ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение предусматривает соединения, которые являются ингибиторами фермента PDE1, и их применение в качестве лекарственного препарата, в частности, для лечения нейродегенеративных расстройств и психических расстройств. Настоящее изобретение также предусматривает фармацевтические композиции, содержащие соединения согласно настоящему изобретению, и способы лечения расстройств с использованием соединений согласно настоящему изобретению.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
В настоящей заявке ссылки на различные публикации приводятся во всей полноте. Раскрытие этих публикаций тем самым включено в настоящую заявку посредством ссылки для более полного описания состояния уровня техники, к которому принадлежит настоящее изобретение.
Вторичные посредники циклические нуклеотиды (cN), как циклический аденозинмонофосфат (cAMP) и циклический гуанозинмонофосфат (cGMP), играют основную роль во внутриклеточном каскаде передачи сигналов, регулируя cN-зависимые протеинкиназы (РКА и PKG), EPAC (активируемые cAMP обменные белки), фосфопротеинфосфатазы и/или cN-активируемые катионные каналы. В нейронах это включает активацию cAMP- и cGMP-зависимых киназ и последующее фосфорилирование белков, участвующих в ранней регуляции синаптической передачи, а также в нейрональной дифференциации и выживании. Внутриклеточные концентрации cAMP и cGMP строго регулируются скоростью биосинтеза при помощи циклаз и скоростью деградации при помощи фосфодиэстераз (PDE, EC 3.1.4.17). PDE представляют собой гидролазы, включающие два металла, которые инактивируют cAMP/cGMP посредством каталитического гидролиза 3'-сложноэфирной связи, образуя неактивный 5-монофосфат. Поскольку PDE являются единственными средствами деградации циклических нуклеотидов cAMP и cGMP в клетках, PDE играют незаменимую роль в передаче сигналов циклическими нуклеотидами. Виды каталитической активности PDE обеспечивают разрушение cN в пределах диапазона концентраций во всех клетках, а их разнообразные регуляторные механизмы обеспечивают интеграцию и взаимовлияние с бесчисленными сигнальными путями. Конкретные PDE направлены на отдельные компартменты в клетках, где они контролируют уровень cN и формируют микросреду для множества сигналосом cN (Sharron H. Francis, Mitsi A. Blount, and Jackie D. Corbin. Physiol Rev 2011, 91: 651-690).
На основе субстратной специфичности семейства PDE можно разделить на три группы: 1) cAMP-специфические PDE, которые включают PDE4, PDE7 и PDE8, 2) cGMP-селективные ферменты - PDE5 и PDE9, и 3) двухсубстратные PDE - PDE1, PDE2, PDE3, а также PDE10 и PDE11.
Ранее обозначаемая как кальмодулин-стимулируемая PDE (CaM-PDE), PDE1 является уникальной в том, что она регулируется в зависимости от Ca2+ посредством кальмодулина (CaM, Ca2+-связывающий белок весом 16 кДа) в комплексе с четырьмя Ca2+ (для ознакомления см. Sharron Н. Francis, Mitsi А. Blount, and Jackie D. Corbin. Physiol Rev 2011, 91: 651-690). Таким образом, это семейство представляет вызывающую интерес регуляторную связь между циклическими нуклеотидами и внутриклеточным Ca2+. Семейство PDE1 кодируется тремя генами: PDE1A (картирован на хромосоме человека 2q32), PDE1B (положение на хромосоме человека, hcl: 12q13) и PDE1C (hcl: 7p14.3). Они характеризуются альтернативными промоторами и посредством альтернативного сплайсинга образуют множество белков, которые отличаются по своим регуляторным свойствам, сродству к субстрату, специфическим активностям, константам активации CaM, распределением в тканях и молекулярным весом. Идентифицировано более 10 изоформ у человека. Их молекулярный вес находится в пределах от 58 до 86 кДа на мономер. N-концевой регуляторный домен, который содержит два Ca2+/CaM-связывающих домена и два сайта фосфорилирования, дифференцирует их соответствующие белки и модулирует их биохимические функции. PDE1 представляет собой двухсубстратную PDE, а подтип PDE1C обладает равной активностью по отношению к cAMP и cGMP (Km ≈ 1-3 мкМ), в то время как подтипы PDE1A и PDE1B характеризуются предпочтением в отношении cGMP (Km для cGMP ≈ 1-3 мкМ, а для cAMP ≈ 10-30 мкМ).
Подтипы PDE1 находятся в большом количестве в мозге и расположены главным образом в полосатом теле (PDE1B), гиппокампе (PDE1A) и коре (PDE1A), и такое расположение консервативно для всех видов (Amy Bernard et al. Neuron 2012, 73, 1083-1099). В коре PDE1A присутствует главным образом в глубоких слоях коры 5 и 6 (выходных слоях), и используется в качестве специфического маркера для глубоких слоев коры. Ингибиторы PDE1 повышают уровни вторичных посредников cN, приводя к повышенной возбудимости нейронов.
Таким образом, PDE1 представляет собой терапевтическую мишень для регуляции внутриклеточных сигнальных путей, предпочтительно, в нервной системе, а ингибиторы PDE1 могут повышать уровни вторичных посредников cAMP/cGMP, приводя к модуляции нейрональных процессов и к экспрессии генов, связанных с нейрональной пластичностью, нейротропных факторов и нейропротекторных молекул. Такие свойства повышения нейрональной пластичности вместе с модуляцией синаптической передачи делают ингибиторов PDE1 хорошими кандидатами в качестве терапевтических средств для многих неврологических и психических состояний. Оценка ингибиторов PDE1 в животных моделях (для ознакомления см., например, Blokland et al. Expert Opinion on Therapeutic Patents (2012), 22(4), 349-354 и Medina, A.E. Frontiers in Neuropharmacology (2011), 5(Feb.), 21) свидетельствовала о потенциале для терапевтического применения ингибиторов PDE1 при неврологических расстройствах, таких как, например, болезни Альцгеймера, Паркинсона и Гентингтона, и при психических расстройствах, таких как, например, синдром дефицита внимания и гиперактивности (ADHD), синдром беспокойных ног, депрессия, нарколепсия, нарушение когнитивных функций и нарушение когнитивных функций, ассоциированное с шизофренией (CIAS). Также были представлены заявки на патент, в которых было заявлено, что ингибиторы PDE1 применимы при болезнях, которые могут быть облегчены усилением передачи сигнала при участии прогестерона, таких как половая дисфункция у женщин, например, WO-2010065153.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Ферменты PDE1 экспрессируются в центральной нервной системе (CNS), что делает это семейство генов перспективным источником новых мишеней для лечения психических и нейродегенеративных расстройств.
Соединения согласно настоящему изобретению могут обеспечивать альтернативы современным зарегистрированным лекарственным средствам для лечения нейродегенеративных и/или психических расстройств, которые не являются эффективными для всех пациентов. Таким образом, сохраняется потребность в альтернативных способах лечения.
Целью настоящего изобретения является получение соединений, которые являются ингибиторами PDE1, и в связи с этим применимы для лечения нейродегенеративных и психических расстройств. Согласно предпочтительному варианту осуществления эти соединения являются селективными ингибиторами PDE1.
Соответственно, настоящее изобретение относится к соединениям формулы (I):
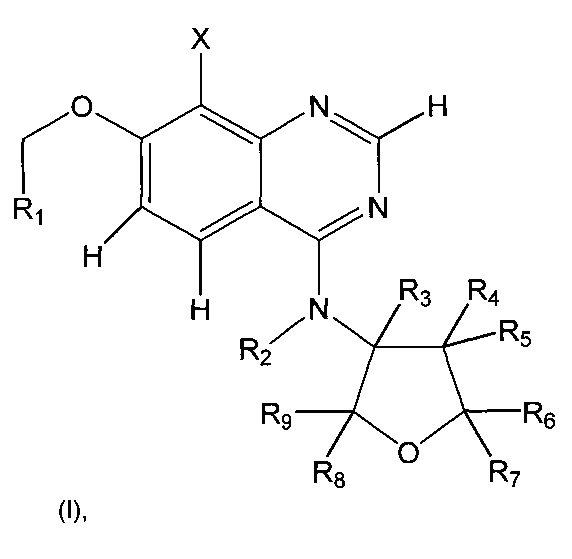
где
X представляет собой галоген, предпочтительно фтор, или хлор, или бром;
R1 выбран из группы, состоящей из H и C1-С3алкила, где алкил необязательно может быть замещен один, два или три раза фтором;
R2 выбран из группы, состоящей из H и С1-С4алкила, где С1-С4алкил необязательно замещен один или несколько раз одним или несколькими заместителями,
R3 выбран из группы, состоящей из H и C1-С6алкила, где C1-С6алкил необязательно замещен один или несколько раз одним или несколькими заместителями,
R4 и R5 независимо друг от друга выбраны из группы, состоящей из H, C1-С6алкила, необязательно замещенного один или несколько раз одним или несколькими заместителями, C3-С6циклоалкила, фтора, хлора, гидрокси и алкокси,
R6 и R7 независимо друг от друга выбраны из группы, состоящей из H и C1-С6алкила, где C1-С6алкил необязательно замещен один или несколько раз одним или несколькими заместителями,
R8 и R9 независимо друг от друга выбраны из группы, состоящей из H и C1-С6алкила, где C1-С6алкил необязательно замещен один или несколько раз одним или несколькими заместителями,
и фармацевтически приемлемые кислотно-аддитивные соли соединения I, рацемические смеси соединения I или соответствующий энантиомер и/или диастереоизомер соединения I и полиморфные формы соединения I, а также таутомерные формы соединения I.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Согласно первому варианту осуществления (E1) настоящее изобретение относится к соединениям формулы (I) (соединение I):
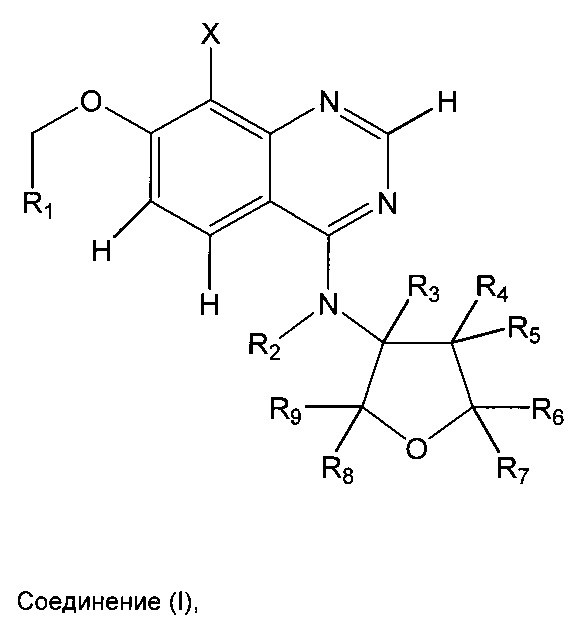
где
R1 выбран из группы, состоящей из H и C1-С3алкила, где алкил необязательно может быть замещен один, два или три раза фтором;
R2 выбран из группы, состоящей из H и С1-С4алкила,
при этом R2, если R2 представляет собой С1-С4алкил, может образовывать насыщенное пятичленное алифатическое кольцо с R9,
при этом С1-С4алкил необязательно замещен один или несколько раз одним или несколькими заместителями, независимо выбранными из группы, состоящей из фенила, моноциклического 5- или 6-членного гетероарила, C3-С6циклоалкила, фтора, хлора и алкокси вида -OR10,
где R10 представляет собой C1-С5алкил;
R3 выбран из группы, состоящей из H и C1-С6алкила,
где C1-С6алкил необязательно замещен один или несколько раз одним или несколькими заместителями, независимо выбранными из группы, состоящей из фенила, моноциклического 5- или 6-членного гетероарила, C3-С6циклоалкила, фтора, хлора и алкокси вида -OR10,
где R10 представляет собой C1-С5алкил;
R4 и R5 независимо друг от друга выбраны из группы, состоящей из H, C1-С6алкила, C3-С6циклоалкила, фтора, хлора, гидрокси и алкокси вида -OR10,
при этом C1-С6алкил необязательно замещен один или несколько раз одним или несколькими заместителями, независимо выбранными из группы, состоящей из фенила, моноциклического 5- или 6-членного гетероарила, C3-С6циклоалкила, фтора, хлора и алкокси вида -OR10,
где R10 представляет собой C1-С5алкил;
R6 и R7 независимо друг от друга выбраны из группы, состоящей из H и C1-С6алкила,
где C1-С6алкил необязательно замещен один или несколько раз одним или несколькими заместителями, независимо выбранными из группы, состоящей из C3-С6циклоалкила, фтора, хлора и алкокси вида -OR10,
где R10 представляет собой C1-С5алкил;
R8 и R9 независимо друг от друга выбраны из группы, состоящей из H и C1-С6алкила,
при этом R9, если R9 представляет собой C1-С6алкил, может образовывать насыщенное алифатическое пятичленное кольцо с R2,
при этом C1-С6алкил необязательно замещен один или несколько раз одним или несколькими заместителями, независимо выбранными из группы, состоящей из C3-С6циклоалкила, фтора, хлора и алкокси вида -OR10,
где R10 представляет собой C1-С5алкил;
и/или фармацевтически приемлемые кислотно-аддитивные соли соединения I, рацемические смеси соединения I или соответствующий энантиомер и/или оптический изомер соединения I и полиморфные формы соединения I, а также таутомерные формы соединения I.
Согласно варианту осуществления (E2) по (E1) R2 представляет собой H или -CH3.
Согласно варианту осуществления (E3) по любому из (E1) и (E2) по меньшей мере один из R6 и R7 представляет собой H.
Согласно варианту осуществления (E4) по (E3) R6 и R7 представляют собой H.
Согласно варианту осуществления (E5) по (E1) по меньшей мере четыре из R3-R9 представляют собой H.
Согласно варианту осуществления (E6) по (E1), если любой из R3, R4 или R5 представляет собой алкил, то не более чем один из них замещен не более одного раза фенилом или моноциклическим 5- или 6-членным гетероарилом.
Согласно варианту осуществления (E7) по (E1) R2 и R9 образуют пятичленную насыщенную алифатическую кольцевую систему.
Согласно варианту осуществления (E8) по (E1) R1 замещен один раз фтором.
Согласно варианту осуществления (E9) по (E1) R1 замещен два раза фтором.
Согласно варианту осуществления (E10) по (E1) R1 замещен три раза фтором.
Согласно варианту осуществления (Е11) по (E1) X представляет собой фтор.
Согласно варианту осуществления (Е12) по (E1) X представляет собой хлор.
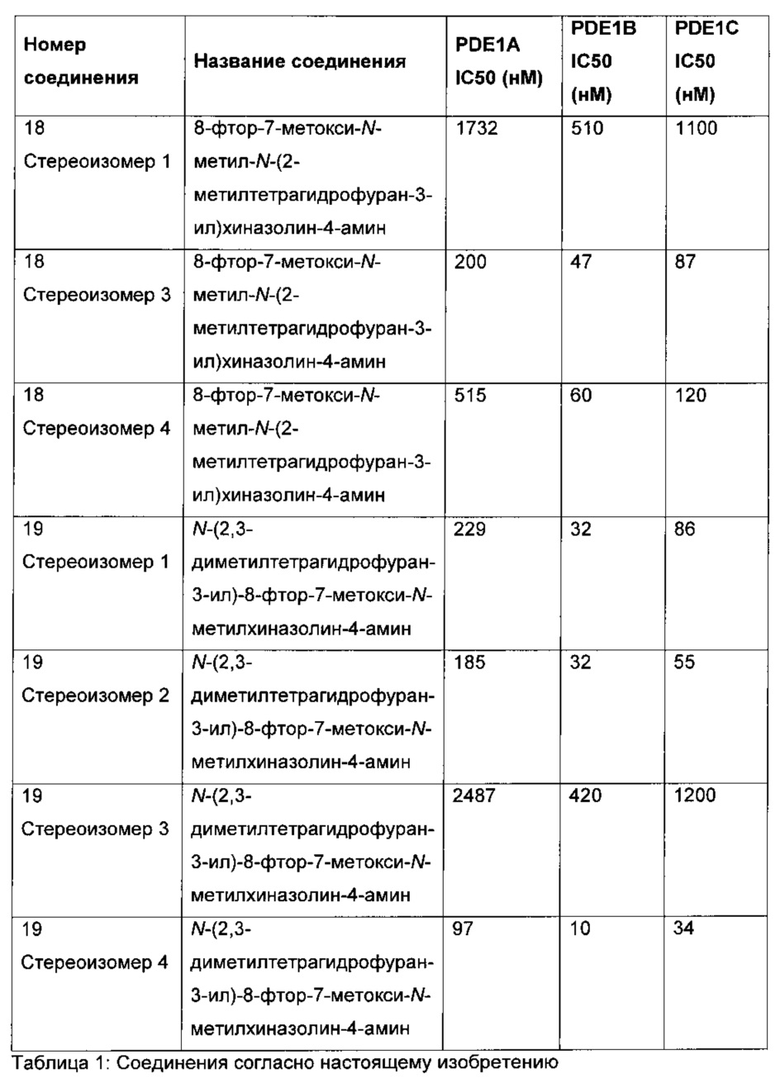
Согласно варианту осуществления (Е13) по любому из (E1)-(E12) соединение выбирают из группы соединений, приведенных в таблице 1.
Согласно варианту осуществления (Е14) по любому из (E1)-(E13) соединение является ингибитором PDE1A.
Согласно варианту осуществления (Е15) по любому из (E1)-(Е13) соединение является ингибитором PDE1B.
Согласно варианту осуществления (Е16) по любому из (E1)-(Е13) соединение является ингибитором PDE1C.
Согласно варианту осуществления (Е17) соединение по любому из (E1)-(Е16) предназначено для использования в качестве лекарственного препарата.
Согласно варианту осуществления (Е18) соединение по любому из (E1)-(Е16) предназначено для использования при лечении синдрома дефицита внимания и гиперактивности (ADHD).
Вариант осуществления (Е19): Фармацевтическая композиция, содержащая соединение по любому из (E1)-(Е16) и один или несколько фармацевтически приемлемых носителей.
Вариант осуществления (Е20): Фармацевтическая композиция по (Е19) для использования в способе лечения синдрома дефицита внимания и гиперактивности (ADHD).
Вариант осуществления (Е21): Соединение по любому из (E1)-(Е16) для использования в способе лечения синдрома дефицита внимания и гиперактивности (ADHD).
Вариант осуществления (Е22): Соединение по любому из (E1)-(Е16) для получения лекарственного препарата для использования в лечении синдрома дефицита внимания и гиперактивности (ADHD).
Вариант осуществления (Е23): Способ лечения субъекта, страдающего синдромом дефицита внимания и гиперактивности (ADHD), причем способ включает введение эффективного количества соединения по любому из (E1)-(Е16).
Вариант осуществления (Е24): Фармацевтическая композиция по (Е19) для использования в способе лечения нейродегенеративного расстройства.
Вариант осуществления (Е25): Соединение по любому из (E1)-(Е16) для использования в способе лечения нейродегенеративного расстройства.
Вариант осуществления (Е26): Соединение по любому из (E1)-(Е16) для получения лекарственного препарата для использования в лечении нейродегенеративного расстройства.
Вариант осуществления (Е27): Способ лечения субъекта, страдающего нейродегенеративным расстройством, причем способ включает введение эффективного количества соединения по любому из (E1)-(Е16).
Согласно варианту осуществления (Е28) по любому из вариантов осуществления (Е24)-(Е27) нейродегенеративное расстройство выбирают из группы, состоящей из болезни Альцгеймера, болезни Паркинсона и болезни Гентингтона, или для лечения психического расстройства, такого как синдром дефицита внимания и гиперактивности (ADHD), депрессия, нарколепсия, нарушение когнитивных функций и нарушение когнитивных функций, ассоциированное с шизофренией (CIAS).
Вариант осуществления (Е29): Применение соединения по любому из пп. 1-7 формула изобретения при производстве лекарственного препарата для лечения нейродегенеративного расстройства, такого как болезни Альцгеймера, болезни Паркинсона и болезни Гентингтона, или для лечения психического расстройства, такого как синдром дефицита внимания и гиперактивности (ADHD), депрессия, нарколепсия, нарушение когнитивных функций и нарушение когнитивных функций, ассоциированное с шизофренией (CIAS), или мозгового нарушения, такого как синдром беспокойных ног.
Вариант осуществления (E30): Согласно варианту осуществления (E30) по (E1) R1 представляет собой H.
ОПРЕДЕЛЕНИЯ
ФЕРМЕНТЫ PDE1
Семейство изозимов PDE1 включает изоформы PDE1, образованные несколькими вариантами сплайсинга. Оно включает три подтипа, PDE1A, PDE1B и PDE1C, которые дополнительно подразделяются на различные изоформы. В контексте настоящего изобретения PDE1 и ферменты PDE1 являются синонимами и относятся к ферментам PDE1A, PDE1B и PDE1C, а также к их изоформам.
ЗАМЕСТИТЕЛИ
Используемые в контексте настоящего изобретения выражения "галогено" и "галоген" используются взаимозаменяемо и относятся к фтору, хлору, брому или йоду.
Выражения "C1-С3алкил", "С1-С4алкил", "С1-С5алкил" и "C1-С6алкил" относятся к насыщенному углеводороду с прямой или разветвленной цепью, который имеет от одного до шести атомов углерода включительно. Примеры таких групп включают без ограничения метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2-пропил, 2-метил-1-бутил и н-гексил.
Выражение "C3-С6циклоалкил" относится к циклопропилу, циклобутилу, циклопентилу и циклогексилу.
Выражение "алкокси" относится к насыщенной алкоксигруппе с прямой или разветвленной цепью, которая имеет от одного до шести атомов углерода включительно, при этом открытая валентность находится на кислороде. Примеры таких групп включают без ограничения метокси, этокси, н-бутокси, 2-метил-пентокси и н-гексилокси.
Выражение "арил" относится к фенильному кольцу, необязательно замещенному галогеном, C1-С6алкилом, C1-С6алкокси или галоген(C1-C6)алкилом, определенными выше.
Выражение "гетероарил" относится к моноциклическому или полициклическому ароматическому кольцу, содержащему атомы углерода, атомы водорода и один или несколько гетероатомов, предпочтительно 1-3 гетероатома, независимо выбранных из азота, кислорода и серы. Типичные примеры гетероарильных групп включают без ограничения пиридинил, пиридазинил, триазинил, пирролил, пиразолил, имидазолил, (1,2,3,)- и (1,2,4)-триазолил, пиразинил, пиримидинил, тетразолил, фуранил, тиофенил, изоксазолил, тиазолил, изоксазолил и оксазолил. Гетероарильная группа может быть незамещенной или замещенной одним или двумя подходящими заместителями. Гетероарил согласно настоящему изобретению предпочтительно представляет собой моноциклический 5- или 6-членный гетероарил, где кольцо содержит 2-5 атомов углерода и 1-3 гетероатома, обозначаемый в данном документе как "моноциклический 5- или 6-членный гетероарил".
ИЗОМЕРНЫЕ ФОРМЫ
Если соединения согласно настоящему изобретению содержат один или несколько хиральных центров, ссылка на любое из соединений будет, если не указано иное, охватывать любые из энантиомерно или диастереомерно чистых соединений, а также смеси энантиомеров или диастереомеров в любом соотношении.
Например, ссылка на соединение 8-фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин без какого-либо дополнительного описания охватывает (R)- 8-фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин, (S)- 8-фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин, а также смеси энантиомеров в любом соотношении, в том числе рацемическую смесь (±)8-фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина.
Соответственно, ссылка на соединение 8-фтор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин без какого-либо дополнительного описания охватывает все четыре стереоизомерных варианта, а также их смеси в любом соотношении, в том числе рацемические смеси.
Вышеупомянутое также применимо, если соединения согласно настоящему изобретению содержат более двух хиральных центров.
ИНГИБИТОРЫ PDE1
В контексте настоящего изобретения соединение считается ингибитором PDE1, если количество, требуемое для достижения уровня IC50 в отношении PDE1B, составляет 5 микромолей или менее, предпочтительно менее 4 микромолей, например, 3 микромоля или менее, более предпочтительно 2 микромоля или менее, например, 1 микромоль или менее, в частности, 500 нМ или менее. Согласно предпочтительным вариантам осуществления количество ингибитора PDE1, необходимое для достижения уровня IC50 в отношении PDE1B, составляет 400 нМ или менее, например, 300 нМ или менее, 200 нМ или менее, 100 нМ или менее, или даже 80 нМ или менее, например, 50 нМ или менее, например, 25 нМ или менее.
ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ
Настоящее изобретение также содержит фармацевтически приемлемые соли соединений. Такие соли включают кислотно-аддитивные соли. Кислотно-аддитивные соли включают соли неорганических кислот, а также органических кислот.
Типичные примеры подходящих неорганических кислот включают хлористоводородную, бромистоводородную, йодистоводородную, фосфорную, серную, сульфаминовую, азотную кислоты и т.п. Типичные примеры подходящих органических кислот включают муравьиную, уксусную, трихлоруксусную, трифторуксусную, пропионовую, бензойную, коричную, лимонную, фумаровую, гликолевую, итаконовую, молочную, метансульфоновую, малеиновую, яблочную, малоновую, миндальную, щавелевую, пикриновую, пировиноградную, салициловую, янтарную, метансульфоновую, этансульфоновую, винную, аскорбиновую, памовую, бисметиленсалициловую, этандисульфоновую, глюконовую, цитраконовую, аспарагиновую, стеариновую, пальмитиновую, EDTA, гликолевую, п-аминобензойную, глутаминовую, бензолсульфоновую, п-толуолсульфоновую кислоты, теофиллин-уксусные кислоты, а также 8-галогентеофиллины, например, 8-бромтеофиллин и т.п. Дополнительные примеры фармацевтически приемлемых аддитивных солей неорганических или органических кислот включают фармацевтически приемлемые соли, приведенные в Berge, S.M. et al., J. Pharm. Sci. 1977, 66, 2, содержание которого включено в данный документ посредством ссылки.
Кроме того, соединения согласно настоящему изобретению могут существовать в несольватированной, а также в сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. Как правило, для целей настоящего изобретения сольватированные формы считаются эквивалентными несольватированным формам.
ТЕРАПЕВТИЧЕСКИ ЭФФЕКТИВНОЕ КОЛИЧЕСТВО
В данном контексте выражение "терапевтически эффективное количество" соединения означает количество, достаточное для излечения, облегчения или частичной остановки клинических проявлений данного заболевания и/или его осложнений при терапевтическом вмешательстве, предусматривающем введение указанного соединения. Количество, достаточное для осуществления этого, определяется как "терапевтически эффективное количество". Эффективные количества для каждой цели будут зависеть от тяжести заболевания или повреждения, а также от веса и общего состояния субъекта. Следует понимать, что определения соответствующей дозировки можно достигнуть с использованием общепринятого эксперимента, путем построения матрицы значений и тестирования различных точек в матрице, что находится в компетенции квалифицированного врача.
