Область изобретения
Настоящее изобретение относится к новым производным гидантоина, к способам их получения, к содержащим их фармацевтическим композициям и их применению в терапии.
Предпосылки изобретения
Металлопротеиназы представляют собой надсемейство протеиназ (ферментов), число которых в последние годы поразительно возросло. На основании структурного и функционального анализов эти ферменты классифицировали в семейства и подсемейства, как описано в N. М. Hooper (1994) FEBS Letters 354:1-6. Примеры металлопротеиназ включают матриксные металлопротеиназы (matrix metalloproteinases, MMP), такие как коллагеназы (ММР1, ММР8, ММР13), желатиназы (ММР2, ММР9), стромелизины (ММР3, ММР10, ММР11), матрилизин (ММР7), металлоэластазу (ММР12), энамелизин (enamelysin, MMP19), МТ-ММР (металлопротеиназы матриксного типа, ММР14, ММР15, ММР16, ММР17); репролизин или адамализин или MDC семейство, которое включает в себя секретазы и шеддазы, такие как TNF (фактор некроза опухоли) - конвертирующие ферменты (ADAM10 и ТАСЕ); семейство астацина, которое включает в себя такие ферменты, как проколлаген-процессирующая протеиназа (РСР); и другие металлопротеиназы, такие как агреканаза, семейство эндотелин-конвертирующих ферментов и семейство ангиотензин-конвертирующих ферментов.
Полагают, что металлопротеиназы важны в развитии многих физиологических болезненных процессов, которое включает в себя ремоделирование тканей, такое как эмбриональное развитие, образование костей и ремоделирование матки во время менструации. Это основано на способности металлопротеиназ расщеплять широкий спектр матриксных субстратов, таких как коллаген, протеогликан и фибронектин. Также полагают, что металлопротеиназы важны для процессинга, или секреции, биологически важных клеточных медиаторов, таких как фактор некроза опухоли (TNF); и для пост-трансляционного протеолитического процессинга, или шеддинга, биологически важных мембранных белков, таких как lgE-рецептор CD23 с низким сродством (для более полного перечня смотри N. М. Hooper et al. (1997) Biochem J. 321:265-279).
Металлопротеиназы связывают со многими заболеваниями или состояниями. Ингибирование активности одной или более металлопротеиназ может быть полезно при таких заболеваниях или состояниях, как например: различные воспалительные и аллергические заболевания, такие как воспаление сустава (особенно ревматоидный артрит, остеоартрит и подагра), воспаление желудочно-кишечного тракта (особенно воспалительное заболевание кишечника, язвенный колит и гастрит), воспаление кожи (особенно псориаз, экзема, дерматит); метастазирование или инвазия опухолей; заболевание, связанное с неконтролируемой деградацией внеклеточного матрикса, такое как остеоартрит; резорбтивное заболевание костей (такое как остеопороз и болезнь Педжета); заболевания, связанные с аберрантным ангиогенезом; ускоренное ремоделирование коллагена, связанное с диабетом, периодонтальным заболеванием (таким как гингивит), изъязвлением роговицы, изъязвлением кожи, послеоперационными состояниями (такими как анастомоз толстой кишки) и заживлением кожных ран; демиелинизирующие заболевания центральной и периферической нервной системы (такие как рассеянный склероз); болезнь Альцгеймера; ремоделирование внеклеточного матрикса, наблюдаемое при сердечно-сосудистых заболеваниях, таких как рестеноз и атеросклероз; астма; ринит; и хроническая обструктивная болезнь легких (ХОБЛ).
ММР12, также известная как макрофагальная эластаза или металлоэластаза, первоначально была клонирована из мыши Shapiro et al. [1992, Journal of Biological Chemistry, 267: 4664] и человека той же группой в 1995 году. ММР12 предпочтительно экспрессируется в активированных макрофагах и, как было показано, выделяется из альвеолярных макрофагов у курильщиков [Shapiro et al., 1993, Journal of Biological Chemistry, 268: 23824], a также в пенистых клетках в атеросклеротических повреждениях [Matsumoto et al., 1998, Am. J. Pathol., 153: 109]. Мышиная модель ХОБЛ основана на введении мышам сигаретного дыма в течение 6 месяцев, по две сигареты в день шесть дней в неделю. У мышей дикого типа развивалась эмфизема легких после этой обработки. Когда по этой модели тестировали мышей с «нокаутированной» ММР12, у них не развивалась какая-либо значительная эмфизема, что с очевидностью указывает на то, что ММР12 является ключевым ферментом в патогенезе ХОБЛ. Роль ММР, таких как ММР12, в ХОБЛ (эмфизема и бронхит), обсуждается в Anderson and Shinagawa, 1999, Current Opinion in Anti-inflammatory and Immunomodulatory Investigational Drugs 1(1): 29-38. Недавно установлено, что курение повышает инфильтрацию макрофагов и экспрессию ММР12 макрофагального происхождения в бляшках Кангавари (Kangavari) сонной артерии человека [Matetzky S., Fishbein М.С. et al., Circulation 102:(18). 36-39 Suppl. S, Oct 31, 2000].
Клинические исследования с ингибиторами матриксных металлопротеиназ часто выявляли серьезные побочные эффекты, именуемые скелетно-мышечным синдромом. Эти побочные эффекты препятствовали дальнейшему развитию лекарственных средств-кандидатов ингибиторов определенных матриксных металлопроиназ. Был предложен ряд гипотез, базирующихся на отсутствии селективности в отношении различных металлопротеиназ для этих лекарственных средств-кандидатов для объяснения скелетно-мышечного синдрома (смотри, например, J. Thomas Peterson, Cardiovascular Research, 69 (2006): 677-687). Для того чтобы свести к минимуму любые возможные вредные скелетно-мышечные побочные эффекты, имеется явное основание для создания селективных ММР-12 ингибиторов для лечения ММР-12 опосредованных заболеваний человека.
Известен ряд ингибиторов металлопротеиназ (смотри, например, обзоры ингибиторов ММР у Beckett R.P. and Whittaker М., 1998, Exp.Opin. Ther. Patents, 8(3):259-282, и Whittaker М. et al., 1999, Chemical Reviews 99(9):2735-2776).
В WO 02/074751 раскрыты производные гидантоина формулы 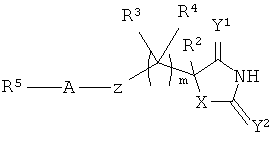
которые являются полезными в качестве ММР ингибиторов.
Авторы настоящего изобретения предлагают еще одну группу производных гидантоина, которые являются ингибиторами металлопротеиназ и представляют особый интерес как мощные и селективные ингибиторы ММР12. Соединения по настоящему изобретению обладают благоприятной эффективностью, селективностью и/или фармакокинетическими свойствами.
Описание изобретения
Согласно настоящему изобретению предложены соединения формулы (I)
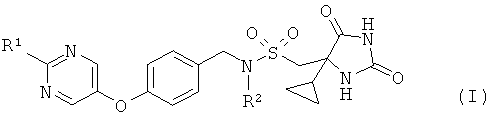
где
R1 представляет собой Н, СН3, СН3СН2, CF3 или циклопропил; и
R2 представляет собой Н или СН3;
и их фармацевтически приемлемые соли.
В одном воплощении R2 представляет собой СН3.
В еще одном воплощении R1 представляет собой циклопропил или CF3.
В еще одном воплощении R1 представляет собой циклопропил, и R2 представляет собой СН3.
В еще одном воплощении R1 представляет собой CF3, и R2 представляет собой СН3.
Примеры соединений по изобретению включают:
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-метил-N-{[4-(пиримидин-5-илокси)фенил]метил}метансульфонамид;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-({4-[(2-циклопропилпиримидин-5-ил)окси]фенил}метил)-N-метилметансульфонамид;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-метил-N-({4-[(2-метилпиримидин-5-ил)окси]фенил}метил)метансульфонамид;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-метил-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}фенил)метил]метансульфонамид;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-({4-[(2-этилпиримидин-5-ил)окси]фенил}метил)-М-метилметансульфонамид;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}бензил)]метансульфонамид;
и их фармацевтически приемлемые соли.
Каждое приведенное в качестве примера соединение представляет собой частный и независимый аспект изобретения.
Соединения формулы (I) могут существовать в энантиомерных формах. Следует понимать, что все энантиомеры, диастереомеры, рацематы и их смеси включены в объем изобретения. Разные оптические изомеры могут быть выделены путем разделения рацемической смеси соединения с помощью традиционных методик, например фракционной кристаллизации или HPLC (высокоэффективная жидкостная хроматография). Альтернативно оптические изомеры могут быть получены посредством ассимметричного синтеза или посредством синтеза из оптически активных исходных веществ.
Если соединение по изобретению существует в виде оптически активных изомеров, все индивидуальные оптически активные формы и их комбинации описываются как индивидуальные конкретные воплощения изобретения так же, как и их соответствующие рацематы.
В одном воплощении соединения формулы (I) имеют (4S)-стереохимию, как показано ниже:
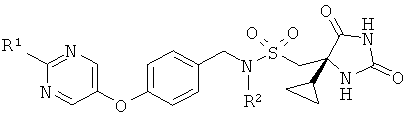
Во избежание неопределенности, (4S)-стереоизомер может быть представлен в виде смеси с (4R)-стереоизомером. Например, (4S)-стереоизомер может быть представлен в смеси 1:1 с (4R)-стереоизомером.
В одном воплощении соединение формулы (I) является оптически чистым. В контексте настоящего описания термин «оптически чистый» определяют в виде энантиомерного избытка (е.е.), который рассчитывают из отношения разницы между количествами соответствующих присутствующих энантиомеров и суммой этих количеств, выраженного в процентах. В качестве иллюстрации, препарат, содержащий 95% одного энантиомера и 5% другого энантиомера, имеет энантиомерный избыток 90% [то есть (95-5)/(95+5)×100]. Оптически чистое соединение по настоящему изобретению имеет е.е. по меньшей мере 90%. В одном воплощении оптически чистое соединение по настоящему изобретению имеет е.е. по меньшей мере 95%. В еще одном воплощении оптически чистое соединение по настоящему изобретению имеет е.е. по меньшей мере 98%.
Если для соединений по изобретению существуют таутомеры, все индивидуальные таутомерные формы и их комбинации включены как индивидуальные конкретные воплощения изобретения.
Настоящее изобретение включает соединения формулы (I) в форме солей. Подходящие соли включают соли, образованные с органическими или неорганическими кислотами либо органическими или неорганическими основаниями. Такие соли обычно представляют собой фармацевтически приемлемые соли, хотя фармацевтически неприемлемые соли могут находить применение в получении и очистке конкретного соединения. Такие соли включают соли присоединения кислот, такие как соли гидрохлорид, гидробромид, цитрат, тозилат и малеат, и соли, образованные с фосфорной кислотой или серной кислотой. В другом аспекте приемлемые соли представляют собой соли с основаниями, такие как соли с щелочными металлами, например натрием или калием, соль с щелочноземельным металлом, например кальцием или магнием, или соль с органическим амином, например триэтиламином.
