Изобретение относится к медицинским токсикологическим исследованиям, в частности к санитарной токсикологии, и может быть использовано для количественного определения бенз(а)пирена в моче для оценки риска здоровью человека и разработки мероприятий по обеспечению химической безопасности.
Бенз(а)пирен является одним из наиболее сильных канцерогенов среди полиароматических углеводородов (далее ПАУ). Концентрация бенз(а)пирена в воздухе на уровне 3-6 нг/м3 при длительном воздействии может привести к увеличению частоты рака легкого у населения. Поэтому в России и многих других странах законодательно введены максимально допустимые пределы для бенз(а)пирена, что, в свою очередь, обусловливает необходимость разработки применения соответствующих методов контроля.
Для определения бенз(а)пирена и других ПАУ в различных средах используют методы газовой и жидкостной хроматографии, масс-спектроскопии и т.д. Наиболее высокую достоверность идентификации и чувствительность имеют методы, основанные на использовании высокоэффективной жидкостной хроматографии с обращенной фазой. Определение бенз(а)пирена в различных объектах окружающей среды сильно осложняется тем, что он присутствует обычно в очень малых концентрациях. При определении возможны потери вещества на различных этапах анализа, в частности в результате улетучивания его в процессе отгонки растворителей.
Анализ литературы показал, что представленные химико-аналитические методы касаются в основном определения метаболитов бенз(а)пирена в биологических средах при его экспозиции различными путями. Это связано, вероятнее всего, с тем, что бенз(а)пирен является «латентным» химическим веществом. При попадании в организм он метаболизирует с образованием более токсичных соединений, которые вызывают канцерогенный и другие опасные для организма эффекты.
Известны различные способы определения бенз(а)пирена: в воде, в биологических объектах, в моче.
Известен способ определения массовой концентрации бенз(а)пирена в питьевой воде и в воде источников хозяйственно-питьевого водоснабжения в диапазоне от 0,002 до 0,5 мкг/дм3 методом высокоэффективной жидкостной хроматографии с флуориметрическим детектированием [1]. Известный метод измерения основан на экстракции бенз(а)пирена из проб воды н-гексаном (хлористым метиленом), концентрировании экстракта, его хроматографическом разделении, регистрации сигнала флуоресценции с использованием флуоресцентного детектора, идентификации пика бенз(а)пирена на хроматограмме по времени удержания и расчете массовой концентрации бенз(а)пирена.
Разработчиками компании Waters предложен метод определения ПАУ, в том числе бенз(а)пирена, в питьевой воде [2]. Анализ методом высокоэффективной жидкостной хроматографии (ВЭЖХ) проводится на ультрафиолетовом и флуоресцентном детекторах. Предварительная подготовка пробы проводится на твердом сорбенте С18 с экстракцией метиленхлоридом.
Также известны: Способ определения ПАУ в биологических объектах (Патент РФ №2187106), Способ количественного определения углеводородов в биологических объектах (Патент РФ №1337764). Однако их недостатком является то, что определение производится только суммы ПАУ, без разделения на составляющие, что делает известные способы неселективными.
Известен способ определения маркерных метаболитов бенз(а)пирена в моче методом спектрально-флуоресцентного анализа [3]. Реализация известного способа производится путем экстракции и спектрально-флуоресцентного анализа, при этом регистрируют избирательное монохроматическое возбуждение люминесценции с помощью света с длиной волны 353 нм для определения 7,8-дигидродиол-бенз(а)пирена и длиной волны 405 нм для определения 3-гидрокси-бенз(а)пирена, затем производят расчет количества метаболитов в мкг на 1 кг массы тела. Данным способом можно определить содержание окисленных метаболитов бенз(а)пирена, а не само вещество.
