Область техники, к которой относится изобретение
Настоящее изобретение относится к лекарственному средству и, конкретно, к 1-замещенному производному тетрагидроизохинолина, которое является применимым в качестве активного ингредиента фармацевтической композиции для предупреждения и/или лечения боли, абдоминальных симптомов, спастической констипации и синдрома раздраженной толстой кишки.
Уровень техники
Боль является важным механизмом биологической защиты, которая отражает появление любого заболевания в организмах. Когда боль или дизестезия все же сохраняется, даже после того, как тканевое повреждение или заболевания, являющиеся причиной появления боли, подвергали лечению, такое состояние идентифицируют как заболевание. Боль в общем классифицируют на ноцицептивную боль и невропатическую боль. Ноцицептивная боль включает боль, вызванную воспалением ткани, сдавливанием нерва, индуцированным раком или тому подобное (воспалительную боль, раковую боль и т.д.). Нестероидные противовоспалительные лекарственные средства (NSAID) или опиоиды являются терапевтически эффективными для лечения ноцицептивной боли.
С другой стороны, невропатическая боль является хронической болью, вызванной повреждением или сдавливанием нервной ткани, или тому подобным. Симптомы невропатической боли включают неприятную дизестезию, такую как непрерывная или внезапная самопроизвольная боль, онемение, ощущение жжения, боль, испытываемую, когда пациента как бы разрезают на маленькие кусочки, и кинжальная боль; состояние, которое является болезненной реакцией на обычно неболезненный слабый стимул (гипералгезия); боль вследствие стимула, который обычно не вызывает боль (аллодиния), такая как вызванная контактированием с одеждой или изменениями в температурах, и тому подобное. Конкретные заболевания невропатической боли включают невралгию тройничного нерва, синдром комплексной региональной боли, синдром после спинальной хирургии, фантомную боль конечностей, боль после повреждения плечевого сплетения, боль после повреждения спинного мозга, боль после удара, болезненную диабетическую невропатию, постгерпетическую невралгию, ВИЧ-индуцированную невропатию и, кроме того, некоторые случаи раковой боли и боли поясницы, на которые не оказывают достаточные аналгезирующие действия опиоиды, в дополнении к невропатии, индуцированной противораковыми лекарственными средствами и лекарственными средствами против ВИЧ.
Невропатическая боль известна как боль, при лечении которой NSAID или опиоидами, которые являются эффективными при лечении ноцицептивной боли, трудно достичь терапевтической эффективности. При практической лекарственной терапии облегчение боли выполняют коноплей, кремом капсаицина или интраспинальным введением опиоидов, а также введением антидепрессантов (дулоксетина, амитриптилина и т.д.), антиэпилептических лекарственных средств (прегабалина, карбамазепина и т.д.) или местных аналгетиков (мексилетина и т.д.). К сожалению, действия этих лекарственных средств ограничены, поскольку многие невропатические боли развиваются посредством перекрытия многочисленных патогенных причин, и отдельные пациенты имеют разные фоны заболеваний. Кроме того, имеются также проблемы, связанные с побочными действиями, свойственными отдельным лекарственным средствам. В результате этого, существует огромная потребность в средстве против невропатической боли, которое является более сильнодействующим и имеет более широкий спектр аналгезирующего действия и более слабые побочные действия.
Синдром раздраженной толстой кишки (IBS) является синдромом, который вызывают абдоминальные симптомы, такие как абдоминальная боль и вздутие живота, и нарушения стула, такие как диарея или острый позыв к дефекации и констипация или трудность в дефекации вследствие дисфункции нижнего пищеварительного тракта около толстой кишки, несмотря на отсутствие органического изменения, такого как воспаление и опухоль. В зависимости от доминирующих особенностей стула, IBS в общем субклассифицируют на IBS типа диареи (IBS-D), IBS типа констипации (IBS-C) и IBS смешанного типа (IBS-M) с чередующейся диареей и констипацией (Gastroenterology 130: 1377-90, 1480-91 (2006)). В качестве средств для лекарственной терапии для IBS здесь можно указать антихолинергические лекарственные средства для абдоминальной боли, трициклические антидепрессанты (ТСА) для улучшения (повышения) пониженного порога болевой чувствительности пищеварительного тракта и в случае нарушения перистальтики кишечника, средства против диареи или кишечные лекарственные средства против диареи и слабительные соли против констипации, которые являются только средствами аллопатических терапий и являются также ненадежными в их действиях (синдром раздраженной толстой кишки ~ связь между головным мозгом и кишечником (ISBN4-521-67671-5, 2006)).
В качестве лекарственных средств, которые за последнее время привлекали внимание, можно указать алосетрон, который является антагонистом 5-НТ3-рецептора, и тегасерод, который является агонистом 5-НТ4-рецептора, их применяют для IBS-D и IBS-C соответственно. Однако применение алосетрона ограничено вследствие распространенности констипации у 30-35% пациентов в сочетании с серьезными побочными действиями ишемического колита (включая смертельные случаи), даже хотя он проявляет относительно высокую степень улучшения, от 40% до 60%, абдоминальных симптомов и диареи (Drug Today 36: 595-607 (2000), FDA information about lotronex, GlaxoSmithKline press release). Кроме того, указывается, что тегасерол обладает незначительным действием на абдоминальные симптомы вследствие незначительных ослабляющих констипацию действий, которые могут привести к риску появления тахифилаксии (феномен индуцирования резистенции к лекарственному средству после многократно введенных доз на протяжении короткого периода времени) (Clinical Therapeutics 25: 1952-1974 (2003)). Кроме того, применение тегасерола сильно ограничивается также с точки зрения побочных действий вследствие имеющихся отрицательных действий на сердечно-сосудистую систему (FDA information about zelnorm, Novartis press release).
Известно, что опиоиды, такие как морфин, которые обычно применяли в качестве ослабляющих боль лекарственных средств, вызывают серьезную дисфункцию пищеварительного тракта, включающую констипацию, которую называют опиоидной дисфункцией пищеварительного тракта (OBD). Среди симптомов OBD имеется очень быстрое появление констипации без проявления резистентности к лекарственному средству, в отличие от других побочных действий на центральную нервную систему, индуцированных опиоидами, поэтому необходимо применять подходящие дозы, чтобы разрешить данную проблему (American J. Surgery 182: 11S-18S (2001), Jpn. Cancer Chemother. 32: 1377-1383 (2005)). По этой причине при лечении опиоидом, особенно пациентов с раковыми болями, существенное значение имеет комбинированное профилактическое лечение слабительным средством с начала введения опиоидного лекарственного средства, но нелегко регулировать дефикацию при помощи слабительного средства (Drugs 63: 649-671 (2003), Pharmacotherapy 22: 240-250 (2002)).
Пищеварительный тракт обеспечен независимой нервной сетью, называемой брюшной нервной системой. В брюшной нервной системе присутствуют различные типы нейронов, они являются ответственными за регулирование соответствующих функций пищеварительного тракта. Среди этих нейронов собственные первичные афферентные нейроны (IPAN) являются нейронами, которые главным образом воспринимают изменения в полости пищеварительного тракта. IPAN детектируют физические или химические изменения в полости пищеварительного тракта и передают информацию двигательным нейронам или сенсорным нейронам. Поэтому лекарственные средства, изменяющие активность IPAN, вызывают изменения в функции пищеварительного тракта, называемой перистальтикой кишечника или висцеральной перцепцией (Progress in Neurobiol. 54: 1-18 (1998)). Кроме того, на основании того факта, что Са2+-канал N-типа экспрессируется в IPAN и содействует активности IPAN (J. Comp. Neurol. 409: 85-104 (1999)), можно считать, что соединение, блокирующее Са2+-каналы N-типа, может быть применимым при функциональных заболеваниях пищеварительного тракта изменением функций пищеварительного тракта.
Кроме того, известно, что сигналы абдоминальной боли, подобно сигналам соматической боли, перемещаются в головной мозг через ганглии задних корешков спинномозговых нервов (DRG) и спинной мозг (Neurogastroentel. Motil. 16: 113-124 (2004)). Этот путь передачи сигналов гиперсенсибилизирован у пациентов с IBS, что позволяет предположить существенные проявления абдоминальных симптомов (Gut 53: 1465-1470 (2004)). Поэтому ожидается, что блокатор Са2+-канала N-типа, участвующий в этом пути передачи болевого сигнала, может быть эффективным терапевтическим агентом против абдоминальных симптомов IBS. В действительности, описано, что габапентин или прегабалин, который является лигандом для α2δ-субъединицы Са2+-канала, оказывает аналгезирующие действия на животных моделях гиперсенсибилизации абдоминальной боли (J. Pharmacol. Exp. Ther. 295: 162-167 (2000), Anesthesiology 98: 729-733 (2003)).
В клетках имеется много типов Са2+-зависимых функциональных белков, и изменения в концентрации внутриклеточного Са2+ играют важную роль в проявлении или регуляции различных физиологических функций, таких как нейрональная жизнеспособность, синаптическая пластичность и экспрессия гена. Среди Са2+-каналов, присутствующих на клеточной мембране, канал, использующий мембранный потенциал в качестве триггера при открытии канала, называют потенциал-зависимым Са2+-каналом (VDCC), который состоит в основном из α1-субъединицы, образующей тело канала, β-субъединицы, регулирующей уровень экспрессии α1-субъединицы или функции канала, и α2δ-субъединицы (Trends Neurosci. 21 148-154 (1998)). Ca2+-каналы классифицируют на Ca2+-каналы с высоким потенциалом порога активации, такие как каналы L-типа (α1S, C, D и F), P/Q-типа (α1A), N-типа (α1B) и R-типа (α1E); и Ca2+-каналы с низким потенциалом порога активации, такие как каналы T-типа (α1G, H, I), в зависимости от типа α1-субъединицы и потенциала порога активации (Rev. Physiol. Biochem. Pharmacol. 139: 33-87 (1999)).
Среди Са2+-каналов с высоким потенциалом порога активации Са2+-каналы P/Q-, N- и R-типа присутствуют в синаптических терминалях нейронов и служат в качестве триггера секреции нейромедиаторов. В частности, Ca2+-канал N-типа значительно экспрессируется в ганглии задних корешков спинномозговых нервов (DRG) (J. Neurosci. 15: 4315-4327 (1995)), который является скоплением тел клеток сенсорных нейронов, или заднем роге спинного мозга (J. Neurosci. 18: 6319-6330 (1998)), который является областью синаптического выступа сенсорных нейронов. Кроме того, задний рог спинного мозга крысиных моделей невропатической боли обнаруживал повышенную экспрессию Са2+-канала N-типа синхронно с развитием гиперплазии (Exp. Brain Res. 147: 456-463 (2002)). На основании этих фактов считается, что Са2+-канал N-типа играет роль триггера, который передает избыток болевых сигналов в головной мозг.
На основании последних обзоров, показывающих, что блокирующий Ca2+-канал N-типа селективный пептид, ω-конотоксин (ω-CTx), проявляет аналгезирующие действия широкого спектра на животных моделях ноцицептивной, воспалительной и невропатической боли, соответственно (J. Pharmacol. Exp. Ther. 279: 1243-1249 (1996), J. Pharmacol. Exp. Ther. 287: 232-237 (1998), J. Pharmacol. Exp. Ther. 269: 1117-1123 (1994)) и что и не имеет место невропатическая боль у мышей с дефицитом α1B (EMBO J. 20: 2349-2356 (2001)), было предположено, что Ca2+-канал N-типа принимает глубинное участие в патогенезе невропатической боли. Действительно, было описано, что постоянное спинальное введение циконотида (ω-конотоксин MVIIA:ω-CTxMVIIA) посредством имплантируемого насоса улучшает состояние гипералгезии и аллодинии у невосприимчивых к морфину пациентов с невропатической болью (Clin. J. Pain 13: 256-259 (1997)). Кроме того, было показано, что габапентин или прегабалин, часто применяемый в качестве средства, действующего против невропатической боли, связывается с высокой аффинностью с α2δ-субъединицей Са2+-канала, тем самым проявляя аналгезирующие действия (J. Pharm. Sci. 100: 471-486 (2006)). На основании вышеуказанных открытий предполагается, что блокатор Ca2+-канала N-типа является превосходным терапевтическим средством против боли, особенно невропатической боли. Кроме того, на основании факта, что Са2+-канал N-типа принимает участие в гиперактивности нейронов, клеточной гибели и тому подобном, предполагается, что блокатор Са2+-канала N-типа является применимым для предупреждения или лечения состояний или заболеваний, связанных с активацией Са2+-канала N-типа, помимо вышеуказанной боли. В общем, считается, что соединение, обладающее действием, блокирующим Са2+-канал N-типа, может быть применимым при различных болях, таких как невропатическая боль и ноцицептивная боль, головные боли, такие как мигрень и кластерная головная боль, заболеваниях центральной нервной системы, таких как тревога, депрессия, эпилепсия, церебральный удар и синдром усталых ног, заболеваниях пищеварительной системы, таких как абдоминальная боль и синдром раздраженной толстой кишки, и заболеваниях мочевых путей, таких как гиперактивный мочевой пузырь и интерстициальный цистит.
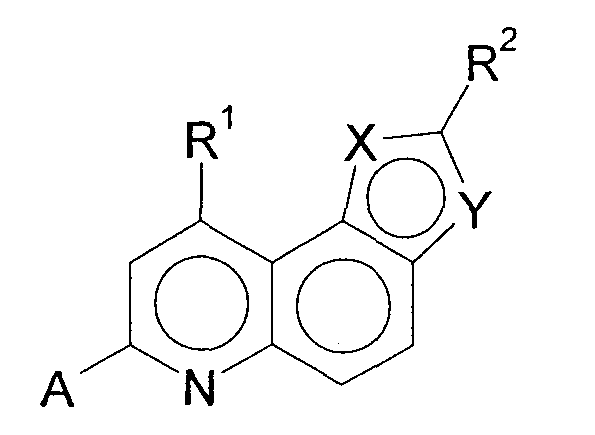
К настоящему времени описаны соединения, блокирующие Са2+-каналы N-типа. Например, было описано, что нижеследующие производные бензазепина обладают блокирующим действием на Са2+-каналы N-типа и являются применимыми в качестве средства для предупреждения и/или лечения церебрального инфаркта, преходящего нарушения мозгового кровообращения, энцефаломиелопатии после хирургической операции на сердце, васкулярных нарушений спинного мозга, индуцированной стрессом гипертензии, невроза, эпилепсии, астмы, частого мочеиспускания и глазных заболеваний, или в качестве лекарственных средств против боли (патентный документ 1).
[Химическое соединение 1]

(См. в указанном выше документе значения для символов в формуле).
Однако не имеется конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению.
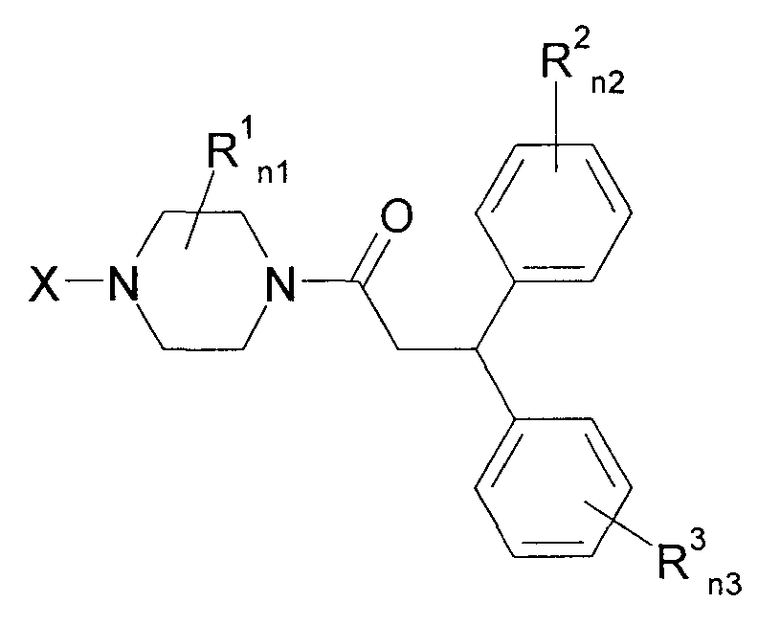
Далее, было описано, что нижеследующие производные диарилалкена или диарилалкана обладают блокирующим действием на Са2+-каналы N-типа и являются применимыми для лечения боли, инфаркта головного мозга, церебральных нарушений, вызванных острой ишемией после начала внутримозгового кровоизлияния, болезни Альцгеймера, связанной со СПИД деменции, болезни Паркинсона, прогрессирующих дегенеративных заболеваний головного мозга, неврологических нарушений, вызванных повреждением головы, бронхиальной астмы, нестабильной стенокардии, воспалительных заболеваний раздраженной толстой кишки и синдромов отмены чрезмерного употребления лекарственных средств (патентный документ 2).
[Химическое соединение 2]

(См. в указанном выше документе значения для символов в формуле).
Однако не имеется конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению.
Далее, было описано, что нижеследующие трициклические гетероароматические соединения обладают блокирующим действием на Са2+-каналы N-типа и являются применимыми в качестве лекарственного средства, особенно аналгезирующего средства (патентный документ 3)
[Химическое соединение 3]
(См. в указанном выше документе значения для символов в формуле).
Однако не имеется конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению.
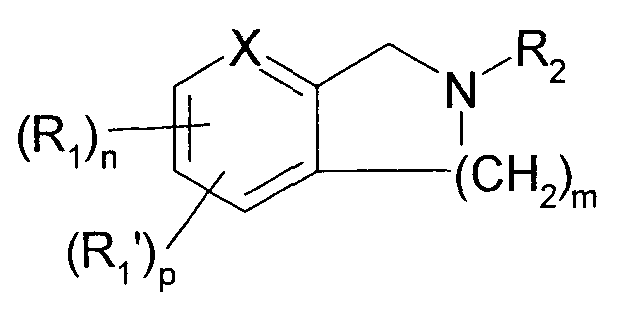
Далее, было описано, что нижеследующие замещенные производные пиперазина обладают блокирующим действием на Са2+-каналы N-типа и являются применимыми для лечения церебрального удара, боли, тревоги, депрессии, желудочно-кишечных нарушений, нарушения мочеполовой системы, сердечно-сосудистого нарушения, эпилепсии, диабета и рака (патентный документ 4).
[Химическое соединение 4]
(См. в указанном выше документе значения для символов в формуле).
Однако не имеется конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению.
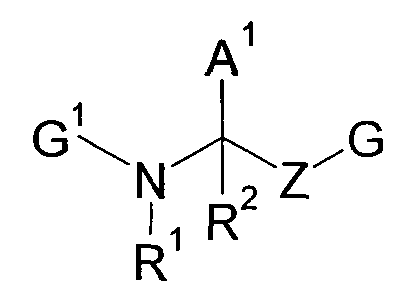
Далее, было описано, что нижеследующие азациклосоединения являются применимыми для лечения или предупреждения заболеваний, связанных с потоком ионов натрия канала сенсорных нейронов, например боли, такой как хроническая и острая боль, аллергических болезней, таких как заболевания мочевого пузыря и синдром раздраженной толстой кишки, и демиелирующих заболеваний (патентный документ 5).
[Химическое соединение 5]
(См. в указанном выше документе значения для символов в формуле).
Однако не имеется конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению.
Далее, было описано, что нижеследующие соединения обладают ингибирующей фарнезилпротеинтрансферазу активностью и являются применимыми в качестве противораковых агентов (патентный документ 6).
[Химическое соединение 6]
(См. в указанном выше документе значения для символов в формуле).
Однако не имеется конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению. Кроме того, не имеется описания или предположения об их блокирующем действии на Са2+-каналы N-типа, их действии на боль, включающую невропатическую боль, и на заболевания пищеварительной системы, включающие синдром раздраженной толстой кишки.
Далее, было описано, что нижеследующие соединения обладают антиаритмическим действием (непатентный документ 1).
[Химическое соединение 7]

(См. в указанном выше документе значения для символов в формуле).
Однако English Abstract, относящийся к вышеуказанному документу, не содержит конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению. Кроме того, не имеется описания или предположения об их блокирующем действии на Са2+-каналы N-типа, их действии на боль, включающую невропатическую боль, и заболевания пищеварительной системы, включающие синдром раздраженной толстой кишки.
Далее, было описано, что нижеследующие соединения обладают антиаритмическим действием (непатентный документ 2).
[Химическое соединение 8]
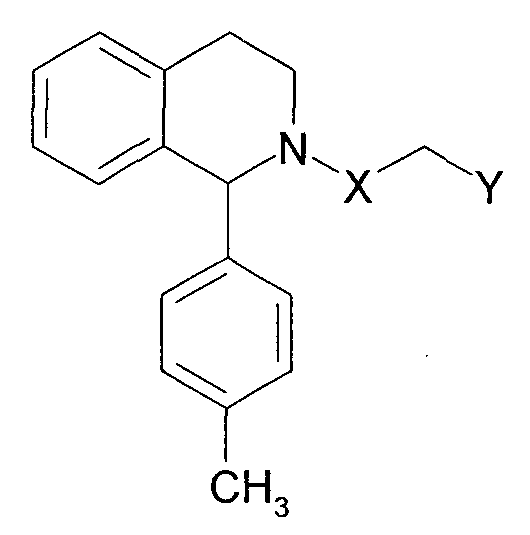
(См. в указанном выше документе значения для символов в формуле).
Однако English Abstract, относящийся к вышеуказанному документу, не содержит конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению. Кроме того, не имеется описания или предположения об их блокирующем действии на Са2+-каналы N-типа, их действии на боль, включающую невропатическую боль, и заболевания пищеварительной системы, включающие синдром раздраженной толстой кишки.
Далее, было описано, что нижеследующие соединения обладают блокирующим действием на Са2+-каналы и являются применимыми в качестве гипотензивных средств и антиаритмических средств (непатентный документ 3).
[Химическое соединение 9]
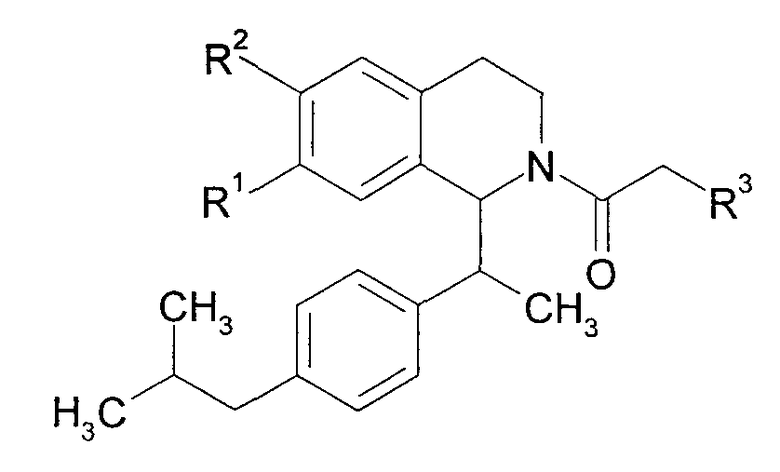
(См. в указанном выше документе значения для символов в формуле).
Однако English Abstract, относящийся к вышеуказанному документу, не содержит конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению. Кроме того, не имеется описания или предположения об их блокирующем действии на Са2+-каналы N-типа, их действия на боль, включающую невропатическую боль, и заболевания пищеварительной системы, включающие синдром раздраженной толстой кишки.
Далее, было описано, что нижеследующие соединения обладают блокирующим действием на Са2+-каналы, блокирующим действием на Na+-каналы и ингибирующей калмодулин активностью и возможно являются применимыми в нейрозащитной терапии (непатентные документы 4 и 5).
[Химическое соединение 10]

(См. в указанном выше документе значения для символов в формуле).
Однако не имеется конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению.
Далее, были описаны нижеследующие соединения в качестве антагониста рецептора орексина-2 (непатентный документ 6). Кроме этого, было также высказано предположение, что рецептор орексина-2 принимает участие в передаче ноцицептивных стимулов.
[Химическое соединение 11]
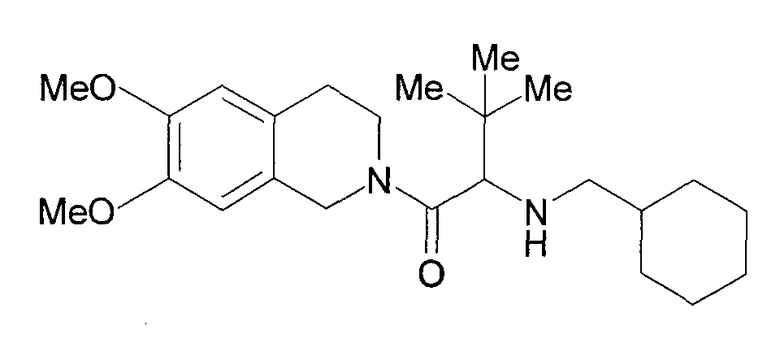
(Ме в формуле представляет собой метил).
Однако не имеется конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению.
В качестве других ссылок, в которых описаны соединения, имеющие скелет тетрагидроизохинолина, имеются патентные документы 7-9. Однако эти документы не содержат конкретного описания 1-замещенного производного тетрагидроизохинолина, которое относится к настоящему изобретению.
[Патентный документ 1] JP-A-2002-363163.
[Патентный документ 2] Бюллетень международной публикации No. WO 03/018538.
[Патентный документ 3] Бюллетень международной публикации No. WO 2004/089950.
[Патентный документ 4] Бюллетень международной публикации No. WO 2005/021523.
[Патентный документ 5] Бюллетень международной публикации No. WO 2005/005392.
[Патентный документ 6] Выложенная публикация заявки на европейский патент No. EP 0696593.
[Патентный документ 7] Бюллетень международной публикации No. WO 01/85693
[Патентный документ 8] Бюллетень международной публикации No. WO 02/079189.
[Патентный документ 9] Бюллетень международной публикации No. WO 03/082828.
[Непатентный документ 1] Fudan University Journal of Medical Science, 1987, 14(1), 15-20.
[Непатентный документ 2] Fudan University Journal of Medical Science, 1989, 16(1), 71-74.
[Непатентный документ 3] Journal of China Pharmaceutical University, 1993, 24(4), 193-201.
[Непатентный документ 4] Biological & Pharmaceutical Bulletin, 2000, 23(3), 375-378.
[Непатентный документ 5] Neurochemical Research, 2003, 28(12), 1813-1818.
[Непатентный документ 6] Bioorganic & Medicinal Chemistry Letters, 2003, 13(24), 4497-4499.
Описание изобретения
Проблема, которая должна быть разрешена изобретением
Задачей настоящего изобретения является обеспечение лекарственного средства, обладающего селективным блокирующим действием на Са2+-каналы N-типа, и особенно соединения, применимого в качестве активного ингредиента фармацевтической композиции для предупреждения и/или лечения боли и синдрома раздраженной толстой кишки.
Соединение настоящего изобретения имеет структурную характеристику, по которой в формуле (1) по меньшей мере один из R1a и R1b представляет собой заместитель, отличающийся от -Н, и R22 представляет собой гидроксилсодержащий заместитель. Кроме того, соединение настоящего изобретения обладает фармакологическими свойствами, проявляющимися в том, что оно обладает действием, блокирующим Са2+-каналы N-типа, действием против ноцицептивной боли, действием против невропатической боли, действием, ингибирующим абдоминальную боль, и действием, улучшающим состояние констипации, индуцированной опиоидами.
Способ разрешения проблемы
В результате интенсивных изучений соединений, обладающих селективным блокирующим действием на Са2+-каналы N-типа, авторами настоящего изобретения обнаружено, что 1-замещенное производное тетрагидроизохинолина настоящего изобретения обладает действием, селективно блокирующим Са2+-каналы N-типа, действием против ноцицептивной боли, действием против невропатической боли, действием, ингибирующим абдоминальную боль, и действием, улучшающим состояние констипации, индуцированной опиоидами. Настоящее изобретение было завершено на основании этих полученных данных.
То есть настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли и фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
[1]
Соединение формулы (I)
[Химическое соединение 12]

где символы в формуле имеют следующие значения:
R1a и R1b являются одинаковыми или разными и представляют собой -H, C1-6алкил, который может быть замещен, циклоалкил, который может быть замещен, арил, который может быть замещен, или ароматический гетероцикл, который может быть замещен, при условии, что оба из R1a и R1b не могут быть -H, и R1a и R1b, взятые вместе с атомом углерода, к которому они присоединены, могут представлять собой циклоалкил, который может быть замещен,
R3a, R3b, R4a и R4b являются одинаковыми или разными и представляют собой -H или C1-6алкил,
R5, R6, R7 и R8 являются одинаковыми или разными и представляют собой -H, C1-6алкил, который может быть замещен, -O-(C1-6алкил), который может быть замещен, циано, карбамоил, который может быть замещен одним или двумя C1-6алкилами, или галоген, и любые две соседние группы R5, R6, R7 и R8, взятые вместе, могут образовывать -O-CH2-O- или -O-(CH2)2-O-,
R11, R12, R13, R14, R15 и R16 являются одинаковыми или разными и представляют собой -H или C1-6алкил,
R21 представляет собой -H, C1-6алкил, который может быть замещен, или циклоалкил, который может быть замещен,
R22 представляет собой
(1) циклоалкил, который замещен одной или несколькими группами, выбранными из группы, состоящей из -OH и -CH2OH, и который может быть дополнительно замещен;
(2) C1-8алкил, замещенный одним или двумя -OH, где C1-8алкил может иметь дополнительный заместитель, и одна или две метиленовые группы (-CH2-), содержащиеся в этой алкильной цепи, могут быть заменены на -O-; или
(3) C1-6алкил, замещенный циклоалкилом, который замещен одной или несколькими группами, выбранными из группы, состоящей из -OH и -CH2OH, и который может быть дополнительно замещен, где C1-6алкил может быть замещен -OH, и одна или две метиленовые группы (-CH2-), содержащиеся в этой алкильной цепи, могут быть заменены на -O-;
n и m являются одинаковыми или разными и равны 0 или 1,
R12 и R21, взятые вместе, могут образовывать метилен, этилен или триметилен и в этом случае R11 может представлять собой -OH, или
R21 и R22, взятые вместе с атомом азота, к которому они присоединены, могут образовывать азетидин, пирролидин, пиперидин, азепан, азокан, морфолин, тетрагидроизохинолин или тиоморфолин, которые замещены -OH или C1-6алкилом, замещенным -OH; или его фармацевтически приемлемая соль.
[2]
Соединение согласно [1], где m равно 0, n равно 0 и каждый из R1a, R3a, R3b, R4a, R4b, R11, R12 и R21 представляет собой -Н, или его фармацевтически приемлемая соль.
[3]
Соединение согласно [2], где R1b представляет собой изопропил, метоксиметил, фенил, 2-(трифторметил)бензил или циклогексил, или его фармацевтически приемлемая соль.
[4]
Соединение согласно [2] или [3], где R5, R6, R7 и R8 являются одинаковыми или разными и независимо выбраны из группы, состоящей из -Н, метила, этила, метокси и фтора, или его фармацевтически приемлемая соль.
[5]
Соединение согласно [2], [3] или [4], где R22 представляет собой 2-гидроксипропан-1-ил, 2-гидрокси-3-метоксипропан-1-ил или (1-гидроксициклогексил)метил, или его фармацевтически приемлемая соль.
[6]
Соединение согласно [1], которое представляет собой
1-[({2-[(1S)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(2S)-1-({2-[(1S)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)-3-метоксипропан-2-ол,
1-({[2-(1(1S)-изопропил-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
(2R)-1-({2-[(1S)-8-метокси-1-фенил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)пропан-2-ол,
1-[({2-[(1R)-7-этил-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(2S)-1-метокси-3-[(2-oxo-2-{1(1S)-[2-(трифторметил)бензил]-3,4-дигидроизохинолин-2(1H)-ил}этил)амино]пропан-2-ол,
1-({[3-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-3-оксопропил]амино}метил)циклогексанол,
(2R)-1-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}пропан-2-ол,
(2R)-1-[(2-оксо-2-{1-[2-(трифторметил)фенил]-3,4-дигидроизохинолин-2(1H)-ил}этил)амино]пропан-2-ол,
(2S)-1-{[2-(1-циклогексил-7-метил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}-3-метоксипропан-2-ол,
(2R)-1-({2-оксо-2-[(1S)-1-фенил-3,4-дигидроизохинолин-2(1H)-ил]этил}амино)пропан-2-ол,
1-[({2-[7-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[7-этил-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-({[2-(1-изопропил-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[5-метокси-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[1-(метоксиметил)-6-метил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(1S,2S)-2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}-1-фенилпропан-1,3-диол,
1-({(2R)-2-[(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)карбонил]пирролидин-1-ил}метил)циклогексанол,
(2R)-1-{[2-(1-циклогексил-1-метил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}пропан-2-ол,
1-({[2-(3',4'-дигидро-2'H-спиро[циклогексан-1,1'-изохинолин]-2'-ил)-2-оксоэтил]амино}метил)циклогексанол,
(2R)-1-[(2-оксо-2-{1-[2-(трифторметокси)фенил]-3,4-дигидроизохинолин-2(1H)-ил}этил)амино]пропан-2-ол,
(2R)-1-{[2-(1-циклогексил-7-этил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}пропан-2-ол,
1-({[2-(6-фтор-1-изопропил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1,1-дициклопропил-2-({2-[6-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)этанол,
1-({[2-(1-трет-бутил-8-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-({[2-(1-изопропил-6-метил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-({[2-(6-фтор-1-пропил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[1-(метоксиметил)-7-метил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-({[2-(5-фтор-1-пропил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[5-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[8-метокси-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[1-(этоксиметил)-7-метил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол или
(1R,2S)-2-({2-[(1R)-1-(2-метоксифенил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)циклопентанол или его фармацевтически приемлемая соль.
[7]
Фармацевтическая композиция, содержащая соединение [1] или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
[8]
Блокатор Са2+-канала N-типа, содержащий соединение [1] или его фармацевтически приемлемую соль.
[9]
Фармацевтическая композиция для предупреждения или лечения боли, невропатической боли, абдоминального симптома, спастической констипации, индуцированной опиоидом констипации, синдрома раздраженной толстой кишки или синдрома раздраженной толстой кишки типа констипации, содержащая соединение [1] или его фармацевтически приемлемую соль.
[10]
Фармацевтическая композиция согласно [9], которая является фармацевтической композицией для предупреждения или лечения боли.
[11]
Фармацевтическая композиция согласно [10], которая является фармацевтической композицией для предупреждения или лечения невропатической боли.
[12]
Фармацевтическая композиция согласно [9], которая является фармацевтической композицией для предупреждения или лечения абдоминального симптома.
[13]
Фармацевтическая композиция по [9], которая является фармацевтической композицией для предупреждения или лечения спастической констипации.
[14]
Фармацевтическая композиция по [13], которая является фармацевтической композицией для предупреждения или лечения индуцированной опиоидом констипации.
[15]
Фармацевтическая композиция по [9], которая является фармацевтической композицией для предупреждения или лечения синдрома раздраженной толстой кишки.
[16]
Фармацевтическая композиция по [15], которая является фармацевтической композицией для предупреждения или лечения синдрома раздраженной толстой кишки типа констипации.
[17]
Фармацевтическая композиция, содержащая соединение [1] или его фармацевтически приемлемую соль и опиоид в качестве активных ингредиентов.
[18]
Фармацевтическая композиция, содержащая соединение [1] или его фармацевтически приемлемую соль в качестве активного ингредиента, где композицию применяют в комбинации с опиоидом.
[19]
Применение соединения [1] или его фармацевтически приемлемой соли для изготовления фармацевтической композиции для предупреждения или лечения боли, невропатической боли, абдоминального симптома, спастической констипации, индуцированной опиоидом констипации, синдрома раздраженной толстой кишки или синдрома раздраженной толстой кишки типа констипации.
[20]
Соединение [1] для применения в качестве активного ингредиента фармацевтической композиции для предупреждения или лечения боли, невропатической боли, абдоминального симптома, спастической констипации, индуцированной опиоидом констипации, синдрома раздраженной толстой кишки или синдрома раздраженной толстой кишки типа констипации.
[21]
Способ предупреждения или лечения боли, невропатической боли, абдоминального симптома, спастической констипации, индуцированной опиоидом констипации, синдрома раздраженной толстой кишки или синдрома раздраженной толстой кишки типа констипации, содержащий введение пациенту эффективного количества соединения [1] или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к фармацевтической композиции для лечения боли, в определенном варианте осуществления невропатической боли; абдоминальных симптомов; спастической констипации, в определенном варианте осуществления индуцированной опиоидом констипации; или синдрома раздраженной толстой кишки, в определенном варианте осуществления синдрома раздраженной толстой кишки типа констипации, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, т.е. фармацевтической композиции для предупреждения и/или лечения боли, в определенном варианте осуществления невропатической боли; абдоминальных симптомов; спастической констипации, в определенном варианте осуществления индуцированной опиоидом констипации; или синдрома раздраженной толстой кишки, в определенном варианте осуществления синдрома раздраженной толстой кишки типа констипации, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Далее, настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления фармацевтической композиции для лечения боли, в определенном варианте осуществления невропатической боли; абдоминальных симптомов; спастической констипации, в определенном варианте осуществления индуцированной опиоидом констипации; или синдрома раздраженной толстой кишки, в определенном варианте осуществления синдрома раздраженной толстой кишки типа констипации, и способу лечения боли, в определенном варианте осуществления невропатической боли; абдоминальных симптомов; спастической констипации, в определенном варианте осуществления индуцированной опиоидом констипации; или синдрома раздраженной толстой кишки, в определенном варианте осуществления синдрома раздраженной толстой кишки типа констипации, содержащему введение пациенту эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Действие патента
Соединение настоящего изобретения можно применять в виде фармацевтической композиции для предупреждения и/или лечения различных болей, таких как невропатическая боль и ноцицептивная боль, головные боли, такие как мигрень и кластерная головная боль, заболеваний центральной нервной системы, таких как тревога, депрессия, эпилепсия, церебральный удар и синдром усталых ног, абдоминальных симптомов, таких как абдоминальная боль и вздутие живота, нарушений стула, таких как диарея и констипация, заболеваний пищеварительной системы, таких как синдром раздраженной толстой кишки, заболеваний мочевых путей, таких как гиперактивный мочевой пузырь и интерстициальный цистит, и т.д.
Лучший способ осуществления изобретения
В дальнейшем настоящее изобретение будет описано подробно.
В определениях настоящего описания “С1-6алкил“ означает неразветвленный или разветвленный алкил, имеющий 1-6 атомов углерода, и примеры его включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и тому подобное. “С1-8алкил“ означает неразветвленный или разветвленный алкил, имеющий 1-8 атомов углерода, и примеры его включают н-гептил, н-октил, диизопропилэтил и тому подобное, помимо вышеописанных С1-6алкилов.
“Галоген“ означает F, Cl, Br или I.
“Циклоалкил” представляет собой группу насыщенного С3-10углеводородного кольца и примеры ее включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, адамантил и тому подобное. Он включает также циклогексенил, циклооктадиенил и тому подобное, который содержит частично ненасыщенную связь. Кроме того, он включает также группы, у которых одна или две метиленовые группы в кольце заменены на -О-, например, тетрагидропиранил, тетрагидрофуранил и тому подобное. Далее, его кольцо может быть конденсировано с кольцом бензола и примеры его включают инденил, инданил, дигидронафтил и тетрагидронафтил.
“Арил” представляет собой группу от моноциклического до трициклического ароматического С6-14углеводородного кольца и примеры его включают фенил, нафтил и тому подобное.
“Ароматический гетероцикл” представляет собой группу 5- или 6-членного моноциклического гетероцикла, содержащего 1-3 гетероатома, выбранные из кислорода, серы и азота, и примеры его включают фурил, тиенил, пирролил, пиразолил, имидазолил, оксазолил, тиазолил, изотиазолил, триазинил, пиридил, пиразил, пиримидинил, пиридазинил и тому подобное.
Выражение “который может быть замещен” означает, что группа является “незамещенной” или “замещенной 1-5 заместителями, которые являются одинаковыми или разными”. Кроме того, если она имеет много заместителей, заместители могут быть одинаковыми или отличными друг от друга.
Примеры заместителя для “С1-6алкила, который может быть замещен”, в определении R1a и R1b включают -OH; -ORz; или фенил, который может быть замещен одной или несколькими одинаковыми или разными группами, выбранными из группы, состоящей из галогена, RY и -ORY. RY представляет собой C1-6алкил, который может быть замещен 1-5 атомами галогена, и Rz представляет собой C1-6алкил, который может быть замещен одной или несколькими одинаковыми или разными группами, выбранными из группы, состоящей из -O-(C1-6алкила) и -ORY (такое же определение будет применяться в контексте в дальнейшем).
Примеры заместителя для “циклоалкила, который может быть замещен”, в определении R1a и R1b включают -OH, галоген, RY и -ORY.
Примеры заместителя для “арила, который может быть замещен”, и “ароматического гетерокольца, которое может быть замещено”, в определении R1a и R1b включают -OH, галоген, RY, -ORY, -SRY, циано и циклоалкил.
Примеры заместителя для “C1-6алкила, который может быть замещен”, и “-O-(C1-6алкила), который может быть замещен”, в определении R5, R6, R7 и R8 включают -OH, галоген, -ORY и -NHCO-(C1-6алкил).
Примеры заместителя для “C1-6алкила, который может быть замещен”, в определении R21 включают -OH, галоген, -ORY и циклоалкил.
Примеры заместителя для “циклоалкила, который может быть замещен”, в определении R21 включают -OH, галоген, RY и -ORY.
“Циклоалкил, который замещен одной или несколькими группами, выбранными из группы, состоящей из -ОН и -СН2ОН, и который может быть дополнительно замещен”, в определении R22 означает, что циклоалкил имеет по меньшей мере одну или несколько одинаковых или разных групп, выбранных из группы, состоящей из -OH
и -CH2OH, в качестве заместителей и может быть дополнительно замещен другими заместителями. Примеры приемлемых дополнительных заместителей включают галоген, RY, -ORY, оксо (=O) и оксо, защищенный этиленгликолем.
“C1-8алкил, замещенный одним или двумя -OH, где C1-8алкил может дополнительно иметь заместитель и одна или две метиленовые группы (-CH2-), содержащиеся в этой алкильной цепи, могут быть заменены на -O-”, в определении R22 означает, что метиленовая группа(ы) в алкильной цепи C1-8алкила может быть заменена на -O-, и C1-8алкил имеет по меньшей мере один или два -OH в качестве заместителей и может быть дополнительно замещен другими заместителями. Примеры приемлемого дополнительного заместителя включают галоген; -ORY; циклоалкил или арил, который может быть замещен одной или несколькими одинаковыми или разными группами, выбранными из группы, состоящей из -OH, галогена, RY и -ORY.
“C1-6алкил, замещенный циклоалкилом, который замещен одной или несколькими группами, выбранными из группы, состоящей из -ОН и -СН2ОН, и который может быть дополнительно замещен, где C1-6алкил может быть замещен -OH, и одна или две метиленовые группы (-CH2-), содержащиеся в этой алкильной цепи, могут быть заменены -O-”, в определении R22 означает, что С1-6алкил может быть замещен -ОН, метиленовая группа(ы) в алкильной цепи может быть заменена -О-, и С1-6алкил имеет по меньшей мере циклоалкил, который может быть замещен, в качестве заместителя. Циклоалкил в качестве заместителя C1-6алкила имеет по меньшей мере один или несколько одинаковых или разных групп, выбранных из группы, состоящей из -OH и -CH2OH, в качестве заместителей, и может быть дополнительно замещен другими заместителями. Примеры приемлемых дополнительных заместителей включают галоген, RY, -ORY, оксо (=O) и оксо, защищенный этиленгликолем.
“Боль” означает различные боли, включающие ноцицептивную боль и невропатическую боль.
“Ноцицептивная боль” является болью, которая вызывается увеличением ноцицептивных стимулов посредством ноцицепторов, и примеры ее включают боль, вызванную повреждением ткани, боль, вызванную воспалением ткани (воспалительная боль), боль, вызванную сжатием нерва, индуцированным раком (раковая боль).
“Невропатическая боль” является хронической болью, которая вызывается повреждением или сжатием нервной ткани или тому подобным, и примеры ее включают невралгию тройничного нерва, синдром комплексной региональной боли, синдром после спинальной хирургии, фантомную боль конечностей, боль после повреждения плечевого сплетения, боль после повреждения спинного мозга, боль после удара, болезненную диабетическую невропатию, постгерпетическую невралгию, ВИЧ-индуцированную невропатию и, кроме того, некоторые случаи раковой боли и боли поясницы, на которые не оказывают достаточные аналгезирующие действия опиоиды, в дополнение к невропатии, индуцированной противораковыми лекарственными средствами и лекарственными средствами против ВИЧ.
“Абдоминальный симптом” означает абдоминальный дискомфорт, такой как абдоминальная боль и абдоминальная дистензия.
“Спастическая констипация” является констипацией, вызванной спастическим нарушением моторики пищеварительного тракта, и примеры ее включают индуцированную опиоидом констипацию и констипацию, обнаруживаемую при синдроме раздраженной толстой кишки типа констипации (IBS-C).
“Индуцированная опиоидом констипация” означает констипацию, вызванную опиоидами, такими как морфин.
“Синдром раздраженной толстой кишки” является заболеванием, которое вызывает абдоминальные симптомы, такие как абдоминальная боль и вздутие живота, и нарушения стула, такие как диарея или острый позыв к дефекации и констипация или трудность в дефекации, вследствие дисфункции нижнего пищеварительного тракта около толстой кишки, несмотря на отсутствие органических изменений, таких как воспаление и опухоль и тому подобное, и является заболеванием, которое классифицируют на IBS типа диареи (IBS-D), IBS типа констипации (IBS-C) и IBS смешанного типа (IBS-M) с чередующейся диареей и констипацией, в зависимости от состояний кишечника.
В дальнейшем будут описаны некоторые варианты осуществления настоящего изобретения.
(1) В одном варианте осуществления соединение формулы (I), в которой R1a представляет собой -Н или С1-6алкил, который может быть замещен. В другом варианте осуществления соединение, у которого R1a представляет собой -Н или метил. Еще в одном варианте осуществления соединение, у которого R1a представляет собой
-Н.
(2) В одном варианте осуществления соединение формулы (I), в которой R1b представляет собой С1-6алкил, который может быть замещен, циклоалкил, который может быть замещен, или арил, который может быть замещен. В другом варианте осуществления соединение, у которого R1b представляет собой н-пропил, изопропил, трет-бутил, метоксиметил, этоксиметил, фенил, 2-метоксифенил, 2-(трифторметил)фенил, 2-(трифторметокси)фенил, 2-(трифторметил)бензил или циклогексил. Еще в одном варианте осуществления соединение, у которого R1b представляет собой изопропил, метоксиметил, фенил, 2-(трифторметил)бензил или циклогексил.
(3) В одном варианте осуществления соединение формулы (I), в которой R1a и R1b вместе с атомом углерода, к которому они присоединены, представляют собой циклоалкил, который может быть замещен. В другом варианте осуществления соединение, у которого R1a и R1b вместе с атомом углерода, к которому они присоединены, представляют собой циклогексил.
(4) Соединение формулы (I), в которой каждый из R3a, R3b, R4a и R4b представляет собой -Н.
(5) В одном варианте осуществления соединение формулы (I), в которой m равно 0 и n равно 0 или 1. В другом варианте осуществления соединение, у которого m равно 0 и n равно 0.
(6) В одном варианте осуществления соединение формулы (I), в которой R5, R6, R7 и R8 являются одинаковыми или разными и независимо выбраны из группы, состоящей из -H, C1-6алкила, -O-(C1-6алкила) и галогена. В другом варианте осуществления, соединение, у которого R5, R6, R7 и R8 являются одинаковыми или разными и выбраны из группы, состоящей из -H, метила, этила, метокси и фтора.
(7) Соединение формулы (I), в которой каждый из R11, R12, R13, R14, R15 и R16 представляет собой -H.
(8) В одном варианте осуществления соединение формулы (I), в которой m равно 0, n равно 0, R11 представляет собой -H и R12 и R21, взятые вместе, представляют собой метилен, этилен или триметилен. В другом варианте осуществления соединение, у которого m равно 0, n равно 0, R11 представляет собой -H и R12 и R21, взятые вместе, представляют собой триметилен.
(9) Соединение, у которого R21 представляет собой -Н.
(10) В одном варианте осуществления соединение, у которого R22 представляет собой циклоалкил, замещенный одной или несколькими группами, выбранными из группы, состоящей из -OH и -CH2OH. В другом варианте осуществления, соединение, у которого R22 представляет собой циклопентил или циклогексил, замещенный одной или несколькими группами, выбранными из группы, состоящей из -OH и -CH2OH. Еще в одном варианте осуществления соединение, у которого R22 представляет собой 2-гидроксициклопентил.
(11) В одном варианте осуществления соединение, у которого R22 представляет собой C1-8алкил, который замещен одним или двумя -OH и дополнительно замещен одной или несколькими одинаковыми или разными группами, выбранными из группы, состоящей из -O-(C1-6алкила), циклоалкила и арила. В другом варианте осуществления соединение, у которого R22 представляет собой C1-8алкил, который замещен одним или двумя -ОН и дополнительно замещен одной или несколькими группами, выбранными из группы, состоящей из метокси, циклопропила и фенила. В следующем варианте осуществления соединение, у которого R22 представляет собой этил или пропил, который замещен одним или двумя -ОН и дополнительно замещен одной или несколькими группами, выбранными из группы, состоящей из метокси, циклопропила и фенила. Еще в одном дополнительном варианте осуществления, соединение, у которого R22 представляет собой 2-гидроксипропан-1-ил, 2-гидрокси-3-метоксипропан-1-ил, 1,3-дигидрокси-1-фенилпропан-2-ил или 2-гидрокси-2,2-дициклопропилэтил. Еще в одном другом варианте осуществления соединение, у которого R22 представляет собой 2-гидроксипропан-1-ил или 2-гидрокси-3-метоксипропан-1-ил.
(12) В одном варианте осуществления соединение, у которого R22 представляет собой C1-6алкил, замещенный циклоалкилом, который замещен одной или несколькими группами, выбранными из группы, состоящей из -OH и -CH2OH. В другом варианте осуществления соединение, у которого R22 представляет собой циклогексилметил, замещенный -OH. Еще в одном варианте осуществления соединение, у которого R22 представляет собой (1-гидроксициклогексил)метил.
(13) В одном варианте осуществления соединение, указываемое в (10), (11) или (12). В другом варианте осуществления соединение, указываемое в (11) или (12).
(14) Соединение, которое является комбинацией любых двух или более соединений, выбранных из группы, состоящей из (1), (2), (4), (5), (6), (7), (9) и (13).
(15) Соединение, которое является комбинацией любых двух или более соединений, выбранных из группы, состоящей из (3), (4), (5), (6), (7), (9) и (13).
(16) Соединение, которое является комбинацией любых двух или более соединений, выбранных из группы, состоящей из (1), (2), (4), (6), (8) и (13).
(17) Соединение, которое является комбинацией любых двух или более соединений, выбранных из группы, состоящей из (3), (4), (6), (8) и (13).
(18) В одном варианте осуществления соединение, которое является любым из соединений (14)-(17). В другом варианте осуществления соединение, указываемое в (14).
(19) Соединение, которое является комбинацией любых двух или более соединений (1)-(12), которые не являются несовместимыми друг с другом.
Примеры соединений, включенных настоящим соединением, включают следующие соединения:
1-[({2-[(1S)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(2S)-1-({2-[(1S)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)-3-метоксипропан-2-ол,
1-({[2-(1(1S)-изопропил-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
(2R)-1-({2-[(1S)-8-метокси-1-фенил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)пропан-2-ол,
1-[({2-[(1R)-7-этил-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(2S)-1-метокси-3-[(2-оксо-2-{1(1S)-[2-(трифторметил)бензил]-3,4-дигидроизохинолин-2(1H)-ил}этил)амино]пропан-2-ол.
В качестве другого варианта осуществления соединений, которые включены в настоящее изобретение, можно указать следующие соединения:
1-({[3-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-3-оксопропил]амино}метил)циклогексанол,
(2R)-1-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}пропан-2-ол,
(2R)-1-[(2-оксо-2-{1-[2-(трифторметил)фенил]-3,4-дигидроизохинолин-2(1H)-ил}этил)амино]пропан-2-ол,
(2S)-1-{[2-(1-циклогексил-7-метил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}-3-метоксипропан-2-ол,
(2R)-1-({2-оксо-2-[(1S)-1-фенил-3,4-дигидроизохинолин-2(1H)-ил]этил}амино)пропан-2-ол,
1-[({2-[7-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[7-этил-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-({[2-(1-изопропил-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[5-метокси-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[1-(метоксиметил)-6-метил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(1S,2S)-2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}-1-фенилпропан-1,3-диол,
1-({(2R)-2-[(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)карбонил]пирролидин-1-ил}метил)циклогексанол,
(2R)-1-{[2-(1-циклогексил-1-метил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}пропан-2-ол,
1-({[2-(3',4'-дигидро-2'H-спиро[циклогексан-1,1'-изохинолин]-2'-ил)-2-оксоэтил]амино}метил)циклогексанол,
(2R)-1-[(2-оксо-2-{1-[2-(трифторметокси)фенил]-3,4-дигидроизохинолин-2(1H)-ил}этил)амино]пропан-2-ол,
(2R)-1-{[2-(1-циклогексил-7-этил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}пропан-2-ол,
1-({[2-(6-фтор-1-изопропил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1,1-дициклопропил-2-({2-[6-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)этанол,
1-({[2-(1-трет-бутил-8-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-({[2-(1-изопропил-6-метил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-({[2-(6-фтор-1-пропил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[1-(метоксиметил)-7-метил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-({[2-(5-фтор-1-пропил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[5-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[8-метокси-1-(метоксиметил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[1-(этоксиметил)-7-метил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(1R,2S)-2-({2-[(1R)-1-(2-метоксифенил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)циклопентанол.
Соединение настоящего изобретения в некоторых случаях может существовать в форме других таутомеров или геометрических изомеров, в зависимости от типа заместителей. В настоящем описании соединение может быть описано только в одной форме изомеров, и настоящее изобретение включает эти изомеры, а также выделенные формы или их смеси.
Кроме того, соединение формулы (I) может иметь в некоторых случаях асимметричные атомы углерода и аксиальные асимметрии и соответственно оно может существовать в форме оптических изомеров, таких как R- и S-формы. Все смеси этих оптических изомеров и выделенные оптические изомеры включены в настоящее изобретение.
Далее, в настоящее изобретение включено также фармацевтически приемлемое пролекарство соединения формулы (I). “Фармацевтически приемлемым пролекарством” является соединение, имеющее группу, которую можно превратить в аминогруппу, гидроксильную группу, карбоксильную группу или тому подобное соединение настоящего изобретения сольволизом или в физиологических условиях. Примеры группы для образования пролекарства включают группы, описываемые, например, в Prog. Med., 5, 2157-2161 (1985) или “Iyakuhin no Kaihatsu (Development of Pharmaceuticals)” (Hirokawa Shoten Ltd., 1990), Vol. 7, “Bunshi Sekkei (Molecular Design)”, pp. 163-198.
Далее, соединение настоящего изобретения может образовывать кислотно-аддитивную соль или соль с основанием, в зависимости от типа заместителей, и такая соль включена в настоящее изобретение при условии, что она является фармацевтически приемлемой солью. В частности, примеры таких солей включают кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота, или с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, миндальная кислота, винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, аспарагиновая кислота и глутаминовая кислота, соли с неорганическими основаниями, такими как натриевое, калиевое, магниевое, кальциевое и алюминиевое основания, или с органическими основаниями, такими как метиламин, этиламин, этаноламин, лизин и орнитин, соли с различными аминокислотами и производными аминокислот, такими как ацетиллейцин, аммониевые соли и тому подобное.
Помимо этого, настоящее изобретение включает также различные гидраты или сольваты и кристаллические полиморфы соединения настоящего изобретения и его фармацевтически приемлемой соли. Кроме того, в настоящее изобретение включены также соединения, меченные различными радиоактивными или нерадиоактивными изотопами.
(Способы получения)
Соединение настоящего изобретения и его фармацевтически приемлемую соль можно получить посредством применения различных известных синтетических способов с использованием характеристик, основанных на его основном скелете или типе заместителей. В этом случае, в зависимости от типа функциональных групп, существует эффективный с точки зрения технологии получения способ для замены функциональной группы подходящей защитной группой (группой, которую можно легко превратить в функциональную группу) на стадии превращения исходных соединений в промежуточные продукты. Примеры таких защитных групп включают группы, описанные, например, в “Protective Groups in Organic Synthesis (3rd edition, 1999)”, edited by Greene and Wuts, и тому подобное, которые можно подходящим образом выбрать и применять в зависимости от условий реакции. Согласно такому способу, требуемое соединение можно получить введением защитной группы и проведением реакции и затем, если нужно, удалением защитной группы.
Кроме того, пролекарство соединения формулы (I) можно получить таким же способом, как в случае вышеуказанных защитных групп, проведением реакции после введения определенной группы на стадии превращения исходных соединений в промежуточные продукты или применением полученного соединения настоящего изобретения. Реакцию можно проводить посредством применения способов, известных специалисту в данной области, таких как обычная этерификация, амидирование, дегидратация и тому подобное.
Далее будут описаны репрезентативные способы получения соединения настоящего изобретения. Каждый из способов получения можно также проводить согласно ссылкам, приложенным к соответствующему описанию. Кроме того, способы получения настоящего изобретения не ограничиваются примерами, показываемыми ниже.
(Способ получения 1)
[Химическое соединение 13]
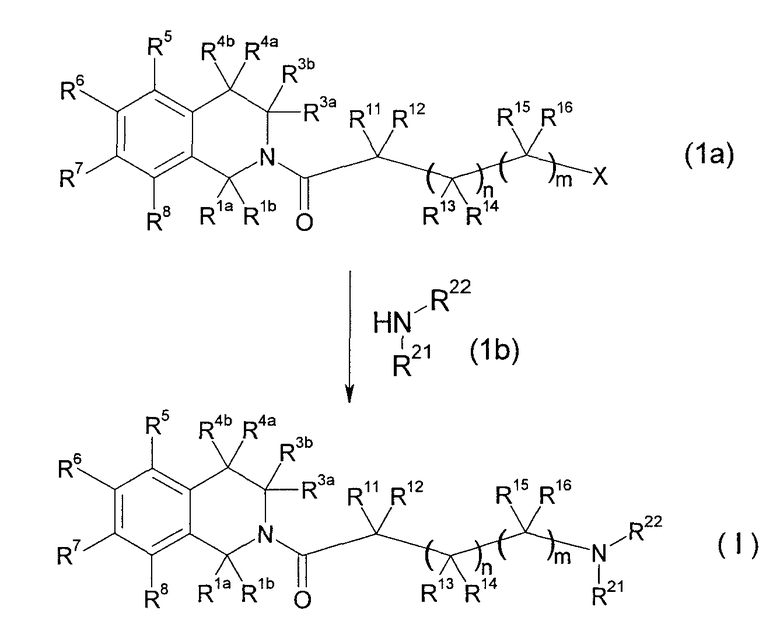
(В формуле Х представляет собой уходящую группу и другие символы имеют значения, указанные выше. Такие же значения будут применяться в дальнейшем).
Данный способ получения является способом, в котором соединение (I) настоящего изобретения получают реакцией соединения (1a), имеющего уходящую группу, с производным амина (1b).
В указанном случае примеры уходящей группы включают галоген, метансульфонилокси и пара-толуолсульфонилокси.
Реакцию можно проводить с применением соединения (1а) и соединения (1b) в эквивалентных количествах или с применением одного из них в избыточном количестве, в условиях от охлаждения до нагревания, например, при температуре от 0°С до 80°С, обычно при перемешивании в течение от 0,1 часа до 5 дней, в инертном в условиях реакции растворителе или без растворителя. Не имеется конкретного ограничения для растворителя, который можно применять в данной реакции. Примеры такого растворителя включают ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый простой эфир, тетрагидрофуран (ТГФ), диоксан и диметоксиэтан (DME); галогенированные углеводороды, такие как дихлорметан (DCM), 1,2-дихлорэтан (DCE) и хлороформ; N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), этилацетат, ацетонитрил и их смеси. В некоторых случаях для спокойного развития реакции может быть благоприятным проведение реакции в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин (DIPEA), 1,8-диазабицикло[5.4.0]-7-ундецен или N-метилморфолин, или неорганического основания, такого как карбонат калия, карбонат натрия, карбонат цезия или гидроксид калия, или в противном случае в присутствии также межфазного катализатора, такого как иодид тетрабутиламмония или 18-краун-6-эфир.
[Ссылочная литература]
S.R. Sandler and W. Karo, Editors, Organic Functional Group Preparations, 2nd edition. Vol. 1, Academic Press Inc., 1991.
Courses in Experimental Chemistry, 5th edition, edited by The Chemical Society of Japan, Vol. 14(2005), Maruzen Co., Ltd.
(Способ получения 2)
[Химическое соединение 14]
(Символы в формулах имеют указанные выше значения).
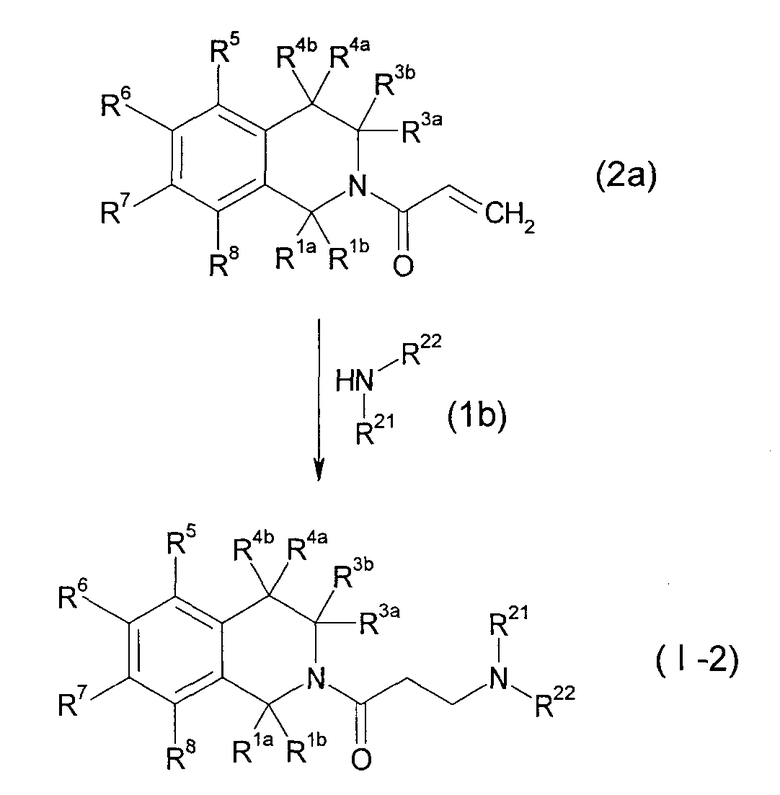
Данный способ получения является способом, в котором соединение (I-2) настоящего изобретения получают реакцией акрилового соединения (2а) с производным амина (1b).
Реакцию можно проводить с применением соединения (2а) и соединения (1b) в эквивалентных количествах или с применением одного их них в избыточном количестве в условиях от охлаждения до нагревания, например, при температуре от 0°С до 120°С, обычно при перемешивании в течение от 0,1 часа до 5 дней, в инертном в условиях реакции растворителе или без растворителя. Не имеется конкретного ограничения для растворителя, который можно применять в данной реакции. Примеры такого растворителя включают ароматические углеводороды, простые эфиры, галогенированные углеводороды, спирты, такие как метанол, этанол и 2-пропанол, N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО), этилацетат, ацетонитрил и их смеси. Когда производное амина находится в форме соли, в некоторых случаях для спокойного развития реакции может быть благоприятным проведение реакции превращения соли в свободный амин в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин (DIPEA), 1,8-диазабицикло[5.4.0]-7-ундецен или N-метилморфолин, или неорганического основания, такого как карбонат калия, карбонат натрия или гидроксид калия.
(Способ получения 3)
[Химическое соединение 15]

