Настоящее изобретение относится к улучшенному способу получения моногидрохлорида 3-[(1R,2R)-3-(диметиламино)-1-этил-2-метилпропил]-фенола.
Тапентадол - это INN (International Non-proprietary Name) моногидрохлорид 3-[(1R,2R)-3-(диметиламино)-1-этил-2-метилпропил]-фенола, который представлен формулой:

Химическая структура тапентадола была раскрыта в EP-A-0,693,475 в качестве соединения (+21). Синтез тапентадола описан в Примере 1 и Примере 24 стадий 1-3 и кратко описан ниже, используя номера соединений, как упомянуто в сказанном EP-A-0, 693, 475.

Синтетический предшественник тапентадола на схеме выше представляет собой (2R,3R)-3-(3-метоксифенил)-N,N,2-триметилпентанамин (интермедиат (+23) на верхней схеме), который может быть получен удалением третичной гидроксигруппы (2S,3R)-1-(диметиламино)-3-(3-метоксифенил)-2-метил-3-пентанола путем последовательного превращения в соответствующий галогенид тионилхлоридом и последующим удалением Cl обработкой борогидридом цинка, цианоборогидридом цинка и/или цианоборогидридом олова.
Этот способ обладает недостатком, что галогеносоединение получено с использованием избыточного количества тионилхлорида, который является огрессивным хлорирующим агентом. Кроме того, восстанавливающие реагенты, такие как борогидрид цинка, цианоборогидрид цинка и цианоборогидрид олова обладают значительной воспламеняемостью и угрозой здоровью, когда используются в промышленном масштабе.
WO-2004/108658 раскрывает альтернативный способ получения (2R,3R)-3-(3-метоксифенил)-N,N,2-триметилпентанамина конвертированием (2S,3S)-1-(диметиламино)-3-(3-метоксифенил)-2-метил-3-пентанола в смесь (2R,3R) и (2R,3S)-3-(3-метоксифенил)-N,N,2-триметилпентанаминов, как описано ниже.

Полученная смесь (2R,3R) и (2R,3S)-3-(3-метоксифенил)-N,N,2-триметилпентанамина была разделена на отдельные стереоизомеры с целью получения желаемого (2R,3R)-3-(3-метоксифенил)-N,N,2-триметилпентанамина, который может быть затем превращен в тапентадол, например нагреванием с концентрированной бромистоводородной кислотой, как описано в EP-A-0,693,475.
WO-2005/000788 раскрывает альтернативный способ получения (2R,3R)-3-(3-метоксифенил)-N,N,2-триметилпентанамина конвертированием (2S,3S)-1-(диметиламино)-3-(3-метоксифенил)-2-метил-3-пентанола в смесь (2R,3R) и (2R,3S)-3-(3-метоксифенил)-N,N,2-триметилпентанаминов, как описано ниже.

Полученная смесь (2R,3R) и (2R,3S)-3-(3-метоксифенил)-N,N,2-триметилпентанамина была разделена на отдельные стереоизомеры с целью получения желаемого (2R,3R)-3-(3-метоксифенил)-N,N,2-триметилпентанамина, который может быть затем превращен в тапентадол, например нагреванием с концентрированной бромистоводородной кислотой, как описано в EP-A-0,693,475.
Оба альтернативных способа WO-2004/108658 и WO-2005/00078 обладают недостатками, что [3-(3-метоксифенил)-N,N,2-триметилпентанамин получен в виде смеси (2R,3R) и (2R,3S) стереоизомеров, которую разделяют с целью получения желаемого (2R,3R) стереоизомера. Нежелательный (2R,3S) стереоизомер не может быть превращен в желаемый (2R,3R) стереоизомер и должен быть удален в качестве химических отходов, что является экономически неприемлемо для любого промышленного производства.
Задача настоящего изобретения относится к улучшенному способу синтеза (2R,3R)-3-(3-гидроксифенил)-N,N,2-триметилпентанамина, который является более удобным и более эффективным, чем известные ранее способы.
Настоящее изобретение обеспечивает эту задачу предоставлением улучшенного способа получения (2R,3R)-3-(3-гидроксифенил)-N,N,2-триметилпентанамина или его соли присоединения кислоты.
Настоящее изобретение относится к способу, который описывается такими стадиями:
a) ацилирование соединение формулы (VI), где R означает C1-6алкил, C3-8циклоалкил, C1-6алкилкарбонил, тетрагидропиранил или C1-3алкил, замещенный фенил или нафтил, при условии, что R=CH3 исключается

