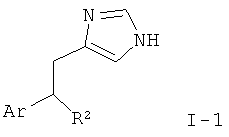
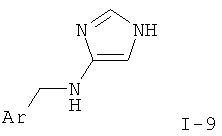
Изобретение относится к соединениям формулы I,
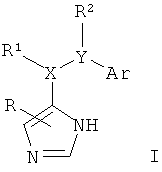
где R представляет собой водород, низший алкил или амино;
X-R1 представляет собой -СН2-, -СН(низший алкокси)- или -СН(ОН)- и
Y-R2 представляет собой -СН2-, -СН(низший алкил)-, -СН(низший алкокси)-, -О-, -S-, -S(O)-, -S(O)2-, -СН(фенил)- или -С(низший алкил)2-;
или
X-R1 представляет собой -NH- и
Y-R2 представляет собой -СН2-, -СН(низший алкил)-, -СН(низший алкокси)-, -СН(фенил)- или -С(низший алкил)2-;
Ar представляет собой фенил, нафтил или бензофуранил, кольца которых не замещены или замещены одним или более заместителями, выбранными из группы, состоящей из низшего алкила, низшего алкила, замещенного галогеном, галогена, низшего алкокси, низшего алкокси, замещенного галогеном, гидрокси, амино, диалкиламино, морфолинила, фенила, бензила или О-бензила;
или фармацевтически приемлемым солям присоединения кислоты, за исключением
5-фенетил-1H-имидазола,
5-(2-фенил-пропил)-1Н-имидазола,
1-(1Н-имидазол-4-ил)-2-фенил-этанола,
5-(2,2-дифенил-этил)-1Н-имидазола,
4-(2-м-толил-этил)-1Н-имидазола,
4-[2-(2,6-диметил-фенил)-этил]-1Н-имидазола,
4-(бифенил-2-илоксиметил)-1Н-имидазола,
5-(2-метил-2-фенил-пропил)-1Н-имидазола,
4-(2-хлор-феноксиметил)-1Н-имидазола,
4-(2-фтор-феноксиметил)-1Н-имидазола,
4-о-толилоксиметил-1Н-имидазола,
4-(3-хлор-феноксиметил)-1Н-имидазола,
4-(2,6-диметил-феноксиметил)-1Н-имидазола и
5-метил-4-фенилсульфанилметил-1Н-имидазола.
Эти известные соединения описаны, например, в упомянутых ниже ссылках или включены в опубликованные библиотеки химических соединений.
А: 5-фенетил-1Н-имидазол (CAS 94714-36-0),
В: 5-(2-фенил-пропил)-1Н-имидазол (CAS 86347-25-3),
С: 1-(1Н-имидазол-4-ил)-2-фенил-этанол (CAS 79928-10-2),
D: 5-(2,2-дифенил-этил)-1Н-имидазол (CAS 102390-63-6),
Е: 4-(2-м-толил-этил)-1Н-имидазол (CAS 79928-27-1),
F: 4-[2-(2,6-диметил-фенил)-этил]-1Н-имидазол (CAS 79924-13-3),
G: 4-(бифенил-2-илоксиметил)-1Н-имидазол (CAS 527696-96-4),
Н: 5-(2-метил-2-фенил-пропил)-1Н-имидазол (Beilstein Registry Number 4407995),
I: 4-(2-хлор-феноксиметил)-1Н-имидазол (CAS 27325-27-5),
J: 4-(2-фтор-феноксиметил)-1Н-имидазол (CAS 401-45-6),
К: 4-о-толилоксиметил-1Н-имидазол (CAS 762177-70-8),
L: 4-(3-хлор-феноксиметил)-1Н-имидазол (CAS 802322-21-0),
М: 4-(2,6-диметил-феноксиметил)-1Н-имидазол (CAS 771450-63-6),
N: 5-метил-4-фенилсульфанилметил-1Н-имидазол (CAS 700355-78-8).
Данное изобретение включает все рацемические смеси, все их соответствующие энантиомеры и/или оптические изомеры.
К тому же настоящим изобретением также охвачены все таутомерные формы соединений формулы I.
Обнаружено, что соединения формулы I обладают высокой аффинностью к рецепторам следовых аминов (trace amine associated receptors, TAAR), особенно к TAAR1.
Соединения могут быть полезны для лечения депрессии, тревожных расстройств, биполярного расстройства, синдрома дефицита внимания и гиперактивности (ADHD), расстройств, вызванных стрессом, психотических расстройств, таких как шизофрения, неврологических заболеваний, таких как болезнь Паркинсона, нейродегенеративных расстройств, таких как болезнь Альцгеймера, эпилепсии, мигрени, гипертензии, злоупотребления веществами, вызывающими зависимость, и метаболических расстройств, таких как расстройства приема пищи, диабет, диабетические осложнения, ожирение, дислипидемия, расстройства потребления и ассимиляции энергии, расстройств и нарушений температурного гомеостаза, нарушений сна и циркадного ритма и сердечно-сосудистых расстройств.
Классические биогенные амины (серотонин, норэпинефрин, эпинефрин, допамин, гистамин) как нейромедиаторы играют важную роль в центральной и периферической нервной системе [1]. Их синтез и хранение, а также их деградация и обратный захват после высвобождения строго регулируются. Известно, что дисбаланс уровней биогенных аминов является ответственным за изменение функции мозга при многих патологических состояниях [2-5]. Соединения, образующие второй класс эндогенных аминов, так называемые следовые амины (trace amines, ТА), очень схожи с классическими биогенными аминами по своей структуре, метаболизму и субклеточной локализации. ТА включают п-тирамин, β-фенилэтиламин, триптамин и октопамин, и их уровень в нервной системе млекопитающих существенно ниже уровня классических биогенных аминов [6].
Нарушение их регуляции связано с различными психическими заболеваниями, такими как шизофрения и депрессия [7], и другими состояниями, такими как синдром дефицита внимания и гиперактивности, головная боль типа мигрени, болезнь Паркинсона, злоупотребление веществами, вызывающими зависимость, и расстройства приема пищи [8, 9].
В течение долгого времени существование ТА-специфических рецепторов являлось всего лишь гипотезой, основанной на присутствии в ЦНС (центральной нервной системе) человека и других млекопитающих анатомически дискретных сайтов связывания, обладающих высокой аффинностью к ТА [10, 11]. Соответственно считалось, что фармакологическое действие ТА опосредовано тем же известным механизмом, что и действие классических биогенных аминов, то есть либо сигналом, вызывающим их высвобождение, либо ингибированием их обратного захвата, либо “перекрестным связыванием” с их рецепторной системой [9, 12, 13]. В последнее время данная точка зрения претерпела значительные изменения в связи с идентификацией нескольких членов нового семейства GPCR (G-белок-связанных рецепторов), рецепторов следовых аминов (TAAR) [7, 14]. Обнаружено 9 TAAR-генов в геноме человека (включая 3 псевдогена) и 16 генов в геноме мыши (включая 1 псевдоген). TAAR-гены не содержат интронов (за одним исключением, TAAR2 содержит 1 интрон) и расположены рядом на одном хромосомном сегменте. Филогенетическое родство генов этих рецепторов, согласующееся с высокой степенью схожести, получаемой при сравнении с GPCR-фармакофором, и фармакологические данные дают возможность предположить, что эти рецепторы образуют три различных подсемейства [7, 14]. TAAR1 относится к первому подклассу, состоящему из четырех генов (TAAR1-4), которые представлены в геномах человека и грызунов высококонсервативными последовательностями. ТА активируют TAAR1 через Gα. Показано, что нарушение регуляции ТА связано с этиологией различных заболеваний, таких как депрессия, психоз, синдром дефицита внимания и гиперактивности, злоупотребление веществами, вызывающими зависимость, болезнь Паркинсона, головная боль типа мигрени, расстройства приема пищи, метаболические расстройства, и поэтому использование TAAR-лигандов в лечении данных заболеваний может являться весьма перспективным.
Поэтому получение новых знаний о рецепторах следовых аминов весьма актуально.
Использованные ссылки


Предметом настоящего изобретения являются новые соединения формулы I и применение соединений формулы I и их фармацевтически приемлемых солей для изготовления лекарств для лечения заболеваний, связанных с аффинностью к рецепторам следовых аминов, новые конкретные соединения, попадающие в объем формулы I, их получение, лекарства на основе соединения по изобретению и их изготовление, а также применение соединений формулы I для контроля или предупреждения таких заболеваний, как депрессия, тревожные расстройства, биполярное расстройство, синдром дефицита внимания и гиперактивности, расстройства, вызванные стрессом, психотические расстройства, такие как шизофрения, неврологические заболевания, такие как болезнь Паркинсона, нейродегенеративные расстройства, такие как болезнь Альцгеймера, эпилепсия, мигрень, гипертензия, злоупотребление веществами, вызывающими зависимость, и метаболические расстройства, такие как расстройства приема пищи, диабет, диабетические осложнения, ожирение, дислипидемия, расстройства потребления и ассимиляции энергии, расстройства и нарушения температурного гомеостаза, нарушения сна и циркадного ритма и сердечно-сосудистые расстройства.
Предпочтительными показаниями к применению соединений по настоящему изобретению являются депрессия, психоз, болезнь Паркинсона, тревога и синдром дефицита внимания и гиперактивности (ADHD).
В контексте данного описания термин “низший алкил” означает группу с насыщенной прямой или разветвленной цепью, содержащую от 1 до 7 атомов углерода, например метил, этил, пропил, изопропил, н-бутил, изобутил, 2-бутил, трет-бутил и тому подобное. Предпочтительными алкильными группами являются группы, содержащие 1-4 атома углерода.
В контексте данного описания термин “низший алкокси” означает группу, где алкильный остаток, такой, как определено выше, присоединен через атом кислорода.
В контексте данного описания термин “низший алкил, замещенный галогеном” означает алкильную группу, такую, как определено выше, где по меньшей мере один атом водорода заменен на галоген, например CF3, CHF2, CH2F, CH2CF3, CH2CH2CF3, CH2CF2CF3 и тому подобное.
Термин “галоген” означает хлор, иод, фтор и бром.
Термин “фармацевтически приемлемые соли присоединения кислот” включает в себя соли с неорганическими и органическими кислотами, такими как соляная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метан-сульфоновая кислота, п-толуолсульфоновая кислота и тому подобное.
Предпочтительными соединениями формулы I являются соединения, где X-R1 и Y-R2 оба представляют собой СН2. Такими соединениями являются
4-[2-(2-хлор-фенил)-этил]-1Н-имидазол,
4-[2-(2-метокси-фенил)-этил]-1Н-имидазол,
4-[2-(3-хлор-фенил)-этил]-1Н-имидазол,
4-[2-(3-фтор-фенил)-этил]-1Н-имидазол,
4-[2-(3-трифторметил-фенил)-этил]-1Н-имидазол,
4-[2-(3-метокси-фенил)-этил]-1Н-имидазол,
4-[2-(4-хлор-фенил)-этил]-1Н-имидазол,
4-[2-(3,5-дихлор-фенил)-этил]-1Н-имидазол,
5-фенетил-1Н-имидазол,
4-(2-м-толил-этил)-1Н-имидазол или
4-[2-(2,6-диметил-фенил)-этил]-1Н-имидазол.
Кроме того, предпочтительны соединения, где X-R1 представляет собой СН2, a Y-R2 представляет собой -СН(низший алкил), например следующие соединения:
4-(2-фенил-бутил)-1Н-имидазол или
5-(2-фенил-пропил)-1Н-имидазол.
Кроме того, предпочтительны соединения, где X-R1 представляет собой СН2, a Y-R2 представляет собой О, например следующие соединения:
4-(2,3-дихлор-феноксиметил)-1Н-имидазол,
4-(2,3-дифтор-феноксиметил)-1Н-имидазол,
4-(3,4-дихлор-феноксиметил)-1Н-имидазол,
4-(4-хлор-3-фтор-феноксиметил)-1Н-имидазол,
5-(бензофуран-6-илоксиметил)-1Н-имидазол,
4-о-толилоксиметил-1Н-имидазол,
4-(3-хлор-феноксиметил)-1Н-имидазол или
4-(2-фтор-феноксиметил)-1Н-имидазол.
Следующим воплощением изобретения являются соединения формулы I, где
X-R1 представляет собой СН2, a Y-R2 представляет собой S, например следующие соединения:
5-(2,3-дихлор-фенилсульфанилметил)-1-имидазол,
4-(4-хлор-фенилсульфанилметил)-5-метил-1Н-имидазол,
4-(нафталин-2-илсульфанилметил)-1Н-имидазол или
5-метил-4-фенилсульфанилметил-1Н-имидазол.
