ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет в соответствии с § 119(е) 35 главы свода законов США согласно заявке на патент США №61/864,215, поданной 9 августа 2013 г., и заявке на патент США №61/936,715, поданной 6 февраля 2014 г., каждая из которых полностью включена посредством ссылки.
ПОЛОЖЕНИЕ ОТНОСИТЕЛЬНО ПЕРЕЧНЯ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Перечень последовательностей, связанный с настоящей заявкой, предложен в текстовом формате вместо бумажной копии и, таким образом, включен в заявку посредством ссылки. Текстовый файл, содержащий перечень последовательностей, называется ARDE_017_01WO_ST25.txt. Размер текстового файла составляет 193 KB, он был создан 8 августа 2014 г. и подан в электронном виде через электронную систему EFS-Web.
УРОВЕНЬ ТЕХНИКИ
Область техники
Настоящее изобретение относится к не связывающимся с NHE3 агентам, проявляющим активность в качестве ингибиторов транспорта фосфатов в желудочно-кишечном тракте, включая тонкий кишечник, способам их применения в качестве терапевтических или профилактических агентов и связанным способам разработки лекарственных средств.
Описание связанного уровня техники
Пациенты с недостаточностью функции почек, гипопаратиреозом или конкретными другими медицинскими состояниями (такими как наследственная гиперфосфатемия, наследственная остеодистрофия Олрайта, амилоидоз и т.д.) часто страдают гиперфосфатемией, или повышенным уровнем фосфора в сыворотке (где указанный уровень составляет, например, более чем примерно 6 мг/дл). Гиперфосфатемия, в частности, присутствующая в течение продолжительного периода времени, приводит к нарушениям метаболизма кальция и фосфора, которые часто проявляются в виде вторичного гиперпаратиреоза, костных заболеваний и эктопической кальцификации в сердечно-сосудистой системе, суставах, легких, глазах и других мягких тканях. Более высокий уровень фосфора в сыворотке сильно связан с прогрессированием почечной недостаточности, кальцификацией в сердечно-сосудистой системе и смертностью у пациентов с конечной стадией заболевания почек (КСЗП). Пограничное увеличение уровня фосфора в сыворотке связано с сердечно-сосудистыми заболеваниями и смертностью среди субъектов, страдающих хроническим заболеванием почек (ХЗП), и субъектов с нормальной функцией почек (см., например, Joy et al., J. Manag. Care Pharm., 13: 397-411, 2007) Прогрессирование заболевания почек можно замедлить путем снижения удержания фосфатов. Таким образом, для пациентов с почечной недостаточностью, страдающих гиперфосфатемией, и для пациентов с хроническими заболеваниями почек, имеющих нормальный или незначительно повышенный уровень фосфора в сыворотке, терапия, направленная на снижение удержания фосфатов, может являться благоприятной.
Соли кальция широко применялись для связывания кишечных фосфатов и предотвращения его всасывания у пациентов, страдающих гиперфосфатемией. Различные типы солей кальция, включая карбонат, ацетат, цитрат, альгинат кальция и соли кетокислот использовали для связывания фосфатов. Однако указанные терапии часто вызывают гиперкальцемию - состояние, которое приводит к всасыванию большого количества потребляемого кальция. Гиперкальцемия вызывает серьезные побочные эффекты, такие как сердечная аритмия, почечная недостаточность и кальцификация кожи и сосудов. При терапии с помощью веществ на основе кальция, связывающих фосфаты, требуется частое наблюдение за уровнем кальция в сыворотке. Другие не содержание кальция и алюминия вещества, связывающие фосфаты, такие как севеламера, поперечно сшитый полиаминный полимер, имеют недостатки, которые включают количество и частоту введения дозы, необходимой для терапевтической активности. Относительно слабая связывающая способность этих лекарственных средств in vivo приводит к необходимости повышения дозы у пациентов (до 7 грамм в сутки или более). Было показано, что такие количества вызывают дискомфорт в желудочно-кишечном тракте, такой как диспепсия, абдоминальная боль и в некоторых крайних случаях перфорацию стенки кишечника.
Альтернативным подходом к предотвращению всасывания фосфатов из кишечника у пациентов с повышенным уровнем фосфатов в сыворотке является подавление системы кишечного транспорта - звена, которое опосредует поглощения фосфатов в кишечнике. Необходимо понимать, что всасывание фосфатов в верхних отделах кишечника обеспечивается по меньшей мере отчасти механизмом, опосредуемым переносчиком, который обеспечивает всасывание фосфатов в сочетании с всасыванием натрия. Подавление кишечного транспорта фосфатов снижает перегрузку организма фосфором. У пациентов с заболеванием почек на поздних стадиях (например, на 4 и 5 стадии) перегрузка организма фосфором проявляется превышающей нормальный уровень концентрацией фосфора в сыворотке, т.е. гиперфосфатемией. Гиперфосфатемия непосредственно связана с заболеваемостью и смертностью. Подавление кишечного транспорта фосфатов снижает концентрацию фосфора в сыворотке и, таким образом, улучшает исход у указанных пациентов. У пациентов, страдающих хроническим заболеванием почек на 2 и 3 стадии, перегрузка организма фосфором не обязательно приводит к гиперфосфатемии, т.е. у некоторых пациентов сохраняется нормальный уровень фосфатов. Однако существует необходимость в снижении или предотвращении перегрузки тела фосфором даже на этих ранних стадиях для предотвращения развития связанных костных и сосудистых расстройств и, в конечном итоге, улучшения показателя смертности. Подобным образом, подавление транспорта фосфатов в кишечнике является особо предпочтительным у пациентов, которые страдают заболеванием, поддающимся лечению путем подавления захвата фосфатов в кишечнике. Более того, подавление транспорта фосфатов может замедлить прогрессирование почечной недостаточности и снизить риск развития сердечно-сосудистых заболеваний.
На люминальной поверхности кишечного эпителия находится так называемый «невозмутимый водный слой» (UWL), в котором транспорт осуществляет по существу посредством диффузии благодаря вязкости указанного слизистого слоя. Указанный «невозмутимый» слой (unstirred layer) определяется как инертный слой, смежный с мембраной апикальной поверхности, действующий как барьер для диффузии таким образом, что может фактически ограничивать скорость диффузии быстро проникающих веществ. Это ограничение диффузии распространяется на H+, и таким образом, UWL вносит вклад в установление микроклимата рН благодаря выходящему потоку протонов и ограничения диффузии, обеспечиваемого слизистым слоем. Кислое окружение на клеточной поверхности поддерживает относительно большой электрохимический градиент эпителиальной мембраны - трансэпителиальный градиент рН или CEPG.
Существуют убедительные доказательства вовлечения указанного CEPG в транспорт питательных веществ через протонные ко-транспортеры и -ОН-антипортеры, такие как РЕРТ1, фолат/ОН- антипортер и β-аланин/Н+ ко-транспортер. См., например, Ikuma, J Med Chem. 50: 1166-1176, 1996. Нарушение микроклимата рН, например, снижение CEPG, может изменять всасывание питательных веществ, что было показано в случае опосредованного протонами всасывания пептида через РЕРТ1. См., например, Thwaites et al., Gastro-enterology. 122: 1322-1333, 2002; и Thwaites and Anderson, Exp. Physiol. 92: 603-619, 2007. Однако роль CEPG в абсорбции фосфат-ионов через кишечную мембрану не была установлена.
Также существует доказательства того, что всасывание воды вовлечено в транспорт ионов через эпителий тонкого кишечника, в частности, тощей кишки. Juan et al., J Clin Endocrinol Metab. 43: 517-22, 1976. Но такие механизмов для терапевтических средств, снижающих уровень фосфатов, мало исследованы.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение в целом относится к не связывающимся с NHE3 соединениям, обладающим активностью в качестве ингибиторов транспорта фосфатов в желудочно-кишечном тракте, в частности, в тонком кишечнике, включая их стереоизомеры, фармацевтически приемлемые соли и пролекарства, и применению указанных соединений для подавления поглощения фосфатов и для лечения, таким образом, любого из различных состояний или заболеваний, при которых модулирование поглощения фосфатов обеспечивает терапевтически благоприятный эффект.
Варианты реализации настоящего изобретения, таким образом, включают способы подавления поглощения фосфатов в желудочно-кишечном тракте пациента, нуждающегося в снижении уровня фосфатов, включающие введение указанному пациенту соединения, которое не связывается с NHE3, где указанное соединение является по существу активным в желудочно-кишечном тракте для подавления транспорта фосфат-ионов (Pi) при введении нуждающемуся в этом пациенту.
Согласно конкретным вариантам реализации изобретения, соединение представляет собой соединение, являющееся агонистом рецептора гуанилатциклазы С.
Согласно конкретным вариантам реализации изобретения, соединения представляют собой рН-модулирующие агенты. Указанные и связанные варианты реализации включают способы подавления поглощения фосфатов в желудочно-кишечном тракте пациента, нуждающегося в снижении содержания фосфатов, включающие введение пациенту соединения, которое снижает трансэпителиальный градиент рН (CEPG) в тонком кишечнике, где CEPG определяется как разница в рН между (i) цитоплазмой эпителиальных клеток поверхности тонкого кишечника, необязательно субапикальной части эпителиальной клетки, и (ii) «невозмутимым» слоем (unstirred layer) на апикальной поверхности клеток тонкого кишечника, где соединение является по существу активным в желудочно-кишечном тракте для подавления транспорта фосфат-ионов (Pi) при введении нуждающемуся в этом пациенту, и где указанное соединение не связывается NHE3.
Согласно некоторым вариантам реализации изобретения, соединения снижают всасывание воды в тонком кишечнике, необязательно тощей кишке. Указанные и связанные варианты реализации изобретения включают способы подавления поглощения фосфатов в желудочно-кишечном тракте пациента, нуждающегося в снижении содержания фосфатов, включающие введение указанному пациенту соединения, которое снижает всасывание воды в тонком кишечнике, необязательно в тощей кишке, где указанное соединение не связывается с NHE3 и является по существу активным в желудочно-кишечном тракте для подавления транспорта фосфат-ионов (Pi) при его введении нуждающемуся в этом пациенту.
Согласно некоторым вариантам реализации изобретения, соединение снижает CEPG в тонком кишечнике, а также снижает всасывание воды в тонком кишечнике. Согласно некоторым вариантам реализации изобретения, соединение снижает CEPG в тонком кишечнике без значительного снижения всасывания воды в тонком кишечнике. Согласно другим вариантам реализации изобретения, соединение снижает всасывание воды в тонком кишечнике без значительного снижения CEPG в тонком кишечнике (например, без значительной стимуляции секреции бикарбоната и/или подавления секреции кислоты).
Согласно некоторым вариантам реализации изобретения, способ относится к способу, выбранному из одного или более из следующих далее:
(a) способ лечения гиперфосфатемии, необязательно гиперфосфатемии после приема пищи;
(b) способ лечения заболевания почек, необязательно хронического заболевания почек (ХЗП) или конечной стадии заболевания почек (КСЗП);
(c) способ снижения уровня креатинина в сыворотке;
(d) способ лечения протеинурии;
(e) способ отсрочки заместительной почечной терапии (ЗПТ), необязательно диализа;
(f) способ снижения уровня FGF23;
(g) способ снижения гиперфосфатемического эффекта активного витамина D;
(h) способ ослабления гиперпаратиреоза, необязательно вторичного гиперпаратиреоза;
(i) способ снижения уровня паратиреоидного гормона (ПТГ) в сыворотке
(j) способ улучшения нарушенной функции эндотелия, необязательно вызванной содержанием фосфора в сыворотке после приема пищи;
(k) способ снижения кальцификации сосудов, необязательно кальцификации во внутренней оболочке сосудов;
(l) способ снижения содержания фосфора в моче;
(m) способ нормализации уровня фосфора в сыворотке;
(n) способ снижения отложения фосфатов у пожилых пациентов;
(о) способ снижения поглощения фосфора из пищи;
(р) способ снижения почечной гипертрофии; и
(q) способ снижения гипертрофии сердца.
Согласно конкретным вариантам реализации изобретения, соединение снижает внутриклеточный уровень рН эпителиальных клеток поверхности тонкого кишечника, необязательно субапикальной части эпителиальной клетки. Согласно конкретным вариантам реализации изобретения, соединение повышает уровень рН «невозмутимого» слоя на (unstirred layer) апикальной поверхности клеток тонкого кишечника. Согласно некоторым вариантам реализации изобретения, соединение (а) стимулирует секрецию бикарбоната в тонком кишечнике или (b) подавляет секрецию кислоты в тонком кишечнике или (с) стимулирует секрецию бикарбоната и подавляет секрецию кислоты в тонком кишечнике.
Согласно конкретным вариантам реализации изобретения, соединение повышает уровень одного или более внутриклеточных вторичных посредников в клетках эпителия поверхности тонкого кишечника. Согласно некоторым вариантам реализации изобретения, один или более внутриклеточных вторичных посредников выбраны из Са++, циклического аденозинмонофосфата (сАМР) и циклического гуанозинмонофосфата (cGMP).
Согласно конкретным вариантам реализации изобретения, соединение по существу не обладает системной биодоступностью при кишечном введении пациенту. Согласно конкретным вариантам реализации изобретения, соединение по существу не проникает через эпителий желудочно-кишечного тракта. Согласно некоторым вариантам реализации изобретения, соединение по существу проникает через эпителий желудочно-кишечного тракта.
Согласно конкретным вариантам реализации изобретения, введение соединения нуждающемуся в этом пациенту приводит к (а) снижению концентрации или уровня фосфора в сыворотке до примерно 150% или менее от нормального уровня фосфора в сыворотке и/или (b) снижению поглощения фосфора из пищи по меньшей мере примерно на 10% по сравнению с состоянием в отсутствие лечения. Согласно некоторым вариантам реализации изобретения, введение соединения нуждающемуся в этом пациенту приводит к повышению уровня фосфатов в фекалиях по меньшей мере примерно на 10% по сравнению с состоянием в отсутствие лечения. Согласно некоторым вариантам реализации изобретения, введение соединения нуждающемуся в этом пациенту снижает концентрацию или уровень фосфатов в моче по меньшей мере примерно на 10% по сравнению с состоянием в отсутствие лечения.
Согласно некоторым вариантам реализации изобретения, пациент, нуждающийся во введении соединения, страдает КСЗП, и введение соединения указанному пациенту приводит к снижению концентрации или уровня фосфора в сыворотке по меньшей мере примерно на 10% по сравнению с состоянием в отсутствие лечения.
Согласно некоторым вариантам реализации изобретения, пациент, нуждающийся во введении соединения, страдает ХЗП, и введение соединения указанному пациенту приводит к снижению уровня FGF23 и уровня интактного паратиреоидного гормона в сыворотке (иПТГ) по меньшей мере примерно на 10% по сравнению с состоянием в отсутствие лечения.
Согласно конкретным вариантам реализации изобретения, соединение выбрано из одного или более из агониста рецептора гуанилатциклазы С (GC-C), агониста P2Y, агониста рецептора аденозина A2b, агониста растворимой гуанилатциклазы, агониста рецептора аденилатциклазы, агониста рецептора имидазолина-1, холинергического агониста, агониста рецептора простагландина ЕР4, агониста дофамина D1, агониста рецептора мелатонина, агониста5НТ4, агониста рецептора предсердного натрийуретического пептида, ингибитора карбоангидразы, ингибитора фосфодиэстеразы и агониста белка DRA (Down-Regulated in Adenoma, или SLC26A3).
Согласно некоторым вариантам реализации изобретения, агонист GC-C представляет собой пептид, необязательно бактериальный термостабильный энтеротоксин, гуанилин, прогуанилин, урогуанилин, проурогуанилин, лимфогуанилин или вариант или аналог любого из перечисленных.
Согласно некоторым вариантам реализации изобретения, пептидный агонист GC-C содержит аминокислотную последовательность (I): Xaa1 Хаа2 Хаа3 Хаа4 Хаа5 Cys6 Cys7 Xaa8 Xaa9 Cys10 Cys11 Xaa12 Xaa13 Xaa14 Cys15 Xaa16 Xaa17 Cys18 Xaa19 Xaa20 Xaa21 (SEQ ID NO: 1) где: Xaa1 Хаа2 Хаа3 Хаа4 Xaa5 представляет собой Asn Ser Ser Asn Tyr (SEQ ID NO: 2) или отсутствует, или Xaa1 Xaa2 Хаа3 Хаа4 отсутствует.
Согласно конкретным вариантам реализации изобретения, Хаа5 представляет собой Asn, Trp, Tyr, Asp или Phe.
Согласно конкретным вариантам реализации изобретения, Xaa5 представляет собой Thr или Ile.
Согласно конкретным вариантам реализации изобретения, Xaa5 представляет собой Tyr, Asp или Trp.
Согласно конкретным вариантам реализации изобретения, Xaa8 представляет собой Glu, Asp, Gln, Gly или Pro.
Согласно конкретным вариантам реализации изобретения, Xaa9 представляет собой Leu, Ile, Val, Ala, Lys, Arg, Trp, Tyr или Phe.
Согласно конкретным вариантам реализации изобретения, Xaa9 представляет собой Leu, Ile, Val, Lys, Arg, Trp, Tyr или Phe.
Согласно конкретным вариантам реализации изобретения, Xaa12 представляет собой Asn, Tyr, Asp или Ala.
Согласно конкретным вариантам реализации изобретения, Хаа13 представляет собой Ala, Pro или Gly.
Согласно конкретным вариантам реализации изобретения, Xaa14 представляет собой Ala, Leu, Ser, Gly, Val, Glu, Gln, Ile, Leu, Lys, Arg или Asp.
Согласно конкретным вариантам реализации изобретения, Xaa16 представляет собой Thr, Ala, Asn, Lys, Arg или Trp.
Согласно конкретным вариантам реализации изобретения, Xaa17 представляет собой Gly, Pro или Ala.
Согласно конкретным вариантам реализации изобретения, Xaa19 представляет собой Trp, Tyr, Phe, Asn или Leu.
Согласно конкретным вариантам реализации изобретения, Xaa19 представляет собой Lys или Arg.
Согласно конкретным вариантам реализации изобретения, Хаа20 Xaa21 представляет собой AspPhe, или Хаа20 представляет собой Asn или Glu, и Xaa21 отсутствует. Согласно конкретным вариантам реализации изобретения, Xaa19 Хаа20 Xaa21 отсутствует.
Согласно конкретным вариантам реализации изобретения, пептидный агонист GC-C содержит аминокислотную последовательность: Asn Ser Ser Asn Tyr Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr (SEQ ID NO: 3) или ее вариант, содержащий 1, 2, 3, 4 или 5 делеций, инсерций и/или замен. Согласно конкретным вариантам реализации изобретения, пептид содержит аминокислотную последовательность: Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr (SEQ ID NO: 4) или ее вариант, содержащий 1, 2, 3, 4 или 5 делеций, инсерций и/или замен.
Согласно конкретным вариантам реализации изобретения, пептидный агонист GC-C содержит аминокислотную последовательность (III): Xaa1 Хаа2 Хаа3 Cys4 Xaa5 Xaa6 Xaa7 Хаа8 Хаа9 Хаа10 Хаа11 Cys12 Хаа13 Хаа14 Xaa15 Xaa16 (SEQ ID NO: 5), где Xaa1 представляет собой: Ser, Asn, Tyr, Ala, Gln, Pro, Lys, Gly или Thr или отсутствует; Xaa2 представляет собой His, Asp, Glu, Ala, Ser, Asn, Gly или отсутствует; Хаа3 представляет собой Thr, Asp, Ser, Glu, Pro, Val или Leu; Xaa5 представляет собой Asp, Ile или Glu; Xaa6 представляет собой Ile, Trp или Leu; Xaa7 представляет собой Cys, Ser или Tyr; Xaa8 представляет собой Ala, Val, Thr, Ile, Met или отсутствует; Хаа9 представляет собой Phe, Tyr, Asn или Trp; Хаа10 представляет собой Ala, Val, Met, Thr или Ile; Хаа11 представляет собой Ala или Val; Хаа13 представляет собой Thr или Ala; Xaa14 представляет собой Gly, Ala или Ser; Xaa15 представляет собой Cys, Tyr или отсутствует; и Xaa16 представляет собой His, Leu или Ser.
Согласно некоторым вариантам реализации изобретения, пептид содержит аминокислотную последовательность: Asn Asp Glu Cys Glu Leu Cys Val Asn Val Ala Cys Thr Gly Cys Leu (SEQ ID NO: 6) или ее вариант, содержащий 1, 2, 3, 4 или 5 делеций, инсерций и/или замен.
Согласно конкретным вариантам реализации изобретения, агонист P2Y выбран из соединения на Фигуре 4 или Фигурах 5А-5С. Согласно конкретным вариантам реализации изобретения, агонист рецептора аденозина A2b выбран из соединения на Фигурах 6А-6С. Согласно некоторым вариантам реализации изобретения, агонист растворимой гуанилатциклазы выбран из соединения на Фигурах 9A-9L. Согласно конкретным вариантам реализации изобретения, агонист рецептора аденилатциклазы выбран из соединения на Фигуре 10. Согласно некоторым вариантам реализации изобретения, агонист рецептора имидазолина-1 выбран из моксонидина и соединения на Фигуре 11. Согласно конкретным вариантам реализации изобретения, холинергический агонист выбран из соединения на Фигуре 12. Согласно конкретным вариантам реализации изобретения, агонист рецептора простагландина ЕР4 выбран из PGE2 или его аналогов/производных и соединения на Фигуре 7 или Фигуре 13. Согласно конкретным вариантам реализации изобретения, агонист дофамина D1 выбран из соединения на Фигуре 14. Согласно некоторым вариантам реализации изобретения, агонист рецептора мелатонина выбран из мелатонина и соединения на Фигуре 15. Согласно некоторым вариантам реализации изобретения, агонист 5НТ4 выбран из серотонина и его аналогов, прукалоприда, метоклопрамида, клеобоприда, мозаприда, прукалоприда, рензаприда, тегасерода, закоприда, норцисаприда, нароноприда и велусетрага.
Согласно некоторым вариантам реализации изобретения, агонист рецептора предсердного натрийуретического пептида содержит или состоит из аминокислотной последовательности, выбранной из: Ser Leu Arg Arg Ser Ser Cys Phe Gly Gly Arg Ile Asp Arg Ile Gly Ala Gln Ser Gly Leu Gly Cys Asn Ser Phe Arg Tyr (SEQ ID NO: 7), Cys Phe Gly Gly Arg Ile Asp Arg Ile Gly Ala Gln Ser Gly Leu Gly Cys (SEQ ID NO: 8) и Ser Ser Cys Phe Gly Gly Arg Ile Asp Arg Ile Gly Ala Gln Ser Gly Leu Gly Cys Asn Ser Phe Arg (SEQ ID NO: 9), включая ее варианты, содержащие 1, 2, 3, 4 или 5 делеций, инсерций и/или замен.
Согласно конкретным вариантам реализации изобретения, ингибитор карбоангидразы выбран из соединения на Фигуре 17. Согласно конкретным вариантам реализации изобретения, ингибитор фосфодиэстеразы выбран из соединения на Фигуре 18. Согласно некоторым вариантам реализации изобретения, агонист DRA выбран из соединения на Фигурах 21А-В.
Согласно некоторым вариантам реализации изобретения, соединение по существу не обладает системной биодоступностью при кишечном введении пациенту, и (i) tPSA указанного соединения составляет по меньшей мере примерно 200  2. Согласно конкретным вариантам реализации изобретения, tPSA соединения составляет по меньшей мере примерно 250 2, tPSA составляет по меньшей мере примерно 270 2, tPSA составляет по меньшей мере примерно 300 2, tPSA составляет по меньшей мере примерно 350 2, tPSA составляет по меньшей мере примерно 400 2 или tPSA составляет по меньшей мере примерно 500 2. Согласно конкретным вариантам реализации изобретения, молекулярная масса соединения составляет по меньшей мере примерно 500 Да, по меньшей мере примерно 1000 Да, по меньшей мере примерно 2500 Да или по меньшей мере примерно 5000 Да или более. Согласно некоторым вариантам реализации изобретения, (i) общее число групп NH и/или ОН и/или других фрагментов, являющихся потенциальными донорами водородных связей, в соединении составляет более чем примерно 5; (ii) общее число атомов О и/или N и/или других потенциальных акцепторов водородных связей составляет более чем примерно 10; и/или (iii) коэффициент распределения Моригучи соединения составляет более чем примерно 105 или менее чем примерно 10. Согласно некоторым вариантам реализации изобретения, коэффициент проникновения соединения, Papp, составляет менее чем примерно 100×10-6 см/с или менее чем примерно 10×10-6 см/с или менее чем примерно 1×10-6 см/с или менее чем примерно 0,1×10-6 см/с.
2. Согласно конкретным вариантам реализации изобретения, tPSA соединения составляет по меньшей мере примерно 250 2, tPSA составляет по меньшей мере примерно 270 2, tPSA составляет по меньшей мере примерно 300 2, tPSA составляет по меньшей мере примерно 350 2, tPSA составляет по меньшей мере примерно 400 2 или tPSA составляет по меньшей мере примерно 500 2. Согласно конкретным вариантам реализации изобретения, молекулярная масса соединения составляет по меньшей мере примерно 500 Да, по меньшей мере примерно 1000 Да, по меньшей мере примерно 2500 Да или по меньшей мере примерно 5000 Да или более. Согласно некоторым вариантам реализации изобретения, (i) общее число групп NH и/или ОН и/или других фрагментов, являющихся потенциальными донорами водородных связей, в соединении составляет более чем примерно 5; (ii) общее число атомов О и/или N и/или других потенциальных акцепторов водородных связей составляет более чем примерно 10; и/или (iii) коэффициент распределения Моригучи соединения составляет более чем примерно 105 или менее чем примерно 10. Согласно некоторым вариантам реализации изобретения, коэффициент проникновения соединения, Papp, составляет менее чем примерно 100×10-6 см/с или менее чем примерно 10×10-6 см/с или менее чем примерно 1×10-6 см/с или менее чем примерно 0,1×10-6 см/с.
Конкретные способы также включают введение одного или более дополнительных биологически активных агентов. Согласно некоторым вариантам реализации изобретения, соединение и один или более дополнительных биологически активных агентов вводят как часть одной фармацевтической композиции. Согласно конкретным вариантам реализации изобретения, соединение и один или более дополнительных биологически активных агентов вводят в виде отдельных фармацевтических композиций. Согласно некоторым вариантам реализации изобретения, отдельные фармацевтические композиции вводят последовательно. Согласно некоторым вариантам реализации изобретения, отдельные фармацевтические композиции вводят одновременно.
Согласно конкретным вариантам реализации изобретения, дополнительный биологически активный агент выбран из витамина D2 (эргокальциферол), витамина D3 (холекальциферол), активного витамина D (кальцитриол) и аналогов активного витамина D (например, доксеркальциферол, парикальцитола).
Согласно конкретным вариантам реализации изобретения, дополнительный биологически активный агент представляет собой вещество, связывающее фосфаты. Согласно некоторым вариантам реализации изобретения, вещество, связывающее фосфаты, выбрано из группы, состоящей из севеламера (например, Renvela® (севеламера карбоната), Renagel® (севеламера гидрохлорида)), лантана карбоната (например, Fosrenol®), кальция карбоната (например, Calcichew®, Titralac®), кальция ацетата (например, PhosLo®, Phosex®), кальция ацетата/магния карбоната (например, Renepho®, OsvaRen®), MCI-196, лимоннокислого железа (например, Zerenex™), магния-железа гидроксикарбоната (например, Fermagate™), алюминия гидроксида (например, Alucaps®, Basaljel®), APS1585, SBR-759 и PA-21.
Согласно конкретным вариантам реализации изобретения, дополнительный биологически активный агент представляет собой ингибитор NaPi2b. Согласно некоторым вариантам реализации изобретения, дополнительный биологически активный агент представляет собой ниацин или никотинамид.
Согласно конкретным вариантам реализации изобретения, субъект страдает ХЗП, и дополнительный активный биологический агент выбран из одного или более из ингибиторов АСЕ, блокаторов рецептора ангиотензина II, бета-блокаторов, блокаторов кальциевых каналов, прямых ингибиторов ренина, диуретиков, вазодилататоров, агентов терапии эритропоэтином, агентов заместительной терапии железа, ингибиторов конечных продуктов усиленного гликозилирования, витамина D и статинов.
Согласно конкретным вариантам реализации изобретения, соединение или композицию вводят перорально, при этом указанное соединение или композицию необязательно вводят перорально один раз в день.
Также включены способы скрининга для поиска ингибитора захвата фосфатов, включающие (а) культивирование клеток кишечника, (b) приведение культивируемых клеток кишечника во взаимодействие с исследуемым соединением и (с) измерение (i) значения рН на апикальной поверхности клеток кишечника, (ii) внутриклеточного рН клеток кишечника и/или (iii) поглощения/захвата фосфатов клетками кишечника и (d) идентификацию исследуемого соединения как ингибитора захвата фосфатов, где значение рН, полученное в результате (c)(i), повышено по сравнению с контролем, внутриклеточное значение рН, полученное в результате (c)(ii), снижено по сравнению с контролем и/или значение поглощения фосфатов, полученное в результате (с)(iii), снижено по сравнению с контролем.
Согласно некоторым вариантам реализации изобретения, этап (а) включает культивирование клеток кишечника до образования монослоя. Согласно конкретным вариантам реализации изобретения, этап (а) включает выделение клеток из кишечной крипты и культивирование в условиях, достаточных для образования энтероидов. Согласно конкретным вариантам реализации изобретения, этап (а) включает культивирование выделенных эмбриональных стволовых клеток, клеток эндодермы или плюрипотентных стволовых клеток в условиях, достаточных для образования органоидов. Согласно некоторым вариантам реализации изобретения, этап (а) включает культивирование кишечного среза (срезов) в камере Уссинга.
Согласно конкретным вариантам реализации изобретения, этап (c)(i) включает приведение клеток во взаимодействие с рН-чувствительным флуоресцентным красителем и измерение флуоресценции указанного красителя. Согласно некоторым вариантам реализации изобретения, этап (c)(ii) включает приведение клеток во взаимодействие с 33P-меченными фосфат-ионами и измерение поглощения меченных фосфат-ионов.
Согласно некоторым вариантам реализации изобретения, повышение и/или снижение (d) является статистически значимым.
Согласно конкретным вариантам реализации изобретения, исследуемое соединение представляет собой малую молекулу или пептид, которые, как известно или как предполагается, стимулируют секрецию бикарбоната и/или подавляют секрецию кислоты в тонком кишечнике.
Согласно конкретным вариантам реализации изобретения, исследуемое соединение выбрано из одного или более из агониста P2Y, агониста рецептора аденозина A2b, агониста рецептора гуанилатциклазы С, агониста растворимой гуанилатциклазы, агониста рецептора аденилатциклазы, агониста рецептора имидазолина-1, холинергического агониста, агониста рецептора простагландина ЕР4, агониста дофамина D1, агониста рецептора мелатонина, агониста 5НТ4, агониста рецептора предсердного натрийуретического пептида, ингибитора карбоангидразы, ингибитора фосфодиэстеразы и агониста белка DRA (Down-Regulated in Adenoma, или SLC26A3), как описано в настоящей заявке и/или известно в данной области техники.
Указанные и другие аспекты изобретения будут очевидны при рассмотрении следующего далее подробного описания.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фигурах 1А-1В показано, что линаклотидин (агонист рецептора GC-C) снижает поглощения фосфатов в желудочно-кишечном тракте у крыс.
На Фигурах 2А-2В показано, то моксонидин (агонист рецептора имидазолина подтипа 1 (I1)) и водорастворимый аналог форсколина кольфорсин (агонист аденилатциклазы) снижают поглощение фосфатов в желудочно-кишечном тракте у крыс.
На Фигуре 3 показано, что агонист рецептора P2Y2 Up4U снижает поглощение фосфатов в желудочно-кишечном тракте у крыс.
На Фигуре 4 показаны примеры низкомолекулярных агонистов рецептора P2Y.
На Фигурах 5А-5С показаны примеры низкомолекулярных агонистов рецептора P2Y.
На Фигурах 6А-6С показаны примеры низкомолекулярного агониста рецептора аденозина A2b, включая типичные аденозин-подобные агонисты A2b (6В) и типичные дицианопиридиновые агонисты A2b (6С).
На Фигуре 7 показан перечень примеров агонистов рецептора простагландина ЕР4.
На Фигурах 8А-8В показаны фотофизические свойства примеров индикаторов почти нейтрального (8А) и индикаторов кислого рН (8В).
На Фигурах 9A-9L показаны примеры агонистов растворимой гуанилатциклазы (sGC), включая гем-зависимые и гем-независимые агонисты (9А).
На Фигуре 10 показаны примеры агонистов рецептора аденилатциклазы.
На Фигуре 11 показаны примеры агонистов рецептора имидазолина.
На Фигуре 12 показаны примеры холинергических агонистов и антагонистов атропина и (-)-гиозина.
На Фигуре 13 показаны примеры агонистов рецептора ЕР4.
На Фигуре 14 показаны примеры агонистов рецептора дофамина D1.
На Фигуре 15 показаны примеры агонистов рецептора мелатонина (МТ2).
На Фигуре 16 показаны структуры примеров пептидных агонистов (SEQ ID №7, 8 и 9) рецептора (рецепторов) NP.
На Фигуре 17 показаны примеры ингибиторов карбоангидразы.
На Фигуре 18 показаны примеры ингибиторов фосфодиэстеразы.
На Фигуре 19 показаны градиенты рН, наблюдаемые в кишечнике, включая трансмембранный градиент рН клеток и градиент рН в непосредственной близости от мембраны эпителиальных клеток и просвета пищеварительного тракта.
На Фигуре 20 показана фазовая диаграмма растворимости кальция и фосфат-ионов в водном окружении (при комнатной температуре) в диапазоне значений рН.
На Фигурах 21А-21В показаны типичные примеры ингибиторов, селективных в отношении подтипов PKC.
На Фигурах 22А-22С показано, что закисление внутриклеточной среды клеток HEK-293 приводит к значительному снижению поглощения фосфатов по результатам измерения захвата Pi, меченного 33Р.
ПОДРОБНОЕ ОПИСАНИЕ
В следующем далее описании конкретные специфические детали перечислены для полного понимания различных вариантов реализации изобретения. Однако специалисту в данной области техники очевидно, что изобретение можно осуществлять на практике без указанных подробностей.
Если иное не следует из контекста, на протяжении всей заявки и в прилагаемой формуле изобретения слово «содержит» и его варианты, такие как, «содержащий» и «включающий», следует рассматривать в их открытом широком смысле, т.е. как «включая, но не ограничиваясь указанными».
Ссылка на «один вариант реализации изобретения» или «вариант реализации изобретения» на протяжении всей заявки означает, что конкретный признак, структура или характеристика, описанная в связи с указанным вариантом реализации изобретения, включена по меньшей мере в один вариант реализации настоящего изобретения. Таким образом, не обязательно все фразы «согласно одному варианту реализации изобретения» или «согласно варианту реализации изобретения», встречающиеся в различных местах в настоящей заявке, относятся к одному и тому же варианту реализации. Более того, конкретные признаки, структуры или характеристики можно объединять любым подходящим образом согласно одному или более вариантам реализации изобретения.
Варианты реализации настоящего изобретения в целом относятся к открытию того, что не связывающиеся с NHE3 соединения, такие как соединения-агонисты гуанилатциклазы, способны подавлять поглощения фосфатов в желудочно-кишечном тракте, например, в тонком кишечнике.
В соответствии с одной неограничивающей теорией, на клеточный захват фосфат-ионов (Pi) может влиять изменение внутриклеточного рН и/или рН прилежащего внеклеточного окружения. Например, как показано в сопутствующих Примерах, повышение внутриклеточной кислотности клеток эмбриона человека (HEK-293) (при поддержании внеклеточного рН примерно при 7,4) приводило к значительному снижению поглощения фосфатов по результатам измерения захвата Pi, меченного 33Р.
В связанных экспериментах, согласно которым переносчик фосфатов NaPi2b (SLC34A2) временно экспрессировался в клетках HEK-293, наблюли такое же явление. Поскольку эндогенные переносчики Pi, Pit-1 и/или Pit-2 (SLC20A2), отвечают за поглощение Pi в нетрансформированных клетках HEK-293 (для удовлетворения требованиям клеточного метаболизма), было предположено, что эффект снижения внутриклеточного рН на поглощение Pi является общим явлением, необязательно связанным с конкретным фосфатным переносчиком. Pit-1 и Pit-2 переносят одноосновную форму фосфата NaH2PO3-, тогда как NaPi2b переносит двухосновную форму NaHPO32-. Наблюдение того, что окисление клеток приводит к нарушению поглощения фосфатов через обе формы переносчиков, не согласуется с механизмом, основанным на изменении только электрохимического градиента Н+.
Указанные наблюдения являются неожиданными по меньшей мере в связи с тем, что ожидалось повышение поглощения Pi. Например, можно было ожидать снижения внутриклеточного рН (например, без какого-либо соответсвующего изменения внеклеточного рН) для создания движущей силы для поглощения оснвных анионов, таких как двухосновные формы фосфатов (NaPO32-).
Тем не менее, наблюдали снижение поглощения фосфатов, что обеспечивает потенциальную возможность использования прямых или не прямых модулирующих рН агентов, в частности, агентов, обладающих активностью как модулирующих рН агентов в желудочно-кишечном тракте (например, тонком кишечнике), для снижения поглощения фосфатов у пациента, нуждающегося в снижении содержания фосфатов. Указанная возможность подтверждается наблюдением, что различные модулирующие рН агенты способны снижать поглощение фосфатов в желудочно-кишечном тракте млекопитающих (см. сопутствующие Примеры). Термин «модулирующие рН» агенты при использовании в настоящей заявке включает агенты или соединения, которые способны прямо или косвенно повышать секрецию бикарбоната (НСО3-) и/или снижать секрецию кислоты/протонов (например, Н+) в просвет желудочно-кишечного тракта, например, тонкого кишечника или двенадцатиперстной кишки. Некоторые модулирующие рН соединения могут функционировать, например, путем модулирования (например, повышения) конкретных внутриклеточных вторичных посредников в клетках эпителия желудочно-кишечного тракта, таких как Са++, cAMP, cGMP и другие посредники. Некоторые примеры соединений, таким образом, прямо или косвенно стимулируют секрецию бикарбоната в просвет тонкого кишечника, подавляют секрецию кислоты в просвет тонкого кишечника или стимулируют секрецию бикарбоната и подавляют секрецию кислоты в просвет тонкого кишечника. Согласно некоторым аспектам, соединение снижает цитоплазмотический или внутриклеточный рН эпителиальных клеток поверхности тонкого кишечника, необязательно субапикальной поверхности эпителиальных клеток, с модулированием или без модулирования рН прилежащего внеклеточного окружения. Согласно конкретным вариантам реализации изобретения, соединение не связывает и подавляет антипортер натрия/водорода 3 (NHE3).
Согласно некоторым аспектам, соединение снижает рН «невозмутимого слоя» (unstirred layer) на апикальной поверхности клеток тонкого кишечника. «Невозмутимый слой» (unstirred layer) относится к инертному слою, прилежащему к мембране апикальной поверхности (например, глубиной примерно 600 мкм), который действует в качестве барьера для диффузии таким образом, что может ограничивать скорость диффузии быстро проникающих веществ (например, 1Н+). Без ограничения какой-либо теорией, такой подход может приводить к потоку бикарбоната через эпителиальные клетки желудочно-кишечного тракта, повышению рН в непосредственной близости от внешней поверхности клеток (UWL) и, таким образом, снижению градиента рН на поверхности слизистой оболочки. В результате непрерывного обмена ионов водорода и бикарбоната через ко-транспортеры, антипортеры и каналы на апикальной поверхности клеток кишечника поддерживается трансмембранный градиент рН. Благодаря «невозмутимому» слою (unstirred layer) между пространством в непосредственной близости от мембраны эпителиальных клеток и просветом кишечника устанавливается другой градиент рН. Схематичное изображение двух градиентов рН представлено на Фигуре 19.
Соответственно, согласно некоторым аспектам, соединение снижает трансэпителиальный градиент рН (СЕРG) в желудочно-кишечном тракте. Термин «CEPG» включает различие в рН между (i) цитоплазмой эпителиальных клеток поверхности тонкого кишечника (т.е. внутриклеточным рН), необязательно на субапикальной поверхности эпителиальных клеток, и (ii) «невозмутимым» слоем (unstirred layer) на апикальной поверхности клеток тонкого кишечника. Согласно конкретным вариантам реализации изобретения, соединения, которые просто повышают рН просвета желудочно-кишечного тракта без модулирования секреции бикарбоната и/или кислоты или без изменения рН «невозмутимого» слоя (unstirred layer) или UWL (например, антациды), исключаются.
Согласно некоторым вариантам реализации изобретения, и не ограничиваясь какой-либо одной теорией, свободные ионы кальция в просвете пищеварительного тракта могут вносить вклад в подавление захвата Pi, вызванного снижением СЕРG. Фазовая диаграмма кальция и фосфат-ионов в водных условиях при комнатной температуре показывает, что растворимость кальция (и, таким образом, фосфата) является рН-зависимой, т.е. растворимость фосфата снижается по мере повышения рН (см. Фигуру 20). Указанное явление предполагает, что при прочих равных условиях вызванное лекарственным средством повышение рН в микроокружении поверхности слизистой оболочки приводит к минимизации доступного свободного Pi, таким образом, вызывая снижение его клеточного поглощения в желудочно-кишечном тракте.
Согласно другой неограничивающей теории, на поглощение фосфат-ионов может влиять всасывание воды в тонком кишечнике, в первую очередь, в тощей кишке. В частности, повышенное всасывание воды в тонком кишечнике связано с повышенным поглощением фосфатов, и наоборот. В таких примерах не связывающиеся с NHE3 соединения, которые снижают всасывание воды в тонком кишечнике, можно использовать для подавления поглощения фосфатов. Конкретные варианты реализации изобретения, таким образом, относятся к способам подавления поглощения фосфатов в желудочно-кишечном тракте пациента, нуждающегося в снижении содержания фосфатов, включающим введение указанному пациенту соединения, снижающего всасывание воды в тонком кишечнике, где указанное соединение не связывается с NHE3 и является по существу активным в желудочно-кишечном тракте для подавления транспорта фосфат-ионов (Pi) при его введении нуждающемуся в этом пациенту. Согласно конкретным вариантам реализации изобретения, соединение снижает «результирующее» всасывание воды, например, путем модулирования баланса между секрецией и всасыванием, например, путем снижения всасывания, повышения секреции или снижения всасывания и повышения секреции. Согласно некоторым вариантам реализации изобретения, соединение снижает всасывание воды в тощей кишке.
Согласно некоторым аспектам, подавление захвата фосфатов в желудочно-кишечном тракте может достигаться путем введения конкретных соединений и/или содержащих их фармацевтических композиций, которые можно предпочтительно получать таким образом, чтобы указанное соединение почти или по существу не всасывалось в кровоток (т.е. было создано таким образом, чтобы оно не обладало или по существу не обладало системным действием). В этом отношении соединения обладают признаками, которые обеспечивают их незначительную системную биодоступность или по существу отсутствие системной биодоступности при кишечном введении, включая пероральное введение. Другими словами, соединения не всасываются в кровоток на значительном уровне и, таким образом, не проявляют в нем активность, при этом они обладают активностью по существу в пределах ЖКТ.
Таким образом, в частности, иллюстративные варианты реализации соединений согласно изобретению, также описанные в настоящей заявке, в целом должны обладать комбинацией структурных и/или функциональных признаков, имеющих отношение или вносящих вклад в их активность в ЖКТ и/или их по существу отсутствие системной биодоступности. Такие признаки могут включать, например, один или более из следующих показателей: (i) конкретные значения tPSA и/или MW (например, составляющие по меньшей мере примерно 190 2 и/или по меньшей мере примерно 736 Дальтон, соответственно), (ii) конкретный уровень восстановления из фекалий соединения и/или его метаболитов после введения (например, более чем 50% за 72 часа); (iii) конкретное количество групп NH и/или ОН и/или фрагментов, являющихся потенциальными донорами водородных связей (например, более чем примерно пять); (iv) конкретное количество вращаемых связей (например, более чем примерно пять); (iv) конкретные свойства проникновения (например, Рарр менее чем примерно 100×10-6 см/с); и/или любой из ряда других признаков и характеристик, описанных в настоящей заявке.
У пациентов, страдающих заболеванием почек на поздней стадии (например, 4 и 5 стадии), перегрузка организма фосфором проявляется повышенной концентрацией фосфора в сыворотке по сравнению с нормальным уровнем, т.е. гиперфосфатемией. Гиперфосфатемия непосредственно связана с заболеваемостью и смертностью. Подавление кишечного транспорта фосфатов снижает концентрацию фосфора в сыворотке и, таким образом, улучшает исход у указанных пациентов. У пациентов, страдающих хроническим заболеванием почек на 2 и 3 стадии, перегрузка организма фосфором не обязательно приводит к гиперфосфатемии, т.е. у указанных пациентов остается нормальный уровень фосфатов, но вызывает повышение уровня FGF-23 - фактора риска заболеваемости и смертности у указанных пациентов. Таким образом, даже на таких ранних стадиях существует необходимость в снижении перегрузки организма фосфором для предотвращения связанных костных и сосудистых нарушений и улучшения в конечном итоге показателя смертности.
Подавление транспорта фосфатов в кишечнике особо предпочтительно у пациентов, страдающих заболеваниями, которые поддаются лечению путем подавления поглощения фосфатов из кишечника. Более того, подавление транспорта фосфатов может замедлять прогрессирование почечной недостаточности и снижать риск развития сердечно-сосудистых заболеваний, среди прочих заболеваний или состояний, связанных с необходимостью снижения содержания фосфатов.
I. Соединения, которые подавляют транспорт фосфатов
Варианты реализации настоящего изобретения относятся к соединениям, которые способны подавлять или снижать транспорт/захват фосфатов в желудочно-кишечном тракте, например, путем модулирования рН в пределах или около мембраны эпителиальных клеток просвета желудочно-кишечного тракта, путем снижения всасывания воды в тонком кишечнике или с помощью обоих механизмов. Примеры рН-модулирующих соединений включают соединения, которые стимулируют секрецию бикарбоната в тонком кишечнике (т.е. секрецию бикарбоната в двенадцатиперстной кишке или СБДК), подавляют секрецию кислоты/протона в тонком кишечнике или стимулируют секрецию бикарбоната и подавляют секрецию кислоты/протона в тонком кишечнике.
Соединения, предложенные в настоящей заявке, могут включать синтетические и природные малые молекулы и пептиды или полипептиды. Термины «пептид» и «полипептид» используются в настоящей заявке взаимозаменяемо; однако в конкретных примерах термин «пептид» может относиться к более коротким полипептидам, например, полипептидам, которые состоят из примерно 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45 или 50 аминокислот, включая все целые числа и диапазоны между ними (например, 5-10, 8-12, 10-15). Полипептиды и пептиды могут состоять из природных аминокислот и/или неприродных аминокислот. Полипептиды также включают антитела.
Согласно некоторым вариантам реализации изобретения, соединение выбрано из одного или более из агониста рецептора P2Y, агониста рецептора аденозина A2b, агониста рецептора гуанилатциклазы С, агониста растворимой гуанилатциклазы, агониста рецептора аденилатциклазы, агониста рецептора имидазолина-1, холинергического агониста, агониста рецептора простагландина ЕР4, агониста дофамина D1, агониста рецептора мелатонина, агониста 5НТ4, агониста рецептора предсердного натрийуретического пептида, ингибитора карбоангидразы, ингибитора фосфодиэстеразы или агониста белка DRA (Down-Regulated in Adenoma, или SLC26A3). Согласно некоторым аспектам, как отмечено выше, такие соединения-агонисты вызывают секрецию бикарбоната и/или подавляют секрецию кислоты в верхних отделах желудочно-кишечного тракта, включая двенадцатиперстную кишку и проксимальные отделы тощей кишки. Согласно некоторым аспектам, механизм действия представляет собой прямое или косвенное модулирование переносчиков протонов и бикарбоната на апикальной поверхности для снижения CEPG или создания относительно основного микроокружения на поверхности слизистой оболочки, что приводит, таким образом, к снижению поглощения/всасывания фосфатов.
Согласно конкретным аспектам, соединение прямо или косвенно стимулирует секрецию бикарбоната в двенадцатиперстной кишке (СБДК). СБДК представляет собой природную защиту слизистой оболочки, которая обеспечивается в сегментах двенадцатиперстной кишки и проксимальных сегментах тощей кишки для нейтрализации кислого желудочного сока. СБДК можно стимулировать с помощью ряда биологических путей, включая сигнальные пути, регулирующие активность антипортеров хлорида/бикарбоната, таких как SLC26A3 (DRA) и SLC26A3 (РАТ-1), каналов CFTR для хлорида и бикарбоната и кальций-активируемых хлоридных каналов, среди прочих. Согласно некоторым аспектам, указанные пути стимулирует повышение уровня одного или более вторичных посредников, таких как внутриклеточный Са++, сАМР и/или cGMP.
Согласно некоторым аспектам, соединение прямо или косвенно снижает всасывание воды в тонком кишечнике. Согласно конкретным аспектам, соединение снижает всасывание воды в тощей кишке. Согласно конкретным аспектам, соединение снижает всасывание воды в тонком кишечнике примерно или по меньшей мере примерно на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100% по сравнению с контрольным соединением или отсутствием соединения.
Термин «агонист» включает соединение, которое связывается с молекулой-мишенью, такой как рецептор, или возбуждает или стимулирует клеточный ответ с помощью этой молекулы-мишени. Включены суперагонисты, полные агонисты, частичные агонисты и селективные агонисты. Суперагонисты взывают более сильный максимальный ответ молекулы-мишени по сравнению с эндогенными агонистом (агонистами), полные агонисты вызывают максимальный ответ молекулы-мишени, сравнимый с эндогенным агонистом (агонистами), и частичные агонисты вызывают значительно более слабый максимальный ответ молекулы-мишени (например, составляющий 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%) по сравнению с эндогенным агонистом (агонистами).
Согласно конкретным вариантам реализации изобретения, соединение не только обладает активностью агониста, но также может характеризоваться «специфическим связыванием» с мишенью. Например, согласно некоторым вариантам реализации изобретения, соединение (например, соединение прямого действия) может, в частности, связываться с мишенью, описанной в настоящей заявке, с аффинностью связывания (Kd), составляющей по меньшей мере примерно 0,01, 0,05, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 40 или 50 нМ. Согласно конкретным вариантам реализации изобретения, мишень выбрана из одного или более из рецептора P2Y, рецептора аденозин A2b, гуанилатциклазного рецептора С, рецептора аденилатциклазы, рецептора имидазолина-1, рецептора ацетилхолина, рецептора простагландина ЕР4, рецептора дофамина D1, рецептора мелатонина, 5НТ4, рецептора предсердного натрийуретического пептида, карбоангидразы, фосфодиэстеразы и DRA (Down-Regulated in Adenoma, или SLC26A3), как описано в настоящей заявке.
А. Агонисты P2Y
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист P2Y (или агонист рецептора P2Y). Рецепторы P2Y относятся к семейству пуринергических сопряженных с G-белком рецепторов. Примеры человеческих рецепторов P2Y включают P2Y1, P2Y2, P2Y4, P2Y5, P2Y6, P2Y8, P2Y9, P2Y10, P2Y11, P2Y12, P2Y13 и P2Y14. Основные нативные или эндогенные лиганды рецепторов P2Y представляют собой аденозин 5'-трифосфат (АТР), аденозин-5'-дифосфат (ADP), уридин-5'-трифосфат (UTP), уридин-5'-дифосфат (UDP) и UDP-глюкозу (или другие UDP-caxapa). Динуклеотиды, такие как Ap4U, также являются природными агонистами P2Y.
Было показано, что рецепторы P2Y опосредуют сигналинг Са++ в клетках двенадцатиперстной кишки и вносят вклад в секрецию бикарбоната в слизистой двенадцатиперстной кишки. См., например, Dong et al., Am J Physiol Gastro-infest Liver Physiol 296: G424-G432, 2009. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора P2Y подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике (также называемой секрецией бикарбоната в двенадцатиперстной кишке, СБДК).
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо механизмом, агонист рецептора P2Y подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Некоторые рецепторы P2Y селективно активируются, например, адениннуклеотидами, такими как АТФ и АДФ, и другими урацилнуклеотидами или UDP-глюкозой. Рецептор P2Y1 отвечает за функциональную активность конкретного P2Y-пуринорецептора. Он работает в различных тканях, включая гладкие мышцы, эндотелий и нервные ткани, а также в тромбоцитах крови. Рецептор P2Y1 является селективным в отношении адениннуклеотидов. ADP является наиболее активным физиологическим агонистом. Согласно некоторым вариантам реализации изобретения, соединение представляет собой агонист рецептора P2Y1, необязательно селективный агонист рецептора P2Y1 по сравнению с другими рецепторами P2Y. Один пример агониста рецептора P2Y1 представляет собой 2-метилтио- ADP.
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист рецептора P2Y2 и/или P2Y4, необязательно селективный агонист рецептора P2Y2 по сравнению с другими рецепторами P2Y. Указанные два рецептора обладают самой высокой идентичностью по последовательностям их доменов ТМ (66,8%) вреди всех подтипов рецептора P2Y. Рецептор P2Y2 может активироваться, например, нуклеотидом урацилом, UDP-сахарными производными и адениновыми нуклеотидами, такими как АТФ. Рецепторы P2Y2 экспрессируются во многих тканях, включая легкие, сердце, скелетные мышцы, селезенку, почки, печень и эпителий. Указанные рецепторы играют важную роль в регуляции транспорта ионов в эпителиальных клетках. Трифосфатнуклеотиды, включая UTP, АТР, UTPγS и ATPγS, действуют как полные агонисты рецептора Р2Y2. Кроме перечисленных выше агонистов рецептор P2Y2 также отвечает на диаденозинтетрафосфат (АР4А) и Up4U (диквафосол, INS365, используемый для лечения заболевания сухих глаз). Аналог Р-(уридин 5')-Р4-(2'-деоксицитидин 5')тетрафосфат (INS37217) представляет собой активный агонист рецептора P2Y2 с некоторым эффектом в качестве агониста рецептора P2Y4. Денуфосол ((3S,5R)-5-(4-амино-2-оксопиримидин-1-ил)-3-гидроксиоксолан-2-ил]метокси-гидроксифосфорил][[[(2R,3S,4R,5R)-5-(2,4-диоксопиримидин-1-ил)-3,4-дигидроксиоксолан-2-ил]метокси-гидроксифосфорил]окси-гидроксифосфорил] гидрофосфат, включая его тетранатриевую соль) также является примером агониста рецептора P2Y2. Также включен PSB1114.
В случае модификации рибозы и урацила оба из соединений 2'-деокси-2'-амино-UTP и 2-тио-UTP сохраняют активность UTP-агониста в отношении рецептора P2Y2. Комбинация указанных двух модификаций приводит к получению 2'-амино-2-тио-UTP с синергичным усилением активности (8 нМ ЕС50) и селективности (300-кратная селективность в отношении P2Y2 по сравнению с P2Y4). Модификации в положении 5, такие как 5-бром-UTP (EC50=0,75 мкМ) и 5-йод-UTP (ЕС50=0,83 мкМ), предполагают, что введение малой гидрофобной группы может оказывать благоприятный эффект в отношении рецептора P2Y2.
Агонисты рецептора P2Y, предложенные в настоящей заявке, включают мононуклеотиды, динуклеотиды и нуклеотид-сахара, в также другие агонисты, известные в данной области техники. См., например, патент США 6,624,150; европейский патент ЕР 1196396; международную публикацию WO 2008/060632; источники Cosyn et al., Bioorg Med Chem Lett. 19: 3002-5, 2009 (в котором описан уридин-5'-(фосфо)фосфонат и 5'-метиленфосфонатный эквивалент UMP); Ko et al., Bioorg Med Chem. 16: 6319-32, 2008 (в котором описаны, например, альфа, бета-метилен-UDP, агонист рецептора P2Y6; сложный Up(4)-фениловый эфир и Up(4)-[1]глюкоза, селективные агонисты рецептора P2Y2; аналоги дигалометиленфосфоната, селективные агонисты рецептора P2Y2; 2-тиоаналог INS37217 (Р(1)-(уридин-5')-Р(4)-(2'-деоксицитидин-5')тетрафосфат), активный и селективный агонист рецептора P2Y2; Ivanov et al., J Med Chem. 50: 1166-76, 2007; Brookings et al., Bioorg Med Chem Lett. 17: 562-5, 2007 (в котором описан синтез и активность ряда нуклеозидтрифосфатов как агонистов в отношении P2Y2); и Jacobson et al., Purinergic Signal. 5: 75-89, 2009; каждый из которых полностью включен посредством ссылки.
Дополнительные примеры агонистов рецептора P2Y включают примеры, описанные в международной публикации WO 1999/09998 и заявках на патент США №2002/0052336 и 2003/0027785, включая Р1,Р4-диаденозинтетрафосфат (A2P4); уридин-5'-дифосфат (UDP); уридин-5'-O-(2-тиодифосфат) (UDPβS); 5-бромуридин-5'-трифосфат (5-BrUTP); 5-(1-фенилэтинил)-уридин-5'-трифосфат (5-(1-фенилэтинил)UTP); 5-метилуридин-5'-дифосфат (5-метилUDP); 4-гексилтиоуридин-5'-трифосфат (4-гексилтиоUTP); 4-тиоуридин-5'-трифосфат (4-тиоUTP); 2-метоксиуридин-5'-трифосфат (2-метоксиUTP); 4-(1-морфолино)уридин-5'-тетрафосфат (4-(1-морфолино))UP4; 4-гексилоксиуридин-5'-дифосфат (4-гексилоксиUDP); 4-(N,N-диметил)цитидин-5'-трифосфат (N,N-диметилСТР); 4-(N-гексил)цитидин-5'-трифосфат (N-гексилСТР); Р1-(цитидин-5')-P4-(уридин-5'-)тетрафосфат (CP4U); Pl-O-(метил)-P4-(уридин-5'-)тетрафосфат (MeP4U) и 4-(N-циклопентил)тимидин-5'-трифосфат (N-циклопентилСТР).
Также включены 5'-аденозинтрифосфат (АТР), 5'-уридинтрифосфат (UTP), уридин-5'-O-(3-тиотрифосфат)(UTPγS), Р1-(уридин-5')-Р.суп.4-(уридин-5')тетрафосфат (U2P4), 5'-[4-(тиоуридин)]-трифосфат (4-тиоUTP) и Р1-(цитидин-5')-Р4-(уридин-5')тетрафосфат (CP4U). Способы идентификации и получения конкретных тиофосфатных аналогов нуклеозиддифосфатов (таких как UDP-β-S) описаны в патенте США №3,846,402 и Goody and Eckstein (J. Am. Chem. Soc. 93: 6252-6257. 1971). Альтернативно, UTP и другие его аналоги также коммерчески доступны от производителей, таких как Sigma (Сент-Луис, Миссури) и Pharmacia (Уппсала, Швеция). Примеры способов идентификации агонистов рецептора P2Y описаны, например, в заявке на патент США №2003/0175810.
Согласно некоторым вариантам реализации изобретения, агонист рецептора P2Y представляет собой неэндогенный низкомолекулярный агонист.Дополнительные примеры агонистов рецептора P2Y показаны на Фигурах 4 и 5А-5С.
В. Агонисты рецептора аденозина A2b
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист рецептора аденозина A2b, необязательно селективный агонист. Большинство физиологических функций аденозина проявляется его локальным модулирующим действием на четыре подтипа рецептора, называемые аденозиновыми рецепторы A1, A2A, А2В и A3 (AR). Рецептор аденозина A2b (или ADORA2B) представляет собой сопряженный с G-белком аденозиновый рецепторный интегральный мембранный белок, который стимулирует аденилатциклазную активность в присутствии аденозина.
Рецептор A2b экспрессируется в различных тканях и, предположительно, имеет высокую плотность распределения в слепой кишке и толстом кишечнике как на поверхности слизистой оболочки, так и на базолатеральной поверхности эпителиальных клеток толстой кишки, см. Baraldi et al., Purinergic Signal. 5: 3-19, 2009. Активация любого его сайта приводит к секреции Cl- в результате непосредственной активации сАМР-активируемого Cl- канала, представляющего собой муковисцидозный трансмембранный регулятор проводимости (CFTR). CFTR модулирует секрецию как хлорида, так и бикарбоната. Например, у крыс с помощью иммунных методов была показана локализация рецептора А2В на мембране щеточной каемки ворсинок двенадцатиперстной кишки, где, ка было показано, аденозин просвета кишечника стимулирует секрецию бикарбоната через рецептора А2В и CFTR. См., например, Ham et al., J Pharmacol Exp Ther. 335: 607-13, 2010. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора аденозина A2b подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике, например, путем снижения СЕРG.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, агонист рецептора аденозина A2b подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Общие примеры агонистов рецептора аденозина A2b включают аденозин, аденозин-подобные соединения и не аденозиновые соединения. Согласно некоторым вариантам реализации изобретения, агонисты рецептора аденозина A2b на основе нуклеозидов включают модифицированные аденозиновые соединения, такие как аденозиновые соединения, замещенные в N(6)-положении пуринового гетероцикла, С(2)-положении пуринового гетероцикла, 5'-положении рибозного фрагмента, и любую комбинацию перечисленных выше соединений. Также включены нерибозные лиганды, такие как замещенные дикарбонитрилпиридины, примером которых является 2-[6-амино-3,5-дициано-4-[4-(циклопропилметокси)фенил]пиридин-2-илсульфанил]ацетамид. См., например, источники Baraldi et al., Purinergic Signal. 4: 287-303, 2008; и Baraldi et al., Purinergic Signal. 5: 3-19, 2009; каждый из которых полностью включен посредством ссылки.
Дополнительные неограничивающие примеры агонистов рецептора аденозина A2b включают BAY 60-6583, CV 1808, AMP579, NECA (N-этилкарбоксамидоаденозин), (S)-PHPNECA, LUF-5835, 6-гуанил NECA и LUF-584. См. также источники Beukers et al., J. Med. Chem. 47:3707-3709, 2004 (в котором описаны, например, неаденозиновые агонисты, такие как LUF5834 (2-амино-4-(4-гидроксифенил)-6-(1H-имидазол-2-илметилсульфанил)пиридин-3,5-дикарбонитрил) и LUF5835 (аналог 3-гидроксифенила)); Beukers et al., Med Res Rev. 26: 667-98, 2006 (в котором описаны, например, (S)PHPNECA и конкретные нерибозные лиганды в качестве агонистов рецептора аденозина A2b); и Liu et al., Basic Res Cardiol. 105: 129-37, 2010. Также включены агонисты рецептора A2b, описанные в заявке на патент США №2002/0156076. Указанные источники полностью включены в настоящую заявку посредством ссылки.
Примеры агонистов рецептора аденозина A2b показаны на Фигурах 6А-6С, а также описаны вместе со способами их синтеза в заявке на патент США №2009/0221649 и международных публикациях согласно РСТ № WO 2006/027142, WO 2007/101531 и WO 2003/008384, каждая из которых полностью включена посредством ссылки.
С. Агонисты рецептора гуанилатциклазы С
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист гуанилилциклазы С (GC-C), необязательно селективный агонист. GC-C представляет собой изоформу семейства гуанилатциклаз, которая имеет высокую плотность распределения на апикальной мембране клеток кишечного эпителия. Она также является рецепторной мишенью для секретируемых бактериями термостабильных энтеротоксинов, которые отвечают за острую секреторную диарею. GC-C также известна как гуанилатциклаза 2С, кишечная гуанилатциклаза, рецептор гуанилатциклазы С и рецептор термостабильного энтеротоксина (hSTAR).
