Эпотилоны A и B (2a и 2b, схема 1) являются встречающимися в природе цитотоксическими макролидами, которые были выделены из разрушающей целлюлозу микобактерии Sorangium cellulosum (Höfle et al. Angew. Chem., Int. Ed. Engl. 1996, 35, 1567 и J. Antibiot. 1996, 49, 560; каждая из публикаций включена в данное описание в виде ссылки). Несмотря на огромное количество различных структур эпотилоны A и B имеют общий механизм действия, такой же, как механизм действия паклитаксела (Taxol®), который вовлечен в ингибирование роста опухолевых клеток в результате полимеризации тубулина и стабилизации сборки из микротрубочек (Bollag et al. Cancer Res. 1995, 55, 2325; включено в виде ссылки). Несмотря на свое бесспорное клиническое значение в качестве химиотерапевтического средства переднего края, Taxol® далек от идеального лекарственного средства. Его незначительная растворимость в воде делает необходимым прибегать к помощи наполнителей для композиции, таких как кремофоры, которые сами по себе создают риск и проблемы при работе с ними (Essayan et al. J. Allergy Clin. Immunol. 1996, 97, 42; публикация включена в данное описание в виде ссылки). Кроме того, Taxol® уязвим по отношению к дезактивации вследствие множественной лекарственной резистентности (MDR) (Giannakakou et al. J. Biol. Chem. 1997, 272, 17118; публикация включена в данное описание в виде ссылки). Однако также показано, что эпотилоны A и B сохраняют заметную эффективность против опухолевых MDR-клеток (Kowalski et al. Mol. Biol. Cell 1995, 6, 2137; публикация включена в данное описание в виде ссылки). Кроме того, повышенная растворимость в воде по сравнению с паклитакселом может быть полезна для возможности готовить композиции эпотилонов. В то время как встречающееся в природе соединение, эпотилон B (2b, EpoB на схеме 1) является эффективным представителем эпотилонового семейства природных продуктов, к сожалению, он обладает, по меньшей мере у мышей с ксенотрансплантатами, вызывающим беспокойство ограниченным терапевтическим индексом (Su et al. Angew. Chem. Int. Ed. Engl. 1997, 36, 1093; Harris et al. J Org. Chem. 1999, 64, 8434; каждая публикация включена в данное описание в виде ссылки).
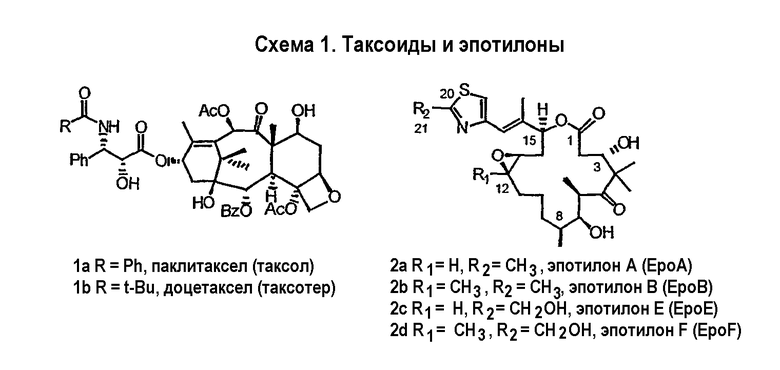
Имея в виду ограниченный терапевтический индекс EpoB, исследовали другие аналоги эпотилона, в частности 12,13-дезоксиэпотилоны, в отношении их способности давать улучшенный терапевтический профиль (см. патенты США №№ 6242469, 6284781, 6300355, 6369234, 6204388, 6316630; каждый из которых включен в данное описание в виде ссылки). Эксперименты in vivo, проведенные на различных мышиных моделях, показали, что 12,13-дезоксиэпотилон B (3b, dEpoB на схеме 2) обладает терапевтическим потенциалом против различных чувствительных и резистентных опухолей человека в ксенотрансплантатах мышей (Chou et al. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 9642 and 15798; публикация включена в данное описание в виде ссылки). Недавно терапевтическое преимущество указанных дезоксиэпотилонов по сравнению с другими противоопухолевыми агентами окончательно показано посредством сравнительных исследований (Chou et al. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8113; публикация включена в данное описание в виде ссылки). Вследствие впечатляющего профиля in vivo dEpoB был далее подвергнут токсикологическим исследованиям на собаках и в настоящее время проходит испытания на человеке в качестве противоопухолевого лекарственного средства.

В свете многообещающей терапевтической пользы 12,13-дезоксиэпотилонов может быть желательным исследовать дополнительные аналоги, а также дополнительные методики синтеза для синтеза существующих эпотилонов, дезоксиэпотилонов и их аналогов, а также их новых аналогов. В частности при наличии интереса к терапевтическому применению данного класса соединений может быть желательна разработка методик, способных обеспечивать значительные количества любых эпотилонов или дезоксиэпотилонов, описанных ранее или описанных в данной заявке, для клинических испытаний и для крупномасштабного получения.
Описание чертежей
Фиг.1 представляет собой таблицу значений IC50 для эпотилонов по отношению к росту клеток CCRF-CEM, CCRF-CEM/VBL и CCRF-CEM/таксол. Ингибирование роста клеток измеряли с помощью анализа на основе тетразония XTT после 72-часовой инкубации в течение роста клеток, как описано ранее (Scudiero et al. Cancer Res. 46: 4827-4833, 1988; публикация включена в данное описание в виде ссылки). Значения IC50 определяли на основе взаимосвязи доза-эффект при шести или семи концентрациях каждого лекарственного средства, используя компьютерную программу (Chou et al. Adv. Enzyme Regul. 22: 27-55, 1984; Chou et al. CalcuSyn for Windows (Biosoft, Cambridge, UK), 1997; каждая из которых включена в данное описание в виде ссылки), как описано ранее (Chou et al. Proc. Natl. Acad. Sci. USA 95: 15798-15802, 1998; публикация включена в данное описание в виде ссылки).
Фиг.2 является 1H-ЯМР-спектром транс-9,10-дегидро-12,13-дезоксиEpoB.
Фиг.3 является 13C-ЯМР-спектром транс-9,10-дегидро-12,13-дезоксиEpoB.
На фиг.4 показана схема синтеза 11-R- и 14-R-эпотилонов с использованием метатезиса олефинов с замыканием цикла LACDAC, и иллюстрируются некоторые замещения, доступные в случае способов синтеза, которые проходят через 9,10-дегидроэпотилон.
На фиг.5 представлены данные относительной цитотоксичности по отношению к лейкозным клеткам человека in vitro для ряда соединений и производных эпотилона, включая некоторые 9,10-дегидросоединения (например, соединение 7 на фиг.5A и соединение 88 и 89 на фиг.5B).
На фиг.6 изображена альтернативная методика синтеза для получения аналогов 9,10-дегидроэпотилона. На фиг.6A показана методика Макро-Стилле, методика сопряжения sp3-sp3 и методика β-Сузуки. На фиг.6B показана методика олефинирования Джулии, методика Вадсворт-Эммонса и методика Макро-Реформатского. На фиг.6C показана методика сочетания МакМурри и синтез аналога лактама.
На фиг.7 показаны различные аналоги 9,10-дегидро-12,13-дезоксиEpoB.
На фиг.8 показано терапевтическое действие 9,10-дегидро-dEpoB и dEpoB у мышей nude, несущих ксенотрансплантат карциномы молочной железы человека MX-1 (в/в-инфузия, Q2Dx3).
На фиг.9 показана стабильность аналогов эпотилона в плазме мышей. Epo 1 означает 12,13-дезоксиEpoB, Epo 2 означает 26-F3-12,13-дезоксиEpoB, Epo 3 означает (E)-9,10-дегидро-12,13-дезоксиEpoB и Epo 4 означает 26-F3-(E)-9,10-дегидро-12,13-дезоксиEpoB.
На фиг.10 изображено терапевтическое действие аналогов эпотилона у мышей nude (голых), несущих ксенотрансплантат HCT-116 (в/в-инфузия, Q2Dx7, n=3). Стрелки указывают введение лекарственного средства. Epo 3 означает (E)-9,10-дегидро-12,13-дезоксиEpoB.
На фиг.11 показаны эффективности различных аналогов эпотилона против роста опухолевых клеток in vitro и терапевтический индекс по сравнению с паклитакселом и винбластином.
Фиг.12 представляет собой таблицу, суммирующую действие dEpoB, таксола и 26-триF-9,10-deH-dEpoB против ксенотрансплантата MX-1 у мышей nude.
На фиг.13 показано терапевтическое действие 26-трифтор-9,10-дегидро-dEpoB и 9,10-дегидро-EpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты MX-1 (6-часовая в/в-инфузия, Q2Dx6 и Q2Dx9, соответственно).
На фиг.14 показаны изменения массы тела мышей nude, несущих ксенотрансплантат опухоли карциномы молочной железы человека MX-1, после лечения 26-трифтор-9,10-дегидро-dEpoB и 9,10-дегидро-EpoB (6-часовая инфузия, Q2Dx6 и Q2Dx9, соответственно).
На фиг.15 показано терапевтическое действие 26-трифтор-9,10-дегидро-dEpoB и 9,10-дегидроEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты MX-1 (6-часовая в/в-инфузия, Q2Dx6 и Q2Dx9, соответственно).
На фиг.16 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат опухоли карциномы молочной железы человека MX-1, после лечения 26-трифтор-9,10-дегидро-dEpoB и 9,10-дегидро-EpoB (6-часовая в/в-инфузия, Q2Dx6 и Q2Dx9, соответственно).
На фиг.17 показано терапевтическое действие 9,10-дегидро-dEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты HCT-116 (в/в-инфузия, Q2Dx7).
На фиг.18 показано действие 9,10-дегидро-dEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты карциномы ободочной кишки человека HCT-116 (в/в-инфузия, Q3Dx5).
На фиг.19 показано действие 9,10-дегидро-dEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты A549/Taxol (6-часовая в/в-инфузия, Q3Dx7).
На фиг.20 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат A549/Taxol, обработанных 26-трифтор-9,10-дегидро-dEpoB и 9,10-дегидро-dEpoB (6-часовая в/в-инфузия, Q3Dx7).
На фиг.21 показано действие 26-трифтор-9,10-дегидро-dEpoB и 9,10-дегидро-dEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты A549/Taxol (6-часовая в/в-инфузия, Q2Dx7).
На фиг.22 показаны изменения массы тела мышей nude, несущих ксенотрансплантаты A549/Taxol, обработанных 26-трифтор-9,10-дегидро-dEpoB и 9,10-дегидро-dEpoB (6-часовая в/в-инфузия, Q2Dx7).
На фиг.23 показано действие 9,10-дегидро-EpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты опухоли HCT-116 карциномы ободочной кишки человека (6-часовая в/в-инфузия).
На фиг.24 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат опухоли HCT-116 карциномы ободочной кишки человека, после лечения 9,10-дегидро-EpoB (6-часовая в/в-инфузия).
На фиг.25 показано образование микротрубочек из тубулина в присутствии различных аналогов эпотилона при 37°C.
На фиг.26 показано образование микротрубочек из тубулина в присутствии различных аналогов эпотилона при 4°C.
На фиг.27 показано действие 9,10-дегидро-dEpoB и dEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты HCT-116 (в/в-инфузия, Q2Dx6).
На фиг.28 показаны изменения массы тела у мышей nude, несущих ксенотрансплантаты HCT-116, после лечения 9,10-дегидро-dEpoB и dEpoB (в/в-инфузия, Q2Dx6).
На фиг.29 показано действие 9,10-дегидро-dEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты HCT-116 карциномы ободочной кишки человека (в/в-инфузия, Q3Dx4).
На фиг.30 показаны изменения массы тела мышей nude, несущих ксенотрансплантаты HCT-116 опухоли карциномы ободочной кишки человека, после лечения 9,10-дегидро-dEpoB (5 мг/кг, в/в-инфузия, X3Dx4).
Фиг.31 представляет собой таблицу со значениями IC50 для аналогов эпотилона по отношению к росту клеток CCRF-CEM.
На фиг.32 показана метаболическая стабильность аналогов эпотилона in vitro.
Фиг.33 представляет собой таблицу, детализирующую терапевтические эффекты различных аналогов эпотилона против ксенотрансплантатов опухолей человека у мышей в случае 6-часовой в/в-инфузии.
На фиг.34 показано действие 9,10-дегидро-EpoB на размер опухолей у мышей nude, несущих ксенотрансплантат опухоли HCT-116 карциномы ободочной кишки человека (6-часовая в/в-инфузия, Q2Dx7).
На фиг.35 показаны изменения массы тела мышей nude, несущих ксенотрансплантаты опухоли HCT-116 карциномы ободочной кишки человека, после лечения 9,10-дегидро-EpoB и оксазол-EpoD (6-часовая инфузия, Q2Dx7).
На фиг.36 показано действие 26-трифтор-9,10-дегидро-dEpoB и 9,10-дегидро-dEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты A549/Taxol (6-часовая в/в-инфузия, Q2Dx4).
На фиг.37 показано действие 9,10-дегидро-dEpoB на размер опухолей у мышей nude, несущих ксенотрансплантаты A549/Taxol (6-часовая в/в-инфузия, Q3Dx3).
На фиг.38 показана стабильность аналогов эпотилона в 20% плазме мышей/PBS.
На фиг.39 показана стабильность аналогов эпотилона в 10% фракции S9 печени человека/PBS.
На фиг.40 показана хроматограмма стабильности EpoD в 10% печени человека S9/PBS.
На фиг.41 представлены таблицы, описывающие действие различных аналогов эпотилона на полимеризацию микротрубочек in vitro при 37°C в отсутствие GTP (A) и цитотоксичность различных аналогов эпотилона в линии клеток легкого человека A549 (B).
На фиг.42 показана стабилизация образования микротрубочек эпотилонами при 35°C и 4°C.
На фиг.43 показано терапевтическое действие 9,10-дегидро-dEpoB у мышей nude, несущих ксенотрансплантат карциномы молочной железы человека T (MX-1) (6-часовая инфузия, Q2Dx5).
На фиг.44 показано изменение массы тела мышей nude, несущих ксенотрансплантат карциномы молочной железы человека (MX-1), после лечения 9,10-дегидро-dEpoB (6-часовая инфузия, Q2Dx8).
На фиг.45 показано изменение массы тела мышей nude, несущих ксенотрансплантат HCT-116, после лечения 9,10-дегидро-dEpoB (в/в-инфузия, Q2Dx7).
На фиг.46 показано терапевтическое действие 9,10-дегидро-dEpoF, dEpoB и таксола на размер опухолей у мышей nude, несущих ксенотрансплантат опухоли карциномы молочной железы человека (MX-1) (6-часовая в/в-инфузия, Q2Dx6).
На фиг.47 показаны изменения массы тела мышей nude, несущих ксенотрансплантат опухоли карциномы молочной железы человека (MX-1), после лечения 9,10-дегидро-dEpoF, dEpoB и таксолом (6-часовая инфузия, Q2Dx6).
На фиг.48 показано терапевтическое действие 9,10-дегидро-dEpoF и dEpoB у мышей nude, несущих ксенотрансплантат HCT-116 карциномы ободочной кишки человека (6-часовая инфузия, Q2Dx8).
На фиг.49 показаны изменения массы тела мышей nude, несущих ксенотрансплантат HCT-116, после лечения 9,10-дегидро-dEpoF и dEpoB (6-часовая инфузия, Q2Dx8).
На фиг.50 показано терапевтическое действие 9,10-дегидро-dEpoF и dEpoB у мышей nude, несущих ксенотрансплантат резистентной к таксолу карциномы легкого человека (A549/Taxol) (6-часовая инфузия, Q2Dx5).
На фиг.51 показаны изменения массы тела мышей nude, несущих ксенотрансплантат резистентной к таксолу карциномы легкого человека (A549/Taxol), после лечения 9,10-дегидро-dEpoF и dEpoB (6-часовая инфузия, Q2Dx5).
Фиг.52 представляет собой таблицу сравнения эффективности различных аналогов эпотилона по отношению к ингибированию опухолевого роста in vitro и относительному терапевтическому индексу.
На фиг.53 показано терапевтическое действие 9,10-дегидро-dEpoB у мышей nude, несущих ксенотрансплантат MX-1 (Q3Dx9, 6-часовая в/в-инфузия).
На фиг.54 показаны изменения массы тела мышей nude, несущих ксенотрансплантат MX-1, после лечения 9,10-дегидро-dEpoB (Q3Dx9, 6-часовая в/в-инфузия).
На фиг.55 показано терапевтическое действие 9,10-дегидроэпотилона B у мышей nude, несущих ксенотрансплантат MX-1 (Q3Dx9, 6-часовая инфузия).
На фиг.56 показаны изменения массы тела мышей nude, несущих ксенотрансплантат MX-1, после лечения 9,10-дегидроэпотилоном B (Q3Dx9, 6-часовая в/в-инфузия).
На фиг.57 показано терапевтическое действие низких доз 26-трифтор-9,10-дегидро-dEpoB у мышей nude, несущих ксенотрансплантат MX-1 (6-часовая в/в-инфузия, Q2Dx12).
На фиг.58 показаны изменения массы тела мышей nude, несущих ксенотрансплантат MX-1, после лечения низкими дозами 26-трифтор-9,10-дегидро-dEpoB (6-часовая в/в-инфузия, Q2Dx12).
На фиг.59 показано химиотерапевтическое действие аналогов эпотилона против ксенотрансплантатов опухолей человека у мышей nude. Ткань опухоли (40-50 мг) имплантировали п/к в день 0. Лечение начинали, когда размер опухоли достигал примерно 100 мм3 или больше, как указано. Все обработки, которые указаны стрелками, проводили с помощью 6-часовой в/в-инфузии через хвостовую вену, используя миникатетер и программируемый насос, как описано ранее (Su, D.-S. et al, Angew. Chem. Int. Ed. 1997, 36, 2093; Chou, T. C. et al. Proc. Natl. Acad. Sci. USA. 1998, 95, 15798; каждая публикация включена в данное описание в виде ссылки). Каждая группа дозирования состояла из четырех или более мышей. Массой тела называли суммарную массу тела минус массу опухоли, полагая, что 1 мм3 опухоли равен 1 мг опухолевой ткани. A. Ксенотрансплантат карциномы молочной железы MX-1, обработанный низкими дозами 25-трифтор-(E)-9,10-дегидро-12,13-дезоксиEpoB (10 мг/кг) при сравнении с обработками в таблице 1 (20 мг/кг и 30 мг/кг). B. Крупные ксенотрансплантаты MX-1 (500 мм3) обрабатывали 25-трифтор-(E)-9,10-дегидро-12,13-дезоксиEpoB (25 мг/кг) и dEpoB (30 мг/кг). C. Медленно растущий ксенотрансплантат карциномы легкого A549, обработанный 25-трифтор-(E)-9,10-дегидро-12,13-дезоксиEpoB (25 мг/кг) и dEpoB (30 мг/кг). D. Ксенотрансплантат A549/Taxol (44-кратная резистентность к паклитакселу in vitro), обработанный 25-трифтор-(E)-9,10-дегидро-12,13-дезоксиEpoB (20 мг/кг) и (E)-9,10-дегидро-12,13-дезоксиEpoB (4 мг/кг). Обработка deH-dEpoB на 28 день была пропущена вследствие заметного и быстрого снижения массы тела.
На фиг.60 изображен синтез C-21-модифицированных 9,10-(E)-дегидроэпотилонов. На фиг.60A показан синтез 26-трифтор-21-метиламино-9,10-(E)-дегидро-12,13-дезоксиэпотилона B. Фиг.60B является схемой синтеза для получения 26-трифтор-21-амино-9,10-(E)-дегидро-12,13-дезоксиэпотилона B в качестве промежуточного продукта синтеза 26-трифтор-21-диметиламино-9,10-(E)-дегидро-12,13-дезоксиэпотилона B.
Фиг.61 представляет собой таблицу со значениями IC50 для C-21-модифицированных эпотилонов по отношению к линии опухолевых клеток CCRF-CEM и ее подлиний, резистентных к лекарственным средствам.
На фиг.62 показано терапевтическое действие 26-трифтор-9,10-дегидро-dEpoB и таксола у мышей nude, несущих ксенотрансплантат T-клеточного лимфобластного лейкоза человека CCRF-CEM (6-часовая в/в-инфузия, Q2Dx8).
На фиг.63 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат T-клеточного лимфобластного лейкоза человека CCRF-CEM, после лечения 25-трифтор-9,10-дегидро-dEpoB и таксолом (6-часовая в/в-инфузия, Q2Dx8).
На фиг.64 показано терапевтическое действие 26-трифтор-9, 10-дегидро-dEpoB и таксола у мышей nude, несущих ксенотрансплантат T-клеточного лимфобластного лейкоза человека CCRF-CEM/Taxol (резистентный к таксолу) (6-часовая в/в-инфузия, Q2Dx7, x5).
На фиг.65 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат T-клеточного лимфобластного лейкоза человека CCRF-CEM/Taxol (резистентный к таксолу), после лечения 26-трифтор-9,10-дегидро-dEpoB и таксолом (6-часовая в/в-инфузия, Q2Dx7, x5).
На фиг.66 показано терапевтическое действие 26-трифтор-9,10-дегидро-dEpoB и таксола у мышей nude, несущих ксенотрансплантат карциномы ободочной кишки человека HCT-116 (Q2Dx4, x2, 6-часовая в/в-инфузия).
На фиг.67 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат карциномы ободочной кишки человека HCT-116, после лечения 26-трифтор-9,10-дегидро-dEpoB и таксолом (Q2Dx4, x2, 6-часовая в/в-инфузия).
На фиг.68 показано терапевтическое действие 9,10-дегидро-EpoB у мышей nude, несущих ксенотрансплантат MX-1 (6-часовая в/в-инфузия).
На фиг.69 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат карциномы молочной железы человека MX-1, после лечения 9,10-дегидро-EpoB (6-часовая инфузия в/в-инфузия).
На фиг.70 показано терапевтическое действие 9,10-дегидро-EpoB у мышей nude, несущих ксенотрансплантат T-клеточного лимфобластного лейкоза человека CCRF-CEM/Taxol (резистентный к таксолу) (6-часовая в/в-инфузия, Q3Dx5, x2).
На фиг.71 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат T-клеточного лимфобластного лейкоза человека CCRF-CEM/Taxol (резистентный к таксолу), после лечения 9,10-дегидро-EpoB (6-часовая в/в-инфузия, Q3Dx5, x2).
На фиг.72 показано терапевтическое действие 26-трифтор-dEpoB и 26-трифтор-9,10-дегидро-dEpoF у мышей nude, несущих ксенотрансплантат карциномы молочной железы человека MX-1 (Q2Dx11, в/в-инъекция).
На фиг.73 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат карциномы молочной железы человека MX-1, после лечения 26-трифтор-dEpoB и 26-трифтор-9,10-дегидро-dEpoF (Q2Dx11, в/в-инъекция).
На фиг.74 показано терапевтическое действие 9,10-дегидро-dEpoB у мышей nude, несущих ксенотрансплантат карциномы молочной железы человека MX-1 (Q3Dx9, 6-часовая в/в-инфузия).
На фиг.75 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат карциномы молочной железы человека MX-1, после лечения 9,10-дегидро-dEpoB (Q3Dx9, 6-часовая в/в-инфузия).
На фиг.76 показано терапевтическое действие 26-трифтор-9,10-дегидро-dEpoF у мышей nude, несущих ксенотрансплантат карциномы легкого человека (MX-1) (6-часовая в/в-инфузия и в/в-инъекция).
На фиг.77 показаны изменения массы тела у мышей nude, несущих ксенотрансплантат MX-1, после лечения 26-трифтор-9,10-дегидро-dEpoF (6-часовая в/в-инфузия и в/в-инъекция).
Определения
Некоторые предлагаемые в данном изобретении соединения и определения конкретных функциональных групп также описаны более подробно ниже. В данном изобретении химические элементы идентифицированы в соответствии с Периодической таблицей элементов, CAS-версия, Handbook of Chemistry and Physics, 75th Ed., внутренняя сторона обложки, и конкретные функциональные группы, как правило, определяют, как описано в данной заявке. Кроме того, общие принципы органической химии, а также конкретные функциональные остатки и химическая активность описаны в «Organic Chemistry», Thomas Sorrel, University Science Books, Sausalito: 1999, полное содержание которой включено в данное описание в виде ссылки. Кроме того, специалисту в данной области будет понятно, что в способах синтеза, которые приведены в данном описании, используется ряд защитных групп. Под термином «защитная группа», используемым в данном описании, подразумевается, что конкретный функциональный остаток, например O, S или N, временно блокируют, так чтобы реакция осуществлялась избирательно в другом химически активном сайте в многофункциональном соединении. В предпочтительных вариантах защитная группа избирательно взаимодействует, с хорошим выходом давая защищенный субстрат, который стабилен по отношению к намеченным реакциям; защитная группа должна избирательно удаляться с хорошим выходом легко доступными, предпочтительно нетоксичными реагентами, которые не воздействуют на другие функциональные группы; защитная группа образует легко отделяемое производное (более предпочтительно без образования новых стереогенных центров); и защитная группа имеет минимум дополнительной функциональности, чтобы избежать дополнительных реакционных сайтов. Как подробно изложено в данном описании, можно использовать защитные группы кислорода, серы, азота и углерода. Примеры защитных групп подробно указаны в данном описании, однако, будет понятно, что это не означает, что данное изобретение ограничивается указанными защитными группами; точнее множество дополнительных эквивалентных защитных групп можно легко идентифицировать, используя указанные выше критерии, и использовать в способе, предлагаемом в данном изобретении. Кроме того, ряд защитных групп описаны в «Protective Groups in Organic Synthesis» Third Ed. Greene, T. W. and Wuts, P.G., Eds., John Wiley and Sons, New York: 1999, полное содержание которой тем самым включено в виде ссылки.
Будет понятно, что соединения, которые указаны в описании, могут быть замещены любым количеством заместителей или функциональных остатков. В общем, термин «замещенный», которому предшествует или не предшествует термин «необязательно», и заместители, входящие в формулы согласно данному изобретению, относятся к замене водородных радикалов в данной структуре радикалом определенного заместителя. В том случае, когда в какой-либо данной структуре могут быть замещены более одного положения более чем одним заместителем, выбранным из определенной группы, заместители во всех положениях могут быть либо одинаковыми, либо разными. В используемом в данном описании смысле предполагается, что термин «замещенный» включает в себя все допустимые заместители органических соединений. В широком аспекте допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. В настоящем изобретении гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанных в данной публикации, которые удовлетворяют валентностям гетероатомов. Кроме того, подразумевается, что данное изобретение никоим образом не ограничено допустимыми заместителями органических соединений. Комбинациями заместителей и переменных, предполагаемых в данном изобретении, предпочтительно являются комбинации, которые приводят к образованию стабильных соединений, применимых для лечения, например, пролиферативных расстройств, включая, но не ограничивая указанным, злокачественную опухоль. Термин «стабильное», используемый в данном описании, предпочтительно относится к соединениям, которые обладают стабильностью, достаточной для того, чтобы обеспечить возможность производства, и которые сохраняют целостность соединения в течение периода времени, достаточного для его регистрации, и предпочтительно в течение периода времени, достаточного для его использования в целях, подробно указанных в данном описании.
Термин «алифатический» в используемом в данном описании смысле включает в себя как насыщенные, так и ненасыщенные, с прямой цепью (т.е. неразветвленные), разветвленные, циклические или полициклические алифатические углеводороды, которые необязательно замещены одной или несколькими функциональными группами. Как будет понятно специалисту в данной области, в данном описании подразумевается, что термин «алифатический» включает, но не ограничен указанным, алкильный, алкенильный, алкинильный, циклоалкильный, циклоалкенильный и циклоалкинильный остатки. Таким образом, используемый в данном описании термин «алкил» включает неразветвленные, разветвленные и циклические алкильные группы. Аналогичное условие применимо к другим общим терминам, таким как «алкенил», «алкинил» и тому подобным. Кроме того, используемые в данном описании термины «алкил», «алкенил», «алкинил» и тому подобные охватывают как замещенные, так и незамещенные группы. В некоторых вариантах используемый в данном описании термин «низший алкил» используют для указания алкильных групп (циклических, ациклических, замещенных, незамещенных, разветвленных или неразветвленных), имеющих 1-6 атомов углерода.
В некоторых вариантах алкильные, алкенильные и алкинильные группы, используемые в изобретении, содержат 1-20 алифатических атомов углерода. В некоторых других вариантах алкильные, алкенильные и алкинильные группы, используемые в изобретении, содержат 1-10 алифатических атомов углерода. В других вариантах алкильные, алкенильные и алкинильные группы, используемые в изобретении, содержат 1-8 алифатических атомов углерода. В следующих вариантах алкильные, алкенильные и алкинильные группы, используемые в изобретении, содержат 1-6 алифатических атомов углерода. Кроме того, в других вариантах алкильные, алкенильные и алкинильные группы, используемые в изобретении, содержат 1-4 атома углерода. Таким образом, иллюстративные алифатические группы включают, но не ограничены указанным, например, остатки метила, этила, н-пропила, изопропила, циклопропила, -CH2-циклопропила, аллила, н-бутила, втор-бутила, изобутила, трет-бутила, циклобутила, -CH2-циклобутила, н-пентила, втор-пентила, изопентила, трет-пентила, циклопентила, -CH2-циклопентила, н-гексила, втор-гексила, циклогексила, -CH2-циклогексила и тому подобные, которые к тому же могут нести один или несколько заместителей. Алкенильные группы включают, но не ограничены указанным, например, этенил, пропенил, бутенил, 1-метил-2-бутен-1-ил и тому подобное. Типичные алкинильные группы включают, но не ограничены указанным, этинил, 2-пропинил (пропаргил), 1-пропинил и тому подобные.
Термин «алкоксигруппа» или «тиоалкил», используемый в данном описании, относится к алкильной группе, которая определена ранее, связанной с остатком родительской молекулы через атом кислорода или через атом серы. В некоторых вариантах алкильная группа содержит 1-20 алифатических атомов углерода. В некоторых других вариантах алкильная группа содержит 1-10 алифатических атомов углерода. В других вариантах алкильные, алкенильные и алкинильные группы, используемые в изобретении, содержат 1-8 алифатических атомов углерода. В следующих вариантах алкильная группа содержит 1-6 алифатических атомов углерода. В других вариантах алкильная группа содержит 1-4 алифатических атома углерода. Примеры алкоксигрупп включают, но не ограничены указанным, метокси-, этокси-, пропокси-, изопропокси-, н-бутокси-, трет-бутокси, неопентокси- и н-гексоксигруппу. Примеры тиоалкила включают, но не ограничены указанным, метилтио-, этилтио-, пропилтио-, изопропилтио-, н-бутилтиогруппу и тому подобные.
Термин «алкиламиногруппа» относится к группе, имеющей структуру -NHR', где R' означает алкил, который определен в данном описании. В некоторых вариантах алкильная группа содержит 1-20 алифатических атомов углерода. В некоторых других вариантах алкильная группа содержит 1-10 алифатических атомов углерода. В других вариантах алкильные, алкенильные и алкинильные группы, используемые в изобретении, содержат 1-8 алифатических атомов углерода. В следующих вариантах алкильная группа содержит 1-6 алифатических атомов углерода. В других вариантах алкильная группа содержит 1-4 алифатических атомов углерода. Примеры алкиламиногрупп включают, но не ограничены, метиламино-, этиламино-, изо-пропиламиногруппой и тому подобными.
Некоторые примеры заместителей описанных выше алифатических (и других) остатков соединений, предлагаемых в изобретении, включают, но не ограничены указанным, алифатическую группу; гетероалифатическую группу; арил; гетероарил; арилалкил; гетероарилалкил; алкоксигруппу; арилоксигруппу; гетероалкоксигруппу; гетероарилоксигруппу; алкилтиогруппу; арилтиогруппу; гетероалкилтиогруппу; гетероарилтиогруппу; F; Cl; Br; I; -OH; -NO2; -CN; -CF3; -CH2CF3; -CHCl2; -CH2OH; -CH2CH2OH; -CH2NH2; -CH2SO2CH3; -C(O)RX; -CO2(RX); -CON(RX)2; -OC(O)RX; -OCO2RX; -OCON(RX)2; -N(RX)2; -S(O)2RX; -NRX(CO)RX, где в каждом случае RX независимо включает, но не ограничен указанным, алифатическую группу, гетероалифатическую группу, арил, гетероарил, арилалкил или гетероарилалкил, где любой из алифатических, гетероалифатических, арилалкильных или гетероарилалкильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным, разветвленным или неразветвленным, циклическим или ациклическим, и где любой из арильных или гетероарильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным. Дополнительные примеры обычно применимых заместителей иллюстрированы конкретными вариантами, показанными в примерах, которые приведены в данном описании.
Термины «арил» и «гетероарил», используемые в данном описании, относятся к стабильным моно- или полициклическим, гетероциклическим, полициклическим и полигетероциклическим ненасыщенным остаткам, имеющим предпочтительно 3-14 атомов углерода, каждый из которых может быть замещенным или незамещенным. Заместители включают без ограничения любой из ранее упомянутых заместителей, т.е. заместителей, перечисленных для алифатических остатков или для других остатков, которые описаны в данной публикации, которые приводят к образованию стабильного соединения. В некоторых вариантах осуществления данного изобретения «арил» относится к моно- или бициклической карбоциклической кольцевой системе, имеющей одно или два ароматических кольца, включая, но не ограничиваясь указанным, фенил, нафтил, тетрагидронафтил, индалил, инденил и тому подобное. В некоторых вариантах осуществления данного изобретения термин «гетероарил», используемый в данном описании, относится к циклическому ароматическому радикалу, имеющему от пяти до десяти атомов в цикле, из которых один атом в цикле выбран из S, O и N; ноль, один или два атома в цикле являются дополнительными гетероатомами, независимо выбранными из S, O и N; и остальные атомы в цикле являются атомами углерода, при этом радикал связан с остальной частью молекулы посредством любого атома в цикле, например, такой как пиридил, пиразинил, пиримидинил, пирролил, пиразолил, имидазолил, тиазолил, оксазолил, изооксазолил, тиадиазолил, оксадиазолил, тиофенил, фуранил, хинолинил, изохинолинил и тому подобное.
Будет понятно, что арильные и гетероарильные группы (включая бициклические арильные группы) могут быть незамещенными или замещенными, при этом замещение включает замену в них одного, двух или трех атомов водорода независимо любым одним или несколькими из следующих остатков, включая без ограничения: алифатическую группу; гетероалифатическую группу; арил; гетероарил; арилалкил; гетероарилалкил; алкоксигруппу; арилоксигруппу; гетероалкоксигруппу; гетероарилоксигруппу; алкилтиогруппу; арилтиогруппу; гетероалкилтиогруппу; гетероарилтиогруппу; F; Cl; Br; I; -OH; -NO2; -CN; -CF3; -CH2CF3; -CHCl2; -CH2OH; -CH2CH2OH; -CH2NH2; -CH2SO2CH3; -C(O)RX; -CO2(RX); -CON(RX)2; -OC(O)RX; -OCO2RX; -OCON(RX)2; -N(RX)2; -S(O)2RX; -NRX(CO)RX, где в каждом случае RX независимо включает, но не ограничен указанным, алифатическую группу, гетероалифатическую группу, арил, гетероарил, арилалкил или гетероарилалкил, где любой из алифатических, гетероалифатических, арилалкильных или гетероарилалкильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным, разветвленным или неразветвленным, циклическим или ациклическим, и где любой из арильных или гетероарильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным. Дополнительные примеры обычно применимых заместителей иллюстрированы конкретными вариантами, показанными в примерах, которые приведены в данном описании.
Термин «циклоалкил», используемый в данном описании, в частности, относится к группам, имеющим от трех до семи, предпочтительно от трех до десяти атомов углерода. Подходящие циклоалкилы включают, но не ограничены указанным, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и тому подобное, которые, как и в случае других алифатических, гетероалифатических или гетероциклических остатков, могут быть необязательно замещены заместителями, включая без ограничения алифатическую группу; гетероалифатическую группу; арил; гетероарил; арилалкил; гетероарилалкил; алкоксигруппу; арилоксигруппу; гетероалкоксигруппу; гетероарилоксигруппу; алкилтиогруппу; арилтиогруппу; гетероалкилтиогруппу; гетероарилтиогруппу; F; Cl; Br; I; -OH; -NO2; -CN; -CF3; -CH2CF3; -CHCl2; -CH2OH; -CH2CH2OH; -CH2NH2; -CH2SO2CH3; -C(O)RX; -CO2(RX); -CON(RX)2; -OC(O)RX; -OCO2RX; -OCON(RX)2; -N(RX)2; -S(O)2RX; -NRX(CO)RX, где в каждом случае RX независимо включает, но не ограничен указанным, алифатическую группу, гетероалифатическую группу, арил, гетероарил, арилалкил или гетероарилалкил, где любой из алифатических, гетероалифатических, арилалкильных или гетероарилалкильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным, разветвленным или неразветвленным, циклическим или ациклическим, и где любой из арильных или гетероарильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным. Дополнительные примеры обычно применимых заместителей иллюстрированы конкретными вариантами, показанными в примерах, которые приведены в данном описании.
Термин «гетероалифатический», используемый в данном описании, относится к алифатическим остаткам, которые содержат один или несколько атомов кислорода, серы, азота, фосфора или кремния, например, вместо атомов углерода. Гетероалифатические остатки могут быть разветвленными, неразветвленными, циклическими или ациклическими и включают насыщенные и ненасыщенные гетероциклы, такие как морфолиногруппу, пирролидинил и т.д. В некоторых вариантах гетероалифатические остатки замещены независимой заменой в них одного или нескольких атомов водорода одним или несколькими остатками, включая без ограничения алифатическую группу; гетероалифатическую группу; арил; гетероарил; арилалкил; гетероарилалкил; алкоксигруппу; арилоксигруппу; гетероалкоксигруппу; гетероарилоксигруппу; алкилтиогруппу; арилтиогруппу; гетероалкилтиогруппу; гетероарилтиогруппу; F; Cl; Br; I; -OH; -NO2; -CN; -CF3; -CH2CF3; -CHCl2; -CH2OH; -CH2CH2OH; -CH2NH2; -CH2SO2CH3; -C(O)RX; -CO2(RX); -CON(RX)2; -OC(O)RX; -OCO2RX; -OCON(RX)2; -N(RX)2; -S(O)2RX; -NRX(CO)RX, где в каждом случае RX независимо включает, но не ограничен указанным, алифатическую группу, гетероалифатическую группу, арил, гетероарил, арилалкил или гетероарилалкил, где любой из алифатических, гетероалифатических, арилалкильных или гетероарилалкильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным, разветвленным или неразветвленным, циклическим или ациклическим, и где любой из арильных или гетероарильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным. Дополнительные примеры обычно применимых заместителей иллюстрированы конкретными вариантами, показанными в примерах, которые приведены в данном описании.
Термин «галоген», используемый в данном описании, относится к атому, выбранному из фтора, хлора, брома и йода.
Термин «галогеналкил» означает алкильную группу, которая определена выше, имеющую один, два или три связанных с ней атомов галогена, примерами которой являются такие группы, как хлорметил, бромэтил, трифторметил и тому подобные.
Термин «гетероциклоалкил» или «гетероцикл», используемый в данном описании, относится к неароматическому 5-, 6- или 7-членному циклу или полициклической группе, включая без ограничения би- или трициклическую группу, содержащую конденсированные шестичленные циклы, имеющие от одного до трех гетероатомов, независимо выбранных из кислорода, серы и азота, где (i) каждый 5-членный цикл имеет от 0 до 1 двойной связи и каждый 6-членный цикл имеет от 0 до 2 двойных связей, (ii) гетероатомы азота и серы необязательно могут быть окислены, (iii) гетероатом азота необязательно может быть четвертичным и (iv) любое из указанных выше гетероциклических колец может быть слито с бензольным кольцом. Типичные гетероциклы включают, но не ограничены указанным, пирролидинил, пиразолинил, пиразолидинил, имидазолинил, имидазолидинил, пиперидинил, пиперазинил, оксазолидинил, изоксазолидинил, морфолинил, тиазолидинил, изотиазолидинил и тетрагидрофурил. В некоторых вариантах используется термин «замещенная гетероциклоалкильная или гетероциклическая» группа и используемый в данном описании термин относится к гетероциклоалкильной или гетероциклической группе, которая определена выше, с замещением в ней одного, двух или трех атомов водорода без ограничения алифатической группой; гетероалифатической группой; арилом; гетероарилом; арилалкилом; гетероарилалкилом; алкоксигруппой; арилоксигруппой; гетероалкоксигруппой; гетероарилоксигруппой; алкилтиогруппой; арилтиогруппой; гетероалкилтиогруппой; гетероарилтиогруппой; F; Cl; Br; I; -OH; -NO2; -CN; -CF3; -CH2CF3; -CHCl2; -CH2OH; -CH2CH2OH; -CH2NH2; -CH2SO2CH3; -C(O)Rx; -CO2(Rx); -CON(Rx)2; -OC(O)Rx; -OCO2Rx; -OCON(Rx)2; -N(Rx)2; -S(O)2Rx; -NRx(CO)Rx, где в каждом случае Rx независимо включает, но не ограничен указанным, алифатическую группу, гетероалифатическую группу, арил, гетероарил, арилалкил или гетероарилалкил, где любой из алифатических, гетероалифатических, арилалкильных или гетероарилалкильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным, разветвленным или неразветвленным, циклическим или ациклическим, и где любой из арильных или гетероарильных заместителей, описанных выше и в данном случае, может быть замещенным или незамещенным. Дополнительные примеры обычно используемых заместителей иллюстрированы конкретными вариантами, показанными в примерах, которые приведены в данном описании.
Термин «меченое», используемый в данном описании, означает, что соединение имеет, по меньшей мере, один связанный элемент, изотоп или химическое соединение, делающее возможной регистрацию соединения. В общем, метки можно отнести к трем классам: a) изотопные метки, которые могут быть радиоактивными или тяжелыми изотопами, включая, но, не ограничиваясь указанным, 2H, 3H, 32P, 35S, 67Ga, 99mTc (Tc-99m), 111In, 123I, 125I, 169Yb и 186Re; b) иммунные метки, которые могут быть антителами или антигенами; и c) окрашенные или флуоресцирующие красители. Будет понятно, что метки могут быть включены в соединение в любом положении, которое не препятствует биологической активности или признаку соединения, которое регистрируют. В некоторых вариантах осуществления изобретения используют фотоаффинное мечение для прямого определения межмолекулярных взаимодействий в биологических системах (например, чтобы исследовать сайт связывания эпотилона в димере тубулина). Можно использовать множество фотофоров, большинство которых основано на фотопревращении диазосоединений, азидов или диазиринов в нитрены или карбены (см. Bayley, H., Photogenerated Reagents in Biochemistry and Molecular Biology (1983), Elsevier, Amsterdam.), полное содержание публикации включено в данное описание в виде ссылки. В некоторых вариантах осуществления изобретения используемыми фотоаффинными метками являются орто-, мета- и пара-азидобензоилы, замещенные одним или несколькими остатками галогена, включая, но не ограничиваясь указанным, 4-азидо-2,3,5,6-тетрафторбензойную кислоту.
«Полимер»: Термин «полимер», используемый в данном описании, относится к композиции, содержащей цепи, которые могут быть открытыми, замкнутыми, линейными, разветвленными или сшитыми из повторяющихся единиц (мономеров), которые могут быть одинаковыми или разными. Будет понятно, что в некоторых вариантах термин «полимер» относится к биополимерам, которые, как подразумевается, относятся к природным полимерным материалам, или основанным на таких материалах, найденных в природе, включая, но не ограничиваясь указанным, нуклеиновые кислоты, пептиды и их миметики. В некоторых других вариантах термин «полимер» относится к синтетическим полимерам, таким как биоразлагаемые полимеры или другие полимерные материалы. Будет понятно, что полимерные твердые носители также включены в полимеры, предлагаемые в данном изобретении. Предлагаемые в изобретении соединения могут быть связаны с полимерными носителями и, следовательно, некоторые синтетические модификации могут осуществляться на твердой фазе. Используемый в данном описании термин «твердый носитель» включает, но не ограничен указанным, гранулы, диски, капилляры, полые волокна, иглы, штыри, твердые волокна, целлюлозные шарики, пористые стеклянные шарики, силикагели, полистироловые шарики, необязательно сшитые с дивинилбензолом, шарики из привитых сополимеров, полиакриламидные шарики, латексные шарики, диметилакриламидные шарики, необязательно перекрестно сшитые с N-N'-бис-акрилоилэтилендиамином, и стеклянные частицы, покрытые гидрофобным полимером. Специалисту в данной области будет понятно, что выбор конкретного твердого носителя будет ограничен совместимостью носителя с используемыми химическими реакциями. Примером твердого носителя является аминосмола Tentagel, композиция 1) полистироловых шариков, перекрестно сшитых с дивинилбензолом, и 2) ПЭГ (полиэтиленгликоль). Tentagel является особенно применимым твердым носителем, так как он обеспечивает универсальную поддержку для использования в анализах на шарикам и без шариков, а также прекрасно набухает в растворителях в диапазоне от толуола до воды.
Описание некоторых вариантов осуществления изобретения
Признавая необходимость в разработке новых и эффективных способов терапии злокачественных опухолей, в данном изобретении предлагаются новые методики синтеза, открывающих доступ к макроциклам, обладающим широким диапазоном биологической и фармакологической активности, а также новые соединения с такой активностью, новые терапевтические композиции и способы применения таких соединения и композиций.
В некоторых вариантах предлагаемые в изобретении соединения применимы при лечении злокачественной опухоли. Некоторые предлагаемые в изобретении соединения оказывают цитотоксическое или ингибирующее рост действие на линии клеток злокачественных опухолей, проявляют способность полимеризовать тубулин и стабилизировать конструкции из микротрубочек и/или приводят к сокращению или исчезновению опухолей в моделях ксенотрансплантатов клеток злокачественной опухоли. В некоторых вариантах соединения могут иметь сниженные или минимальные побочные эффекты, включая токсичность по отношению к жизненно важных органам, тошноту, рвоту, диарею, аллопецию, потерю массы, увеличение массы, токсичность для печени, кожные нарушения и т.д. Соединения также легче могут быть приготовлены в виде композиций вследствие повышенной растворимости в воде, пониженной токсичности, увеличенному терапевтическому диапазону, повышенной эффективности и т.д.
Общее описание предлагаемых в изобретении соединений
Соединения настоящего изобретения включают соединения общей формулы (0) и (0'), которые дополнительно определены ниже:

