Настоящее изобретение относится к способам получения гексагидрофуро[2,3-b]фуран-3-ола и особенно его энантиомера (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола, а также некоторым новым промежуточным продуктам для применения в таких способах.
(3R,3aS,6aR)-Гексагидрофуро[2,3-b]фуран-3-оксирадикал является важной фармакологической частью, присутствующей в структуре ингибиторов ретровирусной протеазы, таких как ингибиторы, описанные Glosh et al. в J. Med. Chem. 1996, 39(17), 3278-3290, а также ингибиторов, описанных в WO 95/24385, WO 99/65870, WO 99/67254, WO 99/67417, WO 00/47551, WO 00/76961, WO 01/25240, US 6127372 и EP 0715618. Указанные публикации включены здесь в качестве ссылки. Один такой ингибитор протеазы, который разрешен в США для клинического применения при лечении ретровирусных инфекций у людей и имеет указанную выше структурную часть, является соединением, имеющим разрешенное USAN название дарунавир и химическое название (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-иловый эфир [(1S,2R)-3-[[(4-аминофенил)сульфонил](2-метоксипропил)амино]-2-гидрокси-1-(фенилметил)пропил]карбаминовой кислоты и структуру формулы (А)
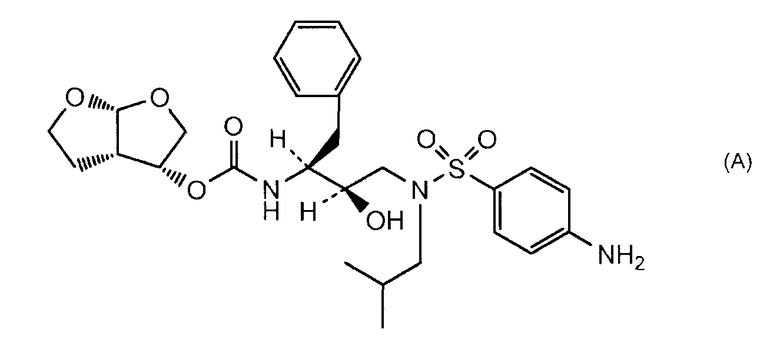
Важным предшественником в синтезе ингибиторов протеазы, описанных выше и содержащих гексагидрофуро[2,3-b]фуран-3-оксирадикал, является соединение гексагидрофуро[2,3-b]фуран-3-ол формулы (I)
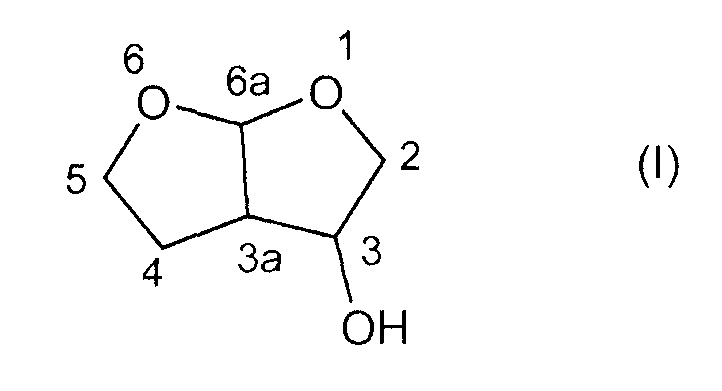
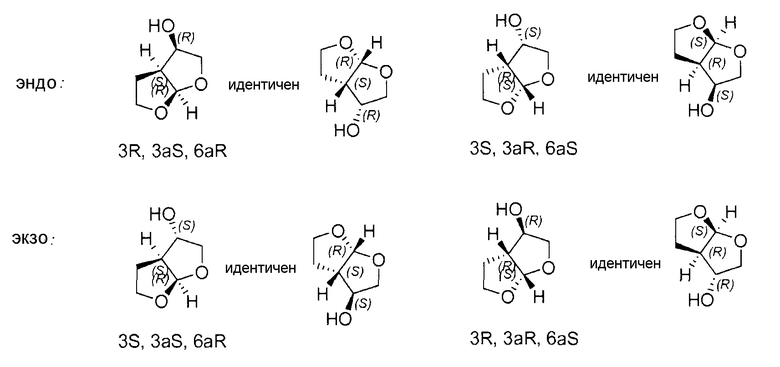
Несмотря на тот факт, что гексагидрофуро[2,3-b]фуран-3-ол имеет три стереогенных центра и теоретически должны иметь место восемь различных стереоизомеров, считают, что существует только четыре стереоизомера. Это обусловлено жесткостью структуры бициклического кольца в гексагидрофуро[2,3-b]фуран-3-оле, которая заставляет его транс-конденсированные изомеры быть термодинамически невыгодными. Только стереоизомеры, имеющие цис-конденсированную конфигурацию, являются термодинамически стабильными, уменьшая число стереоизомеров гексагидрофуро[2,3-b]фуран-3-ола до эндо- и экзо-диастереоизомеров, причем каждый содержит пару энантиомеров, как показано ниже
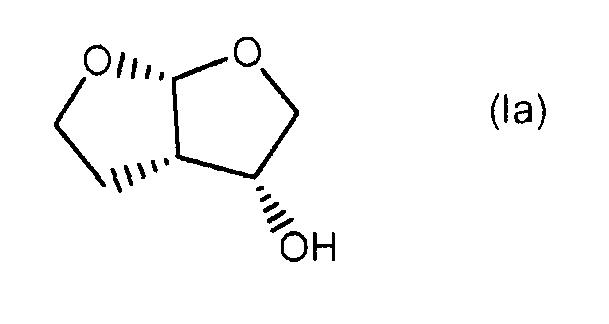
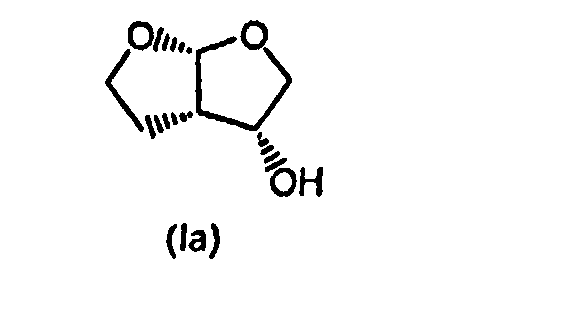
Более конкретно, для получения таких ингибиторов протеазы, содержащих энантиомерный (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-оксирадикал, таких как ингибитор протеазы дарунавир, относящийся к указанному выше (3R,3aS,6aR)-энантиомеру формулы (Ia), особенно применимым является
Ввиду потенциальной важности указанных выше ингибиторов протеазы и являющаяся результатом этого необходимость производства этих соединений в коммерческом масштабе, в литературе были многочисленные предложения по способам, которыми можно получить указанные выше соединения формул (I) и (Ia).
Много таких предложений включали в себя образование структуры бициклического бисфурана, исходя из нециклических предшественников, например включающих в себя промежуточное образование лактонового промежуточного соединения и затем восстановление и циклизацию, такими способами, как способы получения, описанные в WO 03/022853, US 2004/0162340, WO 2004/033462, US 6867321, WO 2005/095410, а также Ghosh et al., J. Org. Chem. 2004, 69, 7822-7829. Эти способы включают в себя относительно большое число стадий, и в некоторых случаях образование нитрометильного промежуточного соединения, требующее применение нитрометана, который является вредным реагентом. Другой подход, описанный в WO 02/060905, включает в себя реакцию 2,3-дигидрофурана с алкинилпроизводным с образованием производного 2-алкинилоксифурана, которое затем циклизуют в присутствии излучения. Применение излучения, однако, является неподходящим для практического применения способа в промышленном масштабе. Применение излучения требуется также в способе, описанном в WO 03/024974, в котором фуран подвергают реакции с карбонильным производным в присутствии излучения. В WO 2004/002975 описан способ, исходящий из 2,3-дигидрофурана, который подвергают реакции, например, с хлорглиоксилатным эфиром для осуществления введения глиоксилатной группы в 3-положение кольца фурана и затем восстановления с образованием 1,2-дигидроксиэтильной боковой цепи и с последующей обработкой, например, галогенирующим агентом с образованием соединения 3а-галогенгексагидро[2,3-b]фуран-3-ола, которое затем восстанавливают. Этот способ также имеет недостаток, заключающийся в том, что требуются многочисленные стадии при получении из исходного фурана, что является неэкономичным для промышленного масштаба.
Аналогичный подход предложен Ghosh et al. in Tetrahedron Letters 40 (1999) 1083-1086, включающий в себя реакцию 2,3-дигидрофурана с этилглиоксилатом и тетрахлоридом титана с получением промежуточного оксониевого иона, который затем подвергают реакции с нуклеофилом, получая при этом производные 3-(β-карбоэтокси-α-гидроксиметил)-2-замещенного тетрагидрофурана. Примеры таких нуклеофилов содержат силилпроизводные и метанол. В единственном описанном примере смесь этилглиоксилата и 2,3-дигидрофурана в дихлорметане добавляли к раствору тетрахлорида титана в дихлорметане при -78°С и перемешивали в течение одного часа. К смеси при -78°С добавляли аллилтриметилсилан и образовавшуюся смесь перемешивали при температуре от -78°С до 23°С в течение одного часа. Реакцию гасили водным раствором гидрокарбоната натрия и экстрагировали этилацетатом и объединенные органические слои сушили, концентрировали и затем очищали флэш-хроматографией. Данный способ страдает некоторыми недостатками, например применением очень низкой температуры реакции -78°С, которая практически невозможна для реакции промышленного масштаба. Кроме того, авторами настоящего изобретения обнаружено, что применение способа с тетрахлоридом титана, описанного Ghosh et al., вызывает проблемы при последующей обработке для обеспечения эффективного удаления соединения титана, причем удаление соли титана является основным условием, чтобы избежать примеси и побочных реакций в последующих стадиях.
Задачей изобретения является обеспечение нового и усовершенствованного синтеза для получения гексагидрофуро[2,3-b]фуран-3-ола. Дополнительной задачей изобретения является обеспечение такого синтеза, в котором применяют легко доступные и экономичные исходные соединения и применяют условия реакции, которые легко достижимы при синтезе в промышленном масштабе. Дополнительной задачей изобретения является обеспечение пригодного способа получения производных 3-(β-карбоэтокси-α-гидроксиметил)-2-замещенного тетрагидрофурана и их аналогов. Дополнительной задачей настоящего изобретения является обеспечение новых и пригодных промежуточных продуктов, применимых в синтезе гексагидрофуро[2,3-b]фуран-3-ола. Следующей задачей изобретения является обеспечение нового и усовершенствованного синтеза (3R,3aR,6aR)-гексагидрофуро[2,3-b]фуран-3-ола, применимого при получении антиретровирусных ингибиторов протеазы.
Обнаружено, что применение некоторых солей титана, отличных от тетрахлорида титана, использованных Ghosh et al. в описанном выше способе, обеспечивает некоторые преимущества, описываемые ниже. Авторами настоящего изобретения обнаружено также, что очистку сырого продукта 3-(β-карбоэтокси-α-гидроксиметил)-2-замещенного тетрагидрофурана можно усовершенствовать применением некоторых агентов, например водорастворимых комплексообразующих агентов (например, сегнетовой соли или диэтаноламина), для гашения реакции и удаления солей титана, которые могут быть вредными для последующих стадий. Так, применение водорастворимых комплексообразующих агентов вместо гидрокарбоната натрия, описанного Ghosh et al., приводит к значительным улучшениям качества образовавшегося продукта.
Применение указанного выше способа и последующее превращение образовавшегося продукта 3-(β-карбоэтокси-α-гидроксиметил)-2-замещенного тетрагидрофурана обеспечивает пригодный синтетический путь получения гексагидрофуро[2,3-b]фуран-3-ола и его (3R,3aR,6aR)-энантиомера с относительно малым числом стадий по сравнению со способами известного уровня техники и с применением экономичных исходных соединений и условий реакции, которые обеспечивают получение конечного продукта и промежуточных продуктов с хорошим выходом и чистотой.
Согласно одному отличительному признаку настоящего изобретения авторами изобретения предложен способ получения соединения формулы (V), который включает в себя реакцию 2,3-дигидрофурана формулы (II) с производным глиоксилата формулы (III) в присутствии соли титана формулы Ti(Hal)n(OR)4-n, в которой Hal представляет собой радикал галогена, n равно 0, 1, 2 или 3 и R представляет собой алкил или арилалкил, и последующую реакцию образовавшегося продукта реакции со спиртом формулы (IV) с образованием соединения формулы (V)
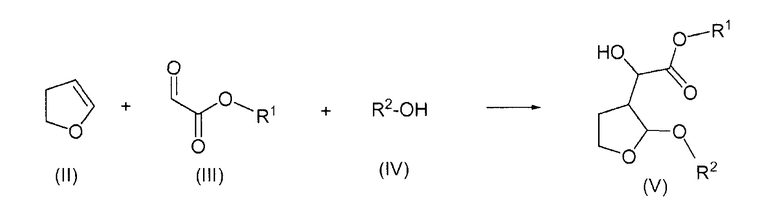
в которой R1 представляет собой алкил или арилалкил и R2 представляет собой алкил или арилалкил.
