Уровень техники изобретения
Деградация белков у эукариотов преимущественно опосредована метаболическим путем убиквитина, в котором белки, нацеленные на деструкцию, присоединены к 76 аминокислоте полипептида убиквитина. После нацеливания убиквитинированные белки затем служат в качестве субстратов для протеасомы 26S, мультикаталитической протеазы, которая разрывает белки на короткие пептиды благодаря действию ее трех основных протеолитических активностей. В то время как, имея основной функцией участие во внутриклеточном белковом обмене, опосредованная протеасомой деградация также играет ключевую роль во многих процессах, таких как включение главного комплекса тканевой совместимости (МНС) класса I, апоптоз, деление клеток и NF-κB-активация.
Протеасома 20S представляет собой 700 kDa комплекс мультикаталитической протеазы цилиндрической формы, состоящей из 28 субъединиц, организованных в четыре цикла, который играет важную роль в регулировании роста клеток, включении главного комплекса тканевой совместимости класса I, апоптозе, обработке антигенов, активации NF-κB и трансдукции провоспалительных сигналов. В дрожжах и других эукариотах 7 различных α-субъединиц образуют внешние циклы и 7 различных β-субъединиц содержат внутренние циклы. α-Субъединицы служат в качестве связующих точек для регуляторных комплексов 19S (PA700) и 11S (PA28), также как и в качестве физического барьера для внутренней протеолитической камеры, образованной двумя циклами β-субъединиц. Таким образом, in vivo, считают, что протеасома существует как 26S частица («протеасома 26S»). Эксперименты in vivo показали, что ингибирование формы 20S протеасомы может быть легко скоррелировано с ингибированием 26S-протеасомы. Расщепление аминоконцевых пропоследовательностей β-субъединиц во время образования частиц действует на аминоконцевые остатки треонина, которые служат в качестве каталитических нуклеофилов. Таким образом, субъединицы, отвечающие за каталитическую активность в протеасоме, обладают аминоконцевыми нуклеофильными остатками, и указанные субъединицы относятся к группе N-концевых нуклеофильных (Ntn) гидролаз (в которых N-концевой остаток представляет собой, например, Cys, Ser, Thr и другие нуклеофильные члены). Указанная группа включает в себя, например, ацилазу пенициллина G (PGA), ацилазу пенициллина V (PVA), амидотрансферазу (GAT) глутамина (PRPP) и бактериальную гликозиласпарагиназу. В дополнение к повсеместно экспрессированным β-субъединицам, более высокие позвоночные также обладают тремя β-субъединицами, индуцируемыми γ-интерфероном (LMP7, LMP2 и MECL1), которые заменяют их обычные включения, X, Y и Z, соответственно, изменяя таким образом каталитическую активность протеасомы. Путем использования различных пептидных субстратов определены три главных протеолитических активности протеасомы 20S эукариотов: химотрипсин-подобная активность (CT-L), которая расщепляет большие гидрофобные остатки; трипсин-подобная активность (Т-L), которая расщепляет основные остатки; и пептидилглутамильная пептидная гидролизующая активность (PGPH), которая расщепляет кислотные остатки. Протеасоме приписывают также две дополнительные менее характеристичные активности: BrAAP-активность, которая расщепляет аминокислоты с разветвленными цепями; и SNAAP-активность, которая расщепляет малые нейтральные аминокислоты. Главная протеолитическая активность протеасомы, по-видимому, обеспечена различными каталитическими точками, поскольку ингибиторы, точечные мутации в β-субъединицах и обмен β-субъединиц, включающих γ-интерферон, в различной степени изменяет указанные активности.
В последние годы протеасома становится привлекательной целью для терапевтических вмешательств при раковых, иммунных и аутоиммунных заболеваниях, воспалениях, ишемических состояниях, нейродегенеративных расстройствах и других заболеваниях. На сегодняшний день одобренным FDA ингибитором протеасомы является только бортезомиб (VELCADETM), однако в настоящее время клинические испытания проходят несколько других ингибиторов протеасом. Пока все указанные терапевтические ингибиторы протеасомы в настоящее время вводят IV (внутривенно). Клиническое применение ингибиторов протеасом при лечении гематологической злокачественности, такой как миелома и лимфома, частично ограничено необходимостью частого введения IV и может быть улучшено пероральным (РО) введением. Однако вследствие природы пептида указанных молекул системное действие, следующее за РО-введением указанных ингибиторов, ограничено некоторыми факторами, включая желудочный рН, желудочные и кишечные пептидазы, аспираторы, желчевыделение и кишечная и печеночная метаболическая активности.
Способы, использованные для преодоления способности пептидов к ферментативному расщеплению и улучшения абсорбции в кровоток из пищеварительного тракта, включают в себя аналоги, которые имеют структуру, менее подобную пептидам, и которые уменьшены в размере. Такие способы полагают успешными, если аналог пептида достигает удовлетворительного уровня в крови после перорального введения, или, в случае ингибиторов протеасомы, если активность протеасомы в крови удовлетворительно снижается.
Вышеуказанная технология применима для получения аналогов пептид-эпоксикетонов, ингибиторов протеасомы, обеспечивая им при этом оральную биодоступность.
Сущность изобретения
Изобретение относится к классу молекул, известному как α',β'-эпоксиды пептидов и α',β'-азиридины пептидов. Подразумевают, что исходные молекулы эффективно, необратимо и селективно связываются с N-концевыми нуклеофильными (Ntn) гидролазами и могут специфически ингибировать отдельные активности ферментов, обладающих множественной каталитической активностью.
В том случае, если полагают только избавиться от денатурированных и нескладчатых белков, протеасому теперь распознают как составляющий протеолитический механизм, который регулирует уровень различных внутриклеточных белков путем их разложения сигнально-зависимым образом. Однако большой интерес представляют собой идентифицирующие реагенты, которые могут специфически влиять на активность протеасомы и других Ntn-гидролаз и таким образом быть применимыми в качестве проб для изучения роли указанных ферментов в биологических процессах. Здесь описаны, синтезированы и исследованы соединения, которые нацелены на Ntn-гидролазы. Предложены и заявлены пептид-эпоксиды и пептид-азиридины, которые могут эффективно, селективно и необратимо ингибировать отдельные активности протеасомы.
В отличие от некоторых других ингибиторов на основе пептидов описанные здесь пептид-эпоксиды и пептид-азиридины не предполагают значительного ингибирования непротеасомных протеаз, таких как трипсин, химотрипсин, катепсин В, папаин и кальпаин, в концентрации до 50 мкМ. При более высоких концентрациях ингибирование может наблюдаться, но скорее как конкурентный и ненеобратимый процесс, если ингибитор только конкурирует с субстратом. Новые пептид-эпоксиды и пептид-азиридины также предполагают ингибирование активации NF-κВ и стабилизацию уровней р53 в клеточной культуре. Более того, предполагается, что указанные соединения имеют противовоспалительную активность. Таким образом, указанные соединения могут представлять собой уникальные молекулярные зонды, которые универсальны для изучения функции Ntn-фермента в нормальных биологических и патологических процессах.
В одном из аспектов изобретение обеспечивает ингибиторы, состоящие из трехчленного цикла, содержащего гетероатом. Указанные ингибиторы могут ингибировать каталитическую активность ферментов N-концевых нуклеофильных гидролаз (например, протеасомы 20S или протеасомы 26S), если указанный ингибитор присутствует в концентрации ниже примерно 50 мкМ. Относительно протеасомы 20S данные ингибиторы гидролаз ингибируют химотрипсин-подобную активность протеасомы 20S, если ингибитор присутствует в концентрации ниже примерно 5 мкМ, и не ингибирует трипсин-подобную активность или PGPH-активность протеасомы 20S, если присутствует в концентрации ниже примерно 5 мкМ. Ингибитор гидролазы может представлять собой, например, пептидо-α',β'-эпоксикетон или пептидо-α',β'-азиридинкетон, а пептид может быть тетрапептидом. Пептид может включать в себя разветвленные или неразветвленные боковые цепи, такие как водород, С1-6-алкил, С1-6-гидроксиалкил, С1-6-алкоксиалкил, арил, С1-6-аралкил, С1-6-алкиламид, С1-6-алкиламин, С1-6-карбоновую кислоту, С1-6-эфир карбоновой кислоты, С1-6-алкилтиол или простой С1-6-алкилтиоэфир, например изобутил, 1-нафтил, фенилметил и 2-фенилэтил. Αтом α'-углерода α',β'-эпоксикетона или α',β'-азиридинкетона может представлять собой хиральный атом углерода, такой как углерод (R) или β-конфигурации, как определено здесь.
В другом аспекте изобретение предоставляет фармацевтические композиции, включая фармацевтически приемлемый носитель и фармацевтически эффективное количество ингибитора гидролазы, которые ослабляют симптомы нейродегенеративного заболевания (такого как болезнь Альцгеймера), мышечной дистрофии, рака, хронических инфекционных заболеваний, лихорадки, атрофии мышц, денервации, нервных заболеваний, повреждений нервов, голодания и иммунно-зависимых состояний, и прочие.
В другом аспекте изобретение предоставляет соединения и фармацевтические композиции, которые биодоступны перорально.
В другом аспекте изобретение предоставляет противовоспалительные композиции.
В другом аспекте изобретение предоставляет способы, предназначенные для: ингибирования или уменьшения ВИЧ-инфекции у субъекта, воздействия на уровень экспрессии гена вируса у субъекта; изменения ряда антигенных пептидов, продуцированных протеасомой в организме; определения, регулируется ли клеточный, эволюционный или физиологический процесс или продукт в организме протеолитической активностью конкретной Ntn-гидролазой; лечения болезни Альцгеймера у субъекта; уменьшения скорости деградации мышечного белка; уменьшения скорости деградации внутриклеточного белка в клетке; уменьшения скорости деградации белка р53 в клетке; ингибирования роста злокачественных новообразований, родственных р53, у субъекта; ингибирования включения антигена в клетке; подавления иммунной системы субъекта; ингибирования деградации IκB-α в организме; уменьшения содержания NF-κB в клетке, мышце, органе или у субъекта; влияния на циклин-зависимые циклы эукариотических клеток; лечения пролиферативных заболеваний у субъекта; воздействия на протеасом-зависимое регулирование онкопротеинов в клетке; лечения роста злокачественного новообразования у субъекта; лечения у субъекта апоптоза, родственного р53; и скрининга белков, продуцируемых N-концевыми нуклеофильными гидролазами. Каждый из указанных способов предусматривает введение или контактирование эффективного количества композиции, содержащей предложенные ингибиторы гидролазы, субъекту, клетке, ткани, органу или организму.
Другие особенности и преимущества изобретения будут понятны из последующего подробного описания и из формулы изобретения.
Подробное описание изобретения
Изобретение включает в себя соединения, применимые в качестве ингибиторов ферментов. Указанные соединения обычно применимы для ингибирования ферментов, содержащих нуклеофильную группу у N-конца. Например, активности ферментов или субъединиц ферментов, содержащих в боковой цепи N-концевые нуклеофильные аминокислотные группы, такие как (группы) треонина, серина или цистеина, могут быть успешно ингибированы описанными здесь ингибиторами ферментов. Активности ферментов или субъединиц ферментов, содержащих неаминокислотные нуклеофильные группы у N-конца, такие как, например, защитные группы или углеводные группы, также могут быть успешно ингибированы описанными здесь ингибиторами ферментов.
Вне связи с конкретной теорией действия считают, что указанные N-концевые нуклеофилы Ntn образуют аддукты с эпоксидной функциональной группой описанных здесь ингибиторов ферментов. Например, полагают, что в субъединице β5/Pre2 протеасомы 20S N-концевой треонин необратимо образует морфолино- или пиперазино-аддукт в результате взаимодействия с пептид-эпоксидом или пептид-азиридином, таким как описано ниже. Образование такого аддукта представляет собой расщепление с разрывом цикла эпоксида или азиридина.
В вариантах осуществления, включающих указанные группы, связанные с α'-углеродами, стереохимия α'-углерода (углерода, образующего часть эпоксидного или азиридинового цикла) может быть (R) или (S). Изобретение основано отчасти на предложенной здесь структурно-функциональной информации, которая доказывает следующие предпочтительные стереохимические отношения. Отмечено, что предпочтительное соединение может иметь несколько стереоцентров, обозначенных сверху-снизу (или β-α, где β, как использовано здесь, показывает (расположение) над плоскостью (молекулы)), или отношением (R)-(S) (то есть не требуется, чтобы каждый стереоцентр соединения соответствовал предпочтительному состоянию). В некоторых предпочтительных вариантах осуществления стереохимия α'-углерода является (R), то есть атом Х является β, или (расположенным) над плоскостью молекулы.
С точки зрения стереохимии для определения абсолютной стереохимии следуют правилам Кана-Ингольда-Прелога (Cahn-Ingold-Prelog). Указанные правила описаны, например, в Organic Chemistry, Fox and Whitesell; Jones and Bartlett Publishers, Boston, MA (1994); раздел 5-6, стр. 177-178, причем раздел 5-6 приведен в качестве ссылки. Пептиды могут иметь повторяющуюся структуру основной цепи с боковыми цепями, отходящими от звеньев основной цепи. Обычно каждое звено основной цепи содержит связанную с ним боковую цепь, хотя в некоторых случаях боковая цепь представляет собой атом водорода. В других вариантах осуществления не каждое звено основной цепи содержит связанную с ним боковую цепь. Пептиды, применимые в пептид-эпоксидах или пептид-азиридинах, имеют два или более звена основной цепи. В некоторых вариантах осуществления, применимых для ингибирования химотрипсин-подобной (CT-L) активности протеасомы, присутствуют от двух до четырех звеньев основной цепи, а в некоторых предпочтительных вариантах осуществления для ингибирования CT-L присутствуют три звена основной цепи.
Боковые цепи, отходящие от звеньев основной цепи, могут представлять собой боковые цепи природной алифатической или ароматической аминокислоты, такие как водород (глицин), метил (аланин), изопропил (валин), втор-бутил (изолейцин), изобутил (лейцин), фенилметил (фенилаланин), и боковую цепь, составляющую аминокислоту пролин. Боковые цепи также могут представлять собой разветвленные или неразветвленные алифатические или ароматические группы, такие как этил, н-пропил, н-бутил, трет-бутил и арилзамещенные производные, такие как 1-фенилэтил, 2-фенилэтил, (1-нафтил)метил, (2-нафтил)метил, 1-(1-нафтил)этил, 1-(2-нафтил)этил, 2-(1-нафтил)этил, 2-(2-нафтил)этил и подобные соединения. Арильные группы могут быть дополнительно замещены разветвленными или неразветвленными С1-6-алкильными группами или замещенными алкильными группами, ацетилом и подобными или дополнительными арильными группами, или замещенными арильными группами, такими как бензоил и подобные. В качестве заместителей боковой цепи также могут быть использованы гетероарильные и гетероциклильные группы. Гетероарильные группы представляют собой азот-, кислород- и серусодержащие арильные группы, такие как тиенил, бензотиенил, нафтотиенил, тиантренил, фурил, пиранил, изобензофуранил, хроменил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, индолил, пуринил, хинолил и подобные. Гетероциклильные группы представляют собой тетрагидрофуран, пиперидин, пиперазин, пирролидин, морфолин, лактоны, лактамы и подобные.
В некоторых вариантах осуществления в пептид-эпоксиды или пептид-азиридины могут быть введены полярные или заряженные остатки. Например, могут быть введены аминокислоты природного происхождения, такие как гидроксисодержащие (Thr, Tyr, Ser) или серусодержащие (Met, Cys), также как не-незаменимые аминокислоты, например, таурин, карнитин, цитруллин, цистин, орнитин, норлейцин и другие. Неприродные заместители боковой цепи с заряженными или полярными членами могут также представлять собой такие, как, например, С1-6-алкильные цепи или С6-12-арильные группы, с одной или более гидрокси-, короткоцепными алкокси-, сульфидными, тио-, карбоксильными, сложноэфирными, фосфоно-, амидо- или аминогруппами, или указанными заместителями, замещенными одним или более атомами галогена. В некоторых предпочтительных вариантах осуществления в боковой цепи пептидного звена присутствует по меньшей мере одна арильная группа.
В некоторых вариантах осуществления звенья основной цепи представляют собой амидные единицы [-NH-CHR-C(=O)-], в которых R представляет собой боковую цепь. Такая конструкция не исключает аминокислоты природного происхождения, пролина, или другой неприродной циклической аминокислоты, которые могут быть определены специалистами в данной области техники.
В других вариантах осуществления звенья основной цепи представляют собой N-алкилированные амидные единицы (например, N-метилированные и подобные), аналоги олефинов (в которых одна или более амидных связей заменены олефиновыми связями), аналоги тетразола (в которых тетразольный цикл использует цис-конфигурацию основной цепи) или комбинации указанных связей основной цепи. Еще в других вариантах осуществления α-углерод аминокислоты модифицирован α-алкильным заместителем, например аминоизомасляной кислотой. В некоторых дополнительных вариантах осуществления боковые цепи местно модифицированы, например, путем ΔЕ или ΔZ дегидро-модификации, в которой между α- и β-атомами боковой цепи присутствует двойная связь, или, например, путем ΔЕ или ΔZ циклопропильной модификации, в которой между α- и β-атомами боковой цепи присутствует циклопропильная группа. В других дополнительных вариантах осуществления при использовании аминокислотных групп могут быть применены D-аминокислоты. Дополнительные варианты осуществления могут включать в себя циклизацию боковых цепей с основной цепью, образование дисульфидной связи, образование лактама, азосвязь и другие модификации, обсужденные в “Peptides and Mimics, Design of Conformationally constraind” by Hruby and Boteju, в “Molecular Biology and Biotechnology: A Comprehensive Desk Reference” изд. Robert A. Meyers, VCH Publishers (1995), стр. 658-664, приведенных здесь в качестве ссылки.
Один из аспектов изобретения относится к соединениям структуры формулы (I) или их фармацевтически приемлемым солям:
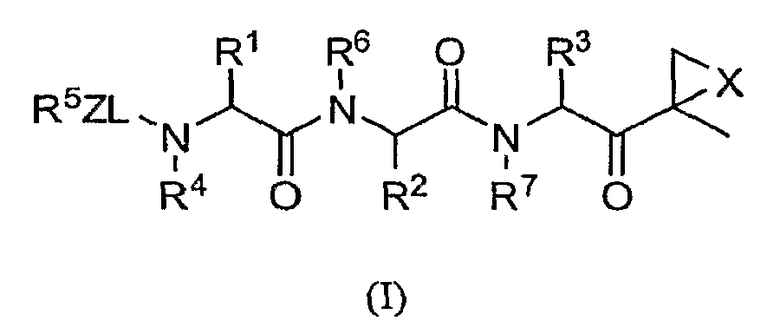
в которой
L выбран из С=О, C=S и SO2, предпочтительно С=О;
Х выбран из О, S, NH и N-C1-6-алкила;
Z отсутствует, С1-6-алкил или С1-6-алкоксигруппа предпочтительно отсутствует;
каждый из R1, R2 и R3 независимо друг от друга выбран из водорода, С1-6-алкила, С1-6-алкенила, С1-6-алкинила, С1-6-гидроксиалкила, С1-6-алкоксиалкила, арила, С1-6-аралкила, гетероарила, гетероциклила, С1-6-гетероциклоалкила, С1-6-гетероаралкила, карбоциклила и С1-6-карбоциклоалкила;
R4 выбран из водорода, С1-6-аралкила и С1-6алкила;
R5 представляет собой гетероарил; и
R6 и R7 независимо друг от друга выбраны из водорода, С1-6-алкила и С1-6-аралкила.
В некоторых вариантах осуществления R1, R2 и R3 независимо друг от друга выбраны из водорода, С1-6-алкила, С1-6-гидроксиалкила, С1-6-алкоксиалкила, С1-6-аралкила, С1-6-гетероциклоалкила, С1-6-гетероаралкила и С1-6-карбоциклоалкила. В некоторых вариантах осуществления любой из R1, R2 и R3 независимо друг от друга представляет собой С1-6-алкил, выбранный из метила, этила, пропила, изопропила, бутила, втор-бутила и изобутила. В некоторых вариантах осуществления любой из R1, R2 и R3 независимо друг от друга представляет собой С1-6-гидроксиалкил. В некоторых таких вариантах осуществления любой из R1, R2 и R3 независимо друг от друга выбран из гидроксиметила и гидроксиэтила, предпочтительно гидроксиметил. В некоторых вариантах осуществления любой из R1, R2 и R3 независимо друг от друга представляет собой С1-6-алкоксиалкил. В некоторых таких вариантах осуществления любой из R1, R2 и R3 независимо друг от друга выбран из метоксиметила и метоксиэтила, предпочтительно метоксиметил. В некоторых вариантах осуществления любой из R1, R2 и R3 независимо друг от друга представляют собой С1-6-гетероаралкил. В некоторых таких вариантах осуществления любой из R1, R2 и R3 независимо друг от друга выбран из имидазолилметила, пиразолилметила, тиазолилметила и пиридилметила, предпочтительно имидазол-4-илметила, тиазол-4-илметила, 2-пиридилметила, 3-пиридилметила или 4-пиридилметила. В некоторых вариантах осуществления любой из R1, R2 и R3 независимо друг от друга представляет собой С1-6-аралкил. В некоторых таких вариантах осуществления любой из R1, R2 и R3 независимо друг от друга выбран из фенилметила (бензила) и фенилэтила, предпочтительно фенилметил. В некоторых вариантах осуществления любой из R1, R2 и R3 независимо друг от друга представляет собой С1-6-карбоциклоалкил. В некоторых таких вариантах осуществления любой R1 представляет собой циклогексилметил. В некоторых вариантах осуществления все R1, R2 и R3 различны. В некоторых вариантах осуществления любые два из R1, R2 и R3 одинаковы. В некоторых вариантах осуществления все R1, R2 и R3 одинаковы.
В некоторых вариантах осуществления по меньшей мере один из R1 и R2 выбран из С1-6-гидроксиалкила и С1-6-алкоксиалкила. В некоторых таких вариантах осуществления по меньшей мере один из R1 и R2 представляет собой алкоксиалкил. В некоторых таких вариантах осуществления по меньшей мере один из R1 и R2 выбран из метоксиметила и метоксиэтила.
В некоторых вариантах осуществления R3 выбран из С1-6-алкила и С1-6-аралкила, предпочтительно С1-6-алкила. В некоторых таких вариантах осуществления R3 выбран из метила, этила, изопропила, втор-бутила и изобутила. В некоторых таких вариантах осуществления R3 представляет собой изобутил. В некоторых альтернативных вариантах осуществления R3 выбран из фенилметила и фенилэтила, предпочтительно фенилметил.
В некоторых вариантах осуществления R4, R6 и R7 независимо друг от друга выбраны из водорода и метила и предпочтительно представляют собой водород.
В некоторых вариантах осуществления R5 представляет собой 5- или 6-членный гетероарил. В некоторых таких вариантах осуществления R5 выбран из изоксазола, изотиазола, фурана, тиофена, оксазола, тиазола, пиразола или имидазола, предпочтительно из изоксазола, фурана или тиазола.
В некоторых вариантах осуществления R5 представляет собой бициклический гетероарил. В некоторых таких вариантах осуществления бициклический гетероарил выбран из бензизоксазола, бензоксазола, бензотиазола и бензизотиазола.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил или изоксазол-5-ил. В некоторых предпочтительных вариантах такого осуществления, если изоксазол-3-ил замещен, он замещен по меньшей мере в 5-положении. В некоторых предпочтительных вариантах осуществления, если изоксазол-5-ил замещен, он замещен по меньшей мере в 3-положении.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой незамещенный изоксазол-3-ил.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой замещенный изоксазол-3-ил. В некоторых таких вариантах осуществления R5 представляет собой изоксазол-3-ил, замещенный заместителем, выбранным из С1-6-алкила, С1-6-алкоксигруппы, С1-6-алкоксиалкила, С1-6-гидроксиалкила, остатка карбоновой кислоты, аминокарбоксилата, С1-6-алкиламинокарбоксилата, (С1-6-алкил)2аминокарбоксилата, С1-6-алкилкарбоксилата, С1-6-гетероаралкила, С1-6-аралкила, С1-6-гетероциклоалкила и С1-6-карбоциклоалкила. В некоторых предпочтительных вариантах такого осуществления R5 представляет собой изоксазол-3-ил, замещенный заместителем, выбранным из метила, этила, изопропила и циклопропилметила.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный от 4- до 6-членным азотсодержащим С1-6-гетероциклоалкилом. В некоторых таких вариантах осуществления R5 представляет собой изоксазол-3-ил, замещенный азетидинилметилом, предпочтительно азетидин-1-илметилом. В некоторых альтернативных вариантах такого осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный радикалом 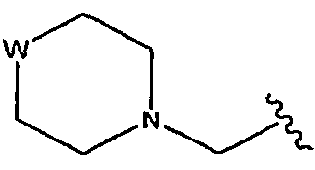 , в котором W представляет собой О, NR или СН2 и R представляет собой Н или С1-6-алкил. В некоторых таких вариантах осуществления W представляет собой O.
, в котором W представляет собой О, NR или СН2 и R представляет собой Н или С1-6-алкил. В некоторых таких вариантах осуществления W представляет собой O.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный 5-членным азотсодержащим С1-6-гетероаралкилом, таким как пиразолилметил, имидазолилметил, триазол-5-илметил, предпочтительно 1,2,4-триазол-5-илметил.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный С1-6-алкоксигруппой или С1-6алкоксиалкилом, предпочтительно метоксигруппой, этоксигруппой, метоксиметилом или метоксиэтилом.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный С1-6-гидроксиалкилом, предпочтительно гидроксиметилом или гидроксиэтилом.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный остатком карбоновой кислоты, аминокарбоксилатом, С1-6-алкиламинокарбоксилатом, (С1-6-алкил)2аминокарбоксилатом или С1-6-алкилкарбоксилатом. В некоторых таких вариантах осуществления R5 замещен метилкарбоксилатом или этилкарбоксилатом, предпочтительно метилкарбоксилатом.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой незамещенный изоксазол-5-ил.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой замещенный изоксазол-5-ил. В некоторых таких вариантах осуществления R5 представляет собой изоксазол-5-ил, замещенный заместителем, выбранным из С1-6-алкила, С1-6-алкоксигруппы, С1-6-алкоксиалкила, С1-6-гидроксиалкила, остатка карбоновой кислоты, аминокарбоксилата, С1-6-алкиламинокарбоксидала, (С1-6-алкил)2аминокарбоксилата, С1-6-алкилкарбоксилата, С1-6-гетероаралкила, С1-6-аралкила, С1-6-гетероциклоалкила и С1-6-карбоциклоалкила. В некоторых предпочтительных вариантах такого осуществления R5 представляет собой изоксазол-3-ил, замещенный заместителем, выбранным из метила, этила, изопропила и циклопропилметила.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный от 4- до 6-членным азотсодержащим С1-6-гетероциклоалкилом. В некоторых таких вариантах осуществления R5 представляет собой изоксазол-5-ил, замещенный азетидинилметилом, предпочтительно азетидин-1-илметилом. В некоторых альтернативных вариантах такого осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный радикалом формулы 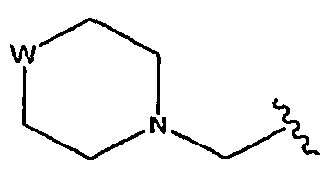 , в которой W представляет собой О, NR или СН2 и R представляет собой Н или С1-6-алкил. В некоторых таких вариантах осуществления W представляет собой O.
, в которой W представляет собой О, NR или СН2 и R представляет собой Н или С1-6-алкил. В некоторых таких вариантах осуществления W представляет собой O.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-5-ил, замещенный 5-членным азотсодержащим С1-6-гетероаралкилом, таким как пиразолилметил, имидазолилметил, триазол-5-илметил, предпочтительно 1,2,4-триазол-5-илметил.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-5-ил, замещенный С1-6-алкоксигруппой или С1-6-алкоксиалкилом, предпочтительно метоксигруппой, этоксигруппой, метоксиметилом или метоксиэтилом.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-5-ил, замещенный С1-6-гидроксиалкилом, предпочтительно гидроксиметилом или гидроксиэтилом.
В некоторых вариантах осуществления L представляет собой С=О, Z отсутствует и R5 представляет собой изоксазол-3-ил, замещенный остатком карбоновой кислоты, аминокарбоксилатом, С1-6-алкиламинокарбоксилатом, (С1-6-алкил)2аминокарбоксилатом или С1-6-алкилкарбоксилатом. В некоторых таких вариантах осуществления R5 замещен метилкарбоксилатом или этилкарбоксилатом, предпочтительно метилкарбоксилатом.
