Предшествующий уровень техники
Белок запрограммированной смерти клеток-1 (PD-1) является представителем надсемейства CD28, который обуславливает негативные сигналы при взаимодействии с двумя своими лигандами, PD-L1 или PD-L2. PD-1 и его лиганды широко экспрессированы и выполняют ряд иммунорегуляторных функций в активации и толерантности Т-клеток. PD-1 и его лиганды участвуют в ослаблении иммунитета к инфекциям и иммунитета к опухолям, и способствуют развитию хронических инфекций и развитию опухоли.
Модулирование PD-1 сигнального пути имеет терапевтический потенциал при различных заболеваниях человека (Hyun-Tak Jin et al., Curr Top Microbiol Immunol. (2011); 350:17-37). Блокада PD-1 сигнального пути стала привлекательной мишенью в терапии рака. Терапевтические антитела, блокирующие иммунную контрольную точку белка запрограммированной смерти клеток-1 (PD-1), предотвращают снижение выработки Т-клеток и промотируют иммунный ответ против рака. Несколько ингибиторов PD-1 пути показали сильную активность на разных фазах клинических испытаний (RD Harvey, Clinical Pharmacology и Therapeutics (2014); 96(2), 214-223).
Есть потребность в агентах, которые блокируют взаимодействие PD-L1 с PD-1 или CD80. Некоторые антитела были разработаны и выпущены на рынок. Было опубликовано несколько патентных заявок, раскрывающих непептидные низкомолекулярные молекулы (WO 2015/160641, WO 2015/034820, и WO 2017/066227 и WO2018/009505 от Bristol-Myers Squibb; WO 2015/033299 и WO 2015/033301 от Aurigene; WO 2017/070089, US 2017/0145025, WO 2017/106634, US2017/0174679, WO2017/192961, WO2017/222976, WO2017/205464, WO2017/112730, WO2017/041899 и WO2018/013789 от Incyte, WO2018/006795 от Maxinovel и WO2018/005374 от Заявителя, ChemoCentryx). Однако все еще есть потребность в альтернативных соединениях, таких как низкомолекулярные молекулы, в качестве ингибиторов PD-L1, которые бы имели улучшенные характеристики в разрезе перорального введения, стабильности, биодоступности, терапевтического индекса и токсичности.
Краткое описание изобретения
В одном аспекте, в настоящем изобретении описаны соединения, имеющие формулу (I):
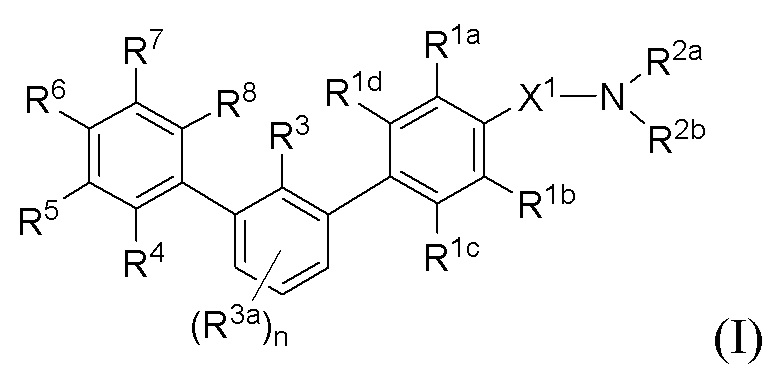
или их фармацевтически приемлемая соль или пролекарство или биоизостер; где R1a, R1b, R1c, R1d, R2a, R2b, R3, R3a, R4, R5, R6, R7, R8 и подстрочный индекс n имеют указанные в настоящем тексте значения.
Помимо описанных в настоящей заявке соединений, настоящее изобретение включает также фармацевтические композиции, содержащие одно или больше из этих соединений, а также способы, связанные с получением и применением этих соединений. В некоторых вариантах осуществления, описанные в настоящем изобретении соединения применяются в терапевтических способах лечения заболеваний, ассоциированных с PD-1/PD-L1 путем.
Подробное описание изобретения
Аббревиатуры и определения
При использовании в настоящем тексте, применение единственного числа охватывает не только аспекты с одним представителем, но также и аспекты с несколькими представителями. Например, формы единственного числа могут означать несколько представителей, если из контекста явно не следует иное. Так, например, слово «клетка» включает случаи с множеством клеток, а «агент» может означать один или несколько агентов, известных в данной области, и т.д.
Термины «около» и «примерно» в целом означают приемлемую степень ошибки для измеренного значения, принимая во внимание природу и точность проведенного измерения. В типичном случае примеры степени погрешности находятся в диапазоне 20 процентов (%), предпочтительно в диапазоне 10%, и более предпочтительно - в диапазоне 5% от указанного значения или диапазона значений. Альтернативно, и особенно в биологических системах, термины «около» и «примерно» могут означать значения, находящиеся в пределах одного порядка, предпочтительно в 5-кратных пределах, и более предпочтительно - в 2-кратных пределах от указанного значения. Приведенные в настоящем тексте числовые значения являются приблизительными, если не указано иное, и это значит, что термин «около» или «примерно» может подразумеваться в тех случаях, когда он не применен в явном виде.
Термин «алкил», сам по себе и как часть другого заместителя, означает, если не указано иное, линейную или разветвленную углеводородную группу, имеющую обозначенное число атомов углерода (например, C1-8 означает 1-8 атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п. Термин «алкенил» означает ненасыщенную алкильную группу, содержащую одну или больше двойных связей. Аналогично, термин «алкинил» означает ненасыщенную алкильную группу, содержащую одну или больше тройных связей. Примеры алкенильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил). Примеры алкинильных групп включают этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры.
Термин «циклоалкил» относится к углеводородному кольцу или кольцам, содержащим указанное число атомов в кольце (например, C3-6 циклоалкил) и являющимся полностью насыщенными или содержащим не более одной двойной связи между вершинами цикла. Термин «циклоалкил» относится также к бициклическим и полициклическим углеводородным кольцам, таким как, например, бицикло[2.2.1]гептан, бицикло[2.2.2]октан и т.д. Бициклические или полициклические кольца могут быть конденсированными, мостиковыми, спиро- или представлять собой их комбинацию.
Термин «гетероциклоалкил» или «гетероциклил» относится к циклоалкильной группе, содержащей 1-5 гетероатомов, выбранных из N, O, и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероциклоалкил может представлять собой моноциклическую, бициклическую или полициклическую кольцевую систему. Бициклические или полициклические кольца могут быть конденсированными, мостиковыми, спиро- или представлять собой их комбинацию. Термин «C4-12 гетероциклил», например, означает группу, содержащую от 4 до 12 членов в цикле, где по меньшей один из членов цикла представляет собой гетероатом. Неограничивающие примеры гетероциклоалкильных групп включают пирролидин, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидинон, тетразолон, гидантоин, диоксолан, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и т.п. Гетероциклоалкильная группа может быть присоединена к остальной части молекулы через атом углерода в цикле или гетероатом в цикле.
Термин «алкилен» в отдельности или как часть другого заместителя означает двухвалентную группу, являющуюся производным алкана, в качестве примера можно привести -CH2CH2CH2CH2-. Алкиленовая группа может быть линейной или разветвленной. Примерами последних являются -CH2C(CH3)2CH2-, -CH2C(CH3)2- или -CH(CH3)CH2CH2-.В типичном случае алкильная (или алкиленовая) группа содержит от 1 до 12 атомов углерода, предпочтительными по настоящему изобретению являются группы, содержащие 8 или меньше атомов углерода. Аналогично, «алкенилен» и «алкинилен» означает ненасыщенные формы «алкилена», содержащие двойные или тройные связи, соответственно.
Термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкокси) применяются в их обычном смысле и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Кроме того, для диалкиламино-групп, алкильные фрагменты могут быть одинаковыми или разными, а также могут объединяться с формированием 3-7-членного цикла с атомом азота, к которому они присоединены. Соответственно, группа, изображаемая как -NRaRb, включает пиперидинил, пирролидинил, морфолинил, азетидинил и т.п.
Термин «галоген» сам по себе или как часть другого заместителя означает, если не указано иное, атом фтора, хлора, брома или иода. Кроме того, такие термины как «галогеналкил» включают моногалогеналкил и полигалогеналкил. Например, термин «C1-4 галогеналкил» включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термин «гидроксиалкил» или «алкил-OH» означает алкильную группу, определение которой приведено выше, в которой по меньшей мере один (и до трех) атомов водорода заменены на гидрокси-группу. Что касается алкильной группы, гидроксиалкильные группы могут содержать любое подходящее число атомов углерода, например, C1-6. Примеры гидроксиалкильных групп включают (но не ограничиваются только ими) гидроксиметил, гидроксиэтил (где гидрокси-группа находится в положении 1 или 2), гидроксипропил (где гидрокси-группа находится в положении 1, 2 или 3) и 2,3-дигидроксипропил.
Термин «арил» означает, если не указано иное, полиненасыщенную, в типичном случае ароматическую, углеводородную группу, которая может представлять собой один цикл или несколько циклов (до трех циклов), сопряженные или связанные ковалентно.
Термин «гетероарил» означает 5-10-членное ароматическое кольцо (или кольца), содержащие от одного до пяти гетероатомов, выбранных из N, O и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероарильная группа может быть присоединена к остальной части молекулы через гетероатом. Под «C5-10 гетероарилом» понимается гетероарильный фрагмент, содержащий 5-10 членов в кольце, где по меньшей мере один из членов кольца представляет собой гетероатом. Неограничивающие примеры арильных групп включают фенил, нафтил и бифенил, а неограничивающие примеры гетероарильных групп включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолил, фталазинил, бензотриазинил, пуринил, бензоимидазолил, бензопиразолил, бензотриазолил, бензизоксазалил, изобензофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, имидазопиридины, бензотиаксолил, бензофуранил, бензотиенил, индолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фурил, тиенил и т.п. Заместители в каждой из перечисленных выше арильных или гетероарильных циклических системах выбраны из группы приемлемых заместителей, описанных ниже.
Когда любой из упомянутых выше терминов (например, «алкил», «арил» и «гетероарил») имеет приставку «замещенный» без дополнительного описания заместителей, то замещенные формы указанных групп будут такими как описано ниже.
Заместителями для алкильных групп (включая группы, часто именуемые алкилен, алкенил, алкинил и циклоалкил) могут быть различные группы, выбранные из следующих: -галоген, -OR’, -NR’R”, -SR’, -SiR’R”R”’, -OC(O)R’, -C(O)R’, -CO2R’, -CONR’R”, -OC(O)NR’R”, -NR”C(O)R’, -NR’-C(O)NR”R”’, -NR”C(O)2R’, -NH-C(NH2)=NH,
-NR’C(NH2)=NH, -NH-C(NH2)=NR’, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, -NR’S(O)2R”, -CN и
-NO2, в количестве от нуля до (2m’+1), где m’ - это общее число атомов углерода в такой группе. R’, R” и R”’ каждый независимо представляют собой атом водорода, незамещенный C1-8 алкил, незамещенный гетероалкил, незамещенный арил, арил, замещенный 1-3 галогенами, незамещенный C1-8 алкил, C1-8 алкокси или C1-8 тиоалкокси группы, или незамещенные арил-C1-4 алкильные группы. Когда R’ и R” присоединены к одном атому азота, они могут объединяться с этим атомом азота, образуя 3-, 4-, 5-, 6- или 7-членное кольцо. Например, -NR’R” включает 1-пирролидинил и 4-морфолинил. Термин «ацил» при использовании в отдельности или как часть другой группы означает алкильную группу, в которой два заместителя у атома углерода, ближайшего к точке присоединения данной группы, заменены на заместитель =O (например, -C(O)CH3, -C(O)CH2CH2OR’ и т.п.).
Аналогично, заместители у арильных и гетероарильных групп варьируются и в целом выбраны из следующих: -галоген, -OR’, -OC(O)R’, -NR’R”, -SR’, -R’, -CN, -NO2,
-CO2R’, -CONR’R”, -C(O)R’, -OC(O)NR’R”, -NR”C(O)R’, -NR”C(O)2R’, -NR’-C(O)NR”R”’, -NH-C(NH2)=NH, -NR’C(NH2)=NH, -NH-C(NH2)=NR’, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, -NR’S(O)2R”, -N3, перфтор(C1-C4)алкокси и перфтор(C1-C4)алкил, в количестве от нуля до общего числа свободных валентностей в ароматической системе; и где R’, R” и R”’ независимо выбраны из атома водорода, C1-8 алкила, C3-6 циклоалкила, C2-8 алкенила, C2-8 алкинила, незамещенного арила и гетероарила, (незамещенный арил)-C1-4 алкила и незамещенного арилокси-C1-4 алкила. Другие подходящие заместители включают все описанные выше арильные заместители, присоединенные к атому кольца через алкиленовый мостик, содержащий 1-4 атомов углерода.
Два заместителя у соседних атомов в арильном или гетероарильном кольце могут быть опционально заменены на заместитель формулы -T-C(O)-(CH2)q-U-, где T и U независимо представляют собой -NH-, -O-, -CH2- или простую связь, и q представляет собой целое число от 0 до 2. Альтернативно, два из заместителей у соседних атомов в арильном или гетероарильном кольце могут быть опционально заменены на заместитель формулы -A-(CH2)r-B-, где A и B независимо представляют собой -CH2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR’- или простую связь, и r представляет собой целое число от 1 до 3. Одна из простых связей в новом образовавшемся таким образом кольце опционально может быть заменена двойной связью. Альтернативно, два из заместителей у соседних атомов в арильном или гетероарильном кольце могут быть опционально заменены на заместитель формулы -(CH2)s-X-(CH2)t-, где s и t независимо представляют собой целые числа от 0 до 3, и X представляет собой -O-, -NR’-,-S-, -S(O)-, -S(O)2- или -S(O)2NR’-. Заместитель R’ в -NR’- и -S(O)2NR’- выбран из атома водорода или незамещенного C1-6 алкила.
При использовании в настоящем тексте, термин «гетероатом» включает кислород (O), азот (N), серу (S) и кремний (Si). В составе циклических систем, гетероатомы в качестве вершин цикла включают N, O и S.
Настоящее описание касается также пролекарств и биоизостеров. Подходящие биоизостеры, например, включают замены карбоксилатов (фосфоновые кислоты, фосфиновые кислоты, сульфокислоты, сульфиновые кислоты и кислые гетероциклические группы, такие как тетразолы). Подходящие пролекарства включают распространенные группы, которые известны как гидролизующиеся и/или окисляющиеся в физиологических условиях с получением соединения формулы I.
Термины «пациент» и «субъект» включают приматов (в особенности человека), домашних животных-компаньонов (таких как собаки, кошки, лошади и т.п.) и сельскохозяйственных животных (таких как крупный рогатый скот, свиньи, овцы и т.п.).
При использовании в настоящем тексте, термин «лечить» или «лечение» охватывает как лечение, модифицирующее заболевание, так и симптоматическое лечение, любое из которых может быть профилактическим (т.е. до появления симптомов, для предотвращения, задержки или уменьшения степени тяжести симптомов) или терапевтическим (т.е. после появления симптомов, для уменьшения степени тяжести и/или длительности симптомов).
Термин «фармацевтически приемлемые соли» включает соли веществ, полученные с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей в описанных в настоящем тексте соединениях. Когда соединения по настоящему изобретению содержат относительно кислые функциональные группы, можно получить основно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количество желаемого основания, даже без растворителя или в подходящем инертном растворителе. Примеры солей, являющихся производными фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа(II), железа (III), лития, магния, марганца, калия, натрия, цинка и т.д. Соли, являющиеся производными фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.д., такие как аргинин, бетаин, кофеин, холин, N,N’-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, можно получить кислотно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количеством желаемой кислоты, без растворителя или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли с неорганическими кислотами, такими как хлористоводородная, бромистоводородная, азотная, угольная, моногидроугольная, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, иодистоводородная или фосфористая кислота и т.п., а также соли с относительно нетоксичными органическими кислотами, такими как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толуолсульфоновая, лимонная, винная, метансульфоновая и т.п. Также охватываются соли с аминокислотами, такие как аргинаты и т.п., и соли таких органических кислот, как глюкуроновая или галактуроновая кислоты и т.п. (см, например, Berge, S.M., et al, «Pharmaceutical Salts», Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые частные соединения по настоящему изобретению содержат и основные, и кислотные функциональные группы, что позволяет таким соединениям образовывать как основно-аддитивные, так и кислотно-аддитивные соли.
Нейтральные формы соединений можно регенерировать путем взаимодействия соли с основанием или кислотой и выделения материнского соединения обычным способом. Материнская форма соединения отличается от различных солевых форм определенными физическими характеристиками, такими как растворимость в полярных растворителях, но во всем остальном соли эквивалентны материнским соединениям, в терминах настоящего изобретения.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. В целом, сольватированные формы эквивалентны несольватированным формам, и все они охватываются настоящим изобретением. Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических или аморфных формах. В целом, все физические формы эквивалентны для областей применения, охватываемых настоящим изобретением, и входят в объем настоящего изобретения.
Некоторые соединения по настоящему изобретению имеют асимметрические атомы углерода (оптические центры) или двойные связи; все рацематы, диастереомеры, геометрические изомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры) входят в объем настоящего изобретения. Когда показаны стереохимические обозначения, это относится к соединению, которое представляет собой один из изомеров и практически не содержит другого изомера. «Практически не содержит» другого изомера означает соотношение двух изомеров по меньшей мере 80/20, более предпочтительно 90/10 или 95/5 или больше. В некоторых вариантах осуществления, один из изомеров присутствует в количестве по меньшей мере 99%.
Соединения по настоящему изобретению могут также иметь неприродные соотношения изотопов по одному или больше атомов, составляющих эти соединения. Например, соединения могут быть радиоактивно мечены радиоактивными изотопами, такими как, например, тритий (3H), иод-125 (125I) или углерод-14 (14C). Все изотопные вариации соединений по настоящему изобретению, радиоактивные и нерадиоактивные, входят в объем настоящего изобретения. Например, соединения могут быть получены так, что какое-то число атомов углерода заменено на изотоп дейтерия (2H). Соединения по настоящему изобретению могут также иметь неприродные соотношения изотопов по одному или больше атомов, составляющих эти соединения. Неприродное содержание изотопа может находиться в диапазоне от природного содержания до 100%. Например, соединения могут содержать радиоактивные изотопы, такие как, например, тритий (3H), иод-125 (125I) или углерод-14 (14C), или нерадиоактивные изотопы, такие как дейтерий (2H) или углерод-13 (13C). Такие изотопные вариации могут придавать дополнительное применение, вдобавок к уже описанному в настоящем тексте. Например, изотопные вариации соединений по настоящему изобретению могут найти дополнительное применение, включая (но не ограничиваясь только ими) применение в качестве диагностических и/или визуализирующих реагентов, или в качестве цитостатических/радиотоксических терапевтических агентов. В дополнение, изотопные вариации соединений по настоящему изобретению могут иметь измененные фармакокинетические и фармакодинамические характеристики, которые могут вносить вклад в повышение безопасности, переносимости или эффективности при лечении ими. Все изотопные вариации соединений по настоящему изобретению, как радиоактивные, так и нерадиоактивные, входят в объем настоящего изобретения.
Соединения
В одном аспекте, в настоящем изобретении описаны соединения, имеющие формулу (I):
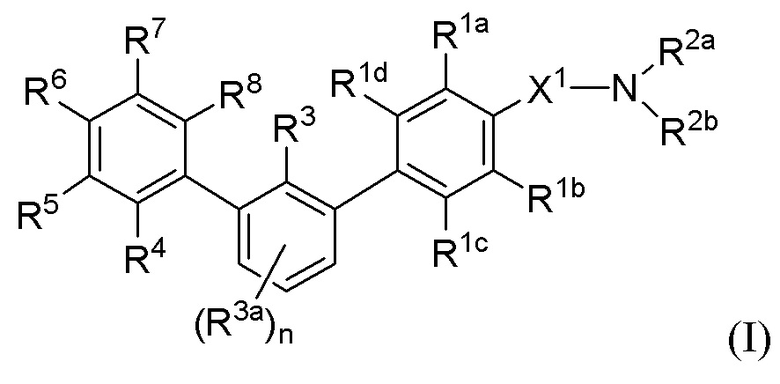
или их фармацевтически приемлемая соль, пролекарство или биоизостер, где:
R1a, R1b, R1c и R1d каждый независимо выбраны из группы, состоящей из H, F, Cl, C1-3 алкила, C1-3 галогеналкила, C1-3 алкокси-группы и CN;
X1 представляет собой C1-3 алкилен, необязательно замещенный одним или двумя C1-2 алкилами или CO2H;
R2a и R2b каждый независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, -Y, -X2-C(O)2Ra, -X2-ORa, -X2NRaRb, -X2-CONRaRb, -X2-SO2Ra,
-X2-SO2NRaRb, -X2-SO3Ra и -X2-Y, где каждый X2 представляет собой C1-6 алкилен, и любой C1-8 алкил или C1-6 алкилен необязательно дополнительно замещен одним или двумя заместителями, независимо выбранными из OH, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8 алкила или CO2H, и каждый Y выбран из группы, состоящей из C3-6 циклоалкила, C4-8 гетероциклила и 5-6-членного гетероарила, каждый из которых необязательно дополнительно замещен 1-4 заместителями, независимо выбранными из группы, состоящей из оксо-группы, OH, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси-группы, C1-4 галогеналкокси-группы, C1-4 гидроксиалкокси-группы, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8 алкила и CO2H;
или R2a и R2b объединены с образованием 4-8-членного кольца или спироциклического кольца, необязательно имеющего один или два дополнительных члена цикла, выбранных из O, N или S;
или R1a и R2a или X1 объединены с формированием 5-7-членного кольца;
или R1b и R2b или X1 объединены с формированием 5-7-членного кольца;
где кольца, образующиеся при объединении R2a и R2b, R1a и R2a или X1, или R1b и R2b или X1, содержат 0-4 заместителей, независимо выбранных из группы, состоящей из оксо-группы, C1-8 алкила, C1-8 галогеналкила, -X3-C(O)2Ra, -X3-ORa, -X3-NRaRb, -X3-CONRaRb, -X3-SO2Ra, -X3-SO2NRaRb и -X3-SO3Ra; где X3 представляет собой связь или C1-6 алкилен;
R3 выбран из группы, состоящей из H, F, Cl, C1-3 алкила, C1-3 алкокси-группы, C1-3 галогеналкила, C1-3 галогеналкокси-группы, C2-3 алкенила и CN;
подстрочный индекс n равен 0, 1 или 2;
каждый R3a независимо выбран из группы, состоящей из F, Cl, C1-3 алкила, C1-3 алкокси-группы, C1-3 галогеналкила, C1-3 галогеналкокси-группы, C2-3 алкенила и CN;
каждый из R4, R6, R7 и R8 независимо выбран из группы, состоящей из H, галогена, CN, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 галогеналкила, C1-6 гидроксиалкила, -Y1,
-X4-C(O)2Ra, -X4-ORa, -X4-NRaRb, -X4-CONRaRb, -X4-SO2Ra, -X4-SO2NRaRb, -X4-SO3Ra,
-O-X4-Y1 и -X4-Y1, где каждый X4 представляет собой связь или C1-6 алкилен и необязательно дополнительно замещен OH, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8алкилом или CO2H, и каждый Y1 выбран из группы, состоящей из C3-6 циклоалкила, C4-8 гетероциклила и 5-6-членного гетероарила, каждый из которых необязательно дополнительно замещен 1-4 заместителями, независимо выбранными из группы, состоящей из оксо-группы, OH, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси-группы, C1-4 галогеналкокси-группы, C1-4 гидроксиалкокси-группы, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8алкила и CO2H;
R5 является представителем, выбранным из группы, состоящей из H, галогена, CN, C1-6 алкила, C1-6 галогеналкила, C1-6 гидроксиалкила, -Y2, -X5-C(O)2Ra, -X5-ORa, -X5-NRaRb, -X5-CONRaRb, -X5-SO2Ra,-X5-SO2NRaRb, -X5-SO3Ra, -X5-Y2, -O-X5-Y2 и -A-Z, где каждый X5 представляет собой связь или C1-6 алкилен и необязательно дополнительно замещен OH, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8алкилом или CO2H, и каждый Y2 выбран из группы, состоящей из C3-6 циклоалкила, C4-8 гетероциклила, C7-9 спирогетероциклила и 5-6-членного гетероарила, каждый из которых необязательно дополнительно замещен 1-4 заместителями, независимо выбранными из группы, состоящей из оксо-группы, галогена, OH, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси-группы, C1-4 галогеналкокси-группы, C1-4 гидроксиалкокси-группы, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8алкила и CO2H;
где
A является представителем, выбранным из группы, состоящей из связи, -O- и -N(Ra)-;
Z выбран из группы, состоящей из следующих:
i) моноциклическое 5- или 6-членное гетероарильное кольцо, необязательно замещенное 1-3 заместителями Rc;
ii) фенил, необязательно замещенный 1-3 заместителями Rc; и
iii) 5- или 6-членное неароматическое гетероциклическое кольцо, необязательно замещенное 1-3 заместителями Rc;
и когда A представляет собой -O- или -N(Ra)-, тогда Z отличается от фенила;
и два из R4, R5, R6, R7 и R8 у соседних атомов углерода необязательно объединены с образованием 5- или 6-членного неароматического гетероциклического кольца, имеющего одну или две вершины цикла, выбранные из группы, состоящей из O, -N(Rb)- и =N-; где указанное неароматическое гетероциклическое кольцо необязательно замещено оксо-группой и необязательно 1-4 заместителями Rc;
и по меньшей мере один из R4, R5, R6, R7 и R8 отличается от H;
каждый Ra независимо выбран из группы, состоящей из H, C1-6 алкила, C3-6 циклоалкила, C1-6 галогеналкила, C1-6 гидроксиалкила, C1-6 алкилен-CO2H, C1-6 алкилен-COO-C1-8алкила, C1-6 алкилен-SO3H;
каждый Rb независимо выбран из группы, состоящей из H, C1-6 алкила, C3-6 циклоалкила, C1-6 галогеналкила, C1-6 гидроксиалкила, C1-6 алкилен-CO2H, C1-6 алкилен-SO3H и C1-6 алкилен-Y3, где Y3 представляет собой C3-6 циклоалкил или C4-8 гетероциклил, и каждый Rb необязательно дополнительно замещен одним или двумя заместителями, независимо выбранными из оксо-группы, OH, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8алкила и CO2H;
и Ra и Rb, когда они присоединены к одному атому азота, необязательно объединены с образованием 4-8-членного кольца или спироциклического кольца, необязательно замещенного галогеном, OH, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8алкилом или CO2H;
каждый Rc независимо выбран из группы, состоящей из H, галогена, CN, C1-6 алкила, C1-6 галогеналкила, -Y4, -X6-C(O)2Ra, -X6-ORa, -X6-NRaRb, -X6-CONRaRb, -X6-SO2Ra, -X6-SO2NRaRb, -X6-SO3Ra и -N(Ra)-X6-C(O)2Ra, где каждый X6 представляет собой связь или C1-6 алкилен, и каждый Y4 выбран из группы, состоящей из C3-6 циклоалкила и C4-8 гетероциклила; и необязательно два Rc у соседних вершин цикла объединены с формированием конденсированного 5- или 6-членного гетероциклического кольца.
В некоторых вариантах осуществления, в настоящем изобретении описаны соединения, имеющие формулу (Ia):
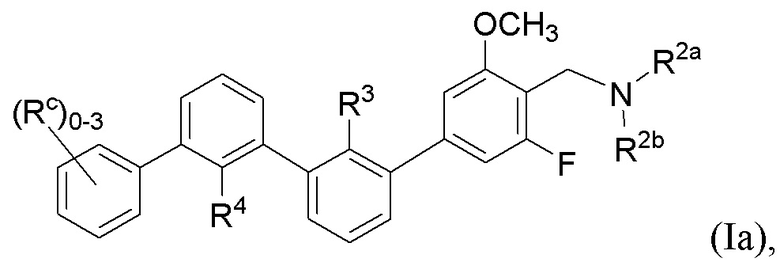
где R2a, R2b, R3, R4 и Rc имеют значения, указанные для формулы (I).
В некоторых вариантах осуществления, в настоящем изобретении описаны соединения, имеющие формулу (Ib):
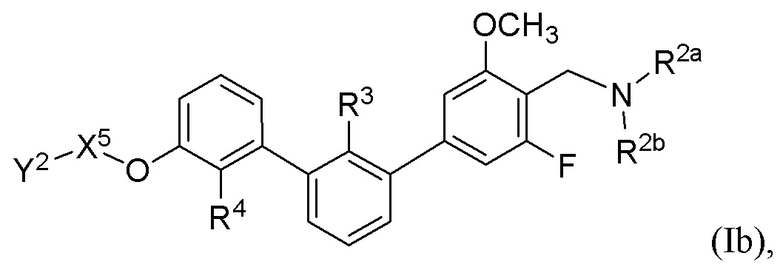
где R2a, R2b, R3, R4, X5 и Y2 имеют значения, указанные для формулы (I). В некоторых частных вариантах осуществления, описаны соединения, имеющие формулу (Ib), где Y2 выбран из группы, состоящей из следующих:
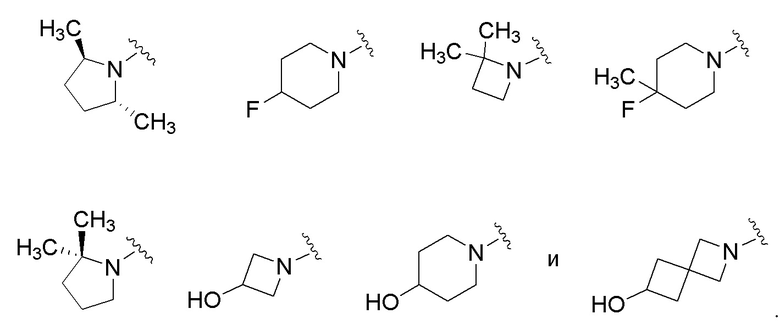 .
.
В некоторых вариантах осуществления, в настоящем изобретении описаны соединения, имеющие формулу (Ic):