В данном контексте выражения "лечение" и "осуществление лечения" означают контроль и уход за пациентом с целью противодействия состоянию, такому как заболевание или расстройство. Выражение предназначено для включения полного спектра видов лечения данного состояния, от которого страдает пациент, таких как введение активного соединения для облегчения симптомов или осложнений, для приостановки прогрессирования заболевания, расстройства или состояния, для смягчения симптомов и осложнений и/или для предотвращения состояния, при этом предотвращение следует понимать как контроль и уход за пациентом с целью противодействия заболеванию, состоянию или расстройству, и оно предусматривает введение активных соединений для предотвращения появления симптомов или осложнений. При этом профилактический (превентивный) и терапевтический (клинический) виды лечения являются двумя отдельными аспектами настоящего изобретения. Подлежащим лечению пациентом предпочтительно является млекопитающее, в частности, человек.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Настоящее изобретение дополнительно предусматривает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель или разбавитель. Настоящее изобретение также предусматривает фармацевтическую композицию, содержащую терапевтически эффективное количество одного из конкретных соединений, раскрытых в экспериментальном разделе в настоящем документе, и фармацевтически приемлемый носитель или разбавитель.
Соединения согласно настоящему изобретению можно вводить отдельно или в комбинации с фармацевтически приемлемыми носителями, разбавителями или наполнителями в однократных или многократных дозах. Фармацевтические композиции согласно настоящему изобретению можно составлять с фармацевтически приемлемыми носителями или разбавителями, а также с любыми другими известными вспомогательными средствами и наполнителями согласно общепринятым методикам, как, например, методикам, раскрытым в Remington: The Science and Practice of Pharmacy, 21th Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 2005.
Фармацевтические композиции можно специально составлять для введения любым подходящим путем, таким как пероральный, ректальный, назальный, легочный, местный (в том числе буккальный и подъязычный), чрескожный, интрацистернальный, внутрибрюшинный, вагинальный и парентеральный (в том числе подкожный, внутримышечный, интратекальный, внутривенный и внутрикожный) пути. Следует принять во внимание, что путь будет зависеть от общего состояния и возраста подлежащего лечению субъекта, природы подлежащего лечению состояния и активного ингредиента.
Фармацевтические композиции для перорального введения включают твердые лекарственные формы, такие как капсулы, таблетки, драже, пилюли, таблетки для рассасывания, порошки и гранулы. При необходимости композиции можно получать с покрытиями, такими как энтеросолюбильные покрытия, или же их можно составлять с обеспечением контролированного высвобождения активного ингредиента, такого как замедленное или пролонгированное высвобождение, согласно способам, хорошо известным из уровня техники. Жидкие лекарственные формы для перорального введения включают растворы, эмульсии, суспензии, сиропы и эликсиры.
Фармацевтические композиции для парентерального введения включают стерильные водные и неводные инъекционные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, подлежащие ресуспендированию в стерильных инъекционных растворах или дисперсиях перед применением. Другие подходящие формы введения включают без ограничения суппозитории, аэрозоли, мази, кремы, гели, ингаляторы, кожные пластыри и имплантаты.
Типичные пероральные дозировки варьируют от приблизительно 0,001 до приблизительно 100 мг/кг массы тела в сутки. Типичные пероральные дозировки также варьируют от приблизительно 0,01 до приблизительно 50 мг/кг веса тела в сутки. Типичные пероральные дозировки дополнительно варьируют от приблизительно 0,05 до приблизительно 10 мг/кг веса тела в сутки. Пероральные дозировки обычно вводят одной или несколькими дозировками, как правило, одной - тремя дозировками в сутки. Точная дозировка будет зависеть от частоты и способа введения, пола, возраста, веса и общего состояния подлежащего лечению субъекта, природы и тяжести подлежащего лечению состояния и каких-либо сопутствующих подлежащих лечению заболеваний, а также других факторов, очевидных специалистам в данной области.
Составы также могут быть представлены в единичной лекарственной форме с помощью способов, известных специалистам в данной области. В качестве примера типичная единичная лекарственная форма для перорального введения может содержать от приблизительно 0,01 до приблизительно 1000 мг, от приблизительно 0,05 до приблизительно 500 мг или от приблизительно 0,5 мг до приблизительно 200 мг.
В случае парентеральных путей, таких как внутривенное, интратекальное, внутримышечное и подобное введение, типичные дозы составляют порядка половины дозы, используемой для перорального введения.
Настоящее изобретение также предусматривает способ получения фармацевтической композиции, включающий смешивание терапевтически эффективного количества соединения формулы (I) и по меньшей мере одного фармацевтически приемлемого носителя или разбавителя. Согласно варианту осуществления настоящего изобретения соединение, используемое в вышеупомянутом способе, является одним из конкретных соединений, раскрытых в экспериментальном разделе в настоящем документе.
Соединения согласно настоящему изобретению, как правило, используют в виде свободного вещества или в виде его фармацевтически приемлемой соли. Одним примером является кислотно-аддитивная соль соединения, которая характеризуется применимостью свободного основания. Если соединение формулы (I) содержит свободное основание, то такие соли получают общепринятым путем при обработке раствора или суспензии свободного основания формулы (I) молярным эквивалентом фармацевтически приемлемой кислоты. Выше описаны типичные примеры подходящих органических и неорганических кислот.
Для парентерального введения можно использовать растворы соединений формулы (I) в стерильном водном растворе, водном растворе пропиленгликоля, водном растворе витамина E или кунжутном или арахисовом масле. Такие водные растворы должны быть подходящим образом забуферены, если необходимо, а жидкий разбавитель сначала делают изотоническим с помощью достаточного количества солевого раствора или глюкозы. Водные растворы являются особенно подходящими для внутривенного, внутримышечного, подкожного и внутрибрюшинного введения. Соединения формулы (I) можно легко включить в известные стерильные водные среды с помощью стандартных методик, известных специалистам в данной области.
Подходящие фармацевтические носители включают инертные твердые разбавители или заполнители, стерильные водные растворы и различные органические растворители. Примеры твердых носителей включают лактозу, сульфат кальция, сахарозу, циклодекстрин, тальк, желатин, агар, пектин, аравийскую камедь, стеарат магния, стеариновую кислоту и низшие алкиловые эфиры целлюлозы. Примеры жидких носителей включают без ограничения сироп, арахисовое масло, оливковое масло, фосфолипиды, жирные кислоты, амины жирных кислот, полиоксиэтилен и воду. Подобным образом, носитель или разбавитель может включать любые материалы для замедленного высвобождения, известные из уровня техники, такие как глицерилмоностеарат или глицерилдистеарат, отдельно или смешанные с воском. Затем фармацевтические композиции, образованные путем объединения соединений формулы (I) с фармацевтически приемлемым носителем, легко вводят в виде множества лекарственных форм, подходящих для раскрытых путей введения. Составы в целях удобства могут быть представлены в единичной лекарственной форме с помощью известных в области фармацевтики способов.
Составы согласно настоящему изобретению, подходящие для перорального введения, могут быть представлены в виде отдельных единиц, таких как капсулы или таблетки, каждая из которых содержит предварительно определенное количество активного ингредиента и необязательно подходящий наполнитель. Кроме того, доступные для перорального введения составы могут быть в форме порошка или гранул, раствора или суспензии в водной или неводной жидкости или жидкой эмульсии по типу масло-в-воде или вода-в-масле.
Если для перорального введения используется твердый носитель, то препарат может быть таблетированным, помещенным в твердую желатиновую капсулу, в форме порошка или пеллеты, или же он может быть в форме пастилки или таблетки для рассасывания. Количество твердого носителя будет существенно варьировать, но будет находиться в диапазоне от приблизительно 25 мг до приблизительно 1 г на дозированную единицу. Если используется жидкий носитель, то препарат может быть в форме сиропа, эмульсии, мягкой желатиновой капсулы или стерильной инъекционной жидкости, такой как водная или неводная жидкая суспензия или раствор.
Фармацевтические композиции согласно настоящему изобретению можно получать с помощью общепринятых в данной области способов. Например, таблетки можно получать путем смешивания активного ингредиента с обычными вспомогательными средствами и/или разбавителями, а затем прессования смеси в стандартной таблетирующей машине для получения таблеток. Примеры вспомогательных средств или разбавителей включают кукурузный крахмал, картофельный крахмал, тальк, стеарат магния, желатин, лактозу, камеди и т.п. Можно использовать любые другие вспомогательные средства или добавки, обычно применяемые для таких целей, как, например, красители, ароматизаторы, консерванты и т.д., при условии, что они совместимы с активными ингредиентами.
ЛЕЧЕНИЕ РАССТРОЙСТВ
Как упоминалось выше, соединения формулы (I) являются ингибиторами фермента PDE1 и в связи с этим применимы для лечения соответствующих неврологических и психических расстройств.
Таким образом, настоящее изобретение предусматривает соединение формулы (I) или его фармацевтически приемлемую кислотно-аддитивную соль, а также фармацевтическую композицию, содержащую такое соединение, для применения при лечении нейродегенеративного расстройства, психического расстройства или наркотической зависимости у млекопитающих, в том числе людей, где нейродегенеративное расстройство выбрано из группы, состоящей из болезни Альцгеймера, мультиинфарктной деменции, алкогольной деменции или другой связанной с наркотиками деменции, деменции, ассоциированной с внутричерепными опухолями или травмой мозга, деменции, ассоциированной с болезнью Гентингтона или болезнью Паркинсона, или деменции, ассоциированной с AIDS; делирия; амнестического расстройства; посттравматического стрессового расстройства; умственной отсталости; нарушения способности к обучению, например, нарушения способности к чтению, нарушения способности к математике или нарушения способности к письменному изложению; синдрома дефицита внимания-гиперактивности; и возрастного снижения когнитивных способностей; и где психическое расстройство выбрано из группы, состоящей из шизофрении, например, параноидального, гебефренического, кататонического, недифференцированного или остаточного типа; шизофреноформного расстройства; шизоаффективного расстройства, например, бредового типа или депрессивного типа; бредового расстройства; интоксикационного психического расстройства, например, психоза, индуцированного алкоголем, амфетамином, марихуаной, кокаином, галлюциногенами, летучими веществами наркотического действия, опиоидами или фенциклидином; расстройства личности параноидального типа и расстройства личности шизоидного типа; и где наркотической зависимостью является алкогольная, амфетаминовая, кокаиновая или опиатная зависимость.
Соединения формулы (I) или их фармацевтически приемлемые соли можно использовать как отдельный активный ингредиент или в комбинации с одним или несколькими другими лекарственными средствами в лечении заболеваний или состояний, при которых полезны соединения в соответствии с настоящим изобретением, при этом объединение лекарственных средств вместе является более безопасным или более эффективным, нежели каждое лекарственное средство по отдельности. Кроме того, соединения согласно настоящему изобретению можно использовать в комбинации с одним или несколькими другими лекарственными средствами, которые лечат, предотвращают, регулируют, облегчают или снижают риск побочных эффектов или токсичности соединений согласно настоящему изобретению. Такие другие лекарственные средства можно вводить путем и в количестве, обычно применяемыми таким образом, одновременно или последовательно с соединениями согласно настоящему изобретению. Следовательно, фармацевтические композиции согласно настоящему изобретению включают фармацевтические композиции, которые содержат один или несколько других активных ингредиентов в дополнение к соединениям согласно настоящему изобретению. Комбинации можно вводить как часть комбинированного препарата в виде единичной лекарственной формы, или как набор, или лечебный протокол, где одно или несколько дополнительных лекарственных средств вводят в отдельных лекарственных формах как часть режима лечения.
Настоящее изобретение предусматривает способ лечения млекопитающего, в том числе человека, страдающего нейродегенеративным расстройством, выбранным из нарушения когнитивных функций или двигательного расстройства, при этом способ включает введение субъекту терапевтически эффективного количества соединения формулы (I).
Настоящее изобретение дополнительно предусматривает способ лечения нейродегенеративного расстройства или состояния у млекопитающего, в том числе человека, при этом способ включает введение упомянутому млекопитающему количества соединения формулы (I), эффективного для ингибирования PDE1.
Настоящее изобретение также предусматривает способ лечения субъекта, страдающего психическим расстройством, при этом способ включает введение субъекту терапевтически эффективного количества соединения формулы (I). Примеры психических расстройств, которые можно лечить согласно настоящему изобретению, включают без ограничения синдром дефицита внимания и гиперактивности (ADHD), шизофрению, например, параноидального, гебефренического, кататонического, недифференцированного или остаточного типа; шизофрениформное расстройство; шизоаффективное расстройство, например, бредового типа или депрессивного типа; бредовое расстройство; интоксикационное психическое расстройство, например, психоз, индуцированный алкоголем, амфетамином, марихуаной, кокаином, галлюциногенами, летучими веществами наркотического действия, опиоидами или фенциклидином; расстройство личности параноидального типа и расстройство личности шизоидного типа; при этом тревожное расстройство выбрано из панического расстройства; агорафобии; специфической фобии; социальной фобии; обсессивно-компульсивного расстройства; посттравматического стрессового расстройства; острого стрессового расстройства и генерализованного тревожного расстройства.
Было обнаружено, что соединения формулы (I) или их фармацевтически приемлемые соли можно преимущественно вводить в комбинации по меньшей мере с одним нейролептическим средством (которым может быть типичное или атипичное антипсихотическое средство) для обеспечения улучшенного лечения психических расстройств, таких как шизофрения. Комбинации, применения и способы лечения согласно настоящему изобретению также могут обеспечивать преимущества при лечении пациентов, которые не способны адекватно отвечать или которые устойчивы к другим известным видам лечения.
Таким образом, настоящее изобретение предусматривает способ лечения млекопитающего, страдающего психическим расстройством, таким как шизофрения, при этом способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I), отдельно или в виде комбинированной терапии совместно по меньшей мере с одним нейролептическим средством.
Используемое в настоящем документе выражение "нейролептическое средство" относится к лекарственным средствам, которые по отношению к когнитивной функции и поведению обладают эффектом антипсихотических лекарственных средств, уменьшающим спутанность сознания, бред, галлюцинации и психомоторное возбуждение у пациентов с психозом. Нейролептические средства, также известные как основные транквилизаторы и антипсихотические лекарственные средства, включают без ограничения типичные антипсихотические лекарственные средства, в том числе фенотиазины, дополнительно разделяемые на алифатические соединения, пиперидины и пиперазины, тиоксантены (например, цисординол), бутирофеноны (например, галоперидол), дибензоксазепины (например, локсапин), дигидроиндолоны (например, молиндон), дифенилбутилпиперидины (например, пимозид), и атипичные антипсихотические лекарственные средства, в том числе бензисоксазолы (например, рисперидон), сертиндол, оланзапин, кветиапин, осанетант и зипрасидон.
Особенно предпочтительными нейролептическими средствами для применения в настоящем изобретении являются сертиндол, оланзапин, рисперидон, кветиапин, арипипразол, галоперидол, клозапин, зипрасидон и осанетант.
Настоящее изобретение дополнительно предусматривает способ лечения субъекта, страдающего нарушением когнитивных функций, при этом способ включает введение субъекту терапевтически эффективного количества соединения формулы (I). Примеры нарушений когнитивных функций, которые можно лечить согласно настоящему изобретению, включают без ограничения болезнь Альцгеймера, мультиинфарктную деменцию, алкогольную деменцию или другую ассоциированную с наркотиками деменцию, деменцию, ассоциированную с внутричерепными опухолями или травмой мозга, деменцию, ассоциированную с болезнью Гентингтона или болезнью Паркинсона, или деменцию, ассоциированную с AIDS; делирий; амнестическое расстройство; посттравматическое стрессовое расстройство; умственную отсталость; нарушение способности к обучению, например, нарушение способности к чтению, нарушение способности к математике или нарушение способности к письменному изложению; синдром дефицита внимания-гиперактивности и возрастное снижение когнитивных способностей.
Настоящее изобретение также предусматривает способ лечения двигательного расстройства, при этом способ включает введение субъекту терапевтически эффективного количества соединения формулы (I). Примеры двигательных расстройств, которые можно лечить согласно настоящему изобретению, включают без ограничения болезнь Гентингтона и дискинезию, ассоциированную с терапией агонистами дофамина. Настоящее изобретение дополнительно предусматривает способ лечения двигательного расстройства, выбранного из болезни Паркинсона и синдрома беспокойных ног, который включает введение субъекту терапевтически эффективного количества соединения формулы (I).
Настоящее изобретение также предусматривает способ лечения расстройства настроения, при этом способ включает введение субъекту терапевтически эффективного количества соединения формулы (I). Примеры расстройств настроения и эпизодов настроения, которые можно лечить согласно настоящему изобретению, включают без ограничения большой депрессивный эпизод слабой, умеренной и тяжелой степени, маниакальный или смешанный эпизод настроения, гипоманиакальный эпизод настроения; депрессивный эпизод с типичными признаками; депрессивный эпизод с меланхолическими признаками; депрессивный эпизод с кататоническими признаками; эпизод настроения с дебютом в послеродовой период; постинсультная депрессия; большое депрессивное расстройство; дистимическое расстройство; малое дистимическое расстройство; предменструальное дисфорическое расстройство; постпсихотическое депрессивное расстройство при шизофрении; большое депрессивное расстройство, развивающееся на фоне психотического расстройства, такого как бредовое расстройство или шизофрения; биполярное расстройство, например, биполярное расстройство I, биполярное расстройство II и циклотимическое расстройство. Подразумевается, что расстройство настроения является психическим расстройством.
Настоящее изобретение дополнительно предусматривает способ лечения расстройства, включающего в качестве симптома дефицит внимания и/или когнитивных функций у млекопитающего, в том числе человека, при этом способ включает введение упомянутому млекопитающему количества соединения формулы (I), эффективного для лечения упомянутого расстройства.
Другие расстройства, которые можно лечить согласно настоящему изобретению, представляют собой обсессивно-компульсивные расстройства, синдром Туретта и другие тиковые расстройства.
Как используется в настоящем документе, и если не указано иное, "нейродегенеративное расстройство или состояние" относится к расстройству или состоянию, которое вызвано дисфункцией и/или гибелью нейронов в центральной нервной системе. Лечение этих расстройств и состояний можно облегчать путем введения средства, которое предотвращает дисфункцию или гибель нейронов, подверженных риску при этих расстройствах или состояниях, и/или усиливает функцию поврежденных или здоровых нейронов таким образом, чтобы компенсировать потерю функции, вызванную дисфункцией или гибелью подверженных риску нейронов. Используемое в настоящем документе выражение "нейротрофическое средство" относится к веществу или средству, которое обладает некоторыми или всеми из этих свойств.
Примеры нейродегенеративных расстройств и состояний, которые можно лечить согласно настоящему изобретению, включают без исключения болезнь Паркинсона; болезнь Гентингтона; деменцию, например, болезнь Альцгеймера, мультиинфарктную деменцию, деменцию, ассоциированную с AIDS, и лобно-височную деменцию; нейродегенерацию, ассоциированную с травмой мозга; нейродегенерацию, ассоциированную с инсультом, нейродегенерацию, ассоциированную с церебральным инфарктом; индуцированную гипогликемией нейродегенерацию; нейродегенерацию, ассоциированную с эпилептическими приступами; нейродегенерацию, ассоциированную с отравлением нейротоксинами; и мультисистемную атрофию.
Согласно одному варианту осуществления нейродегенеративное расстройство или состояние включает нейродегенерацию средних шипиковых нейронов полосатого тела у млекопитающего, в том числе человека.
Согласно дополнительному варианту осуществления настоящего изобретения нейродегенеративным расстройством или состоянием является болезнь Гентингтона.
Все ссылки, включая публикации, патентные заявки и патенты, цитируемые в данном документе, включены при помощи ссылки в данный документ во всей своей полноте и в той же степени, как если бы каждая ссылка была индивидуально и конкретно обозначена для включения при помощи ссылки и приведена во всей своей полноте (в максимальной степени, допускаемой законом).
Заголовки и подзаголовки используют в настоящем документе исключительно для удобства, и их не следует рассматривать как ограничивающие настоящее изобретение каким-либо образом.
Применение всевозможных примеров или типичной фразы (в том числе "так, например", "например", "к примеру" и "как таковой") в настоящем описании предназначено исключительно для лучшего освещения настоящего изобретения и не является ограничением объема настоящего изобретения, если не указано иное.
Цитирование и включение патентных документов в настоящий документ служит исключительно для удобства и не отражает какую-либо оценку действительности, патентоспособности и/или юридической силы таких патентных документов.
Настоящее изобретение включает все модификации и эквиваленты объекта, перечисленного в приложенной к данному документу формулы изобретения согласно действующему законодательству.
СОЕДИНЕНИЯ СОГЛАСНО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ






ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ СОГЛАСНО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Соединения общей формулы I в соответствии с настоящим изобретением можно получать, как описано в следующих схемах реакций. Если иное не указано, на схемах реакций и в следующем обсуждении R1-R10 и X являются такими, как определено выше. На схеме 1 ниже показана реакция связывания между соединением формулы II и производным 3-аминотетрагидрофурана формулы III с получением соединений замещенного галогенированного хиназолин-THF-амина формулы I.
Схема 1

L представляет собой уходящую группу, например, Cl, Br, I, метансульфонил, 4-толуолсульфонил. Эту реакцию обычно проводят в растворителе, таком как, например, толуол, необязательно в присутствии карбонатного основания, в диапазоне температур от приблизительно 0°C до приблизительно 200°C. Другие подходящие растворители включают бензол, хлороформ, диоксан, этилацетат, 2-пропанол и ксилол. Альтернативно, можно использовать смеси растворителей, такие как толуол/2-пропанол. Предпочтительно реагенты нагревают с обратным холодильником в DMSO или DMF в течение периода времени от приблизительно 2 часов до приблизительно 24 часов, необязательно используя микроволновую печь.
Реакцию, показанную на схеме 1, можно также легко проводить с помощью способа, катализируемого палладием. Обычно смесь соединения формулы II, соединения формулы III и источника палладия (II), такого как Pd(OAc)2 или Pd2(dba)3, нагревают в подходящем растворителе, таком как толуол, в присутствии бисфосфинового лиганда, такого как 2,2'-бис(дифенилфосфино)-1,1'-бинафтил "BINAP", и алкоксидного основания, такого как трет-бутоксид натрия. Реакционную смесь перемешивают при 100°C в течение 7 ч. с последующей очисткой продукта при помощи препаративной ВЭЖХ с получением желаемого продукта.
Исходные материалы формулы II, т.е. хиназолины, или коммерчески доступны, или могут быть получены как описано в литературе, например, Dechantsreiter, Michael А. и соавт. из международной заявки PCT 2013192345, поданной 27 декабря 2013 г., Armarego, Wilfred L.F. and Reece, Phillip A. Australian Journal of Chemistry, 34(7), 1561-6; 1981, или как описано в данной патентной заявке.
Исходные материалы формулы III или коммерчески доступны, или могут быть получены способами, аналогичными способам, описанным в литературе, например, Wipf, Peter; Manojlovic, Marija D. Beilstein Journal of Organic Chemistry (2011), 7, 824-830, Yoshimitsu, Y. et al. Journal of Organic Chemistry (2010), 75(11), 3843-3846, Shiau, T.P. et al. Bioorganic & Medicinal Chemistry Letters (2009), 19(4), 1110-1114.
Соединения формулы I, где R2 не является водородом, можно получать алкилированием соединений формулы IV, где R2 является водородом, при помощи алкилгалогенида формулы V, как показано на схеме 2.
Схема 2