GC-C содержит внеклеточный лиганд-связывающий домен, один трансмембранный участок, протеинкиназа-подобный участок и С-концевой гуанилатциклазный домен. Тирозинкиназная активность опосредует сигнальный путь GC-C в клетке. Гуанилин и урогуанилин представляют собой эндогенные пептидные лиганды GC-C. Активация GC-C приводит, например, к внутриклеточному повышению cGMP, PKGII-зависимому фосфорилированию трансмембранного регулятора муковисцедоза (CFTR) и другим последующим сигналам, которые вызывают повышенную секрецию хлорида и бикарбоната в просвет пищеварительного тракта (через CFTR и, возможно, DRA или РАТ-1).
Было показано, что агонисты GC-C, такие как линаклотидин, гуанилин и термостабильные энтеротоксины Е.coli (STa), стимулируют секрецию бикарбоната в двенадцатиперстной кишке. См., например, источники Rao et al., Am J Physiol Gastro-intest Liver Physiol 286: G95-G101, 2004; Busby et al., Eur J Pharmacol. 649: 328-35, 2010; Bryant et al., Life Sci. 86: 760-5, 2010. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист GC-C подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, агонист GC-C подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Общие примеры агонистов GC-C включают пептидные агонисты и их аналоги, включая синтетические аналоги эндогенных пептидных агонистов GC-C. Конкретные примеры агонистов GC-C включают, но не ограничиваются указанными, термостабильные энтеротоксины (пептиды ST или STa), включая энтеротоксины Е.coli, гуанилин, прогуанилин, урогуанилин, проурогуанилин, лимфогуанилин, линаклотидин (Linzess), SP-333 и плеканатид. См., например, DrugDes Devel Ther. 7: 351-60, 2013. Линаклотидин представляет собой синтетический аналог STa, продаваемый для лечения синдрома раздраженного кишечника с преобладающим запором (IBS-С). См., например, источник Bryant et al., Life Sci. 86: 760-5, 2010. Плеканатид представляет собой синтетический аналог урогуанилина, разработанный для лечения IBS-C. См., например, источники Pitari, выше, и Shailubhai et al., dig dis Sci. 2013 Apr 27 [опубликовано в формате Epub до официального издания]. Дополнительные примеры агонистов GC-C описаны в заявках на патент США №2012/0064039, 2004/0258687, 2005/0287067, 2006/0281682, 2006/0258593, 2006/0094658, 2008/0025966, 2003/0073628, 2004/0121961 и 2004/0152868 и в патентах США №5,140,102, 7,041,786 и 7,304,036. Указанные источники полностью включены в настоящую заявку посредством ссылки.
Согласно некоторым вариантам реализации изобретения, агонист GC-C представляет собой бактериальный пептид ST (или STa) или его вариант или аналог или производное. У бактерий пептиды ST или STa образуются из пре-про-белка, который в целом содержит по меньшей мере 70 аминокислот. Пре- и про- участки отщепляются в ходе процесса секреции, и полученный зрелый белок, который в целом содержит менее чем примерно 20 аминокислот, является биологически активным.
Примеры бактериальных пептидов ST включают: ST Ib Е.coli (Moseley et al., Infect. Immun. 39: 1167, 1983), содержащий зрелую аминокислотную последовательность Asn Ser Ser Asn Tyr Cys Cys Glu Leu Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr (SEQ ID NO: 10); ST Ia Е.coli (So and McCarthy, PNAS USA. 77: 4011, 1980), содержащий зрелую аминокислотную последовательность Asn Thr Phe Tyr Cys Cys Glu Leu Cys Cys Asn Pro Ala Cys Ala Gly Cys Tyr (SEQ ID NO: 11); ST I Е.coli (Chan and Giannella, J. Biol. Chem. 256: 7744, 1981), содержащий зрелую аминокислотную последовательность Asn Thr Phe Tyr Cys Cys Glu Leu Cys Cys Tyr Pro Ala Cys Ala Gly Cys Asn (SEQ ID NO: 12); пептид ST С. freundii (Guarino et al., Infect. Immun. 57: 649, 1989), содержащий зрелую аминокислотную последовательность Asn Thr Phe Tyr Cys Cys Glu Leu Cys Cys Asn Pro Ala Cys Ala Gly Cys Tyr (SEQ ID NO: 13); пептиды ST Y. enterocolitica Y-ST(Y-STa), Y-STh и Y-STc (обзор в источнике Huang et al., Microb. Pathog. 22: 89, 1997), содержащие следующие про-формы аминокислотных последовательностей: Gln Ala Cys Asp Pro Pro Ser Pro Pro Ala Glu Val Ser Ser Asp Trp Asp Cys Cys Asp Val Cys Cys Asn Pro Ala Cys Ala Gly Cys (SEQ ID NO: 14) (а также вариант Y-STa с заменой Ser-7 на Leu-7 (SEQ ID NO: 15), (Takao et al., Eur. J. Biochem. 152: 199, 1985); Lys Ala Cys Asp Thr Gln Thr Pro Ser Pro Ser Glu Glu Asn Asp Asp Trp Cys Cys Glu Val Cys Cys Asn Pro Ala Cys Ala Gly Cys (SEQ ID NO: 16); Gln Glu Thr Ala Ser Gly Gln Val Gly Asp Val Ser Ser Ser Thr Ile Ala Thr Glu Val Ser Glu Ala Glu Cys Gly Thr Gln Ser Ala Thr Thr Gln Gly Glu Asn Asp Trp Asp Tip Cys Cys Glu Leu Cys Cys Asn Pro Ala Cys Phe Gly Cys (SEQ ID NO: 17), соответственно; пептид ST Y. kristensenii, содержащий зрелую аминокислотную последовательность Ser Asp Trp Cys Cys Glu Val Cys Cys Asn Pro Ala Cys Ala Gly Cys (SEQ ID NO: 18); пептид ST non-01 V. Cholerae (Takao et al., FEBS Lett. 193: 250, 1985), содержащий зрелую аминокислотную последовательность Ile Asp Cys Cys Glu Ile Cys Cys Asn Pro Ala Cys Phe Gly Cys Leu Asn (SEQ ID NO: 19); и пептид ST V. mimicus (Arita et al., FEMS Microbiol. Lett. 79: 105, 1991), содержащий зрелую аминокислотную последовательность Ile Asp Cys Cys Glu Ile Cys Cys Asn Pro Ala Cys Phe Gly Cys Leu Asn (SEQ ID NO: 20). В приведенной ниже Таблице A1 показаны последовательности примеров зрелых пептидов ST.


Незрелая форма белка ST-IA (ST-P) Е.coli (включая пре- и про-участки) имеет Последовательность: mkklmlaifisvlsfpsfsqstesldsskekitletkkcdvvknnsekksenmnntfyccelccnpacagcy (SEQ ID NO: 41); см. GenBank®, номер доступа Р01559 (gi: 123711). Пре-последовательность состоит из остатков 1-19. Про-последовательность состоит из остатков 20-54. Зрелый белок состоит из остатков 55-72. Незрелая форма белка ST-1B (ST-H) Е.coli (включая пре- и про-участки) имеет последовательность: mkksilfiflsvlsfspfaqdakpvesskekitleskkcniakksnksgpesmnssnyccelccnpactgcy (SEQ ID NO: 42); см. GenBank®, номер доступа Р07965 (gi:3915589)). Незрелая форма белка ST Y. Enterocolitica (включая пре- и про-участки) имеет последовательность: mkkivfvlylmlssfgafgqetvsgqfsdalstpitaevykqacdpplppaevssdwdccdvccnpacagc (SEQ ID NO: 43); см. GenBank®, номер доступа S25659 (gi:282047)). Соответственно, пептидный агонист GC-C может содержать или состоять из любой одной или более последовательностей бактериальных пептидов ST, описанных в настоящей заявке, включая их варианты.
Бактериальные пептиды ST, как правило, содержат шесть остатков Cys. Указанные шесть остатков Cys образуют три дисульфидные связи в зрелой и активной форме пептида. Если шесть остатков Cys идентифицировать как А, В, С, D, Е и F от амино- к карбокс-концу пептида, то дисульфидные связи обычно образуются следующим образом: A-D, В-Е и C-F. Считается, что образование указанных связей вносит вклад в связывание с рецептором GC-C. Таким образом, согласно конкретным вариантам реализации изобретения, пептидный агонист GC-C содержит по меньшей мере одну, две или три дисульфидные связи, выбранные из любой комбинации A-D, В-Е и C-F, как показано выше. Однако согласно некоторым вариантам реализации изобретения, один или более цистеинов пептидных агонистов GC-C, описанных в настоящей заявке, удалены или замещены на другую аминокислоту. Согласно некоторым вариантам реализации изобретения, 1, 2, 3, 4, 5 или 6 цистеинов удалены или замещены на другую аминокислоту. Согласно конкретным аспектам, большинство N-концевых цистеиновых остатков (например. А, В или А и В) и/или С-концевой цистеиновый остаток или большинство С-концевых цистеиновых остатков (например, Е, F или Е и F) удалены или замещены на другую аминокислоту. Согласно конкретным вариантам реализации изобретения, другая аминокислота представляет собой аланин или серин.
Конкретные пептидные агонисты GC-C включают потенциальный функциональный сайт расщепления химотрипсином, например, Trp, Tyr или Phe расположены между Cys B/Cys D или между Cys E/Cys F. Расщепление по любому сайту расщепления химотрипсином может приводить к снижению способности пептида связываться с рецептором GC-C. В теле человека неактивная форма химотрипсина, химотрипсиноген, продуцируется в поджелудочной железе. Когда этот неактивный фермент достигает тонкого кишечника, он превращается в активный химотрипсин в результате отрезания двух дипептидов. Активный химотрипсин может расщеплять пептиды по пептидной связи со стороны карбокси-конца Trp, Tyr или Phe. Присутствие активного химотрипсина в кишечном тракте может приводить к расщеплению конкретных пептидных агонистов GC-C, содержащих расположенный соответствующим образом функциональный сайт расщепления химотрипсином. В некоторых примерах, ожидается, что расщепление химотрипсином ослабляет действие пептидного агониста GC-C, содержащего расположенный соответствующим образом сайт расщепления химотрипсином, по мере прохождения пептида через пищеварительный тракт.
Конкретные пептидные агонисты GC-C включают потенциальный функциональный сайт расщепления трипсином, например, Lys или Arg. Трипсиноген, как и химотрипсин, представляет собой сериновую протеазу, которая продуцируется в поджелудочной железе и присутствует в пищеварительном тракте. Активная форма, трипсин, расщепляет пептиды, содержащие Lys или Arg. Присутствие активного трипсина в кишечном тракте может приводить к расщеплению конкретных пептидных агонистов GC-C, содержащих расположенный подходящим образом функциональный сайт расщепления трипсином. В конкретных примерах, ожидается, что расщепление трипсином ослабляет действие пептидного агониста GC-C, содержащего расположенный подходящим образом сайт расщепления трипсином, по мере прохождения пептида через пищеварительный тракт.
Согласно конкретным вариантам реализации изобретения, пептид содержит по меньшей мере шесть цистеинов, которые могут образовывать три дисульфидные связи. Согласно конкретным вариантам реализации изобретения, дисульфидные связи замещены на другие ковалентные поперечные связи, и в некоторых случаях цистеины замещены на другие остатки для получения альтернативных ковалентных поперечных связей (описанных в другом месте в настоящей заявке). Конкретные пептиды содержат функциональный сайт расщепления химотрипсином или сайт расщепления трипсином, расположенный таким образом, что обеспечивают инактивацию пептида при его расщеплении. Конкретные пептиды, содержащие функциональный сайт расщепления, расщепляются и постепенно инактивируются в пищеварительном тракте, что является желательным в некоторых условиях. В частности, изменение функционального сайта расщепления химотрипсином приводит к повышению стабильности пептида in vivo.
Согласно конкретным вариантам реализации изобретения, пептиды содержат одну или две или более последовательно расположенных отрицательно заряженных аминокислот (например, Asp или Glu) или один или два или более последовательно расположенных положительно заряженных остатков (например, Lys или Arg) или одну или две или более последовательно расположенных положительно или отрицательно заряженных аминокислот на карбокси конце. В указанных и связанных вариантах реализации, все их фланкирующих аминокислот на карбокси конце являются положительно или отрицательно заряженными. Согласно некоторым вариантам реализации изобретения, перед карбокси-концевыми заряженными аминокислотами находится Leu. Например, следующие аминокислотные последовательности могут быть добавлены к карбокси-концу пептида: Asp; Asp Lys; Lys Lys Lys Lys Lys Lys (SEQ ID NO: 44); Asp Lys Lys Lys Lys Lys Lys (SEQ ID NO: 45); Leu Lys Lys и Leu Asp. Согласно конкретным вариантам реализации изобретения, Leu добавлен к карбокси концу.
Согласно некоторым аспектам, пептидный агонист GC-C (аналог бактериального ST) содержит, состоит или по существу состоит из аминокислотной последовательности, показанной ниже (I):

Согласно некоторым вариантам реализации изобретения, Хаа1 Хаа2 Хаа3 Хаа4 Хаа5 представляет собой Asn Ser Ser Asn Tyr (SEQ ID NO: 2) или отсутствует или Xaa1 Хаа2 Хаа3 Хаа4 отсутствует. Согласно конкретным вариантам реализации изобретения, Xaa8, Хаа9, Хаа12, Xaa14, Xaa16, Xaa17 и Xaa15 представляют собой любую аминокислоту. Согласно конкретным вариантам реализации изобретения, Xaa8, Xaa9, Xaa12, Xaa14, Xaa16, Xaa17 и Xaa19 представляют собой любую природную или неприродную аминокислоту или аналог аминокислоты.
Согласно конкретным вариантам реализации изобретения, Хаа5 представляет собой Asn, Trp, Tyr, Asp или Phe. Согласно другим вариантам реализации, Xaa5 представляет собой Thr или Ile. Согласно некоторым вариантам реализации изобретения, Хаа5 представляет собой Tyr, Asp или Trp. Согласно конкретным вариантам реализации изобретения, Xaa5 представляет собой Asn, Trp, Tyr, Asp, Ile, Thr или Phe. Согласно конкретным вариантам реализации изобретения Xaa5 представляет собой Asn.
Согласно конкретным вариантам реализации изобретения, Xaa8 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты. Согласно некоторым вариантам реализации изобретения, Xaa8 представляет собой Glu, Asp, Gln, Gly или Pro. Согласно другим вариантам реализации, Xaa8 представляет собой Glu. Согласно некоторым вариантам реализации изобретения, Xaa8 представляет собой Glu или Asp. Согласно некоторым вариантам реализации изобретения, Хаа8 представляет собой Asn, Glu или Asp. Согласно некоторым вариантам реализации изобретения, Xaa8 представляет собой Glu, His, Lys, Gln, Asn или Asp. Согласно некоторым вариантам реализации изобретения, Xaa8 представляет собой Glu, His, Gln, Asn или Asp. Согласно некоторым вариантам реализации изобретения, Xaa8 представляет собой Glu, Asn, His, Gln, Lys, Asp или Ser. Согласно конкретным вариантам реализации изобретения, Xaa8 представляет собой Pro.
Согласно конкретным вариантам реализации изобретения, Хаа9 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой любую природную или неприродную ароматическую аминокислоту или аналог аминокислоты. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Leu, Ile, Val, Ala, Lys, Arg, Trp, Tyr или Phe. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Leu, Ile, Val, Lys, Arg, Trp, Tyr или Phe. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Leu, Ile, Val, Trp, Tyr или Phe. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Leu, Ile или Val. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Trp, Tyr или Phe. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Leu, Ile, Lys, Arg, Trp, Tyr или Phe. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Leu, Val, Ile или Met. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Leu или Phe. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Leu, Phe или Tyr. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Tyr, Phe или His. Согласно некоторым вариантам реализации изобретения, Хаа9 представляет собой Phe, His, Trp или Tyr. Согласно конкретным вариантам реализации изобретения, Хаа9 не представляет собой Leu. Согласно конкретным вариантам реализации изобретения, Хаа9 представляет собой Tyr.
Согласно конкретным вариантам реализации изобретения, Xaa12 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты. Согласно конкретным вариантам реализации изобретения, Хаа12 представляет собой Asn, Tyr, Asp или Ala. Согласно конкретным вариантам реализации изобретения, Xaa12 Asn. Согласно конкретным вариантам реализации изобретения, Хаа12 представляет собой Asn, Met, Arg, Lys, His или Gln. Согласно конкретным вариантам реализации изобретения, Xaa12 представляет собой Asn, Lys, His или Gln. Согласно конкретным вариантам реализации изобретения, Хаа12 представляет собой Asn, Asp, Glu или Gln. Согласно конкретным вариантам реализации изобретения, Xaa12 представляет собой Asn, Thr, Ser, Arg, Lys, Gln или His. Согласно некоторым вариантам реализации изобретения, Хаа12 представляет собой Asn, Ser или His.
Согласно конкретным вариантам реализации изобретения, Xaa13 представляет собой Ala, Pro или Gly. Согласно конкретным вариантам реализации изобретения, Xaa13 представляет собой Pro или Gly. Согласно конкретным вариантам реализации изобретения, Хаа13 представляет собой Pro. Согласно конкретным вариантам реализации изобретения, Xaa13 представляет собой Gly.
Согласно конкретным вариантам реализации изобретения, Хаа14 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты. Согласно конкретным вариантам реализации изобретения, Xaa14 представляет собой Ala, Leu, Ser, Gly, Val, Glu, Gln, Ile, Leu, Thr, Lys, Arg или Asp. Согласно конкретным вариантам реализации изобретения, Xaa14 представляет собой Ala или Gly. Согласно некоторым вариантам реализации изобретения, Xaa14 представляет собой Val или Ala. Согласно конкретным вариантам реализации изобретения, Xaa14 представляет собой Ala или Thr. Согласно конкретным вариантам реализации изобретения, Xaa14 представляет собой Ala. Согласно конкретным вариантам реализации изобретения, Xaa14 представляет собой Val, Gln, Asn, Glu, Asp, Thr или Ala. Согласно конкретным вариантам реализации изобретения, Xaa14 представляет собой Gly, Cys или Ser.
Согласно конкретным вариантам реализации изобретения, Хаа16 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты. Согласно некоторым вариантам реализации изобретения, Xaa16 представляет собой любую природную или неприродную неароматическую аминокислоту или аналог аминокислоты. Согласно конкретным вариантам реализации изобретения, Хаа16 Thr, Ala, Asn, Lys, Arg, Trp, Gly или Val. Согласно конкретным вариантам реализации изобретения, Xaa16 представляет собой Thr, Ala, Asn, Lys, Arg или Trp. Согласно конкретным вариантам реализации изобретения, Xaa16 представляет собой Thr, Ala, Lys, Arg или Trp. Согласно некоторым вариантам реализации изобретения, Xaa16 представляет собой Thr, Ala или Trp. Согласно некоторым вариантам реализации изобретения, Xaa16 представляет собой Thr. Согласно некоторым вариантам реализации изобретения, Xaa16 представляет собой Trp, Tyr или Phe. Согласно некоторым вариантам реализации изобретения, Xaa16 представляет собой Thr или Ala. Согласно конкретным вариантам реализации изобретения, Xaa16 it представляет собой Val. Согласно конкретным вариантам реализации изобретения, Xaa16 представляет собой Gly. Согласно некоторым вариантам реализации изобретения, Xaa16 представляет собой Thr, Ser, Met или Val. Согласно некоторым вариантам реализации изобретения, Xaa16 представляет собой Val, Ala или Thr. Согласно некоторым вариантам реализации изобретения, Xaa16 представляет собой Ile, Val, Lys, Asn, Glu, Asp или Thr.
Согласно конкретным вариантам реализации изобретения, Xaa17 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты. Согласно некоторым вариантам реализации изобретения, Xaa17 представляет собой Gly, Pro или Ala. Согласно конкретным вариантам реализации изобретения, Xaa17 представляет собой Gly. Согласно конкретным вариантам реализации изобретения, Xaa17 представляет собой Ala. Согласно некоторым вариантам реализации изобретения, Xaa17 представляет собой Gly или Ala. Согласно некоторым вариантам реализации изобретения, Xaa17 представляет собой Gly, Asn, Ser или Ala. Согласно некоторым вариантам реализации изобретения, Xaa17 представляет собой Asn, Glu, Asp, Thr, Ala, Ser или Gly. Согласно некоторым вариантам реализации изобретения, Xaa17 представляет собой Asp, Ala, Ser или Gly.
Согласно конкретным вариантам реализации изобретения, Xaa19 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Trp, Tyr, Phe, Asn, Ile, Val, His, Leu или Arg. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Trp, Tyr, Asn или Leu. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Trp, Tyr или Phe. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Tyr, Phe или His. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Tyr или Trp. Согласно конкретным вариантам реализации изобретения, Xaa19 представляет собой Tyr. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Leu, Ile или Val. Согласно конкретным вариантам реализации изобретения, Xaa19 представляет собой His. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Trp, Tyr, Phe, Asn, Ile, Val, His или Leu. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Trp, Tyr, Phe или Leu. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Tyr или Leu. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой Lys или Arg. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой любую аминокислоту, отличную от Pro, Arg, Lys, Asp или Glu. Согласно некоторым вариантам реализации изобретения, Xaa19 представляет собой любую аминокислоту, отличную от Pro. Согласно некоторым вариантам реализации изобретения, Xaa19 отсутствует.
Согласно конкретным вариантам реализации изобретения Хаа20 представляет собой Asp или Asn. Согласно конкретным вариантам реализации изобретения Xaa20Xaa21 представляет собой AspPhe или отсутствует. Согласно некоторым вариантам реализации изобретения, Хаа20 представляет собой Asn или Glu и Xaa21 отсутствует. Согласно некоторым вариантам реализации изобретения, Xaa19 Xaa20 Xaa21 отсутствует.
Согласно некоторым аспектам, пептидный агонист GC-C содержит, состоит или по существу состоит из аминокислотной последовательности, показанной ниже (II):

где Xaa1 Хаа2 Хаа3 Хаа4 Хаа5 представляет собой Asn Ser Ser Asn Tyr (SEQ ID NO: 2) или отсутствует, или Xaa1 Хаа2 Хаа3 Хаа4 отсутствует, и Хаа5 представляет собой Asn;
Xaa8 представляет собой Glu или Asp;
Хаа9 представляет собой Leu, Ile, Val, Trp, Tyr или Phe;
Хаа16 представляет собой Thr, Ala, Trp;
Xaa19 представляет собой Trp, Tyr, Phe или Leu или отсутствует; и Xaa20Xaa21 представляет собой AspPhe.
Согласно некоторым аспектам, пептидный агонист GC-C содержит, состоит или по существу состоит из аминокислотной последовательности (II): Xaa1 Хаа2 Хаа3 Хаа4 Хаа5 Cys6 Cys7 Xaa8 Хаа9 Cys10 Cys11 Asn12 Pro13 Ala14 Cys15 Xaa16 Gly17 Cys18 Xaa19 Хаа20 Xaa21 (SEQ ID NO: 48), где Xaa9 представляет собой Leu, Ile или Val, и Xaa16 представляет собой Trp, Tyr или Phe; Хаа9 представляет собой Trp, Tyr или Phe, и Xaa16 представляет собой Thr или Ala; Xaa19 представляет собой Trp, Tyr, Phe и, Xaa20Xaa21 представляет собой AspPhe; и Xaa1 Хаа2 Хаа3 Хаа4 отсутствует, и Хаа5 представляет собой Asn; пептид содержит менее чем 50, 40, 30 или 25 аминокислот; или менее чем пять аминокислот предшествует Cys6.
Согласно некоторым аспектам, пептидный агонист GC-C содержит, состоит или по существу состоит из аминокислотной последовательности Xaa1 Хаа2 Хаа3 Хаа4 Хаа5 Cys Cys Glu Хаа9 Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr Xaa20 Xaa21 (II) (SEQ ID NO: 49), где Хаа9 представляет собой любую аминокислоту: где Хаа9 представляет собой любую аминокислоту, отличную от Leu; где Хаа9 выбран из Phe, Trp и Tyr; где Хаа9 выбран из любой другой природной или неприродной ароматической аминокислоты; где Хаа9 представляет собой Tyr; где Хаа9 представляет собой Phe; где Хаа9 представляет собой Trp; где Xaa1 Хаа2 Хаа3 Хаа4 Хаа5 представляет собой Asn Ser Ser Asn Tyr; где Xaa1, Хаа2, Хаа3, Хаа4 и Хаа5 отсутствуют; где Xaa1, Хаа2, Хаа3 и Хаа4 отсутствуют; где Xaa1, Хаа2 и Хаа3 отсутствуют; где Xaa1 и Хаа2 отсутствуют; где Xaa1 отсутствует; где Xaa20Xaa21 представляет собой AspPhe или отсутствует, или Xaa20 представляет собой Asn или Glu, и Xaa21 отсутствует, или Xaa19Xaa20Xaa21 отсутствует; где Xaa1 Хаа2 Хаа3 Хаа4 Xaa5 и Tyr Хаа20 Xaa21 отсутствуют. Согласно некоторым аспектам, пептидный агонист GC-C содержит, состоит или по существу состоит из аминокислотной последовательности Xaa1 Хаа2 Хаа3 Хаа4 Xaa5 Cys6 Cys7 Xaa8 Xaa9 Cys10 Cys11 Xaa12 Xaa13 Xaa14 Cys15 Xaa16 Xaa17 Cys18 Xaa19 Xaa20Xaa21 (I) (SEQ ID NO: 50), где: Xaa1 Xaa2 Хаа3 Хаа4 Xaa5 отсутствует, и/или последовательность Xaa19 Xaa20 Xaa21 отсутствует, где пептид необязательно содержит дополнительные аминокислоты на карбокси-конце и/или амино-конце. В примерах, где у пептида отсутствует одна или более концевых аминокислот, таких как Xaa1 или Xaa21, указанный пептид может необязательно содержать дополнительные карбокси-концевые и/или амино-концевые аминокислоты.
Согласно конкретным вариантам реализации изобретения, пептид содержит дисульфидные связи между Cys6 и Cys11, между Cys7 и Cys15 и между Cys10 и Cys16. Согласно некоторым вариантам реализации изобретения, пептид представляет собой восстановленный пептид, не содержащий дисульфидных связей. Согласно другим вариантам реализации изобретения, пептид содержит одну или две дисульфидные связи, выбранные из дисульфидной связи между Cys6 и Cys11, дисульфидной связи между Cys7 и Cys15 и дисульфидной связи между Cys10 и Cys16.
Согласно конкретным вариантам реализации изобретения, одна или более аминокислот замещены на неприродную аминокислоту или аналог природной или неприродной аминокислоты. Существует много аминокислот помимо стандартных 20 аминокислот. Некоторые являются природными, другие являются неприродными (см., например. Hunt, The Non-Protein Amino Acids: In Chemistry and BioChemistry of the Amino Acids, Barrett, Chapman and Hall, 1985). Например, ароматическая аминокислота может быть замещена на 3,4-дигидрокси-L-фенилаланин, 3-йод-L-тирозин, трийодтиронин, L-тироксин, фенилглицин (Phg) или нортирозин (norTyr). Phg и норTyr и другие аминокислоты, включая Phe и Tyr, могут быть замещены, например, на галоген, -СН3, -ОН, -CH2NH3, -С(O)Н, -СН2СН3, -CN, -СН2СН2СН3, -SH или другую группу. Любая аминокислота может быть замещена на аминокислоту в D-форме.
В пептиде формулы I или пептиде формулы II возможен ряд замен на неприродные аминокислоты или аналоги природных и неприродных аминокислот. Например, согласно некоторым аспектам, Xaa8 может быть замещен на гамма-гидрокси-Glu или гамма-карбокси-Glu. Согласно некоторым аспектам, Xaa9 может быть замещен на альфа-замещенную аминокислоту, такую как L-альфа-метилфенилаланин, или на аналоги, такие как: 3-Амино-Tyr; Tyr(СН3); Tyr(РО3(СН3)2); Tyr(SO3H); бета-циклогексил-Ala; бета-(1-циклопентенил)-Ala; бета-циклопентил-Ala; бета-циклопропил-Ala; бета-хинолил-Ala; бета-2-тиазолил)-Ala; бета-(триазол-1-ил)-Ala; бета-(2-пиридил)-Ala; бета-(3-пиридил)-Ala; амино-Phe; фтор-Phe; циклогексил-Gly; tBu-Gly; бета-(3-бензотиенил)-Ala; бета-2-тиенил)-Ala; 5-метил-Trp и 4-метил-Trp.
Согласно некоторым вариантам реализации изобретения, Xaa13 может представлять собой N(альфа)-С(альфа)циклизованные аналоги аминокислот, имеющие следующую структуру:
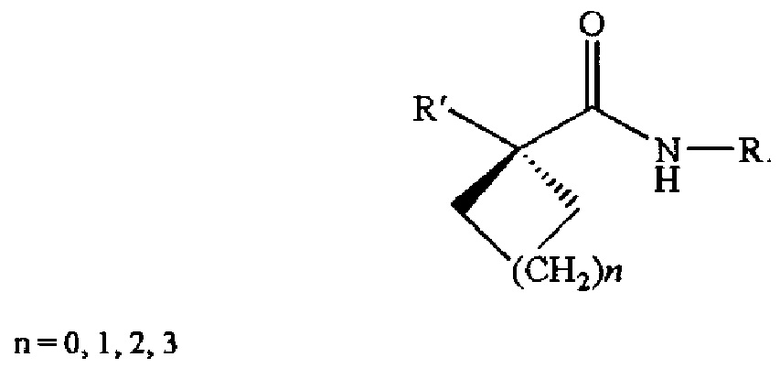
Xaa13 также может представлять собой гомоPro (L-пипеколиновую кислоту); гидрокси-Pro; 3,4-дегидро-Pro; 4-фтор-Pro или альфа-метил-Pro.
Согласно аспектам, где Xaa13 представляет собой Gly, Ala, Leu или Val, Xaa14 может представлять собой:
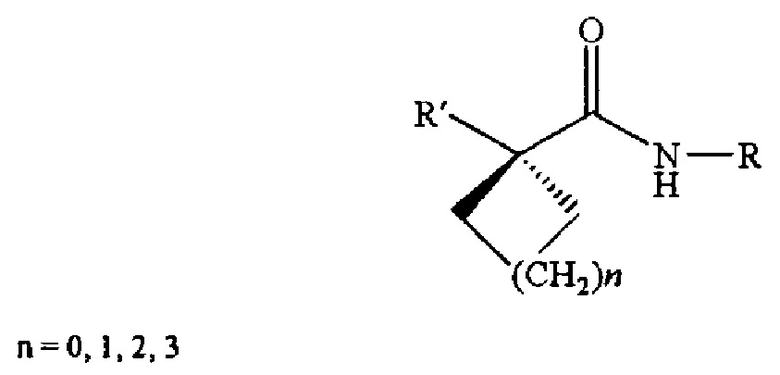
Согласно конкретным аспектам, Xaa14 может представлять собой альфа-замещенную или N-метилированную аминокислоту, такую как альфа-аминоизомасляная кислота (aib), L/D-альфа-этилаланин (L/D-изовалин), L/D-метилвалин или L/D-альфа-метиллейцин, или неприродную аминокислоту, такую как бета-фтор-Ala.
Согласно некоторым аспектам, Xaa17 может представлять собой альфа-аминоизомасляную кислоту (aib) или L/D-альфа-этилаланин (L/D-изовалин).
Дополнительные примеры неприродных аминокислот и аналогов аминокислот известны в данной области техники и описаны в другом месте в настоящей заявке.
В некоторых примерах, например, где Хаа9 представляет собой Trp, Tyr или Phe, или где Xaa16 представляет собой Trp, пептид содержит потенциальный функциональный сайт расщепления химотрипсином, который находится в положении, расщепление по которому может изменять связывание пептида с рецептором GC-C. Когда Хаа9 представляет собой Lys или Arg, или когда Xaa16 представляет собой Lys или Arg, пептид содержит потенциальный функциональный сайт расщепления трипсином, который находится в положении, расщепление по которому может изменять связывание пептида с рецептором GC-C.
В конкретных примерах, например, где Xaa19 представляет собой Trp, Tyr или Phe, пептид содержит сайт расщепления химотрипсином, который находится в положении, расщепление по которому приводит к высвобождению части пептида, карбокси-концевой по отношению к Xaa19. Когда Xaa19 представляет собой Leu, Ile или Val, пептид может содержать сайт расщепления химотрипсином, который находится в положении, расщепление по которому приводит к высвобождению части пептида, амино-концевой по отношению к Xaa19. При относительно высоком рН такой же эффект можно наблюдать, когда Xaa19 представляет собой His. Когда Xaa19 представляет собой Lys или Arg, пептид содержит сайт расщепления трипсином, который находится в положении, расщепление по которому приводит к высвобождению части пептида, карбокси-концевой по отношению к Xaa19.
В некоторых примерах, например, в которых Xaa1 или амино-концевая аминокислота пептида (например, Хаа2 или Хаа3) представляет собой Trp, Tyr или Phe, пептид содержит сайт расщепления химотрипсином, который находится в положении, расщепление по которому приводит к высвобождению части пептида, амино-концевой по отношению к Xaa1 (или Хаа2 или Хаа3), наряду с Xaa1, Хаа2 или Хаа3. Если Xaa1 или амино-концевая аминокислота пептида согласно изобретению (например, Хаа2 или Хаа3) представляет собой Lys или Arg, пептид содержит сайт расщепления трипсином, который находится в положении, расщепление по которому приводит к высвобождению части пептида, амино-концевой по отношению к Xaa1, наряду с Xaa1, Хаа2 или Хаа3. Если Xaa1 или амино-концевая аминокислота пептида согласно изобретению представляет собой Leu, Ile или Val, пептид может содержать сайт расщепления химотрипсином, который находится в положении, расщепление по которому приводит к высвобождению части пептида, амино-концевой по отношению к Xaa1. При относительно высоком значении рН такой же эффект можно наблюдать, когда Xaa1 представляет собой His.
При полностью свернутом состоянии пептида дисульфидные связи могут присутствовать между Cys6 и Cys11; Cys7 и Cys15 и Cys10 и Cys18. Согласно некоторым аспектам, пептидные агонисты GC-C идентичны пептидам ST или имеют последовательность, подобную пептидам ST. Однако согласно некоторым аспектам, пептидные агонисты GC-C содержат аминокислотные замены и/или добавления, которые улучшают функциональную активность. Указанные изменения, например, могут приводить к повышению или снижению активности (например, повышению или снижению способности пептида снижать поглощение фосфатов), изменению способности пептида правильно сворачиваться, изменению стабильности пептида, изменению способности пептида связываться с рецептором GC-C и/или снижению токсичности. В некоторых примерах, пептиды могут обладать более желательными свойствами по сравнению с пептидом ST дикого типа. Например, в конкретных примерах, снижены нежелательные побочные явления, такие как диарея и дегидратация.
В случае пептида, содержащего или состоящего из последовательности (I) Xaa1 Хаа2 Хаа3 Хаа4 Хаа5 Cys6 Cys7 Xaa8 Xaa9 Cys10 Cys11 Xaa12 Xaa13 Xaa14 Cys15 Xaa16 Xaa17 Cys18 Xaa19 Xaa20 Xaa21 (SEQ ID NO: 50) или Xaa1 Xaa2 Хаа3 Хаа4 Хаа5 Cys Cys Glu Xaa9 Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr Хаа20 Xaa21 (II) (SEQ ID NO: 49), где: Xaa1 Хаа2 Хаа3 Хаа4 Хаа5 отсутствует, и/или последовательность Xaa19 Xaa20 Xaa21 отсутствует, указанный пептид может необязательно содержать дополнительные карбокси-концевые и/или амино-концевые аминокислоты. Например, пептид может содержать амино-концевую последовательность, которая облегчает получение пептида с помощью рекомбинации и отщепляется до введения пептида пациенту. Пептид также может содержать другие амино-концевые или карбокси-концевые аминокислоты. В некоторых примерах дополнительные аминокислоты защищают пептид, стабилизируют пептид и/или изменяют активность пептида. В примерах некоторые или все из дополнительных аминокислот удаляют до введения пептида пациенту. Пептид может содержать 1, 2, 3, 4, 5, 10, 15, 20, 25, 30, 40, 50, 60, 70 80, 90, 100 или более дополнительных аминокислот на амино-конце и/или карбокси-конце. Число фланкирующих аминокислот не обязательно должно быть одинаковым. Например, на амино-конце пептида может присутствовать 10 дополнительных аминокислот с отсутствием дополнительных аминокислот на карбокси-конце.
Согласно некоторым вариантам реализации изобретения, пептид содержит аминокислотную последовательность (I): Xaa1 Xaa2 Хаа3 Хаа4 Хаа5 Cys6 Cys7 Xaa8 Xaa9 Cys10 Cys11 Xaa12 Xaa13 Xaa14 Cys15 Xaa16 Xaa17 Cys18 Xaa19 Хаа20 Xaa21 (SEQ ID NO: 50), где: Xaa1 Xaa2 Хаа3 Хаа4 Хаа5 отсутствует; Xaa8 представляет собой Glu; Xaa9 представляет собой Leu, Ile, Lys, Arg, Trp, Tyr или Phe; Xaa12 представляет собой Asn; Xaa13 представляет собой Pro; Xaa14 представляет собой Ala; Xaa16 представляет собой Thr, Ala, Lys, Arg, Trp; Xaa17 представляет собой Gly; Xaa19 представляет собой Tyr или Leu; и Хаа20 Хаа21 представляет собой Asp Phe или отсутствует. В примерах, где Xaa20 Xaa21 и/или Xaa1 Xaa2 Хаа3 Хаа4 Хаа5 отсутствуют, пептид может необязательно содержать дополнительные фланкирующие аминокислоты.
Примеры пептидных агонистов GC-C, которые содержат, состоят или по существу состоят из аминокислотной последовательности Xaa1 Хаа2 Хаа3 Хаа4 Хаа5 Cys Cys Glu Xaa9 Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr Xaa20 Xaa21 (II) (SEQ ID NO: 49), показаны в Таблице А2, ниже.
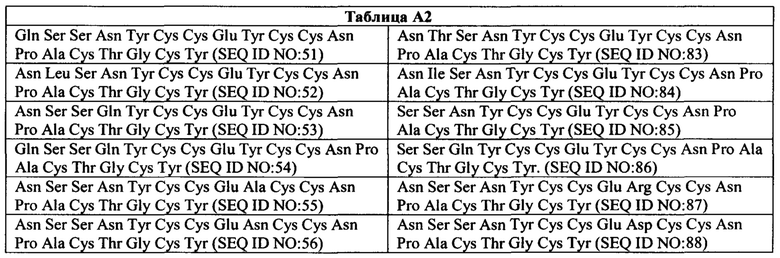
Дополнительные примеры пептидных агонистов GC-C показаны в Таблице A3, ниже.






Согласно конкретным вариантам реализации изобретения, пептидный агонист GС-С содержит, состоит или по существу состоит из аминокислотной последовательности Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr (SEQ ID NO: 4).
Также включены варианты с делениями любых из пептидных агонистов GC-C, описанных в настоящей заявке. Примеры включают варианты с делениями, в которых одна, две, три или четыре аминокислоты (или неприродные аминокислоты или аналога природной или неприродной аминокислоты), отличных от Cys (или аминокислоты, замещающие Cys, например, аминокислоты, способных образовывать ковалентную связь с другой аминокислотой), удалены. Конкретные примеры включают варианты, где две (или более) аминокислоты удалены, и пептид содержит последовательность: Cysa Cysb Xaa Xaa Cysc Cysd Xaa Xaa Xaa Cyse Xaa Xaa Cysf (SEQ ID NO: 526). Согласно некоторым из указанных и связанных вариантов реализации изобретения, две или более делеций могут быть расположены между Cysb и Cysc и/или между Cysd и Cyse и/или между Cyse и Cysf. Однако согласно другим вариантам реализации не более чем одна делеция расположена между каждым из Cysb и Cysc или между Cysd и Cyse или между Cyse и Cysf. Таким образом, включены любые из пептидных агонистов GC-C, описанных в настоящей заявке, содержащих последовательность Cysa Cysb Xaa Xaa Cysc Cysd Xaa Xaa Xaa Cyse Xaa Xaa Cysf (SEQ ID NO: 526), где: а) одна аминокислота между Cysb и Cysc удалена; b) одна аминокислота между Cysd и Cyse удалена; с) одна аминокислота между Cyse и Cysf удалена; d) одна аминокислота между Cysb и Cysc удалена, и одна аминокислота между Cysd и Cyse удалена; е) одна аминокислота между Cysd и Cyse удалена, и одна аминокислота между Cyse и Cysf удалена; f) одна аминокислота между Cysb и Cysc удалена, и одна аминокислота между Cyse и Cysf удалена или g) одна аминокислота между Cysb и Cysc удалена, одна аминокислота между Cysd и Cyse удалена, и одна аминокислота между Cyse и Cysf удалена. Согласно конкретным вариантам реализации изобретения, варианты с делениями представляют собой пептиды, которые связываются с рецептором GC-C и/или являются агонистами рецептора GC-C.
Также включены варианты с инсерциями любых из пептидных агонистов GC-C, описанных в настоящей заявке. Примеры включают варианты инсерций, где одна, две, три или четыре аминокислоты (например, Gly или Ala) встроены до или после любой аминокислоты в пептиде. Согласно некоторым вариантам реализации изобретения, не более чем одна аминокислота встроена между двумя остатками Cys. Конкретные примеры включают варианты, где встроены две или более аминокислот, и пептид содержит последовательность Cysa Cysb Хаа Хаа Cysc Cysd Хаа Хаа Хаа Cyse Xaa Хаа Cysf (SEQ ID NO: 526). Согласно некоторым из указанных и связанных вариантов реализации изобретения, две или более инсерций могут быть расположены между Cysb и Cysc или между Cysd и Cyse или между Cyse и Cysf. Однако согласно другим вариантам реализации, не более чем одна инсерция расположена между Cysb и Cysc или между Cysd и Cyse или между Cyse и Cysf. Таким образом, включены любые из пептидных агонистов GC-С, описанных в настоящей заявке, содержащих последовательность Cysa Cysb Хаа Хаа Cysc Cysd Хаа Хаа Хаа Cyse Хаа Хаа Cysf (SEQ ID NO: 526), где: а) одна аминокислота встроена между Cysb и Cysc; b) одна аминокислота встроена между Cysd и Cyse; с) одна аминокислота встроена между Cyse и Cysf; d) одна аминокислота встроена между Cysb и Cysc, и одна аминокислота встроена между Cysd и Cyse; е) одна аминокислота встроена между Cysd и Cyse, и одна аминокислота встроена между Cyse и Cysf; f) одна аминокислота встроена между Cysb и Cysc, и одна аминокислота встроена между Cyse и Cysf или g) одна аминокислота встроена между Cysb и Cysc, одна аминокислота встроена между Cysd и Cyse, и одна аминокислота встроена между Cyse и Cysf. Кроме того, одна или более аминокислот могут быть встроены до Cysa и/или одна или более аминокислот могут быть встроены после Cysf. Согласно некоторым вариантам реализации изобретения, варианты с инсерциями представляют собой пептиды, которые связываются с рецептором GC-C и/или являются агонистами рецептора GC-C.
Примеры вариантов с инсерциями последовательности Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr (SEQ ID NO: 4) включают такие варианты, в которых до четырех аминокислот (т.е. 0, 1, 2, 3 или 4) встроено после каждой аминокислоты. Таким образом, включены пептиды, содержащие последовательность: Cys Xaa(0-4) Cys Xaa(0-4) Glu Xaa(0-4) Tyr Хаа(0-4) Cys Хаа(0-4) Cys Xaa(0-4) Asn Хаа(0-4) Pro Хаа(0-4) Ala Xaa(0-4) Cys Хаа(0-4) Thr Хаа(0-4) Gly Xaa(0-4) Cys Хаа(0-4) Tyr Хаа(0-4) (SEQ ID NO: 527). Встроенные аминокислоты могут представлять собой любую аминокислоту или аналог аминокислоты (природной или неприродной) и могут быть одинаковыми или разными. Согласно конкретным вариантам реализации изобретения, все встроенные аминокислоты представляют собой Gly, или все встроенные аминокислоты представляют собой Ala или комбинацию Gly and Ala.
Также включены пептидные агонисты GC-C, содержащие или состоящие из последовательности Xaa1 Хаа2 Хаа3 Хаа4 Xaa5 Cys6 Cys7 Xaa8 Xaa9 Cys10 Cys11 Xaa12 Xaa13 Xaa14 Cys15 Xaa16 Xaa17 Cys18 Xaa19 Xaa20 Xaa21 (SEQ ID NO: 46), включая, например, варианты Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr (SEQ ID NO: 4), в которых удалено до четырех аминокислот и/или встроено до четырех аминокислот. В некоторых примерах инсерции и/или делеции могут находиться между Cys6 и Cys18 или могут являться амино-концевыми по отношению к Cys6 и/или карбокси-концевыми по отношению к Cys18.
Согласно конкретным вариантам реализации изобретения, пептидный агонист GC-C создан на основе коровой последовательности: Cys Cys Glu  Cys Cys Asn Pro Ala Cys
Cys Cys Asn Pro Ala Cys  Gly Cys Tyr (SEQ ID NO: 528). Для создания варианта, содержащего потенциальный функциональный сайт расщепления химотрипсином, способный обеспечивать инактивацию пептид. Leu (подчеркнутый) или Thr (подчеркнутый) может быть замещен на Trp, Phe или Tyr; или как Leu, так и Thr, могут быть замещены (независимо) на Trp, Phe или Tyr. Коровая последовательность может необязательно предшествовать Asn Ser Ser Asn Tyr или Asn. Конкретные примеры пептидных агонистов GC-C на основе коровой последовательности включают соединения, представленные в Таблице А4, ниже.
Gly Cys Tyr (SEQ ID NO: 528). Для создания варианта, содержащего потенциальный функциональный сайт расщепления химотрипсином, способный обеспечивать инактивацию пептид. Leu (подчеркнутый) или Thr (подчеркнутый) может быть замещен на Trp, Phe или Tyr; или как Leu, так и Thr, могут быть замещены (независимо) на Trp, Phe или Tyr. Коровая последовательность может необязательно предшествовать Asn Ser Ser Asn Tyr или Asn. Конкретные примеры пептидных агонистов GC-C на основе коровой последовательности включают соединения, представленные в Таблице А4, ниже.
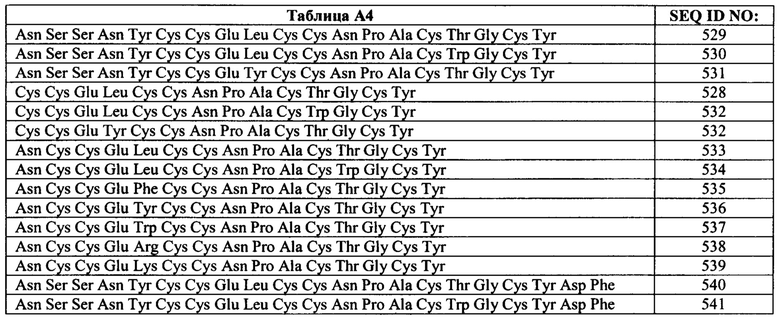
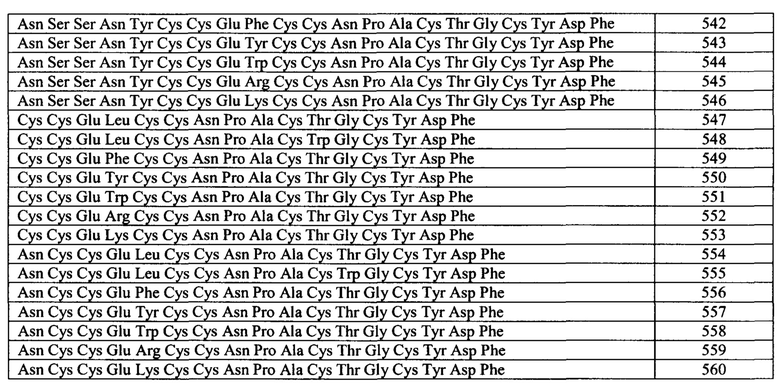
Согласно конкретным вариантам реализации изобретения, пептидный агонист GC представляет собой пептид гуанилин, лимфогуанилин, урогуанилин или реногуанилин, необязательно человеческий пептид или его вариант или производное или аналог. Аминокислотная последовательность человеческого гуанилина представляет собой Pro Gly Thr Cys Glu Ile Cys Ala Tyr Ala Ala Cys Thr Gly Cys (SEQ ID NO: 562). Примеры последовательности аналогов человеческого гуанилина показаны в Таблице А5, ниже.

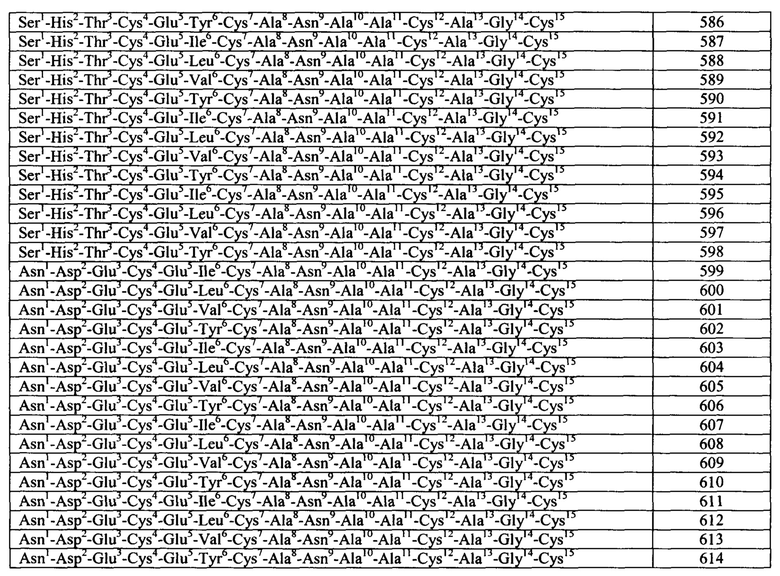
Таким образом, согласно некоторым вариантам реализации изобретения, пептидный агонист GC-C содержит, состоит или по существу состоит из последовательности гуанилина человека или его варианта или производного или аналога.
Аминокислотная последовательность лимфогуанилина представляет собой: Gln-Glu-Glu-Cys-Glu-Leu-Cys-Ile-Asn-Meт-Ala-Cys-Thr-Gly-Tyr. (SEQ ID NO: 615). Примеры аналогов последовательности человеческого лимфогуанилина показаны в Таблице А6, ниже.

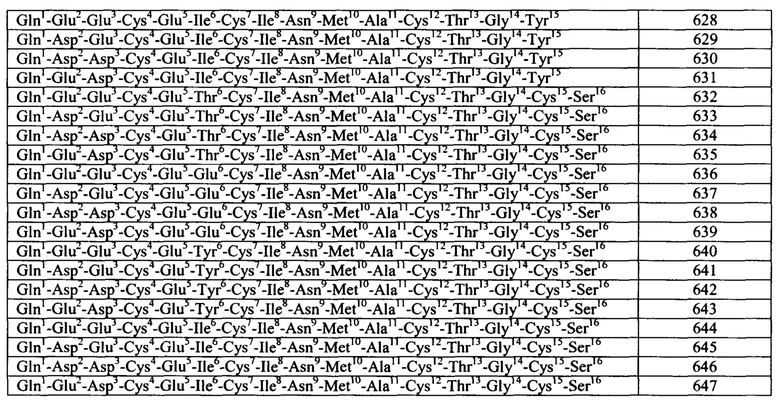
Таким образом, согласно некоторым вариантам реализации изобретения, пептидный агонист GC-C содержит, состоит или по существу состоит из последовательности человеческого лимфогуанилина или его варианта или производного или аналога.
Аминокислотная последовательность человеческого урогуанилина представляет собой Asn Asp Asp Cys Glu Leu Cys Val Asn Val Ala Cys Thr Gly Cys Leu (SEQ ID NO: 648). Согласно некоторым вариантам реализации изобретения, пептидный агонист GC-C содержит, состоит или по существу состоит из последовательности человеческого урогуанилина или его аналога. Согласно конкретным вариантам реализации изобретения, аналог человеческого урогуанилина имеет аминокислотную последовательность Asn Asp Glu Cys Glu Leu Cys Val Asn Val Ala Cys Thr Gly Cys Leu (SEQ ID NO: 6; плеканатид) или Gln Asp Asp Cys Glu Thr Cys Ile Asn Met Ala Cys Thr Gly Tyr (SEQ ID NO: 649). Согласно конкретным вариантам реализации изобретения N-концевой Asn пептида (например, плеканатида) представляет собой пироглутаминовую кислоту. Согласно некоторым вариантам реализации изобретения, С-концевой Leu пептида (например, плеканатида) представляет собой D-аминокислоту (d-Leu).
Согласно конкретным вариантам реализации изобретения, пептид или аналог человеческого урогуанилина содержит, состоит или по существу состоит из аминокислотной последовательности, показанной ниже (III):
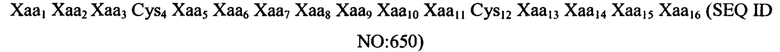
Согласно некоторым вариантам реализации изобретения, пептидный агонист GC-C формулы III определяется следующим образом:
Xaa1 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты или отсутствует;
Хаа2 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты или отсутствует;
Хаа3 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты или отсутствует;
Хаа5 представляет собой Glu;
Хаа6 представляет собой Tyr, Trp, Phe или Leu;
Хаа7 представляет собой Cys;
Xaa8 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты (необязательно любой из 20 природных аминокислот), отличный от Cys, или отсутствует;
Хаа9 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты (необязательно любой из 20 природных аминокислот), отличный от Cys;
Хаа10 представляет собой Pro или Gly;
Хаа11 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты (необязательно любой из 20 природных аминокислот);
Хаа13 представляет собой Thr, Val или Gly;
Xaa14 представляет собой Gly или Ala;
Xaa15 представляет собой Cys; и
Xaa16 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты (необязательно любой из 20 природных аминокислот) или отсутствует.
Согласно конкретным вариантам реализации изобретения: Хаа9 представляет собой Asn; Хаа11 представляет собой Ala или Thr; Xaa8 отсутствует; и Xaa16 представляет собой Tyr.
Согласно некоторым вариантам реализации изобретения перед группой Хаа4 непосредственно расположена аминокислотная последовательность, выбранная из: Ser His Thr; Pro Ser Thr; Thr; Pro Asp Pro; Ile Ala Glu Asp Ser His Thr (SEQ ID NO: 651); Ile Ala Gln Asp Pro Ser Thr (SEQ ID NO: 652); Ala Asn Thr; Asn Thr; Asp Pro Asn Thr (SEQ ID NO: 653); Lys Asn Thr; Pro Asn Thr; Ile Ala Gln Asp Pro Asn Thr (SEQ ID NO: 654); Lys Pro Asn Thr (SEQ ID NO: 655); Asp Pro Gly Thr (SEQ ID NO: 656); Glu Asp Pro Gly Thr (SEQ ID NO: 657); Pro Gly Thr; Pro Ala Thr; Val Ala Ala Arg Ala Asp Leu (SEQ ID NO: 658); Gly Asp Asp; Asn Asp Glu; Gln Glu Asp; Asn Asp Asp; Arg Thr Ile Ala Asn Asp Asp (SEQ ID NO: 659); Thr Ile Ala Asn Asp Asp (SEQ ID NO: 660); Asp Asp; Arg Thr Met Asp Asn Asp Glu (SEQ ID NO: 661); Arg Thr Ile Ala Gly Asp Asp (SEQ ID NO: 662); Arg Thr Ile Ala Asn Asp (SEQ ID NO: 663); Asp; Glu Asp; Arg Ser Ile Ser Gln Glu Asp (SEQ ID NO: 664); Thr Asp Glu; Arg Thr Ile Ala Thr Asp Glu (SEQ ID NO: 665); Glu; Ile Ile Thr Pro Pro Asp Pro (SEQ ID NO: 666); Gln Glu Leu; Lys Asp Asp; Gln Glu Glu; Arg Tyr Ile Asn Gln Glu Glu (SEQ ID NO: 667); Ala Ser Ser Tyr Ala Ser (SEQ ID NO: 668); и Thr Ser Ser Tyr Ala Ser (SEQ ID NO: 669).
Согласно конкретным вариантам реализации изобретения, пептидный агонист GC-C формулы III определяется следующим образом:
Xaa1: а) представляет собой Ser, Asn, Tyr, Ala, Gln, Pro, Lys, Gly или Thr или отсутствует; b) располагается после Lys или Tyr; с) представляет собой любую аминокислоту; d) отсутствует; е) представляет собой любую аминокислоту, отличную от Cys; или f) представляет собой Lys или Arg;
Хаа2 представляет собой: a) His, Asp, Glu, Ala, Ser, Asn, Gly или отсутствует; b) His, Asp, Glu, Ala, Ser, Asn, Gly, Pro или отсутствует; с) Asp, Glu, любую аминокислоту или отсутствует; d) Asp или Glu; е) любую аминокислоту, отличную от Cys; е) Glu; f) отсутствует; g) Trp, Tyr или Phe; или h) Lys или Arg;
Хаа3 представляет собой: a) Thr, Asp, Ser, Glu, Pro, Val или Leu; Asp или Glu; b) любую аминокислоту, отличную от Cys; с) Glu; d) Thr; е) Thr, Asp, Ser, Glu, Pro, Val или Leu или отсутствует; f) Trp, Tyr или Phe; или g) Lys или Arg;
Cys4 представляет собой необязательно Хаа4 и представляет собой Cys, Mpt (меркаптопролин), Pen (пеницилламин), Dpr (диаминопропионовую кислоту), Asp или Glu;
Xaa5 представляет собой: а) любую аминокислоту; b) Glu, Asp, Gln, Gly или Pro; с) Glu; d) Glu или Asp; е) Asp, Ile или Glu; f) любую аминокислоту или g) любую аминокислоту, отличную от Cys;
Хаа6 представляет собой: a) Leu, Ile, Val, Ala, Lys, Arg, Trp, Tyr или Phe; b) Leu, Ile, Val, Lys, Arg, Trp, Tyr или Phe; Leu, Ile, Lys, Arg, Trp, Tyr или Phe; с) Leu, Ile, Val, Trp, Tyr или Phe; d) Trp, Tyr, Phe или Leu; е) Leu, Ile или Val; f) Ile, Trp или Leu; g) Trp, Tyr или Phe; h) Ile или Leu; i) Tyr; j) любую аминокислоту; k) любую аминокислоту за исключением Leu; l) любую природную или неприродную ароматическую аминокислоту или m) любую аминокислоту, отличную от Cys;
Хаа7 представляет собой: а) Cys, Ser или Tyr; Cys; b) Cys, Mpt (меркаптопролин), Pen (пеницилламин), Dpr (диаминопропионовую кислоту), Asp или Glu; с) Ser или d) аминокислоту, отличную от Cys;
Хаа8 представляет собой: а) Ala, Val или Ile; b) Ala, Val, Thr, Ile, Met или отсутствует; с) любую аминокислоту; d) Val; е) любую аминокислоту, отличную от Cys или f) отсутствует;
Хаа9 представляет собой: а) любую аминокислоту; b) любую аминокислоту, отличную от Phe и Tyr; с) любую аминокислоту, отличную от Phe, Tyr и Trp; d) любую аминокислоту, отличную от Phe, Tyr, Trp, Ile, Leu и Val; е) любую аминокислоту, отличную от Phe, Tyr, Trp, Ile, Leu, Val и His; i) любую аминокислоту, отличную от Gln; g) любую аминокислоту, отличную от Lys, Arg, Phe, Tyr и Trp; h) любую аминокислоту, отличную от Lys, Arg, Phe, Tyr, Trp, Ile, Leu и Val; i) любую аминокислоту, отличную от Lys, Arg, Phe, Tyr, Trp, Ile, Leu, Val и His; j) любую неароматическую аминокислоту; k) отсутствует; l) Phe, Tyr, Asn или Trp; m) Asn, Tyr, Asp или Ala; n) Asn, Gln или Tyr; о) Phe или Tyr; р) Asn или q) любую аминокислоту, отличную от Cys;
Хаа10 представляет собой: a) Ala, Pro или Gly; b) Pro или Gly; с) Pro; d) Ala, Val, Met, Thr или Ile; е) любую аминокислоту; f) Val; g) Val или Pro; h) Ala или Val; i) любую аминокислоту, отличную от Cys; j) Pro или k) Gly;
Хаа11 представляет собой: а) любую аминокислоту; b) Ala, Leu, Ser, Gly, Val, Glu, Gln, Ile, Leu, Lys, Arg или Asp; c) Ala или Gly; d) Ala; е) Ala или Val; f) любую аминокислоту; g) Ala или Aib (альфа-аминоизомасляную кислоту); h) любую аминокислоту, отличную от Cys; i) Ala или Thr; или j) Thr;
Cys12 представляет собой необязательно Xaa12 и представляет собой a) Cys, Mpt (меркаптопролин), Pen (пеницилламин), Dpr (диаминопропионовую кислоту), Asp или Glu или b) любую аминокислоту, отличную от Cys;
Хаа13 представляет собой: а) Thr, Ala, Asn, Lys, Arg или Trp; b) Thr, Ala, Lys, Arg или Trp; с) любую аминокислоту; d) любую неароматическую аминокислоту; е) Thr, Ala или Trp; f) Trp, Tyr или Phe; g) Thr или Ala; h) любую аминокислоту; i) Thr; j) любую аминокислоту, отличную от Cys; k) Thr, Val или Gly; 1) Thr или Val, m) Thr или Gly, n) Val или Thr; о) Val; р) Thr или q) Gly;
Xaa14 представляет собой: a) Gly, Pro или Ala; b) Gly; с) любую аминокислоту; d) Gly, Ala или Ser; е) Gly или Ala; f) любую аминокислоту, отличную от Cys; или g) Ala;
Xaa15 представляет собой: а) Cys, Tyr или отсутствует; b) Cys; с) Cys, Mpt (меркаптопролин), Pen (пеницилламин), Dpr (диаминопропионовую кислоту), Asp, Glu или d) любую аминокислоту, отличную от Cys или отсутствует; и
Xaa16 представляет собой: а) Trp, Tyr, Phe, Asn, Ile, Val, His или Leu; b) Trp, Tyr, Phe, Asn или Leu; с) Tip, Tyr, Phe или Leu; d) Trp, Tyr или Phe; е) Leu, Ile или Val; f) His, Leu или Ser; g) Tyr или Leu; Lys или Arg; h) His; i) любую аминокислоту, j) Leu или отсутствует; k) Trp, Tyr, Phe, Lys, Arg или отсутствует; l) отсутствует; m) любую аминокислоту, отличную от Cys; или n) Tyr.
Согласно некоторым вариантам реализации изобретения, пептидный агонист GC-C формулы III определяется следующим образом:
Xaa1 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты или отсутствует;
Xaa2 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты или отсутствует;
Хаа3 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты или отсутствует;
Хаа4 представляет собой Cys, Mpt (меркаптопролин), Pen (пеницилламин), Dpr (диаминопропионовую кислоту), Asp или Glu;
Хаа5 представляет собой Glu;
Хаа6 представляет собой Tyr, Trp, Phe или Leu;
Хаа7 представляет собой Cys, Mpt (меркаптопролин), Pen (пеницилламин), Dpr (диаминопропионовую кислоту), Asp или Glu;
Хаа8 представляет собой любую природную или неприродную аминокислоту или аналог аминокислоты, отличный от Cys, или отсутствует;
Хаа9 представляет собой любую аминокислоту;
Хаа10 представляет собой Pro или Gly;
Хаа11 представляет собой любую аминокислоту;
Хаа12 представляет собой Cys, Mpt (меркаптопролин), Pen (пеницилламин), Dpr (диаминопропионовую кислоту), Asp или Glu;
Xaa13 представляет собой Thr, Val или Gly;
Xaa14 представляет собой Gly или Ala;
Xaa15 представляет собой Cys, Mpt (меркаптопролин), Pen (пеницилламин), Dpr (диаминопропионовую кислоту), Asp или Glu; и
Xaa16 представляет собой любую аминокислоту или отсутствует.