где R2a, R2b, R3, R4, Ra, Rb и X5 имеют значения, указанные для формулы (I).
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R5 представляет собой -A-Z.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R5 представляет собой -O-X5-Y2.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R5 представляет собой -A-Z, A представляет собой связь, и Z представляет собой фенил, необязательно замещенный 1-3 заместителями Rc.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых X1 представляет собой -CH2-.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R1c, R7 и R8 представляют собой H, и R3 выбран из группы, состоящей из F, Cl, CH3, CF3 и OCH3.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых кольцо образуется между одной из пар R4 и R5, R5 и R6, R1b и R2b, или R1a и R2a.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R5 представляет собой -A-Z, и Z выбран из группы, состоящей из пиперидинила, имидазолила и пиридинила.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых n равен 0.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R4 выбран из группы, состоящей из F, Cl, CH3, CF3 и OCH3.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R1a представляет собой OCH3, и R1b представляет собой F.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R2a и R2b каждый представляют собой H.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R2a и R2b объединены с формированием 4-8-членного кольца или спироциклического кольца, необязательно имеющего одну или две вершины цикла, выбранные из O, N или S; где указанное кольцо или спироциклическое кольцо замещено 0-4 заместителями, независимо выбранными из группы, состоящей из оксо-группы, C1-8 алкила, C1-8 галогеналкила, C1-8 гидроксиалкила, -X2-C(O)2Ra, -X2-ORa, -X2-NRaRb, -X2-CONRaRb, -X2-SO2Ra, -X2-SO2NRaRb и -X2-SO3Ra; где X2 представляет собой связь или C1-6 алкилен.
В некоторых частных вариантах осуществления, соединения, имеющие формулу (I), представляют собой соединения, в которых R2a представляет собой H или C1-8 алкил; и R2b представляет собой -Y или -X1-Y. В других частных вариантах осуществления, Y выбран из группы, состоящей из C3-6 циклоалкила и C4-8 гетероциклила, каждый из которых необязательно дополнительно замещен 1-4 заместителями, независимо выбранными из группы, состоящей из оксо-группы, OH, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси-группы, C1-4 галогеналкокси-группы, C1-4 гидроксиалкокси-группы, SO2NH2, CONH2, C(O)NHOH, PO3H2, COO-C1-8алкила и CO2H.
В некоторых вариантах осуществления, для каждой из формул (I), (Ia), (Ib) и (Ic), и описанных выше частных вариантов осуществления, соединения или их фармацевтически приемлемая соль представляют собой выбранные из таблицы 1, имеющие активность ++ или +++.
В дополнение к описанным выше соединениям, настоящее изобретение охватывает также фармацевтически приемлемые соли этих соединений. В некоторых вариантах осуществления, фармацевтически приемлемые соли выбраны из солей аммония, кальция, магния, калия, натрия, цинка, аргинина, бетаина, кофеина, холина, N,N’-дибензилэтилендиамина, диэтиламина, 2-диэтиламиноэтанола, 2-диметиламиноэтанола, этаноламина, этилендиамина, N-этилморфолина, N-этилпиперидина, глюкамина, глюкозамина, гистидина, гидрабамина, изопропиламина, лизина, метилглюкамина, морфолина, пиперазина, пиперидина, прокаина, пуринов, теобромина, триэтиламина, триметиламина, трипропиламина, трометамина, солей с соляной, угольной кислотами, из моногидрокарбонатов, солей с фосфорной кислотой, моногидрофосфатов, дигидрофосфатов, солей с уксусной, пропионовой, изомасляной, малоновой, бензойной, янтарной, субериновой, фумаровой, миндальной, фталевой кислотой, бензолсульфокислотой, п-толуолсульфокислотой, лимонной кислотой, винной кислотой, метансульфокислотой, аргинатов, солей с глюкуроновой кислотой и галактуроновой кислотой. В некоторых вариантах осуществления, фармацевтически приемлемые соли выбраны из солей аммония, кальция, магния, калия, натрия, солей с соляной, угольной кислотами, из моногидрокарбонатов, солей с фосфорной кислотой, моногидрофосфатов, дигидрофосфатов, солей с уксусной, пропионовой, изомасляной, малоновой, бензойной, янтарной, субериновой, фумаровой, миндальной, фталевой кислотой, бензолсульфокислотой, п-толуолсульфокислотой, лимонной кислотой, винной кислотой, метансульфокислотой, аргинатов, солей с глюкуроновой кислотой и галактуроновой кислотой. В некоторых вариантах осуществления, фармацевтически приемлемые соли представляют собой соли натрия или гидрохлориды.
Помимо солевых форм, в настоящем изобретении описаны соединения в форме пролекарства. Пролекарствами описанных в настоящем изобретении соединений являются соединения, которые легко претерпевают химические изменения в физиологических условиях, давая соединения по настоящему изобретению. Кроме того, пролекарства могут быть превращены в соединения по настоящему изобретению химическими или биологическими методами в условиях ex vivo. Например, пролекарства могут медленно превращаться в соединения по настоящему изобретению при помещении в резервуар чрескожного пластыря с подходящим ферментом или химическим реагентом.
Сложный эфир может применяться в качестве пролекарства для соответствующей карбоновой кислоты. Можно применять C1-10 алкиловый эфир или C1-10 галогеналкиловый эфир в качестве пролекарства для соответствующей карбоновой кислоты. Могут применяться следующие сложные эфиры: трет-бутиловый эфир, метиловый эфир, этиловый эфир, изопропиловый эфир.
Фармацевтические композиции
Помимо соединений по настоящему изобретению, композиции этих соединений в типичном случае содержат фармацевтически приемлемый носитель или разбавитель.
Термин «композиция» при использовании в настоящем тексте охватывает продукт, содержащий указанные ингредиенты и указанных количествах, а также любой продукт, который образуется, напрямую или косвенно, из комбинации указанных ингредиентов в указанных количествах. Термин «фармацевтически приемлемый» означает, что носитель, разбавитель или вспомогательное вещество должны быть совместимыми с другими ингредиентами препарата и не вредными для самого пациента.
В другом варианте осуществления описана фармацевтическая композиция, содержащая соединение по настоящему изобретению, включая соединение формул (I), (Ia), (Ib), (Ic) или (Id), или его фармацевтически приемлемую соль, и фармацевтически приемлемое вспомогательное вещество.
В некоторых вариантах осуществления, фармацевтическая композиция дополнительно содержит один или больше дополнительных терапевтических агентов. В некоторых вариантах осуществления, указанные один или больше дополнительных терапевтических агентов выбраны из группы, состоящей из противомикробного агента, противовирусного агента, цитотоксического агента, агента, модулирующего экспрессию гена, химиотерапевтического агента, противоракового агента, антиангиогенного агента, иммунотерапевтического агента, противогормонального агента, противофиброзного агента, лучевой терапии, агента для лучевой терапии, противоопухолевого агента и антипролиферативного агента. В некоторых вариантах осуществления, один или больше дополнительных терапевтических агентов представляют собой антагонист хемокинового и/или хемотаксического рецептора, который включает (но не ограничивается только ими) CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10, CCR11, CCR12, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7, C3aR и/или C5aR. Антагонисты хемокинового и/или хемотаксического рецептора известны в данной области и описаны, например, в WO2007/002667, WO2007/002293, WO/2003/105853, WO/2007/022257, WO/2007/059108, WO/2007/044804, WO2007/115232, WO2007/115231, WO2008/147815, WO2010/030815, WO2010/075257, WO2011/163640, WO2010/054006, WO2010/051561, WO2011/035332, WO2013/082490, WO2013/082429, WO2014/085490, WO2014/100735, WO2014/089495, WO2015/084842, WO2016/187393, WO2017/127409, WO 2017/087607, WO2017/087610, WO2017/176620, WO2018/222598, WO2018/222601, WO2013/130811, WO2006/076644, WO2008/008431, WO2009/038847, WO2008/008375, WO2008/008374, WO2008/010934, WO2009/009740, WO2005/112925, WO2005/112916, WO2005/113513, WO2004/085384, WO2004/046092. Антагонисты хемокинового и/или хемотаксического рецептора включают также CCX354, CCX9588, CCX140, CCX872, CCX598, CCX6239, CCX9664, CCX2553, CCX3587, CCX3624, CCX 2991, CCX282, CCX025, CCX507, CCX430, CCX765, CCX224, CCX662, CCX650, CCX832, CCX168, CCX168-M1, CCX3022 и/или CCX3384.
Фармацевтические композиции для введения соединений по настоящему изобретению можно выпускать в виде дозированных лекарственных форм, и их можно готовить любым из способов, известных в фармакологии и введении лекарственных препаратов. Все способы включают стадию объединения действующего вещества с носителем, который содержит один или несколько вспомогательных ингредиентов. В целом, фармацевтические композиции получают путем однородного и тщательного смешивания действующего вещества с жидким носителем или тонко измельченным твердым носителем, или с обоими, и затем, при необходимости, придание продукту формы желаемого препарата. В фармацевтической композиции действующее вещество присутствует в количестве, достаточном для оказания целевого эффекта на болезнь или на патологическое состояние.
Фармацевтические композиции, содержащие действующее вещество, могут иметь форму, подходящую для перорального применения, например, они могут быть в виде таблеток, саше, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий и самоэмульгирующихся препаратов, как описано в Заявке на Патент США 2002-0012680, твердых или мягких капсул, сиропов, эликсиров, растворов, буккальных пластырей, геля для полости рта, жевательной резинки, жевательных таблеток, шипучих порошков и шипучих таблеток. Композиции, предназначенные для перорального приема, можно готовить любыми способами, известными в области производства фармацевтических композиций. Такие композиции могут содержать один или больше агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей, антиоксидантов и консервантов, для создания фармацевтически привлекательных и приятных на вкус препаратов. Таблетки содержат действующее вещество в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, подходящими для производства таблеток. Такими вспомогательными веществами могут быть, например, инертные разбавители, такие как целлюлоза, диоксид углерода, оксид алюминия, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза, фосфат кальция или фосфат натрия; гранулирующие агенты и агенты, ускоряющие распад таблеток, например кукурузный крахмал или альгиновую кислоту; связующие агенты, например поливинилпирролидон, целлюлозу, крахмал, желатин или смолу акации, и лубриканты, например стеарат магния, стеариновую кислоту или тальк. Таблетки могут не иметь покрытия или они могут быть покрыты кишечнорастворимой оболочкой, или иметь покрытие, нанесенное каким-либо другим известным способом, с целью замедления распадения и всасывания в желудочно-кишечном тракте, тем самым обеспечивая пролонгированное действие в течение более длительного времени. Например, можно применять такие замедляющие распадение материалы как глицерил моностеарат или глицерил дистеарат. На таблетки можно также наносить покрытие способами, описанными в Патентах США № 4,256,108; 4,166,452 и 4,265,874, с образованием осмотических таблеток с замедленным высвобождением.
Препараты, предназначенные для перорального приема, могут также иметь вид твердых желатиновых капсул, где действующее вещество смешано с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, полиэтиленгликолем (ПЭГ) с разным средним размером молекул (например, ПЭГ400, ПЭГ4000), или в виде мягких желатиновых капсул, где действующее вещество смешано с водной или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Кроме того, можно готовить эмульсии с нерастворяющимися в воде ингредиентами, такими как масла, и стабилизировать их поверхностно-активными веществами, такими как моно- или диглицериды, сложные эфиры ПЭГ и т.п.
Водные суспензии содержат действующие вещества в смеси со вспомогательными веществами, подходящими для производства водных суспензий. Такие вспомогательные вещества представляют собой суспендирующие агенты, например натрия карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь, и смолу акации; диспергирующие или увлажняющие агенты могут представлять собой природные фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтилен стеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и гекситола, такие как полиоксиэтилен сорбитмоноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и ангидридами гекситола, например полиэтилен сорбитанмоноолеат. Водные суспензии могут также содержать один или больше консервантов, например этил или н-пропил парагидроксибензоат, один или больше красителей, один или больше ароматизаторов, и один или больше подсластителей, таких как сахароза или сахарин.
Масляные суспензии можно получать путем суспендирования действующего вещества в растительном масле, например в арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загустители, например пчелиный воск, твердый парафин или цетиловые спирты. Можно добавлять подсластители, такие как перечисленные выше, и красители, для получения приятной на вкус композиции для перорального приема. Такие композиции можно стабилизировать добавлением антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии при добавлении воды, содержат действующее вещество в смеси с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или больше консервантами. Примеры диспергирующих или увлажняющих агентов, суспендирующих агентов приведены выше. Могут также присутствовать дополнительные вспомогательные вещества, например подсластители, ароматизаторы и красители.
Фармацевтические композиции по настоящему изобретению могут также иметь форму эмульсий типа масло-в-воде. Масляной фазой может служить растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смесь. Подходящие эмульгаторы могут представлять собой природные смолы, например смолу акации или трагакантовую камедь, природные фосфатиды, например соевое масло, лецитин, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гекситола, например сорбитан моноолеат, и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например полиоксиэтилен сорбитанмоноолеат. Эмульсия может также содержать подсластители и ароматизаторы.
В состав сиропов и эликсиров могут входить подсластители, например глицерин, пропиленгликоль, сорбит или сахароза. Такие препараты могут также содержать средства, уменьшающие раздражение, консервант, ароматизаторы и красители. Растворы для перорального приема можно готовить в комбинации с, например, циклодекстрином, ПЭГ и поверхностно-активными веществами.
Фармацевтические композиции могут иметь форму стерильных инъецируемых водных или масляных суспензий. Такую суспензию можно готовить по известным в данной области методикам, с применением подходящих диспергаторов или увлажняющих агентов и суспендирующих агентов, которые были указаны выше. Стерильные инъецируемые препараты могут также представлять собой стерильные инъецируемые растворы или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например, могут иметь форму раствора в 1,3-бутандиоле. Среди подходящих носителей и растворителей, которые можно использовать, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные масла часто применяют в качестве растворителя или суспендирующей среды. Для этой цели можно использовать любое безвкусное нелетучее масло, включая синтетические моно- и диглицериды. Кроме того, в приготовлении инъецируемых препаратов находят применение жирные кислоты, такие как олеиновая кислота.
Описанные в настоящем тексте соединения можно также вводить в форме суппозиториев для ректального введения лекарственного средства. Такие композиции можно готовить путем смешивания лекарственного средства с подходящим нераздражающим вспомогательным веществом, которое твердое при комнатной температуре, но переходит в жидкое состояние при температуре тела, и поэтому плавится в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао и полиэтиленгликоли. Кроме того, соединения можно вводить в виде глазных препаратов как капли или мази. Кроме того, можно осуществлять чрескожное введение рассматриваемых соединений посредством ионофорезных пластырей и т.п. Для местного нанесения применяют кремы, мази, гели, растворы или суспензии, содержащие соединения по настоящему изобретению. В контексте настоящего изобретения, местное нанесение включает также применение полосканий и растворов для рта.
Соединения по настоящему изобретению можно также соединять с носителем, представляющем собой подходящие полимеры, в качестве целенаправленных носителей лекарственного средства. Такие полимеры могут включать поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный пальмитоильными остатками. Кроме того, соединения по настоящему изобретению можно соединять с носителем, относящимся к классу биоразлагаемых полимеров, которые можно применять для достижения контролируемого высвобождения лекарственного средства, например: полимолочная кислота, полигликолевая кислота, сополимеры полимолочной и полигликолевой кислоты, полиэпсилонкапролактон, полигидроксимасляная кислота, полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты и сшитые или амфипатические блок-сополимеры гидрогелей. Полимеры и полупроницаемые полимерные матриксы можно формовать в изделия, такие как клапаны, стенты, трубки, протезы и т.п. В одном варианте осуществления настоящего изобретения, соединение по настоящему изобретению соединяют с полимером или полупроницаемым полимерным матриксом, сформованным в виде стента или стент-графта.
Способы лечения заболеваний и нарушений
Соединения по настоящему изобретению могут применяться в качестве иммуномодуляторов. Соединения по настоящему изобретению могут применяться в качестве агонистов, антагонистов, частичных агонистов, обратных агонистов и ингибиторов PD-1 и/или PD-L1 в различном контексте, как in vitro, так и in vivo. В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться в качестве ингибиторов PD-1/PD-L1 белок-белкового взаимодействия. В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться в качестве ингибиторов PD-L1. В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться в качестве ингибиторов CD80/PD-L1 белок-белкового взаимодействия. В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для ингибирования взаимодействия между PD-1 и PD-L1 и/или PD-1 и CD80 и/или PD-L1 и PD-L2 in vitro или in vivo. В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для ингибирования VISTA и/или TIM-3. В некоторых вариантах осуществления, соединения по настоящему изобретению могут являться ингибиторами PD-1/PD-L1 белок-белкового взаимодействия и ингибиторами VISTA и/или TIM-3. В некоторых вариантах осуществления, в дополнение к функции ингибиторов PD-1/PD-L1 белок-белкового взаимодействия, соединения по настоящему изобретению могут являться ингибиторами CTLA-4 и/или BTLA и/или LAG-3 и/или KLRG-1 и/или 2B4 и/или CD160 и/или HVEM и/или CD48 и/или E-кадгерина и/или MHC-II и/или галектина-9 и/или CD86 и/или PD-L2 и/или VISTA и/или TIM-3 и/или CD80.
Соединения по настоящему изобретению можно вводить в контакт с рецептором, с которым они взаимодействуют, в водном растворе и в условиях, в остальном подходящих для связывания лиганда с рецептором. Рецептор может находиться в суспензии (например, в выделенной мембране или в препарате клеток), в выращенной или выделенной клетке, или в ткани или органе.
Предпочтительно, количество соединений по настоящему изобретению, контактирующих с рецептором, должно быть достаточно для ингибирования PD-1/PD-L1 связывания in vitro, согласно, например, измерениям методом ELISA. Рецептор может находиться в растворе или суспензии, в препарате выращенных или выделенных клеток, или в организме пациента.
В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для восстановления и усиления активации Т-клеток. В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для усиления иммунного ответа у пациента. В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для лечения, предотвращения или замедления развития заболеваний или нарушений в различных областях терапии, таких как рак и инфекционные заболевания.
В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для лечения пациентов, страдающих от патологических состояний, реагирующих на модулирование PD-1/PD-L1 белок-белкового взаимодействия.
В некоторых вариантах осуществления описан способ модулирования у субъекта иммунного ответа, осуществляемого через PD-1 сигнальный путь, включающий введение субъекту терапевтически эффективного количества соединения по настоящему изобретению, включая соединение формул (I), (Ia), (Ib), (Ic) или (Id) или его фармацевтически приемлемую соль, или композицию, содержащую соединение формул (I), (Ia), (Ib), (Ic) или (Id) или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления описан способ усиления, стимулирования, модулирования и/или повышения иммунного ответа у субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединения по настоящему изобретению, включая соединение формул (I), (Ia), (Ib), (Ic) или (Id) или его фармацевтически приемлемую соль, или композицию, содержащую соединение формул (I), (Ia), (Ib), (Ic) или (Id) или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления описан способ подавления роста, пролиферации или развития метастазов раковых клеток у субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединения по настоящему изобретению, включая соединение формулы (I), (Ia), (Ib), (Ic), или (Id) или его фармацевтически приемлемую соль, или композицию соединения по настоящему изобретению, включая соединение формулы (I), (Ia), (Ib), (Ic), или (Id) или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления описан способ лечения субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединения по настоящему изобретению, включая соединение формул (I), (Ia), (Ib), (Ic) или (Id) или его фармацевтически приемлемую соль, или композицию соединения по настоящему изобретению, включая соединение формул (I), (Ia), (Ib), (Ic) или (Id) или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления, субъект страдает заболеванием или нарушением, выбранным из группы, состоящей из инфекционного заболевания, бактериального инфекционного заболевания, вирусного инфекционного заболевания, грибкового инфекционного заболевания, солидной опухоли, гематологической злокачественной опухоли, иммунного расстройства, воспалительного заболевания и рака. В некоторых вариантах осуществления, заболевание или нарушение выбрано из группы, состоящей из следующих: меланома, глиобластома, рак пищевода, рак носоглотки, увеальная меланома, лимфома, лимфоцитарная лимфома, первичная лимфома ЦНС, Т-клеточная лимфома, диффузная крупноклеточная В-клеточная лимфома, первичная медистианальная В-крупноклеточная лимфома, рак предстательной железы, кастрационно-резистентный рак предстательной железы, хроническая миелоцитарная лейкемия, саркома Капоши, фибросаркома, липосаркома, хондросаркома, остеогенная саркома, ангиосаркома, лимфангиосаркома, синовиома, менингиома, лейомиосаркома, рабдомиосаркома, саркома мягких тканей, саркома, сепсис, рак желчного пузыря, базальноклеточная карцинома, рак вилочковой железы, рак щитовидной железы, рак паращитовидной железы, рак матки, рак надпочечников, инфекция печени, карцинома из клеток Меркеля, рак нервной системы, лимфома из клеток центра фолликула, рак ободочной кишки, лимфома Ходжкина, неходжкинская лимфома, лейкоз, хронический или острый лейкоз, включая острый миелобластный лейкоз, хронический миелобластный лейкоз, острый лимфобластный лейкоз, хронический лимфобластный лейкоз, множественная миелома, рак яичника, миелодиспластический синдром, кожная или внутриглазная злокачественная меланома, почечно-клеточный рак, мелкоклеточный рак легких, рак легких, мезотелиом, рак груди, плоскоклеточный немелкоклеточный рак легких, неплоскоклеточный немелкоклеточный рак легких, колоректальный рак, рак яичника, рак желудка, печеночно-клеточный рак, карцинома поджелудочной железы, рак поджелудочной железы, аденокарцинома протоков поджелудочной железы, плоскоклеточный рак головы и шеи, рак головы и шеи, рак желудочно-кишечного тракта, рак желудка, ВИЧ, гепатит А, гепатит В, гепатит С, гепатит D, вирус герпеса, папилломавирус, грипп, рак костей, рак кожи, рак прямой кишки, рак анального отверстия, рак яичка, рак фаллопиевых труб, рак эндометрия, рак шейки матки, рак вагины, рак вульвы, рак пищевода, рак тонкого кишечника, рак эндокринной системы, рак уретры, рак пениса, рак мочевого пузыря, рак почки, рак уретры, рак почечной лоханки, новообразования в центральной нервной системе (ЦНС), ангиогенез опухоли, рак оси позвоночника, глиома стволовой части мозга, аденома гипофиза, эпидермоидный рак, асбестоз, карционома, аденокарцинома, папиллярная карцинома, цистаденокарцинома, бронхогенная карцинома, почечно-клеточный рак, переходно-клеточный рак, хориокарцинома, семинома, эмбриональный рак, опухоль Вильмса, плеоморфная аденома, папиллома клеток печени, почечная канальцевая аденома, цистаденома, папиллома, аденома, лейомиома, рабдомиома, гемангиома, лимфангиома, остеома, хондрома, липома и фиброма.
В некоторых вариантах осуществления, субъекту дополнительно вводят терапевтически эффективное количество одного или больше дополнительных терапевтических агентов. В некоторых вариантах осуществления, один или больше дополнительных терапевтических агентов выбраны из группы, состоящей из противомикробного агента, противовирусного агента, цитотоксического агента, агента, модулирующего экспрессию гена, химиотерапевтического агента, противоракового агента, антиангиогенного агента, иммунотерапевтического агента, противогормонального агента, противофиброзного агента, лучевой терапии, агента для лучевой терапии, противоопухолевого агента и антипролиферативного агента. В некоторых вариантах осуществления, один или больше дополнительных терапевтических агентов представляют собой антагонист хемокинового и/или хемотаксического рецептора, который включает (но не ограничивается только ими) CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10, CCR11, CCR12, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7, C3aR и/или C5aR. Антагонисты хемокинового и/или хемотаксического рецептора известны в данной области и описаны, например, в WO2007/002667, WO2007/002293, WO/2003/105853, WO/2007/022257, WO/2007/059108, WO/2007/044804, WO2007/115232, WO2007/115231, WO2008/147815, WO2010/030815, WO2010/075257, WO2011/163640, WO2010/054006, WO2010/051561, WO2011/035332, WO2013/082490, WO2013/082429, WO2014/085490, WO2014/100735, WO2014/089495, WO2015/084842, WO2016/187393, WO2017/127409, WO 2017/087607, WO2017/087610, WO2017/176620, WO2018/222598, WO2018/222601, WO2013/130811, WO2006/076644, WO2008/008431, WO2009/038847, WO2008/008375, WO2008/008374, WO2008/010934, WO2009/009740, WO2005/112925, WO2005/112916, WO2005/113513, WO2004/085384, WO2004/046092. Антагонисты хемокинового и/или хемотаксического рецептора включают также CCX354, CCX9588, CCX140, CCX872, CCX598, CCX6239, CCX9664, CCX2553, CCX3587, CCX3624, CCX 2991, CCX282, CCX025, CCX507, CCX430, CCX765, CCX224, CCX662, CCX650, CCX832, CCX168, CCX168-M1, CCX3022 и/или CCX3384.
В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для подавления инфекционного заболевания. Инфекционное заболевание включает (но не ограничивается только ими) ВИЧ, грипп, лямблиоз, малярию, лейшманиоз, патогенные инфекции, вызванные вирусом гепатита (А, В и С), вирусом герпеса (например, VZV, HSV-I, HAV-6, HSV-II и CMV, вирус Эпштейна-Барра), аденовирусом, вирусом гриппа, флавивирусами, эховирусом, риновирусом, вирусом коксаки, коронавирусом, респираторно-синцитиальным вирусом, вирусом свинки, ротавирусом, вирусом кори, вирусом краснухи, карвовирусом, вирусом коровьей оспы, Т-лимфотропным вирусом человека, вирусом денге, папилломавирусом, вирусом моллюска, вирусом полиомиелита, вирусом бешенства, вирусом Джона Каннингема и вирусом арбовирусного энцефалита, патогенные бактериальные инфекции, вызванные хламидиями, риккетсиальными бактериями, микобактериями, стафилококками, стрептококками, пневмококками, менингококками и гонококками, клебсиеллой, протеусом, серратией, синегнойной палочкой, кишечной палочкой, легионеллой, бактерией дифтерии, сальмонеллой, палочковидными бактериями (бациллами), бактериями, вызывающими холеру, столбняк, ботулизм, сибирскую язву, чуму, лептоспироз и болезнь Лайма, патогенные инфекции, вызываемые грибами из рода Candida (albicans, krusei, glabrata, tropicalis и т.д.), дрожжевыми грибами, грибами из рода Aspergillus (fumigatus, niger и т.д.), плесневыми грибами (мукор, абсидия, ризофус), Sporothrix schenkii, Blastomyces dermatitidis, Paracoccidioides brasiliensis, Coccidioides immitis и Histoplasma capsulatum, и патогенные инфекции, вызываемые паразитами Entamoeba histolytica, Balantidium coli, Naegleriafowleri, Acanthamoeba sp., Giardia lambia, Cryptosporidium sp., Pneumocystis carinii, Plasmodium vivax, Babesia microti, Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondi, Nippostrongylus brasiliensis.
В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для подавления ВИЧ инфекции, замедления развития СПИД, уничтожения резервуара вируса ВИЧ или снижения степени тяжести симптомов или ВИЧ-инфекции и СПИД.
Соединения по настоящему изобретению могут применяться для лечения раковых и предраковых состояний у субъекта.
Методы лечения, описанные в настоящем изобретении, включают введение пациенту эффективного количества одного или больше соединений, описанных в настоящем тексте. Подходящие пациенты включают пациентов, страдающих заболеванием или расстройством или подверженных (т.е. профилактическое лечение) заболеванию или расстройству, указанным в настоящем тексте. Типичные пациенты для описанного в настоящем изобретении лечения включают млекопитающих, в частности приматов, в особенности людей. Другие подходящие пациенты включают домашних животных-компаньонов, таких как собаки, кошки, лошади и т.п., или сельскохозяйственных животных, таких как крупный рогатый скот, свиньи, овцы и т.п.
В целом, описанные в настоящем изобретении способы лечения включают введение пациенту эффективного количества одного или больше соединений, описанных в настоящем тексте. В предпочтительном варианте осуществления, соединение (соединения) по настоящему изобретению предпочтительно вводят пациенту (например, человеку) внутривенно, перорально или наружно. Эффективное количество может представлять собой количество, достаточное для модулирования PD-1/PD-L1 взаимодействия, и/или количество, достаточное для уменьшения или ослабления симптомов у пациента. Предпочтительно, вводимое количество достаточно для создания в плазме крови концентрации соединения (или его активного метаболита, если соединение представляет собой пролекарство) достаточно высокой для существенного модулирования PD-1/PD-L1 взаимодействия. Режимы лечения могут варьироваться в зависимости от применяемого соединения и конкретного состояния, подвергающегося лечению; для лечения большинства нарушений, предпочтительная частота введения составляет 4 раза в сутки или меньше. В целом, прием 2 раза в сутки более предпочтителен, и особенно предпочтительно введение 1 раз в сутки. Следует понимать, однако, что конкретный уровень дозировки и режим введения для каждого конкретного пациента зависит от ряда факторов, включая активность конкретного соединения, возраст, вес тела, общее состояние здоровья, пол, диета, время введения, способ введения, скорость выведения, комбинация лекарств (например, другие лекарственные средства, которые вводят пациенту) и степень тяжести конкретного заболевания, подвергающегося лечению, а также мнение лечащего врача. В целом, предпочтительно применение минимальной дозировки, достаточной для обеспечения эффективной терапии. За терапевтической эффективностью для пациентов можно следить с помощью медицинских или ветеринарных критериев, подходящих для конкретного состояния, подвергающегося лечению.
Комбинации
Комбинированный препарат, содержащий соединения по настоящему изобретению и другое лекарственное средство, можно вводить в виде комбинированного препарата, в котором оба компонента содержатся в одной готовой форме, или в виде раздельных препаратов. Введение раздельных препаратов включает одновременное введение и введение с некоторыми интервалами времени. В случае введения с некоторыми интервалами времени, соединение по настоящему изобретению можно вводить первым, и затем вводить другое лекарственное средство, или другое лекарственное средство можно вводить первым, и затем вводить соединение по настоящему изобретению. Способ введения соответствующих лекарственных средств может быть одинаковым или различаться.
Дозировку другого лекарственного средства можно подобрать надлежащим образом на основе клинически применяемых дозировок. Соотношение количеств соединения по настоящему изобретению и другого лекарственного средства можно надлежащим образом подобрать в соответствии с возрастом и весом тела субъекта, которому осуществляется введение, способом введения, временем введения, заболеванием, которое подвергается лечению, его симптомами и их комбинацией. Например, другое лекарственное средство можно применять в количестве от 0,01 до 100 массовых частей относительно 1 массовой части соединения по настоящему изобретению. Другое лекарственное средство может представлять собой комбинацию двух или больше видов независимых лекарственных средств в нужном соотношении.
Соединения по настоящему изобретению можно применять или комбинировать с одним или больше терапевтическими агентами, такими как противомикробный агент, противовирусный агент, цитотоксический агент, агент, модулирующий экспрессию гена, химиотерапевтический агент, противораковый агент, антиангиогенный агент, иммунотерапевтический агент, противогормональный агент, противофиброзный агент, лучевая терапия, агент для лучевой терапии, противоопухолевый агент и антипролиферативный агент. Эти терапевтические агенты могут быть в форме соединений, антител, полипептидов или полинуклеотидов.
Соединения по настоящему изобретению можно применять или комбинировать с одним или больше агентами из следующих: терапевтические антитела, биспецифические антитела и «антителоподобные» терапевтические белки (такие как DARTs®, Duobodies®, Bites®, XmAbs®, TandAbs®, Fab производные), конъюгаты антитело-лекарство, вирус, онколитический вирус, генные модификаторы или редакторы, такие как CRISPR (включая CRISPR Cas9), цинк-пальцевые нуклеазы или синтетические нуклеазы (TALEN), агенты иммунотерапии с использованием Т-клеток с химерными антигенными рецепторами, или любая их комбинация.
Примеры химиотерапевтических средств включают алкилирующий агент, нитрозомочевинный агент, антиметаболит, противораковые антибиотики, алкалоид растительного происхождения, ингибитор топоизомеразы, гормональный лекарственный препарат, антагонист гормонов, ингибитор ароматазы, ингибитор P-гликопротеина, комплексные производные платины, другие иммунотерапевтические агенты и другие противораковые лекарственные средства.
Соединения по настоящему изобретению можно применять или комбинировать со вспомогательным средством противораковой терапии, таким как лекарство для лечения лейкопении (нейтропении), лекарство для лечения тромбоцитопении, противорвотное лекарство и лекарство для лечения раковой боли, в форме сопутствующей или комбинированной терапии.
Соединения по настоящему изобретению можно применять или комбинировать с ингибитором киназ.
В одном варианте осуществления, соединения по настоящему изобретению можно применять с другим иммуномодулятором и/или стимулирующим агентом в форме сопутствующей или комбинированной терапии. Примеры иммуномодулятора включают различные цитокины, вакцины и адъюванты. Примеры таких цитокинов, вакцин и адъювантов, стимулирующих иммунный ответ, включают (но не ограничиваются только ими) GM-CSF, M-CSF, G-CSF, интерферон-альфа, бета или гамма, IL-1, IL-2, IL- 3, IL-12, Poly (I:C) и CPG. Стимулирующие агенты включают циклофосфамид и аналоги циклофосфамида, анти-ТФР и иматиниб (Gleevac), ингибитор митоза, такой как паклитаксел, сунитиниб (Sutent) или другие антиангиогенные агенты, ингибитор ароматазы, такой как летрозол, антагонист A2a аденозинового рецептора (A2AR), ингибитор ангиогенеза, антрациклины, оксалиплатин, доксорубицин, антагонисты TLR4 и антагонисты IL-18.
В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться или комбинироваться с одним или больше модуляторами CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10, CCR11, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7, ChemR23, C5aR, C5a, и C5. В некоторых вариантах осуществления, модулятор является антагонистом.
В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться или комбинироваться с одним или больше антагонистами хемокинового и/или хемотаксического рецептора, описанными, например, в WO2007/002667, WO2007/002293, WO/2003/105853, WO/2007/022257, WO/2007/059108, WO/2007/044804, WO2007/115232, WO2007/115231, WO2008/147815, WO2010/030815, WO2010/075257, WO2011/163640, WO2010/054006, WO2010/051561, WO2011/035332, WO2013/082490, WO2013/082429, WO2014/085490, WO2014/100735, WO2014/089495, WO2015/084842, WO2016/187393, WO2017/127409, WO 2017/087607, WO2017/087610, WO2017/176620, WO2018/222598, WO2018/222601, WO2013/130811, WO2006/076644, WO2008/008431, WO2009/038847, WO2008/008375, WO2008/008374, WO2008/010934, WO2009/009740, WO2005/112925, WO2005/112916, WO2005/113513, WO2004/085384, WO2004/046092. Антагонисты хемокинового и/или хемотаксического рецептора, которые могут применяться по настоящему изобретению, включают также CCX354, CCX9588, CCX140, CCX872, CCX598, CCX6239, CCX9664, CCX2553, CCX3587, CCX3624, CCX 2991, CCX282, CCX025, CCX507, CCX430, CCX765, CCX224, CCX662, CCX650, CCX832, CCX168, CCX168-M1, CCX3022 и/или CCX3384.
Дозировка
Дозировки порядка от примерно 0,1 мг до примерно 140 мг на килограмм веса тела в сутки могут применяться в лечении или профилактики состояний, включающих PD-1/PD-L1 взаимодействие. Количество действующего вещества, которое можно комбинировать с носителями для производства однократной дозированной формы, варьируется в зависимости от конкретного пациента, подвергающегося лечению, и от применяемого способа введения. Дозированные готовые формы обычно содержат от примерно 1 мг до примерно 500 мг действующего вещества. Для соединений, которые вводят перорально, чрескожно, внутривенно или подкожно, предпочтительно, чтобы вводилось достаточное количество соединения для достижения концентрации в плазме крови от 5 нг (нанограмм)/мл до 10 мкг (микрограмм)/мл плазмы крови, более предпочтительно - достаточное количество соединения для достижения концентрации в плазме крови от 20 нг/мл до 1 мкг/мл плазмы крови, наиболее предпочтительно - достаточное количество соединения для достижения концентрации в плазме крови от 50 нг/мл до 200 нг/мл плазмы крови. Для прямого введения в синовиальную оболочку (для лечения артрита), следует вводить достаточное количество соединения для локального достижения примерно 1 микромолярной концентрации.
Частота введения также может варьироваться в зависимости от применяемого соединения и конкретного состояния, подвергающегося лечению. Однако, для лечения большинства заболеваний предпочтителен режим введения 4 раза в сутки, три раза в сутки или меньше, при этом особенно предпочтителен режим введения один раз в сутки или 2 раза в сутки. Следует понимать, однако, что конкретный уровень дозировки для каждого конкретного пациента зависит от ряда факторов, включая активность конкретного соединения, возраст, вес тела, общее состояние здоровья, пол, диета, время введения, способ введения, скорость выведения, комбинация лекарств (например, другие лекарственные средства, которые вводят пациенту), степень тяжести конкретного заболевания, подвергающегося лечению, и другие факторы, включая мнение лечащего врача.
В другом аспекте настоящего изобретения, соединения по настоящему изобретению можно использовать в различных нефармацевтических in vitro и in vivo областях. Соединения по настоящему изобретению можно также применять в качестве положительного контроля в тестах активности в отношении PD-1/PD-L1 взаимодействий, т.е. в качестве стандартов для определения способности кандидата связываться с PD-1 и/или PD-L1, или в качестве радиоактивных индикаторов для позитронно-эмиссионной томографии (ПЭТ) или однофотонной эмиссионной компьютерной томографии (SPECT).
Также в объем настоящего изобретения входят наборы, содержащие соединение по настоящему изобретению или его фармацевтически приемлемую соль и инструкции по применению. Набор может также содержать по меньшей мере один дополнительный реагент. Наборы обычно включают этикетку, на которой указано назначение содержимого набора. Термин «этикетка» включает любые письменные или записанные материалы, находящиеся на упаковке или внутри набора, или иным образом сопровождающие набор.
Примеры
Описанные ниже Примеры иллюстрируют различные способы получения соединений по настоящему изобретению, включая соединения формул (I), (Ia), (Ib), (Ic) или (Id). Следующие далее примеры приведены для иллюстрации, а не для ограничения объема настоящего изобретения.
Описанные ниже реагенты и растворители могут быть получены из коммерческих источников, таких как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). 1H-ЯМР спектры записывали на Varian Mercury 400 МГц ЯМР-спектрометре. Значения хим.сдвигов приведены относительно тетраметилсилана (ТМС) и описаны в порядке: мультиплетность (с, синглет; д, дублет; т, триплет; кв, квартет; м, мультиплет) и число протонов. Результаты масс-спектрометрии приведены в виде значения массы, деленной на заряд. В примерах приведено одно значение m/e для каждого M+H (или, если указано, M-H) иона, содержащего наиболее распространенные изотопы атомов. Во всех случаях изотопное распределение соответствует ожидаемой формуле. Масс-спектрометрию с ионизацией электрораспылением (ESI) проводили на масс-спектрометре Hewlett-Packard MSD с ионизацией электрораспылением, оснащенном ВЭЖХ HP1100 для ввода образца. Обычно аналит растворяли в метаноле или CH3CN в концентрации 0,1 мг/мл, и 1 микролитр полученного раствора вводили в масс-спектрометр, сканирующий ионы в диапазоне от 100 до 1000 дальтон. Все соединения анализировали в режиме ESI с регистрацией положительных или отрицательных ионов, используя смесь ацетонитрил/вода с 1% муравьиной кислоты в качестве растворителя.
В Примерах и в остальном тексте заявки применяются следующие сокращения: ТСХ означает Тонкослойная хроматография; ТГФ означает тетрагидрофуран; ДХЭ означает 1,2-дихлорэтан; ДМФА означает N,N-диметилформамид; ТФУК означает трифторуксусная кислота; Bpin и pinB оба означают 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил.
Соединения по настоящему изобретению можно синтезировать как описано ниже, применяя разнообразные реакции, известные квалифицированному специалисту в данной области техники. Квалифицированному специалисту в данной области будет также понятно, что могут применяться альтернативные методы синтеза целевых соединений, и что подходы, используемые в тексте настоящего документа, не являются исчерпывающими, но дают работающие и практичные способы получения соединений по настоящему изобретению.
Некоторые молекулы, заявленные в настоящем патенте, могут существовать в различных энантиомерных и диастереомерных формах, и все такие варианты соединений входят в объем настоящего изобретения, если только не указан конкретный энантиомер.
Подробное описание экспериментальных методик, используемых для синтеза ключевых соединений в настоящем тексте, ведут к молекулам, которые описаны идентифицирующими их физическими данными, а также описывающими их изображениями химической структуры.
Квалифицированным специалистам в данной области техники будет также понятно, что во время стандартных методик обработки в органической химии часто применяются кислоты и основания. Иногда образуются соли материнских соединений, если они обладают необходимой собственной кислотностью или основностью, во время описанных в настоящем тексте экспериментальных процедур.
Пример 1: 6-(2,2'-диметил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
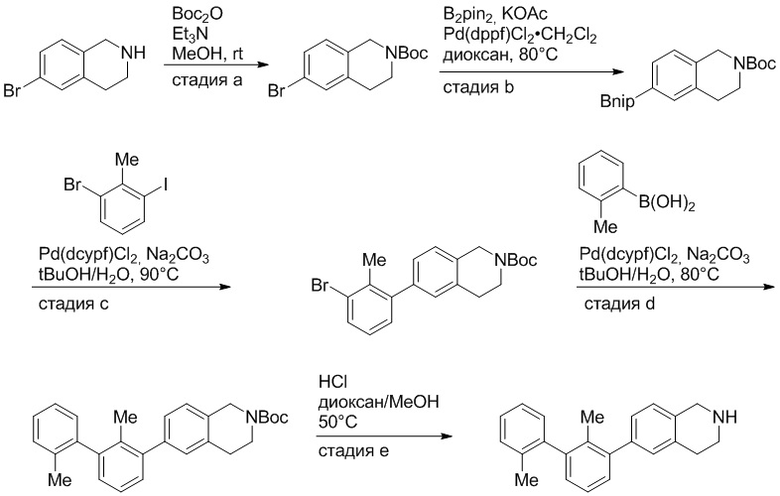
Стадия a. Раствор 6-бром-1,2,3,4-тетрагидроизохинолина (3,18 г, 15,0 ммоль), ди-трет-бутил дикарбоната (6,55 г, 30,0 ммоль) и Et3N (8,4 мл, 60,0 ммоль) в MeOH (40 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь упаривали и очищали методом флешхроматографии на силикагеле (0-40% EtOAc/гексан), получая трет-бутил 6-бром-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия b. Смесь трет-бутил 6-бром-3,4-дигидроизохинолин-2(1H)-карбоксилата (3,12 г, 10,0 ммоль), бис(пинаколато)диборона (3,05 г, 12,0 ммоль) и KOAc (2.94 г, 30,0 ммоль) в диоксане (50 мл) дегазировали (N2) в течение 20 минут. Добавляли комплекс [1,1′-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II) с дихлорметаном (817 мг, 1,0 ммоль), и реакционную смесь дегазировали (N2) еще 5 минут и перемешивали при 80°C в течение ночи в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным раствором NaHCO3 (100 мл) и экстрагировали этилацетатом (3 x 100 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия c. Смесь трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (750 мг, 2,1 ммоль), 1-бром-3-иод-2-метилбензола (802 мг, 2,7 ммоль), дихлор[1,1'-бис(дициклогексилфосфино)ферроцен]палладия(II) (159 мг, 0,21 ммоль) и Na2CO3 (562 мг, 5,3 ммоль) в смеси 3:1 t-BuOH:H2O (12 мл) дегазировали (N2) 10 минут и перемешивали при 90°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали дихлорметаном (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешкроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3-бром-2-метилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия d. Смесь трет-бутил 6-(3-бром-2-метилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (100 мг, 0,25 ммоль), o-толилбороновой кислоты (52 мг, 0,38 ммоль), дихлор[1,1'-бис(дициклогексилфосфино)ферроцен]палладия(II) (19 мг, 0,025 ммоль) и Na2CO3 (67 мг, 0,63 ммоль) в смеси 3:1 t-BuOH:H2O (2 мл) дегазировали (N2) 5 минут и перемешивали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали дихлорметаном (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-100% CH2Cl2/гексан) дала трет-бутил 6-(2,2'-диметил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия e. трет-бутил 6-(2,2'-диметил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (65 мг, 0,16 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 6-(2,2'-диметил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин в виде соли с трифторуксусной кислотой (ТФУК). 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,09 (ушир.с, 2H), 7,34-7,22 (м, 7H), 7,18 (дд, J = 1,4, 7,7 Гц, 1H), 7,10 (тд, J = 1,6, 7,4 Гц, 2H), 4,33 (т, J = 4,2 Гц, 2H), 3,42 (дд, J = 5,1, 7.8 Гц, 2H), 3,06 (т, J = 6,4 Гц, 2H), 2,05 (с, 3H), 1.85 (с, 3H). MS: (ES) m/z вычислено для C23H24N [M+H]+ 314,2, найдено 314,1.
Пример 2. 6-(3'-метокси-2-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
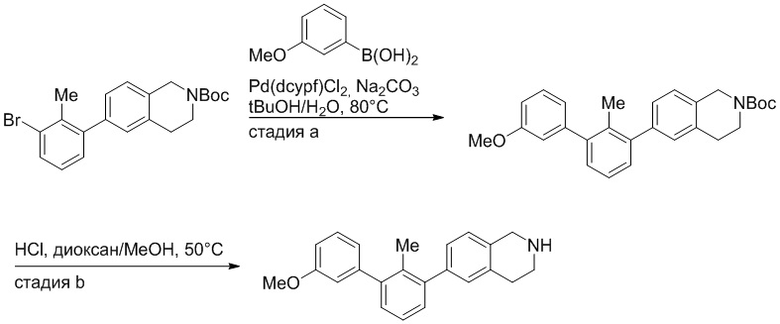
Стадия a. Смесь трет-бутил 6-(3-бром-2-метилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (100 мг, 0,25 ммоль), 3-метоксифенилбороновой кислоты (58 мг, 0,38 ммоль), дихлор[1,1'-бис(дициклогексилфосфино)ферроцен]палладия(II) (19 мг, 0,025 ммоль) и Na2CO3 (67 мг, 0,63 ммоль) в смеси 3:1 t-BuOH:H2O (2 мл) дегазировали (N2) 5 минут и перемешивали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали дихлорметаном (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3'-метокси-2-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия b. трет-бутил 6-(3'-метокси-2-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (75 мг, 0,17 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 6-(3'-метокси-2-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин в виде соли с ТФУК. 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,06 (ушир.с, 2H), 7,39-7,16 (м, 7H), 6.95-6.89 (м, 3H), 4,36-4,30 (м, 2H), 3,79 (с, 3H), 3,47-3,40 (м, 2H), 3,05 (т, J = 6,3 Гц, 2H), 2,06 (с, 3H). MS: (ES) m/z вычислено для C23H24NO [M+H]+ 330,2, найдено 330,1.
Пример 3. 6-(3'-метокси-2,2'-диметил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
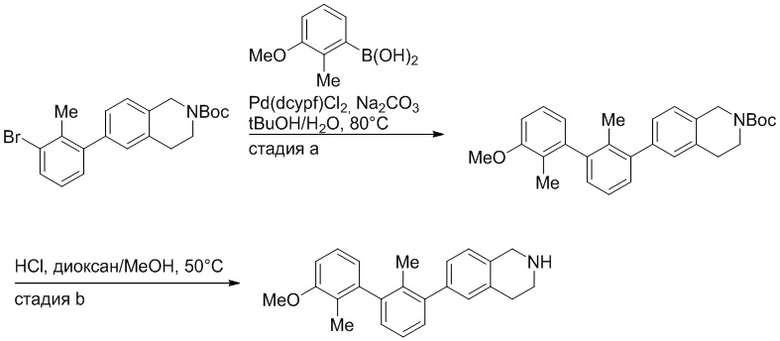
Стадия a. Смесь трет-бутил 6-(3-бром-2-метилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (100 мг, 0,25 ммоль), 3-метокси-2-метилфенилбороновой кислоты (63 мг, 0,38 ммоль), дихлор[1,1'-бис(дициклогексилфосфино)ферроцен]палладия(II) (19 мг, 0,025 ммоль) и Na2CO3 (67 мг, 0,63 ммоль) в смеси 3:1 t-BuOH:H2O (2 мл) дегазировали (N2) 5 минут и перемешивали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали дихлорметаном (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3'-метокси-2,2'-диметил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия b. трет-бутил 6-(3'-метокси-2,2'-диметил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (70 мг, 0,16 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 6-(3'-метокси-2,2'-диметил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин в виде соли с ТФУК. 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,05 (ушир.с, 2H), 7,33-7,20 (м, 5H), 7,18 (дд, J = 1,4, 7.8 Гц, 1H), 7,07 (дд, J = 1,4, 7,6 Гц, 1H), 6.97 (дд, J = 1,1, 8,4 Гц, 1H), 6,72 (дд, J = 1,1, 7,5 Гц, 1H), 4,35-4,30 (м, 2H), 3.83 (с, 3H), 3,47-3,39 (м, 2H), 3,05 (т, J = 6,3 Гц, 2H), 1.88 (с, 3H), 1.85 (с, 3H). MS: (ES) m/z вычислено для C24H26NO [M+H]+ 344,2, найдено 344,2.
Пример 4. 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2,2'-диметил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
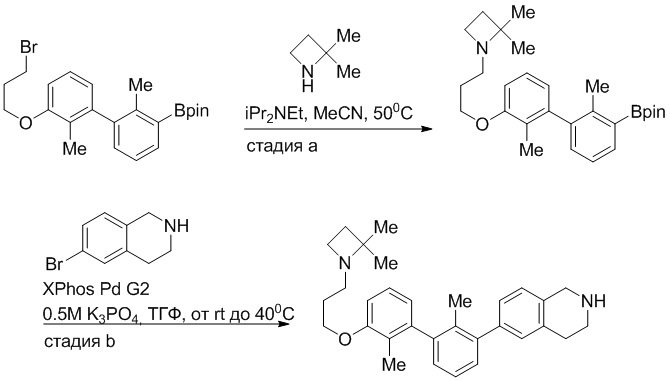
Стадия a. В раствор 2-(3'-(3-бромпропокси)-2,2'-диметил-[1,1'-бифенил]-3-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (130 мг, 0,29 ммоль) и 2,2-диметилазетидина (33 мг, 0,38 ммоль) в MeCN (2 мл) медленно добавляли iPr2NEt (0,15 мл, 0.87 ммоль). Смесь нагревали до 50°C и перемешивали 3 часа. Реакционную смесь упаривали, и сырой продукт очищали методом флешхроматографии на силикагеле (0-15% MeOH/CH2Cl2), получая 1-(3-((2,2'-диметил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-3-ил)окси)пропил)-2,2-диметилазетидин.
Стадия b. В двухфазную смесь 1-(3-((2,2'-диметил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-3-ил)окси)пропил)-2,2-диметилазетидина (45 мг, 0,10 ммоль), 6-бром-1,2,3,4-тетрагидроизохинолина (42 мг, 0,20 ммоль) в 0,5M растворе K3PO4 (0,60 мл 0,30 ммоль) и ТГФ (1,5 мл) добавляли XPhos Pd G2 (16 мг, 0,020 ммоль). Реакционную смесь перемешивали при комнатной температуре 1 час и затем нагревали при 40°C в течение 12 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, органический слой отделяли, фильтровали и очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2,2'-диметил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин. MS: (ES) m/z вычислено для C31H39N2O [M+H]+ 445,3, найдено 455,3. 1H-ЯМР (400 МГц, CDCl3) δ 7,29-7,26 (м, 1H), 7,25-7,22 (м, 1H), 7,22-7,18 (м, 1H), 7,18-7,16 (м, 2H), 7,15 (д, J = 8,0 Гц, 1H), 7,10 (дт, J = 7,5, 1,6 Гц, 1H), 6.82 (д, J = 1.8 Гц, 1H), 6.80 (д, J = 2,5 Гц, 1H), 4,43-4,34 (м, 3H), 4,12-4,02 (м, 2H), 3,62-3,47 (м, 3H), 3,36-3,25 (м, 1H), 3,21-3,08 (м, 3H), 2,67 (кв, J = 9.9 Гц, 1H), 2,23-2,14 (м, 2H), 2,14-2,06 (м, 1H), 1.95 (с, 3H), 1.89 (с, 3H), 1,69 (с, 3H), 1,61 (с, 3H).
Пример 5. 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
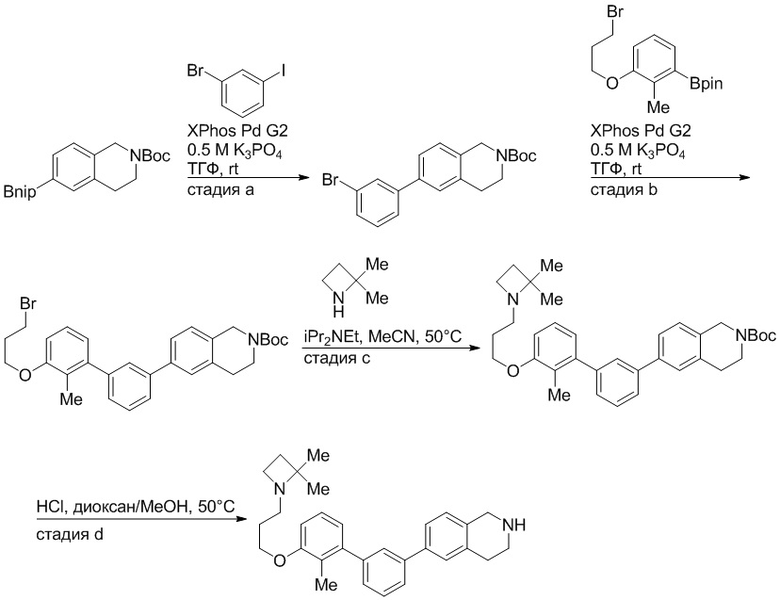
Стадия a. Смесь 1-бром-3-иодбензола (283 мг, 1,0 ммоль), трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (359 мг, 1,0 ммоль), ТГФ (10 мл) и 0,5M раствора K3PO4 (10 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (79 мг, 0,10 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3-бромфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия b. Смесь трет-бутил 6-(3-бромфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (100 мг, 0,26 ммоль), 2-(3-(3-бромпропокси)-2-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (173 мг, 0,39 ммоль), ТГФ (3 мл) и 0,5M раствора K3PO4 (3 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (20 мг, 0,026 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3'-(3-бромпропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия c. Раствор трет-бутил 6-(3'-(3-бромпропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (80 мг, 0,15 ммоль), 2,2-диметилазетидина (26 мг, 0,30 ммоль) и iPr2NEt (79 мкл, 0,45 ммоль) в MeCN (2 мл) перемешивали при 50°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали. Очистка методом флешхроматографии на силикагеле (0-10% MeOH/CH2Cl2) дала трет-бутил 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия d. Трет-бутил 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (55 мг, 0,10 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин в виде соли с двумя молекулами ТФУК. 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,64 (ушир.с, 1H), 9,09 (ушир.с, 2H), 7,67 (д, J = 8,0 Гц, 1H), 7,62-7,58 (м, 2H), 7,56-7,51 (м, 2H), 7,33-7,28 (м, 2H), 7,25 (т, J = 7.9 Гц, 1H), 6.99 (д, J = 8,4 Гц, 1H), 6.91 (д, J = 7,6 Гц, 1H), 4,35-4,29 (м, 2H), 4,13-4,01 (м, 2H), 3.99-3.87 (м, 2H), 3,46-3,38 (м, 2H), 3,33-3,20 (м, 1H), 3,19-3,10 (м, 1H), 3,06 (т, J = 6,3 Гц, 2H), 2,39-2,29 (м, 1H), 2,15-2,06 (м, 4H), 2,03-1.94 (м, 2H), 1,57 (с, 3H), 1,55 (с, 3H). MS: (ES) m/z вычислено для C30H37N2O [M+H]+ 441,3, найдено 441,3.
Пример 6. 3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-3-(1,2,3,4-тетрагидроизохинолин-6-ил)-[1,1'-бифенил]-2-карбонитрил
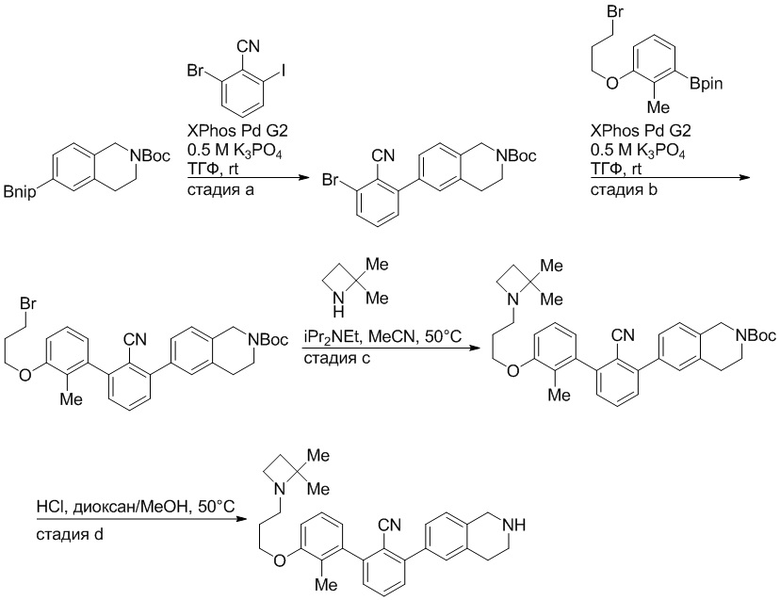
Стадия a. Смесь 2-бром-6-иодбензонитрила (308 мг, 1,0 ммоль), трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (359 мг, 1,0 ммоль), ТГФ (10 мл) и 0,5M раствора K3PO4 (10 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (79 мг, 0,10 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-50% EtOAc/гексан) дала трет-бутил 6-(3-бром-2-цианофенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия b. Смесь трет-бутил 6-(3-бром-2-цианофенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (170 мг, 0,41 ммоль), 2-(3-(3-бромпропокси)-2-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (275 мг, 0,62 ммоль), ТГФ (4 мл), и 0,5M раствора K3PO4 (4 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (32 мг, 0,041 ммоль), реакционную смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3'-(3-бромпропокси)-2-циано-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия c. Раствор трет-бутил 6-(3'-(3-бромпропокси)-2-циано-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (140 мг, 0,25 ммоль), 2,2-диметилазетидина (43 мг, 0,50 ммоль) и iPr2NEt (0,13 мл, 0,75 ммоль) в MeCN (3 мл) перемешивали при 50°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали. Очистка методом флешхроматографии на силикагеле (0-10% MeOH/CH2Cl2) дала трет-бутил 6-(2-циано-3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия d. Трет-бутил 6-(2-циано-3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (120 мг, 0,21 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-3-(1,2,3,4-тетрагидроизохинолин-6-ил)-[1,1'-бифенил]-2-карбонитрил в виде соли с двумя молекулами ТФУК. 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,28 (ушир.с, 1H) 9,16 (ушир.с, 2H), 7.84 (д, J = 7.8 Гц, 1H), 7,61 (дд, J = 1,1, 7.9 Гц, 1H), 7,53-7,47 (м, 2H), 7,45 (дд, J = 1,1, 7.8 Гц, 1H), 7,39 (д, J = 8,0 Гц, 1H), 7,29 (т, J = 7.9 Гц, 1H), 7,07 (д, J = 8,3 Гц, 1H), 6.90 (д, J = 7,6 Гц, 1H), 4,37 (т, J = 4,2 Гц, 2H), 4,17-4,02 (м, 2H), 4,00-3.87 (м, 2H), 3,49-3,41 (м, 2H), 3,34-3,22 (м, 1H), 3,19-3,11 (м, 1H), 3,08 (т, J = 6,2 Гц, 2H), 2,39-2,29 (м, 1H), 2,14-2,05 (м, 1H), 2,05-1.95 (м, 5H), 1,57 (с, 3H), 1,55 (с, 3H). MS: (ES) m/z вычислено для C31H36N3O [M+H]+ 466,3, найдено 466,4.
Пример 7. 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2-фтор-2'-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
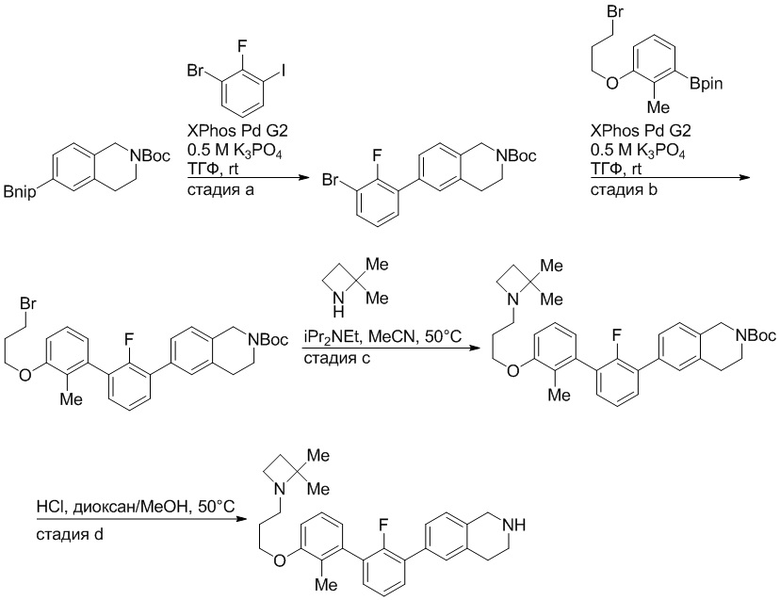
Стадия a. Смесь 1-бром-2-фтор-3-иодбензола (250 мг, 0.83 ммоль), трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (298 мг, 0.83 ммоль), Na2CO3 (180 мг, 1,7 ммоль), ДМЭ (8 мл) и H2O (2 мл) дегазировали (N2) 5 минут. Добавляли Pd(PPh3)4 (197 мг, 0,17 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-100% CH2Cl2/гексан) дала трет-бутил 6-(3-бром-2-фторфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия b. Смесь трет-бутил 6-(3-бром-2-фторфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (90 мг, 0,22 ммоль), 2-(3-(3-бромпропокси)-2-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (146 мг, 0,33 ммоль), ТГФ (2 мл) и 0,5M раствора K3PO4 (2 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (17 мг, 0,022 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3'-(3-бромпропокси)-2-фтор-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия c. Раствор трет-бутил 6-(3'-(3-бромпропокси)-2-фтор-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (60 мг, 0,11 ммоль), 2,2-диметилазетидина (19 мг, 0,22 ммоль) и iPr2NEt (58 мкл, 0,33 ммоль) в MeCN (1 мл) перемешивали при 50°C 3 часа. Реакционную смесь охлаждали до комнатной температуры и упаривали. Очистка методом флешхроматографии на силикагеле (0-10% MeOH/CH2Cl2) дала трет-бутил 6-(2-фтор-3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия d. Трет-бутил 6-(2-фтор-3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (35 мг, 0,063 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2-фтор-2'-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин в виде соли с двумя молекулами ТФУК. 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,64 (ушир.с, 1H) 9,10 (ушир.с, 2H), 7,53 (тд, J = 1.8, 7,6 Гц, 1H), 7,47-7,42 (м, 2H), 7,38 (т, J = 7,6 Гц, 1H), 7,35-7,23 (м, 3H), 7,02 (д, J = 8,0 Гц, 1H), 6.88 (д, J = 7,6 Гц, 1H), 4,36-4,30 (м, 2H), 4,14-4,02 (м, 2H), 3.98-3.86 (м, 2H), 3,47-3,38 (м, 2H), 3,32-3,21 (м, 1H), 3,18-3,09 (м, 1H), 3,05 (т, J = 6,3 Гц, 2H), 2,39-2,29 (м, 1H), 2,14-2,06 (м, 1H), 2,04-1.94 (м, 5H), 1,57 (с, 3H), 1,54 (с, 3H). MS: (ES) m/z вычислено для C30H36FN2O [M+H]+ 459,3, найдено 459,4.
Пример 8. 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2-хлор-2'-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
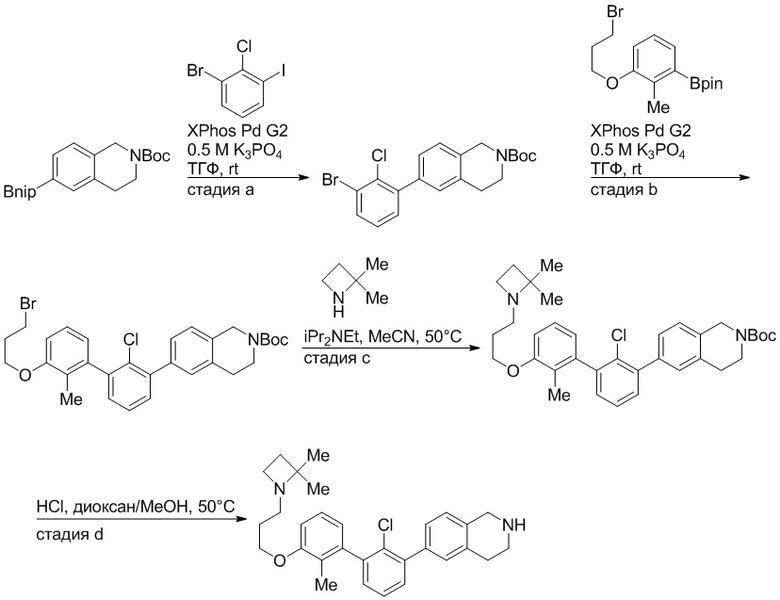
Стадия a. Смесь 1-бром-2-хлор-3-иодбензола (250 мг, 0,79 ммоль), трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (284 мг, 0,79 ммоль), ТГФ (8 мл) и 0,5M раствора K3PO4 (8 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (62 мг, 0,079 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-100% CH2Cl2/гексан) дала трет-бутил 6-(3-бром-2-хлорфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия b. Смесь трет-бутил 6-(3-бром-2-хлорфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (100 мг, 0,23 ммоль), 2-(3-(3-бромпропокси)-2-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (155 мг, 0,35 ммоль), ТГФ (3 мл) и 0,5M раствора K3PO4 (3 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (18 мг, 0,023 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-100% CH2Cl2/гексан) дала трет-бутил 6-(3'-(3-бромпропокси)-2-хлор-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия c. Раствор трет-бутил 6-(3'-(3-бромпропокси)-2-хлор-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (100 мг, 0,18 ммоль), 2,2-диметилазетидина (31 мг, 0,36 ммоль) и iPr2NEt (94 мкл, 0,54 ммоль) в MeCN (2 мл) перемешивали при 50°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали. Очистка методом флешхроматографии на силикагеле (0-10% MeOH/CH2Cl2) дала трет-бутил 6-(2-хлор-3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия d. трет-бутил 6-(2-хлор-3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (70 мг, 0,12 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2-хлор-2'-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин в виде соли с двумя молекулами ТФУК. 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,66 (ушир.с, 1H), 9,10 (ушир.с, 2H), 7,48 (т, J = 7,6 Гц, 1H), 7,39 (дд, J = 1.8, 7,6 Гц, 1H), 7,36-7,29 (м, 3H), 7,29-7,21 (м, 2H), 6.99 (д, J = 8,2 Гц, 1H), 6,78 (д, J = 7,5 Гц, 1H), 4,37-4,31 (м, 2H), 4,13-4,00 (м, 2H), 3.99-3.86 (м, 2H), 3,47-3,39 (м, 2H), 3,33-3,21 (м, 1H), 3,19-3,09 (м, 1H), 3,05 (т, J = 6,3 Гц, 2H), 2,39-2,29 (м, 1H), 2,14-2,06 (м, 1H), 2,03-1.92 (м, 5H), 1,57 (с, 3H), 1,55 (с, 3H). MS: (ES) m/z вычислено для C30H36ClN2O [M+H]+ 475,3, найдено 475,3.
Пример 9. 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2-метокси-2'-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
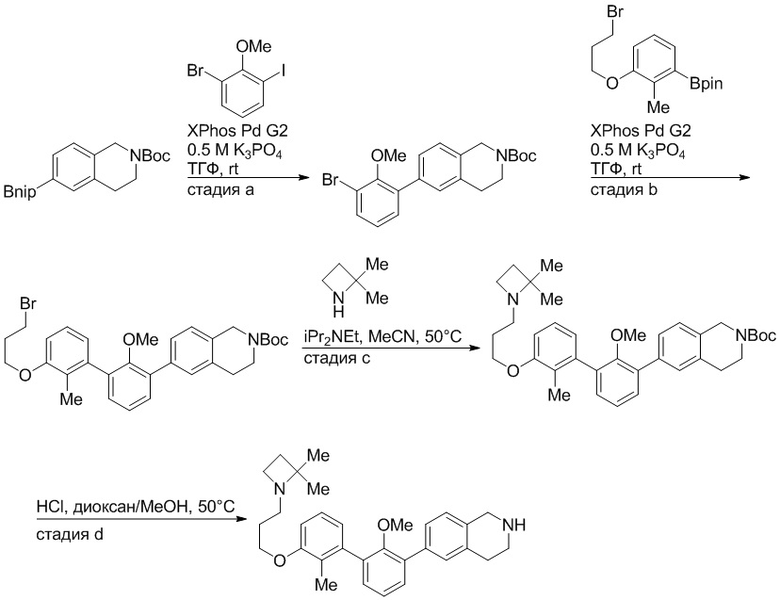
Стадия a. Смесь 2-бром-6-иоданизола (250 мг, 0.80 ммоль), трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (287 мг, 0.80 ммоль), ТГФ (8 мл) и 0,5M раствора K3PO4 (8 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (63 мг, 0,080 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-100% CH2Cl2/гексан) дала трет-бутил 6-(3-бром-2-метоксифенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия b. Смесь трет-бутил 6-(3-бром-2-метоксифенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (80 мг, 0,19 ммоль), 2-(3-(3-бромпропокси)-2-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (128 мг, 0,29 ммоль), ТГФ (2 мл) и 0,5M раствора K3PO4 (2 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (15 мг, 0,019 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-100% CH2Cl2/гексан, затем 0-20% EtOAc/CH2Cl2) дала трет-бутил 6-(3'-(3-бромпропокси)-2-метокси-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия c. Раствор трет-бутил 6-(3'-(3-бромпропокси)-2-метокси-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (60 мг, 0,11 ммоль), 2,2-диметилазетидина (19 мг, 0,22 ммоль) и iPr2NEt (58 мкл, 0,33 ммоль) в MeCN (1 мл) перемешивали при 50°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали. Очистка методом флешхроматографии на силикагеле (0-10% MeOH/CH2Cl2) дала трет-бутил 6-(2-метокси-3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия d. Трет-бутил 6-(2-метокси-3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (40 мг, 0,070 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2-метокси-2'-метил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин в виде соли с двумя молекулами ТФУК. 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,67 (ушир.с, 1H), 9,09 (ушир.с, 2H), 7,47-7,39 (м, 2H), 7,36 (дд, J = 1.8, 7,6 Гц, 1H), 7,32-7,20 (м, 3H), 7,14 (дд, J = 1.8, 7,5 Гц, 1H), 6.97 (д, J = 8,4 Гц, 1H), 6.86 (д, J = 7,6 Гц, 1H), 4,37-4,30 (м, 2H), 4,14-4,01 (м, 2H), 3.99-3.88 (м, 2H), 3,47-3,41 (м, 2H), 3,34-3,22 (м, 1H), 3,19-3,09 (м, 1H), 3,09-3,01 (м, 5H), 2,39-2,29 (м, 1H), 2,15-2,06 (м, 1H), 2,04-1.94 (м, 5H), 1,58 (с, 3H), 1,55 (с, 3H). MS: (ES) m/z вычислено для C31H39N2O2 [M+H]+ 471,3, найдено 471,4.
Пример 10. 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-2-винил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин
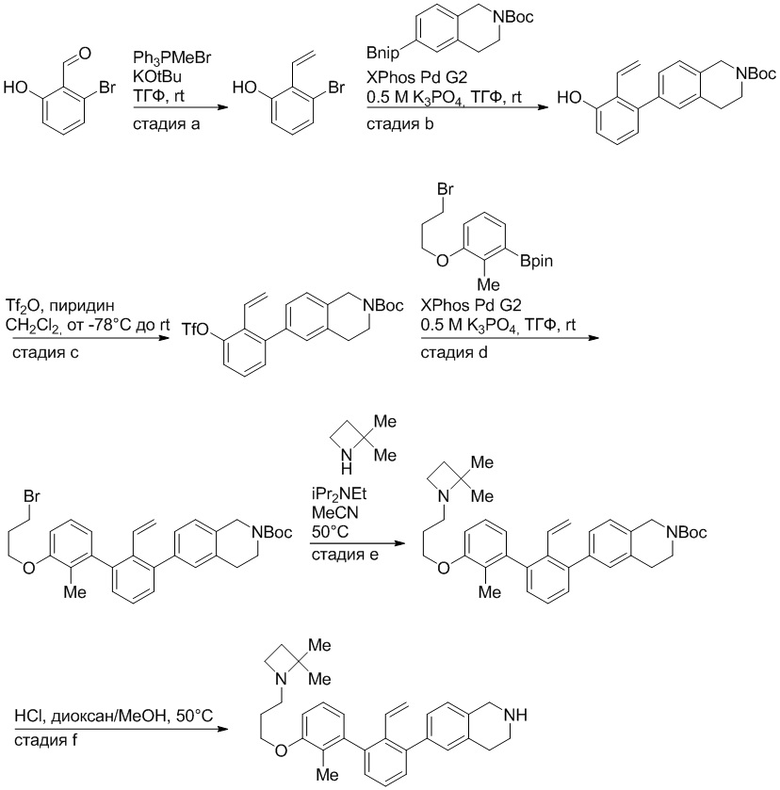
Стадия a. Метилтрифенилфосфоний бромид (4,47 г, 12,5 ммоль) растворяли в ТГФ (20 мл) и добавляли KOtBu (1,0 M раствор в ТГФ, 12,5 мл, 12,5 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Раствор 2-бром-6-гидрокси-бензальдегида (1,01 г, 5,0 ммоль) в ТГФ (5 мл) добавляли по каплям, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили добавлением 1M HCl (50 мл) и экстрагировали этилацетатом (3 x 50 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (50 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-50% EtOAc/гексан) дала 3-бром-2-винилфенол.
Стадия b. Смесь 3-бром-2-винилфенола (299 мг, 1,5 ммоль), трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (647 мг, 1.8 ммоль), ТГФ (15 мл) и 0,5M раствора K3PO4 (15 мл) дегазировали (N2) 10 минут. Добавляли XPhos Pd G2 (118 мг, 0,15 ммоль), смесь дегазировали (N2) еще 10 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (3 x 20 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-60% EtOAc/гексан) дала трет-бутил 6-(3-гидрокси-2-винилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия c. Трет-бутил 6-(3-гидрокси-2-винилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (175 мг, 0,50 ммоль) растворяли в CH2Cl2 (5 мл). Раствор перемешивали при 78°C и добавляли пиридин (0,5 мл), затем по каплям добавляли ангидрид трифторметансульфокислоты (0,17 мл, 1,0 ммоль). Реакционную смесь перемешивали при комнатной температуре 2 часа. Реакцию гасили насыщенным раствором NaHCO3 (10 мл). Слои разделяли, и водный слой экстрагировали дихлорметаном (2 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3-(((трифторметил)сульфонил)окси)-2-винилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия d. Смесь трет-бутил 6-(3-(((трифторметил)сульфонил)окси)-2-винилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (90 мг, 0,19 ммоль), 2-(3-(3-бромпропокси)-2-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (102 мг, 0,23 ммоль), ТГФ (2 мл) и 0,5M раствора K3PO4 (2 мл) дегазировали (N2) 5 минут. Добавляли XPhos Pd G2 (15 мг, 0,019 ммоль), смесь дегазировали (N2) еще 5 минут и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (0-30% EtOAc/гексан) дала трет-бутил 6-(3'-(3-бромпропокси)-2'-метил-2-винил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия e. Раствор трет-бутил 6-(3'-(3-бромпропокси)-2'-метил-2-винил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (80 мг, 0,14 ммоль), 2,2-диметилазетидина (24 мг, 0,28 ммоль) и iPr2NEt (73 мкл, 0,42 ммоль) в MeCN (2 мл) перемешивали при 50°C 3 часа. Реакционную смесь охлаждали до комнатной температуры и упаривали. Очистка методом флешхроматографии на силикагеле (0-10% MeOH/CH2Cl2) дала трет-бутил 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-2-винил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат.
Стадия f. трет-бутил 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-2-винил-[1,1'-бифенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (55 мг, 0,097 ммоль) растворяли в MeOH (0,5 мл), добавляли 4M раствор HCl/диоксан (0,5 мл), и реакционную смесь перемешивали при 50°C в течение 1 часа. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 6-(3'-(3-(2,2-диметилазетидин-1-ил)пропокси)-2'-метил-2-винил-[1,1'-бифенил]-3-ил)-1,2,3,4-тетрагидроизохинолин в виде соли с двумя молекулами ТФУК. 1H-ЯМР (400 МГц, (CD3)2SO) δ 9,51 (ушир.с, 1H), 9,01 (ушир.с, 2H), 7,39 (т, J = 7,6 Гц, 1H), 7,28-7,17 (м, 5H), 7,10 (д, J = 7.9 Гц, 2H), 6.94 (д, J = 8,3 Гц, 1H), 6,77 (д, J = 7,7 Гц, 1H), 6,30 (дд, J = 11,6, 17.8 Гц, 1H), 4.93 (дд, J = 1,6, 11,6 Гц, 1H), 4,59 (дд, J = 1,6, 17.8 Гц, 1H), 4,35-4,29 (м, 2H), 4,12-3.98 (м, 2H), 3.97-3.87 (м, 2H), 3,32-3,18 (м, 2H), 3,18-3,07 (м, 2H), 3,03 (т, J = 6,4 Гц, 2H), 2,38-2,28 (м, 1H), 2,15-2,05 (м, 1H), 2,02-1.92 (м, 2H), 1.87 (с, 3H), 1,56 (с, 3H), 1,54 (с, 3H). MS: (ES) m/z вычислено для C32H39N2O [M+H]+ 467,3, найдено 467,3.
Пример 11. 2,2'-(((3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен))бис(азандиил))бис(этан-1-ол)
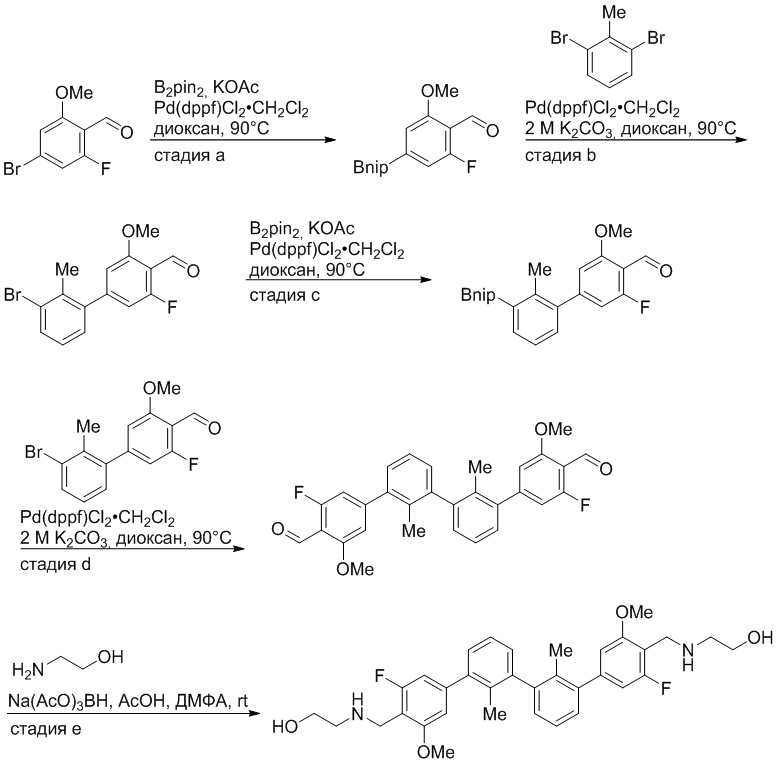
Стадия a. В 2-литровой круглодонной колбе готовили смесь, содержащую 4-бром-2-фтор-6-метоксибензальдегид (22 г, 94 ммоль), бис(пинаколато)диборон (27 г, 106 ммоль), комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (7,4 г, 9,1 ммоль), ацетат калия (32 г, 330 ммоль) и диоксан (460 мл). Смесь тщательно дегазировали (N2) в течение 1 часа, затем перемешивали в атмосфере азота при 90°C в течение 19 часов. После этого диоксан удаляли при пониженном давлении, остаток растворяли в этилацетате и воде и фильтровали через целит. Водную фазу отделяли и отбрасывали. Органическую фазу промывали насыщенным раствором хлорида натрия, наносили на силикагель и очищали методом флешхроматографии на силикагеле (6-15% EtOAc/гексан), получая 2-фтор-6-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегид.
Стадия b. В 200-миллилитровой круглодонной колбе объединяли 2,6-дибромтолуол (6,2 г, 25 ммоль), 2-фтор-6-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегид (4,0 г, 14 ммоль), комплекс [1,1′-бис(дифенилфосфино)ферроцен] дихлорпалладия(II) с дихлорметаном (1,1 г, 1,3 ммоль), 2M раствор K2CO3 (20 мл, 40 ммоль) и диоксан (50 мл). Смесь тщательно дегазировали (N2) 45 минут, затем перемешивали в атмосфере азота при 90°C в течение 2,5 часов. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли диэтиловым эфиром и этилацетатом, и водную фазу отделяли. Полученную органическую фазу очищали методом флешхроматографии на силикагеле (4-16% EtOAc/гексан), получая 3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-карбальдегид.
Стадия c. В 100-миллилитровой круглодонной колбе объединяли 3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-карбальдегид (1,5 г, 4,6 ммоль), бис(пинаколато)диборон (1.9 г, 7,3 ммоль), комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (510 мг, 0,62 ммоль), ацетат калия (1,5 г, 15 ммоль) и диоксан (55 мл). Смесь дегазировали (N2) и перемешивали в атмосфере азота при 90°C в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь разбавляли этилацетатом, эфиром и водой, и фильтровали через целит. Водную фазу отделяли и отбрасывали, и оставшуюся органическую фазу очищали методом флешхроматографии на силикагеле (8-14% EtOAc/гексан), получая 3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-карбальдегид.
Стадия d. В смесь 3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-карбальдегида (500 мг, 1,5 ммоль), 3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-карбальдегида (930 мг, 2,5 ммоль), 2M раствора K2CO3 (2,0 мл, 4,0 ммоль) и диоксана (12 мл) добавляли комплекс [1,1′-бис(дифенилфосфино) ферроцен]дихлорпалладия(II) с дихлорметаном (120 мг, 0,15 ммоль). Смесь дегазировали (N2) и перемешивали при 90°C в течение 5 часов. После охлаждения до комнатной температуры смесь разбавляли этилацетатом, водную фазу отделяли и отбрасывали. Органическую фазу очищали методом флешхроматографии на силикагеле (от 4 до 100% EtOAc/гексан), получая 3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-дикарбальдегид.
Стадия e. В раствор 3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-дикарбальдегида (70 мг, 0,14 ммоль) и уксусной кислоты (0,050 мл, 0.87 ммоль) в ДМФА (2 мл) добавляли 2-аминоэтанол (0,087 мл, 1,4 ммоль). После перемешивания при комнатной температуре в течение 10 минут, добавляли Na(AcO)3BH (180 мг, 0.85 ммоль), и смесь перемешивали при комнатной температуре еще 1,5 часа. Реакционную смесь разбавляли смесью 2:1 хлороформ:изопропанол и водой, водный слой отделяли и отбрасывали. Полученную органическую фазу упаривали при пониженном давлении, и полученный остаток очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая 2,2'-(((3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен))бис(азандиил))бис(этан-1-ол). 1H-ЯМР (400 МГц, CD3OD) δ 7,34 (т, J = 7,6 Гц, 2H), 7,25 (д, J = 7,5 Гц, 2H), 7,18 (д, J = 7,5 Гц, 2H), 6.95-6.83 (м, 4H), 4,36 (с, 4H), 3.98 (с, 6H), 3.88-3.81 (м, 4H), 3,19 (т, J = 5,2 Гц, 4H), 1.97 (с, 6H). MS: (ES) m/z вычислено для C34H39F2N2O4 [M+H]+ 577,3, найдено 577,2.
Пример 12. 2-(((4'''-((диметиламино)метил)-3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4-ил)метил)амино)этан-1-ол

