Область техники, к которой относится изобретение
Настоящее изобретение относится к новым семействам полипептидов и липополипептидов, которые обладают антибактериальной и эндотоксиннейтрализующей активностью. Новые соединения также могут быть использованы в комбинированной терапии с обычными антибиотиками или антиэндотоксиновыми средствами. Кроме того, настоящее изобретение раскрывает способы производства и применения новых соединений.
Уровень техники
Увеличивающееся распространение болезнетворных бактерий, которые являются устойчивыми к коммерчески доступным антибиотикам, привело к растущей заинтересованности в разработке пептидов как антибактериальных лекарственных препаратов. Действительно, большая часть инфекций, приобретаемых в клинике (вплоть до 70%), происходит в настоящее время из-за антибиотикорезистентных бактерий. В дополнение к появлению лекарственной устойчивости, лечение антибиотиками грамотрицательных инфекций может послужить причиной высвобождения эндотоксина, который инициирует септический шок, представляя дополнительную проблему для антибактериальной терапии. Септический шок является главной причиной смертности в блоках интенсивной терапии. Грамотрицательные бактерии, в частности, содержат липополисахариды (или LPS) в своей оболочке (капсуле), которые являются наиболее сильными известными активаторами этого ответа. Кроме того, антибиотики, используемые в настоящее время при грамотрицательных инфекциях, могут уничтожать бактерии, но введение антибиотиков не обезвреживает LPS, высвобожденные из наружных мембран погибающих бактерий. Подобное высвобождение LPS может фактически увеличить повреждение легкого и привести к септическому синдрому. Следовательно, лекарственные препараты, которые обладают антибактериальными свойствами и нейтрализуют высвобожденные эндотоксины, могут иметь огромное значение для лечения бактериальной инфекции.
Так как бактерии изменяются для того, чтобы появилась множественная резистентность к большому числу существующих антибиотиков, новый класс соединений предназначается скорее для сведения к минимуму быстрого появления бактериальной устойчивости. Природа учит нас, что эффекторные молекулы врожденного иммунитета млекопитающих могут обеспечить первую линию обороны от основного набора патогенных микроорганизмов. В частности, считается, что пептиды иммунной защиты являются многофункциональными эффекторными молекулами и представляют новые источники для разработки терапевтических средств, с помощью которых можно преодолеть антибактериальную устойчивость. Тогда как многие обычные антибиотики повреждают или убивают бактерии в течение нескольких дней, большинство антибактериальных пептидов убивают их почти мгновенно, т.е. в течение минут. Целый ряд антибактериальных пептидов также препятствует взаимодействию LPS с его рецепторами, такими как LBP, CD14 и MD-2/TLR4, приводя к ингибированию активации макрофагов, особенность, которая может понизить токсичность LPS.
Лактоферрины представляют собой железо-связывающие эндогенные гликопротеины, обнаруженные в продуктах экзокринной секреции млекопитающих и в гранулах нейтрофилов во время воспалительного ответа, которые обладают антибактериальной и LPS связывающей активностью. Лактоферрины проявляют многофункциональные свойства, которые включают антибактериальные, противогрибковые, противовирусные, противоопухолевые, противовоспалительные и иммунорегулирующие свойства. Известно, что лактоферрин и производные обладают способностью нейтрализовать бактериальный эндотоксин (LPS), таким образом, защищая организмы от вредного действия сепсиса. Таким образом, многие сообщения об антибактериальной и противовоспалительной активности лактоферрина in vitro определяют его как важный в иммунной защите от инфекции и чрезмерного воспаления. Протеолитическое расщепление человеческого лактоферрина in vitro и in vivo дает в результате пептидный фрагмент, называемый лактоферрицин, который обладает повышенной антибактериальной активностью по сравнению с целым лактоферрином. Ряд более коротких синтетических производных лактоферрицина проявляет антибактериальную активность против грамположительных и грамотрицательных бактерий и специфически связывается с LPS (Strϕm et al., J. Peptide Res. 57: 127-139 (2001)). Наряду с тем, что многие пептиды иммунной защиты подходят в качестве кандидатов в новые антибактериальные препараты, человеческий лактоферрин и производные являются уникальными в том, что они обладают многофакторной активностью и, вследствие их человеческой природы, они менее способны вызывать неблагоприятные физиологические эффекты.
Миристоилированный богатый аланином субстрат киназы С (MARCKS) и MARCKS-родственные белки являются главными субстратами протеинкиназы С во многих типах клеток. Было обнаружено, что транскрипция MARCKS подвергается значительной повышающей регуляции стимуляцией макрофагов и микроглиальных клеток в ответ на бактериальный LPS. LPS-связывающий мотив на MARCKS является очень похожим на эффективные антибактериальные гексапептиды, идентифицированные с помощью комбинаторной библиотеки. Так как MARCKS является от природы модифицированным посредством миристоилирования со стороны N-конца, можно ожидать, что включение липофильных групп в более короткие производные MARCKS может дать в результате сильную антибактериальную активность и/или связывание LPS.
Важность гидрофобности для липопептидов и особенно наличия алкильных или ацильных цепей липопептидов для их антибактериальной активности была описана (например, полимиксины, октапептины и даптомицин). Более того, полимиксины имеют высокое сродство к молекулам LPS и проникают через наружную мембрану посредством разрушения отрицательно заряженных групп головок через вытеснение двухвалентных катионов из их мест связывания на LPS. Ацилирование N-конца девятичленного центрального пептида лактоферрицина В приводит к улучшенной антибактериальной активности. Длинноцепочечные N-ациламинокислотные антибиотики недавно были выделены из образцов почвы. Также сообщалось, что липополиамины (DOSPER, DOSPA, и DOGS, содержащие С17 акильную цепь) проявляют активность против эндотоксинов посредством блокирования (связывания) LPS и, по очереди, посредством блокирования в нисходящем направлении событий клеточной активации, что приводит к выработке провоспалительных медиаторов. Несмотря на неэффективность при проверке в одиночку на нейтропенических крысах с инвазивной грамотрицательной бактериемией, вызванной Pseudomonas aeruginosa, эти липополиамины значительно увеличивали выживаемость при применении с антибиотиком цефтазимидином в сравнении с одним цефтазимидином.
Ожидается, что продолжающаяся разработка новых систем доставки увеличит возможности пептидов в области терапии инфекционных болезней. Например, доставка пептидов к ткани мозга теперь возможна вследствие нового развития стратегии слитых пептидов (Bickel et al.. Adv. Drug Deliv. Rev. 46: 247-279 (2001)). Был описан успешный случай пневмонэктомии и последующего лечения с помощью волокна с иммобилизированным полимиксином В и непрерывной гемодиафильтрации для устранения факторов, являющихся причиной сепсиса ("септического шока") у пациента, страдающего туберкулезом легких (Takahashi et al., Ann. Thorac. Cardiovasc. Surg. 9: 319-322 (2003)). Подобным образом, разрабатывается несколько стратегий для увеличения биодоступности пептидных лекарственных препаратов после орального применения. Это включает доставку препарата в форме частиц, таких как наночастицы, микрокапсулы, липосомы или эмульсии, мукоадгезивную доставку и использование веществ, способствующих проникновению (Kompella et al., Adv. Drug Deliv. Rev. 46: 211-245 (2001)).
Работа Japelj В. et al., (J Biol Chem. 280 (17) (2005): 16955-61) относится к эндотоксиннейтрализующему пептиду (LF11), содержащему аминокислотную последовательность FQWQRNIRKVR-NH2, которая получена из лактоферрина. В течение этих исследований был определен аминокислотный остаток LF11, который является ответственным за связывание LPS.
Сопоставимо с работой Japelj В et al. в работе Andrä J. et al. (Biochem J. 385(2005): 135-43) также было исследовано взаимодействие пептида LF11, производного лактоферрина, который был связан с С12-алкильной цепью, с LPS.
В работе Farnaud S. et al. (FEMS Microbiol Lett. 238 (1) (2004): 221-6) раскрыты антибактериальные пептиды, которые получены из коровьего и человеческого лактоферрина. Авторы этой научной работы исследовали связывание этих пептидов с LPS.
США 2003/0022821 A1 относится к модифицированным пептидам лактоферрина, содержащим от 7 до 25 аминокислотных остатков, при этом три или больше из указанных аминокислотных остатков являются катионными. Пептиды согласно США 2003/0022821 A1, кроме того, включают большой и липофильный аминокислотный остаток.
Работа Chen PW et al. (Am J Vet Res. 64 (9) (2003): 1088-92) относится к аналогам лактоферрина с высоким содержанием липофильных и катионных аминокислотных остатков.
Раскрытие изобретения
Целью настоящего изобретения является предоставление пептидов, проявляющих антибактериальные или эндотоксиннейтрализующие свойства.
Следовательно, настоящее изобретение относится к пептидам с антибактериальной или эндотоксиннейтрализующей активностью, имеющим формулу:
(Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S, в которой
Xaa1 представляет собой гидрофобную аминокислоту, предпочтительно выбранную из группы, состоящей из фенилаланина (Phe), аланина (Ala), лейцина (Leu) и валина (Val),
Хаа2 представляет собой основную аминокислоту, предпочтительно выбранную из группы, состоящей из аргинина (Arg) и лизина (Lys),
Хаа3 представляет собой гидрофобную аминокислоту, предпочтительно триптофан (Trp),
Хаа4 является выбранным из группы, состоящей из аланина (Ala), аргинина (Arg), глутамина (Gln), аспарагина (Asn), пролина (Pro), изолейцина (Ile), лейцина (Leu) и валина (Val),
Xaa5 является выбранным из группы, состоящей из изолейцина (Ile), фенилаланина (Phe), триптофана (Trp) и тирозина (Tyr),
Хаа6 является выбранным из группы, состоящей из аргинина (Arg), лизина (Lys), тирозина (Tyr) и фенилаланина (Phe),
Хаа7 представляет собой гидрофобную аминокислоту, предпочтительно выбранную из группы, состоящей из изолейцина (Ile), триптофана (Trp), валина (Val) и лейцина (Leu), и
Xaa8 является выбранным из группы, состоящей из аргинина (Arg), лизина (Lys), изолейцина (Ile) и серина (Ser), и в которой
О является 0,
М является 1 или 2,
Р является 2 или 3,
Q и R являются 1, и
S является 1, 2, 3 или 4.
Настоящее изобретение относится к выделенным пептидам, полипептидам и липопептидам, которые проявляют антибактериальную активность и эндотоксиннейтрализующую активность. Эти молекулы показывают широкий спектр активности против различных патогенов (в том числе бактерий, вирусов, грибов и т.д.). Разработка активных соединений была основана на SAR исследованиях, биофизических, микробиологических, иммунологических и структурных экспериментах с применением новых пептидных соединений.
Данное изобретение предоставляет пептиды и липопептиды, обладающие антибактериальной и/или антиэндотоксической активностью. При использовании здесь термин "аминокислота" относится и к природным аминокислотам и к их производным. Кроме того, имитатор одной или больше аминокислот, по-другому известный как пептидный миметик или пептидомиметик, также может быть использован. При использовании здесь термин "имитатор" означает аминокислоту или аналог аминокислоты, который имеет то же самое или подобное функциональное свойство аминокислоты. Пептидный миметик или пептидомиметик представляет собой органическую молекулу, которая сохраняет такие же фармакофорные группы пептидной цепи как те, которые присутствуют в соответствующем пептиде. Замещение аминокислот аминокислотами неприродного происхождения или пептидомиметиками, как описано выше, может увеличить общую активность или другие свойства отдельного пептида, исходя из модификаций функциональностей боковой цепи. Например, эти типы модификаций в отношении к приведенным в пример пептидам могут усилить стабильность пептида к ферментативному расщеплению или увеличить биологическую активность или уменьшить иммуногенность.
Специалист в данной области техники может легко синтезировать пептиды и полипептиды данного изобретения. Стандартные способы получения синтетических пептидов хорошо известны в данной области техники. Пептиды изобретения могут быть синтезированы с помощью таких обычно используемых способов как t-BOC или FMOC защита альфа-аминогрупп. Оба способа включают ступенчатый синтез, при этом одна аминокислота добавляется на каждой стадии, начиная от карбоксильного конца пептида (см., Coligan et al., Current Protocols in Immunology, Wiley Interscience, 1991, Unit 9). Пептиды изобретения также могут быть синтезированы методами твердофазного синтеза пептидов, хорошо известными в данной области техники (Merrifield, J. Am. Chem. Soc., 85: 2149, 1963), и Stewart и Young, Solid Phase Peptides Synthesis, Pierce, Rockford, I11 (1984)). Пептиды могут быть синтезированы с использованием сополи(стирен-дивинилбензен)содержащего 0,1-1,0 ммоль аминов/г полимера. По окончании химического синтеза пептиды могут быть лишены защиты и отщеплены от полимера обработкой жидким HF-10% анизолом в течение приблизительно от 0,25 до 1 часа при 0°С. После выпаривания реагентов пептиды экстрагируют из полимера с помощью 1% раствора уксусной кислоты, который затем лиофилизируют, чтобы получить необработанный материал. Обычно он может быть очищен с помощью таких методов, как гель-фильтрация на Сефадексе G-15 с использованием 5% уксусной кислоты в качестве растворителя, с помощью высокоэффективной жидкостной хроматографии и тому подобного. Лиофилизация соответствующих фракций колонки будет давать однородный пептид или пептидные производные, которые затем могут быть охарактеризованы с помощью таких стандартных методов, как аминокислотный анализ, тонкослойная хроматография, высокоэффективная жидкостная хроматография, абсорбционная спектроскопия в ультрафиолетовом диапазоне, молярное вращение, растворимость, и оценены с помощью твердофазной деградации по методу Эдмана (см., например, Protein Purification, M.P.Deutscher, ed. Methods in Enzymology, Vol 182, Academic Press, 1990). Автоматический синтез, использующий FMOC твердофазные методы синтеза, можно выполнять с применением автоматизированного синтезатора пептидов (Model 432A, Applied Biosystems, Inc.).