Согласно конкретным вариантам реализации изобретения, пептидный агонист GC-C формулы III определяется следующим образом:
Xaa1 представляет собой Asn, любую аминокислоту или отсутствует;
Хаа2 представляет собой Asp, Glu, любую аминокислоту или отсутствует;
Хаа3 представляет собой Asp или Glu;
Хаа5 представляет собой любую аминокислоту или Glu;
Хаа6 представляет собой любую аминокислоту или Leu;
Хаа7 представляет собой Cys;
Xaa8 представляет собой любую аминокислоту или Val;
Хаа9 представляет собой Asn, Gln или Tyr;
Хаа10 представляет собой любую аминокислоту или Val;
Хаа11 представляет собой любую аминокислоту или Ala;
Xaa13 представляет собой любую аминокислоту или Thr;
Xaa14 представляет собой любую аминокислоту или Gly;
Xaa15 представляет собой Cys;
Xaa16 представляет собой любую аминокислоту, Leu или отсутствует
Согласно некоторым вариантам реализации изобретения, пептидный агонист GC-C формулы III не расщепляется после Хаа9 химотрипсином и определяется следующим образом:
Xaa1 представляет собой Ser, Asn, Tyr, Ala, Gln, Pro, Lys, Gly или Thr или отсутствует;
Хаа2 представляет собой His, Asp, Glu, Ala, Ser, Asn или Gly или отсутствует;
Хаа3 представляет собой Thr, Asp, Ser, Glu, Pro, Val или Leu или отсутствует;
Хаа5 представляет собой Asp, Ile или Glu;
Хаа6 представляет собой Ilе, Trp или Leu;
Хаа7 представляет собой Cys, Ser или Tyr;
Xaa8 представляет собой Ala, Val, Thr, Ile или Met или отсутствует;
Хаа9 представляет собой: а) любую аминокислоту, отличную от Phe и Tyr, b) любую аминокислоту, отличную от Phe, Tyr и Trp, с) любую аминокислоту, отличную от Phe, Tyr, Trp, Ile, Leu и Val; d) любую аминокислоту, отличную от Phe, Tyr, Trp, Ile, Leu, Val и His; d) любую неароматическую аминокислоту или е) отсутствует;
Хаа10 представляет собой Ala, Val, Met, Thr или Ile;
Хаа11 представляет собой Ala или Val;
Xaa13 представляет собой Ala или Thr;
Xaa14 представляет собой Gly, Ala или Ser;
Xaa15 представляет собой Cys, Tyr или отсутствует; и
Xaa16 представляет собой: а) Trp, Tyr или Phe с образованием сайта расщепления химотрипсином; b) Lys или Arg с образованием сайта расщепления трипсином; с) отсутствует или d) His или Leu или Ser.
Согласно конкретным вариантам реализации изобретения, пептид или аналог человеческого урогуанилина содержит, состоит или по существу состоит из аминокислотной последовательности, показанной ниже (IV):
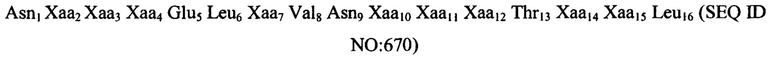
где Хаа2 представляет собой Asp или Glu;
Хаа3 представляет собой Asp или Glu;
Хаа4 представляет собой Cys или Mpt (меркаптопролин) или Pen (пеницилламин) или Dpr (диаминопропионовую кислоту) или Asp или Glu;
Хаа7 представляет собой Cys или Mpt (меркаптопролин) или Pen (пеницилламин) или Dpr (диаминопропионовую кислоту) или Asp или Glu;
Хаа10 представляет собой Val или Pro;
Хаа11 представляет собой Ala или Aib (альфа-аминоизомасляную кислоту);
Xaa12 представляет собой Cys или Mpt (меркаптопролин) или Pen (пеницилламин) или Dpr (диаминопропионовую кислоту) или Asp или Glu;
Xaa14 представляет собой Gly или Ala; и
Xaa15 представляет собой Cys или Mpt (меркаптопролин) или Pen (пеницилламин) или Dpr (диаминопропионовую кислоту) или Asp или Glu.
Согласно конкретным вариантам реализации Формулы IV, Xaa15 не представляет собой Cys или отсутствует, Хаа7 представляет собой Ser или аминокислоту, отличную от Cys.
Согласно конкретным вариантам реализации изобретения, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 из Хаа1, Хаа2, Хаа3, Xaa5, Хаа6, Xaa7, Хаа8, Xaa9, Хаа10, Хаа11, Xaa13, Xaa14 и Xaa16 представляют собой любую аминокислоту, отличную от Cys. Согласно некоторым вариантам реализации изобретения, Xaa9 представляет собой любую аминокислоту, отличную от Gln. Согласно вариантам реализации, где Хаа2 и Хаа3 представляют собой Glu, Xaa9 представляет собой любую аминокислоту, отличную от Gln. Согласно конкретным вариантам реализации изобретения, Xaa1 и Хаа3 отсутствуют; Хаа3 представляет собой Thr; Xaa5 представляет собой Glu; Хаа6 представляет собой Ile или Leu; Xaa8 представляет собой Ala, Val или Ile; Xaa9 представляет собой Phe или Tyr; Хаа10 представляет собой Ala или Val; Хаа11 представляет собой Ala; Xaa13 представляет собой Ala или Thr; Хаа14 представляет собой Gly; и Xaa16 представляет собой Trp, Tyr, Phe, Lys или Arg или отсутствует.
Конкретные примеры аналогов урогуанилина человека представлены в Таблице А7, ниже.

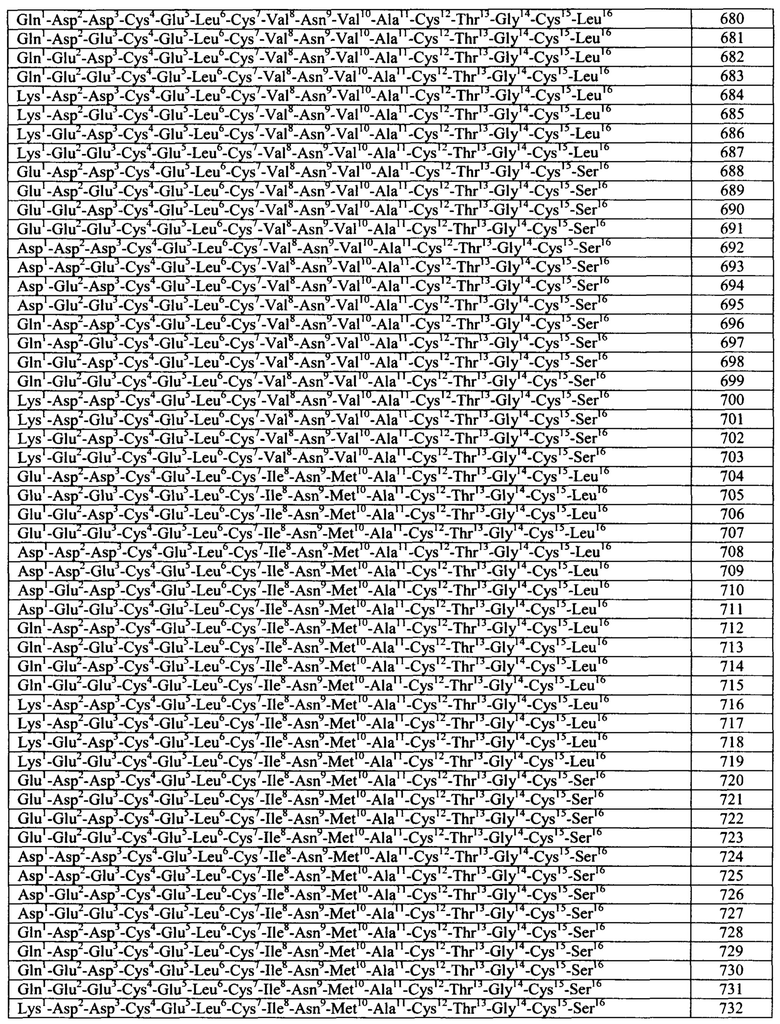
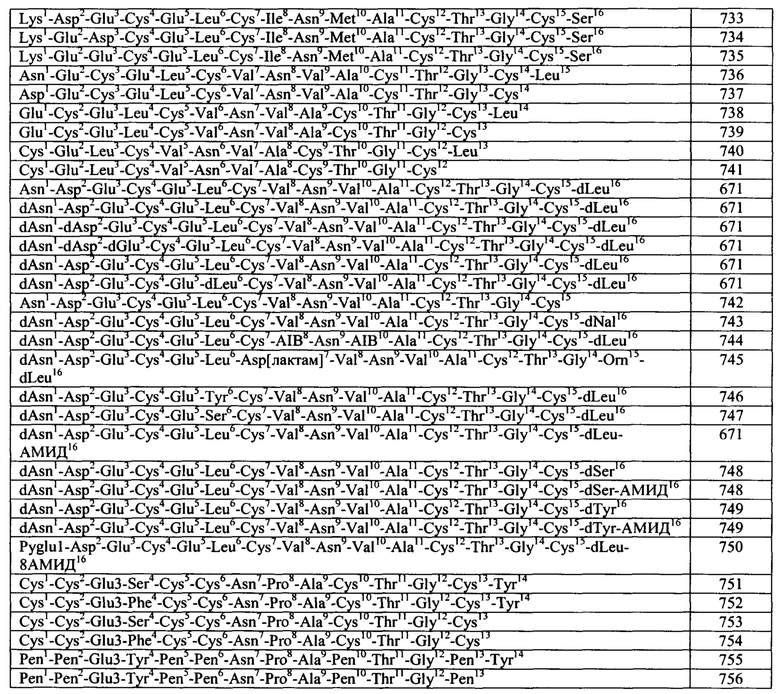
Также включены варианты пептидных агонистов GC-C, описанных в настоящей заявке. Примеры включают варианты пептидов, которые содержат примерно, по меньшей мере примерно или не более чем примерно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 аминокислотных замен, инсерций и/или делеции для любой формулы I, II, III или IV или SEQ ID No. 1, 5, 46-50, 650 и 670 или последовательности в любой из Таблиц А1-А7. Замена (замены) могут быть консервативными или не консервативными. Один пример консервативной аминокислотной замены представляет собой замену, при которой аминокислотный остаток замещен аминокислотным остатком, имеющим сходную боковую цепь. Семейства аминокислотных остатков, имеющих сходные боковые цепи, определены в данной области техники. Указанные семейства включают аминокислоты с основными боковыми цепями (например, лизин, аргинин, гистидин), кислотными боковыми цепями (например, аспарагиновую кислоту, глутаминовую кислоту), незаряженными полярными боковыми цепями (например, глицин, аспарагин, глутамин, серин, треонин, тирозин, цистеин), неполярными боковыми цепями (например, аланин, валин, лейцин, изолейцин, пролин, фенилаланин, метионин, триптофан), бета-разветвленными боковыми цепями (например, треонин, валин, изолейцин) и ароматическими боковыми цепями (например, тирозин, фенилаланин, триптофан, гистидин). Консервативная замена может приводить к замещению природной аминокислоты на неприродную аминокислоту или аналог аминокислоты. Инсерции и/или делеции могут находиться на N-конце, С-конце и/или во внутренних участках пептида (например, инсерция или делеция, состоящая из примерно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 аминокислот на С-конце, N-конце и/или в пределах примерно 2, 3, 4, 5, 6, 7, 8, 9 или 10 аминокислот на N-конце и/или С-конце). В некоторых примерах может являться желательным использование варианта пептида, который связывается и является агонистом кишечного рецептора GC-C, но является менее активным по сравнению с нативной формой пептида. Эта сниженная активность может возникать в результате пониженной аффинности к рецептору или сниженной способности активировать рецептор при связывании или пониженной стабильности указанного пептида.
Пептидные агонисты GC-C могут представлять собой циклические пептиды или линейные пептиды. Кроме того, множество копий одного пептида можно встраивать в один циклический или линейный пептид. Циклические пептиды могут быть получены с помощью способов, известных в данной области техники. Например, макроциклизацию часто осуществляют путем образования амидной связи между N- и С-концами пептида, между боковой цепью и N- или С-концом [например, с K3Fe(CN)6 при рН ~8,5] (Samson et al., Endocrinology, 137: 5182-5185, 1996) или между двумя аминокислотными боковыми цепями, такими как цистеин (DeGrado, Adv Protein Chem, 39: 51-124, 1988).
Пептиды могут включать аминокислотную последовательность пептида, которая встречаются у видов позвоночных (например, млекопитающих) или у видов бактерий. Кроме того, пептиды могут представлять собой частично или полностью неприродные пептиды.
Также включены аналоги пептидов, соответствующие пептидным агонистам GC-C, описанным в настоящей заявке. Аналоги пептидов обычно применяются в фармацевтической промышленности как непептидные лекарственные средства, обладающие свойствами, аналогичными свойствами исходного пептида. Указанные типы непептидных соединений называются «пептидными миметиками» или «пептидомиметиками» (Luthman, et al., A Textbook of Drug Design and Development, 14: 386-406, 2nd Ed., Harwood Academic Publishers, 1996; Joachim Grante, Angew. Chem. Int. Ed. Engl., 33: 1699-1720, 1994; Fauchere, J., Adv. Drug Res., 15: 29 (1986); Veber and Freidinger TINS, p. 392 (1985); и Evans et al., J. Med. Chem. 30: 229, 1987). Пептидомиметик представляет собой молекулу, которая имитирует биологическую активность пептида, но уже не является пептидной по химической природе. Соединения-пептидомиметики известны в данной области техники и описаны, например, в патенте США №6 245 886.
Настоящее изобретение также включает пептоиды. Производные пептидов пептоиды представляют собой другую форму модифицированных пептидов, которые сохраняют структурные детерминанты, важные для биологической активности, но утрачивают пептидные связи, таким образом, приобретая устойчивость к протеолизу (см., например, Simon et al., PNAS USA. 89: 9367-9371, 1992). Пептоиды представляют собой олигомеры N-замещенных глицинов. Был описан ряд N-алкильных групп, каждая из которых соответствует боковой цепи природной аминокислоты. Пептоиды согласно настоящему изобретению включают соединения, в которых по меньшей мере один аминокислотный остаток, несколько аминокислотных остатков или все аминокислотные остатки замещены на соответствующие N-замещенные глицины. Пептоидные библиотеки описаны, например, в патенте США №5 811 387.
Согласно некоторым аспектам, пептидный агонист GC-C содержит или состоит примерно, по меньшей мере примерно или менее чем примерно из 150, 140, 130, 120, 110, 100, 90, 80, 70, 60, 50, 40, 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 8, 7, 6 или 5 аминокислот. Согласно некоторым аспектам, пептид содержит не более 5 аминокислот, к N-концевых по отношению к Cys6 (формулы I или II). Согласно некоторым аспектам, пептид содержит не более 20, 15, 10 или 5 аминокислот, С-концевых по отношению к Cys18 (формулы I или II).
Согласно некоторым аспектам, пептиды являются очищенными. Очищенные пептиды отделяют от других белков, липидов и нуклеиновых кислот или от соединений, из которых они были синтезированы или получены другим способом. Очищенный пептид может составлять по меньшей мере примерно 50, 60, 70, 80, 85, 90, 95, 96, 97 или 98% по сухой массе очищенного препарата.
Как отмечено выше, конкретные пептиды, описанные в настоящей заявке, могут содержать одну или более или все неприродные аминокислоты или аналоги аминокислот. Дополнительные примеры, помимо описанных в другом месте в настоящей заявке (например, выше), включают: неприродный аналог тирозина; неприродный аналог глутамина; неприродный аналог фенилаланина; неприродный аналог серина; неприродный аналог треонина; содержащую в качестве заместителя алкил, арил, ацил, азидо, циано, гало, гидразин, гидразид, гидроксил, алкенил, алкинил, эфир, тиол, сульфонил, селено, сложный эфир, тиокислота, борат, боронат, фосфо, фосфоно, фосфин, гетероцикл, энон, имин, альдегид, гидроксиламин, кето или амино аминокислоту или любую их комбинацию; аминокислоту с фотоактивируемым кросс-линкером; спин-меченную аминокислоту; флуоресцентную аминокислоту; аминокислоту с новой функциональной группой; аминокислоту, которая ковалентно или нековалентно взаимодействует с другой молекулой; металл-связывающую аминокислоту; металл-содержащую аминокислоту; радиоактивно меченую аминокислоту; фотозахваченную и/или фотоизомеризуемую аминокислоту; аминокислоту, содержащую биотин или аналог биотина; гликозилированную или углеводород-модифицированную аминокислоту; кето-содержащую аминокислоту; аминокислоту, содержащую полиэтиленгликоль или полиэфир; замещенную тяжелым атомом аминокислоту (например, аминокислоту, содержащую дейтерий, тритий, 13С, 15N или 18О); поддающуюся химическому расщеплению или поддающуюся фоторасщеплению аминокислоту; аминокислоту с удлиненной боковой цепью; аминокислоту, содержащую токсичную группу; сахар-замещенную аминокислоту, например, сахар-замещенный серин или т.д.; углерод-связанную сахар-содержащую аминокислоту; редокс-активную аминокислоту; α-гидрокси содержащую кислоту; аминокислоту, содержащую аминотиокислоту; α, α дизамещенную аминокислоту; β-аминокислоту; циклическую аминокислоту, отличную от пролина; О-метил-L-тирозин; L-3-(2-нафтил)аланин; 3-метилфенилаланин; п-ацетил-L-фенилаланин; 0-4-аллил-L-тирозин; 4-пропил-L-тирозин; три-О-ацетил-GlcNAcβ-серин; L-допу; фторированный фенилаланин; изопропил-L-фенилаланин; п-азидо-L-фенилаланин; п-ацил-L-фенилаланин; п-бензоил-L-фенилаланин; L-фосфосерин; фосфопосерин; фосфонотирозин; п-йод-фенилаланин; 4-фторфенилглицин; п-бромфенилаланин; п-амино-L-фенилаланин; изопропил-L-фенилаланин; L-3-(2-нафтил)аланин; амино-, изопропил- или O-аллил-содержащий аналог фенилаланина; допу, О-метил-L-тирозин; гликозилированную аминокислоту; п-(пропаргилокси)фенилаланин; диметиллизин; гидроксипролин; меркаптопропионовую кислоту; метиллизин; 3-нитротирозин; норлейцин; пироглумаминовую кислоту; Z (карбобензоксил); ε-ацетиллизин; β-аланин; аминобензоиловое производное; аминомасляную кислоту (Abu); цитруллин; аминогексаноевую кислоту; аминоизомасляную кислоту; циклогексилаланин; d-циклогексилаланин; гидроксипролин; нитроаргинин; нитрофенилаланин; нитротирозин; норвалин; октагидроиндол карбоксилат; орнитин; пеницилламин; тетрагидроизохинолин; ацетамидометил-защищенные аминокислоты и пегилированные аминокислоты. Также примеры неприродных аминокислот и аналогов аминокислот можно найти в заявках на патент США №2003/0108885 и 2003/0082575 и цитированных в указанных заявках ссылках.
Согласно некоторым вариантам реализации изобретения, аминокислота может быть замещена на природную заменимую аминокислоту, например, таурин.
Согласно некоторым вариантам реализации изобретения, 1, 2, 3, 4, 5 или 6 цистеинов удалены или замещены на другие аминокислоты. Согласно конкретным аспектам, большинство N-концевых и/или С-концевых цистеиновых остатков (или остаток) удалено или замещено на другую аминокислоту. Согласно конкретным вариантам реализации изобретения, другая аминокислота представляет собой аланин или серин.
Пептиды могут представлять собой полимеры L-аминокислот, D-аминокислот или их комбинации. Например, согласно конкретным вариантам реализации изобретения, пептиды представляют собой D-ретроинвертированные пептиды. Термин «ретроинвертированный изомер» относится к изомеру линейного пептида, направление последовательности которого обращено, и хиральность каждого аминокислотного остатка инвертирована. См., например, Jameson et al., Nature. 368: 744-746, 1994; Brady et al., Nature. 368: 692-693, 1994. Конечным результатом объединения D-энантиомеров и обращенного синтеза является изменение положения карбонильных и аминогрупп в каждой амидной связи, тогда как положение групп боковых цепей на каждом атоме альфа-углерода сохраняется. Если иное специально не указано, любая данная L-аминокислотная последовательность согласно изобретению может быть создана в D-ретроинвертированном пептиде путем синтеза обратной последовательности соответствующей нативной L-аминокислотной последовательности.
Способы получения пептидов, содержащих неприродные аминокислоты, можно найти, например, в заявках на патент США №2003/0108885 и 2003/0082575, источниках Deiters et al., J Am Chem. Soc. 125: 11782-3, 2003; Chin et al., Science. 301: 964-7, 2003 и цитированных в указанных источниках ссылках.
Согласно некоторым аспектам, пептидные агонисты GC-C могут содержать одну или более стандартных пептидных связей, замещенных на альтернативную связь. Такие замены могут приводить к повышению стабильности пептида. Например, замена пептидной связи между Cys18 и Xaa19 (формулы I или II) на альтернативную связь может приводить к снижению расщепления карбоксипептидазами и увеличивать период пролувыведения из пищеварительного тракта. Связи, которые могут замещать пептидные связи, включают, но не ограничиваются указанными: ретроинвертированные связи (С(O)-NH вместо NH-С(O); восстановленную амидную связь (NH-СН2); тиометиленовую связь (S-СН2 или СН2-S); оксометиленовую связь (О-СН2 или СН2-О); этиленовую связь (CH2-СН2); тиоамидную связь (C(S)-NH); трансолефиновую связь (СН=СН); фтор-замещенную транс-олефиновую связь (CF=CH); кетометиленовую связь (С(O)-CHR или CHR-С(O), где R представляет собой Н или СН3; и фтор-кетометиленовую связь (С(O)-CFR или CFR-C(O), где R представляет собой Н или F или СН3.
В некоторых пептидных агонистах GC-C один или более членов одной или более пар остатков Cys, которые в норме образуют дисульфидный мостик, замещены на гомоцистеин, пеницилламин, 3-меркаптопролин (см., например, Kolodziej et al., Int J Pept Protein Res. 48: 274, 1996); β, β диметилцистеин (см., например, Hunt et al., Int J Pept Protein Res. 42: 249, 1993) или диаминопропионовую кислоту (см., например, Smith et al., J Med Chem. 21: 117, 1978) с образованием альтернативных внутренних поперечных сшивок в положениях нормальных дисульфидных связей.
Согласно некоторым вариантам реализации изобретения, одна или более дисульфидных связей могут быть замещены на альтернативные ковалентные поперечные связи, например, амидную связь (-CH2CH(O)NHCH2- или -CH2NHCH(O)СН2-), сложноэфирную связь, тиоэфирную связь, лактамовый мостик, карбамоильную связь, мочевинную связь, тиомочевинную связь, фосфонат-сложноэфирную связь, алкильную связь (-CH2CH2CH2CH2-), алкенильную связь (-СН2СН=СНСН2-), эфирную связь (-СН2СН2ОСН2- или -CH2OCH2CH2-), тиоэфирную связь (-CH2CH2SCH2- или -CH2SCH2CH2-), аминную связь (-CH2CH2NHCH2- или -CH2NHCH2CH2-) или тиоамидную связь (-CH2CH(S)HNHCH2- или - CH2NHCH(S)CH2-). Например, Леду с соавторами (Ledu et al., PNAS. 100: 11263-78, 2003) описали способы получения лактамовых и амидных поперечных связей. Шафмайстер с соавторами (Schafmeister et al., J. Am. Chem. Soc. 122: 5891, 2000) описали подходящие углеводородные поперечные связи. Углеводородные поперечные связи могут быть получены в результате метатеза (или метатеза с последующей гидрогенизацией в случае насыщенных углеводородных поперечных связей) с использованием какого-либо из катализаторов Граббса (доступных из компаний Materia, Inc. и Sigma-Aldrich и описанных, например, в патентах США №5,831,108 и 6,111,121). В некоторых примерах, образование таких альтернативные поперечных связей требует замещения остатков Cys на другие остатки, такие как Lys или Glu, или неприродные аминокислоты. Кроме того, лактамовые, амидные и углеводородные поперечные связи можно использовать для стабилизации пептида, даже если они связывают аминокислоту в положениях, отличных от положений, занимаемых Cys. Такие поперечные связи могут возникать, например, между двумя аминокислотами, которые разделены двумя аминокислотами, или между двумя аминокислотами, которые разделены шестью аминокислотами (см., например, Schafmeister et al., J. Am. Chem. Soc. 122: 5891, 2000).
Пептидные агонисты GC-C можно модифицировать с помощью стандартных модификаций. Модификации могут располагаться на амино (N-), карбокси (С-) конце, внутри последовательности или в комбинации любых из указанных выше. Согласно некоторым аспектам, пептид может содержать более одного типа модификаций. Модификации включают, но не ограничиваются указанными: ацетилирование, амидирование, биотинилирование, циннамиилирование, фарнезилирование, формилирование, миристоилирование, пальмитоилирование, фосфорилирование (Ser, Tyr или Thr), стеароилирование, сукцинилирование, сульфурилирование и циклизацию (через дисульфидные мостики или амидную циклизацию) и модификацию Cy3 или Cy5. Пептиды согласно изобретению также могут быть модифицированы 2,4-динитрофенилом (DNP), DNP-лизином, 7-амино-4-метилкмарином (АМС), флуоресцеином, NBD (7-нитробенз-2-окса-1,3-диазолом), п-нитроанилидом, родамином В, EDANS (5-((2-аминоэтил)амино)нафталин-1-сульфоновой кислотой), дабцилом, дабсилом, дансилом, техасским красным, FMOC и Tamra (тетраметилродамином). Пептиды согласно изобретению также могут быть конъюгированы, например, с полиэтиленгликолем (ПЭГ); алкильными группами (например, С1-С20 прямыми или разветвленными алкильными группами); жирнокислотными заместителями; комбинациями ПЭГ, алкильными группами и жирнокислотными заместителями (см. патент США №6,309,633; Soltero et al., Innovations in Pharmaceutic Technology. 106-110, 2001); BSA и KLH (Keyhole Limpet Hemocyanin). Например, согласно конкретным вариантам реализации изобретения, N-концевая аминокислота, С-концевая аминокислота или N-концевая аминокислота и С-концевая аминокислота конъюгированы с молекулой ПЭГ.
Согласно конкретным вариантам реализации изобретения, пептидные агонисты GC-C, описанные в настоящей заявке, могут существовать с противоионом. Примеры противоионов включают соли: ацетат, бензолсульфонат, бензоат, эдетат кальция, камзилат, карбонат, цитрат, эдетат (ЭДТА), этисилат, эмбонат, эзилат, фумарат, глюцептат, глюконат, глутамат, гликоллиларсанилат, гексилрезорцинат, йодид, бромид, хлорид, гидроксинафтоат, изетионат, лактат, лактобионат, эстолат, малеат, малат, манделат, мезилат, мукат, напсилат, нитрат, пантотенат, фосфат, салицилат, стеарат, сукцинат, сульфат, тартрат, теослат, ацетамидобензоат, адипат, альгинат, аминосалицилат, ангидрометиленцитрат, аскорбат, аспартат, камфорат, капрат, капроат, каприлат, циннамат, цикламат, дихлорацетат, формат, гентизат, глюкуронат, глицерофосфат, гликолят, гиппурат, фторид, малонат, нападисилат, никотинат, олеат, оротат, оксалат, оксоглутарат, пальмитат, пектинат, пектинатный полимер, фенилэтилбарбитурат, пикрат, пропионат, пидолат, себакат, роданид, тозилат и таннат.
Пептидные агонисты GC-C можно получать в соответствии с различными методами. Например, пептиды можно получать в бактериях, включая, но не ограничиваясь указанными, Е.Coli, или в других системах продукции пептидов или белков (например, Bacillus subtilis, бакуловирусных системах экспрессии с использованием клеток Drosophila Sf9, дрожжей или систем экспрессии на основе мицелиальных грибов, систем экспрессии на основе клеток млекопитающих) или с помощью химического синтеза. Если пептид или вариант пептида должен быть получен в бактерии, например, Е.coli, молекула нуклеиновой кислоты, кодирующая пептид, может необязательно кодировать лидирующую последовательность, которая обеспечивает секрецию зрелого пептида из клетки. Таким образом, последовательность, кодирующая пептид, может включать пре-последовательность и про-последовательность, например, природного бактериального пептида ST. Секретируемый зрелый пептид может быть очищен из культуральной среды.
В некоторых примерах последовательность, кодирующая пептид согласно изобретению, встроена в вектор, способный доставлять и поддерживать молекулу нуклеиновой кислоты в бактериальной клетке. Молекула ДНК может быть встроена в автономно реплицирующийся вектор (подходящие векторы включают, например, pGEM3Z и pcDNA3 и их производные). Векторная нуклеиновая кислота может представлять собой бактериальную ДНК или ДНК бактериофага, такого как бактериофаг лямбда или М13, и их производные. Конструирование вектора, содержащего нуклеиновую кислоту, описанную в настоящей заявке, может сопровождаться последующей трансформацией клетки хозяина, такой как бактерия. Подходящие бактерии-хозяева включают, но не ограничиваются указанными, Е.coli В. subtilis, Pseudomonas, Salmonella. Генетическая конструкция также может содержать помимо кодирующей молекулы нуклеиновой кислоты элементы, которые обеспечивают экспрессию, такие как промоторные и регуляторные последовательности. Векторы экспрессии могут содержать последовательности контроля транскрипции, которые контролируют инициацию транскрипции, такие как промоторные, энхансерные, операторные и репрессорные последовательности. Различные последовательности контроля транскрипции хорошо известны специалистам в данной области техники. Вектор экспрессии также может содержать последовательность регуляции трансляции (например, нетранслируемую 5' последовательность, нетранслируемую 3' последовательность или участок внутренней посадки рибосомы). Вектор может быть способен к автономной репликации или может интегрироваться в ДНК хозяина для обеспечения стабильности во время продукции пептида. Согласно некоторым вариантам реализации изобретения, векторы, системы экспрессии и способы, описанные в патенте США №5,395,490 можно использовать для получения пептидных агонистов GC-C, описанных в настоящей заявке.
Последовательность, кодирующая белок, которая содержит пептид согласно изобретению, также может быть слита с нуклеиновой кислотой, кодирующей полипептидную аффинную метку, например, глутатион-S-трансферазу (GST), белок, связывающийся с мальтозой Е, белок А, метку FLAG, гексагистидин, метку myc или метку НА гриппа для облегчения очистки. Аффинная метка или репортер слияния соединяет рамку считывания интересующего пептида с рамкой считывания гена, кодирующего аффинную метку таким образом, что получается трансляционный гибрид. Экспрессия гибридного гена приводит к трансляции одного полипептида, который содержит как интересующий пептид, так и аффинную метку. В некоторых примерах, в которых используют аффинные метки, последовательность ДНК, кодирующая сайт распознавания протеазы, слита между рамками считывания аффинной метки и интересующего пептида.
Генетические конструкции и способы, подходящие для получения незрелых и зрелых форм пептидов и вариантов согласно изобретению в системах экспрессии белка, отличных от бактерий, и хорошо известные специалистам в данной обалсти техники, также можно использовать для получения пептидов в биологической системе.
Пептиды и их варианты можно синтезировать с помощью твердофазного химического синтеза. Например, пептид можно синтезировать на Cyc(4-CH2Bxl)-ОСН2-4-(оксиметил)-фенилацетамидометиловой смоле с использованием программы двойного сочетания. Необходимо использовать соответствующие защитные группы для достижения правильного распределения дисульфидных связей. Например, следующие защитные группы можно использовать: т-бутилоксикарбонил (для альфа-амино групп); ацетамидометил (для тиоловых групп остатков Cys В и Е); 4-метилбензил (для тиоловых группы остатков Cys С и F); бензил (для у-карбоксила глутаминовой кислоты и гидроксильной группы треонина, при их наличии) и бромбензил (для фенольной группа тирозина, при ее наличии). Осуществляют сочетание с симметричным ангидридом т-бутоксилкарбониламинокислоты или сложным эфиром гидроксибензотриазола (для остатков аспарагина или глутамина), защитную группу удаляют от пептида и пептид отщепляют от твердой подложки в гидрофториде, диметилсульфиде, анизоле и п-тиокрезоле в отношении 8/1/1/0,5 (v/v/v/w) при 0°С в течение 60 мин. После удаления гидрофторида и диметилсульфида с помощью пониженного давления и анизола и п-тиокрезола путем последовательной экстракции этиловым эфиром и этилацетатом неочищенные пептиды экстрагируют смесью 0,5М натрий-фосфатного буфера, рН 8,0, и N,N-диметилформамида в отношении 1/1 (по объему). Дисульфидная связь между остатками Cys В и Е образуется с использованием диметилсульфоксида (Tam et al., J. Am. Chem. Soc. 113: 6657-62, 1991). Полученный в результате пептид можно очищать с помощью обращенно-фазовой хроматографии. Дисульфидную связь между остатками Cys С и F получают путем исходного растворения пептида в 50% уксусной кислоте в воде. Добавляют насыщенный раствор йода в ледяной уксусной кислоте (1 мл раствора йода на 100 мл раствора). После инкубации при комнатной температуре в течение 2 дней в закрытом стеклянном контейнере раствор разбавляют в пять раз деионизированной водой и экстрагируют этилэфиром четыре раза для удаления непрореагировавшего йода. После удаления остаточного этилэфира путем ротационного выпаривания раствор неочищенного продукта лиофилизируют и очищают с помощью последующей обращенно-фазовой хроматографии.
Пептиды также можно синтезировать с помощью многих других способов, включая твердофазный синтез с введением стандартных защитных групп FMOC (т.е. сочетанием с DCC-HOBt и удалением защитной группы с помощью пиперидина в ДМФА). Тиоловые группы Cys могут быть защищены с помощью тритила. Обработку ТФА можно использовать для конечного удаления защитной группы пептида и высвобождения пептида из твердофазной смолы. Во многих случаях для достижения подходящего образования дисульфидной связи достаточным является оксиления на воздухе.
Способность пептидов и других агентов связываться и/или являться агонистом кишечного рецептора GC-C можно проверять, например, в анализах, таких как анализ связывания кишечного рецептора GC-C. В одном примере клетки карциномы толстой кишки человека линии Т84 (Американская коллекция типовых культур (Бетезда, Мэриленд)) растят до конфлюентного состояния в 24-луночных культуральных планшетах в смеси среды Хэма F12 и модифицированной Дальбекко среды Игла (DMEM) (1:1) с добавлением 5% эмбриональной бычьей сыворотки. В анализе используют клетки необязательно на пассажах между 54-60. Вкратце, монослой клеток Т84 на 24-луночных планшетах промывают два раза 1 мл связывающего буфера (DMEM, содержащая 0,05% бычьего сывороточного альбумина и 25 мМ HEPES, рН 7,2), затем инкубируют в течение 30 мин при 37°С в присутствии зрелого радиоактивно меченного пептида ST Е.coli и исследуемого материала в различных концентрациях. Клетки затем промывают четыре раза 1 мл DMEM и солюбилизируют с помощью 0,5 мл/лунку 1н NaOH. Уровень радиоактивности в солюбилизированном материале определяют с использованием стандартных способов.
Дополнительные примеры пептидных агонистов GC-C описаны, например, в патентах США №7,041,786; 7,304,036; 7,371,727; 7,494,979; 7,704,947; 7,799,897; 7,745,409; 7,772,188; 7,879,802; 7,910,546; 8,034,782; 8,080,526; 8,101,579; 8,114,831; 8,110,553; 8,357,775 и 8,367,800; заявках на патент США №2013/0096071; 2013/0190238; 2012/0040892; 2012/0040025; 2012/0213846; 2012/0289460; 2011/0118184; 2010/0152118; 2010/0048489; 2010/0120694; 2010/0261877; 2009/0253634; 2009/0192083; 2009/0305993 и международных публикациях согласно РСТ № WO 2006/086653 и WO 2002/098912, каждая из которых полностью включена в настоящую заявку посредством ссылки.
D. Агонисты растворимой гуанилатциклазы
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист растворимой гуанилатциклазы (sGC). Гуаниннуклеотидил (гуанилил; гуанилат) циклазы (GC) представляют собой имеющие широкое распределение ферменты передачи сигнала, которые в ответ на различные клеточные стимулы превращают GTP во вторичный посредник циклический GMP (cGMP). Существуют как мембран-ассоциированные, так и растворимые гуанилатциклазы, обе из которых могут повышать внутриклеточную концентрацию cGMP.
В энтероцитах кишечника повышенная продукция cGMP подавляет активность обмена Na+/H+ в кишечнике, что приводит к ощелачиванию кишечной слизи. См., например, Fawcus et al., Comp Biochem. Physiol A Physiol. 118: 291-295, 1997. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, активатор растворимой гуанилатциклазы подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте, повышает продукцию cGMP и, таким образом, повышает ощелачивание кишечной слизи.
Общие примеры агонистов sGC включают гем-зависимые и гем-независимые активаторы. См., например, Evgenov et al., Nat. Rev. Drug discov. 5: 755-768, 2006. В соответствии с одной неограничивающей теорией, указанные и другие активаторы GC можно использовать для селективной активации sGC в кишечнике, повышения концентрации cGMP и, таким образом, подавления поглощения фосфатов, как описано в настоящей заявке.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, агонист sGC агонист подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Неограничивающие примеры агонистов sGC включают Bay 41-2271, Bay 58-2667 и соединения, показанные на Фигурах 9A-9L. Дополнительные структуры примеров агонистов sGC описаны вместе со способами их синтеза в патенте США №7,087,644 и международной публикации РСТ № WO 2013/101830, каждый из которых полностью включен в настоящую заявку посредством ссылки.
Е. Агонисты аденилатциклазы
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист аденилатциклазы, необязательно селективный агонист. Аденилатциклаза (или аденилат циклаза) относится к классу ферментов, которые катализируют превращение АТР в 3',5'-циклическую AMP (cAMP) и пирофосфат. В указанной ферментативной активности часто принимают участие дивалентные катионы (например, Mg). cAMP, полученный в результате активности аденилатциклазы, служит регуляторным сигналом, действующим через конкретные сАМР-связывающие белки, включая транскрипционные факторы или другие ферменты (например, сАМР-зависимые киназы).
Аденилатциклаза представляет собой эффекторную молекулу одного из наиболее широко используемых путей передачи сигнала. Ее продукт, cAMP, модулирует клеточный рост и дифференцировку у различных организмов от бактерий до высших эукариот. У животных существуют трансмембранные аденилатциклазы (tmAC) и растворимая аденилатциклаза (sAC). См., например, Tresguerres et al., Kidney Int. 79: 1277-1288, 2011. В отличие от tmAC, sAC не являются трансмембранными белками. Как было обнаружено, они распределены по всей цитоплазме и в конкретных органеллах, где они, как считается, являются источником вторичных посредников, отвечающим за внутриклеточные функций сАМР. См., например, Buck and Levin, Sensors. (Basel) 11: 2112-2128, 2011. Таким образом, tmACs непосредственно модулируют G белки, которые переносят внеклеточные сигналы во внутриклеточные изменения сАМР. Напротив, изоформы sAC регулируются внутриклеточными сигналами, включая бикарбонат, кальций и АТФ.
Трансмембранный регулятор муковисцедоза (CFTR) представляет собой ионный канал хлорида и бикарбоната, активный на эпителии множества тканей. Было показано, что указанный канал отвечает за секрецию НСО3- в тонком кишечнике, где бикарбонат определяет рН на поверхности слизистой оболочки. См., например, Kunzelmann and Mall, Physiol Rev. 82: 245-289, 2002. CFTR регулируется сАМР: фосфорилирование регуляторного домена CFTR сАМР-зависимой протеинкиназой А (PKA) повышает его активность. Селективная активация указанного ионного канала, таким образом, может приводить к ощелачиванию люминальной мембраны и, таким образом, уменьшать или снижать СЕРG. В соответствии с одной неограничивающей теорией, селективная стимуляция tmAC в пищеварительной тракте должна, таким образом, приводить к повышению внутриклеточной концентрации сАМР, стимулировать PKA, повышать активность CFTR и, таким образом, ингибировать захват Pi, опосредованной эффектами СЕРG. Согласно конкретным аспектам, соединение селективно активирует tmAC, например, по сравнению с sAC.
Было показано, что агонисты аденилатциклазы, такие как форсколин, повышают сАМР-опосредованную секрецию бикарбоната в двенадцатиперстной кишке (без повышения желудочной секреции бикарбоната), необязательно через сигнальный путь CFTR. См., например, Takeuchi et al., Am. J. Physiol. 272(3 Pt 1): G646-53, 1997. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист аденилатциклазы подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, без ограничения каким-либо одним механизмом, агонист аденилатциклазы подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист аденилатциклазы III (АС-III), необязательно агонист одной или более из изоформ АС-III ADCY1, ADCY2, ADCY3, ADCY4, ADCY5, ADCY6, ADCY7, ADCY8, ADCY9 и/или ADCY10.
Конкретные примеры агонистов аденилатциклазы включают лабдановые дитерпены, такие как форсколин и его аналоги/производные, включая водорастворимые аналоги форсколина, такие как кольфорсин (NKH477). Форсколин представляет собой дитерпеновое соединение, выделенное из растений, которое активирует все tmAC млекопитающих, за исключением tmAC IX (sAC млекопитающих не чувствительна к форсколину). См., например, Kamenetsky et al., J. Mol. Biol. 362: 623-639, 2006. Форсколиновая стимуляция может вызывать сильные и продолжительные изменения уровня сАМР. См., например, Tresguerres et al., Kidney Int. 79: 1277-1288, 2011. Структура форсколина и некоторых аналогов форсколина показана на Фигуре 10. Водорастворимые производные форсколина включают производные, ацилированные в положении С-6 или С-7 с полярным алифатическим амином. Указанные производные, как правило, являются более селективными в отношении АС и обладают меньшей активностью в отношении других мишеней. См., например, Hartzell and Budnitz, Molecular Pharmacology 41: 880-888, 1992. Таким образом, конкретные аспекты включают применение растворимых аналогов форсколина, которые селективно активируют аденилатциклазы в клетках, выстилающих желудочно-кишечный тракт.
Конкретные примеры аналогов/производных форсколина включают аминоалкилкарбамильные производные форсколина, включая 1-аминоалкилкарбаматы, 9-аминоалкилкарбаматы, 7-аминоалкилкарбаматы, 6-аминоалкилкарбаматы, 6,7-диаминоалкилкарбаматы, 1,6-диаминоалкилкарбаматы, 1,7-диаминоалкилкарбаматы и 1,6,7-триаминоалкилкарбаматы форсколина, которые можно использовать в качестве промежуточных соединений в синтезе производных форсколина. См. патент США 5,350,864. Дополнительные примеры аналогов/производных форсколина включают 12-галогенизированные производные форсколина, включая 12-хлордесацетилфорсколин, 12-хлорфорсколин, 12-бромдесацетилфорсколин, 12-бромдесацетилфорсколин, 12-фтордесацетилфорсколин и 12-фторфорсколин. См. патент США 4,871,764.
Согласно некоторым вариантам реализации изобретения, аналог/производное форсколина представляет собой 6-ацетил-7-деацетилфорсколин, 7-деацетилфорсколин, 7-деацетил-6-(N-ацетилглицил)-форсколин, 7-деацетил-7-β-гемисукцинил-форсколин, 7-деацетил-7-(O-N-метилпиперазино)-γ-бутрилдигидрохлондефорсколин, 7-НРР-форсколин, 6-НРР-форсколин или кольфорсин даропат гидрохлорид (NKH477). См., например, заявку на патент США №2011/0171195, 2006/0004090 и 2011/0077292; Laurenza et al., Mol Pharmacol. 32: 133-9, 1987; Lal et al., BioorgMed Chem. 6: 2075-83, 1998; Mori et al., J. Cardiovasc. Pharmacol. 24: 310-6, 2004. Также примеры способов синтеза аналогов форсколина см. в источнике Levin, Tempahedon Letters. 37: 3079-3082, 1996, и дополнительные примеры водорастворимых аналогов форсколина и способов их синтеза см. в источнике Lal et al., Indian J. Chemistry. 45B: 232-246, 2006. Дополнительные структуры примеров агонистов аденилатциклазы описаны вместе со способами их синтеза в патенте США №4,954,642 и источнике Khandelwal et al., J Med Chem. 31: 1872-9, 1988. Также примеры способов/анализов для выявления стимулированной агонистом аденилатциклазной активности см. в источнике Cunliffe et al., Electrophoresis. 28: 1913-20, 2007. Указанные источники полностью включены в настоящую заявку посредством ссылки.
F. Агонисты рецептора имидазолина-1
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист рецептора имидазолина-1, необязательно селективный агонист. Рецепторы имидазолина включают семейство неадренергических рецепторов с высокоаффинными сайтами связывания для клонидина, идазоксана и других имидазолов. Существуют три класса имидазолиновых рецепторов: рецептор I1, который опосредует симпато-ингибиторное действие имидазолинов, направленное на снижение кровяного давления; рецептор I2, который содержит аллостерический сайт связывания моноаминоксидазы и вовлечен в модулирование боли и нейропротекцию; и рецептор I3, который регулирует секрецию инсулина из бета-клеток поджелудочной железы. Согласно некоторым аспектам, соединение представляет собой селективный агонист рецептора имидазолина-1, например, по сравнению с рецепторами имидазолина-2 и/или имидазолина-3.
Подкласс рецепторов имидазолина-1 отчасти опосредует гипотензивные эффекты клонидин-подобных лекарственных средств центрального действия. В соответствии с одной неограничивающей теорией, активированные рецепторы имидазолина-1 вызывают гидролиз фосфатидилхолина с получением DAG, который затем стимулирует синтез вторичных посредников, таких как арахидоновая кислота и последующие эйкозаноиды, такие как PGE2. См., например, Ernsberger, Ann. NY Acad. Sci. 881: 35-53 1999. PGE2 является эндогенным индуктором СБДК. См., например, Takeuchi et al., Gastro-enterology. 113: 1553-1559, 1997). Без ограничения каким-либо одним механизмом, согласно некоторым аспектам? агонист рецептора имидазолина-1 подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем повышения СБДК.
Солано другой неограничивающей теории, также было показано, что агонисты рецептора имидазолина-1, такие как моксонидин, снижает секрецию кислоты в желудочно-кишечном тракте. См., например, Glavin and Smyth, Br J Pharmacol. 114: 751-4, 1995. Таким образом, без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора имидазолина-1 подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем подавления или снижения секреции кислоты в желудочно-кишечном тракте, например, тонком кишечнике. Согласно конкретных аспектам, и без ограничения каким-либо одним механизмом, агонист рецептора имидазолина-1 подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем повышения СБДК и снижения секреции кислоты в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, агонист рецептора имидазолина-2 подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Неограничивающие примеры агонистов рецептора имидазолина-1 включают агматин, апраклонидин, клонидин, эфароксан, моксонидин, рилменидин, S-23515, S-23757, LNP-509, LNP-911, LNP-509, S-23515, PMS-812, PMS-847, BU-98008 и TVP1022 (S-энантиомер разагилина). См. также источник Head and Mayorov, Cardiovasc Hematol Agents Med Chem. 4: 17-32, 2006, полностью включенный в настоящую заявку посредством ссылки.
Структуры примеров агонистов рецептора имидазолина показаны на Фигуре 11, а также описаны вместе со способами их синтеза, в патентах США №4,323,570; 5,686,477; 3,988,464; 6,300,366; 5,492,912; 5,492,912 и международной публикации РСТ № WO 200141764, каждая из которых полностью включена в настоящую заявку посредством ссылки.
Дополнительные примеры агонистов рецептора имидазолина включают агонисты, описанные в патенте США №7,309,706; патенте США №5,686,477 (ЕР 710,658); патенте США №5,925,665 (ЕР 846,688); международной публикации WO 2001/41764 и международной публикации WO 2000/02878. Производные 5-(арилоксиметил)оксазолина, описанные в патенте США №5,686,477, характеризуются селективной аффинностью в отношении рецептора имидазолина-1. Производные имидазолина, описанные в патенте США №5,925,665, связываются с рецепторами имидазолина без значительного связывания с адренергическими рецепторами. В международной публикации WO 2001/41764 описаны производные изохинолина и хинолина, которые обладают аффинностью к имидазолиновым рецепторам. В международной публикации WO 2000/02878 описаны примеры производных β-карболина в качестве потенциальных лигандов для имидазолиновых рецепторов. Указанные источники включены в настоящую заявку посредством ссылки полностью.
G. Холинергические агонисты
Согласно конкретным вариантам реализации изобретения, соединение представляет собой холинергический агонист, необязательно селективный холинергический агонист. Примеры холинергических агонистов включают непрямые холинергические агонисты, которые стимулируют продукцию или высвобождение ацетилхолина (например, ингибиторы ацетилхолинэстеразы), и прямые холинергические агонисты, которое связываются и стимулируют один или более ацетилхолиновых рецепторов. Нейтротрансмиттер ацетилхолин (2-ацетокси-N,N,N-триметилэтанаминий) представляет собой сложный эфир уксусной кислоты и холина. Общие примеры ацетилхолиновых рецепторов включают никотиновые ацетилхолиновые рецепторы и мускариновые ацетилхолиновые рецепторы. Никотиновые ацетилхолиновые рецепторы представляют собой лиганд-управляемые ионные каналы, состоящие из пяти белковых субъединиц.
Мускариновые ацетилхолиновые рецепторы (т.е. M1, М2, М3, М4 и М5) представляют собой сопряженные с G-белками рецепторы, которые активируют другие ионные каналы через каскад вторичных посредников. Указанные рецепторы экспрессируются в пищеварительном тракте, включая слюнные железы и гладкомышечные и слизистые клетки в желудке и кишечнике. Согласно конкретным вариантам реализации изобретения, соединение представляет собой пан-агонист подтипов мускариновых рецепторов. Эндогенным агонистом всех пяти подтипов мускариновых рецепторов является ацетилхолин, который осуществляет физиологический контроль с помощью гормональных и нейрогенных механизмов. См., например, Eglen, Ann. N. Y. Acad. Sci. 881: 35-53, 2012. Некоторые природные соединения также модулируют мускариновые рецепторы (см. Фигуре 12), включая агонисты, такие как мускарин (токсин из гриба Aminita muscaria, он которого происходит название семейства рецепторов) и пилокарпин, и антагонисты, такие как атропин или (-)-гиосцин (из семейства растений Пасленовые). При введении in vivo мускариновые агонисты вызывают слюноотделение, в то время как мускариновые антагонисты вызывают ксеростомию.
Согласно некоторым вариантам реализации изобретения, соединение представляет собой относительно селективный агонист мускаринового рецептора М3. Секреторный ответ М3 в физиологических условиях стимулируется ацетилхолином (ACh). В частности, Ach связывается с сопряженным с G-белком мускариновым рецептором М3, что приводит к продукции инозитол-1,4,5-трифосфата (InP3) фосфолипазой С. InP3 связывается и вызывает открытие рецептора InP3 на эндоплазматическом ретикулуме, который, согласно одной неограничивающей теории, высвобождает Са2+. Повышенная концентрация [Са2+]i вызывает активацию Cl- каналов на апикальной мембране и K+ каналов на базолатеральной мембране. Отток Cl- в просвет ацинуса вызывает движение Na+ через клетки, и осмотический градиент приводит к секреции жидкости. См., например, Tobin et al., J. Physiol Pharmacol. 60: 3-21, 2009. Эта жидкость является обогащенной бикарбонатом.
Контроль секреции бикарбоната мускариновыми рецепторами был неоднократно показан: при внутрисосудистом или подкожном введении агонистов мускариновых рецепторов повышается высвобождение бикарбоната в просвет кишечника. Этот ответ блокируется мускариновыми антагонистами. Например, было показано, в соответствии с одной неограничивающей теорией, что холинергические агонисты, такие как бетанхол (селективный агонист мускаринового рецептора), карбахол (агонист мускаринового и никотинового ацетилхолинового рецептора) и McN-A-343 (селективный агонист рецептора M1), повышают секрецию бикарбоната в двенадцатиперстной кишке. См., например,  et al., Am J Physiol. 267(1 Pt 1): G10-7, 1994. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, холинергический агонист подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции cекреции бикарбоната в тонком кишечнике.
et al., Am J Physiol. 267(1 Pt 1): G10-7, 1994. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, холинергический агонист подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции cекреции бикарбоната в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, холинергический агонист подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Согласно некоторым аспектам, структура агониста мускоринового рецептора является конформационно-вынужденной в отношении эндогенного лиганда ацетилхолина, такой как структура цис-триметил-(2-метил-[1,3]диоксолан-4-илметил)аммония йодида на Фигуре 12. См., например, Piergentili et al., Bioorganic & Medicinal Chemistry 15: 886-896, 2007. Указанная структура содержит кеталь вместо лабильной сложноэфирной группы ацетилхолина и представляет собой биоизостер, сохраняющий оба акцептора водородной связи Ach, но уже не являющийся стабильным. Подобным образом, карбехол и бетанхол (также показанные на Фигуре 12) являются примерами агонистов, так как в указанных структурах лабильнуая группа сложного эфира Ach замещена на не гидролизуемую карбаматную функциональную группу.
Неограничивающие примеры холинергических агонистов непрямого действия включают ингибиторы ацетилхолинстеразы, такие как карбаматы (например, физостигмин, неостигмин, пиридостигмин), пиперидины (например, динепизил), эдрофоний, гуперзин А, ладостигил, унгеремин, лактукопикрин, такрин, галантамин, транс-дельта-9-тетрагидроканнабинол и фосфаты (например, изофлуорофат, эхотиофат, паратион, малатион). Предпочтительно в способах, предложенных в настоящей заявке, используются обратимые ингибиторы ацетилхолинэстеразы.
Неограничивающие примеры холинергических агонистов прямого действия включают ацетилхолин, никотин, сукцинилхолин, метахолин (ацетил-β-метилхолин), McN-A-343, карбахол (карбамоилхолин), бетанкол (карбамоил-β-метилхолин), мускарин, пилокарпин, оксотреморин, лобелин и диметилфенилпипаразиний.
Н. Агонисты рецептора простагландина ЕР4
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист простаноидного рецептора Е-типа 4 (ЕР4) (или агонист рецептора простагландина ЕР4), необязательно селективный агонист. Рецептор ЕР4 изначально был описан как сопряженный с Gαs-белком рецептор, приводящий к стимуляции аденилатциклазы и повышению внутриклеточного уровня сАМР. Рецептор ЕР4 впервые был клонирован как рецептор простагландина Е2 (PGE2), который стимулировал образование сАМР, и был обозначен как «ЕР2». Однако после того, как был открыт другой сАМР-стимулирующий рецептор PGE2, чувствительный к бутапросту, бутапрост-нечувствительным рецептор, который опосредован расширение сосудов, был переименован в «ЕР4». Он представляет собой один из четырех идентифицированных рецепторов PGE2.
В соответствии с одной неограничивающей теорией, было показано, что агонисты рецептора простагландина ЕР4 стимулируют секрецию бикарбоната в двенадцатиперстной кишке, например, с помощью механизма, опосредованного сАМР. См., например, Aoi et al., Am J Physiol Gastro-intest Liver Physiol. 287: G96-103, 2004; Lundgren, Acta Physiol Scand. 185: 87, 2005; Takeuchi et al., Gastro-enterology. 113: 1553-1559, 1997. Таким образом, без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора простагландина ЕР4 подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, агонист ЕР4 подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Неограничивающие примеры агонистов рецептора простагландина ЕР4 включают PGE2, аналоги PGE2, АЕ1-329, AGN205203, APS-999 Na, Cay10598 (19а), СР-044519-02, CJ-023,423, EP4RAG, ER-819762, L-902688, лубипростон, ONO-4819CD, ONO АЕ1-329, ONO AE1-734, PGE1-ОН, TCS2510, γ-лактамовый аналог PGE 3, 11-докси-PGE1, γ-лактамовый аналог PGE 2а, γ-лактамовый аналог PGE 4. См., например, Konya et al., Pharmacol Ther. 138: 485-502, 2013.
Неограничивающие примеры аналогов PGE2 включают 16,16-диметил PGE2, 16-16 диметилPGE2 сложный п-(п-ацетамидобензамидо)фениловый эфир, 11-деокси-16,16-диметил PGE2, 9-деокси-9-метилен-16, 16-диметил PGE2, 9-деокси-9-метилен PGE2, 9-кетофлупростенол, 5-транс PGE2, 17-фенил-амегатринор PGE2, PGE2 сероноламид, сложный метиловый эфир PGE2, 16-фенилтетранор PGE2, 15(S)-15-метил PGE2, 15(R)-15-метил PGE2, 8-изо-15-кето PGE2, изопропиловый сложный эфир 8-изо PGE2, 20-гидрокси PGE2, 11-деокси PGEi, ноклопрост, сульпростон, бутапрост, 15-кето PGE2 и 19(R) гидроксиPGE2. См., например, заявку на патент США №2012/0202288.
Дополнительные примеры агонистов рецептора простагландина ЕР4 включают агонисты, описанные в заявках на патент США №2001/0056060, 2002/0040149, 2005/0164949 и 2011/0098481. Также включены агонисты рецептора простагландина ЕР4, описанные (наряду со связанными способами синтеза) в патентах США №4,219,479; 4,049,582; 4,423,067; 4,474,802; 4,692,464; 4,708,963; 5,010,065; 5,013,758; 6,747,037 и 7,776,896; европейском патенте № ЕР 0084856; канадском патенте №1248525; заявках на патент США №2004/0102499, 2005/049227, 2005/228185, 2006/106088, 2006/111430, 2007/0010495, 2007/0123568, 2007/0123569, 2005/0020686, 2008/0234337, 2010/0010222, 2010/0216689, 2004/0198701, 2004/0204590, 2005/0227969, 2005/0239872, 2006/0154899, 2006/0167081, 2006/0258726, 2006/0270721, 2009/0105234, 2009/0105321, 2009/0247596, 2009/0258918, 2009/0270395, 2004/0087624, 2004/0102508, 2006/0252799, 2009/0030061, 2009/0170931, 2010/0022650, 2009/0312388, 2009/0318523, 2010/0069457, 2010/0076048, 2007/0066618, 2004/0259921, 2005/0065133 и 2007/0191319 и международных публикациях согласно РСТ №WO 2004/4071428, WO 2006/052630, WO 2006/047476, WO 2006/058080, WO 2004/065365, WO 2003/047513, WO 2004/085421, WO 2004/085430, WO 2005/116010, WO 2005/116010, WO 2007/014454, WO 2006/080323 и WO 2006/137472, каждая из которых полностью включена посредством ссылки.
Конкретные примеры агонистов рецептора ЕР4 показаны на Фигуре 13.
Согласно конкретным вариантам реализации изобретения, агонист рецептора ЕР4 представляет собой лубипростон (также агонист кальций-активируемого хлоридного канала). Лубипростон представляет собой бициклическую жирную кислоту, произошедшую из простагландина Е1, которая действует, в частности, путем активации хлоридных каналов ClC-2 на апикальном поверхности эпителиальных клеток желудочно-кишечного тракта, продуцирующих обогащенный хлоридом жидкий секрет. Этот секрет смягчают стул, повышают перистальтику и обеспечивают самопроизвольную дефекацию (СПД). Лубипростон стимулирует CFTR-зависимую секрецию бикарбоната в двенадцатиперстной кишке без изменения результирующей секреции Cl-. См., например, Muzimori et al., J Physiol. 573: 827-842, 2006. В настоящей заявке перфузионное введение активного антагониста рецептора ЕР4 АН23848 приводила к устранению вызванной лубипростоном секреции бикарбоната в двенадцатиперстной кишке, тогда как антагонист рецептора ЕР1/ЕР2 АН6809 не оказывал эффекта. Указанные результаты дают основания полагать, что лубипростон может повышать секрецию бикарбоната в двенадцатиперстной кишке путем воздействия в качестве агониста на рецептор простагландина ЕР4. Таким образом, согласно конкретным аспектам, лубипростон подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
Как отмечено выше, конкретные аспекты включают селективный агонист рецептора простагландина ЕР4. Селективные агонисты ЕР4 включают соединения, IC50 которых в отношении подтипов рецептора ЕР1, ЕР2 и/или ЕР3 по меньшей мере в 5, по меньшей мере в 10, по меньшей мере в 20, по меньшей мере в 30, по меньшей мере в 40 или по меньшей мере в 50 раз превышает IC50 для рецептора ЕР4.
I. Агонисты рецептора дофамина D1
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист рецептора дофамина D-1, необязательно селективный агонист. Сопряженный с G-белком рецептор дофамина D1 является основным высоко экспрессируемым подтипом дофаминового рецептора среди семейства дофаминовых рецепторов. Он стимулирует аденилатциклазу и активирует цАМФ-зависимые протеинкиназы.
Согласно одной неограничивающей теории, было показано, что агонисты рецептора дофамина D1 и ингибитор катехол-O-метилтрансферазы (СОМТ) периферического действия нитекапон (ингибиторы СОМТ снижают тканевое разрушение катехоламинов, включая дофамин) стимулируют секрецию бикарбоната в кишечнике и повышение продукции циклического AMP в отдельных энтероцитах двенадцатиперстной кишки. См., например, Flemstrom and Safsten, dig  Sci. 39: 1839-42, 1994; Knutson et al., Gastro-enterology. 104: 1409-13 1993; Iwatsuki et al., Eur J Pharmacol. 218: 237-41, 1992; и Fraga et al., Cell Physiol Biochem. 18: 347-60, 2006. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора дофамина D1 подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
Sci. 39: 1839-42, 1994; Knutson et al., Gastro-enterology. 104: 1409-13 1993; Iwatsuki et al., Eur J Pharmacol. 218: 237-41, 1992; и Fraga et al., Cell Physiol Biochem. 18: 347-60, 2006. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора дофамина D1 подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, без ограничения каким-либо одним механизмом, агонист рецептора дофамина D1 подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Неограничивающие примеры агонистов рецептора дофамина D1 включают дофамин (например, дофамин гидрохлорид, NPEC-захваченный дофамин), дигидрексидин (например, дигирексидина гидрохлорид), бензазепаин и его аналоги/производные. Конкретные примеры дигирексидиновых производных включают А86929, динапсолин, диноксилин и доксантрин, и конкретные примеры бензазепиновых производных включают SKF81297, SKF82958, SKF38393, фенолдопам и 6-Br-АРВ. Также включены агонисты рецептора дофамина D1, показанные на Фигуре 14.