Данную реакцию обычно проводят в подходящем растворителе, таком как диметилформамид, диметилацетамид, тетрагидрофуран или ацетонитрил, в присутствии подходящего основания, такого как карбонатное основание, например, карбонат калия, или третичного аминного основания, например, триэтиламина или диизопропилэтиламина, или сильного основания, такого как гидрид натрия, при температуре в диапазоне от приблизительно 0°C до приблизительно 100°C.
Раскрытое в данном документе настоящее изобретение будет дополнительно проиллюстрировано следующими неограничивающими примерами.
Общие способы
Аналитические данные LC-MS получали с использованием способов, определенных ниже.
Способ 1. Использовали систему LCMS Agilent 1200 с детектором ELS. Колонка: Agilent TC-C18, 5 мкм, 2,1×50 мм; температура колонки: 50°C; система растворителей: A = вода/трифторуксусная кислота (99,9:0,1) и B = ацетонитрил/трифторуксусная кислота (99,95:0,05); способ: линейное градиентное элюирование с А:В=99:1-0:100 за 4,0 минут при скорости потока 0,8 мл/минуту.
Способ 2. Использовали систему LCMS Agilent 1200 с детектором ELS. Колонка: XBridge ShieldRP18, 5 мкм, 50×2,1 мм; температура колонки: 40°C; система растворителей: A = вода/NH3*H2O (99,95:0,05) и B = ацетонитрил; способ: линейное градиентное элюирование с А:В=95:5-0:100 за 3,4 минут при скорости потока 0,8 мл/минуту.
Способ 3. Использовали систему LCMS Agilent 1200 с детектором ELS. Колонка: XBridge ShieldRP18, 5 мкм, 50×2,1 мм; температура колонки: 40°C; система растворителей: A = вода/NH3*H2O (99,95:0,05) и B = ацетонитрил; способ: линейное градиентное элюирование с А:В=99:1-0:100 за 3,4 минут при скорости потока 0,8 мл/минуту.
Способ 4. Использовали систему LCMS Agilent 1100 с детектором ELS. Колонка: YMC ODS-AQ, 5 мкм, 50×2,0 мм; температура колонки: 50°C; система растворителей: А=0,1% TFA в воде, а B=0,05% TFA в ацетонитриле; способ: линейное градиентное элюирование с A:B=99:1-5:95 за 3,5 минут при скорости потока 0,8 мл/минуту.
Способ 5. Использовали систему LCMS Agilent 1200 с детектором ELS. Колонка: Agilent TC-C18 5 мкм; 2,1×50 мм; температура колонки: 50°C; система растворителей: A = вода/трифторуксусная кислота (99,9:0,1) и B = ацетонитрил/трифторуксусная кислота (99,95:0,05); способ: элюирование в линейном градиенте с А:В=90:10-0:100 за 4,0 минуты при скорости потока 0,8 мл/мин.
Очистку посредством препаративной LC-MS выполняли на устройстве РЕ Sciex API 150ЕХ с химической ионизацией при атмосферном давлении. Колонка: YMC ODS-A 50X20 мм с размером частиц 5 мкм; система растворителей: A = вода/трифторуксусная кислота (99,965:0,035) и B = ацетонитрил/вода/трифторуксусная кислота (94,965:5:0,035); способ: линейное градиентное элюирование с A:B=80:20-0:100 за 7 минут при скорости потока 22,7 мл/минуту. Сбор фракций выполняли с помощью масс-спектроскопического детектирования MS с разделенным потоком.
Препаративную SFC выполняли на приборе Thar 80. Приведенные в качестве примера условия не ограничиваются приведенными ниже: Колонка AD 250 X 30 мм с размером частиц 20 мкм; температура колонки: 38°C, подвижная фаза: сверхкритический CO2/EtOH (0,2% NH3H2O)=45/55.
Синтез промежуточного соединения I

4-Хлор-8-фтор-7-метоксихиназолин
Стадия 1. Коммерчески доступною (CAS 1180497-45-3) 2-амино-3-фтор-4-метоксибензойную кислоту (8 г, 43,21 ммоль) и ацетат аммония (67 г, 864 моль) в триметоксиметане (250 мл) перемешивали при 100°C в течение 12 ч. Смесь отфильтровывали и промывали водой (3×20 мл), белое твердое вещество сушили под вакуумом с получением 8-фтор-7-метоксихиназолин-4(3H)-она (8 г, 95%).
Стадия 2. К смеси 8-фтор-7-метоксихиназолин-4(3H)-она (4,0 г, 20,6 ммоль) и диизопропилэтиламина (11 г, 82 ммоль) в толуоле (100 мл) добавляли POCl2 (6,32 г, 41,2 ммоль) при 0°C. Реакционную смесь перемешивали при 100°C в течение 12 ч. Смесь затем охлаждали до 20°C и выливали в ледяную воду (100 мл). Водную фазу экстрагировали дихлорметаном (3×100 мл). Объединенные органические фазы промывали солевым раствором (3×10 мл) и концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 3 г 4-хлор-8-фтор-7-метоксихиназолина (70%).
Пример 1

8-Фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин
Смесь 4-хлор-8-фтор-7-метоксихиназолина (560 мг, 2,63 ммоль), (+/-)-3-метилтетрагидрофуран-3-амингидрохлорида (400 мг, 2,92 ммоль) и K2CO3 (800 мг, 5,84 ммоль) в DMSO (30 мл) перемешивали при 100°C в течение 12 часов. Раствор затем выливали в ледяную воду (100 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические фазы промывали солевым раствором (3×10 мл), сушили над MgSO4 и концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 320 мг (+/-)-8-фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина (44%).
Рацемическую смесь (320 мг) очищали разделением посредством SFC (колонка: AY (250 мм*30 мм, 5 мкм)) и нумеровали согласно порядку элюирования:
Стереоизомер 1 (первое элюирование посредством SFC): 140 мг
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,44 (d, J=8,8 Гц, 1Н), 7,19 (t, J=8,4 Гц, 1Н), 5,68 (s, 1Н), 4,16 (d, J=9,2 Гц, 1Н), 4,05 (s, 3H), 4,02-3,98 (m, 2Н), 3,87 (d, J=9,2 Гц, 1Н), 2,66-2,60 (m, 1Н), 2,17-2,09 (m, 1Н), 1,75 (s, 3H).
LC-MS: (масса/заряд) 278,1 (МН+) tR (минуты, Способ 1) = 1,84 минуты.
[α]20D=18° (c=0,1 мг/мл, метанол).
Стереоизомер 2 (второе элюирование посредством SFC): 160 мг
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,44 (dd, J=8,8, 1,6 Гц, 1Н), 7,19 (t, J=8,4 Гц, 1Н), 5,70 (s, 1Н), 4,16 (d, J=9,2 Гц, 1Н), 4,05 (s, 3H), 4,02-3,98 (m, 2Н), 3,87 (d, J=9,2 Гц, 1Н), 2,66-2,60 (m, 1Н), 2,16-2,09 (m, 1Н), 1,75 (s, 3H).
LC-MS: (масса/заряд) 278,1 (МН+) tR (минуты, Способ 1) = 1,84 минуты.
[α]20D=-26° (с=0,1 мг/мл, метанол)
Пример 2
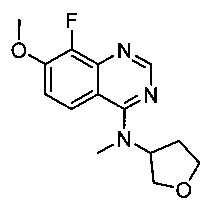
8-Фтор-7-метокси-N-метил-N-(тетрагидрофуран-3-ил)хиназолин-4-амин
Стадия 1. Раствор 4-хлор-8-фтор-7-метоксихиназолина (440 мг, 2,06 ммоль), тетрагидрофуран-3-амина (200 мг, 2,29 ммоль) и диизопропилэтиламина (600 мг, 4,58 ммоль) в DMF (30 мл) перемешивали при 100°C в течение 12 ч. Раствор концентрировали под вакуумом, а остаток очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 500 мг 8-фтор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (83%) в виде белого твердого вещества.
Стадия 2. К раствору 8-фтор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (480 мг, 1,83 ммоль) в THF (20 мл) добавляли 60% суспензию NaH в минеральном масле (100 мг, 2,74 ммоль) при 0°C, затем его перемешивали при 0°C в течение 30 мин., а затем позволяли нагреться до комнатной температуры. Добавляли метилйодид (388 мг, 2,74 ммоль) при 20°C и реакционную смесь перемешивали при 20°C в течение 12 ч. Раствор гасили насыщ. водн. NH4Cl (2 мл), а затем концентрировали под вакуумом. Остаток разбавляли дихлорметаном (100 мл), промывали солевым раствором (3×10 мл), сушили над MgSO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 230 мг 8-фтор-7-метокси-N-метил-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (46%).
Смесь стереоизомеров (230 мг) очищали разделением посредством SFC (колонка: AD-H (250 мм*30 мм, 5 мкм)) и нумеровали согласно порядку элюирования:
Стереоизомер 1 (первое элюирование посредством SFC): 75 мг
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,77-7,74 (m, 1Н), 7,19-7,15 (m, 1Н), 5,29-5,23 (m, 1Н), 4,17-4,12 (m, 1Н), 4,07 (s, 3H), 3,99 (d, J=5,6 Гц, 2Н), 3,75 (q, J=7,6 Гц, 1Н), 3,32 (s, 3H), 2,49-2,44 (m, 1Н), 2,12-2,08 (m, 1Н).
LC-MS: (масса/заряд) 278,1 (МН+) tR (минуты, Способ 1) = 1,78 минуты.
[α]20D=-15° (с=0,1 мг/мл, метанол)
Стереоизомер 2 (второе элюирование посредством SFC): 80 мг
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,76 (dd, J=9,2, 2,0 Гц, 1Н), 7,18 (dd, J=9,2, 8,0 Гц, 1Н), 5,31-5,23 (m, 1Н), 4,17-4,12 (m, 1Н), 4,06 (s, 3H), 3,99 (d, J=5,6 Гц, 2Н), 3,75 (q, J=7,6 Гц, 1Н), 3,32 (s, 3H), 2,48-2,44 (m, 1Н), 2,12-2,08 (m, 1Н).
LC-MS: (масса/заряд) 278,1 (МН+) tR (минуты, Способ 1) = 1,79 минуты.
[α]20D=16° (с=0,1 мг/мл, метанол)
Пример 3

8-Фтор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин
Раствор 4-хлор-8-фтор-7-метоксихиназолина (500 мг, 2,35 ммоль), 2-метилтетрагидрофуран-3-амингидрохлорида (388 мг, 2,82 ммоль) и диизопропилэтиламина (607 мг, 4,70 ммоль) в DMF (20 мл) перемешивали при 100°C в течение 12 часов. Раствор концентрировали под вакуумом, а остаток очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 600 мг 8-фтор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина в виде смеси всех четырех возможных стереоизомеров (84%).
Смесь стереоизомеров (600 мг) очищали разделением посредством SFC (колонка: AD (250 мм*30 мм, 5 мкм)) и нумеровали согласно порядку элюирования:
Стереоизомер 1 (первое элюирование посредством SFC): 180 мг
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,47 (dd, J=8,8, 1,2 Гц, 1Н), 7,21 (t, J=8,8 Гц, 1Н), 5,72 (d, J=6,8 Гц, 1Н), 4,64-4,58 (m, 1Н), 4,09-3,98 (m, 6Н), 2,59-2,50 (m, 1Н), 2,00-1,96 (m, 1Н), 1,36 (d, J=6,4 Гц, 3H).
LC-MS: (масса/заряд) 278,1 (МН+) tR (минуты, Способ 1) = 1,84 минуты.
[α]20D=-23° (с=0,1 мг/мл, метанол)
Стереоизомер 2 (второе элюирование посредством SFC): 80 мг
1H ЯМР (CDCl3, 400 МГц): δ 8,65 (s, 1Н), 7,48 (dd, J=9,2, 1,6 Гц, 1Н), 7,24-7,20 (m, 1Н), 5,68 (d, J=8,4 Гц, 1Н), 5,07-5,02 (m, 1Н), 4,11-4,01 (m, 5Н), 3,85-3,82 (m, 1Н), 2,56-2,50 (m, 1Н), 2,03-1,99 (m, 1Н), 1,26 (d, J=6,0 Гц, 3H).
LC-MS: (масса/заряд) 278,1 (МН+) tR (минуты, Способ 1) = 1,82 минуты.
[α]20D=22° (с=0,1 мг/мл, метанол)
Стереоизомер 3 (третье элюирование посредством SFC): 180 мг
1H ЯМР (CDCl3, 400 МГц): δ 8,64 (s, 1Н), 7,48 (dd, J=9,2, 1,6 Гц, 1Н), 7,24-7,19 (m, 1Н), 5,71 (d, J=8,4 Гц, 1Н), 5,07-5,02 (m, 1Н), 4,11-4,01 (m, 5Н), 3,85-3,82 (m, 1Н), 2,56-2,50 (m, 1Н), 2,03-1,97 (m, 1Н), 1,26 (d, J=6,4 Гц, 3H).
LC-MS: (масса/заряд) 278,1 (МН+) tR (минуты, Способ 1) = 1,81 минуты.
[α]20D=-21° (с=0,1 мг/мл, метанол)
Стереоизомер 4 (четвертое элюирование посредством SFC): 80 мг
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,47 (dd, J=8,8, 1,2 Гц, 1Н), 7,21 (t, J=8,8 Гц, 1Н), 5,72 (d, J=7,2 Гц, 1Н), 4,64-4,60 (m, 1Н), 4,09-3,98 (m, 6Н), 2,59-2,50 (m, 1Н), 2,00-1,93 (m, 1Н), 1,36 (d, J=6,4 Гц, 3H).
LC-MS: (масса/заряд) 278,1 (МН+) tR (минуты, Способ 1) = 1,85 минуты.
[α]20D=34° (с=0,1 мг/мл, метанол)
Синтез промежуточного соединения II
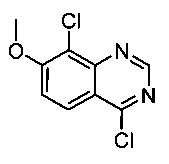
4,8-дихлор-7-метоксихиназолин
Стадия 1. К суспензии коммерчески доступной (CAS 33234-36-5) 2-хлор-3-метоксибензойной кислоты (19,5 г, 104 ммоль) в уксусной кислоте (100 мл) и H2O (100 мл) при комнатной температуре по каплям добавляли бром (10,8 мл, 209 ммоль). Полученную в результате смесь нагревали при 60°C в течение ночи. Затем охлаждали до комнатной температуры и экстрагировали дихлорметаном (3×200 мл). Объединенные органические фазы промывали водой (3×300 мл), сушили над Na2SO4, отфильтровывали и концентрировали с получением 23 г 6-бром-2-хлор-3-метоксибензойной кислоты (83%).
Стадия 2. К суспензии 6-бром-2-хлор-3-метоксибензойной кислоты (10 г, 38 ммоль) в толуоле (200 мл) добавляли дифенилфосфорилазид (12,2 мл, 56,6 ммоль), триэтиламин (15,8 мл, 113 ммоль) и трет-бутанол (18,0 мл, 188 ммоль). Реакционную смесь нагревали при 100°C в течение 2 ч. в N2. Смесь выпаривали и остаток разбавляли этилацетатом. Органическую фазу промывали 5% водным раствором лимонной кислоты, водой, насыщ. водн. NaHCO3, солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 12 г трет-бутил-(6-бром-2-хлор-3-метоксифенил)карбамата (95%).
Стадия 3. К ледяному раствору трет-бутил-(6-бром-2-хлор-3-метоксифенил)карбамата (12 г, 37 ммоль) в дихлорметане (150 мл) добавляли трифторуксусную кислоту (20 мл). Смесь нагревали до комнатной температуры и перемешивали в течение 5 ч. Раствор затем концентрировали, а осадок разбавляли дихлорметаном, доводили до pH=9 при помощи насыщ. водн. NaHCO3, промывали водой, сушили над Na2SO4, отфильтровывали и концентрировали с получением 8,3 г 6-бром-2-хлор-3-метоксианилина (98%).
Стадия 4. К раствору 6-бром-2-хлор-3-метоксианилина (8,3 г, 35 ммоль) в метаноле (300 мл) добавляли 1,3-бис(дифенилфосфино)пропан (2,90 г, 7,02 ммоль), Pd(AcO)2 (1,58 г, 7,02 ммоль) и триэтиламин (4,89 мл, 35,1 ммоль). Реакционную смесь перемешивали при 100°C в атмосфере CO (3 МПа) в течение 2 дней. Смесь охлаждали до комнатной температуры и отфильтровывали. Фильтрат концентрировали, а остаток растворяли в дихлорметане. Полученный в результате раствор промывали водой, сушили над Na2SO4, отфильтровывали и концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 5,0 г метил-2-амино-3-хлор-4-метоксибензоата (65%).
Стадия 5. К раствору метил-2-амино-3-хлор-4-метоксибензоата (4,95 г, 23,0 ммоль) в смеси THF (60 мл) и H2O (30 мл) добавляли LiOH⋅H2O (2,89 г, 68,8 ммоль). Смесь нагревали при 50°C в течение 3 дней. Смесь затем охлаждали до комнатной температуры и экстрагировали этилацетатом. Водную фазу подкисляли водн. KHSO4 до pH=3, отфильтровывали и осадок на фильтре собирали, промывали водой и сушили с получением 3,2 г 2-амино-3-хлор-4-метоксибензойной кислоты (69%).
Стадия 6. К раствору 2-амино-3-хлор-4-метоксибензойной кислоты (700 мг, 3,47 ммоль) в CH(OMe)3 (40 мл) добавляли ацетат аммония (5,35 г, 69,4 ммоль). Смесь затем нагревали при 90°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, отфильтровывали и осадок на фильтре собирали, промывали водой и сушили с получением 630 мг 8-хлор-7-метоксихиназолин-4(3H)-она (86%).
Стадия 7. К ледяному раствору 8-хлор-7-метоксихиназолин-4(3H)-она (630 мг, 2,99 ммоль) в толуоле (15 мл) по каплям добавляли POCl3 (0,56 мл, 6,0 ммоль) и диизопропилэтиламин (2,08 мл, 12,0 ммоль). Смесь нагревали при 100°C в течение ночи, затем охлаждали до комнатной температуры и аккуратно выливали в ледяную воду. Водную фазу экстрагировали дихлорметаном (2×30 мл). Объединенные органические фазы промывали водой, сушили над Na2SO4, отфильтровывали и концентрировали под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле с использованием градиента дихлорметана и этилацетата с получением 580 мг 4,8-дихлор-7-метоксихиназолина (85%).
Пример 4