в которых R0 означает замещенный или незамещенный арильный, гетероарильный, арилалкильный, арилалкенильный, арилалкинильный, гетероарилалкильный, гетероарилалкенильный или гетероарилалкинильный остаток; в некоторых вариантах R0 означает арилалкильный, арилалкенильный, гетероарилалкильный или гетероарилалкенильный остаток; в других вариантах R0 означает гетероарилалкенильный остаток; в некоторых вариантах R0 означает гетероарилалкильный остаток; в других вариантах R0 означает 5-7-членный арильный или гетероарильный остаток; в других вариантах R0 означает 8-12-членный бициклический арильный или гетероарильный остаток; в следующих вариантах R0 означает бициклический остаток, в котором фенильный цикл конденсирован с гетероарильным или арильным остатком; в других вариантах R0 означает бициклический остаток, в котором фенильное кольцо конденсировано с остатком тиазола, оксазола или имидазола; в других вариантах R0 означает замещенный или незамещенный фенильный остаток;
R3 и R4 каждый независимо означают водород; или замещенный или незамещенный, линейный или разветвленный, циклический или ациклический алифатический, гетероалифатический радикал, арильный, гетероарильный, арилалкильный или гетероарилалкильный остаток, необязательно замещенные одним или несколькими группами из гидроксила, защищенного гидроксила, алкоксигруппы, карбоксигруппы, карбоксальдегида, линейного или разветвленного алкила или циклического ацеталя, фтора, аминогруппы, защищенной аминогруппы, аминогруппы, замещенной одним или двумя алкильными или арильными остатками, N-гидроксиминогруппой или N-алкоксииминогруппой; в некоторых вариантах R3 и R4 каждый независимо означают водород, фтор или низший алкил; в других вариантах R3 и R4 каждый независимо означают водород или метил; в других вариантах R3 означает метил, а R4 означает водород;
R5 и R6 каждый независимо означают водород или защитную группу; в некоторых вариантах R5 и R6 оба означают водород;
X означает O, S, C(R7)2 или NR7, где в каждом случае R7 независимо означает водород или низший алкил; в некоторых вариантах X означает O; в других вариантах X означает NH;
Y означает O, S, NH, C(R7)2, CH2, N(R7) или NH, где в каждом случае независимо R7 означает водород или низший алкил; в некоторых вариантах Y означает O; в других вариантах Y означает NH; в других вариантах Y означает CH2;
каждый R8 независимо означает водород; галоген, гидроксил, алкоксигруппу, аминогруппу, диалкиламиногруппу, алкиламиногруппу, фтор, цианогруппу или замещенный или незамещенный, линейный или разветвленный, циклический или ациклический алифатический, гетероалифатический радикал, арильный, гетероарильный, арилалкильный, арилалкенильный, арилалкинильный или гетероарилалкильный, гетероарилалкенильный, гетероарилалкинильный остаток, необязательно замещенные одним или несколькими заместителями из гидроксила, защищенного гидроксила, алкоксигруппы, карбоксигруппы, карбоксальдегида, линейного или разветвленного алкила или циклического ацеталя, фтора, аминогруппы, защищенной аминогруппы, аминогруппы, замещенной одним или двумя алкильными или арильными остатками, N-гидроксиминогруппой или N-алкоксииминогруппой; в некоторых вариантах R8 означает водород; в других вариантах R8 означает гидроксил; в других вариантах R8 означает фтор; в следующих вариантах R8 означает низший алкил, такой как метил; в других вариантах R8 означает -CF3, -CF2H или -CFH2; в других вариантах R8 означает перфторированную или фторированную алкильную группу; в других вариантах R8 означает галогенированную или пергалогенированную алкильную группу;
R9 и R10 каждый независимо означают водород; или замещенный, или незамещенный, линейный или разветвленный, циклический или ациклический алифатический, гетероалифатический радикал, арильный, гетероарильный, арильный, арилалкильный, арилалкенильный, арилалкинильный, гетероарилалкильный, гетероарилалкенильный или гетероарилалкинильный остаток, необязательно замещенные одним или несколькими заместителями из гидроксила, защищенного гидроксила, алкоксигруппы, карбоксигруппы, карбоксальдегида, линейного или разветвленного алкила или циклического ацеталя, фтора, аминогруппы, защищенной аминогруппы, аминогруппы, замещенный одним или двумя алкильными или арильными остатками, N-гидроксиминогруппой или N-алкоксииминогруппой; в некоторых вариантах один из R9 и R10 означает метил; в других вариантах оба R9 и R10 означают метил; в других вариантах один из R9 и R10 означает метил, а другой означает водород; в других вариантах оба R9 и R10 означают водород;
RB независимо для каждого случая означает водород; галоген; -ORB'; -SRB'; -N(RB')2; -C(O)ORB'; -C(O)RB'; -CONHRB'; -O(C=O)RB'; -O(C=O)ORB'; -NRB'(C=O)RB'; N3; N2RB'; циклический ацеталь; или циклический или ациклический, линейный или разветвленный алифатический, гетероалифатический радикал, арил или гетероарил, необязательно замещенные одной или несколькими группами из водорода; галогена; -ORB'; -SRB'; -N(RB')2; -C(O)ORB'; -C(O)RB'; -CONHRB'; -O(C=O)RB'; -O(C=O)ORB'; -NRB'(C=O)RB'; N3; N2RB'; циклического ацеталя; или циклического или ациклического, линейного или разветвленного замещенного или незамещенного алифатического, гетероалифатического радикала, арильного или гетероарильного остатка; или является эпотилоном, дезоксиэпотилоном или их аналогами; или является полимером; углеводом; фотоаффинной меткой; или радиоактивной меткой; в некоторых вариантах RB означает водород, 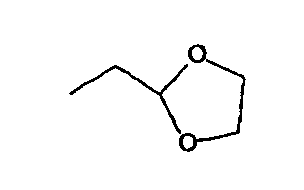 , метил, этил, пропил, бутил, пентил, гексил, циклопропил, циклобутил, циклопентил или циклогексил, каждый незамещен или необязательно замещен встречающимися один или несколько раз группами галогена, -OH, -ORB', NH2 или N(RB')2 или их комбинациями, где в каждом случае независимо RB' означает водород, алкил, арил или защитную группу, в других вариантах RB означает водород, метил или этил, в других вариантах RB означает метил, в других вариантах -CY3, -CHY2, -CH2Y, где Y означает F, Br, Cl, I, ORB', NHRB', N(RB')2 или SRB'; в других вариантах RB означает -CF3, -CH2F или CHF2; в других вариантах RB означает перфторированную или фторированную алкильную группу; в других вариантах RB означает галогенированную или пергалогенированную алкильную группу; в каждом случае RB независимо означает водород; защитную группу; линейный или разветвленный, замещенный или незамещенный, циклический или ациклический, алифатический, гетероалифатический радикал, арильный, гетероарильный, арилалкильный, арилалкенильный, арилалкинильный, гетероарилалкильный, гетероарилалкенильный или гетероарилалкинильный остаток;
, метил, этил, пропил, бутил, пентил, гексил, циклопропил, циклобутил, циклопентил или циклогексил, каждый незамещен или необязательно замещен встречающимися один или несколько раз группами галогена, -OH, -ORB', NH2 или N(RB')2 или их комбинациями, где в каждом случае независимо RB' означает водород, алкил, арил или защитную группу, в других вариантах RB означает водород, метил или этил, в других вариантах RB означает метил, в других вариантах -CY3, -CHY2, -CH2Y, где Y означает F, Br, Cl, I, ORB', NHRB', N(RB')2 или SRB'; в других вариантах RB означает -CF3, -CH2F или CHF2; в других вариантах RB означает перфторированную или фторированную алкильную группу; в других вариантах RB означает галогенированную или пергалогенированную алкильную группу; в каждом случае RB независимо означает водород; защитную группу; линейный или разветвленный, замещенный или незамещенный, циклический или ациклический, алифатический, гетероалифатический радикал, арильный, гетероарильный, арилалкильный, арилалкенильный, арилалкинильный, гетероарилалкильный, гетероарилалкенильный или гетероарилалкинильный остаток;
m равно 1, 2, 3 или 4, m равно 1 или 2 в некоторых вариантах, m равно 1 в других вариантах;
и их фармацевтически приемлемые производные.
Соединения, предлагаемые в изобретении, включают соединения общей формулы (I) или (I'), которые дополнительно определены ниже:
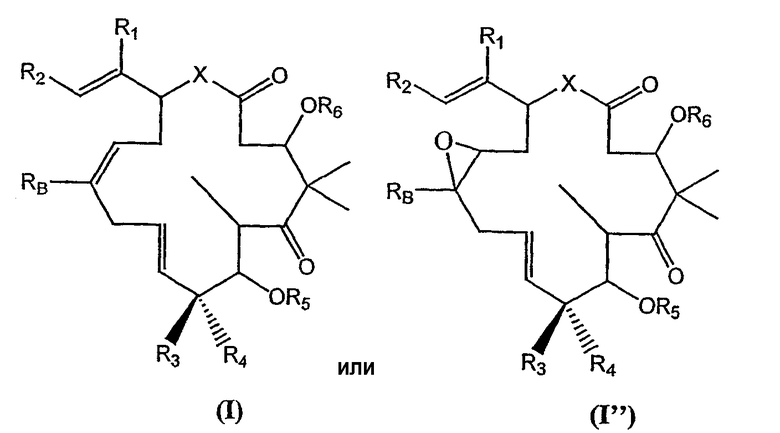
в которых R1 означает водород или низший алкил; в некоторых вариантах R1 означает метил; в некоторых вариантах R1 означает -CF3, -CF2H или CH2F; в других вариантах R1 означает перфторированную или фторированную алкильную группу; в других вариантах R1 означает галогенированную или пергалогенированную алкильную группу;
R2 означает замещенный или незамещенный арильный, гетероарильный, арилалкильный или гетероарилалкильный остаток; в некоторых вариантах R2 означает замещенный или незамещенный оксазол; в других вариантах R2 означает замещенный или незамещенный тиазол;
R3 и R4 каждый независимо означает водород; или замещенный или незамещенный, линейный или разветвленный, циклический или ациклический алифатический, гетероалифатический радикал, арильный, гетероарильный, арилалкильный или гетероарилалкильный остаток, необязательно замещенные одной или несколькими группами из гидроксила, защищенного гидроксила, алкоксигруппы, карбоксигруппы, карбоксальдегида, линейного или разветвленного алкила или циклического ацеталя, фтора, аминогруппы, защищенной аминогруппы, аминогруппы, замещенной одним или двумя алкильными или арильными остатками, N-гидроксиминогруппы или N-алкоксииминогруппы; в некоторых вариантах R3 и R4 каждый независимо означают водород, фтор или низший алкил; в других вариантах R3 и R4 каждый независимо означают водород или метил; в следующих вариантах R3 означает метил, а R4 означает водород;
R5 и R6 каждый независимо означают водород или защитную группу; в некоторых вариантах R5 и R6 оба означают водород;
X означает O, S, C(R7)2 или NR7, где в каждом случае R7 независимо означает водород или низший алкил; в некоторых вариантах X означает O; в других вариантах X означает NH;
RB независимо для каждого случая означает водород; галоген; -ORB'; -SRB'; -N(RB')2; -C(O)ORB'; -C(O)RB'; -CONHRB'; -O(C=O)RB'; -O(C=O)ORB'; -NRB'(C=O)RB'; N3; N2RB'; циклический ацеталь; или циклический или aциклический, линейный или разветвленный алифатический, гетероалифатический радиал, арил или гетероарил, необязательно замещенные одной или несколькими группами из водорода; галогена; -ORB'; -SRB'; -N(RB')2; -C(O)ORB'; -C(O)RB'; -CONHRB'; -O(C=O)RB'; -O(C=O)ORB'; -NRB'(C=O)RB'; N3; N2RB'; циклического ацеталя; или циклического или ациклического, линейного или разветвленного замещенного или незамещенного алифатического, гетероалифатического радикала, арильного или гетероарильного остатка; или является эпотилоном, дезоксиэпотилоном или их аналогами; или является полимером; углеводом; фотоаффинной меткой; или радиоактивной меткой; в некоторых вариантах RB означает водород,  , метил, этил, пропил, бутил, пентил, гексил, циклопропил, циклобутил, циклопентил или циклогексил, каждый из которых незамещен или необязательно замещен встречающимися один или несколько раз группами галогена, -OH, -ORB', NH2 или N(RB')2 или любыми их комбинациями, где в каждом случае независимо RB' означает водород, алкил, арил или защитную группу, в других вариантах RB означает водород, метил или этил, в других вариантах RB означает метил, в других вариантах RB означает -CF3, -CH2F или CHF2;
, метил, этил, пропил, бутил, пентил, гексил, циклопропил, циклобутил, циклопентил или циклогексил, каждый из которых незамещен или необязательно замещен встречающимися один или несколько раз группами галогена, -OH, -ORB', NH2 или N(RB')2 или любыми их комбинациями, где в каждом случае независимо RB' означает водород, алкил, арил или защитную группу, в других вариантах RB означает водород, метил или этил, в других вариантах RB означает метил, в других вариантах RB означает -CF3, -CH2F или CHF2;
и их фармацевтически приемлемые производные.
В некоторых вариантах соединения, предлагаемые в изобретении, включают соединения общей формулы (II) или (II'), имеющие стереохимию, определенную, как показано:
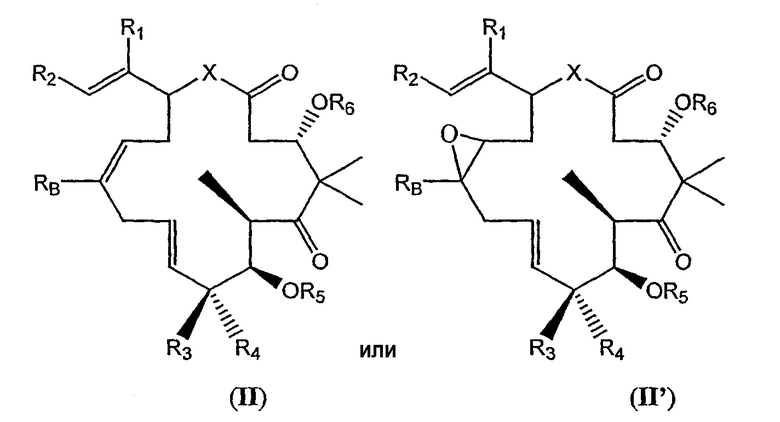
где X, R1, R2, R3, R4, R5, R6, RB и X имеют значения, определенные выше.
В некоторых вариантах X означает O. В других вариантах X означает NH. В других вариантах X означает CH2.
В некоторых вариантах R2 означает замещенный или незамещенный тиазол. В некоторых вариантах R2 означает 2-метилтиазо-4-ил. В других вариантах R2 означает 2-гидроксилметилтиазо-4-ил. В других вариантах R2 означает 2-аминометилтиазо-4-ил. В других вариантах R2 означает 2-тиолметилтиазо-4-ил.
В некоторых вариантах R2 означает замещенный или незамещенный оксазол. В некоторых вариантах R2 означает 2-метилоксазо-4-ил. В других вариантах R2 означает 2-гидроксилметилоксазо-4-ил. В других вариантах R2 означает 2-аминометилоксазо-4-ил. В других вариантах R2 означает 2-тиолметилоксазо-4-ил.
В некоторых вариантах RB означает водород, метил, этил, -CF3, -CH2F, -CF2H. В некоторых вариантах RB означает метил. В других вариантах RB означает -CF3. В некоторых вариантах RB означает водород. В других вариантах RB означает этил.
Некоторые предпочтительные соединения включают, например:

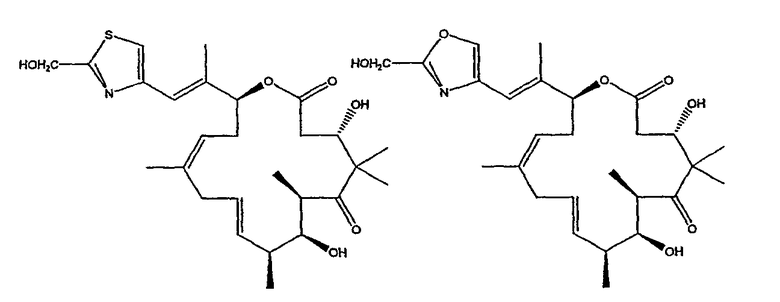
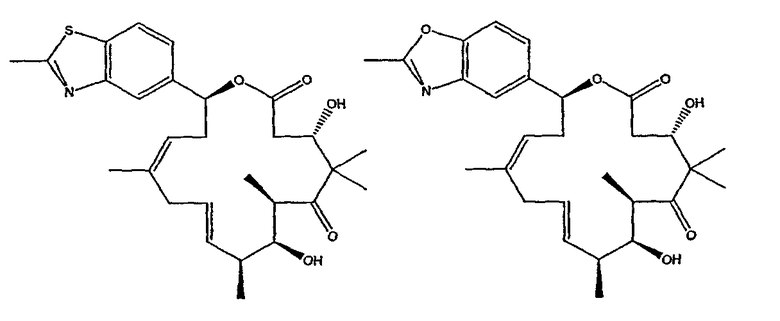


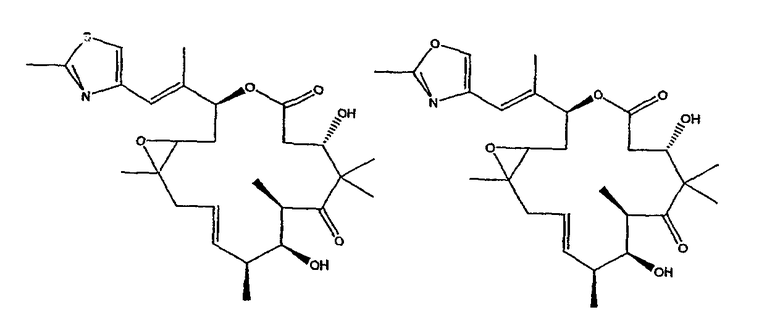
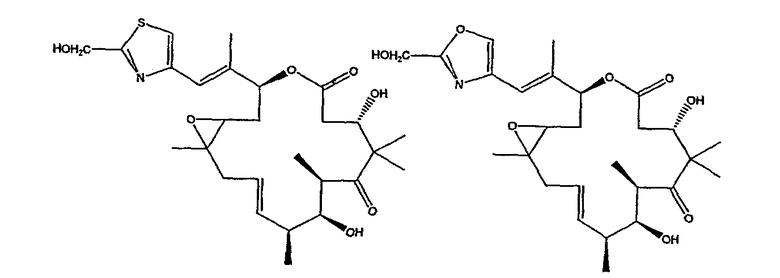
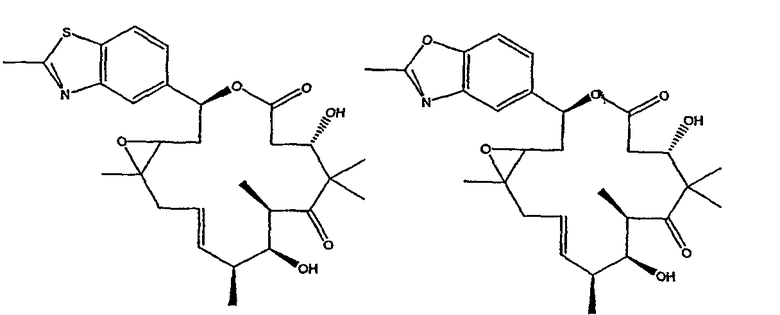
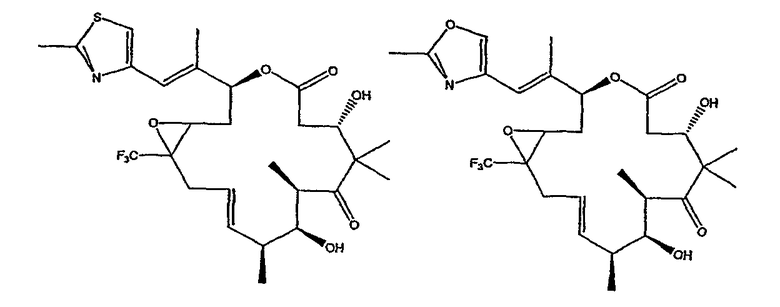
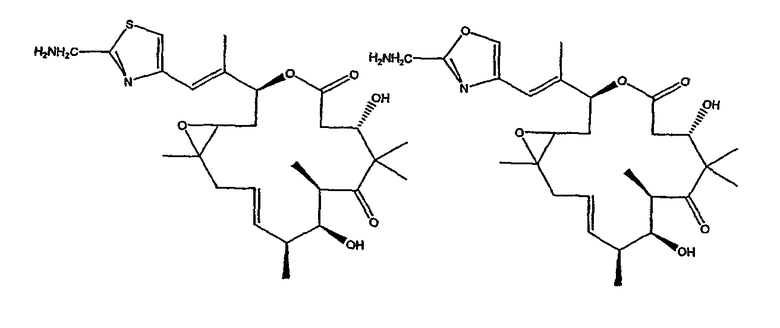
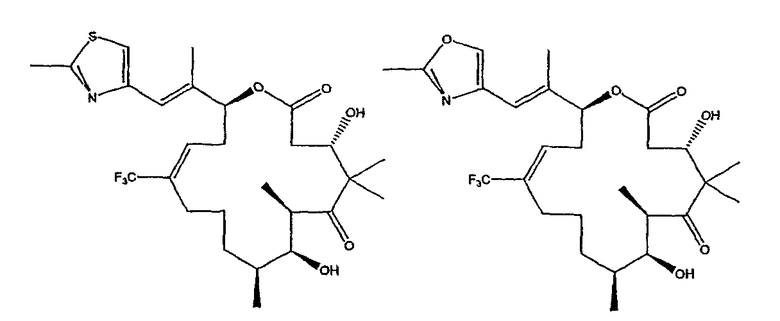
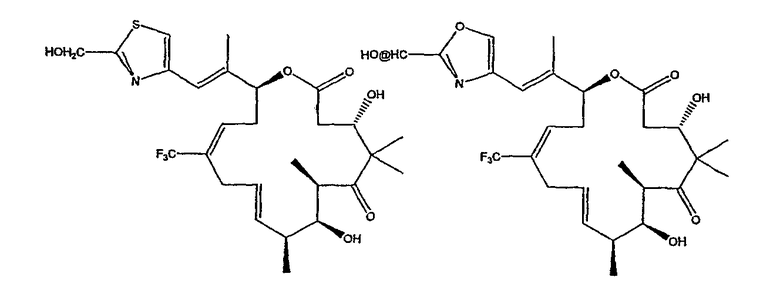
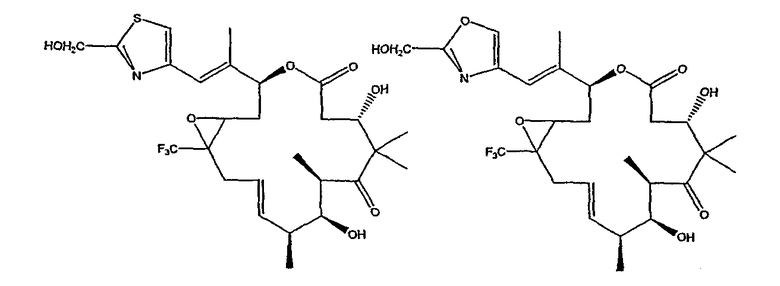
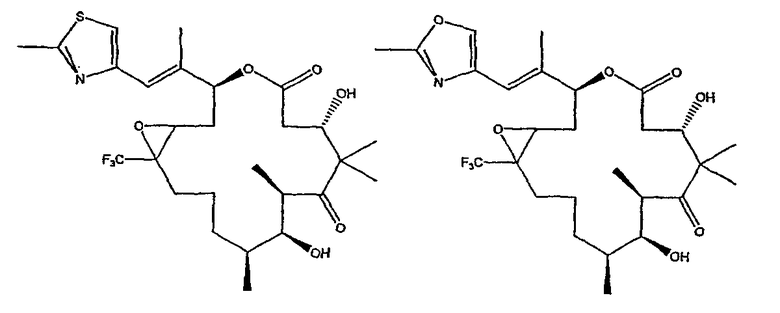

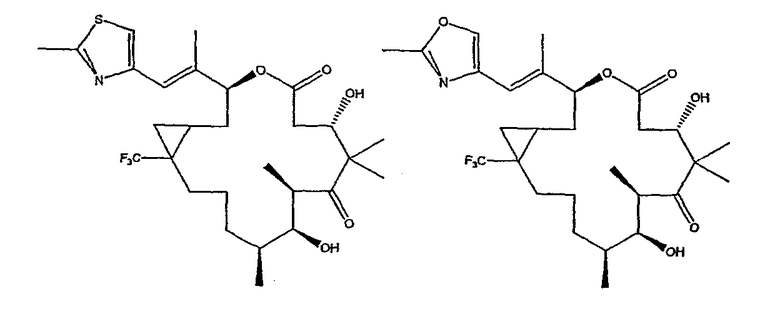
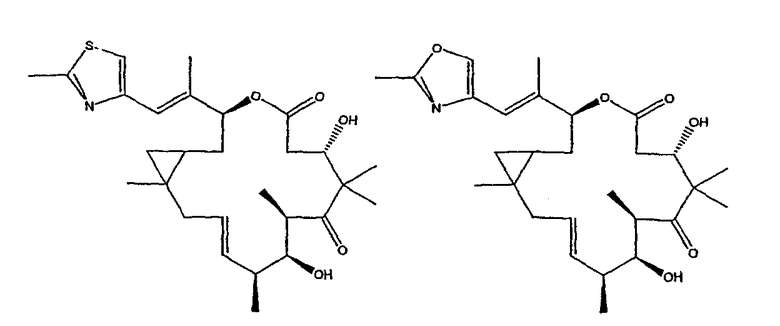

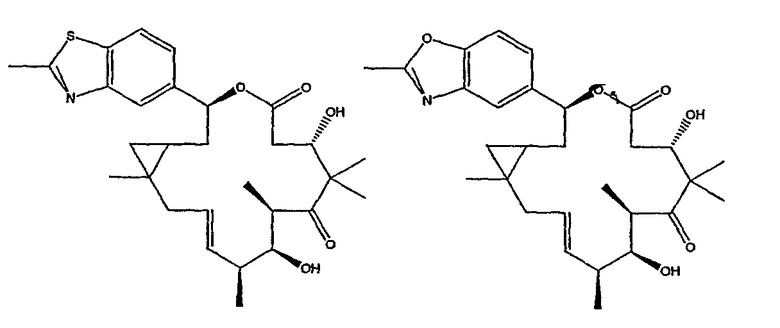
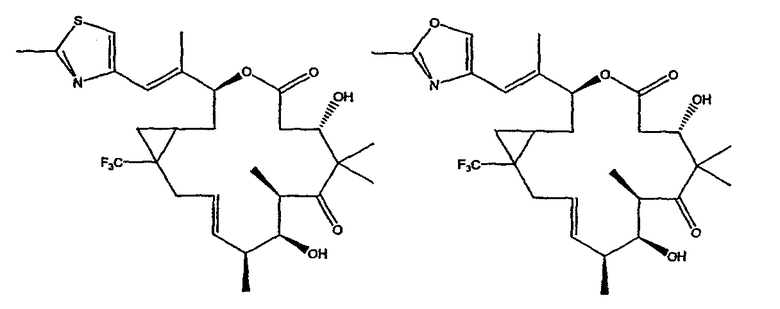
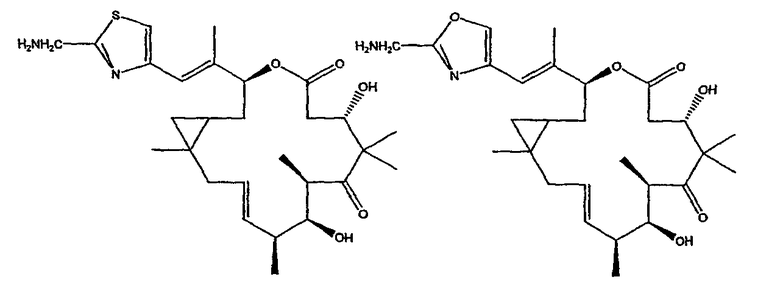
Соединения, предлагаемые в данном изобретении, включают соединения, конкретно указанные выше и описанные в данной публикации, и они частично иллюстрированы различными классами, подродами и видами, раскрытыми в других местах данного описания.
Специалисту в данной области будет понятно, что в соединениях настоящего изобретения могут существовать асимметричные центры. Таким образом, предлагаемые соединения и их фармацевтические композиции могут находиться в форме отдельного энантиомера, диастереомера или геометрического изомера, или могут быть в форме смеси стереоизомеров. В некоторых вариантах предлагаемые в изобретении соединения являются энантиомерно чистыми соединениями. В некоторых других вариантах предлагаются смеси стереоизомеров или диастереоизомеров.
Будет понятно, что некоторые из вышеуказанных классов и подклассов соединений могут существовать в различных изомерных формах. Изобретение включает соединения в виде отдельных изомеров, по существу не содержащих других изомеров, и альтернативно в виде смесей различных изомеров, например, рацемических смесей стереоизомеров. Кроме того, изобретение включает как (Z)-, так и (E)-изомеры по отношению к двойной связи, если не оговорено особо. Таким образом, предлагаемые в изобретении соединения, обычно изображаемые структурами (0), (0'), (I), (I'), (II) и (II') включают такие структуры, в которых двойные связи имеют конфигурацию (Z) или (E). В некоторых предпочтительных вариантах двойная связь в положении C12-C13 находится в цис- или Z-конфигурации. В некоторых вариантах двойная связь в положении C9-C10 находится в транс- или E-конфигурации. В других вариантах двойная связь в положении C12-C13 находится в цис- или Z-конфигурации, а двойная связь в положении C9-C10 находится в транс- или E-конфигурации. Изобретение также охватывает таутомеры конкретных соединений, которые описаны выше.
Кроме того, данное изобретение относится к фармацевтически приемлемым производным предлагаемых в изобретении соединений и способам лечения субъекта с использованием данных соединений, из фармацевтических композиций или любого из указанного в комбинации с одним или несколькими дополнительными терапевтическими средствами. Термин «фармацевтически приемлемое производное», используемый в данном описании, означает любую фармацевтически приемлемую соль, сложный эфир или соль сложного эфира заявленного соединения или любой другой аддукт или производное, которое при введении пациенту способно давать (прямо или опосредованно) соединение, которое во всех других отношениях описано в данной публикации, или его метаболит или остаток. Таким образом, фармацевтически приемлемые производные наряду с прочим включают пролекарства. Пролекарство представляет собой производное соединения обычно с пониженной в значительной степени фармакологической активностью, которое содержит дополнительный остаток, который поддается удалению in vivo, давая исходную молекулу в виде фармакологически активной формы. Примером пролекарства является сложный эфир, который расщепляется in vivo, давая желаемое соединение. Пролекарства ряда соединений и вещества и способы для дериватизации исходного соединения, чтобы создать пролекарства, известны и могут быть адаптированы для данного изобретения. Некоторые примеры фармацевтических композиций и фармацевтически приемлемые производные будут более подробно обсуждаться в данном описании ниже.
Соединения, предлагаемые в изобретении, которые представляют особый интерес, включают соединения, которые:
- оказывают цитотоксическое и ингибирующее рост действие на линии клеток злокачественных опухолей, поддерживаемые in vitro, или в исследованиях на животных с использованием приемлемой для научной цели модели ксенотрансплантата клеток злокачественной опухоли;
- проявляют способность полимеризовать тубулин и стабилизировать конструкции, собранные из микротрубочек;
- проявляют минимальный уровень токсичности по отношению к жизненно важным органам;
- приводят к исчезновению опухолей в приемлемых для научной цели моделях ксенотрансплантатов клеток злокачественных опухолей;
- приводят к уменьшению опухолей в приемлемых для научной цели моделях ксенотрансплантатов клеток злокачественных опухолей;
- приводят к исчезновению опухолей в приемлемых для научной цели моделях ксенотрансплантатов клеток злокачественных опухолей и отсроченному рецидиву/или отсутствию рецидива опухоли после прекращения лечения;
- дают временные и обратимые снижения массы тела и оказывают терапевтическое действие в приемлемых для научной цели моделях ксенотрансплантатов клеток злокачественных опухолей;
- проявляют повышенную растворимость в воде по сравнению с эпотилонами A, B, C или D или паклитакселом, или дополнительно, или альтернативно имеют растворимость, достаточную для приготовления в идее композиций в водной среде с использованием уменьшенной доли кремофора; и/или
- демонстрируют терапевтический профиль (например, оптимальную безопасность и лечебное действие), которые лучше, чем профиль эпотилона B, эпотилона D или паклитаксела.
Множество аналогов эпотилона, которые описаны выше, получены, охарактеризованы и тестированы, как указано в примерах в данном описании. Обнаружено, что аналоги 9,10-дегидроэпотилона применимы для лечения злокачественной опухоли, и, в частности, получены соединения и обнаружено, что они обладают одним или несколькими характерными признаками, перечисленными выше.
Методика синтеза
Синтез некоторых эпотилонов, дезоксиэпотилонов и их аналогов описан ранее (см. патенты США 6242469, 6284781, 6300355, 6204388, 6316630 и 6369234; заявки на выдачу патентов США 09/797027, 09/796959 и 10/236135; и публикации PCT № WO 99/01124, WO 99/43653, и WO 01/64650, полное содержание которых включено в данное описание в виде ссылки). Зная о необходимости усовершенствованных или дополнительных методик синтеза для эффективного образования эпотилонов, дезоксиэпотилонов и их аналогов в больших количествах, в данном изобретении предлагается эффективный и модульный способ синтеза эпотилонов, дезоксиэпотилонов и их аналогов. Хотя синтез некоторых приведенных в качестве примеров соединений описан в иллюстративных примерах в данном описании, будет понятно, что данный способ, в общем, применим к получению аналогов и конъюгатов, обсуждаемых выше, для каждого из классов и подклассов, приведенных в данном описании.
В частности, соединения 9,10-дегидроэпотилонов, предлагаемые в данном изобретении, могут быть получены множеством способов с использованием методик синтеза, применимых при синтезе эпотилонов. В некоторых вариантах соединения получают с использованием конвергентного пути синтеза. Например, эпотилон можно синтезировать, получая два или три промежуточных продукта, которые соединяют вместе, получая требуемое соединение. В одном варианте одним из промежуточных продуктов является ацильная часть, содержащая атомы углерода 1-9, а другой промежуточный продукт содержит атомы углерода 10-15 и также может содержать боковую цепь тиазола. Указанные две примерно равные части эпотилона могут быть соединены вместе сначала с использованием реакции этерификации между C-1 и кислородом у C-15. Затем макроцикл может быть замкнут с использованием реакции образования связи углерод-углерод, такой как реакция сочетания Сузуки или реакции метатезиса с замыканием цикла. В одном варианте конечную стадию замыкания цикла осуществляют с использованием реакции метатезиса с замыканием цикла с образованием 9,10-двойной связи и замыканием макроцикла. Реакцию метатезиса с замыканием цикла осуществляют с использованием металлоорганического катализатора, такого как катализатор Груббса, как показано на схеме 8 ниже. В некоторых вариантах 9,10-двойную связь восстанавливают или окисляют, или 9,10-двойную связь можно дополнительно функционализировать, чтобы получить дополнительные производные эпотилона.
В других вариантах конечную стадию замыкания цикла осуществляют, используя реакцию метатезиса с замыканием цикла, чтобы образовать 12,13-двойную связь и замкнуть макроцикл. В некоторых вариантах 12,13-двойную связь восстанавливают или окисляют. В других вариантах используют реакцию макроальдолизации или макролактонизации, чтобы образовать макроцикл.
Некоторые приведенные в качестве примеров реакции синтезы предлагаемых в изобретении соединений представлены на фигурах и в примерах. Как может быть понятно специалисту, можно получить множество аналогов и производных, используя описанные в данной публикации способы синтеза. Например, можно осуществить много стадий синтеза с разными защитными группами или разными заместителями в 16-членном цикле.
Фармацевтические композиции
Данное изобретение также относится к фармацевтическому препарату, содержащему, по меньшей мере, одно соединение, описанное выше, или его фармацевтически приемлемое производное, и данные соединения способны ингибировать рост или убивать клетки злокачественной опухоли и в некоторых представляющих особый интерес вариантах способны ингибировать рост или убивать клетки злокачественной опухоли с множественной лекарственной резистентностью. В некоторых вариантах фармацевтический препарат также содержит солюбилизирующий или эмульгирующий агент, такой как кремофор (полиоксил 35 касторового масла) или солютол (12-гидроксистеарат полиэтиленгликоля 660).
Как обсуждалось выше, данное изобретение относится к новым соединениям, обладающим противоопухолевой и антипролиферативной активностью, и таким образом, предлагаемые в изобретении соединения применимы для лечения злокачественной опухоли. Соответственно в другом аспекте данного изобретения предлагаются фармацевтические композиции, при этом указанные композиции содержат любое из соединений, описанных в данной заявке, и необязательно содержат фармацевтически приемлемый носитель. В некоторых вариантах указанные композиции необязательно дополнительно содержат одно или несколько дополнительных терапевтических средств. В некоторых других вариантах дополнительным терапевтическим средством является противораковой средство, которое более подробно обсуждается в данном описании.
Некоторые соединения данного изобретения, пригодные для лечения, могут существовать в свободной форме, или в соответствующих случаях в виде их фармацевтически приемлемого производного. Согласно данному изобретению фармацевтически приемлемое производное включает, но не ограничено указанным, фармацевтически приемлемые соли, сложные эфиры, соли таких сложных эфиров или любой другой аддукт или производное, которое при введении нуждающемуся в этом пациенту способно давать, прямо или опосредованно, соединение, которое в другом отношении описано в данной публикации, или его метаболит или остаток, например, пролекарство.
Используемый в данном описании термин «фармацевтически приемлемая соль» относится к таким солям, которые на основании обоснованного медицинского заключения подходят для применения в контакте с тканями человека и более низкоорганизованных животных без чрезмерной токсичности, раздражения, аллергической реакции и тому подобного и соответствуют разумному соотношению польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области. Например, S. M. Berge, et al. подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 66: 1-19 (1977), включенном в данное описание в виде ссылки. Соли могут быть получены in situ в ходе конечного выделения и очистки соединения согласно изобретению или отдельно в результате взаимодействия функции свободного основания с подходящей органической кислотой. Примерами фармацевтически приемлемых нетоксичных кислотно-аддитивных солей являются соли аминогруппы, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с использованием других способов, применяемых в данной области, таких как ионный обмен. Другие фармацевтически приемлемые соли включают такие соли, как адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидройодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, пара-толуолсульфонат, ундеканоат, валерат и тому подобные. Типичные соли щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция, магния и тому подобные. Следующие фармацевтически приемлемые соли включают в подходящем случае нетоксичные катионы аммония, четвертичного аммония и амина, образованные с использованием противоионов, такие как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, низший алкилсульфонат и арилсульфонат.
Кроме того, используемый в данном описании термин «фармацевтически приемлемый сложный эфир» относится к сложным эфирам, которые гидролизуются in vivo и включают эфиры, которые легко распадаются в организме человека, при этом остается исходное соединение или его соль. Подходящие сложноэфирные группы включают, например, группы, полученные из фармацевтически приемлемых алифатических карбоновых кислот, особенно алкановых, алкеновых, циклоалкановых кислот и алкандикислот, в которых алкильный или алкенильный остаток преимущественно имеет не более 6 атомов углерода. Примеры конкретных сложных эфиров включают формиаты, ацетаты, пропионаты, бутираты, акрилаты и этилсукцинаты.
Кроме того, термин «фармацевтически приемлемые пролекарства», используемый в данном описании, относится к таким пролекарствам соединений, предлагаемых в данном изобретении, которые на основании обоснованного медицинского заключения подходят для применения в контакте с тканями человека и более низкоорганизованных животных без чрезмерной токсичности, раздражения, аллергической реакции и тому подобного, соответствуют разумному соотношению польза/риск и эффективны в случае их предполагаемого применения, а также в том случае, когда это возможно, цвиттерионные формы соединений, предлагаемый в изобретении. Термин «пролекарство» относится к соединениям, которые быстро превращаются in vivo, давая исходное соединение указанных выше формул, например, в результате гидролиза в крови. Исчерпывающее обсуждение представлено в T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol.14 of the A. C. S. Symposium Series, и в Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, обе публикации включены в данное описание в виде ссылки.
Как описано выше, фармацевтические композиции, предлагаемые в данном изобретении, дополнительно содержат фармацевтически приемлемый носитель, включающий какой-либо и все растворители, разбавители или другие жидкие наполнители, вспомогательные вещества для образования дисперсий или суспензий, поверхностно-активные агенты, агенты, обеспечивающие изотоничность раствора, загустители или эмульгаторы, консерванты, твердые связующие вещества, смазывающие вещества и тому подобные, которые подходят для конкретной требуемой дозированной форме. В Remington's Pharmaceutical Sciences, Fifteenth Edition, E.W. Martin (Mack Publishing Co., Easton, Pa., 1975) описаны различные носители, используемые для приготовления фармацевтических композиций, и известные способы их получения. За исключением случаев, когда любая обычная среда носителя несовместима с противоопухолевым соединением согласно изобретению, например, оказывая какое-либо нежелательное биологическое действие или иным образом взаимодействуя с каким-либо другим соединением(ями) фармацевтической композиции с нежелательным эффектом, ее применение рассматривается в объеме данного изобретения. Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничены указанным, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразная трагакантовая камедь; солод; желатин; тальк; кремофор; солютол; эксципиенты, такие как масло какао, воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные средства, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотоничный физиологический раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, агенты, способствующие высвобождению, покрывающие агенты, подсластители, корригенты отдушки, консерванты и антиоксиданты также могут присутствовать в композиции в соответствии с решением специалиста, готовящего композицию.
Применения соединений и фармацевтических композиций
Изобретение, кроме того, относится к способу ингибирования роста опухолей и/или метастазирования опухолей. В некоторых представляющих особый интерес вариантах изобретение относится к способу лечения злокачественных опухолей посредством ингибирования роста опухолей и/или метастазирования опухолей в случае клеток злокачественной опухоли с множественной лекарственной резистентностью. Способ заключается во введении терапевтически эффективного количества соединения или его фармацевтически приемлемого производного нуждающемуся в этом субъекту (включая, но не ограничиваясь указанным, человека или животного). В некоторых вариантах, особенно в случае лечения злокачественных опухолей, содержащих злокачественные клетки с множественной лекарственной резистентностью, терапевтически эффективным количеством является количество, достаточное для того, чтобы убивать или ингибировать рост линий клеток злокачественной опухоли с множественной лекарственной резистентностью. В некоторых вариантах предлагаемые в изобретении соединения применимы для лечения солидных опухолей.
Соединения и фармацевтические композиции, предлагаемые в данном изобретении, можно использовать для лечения и профилактики какого-либо заболевания или состояний, включающих пролиферативные заболевания (например, злокачественная опухоль), аутоиммунные заболевания (например, ревматоидный артрит) и инфекции (например, бактериальные, грибковые и т.д.). Соединения и фармацевтические композиции можно вводить животным, предпочтительно млекопитающим (например, одомашненным животным, кошкам, собакам, мышам, крысам) и более предпочтительно человеку. Можно использовать любой способ введения для доставки соединения фармацевтических композиции животному. В некоторых вариантах соединение или фармацевтическую композицию вводят парентерально.
В еще одном аспекте, согласно способам лечения, предлагаемым в данном изобретении, опухолевые клетки убивают или ингибируют их рост в результате контакта опухолевых клеток с предлагаемым соединением или композицией, которые описаны в данной публикации. Таким образом, в еще одном аспекте изобретения предлагается способ лечения злокачественной опухоли, включающий введение терапевтически эффективного количества предлагаемого в изобретении соединения или фармацевтической композиции, содержащей предлагаемое в изобретении соединение, нуждающемуся в этом субъекту в таких количествах и в течение такого периода времени, которое необходимо для достижения требуемого результата. В некоторых вариантах осуществления данного изобретения «терапевтически эффективное количество» предлагаемого в изобретении соединения или фармацевтической композиции представляет собой такое количество, которое эффективно для убивания или ингибирования роста опухолевых клеток. Соединения и композиции согласно способу, предлагаемому в данном изобретении, можно вводить с использованием любого количества и любого пути введения, эффективного для убивания или ингибирования опухолевых клеток. Таким образом, выражение «количество, эффективное для убивания или ингибирования роста опухолевых клеток», используемое в данном описании, относится к количеству агента, достаточного для того, чтобы убить или ингибировать рост опухолевых клеток. Точное требуемое количество будет варьировать в зависимости от вида, возраста и общего состояния субъекта, тяжести инфекции, конкретного противоопухолевого агента, способа его введения и тому подобного. Противоопухолевые соединения, предлагаемые в изобретении, предпочтительно готовят в дозированной лекарственной форме для простоты введения и постоянства дозы. Выражение «дозированная лекарственная форма», используемое в данном описании, относится к физически дискретной единице противоопухолевого агента, подходящей для пациента, подвергаемого лечению. Однако будет понятно, что относительно суммарного суточное применения соединений и композиций, предлагаемых в данном изобретении, решение будет выносить лечащий врач на основании обоснованного медицинского заключения. Конкретный уровень терапевтически эффективной дозы для любого конкретного пациента или организма будет зависеть от множества факторов, включая заболевание, подвергаемое лечению, и тяжесть нарушения; активность конкретного используемого соединения; конкретную используемую композицию; возраст, массу тела, общее состояние здоровья, пол и диету пациента; время введения, путь введения и скорость экскреции конкретного используемого соединения; продолжительность лечения; лекарственные средства, используемые в комбинации или одновременно с конкретным используемым соединением; и тому подобные факторы, хорошо известные в области медицины.
Кроме того, после приготовления состава с подходящим фармацевтически приемлемым носителем в требуемой дозе фармацевтические композиции, предлагаемые в данном изобретении, можно вводить человеку и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), буккально, в виде перорального или назального спрея и тому подобного, в зависимости от тяжести инфекции, подвергаемой лечению. В некоторых вариантах осуществления изобретения предлагаемые соединения, которые описаны в данной публикации, готовят путем конъюгирования с растворимыми в воде хелаторами или растворимыми в воде полимерами, такими как полиэтиленгликоль, как поли(1-глутаминовая кислота) или поли(1-аспарагиновая кислота), которые описаны в патенте США 5977163, полное содержание которого включено в данное описание в виде ссылки. В некоторых вариантах предлагаемые в изобретении соединения можно вводить перорально или парентерально в дозе, достаточной для доставки примерно от 0,001 мг/кг до 100 мг/кг, примерно от 0,01 мг/кг до 50 мг/кг, предпочтительно примерно от 0,1 мг/кг до 40 мг/кг, предпочтительно примерно от 0,5 мг/кг до 30 мг/кг, примерно от 0,01 мг/кг до 10 мг/кг, примерно от 0,1 мг/кг до 10 мг/кг и более предпочтительно примерно от 1 мг/кг до 25 мг/кг массы тела субъекта в сутки, один или несколько раз в сутки, чтобы получить требуемый терапевтический эффект. Требуемая доза может доставляться через день, каждые три дня, каждую неделю, каждые две недели, каждые три недели или каждые четыре недели. В некоторых вариантах требуемая доза может доставляться с использованием многократных введений (например, два, три, четыре, пять, шесть, семь, восемь, девять или десять).
Жидкие дозированные формы для перорального введения включают, но не ограничены указанным, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие дозированные формы могут содержать инертные разбавители, обычно используемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное масла, масло из зародышей пшеницы, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры сорбитана и жирных кислот и их смеси. Кроме инертных разбавителей, пероральные композиции также содержат адъюванты, такие как увлажнители, эмульгаторы и суспендирующие агенты, подсластители, корригенты и отдушки.
Инъекционные препараты, например, стерильные инъекционные водные или масляные суспензии, могут быть приготовлены согласно известной области техники с использованием подходящих диспергирующих и увлажняющих агентов и суспендирующих агентов. Стерильный инъекционный препарат также может представлять собой стерильный инъекционный раствор, суспензию или эмульсию в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. К приемлемым наполнителям и растворителям, которые могут быть использованы, относятся вода, раствор Рингера, U.S.P. и изотоничный раствор хлорида натрия. Кроме того, в качестве растворителя или суспензионной среды обычно используют стерильные нелетучие масла. Для такой цели можно использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для получения инъекционных препаратов используют жирные кислоты, такие как олеиновая кислота.
Инъекционные препараты можно стерилизовать, например, фильтрованием через фильтр, удерживающий бактерии, или внесением стерилизующих агентов в форму стерильных твердых композиций, которые можно растворять или диспергировать в стерильной воде или другой стерильной инъекционной среде перед использованием.
Чтобы продлить действие лекарственного средства, часто желательно замедлить всасывание лекарственного средства, доставленного посредством подкожной или внутримышечной инъекции. Это можно осуществить, используя жидкую суспензию кристаллического или аморфного вещества с плохой растворимостью в воде. Степень всасывания лекарственного средства в таком случае зависит от степени его растворения, которое в свою очередь может зависеть от размера кристаллов и кристаллической формы. Альтернативно замедленное всасывание парентерально введенной формы лекарственного средства достигается растворением или суспендированием лекарственного средства в масляном наполнителе. Инъекционные формы замедленного всасывания готовят путем образования матриц для микроинкапсулирования лекарственных средств в биоразлагаемые полимеры, такие как полилактид-полигликолид. В зависимости от соотношения лекарственного средства к полимеру и природы конкретного используемого полимера скорость высвобождения лекарственного средства можно регулировать. Примеры других биоразлагаемых полимеров включают сложные поли(ортоэфиры) и поли(ангидриды). Инъекционные препараты замедленного всасывания также готовят улавливанием лекарственного средства в липосомах или микроэмульсиях, которые совместимы с тканями организма.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые могут быть приготовлены смешиванием соединений, предлагаемых в данном изобретении, с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела и поэтому плавятся в прямой кишке или вагинальной полости и высвобождают активное соединение.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах активное соединение смешивают, по меньшей мере, с одним инертным фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или фосфат дикальция и/или a) наполнителями или сухими разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими веществами, такими, например, как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и аравийская камедь, c) смачивающими средствами, такими как глицерин, d) дезинтегрирующими агентами, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, e) замедляющими растворение агентов, такими как парафин, f) ускорителями всасывания, такими как соединения четвертичного аммония, g) увлажнителями, такими как, например, цетиловый спирт и глицеринмоностеарат, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смесями. В случае капсул, таблеток и пилюль дозированная форма также может содержать буферные агенты.
Твердые композиции сходного типа также могут быть использованы в качестве наполнителей в мягких и твердых желатиновых капсулах с использованием таких эксципиентов, как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное. Твердые дозированные формы таблеток, драже, капсул, пилюль и гранул можно приготовить с покрытиями и оболочками, такими как кишечнорастворимые покрытия и другие покрытия, хорошо известные в области приготовления фармацевтических композиций. Они необязательно могут содержать контрастные агенты и также могут иметь такой состав, что они высвобождают активный ингредиент(ы) только или предпочтительно в определенной части кишечного тракта необязательно замедленным образом. Примеры инкапсулирующих композиций, которые могут быть использованы, включают полимерные вещества и воски. Твердые композиции сходного типа также можно использовать в качестве наполнителей в мягких и твердых заполняемых желатиновых капсулах с использованием таких эксципиентов как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное.
Активные соединения также могут быть в микроинкапсулированной форме с одним или несколькими эксципиентами, которые указаны выше. Твердые дозированные формы таблеток, драже, капсул, пилюль и гранул можно приготовить с покрытиями и оболочками, такими как кишечнорастворимые покрытия, покрытия для контролируемого высвобождения и другие покрытия, хорошо известные в области приготовления фармацевтических композиций. В таких твердых дозированных формах активное соединение может быть смешано по меньшей мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие дозированные формы могут также содержать, и это является обычной практикой, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие вещества для таблетирования и другие вспомогательные средства для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль дозированные формы также могут содержать буферные агенты. Они необязательно могут содержать контрастные агенты и также могут иметь такой состав, что они высвобождают активный ингредиент(ы) только или предпочтительно в определенной части кишечного тракта необязательно замедленным образом. Примеры инкапсулирующих композиций, которые могут быть использованы, включают полимерные вещества и воски.
Дозированные формы для местного или трансдермального введения соединения, предлагаемого в данном изобретении, включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингаляторы или пластыри. Активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, которые могут требоваться. Офтальмический препарат, ушные капли и глазные капли также считаются входящими в объем данного изобретения. Кроме того, данное изобретение охватывает применение трансдермальных пластырей, которые имеют дополнительное преимущество, обеспечивая контролируемую доставку соединения в организм. Такие дозированные формы можно приготовить растворением или диспергированием соединения в подходящей среде. Также можно использовать усилители всасывания, чтобы увеличить поток соединения через кожу. Скорость можно контролировать либо снабжая контролирующей скорость мембраной, либо диспергируя соединение в полимерном матриксе или геле.
Как обсуждалось выше, соединения, предлагаемые в данном изобретении, применимы в качестве противоопухолевых агентов и таким образом могут быть применимы при лечении злокачественной опухоли посредством осуществления гибели опухолевых клеток или ингибирования роста опухолевых клеток. В общем предлагаемые в изобретении противоопухолевые агенты применимы при лечении злокачественных опухолей и других пролиферативных расстройств, включая без ограничения рак молочной железы, рак головного мозга, рак кожи, рак шейки матки, рак ободочной и прямой кишки, лейкоз, рак легкого, меланому, множественную миелому, неходжкинскую лимфому, рак яичников, рак поджелудочной железы, рак простаты и рак желудка, при этом названы только некоторые. В некоторых вариантах противоопухолевые агенты согласно изобретению активны против лейкозных клеток и клеток меланомы и, следовательно, применимы для лечения лейкозов (например, миелоидного, лимфоцитарного, промиелоцитарного, миелоцитарного и лимфобластного лейкозов, либо в острой, либо в хронической формах) и злокачественных меланом. В других вариантах противоопухолевые агенты согласно изобретению активны против солидных опухолей, а также убивают и/или ингибируют рост клеток с множественной лекарственной резистентностью (MDR-клеток). В некоторых вариантах противоопухолевые агенты согласно изобретению активны против злокачественных опухолей, которые резистентны к другим известным противораковым агентам или которые, как было обнаружено, не отвечают клинически на другие известные противораковые агенты. В других вариантах противоопухолевые агенты согласно изобретению активны против злокачественных опухолей, которые резистентны к другим противораковым стабилизирующим микротрубочки агентам (например, паклитакселу).
Также будет понятно, что соединения и фармацевтические композиции, предлагаемые в данном изобретении, можно использовать в комбинированной терапии, то есть соединения и фармацевтические композиции можно вводить одновременно, перед или после одного или несколько других требуемых терапевтических средств или медицинских процедур. Для конкретной комбинации терапевтических способов (терапевтических средств или процедур), используемых в комбинированной схеме, во внимание будет принята совместимость требуемых терапевтических средств и/или процедур и требуемый терапевтический эффект, который необходимо достичь. Также будет понятно, что применяемые терапевтические способы могут достигать требуемого эффекта в отношении того же самого расстройства (например, предлагаемое в изобретении соединение можно вводить одновременно с другим противоопухолевым агентом) или они могут достигать других эффектов (например, контроль каких-либо вредных эффектов).
Например, другие способы терапии или противоопухолевые агенты, которые можно использовать в комбинации с предлагаемыми в данном изобретении противоопухолевыми агентами, включают хирургию, лучевую терапию (лишь немногими примерами являются гамма-излучение, лучевая терапия пучком нейтронов, лучевая терапия электронным пучком, протонная терапия, брахитерапия и системные радиоактивные изотопы, при этом указаны только некоторые), эндокринную терапию, модификаторы биологического ответа (интерфероны, интерлейкины и фактор некроза опухолей (TNF), при этом указаны только некоторые), гипертермию и криотерапию, агенты для ослабления каких-либо вредных эффектов (например, противорвотные средства) и другие одобренные химиотерапевтические средства, включая, но не ограничиваясь указанным, алкилирующие лекарственные средства (мехлорэтамин, хлорамбуцил, циклофосфамид, мелфалан, ифосамид), антиметаболиты (метотрексат), антагонисты пурина и антагонисты пиримидина (6-меркаптопурин, 5-фторурацил, цитарабил, гемцитабин), яды веретена деления (винбластин, винкристин, винорелбин, паклитаксел, доцетаксел), подофиллотоксины (этопозид, иринотекан, топотекан), антибиотики (доксорубицин, блеомицин, митомицин), нитрозомочевины (кармустин, ломустин), неорганические ионы (цисплатин, карбоплатин), ферменты (аспарагиназа) и гормоны (тамоксифен, леупролид, флутамид и мегестрол), при этом названы только некоторые. Для более полного обсуждения усовершенствованных способов терапии злокачественных опухолей см. , список одобренных FDA лекарственных средств для онкологии в , и в Merck Manual, Seventeenth Ed. 1999, полное содержание которых включено в данное описание в виде ссылки.
В следующем аспекте данное изобретение также относится к фармацевтическому комплекту или набору, содержащему один или несколько емкостей, заполненных одним или несколькими ингредиентами фармацевтических композиций согласно изобретению, и в некоторых вариантах он содержит дополнительное одобренное терапевтическое средство для применения в комбинированной терапии. Необязательно к такой емкости(ям) может прилагаться листок в форме, установленной государственным органом, регламентирующим производство, применение или продажу фармацевтических продуктов, и в данном листке отражено разрешение органом производства, применения или продажи для введения человеку.
Эквиваленты
Типичные примеры, которые следуют далее, предназначены для того, чтобы помочь иллюстрировать изобретение, и не предназначены для ограничения объема изобретения, и не должны считаться таковыми. Действительно, различные модификации изобретения и многие дополнительные варианты его осуществления, кроме тех, которые показаны и описаны в данной публикации, будут очевидны для специалистов в данной области на основании полного содержания данного документа, включая примеры, которые следуют далее, и ссылки на научную и патентную литературу, цитированные в данном описании. Кроме того, должно быть понятно, что содержание указанных цитированных публикаций включено в данное описание в виде ссылки, чтобы помочь проиллюстрировать состояние уровня техники. Следующие примеры содержат важную дополнительную информацию, иллюстративные примеры и руководство, которые могут быть адаптированы по отношению к практике данного изобретения в различных вариантах его осуществления и его эквивалентах.
Иллюстративные примеры
Пример 1
Синтез 9,10-дегидро-12,13-дезоксиэпотилонов
В данном примере описан синтез транс-9,10-дегидро-12,13-дезоксиэпотилона B, 26-трифтор-транс-9,10-дегидро-12,13-дезоксиэпотилона B, 26-трифтор-12,13-дезоксиэпотилона B и 12,13-дезоксиэпотилона B и биологическое тестирование указанных соединений.
Фторированные производные эпотилонов получали и тестировали, учитывая повышенную фармакокинетику и химиотерапевтические показатели других медицинских средств с заместителями в виде фтора (Ojima, I.; Inoue, T.; Chakravarty, S.; J. Fluorine Chem. 1999, 97; Newman, R. A.; Yang, J.; Finlay, M. R. V.; Cabral, F., Vourloumis, D.; Stephens, L. C.; Troncoso, P.; Wu, X.; Logothetis, C. J.; Nicolaou, K. C.; Navone, N. M. Cancer Chemother. Pharmacol. 2001, 48, 319-326; каждая публикация включена в данное описание в виде ссылки).
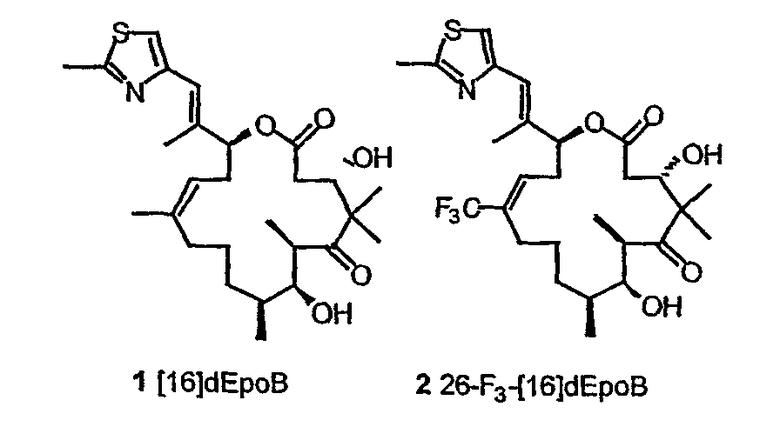
Чтобы получить соединение 2, авторы стремились использовать преимущество высококонвергентного пути, недавно описанного в публикации лаборатории авторов изобретения для синтеза эпотилона 490 (6, дегидродезокси-Epo B) как пути к dEpoB (1, схема 3) (Biswas, K., Lin, H., Njardarson, T., Chappell, M.D., Chou, T. C., Guan, Y., Tong, W.P., He, L., Horwitz, S.B., Danishefsky, S. J. J. Am. Chem. Soc. 2002, 124 (33); 9825-9832; Rivkin, A., Njardarson, J. T., Biswas, K., Chou, T. C., Danishefsky, S. J. J. Org. Chem. 2002, 67, 7737-7740; каждая публикация включена в данное описание в виде ссылки). В случае данного синтеза авторы вводили фланкирующую винильную группу в соединение 4 посредством стереоспецифичного сочетания Стилле предшественника винилйодида 3 с три-н-бутилвинилстаннаном. Метатезис с замыканием цикла с последующим удалением защиты приводил к 6, который затем превращали в dEpoB (1) посредством региоселективного восстановления диимидом.
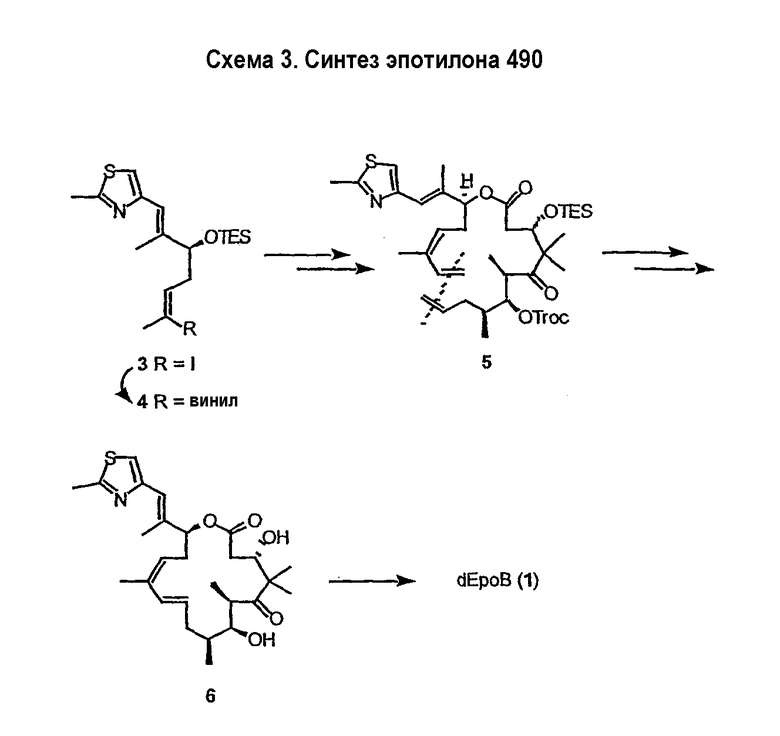
Сначала внимание было направлено на синтез 15 (схема 4). Алкилирование енолята лития 7, о котором сообщалось ранее (Chappell, M. D.; Stachel, S. J.; Lee, C. B.; Danishefsky, S. J. Org. Lett. 2000, 2 (11), 1633-1636; включено в данное описание в виде ссылки), йодидом 8 (синтезированным из известного спирта 16 с использованием TMSI в метиленхлориде) давало 9 с выходом 78% и высокой диастереоселективностью (>25:1 de). Соединение 9 в три стадии превращали в соединение 10, как показано. Попытки осуществить присоединение бромида метилмагния к связи амида Вейнреба 10 были неудачными. Нарушение данной реакции объяснялось присутствием связи йодалкена. Однако авторы смогли осуществить свою цель изменением порядка указанных двух стадий образования связи C-C. Таким образом, после взаимодействия 10 с винилтрибутилоловом в условиях Стилле может следовать присоединение реактива Гриньяра, содержащего метил, с получением требуемого кетона 11. Конденсация кетона 11 с фосфиноксидом 12 с последующим удалением защиты триэтилсилилового эфира давала фрагмент 13 с хорошим выходом. Этерификация полученного в результате 13 C1-C10-фрагментом кислоты 14 (Biswas, K.; Lin, H.; Njardarson, J. T.; Chappell, M. D., Chou, T. C., Guan, Y.; Tong, W. P., He, L.; Horwitz, S. B., Danishefsky, S. J. J. Am. Chem. Soc. 2002, 124 (33); 9825-9832; Rivkin, A.; Njardarson, J. T.; Biswas, K.; Chou, T. C.; Danishefsky, S. J. J. Org. Chem. 2002, 67, 7737-7740; публикации включены в данное описание в виде ссылки) давала требуемое соединение 15 с выходом 75% (схема 4).