Дополнительные неограничивающие примеры агонистов рецептора дофамина D1 включают А68930, А77636, (R)-(-)-апоморфина гидрохлорид, CY208-243, SKF89145, SKF89626, 7,8-Дигидрокси-5-фенил-октагидробензо[h]изохинолин, YM435, АВТ-431, NNC01-0012, SCH23390, SKF7734, SKF81297, SKF38322, SKF83959, каберголин, фенолдапам (например, фенолдапама гидрохлорид), бромкриптин, ропинирол, прамипексол, энтакапон, толкапон, дигексадин, IPX-750 и перголид. См. также Zhang et al., Med Res Rev. 29: 272-94, 2009; Yvonne Connolly Martin, International Journal of Medicinal Chemistry, vol. 2011, Article ID 424535, 8 pages, 2011. doi:10,1155/2011/424535; Salmi et aL, CNS Drug Rev. 10: 230-42, 2004; Bourne, CNS Drug Rev. 7: 399-414, 2001. Более того, агонисты рецептора D1 можно идентифицировать с помощью стандартных способов скрининга, известных в данной области техники. В качестве неограничивающего примера, клеточный функциональный анализ для высокопроизводительного скрининга лекарственных средств для поиска агонистов рецептора дофамина D1 описан в источнике Jiang et al., Acta Pharmacol Sin. 26: 1181-6, 2005. Указанный источник включен в настоящую заявку посредством ссылки полностью.
Как отмечено выше, конкретные аспекты включают селективный агонист рецептора дофамина D1. Селективные агонисты дофамина D1 включают соединения, IC50 которых в отношении подтипов рецептора D2, D3, D4 и/или D5 по меньшей мере в 5, по меньшей мере в 10, по меньшей мере в 20, по меньшей мере в 30, по меньшей мере в 40 или по меньшей мере в 50 раз превышает IC50 для подтипа рецептора D1.
J. Агонисты рецептора мелатонина
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист рецептора мелатонина, необязательно селективный агонист.Рецепторы мелатонина относятся к семейству высокоаффинных сопряженных с G-белком рецепторов, которые связываются с гормоном эпифеза мелатонином. См. Reppert, BiolRhythms, 12: 528-31, 1997.
Примеры рецепторов мелатонина включают рецепторы МТ1 и МТ2. Согласно некоторым аспектам, агонисты рецептора мелатонина связываются как с рецепторами МТ1, так и МТ2. Согласно некоторым вариантам реализации изобретения, агонист рецептора мелатонина связывается селективно с рецептором МТ1 или МТ2, например, связывается с МТ2, но незначительно связывается с МТ1, или связывается с МТ1, но незначительно связывается с МТ2.
Согласно неограничивающей теории, было показано, что агонисты рецептора мелатонина, такие как мелатонин, стимулируют секрецию бикарбоната в двенадцатиперстной кишке, например, путем воздействие на МТ2-рецепторы энтероцитов. См., например,  et al., J Clin Invest. 108: 625-33, 2001; and Flemstrom, J. Pineal Res. 34: 288-293, 2003. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора мелатонина подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
et al., J Clin Invest. 108: 625-33, 2001; and Flemstrom, J. Pineal Res. 34: 288-293, 2003. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора мелатонина подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, агонист рецептора мелатонина подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Примеры агонистов рецептора мелатонина включают мелатонин (N-ацетил-5-метокситриптамин) и аналоги мелатонина, которые связываются и активируют рецептор мелатонина. Общая структура мелатонина содержит индольное кольцо с метокси группой в положении 5 (5-метокси группа) и ациламиноэтиловой боковой цепью в положении 3; две боковые цепи вносят вклад в связывание и активацию рецептора (рецепторов) мелатонина. Исследовали эффект замещения всех положений индольного кольца. См., например, Rivara et al., Curr Top Med Chem. 8: 954-68, 2008; и Sugen et al., Pigment Cell Research. 17: 454-460, 2004.
Конкретные примеры агонистов рецептора мелатонина включают 2-йодмелатонин, 6-хлормелатонин, 6,7-дихлор-2-метилмелатонин и 8-гидроксимелатонин, все из которых содержат в качестве фрагмента 5-метокси индольное, а также циркадин, агомелатин, рамельтеон, тасимельтеон, бета-метил-6-хлормелатонин (TIK-301 или LY156735), TAK-375, VEC-162, GR196429, S20242, S23478, S24268, S25150, GW290569, BMS-214778, 8-метокси-2-хлорацетамидотетралин, 8-метокси-2-пропионамидотетралин, N-ацетилтриптамин, 6-хлормелатонин, 2-йодмелатонин, 8-M-PDOT и 2-фенилмелатонин. См., например, заявку на патент США №2005/0164987, которая полностью включена посредством ссылки. Также включены примеры агонистов рецептора мелатонина (МТ2), показанные на Фигуре 15.
Способы скрининга агонистов рецептора мелатонина описаны, например, в заявках на патент США №2003/0044909, которые полностью включены в настоящую заявку посредством ссылки.
K. Агонисты рецептора 5НТ4
Согласно конкретным вариантам реализации изобретения соединение представляет собой агонист рецептора 5НТ4, необязательно селективный агонист. Рецептор 5-гидрокситриптамина 4 (5НТ4) представляет собой сопряженный с G-белком серотониновый рецептор, который стимулирует продукцию сАМР в ответ на серотонин (5-гидрокситриптамин или 5-НТ) или другой агонист.
Согласно одной неограничивающей теории, было показано, что серотонин повышает защитную секрецию бикарбоната в двенадцатиперстной кишке, например, опосредованную брюшными ганглиями, сАМР- и Са2+-зависимыми сигнальными путями и 5НТ4-зависимым путем. См., например, et al., Scand J Gastro-enterol. 41: 1279-89, 2006; Tuo et al., Am J Physiol Gastro-intest Liver Physiol 286: G444-G451, 2004. Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора 5НТ4 подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, без ограничения каким-либо одним механизмом, агонист 5НТ4 подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Неограничивающие примеры агонистов 5НТ4 включают серотонин и его аналоги, BIMU-8, цисаприд, клеобоприд, CL033466, ML10302, мозаприд, прукалоприд, рензаприд, RS67506, RS67333, SL650155, тегасерод, закоприд, нароноприд (ATI-7505), велусетраг (TD-5108).
Согласно некоторым вариантам реализации изобретения, агонист или частичный агонист рецептора 5НТ4 представляет собой замещенный бензамид, такой как цисаприд, включая отдельные энантиомеры или комбинации энантиомеров цисаприда ((+) цисаприд и (-) цисаприд), мозаприд или рензаприд. Согласно некоторым вариантам реализации изобретения, агонист рецептора 5НТ4 представляет собой бензофурановое производное, такое как прукалоприд, индол, такой как тегасерод или бензимидазолон. Другие неограничивающие примеры агонистов или частичных агонистов рецептора 5НТ4 включают закоприд (CAS RN 90182-92-6), SC-53116 (CAS RN 141196-99-8) и его рацемат SC-49518 (CAS RN 146388-57-0), BIMU1 (CAS RN 127595-43-1), TS-951 (CAS RN 174486-39-6), ML10302 (CAS RN 148868-55-7), метоклопрамид, 5-метокситриптамин, RS67506, 2-[1-(4-пиперонил)пиперазинил]бензотиазол, RS66331, BIMU8, SB 205149 (н-бутиловый четвертичный аналог рензаприда) и индол карбазимидамид, описанный в источнике Buchheit et al., J Med. Chem. 38: 2331-8, 1995. Также включены норцисаприд (CAS RN 102671-04-5), который представляет собой метаболит цисаприда; мозаприда цитрат; малеатная форма тегасерода (CAS RN 189188-57-6); закоприда гидрохлорид (CAS RN 99617-34-2); мезакоприд (CAS RN 89613-77-4); SK-951((+-)-4-амино-N-(2-(1-азабицикло(3,3,0)октан-5-ил)этил)-5-хлор-2,3-дигидро-2-метилбензо[b]фуран-7-карбоксамида гемифумарат); ATI-7505, аналог цисаприда; SDZ-216-454, селективный агонист рецептора 5НТ4, который стимулирует образование сАМР зависимым от концентрации образом (см., например, Markstein et al., Naunyn-Schmiedebergs Arch Pharmacol. 359: 454-9, 1999); SC-54750 или аминометилазадамантан; Y-36912 или 4-амино-N-[1-[3-(бензилсульфонил)пропил]пиперидин-4-илметил]-5-хлор-2-метоксибензамид (см. Sonda et al., Bioorg Med. Chem. 12: 2737-47, 2004); TKS159 или 4-амино-5-хлор-2-метокси-N-[(2S,4S)-1-этил-2-гидроксиметил-4-пирролидинил]бензамид; RS67333 или 1-(4-амино-5-хлор-2-метоксифенил)-3-(1-н-бутил-4-пиперидинил)-1-пропанон; KDR-5169 или 4-амино-5-хлор-N-[1-(3-фтор-4-метоксибензил)пиперидин-4-ил]-2-(2-гидроксиэтокси)бензамида гидрохлорида дигидрат (см. Tazawa, et al., Eur J Pharmacol. 434: 169-76, 2002); SL65,0155 или 5-(8-амино-7-хлор-2,3-дигидро-1,4-бензодиоксин-5-ил)-3-[1-(2-фенилэтил)-4-пиперидинил]-1,3,4-оксадиазол-2(3Н)-она моногидрохлорид; и Y-34959 или 4-амино-5-хлор-2-метокси-N-[1-[5-(1-метилиндол-3-илкарбониламино)пентил]пиперидин-4-илметил]бензамид.
Дополнительные примеры агонистов и частичных агонистов рецептора 5НТ4 включают метоклопрамид (CAS RN 364-62-5), 5-метокситриптамин (CAS RN 608-07-1), RS67506 (CAS RN 168986-61-6), 2-[1-(4-пиперонил)пиперазинил]бензотиазол (CAS RN 155106-73-3), RS66331 (см. Buccafusco et al., Pharmacology. 295: 438-446, 2000); BIMU8 (эндо-N-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,3-дегидро-2-оксо-3-(проп-2-ил)-1Н-бензимидазол-1-карбоксамид) или SB 205149 (н-бутиловый четвертичный аналог рензаприда). Также включены соединения, родственные метоклопрамиду, такие как метоклопрамида дигидрохлорид (CAS RN 2576-84-3), метоклопрамида дигидрохлорид (CAS RN 5581-45-3) и метоклопрамида гидрохлорид (CAS RN 7232-21-5 или 54143-57-6). См., например, заявку на патент США №2009/0325949; De Maeyer et al., Neurogastro-enterology and Motility. 20: 99-112, 2008; Manabe et al., Expert Opin Investig Drugs. 19: 765-75, 2010; Tack et al., Alimentary Pharmacology & Ther. 35: 745-767, 2012. Указанные источники включены в настоящую заявку посредством ссылки полностью.
L. Агонисты рецептора предсердного натрийуретического пептида
Согласно некоторым вариантам реализации изобретения, соединение представляет собой агонист рецептора предсердного натрийуретического пептида (NP). Рецепторы NP представляют собой каталитические рецепторы с единственным трансмембранным доменом, обладающие внутриклеточной гуанилилциклазной (GC) активностью. Существуют три изоформы NP рецепторов; NPR1, NPR2 и NPR3. Указанные рецепторы содержат консервативные каталитические и регуляторные домены и вариабельные лиганд-связывающие домены.
Рецепторы натрийуретического пептида встречаются в мозге, сосудистой системе почек и желудочно-кишечном тракте и связывают α-предсердный натрийуретический пептид, мозговой натрийуретический пептид и натрийуретический пептид типа C с различной аффинностью. Основной физиологической ролью рецепторов NP является гомеостаз объема жидкости тела. В соответствии с одной неограничивающей теорией, экзогенный натрийуретический пептид стимулирует активность GC в желудочно-кишечном тракте. См., например, Rambotti et al., Histochem. J. 29: 117-126, 1997.
Без ограничения каким-либо одним механизмом, согласно конкретным аспектам, агонист рецептора NP подавляет или снижает захват фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната и/или подавления секреции кислоты в тонком кишечнике.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, агонист рецептора NP подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Структуры примеров пептидных агонистов рецептора (рецепторов) NP показаны на Фигуре 16 и описаны, например, в источнике von Geldern et al., J. Med. Chem. 35: 808-816, 1992, который полностью включен в настоящую заявку посредством ссылки.
Согласно конкретным вариантам реализации изобретения, агонист рецептора NP содержит, состоит или по существу состоит из аминокислотной последовательности предсердного натрийуретического пептида: Ser Leu Arg Arg Ser Ser Cys Phe Gly Gly Arg Ile Asp Arg Ile Gly Ala Gln Ser Gly Leu Gly Cys Asn Ser Phe Arg Tyr (SEQ ID NO: 7), включая ее активные варианты, содержащие 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 делеций, инсерций и/или замен. Конкретные примеры мутантных вариантов с делениями включают мутантные варианты, содержащие последовательность: Cys Phe Gly Gly Arg Ile Asp Arg Ile Gly Ala Gln Ser Gly Leu Gly Cys (SEQ ID NO: 8); и Ser Ser Cys Phe Gly Gly Arg Ile Asp Arg Ile Gly Ala Gln Ser Gly Leu Gly Cys Asn Ser Phe Arg (SEQ ID NO: 9). Как описано в другом месте в настоящей заявке, такие пептиды могут состоять из любой комбинации природных и неприродных аминокислот.
М. Ингибиторы карбоангидразы
Согласно некоторым вариантам реализации изобретения, соединение представляет собой ингибитор карбоангидразы. Поглощение бикарбоната эпителиальными клетками происходит путем диффузии СО2 с последующим его превращением в НСО3- и H+ клеточной карбоангидразой (СА). Бикарбонат затем секретируется через апикальную мембрану в ходе анионного обмена. СА представляет собой фермент, который гидратирует CO2 с получением НСО3- и H+ и встречается в большинстве тканей, включая эпителиальные клетки двенадцатиперстной кишки. См., например, Kaunitz and Akiba, 2006. Указанный продуцируемый эндогенный НСО3- является важным источником транспортируемого бикарбоната.
Существует по меньшей мере 15 изоформ карбоангидраз. Карбоангидраза IV (САIV) представляет собой связанную с мембраной изоформу, тогда как CAII является цитозольной, повсеместно распределенной и высоко активной (скорость циркуляции ~106 с-1). См., например, Shandro and Casey, 2007. Оказывается, карбоангидраза II функционально связана, прямо и не прямо, с белками, транспортирующими бикарбонаты, такими как CFTR, SLC26A6 и DRA. См., например, Seidler and  , 2012. В целом, СООН-конец всех белков, транспортирующих бикарбонаты, за исключением DRA, содержит консенсусный связывающийся с карбоангидразой II мотив. См., например, Dudeja and Ramaswamy, 2006.
, 2012. В целом, СООН-конец всех белков, транспортирующих бикарбонаты, за исключением DRA, содержит консенсусный связывающийся с карбоангидразой II мотив. См., например, Dudeja and Ramaswamy, 2006.
Карбоангидразы вовлечены в некоторые физиологические процессы, включая поддержание рН. Было показано, что классические ингибиторы карбоангидразы, такие как ацетазоламид и бензоламид, ингибируют множество изоформ СА, включая CAII и CAIV. См., например, Scozzafava et al., J. Med. Chem. 45: 1466-1476, 2002. В соответствии с одной неограничивающей теорией, ожидается, что подавление карбоангидразы снижает внутриклеточный рНi субапикальной области. Без ограничения каким-либо одним механизмом, селективное подавление СА в энтероцитах двенадцатиперстной кишки могло бы, таким образом, приводить к снижению CEPG, вызывая снижение транспорта фосфатов.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, ингибитор карбоангидразы подавляет или снижает поглощения фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
На Фигуре 17 показаны структуры примеров ингибитора карбоангидразы, включая дорзоламид и бринзоламид, среди прочих. Согласно конкретным аспектам, ингибиторы карбоангидразы можно использовать в комбинации с классами соединений, способных повышать уровень cAMP, cGMP, кальция или других вторичных посредников в апикальных клетках слизистой оболочки желудочно-кишечного тракта.
N. Ингибиторы фосфодиэстеразы
Согласно некоторым вариантам реализации изобретения, соединение представляет собой ингибитор фосфодиэстеразы. Фосфодиэстеразы (PDE) представляют собой семейство родственных фосфогидролиаз, которые селективно катализируют гидролиз 3'-циклических фосфатных связей в аденозин- и/или гуанин-3',5'-циклическом монофосфате (cAMP и/или cGMP). Они регулируют клеточный уровень, локализацию и продолжительность действия указанных вторичных посредников посредством контроля скорости их разрушения.
Существуют 11 подтипов PDE, называемых PDE1-11; PDE4, 7 и 8 селективно разрушают cAMP, PDE5, 6 и 9 селективно разрушают cGMP и PDE1, 2, 3, 10 и 11 разрушают оба циклических нуклеотида. PDE экспрессируются повсеместно, при этом каждый подтип имеет специфическое тканевое распределение. На Фигуре 18 показаны структуры примеров ингибиторов фосфодиэстеразы с различной специфичностью в отношении подтипов, включая теофиллин, цилостазол, винпоцетин, амринон, EHNA, трехинзин, дротаверин, рофлумиласт и силденафил.
В соответствии с одной неограничивающей теорией, ингибиторы фосфодиэстеразы способны модулировать секрецию бикарбоната в двенадцатиперстной кишке (СБДК) отдельно и в комбинации с агентами, которые повышают цитозольное содержание cAMP и cGMP в энтероцитах путем поддержания уровня указанных вторичных посредников. Ингибиторы PDE1 и PDE3, в частности, участвуют в модулировании СБДК. См., например, Hayashi, Biochem. Pharmacol. 74: 1507-1513, 2007. Без ограничения каким-либо одним механизмом, согласно конкретным вариантам реализации изобретения, ингибитор фосфодиэстеразы подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем стимуляции секреции бикарбоната в тонком кишечнике или СБДК.
Согласно некоторым вариантам реализации изобретения, и без ограничения каким-либо одним механизмом, ингибитор фосфодиэстеразы подавляет или снижает поглощение фосфатов в желудочно-кишечном тракте путем снижения всасывания воды в тонком кишечнике.
Согласно конкретным вариантам реализации изобретения, ингибиторы PDE замедляют разрушение циклического AMP (cAMP) и/или циклического GMP (cGMP), что затем может приводить к относительному повышению внутриклеточной концентрации cAMP и/или cGMP. Общие примеры включают ингибиторы PDE1, ингибиторы PDE3, ингибиторы PDE4, ингибиторы PDE5, ингибиторы PDE3/4 и ингибиторы PDE3/4/5. В качестве неограничивающего примера ингибиторы PDE могут включать соединения, описанные в следующих патентных заявках и патентах: DE 1470341, DE 2108438, DE 2123328, DE 2305339, DE 2305575, DE 2315801, DE 2402908, DE 2413935, DE 2451417, DE 2459090, DE 2646469, DE 2727481, DE 2825048, DE 2837161, DE 2845220, DE 2847621, DE 2934747, DE 3021792, DE 3038166, DE 3044568, DE 3142982, DE 1116676, DE 2162096, ЕР 000718, ЕР 0008408, ЕР 0010759, ЕР 0059948, ЕР 0075436, ЕР 0096517, ЕР 0112987, EP 0116948, EP 0150937, EP 0158380, EP 0161632, EP 0161918, EP 0167121, EP 0199127, EP 0220044, EP 0247725, EP 0258191, EP 0272910, EP 0272914, EP 0294647, EP 0300726, EP 0335386, EP 0357788, EP 0389282, EP 0406958, EP 0426180, EP 0428302, EP 0435811, EP 0470805, EP 0482208, EP 0490823, EP 0506194, EP 0511865, EP 0527117, EP 0626939, EP 0664289, EP 0671389, EP 0685474, EP 0685475, EP 0685479, EP 0293063, EP 0463756, EP 0482208, EP 0579496, EP 0667345, EP 0163965, EP 0393500, EP 0510562, EP 0553174, JP 92234389, JP 94329652, JP 95010875, патенты США №4,963,561; 5,141,931 и 6,331,543; международные публикации патентных заявок № WO 9117991, WO 9200968, WO 9212961, WO 9307146, WO 9315044, WO 9315045, WO 9318024, WO 9319068, WO 9319720, WO 9319747, WO 9319749, WO 9319751, WO 9325517, WO 9402465, WO 9406423, WO 9412461, WO 9420455, WO 9422852, WO 9425437, WO 9427947, WO 9500516, WO 9501980, WO 9503794, WO 9504045, WO 9504046, WO 9505386, WO 9508534, WO 9509623, WO 9509624, WO 9509627, WO 9509836, WO 9514667, WO 9514680, WO 9514681, WO 9517392, WO 9517399, WO 9519362, WO 9522520, WO 9524381, WO 9527692, WO 9528926, WO 9535281, WO 9535282, WO 9600218, WO 9601825, WO 9602541, WO 9611917, WO 9307124, WO 9501338 и WO 9603399 и заявка на патент США №2005/0004222 (включая соединения, описанные в формулах I-XIII и параграфах 37-39, 85-0545 и 557-577), каждая из которых полностью включена в настоящую заявку посредством ссылки.
Примеры ингибиторов PDE5 включают RX-RA-69, SCH-51866, KT-734, веснаринон, запринаст, SKF-96231, ER-21355, BF/GP-385, NM-702 и силденафил (Viagra®). Примеры ингибиторов PDE4 включают RO-20-1724, MEM 1414 (R1533/R1500; Pharmacia Roche), денбуфиллин, ролипрам, оксагрелат, нитраквазон, Y-590, DH-6471, SKF-94120, мотапизон, ликсазинон, индолидан, олпринон, атизорам, KS-506-G, дипамфиллин, BMY-43351, атизорам, арофиллин, филаминаст, PDB-093, UCB-29646, CDP-840, SKF-107806, пикламиласт, RS-17597, RS-25344-000, SB-207499, тибенеласт, SB-210667, SB-211572, SB-211600, SB-212066, SB-212179, GW-3600, CDP-840, мопидамол, анагрелид, ибудиласт, амринон, пимобендан, цилостазол, квазинон и N-(3,5-дихлорпирид-4-ил)-3-циклопропилметокси-4-дифторметоксибензамид. Примеры ингибиторов PDE3 включают сульмазол, ампизон, цилостамид, карбазеран, пироксимон, имазодан, CI-930, сигвазодан, адибендан, сатеринон, SKF-95654, SDZ-MKS-492, 349-U-85, эморадан, EMD-53998, EMD-57033, NSP-306, NSP-307, ревизинон, NM-702, WIN-62582 и WIN-63291, эноксимон и милринон. Примеры ингибиторов PDE3/4 включают бенефентрин, трехинизин, ORG-30029, задраверин, L-686398, SDZ-ISQ-844, ORG-20241, EMD-54622 и толафентрин. Другие примеры ингибиторов PDE включают циломиласт, пентоксифиллин, рофлумиласт, тадалафил (Cialis®), теофиллин, варденафил (Levitra®) и запринаст (специфичный в отношении PDE5).
Согласно конкретным аспектам, ингибиторы фосфодиэстераз можно использовать в комбинации с классами соединений, способных повышать уровень cAMP, cGMP, кальция или других вторичных посредников в апикальных клетках слизистой оболочки желудочно-кишечного тракта.
О. Агонисты DRA (SLC26A3)
Согласно конкретным вариантам реализации изобретения, соединение представляет собой агонист антипортера хлорид/бикарбоната SLC26A3, также называемый DRA (Down-Regulated in Adenoma). Одной неограничивающей функцией DRA в кишечнике является всасывание хлорида из просвета кишки и секреция ионов бикарбоната. Предполагается, что фармакологическая стимуляция DRA приводит к снижению pHi, например, путем повышения рН UWL, и обеспечивает снижение эффекта повышенного содержания фосфатов, как описано в настоящей заявке.
Примеры агонистов DRA включают лизофосфатидиловую кислоту (LPA) и структурно родственные соединения. Считается, что указанный класс соединений действует на активность DRA посредством стимуляции сигнального пути рецептора LPA (например LPA2) через путь Pi3K/AKT, который, как считается, не только активирует транскрипцию гена DRA, но также повышает аккумуляцию поверхностного DRA (Singia et al. Am. J. Physiol Gastro-intest. Liver Physiol. 298: G182-G189, 2010; Singia et al. Am. J. Physiol. Gastro-intest. Liver Physiol. 302: G618-G627, 2012). Примеры соединений, родственных LPA, играющих потенциальную роль в стимуляции DRA, описаны в источниках Jiang et al., Bioorg. Med. Chem. Lett. 23: 1865-1869, 2013; Kiss et al., Molecular Pharmacology 82: 1162-1173, 2012; Kozian et al., Bioorg. Med. Chem. Lett. 22: 5239-5243, 2012; Parrill, Expert. Opin. Ther. Pat. 21: 281-286, 2011; Gupte et al., Bioorg. Med. Chem. Lett. 20: 7525-7528, 2010; Liliom et al., Biochim. Biophys. Acta 1761: 1506-1514, 2006; и Durgam et al., Journal of Medicinal Chemistry 48: 4919-4930, 2005.
В соответствии с одной неограничивающей теорией, ингибиторы протеинкиназы С также могут повышать активность DRA и подобным образом создавать трансэпителиальный градиент рН. Например, было показано, что форбол-12-миристат-13-ацетат (РМА), in vitro агонист РКС, непосредственно подавляет активность канала Cl-/НСО3- на апикальной мембране (Gill et al., Physiology of the Gastro-Intestinal Tract, Chapter 67, 2012). Без ограничения каким-либо одним механизмом, подавление соответствующих изоформ PKC может, напротив, приводить к повышению активности каналов Cl-/НСО3- и, таким образом, подавлять поглощения фосфатов с помощью механизмов, описанных согласно изобретению.
На Фигурах 21А-В (Mochly-Rosen et al., Nature Reviews Drug discovery 11, 937-957, 2012) показаны типичные примеры селективных в отношении подтипов ингибиторов РКС, обладающих потенциальной способностью повышать активность Cl-/НСО3-, среди прочих потенциальных механизмов действия. Другие потенциальные агонисты DRA включают полностью транс-ретиноевую кислоту (ATRA) и родственные соединения, более общие соединения, активирующие рецепторы ретиноевой кислоты (RAR) α, β и γ, предпочтительно RAR-β. Считается, что агонисты RAR-β стимулируют DRA на транскрипционном уровне (полностью транс-ретиноевая кислота повышает экспрессию SLC26A3 (DRA) через HNF-1 (Priyamvada et al., DDW 2013, Орландо). Другой пример соединения представляет собой S20787, который, как было показано, стимулирует активность человеческого DRA, экспрессируемого в ооцитах (Chernova et al., J. Physiol., 549, 1, 3-19, 2003). Агонисты рецептора нейропептида Y1 и Y2 стимулируют активность DRA в монослойных культурах клеток Caco2. Было обнаружено, что стимуляция DRA с помощью NPY не зависит от переноса мембран и связана с солокализацией DRA с липидными рафтами (Saksena et al. Am. J. Physiol Gastro-intest Liver Physiol. 299: G1334-G1343, 2010). Примеры типичных агонистов NPY1 и NPY2 включают NPY, [Leu31, Pro34]-NPY, NPY 13-36, пептид YY (3-36) и GR 231118.
II. По существу не обладающие системной биодоступностью соединения
А. Физические свойства и эффективность соединений, локализующихся в ЖКТ
Конкретные соединения, описанные в настоящей заявке, созданы таким образом, чтобы они являлись по существу активным или локализовались в просвете желудочно-кишечного тракта субъекта, представляющего собой человека или животное. Термин «просвет желудочно-кишечного тракта» используется взаимозаменяемо в настоящей заявке с термином «просвет» и относится к пространству или полости в пределах желудочно-кишечного тракта (ЖКТ, который может также называться пищеварительным трактом), ограниченному апикальной мембраной эпителиальных клеток ЖКТ субъекта. Согласно некоторым вариантам реализации изобретения, соединения не всасываются через слой клеток эпителия ЖКТ (также называемый эпителием ЖКТ). «Слизистая оболочка желудочно-кишечного тракта» относится к слою (слоям) клеток, отделяющих просвет желудочно-кишечного тракта от остального тела, и включает слизистую оболочку желудка и кишечника, такую как слизистая оболочка тонкого кишечника. «Эпителиальная клетка желудочно-кишечного тракта» или «эпителиальная клетка пищеварительного тракта» при использовании в настоящей заявке относится к любой эпителиальной клетке на поверхности слизистой оболочки желудочно-кишечного тракта, которая находится со стороны просвета желудочно-кишечного тракта, включая, например, эпителиальную клетку желудка, эпителиальную клетку желудка, эпителиальную клетку толстой кишки и т.д.
Фразы «по существу не обладающий системной биодоступностью» и/или «по существу не проникающий» (а также их варианты) при использовании в настоящей заявке в целом относятся к ситуациям, в которых статистически значимое количество, а согласно некоторым вариантам реализации изобретения, по существу все соединение, остается в просвете желудочно-кишечного тракта. Например, в соответствии с одним или более вариантами реализации настоящего изобретения, предпочтительно по меньшей мере примерно 60%, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90%, примерно 95%, примерно 96%, примерно 97%, примерно 98%, примерно 99% или даже примерно 99,5% соединения остается в просвете желудочно-кишечного тракта. В таких случаях локализация в просвете желудочно-кишечного тракта относится к сниженному суммарному движению соединения через слой клеток эпителия желудочно-кишечного тракта, например, путем как транцеллюлярного, так и парацеллюлярного транспорта, а также путем активного и/или пассивного транспорта. Согласно таким вариантам реализации изобертения, наблюдается ограниченное суммарное проникновение соединения через слой эпителиальных клеток желудочно-кишечного тракта путем трансцеллюлярного транспорта, например, через апикальную мембрану эпителиальной клетки тонкого кишечника. Согласно указанным вариантам реализации изобретения, также ограничено суммарное проникновение соединения через «плотные контакты» эпителиальных клеток, выстилающих просвет желудочно-кишечного тракта путем парацеллюлярного транспорта.
В этом отношении необходимо отметить, что, согласно одному конкретному варианту реализации изобретения, соединение по существу совсем не всасывается в ЖКТ или просвете желудочно-кишечного тракта При использовании в настоящей заявке, термины «по существу не проникающий» или «по существу не обладающий системной биодоступностью» включают варианты реализации, согласно которым с помощью способов, общеизвестных в данной области техники, не наблюдается всасывания или проникновения или системного воздействия поддающегося выявлению количества соединения.
В этом отношении также необходимо отметить, однако, что согласно альтернативным вариантам реализации изобретения, термин «по существу не проникающие» или «по существу не обладающие системной биодоступностью» означает или допускает некоторое ограниченное всасывание в ЖКТ, и более конкретно, в эпителии пищеварительной системы (например, всасывание некоторого поддающегося выявлению количества, такого как, например, по меньшей мере примерно 0,1%, 0,5%, 1% или более и менее чем примерно 30%, 20%, 10%, 5% и т.д, где диапазон уровня всасывания составляет, например, между примерно 1% и 30% или 5% и 20% и т.д); другими словами, «по существу не проникающее» или «по существу не обладающее системной биодоступностью» может относиться к соединениям, которые обладают некоторой поддающейся выявлению проникающей способностью через слой эпителиальных клеток в ЖКТ в количестве, составляющем менее чем примерно 20% от вводимого соединения (например, менее чем примерно 15%, примерно 10% или даже примерно 5%, 4%, 3% или 2%, и например, более чем примерно 0,5% или 1%), но затем вымываются печенью (т.е. путем печеночного выведения) и/или почками (т.е. путем почечного выведения).
В этом отношении следует также отметить, что согласно конкретным вариантам реализации изобретения, в связи с по существу отсутствием проницаемости и/или системной биодоступности соединений согласно настоящему изобретению более чем примерно 50%, 60%, 70%, 80%, 90% или 95% соединений согласно изобретению восстанавливается из фекалий в течение, например, 24, 36, 48, 60, 72, 84 или 96 часов после введения нуждающемуся в этом субъекту. В этом отношении необходимо понимать, что восстановленное соединение может включать смесь исходного соединения и его метаболитов, полученных из исходного соединения, например, в результате гидролиза, конъюгации, восстановления, оксиления, N-алкилирования, глюкуронидирования, ацетилирования, метилирования, сульфатирования, фосфорилирования или любой другой модификации, приводящей к добавлению или удалению атомов от исходного соединения, где указанные метаболиты образуются в результате действия любого фермента или воздействия любого физиологического окружения, включая, рН, температуру, давление или взаимодействие с продуктами питания, присутствующими в пищеварительном тракте.
Восстановление соединения и его метаболитов из фекалий можно измерять с использованием стандартных методов. Например, соединение можно вводить перорально в подходящей дозе (например, 10 мг/кг) и затем собирать фекалии через заранее определенное время после введения дозы (например, через 24 часа, 36 часов, 48 часов, 60 часов, 72 часа, 96 часов). Исходное соединение и его метаболиты можно экстрагировать органическим растворителем и проводить количественный анализ с использованием масс-спектрометрии. Анализ массового баланса исходного соединения и его метаболита (например, исходное соединение = М, метаболит 1 [М+16], и метаболит 2 [М+32]) можно использовать для определения процентного восстановления из фекалий.
(i) Проникающая способность
В этом отношении необходимо отметить, что согласно различным вариантам реализации изобретения, способность соединения по существу не обладать системной биодоступностью основана на заряде, размере и/или других физико-химических параметрах соединения (например, площади полярной поверхности, количестве на ней доноров и/или акцепторов водородных связей, количестве свободно вращающихся связей и т.д). Более конкретно, необходимо отметить, что соединение с определенным характером всасывания можно выбрать с применением принципов фармакокинетики, например, с помощью правила Липински, также известного как «правило пяти». Несмотря на то что это правило скорее представляет собой ряд руководств, Липински показал, что низкомолекулярные лекарственные средства с (i) молекулярной массой, (ii) количеством доноров водородных связей, (iii) количество акцепторов водородных связей и/или (iv) коэффициентом распределения вода/октанол (Log P Моригучи), превышающим конкретное пороговое значение, обычно не достигают значимой концентрации в системном кровотоке (т.е. в целом не всасываются до какого-либо значимого уровня) (см., например, источник Lipinski et al., Advanced Drug Delivery Reviews, 46: 3-26, 2001, включенный в настоящую заявку посредством ссылки). Соответственно, по существу не обладающие системной биодоступностью соединения могут быть созданы таким образом, что их молекулярные структуры превышают одно или более из пороговых значений Липински. (см. также источники Lipinski et al., Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug discovery and Development Settings, Adv. Drug Delivery Reviews, 46: 3-26, 2001; и Lipinski, Drug-like Properties and the Causes of Poor Solubility and Poor Permeability, J. Pharm. & Toxicol. Methods, 44: 235-249, 2000, которые включены в настоящую заявку посредством ссылки полностью.
Согласно некоторым вариантам реализации изобретения, например, соединение, которое по существу не проникает или по существу не обладает системной биодоступностью согласно настоящему изобретению, может быть создано таким образом, что оно обладает одной или более из следующих характеристик: (i) MW составляет более чем примерно 500 Да, примерно 600 Да, примерно 700 Да, примерно 800 Да, примерно 900 Да, примерно 1000 Да, примерно 1200 Да, примерно 1300 Да, примерно 1400 Да, примерно 1500 Да, примерно 1600 Да, примерно 1800 Да, примерно 2000 Да, примерно 2500 Да, примерно 3000 Да, примерно 4000 Да, примерно 5000 Да, примерно 7500 Да, примерно 10,000 Да или более (не для формы соли соединения); (ii) обще число групп NH и/или ОН и/или других потенциальных доноров водородной связи составляет более чем примерно 5, примерно 6, примерно 7, примерно 8, примерно 9, примерно 10, примерно 11, примерно 12, примерно 13, примерно 14, примерно 15, примерно 20 или более; (iii) общее число атомов О и/или атомов N и/или других потенциальных акцепторов водородных связей составляет более чем примерно 5, примерно 6, примерно 7, примерно 8, примерно 9, примерно 10, примерно 11, примерно 12, примерно 13, примерно 14, примерно 15, примерно 20 или более; (iv) коэффициент распределения Моригучи составляет более чем примерно 105 (т.е. Log P составляет более чем примерно 5, примерно 6, примерно 7, примерно 8, примерно 9, примерно 10 и т.д.) или, альтернативно, менее чем примерно 10 (т.е. Log Р составляет менее чем 1 или даже 0); и/или (v) общее число вращаемых связей составляет более чем примерно 5, примерно 10 или примерно 15 или более. Согласно конкретным вариантам реализации изобретения, Log P соединения не составляет 14 или составляет менее чем примерно 14, например, Log P находится в диапазоне примерно 6-7, 6-8, 6-9, 6-10, 6-11, 6-12, 6-13, 7-8, 7-9, 7-10, 7-11, 7-12, 7-13, 8-9, 8-10, 8-11, 8-12, 8-13, 9-10, 9-11, 9-12, 9-13, 10-11, 10-12, 10-13, 11-12, 11-13 или 12-13.
Помимо параметров, отмеченных выше, область поверхности полярных молекул (т.е. «PSA»), которая может характеризоваться как поверхность, принадлежащая полярным атомам, представляет собой характеристику, которая, как было показано, также хорошо коррелирует с пассивным транспортом через мембраны и, таким образом, позволяет предсказывать свойства транспорта лекарственных средств. Эта характеристика успешно применялось для предсказания кишечного всасывания и проницаемости монослоя клеток Сасо2. Пример подробного исследования проницаемости монослоя клеток Сасо2 см., например, в описании модели Сасо2, предложенной в патенте США №6,737,423, включенном в настоящую заявку посредством ссылки, в частности, текст, описывающий модель Сасо2, которую можно применять, например, для оценки или исследования соединений согласно настоящему изобретению. PSA выражается в  2 (квадратных ангстремах) и вычисляется из трехмерного молекулярного изображения. Также доступен быстрый способ расчета (см., например, Ertl et al., Journal of Medicinal Chem. 43: 3714-3717, 2000, полное содержание которого включено в настоящую заявку посредством ссылки во всех уместных и соответствующих целях) с помощью настольного компьютера и коммерчески доступных наборов графических инструментов для химических соединений, таких как ChemDraw. Для этого способа быстрого расчета был предложен термин «топологическая PSA» (tPSA). tPSA хорошо коррелирует с данными всасывания у человека обычных лекарственных средств (см. Таблицу 1, Ertl et al., J. Med. Chem. 43: 3714-3717, 2000):
2 (квадратных ангстремах) и вычисляется из трехмерного молекулярного изображения. Также доступен быстрый способ расчета (см., например, Ertl et al., Journal of Medicinal Chem. 43: 3714-3717, 2000, полное содержание которого включено в настоящую заявку посредством ссылки во всех уместных и соответствующих целях) с помощью настольного компьютера и коммерчески доступных наборов графических инструментов для химических соединений, таких как ChemDraw. Для этого способа быстрого расчета был предложен термин «топологическая PSA» (tPSA). tPSA хорошо коррелирует с данными всасывания у человека обычных лекарственных средств (см. Таблицу 1, Ertl et al., J. Med. Chem. 43: 3714-3717, 2000):
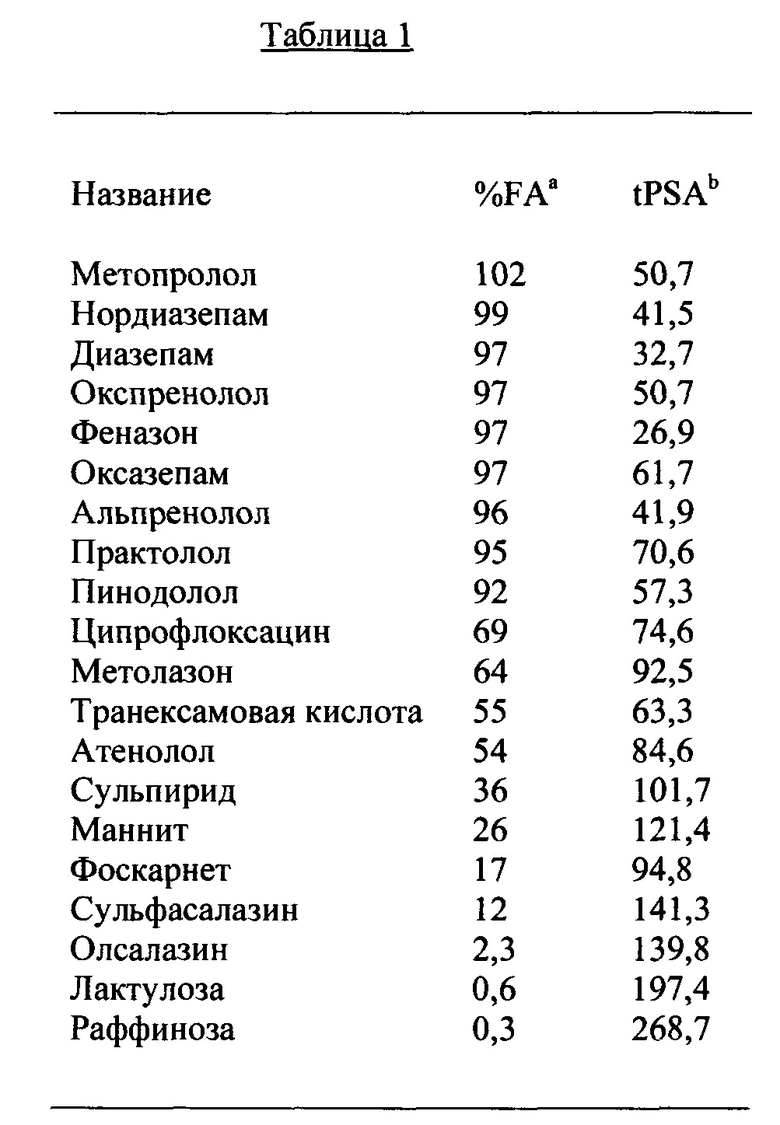
Таким образом, согласно некоторым вариантам реализации изобретения, соединения согласно настоящему изобретению могут быть получены таким образом, что их значение tPSA составляет более чем примерно 100 2, примерно 116 2, примерно 120 2, примерно 130 2 или примерно 140 2, и в некоторых примерах примерно 150 2, примерно 160 2, примерно 170 2, примерно 180 2, примерно 190 2, примерно 200 2, примерно 225 2, примерно 250 2, примерно 270 2, примерно 300 2, примерно 350 2, примерно 400 2, примерно 450 2, примерно 500 2, примерно 750 2 или даже примерно 1000 2 или в диапазоне примерно 100-120 2, 100-130 2, 100-140 2, 100-150 2, 100-160 2, 100-170 2, 100-170 2, 100-190 2, 100-200 2, 100-225 2, 100-250 2, 100-300 2, 100-400 2, 100-500 2, 100-750 2, 100-1000 2, 116-120 2, 116-130 2, 116-140 2, 116-150 2, 116-160 2, 116-170 2, 116-170 2, 116-190 2, 116-200 2, 116-225 2, 116-250 2, 116-300 2, 116-400 2, 116-500 2, 116-750 2, 116-1000 2, 120-130 2, 120-140 2, 120-150 2, 120-160 2, 120-170 2, 120-170 2, 120-190 2, 120-200 2, 120-225 2, 120-250 2, 120-300 2, 120-400 2, 120-500 2, 120-750 2, 120-1000 2, 130-140 2, 130-150 2, 130-160 2, 130-170 2, 130-170 2, 130-190 2, 130-200 2, 130-225 2, 130-250 2, 130-300 2, 130-400 2, 130-500 2, 130-750 2, 130-1000 2, 140-150 2, 140-160 2, 140-170 2, 140-170 2, 140-190 2, 140-200 2, 140-225 2, 140-250 2, 140-300 2, 140-400 2, 140-500 2, 140-750 2, 140-1000 2, 150-160 2, 150-170 2, 150-170 2, 150-190 2, 150-200 2, 150-225 2 или 150-250 2, 150-300 2, 150-400 2, 150-500 2, 150-750 2, 150-1000 2, 200-250 2, 200-300 2, 200-400 2, 200-500 2, 200-750 2, 200-1000 2, 250-250 2, 250-300 2, 250-400 2, 20-500 2, 250-750 2 или 250-1000 2, так что соединения по существу не проникают (например, не проникают через клетку) или по существу не обладают системной биодоступностью (как определено в другом месте в настоящей заявке).
Поскольку существуют исключения из «правила Липински», или модели tPSA, для определения проникающей способности соединений согласно настоящему изобретению можно проводить экспериментальный скрининг. Коэффициент проникновения можно определить с помощью способов, известных специалистам в данной области техники, включая, например, анализ проникновения на клетках Сасо-2 и/или с использованием искусственной мембраны в качестве модели эпителиальной клетки желудочно-кишечного тракта. Синтетическая мембрана, пропитанная, например, лецитином и/или додеканом для имитации характеристик суммарной проницаемости слизистой оболочки желудочно-кишечного тракта можно использовать в качестве модели слизистой оболочки желудочно-кишечного тракта. Мембрану можно использовать для отделения компартмента, содержащего соединение согласно настоящему изобретению, от компартмента для оценки скорости проникновения. Также можно проводить анализы проникновения через параллельные искусственные мембраны (РАМРА). Такие измерения in vitro могут являться надежными показателями действительной проницаемости in vivo (см. Wohnsland et al., J. Med. Chem. 44: 923-930, 2001; Schmidt et al., Millipore Corp. Application Note, 2002, n AN1725EN00, и n AN1728EN00, включенные в настоящую заявку посредством ссылки).
Соответственно, согласно некоторым вариантам реализации изобретения, соединения, используемые в способах согласно настоящему изобретению, могут иметь коэффициент проникновения, Рapp, составляющий менее чем примерно 100×10-6 см/с или менее чем примерно 10×10-6 см/с или менее чем примерно 1×10-6 см/с или менее чем примерно 0,1×10-6 см/с, при измерении с помощью способа, известного в данной области техники (такого как, например, исследование проникающей способности, описанное в источнике Wohnsland et al., 2001, выше).
Как отмечалось ранее, в соответствии с настоящим изобретением, соединения можно модифицировать таким образом, чтобы предотвратить их общее всасывания через слой эпителиальных клеток кишечника, по существу с предотвращением их системной биодоступности. Согласно некоторым конкретным вариантам реализации, соединения согласно настоящему изобретению включают соединение, которое связано, сопряжено или другом образом присоединено к не всасываемой молекуле, которая может представлять собой олигомерный фрагмент, полимерный фрагмент, гидрофобный фрагмент, гидрофильный фрагмент и/или заряженный фрагмент, который обеспечивает по существу отсутствие проникновения или по существу отсутствие системной биодоступности целого соединения. Согласно некоторым предпочтительным вариантам реализации изобертения, соединение объединено с мультимерной или полимерной частью или фрагментом, так что полученная молекула по существу не проникает или по существу не обладает системной биодоступностью. Молекулярная масса мультимерной или полимерной части или фрагмента может составлять более чем примерно 500 Дальтон (Да), примерно 1000 Да, примерно 2500 Да, примерно 5000 Да, примерно 10,000 Да или более, и в частности, может находиться в диапазоне от примерно 1000 Дальтон (Да) до примерно 500,000 Да, предпочтительно в диапазоне от примерно 5000 до примерно 200,000 Да, более предпочтительно может является достаточно высокой для предотвращения по существу любого общего всасывания соединения через слой кишечных эпителиальных клеток. Согласно указанным или другим конкретным вариантам реализации изобретения, соединение модифицировано таким образом, что его общее всасывание через слой кишечных эпителиальные клетки предотвращается.
(ii) Cmax и IC50 или ЕС50
Согласно некоторым вариантам реализации изобретения, максимальная концентрация, выявляемая в сыворотке, по существу не обладающих системной биодоступностью соединений, подробно описанных в настоящей заявке, которая определяется как Cmax, при их введении (например, кишечном введении) отдельно или в комбинации с одним или более дополнительными фармацевтически активными соединениями или агентами нуждающемуся в этом субъекту примерно равна или составляет менее чем ингибирующая концентрация IC50 указанных соединений для транспорта или захвата фосфат-ионов (Pi). Согласно некоторым вариантам реализации изобретения, например, Cmax примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100% меньше чем IC50 для подавления транспорта или поглощения Pi. Согласно некоторым вариантам реализации изобретения, Cmax примерно в 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 раз превышает IC50 для подавления транспорта или поглощения Pi.
Согласно конкретным вариантам реализации изобретения, отношение Cmax : IC50 (для подавления транспорта или захвата Pi) для одного или более из по существу не обладающих системной биодоступностью соединений, подробно описанных в настоящей заявке, при их введении (например, кишечном введении) нуждающемуся в этом субъекту может составлять примерно или менее чем примерно 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 или 1,0 или находиться в диапазоне между примерно 0,01-1,0, 0,01-0,9, 0,01-0,8, 0,01-0,7, 0,01-0,6, 0,01-0,5, 0,01-0,4, 0,01-0,3, 0,01-0,2 или 0,01-0,1 или в диапазоне между примерно 0,1-1,0, 0,1-0,9, 0,1-0,8, 0,1-0,7, 0,1-0,6, 0,1-0,5, 0,1-0,4, 0,1-0,3 или 0,1-0,2, где Cmax и IC50 выражают в одинаковых единицах.
Согласно некоторым вариантам реализации изобретения, максимальная концентрация, выявляемая в сыворотке, по существу не обладающих системной биодоступностью соединений, подробно описанных в настоящей заявке, которая определяется как Cmax, при их введении (например, кишечном введении) отдельно или в комбинации с одним или более дополнительными фармацевтически активными соединениями или агентами нуждающемуся в этом субъекту примерно равна или составляет менее чем ЕС50 указанных соединений для повышения выведения фосфатов с фекалиями, где выход с фекалиями повышен примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100%. Согласно некоторым вариантам реализации изобретения, например, Cmax примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100% меньше чем ЕС50 для повышения выведения фосфатов с фекалиями. Согласно некоторым вариантам реализации изобретения, Cmax примерно в 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 раз превышает EC50 для повышения выведения фосфатов с фекалиями.
Согласно некоторым вариантам реализации изобретения, EC50 одного или более из по существу не обладающих системной биодоступностью соединений, подробно описанных в настоящей заявке, для повышения выведения фосфатов с фекалиями при введении (например, кишечном введении) отдельно или в комбинации с одним или более дополнительными фармацевтически активными соединениями или агентами нуждающемуся в этом субъекту или при измерении на животной модели или в клеточном анализе, может составлять примерно или менее чем примерно 10 мкМ, 9 мкМ, 8 мкМ, 7 мкМ, 7,5 мкМ, 6 мкМ, 5 мкМ, 4 мкМ, 3 мкМ, 2,5 мкМ, 2 мкМ, 1 мкМ, 0,5 мкМ, 0,1 мкМ, 0,05 мкМ или 0,01 мкМ или менее, где IC50 находится, например, в диапазоне, составляющем от примерно 0,01 мкМ до примерно 10 мкМ или от примерно 0,01 мкМ до примерно 7,5 мкМ или от примерно 0,01 мкМ до примерно 5 мкМ или от примерно 0,01 мкМ до примерно 2,5 мкМ или от примерно 0,01 мкМ до примерно 1,0 или от примерно 0,1 мкМ до примерно 10 мкМ или от примерно 0,1 мкМ до примерно 7,5 мкМ или от примерно 0,1 мкМ до примерно 5 мкМ или от примерно 0,1 мкМ до примерно 2,5 мкМ или от примерно 0,1 мкМ до примерно 1,0 или от примерно мкМ 0,5 мкМ до примерно 10 мкМ или от примерно 0,5 мкМ до примерно 7,5 мкМ или от примерно 0,5 мкМ до примерно 5 мкМ или от примерно 0,5 мкМ до примерно 2,5 мкМ или от примерно 0,5 мкМ до примерно 1,0 мкМ.
Согласно конкретным вариантам реализации изобретения, максимальная концентрация, выявляемая в сыворотке, по существу не обладающих системной биодоступностью соединений, подробно описанных в настоящей заявке, которая определяется как Cmax, при их введении (например, кишечном введении) отдельно или в комбинации с одним или более дополнительными фармацевтически активными соединениями или агентами нуждающемуся в этом субъекту примерно равна или составляет менее чем ЕС50 указанных соединений для снижения выведения фосфатов с мочой, где выведение с мочой снижается примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100%. Согласно некоторым вариантам реализации изобретения, например, Cmax примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100% меньше чем ЕС50 для снижения выведения фосфатов с мочой. Согласно некоторым вариантам реализации изобретения, Cmax примерно в 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 раз превышает ЕС50 для снижения выведения фосфатов с мочой.
Согласно некоторым вариантам реализации изобретения, ЕС50 одного или более из по существу не обладающих системной биодоступностью соединений, подробно описанных в настоящей заявке, для снижения выведения фосфатов с мочой при введении (например, кишечном введении) отдельно или в комбинации с одним или более дополнительными фармацевтически активными соединениями или агентами нуждающемуся в этом субъекту или при измерении на животной модели или в клеточном анализе может составлять примерно или менее чем примерно 10 мкМ, 9 мкМ, 8 мкМ, 7 мкМ, 7,5 мкМ, 6 мкМ, 5 мкМ, 4 мкМ, 3 мкМ, 2,5 мкМ, 2 мкМ, 1 мкМ, 0,5 мкМ, 0,1 мкМ, 0,05 мкМ или 0,01 мкМ или менее, где IC50 находится, например, в диапазоне, составляющем от примерно 0,01 мкМ до примерно 10 мкМ или от примерно 0,01 мкМ до примерно 7,5 мкМ или от примерно 0,01 мкМ до примерно 5 мкМ или от примерно 0,01 мкМ до примерно 2,5 мкМ или от примерно 0,01 мкМ до примерно 1,0 или от примерно 0,1 мкМ до примерно 10 мкМ или от примерно 0,1 мкМ до примерно 7,5 мкМ или от примерно 0,1 мкМ до примерно 5 мкМ или от примерно 0,1 мкМ до примерно 2,5 мкМ или от примерно 0,1 мкМ до примерно 1,0 или от примерно мкМ 0,5 мкМ до примерно 10 мкМ или от примерно 0,5 мкМ до примерно 7,5 мкМ или от примерно 0,5 мкМ до примерно 5 мкМ или от примерно 0,5 мкМ до примерно 2,5 мкМ или от примерно 0,5 мкМ до примерно 1,0 мкМ.
Согласно конкретным вариантам реализации изобретения, отношение Cmax : ЕС50 (например, для повышения выведения фосфатов с фекалиями, для снижения выведения фосфатов с мочой) одного или более из по существу не обладающих системной биодоступностью соединений, подробно описанных в настоящей заявке, при введении (например, кишечном введении) нуждающемуся в этом субъекту может составлять примерно или менее чем примерно 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 или 1,0 или находиться в диапазоне между примерно 0,01-1,0, 0,01-0,9, 0,01-0,8, 0,01-0,7, 0,01-0,6, 0,01-0,5, 0,01-0,4, 0,01-0,3, 0,01-0,2 или 0,01-0,1 или в диапазоне между примерно 0,1-1,0, 0,1-0,9, 0,1-0,8, 0,1-0,7, 0,1-0,6, 0,1-0,5, 0,1-0,4, 0,1-0,3 или 0,1-0,2, где Cmax и ЕС50 выражают в одних и тех же единицах.
Дополнительно или альтернативно, Cmax одного или более из по существу не обладающих системной биодоступностью соединений, подробно описанных в настоящей заявке, при введении (например, кишечном введении) отдельно или в комбинации с одним или более дополнительными фармацевтически активным соединениями или агентами нуждающемуся в этом субъекту может составлять примерно или менее чем примерно 10 нг/мл, примерно 7,5 нг/мл, примерно 5 нг/мл, примерно 2,5 нг/мл, примерно 1 нг/мл или примерно 0,5 нг/мл, где указанная Cmax находится, например, в диапазоне, составляющем от примерно 1 нг/мл до примерно 10 нг/мл или от примерно 2,5 нг/мл до примерно 7,5 нг/мл.
III. Фармацевтические композиции и способы лечения
Соединения согласно настоящему изобретению для введения пациенту или субъекту могут быть представлены в виде необработанного химического вещества или в виде фармацевтических композиций. Фармацевтические композиции согласно настоящему изобретению обычно содержат соединения согласно изобретению и фармацевтически приемлемый носитель, разбавитель или наполнитель. Соединение присутствует в композиции в количестве, которое является эффективным для лечения конкретного интересующего заболевания или состояния, как описано в настоящей заявке, и предпочтительно обладает приемлемой токсичностью для субъекта. Специалист в данной области техники может определить активность соединения (соединений), например, как описано в настоящей заявке и в приведенных ниже Примерах. Специалист в данной области техники легко определить подходящие концентрации и дозы.
Соединение или композицию согласно изобретению можно использовать в способе лечения по существу любого заболевания или другого состояния у субъекта, который может иметь благоприятный эффект от подавления поглощения фосфатов в желудочно-кишечном тракте.
Например, в качестве неограничивающего объяснения, повреждение почек приводит к снижению продукции и активности почечной 1-альфа-гидроксилазы, приводя к более низкому уровню 1,25-дигидроксивитамина D. Сниженный уровень витамина D ограничивает всасывание кальция в желудочно-кишечном тракте, приводя к снижению уровня кальция в сыворотке. Более низкий уровень 1,25-дигидроксивитамина D в комбинации с более низким уровнем кальция в сыворотке синергично стимулирует выработку и секрецию ПТГ в ткани паращитовидной железы. Потеря нефронов также нарушает выведение Pi, но уровень Р в сыворотке активно поддерживается действием ПТГ и FGF-23 и более высоким содержанием Р в сыворотке, что значительно усиливает выведение с мочой PO4. Однако действие ПТГ и FGF-23 на тубулярные отделы нефронов не может поддерживать уровень Р в сыворотке из-за непрерывной потери нефронов. При прогрессировании почечной недостаточности до потери почечной функции примерно на 40-50% уменьшение объема функционирующей почечной ткани не позволяет выведение полного количества потребленных фосфатов, необходимое для поддержания гомеостаза. В результате развивается гиперфосфатемия. Кроме того, повышение уровня Р в сыворотке препятствует активности почечной 1-альфагидроксилазы, также подавляя уровень активированного витамина D и стимулируя продукцию ПТГ, что приводит к вторичному гиперпаратиреозу (вГПТ).
Однако нарушенный баланс фосфора не обязательно приравнивается к гиперфосфатемии. Скорее у подавляющего большинства пациентов, страдающих ХЗП, но еще не подвергающихся диализу, наблюдается нормальное содержание фосфатов, но положительный баланс фосфора, где избыточный фосфор откладывается в сосудистой системе в виде эктопической кальцификации, например, кальцификации во внутренней оболочке сосудов. У пациентов, страдающих ХЗП, наблюдается клинически повышенный уровень FGF-23, который сильно связан с ухудшением почечной функции и со сниженным уровнем кальцитриола. Было предположено, что синтез FGF-23 вызван присутствием избыточного Р в теле с последующий почечной недостаточностью.
Более того, эффект фосфатемии после приема пищи, т.е. резкого повышение Р в сыворотке, вторичного по отношению к приему пищи, на сердечно-сосудистые заболевания, не изучен. Также проводились исследования эффекта перегрузки фосфором на функцию эндотелия in vitro и in vivo. Подвержение клеток эндотелия аорты быка перегрузкой фосфора приводило к повышению продукцию активных форм кислорода и снижению содержания оксида азота, известного расширяющего сосуды агента. В описанном выше исследовании острой перегрузки Р у здоровых добровольцев была показана обратная корреляция между опосредованным потоком оксида азота расширением и уровнем Р в сыворотке после еды (см., например, Shuto et al., J. Am. Soc. Nephrol. 20: 1504-12, 2009).
Таким образом, согласно конкретным вариантам реализации изобретения, соединение или композицию согласно изобретению можно использовать в способе, выбранном из одного или более из следующих способов: способ лечения гиперфосфатемии, необязательно гиперфосфатемии после приема пищи; способ лечения заболевания почек (например, хронического заболевания почек (ХЗП), конечной стадии заболевания почек (КСЗП)); способ снижения уровня креатинина в сыворотке; способ лечения протеинурии; способ отсрочки заместительной почечной терапии (ЗПТ), такой как диализ; способ снижения уровня FGF23; способ снижения гиперфосфатемического эффекта активного витамина D; способ ослабления гиперпаратиреоза, такого как вторичный гиперпаратиреоза; способ снижения уровня паратиреоидного гормона (ПТГ или иПТГ) в сыворотке; способ улучшения нарушенной функции эндотелия, необязательно вызванной содержанием фосфора в сыворотке после приема пищи; способ снижения кальцификации сосудов или ослабления кальцификации во внутренней оболочке сосудов; способ снижения уровня фосфора в моче; способ нормализации уровня фосфора в сыворотке; способ снижения отложения фосфатов у пожилых пациентов; способ снижения поглощения фосфора из пищи; способ снижения всасывания кальция после приема пищи; способ снижения почечной гипертрофии и способ снижения гипертрофии сердца. Согласно конкретным вариантам реализации изобретения, субъект, нуждающийся в снижении содержания фосфатов, страдает одним или более из указанных выше состояний. Согласно некоторым вариантам реализации изобретения, способ включает выбор или идентификацию такого субъекта до начала лечения, необязательно на основе одного или более из клинических или диагностических параметров, описанных в настоящей заявке.
Гиперфосфатемия относится к состоянию, при котором наблюдается повышенный уровень фосфатов в крови. Средняя масса фосфора в сыворотке у взрослого человека, как правило, варьирует от примерно 2,5 до 4,5 мг/дл (примерно 0,81-1,45 ммоль/л). Уровень фосфора часто примерно на 50% выше у младенцев и примерно на 30% выше у детей из-за эффекта гормона роста. Таким образом, конкретные способы включают лечения взрослого пациента, представляющего собой человека, страдающего гиперфосфатемией, где у указанного пациента масса фосфора в сыворотке составляет примерно или по меньшей мере примерно 4,5, 4,6, 4,7, 4,8, 4,9, 5,0, 5,1, 5,2, 5,3, 5,4 или 5,5 мг/дл. Согласно некоторым аспектам, лечение приводит к снижению концентрации или уровня фосфора в сыворотке у субъекта, страдающего гиперфосфатемией, до примерно 150%, 145%, 140%, 135%, 130%, 125%, 120%, 115%, 110%, 105% или 100% (нормированный уровень) от нормального уровня фосфора в сыворотке (например, 2,5-4,5 мг/дл или 0,81-1,45 ммоль/л для взрослого). Согласно некоторым аспектам, схема лечения приводит к регуляции и/или включает регуляцию уровня фосфатов таким образом, что указанный уровень остается в диапазоне, составляющем примерно 2,5-4,5 мг/дл (примерно 0,81-1,45 ммоль/л). Согласно некоторым аспектам, лечение приводит к сдвигу баланса поверхностного фосфора относительного суммарного выведения, например, путем повышения суммарного выведения фосфора примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100% или более по сравнению с состоянием в отсутствие лечения со снижением или без снижения концентрации или уровня фосфора в сыворотке.
Также включены способы лечения пациента, представляющего собой ребенка или подростка, где у указанного пациента массу фосфора в сыворотке составляет примерно или по меньшей мере примерно 6,0, 6,1, 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8, 6,9, 7,0, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, 7,9 или 8,0 мг/дл. Как отмечено в настоящей заявке, согласно указанным и связанным вариантам реализации изобретения, введение соединений или композиций, описанных в настоящей заявке, может приводить к снижению массы фосфора в сыворотке у субъекта примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200% или более.
Конкретные варианты реализации изобертения относятся к способам лечения хронического заболевания почек (ХЗП) - состояния, характеризующегося прогрессирующей потерей функции почек. Общие причины ХЗП включают сахарный диабет, гипертонию и гломерулонефрит. Таким образом, конкретные способы включают лечение субъекта, страдающего ХЗП, где указанный субъект необязательно также страдает одним или более из указанных выше состояний.