Настоящие соединения формулы I и их фармацевтически приемлемые соли могут быть получены способами, известными в данной области техники, например способами, описанными ниже, включающими
а) удаление защиты с соединения формулы
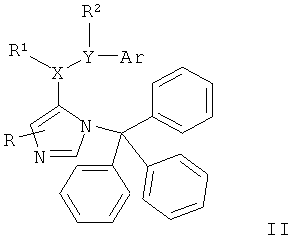
с получением соединения формулы
где определения являются такими, как описано выше, или
b) гидрирование соединения формулы
с получением соединения формулы
и удаление защиты согласно стадии а) с получением соединения формулы
где Ar такой, как определено выше, и R2 представляет собой водород, низший алкил или низший алкокси, или
с) алкилирование соединения формулы
с получением соединения формулы
и удаление защиты согласно стадии а) с получением соединения формулы
где Ar такой, как определено выше; или
d) приведение во взаимодействие соединения формулы
и соединения формулы
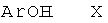
с получением соединения формулы
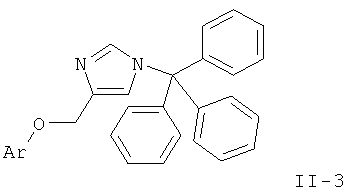
и удаление защиты согласно стадии а) с получением соединения формулы
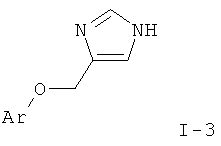
где определения являются такими, как описано выше, или
е) приведение во взаимодействие соединения формулы
с ацетонитрилом с получением соединения формулы
и удаление группы гидрокси с получением соединения формулы
где Ar такой, как определено выше, или
f) приведение во взаимодействие соединения формулы
с соединением формулы
с получением соединения формулы
и удаление защиты с получением соединения формулы
где R представляет собой низший алкил, а Ar такой, как описано выше, или
g) приведение во взаимодействие соединения формулы
и соединения формулы
с получением соединения формулы
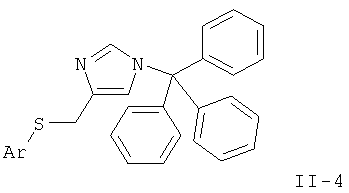
и удаление защиты согласно стадии а) с получением соединения формулы
где Ar такой, как определено выше, или
h) окисление соединения формулы
с получением соединений формул
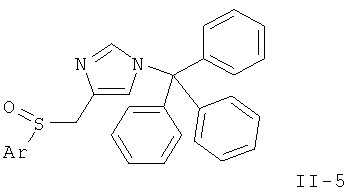 или
или 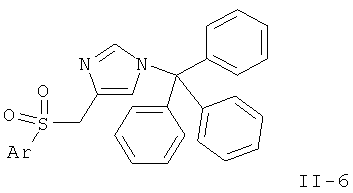
и удаление защиты согласно стадии а) с получением соединений формул
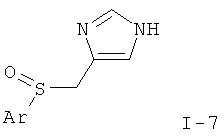 или
или 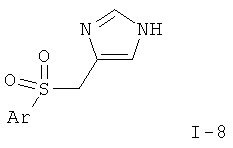
где Ar такой, как определено выше; или
i) восстановление соединения формулы
с получением соединения формулы
и удаление защиты согласно стадии а) с получением соединений формулы
где Ar такой, как описано выше, и PG представляет собой обычную N-защитную группу; и
при желании, превращение полученных соединений в фармацевтически приемлемые соли присоединения кислот.
Соединения формулы I могут быть получены в соответствии с вариантами способа, которые описаны выше, и согласно следующим далее Схемам 1-7. Исходные вещества либо имеются в продаже, либо иным образом известны в химической литературе, либо могут быть получены в соответствии со способами, хорошо известными в данной области техники.
МЕТОДИКА А
Синтез соединений с С-С-связью
Схема 1
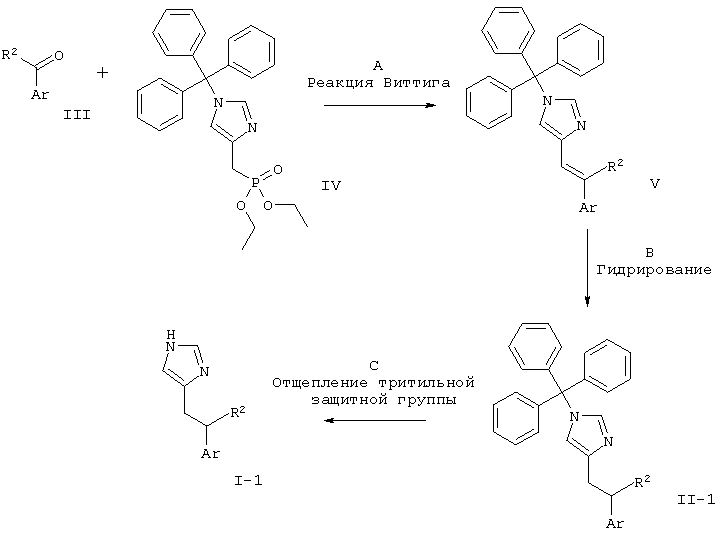
R2 представляет собой низший алкил или водород.
Стадия А: реакция Виттига между альдегидом или кетоном формулы III и (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловым эфиром (IV) может быть выполнена посредством использования основания, такого как NaH, KO-трет-Bu, NaOMe, NaOEt, н-BuLi, LiHMDS (гексаметилдисилазид лития), NaHMDS, KHMDS, LDA (диизопропиламид лития), в растворителе, таком как THF (тетрагидрофуран), диоксан, ацетонитрил, 1,2-диметоксиэтан, DMF (диметилформамид), бензол, толуол или их смеси, при температурах от -78°С до 80°С в течение от 15 мин до 8 ч и, если необходимо, возможного добавления краун-эфира для образования илида и затем конденсации этого илида с карбонильным соединением в том же растворителе при температуре от 0 до 80°С в течение 1-24 ч. Альтернативно к реакционной смеси при температурах от -78°С до 80°С могут быть добавлены одновременно основание, карбонильное соединение и, возможно, краун-эфир без предварительного образования илида.
Предпочтительными условиями для реакций с арилкетонами являются образование илида при к.т. (комнатная температура) с использованием Ko-трет-Bu в качестве основания и THF в качестве растворителя, взаимодействие со сложным эфиром фосфоновой кислоты в течение 15 мин при к.т. и затем конденсация с карбонильным компонентом при 80°С в течение ночи. Предпочтительными условиями для бензальдегидов являются образование илида в присутствии карбонильного соединения с использованием КО-трет-Bu в качестве основания и THF в качестве растворителя при 80°С в течение ночи.
Стадия В: восстановление алкена формулы V может быть осуществлено путем гидрирования водородом при нормальном или повышенном давлении либо посредством гидрирования путем переноса с использованием формиата аммония или циклогексадиена в качестве источника водорода в присутствии такого катализатора, как PtO2, Pd-C или никель Ренея, в таких растворителях, как МеОН, EtOH, H2O, диоксан, THF, HOAc, EtOAc, CH2Cl2, CHCl3, DMF или их смеси. Альтернативно восстановление алкена может быть осуществлено посредством Mg в MeOH или посредством LiAlH4 в THF или диэтиловом эфире.
Предпочтительная методика для тризамещенных алкенов представляет собой гидрирование при нормальном давлении в MeOH/CH2Cl2 с использованием 10% Pd/C в качестве катализатора. Предпочтительная методика для дизамещенных алкенов представляет собой гидрирование при нормальном давлении в MeOH/CHCl3/AcOH с использованием 10% Pd/C в качестве катализатора. И те, и другие условия могут вызывать частичную утрату защитной тритильной группы. В этом случае смесь защищенных и незащищенных продуктов обрабатывают непосредственно в условиях С.
Стадия С: отщепление тритильной группы может быть осуществлено с использованием минеральной кислоты, такой как HCl, H2SO4 или H3PO4, или органической кислоты, такой как CF3COOH, CHCl2COOH, НОАс или п-толуолсульфоновая кислота, в растворителе, таком как CH2Cl2, CHCl3, THF, МеОН, EtOH или H2O, при 0-60°С.
Предпочтительными условиями являются 2 н. HCl в EtOH при температуре дефлегмации в течение 1-3 ч.
МЕТОДИКА В
Синтез соединений с С-С-связью, содержащих заместитель алкокси в α-положении по отношению к имидазолам
Схема 2
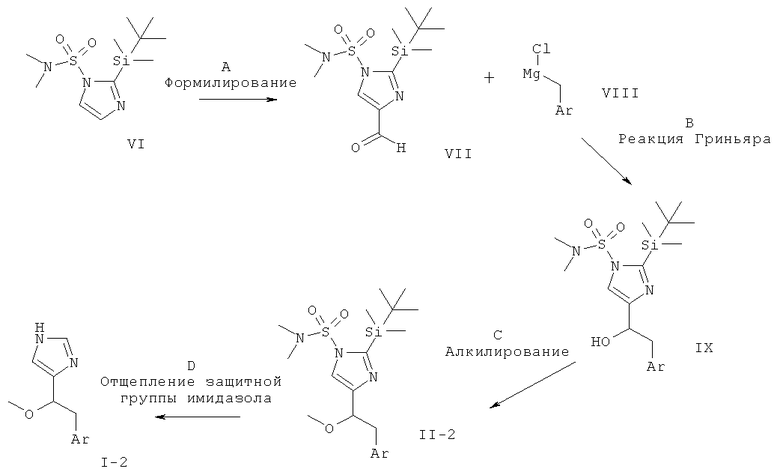
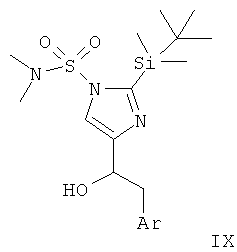
Стадия А: формилирование 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой килоты диметил-амида (VII) может быть осуществлено посредством депротонирования с использованием сильного основания, такого как н-BuLi, втор-BuLi или трет-BuLi, и возможно вспомогательного вещества, такого как тетраметилэтилен-диамин или пентаметилдиэтилен-триамин, в растворителе, таком как THF или диэтиловый эфир, при -78°С - -40°С, с последующим гашением аниона формилирующим электрофилом, таким как DMF, при температуре от -78° до к.т. в течение 1-24 ч.
Предпочтительными условиями являются депротонирование с использованием н-BuLi при -78°С в течение 10 мин, затем взаимодействие с DMF при -78°С в течение 2 ч.
Стадия В: реакция Гриньяра защищенного формилимидазола (VII) с хлоридом или бромидом арилмагния (VIII) может быть осуществлена путем добавления раствора реактива Гриньяра (имеющегося в продаже или полученного из бензил-хлорида или -бромида и Mg стандартными методами) в растворителе, таком как диэтиловый эфир, THF или бензол, к раствору альдегида в одном из ранее упомянутых растворителей при температуре от -20°С до к.т. и предоставления возможности этим двум компонентам взаимодействовать при температуре от к.т. до температуры дефлегмации в течение 1-24 ч.
Предпочтительными условиями являются добавление реактива Гриньяра в диэтиловом эфире к раствору альдегида в THF при к.т. и взаимодействие при к.т. в течение ночи.
Стадия С: алкилирование спирта формулы IX может быть выполнено посредством депротонирования группы гидрокси с использованием основания, такого как NaH, КН, н-BuLi, KO-трет-Bu, КОН или водные NaOH и КОН, в присутствии межфазного катализатора (солей тетраалкиламмония) в подходящем растворителе, таком как THF, DMF, DMSO (диметилсульфоксид), толуол или 1,2-диметоксиэтан, при температуре от -78°С до к.т. в течение от 30 мин до 2 ч и последующего добавления алкилгалогенида.
Предпочтительными условиями являются депротонирование с использованием NaH в THF при к.т. в течение 1 ч и алкилирование алкилиодидом при к.т. в течение ночи.
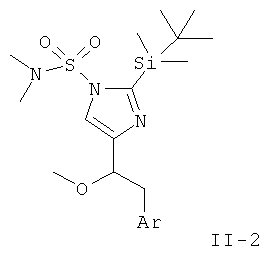
Стадия D: одновременное отщепление обеих защитных групп (II-2) может быть достигнуто в присутствии минеральной кислоты, такой как HCl, HBr или H2SO4, в растворителе, таком как EtOH, МеОН, H2O или THF, при температуре от комнатной до температуры дефлегмации в течение 1-24 ч.
Предпочтительными условиями являются 2 н. HCl в EtOH при температуре дефлегмации в течение 1-3 ч.