Производное глиоксилата формулы (III) предпочтительно является соединением, у которого R1 представляет собой С1-4алкильную группу, особенно этильную группу, или фенил-С1-4алкильную группу, особенно бензильную группу. Солью титана предпочтительно является соль формулы Ti(Hal)n(OR)4-n, в которой Hal представляет собой атом хлора или брома, особенно атом хлора, и R представляет собой С1-4алкильную группу, например пропильную группу, предпочтительно изопропильную группу, или арилалкильную группу, например фенил-С1-4алкильную группу и особенно бензил, и n равно 1 или 2, особенно 2; в частности, предпочтительной солью титана для применения согласно настоящему изобретению является TiCl2(OiPr)2. Будет понятно, что такие соли титана могут быть образованы in situ в реакционной смеси, например, реакцией подходящего галогенида титана с подходящим соединением Ti(OR)4. Образуемая конкретная соль будет зависеть от количества соединения Ti(OR)4, добавленного к галогениду титана, например, добавление одной трети эквивалента соединения Ti(OR)4 приведет к образованию соединения TiHal3(OR). Указанную выше предпочтительную соль TiCl2(OiPr)2 можно получить in situ добавлением TiCl4 и Ti(OiPr)4 к реакционной смеси. Способ получения вышеуказанного соединения TiCl2(OiPr)2 описан Mikami et al., J. Am. Chem. Soc., 1990, 112, 3949-3954. Способ получения TiCl(OiPr)3 описан Reetz et al., Chemische Berichte, 1985, 118, 1421-1440. Аналогичным способом можно получить другие соединения формулы Ti(Hal)n(OR)4-n.
Обнаружено, что обычно требуется присутствие по меньшей мере одной группы Hal в соединении титана, поскольку способ, как было обнаружено, является менее эффективным, если применяют соединение формулы Ti(OR)4. В соединении титана n поэтому предпочтительно равно 1, 2 или 3. Соединение формулы Ti(OR)4 обычно применяют в сочетании с тетрахлоридом титана для образования соединения формулы Ti(Hal)n(OR)4-n, в которой n равно 1, 2 или 3.
Обнаружено, что применение указанных выше соединений титана является особенно подходящим по сравнению с тетрахлоридом титана, применяемым Ghosh et al., поскольку последняя соль является нестабильной, корродирующей жидкостью, тогда как соединения титана, применяемые в способе согласно изобретению, являются обычно стабильными твердыми веществами и поэтому значительно более пригодны для работы с ними в промышленном способе. Кроме того, обнаружено, что применение тетрахлорида титана, описываемое Ghosh et al., приводит к неприемлемо высоким остаточным количествам титановых побочных продуктов в реакционной смеси, которые могут привести к тому, что более поздние стадии синтеза гексагидрофуро[2,3-b]фуран-3-ола не поддаются проведению или имеют очень низкие выходы.
Спиртом формулы (IV) предпочтительно является С1-4алканол, такой как метанол, этанол или пропанол, особенно изопропанол, или фенил-С1-4алканол, такой как бензиловый спирт.
Как первоначальную реакцию 2,3-дигидрофурана с производным глиоксилата, так и последующую реакцию со спиртом формулы (IV) обычно проводят в органическом растворителе, предпочтительно апротонном растворителе, таком как дихлорметан, этилацетат, 1,2-дихлорэтан, тетрагидрофуран (ТГФ), или в 2-метилтетрагидрофуране.
Эти реакции подходящим образом проводят при температуре по меньшей мере -20°С, предпочтительно по меньшей мере -10°С и особенно по меньшей мере -5°С, причем предпочтительной является комнатная температура. Применение таких температур контрастирует с применением температуры -78°С, описанной Ghosh et al. в Tetrahedron Letters методике, указанной выше. Первые, более повышенные температуры, применяемые согласно настоящему изобретению, являются значительно более пригодными для проведения способа в промышленном масштабе.
Следующим преимуществом способа согласно изобретению по сравнению со способом, описанным Ghosh et al., как было обнаружено авторами настоящего изобретения, является то, что соединение титана можно применять в менее чем стехиометрическом количестве, например 0,5 эквивалента или меньше, тогда как в способе Ghosh требуется применение эквивалентного количества соединения титана. Применение меньших количеств титана является более экономичным и приводит к меньшим количествам побочных продуктов, которые требуется удалить, и способ авторов изобретения является поэтому подходящим с точки зрения защиты окружающей среды.
После завершения реакции со спиртом формулы (IV) реакционную смесь обычно обрабатывают щелочным реагентом для гашения или прекращения любых дополнительных реакций и образования побочных продуктов, причем щелочной реагент обычно обеспечивает рН 8-11, предпочтительно приблизительно 10. В способе Ghosh et al. (Tetrahedron Letters) для гашения реакции применяют водный раствор гидрокарбоната натрия. Однако авторами изобретения обнаружено, что применение водорастворимого комплексообразующего агента в качестве альтернативного гасящего реагента обеспечивает значительно улучшенную обработку. В качестве водорастворимого комплексообразующего агента можно применять как ионные соединения, такие как сегнетовую соль (тетрагидрат тартрата натрия-калия), так и нейтральные органические молекулы, такие как диэтаноламин. Так добавление сегнетовой соли или диэтаноламина в водном растворе к органической реакционной смеси, полученной в указанном выше способе, гасит реакцию и дает возможность любому остаточному соединению титана легко отделяться в водной фазе и оставляет органическую фазу, содержащую нужное соединение формулы (V), которое как обычно содержит менее 5 м.д. соединения титана. Образовавшееся соединение формулы (V) получают в виде смеси стереоизомерных форм и может применяться как таковое для следующей стадии в синтезе гексагидрофуро[2,3-b]фуран-3-ола.
Указанный выше способ, начинающийся с 2,3-дигидрофурана, обеспечивает получение требуемого соединения формулы (V) с высокими или даже количественными выходами и хорошего качества с применением реагентов, которые легко доступны из коммерческих источников, и условия реакции, которые можно применять в промышленном масштабе.
Термин «алкил» отдельно или в комбинации с любым другим термином относится, за исключением случаев, когда указывается иначе, к насыщенным алифатическим углеводородным радикалам с неразветвленной или разветвленной цепью или в случае, когда присутствует по меньшей мере три атома углерода, циклическим насыщенным алифатическим углеводородным радикалам, содержащим 1-10 атомов углерода, предпочтительно 1-8 атомов углерода, более предпочтительно 1-6 атомов углерода или еще более предпочтительно 1-4 атома углерода. Примеры таких радикалов включают в себя, но не ограничиваются перечисленным, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, н-гексил, циклопропил, циклобутил, циклопентил, циклогексил и тому подобное.
Термин «арил», отдельно или в комбинации с любым другим термином, относится к карбоциклической ароматической части и включает в себя моноциклические, бициклические и другие полициклические радикалы. Примеры арильных радикалов включают в себя, но не ограничиваются перечисленным, фенильный и нафтильный радикалы.
Термин «галоген» относится к атому фтора, хлора, брома или йода.
Термин «стереоизомерные формы», применяемый здесь, определяет все возможные изомеры, образованные из одинаковых атомов, соединенных одинаковой последовательностью связей, но имеющих разные трехмерные структуры, которые не являются взаимозаменяемыми, которые могут иметь соединения настоящего изобретения. Если не указано или не оговорено особо, химическое обозначение соединения включает в себя смесь всех возможных стереохимически изомерных форм, которые может иметь указанное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры базовой молекулярной структуры указанного соединения. Предполагается, что за исключением случая, когда указано особо, все стереоизомерные формы соединений, применяемых в настоящем изобретении, как в чистой (индивидуальной) форме, так и в смеси друг с другом, включены в объем настоящего изобретения.
Чистые стереоизомерные формы указанных здесь соединений, т.е. когда указывается конкретная стереоизомерная форма, определяют как изомеры, по существу свободные от других энантиомерных или диастереомерных форм одинаковой основной молекулярной структуры указанных соединений или промежуточных продуктов. В частности, термин «стереоизомерно чистый» относится к соединениям или промежуточным продуктам, имеющим стереоизомерный избыток по меньшей мере 80% (т.е. минимум 90% одного изомера и максимум 10% другого возможного изомера) вплоть до стереоизомерного избытка 100% (т.е. 100% одного изомера и никакого другого изомера), более конкретно, соединения, имеющие стереоизомерный избыток от 90% до 100%, еще более конкретно, имеющие стереоизомерный избыток от 94% до 100% и наиболее конкретно, имеющие стереоизомерный избыток от 97% до 100%.
Чистые стереоизомерные формы указанных здесь соединений можно получить применением известных в данной области методик. Например, энантиомеры можно отделить друг от друга селективной кристаллизацией их диастереомерных солей с оптически активными кислотами. Альтернативно, энантиомеры можно разделить хроматографическими способами с применением хиральных неподвижных фаз. Указанные чистые стереохимически изомерные формы можно также получить из соответствующих чистых стереохимически изомерных форм подходящих исходных веществ, при условии, что реакция протекает стереоселективно. Если требуется определенный стереоизомер, указанное соединение предпочтительно будет синтезировано стереоселективными способами получения. В этих способах преимущественно будут применяться энантиомерно чистые исходные вещества.
Образовавшееся соединение формулы (V), полученное указанным выше способом, можно применять на следующей стадии синтеза гексагидрофуро[2,3-b]фуран-3-ола без необходимости отделения или выделения его стереоизомеров.
Указанные выше соединения формулы (V), за исключением тех соединений, у которых R1 представляет собой метил или этил и R2 представляет собой метил, являются новыми соединениями, и поэтому авторами изобретения предложены в качестве дополнительного признака изобретения соединения формулы (Va)
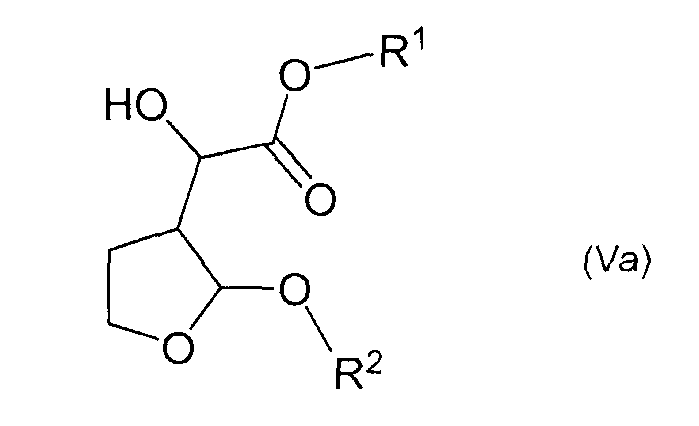
и их стереоизомерные формы и рацемические смеси,
в которых R1 представляет собой алкил или арилалкил и R2 представляет собой алкил или арилалкил, при условии, что когда R2 представляет собой метил, R1 не является метилом или этилом. R1 предпочтительно представляет собой С1-4алкильную группу, такую как пропил, особенно изопропил, или фенил-С1-4алкильную группу, особенно бензильную группу. R2 предпочтительно представляет собой С1-4алкильную группу, такую как этил или пропил, особенно изопропил, или фенил-С1-4алкильную группу, такую как бензил. Соединения формулы (V), в которой R1 представляет собой этил или метил и R2 представляет собой метил, описаны Ghosh et al. в Tetrahedron Letters 40 (1999) 1083-1086, указанной выше.
Соединения формулы (V) таким образом являются применимыми в качестве промежуточных продуктов при получении соединений формулы (I). Особенно применимым соединением формулы (V) является этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат.
