имеющих известную абсолютную конфигурацию.
СОЛИ
Термин "фармацевтически приемлемые соли" относится к солям, получаемым из фармацевтически приемлемых нетоксичных оснований или кислот, включающих неорганические или органические основания и неорганические или органические кислоты. Соли, получаемые из неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа (II), железа (III), лития, магния, марганца (II), марганца (IV), калия, натрия, цинка и подобных им катионов. Особо предпочтительны соли аммония, кальция, лития, магния, калия и натрия. Соли, получаемые из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включающих природные замещенные амины, циклических аминов и основных ионообменных смол, например, аргинина, бетаина, кофеина, холина, N,N-дибензилэтилендиамина, диэтиламина, 2-диэтиламиноэтанола, 2-диметиламиноэтанола, этаноламина, этилендиамина, N-этилморфолина, N-этилпиперидина, глюкамина, глюкозамина, гистидина, гидрабамина, изопропиламина, лизина, метилглюкамина, морфолина, пиперазина, пиперидина, полиаминовых смол, прокаина, пуринов, теобромина, триэтиламина, триметиламина, трипропиламина, трометамина и подобных им веществ.
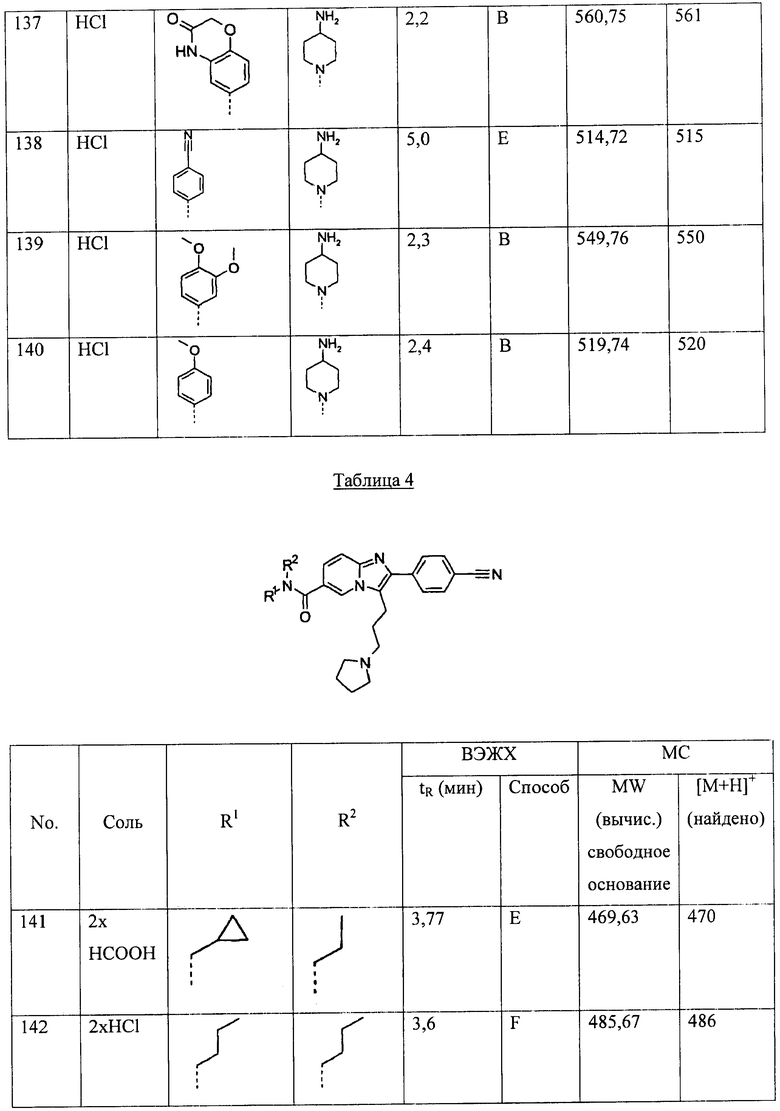
Если соединение, предлагаемое согласно настоящему изобретению, относится к основным, то соли могут быть приготовлены из фармацевтически приемлемых нетоксичных кислот, включающих неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, муравьиную, фумаровую, глюконовую, глютаминовую, бромоводородную, соляную, изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, малоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, пропионовую, янтарную, серную, винную, п-толуолсульфоновую, трифторуксусную кислоты и подобные им кислоты. Особенно предпочтительны лимонная, фумаровая, бромоводородная, соляная, малеиновая, фосфорная, серная и винная кислоты.
Следует понимать, что в соответствии с настоящим описанием, соединения, соответствующие формуле (I), также включают фармацевтически приемлемые соли соединений.
ПРИМЕНИМОСТЬ
Соединения, соответствующие формуле (I), представляют собой антагонисты рецепторов меланокортина и, как таковые, пригодны для лечения, контролирования или предотвращения заболеваний, нарушений или состояний, чувствительных к инактивации одного или более рецепторов меланокортина, неограничивающие примеры которых включают МК-1Р, МК-2Р, МК-3Р, МК-4Р или МК-5Р. Неограничивающие примеры указанных заболеваний, нарушений или состояний включают раковую кахексию, мышечную атрофию, анорексию, боковой амиотрофический склероз, тревожность и депрессию.
Соединения, соответствующие формуле (I), также могут быть использованы для лечения, контролирования или предотвращения заболеваний, нарушений или состояний, чувствительных к инактивации одного или более рецепторов меланокортина, неограничивающие примеры которых включают МК-1Р, МК-2Р, МК-3Р, МК-4Р или МК-5Р. Неограничивающие примеры указанных заболеваний, нарушений или состояний включают гипертонию, гиперлипидемию, остеоартрит, раковое заболевание, заболевание желчного пузыря, синдром ночного апноэ (внезапная остановка дыхания во сне), навязчивость, неврозы, бессонницу/нарушение сна, злоупотребление наркотическими веществами, болевые ощущения, лихорадку, воспаление, иммуномодуляцию, ревматоидный артрит, пигментацию кожных покровов, кожные высыпания и другие нарушения кожных покровов, нейрозащитное воздействие и когнитивные нарушения и нарушения памяти, включающие болезнь Альцгеймера.
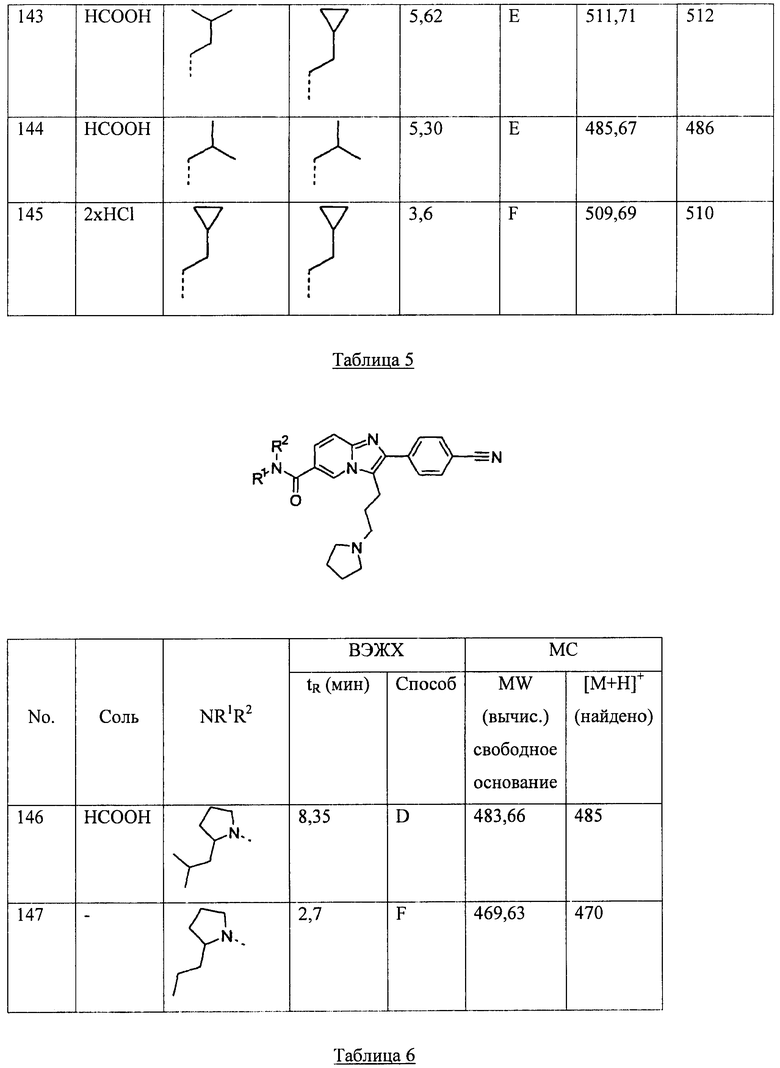
ВВЕДЕНИЕ И ДОЗОВЫЙ ДИАПАЗОН
Для обеспечения млекопитающего, в частности, человека, эффективной дозой соединения, предлагаемого согласно настоящему изобретению, может быть использован любой подходящий способ введения. Например, способ может включать пероральное, перректальное, топическое, парентеральное, глазное, пульмонарное, назальное введение и подобные им способы. Лекарственные формы включают таблетки, драже, дисперсии, суспензии, растворы, капсулы, крема, мази, аэрозоли и подобные им формы. Предпочтительно соединения, соответствующие формуле (I), вводят перорально или топически.
Используемая эффективная доза активного ингредиента может зависеть от типа конкретного используемого соединения, способа введения, состояния, подвергаемого лечению, и тяжести состояния, подвергаемого лечению. Указанная доза может быть легко рассчитана специалистом в данной области техники.
В общем случае, удовлетворительные результаты при лечении раковой кахексии, мышечной атрофии или анорексии были получены при введении соединений, предлагаемых согласно настоящему изобретению, в ежесуточной дозировке, составляющей приблизительно от 0,001 миллиграмма до 100 миллиграммов на килограмм массы тела, предпочтительно в виде однократной дозы или разделенной дозы, вводимой от двух до шести раз в сутки, или в виде лекарственной формы с замедленным высвобождением. При лечении взрослого человека с массой тела, составляющей 70 кг, общая суточная доза, в общем случае, может составлять приблизительно от 0,07 миллиграммов до 3500 миллиграммов. Указанная схема приема может быть уточнена с целью достижения лучшей терапевтической ответной реакции.
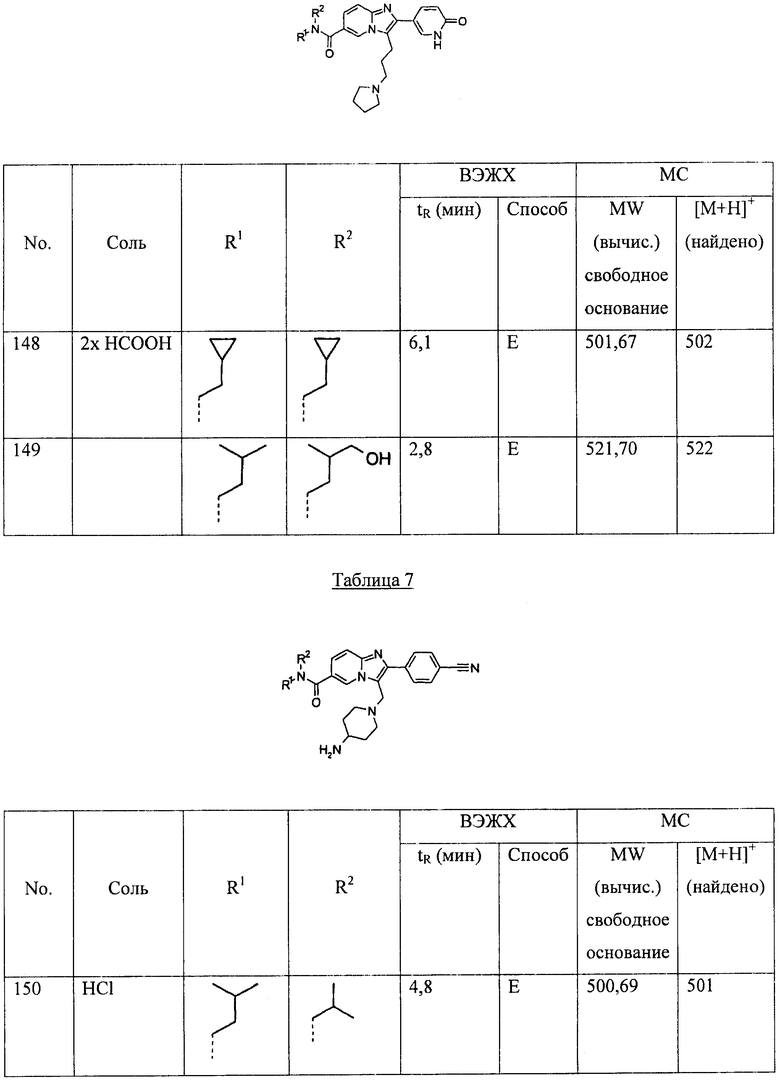
СОСТАВЫ
Предпочтительно, соединения, соответствующие формуле (I), вводят в лекарственные формы до ввода в организм. Соответственно, настоящее изобретение также включает фармацевтическую композицию, включающую соединение, соответствующее формуле (I), и подходящий фармацевтический носитель.
Предлагаемые фармацевтические композиции готовят в соответствии с известными методиками с использованием хорошо известных и легко доступных ингредиентов. При приготовлении составов, применяемых согласно настоящему изобретению, активный ингредиент (соединение, имеющее формулу (I)) обычно смешивают с носителем или разбавляют носителем, или вводят внутрь носителя, который может представлять собой капсулу, пакетик (саше), бумагу или другой контейнер. Если носитель представляет собой разбавитель, то он может представлять собой твердый, полутвердый или жидкий материал, который имеет функцию вспомогательного вещества, наполнителя или среды для активного ингредиента. Таким образом, композиции могут быть приготовлены в виде таблеток, пилюль, порошков, пастилок, саше, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как в твердой, так и в жидкой форме), мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильных упакованных порошков.
Некоторые примеры подходящих носителей, наполнителей и разбавителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, гуммиарабик, фосфат кальция, альгинаты, трагакантовую камедь, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, водный сироп, метилцеллюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. Кроме того, составы могут включать скользящие вещества, смачивающие вещества, эмульгирующие и суспензирующие вещества, консерванты, подсластители или вкусовые добавки. Состав композиций, применяемых согласно настоящему изобретению, может быть подобран таким образом, чтобы обеспечивать быстрое, замедленное или постепенное высвобождение активного ингредиента после введения композиции пациенту.
ПРИГОТОВЛЕНИЕ СОЕДИНЕНИЙ, ПРЕДЛАГАЕМЫХ СОГЛАСНО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Если соединения, соответствующие формуле (I), существуют в виде диастереомерных смесей, то они могут быть разделены на диастереомерные пары энантиомеров фракционной кристаллизацией из подходящего растворителя, например, метанола, этилацетата или смеси указанных растворителей. Полученная таким образом пара энантиомеров может быть разделена на отдельные стереоизомеры традиционными способами, с использованием оптически активной кислоты в качестве агента для оптического расщепления. В альтернативном случае, любой энантиомер соединения, соответствующего формуле (I), может быть получен посредством стереоспецифического синтеза с использованием оптически чистых исходных материалов или реагентов известной конфигурации.
Соединения, соответствующие формуле (I), предлагаемые согласно настоящему изобретению, могут быть получены в соответствии с процедурами, представленными в следующих Схемах и Примерах, с использованием соответствующих материалов и при помощи способов, конкретные примеры которых также рассмотрены ниже. Кроме того, в соответствии с описанными в настоящей заявке процедурами и общими сведениями, известными в данной области техники, могут быть легко приготовлены и другие соединения, охватываемые настоящим изобретением. Тем не менее, не следует считать, что соединения, представленные в примерах, представляют собой единственно возможные варианты соединений, в соответствии с настоящим изобретением. Представленные ниже Примеры даны для пояснения синтеза соединений, предлагаемых согласно настоящему изобретению. Специалистам в данной области техники должны быть известны различные модификации условий и способов, которые допускаются в предлагаемых процессах и позволяющие приготовить указанные соединения. Описываемые соединения в общем случае выделяют в виде их фармацевтически приемлемых солей, например, описанных выше солей. Свободные аминные основания, соответствующие выделенным солям, могут быть получены нейтрализацией подходящим основанием, например, водным раствором гидрокарбоната натрия, карбоната натрия, гидроксида натрия и гидроксида калия, и экстракцией высвобожденного свободного аминного основания органическим растворителем с последующим испарением. Полученные таким образом свободные аминные основания далее могут быть превращены в другие фармацевтически приемлемые соли растворением в органическом растворителе с последующим добавлением соответствующей кислоты и последующим испарением, осаждением или кристаллизацией. Все значения температуры даны в градусах Цельсия.
В схемах, процессах и примерах, представленных ниже, различные обозначения реагентов и аббревиатуры имеют следующие значения:
Схема реакций 1:
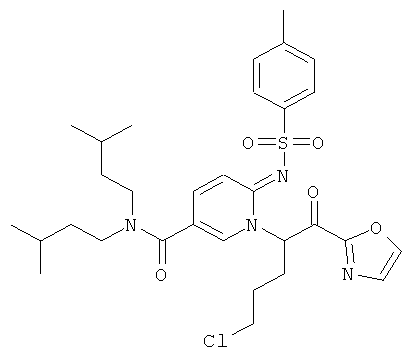
Синтез амидов 2-сульфониламино-пиридин-5-карбоновых кислот
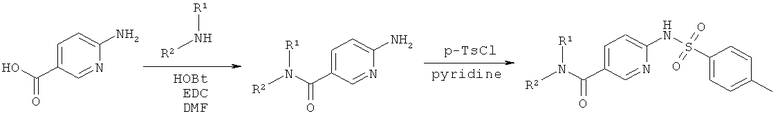
Как показано на Схеме реакций 1, возможно замещенный амин и 2-аминопиридин-5-карбоновую кислоту вводят в реакцию амидного сочетания в присутствии реагента сочетания, например, EDC в органическом растворителе, например, ДМФА или ДХМ при подходящей температуре. Полученный амид затем может быть введен в реакцию с сульфонилхлоридом в растворителе, например, пиридине или любом другом соответствующем растворителе, и с органическим основанием, например, триэтиламином с образованием соответствующих сульфониламино-амидов.
Схема реакций 2:
Синтез метилового эфира 2-сульфониламинопиридин-5-карбоновой кислоты
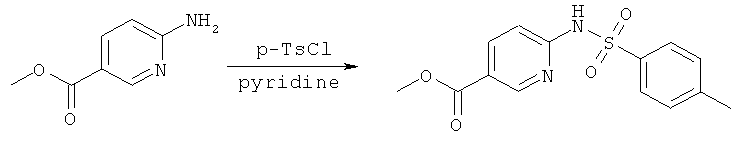
В альтернативном случае, метиловый эфир 2-аминопиридин-5-карбоновой кислоты может быть веден в реакцию в условиях, описанных выше, с образованием соответствующих сульфониламино-эфиров, как показано на Схеме реакций 2.
Схема реакций 3:
Синтез имидазо[1,2-а]пиридинов
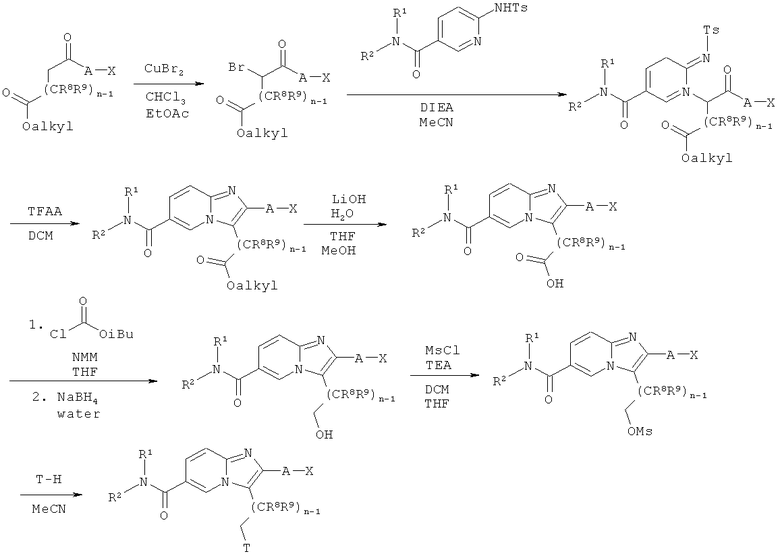
Как показано на Схеме реакций 3, возможно замещенные ω-алкоксикарбонил-α-бромкетоны могут быть получены из соответствующих кетонов по реакции, например с бромидом меди (II) в растворителе, например, смеси этилацетата и хлороформа при соответствующей температуре в течение определенного времени. Полученные α-бромкетоны могут быть введены в реакцию с сульфониламино-амидами в растворителе например, MeCN, в присутствии соответствующего основания, например, DIEA, с образованием N-алкилированных сульфониламино-амидов. Указанные промежуточные соединения затем могут быть подвергнуты циклизации с образованием соответствующих имидазо[1,2-а]пиридинов при обработке TFAA в подходящем растворителе, например, ДХМ или 1,2-дихлорэтане при соответствующей температуре в течение определенного времени. Эфирная функциональная группа возможно замещенных имидазо[1,2-а]пиридинов может быть гидролизована в щелочных условиях с использованием такого реагента, как моногидрат гидроксида лития в подходящем растворителе, например, смеси воды, ТГФ и МеОН.