МЕТОДИКА С
Синтез соединений с С-О-связью
Схема 3
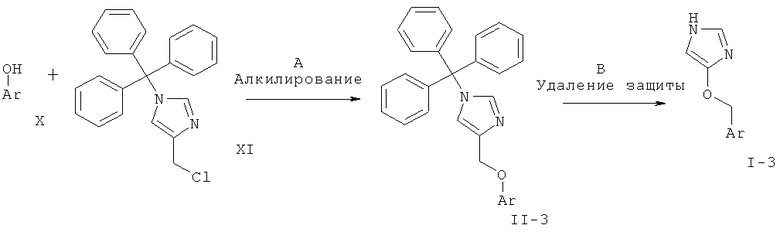
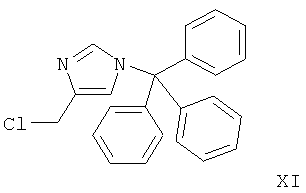
Стадия А: алкилирование замещенного фенола 4-хлорметил-1-тритил-1Н-имидазолом (XI) может быть выполнено с использованием основания, такого как K2CO3, Cs2CO3, Na2CO3, NaHCO3, водный NaOH, КОН, LiOH, NaH, NaOMe, NaOEt или триэтиламин, в растворителе, таком как ацетон, DMF, DMSO, ацетонитрил, толуол, EtOH, МеОН, и, возможно, если необходимо, межфазного катализатора, такого как бромид тетрабутиламмония, или вспомогательного вещества, такого как краун-эфир, иодид тетрабутиламмония или иодид калия, при температуре от к.т. до 120°С в течение 1-24 ч.
Предпочтительными условиями являются K2CO3 в DMF при 80°С в течение 5 ч.
Стадия В: Отщепление тритильной группы может быть осуществлено с использованием минеральной кислоты, такой как HCl, H2SO4 или H3PO4, или органической кислоты, такой как CF3COOH, CHCl2COOH, НОАс или п-толуолсульфоновая кислота, в растворителе, таком как CH2Cl2, CHCl3, THF, МеОН, EtOH или H2O, при 0-60°С.
Предпочтительными условиями являются 2 н. HCl в EtOH при температуре дефлегмации в течение 1-3 ч.
МЕТОДИКА D
Синтез 2-метил-4-имидазолов с С-С-связью
Схема 4
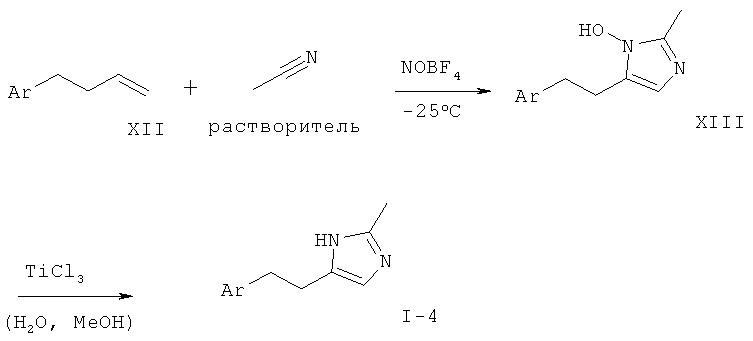
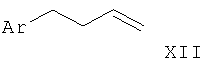
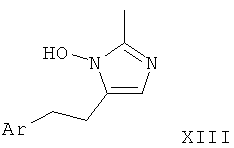
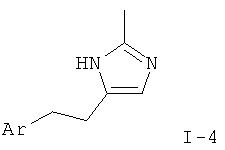
Соответствующий олефин, такой как арил-1-бутен (XII), может быть приведен во взаимодействие при пониженной температуре с нитрилом, таким как ацетонитрил, и фторборатом нитрозония с образованием имидазол-N-оксида в соответствии с Scheinbaum et al., Tetrahedron Lett. 1971, p.2205. Для образования производного имидазола I-4 функциональная группа гидрокси может быть удалена с использованием различных восстанавливающих агентов, таких как Red-Al (бис(2-метоксиэтокси)алюмогидрид натрия), соли титана(III), алюмогидрид лития или другие, как описано в Lipshutz et al. Tetrahedron Lett. 25, 1984, p.1319.
МЕТОДИКА E
Синтез 2-амино-4-имидазолов с С-С-связью
Схема 5
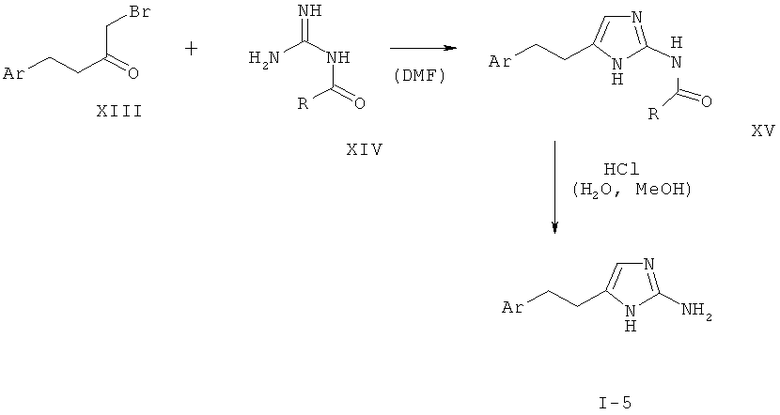
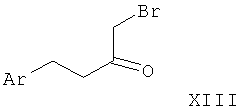
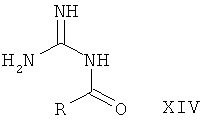
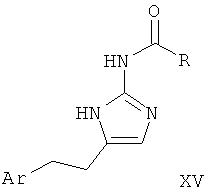
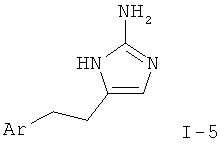
Альфа-бромкетон формулы XIII приводят во взаимодействие с защищенным гуанидином, таким как ацетилгуанидин (XIV), в растворителе, таком как диметилформамид, с последующим удалением защиты с аминогруппы с образованием 2-аминоимидазола I-5. Такое удаление защиты может быть достигнуто, например гидролизом, катализируемым кислотой или основанием, в случае ацетильной группы наиболее предпочтительно путем обработки соляной кислотой в полярном растворителе, таком как вода, спирты или смеси воды и спиртов.
МЕТОДИКА F
Синтез соединений с C-S-связью
Схема 6
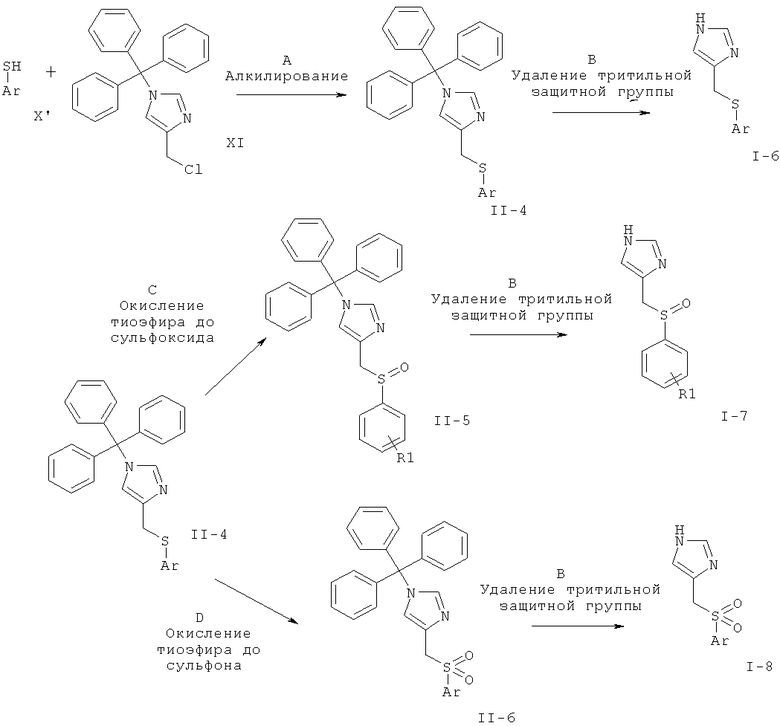
Стадия А: алкилирование замещенного фенола (X) 4-хлорметил-1-тритил-1Н-имидазолом (XI) может быть выполнено с использованием основания, такого как K2CO3, Cs2CO3, Na2CO3, NaHCO3, водный NaOH, КОН, LiOH, NaH, NaOMe, NaOEt или триэтиламин, в растворителе, таком как ацетон, DMF, DMSO, ацетонитрил, толуол, EtOH или МеОН, и, возможно, если необходимо, межфазного катализатора, такого как бромид тетрабутиламмония, или вспомогательного вещества, такого как краун-эфир, иодид тетрабутиламмония иодид или калия, при температуре от к.т. до 120°С в течение 1-24 ч.
Предпочтительными условиями являются K2CO3 в DMF при 80°С в течение 5 ч.
Стадия В: отщепление тритильной группы может быть осуществлено с использованием минеральной кислоты, такой как HCl, H2SO4 или H3PO4, или органической кислоты, такой как CF3COOH, CHCl2COOH, НОАс или п-толуолсульфоновая кислота, в растворителе, таком как CH2Cl2, CHCl3, THF, МеОН, EtOH или H2O, при 0-60°С.
Предпочтительными условиями являются 2 н. HCl в EtOH при температуре дефлегмации в течение 1-3 ч.
Стадия С: окисление простого тиоэфира (II-4) до соответствующего сульфоксида (II-5) может быть выполнено с использованием окислителей, таких как м-СРВА (мета-хлорпероксибензойная кислота), изопропил-2-иодоксибензоат, оксон или периодат натрия, в растворителе, таком как CH2Cl2, дихлорэтан, толуол, ацетонитрил, МеОН, при температурах от 0°С до температуры дефлегмации.
Предпочтительными условиями являются 1 эквивалент м-СРВА в CH2Cl2 при температуре от 0°С до к.т. в течение 1-5 ч.
Стадия D: окисление простого тиоэфира (II-4) до соответствующего сульфона (II-6) может быть выполнено с использованием окислителей, таких как м-СРВА, H2O2, оксон или вольфрамат натрия, в растворителе, таком как CH2Cl2, дихлорэтан, толуол, ацетонитрил, THF, ацетон, МеОН, при температурах от 0°С до температуры дефлегмации.
Предпочтительными условиями являются 2 эквивалента м-СРВА в CH2Cl2 при температуре от 0°С до к.т. в течение 1-5 ч.
МЕТОДИКА G
Синтез соединений с C-N-связью
Схема 7
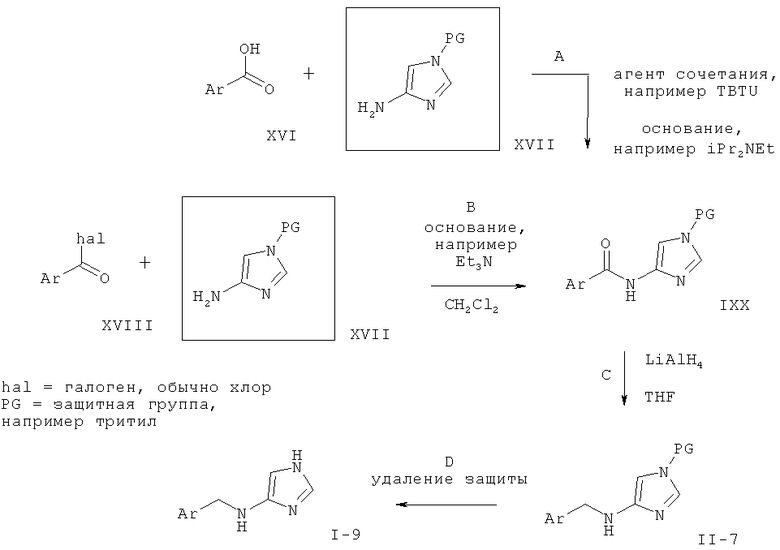
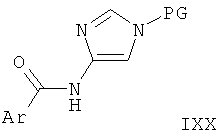
Стадия А: сочетание замещенной арилкарбоновой кислоты (XVI) с соответственно защищенным соединением 4-амино-имидазола (XVII) с целью получения амидного соединения (IXX) может быть выполнено с использованием агента сочетания, такого как 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония тетрафторборат (TBTU) или 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HBTU) или о-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HATU), и основания, такого как триэтиламин или этилдиизопропиламин, в растворителе, таком как THF, DMF или дихлорметан. Подходящие азот-защитные группы включают трет-бутоксикарбамат (ВОС), тритил, диметиламиносульфонил и триметилсилил-этил (SEM).
Предпочтительными условиями являются TBTU и этилдиизопропиламин в DMF при 40°С в течение 16 ч, а предпочтительной защитной группой является тритил.
Стадия В: сочетание замещенного хлорида арилкарбоновой кислоты (XVIII) с соответственно защищенным соединением 4-амино-имидазола (XVII) с целью получения амидного соединения (IXX) может быть выполнено с использованиеим основания, такого как пиридин, триэтиламин или этилдиизопропиламин, в растворителе, таком как THF, DMF или дихлорметан, и, возможно, с использованием катализатора, такого как N,N-диметилформамид или 4-N,N-диметиламинопиридин (DMAP).