Согласно дополнительному признаку настоящего изобретения авторы изобретения предложили способ получения соединений формулы (VI), который включает в себя восстановление соединения формулы (V) с образованием соединения формулы (VI)
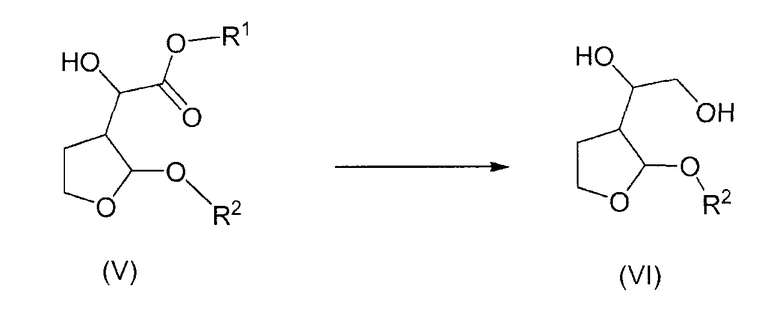
Восстановление соединения формулы (V) обычно проводят с применением гидридного восстанавливающего агента, такого как боргидрид щелочного металла, такой как боргидрид лития, боргидрид натрия, боргидрид калия, ацетоксиборгидрид натрия, триацетоксиборгидрид натрия или цианоборгидрид натрия, восстанавливающий агент на основе гидрида алюминия, такой как литийалюминийгидрид, DibalH (диизобутилалюминийгидрид) или гидрид алюминия, или боргидрид цинка. Альтернативно, восстановление можно проводить каталитическим гидрированием. Гидрирование можно проводить с применением гетерогенного катализатора, такого как активированный никелевый катализатор, например катализатор, коммерчески доступный от Degussa как В 111W, активированный никелевый катализатор, легированный молибденом или сочетанием хром/железо, например катализатор, коммерчески доступный от Degussa как ВК 113W, или активированный медный катализатор, например катализатор, коммерчески доступный от Degussa как В3113. Гидрирование можно также проводить с применением гомогенного катализатора, такого как рутений, согласно методике Milstein, ACIE 2006, 45, 1113. Восстановление можно также проводить гидросилилированием, например, с применением полиметилгидросилоксана (PMHS) или триэтилсилана, например, в комбинации с катализатором Zn(II) (Mimoun, J. Org. Chem., 1999, 64, 2582-2589, рутениевым катализатором (Fuchikami et al., Tetrahedron Letters, 42 (2001), 2149-2151), фторидом тетрабутиламмония (TBAF или тритон B) (Lawrence et al., Synlet, 1997, 989-991), фторид калия или фторид цезия (Coriu et al., Synthesis, 1982, 981 и 1981, 558) или катализатором титан (IV) (Buchwald et al., J. Org. Chem. 1995, 60, 7884-7890).
Боргидрид натрия является особенно предпочтительным в качестве восстанавливающего агента. Восстановление обычно проводят в органическом растворителе, удобно в полярном растворителе, таком как этанол или тетрагидрофуран. Когда применяют боргидридный восстанавливающий агент, после завершения восстановления желательно гасить реакцию комплексообразующим соединением для образования комплекса с любым остаточным соединением бора в реакционной смеси и для того, чтобы избежать дополнительных побочных реакций и образования нежелательных побочных продуктов. Авторами изобретения обнаружено, что обработка реакционной смеси диэтаноламином, например, в форме его гидрохлорида, в качестве реагента для гашения обеспечивает особенно хорошие результаты в смысле чистоты требуемого конечного продукта. Альтернативно для гашения реакции преимущественно можно применять хлорид аммония. Образовавшееся соединение формулы (VI) получают в виде смеси стереоизомерных форм и его можно применять как таковое для следующей стадии в синтезе гексагидрофуро[2,3-b]фуран-3-ола.
Указанные выше соединения формулы (VI) являются новыми соединениями, и поэтому авторами изобретения предложены в качестве дополнительного признака изобретения соединения формулы (VI)
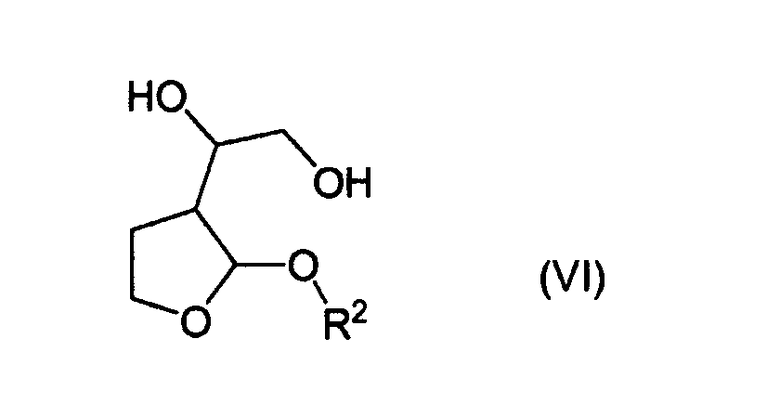
и их стереоизомерные формы и рацемические смеси,
в которых R1 представляет собой алкил или арилалкил, предпочтительно С1-4алкильную группу, такую как метил, этил или пропил, особенно изопропил, или фенил-С1-4алкильную группу, такую как бензил.
Особенно предпочтительным новым соединением формулы (VI) является 1-(2-изопропокситетрагидро-3-фуранил)-1,2-этандиол.
Таким образом, соединения формулы (VI) являются применимыми в качестве промежуточных продуктов в синтезе соединений формул (I) и (Ia).
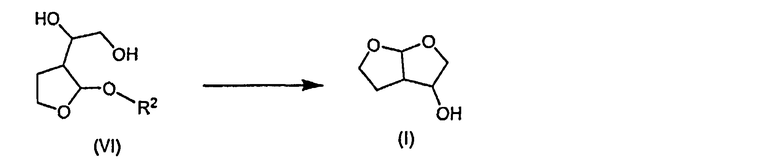
Согласно дополнительному признаку настоящего изобретения авторами изобретения предложен способ получения соединения формулы (I), который содержит циклизацию соединения формулы (VI) с образованием соединения формулы (I)
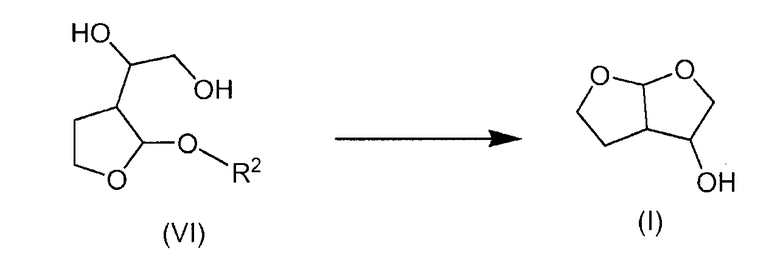
в которой R2 предпочтительно представляет собой С1-4алкильную группу, такую как метил, этил или пропил, особенно изопропил, или фенил-С1-4алкильную группу, такую как бензил.
Циклизацию соединения формулы (VI) можно проводить, например, обработкой кислотой, обычно сильной протонной кислотой, такой как хлористоводородная кислота, п-толуолсульфоновая кислота, метансульфоновая кислота, камфорасульфоновая кислота, смола амберлист, TFA, п-бромбензолсульфоновая кислота или уксусная кислота. Реакцию обычно проводят в органическом растворителе, например полярном растворителе, таком как тетрагидрофуран, дихлорметан, этилацетат, этанол, метанол или ацетон, подходящим образом при температуре от -20°С до 50°С. Когда применяют тетрагидрофуран, предпочтительной температурой является температура от 40°С до 50°С, предпочтительно 45°С. Для нейтрализации реакционной смеси и завершения реакции добавляют основание, такое как триэтиламин или пиридин. Способ приводит к образованию смеси двух диастереоизомеров формулы (I), а именно эндо-диастереизомера, содержащего 3R,3aS,6aR- и 3S,3aR,6aS-энантиомеры, и экзо-диастереоизомера, содержащего 3S,3aS,6aR- и 3R,3aR,6aS-энантиомеры, указанные выше. Два диастереоизомера можно легко разделить общепринятым способом, например хроматографией на силикагеле с применением смеси петролейный эфир/этилацетат (1/9) в качестве элюента.
После разделения указанных выше диастереоизомеров эндо-диастереоизомер можно непосредственно применять при получении ингибиторов протеазы, где требуется эта стереоизомерная часть, если необходимо, после разделения на ее составляющие энантиомеры, общепринятым способом, например, согласно способу, описанному Ghosh et al. в Tetrahedron Letters, Vol. 36, No. 4, 505-508, 1995, или WO 02/060905, ацилированием, например, хлорангидридом или ангидридом кислоты, удобно в апротонном растворителе, таком как тетрагидрофуран или дихлорметан, и в присутствии основания, такого как карбонат натрия или триэтиламин. Образовавшуюся смесь сложных эфиров затем подвергают реакции с подходящим эстеразным ферментом, таким как липаза Ps30, в условиях, которые допускают протекание реакции преимущественно одного из рацемических сложных эфиров с образованием смеси спирта преимущественно одного энантиомера и оставшегося непрореагировавшего сложного эфира соответствующего преимущественно другого энантиомера. Смесь спирта и сложного эфира можно затем разделить общепринятым способом, например хроматографией на силикагеле. Энантиомерный непрореагировавший сложный эфир можно превратить в соответствующий спирт, например, реакцией с метиллитием в тетрагидрофуране.
При желании, экзо-диастереоизомер можно превратить в эндо-диастереоизомер общепринятым способом, например, как описано Ghosh et al. в J. Org. Chem. 2004, 69, 7822-7829, последовательностью окисление/восстановление, включающей в себя промежуточное образование кетона формулы (I')
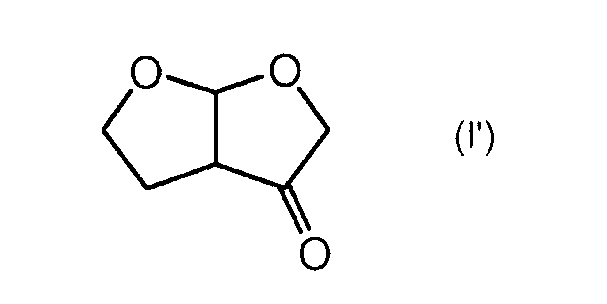
Так, согласно указанному выше способу Ghosh et al., экзо-диастереоизомер окисляют в кетон формулы (I') перрутенатом тетрапропиламмония (ТРАР) и 4-метилморфолино-N-оксидом (NMO) и образовавшийся кетон восстанавливают, например, гидридным восстанавливающим агентом, таким как боргидрид натрия, в органическом растворителе, например полярном растворителе, таком как этанол, получая при этом соответствующий эндо-диастереоизомер. Альтернативно, указанное выше окисление можно проводить системой NaOCl/1-оксид 2,2,6,6-тетраметилпиперидина (ТЕМРО).
Должно быть понятно, что указанную выше методику окисления и восстановления можно также проводить, исходя из смеси эндо- и экзо-форм, что таким образом позволяет избежать необходимости проведения любого предварительного разделения диастереоизомеров.
Согласно дополнительному признаку изобретения авторами изобретения предложен способ получения гексагидро[2,3-b]фуран-3-ола, который включает в себя следующие стадии:
а) реакцию 2,3-дигидрофурана формулы (II) с глиоксилатным производным формулы (III) в присутствии соли титана формулы Ti(Hal)n(OR)4-n, в которой n равно 0, 1, 2 или 3 и R представляет собой алкил или арилалкил, и последующую реакцию образовавшегося продукта реакции со спиртом формулы (IV) с образованием соединения формулы (V)
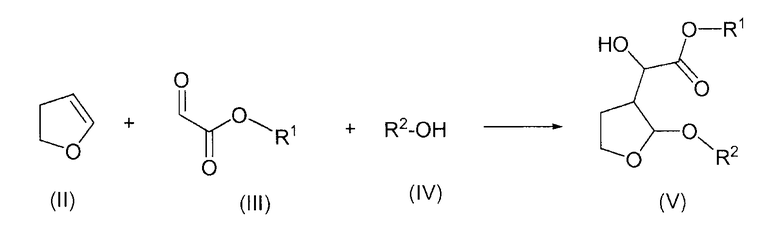
в которой R1 представляет собой алкил или арилалкил и R2 представляет собой алкил или арилалкил, и
b) восстановление образовавшегося соединения формулы (V) с образованием соединения формулы (VI)
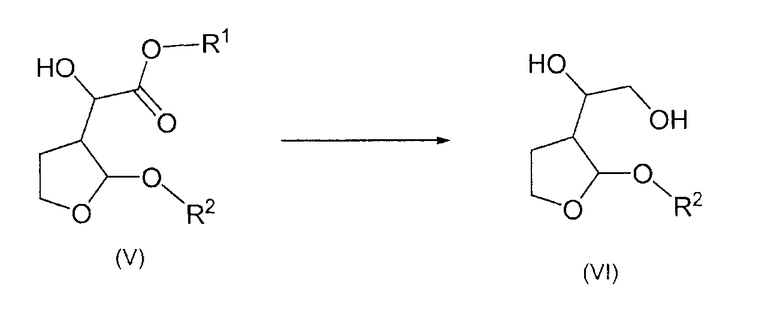
и
с) циклизацию соединения формулы (VI) с образованием соединения формулы (I)

и, при желании, затем (i) разделение образовавшегося соединения формулы (I) для выделения (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола формулы (Ia)
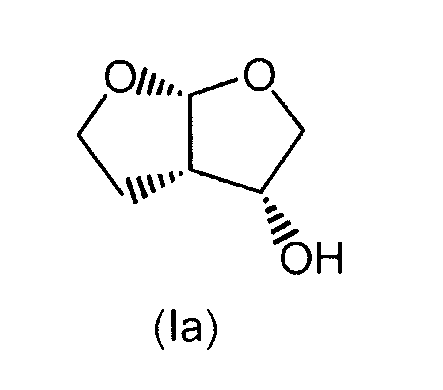
и/или
(ii) окисление образовавшегося соединения формулы (I) с образованием соединения формулы (I')
и затем восстановление соединения формулы (I') в эндо-изомер соединения формулы (I).
