Настоящее изобретение относится к фенилуксусным кислотам и их производным, определенным в настоящем изобретении, которые являются особенно активными и селективными ингибиторами циклооксигеназы-2 (ЦОГ-2), к способам их получения, фармацевтическим композициям, включающим указанные соединения, к способам селективного ингибирования активности ЦОГ-2 и лечения у млекопитающих патологических состояний, которые реагируют на ингибирование ЦОГ-2, с применением указанных соединений или фармацевтических композиций, включающих указанные соединения, предлагаемые в настоящем изобретении.
Настоящее изобретение относится к новым фенилуксусным кислотам и их производным, которые ингибируют ЦОГ-2 без значительного ингибирования циклооксигеназы-1 (ЦОГ-1). Таким образом, настоящее изобретение относится к новым нестероидным противовоспалительным средствам, которые, как неожиданно оказалось, не приводят к нежелательным побочным эффектам, обычно проявляющимся при использовании классических нестероидных противовоспалительных средств, таким как побочные эффекты, связанные с желудочно-кишечным трактом и почками.
Поэтому соединения, предлагаемые в настоящем изобретении, являются особенно подходящими или путем метаболизма могут превратиться в соединения, которые являются особенно подходящими для применения в качестве селективных ингибиторов ЦОГ-2. Поэтому они являются особенно подходящими для лечения зависящих от ЦОГ-2 нарушений у млекопитающих, включая воспаление, нагноения, боли, остеоартрита, ревматоидного артрита, дисменореи, мигрени, рака (такого как рак пищеварительного тракта, например, колоректального рака и меланомы), боли при раке, острой боли, хронической боли, нейродегенеративных заболеваний (таких как рассеянный склероз, болезнь Паркинсона и болезнь Альцгеймера), сердечно-сосудистых заболеваний (таких как атеросклероз, ишемическая болезнь сердца и артериосклероз), остеопороза, подагры, острой подагры, астмы, волчанки и псориаза в основном с исключением нежелательного изъязвления желудочно-кишечного тракта, проявляющегося при использовании обычных ингибиторов циклооксигеназы (ЦОГ). Соединения, предлагаемые в настоящем изобретении, также являются поглотителями УФ-излучения, в особенности поглотителями излучения УФ-В, и применимы для защиты от УФ-излучения или поглощения УФ-излучения, например, для лечения и предупреждения солнечных ожогов, например, в средствах для загара.
Применение соединений, предлагаемых в настоящем изобретении, в офтальмологии включает лечение воспаления глаз, мокрой формы возрастной дегенерации желтого пятна, боли глаз, включая боль после операции глаз, такой как фоторефракционная кератэктомия или операция по поводу катаракты, аллергических заболеваний глаз, фотофобии разной этиологии, повышенного внутриглазного давления (при глаукоме) путем ингибирования продуцирования индуцируемого трабекулярной сетью глюкокортикоидного ответного белка и сухого кератита.
Соединения, предлагаемые в настоящем изобретении, применимы для лечения неоплазии, в особенности неоплазии, при которой продуцируются простагландины или экспрессируется ЦОГ, включая злокачественные и раковые опухоли, наросты и полимы, в частности, неоплазии, образующейся из эпителиальных клеток. Соединения, предлагаемые в настоящем изобретении, в частности, применимы для лечения рака печени, мочевого пузыря, поджелудочной железы, яичников, предстательной железы, шейки матки, легких и молочной железы и в особенности рака желудочно-кишечного тракта, например, рака ободочной кишки и рака кожи, например, плоскоклеточного и базальноклеточного рака и меланомы, как указано выше.
Термин "лечение" при использовании в настоящем изобретении включает и терапевтические, и профилактические режимы лечения, например, лечение неоплазии, лечение с целью предупреждения начала клинически или доклинически диагностируемой неоплазии или предупреждение инициирования злокачественных клеток или остановки или обращения прогрессирования превращения предраковых клеток в раковые, а также предупреждения или подавление роста или метастазирования неоплазии. В этом контексте настоящее изобретение, в частности, следует понимать как включающее применение соединений, предлагаемых в настоящем изобретении, для предупреждения или подавления развития рака кожи, например, плоскоклеточной или базальноклеточной карциномы, вызванной воздействием УФ-излучения, например, вызванной длительным воздействием солнечного света. Соединения можно применять для людей и других млекопитающих.
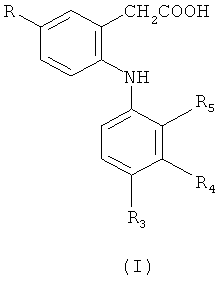
Первым объектом настоящего изобретения является соединение формулы (I)
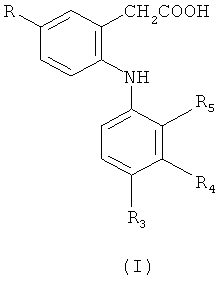
в которой
R обозначает метил или этил;
R3 обозначает галоген или С1-С6-алкил;
R4 обозначает C1-С6-алкил;
R5 обозначает галоген;
указанный выше C1-С6-алкил по R3 и R4 необязательно замещен одной или большим количеством галогенидных групп;
его фармацевтически приемлемые соли; и его фармацевтически приемлемые сложные эфиры.
Предпочтительно, если в соединениях формулы (I) R3 обозначает галоген, метил или этил. Более предпочтительно, если он обозначает галоген. Альтернативно предпочтительно, если он обозначает хлор.
Предпочтительно, если в соединениях формулы (I) R4 обозначает C1-С3 алкил, необязательно замещенный одним или большим количеством галогенидных групп. Более предпочтительно, если он обозначает метил, этил, изопропил или трифторметил. Еще более предпочтительно, если он обозначает метил. Альтернативно предпочтительно, если он обозначает трифторметил.
Предпочтительно, если в соединениях формулы (I) R5 обозначает хлор или фтор.
Еще более предпочтительно, если оба R3 и R5 независимо выбраны из группы, включающей хлор и фтор.
Предпочтительно, если соединение выбрано из следующего перечня соединений:
5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусная кислота
5-этил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусная кислота
5-метил-2-(2'-фтор-3'-трифторметил-4'-этиланилино)фенилуксусная кислота
5-этил-2-(2'-фтор-3'-трифторметил-4'-этиланилино)фенилуксусная кислота
5-метил-2-(2'-фтор-3'-трифторметил-4'-метиланилино)фенилуксусная кислота
5-этил-2-(2'-фтор-3'-трифторметил-4'-метиланилино)фенилуксусная кислота
5-метил-2-(2'-фтор-3'-метил-4'-хлоранилино)фенилуксусная кислота
5-этил-2-(2'-фтор-3'-метил-4'-хлоранилино)фенилуксусная кислота
5-метил-2-(2'-фтор-3'-этил-4'-хлоранилино)фенилуксусная кислота
5-этил-2-(2'-фтор-3'-этил-4'-хлоранилино)фенилуксусная кислота
5-метил-2-(2',4'-дихлор-3'-этиланилино)фенилуксусная кислота
5-этил-2-(2',4'-дихлор-3'-этиланилино)фенилуксусная кислота
5-этил-2-(2'-фтор-3'-изопропил-4'-хлоранилино)фенилуксусная кислота
соль 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты с диэтиламином
5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетат натрия
соль 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты с трометамином
моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата кальция
моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата лизина
моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата холина
5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетат калия.
Вторым объектом настоящего изобретения является фармацевтическая композиция, включающая эффективное количество соединения формулы (I) в комбинации с одним или большим количеством фармацевтически приемлемых носителей.
Третьим объектом настоящего изобретения является способ лечения зависящих от циклооксигеназы-2 (ЦОГ-2) нарушений у млекопитающих, который включает введение нуждающемуся в нем млекопитающему эффективного количества соединения формулы (I).
Четвертым объектом настоящего изобретения является способ селективного ингибирования активности ЦОГ-2 у млекопитающего без значительного ингибирования ингибирования активности циклооксигеназы-1, который включает введение нуждающемуся в нем млекопитающему эффективно ингибирующего ЦОГ-2 количества соединения формулы (I).
Пятым объектом настоящего изобретения является способ лечения ревматоидного артрита, остеоартрита, дисменореи, боли, опухолей или воспаления у млекопитающих, который включает введение нуждающемуся в нем млекопитающему соответствующего эффективного количества соединения формулы (I).
Шестым объектом настоящего изобретения является применение соединения формулы (I) для приготовления лекарственного средства, предназначенного для лечения ревматоидного артрита, остеоартрита, дисменореи, боли, опухолей или воспаления.
Седьмым объектом настоящего изобретения является соединение формулы (I), предназначенное для применения для лечения зависящего от ЦОГ-2 нарушения.
Восьмым объектом настоящего изобретения является способ получения соединения формулы (I) по пункту 1 формулы изобретения, который включает стадии:
(а) проведения сочетания соединения формулы (II) или (III)
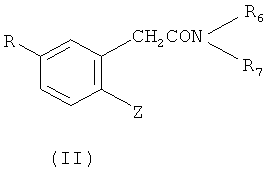 или
или 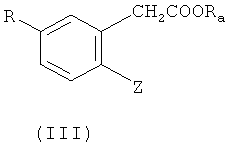
в которой
Z обозначает бром или йод;
R обладает определенным выше значением;
Ra обозначает водород, катион щелочного металла или низш. алкил, предпочтительно - изопропил и
R6 и R7 обозначают низш. алкил или R6 и R7 вместе с атомом азота обозначают морфолиновую, пиперидиновую или пирролидиновую группу,
с соединением формулы (IV)
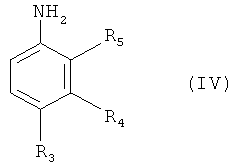
в которой R3-R5 обладают определенными выше значениями, в присутствии меди и йодида меди (I) с получением соединения формулы (V) или (VI)
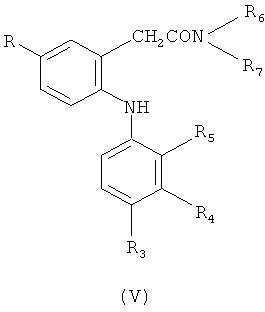 или
или 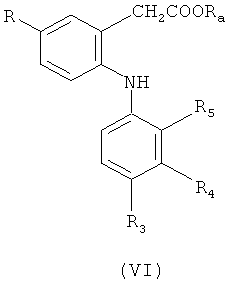
и гидролиза полученного соединения формулы (V) или (VI) с получением соединения формулы (I); или
(b) конденсации соединения формулы (VII)
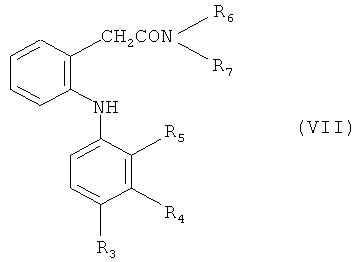
в которой R3-R7 обладают значениями, определенными в настоящем изобретении, с реакционноспособным функциональным производным кислоты, например, уксусной кислоты, таким как ацетилхлорид, по реакции ацилирования Фриделя-Крафтса с получением, например, соединения формулы (VIII)
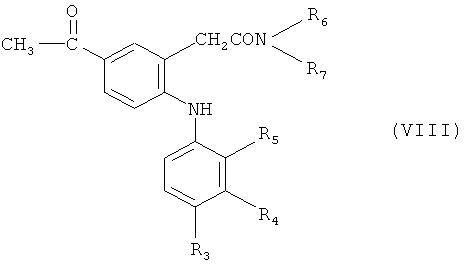
в которой R3-R7 обладают значениями, определенными в настоящем изобретении, которое, в свою очередь, подвергают гидрогенолизу, а затем гидролизу с получением соединения формулы (I), в которой R обозначает, например, этил; или
(с) гидролиза лактама формулы (IX)
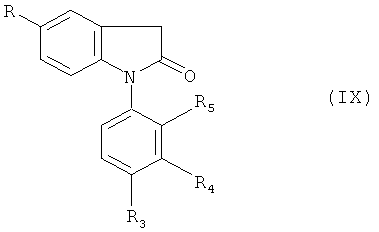
в которой R и R3-R5 обладают значениями, определенными в настоящем изобретении, сильным основанием; и в указанных выше реакциях при необходимости защиты любых мешающих реакционноспособных групп с последующим выделением полученного соединения, предлагаемого в настоящем изобретении; и при необходимости превращения любого полученного соединения в другое соединение, предлагаемое в настоящем изобретении; и/или при необходимости превращения свободной карбоновой кислоты, предлагаемой в настоящем изобретении, в ее фармацевтически приемлемый сложный эфир; и/или при необходимости превращения полученной свободной кислоты в соль или полученной соли в свободную кислоту или в другую соль.