В некоторых вариантах осуществления соединение формулы I выбрано из
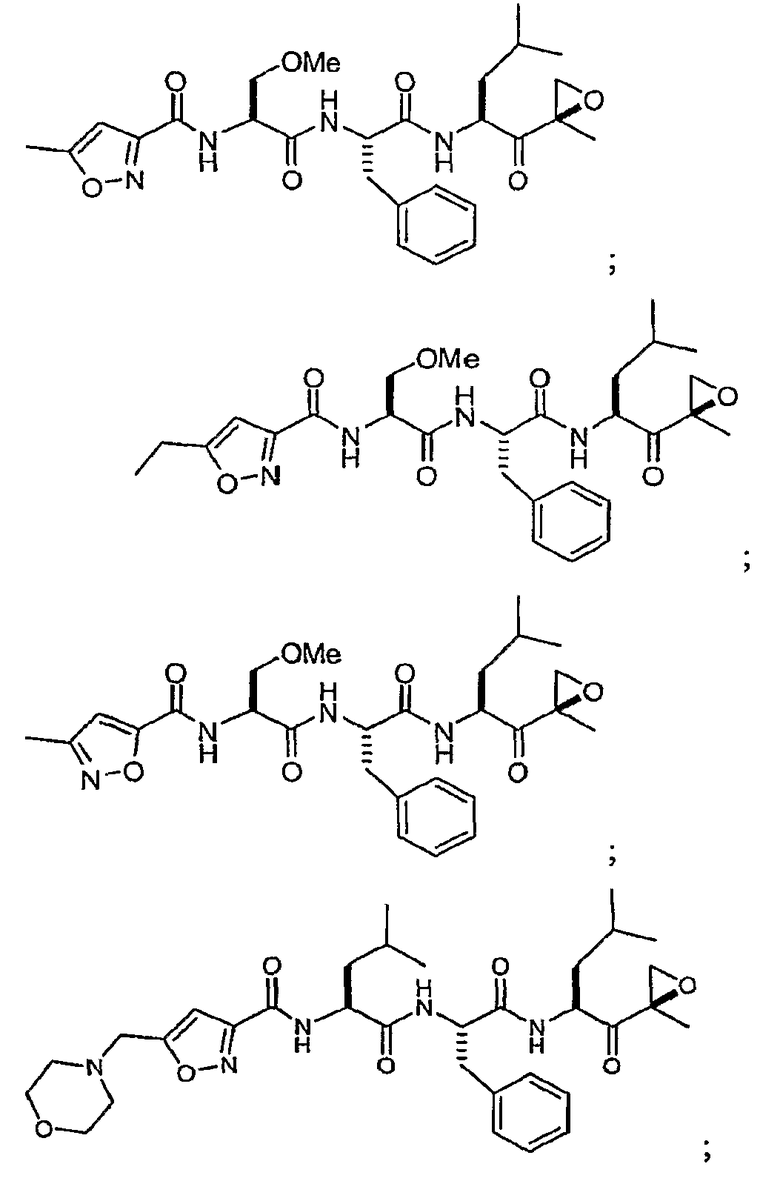
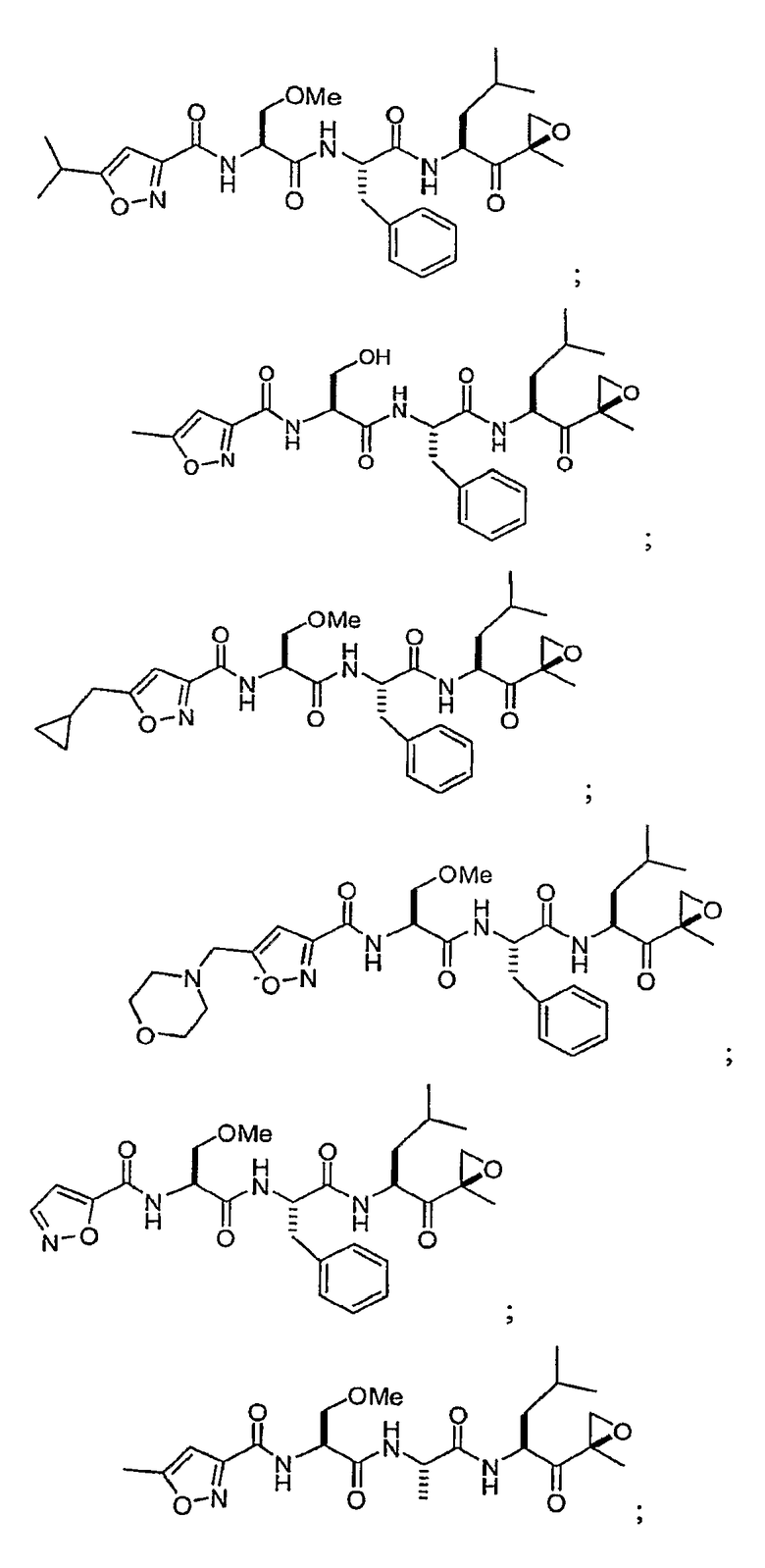
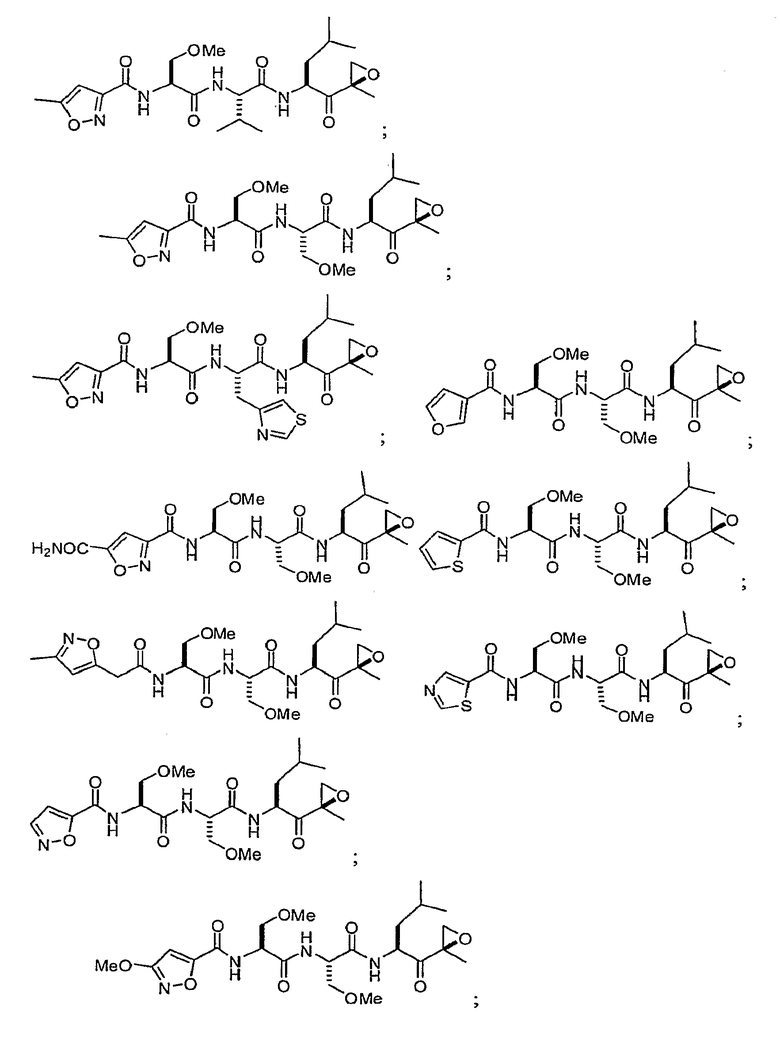
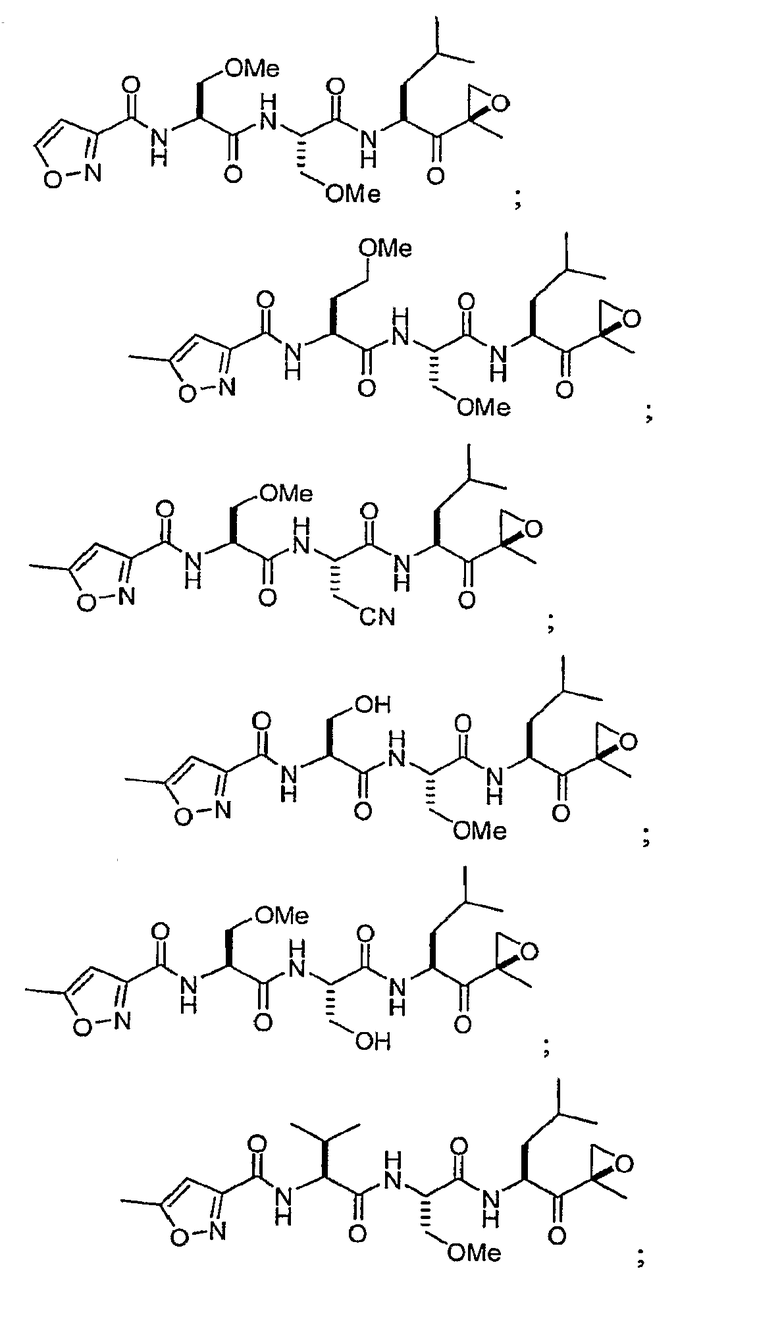
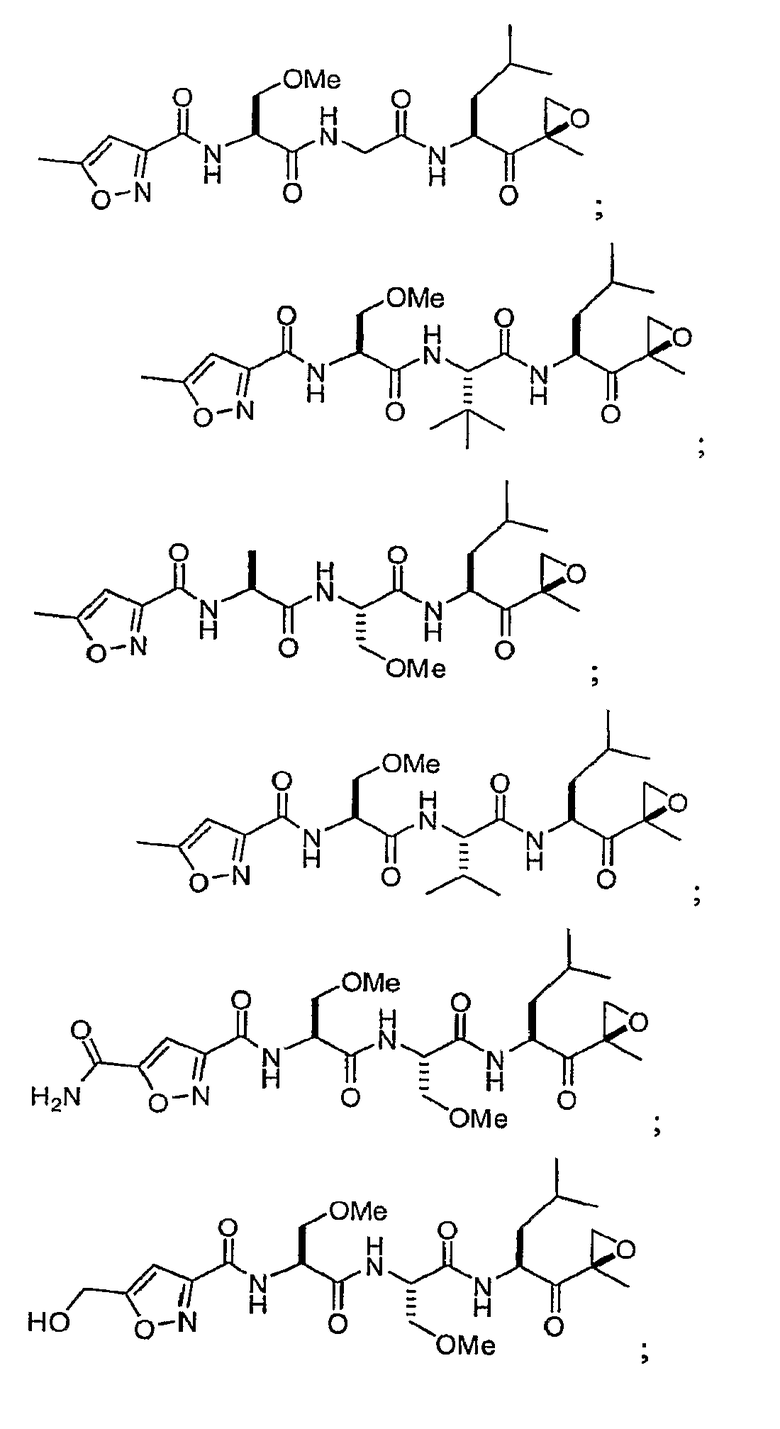
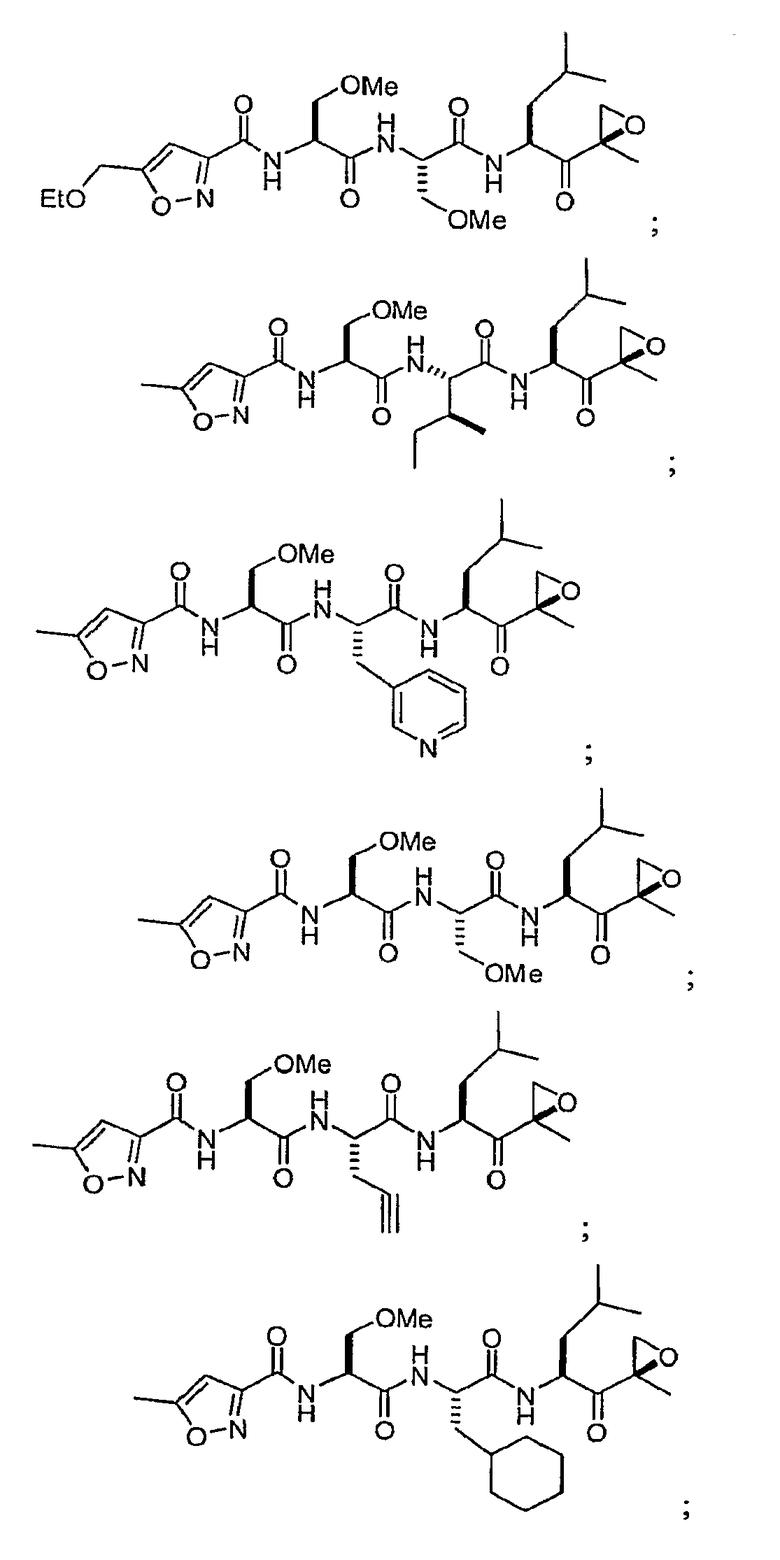
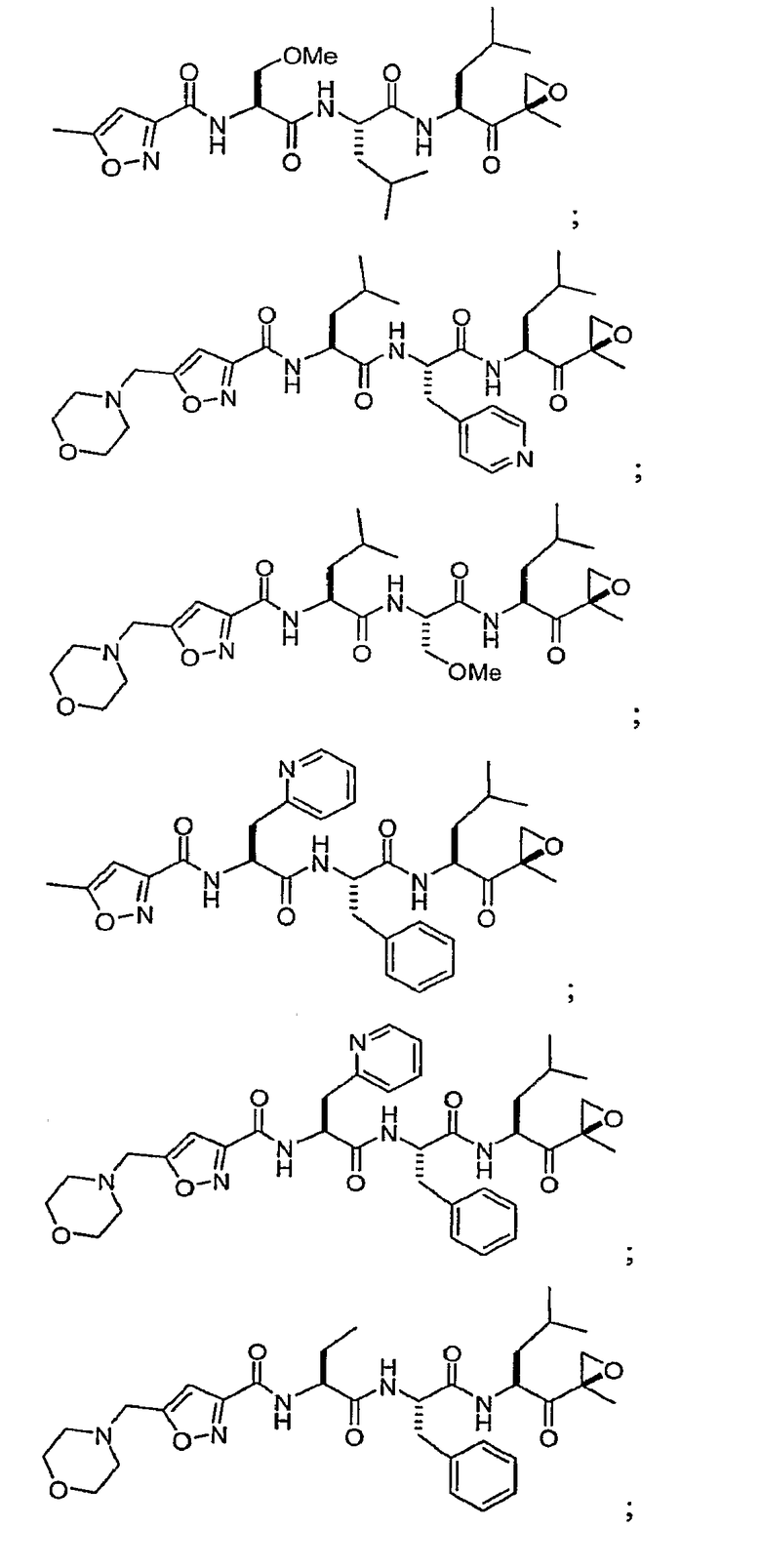
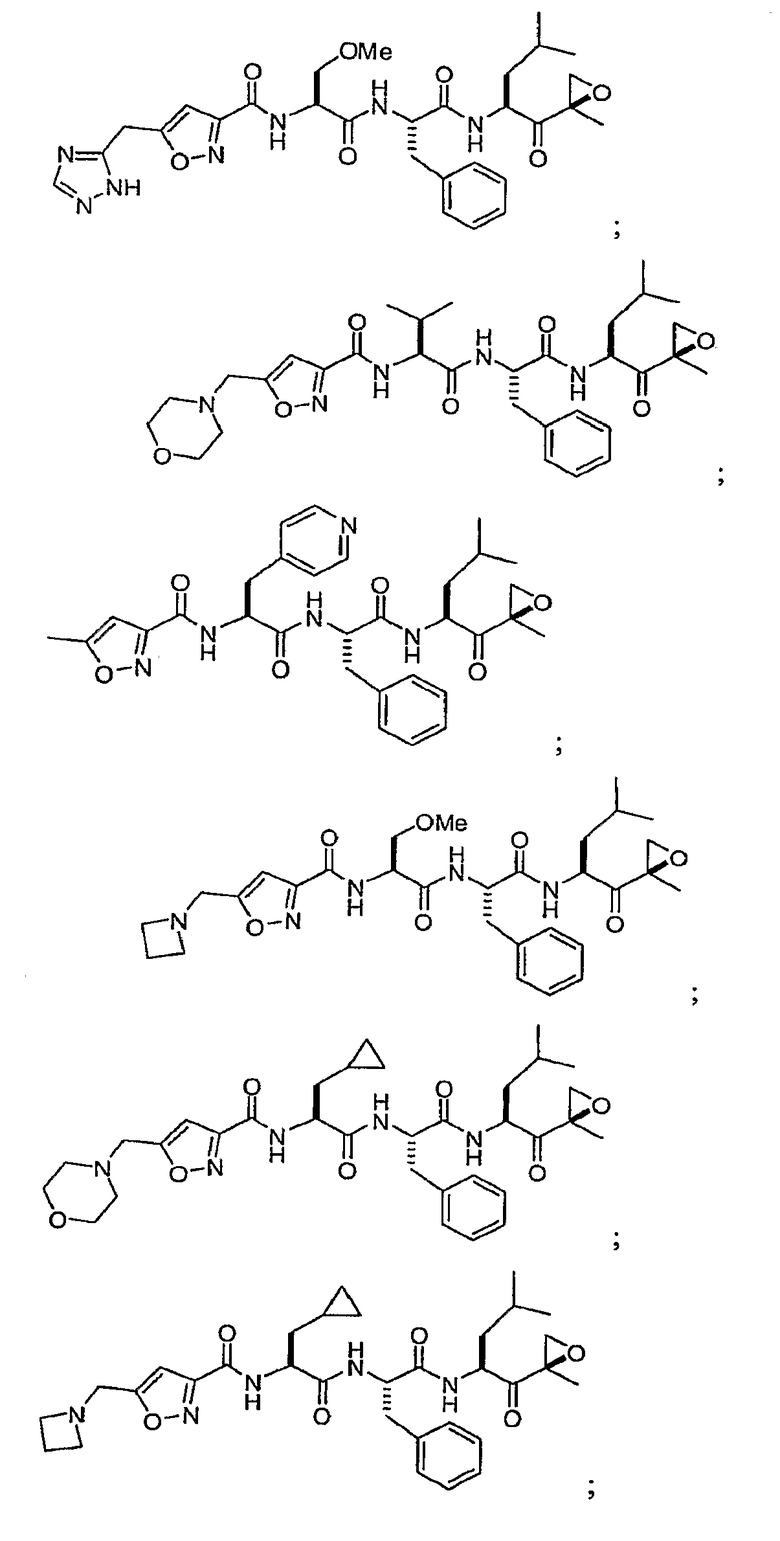
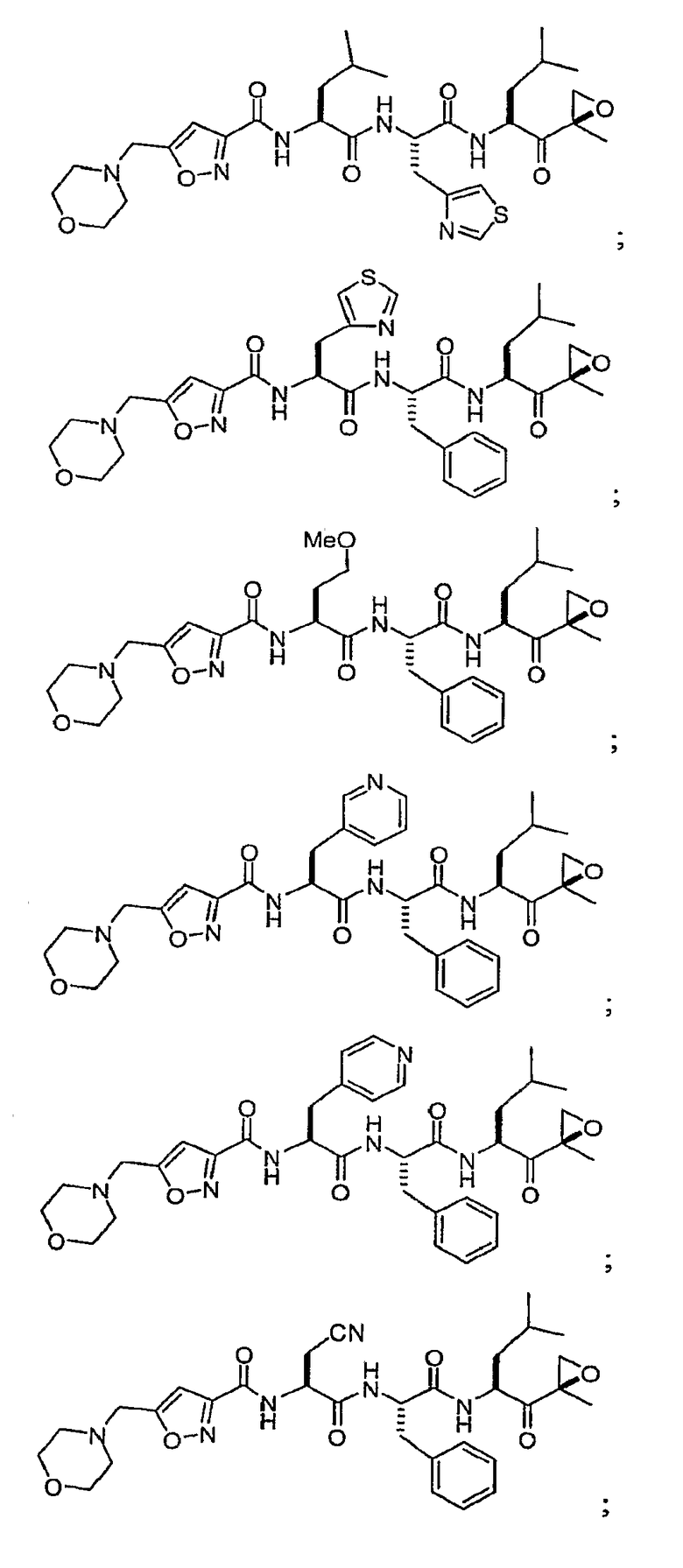
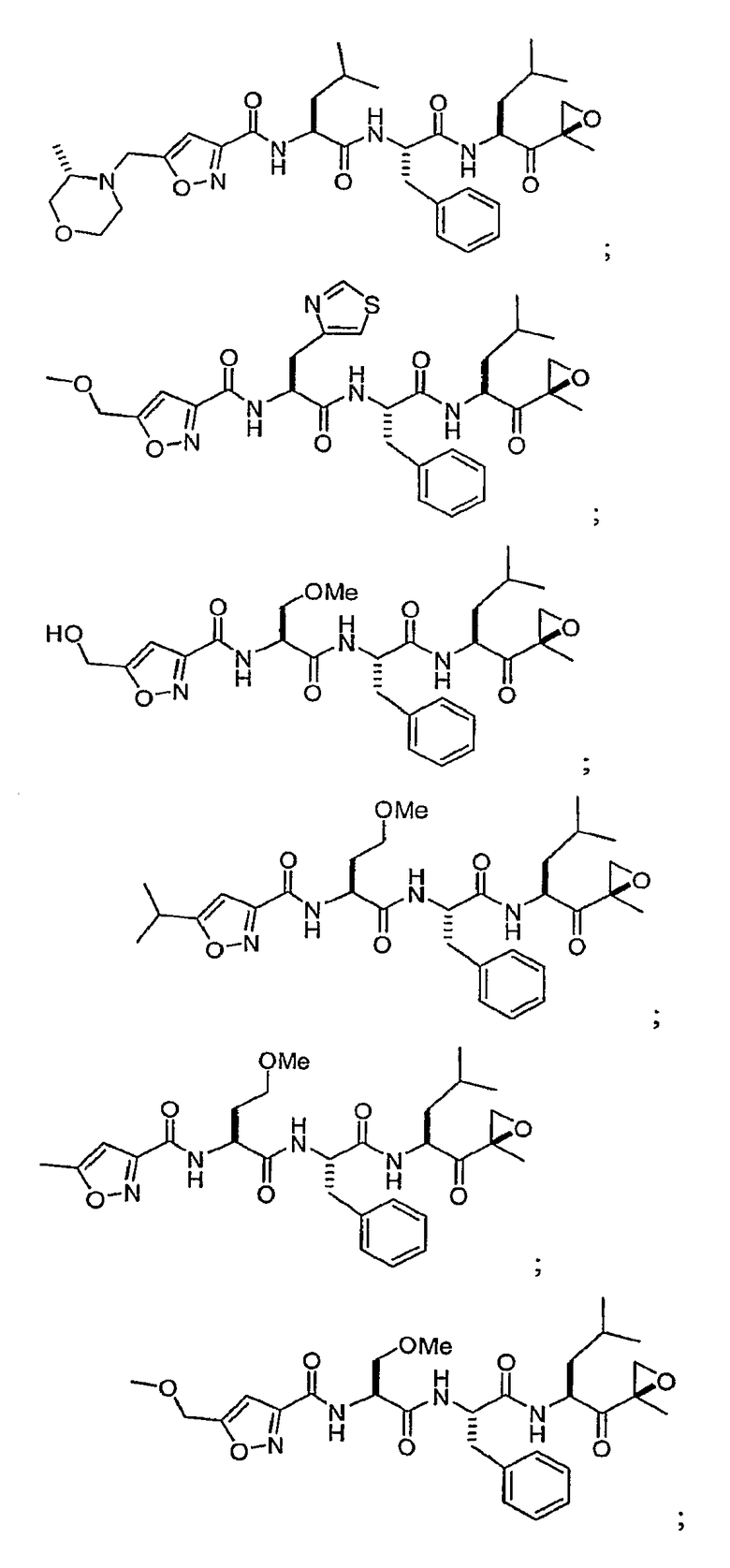
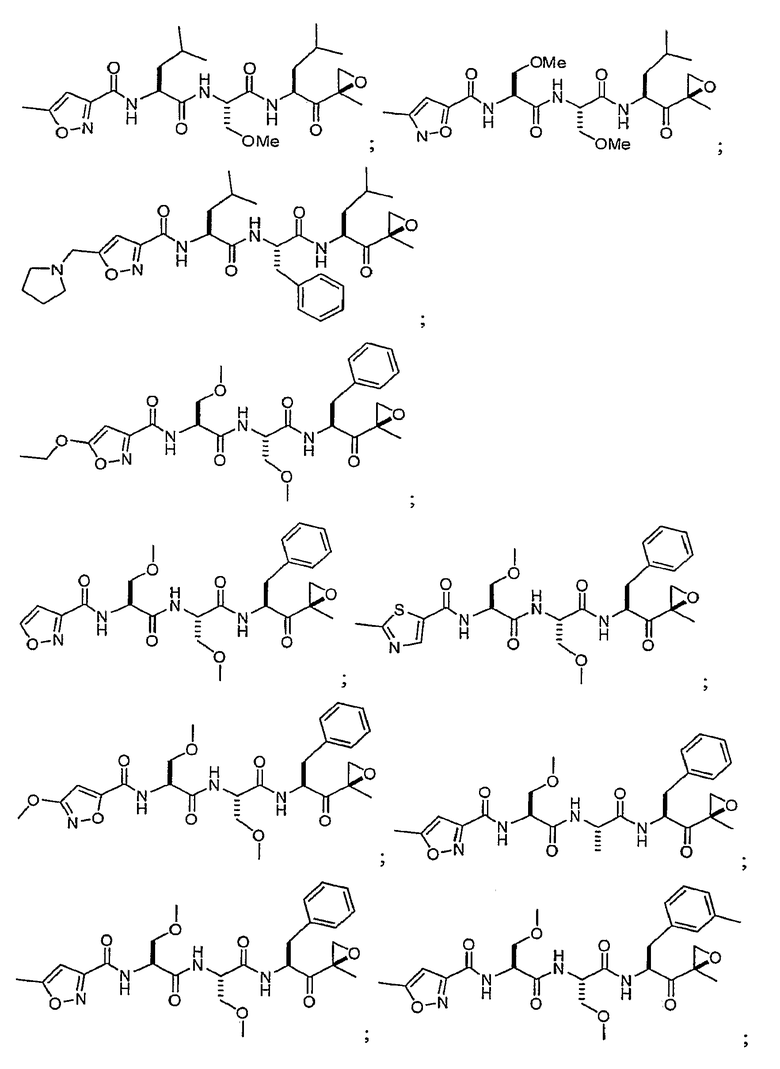
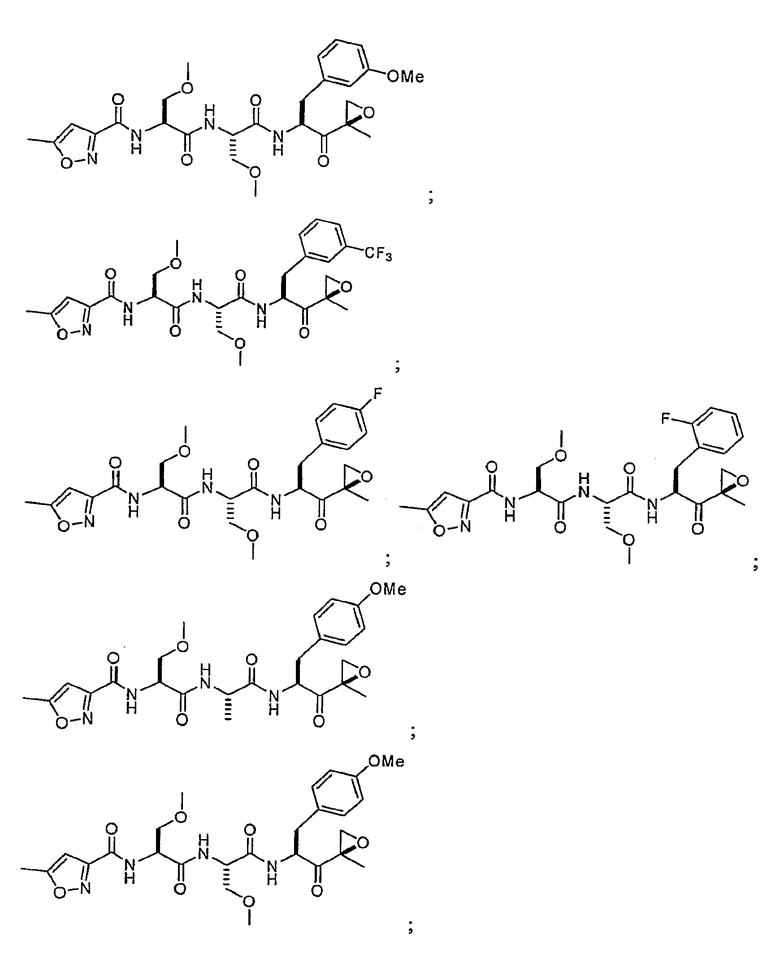
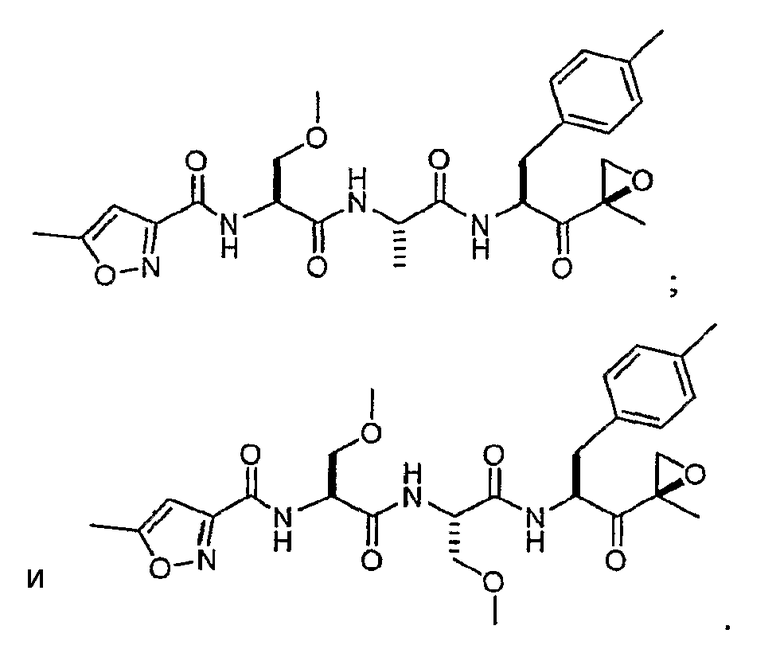
Один из аспектов изобретения относится к медицинскому устройству, включающему описанную здесь композицию, которая содержит ингибитор, имеющий структуру формулы I. В одном из вариантов осуществления композиции вводят в медицинское устройство. В некоторых вариантах осуществления медицинское устройство представляет собой полимерную матрицу, содержащую гель, или керамическую матрицу и ингибитор. Указанный полимер может быть природного происхождения или синтетическим. В некоторых вариантах осуществления указанный гель служит в качестве депо лекарства, адгезива, шва, барьера или пломбы.
