Область техники, к которой относится изобретение
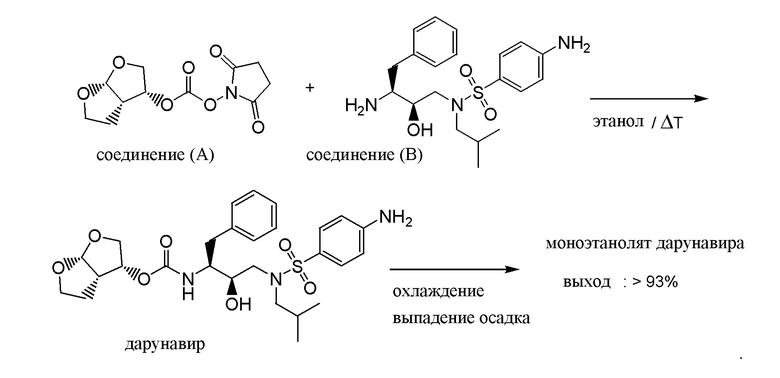
Настоящее изобретение относится к усовершенствованному способу получения (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового сложного эфира [(1S,2R)-3-[[(4-аминофенил)-сульфонил](2-метилпропил)амино]-2-гидрокси-1-(фенилметил)-пропил]-карбаминовой кислоты, это соединение также известно под INN-названием дарунавир, путем взаимодействия 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложного эфира угольной кислоты с 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамидом в этаноле в качестве растворителя. Кроме того, указанный способ позволяет выделять дарунавир сразу в форме этанолята, т.е. как моноэтанолят дарунавира, который является формой дарунавира, поступающей в продажу под торговым названием Prezista™.
Предпосылки создания изобретения
Дарунавир представляет собой непептидный ингибитор протеазы, одобренный для использования при лечении вируса иммунодефицита человека типа 1 (ВИЧ-1), который имеет следующее химическое строение:
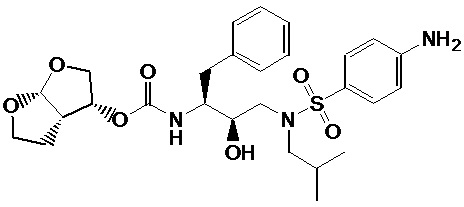
В Примерах 7 в WO-2005/063770 раскрыт способ получения дарунавира взаимодействием 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложного эфира угольной кислоты [в данном тексте обозначен как соединение (A)], который впервые был приготовлен in situ, с 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамидом [в данном тексте обозначен как соединение (B)] в этилацетате в качестве растворителя, к которому добавлен триэтиламин в качестве основания. Реакцию гасили добавлением водного раствора метиламина в этаноле. Неочищенный дарунавир, выделенный из реакционной смеси, переводили в форму этанолата путем кристаллизации из абсолютного этанола с выходом 71%.
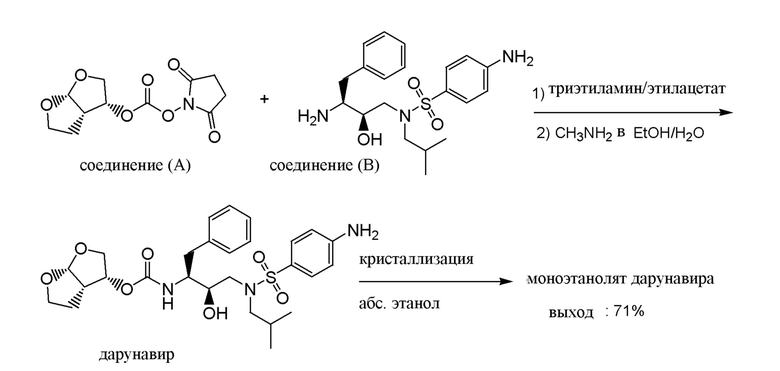
В Примерах 9 в WO-2005/063770 раскрыт способ получения дарунавира взаимодействием соединения (A), который сначала получали in situ, с соединением (B) в ацетонитриле в качестве растворителя, к которому добавлен триэтиламин в качестве основания. Реакцию гасили добавлением водного раствора метиламина в этаноле. Неочищенный дарунавир, выделенный из реакционной смеси, переводили в форму этанолата путем кристаллизации из абсолютного этанола с выходом 81%.