Соли соединений формулы (I) могут быть образованы путем взаимодействия свободного основания или другой его соли с одним или более эквивалентами соответствующей кислоты или основания.
Соединения формулы (I) являются полезными, поскольку они обладают фармакологической активностью у животных, в частности людей, и поэтому потенциально полезны в качестве фармацевтических средств. В частности, соединения по изобретению являются ингибиторами металлопротеиназ и поэтому могут быть использованы в лечении заболеваний или состояний человека, опосредованных ММР12, таких как астма, ринит, хроническая обструктивная болезнь легких (ХОБЛ), артрит (такой как ревматоидный артрит и остеоартрит), атеросклероз и рестеноз, рак, инвазия и метастазирование, заболевания, в которые вовлечена деструкция тканей, расшатывание протезов тазобедренных суставов, периодонтальное заболевание, фиброзное заболевание, инфаркт и болезнь сердца, фиброз печени и почек, эндометриоз, заболевания, связанные с ослаблением внеклеточного матрикса, сердечная недостаточность, аневризмы аорты, заболевания, связанные с ЦНС, такие как болезнь Альцгеймера и рассеянный склероз (PC), и гематологические расстройства.
В общем, соединения по настоящему изобретению являются мощными и селективными ингибиторами человеческой ММР12 (пММР12). Соединения по настоящему изобретению также демонстрируют хорошую селективность в плане относительного отсутствия ингибирования других различных ПММР, таких как hMMP2, hMMP8, hMMP9, hММР14 и hММР19.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в лечении заболеваний респираторного тракта, таких как обструктивные заболевания дыхательных путей, включая: астму, в том числе бронхиальную, аллергическую, наследственную бронхиальную, приобретенную бронхиальную, индуцированную физической нагрузкой, индуцированную приемом лекарств (включая индуцированную приемом аспирина и NSAID (нестероидные противовоспалительные лекарственные средства)) и индуцированную пылью астму, как интермиттирующую, так и персистирующую, и всех степеней тяжести, и другие случаи гиперчувствительности дыхательных путей; хроническую обструктивную болезнь легких (ХОБЛ); бронхит, в том числе инфекционный и эозинофильный бронхит; эмфизему; бронхоэктаз; респираторный дистресс-синдром взрослых (РДСВ); цистический фиброз; саркоидоз; экзогенный аллергический альвеолит и родственные заболевания; гиперчувствительный пневмонит; пневмофиброз, в том числе криптогенный фиброзный альвеолит, идиопатическую интерстициальную пневмонию, фиброз, являющийся осложнением противоопухолевой терапии и хронической инфекции, включая туберкулез и аспергиллез и другие грибковые инфекции; осложнения легочной трансплантации; васкулитоподобные и тромботические расстройства легочной сосудистой сети и легочную гипертензию; противокашлевую активность, в том числе лечение хронического кашля, ассоциированного с воспалительными и секреторными состояниями дыхательных путей, и ятрогенного кашля; острый и хронический ринит, в том числе медикаментозный ринит и вазомоторный ринит; круглогодичный и сезонный аллергический ринит, в том числе нервный ринит (сенная лихорадка); назальный полипоз; острую вирусную инфекцию, в том числе простуду, и инфекцию, вызванную респираторно-синцитиальным вирусом, вирусом гриппа, коронавирусом (включая атипичную пневмонию (SARS)) и аденовирусом.
Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть использованы в лечении заболеваний костей и суставов, таких как артритиды, ассоциированные с остеоартритом/остеоартрозом или включающие остеоартрит/остеоартроз, как первичный, так и вторичный по отношению, например, к врожденной дисплазии тазобедренного сустава; цервикальный и поясничный спондилит и боль в нижней области спины и шеи; ревматоидный артрит и болезнь Стилла; серонегативные спондилоартропатии, в том числе анкилозирующий спондилит, псориатический артрит, реактивный артрит и недифференцируемая спондилоартропатия; септический артрит и другие ассоциированные с инфекцией артропатии и заболевания кости, такие как туберкулез, в том числе болезнь Потта и полиартрит Понсе; острый и хронический индуцируемый отложением кристаллов синовит, в том числе уратная подагра, заболевание вследствие отложения пирофосфата кальция и воспаление сухожилий, синовиальной сумки и синовиальной жидкости, ассоциированное с апатитом кальция; болезнь Бехчета; первичный и вторичный синдром Шегрена; системный склероз и ограниченная склеродермия; системная красная волчанка, заболевание соединительной ткани смешанного типа и недифференцируемое заболевание соединительной ткани; воспалительные миопатии, в том числе дерматомиозит и полимиозит; ревматическая полимиалгия; юношеский артрит, в том числе идиопатические воспалительные поражения кожи ревматического или подагрического происхождения для суставов любой локализации и ассоциированные синдромы, ревматическая лихорадка и ее системные осложнения; васкулитиды, в том числе гигантоклеточный артериит, синдром Такаясу, синдром Чурга-Штраусса (Churg-Strauss), полиартериит нодозный, микроскопический полиартериит и васкулитиды, ассоциированные с вирусной инфекцией, реакциями гиперчувствительности, криоглобулинами и парапротеинами; боль в нижней части спины; семейная средиземноморская лихорадка, синдром Мукла-Вельса (Muckle-WelIs), семейная ирландская лихорадка, болезнь Кикучи (Kikuchi); индуцированные приемом лекарств артралгии, поражения кожи при тендините и миопатии.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении боли и ремоделирования соединительной ткани при скелетно-мышечных нарушениях из-за повреждения (например, спортивной травмы) или следующих заболеваний: артритиды (например, ревматоидный артрит, остеоартрит, подагра или вызванная кристаллами артропатия), другое заболевание суставов (такое как дегенерация межпозвоночного диска или дегенерация височно-челюстного сустава), заболевание с ремоделированием кости (такое как остеопороз, болезнь Педжета или остеонекроз), полихондриты, склеродермия, смешанное нарушение соединительной ткани, спондилоартропатии или заболевание периодонта (такое как периодонтит).
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний кожи, таких как псориаз, атопический дерматит, контактный дерматит или другие экзематозные поражения кожи и реакции гиперчувствительности замедленного типа; фито- и фотодерматит; себорейная экзема, герпетиформная экзема, красный плоский лишай, склеротический и атрофический лишай, гангренозная пиодермия, кожный саркоид, дискоидная красная волчанка, пузырчатка, пемфигоид, врожденный буллезный эпидермолиз, крапивница, ангионевротические отеки, васкулитиды, токсические эритемы, кожные эозинофилии, гнездная алопеция, облысение по мужскому типу, синдром Свита (Sweet), болезнь Вебера-Крисчена, множественная эритема; целлюлит, как инфекционный, так и неинфекционный; панникулит; кожные лимфомы, немеланомный рак кожи и другие диспластические поражения; индуцированные приемом лекарственных средств расстройства, в том числе стойкая лекарственная сыпь.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний глаза, таких как блефарит; конъюнктивит, в том числе круглогодичный и весенний аллергический конъюнктивит; иритит; передний и задний увеит; хореоидит; аутоиммунная реакция; дегенеративные или воспалительные расстройства, влияющие на сетчатку; офтальмия, в том числе симпатическая офтальмия; саркоидоз; инфекции, в том числе вирусные, грибковые и бактериальные.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний желудочно-кишечного тракта, такого как глоссит, гингивит, периодонтит; эзофагит, в том числе рефлюкс-эзофагит; эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона, колит, в том числе неспецифический язвенный колит, проктит, зуд заднего прохода; глютеновая болезнь, синдром раздраженного кишечника, диарея невоспалительного характера и пищевые аллергии, которые проявляют действие, не связанное с кишечником (например, мигрень, ринит и экзема).
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении заболеваний сердечно-сосудистой системы, таких как атеросклероз, влияющий на коронарное и периферическое кровообращение; перикардит; миокардит, воспалительные и аутоиммунные кардиомиопатии, в том числе миокардиальный саркоид; ишемические реперфузионные поражения; эндокардит, вальвулит и аортит, в том числе инфекционный (например, сифилитический); васкулитиды; заболевания проксимальных и периферических вен, в том числе флебит и тромбоз, включая тромбоз глубоких вен и осложнения варикозных вен.
Соединения формулы (I) и их фармацевтически приемлемые соли можно также использовать в лечении онкологических заболеваний, например в лечении обычных видов рака, в том числе опухолей предстательной железы, молочной железы, легкого, яичников, поджелудочной железы, кишечника и толстой кишки, желудка, кожи, и опухолей головного мозга, и злокачественных новообразований, влияющих на костный мозг (включая лейкозы) и лимфопролиферативные системы, например лимфомы Ходжкина или неходжкинских лимфом; включая предупреждение и лечение метастатического заболевания и опухолевых рецидивов и паранеопластических синдромов.
В одном аспекте соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в лечении респираторного дистресс-синдрома взрослых (РДСВ), цистического фиброза, легочной эмфиземы, бронхита, бронхоэктаза, хронической обструктивной болезни легких (ХОБЛ), легочной гипертензии, астмы, ринита, ишемическо-реперфузионного повреждения, ревматоидного артрита, остеоартрита, рака, атеросклероза, рассеянного склероза (PC), заболевания периодонта и повреждения слизистой оболочки желудка.
В другом аспекте соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в лечении или профилактике воспалительных заболеваний или состояний, ассоциированных с неконтролируемой деградацией внеклеточного матрикса и ремоделированием.
В другом аспекте соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в лечении или профилактике воспалительных заболеваний или состояний.
В частности, соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в лечении хронической обструктивной болезни легких (ХОБЛ), астмы и ринита.
Еще более конкретно, соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в лечении хронической обструктивной болезни легких (ХОБЛ).
Таким образом, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь выше, для применения в терапии.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в терапии.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в лечении заболеваний или состояний человека, при которых полезно ингибирование ММР12.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в лечении воспалительного заболевания.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в лечении обструктивного заболевания дыхательных путей, такого как астма или ХОБЛ.
В дополнительном аспекте в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, в изготовлении лекарственного средства для использования в лечении ревматоидного артрита, остеоартрита, атеросклероза, заболевания периодонта или рассеянного склероза.
В дополнительном аспекте в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь выше, для лечения заболеваний или состояний, при которых полезно ингибирование ММР12.
В дополнительном аспекте в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь выше, для лечения воспалительного заболевания.
В дополнительном аспекте в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь выше, для лечения обструктивного заболевания дыхательных путей, такого как астма или ХОБЛ.
В дополнительном аспекте в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь выше, для лечения ревматоидного артрита, остеоартрита, атеросклероза, заболевания периодонта или рассеянного склероза.
В контексте настоящего описания термин «терапия» также включает «профилактику», если нет конкретных указаний на противоположное. Термины «терапевтический» и «терапевтически» следует понимать соответственно.