8-Хлор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амин
К раствору 4,8-дихлор-7-метоксихиназолина (650 мг, 2,84 ммоль) в диметилформамиде (20 мл) добавляли тетрагидрофуран-3-амин (297 мг, 3,41 ммоль) и диизопропилэтиламин (0,99 мл, 5,7 ммоль). Через смесь барботировали N2 в течение 5 минут. Реакционную смесь затем нагревали при 100°C в течение 3 ч. в атмосфере N2. Неочищенную смесь концентрировали и остаток очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 650 мг 8-хлор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (82%).
Рацемическую смесь (650 мг) очищали разделением посредством SFC (колонка: Chiral Рак AD 5 мкм, Daicel Chemical Industries, Ltd, 250×30 мм внутренний диаметр) и нумеровали согласно порядку элюирования:
Стереоизомер 1 (первое элюирование посредством SFC): 200 мг
1H ЯМР (CD3OD, 400 МГц): δ 8,45 (s, 1Н), 8,20 (d, J=9,29 Гц, 1Н), 7,38 (d, J=9,29 Гц, 1Н), 3,99~4,07 (m, 5Н), 3,77~3,89 (m, 2Н), 2,32~2,42 (m, 1Н), 2,04~2,14 (m, 1Н).
LC-MS: (масса/заряд) 280,1 (МН+) tR (минуты, Способ 2) = 1,58 минуты
[α]D20 +38,3° (с=0, 10, метанол).
Стереоизомер 2 (второе элюирование посредством SFC): 200 мг
1H ЯМР (CD3OD, 400 МГЦ): δ 8,45 (s, 1Н), 8,20 (d, J=9,29 Гц, 1Н), 7,38 (d, J=9,05 Гц, 1Н), 3,99~4,07 (m, 5Н), 3,77~3,89 (m, 2Н), 2,32~2,42 (m, 1Н), 2,04~2,14 (m, 1Н).
LC-MS: (масса/заряд) 280,1 (МН+) tR (минуты, Способ 1) = 1,57 минуты
[α]D20 -32,0° (с=0, 10, метанол).
Пример 5

8-Хлор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин
К раствору 4,8-дихлор-7-метоксихиназолина (450 мг, 1,96 ммоль) в DMF (20 мл) добавляли 2-метилтетрагидрофуран-3-амин (смесь всех 4 стереоизомеров) (322 мг, 2,36 ммоль) и диизопропилэтиламин (1,03 мл, 5,89 ммоль). Через смесь барботировали N2 в течение 5 минут, а затем ее нагревали при 100°C в течение ночи. Неочищенную смесь концентрировали и остаток очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением 450 мг 8-хлор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина (78%) в виде смеси всех четырех возможных стереоизомеров.
Смесь стереоизомеров (750 мг) очищали разделением посредством SFC (колонка: Chiral Рак AD 5 мкм, Daicel Chemical Industries, Ltd) и нумеровали согласно порядку элюирования:
Стереоизомер 1 (первое элюирование посредством SFC): 131 мг
1H ЯМР (CD3OD, 400 МГц): δ 8,45 (s, 1Н), 8,19 (d, J=9,6 Гц, 1Н), 7,39 (d, J=9,2 Гц, 1Н), 4,52~4,57 (m, 1Н), 4,04 (s, 4Н), 3,97~4,02 (m, 2Н), 2,41~2,50 (m, 1Н), 1,97~2,04 (m, 1Н), 1,32 (d, J=6,4 Гц, 3H).
LC-MS: (масса/заряд) 294,1 (МН+) tR (минуты, Способ 1) = 1,93 минуты
[α]D20=-59,3° (с=0, 10, метанол).
Стереоизомер 2 (второе элюирование посредством SFC): 97 мг
1H ЯМР (CD3OD, 400 МГц): δ 8,43 (s, 1Н), 8,25 (d, J=9,2 Гц, 1Н), 7,39 (d, J=9,6 Гц, 1Н), 5,01-5,06 (m, 1Н), 4,09~4,14 (m, 2Н), 4,04 (s, 3H), 3,72 (q, J=8,0 Гц, 3H), 2,40~2,46 (m, 1Н), 2,10~2,14 (m, 1Н), 1,09 (d, J=6,0 Гц, 3H).
LC-MS: (масса/заряд) 294,1 (МН+) tR (минуты, Способ 2) = 1,73 минуты
[α]D20=-28,3° (с=0, 10, метанол).
Стереоизомер 3 (третье элюирование посредством SFC): 37 мг
1H ЯМР (CD3OD varian 400): δ 8,43 (s, 1Н), 8,24 (d, J=9,2 Гц, 1Н), 7,38 (d, J=9,2 Гц, 1Н), 5,00~5,05 (m, 1Н), 4,10~4,14 (m, 2Н), 4,04 (s, 3H), 3,72 (q, J=8,0 Гц, 3H), 2,41~2,46 (m, 1Н), 2,09~2,14 (m, 1Н), 1,09 (d, J=6,4 Гц, 3H).
LC-MS: (масса/заряд) 294,1 (МН+) tR (минуты, Способ 1) = 1,73 минуты
[α]D20=+29,3° (с=0, 10, метанол).
Стереоизомер 4 (четвертое элюирование посредством SFC): 50 мг
1H ЯМР (Н000269489 Н20773-029-4А MeOD varian 400): δ 8,46 (s, 1Н), 8,20 (d, J=9,2 Гц, 1Н), 7,39 (d, J=9,2 Гц, 1Н), 4,53~4,57 (m, 1Н), 4,04 (s, 3H), 3,97~4,02 (m, 3H), 2,43~2,48 (m, 1Н), 1,99~2,04 (m, 1Н), 1,32 (d, J=6,4 Гц, 3H).
LC-MS: (масса/заряд) 294,1 (МН+) tR (минуты, Способ 1) = 1,77 минуты
[α]D20=+62,7° (с=0, 10, метанол).
Пример 6

8-Хлор-7-метокси-N-метил-N-(тетрагидрофуран-3-ил)хиназолин-4-амин, стереоизомер 1
К ледяному раствору стереоизомера 1 8-хлор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (150 мг, 0,54 ммоль) в смеси THF (4 мл) и диметилформамида (2 мл) добавляли NaH (32 мг, 0,81 ммоль, 60% в минеральном масле). Смесь перемешивали при 0°C в течение 30 минут. Добавляли метилйодид (100 мг, 0,70 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем гасили насыщ. NH4Cl (водн.) (2 мл). Неочищенную реакционную смесь концентрировали и остаток очищали посредством препаративной TLC (дихлорметан/метанол = 50/1) с получением 8-хлор-7-метокси-N-метил-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (стереоизомер 1).
23 мг (14%).
1H ЯМР (CD3OD, 400 МГц): δ 8,49 (s, 1Н), 8,11 (d, J=9,6 Гц, 1Н), 7,39 (d, J=9,6 Гц, 1Н), 5,24-5,30 (m, 1Н), 4,10-4,13 (m, 1Н), 4,06 (s, 3H), 3,94-3,98 (m, 2Н), 3,73 (q, J=8,0 Гц, 1Н), 3,34 (s, 3H), 2,45-2,49 (m, 1Н), 2,13-2,18 (m, 1Н).
LC-MS: (масса/заряд) 294,0 (МН+) tR (минуты, Способ 3) = 2,55 минуты
[α]D20=19,3° (с=0, 10, CHCl3)
8-Хлор-7-метокси-N-метил-N-(тетрагидрофуран-3-ил)хиназолин-4-амин, стереоизомер 2
К ледяному раствору стереоизомера 2 8-хлор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (150 мг, 0,54 ммоль) в смеси THF (4 мл) и DMF (2 мл) добавляли NaH (32 мг, 0,81 ммоль, 60% в минеральном масле). Реакционную смесь перемешивали при 0°C в течение 30 минут. Затем добавляли CH3I (100 мг, 0,70 ммоль) при 0°C. Обеспечивали нагревание реакционной смеси до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь гасили нас. NH4Cl (водн.) (2 мл). Затем концентрировали и остаток очищали посредством препаративной TLC (дихлорметан/метанол = 50/1) с получением стереоизомера 2 8-хлор-7-метокси-N-метил-N-(тетрагидрофуран-3-ил)хиназолин-4-амина.
25 мг (16%).
1H ЯМР (Н000271637 Н20773-033-2B MeOD varian 400): δ 8,49 (s, 1Н), 8,11 (d, J=9,6 Гц, 1Н), 7,40 (d, J=9,6 Гц, 1Н), 5,24-5,31 (m, 1Н), 4,10-4,13 (m, 1Н), 4,06 (s, 3H), 3,94-3,98 (m, 2Н), 3,73 (q, J=7,6 Гц, 1Н), 3,34 (s, 3H), 2,44-2,49 (m, 1Н), 2,13-2,18 (m, 1Н).
LC-MS: (масса/заряд) 294,0 (МН+) tR (минуты, Способ 1) = 2,11 минуты
[α]D20=-7,7° (с=0, 10, CHCl3).
Пример 7

8-Хлор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин
К раствору 4,8-дихлор-7-метоксихиназолина (350 мг, 1,53 ммоль) в DMSO (30 мл) добавляли 3-метилтетрагидрофуран-3-амин (210 мг, 1,53 ммоль) и NaHCO3 (257 мг, 3,06 ммоль). Смесь нагревали при 100°C в течение 3 часов. Затем охлаждали до комнатной температуры и гасили при помощи H2O (10 мл). Полученную в результате смесь экстрагировали дихлорметаном (3×20 мл). Объединенные органические фазы промывали при помощи H2O (50 мл), сушили над Na2SO4, отфильтровывали и концентрировали. Остаток очищали посредством препаративной HPLC с получением 200 мг 8-хлор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина (45%).
Рацемическую смесь (200 мг) очищали разделением посредством SFC (колонка: Chiralpak AD 250×30 мм внутренний диаметр, 5 мкм) и нумеровали согласно порядку элюирования:
Стереоизомер 1 (первое элюирование посредством SFC): 43 мг
1H ЯМР (CD3OD, 400 МГц): δ 8,46 (s, 1Н), 8,21 (d, J=9,2 Гц, 1Н), 7,39 (d, J=9,6 Гц, 1Н), 4,19 (d, J=9,2 Гц, 1Н), 4,04 (s, 3H), 3,93~3,99 (m, 3H), 2,56~2,62 (m, 1Н), 2,13~2,20 (m, 1Н), 1,67 (s, 3H).
LC-MS: (масса/заряд) 294,0 (МН+) tR (минуты, Способ 4) = 2,16 минуты
[α]D20=+8,3° (с=0, 10, CHCl3).
Стереоизомер 2 (второе элюирование посредством SFC): 39 мг
1H ЯМР (CD3OD, 400): δ 8,46 (s, 1Н), 8,21 (d, J=9,2 Гц, 1Н), 7,39 (d, J=9,2 Гц, 1Н), 4,19 (d, J=9,2 Гц, 1Н), 4,04 (s, 3H), 3,93~3,99 (m, 3H), 2,56~2,62 (m, 1Н), 2,13~2,20 (m, 1Н), 1,67 (s, 3H).
LC-MS: (масса/заряд) 294,0 (МН+) tR (минуты, Способ 4) = 2,17 минуты
[α]D20=-5,7° (с=0, 10, CHCl3).
Пример 8