Другой аспект изобретения относится к медицинскому устройству, содержащему субстрат, имеющий поверхность, на которой расположен ингибитор, имеющий структуру формулы I. В одном из вариантов осуществления ингибитор размещен непосредственно на медицинском устройстве. В другом варианте осуществления покрытие расположено таким образом, чтобы покрытие, состоящее из полимерной матрицы или керамической матрицы с ингибитором, имеющим структуру формулы I, было диспергировано или растворено.
В одном из вариантов осуществления медицинское устройство представляет собой коронарный, сосудистый, периферический или билиарный стент. Более конкретно, стент данного изобретения представляет собой расширяющийся стент. В случае покрытия матрицей, содержащей ингибитор, имеющий структуру формулы I, матрица эластична для приспособления к сжатому или расширенному состоянию указанного расширяющегося стента. В другом варианте осуществления данного изобретения стент имеет по меньшей мере участок, который может вставляться или имплантироваться в тело пациента, причем участок имеет поверхность, которая адаптирована для воздействия на тело пациента и в котором по меньшей мере часть поверхности покрыта ингибитором, имеющим структуру формулы I, или покрытие состоит из матрицы, содержащей диспергированный или растворенный в ней ингибитор, имеющий структуру формулы I. Пример подходящего стента предложен в патенте США № 4733665, который приведен здесь в качестве ссылки.
В другом варианте осуществления медицинское устройство данного изобретения представляет собой хирургический инструмент, такой как сосудистый имплантат, внутрипросветное устройство, хирургический уплотнитель или сосудистая подложка. Более конкретно, медицинское устройство данного изобретения представляет собой катетер, порт доступа имплантируемого сосуда, центральный венозный катетер, артериальный катетер, сосудистый имплантат, внутриаортный баллонный насос, шов, вентрикулярный вспомогательный насос, элюирующий лекарство барьер, адгезив, сосудистую накидку, экстра/перисосудистую подложку, фильтр крови или фильтр, адаптированный для размещения в кровеносном сосуде, покрытом ингибитором, имеющим структуру формулы I, либо непосредственно, либо матрицей, содержащей ингибитор, имеющий структуру формулы I.
В некоторых вариантах осуществления внутрипросветное медицинское устройство покрыто ингибитором, имеющим структуру формулы I, или покрытием, состоящим из биологически толерантной матрицы и ингибитора, имеющего структуру формулы I, диспергированного в полимере, указанное устройство, имеющее внутреннюю поверхность и внешнюю поверхность, имеет покрытие, нанесенное на по меньшей мере часть внутренней поверхности, внешней поверхности или обеих.
В некоторых вариантах осуществления медицинское устройство может быть применимым для предупреждения рестеноза после ангиопластики. Медицинское устройство может также быть применимо для лечения различных заболеваний и состояний осуществлением локализованного введения ингибитора, имеющего структуру формулы I. Указанные заболевания и состояния включают в себя рестеноз, воспаление, ревматоидный артрит, повреждения тканей, вызванные воспалением, гиперпролиферативные заболевания, тяжелый или артритный псориаз, мышечная дистрофия, хронические инфекционные заболевания, аномальная иммунная реакция, состояния, поражающие чувствительные кровяные тельца, повреждения, родственные ишемическим состояниям, и вирусные инфекции и пролиферация. Примерами заболеваний и состояний, которые лечат с применением медицинских устройств данного изобретения, покрытых лекарственным средством, являются атеросклероз, острый коронарный синдром, болезнь Альцгеймера, рак, лихорадка, мышечная слабость (атрофия), денервация, закупорка сосуда, внезапный приступ, ВИЧ-инфекция, повреждения нервов, почечная недостаточность, связанная с ацидозом, и печеночная недостаточность. Смотри, например, Goldberg, патент США № 5340736.
Термин «Сx-y-алкил» относится к замещенным или незамещенным углеводородным группам, включая алкильные группы с нормальной цепью и алкильные группы с разветвленной цепью, которые содержат от х до y атомов углерода в цепи, включая галогеноалкильные группы, такие как трифторметил и 2,2,2-трифторэтил и т.д. С0-алкил означает водород, где группа находится в концевом положении, если связь внутренняя. Термины «С2-y-алкенил» и «С2-y-алкинил» относятся к замещенным или незамещенным ненасыщенным алифатическим группам, аналогичным по длине, и возможному замещению вышеописанным алкилам, но содержащим по меньшей мере одну двойную или тройную связь, соответственно.
Термин «алкоксигруппа» относится к алкильной группе, содержащей присоединенный к ней атом кислорода. Представительные алкоксигруппы включают в себя метоксигруппу, этоксигруппу, пропоксигруппу, трет-бутоксигруппу и подобные. «Простой эфир» представляет собой два углеводорода, ковалентно связанные атомом кислорода. Соответственно, заместитель алкила, который представляет собой простой алкиловый эфир, является или соответствует алкоксигруппе.
Термин «С1-6-алкоксиалкил» относится к С1-6-алкильной группе, замещенной алкоксигруппой, образуя при этом простой эфир.
Термин «С1-6-аралкил», как использовано здесь, относится к С1-6-алкильной группе, замещенной арильной группой.
Термины «амин» и «амино» известны в технике и относятся как к незамещенным, так и к замещенным аминам и их солям, например к членам, которые могут быть представлены общими формулами:
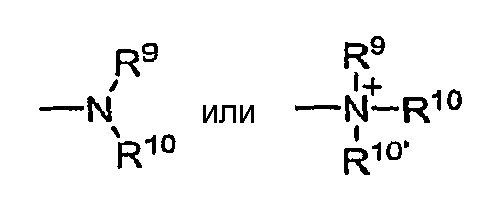
в которых каждый из R9, R10 и R10' независимо друг от друга представляет собой водород, алкил, алкенил, -(СН2)m0R8, или R9 и R10 вместе с атомом N, к которому они присоединены, образуют гетероцикл, содержащий от 4 до 8 атомов в цикле; R8 представляет собой арил, циклоалкил, циклоалкенил, гетероциклил или полициклил и m равно нулю или целому числу от 1 до 8. В предпочтительных вариантах осуществления только один из R9 и R10 может быть карбонилом, например R9, R10 и азот вместе не образуют имид. В еще более предпочтительном варианте осуществления каждый из R9 и R10 (и необязательно R10) независимо друг от друга представляют собой водород, алкил, алкенил или -(СН2)m-R8. В некоторых вариантах осуществления аминогруппа является основной, представляя собой протонированную форму с рКа≥7,00.
Термины «амид» и «амидо» известны в технике как аминозамещенный карбонил и включают в себя группу, которая может быть представлена общей формулой:
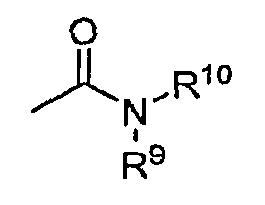
в которой R9 и R10 - как определено выше. Предпочтительные варианты осуществления амида не включают в себя имиды, которые могут быть нестабильными.
Термин «арил», как использовано здесь, включает в себя 5-, 6- и 7-членные замещенные или незамещенные моноциклические ароматические группы, в которых каждый атом цикла представляет собой углерод. Термин «арил» также включает в себя полициклические кольцевые системы, содержащие два или более цикла, в которых два или более (атома) углерода являются общими для двух соседних циклов, в которых по меньшей мере один цикл является ароматическим; например, другие циклы могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Арильные группы включают в себя бензол, нафталин, фенантрен, фенол, анилин и подобные.
Термины «карбоцикл» и «карбоциклил», как использовано здесь, относятся к неароматическим замещенным или незамещенным циклам, в которых каждый атом цикла представляет собой углерод. Термины «карбоцикл» и «карбоциклил» также включают в себя полициклические кольцевые системы, содержащие два или более цикла, в которых два или более атома углерода являются общими для двух соседних циклов, в которых по меньшей мере один из циклов является карбоциклом, например, другие циклы могут быть циклоалкилами, циклоалкенилами, циклоалкинилами, арилами, гетероарилами и/или гетероциклилами.
Термин «карбонил» известен в технике и включает в себя такие группы, которые могут быть представлены общими формулами:
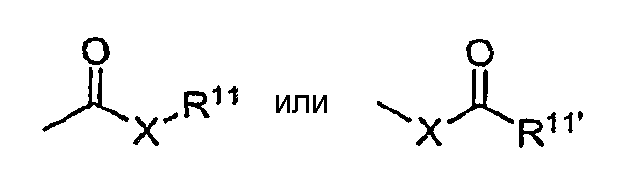
в которых Х представляет собой связь или (атом) кислорода или серы и R11 представляет собой водород, алкил, алкенил, -(СН2)m-R8 или фармацевтически приемлемую соль; R11' представляет собой водород, алкил, алкенил, -(СН2)m-R8, где R8 - как определено выше. Если Х представляет собой кислород и R11 и R11' не являются (атомами) водорода, формула представляет собой «сложный эфир». Если Х представляет собой кислород и R11 означает водород, формула представляет собой «карбоновую кислоту».
Как использовано здесь, «фермент» может быть любой молекулой, частично или полностью напоминающей белок, которая обеспечивает каталитический путь реакции. Такие ферменты могут быть природными ферментами, слитыми ферментами, проферментами, апоферментами, денатурированными ферментами, фарнезилированными ферментами, убиквитинилированными ферментами, ферментами, ацилированными жирными кислотами, геранилгеранилированными ферментами, ферментами, связанными GPI, ферментами, связанными с липидами, пренилированными ферментами, природными или искусственными мутантными ферментами, ферментами с модификациями в боковой или основной цепи, ферментами с ведущими последовательностями и ферменты, закомплексованные с небелковоподобными веществами, такими как протеогликаны, протеолипосомы. Ферменты могут быть получены любыми способами, включая природную экспрессию, промотированную экспрессию, клонирование, различные синтезы пептидов в растворе и в твердой фазе и подобные способы, известные специалистам в данной области техники.
Термин «С1-6-гетероалкил», как использован здесь, относится к С1-6-алкильной группе, замещенной гетероарильной группой.
Термин «гетероарил» включает в себя замещенные или незамещенные от 5- до 7-членные циклические структуры, более предпочтительно от 5- до 6-членные кольца, циклические структуры которых включают в себя от одного до четырех гетероатомов. Термин «гетероарил» также включает в себя полициклические кольцевые системы, содержащие два или более цикла, в которых два или более (атома) углерода являются общими для двух соседних циклов, в которых по меньшей мере один цикл является гетероароматическим, например, другие циклы могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Гетероарильные группы включают в себя, например, пиррол, фуран, тиофен, имидазол, изоксазол, оксазол, тиазол, триазол, пиразол, пиридин, пиразин, пиридазин и пиримидин и подобные.
Термин «гетероатом», как использовано здесь, означает любой элемент, кроме углерода и водорода. Предпочтительные гетероатомы представляют собой азот, кислород, фосфор и серу.
Термины «гетероциклил» или «гетероциклическая группа» относятся к замещенным или незамещенным неароматическим от 3- до 10-членным циклическим структурам, более предпочтительно от 3- до 7-членным циклам, циклические структуры которых включают в себя от одного до четырех гетероатомов. Термины «гетероциклил» или «гетероциклическая группа» также включают в себя полициклические системы, содержащие два или более цикла, в которых два или более (атома) углерода являются общими для двух соседних циклов, в которых по меньшей мере один из циклов является гетероциклическим, например, другие циклы могут быть циклоалкилами, циклоалкенилами, циклоалкинилами, арилами, гетероарилами и/или гетероциклилами. Гетероциклические группы включают в себя, например, тетрагидрофуран, пиперидин, пиперазин, пирролидин, морфолин, лактоны, лактамы и подобные.
Термин «С1-6-гетероциклоалкил», как использовано здесь, относится к С1-6-алкильной группе, замещенной гетероциклильной группой.
Термин «С1-6-гидроксиалкил» относится к С1-6-алкильной группе, замещенной гидроксигруппой.
Как использовано здесь, термин «ингибитор» предназначается для описания соединения, которое блокирует или ослабляет активность фермента (например, ингибирование протеолитического расщепления стандартных флюорогенных пептидных субстратов, таких как suc-LLVY-AMC, Box-LLR-AMC и Z-LLE-AMC, ингибирование различных каталитических активностей протеасомы 20S). Ингибитор может действовать с конкурентным, безконкурентным или неконкурентным ингибированием. Ингибитор может связывать обратимо или необратимо и поэтому термин включает в себя соединения, которые представляют собой суицидальные субстраты фермента. Ингибитор может модифицировать одну или более точек на или вблизи активной точки фермента или он может вызывать конформационное изменение в другом месте фермента.
Как использовано здесь, термин «перорально биодоступный» предназначен для описания соединения, введенного мыши (в количестве) 40 мг/кг или менее, 20 мг/кг или менее или даже 10 мг/кг или менее, которое через час после перорального введения ингибирует активность протеасомы CT-L в крови по меньшей мере примерно на 50%, по меньшей мере примерно на 75% или даже по меньшей мере примерно на 90%.
Как использовано здесь, термин «пептид» включает в себя не только стандартно амидо-связанные (соединения) со стандартными α-заместителями, но все применимые пептидомиметики с другими модифицированными связями, боковыми цепями неприродного происхождения и модификациями боковой цепи, как детализировано ниже.
Термин «полициклил» или «полициклический» относится к от двух до более циклам (например, циклоалкилам, циклоалкенилам, циклоалкинилам, арилам, гетероарилам и/или гетероциклилам), в которых два или более атома углерода являются общими для двух соседних циклов, например циклы являются «конденсированными циклами». Каждый из циклов полицикла может быть замещенным или незамещенным.
Термин «предупреждение» известен в технике и, будучи использован к состоянию, такому как местный рецидив (например, боль), заболеванию, такому как рак, комплексу синдромов, такому как сердечная недостаточность, или к любому другому медицинскому состоянию, хорошо понятен в технике и включает в себя введение композиции, которая уменьшает частоту или отсрочивает наступление симптомов медицинского состояния у субъекта по отношению к субъекту, который не получил композиции. Таким образом, предупреждение рака включает в себя, например, уменьшение числа определяемых раковых опухолей в группе пациентов, получивших профилактическое лечение, по отношению к необработанной контрольной группе и/или отсрочивает появление определяемых раковых опухолей в обработанной группе по сравнению с необработанной контрольной группой, например, при статистически и/или клинически значимом количестве. Предупреждение инфекции включает в себя, например, уменьшение числа диагнозов инфекций в обработанной группе по сравнению с необработанной контрольной группой и/или отсрочивание наступления симптомов инфекции в обработанной группе по сравнению с необработанной контрольной группой. Предупреждение боли включает в себя, например, уменьшение силы или, альтернативно, отсрочка болевых приступов, испытываемых субъектами, в обработанной группе по сравнению с необработанной контрольной группой.
Термин «пролекарство» охватывает соединения, которые в физиологических условиях превращаются в терапевтически активные агенты. Общий способ изготовления пролекарства представляет собой введение выбранных групп, которые гидролизуются в физиологических условиях с высвобождением желаемой молекулы. В других вариантах осуществления пролекарство превращается действием фермента животного-хозяина.
Термин «профилактическое или терапевтическое» лечение известен в технике и включает в себя введение хозяину одной или более надлежащих композиций. Если ее водят до клинического проявления нежелательного состояния (например, заболевания или другого нежелательного состояния животного-хозяина), то лечение является профилактическим (то есть оно защищает хозяина от развития нежелательного состояния), тогда как если ее вводят после проявления нежелательного состояния, лечение является терапевтическим (то есть оно предполагает уменьшить, облегчить или стабилизировать наступающее нежелательное состояние или его побочные эффекты).
Термин «протеасома», как использовано здесь, имеет в виду включение иммунных и конститутивных протеасом.
Термин «замещенный» относится к частям (молекулы), содержащим заместители, заменяющие водород у одного или более атомов углерода основной цепи. Следует понимать, что «замещение» или «замещенный» включает в себя подразумеваемое условие, что указанное замещение идет в соответствии с разрешенной валентностью замещенного атома и заместителя и что результатом замещения является стабильное соединение, например, такое, которое не подвергается самопроизвольной трансформации, такой как перегруппировка, циклизация, элиминирование и т.д. Как использовано здесь, термин «замещенный» рассматривает включение всех допустимых заместителей органических соединений. В широком аспекте допустимые заместители включают в себя ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероцклические, ароматические и неароматические заместители органических соединений. Для соответствующего органического соединения возможны один или более и одинаковые или различные заместители. Для целей данного изобретения гетероатомы, такие как азот, могут иметь заместителями водород и/или любые описанные здесь допустимые заместители органических соединений, которые удовлетворяют валентностям гетероатомов. Заместителями могут быть, например, галоген, гидроксил, карбонил (такой как карбоксил, алкоксикарбонил, формил или ацил), тиокарбонил (такой как сложный тиоэфир, тиоацетат или тиоформиат), алкоксил, фосфорил, фосфат, фосфонат, фосфинат, аминогруппа, амидогруппа, амидиногруппа, иминогруппа, цианогруппа, нитрогруппа, азидогруппа, сульфгидрил, алкилтиогруппа, сульфат, сульфонат, сульфамоил, сульфонамидогруппа, сульфонил, гетероциклил, аралкил или ароматическая или гетероароматическая группа. Специалисту в данной области техники следует понимать, что группы, замещенные на углеводородной цепи, сами, если целесообразно, могут быть замещены.
«Терапевтически эффективное количество» соединения по отношению к данному способу лечения относится к количеству соединения(й) в препарате, если при его введении в качестве части желаемого режима дозирования (млекопитающему, предпочтительно человеку) облегчается симптом, облегчается состояние или замедляется наступление болезненных состояний в соответствии с клинически принятыми стандартами для расстройств или состояний, которые должны быть излечены, или (достигаются) косметические цели, например, при разумном отношении преимущества/риска, присущего любому медицинскому лечению.
Термин «простой тиоэфир» относится к алкильной группе, как определено выше, содержащей присоединенную к ней серу. В предпочтительных вариантах осуществления «простой тиоэфир» представляет собой -S-алкил. Представительные простые тиоэфирные группы включают в себя метилтиогруппу, этилтиогруппу и подобные.
Как использовано здесь, термин «лечение» или «терапия» включает в себя возвращение к прежнему состоянию, уменьшение или купирование симптомов, клинической картины и лежащей в основе патологии состояния таким образом, чтобы улучшить или стабилизировать состояние субъекта.
Селективность для протеасомы 20S
Предложенные здесь протеолитические ферменты применимы отчасти потому, что они ингибируют активность протеасомы 20S. Дополнительно, в отличие от других ингибиторов протеасомы 20S, предложенные здесь соединения высокоселективны по отношению к протеасоме 20S по сравнению с другими ферментами протеаз. То есть данные соединения проявляют селективность для протеасомы 20S, превышающую другие протеазы, такие как катепсины, калпаины, папаин, химотрипсин, трипсин, трипептидилпептидаза II. Селективность ингибиторов ферментов для протеасомы 20S такова, что в концентрации ниже примерно 50 мкМ ингибиторы фермента проявляют ингибирование каталитической активности протеасомы 20S, хотя не проявляют ингибирования каталитической активности других протеаз, таких как катепсины, калпаины, папаин, химотрипсин, трипсин, трипептидилпептидаза II. В предпочтительных вариантах осуществления ингибиторы ферментов проявляют ингибирование каталитической активности протеасомы 20S в концентрации ниже примерно 10 мкМ, хотя не обнаруживают ингибирования каталитической активности других протеаз при указанных концентрациях. В еще более предпочтительных вариантах осуществления ингибиторы ферментов показывают ингибирование каталитической активности протеасомы 20S в концентрации ниже примерно 1 мкМ, хотя не показывают ингибирования каталитической активности других протеаз при указанных концентрациях. Определение кинетики фермента предложено в патентном описании США, сериальный номер 09/569748, пример 2 и Stein и др., Biochem. (1996), 35, 3899-3908.
Селективность химотрипсин-подобной активности
Особые варианты осуществления описанных здесь соединений, ингибирующих фермент, дополнительно применимы вследствие их возможной эффективности и селективности в ингибировании химотрипсин-подобной активности протеасомы 20S по сравнению с трипсин-подобной и PGPH-активностями. Химотрипсин-подобная активность протеасомы 20S характеризуется расщеплением пептидов в непосредственной близости больших гидрофобных остатков. В частности, химотрипсин-подобная активность Ntn-гидролаз может быть определена путем расщепления стандартного субстрата. Примеры таких субстратов известны специалистам в данной области. Например, может быть использовано производное лейцилвалинилтирозина. Определение кинетики фермента предложено в патентном описании США, сериальный номер 09/569748, пример 2 и Stein и др., Biochem. (1996), 35, 3899-3908.
Использование ингибиторов ферментов
Биологические последствия ингибирования протеасомы многочисленны. Предполагают, что ингибирование протеасомы предупреждает или лечит множество заболеваний, включая, но не ограничивая, пролиферативные заболевания, нейротоксично/дегенеративные заболевания, болезнь Альцгеймера, ишемические состояния, воспаления, иммунно-зависимые заболевания, ВИЧ, рак, отторжение трансплантата органа, септический шок, ингибирование включения антигена, снижение экспрессии вирусного гена, паразитарные инфекции, состояния, связанные с ацидозом, дегенерация желтого пятна, легочные заболевания, мышечная дистрофия, фиброзные заболевания, костные заболевания и нарушения роста волос. Поэтому композиции ингибитора протеасомы, такие как перорально доступный класс молекул пептидо-эпокси-кетонов, как описано здесь, дает средства для лечения пациентов с указанными состояниями.
Композиции ингибитора протеасомы могут быть использованы для лечения состояний, непосредственно опосредованных протеолитической функцией протеасомы, такой как мышечная дистрофия, или опосредованных косвенно через белки, которые обработаны протеасомой, такие как NF-κB. Частицы протеасомы при быстром элиминировании и посттрансляционной обработке белков (например, ферментов) приводят к клеточному регулированию (например, клеточного цикла, генной транскрипции и метаболических путей), внутриклеточной коммуникации и иммунной реакции (например, присутствия антигена). Специфические примеры, обсужденные ниже, включают в себя β-амилоидный белок и регуляторные белки, такие как циклины, и фактор транскрипции NF-κB.
На клеточном уровне после обработки клеток различными ингибиторами протеасомы были описаны аккумулирование полиубиквитинированных белков, морфологические изменения клеток и апоптозы. Протеасомы расщепляют многие белки на созревающие ретикулоциты и растущие фибробласты. В клетках, лишенных инсулина или серозной жидкости, скорость протеолиза примерно удваивается. Ингибирование протеасомы уменьшает протеолиз, уменьшая при этом как потерю мышечного белка, так и нагрузку азота на почки или печень. Один из аспектов изобретения относится к лечению кахексии и мышечной дистрофии. Соединения изобретения могут быть использованы для лечения таких состояний, как рак, хронические инфекционные заболевания, лихорадка, омертвение мышц (атрофия) и денервация, повреждения нерва, ожирение, почечная недостаточность, связанная с ацидозом, и печеночная недостаточность. Смотри, например, Goldberg, патент США № 5340736. Поэтому некоторые варианты осуществления изобретения охватывают композиции для: уменьшения скорости деградации мышечного белка в клетке; уменьшения скорости деградации внутриклеточного белка; уменьшения скорости деградации белка р53 в клетке; ингибирования роста рака, родственного р52. Каждый из указанных способов предусматривает контактирование клетки (in vivo или in vitro, например, мышцы у субъекта) с эффективным количеством фармацевтической композиции, содержащей предложенный здесь ингибитор протеасомы.
Внутриклеточный протеолиз генерирует малые пептиды для предоставления в Т-лимфоциты для индуцирования иммунной реакции, опосредованной МСН класса I. Иммунная система экранирует аутогенные клетки, которые инфицированы вирусом или подверглись онкогенной трансформации. В некоторых вариантах осуществления изобретение относится к способу ингибирования включения антигена в клетку, предусматривающему воздействие на клетку описанного здесь соединения. Ингибиторы протеасомы изобретения могут быть использованы для лечения состояний, связанных с иммунитетом, таких как аллергия, астма, отторжение органа/ткани (реакция трансплантат против хозяина) и аутоиммунных заболеваний, включая, но не ограничиваясь, волчанку, ревматоидный артрит, псориаз, рассеянный склероз и воспалительные заболевания кишечника (такие как язвенный колит и болезнь Крона). Таким образом, в некоторых вариантах осуществления изобретение относится к способам подавления иммунной системы субъекта, предусматривающим введение субъекту эффективного количества описанного здесь соединения, ингибитора протеасомы.
В некоторых вариантах осуществления изобретение относится к способу изменения набора антигенных пептидов, продуцированных протеасомой или другим Ntn с мультикаталитической активностью. Например, если PGPH-активность протеасомы 20S селективно ингибирована, другая совокупность антигенных пептидов будет продуцирована протеасомой и представлена в молекулы МСН на поверхностях клеток, чем будет продуцирована и представлена либо в отсутствие любого ингибирования фермента, или, например, с селективным ингибированием химотрипсин-подобной активности протеасомы.
Другой аспект изобретения относится к использованию предложенных здесь композиций ингибитора протеасомы для лечения нейродегенеративных заболеваний и состояний, включая, но не ограничивая, инсульт, ишемические повреждения нервной системы, травмы нервов (например, перкуссивные церебральные нарушения, повреждения спинного мозга и травматические повреждения нервной системы), рассеянный склероз и другие иммунно-опосредованные невропатии (например, синдром Гийена-Барре и его варианты, острая моторная нейритная невропатия, острая воспалительная демиенилирующая полиневропатия и синдром Фишера), комплексная ВИЧ-СПИД-деменция, аксономия, диабетическая невропатия, болезнь Паркинсона, болезнь Хантингтона, рассеянный склероз, бактериальный, паразитарный, грибковый и вирусный менингиты, энцефалит, сосудистая деменция, смешанная инфарктная деменция, деменция тела Леви, деменция лобной доли, такая как болезнь Пика, подкорковая деменция (такая как Хантингтона или прогрессирующий надъядерный паралич), синдром наследственно кортикальной атрофии (такой как первичная афазия), метаболически-токсические деменции (такие как хронический гипотироидизм или дефицит В12) и деменции, вызванные инфекциями (такими как сифилис или хронические менингиты).