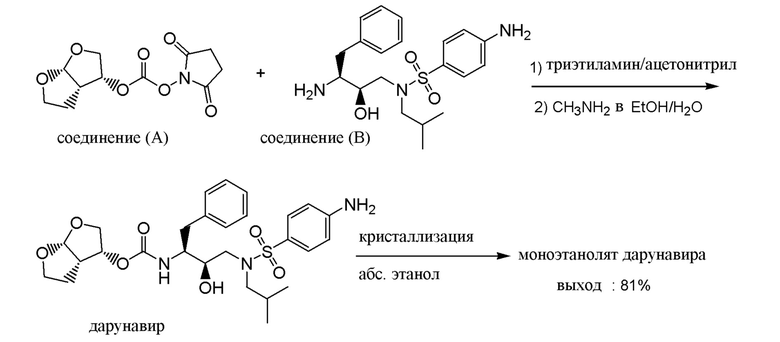
В Journal of Medicinal Chemistry, vol. 48, p. 1813-1822 (2005) раскрыт синтез дарунавира, обозначенного как соединение (1a), взаимодействием соединения (A) с соединением (B) в дихлорметане в качестве растворителя в присутствии триэтиламина в качестве основания с выходом 90%.
В WO-2009/055006 раскрыт способ получения дейтерированной формы дарунавира взаимодействием 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложного эфира угольной кислоты с дейтерированной формой 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамидом в дихлорметане в качестве растворителя в присутствии триэтиламина в качестве основания.
В Примере 3 в WO-2014/016660 раскрыт способ получения дарунавира в сольватированной пропионатной форме взаимодействием 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложного эфира угольной кислоты с 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамидом в двух-фазовой смеси этилацетата и воды, с последующим преобразованием в соль под действием пропионовой кислоты с суммарным выходом 90%.
Нами было обнаружено, что дарунавир можно получать по упрощенной процедуре реакцией 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложного эфира угольной кислоты, т.е. соединения (A), с 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамидом, т.е. соединением (B), при нагревании в отсутствие органического основания и в этаноле в качестве растворителя. По завершении реакции реакционную смесь нагревают пока она не становится однородной, а затем ей дают охладиться до комнатной температуры, при этом дарунавир кристаллизуется в виде моноэтанолята.
Описание изобретения
В одном аспекте настоящее изобретение относится к способу приготовления дарунавира, причем этот способ включает в себя стадии
a) взаимодействия 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложного эфира угольной кислоты с 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамидом в этаноле в качестве растворителя при нагревании реакционной смеси при температуре от 30°C до температуры образования флегмы.
В следующем аспекте настоящее изобретение относится к способу приготовления дарунавира, причем этот способ включает в себя стадии
a) взаимодействия 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложного эфира угольной кислоты с 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамидом в отсутствие органического основания и в этаноле в качестве растворителя при нагревании реакционной смеси при температуре от 30°C до температуры образования флегмы.
В следующем аспекте настоящее изобретение относится к способу приготовления дарунавира в виде моноэтанолята, причем этот способ включает в себя стадии
a) взаимодействия 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложного эфира угольной кислоты с 4-амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамидом в отсутствие органического основания и в этаноле в качестве растворителя при нагревании реакционной смеси при температуре от 30°C до температуры образования флегмы;
b) по завершении реакции, нагревания реакционной смеси до однородного состояния;
c) охлаждения реакционной смеси;
d) выделения выпавшего в осадок продукта;
e) кристаллизации полученного таким образом осажденного продукта из этанола; и
f) выделения кристаллизованного моноэтанолята дарунавира.
На стадии a) реакционную смесь нагревают до температуры в интервале от температуры окружающей среды до температуры образования флегмы, такой как от 20°C до 78°C, или от 30°C до температуры образования флегмы, или от 30°C до 78°C, или от 30°C до 70°C, или от 40° до 60°C. Специалисту в данной области хорошо известно, что скорость реакции возрастает при проведении реакции при повышенной температуре. Специалист может следить за протеканием и завершением реакции путем отбора проб через регулярные промежутки времени и анализирования этих проб, например, используя тонкослойную хроматографию или ВЭЖХ.