Профилактика представляется особенно подходящей для лечения субъектов, которые пострадали от предыдущего эпизода или иным образом считаются имеющими повышенный риск данного заболевания или состояния. Субъекты, имеющие риск развития конкретного заболевания или состояния, обычно включают в себя тех, кто имеет семейную историю этого заболевания или состояния, или тех, кто был идентифицирован генетическим тестированием или скринингом как особенно подверженный развитию этого заболевания или состояния.
В данном изобретении также дополнительно предложен способ лечения или снижения риска заболевания или состояния, при котором полезно ингибирование ММР12, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше.
В данном изобретении также дополнительно предложен способ лечения или снижения риска воспалительного заболевания или состояния, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше.
В данном изобретении также предложен способ лечения или снижения риска обструктивного заболевания дыхательных путей, такого ка астма или ХОБЛ, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше.
Для указанных терапевтических применений вводимые дозировки будут, конечно, варьировать в зависимости от используемого соединения, способа введения, желаемого лечения и указанного расстройства. Суточная дозировка соединения формулы (1)/соли (активный ингредиент) может находиться в диапазоне от 0,001 мг/кг до 75 мг/кг, в частности от 0,5 мг/кг до 30 мг/кг. Эта суточная доза при необходимости может быть введена в разделенных дозах. Обычно формы стандартной дозы содержат приблизительно от 1 мг до 500 мг соединения по данному изобретению.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы сами по себе, но обычно их будут вводить в форме фармацевтической композиции, в которой соединение формулы (1)/соль (активный ингредиент) находится в сочетании с фармацевтически приемлемыми адъювантом, разбавителем или носителем. В зависимости от способа введения фармацевтическая композиция будет предпочтительно содержать от 0,05 до 99 мас.% (массовых процента), более предпочтительно от 0,10 до 70 мас.% активного ингредиента и от 1 до 99,95 мас.%, более предпочтительно от 30 до 99,90 мас.% фармацевтически приемлемых адъюванта, разбавителя или носителя, причем все массовые проценты рассчитаны исходя из общей массы композиции. Общепринятые способы выбора и изготовления подходящих фармацевтических композиций раскрыты, например, в "Pharmaceuticals - The Science of Dosage Form Designs", M.E.Aulton, Churchill Livingstone, 1988.
В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь выше, вместе с фармацевтически приемлемыми адъювантом, разбавителем или носителем.
В этом изобретении дополнительно предложен способ изготовления фармацевтической композиции по изобретению, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь выше, с фармацевтически приемлемыми адъювантом, разбавителем или носителем.
Фармацевтические композиции по данному изобретению могут быть введены стандартным образом для заболевания или состояния, которое желательно лечить, например посредством перорального, местного, парентерального, трансбуккального, интраназального, вагинального или ректального введения или посредством ингаляции. Для этих целей соединения по данному изобретению могут быть приготовлены в виде препарата средствами, известными в уровне техники, например в виде таблеток, капсул, водных или масляных растворов, суспензий, эмульсий, кремов, мазей, гелей, назальных спреев, суппозиториев, мелкодисперсных порошков или аэрозолей для ингаляции, и для парентерального применения (включая внутривенное, внутримышечное введение или инфузию) в виде стерильных водных или масляных растворов или суспензий или стерильных эмульсий.
Помимо соединений по настоящему изобретению фармацевтическая композиция по данному изобретению может также содержать или может быть введена совместно (одновременно или последовательно) с одним или более фармакологических агентов, пригодных для лечения одного или более заболеваний или состояний, упоминаемых здесь ранее, таких как продукт "Symbicort" (товарный знак).
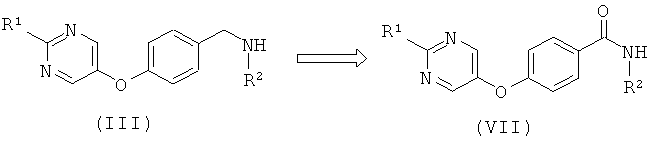
В настоящем изобретении также предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, включающий:
взаимодействие соединения формулы (II)
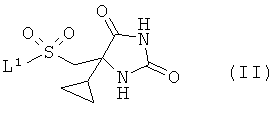
где L1 представляет собой уходящую группу, с соединением формулы (III) или его солью
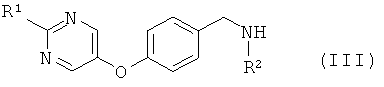
где R1 и R2 являются такими, как определено в формуле (I);
и возможно после этого образование его фармацевтически приемлемой соли.
В вышеописанном способе подходящие уходящие группы L1 включают галоген, в частности хлор, или трифторметилсульфонат. Взаимодействие предпочтительно осуществляют в подходящем растворителе или смеси растворителей, возможно в присутствии добавленного основания, в течение соответствующего периода времени, обычно от 0,1 до 16 ч, при температуре от 0°С до температуры дефлегмации. Обычно используют такие растворители, как N,N-диметилформамид, пиридин, тетрагидрофуран, ацетонитрил, N-метилпирролидин или дихлорметан. Добавленное основание, если оно используется, может представлять собой органическое основание, такое как триэтиламин, N.N-диизопропилэтиламин (DIPEA), N-метилморфолин или пиридин, либо неорганическое основание, такое как карбонат щелочного металла. Взаимодействие обычно проводят при температуре окружающей среды в течение периода времени от 0,5 до 16 ч, либо до достижения завершения реакции, что определяют хроматографическими или спектроскопическими методами. Реакции сульфонилгалогенидов с разными первичными и вторичными аминами хорошо известны из литературы, и вариации условий будут очевидны для специалиста в данной области.
Сульфонилхлориды формулы (II), где L1 представляет собой хлор, описаны в WO 2006/065215 и упомянутых там ссылках.
Амины формулы (III) предпочтительно образуются посредством восстановительного алкилирования первичного амина или аммиака, R2 -NH2, с помощью 4-(пиримидин-5-илокси)-бензальдегида формулы (IV), используя стандартные условия, которые очевидны для специалиста в данной области. Обычно альдегид (IV) кипятят с обратным холодильником с избытком амина R2-NH2 в таком растворителе, как этанол, в течение 1-2 часов. Избыток амина затем выпаривают и промежуточный имин перерастворяют в этаноле. В результате гидрирования при атмосферном давлении с палладием (0) на углероде в течение 0,5-2 часов при температуре окружающей среды получают амин (III).
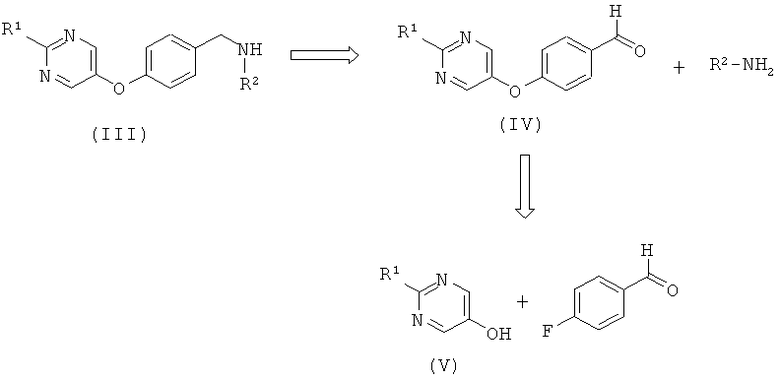
Альдегиды формулы (IV) удобным образом образуют посредством реакции нуклеофильного ароматического замещения между 4-фтор-бензальдегидом и пиримидин-5-олом (V). Условия реакции, которые являются очевидными для специалиста, могут включать нагревание с основанием в полярном апротонном растворителе, таком как тетрагидрофуран, диоксан, ацетонитрил, N,N-диметилформамид, N-метилпирролидин или диметилсульфоксид. Обычная процедура включает смешивание 4-фтор-бензальдегида и пиримидин-5-ола (V) с избытком карбоната калия или трет-бутоксида калия в N,N-диметилформамиде и N-метилпирролидине и нагревание при 120°С в течение приблизительно 16 часов с получением альдегида (IV).
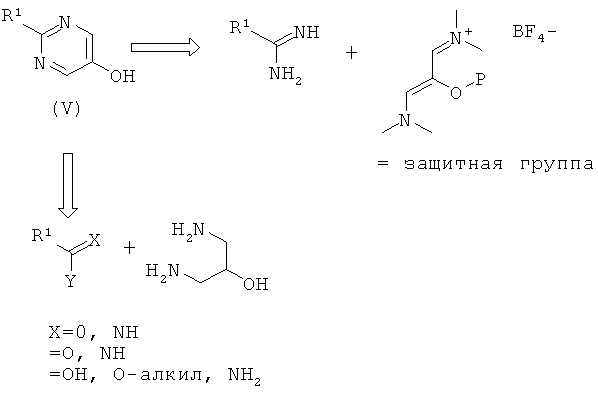
Пиримидин-5-олы формулы (V) могут быть получены различными способами, известными в уровне техники. Комплексный обзор по синтезу пиримидинов смотри в S. Von Angerer, Science of Synthesis, (2004), 16, 379-572. Два таких пути синтеза кратко раскрыты в данном описании.
По первому пути амидин, R1-C(=NH)-NH2, подвергают сочетанию с солью N-[3-(диметиламино)-2-гидроксипроп-2-ен-1-илиден]-N-метилметанаминия по существу так, как описано в US 4558039. Гидроксильную группу предпочтительно защищают, например в виде бензилового эфира. Подходящей солью является тетрафторборат.
По второму пути пиримидин-5-олы, алкиловый сложный эфир, кислоту или амидин подвергают конденсации с 1,3-диамино-пропан-2-олом. Полученное промежуточное соединение с замкнутым кольцом, 1,4,5,6-тетрагидро-пиримидин-5-ол, затем окисляют с получением пиримидин-5-ола (V). Смотри, например, US 5175166 или Hull, J. W. J.; Otterson, K.; Rhubright, D.; J. Org. Chem. 1993, 58, 520-522. Обычно конденсацию осуществляют в толуоле или ксилене при температуре дефлегмации в течение 5-24 часов с азеотропным удалением образовавшейся воды. Промежуточное соединение тетрагидро-пиримидин в конечном итоге выделяют в виде соли, такой как гидрохлорид. Окисление обычно осуществляют, используя избыток нитробензола и основание, такое как метоксид натрия, трет-бутоксид калия или гидроксид калия, при 120° С в течение 1-5 часов. Могут быть использованы сорастворители, такие как толуол или ксилен.
Амины формулы (III), где R2 представляет собой Н, обычно получают восстановлением нитрила (VI). Нитрил (VI) в свою очередь может быть образован посредством реакции нуклеофильного ароматического замещения между 4-замещенным бензонитрилом и пиримидин-5-олом способом, аналогичным способу, описанному для образования альдегида (IV).
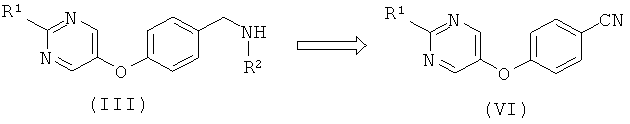
Дополнительный альтернативный путь синтеза аминов (III) включает восстановление амида (VII). Амид (VII) в свою очередь может быть образован посредством восстановления соответствующего нитрила (VI) или его синтетического эквивалента с последующей N-защитой, R2 алкилированием и процедурой снятия защиты.