В исходных соединениях и промежуточных продуктах, которые превращают в соединения, предлагаемые в настоящем изобретении, по методикам, описанным в настоящем изобретении, содержащиеся функциональные группы, такие как аминогруппы, гидроксигруппы и карбоксигруппы, необязательно защищают обычными защитными группами, применяющимися в препаративной органической химии. Защитными группами гидроксигрупп, аминогрупп и карбоксигрупп являются такие, которые в мягких условиях можно превратить в свободные аминогруппы, гидроксигруппы и карбоксигруппы без протекания нежелательных побочных реакций. Например, защитными группами гидроксигрупп предпочтительно являются бензильная или замещенная бензильная группы или ацильные группы, такие как пивалоил.
Получение соединений формул (V) и (VI) способом (а) проводят при условиях модифицированной реакции конденсации Ульмана, использующейся для получения диариламинов, например, в присутствии порошкообразной меди и йодида меди (I) и карбоната калия, необязательно в инертном высококипящем растворителе, таком как нитробензол, толуол, ксилол или N-метилпирролидон, при повышенной температуре, например, в диапазоне 100-200°С, предпочтительно - при температуре кипения растворителя, по общей методологии, описанной в публикации Nohara, Chem Abstr, Vol.94, p.15402x (1951); и Moser et al., J Med Chem, Vol.33, p.2358 (1990). Если Z обозначает бром, то конденсацию проводят в присутствии йодида, например, йодида калия.
Гидролиз полученных орто-анилинофенилацетамидов формулы (V) проводят в водном растворе гидроксида щелочного металла, например, в 6 н. NaOH в присутствии спирта, например, этанола, пропанола и бутанола, при повышенной температуре, такой как температура кипения реакционной смеси.
Гидролиз сложных эфиров формулы (VI) проводят по методикам, известным в данной области техники, например, в щелочной среде, как описано выше для соединений формулы (V) или альтернативно в кислой среде, например, с использованием метансульфоновой кислоты.
Исходные вещества формулы (II) или (III) обычно известны или их можно получить по методикам, известным в данной области техники, например, как описал Nohara в заявке на патент Японии No.78/96434 (1978); в патенте U.S. No.6291523 и как описано в настоящем изобретении.
Например, соответствующую антраниловую кислоту превращают в орто-диазониевое производное с последующей обработкой йодидом щелочного металла в кислоте, например, серной кислоте, и получают 2-йодбензойную кислоту или ее низш. алкиловый эфир. Восстановление соответствующего бензилового спирта, например, дибораном или алюмогидридом лития в случае сложного эфира, превращение спирта сначала в бромид и затем в нитрил, гидролиз нитрила в уксусную кислоту и превращение в N,N-диалкиламида по методикам, известным в данной области техники, дает исходное вещество формулы (II).
Альтернативно, например, исходное вещество формулы (II), в которой Z обозначает Br и R обозначает циклопропил, можно получить сначала путем конденсации по методике, описанной в публикации J Am Chem Soc, Vol.123, p.4155 (2001), например, метилового эфира 2-бром-5-йодбензойной кислоты с циклопропилбромидом в присутствии трихлорида индия с получением метилового эфира 2-бром-5-циклопропилбензойной кислоты, который, как описано выше, превращают в соответствующий 2-бром-5-циклопропилфенилацетамид формулы (II).
Кроме того, исходные вещества формулы (II), в которой R обозначает, например, этил, можно получить ацетилированием по Фриделю-Крафтсу оксиндола, например, ацетилхлоридом в присутствии хлорида алюминия, восстановлением полученного кетона, например, с помощью каталитического гидрогенолиза с последующим гидролитическим расщеплением полученного 5-этилоксиндола в орто-аминофенилуксусную кислоту. Диазотирование в присутствии, например, йодида калия дает орто-йодфенилуксусную кислоту, которую превращают в амид формулы (II).
Сложные эфиры формулы (III) получают из соответствующих кислот путем этерификации по методикам, известным в данной области техники.
Анилины формулы (IV) или известны в данной области техники, или их получают по методикам, хорошо известным в данной области техники, и как показано в настоящем изобретении.
Получение, например, 5-этил- или 5-н-пропилзамещенных соединений способом (b) проводят ацетилированием по Фриделю-Крафтсу, например, в присутствии хлорида алюминия в инертном растворителе, таком как 1,2-дихлорэтан, с последующим гидрогенолизом, например, с использованием палладия на угле в качестве катализатора, предпочтительно - в уксусной кислоте в качестве растворителя, при комнатной температуре и при давлении около 3 атм.
Исходные вещества формулы (VII) обычно получают, как это описано для способа (а), с использованием в качестве исходного вещества амида формулы (II), в которой R обозначает водород, например, как описано в публикации Moser et al. (1990), см. выше.
Получение соединений, предлагаемых в настоящем изобретении, способом (с) можно провести при условиях, известных в данной области техники для гидролитического расщепления лактамов, предпочтительно - водным раствором сильного основания, таким как водный раствор гидроксида натрия, необязательно в присутствии смешивающегося с водой органического растворителя, такого как метанол, при повышенной температуре в диапазоне около 50-100°С, как это описано в патенте US No.3558690.
Исходные оксиндолы формулы (IX) получают N-ацилированием диариламина формулы (X)
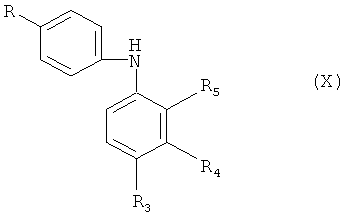
в которой R и R1-R5 обладают определенными выше значениями, галогенацетилхлоридом, предпочтительно - хлорацетилхлоридом, предпочтительно - при повышенной температуре, например, около 100°С, с получением соединения формулы (XI)
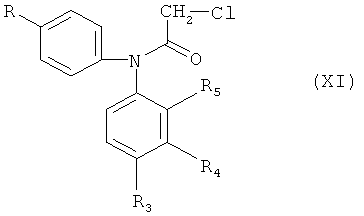
в которой R и R3-R5 обладают значениями, определенными выше в настоящем изобретении. Циклизацию соединения формулы (XI) проводят алкилированием по Фриделю-Крафтсу в инертном растворителе, таком как дихлорбензол, в присутствии катализаторов Фриделя-Крафтса, например, хлорида алюминия или этилалюминийдихлорида, при повышенной температуре, например, при 120-175°С.
Исходные амины формулы (X) можно получить конденсацией по Ульману и другим методикам, известным в данной области техники, например, по реакции сочетания Бухвальда.
Эфиры карбоновых кислот формулы (I) получают конденсацией карбоновой кислоты в виде соли или в присутствии основания галогенидом (бромидом или хлоридом), соответствующим этерифицирующему спирту, таким как бензилхлорацетат, по методикам, хорошо известным в данной области техники, например, в полярном растворителе, таком как N,N-диметилформамид, и при необходимости дополнительно изменяют полученный продукт. Например, если продукт этерификации сам является эфиром, то его можно превратить в карбоновую кислоту, например, с помощью гидрогенолиза полученного бензилового эфира. Если продукт этерификации сам является галогенидом, то его, например, можно превратить в нитрооксипроизводное по реакции, например, с нитратом серебра.
Например, соединения формулы (Ia) предпочтительно получают путем конденсации соли карбоновой кислоты формулы (I), приведенной выше, с соединением формулы
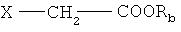
в которой
Х обозначает отщепляющуюся группу и
Rb обозначает защитную группу карбоксигруппы;
с получением соединения формулы (Ia) с защищенной карбоксигруппой и с последующим удалением защитной группы Rb.
Этерификацию можно провести при условиях этерификации, известных в данной области техники, например, в полярном растворителе, таком как N,N-диметилформамид, при температуре в диапазоне от комнатной температуры примерно до 100°С, предпочтительно - в диапазоне 40-60°С, например, по методике, описанной в патенте US №5291523.
Солью кислоты формулы (I) предпочтительно является соль щелочного металла, например, соль натрия, которую можно получить in situ.
Отщепляющейся группой Х предпочтительно является галоген, например, хлор, или бром, или низш. алкилсульфонилоксигруппа, например, метансульфонилоксигруппа.
Защитной группой карбоксигруппы Rb предпочтительно является бензил.
Полученные бензиловые эфиры можно превратить в свободные кислоты формулы (Ia) предпочтительно с помощью гидрогенолиза водородом в присутствии, например, Pd/C в качестве катализатора в уксусной кислоте при атмосферном давлении или гидрированием по Парру при температуре в диапазоне от комнатной температуры примерно до 50°С.
Настоящее изобретение относится к любым новым исходным веществам и способам их получения.
Кроме того, соединения, предлагаемые в настоящем изобретении, получают в свободной форме или в виде их солей, если содержатся солеобразующие группы.