Способ приготовления дарунавира согласно настоящему изобретению можно выполнять без участия органического основания. Органическое основание используют для захвата любого кислотного продукта, образующегося в ходе реакции. Хорошо известными примерами являются, например, триметиламин, триэтиламин, пиридин и т.п.
Эта упрощенная процедура приготовления дарунавира имеет следующие преимущества над известными в области техники процедурами:
- не используются опасные хлорированные растворители, такие как дихлорметан;
- не используются токсичные растворители, такие как ацетонитрил;
- не используется органическое основание, такое как триэтиламин или пиридин, которое чрезвычайно трудно удалять при производстве в промышленном масштабе;
- высокий выход;
- при охлаждении реакционной смеси дарунавир выпадает в осадок в виде моноэтанолята, который можно легко выделять и перекристаллизовывать из этанола с получением дарунавира моноэтанолята, имеющего достаточную для его использования чистоту,
- т.е. нет необходимости в дополнительных стадиях очистки, как этого требует АФИ (активный фармацевтический ингредиент) в таких коммерческих продуктах, как Prezista™.
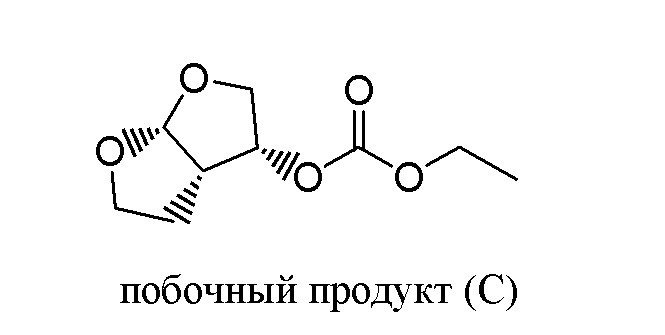
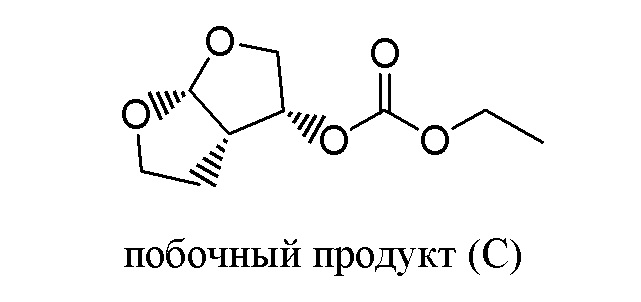
Применение этанола в качестве растворителя для реакции между соединением (A) и соединением (B) не очевидно для специалиста в данной области, поскольку специалист в данной области будет ожидать, что соединение (A) будет вступать в реакцию с протонным растворителем этанолом, присутствующем в большом избытке, по конкурирующей побочной реакции с образованием нежелательного побочного продукта (C). Наблюдали образование только небольших количеств побочного продукта (C) - менее 0,5% по отношению к продукту (A), а практическое отсутствие конкурентной побочной реакции соединения (A) с этанолом совершенно неожиданно для специалиста в данной области.
Когда соединение (A) и соединение (B) вносят в растворитель этанол в реакторе при температуре окружающей среды, реакционная смесь является однородной. Перемешивание и нагрев реакционной смеси продолжают пока не завершится реакция соединения (A) с соединением (B). По достижении температуры образования флегмы растворителя (т.е. 78°C в случае этанола) реакционная смесь становится однородной.
Мольное соотношение соединения (A) и соединения (B) может изменяться в пределах от 0,95 до 1,05.
По завершении реакции между соединением (A) и соединением (B) реакционную смесь медленно охлаждают до температуры в интервале от 15°C до -10°C, при охлаждении твердый продукт выпадает в осадок из реакционной смеси. Выпавший в осадок продукт затем отделяют от реакционной смеси, и его можно дополнительно очищать перекристаллизацией из этанола для получения чистого дарунавира моноэтанолята.