Наиболее близким к предлагаемому изобретению является способ определения бенз(а)пирена и его метаболита в моче методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с флуориметрическим детектированием. Извлечение аналита из биологического образца (моча) проводили в следующем порядке: 7 мл 0,2 М буферного раствора ацетата натрия (рН 5,0) добавляли к 5 мл мочи для подкисления; затем добавляли раствор глюкуронидазы (13 200 единиц) и сульфатазы (220 единиц). Смесь выдерживали при температуре 37°С 16 часов. После центрифугирования (3000 об/мин в течение 10 минут) супернатант загружали на активизированный патрон SepPak C18, промывали 5 см3 40%-ого раствора метанола, элюировали 8 см3 ацетонитрила, а затем 10 см3 раствора гексана и дихлорметана (3:1). Конечный элюат испаряли при 45°С и растворяли в 1 см3 ацетонитрила. Анализ проводили методом ВЭЖХ с флуориметрическим детектором [4]. Недостатком указанного известного способа является низкая чувствительность определения 0,1 нг/см3 (100 нг/дм3), а также длительная пробоподготовка, не приемлемая для проведения массовых анализов.
Технический результат, обеспечиваемый предлагаемым изобретением, заключается в обеспечении высокой чувствительности способа, при одновременном обеспечении селективности и его доступности для серийных анализов.
Указанный технический результат достигается предлагаемым способом количественного определения бенз(а)пирена в моче методом жидкостной хроматографии, включающим отбор пробы мочи, подготовку ее к анализу и проведение определения методом жидкостной хроматографии, при этом новым является то, что пробу мочи подвергают центрифугированию при 2000 об/мин в течение 10 мин, осуществляют твердофазную экстракцию на сорбенте Oasis HLB путем последовательного пропускания через указанный сорбент смеси метиленхлорида с ацетонитрилом в объемном соотношении 1:10 соответственно, дистиллированной воды, пробы мочи после центрифугирования, дистиллированной воды, 50%-ного водного раствора ацетонитрила, далее сорбент высушивают в течение 10 минут под вакуумом, со скоростью потока воздуха 2-5 см3/мин, и пропускают через него экстрагент - 100%-ный метиленхлорид, затем экстракт высушивают в токе воздуха и сухой остаток перерастворяют в 100%-ном ацетонитриле, полученный экстракт анализируют методом жидкостной хроматографии, используя в качестве подвижной фазы смесь ацетонитрила и воды при их изменяющемся соотношении от 60:40 об.% до 90:10 об.% соответственно, а также 100%-ный ацетонитрил в градиентном режиме, который осуществляют при хроматографии путем подачи вначале в течение 2,5 мин подвижной фазы, состоящей из смеси ацетонитрила и воды в соотношении 60:40 об.%, затем путем увеличения в течение 9,5 мин в подвижной фазе ацетонитрила до 90 об.%, а также дальнейшего увеличения его концентрации в течение 8 мин до 100 об.% и пропускания такой подвижной фазы в течение еще 4,5 мин, с последующим снижением объемного количества ацетонитрила за 0,5 мин до 60 об.% и пропусканием такой подвижной фазы через колонку в течение 5 мин, а количество бенз(а)пирена в моче устанавливают по градуировочному графику.
Скорость потока подвижной фазы составляет 1,5 мл/мин.
Жидкостную хроматографию проводят на жидкостном хроматографе «Agilent 1200» с флуориметрическим детектором при длине волны возбуждения 265 нм и длине волны эмиссии 412 нм при температуре колонки 28°С.
Указанный технический результат достигается за счет следующего.
Благодаря тому, что отобранную пробу мочи подвергают центрифугированию со скоростью 2000 об/мин в течение 10 мин, обеспечивается осаждение взвешенных примесей, что исключает засорение картриджа этими примесями при проведении твердофазной экстракции.