Кислотные соединения, предлагаемые в настоящем изобретении, можно превратить в соли металлов путем взаимодействия с фармацевтически приемлемыми основаниями, например, с водным раствором гидроксида щелочного металла, предпочтительно в присутствии простого эфира или спирта в качестве растворителя, такого как низш. алканол. Полученные соли можно превратить в свободные соединения путем обработки кислотами. Эти и другие соли также можно использовать для очистки полученных соединений. Соли аммония получают по реакции с соответствующим амином, например, диэтиламином и т.п.
Соединения, предлагаемые в настоящем изобретении, содержащие основные группы, можно превратить в соли присоединения с кислотами, предпочтительно в фармацевтически приемлемые соли. Они образуются, например, с неорганическими кислотами, например, серной кислотой, фосфорной или галогенводородной кислотой, или с органическими карбоновыми кислотами, такими как (С1-С4)алкаркарбоновые кислоты, которые, например, являются незамещенными или замещены галогеном, например, уксусной кислотой, такими как насыщенные или ненасыщенные дикарбоновые кислоты, например, щавелевая, янтарная, малеиновая или фумаровая кислота, такими как гидроксикарбоновые кислоты, например, гликолевая, молочная, яблочная, винная или лимонная кислота, такими как аминокислоты, например, аспарагиновая или глутаминовая кислота, или с органическими сульфоновыми кислотами, такими как (С1-С4)алкилсульфоновые кислоты, например, метансульфоновая кислота, или арилсульфоновыми кислотами, которые могут быть незамещенными или замещенными, например, галогеном. Предпочтительными являются соли, образованные с хлористоводородной кислотой, метансульфоновой кислотой и малеиновой кислотой.
Ввиду близкого родства между свободными соединениями и соединениями в форме их солей любое указание на соединение в этом контексте следует понимать и как указание на соответствующую соль, если при данных обстоятельствах это является подходящим и целесообразным.
Соединения, включая их соли, также можно получить в форме их гидратов или с включением других растворителей, использовавшихся для их кристаллизации.
Общие определения, использующиеся в настоящем изобретении, если не указано иное, в объеме настоящего изобретения обладают приведенными ниже значениями.
Фармацевтически приемлемые сложные эфиры предпочтительно являются сложными эфирами пролекарств, которые путем сольволиза или при физиологических условиях могут превращаться в свободные карбоновые кислоты, например, формулы (I). Такие сложные эфиры представляют собой, например, низш. алкиловые сложные эфиры, такие как метиловый или этиловый эфир; карбокси-низш. алкиловые сложные эфиры, такие как карбоксиметиловый эфир; нитроокси- или нитрозоокси-низш. алкиловые сложные эфиры, такие как 4-нитрооксибутиловый или 4-нитрозооксибутиловый эфир и т.п.
Предпочтительными являются фенилацетоксиуксусные кислоты формулы (Ia)
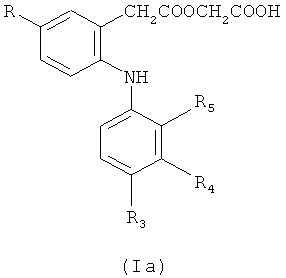
в которой R, R3, R4 и R5 обладают значениями, определенными выше в настоящем изобретении для соединений формулы (I), и его фармацевтически приемлемых солей.
Фармацевтически приемлемые соли представляют собой соли металлов, такие как соли щелочных металлов, например, соли натрия, калия, магния или кальция, а также соли аммония, которые образуются, например, с аммиаком и моно- или диалкиламинами, такими как соли диэтиламмония; и с аминокислотами, такие как соли аргинина и гистидина.
Низш. алкильная группа содержит до 6 атомов углерода, предпочтительно 1-4 атома углерода, может обладать разветвленной или линейной цепью и представляет собой, например, метил, этил, пропил, бутил, изопропил, изобутил и т.п., предпочтительно метил или этил. Низш. алкоксигруппа означает метоксигруппу, этоксигруппу и т.п.
Галоген предпочтительно означает хлор, бром или фтор, более предпочтительно хлор или фтор.
Соединения, предлагаемые в настоящем изобретении, применимы в качестве ингибиторов селективных ЦОГ-2 или в качестве их пролекарств. Селективные ингибиторы ЦОГ-2 и их пролекарства, предлагаемые в настоящем изобретении, особенно полезны для лечения, например, воспаления, нагноения, боли, остеоартрита, дисменореи, ревматоидного артрита и других патологических состояний, реагирующих на ингибирование ЦОГ-2, и обычно практически не приводят к нежелательным побочным эффектам, связанным с желудочно-кишечным трактом, обычно проявляющимся при использовании стандартных нестероидных противовоспалительных средств.
Указанные выше характеристики можно продемонстрировать с помощью проводимых in vitro и in vivo исследований, предпочтительно с использованием млекопитающих, например, крыс, мышей и обезьян и изолированных клеток или ферментных препаратов, полученных от людей или из других источников. Такие соединения можно применять in vitro в воде растворов, например, водных растворов, и in vivo, предпочтительно перорально, местно или парентерально, например, внутривенно. Доза in vitro может составлять примерно 10-5-10-9 М. Доза in vivo в зависимости от пути введения может меняться примерно от 0,1 мг/кг до 100 мг/кг.
Биологические характеристики можно продемонстрировать с помощью исследований, хорошо известных в данной области техники, например, как это описано в патенте US No.6291523 и как описано в настоящем изобретении.
Ингибирование ЦОГ-2 изучают с помощью ферментного исследования in vitro с использованием имеющегося в продаже набора (Cayman Chemical Company).
Исследуемое соединение (исходный раствор в ДМСО, разведенный буфером до разных концентраций) предварительно инкубируют с 30-50 Ед очищенного рекомбинантного ЦОГ-2 человека и гемактином (1 мкМ) в течение 30 мин при 25°С, затем инкубируют с 100 мкМ арахидоновой кислоты и субстратом для колориметрического исследования ТМПД (N,N,N',N'-тетраметил-п-фенилендиамин) в течение 5-7 мин при 25°С с последующим колориметрическим определением окисленного ТМПД при 590 нм. Активность ЦОГ-2 в присутствии исследуемого соединения сопоставляют с активностью ЦОГ-2 для контрольного образца, не содержащего исследуемого соединения.
Ингибирование ЦОГ также исследуют in vitro с использованием методик изучения ингибирования ЦОГ-1 и ЦОГ-2.
Исследования с помощью клеток для изучения ингибиторов ЦОГ хорошо известны в данной области техники и основаны на том, что фермент ЦОГ (синтеза простагландина Н) катализирует лимитирующую стадию синтеза простагландина из арахидоновой кислоты. Два фермента опосредуют эту реакцию: ЦОГ-1 является конститутивной формой фермента, а ЦОГ-2 индуцируется в ответ на различные факторы роста и цитокины.
Ингибирование ЦОГ-1 и ЦОГ-2 in vitro изучают с помощью клеток для оценки активности и селективности ингибирования ЦОГ-2 in vitro с помощью иммунологического анализа простагландина Е2 (набор Cayman PGE2). Используют клетки HEK-293 EBNA, которые трансфицированы и стабильно экспрессируют рекомбинантный ЦОГ-1 человека или рекомбинантный ЦОГ-2 соответственно. Клетки помещают в 96-луночные планшеты, в которых проводят анализ. Клетки обеих линий предварительно обрабатывают разведенными соединениями в течение 30 мин при 37°С, затем прибавляют арахидоновую кислоту (1 мкМ) в качестве экзогенного субстрата. Надосадочный слой собирают через 15 мин и с помощью иммунологического анализа определяют продуцирование PGE2. Для определения IC50 соединения исследуют при 5-9 концентрациях с однократным, двукратным или четырехкратным повтором при каждой концентрации (наибольшая концентрация равна 30 мкМ). Рассчитывают среднее ингибирование PGE2 (по сравнению с клетками, не обработанными соединением) для каждой концентрации и строят зависимость выраженного в % среднего ингибирования от логарифма концентрации соединения и значение IC50 рассчитывают с помощью 4-параметрической логистической аппроксимации. Для оценки ингибирования ЦОГ-2 сопоставляют относительное воздействие каждого фермента.
Ингибирование in vitro ЦОГ-1 и ЦОГ-2 также определяют в цельной крови человека, в которой ЦОГ-1 конститутивно экспрессируется в тромбоцитах, и экспрессирование ЦОГ-2 индуцируют в мононуклеарах путем обработки липополисахаридом (ЛПС) (10 мкг/мл). Для этого исследования гепаринизованную кровь человека делят на 2 аликвоты: одну для определения продуцирования ТхВ2 (имитатор индикатора активности ЦОГ-1) и вторую для определения продуцирования PGE2 (имитатор индикатора активности ЦОГ-2). Перед стимулированием пробы крови предварительно обрабатывают исследуемыми соединениями в течение 1 ч. Соединения исследуют в диапазоне конечных концентраций от 0,1 нМ до 300 мкМ с использованием полулогарифмического увеличения концентраций. Для исследования ингибирования генерации тромбоксана В2 (ТхВ2) прибавляют А23187 (50 мкМ) и кровь инкубируют в течение 1 ч. Продуцирование PGE2 определяют после прибавления ЛПС (10 мкг/мл) с последующим инкубированием в течение ночи. После инкубирования с А23187 или ЛПС образцы центрифугируют при 250×g в течение 10 мин при 4°С для сбора сыворотки. Количества PGE2 и ТхВ2, содержащиеся в сыворотке, определяют с помощью хемилюминесцентного иммуноферментного анализа по методике фирмы Assay Designs Inc. (Ann Arbor, MI). Содержание простагландина в каждом образце нормируют на выраженное в процентах ингибирование для каждой концентрации исследуемого соединения. Данные по выраженному в процентах ингибированию для каждого донора объединяют и аппроксимируют 4-параметричрской логистической функцией с использованием регрессионного анализа.
Значения IC50 для соединений формулы (I), полученные при исследовании ингибирования ЦОГ-2, составляют лишь примерно 0,10 мкМ или даже меньше. Предпочтительными являются соединения, для которых отношение значений IC50 для ингибирования ЦОГ-1 и ЦОГ-2 равно более 50, предпочтительно находится примерно в диапазоне 100-1000 или является более значительным. Например, с помощью описанного выше исследования получены следующие значения IC50, в случае проведения более одного анализа приведены средние значения.
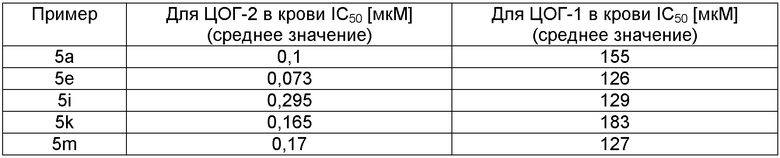
Ингибирование продуцирования простагландина E2, выработанного посредством ЦОГ-2, исследуют in vivo при индуцировании липополисахаридом (ЛПС) в подкожном воздушном кармане у крыс. См. Advances in Inflammation Research, Raven Press (1986); J. Med. Chem., Vol.39, p.1846 (1996); J. Pathol., Vol.141, pp.483-495; и J. Pathol, Vol.134, pp.147-156.