Для специалиста в данной области очевидно, что в способах по настоящему изобретению определенные потенциально реакционноспособные функциональные группы, такие как гидроксильные или аминогруппы, в исходных реагентах или промежуточных соединениях могут нуждаться в защите подходящими защитными группами. Таким образом, получение соединений по изобретению может включать на разных стадиях введение и удаление одной или более защитных групп.
Подходящие защитные группы и подробности способов введения и удаления таких групп описаны в "Protective Groups in Organic Chemistry', edited by J.W.F. McOmie, Plenum Press (1973) и 'Protective Groups in Organic Synthesis', 3rd edition, T.W. Greene and P.G.M. Wuts, Wiley-Interscience (1999).
Конкретные способы получения соединений формулы (I) описаны в разделе «Примеры» настоящего описания. Такие способы составляют один аспект настоящего изобретения.
Необходимые исходные вещества либо имеются в продаже, либо известны из литературы, либо могут быть получены с помощью известных методик. Конкретные способы получения ряда ключевых исходных веществ описаны в разделе «Примеры» настоящего описания, и такие способы составляют один аспект настоящего изобретения.
Определенные новые промежуточные соединения описаны в разделе «Примеры» настоящего описания, и такие промежуточные соединения составляют один аспект настоящего изобретения.
Так, в одном воплощении новые амины формулы (III) и их соли, где R1 и R2 являются такими, как определено выше, описаны как промежуточные соединения, полезные в получении соединений формулы (I).
Соединения формулы (I) могут быть превращены в другие соединения формулы (I) с помощью стандартных процедур.
Соединения по изобретению и их промежуточные соединения могут быть выделены из их реакционных смесей и, если необходимо, дополнительно очищены с использованием стандартных процедур.
Соединения по изобретению можно также вводить в сочетании с другими соединениями, используемыми для лечения указанных выше состояний.
Таким образом, данное изобретение дополнительно относится к комбинированным терапиям, где соединение по изобретению или его фармацевтически приемлемую соль, или фармацевтическую композицию, либо препарат, содержащие соединение по изобретению, вводят одновременно или последовательно либо в виде комбинированного препарата с другим терапевтическим агентом или агентами для лечения одного или более чем одного из перечисленных выше состояний.
В частности, для лечения воспалительных заболеваний, таких как (но без ограничения ими) ревматоидный артрит, остеоартрит, астма, аллергический ринит, хроническая обструктивная болезнь легких (ХОБЛ), псориаз, заболевание периодонта и воспалительное заболевание кишечника, соединения по изобретению можно объединять с агентами, перечисленными ниже.
Нестероидные противовоспалительные агенты (далее NSAID), в том числе неселективные ингибиторы циклооксигеназ СОХ-1 и СОХ-2, применяемые как местно, так и системно (такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флурбипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, азапропазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин); селективные ингибиторы СОХ-2 (такие как мелоксикам, целекоксиб, рофекоксиб, валдекоксиб, лумарококсиб, парекоксиб и эторикоксиб); ингибирующие циклооксигеназу доноры оксида азота (CINOD); глюкокортикостероиды (вводимые как местным, так и пероральным, внутримышечным, внутривенным или внутрисуставным путем); метотрексат; лефлуномид; гидроксихлороквин; d-пеницилламин; ауранофин или другие парентеральные или пероральные препараты золота; анальгетики; диацереин; внутрисуставные лекарственные средства, такие как производные гиалуроновой кислоты; и пищевые добавки, такие как глюкозамин.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с цитокином или агонистом либо антагонистом функции цитокинов (включая агенты, которые действуют на цитокиновые сигнальные пути, такие как модуляторы системы супрессоров цитокиновых сигнальных путей, SOCS), включая альфа-, бета- и гамма-интерфероны; инсулиноподобный фактор роста типа I (IGF-1); интерлейкины (IL), в том числе IL1-23, и антагонисты или ингибиторы интерлейкинов, такие как анакинра; ингибиторы фактора некроза опухолей альфа (TNF-a), такие как моноклональные антитела против TNF (например, инфликсимаб, адалимумаб и CDP-870) и антагонисты рецепторов TNF, в том числе иммуноглобулиновые молекулы (такие как этанерцепт) и низкомолекулярные агенты, такие как пентоксифиллин.
В дополнение, это изобретение относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с моноклональным антителом, направленным на В-лимфоциты (таким как CD20 (ритуксимаб)), MRA-alLI6R, или Т-лимфоциты (CTLA4-lg, HuMax 11-15).
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с модулятором функции рецепторов хемокинов, таким как антагонист CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства С-С); CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства С-Х-С) и СХ3CR1 для семейства С-Х3-С.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором биосинтеза лейкотриенов, ингибитором 5-липоксигеназы (5-LO) или антагонистом белка, активирующего 5-липоксигеназу (FLAP), таким как: зилейтон; АВТ-761; фенлейтон; тепоксалин; Abbott-79175; Abbott-85761; N-(5-замещенный)-тиофен-2-алкилсульфонамид; 2,6-ди-трет-бутилфенол-гидразоны; метокситетрагидропираны, такие как Zeneca ZD-2138; соединение SB-210661; соединение, представляющее собой пиридинил-замещенный 2-цианонафталин, такое как L-739010; соединение, представляющее собой 2-цианохинолин, такое как L-746530; или индольное или хинолиновое соединение, такое как МК-591, МК-886 и BAY×1005.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом рецепторов лейкотриенов (LT) B4, LTC4, LTD4 и LTE4, выбранным из группы, состоящей из фенотиазинов-3-1, таких как L-651392; амидиновых соединений, таких как CGS-25019c; бензоксаламинов, таких как онтазоласт; бензокарбоксимидамидов, таких как BIIL 284/260; и таких соединений, как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст (МК-679), RG-12525, Ro-245913, иралукаст (CGP 45715А) и BAY×7195.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором фосфодиэстеразы (PDE), таким как метилксантанин, включая теофиллин и аминофиллин; селективным ингибитором изофермента PDE, включая ингибитор PDE4, ингибитор изоформы PDE4D или ингибитор PDE5.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом гистаминовых рецепторов типа 1, таким как цетиризин, лоратадин, дезлоратадин, фексофенадин, акривастин, терфенадин, астемизол, азеластин, левокабастин, хлорфенирамин, прометазин, циклизин или мизоластин, применяемым перорально, местно или парентерально.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором протонного насоса (таким как омепразол) или гастропротективным антагонистом гистаминовых рецепторов типа 2.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом гистаминовых рецепторов типа 4.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с сосудосуживающим симпатомиметиком, представляющим собой агонист адренорецепторов альфа-1/альфа-2, таким как пропилгекседрин, фенилэфрин, фенилпропаноламин, эфедрин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид, трамазолина гидрохлорид или этилнорэпинефрина гидрохлорид.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антихолинергическими агентами, в том числе антагонистом мускаринового рецептора (М1, М2 и М3), таким как атропин, хиосцин, гликопирролат, ипратропия бромид, тиотропия бромид, окситропия бромид, пирензепин или телензепин.
Настоящее изобретение также относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли и агониста бета-адренорецептора (включая бета-рецепторы подтипов 1-4), такого как изопреналин, сальбутамол, формотерол, сальметерол, тербуталин, орципреналин, битолтерола мезилат или пирбутерол, либо их хиральный энантиомер.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с хромоном, таким как кромогликат натрия или недокромил натрия.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с глюкокортикоидом, таким как флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат, циклесонид или мометазона фуроат.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом, который модулирует ядерные рецепторы гормона, такие как рецепторы, активируемые пролифератором пероксисом (PPAR).
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с иммуноглобулином (Ig) или препаратом Ig либо антагонистом или антителом, модулирующим функционирование Ig, таким как анти-IgE (например омализумаб).
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с другим системным или местно наносимым противовоспалительным агентом, таким как талидомид или его производное, ретиноид, дитранол или кальципотриол.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с комбинациями аминосалицилатов и сульфапиридина, такого как сульфасалазин, месалазин, бальсалазид и олсалазин; и иммуномодуляторными агентами, такими как тиопурины, и кортикостероидами, такими как будесонид.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с антибактериальным агентом, таким как производное пенициллина, тетрациклин, макролид, бета-лактам, фторхинолон, метронидазол, ингалируемый аминогликозид; противовирусным агентом, включая ацикловир, фамцикловир, валацикловир, ганцикловир, цидофовир, амантадин, римантадин, рибавирин, занамавир и оселтамавир; ингибитором протеазы, таким как индинавир, нелфинавир, ритонавир и саквинавир; нуклеозидным ингибитором обратной транскриптазы, таким как диданозин, ламивудин, ставудин, залцитабин или зидовудин; или ненуклеозидным ингибитором обратной транскриптазы, таким как невирапин или эфавиренц.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с сердечно-сосудистым агентом, таким как блокатор кальциевых каналов, блокатор бета-адренорецепторов, ингибитор ангиотензин-конвертирующего фермента (АСЕ), антагонист рецепторов ангиотензина-2; агентом снижения липидов, таким как статин или фибрат; модулятором морфологии клеток крови, таким как пентоксифиллин; тромболитиком или антикоагулянтом, таким как ингибитор агрегации тромбоцитов.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом, действующим на ЦНС, таким как антидепрессант (такой как сертралин), лекарственное средство против болезни Паркинсона (такое как депренил, L-ДОФА, ропинирол, прамипексол, ингибитор моноаминооксидазы В (МАОВ), такой как селегин и расагилин, ингибитор соединения Р (соmР), такой как тасмар, ингибитор А-2, ингибитор обратного захвата дофамина, антагонист М-метил-D-аспартата (NMDA), никотиновый агонист, дофаминовый агонист или ингибитор нейрональной синтазы оксида азота), или лекарственное средство против болезни Альцгеймера, такое как донепезил, ривастигмин, такрин, ингибитор СОХ-2, пропентофиллин или метрифонат.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом для лечения острой или хронической боли, таким как действующий центрально или периферически анальгетик (например, опиоид или его производное), карбамазепин, фенитоин, вальпроат натрия, амитриптилин или другие антидепрессанты, парацетамол или нестероидный противовоспалительный агент.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с парентерально или местно применяемым (в том числе ингалируемым) локальным обезболивающим средством, таким как лигнокаин или его производное.