Полученная кислота может быть активирована под действием подходящего реагента, например, изобутилхлороформиата или CDI в присутствии подходящего основания, например, N-метилморфолина, в соответствующем растворителе, например, ТГФ, и затем восстановлена с образованием соответствующего спирта под действием восстановителя, например, боргидрида натрия в соответствующем растворителе, например, смеси ТГФ и воды. Спиртовая функциональная группа может быть превращена в уходящую группу под действием подходящего реагента, например, метилсульфонилхлорида или тозилхлорида в соответствующем растворителе, например, смеси ДХМ и ТГФ, в присутствии подходящего основания, такого как ТЭА. Продукт этой реакции может быть обработан амином Т-Н в соответствующем растворителе, например, MeCN, с образованием целевой молекулы.
Схема реакций 4:
Синтез имидазо[1,2-а]пиридинов
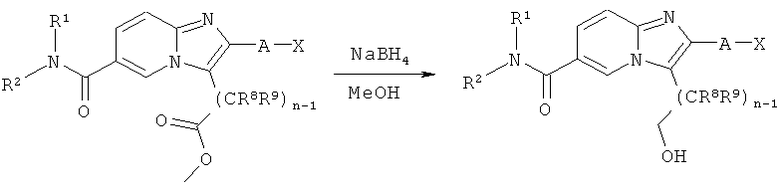
Как показано на Схеме реакций 4, метиловые эфирные функциональные группы возможно замещенных имидазо[1,2-а]пиридинов могут быть восстановлены с образованием соответствующих спиртовых групп под действием подходящего реагента, например, боргидрида натрия, в соответствующем растворителе, например, метаноле. Спирт может быть далее введен в реакцию с получением целевой молекулы, как показано на Схеме реакций 3.
Схема реакций 5:
Синтез имидазо[1,2-а]пиридинов
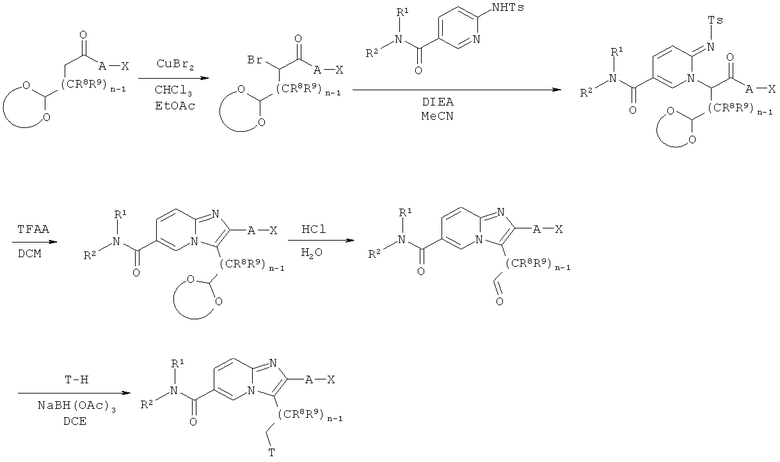
Как показано на Схеме реакций 5, возможно замещенные α-бромкетоны, имеющие ацетальные функциональные группы в ω-положении, могут быть получены из соответствующих кетонов и превращены в соответствующие возможно замещенные имидазо[1,2-а]пиридины, как описано выше. Указанные ацетали могут быть расщеплены с образованием соответствующих альдегидов при использовании соответствующего реагента, например, 6н. НС1 в воде. Возможно замещенные альдегиды могут быть подвергнуты восстановительному аминированию под действием подходящего амина Т-Н в присутствии восстановителя, например, триацетоксиборгидрида натрия в соответствующем растворителе, например, ДХЭ.
Схема реакций 6:
Синтез имидазо[1,2-а]пиридинов
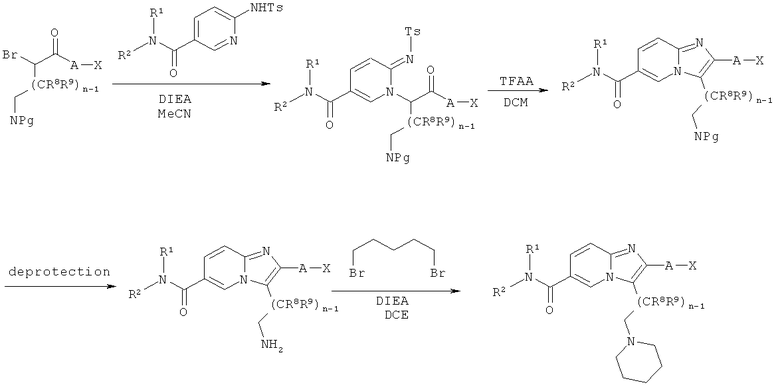
Как показано на Схеме реакций 6, возможно замещенные N-защищенные ω-амино-α-бромкетоны могут быть ведены в реакцию с сульфониламино-амидами в подходящем растворителе, например, MeCN, в присутствии соответствующего основания, например DIEA, с образованием N-алкилированных сульфониламино-амидов. Указанные промежуточные соединения затем могут быть подвергнуты циклизации с образованием соответствующих имидазо[1,2-а]пиридинов при обработке TFAA в подходящем растворителе, например, ДХМ или 1,2-дихлорэтане, при соответствующей температуре в течение определенного времени. Снятие защиты с аминогрупп боковой цепи может быть проведено с использованием соответствующего реагента, например, TMSI, в подходящем растворителе, например, в случае Z-защитных групп, MeCN. Фталимиды могут быть расщеплены под действием гидразингидрата в соответствующем растворителе, например, этилацетате. Возможно замещенные имидазо[1,2-а]пиридины, имеющие первичную аминогруппу в боковой цепи, могут быть непосредственно подвергнуты биологическому анализу или далее превращены в их производные. Например, реакция с 1,5-дибромпентаном в соответствующем растворителе, например, 1,2-дихлорэтане, в присутствии подходящего основания, например, DIEA, приводит к получению соответствующих производных пиперидина.
Схема реакций 7:
Синтез имидазо[1,2-а]пиридинов
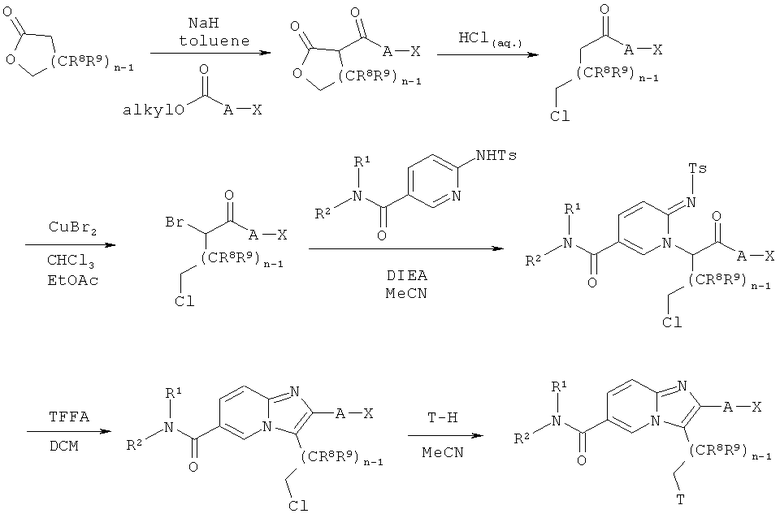
Как показано на Схеме реакций 7, возможно замещенные лактоны могут быть подвергнуты ацилированию алкиловыми эфирами алкил-ОС(O)А-Х в присутствии подходящего основания, например, гидрида натрия, в соответствующем растворителе, например, толуоле, при повышенной температуре. Указанные ацилированные лактоны могут быть превращены в ω-хлоркетоны нагреванием лактонов в концентрированной соляной кислоте. Реакция возможно замещенных ω-хлоркетонов, например, с бромидом меди (II) в подходящем растворителе, например, смеси этилацетата и хлороформа, при соответствующей температуре в течение определенного времени приводит к получению соответствующих ω-хлор-α-бромкетонов. Указанные ω-хлор-α-бромкетоны могут быть введены в реакцию с сульфониламино-амидами в подходящем растворителе, например, MeCN, в присутствии соответствующего основания, например DIEA, с образованием N-алкилированных сульфониламино-амидов. Указанные промежуточные соединения затем могут быть подвергнуты циклизации с образованием соответствующих имидазо[1,2-а]пиридинов при обработке TFAA в подходящем растворителе, например, ДХМ или 1,2-дихлорэтане, при соответствующей температуре в течение определенного времени. Защитная группа Т может быть введена по реакции замещенных хлоралкилом имидазо[1,2-а]пиридинов с подходящей защитной группой Т-Н в соответствующем растворителе, например, MeCN. Если группу Т-Н используют в виде гидрохлорида, то, кроме того, для высвобождения свободного амина Т-Н используют подходящее основание, например, DIEA.
Схема реакций 8:
Синтез имидазо[1,2-а]пиридинов
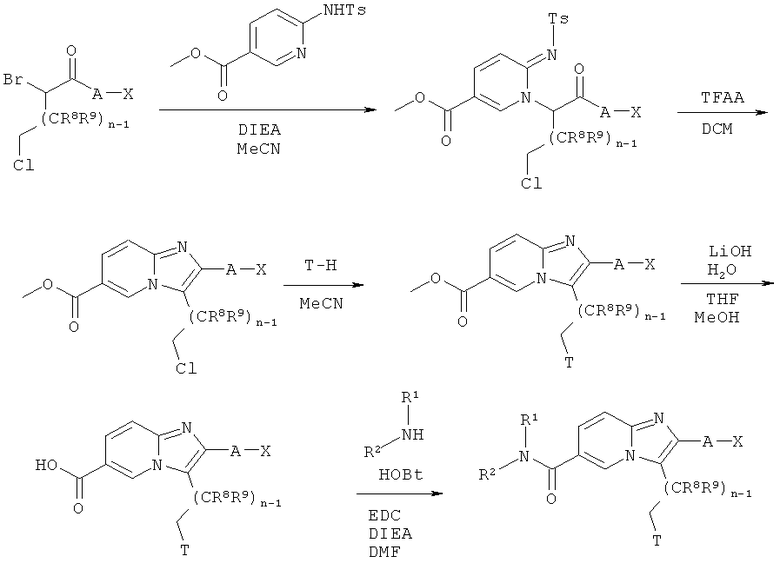
Как показано на Схеме реакций 8, возможно замещенные ω-хлор-α-бромкетоны также могут быть введены в реакцию с подходящим сульфониламиноэфиром в подходящем растворителе, например, MeCN, в присутствии соответствующего основания, например DIEA, с образованием N-алкилированных сульфониламиноэфиров. Указанные промежуточные соединения затем могут быть подвергнуты циклизации с образованием соответствующих имидазо[1,2-а]пиридинов при обработке TFAA в подходящем растворителе, например, ДХМ или 1,2-дихлорэтане, при соответствующей температуре в течение определенного времени. Защитная группа Т может быть введена по реакции имидазо[1,2-а]пиридинов, замещенных хлоралкилом, с защитной группой Т-Н в соответствующем растворителе, например, MeCN. Если группу Т-Н используют в виде гидрохлорида, то, кроме того, для высвобождения свободного амина Т-Н используют подходящее основание, например, DIEA. Эфирная функциональная группа возможно замещенных имидазо[1,2-а]пиридинов может быть гидролизована в щелочных условиях с использованием такого реагента, как моногидрат гидроксида лития в подходящем растворителе, например, смеси воды, ТГФ и МеОН. Продукт омыления может быть выделен в виде литиевой соли или в виде соответствующей кислоты. В альтернативном случае, эфирная функциональная группа может быть расщеплена в кислотных условиях, например, с использованием подходящего реагента, например, водной соляной кислоты. Продукт эфирного расщепления может быть введен в следующую реакцию в виде кислоты или соли лития. Амид может быть образован в соответствии со стандартными процедурами пептидного сочетания. Кислота может быть введена в реакцию с подходящим амином HNR1R2 в присутствии EDC/HOBt, EDC/HOAt, HATU, основания, например, диизопропилэтиламина, и растворителя, например, дихлорметана. Для проведения сочетания может быть использован подходящий растворитель, например, ДХМ, ДМФА, ТГФ или смеси указанных растворителей. Подходящие основания включают триэтиламин (ТЭА), диизопропилэтиламин (DIEA), N-метилморфолин (NMM), коллидин или 2,6-лутидин. Если используют EDC/HOBt, то может не возникать необходимость в использовании основания.
Схема реакций 9:
Гидролиз хлорпиридинов
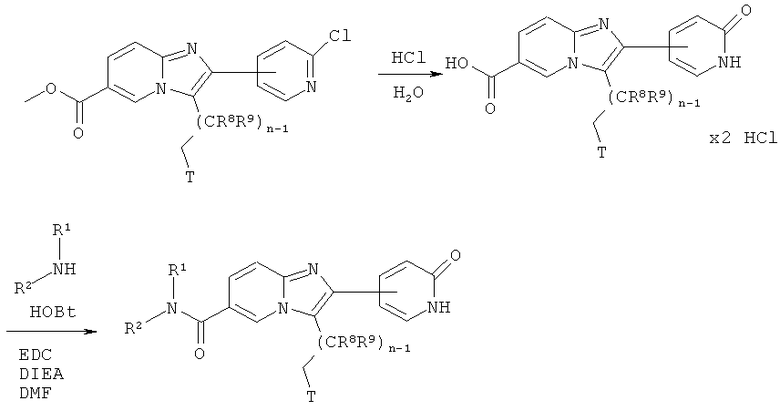
Как показано на Схеме реакций 9, возможно замещенные имидазо[1,2-а]пиридины, включающие хлорпиридин или бромпиридин в виде остатка -А-Х, могут быть превращены в соответствующие пиридоны под действием подходящего реагента, например, водной соляной кислоты при подходящей температуре. Одновременно производят гидролиз эфирной функциональной группы. Кислоты могут быть введены в реакцию с аминами HNR1R2, как описано выше.
Схема реакций 10:
Синтез α-бромкетонов
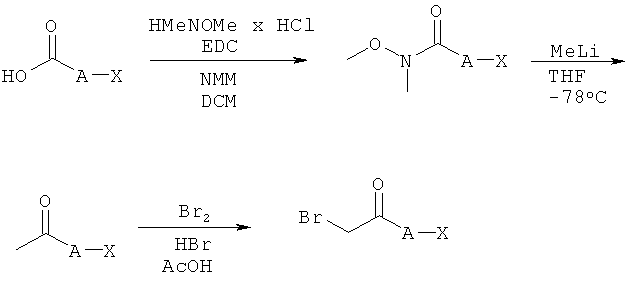
Как показано на Схеме реакций 10, возможно замещенные бромкетоны могут быть получены в соответствии с трехступенчатой реакционной последовательностью, исходя из карбоновых кислот. Указанные карбоновые кислоты могут быть превращены в соответствующие амиды Вайнреба (Weinreb) под действием гидрохлорида N,O-диметилгидроксиламина и подходящего сочетательного реагента, например, EDC, в присутствии подходящего основания, такого как NMM, в соответствующем растворителе, например, ДХМ. Амиды Вайнреба могут быть превращены в соответствующие метилкетоны под действием подходящего реагента, например, метиллития, в инертном растворителе, например, ТГФ, при подходящей температуре. Бромирование может быть произведено с использованием смеси брома и бромоводорода в уксусной кислоте.
Схема реакций 11:
Синтез имидазо[1,2-а]пиридинов
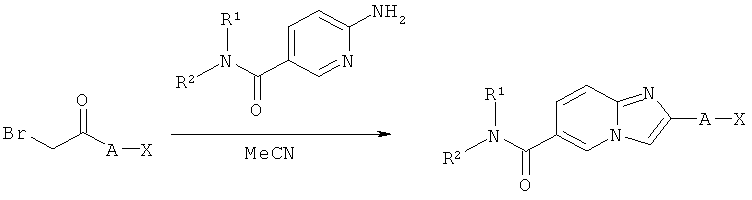
Как показано на Схеме реакций 11, возможно замещенные аминопиридин-амиды, которые могут быть получены, как показано на Схеме реакций 1, могут быть превращены в амиды имидазо[1,2-а]пиридин-6-карбоновой кислоты по реакции с α-бромкетонами в подходящем растворителе, например, MeCN. Указанная реакция может быть проведена либо в колбе в кипящем растворителе или при любой другой подходящей температуре, либо в микроволновом реакторе. Продукты реакции могут быть очищены в соответствии со стандартными процедурами или могут быть осаждены непосредственно из раствора при охлаждении и, таким образом, могут быть использованы в последующих реакциях без дальнейшей очистки.
Схема реакций 12:
Реакция Манниха
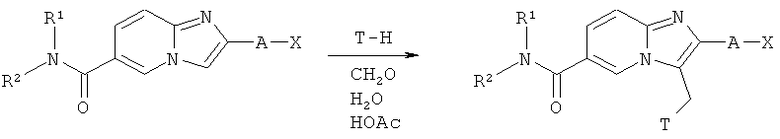
Как показано на Схеме реакций 12, продукты, полученные в соответствии со Схемой реакций 11, т.е. амиды возможно замещенных имидазо[1,2-а]пиридиновых кислот, могут быть использованы в реакции Манниха для получения амидов 3-аминометил-имидазо[1,2-а]пиридин-6-карбоновой кислоты по реакции амидов имидазо[1,2-а]пиридин-6-карбоновых кислот с подходящими аминами и водным раствором формальдегида в подходящем растворителе, например, уксусной кислоте. Снятие защиты с диаминов, содержащих одну защитную группу на азоте, может быть произведено при обработке указанных соединений подходящей кислотой, например, НCl в диоксане или ТФУ в ДХМ. Указанные соединения затем могут быть очищены при помощи стандартных процедур очистки, например, промывной хроматографии или препаративной ВЭЖХ.
Схема реакций 13:
Присоединение α,β-ненасыщенных альдегидов по Михаелю
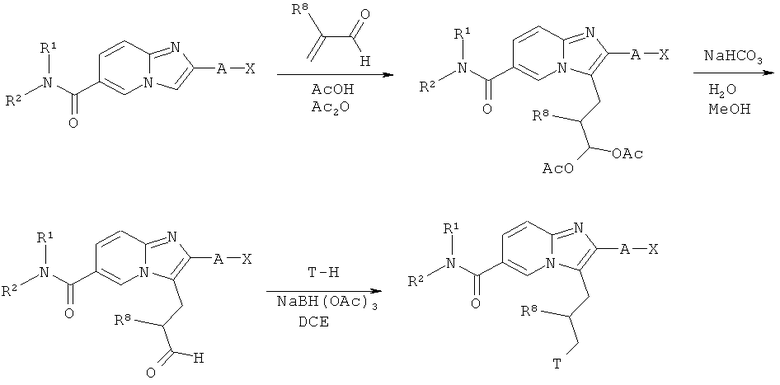
Как показано на Схеме реакций 13, амиды возможно замещенных имидазо[1,2-а]пиридин-6-карбоновых кислот могут быть введены в реакцию присоединения по Михаелю с α,β-ненасыщенными альдегидами в подходящем растворителе, например, смеси уксусной кислоты и уксусного ангидрида при повышенной температуре. Реакции также могут быть проведены в микроволновом реакторе. Продукты указанной реакции могут быть обработаны основанием, например, бикарбонатом натрия в подходящем растворителе, например, смеси воды и метанола, с образованием соответствующих альдегидов, которые могут быть подвергнуты восстановительному аминированию под действием подходящего амина Т-Н в присутствии восстановителя, например, триацетоксиборгидрида натрия, в соответствующем растворителе, например, ДХЭ. В альтернативном случае, в качестве исходных материалов могут быть использованы эфиры возможно замещенных имидазо[1,2-а]пиридин-6-карбоновых кислот. В этом случае, эфирная функциональная группа может быть превращена в амид после введения боковой цепи -CH2CHR8CH2T в соответствии со способами, рассмотренными в Схеме реакций 8.
Схема реакций 14:
Присоединение α,β-ненасыщенных кетонов по Михаелю
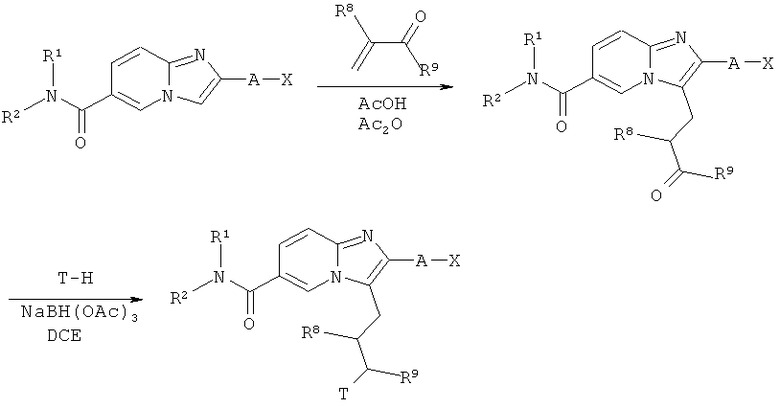
Как показано на Схеме реакций 14, присоединение по Михаелю амидов возможно замещенных имидазо[1,2-а]пиридин-6-карбоновых кислот также может быть проведено с использованием α,β-ненасыщенных кетонов в условиях реакции, рассмотренных в Схеме реакций 13. В этом случае, продукт присоединения по Михаелю может быть непосредственно подвергнут восстановительному аминированию.