Самок крыс линии Lewis анестезируют и с помощью подкожной инъекции 10 мл воздуха через стерильный фильтр с диаметром отверстий 0,45 мкм, встроенный в шприц, формируют дорсальные воздушные карманы. Через 6 или 7 дней после формирования в воздушные карманы путем инъекции вводят ЛПС (5 мкг на карман), суспендированный в стерильном забуференном фосфатом физиологическом растворе. Соединения для исследования вводят через желудочный зонд за 1 ч до или через 2 или большее количество часов после введения ЛПС. Содержимое карманов собирают через 5 с после введения ЛПС и с помощью иммуноферментного анализа определяют содержание PGE2 в жидкости из кармана. Иллюстрирующее настоящее изобретение соединение примера 4(j) ингибирует образование PGE2 примерно на 50% при пероральном введении в количестве 1 мг/кг.
Противовоспалительную активность изучают путем исследования индуцируемой каррагенаном отека лапки у крыс с помощью модификации методики, описанной в публикации Offerness et al., описанной в Nonsteroidal Antiinflammatory Drugs, Lombardino, Ed., John Wiley & Sons, pp.116-128 (1986).
Крысам линии Sprague Dawley (200-225 г) не дают корм в течение ночи и затем им перорально вводят соединение, растворенное в 0,5% металцеллюлозе. Через 1 ч в подподошвенный участок левой задней лапки вводят 0,1 мл 1% каррагенана, который вызывает воспалительную реакцию. Через 3 ч после введения каррагенана крыс умерщвляют и обе задние лапки отрезают по линии границы шерстяного покрова и взвешивают на электронных весах. Степень отечности в воспаленной лапке определяют путем вычитания массы невоспаленной лапки (правой) из массы воспаленной лапки (левой). Выраженное в процентах ингибирование соединением определяют для каждого животного в виде выраженного в процентах увеличения массы лапки по сравнению со средним значением для контроля.
Исследование переносимости в желудке используют для оценки полного изъязвления у крыс, его проводят через 4 ч после перорального введения исследуемого соединения. Исследование проводят следующим образом.
Самцам крыс линии Sprague Dawley не дают корм в течение ночи, затем им через желудочный зонд вводят соединение в 0,5% растворе метилцеллюлозы и через 4 ч их умерщвляют, давая вдыхать диоксид углерода. Удаляют желудки и подсчитывают и измеряют полное поражение желудка, получают полную длину поражения в расчете на 1 крысу. В каждый эксперимент включены следующие группы животных (5-6 крыс в группе): контроль с использованием только растворителя, исследуемые соединения и диклофенак в качестве соединения сравнения.
Данные рассчитывают как среднее количество изъязвлений в группе, среднюю длину изъязвлений (мм) в группе и как показатель изъязвлений (ПИ).
ПИ = средняя длина изъязвлений в группе × частота изъязвлений,
где частота изъязвлений означает долю животных с изъязвлениями в группе (для 100% частота равна 1).
Иллюстрирующие настоящее изобретение соединения примеров при пероральной дозе, равной 30 мг/кг, практически не приводят к изъязвлениям в желудке.
Переносимость в кишечнике можно исследовать путем определения влияния на проницаемость кишечника. Отсутствие увеличения проницаемости свидетельствует о переносимости в кишечнике.
Использующаяся методика является модификацией методики, описанной в публикации Davies et al., Pharm. Res., Vol.11, pp.1652-1656 (1994) и основана на том, что экскреция вводимой перорально 51Cr-ЭДТК (этилендиаминтетрауксусная кислота), маркера проницаемости тонкого кишечника, увеличивается при воздействии НСПС (нестероидные противовоспалительные средства). Группам самцов крыс линии Sprague Dawley (≥12 в каждой группе) перорально через желудочный зонд вводят одну дозу исследуемого соединения или растворителя. Сразу после введения соединения каждой крысе через желудочный зонд вводят 51Cr-ЭДТК (5 мкКи на крысу). Крыс помещают в отдельные клетки для изучения метаболизма и в неограниченном количество дают корм и воду. В течение 24 ч собирают мочу. Через 24 ч после введения 51Cr-ЭДТК крыс умерщвляют. Для количественно изучения влияния соединения на проницаемость кишечника количество выведенного 51Cr-ЭДТК, определенное в моче крыс, которым вводили соединение, сопоставляют с количеством выведенного 51Cr-ЭДТК, определенным в моче крыс, которым соединение растворялось. Относительную проницаемость определяют путем расчета радиоактивности в каждом образце мочи, выраженной в процентах от введенной дозы с поправкой на радиоактивный фон.
Аналгетическую активность соединений, предлагаемых в настоящем изобретении, исследуют путем изучения индуцируемой посредством полного адъюванта Фрейнда (ПАФ) гипералгезии у крыс. 50 мкл ПАФ путем инъекции вводят в левую заднюю лапку. Болевой порог определяют за 24 ч и после инъекции ПАФ с помощью стандартного прибора, нажимающего на лапку (Analgesymeter, Ugo Basile, Milan). Конечную точку определяют как отдергивание или движение лапки. Соединения растворяют в 0,5% метилцеллюлозе и вводят перорально и дополнительные определения болевого порога проводят через 1, 3 и 6 ч после введения соединения. В каждой подопытной группе 6 животным вводили растворитель или одну дозу соединения. Результаты представляют в виде выраженного в процентах обращения гипералгезии до введения дозы. Для каждого соединения рассчитывают дозу, при которой обеспечивается 30% ингибирование гипералгезии до введения дозы (D30), и получают общую оценку активности.
Противоартритное воздействие соединений, предлагаемых в настоящем изобретении, можно исследовать с помощью изучения вызванного длительным введением адъюванта артрита у крыс.
Воздействие на глаза можно продемонстрировать с помощью хорошо известных методик офтальмологических исследований. Аналогичным образом противоопухолевую активность можно продемонстрировать с помощью хорошо известных методик исследований противоопухолевой активности.
Фармацевтические композиции, предлагаемые в настоящем изобретении, применимы для энтерального, такого как пероральное или ректальное, чрескожного, местного и парентерального введения млекопитающим, включая человека, для ингибирования активности ЦОГ-2 и для лечения зависимых от ЦОГ-2 нарушений, и они включают эффективное количество фармакологически активного соединения, предлагаемого в настоящем изобретении, по отдельности или в комбинации с другими терапевтическими средствами и одним или большим количеством фармацевтически приемлемых носителей.
Точнее, фармацевтические композиции включают эффективно ингибирующее ЦОГ-2 количество селективно ингибирующего ЦОГ-2 соединения, предлагаемого в настоящем изобретении, которое практически не обладает ингибирующей активностью по отношению к ЦОГ-1 и не приводит к приписываемым ему побочным эффектам.
Фармакологически активные соединения, предлагаемые в настоящем изобретении, можно применять при изготовлении фармацевтических композиций, включающих их эффективное количество совместно или в смеси с инертными наполнителями или носителями, пригодными для энтерального или парентерального применения. Предпочтительными являются таблетки и желатиновые капсулы, включающие активный ингредиент совместно с:
a) разбавителями, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином;
b) смазывающими веществами, например, диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток также со
c) связующими, например, алюмосиликатом магния, пастой крахмала, желатином, трагакантовой камедью, метилцеллюлозой, натриевой солью карбоксиметилцеллюлозы и/или поливинилпирролидоном; при необходимости с
d) разрыхлителями, например, крахмалами, агар-агаром, альгиновой кислотой, или ее солью, или шипучими смесями.
Композиции для инъекций предпочтительно представляют собой водные изотонические растворы или суспензии, и из эмульсий или суспензий на жировой основе с успехом готовят суппозитории. Указанные композиции могут быть стерилизованы и/или содержать вспомогательные вещества, такие как, консервирующие, стабилизирующие или эмульгирующие агенты, способствующие растворению вещества, соли для регулирования осмотического давления и/или буферы. Кроме того, они также могут содержать другие терапевтически полезные вещества. Указанные композиции получают с помощью обычных методик смешивания, гранулирования или нанесения покрытий соответственно и содержат примерно 0,1-75%, предпочтительно примерно 1-50% активного ингредиента.
На таблетки по методикам, известным в данной области техники, можно нанести пленочное покрытие или энтеросолюбильное покрытие.
Композиции, подходящие для чрескожного введения, включают терапевтически эффективное количество соединения, предлагаемого в настоящем изобретении, с носителем. Предпочтительные носители включают впитывающиеся фармакологически приемлемые растворители, предназначенные для содействия прохождению через кожу пациента. Например, чрескожные устройства представляют собой повязку, включающую изнаночный слой, резервуар, содержащий соединение необязательно с носителями, необязательно регулирующий скорость барьерный элемент для доставки соединения в кожу пациента с регулируемой и заранее заданной скоростью в течение продолжительного периода времени и средства для закрепления устройства на коже.
Композиции, подходящие для местного введения, например, на кожу или в глаза, включают водные растворы, суспензии, мази, кремы, гели или распыляемые композиции, например, для подачи в виде аэрозоля и т.п. Такие устройства местного действия являются особенно подходящими для воздействия на кожу, например, для лечения рака кожи, например, для использования профилактических средств в солнцезащитных кремах, лосьонах, аэрозольных препаратах и т.п. В связи с этим следует отметить, что соединения, предлагаемые в настоящем изобретении, способны поглощать УФ-излучение в диапазоне 290-320 нм, но пропускают способствующее образованию загара излучение с более значительными длинами волн. Поэтому они являются особенно подходящими для использования в средствах местного действия, включая косметические, хорошо известных в данной области техники. Они могут содержать солюбилизаторы, стабилизаторы, агенты, регулирующие тоничность, буферные агенты и консерванты. Препараты, пригодные для местного применения, можно приготовить, например, как описано в патенте US No.4784808. Препараты, пригодные для применения в офтальмологии, можно приготовить, например, как описано в патентах US No. 4829088 и 4960799.
Соединения, предлагаемые в настоящем изобретении, можно применять по отдельности или вместе с другими терапевтическими средствами. Например, дополнительные активные средства, подходящие для лечения неоплазии (злокачественной и доброкачественной) включают, например, противоопухолевые средства или противолучевые средства, указанные в заявке WO 98/16227 и т.п. Другие подходящие терапевтические средства включают анальгетические средства, такие как НСПС, оксикодон, кодеин, парацетамол, ибупрофен, трамадол, левопранол, пропоксифен, кеторолак, пентазоцин, меперидин и т.п.; мышечные релаксанты, например, бензотиадиазолы, например, сирдалуд®; а также антитромбоцитарные средства, такие как аспирин, клопидогрел, тиклопидин и т.п.; а также бисфосфонаты, такие как золедронат, памидронат, ризедронат, алендронат и т.п.; а также статины, такие как флувастатин, аторвастатин, ловастатин, симвастатин, росувастатин, питавастатин, правастатин и т.п.; а также антациды; ингибиторы протонного насоса, например, омепразол, эзомепразол; кальцилитики; кальцитонин, например, кальцитонин для перорального введения; анти-IL-1-бета IgG1/каппа антитела; гипотензивные средства, например, ингибиторы ацетилхолинэстеразы, блокаторы ангиотензина II, ингибиторы ренина.