К сожалению, попытки осуществить реакцию метатезиса с замыканием цикла 15 с использованием катализатора Груббса второго поколения (Обзоры: Grubbs, R. H.; Miller, S. J.; Fu, G. C. Acc. Chem. Res. 1995, 28, 446; Trnka, Т. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18; Alkene Metathesis in Organic Chemistry Ed.: Fürstner, A.; Springer, Berlin, 1998; Fürstner, A. Angew. Chem. Int. Ed. Engl. 2000, 39, 3012; Schrock, R. R. Top. Organomet. Chem. 1998, 1, 1; каждый из которых включен в данное описание в виде ссылки) в метиленхлориде приводили в основном к явной димеризации исходного вещества (уравнение 1). Учитывая тот факт, что RCM работает довольно хорошо в сходных условиях 5 → 6, авторы, естественно, отнесли неудачу в случае 15 к присутствию трифторметильной группы у C12.

Была выдвинута гипотеза, что вредное влияние постоянного 26-трифтор-заместителя на требуемую реакцию может быть ослаблено добавлением углеродного спейсера между реакционным центром RCM и трифторметильной группой. Соответственно авторы предприняли синтез 19 (уравнение 2) посредством метатезиса с замыканием цикла соединения 18, которое может представлять трифторметильную группу в контексте 17-членного цикла, содержащего добавленный (1,4)-диен.

Программа синтеза, направленная на 19, начиналась с получения соединения 21, которое соответствует O-алкильной части предлагаемого авторами субстрата RCM (схема 5). Авторы начинали с аллилирования 10, на этот раз в условиях радикальной реакции, как показано (Keck, G. E.; Yates, J. B. J. Am. Chem. Soc. 1982, 104, 5829; обзор: Curran, D. P. Synthesis 1988, Part 1, pp.417-439; Part 2, p.489; каждая публикация включена в данное описание в виде ссылки). После указанного превращения следовало взаимодействие алкилированного продукта с бромидом метилмагния, таким образом давая требуемый кетон 20. Конденсация данного соединения с фосфиноксидом 12 с последующим удалением защиты функции триэтилсилилового эфира давала 21 с хорошим выходом.
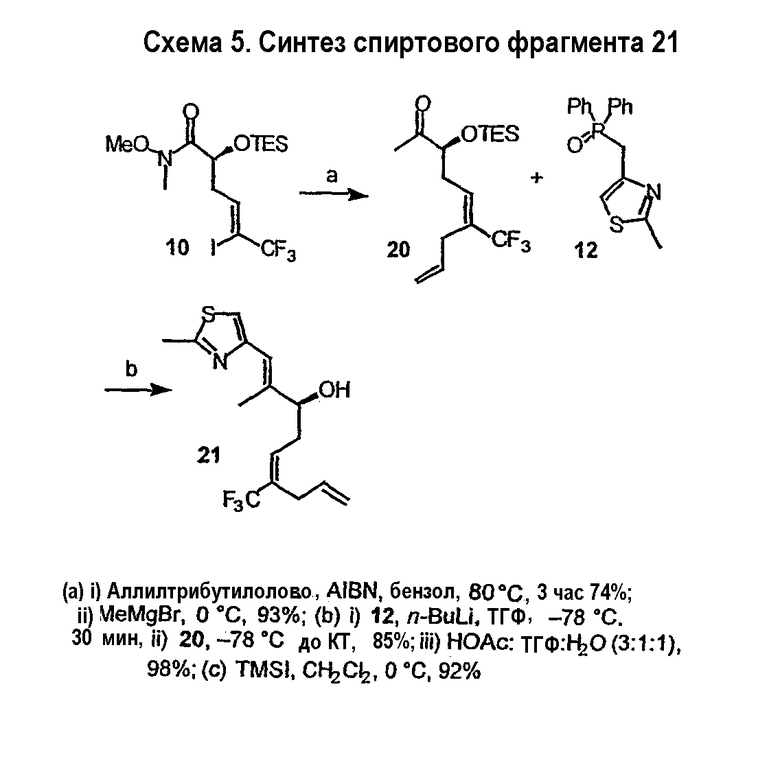
Этерификация 21 C1-C10-кислотным фрагментом 14 давала предлагаемый предшественник RCM 18 с выходом 75% (схема 6). В данном случае реакция метатезиса с замыканием цикла 18 могла быть успешно осуществлена с использованием катализатора Груббса второго поколения в метиленхлориде. Как и в случае превращения 5 → 6, реакция давала исключительно транс-изомер 22 с выходом 57%. Наконец, восстановительное отщепление защитной группы трихлорэтоксикарбонила с использованием цинка и уксусной кислоты с последующим удалением защиты TES-эфира HF-пиридином давало требуемое соединение 19, содержащее трифторметильную функцию у C12, хотя и в контексте ряда 17-членных циклов.

Синтетическое соединение 19 оценивали в отношении его цитотоксической активности. Как показано в таблице 1-1 ниже, прямое сравнение [17]ddEpoB (23), о котором сообщалось ранее, с 27-F3-[17]ddEpoB (19) показало, что новое перфторированное соединение обладало сравнительно высокой цитотоксической эффективностью.
Цитотоксичность in vitro (IC
50
) по отношению к линиям опухолевых клеток
а
(IC
50
(мкМ)
а
)
(IC
50
(мкМ)
а
)
Хотя трифторметильное изостерное замещение мало влияет на общую цитотоксическую активность, предварительные данные исследований метаболической деградации в плазме мышей показали, что 19 является заметно более стабильным, чем исходное соединение 23. Экспозиция эпотилонов 19 и 23 с плазмой мышей nude и человека приводила к деградации 23 в течение 30 минут, тогда как эпотилон 19 оставался по большей части интактным. Так как фармакокинетические проблемы, вероятно, имеют решающее значение при реальном применении эпотилонового агента в качестве лекарственного средства, авторы считают данные наблюдения довольно обнадеживающими.
Синтез 26-F3-dEpoB (2) может быть осуществлен посредством высококонвергентной методики, родственной методике, используемой при синтезе 27-F3-[17]ddEpoB (19). Таким образом, фрагменты сходной сложности могут служить в качестве ключевых строительных блоков (схема 7). Авторы предполагали, что ацильный участок 25 может служить в качестве полипропионатного домена и алкильный участок 21 или 24 может быть получен, как описано ранее во введении. Объединение двух фрагментов 21 (24) и 25 может быть инициировано посредством этерификации и завершено в результате последующего метатезиса с замыканием цикла. Наконец, отщепление защитных групп может привести к требуемым аналогам 28 и 29. Хемоселективное восстановление 9,10-олефина 28 и 29 может давать dEpoB (1) и требуемый 26-F3-12,13-дезоксиEpoB (2).

Синтез 1 и 2 начинался с получения ацильного участка 25. Кетон 30, о котором сообщалось ранее, подвергали альдольной реакции с легкодоступным альдегидом 31. После депротонирования и реакции «литий»-30 с 31, мягкая конденсация приводила к смеси 5,3:1 альдольных продуктов 32 и 33. Основной диастереоизомер 32 легко отделяли флэш-хроматографией и защищали как TBS-силиловый эфир. Гидролиз диизопропилацетальной группы в условиях кислотного катализа давал кетоальдегид 34, определяя стадию второй альдольной реакции. Следуя применяемому ранее на практике способу на основе «титана»-сложного трет-бутилового эфира с использованием нового альдегида 34 в качестве партнера в связывании, получали требуемый альдольный продукт 35 с высокой диастереоселективностью (dr>20:1) и выходом (86%). После защиты C3-спирта 35 TES-силильной следовало удаление защиты бензилового эфира. Окисление полученной в результате первичной гидроксигруппы давало соответствующий альдегид, который затем превращали конечный олефин посредством реакции Виттига, получая 36 с высоким выходом. Наконец, гидролиз сложного трет-бутилового эфира 36 с использованием TESOTf давал ацильный участок 25 (82%) наряду с побочным продуктом 37 (14%), который превращали в ацильный участок 38 с высоким выходом. Спектральные и хроматографические свойства 38 были идентичны свойствам ранее полученного вещества на основе других программ в лаборатории д-ра Sinha (Scripps).
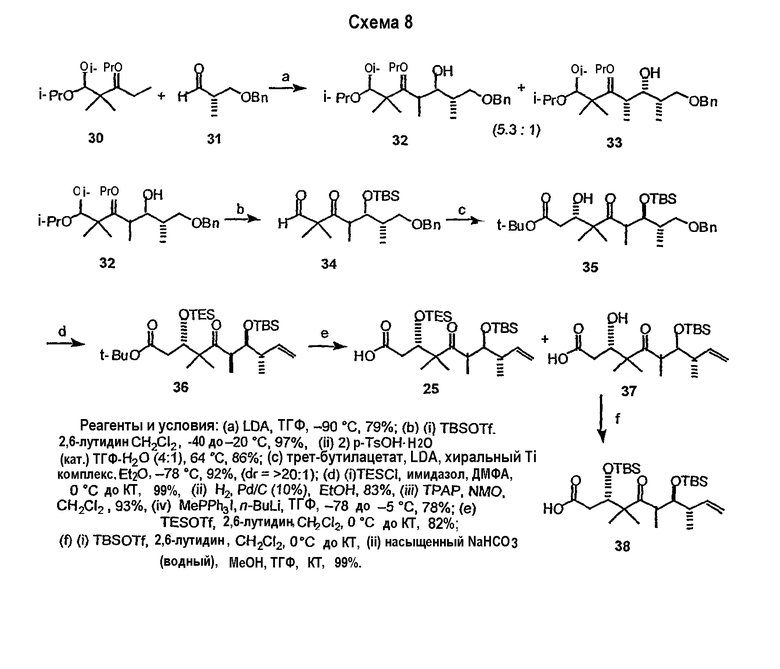
Этерификация аллиловых спиртов 21 и 24 C1-C9-кислотным фрагментом 25 давала соответствующие предшественники RCM-циклизации 26 и 27, соответственно (схема 9).
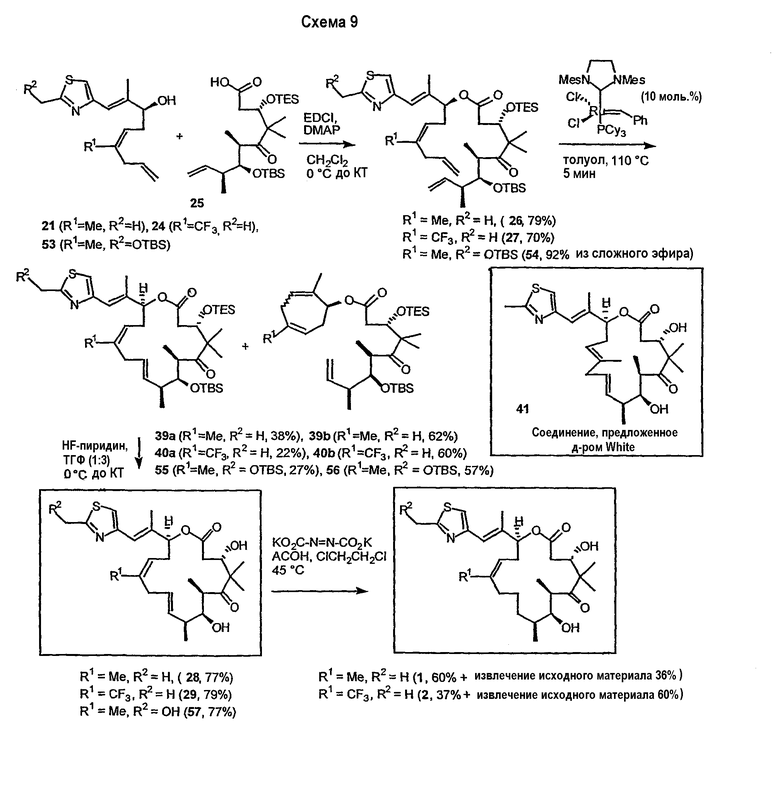
Затем проводили реакции метатезиса с замыканием цикла 26, 27 и 54, используя катализатор Груббса второго поколения в толуоле, которая давала, как и в более раннем исследовании авторов, исключительно транс-изомер 39a, 40a и 55 наряду с соответствующими побочными продуктами 39b, 40b и 56. Наконец, удаление защиты силиловых эфиров с использованием HF-пиридина приводила к требуемым соединениям 28, 29 и 57. Спектральные и хроматографические свойства 28 не были идентичными свойствам ранее полученного вещества на основе программы по эпотилонам в лаборатории д-ра James D. White (Oregon State University). Д-р James D. White, полагая, что он синтезировал 28, однако, вместо этого непреднамеренно он получил 12,13E-изомер 41, что может объяснить плохую биологическую активность, которую он наблюдал. Следовательно, авторы изобретения первыми синтезировали 28 и тестировали данное соединение в отношении его противоопухолевой активности.
Полностью синтетические 28, 29 и 2 оценивали по отношению к ряду типов клеток, чтобы определить их противоопухолевую эффективность. Как показано в таблице 1-2, все три соединения проявляли высокую цитотоксическую активность по отношению к множеству чувствительных и резистентных линий опухолевых клеток. Прямое сравнение 28 с dEpoB (1), о котором сообщалось ранее, показывает, что новое соединение обладает почти в три раза большей эффективностью.
Цитотоксичность in vitro (IC
50
) по отношению к линиям опухолевых клеток
а
Чтобы повысить общий выход синтеза 28, 29 и 2, авторы решили осуществить реакцию RCM в отсутствиe замещенного тиазолом олефина и, поступая таким образом, избежать образования нежелательного побочного продукта 39b и 40b. Удаление защиты силилового эфира 42 и 20, о которых сообщалось ранее, давало гидроксикетоны 43 и 44. Этерификация полученных в результате гидроксикетонов 43 и 44 C1-C9-кислотным фрагментом 25 давала соответствующие предшественники RCM-циклизации 45 и 46, соответственно (схема 10). Затем осуществляли реакцию метатезиса с замыканием цикла 45 и 46, используя катализатор Груббса второго поколения в толуоле, которая давала, как и в более раннем исследовании авторов, исключительно транс-изомер 47 и 48 с высокими выходами. Введение остатка триазола давало 39a, 40a и 55 с высоким выходом. Удаление защиты двух силиловых эфиров HF-пиридином приводило к 28 и 29. Наконец, селективное восстановление C9-C10-олефина давало соответствующие эпотилоны 1 и 2. Структура 28 была строго подтверждена по его превращению в 1 с высоким выходом. Общий синтез 1 был очень существенно упрощен по сравнению с ранее используемыми на практике способами. Таким образом, применение легкодоступного 31, получаемого из хирального пула, безусловно, является большим усовершенствованием по сравнению с тем случаем, когда полагаются на (S)-2-метил-4-пентеналь, синтез которого требует промежуточных хиральных вспомогательных веществ.
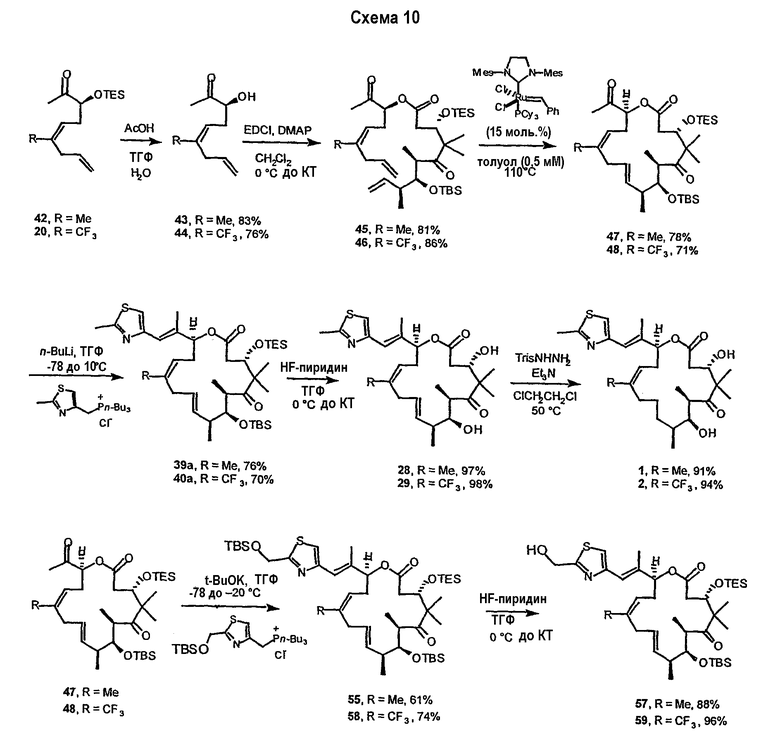
В случае соединения 28 при наличии строго подтвержденной структуры авторы были удивлены, обнаружив, что его спектральные свойства не совпадали со свойствами соединения, о котором сообщалось ранее, предположительно являющегося тем же самым соединением. Однако, обращаясь в прошлое, ясно, что соединение 28 ранее не было получено, в действительности целое семейство (E)-9,10-дегидроэпотилонов, указанных в данном описании, является новым классом соединений.
Исследование синтетических аналогов (2, 28 и 29) в условиях культуры клеток показало более сильное ингибирующее действие на различные чувствительные и MDR-линии опухолевых клеток, чем действие, оказываемое вошедшим в клинику dEpoB (1) (таблица 1-3). Авторы отмечают, что Epo 3 (28) является первым соединением 12,13-дезоксиэпотилона, которое обладает в значительной степени повышенной цитотоксичностью по сравнению с цитотоксичностью dEpoB (1).
Цитотоксичность in vitro (IC
50
) по отношению к линиям опухолевых клеток
a
(мкМ)
(мкМ)
Заметное ингибирование клеточного роста, оказываемое эпотилонами 2, 28 и 29 (Epo 2-4) на ряд различных резистентных к лекарственным средствам опухолей, побудило к определению стабильности указанных новых (E)-9,10-представителей в плазме крови. Например, недавно описанный (E)-10,11-дегидро-dEpoB (в случае 1 с CH3-группой у C-12) проявляет очень плохую стабильность в плазме по отношению к раскрытию лактона. Именно указанная нестабильность в плазме сдерживала продвижение (E)-10,11-дегидро-dEpoB. Напротив, при экспонировании 2, 28 и 29 (Epo 2-4) с плазмой мышей авторы наблюдали намного более медленную деградацию лекарственного средства по сравнению с dEpoB (1), в семь раз. Указанная стабильность представляет собой значительный прогресс с точки зрения доступности лекарственного средства, по сравнению с dEpoB (см. фиг.9).
Объединение данных о цитотоксичности и стабильности в плазме побудило авторов синтезировать значительные количества 28 (Epo 3), чтобы определить его эффективность in vivo у мышей nude, несущих ксенотрансплантаты опухолей человека. Эпотилон 28 (Epo 3) демонстрировал заметно повышенную эффективность в ингибировании роста имплантированных опухолей по сравнению с dEpoB (см. фиг.10). Повышенная эффективность и стабильность в плазме делает возможным очень существенное снижение дозы лекарственного средства (на порядок значений) в контексте ксенотрансплантатов в случае 28 (Epo3).
В более ранних исследованиях авторы обнаружили, что эпотилон B в виде 12,13-эпоксида значительно более цитотоксичен, чем его 12,13-дезоксианалог (dEpoB). Однако с точки зрения терапевтического индекса дезоксисоединение показалось авторам намного более обещающим. Недавно авторы сообщили о синтезе (E)-9,10-дегидро-12,13-дезоксиэпотилона B (28) с использованием стереоселективного метатезиса с замыканием цикла. Авторы показали, что внедрение E-9,10-ненасыщенности в контексте обычного Z-12,13-олефина (см. соединение 1) приводит к сильному увеличении эффективности in vitro. По существу это может быть перенесено на условия in vivo у мышей с ксенотранплантатами. Кроме того, соединение 28 обладает значительными фармацевтическими преимуществами по сравнению с dEpoB (1). Это дает возможность уменьшить уровни доз 28 по сравнению с 1 в экспериментах с ксенотрансплантатами, которые уменьшают на порядок значений.
Соответственно авторам было интересно, может ли внедрение C9-C10-олефина в эпотилон B (51, EpoB) изменить его биологический профиль в том же направлении.

Эпоксидирование 28 2,2'-диметилдиоксираном (DMDO) происходило с высокой хемоселективностью у более замещенного C12-C13-олефина, давая с выходом 87% соотношение 1:2,6 (E)-9,10-дегидроэпотилона B (49) и его диастереомера (50). Стереохимию эпоксидов определяли селективным восстановлением диимидом C9-C10-двойных связей. Исследование спектральных свойств указанных продуктов выявило, что минорный продукт (49) является dEpoB. Предпочтение α-эпоксидирования в случае 28 совершенно не похоже на высоко стереоселективное эпоксидирование dEpoB, которое происходит с β-стороны, приводя к EpoB (Meng, D.; Bertinato, P.; Balog, A.; Su, D.-S.; Kamenecka, T.; Sorensen, E. J.; Danishefsky, S. J. J. Am. Chem. Soc. 1997, 119, 10073; включено в данное описание в виде ссылки).
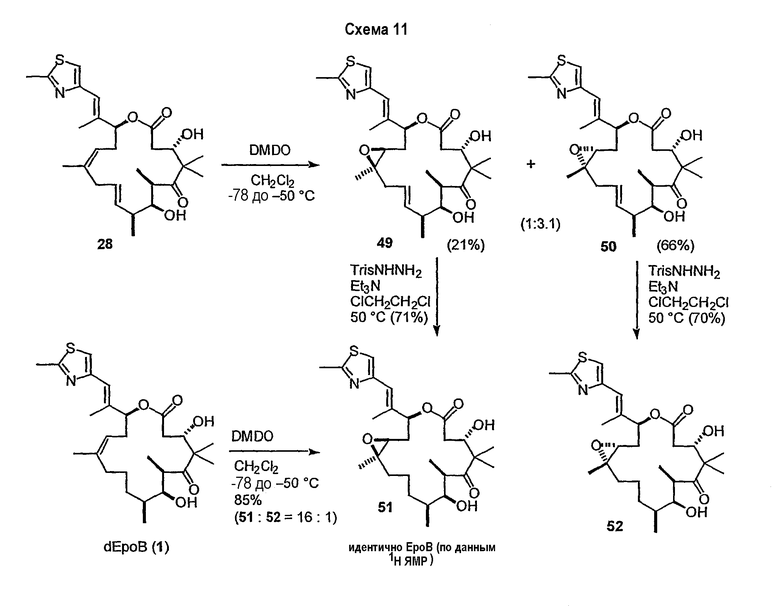
(E)-9,10-дегидроэпотилон B (51) оценивали по отношению к множеству разных типов клеток, чтобы определить их противоопухолевую эффективность. Как показано в таблице 1-4, (E)-9,10-дегидроэпотилон B (49) проявляет высокую цитотоксическую активность против множества чувствительных и резистентных линий опухолевых клеток. Прямое сравнение 49 и EpoB (51) показывает, что новый аналог обладает почти в 3 раза большей эффективностью, чем EpoB (51), что делает его одним из наиболее эффективных аналогов эпотилона, о которых сообщалось до настоящего времени. Интересно, что серия α-эпоксидов (50, 52) проявляет значительно меньшую активность, чем EpoB (51). На приведенном ниже графике показаны данные исследования in vivo соединения 49.
Цитотоксичность in vitro (IC
50
) по отношению к линиям опухолевых клеток
a
Терапевтическое действие 9,10-де-H-EpoB у мышей nude, несущих ксенотрансплантат MX-1 (6-час в/в-инфузия, n=4).
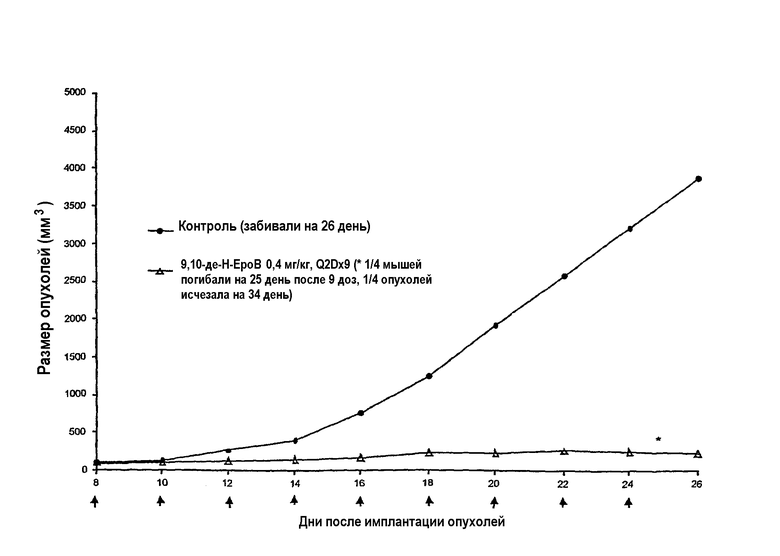
В заключение, описанное выше представляет эффективный стереоселективный общий синтез 28 (Epo 3) и после сайт-селективного восстановления диимидом dEpoB (1) как такового. Приведенную в данном описании методику затем непосредственно применяли к получению соответствующих трифтор-аналогов 2 и 29 (Epo 4). Кроме того, эпоксидирование 28 давало 49 и 50, которые при сайт-селективном восстановлении диимидом давали эпотилон B (51) и 52. Приведенные выше данные свидетельствуют о появлении наиболее многообещающего нового семейства противоопухолевых лекарственных средств, которые целесообразно оценить в отношении пути возможного в таком случае продвижения к условиям клинического испытания на человеке. Кроме того, новая методика синтеза включает в себя существенные практические усовершенствования общего синтеза dEpoB и эпотилона B.
Экспериментальные исследования
Общие способы: Реагенты, полученные от коммерческих поставщиков, использовали без дополнительной очистки, если не оговорено особо. Следующие растворители получали из безводной системы растворителей (пропущенной через предварительно заполненную колонку окиси алюминия) и использовали без дальнейшей сушки: тетрагидрофуран, метиленхлорид, диэтиловый эфир, бензол и толуол. Все чувствительные к атмосфере и воде реакции осуществляли в высушенной в пламени стеклянной посуде при положительном давлении предварительно очищенного газа аргона. Спектры ЯМР (1H и 13C) регистрировали на Bruker AMX-400 МГц или Bruker Advance DRX-500 МГц, которые указаны отдельно, используя в качестве эталона CDCl3 (7,27 м.д. для 1H и 77,0 м.д. для 13C). Инфракрасные спектры (ИК) получали на спектрометре Perkin-Elmer FT-IR модель 1600. Оптические вращения получали на цифровом поляриметре JASCO модели DIP-370 при 22±2°C. Аналитическую тонкослойную хроматографию проводили на пластинах E. Merck с силикагелем 60 F254. Соединения, которые не были УФ-активными, визуализировали, погружая пластины в раствор молибдата церия-аммония или пара-анисальдегида и нагревая. Хроматографию на силикагеле осуществляли, используя указанный растворитель на силикагеле Davisil® (сорт 1740, тип 60A, 170-400 меш).
Акронимы и сокращения
TES, триэтилсилил; TBS, диметилтретбутилсилил; EDCI, 1-этил-3-(3-диметиламинопропил)карбодиимид; HF-PY, фтористый водород в пиридине; DMAP, 4-N,N-диметиламинопиридин; ДХМ, дихлорметан; ДМФА, N,N-диметилформамид; ТГФ, тетрагидрофуран.

Соединение 32: К раствору свежеприготовленного LDA (11,6 ммоль) в ТГФ (25 ммоль) по каплям добавляли раствор кетона 30 (2,40 г, 10,4 ммоль) в ТГФ (6,8 мл) при -78°C. После перемешивания при -40°C в течение 0,5 час смесь охлаждали до -90°C. По каплям добавляли раствор альдегида 31 (1,38 г, 7,72 ммоль) в ТГФ (6,8 мл). После перемешивания при -90°C в течение 35 мин реакцию гасили насыщенным водным NH4Cl (15 мл) и экстрагировали EtOAc (50 мл × 3). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Очистка флэш-хроматографией на колонке (SiO2, гексан/EtOAc = от 15:1 до 12:1) давала 32 (2,09 г, 66%) и изомер 33 (0,39 г, 12%), оба в виде желтых масел. 32: [α]D 25 13,1 (c 1,22, CHCl3); ИК (пленка) ν 3494, 2972, 2932, 1708, 1454, 1380, 1329, 1120, 1038, 998, 734 cм-1; 1H ЯМР (400 МГц, CDCl3): δ 0,98 (3H, д, J=6,9 Гц), 1,06 (3H, д, J=6,9 Гц), 1,10 (3H, д, J=6,1 Гц), 1,14 (3H, д, J=6,9 Гц), 1,15 (3H, c), 1,17 (3H, д, J=6,2 Гц), 1,18 (3H, c), 1,20 (3H, д, J=6,2 Гц), 1,81-1,92 (1H, м), 3,33 (1H, кв.д, J=7,0, 2,2, Гц), 3,51 (1H, дд, J=8,9, 6,3 Гц), 3,64 (1H, д, J=1,8 Гц), 3,66-3,71 (2H, м), 3,78-3,86 (2H, м), 4,51 (1H, д, J=12,0 Гц), 4,54 (1H, д, J=12,0 Гц), 4,58 (1H, c), 7,25-7,35 (5H, м); 13С ЯМР (100 МГц, СDCl3) δ 10,0, 14,3, 20,5, 21,3, 21,9, 22,5, 23,5, 23,6, 36,4, 42,1, 54,1, 69,8, 71,2, 72,8, 73,3, 73,4, 103,8, 127,6, 127,7 (2C), 128,5 (2C), 138,9, 221,6; МС-НР (ESI) вычислено для С24Н40О5Na [M+Na+] 431,3, найдено 431,4.
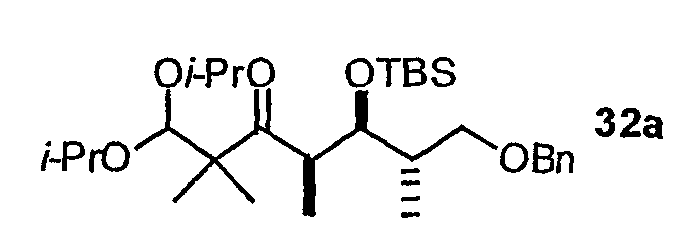
Соединение 32a (не показано): К охлажденному (-40°C) раствору спирта 32 (1,01 г, 2,47 ммоль) и 2,6-лутидина (69 мкл, 5,93 ммоль) добавляли TBSOTf (681 мкл, 3,00 ммоль) и смесь нагревали до -20°C в течение 3,5 час. Реакцию гасили насыщенным водным NaHCO3 (10 мл). После экстракции гексаном (50 мл × 3) объединенные органические экстракты сушили над Na2SO4 и концентрировали. Очистка флэш-хроматографией на колонке (SiO2, гексан/EtOAc = 50:1) давала 32a (1,25 г, 2,39 ммоль, 97%) в виде бесцветного масла; [α]D 25 -19,7 (с 0,58, СНCl3); ИК (пленка) ν 2966, 2931, 1696, 1455, 1378, 1320, 1255, 1091, 1044, 991, 873, 838, 773 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,08 (6H, c), 0,89 (9H, c), 0,99 (3H, д, J=7,0 Гц), 1,04 (3H, д, J=7,0 Гц), 1,07 (3H, д, J=7,0 Гц), 1,07 (3H, c), 1,14 (3H, д, J=6,1 Гц), 1,17 (3H, c), 1,17 (3H, д, J=6,0 Гц), 1,20 (3H, д, J=6,2 Гц), 1,76-1,85 (1H, м), 3,21 (1H, дд, J=9,2, 7,3 Гц), 3,32 (1H, квинт, J=7,4 Гц), 3,62 (1H, дд, J=9,2, 5,7 Гц), 3,78-3,85 (2H, м), 3,87 (1H, дд, J=7,7, 2,0 Гц), 4,46 (1H, д, J=12,1 Гц), 4,50 (1H, д, J=12,1 Гц), 4,73 (1H, c), 7,24-7,37 (5H, м); 13С ЯМР (100 МГц, СDCl3) δ -3,6, -3,3, 15,6, 16,8, 18,7, 18,8, 21,8, 22,1, 22,5, 23,5, 23,7, 26,4 (3С), 39,0, 46,2, 54,0, 69,7, 70,9, 72,1, 73,4, 76,7, 103,1, 127,6, 127,8 (2С), 128,5 (2С), 139,0, 218,9; МС-НР (ESI) вычислено для С30Н54О5SiNa [M+Na+] 545,4, найдено 545,4.

Соединение 34: Смесь 32a (3,03 г, 5,79 ммоль)и п-TsOH·H2O (286 мг) в водном ТГФ (64 мл, ТГФ/H2O = 4:1) кипятили с обратным холодильником в течение 6,5 час. Реакционную смесь охлаждали до КТ и вливали в насыщенный водный NaHCO3 (25 мл). После экстракции EtOAc (100 мл + 50 мл × 2) объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Очистка флэш-хроматографией на колонке (SiO2, гексан/EtOAc = от 50:1 до 30:1) давала 34 (2,37 г, 5,64 ммоль, 98%) в виде бесцветного масла: [α]D 25 -25,8 (с 0,515, СНСl3); ИК (пленка) ν 2955, 2931, 1731, 1696, 1455, 1360, 1255, 1091, 1026, 873, 826, 767 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,06 (3H, c), 0,07 (3H, c), 0,90 (9H, c), 0,95 (3H, д, J=7,1 Гц), 1,03 (3H, д, J=7,0 Гц), 1,28 (3H, c), 1,33 (3H, c), 1,73-1,82 (1H, м), 3,16 (1H, дд, J=9,2, 6,1 Гц), 3,28 (1H, квинт, J=7,3 Гц), 3,55 (1H, дд, J=9,2, 6,7 Гц), 3,91 (1H, дд, J=7,8, 2,1 Гц), 4,46 (2H, c), 7,27-7,36 (5H, м), 9,58 (1H, c); 13С ЯМР (100 МГц, СDCl3) δ -3,6, -3,5, 15,7, 16,3, 18,6, 19,8, 20,1, 26,3 (3C), 39,1, 47,0, 61,1, 71,9, 73,4, 75,8, 127,7, 128,0 (2С), 128,5 (2С), 138,6, 201,3, 213,3; МС-НР (ESI) вычислено для С24Н40О4SiNa [M+Na+] 443,3, найдено 443,2.
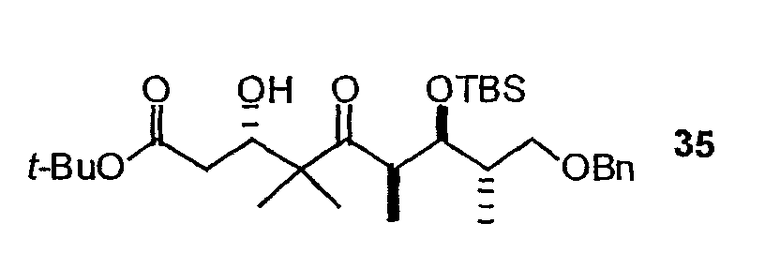
Соединение 35: К раствору свежеприготовленного LDA (18 мл 0,5 М раствора в Et2O, 9,0 ммоль) в Et2O (20 мл) добавляли трет-бутилацетат (1,16 мл, 8,61 ммоль) при -78°C. После перемешивания в течение 50 мин по каплям через шприцевой насос добавляли CpTiCl(OR)2 (100 мл 0,1 М раствора в Et2O, 10,0 ммоль) в течение 65 мин. После перемешивания в течение 20 мин реакционную смесь нагревали до -30°C, перемешивали в течение 50 мин и снова охлаждали до -78°C. По каплям в течение 10 мин добавляли раствор 34 (2,42 г, 5,75 ммоль) в Et2O (9 мл) и полученную в результате смесь перемешивали при -78°C. После перемешивания в течение 2 час реакцию гасили водным ТГФ (5 М H2O, 37 мл) и перемешивали при КТ в течение 2 час. После добавления воды (40 мл) смесь перемешивали еще в течение 1 час. Образовавшийся осадок отфильтровывали на целите (промывка Et2O) и фильтрат промывали водой (40 мл). Водный слой экстрагировали Et2O (100 мл × 2) и объединенные органические слои промывали насыщенным раствором соли (40 мл), сушили над Na2SO4 и концентрировали. Очистка флэш-хроматографией на колонке (SiO2, гексан/EtOAc = 10:1) давала 35 (2,65 г, 4,94 ммоль, 86%) в виде бледно-желтого масла; [α]D 25 -20,3 (с 1,0, СНСl3); ИК (пленка) ν 3523, 2957, 2930, 2856, 1732, 1700, 1472, 1368, 1252, 1152, 1091, 1042, 986, 834, 774 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,07 (3H, c), 0,07 (3H, c), 0,90 (9H, c), 0,99 (3H, д, J=7,0 Гц), 1,07 (3H, д, J=7,0 Гц), 1,10 (3H, c), 1,14 (3H, c), 1,47 (9H, c), 1,77-1,83 (1H, м), 2,26 (1H, дд, J=16,0, 10,0 Гц), 2,34 (1H, дд, J=15,9, 2,7 Гц), 3,23 (1H, дд, J=9,2, 7,1 Гц), 3,35 (1H, д, J=2,7 Гц, -OH), 3,36 (1H, квинт, J=7,0 Гц), 3,61 (1H, дд, J=9,2, 5,9 Гц), 3,88 (1H, дд, J=7,6, 2,0 Гц), 4,17 (1H, дт, J=10,0, 2,7 Гц), 4,48 (2H, c), 7,27-7,36 (5H, м); 13С ЯМР (100 МГц, СDCl3) δ -3,5, -3,4, 16,3, 16,7, 18,7, 20,1, 21,6, 26,4 (3C), 28,3 (3C), 38,0, 39,1, 45,8, 51,8, 72,2, 72,9, 73,5, 76,7, 81,4, 127,7, 128,0 (2C), 128,5 (2C), 138,8, 172,7, 219,6; МС-НР (ESI) вычислено для С30Н52О6SiNa [M+Na+] 559,3, найдено 559,4.

Соединение 35a (не показано): К смеси спирта 35 (10,2 г, 18,9 ммоль) и имидазола (2,70 г, 39,7 ммоль) в ДМФА (25 мл) добавляли TESCl (3,3 мл, 19,8 ммоль) при 0°C и смесь перемешивали при КТ в течение 2 час. Реакцию гасили насыщенным водным NaHCO3 (50 мл). После экстракции гексаном (500 мл + 120 мл × 2) объединенные органические экстракты последовательно промывали водой (30 мл × 2) и насыщенным раствором соли (30 мл), сушили над Na2SO4 и концентрировали. Очистка флэш-хроматографией на колонке (SiO2, гексан/EtOAc = 40:1) давала 35a (12,1 г, 18,5 ммоль, 98%) в виде бесцветного масла: [α]D 25 -38,0 (с 0,46, СНСl3); ИК (пленка) ν 2955, 2877, 1733, 1697, 1456, 1367, 1298, 1251, 1155, 1099, 988, 835, 742 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,05 (6H, c), 0,57-0,68 (6H, м), 0,89 (9H, c), 0,95 (9H, т, J=7,9 Гц), 0,99 (3H, д, J=7,0 Гц), 1,02 (3H, д, J=6,8 Гц), 1,04 (3H, c), 1,18 (3H, c), 1,45 (9H, c), 1,70-1,79 (1H, м), 2,16 (1H, дд, J=17,0, 7,0 Гц), 2,40 (1H, дд, J=17,0, 3,1 Гц), 3,22 (1H, дд, J=9,1, 7,5 Гц), 3,31 (1H, квинт, J=6,9 Гц), 3,61 (1H, дд, J=9,1, 5,4 Гц), 3,83 (1H, дд, J=7,3, 2,3 Гц), 4,30 (1H, дд, J=6,9, 3,1 Гц), 4,48 (2H, c), 7,27-7,36 (5H, м); 13С ЯМР (100 МГц, СDCl3) δ -3,5, -3,4, 5,3 (3C), 7,3 (3C), 15,3, 16,9, 18,7, 20,1, 23,4, 26,4 (3C), 28,3 (3C), 39,1, 41,1, 46,2, 53,4, 72,2, 73,4, 74,3, 76,7, 80,6, 127,6, 127,9 (2C), 128,5 (2C), 138,9, 171,5, 218,4; МС-НР (ESI) вычислено для С36Н66О6Si2Na [M+Na+] 673,4, найдено 673,5.

Соединение 35b (не показано): К перемешиваемому раствору 35a (4,37 г, 6,72 ммоль) в ТГФ (67 мл) добавляли Pd/C (приобретенный из Acros, 10% мас., 437 мг) и смесь перемешивали в атмосфере H2. После перемешивания в течение 2,2 час смесь фильтровали через подушку целита, который промывали ТГФ (120 мл). Фильтрат концентрировали и очищали флэш-хроматографией на колонке (SiO2, гексан/EtOAc = от 30:1 до 10:1), получая 35b (3,53 г, 6,28 ммоль, 94%) в виде бесцветного масла;
[α]D 25 -16,1 (с 0,62, СНСl3); ИК (пленка) ν 3543, 2956, 1732, 1696, 1472, 1368, 1299, 1252, 1155, 1100, 988, 837, 775, 742 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,10 (3Н, c), 0,12 (3H, c), 0,60-0,68 (6H, м), 0,93 (9H, c), 0,96 (9H, т, J=8,0 Гц), 0,99 (3H, д, J=7,1 Гц), 1,10 (3H, д, J=6,9 Гц), 1,14 (3H, c), 1,20 (3H, c), 1,45 (9H, c), 1,46-1,55 (1H, м), 2,21 (1H, дд, J=17,2, 7,1 Гц), 2,39 (1H, дд, J=17,2, 2,8 Гц), 2,54 (1H, т, J=5,8 Гц, -ОН), 3,30 (1H, квинт, J=6,9 Гц), 3,58 (1H, дт, J=11,5, 5,5 Гц), 3,66 (1H, дт, J=11,3, 5,4 Гц), 3,92 (1H, дд, J=8,0, 2,1 Гц), 4,32 (1H, дд, J=7,1, 2,9 Гц); 13С ЯМР (100 МГц, СDCl3) δ -3,6, -3,5, 5,3 (3С), 7,2 (3С), 16,0, 16,1, 18,6, 20,0, 23,4, 26,4 (3С), 28,3 (3С), 40,0, 40,9, 46,9, 53,7, 64,8, 73,3, 78,1, 80,9, 171,7, 218,5; МС-НР (ESI) вычислено для С29Н60О6Si2Na [M+Na+] 583,4, найдено 583,5.
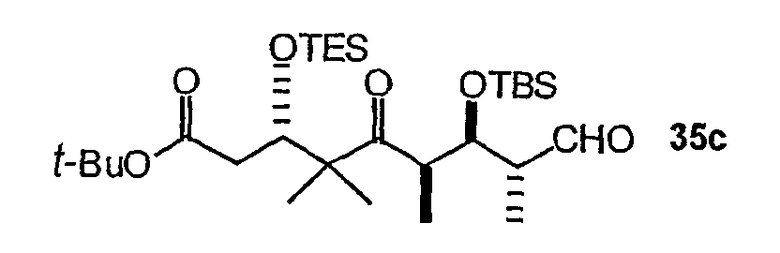
Соединение 35c (не показано): К перемешиваемой смеси спирта 35b (3,53 г, 6,28 ммоль) и порошкообразного MS4A (свежеактивированный, 2,50 г) в CH2Cl2 (32 мл) добавляли NMO (1,17 г, 10,0 ммоль), затем TPAP (132 мг, 0,377 ммоль). После перемешивания при КТ в течение 35 мин смесь фильтровали через колонку с силикагелем (гексан/Et2O = 8:1), получая 35c (3,34 г, 5,98 ммоль, 95%) в виде бесцветного масла; [α]D 25 -69,6 (с 0,25, СНСl3); ИК (пленка) ν 2955, 2878, 1732, 1696, 1472, 1368, 1253, 1155, 1097, 989, 837 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,09 (3H, c), 0,10 (3H, c), 0,59-0,68 (6Н, м), 0,89 (9Н, c), 0,95 (9Н, т, J=8,0 Гц), 1,08 (3H, c), 1,11 (3H, д, J=6,9 Гц), 1,14 (3H, д, J=7,1 Гц), 1,24 (3H, c), 1,45 (9Н, c), 2,19 (1H, дд, J=17,0, 6,7 Гц), 2,33 (1H, кв.т, J=7,1, 2,2 Гц), 2,41 (1H, дд, J=17,0, 3,3 Гц), 3,28 (1H, квинт, J=7,5 Гц), 4,07 (1H, дд, J=7,9, 2,2 Гц), 4,32 (1H, дд, J=6,7, 3,2 Гц), 9,74 (1H, д, J=2,0 Гц); 13С ЯМР (100 МГц, СDCl3) δ -3,8, -3,5, 5,3 (3С), 7,2 (3С), 12,6, 15,6, 18,5, 20,5, 23,3, 26,2 (3С), 28,3 (3С), 41,1, 46,9, 51,1, 53,5, 74,0, 76,5, 80,7, 171,1, 204,3, 218,0 МС-НР (ESI) вычислено для С29Н58О6Si2Na [M+Na+] 581,3, найдено 581,3.
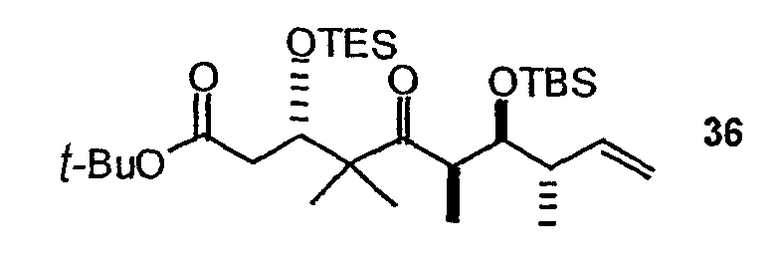
Соединение 36: MePPh3I (2,56 г, 7,18 ммоль) в ТГФ (40,0 мл) обрабатывали t-BuOK (6,57 мл 1,0 М раствора в ТГФ; 6,57 ммоль) при 0°C. После перемешивания при 0°C в течение 20 мин полученную в результате суспензию охлаждали до -78°C и добавляли раствор альдегида 35c (3,34 г, 5,98 ммоль) в ТГФ (14 мл). После перемешивания при -78°C в течение 15 мин смесь перемешивали при 0°C в течение 15 мин и при КТ в течение 15 мин. Реакцию гасили насыщенным водным NH4Cl (20 мл) и экстрагировали Et2O (120 мл + 50 мл × 2). Объединенные органические экстракты промывали насыщенным раствором соли (20 мл), сушили над Na2SO4 и концентрировали. Остаток очищали флэш-хроматографией на колонке (SiO2 ~80 г, гексан/Et2O = 40:1), получая 36 (125,3 мг, 0,225 ммоль, 78%) в виде бесцветного масла; [α]D 25 -33,6 (с 0,250, СНСl3); ИК (пленка) ν 2956, 2878, 1733, 1696, 1472, 1367,1299, 1253, 1156, 1100, 988, 837, 774 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,08 (3H, c), 0,08 (3H, c), 0,60-0,68 (6H, м), 0,93 (9H, c), 0,96 (9H, т, J=8,0 Гц), 1,04 (6H, д, J=7,0 Гц), 1,09 (3H, c), 1,20 (3H, c), 1,45 (9H, c), 2,08-2,15 (1H, м), 2,29 (1H, дд, J=17,0, 7,0 Гц), 2,41 (1H, дд, J=17,0, 3,1 Гц), 3,08 (1H, квинт, J=7,0 Гц), 3,84 (1H, дд, J=7,0, 2,1 Гц), 4,32 (1H, дд, J=7,0, 3,1 Гц), 5,02 (1H, дд, J=17,9, 1,0 Гц), 5,06 (1H, дд, J=10,5, 1,0 Гц), 5,93 (1H, ддд, J=17,9, 10,5, 7,7 Гц); 13С ЯМР (100 МГц, СDCl3) δ -3,6, -3,3, 5,4 (3С), 7,2 (3С), 15,2, 18,7, 19,0, 20,2, 23,6, 26,4 (3С), 28,3 (3С), 41,1, 43,8, 46,4, 53,5, 73,9, 76,6, 80,6, 115,5, 140,2, 171,5, 218,5; МС-НР (ESI) вычислено для С30Н60О5Si2Na [M+Na+] 579,4, найдено 579,4.