Схема реакций 15:
Алкилирование боковой цепи
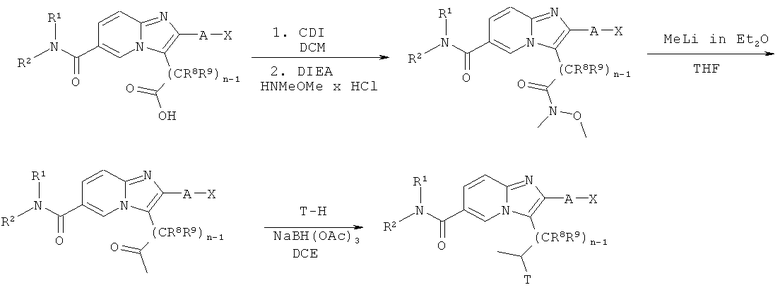
Продукты, полученные в соответствии со Схемой реакций 3, возможно замещенные имидазо[1,2-а]пиридины, имеющие карбоксилатные группы в боковой цепи, могут быть активированы под действием подходящего реагента, например, CDI в соответствующем растворителе, например, ДХМ, и затем введены в реакцию с гидрохлоридом N,O-диметилгидроксиламина в присутствии подходящего основания, например, DIEA. Реакция получаемого продукта с подходящим реагентом, например, метиллитием, в подходящем растворителе, например, ТГФ или диэтиловом эфире, приводит к получению соответствующих кетонов, которые могут быть подвергнуты восстановительному аминированию под действием подходящего амина Т-Н в присутствии восстановителя, например, триацетоксиборгидрида натрия в соответствующем растворителе, например, ДХЭ.
Схема реакций 16:
Реакции хлорпиридинов с аминами
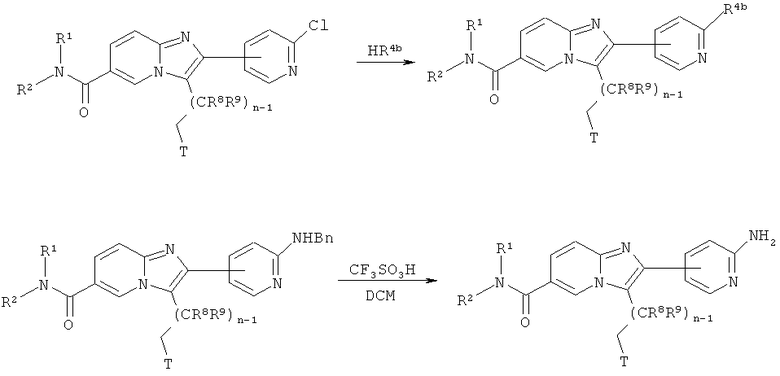
Возможно замещенные имидазо[1,2-а]пиридины, имеющие 2-хлорпиридиновый заместитель, могут быть введены в реакцию с аминами, как показано на Схеме реакций 16. Указанные хлорпиридины могут быть введены в реакцию с чистыми аминами HR4b при повышенной температуре с образованием соответствующих 2-аминопиридинов. Реакция также может быть проведена в микроволновом реакторе. Бензильная защитная группа может быть удалена при обработке N-бензилированных 2-аминопиридинов подходящим реагентом, например, трифторметансульфоновой кислотой, в инертном растворителе, например, ДХМ, при соответствующей температуре.
Схема реакций 17:
Реакции хлорпиридинов со спиртами
Как показано на Схеме реакций 17, возможно замещенные имидазо[1,2-а]пиридины, имеющие 2-хлорпиридиновый заместитель, могут быть введены в реакцию с подходящим алкоголятом с образованием соответствующих алкоксипиридинов. Алкоголят может быть приготовлен из соответствующего спирта HR14 и подходящего основания, например, гидрида натрия, в соответствующем растворителе, например, ДМФА. Реакции алкоголята с хлорпиридином могут быть проведены при повышенных температурах.
Схема реакций 18:
Алкилирование пиридонов
Возможно замещенные имидазо[1,2-а]пиридины, включающие пиридоновый фрагмент, могут быть подвергнуты N-алкилированию, как показано на Схеме реакций 18. Атом азота пиридона может быть подвергнут алкилированию под действием подходящего алкилбромида Br-R14 в присутствии основания, например, карбоната цезия или карбоната калия, в подходящем растворителе, например, ацетоне, при соответствующей температуре. Спиртовые заместители на остатке R14 могут быть защищены, например, превращением в сложноэфирные группы. После проведения алкилирования, свободный спирт может быть получен гидролизом эфира под действием подходящего реагента, например, моногидрата гидроксида лития, в соответствующем растворителе, например, смеси воды и ТГФ.
Аналитическая жидкостная хроматография с масс-спектрометрией (ЖХ-МС)
Соединения, предлагаемые согласно настоящему изобретению, имеющие формулу (I), анализировали при помощи аналитической ЖХ-МС. Условия приведены ниже.
Условия анализа:
Устройство LC 10 Advp-Pump (Shimadzu) с SPD-M 10 Avp (Shimadzu) UV/Vis детектором на диодной матрице с QP 2010 MS-детектором (Shimadzu) в режиме ESI+ с УФ-обнаружением при 214,254 и 275 нм.
Колонка: Waters XTerra MS С18, 3,5 мкм, 2,1·100 мм, линейный градиент ацетонитрила в воде (0,15% НСООН), исключение: способы D и Е (криволинейный градиент).
Скорость потока 0,4 мл/мин
Подвижная фаза А: вода (0,15% НСООН)
Подвижная фаза В: ацетонитрил (0,15% НСООН)
Способы:
А: линейный градиент от 5% до 95% ацетонитрила в воде (0,1% НСООН)
0,00 мин 5% В
5,00 мин 95% В
5,10 мин 99% В
6,40 мин 99% В
6,50 мин 5% В
8,00 мин Остановка насоса
В: линейный градиент от 10% до 90% ацетонитрила в воде (0,1% НСООН)
0,00 мин 10% В
5,00 мин 90% В
5,10 мин 99% В
6,40 мин 99% В
6,50 мин 5% В
8,00 мин Остановка насоса
С: линейный градиент от 5% до 95% ацетонитрила в воде (0,1% НСООН)
0,00 мин 5% В
10,00 мин 95% В
10,10 мин 99% В
11,40 мин 99% В
11,50 мин 5% В
13,00 мин Остановка насоса
D: исходная концентрация 1% ацетонитрила
9,00 В. Конц. 30
10,00 В. Кривая 3
12,00 В. Конц. 99
15,00 В. Конц. 99
15,20 В. Конц. 1
18,00 Остановка насоса
Е: исходная концентрация 10% ацетонитрила
10,00 В. Конц. 60
11,00 В. Кривая 2
12,00 В. Конц. 99
15,00 В. Конц. 99
15,20 В. Конц. 10
18,00 Остановка насоса
F: исходная концентрация 15% ацетонитрила
12,00 В. Конц. 99
15,00 В. Конц. 99
15,20 В. Конц. 15
18,00 Остановка 0
Ниже подробно описаны примеры осуществления изобретения, которые могут быть приготовлены в соответствии со схемами реакций 1-18.
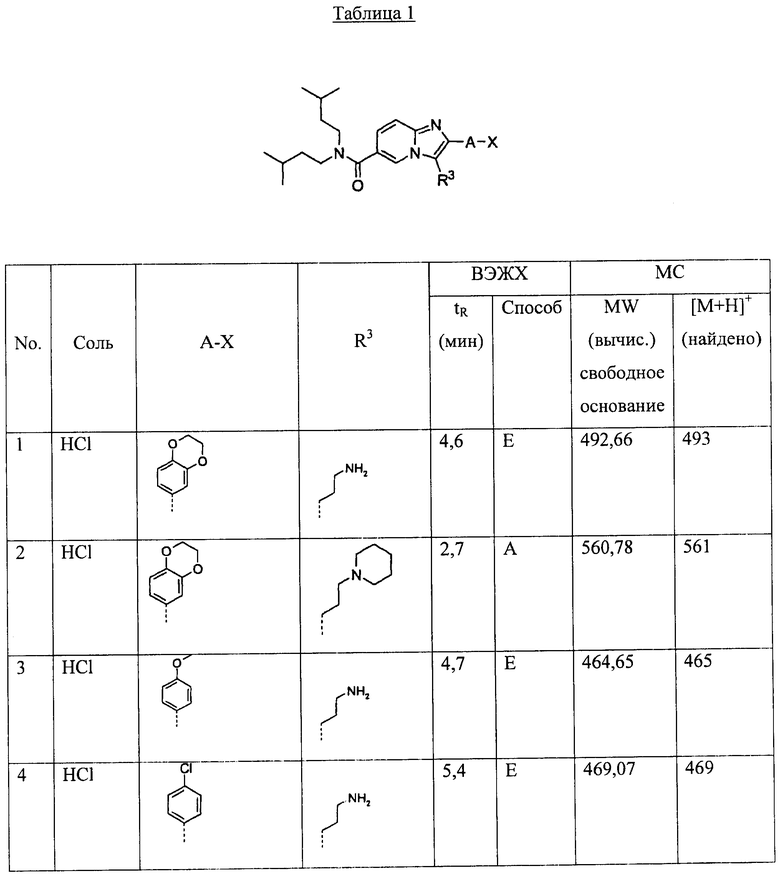
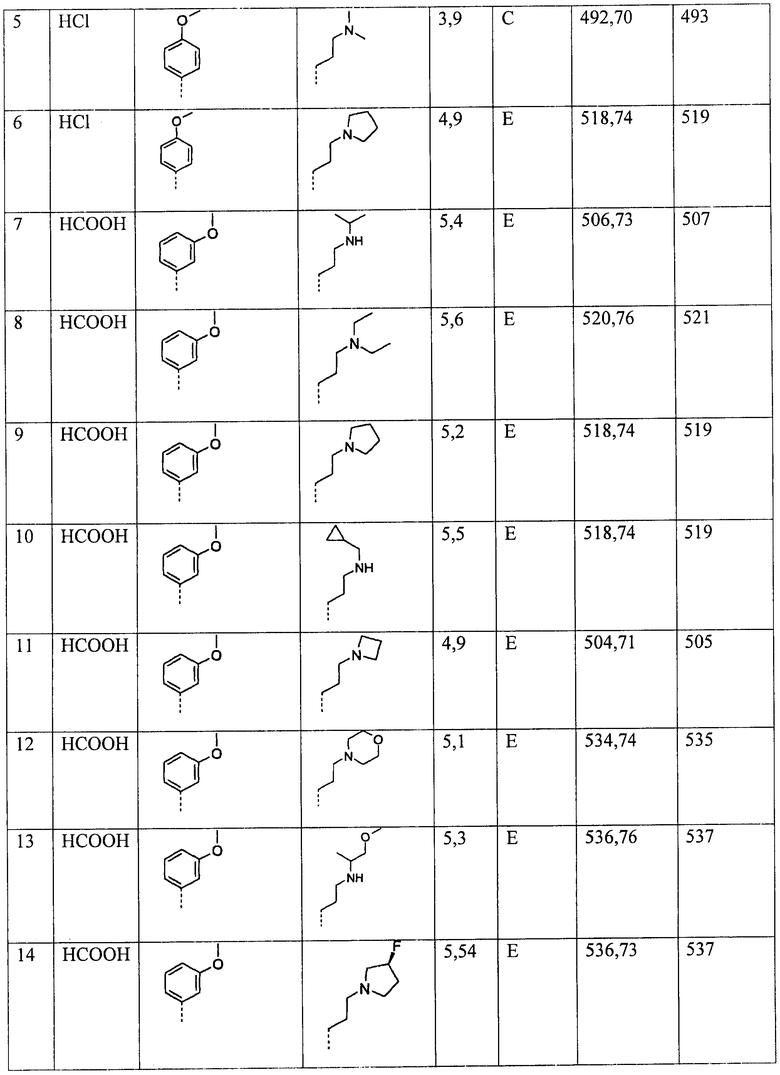
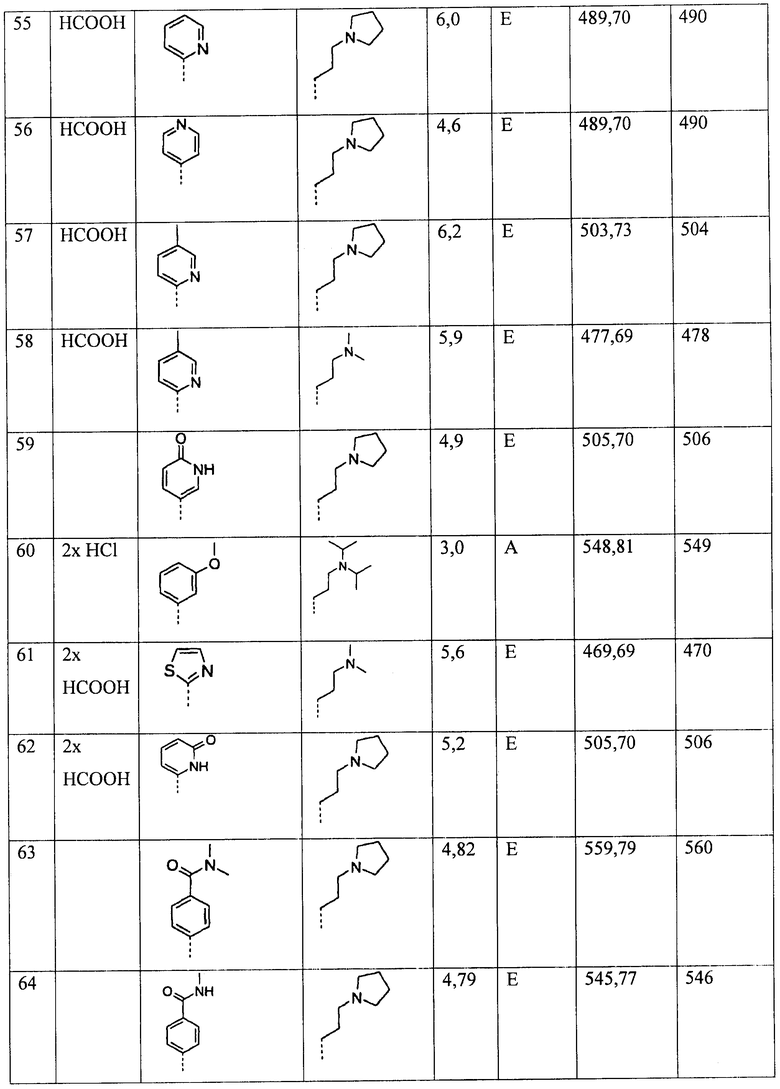
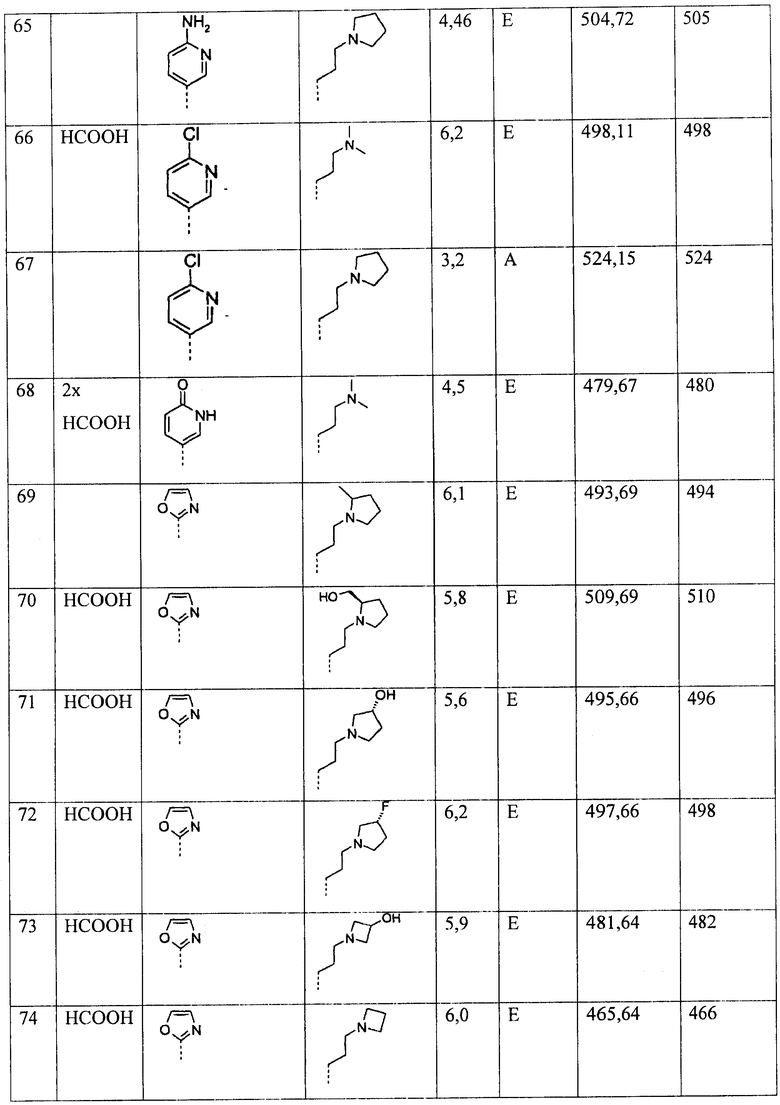
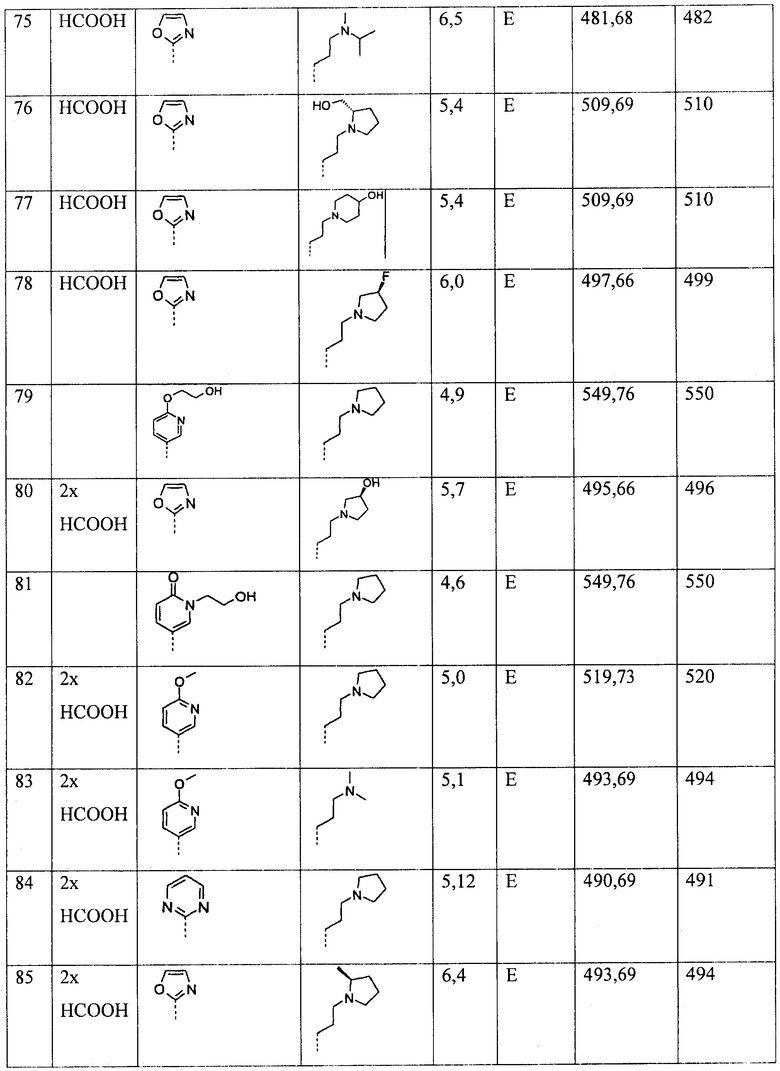
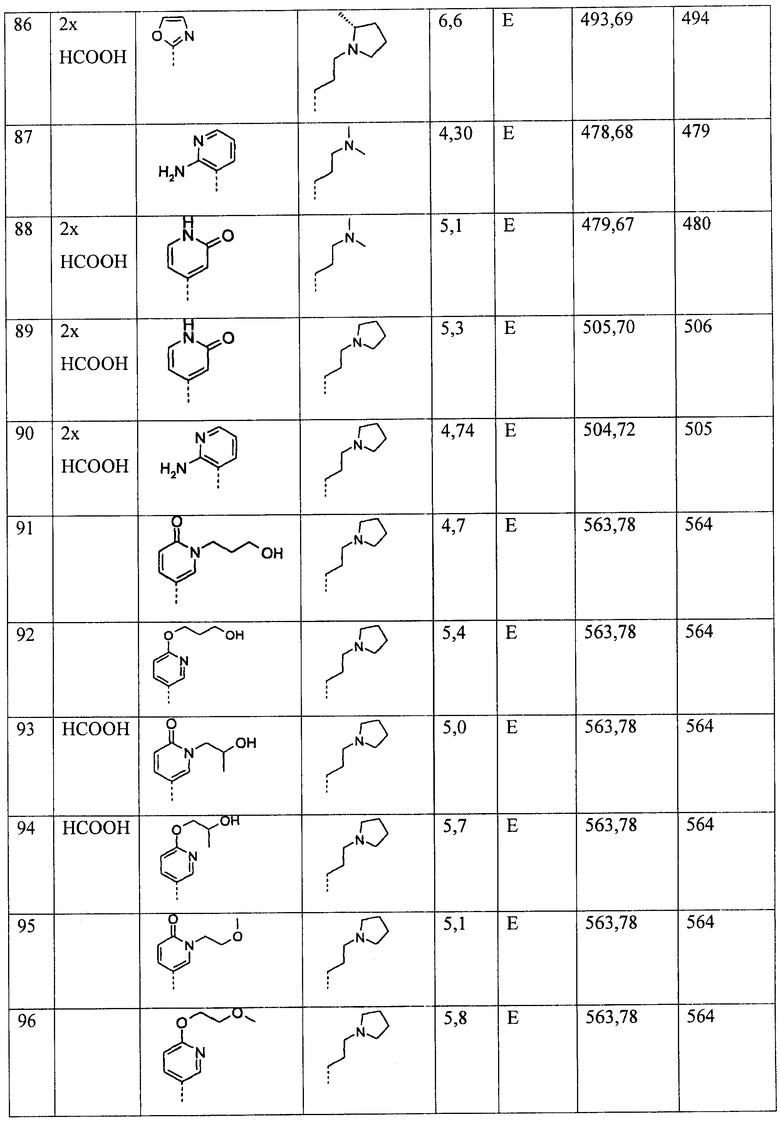
Ниже приведены некоторые иллюстративные неограничивающие примеры осуществления изобретения.