Соединения формул (I) и (Ia) являются особенно применимыми при получении лекарственных средств. Согласно предпочтительному варианту осуществления настоящие соединения формул (I) и (Ia) применяют в качестве предшественников при получении противовирусных лекарственных средств, в частности лекарственных средств против ВИЧ, более конкретно, ингибиторов протеазы ВИЧ.
Соединения формул (I) и (Ia) и все промежуточные продукты, приводящие к образованию этих соединений, представляют собой особый интерес при получении ингибиторов протеазы ВИЧ, как описано Ghosh et al. Bioorganic & Medicinal Chemistry Letters 8 (1998) 687-690 и WO 95/24385, WO 99/65870, WO 99/67254, WO 99/67417, WO 00/47551, WO 00/76961, WO 01/25240, US 6127372 и EP 0715618, все из которых включены здесь в качестве ссылки, и особенно следующих ингибиторов протеазы ВИЧ:
(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового эфира [(1S,2R)-3-[[(4-аминофенил)сульфонил](2-метилпропил)амино]-2-гидрокси-1-(фенилметил)пропил]карбаминовой кислоты, то есть указанного выше дарунавира (ингибитора 1 протеазы ВИЧ);
(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового эфира [(1S,2R)-2-гидрокси-3-[[(4-метоксифенил)сульфонил](2-метилпропил)амино]-1-(фенилметил)пропил]карбаминовой кислоты (ингибитора 2 протеазы ВИЧ) и
(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового эфира [(1S,2R)-3-[(1,3-бензодиоксол-5-илсульфонил)(2-метилпропил)амино]-2-гидрокси-1-(фенилметил)пропил]карбаминовой кислоты (ингибитора 3 протеазы ВИЧ) или любых их фармацевтически приемлемых аддитивных солей.
Таким образом, настоящее изобретение относится также к ингибиторам 1, 2 или 3 протеазы ВИЧ или их любым фармацевтически приемлемым солям или пролекарствам, полученным с применением соединения формулы (I), полученного согласно настоящему изобретению в химическом синтезе указанных ингибиторов протеазы ВИЧ. Такой химический синтез описан в литературе, например, в указанных выше патентных и литературных ссылках.
Соединение указанной выше формулы (Ia) можно применять после образования активированного производного для синтеза ингибитора 1 протеазы, то есть дарунавира указанной выше формулы (А), как описано, например, в WO2005/063770, содержание которой включено здесь в качестве ссылки, нижеследующим способом, который содержит
(i) введение изобутиламиногруппы в соединение формулы (I)
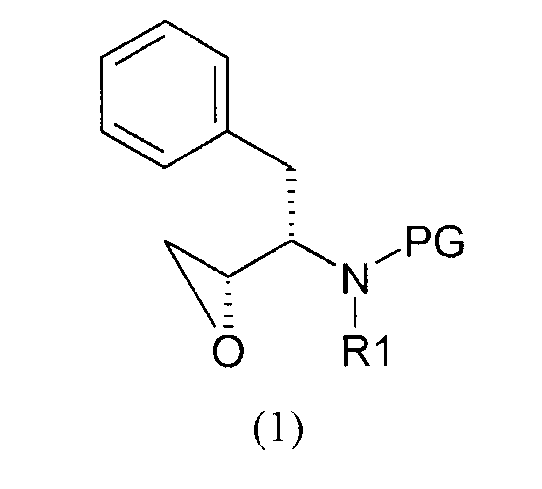
где
PG представляет собой аминозащитную группу;
R1 представляет собой водород или С1-6алкил;
(ii) введение п-нитрофенилсульфонильной группы в образовавшееся соединение стадии (i);
(iii) восстановление нитрогруппы образовавшегося соединения стадии (ii);
(iv) снятие защиты у образовавшегося соединения стадии (iii) и
(v) сочетание образовавшегося соединения стадии (iv) с (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илпроизводным с образованием указанного выше соединения (А).
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (А), отличающегося тем, что указанный способ содержит стадии введения изобутиламиногруппы в соединение формулы (I')
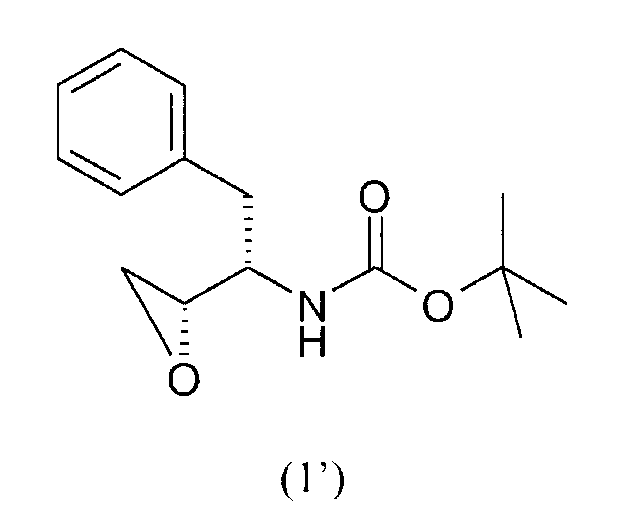
с получением соединения формулы (2')
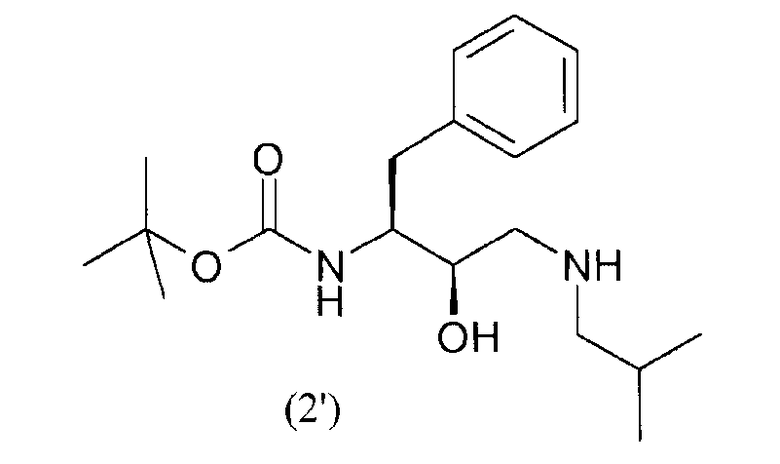
введения п-нитрофенилсульфонильной группы в соединение формулы (2') с получением соединения формулы (3')
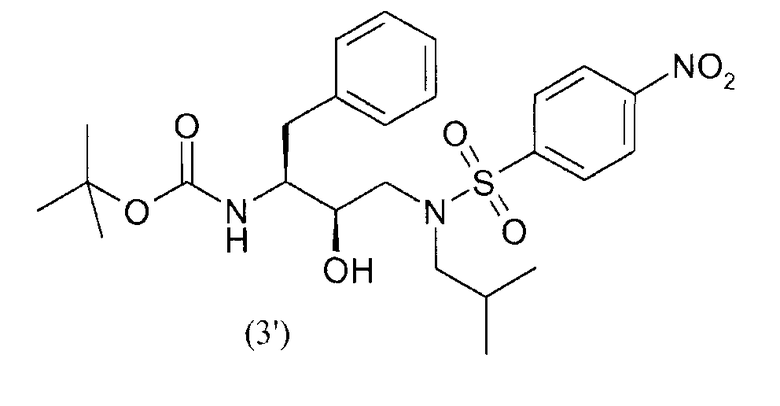
восстановления нитрогруппы соединения формулы (3') с получением соединения формулы (4')
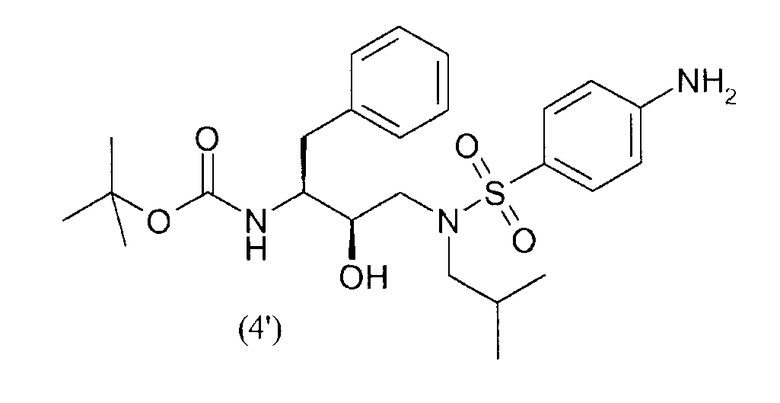
снятия защиты у соединения формулы (4') с получением соединения формулы (5)
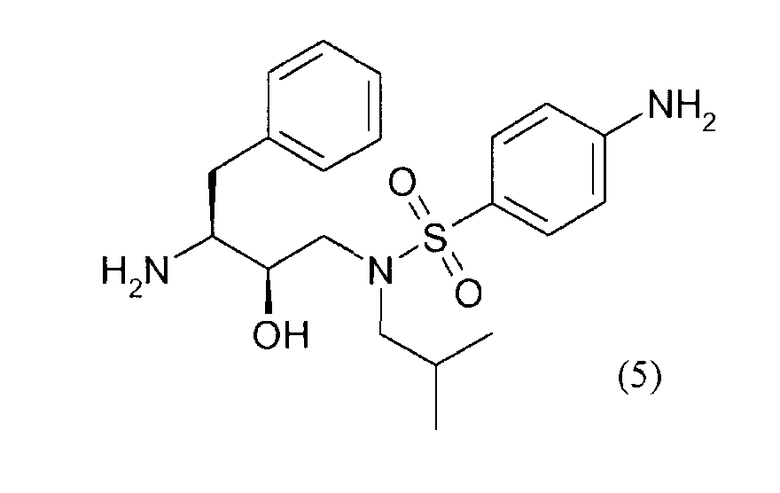
сочетания соединения формулы (5) с (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илпроизводным с образованием соединения (А).
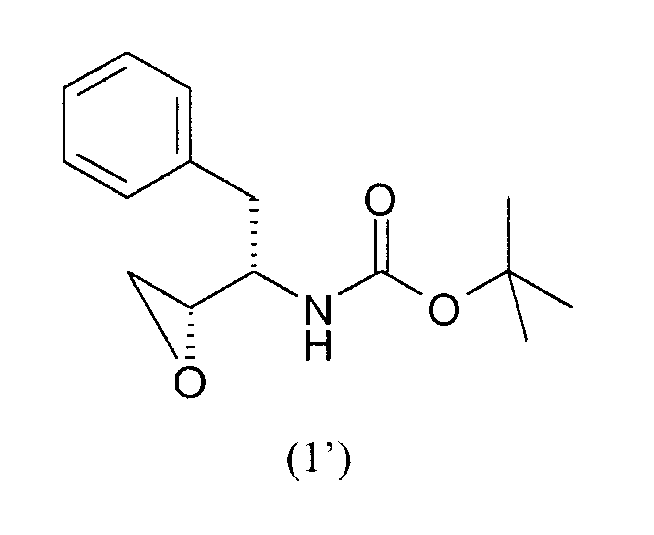
Соединение формулы (1)
Соединение формулы (1) является
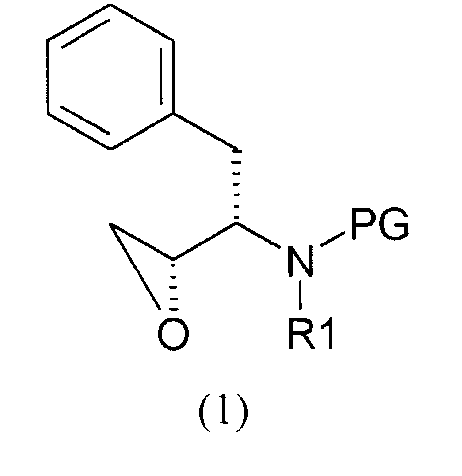
где
PG представляет собой аминозащитную группу;
R1 представляет собой водород или С1-6алкил.
Соединением формулы (1) предпочтительно является соединение формулы (1'), как показано ниже, где PG представляет собой трет-бутилоксикарбонил или “Boc” и R1 представляет собой водород. Соединения формул (1) и (1') являются коммерчески доступными и их можно получить несколькими путями, доступными в литературе, например, как описано в WO95/06030 (Searle & Co.), как описано Kaneka Corporation в EP 0754669, EP 1029856 и EP 1067125, и как описано Ajinomoto KK в EP 1081133 и EP 1215209.
Соединение формулы (2)
Соединение формулы (1) подвергают аминированию у эпоксида с получением соединения формулы (2).