Цис-4-(8-фтор-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррол
Смесь 4-хлор-8-фтор-7-метоксихиназолина (320 мг, 1,50 ммоль), цис-гексагидро-2Н-фуро[3,2-b]пиррола (200 мг, 1,77 ммоль) и диизопропилэтиламина (457 мг, 3,54 ммоль) в DMF (30 мл) перемешивали при 100°C в течение 12 ч. Раствор концентрировали под вакуумом, остаток разбавляли дихлорметаном (100 мл), промывали солевым раствором (3×10 мл), сушили и концентрировали под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле с использованием градиента этилацетата и петролейного эфира с получением рацемического цис-4-(8-фтор-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола (300 мг, 69%).
Рацемат цис-4-(8-фтор-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола (300 мг) очищали разделением посредством SFC (колонка: IC (250 мм*30 мм,10 мкм)) и нумеровали согласно порядку элюирования:
Стереоизомер 1
100 мг (33%).
1H ЯМР (CDCl3, 400 МГц): δ 8,62 (s, 1Н), 7,90 (dd, J=9,2, 1,6 Гц, 1Н), 7,15 (t, J=8,8 Гц, 1Н), 5,15 (t, J=4,9 Гц, 1Н), 4,60 (d, J=4,2 Гц, 1Н), 4,11-4,09 (m, 2Н), 4,06 (s, 3H), 3,95-3,91 (m, 2Н), 2,44-2,33 (m, 2Н), 2,18-2,15 (m, 1Н), 2,04-2,01 (m, 1Н).
LC-MS (масса/заряд) 290,1 (MH+) tR (минуты, Способ 1) = 1,81
[α]D20 +181,3° (с=0, 10, метанол).
Стереоизомер 2
100 мг (33%).
1H ЯМР (CDCl3, 400 МГц): δ 8,63 (s, 1Н), 7,91 (dd, J=9,2, 1,6 Гц, 1Н), 7,15 (t, J=8,8 Гц, 1Н), 5,15 (t, J=4,8 Гц, 1Н), 4,60 (t, J=4,4 Гц, 1Н), 4,12-4,09 (m, 2Н), 4,06 (s, 3H), 3,95-3,91 (m, 2Н), 2,44-2,33 (m, 2Н), 2,18-2,15 (m, 1Н), 2,04-2,01 (m, 1Н).
LC-MS (масса/заряд) 290,1 (MH+) tR (минуты, Способ 1) = 1,80.
[α]D20 -202° (с=0, 10, метанол).
Пример 9
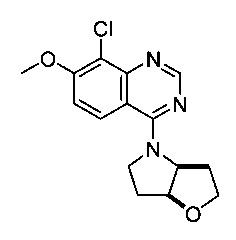
Цис-4-(8-хлор-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррол
К раствору 4,8-дихлор-7-метоксихиназолина (350 мг, 1,53 ммоль) в DMF (12 мл) добавляли гексагидро-2Н-фуро[3,2-b]пиррол (208 мг, 1,84 ммоль) и диизопропилэтиламин (0,54 мл, 3,0 ммоль). Азот барботировали через смесь в течение 2 мин., и ее нагревали при 100°C в течение ночи. Реакционную смесь концентрировали под вакуумом, суспендировали в этилацетате и перемешивали в течение 1 часа при комнатной температуре. Твердое вещество отфильтровывали и промывали этилацетатом с получением 4-(8-хлор-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола (400 мг, 99%). Рацемат 4-(8-хлор-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола (400 мг) очищали разделением посредством SFC (колонка: Chiral Cel OJ 20 мкм, Daicel Chemical Industries, Ltd, 250×30 мм внутренний диаметр) и нумеровали согласно порядку элюирования:
Стереоизомер 1
106 мг (26,5%).
1H ЯМР (CD3OD, 400 МГц): δ 8,47 (s, 1Н), 8,31 (d, J=9,2 Гц, 1Н), 7,40 (d, J=9,6 Гц, 1Н), 5,20 (t, J=4,8 Гц, 1Н), 4,61 (t, J=4,0 Гц, 1Н), 4,14~4,18 (m, 2Н), 4,08 (s, 3H), 3,92~3,95 (m, 2Н), 2,43~2,48 (m, 1Н), 2,29~2,32 (m, 1Н), 2,07~2,13 (m, 1Н).
LC-MS (масса/заряд) 306,1 (MH+) tR (минуты, Способ 1) = 1,88
[α]D20 +280° (с=0, 10, метанол).
Стереоизомер 2
102 мг (25,5%).
1H ЯМР (CD3OD, 400 МГц): δ 8,44 (s, 1Н), 8,28 (d, J=9,6 Гц, 1Н), 7,37 (d, J=9,6 Гц, 1Н), 5,17 (t, J=4,8 Гц, 1Н), 4,59 (t, J=4,0 Гц, 1Н), 4,09~4,15 (m, 2Н), 4,05 (s, 3H), 3,87~3,94 (m, 2Н), 2,37~2,48 (m, 1Н), 2,24~2,32 (m, 1Н), 1,99~2,15 (m, 1Н).
LC-MS (масса/заряд) 306,1 (МН+) tR (минуты, Способ 1) = 1,89
[α]D20 -301° (с=0, 10, метанол).
Промежуточное соединение III

8-Бром-4-хлор-7-метоксихиназолин
Стадия 1. К суспензии 2-амино-3-бром-4-метоксибензойной кислоты (CAS1180497-47-5) (5,50 г, 22,4 ммоль) в триметоксиметане (100 мл) добавляли NH4OAC (17,2 г, 224 ммоль). Смесь нагревали при 90°C в течение 12 ч. Смесь охлаждали до 25°C, твердое вещество отфильтровывали, промывали при помощи H2O (50 мл) и сушили под вакуумом с получением 3,50 г 8-бром-7-метоксихиназолин-4(3H)-она (61,4%).
Стадия 2. К ледяному раствору 8-бром-7-метоксихиназолин-4(3H)-она (3,50 г, 13,7 ммоль) в сухом толуоле (50 мл) по каплям добавляли диизопропилэтиламин (7,09 г, 54,9 ммоль) и POCl3 (18 г, 0,12 моль). Смесь нагревали при 100°C в течение 12 ч., затем охлаждали до 25°C и выливали в H2O (100 мл). Водный слой экстрагировали дихлорметаном (100 мл). Органический слой сушили над Na2SO4, отфильтровывали и концентрировали под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле с использованием градиента дихлорметана и этилацетата с получением 2,4 г 8-бром-4-хлор-7-метоксихиназолина (64%).
Пример 10

Цис-4-(8-бром-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррол
К раствору 8-бром-4-хлор-7-метоксихиназолина (1,30 г, 4,75 ммоль) в сухом диметилформамиде (20 мл) добавляли гексагидро-2Н-фуро[3,2-b]пиррол (860 мг, 7,60 ммоль) и диизопропилэтиламин (1,84 г, 14,3 ммоль). Азот барботировали через смесь в течение 5 мин., и ее нагревали при 100°C в течение 12 ч. в атмосфере N2. Смесь концентрировали под вакуумом, остаток растворяли в дихлорметане (50 мл). Смесь доводили до pH 8 при помощи насыщ. водн. NaHCO3. Водный слой экстрагировали дихлорметаном (50 мл). Объединенные органические фазы сушили над Na2SO4, отфильтровывали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле с использованием градиента дихлорметана и этилацетата с получением 1,6 г 4-(8-бром-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола (95%).
Рацемат цис-4-(8-бром-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола (1,6 г) очищали посредством SFC (колонка: AD 250 мм*50 мм, 10 мкм) разделения и пронумерованы согласно порядку элюирования:
Стереоизомер 1
651 мг (39,3%).
1H ЯМР (CDCl3, 400 МГц): δ 8,70 (s, 1 Н), 8,11 (d, J=9,2 Гц, 1 Н), 7,12 (d, J=9,2 Гц, 1Н), 5,16-5,14 (m, 1Н), 4,58-4,55 (m, 1Н), 4,14-4,08 (m, 2Н), 4,05 (s, 3 Н), 3,94-3,91 (m, 2Н), 2,44-2,31 (m, 2Н), 2,16-2,15 (m, 1Н), 2,03-1,95 (m, 1Н).
LC-MS (масса/заряд) 350,0 (МН+) tR (минуты, Способ 1) = 2,02
[α]D20 +303° (с=0, 10, CHCl3)
Стереоизомер 2
564 мг (35,2%).
1H ЯМР (CDCl3, 400 МГц): δ 8,70 (s, 1 Н), 8,11 (d, J=9,2 Гц, 1 Н), 7,12 (d, J=9,2 Гц, 1Н), 5,16-5,14 (m, 1Н), 4,58-4,56 (m, 1Н), 4,13-4,07 (m, 2Н), 4,05 (s, 3 Н), 3,94-3,90 (m, 2Н), 2,44-2,31 (m, 2Н), 2,16-2,14 (m, 1Н), 2,01-1,98 (m, 1Н).
LC-MS (масса/заряд) 350,0 (МН+) tR (минуты, Способ 1) = 2,02
[α]D20 -233° (с=0, 10, CHCl3)
Пример 11

8-Хлор-7-метокси-N-метил-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин
Стереоизомер 1
К ледяному раствору стереоизомера 2 (пример 7) 8-хлор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина (200 мг, 0,681 ммоль) в THF (10 мл) добавляли NaH (60% дисперсия в минеральном масле) (41 мг, 1,0 ммоль). Смесь перемешивали при 0°C в течение 30 мин. Затем добавляли MeI (126 мг, 0,885 ммоль) при 0°C, и ее перемешивали при 25°C в течение 3 ч. К смеси добавляли H2O (5 мл) и удаляли THF под вакуумом. Остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4, отфильтровывали и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, элюируя дихлорметаном/метанолом = 50/1, с получением 156 мг стереоизомера 1 8-хлор-7-метокси-N-метил-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина (70,7%).
1H ЯМР (CD3OD, 400 МГц): δ 8,49 (s, 1 Н), 8,08 (d, J=9,2 Гц, 1 Н), 7,39 (d, J=9,6 Гц, 1 Н), 4,37 (d, J=8,8 Гц, 1 Н), 4,05 (s, 3 Н), 3,97-3,85 (m, 3 Н), 3,35 (s, 3 Н), 2,47-2,41 (m, 1 Н), 2,31-2,27 (m, 1 Н), 1,69 (s, 3 Н).
LC-MS (масса/заряд) 308,1 (МН+) tR (минуты, Способ 1) = 1,932
[α]D20 +20,33° (с=0, 10, метанол).
Стереоизомер 2
К ледяному раствору стереоизомера 1 (пример 7) 8-хлор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина (200 мг, 0,681 ммоль) в THF (10 мл) добавляли NaH (60% дисперсию в минеральном масле) (41 мг, 1,0 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (126 мг, 0, 885 ммоль). Смесь нагревали до 25°C и перемешивали в течение 3 ч. К реакционной смеси добавляли H2O (5 мл) и удаляли THF под вакуумом. Остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали водой (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, элюируя дихлорметаном/метанолом = 50/1, с получением 141 мг стереоизомера 2 8-хлор-7-метокси-N-метил-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина (63,9%).
1H ЯМР (CD3OD, 400 МГц): δ 8,49 (s, 1 Н), 8,08 (d, J=9,2 Гц, 1 Н), 7,39 (d, J=9,6 Гц, 1 Н), 4,37 (d, J=8,8 Гц, 1 Н), 4,05 (s, 3 Н), 3,97-3,85 (m, 3 Н), 3,35 (s, 3 Н), 2,47-2,44 (m, 1 Н), 2,31-2,6 (m, 1 Н), 1,68 (s, 3 Н).
LC-MS (масса/заряд) 308,1 (МН+) tR (минуты, Способ 1) = 1,950
[α]D20 -26,33° (с=0, 10, метанол).
Пример 12

8-Хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метоксихиназолин-4-амин:
К раствору 4,8-дихлор-7-метоксихиназолина (1,8 г, 7,9 ммоль) в DMSO (30 мл) добавляли 2,3-диметилтетрагидрофуран-3-амин (905 мг, 7,86 ммоль) и NaHCO3 (660 мг, 7,86 ммоль). Смесь нагревали при 100°C в течение 3 ч., а затем охлаждали до 20°C. H2O (30 мл) добавляли в смесь, осадок отфильтровывали и промывали водой (50 мл), сушили и очищали посредством флэш-хроматографии на силикагеле с использованием градиента дихлорметана и метанола с получением 8-хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метоксихиназолин-4-амина (1,2 г, 50
Смесь всех 4 стереоизомеров 8-хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метоксихиназолин-4-амина 2,4 г очищали посредством SFC (колонка: AS 300 мм*50 мм, 10 мкм). Стереоизомеры нумеровали согласно порядку их элюирования.
Стереоизомер 1
400 мг (15,8%).
1H ЯМР (CD3OD, 400 МГц): δ 8,47 (s, 1 Н), 8,17 (d, J=9,2 Гц, 1 Н), 7,38 (d, J=8,8 Гц, 1 Н), 4,52 (q, J=6,4 Гц, 1 Н), 4,04 (s, 3 Н), 4,02-3,96 (m, 1 Н), 3,86 (q, J=8,4 Гц, 1 Н), 2,61-2,55 (m, 1 Н), 2,19-2,13 (m, 1 Н), 1,65 (s, 3 Н), 1,06 (d, J=6,4 Гц, 3 Н).
LC-MS (масса/заряд) 308,1 (МН+) tR (минуты, Способ 5) = 1,267
[α]D20 +20,00° (с=0, 10, метанол).
Стереоизомер 2
300 мг (11,9%).
1H ЯМР (CD3OD, 400 МГц): δ 8,47 (s, 1 Н), 8,16 (d, J=9,2 Гц, 1 Н), 7,38 (d, J=9,6 Гц, 1 Н), 4,51 (q, J=6,4 Гц, 1 Н), 4,04 (s, 3 Н), 4,02-3,97 (m, 1 Н), 3,86 (q, J=8,4 Гц, 1 Н), 2,61-2,55 (m, 1 Н), 2,19-2,13 (m, 1 Н), 1,65 (s, 3 Н), 1,06 (d, J=6,4 Гц, 3 Н).
LC-MS (масса/заряд) 308,1 (МН+) tR (минуты, Способ 5) = 1,272
[α]D20 -13,33° (с=0, 10, метанол).
Стереоизомер 3
600 мг (23,7%).
1H ЯМР (CD3OD, 400 МГц): δ 8,46 (s, 1 Н), 8,20 (d, J=9,2 Гц, 1 Н), 7,36 (d, J=9,2 Гц, 1 Н), 4,47 (q, J=6,4 Гц, 1 Н), 4,03 (s, 3 Н), 3,99-3,90 (m, 2 Н), 2,71-2,66 (m, 1 Н), 2,26-2,21 (m, 1 Н), 1,57 (s, 3 Н), 1,26 (d, J=6,4 Гц, 3 Н).
LC-MS (масса/заряд) 308,1 (МН+) tR (минуты, Способ 5) = 1,295
[α]D20 -25,33° (с=0, 10, метанол).
Стереоизомер 4
700 мг (27,7%).
1H ЯМР (CD3OD, 400 МГц): δ 8,46 (s, 1 Н), 8,21 (d, J=9,2 Гц, 1 Н), 7,36 (d, J=9,2 Гц, 1 Н), 4,47 (q, J=6,4 Гц, 1 Н), 4,04 (s, 3 Н), 3,99-3,90 (m, 2 Н), 2,71-2,66 (m, 1 Н), 2,26-2,21 (m, 1 Н), 1,57 (s, 3 Н), 1,26 (d, J=6,4 Гц, 3 Н).
LC-MS (масса/заряд) 308,1 (МН+) tR (минуты, Способ 5) = 1,30
[α]D20 +43,67° (с=0, 10, метанол).
Пример 13

8-Хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метокси-N-метилхиназолин-4-амин
Стереоизомер 1
К ледяному раствору стереоизомера 1 из примера 12 (200 мг, 0,650 ммоль) в THF (10 мл) добавляли NaH (60% дисперсия в минеральном масле) (39 мг, 0,98 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (120 мг, 0,845 ммоль). Реакционной смеси позволяли нагреться до 25°C и перемешивали в течение 3 ч. К смеси добавляли H2O (5 мл) и удаляли THF под вакуумом. Остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4, концентрировали под вакуумом и очищали посредством препаративной TLC, используя дихлорметан/метанол = 50/1, с получением стереоизомера 1 8-хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метокси-N-метилхиназолин-4-амина (118 мг, 57%).
1H ЯМР (CD3OD, 400 МГц): δ 8,54 (s, 1 Н), 8,12 (d, J=9,2 Гц, 1 Н), 7,46 (d, J=9,2 Гц, 1 Н), 4,09 (s, 3 Н), 4,07-4,04 (m, 1 Н), 3,94-3,88 (m, 1 Н), 3,40 (s, 3 Н), 2,66-2,58 (m, 1 Н), 2,26-2,21 (m, 1 Н), 1,73 (s, 3 Н), 0,96 (d, J=6,4 Гц, 3 Н).
LC-MS (масса/заряд) 322,2 (MH+) tR (минуты, Способ 5) = 1,46
[α]D20 -9,67° (с=0, 10, метанол).
Стереоизомер 2
К ледяному раствору стереоизомера 2 из примера (200 мг, 0,650 ммоль) в THF (10 мл) добавляли NaH (60% дисперсия в минеральном масле) (39 мг, 0,98 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (120 мг, 0,845 ммоль). Реакционную смесь перемешивали при 25°C в течение 3 ч., а затем добавляли H2O (5 мл). THF удаляли под вакуумом, а остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя дихлорметан/метанол = 50/1, с получением стереоизомера 2 8-хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метокси-N-метилхиназолин-4-амина (120 мг, 57%).
1H ЯМР (CD3OD, 400 МГц): δ 8,54 (s, 1 Н), 8,12 (d, J=9,2 Гц, 1 Н), 7,46 (d, J=9,2 Гц, 1 Н), 4,09 (s, 3 Н), 4,07-4,04 (m, 1 Н), 3,94-3,88 (m, 1 Н), 3,40 (s, 3 Н), 2,66-2,58 (m, 1 Н), 2,25-2,21 (m, 1 Н), 1,73 (s, 3 Н), 0,96 (d, J=6,0 Гц, 3 Н).
LC-MS (масса/заряд) 322,2 (MH+) tR (минуты, Способ 5) = 1,46
[α]D20 +5,33° (с=0, 10, метанол).
Стереоизомер 3
К ледяному раствору стереоизомера 3 из примера 12 (200 мг, 0,650 ммоль) в THF (10 мл) добавляли NaH (60% дисперсия в минеральном масле) (39 мг, 0,98 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (120 мг, 0,845 ммоль) при 0°C. Реакционную смесь перемешивали при 25°C в течение 3 ч., а затем добавляли H2O (5 мл). THF удаляли под вакуумом, а остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя дихлорметан/метанол = 50/1, с получением стереоизомера 3 8-хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метокси-N-метилхиназолин-4-амина (78 мг, 37%).
1H ЯМР (CD3OD, 400 МГц): δ 8,61 (s, 1 Н), 8,08 (d, J=9,6 Гц, 1 Н), 7,48 (d, J=9,6 Гц, 1 Н), 4,50-4,47 (m, 1 Н) 4,10 (s, 3 Н), 3,95-3,91 (m, 1 Н), 3,88-3,84 (m, 1 Н), 3,31 (s, 3 Н), 2,67-2,62 (m, 1 Н), 2,32-2,26 (m, 1 Н), 1,65 (s, 3 Н), 1,39 (d, J=6,4 Гц, 3 Н).
LC-MS (масса/заряд) 322,2 (MH+) tR (минуты, Способ 5) = 1,50
[α]D20 -49,33° (с=0, 10, метанол).
Стереоизомер 4
К ледяному раствору стереоизомера 4 из примера 12 (200 мг, 0,650 ммоль) в THF (10 мл) добавляли NaH (60% дисперсия в масле) (39 мг, 0,98 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (120 мг, 0,845 ммоль). Реакционную смесь перемешивали при 25°C в течение 3 ч. Затем добавляли H2O (5 мл) и удаляли THF под вакуумом. Остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя дихлорметан/метанол = 50/1, с получением стереоизомера 4 8-хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метокси-N-метилхиназолин-4-амина (92 мг, 44%).
1H ЯМР (CD3OD, 400 МГц): δ 8,62 (s, 1 Н), 8,08 (d, J=9,6 Гц, 1 Н), 7,48 (d, J=9,6 Гц, 1 Н), 4,50-4,47 (m, 1 Н) 4,10 (s, 3 Н), 3,95-3,91 (m, 1 Н), 3,88-3,84 (m, 1 Н), 3,32 (s, 3 Н), 2,67-2,62 (m, 1 Н), 2,32-2,26 (m, 1 Н), 1,65 (s, 3 Н), 1,39 (d, J=7,6 Гц, 3 Н).
LC-MS (масса/заряд) 322,2 (MH+) tR (минуты, Способ 5) = 1,50
[α]D20 +33,33° (с=0, 10, метанол).
Пример 14