Вместе с другим активным ингредиентом соединение, предлагаемое в настоящем изобретении, можно вводить одновременно, до или после другого активного ингредиента, или отдельно тем же или другим путем введения, или вместе в одном фармацевтическом препарате.
Вводимая доза активного соединения зависит от вида теплокровного животного (млекопитающего), массы тела, возраста и индивидуального состояния и от вводимой формы. Разовая доза для перорального введения млекопитающему массой примерно 50-70 кг может содержать примерно от 5 мг до 500 мг активного ингредиента.
Настоящее изобретение также относится к способам применения соединений, предлагаемых в настоящем изобретении, и их фармацевтически приемлемых солей или их фармацевтическим композициям для млекопитающих для ингибирования ЦОГ-2 и для лечения патологических состояний, описанных в настоящем изобретении, например, воспаления, боли, ревматоидного артрита, остеоартрита, дисменореи, опухолей и других зависящих от ЦОГ-2 нарушений.
Настоящее изобретение предпочтительно относится к способу селективного ингибирования активности ЦОГ-2 у млекопитающего без значительного ингибирования активности ЦОГ-1, который включает введение нуждающемуся в нем млекопитающему эффективно ингибирующего ЦОГ-2 количества соединения, предлагаемого в настоящем изобретении.
Таким образом, настоящее изобретение также относится к способу лечения зависящих от ЦОГ-2 нарушений у млекопитающих, который включает введение нуждающемуся в нем млекопитающему эффективно ингибирующего ЦОГ-2 количества соединения, предлагаемого в настоящем изобретении.
Настоящее изобретение более предпочтительно относится к способу лечения зависящих от ЦОГ-2 нарушений у млекопитающих в основном с исключением нежелательных побочных эффектов, связанных с ингибированием активности ЦОГ-1, который включает введение нуждающемуся в нем млекопитающему эффективно ингибирующего ЦОГ-2 количества селективно ЦОГ-2 ингибирующего соединения, предлагаемого в настоящем изобретении, которое практически не обладает ингибирующей активностью по отношению к ЦОГ-1.
Точнее, оно относится к способу, например, лечения ревматоидного артрита, остеоартрита, боли, дисменореи, подагры или воспаления у млекопитающих с исключением нежелательного изъязвления желудочно-кишечного тракта, способ включает введение нуждающемуся в нем млекопитающему соответствующего эффективного количества соединения, предлагаемого в настоящем изобретении.
Приведенные ниже примеры предназначены для иллюстрации настоящего изобретения и не считаются налагающими на него ограничения. Температуры приведены в градусах Цельсия. Если не указано иное, то выпаривание всегда проводят при пониженном давлении, предпочтительно примерно от 15 до 100 мм рт.ст. (= 20-133 мбар). Структура конечных продуктов, промежуточных продуктов и исходных веществ подтверждена с помощью стандартных методов анализа, например, микроанализа, и спектроскопических характеристик (например, МС (масс-спектроскопия), ИК-спектроскопия, ЯМР). Использующиеся аббревиатуры и торговые названия являются стандартными для данной области техники. Типичными являются приведенные ниже.
ПРИМЕРЫ
Пример 1
Исходные анилины
А. 2,4-Дихлор-3-метиланилин
2,4-Дихлор-3-метиланилин получают путем восстановления 2,4-дихлор-3-метилнитробензола по методике, описанной в публикации Tetrahedron, Vol.53, No.17, p.6145 (1997).
В. 2,4-Дихлор-3-этиланилин
К раствору 2,4-дихлоранилина (42,0 г, 260 ммолей) в АсОН (40,0 мл) прибавляют Ас2О (80 мл). Реакционную смесь нагревают до 50°С и перемешивают при этой температуре в течение 1 ч. Реакционную смесь охлаждают до комнатной температуры и выливают в смесь воды со льдом (500 мл). Твердое вещество выпадает в осадок, и смесь перемешивают в течение еще 1 ч при комнатной температуре. Твердое вещество отфильтровывают и промывают водой, гексанами и сушат на воздухе и получают N-(2,4-дихлорфенил)-ацетамид.
К раствору N-(4-хлор-2-фторфенил)-ацетамида (30,0 г, 147 ммолей) в ТГФ (300 мл) при -70°С по каплям прибавляют (поддерживая температуру реакционной смеси ниже -60°С) n-BuLi (2,0 М раствор в циклогексане, 147 мл, 294 ммоля). Реакционную смесь перемешивают при температуре от -60 до -70°С в течение 2 ч и при -70°С по каплям прибавляют 1,1,1-трифтор-2-йодэтан (46,2 г, 220 ммолей). Реакционную смесь перемешивают при этой температуре в течение еще 1,5 ч, затем медленно прибавляют 3 н. раствор HCl (108 мл). Смеси дают нагреться до комнатной температуры и экстрагируют с помощью EtOAc (200 мл × 3). Органические слои объединяют, промывают водой, рассолом и затем сушат над MgSO4. Растворители удаляют и остаток перемешивают в смеси эфира и гексанов (1:2, 120 мл) в течение 1 ч. Выпавший осадок фильтруют и получают N-(2,4-дихлор-3-йодфенил)-ацетамид.
К раствору N-(2,4-дихлор-3-йодфенил)-ацетамида (40,0 г, 121 ммоль) в МеОН (100 мл) прибавляют концентрированную HCl (50 мл). Смесь перемешивают и кипятят с обратным холодильником в течение 18 ч. Смесь охлаждают и растворители удаляют при пониженном давлении (водяная баня, температура ниже 45°С). Остаток охлаждают в бане со льдом и прибавляют 3 н. раствор NaOH до доведения значения рН, до равного от 9 до 10. Смесь экстрагируют эфиром и сушат над MgSO4. Растворители удаляют и остаток очищают на флэш-хроматографической колонке в градиентном режиме с использованием смеси гексаны/эфир и получают 2,4-дихлор-3-йоданилин.
К раствору 2,4-дихлор-3-йоданилина (9,0 г, 31 ммоля) в смеси ДМЭ/вода (180 мл/60 мл) прибавляют комплекс винилпиридина с трибороксином (5,0 г, 20,8 ммоля) и K2CO3 (8,5 г, 62,0 ммоля). Реакционную смесь перемешивают и через смесь в течение 15 мин пропускают N2. При комнатной температуре прибавляют тетракис(трифенилфосфин)палладий(0) (1,8 г, 1,6 ммоля) и через смесь пропускают N2 в течение еще 20 мин. Реакционную смесь нагревают до 80°С и перемешивают в течение 18 ч до тех пор, пока с помощью метода ГХ-МС (газовая хроматография - масс-спектрометрия) не будет показано, что реакция завершилась. Смесь фильтруют и промывают эфиром (400 мл) и водой (50 мл). Органический слой отделяют и промывают рассолом и сушат над MgSO4. Растворители удаляют и остаток очищают на флэш-хроматографической колонке в градиентном режиме с использованием смеси гексаны/эфир и получают 2,4-дихлор-3-виниланилин.
К раствору 2,4-дихлор-3-виниланилина (4,9 г, 26 ммолей) в EtOAc (60 мл) прибавляют 10% Pd/C (0,50 г). Сосуд высокого давления заполняют с помощью H2 при давлении, равном 55 фунт-сила/дюйм2, и встряхивают в течение 2 ч. Избыток H2 удаляют и смесь фильтруют через слой целита. Растворитель удаляют и полученную смесь очищают на флэш-хроматографической колонке в градиентном режиме с использованием смеси гексаны/эфир и получают 2,4-дихлор-3-этиланилин.
С. 2-Фтор-4-метил-3-трифторанилин
К раствору 2-фтор-3-трифторметиланилина (5,0 г, 28 ммолей) в ДМФ (25 мл) прибавляют раствор NBS (5,0 г, 28 ммолей) в ДМФ (25 мл). Через 2,5 ч реакционную смесь подвергают распределению между эфиром и насыщенным водным раствором NaCl. Органическую фазу отделяют и дважды промывают свежеприготовленным насыщенным водным раствором NaCl, сушат над Na2SO4 и концентрируют при пониженном давлении и получают 4-бром-2-фтор-3-трифторметиланилин в виде масла.
Полученный выше бромид (10,0 г, 38,8 ммоля), триметилбороксин (4,9 г, 38,8 ммоля), K2CO3 (16,1 г, 116 ммоля) и тетракис(трифенилфосфин)палладий (4,5 г, 3,9 ммоля) нагревают в атмосфере азота. Прибавляют еще одну аликвоту триметилбороксина (4,9 г, 38,8 ммоля) для полного израсходования исходного бромида. Через 18 ч охлажденную реакционную смесь подвергают распределению между EtOAc и насыщенным водным раствором NaCl. Органический слой отделяют и промывают свежеприготовленным рассолом (3×), сушат над Na2SO4 и концентрируют при пониженном давлении. Остаток очищают с помощью флэш-хроматографии (Et2O/гексаны) и получают искомый анилин.
D. 2-Фтор-4-этил-3-трифторанилин
4-Бром-2-фтор-3-трифторметиланилин, полученный выше (5,0 г, 19,4 ммоля), и трибутилоловохлорид (7,1 г, 20,4 ммоля) прибавляют к безводному ДМФ (100 мл). Раствор дегазируют с помощью N2, прибавляют тетракис(трифенилфосфин)палладий (1,5 г, 1,3 ммоля) и реакционную смесь нагревают при 120°С в течение 18 ч. После охлаждения реакционную смесь подвергают распределению между Et2O и насыщенным водным раствором NaCl. Эфирный слой отделяют и промывают свежеприготовленным насыщенным водным раствором NaCl (2×), сушат над Na2SO4 и концентрируют при пониженном давлении. Остаток очищают с помощью флэш-хроматографии при элюировании 5, затем 10, затем 20% раствором EtOAc в гексанах и получают искомый продукт, 2-фтор-3-трифторметил-4-виниланилин.
2-Фтор-3-трифторметил-4-виниланилин, полученный выше (4,0 г, 19,4 ммоля), растворяют в EtOH и дегазируют с помощью N2 и прибавляют 10% Pd на древесном угле (0,5 г). Смесь помещают в реактор Парра и обрабатывают с помощью Н2 при комнатной температуре. Реакционную смесь продувают с помощью N2 и фильтруют. Слой фильтрата промывают свежеприготовленным EtOH и фильтраты объединяют. К охлажденному фильтрату (0°С) прибавляют концентрированную HCl (6,5 мл), затем летучие компоненты удаляют при пониженном давлении. Полученное белое твердое вещество промывают с помощью Et2O и собирают фильтрованием и получают 4-этил-2-фтор-3-трифторметиланилингидрохлорид.