Соединение 25: К раствору сложного трет-бутилового эфира 36 (4,87 г, 8,74 ммоль) и 2,6-лутидина (свежеперегнанного, 4,1 мл, 35,0 ммоль) в CH2Cl2 (58 мл) добавляли TESOTf (4,0 мл, 17,5 ммоль) при 0°C. После перемешивания при 0°C в течение 25 мин смесь перемешивали при КТ в течение 3,2 час. Смесь разбавляли Et2O (600 мл), последовательно промывали 5% водным KHSO4 (60 мл × 2) и насыщенным раствором соли (60 мл), сушили над Na2SO4 и концентрировали. Остаток сушили в условиях высокого вакуума в течение 1,5 час, получая неочищенную кислоту 25 (6,30 г, загрязненную TESOH). Неочищенный продукт (6,30 г) растворяли в водном ТГФ (87,5 мл, ТГФ/H2O = 6:1) и обрабатывали насыщенным водным NaHCO3 (12,5 мл). После перемешивания при КТ в течение 20 мин полученную в результате суспензию разбавляли Et2O (500 мл) и подкисляли водным 5% KHSO4 (55 мл). После разделения слоев водный слой экстрагировали Et2O (100 мл × 2) и объединенные органические слои промывали насыщенным раствором соли (50 мл × 2), сушили над Na2SO4 и концентрировали. Остаток сушили в условиях высокого вакуума в течение ночи, получая неочищенную кислоту (5,60 г, загрязненную TESOH) в виде бесцветного масла, которое использовали для следующей реакции без дополнительной очистки. Для характеристики очищали флэш-хроматографией на колонке на силикагеле, элюируя смесью гексан/EtOAc = 4/1. [α]D 25 -30,7 (с 0,985, СНСl3); ИК (пленка) ν 2956, 2936, 2879, 1712, 1472, 1417, 1303, 1253, 1107, 1046, 1003, 988, 872, 837, 775, 741 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,08 (3H, c), 0,09 (3H, c), 0,59-0,67 (6H, м), 0,93 (9H, c), 0,96 (9H, т, J=8,1 Гц), 1,05 (3H, д, J=7,0 Гц), 1,05 (3H, д, J=7,0 Гц), 1,20 (3H, c), 1,21 (3H, c), 2,06-2,13 (1H, м), 2,34 (1H, дд, J=16,4, 7,4 Гц), 2,50 (1H, дд, J=16,4, 3,0 Гц), 3,06 (1H, квинт, J=7,3 Гц), 3,87 (1H, дд, J=7,5, 1,8 Гц), 4,40 (1H, дд, J=7,3, 2,9 Гц), 5,01 (1H, дд, J=18,0, 0,9 Гц), 5,07 (1H, дд, J=10,4, 1,2 Гц), 5,93 (1H, ддд, J=18,0, 10,4, 7,8 Гц); 13С ЯМР (100 МГц, СDCl3) δ -3,6, -3,3, 5,3 (3C), 7,1 (3C), 15,6, 18,7, 19,1, 19,2, 24,1, 26,4 (3C), 39,8, 43,6, 46,4, 53,5, 73,7, 76,6, 115,6, 140,0, 177,9, 218,7; МС-НР (ESI) вычислено для С26Н52О5Si2Na [M+Na+] 523,3, найдено 522,9.

Соединение 45: 3-O-TES-6-O-TBS-защищенную кислоту 25 сушили посредством азеотропной перегонки с бензолом. Свежевысушенный спирт 43 (200 мг, 1,19 ммоль) растворяли в ДХМ (10 мл) и охлаждали до 0°C и в это время добавляли твердый DMAP (167 мг, 1,37 ммоль) и твердый EDCI (261 мг, 1,37 ммоль). После перемешивания реакционной смеси при 0°C в течение 15 мин по каплям добавляли раствор кислоты 25 (425 мг, 0,85 ммоль) в ДХМ (2 мл). Охлаждающую баню убирали и перемешивание продолжали еще в течение 2 часов. Неочищенную реакционную смесь разбавляли ДХМ (10 мл) и очищали хроматографией на силикагеле, используя 10% EtOAc/гексаны в качестве элюента, получая сложный эфир 45 (380 мг, выход 81%, две стадии, начиная с 36) в виде прозрачного масла: [α]D -15,1 (c 1,2, CDCl3); ИК (чистое вещество) 2955, 2932, 2877, 1743, 1732, 1694, 1474, 1461, 1417, 1380, 1360, 1295, 1252, 1169, 1094, 1043, 988,3, 912,9, 871,4, 836,5, 774,8, 741,6 cм-1; 1H ЯМР (500 МГц, CDCl3) 0,08 (3H, c), 0,08 (3H, c), 0,60-0,68 (6H, м), 0,93 (9Н, c), 0,95 (9Н, т, J=8,0 Гц), 1,04 (3H, д, J=6,9 Гц), 1,05 (3H, д, J=6,9 Гц), 1,10 (3H, c), 1,25 (3H, c), 1,69 (3H, c), 2,08-2,15 (2H, м), 2,16 (3H, c), 2,38 (1H, дд, J=17,0,7,0 Гц), 2,48 (2H, т, J=6,5 Гц), 2,57 (1H, дд, J=17,0, 2,7 Гц), 2,71-2,76 (2H, м), 3,07 (1H, квинт, J=7,0 Гц), 3,83 (1H, д, J=7,2 Гц), 4,36 (1H, дд, J=7,0, 2,7 Гц), 4,97-5,07 (4H, м), 5,19 (1H, т, J=7,0), 5,73 (1H, тд, J=15,4, 5,9 Гц), 5,92 (1H, дд, J=15,7, 8,0 Гц); 13С ЯМР (500 МГц, СDCl3) δ 218,4, 205,4, 172,1, 140,1, 137,4, 135,4, 119,1, 115,8, 115,6, 78,7, 76,5, 73,9, 53,3, 46,3, 43,7, 39,6, 36,6, 29,2, 26,7, 26,4, 23,8, 23,7, 19,9, 18,9, 18,7, 15,4, 7,06, 5,30, -3,29, -3,62; МС-НР (ESI) вычислено для С36Н66О6Si2Na [M+Na+] 673,4, найдено 673,5.

Соединение 47: К раствору соединения 45 (20 мг, 0,031 ммоль) в безводном толуоле (60 мл) при кипячении с обратным холодильником одной порцией добавляли раствор дихлорида трициклогексилфосфин[1,3-бис(2,4,6-триметилфенил)-4,5-дигидроимидазол-2-илиден][бензилиден]рутения (IV) (5,2 мг, 0,0061 ммоль) в безводном толуоле (2 мл) и смесь нагревали в течение 10 минут. Реакционную смесь сразу же охлаждали на бане со льдом, наносили на диоксид кремния и очищали, используя хроматографию на силикагеле с применением 4-10% градиента EtOAc/пентан в качестве элюента, чтобы получить соединение 47 (15 мг, выход 78%) в виде масла: [α] -28,6 (с 1,2, СНСl3); ИК (чистое вещество) 2955, 2933, 2878, 1745, 1731, 1695, 1471, 1462, 1380, 1361, 1251, 1159, 1104, 1080, 1019, 985,0, 876,1, 835,5, 774,7, 743,1, 670,1 cм-1; 1H ЯМР (500 МГц, CDCl3) 0,07 (3H, c), 0,10 (3H, c), 0,59-0,68 (6H, м), 0,91 (9H, т, J=8,0 Гц), 0,93 (9H, c), 1,04 (3H, д, J=7,0 Гц), 1,10 (3H, c), 1,11 (3H, д, J=7,0 Гц), 1,17 (3H, c), 1,71 (3H, c), 2,21 (3H, c), 2,27-2,32 (1H), 2,38 (1H, дд, J=14,6, 6,8 Гц), 2,51-2,61 (2H, м), 2,57 (1H, дд, J=15,5, 3,3 Гц), 2,93-3,1 (3H, м), 3,94 (1H, д, J=8,5 Гц), 4,28 (1H, дд, J=8,6, 3,0 Гц), 5,04 (1H, дд, J=8,7, 2,4), 5,16 (1H, т, J=7,5), 5,73 (1H, tдд, J=12,8, 9,94, 6,9 Гц), 5,92 (1H, ддд, J=18,0, 10,3, 7,8 Гц); 13С ЯМР (125 МГц, СDCl3) δ 215,9, 204,8, 171,3, 140,0, 132,7, 129,2, 118,6, 79,1, 78,2, 75,4, 54,0, 48,2, 41,7, 40,3, 35,0, 29,2, 26,6, 26,5, 23,5, 22,8, 20,6,18,8, 17,5, 14,3, 7,19, 5,53, -3,36; МС-НР (ESI) вычислено для С34Н62О6Si2 645,4, найдено 645,4 (M+Na+).

Соединение 39a: К раствору реагента Виттига (19,1 мг, 54,7 мкмоль) в ТГФ (0,4 мл) добавляли KHMDS (109 мкл 0,5 М раствора в толуоле, 54,7 мкмоль) при 0°C. Смесь перемешивали при 0°C в течение 0,5 час и затем охлаждали до -78°C. К смеси по каплям добавляли раствор кетона 47 (5,7 мг, 9,12 мкмоль) в ТГФ (0,3 мл) и полученной в результате смеси давали возможность нагреться до -20°C в течение 1,5 час. Реакцию гасили насыщенным водным NH4Cl (2 мл) и экстрагировали EtOAc (7 мл × 3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали флэш-хроматографией на колонке (SiO2, гексан/Et2O = 10:1), получая 5,6 мг неразделимой смеси E/Z-олефинов (E/Z = 9:1). Смесь очищали препаративной ТСХ (гексан/Et2O = 4:1), получая чистый 39a (5,0 мг, 6,96 мкмоль, 76%) в виде бесцветного масла; [α]D 25 -41,5 (с 0,715, СНСl3); ИК (пленка) ν 2955, 2884, 1737, 1690, 1467, 1378, 1249, 1179, 1102, 1014, 979, 879, 826, 773 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,08 (3H, c), 0,12 (3H, c), 0,57 (6H, кв, J=7,8 Гц), 0,89 (9H, т, J=8,0 Гц), 0,93 (9H, c), 1,04 (3H, c), 1,06 (3H, д, J=7,1 Гц), 1,12 (3H, c), 1,17 (3H, д, J=7,1 Гц), 1,68 (3H, c), 2,15 (3H, д, J=0,8 Гц), 2,14-2,27 (2H, м), 2,45 (1H, дд, J=14,0, 4,8 Гц), 2,50 (1H, дд, J=14,9, 3,2 Гц), 2,64-2,74 (2H, м), 2,72 (3H, c), 3,02 (1H, квинт, J=7,0 Гц), 3,10 (1H, дд, J=14,4, 7,3 Гц), 3,96 (1H, д, J=8,7 Гц), 4,43 (1H, дд, J=8,3, 2,9 Гц), 5,22 (1H, дд, J=9,8, 5,7 Гц), 5,33-5,42 (2H, м), 5,69 (1H, дд, J=15,8, 8,2 Гц), 6,57 (1H, c), 6,96 (1H, c); 13С ЯМР (100 МГц, СDCl3) δ -3,3, -3,2, 5,6 (3C), 7,1 (3C), 15,0, 17,2, 18,8, 19,4, 21,4, 21,7, 23,8, 24,3, 26,5 (3C), 33,2, 35,6, 41,3, 41,8, 48,2, 54,0, 74,4, 77,4, 79,3, 116,4, 120,5, 121,0, 129,3, 132,1, 137,8, 138,0, 152,7, 164,8, 170,7, 216,8; МС-НР (ESI) вычислено для С39Н68NО5SSi2 [M+H+] 718,4, найдено 718,3.
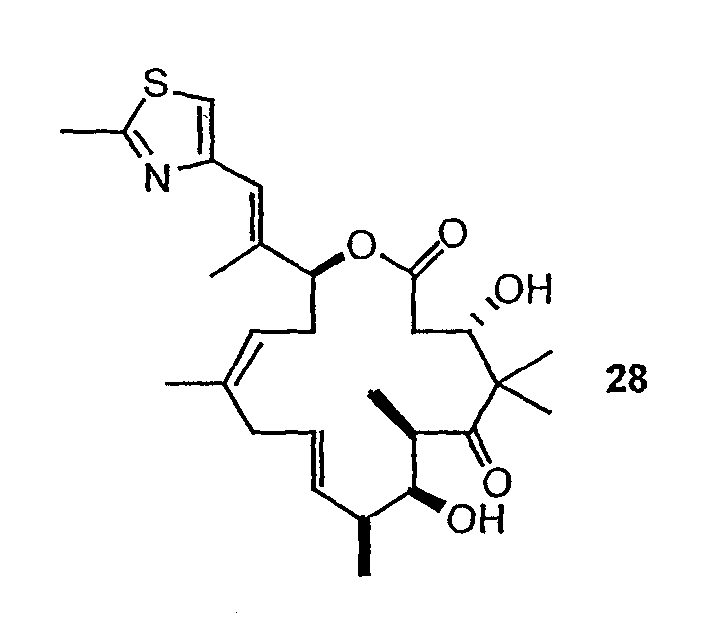
Соединение 28 (Epo 3): К раствору 39a (298,8 мг, 0,416 ммоль) в ТГФ (6,5 мл) добавляли HF·пиридин (3,2 мл) при 0°C и смесь перемешивали при КТ в течение 3 час. Реакцию гасили добавлением по каплям TMSOMe (30 мл) при 0°C. После концентрирования и сушки в условиях высокого вакуума остаток очищали флэш-хроматографией на колонке (SiO2, гексан/EtOAc = 1:1), получая 28 (196,6 мг, 0,402 ммоль, 97%) в виде белого твердого вещества; [α]D 25 -96,6 (с 0,235, СНСl3); ИК (пленка) ν 3502, 2970, 2927, 1733, 1685, 1506, 1456, 1375, 1251, 1152, 1040, 977 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 1,06 (3H, c), 1,11 (3H, д, J=7,0 Гц), 1,22 (3H, д, J=6,8 Гц), 1,28 (3H, c), 1,72 (3H, c), 2,10 (3H, c), 2,31-2,40 (2H, м), 2,43 (1H, дд, J=16,0, 3,7 Гц), 2,49 (1H, дд, J=16,0, 9,2 Гц), 2,55-2,68 (2H, м), 2,71 (3H, c), 2,98 (1H, дд, J=14,4, 6,4 Гц), 3,16 (1H, квинт, J=6,2 Гц), 3,76 (1H, дд, J=5,9, 3,2 Гц), 4,30 (1H, дд, J=9,2, 3,7 Гц), 5,18 (1H, ушир.т, J=7,3 Гц), 5,32 (1H, дд, J=8,4, 2,5 Гц), 5,63 (1H, дд, J=15,7, 6,4 Гц), 5,60 (1H, ддд, J=15,7, 6,9, 5,1 Гц), 6,60 (1H, c), 6,98 (1H, c); 13С ЯМР (100 МГц, СDCl3) δ 15,1, 16,0, 17,7, 19,2, 19,5, 22,5, 23,6, 32,0, 35,0, 39,6, 40,3, 44,8, 53,3, 71,8, 75,6, 78,3, 116,1, 119,6, 120,5, 129,9, 131,3, 137,5, 138,2, 152,2, 165,0, 170,7, 218,8; МС-НР (ESI) вычислено для С27Н40NО5S [M+H+] 490,3, найдено 490,2.
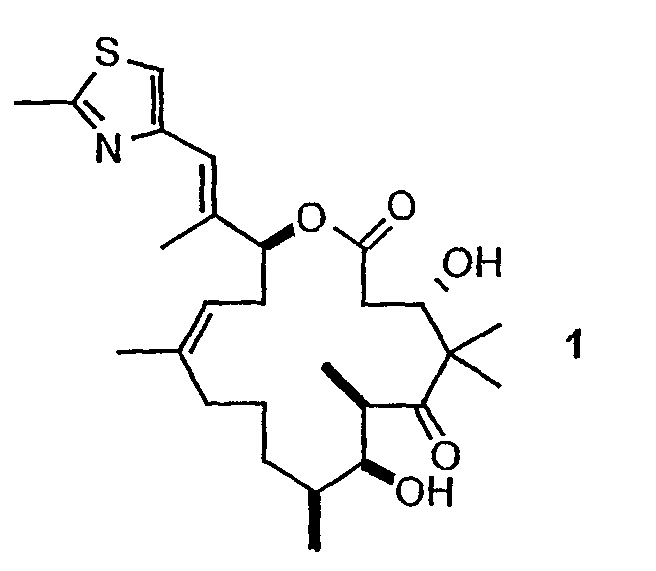
dEpoB (1, Epo 1): К раствору 28 (1,2 мг, 2,5 мкмоль) и ТрисNHNH2 (29,3 мг, 98 мкмоль) в ClCH2CH2Cl (0,7 мл) при 50°C добавляли Et3N (13,7 мкл, 98 мкмоль). Реакцию контролировали с помощью ВЭТСХ (гексан/EtOAc/CH2Cl2 = 1/1/2). После перемешивания в течение 7 час смесь охлаждали до КТ, разбавляли EtOAc и фильтровали через подушку силикагеля, которую промывали EtOAc. После концентрирования остаток очищали препаративной ТСХ (гексан/EtOAc/CH2Cl2 = 1/1/2), получая 1 (1,1 мг, 2,2 мкмоль, 91%) в виде белого твердого вещества. Спектральные данные для 1 были идентичны данным, описанным для dEpoB.
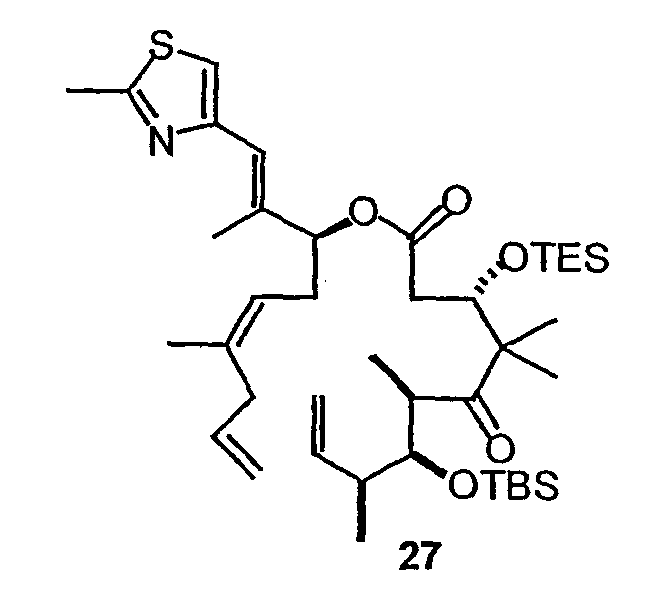
Соединение 27: Перед реакцией кислоту 25 и спирт 24 подвергали азеотропной перегонке с безводным бензолом (5 мл × 2) и сушили в условиях высокого вакуума. К раствору спирта 24 (639 мг, 2,63 ммоль) в CH2Cl2 (13 мл) добавляли EDCI (576 мг, 3,09 ммоль) и DMAP (366 мг, 3,09 ммоль) при 0°C. К смеси по каплям добавляли раствор кислоты 25 (1,11 г, 1,88 ммоль) в CH2Cl2 (промывка 5 мл + 2 мл) в течение 16 мин при 0°C. После перемешивания при 0°C в течение 1,5 час смесь перемешивали при КТ в течение 3,5 час. После концентрирования остаток очищали флэш-хроматографией на колонке (SiO2, гексан/EtOAc = от 30:1 до 20:1), получая 27 (1,20 г, 1,61 ммоль, 86%, исходя из сложного трет-бутилового эфира) в виде бесцветного масла; [α]D 24 -25,1 (с 1,30, СНСl3); ИК (пленка) ν 2955, 2925, 2872, 1732, 1696, 1461, 1378, 1290, 1243, 1173, 1091, 985, 873, 773 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,06 (3H, c), 0,06 (3H, c), 0,58-0,66 (6H, м), 0,92 (9Н, c), 0,95 (9H, т, J=8,0 Гц), 1,02 (3H, д, J=6,5 Гц), 1,03 (3H, д, J=6,5 Гц), 1,07 (3H, c), 1,21 (3H, c), 1,67 (3H, c), 2,07 (3H, c), 2,05-2,12 (1H, м), 2,30 (1H, дд, J=16,9, 7,5 Гц), 2,39 (1H, дт, J=14,8, 6,7 Гц), 2,49 (1H, дд, J=17,0, 3,0 Гц), 2,50 (1H, дт, J=14,8,6,7 Гц), 2,70 (3H, c), 2,74-2,30 (2H, м), 3,07 (1H, дд, J=7,0 Гц), 3,83 (1H, дд, J=7,1, 2,0 Гц), 4,35 (1H, дд, J=7,4, 2,8 Гц), 4,98-5,07 (4H, м), 5,16 (1H, ушир.т, J=7,0 Гц), 5,23 (1H, т, J=6,9 Гц), 5,74 (1H, ддт, J=16,7, 10,2, 6,5 Гц), 5,91 (1H, ддд, J=17,8, 10,5, 7,8 Гц), 6,50 (1H, c), 6,95 (1H, c); 13С ЯМР (100 МГц, СDCl3) δ -3,7, -3,3, 5,3 (3C), 7,2 (3C), 14,8, 15,2, 18,7, 18,9, 19,4, 20,3, 23,6, 23,7, 26,4 (3C), 31,7, 36,7, 40,1, 43,8,46,4, 53,3, 74,2, 76,5, 79,6, 115,5, 115,6, 116,5, 120,5, 121,3, 135,8, 136,1, 137,4, 140,2, 152,9, 164,7, 171,5, 218,4; МС-НР (ESI) вычислено для С41Н71NО5SSi2 [M+Na+] 768,5, найдено 768,5.
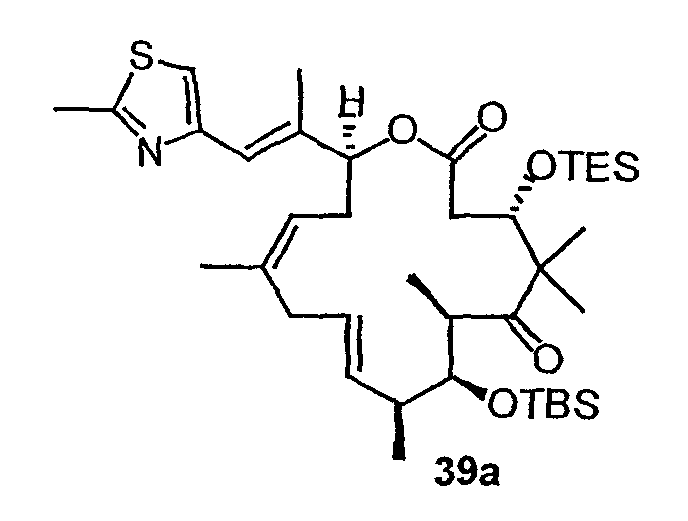
Соединение 39a: Раствор 27 (26,9 мг, 36,1 мкмоль) в толуоле (70 мл) кипятили с обратным холодильником и обрабатывали раствором катализатора Груббса (3,1 мг, 3,61 мкмоль) в толуоле (2 мл). Смесь перемешивали в течение 25 мин, охлаждали до 0°C и фильтровали через подушку силикагеля, которую промывали смесью гексан/EtOAc = 2/1. Объединенные фильтраты концентрировали и очищали флэш-хроматографией на колонке (SiO2, гексан/Et2O = от 40:1 до 5:1), получая 39a (9,9 мг, 13,8 мкмоль, 38%) в виде бесцветного масла; [α]D 25 -41,5 (с 0,715, СНСl3); ИК (пленка) ν 2955, 2884, 1737, 1690, 1467, 1378, 1249, 1179, 1102, 1014, 979, 879, 826, 773 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,08 (3H, c), 0,12 (3H, c), 0,57 (6H, кв, J=7,8 Гц), 0,89 (9H, т, J=8,0 Гц), 0,93 (9H, c), 1,04 (3H, c), 1,06 (3H, д, J=7,1 Гц), 1,12 (3H, c), 1,17 (3H, д, J=7,1 Гц), 1,68 (3H, c), 2,15 (3H, д, J=0,8 Гц), 2,14-2,27 (2H, м), 2,45 (1H, дд, J=14,0, 4,8 Гц), 2,50 (1H, дд, J=14,9, 3,2 Гц), 2,64-2,74 (2H, м), 2,72 (3H, c), 3,02 (1H, квинт, J=7,0 Гц), 3,10 (1H, дд, J=14,4, 7,3 Гц), 3,96 (1H, д, J=8,7 Гц), 4,43 (1H, дд, J=8,3, 2,9 Гц), 5,22 (1H, дд, J=9,8, 5,7 Гц), 5,33-5,42 (2H, м), 5,69 (1H, дд, J=15,8, 8,2 Гц), 6,57 (1H, c), 6,96 (1H, c); 13С ЯМР (100 МГц, СDCl3) δ -3,3, -3,2, 5,6 (3C), 7,1 (3C), 15,0, 17,2, 18,8, 19,4, 21,4, 21,7, 23,8, 24,3, 26,5 (3C), 33,2, 35,6, 41,3, 41,8, 48,2, 54,0, 74,4, 77,4, 79,3, 116,4, 120,5, 121,0, 129,3, 132,1, 137,8, 138,0, 152,7, 164,8, 170,7, 216,8; МС-НР (ESI) вычислено для С39Н68NО5SSi2 [M+H+] 718,4, найдено 718,3.
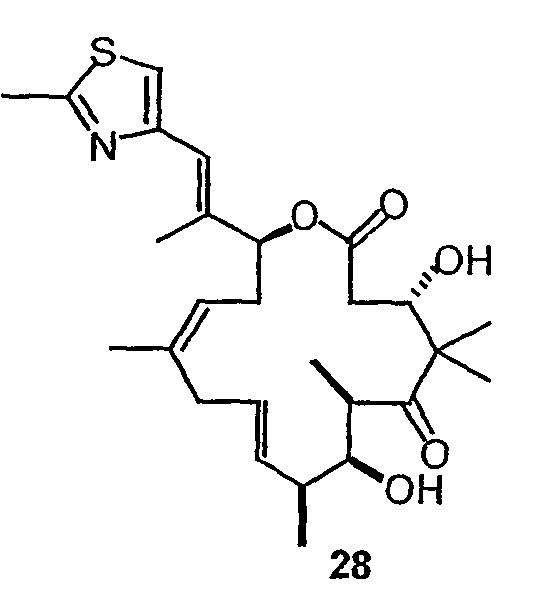
Соединение 28: К раствору 39a (298,8 мг, 0,416 ммоль) в ТГФ (6,5 мл) добавляли HF-пиридин (3,2 мл) при 0°C и смесь перемешивали при КТ в течение 3 час. Реакцию гасили, добавляя по каплям TMSOMe (30 мл) при 0°C, и смесь перемешивали при КТ в течение 3 час. После концентрирования и сушки в условиях высокого вакуума остаток очищали флэш-хроматографией на колонке (SiO2, гексан/EtOAc = 1:1), получая 28 (196,6 мг, 0,402 ммоль, 97%) в виде белого твердого вещества; [α]D 25 -96,6 (c 0,235, CHCl3); ИК (пленка) ν 3502, 2970, 2927, 1733, 1685, 1506, 1456, 1375, 1251, 1152, 1040, 977 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 1,06 (3H, c), 1,11 (3H, д, J=7,0 Гц), 1,22 (3H, д, J=6,8 Гц), 1,28 (3H, c), 1,72 (3H, c), 2,10 (3H, c), 2,31-2,40 (2H, м), 2,43 (1H, дд, J=16,0, 3,7 Гц), 2,49 (1H, дд, J=16,0, 9,2 Гц), 2,55-2,68 (2H, м), 2,71 (3H, c), 2,98 (1H, дд, J=14,4, 6,4 Гц), 3,16 (1H, квинт, J=6,2 Гц), 3,76 (1H, дд, J=5,9, 3,2 Гц), 4,30 (1H, дд, J=9,2, 3,7 Гц), 5,18 (1H, ушир.т, J=7,3 Гц), 5,32 (1H, дд, J=8,4, 2,5 Гц), 5,63 (1H, дд, J=15,7, 6,4 Гц), 5,60 (1H, ддд, J=15,7, 6,9, 5,1 Гц), 6,60 (1H, c), 6,98 (1H, c); 13С ЯМР (100 МГц, СDCl3) δ 15,1, 16,0, 17,7, 19,2, 19,5, 22,5, 23,6, 32,0, 35,0, 39,6, 40,3, 44,8, 53,3, 71,8, 75,6, 78,3, 116,1, 119,6, 120,5, 129,9, 131,3, 137,5, 138,2, 152,2, 165,0, 170,7, 218,8; МС-НР (ESI) вычислено для С27Н40NО5S [M+H+] 490,3, найдено 490,2.

Соединение 26: Перед реакцией кислоту 25 и спирт 21 подвергали азеотропной перегонке с безводным бензолом (5 мл × 2) и сушили в условиях высокого вакуума. К раствору спирта 21 (240 мг, 0,756 ммоль) в CH2Cl2 (5 мл) добавляли EDCI (192,7 мг, 1,01 ммоль) и DMAP (122,8 мг, 1,01 ммоль) при 0°C. К смеси по каплям добавляли раствор кислоты 25 (314,6 мг, 0,628 ммоль) в CH2Cl2 (промывка 2 мл + 1 мл) в течение 15 мин при 0°C. После перемешивания при 0°C в течение 2 час смесь перемешивали при КТ в течение 2 час. После концентрирования остаток очищали флэш-хроматографией на колонке (SiO2, гексан/EtOAc = от 20:1 до 15:1), получая 26 (340,1 мг, 0,425 ммоль, 68% на основе кислоты) в виде бесцветного масла; [α]D 24 -27,5 (с 0,28, СНСl3); ИК (пленка) ν 2956, 2878, 1740, 1692, 1472, 1378, 1317, 1253, 1174, 1118, 988, 915, 872, 837, 775 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,06 (6H, c), 0,57-0,65 (6H, м), 0,92 (9H, c), 0,94 (9H, т, J=7,9 Гц), 1,02 (3H, д, J=6,9 Гц), 1,03 (3H, д, J=6,8 Гц), 1,07 (3H, c), 1,22 (3H, c), 2,07-2,10 (1H, м), 2,09 (3H, c), 2,31 (1H, дд, J=16,9, 7,3 Гц), 2,51 (1H, дд, J=16,8, 3,0 Гц) 2,49-2,65 (2H, м), 2,71 (3H, c), 2,96-2,99 (2H, м), 3,06 (1H, квинт, J=7,1 Гц), 3,83 (1H, дд, J=7,3, 2,1 Гц), 4,35 (1H, дд, J=7,2, 3,0 Гц), 4,98-5,12 (4H, м), 5,30 (1H, т, J=6,7 Гц), 5,76 (1H, ддт, J=16,7,10,2, 6,2 Гц), 5,92 (1H, ддд, J=17,8, 9,9, 7,8 Гц), 6,19 (1H, т, J=7,0 Гц), 6,51 (1H, c), 6,97 (1H, c); МС-НР (ESI) вычислено для С41Н68F3NO5SSi2Na [M+Na+] 822,4, найдено 822,4.
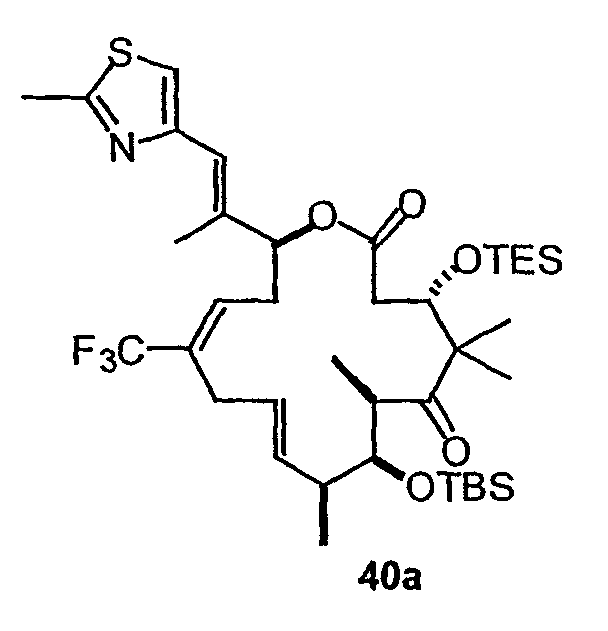
Соединение 40a (посредством RCM 26): Раствор 26 (57,6 мг, 72,0 мкмоль) в толуоле (142 мл) кипятили с обратным холодильником и обрабатывали раствором катализатора Груббса (6,1 мг, 7,20 мкмоль) в толуоле (2 мл). Смесь перемешивали в течение 28 мин, охлаждали до 0°C и фильтровали через подушку силикагеля, которую промывали смесью гексан/EtOAc = 2/1 (300 мл). Объединенные фильтраты концентрировали и очищали флэш-хроматографией на колонке (SiO2, гексан/Et2O = от 40:1 до 15:2), получая 40a (12,0 мг, 15,5 мкмоль, 22%) в виде бесцветного масла; ИК (пленка) ν 2955, 2884, 1743, 1690, 1472, 1320, 1173, 1114,1038, 1008, 873, 832, 773 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,09 (3H, c), 0,12 (3H, c), 0,55 (6H, кв, J=7,7 Гц), 0,88 (9H, т, J=8,0 Гц), 0,96 (9H, c), 1,01 (3H, c), 1,06 (3H, д, J=7,1 Гц), 1,12 (3H, c), 1,20 (3H, д, J=7,1 Гц), 2,07-2,17 (1H, м), 2,19 (3H, c), 2,38 (1H, дд, J=14,3, 3,5 Гц), 2,39-2,49 (1H, м), 2,50 (1H, дд, J=14,3, 7,3 Гц), 2,73 (3H, c), 2,77-2,91 (2H, м), 2,96-3,09 (2H, м), 3,98 (1H, дд, J=8,9 Гц), 4,54 (1H, дд, J=7,3, 3,4 Гц), 5,28-5,38 (1H, м), 5,63 (1H, дд, J=9,6, 2,3 Гц), 5,77 (1H, дд, J=15,9, 8,5 Гц), 6,21-6,28 (1H, м), 6,60 (1H, c), 6,99 (1H, c); МС-НР (ESI) вычислено для С39Н65F3NO5SSi2 [M+H+] 772,4, найдено 772,4.

Соединение 29: К раствору 40a (1,78 г, 2,31 ммоль) в ТГФ (25 мл) медленно добавляли HF·пиридин (12,5 мл) при 0°C и смесь перемешивали при КТ в течение 4 час. Реакцию гасили, добавляя по каплям TMSOMe (80 мл) в течение 10 мин при 0°C. Смесь энергично перемешивали при КТ в течение 2,5 час. После концентрирования и сушки в условиях высокого вакуума в течение 2 час остаток очищали флэш-хроматографией на колонке (SiO2 ~50 г, гексан/EtOAc = 1:1), получая 29 (1,20 г, 2,21 ммоль, 96%) в виде бесцветного порошка; [α]D 25 -54,6 (с 0,28, СНСl3); ИК (пленка) ν 3478, 2974, 2929, 1736, 1689, 1449, 1381, 1318, 1247, 1169, 1113, 1039, 983, 867, 736 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 1,05 (3H, c), 1,12 (3H, д, J=7,0 Гц), 1,23 (3H, д, J=6,8 Гц), 1,37 (3H, c), 2,04 (1H, ушир.д, J=3,8 Гц, -ОН), 2,12 (3H, c), 2,25-2,33 (1H, м), 2,38 (1H, дд, J=15,3, 3,0 Гц), 2,48 (1H, дд, J=15,4, 9,8 Гц), 2,54-2,61 (1H, м), 2,66-2,76 (1H, м), 2,71 (3H, c), 2,96 (1H, дд, J=16,5,4,5 Гц), 3,02 (1H, дд, J=16,3, 6,5 Гц), 3,11 (1H, квинт, J=6,7 Гц), 3,19 (1H, ушир.c, =OH), 3,74 (1H, ушир.c), 4,35 (1H, ушир.д, J=9,5 Гц), 5,42 (1H, дд, J=6,2, 4,1 Гц), 5,60 (1H, ддд, J=15,8, 5,6, 4,5 Гц), 5,66 (1H, дд, J=15,8, 5,8 Гц), 6,24 (1H, т, J=7,2 Гц), 6,64 (1H, c), 7,00 (1H, c); 13C ЯМР (100 МГц, (CDCl3) δ 15,1,16,1, 17,7, 18,5, 19,3, 22,5, 28,8, 31,1, 39,6, 39,7, 45,0, 53,7, 71,4, 75,3, 76,8, 116,7, 120,2, 124,3 [кв, 1J(C,F) = 273,4 Гц], 127,9, 130,2 [кв, 3J(C,F) = 6,0 Гц], 130,6 [кв, 2J(CF) = 28,4 Гц], 132,5, 136,7, 152,0, 165,4, 170,2, 218,4; МС-НР (ESI) вычислено для С27Н37F3NO5S [M+H+] 544,2, найдено 544,1.
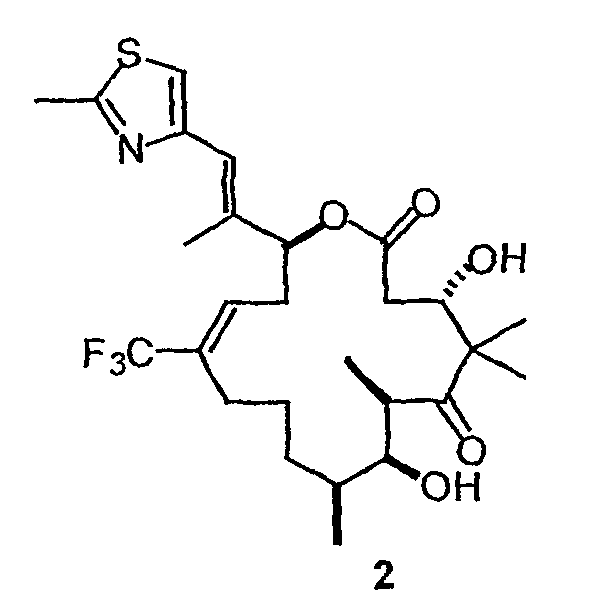
Соединение 2: К раствору 29 (1,22 мг, 2,24 мкмоль) и трисNHNH2 (26,7 мг, 89,6 мкмоль) в ClCH2CH2Cl (1 мл) при 50°C добавляли Et3N (12,5 мкл, 89,6 мкмоль). Реакцию контролировали с помощью ВЭТСХ (гексан/EtOAc/CH2Cl2 = 1/1/2). После перемешивания в течение 6,5 час к смеси дополнительно добавляли трисNHNH2 (26,7 мг, 89,6 мкмоль) и Et3N (12,5 мкл, 89,6 мкмоль). После перемешивания в течение 14 час смесь охлаждали до КТ, разбавляли EtOAc и фильтровали через подушку силикагеля, которую промывали EtOAc. После концентрирования остаток очищали препаративной ТСХ (гексан/EtOAc/CH2Cl2 =1/1/2), получая 2 (1,16 мг, 2,13 мкмоль, 94%) в виде белого твердого вещества; 1H ЯМР (400 МГц, CDCl3) δ 1,03 (3H, д, J=7,0 Гц), 1,08 (3H, c), 1,19 (3H, д, J=6,8 Гц), 1,25-1,35 (2H, м), 1,37 (3H, c), 1,42-1,55 (2H, м), 1,65-1,82 (2H, м), 2,10 (3H, д, J=0,8 Гц), 2,21-2,47 (2H, м), 2,27 (1H, дд, J=14,2, 2,6 Гц), 2,48 (1H, дд, J=14,3, 10,8 Гц), 2,70 (3H, c), 2,70-2,28 (1H, м), 3,02 (1H, д, J=2,0 Гц, -OH), 3,19 (1H, кв.д, J=6,9, 2,2 Гц), 3,65 (1H, д, J=6,2 Гц, -OH), 3,69-3,72 (1H, м), 4,34 (1H, ддд, J=10,8, 6,2, 2,6 Гц), 5,28 (1H, дд, J=10,2, 2,2 Гц), 6,12 (1H, дд, J=10,2, 5,2 Гц), 6,61 (1H, c), 6,98 (1H, c); МС-НР (ESI) вычислено для С27Н39F3NO5S [M+H+] 546,3, найдено 546,2.
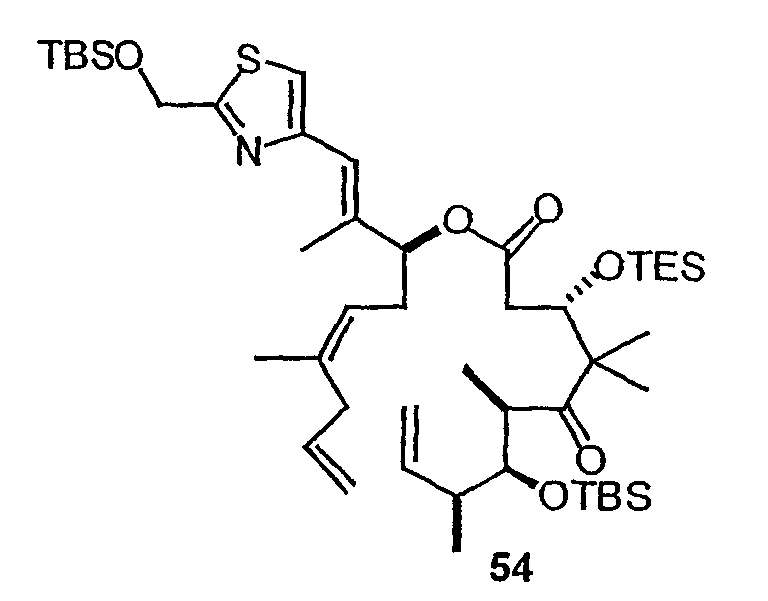
Соединение 54: Перед реакцией кислоту 25 и спирт 53 подвергали азеотропной перегонке с безводным бензолом (3 мл × 2) и сушили в условиях высокого вакуума. К раствору спирта 53 (68,0 мг, 0,173 ммоль) в CH2Cl2 (1,3 мл) добавляли EDCI (37,8 мг, 0,197 ммоль) и DMAP (24,1 мг, 0,197 ммоль) при 0°C. К смеси по каплям добавляли раствор кислоты 25 (72,6 мг, 0,123 ммоль) в CH2Cl2 (0,7 мл) в течение 5 мин при 0°C. После перемешивания при 0°C в течение 1 час смесь перемешивали при КТ в течение 2,5 час. После концентрирования остаток очищали флэш-хроматографией на колонке (SiO2, гексан/EtOAc = 30:1), получая 54 (99,5 мг, 0,114 ммоль, 92%, исходя из сложного трет-бутилового эфира) в виде бесцветного масла; [α]D 25 -23,4 (с 0,56, СНСl3); ИК (пленка) ν 2955, 2931, 2880, 1735, 1696, 1506, 1472, 1386, 1362, 1294, 1254, 1174, 1104, 988, 878, 776, 742 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,06 (3H, c), 0,06 (3H, c), 0,14 (6H, c), 0,63 (6H, кв, J=8,0 Гц), 0,92 (9H, c), 0,94 (9H, т, J=8,0 Гц), 0,97 (9H, c), 1,02 (3H, д, J=6,6 Гц), 1,05 (3H, д, J=6,5 Гц), 1,07 (3H, c), 1,21 (3H, c), 1,67 (3H, c), 2,06 (3H, д, J=0,8 Гц), 2,05-2,14 (1H, м), 2,30 (1H, дд, J=16,9, 7,5 Гц), 2,33-2,53 (2H, м), 2,50 (1H, дд, J=16,9, 2,7 Гц), 2,76-2,80 (2H, м), 3,07 (1H, квинт, J=7,0 Гц), 3,83 (1H, дд, J=7,0, 2,2 Гц), 4,35 (1H, дд, J=7,4, 2,8 Гц), 4,97 (2H, c), 4,97-5,07 (4H, м), 5,16 (1H, т, J=7,2 Гц), 5,24 (1H, т, J=6,9 Гц), 5,74 (1H, ддт, J=16,6, 10,0, 6,5 Гц), 5,91 (1H, ддд, J=17,6, 9,9, 7,7 Гц), 6,50 (1H, c), 7,06 (1H, c); 13C ЯМР (100 МГц, (CDCl3) δ -5,2 (2C), -3,7, -3,3, 5,3 (3C), 7,2 (3C), 14,7, 15,2, 18,5, 18,7, 18,9, 20,3, 23,6, 23,7, 26,0 (3C), 26,4 (3C), 31,7, 36,7, 40,1, 43,8, 46,4, 53,3, 63,4, 74,2, 76,5, 79,6, 115,5, 115,6, 116,6, 120,5, 121,3, 135,8, 136,1, 137,4, 140,1, 153,0, 171,5, 172,2, 218,4; МС-НР (ESI) вычислено для С47Н86NO6SSi3 [M+H+] 876,6, найдено 876,5.

Соединение 55: Раствор 54 (69,7 мг, 79,5 мкмоль) в толуоле (158 мл) кипятили с обратным холодильником и обрабатывали раствором катализатора Груббса (6,7 мг, 7,95 мкмоль) в толуоле (2 мл). Смесь перемешивали в течение 11 мин, охлаждали до 0°C и фильтровали через подушку силикагеля, которую промывали смесью гексан/EtOAc = 3/1 (280 мл). Объединенные фильтраты концентрировали и очищали флэш-хроматографией на колонке (SiO2, гексан/Et2O = от 20:1 до 15:1), получая 55 (18,4 мг, 21,7 мкмоль, 27%) в виде бесцветного масла; [α]D 24 -40,4 (с 0,26, СНСl3); ИК (пленка) ν 2955, 2930, 2879, 1740, 1694, 1472, 1387, 1362, 1253, 1200, 1107, 1007, 838, 776, 742 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 0,08 (3H, c), 0,12 (3H, c), 0,15 (6H, c), 0,57 (6H, кв, J=7,9 Гц), 0,88 (9H, т, J=8,0 Гц), 0,95 (9H, c), 0,97 (9H, c), 1,04 (3H, c), 1,06 (3H, д, J=7,1 Гц), 1,12 (3H, c), 1,17 (3H, д, J=7,0 Гц), 1,69 (3H, c), 2,06-2,30 (2H, м), 2,14 (3H, c), 2,45 (1H, дд, J=15,6, 3,6 Гц), 2,50 (1H, дд, J=14,9, 3,1 Гц), 2,63-2,75 (2H, м), 2,97-3,06 (1H, м), 3,10 (1H, дд, J=14,6, 7,7 Гц), 3,97 (1H, д, J=8,5 Гц), 4,44 (1H, дд, J=8,4, 2,9 Гц), 4,97 (2H, c), 5,22 (1H, дд, J=8,7, 5,2 Гц), 5,33-5,44 (2H, м), 5,70 (1H, дд, J=15,6, 8,1 Гц), 6,57 (1H, c), 7,07 (1H, c); МС-НР (ESI) вычислено для С45Н82NO6SSi3 [M+H+] 848,5, найдено 848,5.
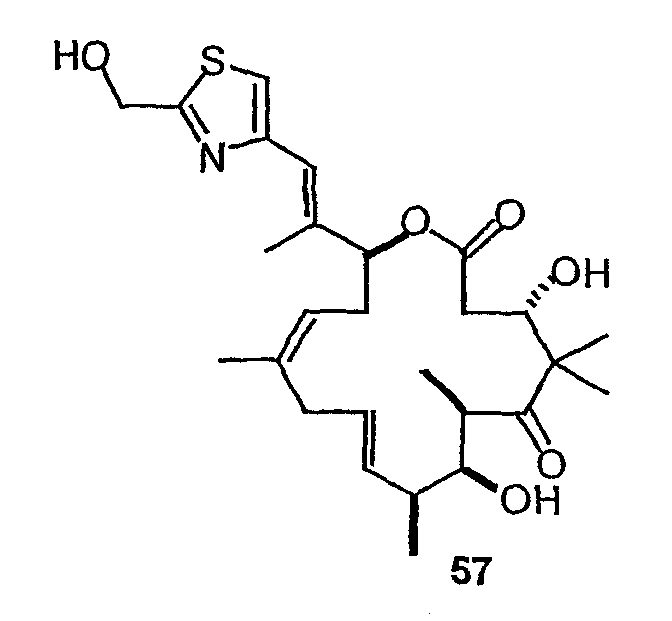
Соединение 57: К раствору 55 (61,8 мг, 72,8 мкмоль) в ТГФ (2 мл) добавляли HF·пиридин (1 мл) при 0°C и смесь перемешивали при КТ в течение 3,2 час. Реакцию гасили, добавляя по каплям TMSOMe (15 мл) при 0°C. Смесь перемешивали при КТ в течение 2 час. После концентрирования и сушки в условиях высокого вакуума остаток очищали флэш-хроматографией на колонке (SiO2, гексан/EtOAc = 1:3), получая 57 (32,4 мг, 64,1 мкмоль, 88%) в виде белого твердого вещества; [α]D 25 -108,4 (с 0,285, СНСl3); ИК (пленка) ν 3422, 2968, 2919, 2729, 1689, 1449, 1377, 1252, 1152, 1064, 978 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 1,05 (3H, c), 1,12 (3H, д, J=6,9 Гц), 1,22 (3H, д, J=6,8 Гц), 1,32 (3H, c), 1,72 (3H, c), 2,08 (3H, c), 2,31-2,40 (3H, м), 2,43 (1H, дд, J=15,5, 3,5 Гц), 2,49 (1H, дд, J=15,5, 9,5 Гц), 2,55-2,67 (2H, м), 2,95 (1H, дд, J=14,6, 6,3 Гц), 3,13 (1H, квинт, J=6,6 Гц), 3,34 (1H, ушир.c, -ОН), 3,75 (1H, дд, J=6,6, 2,4 Гц), 4,06 (1H, ушир.c, -ОН), 4,33 (1H, дд, J=9,4, 3,0 Гц), 4,92 (2H, c), 5,18 (1H, т, J=6,9 Гц), 5,33 (1H, дд, J=8,0, 2,5 Гц), 5,52 (1H, дд, J=15,8, 6,4 Гц), 5,59 (1H, ддд, J=15,8, 6,6, 5,0 Гц), 6,63 (1H, c), 7,13 (1H, c); 13C ЯМР (100 МГц, CDCl3) δ 15,3, 16,3, 17,8, 19,2, 22,8, 23,7, 31,9, 35,1, 39,7, 40,2, 45,0, 53,4, 61,8, 71,7, 75,8, 78,1, 116,7, 119,0, 120,5, 130,0, 131,2, 137,6, 138,9, 152,5, 170,0, 170,7, 218,7; МС-НР (ESI) вычислено для С27Н39NO6SNa [M+Na+] 528,2, найдено 528,0.

Соединение 46: Перед реакцией неочищенную кислоту 25 (4,65 г, 7,27 ммоль) и спирт 44 (2,18 г, 9,84 ммоль) подвергали азеотропной перегонке с безводным бензолом и сушили в условиях высокого вакуума. К раствору спирта 44 (2,18 г, 9,84 ммоль) в CH2Cl2 (65 мл) добавляли EDCI (2,09 г, 10,9 ммоль) и DMAP (1,33 г, 10,9 ммоль) при 0°C. К смеси по каплям добавляли раствор неочищенной кислоты 25 (4,65 г, 7,27 ммоль) в CH2Cl2 (промывка 20 мл + 5 мл) в течение 20 мин при 0°C. После перемешивания при 0°C в течение 40 мин смесь перемешивали при КТ в течение 4 час. После концентрирования остаток очищали флэш-хроматографией на колонке (SiO2 ~160 г; гексан/EtOAc = 20:1), получая 46 (4,85 г, 6,87 ммоль, 94% исходя из сложного трет-бутилового эфира) в виде бесцветного масла; 1H ЯМР (400 МГц, CDCl3) δ 0,08 (3H, c), 0,08 (3H, c), 0,60 (6H, кв, J=7,8 Гц), 0,93 (9H, c), 0,94 (9H, т, J=8,0 Гц), 1,04 (3H, д, J=7,0 Гц), 1,04 (3H, д, J=7,0 Гц), 1,11 (3H, c), 1,23 (3H, c), 2,05-2,14 (1H, м), 2,17 (3H, c), 2,40 (1H, дд, J=16,9, 7,0 Гц), 2,59 (1Н, дд, J=17,0, 3,6 Гц), 2,56-2,64 (2H, м), 2,90-3,01 (2H, м), 3,06 (1H, квинт, J=7,0 Гц), 3,85 (1H, дд, J=7,3, 2,0 Гц), 4,38 (1H, д, J=7,0, 3,4 Гц), 4,97-5,14 (5H, м), 5,75 (1H, ддт, J=16,0, 9,9, 6,2 Гц), 5,92 (1H, ддд, J=17,8, 10,5, 7,8 Гц), 6,21 (1H, тд, J=7,2, 1,5 Гц); МС-НР (ESI) вычислено для С36Н63F3O6Si2Na [M+Na+] 727,4, найдено 727,3.