Предпочтительными условиями являются триэтиламин в дихлорметане при комнатной температуре в течение 1 ч, а предпочтительной защитной группой является тритил.
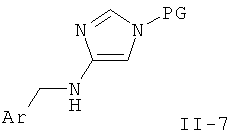
Стадия С: восстановление амида (IXX) до амина (II-7) может быть выполнено с использованием восстанавливающего агента на основе гидрида металла, такого как алюмогидрид лития, или боран-содержащего реагента, такого как боран-тетрагидрофурановый комплекс, в растворителе, таком как диоксан, эфир или тетрагидрофуран, при повышенной температуре.
Предпочтительными условиями являются алюмогидрид лития в тетрагидрофуране при температуре дефлегмации в течение 16 ч.
Стадия D: условия удаления защиты зависят от природы применяемой защитной группы, и в данной области техники хорошо известны многие методы.
В случае тритильной защитной группы предпочтительными условиями удаления защиты являются 4 М водная соляная кислота в диоксане при комнатной температуре в течение 1-2 часов.
Выделение и очистка соединений
Выделение и очистка соединений и промежуточных соединений, описанных в данном изобретении, могут быть выполнены, при желании, с использованием любой подходящей методики разделения или очистки, такой как, например фильтрация, экстракция, кристаллизация, колоночная хроматография, тонкослойная хроматография, хроматография в толстом слое, препаративная жидкостная хроматография низкого или высокого давления, или с использованием комбинации данных методик. Для конкретной иллюстрации подходящих методик разделения или выделения можно сослаться на способы получения и примеры, приведенные в данном описании ниже. Однако, несомненно, также могли бы быть использованы и другие эквивалентные методики разделения или выделения. Рацемические смеси хиральных соединений формулы I могут быть разделены с использованием хиральной ЖХВД.
Соли соединений формулы I
Соединения формулы I являются основными и могут быть превращены в соответствующую соль присоединения кислоты. Такое превращение осуществляют путем обработки по меньшей мере стехиометрическим количеством подходящей кислоты, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, и такие органические кислоты, как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота и тому подобное. Обычно свободное основание растворяют в инертном органическом растворителе, таком как диэтиловый эфир, этилацетат, хлороформ, этанол или метанол и тому подобное, и добавляют кислоту в похожем растворителе. Температуру поддерживают в диапазоне от 0°С до 50°С. Получающаяся соль или спонтанно выпадает в осадок, или может быть осаждена из раствора с использованием менее полярного растворителя.
Соли присоединения кислот основных соединений формулы I могут быть превращены в соответствующие свободные основания путем их обработки по меньшей мере стехиометрическим эквивалентом подходящего основания, такого как гидроксид натрия или калия, карбонат калия, бикарбонат натрия, аммиак и тому подобное.
Соединения формулы I и их фармацевтически приемлемые соли присоединения обладают полезными фармакологическими свойствами. Конкретно, найдено, что соединения по настоящему изобретению обладают высокой аффинностью к рецепторам следовых аминов (TAAR), особенно к TAAR1.
Данные соединения были исследованы в соответствии с тестом, приведенным в данном описании ниже.
Материалы и методы
Конструирование TAAR-экспрессирующих плазмид и стабильно трансфицированных клеточных линий
Для конструирования экспрессирующих плазмид амплифицировали кодирующие последовательности TAAR1 из геномной ДНК человека, крысы и мыши по существу таким способом, как описано Lindemann и др. [14]. Использовали ПЦР-систему Expand High Fidelity (Roche Diagnostics) с 1,5 мМ Mg2+, и очищенные ПЦР-продукты клонировали в клонирующий вектор pCR2.1-TOPO (Invitrogen) в соответствии с инструкциями производителя. ПЦР-продукты субклонировали в вектор plRESneo2 (BD Clontech, Palo Alto, California) и последовательности полученных экспрессирующих плазмид подтверждали путем секвенирования перед введением их в клеточные линии.
Клетки НЕК293 (АТСС (Американская коллекция типовых культур) № CRL-1573) культивировали по существу так, как описано Lindemann и др. (2005). Для получения стабильно трансфицированных клеточных линий клетки НЕК293 трансфицировали экспрессирующими плазмидами plRESneo2, содержащими TAAR-кодирующие последовательности (описанные выше), используя липофектамин 2000 (Lipofectamine 2000, Invitrogen) в соответствии с инструкциями производителя, и через 24 часа после трансфекции в культуральную среду добавляли G418 (Sigma, Buchs, Switzerland) до концентрации 1 мг/мл. После культивирования в течение приблизительно 10 сут клоны выделяли, рассевали и тестировали их отвечаемость на следовые амины (все соединения приобретены в Sigma), используя систему для иммуноферментного анализа (EIA) cAMP Biotrak (Amersham) в соответствии с предоставленной производителем методикой EIA без ацетилирования. Для всех последующих исследований использовали моноклональные клеточные линии, которые показывали стабильную ЕС50 (концентрацию, требуемую для достижения 50% эффекта) в течение периода культивирования, составляющего 15 пассажей.
Приготовление мембран и связывание радиоактивного лиганда
Клетки, находящиеся в состоянии конфлюэнтности, смывали охлажденным во льду забуференным фосфатом физиологическим раствором без Са2+ и Mg2+, содержащим 10 мМ EDTA (этилендиаминтетраацетат), и осаждали путем центрифугирования при 1000 об/мин в течение 5 мин при 4°С. Затем осадок дважды промывали охлажденным во льду забуференным фосфатом физиологическим раствором, клеточный осадок сразу замораживали путем погружения в жидкий азот и хранили до использования при -80°С. Затем клеточный осадок суспендировали в 20 мл буфера HEPES-NaOH (20 мМ), рН 7,4, содержащего 10 мМ EDTA, и гомогенизировали на Polytron (РТ 3000, Kinematica) при 10000 об/мин в течение 10 с. Данный гомогенат центрифугировали при 48000×g в течение 30 мин при 4°С и осадок ресуспендировали в 20 мл буфера HEPES-NaOH (20 мМ), рН 7,4, содержащего 0,1 мМ EDTA, (буфер А) и гомогенизировали на Polytron при 10000 об/мин в течение 10 с. Затем гомогенат центрифугировали при 48000×g в течение 30 мин при 4°С, и осадок ресуспендировали в 20 мл буфера А и гомогенизировали на Polytron при 10000 об/мин в течение 10 с. Концентрацию белка определяли согласно методике Pierce (Rockford, IL). Затем гомогенат центрифугировали при 48000×g в течение 10 мин при 4°С, (осадок) ресуспендировали в буфере HEPES-NaOH (20 мМ), рН 7,0, содержащем MgCl2 (10 мМ) и CaCl2 (2 мМ), (буфер В) (1 г белка в 1 мл) при 200 и гомогенизировали на Polytron при 10000 об/мин в течение 10 с.
Анализ связывания выполняли при 4°С в конечном объеме 1 мл и времени инкубации 30 мин. Радиоактивный лиганд [3Н]-рац-2-(1,2,3,4-тетрагидро-1-нафтил)-2-имидазолин использовали в концентрации, равной рассчитанному значению Kd 60 нМ, для получения связывания приблизительно 0,1% от всего добавленного радиоактивного лиганда и специфического связывания, составляющего приблизительно 70-80% от общего связывания. Неспецифическое связывание определяли как количество [3Н]-рац-2-(1,2,3,4-тетрагидро-1-нафтил)-2-имидазолина, связавшегося в присутствии соответствующего немеченого лиганда (10 мкМ). Конкурирующие лиганды тестировали в широком диапазоне концентраций (10 пМ - 30 мкМ). Конечная концентрация диметилсульфоксида в анализе составляла 2%, и это не оказывало влияния на связывание радиоактивного лиганда. Каждый эксперимент выполняли в двух повторах. Инкубацию всех образцов останавливали путем быстрой фильтрации с использованием планшета UniFilter-96 (Packard Instrument Company) и стеклянного фильтра GF/C, который предварительно вымачивали в течение по меньшей мере 2 ч в 0,3%-ном полиэтиленимине, и с использованием клеточного харвестера Filtermate 96 (Packard Instrument Company). Затем пробирки и фильтры 3 раза промывали аликвотами холодного буфера В объемом 1 мл. Фильтры без предварительной сушки вымачивали в Ultima gold (45 мкл/лунка, Packard Instrument Company) и связанное радиоактивное вещество подсчитывали на сцинтилляционном счетчике для микропланшетов TopCount (Packard Instrument Company).
Ниже в таблице приведены предпочтительные соединения, которые в анализе с использованием мышиных TAAR1 показывают значение Ki (мкМ) в диапазоне <0,1 мкМ.
Соединения формулы I и фармацевтически приемлемые соли соединений формулы I можно применять в качестве лекарственных средств, например в форме фармацевтических композиций. Данные фармацевтические композиции можно вводить перорально, например в форме таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако они также могут быть введены ректально, например в форме суппозиториев, парентерально, например в форме инъекционных растворов.
Для получения фармацевтических композиций соединения формулы I могут быть приготовлены вместе с фармацевтически инертными неорганическими или органическими носителями. Например, в качестве таких носителей для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул могут быть использованы лактоза, кукурузный крахмал или его производные, тальк, стеариновые кислоты или их соли и тому подобное. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное. В случае мягких желатиновых капсул носители обычно не требуются, однако это зависит от природы активного вещества. Подходящими носителями для приготовления растворов и сиропов являются, например, вода, полиолы, глицерин, растительное масло и тому подобное. Подходящими носителями для суппозиториев являются, например, природные или твердые масла, воски, жиры, полужидкие или жидкие полиолы и тому подобное.
Кроме того, данные фармацевтические композиции могут содержать консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красящие вещества, корригенты, соли для изменения осмотического давления, буферы, маскирующие агенты или антиоксиданты. Они также могут дополнительно содержать другие терапевтически полезные вещества.
Лекарства, содержащие соединение формулы I или его фармацевтически приемлемую соль и терапевтически инертный носитель, также являются задачей настоящего изобретения, как и способ их изготовления, который включает приведение одного или более чем одного соединения формулы I и/или фармацевтически приемлемых солей присоединения кислот и, при желании, одного или более чем одного другого терапевтически полезного вещества в форме галенова препарата вместе с одним или более чем одним терапевтически инертным носителем.
Наиболее предпочтительными показаниями согласно настоящему изобретению являются те, которые включают расстройства центральной нервной системы, например лечение или предупреждение депрессии, психоза, болезни Паркинсона, тревоги и синдрома дефицита внимания и гиперактивности (ADHD).
Дозировку можно варьировать в широких пределах, и, конечно, в каждом конкретном случае она должна быть подобрана в соответствии с индивидуальными потребностями. В случае перорального введения дозировка для взрослых может изменяться в диапазоне от приблизительно 0,01 мг до приблизительно 1000 мг в сутки для соединения общей формулы I или соответствующего количества его фармацевтически приемлемой соли. Суточную дозу можно вводить в виде однократной дозы или в виде разделенных доз, и, кроме того, также может быть превышен верхний предел, когда для этого имеются показания.
Таблеточная композиция (влажная грануляция)
Методика приготовления
1. Смешать позиции 1, 2, 3 и 4 и гранулировать их с очищенной водой.
2. Сушить гранулы при 50°С.
3. Пропустить гранулы через подходящее оборудование для помола.
4. Добавить позицию 5 и перемешать в течение трех минут; прессовать на подходящем прессе.
Композиция в виде капсул
Методика приготовления
1. Смешать позиции 1, 2 и 3 в подходящем смесителе в течение 30 минут.
2. Добавить позиции 4 и 5 и смешать в течение 3 минут.
3. Заполнить в подходящую капсулу.
Экспериментальная часть
Следующие далее примеры иллюстрируют данное изобретение, но не предназначены для ограничения его объема.