Согласно некоторым аспектам, субъектов идентифицируют как страдающих ХЗП, если скорость клубочковой фильтрации (GFR) у них составляет менее чем 60 мл/мин/1,73 м2 в течение примерно 3 месяцев с сопутствующим повреждением почек или без него. Конкретные способы, таким образом, включают лечение субъекта GFR которого (например, исходное GFR, до лечения) составляет примерно или менее чем примерно 60, 55, 50, 45, 40, 30, 35, 20, 25, 20, 15 или 10 мл/мин/1,73 м2 или т.п.Согласно конкретным вариантам реализации изобретения, введение соединений или композиций, описанных в настоящей заявке, может приводить к повышению GFR примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200% или более.
ХЗП чаще всего характеризуется в соответствии со стадией заболевания: 1 стадия, 2 стадия, 3 стадия, 4 стадия и 5 стадия. 1 стадия ХЗП включает субъектов с повреждением почек и нормальным или относительно высоким GFR, составляющим примерно или более чем примерно 90 мл/мин/1,73 м2. 2 стадия ХЗП включает субъектов, страдающих повреждением почек, GFR которых составляет примерно 60-89 мл/мин/1,73 м2. 3 стадия ХЗП включает субъектов, страдающих повреждением почек, GFR которых составляет примерно 30-59 мл/мин/1,73 м2. 4 стадия ХЗП включает субъектов, страдающих повреждением почек, GFR которых составляет примерно 15-29 мл/мин/1,73 м2. 5 стадия ХЗП включает субъектов, страдающих установленной почечной недостаточностью, GFR которых составляет менее чем примерно 15 мл/мин/1,73 м2. 5 стадия ХЗП также называется конечной стадией заболевания почек (КСЗП). Соответственно, в конкретных способах субъект страдает ХЗП на 1, 2, 3, 4 или 5 стадии и проявляет одну или более из связанных с ним клинических характеристик (например, определенный GFR, повреждение почек). Согласно некоторым вариантам реализации изобретения, субъект страдает КСЗП и проявляет одну или более из связанных с ним клинических характеристик, как описано в настоящей заявке и известно в данной области техники.
ХЗП может характеризоваться в соответствии с пораженными участками почек. Например, согласно конкретным аспектам, ХЗП включает ассоциированное с сосудами ХЗП, включая заболевания крупных сосудов, такие как двусторонний стеноз почечной артерии, и заболевания малых сосудов, такие как ишемическая нефропатия, гемолитический уремический синдром и васкулит. Согласно конкретным аспектам, ХЗП включает гломерулярно-ассоциированный ХЗП, включая первичные гломерулярные заболевания, такие как фокально-сегментарный гломерулосклероз и IgA-нефрит, и вторичные гломерулярные заболевания, такие как диабетическая нефропатия и волчаночный нефрит. Также включено тубулоинтерстициально-ассоциирвоанное ХЗП, включая полициститные заболевания почек, вызванный лекарственным средством и вызванный токсином хронический тубулоинтерстициальный нефрит и рефлюкс-нефропатию. Конкретные субъекты, подлежащие лечению ХЗП, могут, таким образом, обладать одной или более из указанных выше связанных с ХЗП характеристик.
Конкретные аспекты относятся к способам лечения субъекта, страдающего повреждением почек или одним или более симптомов/клинических признаков повреждения почек. Примеры повреждения почек (например, ХЗП-ассоциированное повреждение почек) и связанных с ним симптома включают патологические нарушения и маркеры повреждения, включая нарушения, идентифицируемые с помощью исследования крови (например, высокий уровень креатинина в крови или сыворотке, клиренс креатинина), исследований мочи (например, протеинурия) и/или визуализирующих исследований.
Креатинин представляет собой продукт распада креатининфосфатов в мышцах и является легко измеряемым и полезным показателем здорового состояния почек. Нормальное референсное значение диапазона креатинина в крови или сыворотке для человека варьирует от примерно 0,5 до 1,0 мг/дл (примерно 45-90 ммоль/л) для женщин и примерно от 0,7 до 1,2 мг/дл (примерно 60-110 мкмоль/л) для мужчин. Таким образом, у конкретных субъектов, подлежащих лечению в соответствии со способами, описанными в настоящей заявке, уровень креатинина в крови или сыворотке (например, изначально, до лечения) может составлять примерно или более чем примерно 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0 мг/дл. В указанных и связанных вариантах реализации изобретения, введение соединения или композиции, описанной в настоящей заявке может приводить к снижению общего уровня креатинина в крови или сыворотке у субъекта примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более.
Скорость клиренса креатинина (CCr или CrCl) относится к объему плазмы крови, который очищается от креатинина в единицу времени; его измеряют путем сравнения уровня креатинина в крови с уровнем креатина в моче в течение определенного периода времени (например, 24 часов). Клиренс креатинина часто измеряют в миллилитрах в минуту (мл/мин) или как функцию массы тела (мл/мин/кг). В зависимости от проводимого исследования, нормальные значения варьируют от примерно 97 до 137 мл/мин для мужчин и примерно от 88 до 128 мл/мин для женщин. Сниженный клиренс креатинина является важным признаком повреждения почек. Таким образом, у конкретных субъектов мужского пола, подлежащих лечению в соответствии со способами, описанными в настоящей заявке, CCr может составлять (например, изначально, до лечения) примерно или менее чем примерно 97, 96, 95, 94, 93, 92, 91, 90, 89, 88, 87, 86, 85, 84, 83, 82, 81, 80, 79, 78, 77, 76, 75, 74, 73, 72, 71, 70, 69, 68, 67, 66, 65, 64, 63, 62, 61, 60, 59, 58, 57, 56, 55, 54, 53, 52, 51, 50 или менее. У конкретных субъектов женского пола, подлежащих лечению в соответствии со способами, описанными в настоящей заявке, CCr (например, изначально, до лечения) может составлять примерно или менее чем примерно 88, 87, 86, 85, 84, 83, 82, 81, 80, 79, 78, 77, 76, 75, 74, 73, 72, 71, 70, 69, 68, 67, 66, 65, 64, 63, 62, 61, 60, 59, 58, 57, 56, 55, 54, 53, 52, 51, 50, 49, 47, 46, 45, 44, 43, 42, 41, 40 или менее. Согласно некоторым вариантам реализации изобретения, введение соединения или композиции, описанной в настоящей заявке, может проводить к поддержанию или повышению CCr у субъекта примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более.
Протеинурия относится к состоянию, характеризующемуся избытком белка в моче. Она связана с различными заболеваниями, включая повреждение почек. Протеинурия часто характеризуется как отношение белок/креатинин в моче, составляющее более чем примерно 45 мг/ммоль, или в конкретных исследованиях как отношение альбумин/креатин, составляющее более чем примерно 30 мг/ммоль. Конкретные субъекты, подлежащие лечению в соответствии со способами, предложенными в настоящей заявке (например, до лечения) страдают протеинурией, отдельно или в комбинации с ХЗП или другим повреждением почек, включая субъектов, у которых отношение белок/креатинин в моче составляет примерно или более чем примерно 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115 или 120 мг/ммоль, и/или отношение альбумин/креатинин в моче составляет примерно или более чем примерно 30, 35, 40, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115 или 120 мг/ммоль. В указанных и связанных вариантах реализации, введение соединения или композиции, описанной в настоящей заявке, может приводить к лечению протеинурии, например, путем снижения отношения белок/креатинин в моче и/или отношения альбумин/креатинин в моче примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более.
ХЗП связано с различными клиническими симптомами. Примеры включают высокое кровяное давление (гипертонию), накопление мочевины, гиперкалемию, анемию, гиперфосфатемию, гипокальцемию, метаболический ацидоз и атеросклероз. Таким образом, в конкретных способах, субъект, страдающий ХЗП, может также страдать или иметь риск развития одного или более из указанных ранее клинических симптомов. Согласно конкретным аспектам, субъект, страдающий ХЗП, страдает или имеет риск развития гиперфосфатемии, как описано в настоящей заявке.
Заместительная почечная терапия (ЗПТ) относится к различным способам жизнеобеспечивающего лечения при почечной недостаточности, включая способы, применимые на поздних стадиях ХЗП и КСЗП. Примеры ЗПТ включают диализ, гемодиализ, гемофильтрацию и почечную трансплантацию. Согласно конкретным вариантам реализации изобретения, субъект, подлежащий лечению в соответствии со способами, предложенными в настоящей заявке, будет подвергаться, подвергается или подвергался одному или более типов ЗПТ. Согласно некоторым вариантам реализации изобретения, субъект еще не подвергался ЗПТ, и введение соединения, описанного в настоящей заявке, обеспечивает отсрочку инициации ЗПТ (например, по сравнению с состоянием в отсутствие лечения) примерно или по меньшей мере примерно на 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 недель или примерно или по меньшей мере примерно на 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 месяцев или примерно или по меньшей мере примерно на 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 лет или более.
Фактор роста фибробластов 23 (FGF23) регулирует метаболизм фосфора и витамина D. Он также вызывает фосфатурию и снижает продукцию кальцитриола. Повышенный уровень FGF23 связан со смертностью, гипертрофией левого желудочка (или индексом массы левого желудочка), производительностью сердечной мышцы, нарушением функции эндотелия и прогрессированием ХЗП. В действительности уровень FGF23 постепенно повышается при раннем ХЗП, предположительно в результате физиологической адаптации к поддержанию нормального уровня фосфора в сыворотке или нормального баланса фосфора. Уровень FGF23 также может вносить вклад непосредственно в тканевое повреждение сердца, сосудов и почек. Конкретные варианты реализации изобретения, таким образом, относятся к лечению субъектов, имеющих повышенный уровень FGF23 в крови или сыворотке (см., например, Kirkpantur et al., Nephrol dial Transplant. 26: 1346-54, 2011), включая субъектов, страдающих ХЗП, и субъектов, подвергавшихся диализу/гемодиализу. Согласно некоторым аспектам, введение соединения или композиции, описанной в настоящей заявке, приводит к снижению логарифма уровня FGF23 в крови или сыворотке примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более.
Витамин D стимулирует, среди прочего, всасывание фосфат-ионов в тонком кишечнике. Таким образом, избыточный уровень или активность витамина D может приводить к повышенному уровню фосфатов и гиперфосфатемии. Конкретные варианты реализации изобретения, таким образом, относятся к способам снижения гиперфосфатемического эффекта активного витамина D, например, у субъекта, имеющего повышенный уровень или активность витамина D. Согласно некоторым аспектам, у субъекта наблюдается токсичное воздействие витамина D из-за избыточного поглощения витамина D.
Гиперпаратиреоз представляет собой расстройство, при котором паращитовидные железы производят слишком много паратиреоидного гормона (ПТГ). Вторичный гиперпаратиреоз характеризуется избыточной секрецией ПТГ в ответ на гипокальцемию и связанной гипертрофией паращитовидных желез. ХЗП является наиболее частой причиной вторичного гиперпаратиреоза, обычно в связи с недостаточной почечной функцией для превращения достаточного количества витамина D в его активную форму и для выведения достаточного количества фосфатов. Нерастворимый фосфат кальция образуется в организме и, таким образом, удаляет кальций из циркуляторного русла, приводя к гипокальцемии. Затем также повышается секреции ПТГ паращитовидными железами для повышения уровня кальция в сыворотке. У конкретных субъектов, подлежащих лечению в соответствии со способами, предложенными в настоящей заявке, таким образом, может наблюдаться (например, изначально, до лечения) гиперпаратиреозом и/или повышенный уровень ПТГ, необязательно в комбинации с ХЗП, гиперфосфатемией, гипокальцемией или другими состояниями или симптомом, описанным в настоящей заявке. Согласно некоторым аспектам, введение соединения или композиции, описанной в настоящей заявке, может приводить к снижению гиперпаратиреоза, включая вторичный гиперпаратиреоз, у нуждающегося в этом субъекта. Согласно некоторым аспектам, введение соединения или композиции, описанной в настоящей заявке, может приводить к снижению уровня ПТГ примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более, например, путем снижения уровня фосфора в сыворотке и связанного образования нерастворимого фосфата кальция, повышения доступного кальция и, таким образом, снижения вызванной гипокальцемией продукции ПТГ.
Согласно конкретным вариантам реализации изобретения, введение соединения, описанного в настоящей заявке, может обеспечивать множество терапевтических эффектов у субъекта, страдающего ХЗП. В некоторых примерах введение соединения приводит к снижению уровня FGF23 и уровня паратиреоидного гормона (ПТГ) в сыворотке примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более по сравнению с состоянием в отсутствие лечения, снижению кровяное давление и снижению протеинурии по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более по сравнению с состоянием в отсутствие лечения.
Согласно конкретным вариантам реализации изобретения, введение соединения, описанного в настоящей заявке, может обеспечивать множество терапевтических эффектов у субъекта, страдающего КСЗП (или ХЗП на 5 стадии). В конкретных примерах, введение соединения приводит к снижению концентрации или уровня фосфора в сыворотке примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более по сравнению с состоянием в отсутствие лечения.
Гиперфосфатемия может приводить к нарушениям функции эндотелия как у здоровых субъектов, так и у субъектов, страдающих заболеваниями почек, независимо от кальцификации сосудов (см., например, di Marco et al., Kidney International. 83: 213-222, 2013). Регуляция уровня фосфора в сыворотке путем ограничения фосфтата в пище или веществ, связывающих фосфаты, может предотвратить развитие у таких субъектов сердечно-сосудистых заболеваний. Исследования также показали, что ограничение фосфатов в пище может приводить к улучшению нарушенной функции эндотелия аорты (например, при ХЗП с гиперфосфатемией) путем повышения активирующего фосфорилирования эндотелиальной синтазы оксида азота и Akt (см., например, Van et al., J Clin Biochem Nutr. 51: 27-32, 2012). Конкретные субъекты, подлежащие лечению в соответствии со способами, предложенными в настоящей заявке, могут страдать или иметь риск развития нарушенной функции эндотелия, необязательно в комбинации с гиперфосфатемией, заболеванием почек или любым другим состоянием, описанным в настоящей заявке. Путем снижения содержания фосфора после еды или поглощения фосфора из пищи, отдельно или в комбинации с ограничением содержания фосфатов в пище, введение соединения или композиции, описанного в настоящей заявке может приводить к снижению риска развития нарушения функции эндотелия или может приводить к улучшению уже существующих нарушений функции эндотелия, включая нарушения функции эндотелия, вызванные содержанием фосфора в сыворотке после приема пищи.
Гиперфосфатемии представляет собой первичный показатель кальцификации сосудов (см. Giachelli, Kidney Int. 75: 890-897, 2009). Отложение фосфатов кальция, в основном в форме апатита, является признаком кальцификации сосудов и может возникать в кровеносных сосудах, миокарде и клапанах сердца. Вместе с пассивным отложением фосфатов кальция во внескелетной ткани неорганический фосфат также может вызывать артериальную кальцификацию непосредственно через «окостенение» средней оболочки сосудов. Более того, гладкомышечные клетки сосудов отвечают на повышенный уровень фосфатов остеохондрогенными фенотипическими изменениями и минерализацией их внеклеточного матрикса, обусловленными эффектами натрий-зависимых ко-транспортеров фосфатов.
Кальцификация внутренней оболочки сосудов обычно наблюдается при атеросклеротических повреждениях. Кальцификация средней оболочки обычно наблюдается при возрастном атеросклерозе и диабете и является основной формой кальцификации, наблюдаемой при КСЗП. Действительно, избыточная кальцификация стенки артерий и мягких тканей часто встречается у пациентов, страдающих ХЗП, включая пациентов, страдающих КСЗП. Кальцификация клапанов является определяющим признаком стеноза аортального клапана и наблюдается как в створках, так и в фиброзном кольце, в основном в участках воспаления и механической нагрузки. Указанные механические изменения связаны с повышением скорости распространения пульсовой волны и пульсового давления артерий и приводят к нарушению расширения сосудов, повышенной вызванной постнагрузкой гипертрофии левого желудочка и нарушенной коронарной перфузии (см. Guerin et al., Circulation. 103: 987-992, 2001). Кальцификация как внутренней оболочки, так и средней оболочки сосудов, таким образом, может вносить вклад в заболеваемость и смертность, связанные с сердечно-сосудистыми заболеваниями и, вероятно, играет основную роль в значительном повышении смертности от сердечно-сосудистых заболеваний у пациентов, страдающих ХЗП и КСЗП. Контроль фосфора в сыворотке, таким образом, может приводить к снижению образования продуктов кальция/фосфатов и, таким образом, снижать кальцификацию сосудов. Соответственно, конкретные субъекты, подлежащие лечению в соответствии со способами, предложенными в настоящей заявке, могут иметь страдать или иметь риск развития кальцификации сосудов, включая кальцификацию внутренней и/или средней оболочки сосудов, необязательно в комбинации с любым заболеванием из гиперфосфатемии, ХЗП и КСЗП. Согласно некоторым вариантам реализации изобретения, введение соединения или композиции, описанной в настоящей заявке, приводит к снижению риска развития или снижению возникновения или уровня кальцификации сосудов у нуждающегося в этом субъекта. Согласно конкретным вариантам реализации изобретения, введение соединения или композиции, описанной в настоящей заявке, может приводить к снижению кальцификации сосудов примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или 200% или более, например, по сравнению с состоянием в отсутствие лечения.
Пожилые пациенты могут быть, в частности, чувствительны к повышенному уровню фосфатов. Например, исследование пищевых и генетических факторов обеспечивают доказательства in vivo того, что токсичность фосфатов ускоряет процесс старения и предполагает новую роль фосфатов в старении млекопитающих (см., например, Ohnishi and Razzaque, FASEB J. 24: 3562-71, 2010). Указанные исследования показывают, что избыточный уровень фосфатов связан со многими признаками преждевременного старения, включая кифоз, нарушенная координация движений, гипогонадизм, бесплодие, потерю скелетно-мышечной массы, эмфизему и остеопению, а также генерализованную атрофию кожи, кишки, тимуса и селезенки. Конкретные варианты реализации изобретения, таким образом, относятся к снижению отложения фосфатов у пожилых пациентов, например, для снижения любого одного или более из признаков преждевременного старения, включая введение пожилым пациентам соединения, описанного в настоящей заявке. В некоторых примерах возраст пожилого пациента составляет примерно или по меньшей мере примерно 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100 или более лет.
Гипертрофия относится к повышению объема органа или ткани из-за увеличения его клеточного компонента. Гиперфосфатемия связана с гипертрофией миокарда, включая гипертрофию левого желудочка (см. Neves et al., Kidney Int. 66: 2237-44, 2004; Achinger and Ayus, Am Soc Nephrol. 17(12 Suppi 3): S255-61, 2006), и компенсаторной почечной гипертрофией, включая гломерулярную гипертрофию, при этом указанная гломерулярная гипертрофия часто наблюдается при ХЗП. У конкретных субъектов, подлежащих лечению в соответствии со способами, предложенными в настоящей заявке, может наблюдаться (например, изначально, до лечения) гипертрофия миокарда, почечная гипертрофию или гипертрофия миокарда и почечная гипертрофия, отдельно или в комбинации с ХЗП или повреждением почек. Согласно некоторым вариантам реализации изобретения, введение соединения, описанного в настоящей заявке может снижать гипертрофию миокарда и/или почечную гипертрофию примерно или по меньшей мере примерно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200% или более по сравнению с состоянием в отсутствие лечения.
Введение соединений согласно изобретению или их фармацевтически приемлемых солей в чистой форме или в подходящей фармацевтической композиции можно осуществлять с помощью любого из общепринятых способов введения агентов для подобного применения. Фармацевтические композиции согласно изобретению могут быть получены путем комбинирования соединений согласно изобретению с подходящим фармацевтически приемлемым носителем, разбавителем или наполнителем и могут быть представлены в виде препаратов в твердой, полужидкой, жидкой или газообразной форме, таких как таблетки, капсулы, порошки, гранулы, мази, растворы, суппозитории, инъекции, ингаляции, гели, микросферы и аэрозоли. Типичные пути введения таких фармацевтических композиций включают, но не ограничиваются указанными, пероральное, местное, трансдермальное, ингаляционное, парентеральное, сублингвальное, буккальное, ректальное, вагинальное и интраназальное. Термин «парентеральное» при использовании в настоящей заявке включает подкожные инъекции, внутривенную, внутримышечную, внутригрудинную инъекцию или методы инфузии. Фармацевтические композиции согласно изобретению представлены таким образом, что они обеспечивают биодоступность содержащихся в них активных ингредиентов при введении композиции пациенту. Композиции, подлежащие введению субъекту или пациенту, могут быть представлены в виде одной или более единиц дозирования, где, например, таблетка может представлять собой одну единицу дозирования, а контейнер, содержащий соединение согласно изобретению в аэрозольной форме, может содержать множество единиц дозирования. Актуальные способы получения таких форм дозирования известны или очевидны специалистам в данной области техники; например, см. Remington: The Science and Practice of Pharmacy, 20th Edition (Philadelphia College of Pharmacy and Science, 2000). Композиция, подлежащая ведению, в любом случае содержит терапевтически эффективное количество соединения согласно изобретению или его фармацевтически приемлемой соли для лечения интересующего заболевания или состояния в соответствии с идеями настоящего изобертения.
Фармацевтическая композиция согласно изобретению может быть представлена в виде твердого вещества или жидкости. В одном аспекте носитель (носители) представлен в виде частиц таким образом, что композиция, например, представлена в виде таблетки или порошка. Носитель (носители) может быть жидким, если композиция представляет собой, например, сироп для перорального введения, жидкость для инъекций или аэрозоль, который применим, например, для ведения путем ингаляции.
Фармацевтическая композиция, предназначенная для перорального введения, предпочтительно представлена в твердой или в жидкой форме, где полутвердые, полужидкие формы, суспензии и гелевые формы включены в понятие формы, рассматриваемой в настоящей заявке как твердая или жидкая.
В случае твердой композиции для перорального введения фармацевтическая композиция может быть представлена в виде порошка, гранул, прессованных таблеток, пилюлей, капсул, жевательной резинки, вафель и т.д. Такая твердая композиция, как правило, содержит один или более инертных разбавителей или съедобных носителей. Кроме того, может присутствовать один или более из следующих компонентов: связующие вещества, такие как карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза, трагантовая камедь или желатин; наполнители, такие как крахмал, лактоза или декстрины, дезинтегрирующие агенты, такие как альгиновая кислота, альгинат натрия, примогель, кукурузный крахмал и т.д.; смазывающие вещества, такие как стеарат магния или Sterotex; вещества, способствующие скольжению, такие как коллоидный диоксид кремния; подсластители, такие как сахароза или сахарин; ароматизатор, такой как мята перечная, метилсалицилат или апельсиновый ароматизатор, и краситель.
Когда фармацевтическая композиция представлена в виде капсулы, например, желатиновой капсулы, она может содержать помимо материалов указанного выше типа жидкий носитель, такой как полиэтиленгликоль или масло.
Фармацевтическая композиция может быть представлена в виде жидкости, например, эликсира, сиропа, раствора, эмульсии или суспензии. Жидкость может быть предназначена, например, для перорального введения или для доставки путем инъекции. Предпочтительные композиции, предназначенные для перорального введения, содержат помимо соединения согласно настоящему изобретению один или более из подсластителя, консервантов, красителя/оттеночного вещества и усилителя вкуса и запаха. В композициях, предназначенных для введения путем инъекции, может содержаться одно или более из поверхностно-активного вещества, консерванта, смачивающего агента, диспергирующего агента, суспендирующего агента, буфера, стабилизатора и изотонического агента.
Жидкие фармацевтические композиции согласно изобретению, представляющие собой растворы, суспензии или другие подобные формы, могут содержать одно или более из следующих вспомогательных веществ: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно физиологический солевой раствор, раствор Рингера, изотонический хлорид натрия, фиксированные масла, такие как синтетические моно- или диглицериды, которые могут служить в качестве растворителя или суспендирующей среды, полиэтиленгликоли, глицерин, пропиленгликоль или другие растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регуляции тоничности, такие как хлорид натрия или декстроза. Препарат для парентерального введения может быть заключен в ампулы, одноразовые шприцы или многодозовые флаконы, сделанные из стекла или пластика. Физиологический солевой раствор представляет собой предпочтительное вспомогательное вещество. Фармацевтическая композиция для инъекций предпочтительно является стерильной.
Жидкая фармацевтическая композиция согласно изобретению, предназначенная для парентерального или перорального введения, должна содержать соединение согласно изобретению в количестве, достаточном для получения подходящей дозы.
Фармацевтическая композиция согласно изобретению может быть предназначена для местного введения, в случае чего носитель может содержать подходящий раствор, эмульсию, мазь или гелевую основу. Основа, например, может содержать один или более из следующих компонентов: вазелин, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирт, и эмульгаторы и стабилизаторы. В фармацевтической композиции для местного введения могут присутствовать загустители. Композиция, предназначенная для трансдермального введения, может содержать трансдермальный пластырь или устройство для ионофореза.
Фармацевтическая композиция согласно изобретению может быть предназначена для ректального введения в форме, например, суппозитория, который тает в прямой кишке с высвобождением лекарственного средства. Композиция для ректального введения может содержать масляную основу в качестве подходящего не вызывающего раздражения наполнителя. Такие основы включают, но не ограничиваются указанными, ланолин, масло какао и полиэтиленгликоль.
Фармацевтическая композиция согласно изобретению может содержать различные материалы, которые модифицируют физическую форму твердой или жидкой единицы дозирования. Например, композиция может содержать материалы, которые образуют покрывающую оболочку вокруг активных ингредиентов. Материалы, которые образуют покрывающую оболочку, как правило, являются инертными и могут быть выбраны, например, из сахара, шеллака и других агентов кишечнорастворимых оболочек. Альтернативно, активные ингредиенты могут быть заключены в желатиновую капсулу.
Фармацевтическая композиция согласно изобретению в твердой или жидкой форме может содержать агент, который связывается с соединениями согласно изобретению и, таким образом, способствует доставке соединения. Подходящие агенты, которые могут обладать указанной способностью, включают моноклональные или поликлональные антитела, белок или липосому.
Фармацевтическая композиция согласно изобретению может состоять из единиц дозирования, которые можно вводить в виде аэрозоля. Термин «аэрозоль» используется для обозначения различных систем, варьирующих от систем коллоидной природы до систем, состоящих из герметизированных упаковок. Доставка может осуществляться с помощью сжиженного или сжатого газа или с помощью подходящей насосной системы, которая распыляет активные ингредиенты. Аэрозоли соединений согласно изобретению можно доставлять в однофазной, двухфазной или трехфазной системе для доставки активного ингредиента (ингредиентов). Доставка аэрозоля включает необходимый контейнер, активаторы, клапаны, субконтейнеры и т.д., которые вместе могут образовывать набор. Специалист в данной области техники может определить предпочтительные аэрозоли без излишней экспериментальной работы.
Фармацевтические композиции согласно изобретению можно получать с помощью способов, хорошо известных в области фармацевтики. Например, фармацевтическую композицию, предназначенную для введения путем инъекции, можно получить путем объединения соединения согласно изобретению со стерильной дистиллированной водой с образованием раствора. Поверхностно-активное вещество можно добавлять для облегчения образования гомогенного раствора или суспензии. Поверхностно-активные вещества представляют собой соединения, которые нековалентно взаимодействуют с соединениями согласно изобретению, облегчая растворение или получение гомогенной суспензии соединения в водной системе доставки.
Соединения согласно изобретению или их фармацевтически приемлемые соли вводят в терапевтически эффективном количестве, которое варьирует в зависимости от различных факторов, включая активность конкретного используемого соединения; метаболическая стабильность и продолжительность действия соединения; возраст, масса тела, общее состояние здоровья, пол и диета пациента; способ и время введения; скорость выведения; комбинация лекарственных средств; тяжесть конкретного нарушения или состояния; и субъект, проходящий терапию.
Согласно конкретным вариантам реализации изобретения, типичная доза по существу не проникающего или по существу не обладающего системной биодоступностью соединения, которое вводят субъекту, нуждающемуся в лечение, может составлять между примерно 0,2 мг в сутки и примерно 2 г в сутки или между примерно 1 мг и примерно 1 г в сутки или между примерно 5 мг и примерно 500 мг или между примерно 10 мг и примерно 250 мг в сутки.
Частота введения соединений и композиций, описанных в настоящей заявке, может варьировать от одного раза в день (QD) до двух раз в день (BID) или трех раз в день (TID) и т.д., при этом точная частота введения варьирует в зависимости, например, от состояния пациента, дозы и т.д.
Соединения согласно изобретению или их фармацевтически приемлемые производные можно также вводить одновременно, до или после введения одного или более других терапевтически или биологически активных агентов, пищевых добавок или любой их комбинации. Такая комбинированная терапия включает введение единичной фармацевтически лекарственной формы дозирования, которая содержит соединение согласно изобретению и одно или более дополнительных активных агентов, а также введение соединения согласно изобретению и каждого активного агента в отдельной фармацевтической лекарственной форме дозирования. Например, соединение согласно изобретению и другой активный агент можно вводить пациенту вместе в виде одной пероральной формы дозирования композиции, такой как таблетка или капсула, или каждый агент вводят в отдельной лекарственной форме дозирования для перорального введения. При использовании раздельных лекарственных форм дозирования соединения согласно изобретению и один или более дополнительных активных агентов можно вводить по существу в одно время, т.е. одновременно, или в разное время, т.е., последовательно; предполагается, что комбинированная терапия включает все указанные режимы.
Например, согласно конкретным вариантам реализации изобретения, дополнительный биологически активный агент, включенный в фармацевтическую композицию (или способ) согласно изобретению выбран, например, из витамина D2 (эргокальциферола), витамина D3 (холекальциферола), активного витамина D (кальцитриола) и аналогов активного витамина D (например, доксеркальциферола, парикальцитола).
Согласно другим вариантам реализации изобретения, дополнительный биологически активный агент, включенный в фармацевтическую композицию (или способ) согласно изобретению, представляет собой вещество, связывающее фосфаты, такое как севеламера (например, Renvela® (севеламера карбонат), Renagel® (севеламера гидрохлорид)), лантана карбонат (например, Fosrenol®), кальция карбонат (например, Calcichew®, Titralac®), кальция ацетат (например, PhosLo®, Phosex®), кальция ацетат/магния карбонат (например, Renepho®, OsvaRen®), MCI-196, лимоннокислое железо (например, Zerenex™), магния-железа гидроксикарбонат (например, Fermagate™), алюминия гидроксид (например, Alucaps®, Basaljel®), APS1585, SBR-759, РА-21 и т.д.
Согласно некоторым вариантам реализации изобретения, дополнительный биологически активный агент представляет собой ингибитор кишечного натрий-зависимого переносчика фосфатов (ингибитор NaPi2b). Примеры ингибиторов NaPi2b можно найти, например, в международных заявках № PCT/US2011/043267; PCT/US2011/043261; PCT/US2011/043232; PCT/US2011/043266 и PCT/US2011/043263 и патенте США №8,134,015, каждый из которых полностью включен в настоящую заявку посредством ссылки.
Согласно конкретным вариантам реализации изобретения, дополнительный биологически активный агент представляет собой ниацин или никотинамид.
Согласно некоторым вариантам реализации изобретения, субъект страдает ХЗП или подвергается лечению от ХЗП, и дополнительный биологически активный агент представляет собой соединение, используемое для лечения или сдерживания ХЗП. Примеры таких соединения включают лекарственные препараты для лечения повышенного кровяного давления, такие как ингибиторы АСЕ, блокаторы рецептора ангиотензина II, бета-блокаторы, блокаторы кальциевых каналов, прямые ингибиторы ренина, диуретики и вазодилататоры; лекарственные препараты для лечения симптомов и осложнений ХЗП, такие как эритропоетиновая терапия и/или заместительная терапия железа при анемии, электролиты при дисбалансе электролитов, диуретики, ингибиторы АСЕ и блокаторы рецептора ангиотензина II, ингибиторы конечных продуктов усиленного гликозилирования (например, аминогуанидин, пиридоксамин) и витамин D; агенты, снижающие содержание липидов, такие как ингибиторы HMG-CoA (3-гидрокси-3-метил-глутарил-СоА)редуктазы или статины (например, аторвастатин, флувастатин, ловастатин, питавастатин, правастатин, розувастатин, симвастатин).
Необходимо понимать, что настоящее описание, комбинации заместителей и/или переменных изображенных формул приемлемы только в том случае, если они приводят к стабильному или довольно стабильному соединению.
Специалисту в данной области техники также очевидно, что процесс, описанный в настоящей заявке, может требовать защиты функциональный группы промежуточного соединения с помощью подходящих защитных групп. Такие функциональные группы включают гидрокси, амино, меркапто и карбоновую кислоту. Подходящие защитные группы для гидрокси-групп включают триалкилсилил или диарилалкилсилил (например, m-бутилдиметилсилил, m-бутилдифенилсилил или триметилсилил), тетрагидропиранил, бензил и т.д. Подходящие защитные группы для амино, амидино и гуанидино включают m-бутоксикарбонил, бензилоксикарбонил и т.д. Подходящие защитные группы для меркапто включают -C(O)-R» (где «R» представляет собой алкил, арил или арилалкил), n-метоксибензил, тритил и т.д. Подходящие защитные группы для карбоновой кислоты включают сложные алкил-, арил- или арилалкилэфиры. Защитные группы можно добавлять или удалять в соответствии со стандартными способами, которое известны специалистам в данной области техники и описаны в настоящей заявке. Применение защитных групп подробно описано в источнике Green, T.W. and P.G.M. Wutz, Protective Groups in Organic Synthesis (1999), 3rd Ed., Wiley. Специалисту в данной обалсти техники очевидно, что защитная группа также может представлять собой полимерную смолу, такую кака смола Ванга, смола Ринка или 2-хлортритилхлоридная смола.
Специалистам в данной области техники также очевидно, что несмотря на то что такие защищенные производные соединений согласно настоящему изобретению могут не обладать фармакологической активностью, они могут вводиться млекопитающему и затем подвергаться метаболизму в организме с образованием соединений согласно изобретению, которые являются фармакологически активными. Такие производные можно, таким образом, описывать как «пролекарства». Все пролекарства соединений согласно настоящему изобретению включены в объем изобертения.
Более того, все соединения согласно изобретению, которые существуют в форме свободных оснований или кислот, можно превращать в их фармацевтически приемлемые соли путем обработки подходящим неорганическим или органическим основанием или кислотой с помощью способов, известных специалистам в данной области техники. Соли соединений согласно изобретению можно превращать в форму их свободного основания или кислоты с помощью стандартных методов.
IV. Разработка лекарственных средств
Также включены способы, относящиеся к разработке соединений, которые могут подавлять поглощение фосфатов в желудочно-кишечном тракте. Конкретные варианты реализации включают способы скрининга лекарственных средств in vitro, в которых используются клеточные культуры, такие как культуры клеток кишечника или клеточные линии, включая клеточные линии млекопитающих.
Конкретные варианты реализации изобретения, таким образом, относятся к способам скрининга для поиска ингибиторов захвата фосфатов, включающим культивирование клеток, приведение культивируемых клеток во взаимодействие с исследуемым соединением и измерение одного или более из следующих показателей: рН на апикальной поверхности клетки, внутриклеточный рН клетки, секреция бикарбоната клетками, секреция кислоты клетками, всасывание воды и/или поглощения фосфатов клетками.
Также включен этап идентификации исследуемого соединения как ингибитора захвата фосфатов, где происходит одно или более из следующих событий: рН на апикальной поверхности клетки повышается по сравнению с контролем, внутриклеточный рН клетки снижается по сравнению с контролем, секреция бикарбоната клетками повышается по сравнению с контролем, секреция кислоты клетками снижается по сравнению с контролем, всасывание воды снижается по сравнению с контролем и/или поглощение фосфатов клетками снижается по сравнению с контролем. Согласно некоторым аспектам, повышение или снижение является статистически значимым. Термины «повышение» и «снижение» и «статистически значимый» описаны в другом месте в настоящей заявке. Контроль может не содержать соединение (например, содержит только носитель) или содержать соединение, которое, как известно, не обладает любой из описанных выше активностей. Контроль также может содержать заранее определенное референсное значение.
Согласно конкретным вариантам реализации изобретения, клетки представляют собой клетки кишечника. Неограничивающие примеры культур клеток кишечника включают монослойные культуры клеток кишечника, энтероиды и органоиды клеток кишечника. Монослойные культуры клеток кишечника могут быть получены в соответствии со стандартными методами в данной области техники. Неограничивающие примеры монослойных культур клеток кишечника включают клеточные линии, такие как клеточные линии Сасо-2, НСТ-8 и Т84 (см., например, Watson et al., Am J Physiol Cell Physiol. 281: C388-9, 2001; Shah et al., Biotechnol Prog. 22: 186-9, 2006) и монослойные культуры клеток тощей кишки новорожденных свиней IPEC-J2 (см., например. Chapman et al., Pediatr Res. 72: 576-82, 2012).
Термин «энтероид» включает культуры клеток кишечника, полученные из сегмента (сегментов) кишечных ворсинок кишечной ткани, которые необязательно сохраняют структурную целостность (например, трехмерную структуру кишечного эпителия) и типы клеток кишечной ткани, и обладают генотипическими и фенотипическими свойствами первичной кишечной ткани. Энтероидные клеточные культуры можно получать в соответствии с методами, известными в данной области техники (см., например, заявку на патент США №2010/0047853; международную публикацию WO 2010/090513; заявку на патент США №2012/0196312 и международную публикацию WO 2012/168930).
Термин «органоид» или «кишечный органоид» включает культуры клеток кишечника, полученные главным образом из клеток-предшественников, таких как выделенные эмбриональные стволовые клетки, клети эндодермы или другие плюрипотентные стволовые клетки. Органоиды могут быть получены, например, путем поэтапной дифференцировки клеток-предшественников в сложные трехмерные ткани кишечника (см., например, международную публикацию WO 2011/140441), включая ткани кишечника, которые могут содержать поляризованный цилиндрический эпителий, окруженный мезенхимой, содержащей подобный гладкомышечному слой. Согласно некоторым аспектам, эпителий организован в подобные криптам пролиферативные зоны и подобные ворсинкам структуры, содержащие все или большинство основных функциональных типов кишечных клеток. Согласно некоторым аспектам, сначала осуществляют отбор или обогащение культы клетками-предшественниками для экспрессии маркеров, таких как LGR5 и/или LGR6.
Также включены культуры, содержащие препараты всей толщины кишки (см., например, Binder et al., Am J Physiol. 225: 1232-1239, 1973) и полученные путем фармакологической обработки и «соскабливания» серозно-мышечного слоя для минимизации влияния собственной нервно-мышечной системы (см., например, Clarke, Am. J. Physiol. Gastro-intestin. Liver Physiol. 296: G1151-66, 2009). «Соскабливание» серозно-мышечного слоя приводит к удалению серозной оболочки (висцеральной брюшины) и продольных/кольцевых мышечных слоев кишечной стенки с сохранением только подлежащих подслизистых элементов, остатков мышц и эпителия. Указанные культуры могут быть, в частности, применимы при использовании камеры Уссинга.
Согласно конкретным вариантам реализации изобретения, можно использовать камеру Уссинга. Камера Уссинга обеспечивает физиологическую систему для измерения транспорта ионов, питательных веществ и лекарственных средств через различные эпителиальные ткани, такие как ткани кишечника (см., например, Clarke et al., выше). Например, в некоторых способах могут использоваться методы pH-stat для измерения трансэпителиальной секреции бикарбоната и/или методы изотопного потока для измерения суммарной секреции или всасывания субстратов. Согласно конкретным вариантам реализации изобретения, камеру Уссинга адаптируют для проведения исследований на мышиной или крысиной кишке, включая препараты всей толщины кишки и полученные путем соскабливания серозно-мышечного слоя (см., например, Clarke et al., выше).
В конкретных способах скрининга могут использоваться различные не кишечные клеточные линии, включая клеточные линии млекопитающих. Примеры клеточных линии млекопитающих включают клеточные линии почки эмбриона человека (например, клетки HEK 293), линию клеток почки обезьяны CV1, трансформированных вирусом SV40 (COS-7, ATCC CRL 1651); клетки почки новорожденного хомяка (BHK, ATCC CCL 10); мышиные клетки Сертоли (ТМ4); клетки почки обезьяны (CV1 ATCC CCL 70); клетки почки зеленой африканской мартышки (VERO-76, ATCC CRL-1587); клетки цервикальной карциномы человека (HELA, ATCC CCL 2); клетки почки собаки (MDCK, ATCC CCL 34); клетки печени крыс линии Buffalo (BRL 3А, ATCC CRL 1442); клетки легкого человека (W138, ATCC CCL 75); клетки печени человека (Нер G2, НВ 8065); клетки опухоли опухоли молочной железы (ММТ 060562, ATCC CCL51); клетки TR1; клетки MRC 5; клетки FS4 и линию клеток гепатомы человека (Нер G2). Другие применимые клеточные линии млекопитающих включают клетки яичника китайского хомячка (СНО), включая клетки DHFR-CHO, и клеточные линии миеломы, такие как NSO и Sp2/0.
Методы для измерения изменений рН, секреции бикарбоната, секреции кислоты, всасывания воды и поглощения фосфатов известны в данной области техники. Например, изменения внутриклеточного рН можно измерить путем приведения клеток или тканей во взаимодействие с рН-чувствительным флуоресцентным красителем или меткой и измерения флуоресценции указанного красителя или метки. Примеры рН-чувствительных красителей включают 2'',7''-бис-(2-карбоксиэтил)-5-(6-)карбоксифлуоресцеин 4 (BCECF), 2'',7''-бис-(2-карбоксипропил)-5-(6-)-карбоксифлуоресцеин (BCPCF 11), 5-(6)-карбоксинафтофлуоресцеин и другие (см., например, Фигуры 8А и 8В; Han and Burgess, Chem Rev. 110:2709-28, 2010). Методы измерения транспорта бикарбоната (in vitro) через отдельные ионные каналы, отдельные клетки и интактные эпителиальные слои описаны, например, в источнике Hug et al., Methods Mol Biol. 741: 489-509, 2011; Feldman et al., Am. J. Physiol. 254: C383-90, 1988. Как отмечено выше, изменения рН, секреции бикарбоната и/или секреции кислоты также можно измерить в камере Уссинга, например, с использованием способов pH-stat или изотопного потока. Поглощение фосфатов можно измерить, например, путем приведения клеток или тканей во взаимодействие с 33Р-меченными фосфат-ионами и измерения поглощения указанных меченных фосфат-ионов (см. Примеры; Matsuo et al., Eur. J. Pharmacol. 517: 111-19, 2005). Другие методы измерения рН, секреции бикарбоната, секреции кислоты и поглощения фосфатов будут очевидны специалистам в данной области техники.
Согласно конкретным аспектам, исследуемое соединение представляет собой малую молекулу или пептид, который, как известно или как предполагается, стимулирует секрецию бикарбоната (например, СБДК), подавляет секрецию кислоты и/или снижает всасывание воды в желудочно-кишечном тракте, включая тонкий кишечник. Примеры таких соединения включают, но не ограничиваются указанными, агонисты P2Y, агонисты рецептора аденозина A2b, агонисты рецептора гуанилатциклазы С (например, пептидные агонисты), агонисты растворимой гуанилатциклазы, агонисты рецептора аденилатциклазы, агонисты рецептора имидазолина-1, холинергические агонисты, агонисты рецептора простагландина ЕР4, агонисты дофамина D1, агонисты рецептора мелатонина, агонисты 5НТ4, агонисты рецептора предсердного натрийуретического пептида, ингибиторы карбоангидразы и ингибиторы фосфодиэстеразы. Неограничивающие примеры таких соединения описаны в другом месте в настоящей заявке. Согласно некоторым вариантам реализации изобретения, соединение представляет собой производное или аналог одного или более из таких соединений. Такие производные или аналоги могут содержать модификации, например, предназначенные для снижения системной биодоступности соединения, как описано в настоящей заявке.
Также включены любые из указанных выше способов или других способов скрининга, известных в данной области техники, которые адаптированы для высокопроизводительного скрининга (ВПС). Для ВПС, как правило, используются автоматизированные системы для проведения скринингового анализа библиотеки агентов-кандидатов, например, анализа, оценивающего повышение или снижение связывания и/или активности, как описано в настоящей заявке.
Любой из способов скрининга, предложенных в настоящей заявке, может использовать библиотеки малых молекул или библиотеки, созданные с помощью комбинаторной химии. В качестве одного примера, такие библиотеки можно использовать для скрининга малых молекул, которые ассоциируют или взаимодействуют с молекулой-мишенью или вызывают желаемый физиологический ответ (например, снижение внутриклеточного рН клеток кишечника, подавление поглощения фосфатов). Библиотеки химические и/или биологических смесей, таких как грибные, бактериальные или водорослевые экстракты, известны в данной области техники. Примеры способов синтеза молекулярных библиотек можно найти в источниках Carell et al., 1994a; Carell et al., 1994b; Cho et al., 1993; DeWitt et al., 1993; Gallop et al., 1994; Zuckermann et al., 1994.
Библиотеки агентов могут быть представлены в растворе (Houghten et al., 1992) или на носителях (Lam et al., 1991), на чипах (Fodor et al., 1993), бактериях, спорах (Ladner et al., патент США №5,223,409, 1993), плазмидах (Cull et al., 1992) или на фаге (Cwiria et al., 1990; Devlin et al., 1990; Felici et al., 1991; Ladner et al., патент США №5,223,409, 1993; Scott and Smith, 1990). Библиотеки, применимые в целях изобретения, включают, но не ограничиваются указанными, (1) химические библиотеки, (2) библиотеки природных продуктов и (3) комбинаторные библиотеки, состоящие из случайных пептидов, олигонуклеотидов и/или органических молекул.
Химические библиотеки состоят из структурных аналогов известных агентов или агентов, которые идентифицируют как «hit» или «lead» путем скрининга природных продуктов. Библиотеки природных продуктов получают из коллекций микроорганизмов, животных, растений или морских организмов, которые используют для создания смесей для скрининга путем: (1) ферментации и экстракции жидкой среды из почвы, растения или морских организмов или (2) экстракции растений или морских организмов. Библиотеки природных продуктов включают полипептиды, нерибосомальные пептиды и их варианты (неприродные). См., например. Саnе et al., Science 282: 63-68, 1998. Комбинаторные библиотеки могут быть составлены из большого числа пептидов или органических соединений в виде смеси. Их можно относительно легко получить с помощью стандартных автоматизированных способов синтеза, ПЦР, клонирования или собственных способов синтеза.
Более конкурентно, химическая комбинаторная библиотека представляет собой коллекцию различных химических агентов, созданных путем химического синтеза или биологического синтеза, путем комбинирования ряда химических «структурных элементов», таких как реагенты. Например, линейная комбинаторная химическая библиотека, такая как полипептидиая библиотека, получена путем комбинирования ряда химических структурных элементов (аминокислот) каждым возможным способом для данной длины соединения (т.е. количества аминокислот в полипептидном агенте). Миллионы химических агентов могут быть синтезированы путем такого комбинаторного смешивание химических структурных элементов.
Обзор способов комбинаторной химии и созданных с их помощью библиотек можно найти, например, в источниках Huc and Nguyen, (2001) Comb. Chem. High Throughput Screen. 4: 53-74; Lepre, (2001) Drug discov. Today 6: 133-140; Peng, (2000) Biomed. Chromatogr. 14: 430-441; Bohm, H.J. и Stahl, M. (2000) Curr. Opin. Chem. Biol. 4: 283-286; Barnes and Balasubramanian, (2000) Curr. Opin. Chem. Biol. 4: 346-350; Lepre et al., (2000) Mass Spectrom Rev. 19: 139-161; Hall, (2000) Nat. Biotechnol. 18: 262-262; Lazo and Wipf, (2000) J. Pharmacol. Exp. Ther. 293: 705-709; Houghten, (2000) Ann. Rev. Pharmacol. Toxicol. 40: 273-282; Kobayashi (2000) Curr. Opin. Chem. Biol. (2000) 4: 338-345; Kopylov Spiridonova, (2000) Mol. Biol. (Mosk) 34: 1097-1113; Weber, (2000) Curr. Opin. Chem. Biol. 4: 295-302; Dolle, (2000) J. Comb. Chem. 2: 383-433; Floyd et al., (1999) Prog. Med. Chem. 36: 91-168; Kundu et al., (1999) Prog. Drug Res. 53: 89-156; Cabilly, (1999) Mol. Biotechnol. 12: 143-148; Lowe, (1999) Nat. Prod. Rep. 16: 641-651; Dolle и Nelson, (1999) J. Comb. Chem. 1: 235-282; Czarnick and Keene, (1998) Curr. Biol. 8: R705-R707; Dolle, (1998) Mol. Divers. 4: 233-256; Myers, (1997) Curr. Opin. Biotechnol. 8: 701-707; и Pluckthun and Cortese, (1997) Biol. Chem. 378: 443.
Устройства для получения комбинаторных библиотек являются коммерчески доступными (см., например, 357 MPS, 390 MPS, Advanced Chem Tech, Луисвилл, Кентукки, Symphony, Rainin, Уоберн, Массачусетс, 433А Applied Biosystems, Фостер Сити, Калифорния, 9050 Plus, Millipore, Бедфорд, Массачусетс). Кроме того, многие комбинаторные библиотек являются сами по себе коммерчески доступными (см., например, ComGenex, Принстон, Нью-Джерси, Asinex, Москва, Россия, Tripos, Inc., Сент-Луис, Миссури, ChemStar, Ltd., Москва, Россия, 3D Pharmaceuticals, Экстон, Пенсильвания, Martek Biosciences, Колумбия, Мэриленд, и т.д.).
Определения и термины
«Амино» относится к заместителю -NH2.
«Аминокарбонил» относится к заместителю -C(=O)NH2.
«Карбокси» относится к заместителю -СО2Н.
«Карбоксилат» относится к его соли или сложному эфиру.
«Циано» относится к заместителю -CN.
«Гидрокси» или «гидроксил» относится к заместителю -ОН.
«Имино» относится к заместителю =NH.
«Нитро» относится к заместителю -NO2.
«Оксо» или «карбонил» относится к заместителю =O.
«Тиоксо» относится к заместителю =S.
«Гуанидинил» (или «гуанидин») относится к заместителю -NHC(=NH)NH2.
«Амидинил» (или «амидин») относится к заместителю -C(=NH)NH2.
«Фосфат» относится к заместителю -ОР(=O)(ОН)2.
«Фосфонат» относится к заместителю -Р(=O)(ОН)2.
«Фосфинат» относится к заместителю -РН(=O)ОН, где каждый Ra независимо представляет собой алкильную группу, как определено в настоящей заявке.
«Сульфат» относится к заместителю -OS(=O)2OH.
«Сульфонат» или «гидроксисульфонил» относится к заместителю -S(=O)2OH.
«Сульфинат» относится к заместителю -S(=O)OH.
«Сульфонил» относится к фрагменту, содержащему группу -SO2-. Например, «алкилсульфонил» или «алкилсульфон» относится к группе -SO2-Ra, где Ra представляет собой алкильную группу, как определено в настоящей заявке.
«Алкил» относится к заместителю с прямой или разветвленной углеводородной цепью, состоящему только из атомов кислорода и водорода, который является насыщенным или ненасыщенным (т.е. содержит одну или более двойных и/или тройных связей) и содержит от одного до двенадцати атомов углерода (С1-12 алкил), предпочтительно от одного до восьми атомов углерода (C1-C8 алкил) или от одного до шести атомов углерода (С1-С6 алкил) и который присоединен к остатку молекулы одинарной связью, например, метилу, этилу, н-пропилу, 1-метилэтилу (изо-пропилу), н-бутилу, н-пентилу, 1,1-диметилэтилу (m-бутил), 3-метилгексилу, 2-метилгексилу, этенилу, проп-1-енилу, бут-1-енилу, пент-1-енилу, пента-1,4-диенилу, этинилу, пропинилу, бутинилу, пентинилу, гексинилу и т.д. Если иное специально не указано в настоящей заявке, алкильная группа может быть необязательно замещенной.
«Алкилен» или «алкиленовая цепь» относится к прямой или разветвленной бивалентной углеводородной цепи, связывающей остаток молекулы с группой заместителя, состоящей только из атомов углерода и водорода, которая является насыщенной или ненасыщенной (т.е. содержит одну или более двойных и/или тройных связей) и содержит от одного до двенадцати атомов углерода, такой как метилен, этилен, пропилен, н-бутилен, этенилен, пропенилен, н-бутенилен, пропинилен, н-бутинилен и т.д. Алкиленовая цепь присоединена к остатку молекулы через одинарную или двойную связь и к группе заместителя через одинарную или двойную связь. Точки присоединения алкиленовой цепи к остатку молекулы и к группе заместителя могут располагаться через один атом углерода или любые два атома углерода в пределах цепи. Если иное специально не указано в заявке, алкиленовая цепь может быть необязательно замещенной.
«Алкокси» относится к заместителю формулы -ORa, где Ra представляет собой алкильный заместитель, как определено выше, содержащему от одного до двенадцати атомов углерода. Если иное специально не указано в заявке, алкокси группа может быть необязательно замещенной.
«Алкиламино» относится к заместителю формулы -NHRa или -NRaRa, где каждый Ra независимо представляет собой алкильный заместитель, как определено выше, содержащему от одного до двенадцати атомов углерода. Если иное специально не указано в заявке, алкиламино группа может быть необязательно замещенной.
«Тиоалкил» относится к заместителю формулы -SRa, где Ra представляет собой алкильный заместитель, как определено выше, содержащему от одного до двенадцати атомов углерода. Если иное специально не указано в заявке, тиоалкильная группа может быть необязательно замещенной.
«Арил» относится к заместителю, представляющему собой углеводородную систему колец, содержащему водород, от 6 до 18 атомов углерода и по меньшей мере одно ароматическое кольцо. В целях настоящего изобретения, арильный заместитель может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую систему колец, которая может содержать конденсированные или мостиковые системы колец. Арильные заместители включают, но не ограничиваются указанными, арильные заместители, произошедшие из ацеантгилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, фторантена, флуорена, as-индацена, s-индацена, индана, индена, нафталина, феналена, фенантрена, плейадена, пирена и трифенилена. Если иное специально не указано в заявке, термин «арил» или префикс «ар-» (например, как в «аралкиле») включает арильные заместители, которые являются необязательно замещенными.
«Аралкил» относится к заместителю формулы -Rb-Rc, где Rb представляет собой алкиленовую цепь, как определено выше, и Rc представляет собой один или более арильных заместителей, как определено выше, например, бензил, дифенилметил и т.д. Если иное специально не указано в заявке, аралкильная группа может быть необязательно замещенной.
«Циклоалкил» или «карбоциклическое кольцо» относится к стабильному неароматическому моноциклическому или полициклическому углеводородному заместителю, состоящему только из атомов углерода и атомов водорода, который может содержать конденсированную или мостиковую систему колец, содержащую от трех до пятнадцати атомов углерода, предпочтительно, от трех до десяти атомов углерода, и который является насыщенным или ненасыщенным и присоединен к остатку молекулы с помощью одинарной связи. Моноциклические заместители включают, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, и циклооктил. Полициклические заместители включают, например, адамантил, норборнил, декалинил, 7,7-диметилбицикло[2,2,1]гептанил и т.д. Если иное специально не указано в заявке, циклоалкильная группа может быть необязательно замещенной.
«Циклоалкилалкил» относится к заместителю формулы -RbRd, где Rd представляет собой алкиленовую цепь, как определено выше, и Rg представляет собой циклоалкильный заместитель, как определено выше. Если иное специально не указано в заявке, циклоалкилалкильная группа может быть необязательно замещенной.
«Конденсированный» относится к любой кольцевой структуре, описанной в настоящей заявке, которая конденсирована с существующей кольцевой структурой в соединениях согласно изобретению. Когда конденсированное кольцо представляет собой гетероциклильное кольцо или гетероарильное кольцо, любой атом углерода на существующей кольцевой структуре, которая стала частью конденсированного гетероциклильного кольца или конденсированного гетероарильного кольца, может содержать в качестве заместители атом азота.
«Гало» или «галоген» относится к брому, хлору, фтору или йоду.
«Галоалкил» относится к алкильному заместителю, как определено выше, который содержит в качестве заместителя один или более галогеновых заместителей, как определено выше, например, трифторметилу, дифторметилу, трихлорметилу, 2,2,2-трифторэтилу, 1,2-дифторэтил, 3-бром-2-фторпропил, 1,2-дибромэтилу и т.д. Если иное специально не указано в заявке, галоалкильная группа может быть необязательно замещенной.
«Гетероциклил» или «гетероциклическое кольцо» относится к стабильному 3-18-членному неароматическому кольцевому заместителю, которой состоит из двух - двенадцати атомов углерода и из от одного до шести гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы. Если иное специально не указано в заявке, гетероциклильный заместитель может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую систему колец, которая может включать конденсированные или мостиковые системы колец; и атомы азота, углерода или серы в гетероциклильном заместителе могут быть необязательно окислены; атом азота может быть необязательно кватернизирован; и гетероциклильный заместитель может быть частично или полностью насыщенным. Примеры таких гетероциклильных заместителей включают, но не ограничиваются указанными, диоксоланил, тиенил[1,3]дитианил, декагидроизохинолил, имидазолинил, имидазолидинил, изотиазолидинил, изоксазолидинил, морфолинил, октагидроиндолил, октагидроизоиндолил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, оксазолидинил, пиперидинил, пиперазинил, 4-пиперидонил, пирролидинил, пиразолидинил, хинуклидинил, тиазолидинил, тетрагидрофурил, тритианил, тетрагидропиранил, тиоморфолинил, тиаморфолинил, 1-оксо-тиоморфолинил и 1,1-диоксо-тиоморфолинил. Если иное специально не указано в заявке в заявке, гетероциклильная группа может быть необязательно замещенной.
«N-гетероциклил» относится к гетероциклильному заместителю, как определено выше, содержащему по меньшей мере один атом азота, где точка присоединения гетероциклильного заместителя к остатку молекулы находится на атоме азота в указанном гетероциклильном заместителе. Если иное специально не указано в заявке, N-гетероциклильная группа может быть необязательно замещенной.
«Гетероциклилалкил» относится к заместителю формулы -RbRe, где Rb представляет собой алкиленовую цепь, как определено выше, и Re представляет собой гетероциклильный заместитель, как определено выше, и если гетероциклил представляет собой азот-содержащий гетероциклил, указанный гетероциклил может быть присоединен к алкильному заместителю на атоме азота. Если иное специально не указано в заявке, гетероциклилалкильная группа может быть необязательно замещенной.
«Гетероарил» относится к заместителю, представляющему собой 5-14-членную систему колец, содержащему атомы водорода, от одного до тринадцати атомов углерода, от одного до шести гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, и по меньшей мере одно ароматическое кольцо. В целях настоящего изобретения, гетероарильный заместитель может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую систему колец, которая может представлять собой конденсированную или мостиковую систему колец; и атомы азота, углерода или серы в гетероарильном заместителе могут быть необязательно окислены; атом азота может быть необязательно кватернизирован. Примеры включают, но не ограничиваются указанными, азепинил, акридинил, бензимидазолил, бензотиазолил, бензиндолил, бензодиоксолил, бензофуранил, бензоксазолил, бензотиазолил, бензотиадиазолил, бензо[b][1,4]диокеепинил, 1,4-бензодиоксанил, бензонафтофуранил, бензоксазолил, бензодиоксолил, бензодиоксинил, бензопиранил, бензопираноил, бензофуранил, бензофураноил, бензотиенил (бензотиофенил), бензотриазолил, бензо[4,6]имидазо[1,2-а]пиридинил, карбазолил, циннолинил, дибензофуранил, дибензотиофенил, фуранил, фураноил, изотиазолил, имидазолил, индазолил, индолил, индазолил, изоиндолил, индолинил, изоиндолинил, изохинолил, индолизинил, изоксазолил, нафтиридинил, оксадиазолил, 2-оксоазепинил, оксазолил, оксиранил, 1-оксидопиридинил, 1-оксидопиримидинил, 1-оксидопиразинил, 1-оксидопиридазинил, 1-фенил-1H-пирролил, феназинил, фенотиазинил, феноксазинил, фталазинил, птеридинил, пуринил, пирролил, пиразолил, пиридинил, пиразинил, пиримидинил, пиридазинил, хиназолинил, хиноксалинил, хинолинил, хинуклидинил, изохинолинил, тетрагидрохинолинил, тиазолил, тиадиазолил, триазолил, тетразолил, триазинил и тиофенил (т.е. тиенил). Если иное специально не указано в заявке, гетероарильная группа может быть необязательно замещенной.
«N-гетероарил» относится к гетероарильному заместителю, как определено выше, содержащему по меньшей мере один атом азота, где точка присоединения гетероарильного заместителя к остатку молекулы находится на атоме азота в гетероарильном заместителе. Если иное специально не указано в заявке, N-гетероарильная группа может быть необязательно замещенной.
«Гетероарилалкил» относится к заместителю формулы -RbRf, где Rb представляет собой алкиленовую цепь, как определено выше, и Rf представляет собой гетероарильный заместитель, как определено выше. Если иное специально не указано в заявке, гетероарилалкильная группа может быть необязательно замещенной.
Термин «замещенный» при использовании в настоящей заявке означает любую из перечисленных выше групп (т.е. алкил, алкилен, алкокси, алкиламино, тиоалкил, арил, аралкил, циклоалкил, циклоалкилалкил, галоалкил, гетероциклил, N-гетероциклил, гетероциклилалкил, гетероарил, N-гетероарил и/или гетероарилалкил), где по меньшей мере один атом водорода замещен на связь с атомами, не представляющими собой атом водорода, включая, но не ограничиваясь указанными: атом галогена, такой как F, Cl, Br, и I; атом кислорода в группах, таких как гидроксильные группы, карбоксильные группы, фосфатные группы, сульфатные группы, алкокси группы и сложноэфирные группы; атом серы в группах, таких как тиоловые группы, тиоалкильные группы, сульфинатные группы, сульфоновые группы, сульфонильные группы и сульфоксидные группы; атом фосфора в группах, таких как фосфинатные группы и фосфонатные группы; атом азота в группах, таких как гуанидиновые группы, амины, амиды, алкиламины, диалкиламины, ариламины, алкилариламины, диариламины, N-оксиды, имиды и енамины; атом кремния в группах, таких как триалкилсилильные группы, диалкиларилсилильные группы, алкилдиарилсилильные группы и триарилсилильные группы; и другие гетероатомы в различных других группах. «Замещенный» также означает любую из перечисленных выше групп, в которой один или более атомов водорода замещены на связь более высокого порядка (например, двойную или тройную связь) с гентероатомом, таким как кислород в оксо, карбонильных, карбоксильных и сложноэфирных группах; и азот в группах, таких как имины, оксимы, гидразоны и нитрилы. Например, «замещенный» включает любую из перечисленных выше групп, в которых один или более атомов водорода замещены на -NRgRh, -NRgC(=O)Rh, -NRgC(=O)NRgRh, -NRgC(=O)ORh, -NRgSO2Rh, -OC(=O)NRgRh, -ORg, -SRg, -SORg, -SO2Rg, -OSO2Rg, -SO2ORg, =NSO2Rg и -SO2NRgRh. «Замещенной» также означает любую из перечисленных выше групп, в которой один или более атомов водорода замещены на -C(=O)Rg, -C(=O)ORg, -C(=O)NRgRh, -CH2SO2Rg, -CH2SO2NRgRh, -(CH2CH2O)1-10Rg, -(CH2CH2O)2-10Rg, -(OCH2CH2)1-10Rg и -(OCH2CH2)2-10Rg. Указанные выше Rg и Rh являются одинаковыми или разными и независимо представляют собой водород, алкил, алкокси, алкиламино, тиоалкил, арил, аралкил, циклоалкил, циклоалкилалкил, галоалкил, гетероциклил, N-гетероциклил, гетероциклилалкил, гетероарил, N-гетероарил и/или гетероарилалкил. Термин «замещенный» также означает любую из перечисленных выше групп, в которой один или более атомов водорода замещены на связь с амино, циано, гидроксильной, имино, нитро, оксо, тиоксо, гало, алкильной, алкокси, алкиламино, тиоалкильной, арильной, аралкильной, циклоалкильной, циклоалкилалкильной, галоалкильной, гетероциклильной, N-гетероциклильной, гетероциклилалкильной, гетероарильной, N-гетероарильной и/или гетероарилалкильной группой. Указанные выше группы, не представляющие собой водород, в целом называются в настоящей заявке «заместителями» или «заместителями, не представляющими собой водород». Кроме того, каждый из перечисленных выше заместителей также может необязательно содержать один или более из указанных выше заместителей.