Синтез Примера 9:
Промежуточное соединение 9а):
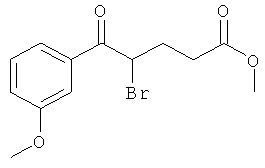
Суспензию CuBr2 (1071 мг) в ЕtOАс (10 мл) нагревали до кипения и добавляли раствор метилового эфира 5-(3-метокси-фенил)-5-оксо-пентановой кислоты (913 мг) в СНСl3. Реакционную смесь кипятили с обратным холодильником в течение ночи. Затем за один раз добавили дополнительное количество CuBr2 (300 мг), и смесь оставляли кипятиться еще 4 ч. Реакционную смесь профильтровали через целит для удаления солей меди, и растворитель испаряли в вакууме досуха. Остаток очищали флэш-хроматографией (ЕtOАс/циклогексан), получая названное соединение.
Промежуточное соединение 9b):
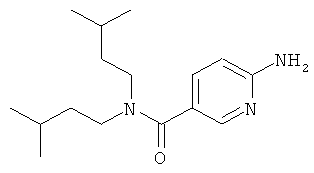
К раствору 6-амино-никотиновой кислоты (3 г) в ДМФА/ДХМ (80/20) добавили диизоамиламин (4.1 г), EDC (5 г), HOBt (3.52 г) и DIEA (4.54 мл). Реакционную смесь перемешивали при 50°С в течение ночи. Раствор испаряли в вакууме досуха. Остаток повторно растворяли в небольшом количестве ДМФА и добавляли буфер (рН 7). Полученный осадок собирали, промывали водой и сушили, получая названное соединение.
Промежуточное соединение 9с):
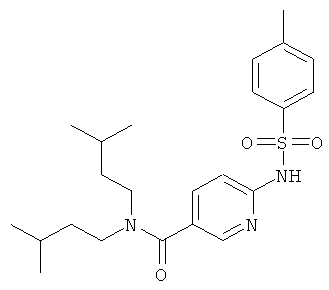
К раствору промежуточного соединения 9b) (1 г) в пиридине (25 мл) добавляли за один раз п-толуолсульфонилхлорид (756 мг). Реакционную смесь нагревали при 85°С в течение 16 ч. Раствор испаряли в вакууме досуха и к остатку добавляли Н2O. Полученную взвесь перемешивали в течение 30 мин, затем отфильтровали и промывали толуолом. Оставшееся твердое вещество собирали и высушивали, получая названное соединение в виде желтого твердого вещества.
Промежуточное соединение 9d):
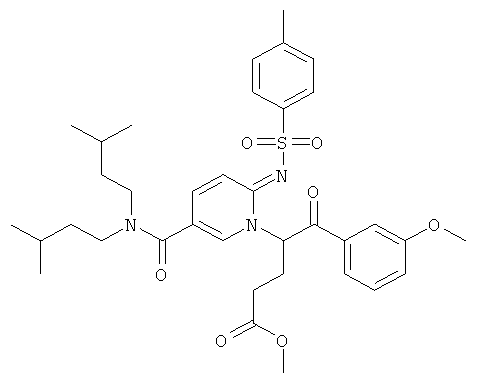
К теплому раствору промежуточного соединения 9с) (1781 мг) и DIEA (1,198 мл) в MeCN (50 мл) добавляли промежуточное соединение 9а) (1084 мг). Реакционную смесь нагревали при 100°С в течение 4 ч. Раствор испаряли в вакууме досуха, и остаток очищали флэш-хроматографией (EtOAc/циклогексан), получая названное соединение.
Промежуточное соединение 9е):
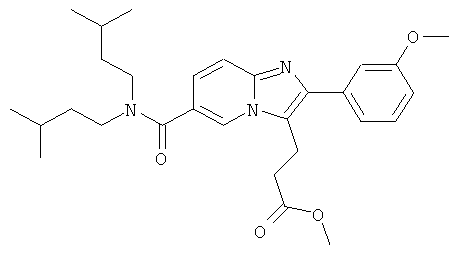
Промежуточное соединение 9d) (1,372 г) растворяли в ДХМ (50 мл), и реакционную смесь продували аргоном. К этому раствору добавляли TFAA (2 мл), и реакционную смесь оставляли перемешиваться в течение 16 ч. Раствор испаряли в вакууме досуха, и остаток очищали флэш-хроматографией (ДХМ/МеОН), получая названное соединение в виде бесцветной пены.
Промежуточное соединение 9f):
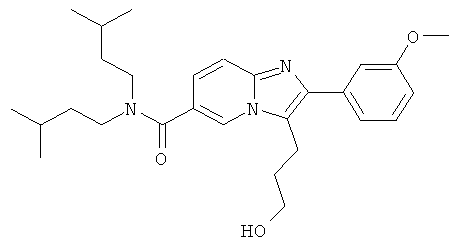
Промежуточное соединение 9е) (568 мг) растворяли в ТГФ (40 мл) и добавляли метанол (4 мл). Раствор нагревали до кипения и затем добавляли боргидрид натрия (87 мг). Далее боргидрид натрия добавляли несколькими порциями, и кипячение продолжали. Реакцию останавливали добавлением ацетона, и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (ДХМ/МеОН 98:2), получая названное соединение.
Промежуточное соединение 9g):
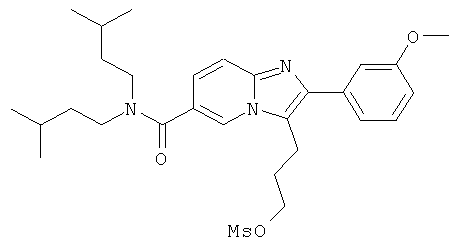
Раствор промежуточного соединения 9f) в сухом ДХМ (10 мл) продували аргоном и охлаждали до 0°С. К этому раствору добавляли ТЭА (170 мкл) и метилсульфонилхлорид (95 мкл), и реакционную смесь оставляли нагреваться. Перемешивание продолжали в течение 3 ч. Реакционную смесь промывали Н2O и насыщенным раствором NaHCO3. Органический слой сушили над Na2SO4 и испаряли в вакууме досуха, получая названное соединение, которое использовали в следующей операции без дальнейшей очистки.
Пример 9:
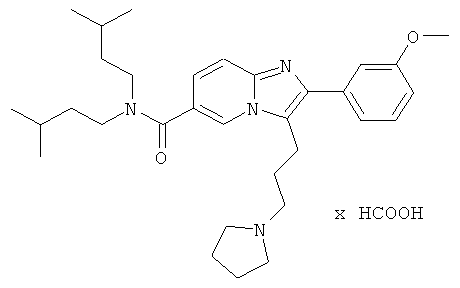
К раствору пирролидина (168 мг) в сухом MeCN (5 мл) добавляли раствор промежуточного соединения 9g) (86 мг) в сухом MeCN. Реакционную смесь нагревали до 75°С в течение 14 ч. Раствор испаряли в вакууме досуха, и остаток очищали препаративной ВЭЖХ, получая названное соединение в виде бесцветного твердого вещества.
Пример синтеза 17:
Промежуточное соединение 17а):
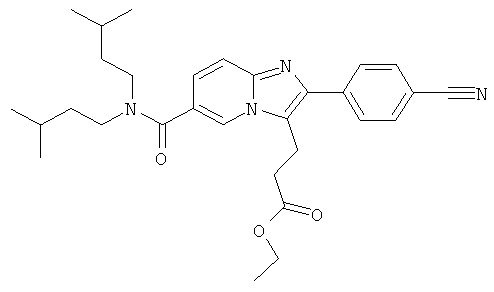
Синтез был проведен, как указано для промежуточного соединения 9е).
Промежуточное соединение 17b):
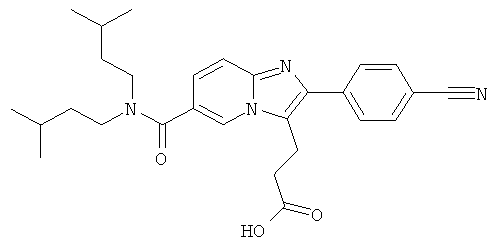
К раствору промежуточного соединения 17а) (370 мг) в ТГФ (15 мл) добавляли 2М раствор гидроксида лития в воде (0.74 мл) при 0°С. Затем ледяную баню удаляли, и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 суток. Смесь разбавляли этилацетатом и солевым раствором, и рН доводили до рН 6 раствором 3%-ной лимонной кислоты. После разделения слоев, органический слой промывали солевым раствором и сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении, получая названное соединение, которое использовали в следующей операции без дальнейшей очистки.
Промежуточное соединение 17с):
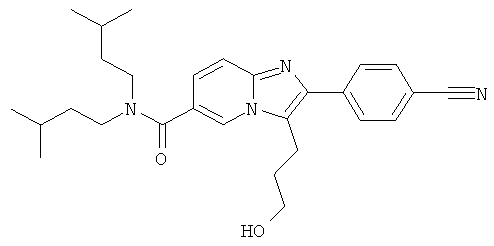
Промежуточное соединение 17b) (315 мг) растворяли в ТГФ (20 мл) и добавляли карбонилдиимидазол (162 мг). Смесь перемешивали при комнатной температуре в течение 1 ч и затем охлаждали до 0°С. Добавляли раствор боргидрида натрия (38 мг) и воде, и перемешивание продолжали еще 10 мин. Реакцию гасили добавлением ацетона, и растворитель удаляли в вакууме. Остаток растворяли в этилацетате и воде. После разделения слоев, органический слой промывали 5% раствором лимонной кислоты. насыщенным раствором бикарбоната натрия и солевым раствором. После сутки над сульфатом натрия, растворитель удаляли в вакууме, и остаток очищали флеш-хроматографией (ДХМ/МеОН), получая названное соединение.
Промежуточное соединение 17d):
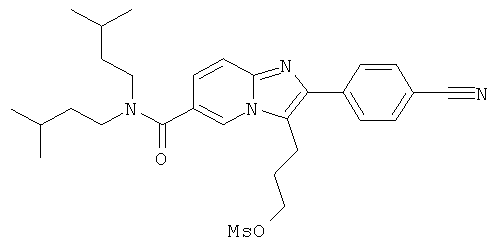
Промежуточное соединение 17с) (100 мг) растворяли в дихлормстапе (10 мл) и при 0°С добавляли метилсульфонилхлорид (0,026 мл) и триэтиламин (0,046 мл). Ледяную баню удаляли, и смесь перемешивали при комнатной температуре, и зачем добавляли дополнителное количество метилсульфонилхлорида (0,007 мл) и триэтиламина (0,012 мл), и перемешивание продолжали еще час. Смесь разбавляли дихлорметаном и промывали водой/насыщенным раствором бикарбоната натрия и водой. Органический слой сушили над сульфатом натрия, и растворитель удаляли к вакууме, получая названное соединение, которое использовали в следующей операции без дальнейшей очистки.
Пример 17:
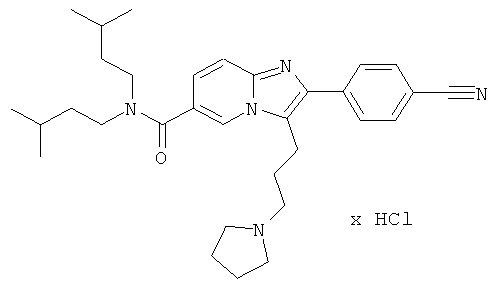
К раствору пирролидина (78 мг) в сухом МеCN (1,5 мл) добавляли раствор промежуточного соединения 17d) (55 мг) в сухом ацетонитриле. Реакционную смесь нагревали до 50°С в течение ночи. Смесь испаряли в вакууме досуха, и остаток очищали флэш-хроматографией (ДХМ/МеОН), получая названное соединение. Свободное основание превращали в соответствующую соль HCl.
Пример синтеза 21:
Промежуточное соединение 21а):
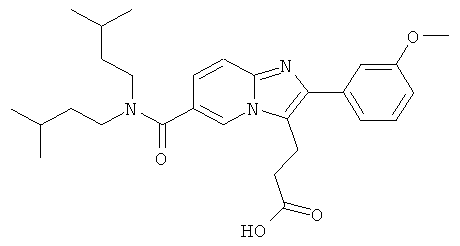
К раствору промежуточного соединения 9е) (300 мг) в ТГФ (12 мл) добавляли при 0°С 2M раствор гидроксида лития в воде (0.61 мл). Затем ледяную баню удаляли, и реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Смесь разбавляли этилацетатом и солевым раствором и pH доводили до pH 6 раствором 3% лимонной кислоты. После разделения слоев, органический слой промывали солевым раствором и сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении, получая названное соединение, которое использовали и следующей операции без дальнейшей очистки.
Промежуточное соединение 21b):
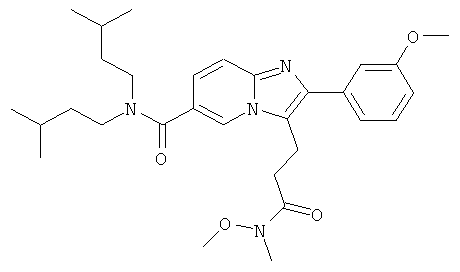
Промежуточное соединение 21а) (275 мг) растворяли в дихлорметане и добавляли карбонилдиимидазол (102 мг). Смесь перемешивали при комнатной температуре в течение 30 мин, и затем добавляли гидрохлорид N,O-диметилгидроксиламина (62 мг) и диизопропилэтиламин (0,110 мл). Перемешивание продолжали в течение ночи. Смесь разбавляли дихлорметаном, и органическую фазу промывали 5% раствором лимонной кислоты, раствором бикарбоната натрия и солевым раствором и затем сушили над сульфатом натрия и удаляли растворитель в вакууме. Очистка флэш-хроматографией (ДХМ/МеОН) позволила получить названное соединение.
Промежуточное соединение 21с):
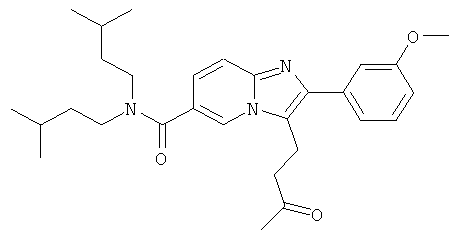
К раствору промежуточного соединения 21b) (100 мг) в сухом тетрагидрофуране добавляли раствор метиллития (1,5М в диэтиловом эфире, 0,12 мл) при -78°С. Смесь перемешивали в течение 30 мин и затем гидролизовали насыщенным раствором хлорида аммония. После разбавления диэтиловым эфиром, слои разделяли, и водный слой дважды экстрагировали диэтиловым эфиром. Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, и растворитель удаляли в вакууме. Очистка флэш-хроматографией позволила получить названное соединение.
Пример 21:
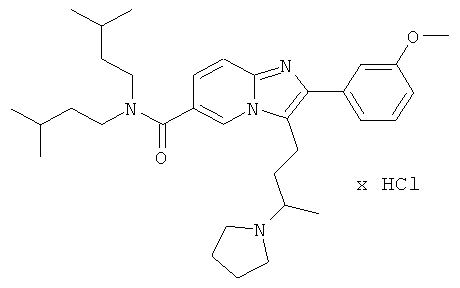
Промежуточное соединение 21с) (65 мг) и пирролидин (0,013 мл) растворяли в дихлорэтане (2 мл) и затем добавляли ледяную уксусную кислоту (0,008 мл) и триацетоксиборгидрид натрия (42 мг). Смесь перемешивали при комнатной температуре в течение 3 суток. Растворитель удаляли при пониженном давлении, и остаток очищали флэш-хроматографией (ДХМ/МеОН), получая названное соединение. Свободное основание превращали в соль НСl.
Пример синтеза 25:
Пример 25:
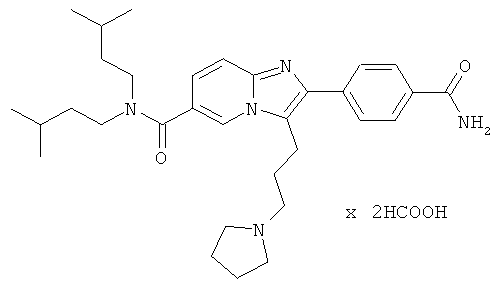
К раствору Примера 17 (407 мг) в трет-бутиловом спирте (8 мл) добавляли мелко раздробленный гидроксид калия (240 мг), и смесь нагревали до 70°С в течение 4 часов. Затем смесь разделяли между солевым раствором и этилацетатом. Водную фазу трижды экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении, получая названное соединение, которое очищали препаративной ВЭЖХ.
Пример синтеза 27:
Промежуточное соединение 27а):
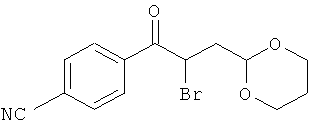
Бромид меди (547 мг) суспендировали в этилацетате (13 мл) и нагревали до кипения. Затем добавляли 4'-циано-3-(1,3-диоксан-2-ил)пропиофенон (500 мг) в хлороформе (13 мл). Реакционную смесь кипятили с обратным холодильником в течение 2 ч. Добавляли еще 340 мг бромида меди двумя порциями и затем кипятили 2 часа. Смесь перемешивали в течение ночи при комнатной температуре и затем фильтровали через целит. Растворители испаряли при пониженном давлении, и продукт очищали хроматографией.
Промежуточное соединение 27b):
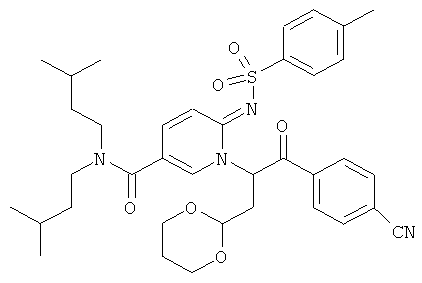
Смесь промежуточного тозилата 9с) (700 мг) и диизопропилэтиламина (0.52 мл) в ацетонитриле (20 мл) нагревали до 50°С. Затем добавляли промежуточное соединение 27а) (480 мг) в ацетонитриле, и реакционную смесь перемешивали при 50°С в течение 30 мин, и при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Смесь очищали флэш-хроматографией, получая названное соединение.
Промежуточное соединение 27с):
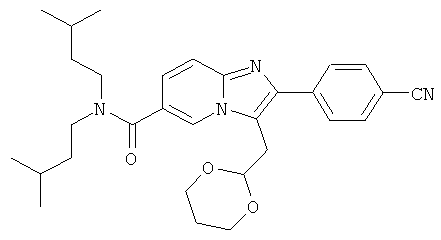
Промежуточное соединение 27b) (620 мг) растворяли в сухом дихлорметане (16 мл). Смесь охлаждали до 0°С на ледяной бане. Затем добавляли трифторуксусный ангидрид (1,62 мл). Смесь перемешивали при 0°С в течение 30 минут и затем при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении. Промежуточный продукт 27с) использовали в следующей операции без очистки.