Болезнь Альцгеймера характеризуется внеклеточными отложениями β-амилоидного белка (β-АР) в сенильных бляшках и церебральных сосудах. Β-АР представляет собой пептидный фрагмент с 39 по 42 аминокислоты, производный от предшественника амилоидного белка (АРР). Известны по меньшей мере три изоформы АРР (695, 751 и 770 аминокислот). Альтернативное соединение мРНК генерирует изоформы; нормальный процесс влияет на часть последовательности β-АР, предупреждая при этом генерирование β-АР. Считают, что аномальное действие протеасомы на белок обеспечивает изобилие β-АР в головном мозге страдающего болезнью Альцгеймера. Фермент, дающий АРР, у крыс содержит около десяти различных субъединиц (22-32 кДа). Субъединица 25 кДа содержит N-концевую последовательность Х-Gln-Asn-Pro-Met-X-Thr-Gly-Thr-Ser, которая идентична β-субъединице чрезмерной боли (макроболи, macropain) человека (Kojima, S. И др., Fed. Eur. Biochem. Soc., (1992) 304:57-60). Фермент, производящий АРР, расщепляет связь Gln15-Lys16; в присутствии иона кальция фермент также расщепляет связь Met1-Asp1 и связи Asp1-Ala2, высвобождая внеклеточный домен β-АР.
Поэтому один из аспектов изобретения относится к способу лечения болезни Альцгеймера, предусматривающему введение субъекту эффективного количества предложенной здесь композиции ингибитора протеасомы. Указанное лечение включает в себя уменьшение скорости продуцирования β-АР, уменьшение скорости образования бляшек β-АР, уменьшение скорости генерирования β-АР и уменьшение клинических симптомов болезни Альцгеймера.
Фиброз представляет собой чрезмерное и устойчивое образование рубцовой ткани в результате гиперпролиферативного роста фибробластов и связанное с активацией TGF-β-сигнального пути. Фиброз вызывает избыточное отложение внеклеточной матрицы и может проявляться фактически в любой ткани или поперек нескольких различных тканей. Обычно уровень внутриклеточного сигнального белка (Smad), который активирует транскрипцию целевых генов после TGF-β-стимулирования, регулируется активностью протеасомы (Xu и др., 2000). Однако при раке и других гиперпролиферативных заболеваниях наблюдали ускоренную деградацию TGF-β-сигнальных компонентов. Таким образом, в некоторых вариантах осуществления изобретение относится к способу лечения гиперпролиферативных заболеваний, таких как диабетическая ретинопатия, дегенерация желтого пятна, диабетическая нефропатия, гломерулосклероз, IgA-нефропатия, цирроз, атрезия желчных протоков, застойная сердечная недостаточность, склеродермия, фиброз, индуцированный облучением, и фиброз легких (идиопатический пневмосклероз, коллагеновая болезнь сосудов, саркоидоз, интерстициальные легочные заболевания и экзогенные легочные заболевания). Лечению пострадавших от ожогов часто мешает фиброз, поэтому в некоторых вариантах осуществления изобретение относится к местному или системному введению ингибиторов для лечения ожогов. Заживление ран после хирургических вмешательств часто связано с обезображивающими рубцами, которые могут быть предупреждены ингибированием фиброза. Таким образом, в некоторых вариантах осуществления изобретение относится к способу предупреждения или уменьшения рубцевания.
Некоторые ингибиторы протеасомы блокируют как деградацию, так и обработку убиквитинированного NF-κB in vivo и in vitro. Ингибиторы протеасомы также блокируют деградацию IκB-A и активацию NF-κB (Palombella и др., Cell (1994) 78:773-785; и Traenckner и др., EMBO J. (1994) 13:5433-5441). Один из аспектов изобретения относится к способу ингибирования деградации IκB-α, предусматривающему контактирование клетки с описанным здесь соединением. В некоторых вариантах осуществления изобретение относится к способу уменьшения клеточного содержания NF-κB в клетке, мышце, органе или у субъекта, предусматривающему контактирование клетки, мышцы, органа или субъекта с описанным здесь соединением, ингибитором протеасомы.
NF-κB является членом семейства Rel-белков. Семейство Rel транскрипторных белков-активаторов может быть разделено на две группы. Первая группа требует протеолитической обработки и включает в себя р50 (NF-κB1, 105 kDa) и р52 (NF-κ2, 100 kDa). Вторая группа не требует протеолитической обработки и включает в себя р65 (RelA, Rel (c-Rel) и RelB). Членами семейства Rel могут быть образованы оба гомо- и гетеродимера; NG-κB, например, представляет собой гетеродимер р59-р65. После фосфорилирования и убиквитинирования IκB и р105 два белка деградируют и подвергаются обработке с образованием активного NF-κB, который перемещается от цитоплазмы к ядру. Убиквитинилированный р105 также подвергается действию очищенных протеасом (Palombella и др., Cell (1994) 78:773-785). Активный NF-κB образует стереоспецифический комплекс гена-усилителя с другими транскрипциональными активаторами и, например, HMGI(Y), индуцируя селективную экспрессию конкретного гена.
NF-κB регулирует гены, включенные в иммунную и воспалительную реакцию, и митотические явления. Например, NF-κB требуется для экспрессии κ-гена легкой цепи иммуноглобулина, гена α-цепи рецептора IL-2, комплексного гена главной биосовместимости класса I и ряда генов, кодирующих цитокины, например, IL-2, IL-6, фактора, стимулирующего колонию гранулоцитов, и IFN-β (Palombella и др., Cell (1994) 78:773-785). В некоторых вариантах осуществления изобретение относится к способам воздействия на уровни экспрессии IL-2, MHC-I, IL-6, TNFα, IFN-β или любых других ранее упомянутых белков, каждый способ предусматривает введение субъекту эффективного количества описанной здесь композиции ингибитора протеасомы. Комплексы, включающие в себя р50, представляют собой быстрые медиаторы острых воспалительных и иммунных реакций (Thanos, D и Maniatis, T., Cell (1995) 80:529-532).
Избыточное образование липополисахарида (LPS), индуцированное цитокинами, такими как TNFα, рассматривают центральными в процессах, связанных с септическим шоком. Более того, обычно полагают, что первая стадия в активации клеток LPS состоит в связывании LPS со специфическими мембранными рецепторами. Субъединицы α- и β комплекса протеасомы 20S идентифицированы как белки, связывающие LPS, доказывая, что сигнальная трансдукция, индуцированная LPS, может быть важной терапевтической мишенью при лечении или предупреждении сепсиса (Qureshi, N. и др., J. Immun. (2003) 171:1515-1525). Поэтому в некоторых вариантах осуществления композиции ингибитора протеасомы могут быть использованы для ингибирования TNFα c целью предупреждения и/или лечения септического шока.
NF-κB также участвует в экспресии генов клеточной адгезии, что кодирует Е-селектин, Р-селектин, ICAM и VCAM-1 (Collins, T., Lab.Invest. (1993) 68:499-508). В некоторых вариантах осуществления изобретение относится к способу ингибирования клеточной адгезии (например, клеточной адгезии, опосредованной Е-селектином, Р-селектином, ICAM или VCAM-1), предусматривающему контактирование клетки с (или введение субъекту) эффективного количества фармацевтической композиции, содержащей предложенный здесь ингибитор протеасомы.
NF-κB также специфически связывается с усилителем/промотором ВИЧ. По сравнению с Nef mac239 регуляторный белок ВИЧ Nef pbj14 отличается двумя аминокислотами в области, которая контролирует связывание протеинкиназы. Считают, что протеинкиназа дает импульс фосфорилированию IκB, вызывая деградацию IκB убиквитин-протеасомным путем. После деградации NF-κB высвобождается в ядро, усиливая транскрипцию ВИЧ (Cohen, J., Science (1995) 267:960). В некоторых вариантах осуществления изобретение относится к способу ингибирования или уменьшения ВИЧ-инфекции у субъекта или к способу снижения уровня экспрессии гена вируса, причем каждый способ предусматривает введение субъекту эффективного количества описанной здесь композиции ингибитора протеасомы.
Вирусные инфекции способствуют патологии многих заболеваний. Состояния сердца, такие как хронические миокардиты и дилатационная кардиомиопатия, связаны с кокссакиевирусом (coxsackievirus) В3. В сравнительных анализах геномной микрообласти сердца инфицированной мыши, которая имела хронический миокардит, были однородно отрегулированы субъединицы специфической протеасомы (Szalay и др., Am. J. Patol. 168;1542-52, 2006). Некоторые вирусы используют убиквитин-протеасомную систему на стадии внедрения вируса, когда вирус высвобождается из ядрышка в цитозоль. Вирус мышиного гепатита (MHV) относится к семейству Coronaviridae, которое также включает в себя некоторые коронавирусы острого респираторного синдрома (SARS). Yu и Lai (J. Virol. 79:644-648, 2005) показали, что лечение клеток, инфицированных MHV, ингибитором протеасомы, приводит к уменьшению репликации вируса, коррелируя с уменьшенным титром вируса по сравнению с необработанными клетками. Вирус гепатита В человека (HBV), член семейства вирусов Hepadnaviridae, подобным образом требует для размножения белки закодированной вирусом оболочки. Ингибирование пути деградации протеасомы вызывает заметное уменьшение количества секретированного белка оболочки (Simsek и др., J. Virol. 79:12914-12920, 2005). В добавление к HBV для секреции, морфогенеза и патогенеза пути убиквитин-протеасомной деградации могут также использовать другие вирусы гепатита (А, С, D и Е). Соответственно, в некоторых вариантах осуществления изобретение относится к способу лечения вирусной инфекции, такой как SARS или гепатиты А, В, С, D и Е, предусматривающему контактирование клеток с (или введение субъекту) эффективным количеством предложенного здесь соединения.
Ишемия и реперфузионное повреждение приводит к гипоксии, состоянию, при котором существует дефицит кислорода, достигающего тканей тела. Указанное состояние вызывает повышенная деградация Iκ-Bα, тем самым приводя к активации NF-κB (Koong и др., 1994). Было показано, что множество повреждений в результате гипоксии может быть уменьшено введением ингибитора протеасомы (Gao и др., 2000; Вао и др., 2001; Pye и др., 2003). Поэтому некоторые варианты осуществления изобретение относится к способу лечения ишемического состояния или реперфузионного повреждения, предусматривающему введение субъекту в случае необходимости такого лечения эффективного количества предложенного здесь соединения, ингибитора протеасомы. Примеры таких состояний или повреждений включают в себя, но не ограничивают, острый коронарный синдром (восприимчивые тромбоциты), окклюзионное поражение артерии (сердечной, церебральной, периферической артерии и окклюзия сосуда), атеросклероз (коронарный склероз, заболевание коронарной артерии), инфаркты, сердечная недостаточность, панкреатиты, гипертрофия миокарда, стенозы и рестенозы.
Другими факторами транскрипции эукариотов, которые требуют протеолитической обработки, являются генеральный фактор транскрипции TFIIA, дополнительный белок простого вируса герпеса VP16 (фактор клетка хозяина), фактор 2 регуляторного белка вируса, индуцирующего IFN, и регуляторный белок 1, связывающий элемент мембраносвязанного стерина.
В некоторых вариантах осуществления изобретение относится к способам возбуждения циклинзависимых циклов клеток эукариота, предусматривающим действие на клетки (in vitro или in vivo) предложенной здесь композиции ингибитора протеасомы. Циклины представляют собой белки, вовлеченные в регуляцию клеточного цикла. Протеасома участвует в деградации циклинов. Примерами циклинов являются митотические циклины, G1-циклины и циклин В. Деградация циклинов дает возможность клетке выходить из одной стадии клеточного цикла (например, митоза) и входить в другую (например, деление). Считают, что все циклины ассоциированы с р34cdc2 протеинкиназой или родственными киназами. Сигнал прицельного протеолиза направлен на аминокислоты 42-RAALGNISEN-50 (бокс деструкции). Очевидно, что циклин превращается в форму, незащищенную от убиквитинлигазы, или что во время митоза активируется циклин-специфическая лигаза (Ciechanover, A., Cell, (1994) 79:13-21). Ингибирование протеасомы ингибирует деградацию циклина и, следовательно, ингибирует пролиферацию клеток, например, при злокачественных новообразованиях, связанных с циклином (Kumatori и др., Proc. Natl. Acad. Sci. USA (1990) 87:7071-7075). Один из аспектов изобретения относится к способу лечения у субъекта пролиферативного заболевания (например, рака, псориаза или рестеноза), предусматривающему введение субъекту эффективного количества предложенной здесь композиции ингибитора протеасомы. Изобретение также относится к способу лечения у субъекта связанных с циклином воспалений, предусматривающему введение субъекту эффективного количества предложенной здесь композиции ингибитора протеасомы.
Дополнительные варианты осуществления изобретения относятся к способам возбуждения зависимой от протеасомы регуляции онкопротеинов и способам лечения или ингибирования роста злокачественных новообразований; каждый способ предусматривает воздействие на клетку (in vivo, например, у субъекта или in vitro) предложенной здесь композиции ингибитора протеасомы. Белки HPV-16 и HPV-18, производные Е6, стимулируют АТР- и убиквитин-зависимое конъюгирование и деградацию р53 в неочищенных лизатах ретикулоцитов. Показано, что рецессивный онкогенный р53 при недопустимой температуре накапливается в клеточной линии с мутированным термолабильным Е1. Повышенные уровни р53 ведут к апоптозу. Примеры прото-онкопротеинов, деградированных системой убиквитина, включают в себя c-Mos, c-Fos и c-Jun. В некоторых вариантах осуществления изобретение относится к способам лечения апоптозов, связанных с р-53, предусматривающим введение субъекту эффективного количества предложенной здесь композиции ингибитора протеасомы.
В некоторых вариантах осуществления предложенные композиции могут быть использованы для лечения паразитарных инфекций, таких как инфекции, вызванные протозойными паразитами. Считают, что протеасома указанных паразитов вовлечена, в основном, в активность дифференциации и репликации клеток (Paugam и др., Trends Parasitol. 2003, 19(2):55-59). Более того, показано, что род дизентерийной амебы теряет способность инцистирования, будучи обработан ингибиторами протеасомы (Gonzales и др., Arch. Med. Res. 1997, 28, Spec №: 139-140). В некоторых указанных вариантах осуществления протоколы введения композиций ингибитора протеосомы применимы для лечения у человека паразитарных инфекций, вызванных протозойными паразитами, выбранными из рода Plasmodium (включая P. falciparum, P. vivax, P. malariae и P. ovale, которые вызывают малярию), рода Trypanosoma (включая T. сruzi, которая вызывает болезнь Шагаса, и T. brucei, которая вызывает африканский трипаносомоз), рода Leishmania (включая L. amazonesis, L. donovani, L. infantum, L. mexicana и т.д.), Pneumocystis carini (простейшее, как известно, вызывающее пневмонию у (больных) СПИДом и других субъектов с подавленным иммунитетом), Toxoplasma gondii, Entamoeba histolytica, Entamoeba invanens и Giardia lamblia. В некоторых вариантах осуществления предложенные композиции ингибитора протеасомы применимы для лечения паразитарных инфекций у животных и скота, вызванных протозойными паразитами, выбранными из Plasmodium hermani, Cryptosporidium sps., Echinococcus granulosus, Eimeria tenella, Sarcocystis neurona и Neurpspora crassa. Другие соединения, применимые в качестве ингибиторов протеасомы при лечении паразитарных заболеваний, описаны в патенте WO 98/10779, который приведен здесь в качестве ссылки.
В некоторых вариантах осуществления композиции ингибитора протеасомы ингибируют активность протеасомы у паразитов без регенерации в эритроциты и лейкоциты. В некоторых таких вариантах осуществления длительный полупериод существования гемоцитов может обеспечивать пролонгированное предохранение против рецидивирующего действия паразитов. В некоторых вариантах осуществления композиции ингибитора протеасомы могут обеспечивать пролонгированную химиопрофилактику против будущих инфекций.
Показано также, что ингибиторы, которые связывают протеасому 20S, стимулируют образование кости в культурах костных органов. Более того, если указанные ингибиторы систематически вводят мышам, некоторые ингибиторы протеасомы увеличивают объем костей и скорость образования костей на более чем 70% (Garrett, I. R. и др., J. Clin. Invest. (2003) 111:1771-1782), доказывая этим, что убиквитин-протеасомный механизм регулирует дифференциацию остеобласта и образование кости. Поэтому предложенные композиции ингибитора протеасомы могут быть применимы при лечении и/или предупреждении заболеваний, связанных с утратой кости, таких как остеопороз.
Ингибирование протеасомы уже обосновано как терапевтическая стратегия для лечения рака, в частности множественной миеломы. Однако на основе обеих моделей, in vitro и in vivo, можно предсказать, что оно может служить в качестве стратегии против других (видов) рака, в частности злокачественностей, связанных с гемом, и солидных опухолей. Поэтому некоторые варианты осуществления изобретения относятся к способу лечения раковых заболеваний, предусматривающему введение субъекту, при необходимости такого лечения, эффективного количества предложенного здесь соединения, ингибитора протеасомы.
Введение
Композиции, полученные, как описано здесь, могут быть введены в различных формах, в зависимости от расстройства, которое должно быть вылечено, и возраста, состояния и массы тела пациента, что хорошо известно в медицине. Например, если соединения вводят перорально, они могут быть изготовлены в виде таблеток, капсул, гранул, порошков или сиропов; или для парентерального введения они могут быть изготовлены в виде раствора для инъекций (внутривенных, внутримышечных или подкожных), препарата для капельного вливания или суппозиториев. Для введения через глазную слизистую мембрану они могут быть изготовлены в виде глазных капель или глазных мазей. Указанные готовые препаративные формы могут быть получены обычными способами, и при желании активный ингредиент может быть смешан с любой обычной добавкой или наполнителем, таким как связующее, дезинтегрирующим агентом, скользящим веществом, корректирующим агентом, солюбилизатором, суспендирующим вспомогательным средством, эмульгатором, покрывающим агентом, циклодекстрином и/или буферной смесью. Хотя дозировка может варьироваться в зависимости от симптомов, возраста и массы тела пациента, природы и тяжести расстройства, которое должно быть вылечено или предупреждено, пути введения и формы лекарственного препарата, обычно для взрослого человека рекомендуют суточную дозу от 0,01 до 2000 мг, и она может быть введена в виде разовой дозы или в виде раздельных доз. Количество активного ингредиента, который может быть скомбинирован с веществом носителя для получения разовой дозированной формы, обычно равно количеству соединения, которое дает терапевтический эффект.
Точное время введения и/или количество композиции, которые дадут наиболее эффективные результаты в смысле эффективности лечения у данного пациента, зависят от активности, фармакокинетики и биодоступности конкретного соединения, физиологического состояния пациента (включая возраст, пол, тип и стадию заболевания, общее физическое состояние, восприимчивость к данной дозе и типа лекарственной терапии), пути введения и т.д. Однако приведенные указания могут быть использованы как основные для тонкой корректировки лечения, например определения оптимального времени и/или количества введения, которая требует не более чем рутинного экспериментирования, состоящего в мониторинге субъекта и регулировании доз и/или времени.
Фраза «фармацевтически приемлемый» использована здесь для обозначения таких лигандов, веществ, композиций и/или дозированных форм, которые, в рамках обоснованной медицинской оценки, подходят для использования в контакте с тканями человека и животных без избыточной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмеримых с разумным отношением польза/риск.
Фраза «фармацевтически приемлемый носитель», как использовано здесь, означает фармацевтически приемлемое вещество, композицию или наполнитель, такой как жидкий или твердый наполнитель, разбавитель, наполнитель, растворитель или инкапсулирующее вещество. Каждый носитель должен быть «приемлемым» в смысле совместимости с другими ингредиентами готовой препаративной формы и не вредить пациенту. Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают в себя; (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как маисовый крахмал, картофельный крахмал и замещенный или незамещенный β-циклодекстрин; (3) целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) наполнители, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) апирогенная вода; (17) изотонический раствор; (18) раствор Рингера (Ringer's); (19) этиловый спирт; (20) фосфатный буферный раствор; и (21) другие нетоксичные совместимые вещества, используемые в фармацевтических готовых препаративных формах. В некоторых вариантах осуществления фармацевтические композиции данного изобретения апирогенны, то есть не вызывают заметного повышения температуры при введении пациенту.
Термин «фармацевтически приемлемая соль» относится к относительно нетоксичным аддитивным солям ингибитора(ов) с неорганической и органической кислотой. Указанные соли могут быть получены in situ во время конечного выделения и очистки ингибитора(ов) или отдельным взаимодействием очищенного(ых) ингибитора(ов) в форме его свободного основания с подходящей органической или неорганической кислотой и выделением образовавшейся при этом соли. Представительные соли включают в себя гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, нафтилат, мезилат, глюкогептонат, лактобионат, соли лаурилсульфоната и соли аминокислот и подобные. (Смотри, например, Berge и др., (1977) “Pharmaceutical Salts”, J. Pharm. Sci, 66: 1-19.)
В других случаях ингибиторы, применимые в способах данного изобретения, могут содержать одну или более кислотных функциональных групп и, таким образом, способны к образованию фармацевтически приемлемых солей с фармацевтически приемлемыми основаниями. Термин «фармацевтически приемлемые соли» в указанных случаях относится к относительно нетоксичным аддитивным солям ингибитора(ов) с неорганическим или органическим основанием. Указанные соли также могут быть получены in situ во время конечного выделения и очистки ингибитора(ов) или отдельным взаимодействием очищенного(ых) ингибитора(ов) в форме его свободной кислоты с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным или третичным амином. Представительные щелочные или щелочноземельные соли включают в себя соли лития, натрия, калия, кальция, магния и алюминия и подобные. Представительные органические амины, применимые для образования аддитивных солей с основаниями, включают в себя этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и подобные (смотри, например, Berge et al., supra).
В композициях могут также присутствовать смачивающие агенты, эмульгаторы и скользящие вещества, такие как лаурилсульфат натрия и стеарат магния, также как и красители, антиадгезивы, покрывные средства, подслащивающие вещества, отдушки и ароматизирующие вещества, консерванты и антиоксиданты.
Примеры фармацевтически приемлемых антиоксидантов включают в себя: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и подобные; (2) маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (ВНА), бутилированный гидрокситолуол (ВНТ), лецитин, пропилгаллат, альфа-токоферол и подобные; и (3) агенты, образующие хелаты с металлами, такие как лимонная кислота, этилендиаминтетрауксусная кислота (EDTA), сорбит, винная кислота, фосфорная кислота и подобные.
Готовые препаративные формы, подходящие для перорального применения, могут быть в форме капсул, крахмальных облаток, пилюль, таблеток, лепешек (с использованием ароматизированных основ, обычно сахарозы и камеди или трагаканта), порошков, гранул, или в виде раствора или суспензии в водной или неводной жидкости, или в виде эмульсии масло-в-воде или вода-в-масле, или в виде эликсира или сиропа, или пастилок (с использованием инертной матрицы, такой как желатин и глицерин или сахароза и камедь), и/или в виде жидкости для полоскания рта и подобные, каждая из которых содержит предварительно определенное количество ингибитора(ов) в качестве активного ингредиента. Композиция может быть введена в виде болюсов, электуарий или пасты.
В твердых дозированных формах для перорального введения (капсулах, таблетках, пилюлях, драже, порошках, гранулах и подобных) активный ингредиент смешан с одним или более фармацевтически приемлемыми носителями, такими как цитрат натрия или дикальцийфосфат и/или одним из следующих: (1) наполнители или сухие разбавители, такие как крахмалы, циклодекстрины, лактоза, сахароза, глюкоза, маннит и/или кремневая кислота; (2) связующие, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или камедь; (3) увлажнители, такие как глицерин; (4) дизинтегрирующие агенты, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал из кассавы, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) агенты, замедляющие растворение, такие как парафин; (6) ускорители абсорбции, такие как четвертичные соединения аммония; (7) увлажнители, такие как, например, ацетилированный спирт (acetyl alcohol) и моностеарат глицерина; (8) абсорбенты, такие как каолин и бентонитовая глина; (9) скользящие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси; и (10) красители. В случае капсул, таблеток и пилюль фармацевтические композиции могут также содержать буферные агенты. Твердые композиции подобного типа могут также быть произведены как наполнители мягких и твердых желатиновых капсул с использованием таких наполнителей, как лактоза или молочный сахар, также как и высокомолекулярные полиэтиленгликоли и подобное.
Таблетки могут быть изготовлены сжатием или прессованием, возможно с одним или более вспомогательными ингредиентами. Прессованные таблетки могут быть получены с использованием связующего (например, желатина или гидроксипропилметилцеллюлозы), скользящего вещества, инертного разбавителя, консерванта, дезинтегрирующего вещества (например, крахмал-гликолята натрия или поперечно сшитой натрийкарбоксиметилцеллюлозы), поверхностно-активного или диспергирующего агента. Прессованные таблетки могут быть получены прессованием в подходящей машине смеси порошкообразного(ых) ингибитора(ов), увлажненного(ных) инертным жидким разбавителем.
Таблетки и другие твердые дозированные формы, такие как драже, капсулы, пилюли и гранулы, могут быть с насечками или получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в технике изготовления фармацевтических готовых препаративных форм. Они также могут быть изготовлены таким образом, чтобы обеспечить замедленное или контролируемое высвобождение активного ингредиента, с использованием, например, гидроксипропилметилцеллюлозы в различных пропорциях для обеспечения желаемого показателя высвобождения, других полимерных матриц, липосом и/или микросфер. Они могут быть стерилизованы, например, фильтрованием через фильтр, удерживающий бактерии, или введением стерилизующих агентов в стерильные твердые композиции, которые могут быть растворены в стерильной воде или какой-либо другой стерильной среде для инъекций непосредственно перед употреблением. Указанные композиции могут также содержать замутняющие агенты и могут представлять собой композицию, которая высвобождает только активный(е) ингредиент(ы), или предпочтительно в некоторой части желудочно-кишечного тракта, возможно, с задержкой. Примеры покрывных композиций, которые могут быть использованы, включают в себя полимерные вещества и воски. Активный ингредиент, если целесообразно, может также быть в микроинкапсулированной форме с одним или более вышеописанными наполнителями.
Жидкие дозированные формы для перорального введения включают в себя фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В добавление к активному ингредиенту жидкие дозированные формы могут содержать инертные разбавители, широко применяемые в технике, такие как, например, вода или другие растворители, солюбилизаторы и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и эфиры сорбита с жирными кислотами и их смеси.
Кроме инертных разбавителей пероральные композиции могут также содержать адъюванты, такие как увлажнители, эмульгаторы и суспендирующие агенты, подслащивающие вещества, ароматизаторы, красители, отдушки и консерванты.
Суспензии, в добавление к активному(ым) ингредиенту(ам), могут содержать суспендирующие агенты, например этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбита, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант и их смеси.
Готовые препаративные формы для ректального или вагинального введения могут быть представлены в виде суппозиториев, которые могут быть получены смешением одного или более ингибитора(ов) с одним или более подходящими нераздражающими наполнителями или носителями, представляющими собой, например, масло какао, полиэтиленгликоль, воск для суппозиториев или салицилат, которые твердые при комнатной температуре, но жидкие при температуре тела и поэтому плавятся в ректальной или вагинальной полости и высвобождают активный агент.
Готовые препаративные формы, которые пригодны для вагинального введения, в случае целесообразности также включают в себя маточные кольца, тампоны, гели, пасты, пены или аэрозольные препараты, содержащие указанные носители, как известно в технике.
Дозированные формы для местного и чрескожного введения ингибитора(ов) включают в себя порошки, аэрозоли, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и формы для ингаляций. Активный компонент может быть смешан в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферными растворами или газами-вытеснителями, которые могут потребоваться.
Мази, пасты, кремы и гели могут содержать дополнительно к ингибитору(ам) наполнители, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремневую кислоту, тальк и оксид цинка или их смеси.
Порошки и аэрозольные препараты могут содержать дополнительно к ингибитору(ам) наполнители, такие как лактоза, тальк, кремневая кислота, гидроксид алюминия, силикаты кальция и порошки полиамидов или смеси указанных веществ. Аэрозольные препараты могут дополнительно содержать обычный газ-вытеснитель, такой как хлорфторуглеводороды, и летучие незамещенные углеводороды, такие как бутан и пропан.
В качестве альтернативы ингибитор(ы) может(гут) быть введены в виде аэрозоля. Это выполняют приготовлением водного аэрозоля, липосомального препарата или твердых частиц, содержащих композицию. Может быть использована неводная суспензия (например, фторуглеродный газ-вытеснитель). Предпочтительны ультразвуковые ингаляторы, поскольку они минимизируют действие агента на сдвиг (shear), что может привести к разложению соединения.
Обычно водный аэрозоль изготавливают составлением водного раствора или суспензии агента вместе с обычными фармацевтически приемлемыми носителями и стабилизаторами. Носители и стабилизаторы варьируются (в соответствии) с требованиями конкретной композиции, но обычно включают в себя твин (Tweens), плюроник (Pluronics), сложные эфиры сорбита, лецитин, кремофор (Cremophors), фармацевтически приемлемые сорастворители, такие как полиэтиленгликоль, безвредные белки, такие как сывороточный альбумин, олеиновую кислоту, аминокислоты, такие как глицин, буферные растворы, соли, сахара или сахарные спирты. Аэрозоли обычно получают из изотонических растворов.
Трансдермальные пластыри имеют дополнительное преимущество в обеспечении контролируемой доставки ингибитора(ов) в тело. Указанные дозированные формы могут быть изготовлены растворением или диспергированием агента в подходящей среде. Для повышения проницания ингибитора(ов) через кожу могут быть также использованы усилители абсорбции. Скорость указанного проницания может контролироваться либо введением мембраны, контролирующей скорость, либо диспергированием ингибитора(ов) в полимерной матрице или геле.
Фармацевтические композиции данного изобретения, пригодные для парентерального введения, состоят из одного или более ингибитора(ов) в сочетании с одним или более фармацевтически приемлемыми стерильными водными или неводными растворами, дисперсиями, суспензиями или эмульсиями или стерильными порошками, которые могут быть превращены в стерильные впрыскиваемые растворы или дисперсии непосредственно перед использованием, которые могут содержать антиоксиданты, буферные агенты, бактериостатические факторы, растворенные вещества, которые способствуют созданию изотоничности с кровью предполагаемого реципиента, или суспендирующие агенты или загустители.
Примеры подходящих водных и неводных носителей, которые могут быть использованы в фармацевтических композициях изобретения, включают в себя воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и подобные) и их подходящие смеси, растительные масла, такие как оливковое масло, и впрыскиваемые органические сложные эфиры, такие как этилолеат. Надлежащая текучесть может поддерживаться, например, использованием покрывных веществ, таких как лецитин, требуемым размером частиц в случае дисперсий и путем использования поверхностно-активных веществ.
Указанные композиции могут также содержать адъюванты, такие как консерванты, увлажнители, эмульгаторы и диспергаторы. Предупреждение действия микроорганизмов может быть обеспечено включением различных противобактериальных и противогрибковых агентов, например парабена, хлорбутанола, фенолсорбиновой кислоты и подобных. Желательно также вводить в композиции агенты, регулирующие тонус, такие как сахара, хлорид натрия и подобные. Дополнительно путем включения агентов, которые задерживают абсорбцию, таких как моностеарат алюминия и желатин, впрыскиваемой фармацевтической форме может быть придано (свойство) пролонгированной абсорбции.
В некоторых случаях для продления действия лекарства желательно замедлить абсорбцию лекарственного препарата из подкожных и внутримышечных инъекций. Например, замедленная абсорбция парентерально введенных лекарственных форм достигается растворением или суспендированием лекарственного средства в масляном наполнителе.
Впрыскиваемые депо-формы изготавливают созданием микроинкапсулированных матриц ингибитора(ов) в биоразлагаемом полимере, таком как полилактид-полигликолид. Скорость высвобождения может регулироваться в зависимости от соотношения лекарственного средства и полимера и природы конкурентных использованных полимерных частиц. Примеры других биоразлагаемых полимеров включают в себя поли(ортоэфиры) и поли(ангидриды). Впрыскиваемые депо-композиции также могут быть получены внедрением лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканью тела.
Препараты агентов могут быть даны перорально, парентерально, местно или ректально. Конечно, они могут быть даны в формах, подходящих для каждого пути введения. Например, их вводят в форме таблеток или капсул, путем инъекций, ингаляций, глазных лосьонов, мазей, суппозиториев, инфузий; местно путем лосьонов и мазей; и ректально путем суппозиториев. Пероральное введение предпочтительно.