Перекристаллизацию полученного выпавшего в осадок продукта выполняют путем суспендирования выпавшего в осадок продукта в этаноле, нагревания полученной смеси до получения однородного раствора с последующим охлаждением до температуры окружающей среды или до температуры в интервале от -5°C до 25°C. Охлаждение кристаллизационной смеси можно выполнять путем естественного охлаждения или согласно определенному температурному профилю охлаждения. Например, температурный профиль охлаждения может представлять собой линейный профиль, например, 0,1°C/мин, 0,3°C/мин, 0,5°C/мин, 0,75°C/мин, 1°C/мин, 2°C/мин или любое другое значение. В качестве альтернативы можно применять кубический профиль охлаждения. В ходе охлаждения можно вносить затравку из кристаллов дарунавира моноэтанолята.
Процедура охлаждения кристаллизационной смеси также может включать процедуру Оствальдовского созревания, также называемую процедурой цикличного изменения температуры, при которой кристаллизационную смесь после внесения затравки охлаждают до определенной температуры, затем снова нагревают и снова охлаждают до определенной температуры перед тем, как кристаллизационной смеси дают охладиться до температуры окружающей среды. Такая процедура Оствальдовского созревания может также улучшать морфологию кристаллов дарунавира моноэтанолята. На практике используют следующую процедуру Оствальдовского созревания:
- выдерживание кристаллизационной смеси при температуре в интервале от 61°C до 63°C в течение периода от 20 до 40 минут,
- охлаждение кристаллизационной смеси до температуры в интервале от 57°C до 59°C в течение периода от 5 до 20 минут,
- выдерживание кристаллизационной смеси при температуре в интервале от 57°C до 59°C в течение периода от 20 до 40 минут,
- нагревание кристаллизационной смеси до температуры в интервале от 66°C до 68°C в течение периода от 5 до 20 минут,
- выдерживание кристаллизационной смеси при температуре в интервале от 66°C до 68°C в течение периода от 20 до 40 минут.
Процедуру Оствальдовского созревания можно повторять до получения желаемой морфологии кристаллов дарунавира моноэтанолята.
Выделение кристаллов дарунавира моноэтанолята можно осуществлять с помощью любых традиционных способов, как например, путем фильтрации или центрифугирования.
Способ приготовления дарунавира моноэтанолята согласно настоящему изобретению можно применять в периодических химических процессах, а также при полунепрерывном получении или непрерывном химическом получении (также известном как непрерывное производство).
Способ приготовления дарунавира моноэтанолята согласно настоящему изобретению можно также осуществлять в другом спирте, отличающемся от этанола, таком как, например, изопропаноле, пропаноле, бутаноле и т.п. Такой способ должен включать последующую дополнительную стадию преобразования полученного дарунавира алкоголята в дарунавир моноэтанолят.
Экспериментальная часть
Аналитический анализ
Присутствие побочного продукта (С)
определяли, используя газовою хроматографию при следующих условиях:
размер частиц 0,1 мкм или эквивалентный
вкладыш : Agilent 210-4004-5
промывочный растворитель В: ацетонитрил
предварительная промывка растворителем А: 6 раз
предварительная промывка растворителем В: 0 раз
промывка образцом: 0 раз
инъекционных насосов: 6 раз
последующая отмывка растворителем А: 0 раз
последующая отмывка растворителем В: 6 раз
запрограммированный прогон: 30°C/мин до 310°C
Пример 1
4-Амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамид (39,2 г; 0,1 моль) и этанол (170 мл) вносили в 1-литровый реактор. Реакционную смесь перемешивали 15 минут при 20°C и добавляли 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового] сложный эфир угольной кислоты (26,6 г; 0,098 моль) при 20°C. Реакционную смесь нагревали до 50°C за период продолжительностью от 30 минут до 1 часа и перемешивали 2 часа при этой температуре. Реакционную смесь нагревали до 78°C (кипячение с обратным холодильником) за 30 минут и перемешивали 10 минут.
Реакционную смесь охлаждали до -5°C за период продолжительностью 4,5 часа (+/- 0,3°C/мин), в ходе которого происходила спонтанная кристаллизация. Реакционную смесь фильтровали и осадок дважды промывали этанолом (2×40 мл).