Это соединение было выделено в качестве побочного продукта при получении Примера 11. 1H-ЯМР (400 МГц, CD3OD) δ 7,37-7,15 (м, 6H), 6.98-6.83 (м, 4H), 4,43 (с, 2H), 4,36 (с, 2H), 3.99 (с, 3H), 3.98 (с, 3H), 3.88-3.81 (м, 2H), 3,19 (т, J = 5,3 Гц, 2H), 2.93 (с, 6H), 1.98 (с, 3H), 1.97 (с, 3H). MS: (ES) m/z вычислено для C34H39F2N2O3 [M+H]+ 561,3, найдено 561,2.
Пример 13. N-(4''-(аминометил)-3''-фтор-5''-метокси-2,2'-диметил-[1,1':3',1''-терфенил]-3-ил)пиперидин-4-амин
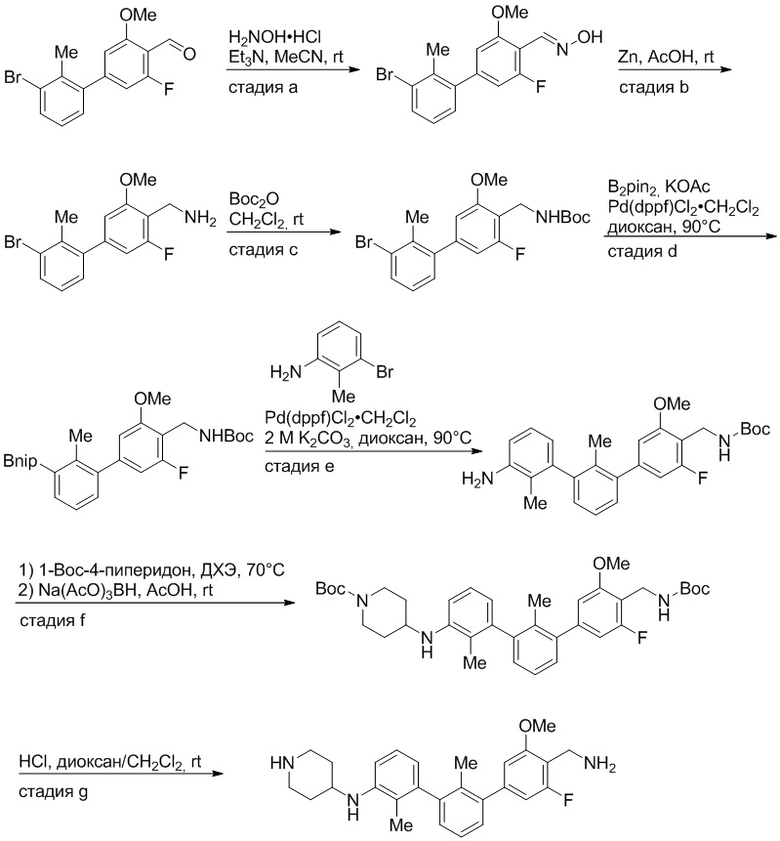
Стадия a. В перемешиваемый раствор 3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-карбальдегида (5,0 г, 15 ммоль) в ацетонитриле (100 мл) добавляли гидроксиламин гидрохлорид (1,6 г, 24 ммоль) и триэтиламин (3,3 мл, 24 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов, затем выливали в насыщенный водный раствор бикарбоната натрия (300 мл). Выпавший в осадок 3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-карбальдегид оксим отделяли фильтрованием частично высушивали и использовали на следующей стадии без очистки.
Стадия b. 3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-карбальдегид оксим перемешивали в уксусной кислоте с образованием суспензии. В нее добавляли цинковую пыль (8,0 г, 120 ммоль), и смесь перемешивали при комнатной температуре 1,5 часа. Реакционную смесь фильтровали через целит для удаления цинка, и уксусную кислоту удаляли при пониженном давлении. Остаток растворяли в смеси 2:1 хлороформ:изопропанол и комбинации насыщенного раствора бикарбоната натрия, 6M NaOH и воды. Водную фазу отделяли, органическую фазу промывали насыщенным раствором хлорида натрия и упаривали, получая вязкое масло неочищенного (3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-ил)метанамина (5,3 г).
Стадия c. В раствор маслянистого остатка с предыдущей стадии в дихлорметане (80 мл) добавляли ди-трет-бутилдикарбонат (6,4 г, 29 ммоль). После перемешивания при комнатной температуре в течение 2 дней, дихлорметан удаляли при пониженном давлении, и остаток очищали методом флешхроматографии на силикагеле (4-80% EtOAc/гексан), получая трет-бутил ((3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-ил)метил)карбамат.
Стадия d. В 500-миллилитровой круглодонной колбе объединяли трет-бутил ((3'-бром-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-ил)метил)карбамат (5,3 г, 13 ммоль), бис(пинаколато)диборон (4,0 г, 16 ммоль), комплекс [1,1′-бис(дифенилфосфино)ферроцен] дихлорпалладия(II) с дихлорметаном (1,3 г, 1,6 ммоль), ацетат калия (3,2 г, 33 ммоль) и диоксан (100 мл). Смесь дегазировали (N2) и перемешивали в атмосфере азота при 90°C 3 часа. После охлаждения до комнатной температуры, примерно половину диоксана удаляли в вакууме, полученную смесь разбавляли этилацетатом и водой и фильтровали через целит. Органическую фазу отделяли, промывали насыщенным раствором хлорида натрия и очищали методом флешхроматографии на силикагеле (10-40% EtOAc/гексан), получая трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-ил)метил)карбамат.
Стадия e. В 40-миллилитровой виале объединяли трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-ил)метил)карбамат (700 мг, 1,5 ммоль), 3-бром-2-метиланилин (340 мг, 1.8 ммоль), комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (140 мг, 0,17 ммоль), 2M раствор K2CO3 (2,0 мл, 4,0 ммоль) и диоксан (12 мл). Смесь тщательно дегазировали (N2), затем перемешивали при 90°C в течение 5 часов. После охлаждения реакционную смесь разбавляли насыщенным раствором хлорида натрия и эфиром. Водную фазу отделяли, органическую фазу очищали методом флешхроматографии на силикагеле (10-34% EtOAc/гексан), получая трет-бутил ((3''-амино-3-фтор-5-метокси-2',2''-диметил-[1,1':3',1''-терфенил]-4-ил)метил)карбамат.
Стадия f. В раствор трет-бутил ((3''-амино-3-фтор-5-метокси-2',2''-диметил-[1,1':3',1''-терфенил]-4-ил)метил)карбамата (40 мг, 0,089 ммоль) в ДХЭ (1 мл) добавляли 1-Boc-4-пиперидон (24 мг, 0,12 ммоль), и реакционную смесь перемешивали при 70°C 1 час. После охлаждения до комнатной температуры добавляли Na(AcO)3BH (87 мг, 0,41 ммоль), затем уксусную кислоту (0,010 мл, 0,17 ммоль). После перемешивания при комнатной температуре в течение 2,5 дней, реакционную смесь растворяли в хлороформе и воде. Водную фазу отделяли, и органическую фазу очищали методом флешхроматографии на силикагеле (4-30% EtOAc/гексан), получая трет-бутил 4-((4''-(((трет-бутоксикарбонил) амино)метил)-3''-фтор-5''-метокси-2,2'-диметил-[1,1':3',1''-терфенил]-3-ил)амино)пиперидин-1-карбоксилат.
Стадия g. В раствор трет-бутил 4-((4''-(((трет-бутоксикарбонил)амино)метил)-3''-фтор-5''-метокси-2,2'-диметил-[1,1':3',1''-терфенил]-3-ил)амино)пиперидин-1-карбоксилата (45 мг, 0,071 ммоль) в дихлорметане (1 мл) добавляли 4M раствор HCl в диоксане (0,40 мл, 1,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Растворители удаляли в вакууме, и полученный остаток очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК в качестве элюента), получая N-(4''-(аминометил)-3''-фтор-5''-метокси-2,2'-диметил-[1,1':3',1''-терфенил]-3-ил)пиперидин-4-амин. 1H-ЯМР (400 МГц, CD3OD) δ 7,29 (т, J = 8,0 Гц, 1H), 7,20 (дд, J = 1,5, 8,0 Гц, 1H), 7,15-7,06 (м, 3H), 6.82 (дд, J = 1,4, 10,0 Гц, 1H), 6,77 (д, J = 8,4 Гц, 1H), 6,52 (д, J = 7,2 Гц, 1H), 4,22 (с, 2H), 3.97 (с, 3H), 3.80-3,69 (м, 1H), 3,48 (д, J = 13,4 Гц, 2H), 3,18 (т, J = 12,1 Гц, 2H), 2,31 (д, J = 14,2 Гц, 2H), 1.91 (с, 3H), 1.88 (с, 3H), 1.83-1,68 (м, 2H). MS: (ES) m/z вычислено для C27H33FN3O [M+H]+ 434,3, найдено 434,2.
Пример 14. 1,1'-((3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен))бис(пиперидин-4-ол)
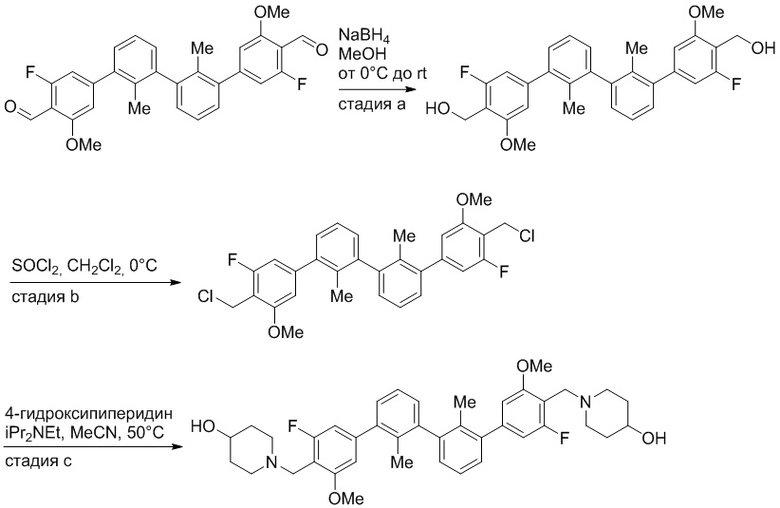
Стадия a. 3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-дикарбальдегид (97 мг, 0,20 ммоль) растворяли в MeOH (2 мл). Раствор перемешивали при 0°C и медленно добавляли боргидрид натрия (30 мг, 0.80 ммоль). Реакционную смесь перемешивали 2,5 часа при постепенно нагревании до комнатной температуры. Реакцию гасили насыщенным раствором NaHCO3 (10 мл) и экстрагировали дихлорметаном (3 x 10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (10 мл), сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле дала (3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)диметанол.
Стадия b. (3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)диметанол (38 мг, 0,077 ммоль) растворяли в CH2Cl2 (2 мл). Раствор перемешивали при 0°C и добавляли по каплям тионилхлорид (34 мкл, 0,46 ммоль). Смесь перемешивали при 0°C в течение 1 часа. Реакционную смесь упаривали, получая 4,4'''-бис(хлорметил)-3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-1,1':3',1'':3'',1'''-кватерфенил, который использовали без дополнительной очистки.
Стадия c. Смесь 4,4'''-бис(хлорметил)-3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-1,1':3',1'':3'',1'''-кватерфенила (40 мг, 0,076 ммоль), 4-гидроксипиперидина (47 мг, 0,46 ммоль), iPr2NEt (0,13 мл, 0,76 ммоль) и MeCN (1 мл) перемешивали при 50°C в течение ночи. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 1,1'-((3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен)) бис(пиперидин-4-ол). 1H-ЯМР (400 МГц, CD3OD) δ 7,32 (т, J = 7,6 Гц, 2H), 7,24 (д, J = 6,6 Гц, 2H), 7,15 (д, J = 7,3 Гц, 2H), 6.80 (с, 2H), 6,73 (д, J = 9,7 Гц, 2H), 3.88 (с, 6H), 3,74 (ушир.с, 4H), 3,61 (ушир.с, 2H), 2.96-2.87 (м, 4H), 2,43-2,32 (м, 4H), 1.98 (с, 6H), 1.90-1.80 (м, 4H), 1,66-1,54 (м, 4H). MS: (ES) m/z вычислено для C40H47F2N2O4 [M+H]+ 657,3, найдено 657,2.
Пример 15. 4,4'-(((3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен))бис(азандиил))дибутановая кислота
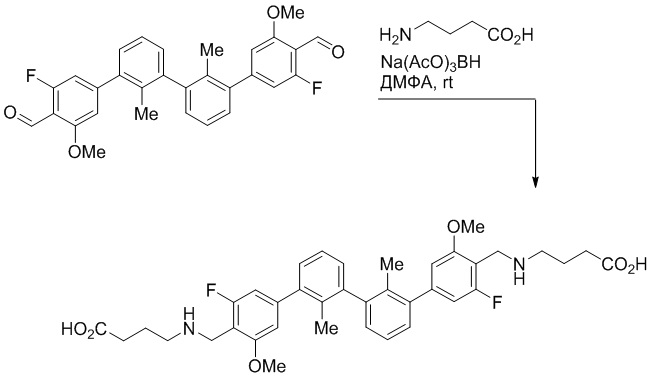
Раствор 3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-дикарбальдегида (36 мг, 0,074 ммоль) и γ-аминомасляной кислоты (46 мг, 0,44 ммоль) перемешивали в ДМФА (3 мл) при комнатной температуре 2 часа, затем добавляли Na(AcO)3BH (50 мг, 0,22 ммоль) порциями в течение 5 минут. Реакционную смесь оставляли перемешиваться в течение ночи при комнатной температуре. Основное количество ДМФА удаляли в вакууме, и сырой продукт снова разбавляли в MeOH и фильтровали. Фильтрат очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая 4,4'-(((3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен))бис(азандиил))дибутановую кислоту. MS: (ES) m/z вычислено для C38H43F2N2O6 [M+H]+ 661,3, найдено 661,2. 1H-ЯМР (400 МГц, CD3OD) δ 7,34 (т, J = 7,6 Гц, 2H), 7,25 (д, J = 7,6 Гц, 2H), 7,22-7,13 (м, 2H), 6.92 (с, 2H), 6.90-6.82 (м, 2H), 4,33 (д, J = 1,2 Гц, 4H), 3.98 (с, 6H), 3,23-3,09 (м, 4H), 2,49 (т, J = 6.9 Гц, 4H), 2,05-1.98 (м, 4H), 1.98 (с, 6H).
Пример 16. (3''-(3-((2R,5R)-2,5-диметилпирролидин-1-ил)пропокси)-2',2''-диметил-[1,1':3',1''-терфенил]-4-ил)метанамин
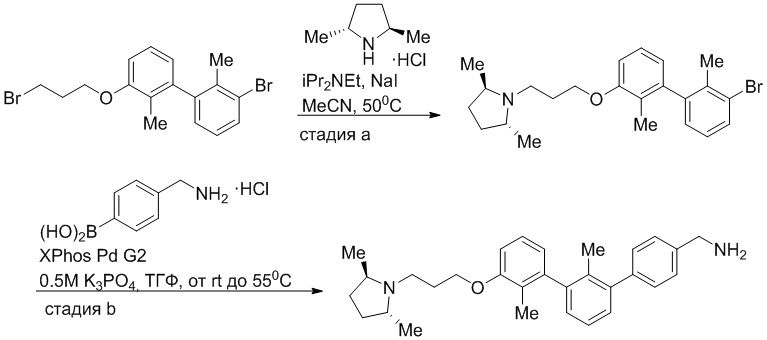
Стадия a. В раствор 3-бром-3'-(3-бромпропокси)-2,2'-диметил-1,1'-бифенила (170 мг, 0,42 ммоль) и (2R, 5R)-2,5-диметилпирролидин гидрохлорида (74 мг, 0,55 ммоль) в MeCN (2 мл) медленно добавляли iPr2NEt (0,30 мл, 1,7 ммоль). Смесь обрабатывали ультразвуком несколько минут, нагревали до 50°C и перемешивали 18 часов. Реакция проходила неполностью, добавляли каталитическое количество иодида натрия (3 мг), и раствор перемешивали еще 8 часов при 50°C. Реакционную смесь упаривали. Сырой продукт очищали методом флешхроматографии на силикагеле (0-5% MeOH/CH2Cl2), получая (2R,5R)-1-(3-((3'-бром-2,2'-диметил-[1,1'-бифенил]-3-ил)окси)пропил)-2,5-диметилпирролидин.
Стадия b. В двухфазную смесь (2R,5R)-1-(3-((3'-бром-2,2'-диметил-[1,1'-бифенил]-3-ил)окси)пропил)-2,5-диметилпирролидина (38 мг, 0,091 ммоль) и гидрохлорида 4-(аминометил)фенилбороновой кислоты (34 мг, 0,18 ммоль) в 0,5M растворе K3PO4 (0.90 мл 0,46 ммоль) и ТГФ (2 мл) добавляли XPhos Pd G2 (14 мг, 0,018 ммоль). Реакционную смесь нагревали при 50°C в течение 12 часов. Смесь оставляли охлаждаться до комнатной температуры, органический слой отделяли, фильтровали и очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая (3''-(3-((2R,5R)-2,5-диметилпирролидин-1-ил)пропокси)-2',2''-диметил-[1,1':3',1''-терфенил]-4-ил)метанамин. MS: (ES) m/z вычислено для C30H39N2O [M+H]+ 443,3, найдено 443,2. 1H-ЯМР (400 МГц, CDCl3) δ 8,70-8,10 (ушир.с, 2H), 7,40 (д, J = 8,0 Гц, 2H), 7,36-7,29 (м, 2H), 7,21-7,04 (м, 4H), 6.83-6,66 (м, 2H), 4,15-3,66 (м, 4H), 3,47-3,22 (м, 2H), 3,22-2.91 (м, 2H), 2,34-2,20 (м, 2H), 2,19 (с, 2H), 1.90 (с, 3H), 1.82 (д, J = 3,0 Гц, 3H), 1,53 (д, J = 6,6 Гц, 2H), 1,30 (д, J = 6.9 Гц, 3H), 1,27 (дд, J = 6.9, 8,6 Гц, 3H).
Пример 17. (3-фтор-5-метокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)диметанамин
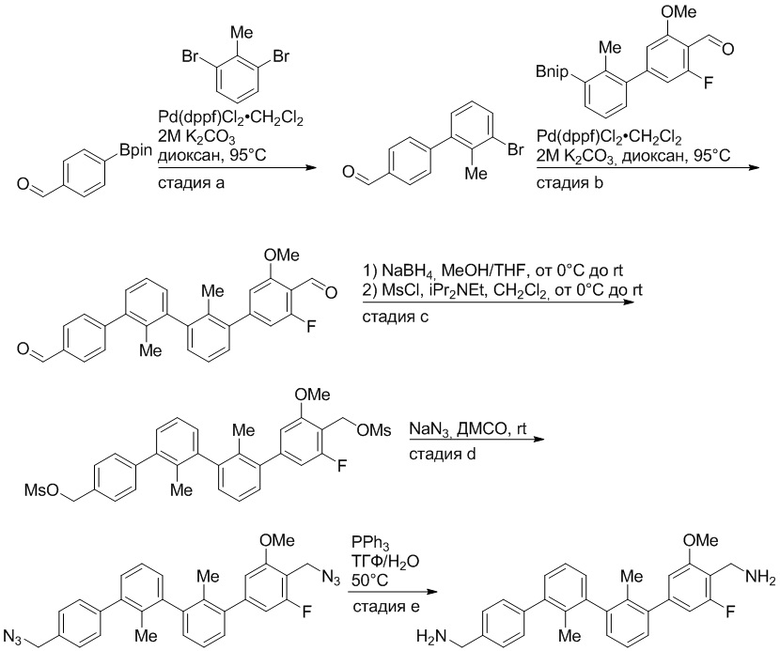
Стадия a. В смесь 1,3-дибром-2-метилбензола (1,6 г, 6,5 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегида (1,0 г, 4,3 ммоль) и 2 M раствора K2CO3 (6,4 мл, 12.9 ммоль) в диоксане (30 мл) добавляли комплекс [1,1′-бис(дифенилфосфино) ферроцен]-дихлорпалладия(II) с дихлорметаном (528 мг, 0,7 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 95°C в течение 5 часов. Смесь разбавляли этилацетатом и фильтровали через целит. Фильтрат промывали насыщенным раствором хлорида натрия, сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (5-40% EtOAc/гексан) дала 3'-бром-2'-метил-[1,1'-бифенил]-4-карбальдегид. 1H-ЯМР (400 МГц, CDCl3) δ 10,07 (с, 1H), 7.95 (дд, J = 6,3, 8,1 Гц, 2H), 7.90-7,68 (м, 1H), 7,67-7,37 (м, 2H), 7,22-7,07 (м, 2H), 2,30 (с, 3H).
Стадия b. В смесь 3'-бром-2'-метил-[1,1'-бифенил]-4-карбальдегида (223 мг, 0.81 ммоль), 3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-карбальдегида (200 мг, 0,54 ммоль) и 2M раствора K2CO3 (0.81 мл, 1,62 ммоль) в диоксане (4 мл) добавляли комплекс [1,1′-бис(дифенилфосфино)ферроцен] дихлорпалладия(II) с дихлорметаном (66 мг, 0,08 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 95°C в течение 5 часов. Реакционную смесь разбавляли этилацетатом и фильтровали через целит. Фильтрат промывали насыщенным раствором хлорида натрия, сушили над MgSO4, фильтровали и упаривали. Очистка методом флешхроматографии на силикагеле (5-40% EtOAc/гексан) дала 3-фтор-5-метокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-дикарбальдегид. 1H-ЯМР (400 МГц, CDCl3) δ 10,46 (с, 1H), 10,08 (с, 1H), 7.99-7.92 (м, 2H), 7,54 (д, J = 8,0 Гц, 2H), 7,37-7,16 (м, 6H), 6,79-6,66 (м, 2H), 3.96 (с, 3H), 2,00 (с, 3H), 1.97 (с, 3H).
Стадия c. 3-фтор-5-метокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-дикарбальдегид (129 мг, 0,29 ммоль) растворяли в 1:1 смеси метанола и тетрагидрофурана (12 мл). Реакционную смесь охлаждали до 0°C и медленно добавляли боргидрид натрия (45 мг, 1,18 ммоль). Реакционную смесь перемешивали при постепенном нагревании до комнатной температуры в течение 2 часов. Реакционную смесь охлаждали до 0°C, разбавляли насыщенным раствором NaHCO3 (50 мл) и экстрагировали хлороформом (3 x 50 мл). Объединенные органические фракции сушили над MgSO4, фильтровали и упаривали, получая (3-фтор-5-метокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)диметанол. Сырой продукт затем растворяли в CH2Cl2 (7 мл) и охлаждали до 0°C, затем добавляли iPr2NEt (0,57 мл, 3,3 ммоль) и метансульфонилхлорид (0,13 мл, 1,6 ммоль). Реакционную смесь перемешивали при постепенном нагревании до комнатной температуры в течение 2 часов. Реакционную смесь охлаждали до 0°C, разбавляли водой (25 мл) и экстрагировали дихлорметаном (3 x 25 мл). Объединенные органические фракции сушили над MgSO4, фильтровали и упаривали, получая (3-фтор-5-метокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен)диметансульфонат.
Стадия d. (3-фтор-5-метокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''- диил)бис(метилен) диметансульфонат (163 мг, 0,27 ммоль) растворяли в диметилсульфоксиде (3 мл) при комнатной температуре. Добавляли азид натрия (88 мг, 1,4 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь охлаждали до 0°C, разбавляли водой (25 мл) и экстрагировали хлорформом (3 x 25 мл). Объединенные органические фракции сушили над MgSO4, фильтровали и упаривали, получая 4,4'''-бис(азидометил)-3-фтор-5-метокси-2',2''-диметил-1,1':3',1'':3'',1'''-кватерфенил.
Стадия e. 4,4'''-бис(азидометил)-3-фтор-5-метокси-2',2''-диметил-1,1':3',1'':3'',1'''-кватерфенил (98 мг, 0,20 ммоль) растворяли в 4:1 смеси тетрагидрофурана и метанола (7,5 мл). Медленно добавляли трифенилфосфин (261 мг, 0.99 ммоль), и полученный раствор перемешивали при 50°C в течение 2,5 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (25 мл) и экстрагировали этилацетатом (3 x 25 мл). Объединенные органические фракции сушили над MgSO4, фильтровали и упаривали. Остаток очищали методом препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая (3-фтор-5-метокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)диметанамин. 1H-ЯМР (400 МГц, CD3OD) δ 7,56-7,49 (м, 2H), 7,43 (д, J = 8,1 Гц, 2H), 7,36-7,28 (м, 2H), 7,25-7,12 (м, 4H), 6.89 (с, 1H), 6.82 (дд, J = 1,4, 9.9 Гц, 1H), 4,23 (с, 2H), 4,18 (с, 2H), 3.97 (с, 3H), 1.97 (с, 3H), 1.94 (с, 3H). MS: (ES) m/z вычислено для C29H27FNO [M-NH2]+ 424,2, найдено 424,2.
Пример 18. (2'',3-дифтор-3'',5-диметокси-2'-метил-[1,1':3',1''-терфенил]-4-ил)метанамин
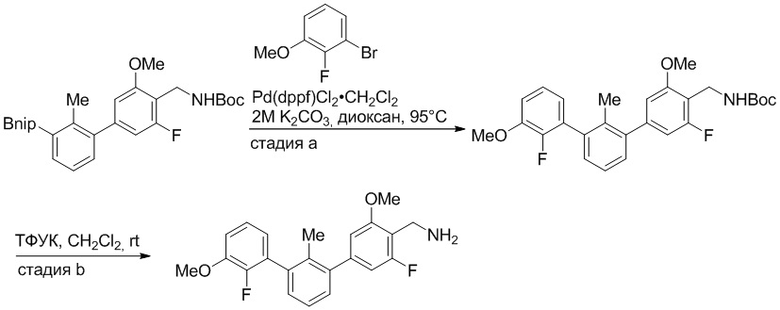
Стадия a. В смесь 1-бром-2-фтор-3-метоксибензола (50 мг, 0,22 ммоль), трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-ил)метил)карбамата (70 мг, 0,15 ммоль), 2M раствора K2CO3 (0,22 мл, 0,45 ммоль) и диоксана (3 мл) добавляли комплекс [1,1′-бис(дифенилфосфино)ферроцен] дихлорпалладия(II) с дихлорметаном (18 мг, 0,02 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 95°C в течение 5 часов. Реакционную смесь разбавляли этилацетатом и фильтровали через целит. Фильтрат промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флешхроматографии на силикагеле (от 5 до 20% до 40% EtOAc/гексан), получая трет-бутил ((2'',3-дифтор-3'',5-диметокси-2'-метил-[1,1':3',1''-терфенил]-4-ил)метил)карбамат. 1H-ЯМР (400 МГц, CDCl3) δ 7,33-7,17 (м, 3H), 7,15-7,10 (м, 1H), 7,03-6.96 (м, 1H), 6.87-6.82 (м, 1H), 6,74-6,63 (м, 2H), 3.93 (с, 3H), 3.86 (с, 3H), 3,70 (с, 2H), 2,04 (с, 3H), 1,45 (с, 9H).
Стадия b. В раствор трет-бутил ((2'',3-дифтор-3'',5-диметокси-2'-метил-[1,1':3',1''-терфенил]-4-ил)метил)карбамата (41 мг, 0,09 ммоль) в CH2Cl2 (4 мл) добавляли трифторуксусную кислоту (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь упаривали, и остаток очищали методом препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая (2'',3-дифтор-3'',5-диметокси-2'-метил-[1,1':3',1''-терфенил]-4-ил)метанамин. 1H-ЯМР (400 МГц, CD3OD) δ 7,35-7,30 (м, 1H), 7,28-7,10 (м, 4H), 6.90 (с, 1H), 6.85-6,77 (м, 2H), 4,23 (с, 2H), 3.97 (с, 3H), 3.91 (с, 3H), 2,03 (с, 3H). MS: (ES) m/z вычислено для C22H19F2O2 [M-NH2]+ 353,1, найдено 353,2.
Пример 19. (3-фтор-5-метокси-2'-метил-3'-(5-метил-2,3-дигидробензо[b][1,4]диоксин-6-ил)-[1,1'-бифенил]-4-ил)метанамин
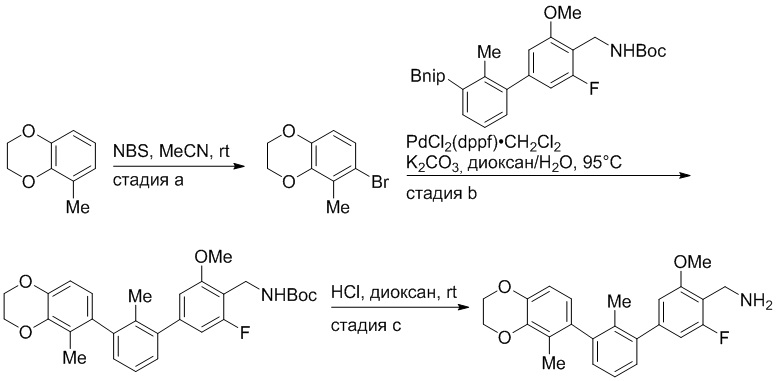
Стадия a. Смесь 5-метил-2,3-дигидробензо[b][1,4]диоксина (0,100 г, 0,67 ммоль), NBS (0,118 г, 0,67 ммоль) в ацетонитриле (2,5 мл) перемешивали при комнатной температуре 30 минут. Растворитель удаляли при пониженном давлении, и остаток очищали методом флешхроматографии на силикагеле (0-30% EtOAc/гексан), получая 6-бром-5-метил-2,3-дигидробензо[b][1,4]диоксин. MS: (ES) m/z вычислено для C9H8BrO2 [M-H]-, 227,0, найдено 227,0.
Стадия b. Смесь 6-бром-5-метил-2,3-дигидробензо[b][1,4]диоксина (0,044 г, 0,19 ммоль), трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-ил)метил)карбамата (0,060 мг, 0,13 ммоль), K2CO3 (0,053 г, 0,38 ммоль), комплекса [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (0,035 г, 0,043 ммоль) в парадиоксане (1,7 мл) и воде (0,30 мл) дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 95°C в течение 1,5 часов Смесь разбавляли этилацетатом и фильтровали через целит/Na2SO4. Фильтрат упаривали при пониженном давлении, и остаток очищали методом флешхроматографии на силикагеле (0-70% EtOAc/гексан), получая трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(5-метил-2,3-дигидробензо[b][1,4]диоксин-6-ил)-[1,1'-бифенил]-4-ил)метил)карбамат. MS: (ES) m/z вычислено для C29H32FNNaO5 [M + Na]+ 516,2, найдено 516,2.
Стадия c. Смесь трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(5-метил-2,3-дигидробензо[b][1,4]диоксин-6-ил)-[1,1'-бифенил]-4-ил)метил)карбамата (0,040 г, 0,080 ммоль) перемешивали в HCl (0,70 мл, 4M/диоксан) в течение 30 минут. Реакционную смесь затем упаривали при пониженном давлении. Остаток очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая (3-фтор-5-метокси-2'-метил-3'-(5-метил-2,3-дигидробензо[b][1,4]диоксин-6-ил)-[1,1'-бифенил]-4-ил)метанамин). 1H-ЯМР (400 МГц, CD3OD) δ 7,27 (т, J = 7,6 Гц, 1H), 7,18 (дд, J = 1,6, 7,2 Гц, 1H), 7,08 (дд, J = 1,6, 7,6, Гц 1H), 6.88 (с, 1H), 6.81 (дд, J = 1,2, 10 Гц, 1H), 6,71 (дд, J = 0.8, 8,4 Гц, 1H), 6,55 (д, J = 8,0 Гц, 1H), 4,32-4,29 (м, 2H), 4,26-4,23 (м, 2H), 4,22 (с, 2H), 3.96 (с, 3H), 1.91 (с, 3H), 1.89 (с, 3H). MS: (ES) m/z вычислено для C24H22FO3 [M-NH2]+ 377,2, найдено 377,2.
Пример 20. (3-фтор-5-метокси-2',2''-диметил-3''-(пиридин-2-илокси)-[1,1':3',1''-терфенил]-4-ил)метанамин
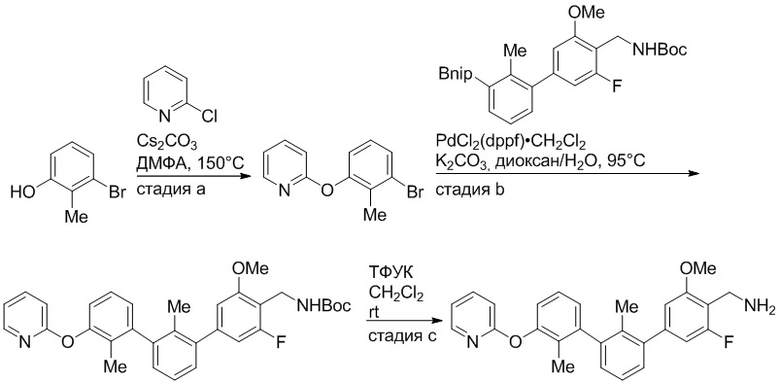
Стадия a. В перемешиваемый раствор 3-бром-2-метилфенола (1,0 г, 5,34 ммоль) в безводном ДМФА (10 мл) добавляли 2-хлорпиридин (0,78 г, 5.88 ммоль) и карбонат цезия (2,6 г, 8,01 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 150°C в течение 24 часов. Реакционную смесь затем выливали в воду (25 мл) и экстрагировали этилацетатом (2 x 50 мл). Объединенные органические слои упаривали, и полученный остаток очищали методом флешхроматографии на силикагеле (10-40% EtOAc/гексан), получая 2-(3-бром-2-метилфенокси)пиридин.
Стадия b. В смесь 2-(3-бром-2-метилфенокси)пиридина (75 мг, 0,24 ммоль), трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-ил)метил)карбамата (133 мг, 0,28 ммоль), 2M раствора K2CO3 (0,35 мл, 0,71 ммоль) и диоксана (10 мл) добавляли комплекс [1,1′-бис(дифенилфосфино)ферроцен] дихлорпалладия(II) с дихлорметаном (23 мг, 0,028 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 95°C в течение 5 часов. Реакционную смесь разбавляли этилацетатом и фильтровали через целит. Фильтрат промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флешхроматографии на силикагеле (5-100% EtOAc/гексан), получая трет-бутил ((3-фтор-5-метокси-2',2''-диметил-3''-(пиридин-2-илокси)-[1,1':3',1''-терфенил]-4-ил)метил)карбамат. MS: (ES) m/z вычислено для C32H33FN2O4 [M + H]+ 529,2, найдено 529,0.
Стадия c. В перемешиваемый раствор трет-бутил ((3-фтор-5-метокси-2',2''-диметил-3''-(пиридин-2-илокси)-[1,1':3',1''-терфенил]-4-ил)метил)карбамата (50 мг, 0,09 ммоль) в безводном дихлорметана (2,5 мл) при комнатной температуре добавляли трифторуксусную кислоту (107 мг, 0.94 ммоль) по каплям в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре 2 часа. После окончания реакции растворитель удаляли в вакууме, и остаток очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая (3-фтор-5-метокси-2',2''-диметил-3''-(пиридин-2-илокси)-[1,1':3',1''-терфенил]-4-ил)метанамин. 1H-ЯМР (400 МГц, CD3OD) δ 8,12 (дд, J = 2,2, 4.9 Гц, 1H), 7.83 (ддд, J = 1.9, 2,2, 6,6 Гц, 1H), 7,45-7,30 (м, 2H), 7,27-7,16 (м, 2H), 7,14-7,02 (м, 3H), 6.95-6,79 (м, 3H), 4,22 (с, 2H), 3.96 (с, 3H), 1.96 (с, 3H), 1.89 (с, 3H). MS: (ES) m/z вычислено для C27H23FNO2 [M-NH2]+ 412,2, найдено 412,0.
Пример 21. (3'-(3-(аминометил)-2,3-дигидробензо[b][1,4]диоксин-6-ил)-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-ил)метанамин