Пептиды/полипептиды настоящего изобретения также могут быть синтезированы с помощью метода бактериальных слитых белков, в котором анионнесущий пептид соединяется с катионным пептидом. Способ такого бактериального производства катионных пептидов, имеющих антибактериальную активность, предоставлен в США 5593866.
Пептид настоящего изобретения, полученный таким образом, может быть очищен с помощью способов выделения/очистки белков, общеизвестных в области химии белков. Конкретнее, можно упомянуть, например, экстракцию, рекристаллизацию, высаливание сульфатом аммония, сульфатом натрия и т.д., центрифугирование, диализ, ультрафильтрацию, адсорбционную хроматографию, ионообменную хроматографию, гидрофобную хроматографию, нормально-фазовую хроматографию, обращеннофазовую хроматографию, способ гель-фильтрации, гельпроникающую хроматографию, аффинную хроматографию, электрофорез, метод противоточного распределения и т.д. и их комбинации. Наиболее эффективным является способ обращеннофазовой высокоэффективной жидкостной хроматографии.
Пептид настоящего изобретения может образовывать соль при добавлении кислоты. Примеры кислоты включают неорганические кислоты (такие как трифторуксусная кислота, соляная кислота, бромисто-водородная кислота, ортофосфорная кислота, азотная кислота и серная кислота) или органические карбоновые кислоты (такие как уксусная кислота, пропионовая кислота, малеиновая кислота, янтарная кислота, яблочная кислота, лимонная кислота, винная кислота и салициловая кислота), кислые сахара, такие как глюкуроновая кислота, галактуроновая кислота, глюконовая кислота, аскорбиновая кислота и т.д., кислые полисахариды, такие как гиалуроновая кислота, хондроитинсульфаты, альгиновая кислота, или органические сульфоновые кислоты (такие как метансульфокислота и п-толуолсульфокислота) и тому подобное. Из этих солей предпочтительной является фармацевтически приемлемая соль.
Пептид настоящего изобретения может образовывать соль с основным веществом. Примеры соли включают, например, фармацевтически приемлемые соли, выбранные из солей с неорганическими основаниями, такими как соли щелочных металлов (соль натрия, соль лития, соль калия и т.д.), соли щелочноземельных металлов, соли аммония и тому подобные, или солей с органическими основаниями, такими как соли диэтаноламина, соли циклогексиламина и тому подобные.
Термин "аминокислота", использованный здесь, означает L-аминокислоту. Однако в производстве пептидов согласно настоящему изобретению также могут быть использованы D-аминокислоты.
"Пептиды", при использовании в данном описании, содержат от 2 до 50 аминокислотных остатков. "Полипептиды" и "белки" содержат больше чем 50 аминокислотных остатков.
"Антибактериальный", при использовании здесь, относится к биологической активности пептидов и полипептидов настоящего изобретения и означает, что пептид/полипептид имеет способность убивать, нарушать размножение или иным способом подрывать развитие микробов, так что полипептид имеет минимальную ингибирующую концентрацию ("MIC" как определено в среде Мюллера-Хинтона, следуя рекомендациям Института клинических и лабораторных стандартов, CLSI - прежде NCCLS) меньше чем 32 мкМ, предпочтительно меньше чем 16 мкМ. Микроорганизмы, подлежащие ингибированию согласно настоящему изобретению, включают бактерии, грибы, дрожжи и т.д. Способы определения MIC антибактериального полипептида известны специалистам в данной области техники и описаны, например, в Powell et al. (Molecular Plant-Microbe Interactions, 8: 792-794 (1995)), Wu и Hancock (J. Biol Chem. 274: 29-35 (1999)) и Lorian V. ("Antimicrobials in laboratory Medicine", 1996 4th ed. pp.330-396, Williams и Wilkins, Baltimore, Md). Изучение MIC дает возможность определения самой низкой концентрации пептида, которая ингибирует размножение и рост микроорганизмов. Для целей настоящего изобретения полагают, что полипептид является антибактериальным, если он обладает вышеупомянутой MIC в отношении микроорганизма, как использовано здесь.
"Эндотоксиннейтрализующая" и/или связывающая активность пептидов настоящего изобретения может быть проверена в исследовании in vitro с использованием, например, клеточной линии макрофагов (Gough et al. (1996) Infect. Immun. 64: 4922-4927).
Пептиды согласно настоящему изобретению также демонстрируют противогрибковую активность. Эта активность была показана для некоторых грибов, например Cryptococcus neoformans.
Формула предпочтительно включает аминокислотную последовательность, выбранную из группы, состоящей из FWQRIRKVR (SEQ ID No. 1), FWQRRIRKVRR (SEQ ID No. 2), FWQRKIRKVRK (SEQ ID No. 3), FWQRNIRIRR (SEQ ID No. 4), FWQRNIRKVR (SEQ ID No. 5), FWQRNIRVR (SEQ ID No. 6), FWQRNIRKVRR (SEQ ID No. 7), FWQRNIRKVKK (SEQ ID No. 8), FWQRNIRKVRRR (SEQ ID No. 9), FWQRNIRKVKKK (SEQ ID No. 10), FWQRNIRKVRRRR (SEQ ID No. 11), FWQRNIRKVRRRI (SEQ ID No. 12), FWQRNIRKVKKKK (SEQ ID No. 13), FWQRNIRKVKKKI (SEQ ID No. 14), FWQRNIRKIR (SEQ ID No. 15), FWQRNIRKLR (SEQ ID No. 16), FWQRNIRKWR (SEQ ID No. 17), FWQRNWRKVR (SEQ ID No. 18), FWQRNFRKVR (SEQ ID No. 19), FWQRNYRKVR (SEQ ID No. 20), FWQRNIRKVS (SEQ ID No. 21), FWQRRIRIRR (SEQ ID No. 22), FWQRPIRKVR (SEQ ID No. 23), FWQRRIRKWR (SEQ ID No. 24), FWQRRIRRWRR (SEQ ID No. 25), FWPRNIRKVR (SEQ ID No. 26), FWARNIRKVR (SEQ ID No. 27), FWIRNIRKVR (SEQ ID No. 28), FWLRNIRKVR (SEQ ID No. 29), FWVRNIRKVR (SEQ ID No. 30), FWQRNIFKVR (SEQ ID No. 31), FWQRNIYKVR (SEQ ID No. 32), FAWQRNIRKVR (SEQ ID No. 33), FLWQRNIRKVR (SEQ ID No. 35) и FVWQRNIRKVR (SEQ ID No. 36).
При использовании в настоящем изобретении маленькие буквы в аминокислотной последовательности означают, что эти конкретные аминокислотные остатки находятся в D-конфигурации, а не в L-конфигурации (заглавные буквы).
Другой аспект настоящего изобретения относится к пептиду с антибактериальной или эндотоксиннейтрализующей активностью, имеющему формулу:
(Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S, в которой
Xaa1 является гидрофобной аминокислотой, предпочтительно выбранной из группы, состоящей из фенилаланина (Phe) и изолейцина (Ilе),
Хаа2 является основной аминокислотой, предпочтительно выбранной из группы, состоящей из аргинина (Arg), лизина (Lys),
Хаа3 является гидрофобной аминокислотой, предпочтительно триптофаном (Trp),
Хаа4 является выбранным из группы, состоящей из глицина (Gly), аспарагина (Asn), изолейцина (Ile) и фенилаланина (Phe),
Хаа5 является изолейцином (Ile) или триптофаном (Trp),
Xaa6 является аргинином (Arg) или лизином (Lys),
Хаа7 является гидрофобной аминокислотой, предпочтительно выбранной из группы, состоящей из изолейцина (Ile), триптофана (Trp) и валина (Val) и
Xaa8 является аргинином (Arg), и в которой
О является 0,
М является 1 или 2,
R является 0 или 1,
Р является 1, 2 или 3,
Q является 1, и
S является 0, 1 или 2.
Формула предпочтительно включает аминокислотную последовательность, выбранную из группы, состоящей из FWRIRKWR (SEQ ID No. 37), FWRIRKVR (SEQ ID No. 38), FWRWRR (SEQ ID No. 39), FWRRWRR (SEQ ID No. 40), FWRRWIRR (SEQ ID No. 41), FWRGWRIRR (SEQ ID No. 42), FWRRFWRR (SEQ ID No. 43), FWRWRWR (SEQ ID No. 44), FWRIWRWR (SEQ ID No. 45), FWRIWRIWR (SEQ ID No. 46), FWRNIRKWR (SEQ ID No. 47) и FWRRRIRIRR (SEQ ID No. 48).
Другой аспект настоящего изобретения относится к пептиду с антибактериальной или эндотоксиннейтрализующей активностью, имеющему формулу:
(Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S, в которой
Xaa1 является гидрофобной аминокислотой, предпочтительно выбранной из группы, состоящей из пролина (Pro) и фенилаланина (Phe),
Хаа2 является основной аминокислотой, предпочтительно выбранной из группы, состоящей из аргинина (Arg), лизина (Lys)
Хаа3 является гидрофобной аминокислотой, предпочтительно триптофаном (Trp),
Хаа4 является выбранным из группы, состоящей из аланина (Ala), аргинина (Arg), глутамина (Gln), лизина (Lys), триптофана (Trp) и изолейцина (Ile),
Хаа5 является выбранным из группы, состоящей из изолейцина (Ilе) и триптофана (Trp),
Хаа6 является выбранным из группы, состоящей из аргинина (Arg) и аспартата (Asp),
Хаа7 является гидрофобной аминокислотой, предпочтительно выбранной из группы, состоящей из изолейцина (Ile), триптофана (Trp), фенилаланина (Phe), валина (Val) и лейцина (Leu), и
Xaa8 является выбранным из группы, состоящей из аргинина (Arg), лизина (Lys), изолейцина (Ile), серина (Ser) и аспартата (Asp), и в которой
О и Q являются 0,
М является 0, 1, 2 или 3,
R является 1 или 2,
Р является 1, 2 или 3, и
S является 1, 2 или 3.
Формула предпочтительно включает аминокислотную последовательность, выбранную из группы, состоящей из PFWRWRIWR (SEQ ID No. 50), PFWRIRIRR (SEQ ID No. 51), PFWRQRIRR (SEQ ID No. 52), PFWRARIRR (SEQ ID No. 53), PFWRKRIRR (SEQ ID No. 54), PFWRKRLRR (SEQ ID No. 55), PFWRKRWRR (SEQ ID No. 56), PFWRRRIRR (SEQ ID No. 57), PFWRRRWRR (SEQ ID No. 58), PFWRIRIRRD (SEQ ID No. 59), PFFWRIRIRR (SEQ ID No. 60), PWRIRIRR (SEQ ID No. 61), PFWRRQIRR (SEQ ID No. 81), PFWRKKLKR (SEQ ID No. 82), PWRRIRR (SEQ ID No. 83), PWRRKIRR (SEQ ID No. 84) и PFWRRIRIRR (SEQ ID No. 85).
Еще один аспект настоящего изобретения относится к пептиду с антибактериальной или эндотоксиннейтрализующей активностью, имеющему формулу:
(Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S, в которой
Xaa1 является гидрофобной аминокислотой, предпочтительно выбранной из группы, состоящей из пролина (Pro) и фенилаланина (Phe),
Хаа2 является основной аминокислотой, предпочтительно аргинином (Arg),
Хаа3 является гидрофобной аминокислотой, предпочтительно триптофаном (Trp),
Хаа4 является выбранным из группы, состоящей из аланина (Ala), аргинина (Arg), глутамина (Gln), аспарагина (Asn) и лизина (Lys),
Хаа5 является выбранным из группы, состоящей из изолейцина (Ile), фенилаланина (Phe) и триптофана (Trp),
Хаа6 является выбранным из группы, состоящей из глутамина (Gln), аргинина (Arg) и аспарагина (Asn),
Хаа7 является гидрофобной аминокислотой, предпочтительно выбранной из группы, состоящей из изолейцина (Ile), триптофана (Trp) и фенилаланина (Phe), и
Xaa8 является аргинином (Arg), и в которой
М является 0, 1, 2 или 3,
О является 0 или 1,
Р является 1, 2 или 3,
Q является 1 или 2, и
R и S являются 0, 1 или 2.
Формула предпочтительно включает аминокислотную последовательность, выбранную из группы, состоящей из FWRNIRIRR (SEQ ID No. 72), FWQRIRIRR (SEQ ID No. 73), FWRWRIWR (SEQ ID No. 74), FWRIRIRR (SEQ ID No. 75), FWRNIRIWRR (SEQ ID No. 76) и FwRNIRIRR (SEQ ID No. 77).