(Символы в формуле имеют значения, указанные выше).
Данный способ получения является способом, в котором соединение (I) настоящего изобретения получают реакцией производного тетрагидроизохинолина (3а) с производным аминокислоты (3b).
Реакцию можно проводить с применением соединения (3а) и соединения (3b) в эквивалентных количествах или с применением одного из них в избыточном количестве в присутствии агента конденсации, в условиях от охлаждения до нагревания, например, при температуре от -20°С до 60°С, обычно при перемешивании в течение от 0,1 часа до 5 дней, в инертном в условиях реакции растворителе. Не имеется конкретного ограничения для растворителя, который можно применять в данной реакции. Примеры такого растворителя включают ароматические углеводороды, галогенированные углеводороды, простые эфиры, N,N-диметилформамид (ДМФА), N-метилпирролидон, этилацетат, ацетонитрил, воду и их смеси. Примеры агента конденсации включают, но не ограничиваются перечисленным, 1-(3-диметиламинопропил)-3-этилкарбодиимид (WSC), дициклогексилкарбодиимид (DCC), 1,1'-карбонилдиимидазол (CDI), дифенилфосфорилазид и оксихлорид фосфора. В некоторых случаях для спокойного развития реакции может быть благоприятным проведение реакции с применением, например, добавки, такой как 1-гидроксибензотриазол (HOBt).
Кроме того, можно также применять способ, в котором производное аминокислоты (3b) (у которого карбоксильная группа служит в качестве места реакции) превращают в реакционноспособное производное и затем реакционноспособное производное подвергают реакции с производным тетрагидроизохинолина (3а). В этом случае примеры реакционноспособного производного включают галогенангидриды кислот, полученные реакцией с галогенирующим агентом, таким как оксихлорид фосфора или тионилхлорид, ангидриды смешанных кислот, полученные реакцией с изобутилхлорформиатом или т.п., и активные сложные эфиры, полученные конденсацией с HOBr или т.п. Реакцию между реакционноспособным производным соединения (3b) и соединением (3а) можно проводить в условиях от охлаждения до нагревания, например, при температуре от -20°С до 60°С, в инертном в условиях реакции растворителе, таком как галогенированные углеводороды, ароматические углеводороды или простые эфиры.
[Ссылочная литература]
S.R. Sandler and W. Karo, Editors, Organic Functional Group Preparations, 2nd edition. Vol. 1, Academic Press Inc., 1991.
Courses in Experimental Chemistry, 5th edition, edited by The Chemical Society of Japan, Vol. 16(2005), Maruzen Co., Ltd.
(Способ получения 4)
[Химическое соединение 16]