Стадия a: В перемешиваемый раствор 4-бромбензол-1,2-диола (5,0 г, 26,4 ммоль) в безводном ацетоне (100 мл) добавляли 2-хлоракрилонитрил (3,0 мл, 26,4 ммоль) и карбонат калия (7,3 г, 52.9 ммоль) при комнатной температуре. Смесь нагревали при 75°C в течение 24 часов. После окончания реакции растворитель удаляли. Остаток выливали в воду (150 мл) и экстрагировали этилацетатом (150 мл). Органический слой упаривали, и сырой продукт очищали методом флешхроматографии на силикагеле (10-40% EtOAc/гексан), получая 7-бром-2,3-дигидробензо[b][1,4]диоксин-2-карбонитрил.
Стадия b. В смесь 7-бром-2,3-дигидробензо[b][1,4]диоксин-2-карбонитрила (125 мг, 0,52 ммоль), трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-ил)метил)карбамата (245 мг, 0,52 ммоль), 2M раствора K2CO3 (0,65 мл, 1,30 ммоль) и диоксана (4 мл) добавляли комплекс [1,1′-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II) с дихлорметаном (42 мг, 0,052 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 95°C в течение 4 часов. Реакционную смесь разбавляли этилацетатом и фильтровали через целит. Фильтрат промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флешхроматографии на силикагеле (5-40% EtOAc/гексан), получая трет-бутил ((3'-(3-циано-2,3-дигидробензо[b][1,4]диоксин-6-ил)-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-ил)метил)карбамат. MS: (ES) m/z вычислено для C29H29FN2O5 [M + H]+ 505,21, найдено 505,2
Стадия c. В раствор трет-бутил ((3'-(3-циано-2,3-дигидробензо[b][1,4]диоксин-6-ил)-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-ил)метил)карбамата (100 мг, 0,19 ммоль) в MeOH (10 мл) добавляли никель Ренея (примерно 200 мг). Реакционную смесь гидрировали под давлением 50 фунт/кв.дюйм в течение5 часов. После окончания реакции смесь фильтровали через целит, промывали смесью 1:1 EtOAc/MeOH (50 мл) и упаривали досуха. Остаток растворяли в безводном дихлорметане (4 мл) при комнатной температуре и добавляли по каплям трифторуксусную кислоту (141 мг, 1,23 ммоль) в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре 2 часа. После окончания реакции растворитель удаляли в вакууме, и остаток очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая (3'-(3-(аминометил)-2,3-дигидробензо[b][1,4]диоксин-6-ил)-3-фтор-5-метокси-2'-метил-[1,1'-бифенил]-4-ил)метанамин. 1H-ЯМР (400 МГц, CD3OD) δ 7,33-7,14 (м, 3H), 7,07-6.92 (м, 2H), 6.91-6,78 (м, 3H), 4,55-4,45 (м, 1H), 4,39 (дд, J = 2,3, 11.9 Гц, 1H), 4,22 (с, 2H), 4,07 (дд, J = 6.9, 11.9 Гц, 1H), 3.96 (с, 3H), 3,37 (дд, J = 3,2, 13,5 Гц, 1H), 3,32 (дд, J = 3,2, 13,5 Гц, 1H), 2,12 (с, 3H). MS: (ES) m/z вычислено для C24H23FNO3 [M-NH2]+ 392,2, найдено 392,1.
Пример 22. 7-(4'-(аминометил)-3'-фтор-5'-метокси-2-метил-[1,1'-бифенил]-3-ил)-3,4-дигидрохинолин-2(1H)-он
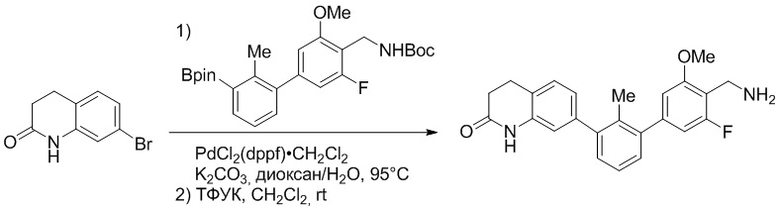
Данное соединение было получено из 7-бром-3,4-дигидрохинолин-2(1H)-она и трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-ил)метил)карбамата по методике из Примера 21 (стадия b и стадия c). Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 7-(4'-(аминометил)-3'-фтор-5'-метокси-2-метил-[1,1'-бифенил]-3-ил)-3,4-дигидрохинолин-2(1H)-он в виде белого твердого вещества. 1H-ЯМР (400 МГц, CD3OD) δ 7.93-7.87 (м, 1H), 7,52-7,45 (м, 1H), 7,43-7,19 (м, 4H), 6.92 (д, J = 1,4 Гц, 1H), 6.85 (д, J = 9.9 Гц, 1H), 4,22 (с, 2H), 3.97 (с, 3H), 3,55 (т, J = 6,7 Гц, 2H), 3,05 (т, J = 6,7 Гц, 2H), 2,10 (с, 3H). MS: (ES) m/z вычислено для C24H24FN2O2 [M+H]+ 391,2, найдено 391,2.
Пример 23. Метил 6-(4'-(аминометил)-3'-фтор-5'-метокси-2-метил-[1,1'-бифенил]-3-ил)-2H-хромен-3-карбоксилат
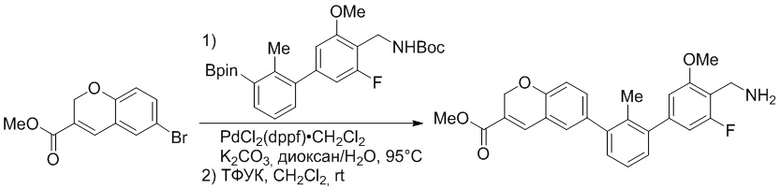
Данное соединение было получено из метил 6-бром-2H-хромен-3-карбоксилата и трет-бутил ((3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-ил)метил)карбамата по методике из Примера 21 (стадия b и стадия c). Продукт очищали методом обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт метил 6-(4'-(аминометил)-3'-фтор-5'-метокси-2-метил-[1,1'-бифенил]-3-ил)-2H-хромен-3-карбоксилат в виде белого твердого вещества. 1H-ЯМР (400 МГц, CD3OD) δ 7,55 (с, 1H). 7,34-7,15 (м, 5H), 6.94-6.80 (м, 3H), 5,00 (с, 2H), 4,22 (с, 2H), 3.97 (с, 3H), 3.81 (с, 3H), 2,10 (с, 3H). MS: (ES) m/z вычислено для C26H25FNO4 [M+H]+ 434,2, найдено 434,1
Пример 24. 2,2'-(((3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен))бис(азандиил))бис(этан-1-сульфокислота)
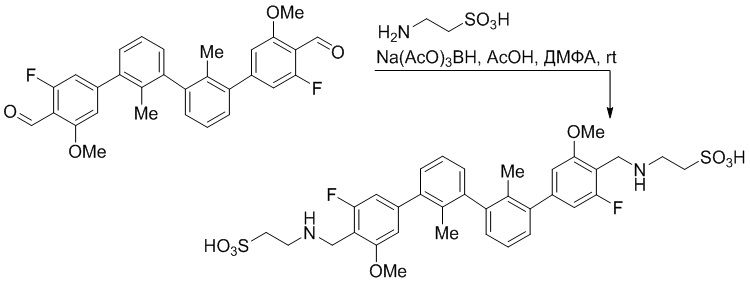
В перемешиваемый раствор 3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-дикарбальдегида (65 мг, 0,133 ммоль), и 2-аминоэтан-1-сульфокислоты (10 мг, 0,16 ммоль) в ДМФА (2 мл) добавляли Na(AcO)3BH (52 мг, 0,24 ммоль) и AcOH (5 капель). Реакционную смесь перемешивали при комнатной температуре 16 часов. Растворитель удаляли при пониженном давлении, и остаток очищали методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК), получая 2,2'-(((3,3'''-дифтор-5,5'''-диметокси-2',2''-диметил-[1,1':3',1'':3'',1'''-кватерфенил]-4,4'''-диил)бис(метилен)) бис(азандиил))-бис(этан-1-сульфокислоту). 1H-ЯМР (400 МГц, CD3OD) δ 7,33 (т, J = 7,6 Гц, 2H), 7,25 (дд, J = 1,5, 7,6 Гц, 2H), 7,21-7,14 (м, 2H), 6.96-6.81 (м, 4H), 4,39 (с, 4H), 3.98 (с, 6H) 3,49 (д, J = 6,3 Гц, 4H) 3,15 (т, J = 6,3 Гц, 4H) 1.97 (с, 6H). MS: (ES) m/z вычислено для C34H39F2N2O8S2 [M+H]+ 705,2, найдено 705,2.
Пример 25. 2-(((3-фтор-5-метокси-2',2''-диметил-3''-(1,2,3,4-тетрагидроизохинолин-6-ил)-[1,1':3',1''-терфенил]-4-ил)метил)амино)этан-1-ол
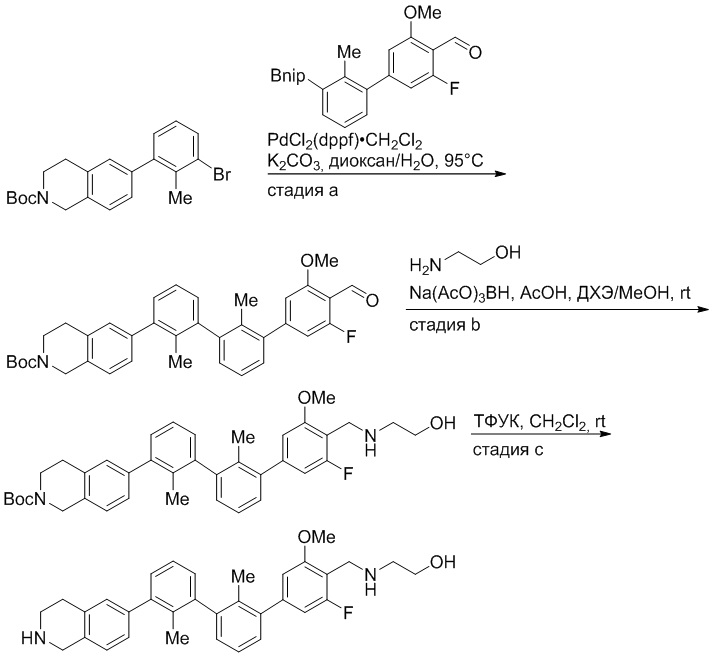
Стадия a. В смесь трет-бутил 6-(3-бром-2-метилфенил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (100 мг, 0,249 ммоль), 3-фтор-5-метокси-2'-метил-3'-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1'-бифенил]-4-карбальдегида (101 мг, 0,27 ммоль), 2M раствора K2CO3 (0,35 мл, 0,024 ммоль) и диоксана (4 мл) добавляли комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1 мг, 0,148 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 95°C в течение 5 часов. Смесь разбавляли этилацетатом и фильтровали через целит. Фильтрат промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флешхроматографии на силикагеле (5-100% EtOAc/гексан), получая трет-бутил 6-(3''-фтор-4''-формил-5''-метокси-2,2'-диметил-[1,1':3',1''-терфенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (100 мг). MS: (ES) m/z вычислено для C36H36FNO4 [M+H]+ 566,3, найдено 566,2
Стадия b. В перемешиваемый раствор трет-бутил 6-(3''-фтор-4''-формил-5''-метокси-2,2'-диметил-[1,1':3',1''-терфенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (70 мг, 0,082 ммоль) и 2-аминоэтанола (30 мг, 0,16 ммоль) в MeOH/ДХЭ (2 мл) добавляли Na(AcO)3BH (65 мг, 0,24 ммоль) и AcOH (5 капель). Реакционную смесь перемешивали при комнатной температуре 2 часа. Растворитель удаляли при пониженном давлении, получая сырой трет-бутил 6-(3''-фтор-4''-(((2-гидроксиэтил)амино)метил)-5''-метокси-2,2'-диметил-[1,1':3',1''-терфенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат, который использовали без дополнительной очистки.
Стадия c. В перемешиваемый раствор трет-бутил 6-(3''-фтор-4''-(((2-гидроксиэтил)амино)метил)-5''-метокси-2,2'-диметил-[1,1':3',1''-терфенил]-3-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (70 мг, 0,114 ммоль) в безводном дихлорметане (2,5 мл) при комнатной температуре добавляли трифторуксусную кислоту (130 мг, 1,14 ммоль) по каплям в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре 2 часа. После окончания реакции растворитель удаляли в вакууме. Очистка методом обращенно-фазовой препаративной ВЭЖХ (H2O/MeCN с 0,1% ТФУК) дала 2-(((3-фтор-5-метокси-2',2''-диметил-3''-(1,2,3,4-тетрагидроизохинолин-6-ил)-[1,1':3',1''-терфенил]-4-ил)метил)амино)этан-1-ол. 1H-ЯМР (400 МГц, CD3OD) δ 7,38-7,09 (м, 9H), 6.94-6.82 (м, 2H), 4,41 (с, 2H), 4,36 (с, 2H), 3.98 (с, 3H), 3.84 (дд, J = 1,4, 6,1 Гц, 2H), 3,54 (т, J = 6,4 Гц, 2H), 3,18 (м, 4H), 1.97 (с, 3H), 1.93 (с, 3H). MS: (ES) m/z вычислено для C33H36FN2O2 [M + H]+ 511,3, найдено 511,2.
Условия, используемые при характеризации соединений
Условия проведения обращенно-фазовой ВЭЖХ для определения времени удерживания, указанного в таблице 1:
Колонка: ZORBAX (SB-C18 2,1 x 50 мм, 5 мкм)
Подвижная фаза A: 95% H2O, 5% MeCN (с 0,1% муравьиной кислоты)
Подвижная фаза B: 5% H2O, 95% MeCN (с 0,1% муравьиной кислоты)
Скорость потока: 1,0 мл/мин
Градиент: от 20 до 100% фазы B за 3,5 мин (Метод A) или от 0 до 100% B за 4,5 мин (Метод B)
Биологический пример: Твердофазный иммуноферментный анализ (ИФА) - ELISA
96-луночные планшеты покрывали 1 мкг/мл раствором человеческого PD-L1 (получено от R&D) в фосфатно-солевом буфере (PBS) на ночь при 4°C. Затем лунки блокировали 2%-ным BSA в PBS (вес/об) с 0,05 % TWEEN-20 в течение 1 часа при 37°C. Затем планшеты промывали 3 раза раствором PBS/0,05% TWEEN-20, тестируемые соединения подвергали серийным разведениям (1:5) в среде и добавляли в ELISA планшеты. Добавляли человеческий PD-1 и биотин 0,3 мкг/мл (ACRO Biosystems) и инкубировали 1 час при 37°C, затем промывали 3 раза раствором PBS/0,05% TWEEN-20. Второе блокирование проводили с помощью 2% BSA в PBS (вес/об)/0,05% TWEEN-20 в течение 10 минут при 37°C, и планшеты промывали 3 раза раствором PBS/0,05% TWEEN-20. Добавляли стрептавидин-HRP на 1 час при 37°C, затем планшеты промывали 3 раза раствором PBS/0,05% TWEEN-20. Добавляли TMB субстрат и проводили реакцию 20 минут при 37°C. Добавляли стоп-раствор (2н. водный раствор H2SO4). Считывали поглощение при 450 нм с помощью спектрофотометра для микропланшетов. Полученные результаты приведены в таблице 1. Значения IC50 приведены в следующем виде: от 1000 до 10000 нM (+); меньше 1000 нM (++).
Таблица 1
ES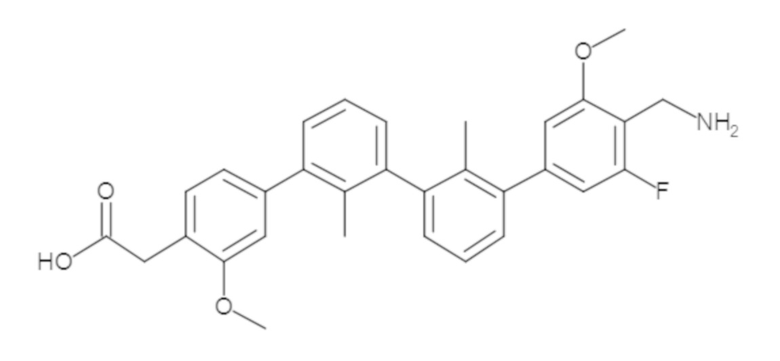
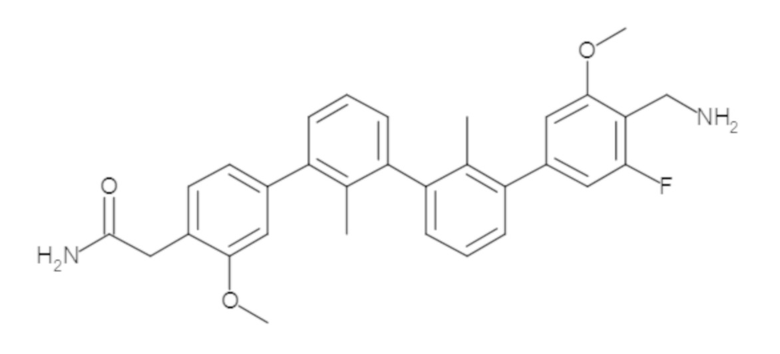
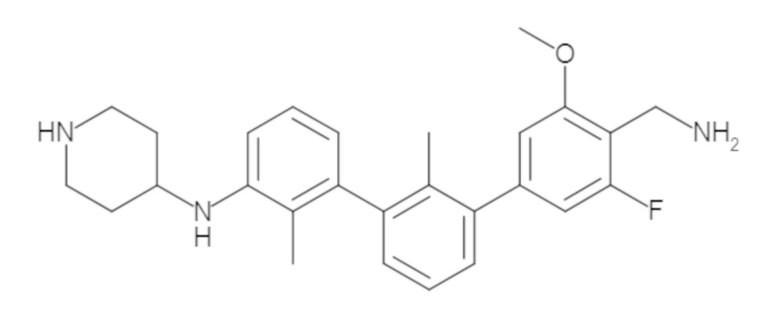
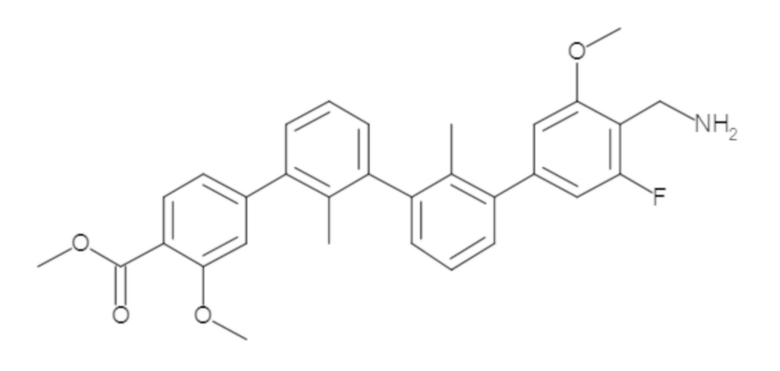
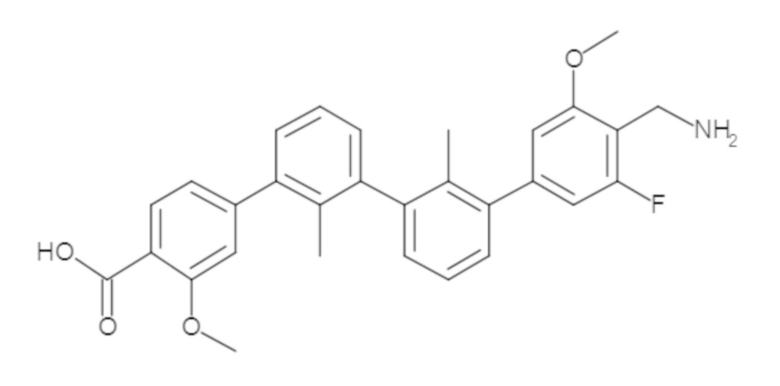
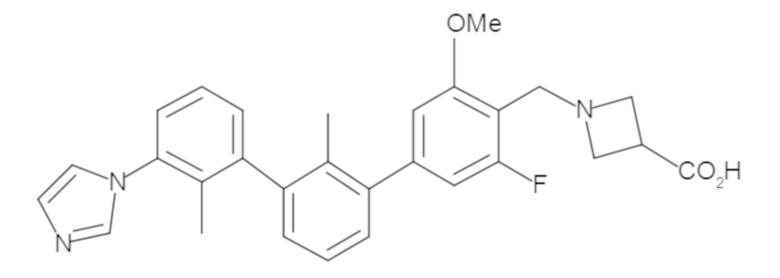
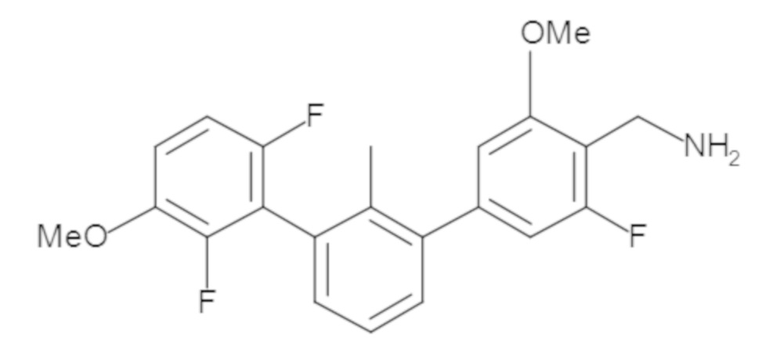
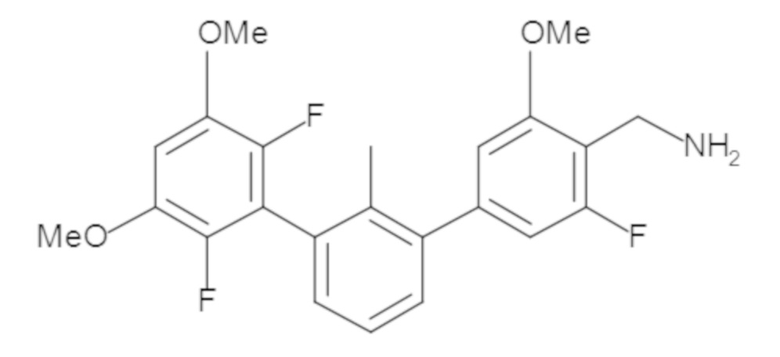
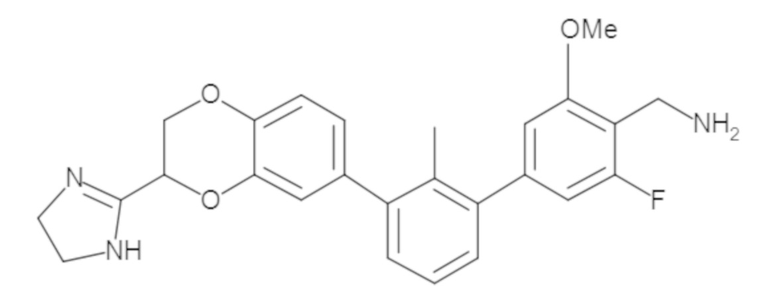
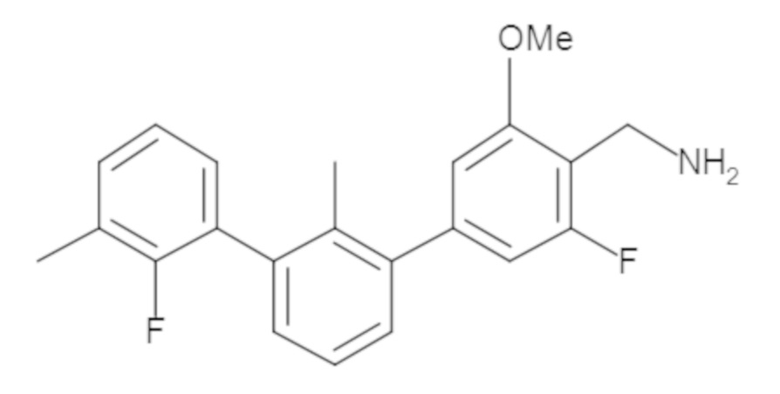
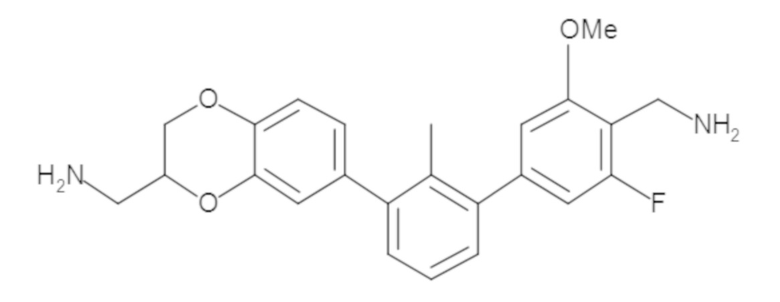
2,0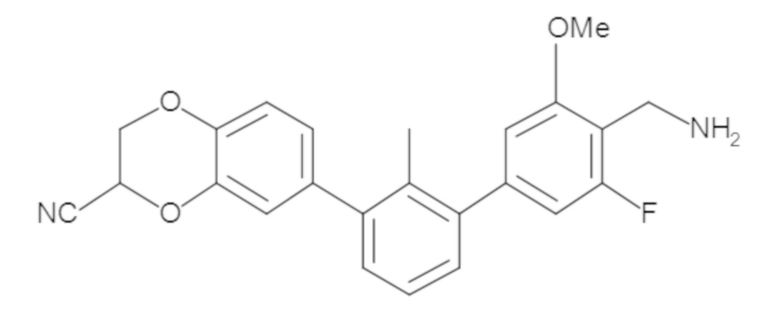
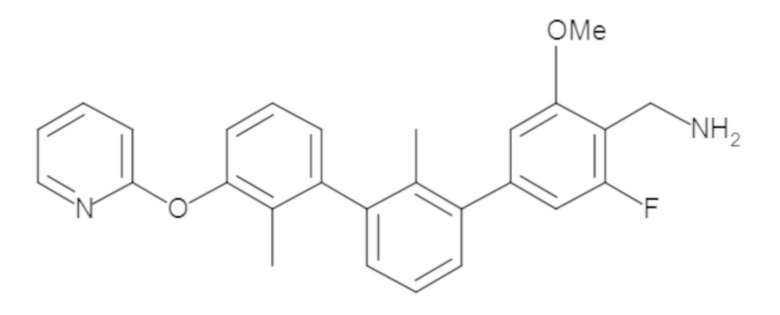
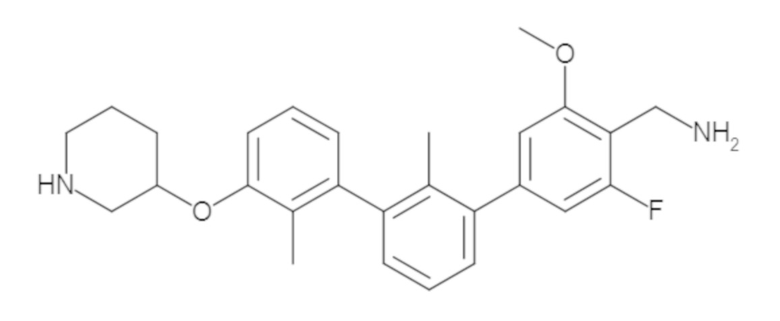
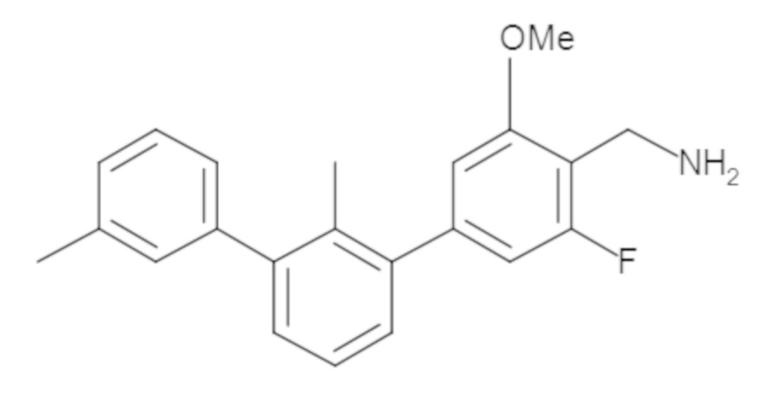
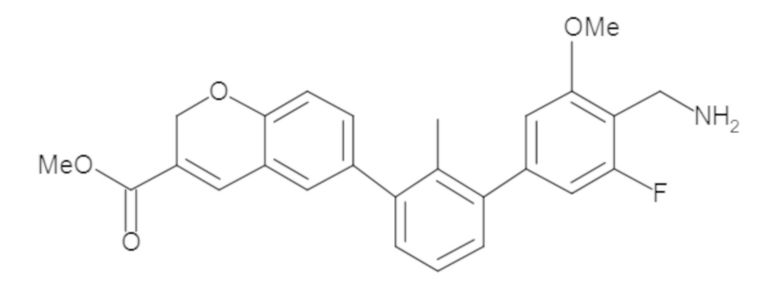
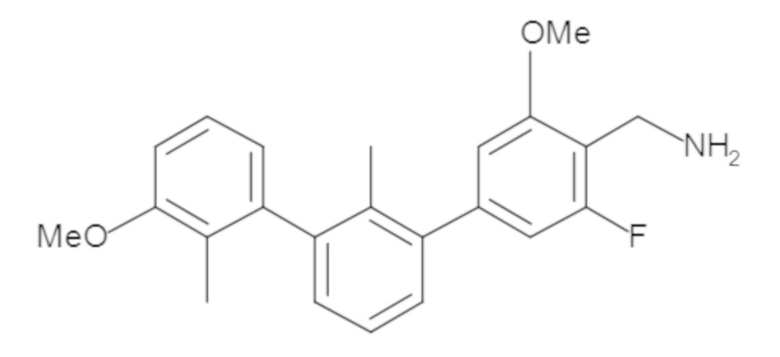
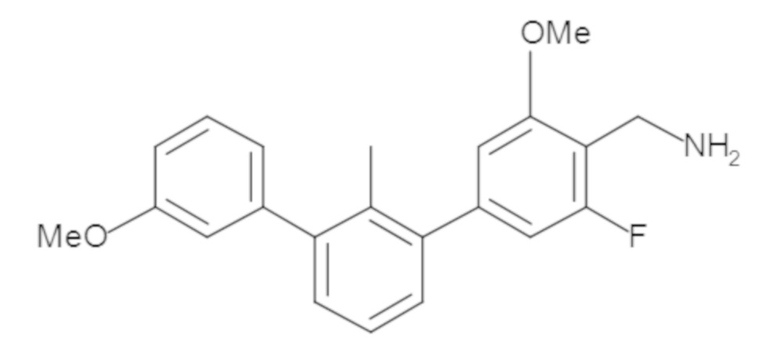