Фразы «парентеральное введение» и «введено парентерально», как использовано здесь, означает способы введения, отличающиеся от энтерального и местного введения, обычно путем инъекции, и включают в себя, не ограничивая, внутривенные, внутримышечные, внутриартериальные, внутриоболочечные, внутрикапсульные, внутриглазничные, внутрисердечные, внутридермальные, внутрибрюшинные, транстрахеальные, подкожные, субэпидермальные, внутрисуставные, субкапсульные, субарахноидальные, интраспинальные и интрагрудинные инъекции и инфузии.
Фразы «системное введение», «введено системно», «периферическое введение» и «введено периферически», как использовано здесь, означает введение лиганда, лекарственного препарата или другого вещества иначе, чем непосредственно в центральную нервную систему, так что оно входит в систему пациента и, таким образом, подвергается метаболизму и другим подобным процессам, например подкожное введение.
Указанный(ые) ингибитор(ы) могут быть введены человеку или животным для терапии любым подходящим путем введения, в том числе перорально, назально, как, например, при помощи аэрозольного препарата, ректально, интравагинально, парентерально, внутриполостно и местно в виде порошков, мазей или капель, в том числе защечно и подъязычно.
Независимо от выбранного способа введения ингибитор(ы), который(е) может(гут) быть использован(ы) в подходящей гидратированной форме и/или (в виде) фармацевтических композиций данного изобретения, сформирован(ы) в фармацевтически приемлемые дозированные формы обычными способами, известными специалистам в данной области техники.
Фактические уровни доз активных ингредиентов в фармацевтических композициях данного изобретения могут варьироваться таким образом, чтобы получить количество активного ингредиента, которое эффективно для достижения желаемого терапевтического ответа у конкретного пациента, композиции и способа введения без токсичности для пациента.
Концентрация предложенного соединения в фармацевтически приемлемой смеси будет варьироваться в зависимости от нескольких факторов, включая дозу соединения, которая должна быть введена, фармакокинетические характеристики использованного(ых) соединения(й) и способ введения. Обычно композиции данного изобретения могут быть представлены в виде водного раствора для парентерального введения, содержащего примерно 0,1-10% мас./об. предложенного здесь соединения наряду с другими веществами. Типичные дозы находятся в интервалах от примерно 0,01 до примерно 50 мг/кг массы тела в день, разделенные ни 1-4 дозы. Каждая раздельная доза может содержать одни и те же или различные соединения изобретения. Дозировка должна быть эффективным количеством в зависимости от нескольких факторов, включая общее состояние здоровья пациента, готовую препаративную форму и способ введения выбранного(ых) соединения(й).
Другой аспект изобретения обеспечивает комплексную терапию, если вводят один или более терапевтических средств с ингибитором протеасомы. Такая комплексная терапия может быть достигнута путем одновременного, последовательного или раздельного дозирования отдельных компонентов терапии.
В некоторых вариантах осуществления соединение изобретения вводят совместно с одним или более другим(и) ингибитором(ами) протеасомы.
В некоторых вариантах изобретения соединение изобретения вводят совместно с химиотерапевтическим срендством. Подходящие химиотерапевтические срендства могут включать в себя природные продукты, такие как алкалоиды барвинка (то есть винбластин, винкристин и винорелбин), паклитаксел, эпидиподофиллотоксины (то есть этопозид, тенипозид), антибиотики (дактиномицин (актиномицин D), даунорубицин, доксорубицин и идарубицин), антрациклины, митоксантроны, блеомицины, пликамицин (митрамицин) и митомицин, ферменты (L-аспарагиназа, которая системно метаболизирует аспарагин и депривирует клетки, которые не обладают способностью синтезировать их собственный аспарагин), антитромбоцитные агенты; антипролиферативные/антимитотические алкилирующие агенты, такие как азотистые иприты (мехлоретамин, циклофосфамид и аналоги, мелфалан, хлорамбуцил), этиленимины и метилмеламины (гексаметилмеламин и тиотепа), алкилсульфонаты (бусульфан), нитрозомочевины (кармустин (BCNU) и аналоги, стрептозоцин), тразены-дакарбазины (DTIC); антипролиферативные/антимитотические антиметаболиты, такие как фолиевая кислота и аналоги (метотрексат), аналоги пиримидина (фторурацил, флоксуридин и цитарабин), аналоги пурина и родственные ингибиторы (меркаптопурин, тиогуанин, пентостатин и 2-хлордезоксиаденозин; ингибиторы ароматазы (анастрозол, эксеместан и летрозол); и координационные комплексы платины (цисплатина, карбоплатина), прокарбазин, гидроксимочевина, митотан, аминоглутетимид; ингибиторы гистондеацетилазы (HDAC); гормоны (то есть эстроген) и агонисты гормонов, такие как агонисты гормона, высвобождающего лютеинизирующий гормон (LHRH) (госерилин, лейпролид и трипторелин). Другими химиотерапевтическими агентами могут быть мехлорметамин, камптотецин, ифосфамид, тамоксифен, ралоксифен, гемцитабин, навелбин или их любые варианты аналогов и производных.
В некоторых вариантах осуществления соединение изобретения вводят совместно с цитокином. Цитокины включают в себя, но не ограничивают, интерфероны-γ, -α и -β, интерлейкины 1-8, 10 и 12, фактор, стимулирующий колонии гранулоцитов моноцитов (GM-CSF), TNF-α и -β и TGF-β.
В некоторых вариантах осуществления соединение изобретения вводят совместно со стероидом. Подходящие стероиды могут включать в себя, но не ограничивают, 21-ацетоксипрегненолон, алклометазон, альгестон, амцинонид, беклометазон, бетаметазон, будезонид, хлорпреднизон, клобетазол, клокортолон, клопреднол, кортикостерон, кортизон, кортивазол, дефлазакорт, дезонид, дезоксиметазон, дексаметазон, дифлоразон, дифлукортолон, дифупреднат, еноксолон, флуазакорт, флуклоронид, флуметазон, флунизолид, ацетонид флуоцинолона, флуоцинонид, бутилфлуокортин, флуокортолон, флуометолон, ацетат флуперолона, ацетат флупреднидена, флупреднизолон, флурандренолид, пропионат флутиказона, формокорталь, хальцинонид, пропионат галобетазола, галометазон, гидрокортизон, этабонат лотепреднола, мазипредон, медризон, мепреднизон, метилпреднизолон, фуроат мометазона, параметазон, предникарбат, преднизолон, 25-диэтиламиноацетат преднизолона, преднизолон-натрийфосфат, преднизон, преднивал, преднилиден, римексолон, тиксокортол, триамцинолон, ацетонид триамцинолона, бенетонид триамцинолона, гексацетонид триамцинолона и их соли и/или производные.
В некоторых вариантах осуществления соединение изобретения вводят совместно с иммунотерапевтическим агентом. Подходящие иммунотерапевтические агенты могут включать в себя, но не ограничивать, модуляторы MDR (верапамил, вальспордар, бирикодар, тариквидар, ланиквидар), рапамицин, микофенилат-мофетил, циклофозомид, циклоспорин, талидомид и моноклональные антитела. Моноклональные антитела могут быть либо свободными, либо конъюгированными, такими как ритуксимаб, тозитумомаб, алемтузумаб, даклизумаб, епратузумаб, ибритумомаб-тиуксетан, гемтузумаб-озогамицин, беквацизумаб, цетуксимаб, эрлотиниб и трастузумаб.
ПРИМЕРЫ
Пример 1
Схема 1: синтез соединения 010
Соединение (003):
К охлажденному до 0°С раствору метилового эфира N-Вос-серина (001) (2,5 г, 11,4 ммоль), гидрохлорида бензилового эфира L-аланина (002) (3,3 г, 11,4 ммоль), НОВТ (2,5 г, 18,2 ммоль) и HBTU (6,9 г, 18,24 ммоль) в тетрагидрофуране (400 мл) за 10 минут прибавляли раствор N,N-диизопропилэтиламина (8,0 мл, 45,6 ммоль) в тетрагидрофуране (50 мл). Следующие 5 часов смесь перемешивали при комнатной температуре. Большую часть растворителя удаляли при пониженном давлении и полученный остаток разбавляли этилацетатом (300 мл). Затем раствор промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (100 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), желаемое соединение (003) (4,4 г) выделяли и охарактеризовывали LC/MS (LCRS (MH) m/z: 457,23).
Соединение (004):
К охлажденному до 0°С раствору (003) (5,14 г, 11,25 ммоль) в тетрагидрофуране (100 мл) прибавляли 10%-ный Pd/C (500 мг). Полученную смесь перемешивали под давлением водорода в 1 атмосферу в течение 4 часов. Смесь фильтровали через целит-545 и лепешку на фильтре промывали тетрагидрофураном. Органический фильтрат концентрировали при пониженном давлении и выдерживали при высоком вакууме в течение 2 часов, получая (004), как было подтверждено LC/MS (LCRS (MH) m/z: 367,18), который использовали без дополнительной очистки.
Соединение (006):
К раствору (005) (синтез (005) смотри патентную заявку США, серийный № 11/131688) (3,9 г, 13 ммоль) в трифторуксусной кислоте (50 мл) прибавляли 10%-ный Pd/C (600 мг). Полученную смесь перемешивали под давлением водорода в 1 атмосферу в течение 6 часов. Смесь фильтровали через целит-545 и лепешку на фильтре промывали дихлорметаном (200 мл). Фильтрат концентрировали при пониженном давлении и выдерживали при высоком вакууме в течение ночи, получая (006), как было подтверждено LC/MS (LCRS (MH) m/z: 172,13), который использовали без дополнительной очистки.
Соединение (007):
К охлажденному до 0°С раствору (004) и (006), HOBT (2,5 г, 18 ммоль) и HBTU (6,9 г, 18 ммоль) в тетрагидрофуране (400 мл) за 10 минут прибавляли раствор N,N-диэтилизопропиламина (8 мл, 46 ммоль) в тетрагидрофуране (50 мл). Следующие 5 часов смесь перемешивали при комнатной температуре. Большую часть растворителей удаляли при пониженном давлении и остаток разбавляли этилацетатом (400 мл). Полученный раствор промывали насыщенным водным (раствором) бикарбоната натрия (2×100 мл) и соляным раствором (100 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (007) (3,5 г), как было показано LC/MS (LCRS(MH) m/z: 520,29).
Соединение (008):
К охлажденному до 0°С раствору (007) (320 мг, 0,616 ммоль) в дихлорметане (10 мл) прибавляли трифторуксусную кислоту (10 мл) и полученный раствор перемешивали при той же температуре в течение следующего часа. Органические слои концентрировали при пониженном давлении и затем выдерживали при высоком вакууме в течение 2 часов, получая (008), что было подтверждено LC/MS (LCRS(MH) m/z: 420,24), который использовали без дополнительной очистки.
Соединение (010):
К охлажденному до 0°С раствору (008), 5-метилизоксазол-3-карбоновой кислоты (009) (94 мг, 0,74 ммоль), НОВТ (135 мг, 1,0 ммоль) и HBTU (350 мг, 1,0 ммоль) в тетрагидрофуране (100 мл) за 5 минут прибавляли раствор N,N-диизопропилэтиламина (0,5 мл, 2,5 ммоль) в тетрагидрофуране (2 мл). Следующие 5 часов смесь перемешивали при комнатной температуре и затем разбавляли этилацетатом (200 мл). Далее промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл) и органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (010) (195 мг), как показано LC/MS (LCRS (MH) m/z: 529,26); ингибирование >90% протеасомы СТ-L при 40 мг/кг РО.
Пример 2
Схема 2; синтез примера 023
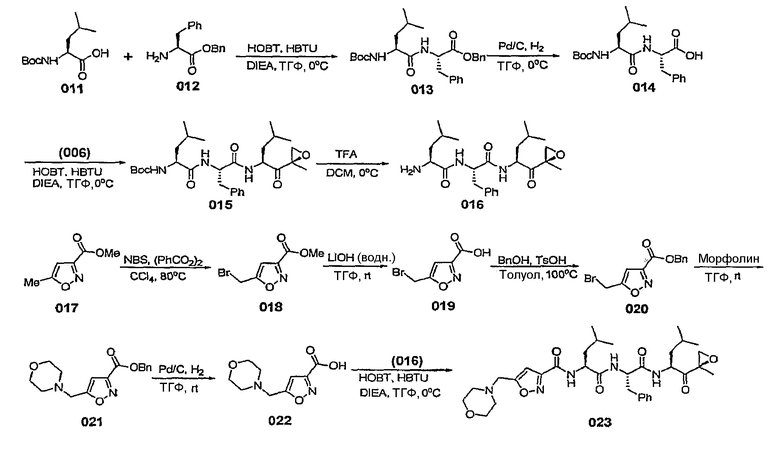
Соединение (013):
К охлажденному до 0°С раствору N-Boc-L-лейцина (011) (2,6 г, 11 ммоль), гидрохлорида бензилового эфира L-фенилаланина (012) (2,9 г, 10 ммоль), НОВТ (1,7 г, 11 ммоль) и HBTU (3,9 г, 11 ммоль) в тетрагидрофуране (200 мл) за 5 минут прибавляли раствор N,N-диизопропилэтиламина (4,9 мл, 30 ммоль) в тетрагидрофуране (10 мл). Следующие 5 часов смесь перемешивали при комнатной температуре, после чего она стала гомогенной. Затем ее разбавляли этилацетатом (300 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (100 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (013) (4,4 г), как показано LC/MS (LCRS (MH) m/z: 469,26).
Соединение (014):
К охлажденному до 0°С раствору (013) (4,32 г, 9,24 ммоль) в тетрагидрофуране (100 мл) прибавляли 10%-ный Pd/C (500 мг). Полученную смесь перемешивали под давлением водорода в 1 атмосферу в течение 4 часов. Смесь фильтровали через целит-545 и лепешку на фильтре промывали тетрагидрофураном. Органический фильтрат концентрировали при пониженном давлении и выдерживали при высоком вакууме, получая (014) (3,5 г), как было подтверждено LC/MS (LCRS (MH) m/z: 378,22), который использовали без дополнительной очистки.
Соединение (015):
К охлажденному до 0°С раствору (014) (3,5 г, 9,24 ммоль) и (006) (2,4 г, 11 ммоль), НОВТ (1,7 г, 11 ммоль) и HBTU (3,9 г, 11 ммоль) в тетрагидрофуране (200 мл) за 5 минут прибавляли раствор N,N-диизопропилэтиламина (4,9 мл, 30 ммоль) в тетрагидрофуране (10 мл). Следующие 5 часов смесь перемешивали при комнатной температуре, в результате она становилась гомогенной. Затем ее разбавляли этилацетатом (400 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×100 мл) и соляным раствором (100 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), и желаемое соединение (015) (5,0 г) выделяли и охарактеризовывали LC/MS (LCRS (MH) m/z: 532,33).
Соединение (016):
К охлажденному до 0°С раствору (015) (5,0 г, 9,40 ммоль) в дихлорметане (50 мл) за 5 минут прибавляли трифторуксусную кислоту (20 мл) и полученный раствор перемешивали при той же температуре в течение следующего часа. Органические слои концентрировали при пониженном давлении и затем выдерживали при высоком вакууме, получая (016), что было подтверждено LC/MS (LCRS (MH) m/z: 432,33), который использовали без дополнительной очистки.
Соединение (018):
К раствору метилового эфира 5-метил-3-изоксазолкарбоновой кислоты (017) (14,1 г, 100 ммоль) в четыреххлористом углероде (500 мл) при комнатной температуре прибавляли N-бромсукцинимид (23 г, 130 ммоль) и пероксид бензоила (2,5 г, 10 ммоль). Полученную смесь перемешивали в течение ночи в атмосфере аргона при 80°С. Реакционную смесь охлаждали и разбавляли 500 мл дихлорметана и промывали насыщенным водным (раствором) бикарбоната натрия (3×100 мл). Водную фазу экстрагировали 200 мл дихлорметана и объединенные органические слои промывали соляным раствором и сушили над MgSO4. Растворители удаляли и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (018) (7,9 г), который охарактеризовывали LC/MS (LCRS (MH) m/z: 219,05).
Соединение (019):
К охлажденному до 0°С раствору (018) (12 г, 55 ммоль) в тетрагидрофуране (20 мл) прибавляли водный гидроксид лития (35 мл, 4н.). Полученную смесь в течение ночи перемешивали при комнатной температуре. Затем подкисляли соляной кислотой (2 н.) до рН = 1 и экстрагировали тетрагидрофураном (3×200 мл). Объединенные органические слои промывали соляным раствором (10 мл), сушили над сульфатом натрия и фильтровали. Растворители удаляли и остаток подвергали лиофилизации, получая (019)(8,2 г), что было подтверждено LC/MS (LCRS (MH) m/z: 205,95), который использовали без дополнительной очистки.
Соединение (020):
Раствор (019) (6,0 г, 30 ммоль), бензилового спирта (3,5 мл) и пара-толуолсульфокислоты (1,1 г, 6 ммоль) в толуоле (100 мл) перемешивали в течение ночи при 100°С. Затем оставляли охлаждаться, разбавляли 300 мл этилацетата и промывали насыщенным водным (раствором) бикарбоната натрия. Водную фазу затем экстрагировали 200 мл этилацетата. Объединенные органические слои промывали соляным раствором, сушили над сульфатом натрия и фильтровали. Растворители удаляли и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (020) (5,8 г), который охарактеризовывали LC/MS (LCRS (MH) m/z: 295,98).
Соединение (021):
Раствор (020) (2,0 г, 6,8 ммоль) и морфолина (3,0 мл) в тетрагидрофуране (50 мл) перемешивали при комнатной температуре в течение двух часов. Затем растворители удаляли и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (021) (820 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 303,13).
Соединение (022):
К раствору (021) (400 мг, 1,32 ммоль) в тетрагидрофуране (40 мл) прибавляли 10%-ный Pd/C (100 мг) и смесь перемешивали при комнатной температуре под давлением водорода в одну атмосферу в течение 2 часов. Затем ее фильтровали через целит и концентрировали, получая (022), что было подтверждено LC/MS (LCRS (MH) m/z: 213,08) и использовали без дополнительной очистки.
Соединение (023)
К охлажденному до 0°С раствору (016) (139 мг, 0,3 ммоль) и (022) (70 мг, 0,4 ммоль), НОВТ (70 мг, 0,5 ммоль) и HBTU (170 мг, 0,5 ммоль) в тетрагидрофуране (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл, 2,5 ммоль) в тетрагидрофуране (5 мл). Следующие 5 часов смесь перемешивали при комнатной температуре, в результате она становилась гомогенной. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая соединение (023) (125 мг), которое охарактеризовывали LC/MS (LCRS (MH) m/z: 626,35); ингибирование >80% протеасомы СТ-L при 40 мг/кг РО.
Пример 3
Схема 3: синтез примера 029
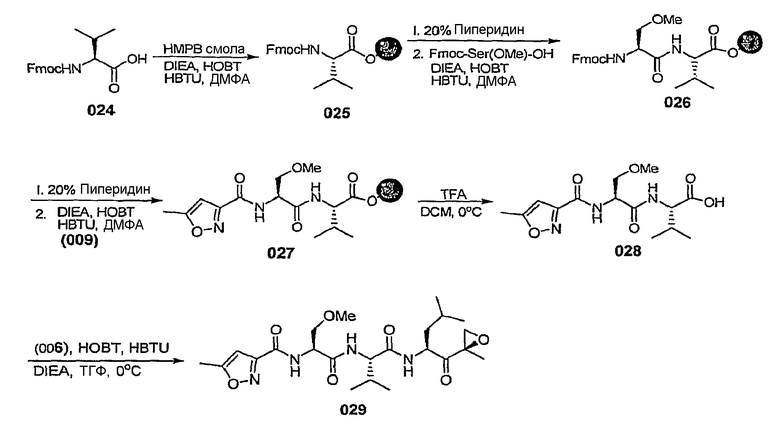
Соединение (025):
К охлажденному до 0°С раствору Fmoc-Val-OH (024) (348 мг, 1,6 ммоль) в дихлорметане (4 мл) прибавляли MSNT (474 мг, 1,6 ммоль) и N-метилимидазол (0,13 мл, 1,6 ммоль). После того как смесь становилась гомогенной, прибавляли смолу НМРВ (400 мг, 0,32 ммоль). Полученную реакционную смесь встряхивали два часа при комнатной температуре. Смолу отфильтровывали, промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл) и оставляли сохнуть на воздухе, получая (025).
Соединение (026):
Смолу (025) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали в течение 1 часа при комнатной температуре. Смолу отфильтровывали и промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл).
К охлажденному до 0°С раствору Fmoc-Ser(OМe)-OH (546 мг, 1,6 ммоль) в N,N-диметилформамиде (4 мл) прибавляли НОВТ (245 мг, 1,6 ммоль), HBTU (606 мг, 1,6 ммоль) и N,N-диизопропилэтиламин (0,6 мл, 3,2 ммоль). После того как смесь становилась гомогенной, прибавляли смолу. Полученную смесь оставляли встряхиваться в течение ночи. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл) и оставляли сохнуть на воздухе, получая (026).
Соединение (027):
Смолу (026) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали в течение 1 часа при комнатной температуре. Смолу отфильтровывали и промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл).
К охлажденному до 0°С раствору 5-метилизоксазол-3-карбоновой кислоты (009) (162 мг, 1,6 ммоль) в N,N-диметилформамиде (4 мл) прибавляли НОВТ (245 мг, 1,6 ммоль), HBTU (606 мг, 1,6 ммоль) и N,N-диизопропилэтиламин (0,6 мл, 3,2 ммоль). После того как смесь становилась гомогенной, прибавляли смолу и полученную реакционную смесь оставляли встряхиваться в течение ночи. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл) и оставляли сохнуть на воздухе, получая (027).
Соединение (028):
К смоле (027) прибавляли 50%-ный раствор трифторуксусной кислоты в дихлорметане (10 мл) и полученную смесь оставляли встряхиваться в течение 30 минут. Смолу затем отфильтровывали и промывали дихлорметаном (3×10 мл). Летучие (вещества) удаляли при пониженном давлении, получая (028), который охарактеризовывали LC/MS (LCRS (MH) m/z: 328,14) и использовали без дополнительной очистки.
Соединение (029):
К охлажденному до 0°С раствору (028) и (006) (117 мг, 0,4 ммоль), НОВТ (70 мг, 0,5 ммоль) и HBTU (170 мг, 0,5 ммоль) в тетрагидрофуране (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл, 2,5 ммоль) в тетрагидрофуране (5 мл). Следующие 5 часов смесь перемешивали при комнатной температуре, в результате чего она становилась гомогенной. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая соединение (029) (125 мг), которое охарактеризовывали LC/MS (LCRS (MH) m/z: 481,26); ингибирование >70% протеасомы СТ-L при 20 мг/кг РО.
Пример 4
Схема 4: синтез примера 035
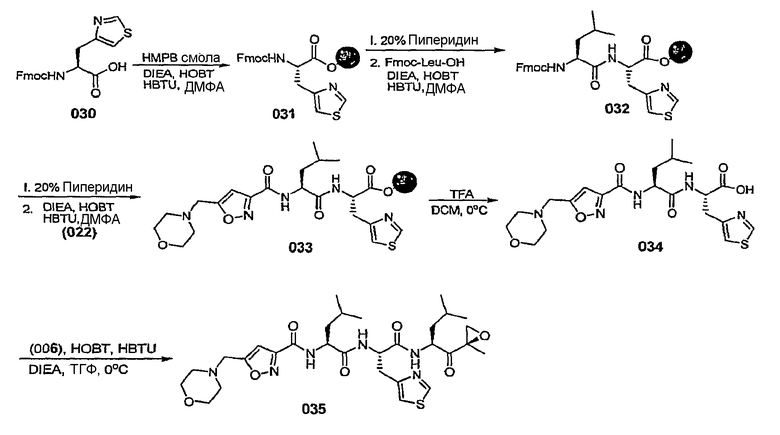
Соединение (031):
К охлажденному до 0°С раствору Fmoc-L-4-тиазолилаланина (030) (1,0 г, 2,5 ммоль) в дихлорметана (4 мл) прибавляли N-метилимидазол (150 мл, 1,9 ммоль), MSNT (755 мг, 2,55 ммоль) и после того, как смесь становилась гомогенной, прибавляли смолу HMPB (800 мг, 0,51 ммоль). Полученную реакционную смесь оставляли встряхиваться в течение двух часов при комнатной температуре. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл) и оставляли сохнуть на воздухе, получая (031).
Соединение (032):
Смолу (031) (360 мг, 0,23 ммоль) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали в течение 1 часа при комнатной температуре. Затем смолу отфильтровывали и промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл).
К охлажденному до 0°С раствору Fmoc-L-лейцина (204 мг, 0,58 ммоль) в N,N-диметилформамиде (4 мл) прибавляли НОВТ (124 мг, 0,92 ммоль), HBTU (349 мг, 0,92 ммоль) и N,N-диизопропилэтиламин (402 мкл, 2,3 ммоль). После того как смесь становилась гомогенной, прибавляли смолу. Полученную смесь оставляли встряхиваться при 5°С в течение 5 часов. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл) и полученную смолу оставляли сохнуть на воздухе, получая (032).
Соединение (033):
Смолу (032) (0,23 ммоль) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали в течение 1 часа при комнатной температуре. Затем смолу отфильтровывали и промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл).
К охлажденному до 0°С раствору (022) (123 мг, 0,58 ммоль) в N,N-диметилформамиде (4 мл) прибавляли НОВТ (124 мг, 0,92 ммоль), HBTU (349 мг, 0,92 ммоль) и N,N-диизопропилэтиламин (402 мкл, 2,3 ммоль). После того как смесь становилась гомогенной, прибавляли смолу и полученную реакционную смесь оставляли встряхиваться при комнатной температуре в течение ночи. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл) и полученную смолу оставляли сохнуть на воздухе, получая (033).
Соединение (034):
К смоле (033) прибавляли 50%-ный раствор трифторуксусной кислоты в дихлорметане (10 мл) и полученную смесь оставляли встряхиваться в течение 30 минут. Затем смолу отфильтровывали и промывали дихлорметаном (3×10 мл). Летучие (вещества) удаляли при пониженном давлении, получая (34), как было определено LC/MS (LCRS (MH) m/z: 480,18), которое использовали без дополнительной очистки.
Соединение (035):
К охлажденному до 0°С раствору (034) и (006) (70 мг, 0,23 ммоль), НОВТ (50 мг, 0,37 ммоль) и HBTU (140 мг, 0,37 ммоль) в тетрагидрофуране (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл, 2,5 ммоль) в тетрагидрофуране (5 мл). Смесь перемешивали при комнатной температуре в течение 5 часов и затем разбавляли этилацетатом (200 мл). Далее промывали ее насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия, фильтровали через целит-545 и растворители удаляли при пониженном давлении. Полученный остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая соединение (035) (15 мг), которое охарактеризовывали LC/MS (LCRS (MH) m/z: 633,3); ингибирование >90% протеасомы СТ-L при 40 мг/кг РО.
Пример 5
Схема 5: синтез примера 039
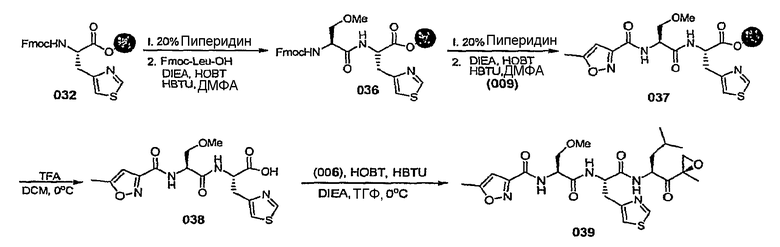
Соединение (036):
Смолу (031) (800 мг, 0,23 ммоль) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали при комнатной температуре в течение 1 часа. Смолу отфильтровывали и промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл).
К охлажденному до 0°С раствору Fmoc-L-Ser(OМe)-OH (435 мг, 1,3 ммоль) в N,N-диметилформамиде (10 мл) прибавляли НОВТ (276 мг, 2,0 ммоль), HBTU (710 мг, 2,0 ммоль) и N,N-диизопропилэтиламин (0,9 мл, 5,1 ммоль). После того как смесь становилась гомогенной, прибавляли смолу. Полученную смесь оставляли встряхиваться при 5°С в течение 5 часов. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл) и оставляли сохнуть на воздухе, получая (036).
Соединение (037)
Смолу (036) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали при комнатной температуре в течение 1 часа. Смолу отфильтровывали и промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл).
К охлажденному до 0°С раствору (009) (162 мг, 1,3 ммоль) в N,N-диметилформамиде (4 мл) прибавляли НОВТ (276 мг, 2,0 ммоль), HBTU (710 мг, 2,0 ммоль) и N,N-диизопропилэтиламин (0,9 мл, 5,1 ммоль). После того как реакционная смесь становилась гомогенной, прибавляли смолу и полученную реакционную смесь оставляли встряхиваться при комнатной температуре в течение ночи. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл) и оставляли сохнуть на воздухе, получая (037).
Соединение (038):
К (037) прибавляли 50%-ный раствор трифторуксусной кислоты в дихлорметане (10 мл) и полученную смесь оставляли встряхиваться в течение 30 минут. Затем смолу отфильтровывали и промывали дихлорметаном (3×10 мл). Летучие (вещества) удаляли при пониженном давлении, получая (38), как было определено LC/MS (LCRS (MH) m/z: 383,09), которое использовали без дополнительной очистки.
Соединение (039):
К охлажденному до 0°С раствору (038) и (006) (156 мг, 0,51 ммоль), НОВТ (111 мг, 0,82 ммоль) и HBTU (311 мг, 0,82 ммоль) в тетрагидрофуране (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл, 2,5 ммоль) в тетрагидрофуране (5 мл). Смесь перемешивали при комнатной температуре в течение 5 часов, и она становилась гомогенной. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (039) (22 мг), которое охарактеризовывали LC/MS (LCRS (MH) m/z: 536,21; ингибирование >75% протеасомы СТ-L при 20 мг/кг РО.
Пример 6
Схема 6: синтез примера 045 (способ А)
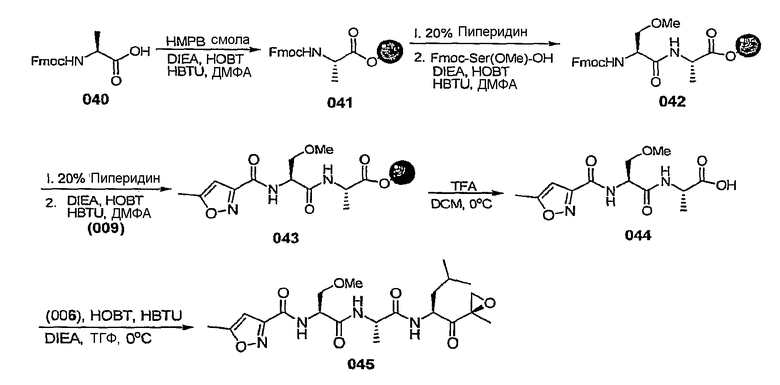
Соединение (041):
К охлажденному до 0°С раствору Fmoc-L-аланина (040) (1,0 г, 3,2 ммоль) в дихлорметане (30 мл) прибавляли N-метилимидазол (190 мкл, 12,4 ммоль), MSNT (950 мг, 3,2 ммоль) и после того, как смесь становилась гомогенной, прибавляли смолу HMPB (1 г, 0,64 ммоль). Полученную реакционную смесь оставляли встряхиваться в течение двух часов при комнатной температуре. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл), получая (041).
Соединение (042):
Смолу (041) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали при комнатной температуре в течение 1 часа. Смолу отфильтровывали и промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл).
К охлажденному до 0°С раствору Fmoc-Ser(OМe)-OH (546 мг, 1,6 ммоль) в N,N-диметилформамиде (10 мл) прибавляли НОВТ (346 мг, 2,6 ммоль), HBTU (970 мг, 2,6 ммоль) и N,N-диизопропилэтиламин (1,1 мл, 6,4 ммоль). После того как реакционная смесь становилась гомогенной, прибавляли смолу. Полученную смесь оставляли встряхиваться при 5°С в течение 5 часов. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл) и оставляли сохнуть на воздухе, получая (042).