Экспериментальным путем было установлено, что высокая чувствительность и точность определения бенз(а)пирена в моче достигается лишь при строго определенной последовательности пробоподготовки мочи. Пропусканием через сорбент Oasis HLB смеси метиленхлорида с ацетонитрилом в объемном соотношении 1:10 соответственно и дистиллированной воды обеспечивает очистку сорбента от возможного присутствия посторонних примесей и увлажнение сорбента перед пропусканием пробы мочи (в целом - это подготовка сорбента к анализу). А промывка сорбента после пропускания мочи дистиллированной водой и 50%-ным водным раствором ацетонитрила необходима для цели удаления с сорбента слабо удерживаемых компонентов, присутствующих в моче, и повышения селективности извлечения. Использование водного раствора ацетонитрила именно такой концентрации обеспечивает максимальное удаление сопутствующих компонентов из пробы, и в то же время не производится экстракция.
Сушка картриджа с сорбентом под вакуумом необходима для удаления следов ацетонитрила и воды.
Сушка экстракта после пропускания через сорбент экстрагента 100%-ного метиленхлорида необходима для удаления из пробы не смешивающегося с водой растворителя (метиленхлорида). А перерастворение сухого остатка в 100%-ном ацетонитриле обеспечивает проведение анализа в режиме обращеннофазной высокоэффективной жидкостной хроматографии, где в качестве подвижной фазы используются вода и водорастворимые органические растворители.
Проведение последующего анализа экстракта мочи методом жидкостной хроматографии с особыми режимами, а именно: используя в качестве подвижной фазы смесь ацетонитрила и воды при их изменяющемся соотношении от 60:40 об% до 90:10 об.% соответственно, а также 100%-ный ацетонитрил в градиентном режиме, который осуществляют при хроматографии путем подачи вначале в течение 2,5 мин подвижной фазы, состоящей из смеси ацетонитрила и воды в соотношении 60:40 об%, затем путем увеличения в течение 9,5 мин в подвижной фазе ацетонитрила до 90 об.%, а также дальнейшего увеличения его концентрации в течение 8 мин до 100 об.% и пропускания такой подвижной фазы в течение еще 4,5 мин, с последующим снижением объемного количества ацетонитрила за 0,5 мин до 60 об.% и пропусканием такой подвижной фазы через колонку в течение 5 мин, соответствует оптимизации элюирования, а значит, обеспечивает высокую чувствительность способа. Следует пояснить, что изменение объемного соотношения ацетонитрила и воды в подвижной фазе осуществляется за счет работы 4-канального градиентного насоса Agilent 1200, смешивающего до 4 компонентов подвижной фазы.
Исследование экстрактов на жидкостном хроматографе «Agilent 1200» с флуориметрическим детектором при длине волны возбуждения 265 нм и длине волны эмиссии 412 нм при температуре колонки 28°С обусловлено достижением максимального сигнала флуориметрического детектора, что влияет на чувствительность и точность определения.
Скорость потока подвижной фазы 1,5 мл/мин является оптимальной при проведении исследований.
Количество бенз(а)пирена в моче устанавливают по градуировочному графику.
Для осуществления предлагаемого способа проводят следующие операции в нижеуказанной последовательности:
- производят отбор пробы мочи, например, в количестве 20 см3;
- переносят указанную пробу в центрифужную пробирку и центрифугируют со скоростью 2000 об/мин в течение 10 мин;
- проводят твердофазную экстракцию по следующему алгоритму:
осуществляют конденционирование картриджа Oasis HLB 1 сс (30 мг) 1 см3 раствора метиленхлорида в ацетонитриле (1:10) и 1 см3 дистиллированной воды, далее загружают 1 см3 мочи на картридж Oasis HLB 1 cc (30 мг), промывают картридж с нанесенной пробой 1 см3 дистиллированной воды, 0,2 см3 50%-ного водного раствора ацетонитрила (для очистки пробы мочи от слабоудерживаемых компонентов), высушивают картридж в токе воздуха посредством вакуумного насоса. Переносят патрон с сорбентом в накопительный сосуд (бюкс) и элюируют 1 см3 100%-ного метиленхлорида. Затем метиленхлорид высушивают в токе воздуха, сухой остаток перерастворяют в 1 см3 100%-ного ацетонитрила. Полученный экстракт анализируют в количестве 20 мм3 на жидкостном хроматографе;
- далее проводят жидкостно-хроматографический анализ, условия проведения которого соответствуют следующим характеристикам:
- а количество бенз(а)пирена в моче устанавливают по градуировочному графику.