Соединение по настоящему изобретению или его фармацевтически приемлемую соль можно также использовать в комбинации с агентом против остеопороза, включая гормональный агент, такой как ралоксифен или бифосфонат, такой как алендронат.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с: (1) ингибитором триптазы; (2) антагонистом фактора активации тромбоцитов (PAF); (3) ингибитором интерлейкин-превращающего фермента (ICE); (4) ингибитором инозинмонофосфатдегидрогеназы IMPDH; (5) ингибитором молекул адгезии, в том числе антагонистами VLA-4 (очень поздний антиген); (6) катепсином; (7) ингибитором киназы, таким как ингибитор тирозинкиназы (такой как Btk, Itk, Jak3 или MAP, например, гефитиниб или мезилат иматиниба), серин/треонинкиназы (таким как ингибитор митоген-активируемой протеинкиназы (МАР-киназы), такой как р38, JNK, протеинкиназа А, В или С, или киназа ингибиторной киназы (IKK)), или киназы, участвующей в регуляции клеточного цикла (такой как циклин-зависимая киназа); (8) ингибитором глюкозо-6-фосфат-дегидрогеназы; (9) антагонистом рецепторов кинин-B1 либо -В2; (10) средством против подагры, например колхицином; (11) ингибитором ксантин-оксидазы, например аллопуринолом; (12) средством, способствующим выведению мочевой кислоты, например пробенецидом, сульфинпиразоном или бензбромароном; (13) стимулятором секреции гормона роста; (14) трансформирующим фактором роста (TGFβ); (15) тромбоцитарным фактором роста (PDGF); (16) фактором роста фибробластов, например основным фактором роста фибробластов (bFGF); (17) гранулоцитарно-макрофагальным колониестимулирующим фактором (GM-CSF); (18) капсаициновым кремом; (19) антагонистом рецепторов тахикинина NK1 или MK3, выбранным из группы, состоящей из NKP-608C, SB-233412 (талнетант) или D-4418; (20) ингибитором эластазы, выбранным из группы, состоящей из UT-77 или ZD-0892; (21) ингибитором THF-альфа-конвертирующего фермента (ТАСЕ); (22) ингибитором индуцибельной синтазы оксида азота (iNOS); (23) рецептор-гомологичной молекулой для хемоаттрактанта, экспрессированной на ТН2-клетках (такой как антагонист CRTH2); (24) ингибитором Р38; (25) агентом модулирования функционирования Toll-подобных рецепторов (TLR); (26) агентом модулирования активности пуринергических рецепторов, таких как Р2Х7; (27) ингибитором фактора активации транскрипции, такого как NF-кВ, API или STATS.
Соединение по изобретению или его фармацевтически приемлемая соль могут также быть использованы в комбинации с существующим терапевтическим агентом для лечения рака, причем подходящие агенты включают, например:
(1) антипролиферативное/антинеопластическое лекарственное средство или их комбинацию, используемые в медицинской онкологии, такое как алкилирующий агент (например, цис-платин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан или нитрозомочевина); антиметаболит (например, антифолат, такой как фторпиримидин, например 5-фторурацил или тегафур, ралтитрексед, метотрексат, цитозина арабинозид, гидроксимочевина, гемцитабин или паклитаксел); противоопухолевый антибиотик (например, антрациклин, такой как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин или митрамицин); антимитотический агент (например, алкалоид барвинка, такой как винкристин, винбластин, виндезин или винорелбин, или таксоид, такой как таксол или таксотере); или ингибитор топоизомеразы (например, эпиподофиллотоксин, такой как этопозид, тенипозид, амсакрин, топотекан или камптотецин);
(2) цитостатический агент, такой как антиэстроген (например, тамоксифен, торемифен, ралоксифен, дролоксифен или иодоксифен), обратный регулятор эстрогенового рецептора (например, фулвестрант), антиандроген (например, бикалутамид, флутамид, нилутамид или ципротерона ацетат), антагонист LHRH (лютеинизирующий гормон роста) или агонист LHRH (например, гозерелин, леупрорелин или бузерелин), прогестоген (например, мерестрола ацетат), ингибитор ароматазы (например, анастрозол, летрозол, воразол или экземестан) или ингибитор 5α-редуктазы, такой как финастерид;
(3) агент, который ингибирует инвазию раковых клеток (например, ингибитор металлопротеиназ, такой как маримастат, или ингибитор рецепторного функционирования урокиназного активатора плазминогена);
(4) ингибитор функционирования ростовых факторов, например: антитело к ростовому фактору (например, анти-erbb2 антитело трастузумаб или анти-erbb1 антитело цетуксимаб [С225]), ингибитор фарнезилтрансферазы, ингибитор тирозинкиназ или ингибитор серин/треонинкиназ, ингибитор семейства эпидермальных факторов роста (например, ингибитор EGFR семейства тирозинкиназ, такой как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, AZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) или 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)-хиназолин-4-амин (CI 1033)), ингибитор семейства факторов роста тромбоцитарного происхождения или ингибитор семейства факторов роста гепатоцитарного происхождения;
(5) антиангиогенный агент, такой как агент, который ингибирует эффекты васкулярно-эндотелиального фактора роста (например, антитело против васкулярно-эндотелиального клеточного фактора роста бевацизумаб, соединение, описанное в WO 97/22596, WO 97/30035, WO 97/32856 или WO 98/13354), или соединение, которое действует по другому механизму (например линомид, ингибитор функционирования интегрина avβ3 или ангиостатин);
(6) агент, поражающий сосуды, такой как комбретастатин А4, или соединение, описанное в WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 или WO 02/08213;
(7) агент, используемый в антисмысловой терапии, например агент, нацеленный на одну из мишеней, указанных выше, такой как ISIS 2503, анти-ras антисмысловой агент;
(8) агент, используемый в методологии генной терапии, например в подходах, заключающихся в замене аберрантных генов, таких как подходы в отношении аберрантного р53 или аберрантного BRCA1 или BRCA2, GDEPT (нацеленная на гены ферментная пролекарственная терапия), такие как подходы с использованием цитозиндезаминазы, тимидинкиназы или бактериальной нитроредуктазы, и подходы, состоящие в увеличении устойчивости пациента к химиотерапии или радиотерапии, такие как генная терапия множественной лекарственной устойчивости; или
(9) агент, используемый в иммунотерапевтическом подходе, например ex-vivo и in-vivo методы для увеличения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, методы снижения вялости Т-клеток, методы с использованием трансфицированных иммунных клеток, таких как цитокин-трансфицированные дендритные клетки, методы с использованием цитокин-трансфицированных опухолевых клеточных линий и методы с использованием анти-идиотипических антител.
В одном аспекте изобретения предложен фармацевтический продукт, содержащий, в комбинации, два или более активных ингредиентов, включая первый активный ингредиент, который представляет собой соединение формулы (I), как оно определено выше, и один или более дополнительных активных ингредиентов, которые выбраны из:
- ингибитора фосфодиэстеразы;
- агониста β2-адренорецептора;
- модулятора функционирования хемокинового рецептора;
- ингибитора функционирования киназы;
- ингибитора протеазы;
- глюкокортикоида;
- антихолинергического агента и
- нестероидного агониста глюкокортикоидного рецептора.
Примерами ингибитора фосфодиэстеразы являются ингибитор PDE4, включая ингибитор изоформы PDE4D, или ингибитор PDE5; примеры селективного агониста β2-адренорецептора включают метапротеренол, изопротеренол, изопреналин, альбутерол, сальбутамол, формотерол, сальметерол, тербуталин, орципреналин, битолтерол мезилат, пирбутерол или индакатерол; примерами антагониста мускариновых рецепторов являются М1-, М2- или М3-антагонист, такого как селективный М3-антагонист, такой как ипратропия бромид, тиотропия бромид, окситропия бромид, пирензепин, телензепин; примером модулятора функционирования хемокинового рецептора является антагонист CCR1-рецептора; примером ингибитора киназы является ингибитор функционирования р38 или IKK2; примером ингибитора протеазы является ингибитор нейтрофильной эластазы; примеры глюкокртикоида включают флунисолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат, циклесонид или мометазона фуроат.
Настоящее изобретение будет дополнительно проиллюстрировано посредством ссылки на следующие иллюстративные примеры.
Общие методы
1-ЯМР спектры записывали на приборе Varian Inova при 400 МГц или Varian Mercury-VX при 300 МГц. Центральные пики хлороформа-d (δн 7,27 м.д.), диметилсульфоксида-d6 (δн 2,50 м.д.) или метанола-d4 (δн 3,31 м.д.) использовали как внутренние стандарты. Использовали следующие аббревиатуры: s, синглет; br s, широкий синглет; d, дублет; dd, двойной дублет; ddd, двойной двойной дублет; t, триплет; dt, двойной триплет; q, квартет; m, мультиплет. Для мультиплетов величина химического сдвига приводится либо для центра сигнала, либо в виде диапазона. Аналитическую тонкослойную хроматографию проводили на пластинах силикагеля 60 (Merck) с флуоресцентным индикатором. Колоночную хроматографию проводили на силикагеле (0,040-0,063 мм, Merck) с приложением к колонке незначительно повышенного давления (0,2-0,4 бар (20-40 кПа)). Для препаративной HPLC использовали колонку Kromasil KR-100-5-C18 (250×20 мм, Akzo Nobel) и смеси ацетонитрила и воды (с 0,1 об.% трифторуксусной кислоты, добавленной, где указано) при скорости потока 10 мл в минуту. Фракции, содержавшие желаемое соединение, объединяли, концентрировали посредством роторного испарителя и в заключение подвергали сублимационной сушке. Если не указано иного, исходные вещества имелись в продаже. Все растворители и коммерческие реагенты имели лабораторную чистоту и использовались в том виде, как были получены. Операции осуществляли при температуре окружающей среды, то есть в диапазоне от 17 до 25°С и в атмосфере инертного газа, такого как аргон, если не указано иного. Время проведения реакций для завершения взаимодействий в примерах может быть больше или меньше, чем указано. Органические фазы, полученные в результате экстракций, сушили над безводным сульфатом натрия, если не указано иного, и концентрировали посредством роторного испарителя. Выходы не оптимизировали.
Для LC-MS анализа использвали следующий метод:
Прибор Agilent 1100; колонка Waters Symmetry 2,1×30 мм; массовая APCI (химическая ионизация при атмосферном давлении); скорость потока 0,7 мл/мин; длина волны 254 или 220 нм; растворитель А: вода +0,1% TFA; растворитель В: ацетонитрил +0,1% TFA; градиент 15-95% /В 2,7 мин, 95% В 0,3 мин.
Для GC-MS анализа использовали следующий метод:
Прибор Hewlett Packard 5890 Series II; колонка Agilent HP-5 (30 м×0,32 мм ID (внутренний диаметр)); массовый селективный детектор Hewlett Packard 5971 Series; давление 55 кПа Не; программа нагрева: от 100°С (3 мин) до 300°С, 25°С/мин.
Сокращения:
Пример 1
1-[(4S)-4-Циклопропил-2.5-диоксоимидазолидин-4-ил]-N-метил-N-{[4-(пиримидин-5-илокси)фенил]метил}метансульфонамид. соль трифторуксусной кислоты
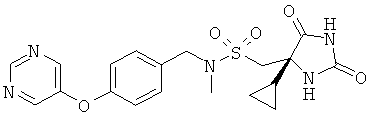
N-Метил-М-{[4-(пиримидин-5-илокси)фенил]метил}амин (0,043 г, 0,20 ммоль) перемешивали в NMP (1,0 мл). Смесь охлаждали с помощью холодной водяной бани и добавляли DIPEA (36 мкл, 0,22 ммоль), затем порциями в течение 5 минут добавляли (4S)-(4-циклопропил-2,5-диоксоимидазолидин-4-ил)метансульфонилхлорид (WO 2006/065215; 0,051 г, 0,20 ммоль). Через 10 минут добавляли воду и продукт экстрагировали три раза EtOAc. Экстракты промывали рассолом, сушили и концентрировали. Продукт очищали посредством препаративной HPLC (0,1% TFA в элюенте) с получением 0,057 г (66%) указанного в заголовке соединения в виде соли трифторуксусной кислоты.