Использование в настоящей заявке предметов в единственном числе относится к одному или более чем одному (т.е. по меньшей мере одному) указанному предмету. Например, «элемент» означает один элемент или более чем один элемент.
Термин «примерно» означает количество, уровень, значение, число, частоту, процент, расстояние, размер, количество, массу или длину, которая варьирует в пределах 30, 25, 20, 15, 10, 9, 8, 7, 6, 5, 4, 3, 2 или 1% от референсного количества, уровня, значения, чиста, частоты, процента, расстояния, размера, количества, массы, длины или другой единицы, описанной в настоящей заявке.
Термин «активировать» относится к приложению физических, химических или биохимических условий, веществ или процессов к рецептору (например, поровому рецептору) для обеспечения его структурного изменения, приводящего к возможности прохождения ионов, молекул или других веществ.
Термин «активное состояние» относится к условию или состоянию рецептора не в его состоянии отдыха.
«Отток» относится к движению или потоку ионов, молекул или других веществ из внутриклеточного пространства во внеклеточное пространство.
«Кишечный» или «кишечное введение» относится к введению через желудочно-кишечный тракт, включая пероральное, сублингвальное, сублабиальное, буккальное и ректальное введение, и включает введение через желудочный или дуоденальный зонд.
Термин «неактивное состояние» относится к состоянию рецептора в его исходном эндогенном состоянии, т.е. в состоянии отдыха.
Термин «модулирование» включает «повышение» или «усиление», а также «снижение» или «уменьшение» по сравнению с контролем, как правило, статистически или физиологически значимое. «Повышенное» или «увеличенное» количество, как правило, является «статистически значимым» количеством и может включать повышение, примерно в 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9, 3,0, 3,2, 3,4, 3,6, 3,8, 4,0, 4,2, 4,3, 4,4, 4,6, 4,8, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50 или более раз (например, в 100, 200, 500, 1000 раз) (включая все целые числа и десятичные дроби и диапазоны между ними, превышающие 1, например, 5,5, 5,6, 5,7. 5,8 и т.д.) превышающее контрольное количество (например, в отсутствие или в присутствии меньшего количества соединения, другого соединения или лечения) или количество в более ранний момент времени (например, до лечения соединением). «Сниженное» или «уменьшенное» количество, как правило, представляет собой «статистически значимое» количество и может включать снижение на 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% или 100% (включая все целые числа и десятичные дроби и диапазоны между ними) контрольного количества или активности (например, в отсутствие соединения или в присутствии меньшего количества соединения, другого соединения или лечения) или количества в более ранний момент времени (например, до лечения соединением).
«Пролекарство» означает соединение, которое может превращаться в физиологических условиях или путем сольволизиса в биологически активное соединение согласно изобретению. Таким образом, термин «Пролекарство» относится к метаболическому предшественнику соединения согласно изобретению, который является фармацевтически приемлемым. Пролекарство может быть неактивным при введении нуждающемуся в этом субъекту, но превращается in vivo в активное соединение согласно изобретению. Пролекарства, как правило, быстро трансформируются in vivo с получением исходного соединения согласно изобретению, например, в результате гидролиза в крови. Пролекарственное соединение часто предоставляет преимущества, связанные с растворимостью, тканевой совместимостью или замедленным высвобождения в организме млекопитающего (См., Bundgard, H., Design of Drugs (1985), pp. 7-9, 21-24 (Elsevier, Амстердам)). Описание лекарственных средств предложено в источниках Higuchi, Т., et al., A.C.S. Symposium Series, Vol. 14, и Bioreversible Carriers in Drug Design, Ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
Также предполагается, что термин «Пролекарство» включает любые ковалентно связанные носители, которые высвобождают активное соединение согласно изобретению in vivo при введении указанного пролекарства субъекту, представляющему собой млекопитающее. Пролекарства соединения согласно изобретению могут быть получены путем модификации функциональной группы, присутствующей в указанном соединении согласно изобретению таким образом, что указанные модифицированные соединения расщепляются либо с помощью стандартных методов, либо in vivo, с получением исходного соединения согласно изобретению. Пролекарства включают соединения согласно изобретению, в которых гидрокси, амино или меркапто группа связана с любой группой, которая при введении пролекарственного соединения согласно изобретению субъекту, представляющему собой млекопитающее, расщепляется с образованием свободной гидрокси-, свободной амино- или свободной меркапто- группы, соответственно. Примеры пролекарств включают, но не ограничиваются указанными, ацетатные, форматные и бензоатные производные спирта или амидные производные аминных функциональных групп соединений согласно изобретению и т.д.
Также предполагается, что изобретение, описанное в настоящей заявке, включает продукты in vivo метаболизма описанных соединений. Такие продукты могут являться результатом, например, окисления, восстановления, гидролиза, амидирования, эстерификации и т.д. вводимого соединения, в первую очередь в результате ферментативных процессов. Соответственно, изобретение включает соединения, полученные с помощью процесса, включающего введение соединения согласно настоящему изобретению млекопитающему в течение периода времени, достаточного для получения его метаболического продукта. Такие продукты, как правило, идентифицируют с помощью введения радиоактивно меченных соединений согласно изобретению в поддающейся выявлению дозе животному, такому как крыса, мышь, морская свинка, обезьяна или человек с предоставлением времени, достаточного для прохождения метаболизма, и выделения продуктов его превращения из мочи, крови или других биологических образцов.
Термин «млекопитающее» включает человека и ручных животных, таких как лабораторные животные, и домашних животных (например, кошек, собак, свиней, коров, овец, коз, лошадей, кроликов), а также не домашних животных, таких как дикие животные и т.д.
«Возможно» или «необязательно» означает, что описанное далее событие или обстоятельство может происходить или не происходить, и что описание включает примеры, в которых указанное событие или обстоятельство происходит, и примеры, в которых оно не происходит. Например, «необязательно замещенной арил» означает, что арильный заместитель может быть замещенным или не быть замещенным, и что описание включает как замещенные арильные заместители, так и арильные заместители, не содержащие заместителей.
«Фармацевтически приемлемый носитель, разбавитель или наполнитель» включает, о не ограничивается указанными, любое вспомогательное вещество, носитель, наполнитель, вещество, способствующее скольжению, подсластитель, разбавитель, консервант, краситель/оттеночное вещество, усилитель вкуса и запаха, поверхностно-активное вещество, смачивающий агент, диспергирующий агент, суспендирующий агент, стабилизатор, изотонический агент, растворитель или эмульгатор, который был одобрен Управлением по контролю качества продуктов питания и лекарственных средств США как приемлемый для применения у человека или домашних животных.
«Фармацевтически приемлемая соль» включает как соли присоединения кислот, так и соли присоединения оснований.
«Фармацевтически приемлемая соль присоединения кислоты» относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются нежелательными с биологической или другой точки зрения и которые образуются с неорганическими кислотами, включая, но не ограничиваюсь указанными, соляную кислоту, бромоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т.д., и органическими кислотами, включая, но не ограничиваясь указанными, уксусную кислоту, 2,2-дихлоруксусную кислоту, адипиновую кислоту, альгиновую кислоту, аскорбиновую кислоту, аспарагиновую кислоту, бензолсульфоновую кислоту, бензойную кислоту, 4-ацетамидобензойную кислоту, камфорную кислоту, камфор-10-сульфоновую кислоту, каприновую кислоту, капроновую кислоту, каприловую кислоту, карбоновую кислоту, коричную кислоту, лимонную кислоту, цикламовую кислоту, додецилсерную кислоту, этан-1,2-дисульфоновую кислоту, этансульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, муравьиную кислоту, фумаровую кислоту, галактаровую кислоту, гентизиновую кислоту, глюкогептоновую кислоту, глюконовую кислоту, глюкуроновую кислоту, глутамовую кислоту, глутаровую кислоту, 2-оксо-глутаровую кислоту, глицерофосфорную кислоту, гликолевую кислоту, гиппуровую кислоту, изомасляную кислоту, молочную кислоту, лактобионовую кислоту, лауровую кислоту, малеиновую кислоту, яблочную кислоту, малоновую кислоту, миндальную кислоту, метансульфоновую кислоту, муциновую кислоту, нафталин-1,5-дисульфоновую кислоту, нафталин-2-сульфоновую кислоту, 1-гидрокси-2-нафтоевую кислоту, никотиновую кислоту, олеиновую кислоту, оротовую кислоту, оксалиновую кислоту, пальмитиновую кислоту, памоевую кислоту, пропионовую кислоту, пироглутаминовую кислоту, пировиноградную кислоту, салициловую кислоту, 4-аминосалициловую кислоту, себациновую кислоту, стеариновую кислоту, сукциновую кислоту, винную кислоту, тиоциановую кислоту, n-толуолсульфоновую кислоту, трифторуксусную кислоту, ундециленовую кислоту и т.д.
«Фармацевтически приемлемая соль присоединения основания» относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются нежелательными с биологической или другой точки зрения. Указанные соли получают путем добавления неорганического или органического основания к свободной кислоте. Соли, полученные с неорганическими основаниями, включают, но не ограничиваются указанными, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т.д. Предпочтительные неорганические соли представляют собой соли аммония, натрия, калия, кальция и магния. Соли, полученные из органических оснований, включают, но не ограничиваются указанными, соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как аммиак, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, диэтаноламин, этаноламин, деанол, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, бенетамин, бензатин, этилендиамин, глюкозамин, метилглюкамин, теобромин, триэтаноламин, трометамин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминные смолы и т.д. Конкретные предпочтительные органические основания представляют собой изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин.
Часто кристаллизация приводит к получению сольвата соединения согласно изобретению. При использовании в настоящей заявке, термин «сольват» относится к агрегату, который содержит одну или более молекул соединения согласно изобретению с одной или более молекул растворителя. Растворитель может представлять собой воду, в случае чего сольват может представлять собой гидрат. Альтернативно, растворитель может представлять собой органический растворитель. Таким образом, соединения согласно настоящему изобретению могут существовать в виде гидрата, включая моногидрат, дигидрат, гемигидрат, секвигидрат, тригидрат, тетрагидрат и т.д., а также соответствующие сольватированные формы. Соединения согласно изобретению могут представлять собой истинные сольваты, однако в других случаях соединения согласно изобретению могут лишь удерживать дополнительные молекулы воды или представлять собой смесь воды с некоторым дополнительным растворителем.
«Фармацевтическая композиция» относится к лекарственной форме, содержащей соединение согласно настоящему изобретению и среду, общепринятую в данной области техники для доставки биологически активного соединения млекопитающим, например, человеку. Такая среда включает все фармацевтически приемлемые носители, разбавители или наполнители.
Соединения согласно изобретению или их фармацевтически приемлемые соли могут содержать один или более центров асимметрии и могут, таким образом, давать начало энантиомерам, диастереомерам и другим стереоизомерическим формам, которые можно определить в терминах абсолютной стереохимии как (R)- или (S)- или как (D)- или (L)- формы в случае аминоксилот. Предполагается, что настоящее изобретение включает все такие возможные изомеры, а также их рацемические и оптически чистые формы. Оптически активные (+) и (-), (R)- и (S)- или (D)- и (L)- изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов или разделены с использованием стандартных методов, например, хроматографии и фракционной кристаллизации. Стандартные методы получения/выделения отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разрешение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ЖХВД). Предполагается, если иное специально не указано, что когда соединения, описанные в настоящей заявке, содержат олефиновые двойные связи или другие центры геометрической асимметрии, они включают как Е, так и Z геометрические изомеры. Подобным образом, также предполагается, что включены все таутомерные формы.
Предполагается, что «стабильное соединение» и «стабильная структура» обозначает соединение, которое является достаточно прочным для возможности его выделения до полезной степени чистоты из реакционной смеси и получения в виде эффективного терапевтического агента.
«Статистически значимый» означает, что результат, вероятно, не является случайным. Статистическую значимость можно определить с помощью любого способа, известного в данной области техники. Обычно используемые показатели значимости включают р-значение, которое подставляет собой частоту или вероятность, с которой наблюдаемое событие произошло бы, если бы нулевая гипотеза была верна. Если полученное р-значение меньше уровня значимости, то нулевая гипотеза отвергается. В простых случаях уровень значимости определяют как р-значение, составляющее 0,05 или менее.
«По существу» или «фактически» включает почти полностью или полностью, например, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% или более от некоторого данного количества.
Термин «вторичный» относится к условию или состоянию, которое может возникнуть при другом заболевании, состоянии или лечении, может являться следствием другого заболевания, состояния или лечения или может быть результатом другого заболевания, состояния или лечения. Термин также относится к случаям, когда указанное заболевание, состояние или лечение может играть лишь незначительную роль в возникновении симптомов или ответа на конечной стадии заболевания, симптома или состояния пациента.
«Субъекты» или «пациенты» (термины используются в настоящей заявке взаимозаменяемо), нуждающиеся в лечение соединением согласно настоящему изобретению, включают, например, субъектов, «нуждающихся в снижении содержания фосфатов», которые могут включать субъектов, нуждающихся в «фосфатной терапии» например, профилактической фосфатной терапии, или терапии уровня фосфатов. Включены млекопитающие, страдающие или имеющие риск развития заболеваний и/или состояний, описанных в настоящей заявке, конкретных заболеваний и/или состояний, которые можно лечить соединениями согласно изобретению в комбинации с другими активными агентами или без них, для достижения благоприятного терапевтического и/или профилактического результата. Благоприятный результат включает снижение тяжести симптомов, задержку возникновения симптомов, поддержание нормального содержания фосфатов, снижение риска развития гиперфосфатемии, модулирование одного или более показателей, описанных в настоящей заявке (таких как сниженный уровень фосфатов в сыворотке или крови пациента, страдающего или имеющего риск развития гиперфосфатемии, повышенное выведение с фекалиями фосфат-ионов у пациентов, страдающих или имеющих риск развития гиперфосфатемии), повышение продолжительности жизни и/или более быстрое или более полное устранение заболевания или состояния.
«Стереоизомер» относится к соединениям, образованным из одинаковых атомов, связанных одинаковыми связями, но имеющим разные трехмерные структуры, которые не являются взаимозаменяемыми. Настоящее изобретение предполагает различные стереоизомеры и их смеси и включает «энантиомеры», которые относятся к двум стереоизомерам, молекулы которых являются неналагающимися зеркальными отражениями друг друга.
Термин «таутомер» относится к сдвигу протона от одного атома молекулы к другому атому той же молекулы. Настоящее изобретение включает таутомеры любого указанного соединения.
«Терапевтически эффективное количество» или «эффективное количество» включает количество соединения согласно изобретению, которое при введении млекопитающему, предпочтительно человеку, является достаточным для подавления или снижения другим образом транспорта фосфат-ионов из просвета желудочно-кишечного тракта, повышения выведения с фекалиями фосфат-ионов, снижения уровня фосфат-ионов в сыворотке, лечения гиперфосфатемии у млекопитающего, предпочтительно человека, и/или лечения любого одного или более других состояний, описанных в настоящей заявке. Количество соединения согласно изобретению, которое составляет «терапевтически эффективное количество», варьирует в зависимости от соединения, состояния и его тяжести, способа введения и возраста млекопитающего, подлежащего лечению, но может быть определено специалистом в данной области техники с помощью стандартных методов с учетом его личных знаний и идей настоящего изобретения.
«Лечить» или «лечение» при использовании в настоящей заявке включает лечение интересующего заболевания или состояния у млекопитающего, предпочтительно человека, страдающего указанным интересующим заболеванием или состоянием, и включает:
(i) предотвращение возникновения заболевания или состояния у млекопитающего, в частности, когда у такого млекопитающего состояние еще не диагностировано, но он имеет предрасположенность к указанному состоянию;
(ii) подавление заболевания или состояния, т.е. сдерживание его развития;
(iii) облегчение заболевания или состояния, т.е. индукцию регрессии заболевания или состояния; или
(iv) облегчение симптомов, вызванных заболеванием или состоянием, т.е. облегчение боли без устранения лежащего в основе заболевания или состояния. При использовании в настоящей заявке термины «заболевание» и «состояние» могут использоваться взаимозаменяемо или могут отличаться тем, что для конкретного расстройства или состояния может отсутствовать известный вызывающий агент (так что этиология еще определена), и, таким образом, указанное расстройство или состояние еще не считается заболеванием, а рассматривается только как нежелательное состояние или синдром, для которого клиницистами был определен более или менее специфичный набор симптомов.
ПРИМЕРЫ
Пример 1
Повышенное значение внутриклеточного рН приводит к снижению поглощения фосфатов в клетках
Проводили эксперименты для исследования взаимоотношений между изменениями внутриклеточного рН и захвата фосфат-ионов (Pi) в клетках почки эмбриона человека (HEK-293).
Клетки HEK-293 высевали на 96-луночные планшеты в концентрации 25,000 клеток на лунку и культивировали в течение ночи. Клетки трансфицировали крысиной или человеческой кДНК NaP2b или подвергали ложной трансфекции (без ДНК) с использованием липофектамина 2000 (Invitrogen). Клеткам позволяли достигать конфлюентного состояния во время второй инкубации в течение ночи.
Метод введения аммония использовали для снижения внутриклеточного значения рН от ~7,4 до ~6,8. Среду отсасывали из лунок, клетки дважды промывали буфером NaCl-HEPES (100 мМ NaCl, 50 мМ HEPES, 10 мМ глюкоза, 5 мМ KCl, 2 мМ CaCl2, 1 мМ MgCl2, рН 7,4), затем инкубировали в течение 30 мин при комнатной температуре с буфером NH4Cl-HEPES (20 мМ NH4Cl, 80 мМ NaCl, 50 мМ HEPES, 5 мМ KCl, 2 мМ CaCl2, 1 мМ MgCl2, рН 7,4), содержащим 5 мкМ BCECF-AM. Клетки дважды промывали безаммоннийным не содержащим Na+HEPES буфером (100 мМ холин, 50 мМ HEPES, 10 мМ глюкоза, 5 мМ KCl, 2 мМ CaCl2, 1 мМ MgCl2, рН 7,4) и инкубировали в том же буфере в течение 10 минут при комнатной температуре для снижения внутриклеточного рН. Снижение внутриклеточного рН до примерно рН 6,8 подтверждали путем отслеживания рН-чувствительных изменений флуоресценции BCECF (λех 505 нм, λem 538 нм) нормированных к рН-нечувствительной флуоресценции BCECF (λех 439 нм, λem 538 нм). Был включен контроль с отсутствием процедуры введения аммония. BCECF использовали для получения нормального внутриклеточного рН 7,4.
Клетки затем промывали не содержащим натрий буфером для захвата (14 мМ трис, 137 мМ холин хлорид, 5,4 мМ KCl, 2,8 мМ CaCl2, 1,2 мМ MgSO4, 100 мкМ KH2PO4, 1 мг/мл бычьего сывороточного альбумина, рН 7,4) и захват 33Р инициировали путем добавления в клетки содержащего натрий буфера для захвата (14 мМ трис, 137 мМ хлорид натрия, 5,4 мМ KCl, 2,8 мМ CaCl2, 1,2 мМ MgSO4, 100 мкМ KH2PO4, 1 мг/мл бычьего сывороточного альбумина, рН 7,4). В клеточных линиях, трансфицированных крысиным или человеческим NaP2b, эндогенную активность PiT подавляли с помощью агента сайленсинга PiT таким образом, что только натрий-зависимый захват 33Р был опосредован NaP2b. Агент сайленсинга PiT не использовали в ложно трансфицированных клетках, в которых, таким образом, наблюдался только натрий-зависимый захват 33Р, обусловленный PiT.
Захват 33Р измеряли в присутствии и в отсутствие 5 мкМ EIPA, специфичного ингибитора NHE1. Через 23 минуты при комнатной температуре аналитические смеси удаляли и клетки промывали два раза ледяным буфером для захвата, не содержащим натрия. Клетки лизировали путем добавления 20 мкл 0,1% раствора Твин 80 с последующим добавлением 100 мкл сцинтилляционной жидкости и подсчет проводили с использованием прибора TopCount (Perkin Elmer).
Как показано на Фигурах 22А-22С, внутриклеточное окисление вызвано снижением на >75% либо PiT (22A), либо NaPi2b (22В-22С)-опосредованного захвата 33Р. EIPA, блокирующий NHE1-опосредованный экспорт протонов из цитоплазмы, также вызывал небольшое, но значимое снижение захвата Pi в клетках, которые не были предварительно обработаны для снижения их внутриклеточного рН.
Пример 2
Агонист рецептора гуанилатциклазы С (GC-C) снижает всасывание фосфатов
Проводили эксперименты для определения способности агонистов рецептора гуанилатциклазы С (GC-C) снижать всасывание/захват в тонком кишечнике по результатам измерения захвата 33Р. Крысам одновременно вводили 33Р и линаклотидин, как показано ниже:
1. Носитель (N=5/группа)
2. Линаклотидин в концентрации 0,1 мг/кг (N=6/группа)
3. Линаклотидин в концентрации 0,3 мг/кг (N=4/группа)
Кровь собирали через 5, 15, 30, 45 и 60 минут после введения 33Р и проводили сцинтилляционный анализ плазмы. Результаты показаны на Фигурах 1А-1В. На Фигуре 1А показаны результаты двухстороннего дисперсионного анализа с повторными измерениями и последующим применением критерия множественного сравнения Даннетта, и на Фигуре 1В показаны результаты одностороннего дисперсионного анализа с последующим применением критерием множественного сравнения Даннетта. Указанные результаты показывают, что обе дозы линаклотидина снижали всасывание фосфатов в желудочно-кишечном тракте.
Пример 3
Агонист рецептора I1 и агонист аденилатциклазы снижает всасывание фосфатов
Проводили эксперименты для определения способности других классов лекарственных средств снижать всасывание/захват фосфатов в тонком кишечник по результатам измерения захвата 33Р. Крысам одновременно вводили 33Р и либо агонист рецептора имидазолина подтипа 1 (I1) (моксонидин), либо агонист аденилатцикслазы (водорастворимый аналог форксколина NKH477), как показано ниже:
1. Носитель
2. Моксонидин в концентрации 2 мг/кг
3. Моксонидин в концентрации 6 мг/кг
4. NKH477 в концентрации 1 мг/кг
5. NKH477 в концентрации 3 мг/кг
Кровь собирали через 5, 15, 30, 45 и 60 минут после введения 33Р и проводили сцинтилляционный анализ плазмы. Результаты показаны на Фигурах 2А-2В. На Фигуре 2А показаны результаты двухстороннего дисперсионного анализа с повторными измерениями и последующим применением критерия множественного сравнения Даннетта, и на Фигуре 2В показаны результаты одностороннего дисперсионного анализа с последующим применением критерия множественного сравнения Даннетта. Указанные результаты показывают, что все исследуемые соединения значительно снижали захват/всасывание 33Р через 15 минут.
Пример 4
Соединения, представляющие собой агонисты А2В и агонисты P2Y2, снижают всасывание фосфатов
Проводили эксперименты для определения способности повышенного уровня внутриклеточного кальция (Са++) также приводить к снижению всасывания фосфатов в тонком кишечнике с помощью различных механизмов по результатам измерения захвата 33Р. Крысам одновременно вводили 33Р и исследуемое соединение, как показано ниже:
1. Носитель, n=6
2. BAY 60-6583 в концентрации 10 мг/кг (агонист аденозина А2В)
3. Up4U в концентрации 15 мг/кг (агонист рецептора P2Y2)
Кровь собирали через 5, 15, 30, 45 и 60 минут после введения 33Р и проводили сцинтилляционный анализ плазмы. На Фигуре 3 показано, что агонист рецептора P2Y2 Up4U (15 мг/кг) значительно снижал захват/всасывание 33Р.
Пример 5
Фармакодинамические эффекты на острое поглощения фосфатов у крыс
Исследовали способность соединений снижать появления в кровотоке радиоактивно меченных фосфатов после их введения в пищеварительный тракт крысам. Скорость накопления радиоактивно меченной фосфатной метки в крови у крыс использовали как показатель скорости кишечного всасывания фосфат-содержащей пищи из желудочно-кишечного тракта. С этой целью после совместного введения в желудок крысам пищи с фосфатной меткой и примера соединения отслеживали циркулирующий радиоактивно меченный фосфат. Однако поскольку некоторые из исследуемых соединений имели потенциальные свойства, препятствующие указанному анализу, такие как предполагаемый эффект на перистальтику желудочно-кишечного тракта (например, задержку опорожнения желудка) или целенаправленная химическая нестабильность в желудочно-кишечном тракте, то также периодически осуществляли введение непосредственно в двенадцатиперстную кишку болюса с фосфатной меткой.
Восьминедельных самцов крыс линии Sprague-Dawley получали из компании Charles River Laboratories (Холлистре, Калифорния). Для возможности отбора образцов крови получали крыс с катетерами, имплантированным в яремную вену хирургическим путем поставщиком. Для исследований, требующих введения в двенадцатиперстную кишку, дополнительный катетер был имплантирован хирургическим путем поставщиком для возможности прямой инфузии в просвет двенадцатиперстной кишки. Крыс кормили нормальным гранулированным кормом (Harlan Teklad, Мэдисон, Висконсин; корм для грызунов 2018 Teklad Global, содержащий 18% белка), содержащим 0,65% Р, 1% Са и 1,5 МЕ/г Витамина D3, и обеспечивали свободный доступ к воде до начала исследования.
Крысам, не получающим пищу в течение ночи, вводили раствор фосфата, содержащий [33Р]ортофосфат (PerkinElmer, Уолтам, Массачусетс) в качестве метки, вместе с исследуемыми изделиями, диспергированными в растворе в указанной дозе, или в отсутствие исследуемых изделий. Этот раствор для дозирования, как правило, содержал 8 мМ одноосновный фосфат натрия (1,25 мкКи [33Р]ортофосфат/мкмоль), 4 мМ хлорид кальция, 0,4% гидроксипропилметилцеллюлозу (масса к объему) и 2% диметилсульфоксид (масса к объему). Растворы для дозирования получали в воде с дозой 10 мл/кг в случае введения в желудок, и в солевом растворе с дозой 5 мл/кг при введении виде болюса в двенадцатиперстную кишку с помощью заранее имплантированного катетера.
Образцы крови отбирали у бодрствующих крыс из яремной вены через имплантированные катетеры после введения дозы. Радиоизотоп в общем объеме плазмы выявляли с помощью сцинтилляционного анализа. Относительное количество поглощенного из вводимой дозы фосфата в плазме определяли путем оценки отношения массы тела к общему объему циркулирующей плазмы. См. Bijsterbosch et al., Experientia. 37: 381-382, 1981 (объема плазмы крови крыс линии Wistar по отношению к массе тела). Сравнительное количество поглощенного фосфата через 15 мин после введения дозы для каждой группы (n=6) выражали как процентное количество по отношению к группе исследования, получающей носитель (n=6), как среднее ± SEM. Статистическое сравнение среднего для каждой исследуемой группы по сравнению со средним для группы носителя определяли с помощью одностороннего дисперсионного анализа с последующим ретроспективным анализом Даннетта, Р<0,05 принимали за статистически значимый уровень (НЗ - не значимый; *, Р<0,05; **, Р<0,01; и ***, Р<0,001).
Результаты исследований примеров соединений при их введении в желудок суммированы в Таблице Е1, ниже.
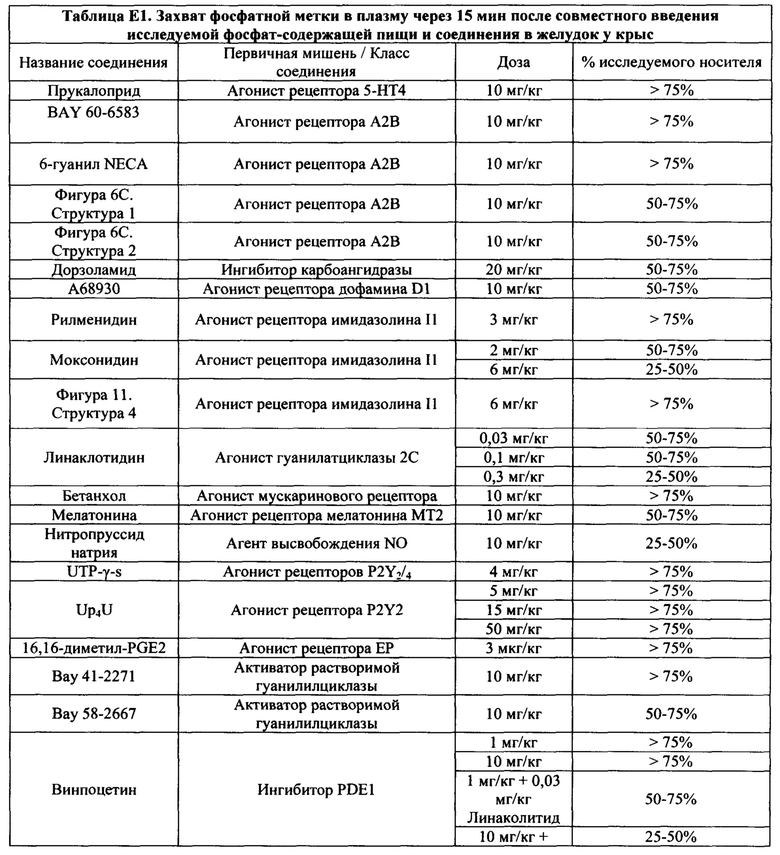
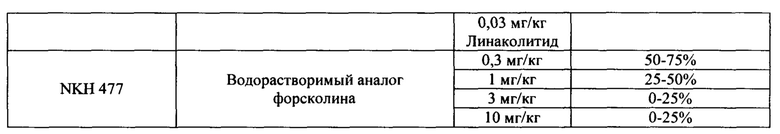
Результаты исследований примеров соединений при их введении в двенадцатиперстную кишку показаны в Таблице Е2, ниже.
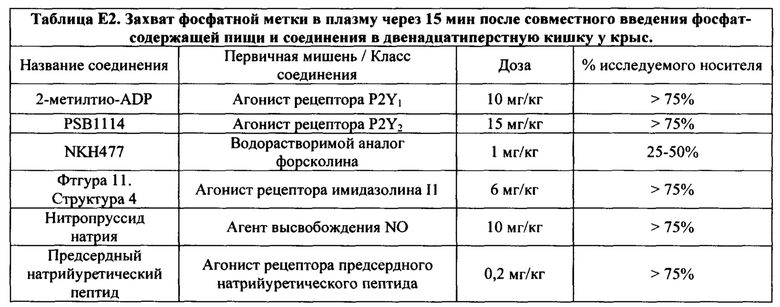
Все отдельные исследуемые соединения, которые являлись примерами агониста рецептора А2В, ингибитора карбоангидразы, агониста рецептора дофамина D1, агониста рецептора имидазолина I1, агониста гуанилатциклазы 2С, агониста рецептора мелатонина МТ2, агента высвобождения NO, активатора растворимой гуанилилциклазы и растворимого аналога форсколина, значительно снижали острый захват фосфатов из доставляемой в желудок пищи. Кроме того, было определено, что растворимый аналог форсколина при его непосредственном введении в двенадцатиперстную кишку тонкого кишечника подавлял поглощение фосфатов из вводимой совместно с ним исследуемого болюса.
Пример 6
Камера Уссинга
Сегменты двенадцатиперстной кишки и тощей кишки быстро извлекали из тела животных, находящихся под анестезией, и вскрывали вдоль мезентериальной линии и фиксировали на планшете Pyrex поверхностью слизистой оболочки вверх. Эпителиальные ткани счищали с мышечных слоев и помещали на камеры Уссинга с компьютерным управлением (National Physiology Instrument, Калифорния) с областью экспозиции 100 мм2. Ткани инкубировали с обеих сторон с 13 мл изотонического буферного раствора (рН 6,0 или рН7,4), содержащего (ммоль/л) NaCl 125,4, KCl 5,4, CaCl2, 1,2, NaHCO3, 21, NaHPO, 0,3, NaH2PO4, 1,2. Функциональную жизнеспособность и целостность ткани в начале и в конце измерения потока подтверждали с помощью измерения тока короткого замыкания (Isc) в ответ на теофиллин (10 мМ для серозной оболочки) или глюкозу (10 мМ для слизистой оболочки) или L-аланин (5 мМ для слизистой оболочки).
Для расчета скорости однонаправленных потоков Pi (Jms: поток из слизистой в серозную оболочку, Jsm: поток в противоположном направлении) на одну сторону ткани добавляли 185 КБк [33Р]-ортофосфата (370 Мбк/мл, Perkin-Elmer) и исследуемые соединения. Образцы (0,1 мл) отбирали с меченной стороны через 20 минут и затем по меньшей мере через три интервала по 10 мин с немеченой стороны (0,5 мл) камеры Уссинга. Все образцы, отобранные с немеченой стороны замещали на равные объемы изоосмотической промывочной жидкости. Суммарные потоки (Jnet) рассчитывали как различия между Jms и Jsm для тканей-партнеров, проводимость которых не отличалась более чем на 25%. В другой серии экспериментов измерения потока проводили до и после добавления арсената (для слизистой оболочки) или оуабаина (для серозной оболочки) в промывочный раствор. Измерения радиоактивности проводили в жидкостном сцинтилляционном счетчике TopCount (Perkin Elmer).
Пример 7
Анализы in vitro - ex vivo
Сегменты двенадцатиперстной кишки и тощей кишки (5 см) извлекали из тела животных, находящихся под анестезией пентобарбитоном натрия, промывали ледяным 0,9% солевым раствором и выворачивали на стеклянные стержни. Образцы надежно фиксировали на стержне и затем инкубировали в течении 5 мин при 37°С в подкисленном буфере, рН 7,4 или 6,0, содержащем 16 мМ гидроксиэтилпиперазин-N'-2-этансульфоновую кислоту, 10 мМ глюкозу, 3,5 мМ KCl, 10 мМ MgSO4, 1 мМ CaCl2, 125 мМ NaCl, с последующей инкубацией в течение 2 мин в том же буфере, содержащем 100 мМ 33Pi (33Pi-специфическая активность 1,85 МБк/мл) и исследуемые соединения. Буфер быстро перемешивали с помощью магнитной мешалки для минимизации воздействия статичных водных слоев на слизистые поверхности.
Захват останавливали путем воздействия на ткань фосфатно-солевым буфером, содержащим 10-кратный избыток нерадиоактивного фосфата, в течение 10 минут при комнатной температуре. Затем проводили дополнительный этап промывки в течение 10 минут в фосфатно-солевом буфере при комнатной температуре. Затем образцы промакивали насухо и записывали их массу. Образцы перерабатывали в течение ночи в смеси Protosol (PerkinElmer). Сцинтилляционный анализ захвата в переработанном образце и исходном растворе позволяет рассчитывать удержание фосфатов в ткани (в нмоль/г).
Пример 8
Скриниговые анализы, связанные с мишенью
Активация кишечных рецепторов может приводить к сигнальному пути, который прямо или косвенно вызывает подавление всасывания фосфатов (например, по результатам изменения локального рН люминальных мембран кишки). Способность соединения взаимодействовать с указанными мишенями можно измерять с использованием коммерчески доступных клеточных линий, в которых по-разному экспрессируется интересующая мишень. Указанные клеточные линии общедоступны из компаний, таких как Perkin Elmer или Multispan. Альтернативно, можно также использовать первичные клетки, экспрессирующие интересующую мишень.
Измерение взаимодействия гипотетического лиганда можно осуществлять с помощью любого из двух подходов (см. Таблица Е3, ниже): (1) перемещение радиоизотопно меченного стандартного лиганда из интактных клеток или мембран, полученных из таких клеток или (2) измерение продукции вторичного посредника при лечении с помощью исследуемого соединения. Для оценки вторичных посредников доступно множество коммерческих наборов для измерения внутриклеточного cAMP, cGMP (например, из компании Cis Bio) и кальция (например, краситель кальция 6 из компании Molecular Devices).
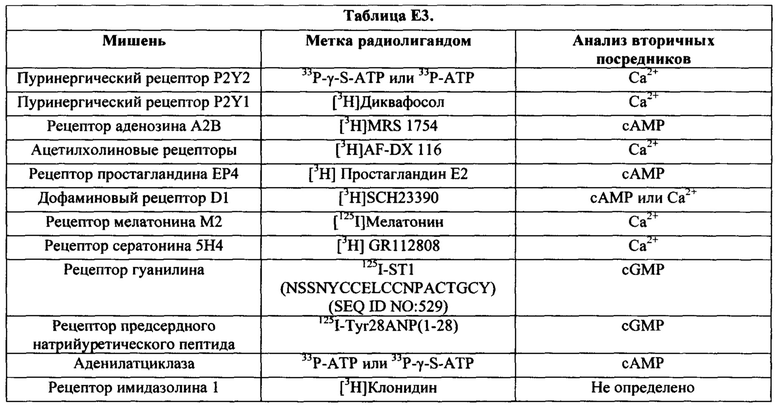
В случаях непосредственного воздействия на активность растворимого фермента можно использовать ферментативный анализ, в котором используется очищенный фермент и отслеживается продукт ферментативной реакции (см. Таблицу Е4, ниже).

Пример 9
Подавление кишечного всасывания натрия и фосфатов
Для оценки способности выбранных примеров соединений подавлять всасывание фосфатов из просвета кишечника измеряли баланс захвата и выведения фосфатов у крыс. Восьминедельных крыс линии Sprague Dawley получали из компании Charles River Laboratories (Холлистер, Калифорния). Животным позволяли акклиматизироваться в течение по меньшей мере 6 дней со свободным доступом к пище и воде. В течение этого времени и на протяжении всего исследования крыс можно кормить стандартным кормом (Harlan Teklad, Мэдисон, Висконсин; корм для грызунов 2018 Teklad Global, содержащий 18% белка) или синтетическим кормом на основе яичного белка, состоящим из 0,6% Са и 0,35 или 0,6% фосфора (Harlan Teklad; TD.84122 и TD. 130318, соответственно).
За день до начала исследования крысам позволяли акклиматизироваться к отдельным метаболическим клеткам со свободным доступом к воде и кормам, описанным выше, в порошкообразном виде. Животным вводили дозу примерно за час 1 до наступления темновой фазы либо путем перорального введения эффективной дозы исследуемого изделия 10 мл/кг, либо путем добавления лекарственного средства в корм (на основе установленной для потребления крысами суточной массы корма). При обеих схемах дозирования каждой крысе в течение каждого дня их содержания в метаболических клетках обеспечивали свободный доступ к воде и аликвоте порошкообразного корма, что представляло собой среднее суточного потребления ad libitum этого типа корма для крыс одного типа (т.е. 8-недельные самцы крыс в среднем потребляют 18 г/сутки очищенного корма, указанного выше). Такой подход обеспечивал снижение вариабельности и стандартизацию измерений потребления и выведения через 24 часа. Измерение суточного потребления воды и корма, а также сбор суточной мочи и кала проводили в течении четырех последовательных дней (с 1 по 4 день).
Содержание фосфата, натрия и калия в образцах мочи определяли с помощью ионной хроматографии. Образцы мочи обрабатывали с определением гравиметрического объема с последующим окислением 6 н HCl. Окисленные образцы быстро центрифугировали (3,600 × g) и супернатанты затем разводили 10 мМ HCl. Разведенные образцы, калибровочные стандарты (Sigma/Fluka Analytical) и образцы ВС (внутренние стандарты) фильтровали до введения в систему ионообменной хроматографии (Dionex ICS-3000). Натрий и калий элюировали в изократическом режиме с использованием 25 мМ метансульфоновой кислоты в качестве подвижной фазы и катионообменной аналитической колонки Dionex CS12A. Фосфат элюировали в изократическом режиме с использованием 35 мМ гидроксида калия в качестве подвижной фазы и анионообменной аналитической колонки Dionex AS18. Количественный анализ проводили с использованием программы Dionex Chromeleon. Все концентрации образцов выводили из калибровочной кривой на основе площади хроматографического пика.
Содержание фосфата, натрия, кальция и калия в образцах кала, полученных через каждые 24 часа, определяли с помощью атомной эмиссионной спектроскопии. Высушенные фекалии или репрезентативные образцы, полученные из высушенных гомогенизированных фекалий, перерабатывали путем многократного добавления концентрированной азотной кислоты и перекиси водорода в течение 2-3 часов при 65-95°С. Растворы образцов затем разбавляли 1% азотной кислотой до проведения анализа с помощью атомной эмиссионной спектроскопии (Agilent 4100 MP-AES) при следующих длинах волн испускания элементов: кальций (422,673 нм), натрий (588,995 нм), калий (766,491 нм) и фосфор (214,915 или 213,618 нм). Раствор цезия использовали в качестве ионизирующего буфера и внутреннего стандарта. Анализ данных проводили с использованием программы Agilent MP Expert.
Рассчитывали суточное выведение фосфатов с мочой или калом относительно Р, потребляемого из корма, для каждого животного и для каждого дня, когда проводились измерения. Процентное подавление всасывания фосфора выражали путем определения снижения указанного отношения по сравнению с контрольной группой (с животными, корм которых не содержал лекарственное средство). Такой способ также применим для других интересующих ионов. Если исследование проводится в разные дни, то оно может предоставлять результаты повторных измерений баланса фосфатов в состоянии покоя для каждой крысы, в случае чего регулярное суточное потребление фосфатов животными является обязательным условием. Повышение содержания фосфатов в фекалиях с почти сопутствующим снижением Р в моче для поддержания нейтрального баланса у крыс является показателем общего снижения всасывания фосфатов у крыс, получающих лечение примером соединения.
Пример 10
Эффекты хронического заболевания почек (ХЗП) на крысиной модели.
Для оценки способности выбранного примера соединения носить вклад в кальцификацию мягких тканей, часто связанную с поздними стадиями ХЗП, использовали модель крыс с 5/6 нефрэктомией (5/6Nx) для оценки минерального гомеостаза при заболевании. У крыс 5/6Nx, обычно используемых в качестве модели для исследования различных аспектов ХЗП, в норме не наблюдается повышенного уровня фосфатов при отсутствии стимуляции фосфатами из пищи (см. Shobeiri et. al., Am J Nephrol. 31: 471-481, 2010, Vascular Calcification in Animal Models of CKD: A Review). Таким образом, для подтверждения эффективности и устойчивого прогрессирования фосфатемической кальцификации сосудов указанным животным давали пищу, содержащую фосфаты с повышенной биодоступностью, в комбинации с витамином D3, что представляло собой адаптированный метод, разработанный группой Лопеза (см. Lopez et al., J Am Soc Nephrol. 17: 795-804, 2006. Calcimimetic R-568 Decreases Extraosseous Calcifications in Uremic Rats Treated with Calcitriol).
Самцов крыс линии Sprague-Dawley с 5/6 нефрэктомией получали из компании Charles River Laboratories (Холлистер, Калифорния), при этом хирургические процедуры осуществлял поставщик. Снижение функциональной почечной массы достигали путем двух хирургических операций: субтотальной нефрэктомии левой почки с последующим восстановлением в течение 1 недели до проведения унинефрэктомии правой почки. После восстановления в течение трех дней после второй хирургической операции крыс перемещали в испытательный центр в возрасте 9 недель.
По прибытии в центр и на протяжении всего исследования крыс кормили очищенной порошкообразной пищей, состоящей из 0,9% неорганического Р (фосфор) и 0,6% Са (TD, 10809, Harlan-Teklad, Мэдисон, Висконсин). Утренние образцы сыворотки получали путем сбора крови из ретроорбитальной области или из хвостовой вены. В исследование включали только животных, имеющих уровень креатинина в сыворотке от 0,9 до 1,2 мг/дл, при этом животных делили на группы (n=12) на основе уровня креатинина в сыворотке и массы тела. Включенным в исследование крысам в лечебных группах вводили лекарственное средство вместе с пищей с использованием того же корма, что и у группы, получающей носитель, описанной выше. Дополнительно инициировали режим введения кальцитриола (активного Витамина D3 80 нг/кг внутрибрюшинно) 3 раза в неделю.
Функцию почек, содержание фосфата, а также другие параметрами отслеживали еженедельно с измерением подходящих сывороточных маркеров с помощью стандартных клинико-химических исследований или ИФА-анализа. Крыс с содержанием креатинина в сыворотке более чем 2 мг/дл или с массой тела, составляющей 80% или менее от средней массы тела группы, удаляли из исследования из-за поздней стадии заболевания. Показатели функции почек в моче также можно измерять при помещении крыс в метаболические клетки, позволяющие сбор выделений.
Через 4 недели крыс усыпляли и органы собирали и взвешивали. Определяли минерализацию фрагментов дуги аорты, сердца, желудка и почки. Цельные образцы ткани перерабатывали путем многократного добавления концентрированной азотной кислоты и перекиси водорода в течение 2-3 часов при температуре 65-95°С. Растворы образцов затем разводили 1% азотной кислотой до проведения анализа с помощью атомно-эмиссионного спектрометра (Agilent 4100 MP-AES) при следующих длинах волн испускания элементов: кальций (422,673 нм), натрий (588,995 нм), калий (766,491 нм) и фосфор (214,915 или 213,618 нм). Раствор цезия использовали в качестве ионизирующего буфера и внутреннего стандарта. Анализ данных осуществляли с помощью программы Agilent MP Expert.