Осадок растворяли в этаноле (212 мл) и смесь нагревали до 78°C (кипячение с обратным холодильником) в течение 60 минут. Реакционную смесь охлаждали до 62°C за период продолжительностью 30 минут (+/- 0,5°C/мин) и вносили затравку из кристаллического дарунавира моноэтанолята (120 мг). Реакционную смесь перемешивали при 62°C в течение 90 минут, охлаждали до 50°C в течение 2 часов (0,1°C/мин), дополнительно охлаждают 15°C за 1 час 30 минут (со скоростью охлаждения 0,3°C/мин) и перемешивают при 15°C в течение периода продолжительностью от 1 часа до 12 часов. Реакционную смесь фильтровали и белый осадок дважды промывали, используя EtOH (2×60 мл). Осадок сушили в вакууме при (40°C, 12 часов) с выходом 55,2 г (93%) дарунавира моноэтанолята.
Присутствие побочного продукта (C) определяли, применяя вышеописанную аналитическую процедуру, и его содержание составляло менее 0,5% по отношению к дарунавиру моноэтаноляту.
Пример 2
4-Амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамид (39,2 г; 0,1 моль) и этанол (170 мл) вносили в 1-литровый реактор. Реакционную смесь перемешивали 15 минут при 20°C и добавляли 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-иловый] сложный эфир угольной кислоты (26,6 г; 0,098 моль) при 20°C. Реакционную смесь нагревали до 50°C за период продолжительностью от 30 минут до 1 часа и перемешивали 2 часа при этой температуре. Реакционную смесь нагревали до 78°C (кипячение с обратным холодильником) за 30 минут и перемешивали 10 минут.
Реакционную смесь охлаждали до 15°C за период продолжительностью 3,5 часа (+/- 0,3°C/мин), в ходе которого происходила спонтанная кристаллизация. Реакционную смесь фильтровали и осадок дважды промывали этанолом (2×40 мл).
Осадок растворяли в этаноле (212 мл) и смесь нагревали до 78°C (кипячение с обратным холодильником) в течение 60 минут. Реакционную смесь охлаждали до 62°C за период продолжительностью 30 минут (+/- 0,5°C/мин) и вносили затравку из кристаллического дарунавира моноэтанолята (120 мг). Реакционную смесь перемешивали при 62°C в течение 90 минут, охлаждали до 50°C в течение 2 часов (0,1°C/мин), дополнительно охлаждали до 15°C за 1 час 30 минут (со скоростью охлаждения 0,3°C/мин) и перемешивали при 15°C в течение периода продолжительностью от 1 часа до 12 часов. Реакционную смесь фильтровали и белый осадок дважды промывали, используя EtOH (2×60 мл). Осадок сушили в вакууме при (40°C, 12 часов) с выходом 55,2 г (93%) дарунавира моноэтанолята.
Присутствие побочного продукта (C) определяли, применяя вышеописанную аналитическую процедуру, и его содержание составляло менее 0,5% по отношению к дарунавиру моноэтаноляту.
Пример 3
4-Амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамид (39,2 г; 0,1 моль) и этанол (170 мл) вносили в 1-литровый реактор. Реакционную смесь перемешивали 15 минут при 20°C и добавляли 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-иловый] сложный эфир угольной кислоты (26,6 г; 0,098 моль) при 20°C. Реакционную смесь нагревали до 50°C за период продолжительностью от 30 минут до 1 часа и перемешивали 2 часа при этой температуре. Реакционную смесь нагревали до 78°C (кипячение с обратным холодильником) за 30 минут и перемешивали 10 минут.
Реакционную смесь охлаждали до 15°C за период продолжительностью 3,5 часа (+/- 0,3°C/мин), в ходе которого происходила спонтанная кристаллизация. Реакционную смесь фильтровали и осадок дважды промывали этанолом (2×40 мл).