Другой аспект настоящего изобретения относится к пептиду с антибактериальной или эндотоксиннейтрализующей активностью, имеющему формулу, включающую аминокислотную последовательность, выбранную из группы, состоящей из RFWQRNIRKVRR (SEQ ID No. 62), RFWQRNIRKYR (SEQ ID No. 63), PFWQRNIRKWR (SEQ ID No. 64), RFRWQRNIRKYRR (SEQ ID No. 65), RWKRINRQWF (SEQ ID No. 66), KRFCFKK (SEQ ID No. 67), KRFSFKKC (SEQ ID No. 68), KRWSWKK (SEQ ID No. 69), FRFSFKK (SEQ ID No. 70), RRFWFRR (SEQ ID No. 71), RFWQRNIRIRR (SEQ ID No. 78), RWQRNIRIRR (SEQ ID No. 79) и RRWFWRR (SEQ ID No. 86).
Другой аспект настоящего изобретения относится к пептиду с антибактериальной или эндотоксиннейтрализующей активностью, имеющему формулу FIWQRNIRKVR (SEQ ID No. 34), FIWRWRWR (SEQ ID No. 49) и RRIRINRQWF (SEQ ID No. 80).
N- и/или С-конец пептидов согласно настоящему изобретению может иметь модификации, такие как ац(ет)илирование, амидирования, эстерификации, восстановления, окисления, (ковалентное) связывание с линкером, пептидные связи, дисульфидные связи и т.д. Пептиды могут быть модифицированы сверх того, например, углеводородами, линкерными молекулами, липидами и т.д.
С-конец пептидов согласно настоящему изобретению состоит предпочтительно из группы, выбранной из группы, состоящей из карбоксильной группы, амидогрупп, в частности состоящей из N-метиламидогруппы, сложного эфира, эфира или кетона, предпочтительно содержащего от 1 до 20 атомов углерода, более предпочтительно от 1 до 10 атомов углерода.
Согласно предпочтительному варианту осуществления настоящего изобретения ацильная группа является связанной с N-концом или С-концом пептида.
Для того чтобы увеличить гидрофобность пептидов согласно настоящему изобретению и в результате увеличить взаимодействие пептидов, например, с гидрофобными частями клеток (например, клеточной мембраной), пептиды предпочтительно модифицируют ацильными группами.
Ацильная группа, которая будет связана с пептидами согласно настоящему изобретению, предпочтительно является гидрофобной цепью, выбранной из группы, состоящей из насыщенных и ненасыщенных линейных и разветвленных ацильных цепей из С2-С20, бензил-производных и F-moc.
Ацильная группа является предпочтительно выбранной из группы, состоящей из додеканоил-группы, деканоил-группы, октаноил-группы, гексаноил-группы, 2-метилгексаноил-группы, 2-этилгексаноил-группы, 2-пропилпентаноил-группы, 2-бутилоктаноил-группы, 2,2-диметилбутаноил-группы, 2-метилпентаноил-группы, 3-метилпентаноил-группы, 4-метилпентаноил-группы, 6-метилоктаноил-группы, бензил-группы и дициклогексилацетил-группы.
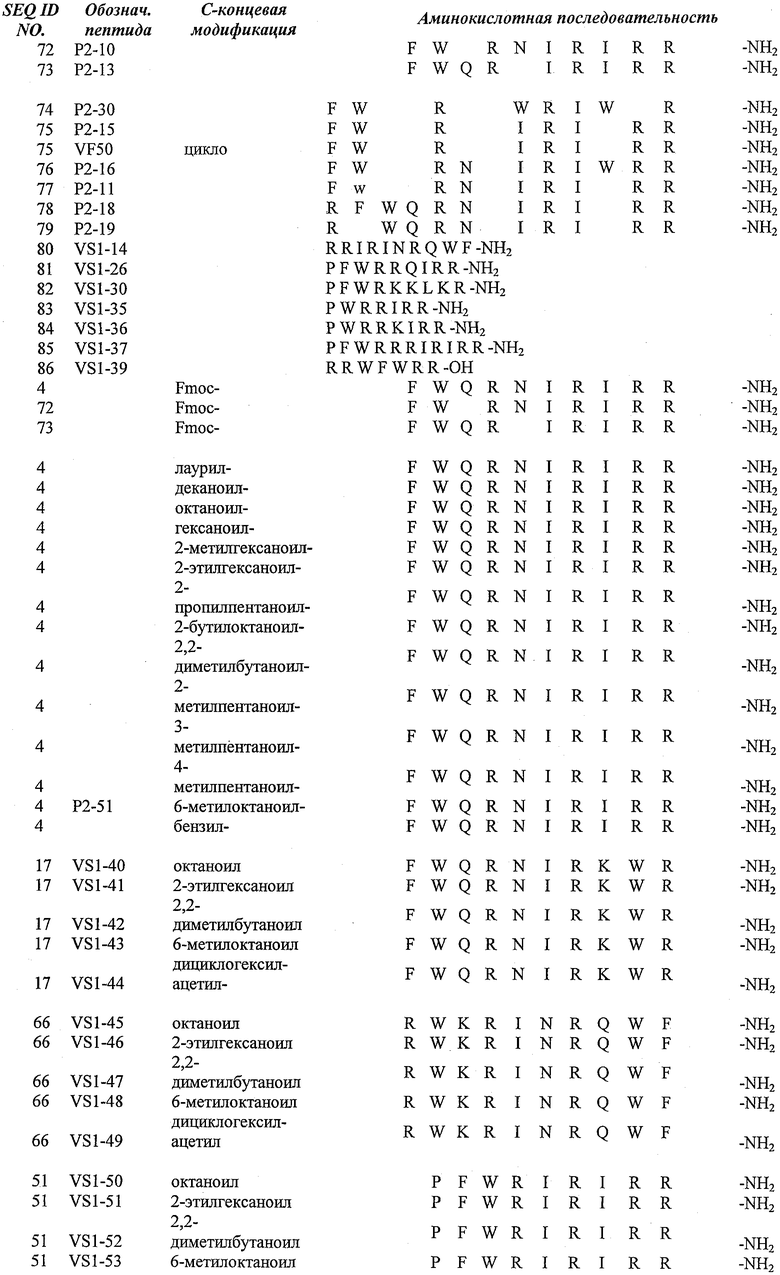
Особенно предпочтительные модифицированные или немодифицированные пептиды согласно настоящему изобретению можно найти в таблице 1.
Таблица 1. Пептиды согласно настоящему изобретению



Следующий аспект настоящего изобретения относится к полипептиду, включающему пептид согласно настоящему изобретению.
Пептиды настоящего изобретения также могут быть частью полипептида, при условии, что (не встречающийся в природе) полипептид, включающий указанный пептид(ы), проявляет ту же самую антибактериальную и/или эндотоксиннейтрализующую активность. Однако слитый полипептид может демонстрировать более низкую или даже более высокую активность, чем пептид.
Различные пептиды согласно настоящему изобретению также могут быть связаны/присоединены друг к другу для образования новых пептидов или полипептидов. То же самое относится к применению пептидов согласно настоящему изобретению в качестве повторяющихся звеньев для того, чтобы получить пептиды или полипептиды с двумя, тремя, четырьмя, пятью, десятью или двадцатью повторяющимися звеньями.
Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей пептид или полипептид согласно настоящему изобретению.
Такая композиция может быть использована для лечения и/или предотвращения, например, бактериальной инфекции или септического шока. Наконец, настоящее изобретение относится к способу совместного введения полипептида или липопептида из данного изобретения с другими антибактериальными или антисептическими препаратами в фармацевтически приемлемом носителе или инертном веществе для улучшения эффективности указанных других антибактериальных или антисептических препаратов.
Согласно предпочтительному варианту осуществления настоящего изобретения композиция включает, кроме того, по меньшей мере, один добавочный антибактериальный или антисептический препарат.
Для получения фармацевтической композиции с еще лучшими антибактериальными и/или эндотоксиннейтрализующими эффектами добавляют дополнительные препараты, проявляющие свойства, сходные со свойствами пептидов согласно настоящему изобретению. Конечно, возможно также добавление веществ, обладающих активностью, помимо пептидов согласно настоящему изобретению. Эти вещества могут быть полезны для увеличения биодоступности, такой как, например, увеличение стабильности пептидов или их доставки.
Примеры отдельных веществ, которые могут быть объединены с пептидами изобретения, включают аминогликозиды (например, тобрамицин), пенициллины (например, пиперациллин), цефалоспорины (например, цефтазидим), фторхинолоны (например, ципрофлоксацин), карбепенемы (например, имипенем), тетрациклины и макролиды (например, эритромицин и кларитромицин). Композиция может также включать дополнение из антибиотиков для комбинированной или синергической терапии. Назначение соответствующего антибиотика обычно зависит от чувствительности микроорганизма, например, является ли микроорганизм грамотрицательным или грамположительным, и легко определяется специалистом в данной области техники. В дополнение к антибиотикам, перечисленным выше, обычные антибиотики включают аминогликозиды (амикацин, гентамицин, канамицин, нетилмицин, тобрамицин, стрептомицин, азитромицин, кларитромицин, эритромицин, эритромицин эстолат/этилсукцинат/глуцептат/лактобионат/стеарат), бета-лактамы, такие как пенициллины (например, пенициллин G, пенициллин V, метициллин, нафциллин, оксациллин, клоксациллин, диклоксациллин, ампициллин, амоксициллин, тикарциллин, карбенициллин, мезлоциллин, азлоциллин и пиперациллин), или цефалоспорины (например, цефалотин, цефазолин, цефаклор, цефамандол, цефокситин, цефуроксим, цефоницид, цефинетазол, цефотетан, цефпрозил, лоракарбеф, цефетамет, цефоперазон, цефотаксим, цефтизоксим, цефтриаксон, цефтазидим, цефепим, цефиксим, цефподоксин и цефсулодин). Другие классы антибиотиков включают карбапемены (например, имипенем), монобактамы (например, азтреонам), хинолоны (например, флероксацин, налидиксовая кислота, норфлоксацин, ципрофлоксацин, офлоксацин, еноксацин, ломефлоксацин и циноксацин), тетрациклины (например, доксициллин, миноциклин, тетрациклин) и гликопептиды (например, ванкомицин, тейкопланин), например. Другие антибиотики включают хлорамфеникол, клиндамицин, триметоприм, сульфаметоксазол, нитрофурантоин, рифампин и мупироцин.
Кроме того, композиция согласно настоящему изобретению может включать предпочтительно фармацевтически приемлемый эксципиент.
Фармацевтическая композиция настоящего изобретения может состоять только из одного пептида настоящего изобретения или может быть в виде композиции, содержащей пептид настоящего изобретения и фармацевтически приемлемый носитель. Фармацевтически приемлемый носитель, который может быть использован, особым образом не ограничивается и включает эксципиент, связующее вещество, смазывающее вещество, краситель, дезинтегрирующее вещество, буфер, изотоническое вещество, консервант, обезболивающее средство и тому подобное, что может быть использовано в медицинской области. Также это может быть использовано в комбинации с другим антибактериальным лекарственным средством, таким как лизоцим, антибиотики и тому подобное.
Композиция настоящего изобретения может быть использована для лечения, например, наружной части тела, инфицированной микроорганизмами, или для лечения бактериальной инфекции внутри тела, причем соответствующий способ введения, следовательно, может быть выбран, в зависимости от цели лечения, из числа инъекции (подкожной, внутрикожной, внутривенной, интраперитонеальной и т.д.), закапывания в глаза, инсталляции, подкожного введения, перорального приема, ингаляции и т.д.
К тому же лекарственная форма, такая как инъецируемые препараты (растворы, суспензии, эмульсии, твердые вещества, которые необходимо растворить при использовании, и т.д.), таблетки, капсулы, гранулы, порошки, жидкости, липосомные включения, мази, гели, наружные порошки, спреи, ингаляционные порошки, глазные капли, глазные мази, суппозитории, вагинальные суппозитории и тому подобное, может быть соответствующим образом выбрана в зависимости от способа введения, и антибактериальное лекарственное средство настоящего изобретения может быть разработано в соответствии с этим.
Другой аспект настоящего изобретения относится к применению пептида или полипептида согласно настоящему изобретению в качестве антибактериального или эндотоксиннейтрализующего средства.
Пептиды, раскрытые в данном описании, проявляют антибактериальную и/или эндотоксиннейтрализующую активность. Следовательно, эти пептиды могут быть соответствующим образом использованы или как антибактериальное средство, или как средство, нейтрализующее эндотоксин.
Другой аспект настоящего изобретения относится к применению пептида или полипептида согласно настоящему изобретению для производства лекарственного средства для лечения или предотвращения инфекций, вызываемых микроорганизмами, предпочтительно бактериями, или сепсиса или септического шока, предпочтительно вызванного эндотоксинами.
Вследствие их биологических характеристик пептиды настоящего изобретения соответствующим образом применяются в лекарственных средствах.
Согласно предпочтительному варианту осуществления настоящего изобретения лекарственное средство затем может включать, по меньшей мере, одно дополнительное антибактериальное или антисептическое средство.
Сверх того лекарственное средство включает предпочтительно фармацевтически приемлемый эксципиент.
Другой аспект настоящего изобретения относится к способу ингибирования роста, по меньшей мере, одного микроорганизма, включая стадию контактирования указанного микроорганизма с эффективным количеством пептида или полипептида согласно настоящему изобретению. Пептиды или полипептиды настоящего изобретения можно использовать для ингибирования роста микроорганизмов. Этот эффект может быть достигнут контактированием указанных молекул с микроорганизмами, которые необходимо подавить.