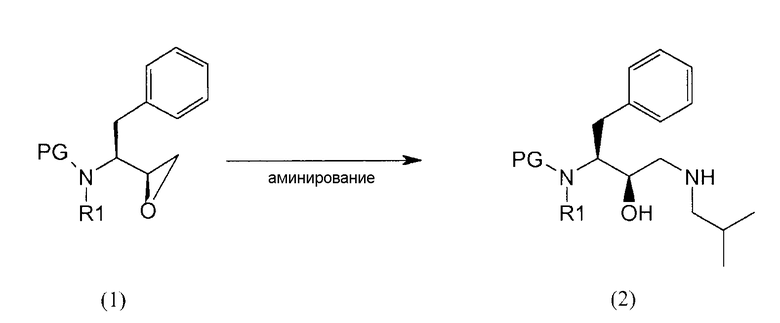
Применяемый здесь термин “аминирование” относится к способу, в котором первичный амин, изобутиламин, вводят в органическую молекулу формулы (1). Аминирование соединения формулы (1) можно выполнить несколькими путями, доступными в литературе, например, как описано в WO95/06030, которая описана здесь в качестве ссылки.
В предпочтительном варианте осуществления соединение формулы (1') подвергают реакции с изобутиламином с получением соединения формулы (2').
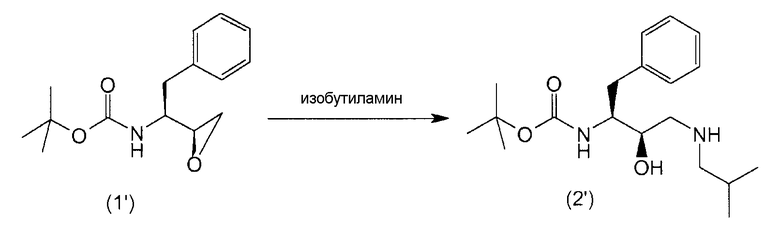
Аминирование эпоксидов описано, например, в March, Advanced Organic Chemistry 368-69 (3rd Ed. 1985) и McManus et al., 3 Synth. Comm. 177 (1973), которые включены здесь в качестве ссылки. Подходящим образом соединения формул (2) и (2') можно получить согласно методике, описанной в WO97/18205.
Агент аминирования, изобутиламин, может функционировать также в качестве растворителя, в этом случае можно добавить избыток изобутиламина. В других вариантах осуществления способ аминирования проводят в присутствии одного или нескольких растворителей, отличных от изобутиламина. В предпочтительном варианте осуществления упомянутые растворители применяют при получении соединений формул (2) и (2').
В варианте осуществления изобретения реакцию аминирования проводят в присутствии приблизительно 15 эквивалентов изобутиламина с применением толуола в качестве растворителя и при нагревании для кипячения с обратным холодильником приблизительно при 79°С.
Соединение формулы (3)
Соединение формулы (3) получают введением сульфонильной части, п-нитробензол-SO2, в промежуточный продукт формулы (2).
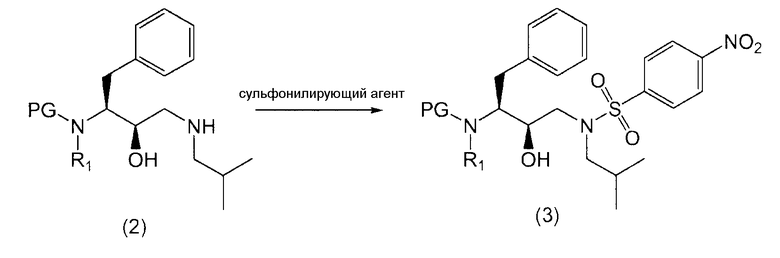
Так, в предпочтительном варианте осуществления соединение формулы (3') можно получить сульфонилированием соединения формулы (2').
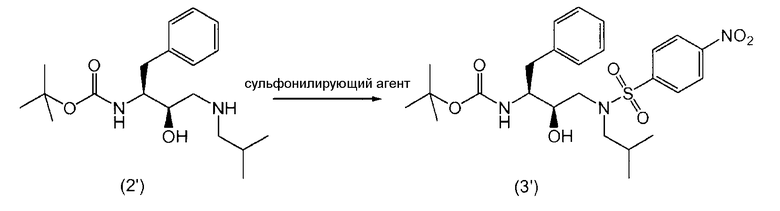
Как таковые, соединения формул (2) и (2') подвергают реакции с сульфонилирующим агентом для превращения в соединения формул (3) и (3').
Термин “сульфонилирующий агент” включает в себя п-нитробензолсульфонилпроизводные, такие как п-нитробензолсульфонилгалогенпроизводные.
Обработку соединений формул (2) и (2') сульфонилирующим агентом можно проводить в присутствии растворителя при нагревании приблизительно от 25°С до 250°С, предпочтительно от 70°С до 100°С, и перемешивании. После сульфонилирования любой оставшийся сульфонилирующий агент или его соли предпочтительно, хотя и необязательно, удаляют из реакционной смеси. Это удаление можно выполнить многократным промыванием водой, изменением рН, разделением органической и водной фаз, ультрафильтрованием, обратным осмосом, центрифугированием и/или фильтрованием или тому подобное.
Соединение формулы (4)
Соединения формул (4) и (4') получают восстановлением нитрогруппы промежуточных соединений формул (3) и (3') соответственно, восстанавливающим агентом, необязательно в атмосфере водорода.
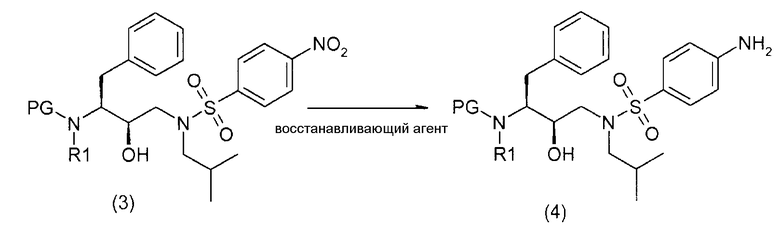
Восстанавливающими агентами, подходящими для восстановления нитрогруппы, являются металлические восстанавливающие реагенты, такие как комплексы боранов, диборан, боргидрид натрия, боргидрид лития, боргидрид натрия-LiCl, литийалюмогидрид или диизобутилалюмогидрид; металлы, такие как железо, цинк, олово и тому подобное, и переходные металлы, такие как палладий-на-угле, оксид платины, никель Ренея, родий, рутений и тому подобное. Когда применяют каталитическое восстановление, в качестве источника водорода можно применять формиат аммония, дигидрофосфат натрия, гидразин.
Соединения формулы (5)
Соединение формулы (5) получают удалением защиты у промежуточных продуктов формул (4) и (4') в общепринятых кислотных условиях. Альтернативно можно применять основные условия.
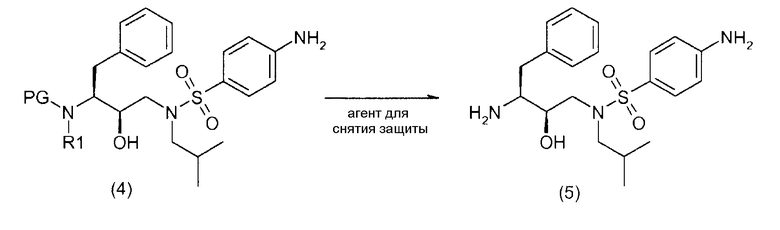
Удаление аминозащитной группы можно осуществить с применением условий, которые не будут затрагивать остальную часть молекулы. Эти способы хорошо известны в данной области и включают в себя кислотный гидролиз, гидрогенолиз и тому подобное, так например, с применением обычно известных кислот в подходящих растворителях.
Примеры реагентов и способов удаления у аминов аминозащитных групп можно дополнительно найти в публикации Protective Groups in Organic Synthesis by Theodora W. Greene, New York, John Wiley and Sons, Inc., 1981, включенной здесь в качестве ссылки.
Как должно быть понятно специалисту в данной области, выбор аминозащитной группы, применяемой в предыдущей стадии способа, будет определять реагенты и методики, применяемые при удалении указанной аминозащитной группы.
Получение дарунавира
(3R,3aS,6aR)-Гексагидрофуро[2,3-b]фуран-3-ол формулы (Ia), полученный, как описано выше, подходящим образом активируют агентом сочетания с образованием (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илпроизводного, которое затем карбамоилируют соединением формулы (5) с получением требуемого ингибитора 1 протеазы, а именно дарунавира.
Примерами агентов сочетания, применяемых в реакциях карбамоилирования, являются карбонаты, такие как бис-(4-нитрофенил)карбонат, дисукцинимидилкарбонат (DSC), карбонилдиимидазол (CDI). Другие агенты сочетания включают в себя хлорформиаты, такие как п-нитрофенилхлорформиат, фосгены, такие как фосген и трифосген.
В частности, когда (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ол подвергают реакции с дисукцинимидилкарбонатом, получают 1-([[(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илокси]карбонил]окси)-2,5-пирролидиндион. Указанное соединение является предпочтительным (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илпроизводным.
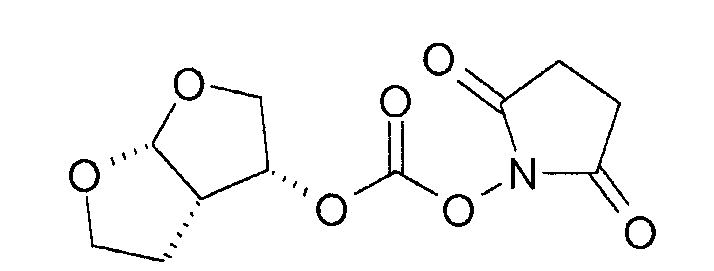
Реакцию (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илпроизводного с соединением формулы (5) можно проводить в присутствии подходящих растворителях, таких как тетрагидрофуран, диметилформамид, ацетонитрил, диоксан, дихлорметан или хлороформ, и необязательно с основаниями, такими как триэтиламин, хотя применяют также дополнительные комбинации указанных выше растворителей и оснований.
Среди растворителей предпочтительными растворителями являются апротонные растворители, такие как тетрагидрофуран, ацетонитрил, диметилформамид, этилацетат и тому подобное.
Указанную выше реакцию карбамоилирования подходящим образом проводят при температуре от -70°С до 40°С, предпочтительно от -10°С до 20°С.
Согласно особенно предпочтительному признаку настоящего изобретения авторами изобретения предложен дарунавир, то есть (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-иловый эфир [(1S,2R)-3-[[(4-аминофенил)сульфонил](2-метилпропил)амино]-2-гидрокси-1-(фенилметил)пропил]карбаминовой кислоты формулы (А), всегда синтезированный с применением промежуточного продукта формулы (I) и особенно промежуточного продукта формулы (Ia), полученного согласно настоящему изобретению.
Примеры
Имеется ввиду, что нижеследующие примеры являются иллюстрацией настоящего изобретения. Эти примеры представлены для иллюстрации изобретения в виде примеров и не должны истолковываться как ограничение объема изобретения.
Газовую хроматографию (ГХ) проводили при следующих условиях: колонка: 5% фенил-, 95% метилполисилоксана, L=25 м; ID=320 мкм; ширина пленки = 0,52 мкм; инжектор с расщеплением при 250°С с отношением 1/50; объем впрыскивания: 1 мкл. Программа: 5 мин при 50°С, затем скорость нагрева 15°С/мин до 240°С в течение 5 мин. Общий поток: 3,0 мл/мин. Качество продукта реакции определяет процентное количество требуемого соединения в таком продукте, определяемое газовой хроматографией с пламенно-ионизационным детектированием (FID) (% площади ГХ).
В нижеследующих примерах “DCM“ относится к дихлорметану, “AcOEt” относится к этилацетату, ТГФ относится к тетрагидрофурану и “TEMPO” относится к 1-оксиду 2,2,6,6-тетраметилпиперидина.
Сравнительный пример
(А) Этилгидрокси-(2-этокситетрагидрофуран-3-ил)ацетат
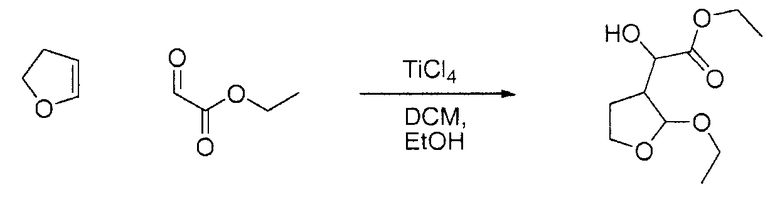
К смеси свежеперегнанного этилглиоксалата (40 ммоль; 1,000 экв.; 3,620 мл; 4,084 г) и 2,3-дигидрофурана (44 ммоль; 1,100 экв.; 3,338 мл; 3,084 г) в сухом дихлорметане (100 мл; 1,560 моль; 132,5 г) по каплям добавляли раствор тетрахлорида титана (44 ммоль; 1,100 экв.; 44,00 мл; 59,84 г) в 1 М DCM при -78°С и образовавшуюся смесь перемешивали в течение 1 ч. Реакционная смесь становится желтой и гетерогенной. К смеси, которая стала гетерогенной, по каплям добавляли этанол (120 ммоль; 3,000 экв.; 6,986 мл; 5,528 г). Охлаждающую баню удаляли, чтобы позволить реакционной смеси нагреться до комнатной температуры в течение 1 ч. Медленно при комнатной температуре добавляли бикарбонат натрия (100 мл; 103,4 ммоль, 104,7 г). Спустя 10 мин реакционную смесь экстрагировали дважды этилацетатом (600 мл; 6,132 моль; 540,2 г). Органический растворитель выпаривали при пониженном давлении, получая при этом зеленое масло, содержащее белые твердые вещества (9,45 г, ГХ: площадь 14%).