Пример 1
4-(2-Фенил-бутил)-1Н-имидазол
а) 4-(2-Фенил-бут-1-енил)-1-тритил-1Н-имидазол
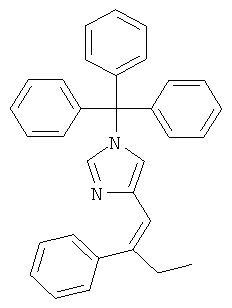
К перемешиваемому раствору (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтилового эфира (1,24 г; CAS 473659-21-1) при к.т. в THF (20 мл) в атмосфере аргона добавляли трет-бутилат калия (301 мг). Через 15 мин перемешивания при к.т. добавляли пропиофенон (0,3 мл) в виде одной порции. Смесь нагревали до 80°С и перемешивание при этой температуре продолжали в течение 2 суток. Плотную суспензию охлаждали до к.т., твердое вещество отфильтровывали и промывали THF. Фильтрат концентрировали, получая темно-фиолетовое вязкое масло. Его переводили в EtOAc и промывали рассолом. Водную фазу вновь экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: циклогексан → циклогексан/EtOAc 3:2), получая 4-(2-фенил-бут-1-енил)-1-тритил-1Н-имидазол (269 мг; неполностью чистый) в виде беловатого твердого вещества. MS (ISP): 243,2 ([Trt]+).
b) 4-(2-Фенил-бутил)-1Н-имидазол
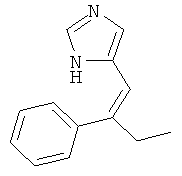
К перемешиваемому раствору 4-(2-фенил-бут-1-енил)-1-тритил-1Н-имидазола (260 мг) при к.т. в метаноле (5 мл) и дихлорметане (2 мл) в атмосфере аргона добавляли 10% Pd/C (26 мг). Затем смесь перемешивали при к.т. в атмосфере водорода в течение 17 ч. Катализатор отфильтровывали и промывали метанолом. Фильтрат концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: CH2Cl2 → CH2Cl2/MeOH 9:1), получая 4-(2-фенил-бутил)-1Н-имидазол (18 мг) в виде бесцветной смолы. MS (ISP): 201,3 ([М+Н]+).
Пример 2
4-(3-Метил-2-фенил-бутил)-1Н-имидазол
а) 4-(3-Метил-2-фенил-бут-1-енил)-1-тритил-1Н-имидазол
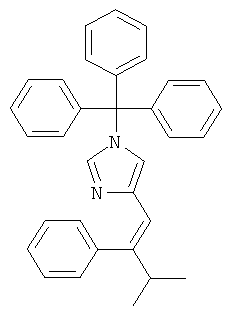
К перемешиваемой суспензии (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтилового эфира (921 мг; CAS 473659-21-1) при к.т. в THF (20 мл) в атмосфере аргона добавляли трет-бутилат калия (241 мг). Затем смесь перемешивали при к.т. в течение 15 мин и добавляли изобутирофенон (0,25 мл) в виде одной порции. Смесь (прозрачный коричнево-оранжевый раствор) нагревали до 80°С в течение 21 ч. Реакционную смесь фильтровали и осадок на фильтре промывали EtOAc. Фильтрат концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: циклогексан → циклогексан/EtOAc 1:1), получая 4-(3-метил-2-фенил-бут-1-енил)-1-тритил-1Н-имидазол (91 мг; неполностью чистый) в виде оранжевого липкого твердого вещества. MS (ISP): 243,2 ([Trt]+).
b) 4-(3-Метил-2-фенил-бутил)-1-тритил-1Н-имидазол
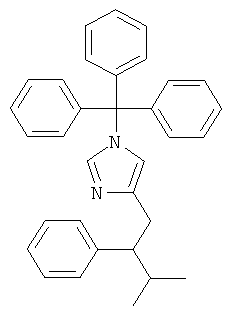
К перемешиваемому раствору 4-(3-метил-2-фенил-бут-1-енил)-1-тритил-1Н-имидазола (87 мг) при к.т. в метаноле (4 мл) и CH2Cl2 (1 мл) в атмосфере аргона добавляли 10% Pd/C (10 мг). Затем смесь перемешивали при к.т. в атмосфере водорода в течение 38 ч. Катализатор отфильтровывали и промывали МеОН. Фильтрат концентрировали, получая 4-(3-метил-2-фенил-бутил)-1-тритил-1Н-имидазол (82 мг) в виде беловатого липкого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS (ISP): 243,2 ([Trt]+).
с) 4-(3-Метил-2-фенил-бутил)-1Н-имидазол

К перемешиваемой суспензии 4-(3-метил-2-фенил-бутил)-1-тритил-1Н-имидазола (80 мг) при к.т. в этаноле (2 мл) в атмосфере аргона добавляли 2 н. HCl (3 мл). Смесь (суспензию) нагревали до температуры дефлегмации (наблюдали превращение в прозрачный светло-желтый раствор при достижении температуры 90°С) и перемешивали в течение 2 ч 30 мин, затем охлаждали до к.т. и концентрировали, получая светло-коричневое липкое твердое вещество. Его переводили в H2O и подщелачивали до рН>12 путем добавления 4 н. NaOH. Продукт экстрагировали CH2Cl2/MeOH 9:1. Объединенные органические фракции сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: CH2Cl2 → CH2Cl2/MeOH 85:15), получая 4-(3-метил-2-фенил-бутил)-1Н-имидазол (8 мг) в виде бесцветной смолы. MS (ISP): 215,4 ([М+Н]+).
Пример 3
4-(3,3-Диметил-2-фенил-бутил)-1Н-имидазол
а) 4-(3,3-Диметил-2-фенил-бут-1-енил)-1-тритил-1Н-имидазол
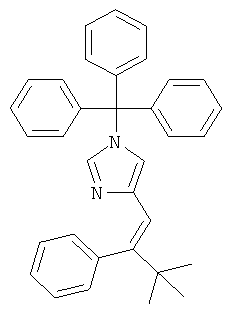
К перемешиваемой суспензии (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтилового эфира (341 мг; CAS 473659-21-1) при к.т. в THF (7,5 мл) в атмосфере аргона добавляли трет-бутилат калия (83 мг). Затем смесь перемешивали при к.т. в течение 15 мин и добавляли 2,2-диметилпропиофенон (0,1 мл) в виде одной порции. Смесь (прозрачный коричнево-оранжевый раствор) нагревали до 80°С и перемешивание при этой температуре продолжали в течение 24 ч. Содержимое реакционной смеси непосредственно адсорбировали на силикагеле. Продукт выделяли хроматографией (градиент: циклогексан → циклогексан/EtOAc 65:35), получая 4-(3,3-диметил-2-фенил-бут-1-енил)-1-тритил-1Н-имидазол (135 мг; неполностью чистый) в виде светло-желтого твердого вещества. MS (ISP): 243,2 ([Trt]+).
b) 4-(3,3-Диметил-2-фенил-бутил)-1Н-имидазол
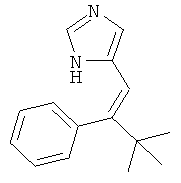
К перемешиваемому раствору 4-(3,3-диметил-2-фенил-бут-1-енил)-1-тритил-1Н-имидазола (121 мг) при к.т. в метаноле (5 мл) и дихлорметане (1 мл) в атмосфере аргона добавляли 10% Pd/C (12 мг). Смесь перемешивали в атмосфере водорода (баллон) в течение 17 ч. Катализатор отфильтровывали и промывали метанолом. Фильтрат концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: CH2Cl2 → CH2Cl2/MeOH 9:1), получая 4-(3,3-диметил-2-фенил-бутил)-1Н-имидазол (25 мг) в виде бесцветной смолы. MS (ISP): 229,4 ([M+H]+).
Пример 4
4-(1-Метокси-2-фенил-этил)-1Н-имидазол
а) 2-(трет-Бутил-диметил-силанил)-4-формил-имидазол-1-сульфоновой килоты диметиламид
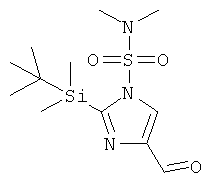
К перемешиваемому охлажденному (-78°С) раствору 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида (1,02 г; CAS 129378-52-5) в THF (20 мл) в атмосфере аргона добавляли по каплям бутиллитий (3,3 мл; 1,6 М раствор в гексанах) в течение периода времени 10 мин. Через 30 мин перемешивания добавляли DMF (1,3 мл) в течение периода времени 5 мин и смесь (прозрачный светло-желтый раствор) перемешивали при -78°С в течение следующих 2 ч. Смесь гасили насыщ. водн. NH4Cl и разбавляли EtOAc. Водную фазу вновь экстрагировали EtOAc. Объединенные органические фазы промывали H2O и рассолом, сушили над MgSO4, фильтровали и концентрировали, получая 2-(трет-бутил-диметил-силанил)-4-формил-имидазол-1-сульфоновой кислоты диметиламид (1,22 г) в виде вязкого оранжевого масла, которое использовали на следующей стадии реакции без дополнительной очистки. MS (ISP): 318,3 ([М+Н]+).
b) 2-(трет-Бутил-диметил-силанил)-4-(1-гидрокси-2-фенил-этил)-имидазол-1-сульфоновой кислоты диметиламид
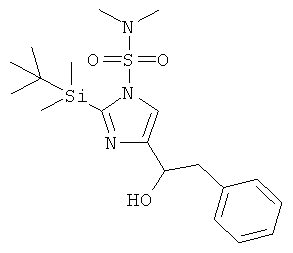
Бензилбромид (4,1 мл) добавляли по каплям к перемешиваемой суспензии магния (1,01 г) в диэтиловом эфире (10 мл). По завершении энергичной экзотермической реакции раствор супернатанта отделяли декантацией от твердого вещества и хранили в холодильнике, готовым для использования. Аликвоту этого раствора (1 мл) добавляли по каплям (экзотермическая реакция!) к охлажденному (0°С, ледяная баня) перемешиваемому раствору 2-(трет-бутил-диметил-силанил)-4-формил-имидазол-1-сульфоновой кислоты диметиламида (725 мг) при к.т. в THF (5 мл) в атмосфере аргона. По завершении добавления перемешивание при к.т. продолжали в течение ночи. Смесь гасили путем добавления насыщ. водн. NH4Cl и экстрагировали EtOAc. Водную фазу вновь экстрагировали EtOAc. Объединенные органические фазы промывали Н2O и рассолом, сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: циклогексан → циклогексан/EtOAc 25:75), получая 2-(трет-бутил-диметил-силанил)-4-(1-гидрокси-2-фенил-этил)-имидазол-1-сульфоновой кислоты диметиламид (168 мг) в виде светло-желтого твердого вещества. MS (ISP): 410,1 ([М+Н]+).
с) 2-(трет-Бутил-диметил-силанил)-4-(1-метокси-2-фенил-этил)-имидазол-1-сульфоновой кислоты диметиламид
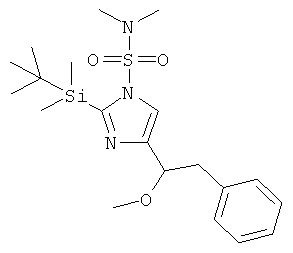
К перемешиваемому раствору 2-(трет-бутил-диметил-силанил)-4-(1-гидрокси-2-фенил-этил)-имидазол-1-сульфоновой кислоты диметиламида (160 мг) при к.т. в THF (5 мл) в атмосфере аргона добавляли NaH (18 мг; 55%-ная дисперсия в минеральном масле) в виде одной порции. Через 1 ч перемешивания при к.т. добавляли Mel (0,04 мл) и перемешивание при к.т. продолжали в течение ночи. Смесь разбавляли EtOAc и промывали H2O. Водную фазу вновь экстрагировали EtOAc. Объединенные органические фазы промывали H2O и рассолом, сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: циклогексан → циклогексан/EtOAc 65:35), получая 2-(трет-бутил-диметил-силанил)-4-(1-метокси-2-фенил-этил)-имидазол-1-сульфоновой кислоты диметиламид (111 мг) в виде светло-желтого вязкого масла. MS (ISP): 424,3 ([М+Н]+).
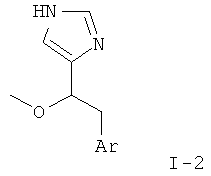
d) 4-(1-Метокси-2-фенил-этил)-1Н-имидазол

К перемешиваемой суспензии 2-(трет-бутил-диметил-силанил)-4-(1-метокси-2-фенил-этил)-имидазол-1-сульфоновой кислоты диметиламида (105 мг) при к.т. в этаноле (3 мл) в атмосфере аргона добавляли 2 н. HCl (3 мл). Смесь нагревали до температуры дефлегмации в течение 3 ч. Смесь охлаждали до к.т. и концентрировали, получая светло-желтое твердое вещество, которое переносили в H2O и рН подводили до 12 путем добавления 4 н. NaOH. Продукт экстрагировали смесью CH2Cl2/MeOH 4:1. Объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: CH2Cl2 → CH2Cl2/MeOH 9:1), получая 4-(1-метокси-2-фенил-этил)-1Н-имидазол (38 мг) в виде белого твердого вещества. MS (ISP): 203,4 ([М+Н]+).