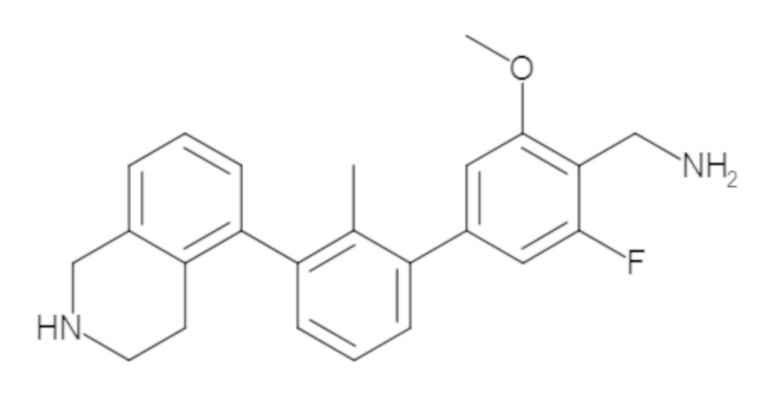
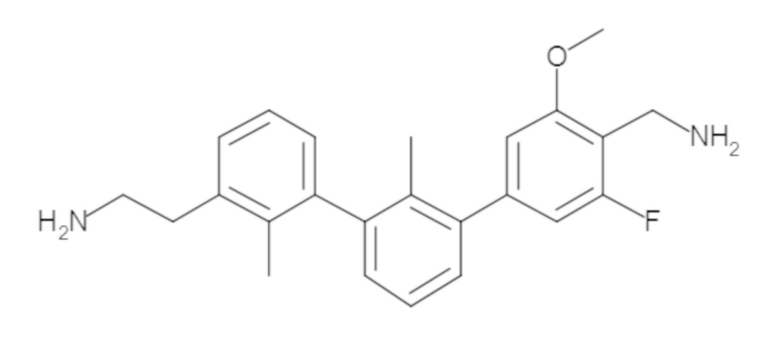
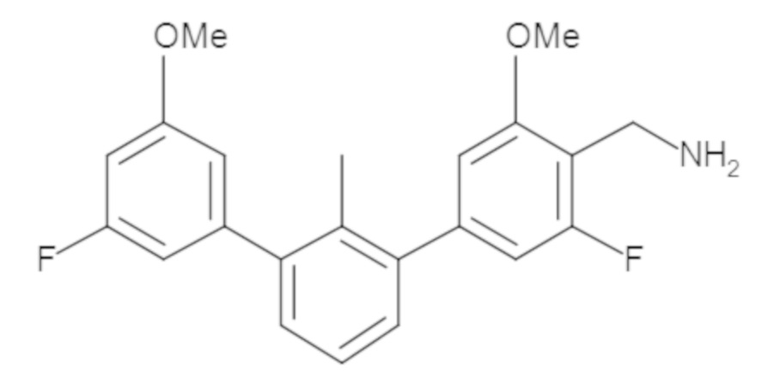
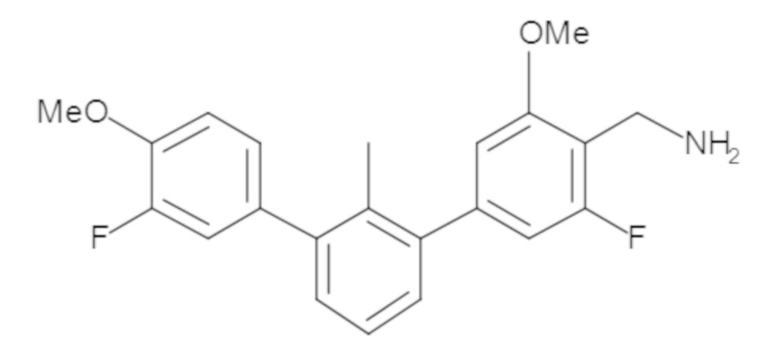
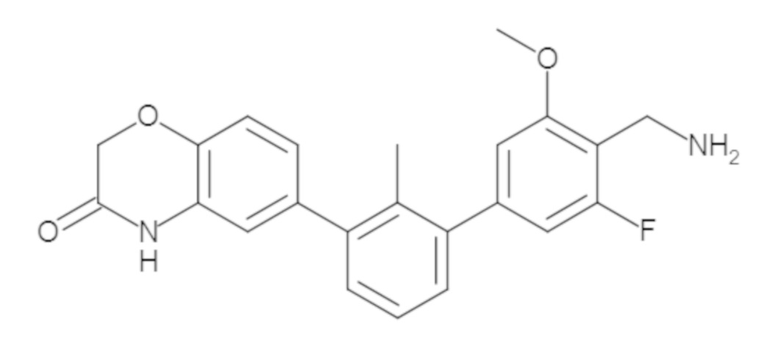
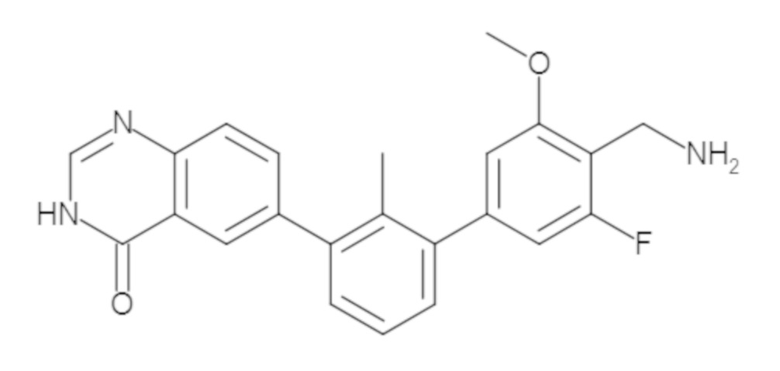
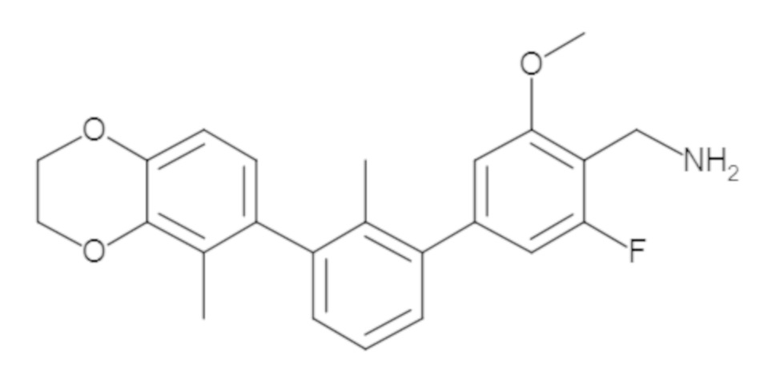
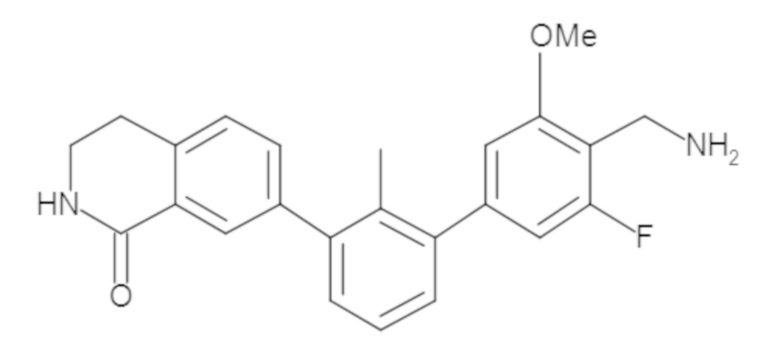
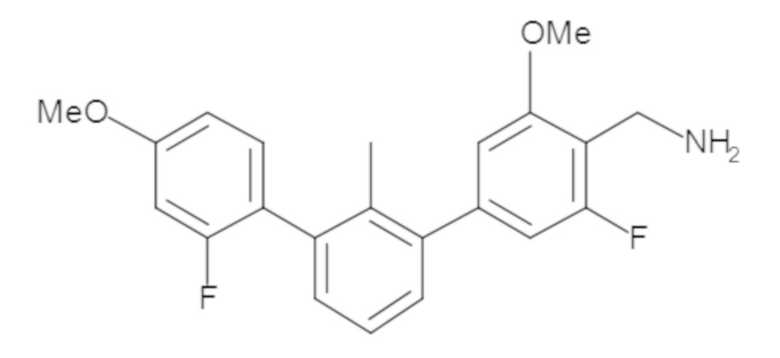
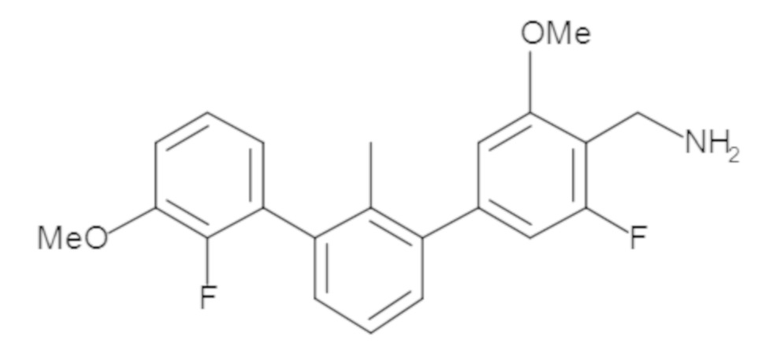
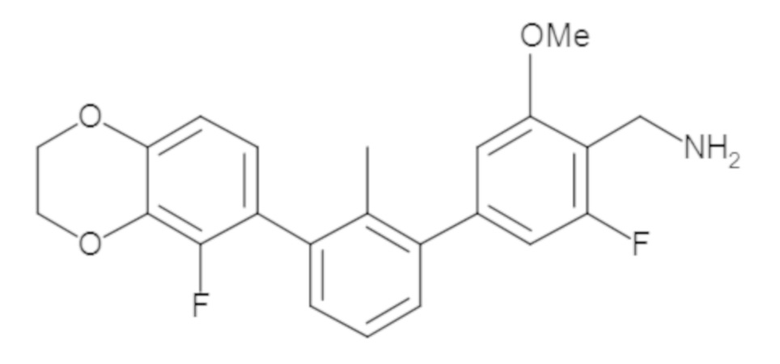
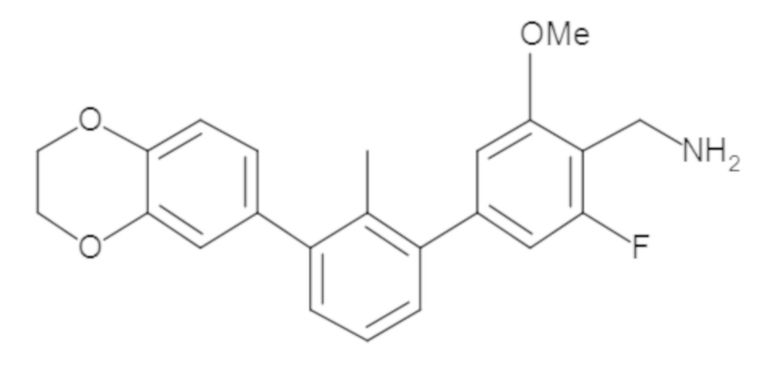
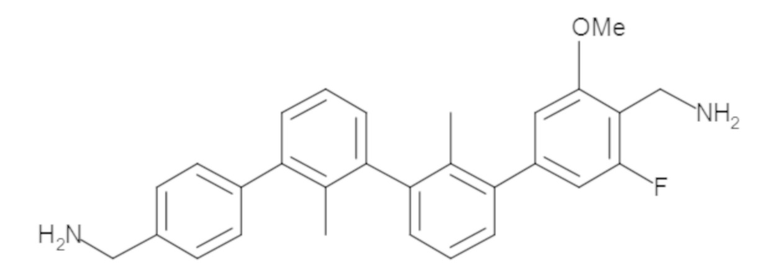
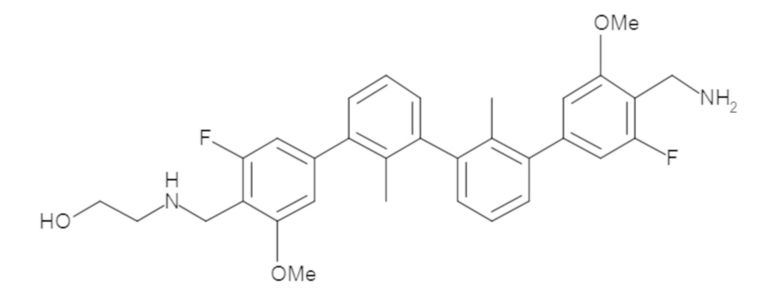

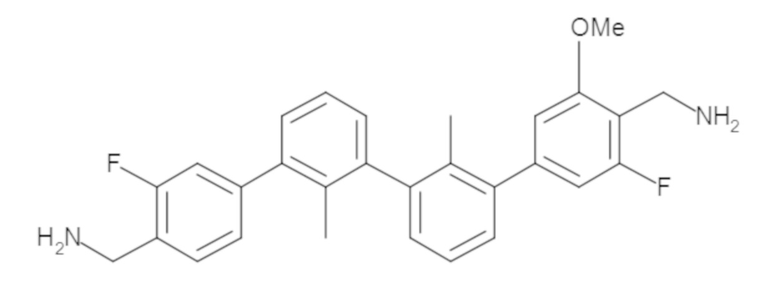

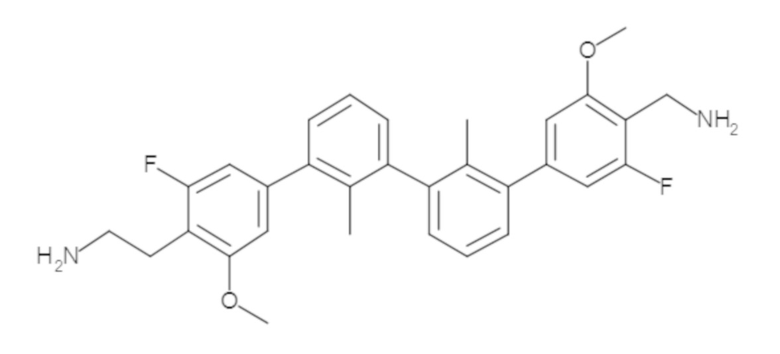
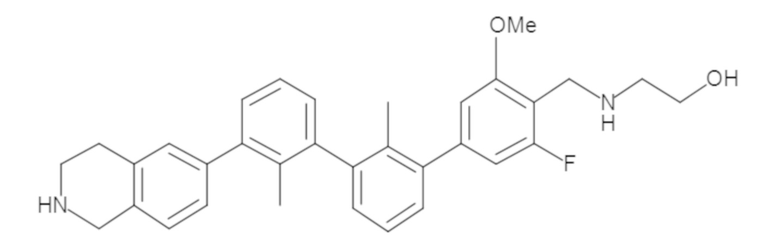
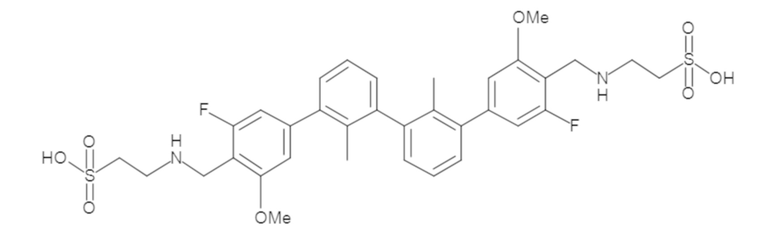
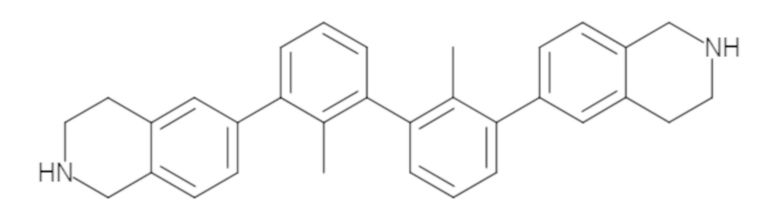
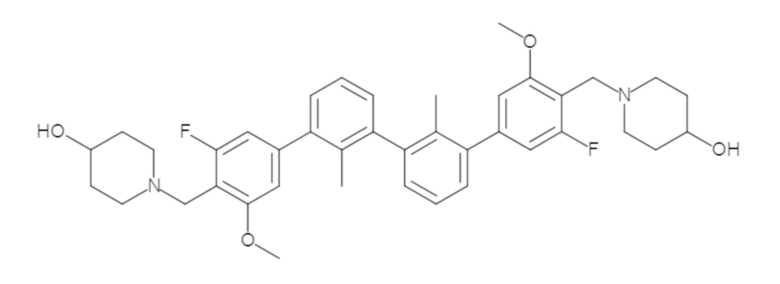
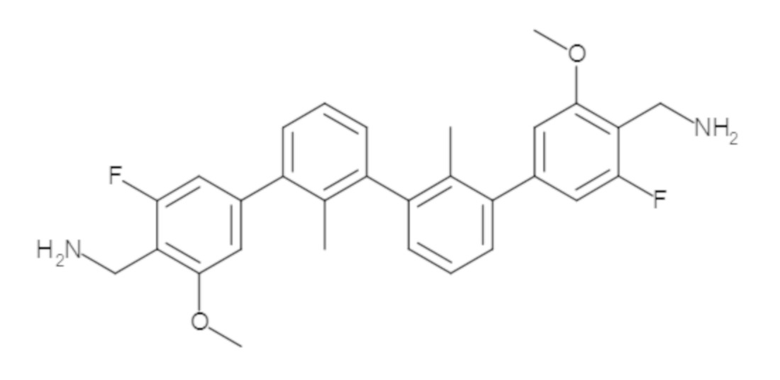
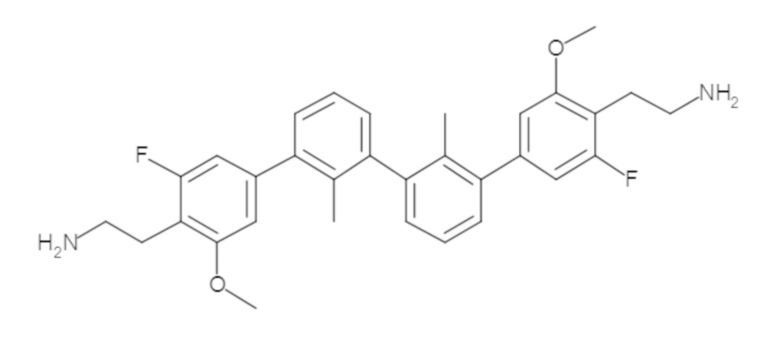
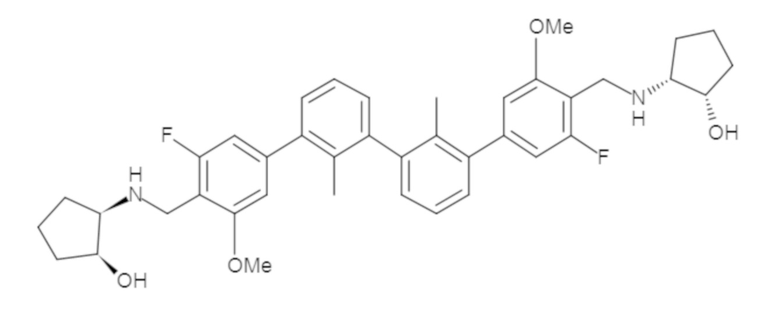
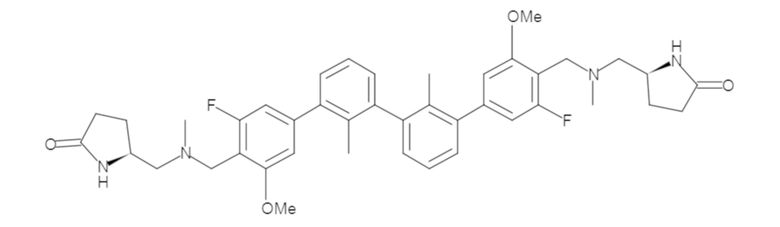
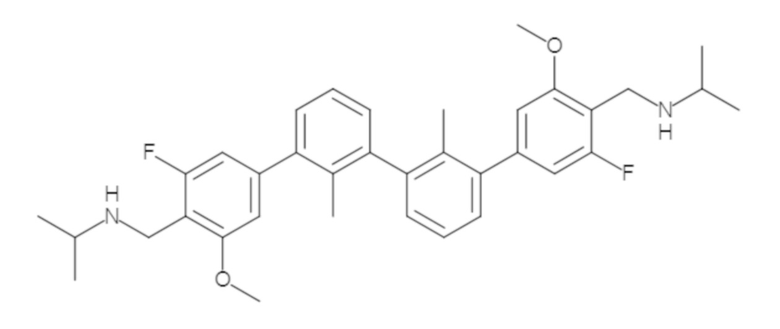
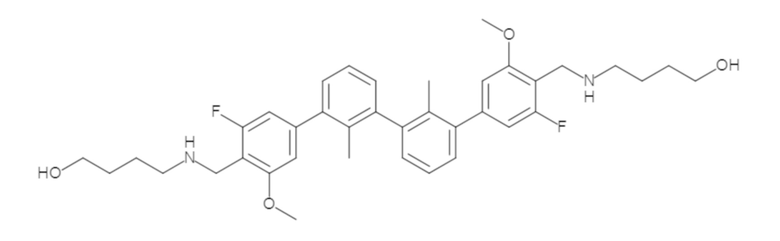