ГХ: время удерживания: 13,4 мин.
МС (Е.I. 70 эВ): 217 (0,5%; M-H); 173 (11%, M-OEt); 155 (59%, 173-H2O); 145 (21%, M-CO2Et); 71 (100%, 173-CHOCO2Et).
Определение титана в этой сырой смеси проводили с применением способа ICP (индуктивно связанной плазмы): 96 м.д. титана.
(В) Попытка восстановления продукта из (А)
В 50-миллилитровую круглодонную колбу, загруженную этанолом (9,6 мл) и тетрагидроборатом натрия (1,1 экв.; 5,46 ммоль; 210 мг) при 0°С, по каплям на протяжении 1 ч добавляли этилгидрокси-(2-этокситетрагидрофуран-3-ил)ацетат (1,44 г; 4,96 ммоль; 1 экв.), растворенный в этаноле (5,8 мл). Реакционной смеси предоставляли возможность нагреться до комнатной температуры и перемешивали на протяжении двух дней. Затем к реакционной смеси при 0°С по каплям добавляли хлорид аммония (1,5 экв.; 7,44 ммоль; 400 мг), растворенный в воде (3,5 мл). Реакционную смесь перемешивали в течение 4 ч при комнатной температуре и растворитель выпаривали при пониженном давлении, получая при этом коричневое твердое вещество. Затем к сырой смеси добавляли этилацетат (7,7 мл) и смесь нагревали при 40°С в течение 30 мин. После фильтрования через дикалит гомогенную смесь упаривали досуха при пониженном давлении, получая при этом исходное вещество.
Пример 1
Этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат
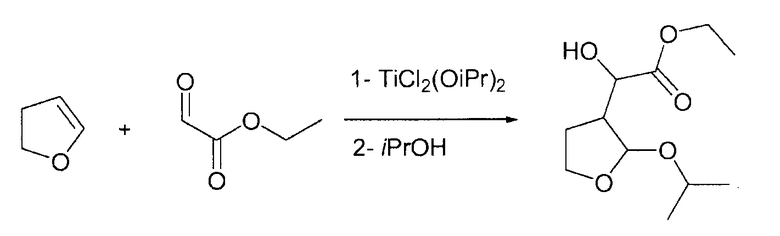
В круглодонной колбе 50% масс./масс. раствор этилглиоксилата в толуоле (4,74 г, 23,21 ммоль, 1,1 экв.) перемешивали при пониженном давлении и при 60°С до полного выпаривания толуола. Затем при комнатной температуре добавляли 80 мл сухого DCM с последующим добавлением TiCl2(OiPr)2 (5 г, 21,1 ммоль). После периода перемешивания 0,5 ч при комнатной температуре по каплям на протяжении 10 мин добавляли 2,3-дигидрофуран (1,48 г, 21,1 ммоль, 1 экв.), растворенного в 15 мл DCM, и смесь перемешивали в течение 5 ч при комнатной температуре. Затем по каплям добавляли изопропанол (16 мл, 211 ммоль, 10 экв.) и смесь перемешивали на протяжении ночи. Наконец, при комнатной температуре по каплям добавляли основную водную смесь сегнетовой соли (20 г в 200 мл воды, 2 г Na2CO3) и смесь перемешивали на протяжении ночи. Два слоя разделяли и органический слой сушили Na2SO4 и концентрировали в вакууме. Полученное масло (3,45 г, ГХ: площадь 85%) можно применять непосредственно на следующей стадии.
Определение титана: <5 м.д.
МС (E.I. 70 эВ): 173 (16%, M-OiPr); 159 (22%, M-CO2Et); 155 (100%, 173-H2O); 71 (98%, 173-CHOCO2Et).
MC (C.I., аммиак): (M+H)+: 233,1353 (теория: 233,1389); (M+NH4)+: 250,1593 (теория: 250,1654).
Пример 2
Этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат
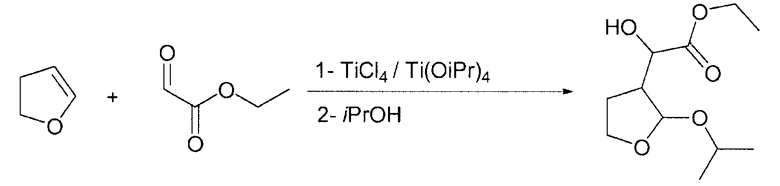
В 1-литровой круглодонной колбе тетрахлорид титана (17,83 ммоль; 1,96 мл; 3,38 г) растворяли в дихлорметане (70 мл; 1,092 моль; 92,75 г) при комнатной температуре. Затем при комнатной температуре по каплям добавляли тетра(изопропоксид) титана (17,83 ммоль; 5,28 мл; 5,07 г), растворенного в дихлорметане (70 мл; 1,092 моль; 92,75 г). После периода перемешивания 1 ч на протяжении 30 мин при комнатной температуре по каплям добавляли этилглиоксалат (1,1 экв.; 39,24 ммоль; 3,55 мл; 4,0 г) без толуола, растворенный в дихлорметане (8,75 мл; 136,5 ммоль, 11,59 г). Спустя 15 мин на протяжении 30 мин при комнатной температуре добавляли 2,3-дигидрофуран (2,5 г; 1,000 экв.; 35,67 ммоль; 2,70 мл), растворенный в дихлорметане (17,5 мл; 273,0 ммоль; 23,19 г). После периода перемешивания 3 ч по каплям добавляли изопропиловый спирт (10 экв.; 356,7 ммоль; 27,26 мл; 21,44 г). Через 3 ч к реакционной смеси добавляли смесь карбоната калия (1,75 г; 12,66 ммоль) и сегнетовой соли (17,5 г; 83,25 ммоль), растворенную в воде (175 мл; 9,72 моль, 175 г). После перемешивания на протяжении двух дней два слоя разделяли и органический слой промывали (2×100 мл) водой (200 мл; 11,10 моль; 200,0 г). Органический растворитель выпаривали при пониженном давлении, получая при этом требуемый продукт (7,48 г; ГХ: 92% площади).
Определение титана:
перед обработкой сегнетовой солью: 14,5%; после обработки сегнетовой солью: <5 м.д.
1H ЯМР (CDCl3, 400 МГц): 5,23 (д, 0,42H, J=4 Гц); 5,20 (д, 0,48H, J=2 Гц); 5,16-5,08 (м, 0,14H); 4,45 (д, 0,42H, J=4 Гц); 4,30-4,15 (м, 3H); 4,11-3,96 (м, 0,8H); 3,96-3,81 (м, 2,85H); 3,54 (ушир. с, 0,4H); 2,97 (ушир. с, 0,60H); 2,60-2,42 (м, 1,1H); 2,28-2,15 (м, 0,49H); 1,97-1,79 (м, 1,79H); 1,30 (т, 3,8H, J=8 Гц); 1,24-1,21 (м, 0,46H); 1,21-1,16 (м, 3,12H); 1,16-1,12 (м, 3H).
13C ЯМР (CDCl3, 400 МГц): 174,1; 172,9; 103,9; 102,2; 70,2; 69,6; 69,4; 66,6; 61,9; 61,2; 49,9; 47,0; 25,1; 23,9; 23,7; 21,9; 21,7; 14,2.
Пример 3
Этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат
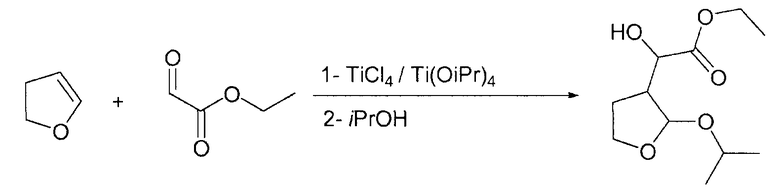
В 1-литровой круглодонной колбе тетрахлорид титана (17,83 ммоль; 1,96 мл; 3,38 г) растворяли в дихлорметане (70 мл; 1,092 моль; 92,75 г) при комнатной температуре. Затем при комнатной температуре по каплям добавляли тетра(изопропоксид) титана (17,83 ммоль, 5,28 мл; 5,07 г), растворенного в дихлорметане (70 мл; 1,092 моль; 92,75 г). После периода перемешивания 1 ч при комнатной температуре на протяжении периода 30 мин по каплям добавляли этилглиоксалат (1,1 экв.; 39,24 ммоль; 3,55 мл; 4,0 г) без толуола, растворенный в дихлорметане (8,75 мл; 136,5 ммоль, 11,59 г). Спустя 15 мин на протяжении 30 мин при комнатной температуре добавляли 2,3-дигидрофуран (2,5 г; 1,000 экв.; 35,67 ммоль; 2,70 мл), растворенный в дихлорметане (17,5 мл; 273,0 ммоль; 23,19 г). После периода перемешивания 3 ч по каплям добавляли изопропиловый спирт (10 экв.; 356,7 ммоль; 27,26 мл; 21,44 г). Через 3 ч к реакционной смеси добавляли смесь карбоната калия (1,75 г; 12,66 ммоль) и диэтаноламина (9,5 г; 90,6 ммоль), растворенную в воде (175 мл; 9,72 моль, 175 г). После перемешивания на протяжении двух дней два слоя разделяли и органический слой промывали (2×100 мл) водой (200 мл; 11,10 моль; 200,0 г). Органический растворитель выпаривали при пониженном давлении, получая при этом требуемый продукт (7,0 г; ГХ: 96% площади).
Определение титана:
после обработки диэтаноламином: <5 м.д.
Пример 4
Этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат
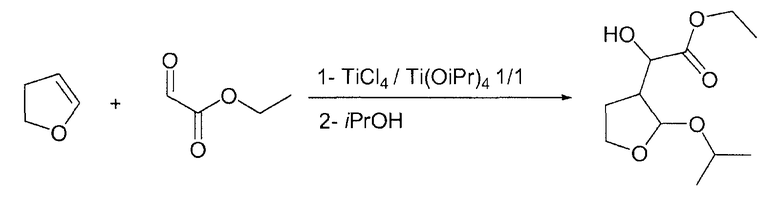
В круглодонную колбу, загруженную 400 мл DCM и Ti(OiPr)4 (56,8 г, 0,2 моль), по каплям при комнатной температуре на протяжении 30 мин добавляли TiCl4 (22 мл, 0,2 моль), растворенный в 400 мл DCM. После периода перемешивания 17 час по каплям при комнатной температуре добавляли этилглиоксилат без толуола (45,2 г, 0,22 ммоль, 1,1 экв.), растворенный в 50 мл DCM. Спустя 15 мин по каплям на протяжении 30 мин добавляли 2,3-дигидрофуран (14 г, 0,2 моль, 1 экв.), растворенный в 100 мл DCM, и смесь перемешивали в течение 3 ч при комнатной температуре. Затем добавляли по каплям изопропанол (153 мл, 2 моль, 10 экв.) и смесь перемешивали в течение 4 ч при комнатной температуре. Наконец, по каплям добавляли основную водную смесь сегнетовой соли (100 г в 1000 мл воды, 10 г K2CO3) и образовавшуюся смесь перемешивали на протяжении ночи при комнатной температуре. Два слоя разделяли и органический слой сушили Na2SO4, фильтровали и упаривали в вакууме. Полученное масло (47,2 г, ГХ: площадь 87%) можно применять непосредственно на следующей стадии.
ГХ: время удерживания: 13,7 мин.
МС (E.I. 70 эВ): 173 (16%, M-OiPr); 159 (22%, M-CO2Et); 155 (100%, 173-H2O); 71 (98%, 173-CHOCO2Et).
МС (C.I., аммиак): (M+H)+: 233,1353 (теория: 233,1389); (M+NH4)+: 250,1593 (теория: 250,1654).
Пример 5
Этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат
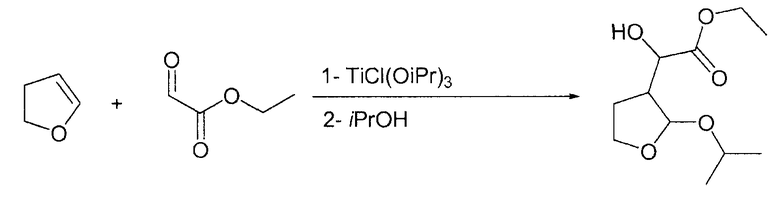
В круглодонную колбу, загруженную TiCl(OiPr)3 (0,1 моль, 1 М в гексане) и 300 мл DCM, по каплям при комнатной температуре добавляли этилглиоксилат без толуола (22,6 г, 0,11 моль, 1,1 экв.), растворенный в 25 мл DCM. После периода перемешивания 0,5 ч при комнатной температуре по каплям при комнатной температуре на протяжении 30 мин добавляли 2,3-дигидрофуран (7 г, 0,1 моль, 1 экв.), растворенный в 50 мл DCM, и смесь перемешивали в течение 5 ч при комнатной температуре. Затем при комнатной температуре добавляли по каплям изопропанол (76 мл, 1 моль, 10 экв.) и смесь перемешивали на протяжении ночи. Наконец, по каплям при комнатной температуре добавляли основную водную смесь сегнетовой соли (50 г в 500 мл воды, 5 г K2CO3) и смесь перемешивали на протяжении ночи. Два слоя разделяли, органический слой сушили Na2SO4, фильтровали и упаривали в вакууме. Полученное масло (18,2 г, ГХ: площадь 85%) можно применять непосредственно на следующей стадии.
МС (E.I. 70 эВ): 173 (16%, M-OiPr); 159 (22%, M-CO2Et); 155 (100%, 173-H2O); 71 (98%, 173-CHOCO2Et).
МС (C.I., аммиак): (M+H)+: 233,1353 (теория: 233,1389); (M+NH4)+: 250,1593 (теория: 250,1654).
Пример 6
Этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат
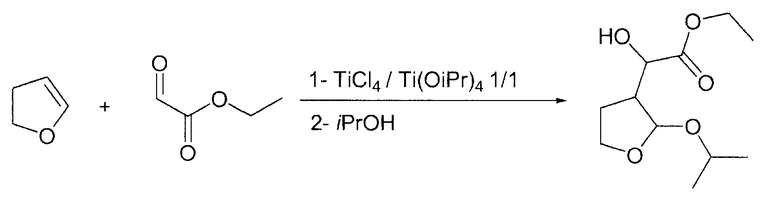
В круглодонную колбу, загруженную 400 мл DCM и Ti(OiPr)4 (28,4 г, 0,1 моль), по каплям при комнатной температуре на протяжении 30 мин добавляли TiCl4 (11 мл, 0,1 моль), растворенный в 400 мл DCM. После периода перемешивания 17 ч по каплям при комнатной температуре на протяжении 30 мин добавляли этилглиоксилат без толуола (90 г, 0,44 ммоль, 1,1 экв.), растворенный в 150 мл DCM. Спустя 15 мин по каплям на протяжении 30 мин при комнатной температуре добавляли 2,3-дигидрофуран (28 г, 0,4 моль, 1 экв.), растворенный в 150 мл DCM, и смесь перемешивали в течение 5 ч. Затем по каплям при комнатной температуре добавляли изопропанол (306 мл, 4 моль, 10 экв.) и смесь перемешивали в течение 3 ч. Наконец, по каплям при комнатной температуре добавляли основную водную смесь сегнетовой соли (100 г в 1000 мл воды, 10 г K2CO3) и смесь перемешивали на протяжении ночи. Два слоя разделяли, органический слой сушили Na2SO4, фильтровали и упаривали в вакууме. Полученное масло (81,5 г, ГХ: площадь 70%) можно применять непосредственно на следующей стадии.
ГХ: время удерживания: 13,7 мин.
МС (E.I. 70 эВ): 173 (16%, M-OiPr); 159 (22%, M-CO2Et); 155 (100%, 173-H2O); 71 (98%, 173-CHOCO2Et).
МС (C.I., аммиак): (M+H)+: 233,1353 (теория: 233,1389); (M+NH4)+: 250,1593 (теория: 250,1654).
Пример 7
а) 1-(2-Изопропокситетрагидро-3-фуранил)-1,2-этандиол
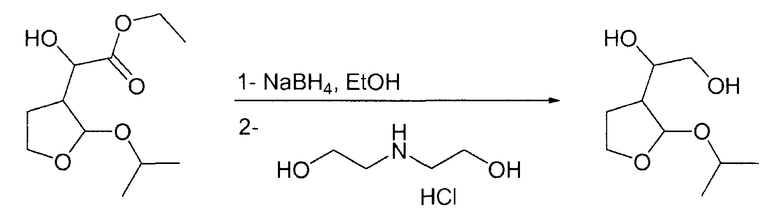
В 2-литровую круглодонную колбу, загруженную 600 мл этанола и NaBH4 (12,55 г, 0,33 моль, 1,1 экв.) при 0°С, по каплям на протяжении 1 ч при 0°С добавляли этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат (70 г, 0,3 моль, 1 экв.), растворенный в 400 мл этанола. Смеси давали возможность нагреться при комнатной температуре и перемешивали в течение 19 ч. После охлаждения при 0°С добавляли на протяжении 10 мин гидрохлорид диэтаноламина (46,7 г, 0,33 моль, 1,1 экв.), растворенный в 100 мл воды, и смесь перемешивали в течение 8 ч. Растворитель выпаривали при пониженном давлении, получая при этом светло-желтое твердое вещество. После разбавления 300 мл этилацетата гетерогенную смесь фильтровали через дикалит. Смесь применяли непосредственно на следующей стадии.
b) 1-(2-Изопропокситетрагидро-3-фуранил)-1,2-этандиол
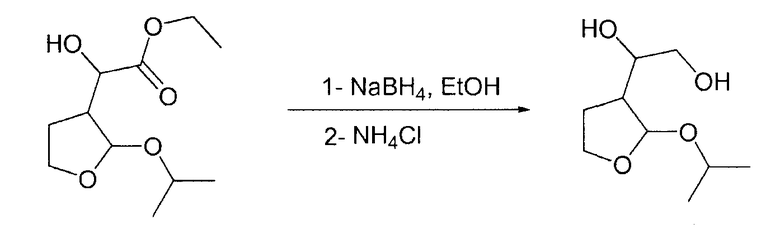
В круглодонную колбу, загруженную этанолом (50 мл) и NaBH4 (1,066 г, 28,19 ммоль, 1,1 экв.) при 0°С, по каплям на протяжении 1 ч при 0°С добавляли этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат (7,44 г, 25,62 ммоль, 1,000 экв.), растворенный в этаноле (30 мл). Смеси давали возможность нагреться до комнатной температуры и перемешивали в течение двух дней. Затем к реакционной смеси при 0°С добавляли хлорид аммония (2,056 г, 38,44 ммоль, 1,5 экв.), растворенный в воде (18 мл). Реакционную смесь перемешивали в течение 4 ч при комнатной температуре и растворитель выпаривали при пониженном давлении, получая при этом коричневое твердое вещество. Затем к сырой смеси добавляли этилацетат (40 мл) и смесь нагревали при 40°С в течение 30 минут. После фильтрования через дикалит гомогенную смесь упаривали досуха при пониженном давлении, получая при этом требуемый продукт (4,39 г, 19,61 ммоль, 0,7655 экв., выход 76,55%).
Смесь диастереомеров:
1H ЯМР (CDCl3, 400 МГц): 5,85 (д, J=8 Гц, 0,11H); 5,11-4,86 (м, 2H); 4,3-3,6 (м, 7,7H); 3,8-3,5 (м, 3H); 3,45 (м, 3,7H); 2,49-2,0 (м, 3,4H); 1,95-1,5 (м, 2,34); 1,28 (м, 1,1H); 1,20-1,12 (м, 6H).
13C ЯМР (CDCl3; 100 МГц): (основные пики) 109,0; 108,2; 105,8; 105,5; 72,6; 69,6; 69,4; 67,8; 66,9; 63,3; 63,1; 63,0; 49,0; 48,7; 32,4; 28,9; 27,6; 26,4; 23,6; 21,8; 21,8; 15,2; 14,2.
ГХ: пики разных изомеров @ 5,7 мин 17%; 6,07 мин 8,29%; 6,32 мин 15,7%; 6,7 мин 20,29%; 6,9 мин 11,0%; 10,6 мин 7,6%; 10,8 мин 5%.
Пример 8
Гексагидрофуро[2,3-b]фуран-3-ол
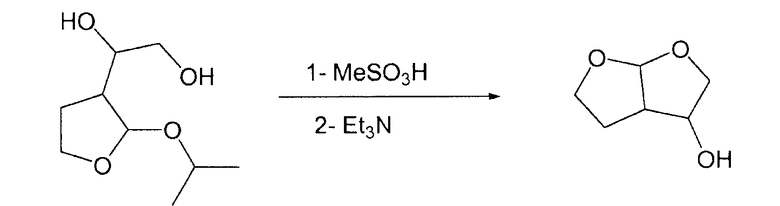
В 50-миллилитровой круглодонной колбе 1-(2-изопропокситетрагидро-3-фуранил)-1,2-этандиол (2,21 г; 8,89 ммоль, 1 экв.) растворяли в тетрагидрофуране (9 мл). После охлаждения при 0°С к смеси добавляли метансульфоновую кислоту (65 мг; 676,33 мкмоль). Реакционную смесь после этого нагревали при 45°С в течение 30 мин. После охлаждения при комнатной температуре к смеси добавляли триэтиламин (0,3 г; 2,96 ммоль). Растворитель упаривали и к смеси при комнатной температуре добавляли этилацетат (9 мл; 91,98 ммоль). Затем смесь фильтровали через дикалит и растворитель упаривали при пониженном давлении, получая при этом эндо/экзо-бис-ТГФ (1,562 г; ГХ: площадь 71%) при диастереомерном отношении эндо/экзо-диастереоизомеры 15/85.
Два диастереоизомера разделяли хроматографией на силикагеле, элюент: смесь 9/1 AcOEt/гексан.
ГХ: экзо-гексагидрофуро[2,3-b]фуран-3-ол: время удерживания 11,36 мин; эндо-гексагидрофуро[2,3-b]фуран-3-ол: время удерживания 11,57 мин.
1Н ЯМР:
экзо-гексагидрофуро[2,3-b]фуран-3-ол: 1,67 (м, 1H); 2,13 (м, 1H); 2,31 (ушир. с, 1H); 2,79 (м, 1H); 3,8-3,9 (м, 3H); 2,95 (дд, 1H, J=3,2 Гц, J=10,3 Гц); 4,2 (д, 1H, J=3,1 Гц); 5,9 (дд, 1H, J=4,9 Гц);
эндо-гексагидрофуро[2,3-b]фуран-3-ол: 1,85 (м, 1H); 1,94 (ушир. с, 1H); 2,27 (м, 1H); 2,84 (м, 1H); 3,6 (дд, 1H, J=7,1 Гц, J=9,2 Гц); 3,89 (м, 1H); 3,97 (м, 1H); 4,43 (дд, 1H, J=6,8 Гц, J=14,5 Гц); 5,68 (д, 1H, J=5,2 Гц).
Пример 9
а) Тетрагидрофуро[2,3-b]фуран-3(2Н)-он

В 250-миллилитровой круглодонной колбе NaOCl (6,15 г, 14% масс./масс.) разбавляли в 100 мл воды. рН раствора регулировали до 9,5 с применением 1 М водного раствора NaHCO3. В отдельной 250-миллилитровой круглодонной колбе гексагидрофуро[2,3-b]фуран-3-ол (1 г, 7,7 ммоль, 1 экв.) растворяли в 15 мл AcOEt при 0°С. Затем добавляли KBr (91 мг, 0,77 ммоль, 0,1 экв.), растворенный в 1 мл воды, с последующим добавлением ТЕМРО (12 мг, 0,08 ммоль, 0,01 экв.). Наконец, по каплям в смесь добавляли NaOCl. После 15 мин перемешивания при 0°С смесь экстрагировали 3 раза 100 мл AcOEt. Собранные органические слои сушили над Na2SO4 и растворитель выпаривали при пониженном давлении, получая при этом 950 мг белого твердого вещества, выход: 96%. Образовавшийся тетрагидрофуро[2,3-b]фуран-3(2Н)-он применяли на следующей стадии без дополнительной очистки.