APCI-MSm/z:432(M+1).
1H ЯМР (DMSO-d6): δ 0.12-0.26 (m, 1H), 0.35-0.58 (m, 3H), 1.12 -1.22 (m, 1H), 2.67 (s, 3H), 3.60 (d, 2H), 4.25 (q, 2H), 7.28 (q, 4H), 7.97 (d, 1H), 8.63 (s, 2H), 9.01 (s, 1Н), 10.74 (s, 1Н) м.д.
Исходные вещества получали следующим образом.
а) 5-(Метилокси)пиримидин
Получали, как в Chem. Eur. J. 2003, 9, 4997-5010, исходя из 31 ммоль, с выходом 47% после очистки.
1H ЯМР (CDCl3): δ 3.93 (s, 3H), 8.42 (s, 2H), 8.86 (s, 1H) м.д.
б) Пиримидин-5-ол
Получали, как в Chem. Eur. J. 2003, 9, 4997-5010, исходя из 15 ммоль, с выходом 27% после очистки.
1H ЯМР (DMSO-d6: δ 8.33 (s, 2Н), 8.66 (s, 1H), 10.45 (s, 1H) м.д.
в) 4-(Пиримидин-5-илокси)бензальдегид
К перемешиваемому раствору пиримидин-5-ола (0,384 г, 4,0 ммоль) в DMF (4,0 мл) добавляли 4-фторбензальдегид (0,429 г, 4,0 ммоль), метансульфинат натрия (0,118 г, 1,0 ммоль) и карбонат калия (0,828 г, 6,0 ммоль). Реакционную смесь нагревали при 120°С в течение 3 часов, охлаждали до температуры окружающей среды, обрабатывали водой и экстрагировали три раза ТВМЕ. Экстракты промывали рассолом, сушили и концентрировали. Колоночная хроматография давала 0,523 г (43%) указанного в подзаголовке соединения.
APCI-MS m/z: 201 (М+1).
1H ЯМР (CDCI3): δ 7.15 (dd, 2Н), 7.93 (dt, 2Н), 8.58 (s, 2Н), 9.13 (s, 1H), 9.98 (s, 1Н) м.д.
г) N-Метил-N-{[4-(пиримидин-5-илокси)фенил]метил}амин
4-(Пиримидин-5-илокси)бензальдегид (0,344 г, 1,7 ммоль) перемешивали с 33%-ным метиламином в ЕtOН (30 мл) с кипячением с обратным холодильником в течение 3 часов и затем концентрировали. Остаток растворяли в МеОН и обрабатывали боргидридом натрия (0,195 г, 5,2 ммоль) на протяжении 1 часа и перемешивали в течение еще 1 часа. Добавляли воду и продукт экстрагировали три раза ЕtOАс. Органические фазы объединяли, промывали рассолом, сушили и концентрировали с получением 0,267 г (100%) продукта, который использовали без дополнительной очистки.
APCI-MS m/z: 216 (М+1).
1H ЯМР (CDCI3): δ 2.38 (s, 3H), 3.68 (s, 2Н), 6.94 (d, 2Н), 7.28 (d, 2Н), 8.38 (d, 2Н), 8.87 (s, 1H) м.д.
Пример 2
1-[(4S)-4-Циклопропил-2.5-диоксоимидазолидин-4-ил]-N-({4-[(2-цикло-пропилпиримидин-5-ил)окси]фенил}метил)-N-метилметансульфонамид
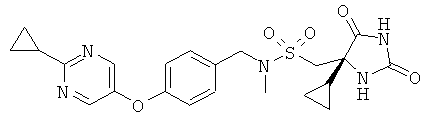
Сырой {4-[(2-циклопропилпиримидин-5-ил)окси]бензил}метиламин дигидрохлорид (0,115 г, 0,35 ммоль) растворяли в NMP (2,0 мл), THF (2,0 мл) и DIPEA (0,30 мл, 1,8 ммоль) с образованием желтого раствора. На протяжении 5 минут порциями добавляли (4S)-(4-циклопропил-2,5-диоксоимидазолидин-4-ил)метансульфонилхлорид (WO 2006/065215; 0,070 г, 0,28 ммоль) и реакционную смесь перемешивали в течение 1 часа. Растворитель удаляли выпариванием и остаток разбавляли водой и дважды экстрагировали ЕtOАс. Объединенные органические фазы промывали водой и концентрировали. Сырой продукт очищали посредством HPLC, используя 35-минутный градиент от 20% до 90% MeCN в воде, с получением 0,081 г (61%-ный выход) указанного в заголовке соединения в виде бесцветного порошка.
APCI-MS m/z 472,1 (М+1); Rt=1,93 мин.
1H-ЯМР (DMSO-d6): δ 0.14-0.24 (m, 1H), 0.33-0.57 (m, 3H), 0.96 (m, 2H), 1.03 (m, 2H), 1,15 (m, 1H), 2.22 (m, 1H), 2.65 (s, 3H), 3.44 (d, 1H), 3.75 (d, 1H), 4.23 (q, 2H), 7.09 (d, 2H), 7.34 (d, 2H), 7.96 (s, 1H), 8.45 (s, 2H), 10.74 (s, 1H) м.д.
Исходные вещества получали следующим образом.
а) 5-(Бензилокси)-2-циклопропилпиримидин
Указанное в подзаголовке соединение синтезировали, используя способ, раскрытый в US 4558039, начиная с циклопропилкарбамидина гидрохлорида.
LC-APCI-MS m/z 227,0 (М+1); Rt=2,36 мин.
1H-ЯМР (DMSO-d6): δ 0.86-0.99 (m, 4H), 2.14 (m, 1H), 5.21 (s, 2H), 7.31-7.47 (m, 5H), 8.44 (s, 2H) м.д.
б) 2-Циклопропилпиримидин-5-ол
5-(Бензилокси)-2-циклопропилпиримидин (4,0 г, 18 ммоль) растворяли в МеОН (100 мл) и добавляли 10% палладий на углероде (0,170 г). Смесь гидрировали в течение ночи при температуре окружающей среды и давлении 1,013 бар (0,1013 МПа). В результате фильтрования и концентрирования получали сырой продукт, который фильтровали через короткую колонку с силикагелем, используя в качестве элюента смесь 5% МеОН-ЕtOАс. В результате выпаривания растворителя получали 1,3 г (54%-ный выход) указанного в подзаголовке соединения в виде бесцветного твердого вещества.
GC-MS m/z 136,0 M+ (относительная интенсивность 41%) 135,0 (относительная интенсивность 100%); Rt=7,36 мин.
1H-ЯМР (DMSO-d6): δ 0.80-0.96 (m, 4H), 2.09 (m, 1H), 8.17 (s, 2H), 10.02 (s, 1Н) м.д.
в) 4-[(2-Циклопропилпиримидин-5-ил)окси]бензальдегид
2-Циклопропилпиримидин-5-ол (0,272 г, 2,0 ммоль), 4-фторбензальдегид (0,22 мл, 2,1 ммоль) и карбонат калия (0,414 г, 3,0 ммоль) в сухом DMF (2,0 мл) нагревали до 120°С в запаянной пробирке в течение 3 часов. Суспензию разбавляли водой и дважды экстрагировали ЕtOAс. Объединенные органические фазы промывали три раза водой и рассолом, сушили, фильтровали и концентрировали с получением желтого масла. В результате очистки посредством колоночной хроматографии с использованием 20 г диоксида кремния и градиента от 0% до 50% ЕtOАс-гептаны в качестве элюента получали 0,478 г (99%-ный выход) указанного в подзаголовке соединения в виде бесцветного масла.
LC-APCI-MS m/z 241,1 (M+1); Rt=1,98 мин.
1H-ЯМР (CDCl3): δ 1.04-1.17 (m, 4H), 2.30 (m, 1H), 7.07 (d, 2H), 7.88 (d, 2H), 8.40 (s, 2H), 9.94 (s, 1Н) м.д.
г) {4-[(2-Циклопропилпиримидин-5-ил)окси]бензил}метиламин дигидрохлорид
К раствору 4-[(2-циклопропилпиримидин-5-ил)окси]бензальдегида (0,240 г, 1,0 ммоль) в MeCN (0,50 мл) добавляли 2 М метиламин в THF (2,0 мл, 4,0 ммоль), а затем боргидрид натрия (0,120 г, 3,2 ммоль) и MeCN (0,50 мл). Суспензию перемешивали в течение 30 минут. Растворители удаляли выпариванием, добавляли воду и смесь экстрагировали ЕtOАс. Органическую фазу промывали рассолом и выпаривали на силикагеле. Этот гель наносили на колонку с 20 г силикагеля. Посредством колоночной хроматографии с градиентом от 10% до 60% ЕtOАс в гептанах элюировали примеси. В результате элюции с 10% МеОН в ЕtOАс (100 мл), а затем 5% концентрированого МН3 в МеОН (100 мл) получали продукт в основных фракциях. Эти фракции объединяли, концентрировали и перерастворяли в воде. Величину рН доводили до 13-14, используя раствор гидроксида натрия, и смесь несколько раз экстрагировали ЕtOАс. Органические экстракты промывали рассолом, сушили над карбонатом калия, фильтровали и концентрировали с получением масляного остатка. Это масло растворяли в ЕtOс и добавляли избыток 1,5 М раствора хлористого водорода в ЕtOАс. Растворитель удаляли выпариванием с получением 0,186 г (56%-ный выход) сырого указанного в подзаголовке соединения. Полученная соль имела чистоту 93,9% и ее использовали без дополнительной очистки.
LC-APCI-MS m/z 256,1 (М+1- 2 HCI); Rt=1,49 мин.
1H-ЯМР (CD3OD): δ 1.21-1.44 (m, 4H), 2.36 (m, 1H), 2.74 (s, 3H), 4.22 (s, 2H), 7.26 (d, 2H), 7.60 (d, 2H), 8.72 (s, 2H) м.д.
Пример 3
1-[(4S)-4-Циклопропил-2.5-диоксоимидазолидин-4-ил]-N-метил-N-({4-[(2-метилпиримидин-5-ил)окси]фенил}метил)метансульфонамид
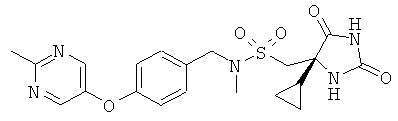
Получали, как в Примере 1, но начиная с N-метил-1-{4-[(2-метилпиримидин-5-ил)окси]-фенил}метанамина, исходя из 0,50 ммоль, с выходом 61% после очистки.