Свободное основание анилина получают с использованием в качестве исходного вещества гидрохлорида с помощью распределения между Et2O и насыщенным водным раствором NaHCO3. Органический слой отделяют, промывают рассолом, сушат (Na2SO4) и концентрируют в вакууме и получают 4-этил-2-фтор-3-трифторметиланилин в виде масла.
Е. 6-Хлор-2-фтор-3-метиланилин
К 6-хлор-2-фтор-3-метилбензойной кислоте (10 г, 53 ммоля) прибавляют CH2Cl2 (60 мл), ДМФ (0,5 мл) и оксалилхлорид (9,25 мл, 106 ммолей). Реакционную смесь перемешивают до гомогенного состояния и затем концентрируют в вакууме. Остаток выливают в смесь 50:50 лед: 36% раствор гидроксида аммония и получают 6-хлор-2-фтор-3-метилбензамид.
6-Хлор-2-фтор-3-метилбензамид, полученный выше, растворяют в МеОН. Прибавляют метоксид натрия (16,1 г, 298 ммолей) и реакционную смесь кипятят с обратным холодильником. Затем порциями прибавляют твердый NBS (21,2 г, 119 ммолей) и реакционную смесь перемешивают при кипячении с обратным холодильником в течение еще 2 ч. После охлаждения смеси летучие компоненты удаляют при пониженном давлении и остаток разбавляют с помощью EtOAc и водой. Органическую фазу отделяют и водный слой экстрагируют свежеприготовленным EtOAc (3×). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют в вакууме и получают метиловый эфир N-6-хлор-2-фтор-3-метилфенил)карбаминовой кислоты.
Карбамат, полученный выше, растворяют в МеОН (220 мл) и воде (22 мл). Прибавляют гидроксид калия (31 г, 560 ммолей) и реакционную смесь кипятят с обратным холодильником в течение 12 ч. После охлаждения реакционной смеси до комнатной температуры МеОН удаляют при пониженном давлении и остаток разбавляют водой и экстрагируют с помощью CH2Cl2 (3×). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии при использовании 10% EtOAc в гексане и получают 6-хлор-2-фтор-3-метиланилин.
F. 4-Хлор-2-фтор-3-этиланилин
К раствору 4-хлор-2-фторанилина (50,0 г, 344 ммоля) в АсОН (30 мл) прибавляют Ac2O (60 мл) и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Смесь выливают в смесь воды со льдом (500 мл) и получают твердый осадок. Смесь перемешивают в течение еще 1 ч при комнатной температуре. Твердое вещество отфильтровывают и промывают водой и гексанами, затем сушат на воздухе и получают N-(4-хлор-2-фторфенил)-ацетамид.
К раствору N-(4-хлор-2-фторфенил)-ацетамида (10,0 г, 53,3 ммоля) и диизопропиламина (7,5 мл, 53,3 ммоля) в ТГФ (300 мл) при -78°С по каплям прибавляют n-BuLi (2,5 М раствор в гексанах, 42,6 мл, 106,6 ммоля), поддерживая температуру реакционной смеси ниже -60°С. Через 2 ч при -78°С по каплям прибавляют йодэтан (12,4 г, 80 ммоля). Реакционную смесь перемешивают при -78°С в течение еще 1,5 ч. Медленно прибавляют 1 н. водный раствор HCl до тех пор, пока значение рН смеси не достигнет величины от 4 до 5. Смесь экстрагируют с помощью EtOAc (200 мл × 3) и органические слои объединяют, промывают водой и рассолом, затем сушат над MgSO4. Растворитель удаляют при пониженном давлении и оставшееся твердое вещество перемешивают в смеси эфира и гексана (1:4, 80 мл) в течение 1 ч. Отфильтрованное твердое вещество сушат и получают N-(4-хлор-3-этил-2-фторфенил)-ацетамид.
К раствору N-(4-хлор-3-этил-2-фторфенил)-ацетамида (11,0 г, 51,0 ммоль) в МеОН (80 мл) прибавляют концентрированную HCl (40 мл), затем кипятят с обратным холодильником в течение 16 ч. Смесь охлаждают и растворитель удаляют при пониженном давлении, поддерживая температуру водяной бани ниже 45°С. Остаток охлаждают с помощью бани со льдом, затем прибавляют 3 н. раствор NaOH, чтобы довести значение рН смеси до величины от 9 до 10. Смесь экстрагируют эфиром и сушат над MgSO4. Растворитель удаляют и остаток очищают на флэш-хроматографической колонке в градиентном режиме с использованием смеси эфир/гексаны и получают 4-хлор-3-этил-2-фторанилин.
Аналогичным способом получают
4-Хлор-3-метил-2-фторанилин.
G. 4-хлор-2-фтор-3-изопропиланилин
К раствору N-(4-хлор-2-фторфенил)-ацетамида, полученному ранее (12,0 г, 64 ммоля), в ТГФ (300 мл) при -78°С по каплям прибавляют n-BuLi (2,0 М в циклогексане, 64 мл, 128 ммолей), поддерживая температуру реакционной смеси ниже -60°С. Реакционную смесь перемешивают при температуре от -60 до -70°С в течение 2 ч и при -78°С по каплям прибавляют 1,1,1-трифтор-2-йодэтан (20,1 г, 96 ммолей). Смесь перемешивают при этой температуре в течение еще 3 ч. Затем при
-78°С медленно прибавляют 1 н. раствор HCl (200 мл). Смеси дают нагреться до комнатной температуры и экстрагируют с помощью EtOAc (200 мл × 3). Объединенные органические слои промывают водой и рассолом, затем сушат над MgSO4. Растворитель удаляют и остаток перемешивают в смеси эфира и гексанов (1:2, 120 мл) в течение 1 ч. Твердое вещество отфильтровывают и получают N-(4-хлор-2-фтор-3-йодфенил)-ацетамид.
К смеси N-(4-хлор-2-фтор-3-йодфенил)-ацетамида (20,0 г, 64 ммоля) с МеОН (80 мл) прибавляют концентрированную HCl (60 мл). Смесь перемешивают при кипячении с обратным холодильником в течение 18 ч, затем охлаждают до комнатной температуры и растворители удаляют при пониженном давлении, поддерживая температуру водяной бани ниже 45°С. Остаток охлаждают с помощью бани со льдом и прибавляют 1 н. раствор NaOH до доведения значения рН, равного от 9 до 10. Водную фазу экстрагируют эфиром и сушат над MgSO4. Растворитель удаляют и остаток очищают на флэш-хроматографической колонке в градиентном режиме с использованием смеси гексаны/эфир и получают 4-хлор-2-фтор-3-йоданилин.
К раствору 4-хлор-2-фтор-3-йоданилина (13,5 г, 50 ммоля) в смеси ДМЭ/вода (150 мл/50 мл) прибавляют 2-пропенбороновую кислоту (6,4 г, 74,6 ммоля) и К2СО3 (20,6 г, 150 ммоля). Через раствор в течение 15 мин пропускают азот, затем при комнатной температуре прибавляют тетракис(трифенилфосфин)палладий(0) (2,8 г, 2,5 ммоля). Азот пропускают в течение еще 20 мин, затем реакционную смесь нагревают при 80°С в течение 18 ч. Смесь фильтруют через слой целита и промывают эфиром (400 мл) и водой (50 мл). Органический слой отделяют и промывают рассолом и сушат над MgSO4. Растворитель удаляют и остаток очищают на флэш-хроматографической колонке в градиентном режиме с использованием смеси гексаны/эфир и получают 4-хлор-3-изопропен-2-фторанилин.
К раствору 4-хлор-3-изопропен-2-фторанилина (4,5 г, 24,2 ммоля) в EtOAc (50 мл) прибавляют 10% Pt/C (0,45 г). Сосуд высокого давления заполняют с помощью H2 при давлении, равном 50 фунт-сила/дюйм2, и встряхивают при комнатной температуре в течение 4 ч. Избыток Н2 удаляют и смесь фильтруют через слой целита. Растворитель удаляют и полученную смесь очищают на флэш-хроматографической колонке в градиентном режиме с использованием смеси гексаны/эфир и получают 4-хлор-3-изопропан-2-фторанилин.
Пример 2
Исходные эфир 2-йодфенилуксусной кислоты и фенилацетамид
Получают по методике, описанной, например, в публикациях J Med Chem, Vol.33, pp.2358-2368 (1990), US Patent No.6291523 и International Application WO 99/11605, используя в качестве исходных веществ соответствующие бензойную кислоту или 2-индолинон, например, получают:
N,N-диметил-5-метил-2-йодфенилацетамид;
N,N-диметил-5-этил-2-йодфенилацетамид;
Пример 3
N,N-Диметиламид 5-этил-2-(2'-фтор-4'-метил-3'-трифторметиланилино)фенилуксусной кислоты.
Смесь N,N-диметил-5-этил-2-йодфенилацетамида (2,0 г, 6,3 ммоля), 2-фторметил-3-трифторметиланилина (2,4 г, 12,6 ммоля), меди (0,2 г, 3,2 ммоля), йодида меди (0,6 г, 3,2 ммоля) и К2СО3 (0,9 г, 6,3 ммоля) в ксилолах (6,0 мл) кипятят с обратным холодильником в течение 24 ч. После охлаждения неочищенную реакционную смесь разбавляют с помощью EtOAc и фильтруют через целит®. Фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью флэш-хроматографии при элюировании гексанами, затем смесями EtOAc (до 25%)/гексан и получают искомое соединение - N,N-диметиламид 5-этил-2-(2'-фтор-4'-метил-3'-трифторметиланилино)фенилуксусной кислоты.
Аналогичным способом получают:
N,N-диметиламид 5-метил-2-(2'-фтор-4'-метил-3'-трифторметиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-метил-2-(2'-фтор-4'-бром-3'-трифторметиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-этил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-метил-2-(2',4'-дихлор-3'-этиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-этил-2-(2',4'-дихлор-3'-этиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-метил-2-(2'-фтор-4'-хлор-3'-метиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-этил-2-(2'-фтор-4'-хлор-3'-метиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-метил-2-(2'-фтор-4'-хлор-3'-этиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-этил-2-(2'-фтор-4'-хлор-3'-этиланилино)фенилуксусной кислоты,
N,N-диметиламид 5-этил-2-(2'-фтор-4'-хлор-3'-изопропиланилино)фенилуксусной кислоты.
Пример 4
N,N-Диметиламид 5-метил-2-(2'-фтор-4'-этил-3'-трифторметиланилино)фенилуксусной кислоты.
