Область изобретения
Настоящее изобретение направлено к способу получения диастереомерно чистой дибензоил-L-тартратной соли соединения формулы (III)
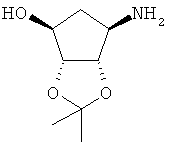
к продуктам указанного способа и их применению.
Предшествующий уровень техники
В Ranganathan, S. and George, K.S.Tetrahedron, 1997, 53, 3347 описан синтез соединения (I).

В Jung, M. et al. Helv. Chim. Acta, 1983, 66, 1915 и в Ranganathan, S. and George, К.S.Tetrahedron, 1997, 53, 3347 раскрыт синтез рацемического соединения (II).

В WO 99/05142, Shireman, В.Т. and Miller, M.J.Tetrahedron Lett., 2000, 41, 9537 и в Rajappan, V.Р. et al. Synth. Commun. 2001, 31, 2849 описаны синтезы или свободного амина, или солянокислой соли соединения (III).
Описание изобретения
Настоящее изобретение направлено на способ получения диастереомерно чистой дибензоил-L-тартратной соли соединения формулы (III)

включающий стадии
(а) смешивания соединения формулы (II)
с энантиомерно чистой дибензоил-L-винной кислотой или ее моногидратом с образованием диастереоизмерной соли и
(б) кристаллизации указанной соли.
Способ по настоящему изобретению особенно полезен для крупномасштабного получения диастереомерно чистой дибензоил-L-тартратной соли соединения формулы (III).
Способ получения диастереомерно чистой дибензоил-L-тартратной соли соединения формулы (III) может начинаться с соединения формулы (II), которое может быть получено, как известно в данной области техники. Затем соединение формулы (II) растворяют с получением желательного (3aS,4R,6S,6aR)-энантиомера посредством кристаллизации диастереомерно чистой соли, используя энантиомерно чистую дибензоил-L-винную кислоту или ее моногидрат с получением соответствующей диастереомерно чистой дибензоил-L-тартратной соли соединения формулы (III).
Альтернативно, способ получения диастереомерно чистой дибензоил-L-тартратной соли соединения формулы (III) может начинаться с соединения формулы (I), которое можно получать так, как известно в данной области техники. Соединение (I) превращают в соединение (II), как известно в данной области техники. Затем соединение формулы (II) растворяют, получая желательный (3aS,4R,6S,6aR)-энантиомер посредством кристаллизации диастереомерно чистой дибензоил-L-тартратной соли с использованием энантиомерно чистой дибензоил-L-винной кислоты или ее моногидрата, с получением соответствующей диастереомерно чистой соли соединения формулы (III).
Следующая схема иллюстрирует способ получения диастереомерно чистой 1:1-соли дибензоил-L-винной кислоты и соединения формулы (III):

Одно воплощение настоящего изобретения представляет собой способ получения дибензоил-L-тартрата соединения формулы (III). Еще одно воплощение настоящего изобретения представляет собой 1:1-соль дибензоил-L-винной кислоты и соединения формулы (III). Указанную соль также можно называть (3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ол-(2R,3R)-2,3-бис(бензоилокси)-3-карбоксипропаноатом, (3aR,4R,6S,6aR)-6-амино-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ол-(2R,3R)-2,3-бис(бензоилокси)сукцинатом или (3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ол-дибензоил-L-тартратом.
Энантиомерно чистая кислота, подходящая для использования на стадии растворения, представляет собой дибензоил-L-винную кислоту, также называемую (2R,3R)-2,3-бис(бензоилокси)-3-карбоксипропановой кислотой или (2R,3R)-2,3-биc(бeнзoилoкcи)янтapнoй кислотой.
Растворители, пригодные для стадии растворения, на которой получают диастереомерно чистую соль соединения формулы (III), могут быть выбраны из алифатических спиртов (таких как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол и трет-бутанол); нитрилов (таких как ацетонитрил); полярных эфиров, таких как тетрагидрофуран, 2-метилтетрагидрофуран, диэтиленгликолевые моноэфиры, подобные целлозольву (этоксиэтанол), метоксиэтанол, изопропоксиэтанол; алифатических эфиров (таких как этилацетат, бутилацетат или изопропилацетат); полярных апротонных растворителей (таких как N-метилпирролидинон, N,N-диметилацетамид или N,N-диметилформамид); и их смесей. Также стадия растворения может быть выполнена в воде или в растворе, содержащем воду, и в любом из вышеперечисленных органических растворителей.