(В формуле Y представляет собой уходящую группу и другие символы имеют значения, указанные выше. Такие же значения будут применяться в дальнейшем).
Данный способ получения является способом, в котором соединение (I) настоящего изобретения получают реакцией производного амина (4а) с соединением (4b) и/или (4с), имеющим уходящую группу.
Реакцию можно проводить таким же способом, как в способе получения 1. Когда R21 представляет собой -Н, стадию А с применением соединения (4b) можно не включать. Кроме того, порядок проведения стадии А с применением соединения (4b) и стадии В с применением соединения (4с) не является критическим.
Помимо N-алкилирования с применением соединения (4b) или (4с), имеющего уходящую группу, в этом способе получения можно также применять N-алкилирование с применением эпоксипроизводного, соответствующего соединению (4b) или (4с), и восстановительное аминирование с применением производного альдегида, соответствующего соединению (4b) или (4с).
N-алкилирование с применением эпоксипроизводного, соответствующего соединению (4b) или (4с), можно проводить таким способом, как в способе получения 1.
Восстановительное аминирование с применением производного альдегида, соответствующего соединению (4b) или (4с), можно проводить с применением соединения (4а) и производного альдегида, соответствующего соединению (4b) или (4с), в эквивалентных количествах или с применением одного из них в избыточном количестве, при температуре от -45°С до нагревания при кипячении с обратным холодильником в присутствии восстанавливающего агента в растворителе, инертном в условиях реакции, например, при температуре от 0°С до комнатной температуры, обычно при перемешивании в течение от 0,1 часа до 5 дней. Не имеется конкретного ограничения для растворителя, который можно применять в данном изобретении. Примеры таких растворителей включают спирты, простые эфиры и их смеси. Примеры восстанавливающего агента включают цианоборгидрид натрия, триацетоксинатрийборгидрид, натрийборгидрид и тому подобное. В некоторых случаях для спокойного развития реакции может быть благоприятным проведение реакции в присутствии дегидратирующего агента, такого как молекулярные сита, или кислоты, такой как уксусная кислота, хлористоводородная кислота, или комплекса изопропоксид титана(IV). В зависимости от реакции, имеется случай, когда аминное соединение можно получить конденсацией соединения (4а) с производным альдегида, соответствующим соединению (4b) или (4С), и затем можно выделить в виде стабильного промежуточного продукта. Кроме того, реакцию можно проводить в растворителе, таком как спирты или этилацетат, в присутствии или в отсутствие кислоты, такой как уксусная кислота или хлористоводородная кислота, с применением катализатора восстановления (такого как Pd, осажденный на уголь (Pd/C), гидроксид палладия или никель Ренея) вместо обработки восстанавливающим агентом. В этом случае реакцию можно проводить в условиях от охлаждения до нагревания в атмосфере водорода при давлении от нормального до 493,46×10-6 Па (50 атмосфер).
[Ссылочная литература]
A.R. Katritzky and R.J.K. Taylor, Editors, Comprehensive Organic Functional Group Transformation II, Vol. 2, Elsevier Pergamon, 2005.
Courses in Experimental Chemistry, 5th edition, edited by The Chemical Society of Japan, Vol. 14(2005), Maruzen Co., Ltd.
Кроме того, исходное соединение (4а) данного способа получения можно получить удалением защиты у амина посредством реакции соединения (1а) с защищенным производным амина таким же способом, как в способе получения 1, или удаление защитной группы у аминогруппы посредством реакции соединения (3а) с аминозащищенным производным аминокислоты таким же способом, как в способе получения 3.
(Синтез исходных соединений)
(1) Получение соединений (1а) и (2а)
[Химическое соединение 17]
(В формуле Hal представляет собой галоген и другие символы имеют значения, указанные выше. Такие же значения будут применяться в дальнейшем).
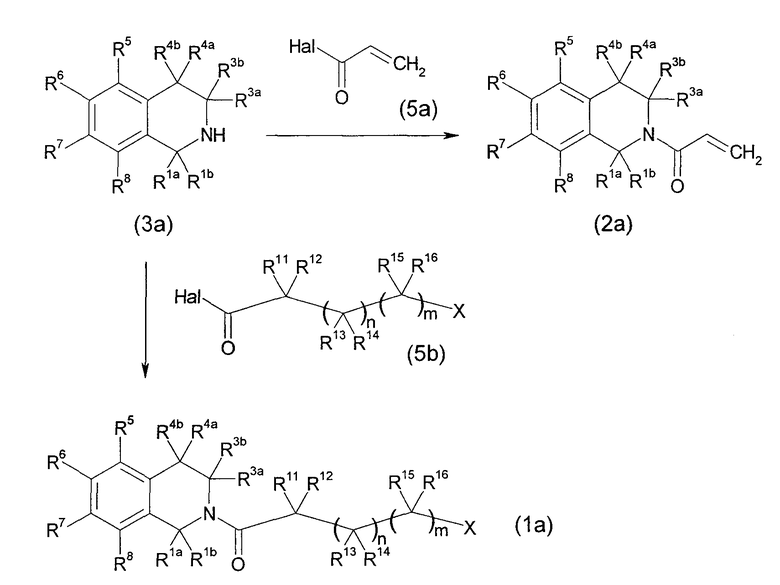
Данный способ получения является способом, в котором соединение (2а) или (1а) получают реакцией производного тетрагидроизохинолина (3а) с галогенангидридом кислоты (5а) или (5b).
Реакцию можно проводить с применением соединения (3а) и соединения (5а) или (5b) в эквивалентных количествах или с применением одного из них в избыточном количестве, в условиях от охлаждения до нагревания, например, при температуре от 0°С до 80°С, обычно при перемешивании в течение от 0,1 часа до 5 дней в инертном в условиях реакции растворителе или без растворителя. Не имеется конкретного ограничения для растворителя, который можно применять в данной реакции. Примеры такого растворителя включают ароматические углеводороды, простые эфиры, галогенированные углеводороды, этилацетат, ацетонитрил и их смеси. В некоторых случаях для спокойного развития реакции может быть благоприятным проведение реакции в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин (DIPEA), пиридин или N-метилморфолин, или неорганического основания, такого как карбонат калия, карбонат натрия, гидрокарбонат натрия или гидроксид калия, или его водного раствора, или в присутствии 0,01-0,2 эквивалентных количеств, предпочтительно 0,05-0,15 эквивалентных количеств катализатора, такого как N,N-диметиламинопиридин.
(2) Получение соединения (3а)-1
[Химическое соединение 18]
(В формуле М представляет собой щелочной металл или щелочноземельный металл и представляет собой анионную соль металла R1b, проявляющего нуклеофильность в форме R1b-M, и другие символы имеют значения, указанные выше. Такие же значения будут применяться в дальнейшем).
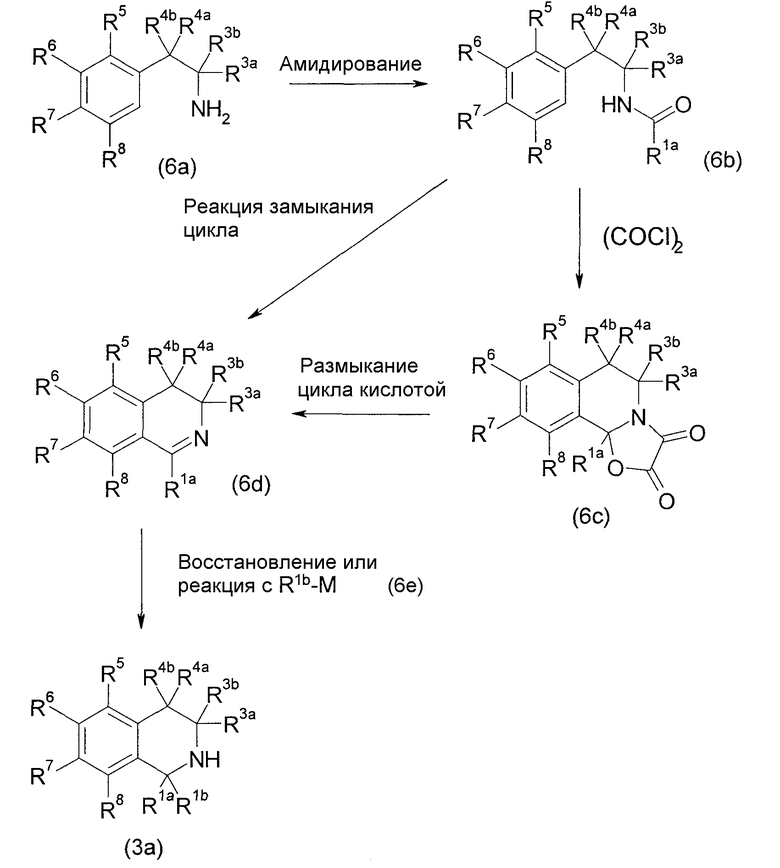
Данный способ получения является способом, в котором соединение (3а) получают реакцией замыкания цикла производного фенетиламина (6b), полученного амидированием производного фенетиламина (6а), с применением производного фосфорной кислоты или реакцией конденсации с применением оксалилхлорида с последующим катализируемым кислотой расщеплением цикла с получением производного дигидроизохинолина (6d) и восстановлением соединения (6d) или добавлением нуклеофильного реагента к соединению (6d).
Стадию амидирования соединения (6а) можно проводить таким же способом, как в способе получения 3.
Стадию замыкания цикла соединения (6b) можно проводить перемешиванием соединения (6b) в инертном в условиях реакции растворителе или без растворителя, в присутствии производного фосфорной кислоты, обычно в течение от 1 часа до 5 дней. Реакцию обычно проводят в условиях от охлаждения до нагревания, например, от комнатной температуры до нагревания при кипячении с обратным холодильником. В некоторых случаях может быть благоприятным проведение реакции в отсутствие растворителя. Растворитель, если его применяют, конкретно не ограничивают, но примеры его включают высококипящие ароматические углеводороды, такие как толуол и ксилол. Примеры производного фосфорной кислоты включают пентоксид дифосфора, смесь пентоксида дифосфора и оксихлорида фосфора, полифосфорную кислоту, этилполифосфат и тому подобное.
Альтернативно, эту стадию можно проводить таким способом, по которому оксалилхлорид подвергают реакции с амидом (6b) для образования кольца 2-хлороксазолона, образовавшийся продукт подвергают конденсации с замыканием цикла в присутствии в качестве катализатора кислоты Льюиса, такой как хлорид железа, с получением производного 6,10b-дигидро-5Н-[1,3]изоксазоло[2,3-a]изохинолин-2,3-диона (6с), и последующим сольволизом производного (6с) в присутствии сильной кислоты, такой как серная кислота, или с применением алкоксида щелочного металла, такого как метоксид натрия, получая при этом соединение (6d).
Когда R1b представляет собой водород, соединение (3а), у которого R1b представляет собой водород, можно получить восстановлением соединения (6d). Реакцию проводят обработкой соединения (6d) эквивалентным или избыточным количеством восстанавливающего агента в условиях от охлаждения до нагревания, например, при температуре от -20°С до 80°С, обычно в течение от 0,1 часа до 3 дней, в инертном в условиях реакции растворителе. Не имеется конкретного ограничения для растворителя, который можно применять в данной реакции. Примеры такого растворителя включают простые эфиры, спирты, ароматические углеводороды, N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО), этилацетат и их смеси. Примеры восстанавливающего агента включают гидридные восстанавливающие агенты, такие как боргидрид натрия, диизобутилалюминийгидрид и литийалюминийгидрид, металлические восстанавливающие агенты, такие как натрий, цинк и железо, и другие восстанавливающие агенты, описываемые в нижеследующей литературе.
[Ссылочная литература]
M. Hulicky, Reductions in Organic Chemistry, 2nd ed (ACS Monograph:188), ACS, 1996.
R.C. Larock, Comprehensive Organic Transformations, 2nd ed, VCH Publishers, Inc., 1999.
T.J. Donohoe, Oxidation and Reduction in Organic Synthesis (Oxford Chemistry Primers 6), Oxford Science Publications, 2000.
Courses in Experimental Chemistry, 5th edition, edited by The Chemical Society of Japan, Vol. 14(2005), Maruzen Co., Ltd.
Когда R1b представляет собой группу, отличающуюся от водорода, можно применять анионное присоединение при помощи нуклеофильного реагента (6е) для соединения (6d). Реакцию можно проводить с применением соединения (6d) и соединения (6е) в эквивалентных количествах или с применением одного из них в избыточном количестве в условиях от охлаждения до нагревания, например, при температуре от -78°С до 0°С, обычно при перемешивании в течение от 0,1 часа до 5 дней, в инертном в условиях реакции растворителе. Не имеется конкретного ограничения для растворителя, который можно применять в данной реакции. Примеры такого растворителя включают простые эфиры, ароматические углеводороды, N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО) и их смеси. Для подбора соответствующего соединения (6е) подходящим образом применяют R1b-магнийгалогенид, R1b-литий, полученный реакцией соответствующего галогенида с магнием.
Помимо этого, положения R1a и R1b в формуле можно заменить одно на другое.
(3) Получение соединения (3а)-2
[Химическое соединение 19]
(Символы в формуле имеют значения, указанные выше).
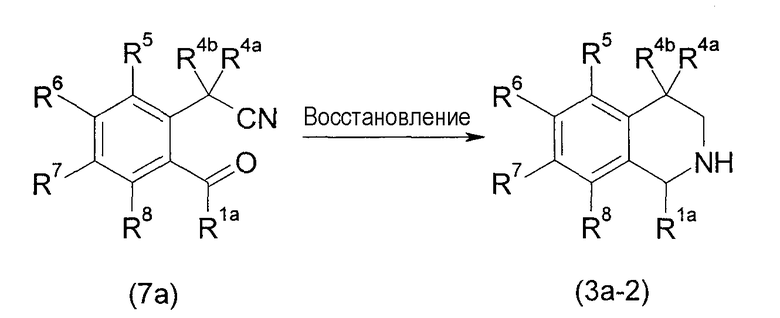
Данный способ получения является способом, в котором соединение (3а-2) получают восстановлением производного ацетонитрила (7а).
Реакцию можно проводить перемешиванием соединения (7а) в инертном в условиях реакции растворителе в атмосфере водорода в присутствии металлического катализатора, обычно в течение от 1 часа до 5 дней. Реакцию обычно проводят в условиях от охлаждения до нагревания, например, при комнатной температуре. Не имеется конкретного ограничения для растворителя, который можно применять в данной реакции. Примеры такого растворителя включают спирты, простые эфиры, воду, этилацетат, N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО) и их смеси. Примеры металлических катализаторов, которые предпочтительно можно применять, включают палладиевые катализаторы, такие как Pd, осажденный на угле (Pd/C), палладиевая чернь и гидроксид палладия, платиновые катализаторы, такие как оксид платины, родиевые катализаторы, такие как тетракистрифенилфосфинхлорродий, никель Ренея, железные катализаторы, такие как восстановленное железо и тому подобное. Вместо применения газообразного водорода в качестве источника водорода можно также применять эквивалентное или избыточное количество, относительно количества соединения (7а), муравьиной кислоты или формиата аммония.
[Ссылочная литература]
M. Hudlicky, Reductions in Organic Chemistry, 2nd ed (ACS Monograph:188), ACS, 1996.
Courses in Experimental Chemistry, 5th edition, edited by The Chemical Society of Japan, Vol. 19(2005), Maruzen Co., Ltd.
Кроме того, R1a в формуле может быть также R1b.
(4) Получение соединения (3а)-3
[Химическое соединение 20]
(Символы в формуле имеют значения, указанные выше).
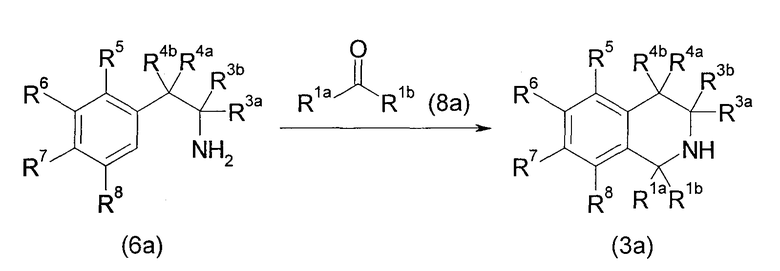
Данный способ получения является способом, в котором соединение (3а) получают конденсацией производного амина (6а) с кетоном (8а).
Реакцию можно проводить с применением соединения (6а) и соединения (8а) в эквивалентных количествах или одного из них в избыточном количестве в инертном в условиях реакции растворителе или без растворителя, в присутствии дегидратирующего агента или в качестве катализатора кислоты Льюиса, в условиях от охлаждения до нагревания, например, при температуре от комнатной до нагревания при кипячении с обратным холодильником, обычно при перемешивании в течение от 0,1 часа до 5 дней. Не имеется конкретного ограничения для растворителя, который можно применять в данной реакции. Примеры такого растворителя включают галогенированные углеводороды, простые эфиры и тому подобное. В некоторых случаях для спокойного развития реакции может быть благоприятным проведение реакции в присутствии сильной кислоты, такой как смесь муравьиная кислота-уксусный ангидрид и трифторуксусная кислота. Примеры дегидратирующего агента включают ангидриды кислот, такие как полифосфорная кислота, уксусный ангидрид и трифторуксусный ангидрид. Примеры кислоты Льюиса, применяемой в качестве катализатора, включают тетраизопропоксид титана и тому подобное.
Соединение настоящего изобретения выделяют и очищают в виде свободного соединения или его фармацевтически приемлемой соли, гидрата, сольвата или кристаллического полиморфа. Фармацевтически приемлемую соль соединения формулы (I) можно также получить согласно общепринятому способу реакции образования соли.
Выделение и очистку проводят посредством применения обычных химических операций, таких как экстракция, фракционная кристаллизация и различные типы хроматографии фракций.
Различные изомеры можно получить выбором подходящего исходного соединения или можно выделить с использованием различия в физико-химических свойствах изомеров. Например, из оптического изомера можно получить оптически чистый изомер при помощи общих способов разделения оптических изомеров (например, фракционной кристаллизацией для получения диастереомеров с оптически активными основаниями или кислотами, хроматографией с применением хиральной колонки и т.д., и тому подобное). Кроме того, изомеры можно также получить из подходящего оптически активного исходного соединения.
Фармакологическую активность соединения настоящего изобретения подтверждали нижеследующими испытаниями.
Пример испытания 1: Испытание соединений на блокирование Са2+-канала N-типа
Культивирование фибробластов человека (клетки IMR-32) и индуцирование дифференцировки проводили модификацией способа, описанного в литературе [Carbone et al., Pflugers Arch. Eur. J. Physiol., 416, 170-179 (1990)]. Клетки IMR-32 субкультивировали в MEM (Invitrogen Corporation, USA), содержащей 10% фетальную бычью сыворотку (FBS), 1% заменимых аминокислот, 1% пирувата натрия, 100 мкг/мл стрептомицина и 100 Е/мл пенициллина. После индуцирования клеточной дифференцировки к культуральной среде добавляли 1 мМ монофосфат циклического дибутириладенина (dbcАМФ) и 2,5 мкМ 5-бромдеоксиуридин (BrdU) и клетки культивировали в течение 10-11 дней, что приводило к экспрессии Са2+-канала N-типа человека.
Клетки IMR-32 с индуцированной 10-11-дневной дифференцировкой засевали при плотности 6×105 клеток на лунку в 96-луночном планшете, покрытом поли-D-лизином. После культивирования клеток в культуральной среде в течение 3 часов или больше в нее добавляли Fluo-3 AM с последующей инкубацией при 37°С в течение 60 минут. Культуру промывали в буфере для анализа (HBSS, 20 мМ HEPES, 2,5 мМ пробенецид, pH 7,4), к которому затем добавляли раствор испытуемого соединения в присутствии 1 мкМ нитрендипина. Спустя 10 минут повышение концентрации внутриклеточного Са2+, индуцированного стимуляцией при высоком содержании К+ 50 мМ раствором KCl, определяли посредством применения набора для анализа кальция FLIPR (Molecular Devices Corporation, USA). Блокирующую активность испытуемого соединения в отношении Ca2+-канала N-типа вычисляли как относительную величину, принимая максимальное повышение концентрации внутриклеточного Ca2+ в контрольной группе за 100%. Затем концентрацию соединения (величина IC50), которая требуется для 50% ингибирования повышения концентрации внутриклеточного Ca2+, вычисляли нелинейным регрессионным анализом.
В результате этого было обнаружено, что соединения настоящего изобретения проявляли блокирующее действие на Са2+-канал N-типа. Величины IC50 для нескольких соединений настоящего изобретения даны ниже в таблице 1.
Пример испытания 2: Влияния соединений на модель ноцицептивной боли (формалиновое испытание)
Формалиновое испытание на мыши проводили модификацией способа, описываемого в литературе [Murakami et al., Eur. J. Pharmacol. 419: 175-181 (2001)]. Когда 20 мкл 2,0% формалина подкожно вводили в подушечки на лапах мышей (ddY, самцы, возраст 5 недель), в обработанных конечностях животных происходило индуцирование болевых поведений (поведения отдергивания и облизывания конечностей). Спустя 15-25 минут после введения формалина измеряли время, принятое за появление болевых поведений, чтобы тем самым оценить ингибирующее действие испытуемого соединения на болевые поведения животных. Испытуемое соединение перорально вводили за 30 минут до введения формалина. Оценку испытуемого соединения проводили вычислением степени ингибирования (%) в группе, обработанной испытуемым соединением, принимая время, взятое за появление болевых поведений в группе, обработанной наполнителем, как 100%.
Степень ингибирования (%) = 100-(среднее время появления болевых поведений в группе, обработанной испытуемым соединением)/(среднее время появления болевых поведений в группе, обработанной наполнителем)×100.
В результате этого было обнаружено, что соединения настоящего изобретения проявляли аналгезирующее действие на боль, индуцированную формалином. Степени ингибирования (%) нескольких соединений настоящего изобретения при дозе 100 мг/кг даны ниже в таблице 2.
Пример испытания 3: Влияния соединений на модель невропатической боли (действия против аллодинии у крыс с лигированными L5/L6 спинномозговыми нервами)
Одним из основных симптомов при невропатической боли является значительно сниженный порог ответной реакции на тактильную стимуляцию (аллодиния). Действия против аллодинии соединений настоящего изобретения подтверждали оценкой аналгезирующего действия у крыс с лигированными L5/L6 спинномозговыми нервами. Оценку проводили способом Кима и Чанга (Pain 50, 355-363, 1992) с некоторыми модификациями.
После анестезии пентобарбиталом левые L5 и L6 спинномозговые нервы самцов крыс SD (возраст 5-6 недель) плотно лигировали шелковой нитью. Для оценки аналгезирующего действия адаптировали тест фон Фрея с применением щетины. То есть подушечку задней лапы прокалывали щетиной и самое слабое действие щетины на реакцию отдергивания конечности обозначали как порог реакции (log грамм) на механическую стимуляцию. Поскольку в предварительных испытаниях было подтверждено, что порог ответной реакции задней лапы животного, расположенной на стороне операции лигирования, был значительно низким в течение 7-14 дней после операции (при состоянии механической аллодинии), действия против аллодинии испытуемого соединения оценивали только в день между днями 7 и 14 после операции. За день перед оценкой испытуемого соединения измеряли порог реакции перед введением испытуемого соединения. Животных делили на 4-5 групп так, чтобы различия средних величин порогов реакции между группами перед введением испытуемого соединения и разброс величин в группе стали небольшими. При оценке испытуемого соединения измеряли порог реакции после введения испытуемого соединения. Испытуемое соединение перорально вводили за 30-60 минут перед измерением порога реакции. Активность против аллодинии испытуемого соединения оценивали как степень возвращения к норме (восстановления) (%) в группе, обработанной испытуемым соединением, принимая пороги ответной реакции лап, расположенных на стороне операции лигирования и на противоположной стороне, в группе, обработанной наполнителем, как 0% и 100%, соответственно.
Степень возвращения к норме (%) = {(среднее значение порога ответной реакции в группе, обработанной испытуемым соединением)-(среднее значение порога ответной реакции лапы, расположенной на стороне операции, в группе, обработанной наполнителем)}/{(среднее значение порога ответной реакции лапы, расположенной на стороне, противоположной стороне операции, в группе, обработанной наполнителем)-(среднее значение порога ответной реакции лапы, расположенной на стороне операции, в группе, обработанной наполнителем)}×100.
В результате этого было обнаружено, что соединения настоящего изобретения проявляли аналгезирующее действие на механическую аллодинию в модели невропатической боли. Степени возвращения к норме крыс (%) для групп с введением нескольких соединений настоящего изобретения даны ниже в таблице 3.
Пример испытания 4: Влияния соединений на модель абдоминальной боли (определение CDR-индуцированной абдоминальной боли у крыс)
Известно, что в ответ на стимуляцию давлением, вызванную колоректальным вздутием (CRD), пациенты с IBS проявляют снижение порога пищеварительного восприятия (аллодинию), которое вызывает дискомфорт по сравнению со слабым стимулом, который не ощущается нормальным индивидуумом, и гипералгезию, которая приводит к более сильной субъективной реакции на пищеварительное восприятие, чем у нормального индивидуума (Gastroenterol. 130: 1377-1390 (2006)), и считается, что такие состояния являются ответственными за абдоминальные симптомы. Улучшающие действия соединений настоящего изобретения на боль пищеварительного тракта подтверждали определением CDR-индуцированной абдоминальной боли у крыс. Определение CDR-индуцированной абдоминальной боли у крыс проводили модификацией способа, описанного в литературе [Neurogastroenterol. Motil. 15: 363-369 (2003)]. Когда стимуляцию постоянным внутренним давлением применяют для прямой кишки животного раздуванием ее баллоном длиной 6 см, вставленным в анус крысы (Вистар, самец, 250-350 г), поведения абдоминального сгибательного рефлекса индуцируются вследствие абдоминальной боли. Частоту рефлекторных поведений, имеющих место во время стимуляции вздутием в течение 5 минут, подсчитывали для оценки ингибирующего действия испытуемого соединения на абдоминальную боль. Испытуемое соединение перорально вводили за 30 минут до начала стимуляции вздутием. Оценку испытуемого соединения проводили вычислением степени ингибирования (%) поведений абдоминальных сгибательных рефлексов относительно группы, обработанной наполнителем.
В результате этого было обнаружено, что соединения настоящего изобретения проявляли ингибирующее действие на абдоминальную боль. Для нескольких соединений настоящего изобретения при дозе 10 мг/кг степени ингибирования (%) поведений абдоминальных сгибательных рефлексов после вздутия при внутреннем давлении 5999,5 Па (45 мм Hg) показаны ниже в таблице 4.
Пример испытания 5: Влияния соединений на модель спастической констипации (испытание на индуцированную лоперамидом задержку переноса шариков в толстой кишке)
Известно, что обычно появление констипации при IBS-C вызывается спастическим нарушением моторики пищеварительного тракта и является аналогичным индуцированной опиоидом констипации в терминах патофизиологии заболевания (Eur. J. Pharmacol. 75: 239-245 (1981), American J. Physiol. 96: 667-676 (1931), Nippon Rinsho 64: 1461-1466 (2006)). Улучшающее действие соединений настоящего изобретения на спастическую констипацию было подтверждено испытанием на индуцированную лоперамидом задержку колоректального переноса шариков у мышей. Испытание на индуцированную лоперамидом задержку колоректального переноса шарика у мышей проводили модификацией способа, описанного в литературе [J. Smooth Muscle Res. 29:47-53 (1993)]. Стеклянный шарик диаметром 3 мм глубоко вставляли при глубине 2 см в анус мыши (ddY, самец, возраст 6 недель) и измеряли время, принятое за экскрецию шарика. Когда 0,3 мг/кг лоперамида подкожно вводили за 30 минут до вставки шарика, имело место индуцирование задержки экскреции шарика. По улучшающим действиям на индуцированную лоперамидом задержку переноса шарика оценивали улучшающее перистальтику кишечника действие испытуемого соединения на спастическую констипацию. Испытуемое соединение перорально вводили одновременно с введением лоперамида (за 30 минут до вставки шарика). Оценку испытуемого соединения осуществляли вычислением степени улучшения времени экскреции шарика обработанной испытуемым соединением/обработанной лоперамидом группы, принимая время экскреции шарика не обработанной испытуемым соединением/не обработанной лоперамидом (обработанной наполнителем/обработанной наполнителем) группы за 100% и принимая время экскреции шарика не обработанной испытуемым соединением/обработанной лоперамидом (обработанной наполнителем/обработанной лоперамидом) группы за 0%.
В результате этого было обнаружено, что соединения настоящего изобретения проявляли улучшающее действие на индуцированную опиоидом констипацию. Для нескольких соединений настоящего изобретения при дозе 3 мг/кг степени улучшения (%) времени экскреции шарика даны ниже в таблице 5.
Пример испытания 6: Действия соединений при комбинированном применении с морфином (1)
Морфин обладает сильным аналгезирующим действием на ноцицептивную боль посредством µ-опиоидных рецепторов. Например, морфин проявляет доза-зависимые аналгезирующие действия в формалиновом испытании, в котором применяют модель ноницептивной боли, вызванной формалином (Pharmacol. Biochem. Behav. 84: 479-486 (2006)). Между тем, известно, что селективный пептид, блокирующий Са2+-канал N-типа, ω-конотоксин (ω-CTx), также независимо проявляет доза-зависимые аналгезирующие действия в формалиновом испытании и его комбинированное применение с морфином повышает аналгезирующие действия по сравнению с аналгезирующими действиями, полученными при применении только морфина (дополнительные действия) (Pain 84: 271-281 (2000)). Следовательно, может быть подтверждено, что когда соединения настоящего изобретения, обладающие блокирующим Са2+-канал N-типа действием, применяли в комбинации с морфином в формалиновом испытании, достигалось сильное действие против ноцицептивной боли, сравнимое с действием или более сильное, чем действие при введении только морфина или действие при введении только соединения настоящего изобретения.
Пример испытания 7: Влияние соединений при комбинированном применении с морфином (2)
Известно, что механическая аллодиния, наблюдаемая у крыс с лигированным L5/L6-спинномозговым нервом, проявляет только частичное возвращение к норме при обработке морфином. С другой стороны, как описано в контексте выше, соединения настоящего изобретения проявляют действия почти со 100% возвращением к норме при механической аллодинии у крыс с лигированным L5/L6-спинномозговым нервом. Поэтому когда соединения настоящего изобретения применяли в комбинации с морфином, сильное действие против невропатической боли, сравнимое с действием или более сильное, чем действие при введении только морфина или при введении только соединений настоящего изобретения, можно подтвердить испытанием на их действия против аллодинии на крысах с лигированным L5/L6-спинномозговым нервом.
Пример испытания 8: Действия соединений при комбинированном применении с морфином (3)
Морфин является агонистом µ-опиоидного рецептора, имеющим такой же механизм действия, как лоперамин, и обладает задерживающим действием на перенос шарика в толстой кишке у мышей, аналогичным действию лоперамида. При введении дозы морфина, которая проявляет ингибирующее действие на абдоминальную боль в испытании абдоминальной боли у крысы, индуцированной CRD, и проявляет действие по задержке переноса в испытании на колоректальный перенос шарика у мыши, и дозы испытуемого соединения, которая ингибирует задержку переноса шарика, вызванной вышеуказанной дозой морфина, можно подтвердить, что такое комбинированное применение проявляет сильное ингибирующее абдоминальную боль действие, сравнимое или более сильное, чем действие при введении только морфина, при определении CRD-индуцированной абдоминальной боли у крыс, а также оказывает ингибирующее действие на индуцированную морфином задержку переноса при испытании на перенос шарика.
Альтернативно, при введении испытуемого соединения с низкой дозой морфина, при которой ингибирующее абдоминальную боль действие является недостаточным в анализе CRD-индуцированной абдоминальной боли у крысы, но задерживающее действие не обнаруживается в испытании на перенос шарика в толстой кишке мыши, можно подтвердить достаточное ингибирующее абдоминальную боль действие, которое не получали при применении низкой дозы одного морфина.
На основании экспериментальных результатов, описанных выше, было подтверждено, что соединения настоящего изобретения обладают блокирующим действием на Ca2+-каналы N-типа. Поэтому очевидно, что соединения настоящего изобретения являются применимыми в качестве активного ингредиента фармацевтической композиции для предупреждения и/или лечения различных болей, таких как невропатическая боль и ноцицептивная боль, головные боли, такие как мигрень и кластерная головная боль, заболеваний центральной нервной системы, таких как тревога, депрессия, эпилепсия, церебральный удар и синдром усталых ног, заболевания пищеварительной системы, такие как абдоминальная боль и синдром раздраженной толстой кишки, и заболеваний мочевых путей, таких как гиперактивный мочевой пузырь и интерстициальный цистит.
На основании результатов испытания с формалином, описываемым выше, было подтверждено, что соединения настоящего изобретения обладают действием против ноцицептивной боли. Кроме того, на основании результатов действий против аллодинии у крыс с лигированным L5/L6 спинномозговым нервом было подтверждено, что соединения настоящего изобретения обладают действием против невропатической боли. После рассмотрения этих фактов становится ясным, что соединения настоящего изобретения являются применимыми в качестве активного ингредиента фармацевтической композиции для предупреждения и/или лечения различных болей, включая невропатическую боль и ноцицептивную боль. Кроме того, клинически показано, что прегабалин, который является лигандом субъединицы α2δ Са2+-канала и который применяют в качестве агента против невропатической боли, оказывает терапевтические действия на синдром фибромиалгии, имеющий много общего с невропатической болью в терминах клинического состояния. На основании этого заключения можно считать, что соединения настоящего изобретения являются также применимыми в качестве активного ингредиента фармацевтической композиции для предупреждения и/или лечения синдрома фибромиалгии.
На основании результатов анализа CRD-индуцированной абдоминальной боли, описываемого выше, было показано, что соединения настоящего изобретения обладают действием, ингибирующим абдоминальную боль. Поэтому очевидно, что соединения настоящего изобретения являются применимыми в качестве активного ингредиента фармацевтической композиции для предупреждения и/или лечения абдоминальных симптомов, особенно абдоминальных симптомов IBS.
На основании результатов испытания на индуцированную лоперамидом задержку колоректального переноса шарика у мыши было показано, что соединения настоящего изобретения обладают улучшающим действием на индуцированную опиоидом констипацию. На основании этого факта становится очевидным, что соединения настоящего изобретения являются применимыми в качестве активного ингредиента фармацевтической композиции для предупреждения и/или лечения спастической констипации, особенно констипации при OBD. Кроме того, на основании того факта, что констипация при IBS-C является спастической констипацией, аналогичной констипации, вызванной опиоидами, становится очевидным, что соединения настоящего изобретения являются применимыми в качестве активного ингредиента фармацевтической композиции для предупреждения и/или лечения констипации при IBS-C.
На основании факта, демонстрирующего, что соединения настоящего изобретения являются эффективными как в испытании на абдоминальную боль у крысы, индуцированную CDR, так и в испытании задержки колоректального переноса шарика у мыши, становится очевидным, что соединения настоящего изобретения являются применимыми в качестве активного ингредиента превосходной фармацевтической композиции для предупреждения и/или лечения IBS-C, обладающей комбинацией действия, ослабляющего абдоминальный симптом, и действия, улучшающего состояние при констипации.
Известно, что применение пептида, селективно блокирующего Са2+-канал N-типа, конотоксина (ω-CTx), в комбинации с морфином повышает аналгезирующие действия по сравнению с аналгезирующим действием, полученным при применении только морфина (дополнительные действия) (Pain 84: 271-281 (2000), Life Science 73: 2873-2881 (2003)). Поэтому можно предположить, что комбинированное применение соединений настоящего изобретения и опиоидов приведет к получению превосходной фармацевтической композиции для предупреждения и/или лечения боли, которая проявляет более сильные аналгезирующие действия, чем действия при применении только опиоидов.
Опиоиды применяют в качестве терапевтического средства против сильной боли, такой как раковая боль, но применение их сталкивается с клиническими проблемами, связанными с доза-зависимыми побочными действиями на пищеварительную систему, такими как рвота и констипация (Eur. J. Pharmaceutical Sci. 20: 357-363 (2003)). Соединения настоящего изобретения проявляют превосходные улучшающие действия на индуцированную опиоидами констипацию (OIC). На основании этого факта можно предположить, что соединения настоящего изобретения при комбинированном применении с опиоидами могут привести к созданию фармацевтической композиции для предупреждения и/или лечения боли, которая ингибирует индуцированную опиоидом констипацию с меньшими побочными действиями. Кроме того, можно предположить, что комбинированное применение соединений настоящего изобретения и низкой дозы опиоидов может привести к созданию превосходной фармацевтической композицию для предупреждения и/или лечения боли, которая способна проявлять достаточные аналгезирующие действия при снижении дозы опиоидов и которая способна также задерживать появление констипации посредством снижения дозы опиоида.
Препарат, содержащий один или два или более типов соединения формулы (I) или его фармацевтически приемлемой соли в качестве активного ингредиента, можно получить согласно обычно применяемому способу, используя фармацевтически приемлемый носитель, эксципиент или тому подобное, которые обычно применяют в данной области.
Введение можно проводить пероральным введением посредством таблеток, пилюль, капсул, гранул, порошков, жидких препаратов или тому подобного, или парентеральным введением посредством инъекций, таких как внутрисуставная инъекция, внутривенная инъекция, внутримышечная инъекция или тому подобное, а также препаратами в виде суппозиториев, глазных капель, глазных мазей, чрескожных жидких препаратов, мазей, чрескожных пластырей, жидких препаратов для введения через слизистую оболочку, пластырей для введения через слизистую оболочку, препаратов для ингаляции и тому подобного.
В качестве твердых композиций для перорального введения согласно настоящему изобретению применяют таблетки, порошки, гранулы или тому подобное. В такой твердой композиции один или два или больше типов активных ингредиентов смешивают по меньшей мере с одним инертным эксципиентом, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон и/или алюминометасиликат магния. Согласно общепринятому способу, композиция может содержать инертные добавки, такие как смазывающее вещество, такое как стеарат магния, дезинтегрирующее вещество, такое как натриевая соль карбоксиметилкрахмала, стабилизирующий агент и солюбилизирующее вспомогательное средство. В зависимости от определенного случая таблетки или пилюли могут быть покрыты пленкой или сахарным покрытием или гастро- или энтеросолюбильным покрытием.
Жидкие композиции для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры или тому подобное и содержат обычно применяемый инертный разбавитель, такой как очищенная вода или этанол. Помимо инертного разбавителя, жидкая композиция может содержать адъювант, такой как солюбилизирующий агент, увлажняющий агент и суспендирующий агент, подслащивающее вещество, корригент, ароматизирующее вещество и консервант.
Инъекции для парентерального введения включают стерильные водные или неводные растворы, суспензии и эмульсии. Водный растворитель включает, например, дистиллированную воду для инъекции и физиологический солевой раствор. Примеры неводного растворителя включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, спирты, такие как этанол, полисорбат 80 (Japanese Pharmacopeia) и тому подобное. Такая композиция может дополнительно содержать агент тоничности, консервант, увлажняющий агент, эмульгирующий агент, диспергирующий агент, стабилизирующий агент или солюбилизирующее вспомогательное средство. Их стерилизуют, например, фильтрованием через задерживающий бактерии фильтр, включением стерилизующего агента или облучением. Кроме того, их можно также применять изготовлением стерильной твердой композиции и растворением или суспендированием ее в стерильной воде или стерильном растворителе для инъекции перед его применением.
Препараты для наружного применения включают мази, пластыри, кремы, желе, адгезивные пластыри для кожи, спреи, лосьоны, глазные капли, глазные мази и тому подобное. Препараты для наружного применения содержат обычно применяемые основы мазей, основы лосьонов, водные или неводные жидкости, суспензии, эмульсии и тому подобное. Примеры основ мазей или лосьонов включают полиэтиленгликоль, пропиленгликоль, белый вазелин, белый пчелиный воск, полиоксиэтилированное гидрогенизированное касторовое масло, глицерилмоностеарат, стеариловый спирт, цетиловый спирт, лауромакрогол, сесквиолеат сорбитана и тому подобное.
Препараты для введения через слизистую оболочку, такие как препараты для ингаляции и препараты для введения через нос, применяют в твердой, жидкой или полутвердой форме, их можно получить согласно общепринятому известному способу. К ним можно подходящим образом добавить, например, известный эксципиент, а также агент регуляции рН, консервант, поверхностно-активное вещество, смазывающее вещество, стабилизирующий агент, загущающий агент или тому подобное. Для их введения можно применять подходящее устройство для ингаляции или вдувания. Например, соединение можно вводить отдельно или в виде порошка приготовленной смеси, или в виде раствора или суспензии в комбинации с фармацевтически приемлемым носителем с применением общепринятого известного устройства или пульверизатора, такого как устройство для ингаляции, применяемое для введения дозированного количества. Ингалятор с сухим порошком или тому подобное можно применять для введения один или много раз, можно применять сухой порошок или капсулу, содержащую порошок. Альтернативно, он может быть в такой форме, как находящийся под давлением аэрозольный спрей или тому подобное, в котором применяют подходящий пропеллент, например подходящий газ, такой как хлорфторалкан, гидрофторалкан или диоксид углерода.
При пероральном введении суточная доза обычно бывает от приблизительно 0,001 до 100 мг/кг, предпочтительно от 0,1 до 30 мг/кг и более предпочтительно 0,1-10 мкг на массу тела, которую вводят в виде одной порции или в виде 2-4 разделенных порций. В случае внутривенного введения суточную дозу подходящим образом вводят в количестве от приблизительно 0,0001 до 10 мг/кг массы тела один раз в день или два или более раз в день. Кроме того, средство для введения через слизистую оболочку вводят при дозе от приблизительно 0,001 до 100 мг/кг массы тела один раз в день или два или более раз в день. Дозу подходящим образом выбирают в зависимости от отдельного случая, принимая во внимание симптомы, возраст и пол и тому подобное.
Соединения настоящего изобретения можно применять в комбинации с различными средствами для лечения или предупреждения заболеваний, для которых, как считается, соединения настоящего изобретения являются эффективными. Примеры лекарственных средств, которые можно применять в комбинации с соединениями настоящего изобретения, включают опиоиды, такие как морфин, антидепрессанты, такие как дулоксетин и амитриптилин, антиэпилептические лекарственные средства, такие как прегабалин и мексилетин, нестероидные противовоспалительные лекарственные средства, такие как диклофенак, и тому подобное. Для такого комбинированного применения соединения настоящего изобретения изготавливают в составе подходящих лекарственных форм, таких как жидкие препараты, капсулы, гранулы, пилюли, порошки, таблетки, препараты для наружного применения, желе, спреи, пластыри, суппозитории и содержащие такие комбинации имплантируемые насосы, и части комбинированного препарата можно вводить одновременно или раздельно и непрерывно или через требуемые интервалы времени посредством перорального, внутривенного, подкожного, трансназального, энтерального, спинального эпидурального или спинального субарахиноидального пути. Препараты, части которых вводят совместно, могут быть смесью или части могут быть изготовлены по отдельности.
ПРИМЕРЫ
Далее в контексте способы получения соединения настоящего изобретения будут описаны более подробно посредством примеров. Настоящее изобретение не ограничивается нижеследующими примерами. Кроме того, в примерах получения показаны способы получения исходных соединений. Способы получения соединения настоящего изобретения не ограничиваются способами получения конкретных примеров, описываемых ниже. Соединение настоящего изобретения можно получить согласно комбинации этих способов получения или согласно способу, известному специалисту в данной области.
В описанных ниже примерах, примерах получения и таблицах будут применяться следующие аббревиатуры.
Rex: номер примера получения, Ех: номер примера, №: номер соединения, СТРУКТУРА: структурная формула, Данные: физико-химические данные (FAB: FAB-MS[M+H]+, FAN: FAB-MS[M-H]-, FA1: FAB-MS[M]+, FA2: FAB-MS[M+2H]+, ES: ESI-MC[M+H]+, ES1: ESI-MC[M]+, ES2: ESI-MC[M+2H]+, ESNa: ESI-MC[M+Na]+, AP: APCI-MC[M+H]+, AP1: APCI-MC[M]+, CI: CI[M+H]+, CIN: CI[M-H]-, CI1: CI[M]+, EI: EI[M+H]+, EIN: EI[M-H]-, EI1: EI[M]+, EIBr: EI[M-Br]-, ЯМР: δ (м.д.) пик 1H-ЯМР, в ДМСО-d6), N/D: не определяли, соль: соль (такой термин в пустом столбце или отсутствие столбца с таким термином означает, что соединение находится в свободной форме), CL: гидрохлорид, BR: гидробромат, OX: оксалат, FM: фумарат, MD: D-манделат, ML: L-миндальная кислота, LL: соль N-ацетил-L-лейцина, T1: L-тартрат, T2: D-тартрат, TX: дибензоил-D-тартрат, TY: дибензоил-L-тартрат, TP: дипаратолуоил-D-тартрат, TQ: дипаратолуоил-L-тартрат, MA: L-яблочная кислота, MB: D-яблочная кислота), Me: метил, Et: этил, nPr: нормальный пропил, iPr: изопропил, tBu: трет-бутил, cPr: циклопропил, cBu: циклобутил, cPen: циклопентил, cHex: циклогексил Admt: адамантил, Ph: фенил, Bn: бензил, Thp: тетрагидропиранил, pipe: пиперидинил, pipa: пиперадинил, CN: циано, boc: трет-бутилоксикарбонил, Ac: ацетил, MOM: метоксиметил, TMS: триметилсилил, di: ди, ТГФ: тетрагидрофуран, ДМФА: N,N-диметилформамид, ДМСО: диметилсульфоксид. Число перед заместителем означает положение заместителя и, например, 6-Cl-2-Py представляет собой 6-хлорпиридин-2-ил и 3,3-ди-F-cHex представляет собой 3,3-дифторциклогексил. Rsyn и Syn: способ получения (числа указывают, что соединения были получены с применением соответствующих исходных соединений способом, аналогичным способу получения соединений, соответственно имеющих числа в качестве чисел примеров получения или чисел примеров). Кроме того, среди соединений примеров получения или примеров в таблицах для соединения, у которого конфигурация заместителя в 1-положении тетрагидроизохинолина не определена, но показана одна конфигурация в любую из сторон, конфигурация в любую из сторон отмечена и тогда номер примера получения или номер примера указывается со значком *. С другой стороны, для соединения, у которого конфигурация заместителя в 1-положении тетрагидроизохинолина определена, или соединение, у которого конфигурация с достаточным основанием аналогизирована на основании поведения при хиральной колоночной хроматографии или активности в испытании на блокирование Са2+-канала N-типа, конфигурация только отмечена.
Кроме того, соединение, у которого такой же номер дается после *, означает, что соединение получают с применением соединения, которому дан тот же номер и у которого конфигурация заместителя в 1-положении тетрагидроизохинолина не определена, но одна конфигурация отмечена в любую из сторон, в качестве исходного соединения.
Пример получения 1
N-(2-Циклогекса-1-ен-1-илэтил)-2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтанамин (431 мг) растворяли в хлороформе (12 мл), в раствор добавляли трифторуксусный ангидрид (0,3 мл) при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 10 часов и затем перемешивали при 60°С в течение 2 часов. Растворитель выпаривали и к реакционной жидкости добавляли насыщенный водный бикарбонат натрия и смесь затем экстрагировали хлороформом. Реакционную жидкость промывали насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ), получая при этом N-(2-циклогекса-1-ен-1-илэтил)-N-[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]-2,2,2-трифторацетамид (419 мг).
Пример получения 2
N-(2-Циклогекса-1-ен-1-илэтил)-N-[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]-2,2,2-трифторацетамид (408 мг) растворяли в смеси 3:1 (8 мл) ацетон-вода. К реакционной жидкости добавляли 4-оксид 4-метилморфолина (200 мг) и раствор 2,5% тетроксида осмия в трет-бутиловом спирте (2,68 мл) с последующим перемешиванием при комнатной температуре в течение 18 часов. Затем растворитель реакции выпаривали при пониженном давлении и к реакционной жидкости добавляли воду с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом N-[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]-N-{2-[цис-1,2-дигидроксициклогексил]этил}-2,2,2-трифторацетамид (276 мг).
Пример получения 3
2-(Хлорацетил)-1-циклогексил-7-метокси-1,2,3,4-тетрагидроизохинолин (700 мг) растворяли в ацетонитриле (15 мл), к раствору затем добавляли карбонат калия (2,1 г), гидрохлорид 2-циклопента-1-ен-1-илэтанамина (1,6 г) и иодид тетра-н-бутиламмония (80 мг) с последующим перемешиванием при 70°С в течение 5 часов. После этого растворитель выпаривали и к реакционной жидкости добавляли воду с последующей экстракцией EtOAc. Экстракт промывали насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН).
Образовавшееся соединение растворяли в хлороформе (10 мл) и к раствору добавляли трифторуксусный ангидрид (0,34 мл) с последующим перемешиванием при комнатной температуре в течение 14 часов. Затем растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом N-[2-(1-циклогексил-7-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]-N-(2-циклопента-1-ен-1-илэтил)-2,2,2-трифторацетамид (450 мг).
Пример получения 4
(L)-Тартрат (1R)-1-циклогексил-1,2,3,4-тетрагидроизохинолина (520 мг) растворяли в EtOAc (10 мл) и к раствору добавляли насыщенный водный бикарбонат натрия (10 мл). К реакционной жидкости на протяжении 5 минут при охлаждении льдом по каплям добавляли раствор хлорацетилхлорида (0,14 мл) в EtOAc (5 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционную жидкость экстрагировали EtOAc и сушили над сульфатом магния, получая при этом (1R)-2-(хлорацетил)-1-циклогексил-1,2,3,4-тетрагидроизохинолин (415 мг).
Пример получения 5
Гидрохлорид 7-хлор-1-циклогексил-1,2,3,4-тетрагидроизохинолина (899 мг) добавляли к насыщенному водному бикарбонату натрия (15 мл), к которому затем дополнительно добавляли EtOAc (10 мл). К реакционной жидкости на протяжении 5 минут по каплям добавляли раствор хлорацетилхлорида (390 мг) в EtOAc (5 мл). Реакционную жидкость перемешивали в течение 1 часа, экстрагировали EtOAc и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 7-хлор-2-(хлорацетил)-1-циклогексил-1,2,3,4-тетрагидроизохинолин (888 мг).
Пример получения 6
Смесь хлорацетилхлорида (1,03 г) и EtOAc (5 мл) добавляли по каплям с перемешиванием к смеси (1S)-1-фенил-1,2,3,4-тетрагидроизохинолина (1,58 г), гидрокарбоната натрия (960 мг), воды (25 мл) и EtOAc (25 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционную жидкость экстрагировали EtOAc и экстракт промывали последовательно насыщенным водным бикарбонатом натрия и насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-AcOEt, 4:1), получая при этом (1S)-2-(хлорацетил)-1-фенил-1,2,3,4-тетрагидроизохинолин (2,14 г).
Пример получения 7
Гидрохлорид 1-циклогексил-1,2,3,4-тетрагидроизохинолина (800 мг) растворяли в метиленхлориде (12 мл), к раствору добавляли при охлаждении льдом акрилоилхлорид (0,28 мл) с последующим перемешиванием при охлаждении льдом в течение 30 минут и затем перемешивали при комнатной температуре в течение 14 часов. К реакционной жидкости добавляли воду и смесь затем экстрагировали хлороформом. Экстракт промывали насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали, получая при этом 2-акрилоил-1-циклогексил-1,2,3,4-тетрагидроизохинолин (856 мг).
Пример получения 8
1-Бензил-4-гидроксипиперидин-4-карбоновую кислоту (951 мг) растворяли в ДМФА (25 мл) и к раствору добавляли N,N'-карбонилдиимидазол (720 мг) с последующим перемешиванием при комнатной температуре в течение 18 часов. После этого к реакционной жидкости добавляли N,N-диизопропилэтиламин (784 мг) и гидрохлорид 1-циклогексил-1,2,3,4-тетрагидроизохинолина (1,22 г) с последующим перемешиванием при 60°С в течение 18 часов. Растворитель выпаривали и к реакционной жидкости добавляли воду и EtOAc. Образовавшиеся нерастворимые вещества отделяли при помощи целита, экстрагировали EtOAc и сушили над сульфатом магния и растворитель выпаривали. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН) и растворяли в 1,4-диоксане (12 мл) и к раствору добавляли ди-трет-бутилдикарбонат (1,3 г) с последующим перемешиванием при комнатной температуре в течение 1 часа. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc и затем хлороформ-МеОН), получая при этом 1-бензил-4-[(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)карбонил]пиперидин-4-ол (115 мг).
Пример получения 9
1-Бензил-4-[(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)карбонил]пиперидин-4-ол (220 мг) растворяли в МеОН (12 мл) и к раствору добавляли 20% гидроксид палладия, осажденного на активированном угле (360 мг) с последующим перемешиванием в атмосфере водорода при комнатной температуре и нормальном давлении в течение 15 часов. После этого катализатор отделяли при помощи целита. Растворитель выпаривали, получая при этом 4-[(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)карбонил]пиперидин-4-ол (154 мг).
Пример получения 10
N-Метилморфолин (0,873 мл) добавляли к раствору 1-(трет-бутоксикарбонил)-L-пролина (1,28 г) в 1,2-дихлорэтане (10 мл) при охлаждении льдом с последующим дополнительным добавлением пивалоилхлорида (0,734 мл). Реакционную жидкость перемешивали в течение 1 часа и затем к ней добавляли N-метилморфолин (1,09 мл) и гидрохлорид 1-циклогексил-1,2,3,4-тетрагидроизохинолина (1,00 г). Смесь перемешивали при комнатной температуре в течение 15 часов. К реакционному раствору добавляли EtOAc и водный 1 М раствор HCl. Органический слой промывали водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, получая при этом трет-бутил-(2S)-2-[(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)карбонил]пирролидин-1-карбоксилат (1,79 г).
Пример получения 11
Гидрохлорид 1-циклогексил-1,2,3,4-тетрагидроизохинолина (1,00 г) растворяли в метиленхлориде (20 мл) и при охлаждении льдом к раствору добавляли пивалоилхлорид (0,98 мл) и 4-метилморфолин (2,2 мл). Реакционную жидкость перемешивали при комнатной температуре в течение 30 минут и затем охлаждали льдом и к ней добавляли [(трет-бутоксикарбонил)амино]уксусную кислоту (1,54 г). Реакционную жидкость перемешивали при комнатной температуре в течение 14 часов и затем к ней добавляли воду с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом трет-бутил-[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]карбамат (1,49 г).
Пример получения 12
4 M HCl/EtOAc (4 мл) добавляли к раствору трет-бутил-(2S)-2-[(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)карбонил]пирролидин-1-карбоксилата (1,79 г) в EtOAc (4 мл). Смесь перемешивали при комнатной температуре в течение 5 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли хлороформ и насыщенный водный раствор гидрокарбоната натрия. Органический слой промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали, получая при этом 1-циклогексил-2-L-пролил-1,2,3,4-тетрагидроизохинолин (1,26 г).
Пример получения 13
трет-Бутил-[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]карбамат (1,5 г) растворяли в EtOAc (20 мл) и к раствору при охлаждении льдом добавляли 4 M HCl/EtOAc (3 мл) с последующим перемешиванием при 50°С в течение 5 часов. Затем растворитель реакции выпаривали. К реакционной жидкости добавляли насыщенный водный бикарбонат натрия и затем экстрагировали хлороформом. Экстракт промывали насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтанамин (1,09 г).
Пример получения 14
2-(1-Циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтанамин (695 мг) растворяли в метиленхлориде (12 мл) и к раствору добавляли тетраизопропоксид титана (1,1 мл) и 1-циклогексен-1-карбоальдегид (309 мг) с последующим перемешиванием при комнатной температуре в течение 3 часов. После этого растворитель выпаривали и к смеси добавляли МеОН (15 мл) и затем цианотригидроборат натрия (190 мг) с последующим перемешиванием смеси в течение 14 часов. Растворитель выпаривали и к смеси добавляли воду и EtOAc. Смесь фильтровали через целит и экстрагировали EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом N-(циклогекса-1-ен-1-илметил)-2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтанамин (585 мг).
Образовавшееся соединение (525 мг) растворяли в 1,4-диоксане (10 мл) и к раствору добавляли ди-трет-бутилдикарбонат (312 мг) с последующим перемешиванием при комнатной температуре в течение 4 часов. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом трет-бутил-N-(циклогекса-1-ен-1-илметил)-N-[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]карбамат (564 мг).
Пример получения 15
Хлорацетилхлорид (0,151 мл) добавляли к раствору 1,1-дифенил-1,2,3,4-тетрагидроизохинолина (339 мг) и моногидрат пара-толуолсульфоната (11,3 мг) в толуоле (5 мл). Смесь нагревали при кипячении с обратным холодильником в течение 3 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли EtOAc и водный 1 М раствор HCl. Органический слой промывали водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, получая при этом 2-(хлорацетил)-1,1-дифенил-1,2,3,4-тетрагидроизохинолин (452 мг).
Пример получения 16
10% Pd, осажденного на угле (900 мг), добавляли к раствору 2-бензил-1,1-дифенил-1,2,3,4-тетрагидроизохинолина (1,81 г) в смеси 2:1 ТГФ-MeOH (30 мл). Смесь перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. Далее к смеси добавляли 10% Pd, осажденного на угле (900 мг), с последующим перемешиванием в течение 8 часов. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 1,1-дифенил-1,2,3,4-тетрагидроизохинолин (339 мг).
Пример получения 17
На ледяной бане при пропускании потока аргона 1,07 M раствор фенилмагнийбромида в ТГФ (33,2 мл) по каплям добавляли к раствору гидробромата 2-бензил-1-фенил-3,4-дигидроизохинолина (8,95 г) в ТГФ (80 мл) на протяжении 1 часа. Смесь перемешивали при комнатной температуре в течение 1 часа. К смеси добавляли насыщенный водный раствор хлорида аммония и затем экстрагировали EtOAc. Экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-бензил-1,1-дифенил-1,2,3,4-тетрагидроизохинолин (1,81 г).
Пример получения 18
Боргидрид натрия (450 мг) добавляли при перемешивании к раствору 6,8-диметокси-1-фенил-3,4-дигидроизохинолина (1,96 г) в EtOH (50 мл) на протяжении 5 минут. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем дополнительно перемешивали при 60°С в течение 1,5 часа. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К образовавшемуся остатку добавляли водный 3 М раствор HCl (60 мл) с последующим нагреванием при кипячении с обратным холодильником в течение 3 минут. После охлаждения к смеси добавляли водный 20% раствор NaOH до достижения сильной щелочности с последующей экстракцией хлороформом. Органический слой промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении, получая при этом 6,8-диметокси-1-фенил-1,2,3,4-тетрагидроизохинолин (1,88 г).
Пример получения 19
1-Циклогексил-6-метил-3,4-дигидроизохинолин (4,84 г) растворяли в МеОН (100 мл) и затем к раствору добавляли боргидрид натрия (966 мг) с последующим перемешиванием при комнатной температуре в течение 3 часов. Растворитель выпаривали при пониженном давлении. К реакционной смеси добавляли воду и затем экстрагировали хлороформом. Экстракт сушили над сульфатом магния и растворитель выпаривали при пониженном давлении.
Образовавшийся остаток растворяли в EtOAc (100 мл) и к раствору при охлаждении льдом добавляли раствор 4 М HCl/EtOAc (8 мл) с последующим перемешиванием при комнатной температуре. Образовавшиеся нерастворимые вещества собирали и промывали EtOAc, получая при этом гидрохлорид 1-циклогексил-6-метил-1,2,3,4-тетрагидроизохинолина (3,6 г).
Пример получения 20
Карбонат калия (92 г) и воду (500 мл) добавляли к гидрохлориду 1-изопропил-6-метокси-1,2,3,4-тетрагидроизохинолина (86 г). Реакционную смесь экстрагировали EtOAc и сушили над сульфатом магния и затем растворитель выпаривали. К образовавшемуся остатку добавляли iPrOH (1100 мл) и (+)-миндальную кислоту (50 г) с последующим перемешиванием при нагревании при 95°С для растворения. Смесь оставляли для охлаждения и перемешивали при комнатной температуре на протяжении ночи. Образовавшееся твердое вещество собирали и перекристаллизовывали три раза с применением iPrOH, получая при этом (+)-манделат 1-изопропил-6-метокси-1,2,3,4-тетрагидроизохинолина (43 г) в виде индивидуального энантиомера.
Пример получения 21
1-Циклогексил-1,2,3,4-тетрагидроизохинолин (31,1 г) растворяли в EtOH (1,26 л) при 80°C и к раствору затем добавляли (D)-винную кислоту (10,83 г). Реакционную смесь оставляли для охлаждения и перемешивали при комнатной температуре на протяжении ночи. Образовавшиеся нерастворимые вещества (16,64 г) собирали и сушили.
Твердое вещество смешивали с твердым веществом, полученным таким же способом, как указано выше, и смесь (33,26 г) растворяли в EtOH (1 л) с последующим перемешиванием при кипячении с обратным холодильником в течение 2 часов и затем перемешивали при 80°С в течение 5 часов. Смесь перемешивали при комнатной температуре на протяжении ночи и затем нерастворимые вещества собирали, получая при этом (D)-тартрат (1S)-1-циклогексил-1,2,3,4-тетрагидроизохинолина (30,8 г).
Пример получения 22
В атмосфере аргона раствор комплекса 1,0 М боран-ТГФ (110 мл) добавляли к смеси (1R,2S)-1-амино-2-инданола (8,17 г) и диэтилового эфира (200 мл) при перемешивании и внутренней температуре 5°С или ниже. Смесь дополнительно перемешивали при комнатной температуре в течение 1,5 часа. Смесь охлаждали до внутренней температуры 4°С. К смеси при внутренней температуре 5°С или ниже постепенно добавляли 1-(2-метоксифенил)-3,4-дигидроизохинолин (10 г) с последующим перемешиванием при такой же температуре в течение 30 минут. Смесь перемешивали при комнатной температуре в течение 3 дней. К реакционной смеси добавляли трифторуксусную кислоту (61 мл) для разложения избытка реагента с последующим нагреванием смеси при кипячении с обратным холодильником в течение 3 часов. После охлаждения диэтиловый эфир выпаривали при пониженном давлении и смесь нагревали при кипячении с обратным холодильником в течение 10 минут. Остаток разбавляли хлороформом и экстрагировали концентрированным водным аммиаком, чтобы сделать его щелочным. Органический слой промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-EtOH-водный аммиак), получая при этом 1-(2-метоксифенил)-1,2,3,4-тетрагидроизохинолин (8,23 г).
1-(2-Метоксифенил)-1,2,3,4-тетрагидроизохинолин (8,227 г) и (2S,3S)-2,3-бис-[(4-метилбензоил)окси]янтарную кислоту (13,282 г) растворяли при перемешивании в ацетонитриле (246 мл) при 70°С. Смесь медленно охлаждали с перемешиванием. Образовавшиеся кристаллы собирали фильтрованием, промывали ацетонитрилом и сушили при пониженном давлении, получая при этом (2S,3S)-2,3-бис-[(4-метилбензоил)окси]сукцинат (1S)-1-(2-метоксифенил)-1,2,3,4-тетрагидроизохинолина (16,193 г).
Пример получения 23
При охлаждении на бане сухой лед-ацетон литийалюминийгидрид (1,03 г) добавляли к ТГФ (30 мл) с получением суспензии. К суспензии в атмосфере аргона по каплям добавляли раствор 1-[2-(трифторметил)фенил]-3,4-дигидроизохинолина (6,22 г) в ТГФ (30 мл). Реакционный раствор перемешивали при комнатной температуре в течение 15 часов. Реакционную жидкость охлаждали на льду и затем для остановки реакции добавляли насыщенный водный раствор сегнетовой соли (1,5 мл). Жидкость перемешивали при комнатной температуре в течение 1 часа и к ней добавляли сульфат магния и целит. Смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 1-[2-(трифторметил)фенил]-1,2,3,4-тетрагидроизохинолин (5,42 г).
Пример получения 24
N-[2-(4-хлорфенил)этил]циклогексанкарбоксамид (2,03 г) растворяли в 1,2-дихлорэтане (15 мл) и к раствору при охлаждении льдом добавляли оксалилхлорид (0,8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем охлаждали до -20°С. К смеси добавляли хлорид железа(III) (1,49 г) с последующим перемешиванием при комнатной температуре в течение 16 часов. К смеси добавляли водный 1 М раствор HCl и затем перемешивали при комнатной температуре в течение 30 минут с последующей экстракцией хлороформом. Экстракт промывали водой и насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток сушили, получая при этом 9-хлор-10b-циклогексил-6,10b-дигидро-5H-[1,3]оксазоло[2,3-a]изохинолин-2,3-дион (2,38 г).
Пример получения 25
9-Хлор-10b-циклогексил-6,10b-дигидро-5H-[1,3]оксазоло[2,3-a]изохинолин-2,3-дион (2,37 г) растворяли в МеОН (16 мл) и к раствору добавляли раствор серной кислоты (8 мл) в МеОН (24 мл) с последующим перемешиванием при кипячении с обратным холодильником в течение 18 часов. Реакционную смесь оставляли для охлаждения и затем растворитель выпаривали. Реакционную смесь нейтрализовали водным 1 М раствором гидроксида натрия, экстрагировали хлороформом, промывали насыщенным раствором соли и затем сушили над сульфатом натрия. Растворитель выпаривали и затем образовавшийся остаток сушили, получая при этом 7-хлор-1-циклогексил-3,4-дигидроизохинолин (1,78 г).
Пример получения 26
N-[2-(2-хлорфенил)этил]циклогексанкарбоксамид (2,55 г) растворяли в 1,2-дихлорэтане (25 мл) и к раствору при охлаждении льдом добавляли оксалилхлорид (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем охлаждали до -20°С. К смеси добавляли хлорид железа (1,87 г) с последующим перемешиванием при комнатной температуре в течение 16 часов. К смеси добавляли водный 1 М раствор HCl с последующим перемешиванием при комнатной температуре в течение 30 минут и экстрагировали хлороформом. Экстракт промывали водой и насыщенным раствором соли и сушили над сульфатом магния. Затем растворитель выпаривали.
Образовавшийся остаток (2,55 г) растворяли в 1,2-дихлорэтане (25 мл) и к раствору при охлаждении льдом добавляли оксалилхлорид (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем охлаждали до
-20°С. К смеси добавляли хлорид железа (1,78 г) с последующим перемешиванием при комнатной температуре в течение 16 часов. К смеси добавляли водный 1 М раствор HCl и затем перемешивали при комнатной температуре в течение 30 минут с последующей экстракцией хлороформом. Экстракт промывали водой и насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток растворяли в МеОН (16 мл) и к раствору добавляли раствор серной кислоты (8 мл) в МеОН (24 мл) с последующим перемешиванием при кипячении с обратным холодильником в течение 18 часов. Реакционную смесь оставляли для охлаждения и затем растворитель выпаривали. Реакционную смесь нейтрализовали водным 1 М раствором гидроксида натрия, экстрагировали хлороформом, промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток сушили, получая при этом 5-хлор-1-циклогексил-3,4-дигидроизохинолин (2,22 г).
Пример получения 27
N-[2-(4-Метоксифенил)этил]циклогексанкарбоксамид (5,56 г) растворяли в толуоле (120 мл) и к раствору последовательно добавляли пентоксид дифосфора (3,0 г) и оксихлорид фосфора (6,0 мл) с последующим перемешиванием смеси при кипячении с обратным холодильником в течение 5,5 часа. Реакционную смесь оставляли для охлаждения и затем растворитель выпаривали. К образовавшемуся остатку добавляли водный 8 М раствор гидроксида калия, воду и хлороформ до полного растворения нерастворимых веществ, чтобы достичь значения рН около 8 с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 1-циклогексил-7-метокси-3,4-дигидроизохинолин (1,87 г).
Пример получения 28
Фосфорную кислоту (11,9 мл) добавляли к пентоксиду дифосфора (20,0 г) на протяжении 5 минут. Смесь перемешивали при 150°С в течение 0,5 часа. К смеси добавляли 3-фтор-N-(2-фенилэтил)бензамид (5,00 г) с последующим перемешиванием при 160°С в течение 2,5 часа. После охлаждения к реакционному раствору добавляли воду и затем добавляли 28% водный аммиак, чтобы раствор был щелочным. Реакционный раствор экстрагировали EtOAc, промывали насыщенным раствором соли и сушили над сульфатом магния. После фильтрования фильтрат концентрировали при пониженном давлении, получая при этом 1-(3-фторфенил)-3,4-дигидроизохинолин (4,87 г).
Пример получения 29
Этилполифосфорную кислоту (50 мл) добавляли к 3,3-дифтор-N-(2-фенилэтил)циклогексанкарбоксамиду (6,4 г) с последующим перемешиванием при нагревании при 120°С в течение 2 часов. Реакционную жидкость добавляли к ледяной воде (150 мл), экстрагировали хлороформом и сушили над сульфатом магния. Растворитель выпаривали, получая при этом 1-(3,3-дифторциклогексил)-3,4-дигидроизохинолин (4,1 г).
Пример получения 30
Этилполифосфорную кислоту (10 мл) добавляли к транс-4-метил-N-(2-фенилэтил)циклогексанкарбоксамиду (2 г) с последующим перемешиванием при нагревании при 120°С в течение 2 часов. К реакционной жидкости добавляли воду и затем смесь экстрагировали EtOAc. Органический слой промывали водой и насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали. К образовавшемуся остатку добавляли EtOH (10 мл) и затем боргидрид натрия (0,31 г) при охлаждении льдом с непосредственным последующим перемешиванием в течение 2 часов. К реакционной жидкости добавляли воду с последующей экстракцией EtOAc. Органический слой промывали водой и насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали, получая при этом 1-(транс-4-метилциклогексил)-1,2,3,4-тетрагидроизохинолин (2 г).
Пример получения 31
N-[2-(2-метилфенил)этил]бутанамид (4,58 г) растворяли в ксилоле (30 мл) и затем к раствору добавляли пентоксид дифосфора (10 г) с последующим перемешиванием при 140°С в течение 4 часов. Реакционную смесь оставляли для охлаждения и затем растворитель выпаривали. Для полного растворения нерастворимых веществ применяли водный 8 М раствор гидроксида калия, воду и хлороформ. рН реакционной смеси регулировали до значения около 8 и экстрагировали хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 5-метил-1-пропил-3,4-дигидроизохинолин (2,14 г).
Пример получения 32
N-[2-(2-Бром-5-метоксифенил)этил]-2-метоксиацетамид (7,8 г) растворяли в ксилоле (80 мл) и к раствору добавляли пентоксид дифосфора (11 г) с последующим перемешиванием при 140°С в течение 4 часов. Затем растворитель выпаривали и к реакционной смеси добавляли водный 6 М раствор гидроксида натрия, чтобы рН смеси был около 8. Реакционную смесь экстрагировали хлороформом, промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc).
Образовавшееся соединение растворяли в EtOH (30 мл) и к раствору добавляли N,N-диизопропилэтиламин и 20% гидроксида палладия, осажденного на активированном угле (400 мг), с последующим перемешиванием в атмосфере водорода при нормальном давлении и комнатной температуре в течение 3 часов. После этого реакционную смесь фильтровали через целит для отделения катализатора и растворитель выпаривали.
К образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия (30 мл) и затем EtOAc (20 мл). К реакционной жидкости на протяжении 5 минут по каплям добавляли раствор хлорацетилхлорида (1,17 мл) в EtOAc (10 мл) с последующим перемешиванием в течение 5 часов. Затем реакционную жидкость экстрагировали EtOAc и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-(хлорацетил)-8-метокси-1-(метоксиметил)-1,2,3,4-тетрагидроизохинолин (367 мг).
Пример получения 33
[2-(4-Хлорфенил)этил]амин (3,5 г) растворяли в смешанном растворе (45 мл) 1:2 EtOAc-насыщенный водный бикарбонат натрия. К реакционной жидкости на протяжении 5 минут добавляли раствор циклогексанкарбонилхлорида (3,35 мл) в EtOAc (18 мл). После перемешивания в течение 1,5 часа реакционную жидкость экстрагировали EtOAc, промывали водным 1 М раствором гидроксида натрия и водой и сушили над сульфатом натрия. Растворитель выпаривали и образовавшийся остаток сушили, получая при этом N-[2-(4-хлорфенил)этил]циклогексанкарбоксамид (5,69 г).
Пример получения 34
4,4-Дифторциклогексанкарбоновую кислоту (1,48 г) растворяли в метиленхлориде (20 мл) и к раствору последовательно добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимид (1,68 г), 1-гидроксибензотриазол (1,21 г) и (2-фенилэтил)амин (1,2 мл) с последующим перемешиванием при комнатной температуре в течение 18 часов. Затем к реакционной жидкости добавляли насыщенный водный бикарбонат натрия и затем смесь экстрагировали хлороформом. Экстракт промывали водой и далее насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 4,4-дифтор-N-(2-фенилэтил)циклогексанкарбоксамид (1,96 г).
Пример получения 35
Раствор 1,64 M трет-бутиллития в н-пентане (12 мл) добавляли к смеси 2-(2,2-диметилпропаноил)-1,2,3,4-тетрагидроизохинолина (3,0 г), тетраметилэтилендиамина (2,2 мл) и ТГФ (40 мл) при -78°С. Реакционную жидкость перемешивали при -78°С в течение 10 минут. Затем к жидкости при -78°С добавляли ацетон (1,8 мл) с последующим перемешиванием при -78°С в течение 1 часа. К реакционной жидкости добавляли уксусную кислоту (2 мл) и температуру повышали до комнатной температуры. Реакционную жидкость выпаривали при пониженном давлении. К остатку добавляли EtOAc и воду с последующим разделением слоев. Органический слой промывали последовательно водным 5% раствором лимонной кислоты, насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над сульфатом магния и выпаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-[2-(2,2-диметилпропаноил)-1,2,3,4-тетрагидроизохинолин-1-ил]пропан-2-ол (2,62 г).
Пример получения 36
Трифторуксусную кислоту (27 мл) добавляли к 2-[2-(2,2-диметилпропаноил)-1,2,3,4-тетрагидроизохинолин-1-ил]пропан-2-олу (2,79 г) с последующим перемешиванием при комнатной температуре в течение 4 часов. После этого растворитель выпаривали и к образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия с последующей экстракцией EtOAc. Экстракт промывали водой и затем сушили над сульфатом магния. Растворитель выпаривали, получая при этом 1-метил-1-(1,2,3,4-тетрагидроизохинолин-1-ил)этилпивалат.
1-Метил-1-(1,2,3,4-тетрагидроизохинолин-1-ил)этилпивалат (2,79 г) растворяли в смешанном растворе (30 мл) 1:4 EtOAc-насыщенный водный бикарбонат натрия. К реакционной жидкости при охлаждении льдом по каплям добавляли раствор хлорацетилхлорида (0,9 мл) в EtOAc (6 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов и экстракцией EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 1-[2-(хлорацетил)-1,2,3,4-тетрагидроизохинолин-1-ил]-1-метилэтилпивалат (3,01 г).
Пример получения 37
Воду (5 мл) и карбонат калия (1,04 г) добавляли к раствору гидрохлорида 6-бром-1-циклогексил-1,2,3,4-тетрагидроизохинолина (1,00 г) в 1,2-дихлорэтане (5 мл). К реакционной смеси добавляли ди-трет-бутилдикарбонат (726 мг) и затем диметиламинопиридин (36,9 мг). Смесь перемешивали при комнатной температуре в течение 4 часов и экстрагировали хлороформом. Экстракт промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении, получая при этом трет-бутил-6-бром-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилат (1,11 г).
Пример получения 38
ДМФА (20 мл) добавляли к смеси трет-бутил-6-бром-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилата (1,41 г), цианида цинка (848 мг) и [1,1'-бис(дифенилфосфино)ферроцен]палладийхлорида (535 мг), которую затем продували газообразным аргоном. Затем к смеси добавляли трис(дибензилиденацетон)дипалладий (458 мг) и смесь затем перемешивали при 120°С в атмосфере аргона в течение 10 часов. Далее к смеси добавляли трис(дибензилиденацетон)дипалладий (200 мг) с последующим перемешиванием в течение 10 часов. Реакционную смесь фильтровали через целит и к фильтрату добавляли EtOAc и воду. Органический слой собирали, промывали водой и насыщенным раствором соли, сушили над сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом трет-бутил-6-циано-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилат (361 мг).
Пример получения 39
4 M HCl/EtOAc (2 мл) добавляли к раствору трет-бутил-6-циано-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилата (361 мг) в EtOAc (1 мл). Смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь вместе с осажденными кристаллами разбавляли диэтиловым эфиром (5 мл). Кристаллы собирали фильтрованием, промывали диэтиловым эфиром и сушили на воздухе, получая при этом гидрохлорид 1-циклогексил-1,2,3,4-тетрагидроизохинолин-6-карбонитрила (259 мг).
Пример получения 40
Трет-бутил-7-(ацетамидметил)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилат (861 мг) растворяли в смешанном растворе (8 мл) 1:1 EtOAc-МеОН и к раствору затем добавляли смесь 4 М HCl/EtOAc (2,8 мл). Реакционную смесь перемешивали при 50°С в течение 6 часов и затем растворитель выпаривали.
К образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия (15 мл) и затем EtOAc (10 мл). К реакционной жидкости на протяжении 5 минут по каплям добавляли раствор хлорацетилхлорида (0,2 мл) в EtOAc (5 мл) с последующим перемешиванием в течение 1 часа. Реакционную жидкость экстрагировали EtOAc и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток сушили, получая при этом N-{[2-(хлорацетил)-1-циклогексил-1,2,3,4-тетрагидроизохинолин-7-ил]метил}ацетамид (655 мг).
Пример получения 41
7-Бром-1-циклогексил-3,4-дигидроизохинолин (9,47 г) растворяли в N-метил-2-пирролидоне (150 мл), к раствору затем добавляли трис(дибензилиденацетон)дипалладий (2,97 г), 1,1'-бис(дифенилфосфино)ферроцен (7,19 г) и цианид цинка (11,5 г) с последующим перемешиванием при 120°С в течение 18 часов. Затем к реакционной жидкости добавляли воду и затем ее фильтровали через целит для отделения нерастворимых веществ. Нерастворимые вещества экстрагировали EtOAc, промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 1-циклогексил-3,4-дигидроизохинолин-7-карбонитрил (7,19 г).
Пример получения 42
7-Бром-1-циклогексил-3,4-дигидроизохинолин (4,01 г) растворяли в 1,4-диоксане (100 мл), к раствору затем добавляли трибутил-(1-этоксивинил)олово (7,43 г), фторид калия (2,39 г) и тетракис(трифенилфосфин)палладий (1,58 г) с последующим перемешиванием смеси при 80°С в течение 5 часов. После этого к реакционной жидкости дополнительно добавляли трибутил-(1-этоксивинил)олово (2,47 г) и тетракис(трифенилфосфин)палладий (1,58 г) с последующим перемешиванием в течение 14 часов. Затем реакционную жидкость фильтровали через целит и нерастворимые вещества отделяли. К нерастворимым веществам добавляли 4 М HCl/диоксан (20 мл) с последующим перемешиванием при 60°С в течение 30 минут. Растворитель выпаривали и к смеси добавляли воду и затем экстрагировали EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 1-(1-циклогексил-3,4-дигидроизохинолин-7-ил)этанон (2,24 г).
Пример получения 43
1-(1-Циклогексил-3,4-дигидроизохинолин-7-ил)этанон (600 мг) растворяли в ТГФ (6 мл). К реакционной жидкости при охлаждении льдом добавляли 0,5 М реагент Теббе в толуоле (4,7 мл) с последующим перемешиванием при комнатной температуре в течение 45 минут. Затем к реакционной жидкости последовательно добавляли диэтиловый эфир и 10 капель водного 1 М раствора NaOH. Реакционную жидкость сушили над сульфатом натрия и фильтровали через целит.
К образовавшемуся раствору добавляли EtOH (8 мл) и 20% гидроксида палладия, нанесенного на активированной уголь (900 мг). Раствор перемешивали в атмосфере водорода при комнатной температуре и нормальном давлении в течение 13 часов. Затем катализатор отделяли фильтрованием через целит и затем растворитель выпаривали.
К образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия (15 мл) и затем EtOAc (10 мл). К реакционной жидкости на протяжении 5 минут добавляли по каплям раствор хлорацетилхлорида (265 мг) в EtOAc (5 мл) с последующим перемешиванием в течение 1 часа. Реакционную жидкость экстрагировали EtOAc и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-(хлорацетил)-1-циклогексил-7-изопропил-1,2,3,4-тетрагидроизохинолин (186 мг).
Пример получения 44
Гидрохлорид 6-бром-1-циклогексил-1,2,3,4-тетрагидроизохинолина (1,0 г) растворяли в ТГФ (20 мл). К реакционной жидкости при -78°С добавляли 1,6 М раствор н-бутиллития в н-гексане (6 мл) с последующим перемешиванием при -78°С в течение 0,5 часа. После этого к реакционной жидкости добавляли ацетон (20 мл) с последующим дополнительным перемешиванием в течение 2 часов. Растворитель выпаривали и к реакционной жидкости добавляли воду с последующей экстракцией смеси хлороформом. Экстракт промывали насыщенным водным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель выпаривали.
К образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия (15 мл) и затем EtOAc (10 мл). К реакционной жидкости на протяжении 5 минут по каплям добавляли раствор хлорацетилхлорида (0,24 мл) в EtOAc (5 мл) с последующим перемешиванием в течение 18 часов. Реакционную жидкость экстрагировали EtOAc и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-[2-(хлорацетил)-1-циклогексил-1,2,3,4-тетрагидроизохинолин-6-ил]пропан-2-ол (646 мг).
Пример получения 45
К гидрохлориду 5-бром-1-изопропил-8-метокси-1,2,3,4-тетрагидроизохинолина (3,0 г) добавляли EtOH (30 мл), триэтиламин (1,3 мл) и 10% палладия, осажденного на угле (0,30 г), с последующим перемешиванием в атмосфере водорода в течение 2 часов. Реакционную жидкость фильтровали через целит и растворитель выпаривали. К реакционной жидкости добавляли водный 1 М раствор NaOH с последующей экстракцией смеси EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток растворяли в EtOAc (30 мл). К смеси добавляли 4 М HCl/EtOAc (5 мл) и осажденное твердое вещество собирали, получая при этом гидрохлорид 1-изопропил-8-метокси-1,2,3,4-тетрагидроизохинолина (2,2 г).
Пример получения 46
(2-Бром-5-метилфенил)ацетонитрил (8,2 г) растворяли в ТГФ (60 мл), к смеси добавляли комплекс боран-диметилсульфид (5 мл) с последующим перемешиванием при 80°С в течение 4 часов. Реакционную жидкость охлаждали на льду и к ней добавляли МеОН (15 мл) с последующим перемешиванием в это время. Затем растворитель выпаривали. К остатку добавляли 4 М HCl/диоксан (30 мл) и затем смесь перемешивали при нагревании при 50°С в течение 1 часа. После оставления для охлаждения к смеси добавляли толуол (100 мл) и осажденное твердое вещество собирали, получая при этом гидрохлорид 2-(2-бром-5-метилфенил)этанамина (5,5 г).
Пример получения 47
Метансульфонилхлорид (3,9 мл) добавляли к смеси (2-бром-5-метилфенил)метанола (9,2 г), дихлорметана (100 мл) и триэтиламина (8 мл) при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 5 часов. К реакционной жидкости добавляли водный 1 М раствор HCl и смесь затем экстрагировали хлороформом. Органический слой сушили над сульфатом магния и фильтровали. После этого растворитель выпаривали.
К образовавшемуся остатку (11 г) добавляли EtOH (60 мл), воду (40 мл) и цианид натрия (2,1 г) с последующим перемешиванием при 80°С в течение 5 часов. К реакционной жидкости добавляли воду с последующей экстракцией EtOAc. Органический слой сушили над сульфатом магния. Растворитель выпаривали, получая при этом (2-бром-5-метилфенил)ацетонитрил (8,3 г).
Пример получения 48
Хлорид алюминия (30 г) добавляли к бензолу (60 мл). К смеси при перемешивании и охлаждении льдом постепенно добавляли 2,6-диметилбензойную кислоту (10 г) с последующим перемешиванием в течение 30 минут. Температуру возвращали к комнатной температуре и смесь дополнительно перемешивали в течение 1 часа с последующим перемешиванием при кипячении с обратным холодильником в течение 4 часов. Реакционную жидкость выливали в ледяную воду (300 мл), фильтровали через целит и экстрагировали хлороформом. Экстракт промывали водным 1 М раствором NaOH и затем сушили над сульфатом магния. Растворитель выпаривали.
Образовавшийся остаток (13 г) растворяли в тетрахлориде углерода (150 мл). К раствору при перемешивании и нагревании при кипячении с обратным холодильником добавляли N-бромсукцинимид (10 г) и 2,2'-азобис(изобутилонитрил) (0,20 г) с последующим перемешиванием при кипячении с обратным холодильником в течение 7 часов. Реакционную жидкость оставляли для охлаждения и фильтровали. Образовавшуюся жидкость промывали насыщенным водным раствором гидрокарбоната натрия и водным раствором тиосульфата натрия и сушили над сульфатом магния. Растворитель выпаривали.
К образовавшемуся остатку (15 г) добавляли EtOH (60 мл), воду (40 мл) и цианид натрия (1,5 г) с последующим перемешиванием при нагревании при 80°С в течение 5 часов. К реакционной жидкости добавляли воду (200 мл) и затем смесь экстрагировали EtOAc и сушили над сульфатом магния. Растворитель выпаривали и образовавшиеся остаток очищали колоночной хроматографией на силикагеле (гексан:хлороформ), получая при этом (2-бензоил-3-метилфенил)ацетонитрил (4,7 г).
Пример получения 49
К (2-бензоил-3-метилфенил)ацетонитрилу (3,3 г) добавляли EtOH (40 мл), 4 M HCl/EtOAc (5 мл) и оксид платины(IV) (0,53 г) с последующим перемешиванием смеси в атмосфере водорода в течение 5 часов. Реакционную жидкость фильтровали через целит и затем концентрировали. К концентрату добавляли толуол с последующей экстракцией водным 1 М раствором HCl. К водному слою добавляли 28% водный раствор аммиака и затем смесь экстрагировали толуолом и сушили над сульфатом магния. Растворитель выпаривали и остаток растворяли в толуоле, к раствору затем добавляли 4 M HCl/EtOAc (5 мл) с последующим концентрированием. К образовавшемуся остатку добавляли iPrOH и диизопропиловый эфир и осажденное твердое вещество собирали, получая при этом гидрохлорид 8-метил-1-фенил-3,4-дигидроизохинолина (1,5 г).
Пример получения 50
К (2-бензоил-3-метилфенил)ацетонитрилу (4,6 г) добавляли EtOH (70 мл), 4 M HCl/EtOAc (15 мл) и оксид платины(IV) (0,40 г) с последующим перемешиванием смеси в атмосфере водорода в течение 3 дней. Реакционную жидкость фильтровали через целит и затем концентрировали. К концентрату добавляли толуол с последующей экстракцией смеси водным 1 М раствором HCl. К водному слою добавляли 28% водный раствор аммиака и затем смесь экстрагировали толуолом и сушили над сульфатом магния. Растворитель выпаривали и остаток растворяли в толуоле. К смеси добавляли 4 M HCl/EtOAc (7 мл) с последующим концентрированием при пониженном давлении. К образовавшемуся остатку добавляли iPrOH и диизопропиловый эфир и осажденное твердое вещество собирали, получая при этом гидрохлорид 1-циклогексил-8-метил-3,4-дигидроизохинолина (2,2 г).
Пример получения 51
Смесь тетралона (1,50 г), 3-метоксфенетиламина (1,86 г) и тетраизопропоксида титана (4,55 мл) перемешивали в атмосфере аргона при 80°С в течение 1 часа. Реакционную смесь охлаждали на бане лед-МеОН. К реакционной смеси при перемешивании и внутренней температуре 0°С или ниже добавляли смесь муравьиной кислоты (39 мл) и уксусного ангидрида (97 мл). После завершения добавления реакционную смесь перемешивали при 80°С в течение 2 часов и к ней добавляли трифторуксусную кислоту (158 мл) с последующим перемешиванием при внутренней температуре 70°С в течение 3 часов. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток делали слабощелочным с применением насыщенного водного раствора гидрокарбоната натрия и экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 6-метокси-3,3',4,4'-тетрагидро-2H,2'H-спиро[изохинолин-1,1'-нафталин]-2-карбальдегид (1,86 г).
Смесь образовавшегося соединения (1,86 г), диоксана (15 мл) и концентрированной хлористоводородной кислоты (3 мл) кипятили с обратным холодильником в течение 2 часов. После охлаждения реакционную жидкость концентрировали при пониженном давлении и образовавшийся остаток делали щелочным добавлением насыщенного водного бикарбоната натрия и экстрагировали хлороформом. Органический слой промывали водой и насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении. Образовавшийся остаток растворяли в EtOAc (40 мл) и к раствору добавляли насыщенный водный бикарбонат натрия (40 мл). К смеси при перемешивании по каплям добавляли раствор хлорацетилхлорида (700 мг) в EtOAc (10 мл) с последующим перемешиванием в течение 1 часа при комнатной температуре. Реакционную смесь разбавляли EtOAc и органический слой промывали насыщенным водным бикарбонатом натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-(хлорацетил)-6-метокси-3,3',4,4'-тетрагидро-2H,2'H-спиро[изохинолин-1,1'-нафталин] (1,28 г).
Пример получения 52
К полифосфорной кислоте, полученной из 80% фосфорной кислоты (25 г) и пентоксида дифосфора (25 г), добавляли смесь 3-метоксифенетиламина (5,2 г) и тетрагидро-4Н-4-пирона (4,13 г) при внутренней температуре 90°С на протяжении 5 минут. Далее реакционную смесь перемешивали в течение 40 минут, охлаждали до комнатной температуры и выливали в ледяную воду (500 мл). К реакционной смеси добавляли концентрированный водный аммиак, чтобы она была сильно щелочной, с последующей экстракцией EtOAc. Экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-EtOH-водный аммиак), получая при этом 6-метокси-2',3,3',4,5',6'-гексагидро-2H-спиро[изохинолон-1,4'-пиран] (2,36 г).
Пример получения 53
При охлаждении на бане лед-МеОН к литийалюминийгидриду (3,03 г) добавляли ТГФ (80 мл) с получением суспензии. К суспензии добавляли дициклопропил[(триметилсилил)окси]ацетонитрил (8,36 г). Смесь перемешивали при комнатной температуре в течение 20 часов и охлаждали на ледяной бане. К смеси добавляли фторид натрия (3,35 г) и далее воду (4,23 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. После этого смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении, получая при этом маслянистое вещество (4,38 г). К маслянистому веществу добавляли EtOAc (80 мл) и смесь затем охлаждали на льду. К смеси добавляли 4 М HCl/EtOAc (8 мл) и затем перемешивали вместе с осажденным твердым веществом при комнатной температуре в течение 1 часа. После этого твердое вещество собирали фильтрованием, промывали EtOAc и сушили при пониженном давлении и 90°С, получая при этом гидрохлорид 2-амино-1,1-дициклопропилэтанола (3,68 г).
Пример получения 54
На ледяной бане в атмосфере аргона иодид цинка (290 мг) добавляли к раствору дициклопропилметанона (5,00 г) в 1,2-дихлорэтане (50 мл). Затем на протяжении 10 минут к смеси по каплям добавляли триметилсилилцианид (6,84 мл). Смесь перемешивали при комнатной температуре в течение 4 часов и к ней дополнительно добавляли триметилсилилцианид (1,71 мл) с последующим перемешиванием при комнатной температуре в течение 20 часов. Реакционную смесь выливали в насыщенный водный раствор гидрокарбоната натрия и экстрагировали EtOAc. Экстракт промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли и сушили над сульфатом магния. К смеси добавляли активированный уголь и затем ее фильтровали через целит. Фильтрат концентрировали при пониженном давлении, получая при этом дициклопропил[(триметилсилил)окси]ацетонитрил (8,36 г).
Пример получения 55
10% палладия, осажденного на угле (300 мг), добавляли к раствору 2-бензил-1-(1-метокси-1-метилэтил)-1,2,3,4-тетрагидроизохинолина (1,17 г) в MeOH (12 мл). Реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 8 часов. Реакционную жидкость фильтровали через целит и фильтрат концентрировали при пониженном давлении, получая при этом 1-(1-метокси-1-метилэтил)-1,2,3,4-тетрагидроизохинолин (770 мг).
Пример получения 56
При охлаждении на бане лед-МеОН в атмосфере аргона раствор 2-(2-бензил-1,2,3,4-тетрагидроизохинолин-1-ил)пропан-2-ола (1,27 г) в ТГФ (7 мл) добавляли по каплям к раствору гидрида натрия (60%, 199 мг) в ТГФ (5 мл) с последующим перемешиванием при комнатной температуре в течение 0,5 часа. Затем реакционную жидкость охлаждали на льду и к ней добавляли метилиодид (0,42 мл). Смесь перемешивали при комнатной температуре в течение 8 часов. К смеси добавляли гидрид натрия (60%, 199 мг) и метилиодид (0,42 мл) с последующим перемешиванием при комнатной температуре в течение 12 часов. К реакционному раствору добавляли воду с последующей экстракцией EtOAc. Экстракт промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-бензил-1-(1-метокси-1-метилэтил)-1,2,3,4-тетрагидроизохинолин (1,17 г).
Пример получения 57
На бане сухой лед-ацетон в атмосфере аргона 1,0 M раствор метиллития в диэтиловом эфире (16,2 мл) добавляли по каплям к раствору этил-2-бензил-1,2,3,4-тетрагидроизохинолин-1-карбоксилата (1,99 г) в ТГФ (20 мл) на протяжении 15 минут. Реакционную жидкость на бане сухой лед-ацетон перемешивали в течение 0,5 часа и затем дополнительно перемешивали на ледяной бане в течение 1 часа. Реакционную жидкость снова охлаждали на бане сухой лед-ацетон и к ней добавляли 1,04 М раствор метиллитий в диэтиловом эфире (3,24 мл). Реакционную жидкость на бане сухой лед-ацетон перемешивали в течение 0,5 часа и затем перемешивали на ледяной бане в течение 1 часа. К реакционной жидкости добавляли воду с последующей экстракцией смеси EtOAc. Экстракт промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-(2-бензил-1,2,3,4-тетрагидроизохинолин-1-ил)пропан-2-ол (1,27 г).
Пример получения 58
Триацетоксиборгидрид натрия (6,11 г) добавляли к раствору гидрохлорида этил-1,2,3,4-тетрагидроизохинолин-1-карбоксилата (4,98 г) и бензальдегида (2,72 г) в уксусной кислоте (50 мл) на ледяной бане. Смесь перемешивали при комнатной температуре в течение 15 часов. К реакционной жидкости добавляли 1 М водный раствор NaOH и смесь экстрагировали хлороформом. Экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом этил-2-бензил-1,2,3,4-тетрагидроизохинолин-1-карбоксилат (1,99 г).
Пример получения 59
Раствор 5-бром-7,8-диметокси-1-фенил-3,4-дигидроизохинолина (450 мг), EtOH (30 мл), 10% палладия, осажденного на угле (80 мг), и 28% раствор метоксида натрия в МеОН (0,1 мл) перемешивали в атмосфере водорода при комнатной температуре на протяжении ночи. Нерастворимые вещества отделяли фильтрованием и фильтрат концентрировали, получая при этом 7,8-диметокси-1-фенил-1,2,3,4-тетрагидроизохинолин (350 мг).
Раствор хлорацетилхлорида (177 мг) в EtOAc (12 мл) добавляли по каплям к смеси 7,8-диметокси-1-фенил-1,2,3,4-тетрагидроизохинолина (350 мг), насыщенного водного раствора гидрокарбоната натрия (50 мл) и EtOAc (50 мл) при перемешивании. После завершения добавления по каплям смесь перемешивали в течение 2 часов и экстрагировали EtOAc. Экстракт промывали последовательно насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-EtOH), получая при этом 2-(хлорацетил)-7,8-диметокси-1-фенил-1,2,3,4-тетрагидроизохинолин (349 мг).
Пример получения 60
1-Циклогексил-N-изобутил-3,4-дигидроизохинолин-7-карбоксамид (689 мг) растворяли в MeOH (12 мл). К реакционной смеси добавляли боргидрид натрия (100 мг) с последующим перемешиванием при комнатной температуре в течение 5 часов. Выпаривали растворитель. К образовавшемуся остатку добавляли воду и хлороформ. Остаток экстрагировали хлороформом и сушили над сульфатом магния. Затем растворитель выпаривали при пониженном давлении, получая при этом 1-циклогексил-N-изобутил-1,2,3,4-тетрагидроизохинолин-7-карбоксамид.
К образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия (10 мл) и затем EtOAc (5 мл). К реакционной жидкости на протяжении 5 минут по каплям добавляли раствор хлорацетилхлорида (0,19 мл) в EtOAc (5 мл) с последующим перемешиванием смеси в течение 1 часа. Затем реакционную жидкость экстрагировали EtOAc и сушили над сульфатом магния. Растворитель выпаривали, получая при этом 2-(хлорацетил)-1-циклогексил-N-изобутил-1,2,3,4-тетрагидроизохинолин-7-карбоксамид (410 мг).
Пример получения 61
5-Метокси-1-(метоксиметил)-3,4-дигидроизохинолин (1,7 г) растворяли в MeOH (15 мл), к раствору затем добавляли боргидрид натрия (376 мг) с последующим перемешиванием при комнатной температуре в течение 5 часов. При пониженном давлении выпаривали растворитель и к смеси добавляли воду и хлороформ. Смесь экстрагировали хлороформом и сушили над сульфатом магния. Растворитель выпаривали.
К образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия (20 мл) и затем EtOAc (15 мл). К реакционной жидкости по каплям на протяжении 5 минут добавляли раствор хлорацетилхлорид (0,66 мл) в EtOAc (5 мл) с последующим перемешиванием смеси в течение 1 часа. Реакционную жидкость экстрагировали EtOAc
и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-(хлорацетил)-5-метокси-1-(метоксиметил)-1,2,3,4-тетрагидроизохинолин (550 мг).
Пример получения 62
5-Бром-8-метокси-1-пропил-1,2,3,4-дигидроизохинолин (5,5 г) растворяли в EtOH (30 мл). К реакционной смеси добавляли ДМФА (3,4 мл) и 10% палладия, осажденного на угле (500 мг), с последующим перемешиванием в атмосфере водорода при нормальном давлении и комнатной температуре в течение 3 часов. После этого катализатор отделяли фильтрованием через целит и к реакционной смеси добавляли боргидрид натрия (740 мг) с последующим перемешиванием смеси при комнатной температуре в течение 2 часов. Растворитель выпаривали при пониженном давлении и к образовавшемуся остатку добавляли воду и хлороформ. Смесь экстрагировали хлороформом и сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении.
Образовавшийся остаток растворяли в EtOAc (10 мл) и затем к раствору при охлаждении льдом добавляли 4 М HCl/EtOAc (15 мл) с последующим перемешиванием смеси при комнатной температуре. Образовавшиеся нерастворимые вещества собирали и промывали EtOAc, получая при этом 8-метокси-1-пропил-1,2,3,4-тетрагидроизохинолин (3,67 г).
Пример получения 63
7-Бром-1-циклогексил-3,4-дигидроизохинолин (24,45 г) растворяли в MeOH (400 мл). Раствор охлаждали до 0°С и к нему добавляли боргидрид натрия (4,8 г) с последующим перемешиванием при комнатной температуре в течение 2 часов. Затем растворитель выпаривали при пониженном давлении. К остатку добавляли воду с последующей экстракцией смеси хлороформом. Органический слой сушили над сульфатом магния и затем растворитель выпаривали при пониженном давлении. Образовавшийся остаток растворяли в EtOAc (200 мл), к раствору затем добавляли 4 М HCl/EtOAc (21 мл). Образовавшееся твердое вещество собирали.
Образовавшийся остаток (1 г) растворяли в ТГФ (30 мл) с последующим охлаждением при -78°С. К реакционной смеси добавляли 2,6 М раствор н-бутиллития в н-гексане (3,7 мл) с последующим перемешиванием в течение 30 минут. К смеси при -78°С добавляли ацетон (30 мл) и температуру повышали до комнатной температуря с последующим перемешиванием в течение 1 часа. Затем растворитель выпаривали при пониженном давлении. К образовавшемуся остатку добавляли воду и смесь экстрагировали хлороформом. Органический слой сушили над сульфатом магния и затем растворитель выпаривали при пониженном давлении.
Образовавшийся остаток растворяли в смешанном растворе EtOAc (10 мл) и насыщенного водного бикарбоната натрия (15 мл). К реакционной смеси по каплям добавляли раствор хлорацетилхлорида (683 мг) в EtOAc (5 мл) с последующим перемешиванием при комнатной температуре в течение двух дней. Затем к смеси добавляли воду с последующей экстракцией EtOAc. Органический слой промывали насыщенным раствором соли и сушили над сульфатом магния. После этого растворитель выпаривали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 2-[2-(хлорацетил)-1-циклогексил-1,2,3,4-тетрагидроизохинолин-7-ил]пропан-2-ол (606 мг).
Пример получения 64
1-Циклогексил-3,4-дигидроизохинолин-7-карбонитрил (1,01 г) растворяли в EtOH (15
мл), к раствору затем добавляли 6 М водный раствор NaOH (7,0 мл) с последующим перемешиванием смеси при кипячении с обратным холодильником в течение 6 часов. К смеси добавляли воду и затем промывали EtOAc. К смеси добавляли 1 М водный раствор HCl для достижения рН около 3 и затем к ней добавляли насыщенный водный раствор сульфата натрия. Смесь экстрагировали смешанным раствором 4:1 хлороформ-iPrOH, промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали, получая при этом 1-циклогексил-3,4-дигидроизохинолин-7-карбоновую кислоту (1,09 г).
Пример получения 65
1-Циклогексил-3,4-дигидроизохинолин-7-карбоновую кислоту (1,15 г) растворяли в метиленхлориде (15 мл). К раствору добавляли гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (2,03 г), N,N-диизопропилэтиламин (1,55 мл) и 2-метил-1-пропанамин (0,87 мл) с последующим перемешиванием смеси при комнатной температуре в течение 18 часов. Затем к смеси добавляли воду с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 1-циклогексил-N-изобутил-3,4-дигидроизохинолин-7-карбоксамид (700 мг).
Пример получения 66
Метоксид натрия (9,46 г) добавляли к суспензии 10b-(хлорметил)-9-этил-6,10b-дигидро-5H-[1,3]оксазоло[2,3-a]изохинолин-2,3-диона (14,0 мл) в MeOH (140 мл) при охлаждении льдом. Смесь перемешивали при комнатной температуре в течение 0,5 часа и затем нагревали при кипячении с обратным холодильником в течение 3 часов. К смеси добавляли EtOAc и воду с последующим фильтрованием. Органический слой фильтрата собирали, промывали насыщенным раствором соли и сушили над сульфатом магния. К нему добавляли активированный уголь и силикагель с последующим фильтрованием смеси. Фильтрат концентрировали при пониженном давлении, получая при этом 7-этил-1-(метоксиметил)-3,4-дигидроизохинолин (4,75 г).
Пример получения 67
Боргидрид натрия (900 мг) добавляли к смешанному раствору раствора 5,8-диметокси-1-фенил-2-трифторацетил-1,2,3,4-тетрагидроизохинолин (4,21 г) в ТГФ (30 мл) и EtOH (100 мл) при комнатной температуре и при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов и затем перемешивали при 40°С в течение 30 минут и концентрировали при пониженном давлении. К образовавшемуся остатку добавляли 3 М водный раствор HCl (30 мл) с последующим кипячением с обратным холодильником в течение 5 минут. После охлаждения смесь делали сильно щелочной с применением 20% водного раствора NaOH и экстрагировали хлороформом. Органический слой промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении, получая при этом 5,8-диметокси-1-фенил-1,2,3,4-тетрагидроизохинолин (3,05 г).
Пример получения 68
В атмосфере аргона раствор 2,5-диметоксифенетиламина (3,175 г) в бензоле (4 мл) добавляли при перемешивании к суспензии бензальдегида (1,86 г) и сульфата магния (3,89 г) в бензоле (10 мл). Имела место экзотермическая реакция, реакционную смесь далее перемешивали на протяжении ночи. После завершения реакции реакционную жидкость фильтровали и фильтрат концентрировали при пониженном давлении, получая при этом 2-(2,5-диметоксифенил)-N-[(1E)-фенилметилен]этанамин (4,72 г).
Пример получения 69
2-(2,5-Диметоксифенил)-N-[(1E)-фенилметилен]этанамин (4,719 г) растворяли в трифторуксусной кислоте с последующим кипячением раствора с обратным холодильником в течение 2 дней. Реакционную смесь охлаждали до комнатной температуры и к ней постепенно добавляли трифторуксусный ангидрид (55 мл). Смесь кипятили с обратным холодильником в течение 3 дней, охлаждали до комнатной температуры и концентрировали при пониженном давлении. Образовавшийся остаток экстрагировали насыщенным водным раствором гидрокарбоната натрия и хлороформом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, сушили над сульфатом магния и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом 5,8-диметокси-1-фенил-2-(трифторацетил)-1,2,3,4-тетрагидроизохинолин (4,168 г).
Пример получения 70
трет-Бутил-7-циано-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилат (2,38 г) растворяли в метиленхлориде (40 мл). К раствору добавляли 0,99 М раствор изобутилалюминийгидрида в н-гексане (7,8 мл) с последующим перемешиванием при -78°С в течение 4 часов. Затем к реакционной смеси дополнительно добавляли 0,99 М раствор изобутилалюминийгидрида в н-гексане (28 мл). Для остановки реакции добавляли насыщенный водный раствор сегнетовой соли с последующим перемешиванием смеси на протяжении ночи. Смесь экстрагировали EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (бензол-EtOAc).
Образовавшийся остаток растворяли в смешанном растворителе (90 мл) 8:1 EtOH-уксусная кислота-вода. К раствору добавляли гидрохлорид гидроксиламина (812 мг) и ацетат натрия (930 мг) с последующим перемешиванием раствора при комнатной температуре в течение 28 часов. Затем растворитель выпаривали. К смеси добавляли воду и смесь затем экстрагировали хлороформом, промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали, получая при этом трет-бутил-1-циклогексил-7-[(гидроксиимино)метил]-3,4-дигидроизохинолин-2(1H)-карбоксилат (2,03 г).
Пример получения 71
трет-Бутил-1-циклогексил-7-[(гидроксиимино)метил]-3,4-дигидроизохинолин-2(1H)-карбоксилат (1,04 г) растворяли в смешанном растворителе (20 мл) 8:1:1 EtOH-уксусная кислота-вода. К раствору добавляли 10% палладия, осажденного на активированный уголь (500 мг), с последующим перемешиванием смеси в атмосфере водорода при комнатной температуре и нормальном давлении в течение 4 часов. Затем реакционную смесь фильтровали через целит и растворитель выпаривали.
Образовавшийся остаток растворяли в метиленхлориде (12 мл), к раствору затем добавляли триэтиламин (880 мг), уксусный ангидрид (385 мг) и 4-диметиламинопиридин (70 мг) с последующим перемешиванием смеси при комнатной температуре в течение 16 часов. Затем к смеси добавляли воду и экстрагировали хлороформом. Экстракт промывали насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ), получая при этом трет-бутил-7-(ацетамидометил)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилат (875 мг).
Пример получения 72
трет-Бутил-1-циклогексил-7-[(гидроксиимино)метил]-3,4-дигидроизохинолин-2(1H)-карбоксилат (995 мг) растворяли в смеси EtOH-уксусная кислота-вода (8:1:1, 20 мл). К реакционной смеси добавляли 10% палладия, осажденного на уголь (480 мг), с последующим перемешиванием в атмосфере водорода при комнатной температуре и нормальном давлении в течение 4 часов. Затем реакционную смесь фильтровали через целит и растворитель выпаривали, получая при этом трет-бутил-7-(аминометил)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилат (956 мг).
Пример получения 73
трет-Бутил-7-(аминометил)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-карбоксилат (999 мг) растворяли в метиленхлориде. К раствору добавляли изомасляную кислоту (0,33 мл), триэтиламин (1,2 мл) и гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметиламмония (1,32 г) с последующим перемешиванием при комнатной температуре в течение 18 часов. К смеси добавляли воду и затем экстрагировали хлороформом. Экстракт промывали 1 М водным раствором NaOH и насыщенным раствором соли и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле, получая при этом трет-бутил-1-циклогексил-7-[(изобутиламино)метил]-3,4-дигидроизохинолин-2(1H)-карбоксилат (468 мг).
Пример получения 74
1-Циклогексил-3,4-дигидроизохинолин-7-ол (2 г) растворяли в MeOH (40 мл), к раствору затем добавляли боргидрид натрия (396 мг) с последующим перемешиванием при комнатной температуре в течение 4 часов. Затем растворитель выпаривали. К смеси добавляли воду и экстрагировали хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали.
Образовавшийся остаток растворяли в диоксане (40 мл). К раствору добавляли ди-трет-бутилдикарбонат (2,28 г) с последующим перемешиванием при комнатной температуре в течение 2 дней. Затем растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом трет-бутил-1-циклогексил-7-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилат (2,66 г).
Пример получения 75
трет-Бутил-1-циклогексил-7-гидрокси-3,4-дигидроизохинолин-2(1H)-карбоксилат (700 мг) растворяли в ацетонитриле (12 мл). К раствору добавляли 1-хлорацетон (0,2 мл), карбонат калия (438 мг) и иодид тетра-н-бутиламмония (78 мг) с последующим перемешиванием смеси при 60°С в течение 16 часов. Затем к смеси добавляли воду и затем экстрагировали EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом трет-бутил-1-циклогексил-7-(2-оксопропокси)-3,4-дигидроизохинолин-2(1H)-карбоксилат (818 мг).
Пример получения 76
трет-Бутил-1-циклогексил-7-(2-оксопропокси)-3,4-дигидроизохинолин-2(1H)-карбоксилат (808 мг) растворяли в метиленхлориде (15 мл). К реакционной смеси по каплям при -78°С добавляли бис-(2-метоксиэтил)аминосератрифторид (0,65 мл) в метиленхлориде (5 мл) с последующим перемешиванием при комнатной температуре в течение 14 часов. К реакционной смеси добавляли насыщенный водный бикарбонат натрия и затем экстрагировали хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc), получая при этом трет-бутил-1-циклогексил-7-(2,2-дифторпропокси)-3,4-дигидроизохинолин-2(1H)-карбоксилат (747 мг).
Пример получения 77
1 M водный раствор NaOH добавляли к гидрохлориду (1S)-1-изопропил-8-метокси-1,2,3,4-тетрагидроизохинолина (808 мг). Реакционную смесь экстрагировали хлороформом и органический слой сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении. Смешанный раствор 1 М раствора трибромида бора в дихлорметане (13,4 мл) и дихлорметана (10 мл) охлаждали до -78°С и к нему по каплям добавляли раствор остатка экстракции в дихлорметане (10 мл). Температуру постепенно повышали и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Затем к реакционной смеси добавляли насыщенный водный бикарбонат натрия и хлороформ. После завершения разделения жидкости на слои водный слой применяли в следующей реакции.
К образовавшемуся водному слою добавляли ди-трет-бутилдикарбонат с последующим перемешиванием при комнатной температуре в течение 5 часов. Смесь нейтрализовали 1 М водным раствором HCl и экстрагировали хлороформом. Органический слой промывали водой и сушили над сульфатом магния. После этого растворитель выпаривали при пониженном давлении, получая при этом трет-бутил-(1S)-8-гидрокси-1-изопропил-3,4-дигидроизохинолин-2(1H)-карбоксилат (973 мг).
Пример получения 78
трет-Бутил-(1S)-8-гидрокси-1-изопропил-3,4-дигидроизохинолин-2(1H)-карбоксилат (973 мг) растворяли в смешанном растворе iPrOH (6 мл) и 30% водного раствора гидроксида калия (3 мл). К реакционной смеси добавляли хлордифторметан посредством вдувания с последующим перемешиванием при 70°С в течение 20 часов. К смеси добавляли воду и затем экстрагировали хлороформом. Органический слой сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении и затем остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc).
Образовавшийся остаток (790 мг) растворяли в EtOAc. К раствору добавляли 4 М HCl/EtOAc (5,8 мл) с последующим перемешиванием при 60°С в течение 18 часов. Затем растворитель выпаривали при пониженном давлении, получая при этом гидрохлорид (1S)-8-(дифторметокси)-1-изопропил-1,2,3,4-тетрагидроизохинолина (642 мг).
Пример получения 79
1 M водный раствор NaOH добавляли к гидрохлориду (1S)-8-метокси-1-фенил-1,2,3,4-тетрагидроизохинолина (1,81 г). Реакционную смесь экстрагировали хлороформом. Органический слой сушили над сульфатом магния и растворитель выпаривали при пониженном давлении. Смешанный раствор раствора 1 М трибромида бора в дихлорметане (26,3 мл) и дихлорметана (30 мл) охлаждали до -78°С и к нему по каплям добавляли раствор остатка экстракции в дихлорметане (10 мл). Температуру постепенно повышали и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Затем к реакционной смеси добавляли насыщенный водный бикарбонат натрия и хлороформ. После завершения разделения жидкости на слои органический слой сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении.
Образовавшийся остаток (1,48 г) растворяли в ТГФ (50 мл). К раствору добавляли 1 М водный раствор NaOH (8 мл) и ди-трет-бутилдикарбонат (2,87 г) с последующим перемешиванием смеси при комнатной температуре в течение 5 часов. Затем растворитель выпаривали при пониженном давлении. К образовавшемуся остатку добавляли воду и 1 М водный раствор HCl и смесь экстрагировали хлороформом. Органический слой промывали водой и сушили над сульфатом магния. Затем растворитель выпаривали при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc).
Образовавшийся остаток (1,24 г) растворяли в смешанном растворе iPrOH (20 мл) и 50% водного раствора гидроксида натрия (10 мл). К реакционной смеси добавляли хлордифторметан вдуванием с последующим перемешиванием при 70°С в течение 14 часов. К смеси добавляли воду и затем ее экстрагировали хлороформом. Органический слой сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении и затем образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан-EtOAc).
Образовавшийся остаток (927 мг) растворяли в EtOAc (25 мл) и к раствору затем добавляли 4 М HCl/EtOAc (6,2 мл) с последующим перемешиванием при 60°С в течение 18 часов. Затем растворитель выпаривали при пониженном давлении, получая при этом гидрохлорид (1S)-8-(дифторметокси)-1-фенил-1,2,3,4-тетрагидроизохинолина (770 мг).
Пример получения 80
28% водный аммиак добавляли к 1-оксиран-2-илциклогексанолу с последующим перемешиванием в течение 11 часов. Растворитель выпаривали при пониженном давлении и затем воду удаляли азеотропной перегонкой с толуолом.
Образовавшийся остаток растворяли в смешанном растворе EtOH-диэтиловый эфир. К раствору добавляли щавелевую кислоту с последующим перемешиванием в течение некоторого времени. Образовавшиеся нерастворимые вещества собирали, получая при этом оксалат 1-(2-амино-1-гидроксиэтил)циклогексанола (853 мг).
Пример получения 81
(1R,2S)-1-амино-2-инданол (511 мг) растворяли в толуоле (60 мл). К раствору добавляли при охлаждении льдом раствор комплекса 1 М боран-ТГФ в ТГФ (8,16 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем к смеси добавляли 7-бром-1-циклогексил-3,4-дигидроизохинолин (1 г) с последующим перемешиванием при комнатной температуре в течение 3 дней. Реакцию останавливали добавлением трифторуксусной кислоты с последующим дополнительным перемешиванием при 60°С в течение 1 часа. Растворитель выпаривали. К смеси добавляли 1 М водный раствор гидроксида натрия и затем экстрагировали хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом сырой (1S)-7-бром-1-циклогексил-3,4-тетрагидроизохинолин (1,06 г). Образовавшийся сырой продукт (203 мг) растворяли в EtOH (9 мл). К смеси добавляли D-(-)-винную кислоту (104 мг) при 80°C. Смесь постепенно охлаждали до комнатной температуры с последующим перемешиванием в течение 12 часов. Образовавшиеся нерастворимые вещества собирали, получая при этом (1S)-7-бром-1-циклогексил-3,4-тетрагидроизохинолин (72 мг).
Пример получения 82
В атмосфере аргона к суспензии (1R,2S)-1-аминоиндан-2-ола (3,04 г) в толуоле (60 мл) при охлаждении льдом добавляли раствор 1,09 M боран-ТГФ (18,7 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем к реакционной смеси при охлаждении льдом добавляли раствор 1-[2-[(трифторметил)бензил]-3,4-дигидроизохинолина (4,00 г) в толуоле (20 мл). Смесь перемешивали при 4°С в течение 45 часов. Для остановки реакции добавляли трифторуксусную кислоту (20 мл). Смесь нагревали при кипячении с обратным холодильником в течение 1 часа и затем охлаждали. К смеси добавляли 28% водный аммиак (30 мл), чтобы сделать ее щелочной. Смесь экстрагировали EtOAc. Экстракт промывали 3 раза и дополнительно промывали насыщенным раствором соли, сушили над сульфатом магния и затем фильтровали. Фильтрат концентрировали, получая при этом желтое маслянистое вещество (4,11 г). Маслянистое вещество растворяли в ацетонитриле (80 мл). К смеси добавляли N-ацетил-L-лейцин (2,39 г) при 80°С. Смесь постепенно охлаждали с последующим перемешиванием при 60°С в течение 2 часов и при комнатной температуре в течение 12 часов. Когда кристаллы осадили, кристаллы собирали фильтрованием, охлаждали на льду, промывали ацетонитрилом и сушили на воздухе, получая при этом кристаллы (2,48 г). Кристаллы перекристаллизовывали из ацетонитрила (50 мл), получая при этом соль N-ацетил-L-лейцина 1-[2-(трифторметил)бензил]-1,2,3,4-тетрагидроизохинолина (1,72 г).
Пример получения 83
В атмосфере аргона раствор 1,09 M боран-ТГФ (48,8 мл) добавляли к суспензии (1R,2S)-1-аминоиндан-2-ола (3,79 г) в толуоле (60 мл) при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем к реакционной смеси при охлаждении льдом добавляли раствор 7-этил-1-(метоксиметил)-3,4-дигидроизохинолина (4,70 г) в ТГФ (40 мл). Смесь перемешивали при 4°С в течение 8 часов. Для остановки реакции добавляли трифторуксусную кислоту (15 мл). Смесь нагревали при кипячении с обратным холодильником в течение 1 часа. К смеси добавляли хлороформ и 28% водный аммиак. Органический слой промывали 3 раза водой и экстрагировали 5% водным раствором уксусной кислоты. Экстракт делали щелочным добавлением 28% водного аммиака и экстрагировали EtOAc. Экстракт промывали два раза водой и далее промывали насыщенным раствором соли, сушили над сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении, получая при этом желтое маслянистое вещество (3,15 г). Маслянистое вещество растворяли в iPrOH (63 мл), к раствору постепенно при 90°С добавляли (2S,3S)-2,3-бис(бензоилокси)янтарную кислоту (4,14 г) при 90°C. Смесь нагревали при кипячении с обратным холодильником в течение 1 часа и постепенно охлаждали до комнатной температуры с последующим перемешиванием при комнатной температуре в течение 3 часов. Когда кристаллы были осаждены, кристаллы собирали фильтрованием, промывали iPrOH и простым эфиром и сушили при пониженном давлении, получая при этом (2S,3S)-2,3-бис(бензоилокси)сукцинат (7-этил-1-(метоксиметил)-1,2,3,4-тетрагидроизохинолина (5,54 г).
Пример получения 84
Пентоксид фосфора (15,85 г) добавляли к раствору 2-хлор-N-[2-(4-этилфенил)этил]ацетамида (7,87 г) в ксилоле (140 мл) при перемешивании при 90°C на протяжении 5 минут. Реакционную смесь нагревали при 120°С и перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. Супернатант удаляли и остаток промывали последовательно толуолом и простым эфиром. К остатку добавляли измельченный лед (150 г) с последующим перемешиванием смеси. К смеси дополнительно добавляли 20% водный раствор гидроксида натрия до рН 10 или более и смесь экстрагировали хлороформом. Органический слой собирали, промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния. К органическому слою добавляли раствор 4 М HCl/EtOAc (15 мл) и смесь концентрировали при пониженном давлении, получая при этом гидрохлорид 1-(хлорметил)-7-этил-3,4-дигидроизохинолина (8,5 г).
Пример получения 85
Трет-бутил-(1S)-8-гидрокси-1-фенил-3,4-дигидроизохинолин-2(1H)-карбоксилат (1,53 г) растворяли в дихлорметане (20 мл). К раствору при -78°С добавляли 2,6-лутидин (1,1 мл) и ангидрид трифторметансульфоновой кислоты (0,9 мл) с последующим перемешиванием при комнатной температуре в течение 16 часов. Затем к смеси добавляли насыщенный водный бикарбонат натрия и затем экстрагировали хлороформом. Экстракт промывали насыщенным водным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (этилацетат:гексан), получая при этом трет-бутил (1S)-1-фенил-8-{[(трифторметил)сульфонил]окси}-3,4-дигидроизохинолин-2(1H)-карбоксилат (2,08 г).
Пример получения 86
К N-[2-(2-бром-5-метилфенил)этил]-2-метоксиацетамиду (3,9 г) добавляли ксилол (50 мл) с последующим перемешиванием при нагревании при 60°С. К реакционной смеси при перемешивании добавляли пентоксид фосфора (7,0 г) с последующим перемешиванием при 140°С в течение 3 часов. После оставления реакционной смеси для охлаждения супернатант реакционной смеси выгружали. Смесь растворяли в смеси воды, толуола и водного раствора гидроксида натрия и раствор экстрагировали толуолом. Экстракт дополнительно экстрагировали 1 М водным раствором HCl. Выделенный водный слой нейтрализовали, экстрагировали толуолом и сушили над сульфатом магния. После завершения фильтрования к слою добавляли раствор 4 М HCl/EtOAc (5 мл) и растворитель выпаривали при пониженном давлении. К образовавшемуся остатку добавляли EtOH (50 мл), толуол (10 мл) и боргидрид натрия (1,0 г) с последующим перемешиванием смеси в течение 4 дней. К реакционной смеси добавляли 1 М водный раствор HCl с последующим перемешиванием в течение 5 часов. После этого к смеси добавляли водный раствор гидроксида натрия и затем экстрагировали хлороформом. Растворитель выпаривали. К образовавшемуся остатку добавляли карбонат натрия (1,0 г), воду (30 мл), толуол (30 мл) и хлорацетилхлорид (0,3 мл) при охлаждении льдом с последующим перемешиванием смеси при комнатной температуре в течение 17 часов. К реакционной жидкости добавляли воду и затем смесь экстрагировали хлороформом. Экстракт промывали 1 М водным раствором HCl и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали хроматографией на силикагеле, получая при этом 5-бром-2-(хлорацетил)-1-(метоксиметил)-8-метил-1,2,3,4-тетрагидроизохинолин (0,323 г).
Пример получения 87
Гидрохлорид 8-этил-5-метокси-1-фенил-1,2,3,4-тетрагидроизохинолина (2,06 г) растворяли в метиленхлориде (40 мл). К реакционной смеси добавляли раствор трибромида бора в дихлорметане (13,6 мл) при -78°С с последующим перемешиванием при комнатной температуре в течение 16 часов. После этого к реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, чтобы реакционную смесь сделать щелочной. Затем к реакционному раствору добавляли ди-трет-бутилдикарбонат (2,96 г) с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционную жидкость экстрагировали хлороформом, промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток растворяли в дихлорметане (20 мл). К смеси добавляли 2,6-лутидин (1,8 мл) и ангидрид трифторметансульфоновой кислоты (1,55 мл) с последующим перемешиванием при комнатной температуре в течение 18 часов. После этого к смеси добавляли воду и затем экстрагировали хлороформом. Экстракт промывали насыщенным водным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (EtOAc:гексан). Образовавшийся остаток растворяли в ДМФА (30 мл). К смеси добавляли ацетат палладия(II) (305 мг), триэтилсилан (5,4 мл) и 1,1'-бис(дифенилфосфино)ферроцен (750 мг) с последующим перемешиванием при 70°С в течение 20 часов. Затем к смеси добавляли воду и смесь затем фильтровали через целит и экстрагировали диэтиловым эфиром. Экстракт промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (EtOAc:гексан), получая при этом трет-бутил-8-этил-1-фенил-3,4-дигидроизохинолин-2(1H)-карбоксилат (2,29 г).
Пример получения 88
При охлаждении на ледяной бане к метоксиэтанолу (100 мл) при перемешивании на протяжении 20 минут добавляли смесь гидрида натрия (суспензия 8 г гидрида натрия в минеральном масле (60%), промытая гексаном) и ТГФ (10 мл) с получением 2-метоксиэтоксида натрия и последующим дополнительным перемешиванием в течение 2 часов. При перемешивании раствор 2-метоксиэтоксида натрия в 2-метоксиэтаноле (55 мл) добавляли к раствору гидрохлорида 1-(хлорметил)-7-этил-3,4-дигидроизохинолина (8,5 г) в метоксиэтаноле (50 мл) при охлаждении на ледяной бане в течение 5 минут. Реакционную смесь нагревали при 60°С с последующим перемешиванием в течение 3 часов в атмосфере аргона. Реакционную смесь охлаждали до комнатной температуры и разбавляли ТГФ (150 мл) с последующим фильтрованием. Фильтрат концентрировали при пониженном давлении. К образовавшемуся остатку добавляли насыщенный водный раствор хлорида аммония и остаток экстрагировали EtOAc. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния и концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:EtOAc), получая при этом 7-этил-1-[(2-метоксиэтил)метил]-3,4-дигидроизохинолин (2,13 г).
Пример получения 89
трет-Бутил-(1S)-1-фенил-8-{[(трифторметил)сульфонил]окси}-3,4-дигидроизохинолин-2(1H)-карбоксилат (3,75 г) растворяли в N,N-диметилацетамиде (40 мл). К реакционной смеси добавляли цинк (537 мг), цианид цинка (1,15 г), трифторацетат палладия(II) (682 мг) и бифенил-2-ил(ди-трет-бутил)фосфин (1,22 г). Температуру повышали от комнатной температуры до 95°С на протяжении 45 минут и затем смесь перемешивали при 95°С в течение 18 часов. К смеси добавляли воду и затем фильтровали через целит. После этого смесь экстрагировали диэтиловым эфиром. Экстракт промывали насыщенным водным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (EtOAc-гексан), получая при этом трет-бутил-(1S)-8-циано-1-фенил-3,4-дигидроизохинолин-2(1H)-карбоксилат (796 мг).
Пример получения 90
1,55 M раствор н-бутиллития в гексане (10,94 мл) добавляли при перемешивании к раствору 1-(метоксиметил)-5-метил-1,2,3,4-тетрагидроизохинолина (3,07 г) в ТГФ (60 мл) в атмосфере аргона на протяжении около 8 минут при -70°С или ниже с последующим дополнительным перемешиванием в течение 30 минут. К реакционной смеси добавляли при перемешивании раствор (1R,2S,5R)-2-изопропил-5-метилциклогексил-4-метилбензолсульфината (3,375 г) в ТГФ (25 мл) на протяжении 5 минут при -70°C или ниже с последующим дальнейшим перемешиванием в течение 1 часа. После этого к смеси при такой же температуре добавляли насыщенный динатрийфосфат и температуру повышали до комнатной температуры. Смесь экстрагировали простым эфиром. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом магния и затем растворитель выпаривали. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:EtOAc), получая при этом (1R)-1-(метоксиметил)-5-метил-2-[(R)-(4-метилфенил)сульфинил]-1,2,3,4-тетрагидроизохинолин (2,144 г) (величина Rf = 0,14).
Пример получения 91
К смешанному раствору (1R)-1-(метоксиметил)-5-метил-2-[(R)-(4-метилфенил)сульфинил]-1,2,3,4-тетрагидроизохинолина (2,47 г) в EtOH (45 мл) и ТГФ (10 мл) добавляли концентрированную хлористоводородную кислоту (3,1 мл) при перемешивали при 0°С с последующим дополнительным перемешиванием в течение 10 минут. К смеси добавляли насыщенный водный раствор карбоната натрия (50 мл) с последующей экстракцией EtOAc. Органический слой промывали 1 М водным раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния и затем концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ:EtOH:водный аммиак), получая при этом (1R)-1-(метоксиметил)-5-метил-1,2,3,4-тетрагидроизохинолин (1,276 г).
Химические структуры соединений, полученных способами вышеуказанных примеров получения, показаны в таблицах 6-12 (см. в конце описания). Кроме того, таким же способом, как способы вышеуказанных примеров получения, соединения примеров получения, показанные в таблицах 13-35 (см. в конце описания), получают с применением соответствующих исходных соединений. Данные инструментального анализа этих соединений примеров получения показаны в таблицах 36-42 (см. в конце описания).
Пример 1
(1S)-2-(хлорацетил)-1-циклогексил-1,2,3,4-тетрагидроизохинолин (4,496 г) растворяли в ацетонитриле (100 мл). К смеси добавляли карбонат калия (6,25 г), иодид тетра-н-бутиламмония (679 мг) и гидрохлорид 1-(аминометил)циклогексанола (4,50 г) с последующим перемешиванием при 60°С в течение 6 часов. После этого растворитель выпаривали и к реакционной смеси добавляли воду с последующей экстракцией смеси хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле, получая при этом 1-[({2-[(1S)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол (3,02 г).
Образовавшееся соединение (3,02 г) растворяли в EtOH, к раствору добавляли щавелевую кислоту (777 мг). После достижения полного растворения смесь перемешивали в течение некоторого времени и образовавшиеся нерастворимые вещества собирали, получая при этом оксалат 1-[({2-(1S)-1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанола (2,985 г).
Пример 2
2-Акрилоил-1-циклогексил-1,2,3,4-тетрагидроизохинолин (516 мг) растворяли в iPrOH (15 мл). К реакционной смеси добавляли гидрохлорид 1-(аминометил)циклогексанола (635 мг) и триэтиламин (0,59 мл) с последующим перемешиванием при кипячении с обратным холодильником в течение 16 часов. После этого растворитель выпаривали и к смеси добавляли воду с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН) и затем дополнительно очищали колоночной хроматографией на щелочном силикагеле (хлороформ), получая при этом 1-({[3-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-3-оксопропил]амино}метил)циклогексанол (214 мг).
Образовавшееся соединение (214 мг) растворяли в EtOH (8 мл). К реакционной смеси добавляли щавелевую кислоту (51 мг), получая при этом оксалат 1-({[3-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-3-оксопропил]амино}метил)циклогексанола (229 мг).
Пример 3
1-Циклогексил-7-изопропоксид-3,4-дигидроизохинолин (245 мг) растворяли в MeOH (6 мл). После этого к реакционной жидкости добавляли боргидрид натрия (40 мг) с последующим перемешиванием смеси при комнатной температуре в течение 4 часов. Растворитель выпаривали при пониженном давлении и к смеси добавляли воду и хлороформ. Реакционную жидкость экстрагировали хлороформом и сушили над сульфатом магния. После этого растворитель выпаривали при пониженном давлении.
К образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия (6 мл), к смеси добавляли EtOAc (3 мл). К реакционной жидкости на протяжении 5 минут по каплям добавляли раствор хлорацетилхлорида (102 мг) в EtOAc (3 мл) с последующим перемешиванием смеси в течение 1 часа. После этого реакционную жидкость экстрагировали EtOAc и сушили над сульфатом магния и затем растворитель выпаривали.
Образовавшийся остаток растворяли в 1,4-диоксане (8 мл). К раствору добавляли (2R)-1-амино-2-пропанол (180 мг) и 1,8-диазабицикло[5.4.0]ундека-7-ен (146 мг) с последующим перемешиванием при 50°С в течение 3 часов. После этого растворитель выпаривали и к реакционной жидкости добавляли воду с последующей экстракцией смеси EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН).
Образовавшийся остаток (211 мг) растворяли в смешанном растворе 1:4 iPrOH-диэтиловый эфир. К реакционной жидкости добавляли щавелевую кислоту (49 мг), получая при этом оксалат (2R)-1-{[2-(1-циклогексил-7-изопропоксид-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}пропан-2-ола (223 мг).
Пример 4
N-(2-Циклогекса-1-ен-1-илэтил)-N-[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]-2,2,2-трифторацетамид (210 мг) растворяли в метиленхлориде (5 мл). К раствору добавляли 75% 3-хлорпербензойную кислоту (152 мг) с последующим перемешиванием при комнатной температуре в течение 18 часов. После этого к реакционной жидкости добавляли насыщенный водный раствор сульфита натрия с последующим перемешиванием в течение некоторого времени. Реакционную жидкость экстрагировали хлороформом и экстракт промывали 1 М водным раствором NaOH и насыщенным раствором соли и затем сушили над сульфатом магния.
Растворитель выпаривали и образовавшийся остаток растворяли в смешанном растворителе (7,5 мл) 4:1 ТГФ-1,5% водный раствор серной кислоты с последующим перемешиванием при кипячении с обратным холодильником в течение 5 часов. К реакционной жидкости добавляли воду и затем экстрагировали EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния.
Растворитель выпаривали и образовавшийся остаток растворяли в МеОН (6 мл). К реакционной жидкости добавляли карбонат калия (304 мг) с последующим перемешиванием при 60°С в течение 5 часов. После этого растворитель выпаривали и к реакционной жидкости добавляли воду и затем экстрагировали хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом транс-1-(2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этил)циклогексан-1,2-диол (93 мг).
Образовавшееся соединение (93 мг) растворяли в смешанном растворителе хлороформ-EtOH. К реакционной жидкости добавляли щавелевую кислоту (22 мг), получая при этом оксалат транс-1-(2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этил)циклогексан-1,2-диола (66 мг).
Пример 5
2-(1-Циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-N-(циклогексилметил)-2-оксоэтанамин (296 мг) растворяли в ацетонитриле (10 мл). К реакционной жидкости добавляли 2-бромэтанол (400 мг), карбонат калия (555 мг) и иодид калия (133 мг) с последующим перемешиванием смеси при кипячении с обратным холодильником в течение 16 часов. После этого растворитель выпаривали и к реакционной жидкости добавляли воду с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил](циклогексилметил)амино}этанол (155 мг).
Образовавшееся соединение (155 мг) растворяли в EtOH. К реакционной жидкости добавляли щавелевую кислоту (36 мг), получая при этом оксалат 2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил](циклогексилметил)амино}этанола (159 мг).
Пример 6
Смесь 1-({[2-(5-бром-1-изопропил-8-метил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанола (0,14 г), EtOH (10 мл), триэтиламина (0,05 мл) и 10% палладия, осажденного на угле (10 мг), перемешивали в атмосфере водорода в течение 6 часов. Реакционную жидкость фильтровали и растворитель выпаривали. К образовавшемуся остатку добавляли 1 М водный раствор NaOH с последующей экстракцией смеси хлороформом. Органический слой сушили над сульфатом магния и затем растворитель выпаривали. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН). Образовавшийся остаток растворяли в 2-пропаноле (0,8 мл). К реакционной жидкости добавляли щавелевую кислоту (23 мг) и диэтиловый эфир (5 мл) и осажденное твердое вещество собирали фильтрованием и сушили, получая при этом оксалат 1-({[2-(1-изопропил-8-метил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексанола (0,066 г).
Пример 7
2-(1-Циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтанамин (400 мг) растворяли в EtOH (10 мл). К реакционной жидкости добавляли 2-метил-1-оксаспиро[2.5]октан (555 мг) и воду (5 мл) с последующим перемешиванием при кипячении с обратным холодильником в течение 2 дней. Растворитель выпаривали и к реакционной жидкости добавляли воду с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 1-(1-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этил)циклогексанол (585 мг).
Образовавшееся соединение (325 мг) растворяли в смешанном растворителе EtOH-ацетонитрил. К реакционной жидкости добавляли щавелевую кислоту (80 мг), получая при этом оксалат 1-(1-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этил)циклогексанола (292 мг).
Пример 8
2-{[2-(1-Циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этанол (342 мг) растворяли в EtOH (5 мл). К раствору добавляли 1-оксаспиро[2.5]октан (363 мг) и воду (5 мл) с последующим перемешиванием при кипячении с обратным холодильником в течение 2 дней. Растворитель выпаривали и к реакционной жидкости добавляли воду с последующей экстракцией смеси хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 1-({[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил](2-гидроксиэтил)амино}метил)циклогексанол (346 мг).
Образовавшееся соединение (346 мг) растворяли в EtOH (10 мл). К реакционной жидкости добавляли щавелевую кислоту (76 мг), получая при этом оксалат 1-({[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил](2-гидроксиэтил)амино}метил)циклогексанол (210 мг).
Пример 9
N-[2-(1-Циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]-N-{2-[цис-1,2-дигидроксициклогексил]этил}-2,2,2-трифторацетамид (255 мг) растворяли в MeOH (10 мл). К раствору добавляли карбонат калия (345 мг) с последующим перемешиванием при 60°С в течение 4 часов. Растворитель выпаривали и к реакционной жидкости добавляли воду с последующей экстракцией смеси хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом цис-1-(2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этил)циклогексан-1,2-диол (212 мг).
Образовавшееся соединение (212 мг) растворяли в EtOH. К реакционной жидкости добавляли щавелевую кислоту (46 мг), получая при этом оксалат цис-1-(2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этил)циклогексан-1,2-диола (170 мг).
Пример 10
трет-Бутил-[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]{[цис-1,2-дигидроксициклогексил]метил}карбамат (92 мг) растворяли в EtOAc (4 мл). К раствору добавляли 4 M HCl/EtOAc (0,45 мл) с последующим перемешиванием при комнатной температуре в течение 14 часов. Растворитель выпаривали и к образовавшемуся остатку добавляли 1 М водный раствор NaOH с последующей экстракцией смеси хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ), получая при этом цис-1-({[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексан-1,2-диол (174 мг).
Образовавшееся соединение (174 мг) растворяли в iPrOH. К реакционной жидкости добавляли щавелевую кислоту (43 мг), получая при этом оксалат цис-1-({[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)циклогексан-1,2-диола (88 мг).
Пример 11
трет-Бутил-[2-(6-карбамоил-1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил][(1-гидроксициклогексил)метил]карбамат (357 мг) растворяли в EtOAc (8 мл). К реакционной жидкости при охлаждении льдом добавляли 4 М HCl/EtOAc (0,85 мл) с последующим перемешиванием при 60°С в течение 5 часов. Растворитель выпаривали и к образовавшемуся остатку добавляли воду с последующим промыванием смеси хлороформом. К водному слою добавляли насыщенный водный бикарбонат натрия и затем рН смеси регулировали до величины около 8 с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния, получая при этом 1-циклогексил-2-{N-[(1-гидроксициклогексил)метил]глицил}-1,2,3,4-тетрагидроизохинолин-6-карбоксамид (116 мг).
Образовавшееся соединение (116 мг) растворяли в смешанном растворителе iPrOH-диэтиловый эфир. К реакционной жидкости добавляли щавелевую кислоту (24 мг), получая при этом оксалат 1-циклогексил-2-{N-[(1-гидроксициклогексил)метил]глицил}-1,2,3,4-тетрагидроизохинолин-6-карбоксамида (70 мг).
Пример 12
1-[2-(Хлорацетил)-1,2,3,4-тетрагидроизохинолин-1-ил]-1-метилэтилпивалат (1,2 г) растворяли в ацетонитриле (20 мл). К раствору добавляли карбонат калия (2,36 г), гидрохлорид 1-(аминометил)циклогексанола (2,26 г) и иодид тетра-н-бутиламмония (126 мг) с последующим перемешиванием при 60°С в течение 5 часов. К реакционной жидкости добавляли воду с последующей экстракцией EtOAc. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом натрия. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН).
Образовавшийся остаток (1,43 г) растворяли в метиленхлориде (20 мл). К реакционной жидкости добавляли раствор 1,01 М диизобутилалюминийгидрид/н-гексан (9,55 мл) при -78°С с последующим перемешиванием при -78°С в течение 5 часов. После этого температуру повышали до 0°С на протяжении 2 часов. К реакционной жидкости добавляли насыщенный водный раствор сегнетовой соли с последующим перемешиванием смеси в течение 20 минут. Затем к реакционной жидкости добавляли целит и затем ее подвергали разделению фильтрованием с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 1-[({2-[1-(1-гидрокси-1-метилэтил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанол (118 мг).
Образовавшееся соединение (150 мг) растворяли в ацетонитриле. К реакционной жидкости добавляли щавелевую кислоту (41 мг), получая при этом оксалат 1-[({2-[1-(1-гидрокси-1-метилэтил)-3,4-дигидроизохинолин-2(1H)-ил]-2-оксоэтил}амино)метил]циклогексанола (151 мг).
Пример 13
2-(1-Циклогексил-7-метокси-3,4-дигидроизохинолин-2(1H)-ил)-N-(2-{[цис-2-(метоксиметокси)циклопентил]окси}этил)-2-оксоэтанамин (500 мг) растворяли в MeOH (8 мл). К реакционной жидкости при охлаждении льдом добавляли 4 М HCl/EtOAc (0,8 мл) с последующим перемешиванием смеси при 60°С в течение 5 часов. Растворитель выпаривали и к образовавшемуся остатку добавляли насыщенный водный бикарбонат натрия с последующей экстракцией хлороформом. Экстракт промывали насыщенным раствором соли и затем сушили над сульфатом магния. Растворитель выпаривали и образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом цис-2-(2-{[2-(1-циклогексил-7-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этокси)циклопентанол (380 мг).
Образовавшееся соединение (380 мг) растворяли в iPrOH. К реакционной жидкости добавляли щавелевую кислоту (80 мг), получая при этом оксалат цис-2-(2-{[2-(1-циклогексил-7-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}этокси)циклопентанола (384 мг).
Пример 14
Гидрохлорид 1-(4-хлорпиридин-2-ил)-1,2,3,4-тетрагидроизохинолина (200 мг) и карбонат калия (344 мг) растворяли в смеси EtOAc-вода (1:1, 4 мл) при охлаждении льдом. К раствору добавляли хлорацетилхлорид (0,85 мл) и гидробромат бензилтриэтиламина (9,2 мг) с последующим перемешиванием при комнатной температуре в течение 1 часа. К смеси добавляли гидрохлорид 2-амино-1,1-дициклопропилэтанола (190 мг) и карбонат калия (246 мг). Смесь перемешивали при 50°С в течение 8 часов. Органический слой собирали, промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом 2-({2-[1-(4-хлорпиридин-2-ил)-3,4-дигидроизохинолин-2(1H)-ил-2-оксоэтил]амино}-1,1-дициклопропилэтанол (143 мг) в виде желтого маслянистого вещества. Маслянистое вещество растворяли в смешанной жидкости (4 мл) 3:1 диэтиловый эфир-iPrOH. К реакционной смеси добавляли щавелевую кислоту (30,2 мг), получая при этом оксалат 2-({2-[1-(4-хлорпиридин-2-ил)-3,4-дигидроизохинолин-2(1H)-ил-2-оксоэтил]амино}-1,1-дициклопропилэтанола (128 мг).
Пример 15
К раствору 8-({[2-(1-циклогексил-3,4-дигидро-2(1H)-изохинолинил)-2-оксоэтил]амино}метил)-1,4-диоксаспиро[4.5]декан-8-ола (324 мг) в ТГФ (2 мл) добавляли воду (1 мл) и концентрированную хлористоводородную кислоту (1 мл). Реакционную смесь кипятили с обратным холодильником в течение 5 часов. Температуру снижали до комнатной температуры и к смеси добавляли гидрокарбонат натрия, чтобы сделать ее щелочной, с последующей экстракцией хлороформом. Экстракт очищали колоночной хроматографией на силикагеле (хлороформ-МеОН), получая при этом требуемый амин (176 мг).
Амин растворяли в iPrOH (3 мл), к раствору добавляли щавелевую кислоту (41,7 мг) с последующим перемешиванием при комнатной температуре в течение 2 часов. Образовавшиеся кристаллы собирали фильтрованием, промывали простым эфиром и сушили при 90°С и при пониженном давлении, получая при этом оксалат 4-({[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}метил)-4-гидроксициклогексанона (139 мг).
Пример 16
К раствору 2-(хлорацетил)-1-циклогексил-1,2,3,4-тетрагидроизохинолина (12 мг) в ацетонитриле (0,5 мл) добавляли карбонат калия (3 мг) и гидрохлорид (S)-(+)-2-амино-3-циклогексил-1-пропанола (15 мг) с последующим перемешиванием смеси при 80°C в течение 4 часов. После этого к реакционной жидкости добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией хлороформом. Растворитель выпаривали и образовавшийся остаток очищали препаративной ВЭЖХ, получая при этом (2S)-3-циклогексил-2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-2-оксоэтил]амино}пропан-1-ол (4,1 мг).
Пример 17
2-(Изопентиламино)этанол (26 мг) добавляли к раствору 2-акрилоил-1-циклогексил-1,2,3,4-тетрагидроизохинолина (7 мг) в iPrOH (0,1 мл) с последующим перемешиванием при 90°С в течение 10 часов. После этого реакционную жидкость очищали препаративной ВЭЖХ, получая при этом 2-[[3-(1-циклогексил-3,4-дигидроизохинолин-2(1H)-ил)-3-оксопропил]-(3-метилбутил)амино]этанол (3 мг).
Химические структуры соединений, полученных вышеуказанными примерами, показаны в таблицах 43 и 44 (см. в конце описания). Кроме того, таким же способом, как в способах вышеуказанных примеров, соединения примеров, показанные в таблицах 45-110, получали с применением соответствующих исходных соединений. Данные инструментального анализа этих соединений примеров показаны в таблицах 111-125 (см. в конце описания).
Помимо этого, в таблицах 126 и 127 (см. в конце описания) показаны структуры других соединений настоящего изобретения. Эти соединения можно легко получить согласно вышеуказанным способам, описанным в способах получения и примерах, и способам, известным специалисту данной области, или их модификациям.
Далее, в таблицах № означает номер соединения.
Результаты анализа нескольких соединений примеров получения хиральной колоночной хроматографией показаны в таблицах 128 и 129 (см. в конце описания).
Помимо этого, в таблицах RT означает время удерживания (мин) и ОР означает оптическую чистоту (% ее).
Промышленная применимость
Соединение настоящего изобретения можно применять в составе фармацевтической композиции для предупреждения и/или лечения различных болей, включающих невропатическую боль и ноцицептивную боль, головные боли, такие как мигрень и кластерная головная боль, заболеваний центральной нервной системы, таких как тревога, депрессия, эпилепсия, церебральный удар и синдром усталых ног, абдоминальных симптомов, таких как абдоминальная боль и вздутие живота, нарушений стула, таких как диарея и констипация, заболеваний пищеварительной системы, таких как синдром раздраженной толстой кишки, и заболеваний мочевых путей, таких как гиперактивный мочевой пузырь и интерстициальный цистит.
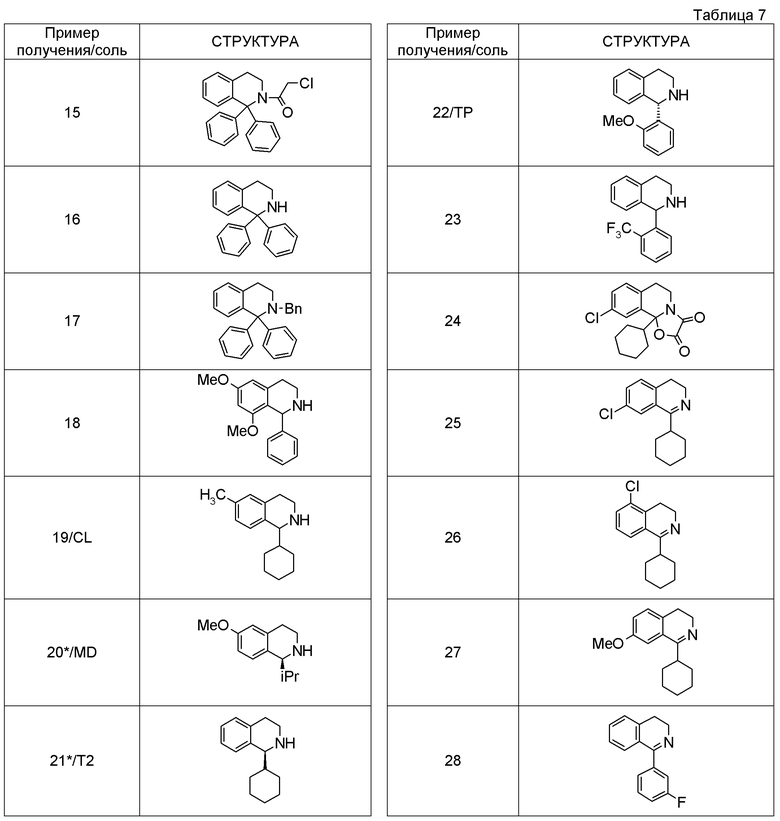