N,N-Диметиламид 5-метил-2-(2'-фтор-4'-бром-3'-трифторметиланилино) фенилуксусной кислоты, полученный по методике, описанной в примере 3 (0,8 г, 1,7 ммоля), Pd(PPh3)4 (0,1 г, 0,09 ммоля) и винилтрибутилстаннан (0,6 г, 1,9 ммоля) в ДМФ (5 мл) нагревают при 120°С в атмосфере азота в течение ночи. После охлаждения реакционную смесь подвергают распределению между EtOAc и насыщенным водным раствором NaCl. Отделенный органический слой дважды промывают свежеприготовленным насыщенным водным раствором NaCl. Слой EtOAc сушат и концентрируют при пониженном давлении. Остаток очищают с помощью флэш-хроматографии и получают N,N-диметиламид 5-метил-2-(2'-фтор-4'-винил-3'-трифторметиланилино)фенилуксусной кислоты.
Полученный выше стирол восстанавливают в описанных условиях. Стирол растворяют в i-PrOH и толуоле и дегазируют в течение 10 мин. Затем прибавляют Cs2CO3 (22 мг, 0,07 ммоля), [Ir(ЦОД)Cl]2 (44 мг, 0,07 ммоля) и ДФФП (27 мг, 0,07 ммоля) и реакционную смесь нагревают при 80°С в течение ночи. Охлажденный раствор концентрируют в вакууме и остаток очищают с помощью флэш-хроматографии при элюировании смесью 10, затем 20, затем 30% EtOAc/гексаны и получают искомый N,N-диметиламид 5-метил-2-(2'-фтор-4'-этил-3'-трифторметиланилино)фенилуксусной кислоты.
Пример 5
5а) 5-Этил-2-(2'-фтор-4'-метил-3'-трифторметиланилино)фенилуксусная кислота
Раствор полученного выше N,N-диметиламида 5-этил-2-(2'-фтор-4'-метил-3'-трифторметиланилино)фенилуксусной кислоты, описанного в примере 3 (0,9 г, 2,4 ммоля), в EtOH (25 мл) и 4 н. раствор NaOH (12 мл) нагревают при 80°С в течение ночи. После охлаждения реакционную смесь концентрируют при пониженном давлении и разбавляют ледяным EtOAc. Значение рН водного слоя доводят до величины, равной 1-2, с помощью 2,5 н. холодного раствора HCl. Отделенный органический слой сушат над Na2SO4 и концентрируют до твердого состояния. Твердое вещество очищают путем растирания со смесью Et2O/гексан и получают искомую кислоту, 5-этил-2-(2'-фтор-4'-метил-3'-трифторметиланилино)фенилуксусную кислоту.
МС m/z 356 (ЭР+), 354 (ЭР-)
CHN найдено С 60,47, Н 4,47, N 3,73
Аналогичным способом получают:
5b) 5-Метил-2-(2'-фтор-4'-метил-3'-трифторметиланилино)фенилуксусная кислота.
МС m/z 342 (ЭР+), 340 (ЭР-)
CHN найдено С 59,56, Н 4,36, N 3,91
5с) 5-Метил-2-(2'-фтор-4'-этил-3'-трифторметиланилино)фенилуксусная кислота.
1Н ЯМР (400 МГц, ДМСО-d6) δ част./млн 1,13 (t, J=7,3 Гц, 3 Н), 2,27 (s, 3 Н), 2,63-2,70 (m, 2 Н), 3,56 (s, 2 Н), 6,87 (t, J=8,6 Гц, 1 Н), 7,02-7,09 (m, 2 Н), 7,12 (s, 1 Н), 7,43 (s, 1 H).
МС m/z 356 (ЭР+), 354 (ЭР-)
5d) 5-Этил-2-(2'-фтор-4'-этил-3'-трифторметиланилино)фенилуксусная кислота.
1Н ЯМР (400 МГц, ДМСО-d6) δ част./млн 1,17 (t, J=7,5 Гц, 3 Н), 1,26 (t, J=7,5 Гц, 3 Н), 2,59 (q, J=7,5 Гц, 2 Н), 2,89 (q, J=7,1 Гц, 2 Н), 3,75 (d, J=22,7 Гц 1 Н), 3,83 (d, J=22,7 Гц, 1 Н), 6,52 (d, J=8,1 Гц, 1 Н), 7,06 (d, J=8,1 Гц, 1 Н), 7,23 (s, 1 H), 7,50 (d, J=8,3 Гц, 1 Н), 7,79 (t, J=7,8 Гц, 1 Н).
MC m/z 370 (ЭР+), 368 (ЭР-)
5е) 5-Метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусная кислота.
1Н ЯМР (400 МГц, ДМСО-d6) δ част./млн 2,29 (s, 3 Н), 2,41 (s, 3 Н), 3,51 (s, 2 Н), 6,51 (d, J=8,8 Гц, 1 Н), 7,07-7,22 (m, 4 H), 12,46 (s, 1 H)
MC m/z 324, 326, 328 (ЭР+), 322, 324, 326 (ЭР-)
5f) 5-Этил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусная кислота.
MC m/z 338, 340, 342 (ЭР+), 336, 338, 340 (ЭР-)
CHN найдено С 59,97, Н 4,71, N 4,12
5g) 5-Метил-2-(2',4'-дихлор-3'-этиланилино)фенилуксусная кислота.
1Н ЯМР (400 МГц, ДМСО-d6) δ част./млн 1,14 (t, J=7,5 Гц, 3 Н), 2,29 (s, 3 H), 2,88 (q, J=7,5 Гц, 2 Н), 3,52 (s, 2 H), 6,52 (d, J=9,0 Гц, 1 Н), 7,07-7,18 (m, 4 H), 7,25 (s, 1 H), 12,49 (br.s., 1 H)
CHN найдено С 60,53, H 5,19, N 3,98
5h) 5-Этил-2-(2',4'-дихлор-3'-этиланилино)фенилуксусная кислота.
1H ЯМР (400 МГц, ДМСО-d6) δ част./млн 1,11-1,22 (m, 6Н), 2,52-2,63 (m, 4 H), 2,88 (q, J=7,5 Гц, 2 H), 3,54 (s, 2 H), 6,55 (d, J=9,0 Гц, 1 H), 7,11-7,15 (m, 3 H), 7,18 (s, 1 H), 7,26 (s, 1 H), 12,50 (br.s., 1 H)
CHN найдено С 61,39, Н 5,22, N 4,24
5i) 5-Метил-2-(2'-фтор-4'-хлор-3'-метиланилино)фенилуксусная кислота.
MC m/z 308 (ЭР+), 306 (ЭР-)
CHN найдено С 62,16, Н 4,79, N 4,49
5j) 5-Этил-2-(2'-фтор-4'-хлор-3'-метиланилино)фенилуксусная кислота.
MC m/z 322 (ЭР+), 320 (ЭР-)
CHN найдено С 63,20, Н 5,22, N 4,32
5k) 5-Метил-2-(2'-фтор-4'-хлор-3'-этиланилино)фенилуксусная кислота.
1Н ЯМР (400 МГц, ДМСО-d6) δ част./млн 1,15 (t, J=7,5 Гц, 3 Н), 2,27 (s, 3 H), 2,72 (qd, J=7,5, 2,1 Гц, 2 Н), 3,54 (s, 2 H), 6,55 (t, J=9,0 Гц, 1 H), 6,98-7,07 (m, 3 H), 7,10 (s, 1 H), 7,30 (br.s., 1 H), 12,35 (br.s., 1 H)
MC m/z 322 (ЭР+), 320 (ЭР-)
CHN найдено С 63,54, Н 5,28, N 4,38
5l) 5-Этил-2-(2'-фтор-4'-хлор-3'-этиланилино)фенилуксусная кислота.
1Н ЯМР (400 МГц, ДМСО-d6) δ част./млн 1,12-1,20 (m, 6 Н), 2,57 (q, J=7,7 Гц, 2 Н), 2,68-2,76 (m, 2 Н), 3,56 (s, 2 Н), 6,58 (t, J=9,0 Гц, 1 Н), 7,00 (dd, J=9,0, 1,5 Гц, 1 Н), 7,04-7,10 (m, 2 Н), 7,13 (s, 1 Н), 7,31 (s, 1 Н), 12,36 (s, 1 Н)
CHN найдено С 64,20, Н 5,61, N 4,37
5m) 5-Этил-2-(2'-фтор-4'-хлор-3'-изопропиланилино)фенилуксусная кислота.
1Н ЯМР (400 МГц, CDCl3) δ част./млн 1,23 (t, J=7,7 Гц, 3 Н), 1,37 (dd, J=7,0, 1,3 Гц, 6 Н), 2,62 (q, J=7,7 Гц, 2 Н), 3,46-3,60 (m, 1 Н), 3,68 (s, 2 Н), 6,72 (t, J=8,8 Гц, 1 Н), 6,92 (dd, J=8,8, 1,8 Гц, 1 Н), 7,07-7,14 (m, 2 Н), 7,23 (d, J=8,1 Гц, 1 Н)
CHN найдено С 65,08, Н 6,16, N 3,84
Пример 6
6а) Соль 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты с диэтиламином
Суспензию 600 мг 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты (1,85 ммоля) в 6 мл трет-бутилметилового эфира (ТБМЭ) нагревают до 40°С. По каплям прибавляют 137 мг диэтиламина (1,85 ммоля). Полученный немного мутный раствор очищают горячим фильтрованием через стекловолоконный фильтр Whatman. Фильтр промывают с помощью 2 мл ТБМЭ при 40°С. Фильтрату дают медленно охладиться.
В раствор при 30°С вносят затравку, и происходит кристаллизация. Суспензию перемешивают при комнатной температуре в течение примерно 15 ч и затем при 0°С в течение 2 ч. Взвесь фильтруют и отфильтрованный осадок промывают с помощью 4,5 мл ТВМЕ при 0°С. Кристаллы сушат в течение 4 ч при 50°С и давлении, равном примерно 10 мбар, и получают белый порошок, температура плавления 115-116°С.
6b) 5-Метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетат натрия
Суспензию 400 мг 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты (1,23 ммоля) в 4 мл ацетона нагревают до 50°С. Полученный почти прозрачный раствор очищают горячим фильтрованием через стекловолоконный фильтр Whatman. Фильтр промывают с помощью 2 мл ацетона при 50°С. При 50°С прибавляют 165 мг 30% водного раствора гидроксида натрия (1,23 ммоля). Раствору дают охладиться до комнатной температуры, и при температуре, равной примерно 25°С, в него вносят затравку. Затем медленно происходит кристаллизация. Суспензию перемешивают при комнатной температуре в течение примерно 15 ч и затем при 0°С в течение 2 ч. После фильтрования получают только 97 мг влажных кристаллов. Кристаллы и маточный раствор объединяют и при перемешивании по каплям прибавляют 6 мл изопропилацетата. Густую суспензию фильтруют и твердое вещество промывают изопропилацетатом. Соль сушат сначала при 50°С/примерно 10 мбар в течение 2 ч и затем при 80°С и примерно 10 мбар в течение 16 ч и получают соль в виде белого порошка, температура плавления 254-255°С. Содержание воды: 2,31% мас./мас.