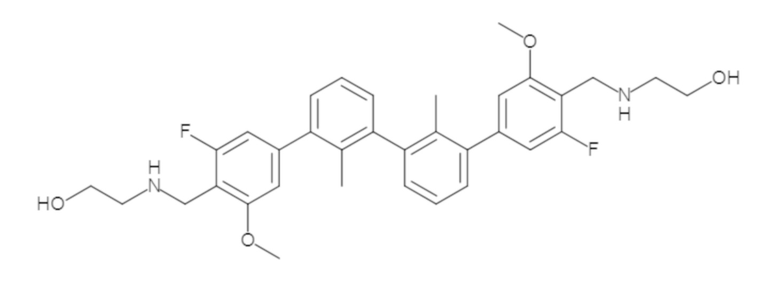
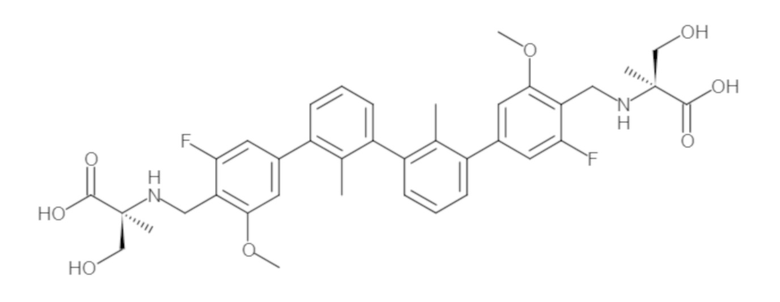
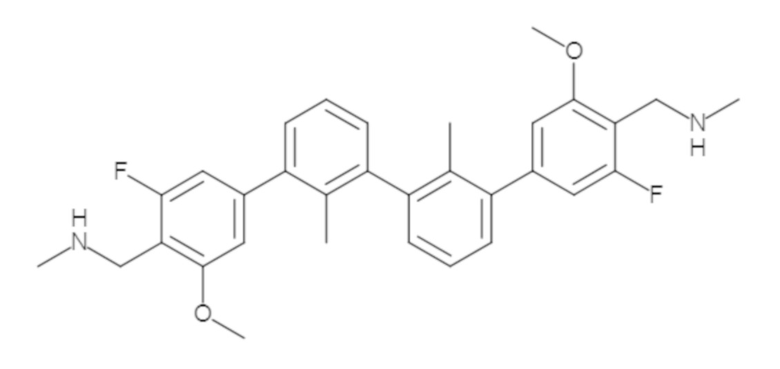
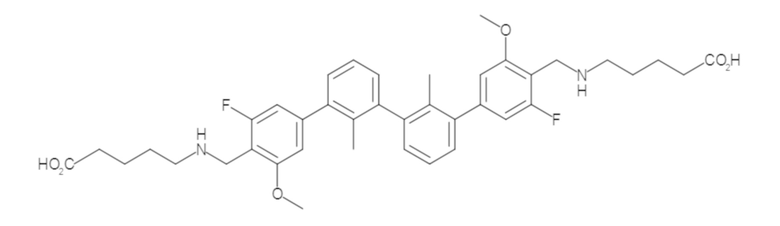
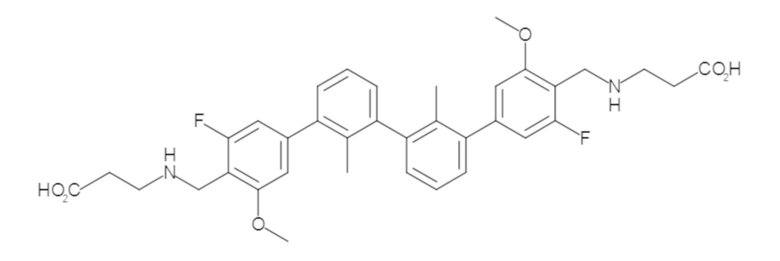
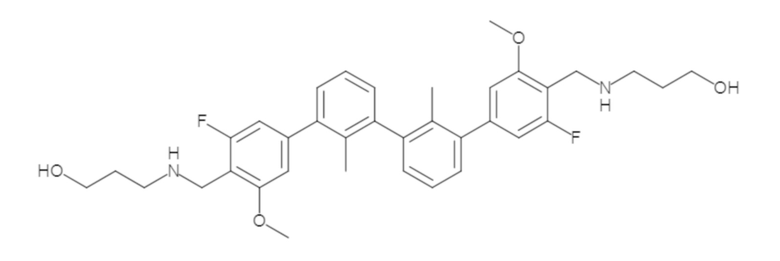
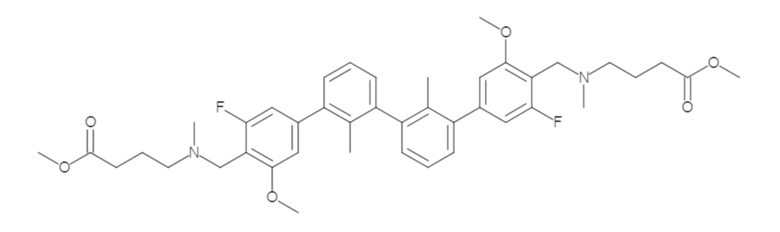
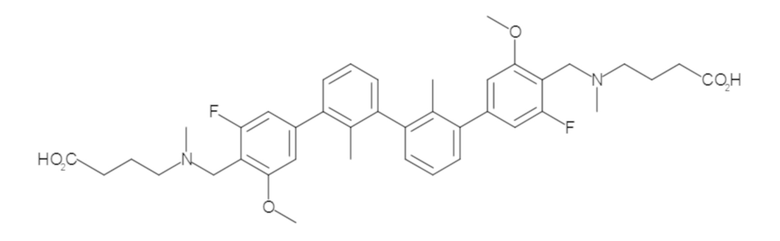
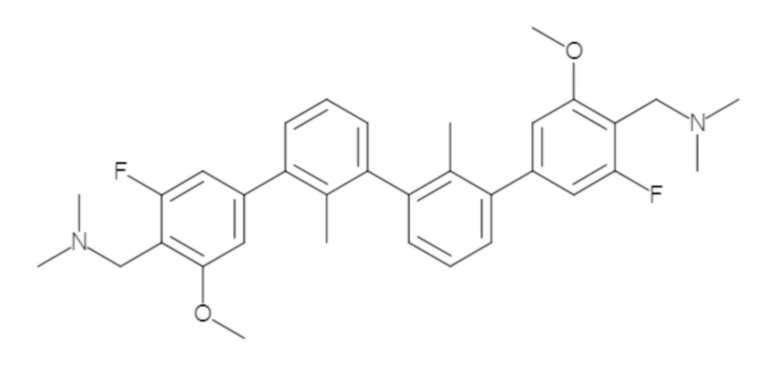
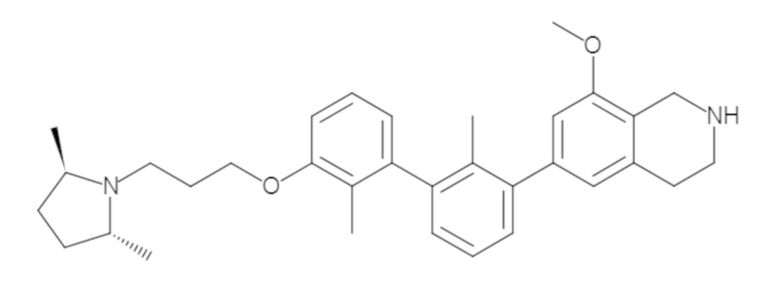
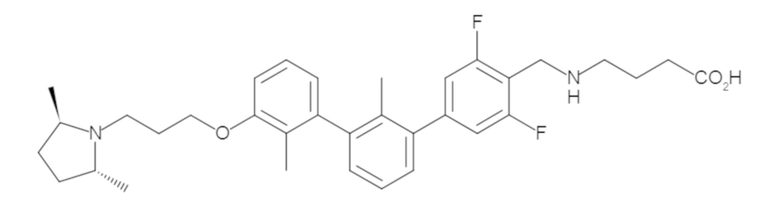

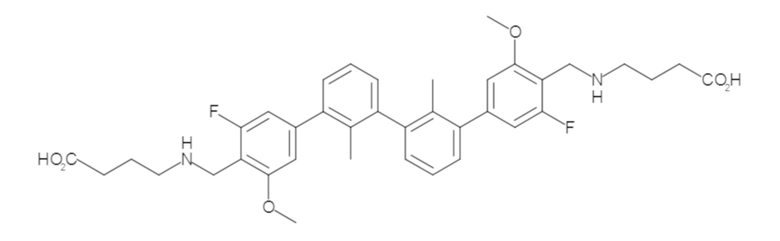
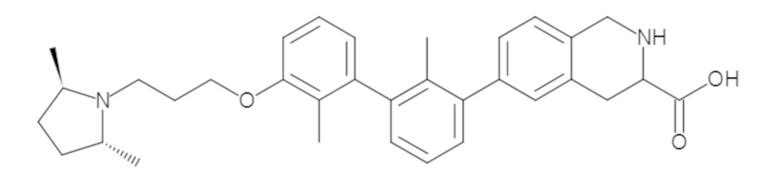
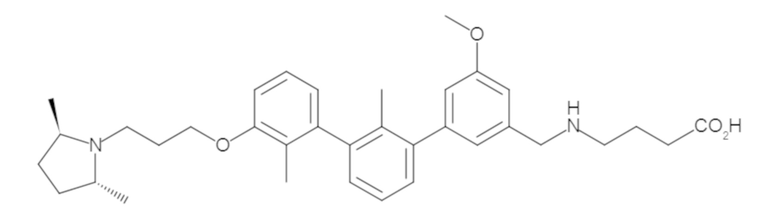
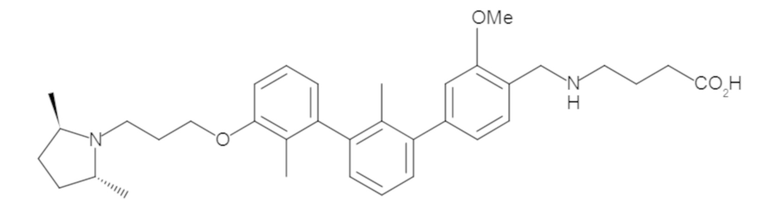
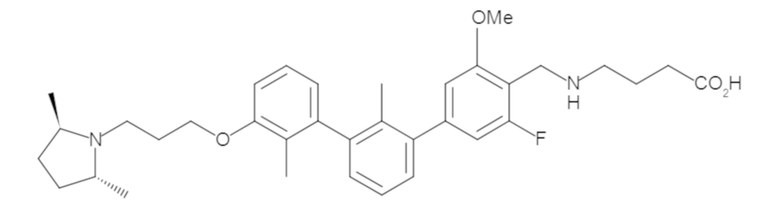
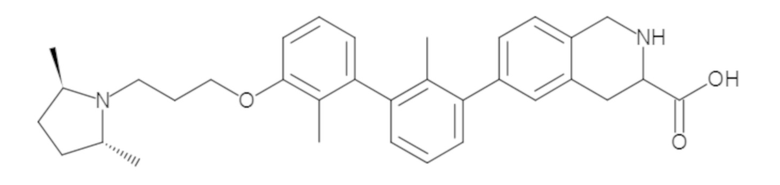

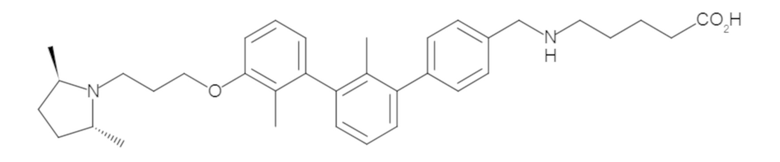
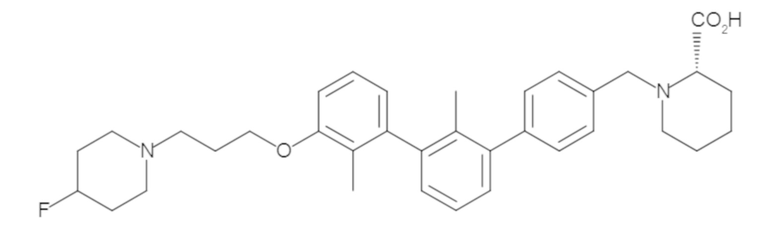
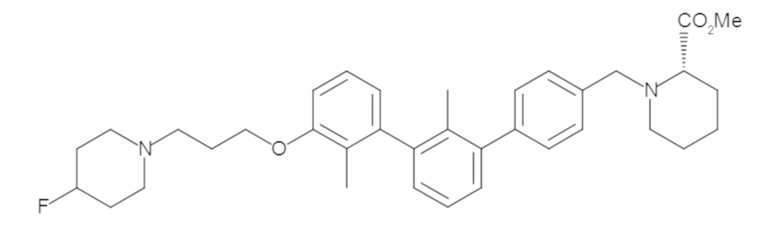
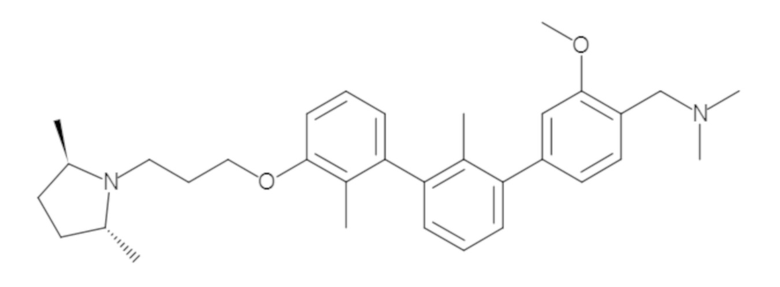
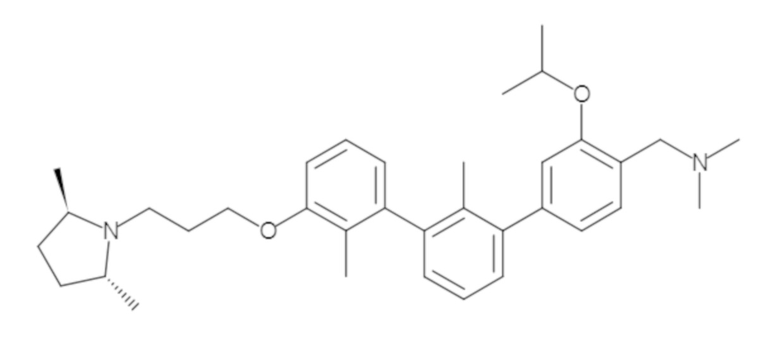
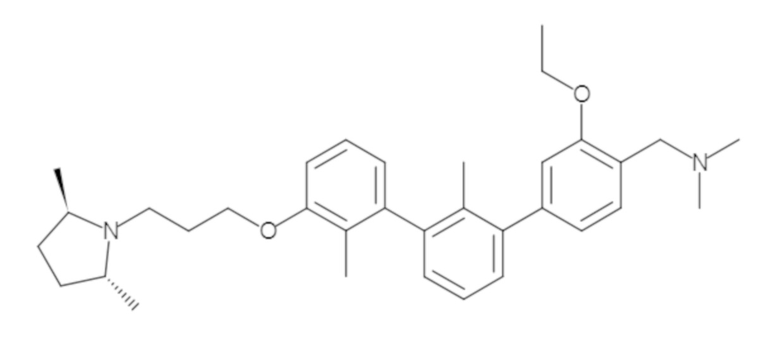
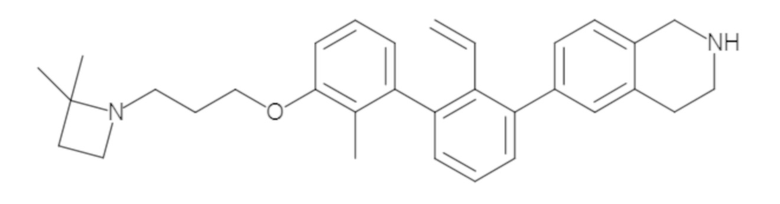
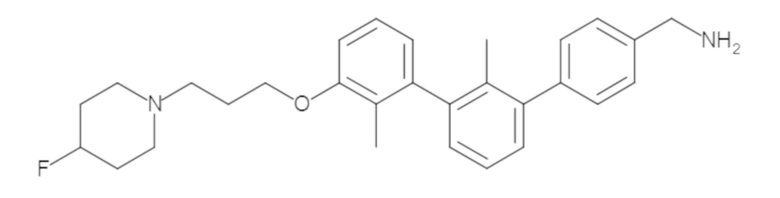
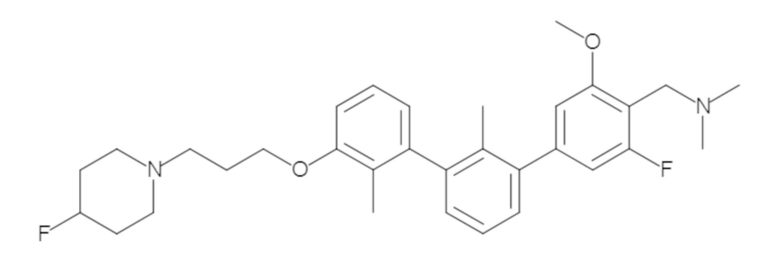
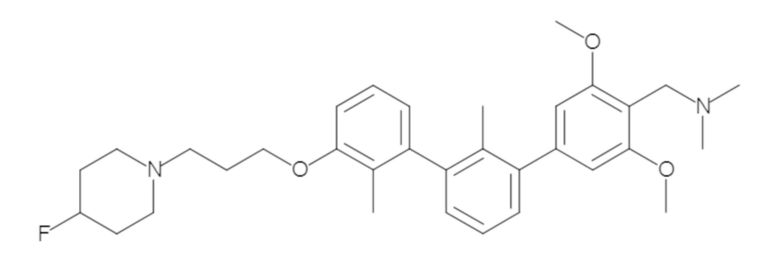
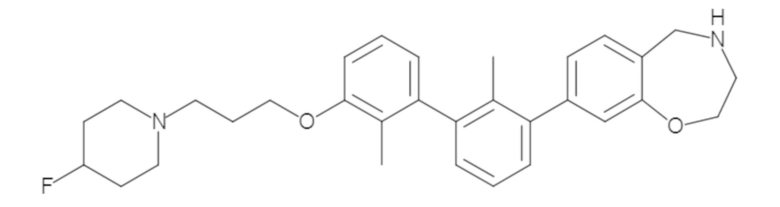
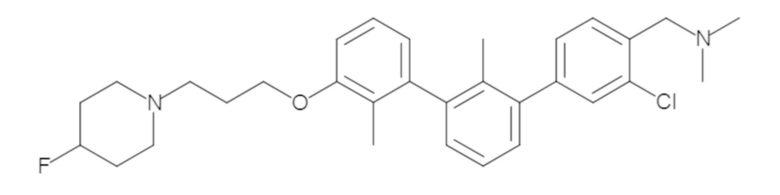
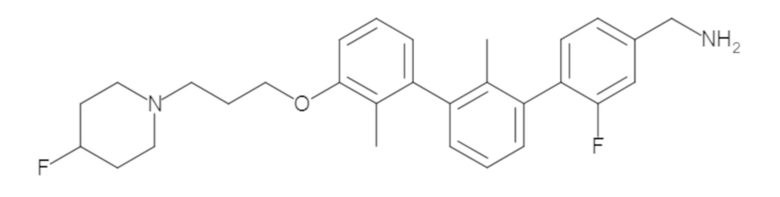
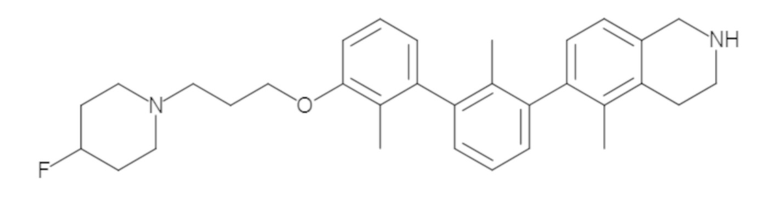
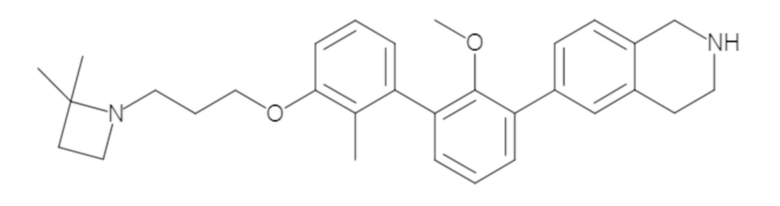
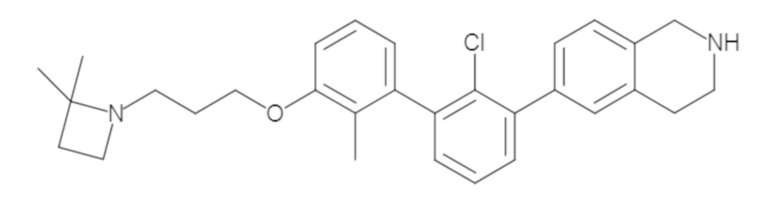
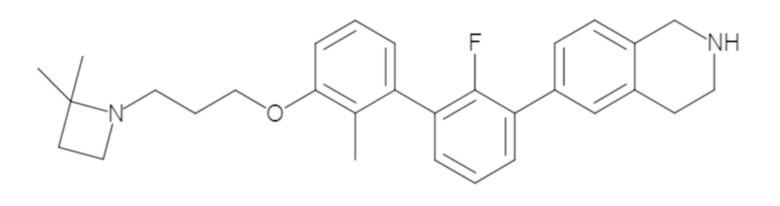
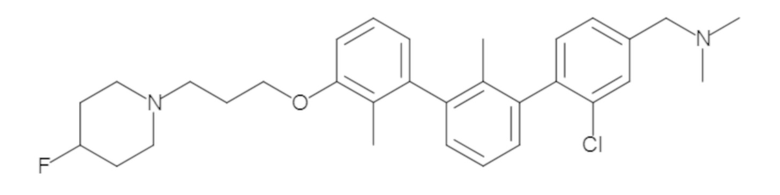
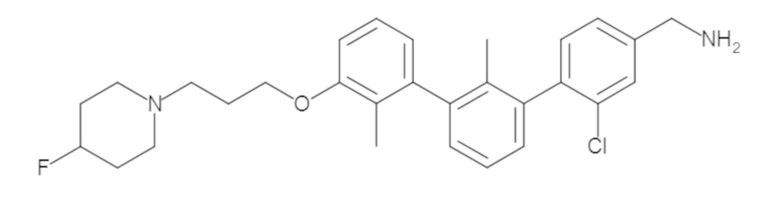
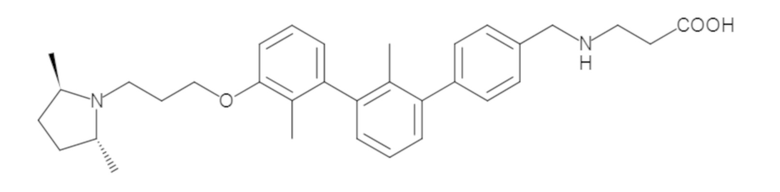
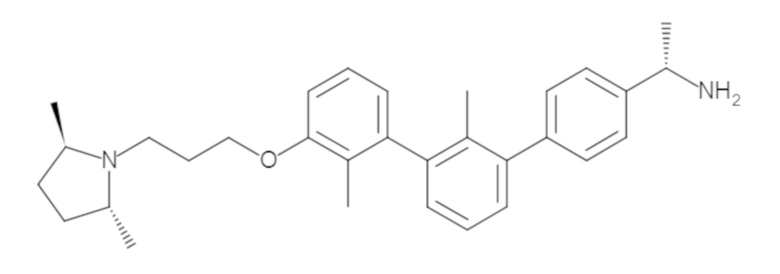
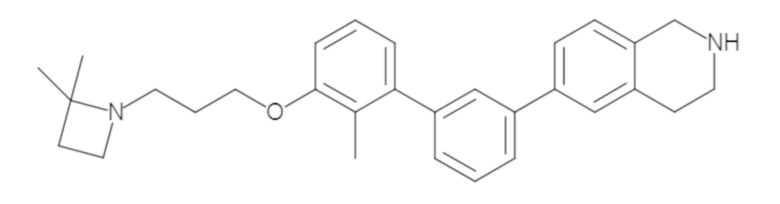
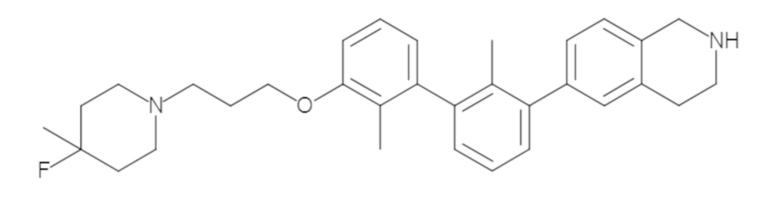
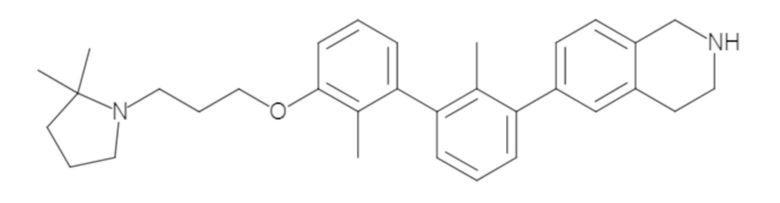
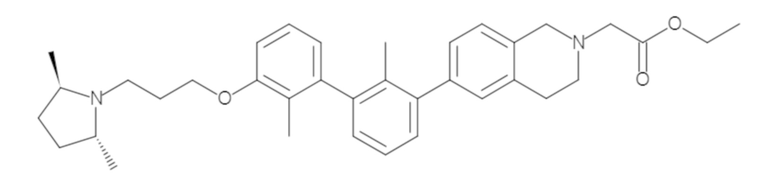
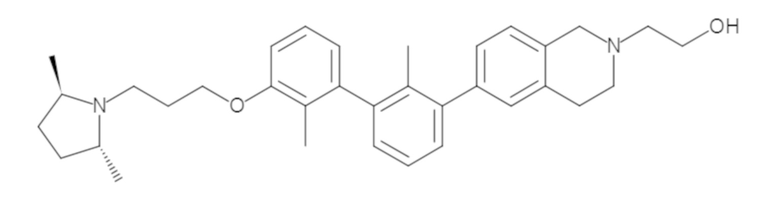
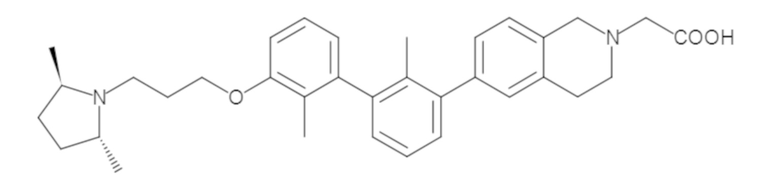
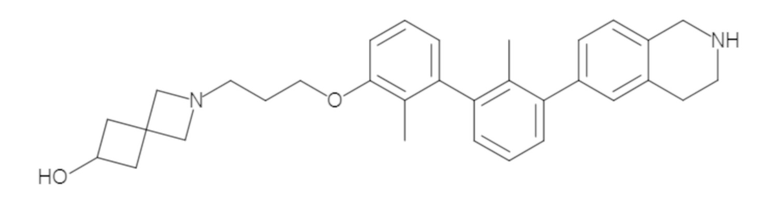
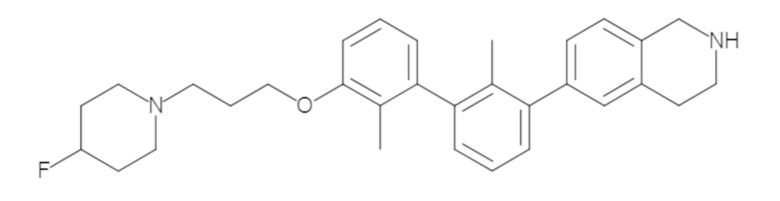
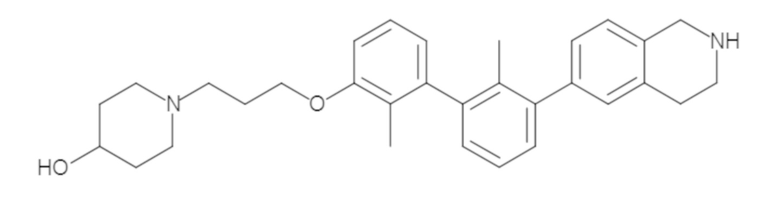
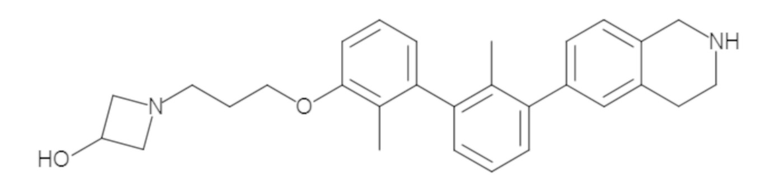
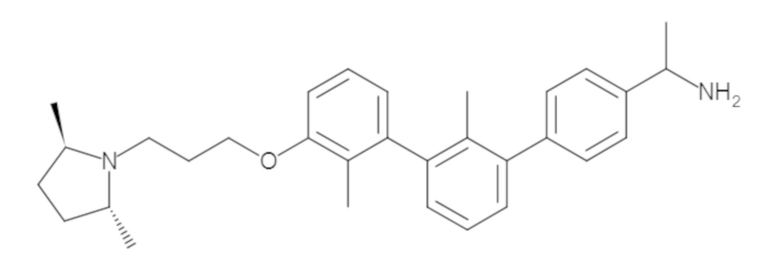
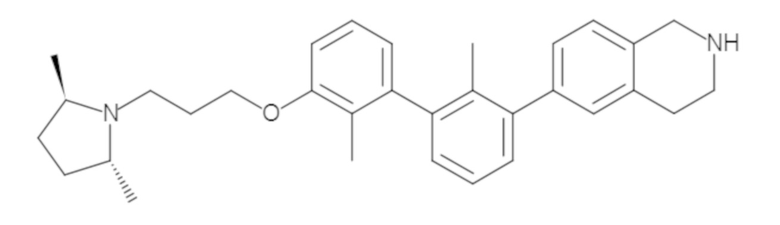
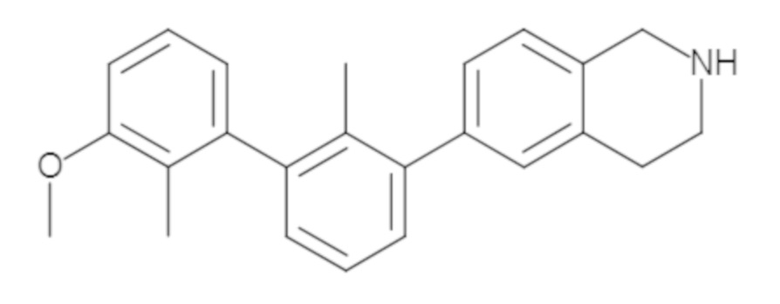
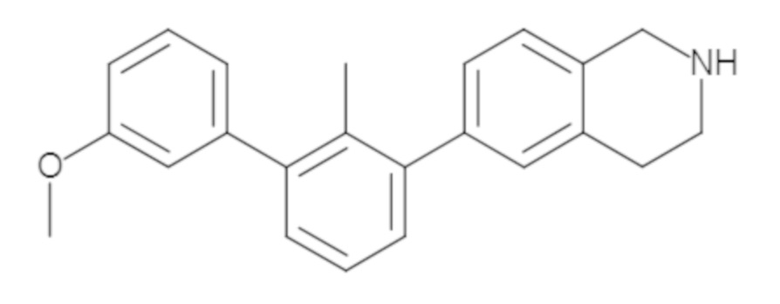
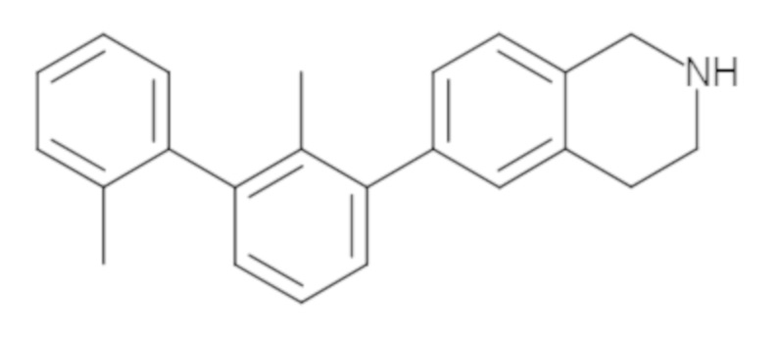
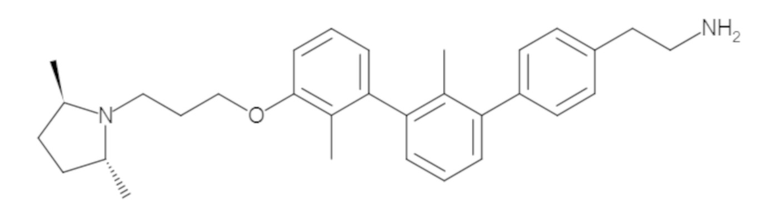
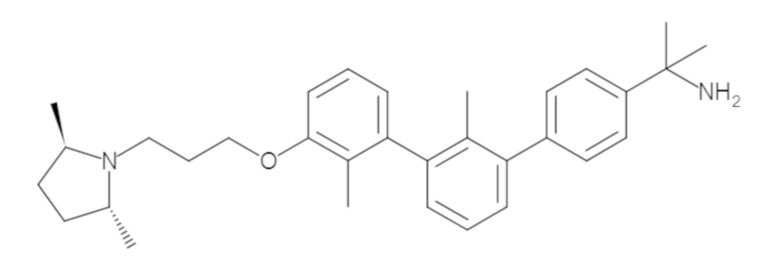
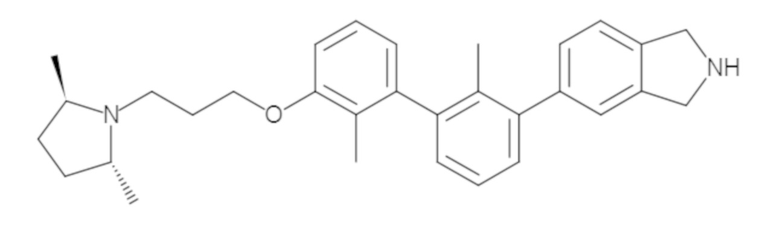
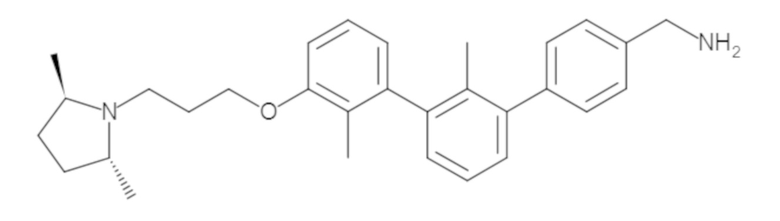
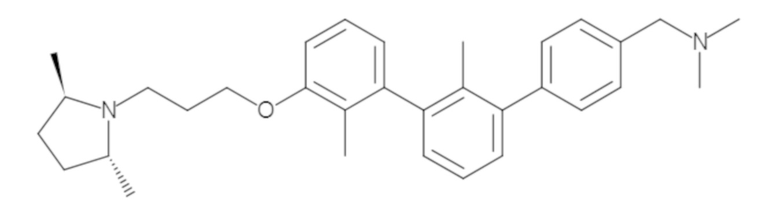
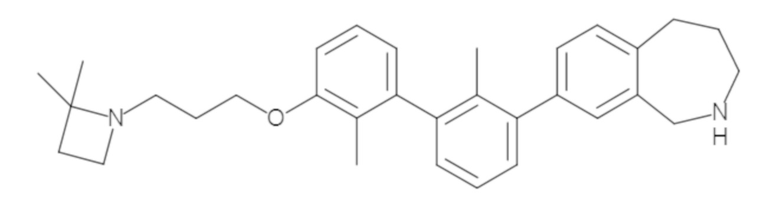
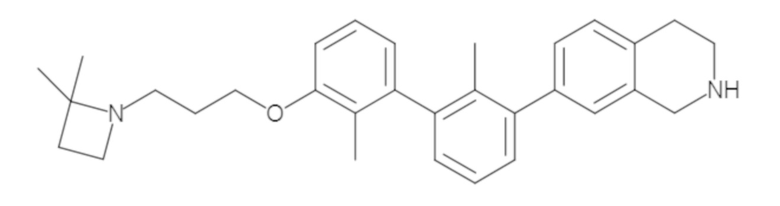
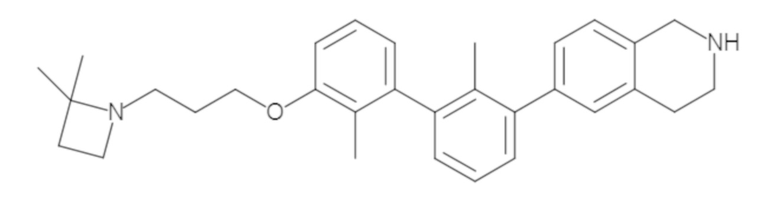
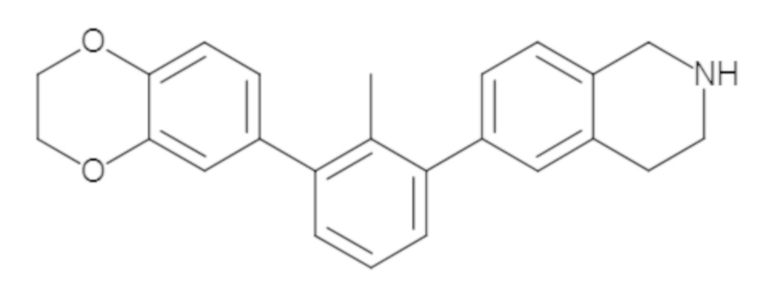
В настоящем тексте описаны частные варианты осуществления изобретения, включая наилучший способ реализации изобретения, известный авторам изобретения. При чтении приведенного выше описания, квалифицированным специалистам в данной области могут быть очевидны вариации описанных вариантов осуществления, и квалифицированный специалист ожидаемо способен применить такие вариации надлежащим образом. Соответственно, настоящее изобретение может быть реализовано на практике способом, отличным от описанных в настоящем тексте частных вариантов, и настоящее изобретение включает все модификации и эквиваленты предмета изобретения, охарактеризованного в прилагающейся Формуле изобретения, в рамках действующего законодательства. Кроме того, любая комбинация описанных выше элементов в их любых возможных вариациях входит в настоящее изобретение, если иное не указано особо или явным образом противоречит контексту.
Все публикации, патентные публикации, учетные номера и другие ссылки, приведенные в настоящей спецификации, включены в настоящий текст посредством ссылки, как если бы каждая отдельная публикация или патентная публикация были конкретно и индивидуально обозначены как включенные в настоящий текст посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ БЕЛКОВЫХ ТИРОЗИНФОСФАТАЗ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2833220C2 |
| ГЕТЕРОАРИЛ-БИФЕНИЛ АМИНЫ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2837843C1 |
| ТРИАРИЛЬНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2840345C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| ИНГИБИТОРЫ HPK1 И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2839132C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ/АКТИВАЦИИ PD1/PD-L1 | 2019 |
|
RU2777980C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ИНГИБИТОРЫ ПРОТЕИН-ТИРОЗИНФОСФАТАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2799446C2 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
Изобретение относится к соединению формулы (I), где R1a, R1b, R1c и R1d каждый независимо выбраны из H, F, Cl, C1-3 алкила, C1-3 галогеналкила и C1-3 алкоксигруппы; X1 представляет собой C1-3 алкилен, необязательно замещенный одним или двумя C1-2 алкилами; R2a и R2b каждый независимо выбраны из H, C1-8 алкила, -X2-C(O)2Ra, -X2-ORa, где каждый X2 представляет собой C1-6 алкилен; или R2a и R2b объединены с формированием 4-8-членного кольца, имеющего одну вершину цикла, выбранную из N; или R1a и R2a объединены с формированием 5-7-членного кольца, имеющего одну или две вершины цикла, выбранные из O и N; или R1b и R2b объединены с формированием 5-7-членного кольца, имеющего одну или две вершины цикла, выбранные из O и N; где кольца, образующиеся при объединении R2a и R2b, R1a и R2a или R1b и R2b, содержат 0 или 1 заместитель, независимо выбранный из -X3-C(O)2Ra и -X3-ORa, где X3 представляет собой связь или C1-6 алкилен; R3 выбран из F, Cl, C1-3 алкила, C1-3 алкоксигруппы, C1-3 галогеналкила, C2-3 алкенила и CN; n равен 0; каждый из R4, R6, R7 и R8 независимо выбран из H, галогена, C1-6 алкилаи -X4-ORa, где каждый X4 представляет собой связь; R5 выбран из H, галогена, C1-6 алкила, -Y2, -X5-ORa, -X5-NRaRb, -O-X5-Y2 и -A-Z, где каждый X5 представляет собой связь или C1-6 алкилен и каждый Y2 выбран из C4-8 гетероциклила, содержащего один гетероатом N, C7-9 спирогетероциклила, содержащего один гетероатом N, и 5-6-членного гетероарила, содержащего один гетероатом N, каждый из которых необязательно дополнительно замещен 1-4 заместителями, независимо выбранными из галогена, OH и C1-4 алкила; где A выбран из связи, -O, и -N(Ra)-; Z выбран из: i) моноциклическое 6-членное гетероарильное кольцо, содержащее один гетероатом N; и ii) 6-членное неароматическое гетероциклическое кольцо, содержащее один гетероатом N; и два из R4, R5 и R6 у соседних атомов углерода необязательно объединены с образованием 6-членного неароматического гетероциклического кольца, имеющего одну или две вершины цикла, выбранные из O и -N(Rb)-; где указанное неароматическое гетероциклическое кольцо необязательно замещено оксогруппой и необязательно 1 заместителем Rc; и по меньшей мере один из R4, R5, R6, R7 и R8 отличается от H; каждый Ra независимо выбран из H и C1-6 алкила; каждый Rb независимо выбран из H; и каждый Rc независимо выбран из H, CN, -Y4, -X6-C(O)2Ra и -X6-NRaRb, где каждый X6 представляет собой связь или C1-6 алкилен и каждый Y4 выбран из C4-8 гетероциклила, содержащего один или два гетероатома N. Также изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), способу лечения заболевания или нарушения, связанного с PD-1, при помощи соединения формулы (I), применению соединения формулы (I) для лечения заболевания или нарушения, связанного с PD-1, и соединению 1.006, указанному в формуле изобретения. Технический результат – соединения, обладающие ингибирующей активностью в отношении PD-1 для лечения заболеваний, опосредованных PD-1. 5 н. и 14 з.п. ф-лы, 1 табл., 25 пр.
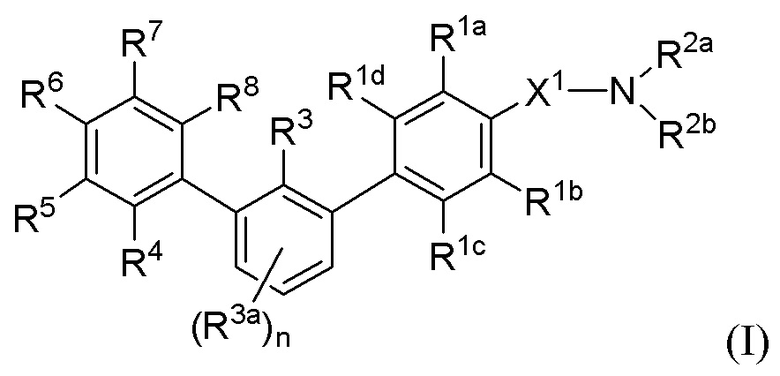
1. Соединение, имеющее формулу (I)