В одном воплощении настоящего изобретения растворитель на стадии (а) выбирают из алифатических спиртов; нитрилов; простых эфиров; алифатических сложных эфиров; полярных апротонных растворителей; воды и их смесей.
В дополнительном воплощении настоящего изобретения растворитель на стадии (а) представляет собой смесь воды и алифатического спирта или воды и полярного эфирного растворителя или нитрила.
В дополнительном воплощении настоящего изобретения растворитель на стадии (а) представляет собой смесь воды и метанола, этанола, н-пропанола, изопропанола, н-бутанола, изобутанола, целлозольва (этоксиэтанола), метоксиэтанола, изопропоксиэтанола, тетрагидрофурана или ацетонитрила.
В дополнительном воплощении настоящего изобретения растворитель на стадии (а) представляет собой смесь воды и этанола.
Стадию растворения с получением диастереомерно чистой соли соединения формулы (III) сначала выполняют при температурах от 0°С до точки кипения растворителя, чтобы полностью растворить компоненты или образовавшиеся диастереоизомерные соли. Когда компоненты растворены, температуру раствора доводят до температуры от -50°С до +50°С с получением кристаллической соли соединения (III). Эту соль затем можно перекристаллизовывать из растворителя, аналогичного использованным выше или отличного от них, для улучшения оптической и химической чистоты.
Еще одно воплощение настоящего изобретения представляет собой применение дибензоил-L-тартрата соединения формулы (III) в получении (1S-[1α,2α,3β(1S*,2R*),5β]}-3-(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3H-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)-циклопентан-1,2-диола.
Термин "диастереомерно чистая соль" определяет соль энантиомерно чистого катиона (амин III в настоящем изобретении) и энантиомерно чистого аниона (моноанион III дибензоил-L-винной кислоты в настоящем изобретении).
ПРИМЕРЫ
Пример 1. Получение (3aS,4R,6S,6aR)-6-гидрокси-2,2-диметилтетрагидро-3aH-цикло-пента[(d][1,3]диоксол-4-аминия (2R,3R)-2,3-бис(бензоилокси)-3-карбоксипропаноата (дибензоил-L-тартрат соединения (III)).
Методика через выделение соединения (III)
Соединение (I) (415 г, 1,36 моль) растворяли в 1,8 л метанола, и полученный раствор перемещали в реактор вместе с суспензией Pd/C (25 г пасты, содержащей 62% воды мас./мас.) в воде (50 мл). Устанавливали температуру 50°С, и реактор продували азотом. Применяли давление водорода (3 бар (3×105 Па)). За реакцией наблюдали с помощью HPLC. Через 3 ч реакция закончилась. Метанольную суспензию фильтровали и концентрировали при пониженном давлении с получением 230 г (98% выход) соединения (II) в виде бежевато-белого твердого вещества, которое использовали непосредственно на следующей стадии. ГХ (газовая хроматография) - чистота данного вещества составляла более 97%, и количественный анализ посредством титрования соответствовал 95% мас./мас.
Соединение (II) (227 г, 1,31 моль) растворяли в 1641 г смеси этанол/вода (70/30 по объему) при 26°С. Добавляли моногидрат дибензоил-L-винной кислоты (493 г, 1,31 моль), позволяя во время добавления внутренней температуре достичь 32°С. Кристаллизацию оставляли на 18 часов при комнатной температуре. Полученные кристаллы отфильтровывали и промывали смесью 2×300 мл этанол/вода (70/30 по объему). После сушки при 44°С под вакуумом в течение примерно 5 ч, 272 г (39%-ный выход или 78% от теоретического выхода) дибензоил-L-тартрата соединения (III) получали в виде белого кристаллического твердого вещества. Оптическая чистота при определении посредством газовой хроматографии на свободном амине составляла 99% д.и. (диастереомерный избыток).