Соединение 48: Раствор 46 (510,0 мг, 0,723 ммоль) в толуоле (500 мл) кипятили с обратным холодильником и обрабатывали раствором катализатора Груббса (92,1 мг, 0,109 ммоль) в толуоле (10 мл). Смесь перемешивали в течение 17 мин при кипячении с обратным холодильником и сразу же охлаждали до 0°C и хранили при 0°C перед фильтрованием через подушку силикагеля. Вторую партию диена (510,0 мг, 0,723 ммоль) обрабатывали идентично и одновременно. Объединенные реакционные смеси фильтровали через подушку силикагеля (100 г), которую промывали смесью гексан/EtOAc = 3/1 (1,4 л). Объединенные фильтраты концентрировали и очищали флэш-хроматографией на колонке (SiO2 ~65 г, гексан/Et2O = от 10:1 до 5:1), получая 48 (742,4 мг, 1,10 ммоль, 76%) в виде бесцветного масла; 1H ЯМР (400 МГц, CDCl3) δ 0,08 (3Н), 0,10 (3Н, c), 0,60 (6H, кв, J=7,8 Гц), 0,93 (9H, c), 0,94 (9H, т, J=7,8 Гц), 1,03 (3Н, д, J=7,1 Гц), 1,08 (3Н, c), 1,13 (3Н, д, J=7,0 Гц), 1,17 (3Н, c), 2,26 (3Н, c), 2,25-2,34 (1H, м), 2,64 (1H, дд, J=15,5, 5,0 Гц), 2,68-2,75 (2H, м), 2,76 (1H, дд, J=15,6, 6,4 Гц), 2,85 (1H, дд, J=15,6, 5,7 Гц), 2,97 (1H, кв.д, J=8,3, 6,9 Гц), 3,04 (1H, дд, J=15,6, 6,3 Гц), 3,92 (1H, дд, J=8,3, 1,2 Гц), 4,36 (1H, т, J=5,3 Гц), 5,30-5,39 (2H, м), 5,58 (1H, дд, J=15,5, 8,0 Гц), 6,13 (1H, ушир.т, J=7,2 Гц); 13C ЯМР (100 МГц, CDCl3) δ -3,6, -3,6, 5,4 (3С), 7,0 (3С), 17,5, 18,5, 19,0, 21,6, 23,5, 26,3 (3С), 26,5, 28,6, 29,1, 41,0, 42,3, 47,3, 54,1, 74,2, 76,8, 77,7, 124,0 [1J(C,F)=273,7 Гц], 126,0, 128,7 [3J(C,F)=5,9 Гц], 132,2 [2J(C,F)=28,1 Гц], 133,8, 170,5, 204,1, 216,1; МС-НР (ESI) вычислено для С34Н59F3O6Si2Na [M+Na+] 699,4, найдено 699,4.
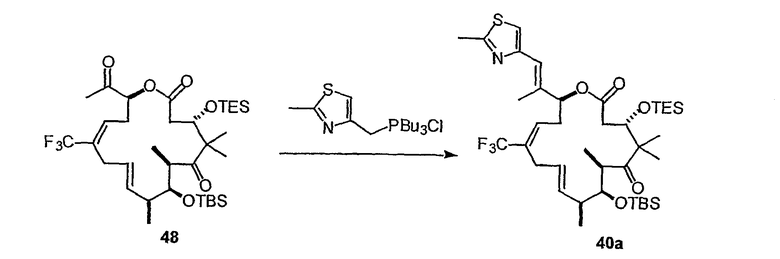
Соединение 40a (посредством реакции Виттига из кетона 48): Кетон 48 подвергали азеотропной перегонке с бензолом (5 мл × 2) и затем сушили в условиях высокого вакуума в течение 0,5 час. К раствору соли Виттига (907 мг, 2,59 ммоль) в ТГФ (19 мл) по каплям добавляли t-BuOK (2,4 мл 1,0 М раствора в ТГФ, 2,43 ммоль) в течение 5 мин при 0°C. Смесь перемешивали при 0°C в течение 0,5 час и затем охлаждали до
-78°C. К смеси по каплям добавляли раствор кетона 48 (1,10 г, 1,62 ммоль) в ТГФ (13 мл) в течение 10 мин и полученной в результате смеси давали возможность нагреться до -20°C в течение 2 час. Реакцию гасили насыщенным водным NH4Cl (15 мл) и экстрагировали EtOAc (50 мл × 3). Объединенные органические слои промывали насыщенным раствором соли (20 мл), сушили над Na2SO4 и концентрировали. Остаток очищали флэш-хроматографией (SiO2, гексан/Et2O = от 20:1 до 10:1), получая требуемый 16(E)-изомер 40a (940 мг, 1,22 ммоль, 75%) вместе с нежелательным 16(Z)-изомером 40b (140,9 мг, 0,182 ммоль, 11%), оба в виде бесцветных масел;
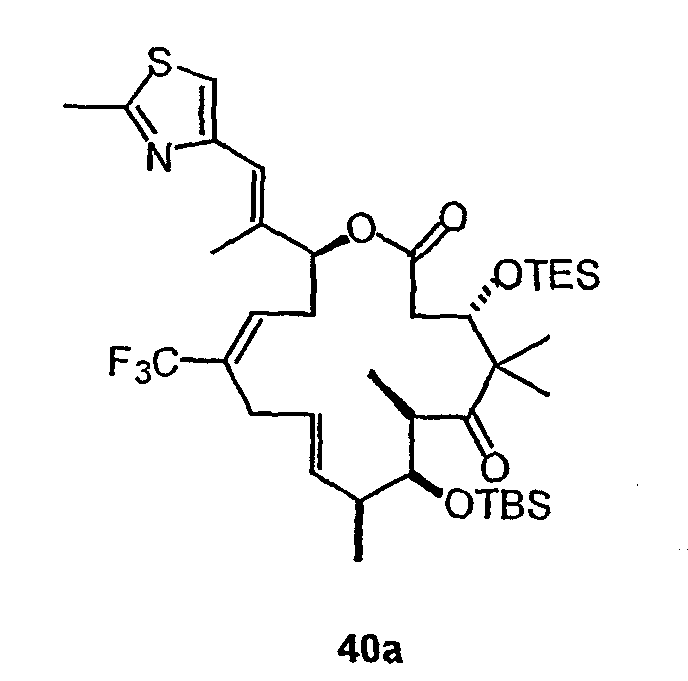
[α]D 26 -17,1 (с 0,14, СНСl3); 1H ЯМР (400 МГц, CDCl3) δ 0,09 (3Н, c), 0,12 (3H, c), 0,55 (6H, кв, J=7,7 Гц), 0,88 (9H, т, J=8,0 Гц), 0,96 (9H, c), 1,01 (3H, c), 1,06 (3Н, д, J=7,1 Гц), 1,12 (3Н, c), 1,20 (3Н, д, J=7,1 Гц), 2,07-2,17 (1H, м), 2,19 (3Н, c), 2,38 (1H, дд, J=14,3, 3,5 Гц), 2,39-2,49 (1H, м), 2,50 (1H, дд, J=14,3, 7,3 Гц), 2,73 (3Н, c), 2,77-2,91 (2H, м), 2,96-3,09 (2H, м), 3,98 (1H, дд, J=8,9 Гц), 4,54 (1H, дд, J=7,3, 3,4 Гц), 5,28-5,38 (1H, м), 5,63 (1H, дд, J=9,6, 2,3 Гц), 5,77 (1H, дд, J=15,9, 8,5 Гц), 6,21-6,28 (1H, м), 6,60 (1H, c), 6,99 (1H, c); 13C ЯМР (100 МГц, CDCl3) δ -3,4,-3,3, 5,5 (3C), 7,0 (3C), 14,6, 17,1, 18,7, 19,4, 19,9, 21,3, 24,8, 26,4 (3C), 29,6, 32,8, 42,0, 42,1, 48,2, 54,1, 73,4, 76,9, 77,8, 117,0, 121,6, 124,3 [1J(C,F) = 273,5 Гц], 127,2, 130,6 [2J(С,F) = 28,2 Гц], 130,8 [3J(C,F) = 6,1 Гц], 133,2, 136,5, 152,3, 165,0, 170,1, 217,1; МС-НР (ESI) вычислено для С39Н65F3NO5SSi2[M+H+] 772,4074, найдено 772,4102.
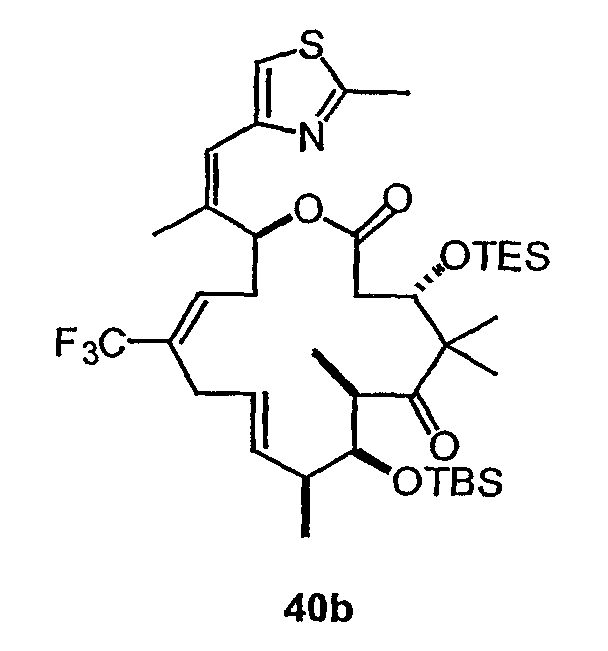
[α]D 25 62,7 (с 0,33, СНСl3); 1H ЯМР (400 МГц, CDCl3) δ 0,09 (3H, c), 0,13 (3H, c), 0,49 (6H, кв, J=7,8 Гц), 0,85 (9H, т, J=7,8 Гц), 0,97 (9H, c), 0,99 (3H, c), 1,06 (3H, д, J=7,1 Гц), 1,11 (3H, c), 1,20 (3H, д, J=7,1 Гц), 2,00 (3H, c), 2,03-2,13 (1H, м), 2,35 (1H, дд, J=14,3, 3,0 Гц), 2,46 (1H, дд, J=14,3, 7,8 Гц), 2,41-2,50 (1H, м), 2,73 (3H, c), 2,71-2,90 (2H, м) 2,98-3,12 (2H, м), 3,99 (1H, д, J=9,2 Гц), 4,56 (1H, дд, J=7,7, 2,8 Гц), 5,33 (1H, ддд, J=15,6, 8,9, 4,1 Гц), 5,82 (1H, дд, J=15,6, 8,4 Гц), 6,29 (1H, c), 6,33-6,40 (1H, м), 6,94 (1H, м), 7,09 (1H, ушир.д, J=8,4 Гц); 13C ЯМР (100 МГц, CDCl3) δ -3,2, -3,2, 5,5 (3С), 7,0 (3С), 17,2, 18,7, 19,3, 19,6, 20,0, 22,3, 24,9, 26,4 (3С), 29,7, 32,9, 41,9, 42,0, 48,6, 54,0, 72,2, 73,3, 77,0, 116,7, 120,7, 124,5 [1J(C,F) = 273,3 Гц], 127,9, 129,7 [2J (C,F) = 28,0 Гц], 131,9 [3J (C,F) = 6,1 Гц], 132,9, 136,6, 152,1, 165,4, 170,2, 217,4; МС-НР (ESI) вычислено для С39Н65F3NO5SSi2[M+H+] 772,4, найдено 772,4.

Соединение 58 (посредством реакции Виттига из кетона 48): Кетон 48 подвергали азеотропной перегонке с бензолом (5 мл × 2) и затем сушили в условиях высокого вакуума в течение 0,5 час. К раствору соли Виттига (1,19 г, 2,27 ммоль) в ТГФ (18 мл) по каплям добавляли t-BuOK (2,2 мл 1,0 М раствора в ТГФ, 2,20 ммоль) в течение 5 мин при 0°C. Смесь перемешивали при 0°C в течение 20 мин и затем охлаждали до
-78°C. К смесь по каплям добавляли раствор кетона (1,06 г, 1,51 ммоль) в ТГФ (промывка 10 мл + 2 мл) в течение 10 мин и полученной в результате смеси давали возможность нагреться до -20°C в течение 2 час. Реакцию гасили насыщенным водным NH4Cl (15 мл) и экстрагировали EtOAc (50 мл × 3). Объединенные органические слои промывали насыщенным раствором соли (20 мл), сушили над Na2SO4 и концентрировали. Остаток очищали флэш-хроматографией на колонке (SiO2 ~65 г, гексан/Et2O = от 30:1 до 20:1), получая требуемый 16(E)-изомер 58 (1,01 г, 1,11 ммоль, 74%) вместе с нежелательным 16(Z)-изомером 58a (154,5 мг, 0,182 ммоль, 11%), оба в виде бесцветных масел;
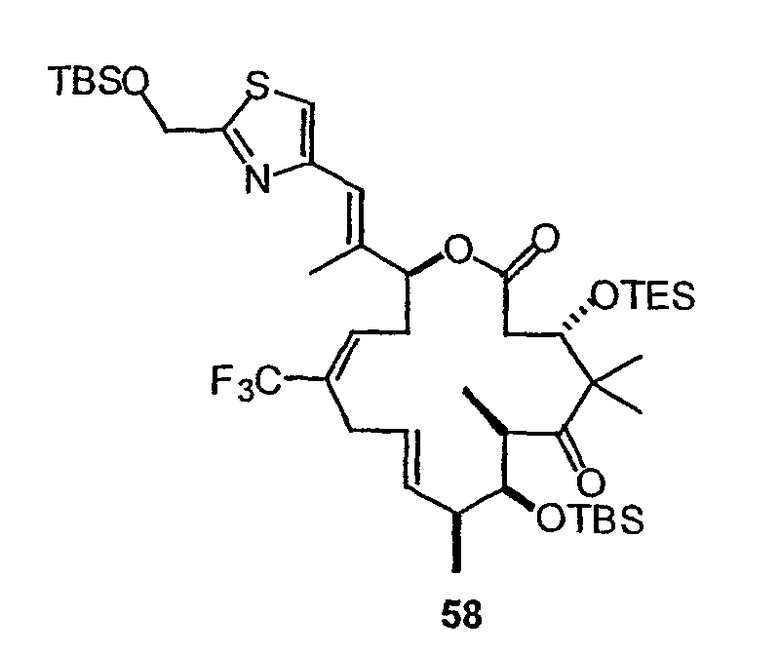
1H ЯМР (400 МГц, CDCl3) δ 0,09 (3H, c), 0,12 (3H, c), 0,15 (6H, c), 0,55 (6H, кв, J=7,8 Гц), 0,87 (9H, т, J=8,0 Гц), 0,96 (9H, c), 0,97 (9H, c), 1,01 (3H, c), 1,06 (3H, д, J=7,1 Гц), 1,12 (3H, c), 1,20 (3H, д, J=7,1 Гц), 2,07-2,16 (1H, м), 2,18 (3H, д, J=1,0 Гц), 2,38 (1H, дд, J=14,4, 3,3 Гц), 2,34-2,46 (1H, м), 2,49 (1H, дд, J=14,4, 7,4 Гц), 2,78-2,90 (2H, м), 2,97-3,09 (2H, м), 3,98 (1H, д, J=8,9 Гц), 4,54 (1H, дд, J=7,3, 3,3 Гц), 4,97 (2H, c), 5,33 (1H, ддд, J=15,8, 8,6, 4,9 Гц), 5,63 (1H, дд, J=9,6, 2,4 Гц), 5,78 (1H, дд, J=15,8, 8,2 Гц), 6,22-6,27 (1H, м), 6,60 (1H, c), 7,09 (1H, c); 13C ЯМР (100 МГц, CDCl3) δ -5,3 (2C), -3,4, -3,3, 5,5 (3C), 7,0 (3C), 14,6, 17,1, 18,4, 18,7, 19,8, 21,3, 24,8, 25,9 (3C), 26,4 (3C), 29,6, 32,9, 42,0, 42,1, 48,2, 54,1, 63,4, 73,4, 76,9, 77,8, 117,2, 121,7, 124,3 [кв, 1J(C,F)=273,6 Гц], 127,2, 130,7 [кв, 2J(C,F) = 27,5 Гц], 130,8 [кв, 3J(C,F) = 6,2 Гц], 133,2, 136,4, 152,6, 170,1, 172,4, 217,1; МС-НР (ESI) вычислено для С45Н78F3NO6SSi3Na[M+Na+] 924,5, найдено 924,5.
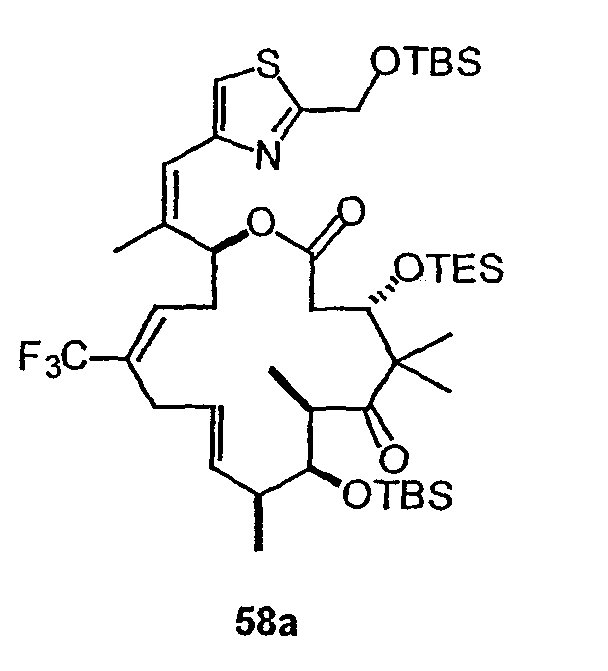
1H ЯМР (400 МГц, CDCl3) δ 0,07 (3H, c), 0,13 (3H, c), 0,16 (6H, c), 0,48 (6H, кв, J=7,8 Гц), 0,84 (9H, т, J=7,9 Гц), 0,97 (18H, c), 0,98 (3H, c), 1,06 (3H, д, J=7,1 Гц), 1,11 (3H, c), 1,20 (3H, д, J=7,2 Гц), 2,00 (3H, c), 2,03-2,11 (1H, м), 2,33 (1H, дд, J=14,1, 2,8 Гц), 2,43 (1H, дд, J=14,0, 7,8 Гц), 2,40-2,48 (1H, м), 2,76-2,89 (2Н, м), 2,97-3,10 (2Н, м), 3,99 (1H, д, J=9,3 Гц), 4,57 (1H, дд, J=7,8, 2,6 Гц), 4,95 (1H, д, J=14,6 Гц), 5,00 (1H, д, J=14,6 Гц), 5,33 (1H, ддд, J=15,6, 9,1, 3,8 Гц), 5,82 (1H, дд, J=15,6, 8,3 Гц), 6,30 (1H, c), 6,32-6,38 (1H, м), 7,04 (1H, c), 7,11 (1H, дд, J=11,0, 2,3 Гц); МС-НР (ESI) вычислено для С45Н78F3NO6SNa[M+Na+] 924,5, найдено 924,5.
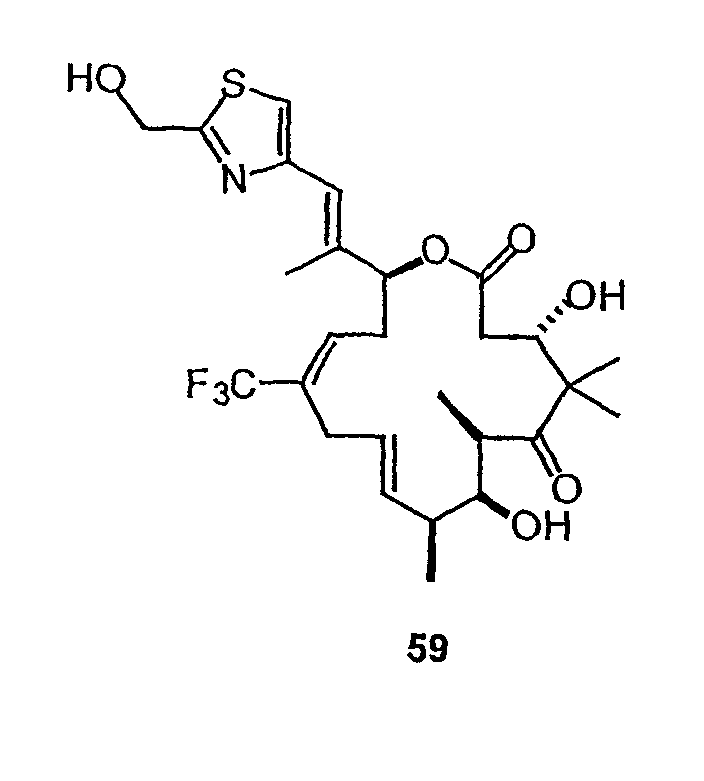
Соединение 59: К раствору 58 (1,04 г, 2,25 ммоль) в ТГФ (22 мл) медленно добавляли HF·пиридин (11 мл) при 0°C и смесь перемешивали при КТ в течение 4,3 час. Реакцию гасили, добавляя по каплям TMSOMe (75 мл) в течение 10 мин при 0°C. Смесь энергично перемешивали при КТ в течение 4,2 час. После концентрирования и сушки в условиях высокого вакуума в течение 1 час остаток очищали флэш-хроматографией на колонке (SiO2 ~25 г; гексан/EtOAc = от 3:4 до 1:2), получая 59 (615,7 мг, 1,00 ммоль, 96%) в виде бесцветного порошка; [α]D 25 -57,7 (с 1,20, СНСl3); 1H ЯМР (400 МГц, CDCl3) δ 1,04 (3Н, c), 1,12 (3H, д, J=6,9 Гц), 1,25 (3H, д, J=6,8 Гц), 1,36 (3H, c), 1,90 (1H, д, J=6,6 Гц, OH), 2,08 (3H, c), 2,23-2,32 (1H, м), 2,34 (1H, дд, J=15,7, 2,4 Гц), 2,49 (1H, дд, J=15,7, 10,1 Гц), 2,59-2,69 (2H, м), 2,95-3,01 (2Н, м), 3,04 (1H, квинтeт, J=6,8 Гц), 3,72 (1H, тд, J=7,0, 3,0 Гц), 3,78 (1H, д, J=5,7 Гц, ОН), 4,38 (1H, ддд, J=10,1, 5,7, 2,4 Гц), 4,90 (2H, д, J=6,1 Гц), 5,10 (1H, т, J=6,1 Гц, ОН), 5,44 (1H, т, J=4,7 Гц), 5,60 (1H, дд, J=15,9, 4,4 Гц), 5,66 (1H, дд, J=15,9, 5,0 Гц), 6,28 (1H, т, J=6,7 Гц), 6,73 (1H, c), 7,16 (1H, c); МС-НР (ESI) вычислено для С27Н37F3NO6SNa[M+Н+] 560,2, найдено 560,1.

Соединения 49 и 50: Раствор 28 (12,2 мг, 24,9 мкмоль) в CH2Cl2 (1,25 мл) охлаждали до -78°C и обрабатывали охлажденным раствором DMDO (-78°C, 0,06 М в ацетоне; 914 мкл, 54,8 мкмоль). Смеси давали возможность нагреться до -50°C и перемешивали при -50°C в течение 2,7 час. Избыток DMDO гасили при -50°C добавлением диметилсульфида (117 мкл) и смесь перемешивали при данной температуре в течение 0,5 час. Растворитель удаляли в вакууме. Очистка препаративной тонкослойной хроматографией (гексан/EtOAc = 1/2) давала β-эпоксид 49 (3,0 мг, 5,93 мкмоль, 24%) и α-эпоксид 50 (7,9 мг, 15,6 мкмоль, 63%), оба в виде бесцветного твердого вещества.

Соединение 49 : 1H ЯМР (400 МГц, CDCl3) δ 1,03 (3Н, с), 1,11 (3Н, д, J=7,0 Гц), 1,14 (3Н, д, J=6,9 Гц), 1,34 (3Н, c), 1,36 (3Н, c), 2,00 (1H, ддд, J=15,1, 7,3, 4,0 Гц), 2,14 (1H, дт, J=15,1, 5,2 Гц), 2,14 (3Н, c), 2,21 (1H, дд, J=14,6, 8,0 Гц), 2,33 (1H, дд, J=14,7, 4,8 Гц), 2,47 (1H, дд, J=13,8, 3,3 Гц), 2,59 (1H, дд, J=13,8, 9,4 Гц), 2,73 (3Н, c), 2,77 (1H, ушир.c, ОН), 2,93 (1H, дд, J=7,3, 4,8 Гц), 3,34 (1H, кв.д, J=6,9, 3,7 Гц), 3,75-3,82 (1H, м), 4,12-4,24 (2H, м, включая ОН), 5,54 (1H, ддд, J=15,7, 7,4, 5,0 Гц), 5,54-5,60 (1H, м), 5,64 (1H, дд, J=15,7, 5,6 Гц), 6,94 (1H, c), 7,01 (1H, c); МС-НР (ESI) вычислено для С27Н40NO6S[M+Н+] 506,3, найдено 506,3.

Соединение 50: 1H ЯМР (400 МГц, CDCl3) δ 1,00 (3H, c), 1,04 (3H, д, J=6,9 Гц), 1,12 (3H, д, J=7,0 Гц), 1,35 (3H, c), 1,35 (3H, c), 1,87 (1H, дт, J=15,0, 9,2 Гц), 2,03 (1H, дд, J=13,9, 9,2 Гц), 2,13 (3H, c), 2,13-2,19 (1H, м), 2,36 (1H, дд, J=13,9, 3,4 Гц), 2,39 (1H, дд, J=12,2, 2,1 Гц), 2,42-2,51 (1H, м), 2,49 (1H, дд, J=12,4, 10,9 Гц), 2,69 (1H, д, J=2,7 Гц), 2,72 (3H, c), 3,06 (1H, дд, J=9,7, 3,1 Гц), 3,54 (1H, кв.д, J=7,0, 2,0 Гц), 3,76-3,80 (1H, м), 4,07-4,14 (1H, м), 4,31 (1Н, д, J=4,1 Гц), 5,52 (1H, дд, J=15,5, 8,7 Гц), 5,60 (1H, ддд, J=15,1, 9,4, 3,4 Гц), 5,71 (1H, д, J=8,4 Гц), 6,63 (1H, c), 6,99 (1H, c); МС-НР (ESI) вычислено для С27Н39NO6SNa[M+Na+] 528,2, найдено 528,2.
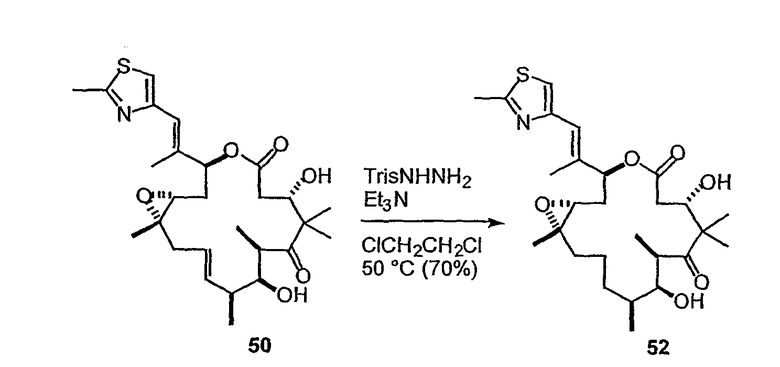
Соединение 52: К раствору 50 (1,7 мг, 3,4 мкмоль) и трисNHNH2 (40,1 мг, 0,134 ммоль) в ClCH2CH2Cl (0,8 мл) при 50°C добавляли Et3N (18,7 мкл, 0,134 ммоль). Реакцию контролировали с помощью ВЭТСХ (гексан/EtOAc = 1/2). После перемешивания в течение 4 час смесь охлаждали до КТ, разбавляли EtOAc и фильтровали через подушку силикагеля, которую промывали EtOAc. После концентрирования остаток очищали препаративной ТСХ (гексан/EtOAc = 1/2), получая 52 (1,2 мг, 2,4 мкмоль, 70%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 0,95 (3Н, д, J=7,1 Гц), 1,04 (3H, c), 1,11 (3H, д, J=7,0 Гц), 1,28 (3H, c), 1,37 (3H, c), 1,35-1,44 (1H, м), 1,45-1,59 (4H, м), 1,71-1,82 (2H, м), 1,86 (1H, дт, J=15,3, 9,5 Гц), 2,10 (1H, дд, J=15,3, 3,6 Гц), 2,13 (3H, c), 2,40 (1H, дд, J=12,5, 2,5 Гц), 2,49 (1H, дд, J=12,5, 11,0 Гц), 2,74 (3H, c), 2,80 (1H, ушир.c, ОН), 3,07 (1H, дд, J=10,3, 3,3 Гц), 3,34 (1H, кв.д, J=7,0, 1,0 Гц), 3,89 (1H, ушир.c, ОН), 4,03-4,09 (1H, м), 4,12-4,17 (1H, м), 5,69 (1H, д, J=9,1 Гц), 6,63 (1H, c), 7,00 (1H, c); МС-НР (ESI) вычислено для С27Н41NO6SNa[M+Na+] 530,3, найдено 530,2.
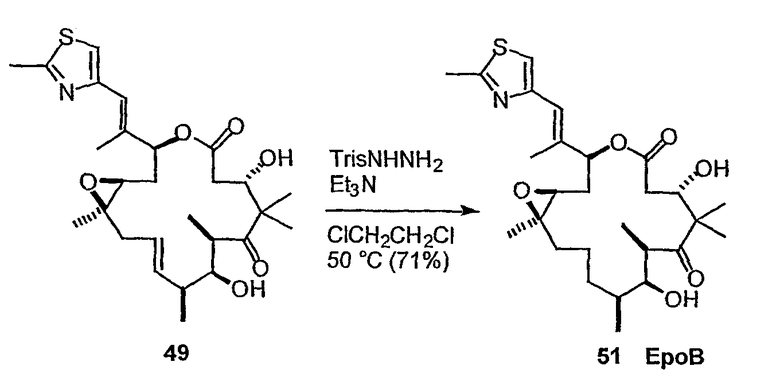
Соединение 51: К раствору 49 (0,7 мг, 1,38 мкмоль) и трисNHNH2 (20,6 мг, 69 мкмоль) в ClCH2CH2Cl (0,4 мл) при 50°C добавляли Et3N (9,6 мкл, 69 мкмоль). Реакцию контролировали с помощью ВЭТСХ (гексан/EtOAc = 1/2). После перемешивания в течение 6 час смесь охлаждали до КТ, разбавляли EtOAc и фильтровали через подушку силикагеля, которую промывали EtOAc. После концентрирования остаток очищали препаративной ТСХ (гексан/EtOAc = 1/2), получая 51 (0,5 мг, 0,985 мкмоль, 71%) в виде белого твердого вещества. Спектральные данные 51 были идентичны данным, описанным для EpoB.
Пример 2
Альтернативная методика синтеза для синтеза промежуточных продуктов эпотилонов
В следующих примерах предлагаются способы получения различных промежуточных продуктов синтеза аналогов эпотилонов.
Оптимизация синтеза 9,10-дегидроэпотилонов
Пример 1
Пример 2
Восстановления Нойори

Пример 3
Восстановления Нойори
Пример 4
Альтернативный синтез ключевого дикетона
Пример 5
Способ 1. Миграция силильной группы - декарбоксилирование
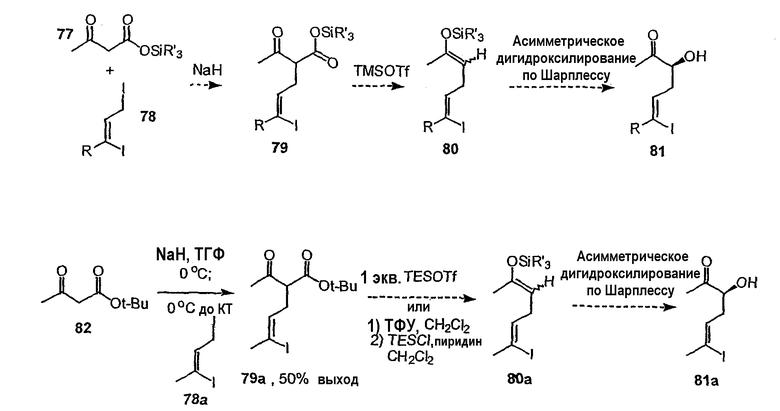
Способ 2. Декарбоксилирование - введение силильной группы
Пример 6
Дополнительный способ синтеза 2-гидроксикетона Эванса
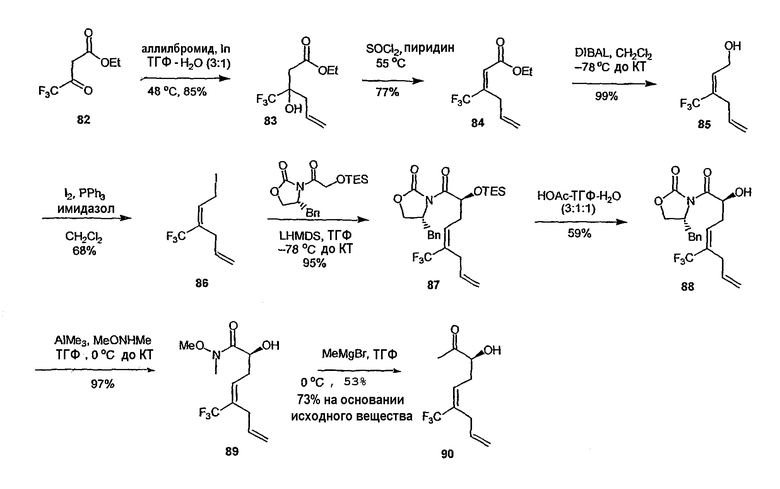
Пример 7
Способ синтеза 2-гидроксикетона Ковальского-Шарплесса

Эксперименты

1-(2-Бензилокси-1-метилэтил)-5,5-диизопропокси-2,4,4-триметил-3-оксопентиловый; 2,2,2-трихлорэтиловый диэфир карбоновой кислоты (32a)
К раствору 7-бензилокси-5-гидрокси-1,1-диизопропокси-2,2,4,6-тетраметилгептан-3-она 32 (1,0 г, 2,4 ммоль) и пиридина (0,8 мл, 7,3 ммоль) в CH2Cl2 (10,0 мл) при 0°C добавляли 2,2,2-трихлорэтилхлорформиат (668,0 мкл, 4,9 ммоль) и затем смеси давали возможность нагреться до КТ. Спустя 1 час реакционную смесь гасили насыщенным раствором соли и затем экстрагировали CH2Cl2. Объединенные органические слои сушили над MgSO4 и концентрировали при пониженном давлении. Грубый продукт очищали флэш-хроматографией (градиент от гексана до смеси гексан/EtOAc 93:7), получая 32a (1,285 г, 92%) в виде прозрачного масла: 1H ЯМР (400 МГц, CDCl3) δ 1,03-1,09 (м, 12H), 1,15 (д, J=1,8 Гц, 3H), 1,17 (д, J=1,9 Гц, 3H), 1,19-1,21 (м, 6H), 1,97-2,11 (м, 1H), 3,2 (дд, J=6,2 и 9,0 Гц, 1H), 3,54 (дд, J=4,8 и 9,1 Гц, 1H), 3,57-3,60 (м, 1H), 3,82 (кв.д, J=3,6 и 5,9 Гц, 2H), 4,47 (c, 2H), 4,57 (c, 1H), 4,72 (д, J=11,9 Гц, 1H), 4,81 (д, J=11,9 Гц, 1H), 5,08 (т, J= 6,0 Гц, 1H), 7,29-7,35 (м, 5H); 13С ЯМР (100 МГц, СDCl3) δ 11,9, 15,0, 18,8, 21,4, 21,7, 22,3, 23,2, 23,4, 35,7, 42,5, 53,4, 53,9, 69,4, 70,9, 71,4, 73,3, 81,3, 94,7, 103,4, 127,5, 127,6, 128,2, 138,2, 154,0, 215,6; ИК (пленка, NaCl, cм-1) 2966, 1760, 1698, 1247; МС-НР (ESI) вычислено для С27Н41О7Cl3Na [M+Na+] 605,2, найдено 605,2; [α]23 D = -20,4 (c = 1,0, CHCl3).
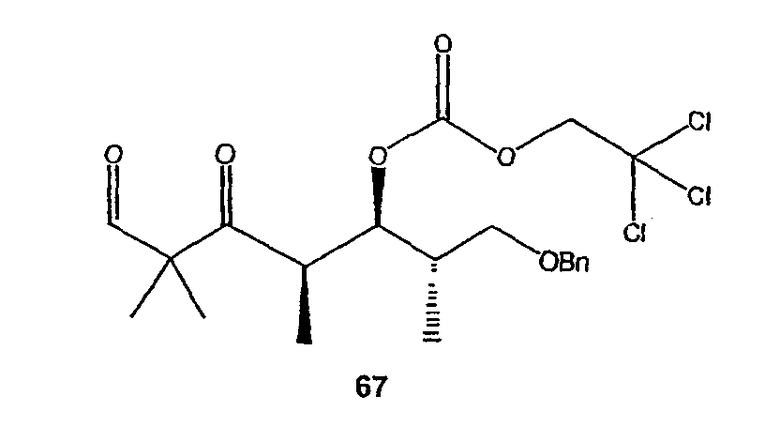
1-(2-Бензилокси-1-метилэтил)-2,4,4-триметил-3,5-диоксопентиловый; 2,2,2-трихлорэтиловый диэфир карбоновой кислоты (67)
К раствору 32a (1,28 г, 2,25 ммоль) в 4:1 ТГФ/H2O (25 мл) добавляли п-TsOH (111,0 мг, 0,6 ммоль). После нагревания при 70°C в течение 5 час реакционную смесь вливали в холодный (0°C) насыщенный водный раствор NaHCO3 (12 мл) и затем экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали при пониженном давлении. Грубый продукт очищали флэш-хроматографией (градиент от гексана до смеси гексан/EtOAc 84:16), получая 67 (793,2 мг, 76%) в виде прозрачного масла: 1H ЯМР (400 МГц, CDCl3) δ 0,90 (д, J=5,8 Гц, 3H), 1,0 (д, J=6,9 Гц, 3H), 1,24 (c, 6H), 1,97-2,04 (м, 1H), 3,24 (дд, J=4,8 и 9,2 Гц, 1H), 3,34 (м, 1H), 3,42 (дд, J=5,8 и 9,2 Гц, 1H), 4,35 (д, J=11,9 Гц, 1H), 4,39 (д, J=11,9 Гц, 1H), 4,64 (д, J=11,9 Гц, 1H), 4,69 (д, J=11,9 Гц, 1H), 4,96 (т, J=6,0 Гц, 1H), 7,19-7,28 (м, 5H), 9,49 (c, 1H); 13С ЯМР (100 МГц, СDCl3) -12,0, 14,8, 19,5, 19,6, 35,4, 43,3, 60,9, 71,1, 73,3, 80,37, 94,5, 127,7, 127,8, 128,3, 137,9, 154,1, 201,0, 210,1; ИК (пленка, NaCl, cм-1) 2973, 2880, 1758, 1701, 1453, 1380, 1248; МС-НР (ESI) вычислено для С21Н27О6Cl3Na [M+Na+] 503,0, найдено 503,0; [α]23 D = -18,5 (c = 0,8, CHCl3).

Трет-бутиловый эфир 9-бензилокси-4,4,6,8-тетраметил-3,5-диоксо-7-(2,2,2-трихлорэтоксикарбонилокси)нонановой кислоты (69)
К раствору LDA (1,17 ммоль, 0,3 М в Et2O) при -78°C добавляли трет-бутилацетат (1,0 ммоль, 135,0 мкл). Через 30 мин медленно в течение 15 мин добавляли раствор 67 (464,0 мг, 1 ммоль) в Et2O (2 мл). После перемешивания в течение 1 час реакцию гасили насыщенным водным раствором NH4Cl и затем экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали при пониженном давлении. Грубый продукт очищали флэш-хроматографией (градиент от гексана до смеси гексан/EtOAc 86:14), получая 68 (1:1 смесь эпимеров, 461,4 мг, 80%) в виде прозрачного масла: 1H ЯМР (400 МГц, CDCl3) δ 0,87 (д, J=5,3 Гц, 3H), 0,89 (д, J=5,5 Гц, 3H), 1,02-1,10 (м, 18H), 1,38 (c, 18H), 1,97-2,2 (м, 2H), 2,27-2,31 (м, 2H), 3,22-3,27 (м, 3H), 3,39-3,48 (м, 5H), 4,03-4,06 (м, 1H), 4,11-4,14 (м, 1H), 4,38-4,45 (м, 4H), 4,58-4,73 (м, 4H), 4,97 (т, J=5,8 Гц, 1H), 5,02 (т, J=5,8 Гц, 1H), 7,18-7,27 (м, 10H); 13С ЯМР (100 МГц, СDCl3) δ 11,9, 12,7, 14,9, 15,2, 18,7, 19,3, 21,4, 21,6, 28,0, 35,6, 37,4, 41,7, 42,0, 51,8, 51,9, 71,3, 71,3, 72,5, 73,0, 73,3, 73,3, 80,6, 81,2, 81,3, 94,6, 127,5, 127,7, 127,8, 128,3, 138,0, 138,1, 154,0, 154,1, 172,3, 172,4, 216,0, 216,3; ИК (пленка, NaCl, cм-1) 3509, 2975, 1759, 1707, 1368, 1248, 1152; МС-НР (ESI) вычислено для С27Н39О8Cl3Na [M+Na+] 619,1, найдено 619,2.
К раствору 68 при 0°C (350,0 мг, 0,6 ммоль) в CH2Cl2 (10 мл) добавляли периодинан Десс-Мартина (398,0 мг, 0,9 ммоль). Смесь перемешивали при КТ в течение 1 час и затем вливали в хорошо перемешанную смесь 1:1 насыщенный Na2S2O3/насыщенный NaHCO3. Слои разделяли через 30 мин. Водный слой три раза экстрагировали Et2O. Объединенные органические экстракты промывали насыщенным NaHCO3, насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Грубый продукт очищали флэш-хроматографией (градиент от гексана до смеси гексан/EtOAc 91:9), получая 69 (258,4 мг, 74%) в виде прозрачного масла: 1H ЯМР (400 МГц, CDCl3) δ 0,80 (д, J=6,9 Гц, 3H), 0,87 (д, J=6,9 Гц, 3H), 1,13 (c, 3H), 1,19 (c, 3H), 1,23 (c, 9H), 2,04-2,12 (м, 1H), 3,09-3,28 (м, 5H), 4,23 (c, 2H), 4,48(д, J=11,9 Гц, 1H), 4,55 (д, J=11,9 Гц, 1H), 4,79 (дд, J=4,6 и 7,3 Гц, 1H), 7,04-7,13 (м, 5H); 13С ЯМР (100 МГц, СDCl3) δ 11,7, 14,6, 20,7, 21,5, 27,9, 35,5, 42,2, 43,4, 63,3, 71,3, 73,3, 79,9, 81,5, 90,5, 94,5, 127,6, 127,7, 128,2, 138,0, 154,0, 166,2, 202,9, 210,0; ИК (пленка, NaCl, cм-1) 2977, 1758, 1697, 1368, 1248, 1154; МС-НР (ESI) вычислено для С27Н37О8Cl3Na [M+Na+] 617,1, найдено 617,1; [α]23 D = -49,1 (c = 0,9, CHCl3).
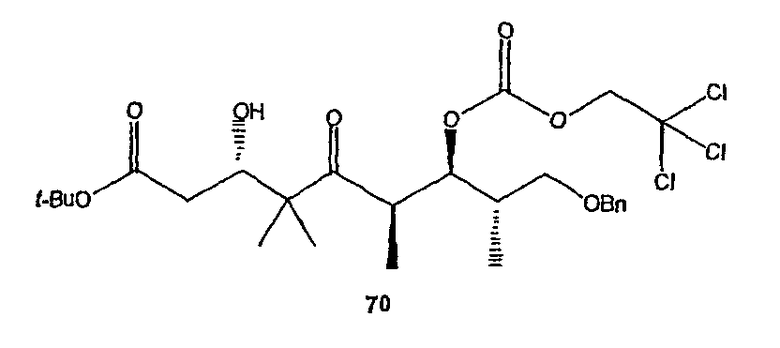
Трет-бутиловый эфир 9-бензилокси-3-гидрокси-4,4,6,8-тетраметил-5-оксо-7-(2,2,2-трихлорэтоксикарбонилокси)нонановой кислоты (70)
Гильзу высокого давления загружали катализатором (R)-RuBINAP (16,8 мг, 10,0 мкмоль). Добавляли HCl (555 мкл, 0,2N в MeOH) и затем смесь обрабатывали ультразвуком в течение 15 сек. Затем добавляли раствор 69 (59,4 мг, 0,1 ммоль) в MeOH (555 мкл) и смесь переносили в аппарат Парра. Сосуд продували H2 в течение 5 мин и затем создавали давление до 1200 фунтов/кв.дюйм. Через 17 час реакционную смесь возвращали в условия атмосферного давления и вливали в насыщенный водный раствор NaHCO3. Водный слой три раза экстрагировали EtOAc. Объединенные органические экстракты сушили над MgSO4 и концентрировали при пониженном давлении. Грубый продукт очищали флэш-хроматографией (градиент от гексана до смеси гексан/EtOAc 88:12), получая 70 (dr>20:1 на основании 1H-ЯМР-анализа) (47,6 мг, 80%) в виде бесцветного масла: 1H ЯМР (400 МГц, CDCl3) δ 1,06 (д, J=6,9 Гц, 3Н), 1,11 (д, J=6,8 Гц, 3Н), 1,14 (c, 3H), 1,18 (c, 3H), 1,47 (c, 9H), 2,05-2,12 (м, 1H), 2,35-2,40 (м, 1H), 3,31-3,37 (м, 2H), 3,51-3,54 (м, 2H), 4,11-4,14 (м, 1H), 4,46 (c, 2H), 4,72 (д, J=11,9 Гц, 1H), 4,80 (д, J=11,9 Гц, 1H), 5,05 (дд, J=5,0 и 6,7 Гц, 1H), 7,27-7,35 (м, 5H); 13С ЯМР (100 МГц, СDCl3) δ 12,0, 15,0, 19,3, 21,7, 28,0, 35,6, 37,5, 41,7, 51,8, 71,3, 73,0, 73,3, 80,6, 81,3, 94,7, 127,5, 127,7, 128,3, 138,2, 154,1, 172,4, 216; ИК (пленка, NaCl, cм-1) 3849, 2974, 2879, 1758, 1701, 1454, 1368, 1248, 1152, 926, 734; МС-НР (ESI) вычислено для С27Н39О8Cl3Na [M+Na+] 619,1, найдено 619,2; [α]23 D = -13,0 (c = 0,4, CHCl3).

Трет-бутиловый эфир 9-бензилокси-4,4,6,8-тетраметил-5-оксо-7-(2,2,2-трихлорэтоксикарбонилокси)-3-(триэтилсиланилокси)нонановой кислоты (71)
К раствору 70 (37,6 мг, 6,3 мкмоль) и имидазола (9,4 мг, 13,8 мкмоль) в ДМФА (0,4 мл) при 0°C добавляли TESCl (11,6 мкл, 69,3 мкмоль). Через 3 час смесь разбавляли насыщенным водным NaHCO3. Водный слой три раза экстрагировали гексанами. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали при пониженном давлении. Грубый продукт очищали флэш-хроматографией (градиент от гексана до смеси гексан/EtOAc 93:7), получая в порядке элюирования 71 (22,9 мг, 51%) и извлекаемый 70 (12,9 мг, 34%) в виде бесцветных масел. 7: 1H ЯМР (400 МГц, CDCl3) δ 0,66 (кв, J=7,9 Гц, 6H), 0,96 (т, J=7,9 Гц, 9H), 1,01 (c, 3H), 1,05 (д, J=5,2 Гц, 3H), 1,07 (д, J=5,3 Гц, 3H), 1,35 (c, 3H), 1,44 (c, 9H), 2,05-2,11 (м, 2H), 2,50 (дд, J=3,5 и 17,2 Гц, 1H), 3,35 (дд, J=5,9 и 9,0 Гц, 1H), 3,49 (дд, J=4,0 и 9,0 Гц, 1H), 3,53 (дд, J=3,8 и 6,7 Гц, 1H), 4,18 (дд, J=3,5 и 6,5 Гц, 1H), 4,45 (c, 2H), 4,65 (д, J=11,9 Гц, 1H), 4,79 (д, J=11,9 Гц, 1H), 4,97 (дд, J=3,7 и 8,1 Гц, 1H), 7,29-7,52 (м, 5H); 13С ЯМР (125 МГц, СDCl3) δ 5,3, 7,3, 10,9, 14,9, 21,3, 22,6, 28,4, 35,9, 41,1, 42,7, 53,7, 71,9, 73,7, 75,7, 80,1, 80,9, 95,1, 127,9, 128,0, 128,7, 138,6, 154,3, 171,7, 215,7; ИК (пленка, NaCl, cм-1) 2956, 2876, 1732, 1694, 1456, 1366, 1257, 1154, 1098, 988, 835, 774, 741; МС-НР (ESI) вычислено для С33Н53О8SiCl3Na [M+Na+] 733,2, найдено 733,3; [α]23 D = -16,1 (c = 0,1, CHCl3).

Трет-бутиловый эфир 9-бензилокси-3-(диэтилметилсиланилокси)-7-гидрокси-4,4,6,8-тетраметил-5-оксононановой кислоты (71a)
К раствору 71 (22,9 мг, 3,2 мкмоль) в смеси 1:1 ТГФ/AcOH (1,4 мл) добавляли Zn (5,0 мг, 7,8 мкмоль, наноразмер). Смесь обрабатывали ультразвуком в течение 15 мин. Добавляли еще Zn (5,0 мг, 7,8 мкмоль, наноразмер), после чего обрабатывали ультразвуком еще в течение 15 мин. Суспензию фильтровали через подушку целита, несколько раз промывая EtOAc. Фильтраты промывали насыщенным NaHCO3, насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Грубый продукт пропускали через небольшой слой силикагеля, элюируя смесью гексан/EtOAc 4:1, получая 17,1 мг (выход 99%) 71a в виде бесцветного масла: 1H ЯМР (400 МГц, CDCl3) δ (м, 6H), 0,96 (т, J=7,9 Гц, 9H), 0,97 (д, J=6,8 Гц, 3H), 1,05 (д, J=6,8 Гц, 3H), 1,11 (c, 3H), 1,26 (c, 3H), 1,44 (c, 9H), 1,84-1,90 (м, 1H), 2,21 (дд, J=6,7 и 17,0 Гц, 1H), 2,36 (дд, J=6,7 и 17,0 Гц, 1H), 3,24-3,29 (м, 1H), 3,44-3,52 (м, 2H), 3,67 (дд, J=3,9 и 8,9 Гц, 1H), 4,36 (дд, J=3,5 и 6,5 Гц, 1H), 4,50 (д, J=12,0 Гц, 1Н), 4,54 (д, J=12,0 Гц, 1H), 7,32-7,36 (м, 5H); 13С ЯМР (100 МГц, СDCl3) δ 5,0, 6,9, 9,7,13,9, 20,2, 21,8, 28,0, 36,3, 40,8, 41,5, 53,7, 72,5, 72,9, 73,2, 73,6, 80,7, 127,4, 127,5, 128,2, 138,6, 171,0, 221,4; ИК (пленка, NaCl, cм-1) 3502, 2959, 2875, 1731, 1683, 1456, 1366, 1154, 1098, 996, 739; МС-НР (ESI) вычислено для С30Н52О6SiCl3Na [M+Na+] 559,3, найдено 559,3; [α]23 D = -41,0 (c = 0,4, CHCl3).
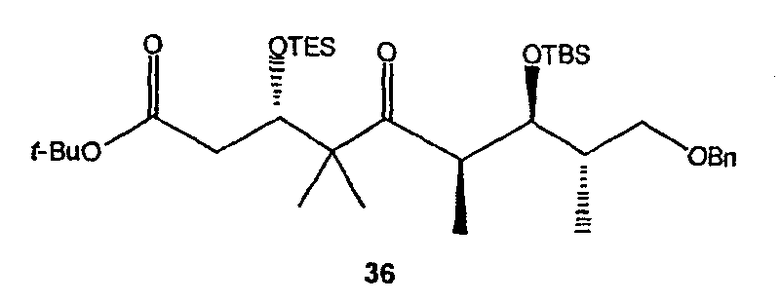
Трет-бутиловый эфир 9-бензилокси-7-(трет-бутилдиметилсиланилокси)-3-(диэтилметилсиланилокси)-4,4,6,8-тетраметил-5-оксононановой кислоты (36)
К раствору 71a (4,1 мг, 7,6 мкмоль) и 2,6-лутидина (10,0 мкл, 43,5 ммоль) в CH2Cl2 (0,2 мл) при -78°C добавляли TBSOTf (10,0 мкл, 85,8 ммоль). Через 2 час дополнительно добавляли 2,6-лутидин (10,0 мкл, 43,5 ммоль) и TBSOTf (10,0 мкл, 85,8 ммоль). Через 6 час смесь разбавляли насыщенным водным NaHCO3. Водный слой три раза экстрагировали EtOAc. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали при пониженном давлении. Грубый продукт очищали флэш-хроматографией (градиент от гексана до смеси гексан/EtOAc 91:9), получая 36 (5,4 мг, 82%) в виде прозрачного масла. Спектроскопические данные хорошо согласуются с описанными значениями.
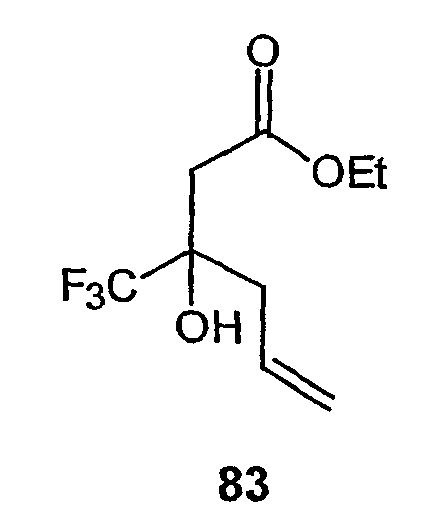
Спирт 83. К раствору этил 4,4,4-трифторацетоацетата (24,0 мл, 0,164 моль) в смеси ТГФ-вода (3:1 = V:V, 320 мл) при комнатной температуре добавляли аллилбромид (20,0 мл, 1,4 экв.) и индий (порошок, -100 меш, 25 г, 1,3 экв.) и полученную в результате смесь перемешивали при 48°C в течение 15 час. Реакционную смесь охлаждали до комнатной температуры, гасили 2N водным HCl (400 мл) и экстрагировали CH2Cl2 (400 мл, 2×200 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Флэш-хроматография (гексаны → гексаны-эфир 10:1 → 8:1 → 6:1 → 4:1) давала спирт 83 в виде прозрачного масла (31,64 г, выход 85%): ИК (пленка) 3426 (ушир.м), 2986 (м), 1713 (c), 1377 (м), 1345 (м), 1301 (м), 1232 (м), 1173 (c), 1095 (м), 1023 (м), 927 (м) cм-1; 1H ЯМР (400 МГц, CDCl3) δ 5,82 (м, 1H), 5,15 (м, 3H), 4,17 (м, 2H), 2,59 (м, 1H), 2,58 (д, J=3,4 Гц, 2H), 2,29 (дд, J=14,2, 8,6 Гц, 1H), 1,24 (т, J=7,2 Гц, 3H); 13С ЯМР (100 МГц, СDCl3) δ 172,08, 130,89, 125,65 (кв, J=280 Гц), 120,27, 73,79 (кв, J=28 Гц), 61,55, 38,97, 35,65, 13,82; масс-спектр высокого разрешения m/z 227,0895 [(M+H)+; вычислено для С9Н14О3F3: 227,0895].