или его фармацевтически приемлемая соль,
где R1a, R1b, R1c и R1d, каждый независимо, выбраны из группы, состоящей из H, F, Cl, C1-3 алкила, C1-3 галогеналкила и C1-3 алкоксигруппы;
X1 представляет собой C1-3 алкилен, необязательно замещенный одним или двумя C1-2 алкилами;
R2a и R2b, каждый независимо, выбраны из группы, состоящей из H, C1-8 алкила, -X2-C(O)2Ra, -X2-ORa, где каждый X2 представляет собой C1-6 алкилен;
или R2a и R2b объединены с формированием 4-8-членного кольца, имеющего одну вершину цикла, выбранную из N;
или R1a и R2a объединены с формированием 5-7-членного кольца, имеющего одну или две вершины цикла, выбранные из O и N;
или R1b и R2b объединены с формированием 5-7-членного кольца, имеющего одну или две вершины цикла, выбранные из O и N;
где кольца, образующиеся при объединении R2a и R2b, R1a и R2a или R1b и R2b, содержат 0 или один заместитель, независимо выбранный из группы, состоящей из -X3-C(O)2Ra и -X3-ORa, где X3 представляет собой связь или C1-6 алкилен;
R3 выбран из группы, состоящей из F, Cl, C1-3 алкила, C1-3 алкоксигруппы, C1-3 галогеналкила, C2-3 алкенила и CN;
подстрочный индекс n равен 0;
каждый из R4, R6, R7 и R8 независимо выбран из группы, состоящей из H, галогена, C1-6 алкила и -X4-ORa, где каждый X4 представляет собой связь;
R5 является представителем, выбранным из группы, состоящей из H, галогена, C1-6 алкила, -Y2, -X5-ORa, -X5-NRaRb, -O-X5-Y2 и -A-Z, где каждый X5 представляет собой связь или C1-6 алкилен и каждый Y2 выбран из группы, состоящей из C4-8 гетероциклила, содержащего один гетероатом N, C7-9 спирогетероциклила, содержащего один гетероатом N, и 5-6-членного гетероарила, содержащего один гетероатом N, каждый из которых необязательно дополнительно замещен 1-4 заместителями, независимо выбранными из группы, состоящей из галогена, OH и C1-4 алкила;
где A выбран из группы, состоящей из связи, -O и -N(Ra)-;
Z выбран из группы, состоящей из следующих:
i) моноциклическое 6-членное гетероарильное кольцо, содержащее один гетероатом N; и
ii) 6-членное неароматическое гетероциклическое кольцо, содержащее один гетероатом N;
и два из R4, R5 и R6 у соседних атомов углерода необязательно объединены с образованием 6-членного неароматического гетероциклического кольца, имеющего одну или две вершины цикла, выбранные из группы, состоящей из O и -N(Rb)-; где указанное неароматическое гетероциклическое кольцо необязательно замещено оксогруппой и необязательно 1 заместителем Rc;
и по меньшей мере один из R4, R5, R6, R7 и R8 отличается от H;
каждый Ra независимо выбран из группы, состоящей из H и C1-6 алкила;
каждый Rb независимо выбран из группы, состоящей из H; и
каждый Rc независимо выбран из группы, состоящей из H, CN, -Y4, -X6-C(O)2Ra и -X6-NRaRb, где каждый X6 представляет собой связь или C1-6 алкилен и каждый Y4 выбран из группы, состоящей из C4-8 гетероциклила, содержащего один или два гетероатома N.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R5 представляет собой -A-Z.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где R5 представляет собой -O-X5-Y2.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, где X1 представляет собой -CH2-.
5. Соединение по п. 1 или его фармацевтически приемлемая соль,
где R1c, R7 и R8 представляет собой H и
R3 выбран из группы, состоящей из F, Cl, CH3, CF3 и OCH3.
6. Соединение по п. 1 или его фармацевтически приемлемая соль, где кольцо образуется между одной из пар R4 и R5, R5 и R6, R1b и R2b или R1a и R2a.
7. Соединение по п. 1 или его фармацевтически приемлемая соль,
где R5 представляет собой -A-Z и
Z выбран из группы, состоящей из пиперидинила и пиридинила.
8. Соединение по п. 1 или его фармацевтически приемлемая соль, где R4 выбран из группы, состоящей из F, Cl, CH3 и OCH3.
9. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1a представляет собой OCH3 и R1b представляет собой F.
10. Соединение по п. 1 или его фармацевтически приемлемая соль, где R2a и R2b каждый представляют собой H.
11. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R2a и R2b объединены с формированием 5-7-членного кольца, имеющего одну вершину цикла, выбранную из N, где указанное кольцо замещено 0 или 1 заместителем, независимо выбранными из группы, состоящей из -X3-C(O)2Ra и -X3-ORa, где X3 представляет собой связь или C1-6 алкилен.
12. Соединение по п. 1 или его фармацевтически приемлемая соль, имеющее формулу (Ib)
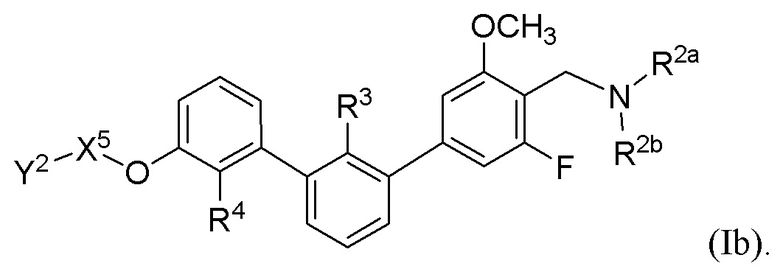
13. Соединение по п. 1 или его фармацевтически приемлемая соль, имеющее формулу (Ic)
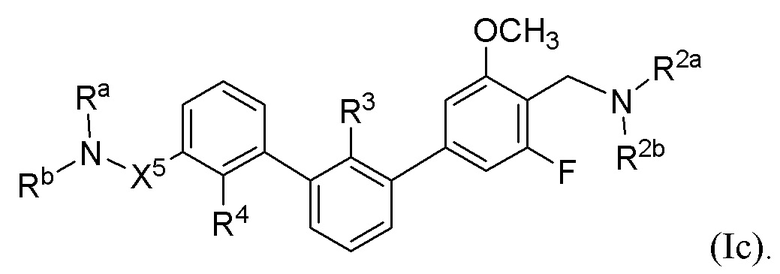
14. Соединение по п. 12 или его фармацевтически приемлемая соль, где Y2 выбран из группы, состоящей из следующих:
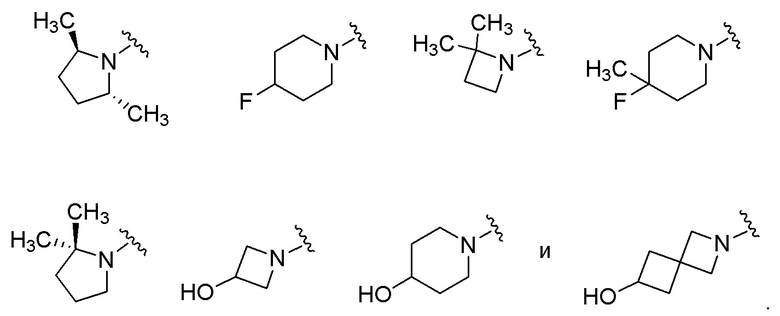
15. Соединение по п. 1 или его фармацевтически приемлемая соль, выбранное из:
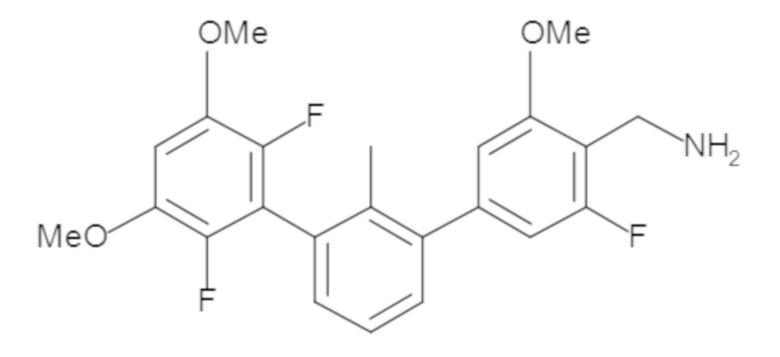
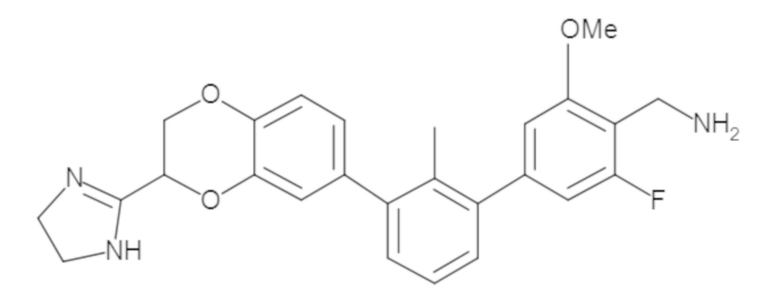
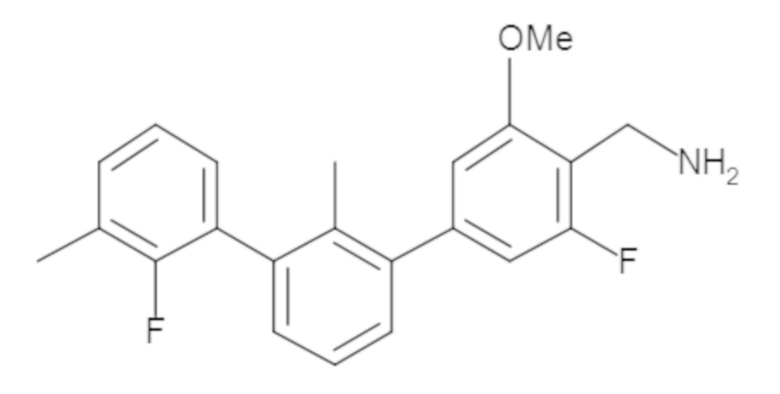
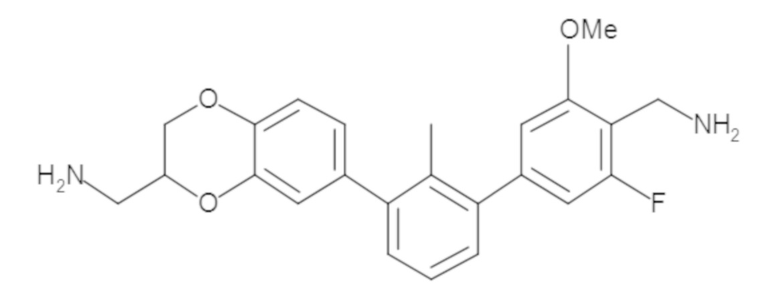
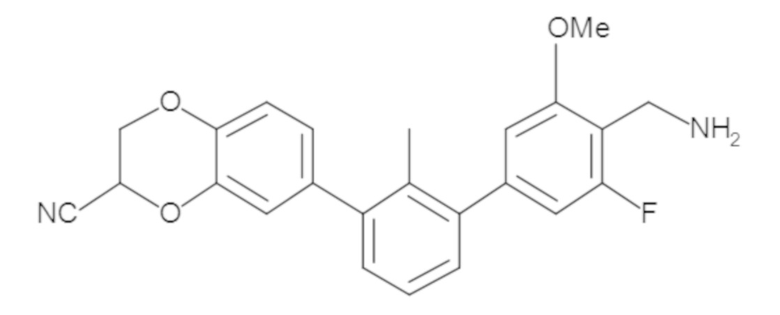
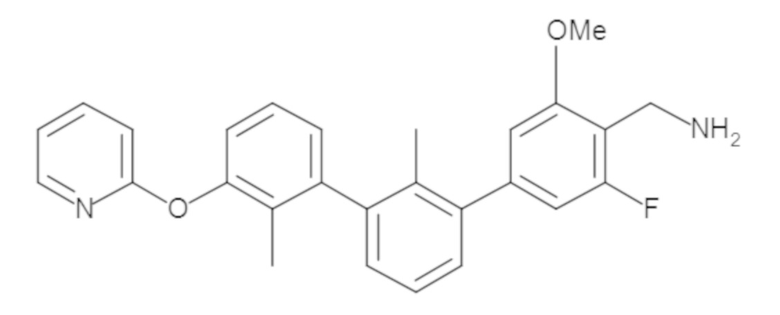
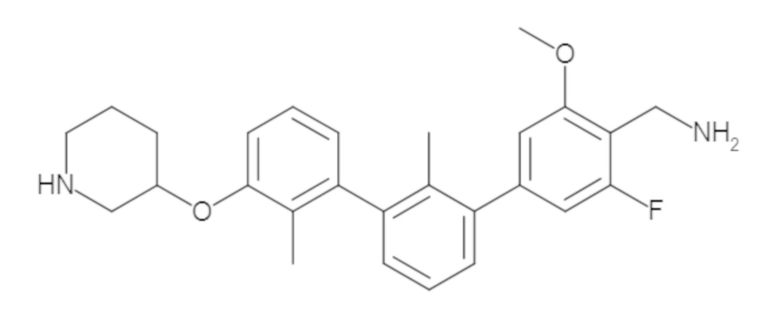
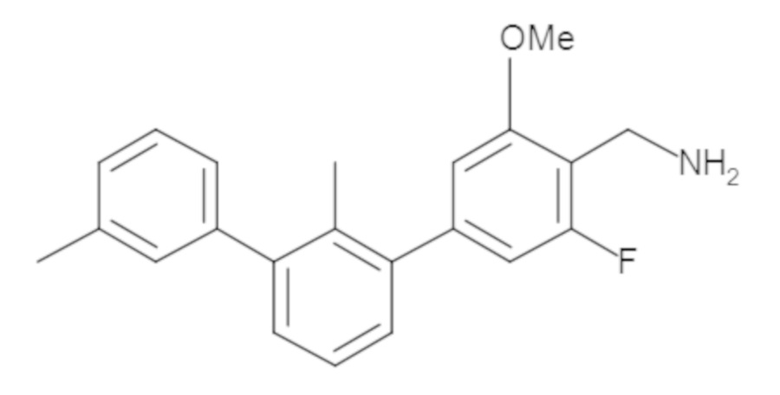
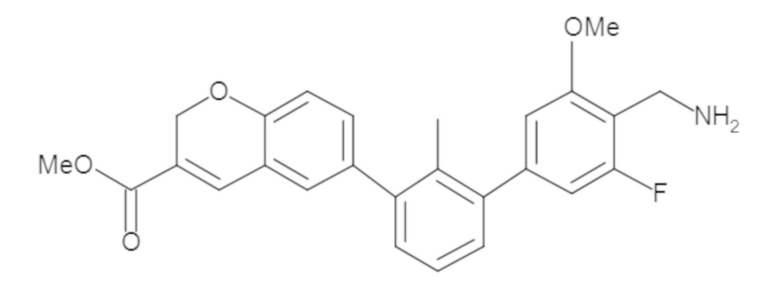

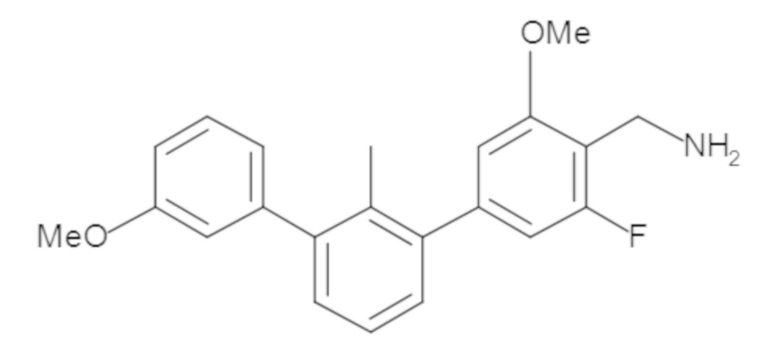

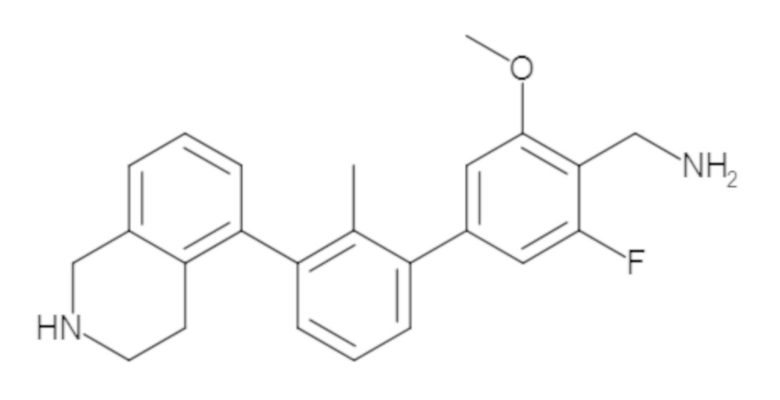

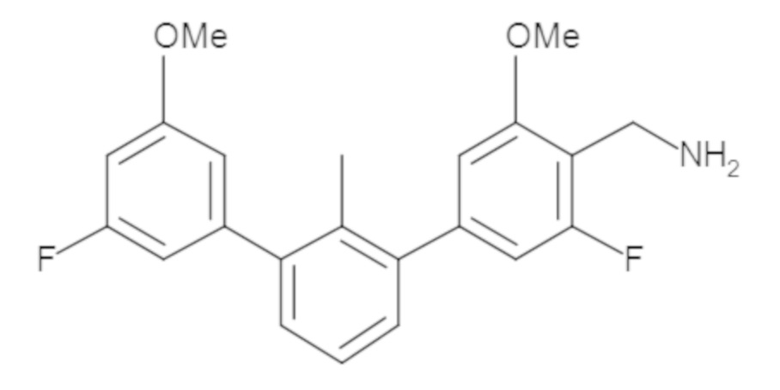
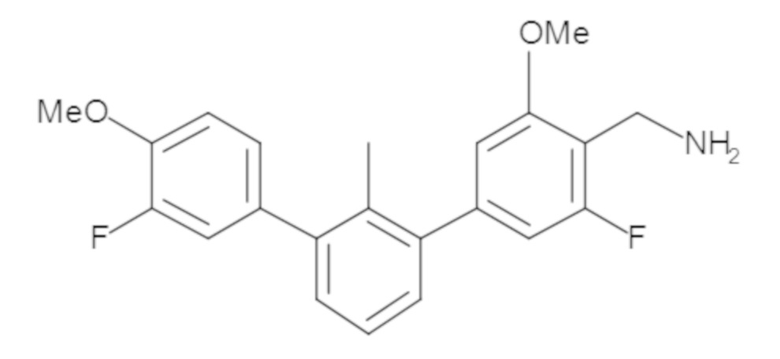

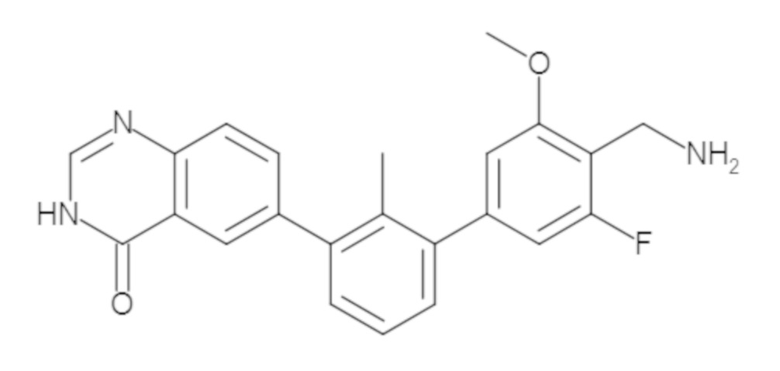
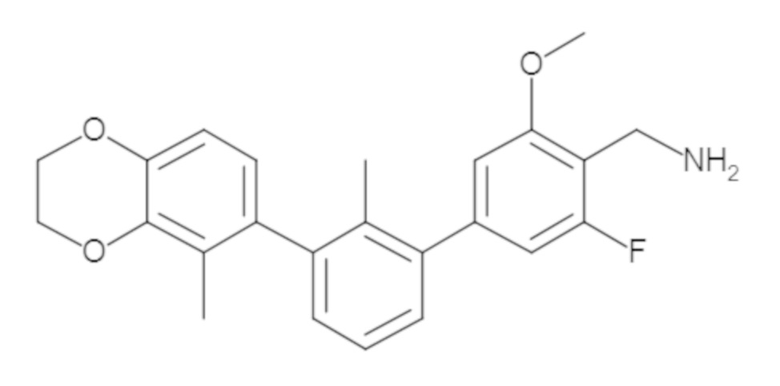
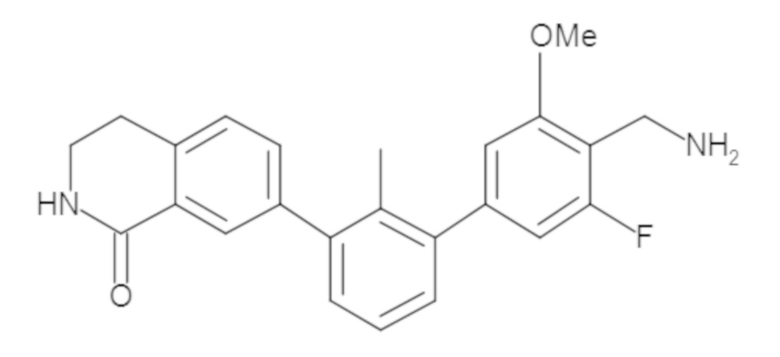
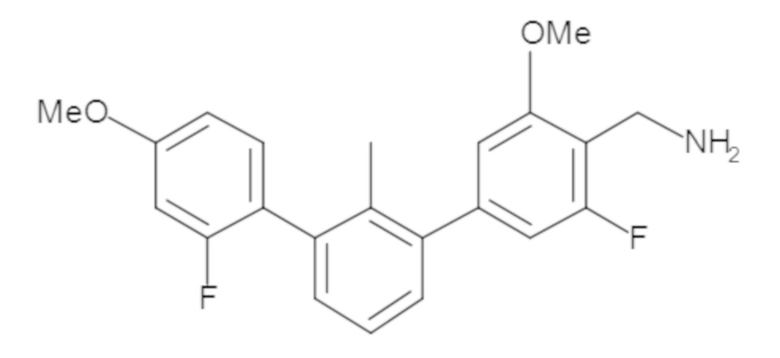
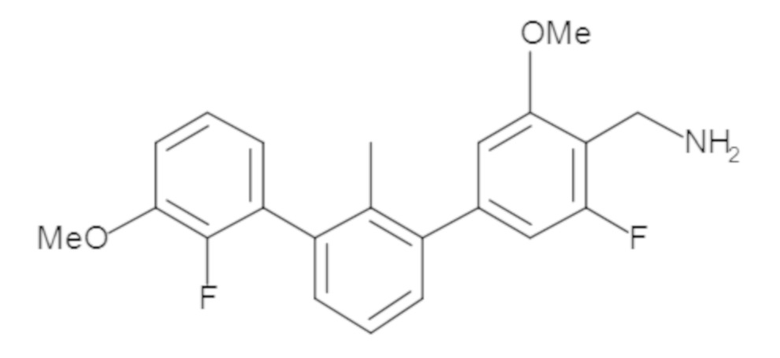
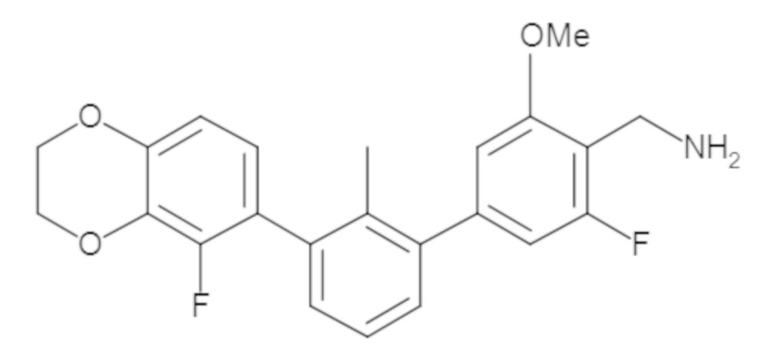
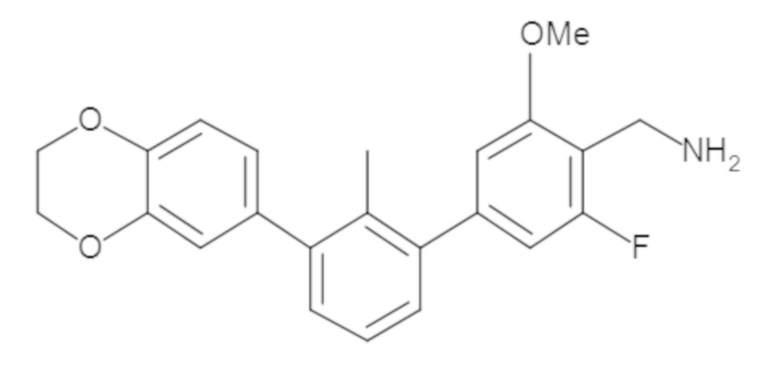
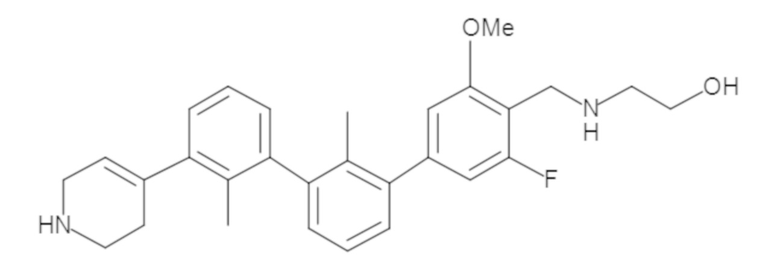