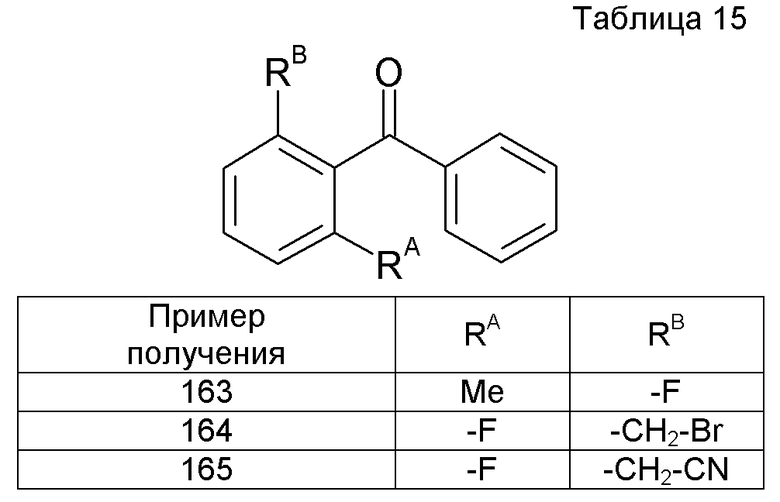
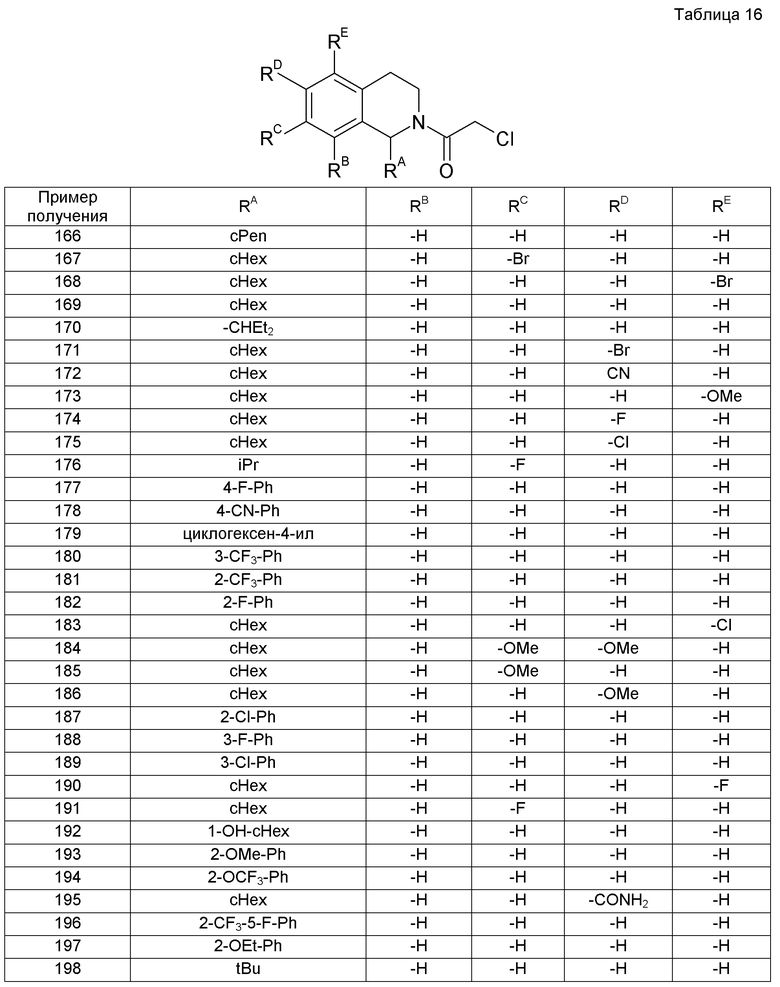
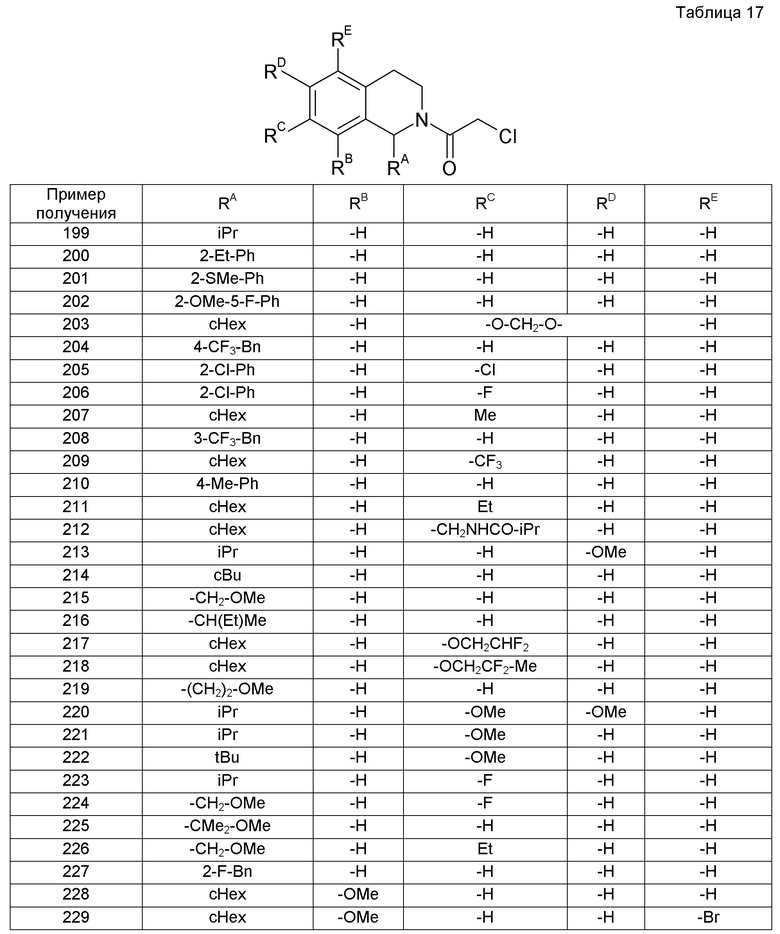
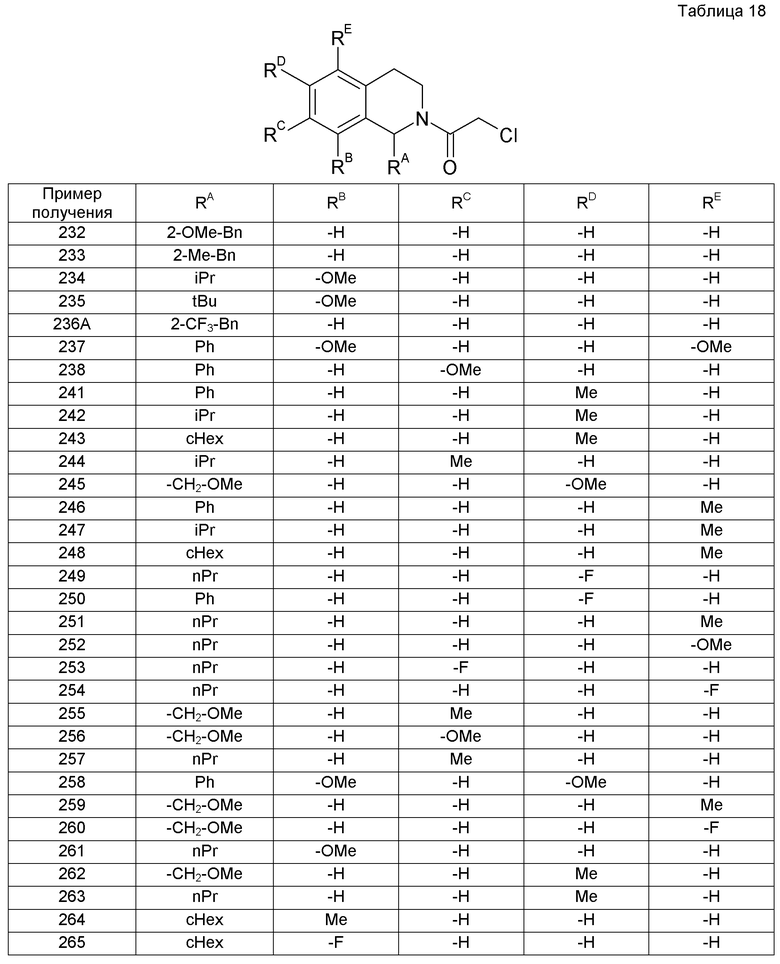
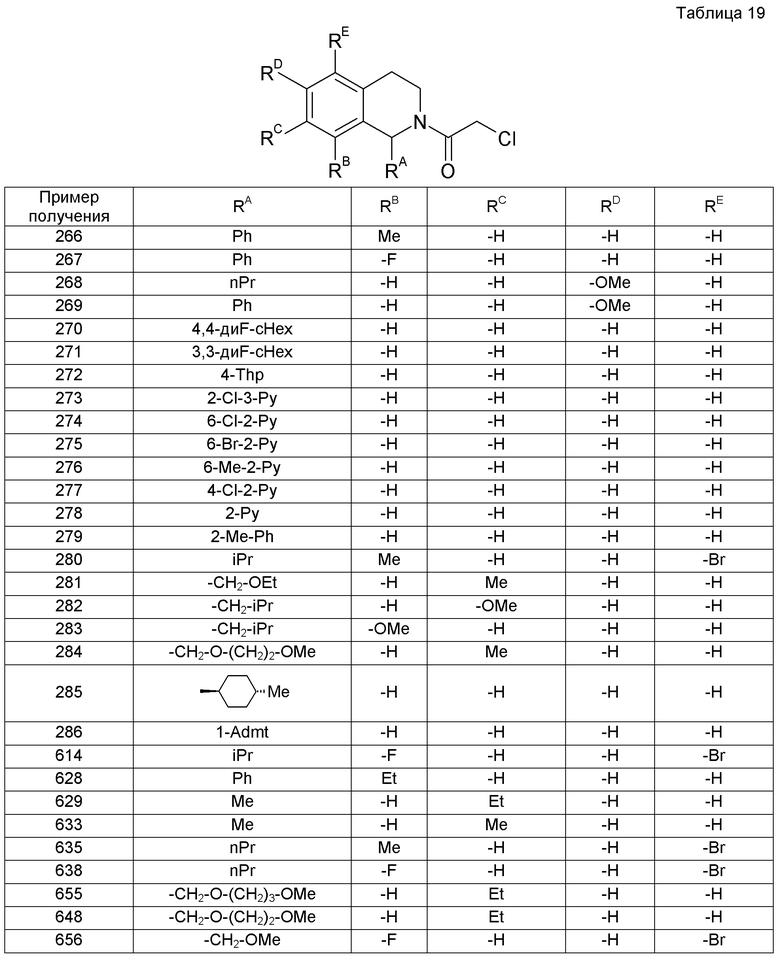