Осадок растворяли в этаноле (212 мл) и смесь нагревали до 78°C (кипячение с обратным холодильником) в течение 60 минут. Реакционную смесь охлаждали до 62°C за период продолжительностью 30 минут (+/- 0,5°C/мин) и вносили затравку из кристаллического дарунавира моноэтанолята (120 мг). Реакционную смесь перемешивали при 62°C в течение 30 минут, охлаждали до 58°C в течение 10 минут и выдерживали в течение 30 минут, нагревали до 67°C за период продолжительностью 10 минут и выдерживали в течение 30 минут, а затем охлаждали до 50°C в течение 2 часов (0,1°C/мин), дополнительно охлаждали до 15°C за 1 час 30 минут (со скоростью охлаждения 0,3°C/мин) и перемешивали при 15°C в течение периода продолжительностью от 1 часа до 12 часов. Реакционную смесь фильтровали и белый осадок дважды промывали, используя EtOH (2×60 мл). Осадок сушили в вакууме при (40°C, 12 часов) с выходом 55,2 г (93%) дарунавира моноэтанолята.
Присутствие побочного продукта (C) определяли, применяя вышеописанную аналитическую процедуру, его содержание составляло менее 0,5% по отношению к дарунавиру моноэтаноляту.
Пример 4
4-Амино-N-[(2R,3S)-3-амино-2-гидрокси-4-фенилбутил]-N-(2-метилпропил)-бензолсульфамид (39,2 г; 0,1 моль) и этанол (220 мл) вносили в 1-литровый реактор. После перемешивания в течение 10 минут при 10°C вносили 2,5-диоксо-1-пирролидинил [(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-иловый] сложный эфир угольной кислоты (26,6 г; 0,098 моль). Реакционную смесь гомогенизировали, используя смеситель с высоким усилием сдвига в течение 4 минут при 10°C. Эту взвесь использовали в качестве исходного материала для трубчатого проточного реактора. Реакция завершалась через 30 секунд при 80°C. Качество in situ идентично таковому в периодическом процессе в примерах 1-3.
Присутствие побочного продукта (C) определяли, применяя вышеописанную аналитическую процедуру, и его содержание составляло менее 0,5% по отношению к дарунавиру моноэтаноляту.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (3R, 3aS,6aR)-ГЕКСАГИДРОФУРО[2,3-b]ФУРАН-3-ИЛ(1S,2R)-3-[[(4-АМИНОФЕНИЛ)СУЛЬФОНИЛ](ИЗОБУТИЛ)АМИНО]-1-БЕНЗИЛ-2-ГИДРОКСИПРОПИЛКАРБАМАТА | 2004 |
|
RU2376308C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ГЕКСАГИДРОФУРО[2,3-b] ФУРАН-3-ОЛА | 2007 |
|
RU2464266C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (3R,3aS,6aR) ГЕКСАГИДРОФУРО[2,3-b] ФУРАН-3-ОЛА | 2005 |
|
RU2421458C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ВИРУСНОЙ ПОЛИМЕРАЗЫ | 2013 |
|
RU2654482C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ЭЛЕКТРОПРЯДЕНЫЕ АМОРФНЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2003 |
|
RU2331411C2 |
| ТЕТРАГИДРОФУРО(3,2-b)ПИРРОЛ-3-ОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА К | 2007 |
|
RU2456290C2 |
| СПОСОБ ПОЛУЧЕНИЯ (3aR,4S,6R,6aS)-6-АМИНО-2,2-ДИМЕТИЛТЕТРАГИДРО-3aН-ЦИКЛОПЕНТА[d][1,3]ДИОКСОЛ-4-ОЛ-ДИБЕНЗОИЛ-L-ТАРТРАТА И ПРОДУКТЫ УКАЗАННОГО СПОСОБА | 2008 |
|
RU2477277C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
Изобретение относится к способу получения (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илового сложного эфира [(1S,2R)-3-[[(4-аминофенил)-сульфонил](2-метилпропил)амино]-2-гидрокси-1-(фенилметил)-пропил]-карбаминовой кислоты (дарунавир), который заключается во взаимодействии 2,5–диоксо–1–пирролидинил[(3R,3aS,6aR)–гексагидрофуро[2,3–b]фуран–3–илового] сложного эфира угольной кислоты с 4–амино–N–[(2R,3S)–3–амино–2–гидрокси–4–фенилбутил]–N–(2–метилпропил)–бензолсульфамидом в этаноле в качестве растворителя при нагревании реакционной смеси при температуре от 30°C до температуры образования флегмы.