ацилирующим агентом;
b) стереоселективный гидрогенолиз таким образом полученного соединения формулы (VII)

c) использование подходящего катализатора в реакционном инертном растворителе в присутствии водорода с образованием продукта VIII, который имеет R1=H (уже со снятой защитой на стадии b) или защитная группа R все еще является частью продукта VIII. В этом случае (R1≠H в соединении VIII) группа R полученного соединения формулы VIII может быть снята на стадии c)

и d) необязательно конвертирование полученного незащищенного продукта в соль присоединения кислоты.
Предпочтительно R означает этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, н-гексил, циклопропил, циклобутил, циклобутил, циклопентил, циклогексил, циклогептил, бензил, фенилэтил, тетрагидропиранил, -(C=O)-CH3, -(C=O)-CH2CH3 или -(C=O)-C(CH3)3, в соединениях формулы (VI), (VII) и (VIII). Более предпочтительно R означает этил, циклопропил, циклобутил, циклопентил, циклогексил, бензил, фенилэтил, тетрагидропиранил или -(C=O)-CH3 в соединениях формулы (VI), (VII) и (VIII). Еще более предпочтительно R означает бензил или тетрагидропиранил в соединениях формулы (VI), (VII) и (VIII).
Например, в случае R=бензил
стадия снятия защиты с) не является обязательной, поскольку соединение VII непосредственно превращается в VIII с R1=H через стадию восстановления. Бензильная группа является очень предпочтительной как заместитель R, который может быть необязательно замещен например галогенами или/и нитрогруппами.
Ацилирующий агент стадии а) является органическим ацилхлоридом или ангидридом органической кислоты, выбранным из уксусного ангидрида, ацетилхлорида, трифторуксусного ангидрида, хлоруксусного ангидрида, хлорацетилхлорида, дихлоруксусного ангидрида, трихлоруксусного ангидрида, бензойного ангидрида, бензоилхлорида, фталевого ангидрида, фталоилдихлорида, терефталоилдихлорида, янтарного ангидрида, сукцинилхлорида, этилоксалилхлорида, метилоксалилхлорида, кислоты Мельдрума, этилхлороформата, метилхлороформата, ацетилсалицилоилхлорида или любого другого подходящего ацилирующено агента.
Катализатор стадии b) выбран из палладиевого катализатора или любого другого подходящего катализатора, такого как, например, никель Реннея, платина, платина на угле, рутений или родий на угле.
Палладиевым (Pd) катализатором может быть гомогенный Pd катализатор, такой как например Pd(OAc)2, PdCl2, Pd(PPh3)4, Pd(PPh3)2Cl2, Pd2(dba)3 (трис(дибензилиденацетон)дипалладий), палладий
тиометилфенилглутарамид металлацикл и т.п., или гетерогенный Pd катализатор, такой как, например, палладий на угле, палладий на оксидах металлов, палладий на цеолитах. Предпочтительно палладиевый катализатор представляет собой гетерогенный Pd катализатор, более предпочтительно палладий на угле или палладий на углероде (Pd/C). Pd/C является регенеруемым катализатором, стабилен и относительно недорогой. Он может быть легко отделен (фильтрованием) от реакционной смеси, таким образом уменьшая возможность наличия следов Pd в конечном продукте. Применение Pd/C также лишено использования лигандов, таких как например фосфиновые лиганды, которые являются дорогими, токсическими и загрязняют синтезированные продукты.
Реакционно-инертный растворитель стадии b) выбран из диэтилового эфира, тетрагидрофурана, 2-метилтетрагидрофурана или их смесей.
Предпочтительными агентами для снятия защиты на стадии c) являются йодтриметилсилан, этилсульфид натрия, йодид лития, бромистоводородная кислота; более предпочтительно бромистоводородная кислота.
В одном варианте воплощения настоящего изобретения стадии a) и b) проведены как "однореакторная синтетическая" процедура.
Настоящее изобретение также относится к новым соединениям формулы (VII)
Ацильная группа в соединениях формулы (VII) означает СН3-СО-, CF3-СО-, CH2Cl-CO-, CHCl2-CO-, CCl3-СО-, CH3O-СО-СО-, CH3O-СО-, CH3CH2O-СО-, CH3CH2O-СО-СО, фенил-СО- или мета-СН3СОО-фенил-СО-, когда ацилирующий агент, используемый для получения соединения формулы (III), как изложено выше, выбран из уксусного ангидрида, ацетилхлорида, трифторуксусного ангидрида, хлоруксусного ангидрида, хлорацетилхлорида, дихлоруксусного ангидрида, трихлоруксусного ангидрида, метилоксалилхлорида, этилоксалилхлорида, метилхлороформата, этилхлороформата, бензойного ангидрида, бензоилхлорида или ацетилсалицилоилхлорида. Группа R в соединениях формулы (VII) означает C1-6алкил, C3-8циклоалкил, C1-6алкилкарбонил, тетрагидропиранил или C1-3алкил, замещенный фенил или нафтил, при условии, что R=метил исключается.
Предпочтительный вариант воплощения изобретения (R=бензил) детально рассмотрен для примера в следующих параграфах.
Исходное соединение в способе настоящего изобретения, т.е. (2S,3R)-1-(диметиламино-3-(3-(бензилокси)-фенил)))-2-метил-3-пентанол (соединение 4), получено взаимодействием (2S)-3-(диметиламино)-1-(3-(бензилокси)-фенил)-2-метил-1-пропанона (соединение 3) с этилмагнийбромидом в THF в условиях реакции Гриньяра.