При использовании здесь термин "терапевтически эффективное количество" или "эффективное количество" для ингибирования роста микроорганизма относится к количеству пептида, достаточного для снижения ответа субъекта на LPS и уменьшения симптомов сепсиса. Термин "терапевтически эффективный" поэтому включает, что количество пептида является достаточным для предотвращения, и предпочтительно для снижения, по меньшей мере, до 50%, и более предпочтительно достаточно для снижения до 90%, клинически важного увеличения уровня TNF в плазме. Диапазоны доз для применения пептида являются достаточно большими, для того чтобы вызвать необходимый эффект. В большинстве случаев дозировка будет изменяться в зависимости от возраста, состояния, пола и степени зараженности пациента бактериями или другим агентом, как описано выше, и может быть определена специалистом в данной области техники. Дозировка может быть установлена отдельным врачом в случае любых противопоказаний. В любом случае эффективность лечения может быть определена мониторингом уровня LPS и TNF у пациента. Снижение сывороточных уровней LPS и TNF должно коррелировать с восстановлением пациента.
Согласно предпочтительному варианту осуществления настоящего изобретения указанный микроорганизм является грамположительной или грамотрицательной бактерией.
Пептиды или полипептиды, раскрытые здесь, в частности, являются эффективными против бактерий. Следовательно, предпочтительным микроорганизмом для контактирования предпочтительно являются микроорганизмы из семейства enterobactericeae, в частности Escherichia coli, Salmonella spp., Yersinia pestis, Yersinia enterocolitica или Klebsiella spp., предпочтительно из семейства Pseudomonadaceae, в частности Pseudomonas aeruginosa, предпочтительно из семейства Alcaligenaceae, в частности Bordetella bronchiseptica or Bordetella pertussis, предпочтительно из семейства Brucellaceae, в частности Brucella abortus, предпочтительно из семейства Moraxellaceae, в частности Acinetobacter baumanii, предпочтительно из семейства Xanthomonadaceae, в частности Stenotrophomonas maltophilia, предпочтительно из семейства Pasteuerellaceae, в частности Haemophilus Influenzae, предпочтительно из семейства Neisseriaceae, в частности Neisseria meningitidis, предпочтительно из семейства Staphylococcaceae, в частности Staphylococcus aureus or Staphylococcus epidermidis, предпочтительно из семейства Enterococcaceae, в частности Enterococcus faecalis, предпочтительно из семейства Streptococcaceae, в частности Streptococcus agalactiae и предпочтительно из семейства Chlamydiaceae, в частности Chlamydia pneumoniae.
Применение пептидов согласно настоящему изобретению особенно подходит, если указанный микроорганизм проявляет множественную лекарственную устойчивость.
Множественная лекарственная устойчивость (т.е. устойчивость микроорганизмов к некоторому количеству препаратов, в частности антибиотиков) является одной из основных проблем в клинической практике. Поэтому важно предоставить новые препараты, которые могут оказывать влияние на рост микроорганизмов.
Другой аспект настоящего изобретения относится к способу нейтрализации биологической активности бактериальных компонентов, предпочтительно компонентов клеточной стенки, более предпочтительно липополисахаридов микроорганизмов введением эффективного количества пептида или полипептида, или фармацевтической композиции согласно настоящему изобретению.
Пептиды или полипептиды согласно настоящему изобретению проявляют эндотоксиннейтрализующую активность. Следовательно, эти вещества могут применяться для связывания бактериальных компонентов, в частности компонентов клеточной стенки, и в результате нейтрализовать их биологическую активность.
Еще один аспект настоящего изобретения относится к способу нейтрализации биологической активности бактериальных компонентов, предпочтительно компонентов клеточной стенки, более предпочтительно липополисахарида микроорганизмов, или способу лечения млекопитающих, в частности человека, страдающего от бактериальной инфекции или септического шока, введением эффективного количества пептида или полипептида, или фармацевтической композиции согласно настоящему изобретению.
Терапевтически и профилактически эффективное количество составляет предпочтительно приблизительно от 0,5 мг/кг до около 100 мг/кг веса тела, более предпочтительно приблизительно от 1 мг/кг до около 20 мг/кг и наиболее предпочтительно приблизительно от 2 мг/кг до около 10 мг/кг. Для кожного применения соединения могут быть введены в концентрации достаточно высокой для того, чтобы быстро убить организм-мишень (по меньшей мере, 10-100 MIC или 100-1000 мкг/мл). Для интраперитонеального применения терапевтический предел составляет предпочтительно от около 7,5 мг/кг до около 75 мг/кг. В случае совместного введения с обычными антибиотиками терапевтически эффективное количество уменьшают на коэффициент от 10 до 100.
Другой аспект настоящего изобретения относится к способу производства пептида согласно настоящему изобретению, имеющего N-концевой пролиновый остаток, включающий стадии:
- обеспечение клетки-хозяина, содержащей молекулу нуклеиновой кислоты, кодирующей слитый полипептид или белок, содержащий пептид согласно настоящему изобретению, имеющий N-концевой остаток пролина, в котором пептид слит на С-конце с указанным полипептидом или белком, имеющим С-концевой аспартат,
- выработки (стадию экспрессии) и выделения указанного слитого полипептида или белка,
- стадию обработки выделенного слитого полипептида или белка при значении рН между 0,5 и 4 (Skribanek Z. et al., J. Pept. Sci. 8: 398-406 (2002)).
В ходе снижения значения рН полипептид или белок предпочтительно инкубируют при 85°С в течение одного часа, например, в 90 мМ HCl. Полученные пептиды предпочтительно очищают с помощью обращеннофазовой высокоэффективной жидкостной хроматографии (RP-HPLC) и необязательно подтверждают с помощью масс-спектрального анализа.
Другой аспект настоящего изобретения относится к способу адсорбции и удаления или инактивации бактерий или бактериальных компонентов из образцов, включающий стадии контактирования указанного образца с иммобилизованным пептидом согласно настоящему изобретению.
Антибактериальный и эндотоксиннейтрализующий/-связывающий агент настоящего изобретения может быть нанесен на поверхность подходящего материала или смешан с подходящим материалом с целью получения антибактериального материала. Такой антибактериальный материал может быть использован в различных формах: в грануле, пленке, пластинке, в хирургической мононити, нетканом материале, тампоне, ткани, в форме трикотажного полотна, короткого волокна, трубки, полого волокна и тому подобного. Конкретнее, он может быть использован для искусственного органа, катетера, шовного материала (соединительного волокна) для хирургической операции, диализной мембраны и тому подобного, а также в гигиенических товарах, антибактериальном фильтре и тому подобном.
Устройство или имплантат может быть использован в качестве эндотоксинудаляющего средства, содержащего пептид настоящего изобретения, иммобилизованный на нерастворимом носителе. Эндотоксинудаляющее средство настоящего изобретения основывается на применении высокой способности пептида настоящего изобретения связывать эндотоксин для адсорбции и удаления эндотоксина.
Форма нерастворимого носителя, на котором иммобилизован пептид настоящего изобретения, не является особым образом ограниченной, и здесь можно упомянуть различные формы, например формы мембраны (фильтрующий тип, полый тип, трубчатый тип, плоский тип мембран и т.д.), гранула, латекс, чип, порошок и микропластинка.
Материал нерастворимого носителя также не является ограниченным особенным образом, и здесь можно упомянуть различные материалы, например, полистирольные материалы, полипропиленовые материалы, полиамидные материалы, целлюлозные материалы, агарозные материалы, полиакриламидные материалы, декстрановые материалы и виниловые полимерные материалы.
Способ иммобилизации пептида настоящего изобретения на нерастворимом носителе также не является ограниченным особым образом, и иммобилизация пептида настоящего изобретения может быть достигнута применением общих методов, используемых для получения иммобилизованных ферментов, таких как метод физической адсорбции, метод ионного связывания, метод ковалентного связывания, метод включения.
Например, для нерастворимых носителей, изготовленных из полистироловых материалов или полипропиленовых материалов пептид настоящего изобретения может быть физически иммобилизован. К тому же, например, для нерастворимых носителей, изготовленных из полиамидных материалов, целлюлозных материалов, агарозных материалов, полиакриламидных материалов, декстрановых материалов или виниловых полимерных материалов, пептид настоящего изобретения может быть химически иммобилизован. В качестве способа химической иммобилизации (связывания) здесь можно упомянуть, например, метод диазотирования, в котором диазосоединение осуществляется с использованием ароматической аминогруппы в нерастворимом носителе; метод CNBr, в котором пептидная связь образуется посредством активирования гидроксильной группы в нерастворимом носителе с CNBr; кислотно-азидный метод, в котором пептидная связь образуется с использованием производного гидразина нерастворимого носителя; метод алкилирования, в котором пептид алкилируется с использованием реакционноспособной функциональной группы, такой как галоген в нерастворимом носителе; метод образования поперечных связей, в котором сшивающий агент реагирует со свободной аминогруппой, например, так, как глутаральдегид сшивает нерастворимый носитель со свободной аминогруппой в пептиде; карбодиимидный метод; метод эпоксиактивирования и методы, в которых связь образуется через спэйсер с использованием одного их вышеописанных методов. Подходящий метод может быть выбран из этих известных методов в зависимости от вида нерастворимого носителя для применения в связывании пептида настоящего изобретения.
Нерастворимый носитель, на котором иммобилизован пептид настоящего изобретения, приводится в контакт с раствором, из которого желательно удалить эндотоксин, для формирования комплекса эндотоксина в растворе, и нерастворимый носитель, на котором иммобилизован пептид настоящего изобретения, и затем образованный таким образом комплекс удаляется, посредством чего эндотоксин в растворе может быть удален.
Способ контактирования нерастворимого носителя, на котором иммобилизован пептид настоящего изобретения, с раствором, из которого желательно удалить эндотоксин, не является ограниченным особенным образом, и могут быть использованы известные способы контактирования твердого вещества и жидкости. Например, метод, в котором раствор проходит через имеющий форму фильтра или имеющий форму полого волокна нерастворимый носитель или через имеющий форму плоской мембраны нерастворимый носитель; метод, в котором раствор пропускается через колонку, заполненную гранулированным нерастворимым носителем; метод, в котором раствор загружается в лунки в форме микроплат и раствор остается стоять в течение определенного времени и затем раствор отделяется; метод, в котором раствор заливают на нерастворимый носитель любой формы и встряхивают или оставляют стоять в течение определенного времени, и, кроме того, могут быть использованы обычные способы разделения жидкости и твердого вещества (фильтрация, центрифугирование, аспирация, декантация и т.д.) для получения раствора, который свободен от эндотоксина, или тому подобное.
Раствор, из которого желательно удалить эндотоксин, не является ограниченным особенным образом, и примеры его включает растворы, используемые в фармацевтической промышленной установке, медицинской аппаратуре и тому подобном, конкретнее, диализатную жидкость, парентеральную жидкость, кровь, фармацевтические препараты, сверхчистую воду и тому подобное, но без ограничения.
Одним аспектом изобретения является антибактериальное соединение, т.е. которое ингибирует, предотвращает или уничтожает рост или пролиферацию микробов, таких как бактерии, грибы или тому подобного. Эти соединения являются пептидами или липопептидами общей формулы как приведено в данном описании.
Другим аспектом изобретения является способ лечения эндотоксемии (наличия в крови токсинов) посредством нейтрализации биологической активности бактериальных компонентов, предпочтительно из клеточных стенок, таких как эндотоксин, применением пептидов или липопептидов общей формулы как приведено в данном описании.
Следующие примеры и фигуры предоставлены в качестве руководства для средних специалистов в данной области техники, и не имеют целью ограничивать рамки заявленного изобретения в любом случае.
Краткий перечень фигур
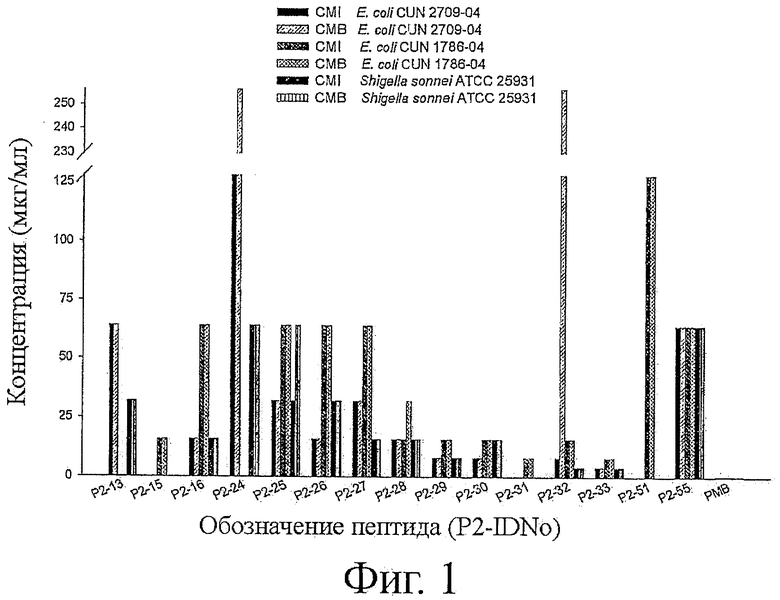
Фигура 1 показывает минимальную ингибирующую концентрацию (MIC) и минимальную бактерицидную концентрацию (МВС) выбранных пептидов (см. обозначение пептидов) и полимиксина (РМВ) для двух штаммов Е. coli и Shigella sonnei как указано на фигуре.