Сложный эфир 84. Смесь спирта 83 (16,71 г, 0,07386 моль) и пиридина (15,0 мл, 2,5 экв.) охлаждали до -10°C и медленно в течение 11 мин обрабатывали тионилхлоридом (11,3 мл, 2,1 экв.). Полученную в результате смесь нагревали до 55°C и перемешивали в течение 12 час. Реакционную смесь охлаждали до -5°C, гасили водой (200 мл) и экстрагировали CH2Cl2 (2×200 мл, 2×150 мл). Объединенные органические слои промывали насыщенным NaHCO3 (2×200 мл) и насыщенным раствором соли (200 мл), сушили (MgSO4) и концентрировали в вакуум. Флэш-хроматография (пентан:эфир, 15:1) давала сложный эфир 84 (11,90 г, выход 77%) в виде желтого масла: ИК (пленка) 2986 (w), 1731 (c), 1308 (c), 1265 (w), 1227 (м), 1197 (c), 1133 (c), 1025 (м), 920 (w), 896 (w) cм-1; 1H ЯМР (400 МГц, CDCl3) δ 6,36 (c, 1H), 5,79 (ддт, J=16,9, 10,2, 6,6 Гц, 1H), 5,15 (дд, J=17,1, 1,5 Гц, 1H), 5,08 (дд, J=10,0, 1,4 Гц, 1H), 4,22 (кв, J=7,1 Гц, 2H), 3,44 (д, J=6,5 Гц, 2H), 1,29 (т, J=7,1 Гц, 3H); 13С ЯМР (100 МГц, СDCl3) δ 164,22, 143,37 (кв, J=29 Гц), 132,71, 123,21 (кв, J=274 Гц), 122,60 (кв, J=6 Гц), 117,32, 60,85, 30,54, 13,85; масс-спектр высокого разрешения m/z 209,0788 [(M+H)+; вычислено для С9Н12О2F3: 209,0789].

Спирт 85. К охлажденному (-75°C) раствору сложного эфира 84 (7,12 г, 0,0342 моль) в CH2Cl2 (120 мл) добавляли раствор DIBAL-H (75 мл, 2,2 экв.) в CH2Cl2 (1,0 М) и полученную в результате смесь нагревали до комнатной температуры в течение 3 час. Реакционную смесь охлаждали до 0°C, гасили насыщенным NH4Cl (12 мл) и перемешивали при комнатной температуре в течение 20 мин. Реакционную смесь разбавляли эфиром (200 мл), сушили (MgSO4) и концентрировали в вакууме. Флэш-хроматография (пентан:эфир, 3:1-1:1) давала спирт 85 (5,68 г, 99%) в виде прозрачного масла: ИК (пленка) 3331 (ушир.c), 2929 (м), 1642 (м), 1445 (м), 1417 (w), 1348 (c), 1316 (c), 1217 (c), 1175 (c), 1119 (c), 1045 (м), 985 (c), 921 (м), 831 (w) cм-1; 1H ЯМР (400 МГц, CDCl3) δ 6,33 (тд, J=6,1, 1,6 Гц, 1H), 5,75 (ддт, J=17,2, 10,0, 6,2 Гц, 1H), 5,07 (м, 2 H), 4,29 (ддд, J=6,3, 4,3, 2,1 Гц, 2H), 2,95 (д, J=6,2 Гц, 2H); 13С ЯМР (100 МГц, СDCl3) δ 134,45 (кв, J=6 Гц), 133,38, 127,97 (кв, J=29 Гц), 123,76 (кв, J=271 Гц), 116,25, 57,87, 29,79.
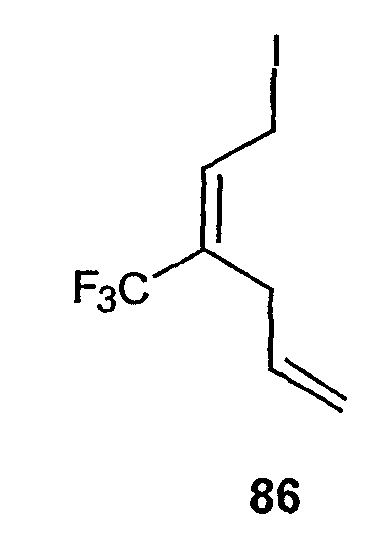
Йодид 86. Охлажденный (0°C) раствор спирта 85 (5,97 г, 0,0358 моль) в CH2Cl2 (50 мл) обрабатывали PPh3 (11,17 г, 1,2 экв.), имидазолом (3,55 г, 1,5 экв.) и I2 (9,10 г, 1,1 экв.) и полученную в результате смесь перемешивали при 0°C в течение 10 мин. Реакционную смесь гасили смесью насыщенного Na2S2O3 - насыщенного NaHCO3 (1:1 = V:V, 200 мл) и экстрагировали пентаном (3×200 мл). Объединенные органические слои промывали смесью насыщенного Na2S2O3 - насыщенного NaHCO3 (1:1 = V:V, 200 мл) и насыщенным раствором соли (100 мл), сушили (MgSO4) и концентрировали в вакууме. Флэш-хроматография (пентан) давала йодид 86 (6,69 г, 68%) в виде бледно-красного масла: ИК (пленка) 3083 (w), 2982 (w), 1636 (w), 1558 (w), 1456 (w), 1367 (w), 1317 (c), 1216 (м), 1181 (c), 1151 (c), 1120 (c), 989 (м), 921 (м), 896 (м) cм-1; 1H ЯМР (400 МГц, CDCl3) δ 6,45 (тд, J=8,9, 1,5 Гц, 1H), 5,79 (ддт, J=16,8, 10,3, 6,2 Гц, 1H), 5,12 (м, 2H), 3,85 (ддд, J=8,9, 2,9, 1,4 Гц, 2H), 3,00 (дт, J=6,1, 1,4 Гц, 2H); 13С ЯМР (100 МГц, СDCl3) δ 132,42, 131,64 (кв, J=6 Гц), 129,63 (кв, J=29 Гц), 123,64 (кв, J=272 Гц), 117,00, 29,32, -4,27; масс-спектр высокого разрешения m/z 298,7 [(M+Na)+; вычислено для С7Н8F3INa: 299,0].
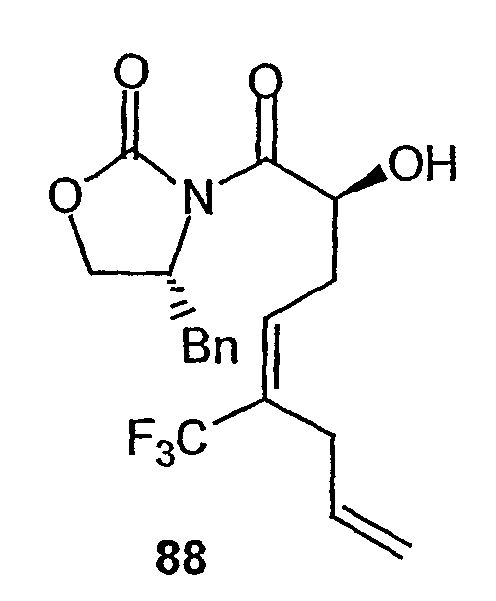
α-Гидроксиоксазолидинон 88. К охлажденному (-78°C) TES-защищенному 4-бензил-3-гидроксиацетилоксазолидин-2-ону 7 (16,28 г, 1,92 экв.) в ТГФ (160 мл) по каплям в течение 51 мин добавляли раствор LHMDS (42,0 мл, 1,73 экв.) в ТГФ (1,0 М) и полученную в результате смесь перемешивали при -78°C в течение 35 мин. Реакционную смесь обрабатывали раствором йодида 86 (6,69 г, 24,2 ммоль) в ТГФ (10 мл) и полученной в результате смеси давали возможность медленно нагреться до комнатной температуры в течение ночи. Реакционную смесь гасили насыщенным NAHCO3 (200 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным NH4Cl (150 мл), насыщенным раствором соли (150 мл), сушили (MgSO4) и концентрировали в вакууме. Флэш-хроматография (гексаны-EtOAc 6:1 → 3:1) давала смесь продуктов алкилирования (13,6 г), которые использовали в следующей реакции без дополнительной очистка. Раствор продуктов алкилирования в смеси HOAc-вода-ТГФ (3:1:1 = V:V:V, 200 мл) перемешивали при комнатной температуре в течение 4 час. Реакционную смесь концентрировали в вакууме, чтобы удалить HOAc, гасили насыщенным NaHCO3 (400 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои сушили (MgSO4) и концентрировали в вакууме. Флэш-хроматография (гексаны : EtOAc, 3:1 → 2:1) давала α-гидроксиоксазолидинон 88 (7,55 г, выход 81% для двух стадий) в виде прозрачного масла: [α]D 25 -48,2 (с 1,08, СНСl3); ИК (пленка) 3486 (ушир.с), 3030 (м), 2983 (c), 2925 (м), 1790 (c), 1682 (c), 1481 (м), 1393 (м), 1360 (м), 1217 (м), 1171 (м), 1113 (м), 992 (м), 919 (м), 847 (w) cм-1; 1H ЯМР (400 МГц, CDCl3) δ 7,32 (м, 3H), 7,17 (м, 2Н), 6,33 (тд, J=7,2, 1,5 Гц, 1H), 5,77 (ддт, J=16,6, 10,1, 6,2 Гц, 1H), 5,08 (м, 3H), 4,74 (ддт, J=4,8, 3,7, 4,4 Гц, 1H), 4,33 (дд, J=8,6, 8,6 Гц, 1H), 4,26 (дд, J=9,2, 3,4 Гц, 1H), 3,42 (ушир.д, J=6,4 Гц, 1H), 3,24 (дд, J=13,5, 3,4 Гц, 1H), 2,99 (м, 2H), 2,79 (дд, J=13,5, 9,4 Гц, 1H), 2,70 (м, 1H), 2,50 (м, 1H); 13С ЯМР (125 МГц, СDCl3) δ 173,93, 153,05, 134,43, 133,64, 129,98 (кв, J=6 Гц), 129,82 (кв, J=28 Гц), 129,29, 120,01, 127,58, 124,00 (кв, J=272 Гц), 116,34, 69,60, 67,31, 54,95, 37,78, 32,29, 29,84; масс-спектр высокого разрешения m/z 384,1421 [(M+Н)+; вычислено для С19Н21NO4F3: 384,1423].
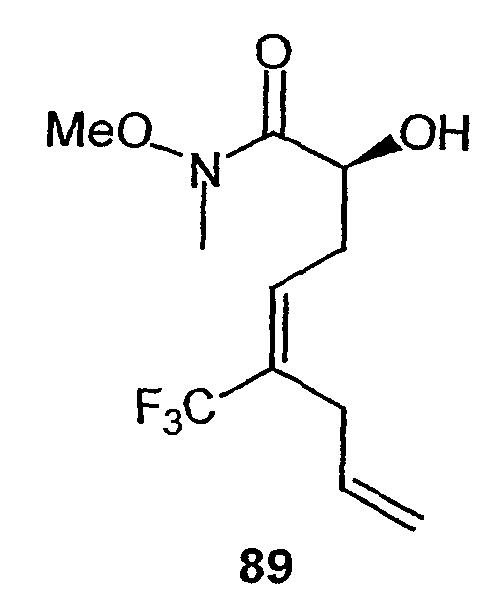
α-Гидроксиамид 89. Суспензию (MeO)NHMe·HCl (10,1 г, 5,25 экв.) в ТГФ (100 мл) при 0°C по каплям обрабатывали раствором AlMe3 (50 мл, 5,1 экв.) в толуоле (2,0 М) и полученный в результате прозрачный раствор перемешивали при комнатной температуре в течение 34 мин, затем добавляли к охлажденному (0ºC) раствору α-гидроксиоксазолидинона 88 (7,55 г, 19,7 ммоль) в ТГФ (70 мл). Полученную в результате смесь нагревали до комнатной температуры и перемешивали в течение 12 час. Реакционную смесь охлаждали до 0°C, гасили медленным добавлением 1N водной винной кислоты (100 мл), перемешивали при комнатной температуре в течение 25 мин и экстрагировали EtOAc (3×200 мл). Объединенные органические слои сушили (MgSO4) и концентрировали в вакууме. Флэш-хроматография (гексаны:EtOAc, 2:1 → 1:1) давала α-гидроксиамид 89 (5,12 г, выход 97%) в виде прозрачного масла: [α]D 25 -57,2 (с 1,03, СНСl3); ИК (пленка) 3432 (ушир.c), 3084 (w), 2980 (м), 2943 (м), 1652 (c), 1464 (м), 1373 (м), 1318 (м), 1214 (м), 1171 (м), 1112 (м), 991 (м), 919 (м), 818 (w) cм-1; 1H ЯМР (400 МГц, CDCl3) δ 6,32 (тд, J=7,3, 1,5 Гц, 1H), 5,74 (ддт, J=16,9, 10,3, 6,1 Гц, 1H), 5,05 (м, 2H), 4,43 (дд, J=7,6, 3,5 Гц, 1H), 3,70 (c, 3H), 3,35 (ушир.c, 1H), 3,24 (c, 3H), 2,94 (д, J=6,1 Гц, 2H), 2,59 (м, 1H), 2,36 (м, 1H); 13С ЯМР (100 МГц, СDCl3) δ 173,43, 133,68, 130,59 (кв, J=6 Гц), 129,25 (кв, J=28 Гц), 124,05 (кв, J=271 Гц), 116,17, 67,57, 61,44, 32,56, 32,38, 29,75; масс-спектр высокого разрешения m/z 268,1161 [(M+Н)+; вычислено для С11Н17NO3F3: 268,1161].
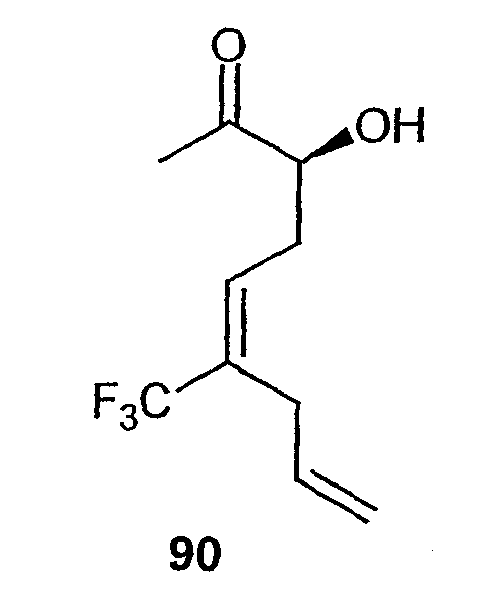
α-Гидроксикетон 90. К охлажденному (0°C) раствору α-гидроксиамида 89 (4,87 г, 18,2 ммоль) в ТГФ (150 мл) добавляли раствор MeMgBr (75 мл, 12 экв.) в простом эфире (3,0 М). Через 5 мин реакционную смесь гасили насыщенным NH4Cl (250 мл) и экстрагировали EtOAc (5×200 мл). Объединенные органические слои сушили (MgSO4) и концентрировали в вакууме. Флэш-хроматография (гексаны:EtOAc 4:1 → 2:1 → 1:2) давала α-гидроксикетон 90 (2,16 г, выход 53%, выход 73% на основании извлечения исходного вещества) в виде прозрачного масла и исходное вещество α-гидроксиамид 89 (1,30 г, выход 27%): [α]D 25 +58,5 (с 1,30, СНСl3); ИК (пленка) 3460 (ушир.c), 3085 (w), 2984 (м), 2926 (м), 1716 (c), 1679 (м), 1641 (м), 1417 (м), 1361 (м), 1319 (c), 1247 (м), 1216 (c), 1172 (c), 1113 (c), 1020 (м), 994 (м), 968 (w), 919 (м) cм-1; 1H ЯМР (500 МГц, CDCl3) δ 6,21 (т, J=7,0 Гц, 1H), 5,75 (ддт, J=16,7, 10,4, 6,2 Гц, 1H), 5,07 (м, 2H), 4,26 (дт, J=7,1, 4,5 Гц, 1H), 3,51 (д, J=4,7 Гц, 1H), 2,96 (д, J=6,1 Гц, 2H), 2,66 (м, 1H), 2,42 (м, 1H), 2,19 (c, 3H); 13С ЯМР (100 МГц, СDCl3) δ 208,53, 133,43, 129,80 (кв, J=28 Гц), 129,76 (кв, J=6 Гц), 123,85 (кв, J=271 Гц), 116,32, 75,36, 31,22, 29,81, 25,11; масс-спектр высокого разрешения m/z 223,0945 [(M+Н)+; вычислено для С10Н14NO2F3: 223,0946].
Пример 8
Способ каталитического асимметрического окисления
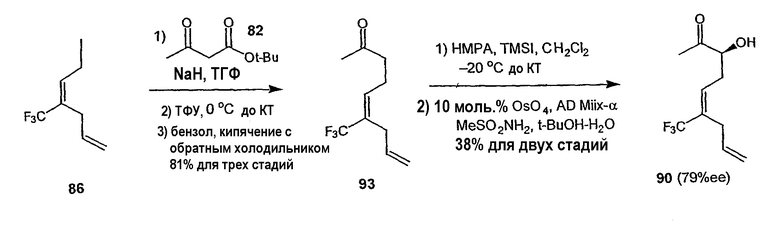
Пример 9
Синтез 21-амино-26-трифтор-(E)-9,10-дегидро-dEpoB
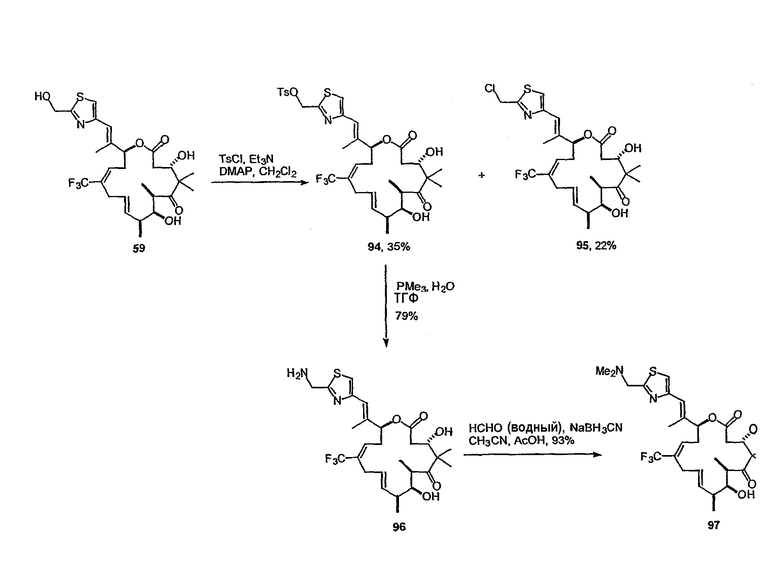

Соединение 98: К раствору 59 (50,4 мг, 90,1 мкмоль) в ТГФ (1 мл) добавляли (PhO)2PON3 (27,2 мкл, 126 мкмоль) при 0°C. После перемешивания при 0°C в течение 5 мин добавляли DBU (16,2 мкл, 108 мкмоль). После перемешивания при 0°C в течение 2 час смесь перемешивали при КТ в течение 20,5 час. Реакционную смесь разбавляли EtOAc и гасили добавлением воды (2 мл). После разделения слоев водный слой экстрагировали EtOAc (три раза) и объединенные органические слои сушили над Na2SO4. После концентрирования остаток сушили в условиях высокого вакуума в течение 10 мин, чтобы удалить DBU. Очистка флэш-хроматографией на колонке (SiO2, гексан/EtOAc = 3:2) давала азид 98 (45,6 мг, 78,0 мкмоль, 87%) в виде бесцветного твердого вещества; 1H ЯМР (400 МГц, CDCl3) δ 1,05 (3H, c), 1,12 (3H, д, J=7,0 Гц), 1,23 (3H, д, J=6,8 Гц), 1,33 (3H, c), 2,01 (1H, д, J=5,5 Гц, OH), 2,17 (3H, c), 2,25-2,35 (1H, м), 2,41 (1H, дд, J=15,5, 3,2 Гц), 2,49 (1H, дд, J=15,5, 9,5 Гц), 2,54-2,60 (1H, м), 2,66 (1H, д, J=6,0 Гц), 2,65-2,76 (1H, м), 2,96 (1H, дд, J=16,0, 4,2 Гц), 3,03 (1H, дд, J=16,1, 6,7 Гц), 3,11 (1H, квинтeт, J=6,8 Гц), 3,71-3,76 (1H, м), 4,31 (1H, ддд, J=9,2, 5,9, 3,2 Гц), 4,65 (2H, c), 5,43 (1H, дд, J=6,0, 4,3 Гц), 5,58 (1H, ддд, J=15,8, 6,4, 4,6 Гц), 5,66 (1H, дд, J=15,8, 6,1 Гц), 6,23 (1H, т, J=7,3 Гц), 6,63 (1H, c), 7,18 (1H, c); МС-НР (ESI) вычислено для С27Н35F3N4O5SNa[M+Na+] 607,2, найдено 607,2.
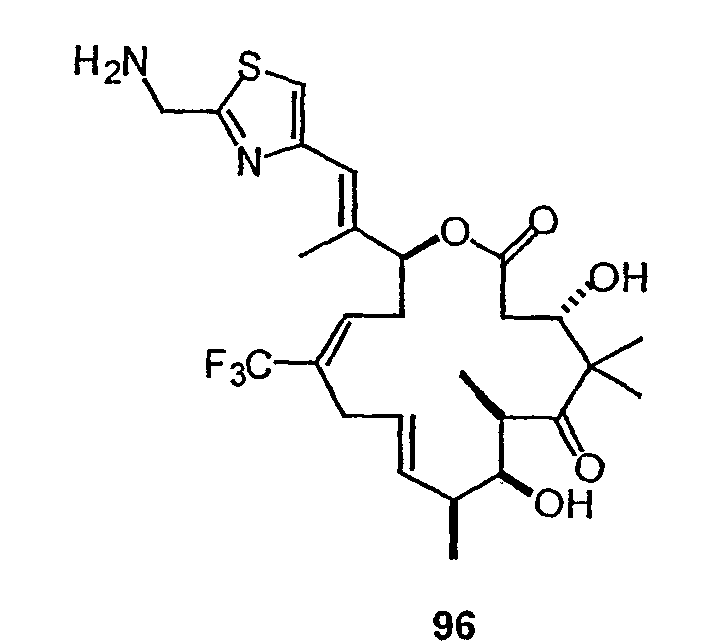
Соединение 96: К раствору азида 98 (21,0 мг, 35,9 мкмоль) в ТГФ (0,6 мл) добавляли PMe3 (1,0 М в ТГФ, 43,1 мкл, 43,1 мкмоль). После перемешивания при КТ в течение 2 мин добавляли воду (0,1 мл) и смесь перемешивали при КТ в течение 3 час. Добавляли PMe3 (1,0 М в ТГФ, 7,2 мкл, 7,2 мкмоль) и смесь перемешивали при КТ в течение 1,5 час. К смеси добавляли 28% NH4OH (водный) (54,5 мкл). После перемешивания в течение 1 час смесь непосредственно очищали препаративной ТСХ (CH2Cl2/MeOH = 100:7,5), получая амин 96 (15,9 мг, 28,5 мкмоль, 79%) в виде бесцветного твердого вещества; 1H ЯМР (400 МГц, CDCl3) δ 1,05 (3H, c), 1,12 (3H, д, J=7,0 Гц), 1,23 (3H, д, J=6,8 Гц), 1,34 (3H, c), 2,12 (3H, д, J=0,7 Гц), 2,24-2,35 (1H, м), 2,39 (1H, дд, J=15,4, 3,0 Гц), 2,49 (1H, дд, J=15,4, 9,8 Гц), 2,54-2,63 (1H, м), 2,66-2,76 (1H, м), 2,97 (1H, дд, J=16,2, 4,2 Гц), 3,03 (1H, дд, J=16,3, 6,5 Гц), 3,10 (1H, квинтeт, J=6,8 Гц), 3,74 (1H, дд, J=6,7, 3,5 Гц), 4,18 (2H, c), 4,34 (1H, дд, J=9,8, 2,9 Гц), 5,43 (1H, дд, J=6,0, 4,3 Гц), 5,55-5,64 (1H, м), 5,67 (1H, дд, J=15,9, 5,8 Гц), 6,24 (1H, ушир.т, J=7,3 Гц), 6,66 (1H, c), 7,10 (1H, c); МС-НР (ESI) вычислено для С27Н38F3N2O5S [M+Н+] 559,2, найдено 559,2.

Соединение 97: К раствору амина 96 (15,9 м, 28,5 мкмоль) в CH3CN (0,78 мл) добавляли 37% HCHO (водный) (31,4 мкл, 0,143 ммоль), затем NaBH3CN (1,0 М в ТГФ, 85,5 мкл, 85,5 мкмоль) и смесь перемешивали при КТ в течение 20 мин. Добавляли AcOH (1 каплю) и смесь перемешивали при КТ в течение 40 мин. Смесь очищали непосредственно препаративной ТСХ (CH2Cl2/MeOH = 100:8), получая продукт 97 (15,6 мг, 26,6 мкмоль, 93%) в виде бесцветного твердого вещества; 1H ЯМР (400 МГц, CDCl3) δ 1,05 (3H, c), 1,12 (3H, д, J=6,9 Гц), 1,23 (3H, д, J=6,8 Гц), 1,33 (3H, c), 2,17 (3H, c), 2,24-2,35 (1H, м), 2,43 (1H, дд, J=15,7, 3,6 Гц), 2,49 (1H, дд, J=15,6, 9,1 Гц), 2,55-2,64 (2H, м, включая ОН), 2,68-2,77 (1H, м), 2,80 (3H, c), 2,81 (3H, c), 2,92-3,06 (2H, м), 3,10 (1H, квинтeт, J=6,8 Гц), 3,69-3,76 (1H, м), 4,25-4,34 (1H, м), 4,33 (2H, c), 5,42 (1H, т, J=5,5 Гц), 5,57 (1H, дт, J=15,8, 6,3 Гц), 5,66 (1H, дд, J=15,7, 6,4 Гц), 6,22 (1H, ушир.т, J=7,2 Гц), 6,64 (1H, c), 7,30 (1H, c); МС-НР (ESI) вычислено для С29Н42F3N2O5S [M+H+] 580,2, найдено 580,2.
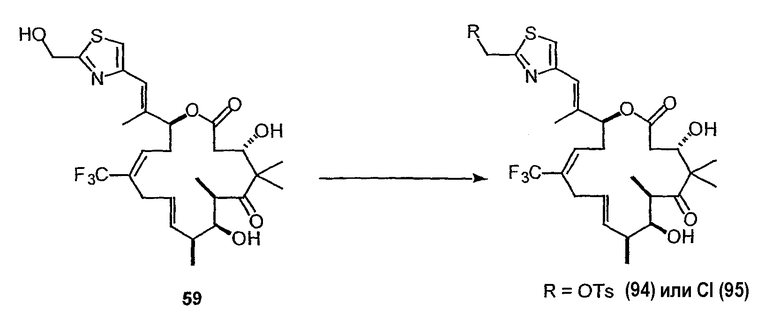
Соединения 94 и 95: К смеси 59 (18,9 мг, 33,8 мкмоль) и Et3N (18,8 мкл, 0,135 ммоль) в CH2Cl2 (1 мл) добавляли TsCl (12,9 мг, 67,5 мкмоль) и DMAP (2,1 мг, 16,9 мкмоль) при 0°C. После перемешивания при КТ в течение 1,5 час смесь разбавляли EtOAc и фильтровали через подушку силикагеля (промывка EtOAc). После концентрирования остаток очищали препаративной ТСХ (гексан/EtOAc = 1:1), получая тозилат 94 (8,5 мг, 11,9 мкмоль, 35%) и хлорид 95 (4,3 мг, 7,44 мкмоль, 22%); оба в виде бесцветного твердого вещества;
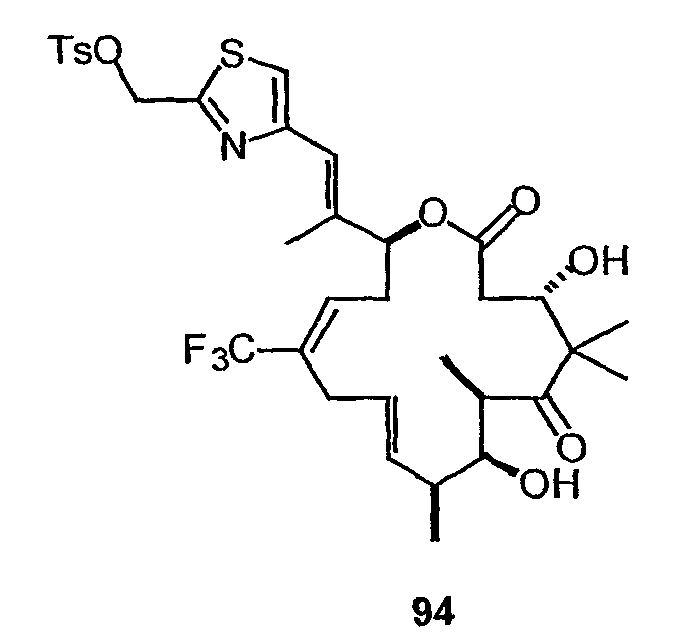
1H ЯМР (400 МГц, CDCl3) δ 1,06 (3H, c), 1,12 (3H, д, J=7,0 Гц), 1,23 (3H, д, J=6,7 Гц), 1,33 (3H, c), 1,99 (1H, д, J=5,5 Гц), 2,10 (3H, c), 2,25-2,34 (1H, м) 2,41 (1H, дд, J=15,5, 3,3 Гц), 2,47 (3H, c), 2,48 (1H, дд, J=15,7, 9,4 Гц), 2,51-2,63 (1H, м), 2,63 (1H, д, J=6,1 Гц, OH), 2,64-2,75 (1H, м), 2,91-3,05 (2H, м), 3,10 (1H, квинтeт, J=6,8 Гц), 3,70-3,75 (1H, м), 4,30 (1H, ддд, J=9,3, 6,1, 3,2 Гц), 5,32 (2H, c), 5,41 (1H, дд, J=5,8,4,5 Гц), 5,57 (1H, ддд, J=15,8, 6,4, 4,6 Гц), 5,65 (1H, дд, J=15,8, 6,0 Гц), 6,21 (1H, т, J=7,1 Гц), 6,59 (1H, c), 7,18 (1H, c), 7,37 (2H, д, J=8,1 Гц), 7,84 (2H, д, J=8,3 Гц); МС-НР (ESI) вычислено для С34Н42F3NO8S2Na[M+Na+] 736,2, найдено 736,3.

1H ЯМР (400 МГц, CDCl3) δ 1,06 (3Н, c), 1,12 (3H, д, J=6,9 Гц), 1,23 (3H, д, J=6,7 Гц), 1,34 (3H, c), 2,00 (1H, д, J=5,6 Гц, ОН), 2,15 (3Н, c), 2,25-2,35 (1H, м), 2,41 (1H, дд, J=15,5, 3,2 Гц), 2,49 (1H, дд, J=15,5, 9,4 Гц), 2,53-2,62 (1H, м), 2,69 (1H, д, J=6,1 Гц, ОН), 2,66-2,76 (1H, м), 2,92-3,05 (2Н, м), 3,11 (1H, квинтeт, J=6,4 Гц), 3,70-3,76 (1H, м), 4,32 (1H, ддд, J=9,2, 5,9, 3,1 Гц), 4,85 (2Н, c), 5,43 (1H, дд, J=6,0, 4,4 Гц), 5,59 (1H, ддд, J=15,9, 6,4, 4,5 Гц), 5,66 (1H, дд, J=15,9, 6,1 Гц), 6,23 (1H, т, J=6,8 Гц), 6,63 (1H, c), 7,20 (1H, c); МС-НР (ESI) вычислено для С27Н35ClF3NO5Sna [M+Na+] 600,2, найдено 600,2.

Соединение 99: К раствору 59 (6,9 мг, 12,3 мкмоль) в CH2Cl2 (0,4 мл) добавляли активированный MnO2 (приобретенный из Acros, 26,8 мг, 0,308 ммоль). После энергичного перемешивания при КТ в течение 4 час смесь фильтровали через подушку целита, которую промывали EtOAc. После концентрирования остаток очищали препаративной ТСХ (гексан/EtOAc = 1:1), получая альдегид 99 (2,7 мг, 4,84 мкмоль, 39%) в виде бесцветного твердого вещества; 1H ЯМР (400 МГц, CDCl3) δ 1,06 (3H, c), 1,13 (3H, д, J=7,2 Гц), 1,24 (3H, д, J=6,9 Гц), 1,35 (3H, c), 1,96 (1Н, д, J=5,6 Гц, ОН), 2,22 (3H, д, J=0,7 Гц), 2,25-2,35 (1H, м), 2,44 (1H, дд, J=15,4, 3,5 Гц), 2,46 (1H, д, J=5,9 Гц, OH), 2,51 (1H, дд, J=15,7, 9,3 Гц), 2,57-28 (1H, м), 2,68-2,79 (1H, м), 2,96-3,03 (2H, м), 3,10 (1H, квинтeт, J=6,8 Гц), 3,71-3,76 (1H, м), 4,31 (1H, ддд, J=9,4, 6,3, 3,5 Гц), 5,45 (1H, т, J=5,0 Гц), 5,53-5,63 (1H, м), 5,67 (1H, дд, J=15,7, 6,2 Гц), 6,24 (1H, т, J=6,6 Гц), 6,72 (1H, c), 7,57 (1H, д, J=0,9 Гц), 10,01 (1H, д, J=1,2 Гц).

Соединение 100: К раствору альдегида 99 (4,6 мг, 8,25 мкмоль) в CH3CN (0,5 мл) при 0°C добавляли MeNH2 (2,0 М в ТГФ, 41,3 мкл, 41,3 мкмоль). После перемешивания при 0°C в течение 15 мин добавляли NaBH3CN (1,0 М в ТГФ; 25 мкл; 25 мкмоль). После перемешивания при 0°C в течение 0,5 час добавляли AcOH (3 капли). После перемешивания при 0°C в течение 2 час добавляли 28% NH4OH (водный)(40 мкл) и смесь перемешивали при КТ в течение 10 мин. Смесь непосредственно дважды очищали препаративной ТСХ (CH2Cl2/MeOH = 100:9), получая 100 (2,4 мг, 4,19 мкмоль, 51%) в виде бесцветного твердого вещества; 1H ЯМР (400 МГц, CDCl3) δ 1,05 (3H, c), 1,12 (3H, д, J=7,0 Гц), 1,23 (3H, д, J=6,8 Гц), 1,34 (3H, c), 2,13 (3H, c), 2,25-2,34 (1H, м), 2,39 (1H, дд, J=15,3, 3,0 Гц), 2,49 (1H, дд, J=15,3, 9,7 Гц), 2,56 (3H, c), 2,54-2,64 (1H, м), 2,66-2,75 (1H, м), 2,89 (1H, д, J=5,1 Гц), 2,94-3,05 (2H, м), 3,11 (1H, квинтeт, J=6,8 Гц), 3,74 (1H, дд, J=6,6, 3,5 Гц), 4,08 (2H, c), 4,34 (1H, дд, J=9,6, 2,9 Гц), 5,43 (1H, дд, J=6,2, 4,1 Гц), 5,56-5,63 (1H, м), 5,66 (1H, дд, J=15,9, 5,7 Гц), 6,24 (1H, т, J=7,3 Гц), 6,66 (1H, c), 7,11 (1H, c); МС-НР (ESI) вычислено для С28Н40F3N2O5S [M+H+] 573,3, найдено 573,3.
Пример 10
Аналоги эпотилона, которые приводят к абляции ксенотрансплантированных опухолей до нерецидивирующего состояния
Комбинируя химический синтез, молекулярное моделирование и спектроскопические анализы, авторы обнаружили, что введение E-9,10-двойной связи (см. соединение 28 ниже) дает примерно 10-кратное повышение эффективности лекарственного средства в экспериментах на основе ксенотрансплантатов с использованием опухолей MX-1, резистентных к лекарственным средствам (A. Rivkin et al. J. Am. Chem. Soc. 2003, 125, 2899; включено в данное описание в виде ссылки). После сопоставления экспериментов in vitro и in vivo, направленных на опухоли типа MX-1, стало очевидным, что 28 по сути более цитотоксичен, чем 2b. Однако другим фактором, который вносит свой вклад, является то, что остаток лактона в ряду 9,10-дегидросоединений является значительно более стабильным в плазме мышей и человека, чем в случае с 9,10-дегидропредставителями того же ряда. Итогом указанных двух дополнительных эффектов было придание 28 способности осуществлять полное подавление опухоли в случае множества ксенотрансплантатов в дозе 3 мг/кг, в отличие от схемы с дозой 30 мг/кг в случае 1.

При прекращении обработки пальпируемые опухоли появляются снова у некоторой части животных. Таким образом, по меньшей мере в настоящее время полностью синтетический 28 не полностью соответствует строгим стандартам высоко предпочтительного эффективного терапевтического индекса и элиминации опухолей до нерецидивирующего состояния.
Указанные данные обратили внимание авторов на последствия замещения трех атомов водорода 26-метильной группы 28 тремя атомами фтора. Введение атомов фтора в данном положении приводит к повышенной стабильности 12,13-двойной связи по отношению к окислению (Smart, B. E. J. Fluorine Chem. 2001, 109, 3; публикация включена в данное изобретение в виде ссылки). Предшествующий опыт свидетельствовал о некотором ослаблении цитотоксичности при замене полярных групп в области C12-C13-двойной связи (A. Rivkin et al. J. Am. Chem. Soc. 2003, 125, 2899; публикация включена в данное описание в виде ссылки). В данном описании авторы сообщают о получении посредством общего химического синтеза 9,10-дегидро-26-трифторэпотилонов, особенно фокусируя внимание на уникальной биологической эффективности исходной структуры 29.
Внимательно изучали терапевтическую эффективность dEpoB (30 мг/кг), паклитаксела (20 мг/кг) и F3-deH-dEpoB (29; 20 и 30 мг/кг) против ксенотрансплантатов карциномы молочной железы человека MX-1 в отношении исчезновения опухолей и рецидива, и результаты показаны в таблице 10-1. Каждая группа дозирования состояла из четырех или более мышей nude. Масса тела относится к общей массе тела минус масса опухоли. В случае всех трех соединений достигалось исчезновение опухолей. На 10 день после прекращения лечения рецидивы происходили у 5/10 (dEpoB),
2/7 (паклитаксел) и 0/4 (соединение 29) мышей. Продленные наблюдения после прекращения лечения дозами 29, составляющими 20 мг/кг, показали долговременное отсутствие опухолей вплоть до 27 дня, и в это время 2 из 4 опухолей мышей рецидивировали. Примечательно, что лечение дозами 29, составляющими 30 мг/кг, приводило к полному исчезновению опухолей и отсутствию какого-либо рецидива в течение двух месяцев после прекращения лечения.
Терапевтическое действие dEpoB, паклитаксела и F
3
-deH-dEpoB против ксенотрансплантата MX-1 у мышей nude
[a]
средство
(мг/кг)
на 10 день после введения
после прекращения введения
[b] Регистрируемое повторное появление опухоли у 2/4 на 27-й день после прекращения лечения. Не наблюдалось дальнейшего повторного появления опухолей в течение 28-64-го дней после прекращения лечения.
[c] Отсутствие повторного появления опухолей в течение 64 дней после прекращения лечения, когда был закончен эксперимент.
Снижение дозы агента 29 до 10 мг/кг (Q2D) также приводило к исчезновению опухолей MX-1, но для достижения данного результата требовалось девять доз (фиг.57, 58 и 59A). В качестве дополнительной проверки химиотерапевтическое лечение задерживали вплоть до того, как размер опухоли достигал 0,5 г (~2,3% массы тела). Лечение дозами 29, составляющими 25 мг/кг (Q2Dx7), вызывало исчезновение 4/4 опухолей мышей. В отличие от этого требовались дозы dEpoB, составляющие 30 мг/кг (Q2Dx8), для того, чтобы индуцировать исчезновение опухолей у 3 из 4 мышей. Однако в отличие от случая с применением 29 кажущееся исчезновение, которое происходило после лечения dEpoB, подвергалось рецидивам с течением времени (фиг.59B).
Тот факт, что агент 29 полностью подавлял рост ксенотрансплантатов карциномы молочной железы человека MX-1, сокращал опухоли и вызывал их исчезновение в течение такого длительного периода времени как 64 дня, производит глубокое впечатление. Более того, после курсов лечения, проводимых с помощью 29 (20 мг/кг или 30 мг/кг Q2Dx6, 6-часовая в/в-инфузия, таблица 1 выше) масса тела организмов с ксенотрансплантатами возвращалась к контрольному уровню до лечения в течение 12-18 дней после прекращения лечения. Указанное наблюдение свидетельствует об отсутствии повреждения жизненно важных органов. При низких лечебных дозах 10 мг/кг, Q2DX12 (фиг.59B) максимальное снижение массы тела составляло только 12% с приростом массы тела на 6% во время последних трех доз. Масса тела возвращалась к контрольному уровню до лечения всего за три дня после прекращения лечения. В таблице 1 выше показано, что животные могут переживать потери массы тела до 27%. Достигаемый в данном случае запас терапевтической безопасности удивительно широк для лечебного противоопухолевого терапевтического средства.
Также оценивали терапевтическую эффективность 29 против ксенотрансплантата карциномы легкого человека (A549) и ксенотрансплантатов резистентной к паклитакселу карциномы легкого человека A549/Taxol (фиг.59C и 59D). Медленно растущие ксенотрансплантаты карциномы легкого A549 лечили 29 (25 мг/кг, Q2DX6, дважды, восемь дней отдельно), и это приводило к 99,5% подавлению опухолей в конце концов с полным искоренением 4 из 4 опухолей после дополнительных двух доз (фиг.59C). Интересно, что масса тела мышей снижалась до 35% без какой-либо летальности и прекращение лечения приводило к быстрому восстановлению массы тела почти до контрольного уровня до лечения (фиг.59C). В отличие от этого параллельное исследование с применением dEpoB (30 мг/кг, Q2Dx6) приводило к подавлению 97,6% опухолей, но не приводило к искоренению опухолей. В дополнительном исследовании 29 (доза 20 мг/кг) по отношению к резистентному ксенотрансплантату A549/Taxol (фиг.59D) рост опухолей полностью подавлялся и опухоли в конечном итоге уменьшались на 24,4% от контроля до лечения. В ходе данного исследования максимальная масса тела снижалась на 24%, однако после прекращения лечения лекарственным средством масса тела возвращалась до уровня 90% от контроля до лечения. В сравнительном исследовании (E)-9,10-дегидро-dEpoB (28, группа 4 мг/кг) рост опухолей подавлялся на 41,6%.
Данные, имеющие отношение к анализу того, какие факторы обеспечивают поразительный терапевтический индекс соединения 29, наряду с сопоставимыми данными, имеющими отношение к близко родственным представителям данного ряда, представлены в таблице 10-2. Отмечается, что в отношении присущей цитотоксичности при переходе от EpoB (2b) к dEpoB (1) теряется целый порядок значений. Примерно 60% указанной потери восстанавливается в случае 9,10-дегидро-dEpoB (28). Часть указанной присущей цитотоксичности утрачивается при переходе к соединению 29, которое в клетке имеет токсичность по меньшей мере в ~1,8 раз выше, чем в случае эталонного соединения dEpoB.
Авторы отмечают, что среди 12,13-дегидроэпотилонов, 29 проявляет намного лучшую стабильность в плазме мышей, а также является наиболее стабильным в плазме печени человека S9. Авторы также отмечают, что в 2-наборах 12,13-дегидроизомеров 26-трифтор-содержащий образец привносит пониженную липофильность и в некоторой степени повышенную растворимость в воде (таблица 10-2, ниже). Теперь может быть ясно, что большое преимущество 29 является результатом повышения стабильности в сыворотке и биодоступности.
Профиль производных dEpoB
снижение массы тела в % без гибели
[b] Ранжированный относительный терапевтический индекс (TI) при MTD (максимальной допустимой дозе):
+ Рост опухоли подавлен на 25-50%.
++ Рост опухоли подавлен на 50-100%.
+++ Опухоль сокращается, но не исчезает.
++++ опухоль исчезает у некоторых или у всех мышей nude с медленным восстановлением массы тела и/или с рецидивом у некоторых мышей в течение одной недели после прекращения лечения.
+++++ Опухоль исчезала у всех мышей nude, масса тела быстро восстанавливалась и/или не наступал рецидив.
Терапевтическое испытание эпотилонов против ксенотрансплантатов человека у мышей nude, таких как MX-1, исследовали в Chou, T. C. et al. Proc. Natl. Acad. Sci. USA. 1998, 95, 15798 и в 2001, 98, 8113.
Все агенты 1-2 и 28-29 впервые получены с использованием общего синтеза. Практический синтез 1 описан ранее (Rivkin et al. J. Am. Chem. Soc. 2003, 125, 2899; White et al. J. Am. Chem. Soc. 2001, 123, 5407; Yoshimura et al. Angew. Chem. 2003, 42, 2518; Rivkin et al., J. Org. Chem. 2002, 124, 7737; каждая публикация включена в данное описание в виде ссылки). Также впервые описаны пути создания 28 и 29, относящиеся к изобретательскому уровню. Избирательное восстановление 9,10-двойной связи 29 давало 2. Поразительные результаты, полученные на основе описанных выше исследований ксенотрансплантатов в отношении наиболее многообещающего в настоящее время соединения 29, несомненно требуют дальнейшего продвижения его подробного токсикологического и фармакокинетического исследований у более высокоорганизованных животных и от них, если это целесообразно, продвижения к клиническим испытаниям на человеке. Такие перспективы полностью изменили природу задачи синтеза от получения пробных образцов к задаче получения количеств указанных новых эпотилоновых агентов, измеряемых многими граммами. Осуществлена важная модернизация предыдущих способов авторов, впервые задуманная и показанная в условиях осуществления изобретения. В частности, в новых протоколах авторов осуществлены большие упрощения в стереоспецифическом конструировании в отношении атомов углерода 3 и 26. Спирт 32 получают, как описано ранее (Rivkin et al. J. Am. Chem. Soc. 2003, 125, 2899; включено в данное описание в виде ссылки). Будет отмечено, что в случае нового синтеза стереоцентры 6, 7 и 8 получают из заведомо доступных кетона 30 и альдегида 31. После защиты спирта и гидролиза ацеталя соответствующий альдегид конденсировали с трет-бутилацетатом, получая продукт, подобный альдолю. Так как данная конденсация не контролируется в отношении диастереомеров, требовалась и осуществлена корректирующая мера. Окисление данной смеси 1:1 C3-эпимеров давало кетон 69. После весьма успешного восстановлении Нойори (Noyori et al. J. Am. Chem. Soc. 1987, 109, 5856; публикация включена в данное описание в виде ссылки) в указанных условиях в наличии был спирт 70. Затем осуществляли получение кислоты 25 в несколько дополнительных простых стадий, которые показаны.
Схема 12. Синтез ацильного участка 25
Реагенты и условия: (a) (i) TrocCl, пиридин, 92%; (ii) п-TsOH·H2O, 76%; (iii) LDA, трет-бутилацетат, ТГФ, 80%; (iv) периодинан Десс-Мартина, 74%; (b) катализатор Нойори (10 моль.%), MeOH/HCl, H2, 1200 фунт./кв.дюйм, 80%. (c) (i) TESCl, имидазол, 77%; (ii) Zn, AcOH, ТГФ, 99%; (iii) TBSOTf, 2,6-лутидин, 82%; для остальных стадий см. Rivkin et al. J. Am. Chem. Soc. 2003, 125, 2899.
Новый прямой и легко масштабируемый синтез также разработан для 90 (схема 13). Синтез начинается с взаимодействия коммерчески доступного трифторкетоэфира 82 с аллилбромидом индия. Ключевой стадией в синтезе является специфичная по положению и стереоспецифичная дегидратация полученного в результате третичного спирта с получением 84 (с общим выходом для двух стадий 65%). Стерическое регулирование данной реакции является результатом «биполярного эффекта», при котором сильно оттягивающие электрон группы CF3 и CO2Et лучше всего представлены в транс-положении по отношению к возникающей двойной связи. Требуемый йодид 86 получали в две стадии из 84. Алкилирование енолята лития 7, описанного ранее, йодидом 86 в ТГФ давало 88 с выходом 81% и высокой диастереоселективностью (>25:1 de). После удаления защиты вторичного спирта соединение 88 в три стадии превращали в 90, как показано.
Схема 13. Синтез алкильного участка 17