Соединение (043):
Смолу (042) (0,23 ммоль) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали при комнатной температуре в течение 1 часа. Смолу отфильтровывали и промывали N,N-диметилформамидом (3×10 мл) и дихлорметаном (3×10 мл).
К охлажденному до 0°С раствору (009) (203 мг, 1,6 ммоль) в N,N-диметилформамиде (10 мл) прибавляли НОВТ (346 мг, 2,6 ммоль), HBTU (970 мг, 2,6 ммоль) и N,N-диизопропилэтиламин (1,1 мл, 6,4 ммоль). После того как реакционная смесь становилась гомогенной, прибавляли смолу и полученную реакционную смесь оставляли встряхиваться при комнатной температуре в течение ночи. Затем смолу отфильтровывали, промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл) и оставляли сохнуть на воздухе, получая (043).
Соединение (044):
К (043) прибавляли 50%-ный раствор трифторуксусной кислоты в дихлорметане (10 мл) и полученную смесь оставляли встряхиваться в течение 30 минут. Затем смолу отфильтровывали и промывали дихлорметаном (3×10 мл). Летучие (вещества) удаляли при пониженном давлении, получая (044), как было определено LC/MS (LCRS (MH) m/z: 300,11), которое использовали без дополнительной очистки.
Соединение (045):
К охлажденному до 0°С раствору вышеприведенного промежуточного продукта (044) и (006) (195 мг, 0,64 ммоль), НОВТ (137 мг, 1,0 ммоль) и HBTU (357 мг, 1,0) в тетрагидрофуране (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл, 2,5 ммоль) в тетрагидрофуране (5 мл). Смесь перемешивали при комнатной температуре в течение 4 часов. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (045) (84 мг), которое охарактеризовывали LC/MS (LCRS (MH) m/z: 453,23; ингибирование >80% протеасомы СТ-L при 20 мг/кг РО.
Пример 7
Схема 7: синтез примера 045 (способ В)
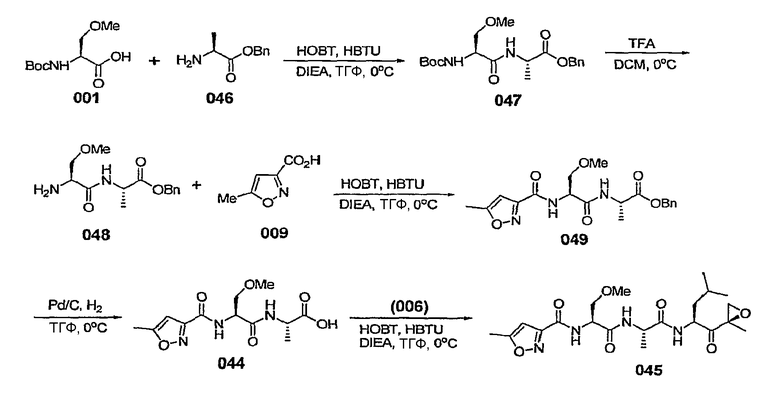
Соединение (047):
К охлажденному до 0°С раствору метилового эфира N-Boc-серина (001) (6,57 г, 33 ммоль), гидрохлорида бензилового эфира L-аланина (046) (6,45 г, 30 ммоль), НОВТ (5,05 г, 33 ммоль) и HBTU (11,8 г, 33 ммоль) в тетрагидрофуране (400 мл) за 10 минут прибавляли раствор N,N-диизопропилэтиламина (9,0 г, 70 ммоль) в тетрагидрофуране (50 мл). Смесь становилась гомогенной и ее перемешивали при комнатной температуре в течение следующих 5 часов. Затем большую часть растворителя удаляли при пониженном давлении и остаток разбавляли этилацетатом (500 мл). Промывали насыщенным водным (раствором) бикарбоната натрия (2×150 мл) и соляным раствором (200 мл) и органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (047) (11,8 г), которое охарактеризовывали LC/MS (LCRS (MH) m/z: 381,19.
Соединение (048):
К охлажденному до 0°С раствору (047) (11,8 г, 31,0 ммоль) в дихлорметане (100 мл) за 10 минут прибавляли трифторуксусную кислоту (50 мл) и полученную смесь перемешивал и при той же температуре еще в течение 3 часов. Затем растворители удаляли при пониженном давлении и остаток помещали на ночь в высокий вакуум, получая соль TFA (048), которую охарактеризовывали LC/MS (LCRS (MH) m/z: 281,15), и использовали без дополнительной очистки.
Соединение (049):
К охлажденному до 0°С раствору (048), 5-метилизоксазол-3-карбоновой кислоты (009) (3,93 г, 31 ммоль), НОВТ (4,7 г, 35 ммоль) и HBTU (12,5 г, 35 ммоль) в тетрагидрофуране (400 мл) за 10 минут прибавляли раствор N,N-диизопропилэтиламина (20 мл) в тетрагидрофуране (100 мл); рН полученной смеси был равен ~8. Смесь перемешивали при комнатной температуре в течение следующих 5 часов. Затем большую часть растворителя удаляли при пониженном давлении и разбавляли этилацетатом (1,0 л). Далее промывали насыщенным водным (раствором) бикарбоната натрия (2×100 мл) и соляным раствором (100 мл) и органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (049) (10,8 г), которое охарактеризовывали LC/MS (LCRS (MH) m/z: 390,16.
Соединение (044):
К охлажденному до 0°С раствору (049) (3,28 г, 8,4 ммоль) в тетрагидрофуране (100 мл) прибавляли 10%-ный Pd/C (500 мг). Полученную смесь перемешивали под давлением водорода в 1 атмосферу в течение 4 часов. Затем смесь фильтровали через целит-545 и лепешку на фильтре промывали тетрагидрофураном. Органический фильтрат концентрировали при пониженном давлении и помещали в высокий вакуум на 2 часа, получая (044), который охарактеризовывали LC/MS (LCRS (MH) m/z: 281,15) и использовали без дополнительной очистки.
Соединение (045):
К охлажденному до 0°С раствору (044) и (006) (1,9 г, 8,5 ммоль), НОВТ (2,0 г, 13 ммоль) и HBTU (5,4 г, 14 ммоль) в тетрагидрофуране (200 мл) прибавляли раствор N,N-диизопропилэтиламина (5,4 г, 42 ммоль) в тетрагидрофуране (10 мл). Смесь перемешивали при комнатной температуре в течение еще 5 часов. Затем большую часть растворителя удаляли при пониженном давлении и полученный остаток разбавляли этилацетатом (400 мл). Далее промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (50 мл) и органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (045) (1,35 г), которое охарактеризовывали LC/MS (LCRS (MH) m/z: 453,23).
Пример 8
Схема 8: синтез примера 055
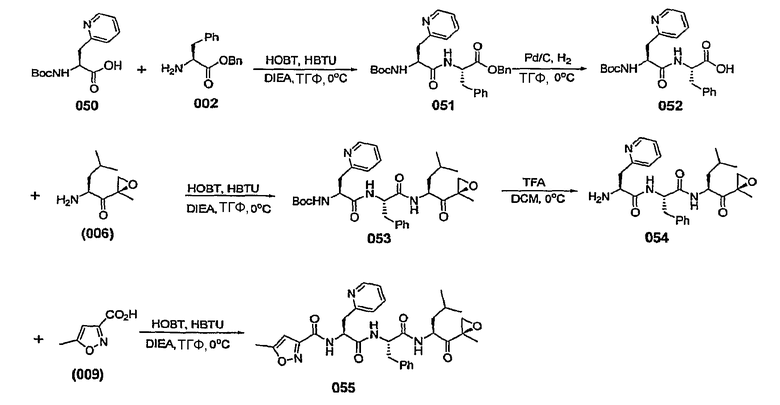
Соединение (051)
К охлажденному до 0°С раствору N-Boc-L-пиридилаланина (050) (1,0 г, 3,76 ммоль), гидрохлорида бензилового эфира L-фенилаланина (002) (1,3 г, 3,76 ммоль), НОВТ (0,68 г, 5,0 ммоль) и HBTU (1,8 г, 5,0 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (1,6 мл) в тетрагидрофуране (10 мл). Смесь перемешивали при комнатной температуре в течение следующих 3 часов и затем разбавляли этилацетатом (200 мл), промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (100 мл); органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворитель удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (051), который охарактеризовывали LC/MS (LCRS (MH) m/z: 504,24).
Соединение (052):
К охлажденному до 0°С раствору (051) в тетрагидрофуране (100 мл) прибавляли 10%-ный Pd/C (100 мг). Полученную смесь перемешивали под давлением водорода в 1 атмосферу в течение 4 часов. Затем смесь фильтровали через целит-545 и лепешку на фильтре промывали тетрагидрофураном. Органический фильтрат затем концентрировали при пониженном давлении и помещали в высокий вакуум, получая (052), (по данным) LC/MS (LCRS (MH) m/z: 414,2), который использовали без дополнительной очистки.
Соединение (053):
К охлажденному до 0°С раствору (052) и (006) (0,85 г, 3,9 ммоль), НОВТ (0,70 г, 5,3 ммоль) и HBTU (1,70 г, 4,9 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (3 мл) в тетрагидрофуране (10 мл) и смесь перемешивали при комнатной температуре в течение ночи. Затем ее разбавляли этилацетатом (200 мл), промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (50 мл). Органические слои сушили над сульфатом натрия, фильтровали через целит-545, растворитель удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (053) (1,51 г), который охарактеризовывали LC/MS (LCRS (MH) m/z: 567,21).
Соединение (054):
К охлажденному до 0°С раствору (053) (200 мг, 0,352 ммоль) в дихлорметане (10 мл) прибавляли трифторуксусную кислоту (10 мл) и полученный раствор перемешивали при той же температуре в течение следующего часа. Раствор концентрировали при пониженном давлении и помещали в высокий вакуум, получая (054), как подтверждено LC/MS (LCRS (MH) m/z: 467,26), который использовали без дополнительной очистки.
Соединение (055):
К охлажденному до 0°С раствору (054), 5-метилизоксазол-3-карбоновой кислоты (009) (127 мг, 1,0 ммоль), НОВТ (135 мг, 1,0 ммоль) и HBTU (350 г, 1,0 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл) в тетрагидрофуране (2 мл). Смесь перемешивали при комнатной температуре в течение 5 часов. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545; растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (055) (40 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 576,27; ингибирование >80% протеасомы CT-L при 20 мг/кг РО.
Пример 9
Схема 9: синтез примера 061
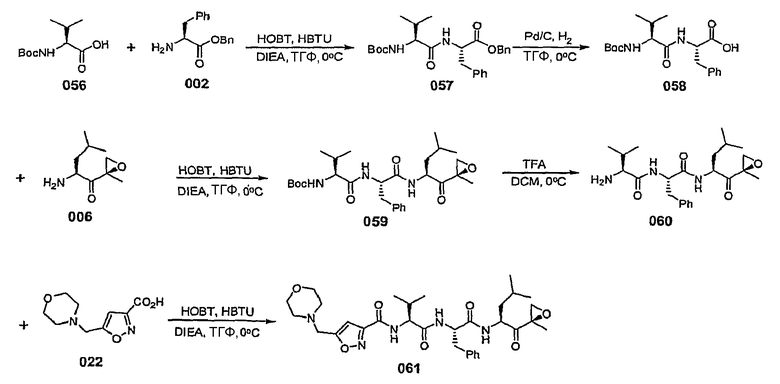
Соединение (057):
К охлажденному до 0°С раствору N-Boc-L-н-валина (056) (1,0 г, 4,6 ммоль), гидрохлорида бензилового эфира L-фенилаланина (002) (1,4 г, 4,6 ммоль), НОВТ (1,0 г, 7,4 ммоль) и HBTU (2,8 г, 7,4 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (3,2 мл, 18,4 ммоль) в тетрагидрофуране (10 мл). Смесь перемешивали при комнатной температуре в течение 3 часов и затем разбавляли этилацетатом (200 мл), промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (100 мл) и органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворитель удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (057), который охарактеризовывали LC/MS (LCRS (MH) m/z: 455,25.
Соединение (058):
К охлажденному до 0°С раствору (057) (1,30 г, 2,875 ммоль) в тетрагидрофуране (100 мл) прибавляли 10%-ный Pd/C (100 мг). Полученную смесь перемешивали под давлением водорода в 1 атмосферу в течение 4 часов. Затем смесь фильтровали через целит-545 и лепешку на фильтре промывали тетрагидрофураном. Затем фильтрат концентрировали при пониженном давлении и помещали в высокий вакуум, получая (058), как подтверждено LC/MS (LCRS (MH) m/z: 365,2), который использовали без дополнительной очистки.
Соединение (059):
К охлажденному до 0°С раствору (058) и (006) (0,99 г, 4,6 ммоль), НОВТ (0,62 г, 4,6 ммоль) и HBTU (1,70 г, 4,9 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (2,4 г) в тетрагидрофуране (10 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (50 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (059) (1,21 г), который охарактеризовывали LC/MS (LCRS (MH) m/z: 518,32).
Соединение (060):
К охлажденному до 0°С раствору (059) (250 мг, 0,48 ммоль) в дихлорметане (10 мл) прибавляли трифторуксусную кислоту (10 мл) и полученный раствор перемешивали при той же температуре в течение следующего часа. Органические слои концентрировали при пониженном давлении и помещали в высокий вакуум, получая (060), как подтверждено LC/MS (LCRS (MH) m/z: 418,26), который использовали без дополнительной очистки.
Соединение (061):
К охлажденному до 0°С раствору (060) и (022) (122 мг, 0,58 ммоль), НОВТ (104 мг, 0,77 ммоль) и HBTU (282 мг, 0,72 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (0,35 мл) в тетрагидрофуране (2 мл) и смесь перемешивали при комнатной температуре в течение следующих 4 часов. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (061) (88,4 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 612,33); ингибирование >80% протеасомы СТ-L при 40 мг/кг РО.
Пример 10
Схема 10: синтез примера 068
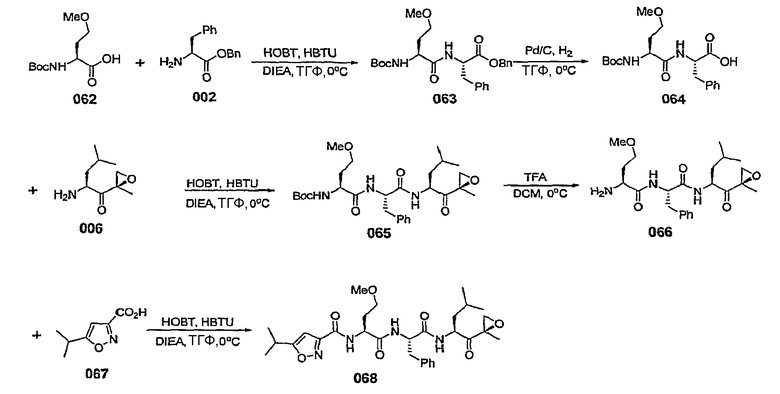
Соединение (063):
К охлажденному до 0°С раствору N-Boc-HoSer(OMe)-OH (062) (1,0 г, 4,3 ммоль), гидрохлорида бензилового эфира L-фенилаланина (002) (1,3 г, 4,3 ммоль), НОВТ (0,88 г, 6,5 ммоль) и HBTU (2,3 г, 6,5 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (2,0 мл) в тетрагидрофуране (5 мл). Смесь перемешивали при комнатной температуре в течение следующих 3 часов, затем разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (100 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (063) (1,81 г), который охарактеризовывали LC/MS (LCRS (MH) m/z: 471,24).
Соединение (064):
К охлажденному до 0°С раствору (063) (1,35 г, 2,875 ммоль) в тетрагидрофуране (100 мл) прибавляли 10%-ный Pd/C (100 мг). Полученную смесь перемешивали под давлением водорода в 1 атмосферу в течение 4 часов. Затем смесь фильтровали через целит-545 и лепешку на фильтре промывали тетрагидрофураном. Органический фильтрат концентрировали при пониженном давлении и помещали в высокий вакуум, получая (064), как подтверждено LC/MS (LCRS (MH) m/z: 381,19), который использовали без дополнительной очистки.
Соединение (065):
К охлажденному до 0°С раствору (065) и (006) (0,99 г, 4,6 ммоль), НОВТ (0,62 г, 4,6 ммоль) и HBTU (1,70 г, 4,9 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (2,4 мл) в тетрагидрофуране (10 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×50 мл) и соляным раствором (50 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (065) (1,11 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 534,31).
Соединение (066):
К охлажденному до 0°С раствору (065) (230 мг, 0,43 ммоль) в дихлорметане (20 мл) прибавляли трифторуксусную кислоту (10 мл) и полученный раствор перемешивали при той же температуре в течение следующего часа. Затем реакционную смесь концентрировали при пониженном давлении и помещали в высокий вакуум, получая (066), как подтверждено LC/MS (LCRS (MH) m/z: 434,26), который использовали без дополнительной очистки.
Соединение (068):
К охлажденному до 0°С раствору (066) и 5-изопропилизоксазол-3-карбоновой кислоты (067) (81 мг, 0,52 ммоль), НОВТ (93 мг, 0,69 ммоль) и HBTU (262 мг, 0,69 ммоль) в тетрагидрофуране (100 мл) прибавляли раствор N,N-диизопропилэтиламина (0,30 мл) в тетрагидрофуране (2 мл) и смесь перемешивали при комнатной температуре в течение следующих 4 часов. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (068) (75,7 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 571,31); ингибирование >70% протеасомы СТ-L при 40 мг/кг РО.
Пример 11
Схема 11: синтез примера 075 (способ А)
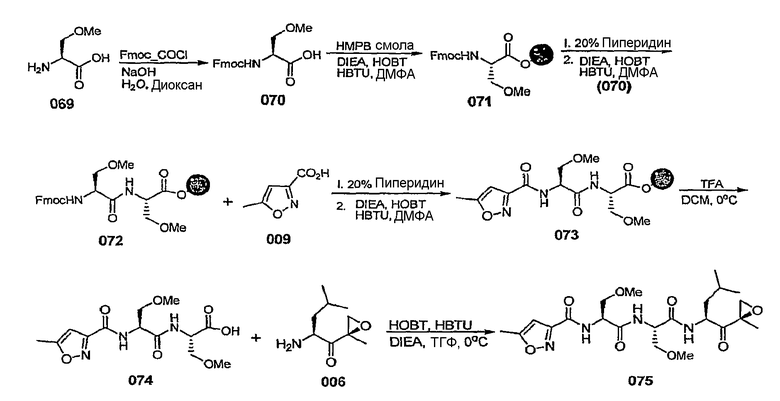
Соединение (070):
К раствору гидрохлорида метилового эфира L-серина (069) (1,0 г, 6,4 ммоль) в смеси вода/диоксан (1:1, 80 мл) прибавляли гидроксид натрия (768 мг, 19,2 ммоль). После перемешивания смеси при комнатной температуре в течение 30 минут ее охлаждали до 0°С и прибавляли по каплям раствор 9-флуоренилметилхлорформиата (1,65 г, 6,4 ммоль) в диоксане (16 мл). Следующие 4 часа реакционную смесь оставляли перемешиваться при комнатной температуре. Затем растворители удаляли, остаток разбавляли водой и 1 н. HCl доводили рН до ~1; водный слой экстрагировали этилацетатом (4×100 мл). Органические слои концентрировали при пониженном давлении и помещали в высокий ваккуум, получая (070) (1,8 г), что было подтверждено LC/MS (LCRS (MH) v/z: 342,13), который использовали без дополнительной очистки.
Соединение (071):
Смолу НМРВ-ВНА (500 мг, 0,32 ммоль) промывали дихлорметаном. В сухой колбе растворяли в дихлорметане Fmoc-Ser(Me)-OH (070) (546 мг, 1,6 ммоль), к раствору прибавляли 1-метилимидазол (95 мкл, 1,2 ммоль) и затем MSNT (474 мг, 1,6 ммоль). После того как реакционная смесь становилась гомогенной (10 минут), ее прибавляли к смоле НМРВ-ВНА в виде суспензии в дихлорметане (5 мл). Полученную реакционную смесь оставляли встряхиваться в течение ночи. Затем смолу отфильтровывали и промывали ДМФА (3×20 мл), МеОН (3×20 мл), DCM (3 x 20 мл) и оставляли сохнуть на воздухе, получая (071).
Соединение (072):
Смолу (071) (300 мг, 0,192 ммоль) помещали в 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали при комнатной температуре в течение 30 минут. Смолу отфильтровывали и дважды промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл).
К охлажденному до 0°С раствору Fmoc-Ser(Me)-OH (070) (0,48 ммоль, 163 мг) в N,N-диметилформамиде (10 мл) прибавляли НОВТ (104 мг, 0,77 ммоль), HBTU (291 мг, 1,92 ммоль) и диизопропилэтиламин (0,34 мл 1,92 ммоль). После того как полученная смесь становилась гомогенной, прибавляли смолу (0,13 ммоль, 200 мг) и полученную реакционную смесь оставляли встряхиваться в течение ночи. Затем смолу отфильтровывали, промывали ДМФА (10 мл), DCM (10 мл), МеОН (10 мл), Н2О (10 мл), ДМФА (10 мл), МеОН (10 мл) и DCM (10 мл) и оставляли сохнуть на воздухе, получая (072).
Соединение (073):
К (072) (300 мг, 0,19 ммоль) прибавляли 20%-ный раствор пиперидина в N,N-диметилформамиде (20 мл) и полученную смесь встряхивали при комнатной температуре в течение 30 минут. Смолу отфильтровывали и дважды промывали N,N-диметилформамидом (3×20 мл) и дихлорметаном (3×20 мл).
К охлажденному до 0°С раствору 009) (61 мг, 0,48 ммоль) в N,N-диметилформамиде (2 мл) прибавляли НОВТ (104 мг, 0,77 ммоль), HBTU (291 мг, 1,77 ммоль) и N,N- диизопропилэтиламин (0,34 мл 1,92 ммоль). После того как полученная смесь становилась гомогенной, прибавляли смолу (300 мг, 0,192 ммоль) и полученную реакционную смесь оставляли встряхиваться при комнатной температуре в течение ночи. Затем смолу отфильтровывали, промывали ДМФА (10 мл), DCM (10 мл), МеОН (10 мл), Н2О (10 мл), ДМФА (10 мл), МеОН (10 мл) и DCM (10 мл) и оставляли сохнуть на воздухе, получая (073).
Соединение (074):
К (073) прибавляли 50%-ный раствор трифторуксусной кислоты в дихлорметане (10 мл) и полученную смесь оставляли встряхиваться в течение 30 минут. Затем смолу отфильтровывали и промывали дихлорметаном (3×10 мл). Летучие (вещества) удаляли при пониженном давлении и желаемое соединение (074) охарактеризовывали LC/MS (LCRS (MH) m/z: 330,12) и использовали без дополнительной очистки.
Соединение (075):
К охлажденному до 0°С раствору (074) и (006) (78 мг, 0,38 ммоль), НОВТ (41 мг, 0,30 ммоль) и HBTU (116 мг, 0,30 ммоль) в ацетонитриле (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,1 мл, 0,6 ммоль). Смесь перемешивали при 0-4°С в течение ночи и затем разбавляли этилацетатом (200 мл). Далее промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл), органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (075) (29 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 483,24); ингибирование >80% протеасомы СТ-L при 20 мг/кг РО.
Пример 12
Схема 12: синтез примера 075 (способ В)
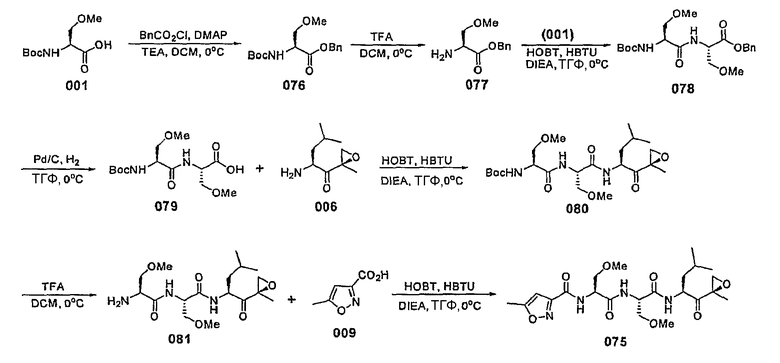
Соединение (076):
К охлажденному до 0°С раствору метилового эфира N-Boc-серин-ОН (43,8 г, 200 ммоль), триэтиламина (26,5 г, 260 ммоль) и 4-(диметиламино)пиридина в дихлорметане (1,2 л) за 30 минут прибавляли раствор бензилхлорформиата (41 г, 240 ммоль) в дихлорметане (250 мл) и полученную смесь перемешивали при той же температуре в течение следующих 3 часов. Затем прибавляли насыщенный водный (раствор) бикарбоната натрия (200 мл) и органический слой промывали насыщенным водным (раствором) бикарбоната натрия (200 мл) и соляным раствором (200 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (076) (54 г), который охарактеризовывали LC/MS (LCRS (MH) m/z: 310,16).
Соединение (077):
К охлажденному до 0°С раствору (076) (54 г, 174,6 ммоль) в дихлорметане (200 мл) за 10 минут прибавляли трифторуксусную кислоту (200 мл) и полученную смесь перемешивали при той же температуре еще в течение 3 часов. Затем растворители удаляли при пониженном давлении и остаток помещали в высокий вакуум, получая соль TFA c (077), как подтверждено LC/MS (LCRS (MH) m/z: 210,11), которую использовали без дополнительной очистки.
Соединение (078):
К охлажденному до 0°С раствору (077) (43,8 г, 200 ммоль), метилового эфира N-Boc-серин-OH (36,7 г, 167 ммоль), НОВТ (27 г, 200 ммоль) и HBTU (71,4 г, 200 ммоль) в тетрагидрофуране (1,2 л) за 10 минут прибавляли раствор N,N-диизопропилэтиламина (75 г, 600 ммоль) в тетрагидрофуране (250 мл); рН полученной смеси равнялось ~8. Смесь перемешивали при комнатной температуре в течение еще 5 часов. Затем большую часть растворителя удаляли при пониженном давлении и полученное вещество разбавляли этилацетатом (1,0 л). Далее промывали насыщенным водным (раствором) бикарбоната натрия (2×150 мл) и соляным раствором (200 мл); органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (078) (65 г), который охарактеризовывали LC/MS (LCRS (MH) m/z: 411,21).
Соединение (079):
К охлажденному до 0°С раствору (079) (13,4 г, 32,7 ммоль), в тетрагидрофуране (300 мл) прибавляли 10%-ный Pd/C (2,7 г) и полученную смесь перемешивали в течение 4 часов под давлением водорода в 1 атмосферу. Смесь фильтровали через целит-545 и лепешку на фильтре промывали тетрагидрофураном. Органические слои концентрировали при пониженном давлении и помещали в высокий вакуум, получая (079), что подтверждено LC/MS (LCRS (MH) m/z: 321,16), который использовали без дополнительной очистки.
Соединение (080):
К охлажденному до 0°С раствору (079) и (006) (5,6 г, 26 ммоль), НОВТ (6 г, 41,4 ммоль) и HBTU (14,8 г, 41,4 ммоль) в тетрагидрофуране (400 мл) прибавляли раствор N,N-диизопропилэтиламина (23 мл) в тетрагидрофуране (40 мл) и смесь перемешивали при комнатной температуре в течение ночи. Затем большую часть растворителя удаляли при пониженном давлении, остаток разбавляли этилацетатом (500 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×100 мл) и соляным раствором (100 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали флэш-хроматографией (гексан и этилацетат), получая (080) (9,2 г), который охарактеризовывали LC/MS (LCRS (MH) m/z: 474,27).
Соединение (081):
К охлажденному до 0°С раствору (080) (200 мг, 0,43 ммоль) в дихлорметане (10 мл) прибавляли трифторуксусную кислоту (10 мл) и полученный раствор перемешивали при той же температуре еще в течение часа. Органические слои концентрировали при пониженном давлении и помещали в высокий вакуум, получая (081), как подтверждено LC/MS (LCRS (MH) m/z: 374,22), который использовали без дополнительной очистки.
Соединение (075):
К охлажденному до 0°С раствору (081) и 5-метилизоксазол-3-карбоновой кислоты (009) (65 мг, 0,5 ммоль), НОВТ (65 мг, 0,5 ммоль) и HBTU (175 мг, 0,5 ммоль) в тетрагидрофуране (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл) в тетрагидрофуране (2 мл) и смесь перемешивали при комнатной температуре в течение еще 5 часов. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (075) (85 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 483,24).
Пример 13
Схема 13: синтез примера 083
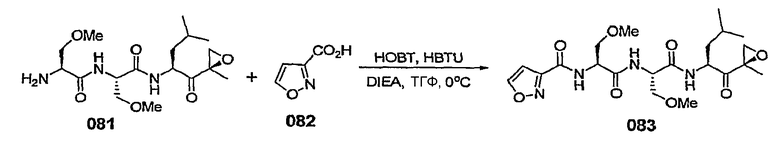
Соединение (083):
К охлажденному до 0°С раствору (081) (160 мг, 0,43 ммоль) и изоксазол-3-карбоновой кислоты (082) (60 мг, 0,5 ммоль), НОВТ (65 мг, 0,5 ммоль) и HBTU (175 мг, 0,5 ммоль) в тетрагидрофуране (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл) в тетрагидрофуране (2 мл) и смесь перемешивали при комнатной температуре в течение еще 5 часов. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (083) (74 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 469,22); ингибирование >80% протеасомы CT-L при 20 мг/кг РО.
Пример 14
Схема 14: синтез примера 085
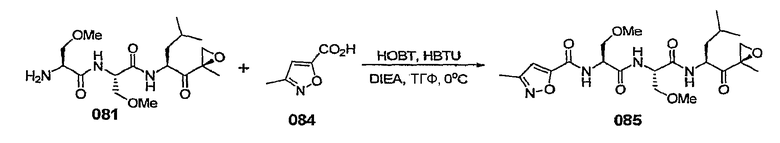
Соединение (085):
К охлажденному до 0°С раствору (081) (160 мг, 0,43 ммоль) и изоксазол-3-карбоновой кислоты (084) (65 мг, 0,5 ммоль), НОВТ (65 мг, 0,5 ммоль) и HBTU (175 мг, 0,5 ммоль) в тетрагидрофуране (50 мл) прибавляли раствор N,N-диизопропилэтиламина (0,5 мл) в тетрагидрофуране (2 мл) и смесь перемешивали при комнатной температуре в течение еще 5 часов. Затем реакционную смесь разбавляли этилацетатом (200 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия и фильтровали через целит-545. Растворители удаляли при пониженном давлении и остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (085) (71 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 483,24); ингибирование >50% протеасомы CT-L при 20 мг/кг РО.
Схема 15: синтез примера 088
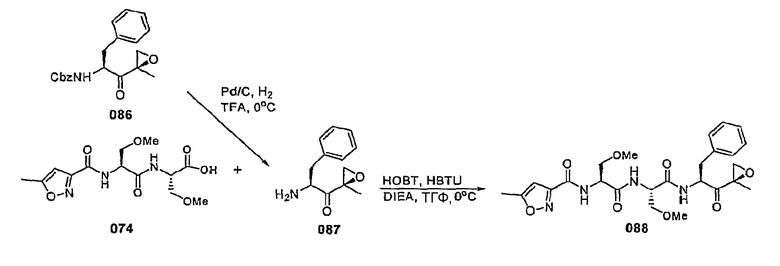
Соединение (087):
К раствору (086) (полученному тем же способом, что и (005), за исключением того, что Cbz-фенилаланин был заменен на Cbz-лейцин) (0,100 г, 0,295 ммоль) в трифторуксусной кислоте (10 мл) прибавляли 10%-ный Pd/C (20 мг). Полученную смесь оставляли перемешиваться под давлением водорода в 1 атмосферу в течение 6 часов. Затем смесь фильтровали через целит-545 и лепешку на фильтре промывали дихлорметаном (50 мл). Фильтрат концентрировали при пониженном давлении и помещали в высокий вакуум на ночь, получая (087), как подтверждено LC/MS (LCRS (MH) m/z: 206,1), который использовали в последующем превращении без дополнительной очистки.