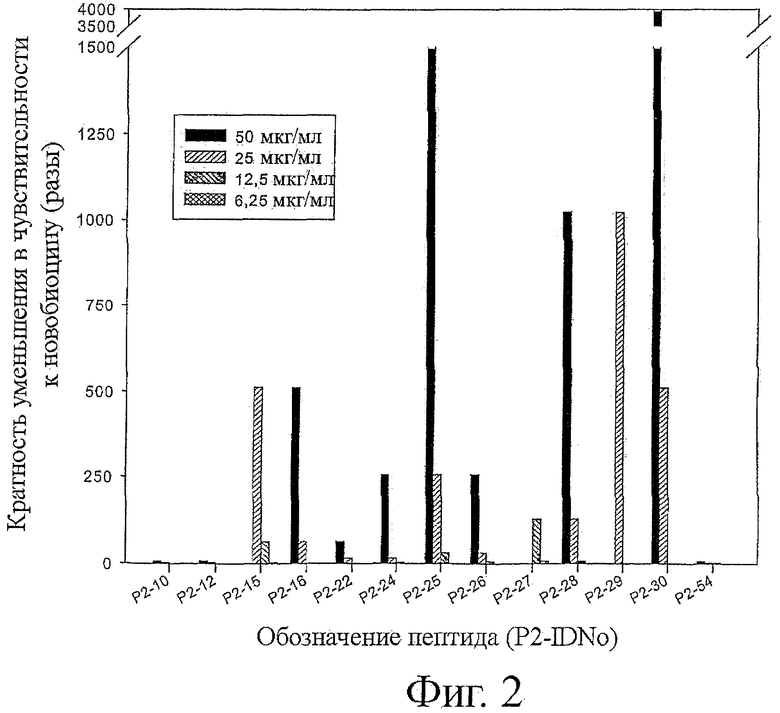
Фигура 2 показывает уменьшение минимальной ингибирующей концентрации новобиоцина после добавления определенных количеств выбранных пептидов.
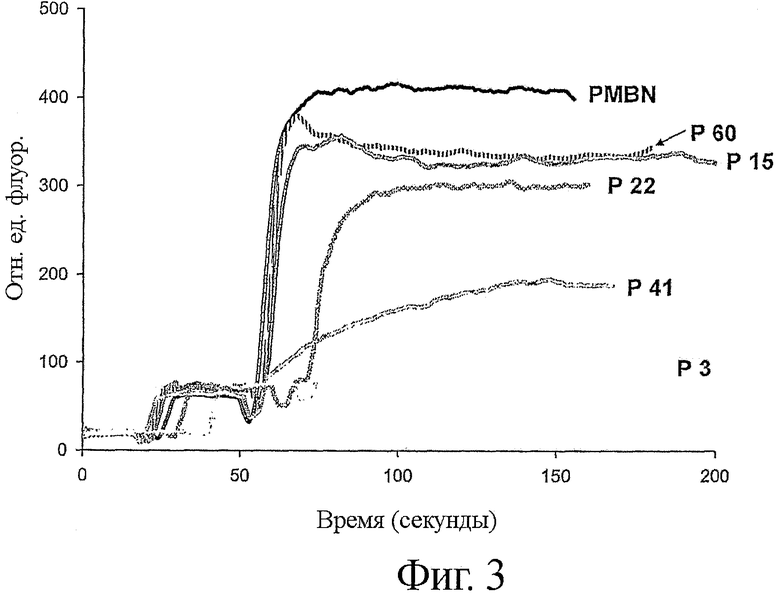
Фигура 3 показывает пермеабилизирующее действие выбранных пептидов полимиксин В нонапептида (PMBN) и не-пермеабилизирующего пептида (Р3), измеренное по увеличению интенсивности флуоресценции вследствие перераспределения N-фенилнафтиламина (NPN) в клеточную оболочку Е. coli. Последовательность веществ, перечисленных в надписи на правой стороне графика, соответствует последовательности кривых на графиках в их конечных точках.
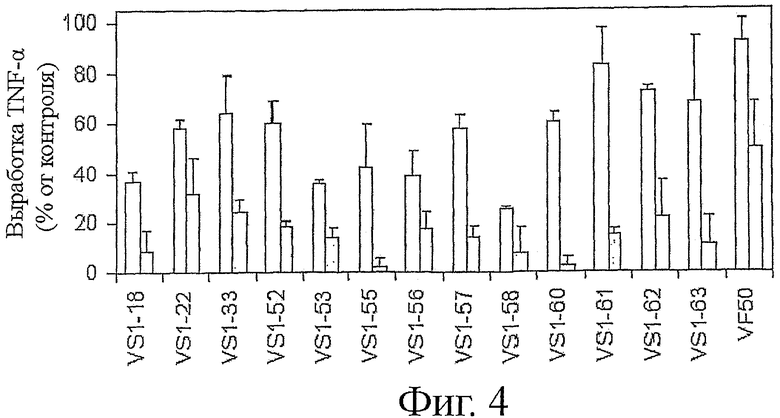
Фигура 4 показывает нейтрализацию TNF-α секреции моноцитов, стимулированных LPS в присутствии выбранных пептидов.
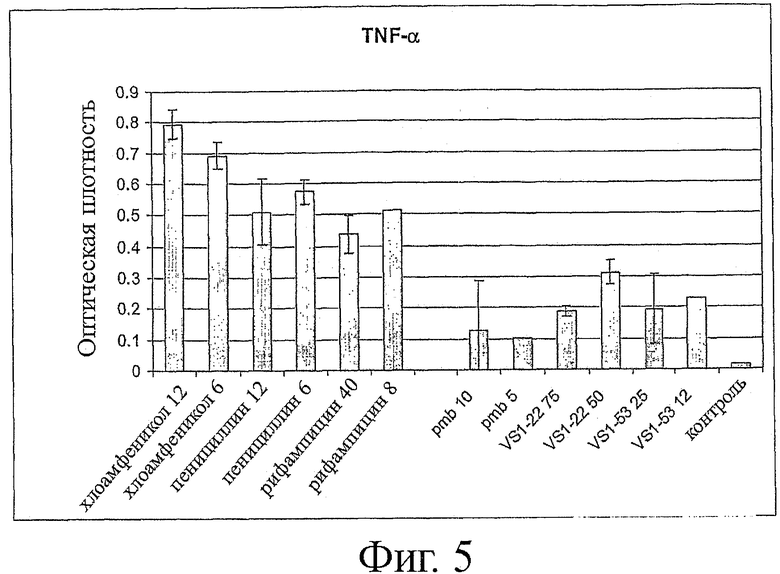
Фигура 5 показывает высвобождение TNF-α в присутствии различных антибиотиков, полимиксина В (РМВ) и выбранных пептидов (см. обозначение пептидов).
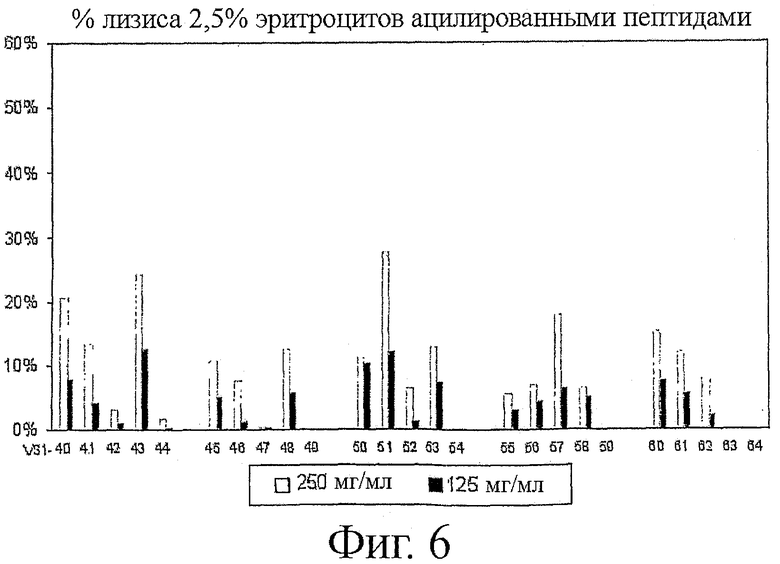
Фигура 6 показывает гемолитическую активность пептидов, содержащих N-ацильные цепи. Количество пептидов, добавленных к 2,5% красным клеткам крови (эритроцитам) человека, указано на фигуре.
Фигура 7 показывает ВЭЖХ-хроматограмму разделения продуктов расщепления рекомбинантного пептида. Фракция со временем удержания 8,175 минут содержала пептид.
Осуществление изобретения
ПРИМЕРЫ.
Пример 1. Синтез пептидов и полипептидов
Пептиды синтезировали с помощью синхронного множественного пептидного синтеза, следуя стандартным протоколам синтеза Fmoc (Houghten, Proc. Natl. Acad. Sci. USA 82: 5131-5135 (1985)). Смола для каждого пептида помещена в различные полипропиленовые сетчатые пакеты, что позволяет проводить все общие синтетические стадии в общем реакционном сосуде (т.е. стадии промывки, снятия защиты и нейтрализации), тогда как необходимые стадии присоединения были выполнены обработкой каждого пакета отдельными соответствующими растворами аминокислот. Липофильная кислота присоединялась к N-концу таким же образом как при связывании защищенной аминокислоты. Боковые цепи лизина и триптофана были защищены tBoc группой, аргинин - пентаметилбензофуран-5-сульфонил группой, цистеин, глутамин и аспарагин - тритил, аспарагиновая кислота, глутаминовая кислота, тирозин, серин и треонин - трет-бутил. Окончательное расщепление было выполнено обработкой трифторуксусной кислотой (Fields et al., Int. J. Peptide Prot. Res. 35: 161-214 (1990)). Идентичность и чистоту пептидов определяли с помощью масс-спектрального анализа, сопряженного с жидкостной хроматографической системой (Finnigan LCQ), и аналитической обращеннофазовой высокоэффективной жидкостной хроматографии (ОФ-ВЭЖХ) с использованием Beckman System Gold HPLC. Пептиды и липопептиды очищали с помощью препаративной ОФ-ВЭЖХ с использованием системы препаративной ВЭЖХ Waters Milliprep 300 с коллектором фракций Foxy. При растворении липопептидов для очистки использовали раствор уксусной кислоты (вплоть до 95%) или ацетонитрила (вплоть до 50%).
Пример 2. Антибактериальные исследования
Каждый пептид и липопептид прошел испытания на минимальную ингибирующую концентрацию (MIC) против перечня бактерий:
Escherichia coli ATCC 25922, Escherichia coli DC2, Klebsiella oxytoca ATCC 8724, Acinetobacter baumanii CUN 10817 - 01, Pseudomonas aeruginosa: CUN 4158- 02, Stenotrophomonas maltophilia: CUN 3998 - 00, Brucella abortus 9.49 per-, Yersinia pestis KIM pYV -, Escherichia coli CUN 2709-04, Escherichia coli CUN 1786-04, Shigella sonnet ATCC 25931, Salmonella minnesota HL63 (S), Salmonella minnesota R60 HL100 (Ra), Salmonella Minnesota R7 HL44 (Rdl), Salmonella minnesota R595 HL111 (Re) Bordetella bronquiseptica: CUN 11844 - 99, Bordetella bronquiseptica RB50, Haemophilus influenzae CUN 6277-04, Neisseria meningitidis CUN 6395-04, Enterococcus faecalis ATCC 29212, Staphylococcus aureus ATCC 25923, Streptococcus agalactiae CUN 4783-03, Enterococcus faecalis ATCC 51299, Staphylococcus aureus CUN 3792-99, Staphylococcus epidermidis ATCC 12228, Staphylococcus epidermidis CUN 5-93, Streptococcus pneumoniae ATCC 49619.
Свежие выращенные бактериальные культуры были инокулированы и разведены в среде Мюллера-Хинтона (МН) до приблизительной конечной концентрации 1-5×105 к.о.е./мл. Количество жизнеспособных микроорганизмов в бактериальной суспензии определяли посредством разбавления культуры средой МН и посева 100 мкл соответствующего 10-кратного разведения на МН агаровую пластинку. MIC была определена после инкубации в течение ночи при 37°С в 96-луночных чашках для культур тканей с помощью метода микроразведений с питательной средой согласно рекомендациям Национального Комитета по Клиническим лабораторным стандартам. Таким образом, 100 мкл бактериальной суспензии смешивали со 100 мкл раствора пептида или липопептида в МН среде в 96-луночных чашках с плоским дном и инкубировали в течение ночи при 37°С. Оптическую плотность каждой лунки при 620 нм измеряли до и после инкубации. Все пептиды и липопептиды были проверены при последовательных двукратных разведениях, начиная с 250 мкг/мл, в двух экземплярах. Активность пептидов сравнивали с клетками в среде МН (ингибирование 0%) и одной средой МН (100% ингибирование). MIC определяли как самую низкую концентрацию пептида или липопептида, при которой не наблюдалось изменения OD между временем О и инкубацией в течение ночи. В качестве стандартных контролей в каждом исследовании были использованы коммерчески доступные антибиотики.