APCI-MS m/z 446(M+1).
1H ЯМР (DMSO-d6): δ 0.13 - 0.24 (m, 1H), 0.33 - 0.57 (m, 3H), 1.15 (ddd, 1H), 2.61 (d, 3H), 2.66 (s, 3H), 3.60 (dd, 2H), 4.23 (dd, 2H), 7.11 (dd, 2H), 7.35 (dd, 2H), 7.97 (s, 1H), 8.52 (s, 2H), 10.74 (s, 1H) м.д.
Исходные вещества получали следующим образом.
а) 2-Метил-5-[(Фенилметил)окси]пиримидин
Получали, как в Примере 2(а), исходя из 15 ммоль, с выходом 73% после очистки.
APCI-MS m/z: 201 (М+1).
1H ЯМР (CDCI3): δ 2.67 (s, 3H), 5.13 (s, 2H), 7.31-7.50 (m, 5H), 8.37 (s, 2H) м.д.
б) 2-Метилпиримидин-5-ол
Получали, как в Примере 2(б), исходя из 11 ммоль, с выходом 100% и использовали без дополнительной очистки.
1H ЯМР (DMSO-d6): δ 8.10 (s, 2H), 2.50 (s, 3H) м.д.
в) 4-[(2-Метилпиримидин-5-ил)окси]бензальдегид
Получали, как в Примере 1(в), исходя из 11 ммоль, с выходом 22% после очистки.
APCI-MS m/z 214(M+1).
1H ЯМР (CDCl3): δ 2.78 (s, 3Н), 7.11 (dd, 2H), 7.91 (dd, 2H), 8.49 (s, 2H), 9.97 (s, 1H) м.д.
г) N-Метил-1-{4-[(2-метилпиримидин-5-ил)окси]фенил}метанамин
Получали, как в Примере 1(г), исходя из 2,4 ммоль, с выходом 82% после очистки.
APCI-MS m/z 228(M+1).
1H ЯМР (CDCl3): δ 2.46 (s, 3Н), 2.72 (s, 3Н), 3.74 (s, 2H), 6.98 (d, 2H), 7.33 (d, 2H), 8.38 (s, 2H) м.д.
Пример 4
1-[(4S)-4-Циклопропил-2.5-диоксоимидазолидин-4-ил]-N-метил-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}фенил)метил]метансульфонамид
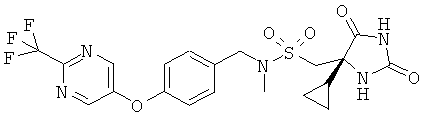
Получали, как в Примере 1, но начиная с N-метил-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}фенил)метил]амина, исходя из 0,60 ммоль, с выходом 7,5% после очистки.
APCI-MS m/z 500(M+1).
1H ЯМР (DMSO-d6) δ 0.14-0.25 (m, 1H), 0.46 (m, 3Н). 1.16 (m, 1H), 2.68 (s, 3Н), 3.47 (d, 3Н), 3.77 (d, 1H), 4.28 (m, 2H), 7.30 (d, 2H), 7.42 (d, 2H), 7.98 (br s, 1H), 8.81 (s, 2H), 10.75 (br s, 1H) м.д.
Исходные вещества получали следующим образом.
а) 2-(Трифторметил)-1.4.5.б-тетрагидропиримидин-5-ол гидрохлорид
Свободное основание получали, как описано в US 5175166, исходя из 114 ммоль. Сырой продукт растворяли в пропан-2-оле, обрабатывали 6 М хлористым водородом в пропан-2-оле и продукт отфильтровывали в виде белых кристаллов с выходом 86%.
APCI-MS m/z 169(M+1).
1H ЯМР (DMSO-d6): δ 3.39 (d, 2H), 3.51 (d, 2H), 4.25 (q, 1H), 6.32 (s, 1H), 12.11 (s,1Н)м.д.
б) 2-(Трифторметил)пиримидин-5-ол
2-(Трифторметил)-1,4,5,6-тетрагидропиримидин-5-ол (4,20 г, 25 ммоль) перемешивали в нитробензоле при 90°С. Метоксид натрия (5,4 г, 100 ммоль) растворяли в метаноле (75 мл) и порциями добавляли к реакционной смеси, отгоняя метанол перед последующим добавлением. Реакционную смесь затем нагревали до 121°С в течение одного часа, охлаждали, встряхивали с водой (150 мл), органическую фазу отделяли, а водную фазу промывали этилацетатом (2×100 мл). Водную фазу доводили до рН 4,0 с помощью 6 М водной соляной кислоты, экстрагировали этилацетатом (2×100 мл), сушили и выпаривали с получением 2,53 г (61,7%) окрашенного в оранжевый цвет продукта, который использовали без дополнительной очистки.
APCI-MS m/z 165(M+1).
1H ЯМР (DMSO-d6): δ 8.54 (s, 2H), 11.48 (s, 1H) м.д.
в) 4-{[2-(Трифторметил)пиримидин-5-ил]окси}бензальдегид
Получали, как в Примере 1(в), исходя из 5,0 ммоль, с выходом 74%.
GC-MS m/z=268 (М+).
1H ЯМР (DMSO-d6): δ 7.44 (d, 2H), 7.99 (d, 2H), 8.95 (s, 2H), 9.97 (d, 1H) м.д.
г) N-Метил-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}фенил)-метил]амин
4-{[2-(Трифторметил)пиримидин-5-ил]окси}бензальдегид перемешивали с 33%-ным метиламином в 95%-ном этаноле (30 мл) при кипячении с обратным холодильником в течение 1 часа и затем концентрировали. Остаток перерастворяли в 95%-ном этаноле, добавляли 10% палладий/углерод и смесь гидрировали при комнатной температуре при атмосферном давлении в течение 30 минут. Взаимодействие осуществляли, исходя из 5,0 ммоль, с выходом 95%.
APCI-MS m/z 284 (M+1).
1H ЯМР (CDCl3): δ 2.49 (s, 3H), 3.80 (s, 2H), 5.30 (s, 1H), 7.08 (dd, 2H), 7.43 (dd, 2H), 8.53 (s, 2H) м.д.
Пример 5
1-[(4S)-4-Циклопропил-2.5-диоксоимидазолидин-4-ил]-N-({4-[(2-этилпиримидин-5-ил)окси]фенил}метил)-N-метилметансульфонамид
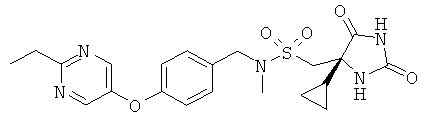
Получали, как в Примере 1, но начиная с [4-(2-этилпиримидин-5-илокси)бензил]метиламина, исходя из 1,6 ммоль, с выходом 37% после очистки.
APCI-MS m/z 460(M+1).
1H ЯМР (DMSO-d6): δ 0.12-0.28 (m, 1H), 0.35-0.58 (m, 3H), 1.08-1.20 (m, 1H), 1.28 (t, 3H), 2.66 (s, 3H), 2.90 (q, 2H), 3.44 (d, 1H), 3.75 (d, 1H), 4.23 (dd, 2H), 7.14 (d, 2H), 7.35 (d, 2H), 7.97 (s, 1H), 8.55 (s, 2H), 10.74 (s, 1H) м.д.
Исходные вещества получали следующим образом.
а) 2-Этилпиримидин-5-ол
Получали, как в Примерах 2(а) и 2(б), исходя из 11 ммоль, с общим выходом 69%, и использовали без дополнительной очистки.
APCI-MS m/z 125(M+1).
1H ЯМР (CDCl6): δ 1.25 (t, 3H), 2.8 (q, 2H), 8.28 (s, 2H), 11.3 (br s, 1H) м.д.
б) 4-(2-Этилпиримидин-5-илокси)бензальдегид
Получали, как в Примере 1(в), исходя из 2,0 ммоль, с выходом 83% после очистки.
APCI-MS m/z 229,1 (М+1).
1H ЯМР (DMSO-d6): δ 1.31 (t, 3H), 2.94 (q, 2H), 7.24 (dd, 2H), 7.96 (d, 2H), 8.70 (s, 2H), 9.96 (s, 1H) м.д.
в) [4-(2-Этилпиримидин-5-илокси)-бензил] метиламин
Получали, как в Примере 1(г), исходя из 1,6 ммоль, с выходом 83% после очистки.
APCI-MS m/z: 244,1 (М+1).
1H ЯМР (DMSO-d6): δ 1.25 (t, 3H), 2.22 (s, 3H), 2.86 (q, 2H), 3.58 (s, 2H), 7.01 (dd, 2H), 7.31 (d, 2H), 8.48 (s, 2H) м.д.
Пример 6
1-[(4S)-4-1-Циклопропил-2.5-диоксоимидазолидин-4-ил]-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}бензил)]метансульсфонамид
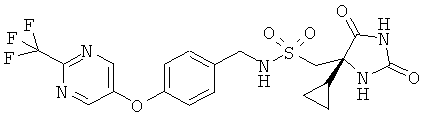
Получали, как в Примере 1, но начиная с (4-{[2-(трифторметил)пиримидин-5-ил]окси}фенил)метил]амина, исходя из 0,26 ммоль, с выходом 75% после очистки.
APCI-MS m/z 486,1 (М+1).
1H ЯМР (DMSO-d6): δ 0.14 - 0.25 (m, 1H), 0.46 (m, 3H), 1.16 (m, 1H), 3.25 (d, 1H), 3.62 (d, 1H), 4.28 (m, 2H), 7.30 (d, 2H), 7.42 (d, 2H), 7.76 (t, 1H), 7.85 (s, 1H), 8.81 (s, 2H), 10.75(br s, 1H) м.д.
Исходные вещества получали следующим образом.
а) 4-{[2-(Трифторметил)пиримидин-5-ил]окси}бензонитрил
Получали, как в Примере 1(в), из 2-циклопропилпиримидин-5-ола и 4-фтор-бензонитрила, исходя из 6,1 ммоль, с выходом 55%.
GC-MS m/z=265,1 (M+).
б) 4-{[2-(Трифторметил)пиримидин-5-ил]окси}бензиламин гидрохлорид
4-{[2-(Трифторметил)пиримидин-5-ил]окси}бензонитрил гидрировали в смеси 1:1 НОАс:ЕtOН, содержащей 10% Pd/C. Сырой продукт очищали посредством HPLC, используя 25-минутный градиент от 10% до 70% MeCN-вода/TFA 0,1%, с получением указанного в подзаголовке соединения.
APCI-MS m/z 270,1 (М+1).
1H ЯМР (DMSO-d6): δ 4.8 (q, 2H), 7.35 (d, 2H), 7.56 (d, 2H), 8.18 (b, 3H), 8.79 (s, 1Н) м.д.