Промежуточное соединение 27d):
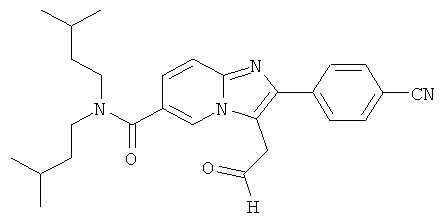
Промежуточное соединение 27с) (460 мг) растворяли в тетрагидрофуране (16 мл), и раствор охлаждали до 0°С. Добавляли 6 М НСl (0.46 мл), и затем реакционную смесь перемешивали при 60°С в течение ночи. Добавляли еще 3 эквивалента 6н. HСl, и смесь продолжали перемешивать при 60°С. Смесь нейтрализовали карбонатом натрия, и продукт экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении, и смесь очищали флэш-хроматографией, получая названное соединение.
Пример 27:
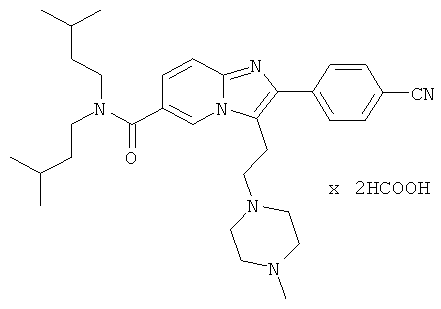
Промежуточное соединение 27d) (19 мг) растворяли в дихлорэтане (0.3 мл), добавляли 1-метилпиперазин (5 мкл), и реакционную смесь перемешивали в течение 30 мин. После добавления триацетоксиборгидрида натрия (12 мг), смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду, и водную фазу дважды экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия, и растворитель удаляли при пониженном давлении. Названное соединение очищали препаративной ВЭЖХ.
Пример синтеза 47:
Промежуточное соединение 47а):
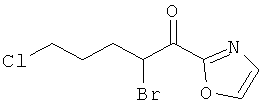
В атмосфере аргона, к перемешиваемой суспензии бромида меди (II) (9.523 г) в этилацетате (100 мл) добавляли 2-(5-хлорвалерил)оксазол (4.000 г) в хлороформе (100 мл). Полученную смесь перемешивали при температуре кипения в течение ночи. Реакционную смесь фильтровали через целит, и фильтрат испаряли досуха. Сырой продукт очищали колоночной хроматографией.
Промежуточное соединение 47b):
При 50°С к перемешиваемому раствору промежуточного соединения 47а) (2665 мг) в ацетонитриле (75 мл) добавляли DIEA (3658 мкл). Полученный раствор перемешивали в течение 15 минут, и затем добавляли промежуточное соединение 9с) (4532 мг) в ацетонитриле (75 мл). Полученный раствор перемешивали при 50°С в течение 3 ч. Летучие вещества удаляли, и продукт очищали колоночной хроматографией.
Промежуточное соединение 47с):
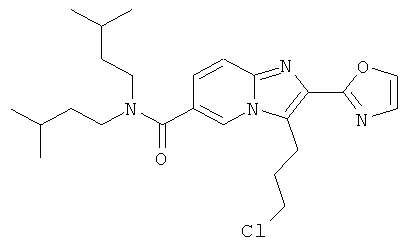
При 0°С, к перемешиваемому раствору промежуточного соединения 47b) (4.65 г) в сухом ДХМ (45 мл) добавляли TFAA (5 мл). Реакционную смесь затем оставляли нагреваться до к.т. и перемешивали в течение 3 ч. Реакционную смесь нейтрализовали насыщенным раствором NaHCO3, и затем фазы разделяли. Органический слой дважды экстрагировали насыщенным раствором NаНСО3. Объединенные водные слои снова экстрагировали ДХМ. Объединенные органические слои промывали солевым раствором, сушили над Nа2СО3, фильтровали, и летучие вещества удаляли. Сырой продукт очищали колоночной хроматографией.
Пример 47:
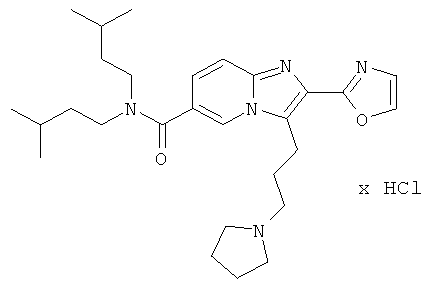
Промежуточное соединение 47с) (1068 мг) растворяли в ацетонитриле (100 мл). Добавляли пирролидин (2003 мкл), и реакционную смесь перемешивали при 70°С в течение 8 ч. Летучие вещества удаляли, и сырой продукт очищали препаративной ЖХ-МС. Очищенное соединение растворяли в этилацетате и промывали насыщенным водным раствором бикарбоната натрия. Водную фазу трижды экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, и растворитель удаляли. Полученное масло растворяли в этилацетате (10 мл) и добавляли 1 М НСl в диэтиловом эфире (2 мл). Летучие вещества удаляли, и продукт получали в виде беловатого порошка.
Пример синтеза 50:
Пример 50:
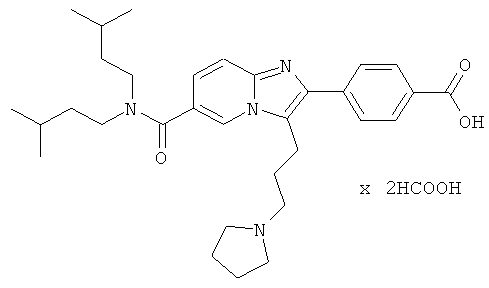
Вещество Примера 25 (422 мг) растворяли в концентрированной соляной кислоте и кипятили с обратным холодильником в течение 2 часов. Растворитель удаляли при пониженном давлении, получая названное соединение, которое очищали препаративной ВЭЖХ.
Пример синтеза 54:
Промежуточное соединение 54а):
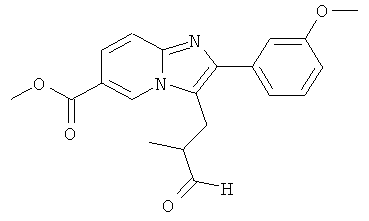
Смесь метилового эфира 2-(3-метокси-фенил)-имидазо[1,2-а]пиридин-6-карбоновой кислоты (1000 мг), метакролеина (990 мг), уксусного ангидрида (5.5 мл) и ледяной уксусной кислоты (14.5 мл) нагревали в микроволновом реакторе при 180°С в течение 75 минут. Затем летучие вещества удаляли при пониженном давлении. Добавляли метанол и 1н. водный раствор бикарбоната натрия, и смесь перемешивали в течение 2 ч. Растворители удаляли, и остаток растворяли в этилацетате и воде. Органический слой разделяли и сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении, получая названное соединение, которое использовали в следующей операции без дальнейшей очистки.
Промежуточное соединение 54b):
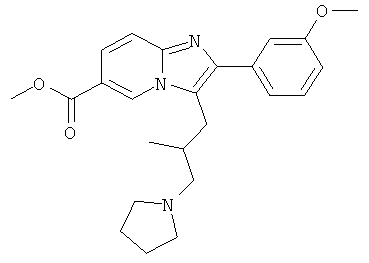
Промежуточное соединение 54а) (450 мг) растворяли в дихлормстаис (27 мл), добавляли пирролидин (0,11 мл), и смесь перемешивали в течение 30 мин при комнатной температуре. Затем добавляли триацетоксиборгидрид натрия (360 мг), и реакционную смесь перемешивали в течение ночи. Добавляли воду, и водную фазу дважды экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия, и растворитель удаляли при пониженном давлении. Продукт очищали флэш-хроматографией.
Промежуточное соединение 54с):
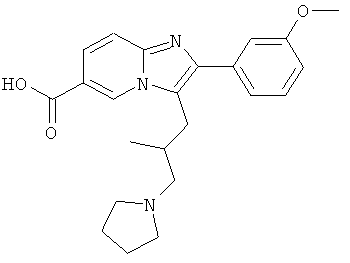
Промежуточное соединение 54b) (78 мг) растворяли в тетрагидрофуране (3.5 мл) и охлаждали до 0°С. Затем добавляли гидроксид лития (0,19 мл, 2н. в воде), и смесь оставляли нагреваться до комнатной температуры, и перемешивали в течение 2 суток. Добавляли этилацетат и солевой раствор. Белый осадок растворяли, добавляя по каплям лимонную кислоту (5%). Органический слой разделяли и сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении, получая промежуточное соединение 54с).
Пример 54:
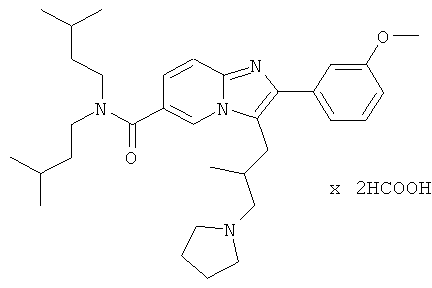
Промежуточное соединение 54с) (38 мг) растворяли в ДМФА (5 мл). Затем добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 44 мг), диизопропилэтиламин (20 мкл) и диизоамиламин (24 мкл), и смесь перемешивали в течение ночи при комнатной температуре. Растворители испаряли. Продукт затем растворяли в этилацетате. Органический слой промывали солевым раствором, затем насыщенным раствором бикарбоната натрия и вновь солевым раствором. Органический слой сушили над сульфатом натрия, отфильтровывали, и растворитель испаряли. Продукт очищали препаративной ВЭЖХ, получая соединение Примера 54.
Пример синтеза 58:
Промежуточное соединение 58а):
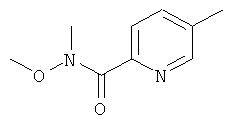
При 0°С, к S-метилпиридин-карбоновой кислоте (1000 мг) в ДХМ (25 мл) добавляли EDC (1608 мг). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем добавляли гидрохлорид N,O-диметилгидроксиламина (818 мг), а затем NMM (922 мкл). Реакционную смесь оставляли нагреваться до к.т. и перемешивали в течение ночи. Реакционную смесь разбавляли ДХМ (25 мл), и затем экстрагировали насыщенным раствором NaHCO3 (2×25 мл). Водный слой снова экстрагировали ДХМ (25 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, отфильтровывали, и растворители удаляли. Сырой продукт очищали колоночной хроматографией.
Промежуточное соединение 58b):
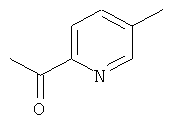
При -78°С, в атмосфере аргона, к перемешиваемому раствору промежуточного соединения 58а) (1270 мг) в сухом ТГФ (10 мл) осторожно добавляли метиллитий (1,6 М в Et2O, 13.2 мл). Реакционную смесь перемешивали при -78°С в течение 90 минут, и затем гидролизовали насыщенным раствором NH4Cl (10 мл). Реакционную смесь разбавляли диэтиловым эфиром (50 мл). Водный слой снова экстрагировали диэтиловым эфиром (2×10 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, отфильтровывали, и растворитель осторожно удаляли. Продукт очищали перегонкой в вакууме в аппарате Kugelrohr (10 мбар, 130°С).
Промежуточное соединение 58с):
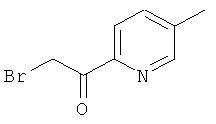
При к.т., к перемешиваемому раствору промежуточного соединения 58b) (541 мг) в АсОН (10 мл) добавляли 33% НВr в АсОН (2 мл), а затем бром (53 мкл). Спустя 30 минут добавляли дополнительное количество брома (50 мкл), и реакционную смесь перемешивали в течение 90 минут. Реакционную смесь концентрировали в вакууме и затем выливали в насыщенный водный раствор NаНСО3. Смесь трижды экстрагировали этилацетатом. Объединенные органические слои сушили над Nа2SO4, отфильтровывали, и растворители осторожно удаляли. Сырой продукт очищали колоночной хроматографией.
Промежуточное соединение 58d):
Промежуточное соединение 58с) (350 мг) и промежуточное соединение 9b) (522 мг) растворяли в MeCN (10 мл) и затем нагревали до 180°С в течение 30 мин под действием микроволнового излучения. Летучие вещества удаляли, и сырой продукт очищали колоночной хроматографией.
Промежуточное соединение 58е):
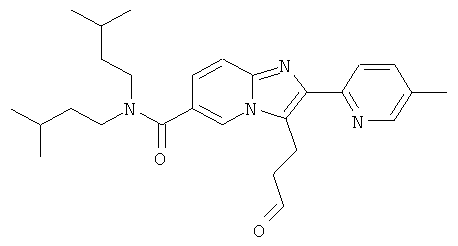
К раствору промежуточного соединения 58d) (381 мг) в ледяной уксусной кислоте (6 мл) добавляли акролеин (233 мкл), затем уксусный ангидрид (2 мл), и смесь нагревали при 180°С в микроволновом реакторе в течение 30 мин. Реакционную смесь вливали в смесь насыщенного водного бикарбоната натрия (100 мл) и насыщенного водного карбоната натрия (50 мл) до получения щелочного pH, и затем экстрагировали этилацетатом (2×50 мл). Органический слой промывали солевым раствором, сушили над Na2SO4, отфильтровывали, и растворитель удаляли. К сырому продукту в метаноле (50 мл) добавляли 1М раствор бикарбоната натрия в воде (10 мл). Реакционную смесь перемешивали при к.т. в течение ночи. Реакционную смесь концентрировали в вакууме и затем разделяли между насыщенным водным раствором NaНСО3 и ДХМ. Водный слой дважды экстрагировали ДХМ. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, отфильтровали, и растворитель удаляли.
Пример 58:
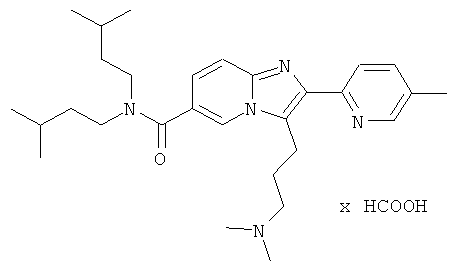
Промежуточное соединение 58е) (83 мг) и 2М диметиламин в ТГФ (463 мкл) растворяли в 1,2-дихлорэтане (5 мл). Перемешивали 1 ч при к.т. и добавляли триацетоксиборгидрид натрия (782 мг). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь экстрагировали 1 М NaHCO3 (2×2 мл). Водный слой снова экстрагировали ДХЭ (2 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, отфильтровали и испаряли досуха в вакууме. Сырой продукт очищали препаративной ЖХ-МС.
Пример синтеза 59:
Промежуточное соединение 59а):
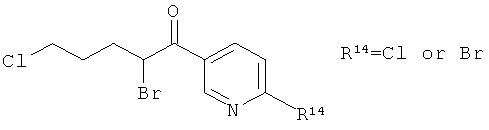
В атмосфере аргона, к перемешиваемой суспензии бромида меди (II) (19.36 г) в этилацетате (200 мл) добавляли 6-хлор-3-(5-хлорвалерил)-пиридин (10.06 г) в хлороформе (200 мл). Полученную смесь перемешивали при кипячении в течение 20 ч. Реакционную смесь фильтровали на целите и концентрировали в вакууме.
Остаток от фильтрования промывали ацетонитрилом, и фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате (500 мл) и промывали насыщенным раствором бикарбоната натрия (500 мл). Водный слой дважды экстрагировали этилацетатом. Органические слои объединяли с первой выделенной порцией, промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором, сушили над MgSO4, отфильтровывали, и растворитель удаляли при пониженном давлении.
Промежуточное соединение 59b):
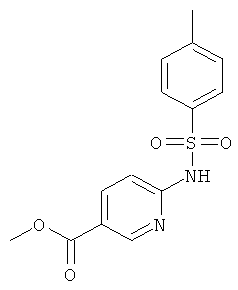
К раствору метилового эфира 6-аминоникотиновой кислоты (20,09 г) в сухом пиридине (400 мл) в атмосфере аргона добавляли тозилхлорид (28.94 г). Реакционную смесь перемешивали при 85°С в течение 16 ч. Растворитель удаляли при пониженном давлении, остаток растворяли в воде и перемешивали в течение 2 ч. Образовывался бежевый осадок. Его отфильтровывали, дважды промывали водой и сушили над Sicapent.
Промежуточное соединение 59с):
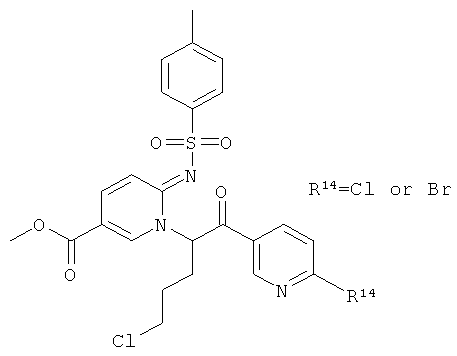
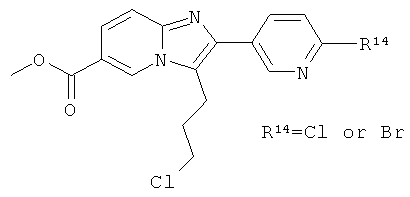
Смесь промежуточного соединения 59b) (9.74 г) и промежуточного соединения 59а) (10.24 г) в ацетонитриле (300 мл) обрабатывали этилдиизопропиламином (6654 мкл) и перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Продукт очищали флэш-хроматографией.
Промежуточное соединение 59d):
Промежуточное соединение 59с) (15,07 г) растворяли в сухом ДХМ (180 мл) и охлаждали до 0°С. Добавляли ангидрид трифторуксусной кислоты (20 мл), и реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь разбавляли ДХМ (200 мл) и осторожно экстрагировали насыщенным раствором бикарбоната натрия. Водный слой дважды экстрагировали ДХМ. Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия и растворитель удаляли при пониженном давлении.
Промежуточное соединение 59е):
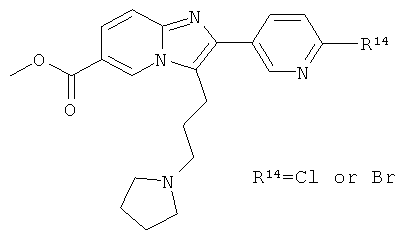
Промежуточное соединение 59d) (5,0 г), ацетонитрил (120 мл) и пирролидин (10.9 мл) перемешивали при 70°С в течение ночи. Растворитель удаляли при пониженном давлении. Продукт обрабатывали добавлением ацетона, отфильтровывали и сушили в вакуумной печи в течение ночи.
Промежуточное соединение 59f):
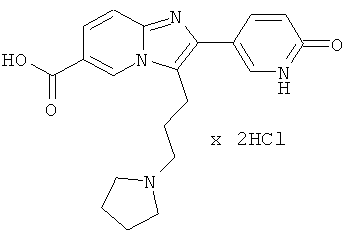
Промежуточное соединение 59е) (4.6 г) растворяли в 3М НСl в воде (150 мл), и реакционную смесь перемешивали при 120°С в течение 36 ч. Растворитель удаляли при пониженном давлении, остаток дважды совместно испаряли с толуолом, и продукт сушили в высоком вакууме в течение 2 суток. Продукт непосредственно использовали в следующей операции без дальнейшей очистки.
Пример 59:
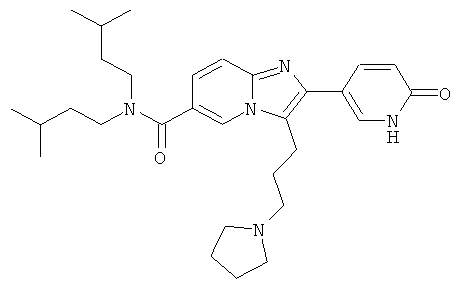
Промежуточное соединение 59f) (1000 мг) растворяли в ДМФА (10 мл) и затем добавляли EDC (525 мг), HOAt (373 мг) и DIEA (1431 мкл). Реакционную смесь перемешивали в течение 1 ч. Добавляли диизоамиламин (562 мкл), и реакционную смесь перемешивали в течение ночи. Летучие вещества удаляли, и остаток растворяли в этилацетате (200 мл). Органический слой промывали насыщенным раствором NaHCO3 (2×100 мл), и затем объединенные водные слои снова экстрагировали этилацетатом (100 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали препаративной ЖХ-МС.
Пример синтеза 64:
Пример 64:
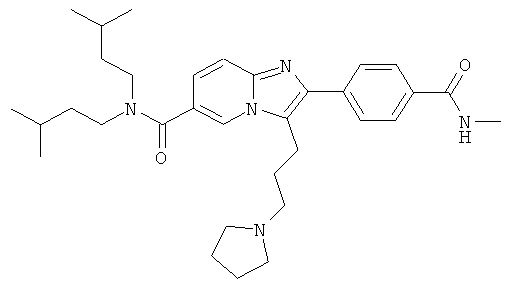
Вещество Примера 50 (200 мг), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 185 мг) и диизопропилэтиламин (1,95 мл) в диметилформамиде (4 мл) перемешивали в течение 15 минут при комнатной температуре, затем добавляли 2н. раствор метиламина в тетрагидрофуране (0.28 мл), и перемешивание продолжали в течение ночи. Добавляли этилацетат. Органический слой промывали насыщенным раствором бикарбоната натрия и солевым раствором и сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении, получая названное соединение, которое очищали препаративной ВЭЖХ.