8-Хлор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин
Стереоизомер 1
К ледяному раствору стереоизомера 1 из примера 5 (8-хлор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин) (250 мг, 0,851 ммоль) в THF (10 мл) добавляли 60% суспензию NaH в минеральном масле (61 мг, 1,5 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (181 мг, 1,28 ммоль) при 0°C. После перемешивания при 30°C в течение 3 ч. к смеси добавляли H2O (5 мл). THF удаляли под вакуумом, а остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя дихлорметан/метанол = 50/1, с получением стереоизомера 1 8-хлор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина (192 мг, 70.
1H ЯМР (CDCl3, 400 МГц): δ 8,75 (s, 1 Н), 7,92 (d, J=8,8 Гц, 1 Н), 7,17 (d, J=9,6 Гц, 1 Н), 4,82-4,76 (m, 1 Н), 4,18 (q, J=6,4 Гц, 1 Н), 4,08 (s, 3 Н), 4,05-4,02 (m, 2 Н), 3,29 (s, 3 Н), 2,54-2,49 (m, 1 Н), 2,18-2,11 (m, 1 Н), 1,28 (d, J=6,0 Гц, 3 Н).
LC-MS (масса/заряд) 308,1 (MH+) tR (минуты, Способ 1) = 1,96
[α]D20 -46,00° (с=0, 10, метанол).
Стереоизомер 2
К ледяному раствору стереоизомера 2 из примера 5 (8-хлор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин) (250 мг, 0,851 ммоль) в THF (10 мл) добавляли 60% суспензию в минеральном масле NaH (61 мг, 1,5 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (181 мг, 1,28 ммоль). Реакционную смесь перемешивали при 30°C в течение 3 ч. перед добавлением H2O (5 мл). THF удаляли под вакуумом, а остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя дихлорметан/метанол = 50/1, с получением стереоизомера 2 8-хлор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина (217 мг, 80%).
1H ЯМР (CDCl3, 400 МГц): δ 8,70 (s, 1 Н), 7,95 (d, J=9,6 Гц, 1 Н), 7,14 (d, J=9,6 Гц, 1 Н), 5,47-5,42 (m, 1 Н), 4,20-4,17 (m, 1 Н), 4,07 (s, 3 Н), 4,05-4,02 (m, 2 Н), 3,73 (q, J=8,8 Гц, 1 Н), 3,38 (s, 3 Н), 2,47-2,42 (m, 1 Н), 2,33-2,30 (m, 1 Н), 1,28 (d, J=6,4 Гц, 3 Н).
LC-MS (масса/заряд) 308,1 (MH+) tR (минуты, Способ 1) = 1,94
[α]D20 -48,00° (с=0, 10, метанол).
Стереоизомер 3
К ледяному раствору стереоизомера 3 из примера 5 (8-хлор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин) (160 мг, 0,545 ммоль) в THF (10 мл) добавляли 60% суспензию NaH в минеральном масле (39 мг, 0,98 ммоль) и смесь перемешивали при 0°C в течение 30 мин. Добавляли MeI (116 мг, 0,817 ммоль) при 0°C. Реакционную смесь перемешивали при 30°C в течение 3 ч., а затем добавляли H2O (5 мл). THF удаляли под вакуумом, а остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя дихлорметан/метанол = 50/1, с получением стереоизомера 3 8-хлор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина
139 мг (81%).
1H ЯМР (CDCl3, 400 МГц): δ 8,70 (s, 1 Н), 7,95 (d, J=9,6 Гц, 1 Н), 7,14 (d, J=9,2 Гц, 1 Н), 5,47-5,42 (m, 1 Н), 4,20-4,16 (m, 1 Н), 4,07 (s, 3 Н), 4,05-4,02 (m, 2 Н), 3,75-3,73 (m, 1 Н), 3,38 (s, 3 Н), 2,47-2,42 (m, 1 Н), 2,34-2,30 (m, 1 Н), 1,28 (d, J=6,0 Гц, 3 Н).
LC-MS (масса/заряд) 308,1 (MH+) tR (минуты, Способ 1) = 1,96
[α]D20 +52,67° (с=0, 10, метанол).
Стереоизомер 4
К ледяному раствору стереоизомера 4 из примера 5 (8-хлор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин) (200 мг, 0,681 ммоль) в THF (10 мл) добавляли 60% суспензию NaH в минеральном масле (49 мг, 1,2 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (145 мг, 1,02 ммоль). Реакционную смесь перемешивали при 30°C в течение 3 ч., а затем добавляли H2O (5 мл). THF удаляли под вакуумом, а остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, дихлорметан/метанол = 50/1, с получением стереоизомера 4 8-хлор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина 184 мг (85%).
1H ЯМР (CDCl3, 400 МГц): δ 8,75 (s, 1 Н), 7,92 (d, J=9,2 Гц, 1 Н), 7,17 (d, J=9,2 Гц, 1 Н), 4,82-4,76 (m, 1 Н), 4,19-4,16 (m, 1 Н), 4,08 (s, 3 Н), 4,05-4,02 (m, 2 Н), 3,29 (s, 3H), 2,54-2,49 (m, 1 Н), 2,18-2,11 (m, 1 Н), 1,28 (d, J=6,4 Гц, 3 Н).
LC-MS (масса/заряд) 308,1 (MH+) tR (минуты, Способ 1) = 1,94
[α]D20 +35,33° (с=0, 10, метанол).
Пример 15
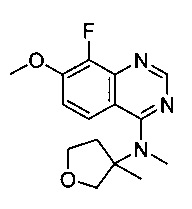
8-Фтор-7-метокси-N-метил-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин
Стереоизомер 1
К раствору стереоизомера 1 из примера 1 (8-фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин) (200 мг, 0,721 ммоль) в THF (4 мл) добавляли 60% суспензию NaH в минеральном масле (43 мг, 1,1 ммоль) при 0°C. После перемешивания в течение 30 мин. реакционную смесь нагревали до 20°C. Добавляли MeI (154 мг, 1,08 ммоль) и реакционную смесь перемешивали в течение 12 ч. Реакционную смесь гасили насыщ. водн. NH4Cl (0,5 мл) и концентрировали под вакуумом. Остаток разбавляли дихлорметаном (20 мл), промывали солевым раствором (10 мл), сушили и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя этилацетат, с получением 150 мг стереоизомера 1 8-фтор-7-метокси-N-метил-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина (70%).
1H ЯМР (CDCl3, 400 МГц): δ 8,65 (s, 1Н), 7,73 (dd, J=9,6, 2,0 Гц, 1Н), 7,19 (dd, J=9,6, 8,0 Гц, 1Н), 4,35 (d, J=8,8 Гц, 1Н), 4,07 (s, 3H), 4,02-3,88 (m, 3H), 3,31 (s, 3H), 2,41-2,35 (m, 1Н), 2,28-2,24 (m, 1Н), 1,71 (s, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 1) = 1,83
[α]D20 +38,33° (с=0, 10, метанол).
Стереоизомер 2
К раствору стереоизомера 2 из примера 1 (8-фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амин) (200 мг, 0,721 ммоль) в THF (4 мл) добавляли 60% суспензию в минеральном масле NaH (43 мг, 1,1 ммоль) при 0°C и реакционную смесь перемешивали в течение 30 мин. перед нагревом до 20°C. Добавляли MeI (154 мг, 1,08 ммоль) и перемешивание продолжали в течение 12 ч. Раствор гасили при помощи насыщ. водн. NH4Cl (1 мл) и концентрировали под вакуумом. Остаток разбавляли дихлорметаном (20 мл), промывали солевым раствором (3×8 мл), сушили над MgSO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя этилацетат, с получением 160 мг стереоизомера 2 8-фтор-7-метокси-N-метил-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина (75%).
1H ЯМР (CDCl3, 400 МГц): δ 8,65 (s, 1Н), 7,73 (dd, J=9,6, 1,6 Гц, 1Н), 7,18 (dd, J=9,2, 8,0 Гц, 1Н), 4,35 (d, J=9,2 Гц, 1Н), 4,02 (s, 3H), 4,00-3,88 (m, 3H), 3,31 (s, 3H), 2,43-2,35 (m, 1Н), 2,28-2,24 (m, 1Н), 1,71 (s, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 1) = 1,83
[α]D20 -30,00° (с=0, 10, метанол).
Пример 16

8-Фтор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амин
Раствор 4-хлор-8-фтор-7-метоксихиназолина (400 мг, 1,88 ммоль), тетрагидрофуран-3-амина (192 мг, 2,26 ммоль) и диизопропилэтиламина (486 мг, 3,76 ммоль) в DMF (10 мл) перемешивали при 100°C в течение 3 ч. Реакционную смесь концентрировали под вакуумом и очищали посредством флэш-хроматографии на силикагеле с использованием градиента дихлорметана/этилацетата с получением 360 мг 8-фтор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (72%).
Рацемат 8-фтор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина (360 мг) очищали разделением посредством SFC (колонка: AD-H (250 мм*30 мм, 5 мкм)) и нумеровали согласно порядку элюирования:
Стереоизомер 1
150 мг (42%).
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,48 (dd, J=9,2, 1,6 Гц, 1Н), 7,22-7,18 (m, 1Н), 5,86 (d, J=6,4 Гц, 1Н), 4,99-4,94 (m, 1 Н), 4,05-3,99 (m, 1Н), 3,99 (s, 3H), 3,97-3,96 (m, 1Н), 3,91-3,86 (m, 2Н), 2,49-2,43 (m, 1Н), 2,05-2,00 (m, 2Н).
LC-MS (масса/заряд) 264,1 (MH+) tR (минуты, Способ 1) = 1,72
[α]D20 +29,33° (с=0, 10, метанол).
Стереоизомер 2
150 мг (42%).
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,48 (d, J=8,8 Гц, 1Н), 7,22-7,18 (m, 1Н), 5,86 (d, J=6,4 Гц, 1Н), 4,99-4,94 (m, 1Н), 4,05-4,00 (m, 1Н), 4,00 (s, 3H), 3,97-3,96 (m, 1Н), 3,91-3,88 (m, 2Н), 2,49-2,44 (m, 1Н), 2,05-2,00 (m, 2Н).
LC-MS (масса/заряд) 264,1 (MH+) tR (минуты, Способ 1) = 1,72
[α]D20 -34,67° (с=0, 10, метанол).
Пример 17
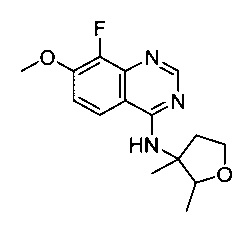
N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин
К раствору 4-хлор-8-фтор-7-метоксихиназолина (1,00 г, 4,70 ммоль) в DMSO (15 мл) добавляли 2,3-диметилтетрагидрофуран-3-амин (541 мг, 4,70 ммоль) и NaHCO3 (395 мг, 4,70 ммоль). Смесь нагревали при 100°C в течение 3 ч., а затем охлаждали до 25°C. Добавляли H2O (50 мл) и смесь экстрагировали дихлорметаном (2×50 мл). Объединенные органические фазы промывали водой (2×50 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле с использованием градиента дихлорметана и метанола с получением 700 мг N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амина (51%).
2,3 г смеси всех 4 стереоизомеров N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амина очищали посредством SFC (колонка: Chiral Рак AD, 5 мкм, Daicel Chemical Industries, Ltd, 250×30 мм внутренний диаметр).
Стереоизомер 3
300 мг (13%).
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,43-7,41 (m, 1Н), 7,21-7,17 (m, 1Н), 5,74 (s, 1Н), 4,05 (s, 3H), 3,92-3,85 (m, 3H), 2,97-2,91 (m, 1Н), 2,10-2,03 (m, 1Н), 1,71 (s, 3H), 1,36 (d, J=6,4 Гц, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 1) = 1,88
[α]D20 -25,67° (с=0, 10, метанол).
Стереоизомер 4
300 мг (13%).
1H ЯМР (CDCl3, 400 МГц): δ 8,64 (s, 1Н), 7,42-7,39 (m, 1Н), 7,19-7,15 (m, 1Н), 5,71 (s, 1Н), 4,02 (s, 3H), 3,89-3,82 (m, 3H), 2,94-2,88 (m, 1Н), 2,08-2,01 (m, 1Н), 1,69 (s, 3H), 1,33 (d,J=6,4 Гц, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 1) = 1,86
[α]D20 +27,33° (с=0, 10, метанол)
Посредством первой очистки при помощи SFC получали смесь стереоизомеров 1 и 2. Эту смесь подвергали второй очистке при помощи SFC (колонка: Chiral Рак AS, 5 мкм, Daicel Chemical Industries, Ltd, 250×30 мм внутренний диаметр) хроматографии с получением:
Стереоизомер 2
600 мг (26%).
1H ЯМР (CDCl3, 400 МГц): δ 8,60 (s, 1Н), 7,45-7,42 (m, 1Н), 7,15-7,11 (m, 1Н), 5,63 (s, 1Н), 4,27 (q, J=6,4 Гц, 1Н), 3,99 (s, 3H), 3,95-3,85 (m, 2Н), 2,72-2,66 (m, 1Н), 2,22-2,17 (m, 1Н), 1,56 (s, 3H), 1,23 (d, J=6,4 Гц, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 1) = 1,90
[α]D20 +41,33° (с=0, 10, метанол).
Стереоизомер 1
600 мг (26%).
1H ЯМР (CDCl3, 400 МГц): δ 8,60 (s, 1Н), 7,44-7,42 (m, 1Н), 7,16-7,12 (m, 1Н), 5,61 (s, 1Н), 4,27 (q, J=6,4 Гц, 1Н), 4,00 (s, 3H), 3,99-3,87 (m, 2Н), 2,73-2,66 (m, 1Н), 2,22-2,18 (m, 1Н), 1,57 (s, 3H), 1,23 (d, J=6,0 Гц, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 1) = 1,89
[α]D20 -23,67° (с=0, 10, метанол).
Пример 18