Пример 5
4-[2-(2-Хлор-фенил)-этил]-1Н-имидазол
а) 4-[2-(2-Хлор-фенил)-винил]-1-тритил-1Н-имидазол
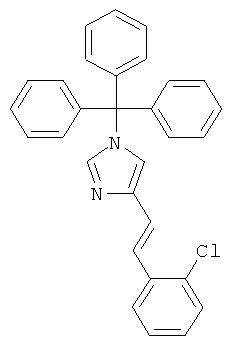
К перемешиваемой суспензии (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтилового эфира (448 мг; CAS 473659-21-1) при к.т. в THF (7 мл) в атмосфере аргона добавляли трет-бутилат калия (109 мг) и 2-хлорбензальдегид (114 мг). Смесь (прозрачный коричнево-оранжевый раствор) нагревали до 80°С в течение ночи. Реакционную смесь охлаждали до к.т. и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: циклогексан → циклогексан/EtOAc 3:2), получая 4-[2-(2-хлор-фенил)-винил]-1-тритил-1Н-имидазол (329 мг) в виде беловатого твердого вещества. MS (ISP): 243,3 ([Trt]+).
b) 4-[2-(2-Хлор-фенил)-этил]-1Н-имидазол
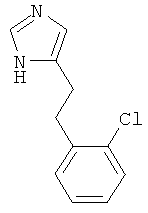
К перемешиваемой смеси 4-[2-(2-хлор-фенил)-винил]-1-тритил-1Н-имидазола (329 мг) при к.т. в этаноле (7 мл) и хлороформе (3 мл) в атмосфере аргона добавляли уксусную кислоту (0,2 мл) и 10% Pd/C (30 мг). Смесь гидрировали (давление окружающей среды) в течение ночи. Катализатор отфильтровывали и промывали этанолом. Смесь концентрировали, получая светло-коричневое смолообразное вещество. Это вещество переносили в этанол (3 мл) и 2 н. HCl (3 мл) и нагревали до температуры дефлегмации в течение 3 ч. Затем смесь охлаждали до к.т., концентрировали. Оставшееся твердое вещество переносили в 1 н. NaOH (10 мл) и экстрагировали CH2Cl2/MeOH 4:1. Объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: CH2Cl2 → CH2Cl2/MeOH 9:1), получая 4-[2-(2-хлор-фенил)-этил]-1Н-имидазол (44 мг) в виде светло-коричневого смолообразного вещества. MS (ISP): 207,1 ([М+Н]+).
Пример 6
4-[2-(2-Этил-фенил)-этил]-1Н-имидазол
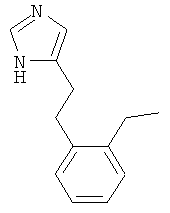
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 2-этилбензальдегидом и затем превращали в 4-[2-(2-этил-фенил)-этил]-1Н-имидазол. Бесцветное вязкое масло. MS (ISP): 201,3 ([М+Н]+).
Пример 7
4-[2-(2-Трифторметил-фенил)-этил]-1Н-имидазол
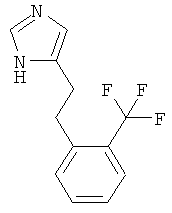
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 2-(трифторметил)бензальдегидом и затем превращали в 4-[2-(2-трифторметил-фенил)-этил]-1Н-имидазол. Бесцветное вязкое масло. MS (ISP): 241,3 ([М+Н]+).
Пример 8
4-[2-(2-Метокси-фенил)-этил]-1Н-имидазол
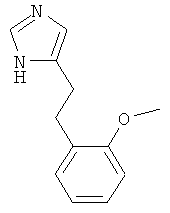
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 2-метоксибензальдегидом и затем превращали в 4-[2-(2-метокси-фенил)-этил]-1Н-имидазол. Бесцветное вязкое масло. MS (ISP): 203,1 ([М+Н]+).
Пример 9
{2-[2-(1Н-Имидазол-4-ил)-этил]-фенил}-диметил-амин
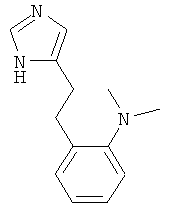
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 2-(N,N-диметиламино)бензальдегидом и затем превращали в {2-[2-(1Н-имидазол-4-ил)-этил]-фенил}-диметил-амин. Светло-желтое вязкое масло. MS (ISP): 216,3 ([M+H]+).
Пример 10
4-{2-[2-(1Н-Имидазол-4-ил)-этил]-фенил}-морфолин
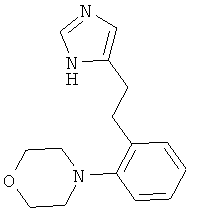
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 2-морфолинобензальдегидом и затем превращали в 4-{2-[2-(1Н-имидазол-4-ил)-этил]-фенил}-морфолин. Светло-желтое вязкое масло. MS (ISP): 258,3 ([М+Н]+).
Пример 11
4-[2-(3-Хлор-фенил)-этил]-1Н-имидазол

По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 3-хлорбензальдегидом и затем превращали в 4-[2-(3-хлор-фенил)-этил]-1Н-имидазол. Беловатое твердое вещество. MS (ISP): 207,1 ([М+Н]+).
Пример 12
4-[2-(3-Фтор-фенил)-этил]-1Н-имидазол
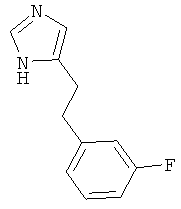
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 3-фторбензальдегидом и затем превращали в 4-[2-(3-фтор-фенил)-этил]-1Н-имидазол. Беловатое твердое вещество. MS (ISP): 191,1 ([М+Н]+).
Пример 13
4-[2-(3-Трифторметил-фенил)-этил]-1Н-имидазол
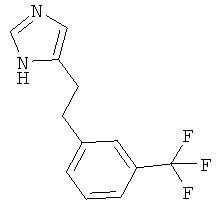
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 3-(трифторметил)бензальдегидом и затем превращали в 4-[2-(3-трифторметил-фенил)-этил]-1Н-имидазол. Беловатое вязкое масло. MS (ISP): 241,1 ([М+Н]+).
Пример 14
4-[2-{3-Метокси-фенил)-этил]-1Н-имидазол
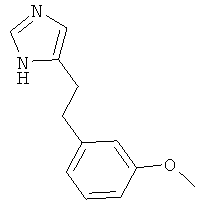
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 3-метоксибензальдегидом и затем превращали в 4-[2-(3-метокси-фенил)-этил]-1Н-имидазол. Беловатое твердое вещество. MS (ISP): 203,3 ([М+Н]+).
Пример 15
4-[2-(3-Трифторметокси-фенил)-этил]-1Н-имидазол
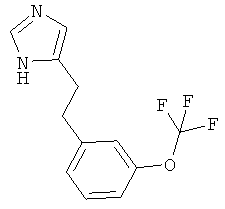
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 3-(трифторметокси)бензальдегидом и затем превращали в 4-[2-(3-трифторметокси-фенил)-этил]-1Н-имидазол. Светло-желтое вязкое масло. MS (ISP): 257,3 ([М+Н]+).
Пример 16
4-[2-(4-Хлор-фенил)-этил]-1Н-имидазол
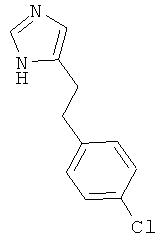
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 4-хлорбензальдегидом и затем превращали в 4-[2-(4-хлор-фенил)-этил]-1Н-имидазол. Беловатое твердое вещество. MS (ISP): 207,1 ([М+Н]+).
Пример 17
4-[2-(3,5-Дихлор-фенил)-этил]-1Н-имидазол
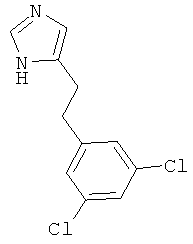
По аналогии с примером 5 (1-тритил-1Н-имидазол-4-илметил)-фосфоновой кислоты диэтиловый эфир (CAS 473659-21-1) приводили во взаимодействие с 3,5-дихлорбензальдегидом и затем превращали в 4-[2-(3,5-дихлор-фенил)-этил]-1Н-имидазол. Беловатое твердое вещество. MS (ISP): 241,1 ([М+Н]+).
Пример 18
2-Метил-5-фенетил-1Н-имидазол
а) 2-Метил-5-фенетил-имидазол-1-ол
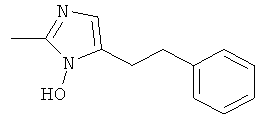
К раствору тетрафторбората нитрозония (0,564 г; 4,83 ммоль) в ацетонитриле (8 мл) добавляли 4-фенил-1-бутен при -30°С. Смесь перемешивали в течение 1 часа при этой температуре, затем осторожно добавляли 0,5 мл воды. При комнатной температуре добавляли насыщенный раствор хлорида аммония и ацетонитрил выпаривали в вакууме. рН оставшегося водного раствора подводили до нейтрального значения с использованием небольшого количества гидроксида натрия и экстрагировали дихлорметаном. Органический слой отделяли, сушили над сульфатом магния и упаривали. Остаток очищали, используя флэш-хроматографию (SiO2, дихлорметан/метанол = 9:1) и получая беловатое твердое вещество (0,245 г; 17%); MS (ISP): 202,9 ([M+H]+).
b) 2-Метил-5-фенетил-1Н-имидазол
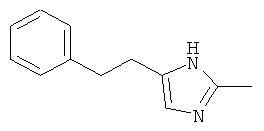
К раствору 2-метил-5-фенетил-имидазол-1-ола (0,20 г; 1,0 ммоль) в метаноле (3,5 мл) добавляли раствор хлорида титана(III) (2,5 мл; 15%) и смесь перемешивали в течение ночи при комнатной температуре. Для достижения основного значения рН сначала добавляли насыщенный раствор бикарбоната натрия, затем разбавленный раствор гидроксида натрия. Смесь дважды экстрагировали дихлорметаном, объединенные органические слои сушили над сульфатом магния и упаривали. Остаток очищали колоночной хроматографией (дихлорметан/метанол = 9:1), получая белое твердое вещество (0,14 мг; 75%); MS (El): 186,1 (М+).
Пример 19
5-Фенетил-1Н-имидазол-2-иламин
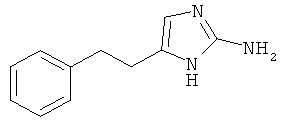
К раствору 1-ацетилгуанидина (1,34 г; 13,2 ммоль) в диметилформамиде (7 мл) добавляли раствор 1-бром-4-фенил-бутан-2-она (1,5 г; 6,6 ммоль) в диметилформамиде (7 мл) при 0°С. Смесь перемешивали в течение ночи при комнатной температуре и затем растворитель выпаривали. После добавления смеси этилацетат/гептан (1:1) образовывалось белое твердое вещество, которое отфильтровывали и промывали смесью этилацетат/гептан (1:1). После сушки в вакууме твердое вещество растворяли в смеси концентрированной соляной кислоты (2 мл) и метанола (4 мл) и перемешивали в течение 2,5 часа при 85°С. Растворитель выпаривали и остаток очищали хроматографией (колонка: Isolute® Flash-NH2 от Separtis; элюент: этилацетат/метанол = 1:1), получая светло-желтое твердое вещество (0,063 мг; 5%); MS (El): 187,2 (М+).
Пример 20
4-(2,3-Дихлор-феноксиметил)-1Н-имидазол
а) 4-(2,3-Дихлор-феноксиметил)-1-тритил-1Н-имидазол
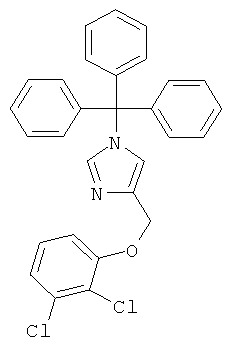
К перемешиваемому раствору 4-хлорметил-1-тритил-1Н-имидазола (400 мг; CAS 103057-10-9) при к.т. в DMF (5 мл) в атмосфере аргона добавляли 2,3-дихлорфенол (273 мг) и K2CO3 (385 мг). Реакционную смесь нагревали до 80°С в течение 5 ч, затем охлаждали до к.т., разбавляли EtOAc и промывали 1 н. NaOH. Водную фазу вновь экстрагировали EtOAc. Объединенные органические фазы промывали H2O и рассолом, сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: циклогексан → циклогексан/EtOAc 1:1), получая 4-(2,3-дихлор-феноксиметил)-1-тритил-1Н-имидазол (360 мг) в виде белого твердого вещества. MS (ISP): 243,3 ([Trt]+)
b) 4-(2,3-Дихлор-феноксиметил)-1Н-имидазол
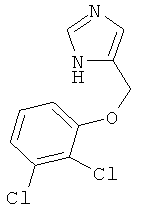
К перемешиваемой суспензии 4-(2,3-дихлор-феноксиметил)-1-тритил-1Н-имидазола (150 мг) при к.т. в этаноле (2 мл) в атмосфере аргона добавляли 2 н. HCl (3 мл). Смесь нагревали до температуры дефлегмации в течение 6 ч, затем концентрировали, получая беловатое твердое вещество. Его переносили в насыщенный водный Na2CO3 и экстрагировали CH2Cl2/MeOH 4:1. Объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: CH2Cl2 → CH2Cl2/МеОН 4:1), получая 4-(2,3-дихлор-феноксиметил)-1Н-имидазол (65 мг) в виде белого твердого вещества. MS (ISP): 243,4 ([М+Н]+).