Реагенты и условия: (a) (i) аллилбромид, In, ТГФ-вода (3:1) 48°C, 85%; SOCl2, пиридин 55°C, 77%; (b) (i) DIBAL-H, CH2Cl2, от -78°C до КТ 99%; (ii) I2, PPh3, имидазол, CH2Cl2, 74%; (c) (i) LHMDS, ТГФ, от -78°C до КТ; (ii) HOAc-ТГФ-H2O (3:1:1), 81% для двух стадий; (d) (i) AlMe3, MeONHMe, ТГФ, от 0°C до КТ, 97%; (ii) MeMgBr, ТГФ, 0°C, 53% (73% на основании извлеченного исходного вещества).
При наличии 25 и 90 в результате легко осуществляемой химии путь к 29 был понятен, если следовать протоколам, впервые разработанным авторами в фазе изобретения (A. Rivkin et al. J. Am. Chem. Soc. 2003, 125, 2899; включено в данное описание в виде ссылки). Ключевую реакцию метатезиса с замыканием цикла 25 осуществляли в толуоле, используя катализатор Груббса второго поколения (Grubbs, R. H.; Miller, S. J.; Fu, G. C. Acc. Chem. Res. 1995, 28, 446; Trnka, T.M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18; Alkene Metathesis in Organic Chemistry Ed.: Fürstner, A.; Springer, Berlin (1998); Fürstner, А. Angew. Chem. Int. Ed. Engl. 2000, 39, 3012; Schrock, R. R. Top. Organomet. Chem. 1998, 1, 1; каждая публикация включена в данное описание в виде ссылки). Реакция давала исключительно транс-изомер 48 с выходом 71%. После введения остатка тиазола посредством протокола, показанного на схеме 14, следовало удаление двух защитных групп силила с использованием HF-пиридина, тем самым приводя к соединению 29, которое затем превращали посредством восстановления 9,10-олефина в 2 с высоким выходом. Граммовые количества структурно новых эпотилонов получали общим синтезом в условиях лаборатории академического уровня.
Схема 14. Конечные стадии синтеза 26-CF 3 -(E)-9,10-дегидро-dEpoB (29)
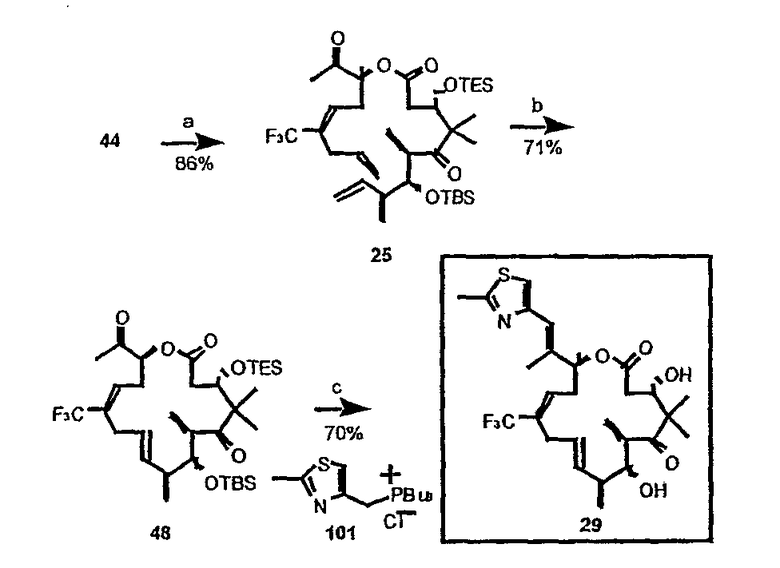
Реагенты и условия: (a) EDCI, DMAP, CH2Cl2, 25, от 0°C до КТ, 86% исходя из сложного трет-бутилового эфира; (b) катализатор Груббса, толуол, 110°C, 20 мин, 71%; (c) (i) KHMDS, 101, ТГФ, от -78°C до -20°C, 70%; (ii) HF-пиридин, ТГФ, 98%.
Эксперименты
Общие способы: Реагенты, полученные от коммерческих поставщиков, использовали без дополнительной очистки, если не оговорено особо. Метиленхлорид получали из безводной системы растворителей (пропущенной через колонку, предварительно заполненную окисью алюминия) и использовали без дальнейшей сушки. Все чувствительные к атмосфере и воде реакции осуществляли в высушенной в пламени стеклянной посуде при положительном давлении предварительно очищенного газа аргона. Спектры ЯМР (1H и 13C) регистрировали на Bruker AMX-400 МГц или Bruker Advance DRX-500 МГц, которые указаны отдельно, используя в качестве эталона CDCl3 (7,27 м.д. для 1H и 77,0 м.д. для 13C) или CD2Cl2 (5,32 м.д. для 1H и 53,5 м.д. для 13C). Инфракрасные спектры (ИК) получали на спектрометре Perkin-Elmer FT-IR модель 1600. Оптические вращения получали на цифровом поляриметре JASCO модели DIP-370. Аналитическую тонкослойную хроматографию проводили на пластинах E. Merck с силикагелем 60 F254. Соединения, которые не были УФ-активными, визуализировали, погружая пластины в раствор пара-анисальдегида и нагревая. Препаративную тонкослойную хроматографию осуществляли, используя указанный растворитель, на пластине для ТСХ Whatman® (LK6F силикагель 60A).
Химические вещества. Все эпотилоны синтезировали собственными силами (C. R. Harris, S. J. Danishefsky, J. Org. Chem. 1999, 64, 8434; D.-S. Su et al. Angew. Chem. Int. Ed. Engl. 1997, 36, 2093; Smart, B. E. J. Fluorine Chem. 2001, 109, 3; F. Yoshimura, et al. Angew. Chem. 2003, 42, 2518; Rivkin et al. J. Org. Chem. 2002, 124, 7737; каждая публикация включена в данное описание в виде ссылки). Паклитаксел (Taxol®) и сульфат винбластина (VBL) приобретали из Sigma. Все указанные соединения растворяли в диметилсульфоксиде для анализов in vitro (за исключением VBL, который растворяли в физиологическом растворе). Для исследование in vivo все эпотилоны и паклитаксел растворяли в наполнителе кремофор/этанол (1:1) и затем разбавляли физиологическим раствором для в/в-инфузии в течение 6 час через хвостовую вену, используя изготовленный по заказу миникатетер (T.-C. Chou et al. Proc. Natl. Acad. Sci. USA. 2001, 98, 8113-8118; T.-C. Chou et al. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 15798-15802; каждая публикация включена в данное описание в виде ссылки).
Опухоли и линии клеток. Клетки лимфобластного лейкоза человека CCRF-CEM получали от Dr. William Beck, the University of Illinois, Chicago. Клетки карциномы молочной железы человека (MX-1) и карциномы легкого человека (A549) получали из американской коллекции типов культур (ATCC, Rockville, MD). Резистентные к паклитакселу клетки A549/taxol (44-кратная резистентность) создавали способом, описанным выше (T.-C. Chou et al. Proc. Natl. Acad. Sci. USA. 2001, 98, 8113-8118; публикация включена в данное описание в виде ссылки).
Животные. Бестимусных мышей nude, несущих ген nu/nu, получали из NCI, Frederick, MD и использовали для всех ксенотрансплантатов опухолей человека. Использовали самцов мышей 6-недельного возраста или старше, весящих 20-22 г. Лекарственные средства вводили через хвостовую вену в течение 6 часов в/в-инфузией, используя миникатетер для инфузии собственного изготовления и герметичную пробирку (T.-C. Chou et al. Proc. Natl. Acad. Sci. USA. 2001, 98, 8113-8118; публикация включена в данное описание в виде ссылки). Для в/в-инфузии использовали многоканальный программируемый шприцевый насос. Обычный объем при 6-часовой инфузии для каждого лекарственного средства в смеси кремофор/этанол (1:1) составлял 100 мл в 2,0 мл физиологического раствора. Объем опухоли оценивали измерением длины × ширину × высоту (или ширину), используя штангенциркуль. В случае несущих опухоли мышей nude в ходе эксперимента масса тела относится к общей массе минус масса опухоли. Все исследования на животных проводились в соответствии с правилами руководства по уходу и использованию животных Национального института здравоохранения и протоколом, одобренным Институтским комитетом по уходу и использованию животных Memorial Sloan-Kettering Cancer Center.
Анализы цитотоксичности. В ходе подготовки для анализов цитотоксичности in vitro клетки культивировали с начальной плотностью 2-5×104 клеток в миллилитре. Их поддерживали во влажной атмосфере с 5% CO2 при 37°C в среде RPMI 1640 (GIBCO/BRL), содержащей пенициллин (100 единиц/мл), стрептомицин (100 мкг/мл, GIBCO/BRL), и 5% инактивированной нагреванием FBS. Для растущих в монослое клеток солидных опухолей (таких как A549) цитотоксичность лекарственного средства определяли в 96-луночных планшетах для микротитрования, используя способ на основе сульфородамина B (P. Skehan et al. J. Natl. Cancer. Inst. 1990, 82, 1107-1112; включено в данное описание в виде ссылки). Для клеток, выращенных в суспензии (таких как CCRF-CEM и их сублинии) цитотоксичность измеряли в двух повторах, используя способ для микрокультуры на основе гидроксида 2,3-бис-(2-метокси-4-нитро-5-сульфофенил)-5-карбоксанилид)-2H-тетразония (XTT) (D. A. Scudiero et al. Cancer Res. 1988, 48, 4827-4833; публикация включена в данное описание в виде ссылки), в 96-луночных планшетах для микротитрования. В случае обоих способов оптическую плотность каждой лунки измеряли с помощью считывающего устройства для микропланшетов (Power Wave XS, Bio-Tek, Winooski, VT). Данные зависимости доза-эффект, полученные для 6-7 концентраций каждого лекарственного средства в двух повторах, анализировали на основе графика медиан эффекта, используя компьютерную программу (T.-C. Chou, M. Hayball. CalcuSyn for Windows, Multiple-drug dose effect analyzer and manual. Biosoft, Cambridge Place, Cambridge, UK (1997); включено в данное описание в виде ссылки).
Стабильность эпотилонов у мышей и во фракции S9 печени человека. Исследование стабильности осуществляли с использованием полностью автоматизированной системы ВЭЖХ, которая состояла из системы приготовления образца Prospekt-2 (Spark Holland, Netherlands) и системы ВЭЖХ Agilent 1100. Коротко, Prospekt 2 захватывал картридж для экстракции C8 и промывал его ацетонитрилом и водой. Автоматический пробоотборник Agilent, установленный на 37°C, отбирал 20 мкл образца, наносил его на картридж, промывал его водой, затем Prospekt-2 направлял поток подвижной фазы через картридж для экстракции на аналитическую колонку Reliance Stable Bond C8 4×80 мм с защитной колонкой (MacMod, Chadds Ford, PA) и элюент контролировали при 250 нм. Подвижная фаза состояла из смеси 53 или 65% ацетонитрила/0,1% муравьиной кислоты при расходе 0,4 мл/мин, так что время удержания представляющего интерес соединения составляло примерно 6 минут. Приготовление образца заключалось в добавлении равных объемов плазмы к PBS до общего объема 300-400 мкл, фильтрование и добавление 0,5-2 мкл субстрата (20 мМ) до достижения примерно 30-50 мАU при 250 нм в анализе ВЭЖХ. Для объединенной фракции микросом печени человека S9 (Xeno Tech, Lenex, KS), 20 мкл (400 мкг) фракции S9 смешивали с 280 мкл PBS, затем действовали, как описано выше. Период отбора образца контролировали автоматическим пробоотборником и собирали данные площади пика, чтобы сравнить скорость исчезновения исходного соединения.
Определение коэффициента распределения октанол-вода (POW). Используют способ ВЭЖХ, чтобы оценить распределение октанол-вода. Используют систему ВЭЖХ Agilent 1100 с колонкой Eclipse XDB C18 4,6×250 мм с подвижной фазой в виде смеси 60% ацетонитрил/40% 25 мМ калий-фосфатный буфер при pH 7,4 и скоростью потока 0,8 мл в мин, и элюент контролируют при 250 нм. Используемыми стандартами являются бензиловый спирт, ацетофенон, бензофенон, нафталин, дифениловый эфир и дибензил с известными POW 1,1; 1,7; 3,2; 4,2 и 4,8, соответственно. Используют дихромат натрия, чтобы оценить начало отсчета, которое составляет 2,5 мин, и время удержания для стандартов составляет 3,9; 5,4; 10,6; 14, 18,7 и 19,8 мин, соответственно. Значение k value рассчитывают по формуле k=(trt-t0)/t0. Линейная регрессия log k против log POW дает прямую линию с r2 = 0,966. Данный график используют для оценки значения POW аналогов эпотилона.
Спектроскопические данные для 29 (26-трифтор-(E)-9,10-дегидро-dEpoB):
[α]D 25 -54,6 (с 0,28, СНСl3); ИК (пленка) ν 3478, 2974, 2929, 1736, 1689, 1449, 1381, 1318, 1247, 1169, 1113, 1039, 983, 867, 736 cм-1; 1H ЯМР (400 МГц, CDCl3) δ 1,05 (3H, c), 1,12 (3H, д, J=7,0 Гц), 1,23 (3H, д, J=6,8 Гц), 1,37 (3H, c), 2,04 (1H, ушир.д, J=3,8 Гц, -OH), 2,12 (3H, c), 2,25-2,33 (1H, м), 2,38 (1H, дд, J=15,3 и 3,0 Гц), 2,48 (1H, дд, J=15,4 и 9,8 Гц), 2,54-2,61 (1H, м), 2,66-2,76 (1H, м), 2,71 (3H, c), 2,96 (1H, дд, J=16,5 и 4,5 Гц), 3,02 (1H, дд, J=16,3 и 6,5 Гц), 3,11 (1H, квинтeт, J=6,7 Гц), 3,19 (1H, ушир.c, =OH), 3,74 (1H, ушир.c), 4,35 (1H, ушир.д, J=9,5 Гц), 5,42 (1H, дд, J=6,2 и 4,1 Гц), 5,60 (1H, ддд, J=15,8, 5,6, и 4,5 Гц), 5,66 (1H, дд, J=15,8 и 5,8 Гц), 6,24 (1H, т, J=7,2 Гц), 6,64 (1H, c), 7,00 (1H, c); 13C ЯМР (100 МГц, CDCl3) δ 15,1, 16,1, 17,7, 18,5, 19,3, 22,5, 28,8, 31,1, 39,6, 39,7, 45,0, 53,7, 71,4, 75,3, 76,8, 116,7, 120,2, 124,3 [кв, 1 J(C,F) = 273,4 Гц], 127,9, 130,2 [кв, 3 J(C,F) = 6,0 Гц], 130,6 [кв, 2 J(C,F) = 28,4 Гц], 132,5, 136,7, 152,0, 165,4, 170,2, 218,4; МС-НР (ESI) вычислено для С27Н37F3NO5S [M+H+] 544,2, найдено 544,1.
Пример 11
Исследования in vitro
Типичный эксперимент включает в себя культивирование клеток (например, CCRF-CEM) с начальной плотностью 2-5×104 клеток в мл. Их поддерживают во влажной атмосфере с 5% CO2 при 37°C в среде RPMI 1640 (GIBCO/BRL), содержащей пенициллин (100 единиц/мл), стрептомицин (100 мкг/мл) (GIBCO/BRL) и 5% инактивированной нагреванием фетальной сыворотки теленка. В случае клеток, которые выращивали в суспензии (таких как CCRF-CEM и ее сублинии), цитотоксичность измеряют, используя тетразониевый способ для микрокультур на основе гидроксида 2,3-бис(2-метокси-4-нитро-5-сульфофенил)-5-карбоксанилид)-2H-тетразония (XTT), в двух повторах в 96-луночных планшетах для микротитрования. В случае обоих способов оптическую плотность измеряют с использованием считывающего устройства для микропланшетов (EL-340, Bio-Tek, Burlington, VT). Каждая серия содержит шесть или семь концентраций тестируемых лекарственных средств. Данные взаимосвязи доза-эффект анализируют на основе графика медиан эффекта.
T-клетки человека CCRF-CEM, клетки острого лимфобластного лейкоза, их резистентную к тенипозиду подлинию (CCRF-CEM/VM1) и резистентную к винбластину подлинию (CCRF-CEM/VBL100) получены от W. T. Beck (University of Illinois, Chicago, I1).
В типичном эксперименте, который в общем описан выше, некоторые предлагаемые в изобретении соединения (например, 9,10-дегидро-EpoD) проявляли активность в линиях клеток CCRF-CEM и линиях клеток CCRF-CEM, резистентных к таксолу. Некоторые из указанных соединений проявляют IC50 в диапазоне от 0,0015 до примерно 0,120 на линиях клеток CCRF-CEM. Некоторые другие соединения проявляют IC50 в диапазоне от 0,0015 до примерно 10,5. Некоторые из указанных соединений также проявляют IC50 в диапазоне от 0,011 до примерно 0,80 на линия резистентных клеток CCRF-CEM/Taxol, а некоторые другие соединения проявляют IC50 в диапазоне примерно от 0,011 до 13,0 мкМ. В некоторых вариантах 26F-EpoD проявляет активности в пределах 0,0015 мкМ на линиях клеток CCRF-CEM и в пределах 0,011 мкМ на линиях резистентных клеток CCRF-CEM/Taxol (фиг.11).
Пример 12
Исследования in vivo
Обычно для ксенотрансплантатов опухолей использовали бестимусных мышей nude, несущих ген nu/nu. Аутбредных мышей, происходящих от Swiss, получали из Charles River Laboratories. Для большинства экспериментов использовали самцов мышей 8-недельного возраста или старше, весящих 22 г. Лекарственное средство вводили через хвостовую вену посредством 6-часовой в/в-инфузии. Каждую отдельную мышь удерживали сосуде для иммобилизации из полипропиленовой пробирке Falcon с отверстиями для введения лекарственного средства. Объем опухоли оценивали измерением длины × ширину × высоту (или ширину), используя штангенциркуль. Для в/в-инфузии использовали многоканальный программируемый шприцевый насос Harvard PHD2000 (Harvard Apparatus). Все исследования на животных проводили в соответствии с правилами «Руководства по уходу и использованию животных» Национального института здравоохранения и протоколом, одобренным Институтским комитетом по уходу и использованию животных Memorial Sloan-Kettering Cancer Center. В соответствии с нормативами комитета в отношении гуманной обработки несущих опухоли животных мышей подвергали эвтаназии, когда опухоли достигали >10% их общей массы тела.
Как изображено на фиг.8, 9,10-дегидро-EpoB тестировали у мышей nude, несущих карциному молочной железы человека MX-1. В общем 9,10-дегидро-EpoB готовили следующим образом: 9,10-дегидро-EpoB растворяли в этаноле и добавляли кремофор (1:1) в концентрации 20 мг/мл. Данный раствор разбавляли физиологическим раствором для в/в-инфузии. Разбавленный раствор использовали для в/в-инфузии в течение одного часа. Затем измеряли размер опухоли и массу тела, используя дозы 10 мг/кг, 20 мг/кг и 30 мг/кг на протяжении 15 дней. Размер опухоли и массу тела также измеряли, используя схему дозирования 0,4 мг/кг Q3Dx2, 0,5 мг/кг Q3Dx2 и 0,6 мг/кг Q3Dx5 (см. фиг.33, 34, 55 и 56). Использовали схему введения дозы на каждый третий день, чтобы уменьшить токсичность. Другие терапевтические исследования 9,10-дегидро-EpoB показаны на фиг.70 и 71 (CCRF-CEM/Taxol Q3Dx5) и на фиг.23 и 24 (HCT-116, Q2Dx7).
Соединение 9,10-дегидро-12,13-дезоксипотилон B (изо-490-эпотилон) является в три раза более эффективным, чем dEpoB. Показано, что 9,10-дегидро-12,13-дезоксиэпотилон D тормозит рост опухоли после двух-трех инфузий в дозах 10 мг/кг или 20 мг/кг, каждую из которых вводили через день. Лучше результаты получены у мышей с использованием дозы 30 мг/кг 9,10-дегидро-12,13-дезоксиэпотилона B с использованием 6-часовой инфузии в/в через день. Также показано, что в случае 9,10-дегидро-dEpoB в дозе 5 мг/кг, Q3Dx9, 6-часовой в/в-инфузии, достигается исчезновение опухоли у мышей nude, несущих ксенотрансплантат MX-1, без гибели мышей и только с умеренной потерей массы тела (фиг.74 и 75). По-видимому, это осуществлено благодаря введению аналогов эпотилона каждый третий день, чтобы уменьшить токсичность (см. фиг.53 и 54). В заключение, 9,10-дегидро-12,13-дезоксиэпотилон B проявляет пониженную токсичность по сравнению с другими эпотилонами, более высокую эффективность в задержке опухолевого роста и более высокую стабильность в сыворотке. Другие терапевтические исследования показаны на фиг.17 и 18 (HCT-116, Q2Dx5 и Q3Dx5); на фиг.19 и 20 (A549/Taxol, Q3Dx7); и на фиг.21 и 22 (A549/Taxol, Q2Dx7).
9,10-дегидро-Epo B при введении каждый третий день 9-11 раз 6-часовой в/в-инфузией в дозе 0,4-0,6 мг/кг приводил к сокращению и исчезновению опухоли у мышей nude с имплантированными ксенотрансплантатами карциномы молочной железы человека MX-1 (фиг.68 и 69). Введение через день 8 доз приводило к подавлению роста опухоли, но не к сокращению опухоли. Когда 9,10-дегидро-Epo B вводили через день в количестве 9 доз, имплантированная опухоль продолжала умеренно сокращаться со второго по восьмой день, но масса тела восстанавливалась очень медленно от 76% до 82% от контроля в течение того же периода времени. На десятый день одна четверть опухоли исчезала. Когда вводили дозу 0,6 мг/кг 9,10-дегидро-EpoB, Q2Wx6, 6-часовой инфузией мышам nude с ксенотрансплантатами HCT-116, четыре из четырех мышей погибали от токсичности в течение трех дней после шести доз. 9,10-дегидро-EpoB прекращал рост опухоли CCRF-CEM/Taxol при использовании 0,6 мг/кг, схема Q3Dx5, x2 (фиг.70 и 71).
26-Трифтор-9,10-дегидро-12,13-дезоксиэпотилон B (F3-deH-dEpoB), как показано на фигурах, оказывает целебное действие в дозах 20мг/кг и 30 мг/кг, Q2Dx6, 6-часовая инфузия, у модели на мышах nude, имплантированных ксенотрансплантатами карциномы молочной железы человека MX-1. Данные также свидетельствуют о том, что 30 мг/кг, Q2Dx6 приближенно является максимальной переносимой дозой. В дозе 20 мг/кг, Q2Dx6, 6-часовая инфузия, 26-трифтор-9,10-дегидро-12,13-дезоксиэпотилон B приводил к сокращению и исчезновению опухоли у четырех из четырех мышей nude с ксенотрансплантатами карциномы молочной железы человека MX-1. Не наблюдалось повторного появления опухоли на 20-й день после прекращения лечения. На 27-й день после прекращения лечения повторно появлялись 2/4. Не было дальнейшего повторного появления опухолей в течение 28-64 дней после прекращения лечения. В сравнении в случае dEpoB в дозе 30 мг/кг достигалось исчезновение опухоли в такой же мышиной модели у семи из семи мышей; однако опухоль повторно появлялась у 2 из пяти мышей на 8 день после прекращения лечения. Введение 26-трифтор-9,10-дегидро-12,13-дезоксиэпотилона B в дозе 20 мг/кг, Q2Dx6, 6-часовая в/в-инфузия, приводило к временному снижению массы тела мышей до 26%. Это уменьшение массы тела не приводило к гибели, свидетельствуя об отсутствии тяжелой токсичности по отношению к жизненно важным органам. Через два дня после последней обработки масса тела начинала восстанавливаться. На 16 день после обработки масса тела восстанавливалась до 109% от контроля до лечения, что свидетельствует о том, что токсичность, если она имеется, полностью обратима. В сравнении dEpoB, вводимый в дозе 30 мг/кг, приводил к 31% снижению массы тела без летальности.
В том случае, когда 26-трифтор-9,10-дегидро-12,13-дезоксиэпотилон B вводили в дозе 30 мг/кг, Q2Dx6, 6-часовая в/в-инфузия, исчезновение опухоли происходило на 2-3 дня раньше, чем при дозе 20 мг/кг. В случае данной более высокой дозы масса тела уменьшалась на 27% и удерживалась 4 дня, не приводя к летальности, что подтверждает отсутствие тяжелой токсичности по отношению к жизненно важным органам. Через четыре дня после последней обработки в дозе 30 мг/кг масса тела начинала восстанавливаться. На 16-й день после обработки масса тела восстанавливалась до 98% от контроля до лечения, что снова подтверждает обратимость токсичности. Лечение 26-трифтор-9,10-дегидро-dEpoB в дозе 20 мг/кг и 30 мг/кг приводило к полному исчезновению опухолей, и при дозе 30 мг/кг не наблюдалось рецидива спустя 62 дня. Исчезновение опухолей также достигалось в случае 10 мг/кг при введении 9 доз, когда давали три дополнительные дозы (фиг.57). При дозе 26-трифтор-9,10-дегидро-dEpoB в дозе 10 мг/кг наблюдалась только небольшая потеря массы тела (фиг.58). Не наблюдалось дальнейшей потери массы тела при продолжительном лечении.
На фиг.59 суммированы эффекты 26-F3-9,10-deH-dEpo B (и других эпотилонов) против ксенотрансплантата MX-1, A. При низкой дозе; B. Против крупной опухоли; против ксенотрансплантата карциномы легкого A549, C; и против резистентного к таксолу ксенотрансплантата карциномы легкого A549/Taxol, D.
На фиг.61 приведен список, указывающий эффективность in vitro C-21-модифицированных эпотилонов в отношении CCRF-CEM, CCRF-CEM/VBL и CCRF-CEM/Taxol.
На фиг.62 показано терапевтическое действие 26-F3-9,10-deH-dEpoB (15 мг/кг и 30 мг/кг) и таксола (20 мг/кг) Q2Dx8, 6-часовая в/в-инфузия, против ксенотрансплантата T-клеточного лимфобластного лейкоза человека CCRF-CEM. Сходное снижение массы тела наблюдали во всех трех группах обработки (фиг.63).
При лечении ксенотрансплантата CCRF-CEM/Taxol (резистентного к таксолу) 26-F3-9,10-deH-dEpo B в дозе 15 мг/кг достигалось исчезновение 1/3 опухолей, а в дозе 30 мг/кг достигалось исчезновение 3/4 опухолей. Такое же лечение таксолом в дозе 20 мг/кг давало только частичное подавление роста опухолей и не удавалось достичь сокращения опухоли (фиг.64). Изменения массы тела в ходе данного эксперимента показаны на фиг.65.
При лечении ксенотрансплантата карциномы ободочной кишки человека HCT-116 26-F3-9,10-deH-dEpo B (20 мг/кг) достигали сходной эффективности как и в случае таксола (20 мг/кг). Однако F3-deH-dEpo B в дозе 30 мг/кг оказывал терапевтический эффект лучше с исчезновением 2/4 опухолей после 5 доз (фиг.66). Изменения массы тела в ходе данного эксперимента показаны на фиг.67.
Терапевтические эффекты F3-9,10-дегидро-dEpoF против ксенотрансплантатов MX-1 при различных дозах (5-30 мг/кг) в случае 6-часовой в/в-инфузии и в/в-инъекции показаны на фиг.76 и 77.
Заключение. 9,10-дегидро-, 26-трифтор- или обе модификации dEpoB приводят к 1,5-5-кратному увеличению цитотоксичности in vitro и 2-5-кратному увеличению периода полужизни в плазме мышей in vitro. Используя модели ксенотрансплантатов солидных опухолей человека у мышей nude и используя методику Q2Dx5~9, 6-часовой в/в-инфузии через хвостовую вену при максимальных переносимых дозах, оценивали противоопухолевую эффективность и токсичность 9,10-дегидроэпотилонов. Способность достигать полного подавления роста опухолей, сокращения и исчезновения опухолей способствовала проведению дальнейших исследований для того, чтобы определить частоту рецидивов и показатель эффективности лечения после прекращения лечения. 9,10-дегидро-EpoB, наиболее эффективный in vitro известный эпотилон, хотя и был высоко эффективным, но показал узкие пределы терапевтической безопасности in vivo. 9,10-дегидро-dEpoB в дозе 4 мг/кг, 9,10-дегидро-EpoB в дозе 0,4 мг/кг и 21-гидрокси-9,10-дегидро-dEpoB в дозе 3 мг/кг - все сильно подавляли рост опухоли в течение длительного периода времени и достигали некоторого сокращения опухолей, а иногда достигали исчезновения опухолей. dEpoB в дозе 30 мг/кг, 26-трифтор-9,10-дегидро-dEpoB в дозе 20 мг/кг и паклитаксел в дозе 20 мг/кг - все демонстрировали сильное подавление роста опухолей и достигали сокращения и исчезновения опухолей ксенотрансплантатов карциномы молочной железы человека MX-1 у всех тестированных мышей. 26-трифтор-9,10-дегидро-dEpoB по сравнению с dEpoB или паклитакселом достигал долговременного излечения без рецидива опухоли и в равной мере демонстрировал быстрое восстановление массы тела до контрольного уровня до лечения.
Пример 13
Синтез аналогов циклопропилэпотилона
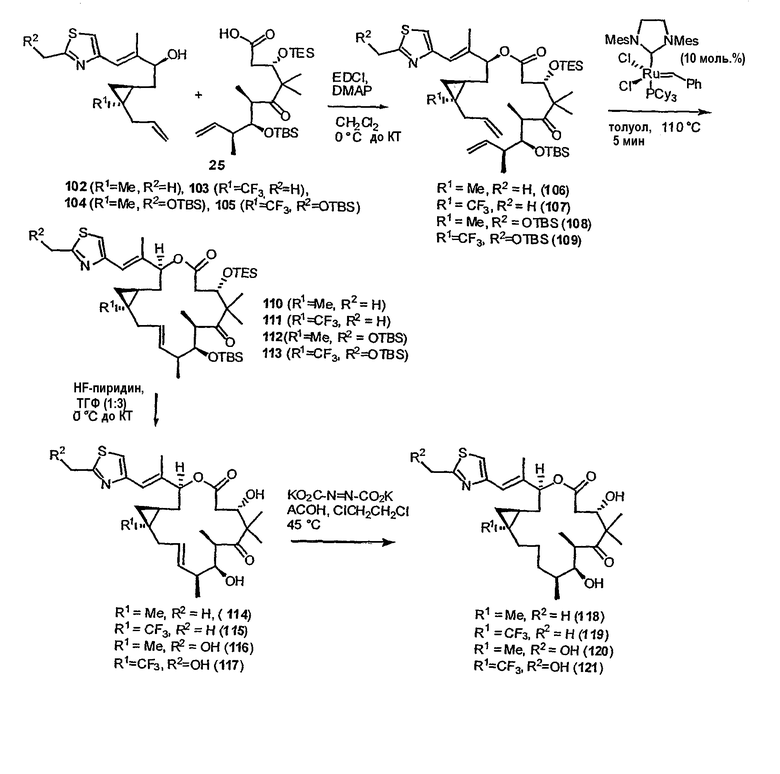
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ ЭПОТИЛОНОВ, ИХ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ, АНАЛОГОВ И ИХ ПРИМЕНЕНИЯ | 2003 |
|
RU2311415C2 |
| ПРОИЗВОДНЫЕ ТАКСОЛА С ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2419622C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2016 |
|
RU2718915C2 |
| ПРОИЗВОДНОЕ ХИНАЗОЛИНОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЯ | 2017 |
|
RU2730500C2 |
| ПРОИЗВОДНЫЕ СУЛЬФОНИЛМОЧЕВИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ПРОИЗВОДНЫХ СУЛЬФОНИЛМОЧЕВИНЫ, ОБЛАДАЮЩАЯ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1994 |
|
RU2127259C1 |
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСИВЕ ТЕРАПЕВТИЧЕСКИХ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2011 |
|
RU2518069C1 |
| ПИРРОЛЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ИНДОЛОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ВКЛЮЧАЮЩАЯ ЕГО КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2015 |
|
RU2650682C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| 6,7-МОДИФИЦИРОВАННЫЕ ПАКЛИТАКСЕЛЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1993 |
|
RU2125998C1 |
| ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА, НАЦЕЛЕННЫЕ НА УКОРОЧЕННЫЕ БЕЛКИ АДЕНОМАТОЗНОГО ПОЛИПОЗА ТОЛСТОГО КИШЕЧНИКА (АРС) | 2014 |
|
RU2727516C2 |
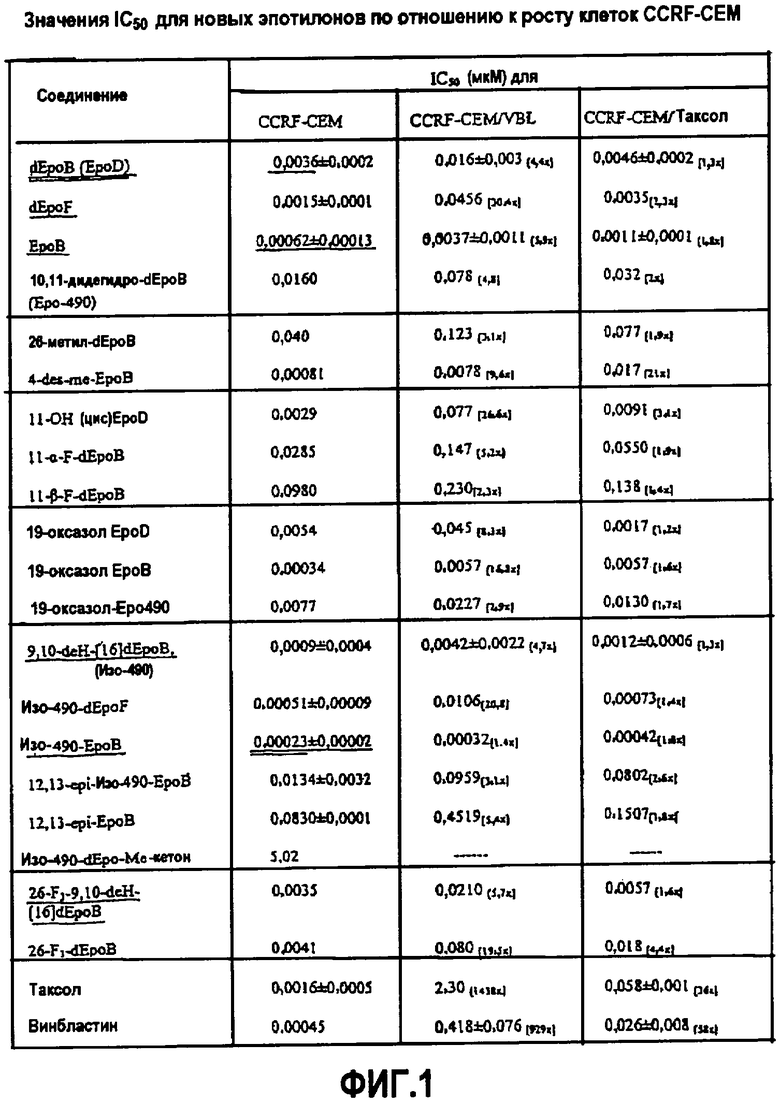


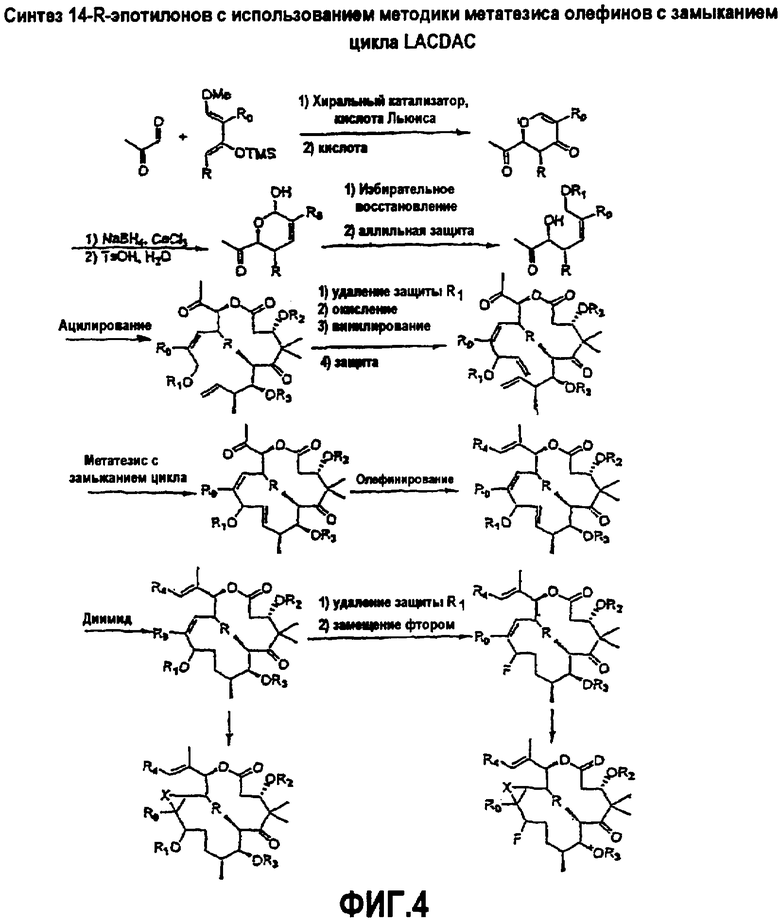

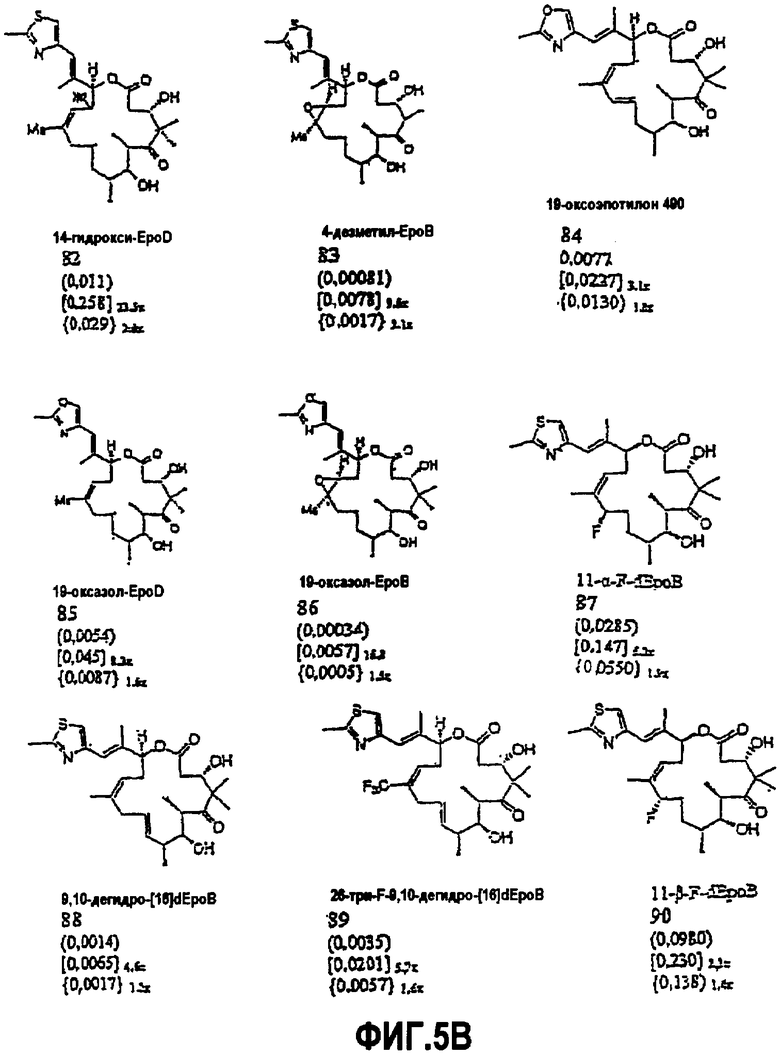
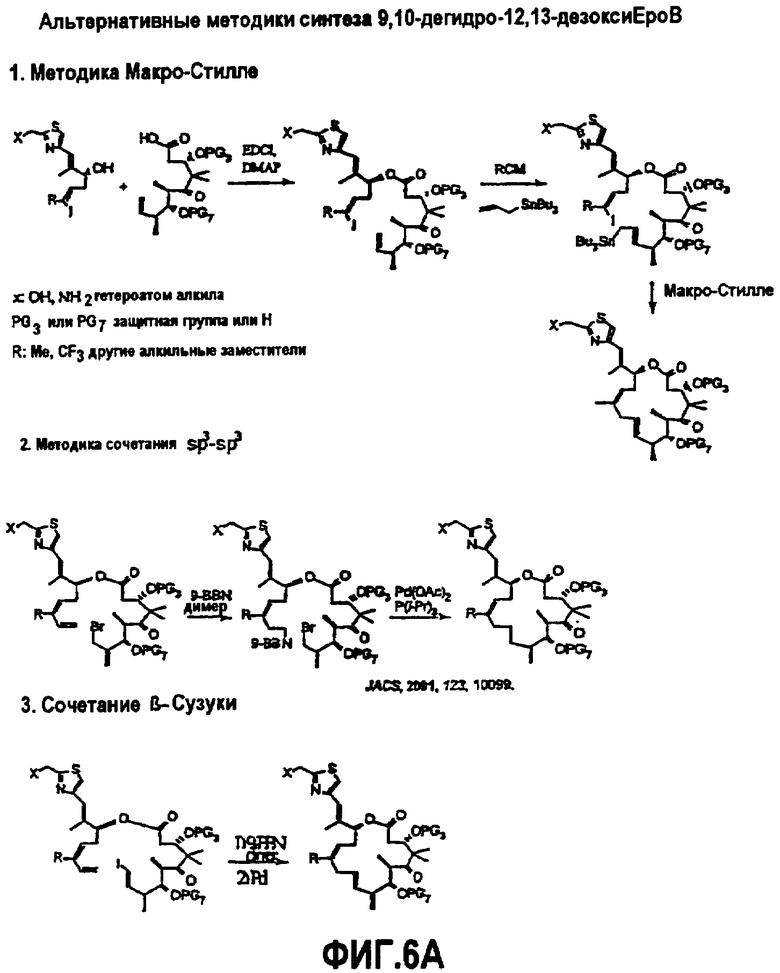
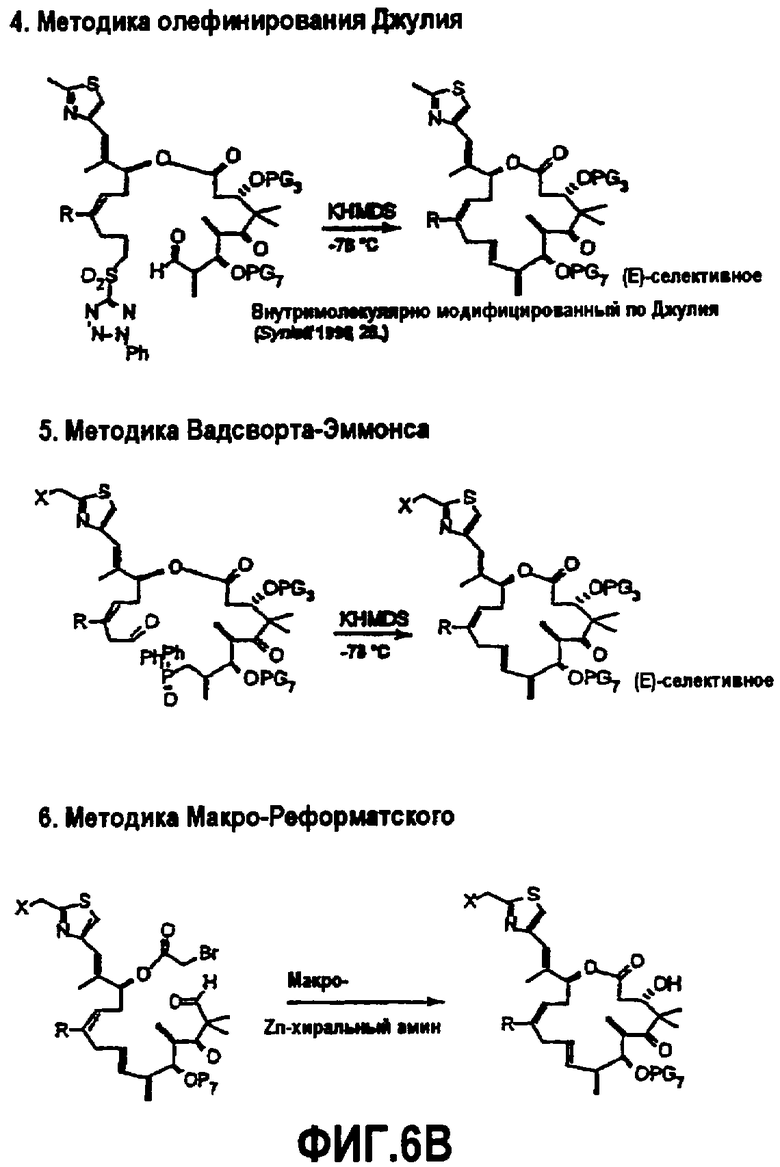
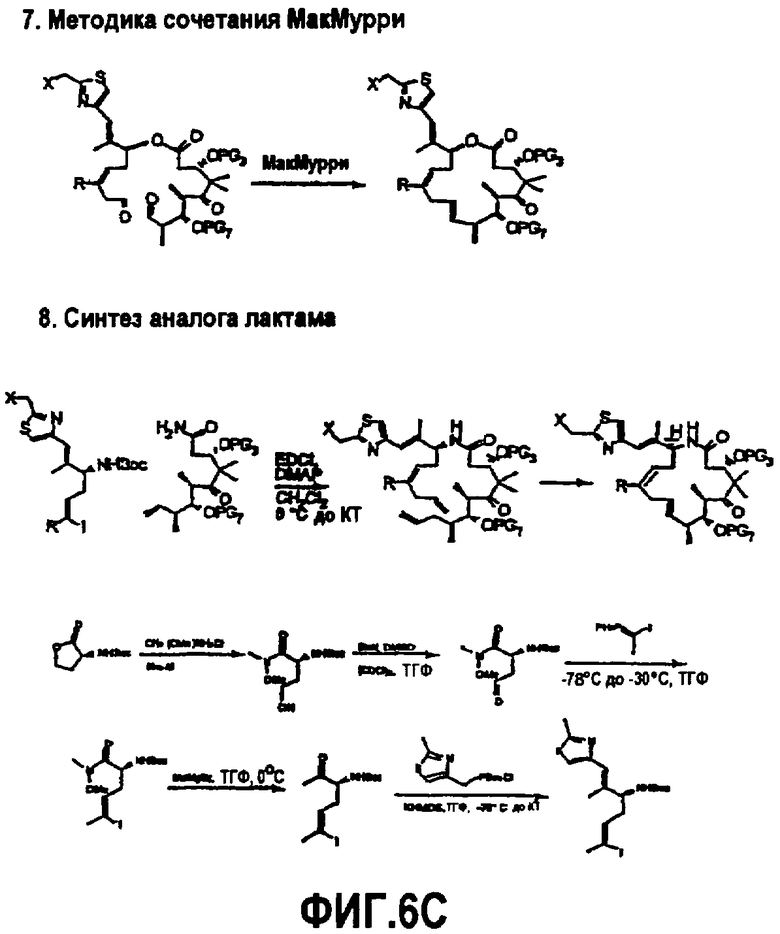

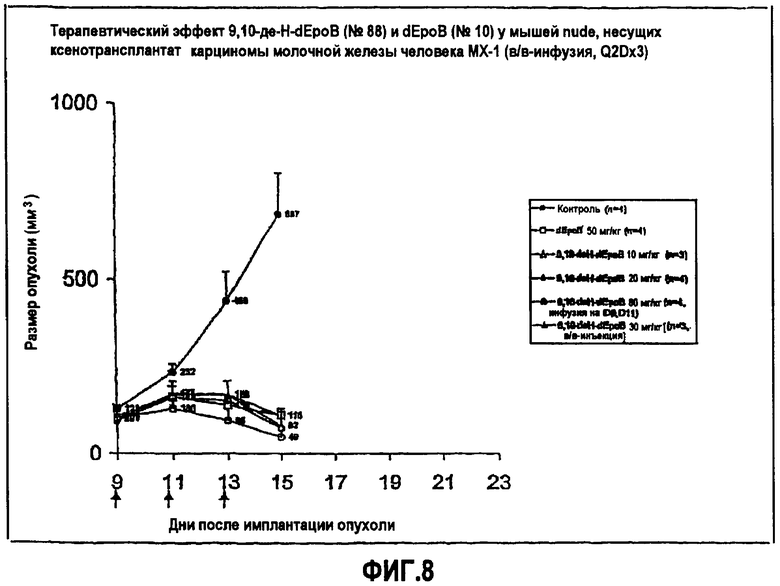

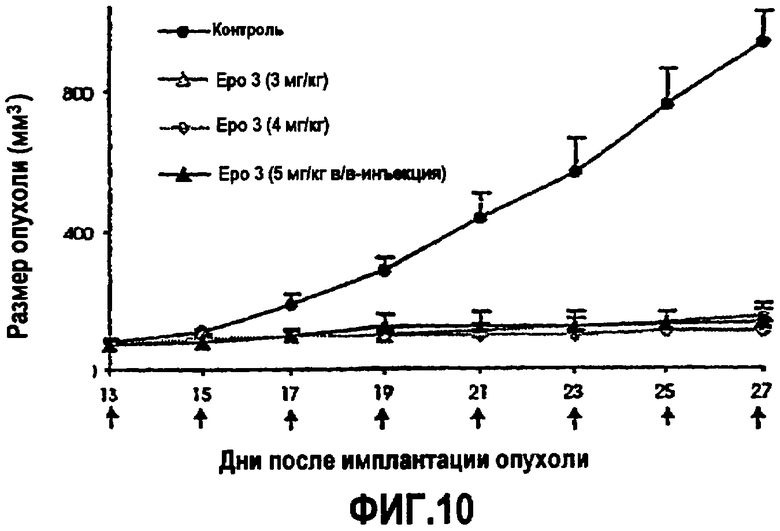
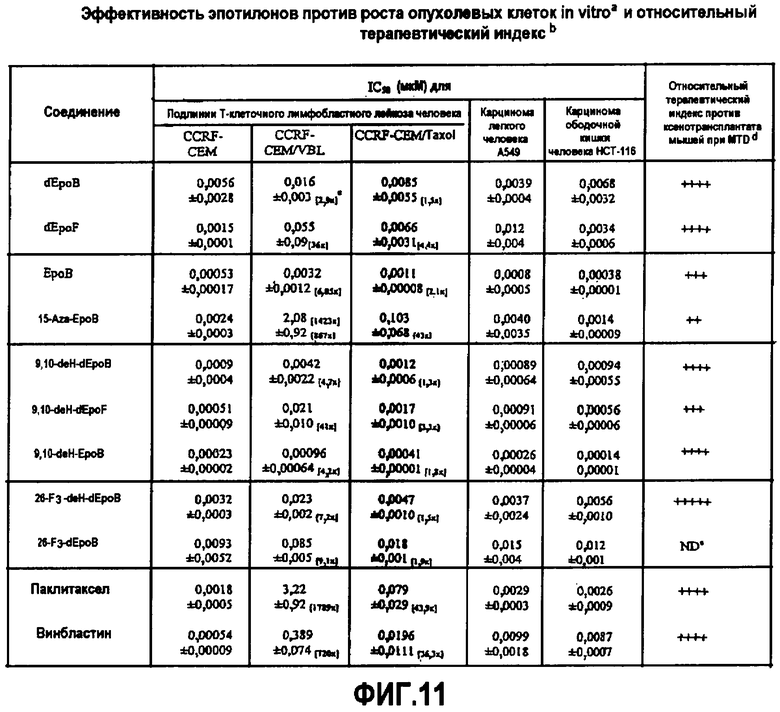

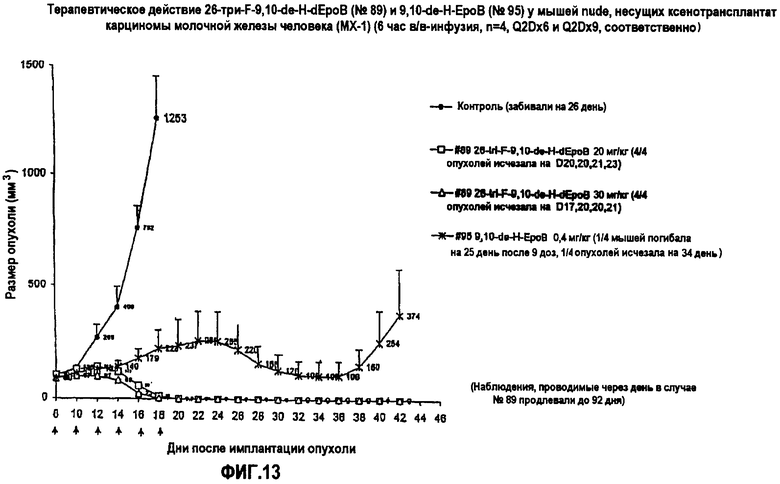
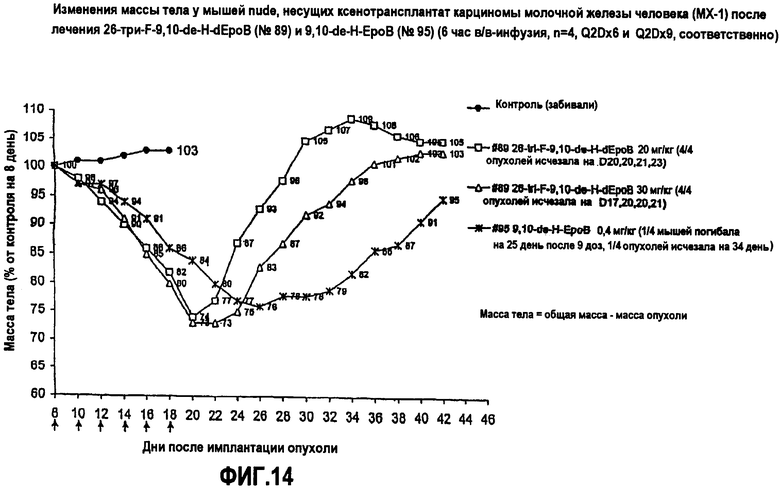