Для построения градуировочного графика в мерную колбу вместимостью 100 см3 вносят 1 см3 стандартных образцов (ГСО) бенз(а)пирена в ацетонитриле с концентрацией 100 мкг/см3 и доводят ацетонитрилом до метки. Концентрация бенз(а)пирена в исходном растворе (раствор №1) составляет 1 мкг/см3. Затем 2,5 см3 раствора №1 вносят в мерную колбу вместимостью 100 см3 и доводят ацетонитрилом до метки. Концентрация бенз(а)пирена в разбавленном растворе (раствор №2) составляет 25 нг/дм3. Используют свежеприготовленный раствор.
Градуировочную характеристику устанавливают методом абсолютной градуировки. Она выражает зависимость площади пика на хроматограмме (мм2) от концентрации бенз(а)пирена в моче (мг/дм3) и строится по 5 сериям рабочих стандартных растворов. Каждую серию, состоящую из 3 стандартных растворов, готовят в мерных колбах объемом 25 см3. Для этого в каждую колбу вносят растворы бенз(а)пирена для градуировки №1 и 2 в соответствии с таблицей 1, доводят объем колбы до метки мочой и перемешивают. По 10 см3 свежеприготовленных стандартных растворов переносят в центрифужные пробирки и центрифугируют со скоростью 2000 об/мин в течение 10 мин. Отбирают 1 см3 верхнего слоя мочи и проводят твердофазную экстракцию на сорбенте Oasis HLB 3сс, как в предлагаемом способе. Полученные экстракты хроматографируют на жидкостном хроматографе «Agilent 1200» с флуориметрическим детектором.
Отработка оптимальных жидкостно-хроматографических параметров для определения бенз(а)пирена в моче предлагаемым способом осуществлялась с использованием жидкостного хроматографа «Agilent 1200», оснащенного термостатом колонок, градиентным насосом и флуориметрическим детектором.
Полнота разделения бенз(а)пирена в присутствии мешающих компонентов биологической пробы достигнута на колонке 4,6×150 мм с сорбентом Zorbax Eclipse XDB-C18 при температуре колонки 28°С, при использовании в качестве подвижной фазы вначале смеси ацетонитрила и воды в объемном соотношении 60:40 со скоростью потока 1,5 см3/мин и оптимизации элюирования в градиентном режиме (2,5 мин подача подвижной фазы 60 об.% ацетонитрила и 40 об.% воды, увеличение ацетонитрила с 60 об.% до 90 об.% в течение 9,5 мин, увеличение ацетонитрила с 90 до 100% в течение 8 мин, 4,5 мин подача 100% ацетонитрила, снижение ацетонитрила до 60% в течение 2,5 мин, подача 60% ацетонитрила до уравновешивания колонки (5 мин). Максимальный сигнал флуориметрического детектора получен при длине волны возбуждения 265 нм и длине волны эмиссии 412 нм. В случае, если не был применен градиентный режим элюирования, чувствительность способа резко изменялась.
В процессе исследований по выбору оптимальных условий проведения твердофазной экстракции (ТФЭ) бенз(а)пирена (БаП) из мочи, включающей конденционирование сорбента Oasis HLB раствором метиленхлорида в ацетонитриле (1:10) и дистиллированной водой, промывку картриджа от мешающих компонентов мочи дистиллированной водой и 50%-ным водным раствором ацетонитрила после нанесения анализируемой пробы мочи на указанный сорбент, высушивание картриджа в токе воздуха посредством вакуумного насоса и последующую десорбцию бенз(а)пирена 100%-ным метиленхлоридом, были апробированы режимы 1-12. В режимах 1-10 конденционирование картриджа проводили пропусканием 1 см3 ацетонитрила и 1 см3 дистиллированной воды. В режимах 1-7 и 10-12 промывку картриджа с нанесенной пробой проводили 1 см3 воды, 0,2 см3 50%-ного раствора ацетонитрила в воде. Обработку экстракта и анализ проводили путем его высушивания в токе воздуха, растворения сухого остатка в 1 см3 ацетонитрила. Полученные экстракты анализировали отдельно, а результаты анализов суммировали.