Фармакологический пример
Анализы с выделенными ферментами
ММР12
Каталитический домен рекомбинантной человеческой ММР12 может быть экспрессирован и очищен, как описано Parkar A.A. et al., (2000), Protein Expression and Purification, 20, 152. Очищенный фермент может быть использован для мониторинга ингибиторов его активности следующим образом: ММР12 (конечная концентрация 50 нг/мл) инкубируют в течение 60 минут при комнатной температуре с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (10 мкМ) в буфере для анализа (0,1М "Tris-HCI" (товарный знак) буфер, рН 7,3, содержащий 0,1 М NaCI, 20 мМ CaCl2, 0,020 мМ ZnCI и 0,05% (мас./об.) детергента "Brij 35" (товарный знак)) в присутствии (10 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресциенции при λех 320 нм и λеm 405 нм. Процент ингибирования рассчитывают следующим образом:
% ингибирования равен частному от деления [флуоресценцияплюс ингибитор-флуоресценцияфон] на [флуоресценцияминус ингибитор-флуоресценцияфон].
ММР8
Очищенную про-ММР8 получали от Calbiochem. Фермент (в концентрации 10 мкг/мл) активируют ацетатом п-амино-фенил-ртути (II) (АРМА) в концентрации 1 мМ в тчение 2,5 ч, 35°С. Активированный фермент может быть использован для мониторинга ингибиторов его активности следующим образом: ММР8 (конечная концентрация 200 нг/мл) инкубируют в течение 90 минут при 35°С (80% H2O) с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (12,5 мкМ) в буфере для анализа (0,1 М "Tris-HCI" (товарный знак) буфер, рН 7,5, содержащий 0,1 М NaCI, 30 мМ СаСl2, 0,040 мМ ZnCI и 0,05% (мас./об.) детергента "Brij 35" (товарный знак)) в присутствии (10 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при λех 320 нм и λет 405 нм. Процент ингибирования рассчитывают следующим образом:
% ингибирования равен частному от деления [флуоресценцияплюс ингибитор-флуоресценцияфон] на [флуоресценцияминус ингибитор-флуоресценцияфон].
ММР9
Каталитический домен рекомбинантной человеческой ММР9 экспрессировали и затем очищали посредством Zn-хелатной колоночной хроматографии, а затем посредством колоночной хроматографии со сродством к гидроксамату. Фермент может быть использован для мониторинга ингибиторов его активности следующим образом: ММР9 (конечная концентрация 5 нг/мл) инкубируют в течение 30 минут при КТ с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (5 мкМ) в буфере для анализа (0,1 М "Tris-HCI" (товарный знак) буфер, рН 7,3, содержащий 0,1 М NaCI, 20 мМ CaCl2, 0,020 мМ ZnCI и 0,05% (мас./об.) детергента "Brij 35" (товарный знак)) в присутствии (10 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при λех 320 нм и λеm 405 нм. Процент ингибирования рассчитывают следующим образом:
% ингибирования равен частному от деления [флуоресценцияплюс ингибитор-флуоресценцияфон] на [флуоресценцияминус ингибитор-флуоресценцияфон].
ММР14
Каталитический домен рекомбинантной человеческой ММР14 может быть экспрессирован и очищен, как описано Parkar A.A. et al., (2000), Protein Expression and Purification, 20, 152. Очищенный фермент может быть использован для мониторинга ингибиторов его активности следующим образом:ММР14 (конечная концентрация 10 нг/мл) инкубируют в течение 60 минут при комнатной температуре с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (10 мкМ) в буфере для анализа (0,1 М "Tris-HCI" (товарный знак) буфер, рН 7,5, содержащий 0,1 М NaCI, 20 мМ CaCl2, 0,020 мМ ZnCI и 0,05% (мас./об.) детергента "Brij 35" (товарный знак)) в присутствии (5 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при λех 320 нм и λеm 405 нм. Процент ингибирования рассчитывают следующим образом:
% ингибирования равен частному от деления [флуоресценцияминус ингибитор-флуоресценцияфон] на [флуоресценцияминус ингибитор-флуоресценцияфон].
Протокол тестирования в отношении других матричных металлопротеиназ, включая ММР9, с использованием экспрессированной и очищенной про-ММР описан, например, С.Graham Knight et al., (1992) FEBS Lett., 296(3), 263-266.
MMP19
Каталитический домен рекомбинантной человеческой MMP19 может быть экспрессирован и очищен, как описано Parkar A.A. et al., (2000), Protein Expression and Purification, 20, 152. Очищенный фермент может быть использован для мониторинга ингибиторов его активности следующим образом: MMP19 (конечная концентрация 40 нг/мл) инкубируют в течение 120 минут при 35°С с синтетическим субстратом Mca-Pro-Leu-Ala-Nva-Dpa-Ala-Arg-NH2 (5 мкМ) в буфере для анализа (0,1 М "Tris-HCI" (товарный знак) буфер, рН 7,3, содержащий 0,1 М NaCI, 20 мМ CaCl2, 0,020 мМ ZnCI и 0,05% (мас./об.) детергента "Brij 35" (товарный знак)) в присутствии (5 концентраций) или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при λех 320 нм и λеm 405 нм. Процент ингибирования рассчитывают следующим образом:
% ингибирования равен частному от деления [флуоресценцияплюс ингибитор-флуоресценцияфон] на [флуоресценцияминус ингибитор-флуоресценцияфон].
В следующей далее таблице представлены данные для ряда репрезентативных соединений по настоящему изобретению в сравнении со структурно наиболее близким соединением, описанным в WO 02/074751. Селективность ингибирования hMMP12 относительно hMMPx определяют как величину, равную частному от деления IC50 (50%-ная ингибирующая концентрация, ММРх) на IC50 (ММР12).
IC50 (НМ)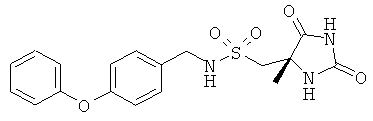
Как ясно видно из данных, представленных в Таблице, соединения по настоящему изобретению по сравнению с 1-[(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]-М-[(4-фенокси)фенил)метил]метансульфонамидом являются, с одной стороны, существенно более активными ингибиторами hMMP12 и, с другой стороны, значительно более селективны в отношении ингибирования других hMMPs, в частности hMMP9.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2376301C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2378269C2 |
| НОВЫЕ ГИДАНТОИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОБСТРУКТИВНЫХ ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2005 |
|
RU2386629C2 |
| 2, 5-ДИОКСОИМИДАЗОЛИДИН-4-ИЛ-МЕТИЛКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2003 |
|
RU2326117C2 |
| ПРОИЗВОДНЫЕ 2-ПИРАЗИНОНА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ ИЛИ СОСТОЯНИЯ, ПРИ КОТОРЫХ ПОЛЕЗНО ИНГИБИРОВАНИЕ АКТИВНОСТИ НЕЙТРОФИЛЬНОЙ ЭЛАСТАЗЫ | 2007 |
|
RU2448098C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2288228C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2293729C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE10 | 2011 |
|
RU2543386C2 |
| ФЕНИЛТЕТРАГИДРОИЗОХИНОЛИНОВОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ ГЕТЕРОАРИЛОМ | 2015 |
|
RU2703456C2 |
Изобретение относится к соединениям формулы (I) и их фармацевтически приемлемым солям, обладающим свойствами ингибитора ММР12, способу их получения, промежуточному соединению формулы (III), фармацевтической композиции, способу ее изготовления, применению соединений формулы (I) и вариантам способов лечения с использованием соединений формулы (I). Соединения могут быть использованы для лечения заболеваний, опосредованных ММР12, таких как хроническая обструктивная болезнь легких. В формуле (I) и (III)
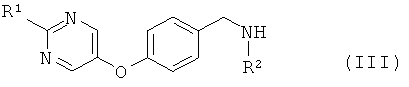
R1 представляет собой Н, СН3, СН3СН2, CF3 или циклопропил; и R2 представляет собой Н или СН3. 8 н. и 7 з.п. ф-лы, 1 табл., 6 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль
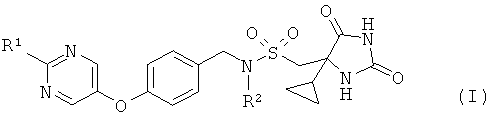
где R1 представляет собой Н, СН3, СН3СН2, CF3 или циклопропил; и R2 представляет собой Н или СН3.
2. Соединение по п.1, имеющее (48)-стереохимию, как показано ниже
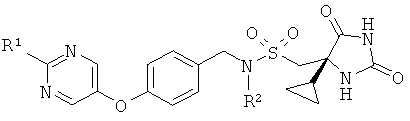
3. Соединение по п.1, где R представляет собой метил.
4. Соединение по п.1, где R1 представляет собой циклопропил или CF3.
5. Соединение по п.1, где R1 представляет собой циклопропил.
6. Соединение по п.1, где R1 представляет собой CF3.
7. Соединение по п.1, которое выбрано из группы, состоящей из:
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-метил-N-{[4-(пиримидин-5-илокси)фенил]метил}метансульфонамида;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-({4-[(2-циклопропилпиримидин-5-ил)окси]фенил}метил)-N-метилметансульфонамида;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-метил-N-({4-[(2-метилпиримидин-5-ил)окси]фенил}метил)метансульфонамида;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-метил-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}фенил)метил]метансульфонамида;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-({4-[(2-этилпиримидин-5-ил)окси]фенил}метил)-N-метилметансульфонамида;
1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}бензил)]метансульфонамида;
и их фармацевтически приемлемых солей.
8. Соединение, представляющее собой 1-[(4S)-4-циклопропил-2,5-диоксоимидазолидин-4-ил]-N-метил-N-[(4-{[2-(трифторметил)пиримидин-5-ил]окси}фенил)метил]метансульфонамид или его фармацевтически приемлемые соли.
9. Способ получения соединения формулы (I), как оно определено по п.1, или его фармацевтически приемлемой соли, включающий:
взаимодействие соединения формулы (II)
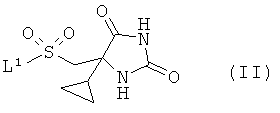
где L1 представляет собой уходящую группу, с соединением формулы (III) (или его солью)
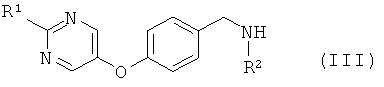
где R1 и R2 являются такими, как определено в формуле (I); и возможно после этого образование его фармацевтически приемлемой соли.
10. Соединение формулы (III) или его соль,
где R1 и R2 являются такими, как определено по п.1, для применения в качестве промежуточного соединения в получении соединений формулы (I).
11. Фармацевтическая композиция, обладающая свойствами ингибитора ММР12, содержащая соединение формулы (I) или его фармацевтически приемлемую соль по любому из пп.1-8 вместе с фармацевтически приемлемыми адъювантом, разбавителем или носителем.
12. Способ изготовления фармацевтической композиции по п.11, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли, как определено по любому из пп.1-8, с фармацевтически приемлемыми адъювантом, разбавителем или носителем.
13. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-8 в изготовлении лекарственного средства для использования в лечении хронической обструктивной болезни легких.
14. Способ лечения заболевания или состояния, опосредованных ММР12 (матриксная металлопротеиназа 12), включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-8.
15. Способ лечения хронической обструктивной болезни легких, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-8.
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