Снижение кальцификации сосудов у животных, получавших лечение исследуемыми изделиями, по сравнению с партнерами, не получавшими указанное лечение, согласуется с заявленным подавлением всасывания фосфатов из пищи, необходимого для развития заболевания в этой крысиной модели ХЗП.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ | 2004 |
|
RU2543350C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ | 2004 |
|
RU2353383C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ | 2014 |
|
RU2763796C2 |
| ПЕПТИДНЫЕ АНТАГОНИСТЫ ПЕПТИДНЫХ ГОРМОНОВ ИЗ СЕМЕЙСТВА КАЛЬЦИТОНИНА (CGRP) И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2742826C2 |
| ПЕПТИДНЫЕ АНТАГОНИСТЫ ПЕПТИДНЫХ ГОРМОНОВ ИЗ СЕМЕЙСТВА КАЛЬЦИТОНИНА (КАЛЬЦИТОНИН ГЕН-РОДСТВЕННЫХ ПЕПТИДОВ (CGRP)) И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2624016C2 |
| РУКОВОДСТВО ПО ТЕРАПИИ И/ИЛИ МОНИТОРИНГ ТЕРАПИИ ДЛЯ ЛЕЧЕНИЯ ШОКА | 2020 |
|
RU2835616C1 |
| СОЕДИНЕНИЯ-АГОНИСТЫ GIP И СПОСОБЫ | 2015 |
|
RU2716985C2 |
| МНОГОФУНКЦИОНАЛЬНЫЕ СЛИТЫЕ БЕЛКИ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2815388C2 |
| Аналоги инсулина | 2017 |
|
RU2769476C2 |
| ВАРИАНТЫ, КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ ЭНДОНУКЛЕАЗЫ CBLB | 2018 |
|
RU2779097C2 |
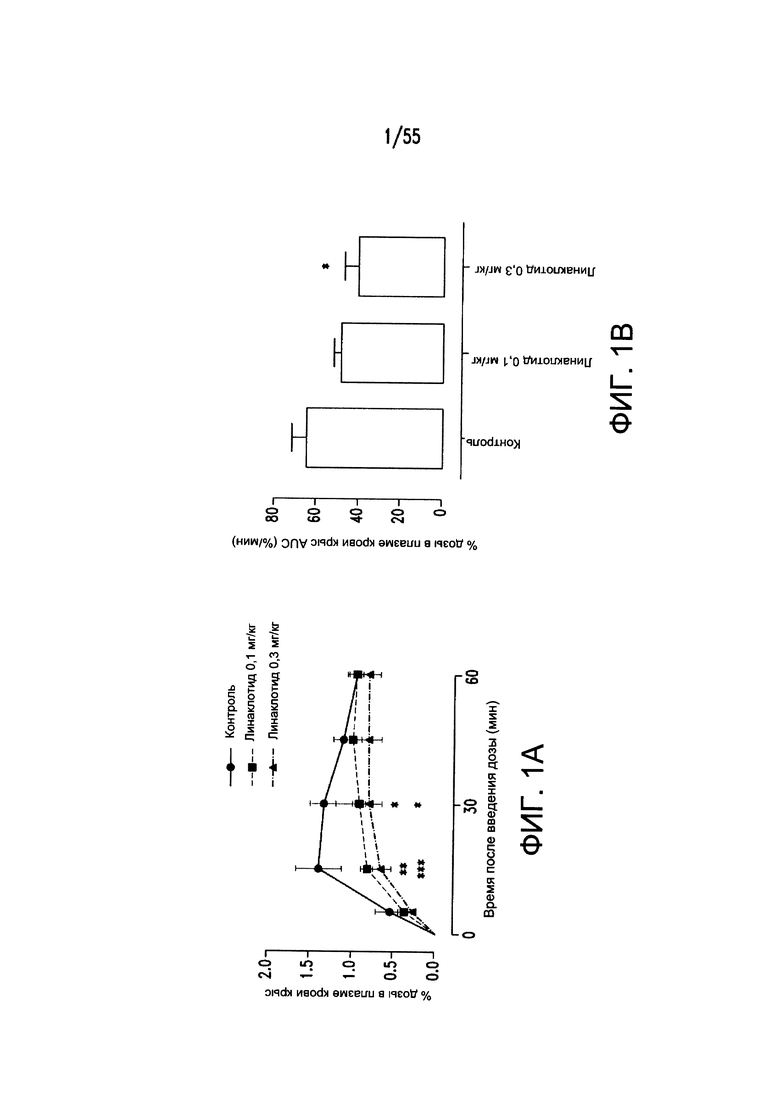
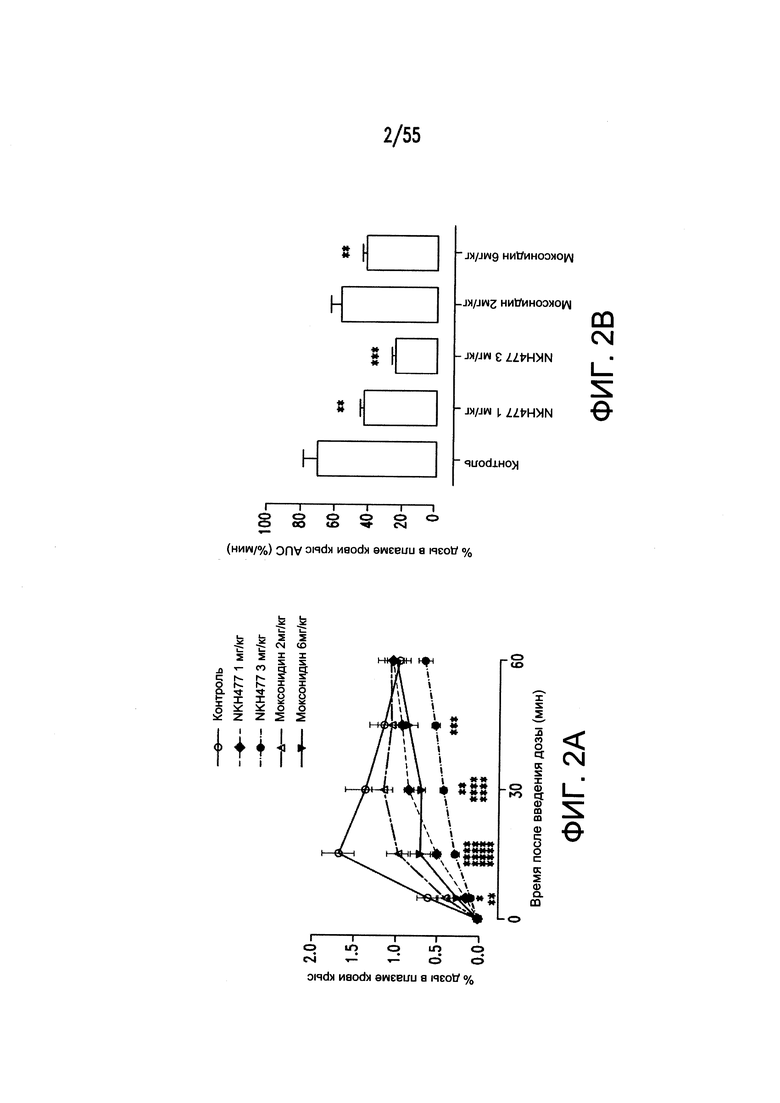
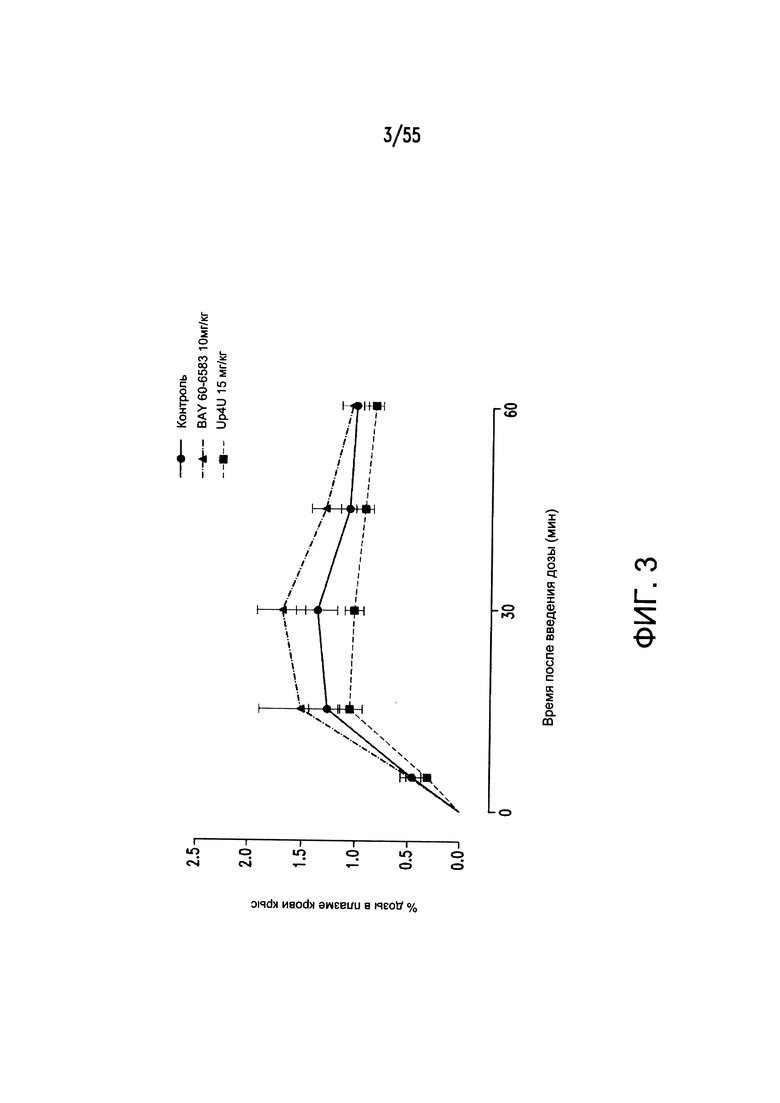
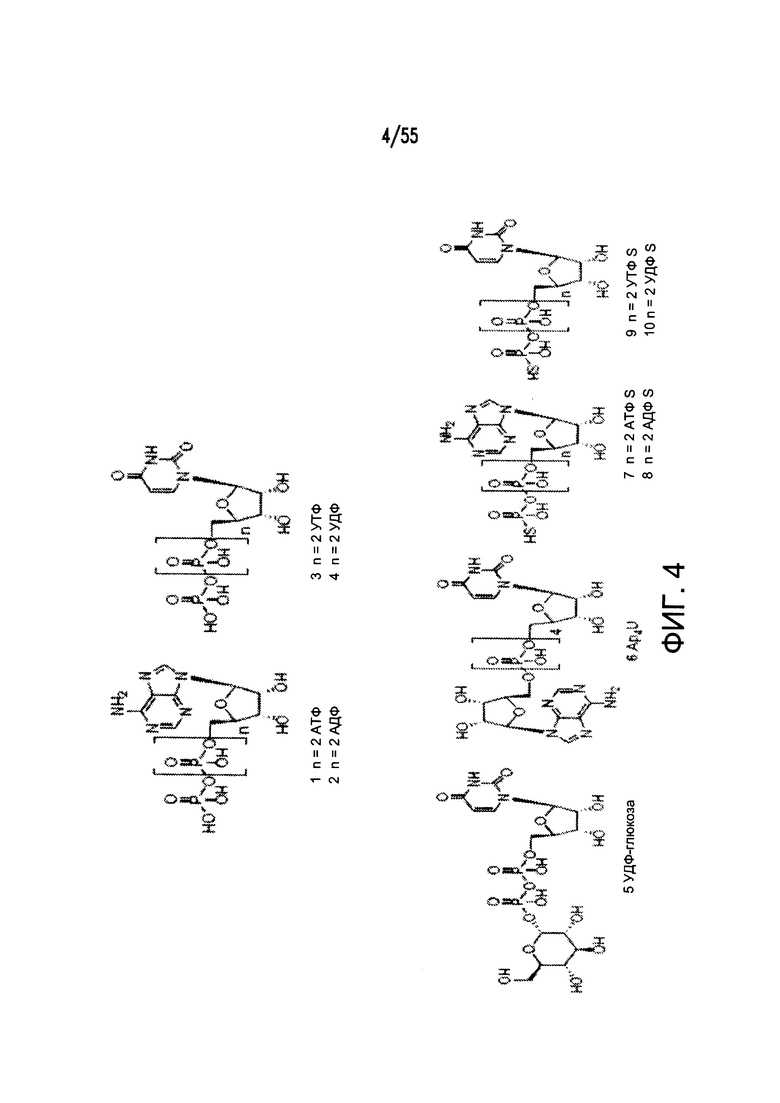
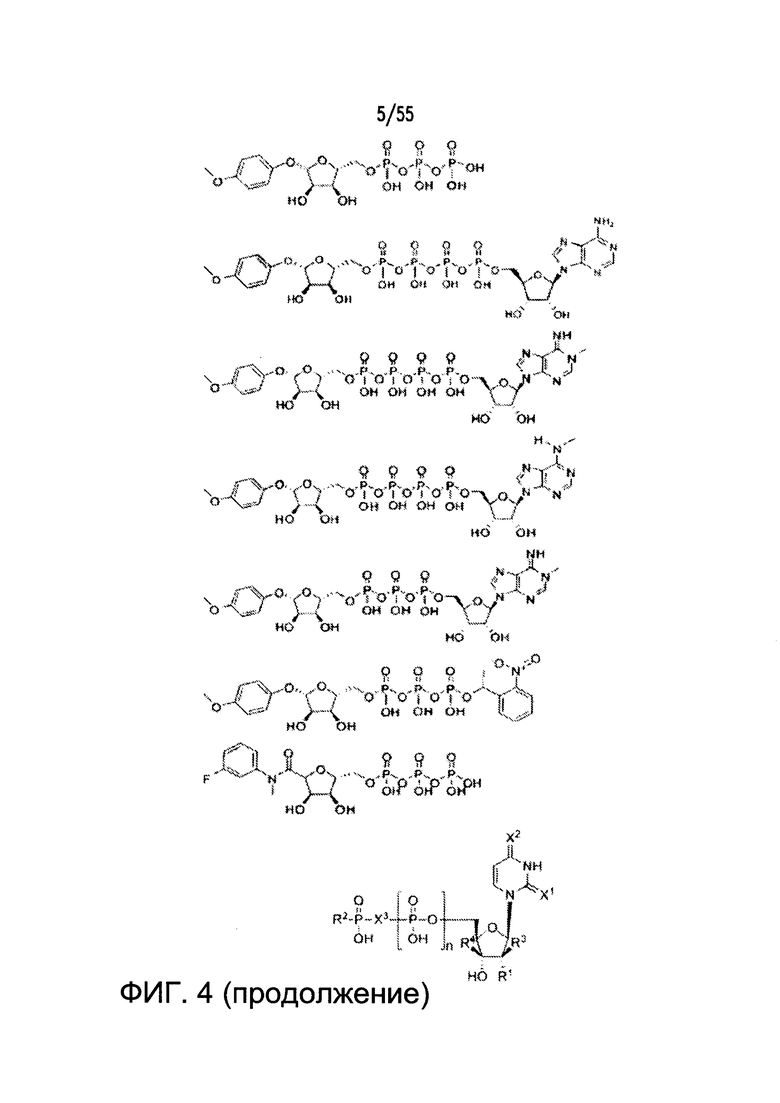
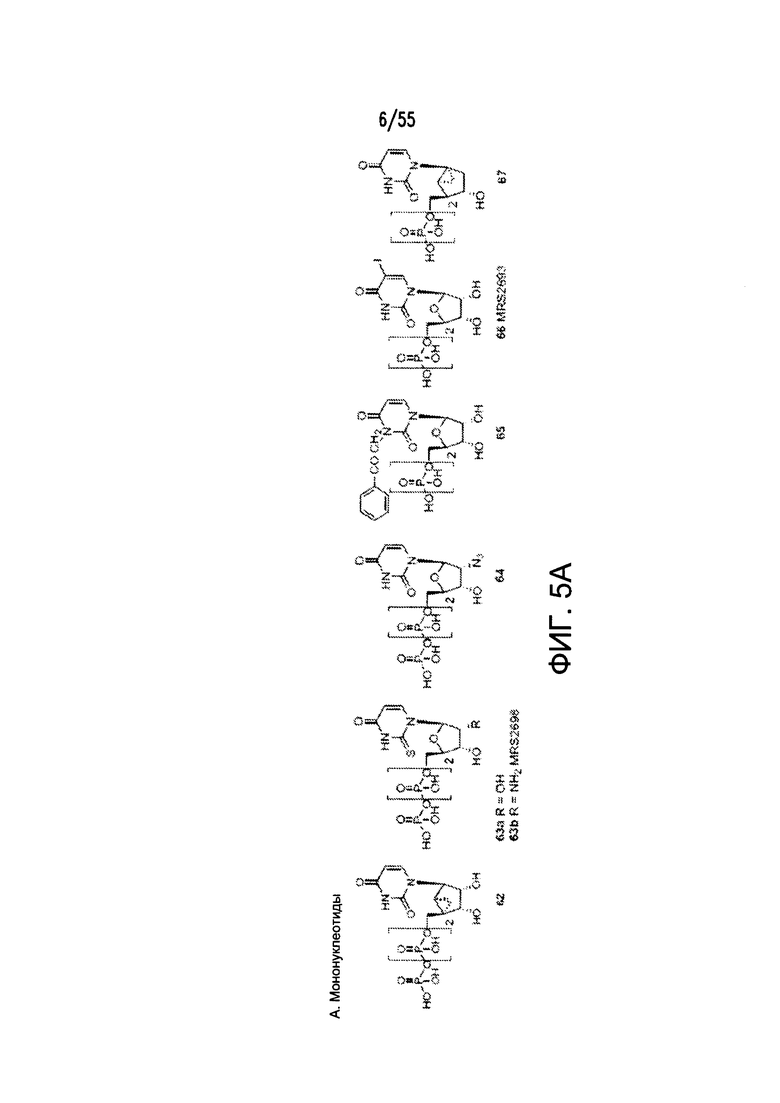

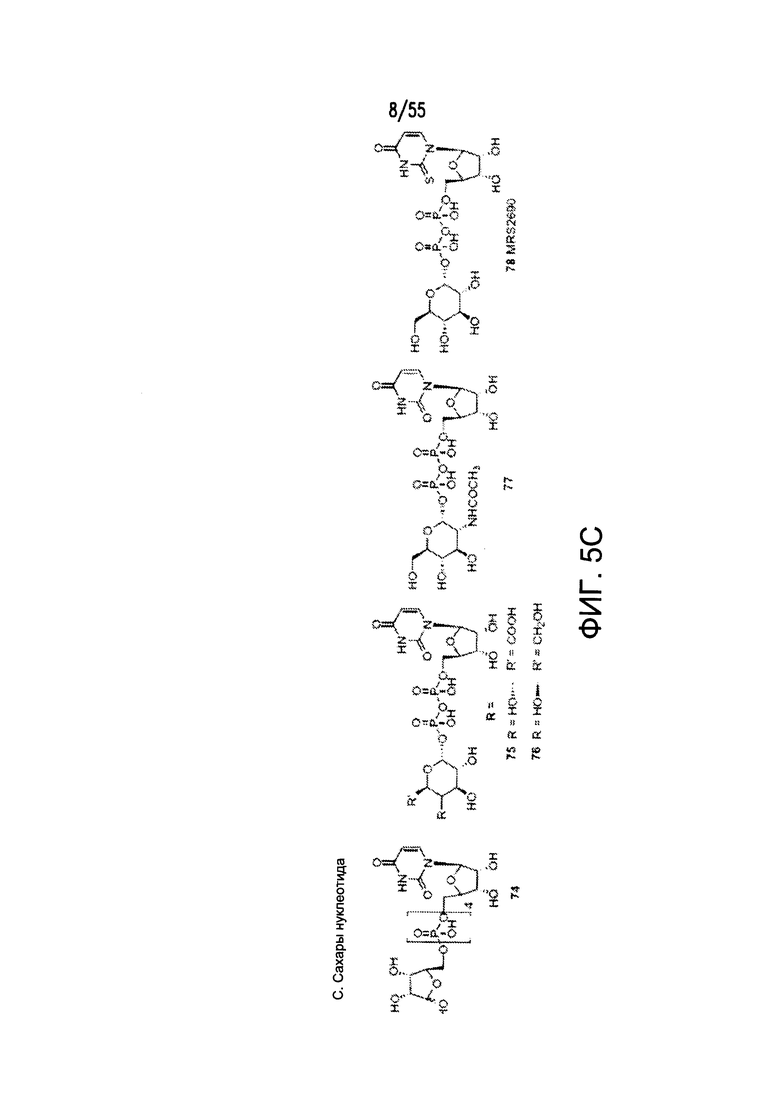
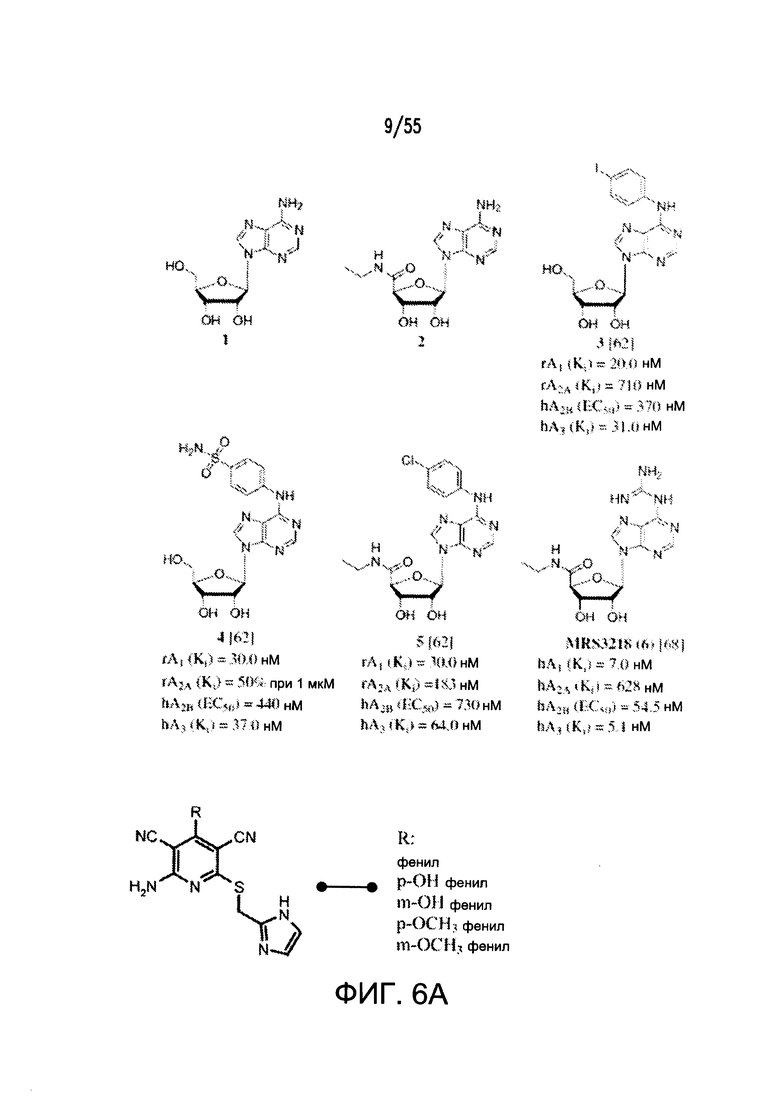
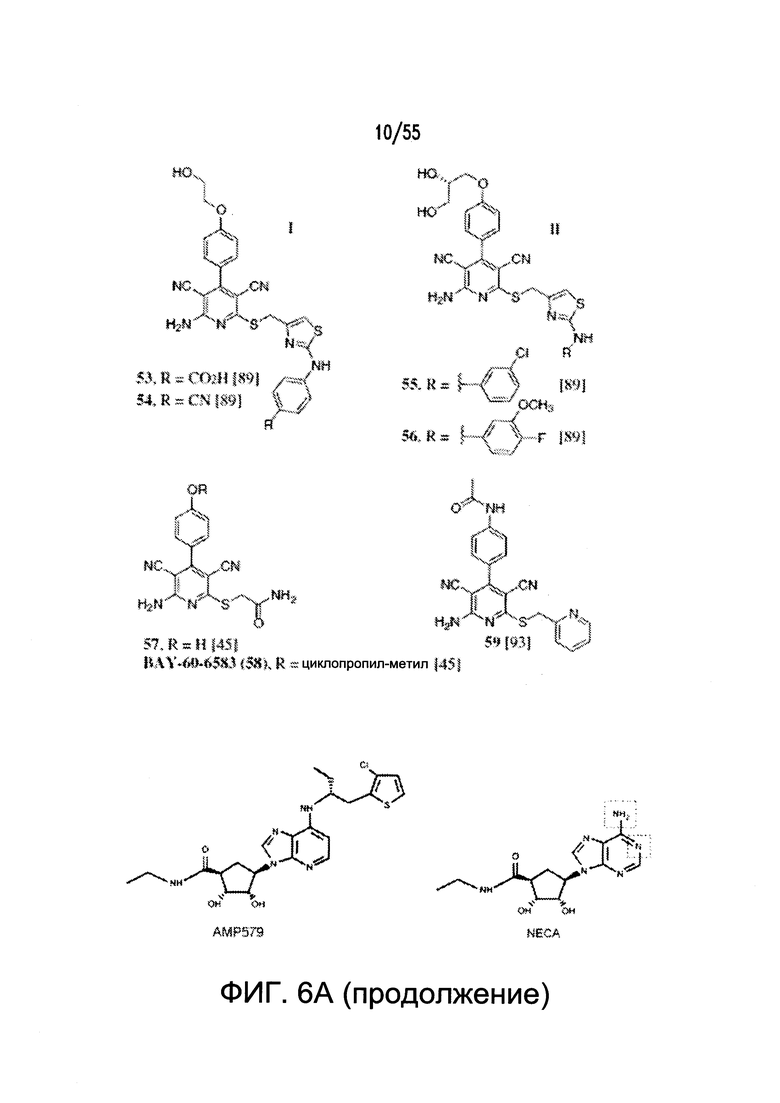
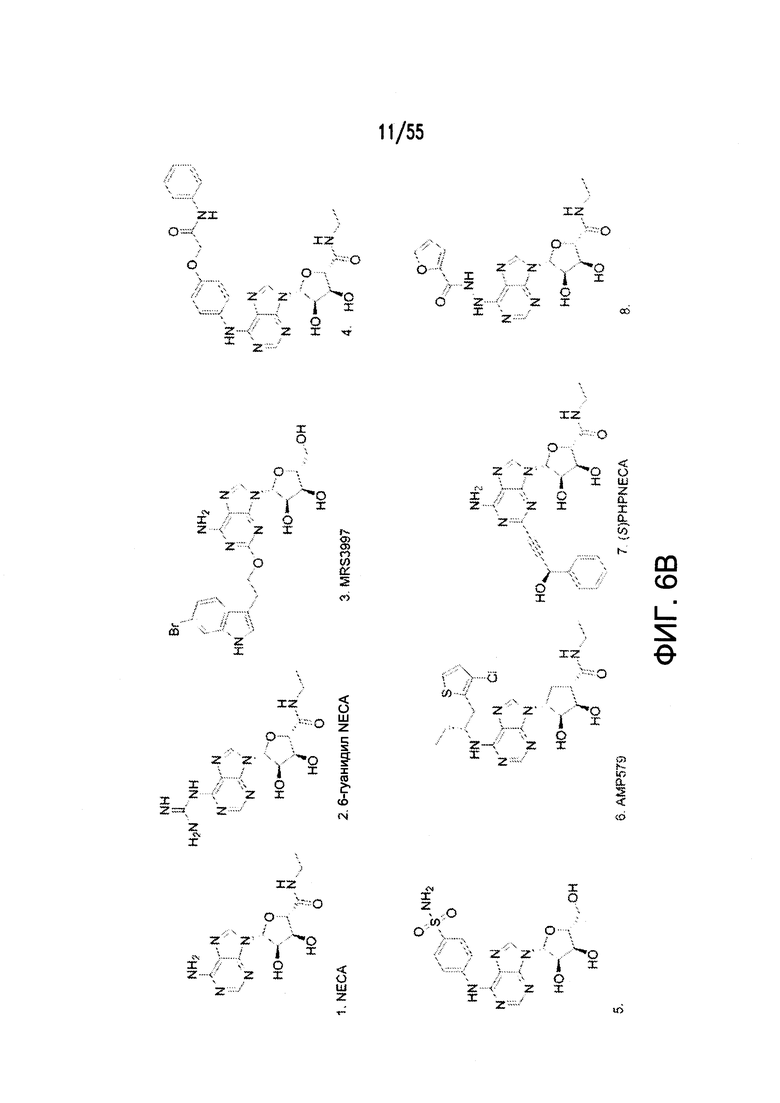
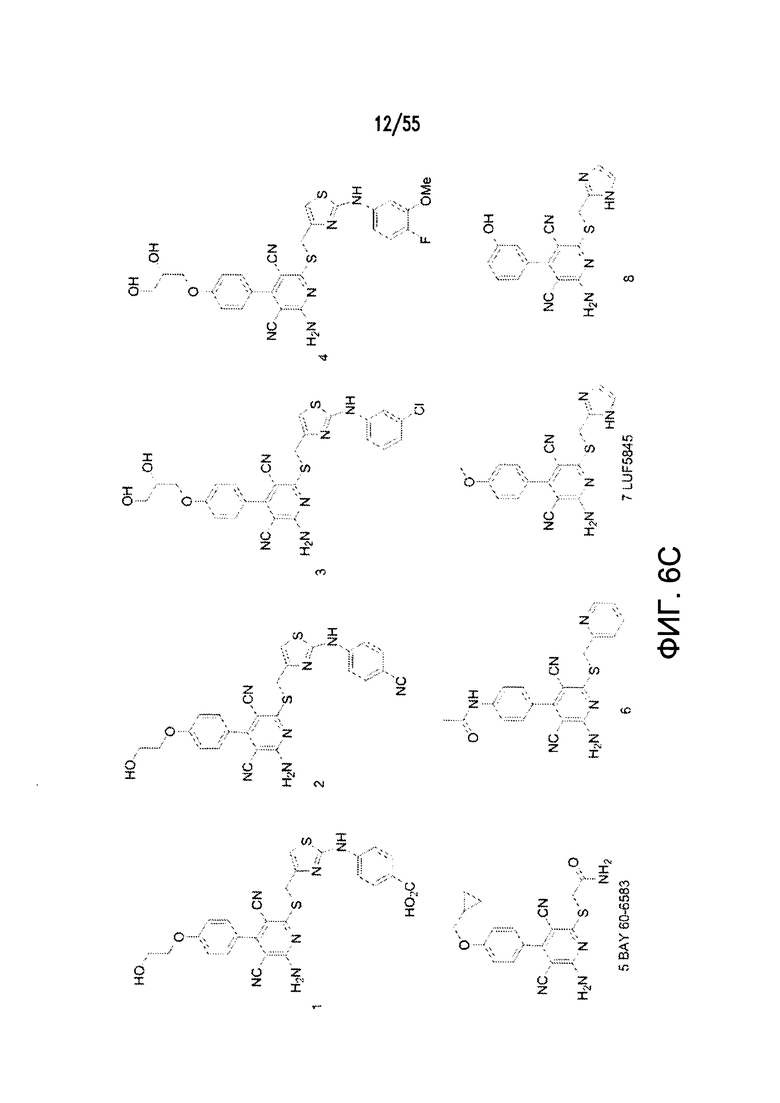
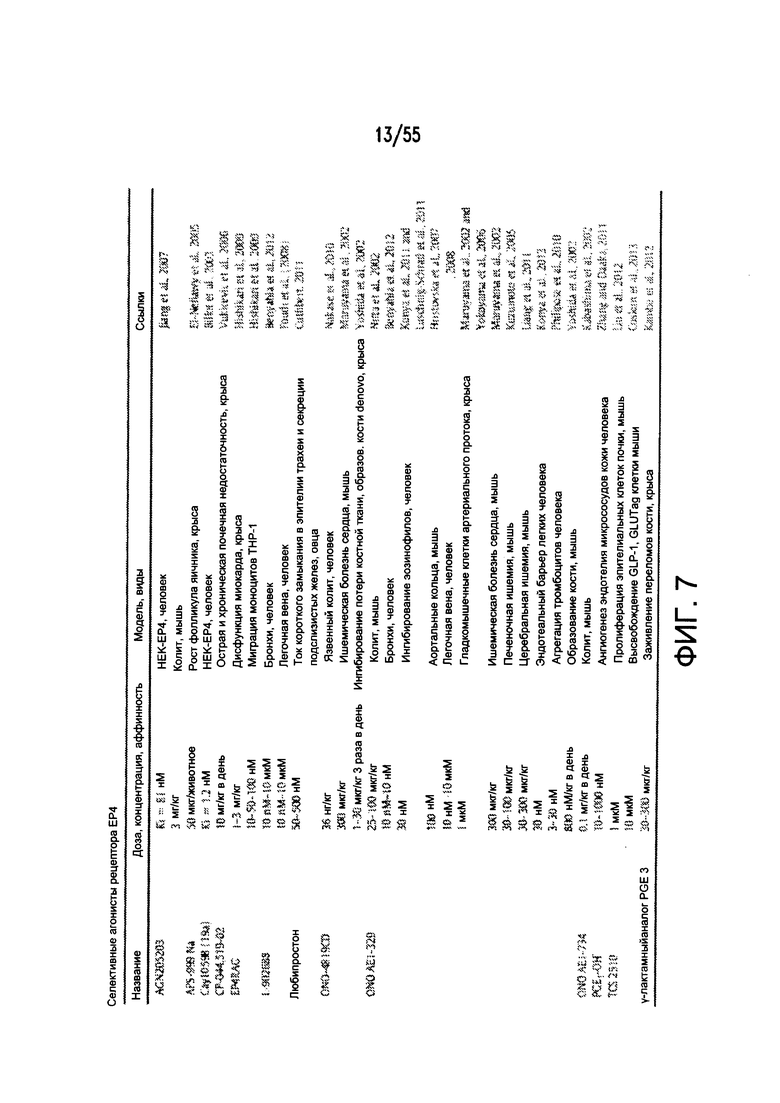


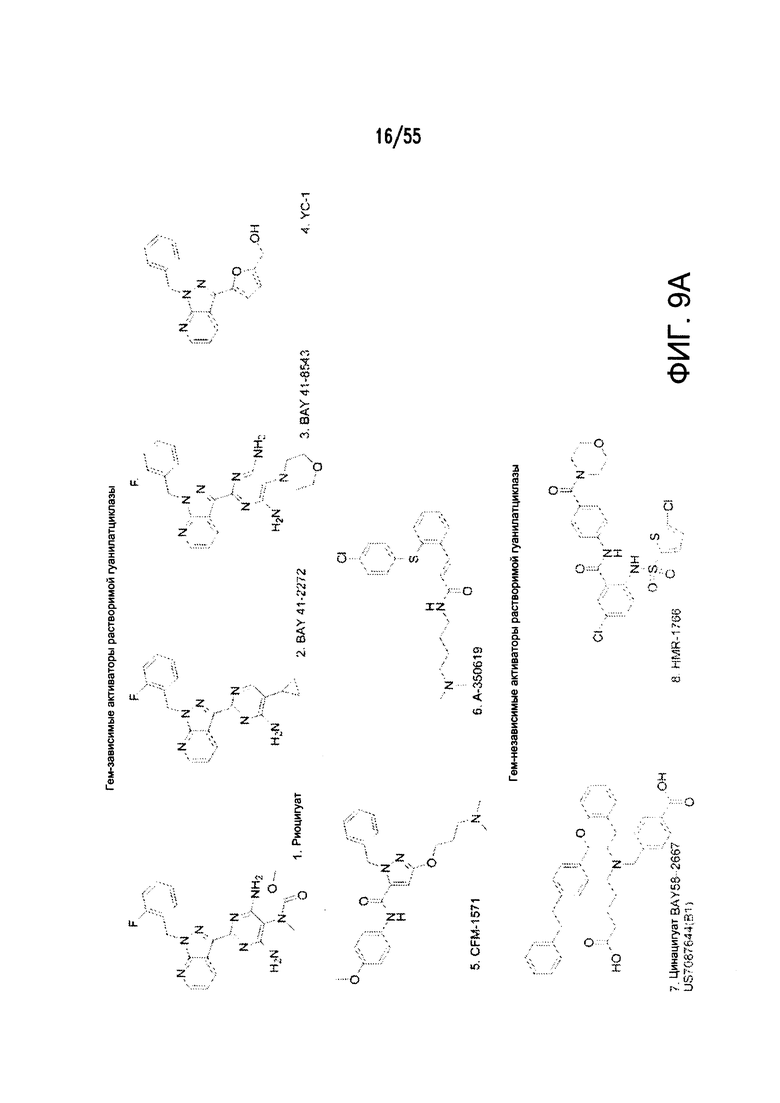
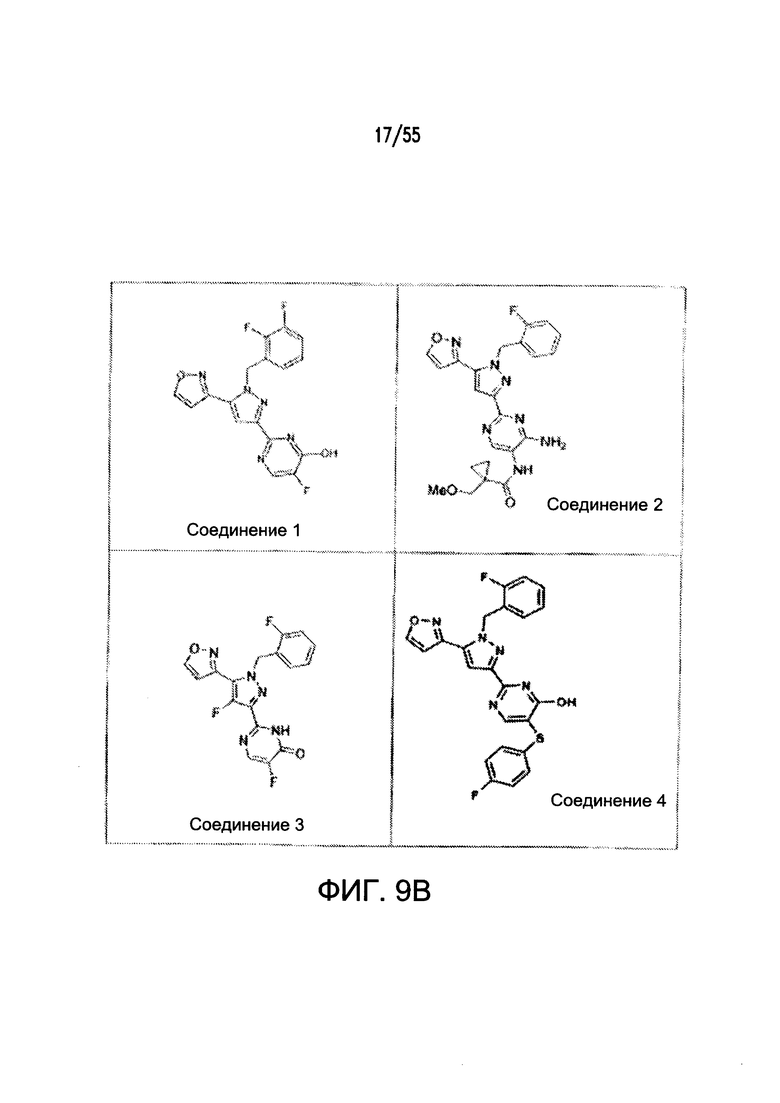
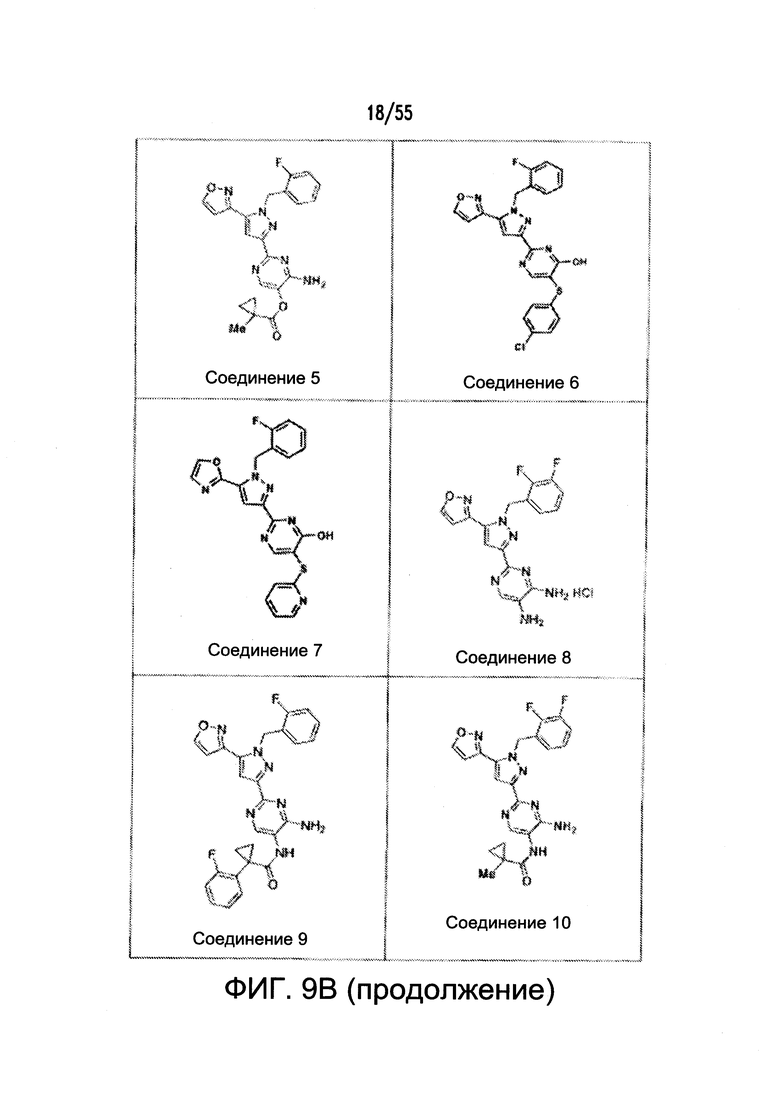
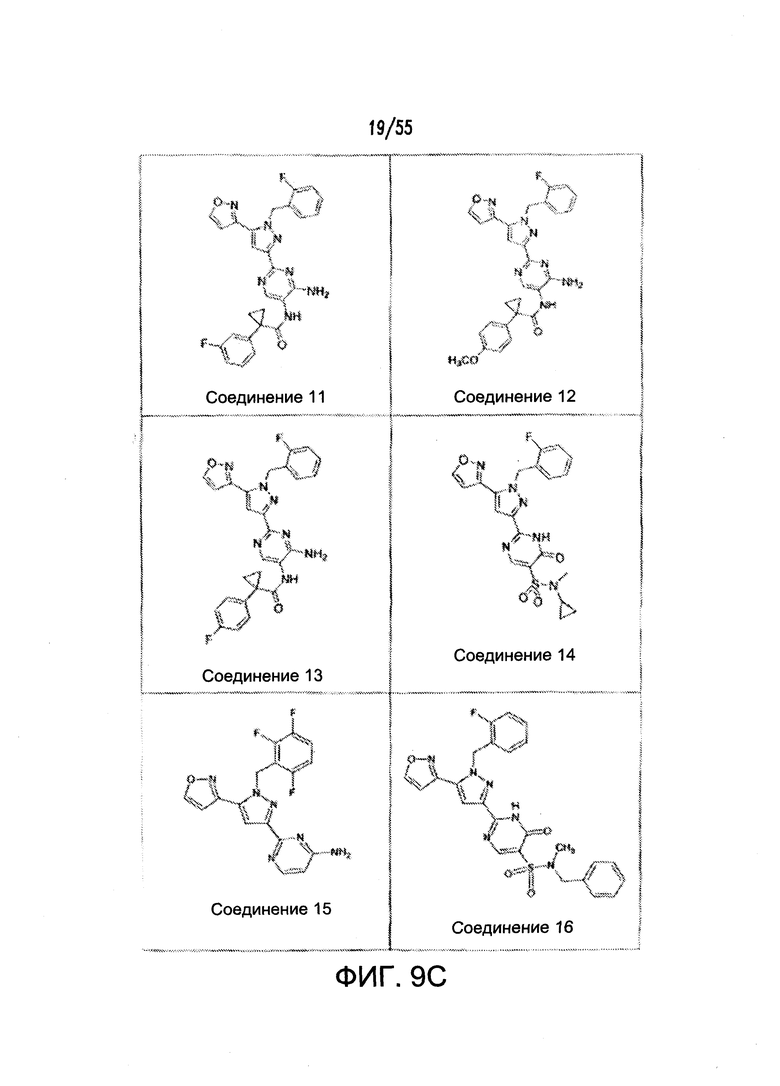
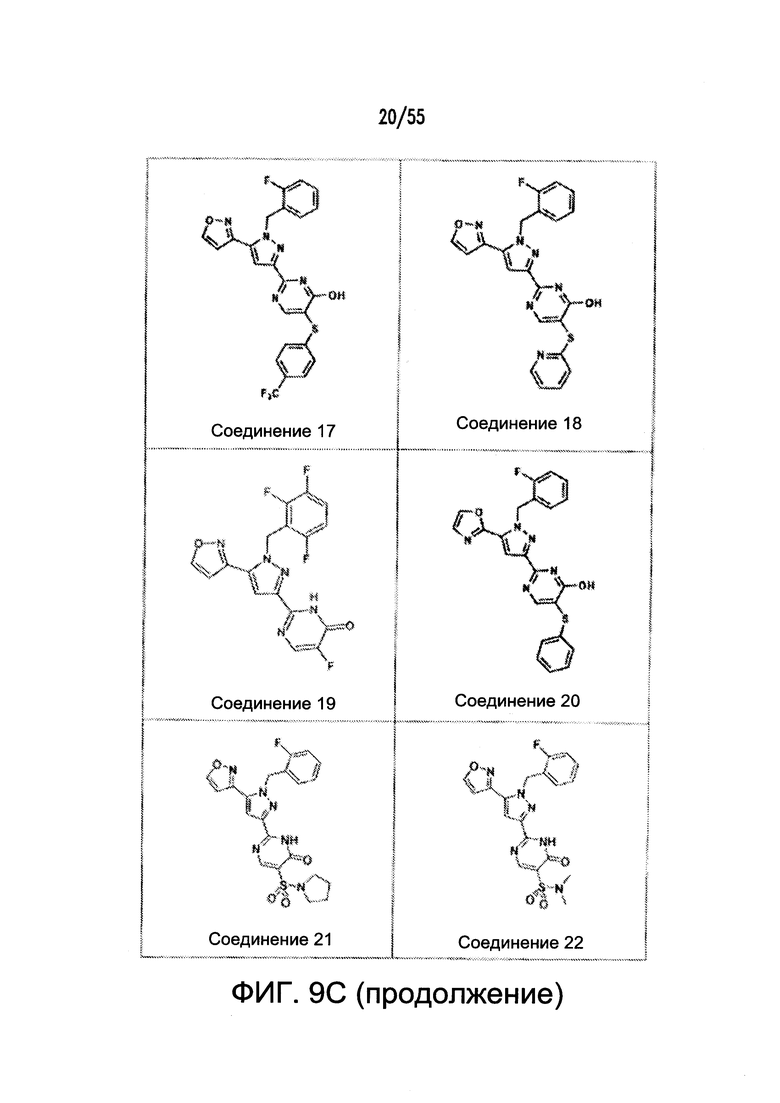
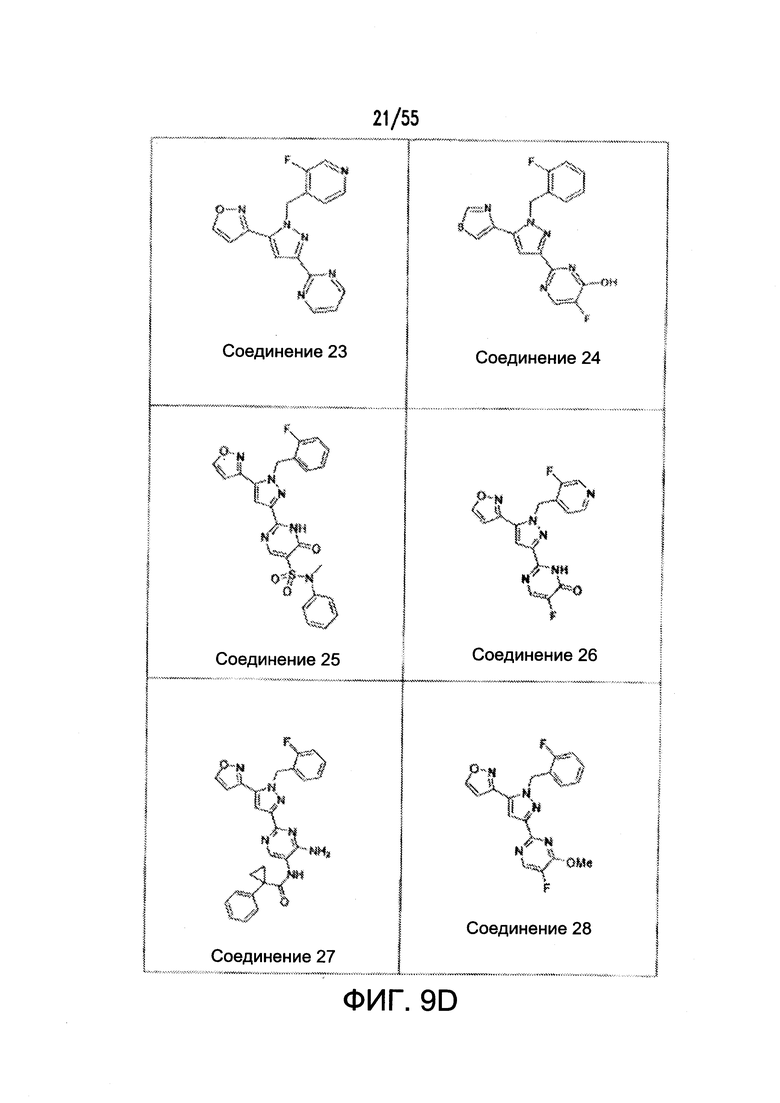
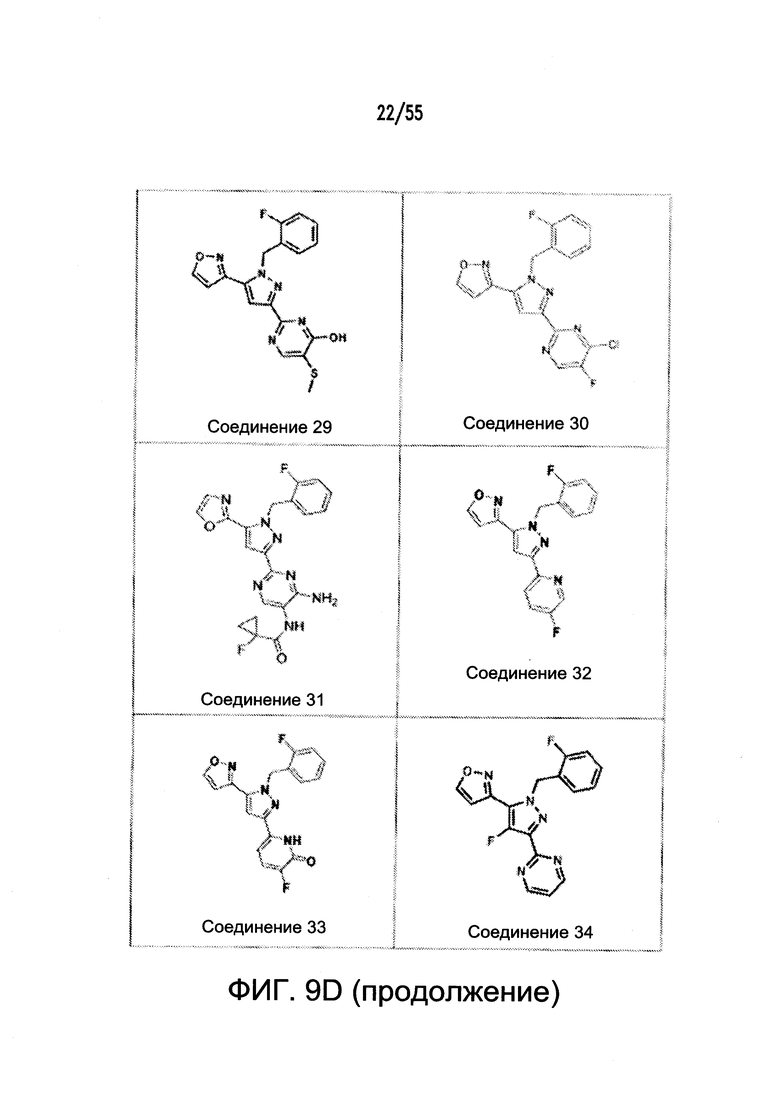
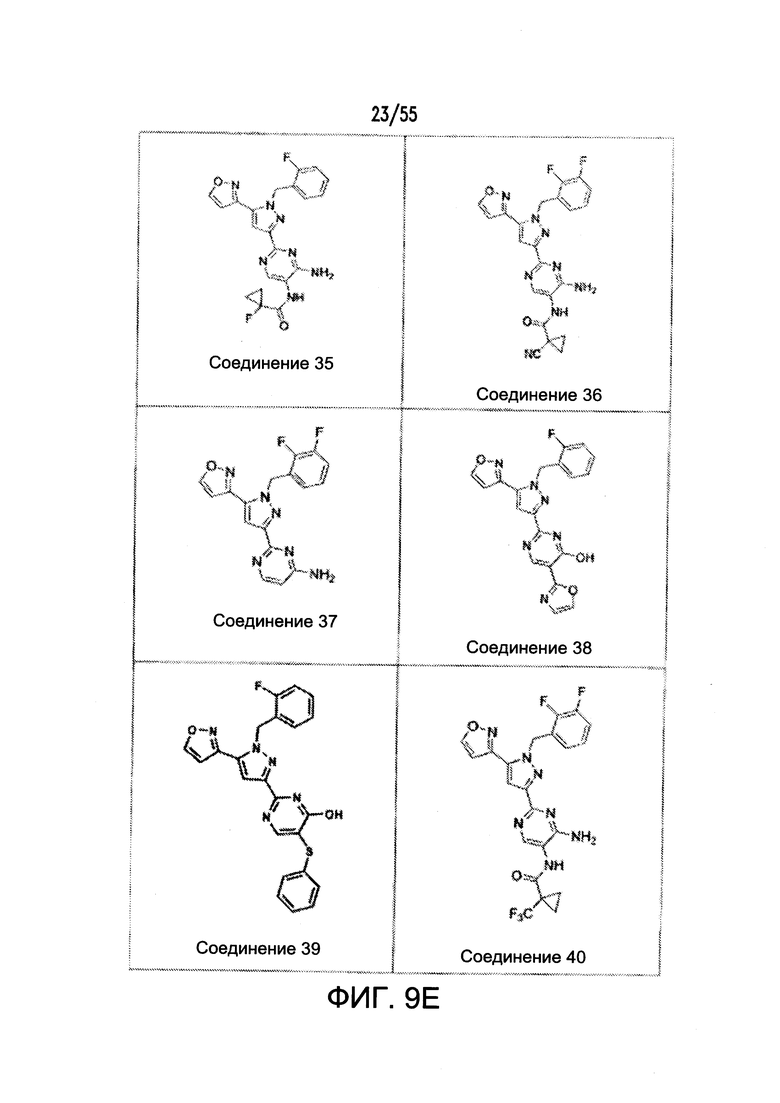
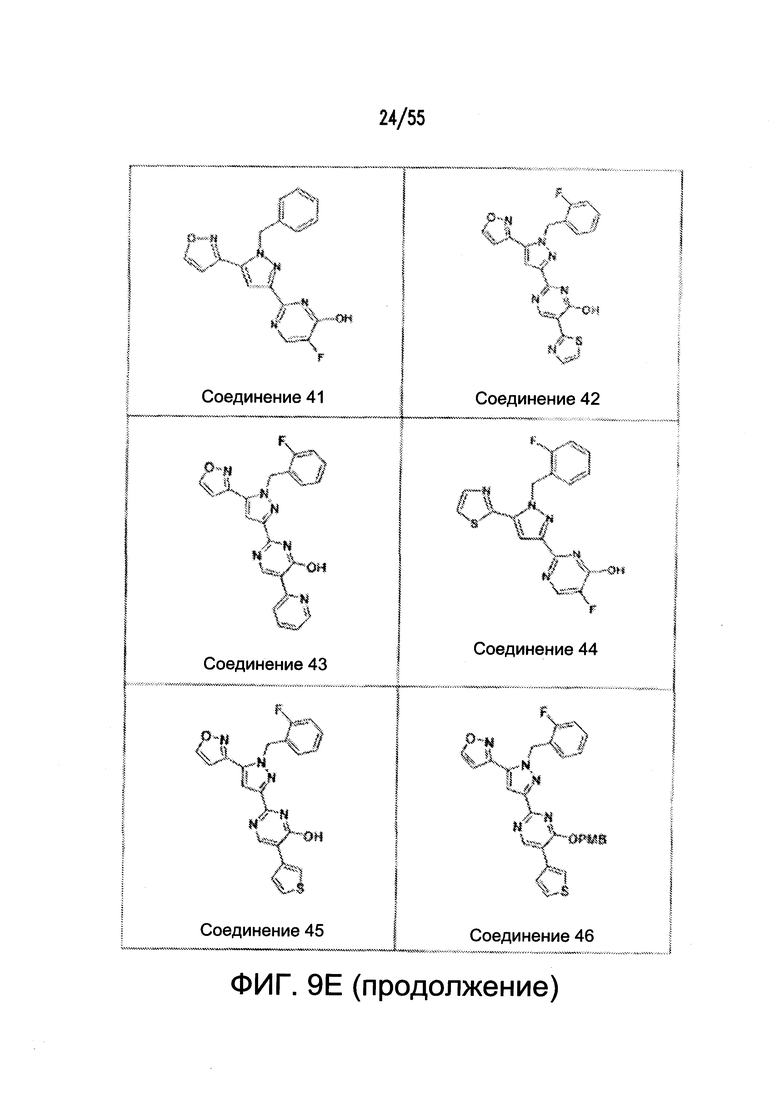
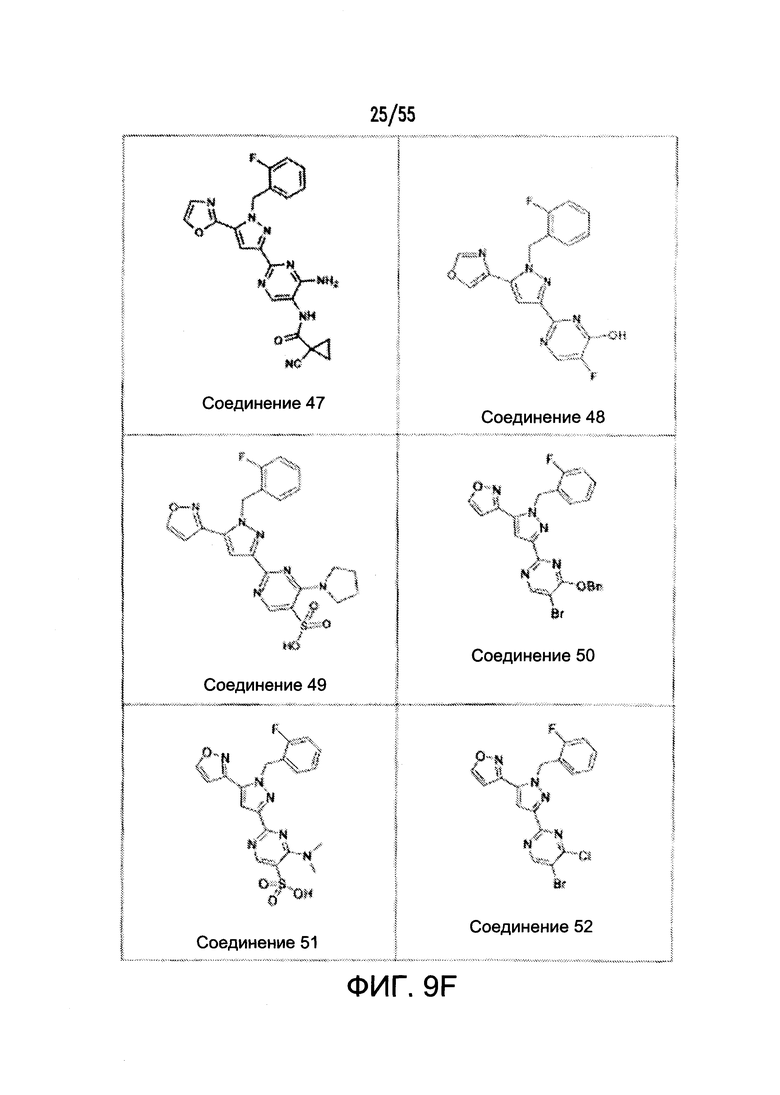
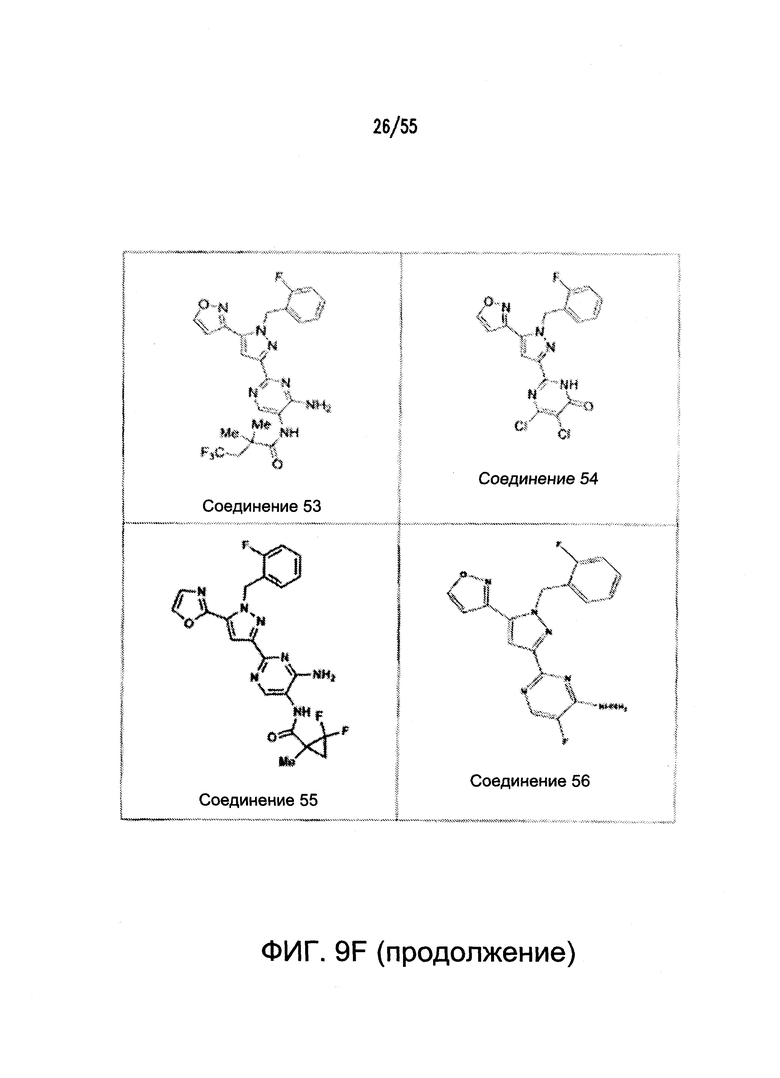
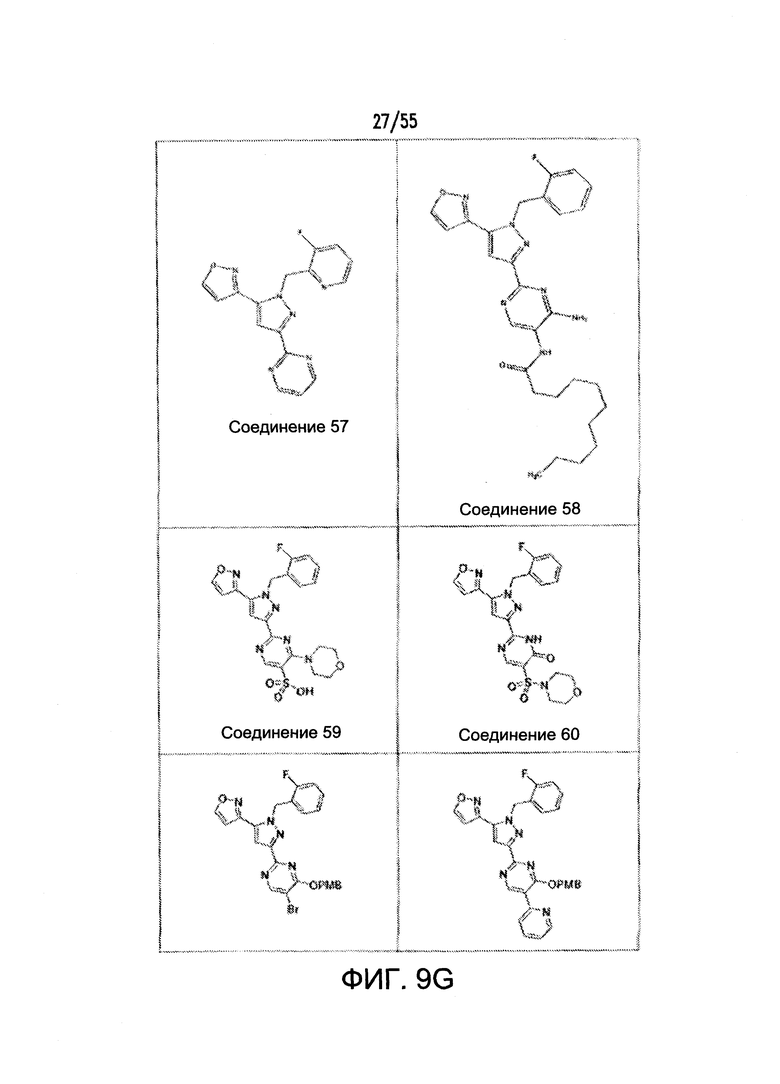
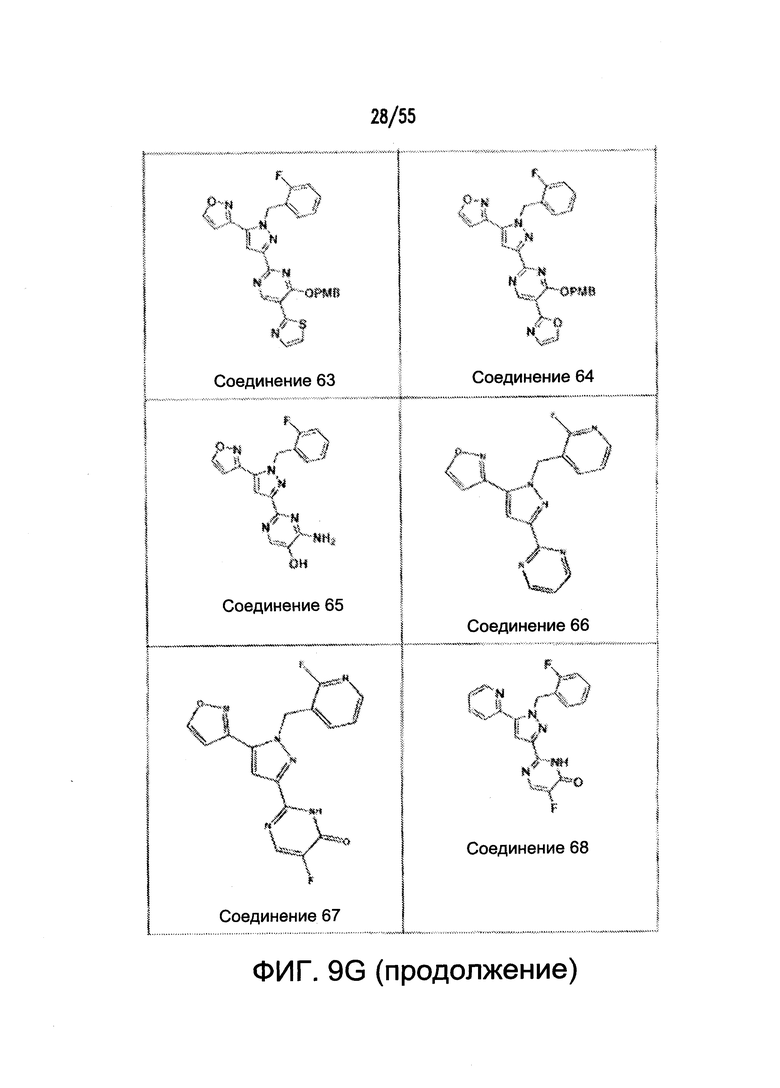
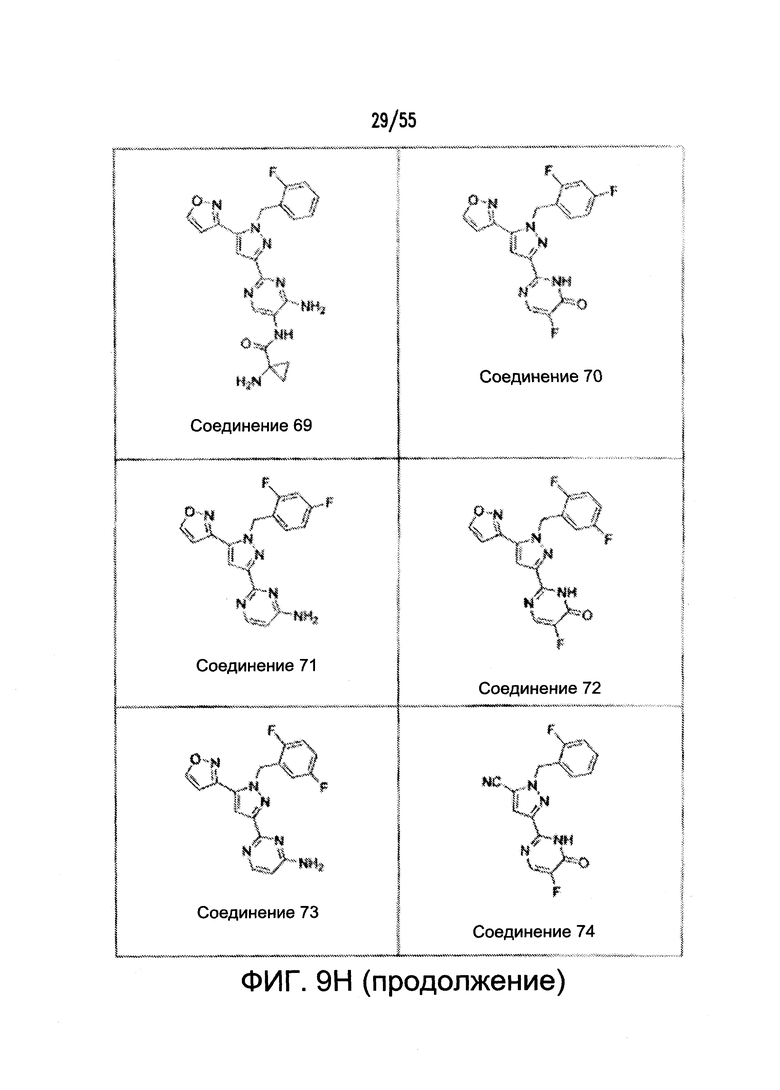
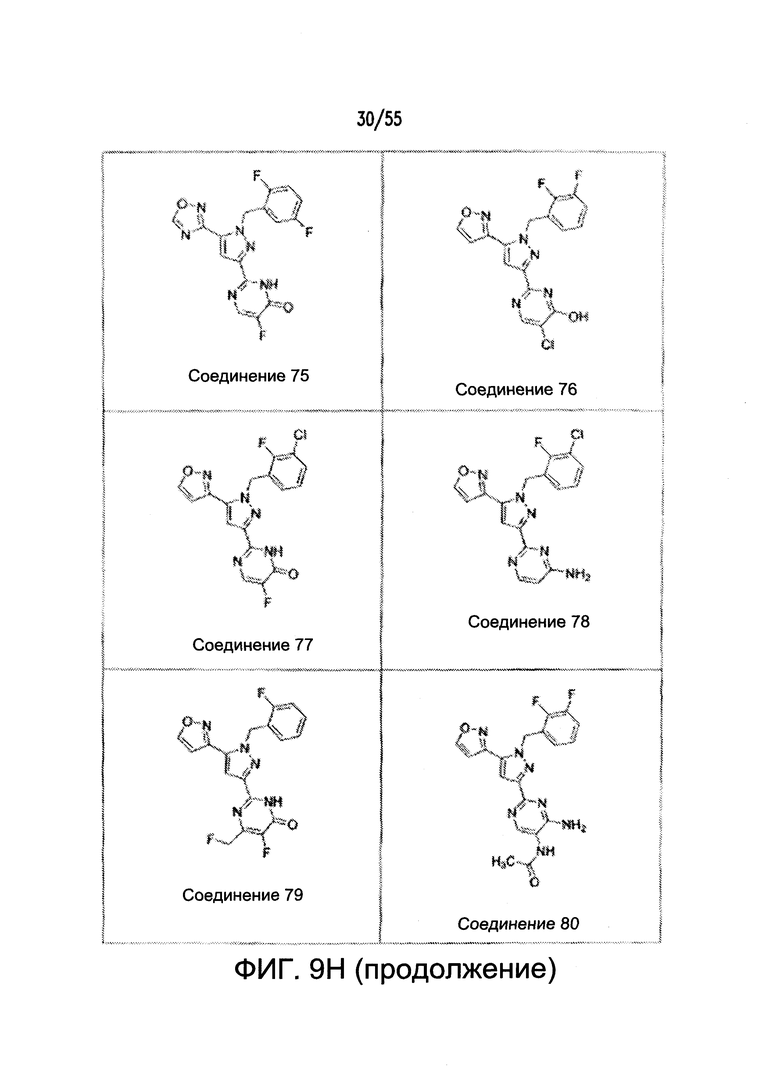
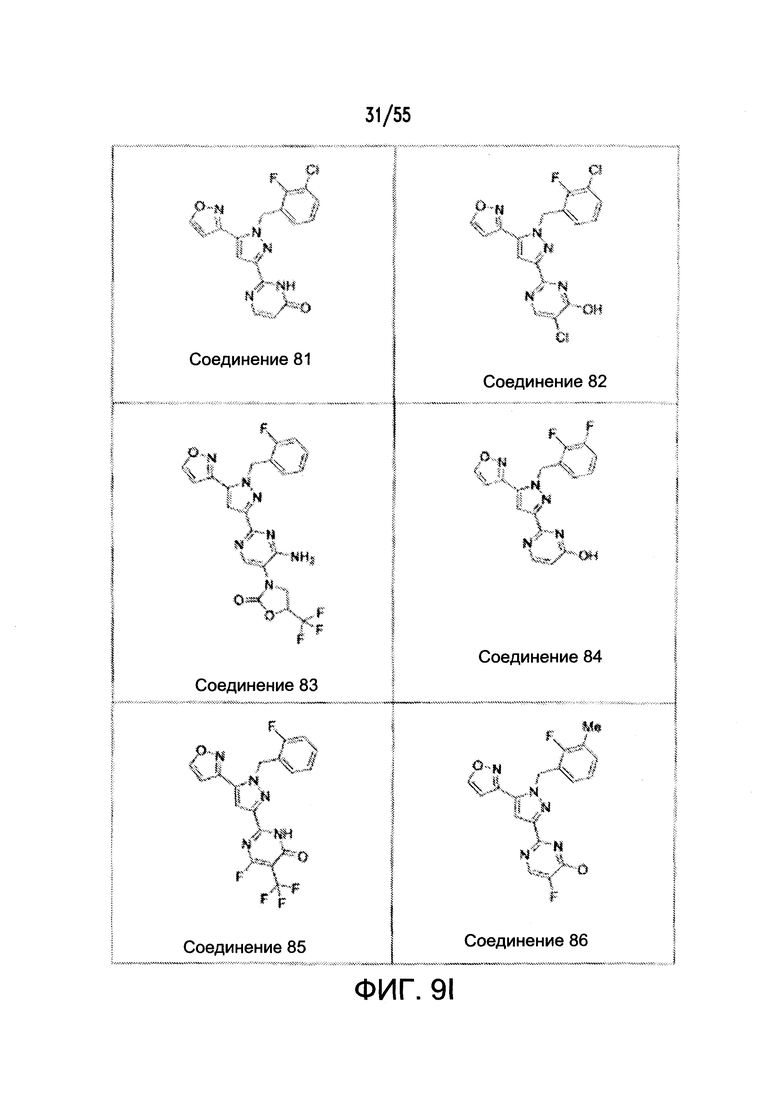
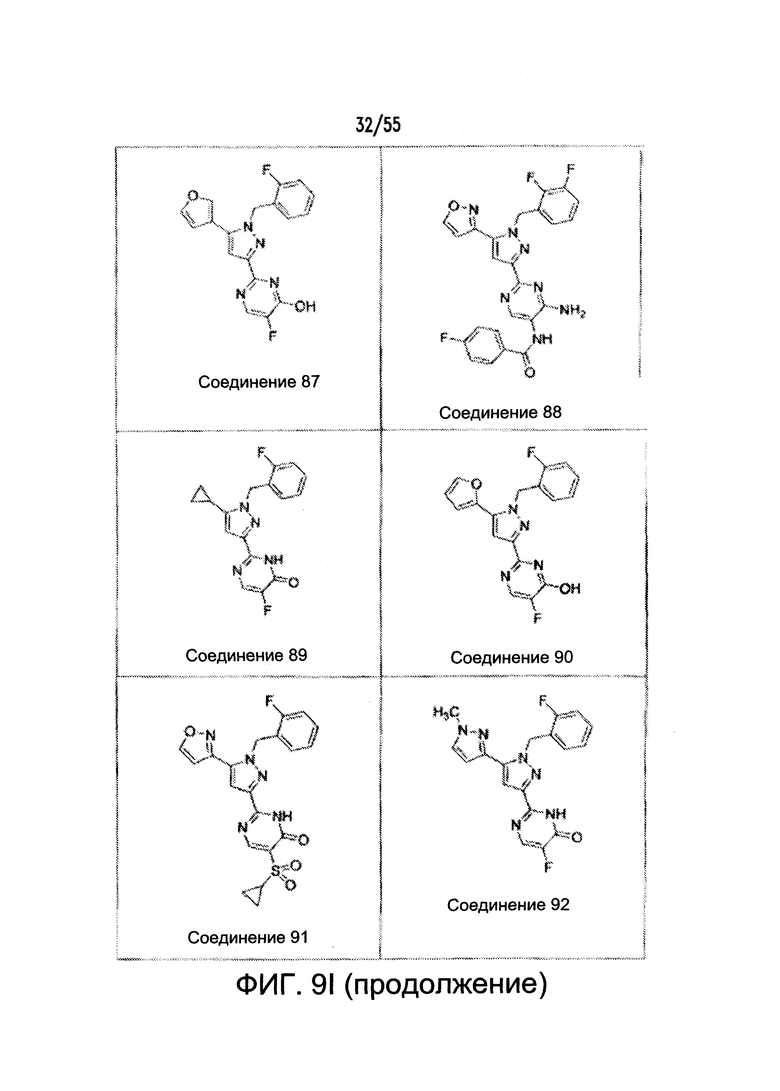
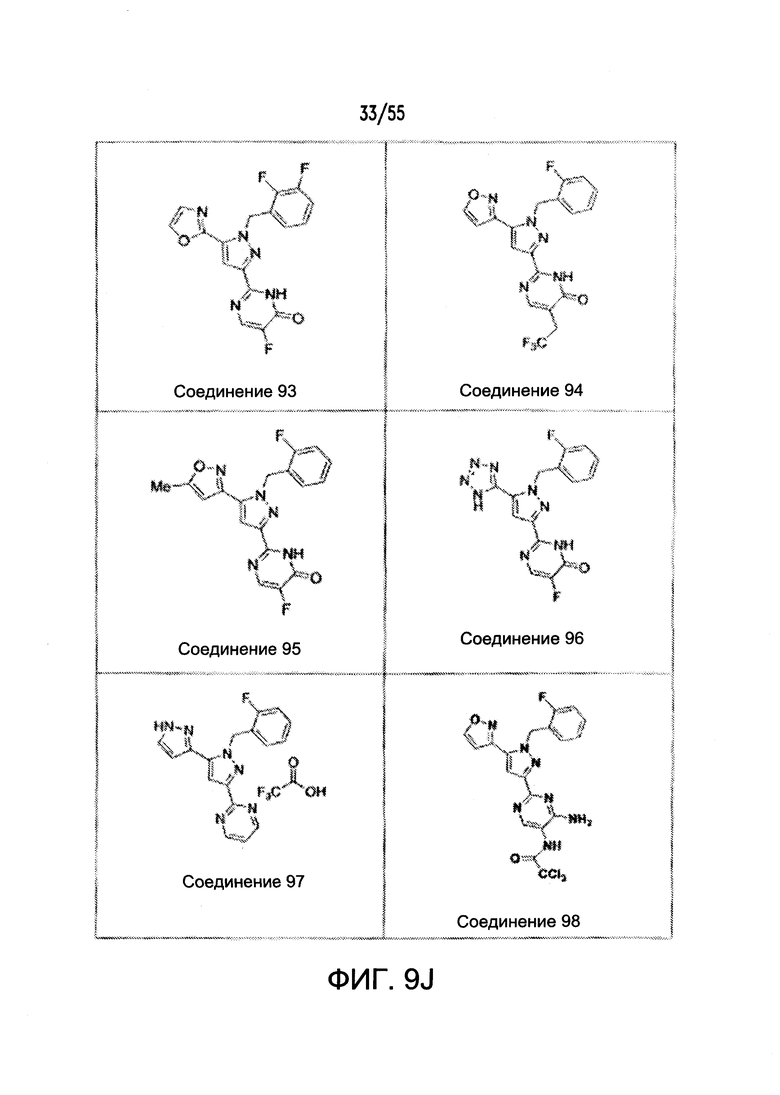
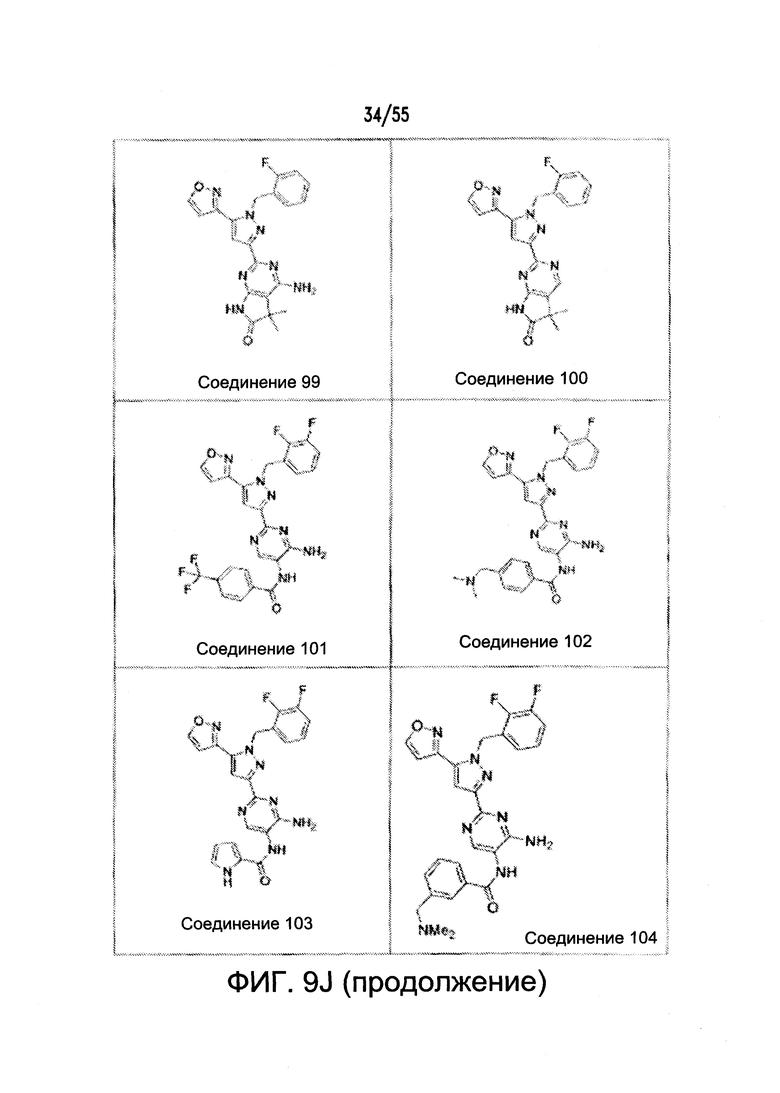
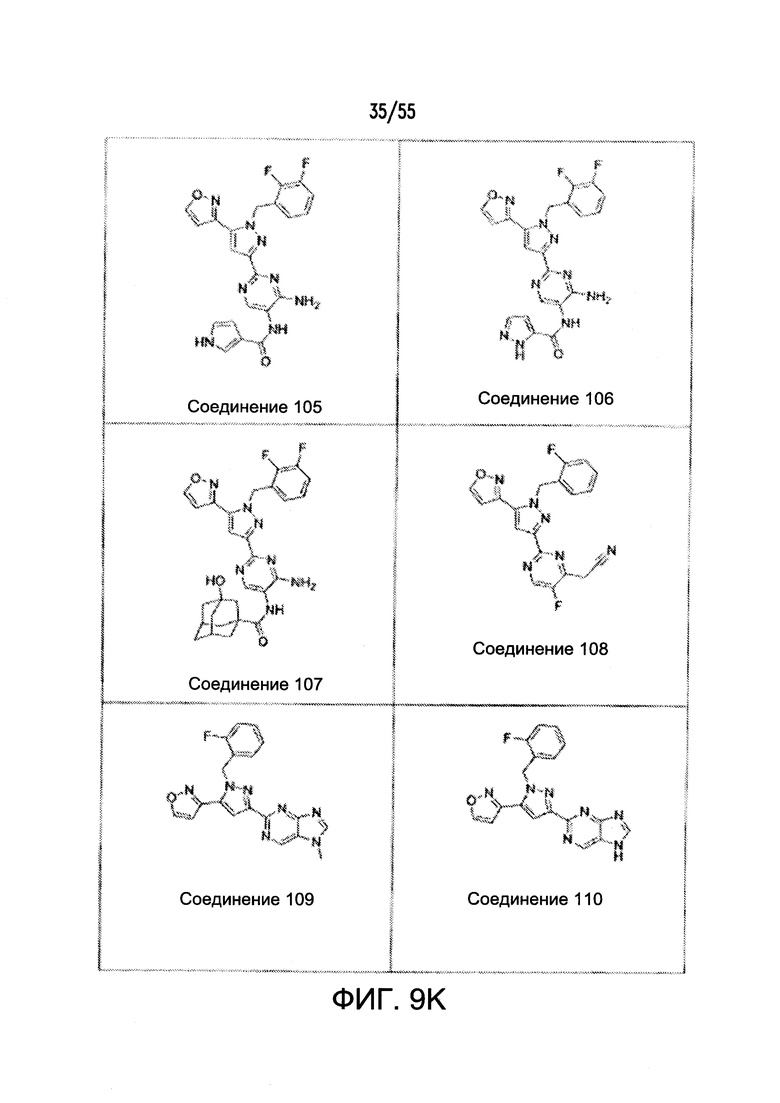
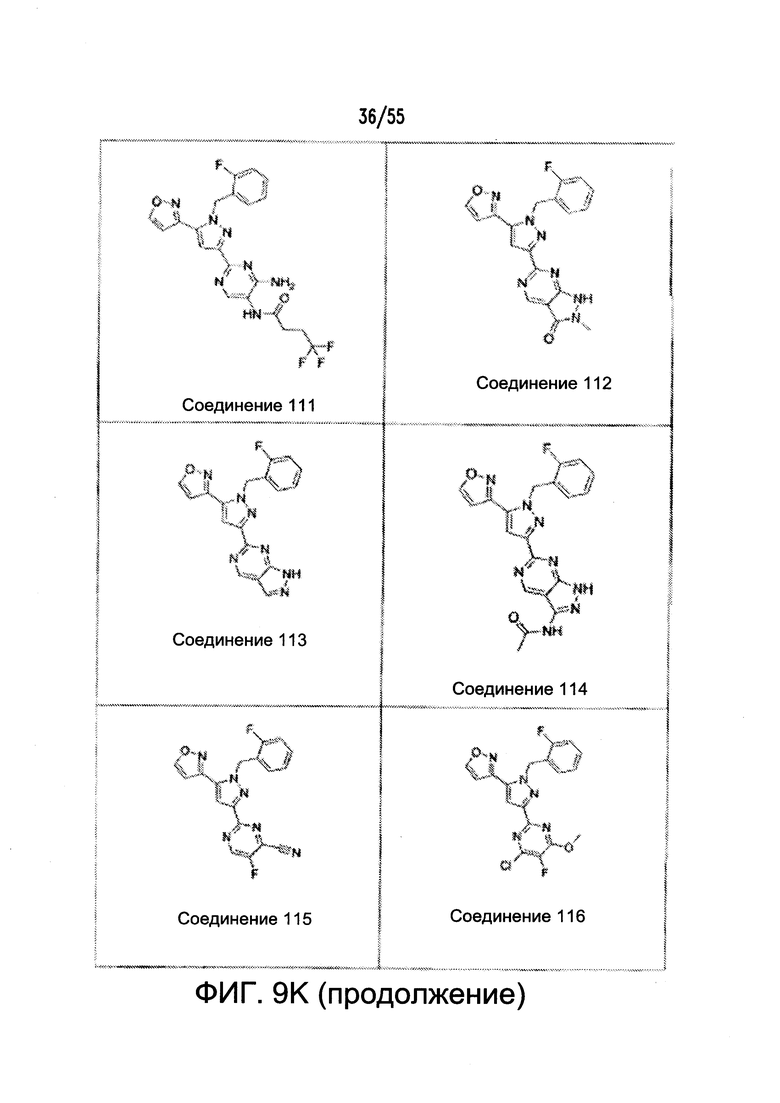
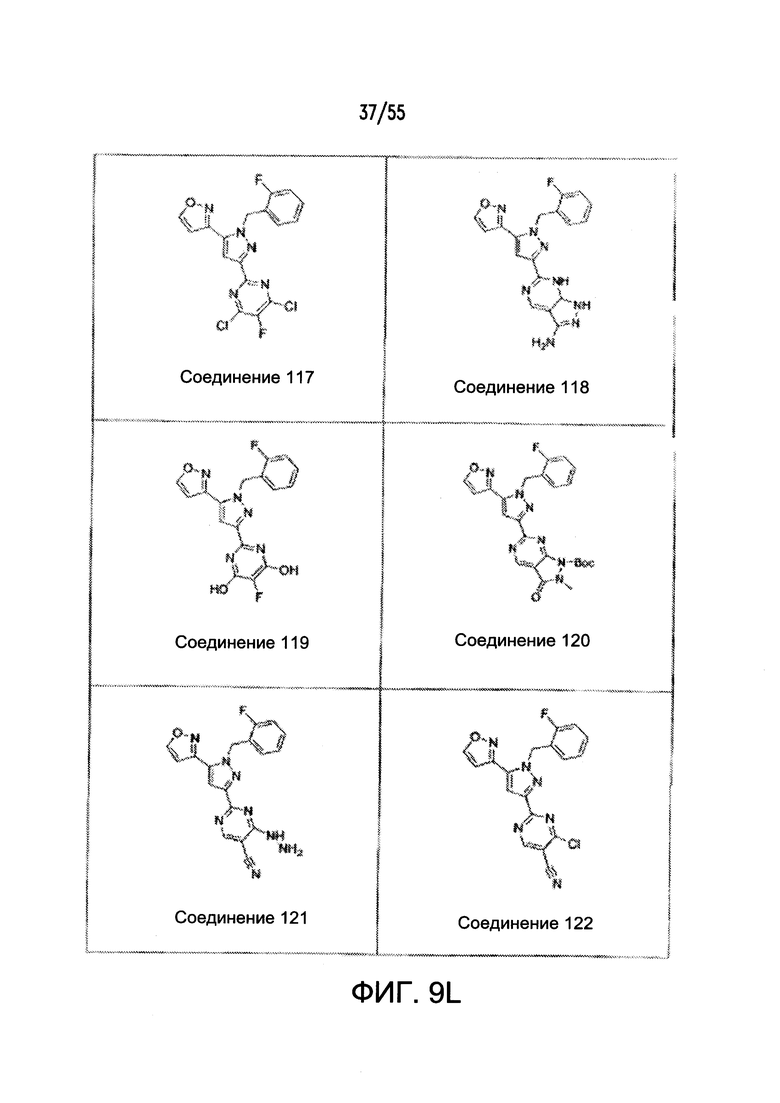
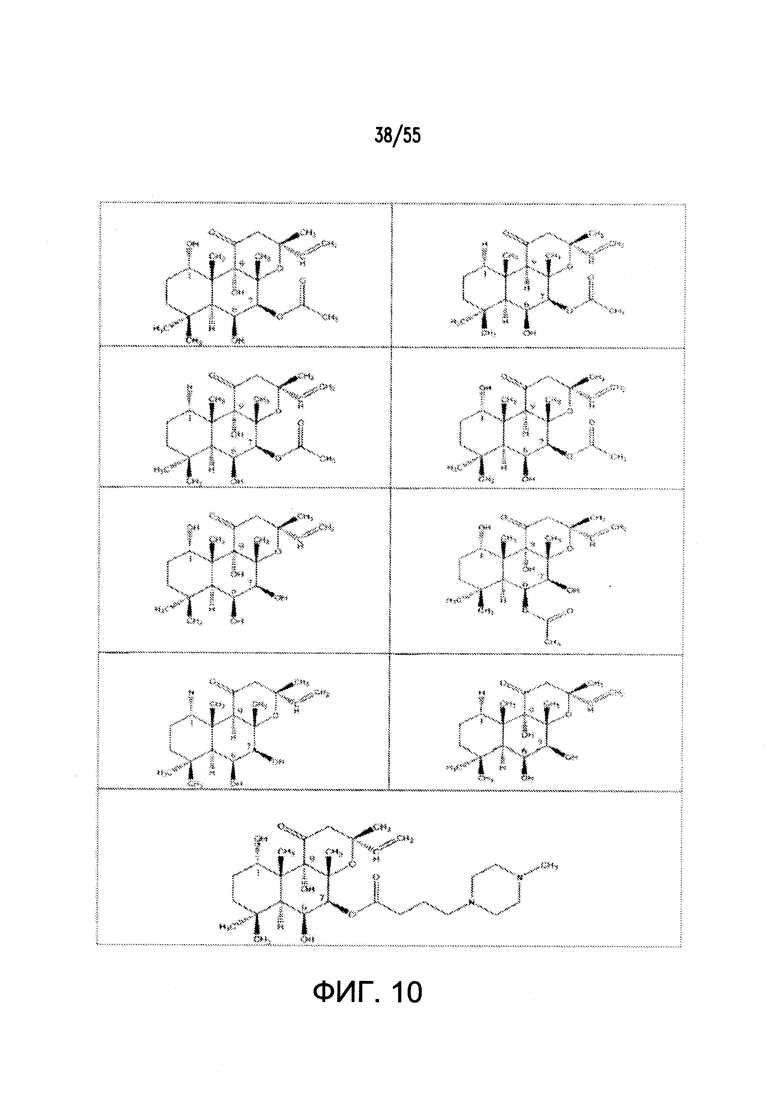
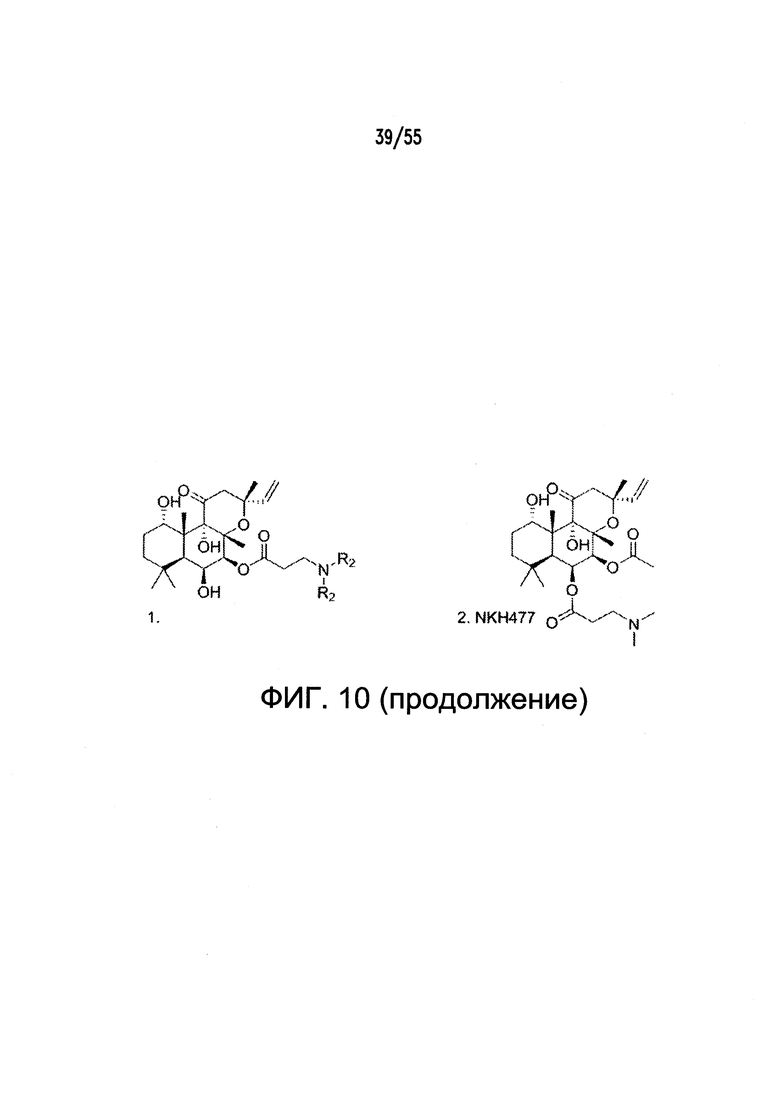
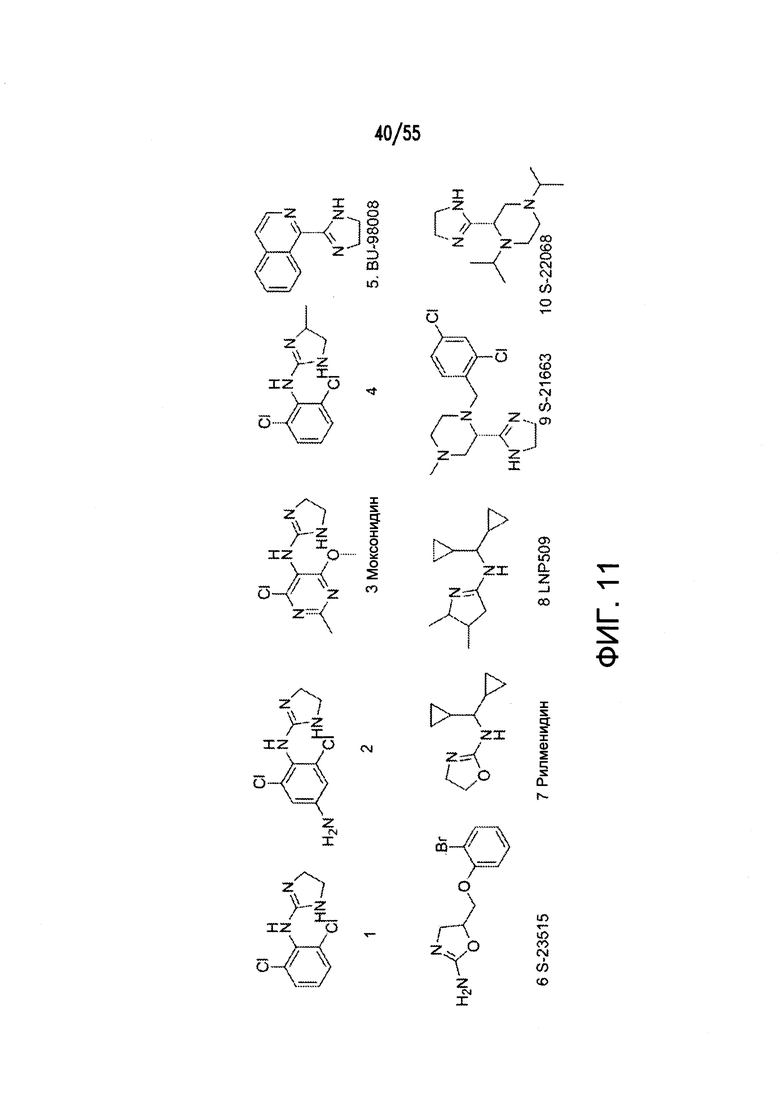
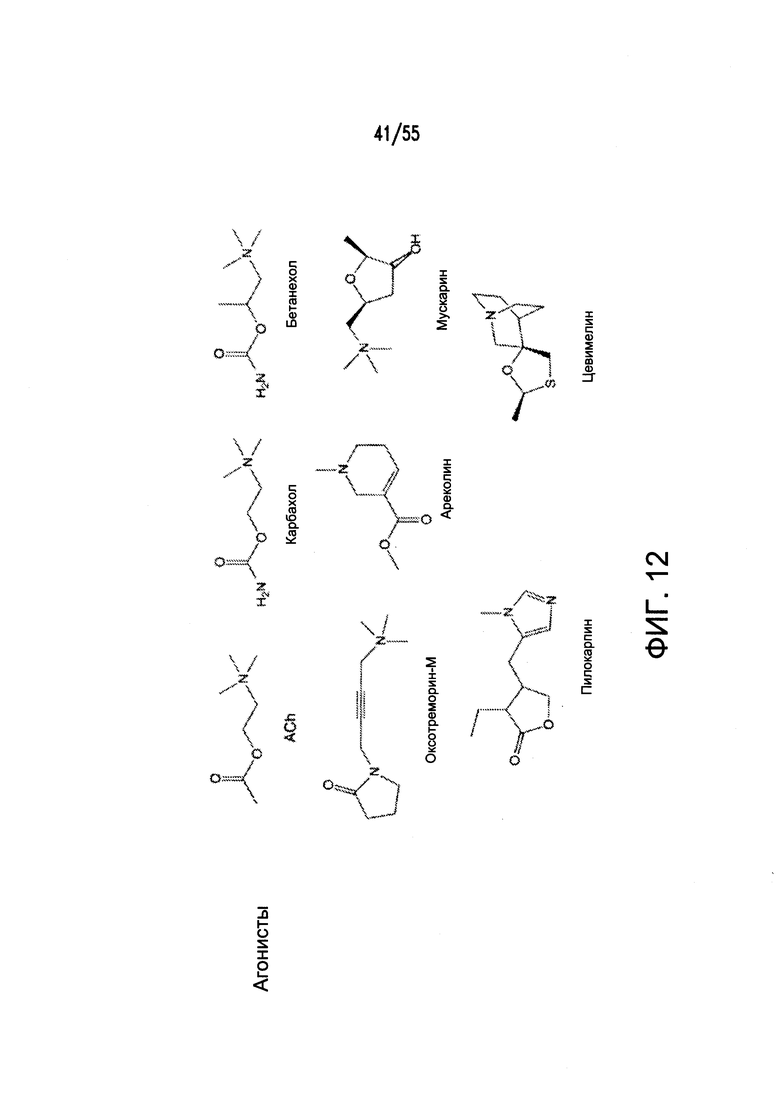
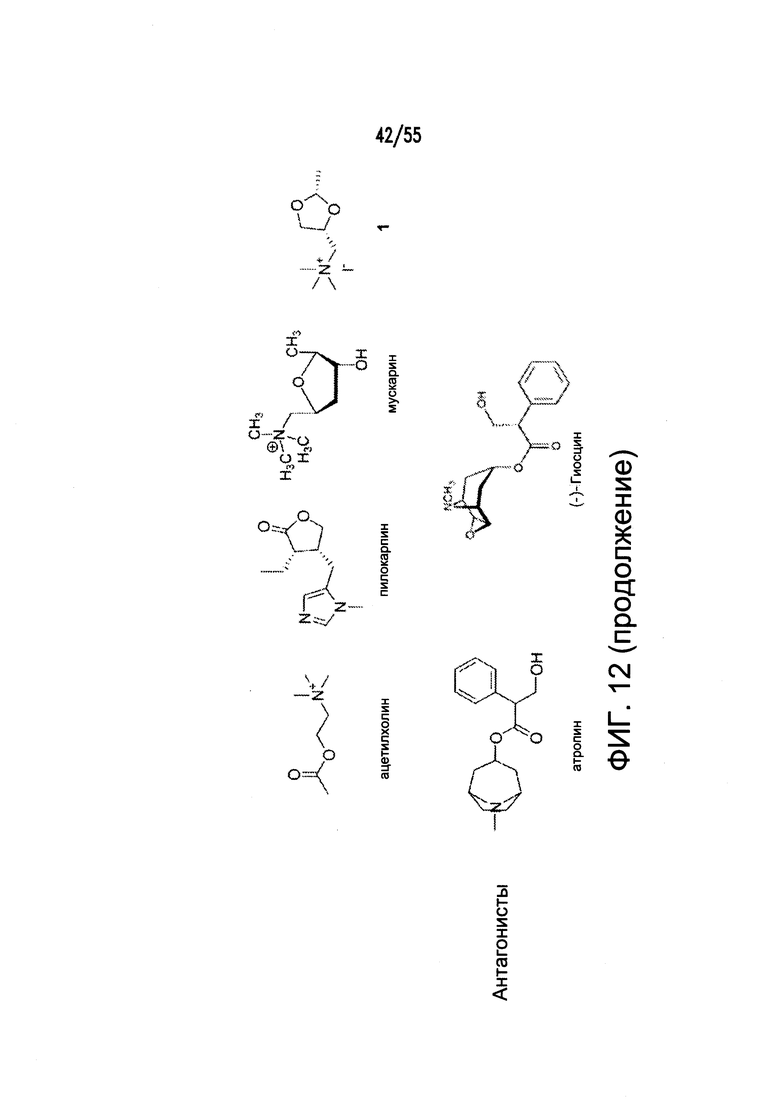
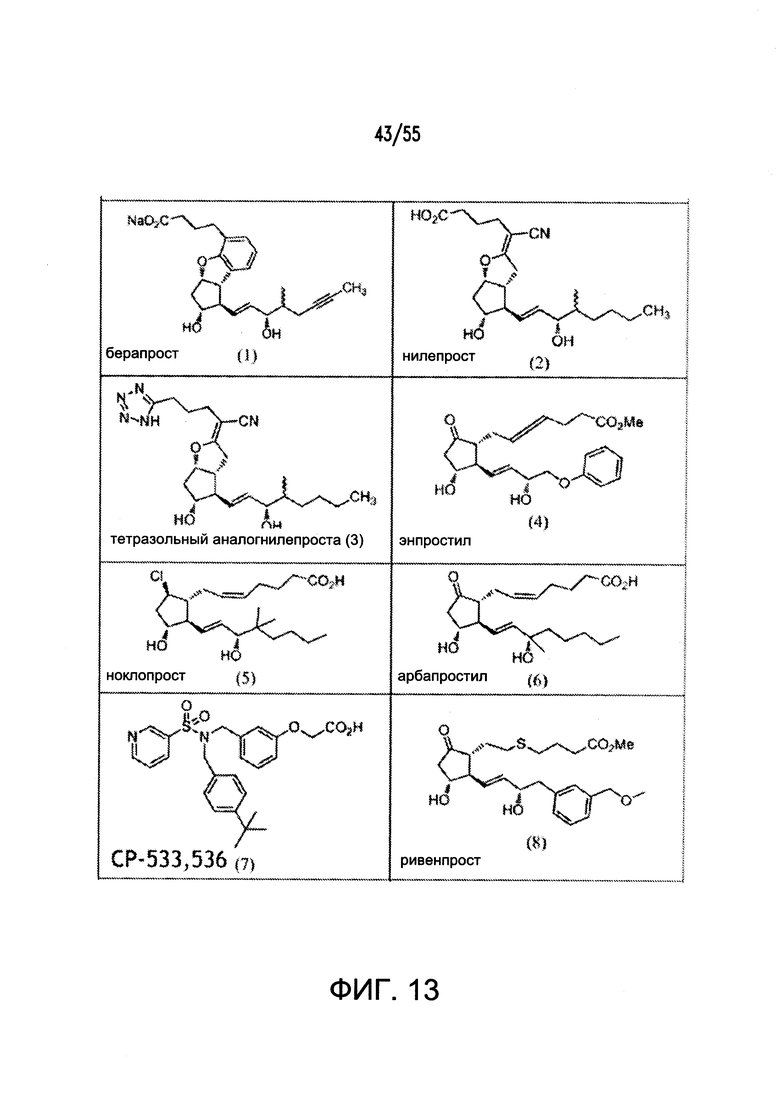
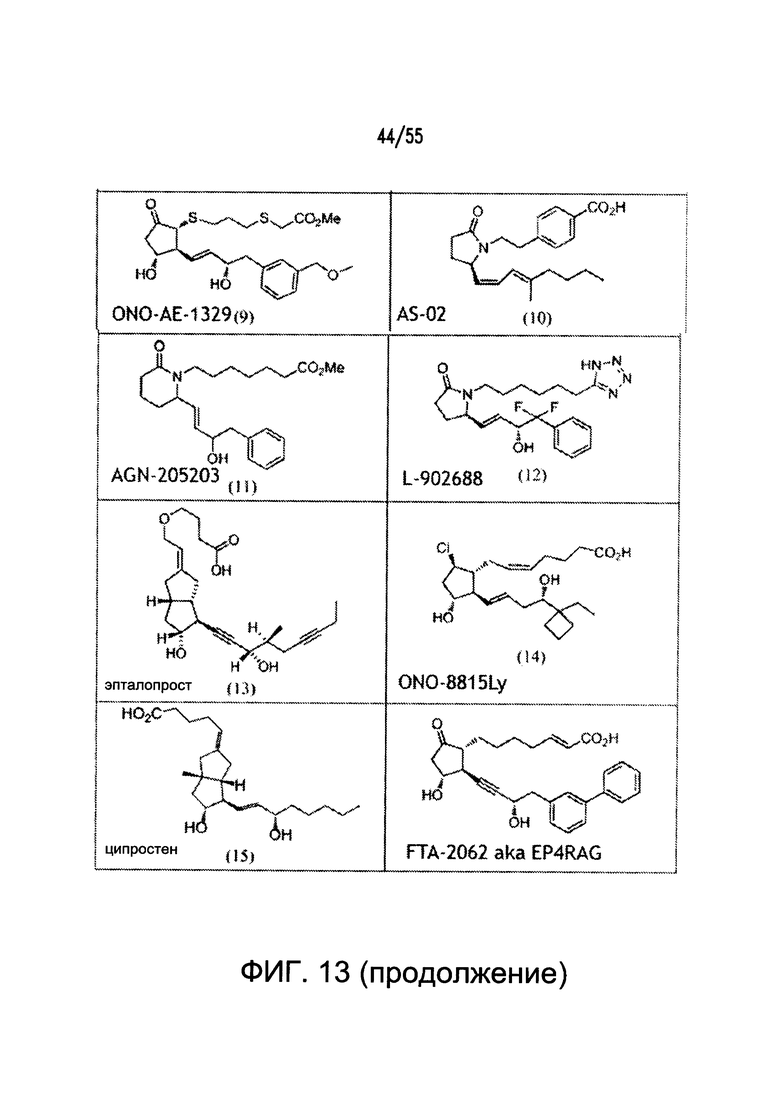
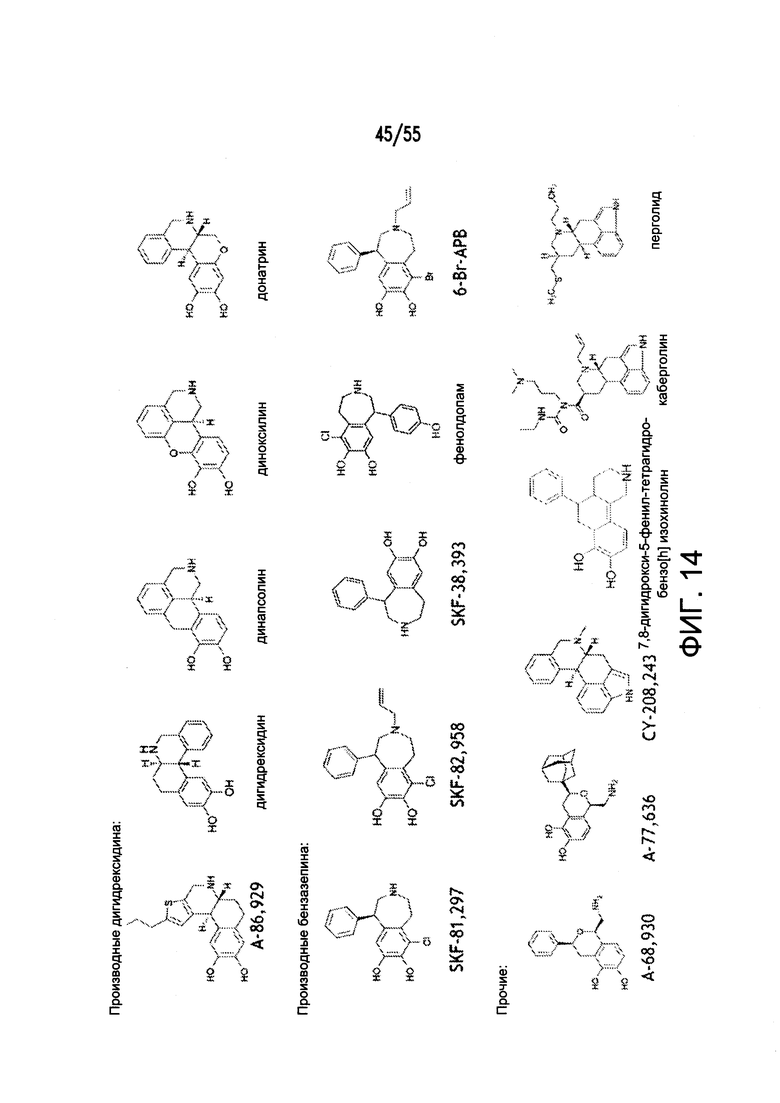
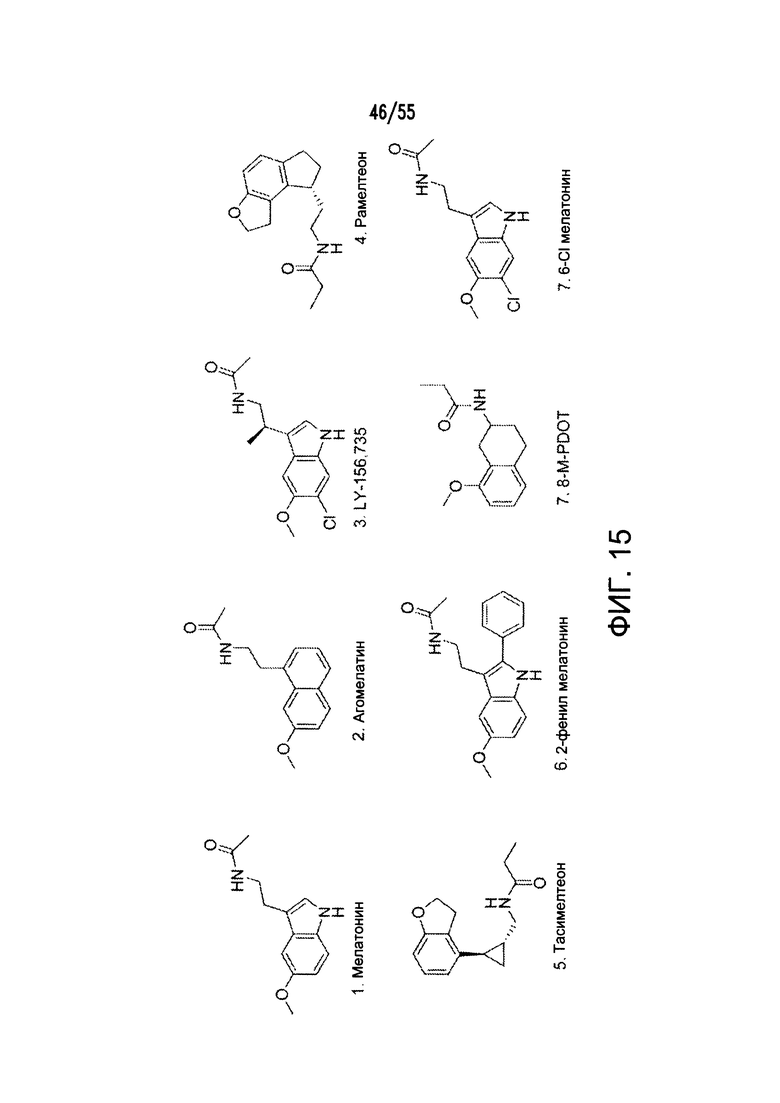
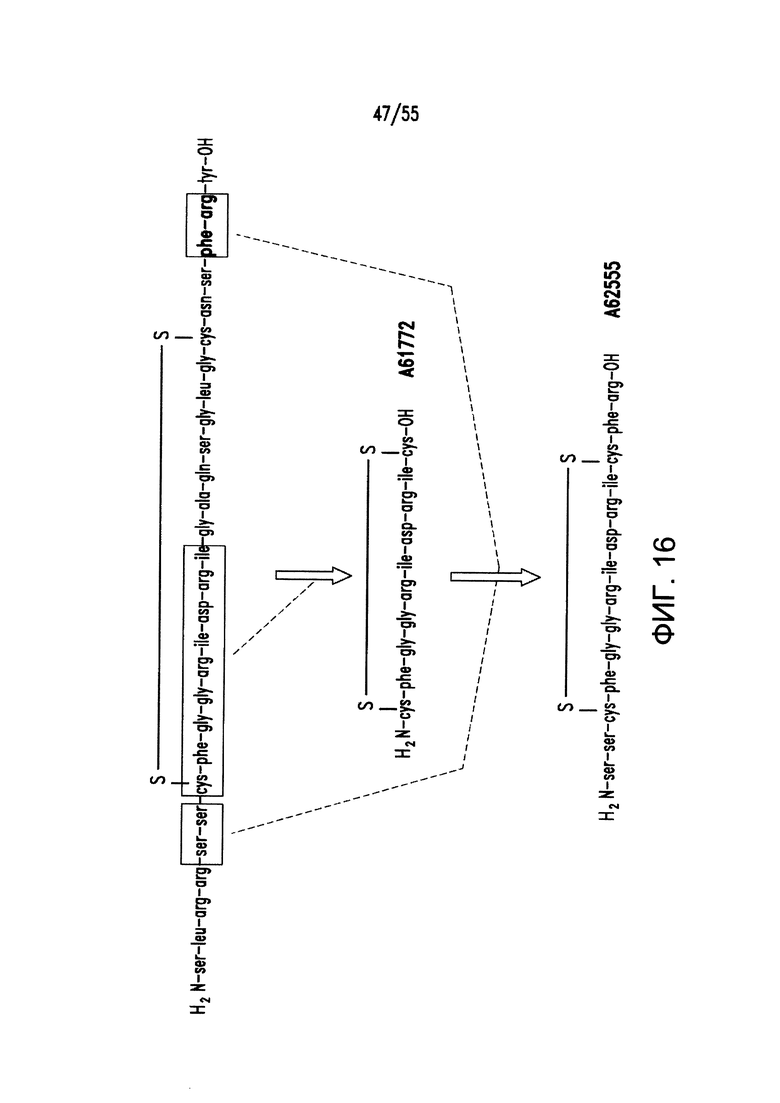
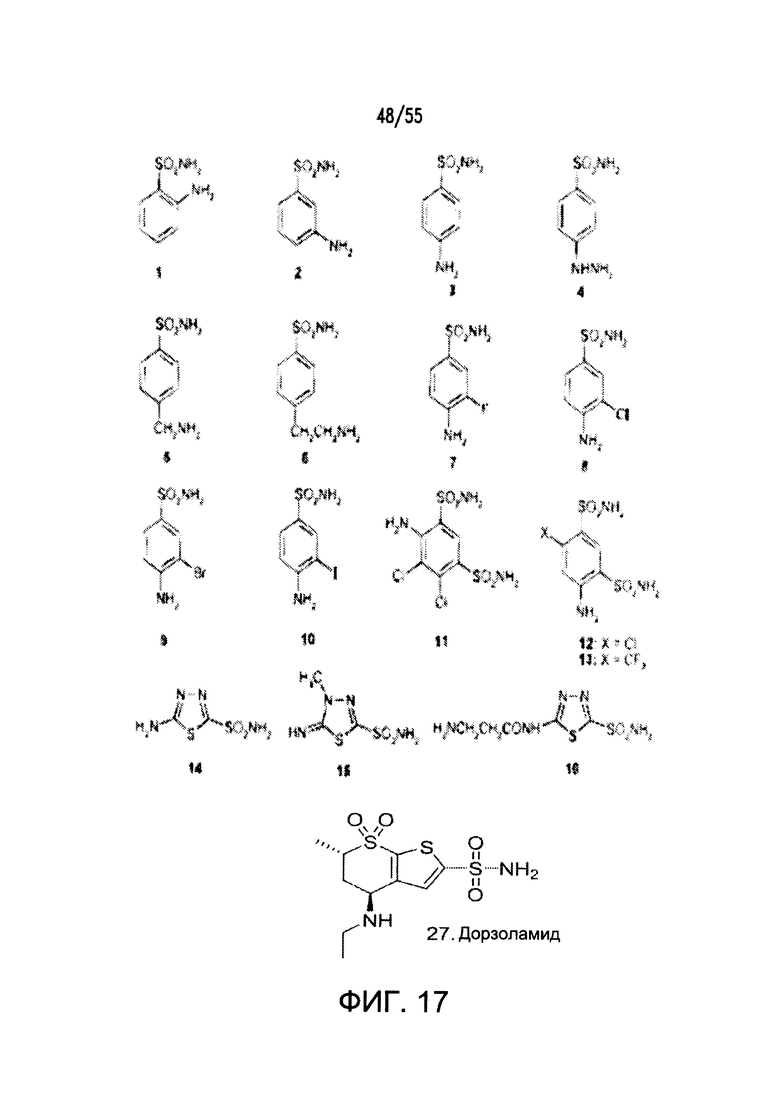
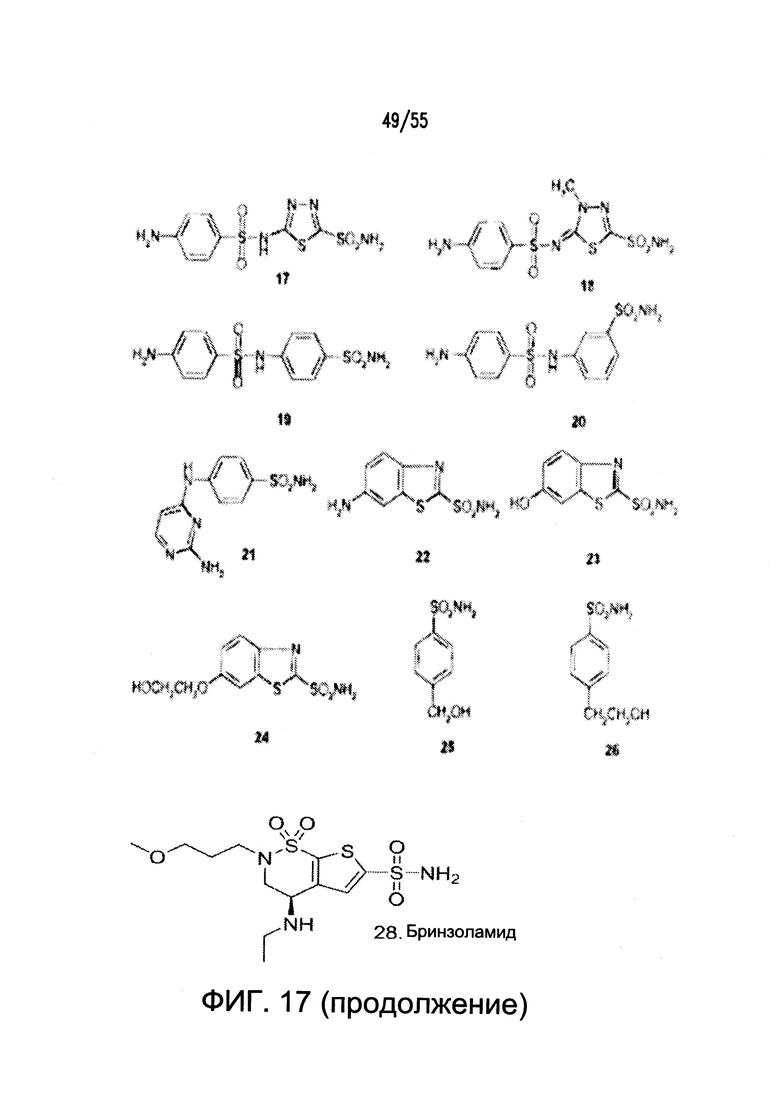
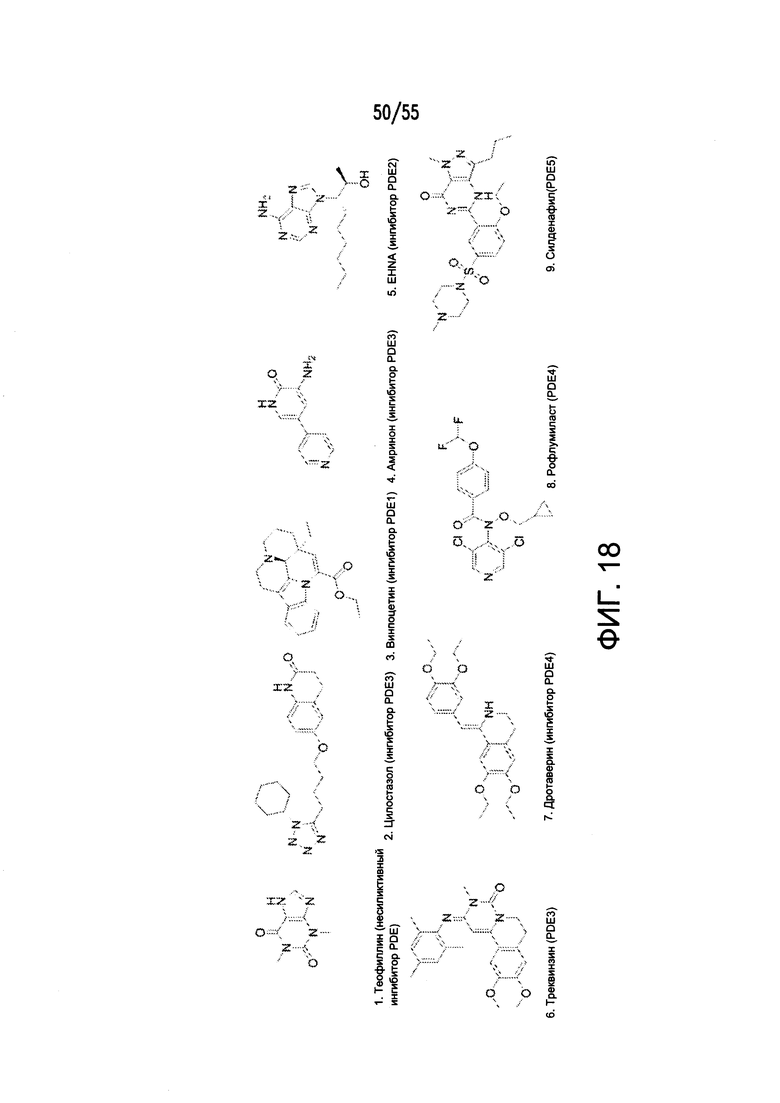
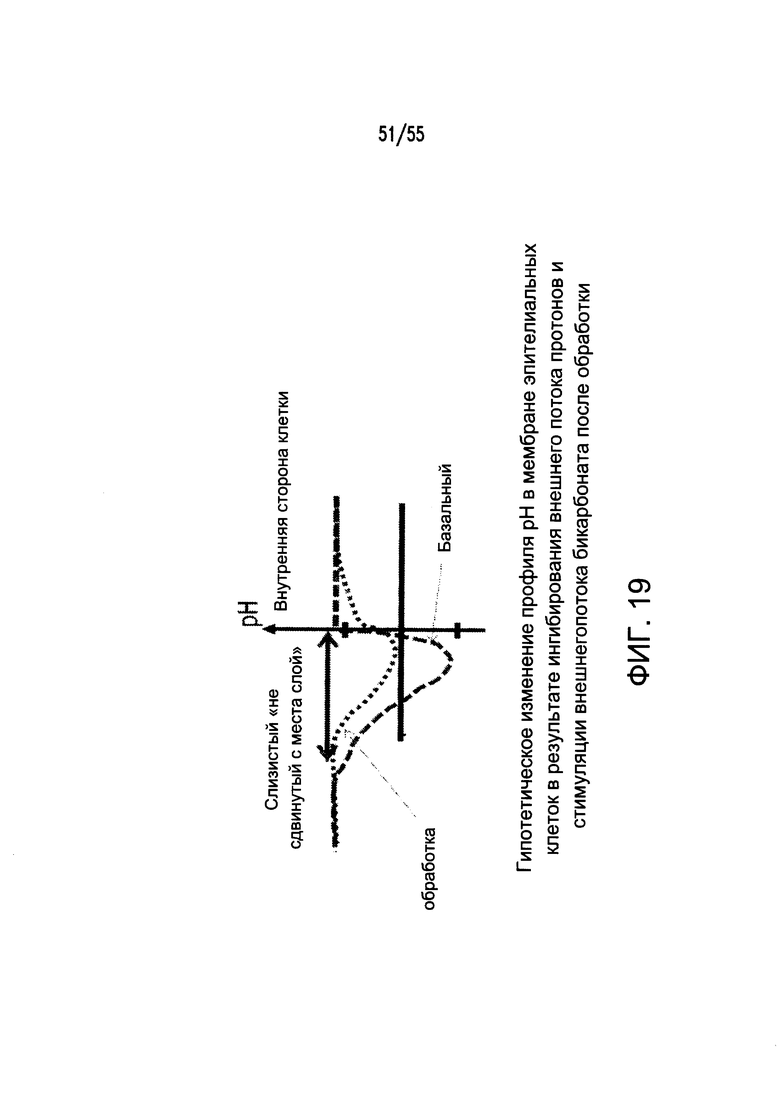
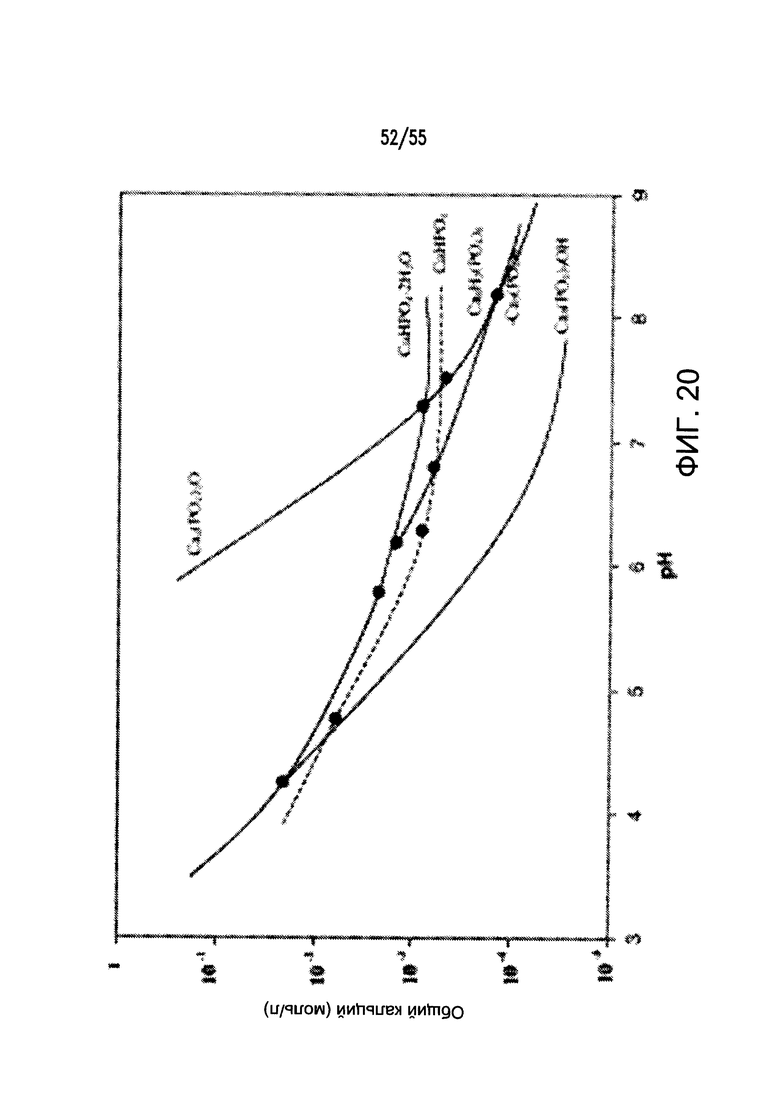
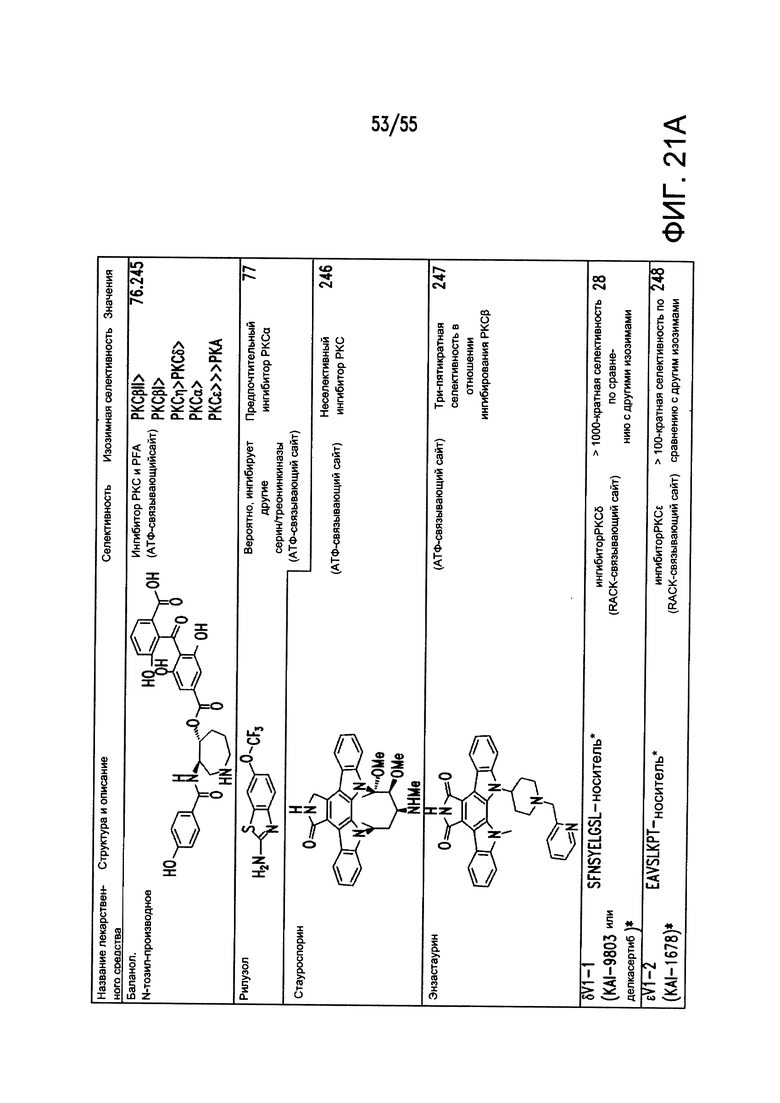

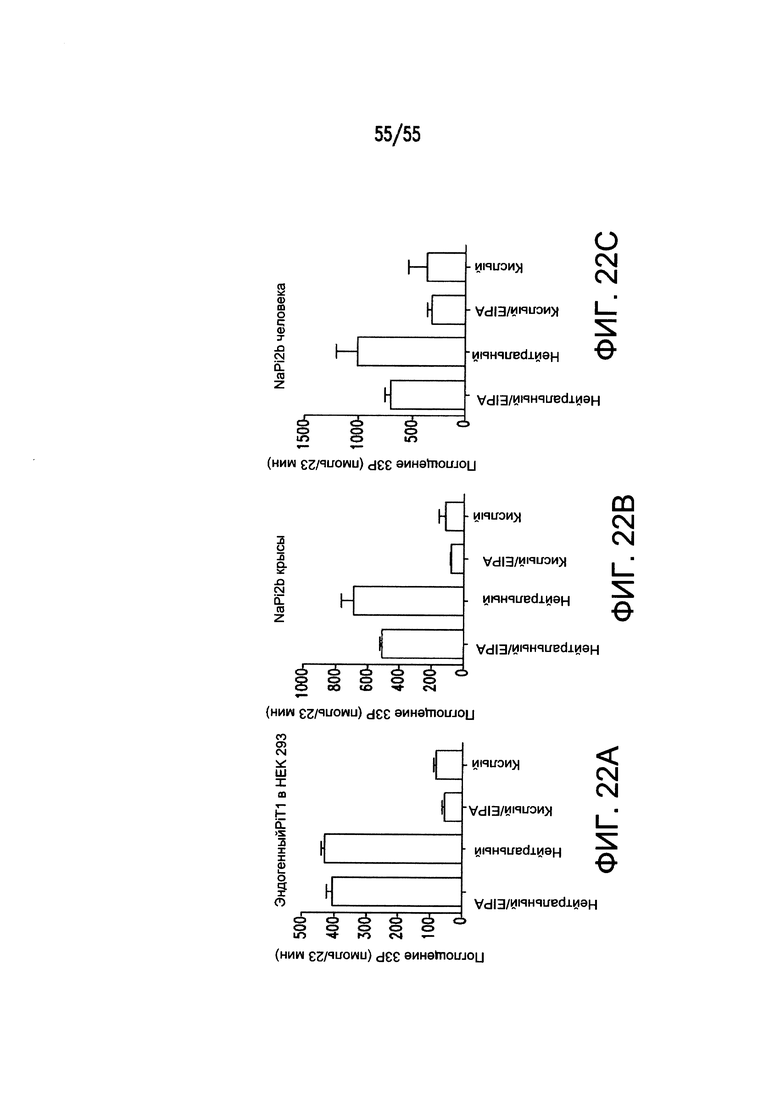
Изобретение относится к ингибиторам транспорта/поглощения фосфатов в желудочно-кишечном тракте. Раскрыто применение агониста рецептора гуанилатциклазы C (GC-C) в лечении гиперфосфатемии у пациента, нуждающегося в этом. Изобретение обеспечивает расширение арсенала средств определенного назначения. 10 з.п. ф-лы, 22 ил., 12 табл., 10 пр.
1. Применение агониста рецептора гуанилатциклазы C (GC-C) в лечении гиперфосфатемии у пациента, нуждающегося в этом.
2. Применение по п. 1, отличающееся тем, что соединение не связывается с NHE3.
3. Применение по п. 1 или 2, отличающееся тем, что гиперфосфатемия связана с заболеванием почек.
4. Применение по любому из предыдущих пунктов, отличающееся тем, что агонист GC-C представляет собой пептид, выбранный из группы, включающей бактериальный термостабильный энтеротоксин, гуанилин, прогуанилин, урогуанилин, проурогуанилин, лимфогуанилин или вариант или аналог любого из перечисленных.