Режим 1: конденционирование картриджа, добавка к 0,5 см3 анализируемого образца мочи 0,5 см3 70%-ного раствора этанола в воде, загрузка образца, промывка картриджа, элюирование 1 см3 ацетонитрила, 1 см3 метиленхлорида. Обработка и анализ экстракта.
Режим 2: конденционирование картриджа, добавка к 0,5 см3 образца мочи 0,5 см3 70%-ного раствора этанола в воде, загрузка образца, промывка картриджа, элюирование 0,2 см3 ацетонитрила, 1 см3 метиленхлорида. Обработка и анализ экстракта.
Режим 3: конденционирование картриджа, добавка к 0,5 см3 анализируемого образца мочи 0,5 см3 70%-ного раствора этилового спирта в воде, загрузка образца 1 см3, промывка картриджа, элюирование 0,2 см3 ацетонитрила, 1 см3 метиленхлорида. Элюирование метиленхлоридом сопровождалось воздействием УЗ-волн частотой 37 КГц в У3-бане Elmasonic в течение 10 мин. Обработка и анализ экстракта.
Режим 4: конденционирование картриджа, добавка к 5 см3 анализируемого образца мочи 0,25 см3 ацетонитрила, загрузка образца 1 см3, промывка картриджа, трижды элюирование по 1 см3 ацетонитрила. Полученные экстракты анализировали отдельно, а результаты анализов суммировали.
Режим 5: конденционирование картриджа, добавка к 5 см3 анализируемого образца мочи 0,5 см3 ацетонитрила, загрузка образца 1 см3, промывка картриджа, трижды элюирование по 1 см3 ацетонитрила. Полученные экстракты анализировали отдельно, а результаты анализов суммировали.
Режим 6: конденционирование картриджа, добавка к 5 см3 анализируемого образца мочи 0,25 см3 ацетонитрила, загрузка образца 1 см3, промывка картриджа, трижды элюирование по 1 см3 ацетонитрила. Элюирование ацетонитрилом с УЗ-обработкой по 6 мин на каждый экстракт. Полученные экстракты анализировали отдельно, а результаты анализов суммировали.
Режим 7: конденционирование картриджа, добавка к 5 см3 анализируемого образца мочи 0,25 см3 ацетонитрила, загрузка образца 1 см3, промывка картриджа, трижды элюирование по 1 см3 ацетонитрила. Элюирование проводили ацетонитрилом с УЗ-обработкой в течение 10 минут. Полученные экстракты анализировали отдельно, а результаты анализов суммировали.
Режим 8: конденционирование картриджа, добавка к 5 см3 анализируемого образца мочи 0,25 см3 ацетонитрила, загрузка образца 1 см3, элюирование 0,2 см3 ацетонитрила, 0,85 см3 раствора метиленхлорида в гексане (1:9). Обработка и анализ экстракта.
Режим 9: конденционирование картриджа, добавка к 5 см3 анализируемого образца мочи 0,25 см3 ацетонитрила, загрузка образца 1 см3, элюирование 0,2 см3 ацетонитрила, 0,9 см3 раствора метиленхлорида в гексане (2:8). Обработка и анализ экстракта.
Режим 10: конденционирование картриджа, загрузка образца 1 см3, промывка картриджа, элюирование 0,2 см3 ацетонитрила, 0,9 см3 раствора метиленхлорида в гексане (1:9). Обработка и анализ экстракта.
Режим 11: конденционирование 1 см3 раствора метиленхлорида в ацетонитриле (0,3:1), 1 см3 дистиллированной воды, загрузка образца мочи 1 см3, промывка картриджа, элюирование 0,2 см3 ацетонитрила, 1 см3 метиленхлорида. Обработка и анализ экстракта.