Соединение (088):
К охлажденному до 0°С раствору (087) и (074) (166 мг, 0,354 ммоль), НОВТ (54 мг, 0,354 ммоль) и HBTU (134 мг, 0,354 ммоль) в тетрагидрофуране (20 мл) прибавляли раствор N,N-диизопропилэтиламина (0,2 мл, 1,18 ммоль). Смесь перемешивали при 0°С в течение ночи, и она становилась гомогенной. Затем ее разбавляли этилацетатом (20 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия, фильтровали через целит-545 и концентрировали при пониженном давлении; остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (088) (10 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 517,69); ингибирование >80% протеасомы CT-L при 20 мг/кг РО.
Схема 16: синтез примера 091
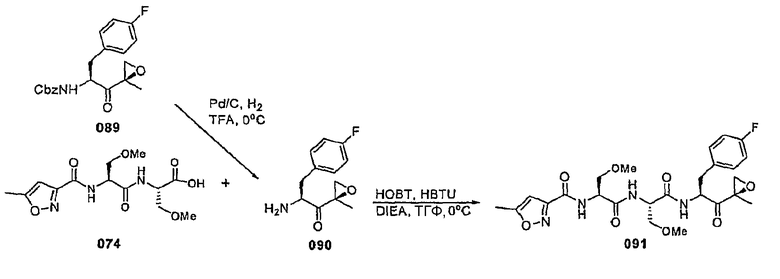
Соединение (090):
К раствору (089) (полученному тем же способом, что и (005), за исключением того, что Cbz-4-фторфенилаланин был заменен на Cbz-лейцин) (0,100 г, 0,28 ммоль) в трифторуксусной кислоте (10 мл) прибавляли 10%-ный Pd/C (20 мг). Полученную смесь оставляли перемешиваться под давлением водорода в 1 атмосферу в течение 6 часов. Затем смесь фильтровали через целит-545 и лепешку на фильтре промывали дихлорметаном (50 мл). Фильтрат концентрировали при пониженном давлении и помещали в высокий вакуум на ночь, получая (090), как подтверждено LC/MS (LCRS (MH) m/z: 224,1), который использовали в последующем превращении без дополнительной очистки.
Соединение (091):
К охлажденному до 0°С раствору (090) и (074) (110 мг, 0,336 ммоль), НОВТ (51 мг, 0,336 ммоль) и HBTU (127 мг, 0,336 ммоль) в тетрагидрофуране (20 мл) прибавляли N,N-диизопропилэтиламин (0,2 мл, 1,18 ммоль). Смесь перемешивали при 0°С в течение ночи, и она становилась гомогенной. Затем ее разбавляли этилацетатом (20 мл) и промывали насыщенным водным (раствором) бикарбоната натрия (2×10 мл) и соляным раствором (10 мл). Органические слои сушили над сульфатом натрия, фильтровали через целит-545 и концентрировали при пониженном давлении. Затем остаток очищали ВЭЖХ (водный ацетат аммония и ацетонитрил), получая (091) (60 мг), который охарактеризовывали LC/MS (LCRS (MH) m/z: 535,69); ингибирование >80% протеасомы CT-L при 20 мг/кг РО.
Биологическая активность
Соединения готовили в носителе 10%-ном PS80/цитрат Na (рН 3) и вводили перорально (РО) мышам (3 особи на группу). Через один час после дозирования животных убивали и отбирали следующие ткани: кровь, мозг, надпочечник, сердце и печень. Цельную кровь (~200 мкл) дважды промывали PBS и лизировали гипотоническим шоком (hypotonic shock) (300 мкл 50 мМ Tris рН 8, 5 мМ EDTA). Лизаты крови хранили при -80°С до анализа. Лизаты крови очищали центрифугированием в микроцентрифуге. Специфическую активность протеасомы CT-L в каждом лизате оценивали определением: а) концентрации белка пробой Бредфорда (Bradford) c бычьим гамма-глобулином в качестве стандарта и b) cкорости расщепления субстрата флуорогенной протеасомы LLVY-AMC. Процент активности протеасомы для аналогично обработанных животных вычисляли делением средней специфической активности каждой аналогично дозированной группы на среднюю специфическую активность группы, дозированной носителем. Процент ингибирования протеасомы вычисляли вычитанием процента активности протеасомы из 100.
Эквиваленты
Специалисты в данной области способны выявить и установить, используя не более чем обычное экспериментирование, многочисленные эквиваленты описанных здесь соединений и способов их использования. Такие эквиваленты рассматриваются как (входящие) в рамки изобретения и охватываемые следующей формулой изобретения.
Все процитированные выше ссылки и публикации приведены здесь в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЕ ЭПОКСИКЕТОНЫ ДЛЯ ИНГИБИРОВАНИЯ ПРОТЕАСОМЫ | 2007 |
|
RU2450016C2 |
| ГАЛОГЕНИРОВАННЫЕ ХИНАЗОЛИН-THF-АМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2015 |
|
RU2692808C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ХИМИЧЕСКИЕ СОСТАВЫ, КОМПОЗИЦИИ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2005 |
|
RU2413720C2 |
| ИНГИБИТОРЫ МАТРИКСНЫХ МЕТАЛЛОПРОТЕИНАЗ | 2004 |
|
RU2370488C2 |
| ЗАМЕЩЕННЫЕ АМИНО ШЕСТИЧЛЕННЫЕ НАСЫЩЕННЫЕ ГЕТЕРОАЛИЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ DPP-IV ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2016 |
|
RU2720488C2 |
| ЗАМЕЩЕННЫЕ АРИЛПИРАЗОЛЫ В КАЧЕСТВЕ АГЕНТОВ ДЛЯ УНИЧТОЖЕНИЯ ПАРАЗИТОВ | 2006 |
|
RU2381218C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ КИНАЗЫ LRRK2 | 2012 |
|
RU2622104C2 |
| ЛИГАНДЫ ЦЕРЕБЛОНА И БИФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ | 2018 |
|
RU2795146C2 |
Изобретение относится к соединениям на основе пептидов, включающим в себя трехчленные циклы, содержащие гетероатом, которые эффективно и селективно ингибируют специфические активности N-концевых нуклеофильных (Ntn) гидролаз, связанных с протеасомой. Соединения на основе пептидов содержат эпоксид и функционализированы у N-конца. Соединения на основе пептидов проявляют противовоспалительные свойства и ингибирование пролиферации клеток. Возможно пероральное введение указанных ингибиторов протеасомы на основе пептидов благодаря их биодоступности. 7 н. и 27 з.п. ф-лы, 14 пр.
1. Соединение, имеющее структуру формулы (I), или его фармацевтически приемлемая соль:
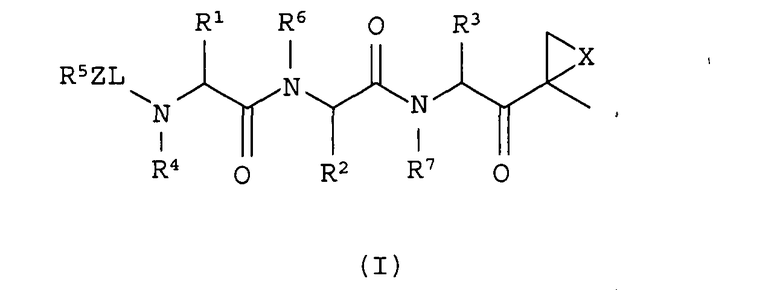
в которой L выбран из С=O;
Х выбран из О;
Z отсутствует;
каждый из R1 и R2 независимо друг от друга выбран из водорода, С1-6-алкила, С1-6-карбоциклоалкила, С1-6-алкоксиалкила, С1-6-гидроксиалкила, С1-6-цианоалкила, C1-6-аралкила, С2-6-алкинила, и С1-6-гетероаралкила, где гетероарил представляет собой 5- или 6-членное кольцо, содержащее один или два гетероатома, выбранных из N или S;