5. Применение по п. 4, отличающееся тем, что пептидный агонист GC-C содержит аминокислотную последовательность (I): Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 Cys6 Cys7 Xaa8 Xaa9 Cys10 Cys11 Xaa12 Xaa13 Xaa14 Cys15 Xaa16 Xaa17 Cys18 Xaa19 Xaa20 Xaa21 (SEQ ID NO:1), где: Xaa1 Xaa2 Xaa3 Xaa4 Xaa5 представляет собой Asn Ser Ser Asn Tyr (SEQ ID NO:2) или отсутствует, или Xaa1 Xaa2 Xaa3 Xaa4 отсутствует; или аминокислотную последовательность (III): Xaa1 Xaa2 Xaa3 Cys4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 Xaa11 Cys12 Xaa13 Xaa14 Xaa15 Xaa16 (SEQ ID NO:5), где Xaa1 представляет собой Ser, Asn, Tyr, Ala, Gln, Pro, Lys, Gly или Thr или отсутствует; Xaa2 представляет собой His, Asp, Glu, Ala, Ser, Asn, Gly или отсутствует; Xaa3 представляет собой Thr, Asp, Ser, Glu, Pro, Val или Leu; Xaa4 представляет собой Asn или отсутствует; Xaa5 выбирают из группы, включающей Asn, Trp, Tyr, Asp, Phe, Thr и Ile; Xaa6 представляет собой Ile, Trp или Leu; Xaa7 представляет собой Cys, Ser или Tyr; Xaa8 выбирают из группы, включающей Glu, Asp, Gln, Gly и Pro; Xaa9 представляет собой Leu, Ile, Val, Ala, Lys, Arg, Trp, Tyr и Phe; Xaa10 представляет собой Ala, Val, Met, Thr или Ile; Xaa11 представляет собой Ala или Val; Xaa12 представляет собой Cys; Xaa13 представляет собой Thr или Ala; Xaa14 выбирают из группы, включающей Ala, Leu, Ser, Gly, Val, Glu, Gln, Ile, Leu, Lys, Arg и Asp; Xaa15 представляет собой Cys, Tyr или отсутствует; и Xaa16 выбирают из группы, включающей Thr, Ala, Asn, Lys, Arg и Trp; Xaa17 выбирают из группы, включающей Gly, Pro и Ala; Xaa18 представляет собой Cys; Xaa19 выбирают из группы, включающей Trp, Tyr, Phe, Asn, Leu, Lys и Arg; Xaa20 Xaa21 представляет собой Asp, Phe, или Xaa20 представляет собой Asn или Glu и Xaa21 отсутствует; или Xaa19 Xaa20 Xaa21 отсутствует.
6. Соединение для применения по п. 5, отличающееся тем, что аминокислотная последовательность представляет собой
Asn Ser Ser Asn Tyr Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr (SEQ ID NO:3) или ее вариант, содержащий 1, 2, 3, 4 или 5 делеций, инсерций и/или замен;
Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr (SEQ ID NO:4) или ее вариант, содержащий 1, 2, 3, 4 или 5 делеций, инсерций и/или замен; или
Asn Asp Glu Cys Glu Leu Cys Val Asn Val Ala Cys Thr Gly Cys Leu (SEQ ID NO:6) или ее вариант, содержащий 1, 2, 3, 4 или 5 делеций, инсерций и/или замен.
7. Применение по п. 1, отличающееся тем, что соединение представляет собой линаклотидин.
8. Применение по п. 1, отличающееся тем, что соединение представляет собой плеканатид.
9. Применение по любому из предыдущих пунктов, где агонист используется в комбинации с одним или более дополнительных биологически активных агентов.
10. Применение по п. 9, отличающееся тем, что соединение и один или более дополнительных биологически активных агентов вводят в форме одной фармацевтической композиции или в форме отдельных фармацевтических композиций, которые вводят последовательно или одновременно.
11. Применение по п. 9 или 10, отличающееся тем, что дополнительный биологически активный агент представляет собой витамин D2 (эргокальциферол), витамин D3 (холекальциферол), активный витамин D (кальцитриол), активный аналог витамина D (доксеркальциферол, парикальцитол), фосфатсвязывающий агент, ингибитор NaPi2b, ниацин, никотинамид, один или несколько ингибиторов АПФ, блокаторы рецепторов антиогензина II, бета-блокаторы, блокаторы кальциевых каналов, прямые ингибиторы ренина, диуретики, вазодилататоры, терапия эритропоэтином, заместительная терапия железом, ингибиторы конечных продуктов гликирования, витамин D или статины.
| WO2012006475 A1, 12.01.2012 | |||
| WO2012006477 A1, 12.01.2012 | |||
| МИЛОВАНОВ Ю.С | |||
| и др., Выбор фосфат-связывающего препарата для лечения гиперфосфатемии при хронической болезни почек: эффекты на кальцификацию артерий и смертность, Клиническая Фармакология и Терапия, 2012, N 21, с | |||
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| LANASPA MA et al, Interaction of MAP17 with NHERF3/4 induces | |||