Режим 12: конденционирование 1 см3 раствора метиленхлорида в ацетонитриле (1:10), 1 см3 дистиллированной воды, загрузка образца мочи 1 см3, промывка картриджа. Высушивание картриджа в токе воздуха посредством вакуумного насоса. Экстракция 1 см3 метиленхлорида. Обработка и анализ экстракта. Данные, полученные в ходе испытаний, приведены в таблице 2.
Данные, приведенные в таблице 2, показывают, что максимальная степень экстракции бенз(а)пирена из проб мочи была получена в условиях режима №12 и составляет 92,0%. В дальнейших исследованиях использовали подготовку образцов мочи к анализу по режиму №12.
Экспериментальным путем также были установлены, наряду с необходимостью применения в качестве экстрагента 100%-ного метиленхлорида, и другие характеристики и режимы предлагаемого способа, обеспечивающие эффективное и селективное извлечение бенз(а)пирена и повышение чувствительности и точности определения, а именно: центрифугирование пробы мочи при 2000 об/мин, использование при твердофазной экстракции 1 см3 раствора метиленхлорида в ацетонитриле (1:10) и 1 см3 дистиллированной воды для конденционирования сорбента, промывание картриджа 1 см3 дистиллированной воды и 0,2 см3 50%-ного водного раствора ацетонитрила после пропускания анализируемой пробы мочи, высушивание картриджа в токе воздуха посредством вакуумного насоса и экстракции 1,0 см3 100%-ного метиленхлорида.
Пример конкретного выполнения предлагаемого способа.
Анализируют пробы мочи (обследуемая группа детей). Каждую пробу мочи анализируют дважды. Свежеотобранную пробу мочи объемом 10 см3 центрифугируют со скоростью 2000 об/мин в течение 10 мин. Отбирают 1 см3 верхнего слоя и проводят твердофазную экстракцию, последовательно пропуская под вакуумом через картридж с сорбентом Oasis HLB 1 сс 1 см3 раствора метиленхлорида в ацетонитриле (1:10), 1 см3 дистиллированной воды, 1 см3 мочи, 1 см3 дистиллированной воды, 0,2 см3 50-% водного раствора ацетонитрила. Высушивают картридж в токе воздуха посредством вакуумного насоса. Переносят патрон с сорбентом в накопительный сосуд (бюкс) и пропускают через сорбент 1,0 см3 100%-ного метиленхлорида. Скорость экстракции (скорость потока) 1,5 см3/мин. Аликвотную часть полученного экстракта отбирают и хроматографируют на жидкостном хроматографе «Agilent серии 1200» с флуориметрическим детектором на колонке 4,6×150 мм с сорбентом Zorbax Eclipse XDB-C18 при температуре колонки 28°С, при использовании в качестве подвижной фазы смеси ацетонитрила и воды со скоростью потока 1,5 см3/мин и оптимизации элюирования в градиентном режиме (2,5 мин подача подвижной фазы 60 об.% ацетонитрила и 40 об.% воды, увеличение ацетонитрила с 60 до 90% в течение 9,5 мин, увеличение ацетонитрила с 90 до 100% в течение 8 мин, 4,5 мин подача 100% ацетонитрила, снижение ацетонитрила до 60% в течение 2,5 мин, подача 60% ацетонитрила в течение 5 мин до уравновешивания колонки). При этом длина волны возбуждения флуориметрического детектора составляла 265 нм и длина волны эмиссии - 412 нм.
По градуировочному графику определяют содержание бенз(а)пирена в пробах 1-5. Полученные результаты приведены в таблице 3.
Чувствительность определения бенз(а)пирена в анализируемом объеме пробы мочи (20 мм3) составила 0,2 пг (или 0,01 мкг/дм3). Погрешность определения не превышает 23%.
Методика выполнения измерений обеспечивает получение результатов измерений с погрешностью, не превышающей значений, приведенных в таблицах 4 и 5.