Пример синтеза 79:
Промежуточное соединение 79а):
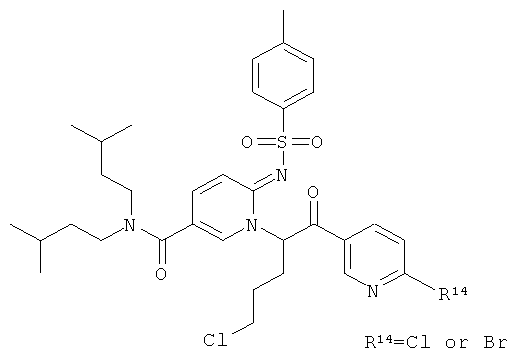
При 50°С к перемешиваемой суспензии промежуточного соединения 59а) (679 мг) в ацетонитриле (15 мл) добавляли DIEA (806 мкл). Полученный раствор перемешивали в течение 10 минут, и затем добавляли промежуточное соединение 9с) (993 мг) в ацетонитриле (15 мл). Полученный раствор перемешивали при 50°С в течение 2 ч. Растворители удаляли, и полученное масло очищали колоночной хроматографией.
Промежуточное соединение 79b):
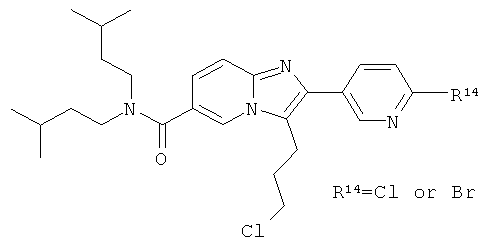
При 0°С, к перемешиваемому раствору промежуточного соединения 79а) (1,35 г) в сухом ДХМ (18 мл) добавляли TFAA (2 мл). Реакционную смесь затем оставляли нагреваться до к.т. и перемешивали в течение ночи. Летучие вещества удаляли, и полученное масло очищали колоночной хроматографией.
Промежуточное соединение 79с):
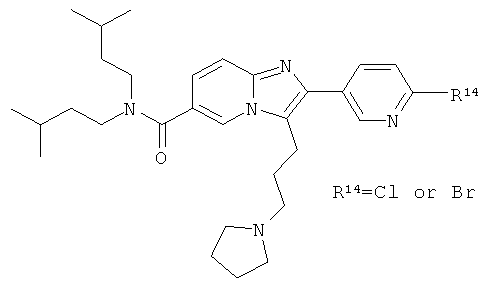
Промежуточное соединение 79b) (100 мг), ацетонитрил (5 мл) и пирролидин (167 мкл) перемешивали при 70°С в течение ночи. Растворитель удаляли при пониженном давлении. Продукт очищали колоночной хроматографией.
Пример 79:
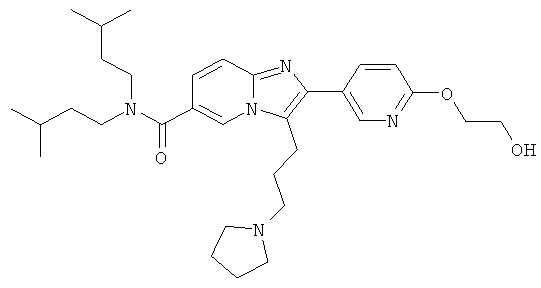
К суспензии гидрида натрия (31 мг) в ДМФА (5 мл) в атмосфере аргона добавляли этиленгликоль (22 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, и затем добавляли промежуточное соединение 79 с) (70 мг) в ДМФА (5 мл). Смесь перемешивали при 140°С в течение ночи. Смесь охлаждали и гидролизовали водой. Растворитель испаряли, и остаток растворяли в этилацетате. Органический слой экстрагировали водой и солевым раствором, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали с использованием препаративной ЖХ-МС.
Пример синтеза 90:
Промежуточное соединение 90а):
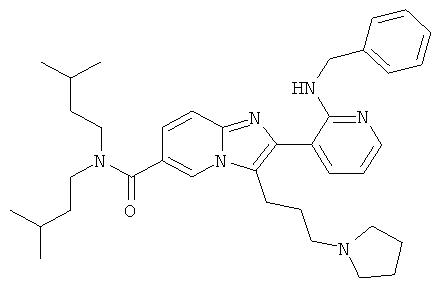
Бис-(3-метил-бутил)-амид 2-(2-хлорпиридин-3-ил)-3-(3-пирролидии-1-ил-пропил)-имидазо[1,2-а]пиридин-6-карбоновой кислоты (100 мг) и бензиламин (0.5 мл) нагревали в микроволновом реакторе при 150°С в течение 2 ч. Смесь очищали флэш-хроматографией, получая названное соединение.
Пример 90:
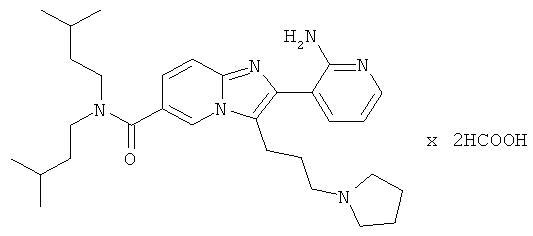
К промежуточному соединению 90а) (78 мг) в сухом дихлорметане (1,5 мл) при 0°С добавляли по каплям трифторметансульфоновую кислоту (300 мг). Затем смесь нагревали до 40°С в течение 3 часов. Смесь снова охлаждали до 0°С и добавляли еще 300 мг трифторметансульфоновой кислоты, а затем нагревали до кипения в течение 2 ч. Растворители испаряли при пониженном давлении. Названное соединение получали после очистки препаративной ВЭЖХ.
Примеры синтеза 91 и 92:
Примеры 91 и 92:
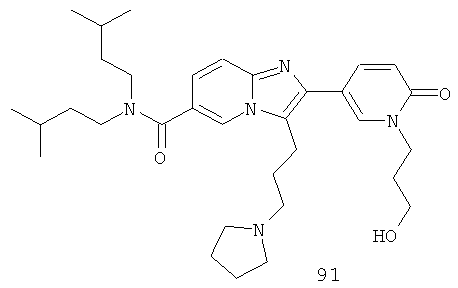
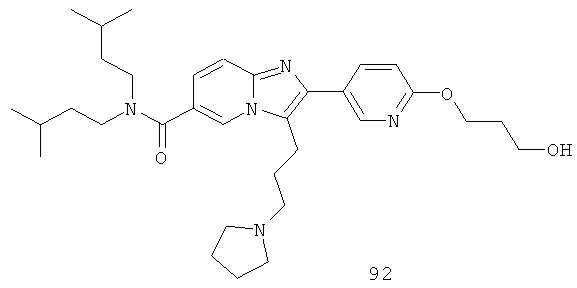
Вещество Примера 59) (100 мг) растворяли в ацетоне (5 мл) и затем добавляли карбонат калия (41 мг) и 3-бром-1-пропанол (20 мкл). Реакционную смесь перемешивали при 60°С в течение ночи, отфильтровывали, и растворитель удаляли при пониженном давлении. Продукты разделяли препаративной ЖХ-МС.
Пример синтеза 98
Промежуточное соединение 98а):
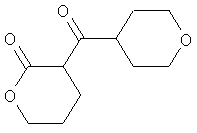
Перед использованием δ-валеролактон перегоняли при пониженном давлении (т.кип. 106°С/22 мбар). К перемешиваемой суспензии гидрида натрия (60% в масле, 2.66 г) в сухом толуоле (100 мл) при кипячении добавляли по каплям смесь метил тетрагидро-2Н-пиран-4-карбоксилата (8.87 мл) и δ-валеролактона (6.34 г) в сухом толуоле (50 мл). После добавления, реакционную смесь перемешивали при температуре кипения в течение ночи. После охлаждения до комнатной температуры, реакционную смесь выливали в ледяную воду (300 мл). Твердое вещество в колбе извлекали водой (150 мл) и толуолом (50 мл). После разделения фаз, водный слой подкисляли АсОН (5 мл) и экстрагировали этилацетатом (3×150 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали, и летучие вещества удаляли. Неочищенный продукт очищали колоночной хроматографией.
Промежуточное соединение 98b):
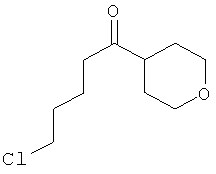
Промежуточное соединение 98а) (1,86 г) перемешивали в концентрированной соляной кислоте (10 мл) при 80°С в течение 1 ч. Реакционную смесь вливали в насыщенный раствор Na2CO3 (300 мл) и экстрагировали ДХМ (3×100 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4. фильтровали, и летучие вещества удаляли.
Промежуточное соединение 98с):
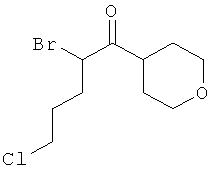
В атмосфере аргона, к перемешиваемой суспензии бромида меди (II) (1965 мг) в этилацетате (25 мл) добавляли промежуточное соединение 98b) (934 мг) в хлороформе (25 мл). Полученную смесь перемешивали при кипячении в течение ночи. Реакционную смесь фильтровали через целит, и слой целита тщательно промывали этилацетатом. Органический слой дважды экстрагировали насыщенным раствором NaHCO3. Водный слой снова экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали, и летучие вещества удаляли.
Промежуточное соединение 98d):
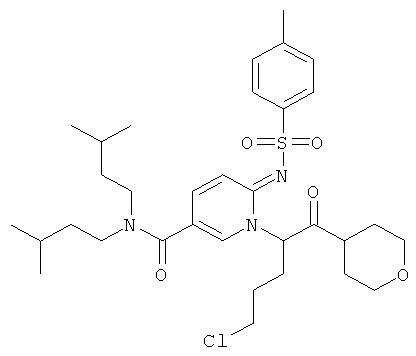
При 50°С к перемешиваемой суспензии 9с) (1994 мг) в ацетонитриле (50 мл) добавляли DIEA (1610 мкл). Полученный раствор перемешивали при 50°С в течение 15 минут, и затем добавляли промежуточное соединение 98с) (1,25 г) в ацетонитриле (50 мл). Полученную реакционную смесь перемешивали при 50°С в течение ночи. Летучие вещества удаляли, и неочищенный продукт очищали колоночной хроматографией.
Промежуточное соединение 98е):
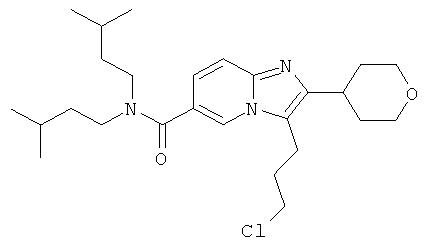
В атмосфере аргона, к раствору промежуточного соединения 98d) (525 мг) в ДХЭ (9 мл) добавляли TFAA (1 мл), и реакционную смесь перемешивали при к.т. в течение 4 суток. Добавляли дополнительное количество TFAA (1 мл) и ДХЭ (9 мл), и реакционную смесь нагревали до кипения в течение ночи. Летучие вещества удаляли, и остаток растворяли в этилацетате (50 мл). Органический слой дважды экстрагировали насыщенным раствором Na2CO3 (25 мл). Объединенные водные слои снова экстрагировали этилацетатом (25 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали, и летучие вещества удаляли. Неочищенный продукт очищали колоночной хроматографией.
Пример 98:
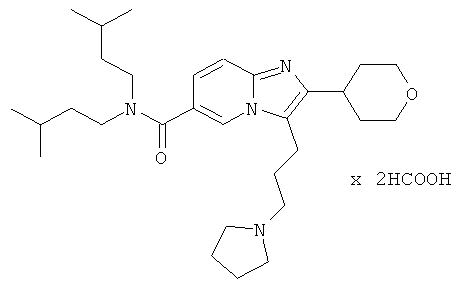
К перемешиваемому раствору промежуточного соединения 98е) (85 мг) в MeCN (10 мл) добавляли пирролидин (172 мкл), и реакционную смесь перемешивали в запаянной пробирке при 50°С в течение 2 суток. Летучие вещества удаляли, и неочищенный продукт очищали препаративной ЖХ-МС.
Пример синтеза 109:
Промежуточное соединение 109а):
К раствору циклопропилуксусной кислоты (2002 мг) в ДХМ (100 мл) при 0°С добавляли EDC (3834 мг) и HOBt (3063 мг). Реакционную смесь перемешивали при 0°С в течение 30 мин. Добавляли циклопропилэтиламин (1703 мг) и DIEA (10.45 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь экстрагировали 1.0 н. НСl (100 мл), насыщенным раствором NaHCO3 (100 мл), водой (100 мл) и солевым раствором (100 мл). Органический слой сушили над Na2SO4 и испаряли в вакууме досуха.
Промежуточное соединение 109b):
Раствор промежуточного соединения 109а) (2448 мг) в безводном ТГФ (40 мл) добавляли по каплям при комнатной температуре в атмосфере аргона к суспензии литийалюминийгидрида (1389 мг) в безводном ТГФ (60 мл). Реакционную смесь нагревали до температуры кипения и перемешивали еще 2 суток. Реакционную смесь гидролизовали добавлением 10% водного КОН (100 мл) при 0°С. После перемешивания в течение 20 мин при комнатной температуре, реакционную смесь профильтровали, и твердое вещество промывали диэтиловым эфиром (100 мл). Двухфазный фильтрат переносили в делительную воронку, и органический слой промывали водой и солевым раствором. Объединенные водные слои дважды промывали диэтиловым эфиром (2×100 мл). Объединенные органические слои промывали солевым раствором, сушили над сульфатом магния, и растворитель удаляли при пониженном давлении.
Промежуточное соединение 109с):
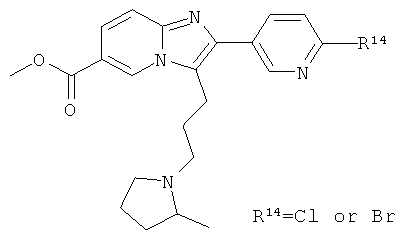
Промежуточное соединение 59d) (2.0 г), ацетонитрил (30 мл) и 2-метил-пирролидин (1,27 мл) перемешивали при 70°С в течение ночи. Добавляли DIEA (500 мкл), и смесь перемешивали при 70°С в течение 20 ч. Растворитель удаляли при пониженном давлении, и остаток растворяли в этилацетате и дважды промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором. Органический слой сушили над сульфатом натрия, отфильтровывали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией.
Промежуточное соединение 109d):
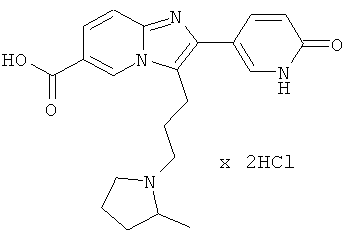
Промежуточное соединение 109с) (1,7 г) растворяли в 3М НСl в воде (100 мл), и реакционную смесь перемешивали при 120°С в течение 24 ч. Растворитель удаляли при пониженном давлении, остаток дважды совместно испаряли с толуолом, и продукт сушили в высоком вакууме в течение 3 ч, а затем в печи при 40°С при пониженном давлении в течение 3 суток. Продукт использовали как таковой в следующей операции без дальнейшей очистки.
Пример 109:
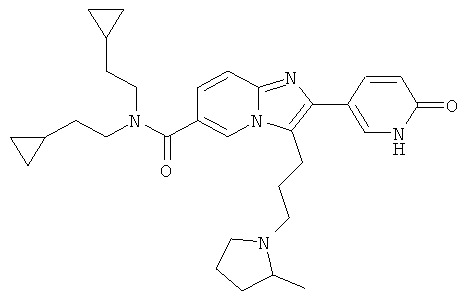
Промежуточное соединение 109d) (600 мг) растворяли в ДМФА (5 мл) и затем добавляли EDC (299 мг), НОAt (212 мг) и DIEA (815 мкл). Реакционную смесь перемешивали в течение 1 ч. Добавляли промежуточное соединение 109b) (239 мг), и реакционную смесь перемешивали в течение ночи. Летучие вещества удаляли, и затем остаток растворяли в этилацетате (100 мл). Органический слой промывали насыщенным раствором NаНСО3 (2×50 мл), затем объединенные водные слои снова экстрагировали этилацетатом (50 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали препаративной ЖХ-МС.
Пример синтеза 110:
Пример 110:
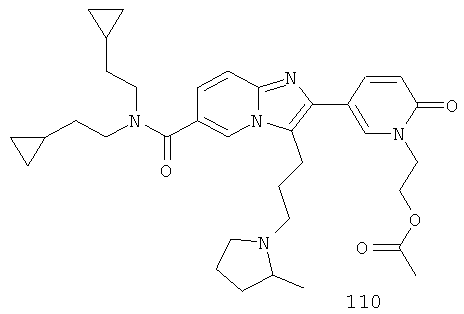
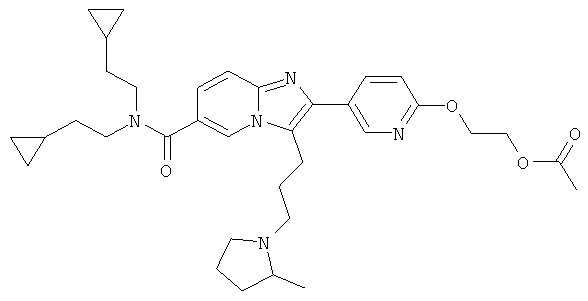
Вещество Примера 109 (230 мг) растворяли в ацетоне (7 мл) и затем добавляли карбонат цезия (222 мг) и 2-бромэтилацетат (55 мкл). Реакционную смесь перемешивали при 60°С в течение ночи, отфильтровывали, и растворитель удаляли при пониженном давлении. N- и О-алкилированные продукты разделяли препаративной ЖХ-МС.
Примеры синтеза 111 и 112:
Примеры 111 и 112:
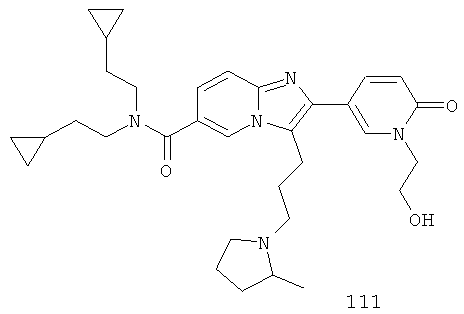
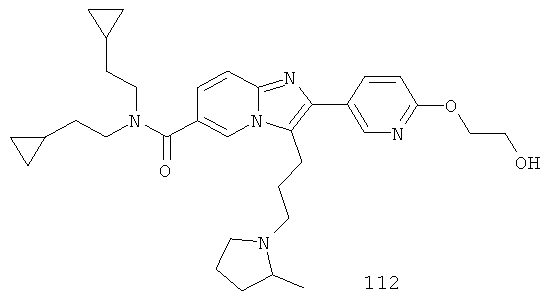
Смесь вещества Примера 110 и О-алкилированный побочный продукт (неочищенный продукт до очистки) (116 мг) растворяли в ТГФ (10 мл) и охлаждали до 0°С. Добавляли моногидрат гидроксида лития (30 мг) в воде (2 мл), и реакционную смесь перемешивали при 0°С в течение 30 мин и при комнатной температуре в течение 2.5 ч. Реакционную смесь концентрировали и затем разделяли между этилацетатом и водой. Водный слой трижды экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, и растворитель удаляли при пониженном давлении. Продукты разделяли препаративной ЖХ-МС.
Пример синтеза 114:
Промежуточное соединение 114а):
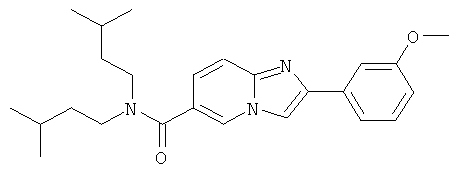
К раствору 6-амино-N,N-бис-(3-метил-бутил)-никотинамид (100 мг) в MeCN (2 мл) добавляли 2-бром-3'-метоксиацетофенон в MeCN (2 мл) и смесь нагревали при 170°С в микроволновом реакторе в течение 40 мин. Растворитель испаряли, и неочищенную смесь очищали флэш-хроматографией (EtOAc/циклогексап). получая названное соединение.
Пример 114:
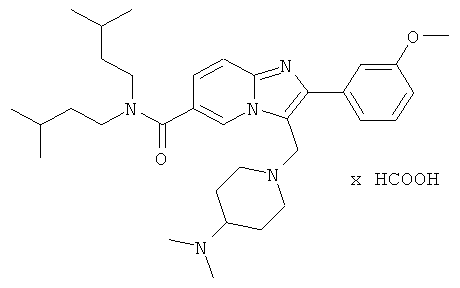
Раствор 4-диметиламинопиперидина (14.1 мг) в НОАс и формальдегида (37% водный раствор, 8.2 мкл) добавляли к промежуточному соединению 114а) (30 мг) в НОАс, и смесь нагревали при 60°С в течение 16 ч. После испарения растворителя, неочищенную реакционную смесь очищали препаративной ВЭЖХ, получая названное соединение.
Пример синтеза 119:
Промежуточное соединение 119а):
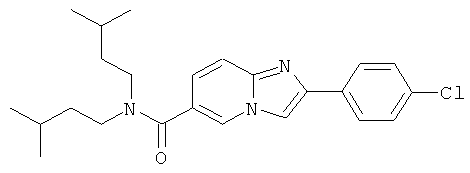
К раствору 6-амино-N,N-бис-(3-метилбутил)-никотинамида (1000 мг) в MeCN (10 мл) добавляли 2-бром-4'-хлорацетофенон в MeCN (10 мл), и смесь нагревали при 160°С в микроволновом реакторе в течение 15 мин. При охлаждении образовался осадок, который отделяли и высушивали, получая названное соединение.