R3 выбран из С1-6-алкила и С1-6-аралкила, необязательно замещенного галогеном, С1-6-алкилом, С1-6-алкокси или СF3;
R4 представляет собой водород;
R5 представляет собой изоксазол, замещенный амидом, C1-6-алкилом, C1-6-карбоциклоалкилом, С1-6-алкокси, С1-6-гидроксиалкилом, C1-6-алкоксиалкилом, C1-6-гетероциклоалкилом, где гетероцикл представляет собой неароматическое 6-членное кольцо, содержащее N и О и дополнительно необязательно замещенное C1-6-алкилом, неароматическим 4-членным кольцом, содержащим один N,
или неароматическим 5-членным кольцом, содержащим один N, или С1-6-гетероаралкилом, где гетероарил содержит три атома N; фуран или тиазол, необязательно замещенный С1-6-алкилом; и R6 и R7 представляет собой водород.
2. Соединение по п.1, в котором любой из R1, R2 и R3 независимо друг от друга представляет собой C1-6-алкил.
3. Соединение по п.2, в котором любой из R1, R2 и R3 независимо друг от друга выбран из метила, этила, пропила, изопропила, бутила, втор-бутила и изобутила.
4. Соединение по п.1, в котором любой из R1, R2 и R3 независимо друг от друга представляет собой пропаргил.
5. Соединение по п.1, в котором любой из R1, и R2 независимо друг от друга представляет собой C1-6-гидроксиалкил.
6. Соединение по п.5, в котором любой из R1 и R2 независимо друг от друга выбран из гидроксиметила и гидроксиэтила.
7. Соединение по п.1, в котором любой из R' и R2 независимо друг от друга представляет собой С1-6-алкоксиалкил.
8. Соединение по п.7, в котором любой из R1 и R2 независимо друг от друга выбран из метоксиметила и метоксиэтила.
9. Соединение по п.1, в котором любой из R1 и R2 независимо друг от друга представляет собой С1-6-гетероаралкил.
10. Соединение по п.1, в котором по меньшей мере один из R1 и R2 выбран из С1-6-гидроксиалкила и С1-6-алкоксиалкила.
11. Соединение по п.10, в котором по меньшей мере один из R1 и R2 представляет собой С1-6-алкоксиалкил.
12. Соединение по п.11, в котором по меньшей мере один из R1 и R2 выбран из метоксиметила и метоксиэтила.
13. Соединение по п.1, в котором R3 выбран из C1-6-алкила и C1-6-аралкила.
14. Соединение по п.13, в котором R3 представляет собой С1-6-алкил.
15. Соединение по п.14, в котором R3 выбран из метила, этила, изопропила, втор-бутила и изобутила.
16. Соединение по п.15, в котором R3 представляет собой изобутил.
17. Соединение по п.13, в котором R3 представляет собой C1-6-аралкил.
18. Соединение по п.17, в котором R представляет собой фенилметил.
19. Соединение по п.1, в котором R5 представляет собой изоксазол-3-ил или изоксазол-5-ил.
20. Соединение по п.19, в котором R5 представляет собой изоксазол-3-ил, содержащий заместитель в 5-положении.
21. Соединение по п.19, в котором R5 представляет собой изоксазол-5-ил, содержащий заместитель в 3-положении.
22. Соединение по п.20 или 21, в котором заместитель выбран из метила, этила, изопропила и циклопропилметила.
23. Соединение по п.20 или 21, в котором заместитель выбран из C1-6-гетероаралкила и С1-6-гетероциклоалкила.
24. Соединение по п.23, в котором заместитель представляет собой 1,2,4-триазол-5-илметил.
25. Соединение по п.23, в котором заместитель представляет собой азетидин-1-илметил.
26. Соединение по п.23, в котором заместитель выбран из C1-6-алкоксигруппы и C1-6-алкоксиалкила.
27. Соединение по п.26, в котором заместитель выбран из метоксигруппы, этоксигруппы, метоксиметила и метоксиэтила.
28. Соединение, выбранное из
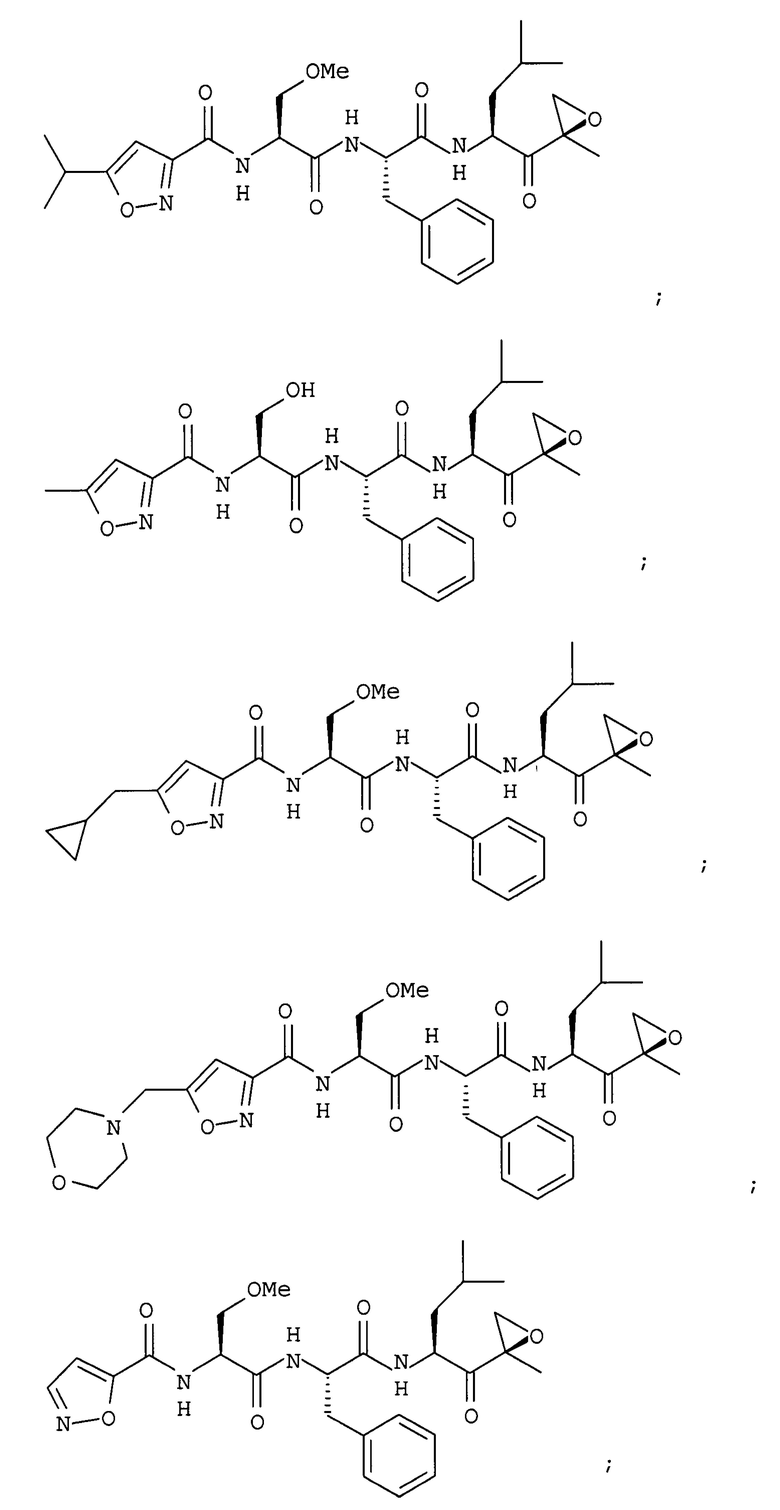
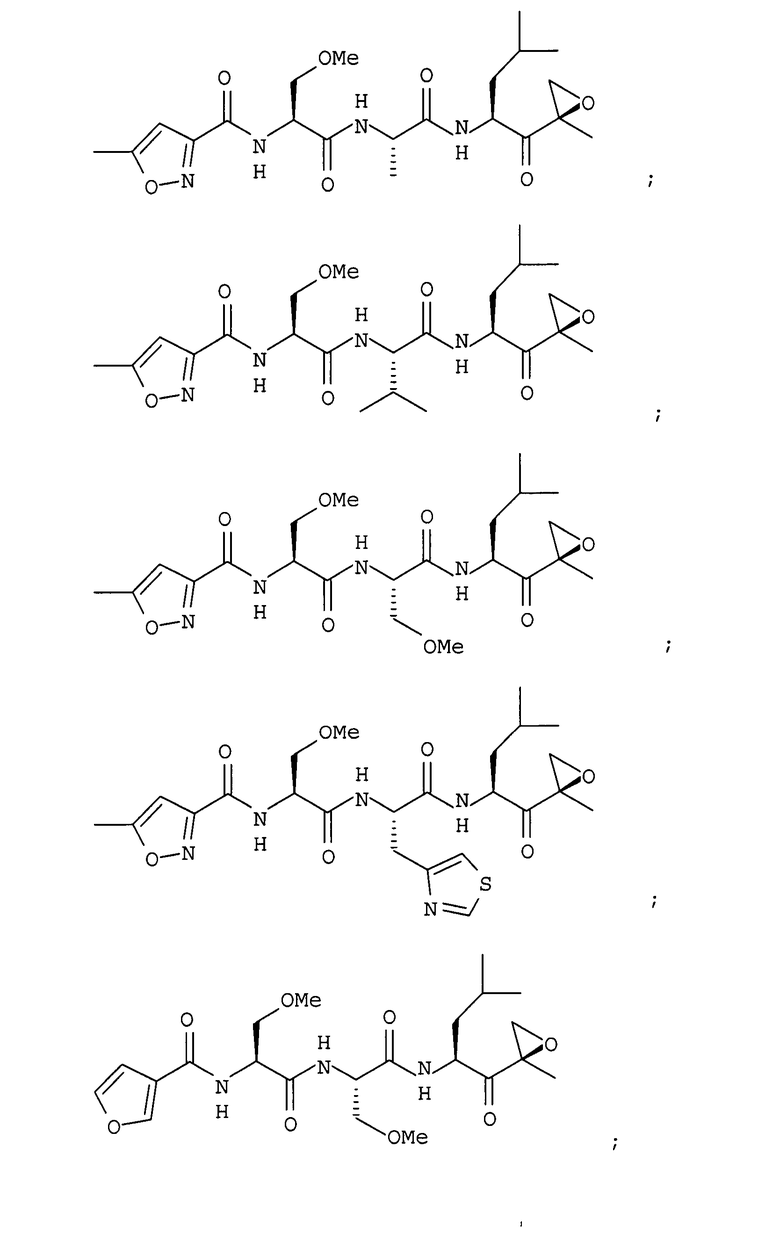
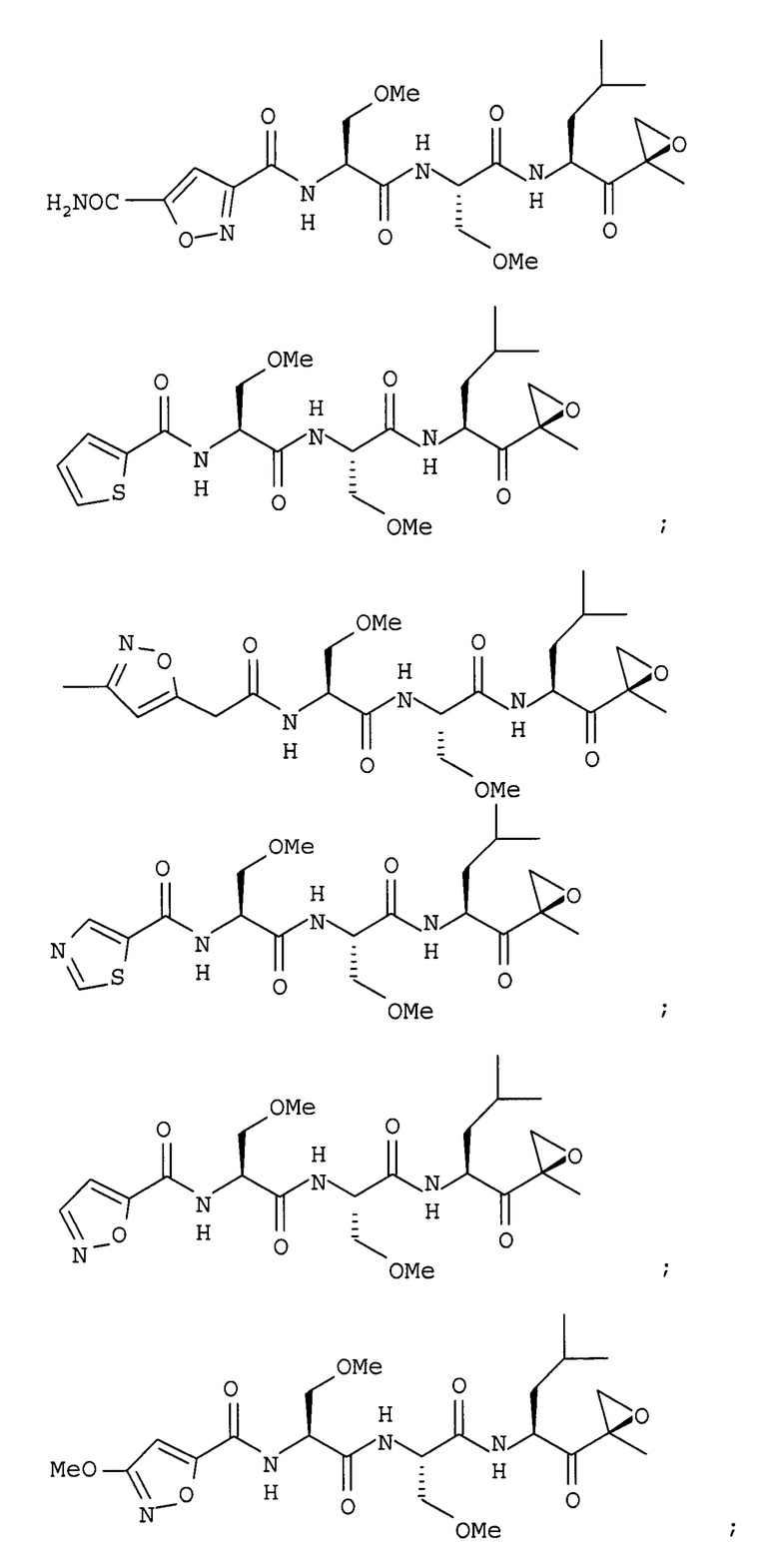
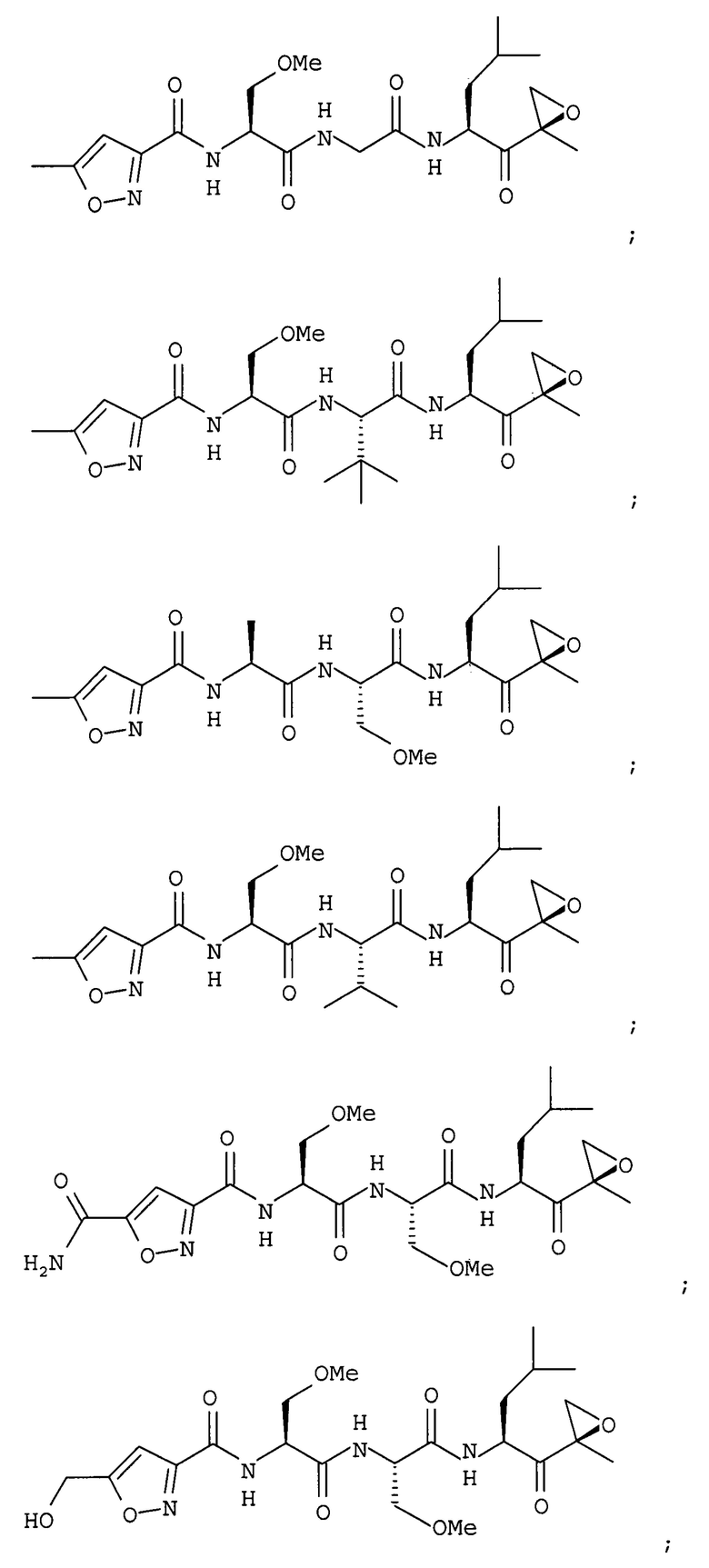
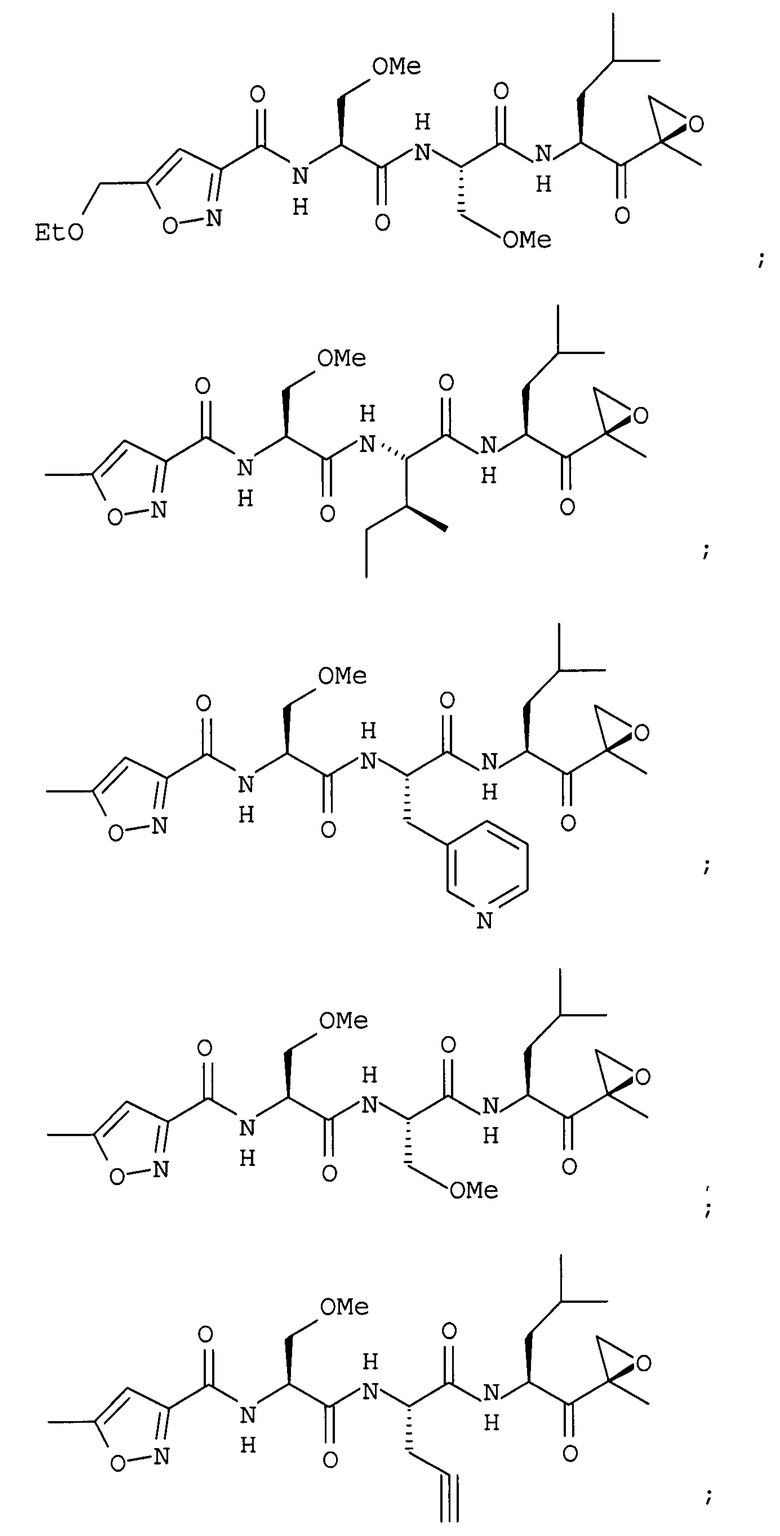
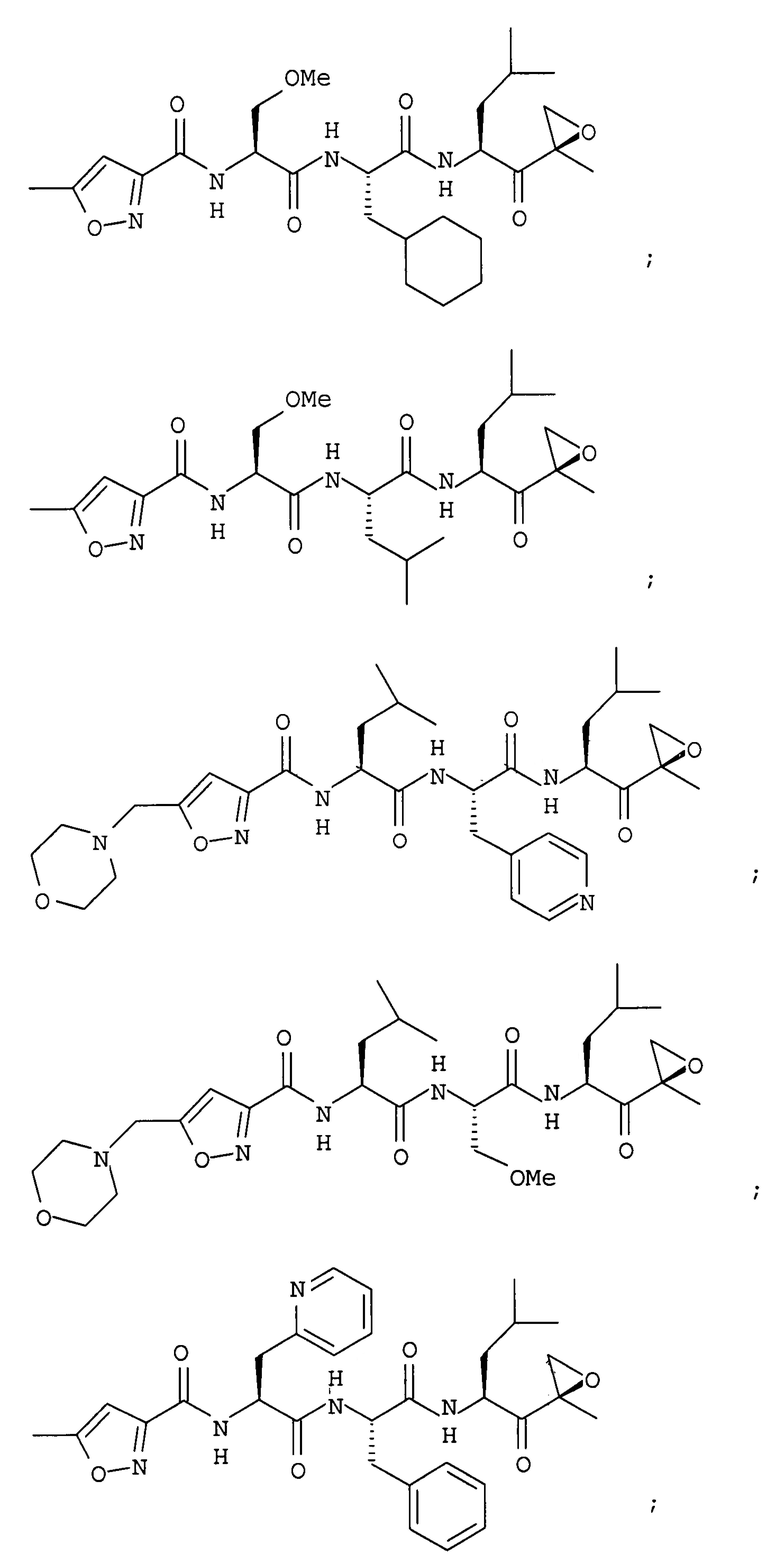
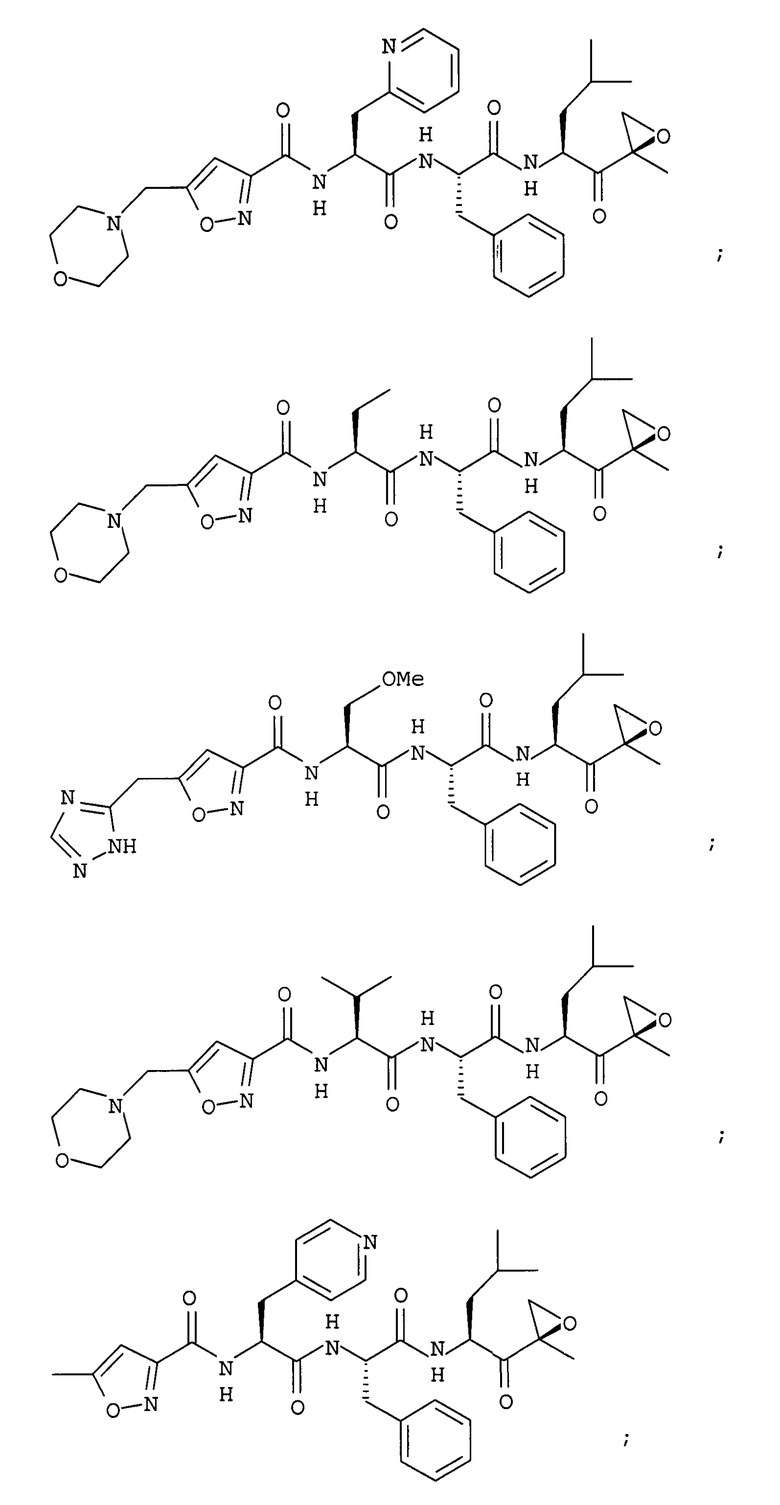
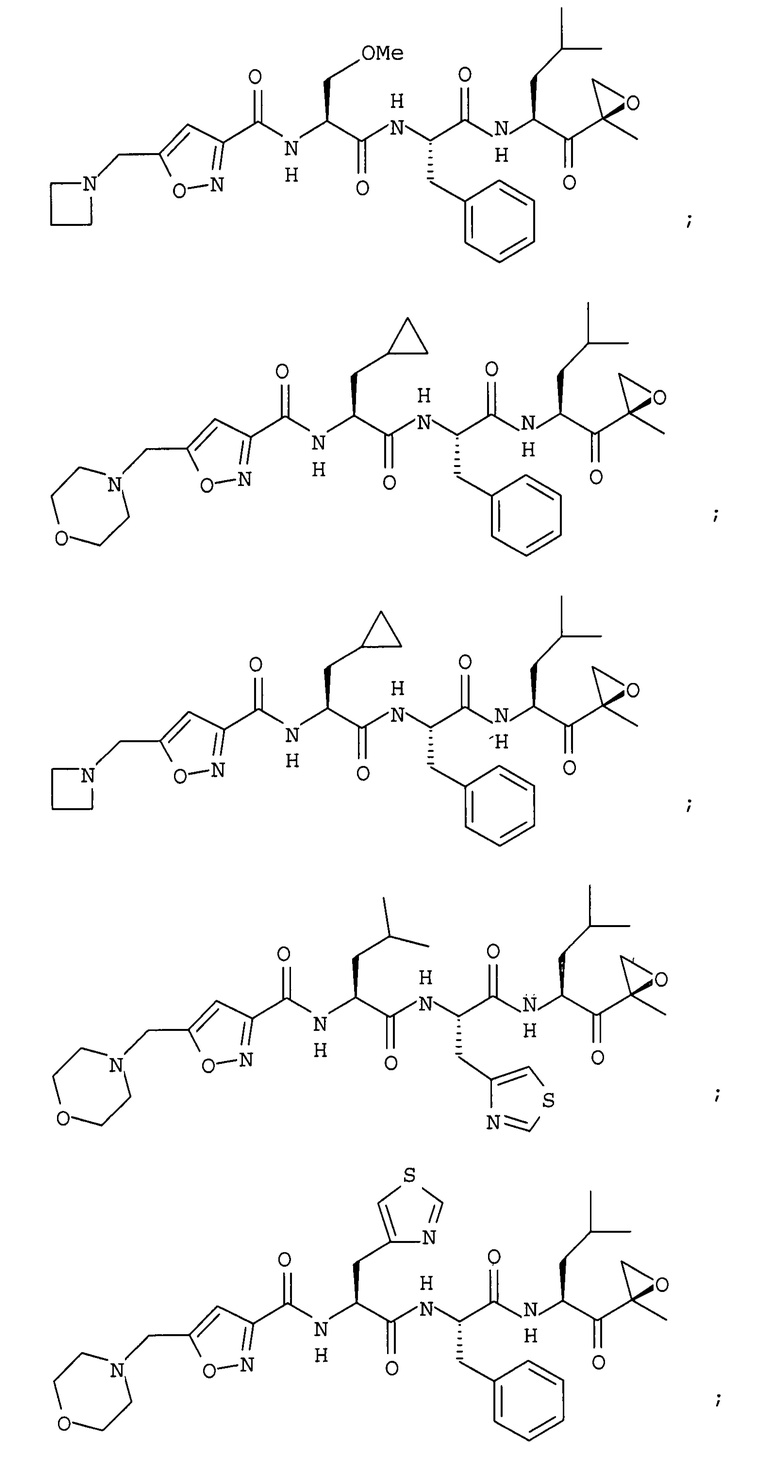
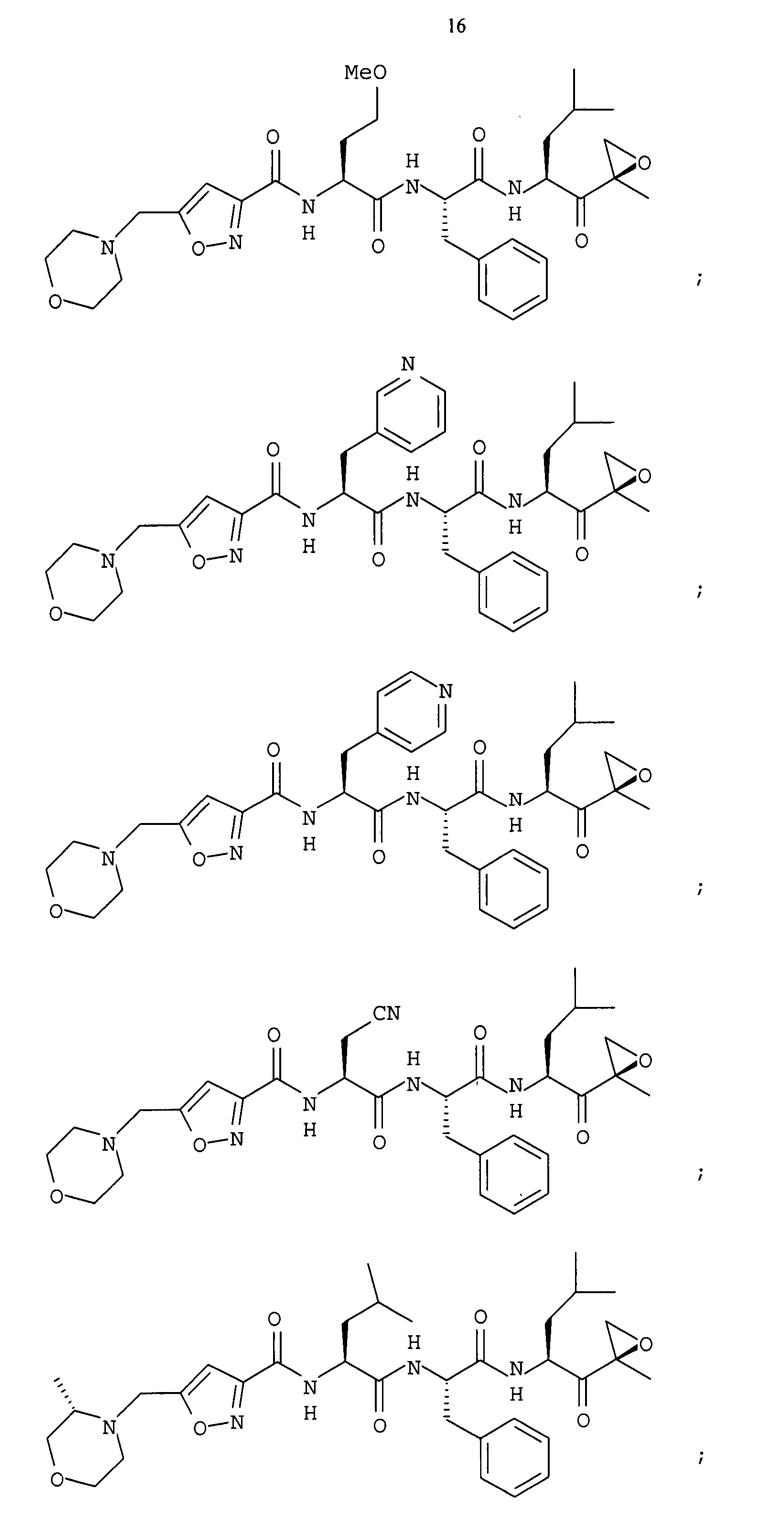
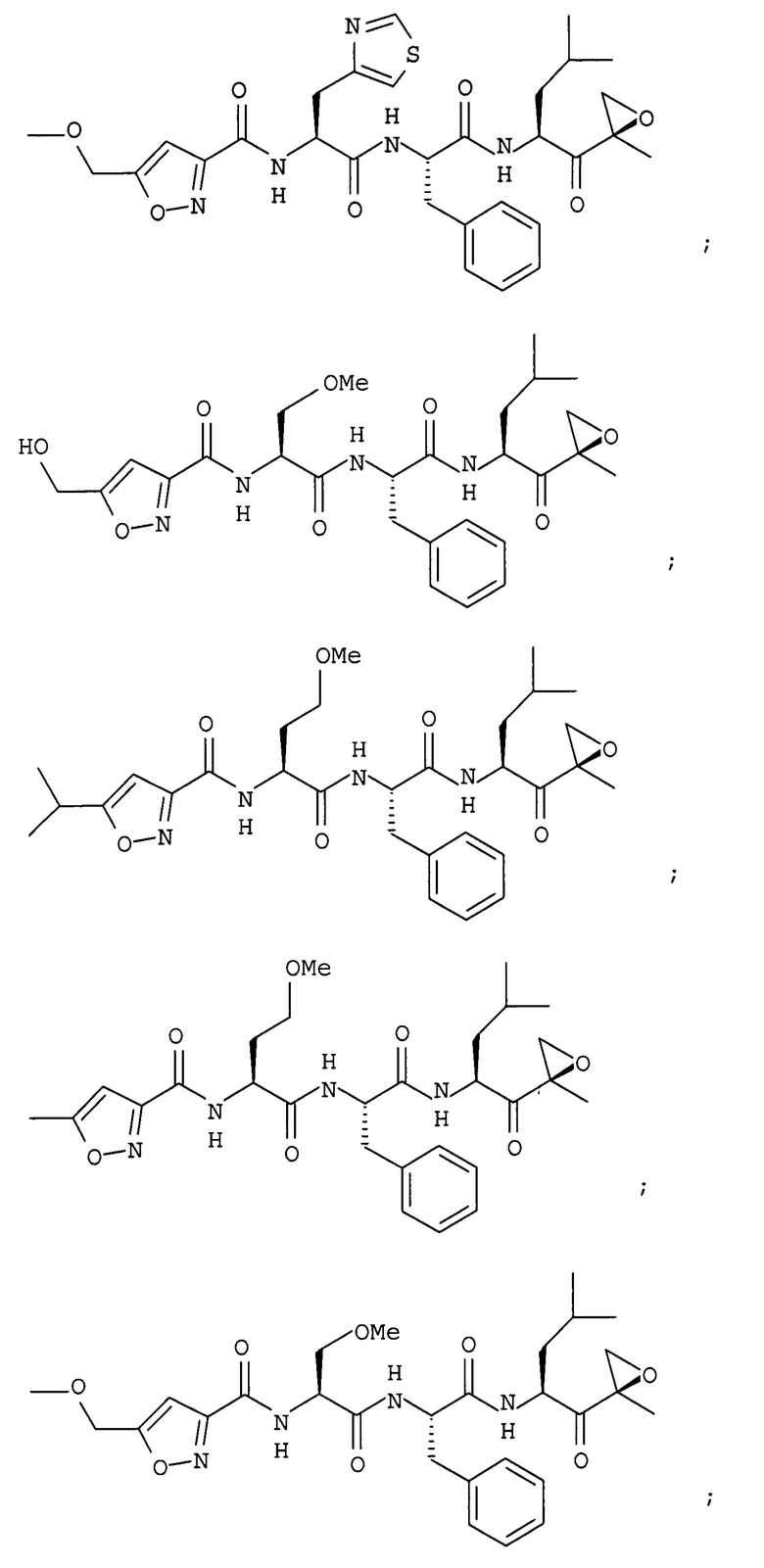
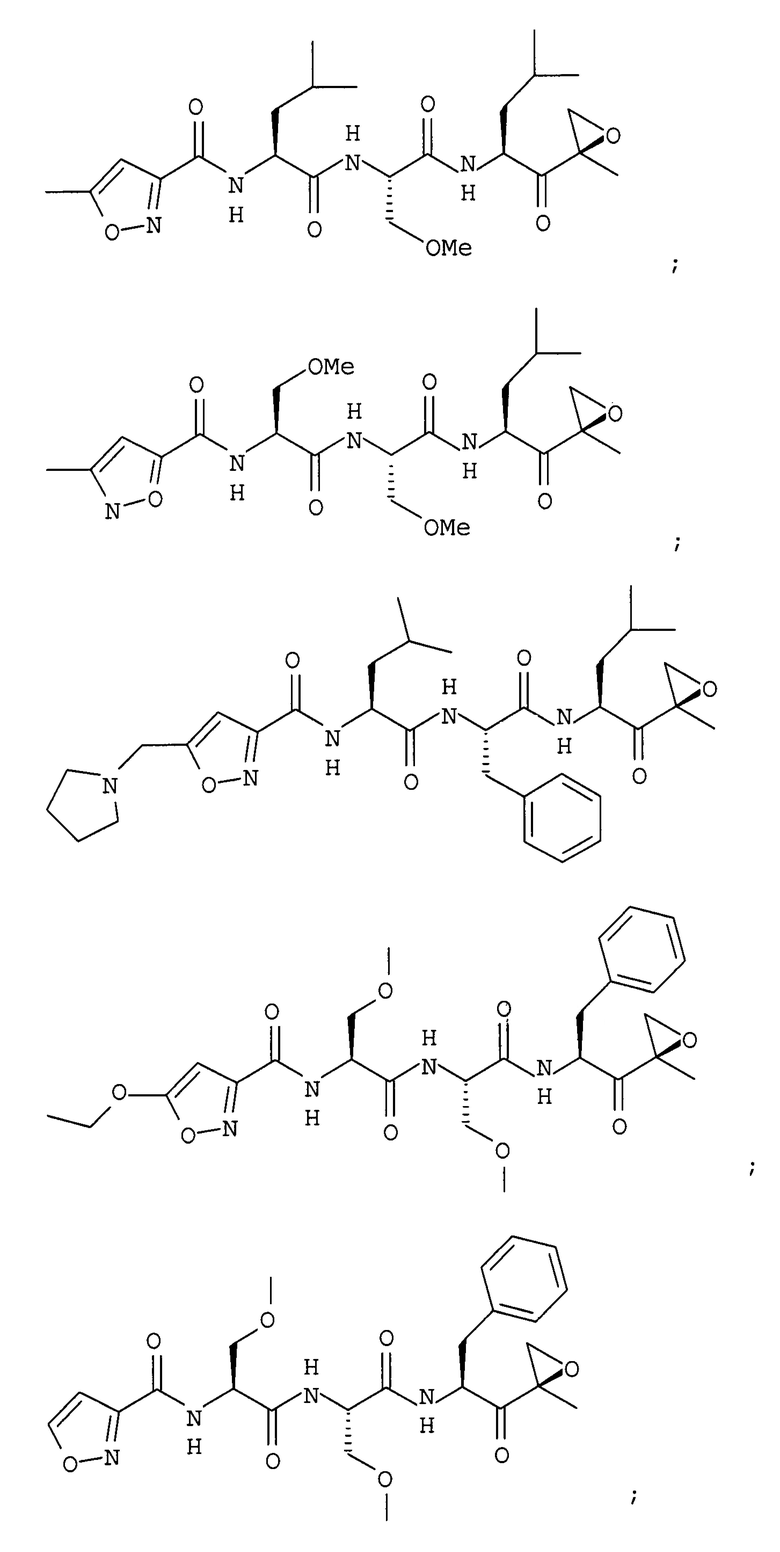
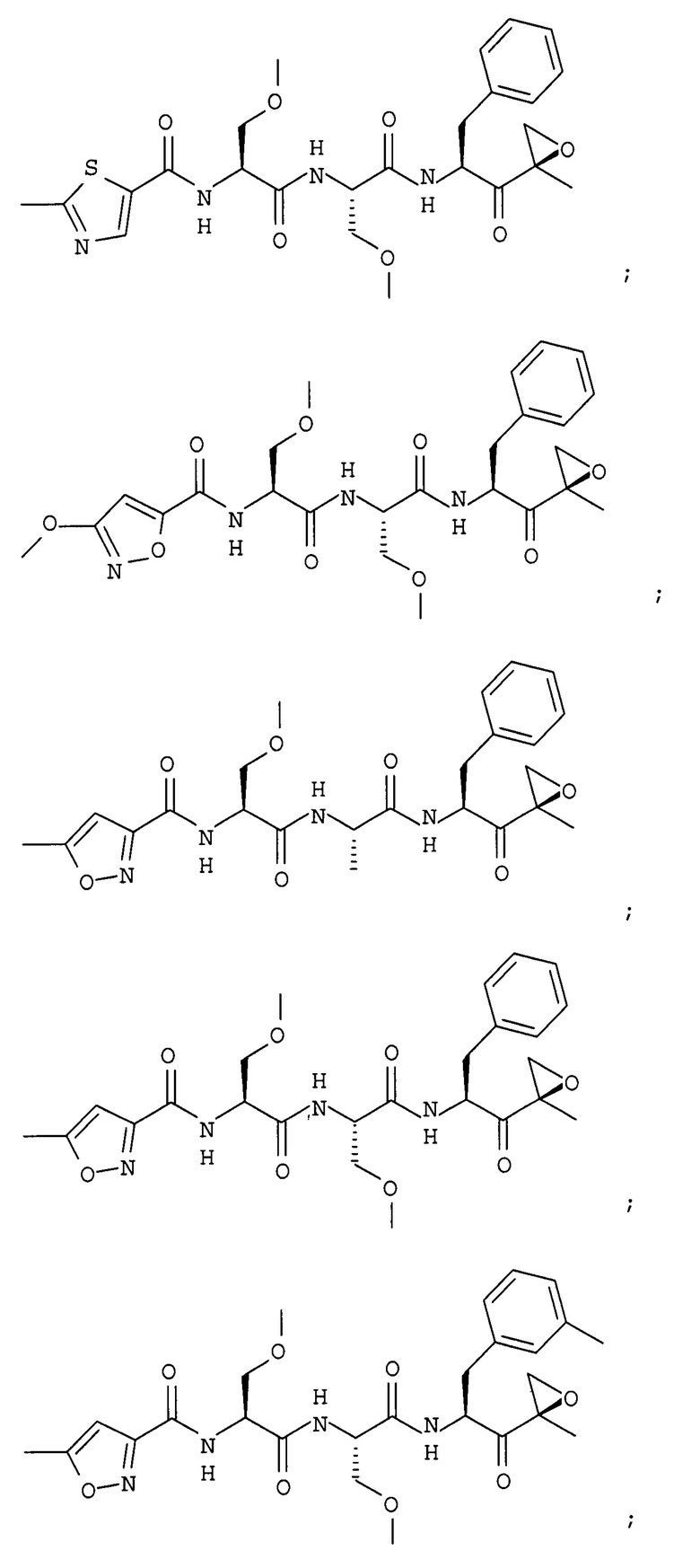
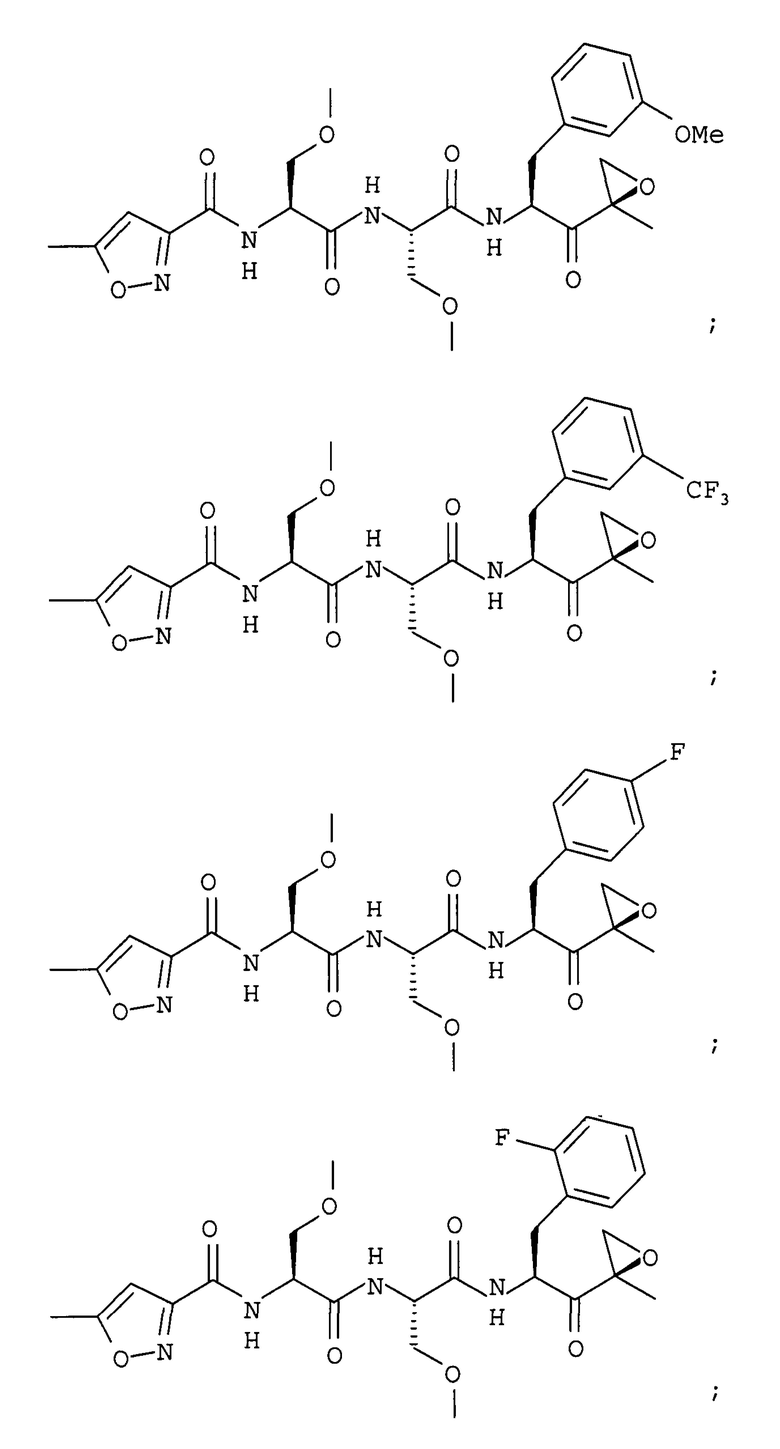
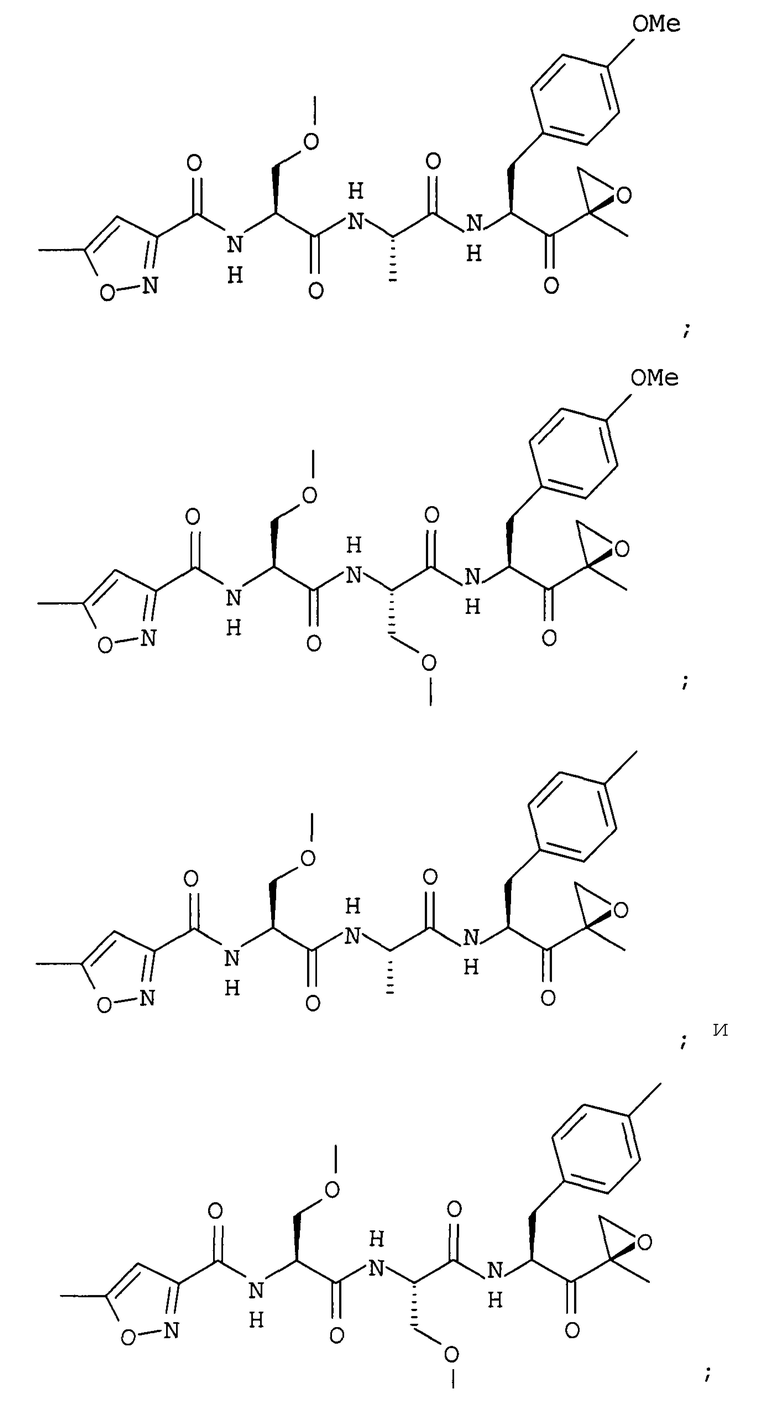
29. Фармацевтическая композиция, обладающая ингибирующим действием в отношении 20S протеасомы и содержащая терапевтически эффективное количество соединения по любому из пп.1-28 и фармацевтически приемлемый разбавитель или носитель.
30. Фармацевтическая композиция по п.29, которая биодоступна перорально.
31. Способ лечения рака, предусматривающий введение терапевтически эффективного количества фармацевтической композиции по п.30.
32. Соединение формулы
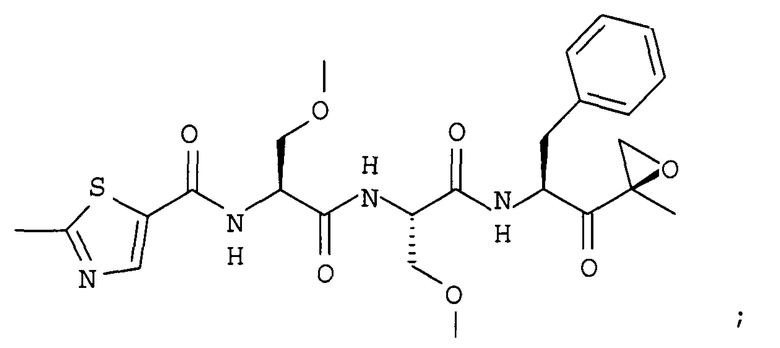
или его фармацевтически приемлемая соль.
33. Фармацевтическая композиция, обладающая ингибирующим действием в отношении 2OS протеасомы и содержащая терапевтически эффективное количество соединения по п.32 и фармацевтически приемлемый разбавитель или носитель.
34. Способ лечения рака, предусматривающий введение терапевтически эффективного количества фармацевтической композиции по п.33.
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ ФЕРМЕНТОВ | 2005 |
|
RU2384585C2 |
| α-КЕТОАМИДНЫЕ ИНГИБИТОРЫ 20S ПРОТЕАСОМЫ | 1999 |
|
RU2192429C2 |
| US 6831099 B1, 14.12.2004 | |||
| MYUNG, JAYHYUK ET AL.: "Lack of proteasome active site allostery as revealed by subunit-specific inhibitors" MOLECULAR CELL, 2001, vol.7, no.2, pages 411-420 | |||
| ELOFSSON M | |||
| ET AL.: "TOWARDS SUBUNTT-SPECIFIC PROTEASOME INHIBITORS: SYNTHESIS AND EVALUATION OF PEPTIDE | |||