Минимальная бактерицидная концентрация (МВС) была определена как самая низкая концентрация антибактериального препарата, которая убивала 99,9% исходного инокулума, и была определена, как рекомендовано CLSI/NCCLS. Коротко, суспензия (100 мкл) была взята из тех лунок, где рост не был обнаружен, и посеяна на МН чашки. Чашки инкубировали при 37°С в течение 24 ч (фиг.1).
Пример 3. Синергическая активность с обычными антибиотиками
Внешняя мембрана грамотрицательной бактерии действует как барьер проницаемости в отношении гидрофобных соединений. Для измерения пермеабилизирующей активности пептидов были использованы два метода. Оба способа анализа имеют одинаковые основы: мембрана, пермеабилизированная пептидами, позволяет гидрофобным веществам (NPN) достигать липидного бислоя, а новобиоцину - достигать его внутреннюю мишень (ДНК-гираза). Такие тесты были выполнены на P. aeruginosa 4158-02 (CUN) вследствие более низкой проницаемости, свойственной этим бактериям.
Исследование синергизма новобиоцин-пептид:
Пермеабилизирующая активность пептидов была измерена посредством сравнения MIC каждой комбинации пептид-новобиоцин с активностью одного новобиоцина в соответствии с уже опубликованным способом титрования методом «шахматной доски» (Lorian V. Antimicrobials in laboratory Medicine", 1996 4th ed. pp.330-396, Williams и Wilkins, Baltimore, Md). Для сравнения пермеабилизирующих активностей пептидов было определено два показателя: (i), показатель фракционной ингибиторной концентрации (FIC) был вычислен в соответствии со следующим уравнением: показатель FIC=(MIC проверенной комбинации новобиоцина)/(М1С одного новобиоцина)+(М1С пептида в комбинации)/(М1С одного пептида). Взаимодействие было определено как синергическое, если показатель FIC составлял ≤0,5; (b), MIC-Drop было определено как отношение концентраций новобиоцина MIC в отсутствие и в присутствии данного пептида. Одна комбинация была рассмотрена как синергическая, когда ее MIC-Drop было ≥4 (фиг.2).
Флуоресцентный анализ:
Эксперименты по флуоресценции были выполнены, как описано Loh и соавторами (1984. Antimicrob. Agents Chemother. 26: 546-551), с некоторыми модификациями. Коротко, бактерии выращивали в среде LB до логарифмической фазы, отмывали в 5 мМ буфере HEPES (рН 7,2) и ресуспендировали в том же самом буфере с 0,1% глюкозы до конечной оптической плотности 0,5 при λ=600 нм. Флуоресценцию измеряли при 37°С на флюориметре LS-50 Perkin-Elmer при длине волны возбуждения 350 нм и длине волны излучения 420 нм. К суспензии был добавлен NPN до конечной концентрации 10 мкМ и потом были добавлены пептиды до конечной концентрации 50 мкг/мл (фиг.3).
Пример 4. Нейтрализация TNF-α секреции моноцитов, стимулированных LPS
Ингибирование LPS-вызванной активации мононуклеарных клеток человека пептидами-производными лактоферрина было измерено с использованием LPS Ra от шероховатого мутантного штамма R60 Salmonella enterica (серовар minnesota). Липополисахарид инкубировали с пептидами (открытые столбики, 0,1 мкг/мл; заполненные столбики, 1 мкг/мл) в течение 30 мин при 37°С и добавляли к только что выделенным клеткам от здоровых доноров (конечная концентрация: 1 нг/мл LPS). Количество TNFα в супернатанте клеточной культуры, вызванное одним LPS, использовали в качестве необработанного контроля (фиг.4).
Пример 5. Нейтрализация стимулирования иммунных клеток убитыми бактериями
Бактерии могут быть уничтожены различными антибиотиками, нацеленными на различные молекулы, существенные для выживания бактерий. Когда грамотрицательные бактерии убиты, высвобожденный LPS может стимулировать выработку цитокинов, таких как TNFα, или других медиаторов воспаления. Сравнение твердо установленных антибактериальных препаратов, действующих через различные клеточные мишени, с соединениями данного изобретения было выполнено следующим образом: бактерии (штамм Е. coli 0:111) выращивали в среде LB до оптической плотности 0,4 при 600 нм и разводили в 2500 раз в среде RPMI с добавлением глюкозы. Добавляли различные концентрации антибиотиков или пептидов из данного изобретения и инкубировали в течение ночи. 80 мкл клеточной суспензии добавляли к 100 мкл, содержащим 105 МоnоМас6 клеток, и через 15 часов определяли высвобождение TNFα в среду с помощью теста ELISA. Результаты явно показали, что хлорамфеникол, пенициллин и рифампицин, которые убивали бактериальные клетки, приводили к сильному стимулированию моноцитов, тогда как пептиды VS1-22 и VS1-53 значительно ингибировали высвобождение TNFα, подобно токсическому липопептиду полимиксина В (фиг.5).
Пример 6. Исследования in vivo
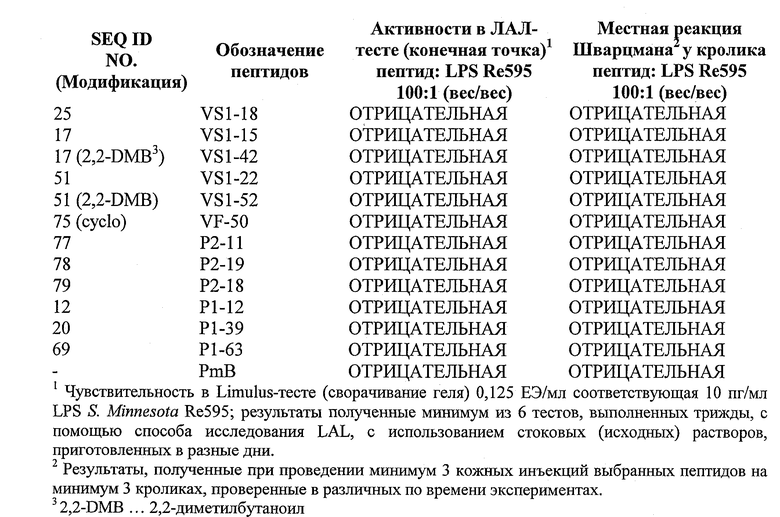
Модель острой эндотоксемии на мышах для определения антиэндотоксической активности пептидов:
Мыши необыкновенно устойчивы к LPS-опосредованному септическому шоку. Однако чувствительность мышей к эндотоксину может быть усилена совместным введением LPS с галактозамином. Самкам мышей ICR-CD1 весом 20-25 г (группы из 16-18 мышей) интраперитонеально вводили 200 мкл апирогенного физиологического раствора, содержащего смесь из 0,3 мкг LPS Е. coli и 18 мг галактозамина. Предыдущие эксперименты дали возможность определить, что такая комбинация была летальной для 90% (LD9o) животных через 48 ч после инъекции. Немедленно после этой инъекции мыши получали вторую интраперитонеальную инъекцию в другое место брюшка, содержащую 150 мкг пептида, растворенного в 150 мкл 10% DMF апирогенного физиологического раствора. Во всех экспериментах была оставлена необработанная группа мышей, тогда как другая группа получала 150 мкг полимиксина В. Смертность животных регистрировали ежедневно вплоть до 168 часов после теста. При наших экспериментальных условиях полимиксин В не давал значительной защиты от эндотоксического шока.
Модель на кроликах для определения антиэндотоксической активности пептидов:
Принцип индуцированной липидом А активности провоспалительных цитокинов, приводящей к геморрагическому кожному некрозу (классическая реакция Шварцмана), был проверен на кролике, животной модели, очень близкой к человеческой в отношении чувствительности к активности LPS, и был сравнен с ингибированием индуцированной липидом А активации ферментного каскада LAL, приводящего к образованию сгустка. В связи с этим новозеландским белым кроликам в выбритую область спины вводили липид A S.minnesota Re595 отдельно или с пептидом 1:100 (вес./вес.) (5 мкг в 0,2 мл солевого буфера; интрадермально, i.d.). Через 72-96 часов после инъекции кожу животных осматривали на наличие открытого некроза или его ингибирования. Полимиксин В (PmB) использовали в качестве контроля.
Таблица 4. Пептиды из различных пунктов патентной формулы, демонстрирующие положительную корреляцию между "in vitro" против "in vivo" при анализе результатов испытаний.
Пример 7. Проверка токсичности на клетки млекопитающих
Гемолитическая активность пептидов по отношению к красным клеткам крови, которые были получены из гепаринизированной крови человека, была определена посредством высвобождения гемоглобина после часовой инкубации при 37°С. Общее высвобождение гемоглобина (оптическая плотность измерена при 414 нм) достигалось при добавлении Тритона Х-100 (0,5% конечная концентрация). Показаны данные для ацилированных пептидов при концентрации выше их MIC (в 5-50 раз в зависимости от пептида и видов бактерий) (фиг.6).
Кроме того, были выбраны пептиды, показывающие наивысшую мембран-пермеабилизирующую активность, и их токсичность по отношению к линии клеток человека Hela была оценена методом исключения трипанового синего (Mishell, В.В., и S.М. Shiigi. 1980. Selected methods in cellular immunology. Freeman и Co., San Francisco. 14-17). При проверке при 100 мкг/мл все пептиды (n=16) показали отсутствие эффектов или незначительные эффекты на способность клеток исключать краситель.
Пример 8. Очистка выбранных пептидов
Для очистки рекомбинантных белков осадок бактериальных клеток из 1 литра ресуспендировали в 20 мл лизирующего буфера (10 мМ Трис рН=8,0, 1 мМ ЭДТА, 0,1% DOC) и диспергировали с помощью обработки ультразвуком. Смесь центрифугировали при 12000 об/мин в течение 15 мин при 4°С для разделения растворимого супернатанта и фракции нерастворимого осадка, содержащего тельца включения. Нерастворимую фракцию телец включения, содержащую слитый белок KSI-P2-33, промывали дважды с 20 мл промывочного буфера, содержащего 10 мМ Трис рН=8,0, 1 мМ ЭДТА и 0,1% DOC, дважды с 10 мМ Трис рН=8,0, 1 мМ ЭДТА и 2 М мочевины и три раза с 20 мМ Трис рН=8,0. Нерастворимую фракцию телец включения растворяли в 10 мл 6М гуанидин-HCl, центрифугировали и растворимый супернатант диализовали 2 литрами дианизованной воды, что вызывало осаждение KSI-P2-33. Слитые белки (10 мг) растворяли в 10 мл 90 мМ HCl, смеси перемешивали 2 часа при 85°С, для того чтобы расщепить связь аспартил-пролил между слитым белком и пептидами. Пептид, высвобожденный кислотным расщеплением, очищали с помощью ВЭЖХ: реакционную смесь высушивали, растворяли в дианизованной воде и вводили в колонку С5 ОФ-ВЭЖХ (Sephasil) и элюировали градиентом от 5% ацетонитрила, 5 мМ HCl до 95% ацетонитрила, 5 мМ HCl. Пик пептида (фиг.7) определяли по положению в УФ при 280 нм. Идентичность пептида устанавливали с помощью масс-спектрометрии.
Пример 9. Антибактериальные исследования с иммобилизованными пептидами
Пептид Р2-32 (500 мкг) ковалентно связывали с активированными цианурхлоридом магнитными частицами (10 мг) (Chemicell, Продукт номер 1314) с использованием фосфатно-солевого буфера рН=7,5. После перемешивания суспензии на шейкере в течение 2 часов при комнатной температуре добавляли блокирующий буфер (PBS рН=7,5) и 2% этаноламин и перемешивали суспензию с помощью шейкера в течение 30 минут при комнатной температуре. Частицы дважды промывали PBS. Иммобилизованные пептиды проверяли против штамма E. coli (штамм 0:111), выросшего в среде LB до оптической плотности 0,4 при 600 нм и разбавленного в 2500 раз в среде LB. Добавляли различные концентрации магнитных частиц с иммобилизованными пептидами и инкубировали в течение ночи. Бактериальный рост был предотвращен при концентрациях иммобилизованных магнитных частиц 50, 25 и 10 мг/мл.