Пример 2. Получение (3aS,4R,6S,6aR)-6-гидрокси-2,2-диметилтетрагидро-3aH-цикло-пента[(d][1,3]диоксол-4-аминия (2R,3R)-2,3-бис(бензоилокси)-3-карбоксипропаноата (дибензоил-L-тартрат соединения (III))
Методика через выделение соединения (II)
Соединение (II) (3,21 г, чистота 92%, 17,0 ммоль) растворяли в смеси этанол/вода (21,6 г, 70% об./об. этанола в воде) при 22°С. К прозрачному раствору добавляли дибензоил-L-винную кислоту (6,23 г, 17,4 ммоль). Сначала образовался прозрачный раствор, а кристаллизация началась примерно через 10 минут. Полученную суспензию оставляли на 2 ч, затем кристаллы выделяли фильтрацией и промывали смесью этанол/вода (70% об./об. этанол в воде, 2×5 мл). Кристаллы сушили при 40°С под вакуумом, получая в 3,31 г (37% выход) чистого дибензоил-L-тартрата соединения (III). Оптическая чистота, определенная посредством газовой хроматографии на свободном амине, составляла 97,6% д.и.
Пример 3. Получение (3aS,4R,6S,6aR)-6-гидрокси-2,2-диметилтетрагидро-3aH-цикло-пента[d][1,3]диоксол-4-аминия (2R,3R)-2,3-бис(бензоилокси)-3-карбоксипропаноата (дибензоил-L-тартрат соединения (III))
Методика через выделение соединения (II)
Соединение (II) (3,22 г, чистота 92%, 17,1 ммоль) растворяли в смеси этанол/вода (21,6 г, 70% об./об. этанола в воде) при 22°С. Дибензоил-L-винную кислоту (6,50 г, 18,1 ммоль) добавляли к прозрачному раствору. Полученную суспензию нагревали до 70°С для растворения осадка. Затем раствор оставляли охлаждаться до комнатной температуры в течение 3 ч, затем полученные кристаллы выделяли фильтрацией. Кристаллы промывали смесью этанол/вода (70% об./об. этанола в воде, 3×5 мл). Кристаллы сушили при 40°С под вакуумом, получая 3,19 г (35% выход) чистого дибензоил-L-тартрата соединения (III). Оптическая чистота, определенная посредством газовой хроматографии на свободном амине, составляла 98,4% д.и.
Пример 4. Получение (3aS,4R,6S,6aR)-6-гидрокси-2,2-диметилтетрагидро-3aH-цикло-пента[d][1,3]диоксол-4-аминия (2R,3R)-2,3-бис(бензоилокси)-3-карбоксипропаноата (дибензоил-L-тартрат соединения (III))
Методика через невыделенное соединение (II)
Соединение (I) (500 г; 1,64 моль) и Pd/C (25 г, 60% водная паста) добавляли в 5 л стальной реактор с рубашкой при температуре окружающей среды. Реактор продували азотом (3 бар (3×105 Па)). Добавляли смесь этанола и воды (1750 г, 70/30 по объему), и реактор еще раз продували азотом (3 бар (3×105 Па)) при перемешивании. Использовали газообразный водород (3 бар (3×105 Па)) и температуру рубашки повышали до 50°С. Через 2 ч при 50°С исходное вещество не определялось, и реакционную смесь фильтровали для удаления катализатора Pd. Твердый катализатор промывали смесью этанол/вода (300 г, 70/30 по объему), и промывную жидкость объединяли с остальным раствором. Дибензоил-L-винную кислоту (588 г, 1,64 моль) добавляли в стеклянный сосуд с рубашкой. Указанный выше раствор соединения (II) добавляли при 24°С и при медленном перемешивании. Полученную смесь оставляли примерно на 16 ч при 22°С, и затем полученные кристаллы отфильтровали. Осадок на фильтре дважды промывали смесью этанол/вода (2×375 мл, 70/30 по объему). Затем кристаллы сушили вплоть до постоянной массы при 50°С в вакуумной сушильной печи. Получали 324 г (37% выход, 74% от теоретического максимума) дибензоил-L-тартрата соединения (III) в виде белого твердого вещества. Оптическая чистота, которую определяли газовой хроматографией на свободном амине, составляла 99,6% д.и. Точка плавления 150-151°С (нескорректированная); 1H ЯМР (400 МГц, MeOH-d4) δ 7.51 (каж d, J=8 Гц, 1Н), 7.50 (каж d, J=8 Гц, 1Н), 7.24 (каж t, J=8 Гц, 2Н), 4.50 (каж dd, J1=6 Гц, J2=8 Гц, 1Н), 3.02 (каж dd, J1=8 Гц, J2=16 Гц, 1Н), 2.86 (каж dd, J1=6 Гц, J2=16 Гц, 1Н), 1.36 (s, 9Н); 13С ЯМР (100 МГц, MeOH-d4) δ 171.6, 167.4, 134.5, 131.1, 131.0, 129.6, 112.4, 86.8, 84.2, 76.7, 75.0, 58.1, 35.0, 26.4, 24.0. MS [М]+ 173; [α]D (с 1,0 в метаноле, 25°С) - 76,6°.