Реакция реактива Гриньра с кетосоединением (3) привносит второй асимметрический атом углерода. Реакция Гриньра (2S)-3-(диметиламино)-1-(3-(бензилокси)-фенил)-2-метил-1-пропанона (соединение 3) с этилмагнийгалидом является высокостереспецифической, потому что оптическая чистота моногидрохлорида 3-[(1R,2R)-3-(диметиламино)-1-этил-2-метилпропил]-фенола составляет 99%.
Соединение (4) может быть превращено в соединение (5)
1) ацилированием соединения (4) трифторуксусным ангидридом и
2) последующим гидролизом и расщеплением бензильной эфирной группы на палладиевом катализаторе с использованием 2-метилтетрагидрофурана как растворителя и
3) обработкой хлороводородом как осаждающим агентом, в "однореакторной синтетической" процедуре.

Эта стадия реакции является высокостереоселективной, поскольку оптическая чистота моногидрохлорида 3-[(1R,2R)-3-(диметиламино)-1-этил-2-метилпропил]-фенола составляет 99%.
Образование соли соединением (5) в дальнейшем улучшает оптическую чистоту соединения (5).
Экспериментальная часть
Пример 1: Синтез 3-(диметиламино)-1-(3-(бензилокси)-фенил)-2-метил-1-пропанона (1)

При комнатной температуре 1-(3-(бензилокси)-фенил)-пропан-1-он (145.0 г; 0.6 моль) растворяли в ацетонитриле (375 мл) в 500 мл 3-горлой круглодонной колбе, оснащенной верхнеприводной мешалкой и термометром, и при перемешивании прибавляли N-метил-N-метилен-метанаминийхлорид (57.0 г; 0.61 моль) и ацетилхлорид (5 мл). После прибавления температура поднимается до 10°C. Реакционную смесь перемешивали при комнатной температуре всю ночь, прибавляли диэтиловый эфир (375 мл) и продукт кристаллизовали охлаждением до 5-10°C в течение 3 часов. Полученный осадок фильтровали с отсасыванием и сушили при 45°C и 100 мбар. Продукт получали в виде бесцветного порошка с 50% выходом (94 г).
Пример 2: Синтез и отделение (2S)-3-(диметиламино)-1-(3-(бензилокси)-фенил)-2-метил-1-пропанона (3)

Моногидрат дибензоилвинной кислоты (78.0 г; 0.2 моль) растворяли в абсолютном этаноле (360 мл) нагреванием до 35-40°C в 500 мл реакторе, оснащенном термометром. Смесь охлаждали до комнатной температуры и прибавляли к раствору (2RS)-3-(диметиламино)-1-(3-(бензилокси)-фенил)-2-метил-1-пропанона (1) в абсолютном этаноле (230 мл). Для завершения кристаллизации смесь перемешивали 16 часов при 5-8°C. Полученные кристаллы отфильтровывали, промывали этанолом и сушили при 45°C/100 мбар 16 часов. Продукт получали в виде бесцветного порошка с 65% выходом (85.0 г).
(2S)-3-(диметиламино)-1-(3-(бензилокси)-фенил)-2-метил-1-пропанон (L)-(-)-дибензоилтартрат (85.5 г; 0.13 моль) растворяли в воде в 1000 мл реакторе, оснащенном термометром, и прибавляли 3-пентанон (200 мл). pH доводили до 12-13 водным гидроксидом натрия (32%; 25 мл; 0.28 моль). Слои разделяли и органический слой сушили над сульфатом натрия и растворитель полностью удаляли в вакууме при 45-50°C и 5-10 мбар. Продукт получали в виде масла с 87% выходом (33.6 г). [α]=+17°.
Пример 3: Синтез (2S,3R-1-(диметиламино)-3-(3-(бензоилокси)-фенил)-2-метил-3-пентанола(4)