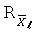 , %
, %
Применение предлагаемого способа позволяет повысить чувствительность определения бенз(а)пирена как минимум в 10 раз, например, по сравнению со способом, описанным в источнике информации [4].
Способ прост и может быть рекомендован для серийных анализов.
Источники информации
1. ГОСТ Р 51310-99 Вода питьевая. Метод определения содержания бенз(а)пирена. М. - 1999. - 10 с.
2. Determination of Polycyclic Aromatic Hydrocarbons in drinking water by HPLC liquid-solid extraction. EPA Method 550.1. 1990/ ЕРА Environ. Monitoring systems laboratory.
3. Способ определения маркерных метаболитов бенз(а)пирена в моче методом спектрально-флуоресцентного анализа. Заявка на патент РФ №2006101613.
4. Kang H., Jeong S., Cho М., Cho J. Changes of biomarkers with oral exposure to benzo(a)pyrene, phenanthrene and pyrene in rats // J. Vet. Sci. 2007. - 8(4). - p.361-368.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БЕНЗ(А)ПИРЕНА В КРОВИ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2014 |
|
RU2546530C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДИМЕТИЛТЕРЕФТАЛАТА В МОЧЕ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2010 |
|
RU2425380C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ АКРОЛЕИНА В АТМОСФЕРНОМ ВОЗДУХЕ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2014 |
|
RU2556294C1 |
| Способ подготовки пробы мочи для определения монометилфталата, моноэтилфталата, монобутилфталата, монобензилфталата, моноэтилгексилфталата методом высокоэффективной жидкостной хроматографии/масс-спектрометрии | 2019 |
|
RU2687738C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ФУМАРОВОЙ И МАЛЕИНОВОЙ КИСЛОТ В ПЛАЗМЕ КРОВИ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2018 |
|
RU2677341C1 |
| Способ определения концентрации стирола в атмосферном воздухе методом высокоэффективной жидкостной хроматографии | 2017 |
|
RU2648018C1 |
| Способ определения массовых концентраций фенола и пирокатехина в крови методом высокоэффективной жидкостной хроматографии | 2022 |
|
RU2786509C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ПЕНТАХЛОРФЕНОЛА В КРОВИ МЕТОДОМ ГАЗОХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА | 2014 |
|
RU2546527C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 2,4-ДИХЛОРФЕНОЛА В КРОВИ МЕТОДОМ ГАЗОХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА | 2013 |
|
RU2521277C1 |
| Способ количественного определения N-нитрозаминов в детских кашах | 2015 |
|
RU2613303C1 |
Изобретение относится к медицинским токсикологическим исследованиям, в частности к санитарной токсикологии. Свежеотобранную пробу мочи объемом 10 см3 центрифугируют со скоростью 2000 об/мин в течение 10 мин. Отбирают 1 см3 верхнего слоя и проводят твердофазную экстракцию, последовательно пропуская под вакуумом через картридж с сорбентом Oasis HLB 1 cc 1 см3 1 см3 раствора метиленхлорида в ацетонитриле (1:10), 1 см3 дистиллированной воды, 1 см3 мочи, 1 см3 дистиллированной воды, 0,2 см3 50-% водного раствора ацетонитрила. Высушивают картридж в токе воздуха посредством вакуумного насоса. Затем переносят патрон с сорбентом в накопительный сосуд (бюкс) и пропускают через сорбент 1,0 см3 100%-ного метиленхлорида. Скорость экстракции 1 см3/мин. Аликвотную часть полученного экстракта отбирают и хроматографируют на жидкостном хроматографе «Agilent серии 1200» с флуориметрическим детектором. Скорость экстракции (скорость потока) 1,5 см3/мин. Аликвотную часть полученного экстракта отбирают и хроматографируют на жидкостном хроматографе «Agilent серии 1200» с флуориметрическим детектором на колонке 4,6×150 мм с сорбентом Zorbax Eclipse XDB-C18 при температуре колонки 28°С, при использовании в качестве подвижной фазы смеси ацетонитрила и воды со скоростью потока 1,5 см3/мин и оптимизации элюирования в градиентном режиме, а именно: 2,5 мин подача подвижной фазы, состоящей из 60 об.% ацетонитрила и 40 об.% воды, увеличение ацетонитрила с 60 об.% до 90 об.% в течение 9,5 мин, увеличение ацетонитрила с 90 об.% до 100 об.% в течение 8 мин, 4,5 мин подача 100% ацетонитрила, снижение ацетонитрила до 60% в течение 2,5 мин, подача 60% ацетонитрила в течение 5 мин до уравновешивания колонки. При этом длина волны возбуждения флуориметрического детектора составляла 265 нм и длина волны эмиссии - 412 нм. Достигается высокая чувствительность способа при одновременном обеспечении селективности и его доступности для серийных анализов. 2 з.п. ф-лы, 5 табл.