6с) Соль 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты с трометамином
Суспензию, содержащую 1,30 г 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты (4,01 ммоль) в 13 мл ацетона, нагревают до 55°С. Полученный почти прозрачный раствор очищают горячим фильтрованием через стекловолоконный фильтр Whatman. Фильтр промывают с помощью 2,6 мл ацетона при 50°С. При 50°С по каплям прибавляют раствор 0,487 г трометамина (основание Trisma, 2-амино-2-(гидроксиметил)-1,3-пропандиол) (4,01 ммоля) в 1 мл воды. Капельную воронку промывают с помощью 0,5 мл воды. Раствору дают охладиться до комнатной температуры, и при температуре, равной примерно 40°С, в него вносят затравку. Происходит кристаллизация. Суспензию перемешивают при комнатной температуре в течение примерно 15 ч и затем при 0°С в течение 2 ч. Взвесь фильтруют и твердое вещество промывают с помощью 6 мл ацетона. Соль сушат сначала при 50°С/примерно 10 мбар в течение 2 ч и затем при 80°С и примерно 10 мбар в течение 16 ч и получают соль в виде белого порошка, температура плавления 156-157°С, ДСК 162,0°С, энтропия плавления 132 Дж/г.
Основные пики на рентгенограмме:
6d) Моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата кальция
Суспензию, содержащую 1,30 г 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты (4,01 ммоля) в 13 мл ацетона, нагревают до 55°С. Прибавляют 0,535 г 30% водного раствора гидроксида натрия (4,01 ммоля). Полученный почти прозрачный раствор очищают горячим фильтрованием через стекловолоконный фильтр Whatman. Фильтр промывают с помощью 3,9 мл ацетона при 50°С. Затем при 50°С по каплям прибавляют раствор 0,51 г хлорида кальция (4,41 ммоля) в 1 мл воды. Капельную воронку промывают с помощью 0,5 мл воды. Полученной густой суспензии дают охладиться до комнатной температуры. Затем при 25°С прибавляют 7 мл воды. Смесь перемешивают при комнатной температуре в течение 1 ч и фильтруют. Отфильтрованный осадок промывают с помощью 20 мл смеси ацетон/вода 2:1 об./об. и затем с помощью 5 мл ацетона. Закристаллизовавшееся вещество сушат при 50°С/примерно 10 мбар в течение 2 ч и затем при 80°С и примерно 10 мбар в течение 16 ч. Т.к. соль все еще содержит примесь некоторого количества хлорида натрия, кристаллы суспендируют в 13,5 мл воды и 1,5 мл ацетона и суспензию перемешивают при комнатной температуре в течение 1 ч. После фильтрования отфильтрованный осадок промывают с помощью 8 мл (в четыре порции) смеси вода/ацетон 9:1 об./об. Соль сушат сначала при 50°С/примерно 10 мбар в течение 2 ч и затем при 80°С и примерно 10 мбар в течение 16 ч и получают соль в виде белого порошка, температура плавления 278-279°С, ДСК 147,1/224,2°С, энтропия плавления 121/2 Дж/г.
Содержание воды: 2,63% мас./мас.
Основные пики на рентгенограмме:
6е) Моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата лизина
Суспензию, содержащую 400 мг 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты (1,23 ммоля) в 4 мл ацетона, нагревают до 50°С. Полученный почти прозрачный раствор очищают горячим фильтрованием через стекловолоконный фильтр Whatman. Фильтр промывают с помощью 4 мл ацетона при 50°С. По каплям прибавляют раствор 184 мг лизина (1,23 ммоля) в 1 мл воды. Полученную густую суспензию выдерживают при 50°С в течение 30 мин и затем ей дают охладиться до комнатной температуры. Затем смесь перемешивают при температуре, примерно равной 25°С, в течение ночи и фильтруют. Отфильтрованный осадок промывают с помощью 6 мл ацетона. Кристаллы сушат сначала при 50°С/примерно 10 мбар в течение 2 ч и затем при 80°С и примерно 10 мбар в течение 16 ч и получают соль в виде белого порошка, температура плавления 162-163°С.
Содержание воды: 3,80% мас./мас.
6f) Моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата холина
Суспензию, содержащую 400 мг 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты (1,23 ммоля) в 4 мл ацетона, нагревают до 50°С. Прибавляют 305 мг 50% раствора холина в воде (1,23 ммоля). Полученный почти прозрачный раствор очищают горячим фильтрованием через стекловолоконный фильтр Whatman. Фильтр промывают с помощью 2 мл ацетона при 50°С. К фильтрату при 40°С медленно прибавляют 6 мл трет-бутилметилового эфира (ТБМЭ). Слегка мутному раствору дают охладиться, и при 25°С в него вносят затравку. Медленно происходит кристаллизация. Смесь перемешивают при комнатной температуре в течение ночи. Через 16 ч по каплям прибавляют еще 12 мл ТБМЭ. Затем густую суспензию перемешивают при 25°С в течение 2 ч и фильтруют.
Твердое вещество промывают с помощью 6 мл смеси ТБМЭ/ацетон 9/1 об./об. и сушат при 50°С и примерно 10 мбар в течение 4 ч и получают белый порошок, температура плавления 105-106°С.
Содержание воды: 4,55% мас./мас.
6g) 5-Метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетат калия
Суспензию 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты (15,42 ммоля) в ацетоне кипятят с обратным холодильником и перемешивают при температуре, равной примерно 55°С, в течение 10 мин. Полученный раствор немного охлаждают до 50°С и горячий раствор фильтруют через стекловолоконный фильтр Whatman. Фильтр промывают ацетоном при 50°С. Фильтрат нагревают до 50°С и по каплям прибавляют в течение 15 мин смесь 45% раствора гидроксида калия (14,65 ммоля) (0,95 экв.) и воды. Капельную воронку промывают водой и ацетоном. Полученную суспензию перемешивают при 50°С в течение 30 мин. Затем взвеси дают охладиться до 25°С в течение примерно 2 ч. Суспензию перемешивают при комнатной температуре в течение ночи и фильтруют. Отфильтрованный осадок три раза промывают. Соль сушат сначала при 50°С/примерно 10 мбар в течение 3 ч и затем при 80°С и примерно 10 мбар в течение 17 ч и получают соль в виде белого порошка, температура плавления 315-317°С, ДСК 326,9°С, энтропия плавления 29 Дж/г.
Основные пики на рентгенограмме:
Изобретение относится к новым соединениям формулы (I) и их фармацевтически приемлемым солям, обладающим ингибирующей активностью в отношении циклооксигеназы-2 (ЦОГ-2). В формуле (I)
R обозначает метил или этил; R3 обозначает галоген; R4 обозначает C1-С6-алкил; R5 обозначает галоген; указанный выше C1-С6-алкил в качестве R4 необязательно замещен одной или более галогенидными группами. Изобретение относится также к фармацевтической композиции, способу лечения нарушений, зависящих от ЦОГ-2, в частности, ревматоидного артрита, остеоартрита, дисменореи, боли, опухолей или воспаления, способу селективного ингибирования активности ЦОГ-2 и применению соединений формулы (I) для приготовления лекарственного средства, предназначенного для лечения указанных выше заболеваний. 6 н. и 4 з.п. ф-лы, 6 пр.
1. Соединение формулы (I)
в которой
R обозначает метил или этил;
R3 обозначает галоген;
R4 обозначает С1-С6-алкил;
R5 обозначает галоген;
указанный выше С1-С6-алкил в качестве R4, необязательно замещен одной или более галогенидных групп;
и его фармацевтически приемлемые соли.
2. Соединение по п.1, где R4 обозначает метил или этил.
3. Соединение по п.2, где R3 и R5 каждый независимо выбран из хлора и фтора.
4. Соединение по п.1, выбранное из следующих соединений:
5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусная кислота,
5-этил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусная кислота,
5-метил-2-(2'-фтор-3'-метил-4'-хлоранилино)фенилуксусная кислота,
5-этил-2-(2'-фтор-3'-метил-4'-хлоранилино)фенилуксусная кислота,
5-метил-2-(2'-фтор-3'-этил-4'-хлоранилино)фенилуксусная кислота,
5-этил-2-(2'-фтор-3'-этил-4'-хлоранилино)фенилуксусная кислота,
5-метил-2-(2',4'-дихлор-3'-этиланилино)фенилуксусная кислота,
5-этил-2-(2',4'-дихлор-3'-этиланилино)фенилуксусная кислота,
5-этил-2-(2'-фтор-3'-изопропил-4'-хлоранилино)фенилуксусная кислота,
соль 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты с диэтиламином,
5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетат натрия,
соль 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилуксусной кислоты с трометамином,
моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата кальция,
моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата лизина, и
моногидрат 5-метил-2-(2',4'-дихлор-3'-метиланилино)фенилацетата холина.
5. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении циклооксигеназы-2 (ЦОГ-2), включающая соединение по п.1 или 4 в эффективном количестве, в комбинации с одним или более фармацевтически приемлемых носителей.
6. Способ лечения нарушений, зависящих от циклооксигеназы-2 (ЦОГ-2), у млекопитающих, который включает введение нуждающемуся в этом млекопитающему соединения по п.1 или 4 в эффективном количестве.
7. Способ селективного ингибирования активности ЦОГ-2 у млекопитающего без значительного ингибирования активности циклооксигеназы-1, который включает введение нуждающемуся в этом млекопитающему соединения по п.1 или 4 в количестве, эффективно ингибирующем ЦОГ-2.
8. Способ лечения ревматоидного артрита, остеоартрита, дисменореи, боли, опухолей или воспаления у млекопитающих, который включает введение нуждающемуся в этом млекопитающему соединения по п.1 или 4 в соответствующем эффективном количестве.
9. Применение соединения по любому из пп.1 или 4 для приготовления лекарственного средства, предназначенного для лечения ревматоидного артрита, остеоартрита, дисменореи, боли, опухолей или воспаления.
10. Соединение по любому из пп.1 или 4, предназначенное для применения для лечения зависящего от ЦОГ-2 нарушения.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| ОПРЕДЕЛЕННЫЕ 5-АЛКИЛ-2-АРИЛАМИНОФЕНИЛУКСУСНЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ | 1998 |
|
RU2186762C2 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| L.A.SORBERA et al, Lumiracoxib, DRUGS OF THE FUTURE, 2002, vol.27, no.8, 740-747 | |||
| ПРОИЗВОДНЫЕ ПИРАЗОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2233272C2 |