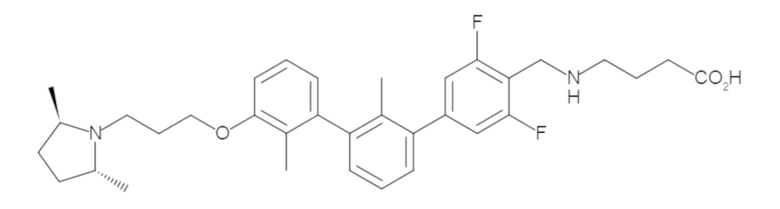
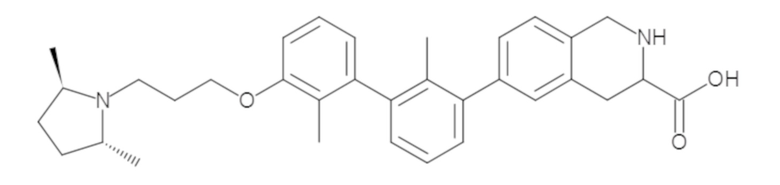
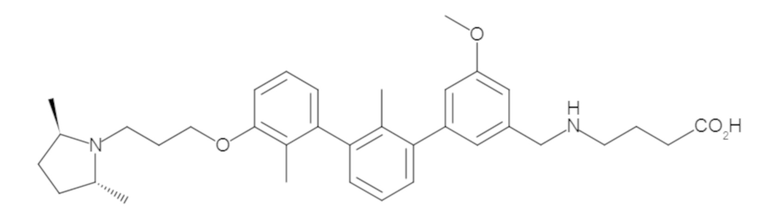
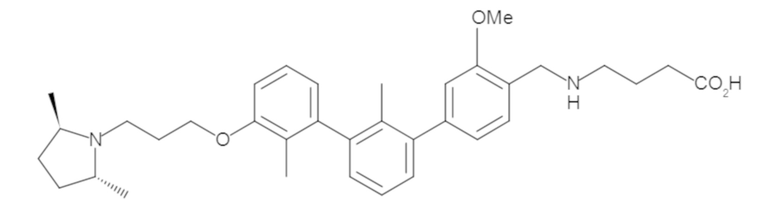
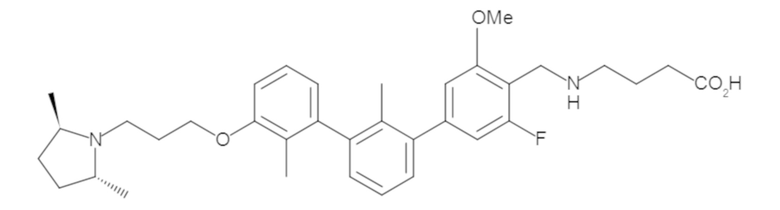
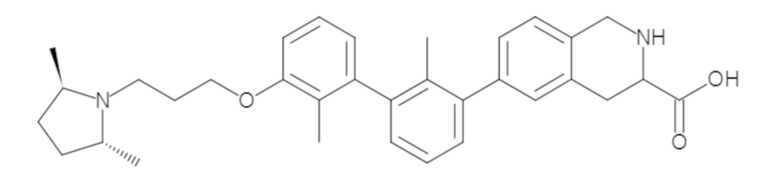
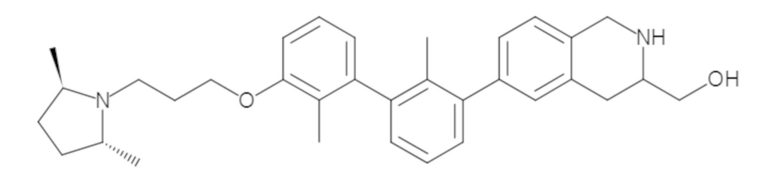
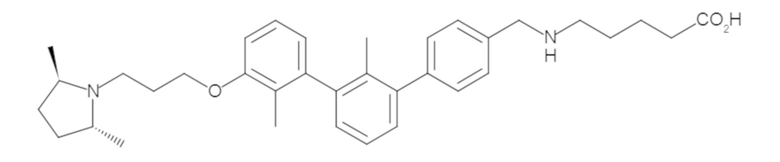
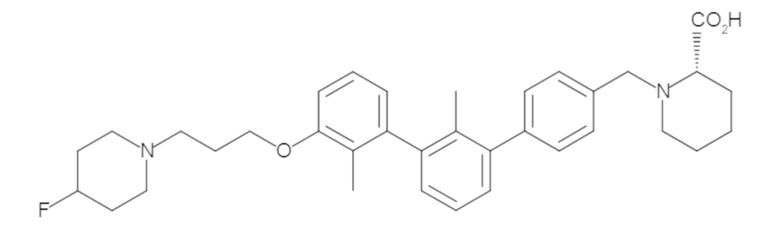
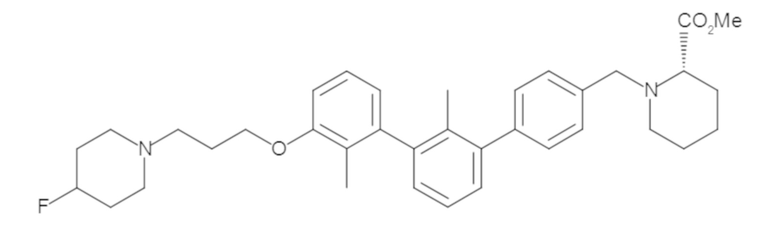
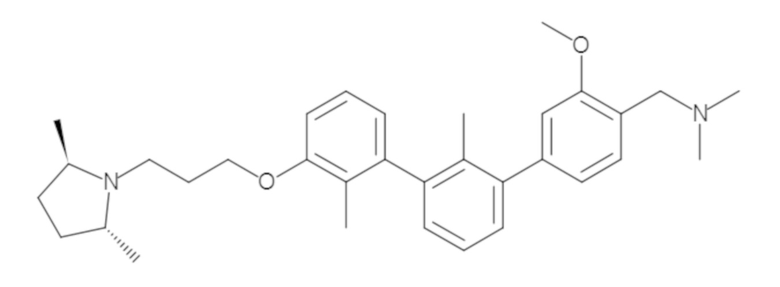
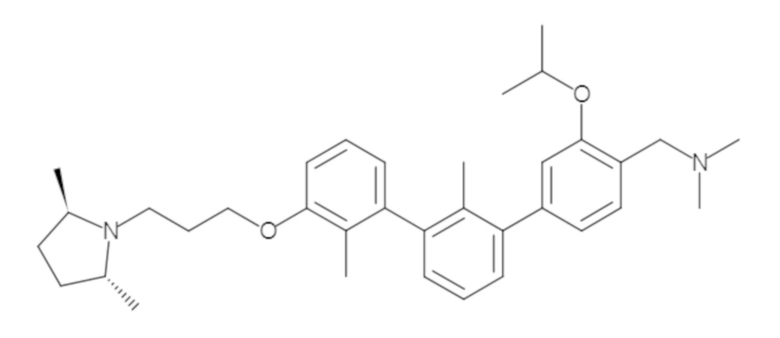
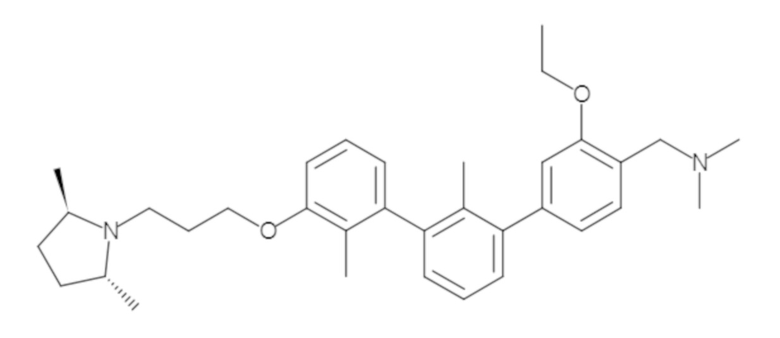
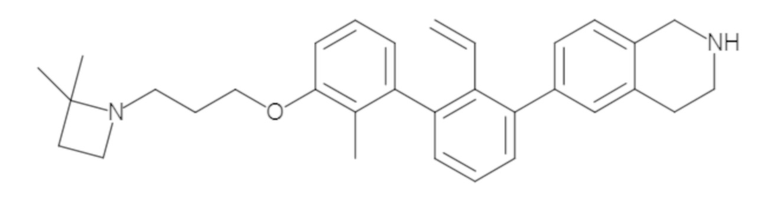
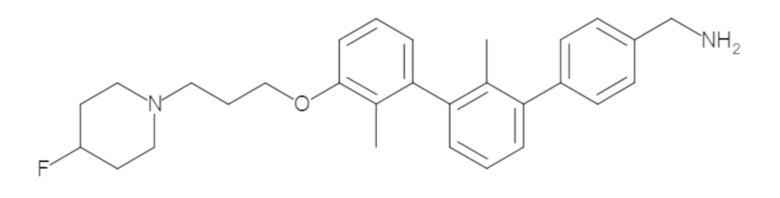
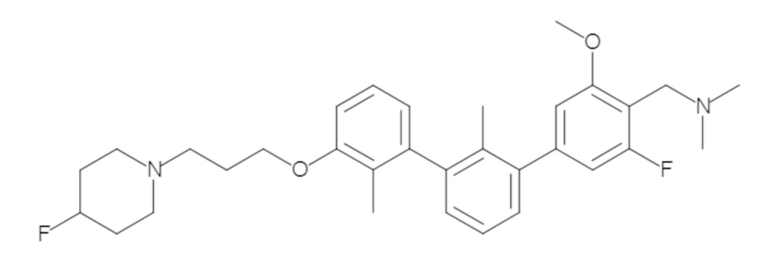
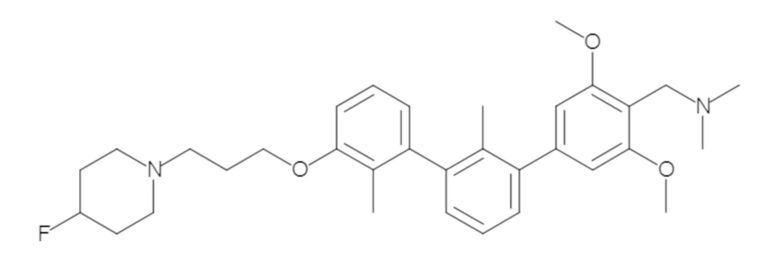
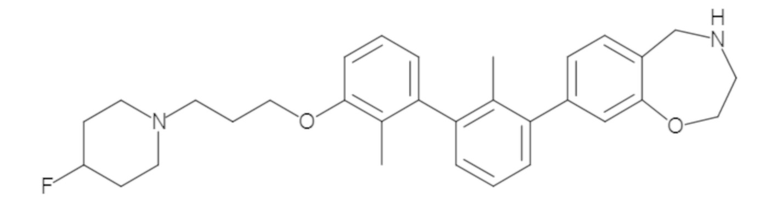
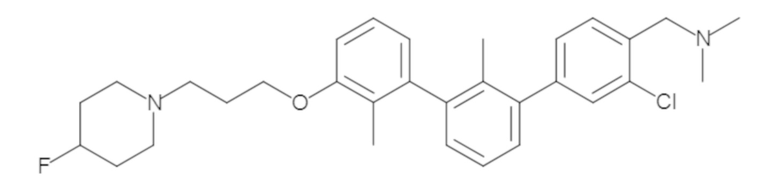
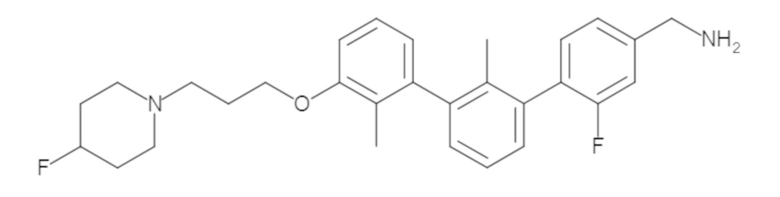
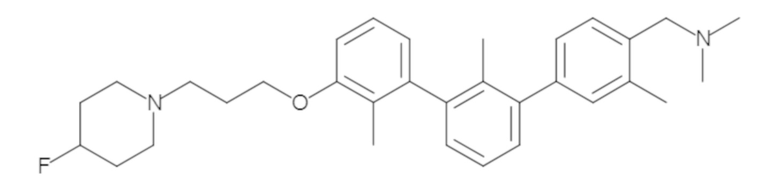
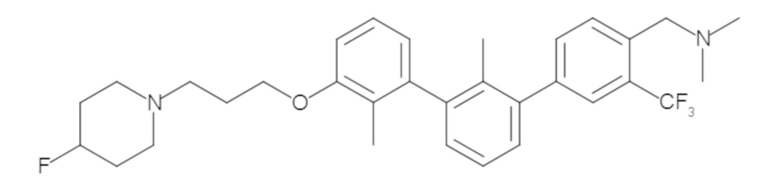
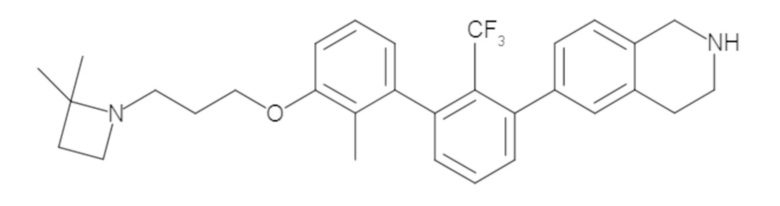
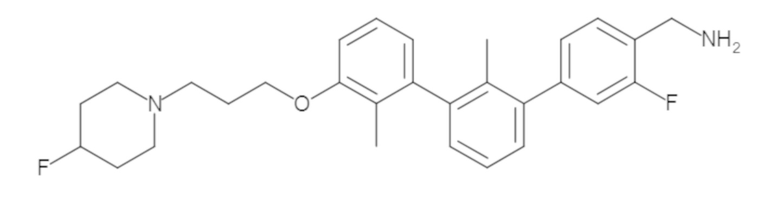
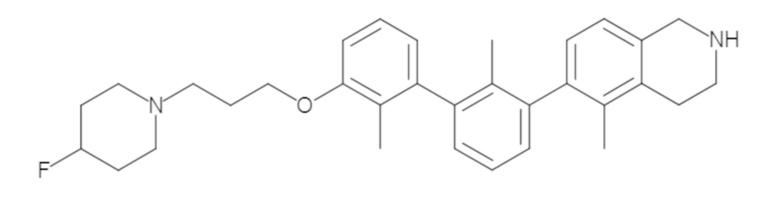
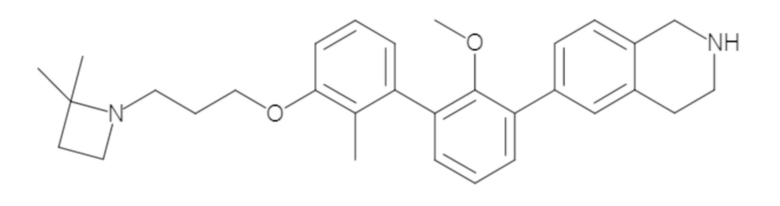
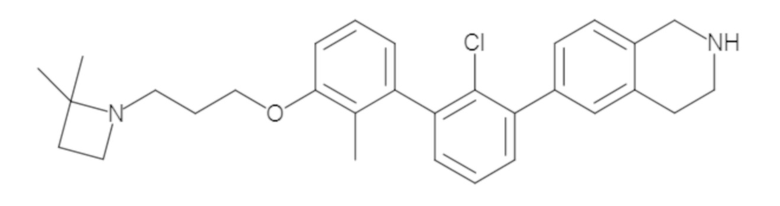
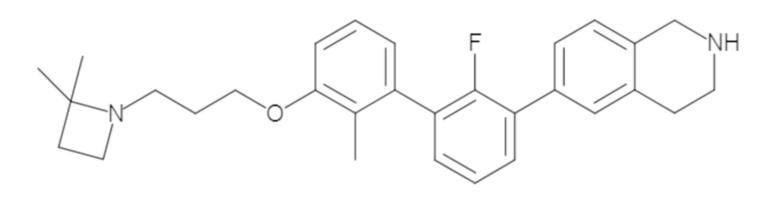
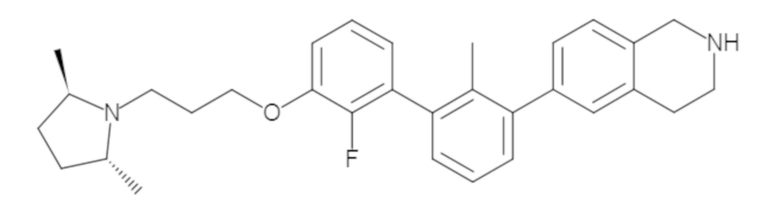
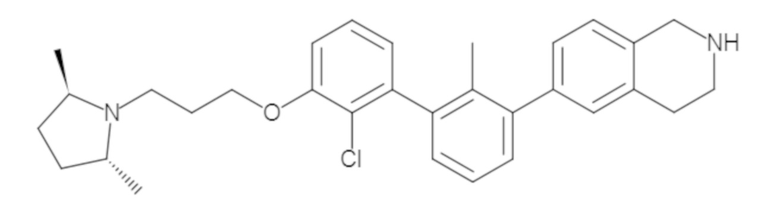
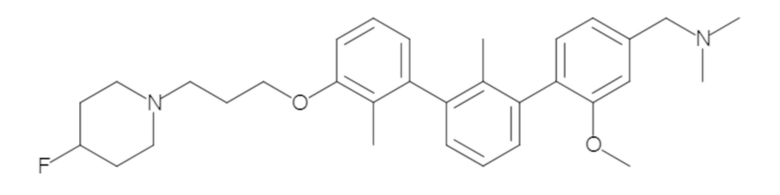
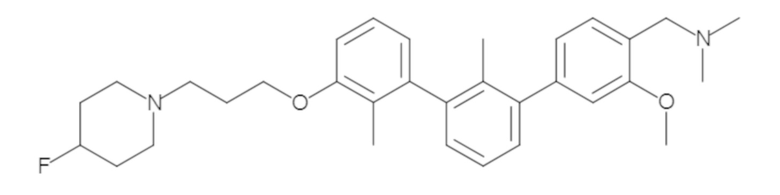
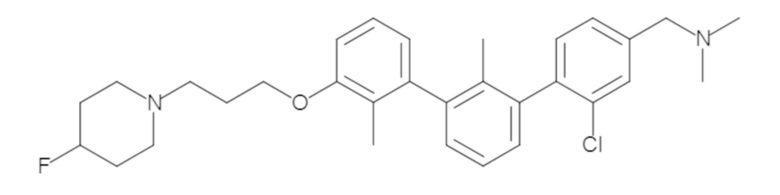
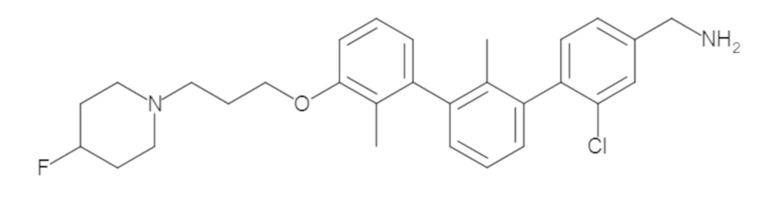

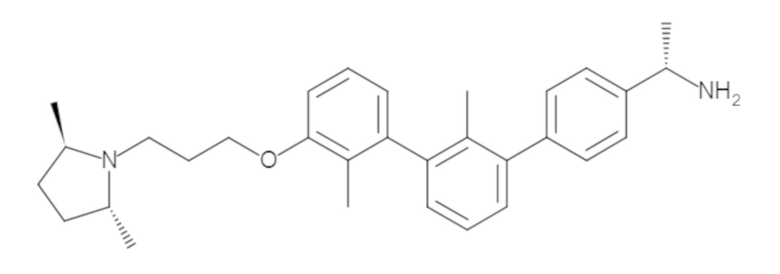
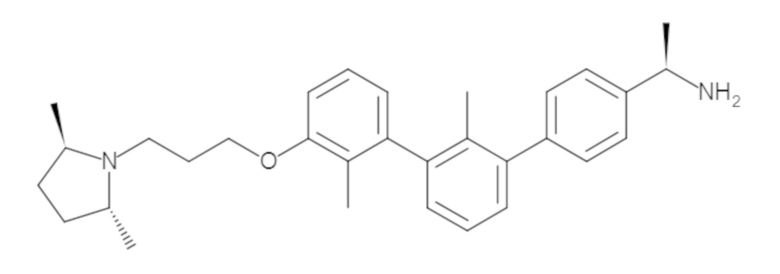
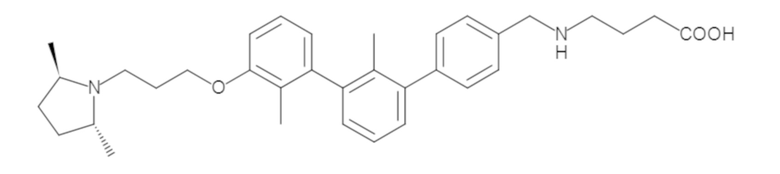
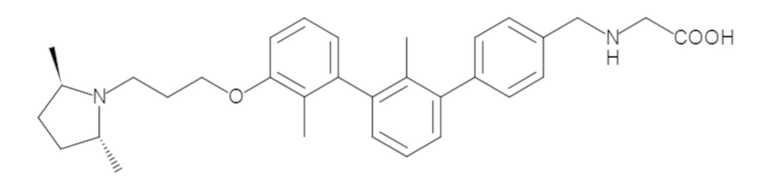
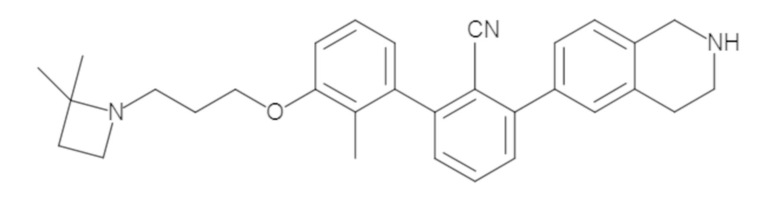
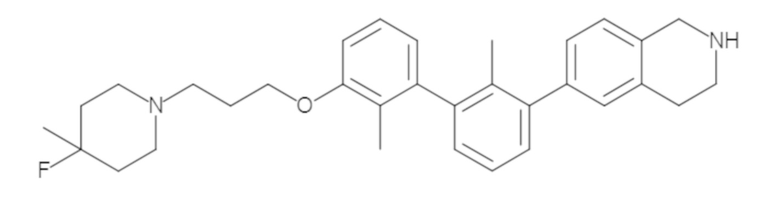
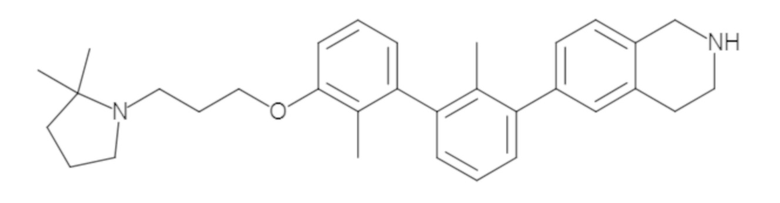
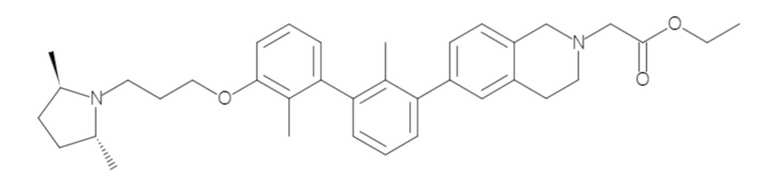
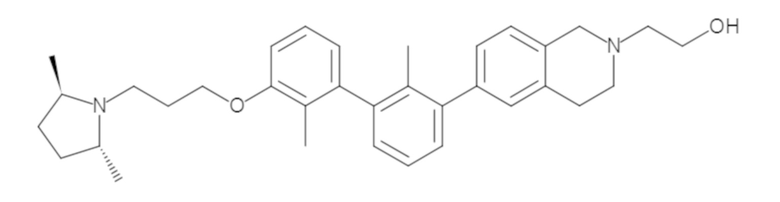
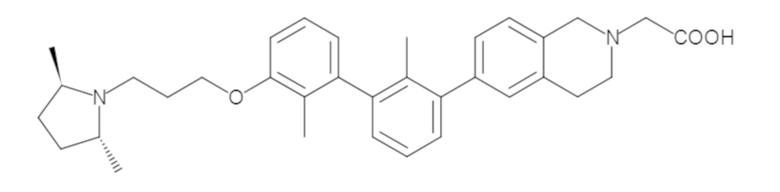
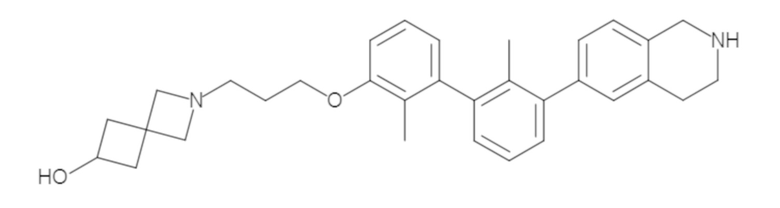
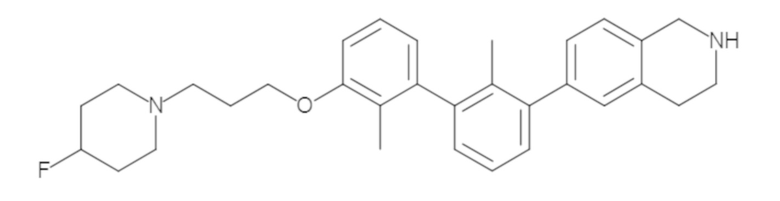
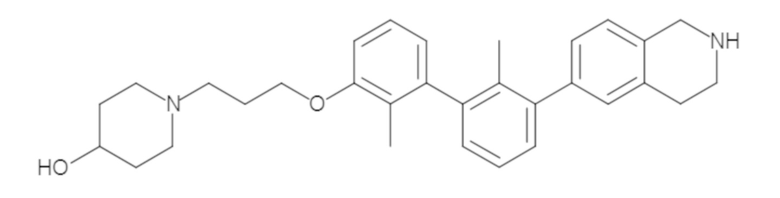
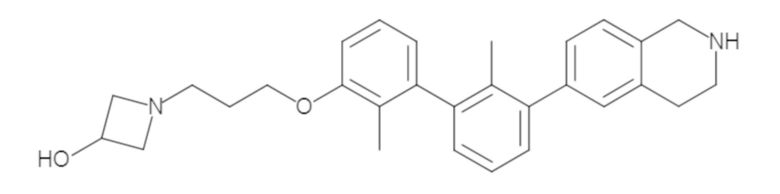
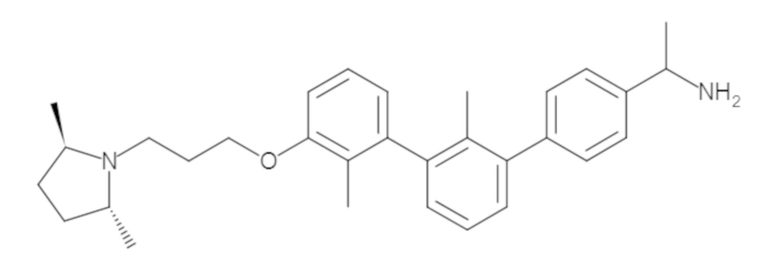
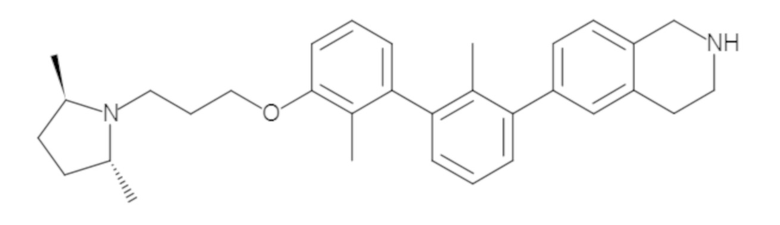
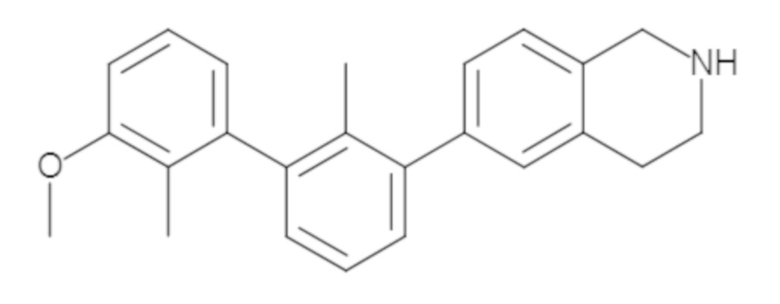
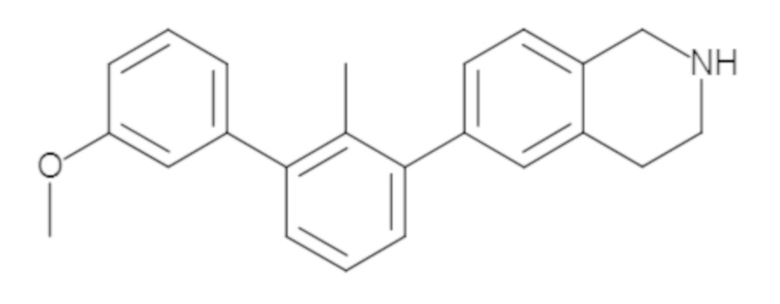
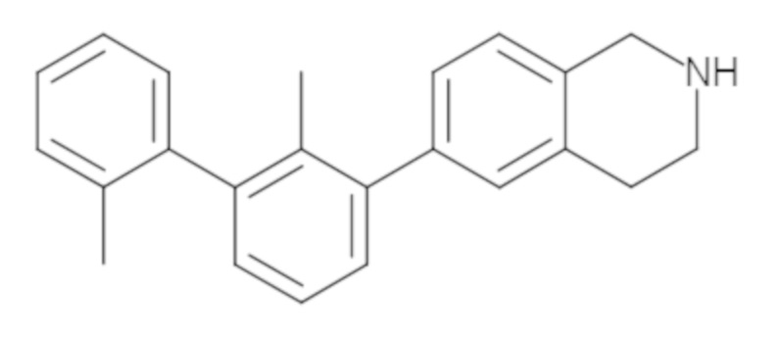
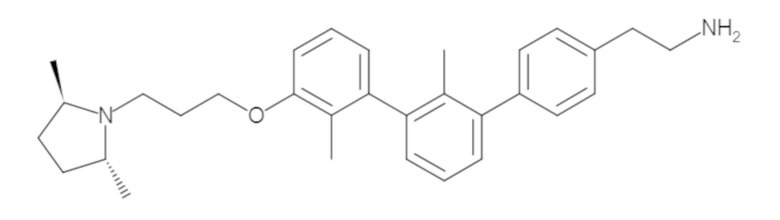
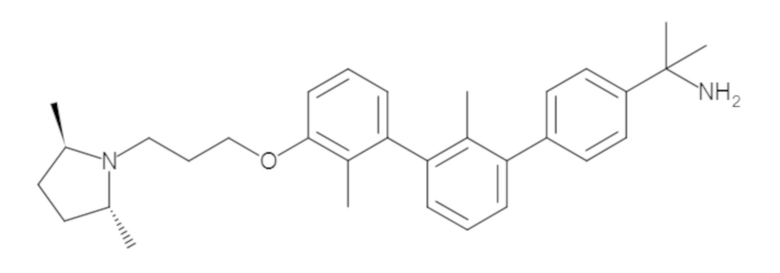
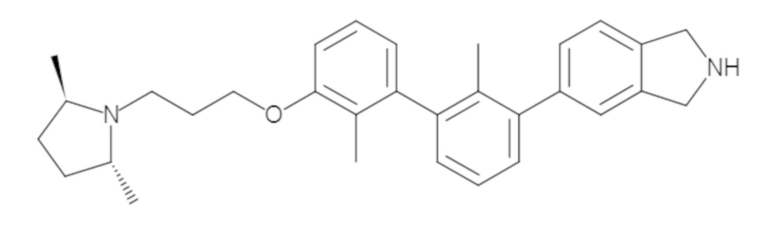
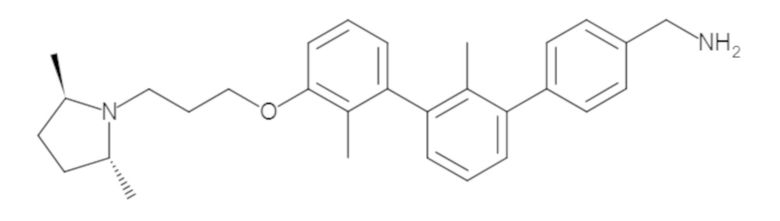
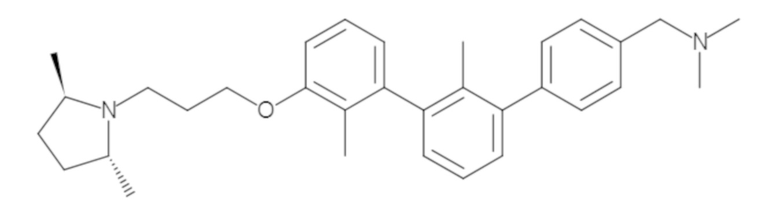
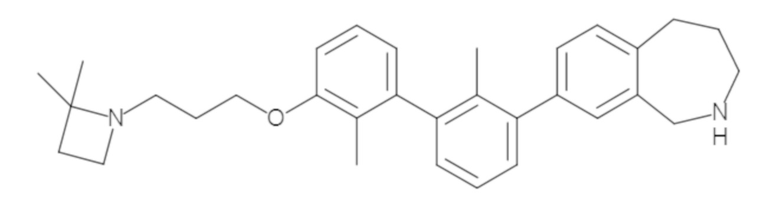
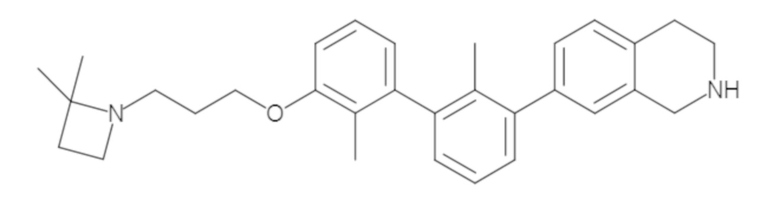
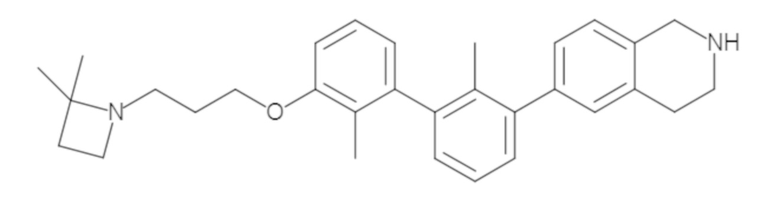
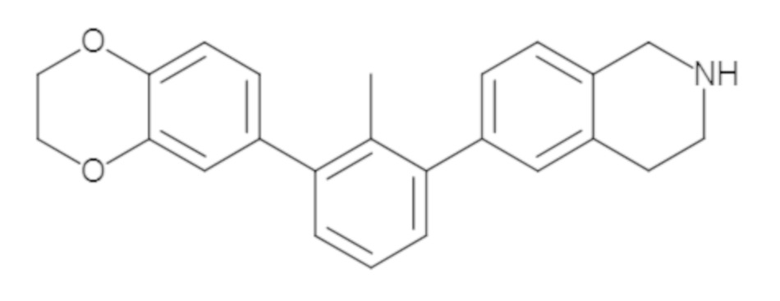
16. Фармацевтическая композиция для лечения заболевания или нарушения с участием PD-1 сигнального пути, содержащая терапевтически эффективное количество соединения по любому из пп. 1-15 или его фармацевтически приемлемой соли и фармацевтически приемлемое вспомогательное вещество, где заболевание или нарушение выбрано из группы, состоящей из следующих: меланома, глиобластома, рак пищевода, рак носоглотки, увеальная меланома, лимфома, лимфоцитарная лимфома, первичная лимфома ЦНС, Т-клеточная лимфома, диффузная крупноклеточная В-клеточная лимфома, первичная медистианальная В-крупноклеточная лимфома, рак предстательной железы, кастрационно-резистентный рак предстательной железы, хроническая миелоцитарная лейкемия, саркома Капоши, фибросаркома, липосаркома, хондросаркома, остеогенная саркома, ангиосаркома, лимфангиосаркома, синовиома, менингиома, лейомиосаркома, рабдомиосаркома, саркома мягких тканей, саркома, сепсис, рак желчного пузыря, базальноклеточная карцинома, рак вилочковой железы, рак щитовидной железы, рак паращитовидной железы, рак матки, рак надпочечников, инфекция печени, карцинома из клеток Меркеля, рак нервной системы, лимфома из клеток центра фолликула, рак ободочной кишки, лимфома Ходжкина, неходжкинская лимфома, лейкоз, хронический или острый лейкоз, острый миелобластный лейкоз, хронический миелобластный лейкоз, острый лимфобластный лейкоз, хронический лимфобластный лейкоз, множественная миелома, рак яичника, миелодиспластический синдром, кожная или внутриглазная злокачественная меланома, почечно-клеточный рак, мелкоклеточный рак легких, рак легких, мезотелиом, рак груди, плоскоклеточный немелкоклеточный рак легких, неплоскоклеточный немелкоклеточный рак легких, колоректальный рак, рак яичника, рак желудка, печеночно-клеточный рак, карцинома поджелудочной железы, рак поджелудочной железы, аденокарцинома протоков поджелудочной железы, плоскоклеточный рак головы и шеи, рак головы и шеи, рак желудочно-кишечного тракта, рак желудка, ВИЧ, гепатит А, гепатит В, гепатит С, гепатит D, вирус герпеса, папилломавирус, грипп, рак костей, рак кожи, рак прямой кишки, рак анального отверстия, рак яичка, рак фаллопиевых труб, рак эндометрия, рак шейки матки, рак вагины, рак вульвы, рак пищевода, рак тонкого кишечника, рак эндокринной системы, рак уретры, рак пениса, рак мочевого пузыря, рак почки, рак уретры, рак почечной лоханки, новообразования в центральной нервной системе (ЦНС), ангиогенез опухоли, рак оси позвоночника, глиома стволовой части мозга, аденома гипофиза, эпидермоидный рак, асбестоз, карцинома, аденокарцинома, папиллярная карцинома, цистаденокарцинома, бронхогенная карцинома, почечно-клеточный рак, переходно-клеточный рак, хориокарцинома, семинома, эмбриональный рак, опухоль Вильмса, плеоморфная аденома, папиллома клеток печени, почечная канальцевая аденома, цистаденома, папиллома, аденома, лейомиома, рабдомиома, гемангиома, лимфангиома, остеома, хондрома, липома и фиброма.
17. Способ лечения субъекта, страдающего или подверженного заболеванию или нарушению с участием PD-1 сигнального пути, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-15 или его фармацевтически приемлемой соли, где заболевание или нарушение выбрано из группы, состоящей из следующих: меланома, глиобластома, рак пищевода, рак носоглотки, увеальная меланома, лимфома, лимфоцитарная лимфома, первичная лимфома ЦНС, Т-клеточная лимфома, диффузная крупноклеточная В-клеточная лимфома, первичная медистианальная В-крупноклеточная лимфома, рак предстательной железы, кастрационно-резистентный рак предстательной железы, хроническая миелоцитарная лейкемия, саркома Капоши, фибросаркома, липосаркома, хондросаркома, остеогенная саркома, ангиосаркома, лимфангиосаркома, синовиома, менингиома, лейомиосаркома, рабдомиосаркома, саркома мягких тканей, саркома, сепсис, рак желчного пузыря, базальноклеточная карцинома, рак вилочковой железы, рак щитовидной железы, рак паращитовидной железы, рак матки, рак надпочечников, инфекция печени, карцинома из клеток Меркеля, рак нервной системы, лимфома из клеток центра фолликула, рак ободочной кишки, лимфома Ходжкина, неходжкинская лимфома, лейкоз, хронический или острый лейкоз, острый миелобластный лейкоз, хронический миелобластный лейкоз, острый лимфобластный лейкоз, хронический лимфобластный лейкоз, множественная миелома, рак яичника, миелодиспластический синдром, кожная или внутриглазная злокачественная меланома, почечно-клеточный рак, мелкоклеточный рак легких, рак легких, мезотелиом, рак груди, плоскоклеточный немелкоклеточный рак легких, неплоскоклеточный немелкоклеточный рак легких, колоректальный рак, рак яичника, рак желудка, печеночно-клеточный рак, карцинома поджелудочной железы, рак поджелудочной железы, аденокарцинома протоков поджелудочной железы, плоскоклеточный рак головы и шеи, рак головы и шеи, рак желудочно-кишечного тракта, рак желудка, ВИЧ, гепатит А, гепатит В, гепатит С, гепатит D, вирус герпеса, папилломавирус, грипп, рак костей, рак кожи, рак прямой кишки, рак анального отверстия, рак яичка, рак фаллопиевых труб, рак эндометрия, рак шейки матки, рак вагины, рак вульвы, рак пищевода, рак тонкого кишечника, рак эндокринной системы, рак уретры, рак пениса, рак мочевого пузыря, рак почки, рак уретры, рак почечной лоханки, новообразования в центральной нервной системе (ЦНС), ангиогенез опухоли, рак оси позвоночника, глиома стволовой части мозга, аденома гипофиза, эпидермоидный рак, асбестоз, карцинома, аденокарцинома, папиллярная карцинома, цистаденокарцинома, бронхогенная карцинома, почечно-клеточный рак, переходно-клеточный рак, хориокарцинома, семинома, эмбриональный рак, опухоль Вильмса, плеоморфная аденома, папиллома клеток печени, почечная канальцевая аденома, цистаденома, папиллома, аденома, лейомиома, рабдомиома, гемангиома, лимфангиома, остеома, хондрома, липома и фиброма.
18. Применение соединения по любому из пп. 1-15 или его фармацевтически приемлемой соли для лечения субъекта, страдающего или подверженного заболеванию или нарушению с участием PD-1 сигнального пути, где заболевание или нарушение выбрано из группы, состоящей из следующих: меланома, глиобластома, рак пищевода, рак носоглотки, увеальная меланома, лимфома, лимфоцитарная лимфома, первичная лимфома ЦНС, Т-клеточная лимфома, диффузная крупноклеточная В-клеточная лимфома, первичная медистианальная В-крупноклеточная лимфома, рак предстательной железы, кастрационно-резистентный рак предстательной железы, хроническая миелоцитарная лейкемия, саркома Капоши, фибросаркома, липосаркома, хондросаркома, остеогенная саркома, ангиосаркома, лимфангиосаркома, синовиома, менингиома, лейомиосаркома, рабдомиосаркома, саркома мягких тканей, саркома, сепсис, рак желчного пузыря, базальноклеточная карцинома, рак вилочковой железы, рак щитовидной железы, рак паращитовидной железы, рак матки, рак надпочечников, инфекция печени, карцинома из клеток Меркеля, рак нервной системы, лимфома из клеток центра фолликула, рак ободочной кишки, лимфома Ходжкина, неходжкинская лимфома, лейкоз, хронический или острый лейкоз, острый миелобластный лейкоз, хронический миелобластный лейкоз, острый лимфобластный лейкоз, хронический лимфобластный лейкоз, множественная миелома, рак яичника, миелодиспластический синдром, кожная или внутриглазная злокачественная меланома, почечно-клеточный рак, мелкоклеточный рак легких, рак легких, мезотелиома, рак груди, плоскоклеточный немелкоклеточный рак легких, неплоскоклеточный немелкоклеточный рак легких, колоректальный рак, рак яичника, рак желудка, печеночно-клеточный рак, карцинома поджелудочной железы, рак поджелудочной железы, аденокарцинома протоков поджелудочной железы, плоскоклеточный рак головы и шеи, рак головы и шеи, рак желудочно-кишечного тракта, рак желудка, ВИЧ, гепатит А, гепатит В, гепатит С, гепатит D, вирус герпеса, папилломавирус, грипп, рак костей, рак кожи, рак прямой кишки, рак анального отверстия, рак яичка, рак фаллопиевых труб, рак эндометрия, рак шейки матки, рак вагины, рак вульвы, рак пищевода, рак тонкого кишечника, рак эндокринной системы, рак уретры, рак пениса, рак мочевого пузыря, рак почки, рак уретры, рак почечной лоханки, новообразования в центральной нервной системе (ЦНС), ангиогенез опухоли, рак оси позвоночника, глиома стволовой части мозга, аденома гипофиза, эпидермоидный рак, асбестоз, карцинома, аденокарцинома, папиллярная карцинома, цистаденокарцинома, бронхогенная карцинома, почечно-клеточный рак, переходно-клеточный рак, хориокарцинома, семинома, эмбриональный рак, опухоль Вильмса, плеоморфная аденома, папиллома клеток печени, почечная канальцевая аденома, цистаденома, папиллома, аденома, лейомиома, рабдомиома, гемангиома, лимфангиома, остеома, хондрома, липома и фиброма.
19. Соединение со структурой
или его фармацевтически приемлемая соль.
| RAJAKUMAR, P | |||
| et al | |||
| Synthesis, Antimicrobial Activity, and Molecular Docking Study of Some Novel Cyclophanes with Imino Intra-Annular Functionality | |||
| Australian Journal of Chemistry, 2013, 66(1), p.84-92 | |||
| UPPADINE, L | |||
| et al | |||
| Metal-directed self-assembly of terphenyl based dithiocarbamate ligands | |||
| Journal of the Chemical Society, Dalton |