1. Способ количественного определения бенз(а)пирена в моче методом жидкостной хроматографии, включающий отбор пробы мочи, подготовку ее к анализу и проведение определения методом жидкостной хроматографии, отличающийся тем, что пробу мочи подвергают центрифугированию при 2000 об/мин в течение 10 мин, осуществляют твердофазную экстракцию на сорбенте Oasis HLB путем последовательного пропускания через указанный сорбент смеси метиленхлорида с ацетонитрилом в объемном соотношении 1:10 соответственно, дистиллированной воды, пробы мочи после центрифугирования, дистиллированной воды, 50%-ного водного раствора ацетонитрила, далее сорбент высушивают в течение 10 мин под вакуумом со скоростью потока воздуха 2-5 см3/мин и пропускают через него экстрагент - 100%-ный метиленхлорид, затем экстракт высушивают в токе воздуха и сухой остаток перерастворяют в 100%-ном ацетонитриле, полученный экстракт анализируют методом жидкостной хроматографии, используя в качестве подвижной фазы смесь ацетонитрила и воды при их изменяющемся соотношении от 60:40 об.% до 90:10 об.% соответственно, а также 100%-ный ацетонитрил в градиентном режиме, который осуществляют при хроматографии путем подачи вначале в течение 2,5 мин подвижной фазы, состоящей из смеси ацетонитрила и воды в соотношении 60:40 об.%, затем путем увеличения в течение 9,5 мин в подвижной фазе ацетонитрила до 90 об.%, а также дальнейшего увеличения его концентрации в течение 8 мин до 100 об.% и пропускания такой подвижной фазы в течение еще 4,5 мин, с последующим снижением объемного количества ацетонитрила за 0,5 мин до 60 об.% и пропусканием такой подвижной фазы через колонку в течение 5 мин, а количество бенз(а)пирена в моче устанавливают по градуировочному графику.
2. Способ по п.1, отличающийся тем, что скорость потока подвижной фазы составляет 1,5 мл/мин.
3. Способ по п.1, отличающийся тем, что жидкостную хроматографию проводят на жидкостном хроматографе «Agilent 1200» с флуориметрическим детектором при длине волны возбуждения 265 нм и длине волны эмиссии 412 нм при температуре колонки 28°С.
| KANG H | |||
| et | |||
| al | |||
| J | |||
| Vet | |||
| Sci | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ газохроматографического анализа полиядерных соединений | 1988 |
|
SU1539654A1 |
| Способ определения 3,4-бензпирена в водной среде | 1981 |
|
SU1010522A1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 3,4-БЕНЗПИРЕНА | 0 |
|
SU343212A1 |
| Способ отбора пробы продуктов сгорания и подготовки ее к анализу на бензапирен | 1987 |
|
SU1511627A2 |
| Способ отбора пробы продуктов сгорания и подготовки ее к анализу на бензапирен | 1987 |
|
SU1435991A1 |
| CN 102087280 A, 08.06.2011 | |||
| KR 20030084841 A, 01.11.2003. | |||