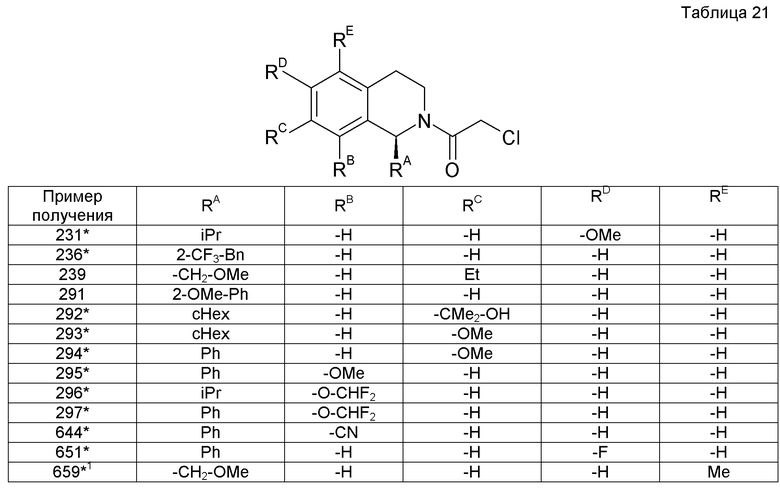
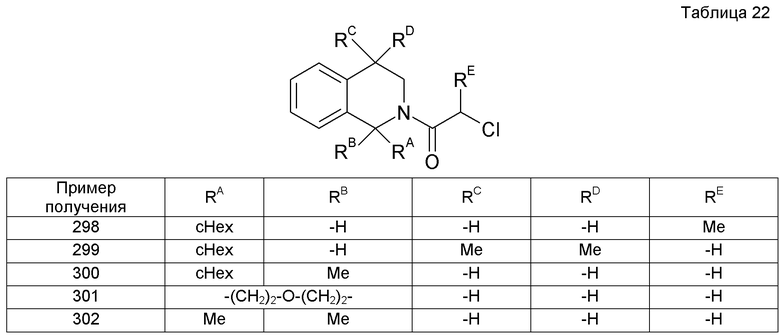

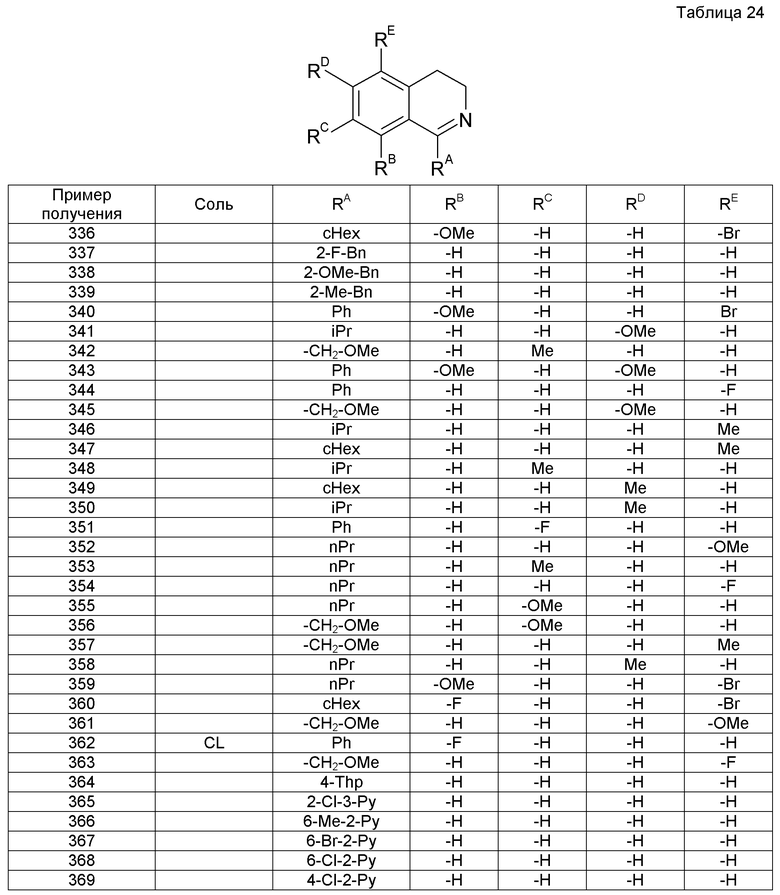


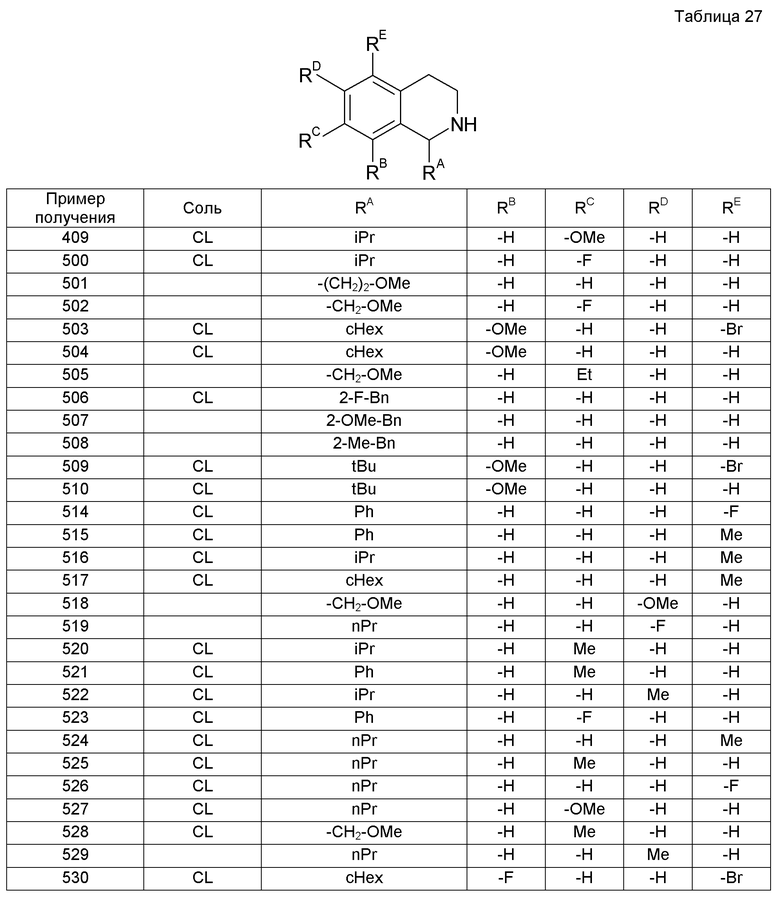
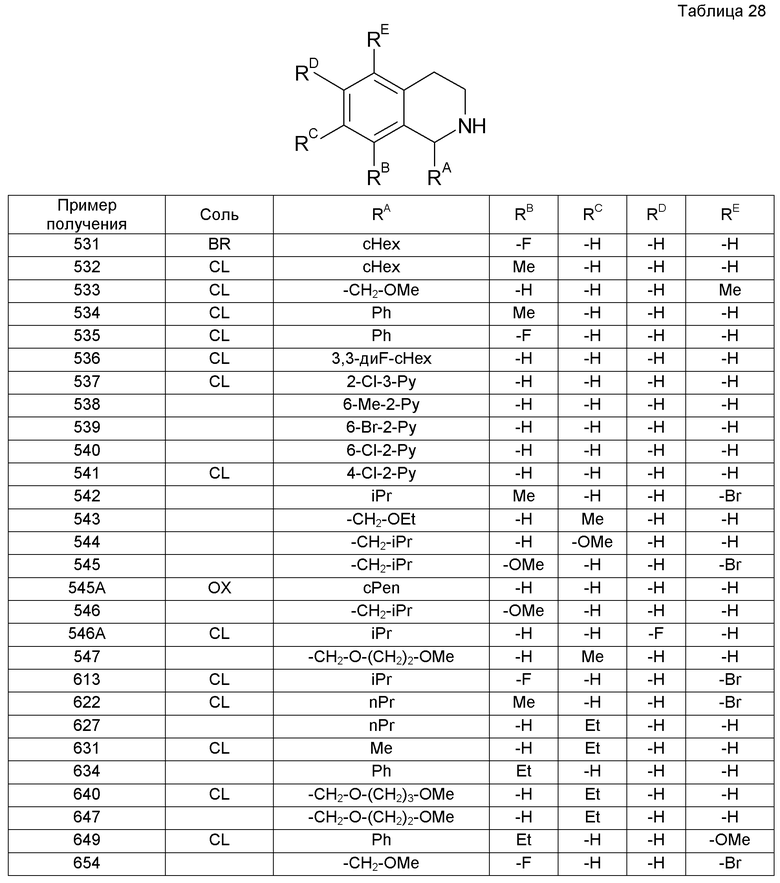
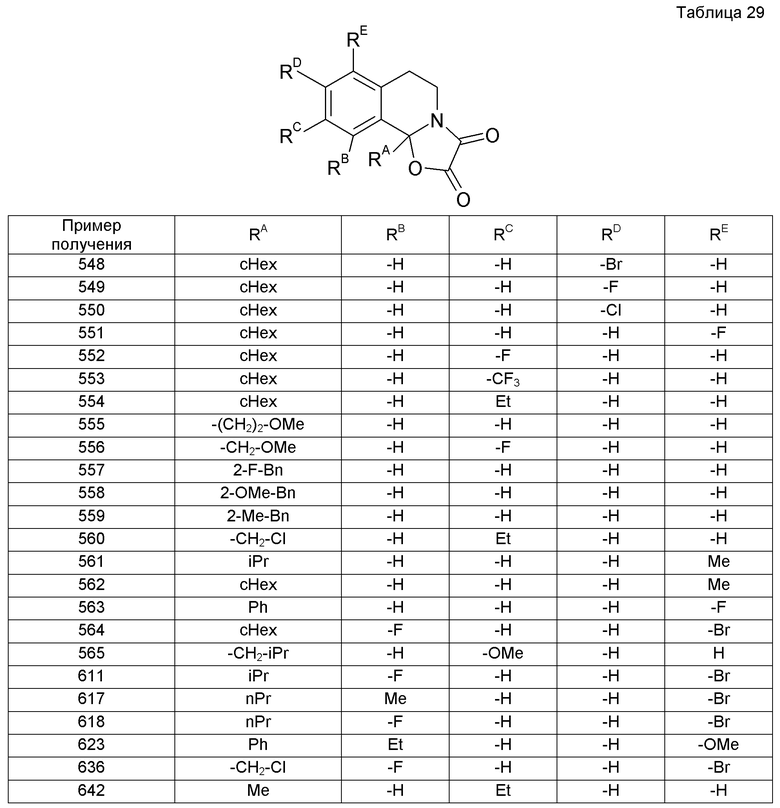
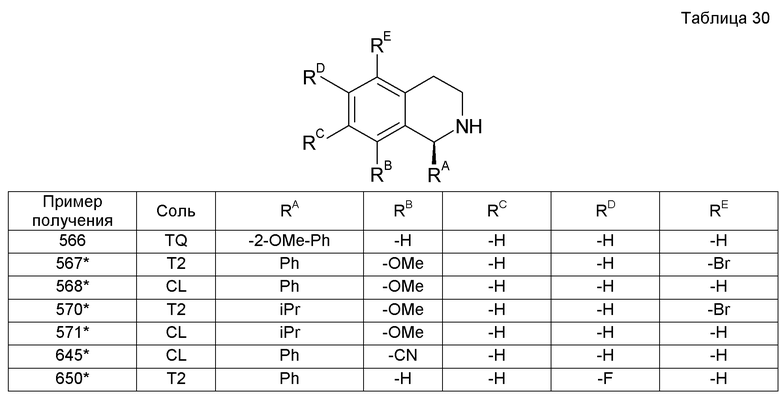

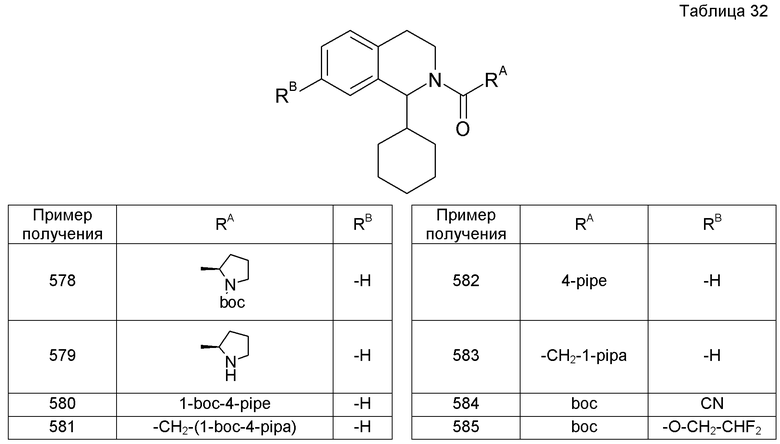
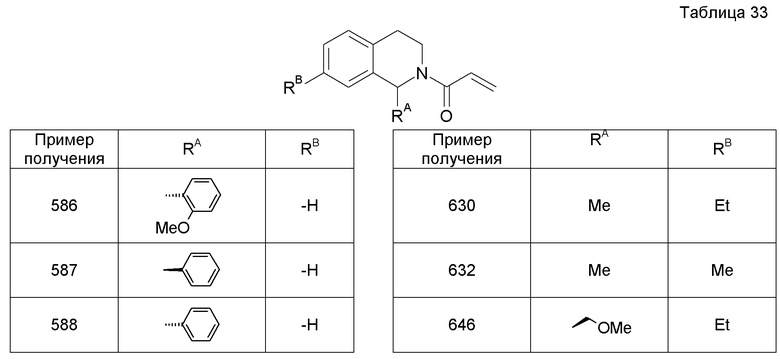
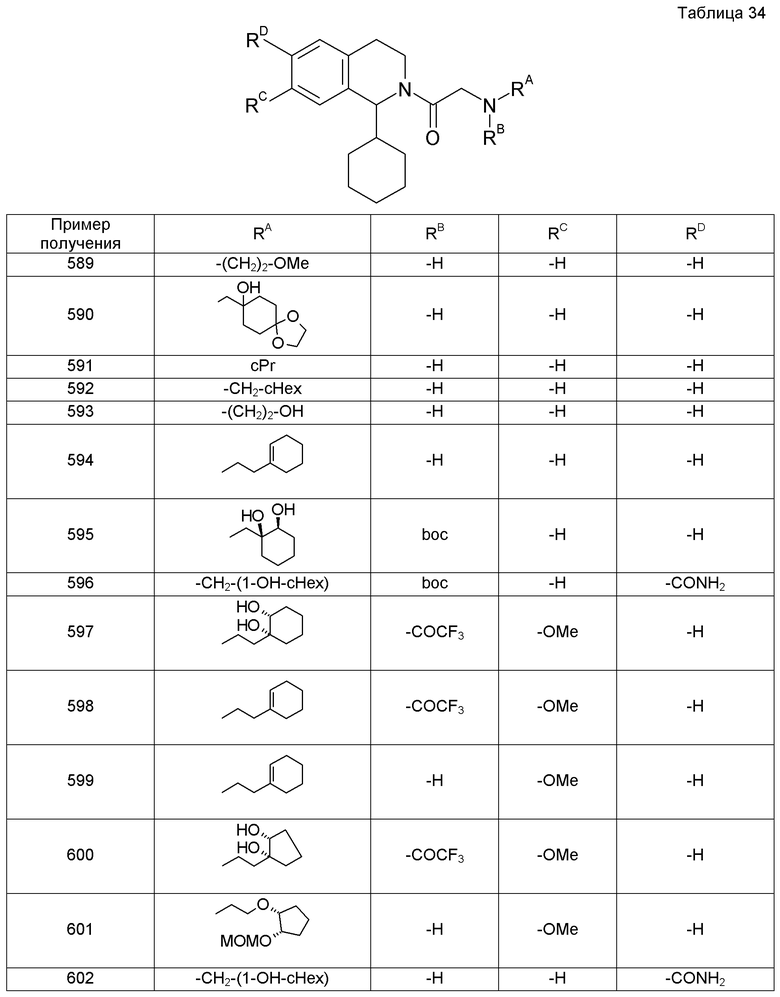
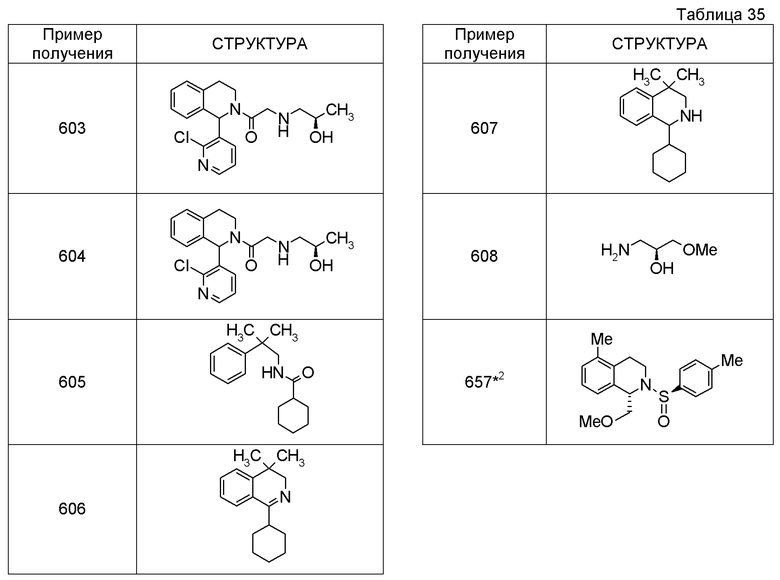


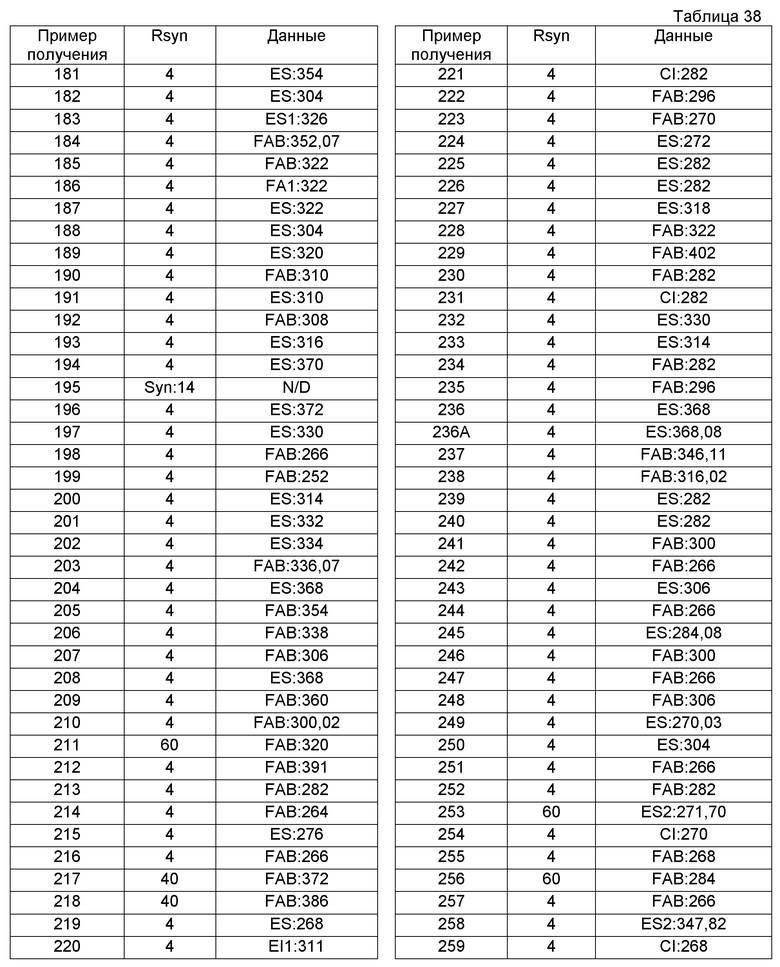
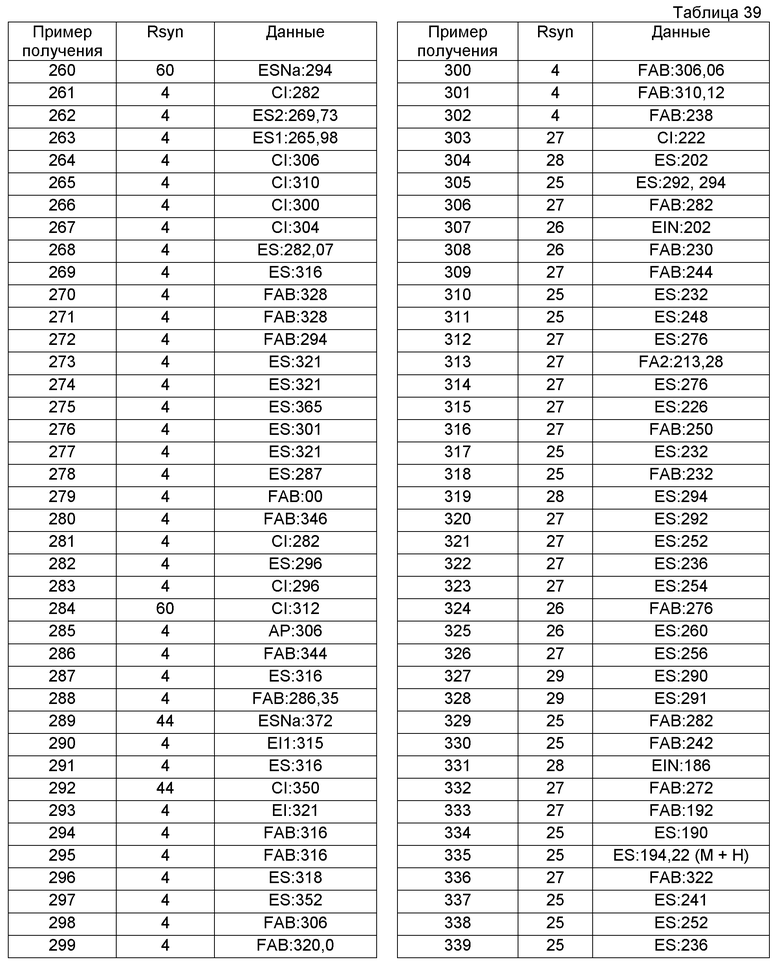
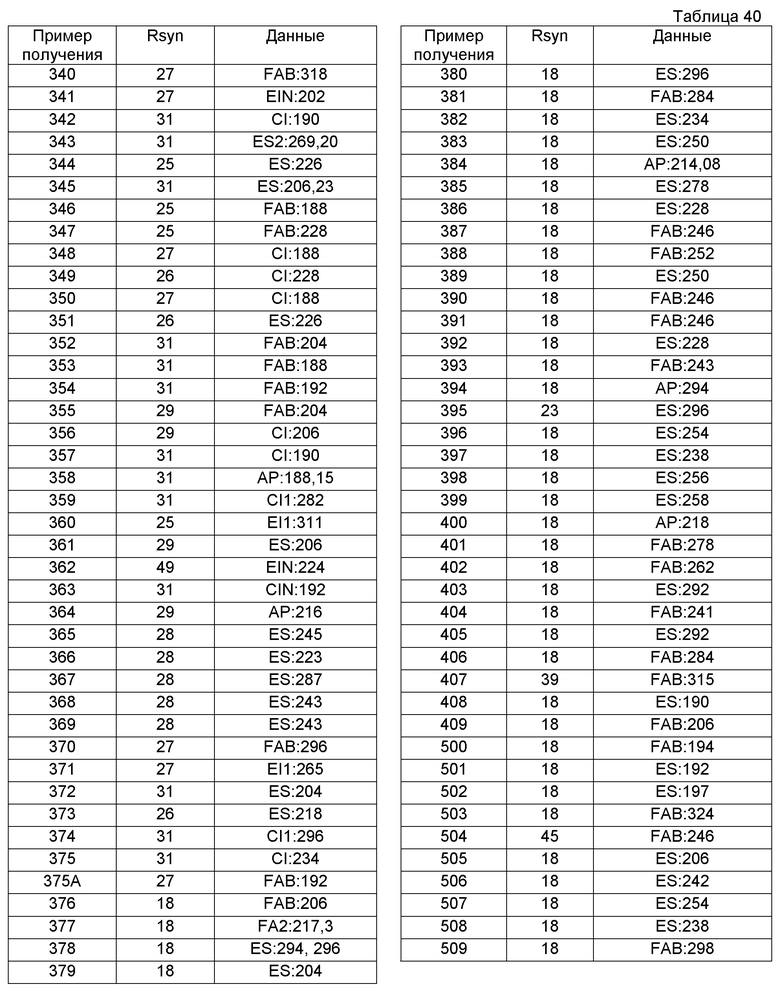
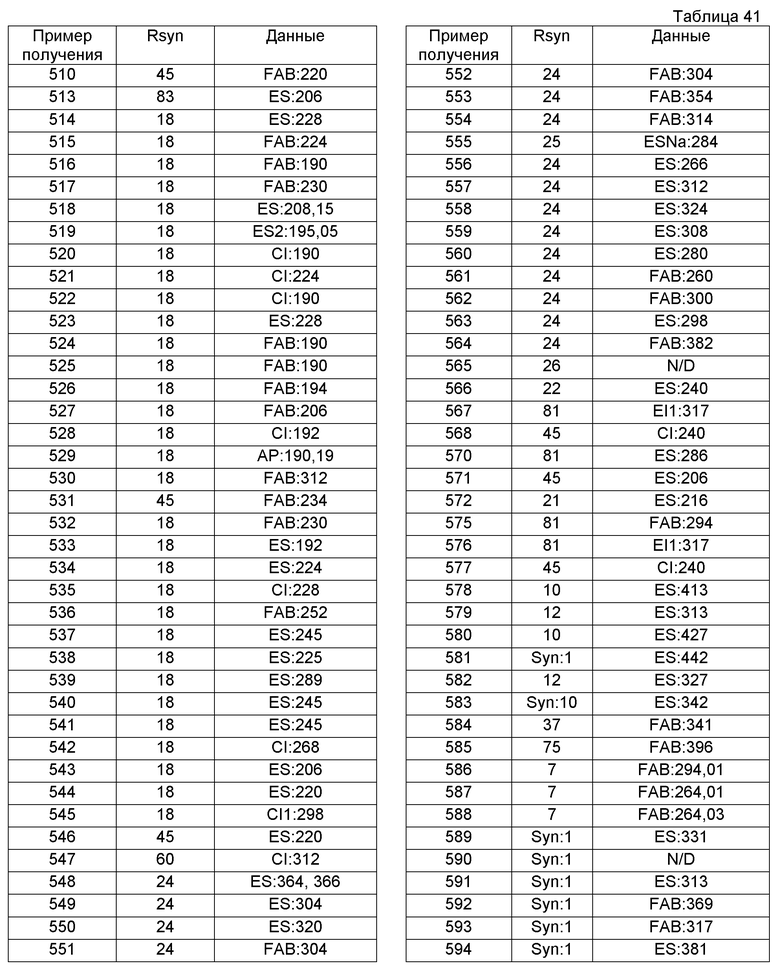
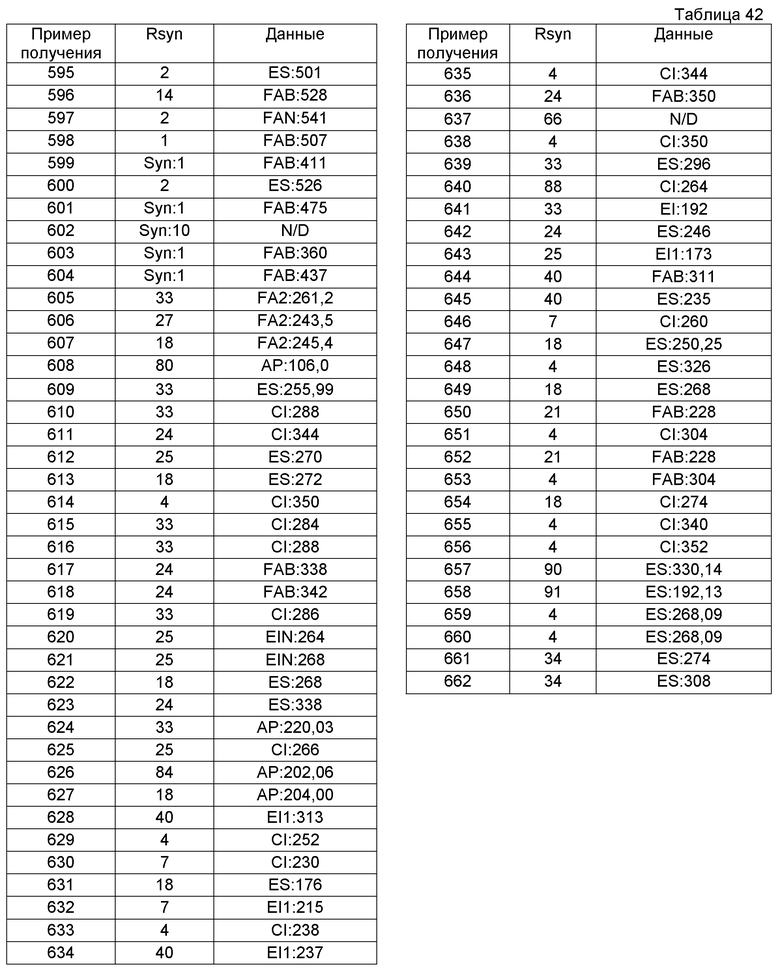

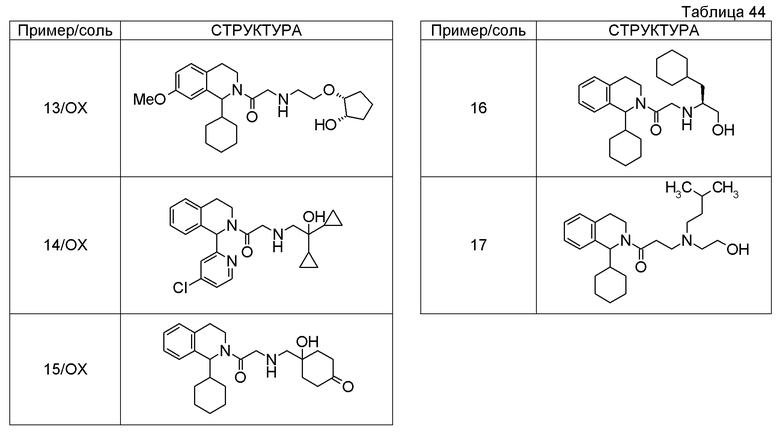
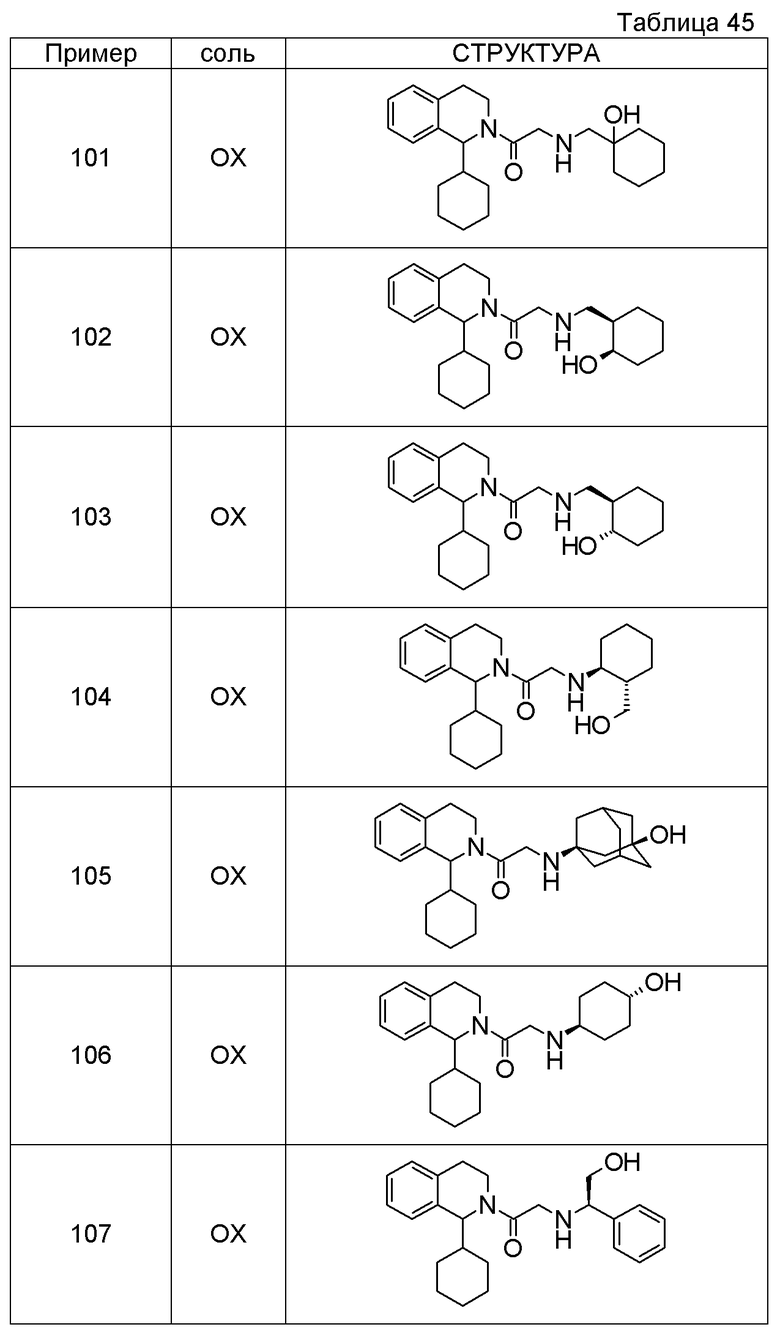

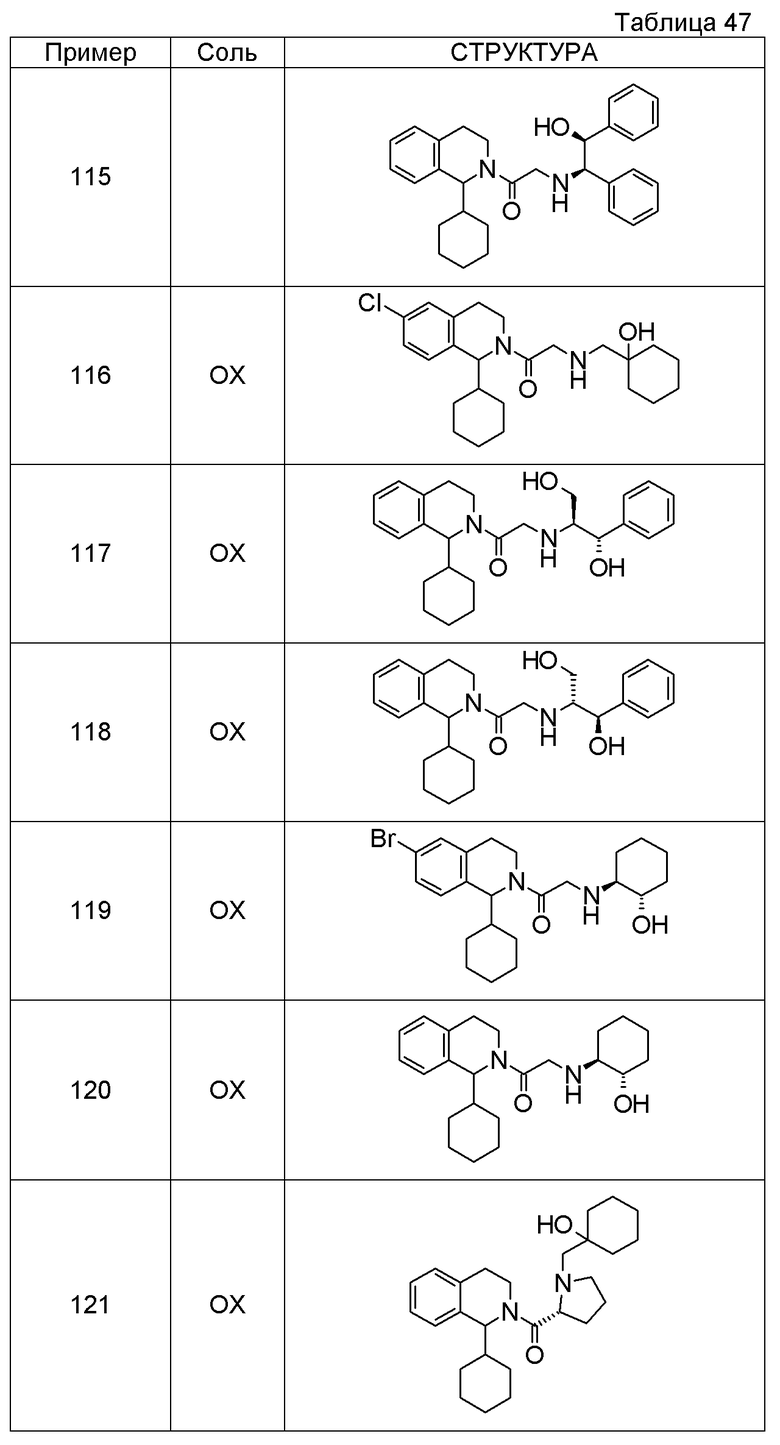
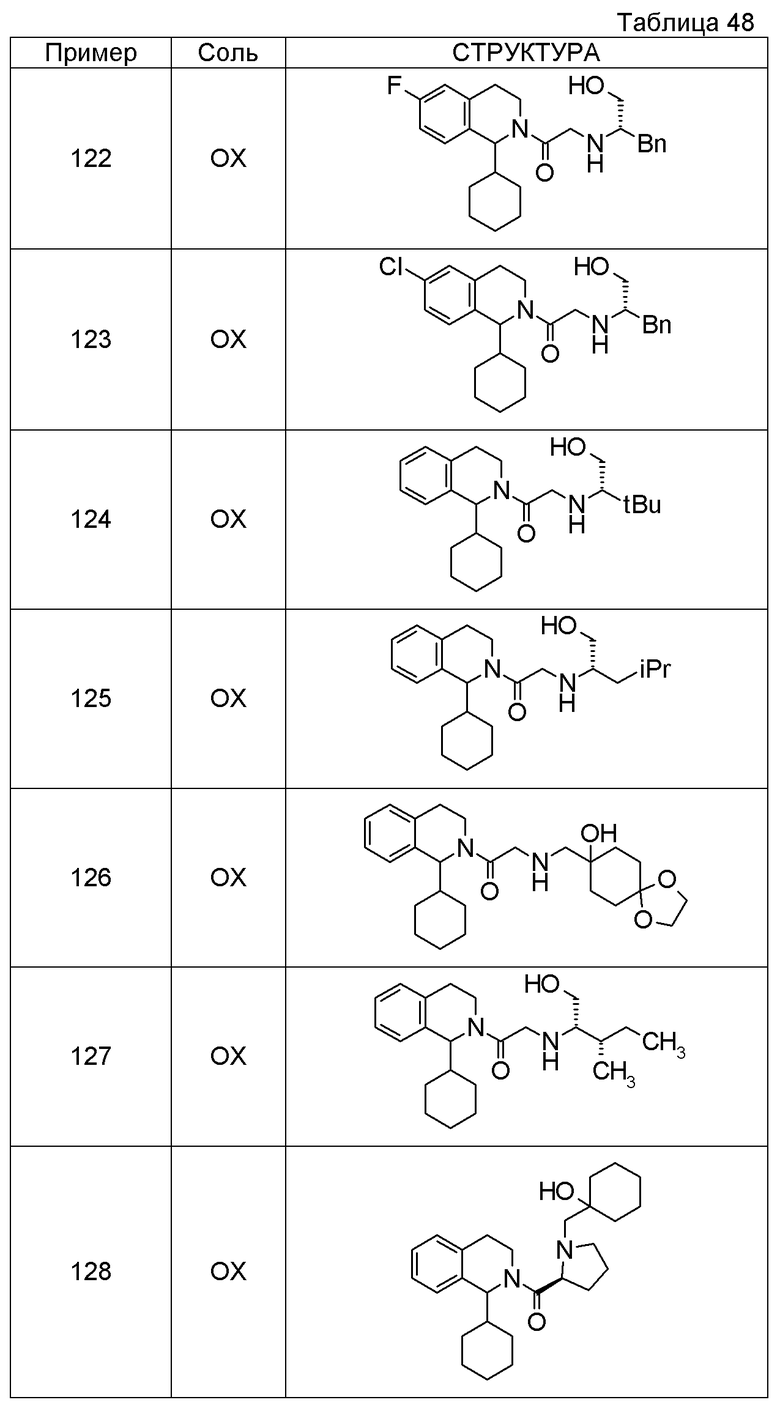