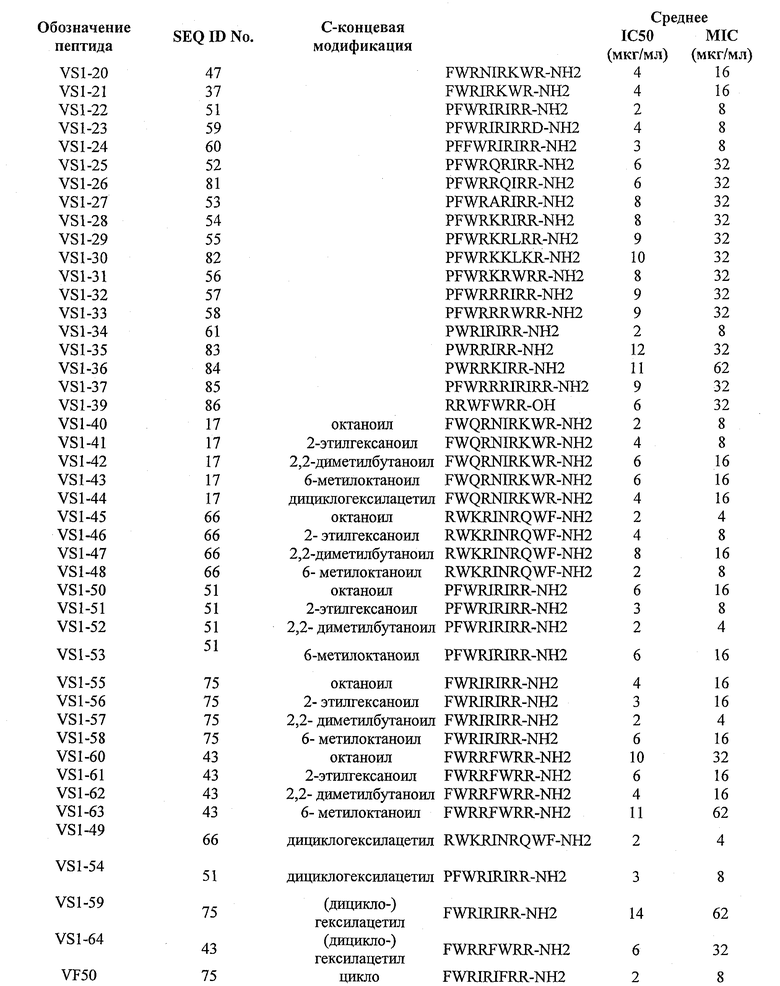
Пример 10. Противогрибковая активность
Культуры С. neoformans ATCC 32045 поддерживали на агаровых чашках с дрожжевой средой (YM; Difco Laboratories, Detroit, Mich.) при 4°С. Перед исследованием культуры выращивали на агаровых чашках и инкубировали в течение 72 часов при 26°С. Затем две колонии этих только что выросших грибковых культур были инокулированы в 5 мл среды 2Х YM, перемешаны на вортексе и разведены в 10 раз в среде 2Х YM до приблизительной конечной концентрации от 1×105 до 5×105 к.о.е./мл. В 96-луночные чашки для культуры ткани к пептидам, добавленным в концентрациях в пределах от 1 мг/мл до 1 мкг/мл, полученных при серийном двукратном разведении в стерильной воде, были добавлены грибковые суспензии в среде 2Х YM. Затем чашки инкубировали в течение 72 часов при 26°С. Относительный процент роста грибков для каждого тестового образца определяли по оптической плотности при 620 нм (OD620) с помощью прибора Titertek Multiskan Plus. MIC определяли как самую низкую концентрацию тестового образца, которая приводила к 2% росту, и IC50 была определена как концентрация тестового образца, которая приводила к 50% ингибированию роста. IC50 была вычислена с использованием компьютерной программы сигмоидальной аппроксимации кривой по точкам (Graphpad Prism; ISI Software, Сан Диего, СА). Результаты, полученные для выбранных пептидов, показаны в таблице 6.

| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИД, ОБЛАДАЮЩИЙ СВОЙСТВАМИ АМИЛИНА (ВАРИАНТЫ), И ЕГО ПРИМЕНЕНИЕ (ВАРИАНТЫ) | 2005 |
|
RU2385878C2 |
| КОМПОЗИЦИЯ И МИКРОСФЕРА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ ЭКЗЕНДИНА И СПОСОБ ПОЛУЧЕНИЯ МИКРОСФЕРЫ | 2008 |
|
RU2463040C2 |
| СПОСОБЫ ПОДАВЛЕНИЯ ГЛЮКАГОНА | 2000 |
|
RU2247575C9 |
| ГУМАНИЗИРОВАННЫЕ АНТИТЕЛА К АМИЛОИДУ БЕТА | 2008 |
|
RU2567151C2 |
| ПРИМЕНЕНИЕ АНТИТЕЛА ПРОТИВ АМИЛОИДА-БЕТА ПРИ ГЛАЗНЫХ ЗАБОЛЕВАНИЯХ | 2008 |
|
RU2571859C2 |
| ГУМАНИЗИРОВАННОЕ АНТИТЕЛО К АМИЛОИДУ БЕТА | 2007 |
|
RU2498999C2 |
| ГУМАНИЗИРОВАННОЕ АНТИТЕЛО | 2008 |
|
RU2538709C2 |
| ПРИМЕНЕНИЕ АНТИТЕЛА ПРОТИВ АМИЛОИДА БЕТА ПРИ ГЛАЗНЫХ ЗАБОЛЕВАНИЯХ | 2008 |
|
RU2542967C2 |
| ГУМАНИЗИРОВАННОЕ АНТИТЕЛО К АМИЛОИДУ БЕТА | 2013 |
|
RU2668161C2 |
| Соединения и способы для подавления транспорта фосфатов | 2014 |
|
RU2771808C2 |




































Настоящее изобретение относится к пептидам с антибактериальной и эндотоксиннейтрализующей активностью, имеющим общую формулу (Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S. 12 н. и 31 з.п. ф-лы, 7 ил., 6 табл., 10 пр.
1. Пептид с антибактериальной и эндотоксиннейтрализующей активностью, имеющий формулу:
(Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S, в которой
Xaa1 является выбранным из группы, состоящей из фенилаланина (Phe), аланина (Ala), лейцина (Leu) и валина (Val),
Xaa2 является выбранным из группы, состоящей из аргинина (Arg) и лизина (Lys),
Хаа3 представляет собой триптофан (Trp),
Хаа4 является выбранным из группы, состоящей из аланина (Ala), аргинина (Arg), глутамина (Gln), аспарагина (Asn), пролина (Pro), изолейцина (Ile), лейцина (Leu) и валина (Val),
Хаа5 является выбранным из группы, состоящей из изолейцина (Ile), фенилаланина (Phe), триптофана (Trp) и тирозина (Tyr),
Хаа6 является выбранным из группы, состоящей из аргинина (Arg), лизина (Lys), тирозина (Tyr) и фенилаланина (Phe),
Хаа7 является выбранным из группы, состоящей из изолейцина (Ile), триптофана (Trp), валина (Val) и лейцина (Leu), и
Xaa8 является выбранным из группы, состоящей из аргинина (Arg), лизина (Lys), изолейцина (Ile) и серина (Ser), и в которой
О является 0,
М является 1 или 2,
Р является 2 или 3,
Q и R являются 1, и
S является 1, 2, 3 или 4,
где, необязательно, N- и/или С-конец пептида может быть модифицирован.
2. Пептид по п.1, характеризующийся тем, что формула включает аминокислотную последовательность, выбранную из группы, состоящей из FWQRIRKVR (SEQ ID No. 1), FWQRRIRKVRR (SEQ ID No. 2), FWQRKIRKVRK (SEQ ID No. 3), FWQRNIRIRR (SEQ ID No. 4), FWQRNIRKVR (SEQ ID No. 5), FWQRNIRVR (SEQ ID No. 6), FWQRNIRKVRR (SEQ ID No. 7), FWQRNIRKVKK (SEQ ID No. 8), FWQRNIRKVRRR (SEQ ID No. 9), FWQRNIRKVKKK (SEQ ID No. 10), FWQRNIRKVRRRR (SEQ ID No. 11), FWQRNIRKVRRRI (SEQ ID No. 12), FWQRNIRKVKKKK (SEQ ID No. 13), FWQRNIRKVKKKI (SEQ ID No. 14), FWQRNIRKIR (SEQ ID No. 15), FWQRNIRKLR (SEQ ID No. 16), FWQRNIRKWR (SEQ ID No. 17), FWQRNWRKVR (SEQ ID No. 18), FWQRNFRKVR (SEQ ID No. 19), FWQRNYRKVR (SEQ ID No. 20), FWQRNIRKVS (SEQ ID No. 21), FWQRRIRIRR (SEQ ID No. 22), FWQRPIRKVR (SEQ ID No. 23), FWQRRIRKWR (SEQ ID No. 24), FWQRRIRRWRR (SEQ ID No. 25), FWPRNIRKVR (SEQ ID No. 26), FWARNIRKVR (SEQ ID No. 27), FWIRNIRKVR (SEQ ID No. 28), FWLRNIRKVR (SEQ ID No. 29), FWVRNIRKVR (SEQ ID No. 30), FWQRNIFKVR (SEQ ID No. 31), FWQRNIYKVR (SEQ ID No. 32), FAWQRNIRKVR (SEQ ID No. 33), FLWQRNIRKVR (SEQ ID No. 35) и FVWQRNIRKVR (SEQ ID No. 36).
3. Пептид по п.1, характеризующийся тем, что С-конец состоит предпочтительно из группы, выбранной из группы, состоящей из N-метиламидо группы или карбоксильной группы, или выбранной из числа амидных групп, сложных эфиров, эфиров или кетонов, предпочтительно содержащих в своем составе от 1 до 120 атомов углерода, более предпочтительно от 1 до 10 атомов углерода, в особенности состоящей из N-метиламидо группы.
4. Пептид по п.1, характеризующийся тем, что ацильная группа связана с N-концом или С-концом пептида.
5. Пептид по п.4, характеризующийся тем, что ацильная группа представляет собой гидрофобную цепь, выбранную из группы, состоящей из насыщенных и ненасыщенных линейных и разветвленных ацильных цепей С2-С20, производных бензила и F-moc.
6. Пептид по любому из пп.4 и 5, характеризующийся тем, что ацильная группа выбрана из группы, состоящей из додеканоил-группы, деканоил-группы, октаноил-группы, гексаноил-группы, 2-метилгексаноил-группы, 2-этилгексаноил-группы, 2-пропил-пентаноил-группы, 2-бутилоктаноил-группы, 2,2-диметилбутаноил-группы, 2-метилпентаноил-группы, 3-метилпентаноил-группы, 4-метилпентаноил-группы, 6-метилоктаноил-группы, бензил-группы и дициклогексилацетил-группы.
7. Пептид с антибактериальной и эндотоксиннейтрализующей активностью, имеющий формулу:
(Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S, в которой
Xaa1 является выбранным из группы, состоящей из фенилаланина (Phe) и изолейцина (Ile),
Хаа2 является выбранным из группы, состоящей из аргинина (Arg), лизина (Lys),
Хаа3 является триптофаном (Trp),
Хаа4 является выбранным из группы, состоящей из глицина (Gly), аспарагина (Asn), изолейцина (Ile) и фенилаланина (Phe),
Хаа5 является изолейцином (Ile) или триптофаном (Trp),
Хаа6 является аргинином (Arg) или лизином (Lys),
Хаа7 является выбранным из группы, состоящей из изолейцина (Ile), триптофана (Trp) и валина (Val) и
Xaa8 является аргинином (Arg), и в которой
О является 0,
М является 1 или 2,
R является 0 или 1,
Р является 1, 2 или 3,
Q является 1, и
S является 0, 1 или 2,
где, необязательно, N- и/или С-конец пептида может быть модифицирован.
8. Пептид по п.7, характеризующийся тем, что формула включает аминокислотную последовательность, выбранную из группы, состоящей из FWRIRKWR (SEQ ID No. 37), FWRIRKVR (SEQ ID No. 38), FWRWRR (SEQ ID No. 39), FWRRWRR (SEQ ID No. 40), FWRRWIRR (SEQ ID No. 41), FWRGWRIRR (SEQ ID No. 42), FWRRFWRR (SEQ ID No. 43), FWRWRWR (SEQ ID No. 44), FWRIWRWR (SEQ ID No. 45), FWRIWRIWR (SEQ ID No. 46), FWRNIRKWR (SEQ ID No. 47) и FWRRRIRIRR (SEQ ID No. 48).
9. Пептид по п.7, характеризующийся тем, что С-конец состоит предпочтительно из группы, выбранной из группы, состоящей из N-метиламидо группы или карбоксильной группы, или выбранной из числа амидных групп, сложных эфиров, эфиров или кетонов, предпочтительно содержащих в своем составе от 1 до 120 атомов углерода, более предпочтительно от 1 до 10 атомов углерода, в особенности состоящей из N-метиламидо группы.
10. Пептид по п.7, характеризующийся тем, что ацильная группа связана с N-концом или С-концом пептида.
11. Пептид по п.10, характеризующийся тем, что ацильная группа представляет собой гидрофобную цепь, выбранную из группы, состоящей из насыщенных и ненасыщенных линейных и разветвленных ацильных цепей С2-С20, производных бензила и F-moc.
12. Пептид по любому из пп.10 и 11, характеризующийся тем, что ацильная группа выбрана из группы, состоящей из додеканоил-группы, деканоил-группы, октаноил-группы, гексаноил-группы, 2-метилгексаноил-группы, 2-этилгексаноил-группы, 2-пропил-пентаноил-группы, 2-бутилоктаноил-группы, 2,2-диметилбутаноил-группы, 2-метилпентаноил-группы, 3-метилпентаноил-группы, 4-метилпентаноил-группы, 6-метилоктаноил-группы, бензил-группы и дициклогексилацетил-группы.