Раствор этилмагнийбромида (1M в THF; 0.15 моль; 150 мл) помещали в 500 мл 3-горлую круглодонную колбу с верхнеприводной мешалкой, термометром, подачей инертного газа и капельной воронкой при 10°C под азотом. К раствору (2S)-3-(диметиламино)-1-(3-(бензилокси)-фенил)-2-метил-1-пропанона (3) (33.0 г; 0.11 моль), растворенного в THF (150 мл), прибавляли по каплям при 10-15°C. После окончания прибавления смесь перемешивали при комнатной температуре 16 ч и гасили раствором гидросульфата аммония (150 мл). Слои разделяли и водный слой реэкстрагировали 3-пентаноном (150 мл). Объединенные органические фазы сушили над сульфатом натрия и растворитель полностью удаляли на роторном испарителе при 45-50°C и <10 мбар. Получали желтое масло с 89% выходом (32.0 г). [α]=-10.5°C
Пример 4: Синтез моногидрохлорида 3-[(1R,2R)-3-(диметиламино)-1-этил-2-метилпропил]-фенола (5)
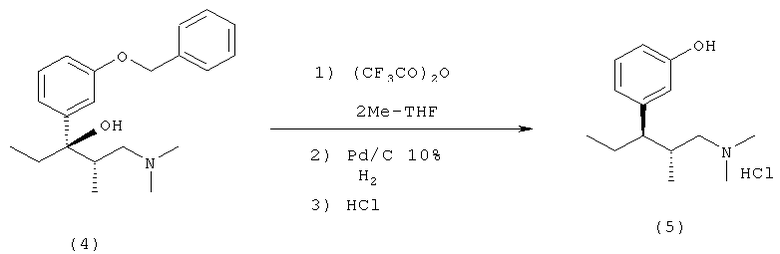
В стандартном лабораторном реакторе, оснащенном мешалкой и термометром, растворяли (2S,3R)-1-(диметиламино)-3-(3-(бензилокси)-фенил)-2-метил-3-пентанол (4) (21.0 г; 0.064 моль) в метилтетрагидрофуране (125 мл) и прибавляли трифторуксусный ангидрид (20 г, 0.095 моль). Смесь нагревали до 40-45°C 4 часа при перемешивании. Смесь затем охлаждали до комнатной температуры и прибавляли Pd/C (5%; 2.5 г; 1.9 мол.%) в атмосфере азота. Смесь переносили в аппарат для гидрирования и гидрировали при 3 бар / 800 об/мин 16 часов. Катализатор отфильтровывали и полученный раствор охлаждали до 5-10°C в бане со льдом. Прибавляли воду (1.1 г; 0.06 моль) и по каплям триметилхлорсилан (6.95 г; 0.064 моль). Для кристаллизации смесь перемешивали при 5-8°C 16 часов. Кристаллы отфильтровывали, промывали ацетоном и сушили в сушильном шкафу при 40-45°C и 100 мбар 16 часов. Продукт получали в виде бесцветного кристаллического вещества с 89% выходом (14.7 г; Тпл. 201°C, энантиомерная чистота: 99%, чистота: 97.7%; (HPLC); анализ: 95.5%(HPLC)).
Соединение (5), полученное согласно процедуре Примера 5, содержит 96.9% желаемого (2R,3R) энантиомера, 1% (2S,3S) энантиомера и 2.1% (2R,3S) энантиомера.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (1R,2R)-3-(3-ДИМЕТИЛАМИНО-1-ЭТИЛ-2-МЕТИЛ-ПРОПИЛ)-ФЕНОЛА | 2007 |
|
RU2466124C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 3-АРИЛБУТИЛАМИНОВЫХ СОЕДИНЕНИЙ | 2004 |
|
RU2380355C2 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРРОЛИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ | 2003 |
|
RU2355679C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИГИДРОХИНАЗОЛИНОВ | 2006 |
|
RU2419617C2 |
| НОВЫЕ ИЗОИНДОЛИНОВЫЕ ИЛИ ИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2689305C2 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА ИЛИ БЕНЗОТИАЗЕПИНА | 1989 |
|
RU2090562C1 |
| БЕТА-ЛАКТАМЫ, СПОСОБ ПОЛУЧЕНИЯ УКАЗАННЫХ СОЕДИНЕНИЙ И СЫВОРОТОЧНЫЕ ГИПОХОЛЕСТЕРИНЕМИЧЕСКИЕ СРЕДСТВА, СОДЕРЖАЩИЕ ТАКИЕ СОЕДИНЕНИЯ | 2002 |
|
RU2301799C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОНДЕНСИРОВАННОГО ТРИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ И СООТВЕТСТВУЮЩЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2020 |
|
RU2785963C1 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2014 |
|
RU2678327C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
Настоящее изобретение относится к улучшенному способу получения 3-[(lR,2R)-3-(диметиламино)-1-этил-2-метилпропил]-фенола формулы VIII или его соли присоединения кислоты, предпочтительно хлористоводородной кислоты. Способ характеризуется следующими стадиями: а) ацилирование соединения формулы (VI), где R означает C1-3алкил, замещенный фенилом