Пример 21
4-(2-Этил-феноксиметил)-1Н-имидазол

По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2-этилфенолом и затем превращали в 4-(2-этил-феноксиметил)-1Н-имидазол. Воскообразное беловатое твердое вещество. MS (ISP): 203,1 ([М+Н]+).
Пример 22
4-(2-Изопропил-феноксиметил)-1Н-имидазол
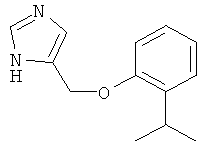
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2-изопропилфенолом и затем превращали в 4-(2-изопропил-феноксиметил)-1Н-имидазол. Воскообразное беловатое твердое вещество. MS (ISP): 217,4 ([М+Н]+).
Пример 23
4-(2-Трифторметил-феноксиметил)-1Н-имидазол
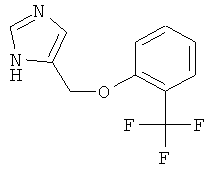
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2-трифторметилфенолом и затем превращали в 4-(2-трифторметил-феноксиметил)-1Н-имидазол. Белое твердое вещество. MS (ISP): 243,4 ([М+Н]+).
Пример 24
4-(2-Бензил-феноксиметил)-1Н-имидазол
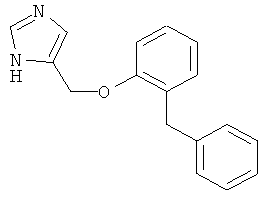
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2-бензилфенолом и затем превращали в 4-(2-бензил-феноксиметил)-1Н-имидазол. Воскообразное белое твердое вещество. MS (ISP): 265,1 ([М+Н]+).
Пример 25
4-(2-Метокси-феноксиметил)-1Н-имидазол
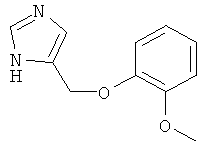
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2-метоксифенолом и затем превращали в 4-(2-метокси-феноксиметил)-1Н-имидазол. Беловатое аморфное твердое вещество. MS (ISP): 205,1 ([М+Н]+).
Пример 26
4-(2-Изопропокси-феноксиметил)-1Н-имидазол
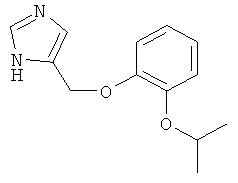
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2-изопропоксифенолом и затем превращали в 4-(2-изопропокси-феноксиметил)-1Н-имидазол. Беловатое твердое вещество. MS (ISP): 233,3 ([М+Н]+).
Пример 27
4-(2-Трифторметокси-феноксиметил)-1Н-имидазол
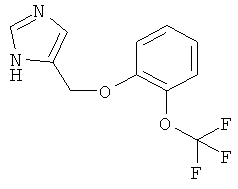
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2-трифторметоксифенолом и затем превращали в 4-(2-трифторметокси-феноксиметил)-1Н-имидазол. Беловатое твердое вещество. MS (ISP): 259,1 ([М+Н]+).
Пример 28
4-(2-Бензилокси-феноксиметил)-1Н-имидазол
а) 4-(2-Бензилокси-феноксиметил)-1-тритил-1Н-имидазол
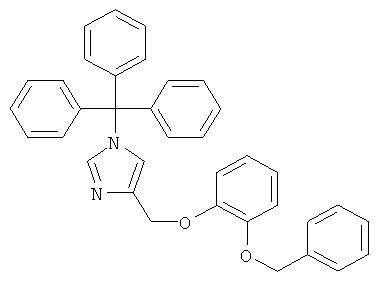
По аналогии с примером 20.а 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2-бензилоксифенолом, получая 4-(2-бензилокси-феноксиметил)-1-тритил-1Н-имидазол. Желтое вязкое масло. MS (ISP): 523,5 ([М+Н]+).
b) 4-(2-Бензилокси-феноксиметил)-1Н-имидазол
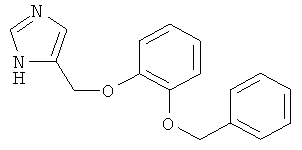
Раствор 4-(2-бензилокси-феноксиметил)-1-тритил-1Н-имидазола (34 мг) в МеОН (2 мл) обрабатывали АсОН (0,1 мл) и нагревали до 70°С в течение 5 ч. Смесь концентрировали. Неочищенный продукт очищали колоночной хроматографией, получая 4-(2-бензилокси-феноксиметил)-1Н-имидазол (11 мг) в виде бесцветного аморфного твердого вещества. MS (ISP): 281,4 ([М+Н]+).
Пример 29
2-(1Н-Имидазол-4-илметокси)-фенол
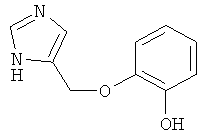
В условиях, которые описаны в примере 20.b, 4-(2-бензилокси-феноксиметил)-1-тритил-1Н-имидазол (пример 28.а) превращали в 2-(1Н-имидазол-4-илметокси)-фенол. Беловатое твердое вещество. MS (ISP): 191,4 ([М+Н]+).
Пример 30
4-(3-Трифторметил-феноксиметил)-1Н-имидазол
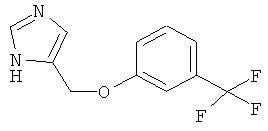
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 3-трифторметилфенолом и затем превращали в 4-(3-трифторметил-феноксиметил)-1Н-имидазол. Белое твердое вещество. MS (ISP): 243,3 ([М+Н]+).
Пример 31
4-(3-Трифторметокси-феноксиметил)-1Н-имидазол
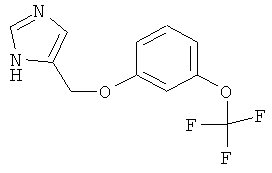
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 3-трифторметоксифенолом и затем превращали в 4-(3-трифторметокси-феноксиметил)-1Н-имидазол. Бесцветное масло. MS (ISP): 259,0 ([M+H]+).
Пример 32
[3-(1Н-Имидазол-4-илметокси)-фенил]-диметил-амин
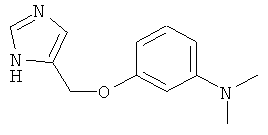
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 3-диметиламинофенолом и затем превращали в [3-(1Н-имидазол-4-илметокси)-фенил]-диметил-амин. Беловатое твердое вещество. MS (ISP): 218,4 ([М+Н]+).
Пример 33
4-[-(1Н-Имидазол-4-илметокси)-фенил]-морфолин
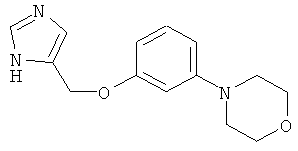
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 3-морфолинофенолом и превращали в 4-[3-(1Н-имидазол-4-илметокси)-фенил]-морфолин. Белое твердое вещество. MS (ISP): 260,3 ([М+Н]+).
Пример 34
4-(2,6-Диэтил-феноксиметил)-1Н-имидазол
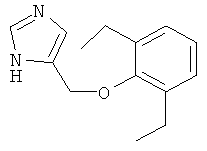
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2,6-диэтилфенолом и превращали в 4-(2,6-диэтил-феноксиметил)-1Н-имидазол. Бесцветное масло. MS (ISP): 231,4 ([М+Н]+).
Пример 35
4-(2,3-Дифтор-феноксиметил)-1Н-имидазол
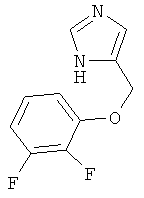
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 2,3-дифторфенолом и превращали в 4-(2,3-дифтор-феноксиметил)-1Н-имидазол. Белое твердое вещество. MS (ISP): 211,1 ([М+Н]+).
Пример 36
4-(3,4-Дихлор-феноксиметил)-1Н-имидазол
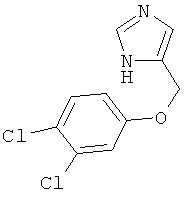
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 3,4-дихлорфенолом и превращали в 4-(3,4-дихлор-феноксиметил)-1Н-имидазол. Белое твердое вещество. MS (ISP): 243,1 ([М+Н]+).
Пример 37
4-(4-Хлор-3-фтор-феноксиметил)-1Н-имидазол
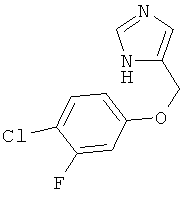
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 4-хлор-3-фторфенолом и превращали в 4-(4-хлор-3-фтор-феноксиметил)-1Н-имидазол. Белое твердое вещество. MS (ISP): 227,1 ([М+Н]+).
Пример 38
4-(3,4-Дифтор-феноксиметил)-1Н-имидазол
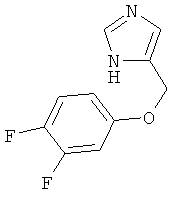
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 3,4-дифторфенолом и превращали в 4-(3,4-дифтор-феноксиметил)-1Н-имидазол. Белое твердое вещество. MS (ISP): 211,1 ([М+Н]+).
Пример 39
5-(Бензофуран-6-илоксиметил)-1Н-имидазол

По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 6-гидроксибензофураном и превращали в 5-(бензофуран-6-илоксиметил)-1Н-имидазол.
Пример 40
4-(3-Хлор-5-фтор-феноксиметил)-1Н-имидазол
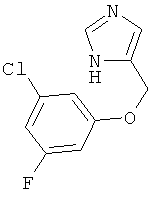
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 3-хлор-5-фторфенолом и превращали в 4-(3-хлор-5-фтор-феноксиметил)-1Н-имидазол. Беловатое аморфное твердое вещество. MS (ISP): 227,1 ([М+Н]+).
Пример 41
5-(4-Бром-2,6-диметил-феноксиметил)-1Н-имидазол
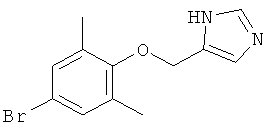
По аналогии с примером 20 4-хлорметил-1-тритил-1Н-имидазол (CAS 103057-10-9) приводили во взаимодействие с 4-бром-2,6-диметилфенолом и превращали в 5-(4-бром-2,6-диметил-феноксиметил)-1Н-имидазол.
Пример 42
5-(2,3-Дихлор-фенилсульфанилметил)-1-имидазол
а) 5-(2,3-Дихлор-фенилсульфанилметил)-1-тритил-1-имидазол
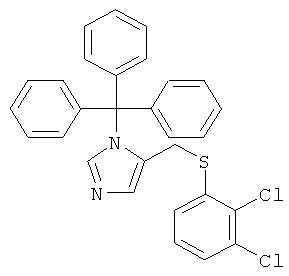
Раствор 4-хлорметил-1-тритил-1Н-имидазола (600 мг; CAS 103057-10-9) в DMF (12 мл) обрабатывали в атмосфере аргона карбонатом калия (578 мг) и 2,3-дихлорбензолтиолом (449 мг). Реакционную смесь нагревали до 80°С в течение 5 ч, затем охлаждали до к.т., переносили в воду и экстрагировали EtOAc. Органический слой промывали водой, сушили над MgSO4 и концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: циклогексан → циклогексан/EtOAc 1:1), получая 5-(2,3-дихлор-фенилсульфанилметил)-1-тритил-1-имидазол (564 мг) в виде беловатого твердого вещества. MS (ISP): 243,3 ([Trt]+).
b) 5-(2,3-Дихлор-фенилсульфанилметил)-1-имидазол
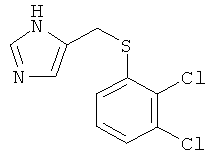
По аналогии с примером 20.b 5-(2,3-дихлор-фенилсульфанилметил)-1-тритил-1-имидазол превращали в 5-(2,3-дихлор-фенилсульфанилметил)-1-имидазол. Беловатое твердое вещество. MS (ISP): 259,0 ([М+Н]+).