13. Пептид с антибактериальной и эндотоксиннейтрализующей активностью, имеющий формулу:
(Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S, в которой
Xaa1 является выбранным из группы, состоящей из пролина (Pro) и фенилаланина (Phe),
Хаа2 является выбранным из группы, состоящей из аргинина (Arg), лизина (Lys)
Хаа3 является триптофаном (Trp),
Хаа4 является выбранным из группы, состоящей из аланина (Ala), аргинина (Arg), глутамина (Gln), лизина (Lys), триптофана (Trp) и изолейцина (Ile),
Хаа5 является выбранным из группы, состоящей из изолейцина (Ile) и триптофана (Trp),
Хаа6 является выбранным из группы, состоящей из аргинина (Arg) и аспартата (Asp),
Хаа7 является выбранным из группы, состоящей из изолейцина (Ile), триптофана (Trp), фенилаланина (Phe), валина (Val) и лейцина (Leu), и
Xaa8 является выбранным из группы, состоящей из аргинина (Arg), лизина (Lys), изолейцина (Ile), серина (Ser) и аспартата (Asp), и в которой
О и Q являются 0,
М является 0, 1, 2 или 3,
R является 1 или 2,
Р является 1, 2 или 3, и
S является 1, 2 или 3,
где, необязательно, N- и/или С-конец пептида может быть модифицирован.
14. Пептид по п.13, характеризующийся тем, что формула включает аминокислотную последовательность, выбранную из группы, состоящей из PFWRWRIWR (SEQ ID No. 50), PFWRIRIRR (SEQ ID No. 51), PFWRQRIRR (SEQ ID No. 52), PFWRARIRR (SEQ ID No. 53), PFWRKRIRR (SEQ ID No. 54), PFWRKRLRR (SEQ ID No. 55), PFWRKRWRR (SEQ ID No. 56), PFWRRRIRR (SEQ ID No. 57), PFWRRRWRR (SEQ ID No. 58). PFWRIRIRRD (SEQ ID No. 59), PFFWRIRIRR (SEQ ID No. 60), PWRIRIRR (SEQ ID No. 61), PFWRRQIRR (SEQ ID No. 81), PFWRKKLKR (SEQ ID No. 82), PWRRIRR (SEQ ID No. 83), PWRRKIRR (SEQ ID No. 84) и PFWRRIRIRR (SEQ ID No. 85).
15. Пептид по п.13, характеризующийся тем, что С-конец состоит предпочтительно из группы, выбранной из группы, состоящей из N-метиламидо группы или карбоксильной группы, или выбранной из числа амидных групп, сложных эфиров, эфиров или кетонов, предпочтительно содержащих в своем составе от 1 до 120 атомов углерода, более предпочтительно от 1 до 10 атомов углерода, в особенности состоящей из N-метиламидо группы.
16. Пептид по п.13, характеризующийся тем, что ацильная группа связана с N-концом или С-концом пептида.
17. Пептид п.16, характеризующийся тем, что ацильная группа представляет собой гидрофобную цепь, выбранную из группы, состоящей из насыщенных и ненасыщенных линейных и разветвленных ацильных цепей С2-С20, производных бензила и F-moc.
18. Пептид по любому из пп.16 или 17, характеризующийся тем, что ацильная группа выбрана из группы, состоящей из додеканоил-группы, деканоил-группы, октаноил-группы, гексаноил-группы, 2-метилгексаноил-группы, 2-этилгексаноил-группы, 2-пропил-пентаноил-группы, 2-бутилоктаноил-группы, 2,2-диметилбутаноил-группы, 2-метилпентаноил-группы, 3-метилпентаноил-группы, 4-метилпентаноил-группы, 6-метилоктаноил-группы, бензил-группы и дициклогексилацетил-группы.
19. Пептид с антибактериальной и эндотоксиннейтрализующей активностью, имеющий формулу:
(Хаа1)M-(Хаа2)O-Хаа3-(Хаа4)P-(Хаа5)Q-(Хаа6)M-(Хаа7)R-(Хаа8)S, в которой
Xaa1 является выбранным из группы, состоящей из пролина (Pro) и фенилаланина (Phe),
Xaa2 является аргинином (Arg),
Хаа3 является триптофаном (Trp),
Хаа4 является выбранным из группы, состоящей из аланина (Ala), аргинина (Arg), глутамина (Gln), аспарагина (Asn) и лизина (Lys),
Хаа5 является выбранным из группы, состоящей из изолейцина (Ile), фенилаланина (Phe) и триптофана (Trp),
Хаа6 является выбранным из группы, состоящей из глутамина (Gln), аргинина (Arg) и аспарагина (Asn),
Хаа7 является о выбранным из группы, состоящей из изолейцина (Ile), триптофана (Trp) и фенилаланина (Phe), и
Xaa8 является аргинином (Arg), и в которой
М является 1, 2 или 3,
О является 0 или 1,
Р является 1, 2 или 3,
Q является 1 или 2, и
R и S являются 0, 1 или 2,
где, необязательно, N- и/или С-конец пептида может быть модифицирован.
20. Пептид по п.19, характеризующийся тем, что формула включает аминокислотную последовательность, выбранную из группы, состоящей из FWRNIRIRR (SEQ ID No. 72), FWQRIRIRR (SEQ ID No. 73), FWRWRIWR (SEQ ID No. 74), FWRIRIRR (SEQ ID No. 75), FWRNIRIWRR (SEQ ID No. 76) и FWRNIRIRR (SEQ ID No. 77).
21. Пептид по п.19, характеризующийся тем, что С-конец состоит предпочтительно из группы, выбранной из группы, состоящей из N-метиламидо группы или карбоксильной группы, или выбранной из числа амидных групп, сложных эфиров, эфиров или кетонов, предпочтительно содержащих в своем составе от 1 до 120 атомов углерода, более предпочтительно от 1 до 10 атомов углерода, в особенности состоящей из N-метиламидо группы.
22. Пептид по п.19, характеризующийся тем, что ацильная группа связана с N-концом или С-концом пептида.
23. Пептид по п.22, характеризующийся тем, что ацильная группа представляет собой гидрофобную цепь, выбранную из группы, состоящей из насыщенных и ненасыщенных линейных и разветвленных ацильных цепей С2-С20, производных бензила и F-moc.
24. Пептид по любому из пп.22 и 23, характеризующийся тем, что ацильная группа выбрана из группы, состоящей из додеканоил-группы, деканоил-группы, октаноил-группы, гексаноил-группы, 2-метилгексаноил-группы, 2-этилгексаноил-группы, 2-пропил-пентаноил-группы, 2-бутилоктаноил-группы, 2,2-диметилбутаноил-группы, 2-метилпентаноил-группы, 3-метилпентаноил-группы, 4-метилпентаноил-группы, 6-метилоктаноил-группы, бензил-группы и дициклогексилацетил-группы.
25. Пептид с антибактериальной и эндотоксиннейтрализующей активностью, представляющий собой аминокислотную последовательность, выбранную из группы, состоящей из RFWQRNIRKVRR (SEQ ID No. 62), RFWQRNIRKYR (SEQ ID No. 63), PFWQRNIRKWR (SEQ ID No. 64), RFRWQRNIRKYRR (SEQ ID No. 65), RWKRINRQWF (SEQ ID No. 66), KRFCFKK (SEQ ID No. 67), KRFSFKKC (SEQ ID No. 68), KRWSWKK (SEQ ID No. 69), FRFSFKK (SEQ ID No. 70), RRFWFRR (SEQ ID No. 71), RFWQRNIRIRR (SEQ ID No. 78), RWQRNIRIRR (SEQ ID No. 79) и RRWFWRR (SEQ ID No. 86), где, необязательно, N- и/или С- конец пептида может быть модифицирован.
26. Пептид по п.25, характеризующийся тем, что С-конец состоит предпочтительно из группы, выбранной из группы, состоящей из N-метиламидо группы или карбоксильной группы, или выбранной из числа амидных групп, сложных эфиров, эфиров или кетонов, предпочтительно содержащих в своем составе от 1 до 120 атомов углерода, более предпочтительно от 1 до 10 атомов углерода, в особенности состоящей из N-метиламидо группы.
27. Пептид по п.25, характеризующийся тем, что ацильная группа связана с N-концом или С-концом пептида.
28. Пептид по п.27, характеризующийся тем, что ацильная группа представляет собой гидрофобную цепь, выбранную из группы, состоящей из насыщенных и ненасыщенных линейных и разветвленных ацильных цепей С2-С20, производных бензила и F-moc.
29. Пептид по любому из пп.27 и 28, характеризующийся тем, что ацильная группа выбрана из группы, состоящей из додеканоил-группы, деканоил-группы, октаноил-группы, гексаноил-группы, 2-метилгексаноил-группы, 2-этилгексаноил-группы, 2-пропил-пентаноил-группы, 2-бутилоктаноил-группы, 2,2-диметилбутаноил-группы, 2-метилпентаноил-группы, 3-метилпентаноил-группы, 4-метилпентаноил-группы, 6-метилоктаноил-группы, бензил-группы и дициклогексилацетил-группы.
30. Пептид с антибактериальной и эндотоксиннейтрализующей активностью, имеющий формулу FIWQRNIRKVR (SEQ ID No. 34), FIWRWRWR (SEQ ID No. 49) и RRIRINRQWF (SEQ ID No. 80),
где, необязательно, N- и/или С-конец пептида может быть модифицирован.
31. Пептид по п.30, характеризующийся тем, что С-конец состоит предпочтительно из группы, выбранной из группы, состоящей из N-метиламидо группы или карбоксильной группы, или выбранной из числа амидных групп, сложных эфиров, эфиров или кетонов, предпочтительно содержащих в своем составе от 1 до 120 атомов углерода, более предпочтительно от 1 до 10 атомов углерода, в особенности состоящей из N-метиламидо группы.
32. Пептид по п.30, характеризующийся тем, что ацильная группа связана с N-концом или С-концом пептида.
33. Пептид по п.32, характеризующийся тем, что ацильная группа представляет собой гидрофобную цепь, выбранную из группы, состоящей из насыщенных и ненасыщенных линейных и разветвленных ацильных цепей С2-С20, производных бензила и F-moc.
34. Пептид по любому из пп.32 и 33, характеризующийся тем, что ацильная группа выбрана из группы, состоящей из додеканоил-группы, деканоил-группы, октаноил-группы, гексаноил-группы, 2-метилгексаноил-группы, 2-этилгексаноил-группы, 2-пропил-пентаноил-группы, 2-бутилоктаноил-группы, 2,2-диметилбутаноил-группы, 2-метилпентаноил-группы, 3-метилпентаноил-группы, 4-метилпентаноил-группы, 6-метилоктаноил-группы, бензил-группы и дициклогексилацетил-группы.
35. Применение пептида по любому из пп.1-34 в качестве антибактериального, эндотоксиннейтрализующего или противогрибкового средства.
36. Применение пептида по любому из пп.1-34 для производства лекарственного средства обладающего антибактериальной и эндотоксиннейтрализующей активностью для лечения или предотвращения инфекций, вызванных микроорганизмами, предпочтительно бактериями и/или грибками, или сепсиса или септического шока, вызванного предпочтительно эндотоксинами.
37. Способ ингибирования роста, по меньшей мере, одного микроорганизма, включающий стадию контактирования указанного микроорганизма с эффективным количеством пептида по любому из пп.1-34.
38. Способ по п.37, отличающийся тем, что указанный микроорганизм является грамположительной или грамотрицательной бактерией.
39. Способ по п.37, отличающийся тем, что указанным микроорганизмом для контактирования предпочтительно являются микроорганизмы из семейства Enterobactericeae, в частности Escherichia coli, Salmonella spp., Yersinia pestis, Yersinia enterocolitica или Klebsiella spp., предпочтительно из семейства Pseudomonadaceae, в частности Pseudomonas aeruginosa, предпочтительно из семейства Alcaligenaceae, в частности Bordetella bronchiseptica или Bordetella pertussis, предпочтительно из семейства Brucellaceae, в частности Brucella abortus, предпочтительно из семейства Moraxellaceae, в частности Acinetobacter baumanii, предпочтительно из семейства Xanthomonadaceae, в частности Stenotrophomonas maltophilia, предпочтительно из семейства Pasteuerellaceae, в частности Haemophilus influenzae, предпочтительно из семейства Neisseriaceae, в частности Neisseria meningitidis, предпочтительно из семейства Staphylococcaceae, в частности Staphylococcus aureus or Staphylococcus epidermidis, предпочтительно из семейства Enterococcaceae, в частности Enterococcus faecalis, предпочтительно из семейства Streptococcaceae, в частности Streptococcus agalactiae и предпочтительно из семейства Chlamydiaceae, в частности Chlamydia pneumoniae.
40. Способ по любому из пп.37-39, отличающийся тем, что указанный микроорганизм проявляет множественную лекарственную устойчивость.
41. Способ нейтрализации биологической активности бактериальных компонентов, предпочтительно компонентов клеточной стенки, более предпочтительно липополисахарида микроорганизмов введением эффективного количества пептида по любому из пп.1-34.
42. Способ производства пептида по любому из пп.1-34, имеющего N-концевой пролиновый остаток, включающий стадии:
- обеспечения клетки-хозяина, содержащей молекулу нуклеиновой кислоты, кодирующей слитый полипептид или белок, содержащий пептид по любому из пп.1-34, имеющий N-концевой остаток пролина, в котором пептид слит на С-конце с указанным полипептидом или белком, имеющим С-концевой аспартат,
- экспрессии и выделения указанного слитого полипептида или белка,
- стадию обработки выделенного слитого полипептида или белка при значении рН между 0,5 и 4.
43. Способ адсорбции и удаления или инактивации бактерий или бактериальных компонентов из образцов, включающий стадии контактирования указанного образца с иммобилизованным пептидом по любому из пп.1-34.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Транспортировочное устройство | 1973 |
|
SU503939A1 |
| US 5424396 A, 13.06.1995 | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |