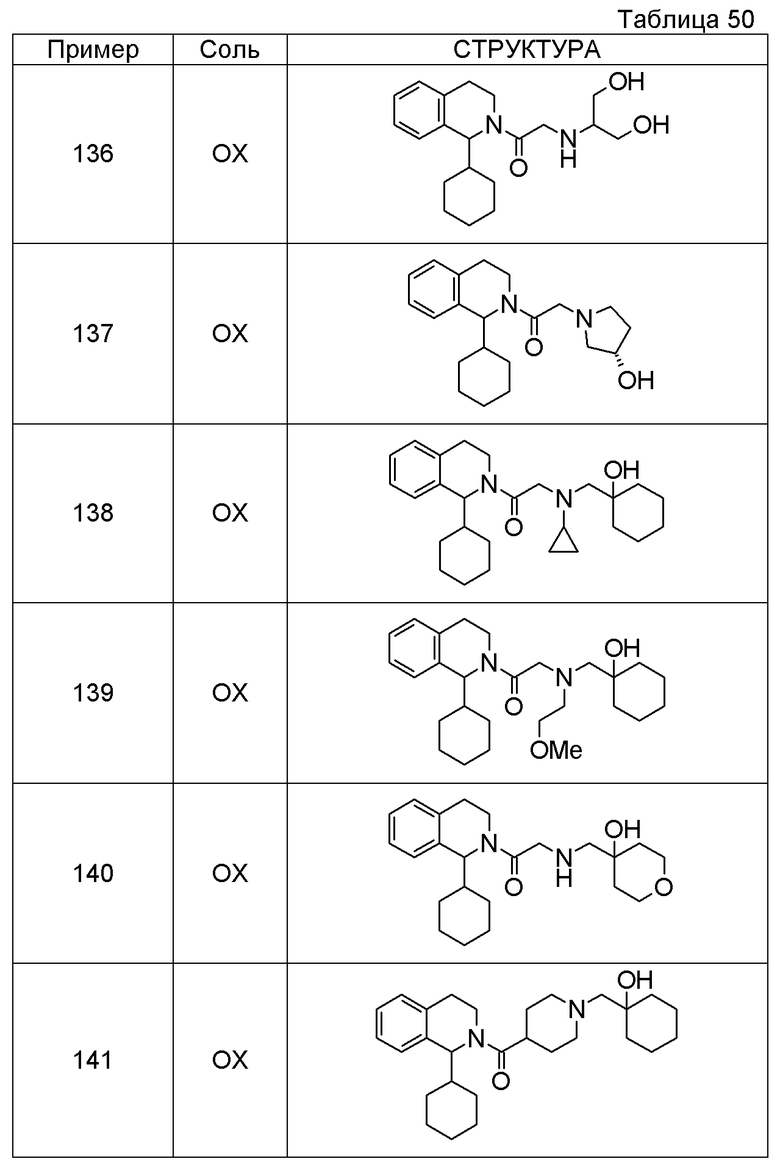
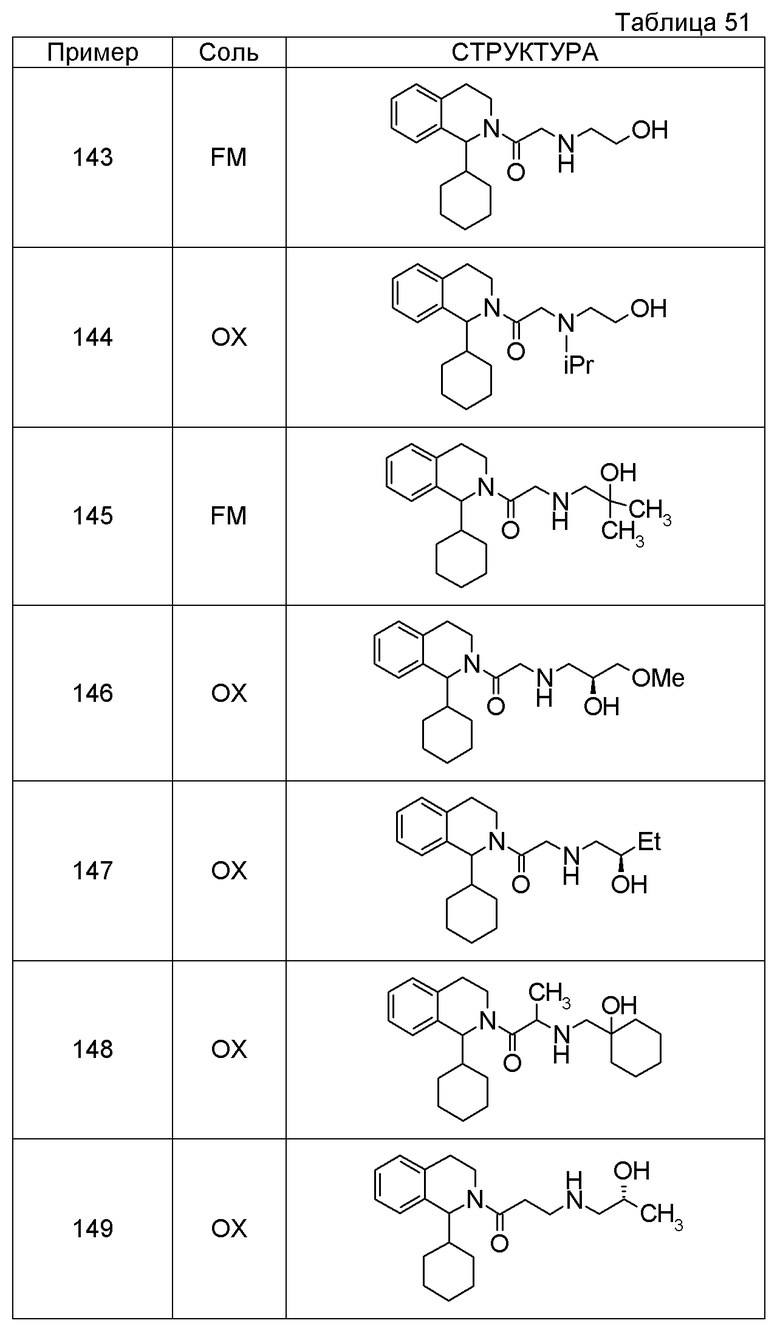
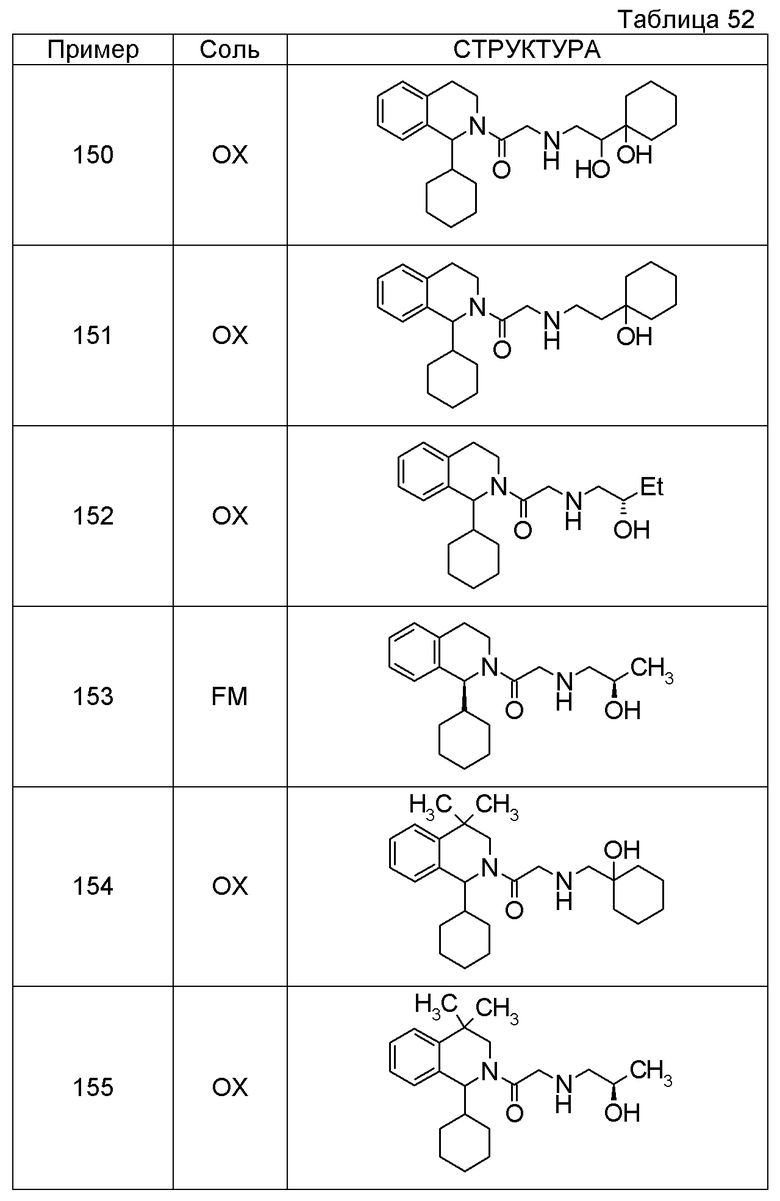
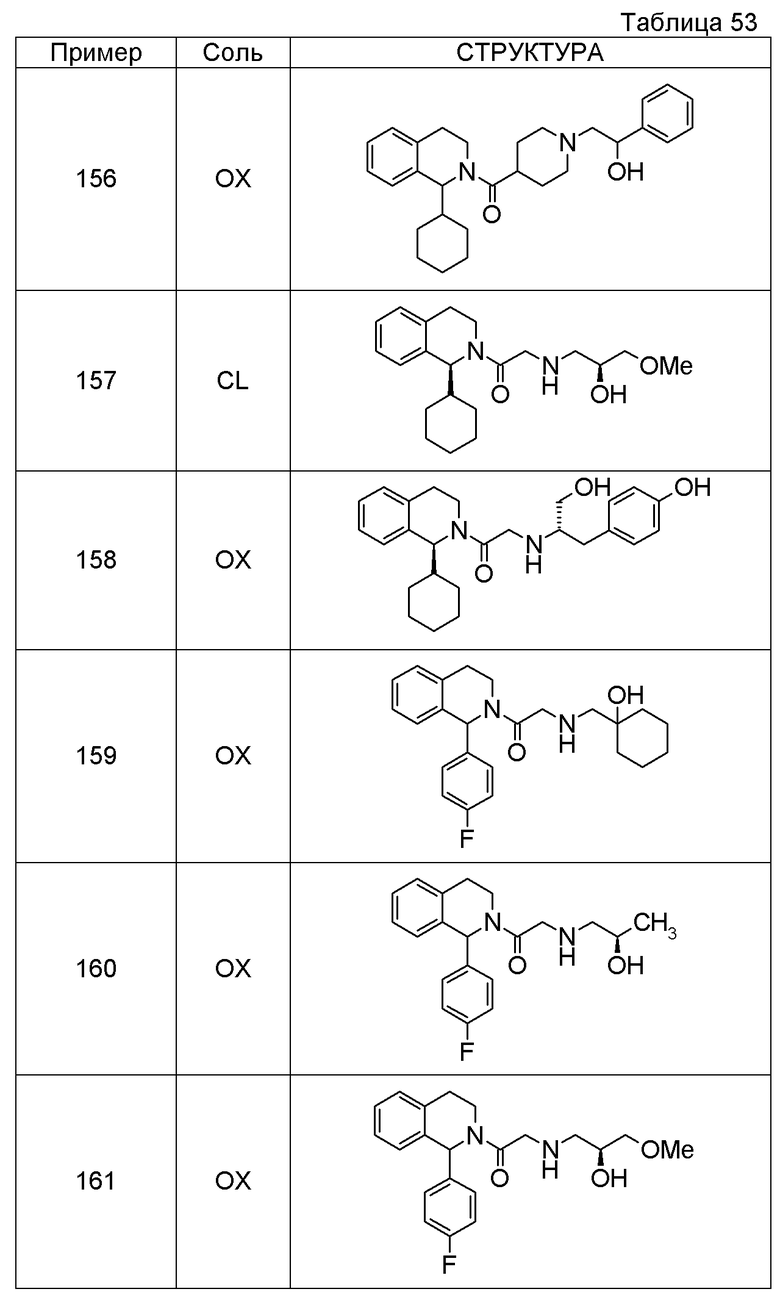
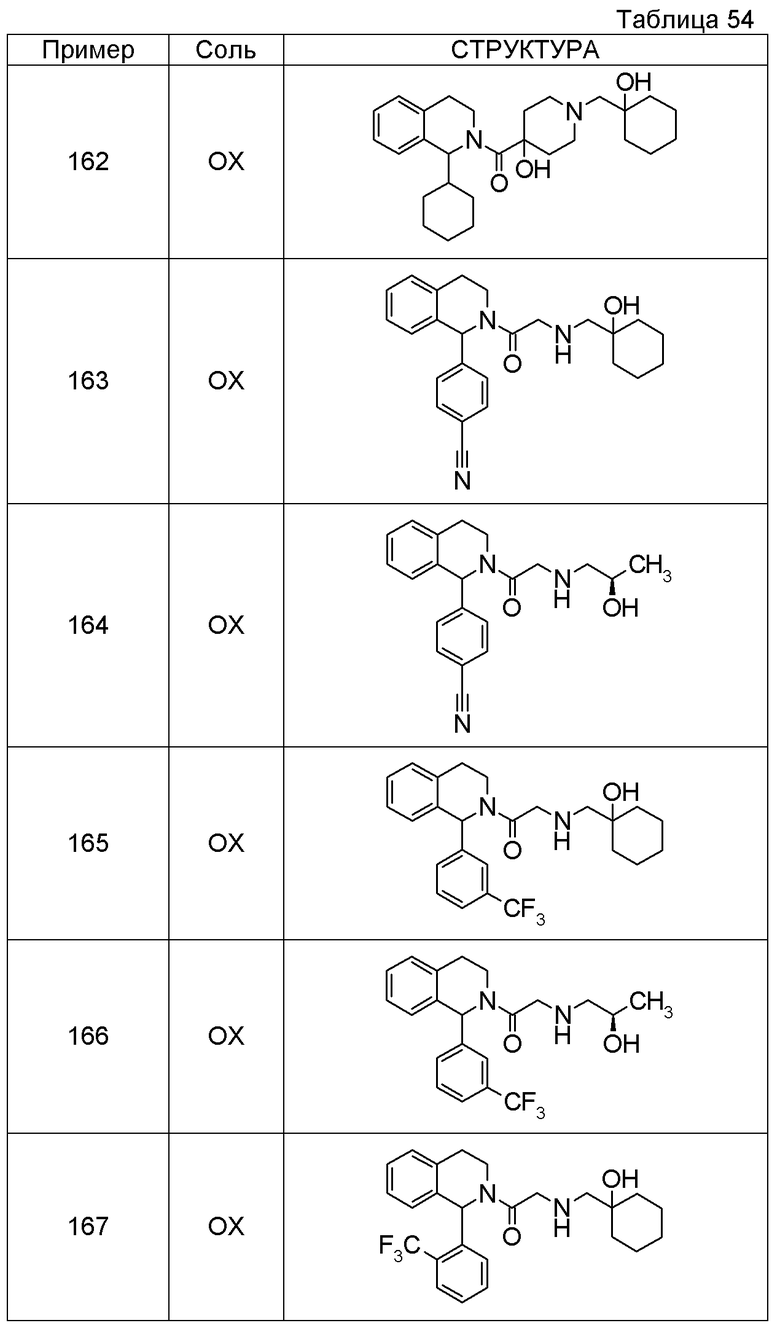
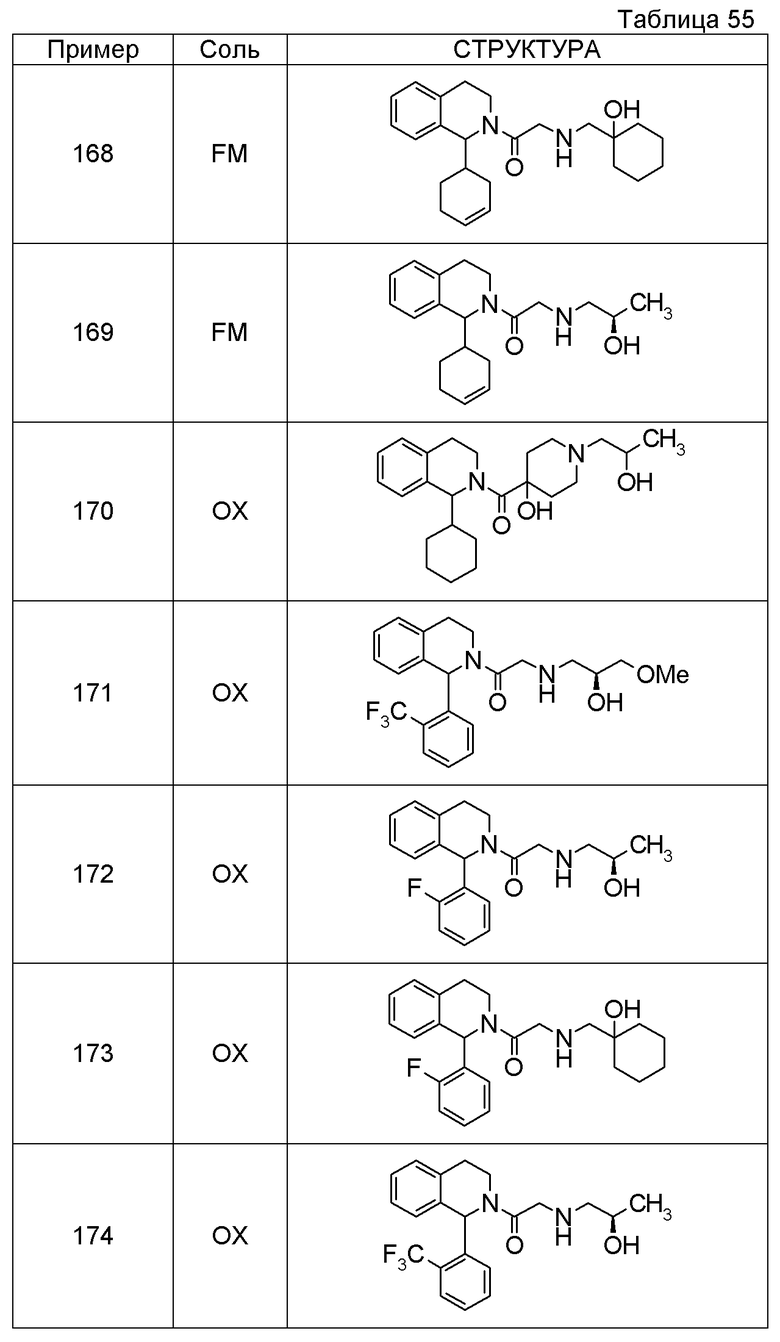

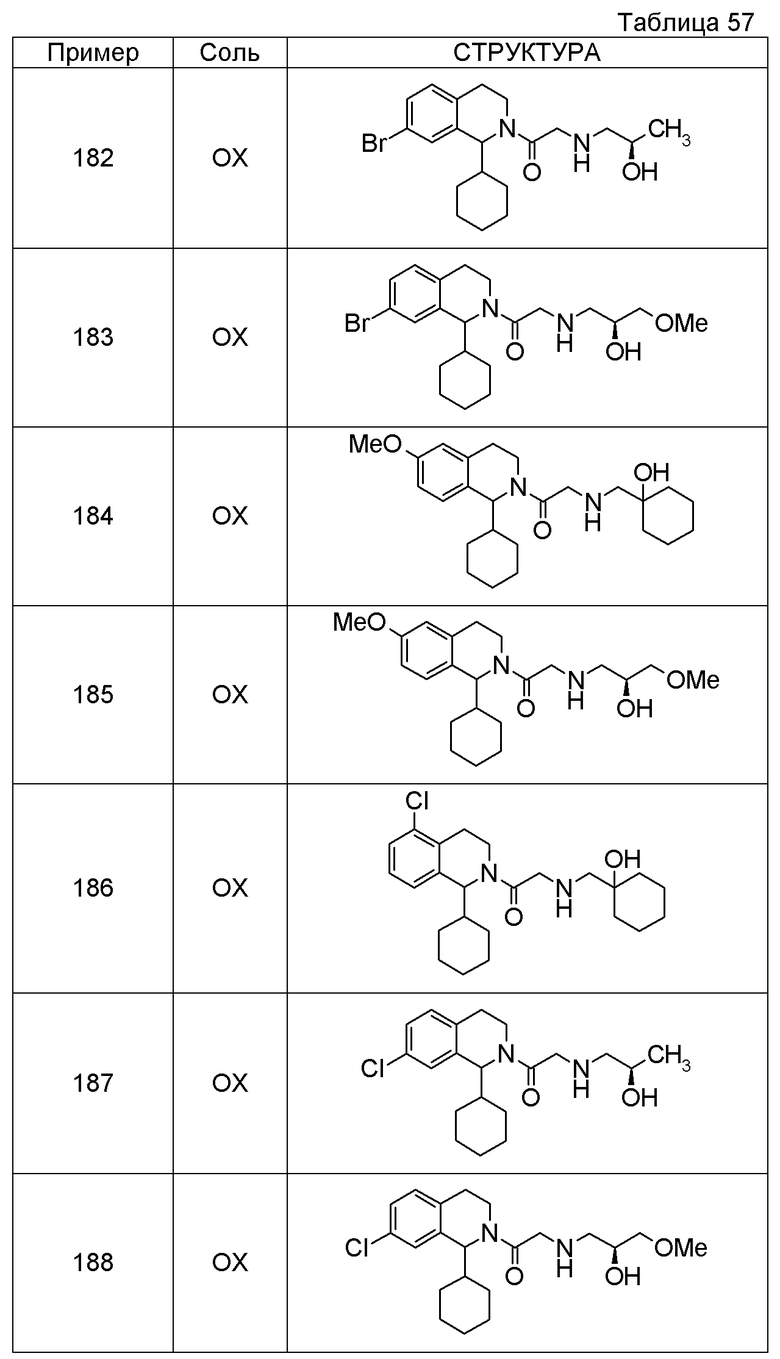
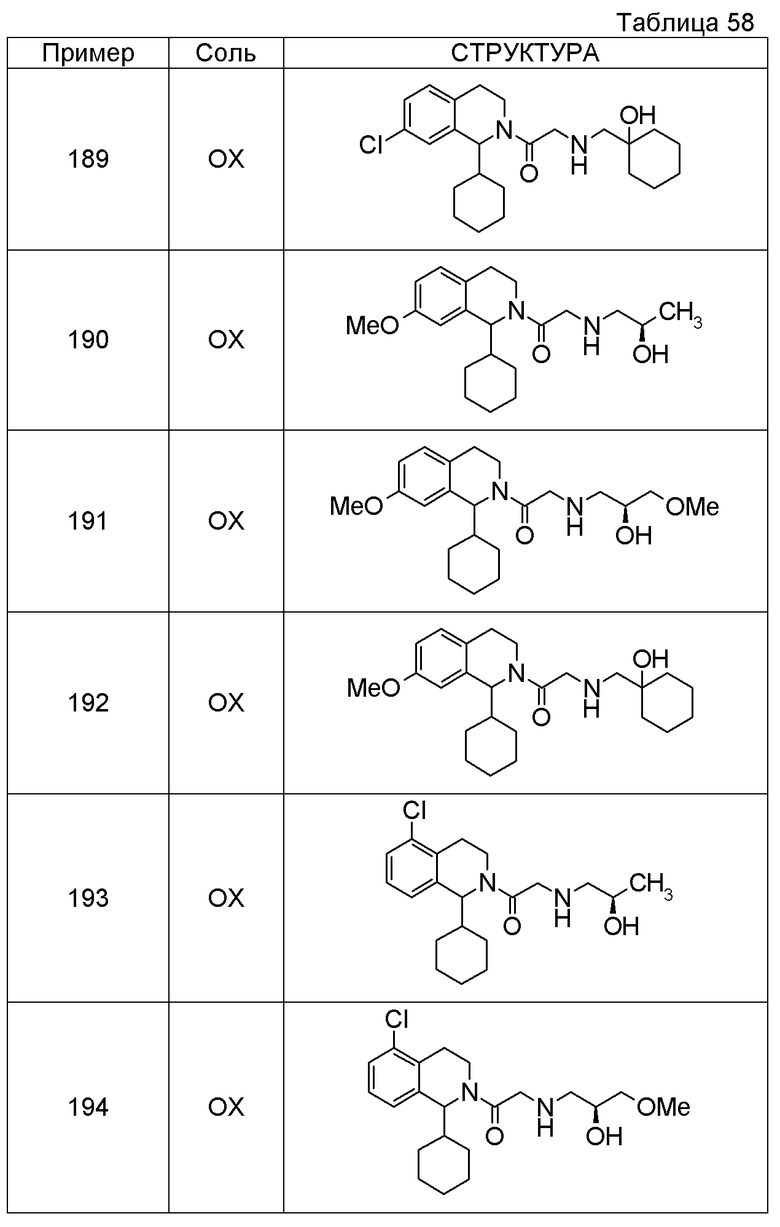
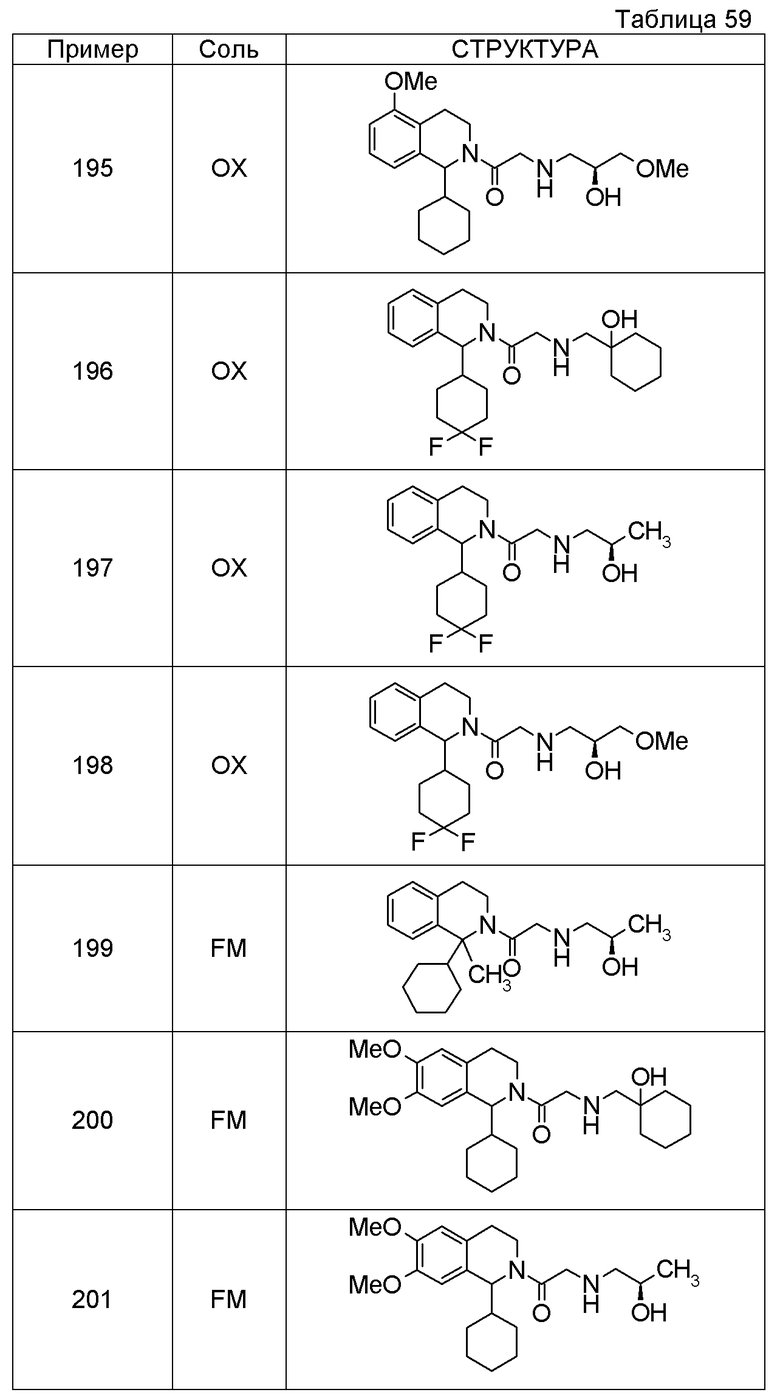
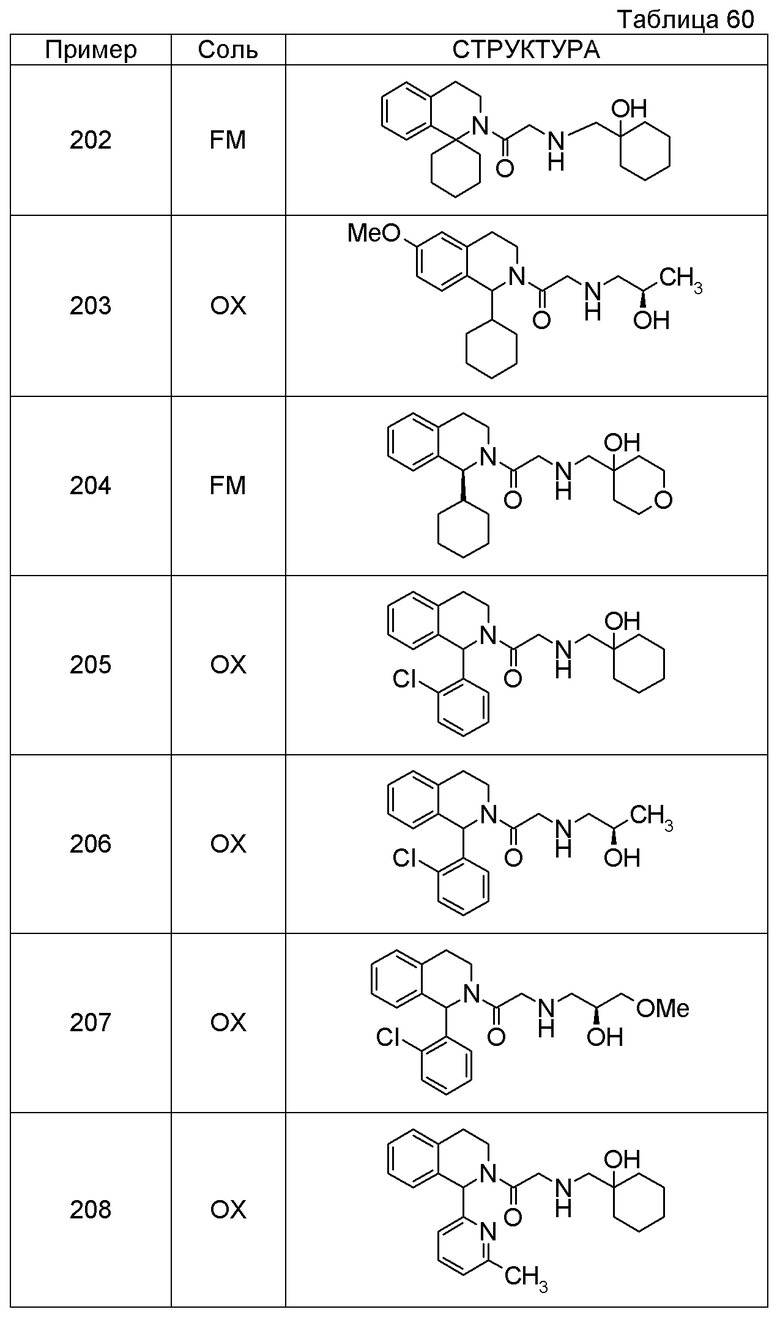

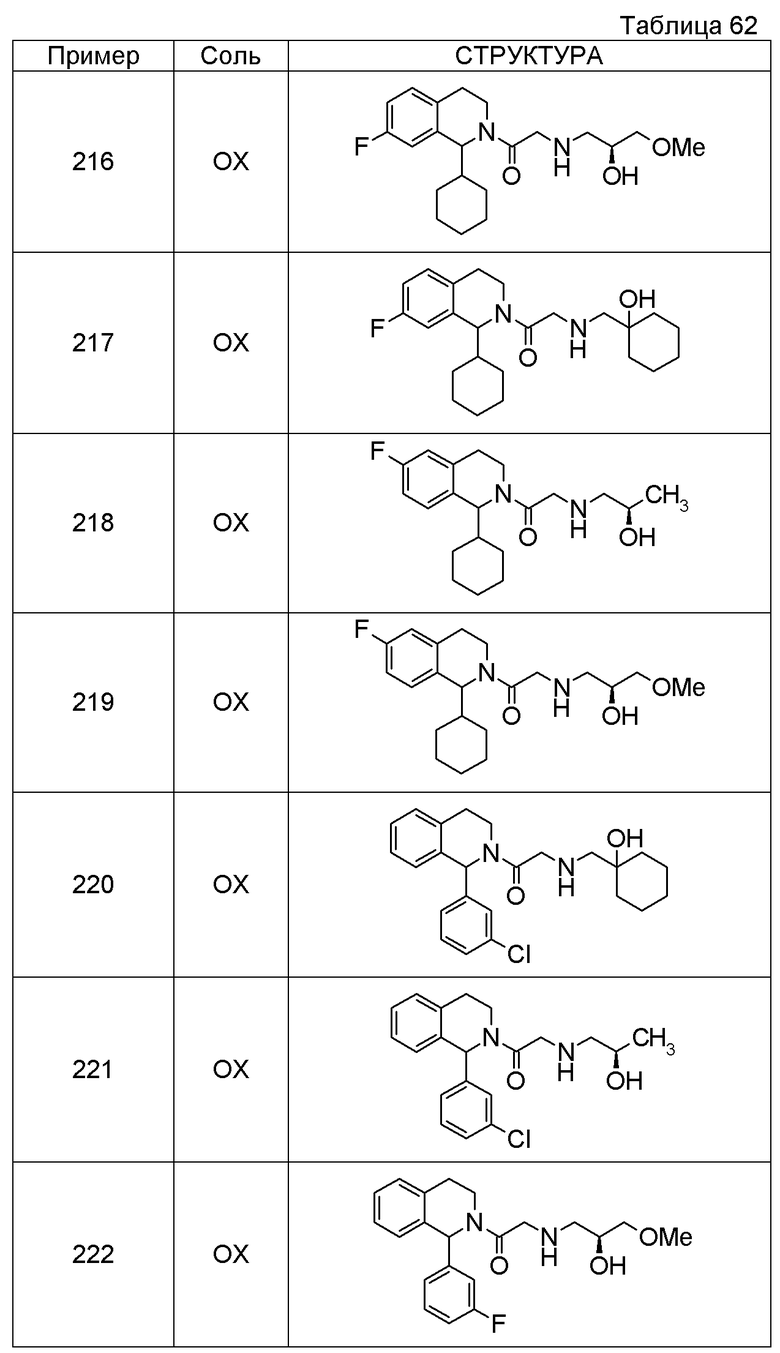
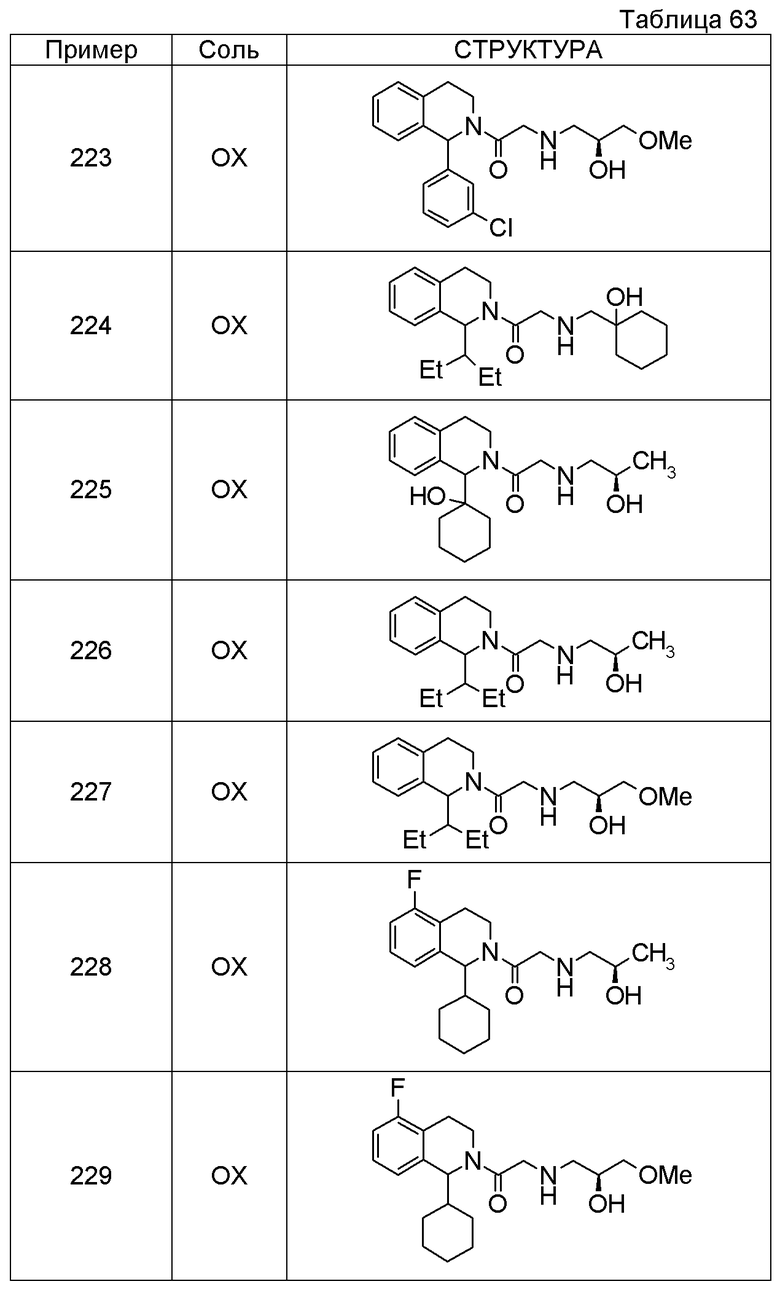
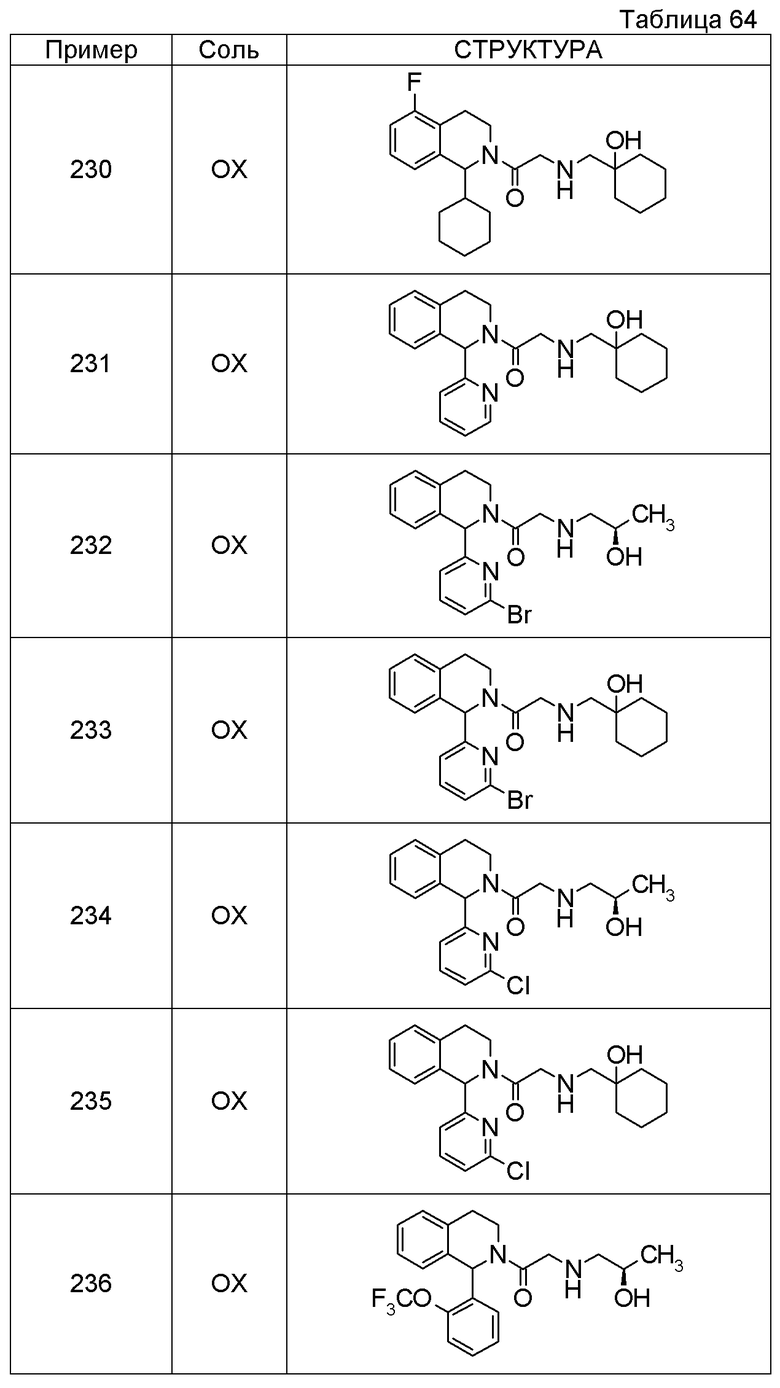
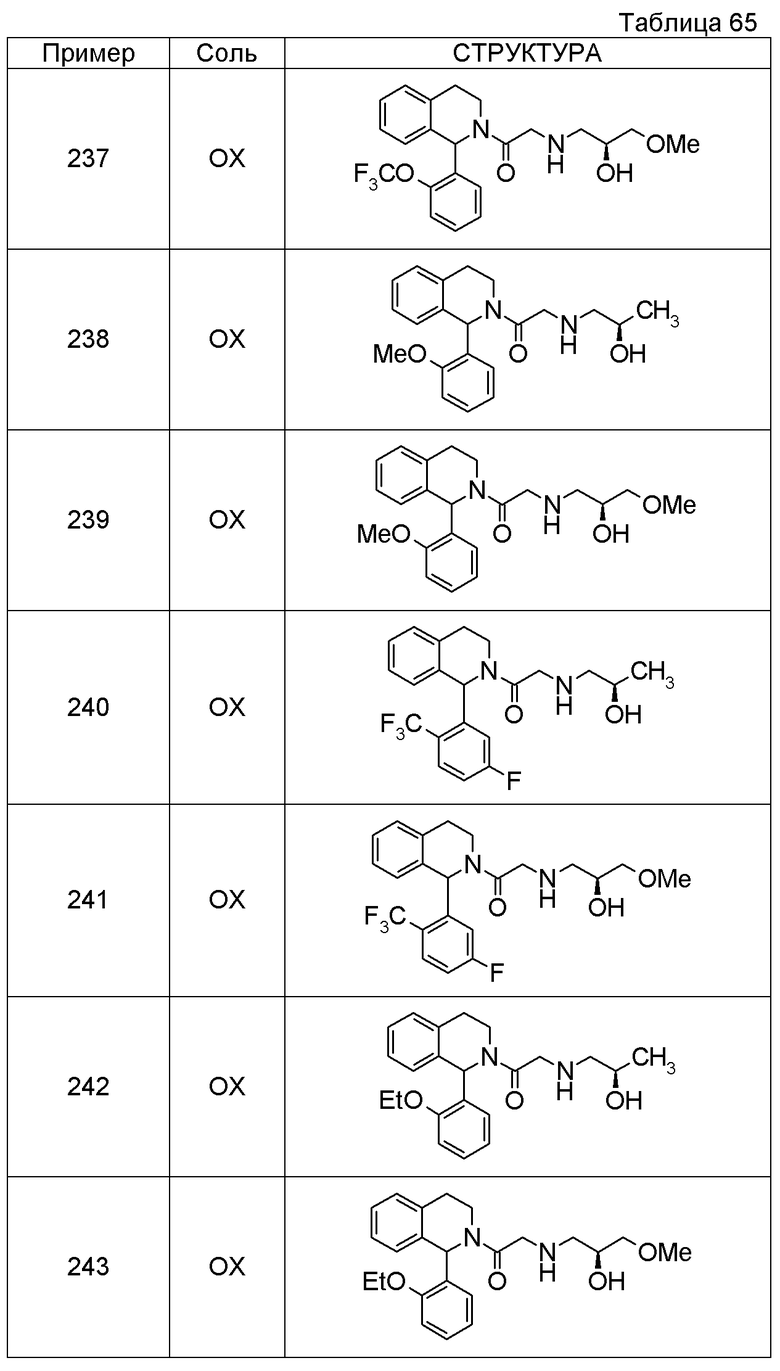
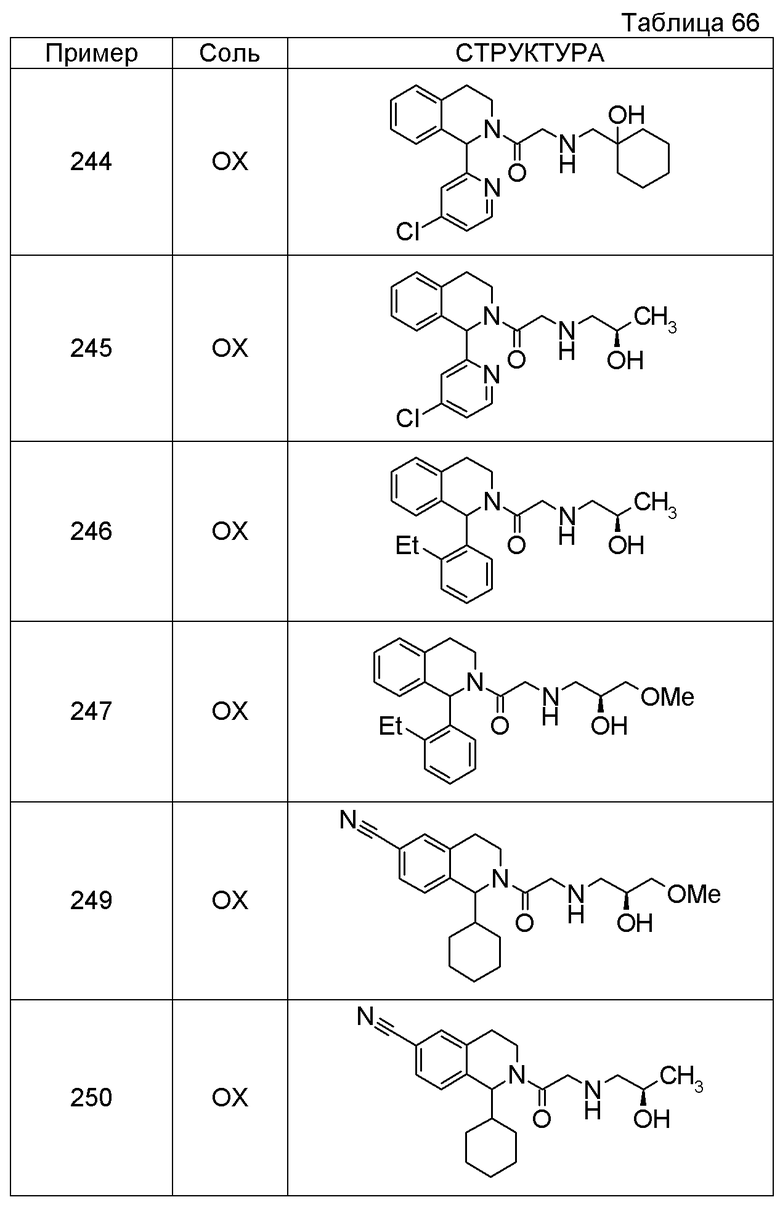
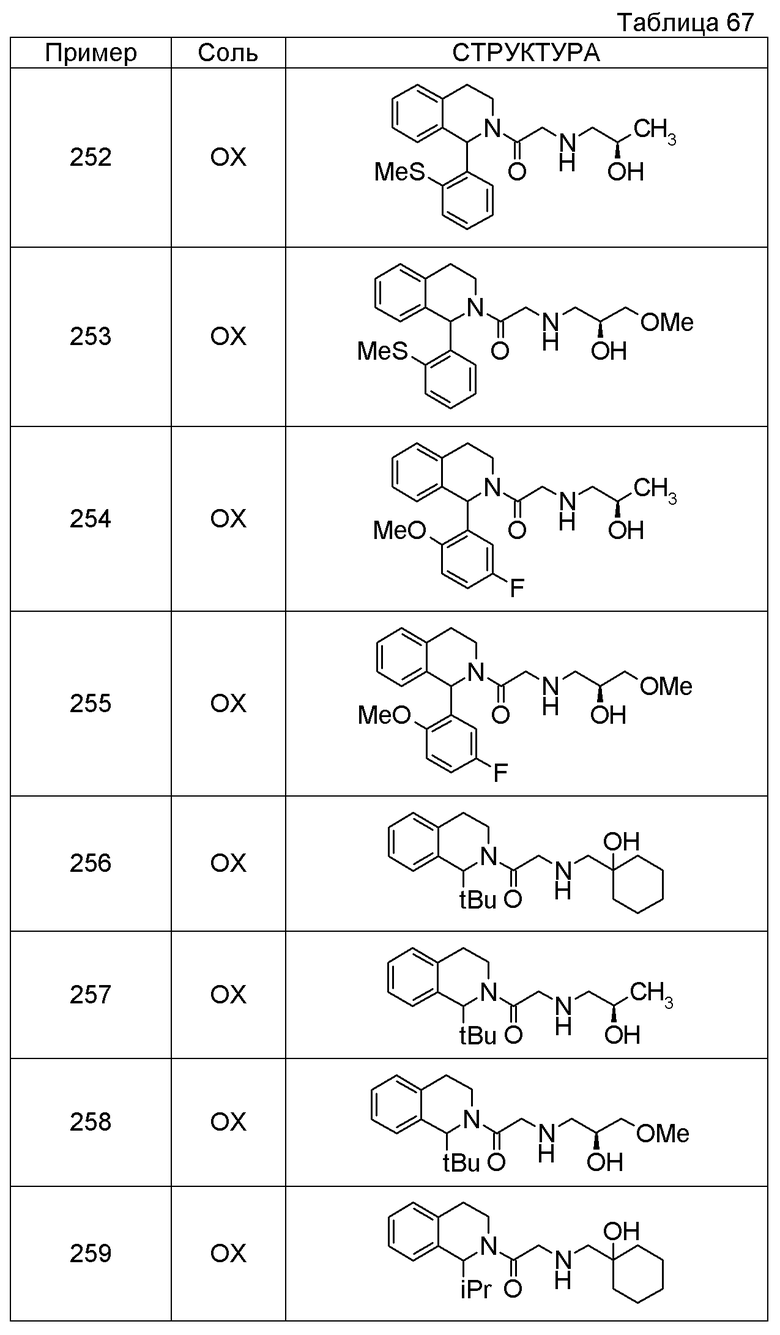
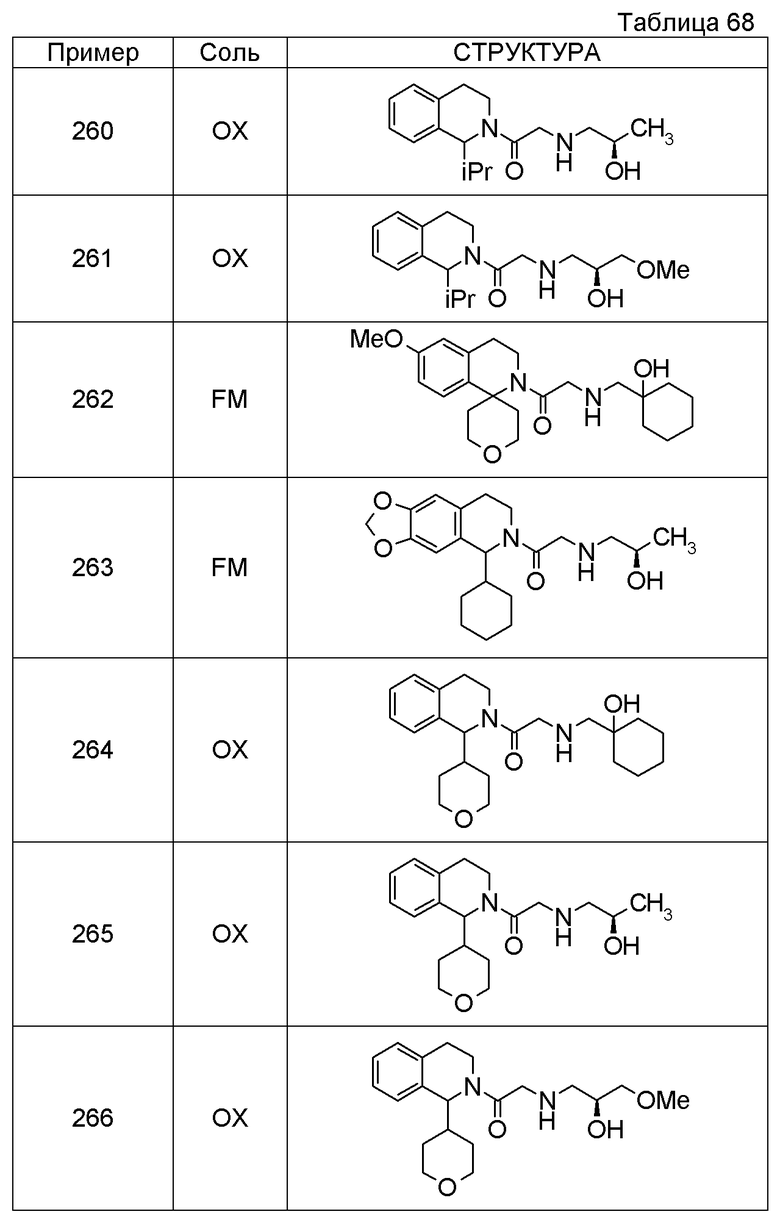