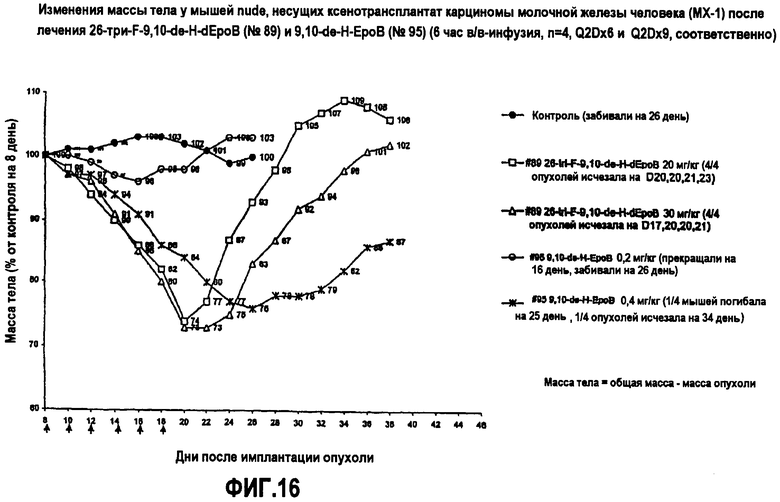
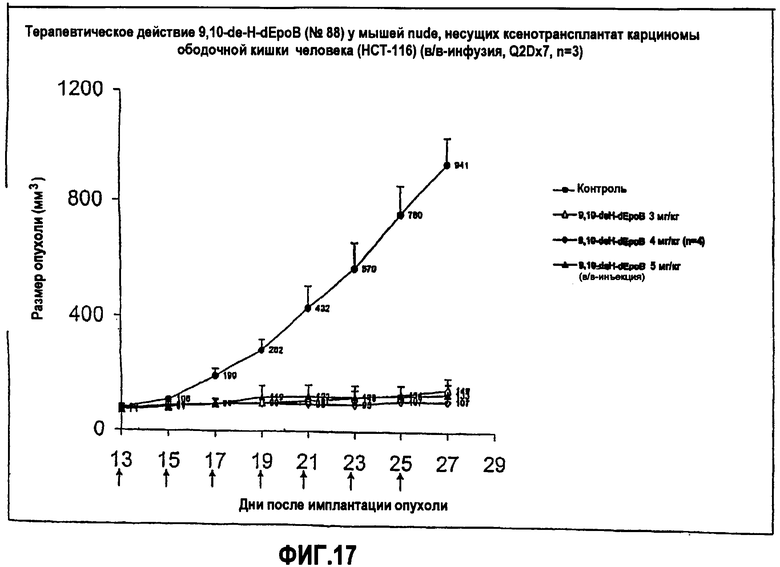





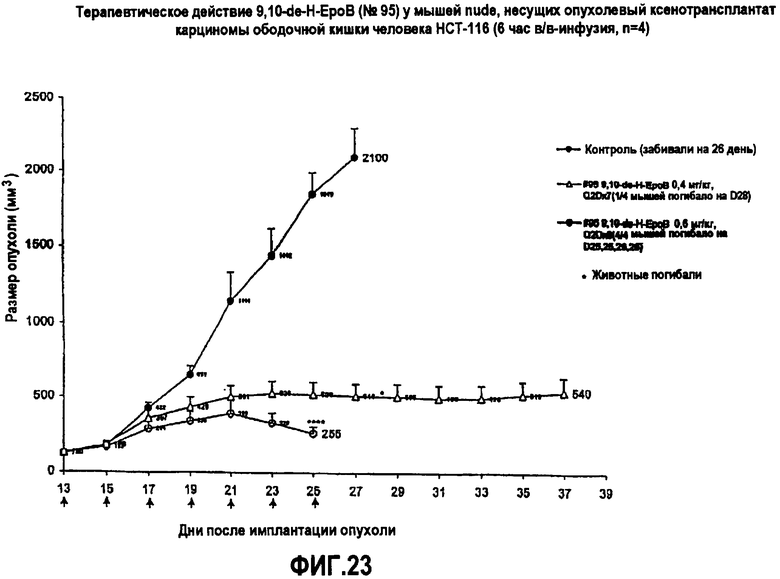



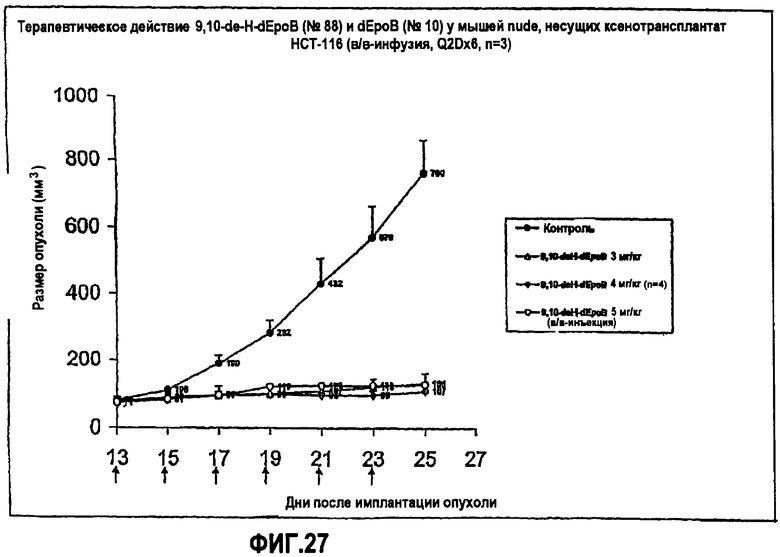


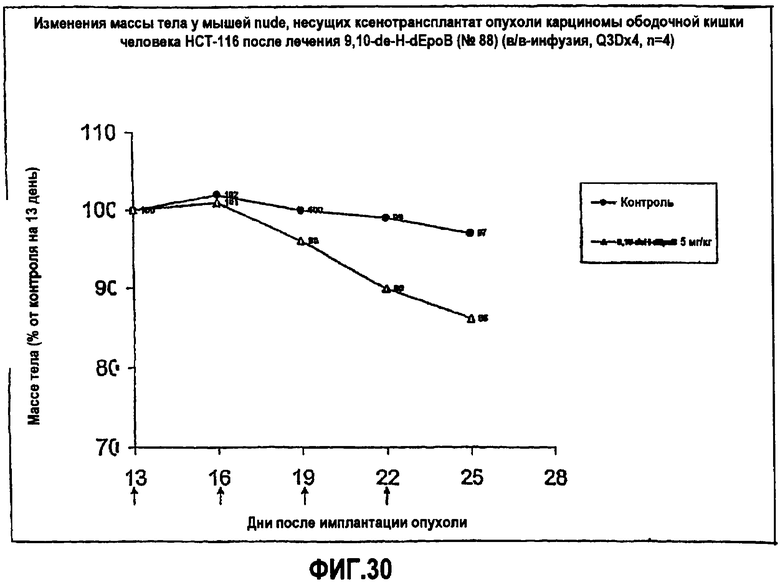
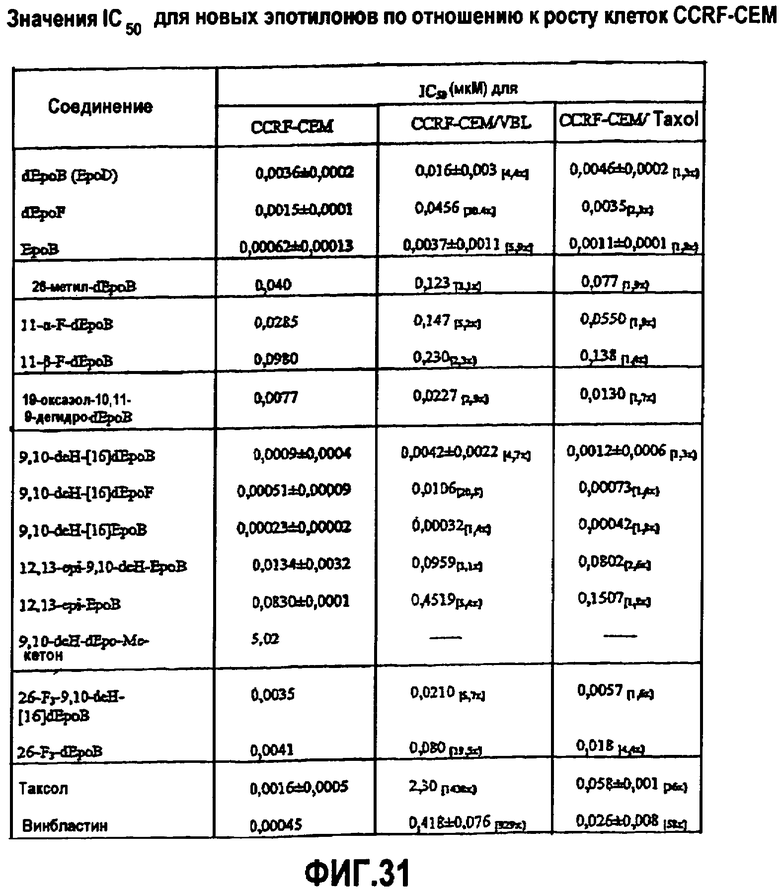
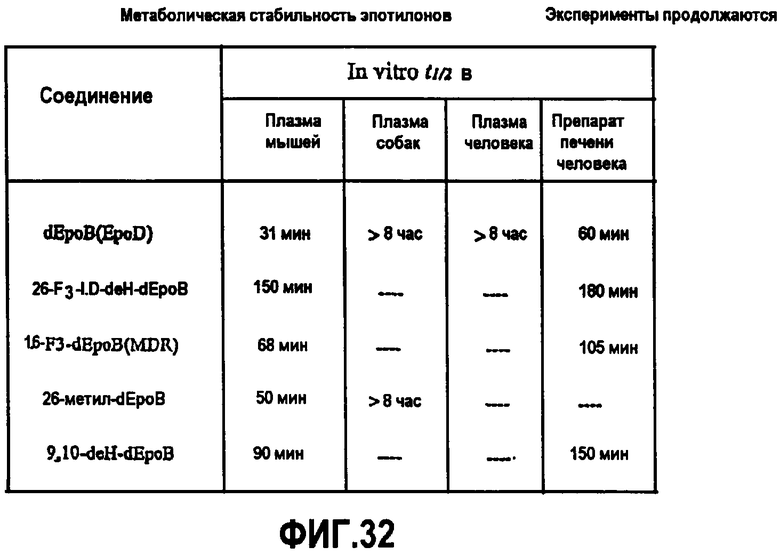


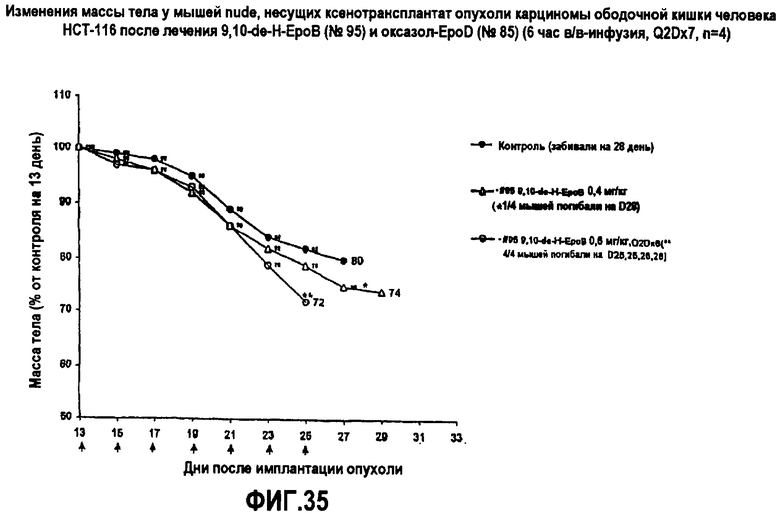

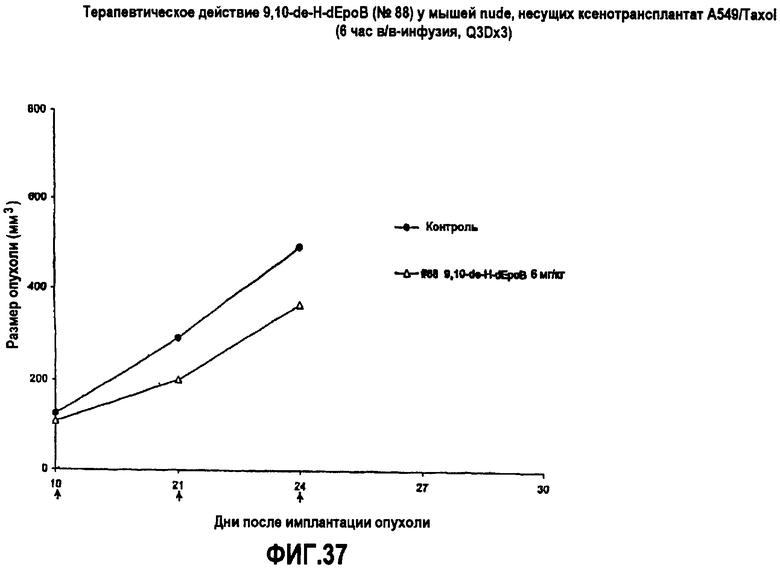
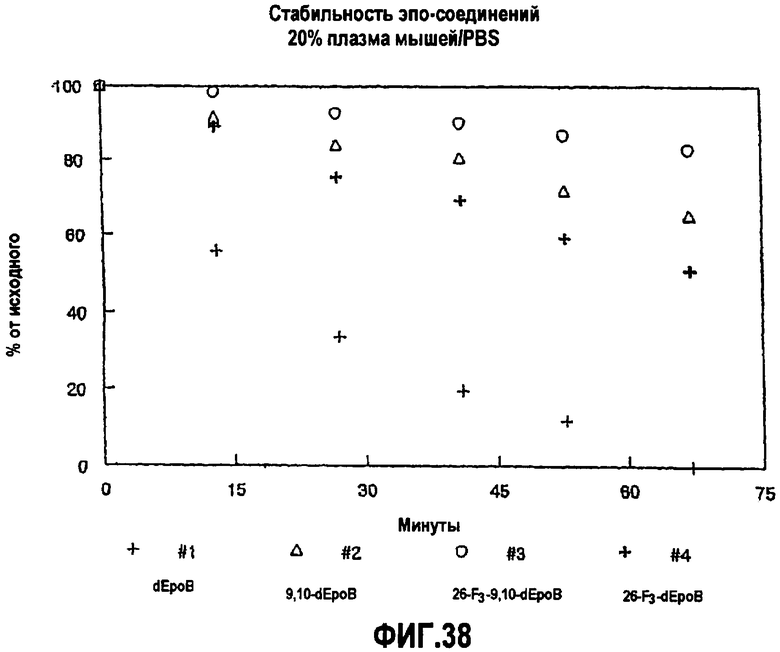
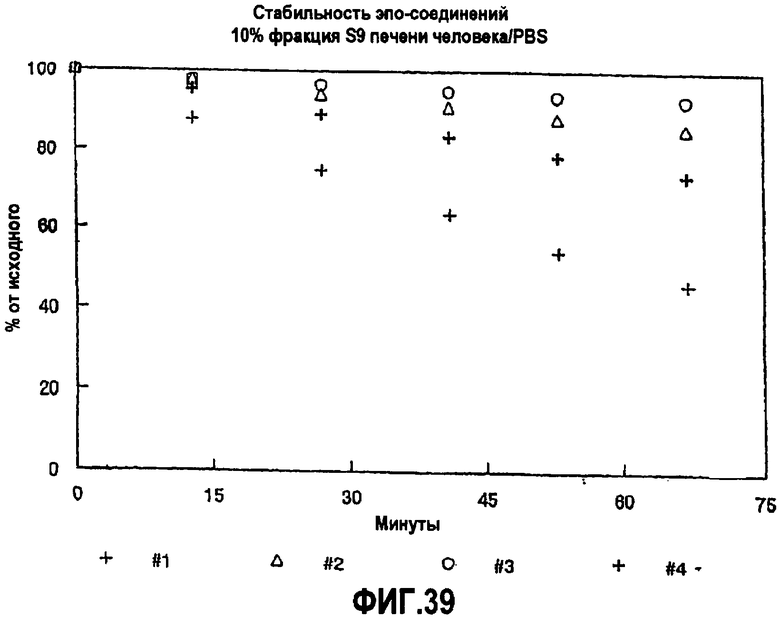
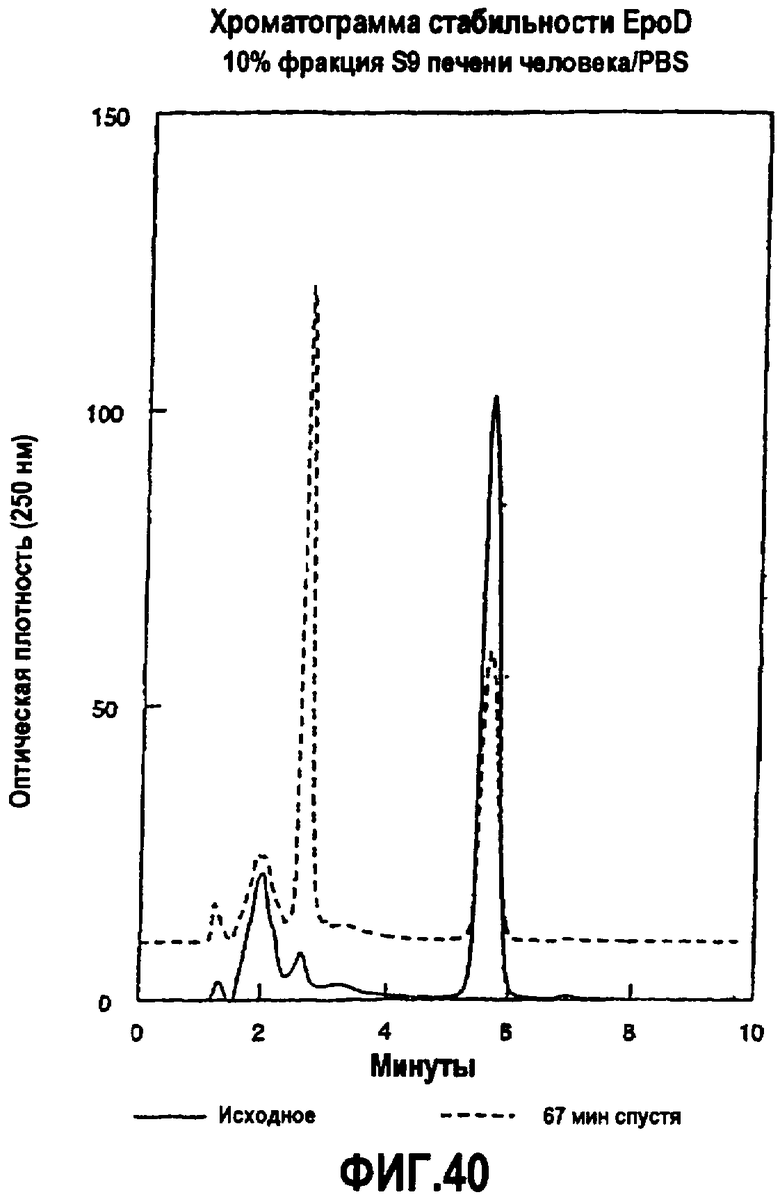
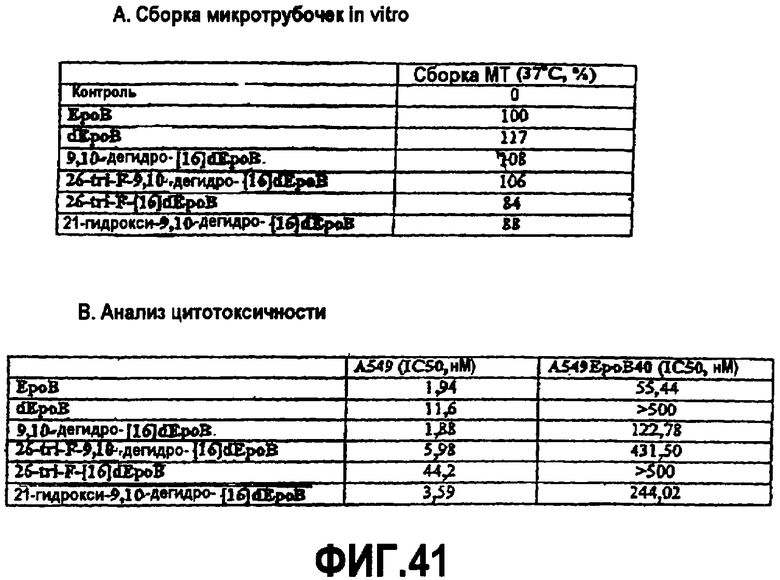
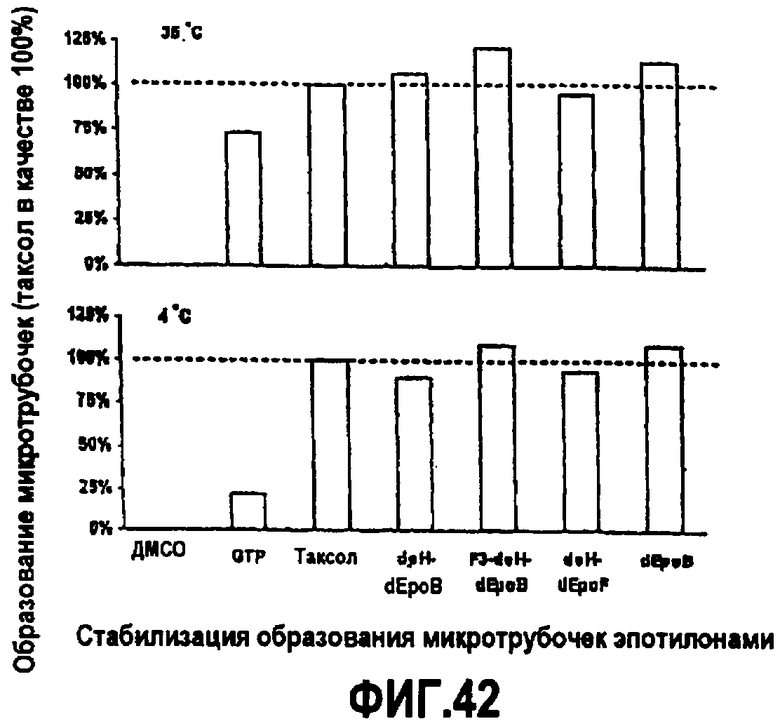

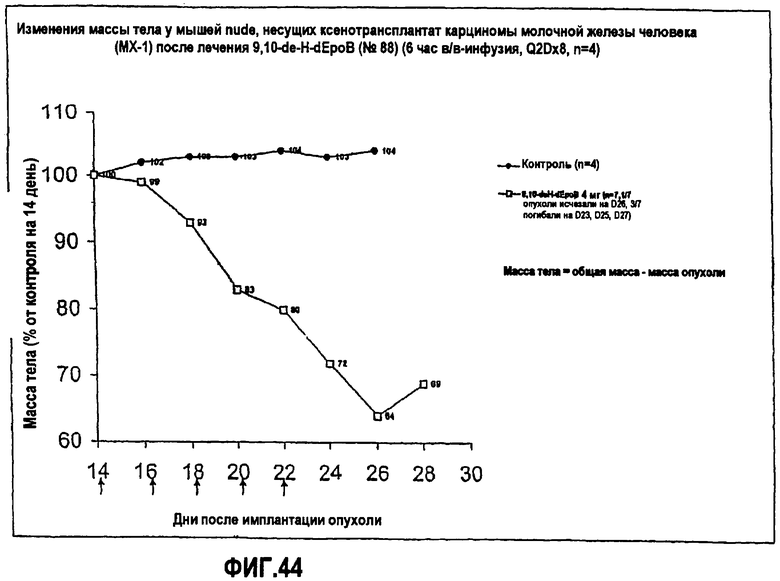
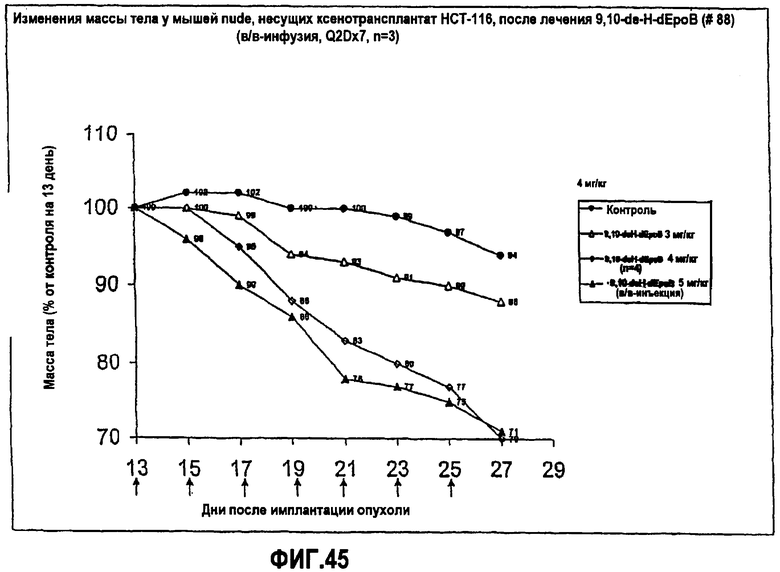
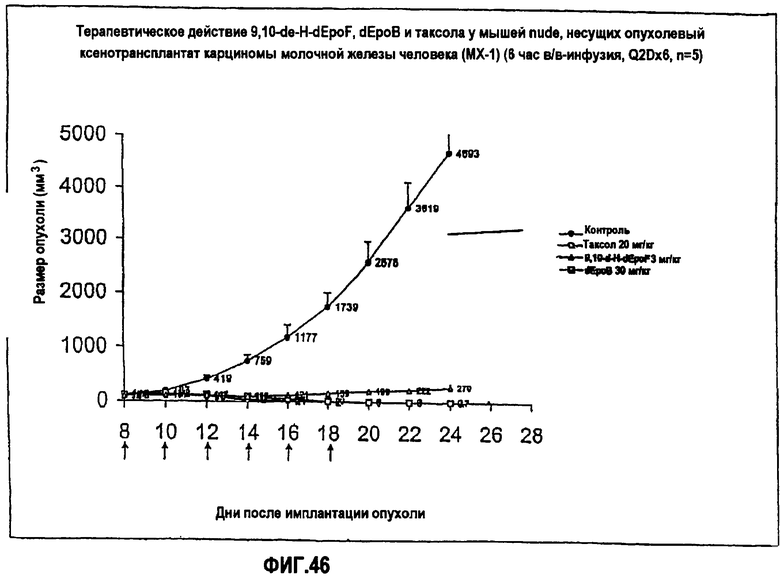
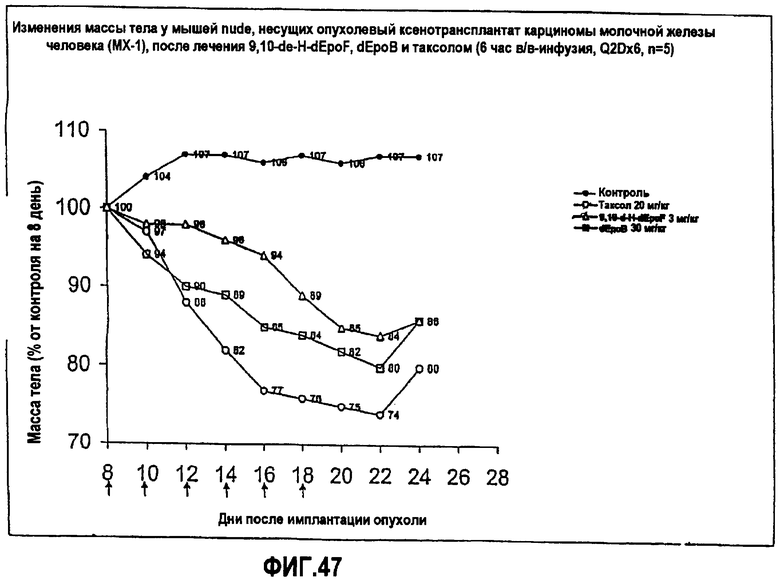
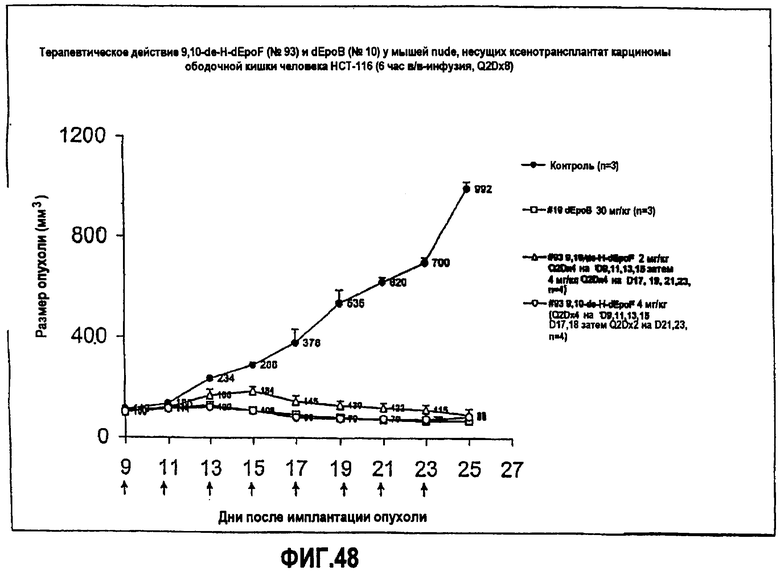
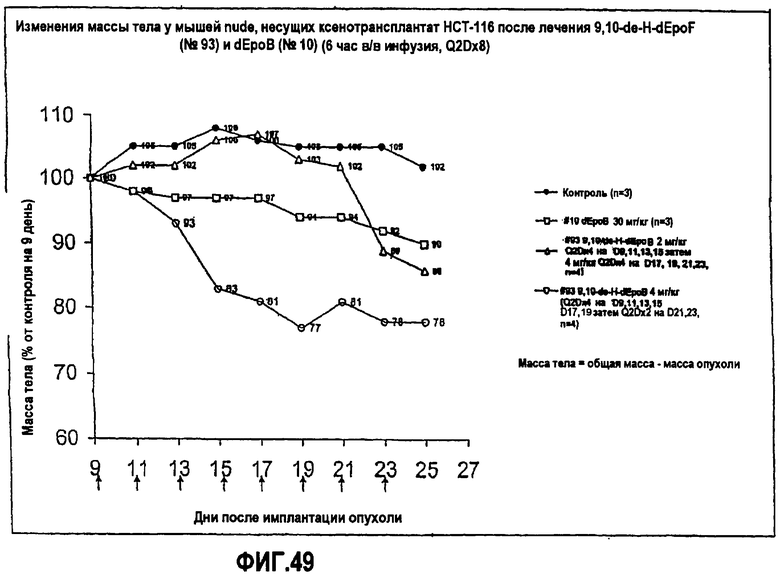
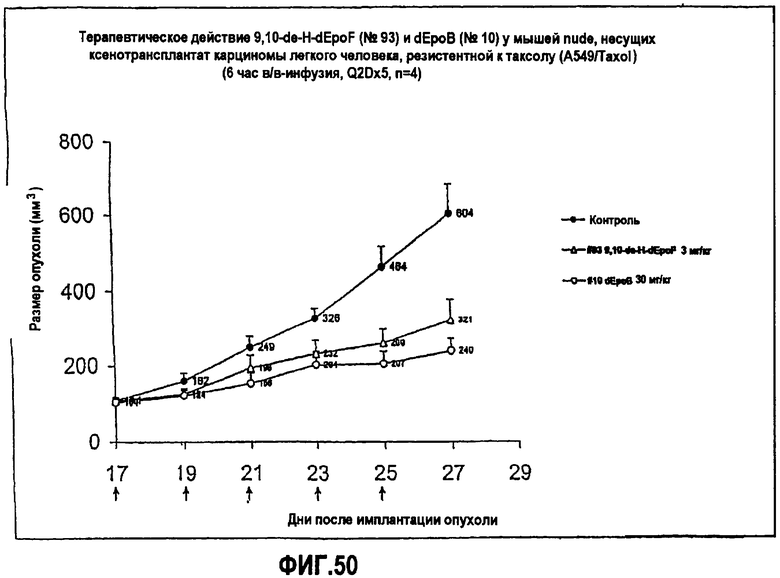

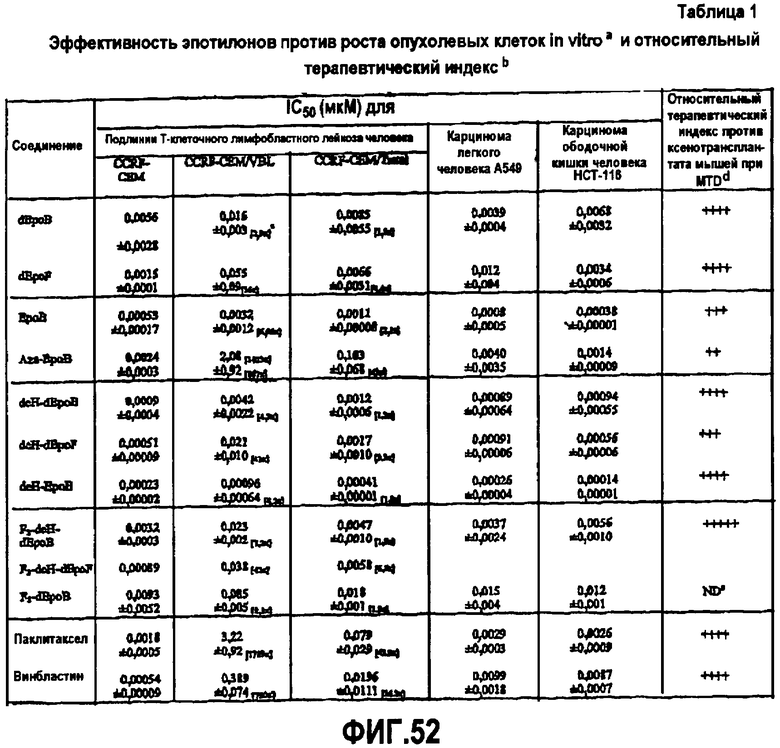
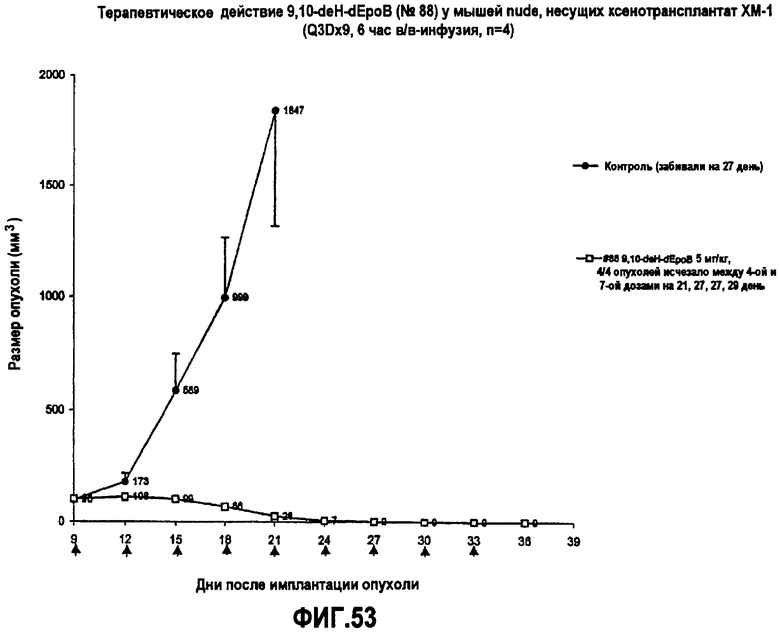

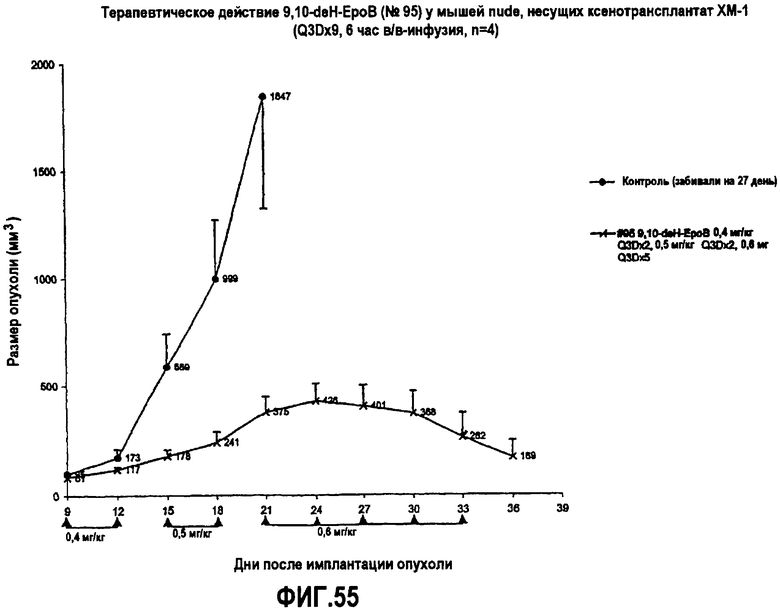
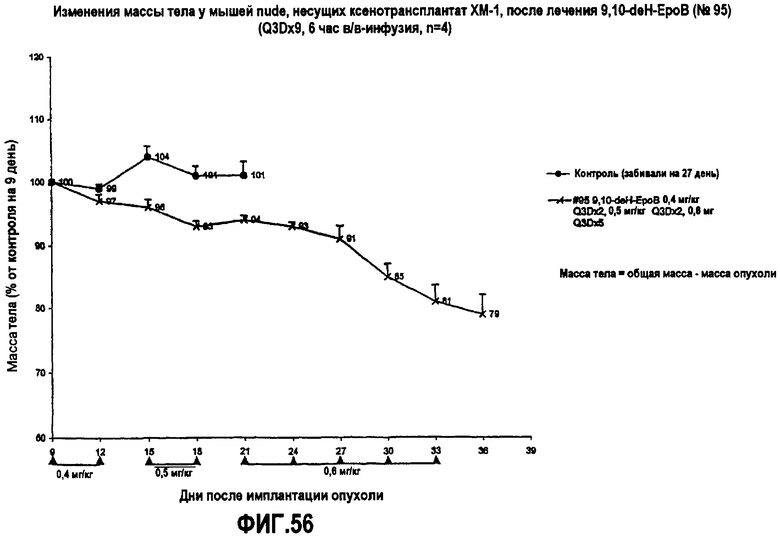
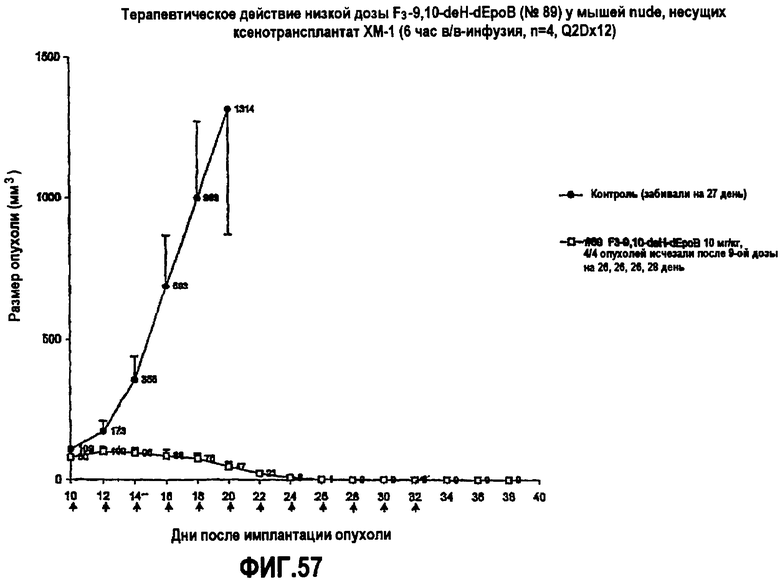







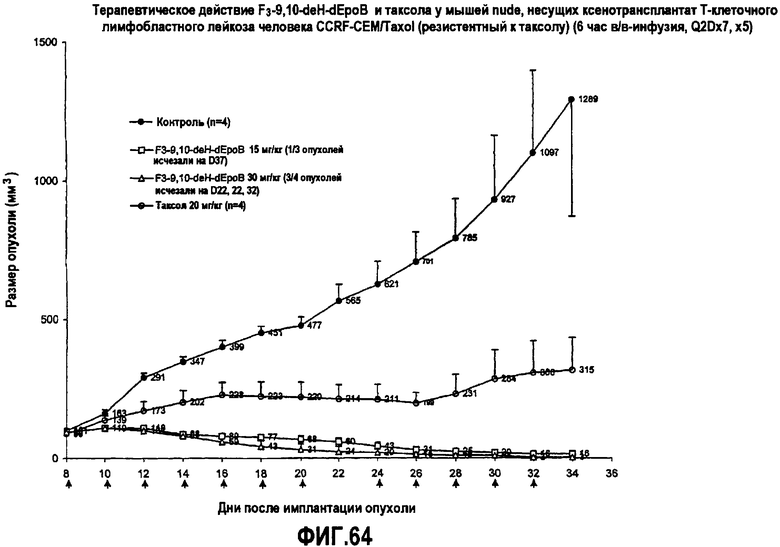
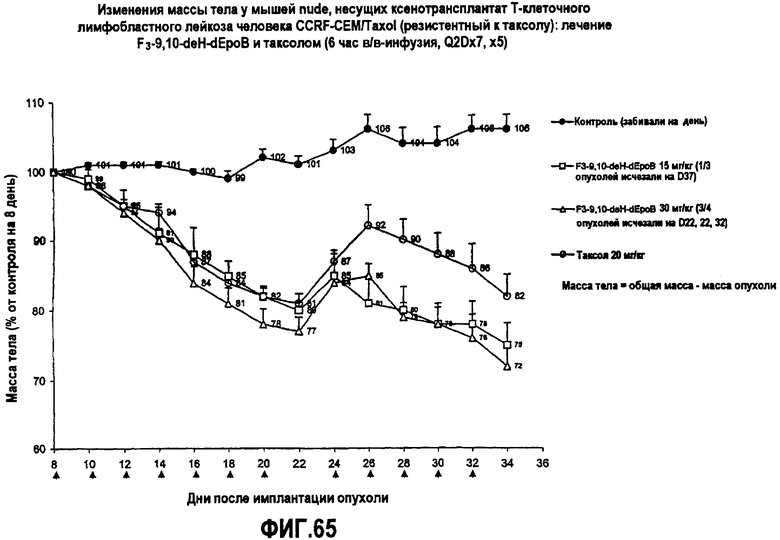

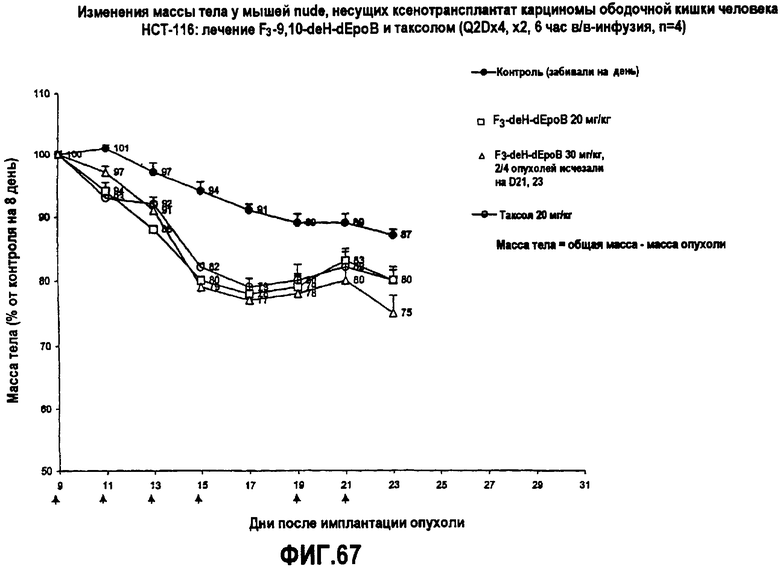


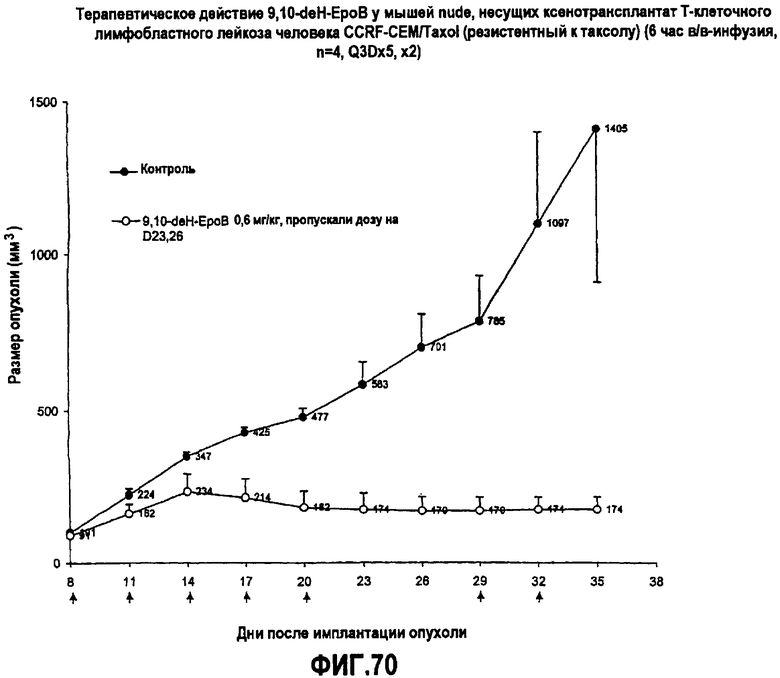


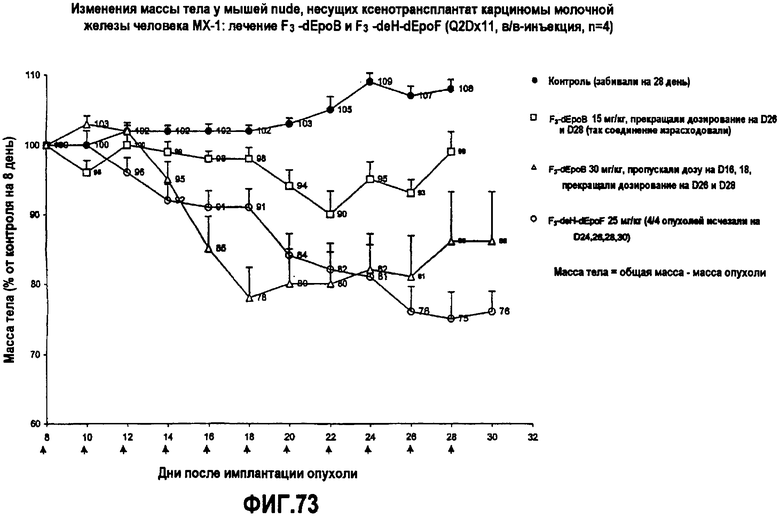

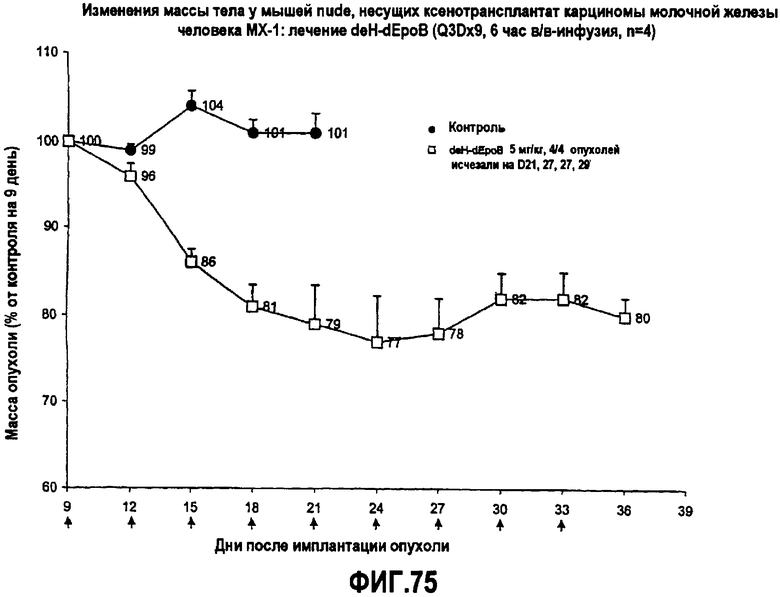
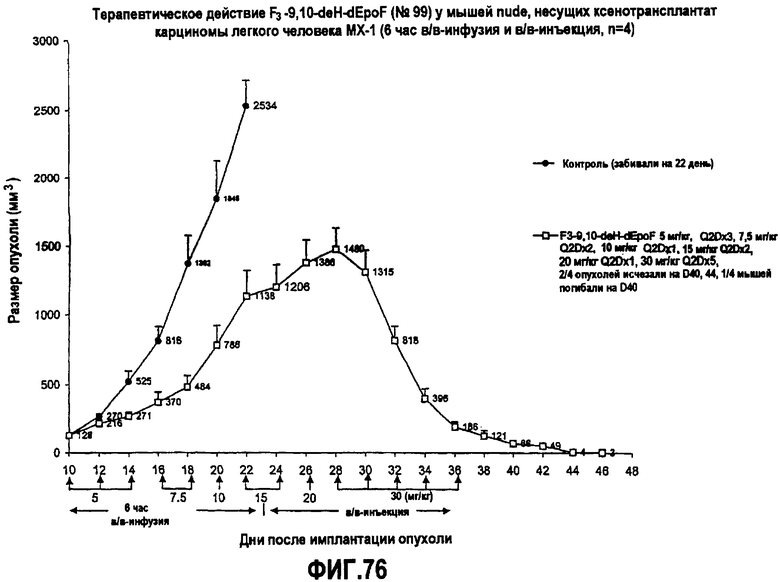
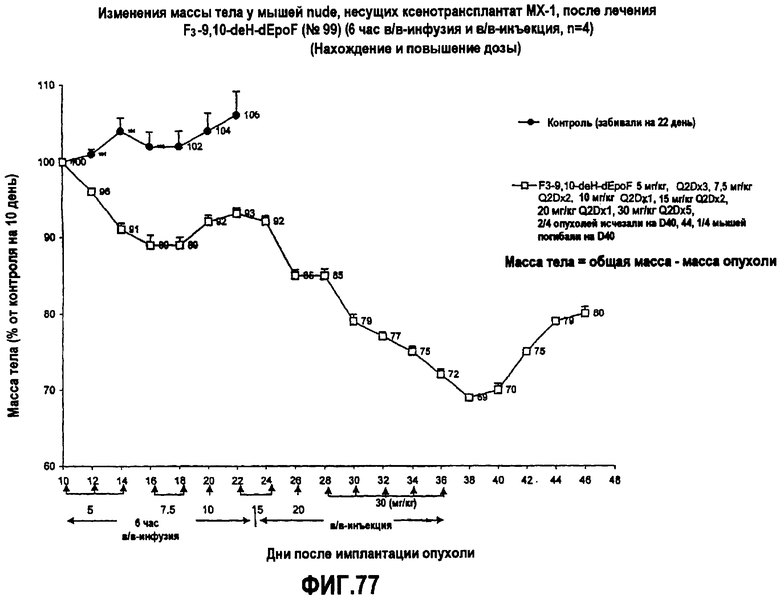
Изобретение относится к соединению формулы

или к его фармацевтически приемлемой соли, в которой R1 означает водород или С1-6лкил; R2 означает изооксазолильная группа, замещенная С1-6алкилом; RB означает -CF3, -CHF2, -CH2F, или C1-6алкил. Изобретение также относится к фармацевтическим композициям для лечения рака, содержащим заявленные соединения. Технический результат - получены новые соединения и фармацевтические композиции на их основе, которые могут найти применение в медицине для лечения раковых заболеваний. 4 н. и 11 з.п. ф-лы, 77 ил., 10 табл., 13 пр.
1. Соединение формулы:
или его фармацевтически приемлемая соль,
в которой R1 означает водород или C1-6алкил;
R2 означает изооксазолильная группа, замещенная C1-6алкилом;
RB означает -CF3, -CHF2, -CH2F, или С1-6алкил.
2. Соединение по п.1, в котором R1 означает C1-6алкил.
3. Соединение по п.2, в котором R1 означает метил.
4. Соединение по п.3, в котором RB означает -CF3.
5. Соединение по п.3, в котором RB означает метил.
6. Соединение по п.1, в котором R2 замещен метилом.
7. Фармацевтическая композиция для лечения рака, содержащая соединение по любому из пп.1-6 и фармацевтически приемлемый эксципиент.
8. Фармацевтическая композиция по п.7, дополнительно содержащая кремофор.
9. Фармацевтическая композиция по п.7, дополнительно содержащая кремофор и этанол.
10. Фармацевтическая композиция по п.7, где соединение суспендировано в смеси кремофор/этанол в соотношении 1:1.
11. Фармацевтическая композиция по п.7, дополнительно содержащая дополнительный цитотоксический агент.
12. Фармацевтическая композиция для лечения рака, содержащая терапевтически эффективное количество соединения по любому из пп.1-6 или его фармацевтически приемлемой соли; и фармацевтически приемлемый носитель или разбавитель,
причем терапевтически эффективное количество соединения является количеством, достаточным для доставки около 0,001 мг до около 40 мг соединения на кг веса субъекта.
13. Применение соединения по любому из пп.1-6 в производстве лекарственного средства для лечения рака.
14. Применение по п.13, где терапевтически эффективное количество соединения является количеством, достаточным для доставки около 0,001 мг до около 40 мг соединения на кг веса субъекта.
15. Применение по п.13, где терапевтически эффективное количество соединения является количеством, достаточным для доставки около 0,1 мг до около 25 мг соединения на кг веса субъекта.
| US 20020058817 А1, 16.05.2002 | |||
| WO 9901124 A1, 14.01.1999 | |||
| Прибор для измерения и записи колебаний кровяного давления и артериального пульса | 1934 |
|
SU49021A1 |
| RU 2000102893 A, 10.12.2001 | |||
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ФАРНЕЗИЛТРАНСФЕРАЗЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2179975C1 |