Пример 119:
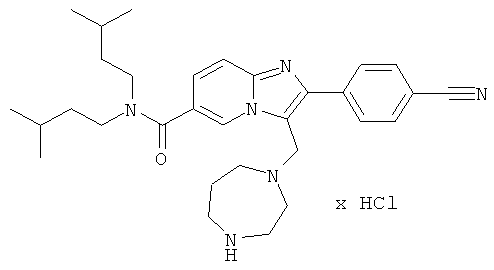
Раствор трет-бутилового эфира [1,4]диазепан-1-карбоновой кислоты (22.3 мг) в НОАс и формальдегид (37% водный раствор, 13.5 мкл) добавляли к промежуточному соединению 119а) (30 мг) в НОАс, и смесь нагревали при 60°С в течение 16 ч. После испарения растворителя, неочищенную реакционную смесь очищали флэш-хроматографией (ДХМ/МеОН). Полученное Вос-защищенное промежуточное соединение затем обрабатывали смесью 4М НСl/диоксан в течение 1 ч, получая названное соединение.
Пример синтеза 143:
Промежуточное соединение 143а):
2-Циклопропил-этиламин гидрохлорид (700 мг) суспендировали в дихлорметане (14 мл). Раствор охлаждали до 0°С, и затем добавляли изовалерилхлорид (0.84 мл) и триэтиламин (1,60 мл). Реакционную смесь оставляли нагреваться до комнатной температуры, и смесь перемешивали в течение 4 ч. Растворитель испаряли, и остаток разделяли между этилацетатом (50 мл) и насыщенным раствором NaHCO3 (40 мл). Водный слой дважды экстрагировали этилацетатом (50 мл), и объединенные органические фазы промывали солевым раствором (40 мл). Органический слой сушили над Na2SO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали препаративной ЖХ-МС.
Промежуточное соединение 143b):
Промежуточное соединение 143а) (838 мг) обрабатывали комплексом борантетрагидрофурана (1М раствор в ТГФ, 14.9 мл), и реакционную смесь перемешивали при кипячении в течение 6 ч. Затем реакционную смесь охлаждали до 0°С и осторожно добавляли метанол (7 мл). Реакционную смесь кипятили с обратным холодильником в течение 5 ч и охлаждали до 0°С. Добавляли ди-трет-бутилдикарбонат, растворенный в ДХМ (7 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли, и остаток растворяли в этилацетате (100 мл). Органический слой промывали водой (60 мл) и солевым раствором (60 мл). Органический слой сушили над Na2SO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт использовали без очистки.
Промежуточное соединение 143с):
Промежуточное соединение 143b) (1,26 г) растворяли в ДХМ (30 мл) и добавляли 4М НСl в диоксане (1,48 мл). Реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Растворитель испаряли, полученное белое твердое вещество обрабатывали эфиром, и продукт отделяли фильтрованием, промывали эфиром и сушили в высоком вакууме. Полученное твердое вещество использовали без дальнейшей очистки.
Промежуточное соединение 143d):
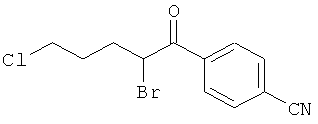
В атмосфере аргона, к перемешиваемой суспензии бромида меди (II) (3.62 г) в этилацетате (60 мл) добавляли 5-хлор-1-(4-цианофенил)-1-оксопентан (3.00 г) в хлороформе (60 мл). Полученную смесь перемешивали при кипячении в течение ночи. Добавляли дополнительное количество бромида меди (II) (0.60 г), и реакционную смесь кипятили с обратным холодильником в течение 3 ч. Реакционную смесь фильтровали через целит, промывали этилацетатом и концентрировали в вакууме. Продукт очищали флэш-хроматографией.
Промежуточное соединение 143е):
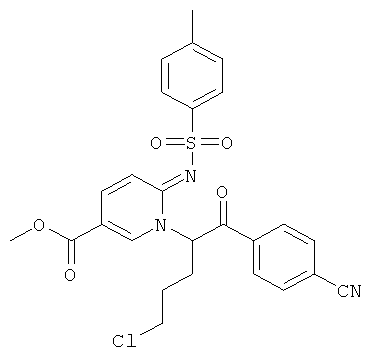
Промежуточное соединение 143d) (3.00 г) растворяли в ацетонитриле (80 мл). Добавляли этилдиизопропиламин (3.2 мл), и реакционную смесь нагревали при 85°С. Добавляли промежуточное соединение 59b) (2.80 г), растворенное в ацетонитриле (20 мл), и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Растворитель удаляли при пониженном давлении. Продукт очищали флэш-хроматографией.
Промежуточное соединение 143f):
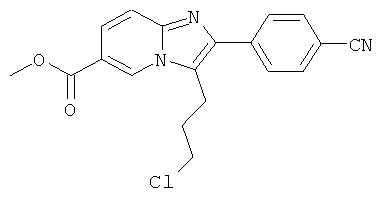
Промежуточное соединение 143е) (4.67 г) растворяли в сухом ДХМ (90 мл) и охлаждали до 0°С. Добавляли ангидрид трифторуксусной кислоты (12 мл), и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 6 ч. Растворитель удаляли при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и выливали в насыщенный раствор бикарбоната натрия, полученный белый осадок отфильтровывали, промывали водой и сушили в высоком вакууме.
Промежуточное соединение 143g):
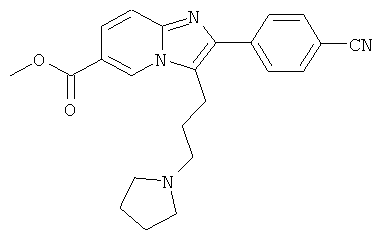
Промежуточное соединение 143f) (1,50 г), ацетонитрил (40 мл) и пирролидин (3.5 мл) перемешивали при 70°С в течение 6 ч. Половину растворителя удаляли при пониженном давлении, и оставшийся раствор охлаждали на ледяной бане в течение 2 ч. Полученное твердое вещество отфильтровали и промывали охлажденным ацетонитрилом. Продукт сушили в высоком вакууме.
Промежуточное соединение 143h):
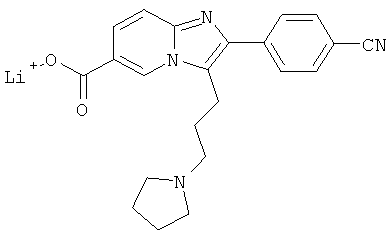
Промежуточное соединение 143g) (313 мг) растворяли в ТГФ (10 мл) и добавляли 2М LiOH в воде (0.92 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли дополнительное количество ТГФ (1,5 мл) и 2М LiOH в воде (0,1 мл), и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Полученное твердое вещество отфильтровали, промывали охлажденным ТГФ и сушили в высоком вакууме. Продукт использовали как таковой в следующей операции без дальнейшей очистки.
Пример 143:
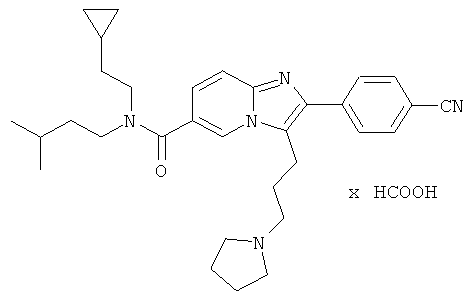
Промежуточное соединение 143h) (40 мг) растворяли в ДМФА (3 мл) и затем добавляли HATU (49 мг), DIEA (22 мкл) и промежуточное соединение 143с) (23 мг). Реакционную смесь перемешивали в течение 1 ч. Реакция оказалась незавершенной, поэтому добавляли дополнительное количество промежуточного соединения 143с) (10 мг) и DIEA (22 мкл), и реакционную смесь перемешивали в течение 2 ч. Летучие вещества удаляли, и остаток растворяли в этилацетате (50 мл). Органический слой промывали солевым раствором (40 мл), насыщенным раствором NaHCO3 (40 мл) и вновь солевым раствором (40 мл). Органический слой сушили над Na2SO4. фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали препаративной ЖХ-МС.
БИОЛОГИЧЕСКИЕ ИССЛЕДОВАНИЯ
А. Анализ связывания
Для идентификации конкурентных ингибиторов, связывающих NDP-альфа-МСГ, меченный флуоресцентной меткой, с препаратами мембран клеток линии НEК293, экспрессирующих человеческие рецепторы меланокортина, использовали мембранный анализ связывания.
Тестируемые соединения или немеченые NDP-альфа-МСГ готовили, используя различные концентрации, на 384-луночных микротитровальных планшетах. NDP-альфа-МСГ, меченные флуоресцентной меткой, готовили в одной концентрации, после чего добавляли препараты мембран. Планшеты инкубировали в течение 5 ч при комнатной температуре.
Степень флуоресцентной поляризации определяли при помощи флуоресцентного поляризационного ридера микропланшетов.
В. Функциональный анализ
Агонистическую активность человеческих рецепторов меланокортина определяли посредством анализа с использованием гомогенизированных мембран. Конкуренция между немеченым цАМФ и фиксированным количеством меченных флуоресцентной меткой цАМФ за ограниченное количество центров связывания на цАМФ-специфическом антителе определяли при помощи флуоресцентной поляризации.
Тестируемые соединения или немеченые NDP-альфа-МСГ готовили, используя различные концентрации, на 384-луночных микротитровальных планшетах. Добавляли препараты мембран клеток НЕК293, экспрессирующих человеческие рецепторы меланокортина. После короткого периода предварительного инкубирования, добавляли соответствующее количество АТФ, GTP и антитела цАМФ, и планшет вновь инкубировали, а затем дозировали конъюгат цАМФ, меченный флуоресцентной меткой. Планшеты инкубировали в течение 2 ч при 4°С и затем считывали на флуоресцентном поляризационном ридере микропланшетов. Количество цАМФ, выработанное в ответ на воздействие тестируемого соединения, сравнивали с выработкой цАМФ, полученной при стимуляции под действием NDP-альфа-МСГ.
Были проанализированы некоторые соединения, предлагаемые согласно настоящему изобретению, и было обнаружено, что они связывают рецептор меланокортина-4. В общем случае было обнаружено, что значения IС50 этих соединений составляют менее 2 мкМ (микромоль). Кроме того, некоторые соединения, предлагаемые согласно настоящему изобретению, были проанализированы в функциональном анализе, и было обнаружено, что они не активируют рецептор меланокортина-4.
В таблице перечислены значения IC50, полученные в анализе связывания чМК-4Р (человеческий рецептор МК-4Р), и значения IC50, полученные в функциональном анализе. Значения IC50 и ЕС50 были объединены в три класса:
а<0,1 мкМ; b>0,1 мкМ и <1,0 мкМ; с>1,0 мкМ
С. Модели потребления пищи in vivo
1. Модель спонтанного питания
Исследовали потребление пищи крысами после внутрибрюшинного и перорального введения тестируемых соединений (см., например, Chen, A.S. et al. Transgenic Res. 2000 Apr; 9(2):145-54).
2. Модели анорексии, вызванной ЛПС, и истощения, вызванного наличием опухоли
Исследовали предотвращение или снижение анорексии, вызванной введением липополисахаридов (ЛПС), или истощения, вызванного ростом опухоли, после внутрибрюшинного и перорального введения крысам тестируемых соединений (см., например, Marks, D.L.; Ling, N и Cone, R.D.Cancer Res. 2001 Feb. 15; 61(4):1432-8).
D. Определения всасывания, распределения, метаболизма и выведения (ADME) in vitro
1. Микросомальая стабильность
Экспериментальная методика
Готовили объединенные микросомы человеческой печени (содержащие микросомы мужчин и женщин) и объединенные микросомы печени крыс (самцов крыс Sprague Dawley). Перед использованием микросомы хранили при -80°С.
Микросомы (конечная концентрация 0,5 мг/мл), 0,1 М фосфатный буфер. рH 7.4, и тестируемое соединение (конечная концентрация субстрата 3 мкМ; конечная концентрация ДМСО=0,25%) подвергали предварительной инкубации при 37°С, после чего для инициирования реакции добавляли NADPH (восстановленный никотинамидадениндинуклеотидфосфат) (конечная концентрация 1 мМ). Конечный инкубационный объем составлял 25 мкл. Кроме того, проводили контрольную инкубацию каждого тестируемого соединения, при проведении которой вместо NADPH добавляли 0,1 М фосфатный буфер с pH 7.4 (минус NADPH). Для каждого вида испытывали два контрольных соединения. Для каждого тестируемого соединения инкубацию проводили один раз.
Каждое соединение инкубировали в течение 0, 5, 15, 30 и 45 мин. Контроль (минус NADPH) инкубировали только в течение 45 мин. Реакции останавливали в соответствующие моменты времени добавлением 50 мкл метанола, содержащего внутренний стандарт. Инкубируемые планшеты центрифугировали при 2500 об/мин в течение 20 мин при 4°С для осаждения протеина.
Количественный анализ
После осаждения протеина, жидкости над осадками объединяли в кассеты, содержащие до 4 соединений, и анализировали с использованием обычных условий ЖХ-MC/MS.
Анализ данных
Линейный градиент определяли из графика зависимости отношения площади пика от времени (площадь под пиком для соединения / площадь под пиком для внутреннего стандарта). Затем, при помощи следующих уравнений вычисляли период полувыведения и собственный клиренс:
Постоянная скорости элиминирования (k)=(-градиент)
Период полувыведения (t1/2)(мин)=0,693/k
Собственный клиренс (CLint) (мкл/мин/мг протеина)=(V×0,693)/t1/2
где V=Инкубационный объем мкл/мг микросомального протеина.
В анализе производили инкубацию двух контрольных соединений, и если значения, полученные для этих соединений, не укладывались в заданные пределы, то результаты отбрасывали, и эксперимент повторяли.
2. Стабильность гепатоцитов
Экспериментальная методика
Для определения стабильности человеческих гепатоцитов использовали суспензии гепатоцитов (объединенные клетки, полученные из организмов грех человек), сохраняемых при криотемпературах. Все гепатоциты, сохраняемые при криотемпературах, были предоставлены Компанией In Vitro Technologies, Xenotech или TCS.
Инкубацию проводили при концентрациях тестируемых или контрольных соединений, составляющих 3 мкМ, при клеточной плотности, составляющей 0,5×106 живых клеток/мл. Конечная концентрация ДМСО при инкубации составляла 0,25%. Для обнаружения неферментного разложения также проводили контрольные инкубации в отсутствие клеток.
Из инкубационной смеси отбирали по 2 образца (50 мкл) каждого вида в моменты времени 0, 5, 10, 20, 40 и 60 мин (контрольный образец отбирали только при 60 мин) и добавляли в метанол, содержащий внутренний стандарт (100 мкл), для остановки реакции.
В качестве контрольных соединений использовали толбутамид, 7-гидроксикумарин и тестостерон.
Образцы центрифугировали (2500 об/мин при 4°С в течение 20 мин) и жидкости над осадком, полученные в каждый момент времени, объединяли для проведения кассетного анализа обычным способом ЖХ-МС/МС.
Анализ данных
Линейный градиент определяли из графика зависимости отношения площади пика от времени (площадь под пиком для соединения/площадь под пиком для внутреннего стандарта). Затем, при помощи следующих уравнений вычисляли период полувыведения и собственный клиренс:
Постоянная скорости элиминирования (k)=(-градиент)
Период полувыведения (t1/2) (мин)=0,693/k
Собственный клиренс (CLint) (мкл/мин/миллион клеток)=(V×0,693)/t1/2
где V=Инкубационный объем мкл/количество клеток.
3. Проницаемость Сасо-2 (в двух направлениях)
Экспериментальная методика
Для анализа использовали клетки Сасо-2, полученные из АТСС при количестве пассажей, равном 27. Клетки (количество пассажей 40-60) высевали па планшеты Millipore Multiscreen Сасо-2 при плотности 1×105 клеток/см2. Клетки культивировали в течение 20 суток в DMEM (модифицированная по способу Дульбекко среда Игла), заменяя среду каждые двое или трое суток. Анализ проницаемости проводили на 20 сутки.
В анализе проницаемости в качестве среды использовали сбалансированный солевой раствор Хенкса (Hanks Balanced Salt Раствор (HBSS)), буфер pH 7.4, с добавлением 25 мМ HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновой кислоты) и 10 мМ глюкозы при 37°С. Инкубацию производили в атмосфере 5% СO2 и относительной влажности 95%.
На 20 сутки приготавливали монослои, дважды промывая базолатеральную (В) и апикальную (А) поверхности средой HBSS при 37°С. Клетки затем инкубировали с HBSS в базолатеральном и апикальном отделениях в течение 40 мин для стабилизации физиологических параметров.
Затем HBSS удаляли из апикального отделения и заменяли дозировочными растворами тестируемых соединений. Растворы готовили разбавлением 10 мМ тестируемого соединения в ДМСО, содержащем HBSS, получая конечную концентрацию тестируемого соединения, равную 10 мкМ (конечная концентрация ДМСО составляла 1%). Дозировочный раствор также включал флуоресцентный маркер чистоты эксперимента люцифер желтый. Аналитические стандарты готовили из дозировочных растворов. Проницаемость каждого из испытуемых соединений определяли по 2 раза. В качестве контроля на каждом планшете также использовали соединения с известной проницаемостью.
Вставки апикального отделения затем вводили в «соответствующие» планшеты, содержащие свежий HBSS. Для определения перехода от базолатсралыной к апикальной (В-А) проницаемости, начинали эксперимент, заменяя буфер во вставках и затем помещая их в соответствующие планшеты, содержащие дозировочные растворы. Спустя 120 минут, соответствующие планшеты удаляли, и апикальные и базолатеральные образцы разбавляли для проведения ЖХ-МС/МС анализа. Исходные концентрации (С0) и экспериментальные выходы вычисляли, исходя из концентраций в апикальных и базолатеральных отделениях.
Целостность монослоев на протяжении эксперимента проверяли по проницаемости люцифера желтого с использованием флуоресцентного анализа. Если монослои не повреждены, то проницаемость люцифера желтого низка. Испытуемые и контрольные соединения оценивали количественно при помощи ЖХ-МС/МС кассетного анализа с использованием 5-точечной калибровки при соответствующем разбавлении образцов. Применяли обычные условия анализа.
Если значение Рарр для люцифера желтого превышает QC-предел в одной из индивидуальных лунок, содержащих тестируемое соединение, то принимают значение n=1. Если значения Рарр для люцифера желтого превышают QC-предел в обеих лунках, содержащих одно и то же тестируемое соединение, то проводят повторное испытание соединения. Постоянно высокие значения проницаемости люцифера желтого для конкретного соединения в обеих лунках указывают на наличие токсичности. В этом случае, дальнейших экспериментов не проводят.
Анализ данных
Коэффициент проницаемости для каждого соединения (Рарр) вычисляют из следующего уравнения:
Рарр=(dQ/dt)/C0×A,
где dQ/dt представляет собой скорость проникновения медикамента через клетку, С0 представляет собой концентрацию в донорном отделении в момент времени ноль, и А представляет собой площадь монослоя клеток. Значение С0 получают из анализа донорного и приемного отделений после окончания инкубации. Полагают, что все тестируемые соединения, обнаруживаемые после 120-минутной инкубации, изначально присутствовали в донорном отделении в момент времени 0 минут. Показатель асимметричности определяют следующим образом:
АI=(Рарр(В-А))/(Рapp(А-В)).
Показатель асимметричности, превышающий единицу, указывает на наличие потока из клеток Сасо-2, что указывает на то, что соединение потенциально может иметь плохую абсорбцию in vivo.
Значения эффективной проницаемости (Рарр (А-В)) тестируемых соединений сравнивали с соответствующими значениями, полученными для контрольных соединений, атенолола и пропранолола, абсорбция которых в организме человека составляет 50 и 90%, соответственно (Zhao, Y.H., et al., (2001). Evaluation of Human Intestinal Absorption Data and Subsequent Derivation of a Quantitative Structure-Activity Relationship (QSAR) with the Abraham Descriptors. Journal of Pharmaceutical Sciences. 90 (6), 749-784). Талинолол (известный субстрат P-gp (Deferme, S., Mols, R., Van Driessche, W., Augustijns, P. (2002). Apricot Extract Inhibits the P-gp-Mediated Efflux of Talinolol. Journal of Pharmaceutical Sciences. 91(12), 2539-48)) также включали в виде контрольного соединения, которое позволяло обнаруживать возможное присутствие функционального P-gp в монослое клеток Сасо-2.
4. Ингибирование цитохрома Р450 (IС50 опеределение на 5 изоформах)
Экспериментальная методика
Ингибирование CYP1A
Шесть концентраций тестируемого соединения (0,05, 0,25, 0,5, 2,5, 5,25 мкМ в ДМСО; конечная концентрация ДМСО=0,35%) инкубировали с микросомами человеческой печени (0,25 мг/мл) и NADPH (1 мМ) в присутствии маркерного субстрата этоксирезоруфина (0,5 мкМ) в течение 5 мин при 37°С. В качестве положительного контроля при испытаниях соединений использовали селективный ингибитор CYP1A, альфа-нафтофлавон.