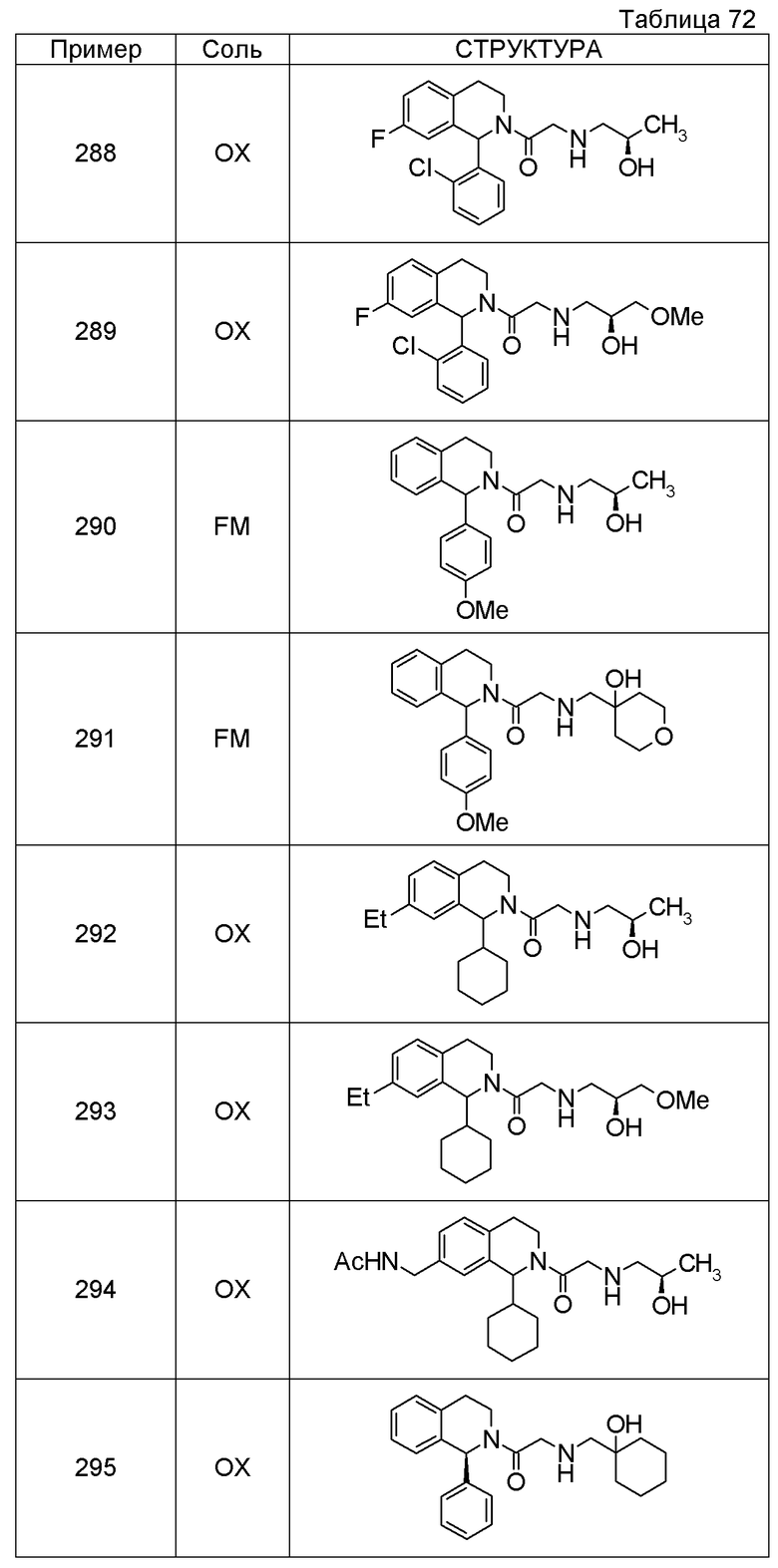

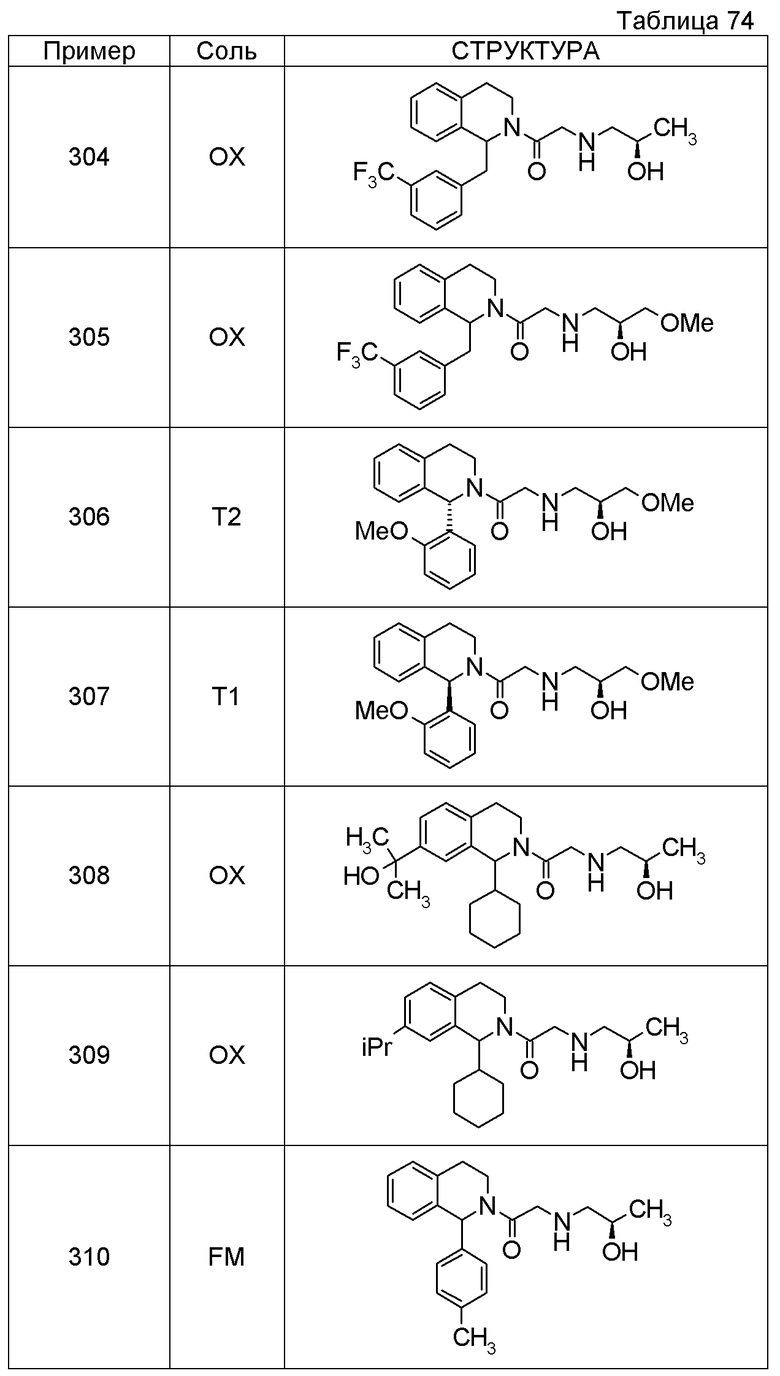
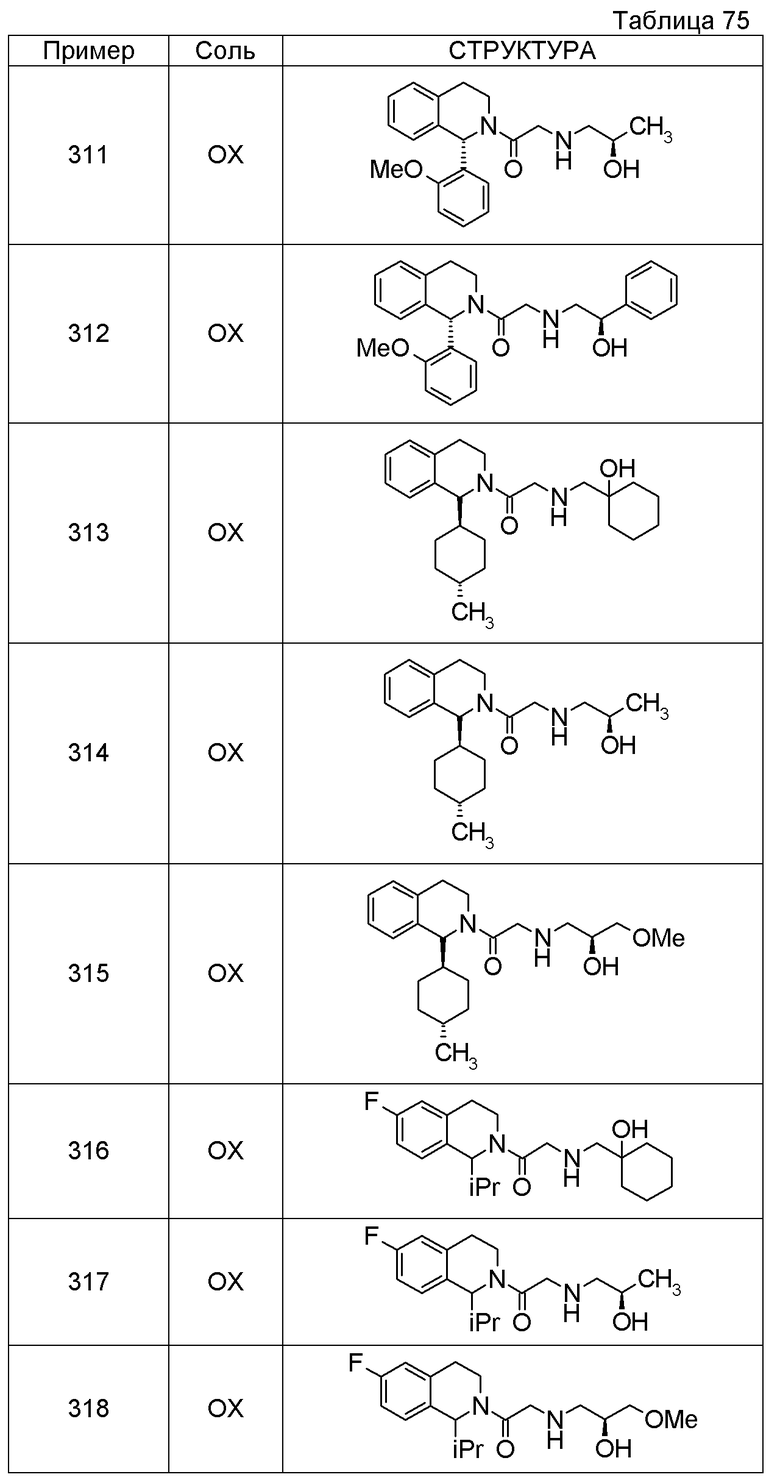
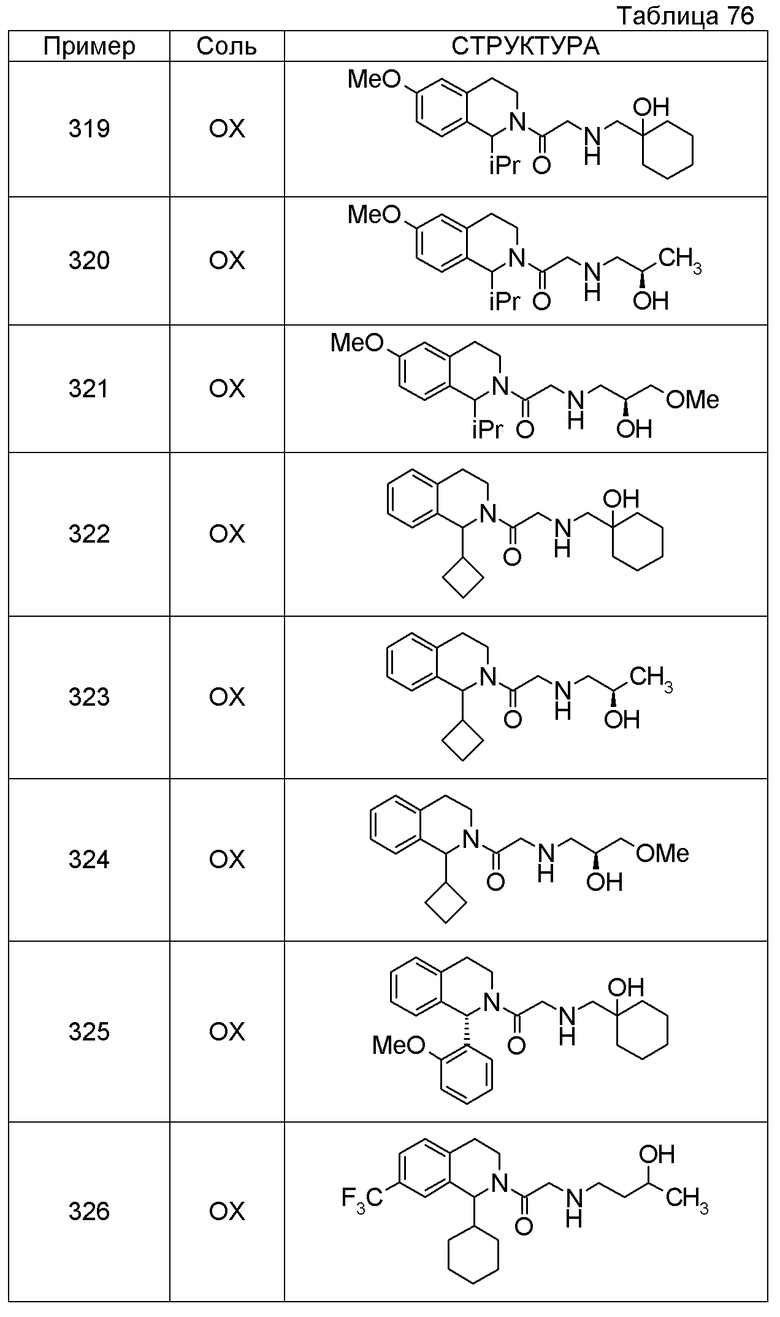
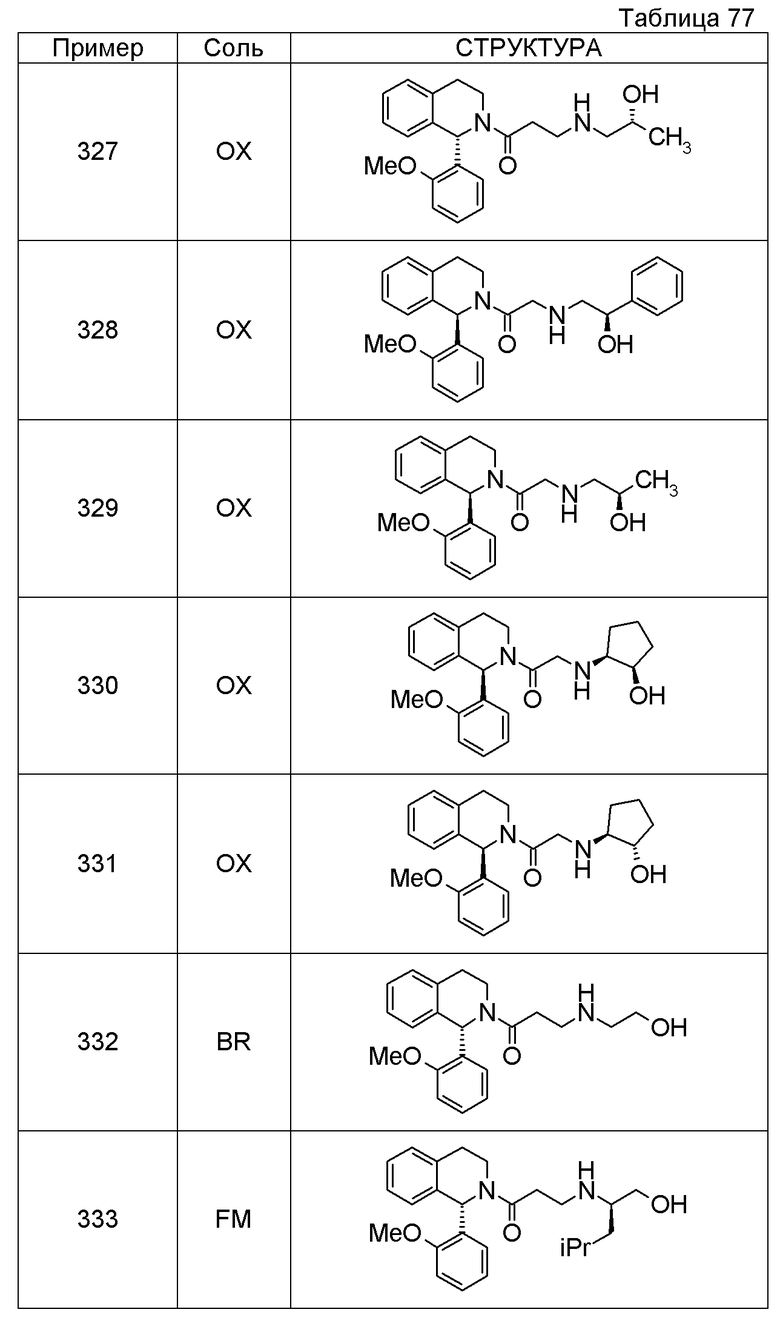

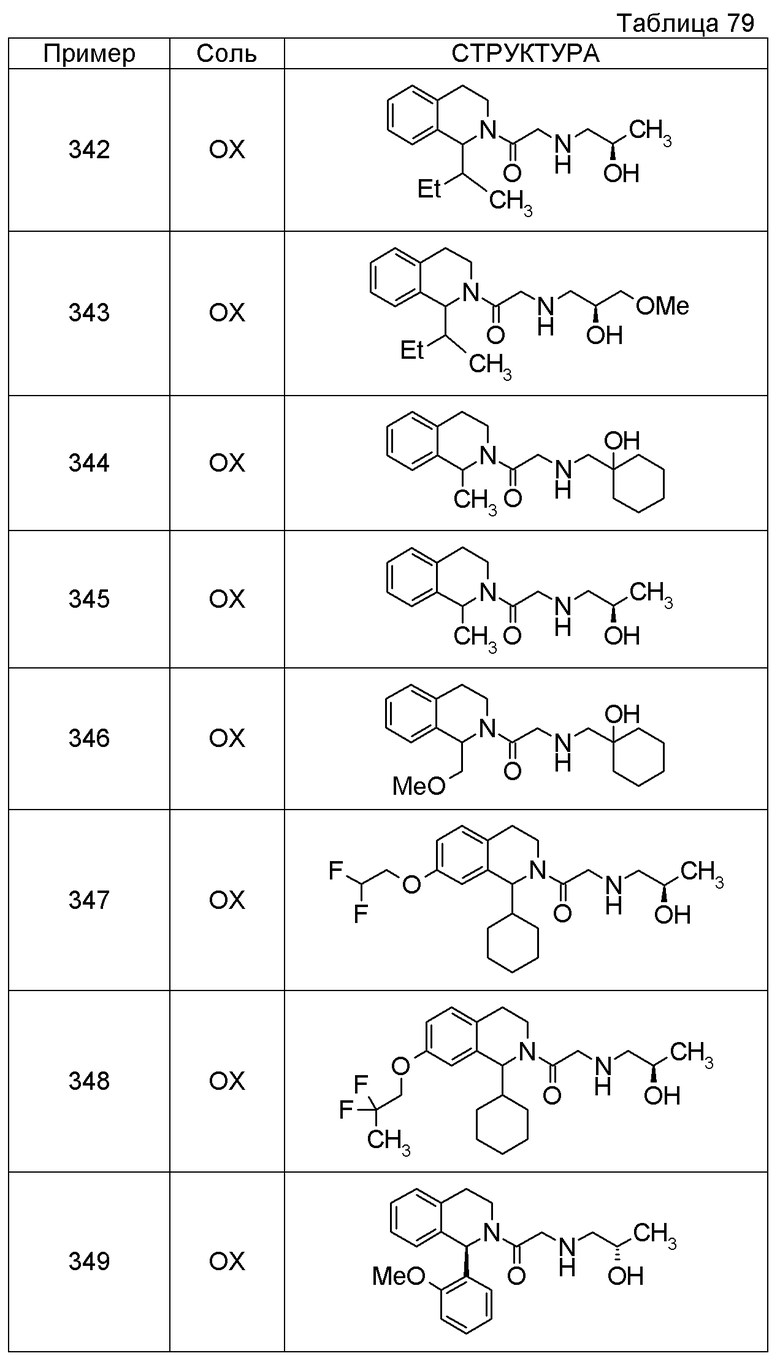
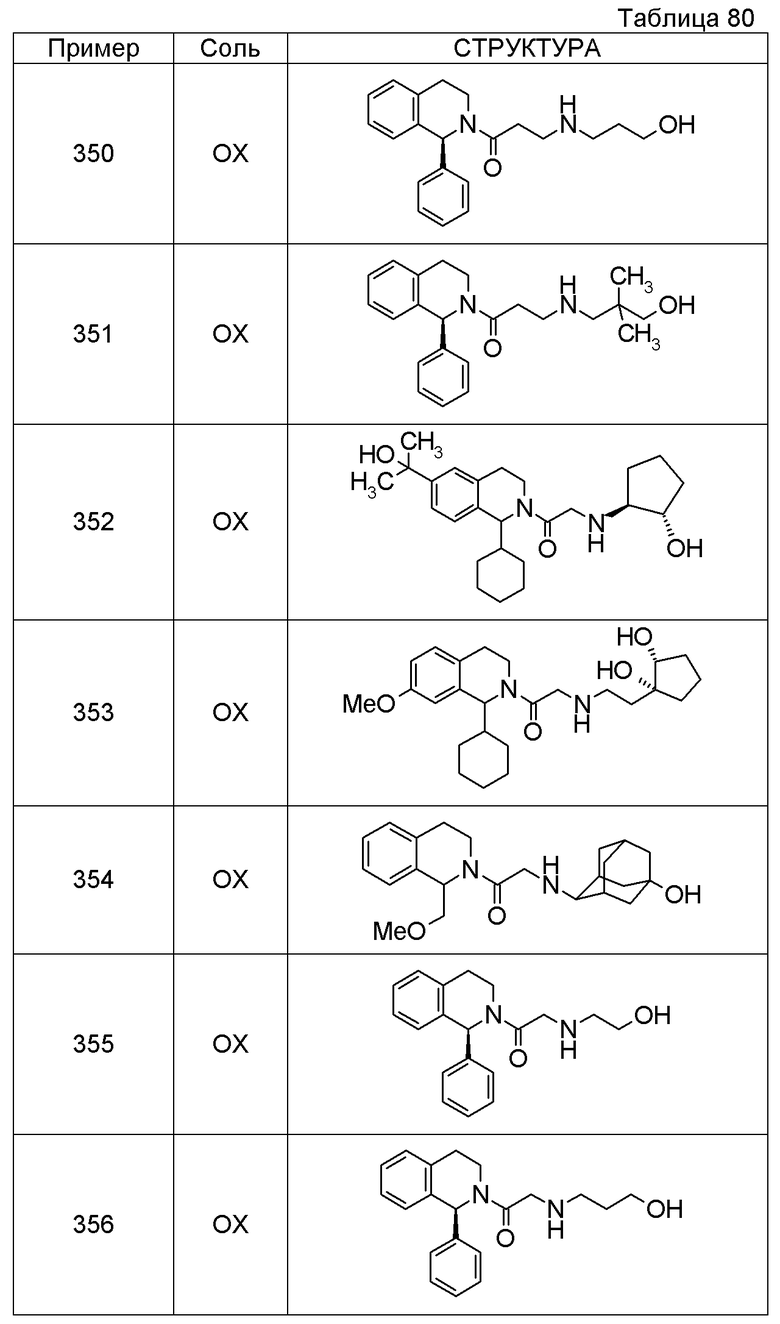
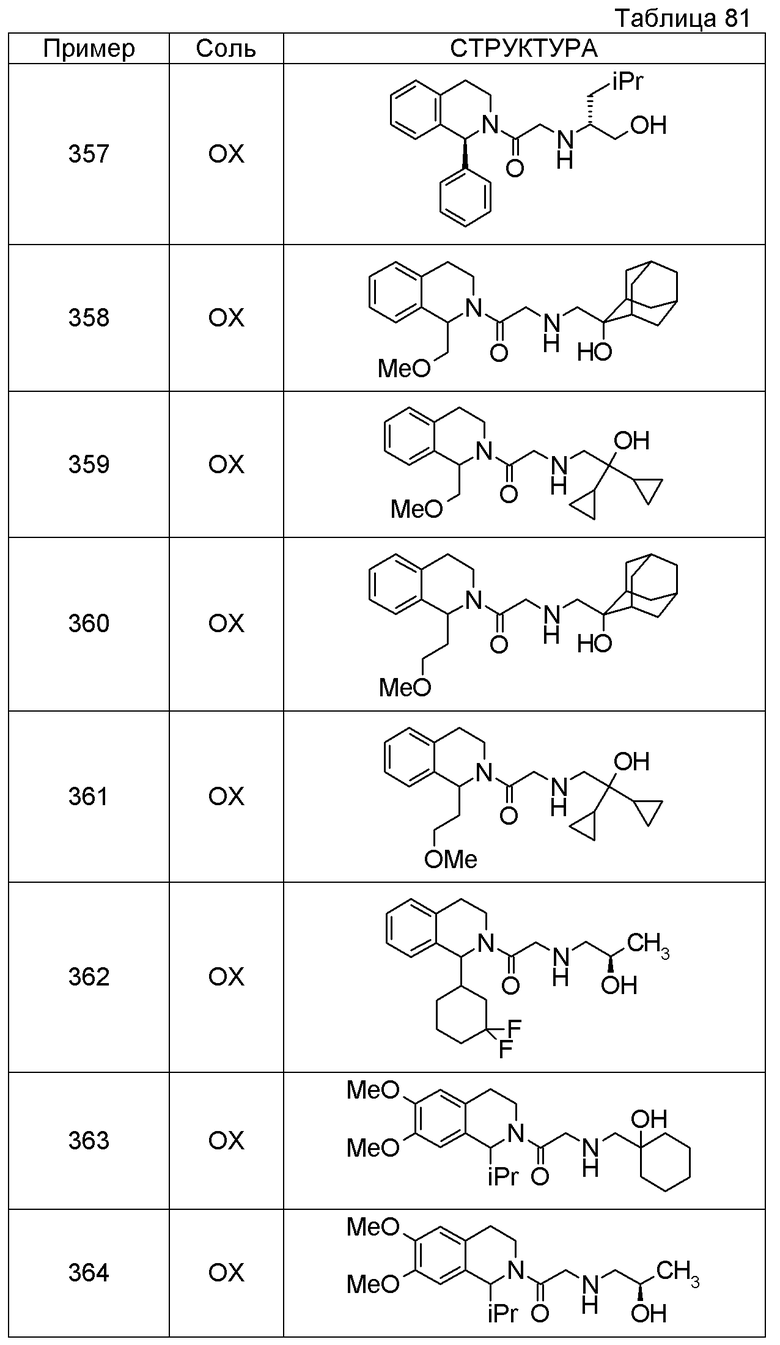
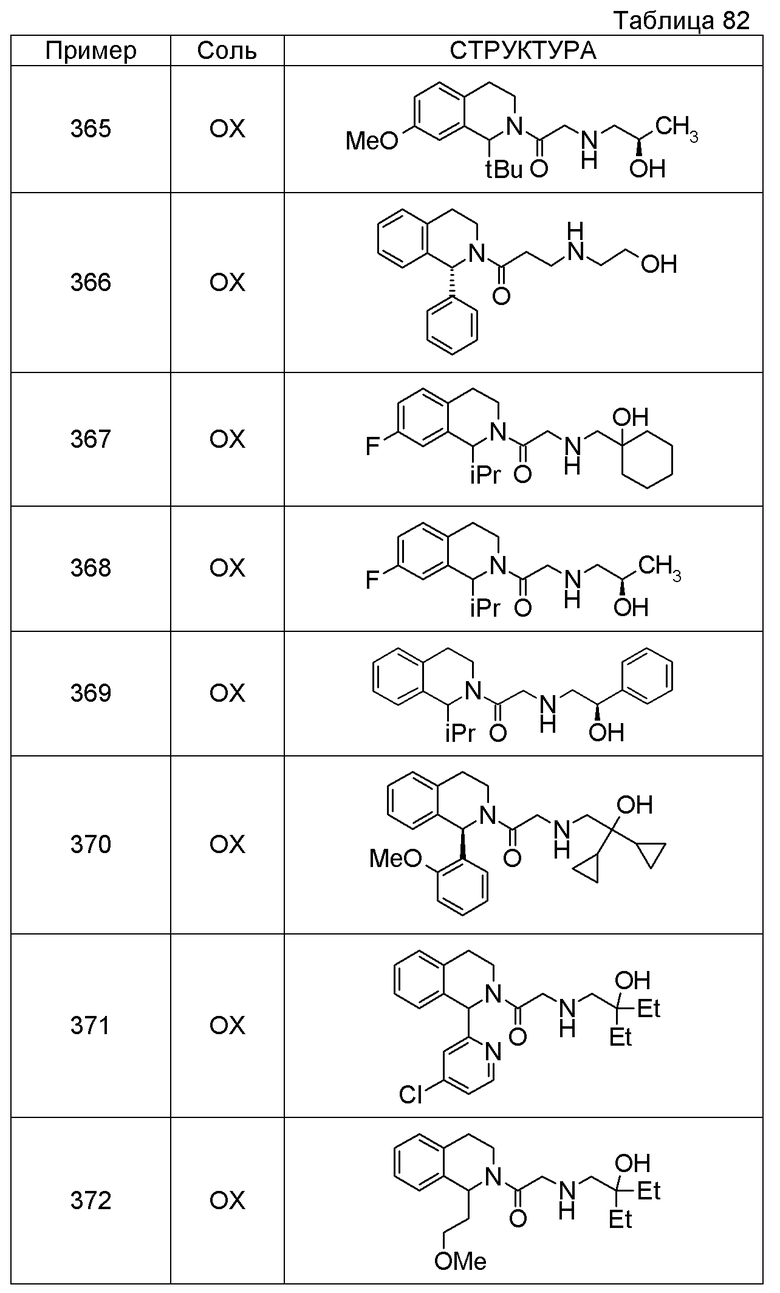

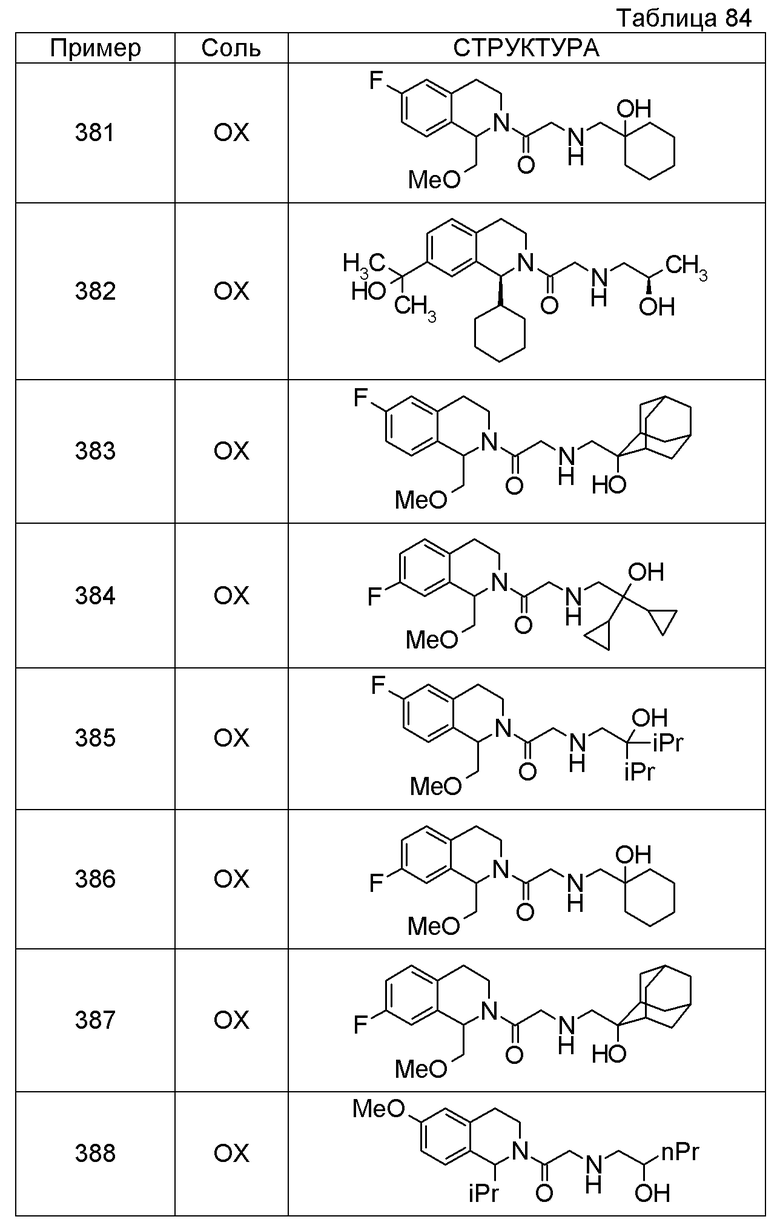

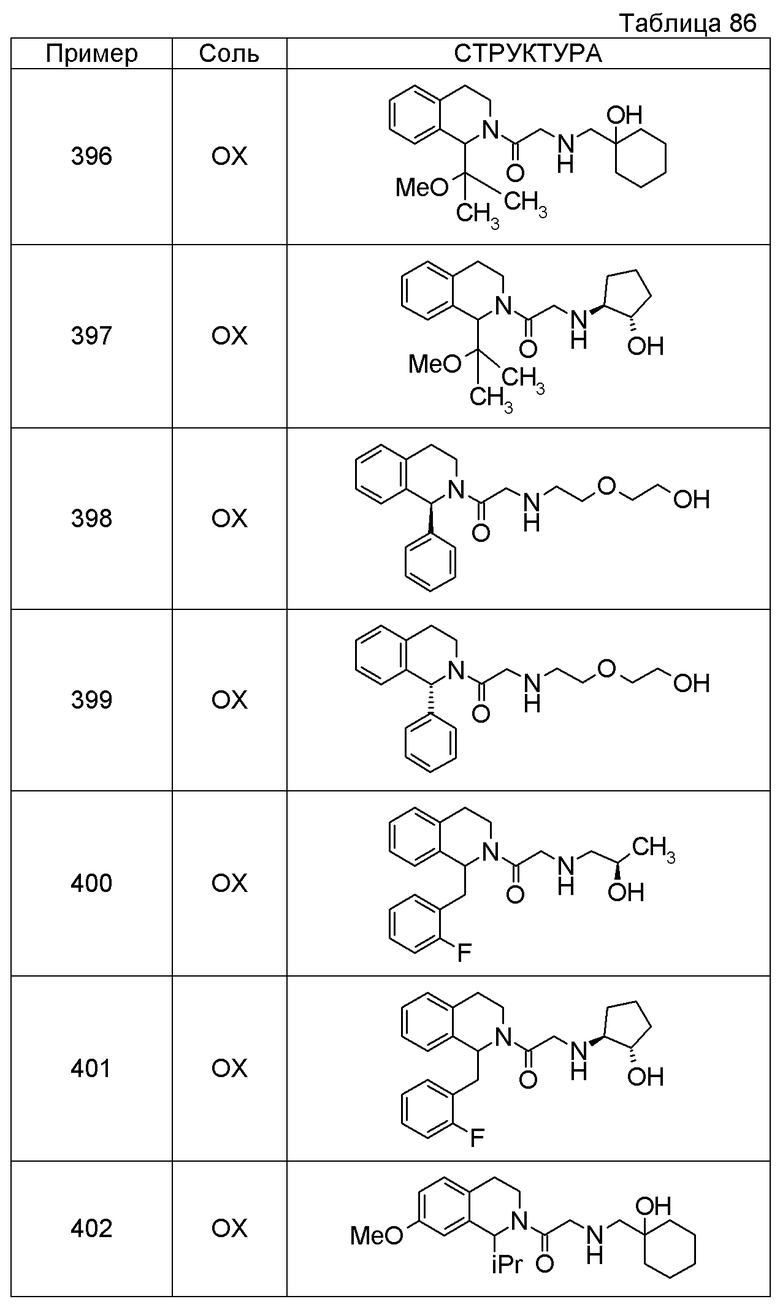
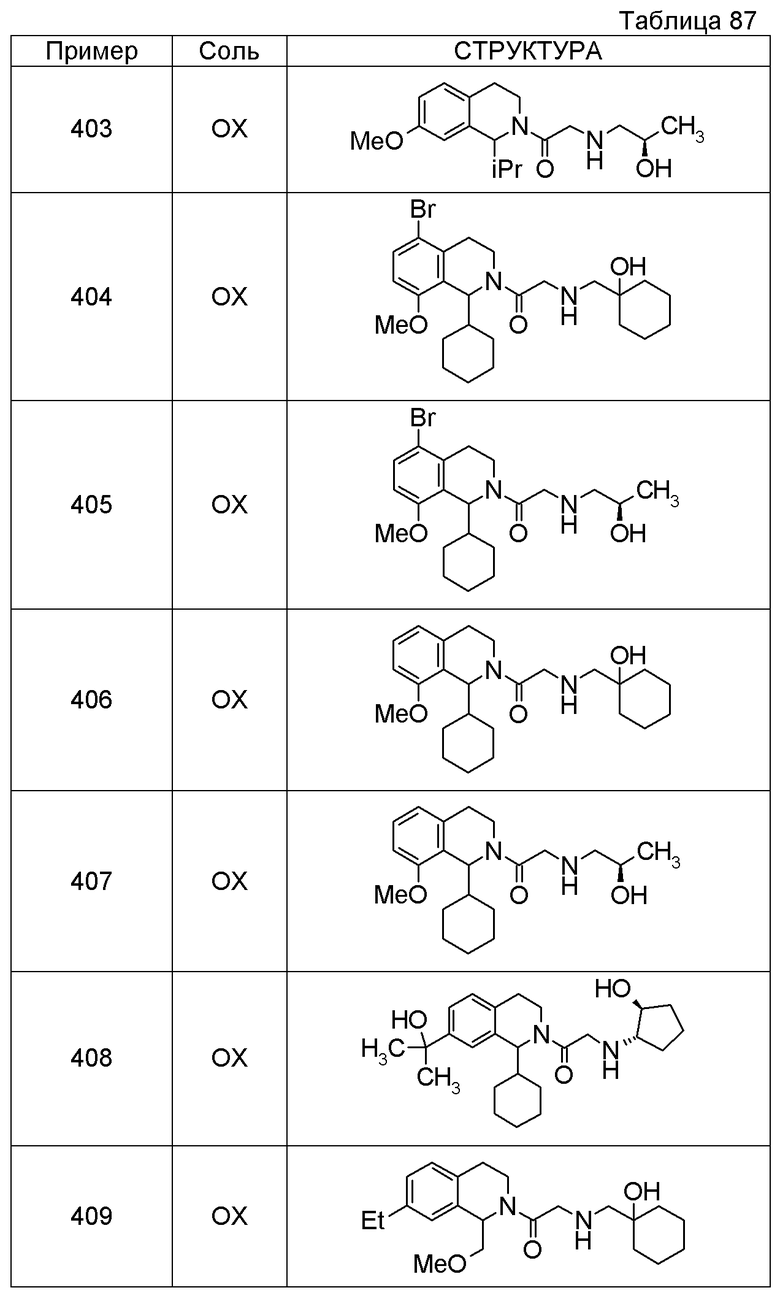
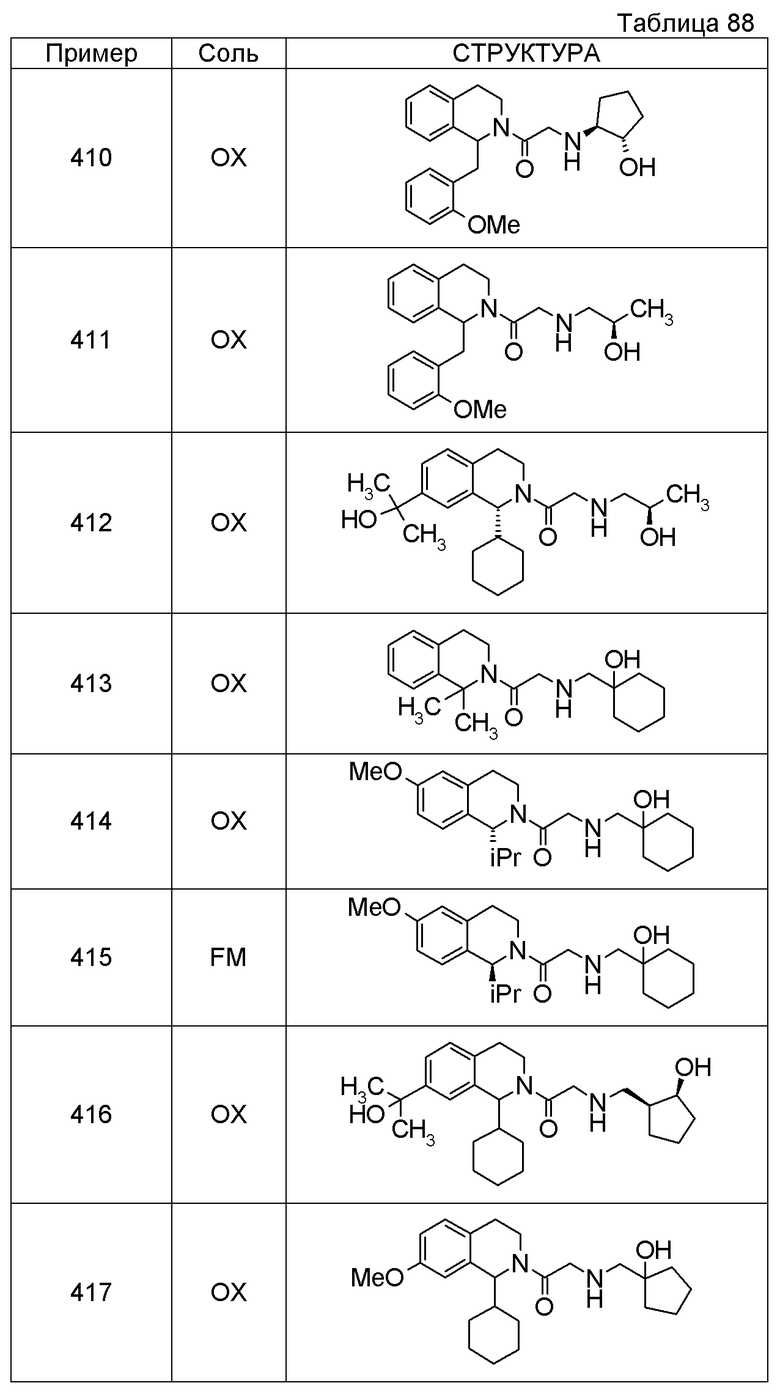
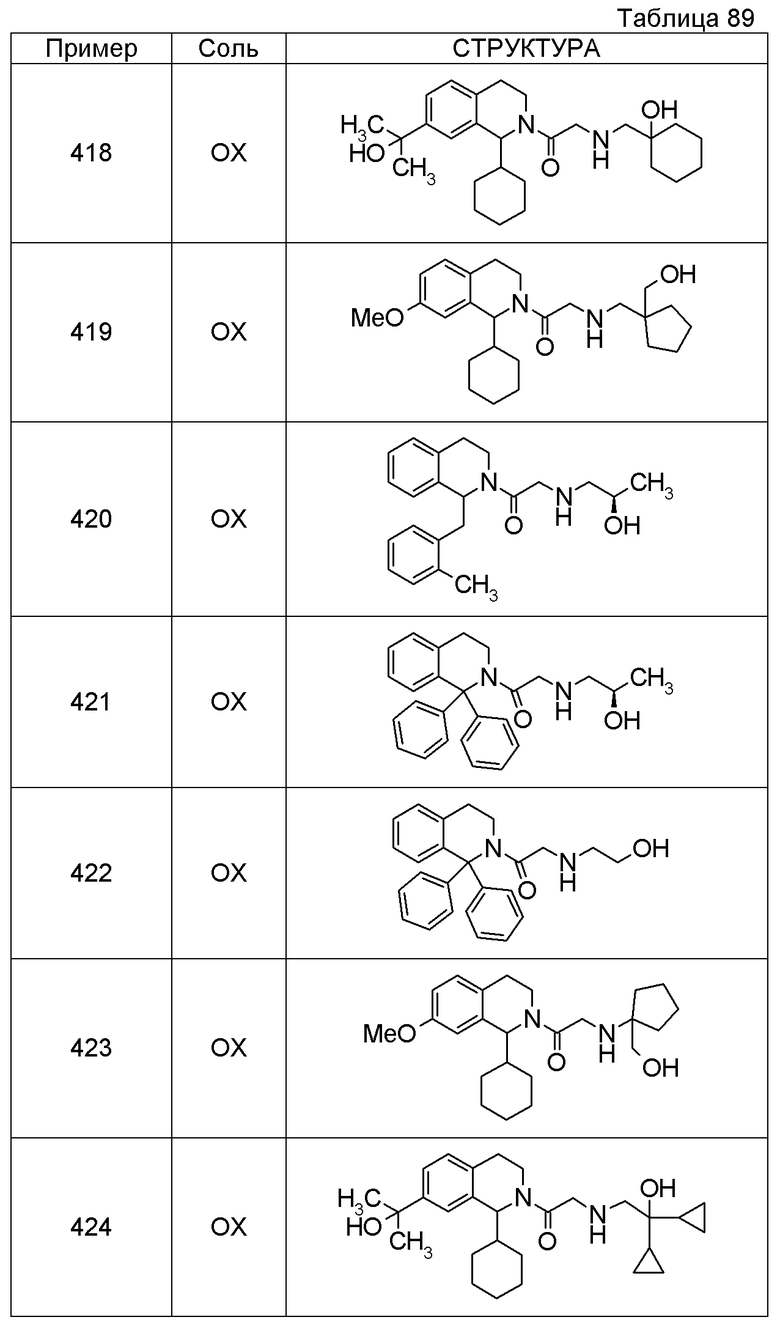
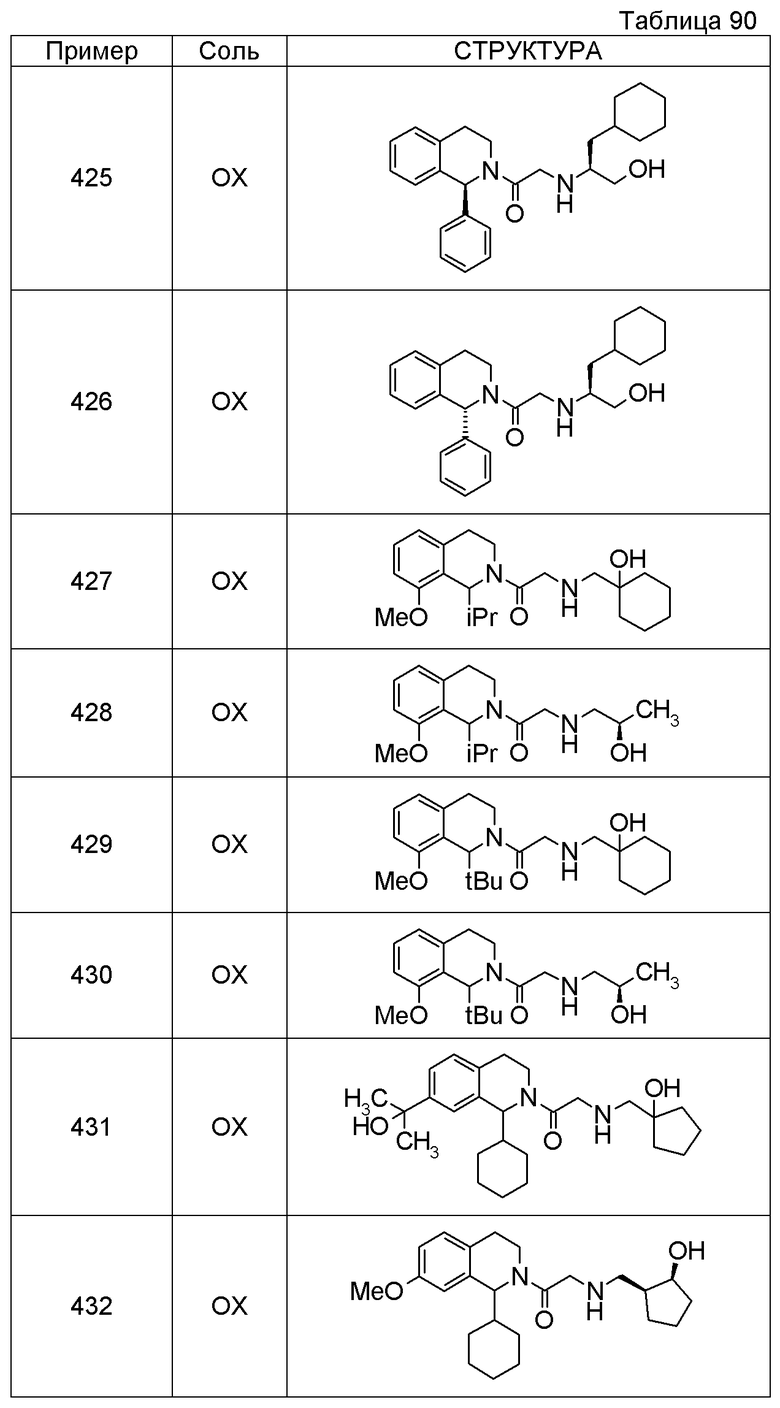

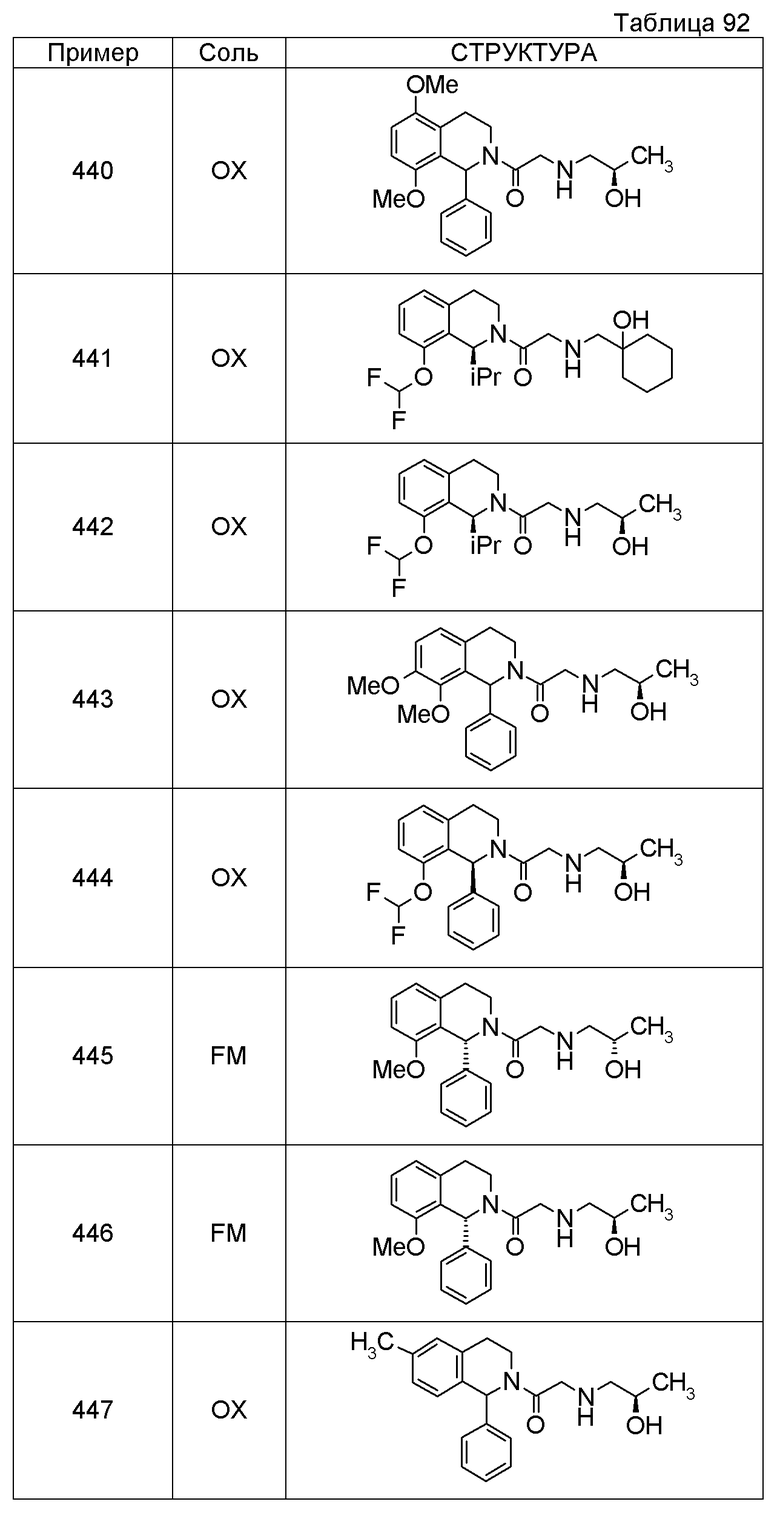
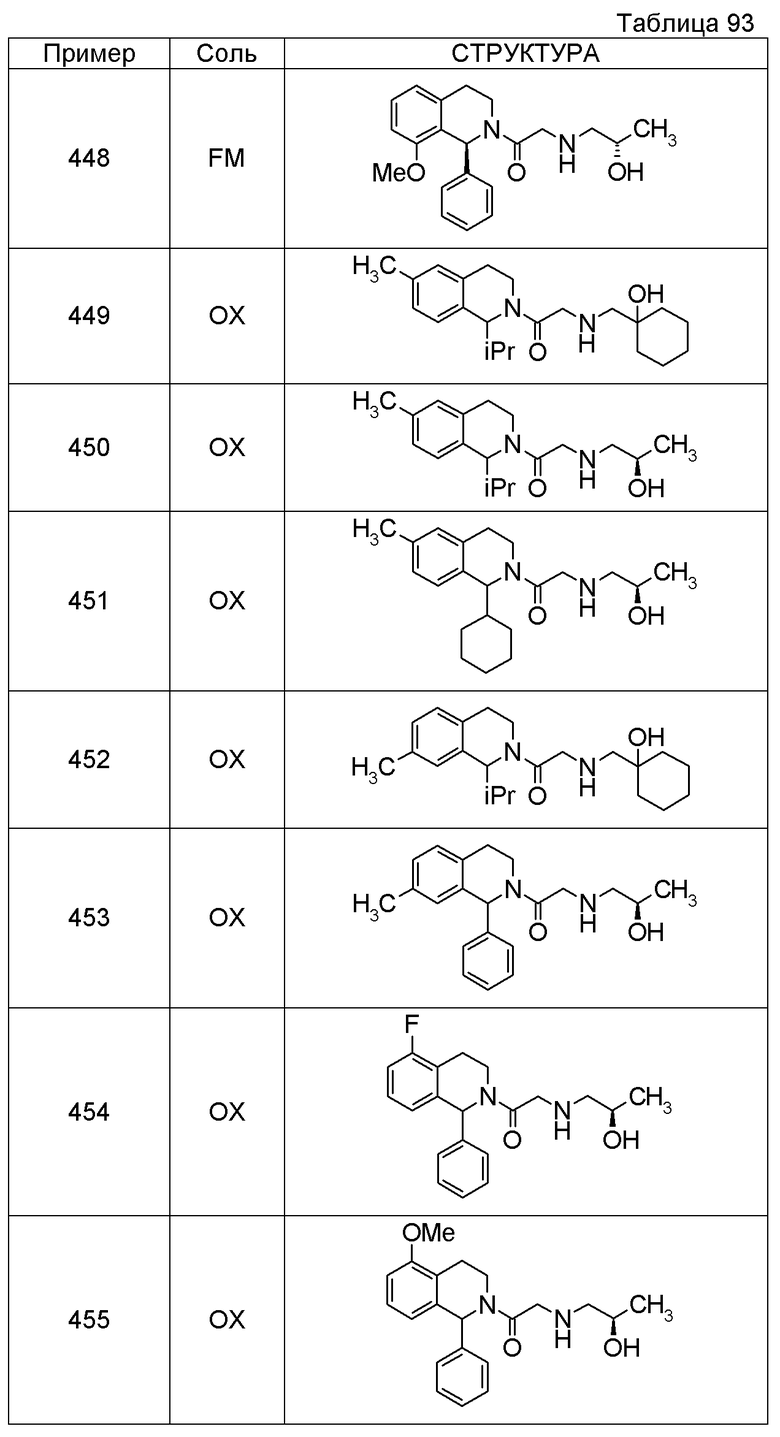

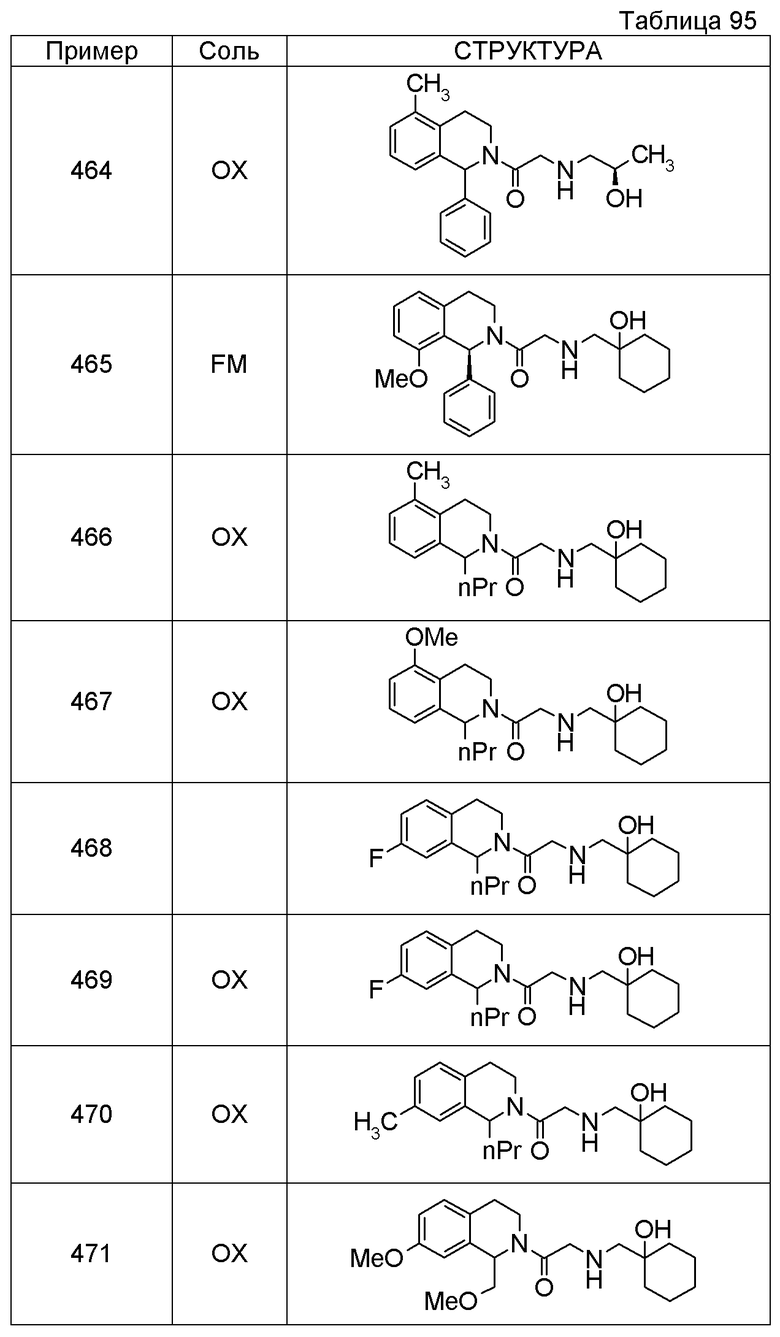
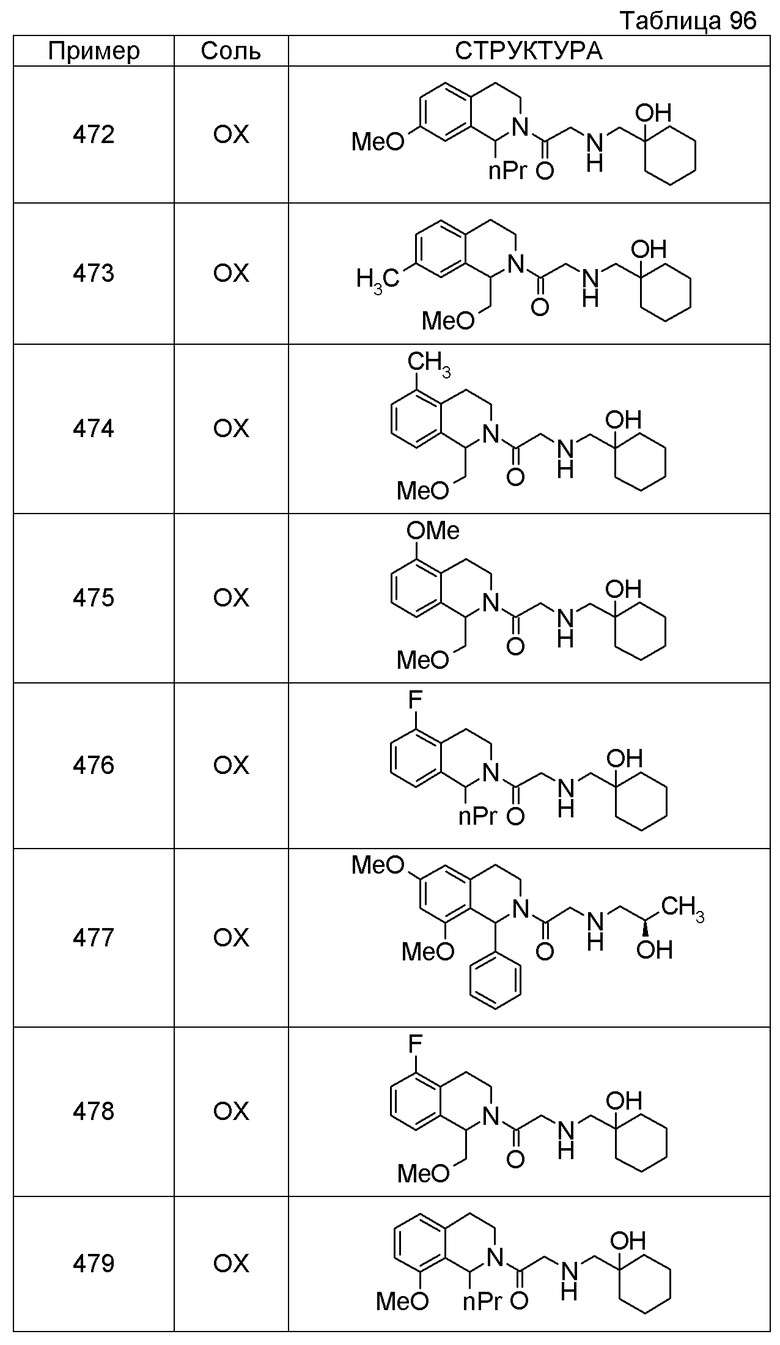