8-Фтор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амин
Стереоизомер 1
К раствору стереоизомера 2 из примера 3 (N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин) (150 мг, 0,540 ммоль) в сухом THF (5 мл) добавляли 60% суспензию NaH в минеральном масле (32 мг, 0,81 ммоль) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 30 мин., затем добавляли CH3I (92 мг, 0,65 ммоль). Реакционную смесь перемешивали в течение 2 ч. при 0°C перед добавлением насыщ. водн. NH4Cl (5 мл). Неочищенную реакционную смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии с использованием градиента петролейного эфира и этилацетата с получением 135 мг стереоизомера 1 8-фтор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина (86%).
1H ЯМР (CDCl3, 400 МГц): δ 8,62 (s, 1Н), 7,79 (dd, J=9,6, 2,0 Гц, 1Н), 7,17-7,13 (m, 1Н), 5,50-5,45 (m, 1Н), 4,20-4,16 (m, 1Н), 4,07 (s, 3H), 4,04-4,03 (m, 1Н), 3,77-3,71 (m, 1Н), 3,39 (s, 3H), 2,46-2,42 (m, 1Н), 2,33-2,29 (m, 1Н), 1,29 (d, J=6,8 Гц, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 2) = 1,80
[α]D20 +43,00° (с=0, 10, метанол).
Стереоизомер 2
К раствору стереоизомера 1 из примера 3 (N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин) (150 мг, 0,540 ммоль) в THF (5 мл) добавляли 60% суспензию NaH в минеральном масле (32 мг, 0,81 ммоль) при 0°C в атмосфере N2. Смесь перемешивали в течение 30 мин. при 0°C, а затем добавляли метилйодид (92 мг, 0,65 ммоль). Перемешивание продолжали в течение 2 ч. при 0°C, а затем добавляли насыщ. водн. NH4Cl (5 мл). Смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии с использованием градиента петролейного эфира и этилацетата с получением 130 мг стереоизомера 2 8-фтор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина (82,5%).
1H ЯМР (CDCl3, 400 МГц): δ 8,67 (s, 1Н), 7,76 (d, J=9,6 Гц, 1Н), 7,20-7,16 (m, 1Н), 4,83 (brs, 1Н), 4,19-4,16 (m, 1Н), 4,07-4,02 (m, 5Н), 3,31 (s, 3H), 2,50 (brs, 1Н), 2,13 (brs, 1Н), 1,30 (d, J=6,0 Гц, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 2) = 1,82
[α]D20 -30,00° (с=0, 10, метанол).
Стереоизомер 3
К раствору стереоизомера 3 из примера 3 (N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин) (150 мг, 0,540 ммоль) в THF (5 мл) добавляли 60% суспензию NaH в минеральном масле (32 мг, 0,81 ммоль) при 0°C в атмосфере N2 и смесь перемешивали в течение 30 мин. Добавляли метилйодид (77 мг, 0,54 ммоль) при 0°C и перемешивание продолжали в течение 2 ч. при 0°C. Реакционную смесь гасили при помощи насыщ. водн. NH4Cl (5 мл), а затем экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии с использованием градиента петролейного эфира и этилацетата с получением стереоизомера 3 8-фтор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина (110 мг(69,8%)).
1H ЯМР (CDCl3, 400 МГц): δ 8,61 (s, 1Н), 7,79 (dd, J=9,6, 2,0 Гц, 1Н), 7,17-7,13 (m, 1Н), 5,49-5,45 (m, 1Н), 4,21-4,16 (m, 1Н), 4,06 (s, 3H), 4,04-4,02 (m, 1Н), 3,77-3,73 (m, 1Н), 3,39 (s, 3H), 2,46-2,42 (m, 1Н), 2,33-2,29 (m, 1Н), 1,29 (d, J=6,4 Гц, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 2) = 1,81
[α]D20 -73,00° (с=0, 10, метанол).
Стереоизомер 4
К раствору стереоизомера 4 из примера 3 (N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин) (150 мг, 0,541 ммоль) в THF (5 мл) добавляли 60% суспензию NaH в минеральном масле (32 мг, 0,81 ммоль) при 0°C в атмосфере N2 и смесь перемешивали в течение 30 мин. Затем перемешивание с метилйодидом (92 мг, 0,65 ммоль) продолжали в течение 2 ч. при 0°C. Реакционную смесь гасили добавлением насыщ. водн. NH4Cl (5 мл). Реакционную смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством флэш-хроматографии с использованием градиента петролейного эфира и этилацетата с получением 120 мг стереоизомера 4 8-фтор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина (76,2%).
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,76 (dd, J=9,6, 2,0 Гц, 1Н), 7,20-7,16 (m, 1Н), 4,86-4,81 (m, 1Н), 4,19-4,14 (m, 1Н), 4,07 (s, 3H), 4,06-4,02 (m, 2Н), 3,31 (s, 3H), 2,54-2,48 (m, 1Н), 2,17-2,10 (m, 1Н), 1,30 (d, J=6,4 Гц, 3H).
LC-MS (масса/заряд) 292,1 (MH+) tR (минуты, Способ 2) = 1,82
[α]D20 +90,33° (с=0, 10, метанол).
Пример 19
N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метокси-N-метилхиназолин-4-амин

Стереоизомер 1
К ледяному раствору стереоизомера 3 из примера 17 (N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин) (200 мг, 0,687 ммоль) в THF (10 мл) добавляли 60% дисперсию NaH в минеральном масле (55 мг, 1,4 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (146 мг, 1,03 ммоль). Реакционной смеси позволяли нагреться до 30°C и перемешивали в течение 2 ч. Добавляли H2O (5 мл) и удаляли THF под вакуумом. Остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя петролейный эфир и этилацетат 1/1, с получением стереоизомера 1 N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метокси-N-метилхиназолин-4-амина
127 мг (60,4%).
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,71 (dd, J=9,2, 2,0 Гц, 1Н), 7,21-7,17 (m, 1Н), 4,81 (q, J=6,4 Гц, 1Н), 4,06 (s, 3H), 4,04-4,01 (m, 1Н), 3,94-3,87 (m, 1Н), 3,31 (s, 3H), 2,48-2,40 (m, 1Н), 2,17-2,13 (m, 1Н), 1,71 (s, 3H), 0,93 (d, J=6,4 Гц, 3H).
LC-MS (масса/заряд) 306,2 (MH+) tR (минуты, Способ 1) = 1,89
[α]D20 -8,00° (с=0, 10, метанол).
Стереоизомер 2
К ледяному раствору стереоизомера 2 из примера 17 (N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин) (200 мг, 0,687 ммоль) в THF (10 мл) добавляли 60% суспензию NaH в минеральном масле (55 мг, 1,3 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 мин., а затем добавляли MeI (146 мг, 1,03 ммоль). Реакционную смесь затем нагревали до 30°C и перемешивали в течение 2 ч. К смеси добавляли H2O (5 мл) и удаляли THF под вакуумом. Остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя петролейный эфир и этилацетат 1/1, с получением стереоизомера 2 N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метокси-N-метилхиназолин-4-амина.
70 мг (33%).
1H ЯМР (CDCl3, 400 МГц): δ 8,72 (s, 1Н), 7,71 (dd, J=9,6, 2,0 Гц, 1Н), 7,22-7,18 (m, 1Н), 4,38-4,35 (m, 1Н), 4,06 (s, 3H), 3,93-3,89 (m, 1Н), 3,84-3,80 (m, 1Н), 3,22 (s, 3H), 2,54-2,50 (m, 1Н), 2,30-2,25 (m, 1Н), 1,60 (s, 3H), 1,37 (d, J=6,0 Гц, 3H).
LC-MS (масса/заряд) 306,2 (MH+) tR (минуты, Способ 1) = 1,93
[α]D20 +38,00° (с=0, 10, метанол).
Стереоизомер 3
К ледяному раствору стереоизомера 4 из примера 17 (N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин) (200 мг, 0,687 ммоль) в THF (10 мл) добавляли 60% суспензию NaH в минеральном масле (55 мг, 1,4 ммоль). Смесь перемешивали при 0°C в течение 30 мин., а затем добавляли метилйодид (146 мг, 1,03 ммоль). Реакционную смесь затем нагревали до 30°C и перемешивали в течение 2 ч. К смеси добавляли H2O (5 мл) и удаляли THF под вакуумом. Остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали водой (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя петролейный эфир и этилацетат 1/1, с получением стереоизомера 3 N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метокси-N-метилхиназолин-4-амина.
103 мг (49,2%).
1H ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1Н), 7,72 (dd, J=9,2, 1,6 Гц, 1Н), 7,21-7,17 (m, 1Н), 4,84-4,80 (m, 1Н), 4,06 (s, 3H), 4,04-4,01 (m, 1Н), 3,94-3,89 (m, 1Н), 3,31 (s, 3H), 2,48-2,40 (m, 1Н), 2,17-2,13 (m, 1Н), 1,71 (s, 3H), 0,93 (d, J=6,4 Гц, 3H).
LC-MS (масса/заряд) 306,2 (MH+) tR (минуты, Способ 1) = 1,88
[α]D20 +13,00° (с=0, 10, метанол).
Стереоизомер 4
К ледяному раствору стереоизомера 1 из примера 17 (N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амин) (200 мг, 0,687 ммоль) в THF (10 мл) добавляли 60% суспензию NaH (55 мг, 1,4 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 мин., а затем добавляли метилйодид (146 мг, 1,03 ммоль). Смеси позволяли нагреться до 30°C и перемешивали в течение 2 ч. К смеси добавляли H2O (5 мл) и удаляли THF под вакуумом. Остаток экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы промывали при помощи H2O (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством препаративной TLC, используя петролейный эфир и этилацетат = 1/1, с получением стереоизомера 4 N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метокси-N-метилхиназолин-4-амина.
73 мг (35%).
1H ЯМР (CDCl3, 400 МГц): δ 8,72 (s, 1Н), 7,72 (dd, J=9,2, 2,0 Гц, 1Н), 7,22-7,18 (m, 1Н), 4,39-4,35 (m, 1Н), 4,06 (s, 3H), 3,92-3,89 (m, 1Н), 3,84-3,80 (m, 1Н), 3,22 (s, 3H), 2,54-2,50 (m, 1Н), 2,30-2,25 (m, 1Н), 1,60 (s, 3H), 1,37 (d, J=6,4 Гц, 3H).
LC-MS (масса/заряд) 306,2 (MH+) tR (минуты, Способ 1) = 1,90
[α]D20 -54,33° (с=0, 10, метанол).
АНАЛИЗ ИНГИБИРОВАНИЯ PDE1
Анализы в отношении PDE1A, PDE1B и PDE1C проводили следующим образом: анализы проводили в 60 мкл образцов, содержащих фиксированное количество фермента PDE1 (достаточное для превращения 20-25% субстрата циклического нуклеотида), буфер (50 мМ HEPES с pH 7,6; 10 мМ MgCl2; 0,02% Tween20), 0,1 мг/мл BSA, 15 нМ меченого тритием cAMP и различные количества ингибиторов. Реакции инициировали путем добавления субстрата циклического нуклеотида, и обеспечивали протекание реакций в течение 1 ч. при комнатной температуре перед их завершением посредством смешивания с 20 мкл (0,2 мг) гранул SPA из силиката иттрия (PerkinElmer). Обеспечивали осаждение гранул в течение 1 ч. в темноте перед подсчетом планшетов на устройстве для подсчета Wallac 1450 Microbeta. Измеренные сигналы переводили в активность относительно неингибированного контроля (100%), и значения IC50 рассчитывали при помощи XIFit (модель 205, IDBS).
| название | год | авторы | номер документа |
|---|---|---|---|
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| Применение 1H-пиразоло[4,3-b]пиридинов в качестве ингибиторов PDE1 | 2017 |
|
RU2759380C2 |
| C-Анилинохиназолиновые соединения и их использование в лечении рака | 2018 |
|
RU2769694C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АНГИОГЕНЕЗА | 2000 |
|
RU2262935C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2827641C1 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
Изобретение относится к соединению (I), где X представляет собой галоген; R1 выбран из Н; R2 выбран из группы, состоящей из Н и C1-С4алкила; или R2 вместе с R9 и атомами, соединяющими их, образуют насыщенное пятичленное кольцо; R3 выбран из группы, состоящей из Н и C1-С6алкила; R4 и R5 независимо друг от друга выбраны из Н; R6 и R7 независимо друг от друга выбраны из Н; R8 и R9 независимо друг от друга выбраны из группы, состоящей из Н и C1-С6алкила, или к фармацевтически приемлемой кислотно-аддитивной соли соединения I, рацемической смеси соединения I или к соответствующему энантиомеру и/или оптическому изомеру соединения I. Изобретение также относится к фармацевтической композиции, ингибирующей PDE1, на основе соединения I. Технический результат – получены новые соединения и фармацевтические композиции на их основе, которые могут найти применение в медицине для лечения нейродегенеративных и психических расстройств, таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, депрессии, гиперактивности с дефицитом внимания (ADHD) и др. 4 н. и 9 з.п. ф-лы, 1 табл., 19 пр.

1. Соединение со структурой:

где
X представляет собой галоген;
R1 выбран из Н;
R2 выбран из группы, состоящей из Н и C1-С4алкила;
или R2 вместе с R9 и атомами, соединяющими их, образуют насыщенное пятичленное кольцо;
R3 выбран из группы, состоящей из Н и C1-С6алкила;
R4 и R5 независимо друг от друга выбраны из Н;
R6 и R7 независимо друг от друга выбраны из Н;
R8 и R9 независимо друг от друга выбраны из группы, состоящей из Н и C1-С6алкила,
или фармацевтически приемлемая кислотно-аддитивная соль соединения I, рацемическая смесь соединения I или соответствующий энантиомер и/или оптический изомер соединения I.
2. Соединение по п. 1, где R2 представляет собой Н или -СН3.
3. Соединение по п. 1, где по меньшей мере один из R6 и R7 представляет собой Н.
4. Соединение по п. 3, где как R6, так и R7 представляют собой Н.
5. Соединение по п. 1, где по меньшей мере четыре из R3 - R9 представляют собой Н.
6. Соединение по п. 1, где R2 и R9 образуют пятичленное насыщенное алифатическое кольцо.
7. Соединение по п. 1, где R3 представляет собой C1-С6 алкил.
8. Соединение по п. 1, где X представляет собой фтор.
9. Соединение по п. 1, где X представляет собой хлор.
10. Соединение по п. 1, выбранное из группы, состоящей из
8-фтор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина;
8-фтор-7-метокси-N-метил-N-(тетрагидрофуран-3-ил)хиназолин-4-амина;
8-хлор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина;
8-фтор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина;
8-хлор-7-метокси-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина;
8-хлор-7-метокси-N-метил-N-(тетрагидрофуран-3-ил)хиназолин-4-амина;
8-хлор-7-метокси-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина;
цис-4-(8-фтор-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола;
цис-4-(8-хлор-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола;
цис-4-(8-бром-7-метоксихиназолин-4-ил)гексагидро-2Н-фуро[3,2-b]пиррола;
8-хлор-7-метокси-N-метил-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина;
8-хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метоксихиназолин-4-амина;
8-хлор-N-(2,3-диметилтетрагидрофуран-3-ил)-7-метокси-N-метилхиназолин-4-
амина;
8-хлор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина;
8-фтор-7-метокси-N-метил-N-(3-метилтетрагидрофуран-3-ил)хиназолин-4-амина;
8-фтор-7-метокси-N-(тетрагидрофуран-3-ил)хиназолин-4-амина;
N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метоксихиназолин-4-амина;
8-фтор-7-метокси-N-метил-N-(2-метилтетрагидрофуран-3-ил)хиназолин-4-амина и
N-(2,3-диметилтетрагидрофуран-3-ил)-8-фтор-7-метокси-N-метилхиназолин-4-амина.
11. Фармацевтическая композиция для лечения нейродегенартивного заболевания, такого как болезнь Альцгеймера, болезнь Паркинсона и болезнь Хантингтона, или психического расстройства, такого как гиперактивность с дефицитом внимания, депрессии, нарколепсии, когнитивного нарушения и когнитивного нарушения, связанного с шизофренией (CIAS), или заболевания мозга, такого как синдром беспокойных ног, содержащая эффективное количество соединения по любому из пп. 1-10 и один или несколько фармацевтически приемлемых носителей или разбавителей.
12. Применение соединения по любому из пп. 1-10 для лечения нейродегенартивного заболевания, такого как Болезнь Альцгеймера, болезнь Паркинсона и болезнь Хантингтона, или психического расстройства, такого как гиперактивность с дефицитом внимания, депрессии, нарколепсии, когнитивного нарушения и когнитивного нарушения, связанного с шизофренией (CIAS), или заболевания мозга, такого как синдром беспокойных ног.
13. Применение соединения по любому из пп. 1-10 для лечения гиперактивности с дефицитом внимания (ADHD).
| WO 2007096743 A1, 30.08.2007 | |||
| WO 2007103370 A2, 13.09.2007 | |||
| ДОЖДЕМЕР | 1926 |
|
SU5679A1 |