Пример 43
5-(2,3-Дихлор-бензолсульфинилметил)-1-имидазол
а) 5-(2,3-Дихлор-бензолсульфинилметил)-1-тритил-1-имидазол
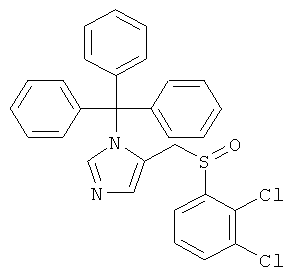
Раствор 5-(2,3-дихлор-фенилсульфанилметил)-1-тритил-1-имидазола (250 мг; пример 42.а) в CH2Cl2 (20 мл) охлаждали в атмосфере аргона до 0° и обрабатывали мета-хлорпербензойной кислотой (86 мг). Реакционную смесь перемешивали в течение 3 ч при 0°С, затем концентрировали. Неочищенный продукт очищали колоночной хроматографией (силикагель; градиент: CH2Cl2 → CH2Cl2/MeOH 98:2), получая 5-(2,3-дихлор-бензолсульфинилметил)-1-тритил-1-имидазол (121 мг) в виде белого твердого вещества. MS (ISP): 517,3 ([М+Н]+).
b) 5-(2,3-Дихлор-бензолсульфинилметил)-1-имидазол

По аналогии с примером 20.b 5-(2,3-дихлор-бензолсульфинилметил)-1-тритил-1-имидазол превращали в 5-(2,3-дихлор-бензолсульфинилметил)-1-имидазол. Белое твердое вещество. MS (ISP): 275,1 ([М+Н]+).
Пример 44
5-(2,3-Дихлор-бензолсульфонилметил)-1Н-имидазол
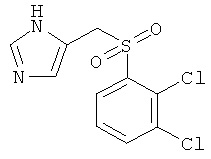
По аналогии с примером 43, но используя 2 эквивалента мета-хлорпербензойной кислоты на первой стадии реакции, 5-(2,3-дихлор-фенилсульфанилметил)-1-тритил-1-имидазол (250 мг; пример 42.а) превращали в 5-(2,3-дихлор-бензолсульфонилметил)-1Н-имидазол. Белое твердое вещество. MS (ISP): 291,0 ([М+Н]+).
Пример 45
4-Бензолсульфинилметил-5-метил-1Н-имидазол
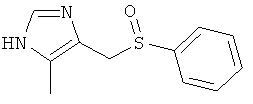
Указанное в заголовке соединение получали по аналогии с примером 43, используя 4-хлорметил-5-метил-1-тритил-1Н-имидазол (CAS 106147-85-7) для алкилирования бензолтиола.
Пример 46
4-(4-Хлор-фенилсульфанилметил)-5-метил-1Н-имидазол
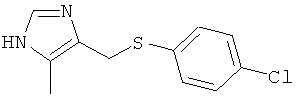
Указанное в заголовке соединение получали по аналогии с примером 42, используя 4-хлорметил-5-метил-1-тритил-1Н-имидазол (CAS 106147-85-7) для алкилирования 4-хлорбензолтиола.
Пример 47
4-(Нафталин-2-илсульфанилметил)-1Н-имидазол
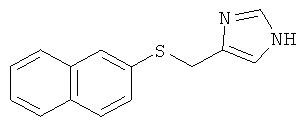
Указанное в заголовке соединение получали по аналогии с примером 42, начиная с нафталин-2-тиола.
Пример 48
Бензил-(1Н-имидазол-4-ил)-амина гидрохлорид
а) N-(1-Тритил-1Н-имидазол-4-ил)-бензамид
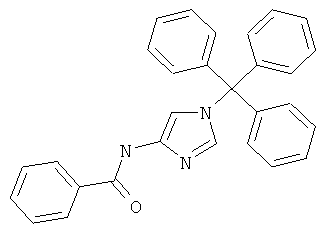
К раствору 4-амино-1-тритилимидазола (0,30 г; 0,92 ммоль) в дихлорметане (4 мл) добавляли последовательно триэтиламин (0,19 мл; 1,37 ммоль) и бензоилхлорид (0,13 мл; 1,12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем разбавляли дихлорметаном и промывали последовательно водой, насыщенным водным раствором NaHCO3, водой и насыщенным рассолом. Органический слой отделяли, сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: метанол/дихлорметан от 0:100 до 10:90), получая указанное в заголовке соединение в виде оранжевого твердого вещества (0,36 г; 92%); MS (ISP): 430,3 ([М+Н]+).
b) Бензил-(1-тритил-1Н-имидазол-4-ил)-амин
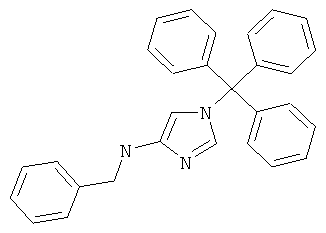
К раствору N-(1-тритил-1Н-имидазол-4-ил)-бензамида (0,36 г; 0,83 ммоль) в тетрагидрофуране (10 мл) добавляли порциями алюмогидрид лития (0,16 г; 4,14 ммоль). Реакционную смесь перемешивали при 80°С в течение 16 часов, затем охлаждали до комнатной температуры и по каплям добавляли воду. Смесь перемешивали при комнатной температуре в течение 20 минут и затем экстрагировали этилацетатом. Органический слой отделяли, промывали водой, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: метанол/дихлорметан от 0:100 до 10:90), получая указанное в заголовке соединение в виде белого твердого вещества (0,15 г; 44%); MS (ISP): 416,5 ([М+Н]+).
с) Бензил-(1Н-имидазол-4-ил)-амина гидрохлорид
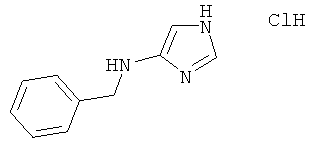
Бензил-(1-тритил-1Н-имидазол-4-ил)-амин (0,15 г; 0,35 ммоль) растворяли в 4 М растворе HCl в диоксане (5 мл). Смесь перемешивали при комнатной температуре в течение 90 мин и затем концентрировали в вакууме. Остаток растирали в эфире, получая указанное в заголовке соединение в виде беловатого твердого вещества (73 мг; 100%); MS (ISP): 174,4 ([М+Н]+).
Соединения примеров 1-48 являются новыми. Соединения примеров A-N известны.
Примеры A-N
В дополнение к этому следующие известные соединения получали как агонисты TAAR1, используя методики, аналогичные описанным выше:
А: 5-фенетил-1Н-имидазол (CAS 94714-36-0),
В: 5-(2-фенил-пропил)-1Н-имидазол (CAS 86347-25-3),
С: 1-(1Н-имидазол-4-ил)-2-фенил-этанол (CAS 79928-10-2),
D: 5-(2,2-дифенил-этил)-1Н-имидазол (CAS 102390-63-6),
Е: 4-(2-м-толил-этил)-1Н-имидазол (CAS 79928-27-1),
F: 4-[2-(2,6-диметил-фенил)-этил]-1Н-имидазол (CAS 79924-13-3),
G: 4-(бифенил-2-илоксиметил)-1Н-имидазол (CAS 527696-96-4),
Н: 5-(2-метил-2-фенил-пропил)-1Н-имидазол (Beilstein Registry Number 4407995),
I: 4-(2-хлор-феноксиметил)-1Н-имидазол (CAS 27325-27-5),
J: 4-(2-фтор-феноксиметил)-1Н-имидазол (CAS 401-45-6),
К: 4-о-толилоксиметил-1Н-имидазол (CAS 762177-70-8),
L: 4-(3-хлор-феноксиметил)-1Н-имидазол (CAS 802322-21-0),
М: 4-(2,6-диметил-феноксиметил)-1Н-имидазол (CAS 771450-63-6),
N: 5-метил-4-фенилсульфанилметил-1Н-имидазол (CAS 700355-78-8).
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ | 2010 |
|
RU2595902C2 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИНОВ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ ЦНС | 2010 |
|
RU2569887C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ РЕЦЕПТОРОВ, АССОЦИИРОВАННЫХ СО СЛЕДОВЫМИ АМИНАМИ (TAARS) | 2013 |
|
RU2621050C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛКАРБОКСАМИДА | 2013 |
|
RU2637938C2 |
| ПРОИЗВОДНЫЕ ДИГИДРООКСАЗОЛ-2-АМИНА | 2011 |
|
RU2587512C2 |
| ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА, ЛЕКАРСТВЕННОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ИХ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2000 |
|
RU2257382C2 |
| 2,5,6,7-ТЕТРАГИДРО-[1,4]ОКСАЗЕПИН-3-ИЛАМИНЫ ИЛИ 2,3,6,7-ТЕТРАГИДРО-[1,4]ОКСАЗЕПИН-5-ИЛАМИНЫ | 2011 |
|
RU2570796C2 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
Настоящее изобретение относится к применению соединения формулы I, где R представляет собой водород, низший алкил или амино; X-R1 представляет собой -СН2- или -СН(низший алкокси)- и Y-R2 представляет собой -СН2-, -CH(низший алкил)-, -O-, -S-, -S(O)- или -S(O)2-; Ar представляет собой фенил, нафтил или бензофуранил, кольца которых замещены одним или более заместителями, выбранными из группы, состоящей из низшего алкила, низшего алкила, замещенного галогеном, галогена, низшего алкокси, низшего алкокси, замещенного галогеном, гидрокси, диалкиламино, морфолинила, бензила или O-бензила; или фармацевтически приемлемых солей присоединения кислоты для изготовления лекарства для лечения депрессии, тревожных расстройств, биполярного расстройства, шизофрении, болезни Паркинсона, эпилепсии, мигрени, диабета, диабетических осложнений или расстройств и нарушений температурного гомеостаза организма. Также изобретение относится к конкретным соединениям, к лекарственному средству на основе заявленных соединений. Технический результат: исследованы известные производные имидазола, а также получены новые производные имидазола, обладающие высокой аффиностью к рецепторам следовых аминов (TAAR). 3 н. и 3 з.п. ф-лы, 1 табл., 48 пр.
1. Применение соединения формулы I
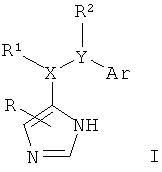
где R представляет собой водород, низший алкил или амино;
X-R1 представляет собой -СН2- или -СН(низший алкокси)- и
Y-R2 представляет собой -CH2-, -СН(низший алкил)-, -O-, -S-, -S(O)- или -S(O)2-;
Ar представляет собой фенил, нафтил или бензофуранил, кольца которых замещены одним или более заместителями, выбранными из группы, состоящей из низшего алкила, низшего алкила, замещенного галогеном, галогена, низшего алкокси, низшего алкокси, замещенного галогеном, гидрокси, диалкиламино, морфолинила, бензила или O-бензила;
или фармацевтически приемлемых солей присоединения кислоты для изготовления лекарства для лечения депрессии, тревожных расстройств, биполярного расстройства, шизофрении, болезни Паркинсона, эпилепсии, мигрени, диабета, диабетических осложнений или расстройств и нарушений температурного гомеостаза организма.
2. Соединения формулы I, где данными соединениями являются
4-[2-(2-хлор-фенил)-этил]-1Н-имидазол,
4-[2-(2-метокси-фенил)-этил]-1Н-имидазол,
4-[2-(3-хлор-фенил)-этил]-1Н-имидазол,
4-[2-(3-фтор-фенил)-этил]-1Н-имидазол,
4-[2-(3-трифторметил-фенил)-этил]-1Н-имидазол,
4-[2-(3-метокси-фенил)-этил]-1Н-имидазол,
4-[2-(4-хлор-фенил)-этил]-1Н-имидазол,
4-[2-(3,5-дихлор-фенил)-этил]-1Н-имидазол,
4-(2-фенил-бутил)-1Н-имидазол или
4-(2,3-дихлор-феноксиметил)-1Н-имидазол,
4-(2,3-дифтор-феноксиметил)-1Н-имидазол,
4-(3,4-дихлор-феноксиметил)-1Н-имидазол,
4-(4-хлор-3-фтор-феноксиметил)-1Н-имидазол,
5-(бензофуран-6-илоксиметил)-1Н-имидазол,
5-(2,3-дихлор-фенилсульфанилметил)-1-имидазол,
4-(4-хлор-фенилсульфанилметил)-5-метил-1Н-имидазол или
4-(нафталин-2-илсульфанилметил)-1Н-имидазол.
3. Соединение по п.2, обладающее аффинностью к TAAR1.
4. Лекарственное средство, обладающее аффинностью к TAAR1, содержащее соединение формулы I по п.1 или 2.
5. Лекарственное средство по п.4 для лечения депрессии, тревожных расстройств, биполярного расстройства, шизофрении, болезни Паркинсона, эпилепсии, мигрени, диабета, диабетических осложнений или расстройств и нарушений температурного гомеостаза организма.
6. Лекарственное средство по п.5 для лечения депрессии, болезни Паркинсона и тревоги.
| US 4639464 А, 27.01.1987 | |||
| Kitbunnadaj R | |||
| Et al: "New high affinity Н receptor agonists without a basic side chain", | |||
| Bioorganic & Medicinal Chemistry, 13(23), 2005, pp.6309-6323 | |||
| Bagley J.R | |||
| et al: "Synthesis and α-adrenergic activities of imidazole and imidazolidine analogues: in vitro and in vivo selectivity", |