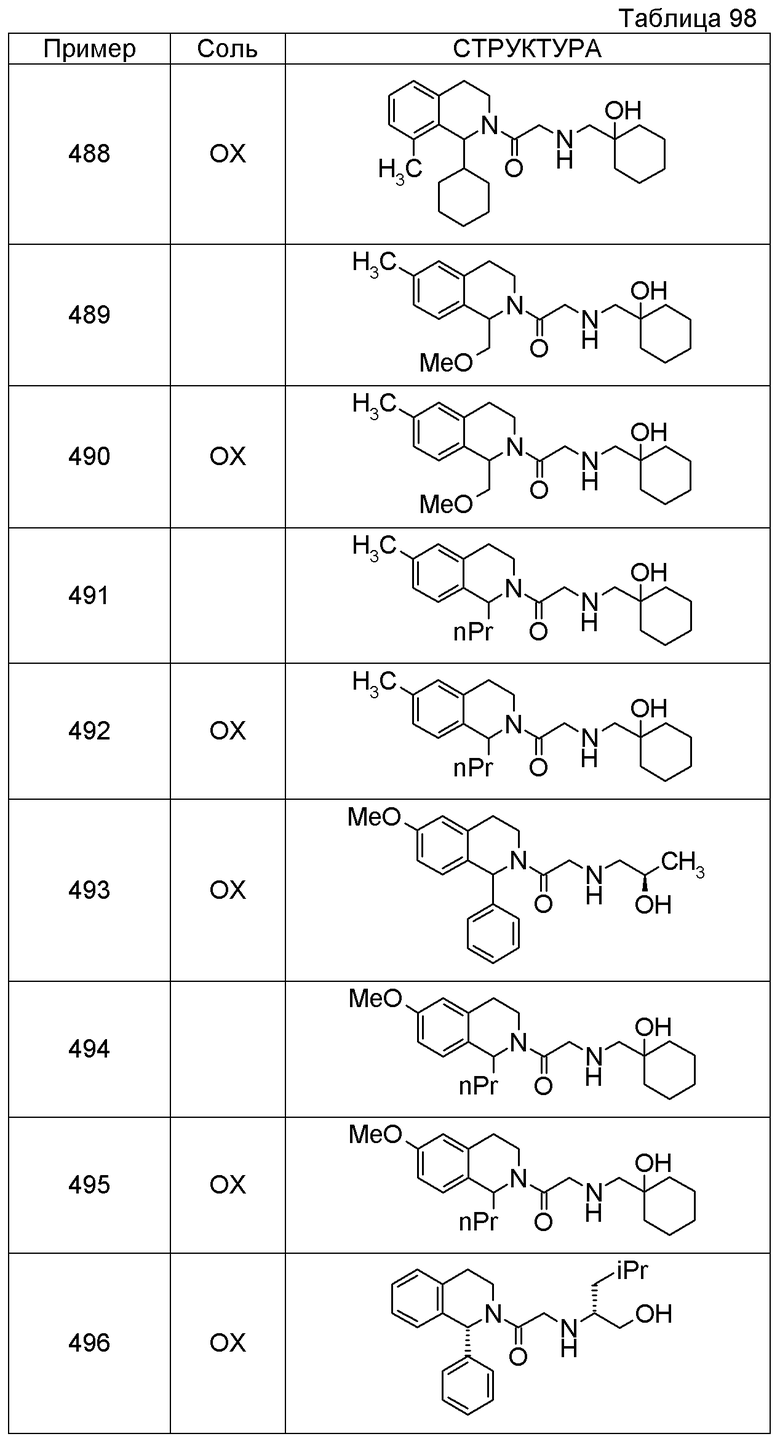
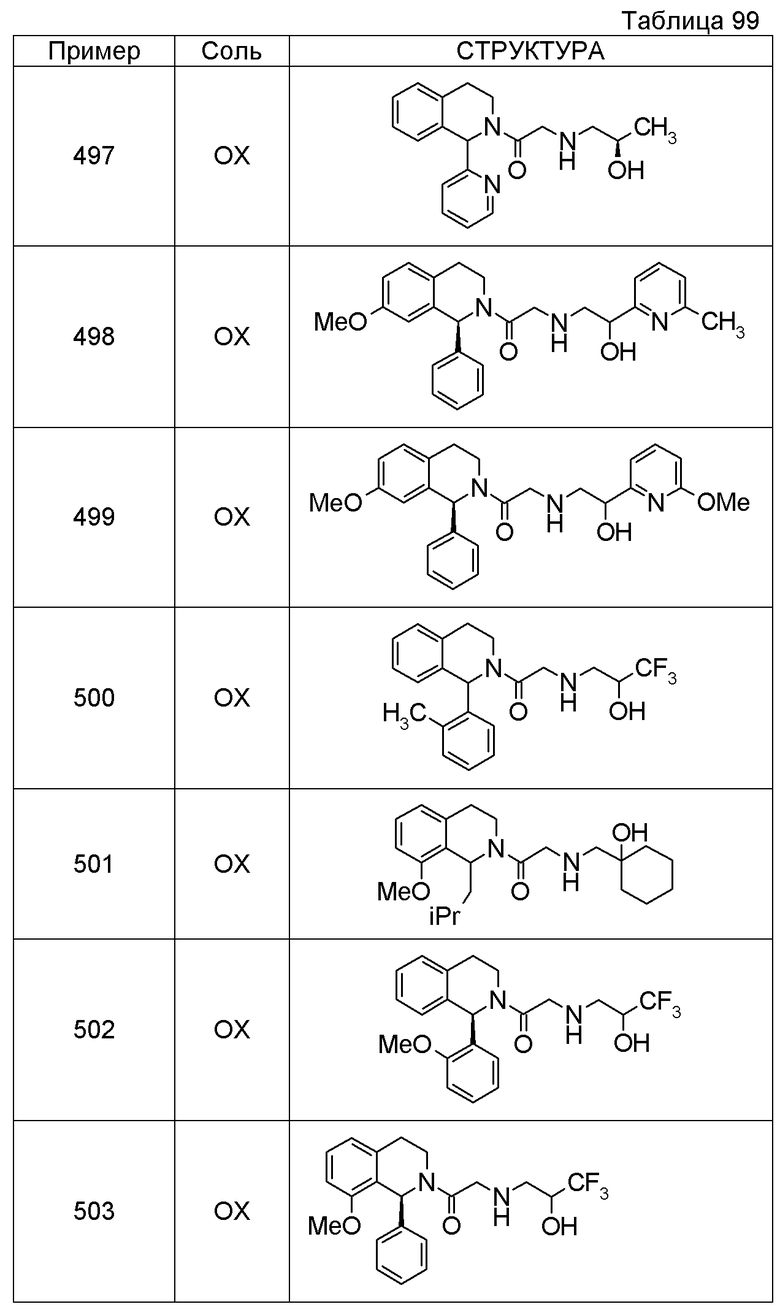

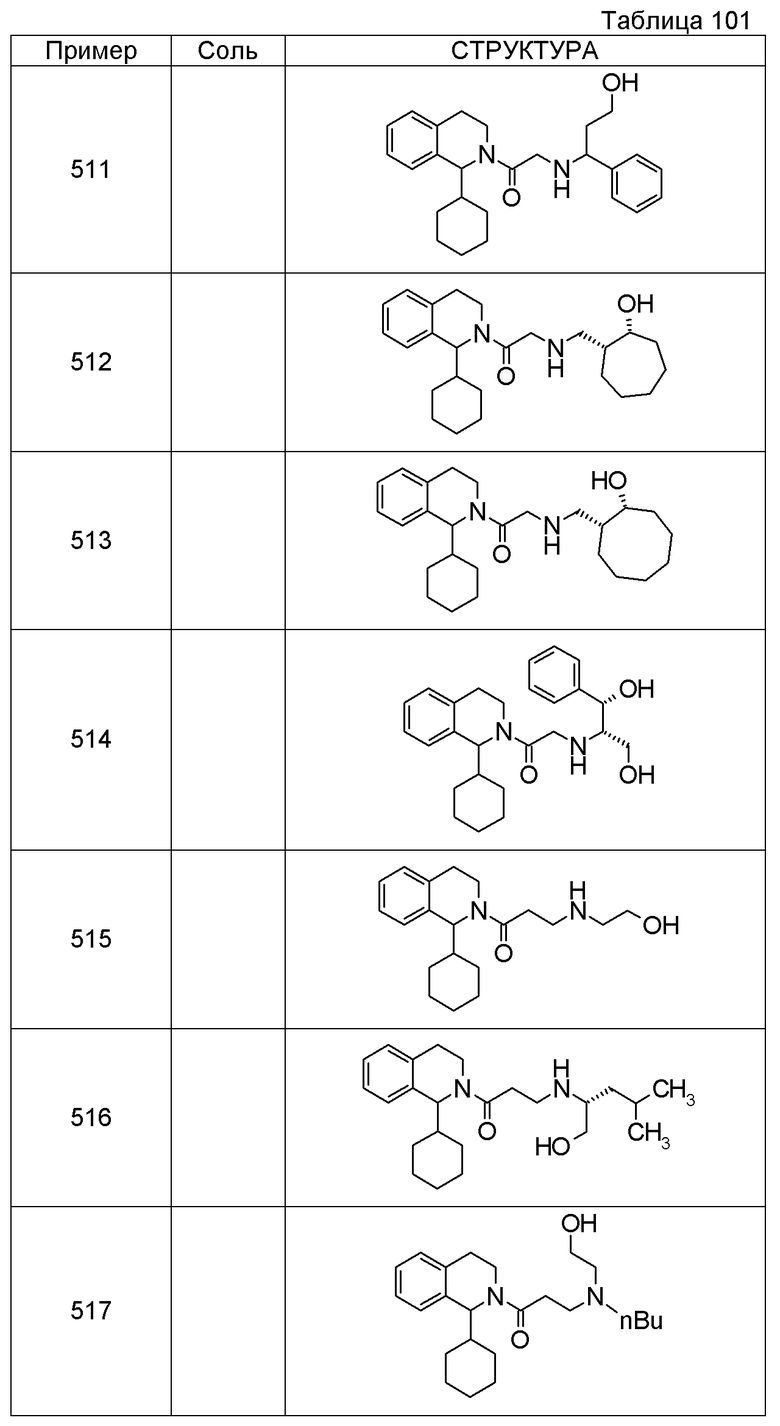
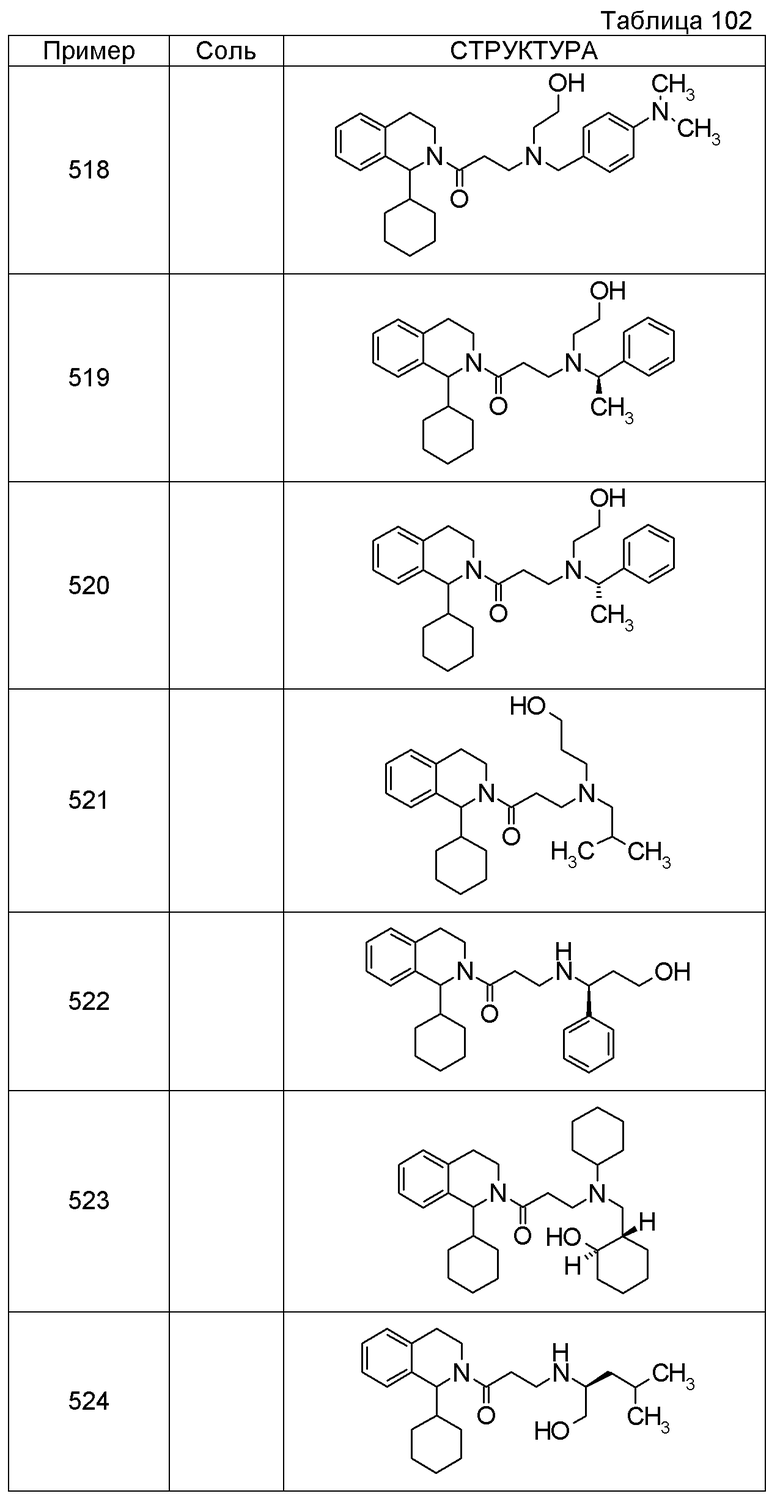

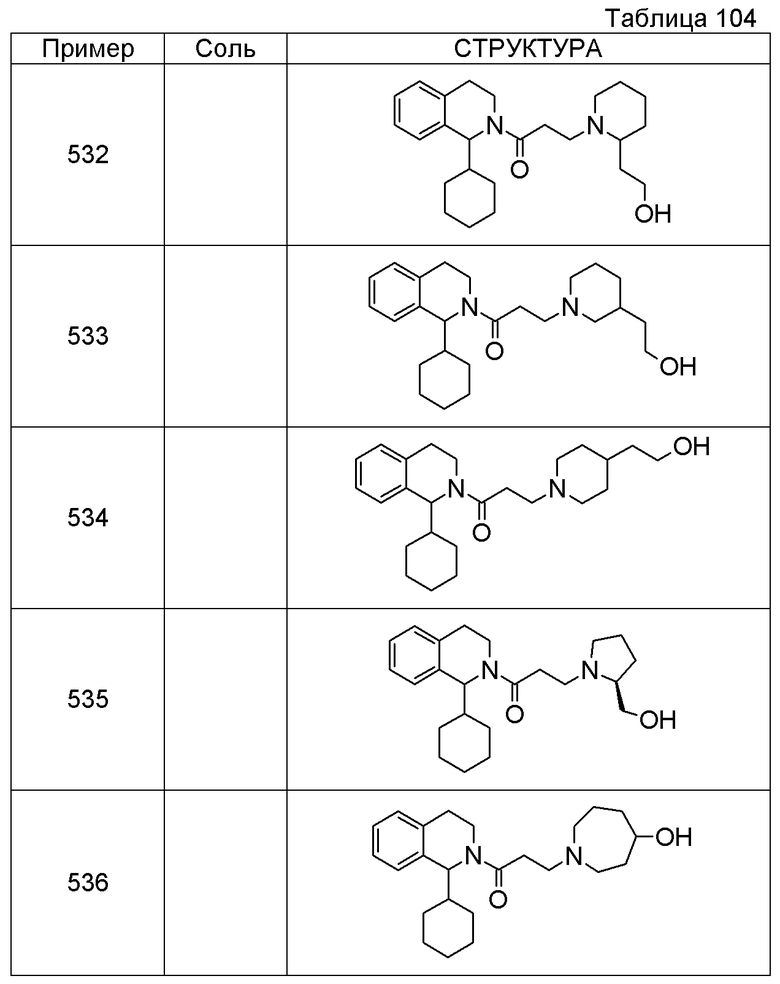
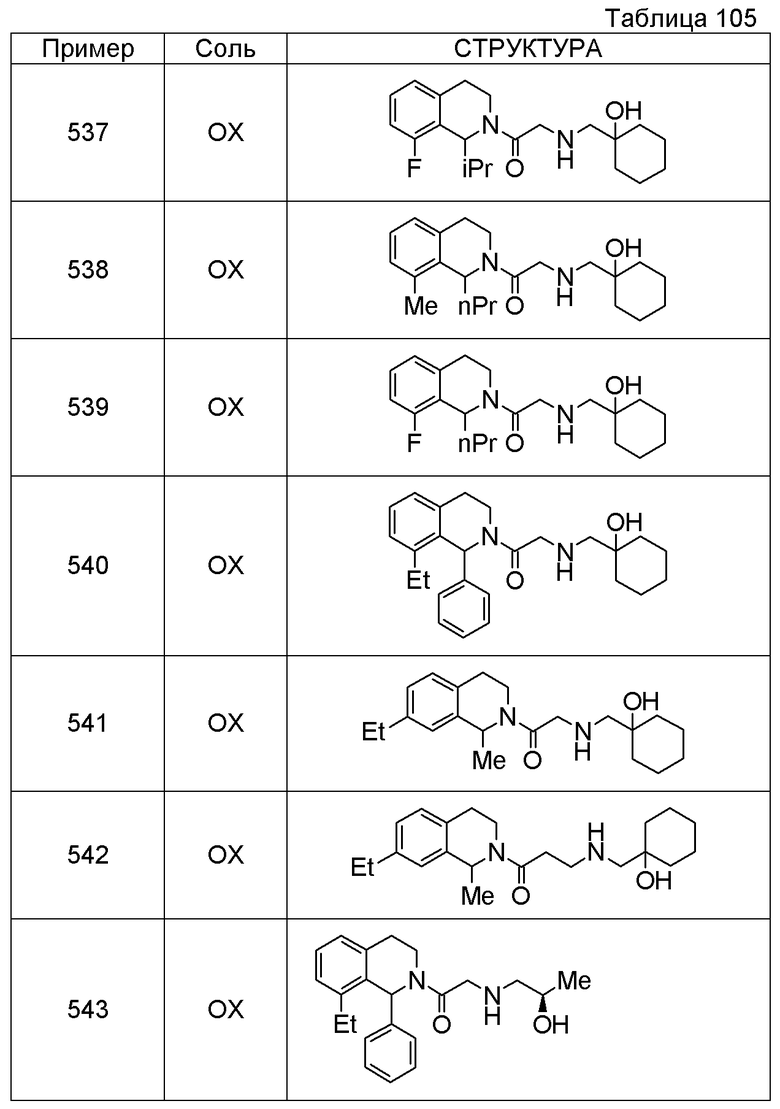
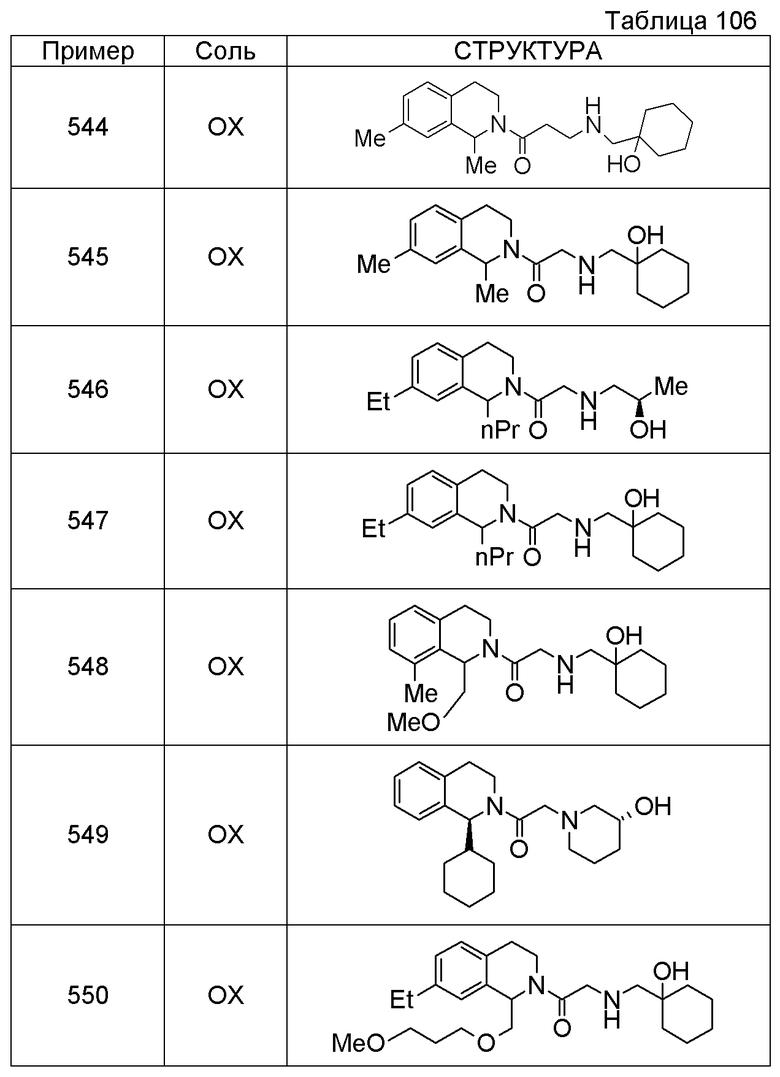

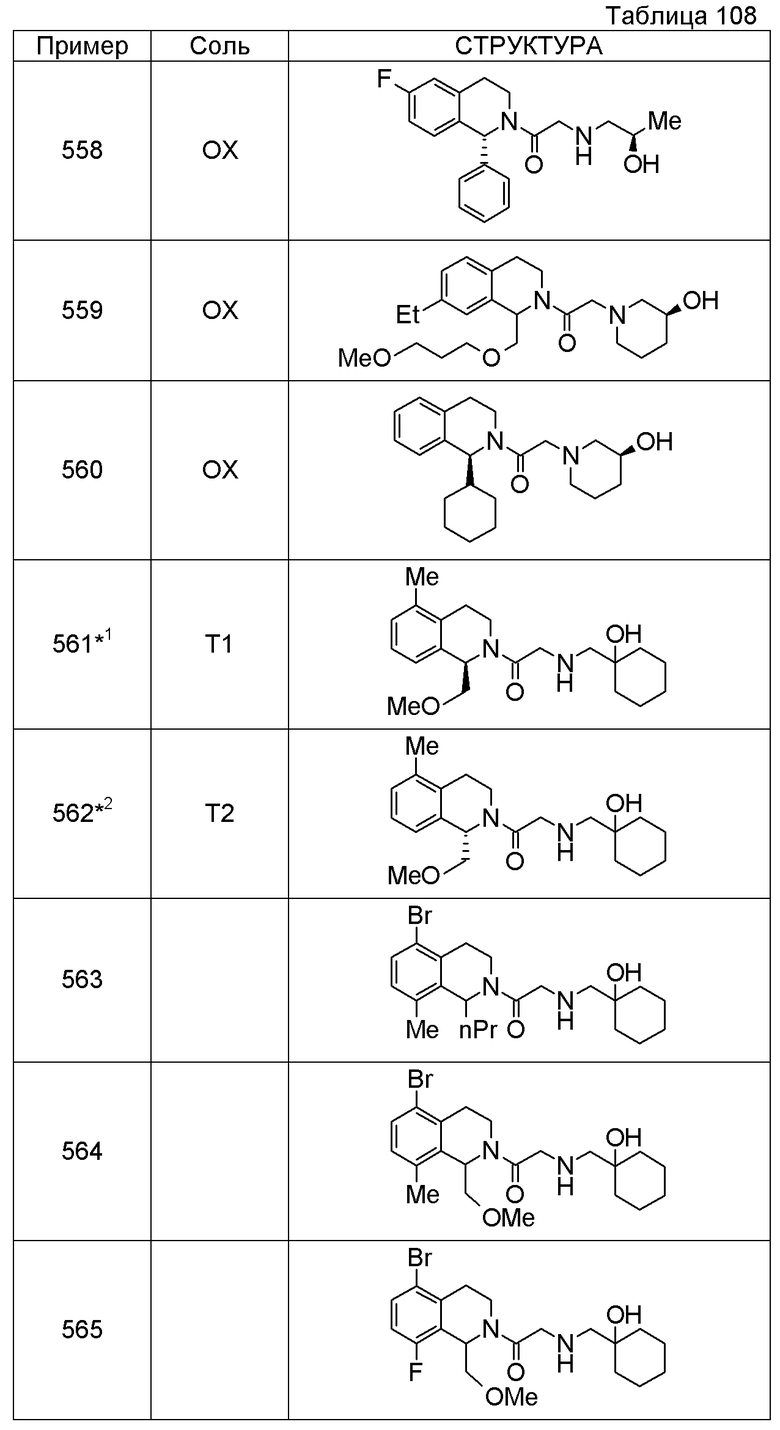
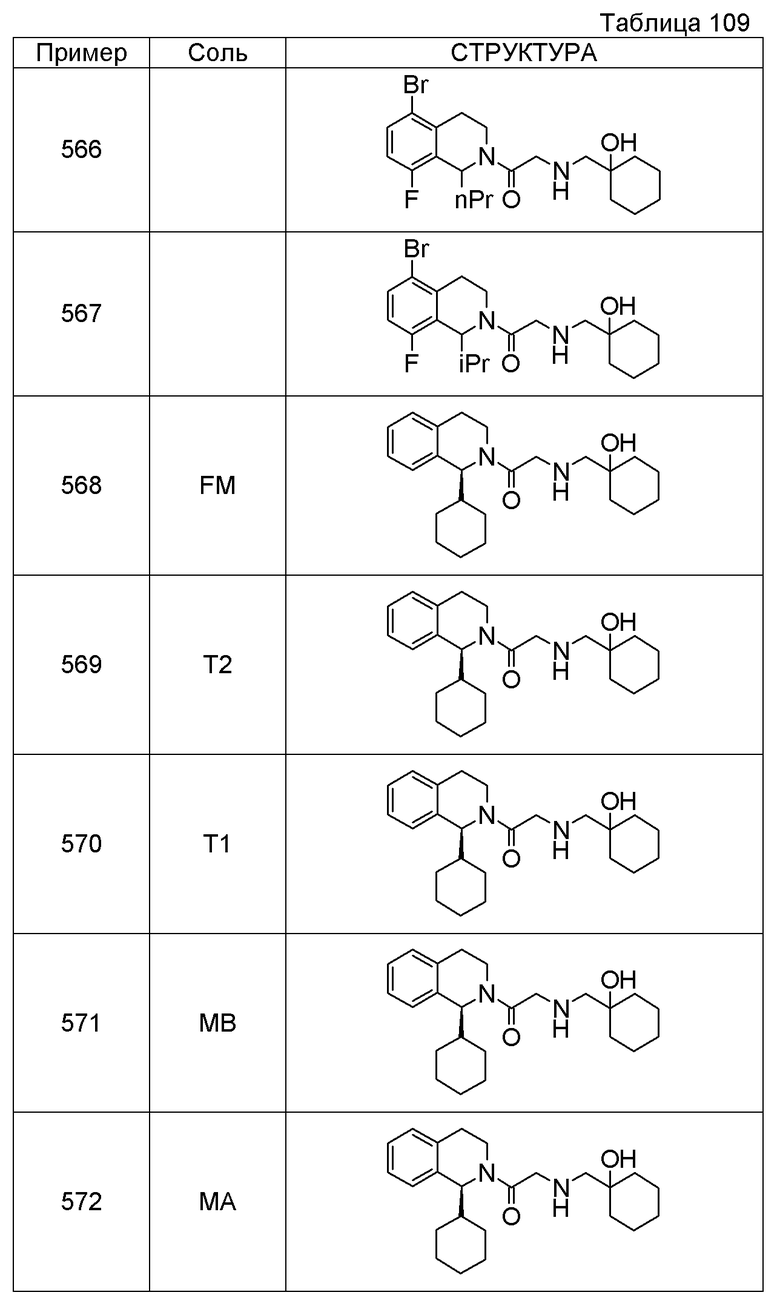




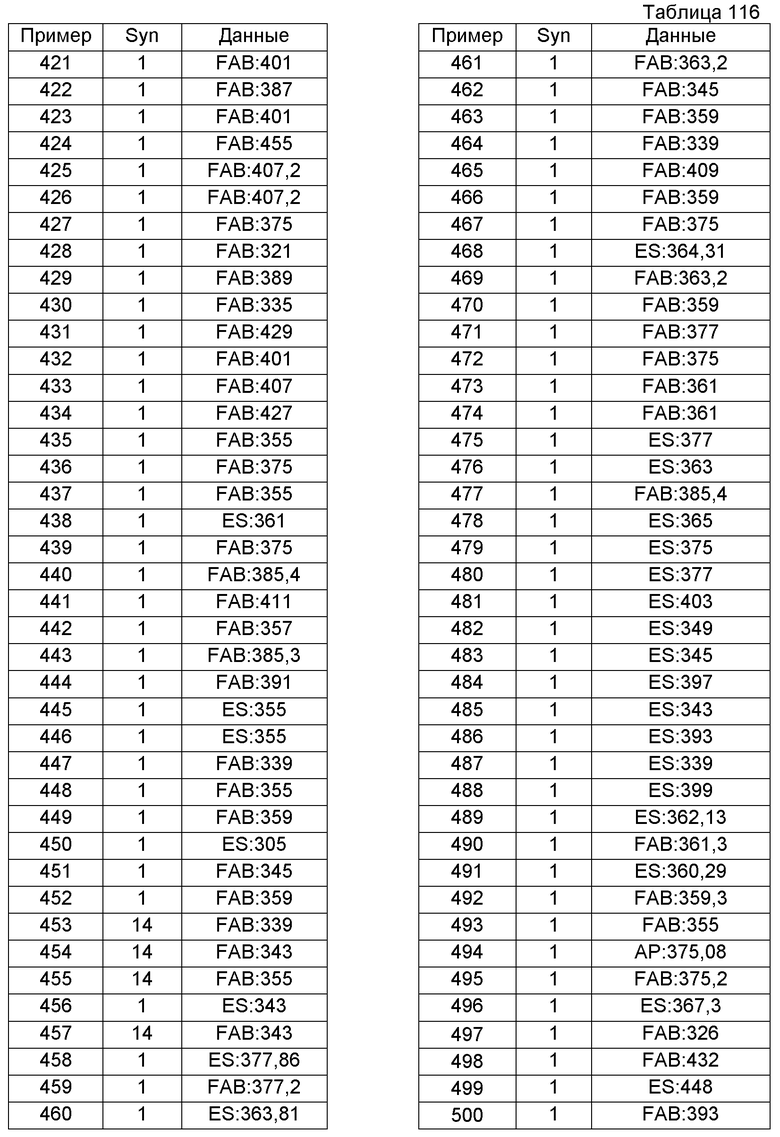

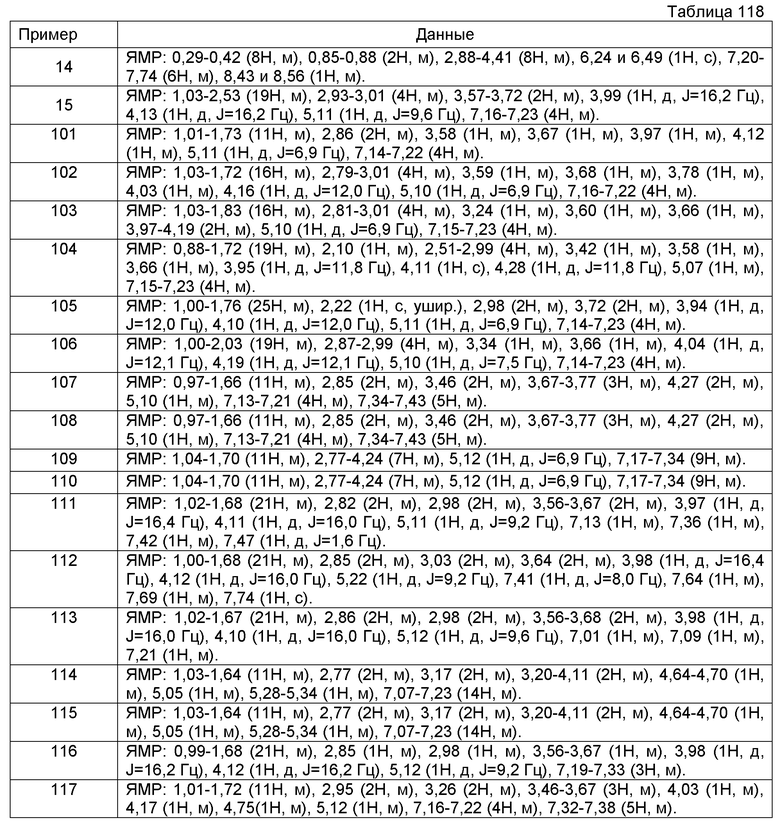

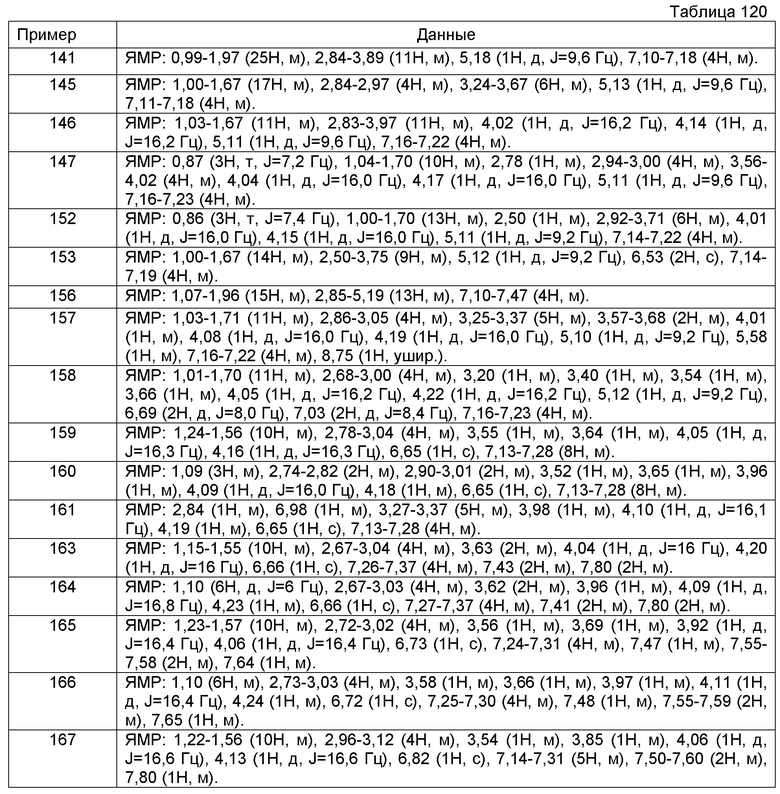

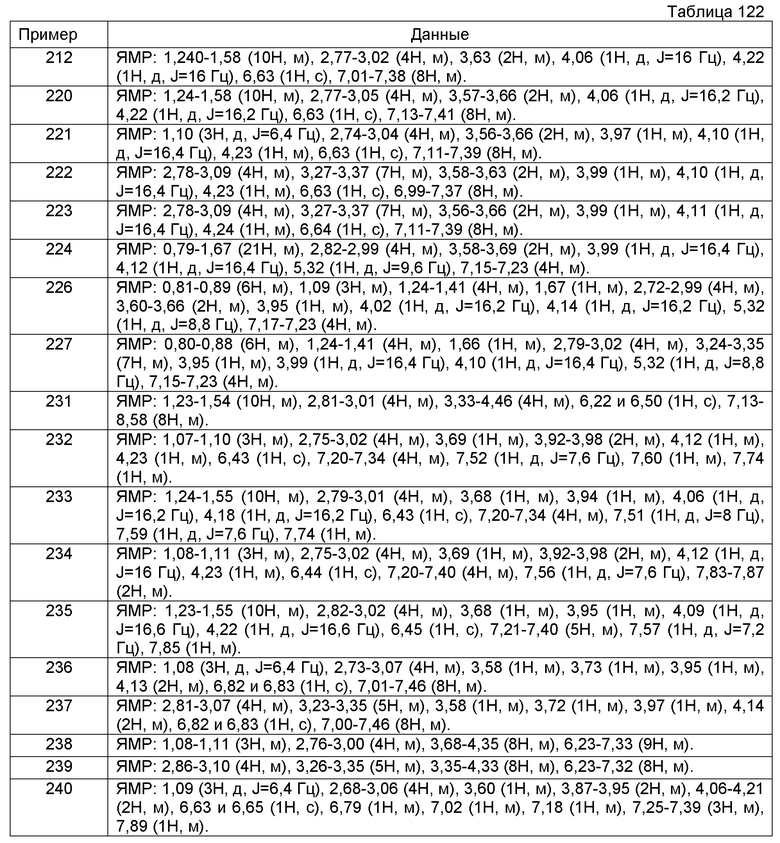
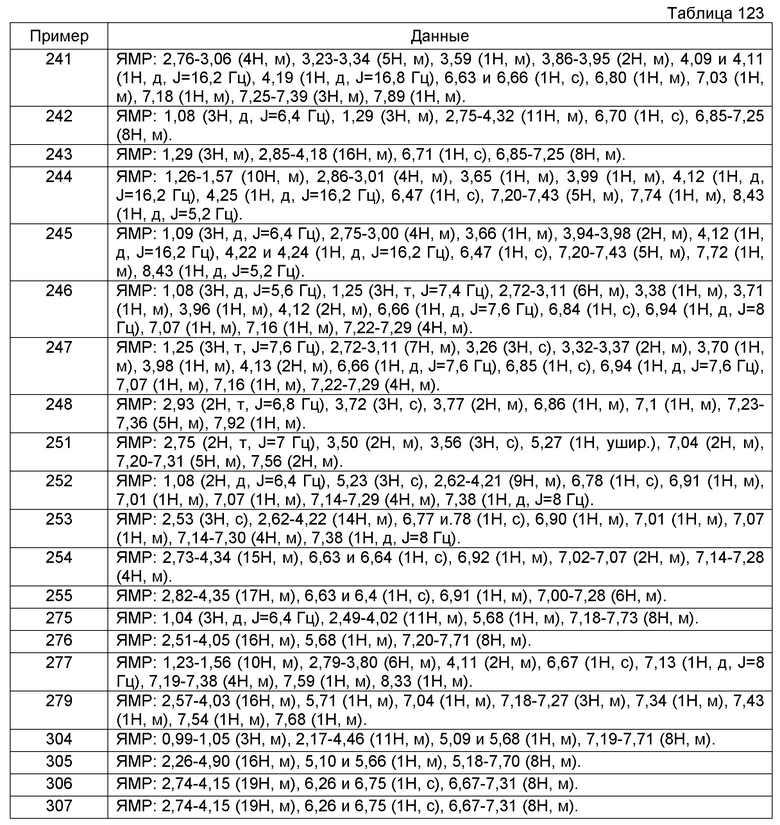

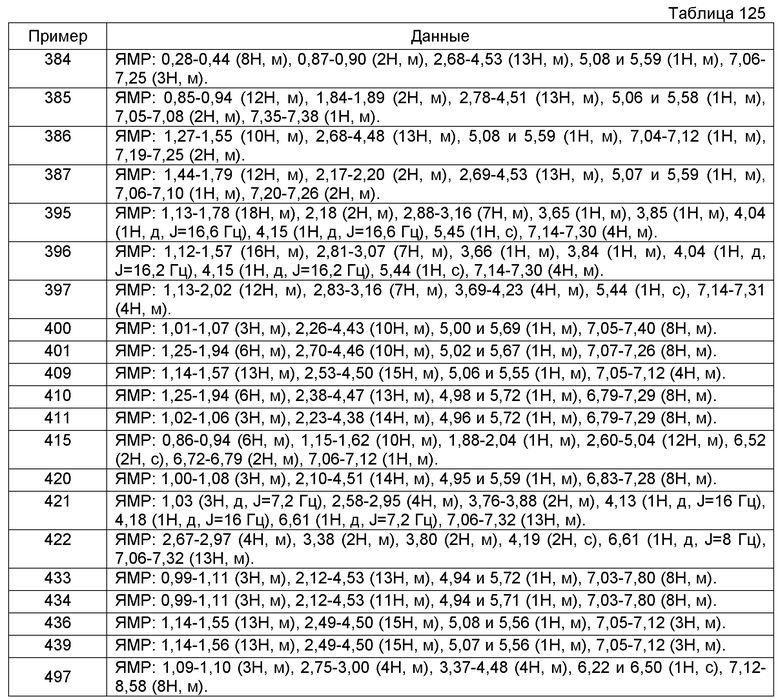
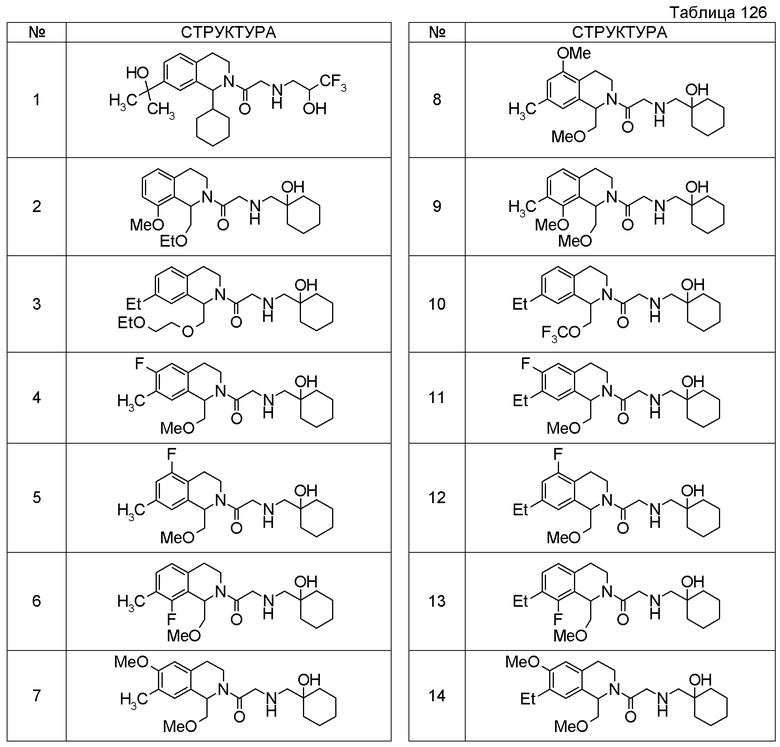

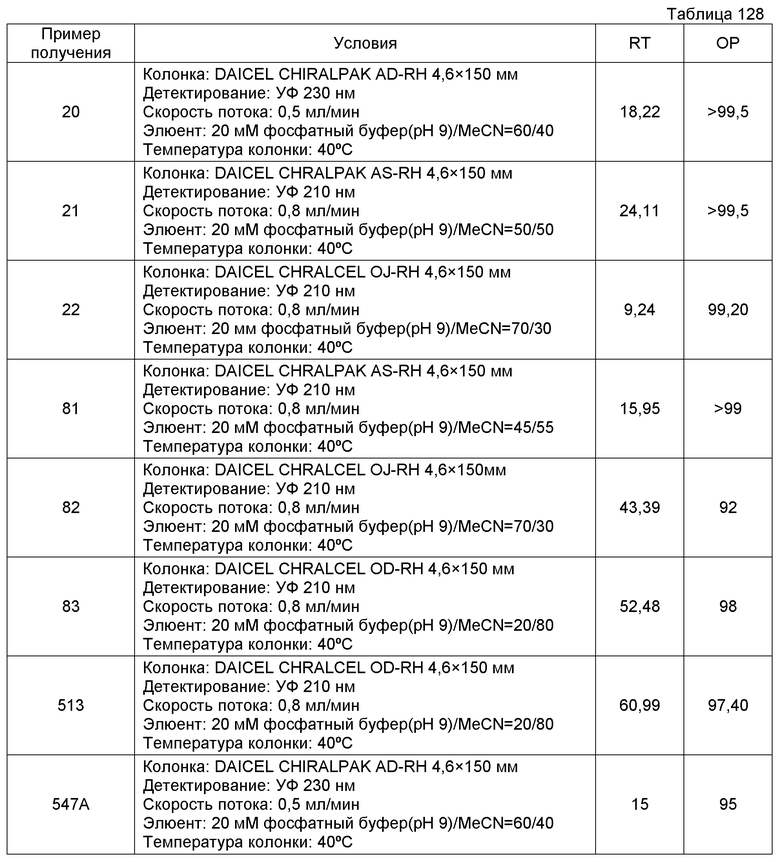
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИН-1-ОНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТА ВВ2 | 2008 |
|
RU2479578C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2378269C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2376301C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| НОВЫЕ ИЗОИНДОЛИНОВЫЕ ИЛИ ИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2689305C2 |
| СОЕДИНЕНИЕ СЛОЖНОГО ЭФИРА ГУАНИДИНОБЕНЗОЙНОЙ КИСЛОТЫ | 2014 |
|
RU2661895C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2838028C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ ТЕТРАЗОЛМЕТАНСУЛЬФОНОВОЙ КИСЛОТЫ И НОВОЕ СОЕДИНЕНИЕ, ИСПОЛЬЗУЕМОЕ В НЕМ | 2011 |
|
RU2509769C1 |
| МОДУЛЯТОРЫ РЕЦЕПТОРОВ МЕЛАНОКОРТИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ И КОСМЕТИЧЕСКИХ ПРЕПАРАТАХ ДЛЯ ЧЕЛОВЕКА | 2009 |
|
RU2589056C2 |
| НОВЫЕ ИНДОЛЬНЫЕ И ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693404C2 |
Настоящее изобретение относится к конкретным соединениям 1-замещенного производного 3,4-тетрагидроизохинолина. Также изобретение относится к фармацевтической композиции на основе заявленных соединений, к блокатору Са2+-канал N-типа на основе заявленных соединений, к применению заявленных соединений, а также к способу предупреждения или лечения некоторых патологических состояний. Технический результат: получены новые производные 3,4-тетрагидроизохинолина, имеющие заместитель в 1-положении и обладающие блокирующим действием на Са2+-каналы N-типа. 7 н. и 8 з. п. ф-лы, 129 табл., 17 пр.
1. Соединение, выбранное из группы, состоящей из:
1-[({2-[(1S)-1-циклогексил-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(2S)-1-({2-[(1S)-1-циклогексил-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)-3-метоксипропан-2-ол,
1-({[2-(1(1S)-изопропил-6-метокси-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}метил)циклогексанол,
(2R)-1-({2-[(1S)-3-метокси-1-фенил-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)пропан-2-ол,
1-[({2-[(1R)-7-этил-1-(метоксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(2S)-1-метокси-3-[(2-охо-2-{1(1S)-[2-(трифторметил)бензил]-3,4-дигидроизохинолин-2(1Н)-ил}этил)амино]пропан-2-ол,
1-({[3-(1-циклогексил-3,4-дигидроизохинолин-2(1Н)-ил)-3-оксопропил]амино}метил)циклогексанол,
(2R)-1-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}пропан-2-ол,
(2R)-1-[(2-оксо-2-{1-[2-(трифторметил)фенил]-3,4-дигидроизохинолин-2(1Н)-ил}этил)амино]пропан-2-ол,
(2S)-1-{[2-(1-циклогексил-7-метил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}-3-метоксипропан-2-ол,
(2R)-1-({2-оксо-2-[(1S)-1-фенил-3,4-дигидроизохинолин-2(1Н)-ил]этил}амино)пропан-2-ол,
1-[({2-[7-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[7-этил-1-(метоксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-({[2-(1-изопропил-6-метокси-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[5-метокси-1-(метоксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[1-(метоксиметил)-6-метил-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
(1S,2S)-2-{[2-(1-циклогексил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}-1-фенилпропан-1,3-диол,
1-({(2R)-2-[(1-циклогексил-3,4-дигидроизохинолин-2(1Н)-ил)карбонил]пирролидин-1-ил}метил)циклогексанол,
(2R)-1-{[2-(1-циклогексил-1-метил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}пропан-2-ол,
1-({[2-(3',4'-дигидро-2'Н-спиро[циклогексан-1,1'-изохинолин]-2'-ил)-2-оксоэтил]амино}метил)циклогексанол,
(2R)-1-[(2-оксо-2-{1-[2-(трифторметокси)фенил]-3,4-дигидроизохинолин-2(1Н)-ил}этил)амино]пропан-2-ол,
(2R)-1-{[2-(1-циклогексил-7-этил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}пропан-2-ол,
1-({[2-(6-фтор-1-изопропил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1,1-дициклопропил-2-({2-[6-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)этанол,
1-({[2-(1-трет-бутил-8-метокси-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-({[2-(1-изопропил-6-метил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-({[2-(6-фтор-1-пропил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[1-(метоксиметил)-7-метил-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-({[2-(5-фтор-1-пропил-3,4-дигидроизохинолин-2(1Н)-ил)-2-оксоэтил]амино}метил)циклогексанол,
1-[({2-[5-фтор-1-(метоксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[8-метокси-1-(метоксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол,
1-[({2-[1-(этоксиметил)-7-метил-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)метил]циклогексанол или
(1R,2S)-2-({2-[(1R)-1-(2-метоксифенил)-3,4-дигидроизохинолин-2(1Н)-ил]-2-оксоэтил}амино)циклопентанол, или его фармацевтически приемлемая соль.
2. Соединение по п.1, которое представляет собой

или его фармацевтически приемлемая соль.
3. Фармацевтическая композиция, обладающая блокирующей Са2+-каналы N-типа активностью, содержащая соединение по п.1 или 2 или его фармацевтически приемлемую соль в смеси с фармацевтически приемлемым носителем и эксципиентом.
4. Блокатор Са2+-канала N-типа, содержащий соединение по п.1 или 2 или его фармацевтически приемлемую соль.
5. Фармацевтическая композиция для предупреждения или лечения боли, невропатической боли, абдоминального симптома, спастической констипации, индуцированной опиоидом констипации, синдрома раздраженной толстой кишки или синдрома раздраженной толстой кишки типа констипации, содержащая соединение по п.1 или 2 или его фармацевтически приемлемую соль.
6. Фармацевтическая композиция по п.5, которая является фармацевтической композицией для предупреждения или лечения боли.
7. Фармацевтическая композиция по п.6, которая является фармацевтической композицией для предупреждения или лечения невропатической боли.
8. Фармацевтическая композиция по п.5, которая является фармацевтической композицией для предупреждения или лечения абдоминального симптома.
9. Фармацевтическая композиция по п.5, которая является фармацевтической композицией для предупреждения или лечения спастической констипации.
10. Фармацевтическая композиция по п.9 которая является фармацевтической композицией для предупреждения или лечения индуцированной опиоидом констипации.
11. Фармацевтическая композиция по п.5, которая является фармацевтической композицией для предупреждения или лечения синдрома раздраженной толстой кишки.
12. Фармацевтическая композиция по п.11, которая является фармацевтической композицией для предупреждения или лечения синдрома раздраженной толстой кишки типа констипации.
13. Применение соединения по п.1 или 2 или его фармацевтически приемлемой соли для изготовления фармацевтической композиции для предупреждения или лечения боли, невропатической боли, абдоминального симптома, спастической констипации, индуцированной опиоидом констипации, синдрома раздраженной толстой кишки или синдрома раздраженной толстой кишки типа констипации.
14. Применение соединение по п.1 или 2 или его фармацевтически приемлемой соли для предупреждения или лечения боли, невропатической боли, абдоминального симптома, спастической констипации, индуцированной опиоидом констипации, синдрома раздраженной толстой кишки или синдрома раздраженной толстой кишки типа констипации.
15. Способ предупреждения или лечения боли, невропатической боли, абдоминального симптома, спастической констипации, индуцированной опиоидом констипации, синдрома раздраженной толстой кишки или синдрома раздраженной толстой кишки типа констипации, включающий введение пациенту эффективного количества соединения по п.1 или 2 или его фармацевтически приемлемой соли.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ZHANG, Y | |||
| et al | |||
| Inhibitory effects of tetrahydroisoquinoline derivatives on Ca and Na channels in crude nerve, endings // Biological & Pharmaceutical Bulletin | |||
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| RU 2006101053 A, 10.06.2006. | |||