Ингибирование CYP2C9
Шесть концентраций тестируемого соединения (0,05, 0,25, 0,5, 2,5, 5,25 мкМ в ДМСО; конечная ДМСО концентрация=0,25%) инкубировали с микросомами человеческой печени (1 мг/мл) и NADPH (1 мМ) в присутствии маркерного субстрата толбутамида (120 мкМ) в течение 60 мин при 37°С. В качестве положительного контроля при испытаниях соединений использовали селективный ингибитор CYP2C9. сульфафеназол.
Ингибирование CYP2C19
Шесть концентраций тестируемого соединения (0,05, 0,25, 0,5, 2,5, 5,25 мкМ в ДМСО; конечная ДМСО концентрация=0,25%) инкубировали с микросомами человеческой печени (0,5 мг/мл) и NADPH (1 мМ) в присутствии маркерного субстрата мефенитоина (25 мкМ) в течение 60 мин при 37°С. В качестве положительного контроля при испытаниях соединений использовали селективный ингибитор СYP2C19. транилципромин.
Ингибирование CYP2D6
Шесть концентраций тестируемого соединения (0,05, 0,25, 0,5, 2,5, 5,25 мкМ в ДМСО; конечная ДМСО концентрация=0,25%) инкубировали с микросомами человеческой печени (0,5 мг/мл) и NADPH (1 мМ) в присутствии маркерного субстрата декстрометорфана (5 мкМ) в течение 30 мин при 37°С. В качестве положительного контроля при испытаниях соединений использовали селективный ингибитор CYP2D6, хинидин.
Ингибирование CYP3A4
Шесть концентраций тестируемого соединения (0,05, 0,25, 0,5, 2,5, 5,25 мкМ в ДМСО; конечная ДМСО концентрация 0,26%) инкубировали с микросомами человеческой печени (0,25 мг/мл) и NADPH (1 мМ) в присутствии маркерного субстрата мидазолама (2.5 мкМ) в течение 5 мин при 37°С. В качестве положительного контроля при испытаниях соединений использовали селективный ингибитор CYP3A4, кетоконазол.
При инкубациях CYP1A, реакции останавливали добавлением метанола, и образование метаболита, резоруфина, отслеживали при помощи флуоресценции (длина волны возбуждения=535 нм, длинна волны эмиссии = 595 нм). При инкубациях CYP2C9, CYP2C19, CYP2D6 и СYP3A4, реакции останавливали добавлением метанола, содержащего внутренний стандарт. Образцы затем центрифугировали, и жидкость над осадком объединяли для проведения одновременного анализа 4-гидрокситолбутамида, 4-гидроксимефенитоина, декстрорфана и 1-гидроксимидазолама плюс внутренний стандарт при помощи ЖХ-МС/МС. Использовали обычные условия ЖХ-МС/МС. К конечному образцу перед проведением анализа добавляли муравьиную кислоту в деионизованной воде (конечная концентрация=0,1%). Для вычисления значения IС50 (концентрации испытуемого соединения, которая вызывает 50% ингибирование) использовали снижение скорости образования метаболитов по сравнению с контрольными образцами, содержащими носитель.
5. Связывание протеина плазмы (10%)
Экспериментальная методика
Растворы тестируемого соединения (5 мкМ, конечная концентрация ДМСО 0,5%) приготавливали в буфере (pH 7.4) и 10% плазме (об./об. в буфере). Эксперимент проводили с использованием равновесного диализа между двумя отделениями, разделенными полупроницаемой мембраной. По одну сторону мембраны добавляли буферный раствор, а по другую сторону - раствор плазмы. Стандарты готовили в плазме и в буфере, и инкубировали их при 37°С. Соответствующие растворы для каждого соединения анализировали в кассетах при помощи ЖХ-МС/МС.
Количественный анализ
После уравновешивания, с обеих сторон мембраны отбирали образцы. Раствор каждой партии соединений объединяли в две группы (содержащие и не содержащие плазму) и анализировали на кассетах при помощи ЖХ-МС/МС с использованием двух наборов калибровочных стандартов для не содержащих плазму (7 точек) и содержащих плазму растворов (6 точек). Использовали обычные условия ЖХ-МС/МС. Образцы оценивали с использованием стандартных кривых, полученных в эквивалентной матрице. Каждое соединение испытывали по два раза. В каждый эксперимент включали контрольное соединение.
Анализ данных
fu=(1-(PC-PF))/(PC)
fu=несвязанная фракция
PC=концентрация образца в протеин-содержащей стороне
PF=концентрация образца в стороне, не содержащей протеина
Значения fu, полученные при 10% плазмы, переводили в значения fu, полученные при 100% плазмы, в соответствии со следующим уравнением:
fu100%=fu10%/(10-(9·fu10%)).
Примеры фармацевтических композиций
Один из специфических примеров осуществления композиции для перорального введения, включающей соединение, предлагаемое согласно настоящему изобретению, готовят из 33 мг соединения Примера 9, которое смешивают с достаточно тонко размолотой лактозой в таком соотношении, что композиция содержит в жесткой желатиновой капсуле размера 0 общее количество соединения, составляющее от 580 до 590.
Другой конкретный пример осуществления композиции для перорального введения, включающей соединение, предлагаемое согласно настоящему изобретению, готовят из 37 мг соединения Примера 17, которое смешивают с достаточно тонко размолотой лактозой в таком соотношении, что композиция содержит в жесткой желатиновой капсуле размера 0 общее количество соединения, составляющее от 580 до 590.
Несмотря на то что настоящее изобретение было описано и проиллюстрировано при помощи некоторых предпочтительных примеров осуществления, специалистам в данной области техники известно, что в этих примерах могут быть произведены различные изменения, модификации и замены, которые также включены в область, защищаемую настоящим изобретением. Например, в зависимости от конкретной фармакологической ответной реакции, могут быть использованы эффективные дозировки, отличные от дозировок, указанных в настоящем описании, которые также могут быть выбраны в зависимости от конкретного выбранного активного соединения, а также от типа композиции и применяемого способа введения, и такие ожидаемые варианты и отличия в результатах также включены в цели настоящего изобретения и область, защищаемую настоящим изобретением. Таким образом, область, защищаемая настоящим изобретением, ограничена лишь Формулой настоящего изобретения, которой должна быть дана максимально широкая интерпретация.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИННАМОИЛАМИНОАЛКИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БЕНЗОСУЛЬФОНАМИДА | 2000 |
|
RU2242462C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800153C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2678305C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗО-ПИРИДИНА, ОБЛАДАЮЩИЕ СРОДСТВОМ К РЕЦЕПТОРУ МЕЛАНОКОРТИНА | 2004 |
|
RU2358974C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2625309C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2801190C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| ИНГИБИТОРЫ JAK | 2010 |
|
RU2538204C2 |
| Амидные соединения, содержащие их фармацевтические композиции и способы их применения | 2017 |
|
RU2759913C2 |
| ПРОИЗВОДНЫЕ И СПОСОБЫ ЛЕЧЕНИЯ ИНФЕКЦИЙ ГЕПАТИТА В | 2015 |
|
RU2742305C2 |
Изобретение относится к замещенным производным имидазопиридина общей формулы (I) и к его энантиомерам, диастереомерам, таутомерам, и фармацевтически приемлемым солям, в котором А представляет собой -NH-, -СН2-, -СН2-СН2- или связь; Х представляет собой фенил, фенил, конденсированный с насыщенным гетероциклическим 5- или 6-членным кольцом, при этом гетероциклическое кольцо может содержать один или два гетероатома, выбранных из О и N, и при этом гетероциклическое кольцо дополнительно возможно замещено оксогруппой, 6-членный насыщенный гетероциклил, содержащий О в качестве гетероатома, 5-6-членный гетероарил, содержащий 1 или 2 гетероатома, выбранные из N, О и S, и при этом каждый фенил и гетероарил возможно замещен от 1 до 2 R14 и/или 1 заместителем R4b и/или 1 заместителем R5; R1 и R2 независимо друг от друга выбирают из следующих групп: C1-6-алкила и C1-6-алкилен-С3-7-циклоалкила, и при этом каждый алкил возможно замещен группой ОН, или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют 5-6-членное кольцо, которое возможно замещено одним заместителем, выбранным из C1-6-алкила и О-С1-6-алкила; R4b представляет собой C(O)NH2, C(O)OH, С(O)NН-С1-6-алкил, C(O)N-(C1-6-алкил)2, SO2-C1-6-алкил, оксогруппу, и при этом цикл по меньшей мере частично насыщен, NH2, NH-C1-6-алкил, N-(С1-6-алкил)2; R5 представляет собой 6-членный гетероарил, содержащий N в качестве гетероатома; R3 представляет собой -(CR8R9)n-T; R8 и R9 независимо друг от друга выбирают из следующих групп: Н и С1-6-алкил; n равен 1, 2, 3, 4, 5 или 6; Т представляет собой 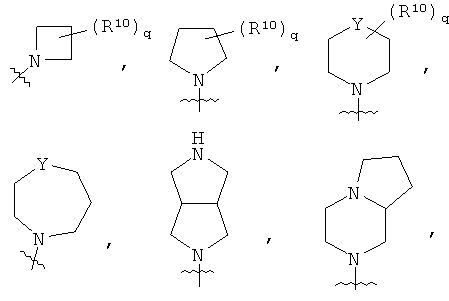 или NR12R13; R10 представляет собой Н, NH2, ОН, С1-6-алкил, возможно замещенный одним ОН, атом галогена, NH(C1-6-алкил) или N(С1-6-алкил)2; q равен 1 или 2; Y представляет собой СН2, NR11 или О; R11 представляет собой Н, или C1-6-алкил; R12 и R13 независимо друг от друга выбирают из следующих групп: Н, С1-6-алкила, C1-6-алкинила, (СН2)0-2-С3-7-циклоалкила, и С1-6-алкилен-О-С1-6-алкил, при этом C1-6-алкил возможно замещен одним галогеном; R14 представляет собой атом галогена, CN, С1-6-алкил, возможно замещенный от одного до трех заместителями, выбранными из атома галогена, ОН, O-C1-6-алкила, O-С(O)С1-6-алкила, O-С1-6-алкил, возможно замещенный одним заместителем, выбранным из ОН, O-С1-6-алкила, и O-С(O)С1-6-алкила, или ОН. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I). Технический результат: получены новые производные имидазопиридина, которые могут быть применимы в качестве модуляторов рецептора меланокортина-4 (МК-4Р). 2 н. и 15 з.п. ф-лы, 8 табл., 22 пр.
или NR12R13; R10 представляет собой Н, NH2, ОН, С1-6-алкил, возможно замещенный одним ОН, атом галогена, NH(C1-6-алкил) или N(С1-6-алкил)2; q равен 1 или 2; Y представляет собой СН2, NR11 или О; R11 представляет собой Н, или C1-6-алкил; R12 и R13 независимо друг от друга выбирают из следующих групп: Н, С1-6-алкила, C1-6-алкинила, (СН2)0-2-С3-7-циклоалкила, и С1-6-алкилен-О-С1-6-алкил, при этом C1-6-алкил возможно замещен одним галогеном; R14 представляет собой атом галогена, CN, С1-6-алкил, возможно замещенный от одного до трех заместителями, выбранными из атома галогена, ОН, O-C1-6-алкила, O-С(O)С1-6-алкила, O-С1-6-алкил, возможно замещенный одним заместителем, выбранным из ОН, O-С1-6-алкила, и O-С(O)С1-6-алкила, или ОН. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I). Технический результат: получены новые производные имидазопиридина, которые могут быть применимы в качестве модуляторов рецептора меланокортина-4 (МК-4Р). 2 н. и 15 з.п. ф-лы, 8 табл., 22 пр.
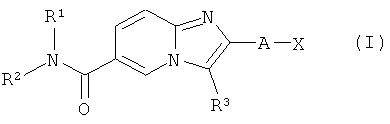
1. Соединение, соответствующее формуле (I)
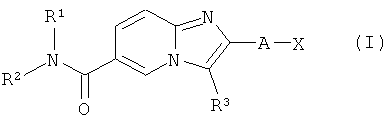
и энантиомеры, диастереомеры, таутомеры, и фармацевтически приемлемые соли указанного соединения,
в котором
А представляет собой -NH-, -CH2-, -СН2-СН2- или связь;
Х представляет собой
фенил,
фенил, конденсированный с насыщенным гетероциклическим 5- или 6-членным кольцом, при этом гетероциклическое кольцо может содержать один или два гетероатома, выбранных из О и N, и при этом гетероциклическое кольцо дополнительно возможно замещено оксогруппой, 6-членный насыщенный гетероциклил, содержащий О в качестве гетероатома, 5-6-членный гетероарил, содержащий 1 или 2 гетероатома, выбранные из N, О и S,
и при этом каждый фенил и гетероарил возможно замещен от 1 до 2 R14 и/или 1 заместителем R4b и/или 1 заместителем R5;
R1 и R2 независимо друг от друга выбирают из следующих групп:
C1-6-алкила и
С1-6-алкилен-С3-7-циклоалкила, и
при этом каждый алкил возможно замещен группой ОН, или
R1 и R2 вместе с атомом азота, к которому они присоединены, образуют 5-6-членное кольцо, которое возможно замещено одним заместителем, выбранным из C1-6-алкила и О-С1-6-алкила;
R4b представляет собой
C(O)NH2,
С(O)ОН,
С(O)NH-С1-6-алкил,
С(O)N-(С1-6-алкил)2,
SO2-С1-6-алкил,
оксогруппу, и при этом цикл по меньшей мере частично насыщен,
NH2,
NH-C1-6-алкил,
N-(C1-6-алкил)2;
R5 представляет собой 6-членный гетероарил, содержащий N в качестве гетероатома;
R3 представляет собой -(CR8R9)n-T;
R8 и R9 независимо друг от друга выбирают из следующих групп: Н и C1-6-алкил;
n равен 1, 2, 3, 4, 5 или 6;
Т представляет собой
или NR12R13;
R10 представляет собой
Н,
NH2,
ОН,
C1-6-алкил, возможно замещенный одним ОН,
атом галогена,
NН(С1-6-алкил) или
N(С1-6-алкил)2;
q равен 1 или 2;
Y представляет собой СН2, NR11 или О;
R11 представляет собой
Н, или C1-6-алкил;
R12 и R13 независимо друг от друга выбирают из следующих групп:
Н,
C1-6-алкила,
C1-6-алкинила,
(СН2)0-2-С3-7-циклоалкила, и
С1-6-алкилен-O-С1-6-алкил,
при этом C1-6-алкил возможно замещен одним галогеном;
R14 представляет собой
атом галогена,
CN,
С1-6-алкил, возможно замещенный от одного до трех заместителями, выбранными из атома галогена, ОН, O-C1-6-алкила, O-С(O)С1-6-алкила,
O-C1-6-алкил, возможно замещенный одним заместителем, выбранным из ОН, O-C1-6-алкила, и O-С(O)С1-6-алкила, или
ОН.
2. Соединение по п.1, в котором А представляет собой -NH-, -СН2-, -СН2-СН2- или связь;
Х представляет собой
фенил,
фенил, конденсированный с насыщенным гетероциклическим 6-членным кольцом, и при этом гетероциклическое кольцо может содержать 1 или 2 гетероатома, выбранных из О и N, и при этом гетероциклическое кольцо дополнительно возможно замещено оксогруппой,
6-членный насыщенный гетероциклил, содержащий О в качестве гетероатома,
5-6-членный гетероарил, содержащий один или два гетероатома, выбранных из N, О и S,
при этом каждый фенил и гетероарил возможно замещен от 1 до 2 R14 и/или 1 заместителем R4b и/или 1 заместителем R5;
R1 и R2 независимо друг от друга выбирают из следующих групп:
C1-6-алкила, или
С1-6-алкилен-С3-7-циклоалкила, или
R1 и R2 вместе с атомом азота, к которому они присоединены, образуют 5-6-членное кольцо, которое возможно замещено одним заместителем, выбранным из C1-6-алкила,
O-C1-6-алкила;
R14 выбирают из:
атома галогена,
CN,
С1-6-алкила, возможно замещенного от одного до трех атомами галогена,
O-C1-6-алкила, возможно замещенного
ОН;
R4b представляет собой
C(O)NH2,
C(O)NH-C1-6-алкил,
С(O)N-(С1-6-алкил)2,
SО2-С1-6-алкил,
NH2.
NH-C1-6-алкил, или N-(С1-6-алкил)2;
R5 представляет собой 6-членный гетероарил, содержащий N в качестве гетероатома;
R3 представляет собой -(CR8R9)n-Т;
R8 и R9 независимо друг от друга выбирают из следующих групп: Н и C1-6-алкил;
n равен 1, 2, 3, 4, 5 или 6;
Т представляет собой
или NR12R13;
R10 представляет собой
Н,
NH2,
ОН,
C1-6-алкил,
атом галогена,
NН(С1-6-алкил) или
N(С1-6-алкил)2;
Y представляет собой СН2, NR11 или О;
R11 представляет собой
Н, или
C1-6-алкил;
R12 и R13 независимо друг от друга выбирают из следующих групп:
H,
C1-6-алкила,
(СН2)0-2-С3-7-циклоалкила, и
С1-6-алкилен-O-С1-6-алкил,
при этом C1-6-алкил возможно замещен одним галогеном.
3. Соединение по п.1 или 2, в котором А представляет собой -NH- или связь.
4. Соединение по п.1 или 2, в котором R1 и R2 представляют собой С3-6-алкил, или
R1 и R2 вместе с атомом азота, к которому они присоединены, образуют 5-6-членное кольцо, которое возможно замещено одним заместителем, выбранным из C1-6-алкила и O-C1-6-алкила.
5. Соединение по п.1 или 2, в котором Т представляет собой NR12R13.
6. Соединение по п.5, в котором R12 и R13 независимо друг от друга выбраны из Н, C1-3-алкила и (СН2)0-2-С3-6-циклоалкила, и при этом алкил возможно замещен одним галогеном.
7. Соединение по п.1 или 2, в котором Т выбирают из
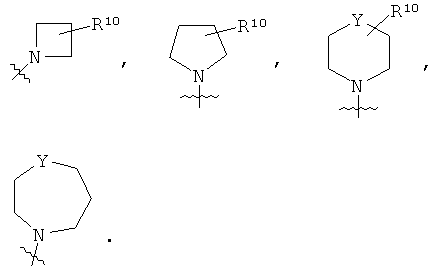
8. Соединение по п.7, в котором Y представляет собой СН2 или NR11, и R10 представляет собой Н, NH2, С1-6-алкил, NH(C1-6-алкил) или N(C1-6-алкил)2.
9. Соединение по любому из пп.1, 2, 6 и 8, в котором
Х представляет собой фенил, конденсированный с насыщенным 6-членным гетероциклическим кольцом, при этом гетероциклическое кольцо может содержать 1 или 2 гетероатома, выбранных из О и N, и при этом гетероциклическое кольцо дополнительно может быть замещено оксогруппой, или
6-членный гетероарил, содержащий О в качестве гетероатома, при этом каждый фенил или гетероарил возможно замещен от 1 до 2 заместителями R14 и/или 1 заместителем R5.
10. Соединение по любому из пп.1, 2, 6 и 8, в котором Х представляет собой
фенил, или
5-6-членный гетероарил, содержащий один или два гетероатома, выбранных из N, О и S, и при этом
каждый фенил и гетероарил возможно замещен от 1 до 2 R14, и/или 1 R4b, и/или 1 R5.
11. Соединение по п.10, в котором Х представляет собой фенил.
12. Соединение по п.10, в котором Х представляет собой пиридил.
13. Соединение по любому из пп.1, 2, 6, 8, 11 и 12, применяемое в качестве медикамента для лечения и профилактики расстройств, заболеваний и состояний, в развитии которых участвует рецептор меланокортина-4.
14. Соединение по любому из пп.1, 2, 6, 8, 11 и 12, применяемое в качестве антагониста рецептора меланокортина-4.
15. Соединение по любому из пп.1, 2, 6, 8, 11 и 12, применяемое для лечения или профилактики нарушений, заболеваний или состояний млекопитающего, чувствительных к инактивации рецептора меланокортина-4.
16. Соединение по п.15 для лечения или профилактики раковой кахексии, мышечной атрофии, анорексии, бокового амиотрофического склероза (БАС), тревожности и/или депрессии.
17. Фармацевтическая композиция, содержащая эффективное количество соединения по любому из пп.1-12 и фармацевтически приемлемый носитель для лечения и профилактики расстройств, заболеваний и состояний млекопитающего, чувствительных к инактивации рецептора меланокортина-4.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗО-3-ИЛАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2000 |
|
RU2264402C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ (ВАРИАНТЫ), СПОСОБ ИНГИБИРОВАНИЯ СЕКРЕЦИИ ЖЕЛУДОЧНОЙ КИСЛОТЫ, СПОСОБ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И СПОСОБ ЛЕЧЕНИЯ СОСТОЯНИЙ, В КОТОРЫХ ВОВЛЕЧЕНО ИНФИЦИРОВАНИЕ H.PYLORI | 1999 |
|
RU2238271C2 |