Пример 5. Получение (3aS,4R,6S,6aR)-6-гидрокси-2,2-диметилтетрагидро-3аH-цикло-пента[d][1,3]диоксол-4-аминия (2R,3R)-2,3-бис(бензоилокси)-3-карбоксипропаноата (дибензоил-L-тартрат соединения (III))
Методика через невыделенное соединение (II)
Раствор соединения (II) (50,0 г, 0,164 моль), воды (58 г) и этанола (104 г) обрабатывали 5%-ным Pd/C (1,3 г) в атмосфере водорода (8 бар (8×105 Па)) при 50°С в течение 18 ч. Реакционную смесь охлаждали до 30°С, фильтровали, и фильтр промывали смесью воды (10,5 г) и этанола (19,5 г). Добавляли моногидрат дибензоил-L-винной кислоты (61,6 г, 0,164 моль). Смесь перемешивали в течение 2 ч при 28°С, охлаждали до 18°С и перемешивали в течение еще 2 ч. В результате фильтрации, промывания смесью воды (26,3 г) и этанола (48,8 г) и сушки получали дибензоил-L-тартрат соединения (III) в виде белого твердого вещества (31,7 г, 36% выход).
Настоящее изобретение относится к способу получения диастереомерно чистой дибензоил-L-тартратной соли соединения формулы (III)

включающему стадии:
(а) смешивания соединения формулы (II)

с энантиомерно чистой дибензоил-L-винной кислотой или ее моногидратом с образованием диастереоизомерной соли, где растворитель представляет собой смесь воды и этанола; и
(б) кристаллизации указанной соли, а также к моно-дибензоил-L-тартратной соли соединения формулы (III), которая является промежуточным продуктом в синтезе (1S-[1α,2α,3β(1S*,2R*),5β]}-3(7-{[2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил)-5-(2-гидроксиэтокси)-циклопентан-1,2-диола, который обладает антитромбозным действием. 2 н.п. ф-лы, 5 пр.
1. Способ получения диастереомерно чистой дибензоил-L-тартратной соли соединения формулы (III)
включающий стадии:
(а) смешивания соединения формулы (II)
с энантиомерно чистой дибензоил-L-винной кислотой или ее моногидратом с образованием диастереоизомерной соли, где растворитель представляет собой смесь воды и этанола; и
(б) кристаллизации указанной соли.
2. Моно-дибензоил-L-тартратная соль соединения формулы (III)
| US 6525060 B1, 25.02.2003 | |||
| СПОСОБ ВЫРАВНИВАНИЯ ЭЛЕКТРИЧЕСКОГО ПОТЕНЦИАЛА В ЗОНЕ ЗАЗЕМЛЕНИЯ ЭЛЕКТРОУСТАНОВОК | 0 |
|
SU192263A1 |
| US 6156756 A, 05.12.2000 | |||
| US 6251910 B1, 26.06.2001 | |||
| US 20030181469 A1, 25.09.2003 | |||
| 1,3-ДИОКСОЛАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2276149C2 |