ацилирующим агентом; b) гидрогенолиз полученного таким образом соединения (VII), где ацил выбран из СН3-СО-, CF3-CO-, CH2Cl-СО-, CHCl2-СО- и CCl3-CO-, при использовании палладиевого катализатора в инертном в условиях реакции растворителе в присутствии водорода с образованием продукта VIII с R1=H:
 и
и
d) необязательное конвертирование полученного незащищенного продукта в соль присоединения кислоты. В предлагаемом способе получены новые промежуточные соединения формулы (VII), которые также являются предметом настоящего изобретения. При этом снижается образование нежелательных изомеров, не используются агрессивные легковоспламеняющиеся вещества. 2 н. и 11 з.п. ф-лы, 4 пр.
1. Способ получения соединения формулы (VIII) или его соли присоединения кислоты, который характеризуется следующими стадиями
a) ацилирование соединения формулы (VI), где R означает С1-3алкил, замещенный фенилом

ацилирующим агентом;
b) гидрогенолиз полученного таким образом соединения (VII)
где ацил выбран из СН3-СО-, CF3-CO-, СН2Сl-СО-, СНСl2-СО- и CCl3-CO-,
при использовании палладиевого катализатора в инертном в условиях реакции растворителе в присутствии водорода с образованием продукта VIII с R1=H
и необязательное конвертирование полученного незащищенного продукта в соль присоединения кислоты.
2. Способ по п.1, в котором ацилирующий агент стадии а) является соответствующим органическим ацилхлоридом или ангидридом органической кислоты.
3. Способ по п.2, в котором органический ацилхлорид или ангидрид органической кислоты выбран из уксусного ангидрида, ацетилхлорида, трифторуксусного ангидрида, хлоруксусного ангидрида, хлорацетилхлорида, дихлоруксусного ангидрида, трихлоруксусного ангидрида.
4. Способ по п.3, в котором ангидридом кислоты является трифторуксусный ангидрид.
5. Способ по любому из пп.1-4, в котором катализатор стадии b) представляет собой палладий на угле.
6. Способ по п.1, в котором инертный в условиях реакции растворитель стадии b) выбран из диэтилового эфира, тетрагидрофурана и 2-метилтетрагидрофурана или их смесей.
7. Способ по п.1, в котором R означает бензил или фенилэтил.
8. Способ по п.7, в котором R означает бензил.
9. Способ по п.8, в котором (2R,3R)-3-(3-гидpoкcифeнил)-N,N,2-тpимeтилпeнтaнaмин превращают в его соответствующую хлористоводородную соль присоединения кислоты.
10. Способ по п.1, в котором стадии а) и b) проводятся как однореакторный процесс.
11. Соединение формулы (VII), в котором R означает С1-3алкил, замещенный фенилом, а ацил выбран из СН3-СО-, CF3-CO-, СН2Сl-СО-, СНСl2-СО- и ССl3-СО-
12. Соединение формулы (VII) по п.11, в котором R означает бензил или фенилэтил.
13. Соединение формулы (VII) по п.12, в котором ацил означает CF3-CO- и R означает бензил.
| Зонд для измерения параметров электронных пучков электроннолучевых приборов | 1977 |
|
SU693475A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЗАМЕЩЕННЫЕ АМИНОСОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197474C2 |