Технический результат – разработан новый способ получения дарунавира с высоким выходом, который позволяет выделять дарунавир сразу в форме моноэтанолята, который находит свое применение в медицине при лечении вируса иммунодефицита человека типа 1 (ВИЧ–1). 2 н. и 12 з.п. ф-лы, 1 табл., 4 пр.
1. Способ получения дарунавира, включающий стадии:
a) взаимодействия 2,5–диоксо–1–пирролидинил[(3R,3aS,6aR)–гексагидрофуро[2,3–b]фуран–3–илового] сложного эфира угольной кислоты с 4–амино–N–[(2R,3S)–3–амино–2–гидрокси–4–фенилбутил]–N–(2–метилпропил)–бензолсульфамидом в этаноле в качестве растворителя при нагревании реакционной смеси при температуре от 30°C до температуры образования флегмы.
2. Способ по п. 1, где стадию а) выполняют в отсутствие органического основания.
3. Способ по п. 2, где мольное соотношение 2,5–диоксо–1–пирролидинил [(3R,3aS,6aR)–гексагидрофуро[2,3–b]фуран–3–илового] сложного эфира угольной кислоты и 4–амино–N–[(2R,3S)–3–амино–2–гидрокси–4–фенилбутил]–N–(2–метилпропил)–бензолсульфамида находится в интервале от 0,95 до 1,05.
4. Способ по п. 3, где стадию а) выполняют при температуре в интервале от 30°C до 70°C.
5. Способ по п. 4, где применяют температуру от 40°C до 60°C.
6. Способ по любому одному из предыдущих пунктов, где за стадией a) следуют
b) нагревание до получения однородной реакционной смеси;
c) охлаждение реакционной смеси;
d) выделение осажденного продукта;
e) кристаллизация полученного осаждением продукта из этанола; и
f) выделение кристаллизованного моноэтанолята дарунавира.
7. Способ по п. 6, где реакционную смесь на стадии b) охлаждают до температуры в интервале от 15°C до –10°C.
8. Способ по п. 6 или 7, где кристаллизацию полученного продукта выполняют путем суспендирования осажденного продукта в этаноле, нагрева получаемой смеси до получения однородного раствора и последующего охлаждения.
9. Способ по п. 8, где кристаллизация включает в себя процедуру Оствальдовского созревания.
10. Способ согласно п. 9, где процедура Оствальдовского созревания включает в себя стадии:
выдерживания кристаллизационной смеси при температуре в интервале от 61°C до 63°C в течение периода от 20 до 40 минут,
охлаждения кристаллизационной смеси до температуры в интервале от 57°C до 59°C в течение периода от 5 до 20 минут,
выдерживания кристаллизационной смеси при температуре в интервале от 57°C до 59°C в течение периода от 20 до 40 минут,
нагревания кристаллизационной смеси до температуры в интервале от 66°C до 68°C в течение периода от 5 до 20 минут,
выдерживания кристаллизационной смеси при температуре в интервале от 66°C до 68°C в течение периода от 20 до 40 минут,
11. Способ согласно любому одному из пп. 8–10, где охлаждение выполняют путем естественного охлаждения.
12. Способ согласно любому одному из пп. 8–10, где охлаждение выполняют следуя линейному профилю охлаждения.
13. Способ согласно п. 11 или 12, где кристаллизационную смесь охлаждают до температуры в интервале от –5°C до 25°C перед отделением кристаллов дарунавира моноэтанолята.
14. Применение способа, заявленного в любом одном из пп. 1–13 для периодического химического производства, полунепрерывного производства или непрерывного производства дарунавира моноэтанолята.
| СПОСОБ ПОЛУЧЕНИЯ (3R, 3aS,6aR)-ГЕКСАГИДРОФУРО[2,3-b]ФУРАН-3-ИЛ(1S,2R)-3-[[(4-АМИНОФЕНИЛ)СУЛЬФОНИЛ](ИЗОБУТИЛ)АМИНО]-1-БЕНЗИЛ-2-ГИДРОКСИПРОПИЛКАРБАМАТА | 2004 |
|
RU2376308C2 |
| WO 2014016660 A2, 30.01.2014 | |||
| Ветроводяной цепной двигатель | 1928 |
|
SU17138A1 |