Продукт идентифицировали по точной массе: m/z 128,0473 (теоретическая масса: 128,0473).
b) Стереоселективное получение (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ола
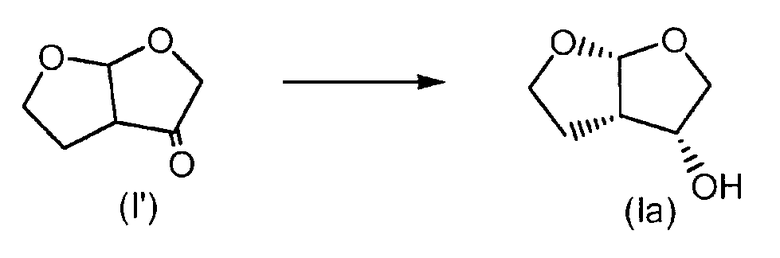
В общем согласно методике, описанной Ghosh et al. в J. Org. Chem. 2004, 69, 7822-7829, тетрагидрофуро[2,3-b]фуран-3(2Н)-он (950 мг, 7,42 ммоль, 1 экв.) растворяли при 0°С в 50 мл этанола. В виде одной порции к смеси добавляли NaBH4 (302,4 мг, 8 ммоль, 1,07 экв.). После периода перемешивания 1 ч добавляли гидрохлоридную соль диэтаноламина (3,2 г, 8 ммоль, 1,07 экв.) и смесь перемешивали на протяжении ночи при комнатной температуре. Гетерогенную смесь фильтровали через дикалит и промывали 20 мл горячего AcOEt. После выпаривания органических растворителей при пониженном давлении получали 1500 мг гексагидрофуро[2,3-b]фуран-3-ола с выходом (% площади) 40%; максимальный выход: 60%, диастереоизомерный избыток: экзо-(3S,3aS,6aR)/эндо-(3R,3aS,6aR): 18,5/81,5.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ (3R,3aS,6aR) ГЕКСАГИДРОФУРО[2,3-b] ФУРАН-3-ОЛА | 2005 |
|
RU2421458C2 |
| СПОСОБ ПОЛУЧЕНИЯ (3R, 3aS,6aR)-ГЕКСАГИДРОФУРО[2,3-b]ФУРАН-3-ИЛ(1S,2R)-3-[[(4-АМИНОФЕНИЛ)СУЛЬФОНИЛ](ИЗОБУТИЛ)АМИНО]-1-БЕНЗИЛ-2-ГИДРОКСИПРОПИЛКАРБАМАТА | 2004 |
|
RU2376308C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| УПРОЩЕННАЯ ПРОЦЕДУРА ПОЛУЧЕНИЯ ДАРУНАВИРА | 2017 |
|
RU2745740C2 |
| ТЕТРАГИДРОФУРО(3,2-b)ПИРРОЛ-3-ОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА К | 2007 |
|
RU2456290C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| СИНТЕЗ ПРЕДШЕСТВЕННИКА ИНГИБИТОРА ПРОТЕАЗЫ | 2006 |
|
RU2421459C2 |
| ГЕКСАГИДРОФУРО [2,3-B] ФУРАН-3-ИЛ-N-{3-[(1,3-БЕНЗОДИОКСОЛ-5- ИЛСУЛЬФОНИЛ)(ИЗОБУТИЛ)АМИНО]-1-БЕНЗИЛ -2-ГИДРОКСИПРОПИЛ}КАРБАМАТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ИНГИБИРОВАНИЯ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2247123C2 |
| ЭЛЕКТРОПРЯДЕНЫЕ АМОРФНЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2003 |
|
RU2331411C2 |
| СПОСОБ ПОЛУЧЕНИЯ (3aR,4S,6R,6aS)-6-АМИНО-2,2-ДИМЕТИЛТЕТРАГИДРО-3aН-ЦИКЛОПЕНТА[d][1,3]ДИОКСОЛ-4-ОЛ-ДИБЕНЗОИЛ-L-ТАРТРАТА И ПРОДУКТЫ УКАЗАННОГО СПОСОБА | 2008 |
|
RU2477277C2 |
Настоящее изобретение относится к способу получения соединения формулы (V), который может быть использован в фармацевтической промышленности
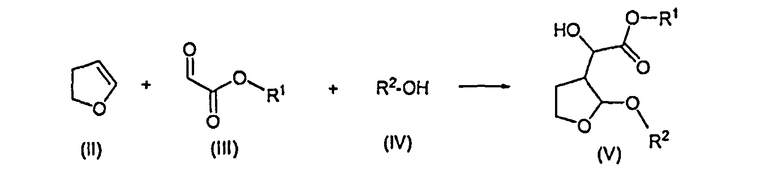
Способ включает реакцию соединения формулы (II) с соединением формулы (III) в присутствии соли титана формулы Ti(Hal)n(OR)4-n, где Hal представляет собой радикал галогена, n равно 0, 1, 2 или 3, R представляет собой алкил или арилалкил, и последующую реакцию образовавшегося продукта реакции со спиртом формулы (IV), где R1 и R2 представляют собой алкил или арилалкил, где арил представляет собой фенил или нафтил. Предложен новый эффективный способ получения соединений формулы (V) с использованием новых промежуточных соединений. 8 н. и 8 з.п. ф-лы, 9 пр.
1. Способ получения соединения формулы (V), который включает в себя реакцию 2,3-дигидрофурана формулы (II) с производным глиоксилата формулы (III) в присутствии соли титана формулы Ti(Hal)n(OR)4-n, в которой Hal представляет собой радикал галогена, n равно 0, 1, 2 или 3 и R представляет собой алкил или арилалкил, где арил представляет собой фенил или нафтил, и последующую реакцию образовавшегося продукта реакции со спиртом формулы (IV) с образованием соединения формулы (V):
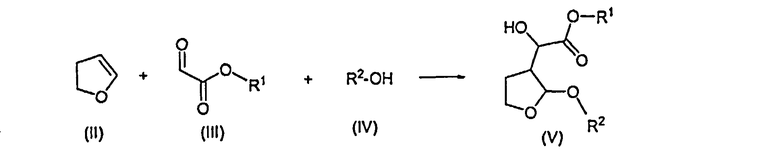
где R1 представляет собой алкил или арилалкил, где арил представляет собой фенил или нафтил, и R2 представляет собой алкил или арилалкил, где арил представляет собой фенил или нафтил.
2. Способ по п.1, в котором солью титана является соединение формулы Ti(Hal)n(OR)4-n, в которой n равно 1, 2 или 3.
3. Способ по п.1, в котором R1 представляет собой С1-4алкил.
4. Способ по п.1, в котором R2 представляет собой С1-4алкил.
5. Способ по любому из пп.1-4, в котором 2,3-дигидрофуран формулы (II) подвергают реакции с производным глиоксилата формулы (III) в присутствии соли титана и затем продукт реакции подвергают реакции со спиртом формулы (IV) с образованием соединения формулы (V).
6. Способ по любому из пп.1-4, в котором образовавшуюся реакционную смесь, содержащую соединение формулы (V), обрабатывают сегнетовой солью для осуществления удаления остаточного соединения титана.
7. Способ по п.6, в котором обработку сегнетовой солью проводят в щелочной среде.
8. Соединения формулы (Va):
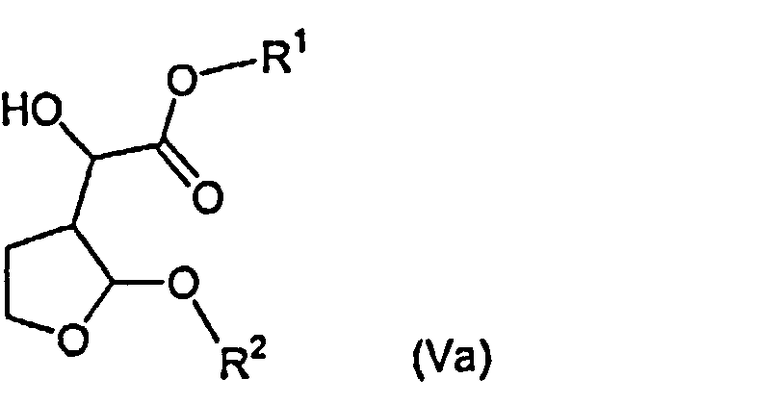
и их стереоизомерные формы и рацемические смеси,
у которых R1 представляет собой алкил или арилалкил, где арил представляет собой фенил или нафтил, и R2 представляет собой алкил или арилалкил, где арил представляет собой фенил или нафтил, при условии, что, когда R2 представляет собой метил, R1 не является метилом или этилом.
9. Этилгидрокси-(2-изопропокситетрагидро-3-фуранил)ацетат.
10. Способ получения соединений формулы (VI), который содержит восстановление соединения формулы (V) с образованием соединения формулы (VI):
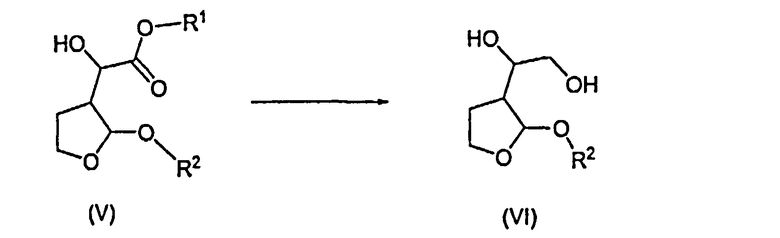
в которой R1 и R2 имеют значения, указанные в п.1.
11. Способ по п.10, в котором восстановление проводят боргидридным восстанавливающим агентом.
12. Соединения формулы (VI):
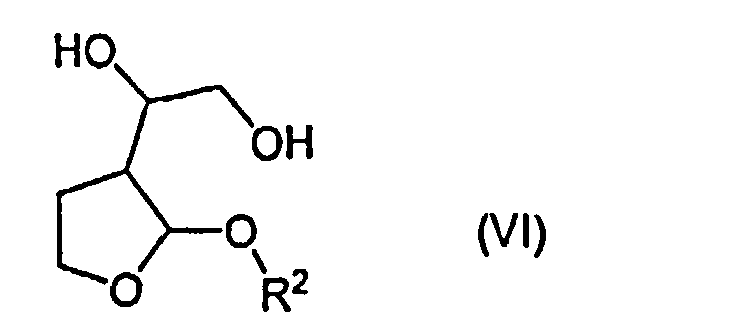
в которой R имеет значения, указанные в п.1.
13. 1-(2-Изопропокситетрагидро-3-фуранил)-1,2-этандиол.
14. Способ получения соединения формулы (I), который содержит циклизацию соединения формулы (VI) с образованием соединения формулы (I):

15. Способ по п.14, в котором циклизацию соединения формулы (VI) проводят обработкой сильной протонной кислотой.
16. Способ получения гексагидрофуро[2,3-b]фуран-3-ола формулы (I), который содержит стадии:
а) реакции 2,3-дигидрофурана формулы (II) с производным глиоксилата формулы (III) в присутствии соли титана формулы Ti(Hal)n(OR)4-n, в которой n равно 0, 1, 2 или 3 и R представляет собой алкил или арилалкил, где арил представляет собой фенил или нафтил, и последующую реакцию образовавшегося продукта реакции со спиртом формулы (IV) с образованием соединения формулы (V):
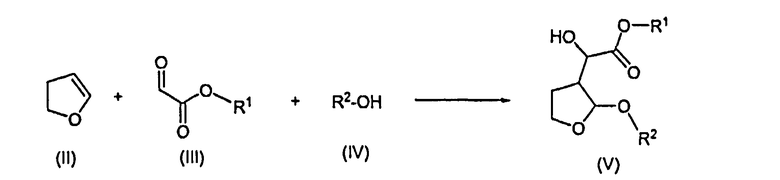
в которой R1 представляет собой алкил или арилалкил, где арил представляет собой фенил или нафтил, и R2 представляет собой алкил или арилалкил, где арил представляет собой фенил или нафтил; и
b) восстановления образовавшегося соединения формулы (V) с образованием соединения формулы (VI):
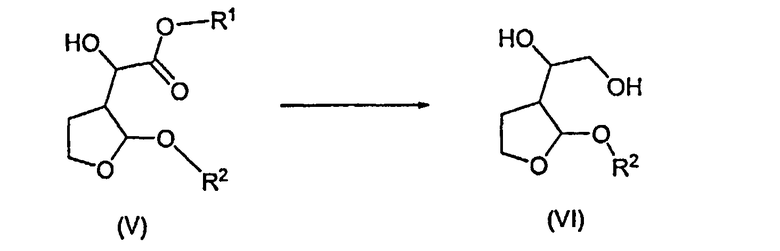
и
с) циклизации соединения формулы (VI) с образованием соединения формулы (I):
и, при желании, затем (i) хиральное разделение образовавшегося соединения формулы (I) для выделения (3R,3аS,6аR)-гексагидрофуро[2,3-b]фуран-3-ола формулы (Iа):
и/или
(ii) окисление образовавшегося соединения формулы (I) с образованием соединения формулы (I'):
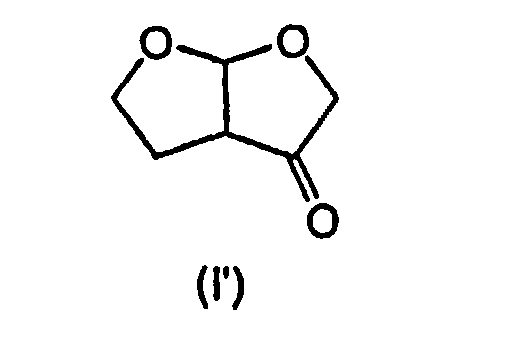
и затем восстановление соединения формулы (I') в соединение формулы (Iа).
| GHOSH A.K | |||
| ЕТ AL | |||
| Bioorganic & Medicinal chemistry Letters, 1998, с.687-690 | |||
| СПОСОБ ПОЛУЧЕНИЯ (3R, 3aS,6aR)-ГЕКСАГИДРОФУРО[2,3-b]ФУРАН-3-ИЛ(1S,2R)-3-[[(4-АМИНОФЕНИЛ)СУЛЬФОНИЛ](ИЗОБУТИЛ)АМИНО]-1-БЕНЗИЛ-2-ГИДРОКСИПРОПИЛКАРБАМАТА | 2004 |
|
RU2376308C2 |
| GHOSH A.K | |||
| ЕТ AL | |||
| Tetrahedron Letters, 1999, vol | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| WO 2004002975 A, 08.01.2004 | |||
| WO 03022853 A, 20.03.2003. | |||

